Job Results:
Ligand
Structure
Job ID
d1681d1a563fba4481a5af742fb3bb0c
Job name
NA
Time
2026-02-27 11:58:22
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | 5,10-methylenetetrahydrofolate reductase | 3FST | 4.59 | |
Target general information Gen name metF Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b3941;JW3913 Protein family Methylenetetrahydrofolate reductase family Biochemical class Oxidoreductase Function FAD binding.Methylenetetrahydrofolate reductase (NAD(P)H) activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.5.1.54 Uniprot keywords 3D-structure; Amino-acid biosynthesis; FAD; Flavoprotein; Methionine biosynthesis; NAD; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A,C,E Molecular weight (Da) 30855.9 Length 274 Aromaticity 0.09 Instability index 27.54 Isoelectric point 5.84 Charge (pH=7) -4.61 2D Binding mode Binding energy (Kcal/mol) -6.26  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence FHASQRDALNQSLAEVQGQINVSFEFFPPRTSEMEQTLWNSIDRLSSLKPKFVSVTYTHSIIKGIKDRTGLEAAPHLTCIDATPDELRTIARDYWNNGIRHIVALRGDEMYASDLVTLLKEVADFDISVAAYPEVHPEAKSAQADLLNLKRKVDAGANRAITQFFFDVESYLRFRDRCVSAGIDVEIIPGILPVSNFKQAKKLADMTNVRIPAWMAQMFDGLDDDAETRKLVGANIAMDMVKILSREGVKDFHFYTLNRAEMSYAICHTLGVRP Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | 30S ribosomal protein S3 | 4ODQ | 4.59 | |
Target general information Gen name rpsC Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW3276;b3314 Protein family Universal ribosomal protein uS3 family Biochemical class Isomerase Function RRNA binding.Structural constituent of ribosome. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09093; DB12455; DB00759 Interacts with NA EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Reference proteome; Ribonucleoprotein; Ribosomal protein; RNA-binding; rRNA-binding Protein physicochemical properties Chain ID B Molecular weight (Da) 12457.7 Length 112 Aromaticity 0.05 Instability index 12.49 Isoelectric point 5.91 Charge (pH=7) -7.24 2D Binding mode Binding energy (Kcal/mol) -6.26 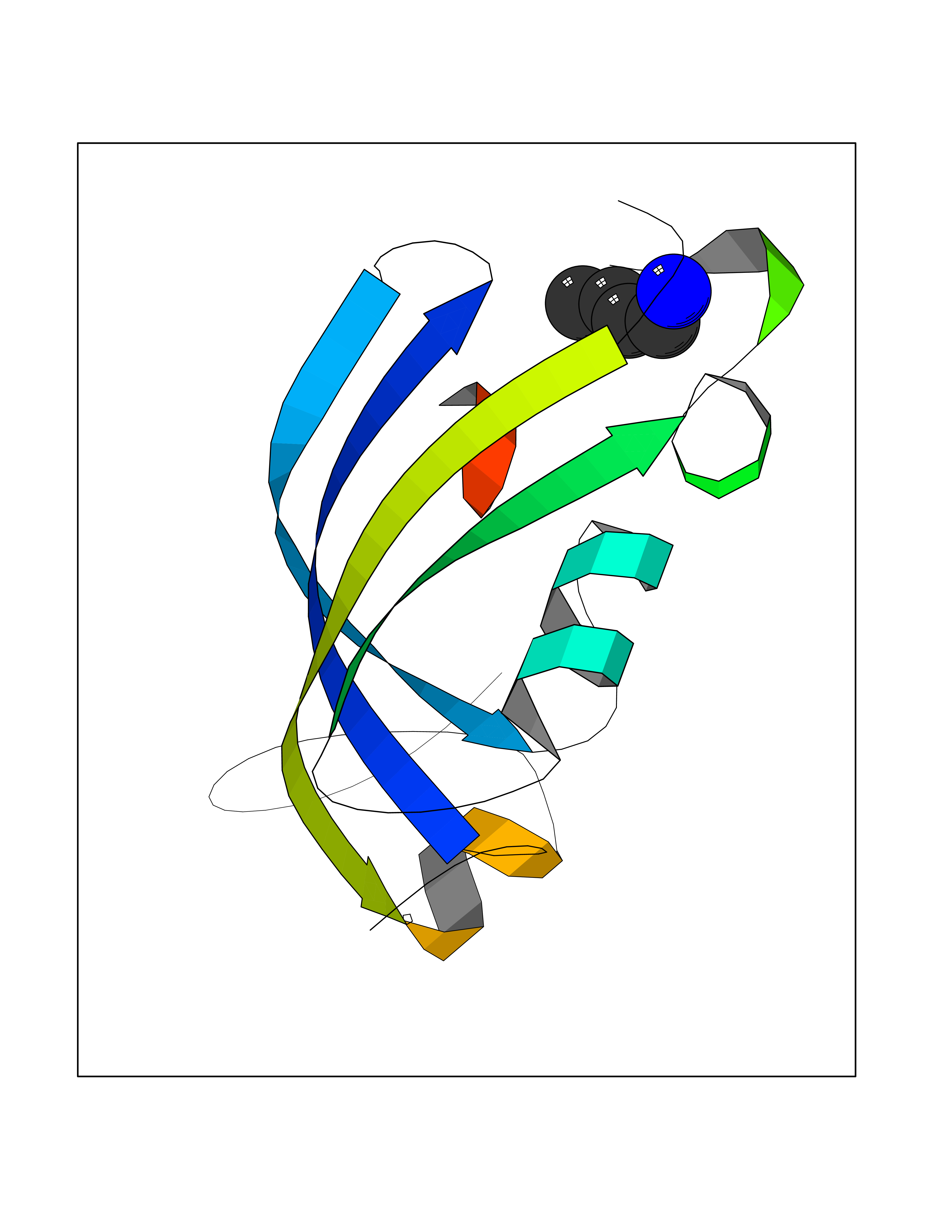 Molscript Map 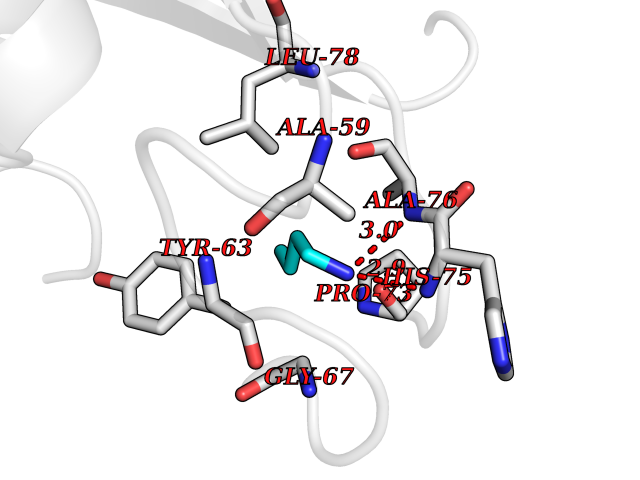 Pymol Map 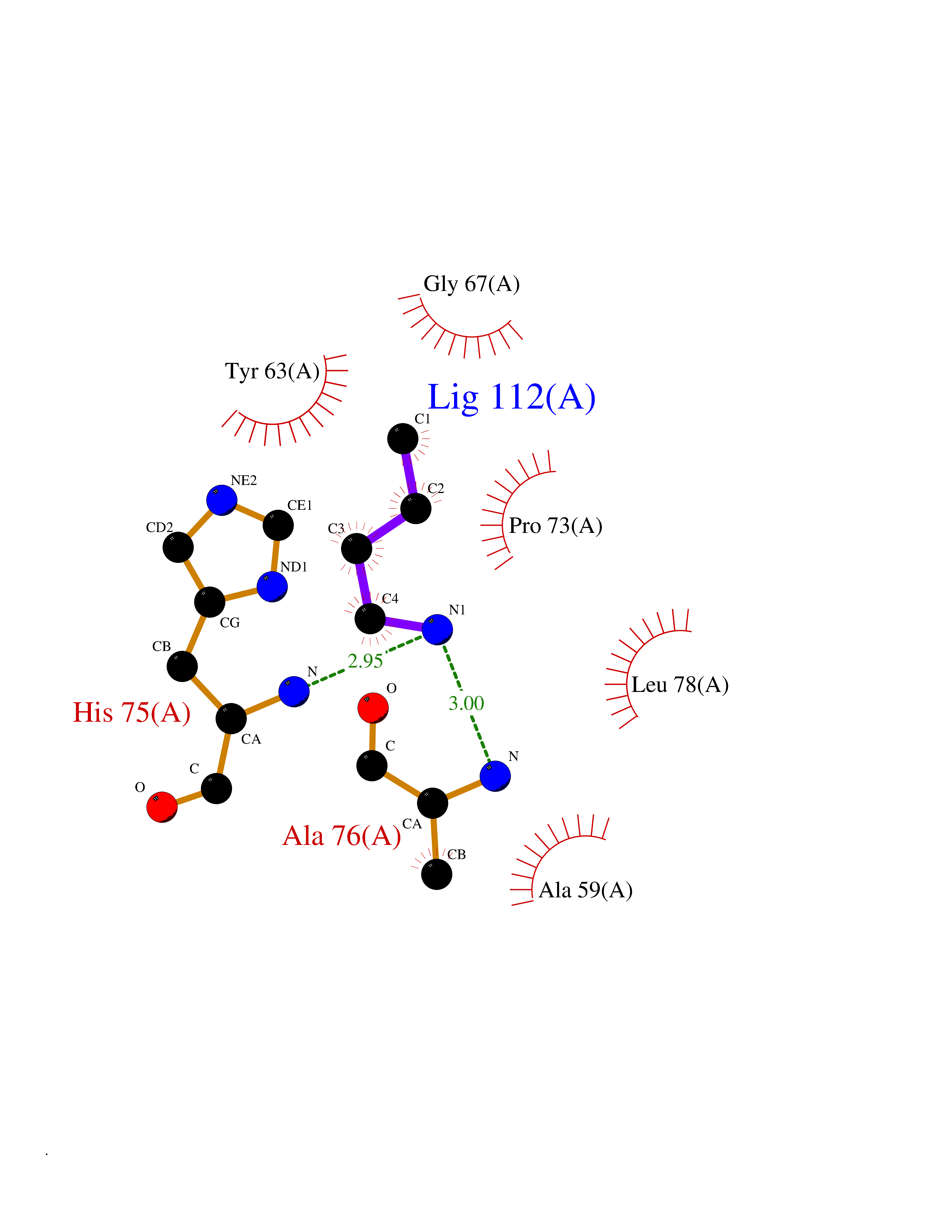 Ligplot Map 3D Binding mode Sequence MKVGQDKVVTIRYTLQVEGEVLDQGELSYLHGHRNLIPGLEEALEGREEGEAFQAHVPAEKAYGATGHPPHATLDFQVEVVKVREATPEELLHGHAHPSGHHHHHHGIVKPW Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Gamma-aminobutyric acid receptor subunit beta-3 | 4COF | 4.59 | |
Target general information Gen name GABRB3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Ligand-gated ion channel (TC 1.A.9) family, Gamma-aminobutyric acid receptor (TC 1.A.9.5) subfamily, GABRB3 sub-subfamily Biochemical class Transport protein Function GABA-A receptor activity.GABA-gated chloride ion channel activity.Identical protein binding. Related diseases Epilepsy, childhood absence 5 (ECA5) [MIM:612269]: A subtype of idiopathic generalized epilepsy characterized by an onset at age 6-7 years, frequent absence seizures (several per day) and bilateral, synchronous, symmetric 3-Hz spike waves on EEG. Tonic-clonic seizures often develop in adolescence. Absence seizures may either remit or persist into adulthood. {ECO:0000269|PubMed:18514161, ECO:0000269|PubMed:22303015}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 43 (DEE43) [MIM:617113]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE43 inheritance is autosomal dominant. {ECO:0000269|PubMed:23934111, ECO:0000269|PubMed:25356899, ECO:0000269|PubMed:26950270, ECO:0000269|PubMed:26993267, ECO:0000269|PubMed:27476654, ECO:0000269|PubMed:27864847}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12537; DB00546; DB00404; DB00543; DB11901; DB14719; DB11859; DB01558; DB09017; DB00237; DB00241; DB01489; DB00475; DB14715; DB01594; DB00349; DB01068; DB00628; DB01559; DB01553; DB01511; DB01189; DB00829; DB13837; DB00228; DB01215; DB00402; DB00898; DB00189; DB01545; DB09166; DB00292; DB01567; DB01205; DB01544; DB00690; DB06716; DB05087; DB01437; DB00801; DB01159; DB00753; DB01587; DB00555; DB00431; DB13643; DB00186; DB13872; DB13437; DB00603; DB01043; DB00371; DB00463; DB01028; DB01107; DB15489; DB00683; DB12458; DB01595; DB14028; DB00842; DB14672; DB00312; DB00252; DB13335; DB00592; DB01708; DB01588; DB00794; DB00818; DB01589; DB12404; DB01236; DB09118; DB00306; DB01956; DB00231; DB11582; DB00897; DB15490 Interacts with P28472 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Chloride; Chloride channel; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 76970.6 Length 663 Aromaticity 0.15 Instability index 39.4 Isoelectric point 8.37 Charge (pH=7) 4.12 2D Binding mode Binding energy (Kcal/mol) -6.26  Molscript Map 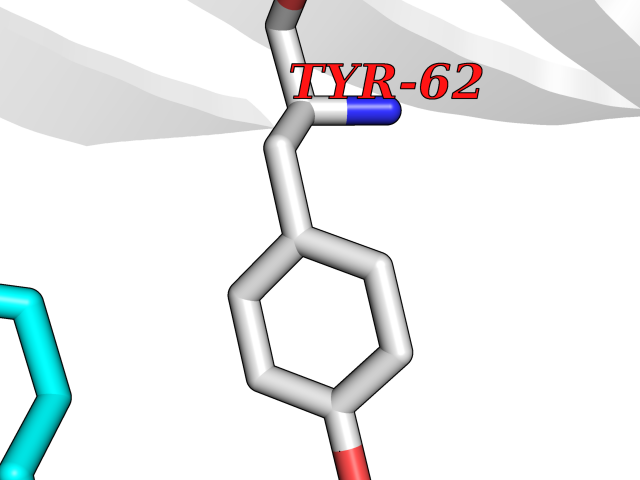 Pymol Map  Ligplot Map 3D Binding mode Sequence SFVKETVDKLLKGYDIRLRPDFGGPPVCVGMNIDIASIDMVSEVNMDYTLTMYFQQYWRDKRLAYSGIPLNLTLDNRVADQLWVPDTYFLNDKKSFVHGVTVKNRMIRLHPDGTVLYGLRITTTAACMMDLRRYPLDEQNCTLEIESYGYTTDDIEFYWRGGDKAVTGVERIELPQFSIVEHRLVSRNVVFATGAYPRLSLSFRLKRNIGYFILQTYMPSILITILSWVSFWINYDASAARVALGITTVLTMTTINTHLRETLPKIPYVKAIDMYLMGCFVFVFLALLEYAFVNYIFFSQPARAAAIDRWSRIVFPFTFSLFNLVYWLYYVNSFVKETVDKLLKGYDIRLRPDFGGPPVCVGMNIDIASIDMVSEVNMDYTLTMYFQQYWRDKRLAYSGIPLNLTLDNRVADQLWVPDTYFLNDKKSFVHGVTVKNRMIRLHPDGTVLYGLRITTTAACMMDLRRYPLDEQNCTLEIESYGYTTDDIEFYWRGGDKAVTGVERIELPQFSIVEHRLVSRNVVFATGAYPRLSLSFRLKRNIGYFILQTYMPSILITILSWVSFWINYDASAARVALGITTVLTMTTINTHLRETLPKIPYVKAIDMYLMGCFVFVFLALLEYAFVNYIFFSQPARAAAIDRWSRIVFPFTFSLFNLVYWLYYV Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Vascular endothelial growth factor receptor 3 | 4BSJ | 4.59 | |
Target general information Gen name FLT4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms VEGFR3 Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Transferase Function ATP binding.Growth factor binding.Protein homodimerization activity.Protein phosphatase binding.Transmembrane receptor protein tyrosine kinase activity.Vascular endothelial growth factor-activated receptor activity.VEGF-C-activated receptor activity. Related diseases Lymphatic malformation 1 (LMPHM1) [MIM:153100]: A form of primary lymphedema, a disease characterized by swelling of body parts due to developmental anomalies and functional defects of the lymphatic system. Patients with lymphedema may suffer from recurrent local infections. LMPHM1 is an autosomal dominant form with variable expression and severity. Onset is usually at birth or in early childhood but can occur later. Affected individuals manifest lymphedema, predominantly in the lower limbs, and hypoplasia of lymphatic vessels. Additional features are hemangioma and nail dysplasia or papillomatosis. {ECO:0000269|PubMed:10835628, ECO:0000269|PubMed:10856194, ECO:0000269|PubMed:12881528, ECO:0000269|PubMed:15102829, ECO:0000269|PubMed:16924388, ECO:0000269|PubMed:16965327, ECO:0000269|PubMed:17458866, ECO:0000269|PubMed:19289394, ECO:0000269|PubMed:26091405, ECO:0000269|PubMed:9817924}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hemangioma, capillary infantile (HCI) [MIM:602089]: A condition characterized by dull red, firm, dome-shaped hemangiomas, sharply demarcated from surrounding skin, usually presenting