Job Results:
Ligand
Structure
Job ID
1aab99f222a82120b630ad6d44e2ad5f
Job name
NA
Time
2026-02-27 11:58:02
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | "Periplasmic trehalase (EC 3.2.1.28) (Alpha,alpha-trehalase) (Alpha,alpha-trehalose glucohydrolase) (Tre37A)" | 2JG0 | 4.54 | |
Target general information Gen name treA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1186;osmA;b1197 Protein family Glycosyl hydrolase 37 family Biochemical class NA Function Provides the cells with the ability to utilize trehalose at high osmolarity by splitting it into glucose molecules that can subsequently be taken up by the phosphotransferase-mediated uptake system. Related diseases SRC kinase activity has been shown to be increased in several tumor tissues and tumor cell lines such as colon carcinoma cells. {ECO:0000269|PubMed:2498394, ECO:0000269|PubMed:3093483}.; DISEASE: Thrombocytopenia 6 (THC6) [MIM:616937]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC6 is an autosomal dominant form. Affected individuals may also have bone abnormalities and an increased risk for myelofibrosis. {ECO:0000269|PubMed:26936507}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.2.1.28 Uniprot keywords 3D-structure; Direct protein sequencing; Glycosidase; Hydrolase; Periplasm; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 57508.9 Length 507 Aromaticity 0.11 Instability index 48.32 Isoelectric point 5.48 Charge (pH=7) -10.13 2D Binding mode Binding energy (Kcal/mol) -6.19  Molscript Map 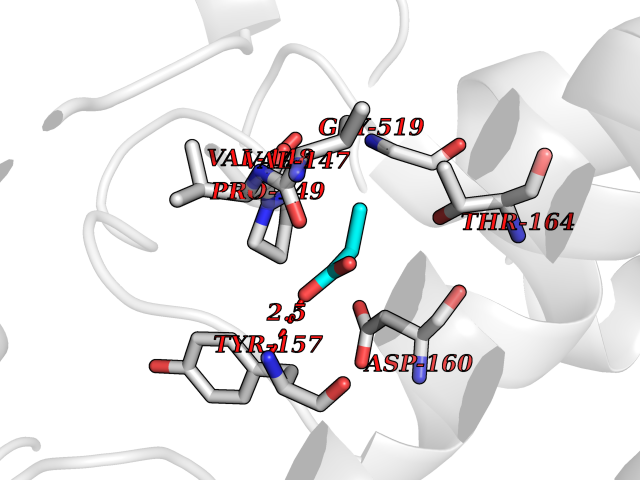 Pymol Map 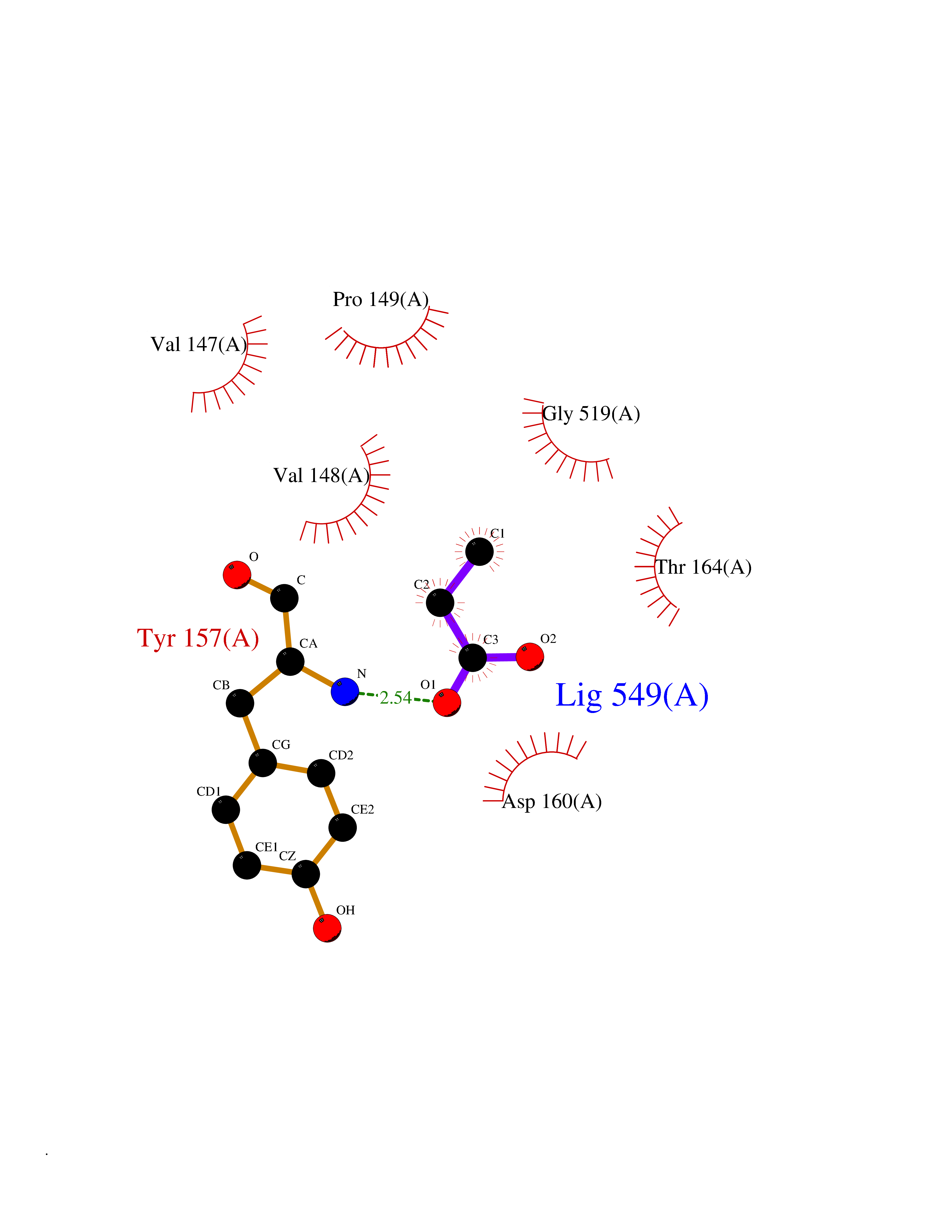 Ligplot Map 3D Binding mode Sequence PQPPDILLGPLFNDVQNAKLFPDQKTFADAVPNSDPLMILADYRMQQNQSGFDLRHFVNVNFTLPKYVPPEGQSLREHIDGLWPVLTRSTENTEKWDSLLPLPEPYVVPGGRFREVYYWDSYFTMLGLAESGHWDKVADMVANFAHEIDTYGHIPNGNRSYYLSRSQPPFFALMVELLAQHEGDAALKQYLPQMQKEYAYWMDGVENLQAGQQEKRVVKLQDGTLLNRYWDDRDTPRPESWVEDIATAKSNPNRPATEIYRDLRSAAASGWDFSSRWMDNPQQLNTLRTTSIVPVDLNSLMFKMEKILARASKAAGDNAMANQYETLANARQKGIEKYLWNDQQGWYADYDLKSHKVRNQLTAAALFPLYVNAAAKDRANKMATATKTHLLQPGGLNTTSVKSGQQWDAPNGWAPLQWVATEGLQNYGQKEVAMDISWHFLTNVQHTYDREKKLVEKYDVSTTGTGGGGGEYPLQDGFGWTNGVTLKMLDLICPKEQPCDNVPATRP Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Tryptophan 5-hydroxylase 1 (TPH1) | 5TPG | 4.54 | |
