Job Results:
Ligand
Structure
Job ID
5c75d0793a5c7fa3ef7fd4eba45db829
Job name
NA
Time
2026-02-27 11:58:42
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Adrenergic receptor alpha-2A (ADRA2A) | 7EJ8 | 5.02 | |
Target general information Gen name ADRA2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Alpha-2AAR; Alpha-2A adrenoreceptor; Alpha-2A adrenoceptor; Alpha-2A adrenergic receptor; Alpha-2 adrenergic receptor subtype C10; ADRAR; ADRA2R Protein family G-protein coupled receptor 1 family, Adrenergic receptor subfamily, ADRA2A sub-subfamily Biochemical class GPCR rhodopsin Function The rank order of potency for agonists of this receptor is oxymetazoline > clonidine > epinephrine > norepinephrine > phenylephrine > dopamine > p-synephrine > p-tyramine > serotonin = p-octopamine. For antagonists, the rank order is yohimbine > phentolamine = mianserine > chlorpromazine = spiperone = prazosin > propanolol > alprenolol = pindolol. Alpha-2 adrenergic receptors mediate the catecholamine-induced inhibition of adenylate cyclase through the action of G proteins. Related diseases Lipodystrophy, familial partial, 8 (FPLD8) [MIM:620679]: An autosomal dominant form of partial lipodystrophy, a disorder characterized by abnormal subcutaneous fat distribution. FPLD8 patients show selective loss of subcutaneous adipose tissue from the limbs, beginning around 13 to 15 years of age, and abnormal accumulation of subcutaneous adipose tissue in the dorsal neck and face, as well as in the posterior thoracic and abdominal regions. The disorder is associated with metabolic abnormalities, including diabetes mellitus and hyperlipidemia. {ECO:0000269|PubMed:27376152}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01472; DB00321; DB00543; DB00182; DB00714; DB00964; DB09229; DB01238; DB14185; DB06216; DB00865; DB00217; DB00484; DB01200; DB00248; DB01136; DB04846; DB00477; DB09202; DB00575; DB00363; DB01151; DB00633; DB01576; DB11273; DB13345; DB00320; DB00449; DB11278; DB09167; DB04855; DB06262; DB01363; DB05492; DB00751; DB00668; DB01049; DB00696; DB01175; DB06678; DB09194; DB00800; DB06623; DB00629; DB01018; DB00502; DB11577; DB00555; DB06707; DB00589; DB04948; DB09195; DB00408; DB08815; DB00934; DB01365; DB01577; DB01403; DB00968; DB06148; DB00370; DB09205; DB09242; DB06711; DB01149; DB00368; DB00540; DB06229; DB00935; DB01267; DB00715; DB01186; DB01608; DB00925; DB00692; DB00397; DB09286; DB09244; DB06153; DB00413; DB00457; DB00433; DB01069; DB00852; DB01224; DB11124; DB11738; DB00268; DB09304; DB06764; DB13025; DB00697; DB00797; DB00193; DB00656; DB00726; DB11477; DB06694; DB01392; DB00246; DB01624 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Direct protein sequencing; Disulfide bond; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Methylation; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 30303.9 Length 263 Aromaticity 0.16 Instability index 35.08 Isoelectric point 9.66 Charge (pH=7) 16.82 2D Binding mode Binding energy (Kcal/mol) -6.85 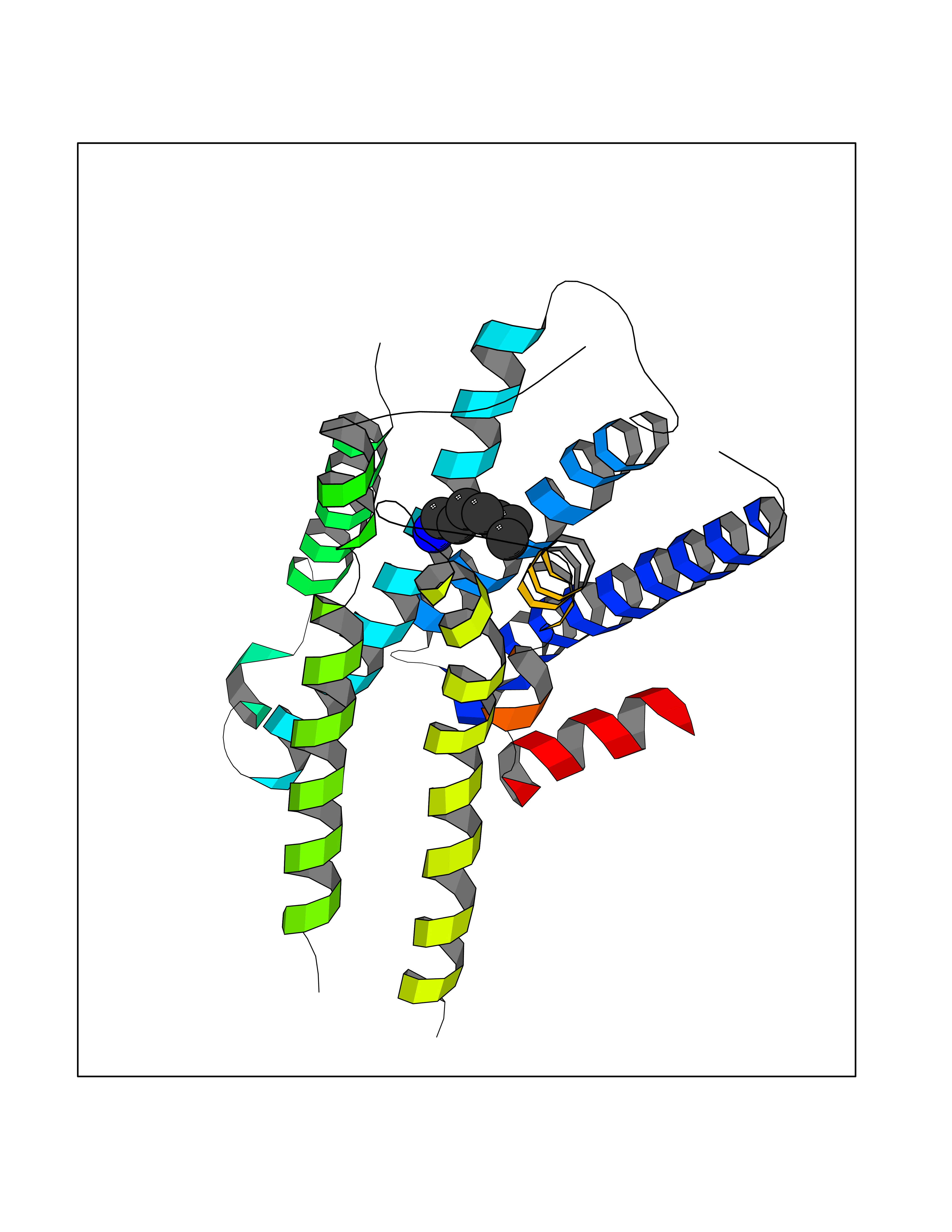 Molscript Map 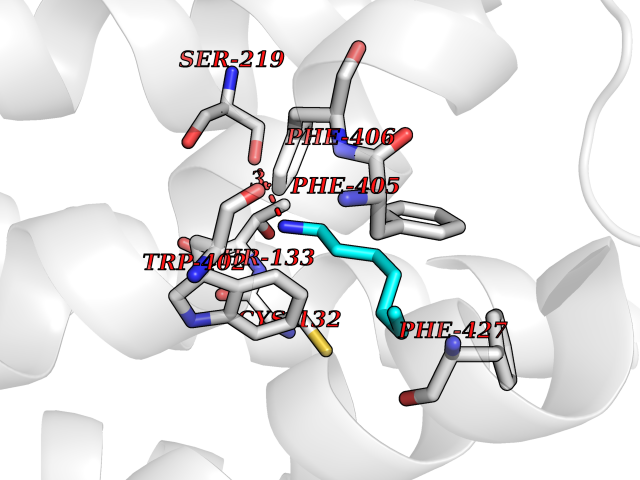 Pymol Map 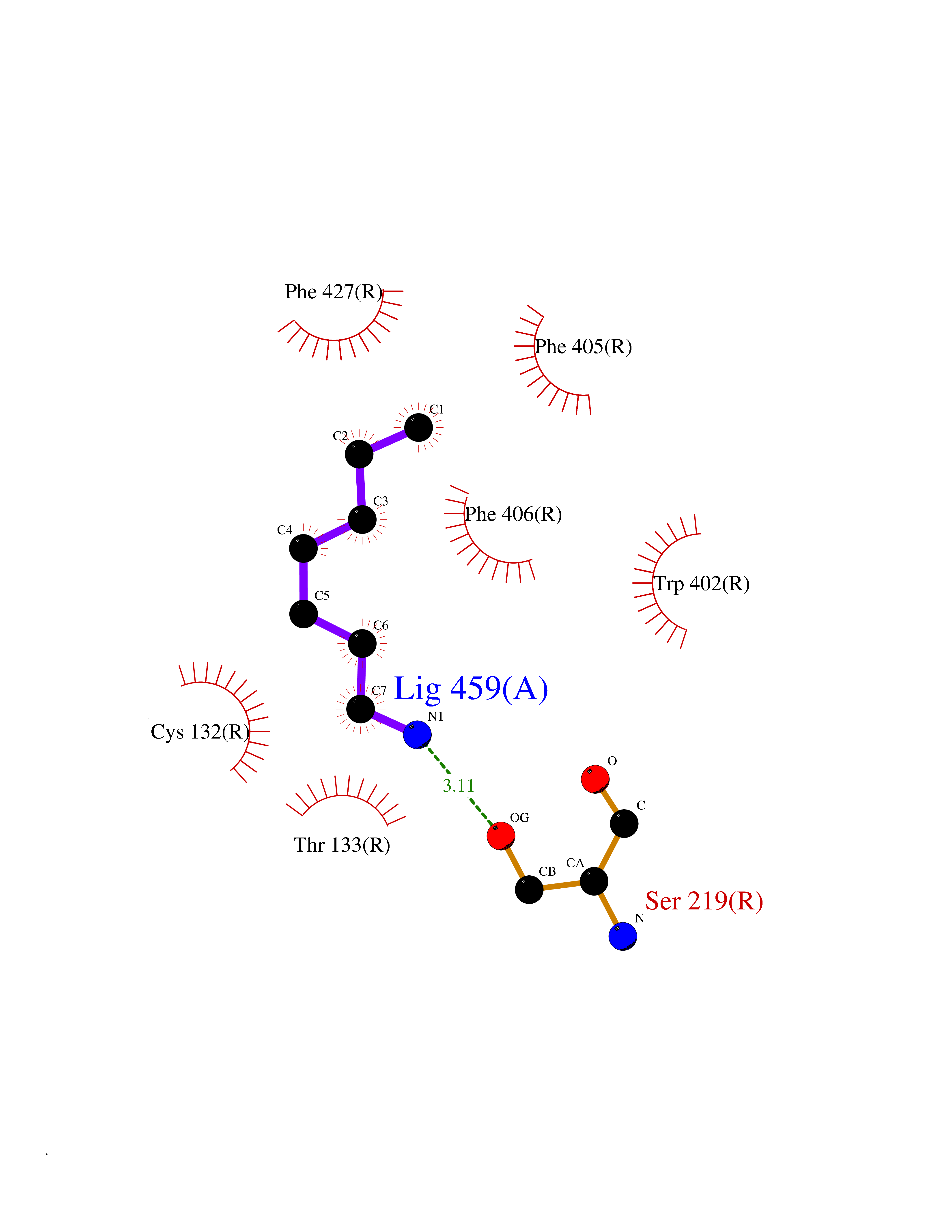 Ligplot Map 3D Binding mode Sequence YSLQVTLTLVCLAGLLMLLTVFGNVLVIIAVFTSRALKAPQNLFLVSLASADILVATLVIPFSLANEVMGYWYFGKAWCEIYLALDVLFCTSSIVHLCAISLDRYWSITQAIEYNLKRTPRRIKAIIITVWVISAVISFPPRCEINDQKWYVISSCIGSFFAPCLIMILVYVRIYQIAKRRTRRGRQNREKRFTFVLAVVIGVFVVCWFPFFFTYTLTAVGCSVPRTLFKFFFWFGYCNSSLNPVIYTIFNHDFRRAFKKILC Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Diacylglycerol acyltransferase 1 (DGAT1) | 6VP0 | 5.02 | |
Target general information Gen name DGAT1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Retinol O-fatty-acyltransferase; Diglyceride acyltransferase; Diacylglycerol O-acyltransferase 1; DGAT; Acyl-CoA retinol O-fatty-acyltransferase; ARAT; AGRP1; ACAT-related gene product 1 Protein family Membrane-bound acyltransferase family, Sterol o-acyltransferase subfamily Biochemical class Acyltransferase Function Catalyzes the terminal and only committed step in triacylglycerol synthesis by using diacylglycerol and fatty acyl CoA as substrates. In contrast to DGAT2 it is not essential for survival. May be involved in VLDL (very low density lipoprotein) assembly. In liver, plays a role in esterifying exogenous fatty acids to glycerol. Functions as the major acyl-CoA retinol acyltransferase (ARAT) in the skin, where it acts to maintain retinoid homeostasis and prevent retinoid toxicity leading to skin and hair disorders. Related diseases Diarrhea 7, protein-losing enteropathy type (DIAR7) [MIM:615863]: A life-threatening disease characterized by severe, intractable, watery diarrhea. {ECO:0000269|PubMed:23114594, ECO:0000269|PubMed:30237576}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O75907; PRO_0000045602 [Q99IB8] EC number EC 2.3.1.20 Uniprot keywords 3D-structure; Acyltransferase; Disease variant; Endoplasmic reticulum; Lipid metabolism; Membrane; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID C Molecular weight (Da) 47494.5 Length 404 Aromaticity 0.17 Instability index 37.65 Isoelectric point 9.51 Charge (pH=7) 15.57 2D Binding mode Binding energy (Kcal/mol) -6.84 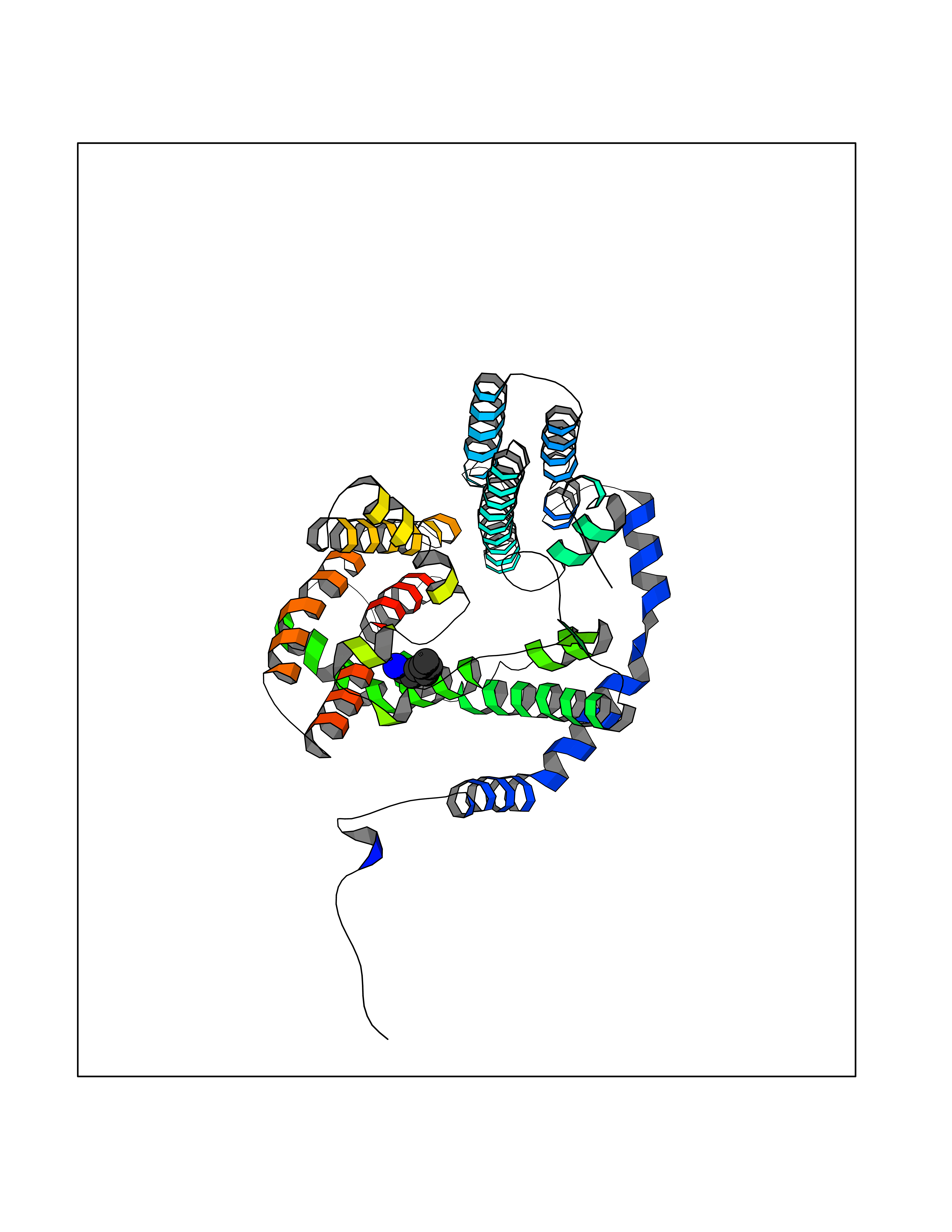 Molscript Map 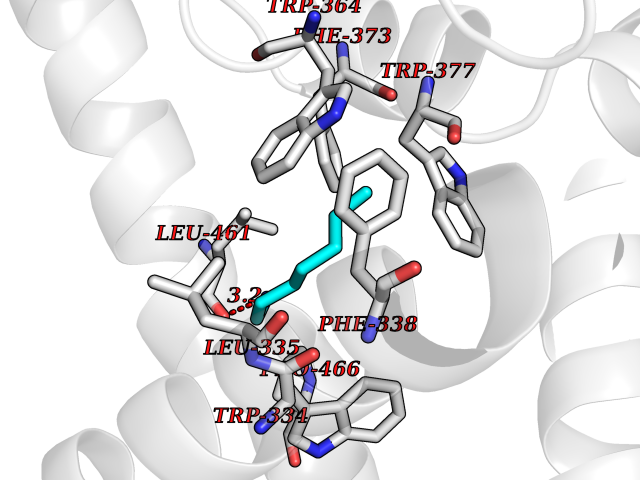 Pymol Map 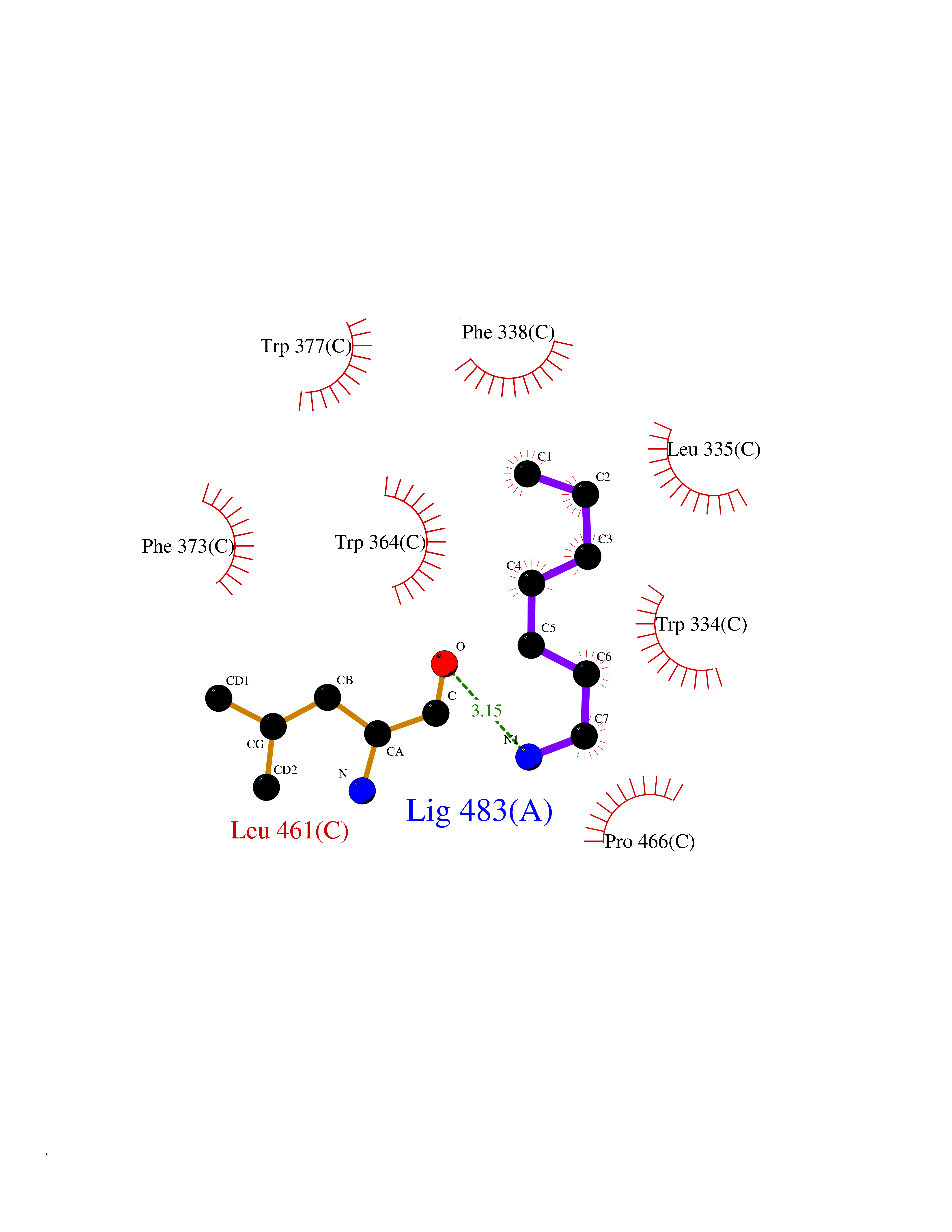 Ligplot Map 3D Binding mode Sequence WELRCHRLQDSLFSSDSGFSNYRGILNWCVVMLILSNARLFLENLIKYGILVDPIQVVSLFLKDPYSWPAPCLVIAANVFAVAAFQVEKRLAVGALTEQAGLLLHVANLATILCFPAAVVLLVESITPVGSLLALMAHTILFLKLFSYRDVNSWCRRARAKHTVSYPDNLTYRDLYYFLFAPTLCYELNFPRSPRIRKRFLLRRILEMLFFTQLQVGLIQQWMVPTIQNSMKPFKDMDYSRIIERLLKLAVPNHLIWLIFFYWLFHSCLNAVAELMQFGDREFYRDWWNSESVTYFWQNWNIPVHKWCIRHFYKPMLRRGSSKWMARTGVFLASAFFHEYLVSVPLRMFRLWAFTGMMAQIPLAWFVGRFFQGNYGNAAVWLSLIIGQPIAVLMYVHDYYVLNY Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Influenza Polymerase acidic endonuclease (Influ PA) | 4ZI0 | 5.02 | |
Target general information Gen name Influ PA Organism Influenza A virus (strain A/Puerto Rico/8/1934 H1N1) Uniprot ID TTD ID Synonyms RNA-directed RNA polymerase subunit P2; Polymerase acidic protein Protein family Influenza viruses PA family Biochemical class NA Function Plays an essential role in viral RNA transcription and replication by forming the heterotrimeric polymerase complex together with PB1 and PB2 subunits. The complex transcribes viral mRNAs by using a unique mechanism called cap-snatching. It consists in the hijacking and cleavage of host capped pre-mRNAs. These short capped RNAs are then used as primers for viral mRNAs. The PB2 subunit is responsible for the binding of the 5' cap of cellular pre-mRNAs which are subsequently cleaved after 10-13 nucleotides by the PA subunit that carries the endonuclease activity. Related diseases Lymphatic malformation 8 (LMPHM8) [MIM:618773]: A form of primary lymphedema, a disease characterized by swelling of body parts due to developmental anomalies and functional defects of the lymphatic system. Adult patients with lymphedema may suffer from recurrent local infections. Impaired lymphatic drainage in the fetus can develop into hydrops fetalis, a severe condition characterized by excessive fluid accumulation in more than two fetal extra-vascular compartments and body cavities, placental enlargement and edema, pericardial or pleural effusion, or ascites. LMPHM8 is an autosomal recessive form characterized by onset in utero and fetal death due to non-immune hydrops fetalis. {ECO:0000269|PubMed:30115739}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB13997 Interacts with P03485; P03466; P03431 EC number EC 3.1.