at birth or occurring within the first two or three months of life. They result from highly proliferative, localized growth of capillary endothelium and generally undergo regression and involution without scarring. {ECO:0000269|PubMed:11807987}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Plays an important role in tumor lymphangiogenesis, in cancer cell survival, migration, and formation of metastases.; DISEASE: Congenital heart defects, multiple types, 7 (CHTD7) [MIM:618780]: An autosomal dominant disorder with incomplete penetrance characterized by congenital developmental abnormalities involving structures of the heart. Common defects include tetralogy of Fallot, pulmonary stenosis or atresia, absent pulmonary valve, right aortic arch, double aortic arch, and major aortopulmonary collateral arteries. {ECO:0000269|PubMed:28991257, ECO:0000269|PubMed:30232381}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06626; DB05932; DB12010; DB11679; DB06101; DB09078; DB06080; DB09079; DB06589; DB08896; DB15685; DB00398; DB01268; DB05075; DB11800; DB04879 Interacts with P08238; P35968; P49767 EC number 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; Angiogenesis; ATP-binding; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Secreted; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 24029.2 Length 213 Aromaticity 0.1 Instability index 47.75 Isoelectric point 8.34 Charge (pH=7) 2.35 2D Binding mode Binding energy (Kcal/mol) -6.26  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence DHNPFISVEWLKGPILEATAGDELVKLPVKLAAYPPPEFQWYKDGKALSGRHSPHALVLKEVTEASTGTYTLALWNSAAGLRRNISLELVVNVPPQIHEKEASSPSIYSRHSRQALTCTAYGVPLPLSIQWHWRPWTPCKMFPQCRDWRAVTTQDAVNPIESLDTWTEFVEGKNKTVSKLVIQNANVSAMYKCVVSNKVGQDERLIYFYVTTH Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | 2-iminobutanoate/2-iminopropanoate deaminase | 1ONI | 4.59 | |
Target general information Gen name RIDA Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms HRSP12 Protein family RutC family Biochemical class Translation Function Deaminase activity.Endoribonuclease activity, producing 3'-phosphomonoesters.Long-chain fatty acid binding.Platinum binding.Protein homodimerization activity.RNA binding.Transition metal ion binding.Xenon atom binding. Related diseases Congenital bile acid synthesis defect 2 (CBAS2) [MIM:235555]: A condition characterized by jaundice, intrahepatic cholestasis and hepatic failure. Patients with this liver disease show absence or low levels of chenodeoxycholic acid and cholic acid in plasma and urine. {ECO:0000269|PubMed:12970144, ECO:0000269|PubMed:15030995, ECO:0000269|PubMed:19175828, ECO:0000269|PubMed:20522910}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q8N9N5-2 EC number 3.5.99.10 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Hydrolase; Lipid metabolism; Mitochondrion; Nucleus; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; RNA-binding Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H,I Molecular weight (Da) 42624.3 Length 404 Aromaticity 0.07 Instability index 36.76 Isoelectric point 8.99 Charge (pH=7) 5.46 2D Binding mode Binding energy (Kcal/mol) -6.26 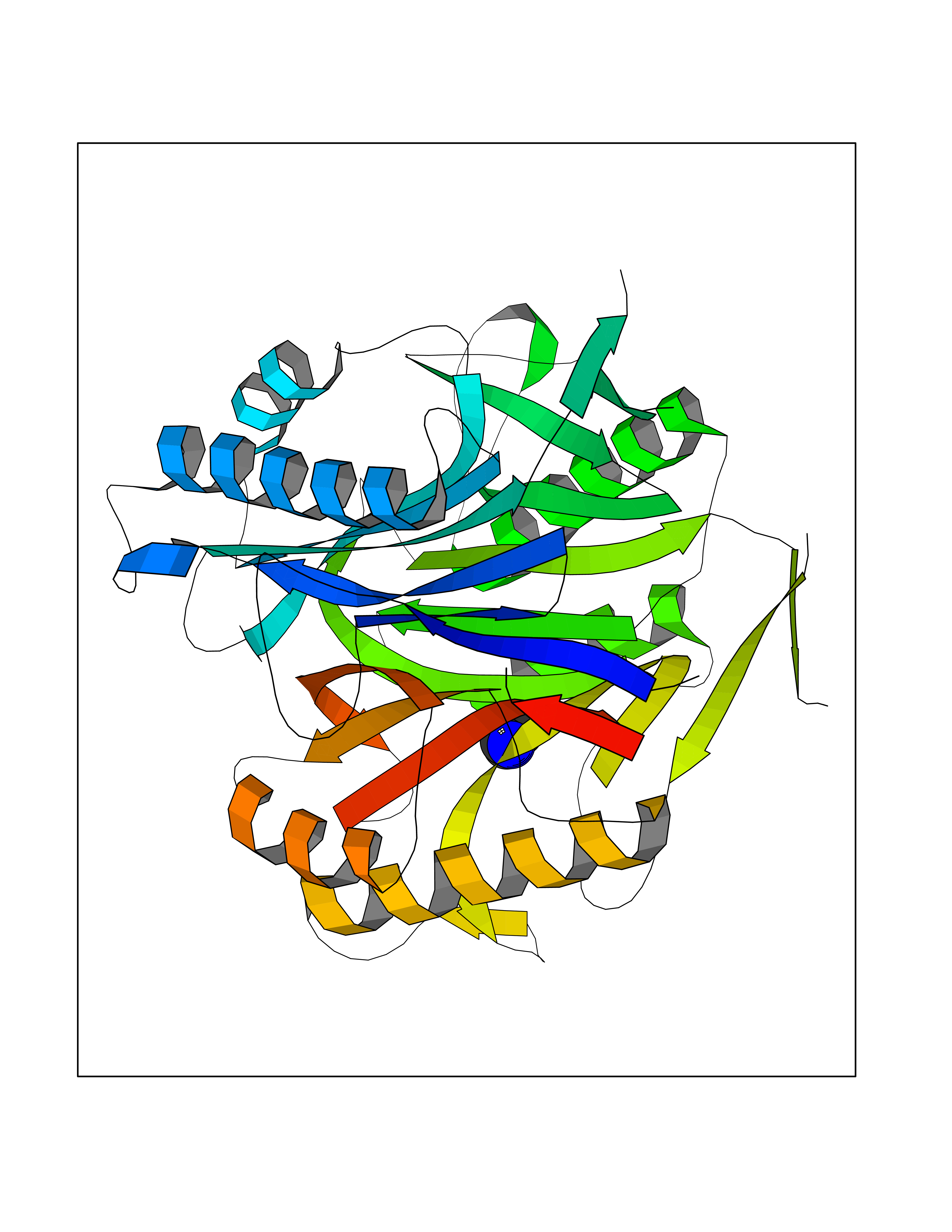 Molscript Map 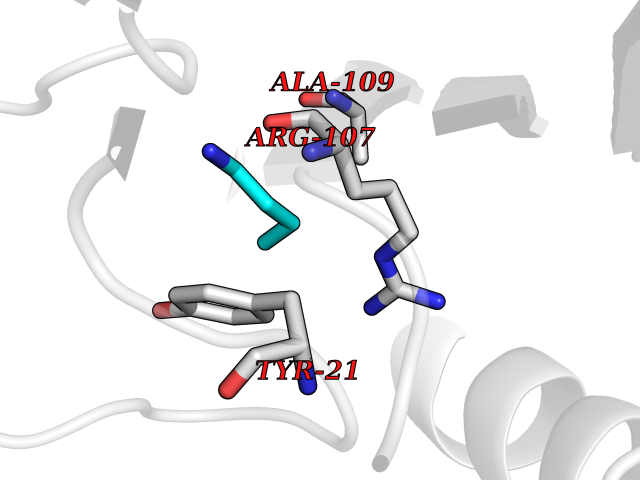 Pymol Map 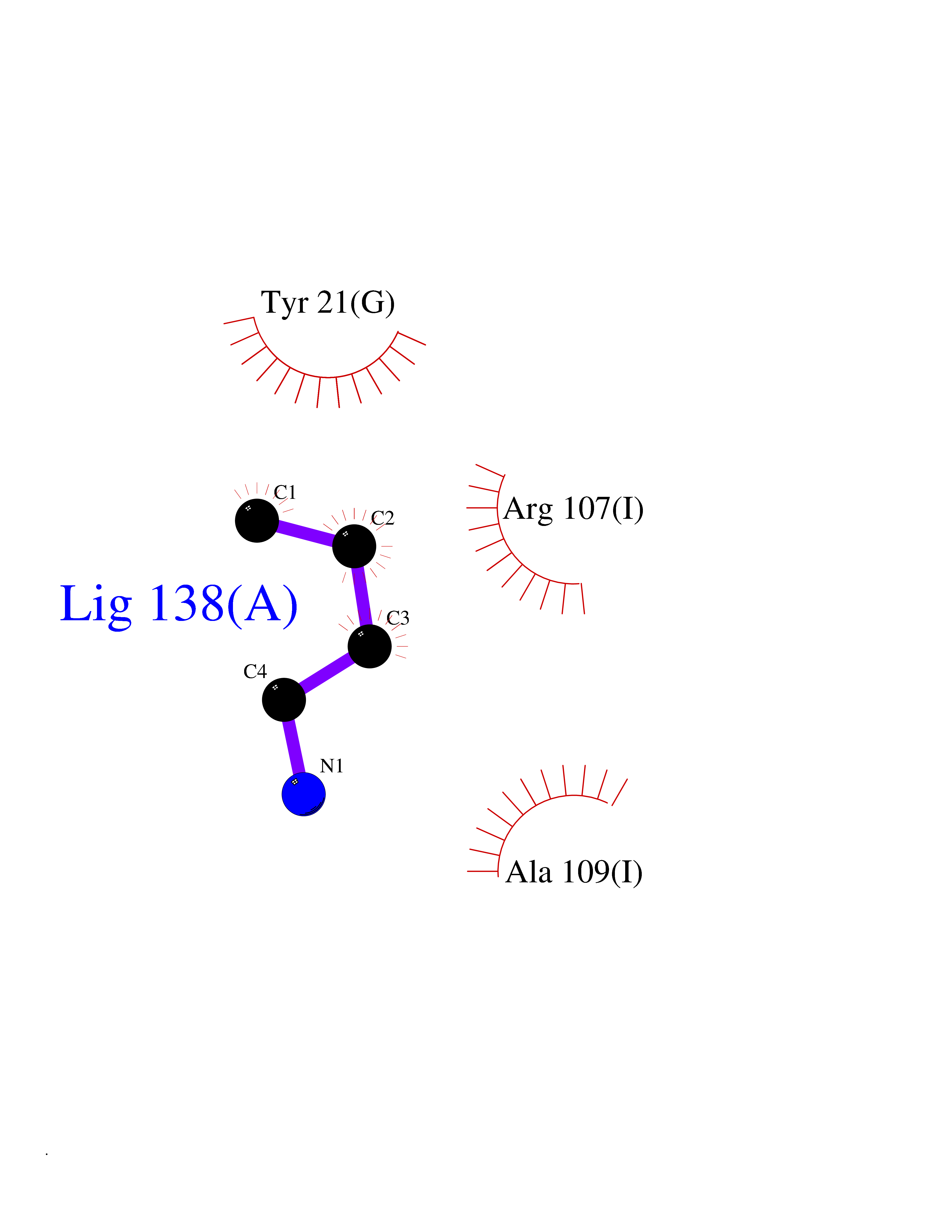 Ligplot Map 3D Binding mode Sequence SSLIRRVISTAKAPGAIGPYSQAVLVDRTIYISGQIGMDPSSGQLVSGGVAEEAKQALKNMGEILKAAGCDFTNVVKTTVLLADINDFNTVNEIYKQYFKSNFPARAAYQVAALPKGSRIEIEAVAIQGPLTTASSSLIRRVISTAKAPGAIGPYSQAVLVDRTIYISGQIGMDPSSGQLVSGGVAEEAKQALKNMGEILKAAGCDFTNVVKTTVLLADINDFNTVNEIYKQYFKSNFPARAAYQVAALPKGSRIEIEAVAIQGPLTTASSSLIRRVISTAKAPGAIGPYSQAVLVDRTIYISGQIGMDPSSGQLVSGGVAEEAKQALKNMGEILKAAGCDFTNVVKTTVLLADINDFNTVNEIYKQYFKSNFPARAAYQVAALPKGSRIEIEAVAIQGPLTTA Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Phosphodiesterase 4B (PDE4B) | 4KP6 | 4.59 | |
Target general information Gen name PDE4B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cAMP-specific 3',5'-cyclic phosphodiesterase 4B; Type 4B cAMP phosphodiesterase; Type 4 cyclic adenosine monophosphate phosphodiesterase (type 4 PDE); PDE32; DPDE4 Protein family Cyclic nucleotide phosphodiesterase family, PDE4 subfamily Biochemical class Phosphoric diester hydrolase Function May be involved in mediating central nervous system effects of therapeutic agents ranging from antidepressants to antiasthmatic and anti-inflammatory agents. Hydrolyzes the second messenger cAMP, which is a key regulator of many important physiological processes. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04149; DB03606; DB03807; DB06909; DB01959; DB08299; DB03349; DB00131; DB01427; DB00201; DB03849; DB05219; DB01647; DB00651; DB00824; DB02660; DB05266; DB01088; DB01113; DB01791; DB01656; DB01954; DB04530; DB01412; DB00277; DB09283 Interacts with Q13936; Q08499 EC number EC 3.1.4.53 Uniprot keywords 3D-structure; Alternative splicing; cAMP; Cell membrane; Cytoplasm; Hydrolase; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 37367.1 Length 324 Aromaticity 0.08 Instability index 36.36 Isoelectric point 5 Charge (pH=7) -20.82 2D Binding mode Binding energy (Kcal/mol) -6.26 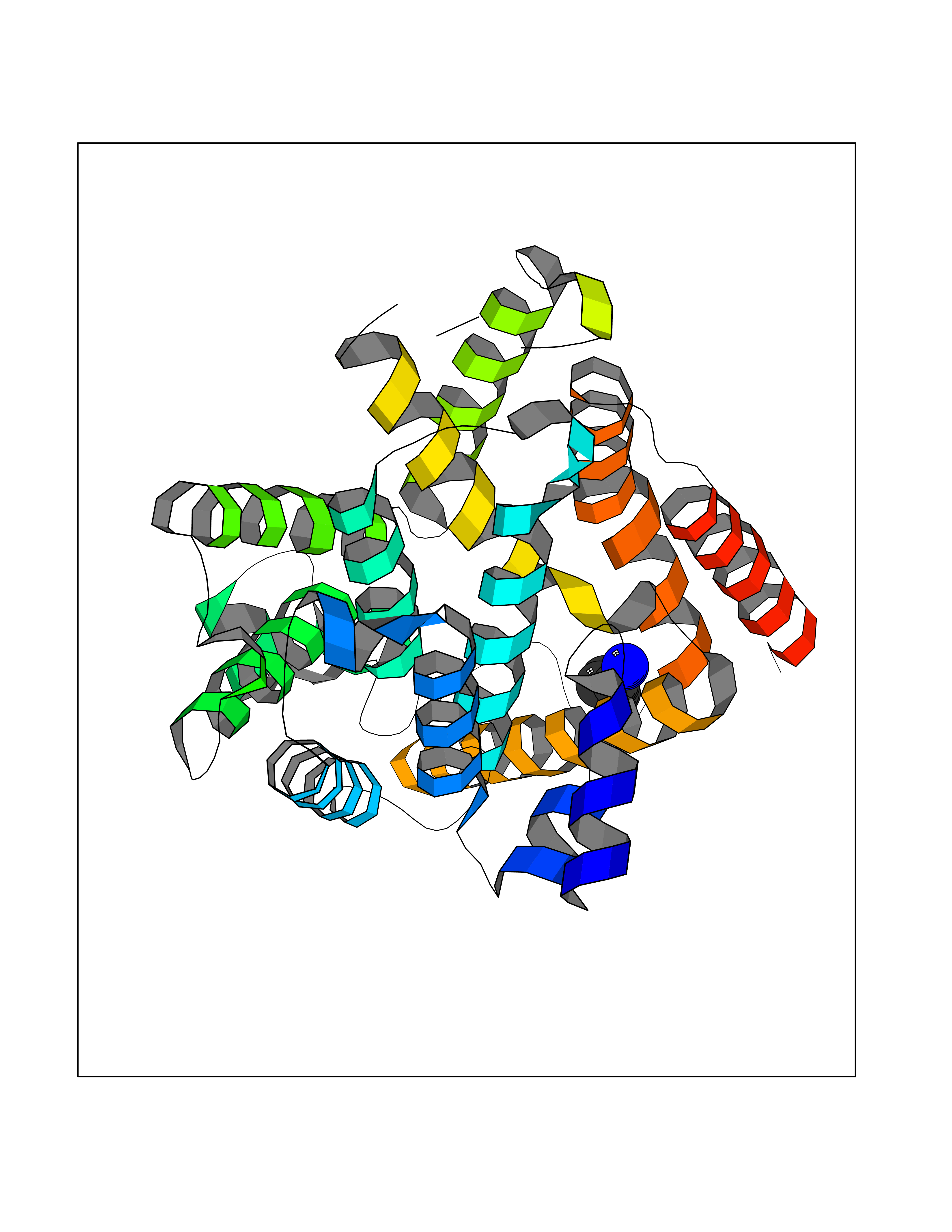 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence NEDHLAKELEDLNKWGLNIFNVAGYSHNRPLTCIMYAIFQERDLLKTFRISSDTFITYMMTLEDHYHSDVAYHNSLHAADVAQSTHVLLSTPALDAVFTDLEILAAIFAAAIHDVDHPGVSNQFLINTNSELALMYNDESVLENHHLAVGFKLLQEEHCDIFMNLTKKQRQTLRKMVIDMVLATDMSKHMSLLADLKTMVETKKVTSSGVLLLDNYTDRIQVLRNMVHCADLSNPTKSLELYRQWTDRIMEEFFQQGDKERERGMEISPMCDKHTASVEKSQVGFIDYIVHPLWETWADLVQPDAQDILDTLEDNRNWYQSMIP Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Prostaglandin reductase 2 | 2ZB4 | 4.59 | |
Target general information Gen name PTGR2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ZADH1 Protein family NADP-dependent oxidoreductase L4BD family Biochemical class Oxidoreductase Function 13-prostaglandin reductase activity.15-oxoprostaglandin 13-oxidase activity. Related diseases Long QT syndrome 10 (LQT10) [MIM:611819]: A heart disorder characterized by a prolonged QT interval on the ECG and polymorphic ventricular arrhythmias. They cause syncope and sudden death in response to exercise or emotional stress, and can present with a sentinel event of sudden cardiac death in infancy. {ECO:0000269|PubMed:17592081}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Atrial fibrillation, familial, 17 (ATFB17) [MIM:611819]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:23604097}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07177; DB00328 Interacts with Q7L4P6 EC number 1.3.1.48 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Lipid metabolism; NADP; Oxidoreductase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 38385.4 Length 350 Aromaticity 0.07 Instability index 36.68 Isoelectric point 5.27 Charge (pH=7) -9 2D Binding mode Binding energy (Kcal/mol) -6.26  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MIVQRVVLNSRPGKNGNPVAENFRMEEVYLPDNINEGQVQVRTLYLSVDPYMRCRMNEDTGTDYITPWQLSQVVDGGGIGIIEESKHTNLTKGDFVTSFYWPWQTKVILDGNSLEKVDPQLVDGHLSYFLGAIGMPGLTSLIGIQEKGHITAGSNKTMVVSGAAGACGSVAGQIGHFLGCSRVVGICGTHEKCILLTSELGFDAAINYKKDNVAEQLRESCPAGVDVYFDNVGGNISDTVISQMNENSHIILCGQISQYNKDVPYPPPLSPAIEAIQKERNITRERFLVLNYKDKFEPGILQLSQWFKEGKLKIKETVINGLENMGAAFQSMMTGGNIGKQIVCISEEIS Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Mitogen-activated protein kinase kinase kinase 2 | 5HQ8 | 4.59 | |