Target general information Gen name TPH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tryptophan 5-monooxygenase 1; TRPH; TPRH Protein family Biopterin-dependent aromatic amino acid hydroxylase family Biochemical class Paired donor oxygen oxidoreductase Function Responsible for addition of the -HO group (hydroxylation) to the 5 position to form the amino acid 5-hydroxytryptophan (5-HTP), which is the initial and rate-limiting step in the synthesis of the neurotransmitter serotonin. Related diseases Tyrosinemia 2 (TYRSN2) [MIM:276600]: An inborn error of metabolism characterized by elevations of tyrosine in the blood and urine, and oculocutaneous manifestations. Typical features include palmoplantar keratosis, painful corneal ulcers, and intellectual disability. {ECO:0000269|PubMed:1357662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05199; DB00360; DB12095; DB00150 Interacts with Q14457; Q96IK1-2; Q9UKB3; Q9H8Y8; O43586; O95789-4 EC number EC 1.14.16.4 Uniprot keywords 3D-structure; Alternative splicing; Iron; Metal-binding; Monooxygenase; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Serotonin biosynthesis; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 31138.2 Length 271 Aromaticity 0.13 Instability index 43.43 Isoelectric point 6.73 Charge (pH=7) -0.86 2D Binding mode Binding energy (Kcal/mol) -6.2 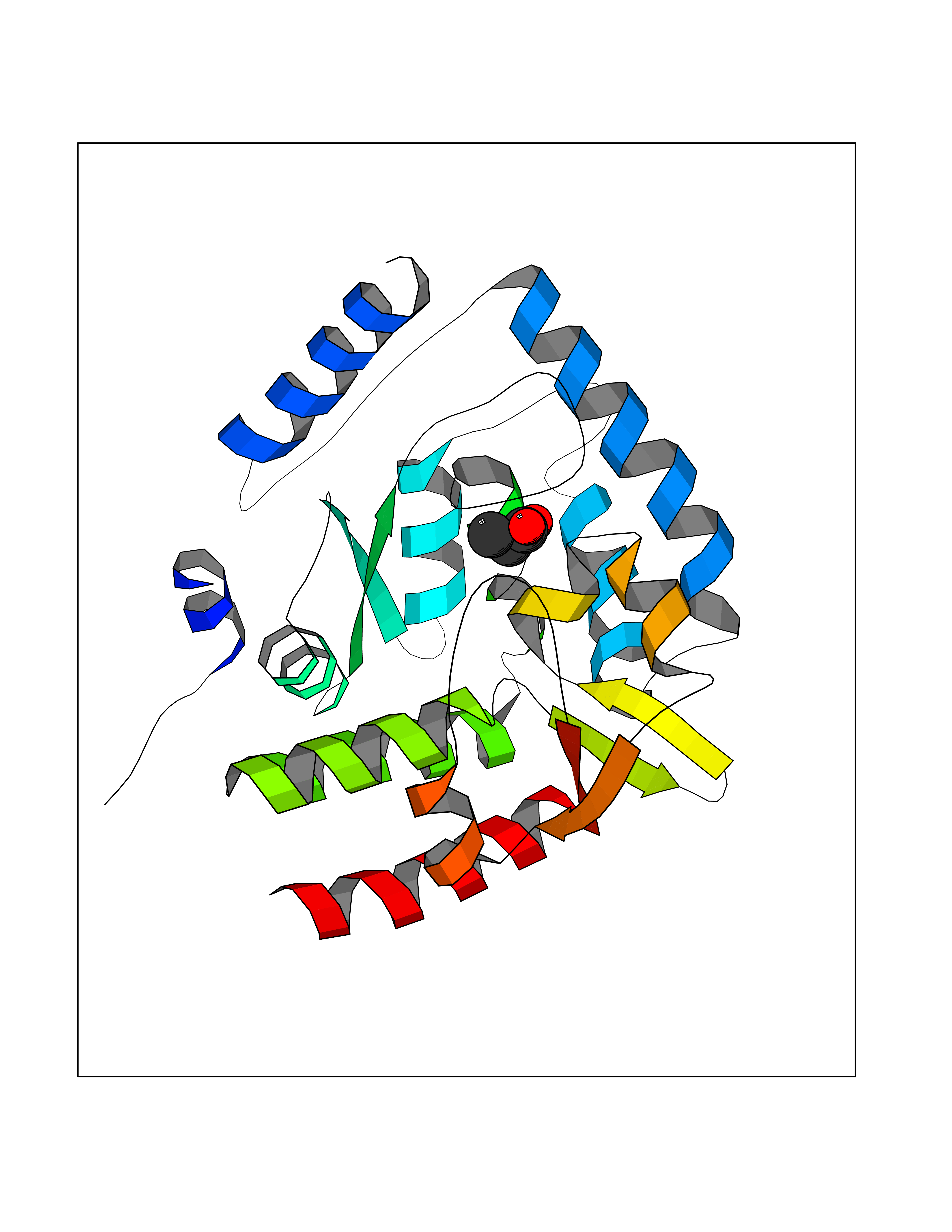 Molscript Map 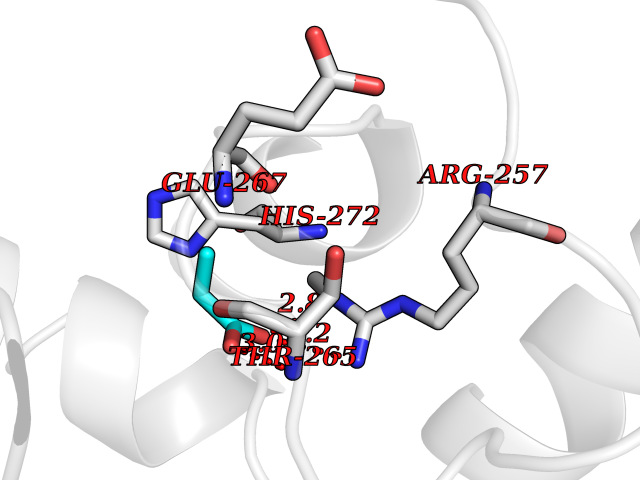 Pymol Map 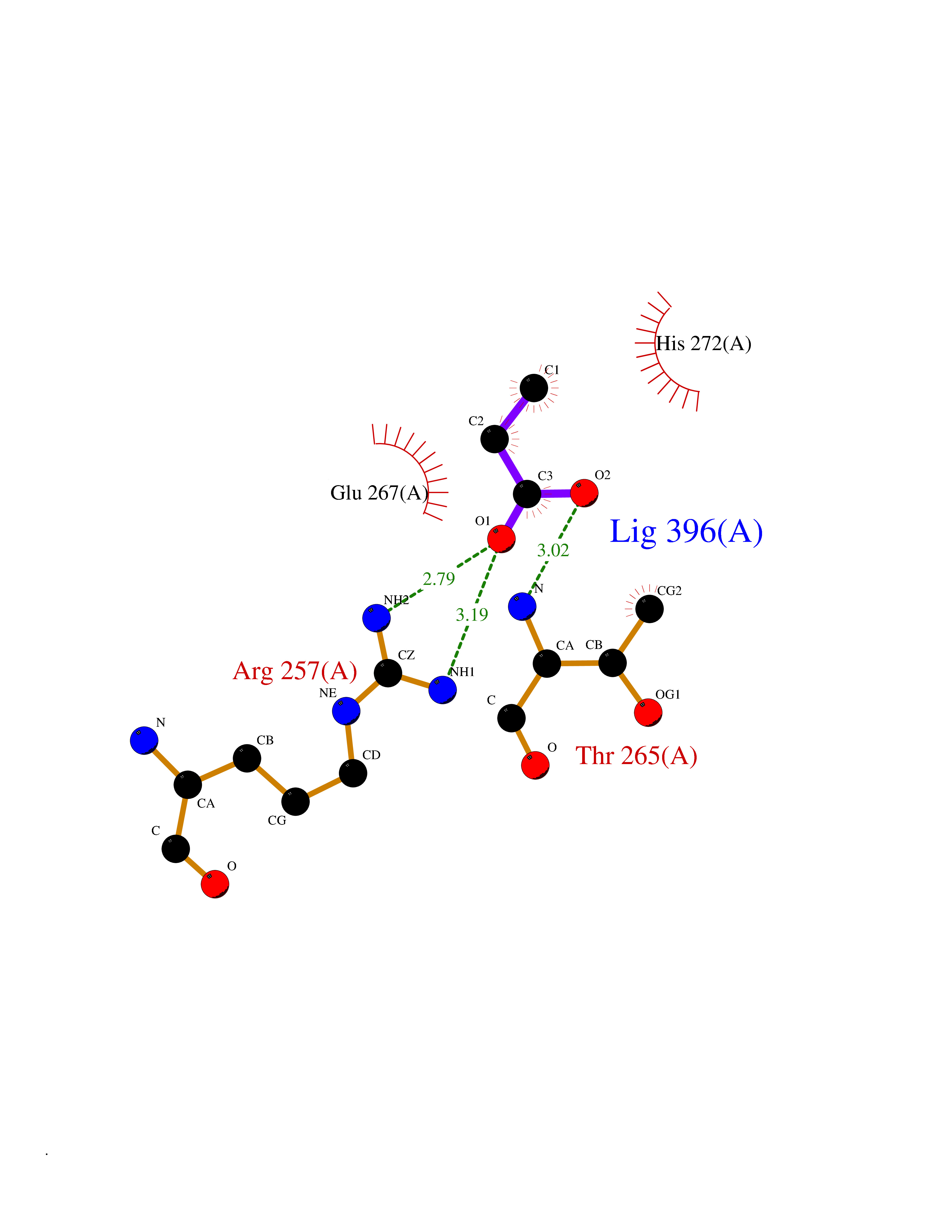 Ligplot Map 3D Binding mode Sequence TVPWFPKKISDLDHCNVYRKRRKYFADLAMNYKHGDPIPKVEFTEEEIKTWGTVFQELNKLYPTHACREYLKNLPLLSKYCGYREDNIPQLEDVSNFLKERTGFSIRPVAGYLSPRDFLSGLAFRVFHCTQYVRHSSDPFYTPEPDTCHELLGHVPLLAEPSFAQFSQEIGLASLGASEEAVQKLATCYFFTVEFGLCKQDGQLRVFGAGLLSSISELKHALSGHAKVKPFDPKITCKQECLITTFQDVYFVSESFEDAKEKMREFTKTIK Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Serine--pyruvate aminotransferase | 5HHY | 4.54 | |
Target general information Gen name AGXT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms AGT1;SPAT Protein family Class-V pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transferase Function Alanine-glyoxylate transaminase activity.Amino acid binding.Identical protein binding.Protein homodimerization activity.Protein self-association.Pyridoxal phosphate binding.Receptor binding.Serine-pyruvate transaminase