-.- Uniprot keywords 3D-structure; Cap snatching; Endonuclease; Eukaryotic host gene expression shutoff by virus; Eukaryotic host transcription shutoff by virus; Host cytoplasm; Host gene expression shutoff by virus; Host nucleus; Host-virus interaction; Hydrolase; Inhibition of host RNA polymerase II by virus; Manganese; Metal-binding; Nuclease; Phosphoprotein; Reference proteome; Ribosomal frameshifting Protein physicochemical properties Chain ID A Molecular weight (Da) 21288.1 Length 181 Aromaticity 0.11 Instability index 49.23 Isoelectric point 6.27 Charge (pH=7) -1.8 2D Binding mode Binding energy (Kcal/mol) -6.85 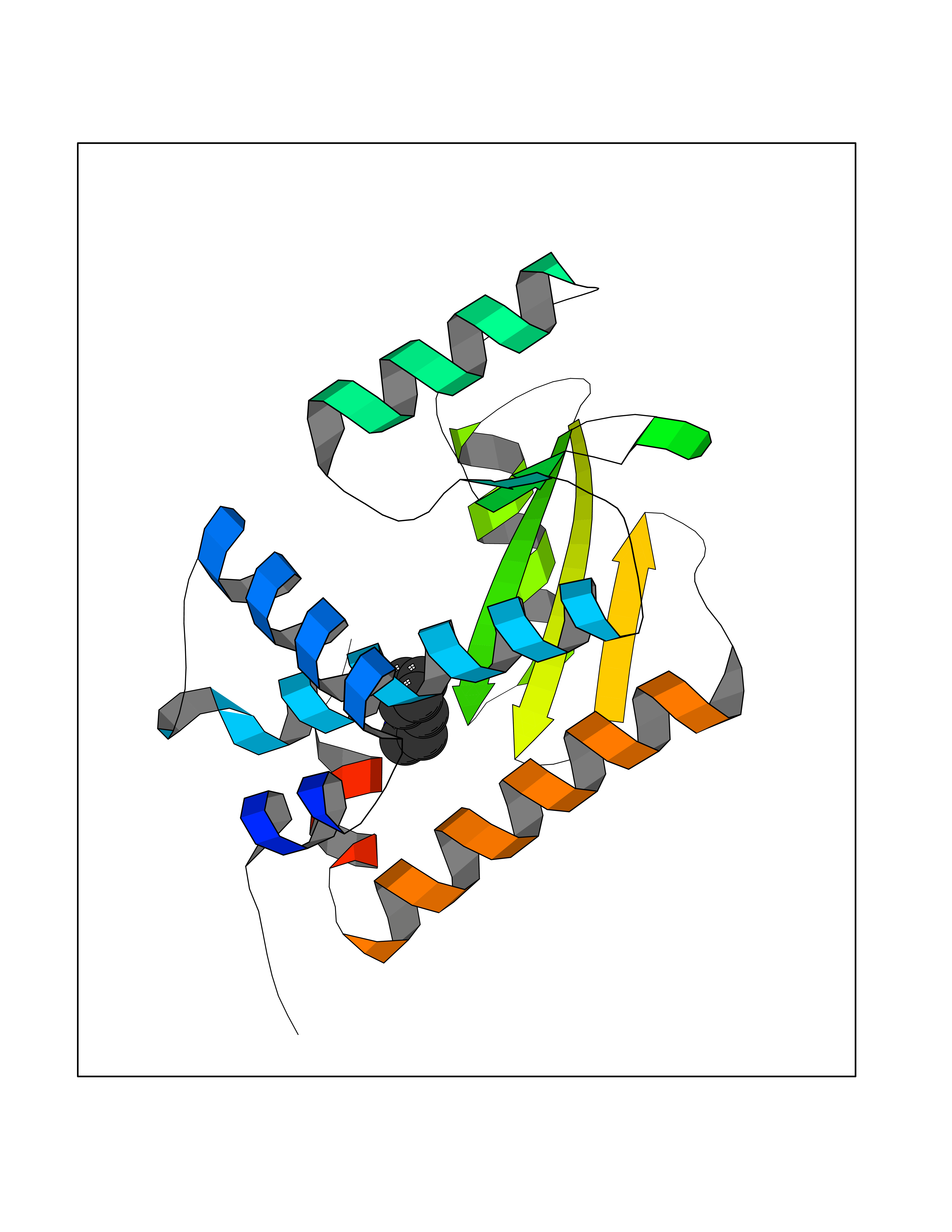 Molscript Map 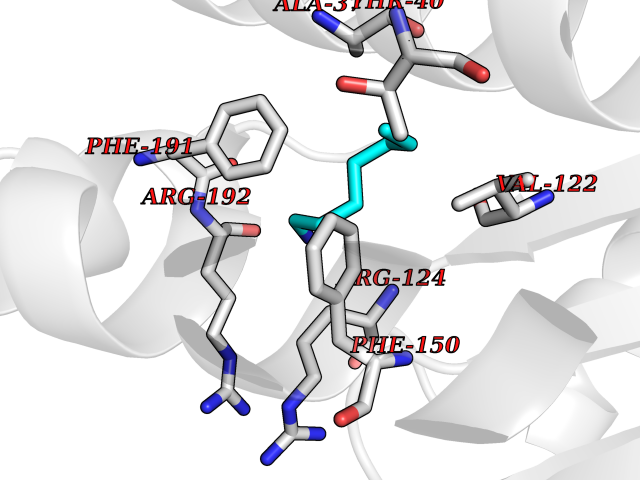 Pymol Map 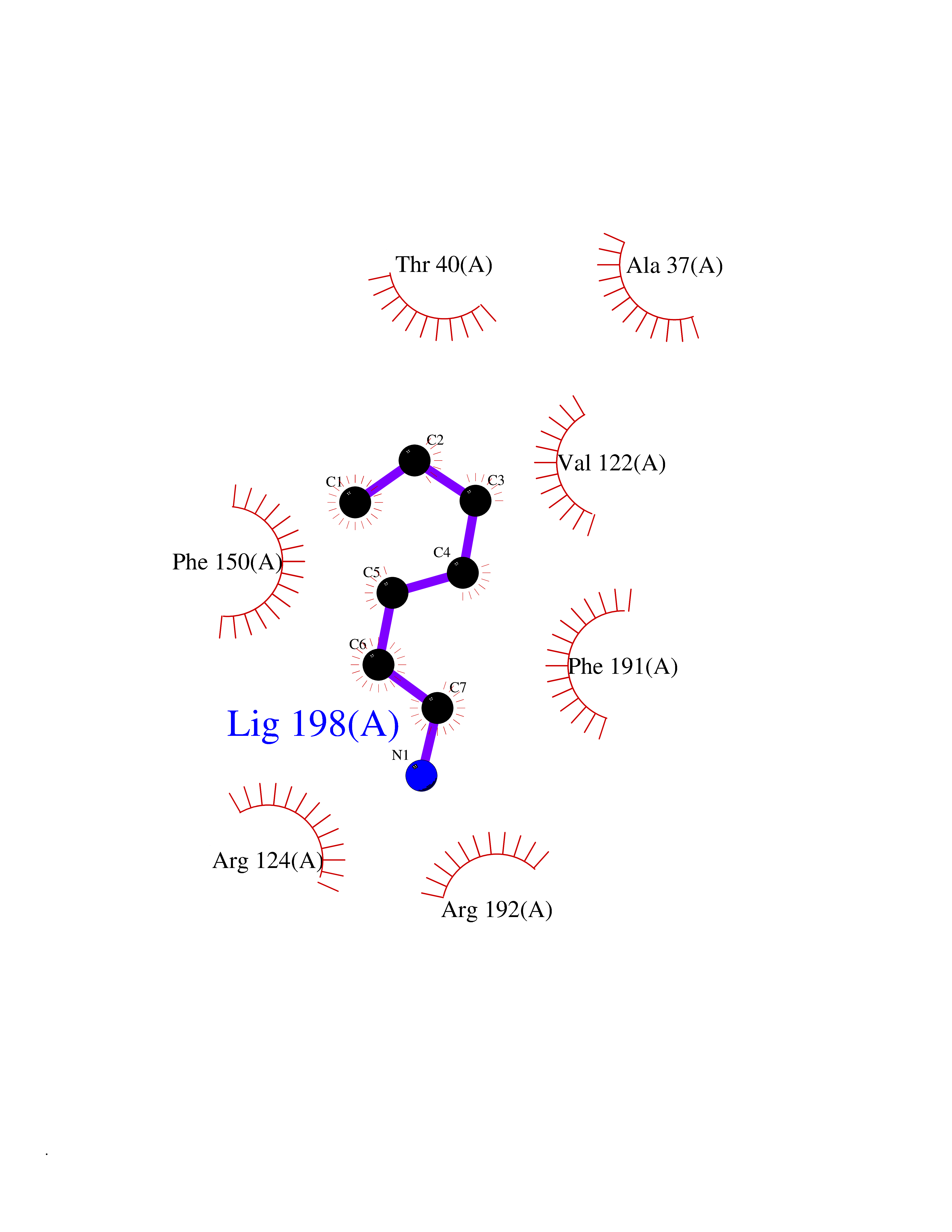 Ligplot Map 3D Binding mode Sequence GPLGSMEDFVRQCFNPMIVELAEKTMKEYGEDLKIETNKFAAICTHLEVCFMYSDASKHRFEIIEGRDRTMAWTVVNSICNTTGAEKPKFLPDLYDYKENRFIEIGVTRREVHIYYLEKANKIKSEKTHIHIFSFTGEEMATKADYTLDEESRARIKTRLFTIRQEMASRGLWDSFRQSER Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Tumor-associated calcium signal transducer 1 (EPCAM) | 4MZV | 5.02 | |
Target general information Gen name EPCAM Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hEGP314; TROP1; TACSTD1; Major gastrointestinal tumor-associated protein GA733-2; MIC18; M4S1; M1S2; KSA; KS 1/4 antigen; Gastrointestinal carcinoma antigen GA733; GA733-2; Epithelial glycoprotein 314 Protein family EPCAM family Biochemical class NA Function Plays a role in embryonic stem cells proliferation and differentiation. Up-regulates the expression of FABP5, MYC and cyclins A and E. May act as a physical homophilic interaction molecule between intestinal epithelial cells (IECs) and intraepithelial lymphocytes (IELs) at the mucosal epithelium for providing immunological barrier as a first line of defense against mucosal infection. Related diseases Diarrhea 5, with tufting enteropathy, congenital (DIAR5) [MIM:613217]: An intractable diarrhea of infancy characterized by villous atrophy and absence of inflammation, with intestinal epithelial cell dysplasia manifesting as focal epithelial tufts in the duodenum and jejunum. {ECO:0000269|PubMed:18572020, ECO:0000269|PubMed:24142340}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Lynch syndrome 8 (LYNCH8) [MIM:613244]: A form of Lynch syndrome, an autosomal dominant disease associated with marked increase in cancer susceptibility. It is characterized by a familial predisposition to early-onset colorectal carcinoma (CRC) and extra-colonic tumors of the gastrointestinal, urological and female reproductive tracts. Lynch syndrome is reported to be the most common form of inherited colorectal cancer in the Western world. Clinically, it is often divided into two subgroups. Type I is characterized by hereditary predisposition to colorectal cancer, a young age of onset, and carcinoma observed in the proximal colon. Type II is characterized by increased risk for cancers in certain tissues such as the uterus, ovary, breast, stomach, small intestine, skin, and larynx in addition to the colon. Diagnosis of classical Lynch syndrome is based on the Amsterdam criteria: 3 or more relatives affected by colorectal cancer, one a first degree relative of the other two; 2 or more generation affected; 1 or more colorectal cancers presenting before 50 years of age; exclusion of hereditary polyposis syndromes. The term 'suspected Lynch syndrome' or 'incomplete Lynch syndrome' can be used to describe families who do not or only partially fulfill the Amsterdam criteria, but in whom a genetic basis for colon cancer is strongly suspected. {ECO:0000269|PubMed:19098912}. The disease is caused by variants affecting the gene represented in this entry. LYNCH8 results from heterozygous deletion of 3-prime exons of EPCAM and intergenic regions directly upstream of MSH2, resulting in transcriptional read-through and epigenetic silencing of MSH2 in tissues expressing EPCAM. Drugs (DrugBank ID) DB06607; DB11075; DB05831; DB05319; DB09336 Interacts with P27797; P12830; Q15078; P36957; Q8TDX7 EC number NA Uniprot keywords 3D-structure; Cell junction; Cell membrane; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hereditary nonpolyposis colorectal cancer; Membrane; Proteomics identification; Reference proteome; Repeat; Signal; Tight junction; Transmembrane; Transmembrane helix; Tumor antigen Protein physicochemical properties Chain ID A Molecular weight (Da) 27439.8 Length 243 Aromaticity 0.07 Instability index 33.77 Isoelectric point 6.03 Charge (pH=7) -1.99 2D Binding mode Binding energy (Kcal/mol) -6.85 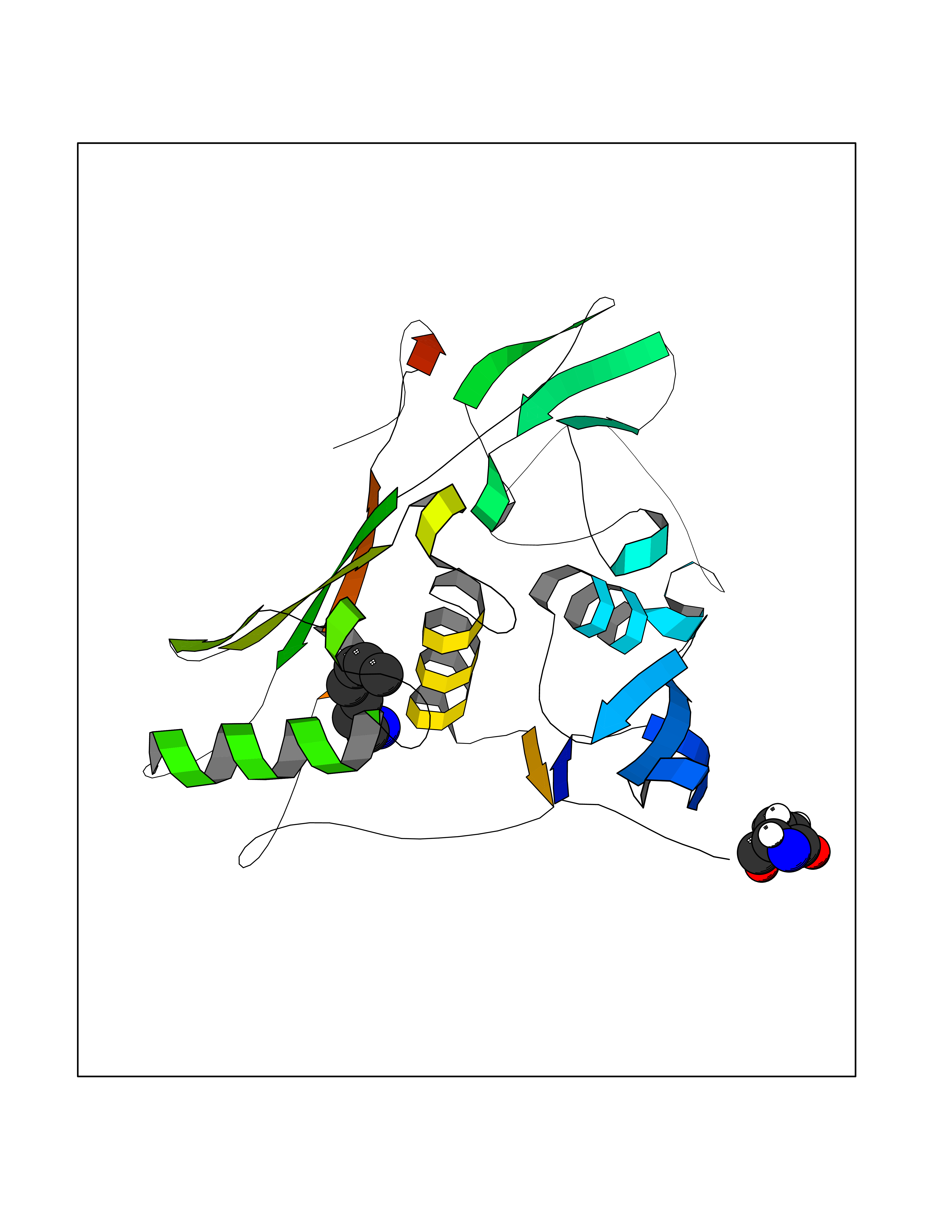 Molscript Map 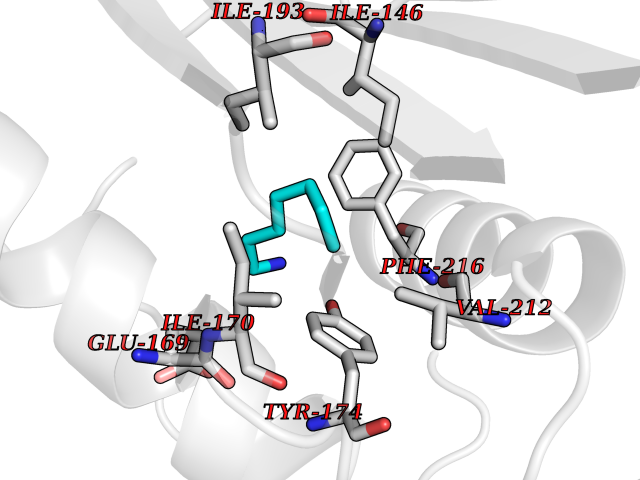 Pymol Map 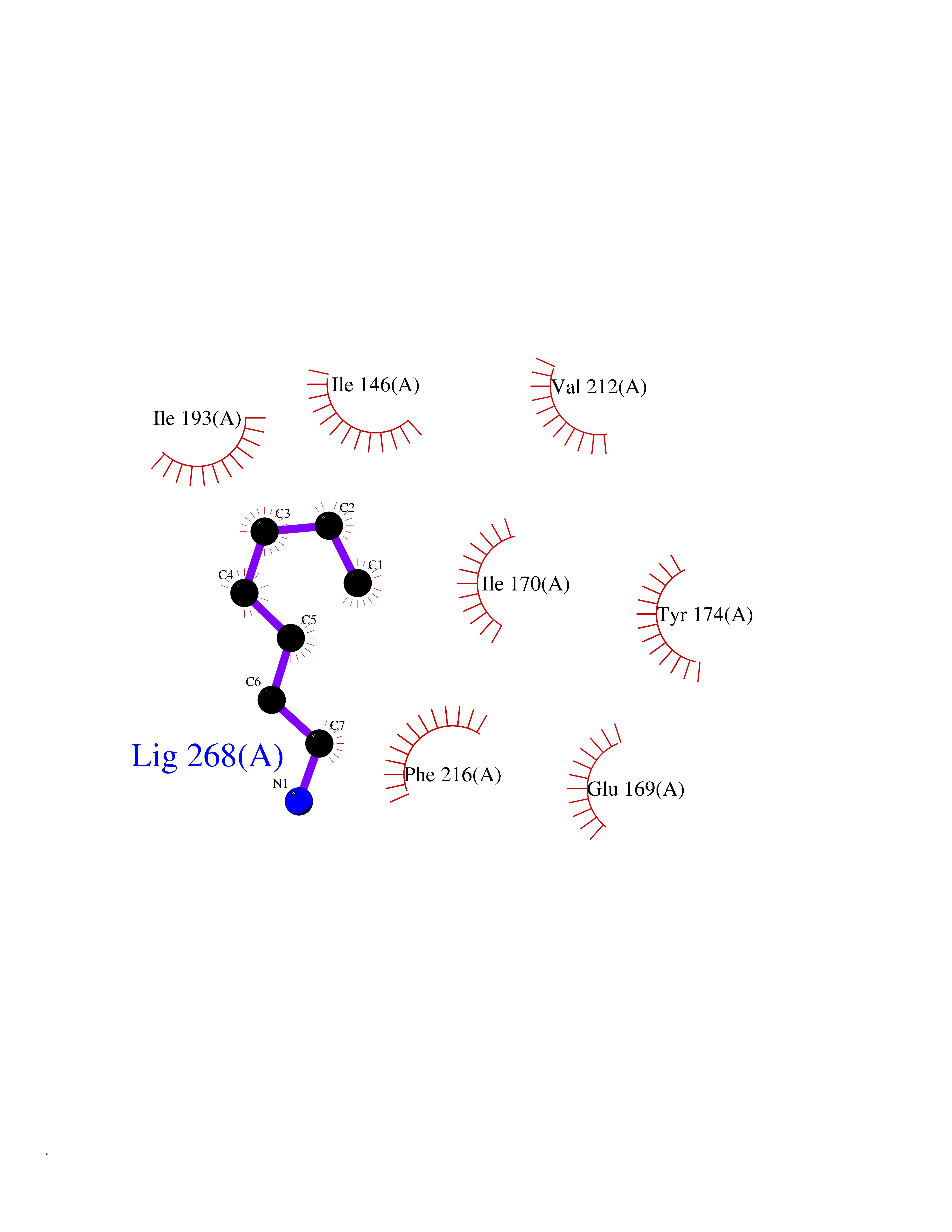 Ligplot Map 3D Binding mode Sequence XEECVCENYKLAVNCFVNNNRQCQCTSVGAQNTVICSKLAAKCLVMKAEMQGSKLGRRAKPEGALQNNDGLYDPDCDESGLFKAKQCQGTSTCWCVNTAGVRRTDKDTEITCSERVRTYWIIIELKHKAREKPYDSKSLRTALQKEITTRYQLDPKFITSILYENNVITIDLVQQSSQKTQNDVDIADVAYYFEKDVKGESLFHSKKMDLTVNGEQLDLDPGQTLIYYVDEKAPEFSMQGLKH Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Macrophage migration inhibitory factor (MIF) | 3IJJ | 5.02 | |
Target general information Gen name MIF Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Phenylpyruvate tautomerase; MMIF; L-dopachrome tautomerase; L-dopachrome isomerase; Glycosylation-inhibiting factor; GLIF; GIF Protein family MIF family Biochemical class Intramolecular oxidoreductase Function Involved in the innate immune response to bacterial pathogens. The expression of MIF at sites of inflammation suggests a role as mediator in regulating the function of macrophages in host defense. Counteracts the anti-inflammatory activity of glucocorticoids. Has phenylpyruvate tautomerase and dopachrome tautomerase activity (in vitro), but the physiological substrate is not known. It is not clear whether the tautomerase activity has any physiological relevance, and whether it is important for cytokine activity. Pro-inflammatory cytokine. Related diseases Rheumatoid arthritis systemic juvenile (RASJ) [MIM:604302]: An inflammatory articular disorder with systemic onset beginning before the age of 16. It represents a subgroup of juvenile arthritis associated with severe extraarticular features and occasionally fatal complications. During