Target general information Gen name MAP3K2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms MEKK2;MAPKKK2 Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, MAP kinase kinase kinase subfamily Biochemical class Transferase Function ATP binding.MAP kinase kinase kinase activity.Metal ion binding.Protein kinase activity.Protein kinase binding.Protein serine/threonine kinase activity. Related diseases Dyskinesia, limb and orofacial, infantile-onset (IOLOD) [MIM:616921]: An autosomal recessive, early-onset hyperkinetic movement disorder characterized by axial hypotonia, dyskinesia of the limbs and trunk, orofacial dyskinesia, drooling, and dysarthria. The severity of the hyperkinesis is variable. {ECO:0000269|PubMed:27058446}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Striatal degeneration, autosomal dominant 2 (ADSD2) [MIM:616922]: An autosomal dominant disorder characterized by striatal degeneration and dysfunction of basal ganglia, resulting in hyperkinesis. {ECO:0000269|PubMed:27058447}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06616; DB12010 Interacts with Q13163; P31947; Q9H7B4-1; Q9UNE7; P62258 EC number 2.7.11.25 Uniprot keywords 3D-structure; ATP-binding; Cytoplasm; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID I,J Molecular weight (Da) 48735.2 Length 425 Aromaticity 0.08 Instability index 44.28 Isoelectric point 7.24 Charge (pH=7) 0.6 2D Binding mode Binding energy (Kcal/mol) -6.26  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence PLKVEKFATANRGNGLRAVTPLRPGELLFRSDPLAYTVCKGSRGVVCDRCLLGKEKLMRCSQCRVAKYCSAKCQKKAWPDHKRECKCLKSCKPRYPPDSVRLLGRVVFKLMDGAPSESEKLYSFYDLESNINKLTEDKKEGLRQLVMTFQHFMREEIQDASQLPPAFDLFEAFAKVICNSFTICNAEMQEVGVGLYPSISLLNHSCDPNCSIVFNGPHLLLRAVRDIEVGEELTICYLDMLMTSEERRKQLRDQYCFECDCFRCQTQDKDADMLTGDEQVWKEVQESLKKIEELKAHWKWEQVLAMCQAIISSNSERLPDINIYQLKVLDCAMDACINLGLLEEALFYGTRTMEPYRIFFPGSHPVRGVQVMKVGKLQLHQGMFPQAMKNLRLAFDIMRVTHGREHSLIEDLILLLEECDANIRA Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Transient receptor potential cation channel V6 (TRPV6) | 6D7T | 4.59 | |
Target general information Gen name TRPV6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Transient receptor potential cation channel subfamily V, member 6; TRPV6; HTRP8; ECaC2; Calcium transport protein CaT1; Calcium channel CAT1 Protein family Transient receptor (TC 1.A.4) family, TrpV subfamily, TRPV6 sub-subfamily Biochemical class Transient receptor potential catioin channel Function Calcium selective cation channel probably involved in Ca(2+) uptake in various tissues, including Ca(2+) reabsorption in intestine. The channel is activated by low internal calcium level, probably including intracellular calcium store depletion, and the current exhibits an inward rectification. Inactivation includes both, a rapid Ca(2+)-dependent and a slower Ca(2+)-calmodulin- dependent mechanism, the lattermay be regulated by phosphorylation. In vitro, is slowly inhibited by Mg(2+) in a voltage-independent manner. Heteromeric assembly with TRPV5 seems to modify channel properties. TRPV5-TRPV6 heteromultimeric concatemers exhibit voltage-dependent gating. Related diseases Hyperparathyroidism, transient neonatal (HRPTTN) [MIM:618188]: An autosomal recessive disease characterized by impaired transplacental maternal-fetal transport of calcium, high serum PTH levels and signs of metabolic bone disease in the neonatal period. Skeletal anomalies include generalized osteopenia, narrow chest, short ribs with multiple healing fractures, and bowing or fractures of long bones. Affected individuals experience postnatal respiratory and feeding difficulties. The condition improves within a short time after birth once calcium is provided orally. {ECO:0000269|PubMed:29861107, ECO:0000269|PubMed:30820485}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11093; DB11348; DB14481 Interacts with P18031 EC number NA Uniprot keywords 3D-structure; Alternative splicing; ANK repeat; Calcium; Calcium channel; Calcium transport; Calmodulin-binding; Cell membrane; Disease variant; Glycoprotein; Ion channel; Ion transport; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 69726.9 Length 611 Aromaticity 0.1 Instability index 43.63 Isoelectric point 6.33 Charge (pH=7) -4.27 2D Binding mode Binding energy (Kcal/mol) -6.26  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SWAQSRDEQNLLQQKRIWESPLLLAAKDNDVQALNKLLKYEDCKVHQRGAMGETALHIAALYDNLEAAMVLMEAAPELVFEPMTSELYEGQTALHIAVVNQNMNLVRALLARRASVSARATGTAFRRSPCNLIYFGEHPLSFAACVNSEEIVRLLIEHGADIRAQDSLGNTVLHILILQPNKTFACQMYNLLLSYDRHGDHLQPLDLVPNHQGLTPFKLAGVEGNTVMFQHLMQKRKHTQWTYGPLTSTLYDLTEIDSSGDEQSLLELIITTKKREARQILDQTPVKELVSLKWKRYGRPYFCMLGAIYLLYIICFTMCCIYRPLKPRTNNRTSPRDNTLLQQKLLQEAYMTPKDDIRLVGELVTVIGAIIILLVEVPDIFRMGVTRFFGQTILGGPFHVLIITYAFMVLVTMVMRLISASGEVVPMSFALVLGWCNVMAFARGFQMLGPFTIMIQKMIFGDLMRFCWLMAVVILGFASAFYIIFQTEDPEELGHFYDYPMALFSTFELFLTIIDGPANYNVDLPFMYSITYAAFAIIATLLMLNLLIAMMGDTHWRVAHERDELWRAQIVATTVMLERKLPRCLWPRSGICGREYGLGDRWFLRVEDRQD Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Caterpiller protein 1.1 (NLRP3) | 7PZC | 4.59 | |
Target general information Gen name NLRP3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PYRIN-containing APAF1-like protein 1; PYPAF1; NALP3; NACHT, LRR and PYD domains-containing protein 3; Cryopyrin; Cold-induced autoinflammatory syndrome 1 protein; CLR1.1; CIAS1; C1orf7; Angiotensin/v Protein family NLRP family Biochemical class NA Function In response to pathogens and other damage-associated signals, initiates the formation of the inflammasome polymeric complex, made of NLRP3, PYCARD and CASP1 (and possibly CASP4 and CASP5). Recruitment of proCASP1 to the inflammasome promotes its activation and CASP1-catalyzed IL1B and IL18 maturation and secretion in the extracellular milieu. Activation of NLRP3 inflammasome is also required for HMGB1 secretion. The active cytokines and HMGB1 stimulate inflammatory responses. Inflammasomes can also induce pyroptosis, an inflammatory form of programmed cell death. Under resting conditions, NLRP3 is autoinhibited. NLRP3 activation stimuli include extracellular ATP, reactive oxygen species, K(+) efflux, crystals of monosodium urate or cholesterol, amyloid-beta fibers, environmental or industrial particles and nanoparticles, cytosolic dsRNA, etc. However, it is unclear what constitutes the direct NLRP3 activator. Activation in presence of cytosolic dsRNA is mediated by DHX33. Independently of inflammasome activation, regulates the differentiation of T helper 2 (Th2) cells and has a role in Th2 cell-dependent asthma and tumor growth. During Th2 differentiation, required for optimal IRF4 binding to IL4 promoter and for IRF4-dependent IL4 transcription. Binds to the consensus DNA sequence 5'-GRRGGNRGAG-3'. May also participate in the transcription of IL5, IL13, GATA3, CCR3, CCR4 and MAF. As the sensor component of the NLRP3 