active phases of the disorder, patients display a typical daily spiking fever, an evanescent macular rash, lymphadenopathy, hepatosplenomegaly, serositis, myalgia and arthritis. {ECO:0000269|PubMed:11508429}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01880; DB07888; DB08334; DB08335; DB08333; DB07718; DB08765; DB02728 Interacts with O43521-2; P00533; Q92743; P14174; Q96HA8 EC number EC 5.3.2.1 Uniprot keywords 3D-structure; Acetylation; Cytokine; Cytoplasm; Direct protein sequencing; Immunity; Inflammatory response; Innate immunity; Isomerase; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,C Molecular weight (Da) 24671.9 Length 228 Aromaticity 0.09 Instability index 31.45 Isoelectric point 8.37 Charge (pH=7) 2.26 2D Binding mode Binding energy (Kcal/mol) -6.85 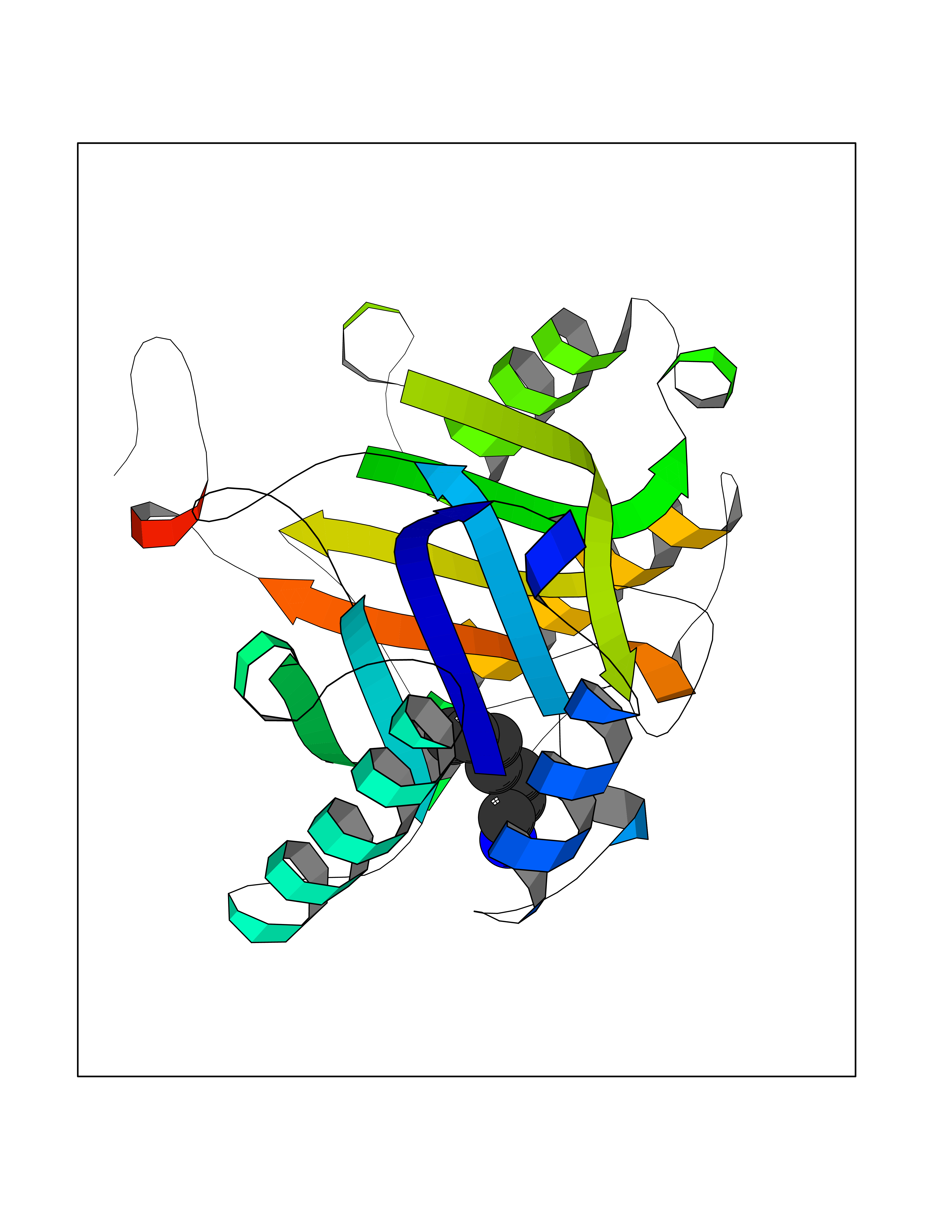 Molscript Map 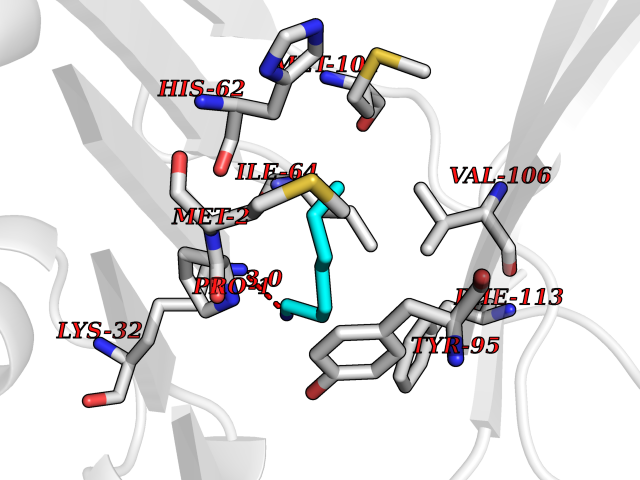 Pymol Map 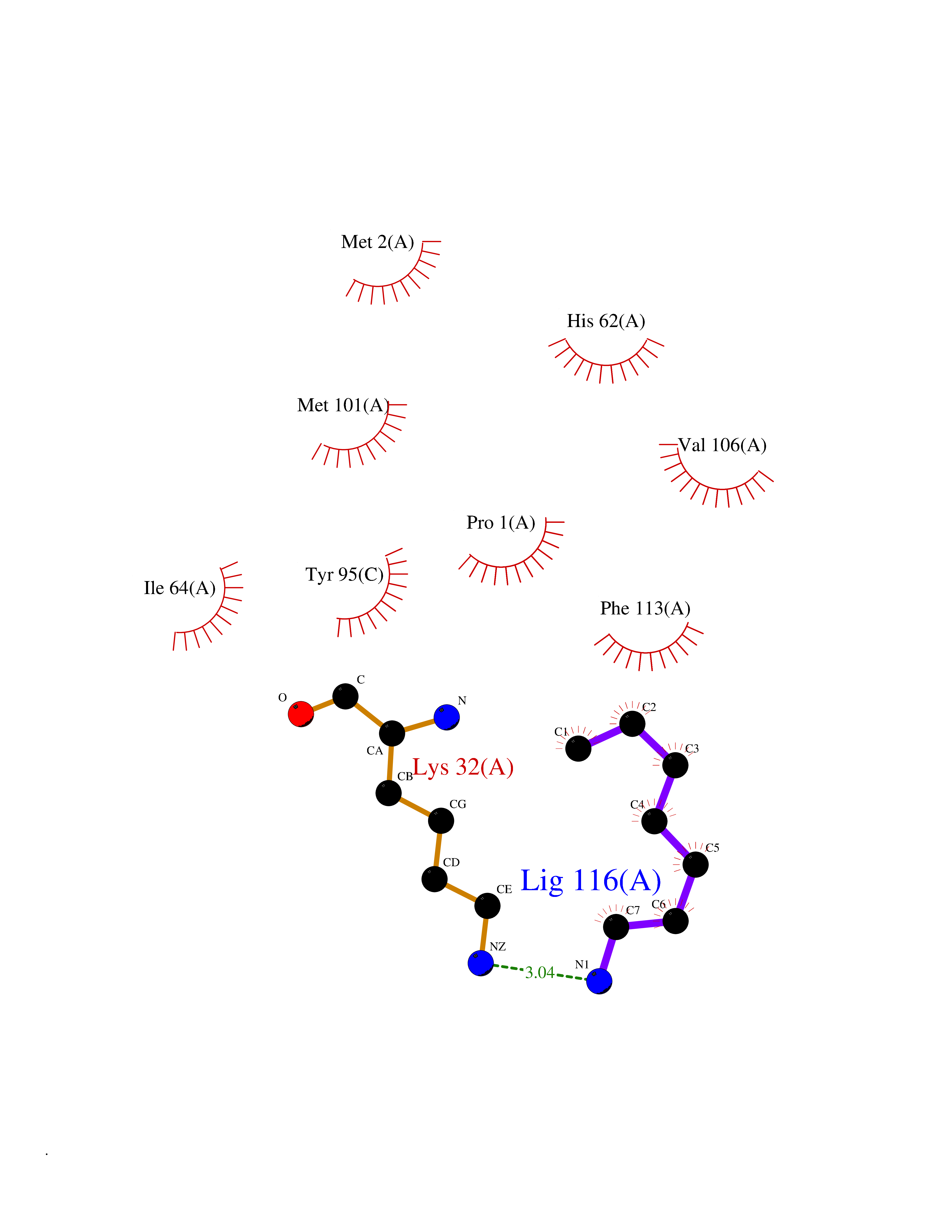 Ligplot Map 3D Binding mode Sequence PMFIVNTNVPRASVPDGFLSELTQQLAQATGKPPQYIAVHVVPDQLMAFGGSSEPCALCSLHSIGKIGGAQNRSYSKLLCGLLAERLRISPDRVYINYYDMNAANVGWNNSTFAPMFIVNTNVPRASVPDGFLSELTQQLAQATGKPPQYIAVHVVPDQLMAFGGSSEPCALCSLHSIGKIGGAQNRSYSKLLCGLLAERLRISPDRVYINYYDMNAANVGWNNSTFA Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Cardiac phospholamban (PLN) | 7E0Z | 5.02 | |
Target general information Gen name PLN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Phospholamban; PLN; PLB Protein family Phospholamban family Biochemical class NA Function Reversibly inhibits the activity of ATP2A2 in cardiac sarcoplasmic reticulum by decreasing the apparent affinity of the ATPase for Ca(2+). Modulates the contractility of the heart muscle in response to physiological stimuli via its effects on ATP2A2. Modulatescalcium re-uptake during muscle relaxation and plays an important role in calcium homeostasis in the heart muscle. The degree of ATP2A2 inhibition depends on the oligomeric state of PLN. ATP2A2 inhibition is alleviated by PLN phosphorylation. Related diseases Cardiomyopathy, dilated, 1P (CMD1P) [MIM:609909]: A disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death. {ECO:0000269|PubMed:12610310, ECO:0000269|PubMed:16432188, ECO:0000269|PubMed:22137083, ECO:0000269|PubMed:22427649, ECO:0000269|PubMed:22707725}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Cardiomyopathy, familial hypertrophic, 18 (CMH18) [MIM:613874]: A hereditary heart disorder characterized by ventricular hypertrophy, which is usually asymmetric and often involves the interventricular septum. The symptoms include dyspnea, syncope, collapse, palpitations, and chest pain. They can be readily provoked by exercise. The disorder has inter- and intrafamilial variability ranging from benign to malignant forms with high risk of cardiac failure and sudden cardiac death. {ECO:0000269|PubMed:12705874}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q3SXY8; P07307-3; O15342; Q9BXK5; Q13323; P19397; O95471; Q9UHP7-3; Q7Z7G2; O43889-2; Q96BA8; Q09013; Q92838; Q9GZR5; Q5JX71; Q14318; Q8TBE3; P48165; Q8TDT2; O60883; Q8TED1; P31937; Q7Z5P4; P43628; Q5T700; Q8N112; Q9GZY8-5; Q6N075; Q99735; O14880; Q9GZW8; Q9H2K0; P15941-11; Q8TBJ4; O95197-3; Q9NR31; A0A0S2Z4U3; Q9Y3P8; Q15849; Q8IWU4; O95436-2; Q9NQQ7-3; Q9NP94; Q9HBV2; Q9NPE6; Q16623; P32856-2; Q9BVX2; Q7Z7N9; Q6UW68; Q9NWC5; Q96B21; Q4KMG9; Q8N661; O15393-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Cardiomyopathy; Disease variant; Endoplasmic reticulum; Lipoprotein; Membrane; Mitochondrion; Palmitate; Phosphoprotein; Proteomics identification; Reference proteome; Sarcoplasmic reticulum; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 40393 Length 349 Aromaticity 0.13 Instability index 35.1 Isoelectric point 8.98 Charge (pH=7) 6.56 2D Binding mode Binding energy (Kcal/mol) -6.85 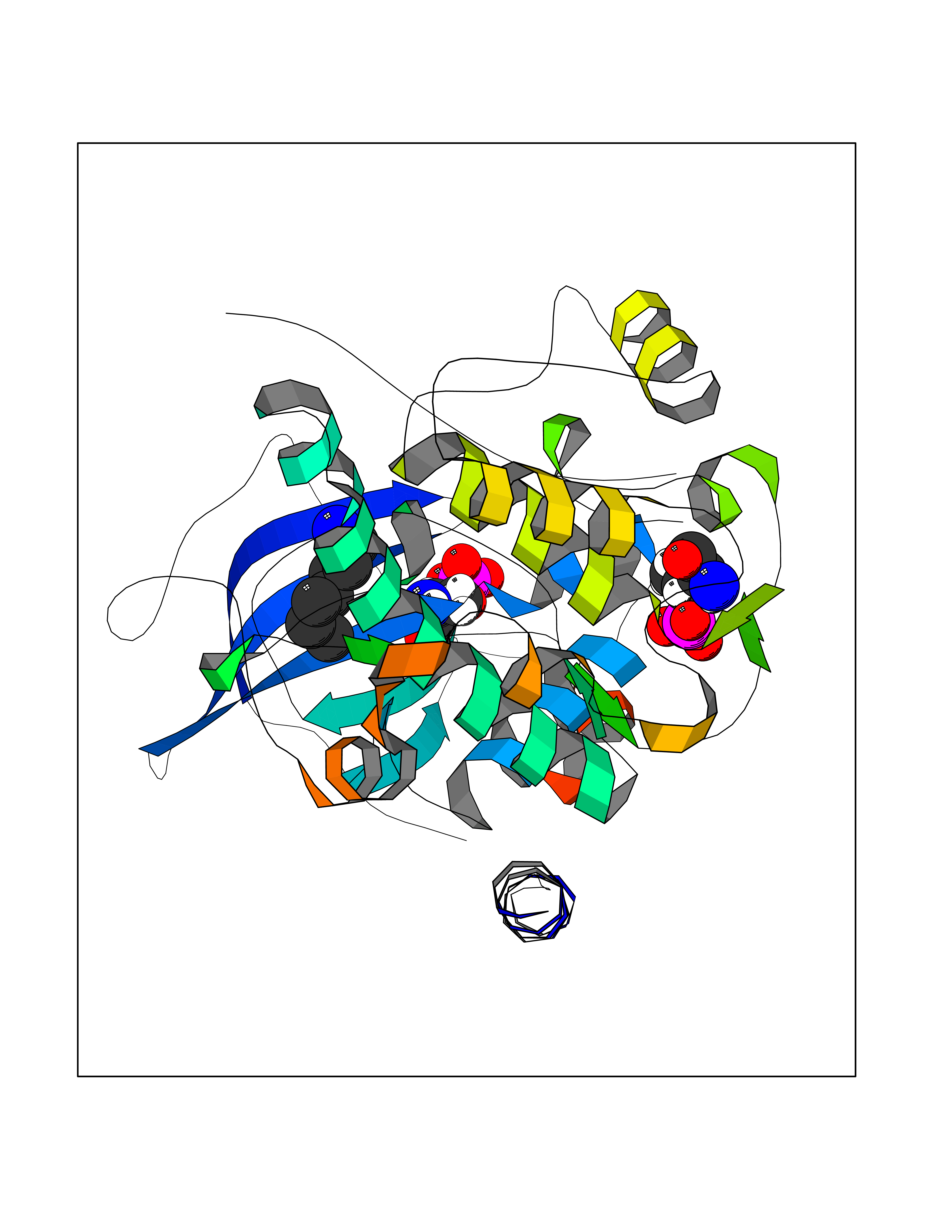 Molscript Map 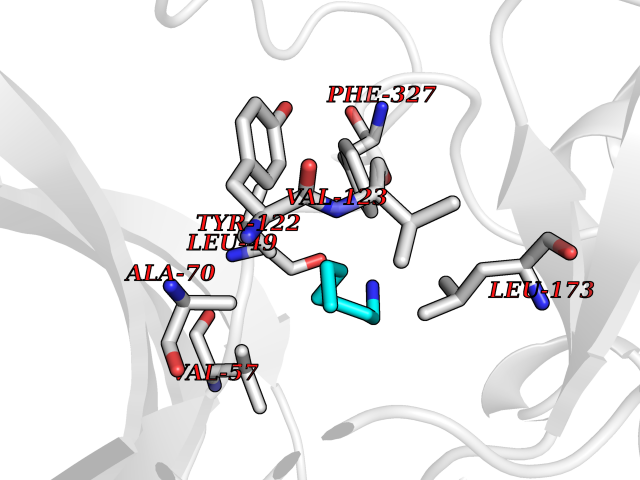 Pymol Map 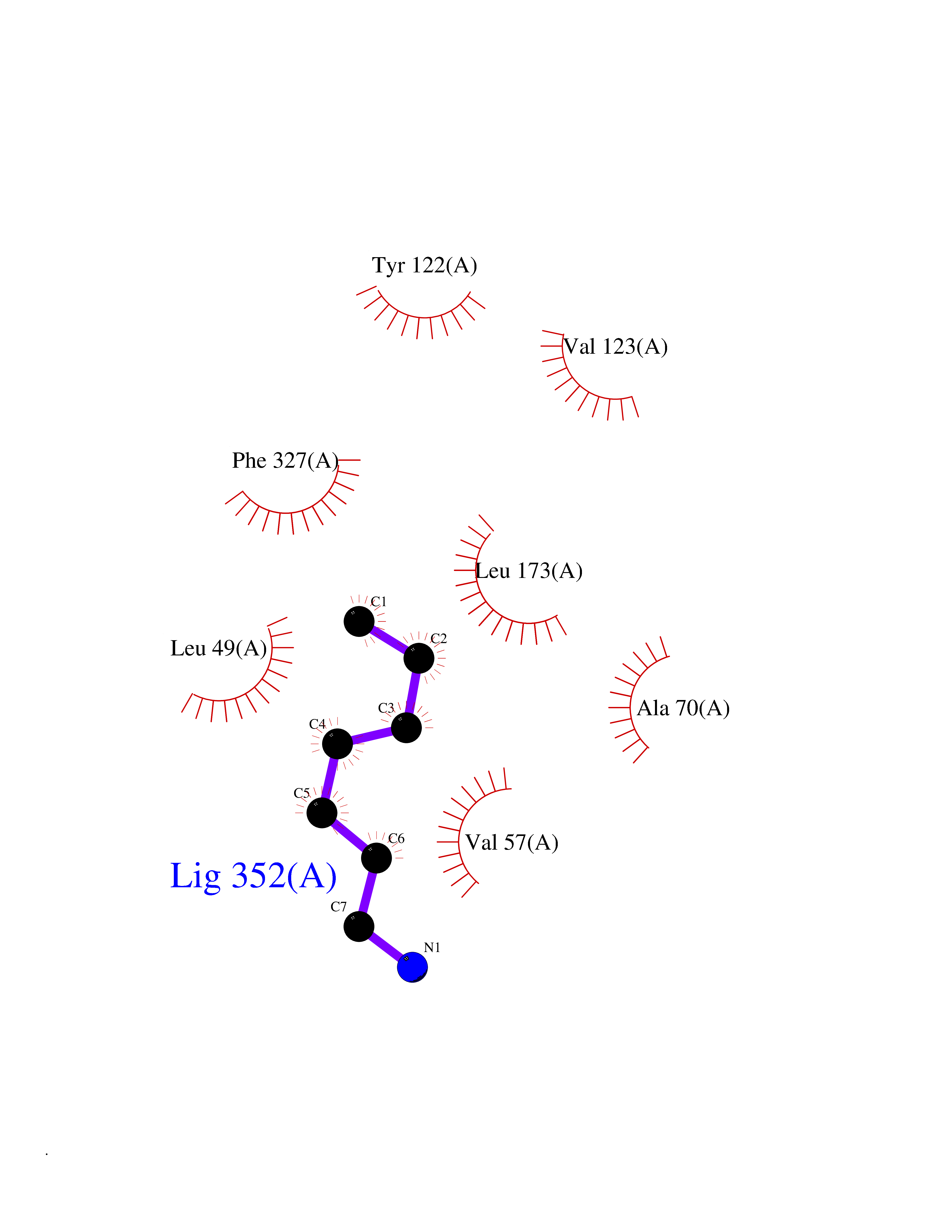 Ligplot Map 3D Binding mode Sequence ESVKEFLAKAKEDFLKKWETPQNTAQLDQFDRIKTLGTGSFGRVMLVKHKESGNHYAMKILDKQKVVKLKQIEHTLNEKRILQAVNFPFLVKLEFSFKDNSNLYMVMEYVAGGEMFSHLRRIGRFSEPHARFYAAQIVLTFEYLHSLDLIYRDLKPENLLIDQQGYIQVTDFGFAKRVKGRTWXLCGTPEYLAPEIILSKGYNKAVDWWALGVLIYEMAAGYPPFFADQPIQIYEKIVSGKVRFPSHFSSDLKDLLRNLLQVDLTKRFGNLKNGVNDIKNHKWFATTDWIAIYQRKVEAPFIPKFKGPGDTSNFDDYEEEEIRVXINEKCGKEFTEFTRSAIRRASTIE Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Gamma-aminobutyric acid type B receptor subunit 1 | 4MS4 | 5.01 | |
Target general information Gen name GABBR1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms GPRC3B;GPR51 Protein family G-protein coupled receptor 3 family, GABA-B receptor subfamily Biochemical class Signaling protein / antagonist Function G-protein coupled GABA receptor activity. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08891; DB08892; DB00181; DB00363; DB02530; DB05010; DB09072 Interacts with Q9UBS5; Q9UBS5-2; P46459; Q86UR5 EC number NA Uniprot keywords 3D-structure; Cell membrane; Coiled coil; Direct protein sequencing; Disease variant; Disulfide bond; Epilepsy; G-protein coupled receptor; Glycoprotein; Intellectual disability; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 46502.1 Length 408 Aromaticity 0.12 Instability index 50.05 Isoelectric point 5.78 Charge (pH=7) -5.62 2D Binding mode Binding energy (Kcal/mol) -6.84 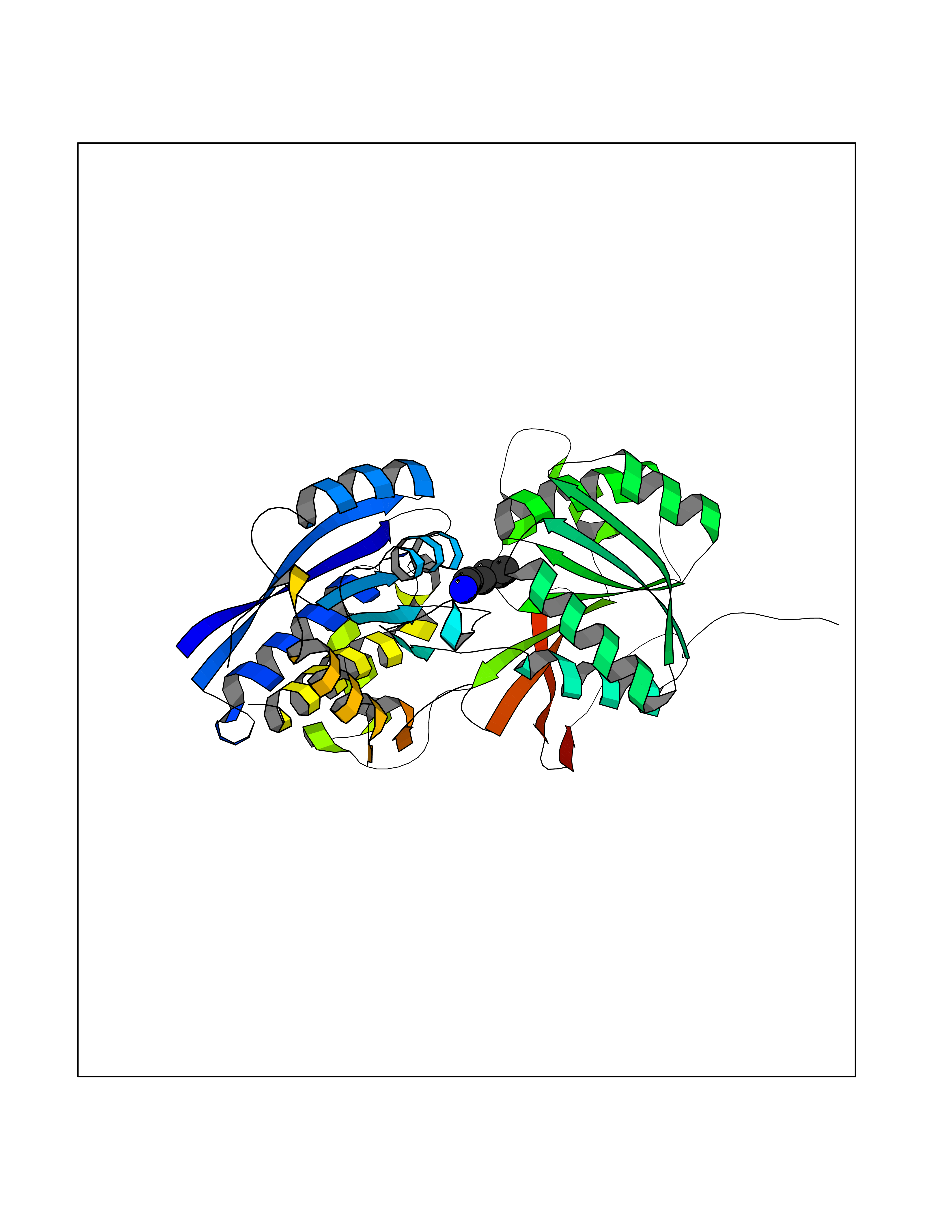 Molscript Map 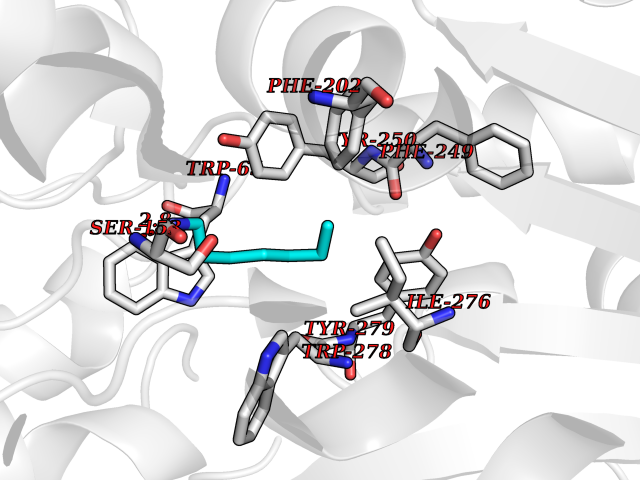 Pymol Map 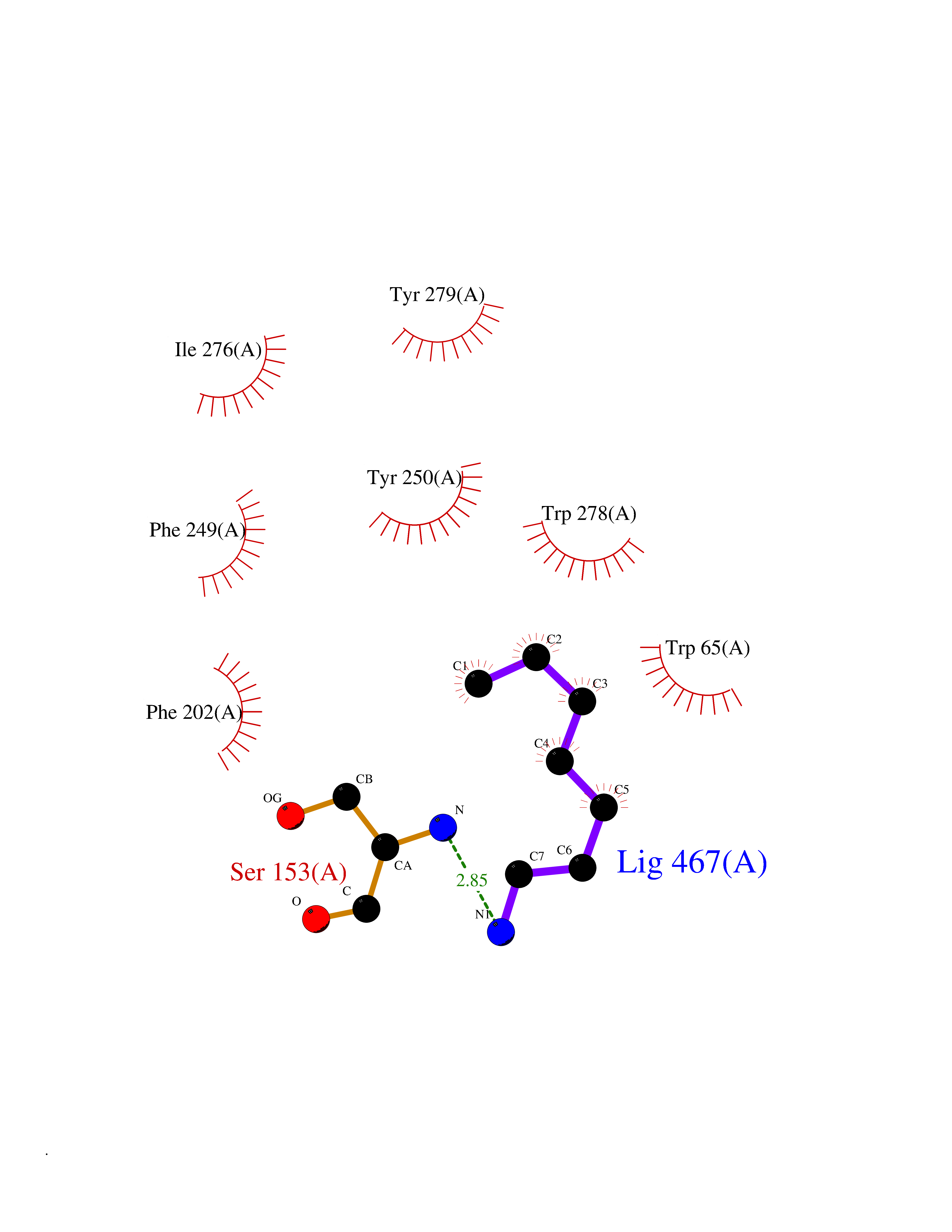 Ligplot Map 3D Binding mode Sequence RRAVYIGALFPMSGGWPGGQACQPAVEMALEDVNSRRDILPDYELKLIHHDSKCDPGQATKYLYELLYNDPIKIILMPGCSSVSTLVAEAARMWNLIVLSYGSSSPALSNRQRFPTFFRTHPSATLHNPTRVKLFEKWGWKKIATIQQTTEVFTSTLDDLEERVKEAGIEITFRQSFFSDPAVPVKNLKRQDARIIVGLFYETEARKVFCEVYKERLFGKKYVWFLIGWYADNWFKIYDPSINCTVDEMTEAVEGHITTEIVMLNPANTRSISNMTSQEFVEKLTKRLKRHPEETGGFQEAPLAYDAIWALALALNKTSRLEDFNYNNQTITDQIYRAMNSSSFEGVSGHVVFDASGSRMAWTLIEQLQGGSYKKIGYYDSTKDDLSWSKTDKWIGGSPPADDYKDDD Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Aldose reductase (AKR1B1) | 1US0 | 5.01 | |
Target general information Gen name AKR1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aldehyde reductase; AKR1B1 Protein family Aldo/keto reductase family Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the NADPH-dependent reduction of a wide variety of carbonyl-containing compounds to their corresponding alcohols with a broad range of catalytic efficiencies. Related diseases Glutamine deficiency, congenital (GLND) [MIM:610015]: An autosomal recessive disorder characterized by variable brain malformations, encephalopathy, severe developmental delay, seizures, and decreased glutamine levels in bodily fluids. Death in early infancy may occur. {ECO:0000269|PubMed:16267323, ECO:0000269|PubMed:26711351, ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 116 (DEE116) [MIM:620806]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE116 is autosomal dominant form characterized by severe developmental delay, seizures, and white matter abnormalities. {ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry. DEE116 is caused by variants that disrupt the canonical translation start codon in GLUL resulting in initiation of translation at Met-18 (PubMed:38579670). The resulting protein is enzymatically competent but insensitive to negative feedback regulation via glutamine-induced degradation. {ECO:0000269|PubMed:38579670}. Drugs (DrugBank ID) DB07028; DB07030; DB07450; DB02101; DB08449; DB08000; DB07139; DB07498; DB02007; DB02020; DB11859; DB02994; DB04272; DB07187; DB00694; DB00997; DB06246; DB01039; DB02021; DB16707; DB00143; DB02834; DB08084; DB01689; DB07063; DB06077; DB02518; DB00157; DB03461; DB05383; DB05533; DB05327; DB02712; DB00605; DB02383; DB02132; DB08772; DB07093; DB07999; DB08098 Interacts with Q9BUY7 EC number EC 1.1.1.300 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Lipid metabolism; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 35447.6 Length 313 Aromaticity 0.09 Instability index 36.41 Isoelectric point 7.1 Charge (pH=7) 0.26 2D Binding mode Binding energy (Kcal/mol) -6.83  Molscript Map  Pymol Map 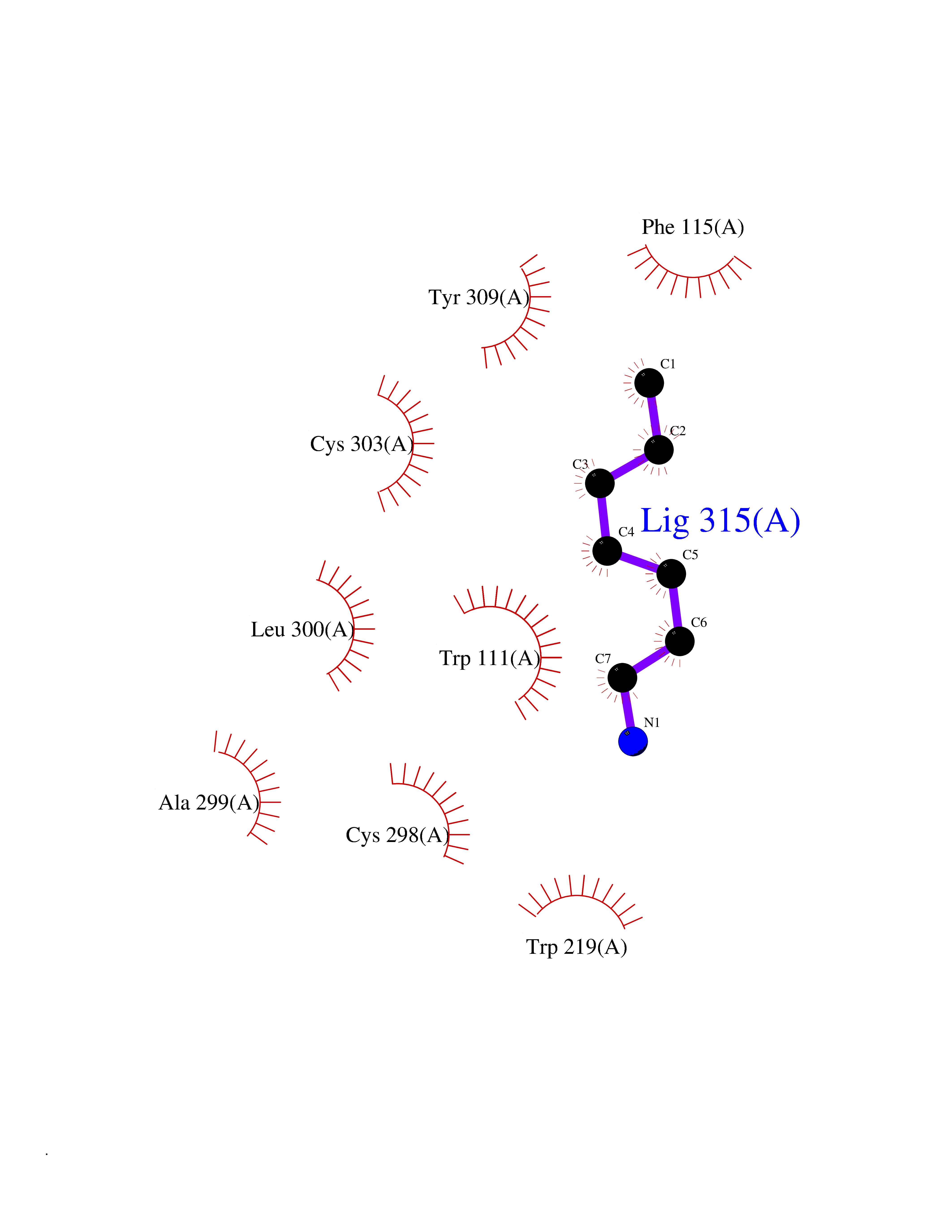 Ligplot Map 3D Binding mode Sequence MASRILLNNGAKMPILGLGTWKSPPGQVTEAVKVAIDVGYRHIDCAHVYQNENEVGVAIQEKLREQVVKREELFIVSKLWCTYHEKGLVKGACQKTLSDLKLDYLDLYLIHWPTGFKPGKEFFPLDESGNVVPSDTNILDTWAAMEELVDEGLVKAIGISNFNHLQVEMILNKPGLKYKPAVNQIECHPYLTQEKLIQYCQSKGIVVTAYSPLGSPDRPWAKPEDPSLLEDPRIKAIAAKHNKTTAQVLIRFPMQRNLVVIPKSVTPERIAENFKVFDFELSSQDMTTLLSYNRNWRVCALLSCTSHKDYPFH Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Aromatase (CYP19A1) | 3S79 | 5.01 | |
Target general information Gen name CYP19A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms P-450AROM; Estrogen synthetase; Estrogen synthase; Cytochrome P450 19A1; Cytochrome P-450AROM; CYPXIX; CYP19; CYAR; ARO1 Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function Catalyzes the formation of aromatic C18 estrogens from C19 androgens. Related diseases Aromatase excess syndrome (AEXS) [MIM:139300]: An autosomal dominant disorder characterized by increased extraglandular aromatization of steroids that presents with heterosexual precocity in males and isosexual precocity in females. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Aromatase deficiency (AROD) [MIM:613546]: A rare disease in which fetal androgens are not converted into estrogens due to placental aromatase deficiency. Thus, pregnant women exhibit a hirsutism, which spontaneously resolves after post-partum. At birth, female babies present with pseudohermaphroditism due to virilization of extern genital organs. In adult females, manifestations include delay of puberty, breast hypoplasia and primary amenorrhoea with multicystic ovaries. {ECO:0000269|PubMed:24705274, ECO:0000269|PubMed:8265607, ECO:0000269|PubMed:8530621, ECO:0000269|PubMed:9211678}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02342; DB00357; DB01217; DB00443; DB04794; DB06719; DB13009; DB00389; DB00269; DB00856; DB04839; DB01406; DB00255; DB00858; DB01127; DB14598; DB14600; DB00974; DB06423; DB00783; DB00655; DB00926; DB00990; DB04539; DB01026; DB01006; DB05667; DB00358; DB01065; DB00333; DB06710; DB01110; DB16236; DB05749; DB08804; DB03467; DB00184; DB09389; DB01229; DB05804; DB00481; DB05875; DB02901; DB06147; DB00675; DB00894; DB00624; DB13943; DB13944; DB13946; DB01007; DB00197 Interacts with NA EC number EC 1.14.14.14 Uniprot keywords 3D-structure; Alternative splicing; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Heme; Iron; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Pseudohermaphroditism; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 52141.6 Length 452 Aromaticity 0.1 Instability index 36.02 Isoelectric point 8.45 Charge (pH=7) 4.41 2D Binding mode Binding energy (Kcal/mol) -6.83 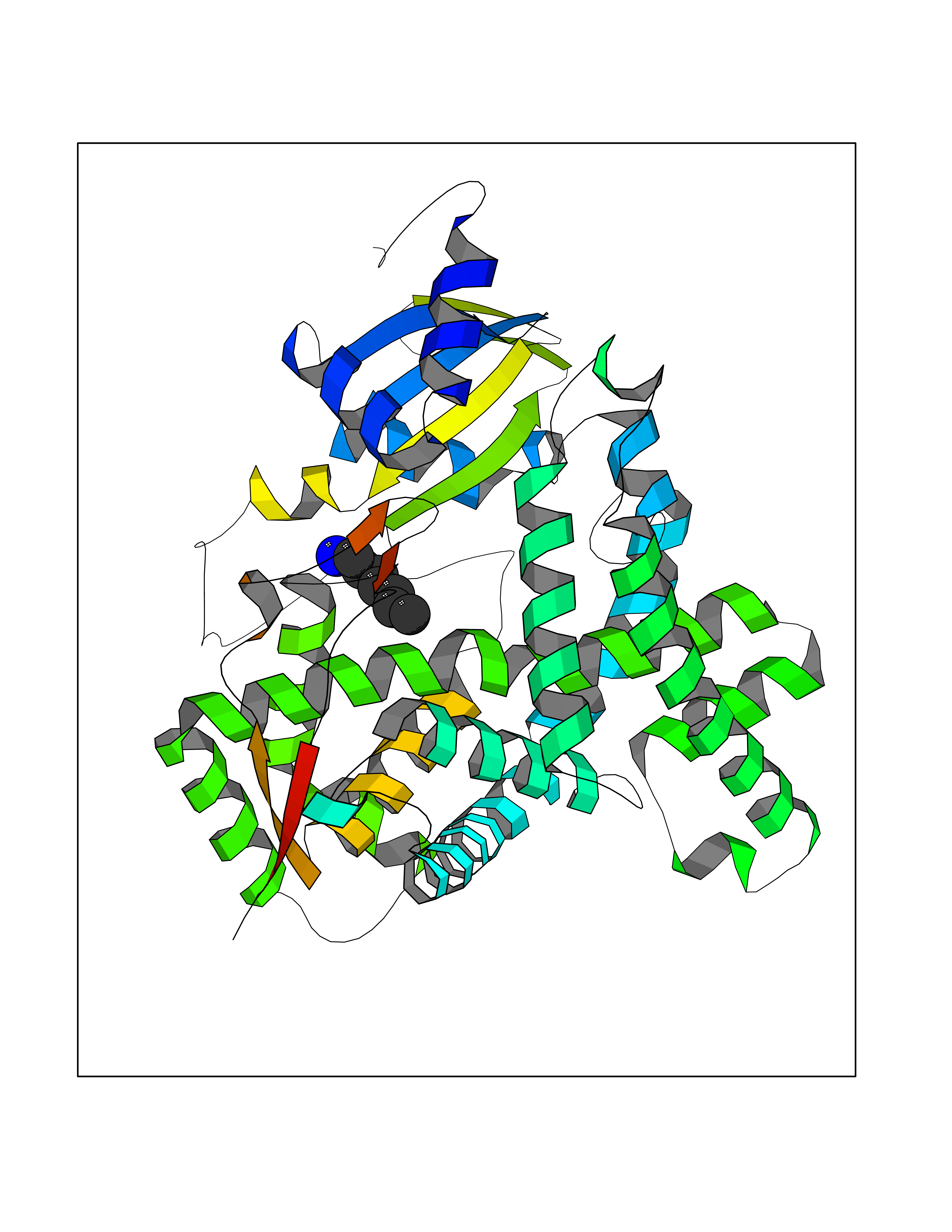 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SSIPGPGYCMGIGPLISHGRFLWMGIGSACNYYNRVYGEFMRVWISGEETLIISKSSSMFHIMKHNHYSSRFGSKLGLQCIGMHEKGIIFNNNPELWKTTRPFFMKALSGPGLVRMVTVCAESLKTHLDRLEEVTNESGYVDVLTLLRRVMLDTSNTLFLRIPLDESAIVVKIQGYFDAWQALLIKPDIFFKISWLYKKYEKSVKDLKDAIEVLIAEKRRRISTEEKLEECMDFATELILAEKRGDLTRENVNQCILEMLIAAPDTMSVSLFFMLFLIAKHPNVEEAIIKEIQTVIGERDIKIDDIQKLKVMENFIYESMRYQPVVDLVMRKALEDDVIDGYPVKKGTNIILNIGRMHRLEFFPKPNEFTLENFAKNVPYRYFQPFGFGPRGCAGKYIAMVMMKAILVTLLRRFHVKTLQGQCVESIQKIHDLSLHPDETKNMLEMIFTPRN Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Acetylcholinesterase (AChE) | 4M0E | 5.01 | |
Target general information Gen name ACHE Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms YT; N-ACHE; ARACHE Protein family Type-B carboxylesterase/lipase family Biochemical class Carboxylic ester hydrolase Function Role in neuronal apoptosis. Terminates signal transduction at the neuromuscular junction by rapid hydrolysis of the acetylcholine released into the synaptic cleft. Related diseases Phosphoribosylaminoimidazole carboxylase deficiency (PAICSD) [MIM:619859]: An autosomal recessive inborn error of purine metabolism, clinically characterized by multiple congenital anomalies and early neonatal death. {ECO:0000269|PubMed:31600779}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07846; DB02673; DB04617; DB04614; DB04615; DB07756; DB07701; DB02404; DB03814; DB08615; DB02343; DB02226; DB03005; DB04114; DB03128; DB01122; DB03283; DB00411; DB00122; DB14006; DB01245; DB00944; DB08357; DB08996; DB00449; DB00843; DB01010; DB01364; DB00898; DB00674; DB00483; DB06525; DB04864; DB03348; DB07555; DB00677; DB04924; DB03359; DB00358; DB00940; DB02825; DB02845; DB08167; DB04021; DB00805; DB01805; DB03740; DB04556; DB01400; DB04892; DB00981; DB00733; DB02166; DB00545; DB00863; DB00989; DB00382; DB04616; DB12816; DB01199; DB13503; DB04859 Interacts with Q9Y215; P06733; P63244 EC number EC 3.1.1.7 Uniprot keywords 3D-structure; Alternative splicing; Blood group antigen; Cell membrane; Direct protein sequencing; Disulfide bond; Glycoprotein; GPI-anchor; Hydrolase; Lipoprotein; Membrane; Neurotransmitter degradation; Nucleus; Proteomics identification; Reference proteome; Secreted; Serine esterase; Signal; Synapse Protein physicochemical properties Chain ID A,B Molecular weight (Da) 58804.1 Length 537 Aromaticity 0.11 Instability index 40.85 Isoelectric point 5.73 Charge (pH=7) -8.18 2D Binding mode Binding energy (Kcal/mol) -6.84 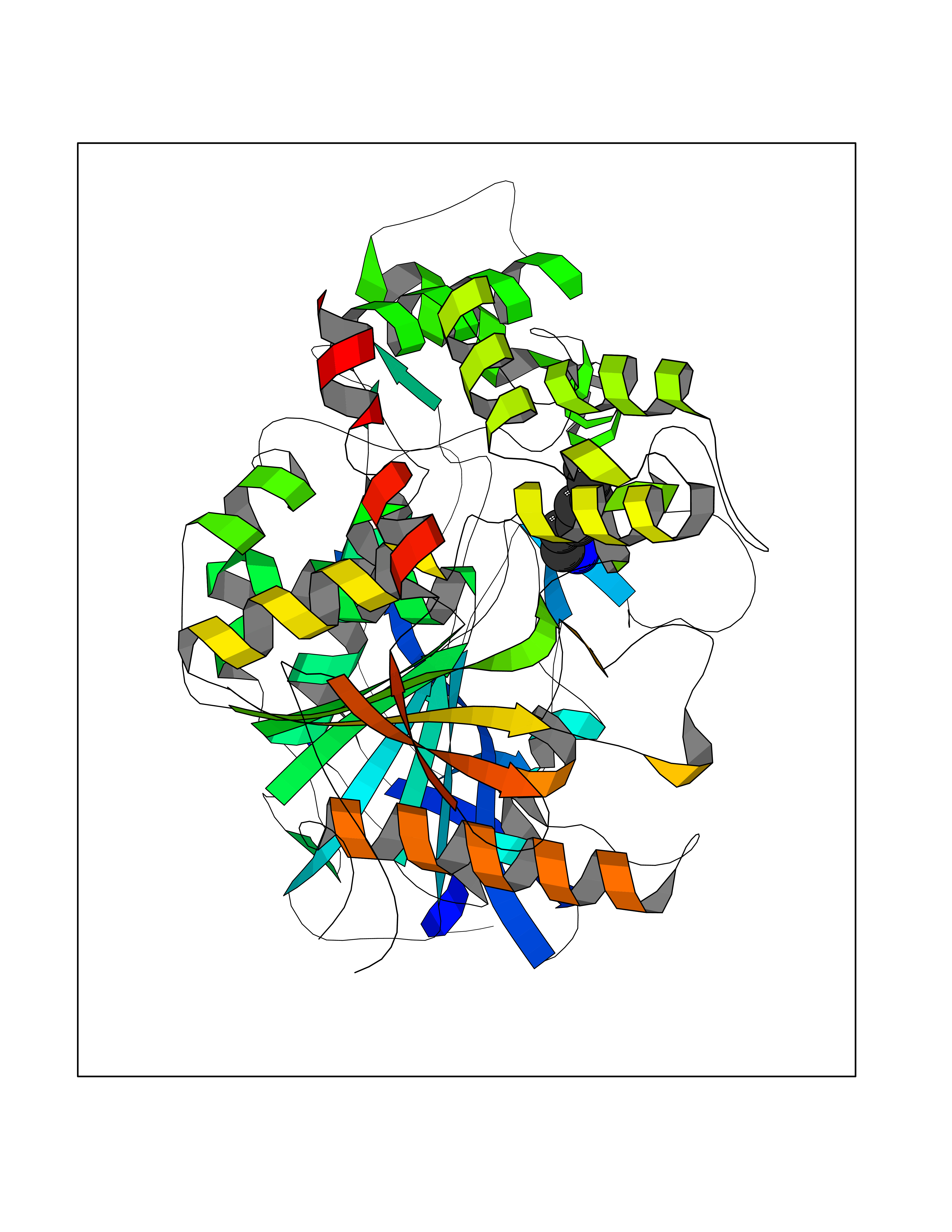 Molscript Map 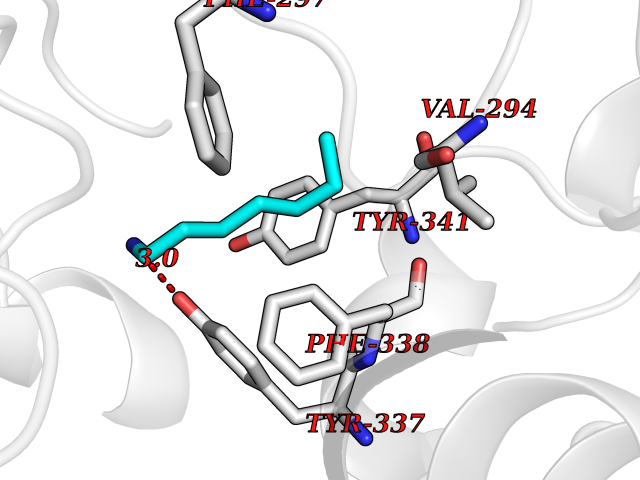 Pymol Map 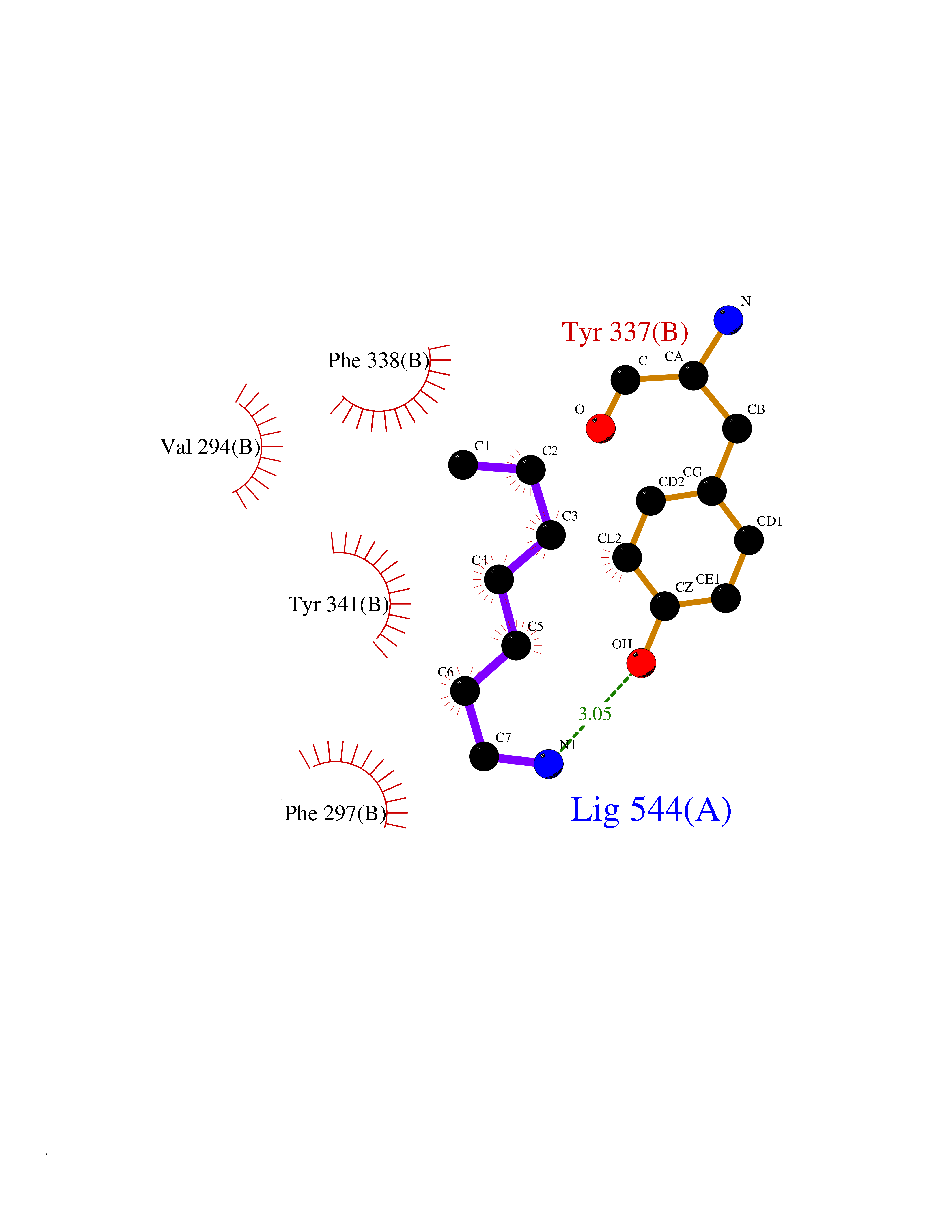 Ligplot Map 3D Binding mode Sequence EDAELLVTVRGGRLRGIRLKTPGGPVSAFLGIPFAEPPMGPRRFLPPEPKQPWSGVVDATTFQSVCYQYVDTLYPGFEGTEMWNPNRELSEDCLYLNVWTPYPRPTSPTPVLVWIYGGGFYSGASSLDVYDGRFLVQAERTVLVSMNYRVGAFGFLALPGSREAPGNVGLLDQRLALQWVQENVAAFGGDPTSVTLFGESAGAASVGMHLLSPPSRGLFHRAVLQSGAPNGPWATVGMGEARRRATQLAHLVGCPPGGTGGNDTELVACLRTRPAQVLVNHEWHVLPQESVFRFSFVPVVDGDFLSDTPEALINAGDFHGLQVLVGVVKDEGSYFLVYGAPGFSKDNESLISRAEFLAGVRVGVPQVSDLAAEAVVLHYTDWLHPEDPARLREALSDVVGDHNVVCPVAQLAGRLAAQGARVYAYVFEHRASTLSWPLWMGVPHGYEIEFIFGIPLDPSRNYTAEEKIFAQRLMRYWANFARTGDPNEPPKAPQWPPYTAGAQQYVSLDLRPLEVRRGLRAQACAFWNRFLPKLLSA Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Estrogen-related receptor-gamma (ESRRG) | 2E2R | 5.01 | |