inflammasome, plays a crucial role in innate immunity and inflammation. Related diseases Familial cold autoinflammatory syndrome 1 (FCAS1) [MIM:120100]: A rare autosomal dominant systemic inflammatory disease characterized by recurrent episodes of maculopapular rash associated with arthralgias, myalgias, fever and chills, swelling of the extremities, and conjunctivitis after generalized exposure to cold. Rarely, some patients may also develop late-onset renal amyloidosis. {ECO:0000269|PubMed:11687797, ECO:0000269|PubMed:11992256, ECO:0000269|PubMed:12355493, ECO:0000269|PubMed:12522564, ECO:0000269|PubMed:15593220, ECO:0000269|PubMed:17284928, ECO:0000269|PubMed:24952504}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Muckle-Wells syndrome (MWS) [MIM:191900]: A hereditary periodic fever syndrome characterized by fever, chronic recurrent urticaria, arthralgias, progressive sensorineural deafness, and reactive renal amyloidosis. The disease may be severe if generalized reactive amyloidosis occurs. {ECO:0000269|PubMed:11687797, ECO:0000269|PubMed:11992256, ECO:0000269|PubMed:12355493, ECO:0000269|PubMed:15593220, ECO:0000269|PubMed:24952504}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Chronic infantile neurologic cutaneous and articular syndrome (CINCA) [MIM:607115]: Rare congenital inflammatory disorder characterized by a triad of neonatal onset of cutaneous symptoms, chronic meningitis, and joint manifestations with recurrent fever and inflammation. {ECO:0000269|PubMed:12032915, ECO:0000269|PubMed:12483741, ECO:0000269|PubMed:14630794, ECO:0000269|PubMed:15231984, ECO:0000269|PubMed:15334500, ECO:0000269|PubMed:15593220, ECO:0000269|PubMed:24952504, ECO:0000269|PubMed:31086329}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Keratoendothelitis fugax hereditaria (KEFH) [MIM:148200]: An autosomal dominant corneal disease that periodically, and fleetingly, affects the corneal endothelium, stroma, and vision, eventually leading to central corneal stromal opacities in some patients. The disease is characterized by unilateral attacks of ocular pain, pericorneal injection, and photophobia. The acute symptoms vanish in 1-2 days but vision remains blurry for several weeks. The attacks start at the age of 3-12 years and can affect either eye. They generally decrease in frequency and get milder with age. {ECO:0000269|PubMed:29366613, ECO:0000269|PubMed:35559676}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, autosomal dominant, 34, with or without inflammation (DFNA34) [MIM:617772]: A form of sensorineural hearing loss. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. DFNA34 is a postlingual, slowly progressive form with variable severity and variable additional features. Some DFNA34 patients have autoinflammatory manifestations. {ECO:0000269|PubMed:28847925}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P27797; P36957; P19525; Q7Z434; Q8TDX7; Q96P20; Q9ULZ3; Q80H93; P0DTC9; Q9ES74; Q8TDX7-1 EC number NA Uniprot keywords 3D-structure; Activator; ADP-ribosylation; Alternative splicing; Amyloidosis; ATP-binding; Cytoplasm; Cytoskeleton; Deafness; Disease variant; Disulfide bond; Endoplasmic reticulum; Golgi apparatus; Hydrolase; Immunity; Inflammasome; Inflammatory response; Innate immunity; Isopeptide bond; Leucine-rich repeat; Lipoprotein; Membrane; Mitochondrion; Non-syndromic deafness; Nucleotide-binding; Nucleus; Palmitate; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Secreted; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 115171 Length 1010 Aromaticity 0.08 Instability index 45.92 Isoelectric point 6.17 Charge (pH=7) -11.22 2D Binding mode Binding energy (Kcal/mol) -6.26  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MASTRCKLARYLEDLEDVDLKKFKMHLEDYPPQKGCIPLPRGQTEKADHVDLATLMIDFNGEEKAWAMAVWIFAAINRRDLYEKAKRDEPKWGSDNARVSNPTVICQEDSIEEEWMGLLEYLSRISICKMKKDYRKKYRKYVRSRFQCIEESVSLNKRYTRLRLIKEHRSQSPVSPIKMELLFDPDDEHSEPVHTVVFQGAAGIGKTILARKMMLDWASGTLYQDRFDYLFYIHCREVSLVTQRSLGDLIMSCCPDPNPPIHKIVRKPSRILFLMDGFDELQGAFDEHIGPLCTDWQKAERGDILLSSLIRKKLLPEASLLITTRPVALEKLQHLLDHPRHVEILGFSEAKRKEYFFKYFSDEAQARAAFSLIQENEVLFTMCFIPLVCWIVCTGLKQQMESGKSLAQTSKTTTAVYVFFLSSLLQPRGGSQEHGLCAHLWGLCSLAADGIWNQKILFEESDLRNHGLQKADVSAFLRMNLFQKEVDCEKFYSFIHMTFQEFFAAMYYLLEEEKEGRTNVPGSRLKLPSRDVTVLLENYGKFEKGYLIFVVRFLFGLVNQERTSYLEKKLSCKISQQIRLELLKWIEVKAKAKKLQIQPSQLELFYCLYEMQEEDFVQRAMDYFPKIEINLSTRMDHMVSSFCIENCHRVESLSLGFLHNMPKEEEEEEKEGRHLDMVQCVLPSSSHAACSHGLVNSHLTSSFCRGLFSVLSTSQSLTELDLSDNSLGDPGMRVLCETLQHPGCNIRRLWLGRCGLSHECCFDISLVLSSNQKLVELDLSDNALGDFGIRLLCVGLKHLLCNLKKLWLVSCCLTSACCQDLASVLSTSHSLTRLYVGENALGDSGVAILCEKAKNPQCNLQKLGLVNSGLTSVCCSALSSVLSTNQNLTHLYLRGNTLGDKGIKLLCEGLLHPDCKLQVLELDNCNLTSHCCWDLSTLLTSSQSLRKLSLGNNDLGDLGVMMFCEVLKQQSCLLQNLGLSEMYFNYETKSALETLQEEKPELTVVFEPSW Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Interleukin 21 receptor (IL21R) | 6PLH | 4.59 | |
Target general information Gen name IL21R Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UNQ3121/PRO10273; Novel interleukin receptor; NILR; Interleukin-21 receptor; IL21 receptor; IL-21R; IL-21 receptor; CD360 Protein family Type I cytokine receptor family, Type 4 subfamily Biochemical class Cytokine receptor Function This is a receptor for interleukin-21. Related diseases Immunodeficiency 56 (IMD56) [MIM:615207]: An autosomal recessive primary immunodeficiency characterized by B- and T-cell defects and variable dysfunction of NK cells. Patients tend to have normal numbers of lymphocytes, but show defective class-switched B-cells, low IgG, defective antibody response, and defective T-cell responses to certain antigens. {ECO:0000269|PubMed:23440042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Chromosomal aberrations involving IL21R is a cause of B-cell non-Hodgkin lymphomas (B-cell NHL). Translocation t(3;16)(q27;p11), with BCL6. Drugs (DrugBank ID) NA Interacts with P29972 EC number NA Uniprot keywords 3D-structure; Chromosomal rearrangement; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,C,B Molecular weight (Da) 48376.5 Length 446 Aromaticity 0.1 Instability index 43.94 Isoelectric point 8.24 Charge (pH=7) 3.56 2D Binding mode Binding energy (Kcal/mol) -6.27 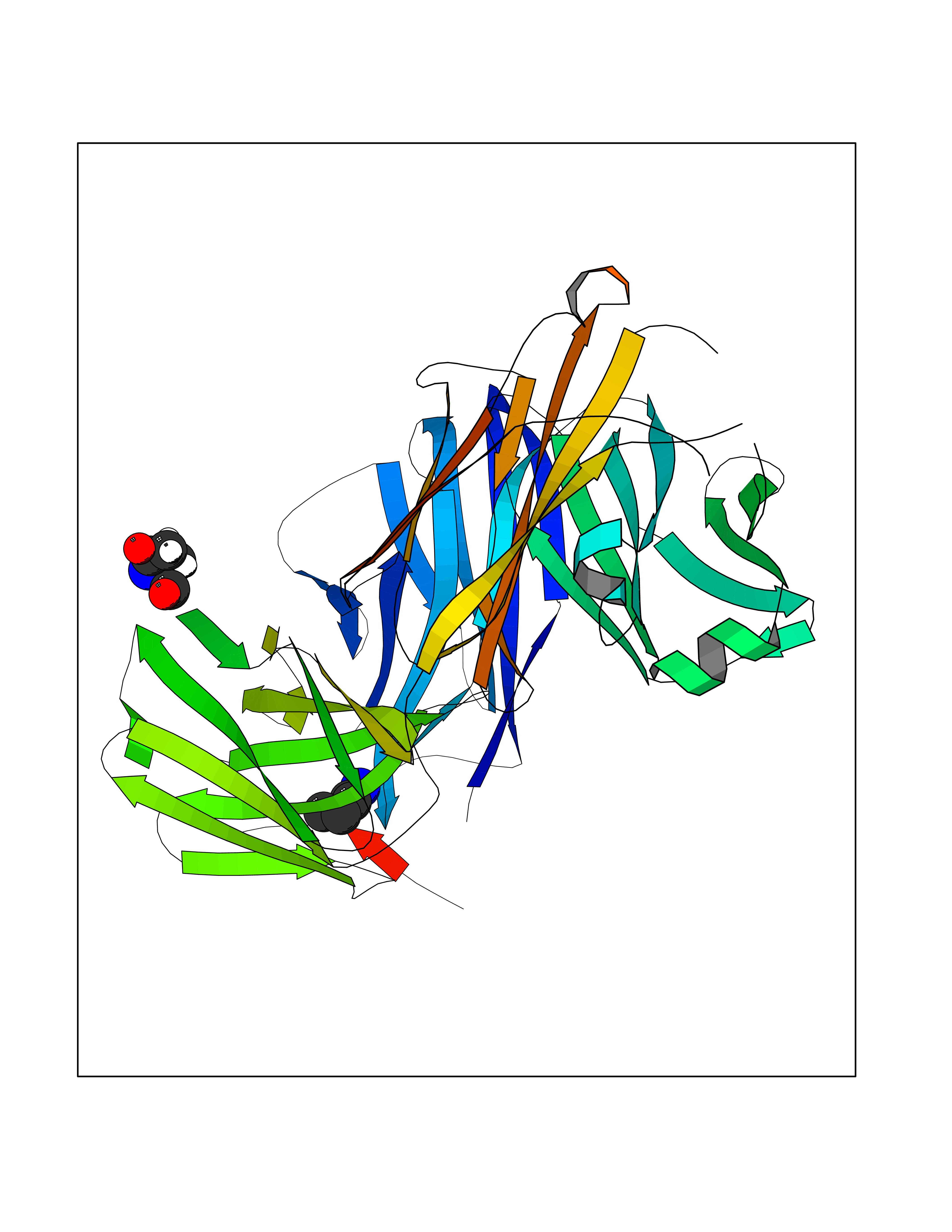 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence DVVMTHTPLSLPVSLGDQASISCRSSQSLVHSNGNTYLHWYLQKPGQSPKLLIYKVSNRFSGVPDRFSGSGSGADFTLKISRVEAEDLGVYFCSQSTHVPRTFGGGTKLEIKRADAAPTVSIFPPSSEQLTSGGASVVCFLNNFYPKDINVKWKIDGSERQNGVLNSWTDQDSKDSTYSMSSTLTLTKDEYERHNSYTCEATHKTSTSPIVKSFNRNECXVHLQQPGADLVKPGASVKMSCKASGYTFTSYWITWVKLRPGQGLEWIGDIYPGSGSTNFIEKFKSKATLTVDTSSSTAYMQLRSLTSEDSAVYYCARRGHGNYEDYWGQGTTLIVSSAKTTAPSVYPLAPVCGTGSSVTLGCLVKGYFPEPVTLTWNSGSLSSGVHTFPAVLQSDLYTLSSSVTVTSSTWPSQSITCNVAHPASSTKVDKKIEPRGPTTWSEWSDP Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Pseudomonas Transcriptional activator protein LasR (Pseudo LasR) | 3IX3 | 4.59 | |