Target general information Gen name ESRRG Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nuclear receptor subfamily 3 group B member 3; NR3B3; KIAA0832; Estrogen-related receptor gamma; Estrogen receptor-related protein 3; ERRG2; ERR3; ERR gamma-2 Protein family Nuclear hormone receptor family, NR3 subfamily Biochemical class Nuclear hormone receptor Function Binds specifically to an estrogen response element and activates reporter genes controlled by estrogen response elements. Induces the expression of PERM1 in the skeletal muscle. Orphan receptor that acts as transcription activator in the absence of bound ligand. Related diseases WHIM syndrome 1 (WHIMS1) [MIM:193670]: An autosomal dominant immunologic disease characterized by neutropenia, hypogammaglobulinemia and extensive human papillomavirus (HPV) infection. Despite the peripheral neutropenia, bone marrow aspirates from affected individuals contain abundant mature myeloid cells, a condition termed myelokathexis. {ECO:0000269|PubMed:12692554, ECO:0000269|PubMed:15536153}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: CXCR4 mutations play a role in the pathogenesis of Waldenstroem macroglobulinemia (WM) and influence disease presentation and outcome, as well as response to therapy. WM is a B-cell lymphoma characterized by accumulation of malignant lymphoplasmacytic cells in the bone marrow, lymph nodes and spleen, and hypersecretion of monoclonal immunoglobulin M (IgM). Excess IgM production results in serum hyperviscosity, tissue infiltration, and autoimmune-related pathology. {ECO:0000269|PubMed:24366360, ECO:0000269|PubMed:24553177}. Drugs (DrugBank ID) DB06884; DB04468; DB06973; DB07485; DB02659; DB00255; DB13952; DB13953; DB13954; DB13955; DB13956; DB06902; DB00675; DB00197 Interacts with Q05D60; Q9BVG8; P50222; P51843; Q12769; Q9UBK2; A0MZ66; G2XKQ0; Q8NFM4; Q13315; Q86WA6-2; Q9BZE7; Q13555-5; Q05D60; Q5JST6; P11474; O95718-2; P62508-3; Q15024; O95990-4; Q8IZU1; Q14296; P23508; Q6IN84; P51843; Q15466; P48552; P26367; Q9NPJ4; P01189; Q9UBK2; P62195; Q8N0T1-2; Q04864-2; Q6NUQ1; A0MZ66-4; Q8TAD8; P19237; P48788; Q96PN7; Q96S82; Q5SQQ9-2; Q7Z4V0 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 25755.7 Length 227 Aromaticity 0.07 Instability index 55.31 Isoelectric point 5.09 Charge (pH=7) -10.6 2D Binding mode Binding energy (Kcal/mol) -6.84 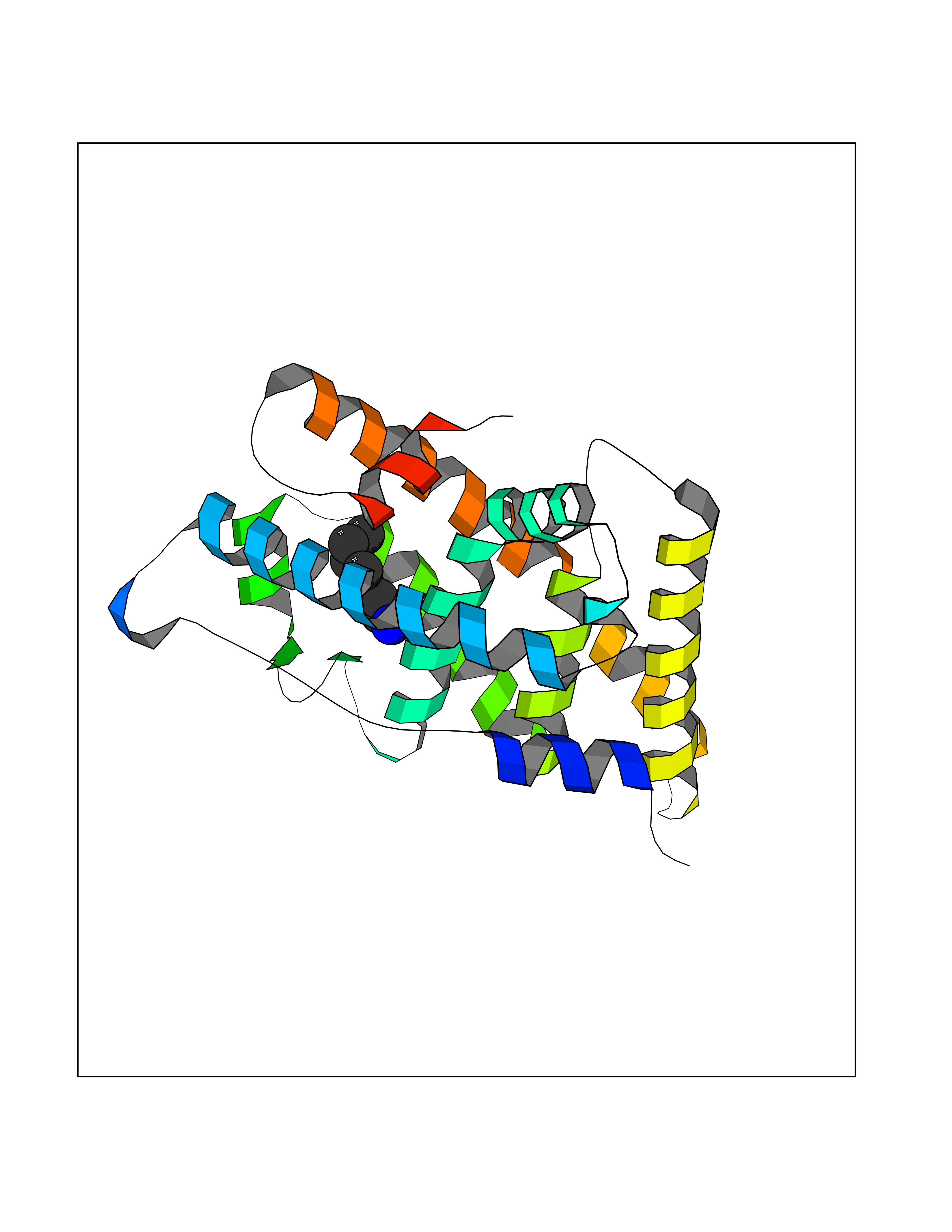 Molscript Map 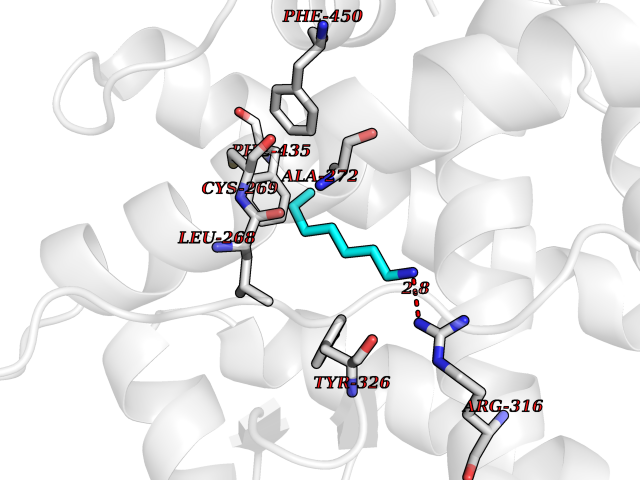 Pymol Map 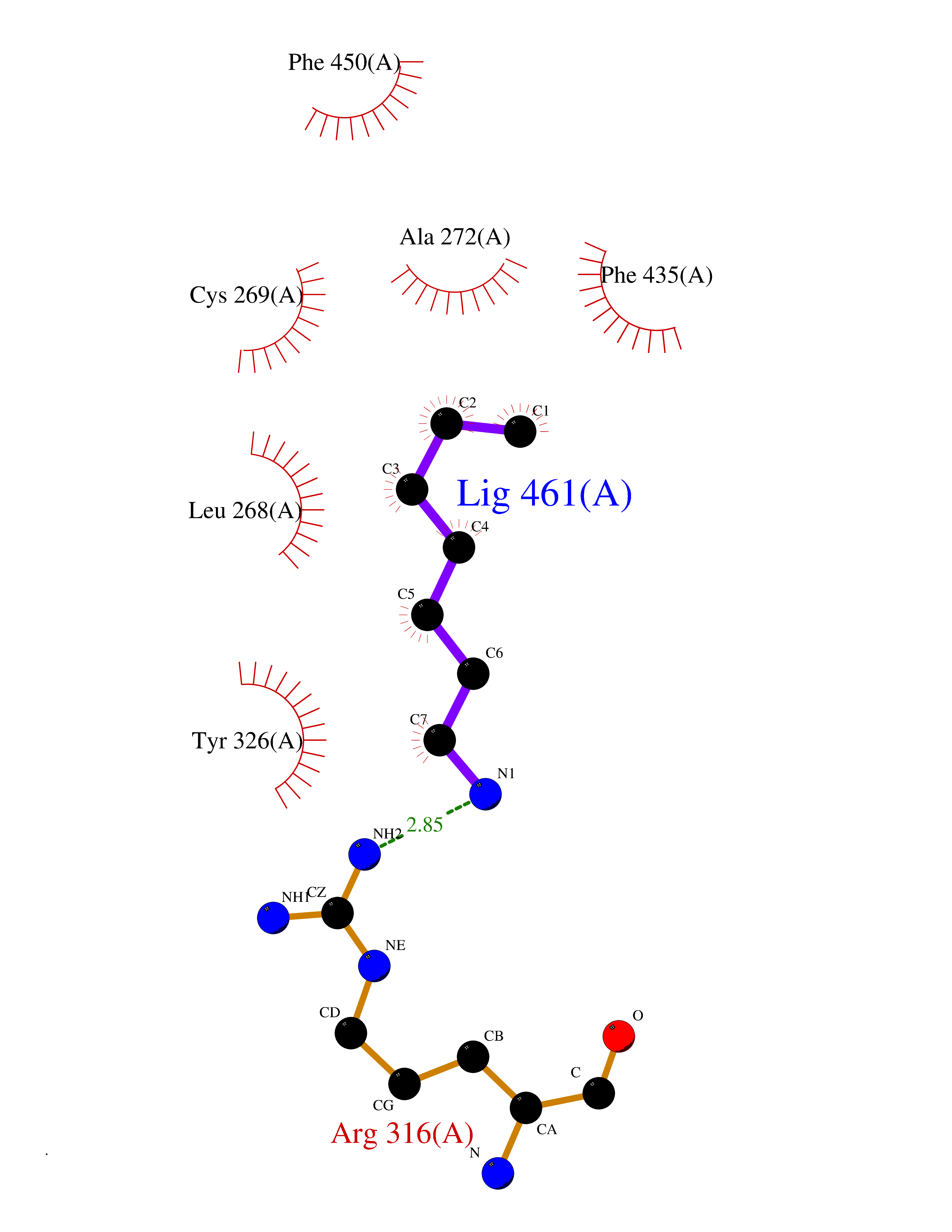 Ligplot Map 3D Binding mode Sequence KPYNKIVSHLLVAEPEKIYAMPDPTVPDSDIKALTTLCDLADRELVVIIGWAKHIPGFSTLSLADQMSLLQSAWMEILILGVVYRSLSFEDELVYADDYIMDEDQSKLAGLLDLNNAILQLVKKYKSMKLEKEEFVTLKAIALANSDSMHIEDVEAVQKLQDVLHEALQDYEAGQHMEDPRRAGKMLMTLPLLRQTSTKAVQHFYNIKLEGKVPMHKLFLEMLEAKV Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | 2-oxoglutarate dehydrogenase, mitochondrial | 3ERY | 5.01 | |
Target general information Gen name OGDH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Alpha-ketoglutarate dehydrogenase family Biochemical class Immune system Function Chaperone binding.Heat shock protein binding.Metal ion binding.Oxoglutarate dehydrogenase (NAD+) activity.Oxoglutarate dehydrogenase (succinyl-transferring) activity.Thiamine pyrophosphate binding. Related diseases Postaxial acrofacial dysostosis (POADS) [MIM:263750]: POADS is characterized by severe micrognathia, cleft lip and/or palate, hypoplasia or aplasia of the posterior elements of the limbs, coloboma of the eyelids and supernumerary nipples. POADS is a very rare disorder: only 2 multiplex families, each consisting of 2 affected siblings born to unaffected, nonconsanguineous parents, have been described among a total of around 30 reported cases. {ECO:0000269|PubMed:19915526}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00157; DB00313; DB09092 Interacts with P54253; P42858 EC number 1.2.4.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Calcium; Glycolysis; Isopeptide bond; Magnesium; Metal-binding; Mitochondrion; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Thiamine pyrophosphate; Transit peptide; Ubl conjugation Protein physicochemical properties Chain ID P,Q Molecular weight (Da) 18311.1 Length 158 Aromaticity 0.15 Instability index 37.26 Isoelectric point 5.64 Charge (pH=7) -2.98 2D Binding mode Binding energy (Kcal/mol) -6.84  Molscript Map 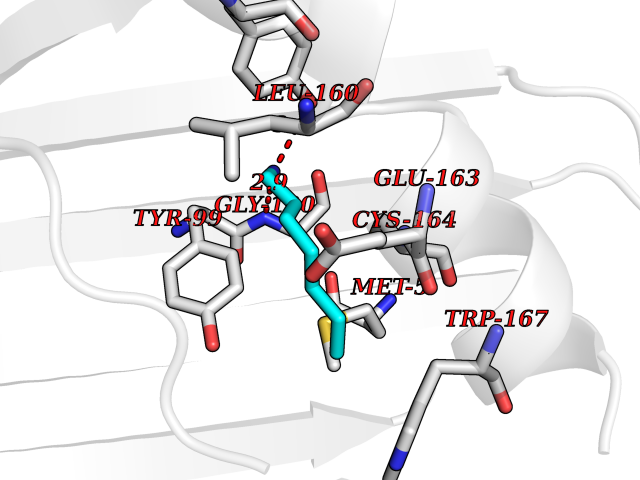 Pymol Map 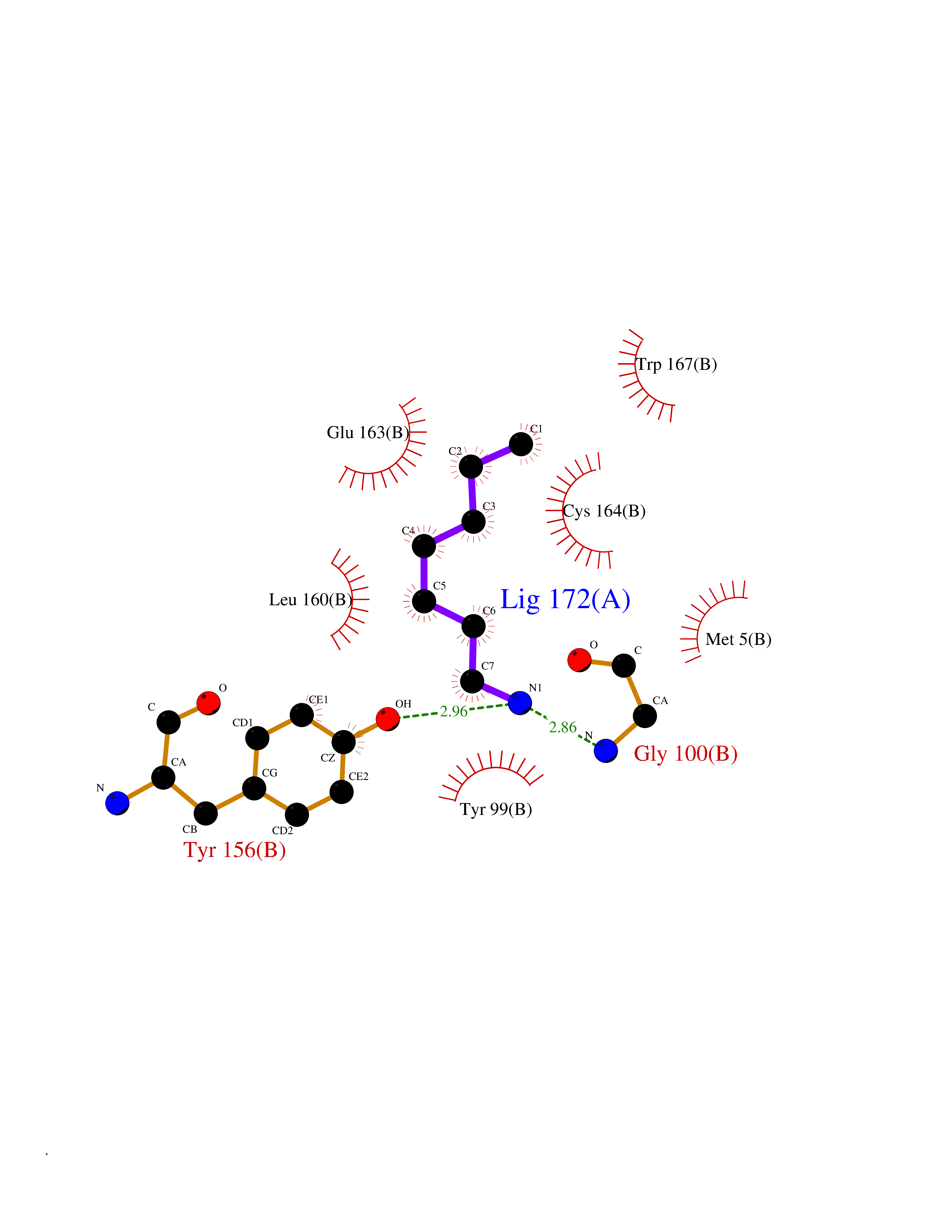 Ligplot Map 3D Binding mode Sequence QLSPFPFDLGPHSMRYYETATSRRGLGEPRYTSVGYVDDKEFVRFDSDARITQVAKGQEQWFRVNLRTLLGYYNQSAGGTHTLQRMYGCDVGSDGRLLRGYEQFAYDGCDYIALNEDLRTWTAADMAAQITRRKWEQAGAAEYYRAYLEGECVEWLHR Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Riboflavin kinase | 1NB0 | 5.01 | |
Target general information Gen name RFK Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Transferase Function ATP binding.Metal ion binding.Riboflavin kinase activity. Related diseases Glutaric aciduria 1 (GA1) [MIM:231670]: An autosomal recessive metabolic disorder characterized by progressive dystonia and athetosis due to gliosis and neuronal loss in the basal ganglia. {ECO:0000269|PubMed:14707522, ECO:0000269|PubMed:18775954, ECO:0000269|PubMed:24973495, ECO:0000269|PubMed:8541831, ECO:0000269|PubMed:8900227, ECO:0000269|PubMed:8900228, ECO:0000269|PubMed:9600243, ECO:0000269|PubMed:9711871}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03247; DB00140 Interacts with Q9NXG0-2; P19438; P19438-1 EC number 2.7.1.26 Uniprot keywords 3D-structure; ATP-binding; Cytoplasm; Flavoprotein; FMN; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Proteomics identification; Reference proteome; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 16749.9 Length 147 Aromaticity 0.12 Instability index 41.55 Isoelectric point 7.09 Charge (pH=7) 0.12 2D Binding mode Binding energy (Kcal/mol) -6.83  Molscript Map 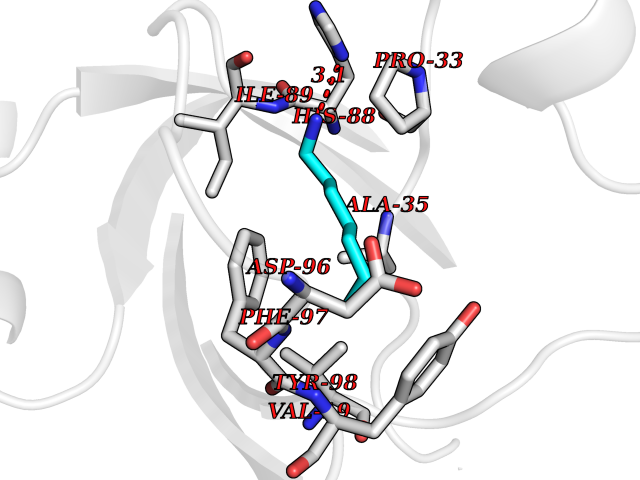 Pymol Map 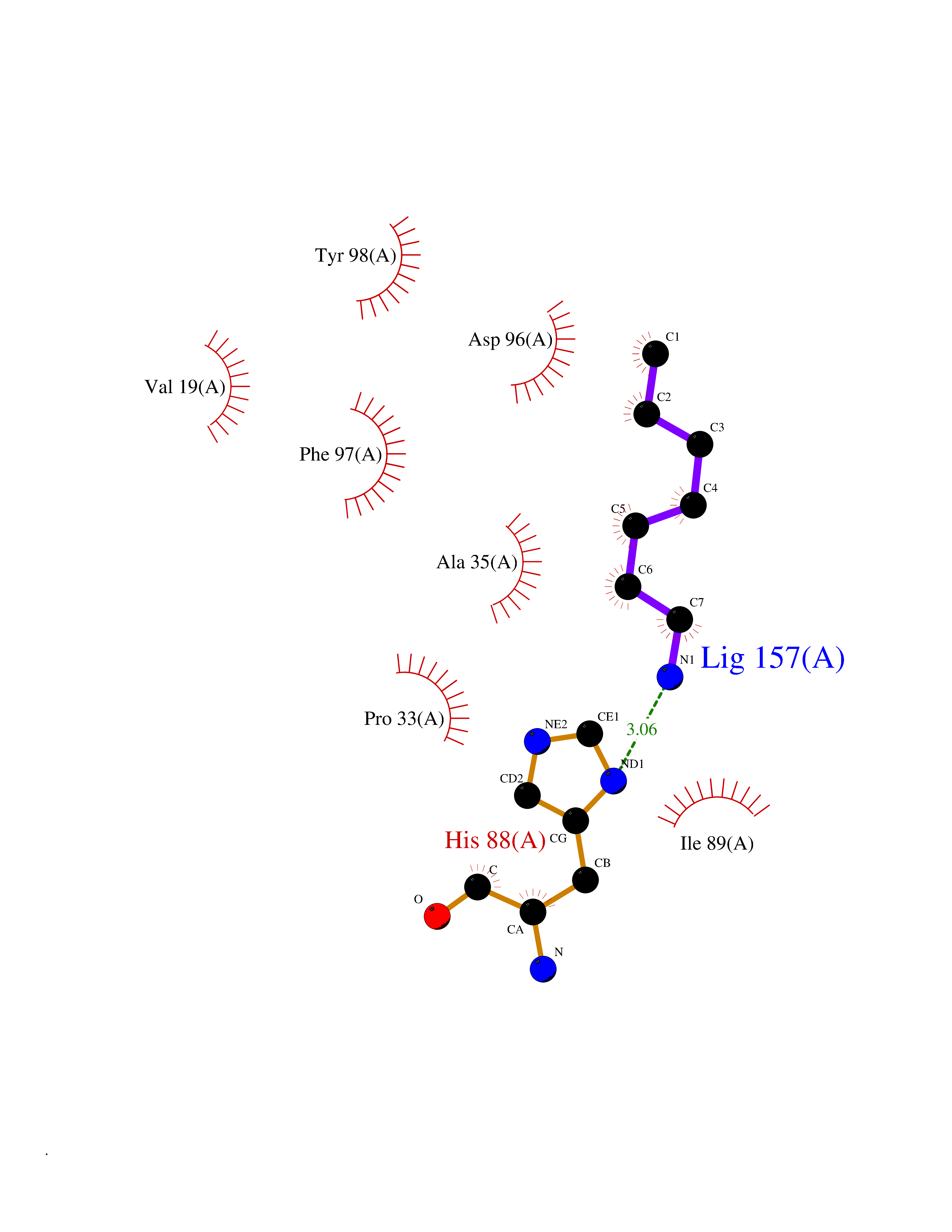 Ligplot Map 3D Binding mode Sequence RHLPYFCRGQVVRGFGRGSKQLGIPTANFPEQVVDNLPADISTGIYYGWASVGSGDVHKMVVSIGWNPYYKNTKKSMETHIMHTFKEDFYGEILNVAIVGYLRPEKNFDSLESLISAIQGDIEEAKKRLELPEYLKIKEDNFFQVSK Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Phosphodiesterase 7A (PDE7A) | 1ZKL | 5.01 | |
Target general information Gen name PDE7A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms TM22; High affinity cAMPspecific 3',5'cyclic phosphodiesterase 7A; High affinity cAMP-specific 3',5'-cyclic phosphodiesterase 7A Protein family Cyclic nucleotide phosphodiesterase family, PDE7 subfamily Biochemical class Phosphoric diester hydrolase Function May have a role in muscle signal transduction. Hydrolyzes the second messenger cAMP, which is a key regulator of many important physiological processes. Related diseases Timothy syndrome (TS) [MIM:601005]: Disorder characterized by multiorgan dysfunction including lethal arrhythmias, webbing of fingers and toes, congenital heart disease, immune deficiency, intermittent hypoglycemia, cognitive abnormalities and autism. {ECO:0000269|PubMed:15454078, ECO:0000269|PubMed:15863612, ECO:0000269|PubMed:25260352, ECO:0000269|PubMed:26253506, ECO:0000269|PubMed:30023270, ECO:0000269|PubMed:30172029, ECO:0000269|PubMed:31430211}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 3 (BRGDA3) [MIM:611875]: A heart disease characterized by the association of Brugada syndrome with shortened QT intervals. Brugada syndrome is a tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:17224476}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Long QT syndrome 8 (LQT8) [MIM:618447]: A form of long QT syndrome, a heart disorder characterized by a prolonged QT interval on the ECG and polymorphic ventricular arrhythmias. They cause syncope and sudden death in response to exercise or emotional stress, and can present with a sentinel event of sudden cardiac death in infancy. LQT8 transmission pattern is consistent with autosomal dominant inheritance with incomplete penetrance. {ECO:0000269|PubMed:23677916, ECO:0000269|PubMed:24728418, ECO:0000269|PubMed:25633834, ECO:0000269|PubMed:30345660}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurodevelopmental disorder with hypotonia, language delay, and skeletal defects with or without seizures (NEDHLSS) [MIM:620029]: An autosomal dominant disorder characterized by global developmental delay apparent from infancy, intellectual disability, poor or absent speech, behavioral abnormalities, and hypotonia with delayed walking or inability to walk. Additional features include epilepsy, mild skeletal defects, and non-specific dysmorphic features. {ECO:0000269|PubMed:30513141, ECO:0000269|PubMed:34163037}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08602; DB07954; DB00201; DB09283 Interacts with NA EC number EC 3.1.4.53 Uniprot keywords 3D-structure; Alternative splicing; cAMP; Cytoplasm; Hydrolase; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 36692.5 Length 317 Aromaticity 0.1 Instability index 38.24 Isoelectric point 6.47 Charge (pH=7) -3.52 2D Binding mode Binding energy (Kcal/mol) -6.84 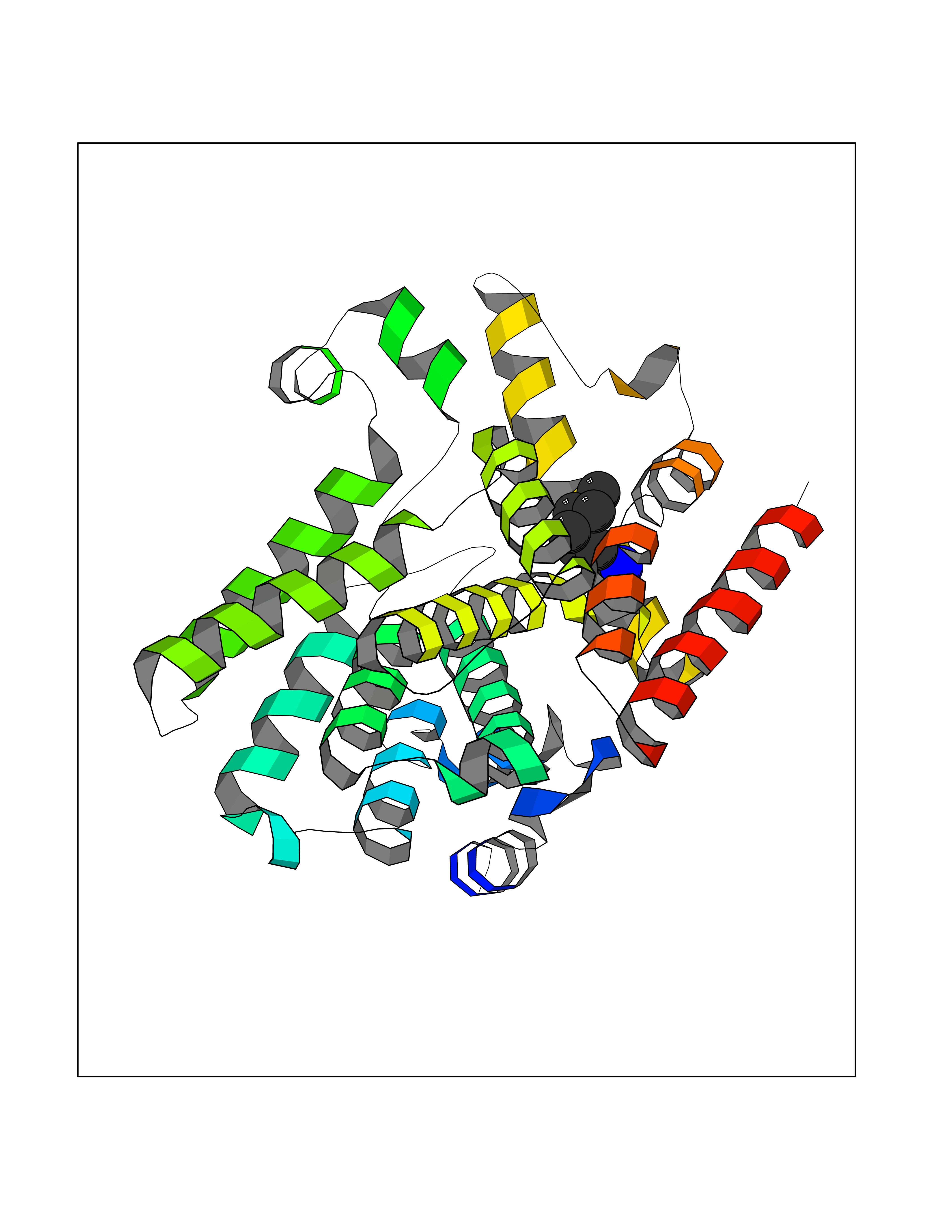 Molscript Map 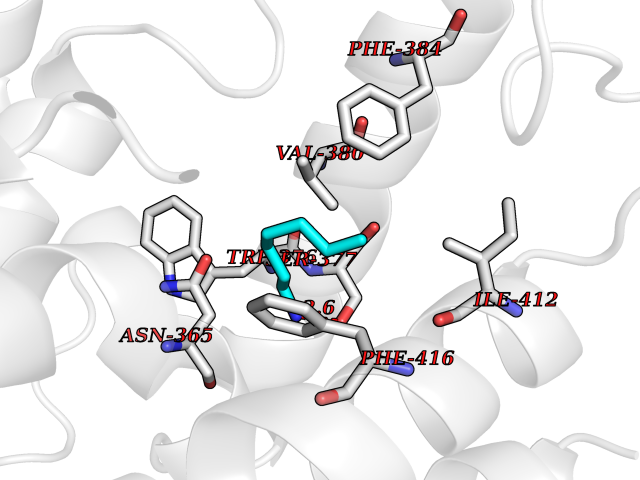 Pymol Map 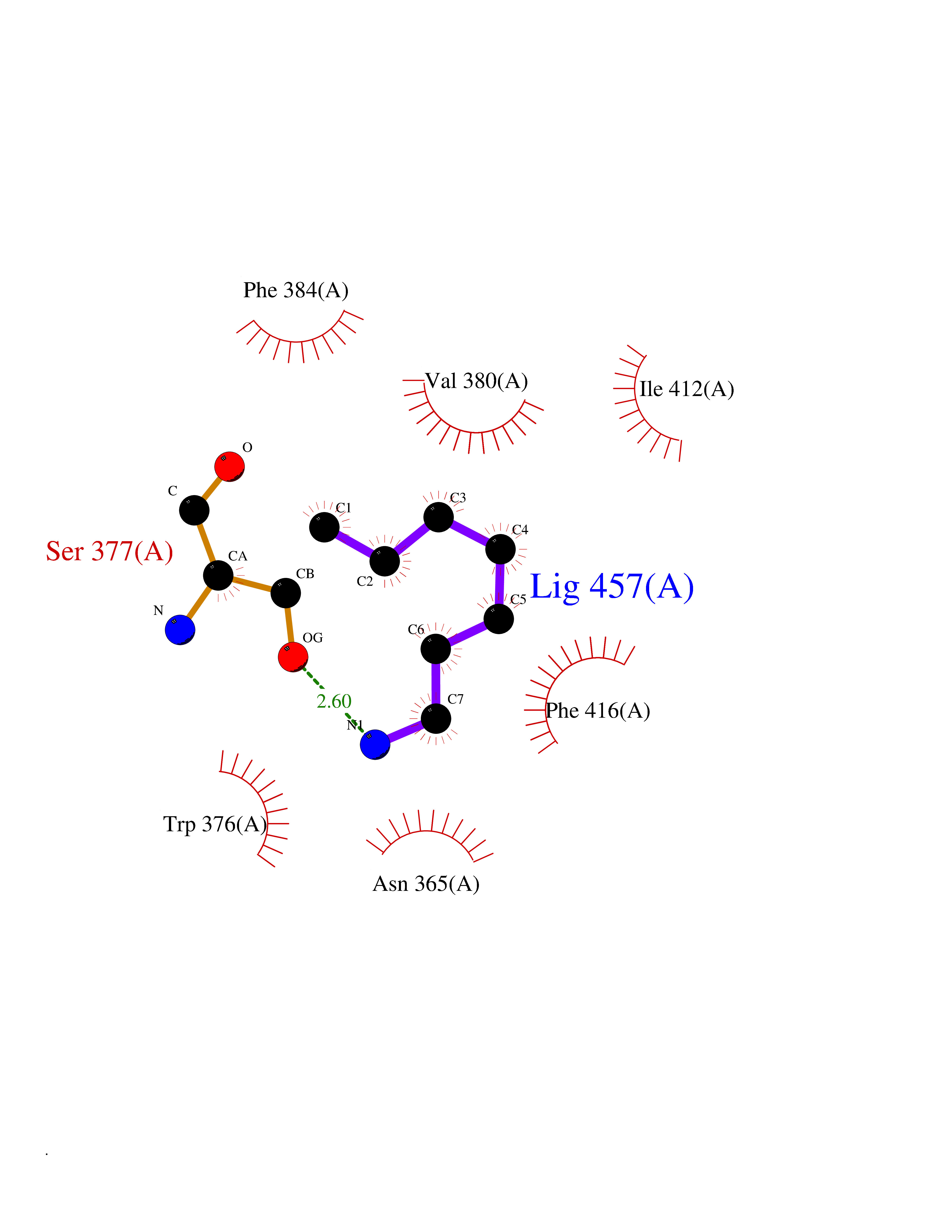 Ligplot Map 3D Binding mode Sequence DYNGQAKCMLEKVGNWNFDIFLFDRLTNGNSLVSLTFHLFSLHGLIEYFHLDMMKLRRFLVMIQEDYHSQNPYHNAVHAADVTQAMHCYLKEPKLANSVTPWDILLSLIAAATHDLDHPGVNQPFLIKTNHYLATLYKNTSVLENHHWRSAVGLLRESGLFSHLPLESRQQMETQIGALILATDISRQNEYLSLFRSHLDRGDLCLEDTRHRHLVLQMALKCADICNPCRTWELSKQWSEKVTEEFFHQGDIEKKYHLGVSPLCDRHTESIANIQIGFMTYLVEPLFTEWARFSNTRLSQTMLGHVGLNKASWKGLQ Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Monoglyceride lipase (MAGL) | 3PE6 | 5.01 | |
Target general information Gen name MGLL Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Monoacylglycerol lipase; MGL; Lysophospholipaselike; Lysophospholipase-like; Lysophospholipase homolog; HUK5; HU-K5 Protein family AB hydrolase superfamily, Monoacylglycerol lipase family Biochemical class Carboxylic ester hydrolase Function Hydrolyzes the endocannabinoid 2-arachidonoylglycerol, and thereby contributes to the regulation of endocannabinoid signaling, nociperception and perception of pain. Regulates the levels of fatty acids that serve as signaling molecules and promote cancer cell migration, invasion and tumor growth. Converts monoacylglycerides to free fatty acids and glycerol. Related diseases Systemic lupus erythematosus 9 (SLEB9) [MIM:610927]: A chronic, relapsing, inflammatory, and often febrile multisystemic disorder of connective tissue, characterized principally by involvement of the skin, joints, kidneys and serosal membranes. It is of unknown etiology, but is thought to represent a failure of the regulatory mechanisms of the autoimmune system. The disease is marked by a wide range of system dysfunctions, an elevated erythrocyte sedimentation rate, and the formation of LE cells in the blood or bone marrow. {ECO:0000269|PubMed:17360460}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Immunodeficiency, common variable, 7 (CVID7) [MIM:614699]: A primary immunodeficiency characterized by antibody deficiency, hypogammaglobulinemia, recurrent bacterial infections and an inability to mount an antibody response to antigen. The defect results from a failure of B-cell differentiation and impaired secretion of immunoglobulins; the numbers of circulating B-cells is usually in the normal range, but can be low. {ECO:0000269|PubMed:22035880}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07550; P37235 EC number EC 3.1.1.23 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Lipid biosynthesis; Lipid degradation; Lipid metabolism; Membrane; Nitration; Phosphoprotein; Proteomics identification; Reference proteome; Serine esterase Protein physicochemical properties Chain ID A Molecular weight (Da) 31808.4 Length 289 Aromaticity 0.08 Instability index 29.7 Isoelectric point 6.73 Charge (pH=7) -0.91 2D Binding mode Binding energy (Kcal/mol) -6.83 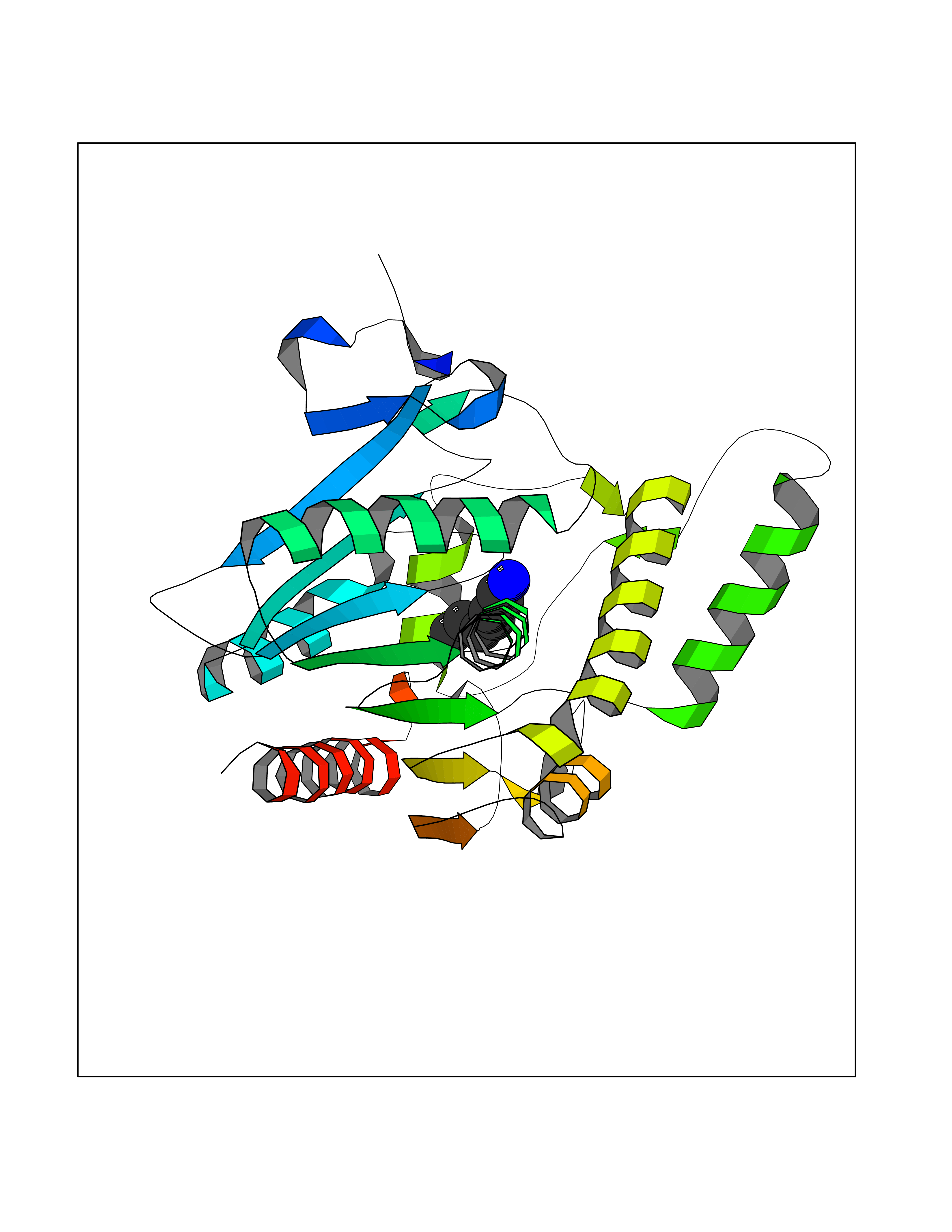 Molscript Map 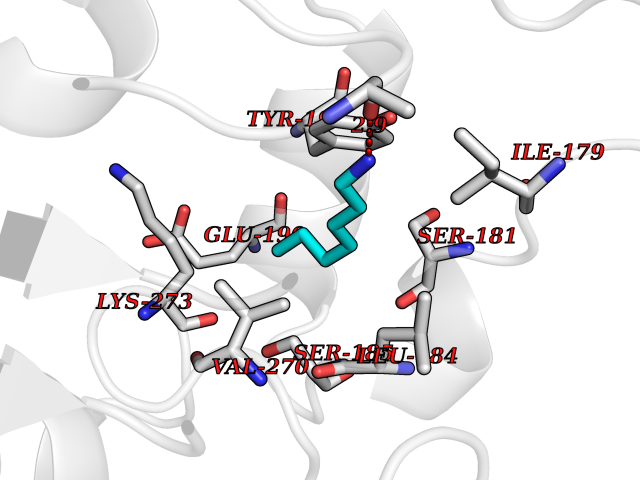 Pymol Map 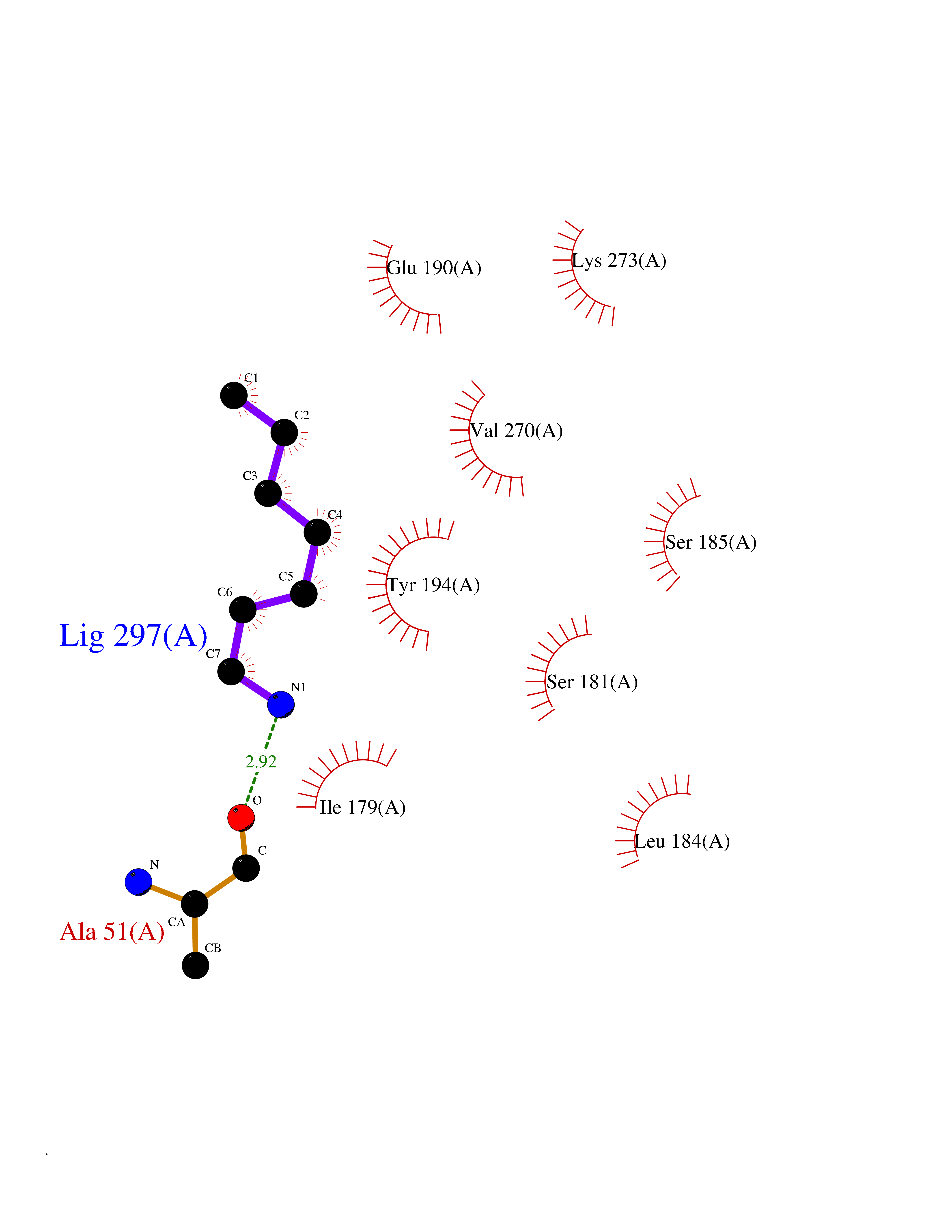 Ligplot Map 3D Binding mode Sequence PRRTPQSIPYQDLPHLVNADGQYLFCRYWAPTGTPKALIFVSHGAGEHSGRYEELARMLMGLDLLVFAHDHVGHGQSEGERMVVSDFHVFVRDVLQHVDSMQKDYPGLPVFLLGHSMGGAIAILTAAERPGHFAGMVLISPLVLANPESATTFKVLAAKVLNSVLPNLSSGPIDSSVLSRNKTEVDIYNSDPLICRAGLKVCFGIQLLNAVSRVERALPKLTVPFLLLQGSADRLCDSKGAYLLMELAKSQDKTLKIYEGAYHVLHKELPEVTNSVFHEINMWVSQRTA Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Bromodomain-containing protein 1 (BRD1) | 5FG6 | 5.01 | |
Target general information Gen name BRD1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Bromodomain and PHD finger-containing protein 2; BRPF2; BRL; BR140-like protein Protein family NA Biochemical class Bromodomain Function Component of the MOZ/MORF complex which has a histone H3 acetyltransferase activity. Related diseases Lynch syndrome 2 (LYNCH2) [MIM:609310]: A form of Lynch syndrome, an autosomal dominant disease associated with marked increase in cancer susceptibility. It is characterized by a familial predisposition to early-onset colorectal carcinoma (CRC) and extra-colonic tumors of the gastrointestinal, urological and female reproductive tracts. Lynch syndrome is reported to be the most common form of inherited colorectal cancer in the Western world. Clinically, it is often divided into two subgroups. Type I is characterized by hereditary predisposition to colorectal cancer, a young age of onset, and carcinoma observed in the proximal colon. Type II is characterized by increased risk for cancers in certain tissues such as the uterus, ovary, breast, stomach, small