Target general information Gen name Pseudo LasR Organism Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Uniprot ID TTD ID Synonyms NA Protein family Autoinducer-regulated transcriptional regulatory protein family Biochemical class NA Function Transcriptional activator of elastase structural gene (LasB). Binds to the PAI autoinducer. Related diseases Growth hormone deficiency, isolated, 1A (IGHD1A) [MIM:262400]: An autosomal recessive, severe deficiency of growth hormone leading to dwarfism. Patients often develop antibodies to administered growth hormone. {ECO:0000269|PubMed:8364549}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 1B (IGHD1B) [MIM:612781]: An autosomal recessive deficiency of growth hormone leading to short stature. Patients have low but detectable levels of growth hormone, significantly retarded bone age, and a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:12655557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Kowarski syndrome (KWKS) [MIM:262650]: A syndrome clinically characterized by short stature associated with bioinactive growth hormone, normal or slightly increased growth hormone secretion, pathologically low insulin-like growth factor 1 levels, and normal catch-up growth on growth hormone replacement therapy. {ECO:0000269|PubMed:17519310, ECO:0000269|PubMed:8552145, ECO:0000269|PubMed:9276733}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 2 (IGHD2) [MIM:173100]: An autosomal dominant deficiency of growth hormone leading to short stature. Clinical severity is variable. Patients have a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:11502836, ECO:0000269|PubMed:9152628}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08324 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; DNA-binding; Quorum sensing; Reference proteome; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 18305.5 Length 163 Aromaticity 0.12 Instability index 46.52 Isoelectric point 5.19 Charge (pH=7) -6.78 2D Binding mode Binding energy (Kcal/mol) -6.26  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence FLELERSSGKLEWSAILQKMASDLGFSKILFGLLPKDSQDYENAFIVGNYPAAWREHYDRAGYARVDPTVSHCTQSVLPIFWEPSIYQTRKQHEFFEEASAAGLVYGLTMPLHGARGELGALSLSVEAENRAEANRFMESVLPTLWMLKDYALQSGAGLAFEH Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | SET domain containing 8 (KMT5A) | 5TEG | 4.59 | |
Target general information Gen name KMT5A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SETD8; SET8; SET07; SET domain-containing protein 8; PRSET7; PR/SET07; PR/SET domain-containing protein 07; PR-Set7; N-lysine methyltransferase KMT5A; Lysine-specific methylase 5A; Lysine N-methyltran Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, PR/SET subfamily Biochemical class Methyltransferase Function Specifically monomethylates 'Lys-20' of histone H4 (H4K20me1). H4K20me1 is enriched during mitosis and represents a specific tag for epigenetic transcriptional repression. Mainly functions in euchromatin regions, thereby playing a central role in the silencing of euchromatic genes. Required for cell proliferation, probably by contributing to the maintenance of proper higher-order structure of DNA during mitosis. Involved in chromosome condensation and proper cytokinesis. Nucleosomes are preferred as substrate compared to free histones. Mediates monomethylation of p53/TP53 at 'Lys-382', leading to repress p53/TP53-target genes. Plays a negative role in TGF-beta response regulation and a positive role in cell migration. Protein-lysine N-methyltransferase that monomethylates both histones and non-histone proteins. Related diseases Sick sinus syndrome 2 (SSS2) [MIM:163800]: The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS2 onset is in utero or at birth. {ECO:0000269|PubMed:15123648, ECO:0000269|PubMed:16407510, ECO:0000269|PubMed:20662977, ECO:0000269|PubMed:23103389}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 8 (BRGDA8) [MIM:613123]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:19165230}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Epilepsy, idiopathic generalized 18 (EIG18) [MIM:619521]: An autosomal dominant form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. EIG18 is characterized by onset of myoclonic seizures in infancy. Although the seizures remit, some patients may have later speech or cognitive impairment. {ECO:0000269|PubMed:30127718}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P62805; P07910; Q15672 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell cycle; Cell division; Chromatin regulator; Chromosome; Coiled coil; Direct protein sequencing; Methyltransferase; Mitosis; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A,D Molecular weight (Da) 19129.4 Length 167 Aromaticity 0.08 Instability index 49.18 Isoelectric point 7.88 Charge (pH=7) 1.37 2D Binding mode Binding energy (Kcal/mol) -6.27 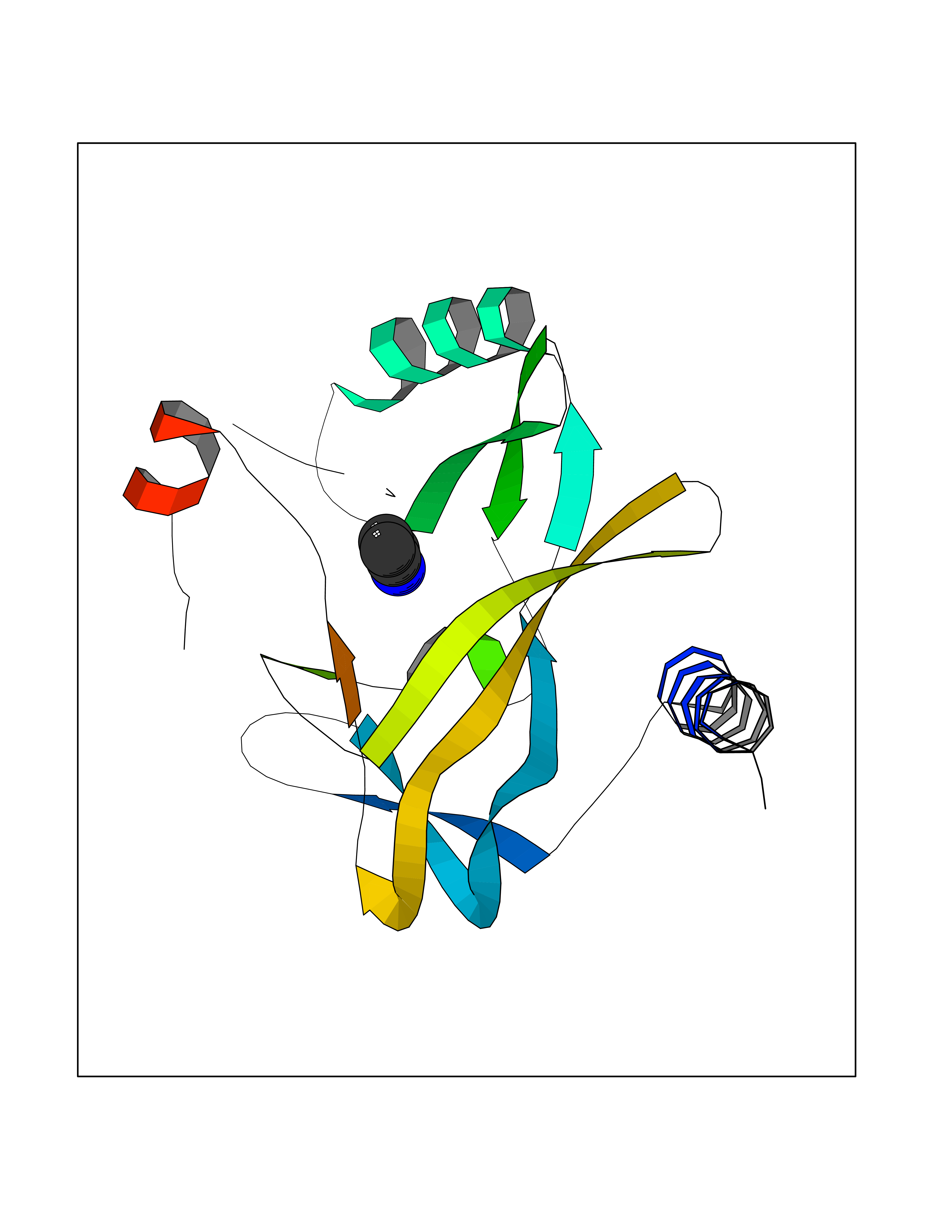 Molscript Map 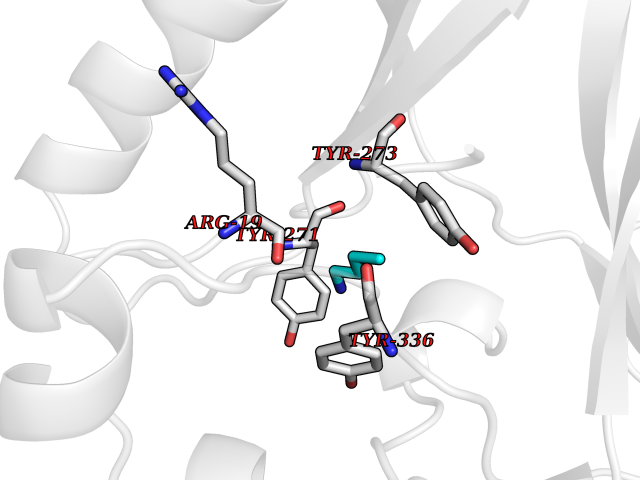 Pymol Map 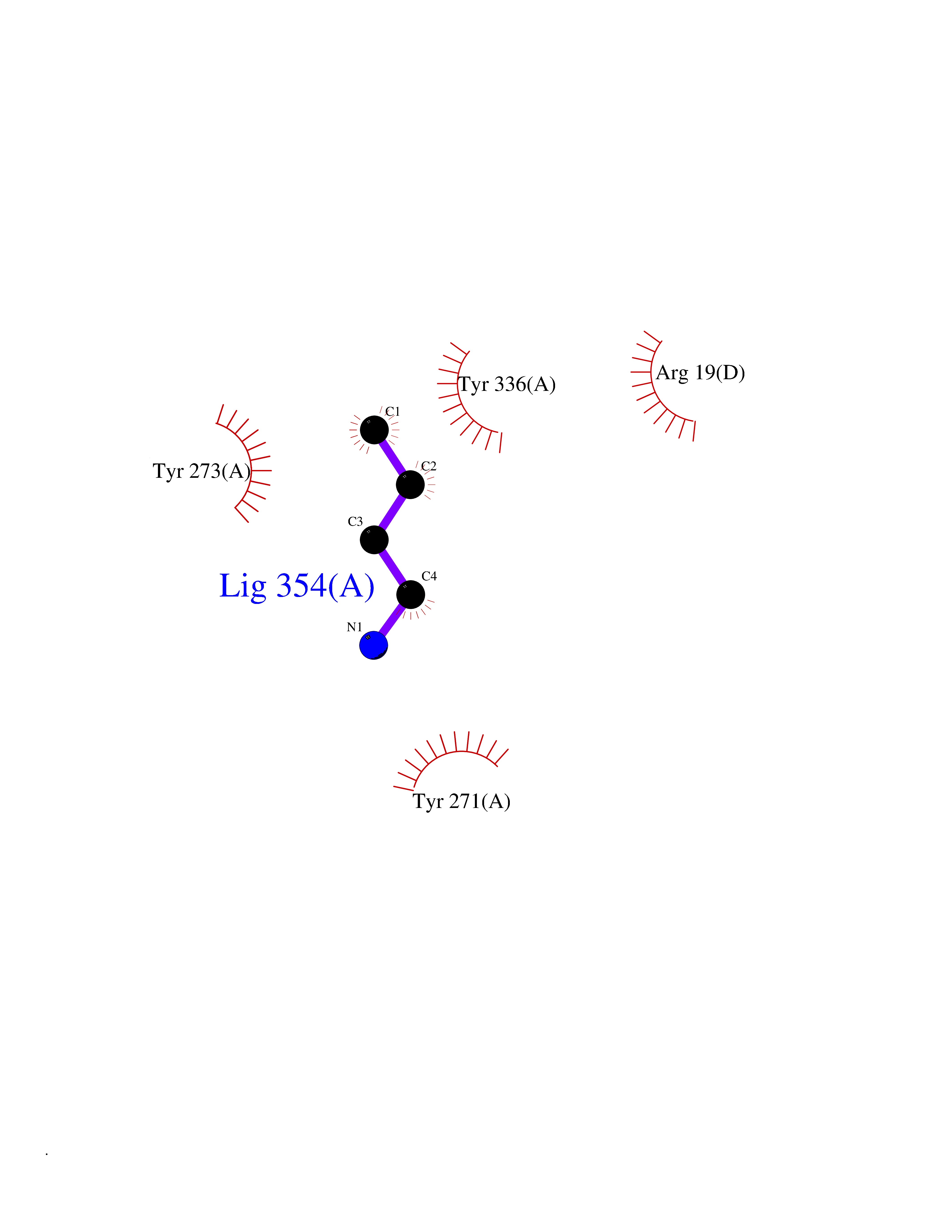 Ligplot Map 3D Binding mode Sequence KSKAELQSEERKRIDELIESGKEEGMKIDLIDGKGRGVIATKQFSRGDFVVEYHGDLIEITDAKKREALYAQDPSTGCYMYYFQYLSKTYCVDATRETNRLGRLINHSKSGNCQTKLHDIDGVPHLILIASRDIAAGEELLYDYGDRSKASIEAHPWLKHKRHRVLR Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Euchromatic histone-lysine N-methyltransferase 1 (EHMT1) | 5TTG | 4.59 | |
Target general information Gen name EHMT1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms EHMT1 Protein family Class V-like SAM-binding methyltransferase superfamily Biochemical class NA Function Histone methyltransferase that specifically mono- and dimethylates 'Lys-9' of histone H3 (H3K9me1 and H3K9me2, respectively) in euchromatin. H3K9me represents a specific tag for epigenetic transcriptional repression by recruiting HP1 proteins to methylated histones. Also weakly methylates 'Lys-27' of histone H3 (H3K27me). Also required for DNA methylation, the histone methyltransferase activity is not required for DNA methylation, suggesting that these 2 activities function independently. Probably targeted to histone H3 by different DNA-binding proteins like E2F6, MGA, MAX and/or DP1. During G0 phase, it probably contributes to silencing of MYC- and E2F-responsive genes, suggesting a role in G0/G1 transition in cell cycle. In addition to the histone methyltransferase activity, also methylates non-histone proteins: mediates dimethylation of 'Lys-373' of p53/TP53. Related diseases Kleefstra syndrome 1 (KLEFS1) [MIM:610253]: A form of Kleefstra syndrome, an autosomal dominant disease characterized by variable intellectual disability, psychomotor developmental delay, seizures, behavioral abnormalities, and facial dysmorphisms. KLEFS1 patients additionally manifest brachy(micro)cephaly, congenital heart defects, and urogenital defects. {ECO:0000269|PubMed:16826528, ECO:0000269|PubMed:19264732}. The disease is caused by variants affecting the gene represented in this entry. The syndrome can be either caused by intragenic EHMT1 mutations leading to haploinsufficiency of the EHMT1 gene or by a submicroscopic 9q34.3 deletion. Although it is not known if and to what extent other genes in the 9q34.3 region contribute to the syndrome observed in deletion cases, EHMT1 seems to be the major determinant of the core disease phenotype (PubMed:19264732). {ECO:0000269|PubMed:16826528, ECO:0000269|PubMed:19264732}. Drugs (DrugBank ID) NA Interacts with Q99549; Q04206; Q04207 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ANK repeat; Chromatin regulator; Chromosome; Disease variant; Intellectual disability; Isopeptide bond; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; S-adenosyl-L-methionine; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 30066.9 Length 260 Aromaticity 0.11 Instability index 49.19 Isoelectric point 5.73 Charge (pH=7) -4.88 2D Binding mode Binding energy (Kcal/mol) -6.26 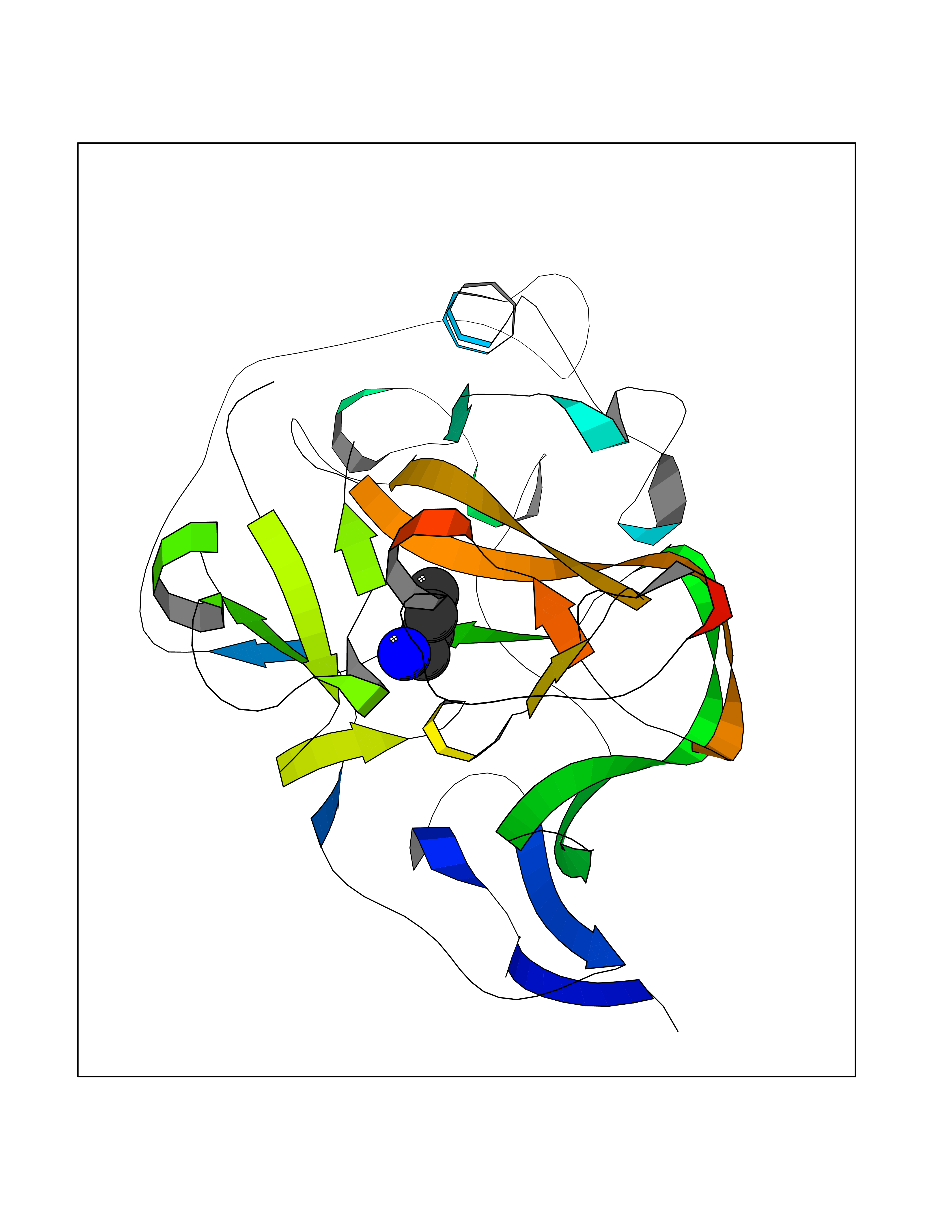 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence VERIVSRDIARGYERIPIPCVNAVDSEPCPSNYKYVSQNCVTSPMNIDRNITHLQYCVCIDDCSSSNCMCGQLSMRCWYDKDGRLLPEFNMAEPPLIFECNHACSCWRNCRNRVVQNGLRARLQLYRTRDMGWGVRSLQDIPPGTFVCEYVGELISDSEADVREEDSYLFDLDNDGEVYCIDARFYGNVSRFINHHCEPNLVPVRVFMAHQDLRFPRIAFFSTRLIEAGEQLGFDYGERFWDIKGKLFSCRCGSPKCRHS Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Dipeptidyl-peptidase 7 (DPP7) | 3N0T | 4.59 | |