intestine, skin, and larynx in addition to the colon. Diagnosis of classical Lynch syndrome is based on the Amsterdam criteria: 3 or more relatives affected by colorectal cancer, one a first degree relative of the other two; 2 or more generation affected; 1 or more colorectal cancers presenting before 50 years of age; exclusion of hereditary polyposis syndromes. The term 'suspected Lynch syndrome' or 'incomplete Lynch syndrome' can be used to describe families who do not or only partially fulfill the Amsterdam criteria, but in whom a genetic basis for colon cancer is strongly suspected. {ECO:0000269|PubMed:10323887, ECO:0000269|PubMed:10375096, ECO:0000269|PubMed:10386556, ECO:0000269|PubMed:10413423, ECO:0000269|PubMed:10480359, ECO:0000269|PubMed:10598809, ECO:0000269|PubMed:10627141, ECO:0000269|PubMed:10660333, ECO:0000269|PubMed:10671064, ECO:0000269|PubMed:10713887, ECO:0000269|PubMed:10777691, ECO:0000269|PubMed:10882759, ECO:0000269|PubMed:11139242, ECO:0000269|PubMed:11427529, ECO:0000269|PubMed:11726306, ECO:0000269|PubMed:11748856, ECO:0000269|PubMed:11754112, ECO:0000269|PubMed:11781295, ECO:0000269|PubMed:11793442, ECO:0000269|PubMed:11839723, ECO:0000269|PubMed:11870161, ECO:0000269|PubMed:12095971, ECO:0000269|PubMed:12132870, ECO:0000269|PubMed:12200596, ECO:0000269|PubMed:12362047, ECO:0000269|PubMed:12373605, ECO:0000269|PubMed:12655562, ECO:0000269|PubMed:12658575, ECO:0000269|PubMed:14635101, ECO:0000269|PubMed:14961575, ECO:0000269|PubMed:15064764, ECO:0000269|PubMed:15139004, ECO:0000269|PubMed:15365995, ECO:0000269|PubMed:15365996, ECO:0000269|PubMed:16083711, ECO:0000269|PubMed:16451135, ECO:0000269|PubMed:17301300, ECO:0000269|PubMed:17510385, ECO:0000269|PubMed:18561205, ECO:0000269|PubMed:20020535, ECO:0000269|PubMed:21120944, ECO:0000269|PubMed:22753075, ECO:0000269|PubMed:7757073, ECO:0000269|PubMed:8566964, ECO:0000269|PubMed:8571956, ECO:0000269|PubMed:8574961, ECO:0000269|PubMed:8797773, ECO:0000269|PubMed:8872463, ECO:0000269|PubMed:8993976, ECO:0000269|PubMed:9048925, ECO:0000269|PubMed:9067757, ECO:0000269|PubMed:9218993, ECO:0000269|PubMed:9272156, ECO:0000269|PubMed:9298827, ECO:0000269|PubMed:9311737, ECO:0000269|PubMed:9326924, ECO:0000269|PubMed:9399661, ECO:0000269|PubMed:9559627, ECO:0000269|PubMed:9718327, ECO:0000269|PubMed:9833759, ECO:0000269|PubMed:9927034, ECO:0000269|Ref.5}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mismatch repair cancer syndrome 1 (MMRCS1) [MIM:276300]: An autosomal recessive form of mismatch repair cancer syndrome, a childhood cancer predisposition syndrome encompassing a broad tumor spectrum. This includes hematological malignancies, central nervous system tumors, Lynch syndrome-associated malignancies such as colorectal tumors as well as multiple intestinal polyps, embryonic tumors and rhabdomyosarcoma. Multiple cafe-au-lait macules, a feature reminiscent of neurofibromatosis type 1, are often found as first manifestation of the underlying cancer. {ECO:0000269|PubMed:11427529, ECO:0000269|PubMed:17440981, ECO:0000269|PubMed:7661930}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Muir-Torre syndrome (MRTES) [MIM:158320]: Rare autosomal dominant disorder characterized by sebaceous neoplasms and visceral malignancy. {ECO:0000269|PubMed:8751876}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in MLH1 may contribute to lobular carcinoma in situ (LCIS), a non-invasive neoplastic disease of the breast.; DISEASE: Endometrial cancer (ENDMC) [MIM:608089]: A malignancy of endometrium, the mucous lining of the uterus. Most endometrial cancers are adenocarcinomas, cancers that begin in cells that make and release mucus and other fluids. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Some epigenetic changes can be transmitted unchanged through the germline (termed 'epigenetic inheritance'). Evidence that this mechanism occurs in humans is provided by the identification of individuals in whom 1 allele of the MLH1 gene is epigenetically silenced throughout the soma (implying a germline event). These individuals are affected by Lynch syndrome but does not have identifiable mutations in MLH1, even though it is silenced, which demonstrates that an epimutation can phenocopy a genetic disease.; DISEASE: Colorectal cancer (CRC) [MIM:114500]: A complex disease characterized by malignant lesions arising from the inner wall of the large intestine (the colon) and the rectum. Genetic alterations are often associated with progression from premalignant lesion (adenoma) to invasive adenocarcinoma. Risk factors for cancer of the colon and rectum include colon polyps, long-standing ulcerative colitis, and genetic family history. {ECO:0000269|PubMed:10598809, ECO:0000269|PubMed:10882759, ECO:0000269|PubMed:12132870, ECO:0000269|PubMed:12655564, ECO:0000269|PubMed:14504054, ECO:0000269|PubMed:15184898, ECO:0000269|PubMed:18033691, ECO:0000269|PubMed:8872463, ECO:0000269|PubMed:9032648, ECO:0000269|PubMed:9087566, ECO:0000269|PubMed:9611074}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q6NVI2; Q8NHQ1; Q9NRI5-2; Q6NT76; P43364; P23508; Q9NRD5; Q92622; Q9UBB9; Q9UGI0; Q86U86; Q86U86 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Bromodomain; Chromatin regulator; Chromosome; Erythrocyte maturation; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 13688.5 Length 118 Aromaticity 0.08 Instability index 48.3 Isoelectric point 5.92 Charge (pH=7) -1.96 2D Binding mode Binding energy (Kcal/mol) -6.83 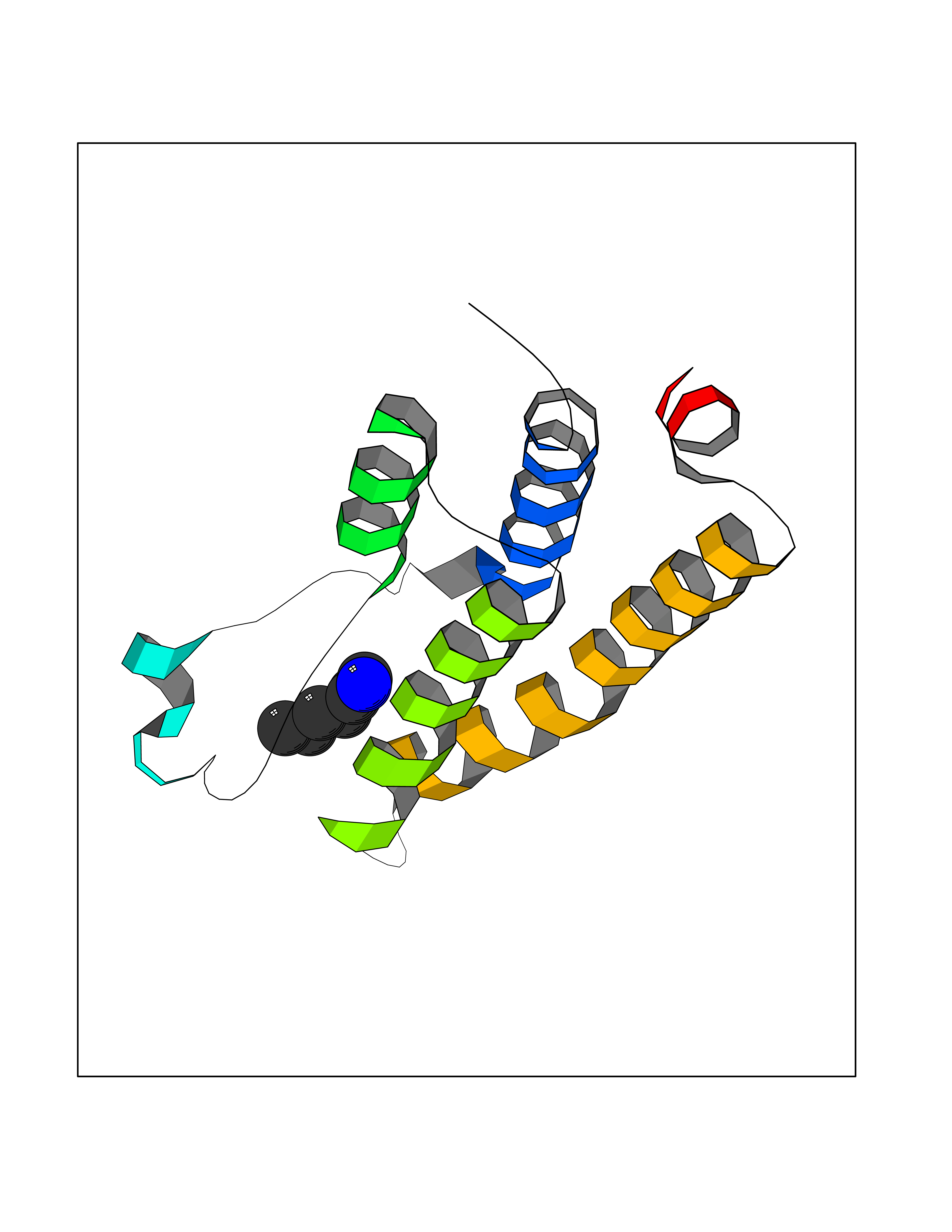 Molscript Map 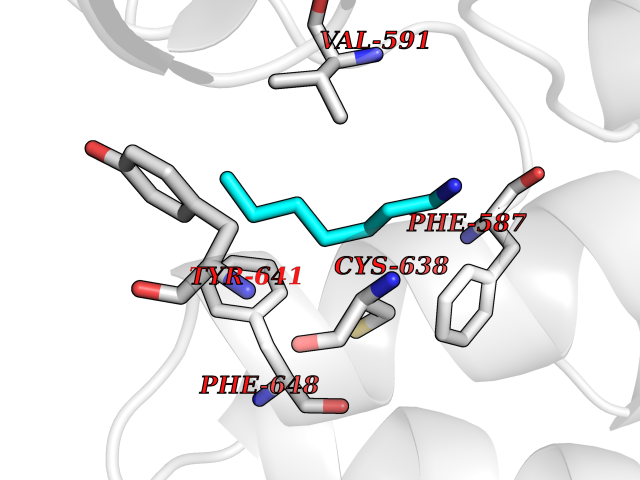 Pymol Map 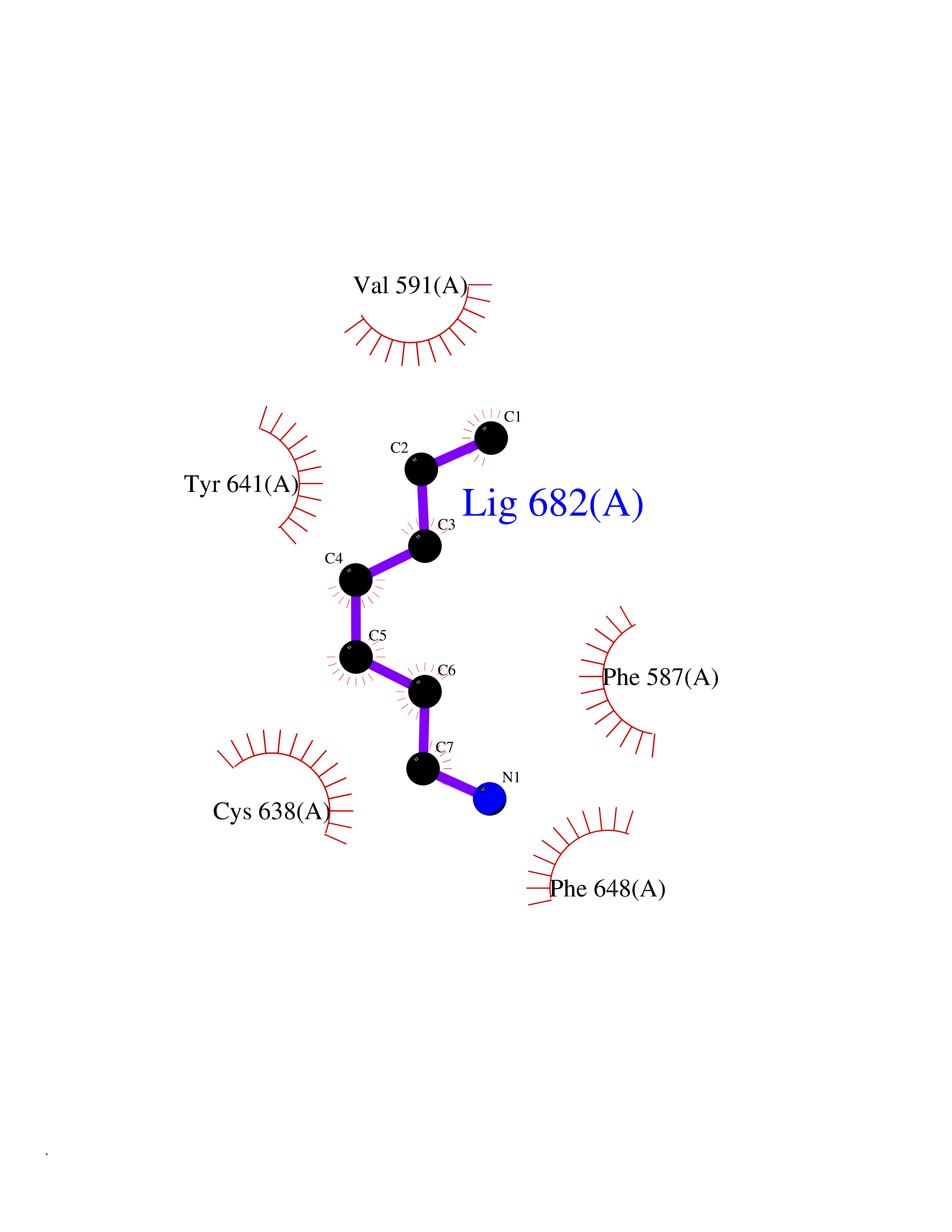 Ligplot Map 3D Binding mode Sequence RLTPLTVLLRSVLDQLQDKDPARIFAQPVSLKEVPDYLDHIKHPMDFATMRKRLEAQGYKNLHEFEEDFDLIIDNCMKYNARDTVFYRAAVRLRDQGGVVLRQARREVDSIGLEEASG Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Phenylethanolamine N-methyltransferase (PNMT) | 2G72 | 5.01 | |
Target general information Gen name PNMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNMTase; PENT; Noradrenaline N-methyltransferase Protein family Class I-like SAM-binding methyltransferase superfamily, NNMT/PNMT/TEMT family Biochemical class NA Function Converts noradrenaline to adrenaline. Related diseases A chromosomal aberration involving TRIM24/TIF1 is found in papillary thyroid carcinomas (PTCs). Translocation t(7;10)(q32;q11) with RET. The translocation generates the TRIM24/RET (PTC6) oncogene. {ECO:0000269|PubMed:10439047}. Drugs (DrugBank ID) DB08129; DB08128; DB07739; DB07798; DB07747; DB03468; DB08550; DB03824; DB04273; DB07906; DB07597; DB09571; DB00968; DB08631; DB01752; DB08654 Interacts with Q9P2G9-2; Q8TBB1 EC number EC 2.1.1.28 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 29198.9 Length 264 Aromaticity 0.09 Instability index 54.33 Isoelectric point 5.91 Charge (pH=7) -3.69 2D Binding mode Binding energy (Kcal/mol) -6.84  Molscript Map 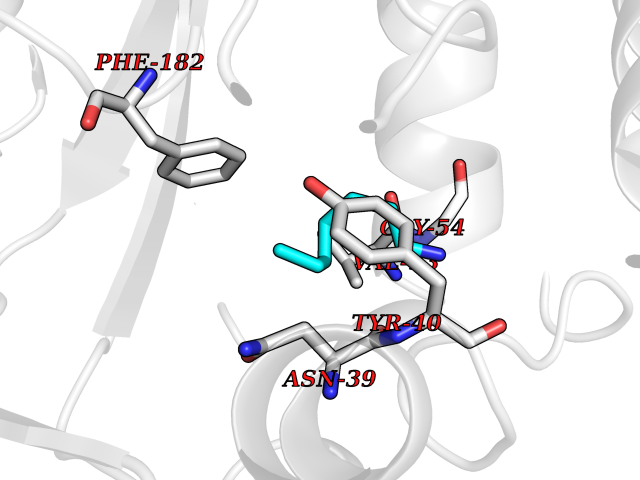 Pymol Map 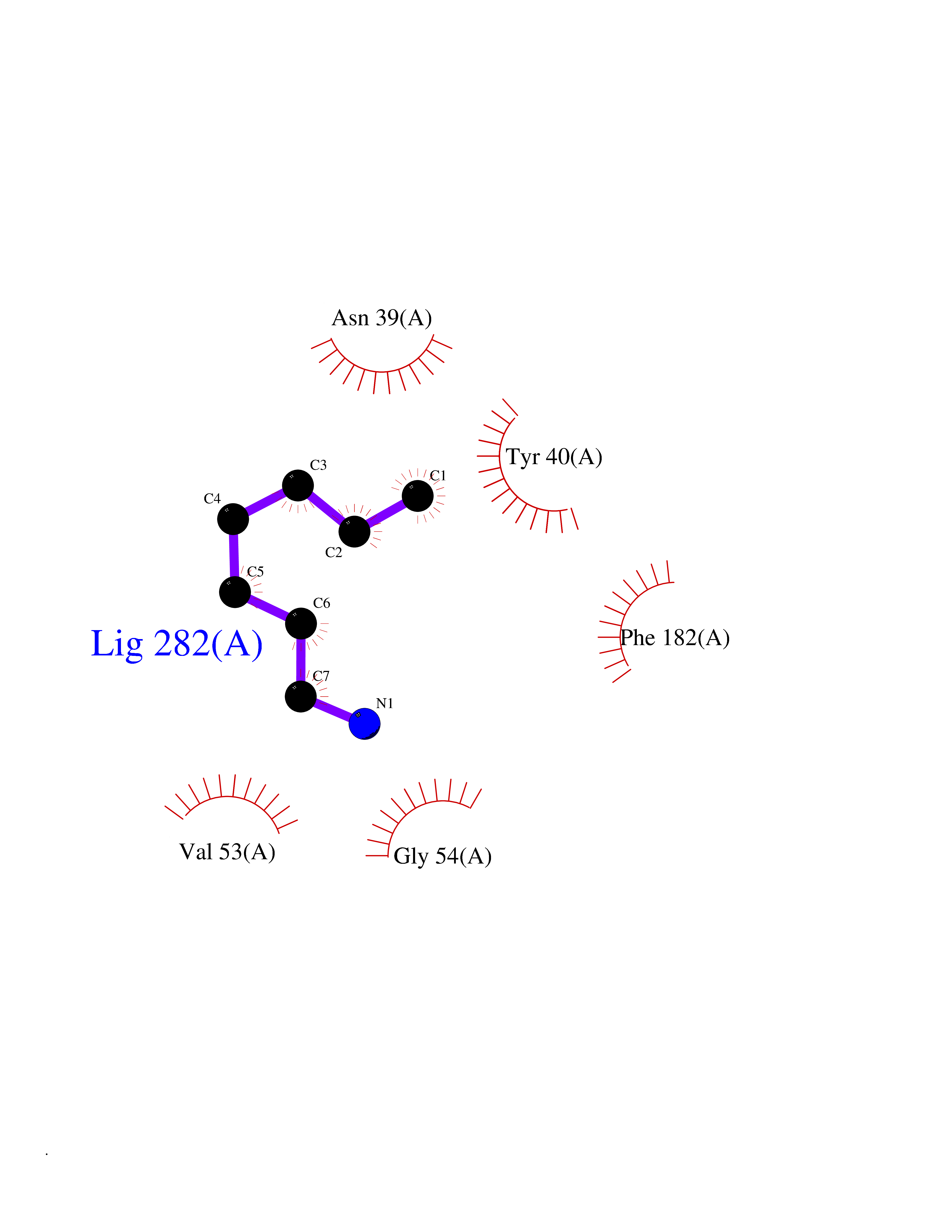 Ligplot Map 3D Binding mode Sequence APGQAAVASAYQRFEPRAYLRNNYAPPRGDLCNPNGVGPWKLRCLAQTFATGEVSGRTLIDIGSGPTVYQLLSACSHFEDITMTDFLEVNRQELGRWLQEEPGAFNWSMYSQHACLIEGKGECWQDKERQLRARVKRVLPIDVHQPQPLGAGSPAPLPADALVSAFCLEAVSPDLASFQRALDHITTLLRPGGHLLLIGALEESWYLAGEARLTVVPVSEEEVREALVRSGYKVRDLRTYIMPAHLQTGVDDVKGVFFAWAQKV Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Neuronal acetylcholine receptor alpha-2 (CHRNA2) | 5FJV | 5.01 | |