Target general information Gen name DPP7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Quiescent cell proline dipeptidase; QPP; Dipeptidyl peptidase II; Dipeptidyl peptidase 7; Dipeptidyl peptidase 2; Dipeptidyl aminopeptidase II; DPP2; DPP II Protein family Peptidase S28 family Biochemical class NA Function Plays an important role in the degradation of some oligopeptides. Related diseases Hereditary non-polyposis colorectal cancer 6 (HNPCC6) [MIM:614331]: An autosomal dominant disease associated with marked increase in cancer susceptibility. It is characterized by a familial predisposition to early-onset colorectal carcinoma (CRC) and extra-colonic tumors of the gastrointestinal, urological and female reproductive tracts. HNPCC is reported to be the most common form of inherited colorectal cancer in the Western world. Clinically, HNPCC is often divided into two subgroups. Type I is characterized by hereditary predisposition to colorectal cancer, a young age of onset, and carcinoma observed in the proximal colon. Type II is characterized by increased risk for cancers in certain tissues such as the uterus, ovary, breast, stomach, small intestine, skin, and larynx in addition to the colon. Diagnosis of classical HNPCC is based on the Amsterdam criteria: 3 or more relatives affected by colorectal cancer, one a first degree relative of the other two; 2 or more generation affected; 1 or more colorectal cancers presenting before 50 years of age; exclusion of hereditary polyposis syndromes. The term 'suspected HNPCC' or 'incomplete HNPCC' can be used to describe families who do not or only partially fulfill the Amsterdam criteria, but in whom a genetic basis for colon cancer is strongly suspected. {ECO:0000269|PubMed:9590282}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Esophageal cancer (ESCR) [MIM:133239]: A malignancy of the esophagus. The most common types are esophageal squamous cell carcinoma and adenocarcinoma. Cancer of the esophagus remains a devastating disease because it is usually not detected until it has progressed to an advanced incurable stage. {ECO:0000269|PubMed:10789724}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Loeys-Dietz syndrome 2 (LDS2) [MIM:610168]: An aortic aneurysm syndrome with widespread systemic involvement, characterized by arterial tortuosity and aneurysms, hypertelorism, and bifid uvula or cleft palate. Physical findings include prominent joint laxity, easy bruising, wide and atrophic scars, velvety and translucent skin with easily visible veins, spontaneous rupture of the spleen or bowel, and catastrophic complications of pregnancy, including rupture of the gravid uterus and the arteries, either during pregnancy or in the immediate postpartum period. Some patients have craniosynostosis, exotropy, micrognathia and retrognathia, structural brain abnormalities, and intellectual deficit. {ECO:0000269|PubMed:15235604, ECO:0000269|PubMed:15731757, ECO:0000269|PubMed:16027248, ECO:0000269|PubMed:16251899, ECO:0000269|PubMed:19533785, ECO:0000269|PubMed:19883511, ECO:0000269|PubMed:20101701, ECO:0000269|PubMed:20358619, ECO:0000269|PubMed:21949523, ECO:0000269|PubMed:22113417}. The disease is caused by variants affecting the gene represented in this entry. TGFBR2 mutations Cys-460 and His-460 have been reported to be associated with thoracic aortic aneurysms and dissection (TAAD). This phenotype, also known as thoracic aortic aneurysms type 3 (AAT3), is distinguised from LDS2 by having aneurysms restricted to thoracic aorta. As individuals carrying these mutations also exhibit descending aortic disease and aneurysms of other arteries (PubMed:16027248), they have been considered as LDS2 by the OMIM resource. {ECO:0000269|PubMed:16027248}. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.14.2 Uniprot keywords 3D-structure; Aminopeptidase; Cleavage on pair of basic residues; Cytoplasmic vesicle; Direct protein sequencing; Glycoprotein; Hydrolase; Lysosome; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 50240.9 Length 452 Aromaticity 0.13 Instability index 31.75 Isoelectric point 5.39 Charge (pH=7) -10.46 2D Binding mode Binding energy (Kcal/mol) -6.26  Molscript Map  Pymol Map 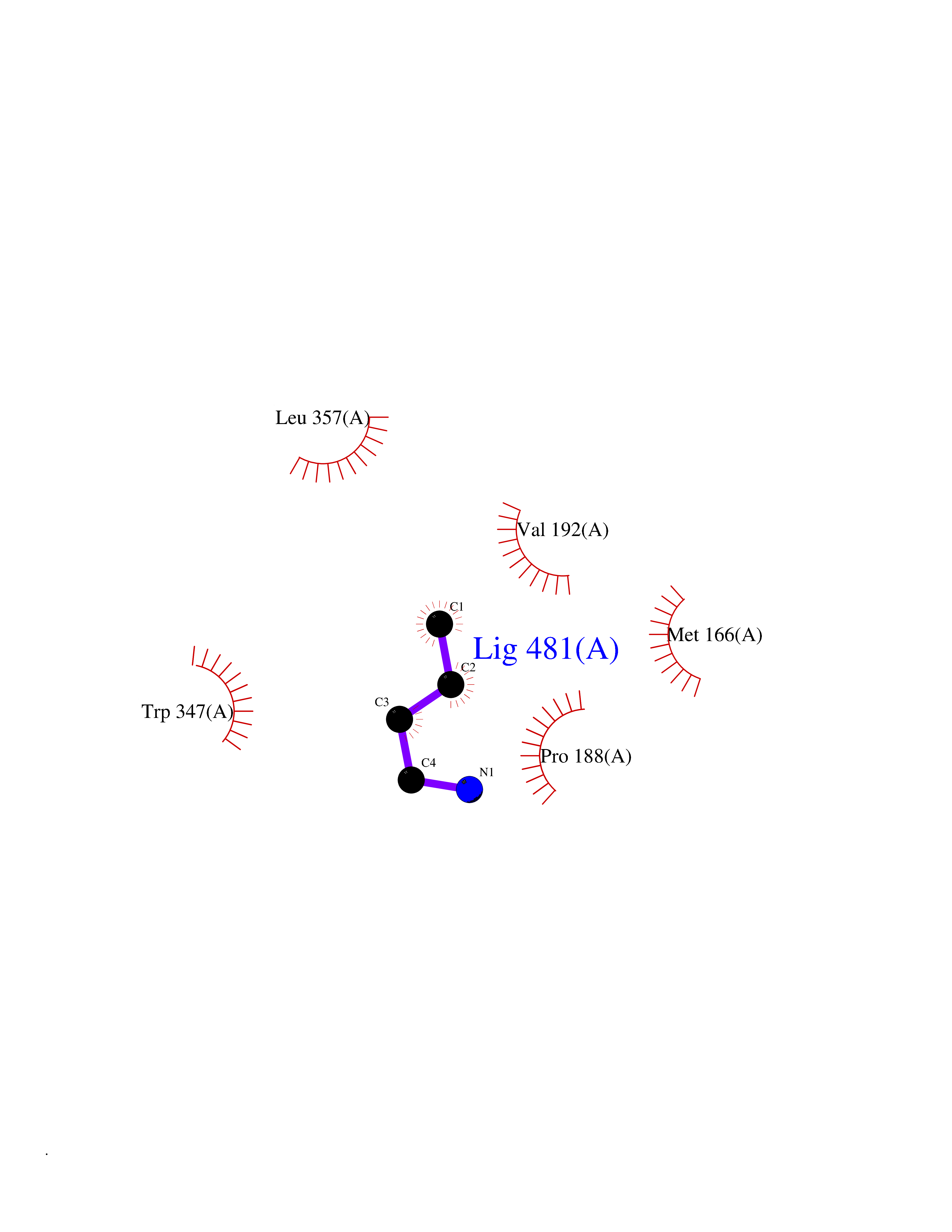 Ligplot Map 3D Binding mode Sequence DPGFQERFFQQRLDHFNFERFGNKTFPQRFLVSDRFWVRGEGPIFFYTGNEGDVWAFANNSAFVAELAAERGALLVFAEHRYYGKSLPFGAQSTQRGHTELLTVEQALADFAELLRALRRDLGAQDAPAIAFGGSYGGMLSAYLRMKYPHLVAGALAASAPVLAVAGLGDSNQFFRDVTADFEGQSPKCTQGVREAFRQIKDLFLQGAYDTVRWEFGTCQPLSDEKDLTQLFMFARNAFTVLAMMDYPYPTDFLGPLPANPVKVGCDRLLSEAQRITGLRALAGLVYNASGSEHCYDIYRLYHSCADPTGCGTGPDARAWDYQACTEINLTFASNNVTDMFPDLPFTDELRQRYCLDTWGVWPRPDWLLTSFWGGDLRAASNIIFSNGNLDPWAGGGIRRNLSASVIAVTIQGGAHHLDLRASHPEDPASVVEARKLEATIIGEWVKAARRE Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Histamine H1 receptor (H1R) | 7DFL | 4.59 | |
Target general information Gen name HRH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HH1R; H1R Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function In peripheral tissues, the H1 subclass of histamine receptors mediates the contraction of smooth muscles, increase in capillary permeability due to contraction of terminal venules, and catecholamine release from adrenal medulla, as well as mediating neurotransmission in the central nervous system. Related diseases Hypertriglyceridemia, transient infantile (HTGTI) [MIM:614480]: An autosomal recessive disorder characterized by onset of moderate to severe transient hypertriglyceridemia in infancy that normalizes with age. The hypertriglyceridemia is associated with hepatomegaly, moderately elevated transaminases, persistent fatty liver, and the development of hepatic