Target general information Gen name CHRNA2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CHRNA2 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-2/CHRNA2 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane. Related diseases Epilepsy, nocturnal frontal lobe, 4 (ENFL4) [MIM:610353]: An autosomal dominant focal epilepsy characterized by nocturnal seizures associated with fear sensation, tongue movements, and nocturnal wandering, closely resembling nightmares and sleep walking. {ECO:0000269|PubMed:16826524}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Seizures, benign familial infantile, 6 (BFIS6) [MIM:610353]: A form of benign familial infantile epilepsy, a neurologic disorder characterized by afebrile seizures occurring in clusters during the first year of life, without neurologic sequelae. BFIS6 inheritance is autosomal dominant. {ECO:0000269|PubMed:25847220}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00732; DB00237; DB00411; DB00565; DB01245; DB00514; DB01135; DB07720; DB00898; DB00472; DB00483; DB08960; DB00657; DB01336; DB00416; DB01226; DB00184; DB01337; DB01338; DB00721; DB00728; DB05740; DB00202; DB01199; DB01339 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 48323.4 Length 413 Aromaticity 0.15 Instability index 32 Isoelectric point 5.69 Charge (pH=7) -6.58 2D Binding mode Binding energy (Kcal/mol) -6.83 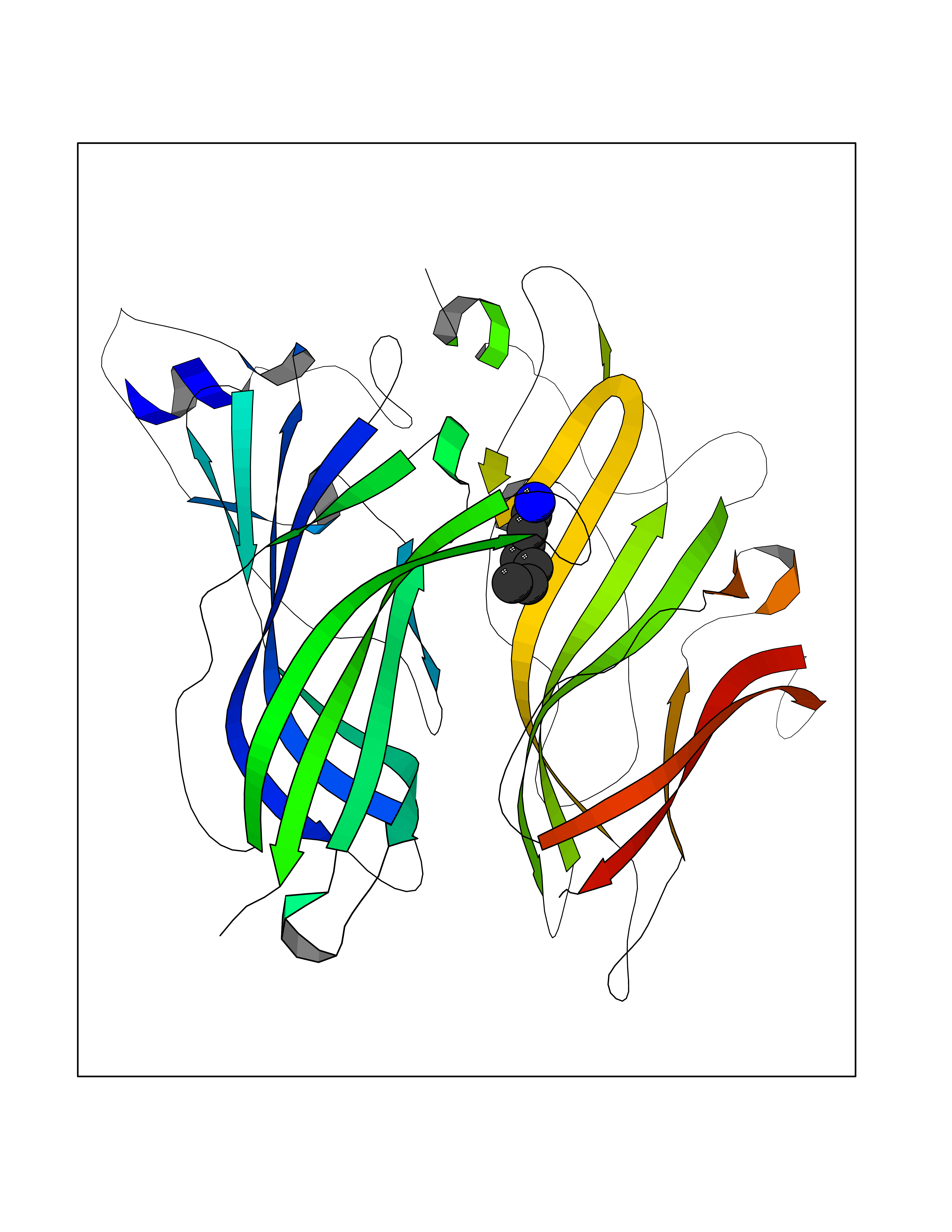 Molscript Map 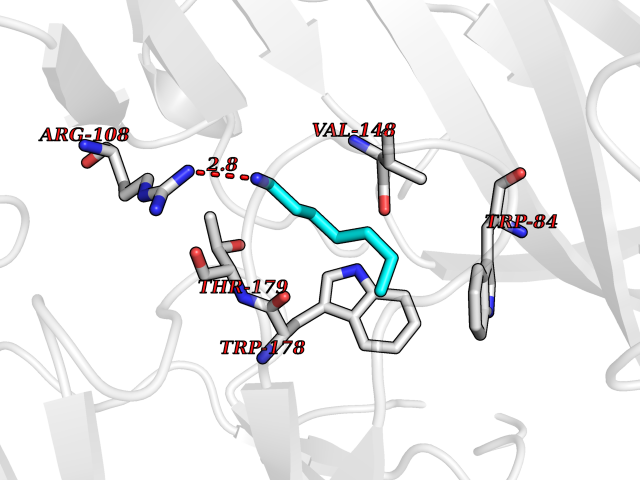 Pymol Map 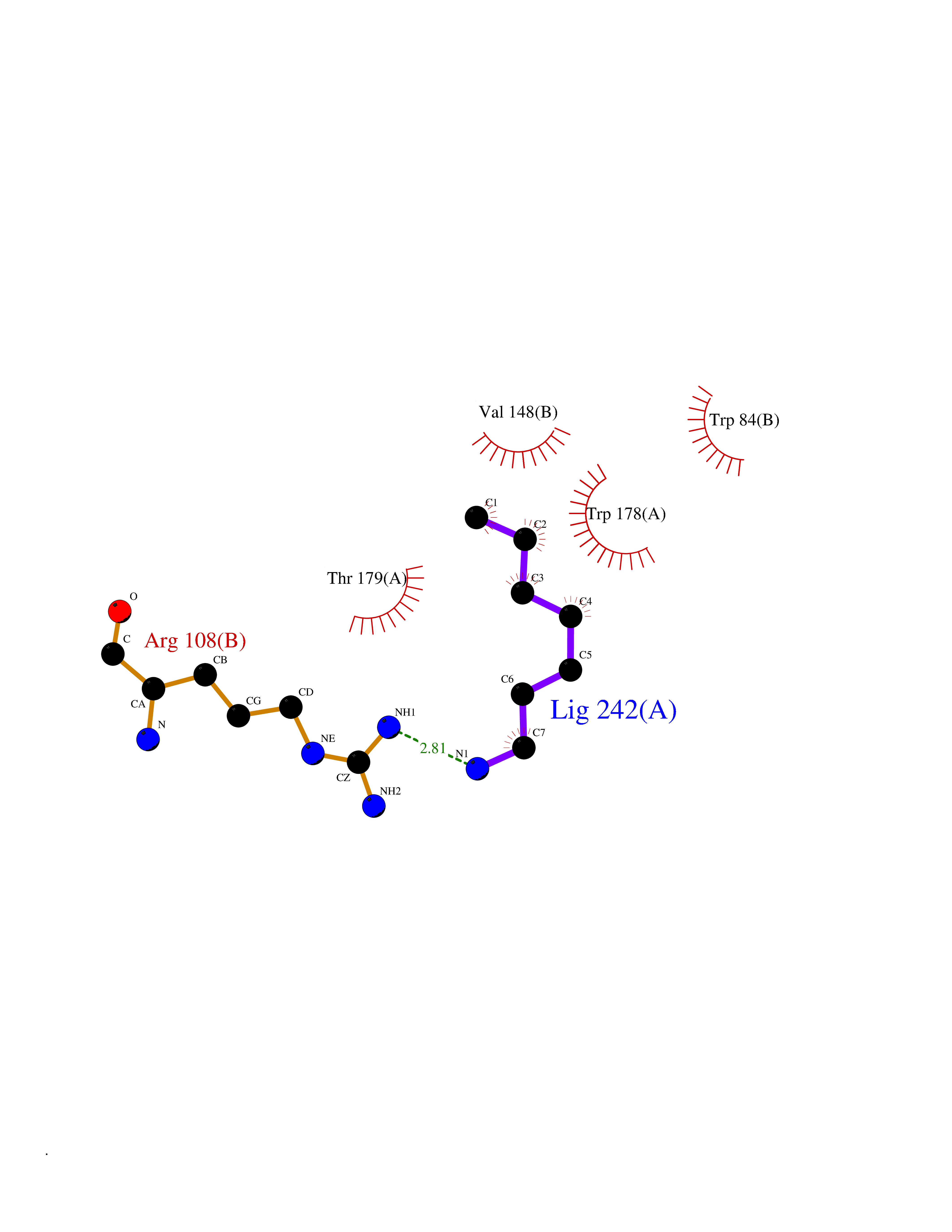 Ligplot Map 3D Binding mode Sequence DRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLP Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | ERK activator kinase 2 (MEK2) | 1S9I | 5.01 | |
Target general information Gen name MAP2K2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PRKMK2; MKK2; MEK 2; MAPKK 2; MAPK/ERK kinase 2; MAP kinase kinase 2; Dual specificity mitogenactivated protein kinase kinase 2; Dual specificity mitogen-activated protein kinase kinase 2 Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, MAP kinase kinase subfamily Biochemical class Kinase Function Activates the ERK1 and ERK2 MAP kinases. Catalyzes the concomitant phosphorylation of a threonine and a tyrosine residue in a Thr-Glu-Tyr sequence located in MAP kinases. Related diseases Cardiofaciocutaneous syndrome 4 (CFC4) [MIM:615280]: A form of cardiofaciocutaneous syndrome, a multiple congenital anomaly disorder characterized by a distinctive facial appearance, heart defects and intellectual disability. Heart defects include pulmonic stenosis, atrial septal defects and hypertrophic cardiomyopathy. Some affected individuals present with ectodermal abnormalities such as sparse, friable hair, hyperkeratotic skin lesions and a generalized ichthyosis-like condition. Typical facial features are similar to Noonan syndrome. They include high forehead with bitemporal constriction, hypoplastic supraorbital ridges, downslanting palpebral fissures, a depressed nasal bridge, and posteriorly angulated ears with prominent helices. {ECO:0000269|PubMed:16439621, ECO:0000269|PubMed:18042262, ECO:0000269|PubMed:20358587}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11967; DB06616; DB12010; DB14904; DB11689; DB08911 Interacts with P05067; P10398; Q96II5; P15056; O95273; Q12959; P61978-2; Q8IVT5; P00540; P04049 EC number EC 2.7.12.2 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Cardiomyopathy; Cytoplasm; Direct protein sequencing; Disease variant; Ectodermal dysplasia; Intellectual disability; Kinase; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 33960.9 Length 303 Aromaticity 0.07 Instability index 45.61 Isoelectric point 6.29 Charge (pH=7) -2.53 2D Binding mode Binding energy (Kcal/mol) -6.83 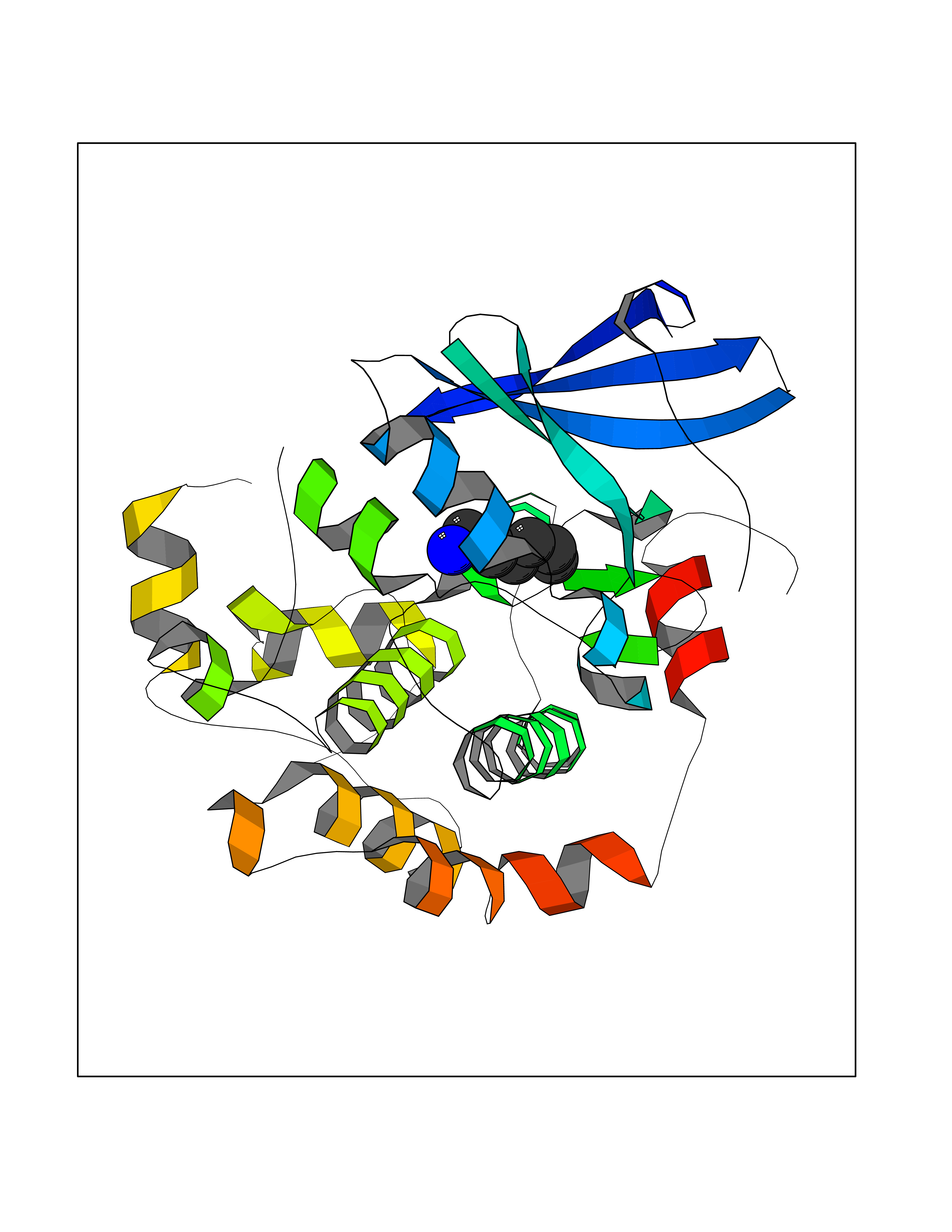 Molscript Map 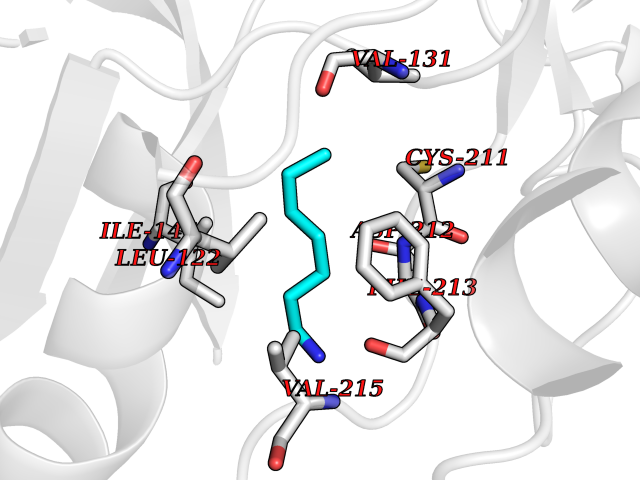 Pymol Map 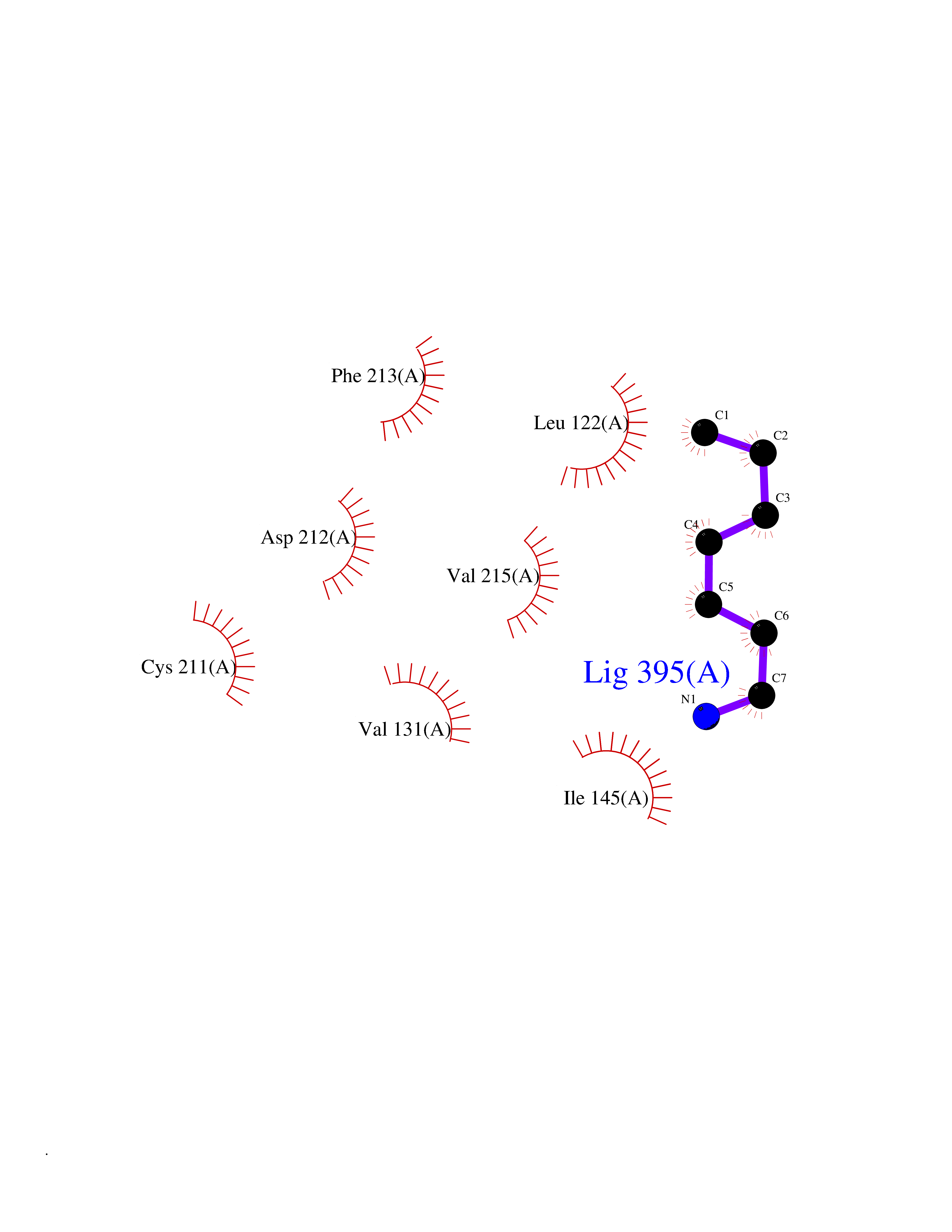 Ligplot Map 3D Binding mode Sequence QKAKVGELKDDDFERISELGAGNGGVVTKVQHRPSGLIMARKLIHLEIKPAIRNQIIRELQVLHECNSPYIVGFYGAFYSDGEISICMEHMDGGSLDQVLKEAKRIPEEILGKVSIAVLRGLAYLREKHQIMHRDVKPSNILVNSRGEIKLCDFGVSGQLIDSMVGTRSYMAPERLQGTHYSVQSDIWSMGLSLVELAVGRYPIPPPDAKELEAIFGRPVVDRPAMAIFELLDYIVNEPPPKLPNGVFTPDFQEFVNKCLIKNPAERADLKMLTNHTFIKRSEVEEVDFAGWLCKTLRLNQPG Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Histone-lysine N-methyltransferase (HLNM) | 3QOW | 5.01 | |
Target general information Gen name DOT1L Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine N-methyltransferase 4; KMT4; KIAA1814; Histone-lysine N-methyltransferase, H3 lysine-79 specific; Histone H3-K79 methyltransferase; H3-K79-HMTase; DOT1-like protein Protein family Class I-like SAM-binding methyltransferase superfamily, DOT1 family Biochemical class Methyltransferase Function Histone methyltransferase. Methylates 'Lys-79' of histone H3. Nucleosomes are preferred as substrate compared to free histones. Binds to DNA. Related diseases Defects in DOTL1 are associated with an autosomal dominant form of global developmental delay and intellectual disability, with or without one or more major congenital anomalies (PubMed:37827158). The patient phenotypes are characterized by central nervous system (CNS) dysfunction, such as mild motor delay and significant speech and language delay, and a range of congenital anomalies, including brain structural anomalies, cardiac defects, varied urogenital features and growth restriction (PubMed:37827158). Variants may cause a gain-of-function effect leading to an increase in cellular H3K79 methylation levels (PubMed:37827158). {ECO:0000269|PubMed:37827158}. Drugs (DrugBank ID) NA Interacts with Q03111; P42568 EC number EC 2.1.1.43 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Disease variant; DNA-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 37367.1 Length 322 Aromaticity 0.11 Instability index 32.71 Isoelectric point 6.03 Charge (pH=7) -5.25 2D Binding mode Binding energy (Kcal/mol) -6.84 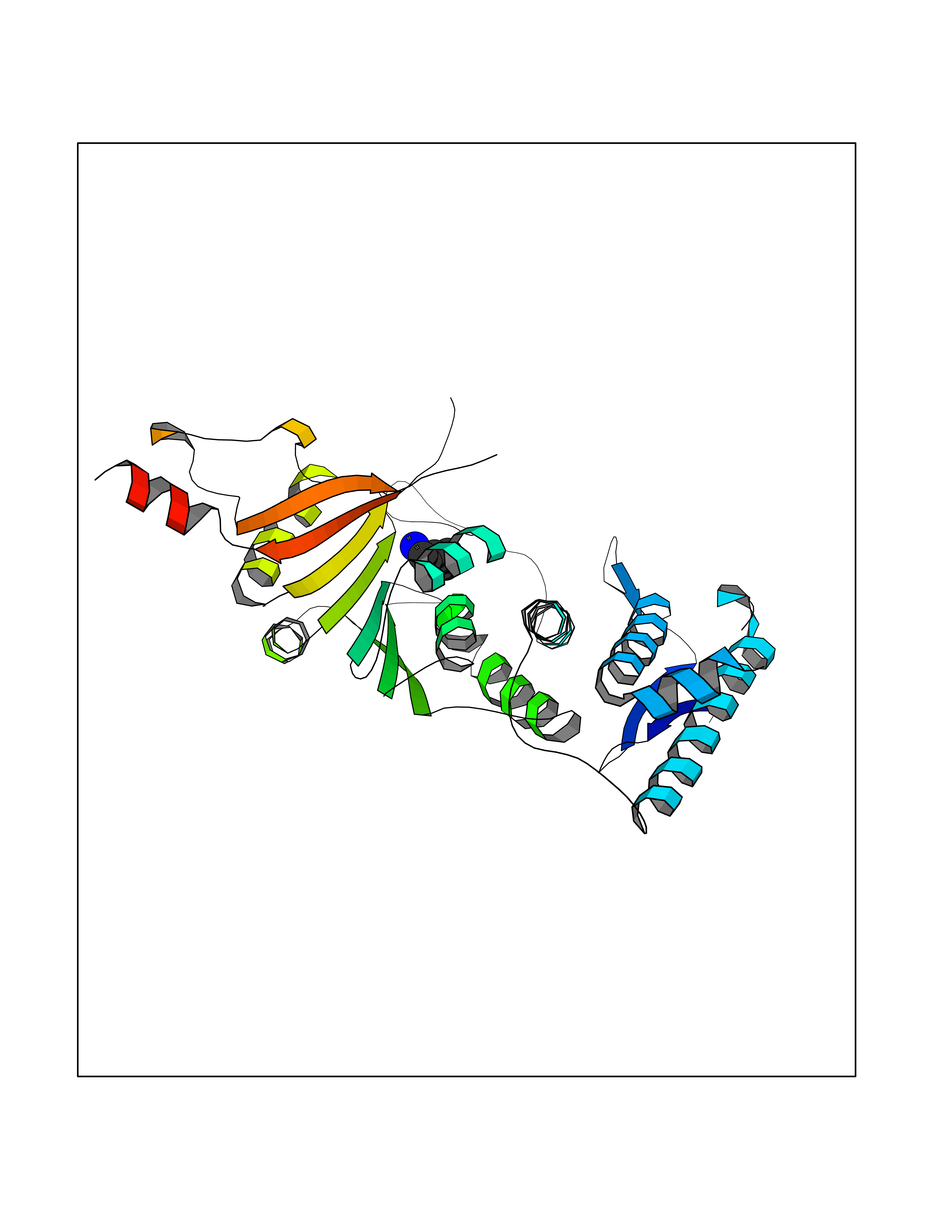 Molscript Map 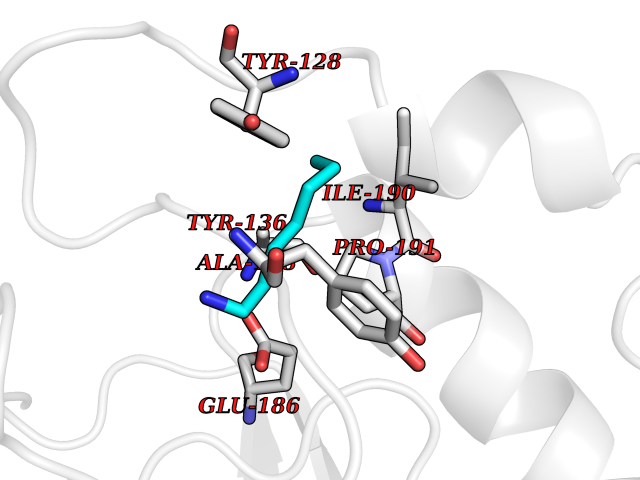 Pymol Map 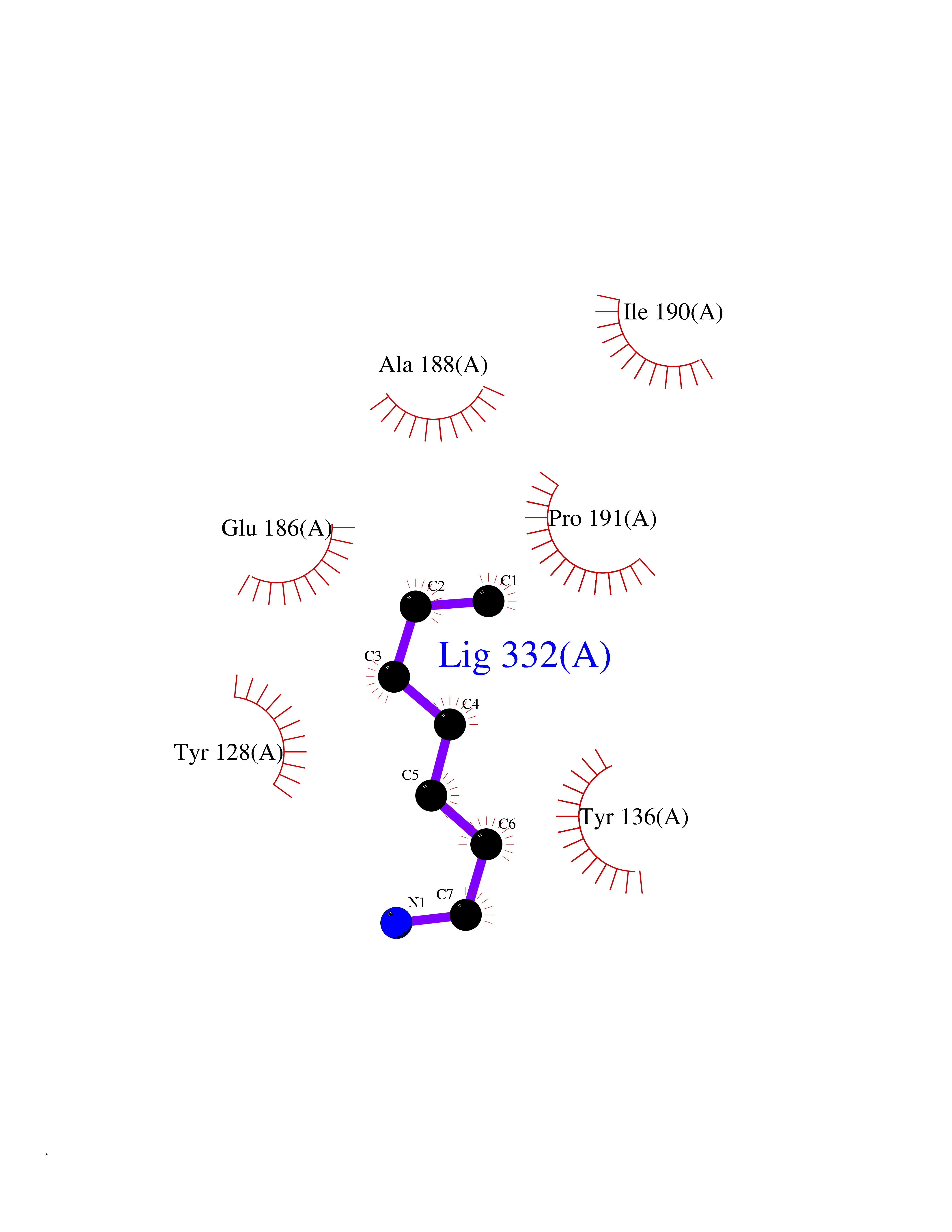 Ligplot Map 3D Binding mode Sequence KLELRLKSPVGAEPAVYPWPLPVYDKHHDAAHEIIETIRWVCEEIPDLKLAMENYLIDYDTKSFESMQRLCDKYNRAIDSIHQLWKGTTQPMKLNTRPSTGLLRHILQQVYNHSVTDPEKLNNYEPFSPEVYGETSFDLVAQMIDEIKMTDDDLFVDLGSGVGQVVLQVAAATNCKHHYGVEKADIPAKYAETMDREFRKWMKWYGKKHAEYTLERGDFLSEEWRERIANTSVIFVNNFAFGPEVDHQLKERFANMKEGGRIVSSKPFAPLNFRINSRNLSDIGTIMRVVELSPLKWTGKPVSYYLHTIDRTILENYFSSLK Hydrogen bonds contact Hydrophobic contact | ||||