fibrosis. {ECO:0000269|PubMed:22226083, ECO:0000269|PubMed:24549054}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01615; DB09488; DB06766; DB01246; DB00321; DB00543; DB08799; DB01238; DB14185; DB06216; DB00637; DB00719; DB00972; DB00245; DB00767; DB04890; DB06698; DB11591; DB09128; DB01237; DB00835; DB00354; DB09016; DB00748; DB06016; DB00341; DB08936; DB08800; DB01114; DB00477; DB01239; DB00568; DB00215; DB00283; DB04837; DB00363; DB01176; DB00434; DB01151; DB00967; DB00405; DB09555; DB00985; DB08801; DB01075; DB01146; DB09167; DB01142; DB00366; DB01084; DB05492; DB00751; DB01175; DB06678; DB00950; DB04841; DB00502; DB05381; DB05079; DB00557; DB04946; DB00458; DB08802; DB00920; DB00555; DB01106; DB06282; DB00455; DB09195; DB00408; DB00934; DB00737; DB06691; DB01071; DB00902; DB01403; DB06148; DB00370; DB00540; DB05080; DB06229; DB00334; DB00768; DB01173; DB01267; DB00715; DB08922; DB01619; DB01620; DB06153; DB00433; DB00420; DB01069; DB00777; DB01224; DB00912; DB00734; DB11614; DB05345; DB00342; DB04905; DB11235; DB00797; DB00656; DB00726; DB00792; DB00427; DB09185; DB00246; DB01624 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32298.5 Length 275 Aromaticity 0.15 Instability index 36.59 Isoelectric point 9.54 Charge (pH=7) 16.01 2D Binding mode Binding energy (Kcal/mol) -6.26 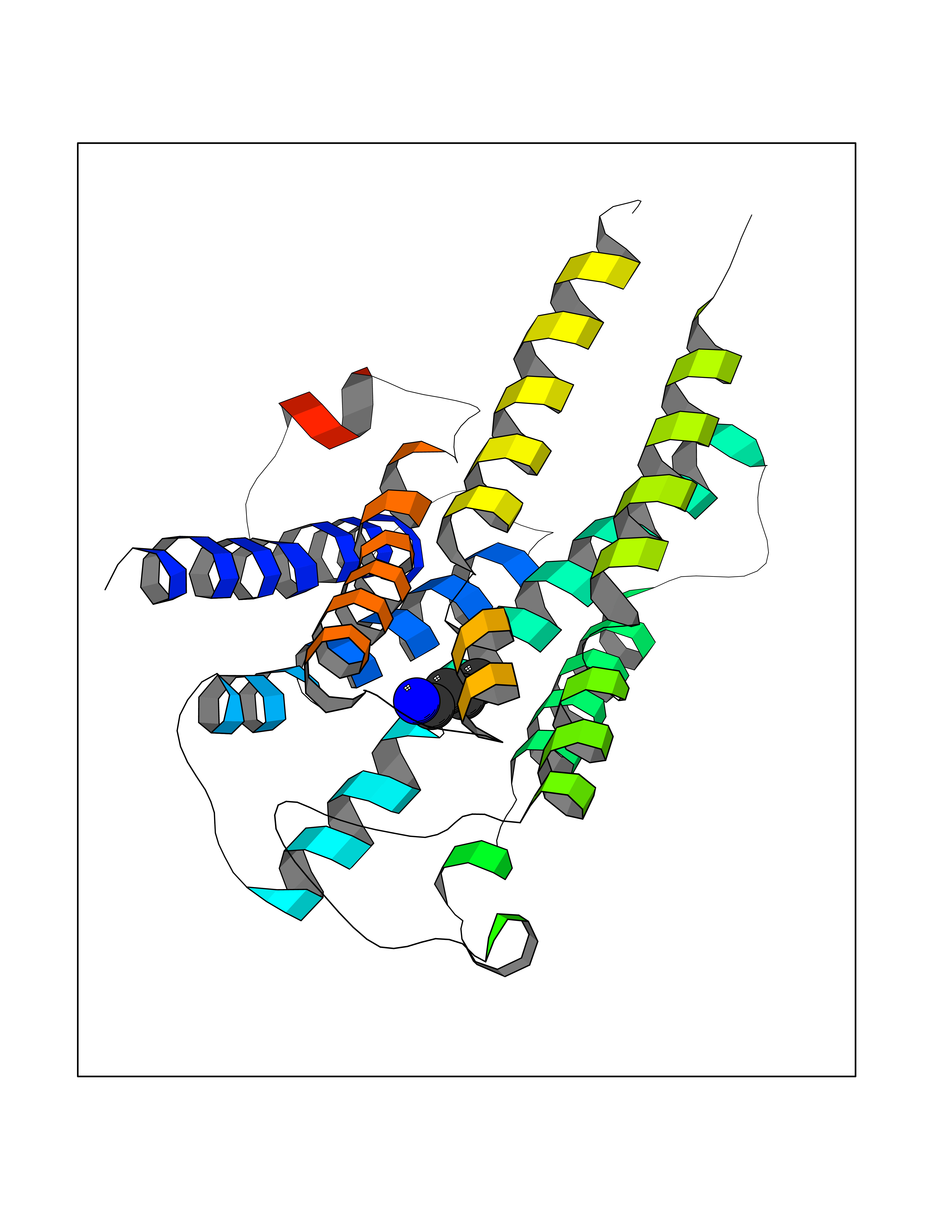 Molscript Map 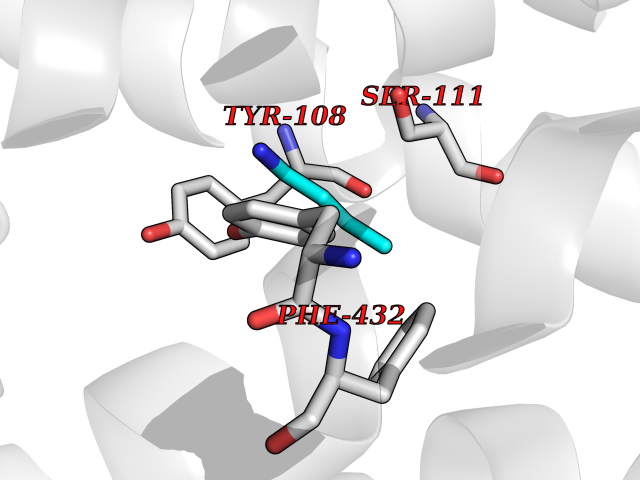 Pymol Map 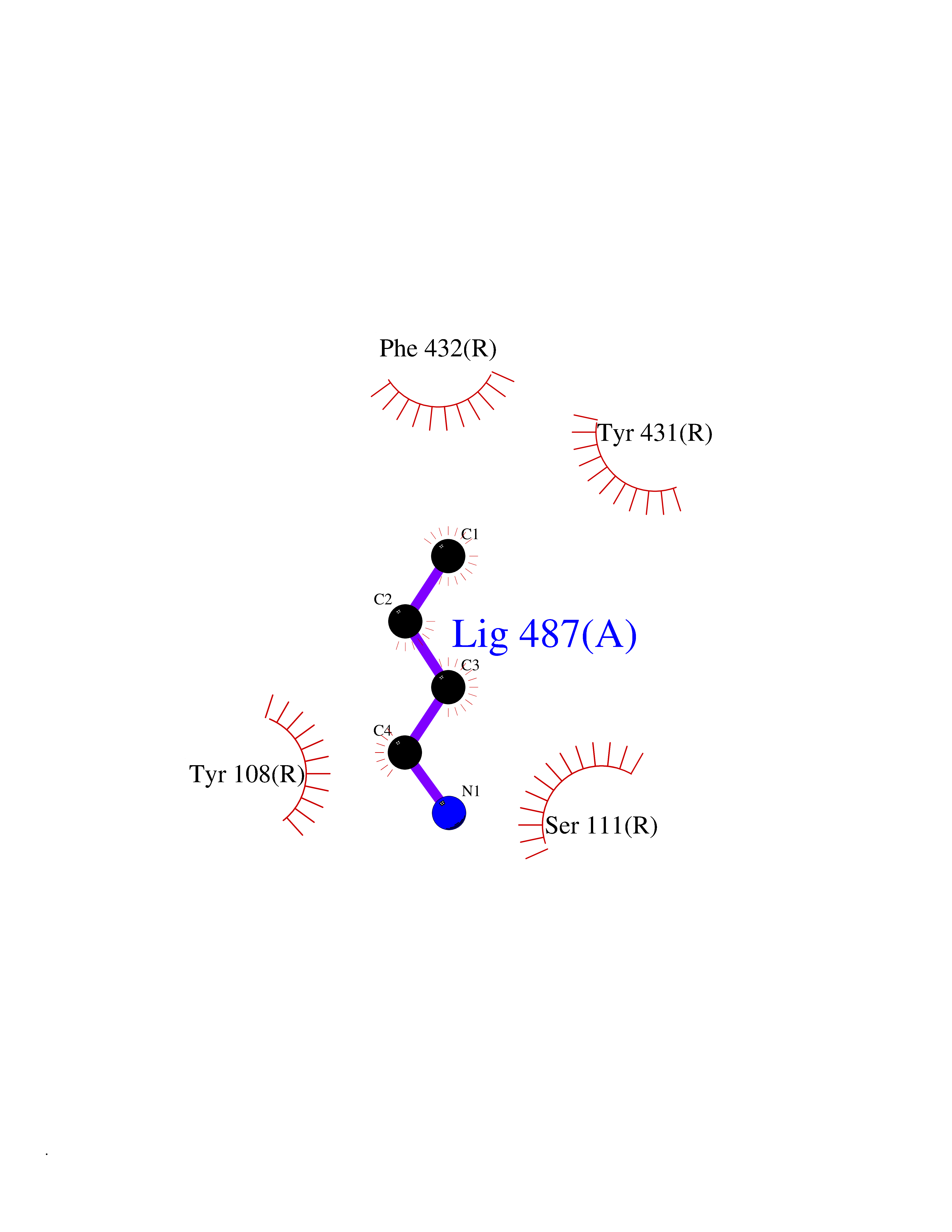 Ligplot Map 3D Binding mode Sequence MPLVVVLSTICLVTVGLNLLVLYAVRSERKLHTVGNLYIVSLSVADLIVGAVVMPMNILYLLMSKWSLGRPLCLFWLSMDYVASTASIFSVFILCIDRYRSVQQPLRYLKYRTKTRASATILGAWFLSFLWVIPILGWNHFMQQTSVRREDKCETDFYDVTWFKVMTAIINFYLPTLLMLWFYAKIYKAVRQHCLHMNRERKAAKQLGFIMAAFILCWIPYFIFFMVIAFCKNCCNEHLHMFTIWLGYINSTLNPLIYPLCNENFKKTFKRILHI Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Ubiquitin carboxyl-terminal hydrolase 2 (USP2) | 5XU8 | 4.59 | |
Target general information Gen name USP2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ubiquitin-specific-processing protease 2; Ubiquitin thioesterase 2; UBP41; Deubiquitinating enzyme 2; 41 kDa ubiquitin-specific protease Protein family Peptidase C19 family, USP2 subfamily Biochemical class Peptidase Function Isoform 1 and isoform 4 possess both ubiquitin-specific peptidase and isopeptidase activities. Deubiquitinates MDM2 without reversing MDM2-mediated p53/TP53 ubiquitination and thus indirectly promotes p53/TP53 degradation and limits p53 activity. Has no deubiquitinase activity against p53/TP53. Prevents MDM2-mediated degradation of MDM4. Plays a role in the G1/S cell-cycle progression in normal and cancer cells. Regulates the circadian clock by modulating its intrinsic circadian rhythm and its capacity to respond to external cues. Associates with clock proteins and deubiquitinates core clock component PER1 but does not affect its overall stability. Regulates the nucleocytoplasmic shuttling and nuclear retention of PER1 and its repressive role on the clock transcription factors CLOCK and ARNTL/BMAL1. Plays a role in the regulation of myogenic differentiation of embryonic muscle cells. Hydrolase that deubiquitinates polyubiquitinated target proteins such as MDM2, MDM4 and CCND1. Related diseases Defects in AKT2 are a cause of susceptibility to breast cancer (BC). AKT2 promotes metastasis of tumor cells without affecting the latency of tumor development. May play a role in glioblastoma cell survival (PubMed:20167810). {ECO:0000269|PubMed:20167810}.; DISEASE: Type 2 diabetes mellitus (T2D) [MIM:125853]: A multifactorial disorder of glucose homeostasis caused by a lack of sensitivity to insulin. Affected individuals usually have an obese body habitus and manifestations of a metabolic syndrome characterized by diabetes, insulin resistance, hypertension and hypertriglyceridemia. The disease results in long-term complications that affect the eyes, kidneys, nerves, and blood vessels. {ECO:0000269|PubMed:15166380, ECO:0000269|PubMed:19164855}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Hypoinsulinemic hypoglycemia with hemihypertrophy (HIHGHH) [MIM:240900]: A disorder characterized by hypoglycemia, low insulin levels, low serum levels of ketone bodies and branched-chain amino acids, left-sided hemihypertrophy, neonatal macrosomia, reduced consciousness and hypoglycemic seizures. {ECO:0000269|PubMed:21979934}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NYB9-2; P12814; P35609; Q08043; Q86U10; Q86V38; P56945; Q8TD16-2; Q96CA5; A2RRN7; Q13137; Q9H257-2; Q96JN2-2; Q2TAC2; A6NC98; Q96MT8-3; Q8NHQ1; Q9BSW2; Q8N4Y2-3; Q8WTU0; O75140-2; Q9NRI5-2; Q8N9I9; Q9H596; Q8WWB3; Q5JST6; Q9NRA8; O00471; Q96B26; P57678; Q08379; Q9NYA3; A6NEM1; Q6PI77; Q14451-3; Q4V328; Q9NSC5; Q9UJC3; Q96ED9-2; Q8IYA8; Q9UKT9; Q5TA45; Q96N16; O75564-2; Q674X7-2; Q9BVG8; Q9BVG8-5; P19012; Q7Z3Y8; Q15323; Q14525; O76011; Q92764; Q6A162; Q9UBR4-2; Q969G2; Q03252; Q9BRK4; Q00987; Q9UJV3-2; Q5VZ52; Q13084; Q5JR59; Q5JR59-3; Q15742; Q9GZM8; I6L9F6; P07196; O43482; Q96CV9; Q4G0R1; Q9NRD5; Q58EX7; Q8ND90; Q16633; Q9GZV8; Q6MZQ0; Q15276; Q8HWS3; Q59EK9-3; P60903; O14492-2; O60504; Q99932-2; A6NLX3; P51692; Q86VP1; Q8WW24; Q9UBB9; Q08117-2; Q03169; Q13077; Q12933; Q9Y4K3; P36406; P14373; Q86XT4; Q15654; Q8N6Y0; Q70EL1-9; Q9UK41-2; Q8N1B4; O96006; Q9NZV7; Q9UGI0; P05067; P54253; G5E9A7; Q01658; Q00403; Q9Y5Q9; P04792; O43464; P42858; Q8WXH2; O60333-2; A0A6Q8PF08; O60260-5; P60891; Q9Y3C5; Q7Z333; P37840; P00441; Q7Z699; Q13148; O76024 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Alternative splicing; Biological rhythms; Cell cycle; Cytoplasm; Hydrolase; Membrane; Metal-binding; Myogenesis; Nucleus; Protease; Proteomics identification; Reference proteome; Thiol protease; Ubl conjugation pathway; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 37785.5 Length 327 Aromaticity 0.11 Instability index 42.45 Isoelectric point 8.23 Charge (pH=7) 3.56 2D Binding mode Binding energy (Kcal/mol) -6.27  Molscript Map 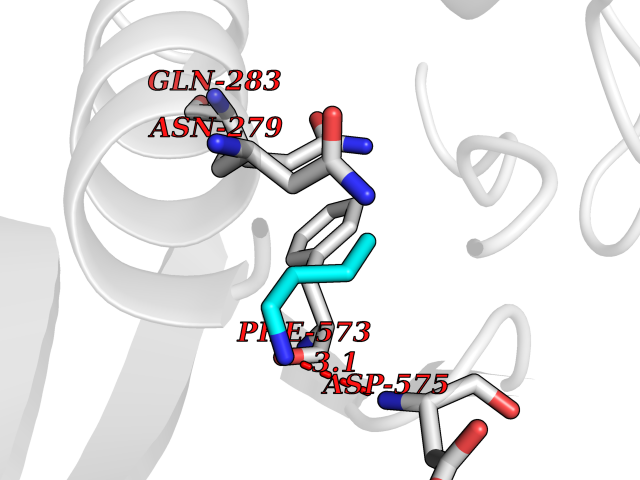 Pymol Map 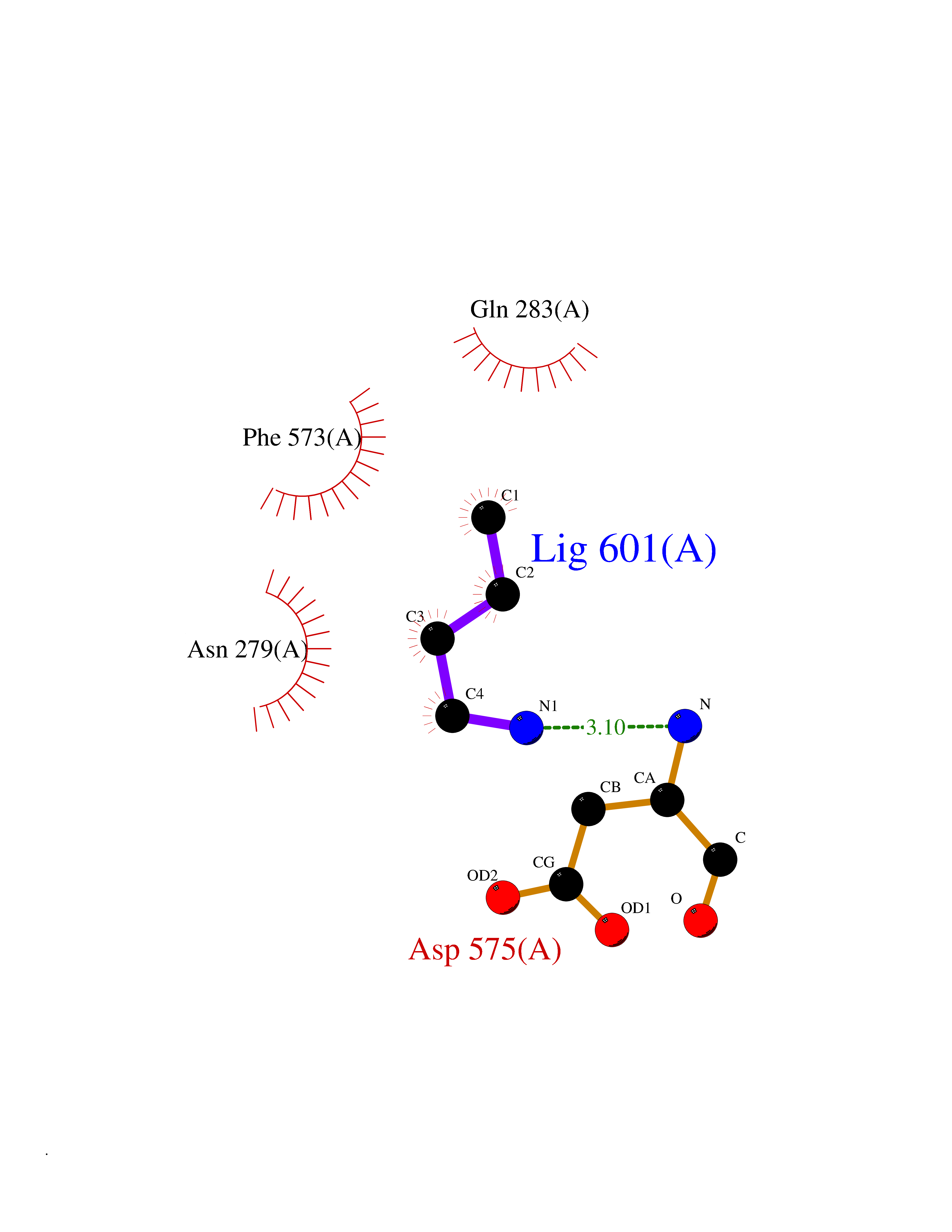 Ligplot Map 3D Binding mode Sequence QGLAGLRNLGNTCFMNSILQCLSNTRELRDYCLQRLYMRDLHHGSNAHTALVEEFAKLIQTIWTSSPNDVVSPSEFKTQIQRYAPRFVGYNQQDAQEFLRFLLDGLHNEVNRVNLDHLPDDEKGRQMWRKYLEREDSRIGDLFVGQLKSSLTCTDCGYCSTVFDPFWDLSLPIAKRGYPEVTLMDCMRLFTKEDVLDGDEKPTCCRCRGRKRCIKKFSIQRFPKILVLHLKRFSESRIRTSKLTTFVNFPLRDLDLREFASENTNHAVYNLYAVSNHSGTTMGGHYTAYCRSPGTGEWHTFNDSSVTPMSSSQVRTSDAYLLFYELA Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Chromodomain-helicase-DNA-binding protein 1 | 4O42 | 4.58 | |
Target general information Gen name CHD1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family SNF2/RAD54 helicase family Biochemical class Dna binding protein / viral protein Function ATP binding.ATP-dependent DNA helicase activity.DNA binding.Methylated histone binding. Related diseases Pilarowski-Bjornsson syndrome (PILBOS) [MIM:617682]: An autosomal dominant disorder characterized by developmental delay, speech apraxia, intellectual disability, autism, and facial dysmorphic features. Some patients may have seizures. {ECO:0000269|PubMed:28866611}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O60341-1; B2BUF1; P28799; O76024 EC number 3.6.4.12 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Chromatin regulator; Cytoplasm; Disease variant; DNA-binding; Helicase; Hydrolase; Intellectual disability; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 20969.1 Length 180 Aromaticity 0.12 Instability index 46.35 Isoelectric point 5.88 Charge (pH=7) -2.83 2D Binding mode Binding energy (Kcal/mol) -6.24  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence EFETIERFMDCRIGRKGATGATTTIYAVEADGDPNAGFEKNKEPGEIQYLIKWKGWSHIHNTWETEETLKQQNVRGMKKLDNYKKKDQETKRWLKNASPEDVEYYNCQQELTDDLHKQYQIVERIIAHSNQKSAAGYPDYYCKWQGLPYSECSWEDGALISKKFQACIDEYFSRTARSXV Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Natriuretic peptides B | 1YK1 | 4.58 | |
Target general information Gen name NPPB Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Natriuretic peptide family Biochemical class Hormone / growth factor receptor Function Diuretic hormone activity.Hormone activity.Peptide hormone receptor binding.Receptor binding. Related diseases Multiple fibroadenomas of the breast (MFAB) [MIM:615554]: A benign breast disease marked by lobuloalveolar growth with abnormally high proliferation of the epithelium, and characterized by the presence of more than 3 fibroadenomas in one breast. Fibroadenomas are adenomas containing fibrous tissue. {ECO:0000269|PubMed:18779591}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperprolactinemia (HPRL) [MIM:615555]: A disorder characterized by increased levels of prolactin in the blood not associated with gestation or the puerperium. HPRL may result in infertility, hypogonadism, and galactorrhea. {ECO:0000269|PubMed:24195502}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01136; DB06412 Interacts with A8MQ03; P57678; Q6A162; P60411; Q7Z3S9; P25788; Q9UJW9 EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Disulfide bond; Glycoprotein; Hormone; Pharmaceutical; Proteoglycan; Proteomics identification; Reference proteome; Secreted; Signal; Vasoactive; Vasodilator Protein physicochemical properties Chain ID E Molecular weight (Da) 46353.1 Length 415 Aromaticity 0.1 Instability index 37.91 Isoelectric point 5.51 Charge (pH=7) -12.09 2D Binding mode Binding energy (Kcal/mol) -6.24  Molscript Map  Pymol Map 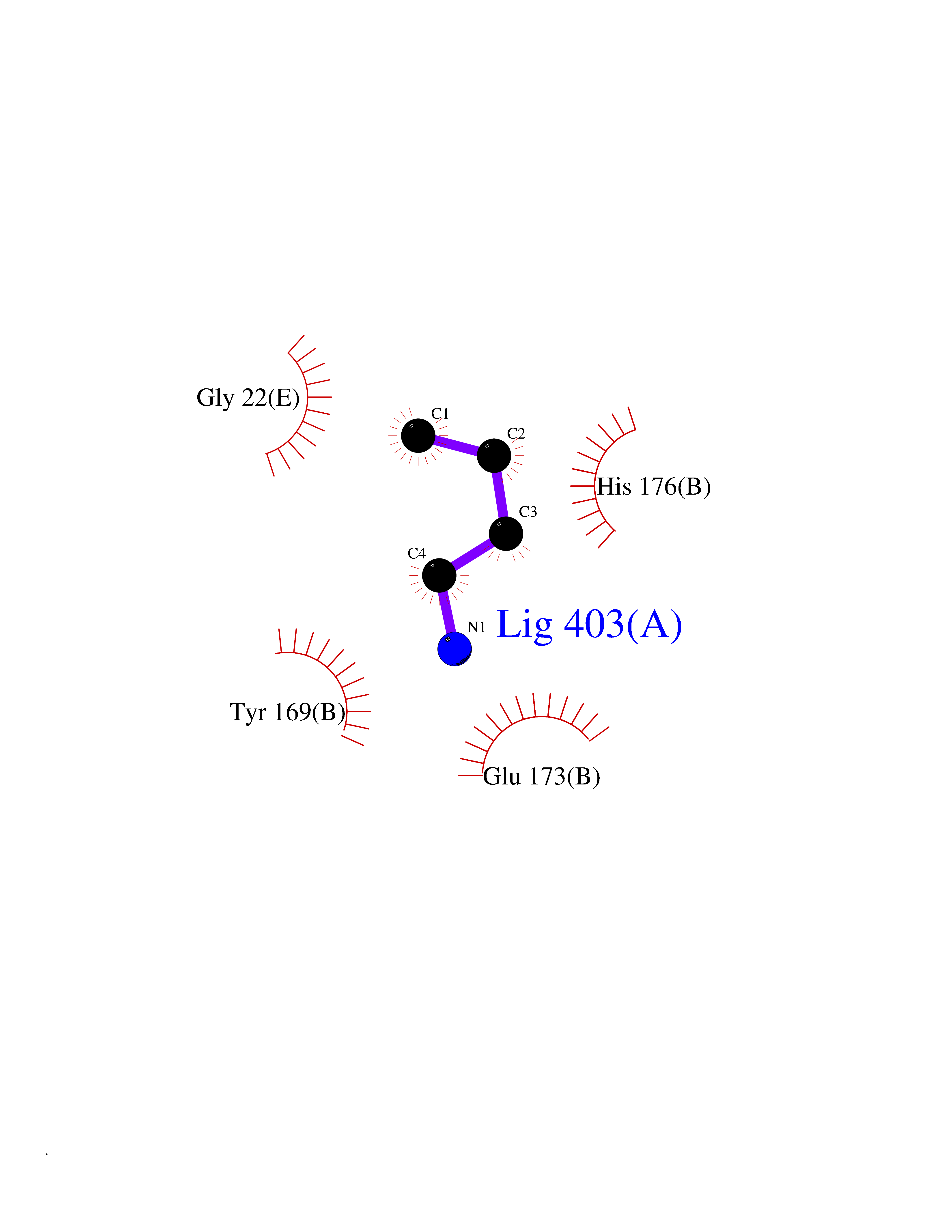 Ligplot Map 3D Binding mode Sequence GCFGRKMDRISSSSGLGCKVLALPPQKIEVLVLLPQDDSYLFSLTRVRPAIEYALRSVEGLLPPGTRFQVAYEDSDCGNRALFSLVDRVAAARGAKPDLILGPVCEYAAAPVARLASHWDLPMLSAGALAAGFQHKDSEYSHLTRVAPAYAKMGEMMLALFRHHHWSRAALVYSDDKLERNCYFTLEGVHEVFQEEGLHTSIYSFDETKDLDLEDIVRNIQASERVVIMCASSDTIRSIMLVAHRHGMTSGDYAFFNIELFNSSSYGDGSWKRGDKHDFEAKQAYSSLQTVTLLRTVKPEFEKFSMEVKSSVEKQGLNMEDYVNMFVEGFHDAILLYVLALHEVLRAGYSKKDGGKIIQQTWNRTFEGIAGQVSIDANGDRYGDFSVIAMTDVEAGTQEVIGDYFGKEGRFEMRP Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Cytochrome b (Complex III subunit 3) (Complex III subunit III) (Cytochrome b-c1 complex subunit 3) (Ubiquinol-cytochrome-c reductase complex cytochrome b subunit) | 1SQB | 4.58 | |
Target general information Gen name MT-CYB Organism Bos taurus (Bovine) Uniprot ID TTD ID NA Synonyms CYTB;MTCYB;COB Protein family Cytochrome b family Biochemical class NA Function Component of the ubiquinol-cytochrome c reductase complex (complex III or cytochrome b-c1 complex) that is part of the mitochondrial respiratory chain. The b-c1 complex mediates electron transfer from ubiquinol to cytochrome c. Contributes to the generation of a proton gradient across the mitochondrial membrane that is then used for ATP synthesis. {ECO:0000269|PubMed:1327781, ECO:0000269|PubMed:20025846, ECO:0000269|PubMed:9485330, ECO:0000305|PubMed:189810}." Related diseases Combined oxidative phosphorylation deficiency 6 (COXPD6) [MIM:300816]: A mitochondrial disease resulting in a neurodegenerative disorder characterized by psychomotor delay, hypotonia, areflexia, muscle weakness and wasting. Some patients manifest prenatal ventriculomegaly and severe postnatal encephalomyopathy. {ECO:0000269|PubMed:20362274, ECO:0000269|PubMed:22019070, ECO:0000269|PubMed:25583628, ECO:0000269|PubMed:26004228, ECO:0000269|PubMed:26173962, ECO:0000269|PubMed:27178839}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, X-linked recessive, 4, with or without cerebellar ataxia (CMTX4) [MIM:310490]: A neuromuscular disorder characterized by progressive sensorimotor axonal neuropathy, distal sensory impairment, difficulty walking due to peripheral neuropathy and/or cerebellar ataxia, and deafness due to auditory neuropathy. Additional features include cognitive impairment, cerebellar atrophy, dysarthria, abnormal extraocular movements, tremor, dysmetria and spasticity. The age at onset ranges from infancy to young adulthood. {ECO:0000269|PubMed:23217327, ECO:0000269|PubMed:26004228}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, X-linked, 5, with peripheral neuropathy (DFNX5) [MIM:300614]: A form of hearing loss characterized by absent or severely abnormal auditory brainstem response, abnormal middle ear reflexes, abnormal speech discrimination, loss of outer hair cell function, and cochlear nerve hypoplasia. DFNX5 patients manifest auditory neuropathy with childhood onset, associated with distal sensory impairment affecting the peripheral nervous system. {ECO:0000269|PubMed:25986071}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepimetaphyseal dysplasia, X-linked, with hypomyelinating leukodystrophy (SEMDHL) [MIM:300232]: An X-linked recessive developmental disorder characterized by slowly progressive skeletal and neurologic abnormalities, including short stature, large and deformed joints, significant motor impairment, visual defects, and sometimes cognitive deficits. Affected individuals typically have normal early development in the first year or so of life, followed by development regression and the development of symptoms. Brain imaging shows white matter abnormalities consistent with hypomyelinating leukodystrophy. {ECO:0000269|PubMed:28842795}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Electron transport; Heme; Iron; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Reference proteome; Respiratory chain; Transmembrane; Transmembrane helix; Transport; Ubiquinone Protein physicochemical properties Chain ID C,D,F,G,H Molecular weight (Da) 99281.2 Length 866 Aromaticity 0.12 Instability index 43.81 Isoelectric point 8.32 Charge (pH=7) 7.12 2D Binding mode Binding energy (Kcal/mol) -6.24 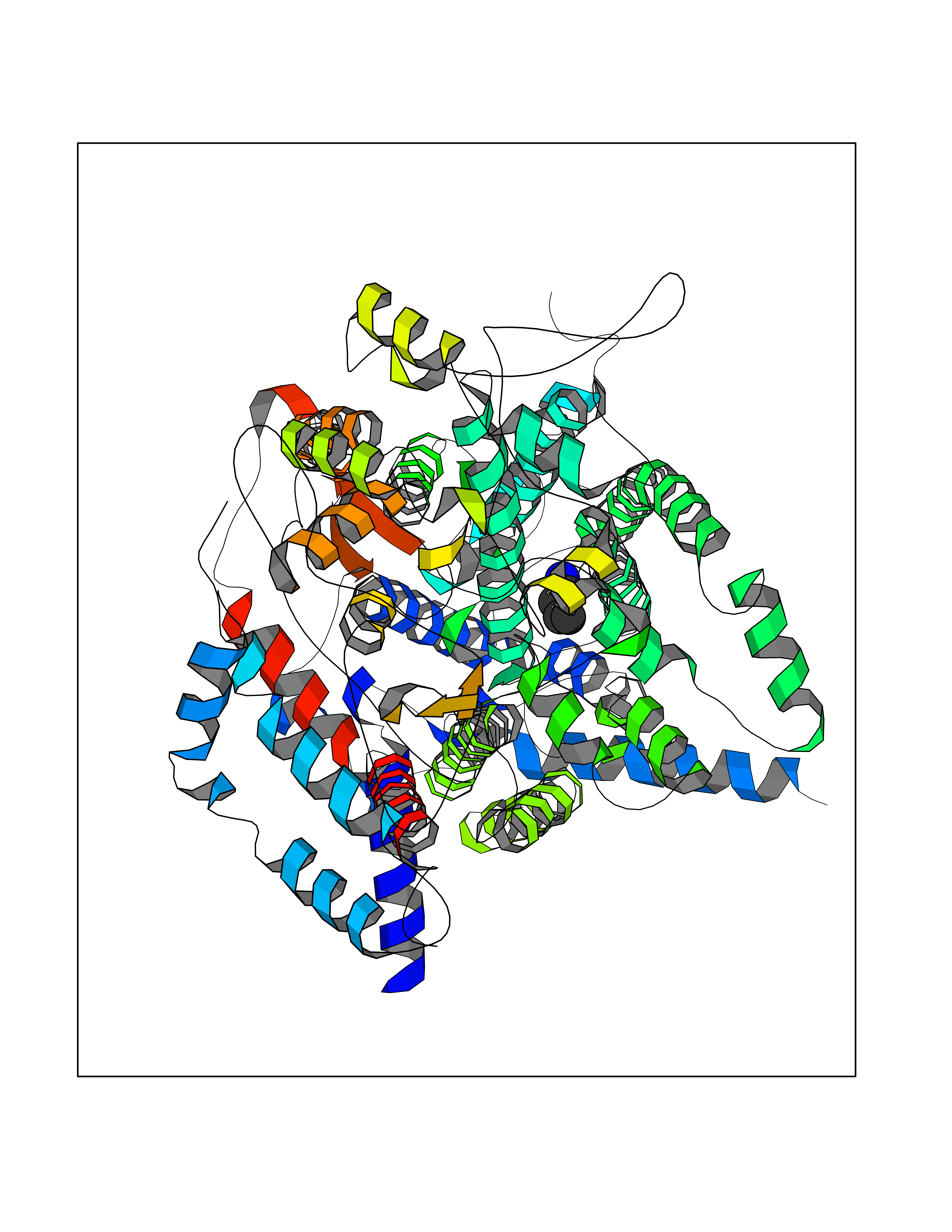 Molscript Map 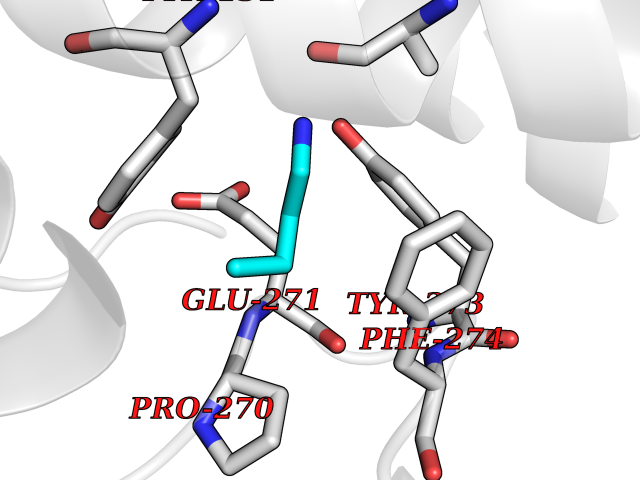 Pymol Map 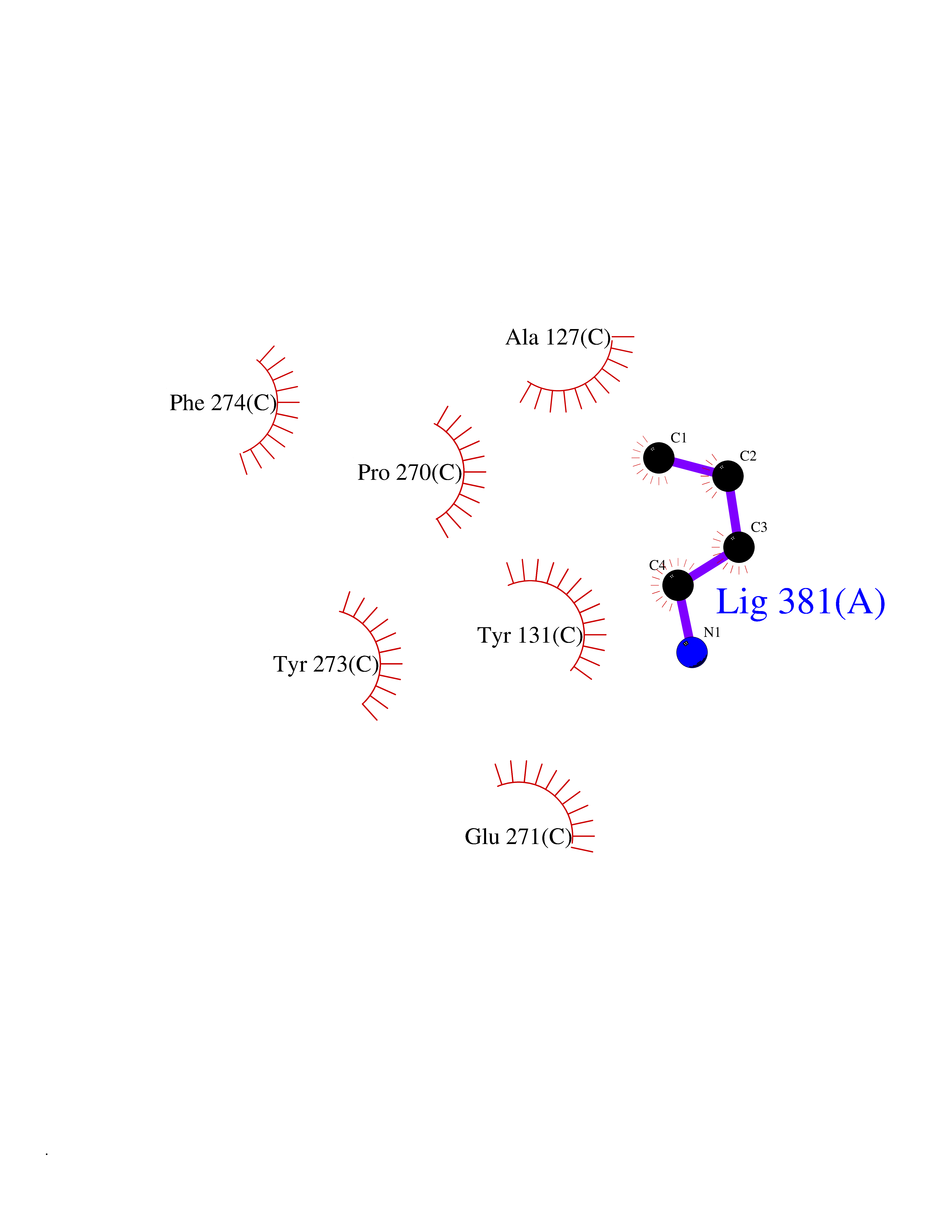 Ligplot Map 3D Binding mode Sequence VSASSRWLEGIRKWYYNAAGFNKLGLMRDDTIHENDDVKEAIRRLPENLYDDRVFRIKRALDLSMRQQILPKEQWTKYEEDKSYLEPYLKEVIRERKEREEWAKKELVDPLTTVREQCEQLEKCVKARERLELCDERVSSRSQTEEDCTEELLDFLHARDHCVAHKLFNSLKTNIRKSHPLMKIVNNAFIDLPAPSNISSWWNFGSLLGICLILQILTGLFLAMHYTSDTTTAFSSVTHICRDVNYGWIIRYMHANGASMFFICLYMHVGRGLYYGSYTFLETWNIGVILLLTVMATAFMGYVLPWGQMSFWGATVITNLLSAIPYIGTNLVEWIWGGFSVDKATLTRFFAFHFILPFIIMAIAMVHLLFLHETGSNNPTGISSDVDKIPFHPYYTIKDILGALLLILALMLLVLFAPDLLGDPDNYTPANPLNTPPHIKPEWYFLFAYAILRSIPNKLGGVLALAFSILILALIPLLHTSKQRSMMFRPLSQCLFWALVADLLTLTWIGGQPVEHPYITIGQLASVLYFLLILVLMPTAGTIENKLLKWSDLELHPPSYPWSHRGLLSSLDHTSIRRGFQVYKQVCSSCHSMDYVAYRHLVGVCYTEDEAKALAEEVEVQDGPNEDGEMFMRPGKLSDYFPKPYPNPEAARAANNGALPPDLSYIVRARHGGEDYVFSLLTGYCEPPTGVSLREGLYFNPYFPGQAIGMAPPIYNEVLEFDDGTPATMSQVAKDVCTFLRWAAEPEHDHRKRMGLKMLLMMGLLLPLVYAMKRHKWSVLKSRKLAYRPPKGRQFGHLTRVRHVITYSLSPFEQRAFPHYFSKGIPNVLRRTRACILRVAPPFVAFYLVYTWGTQEFEKSKRKNPA Hydrogen bonds contact Hydrophobic contact | ||||