activity.Transaminase activity. Related diseases Hyperoxaluria primary 1 (HP1) [MIM:259900]: An inborn error of glyoxylate metabolism characterized by increased excretion of oxalate and glycolate, and progressive tissue accumulation of insoluble calcium oxalate. Affected individuals are at risk for nephrolithiasis, nephrocalcinosis and early onset end-stage renal disease. {ECO:0000269|PubMed:10394939, ECO:0000269|PubMed:10453743, ECO:0000269|PubMed:10541294, ECO:0000269|PubMed:10862087, ECO:0000269|PubMed:10960483, ECO:0000269|PubMed:12559847, ECO:0000269|PubMed:12777626, ECO:0000269|PubMed:1301173, ECO:0000269|PubMed:1349575, ECO:0000269|PubMed:15253729, ECO:0000269|PubMed:15849466, ECO:0000269|PubMed:15961946, ECO:0000269|PubMed:15963748, ECO:0000269|PubMed:16971151, ECO:0000269|PubMed:1703535, ECO:0000269|PubMed:17495019, ECO:0000269|PubMed:2039493, ECO:0000269|PubMed:23229545, ECO:0000269|PubMed:24055001, ECO:0000269|PubMed:24934730, ECO:0000269|PubMed:26149463, ECO:0000269|PubMed:8101040, ECO:0000269|PubMed:9192270, ECO:0000269|PubMed:9604803}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08060; DB00160; DB02079; DB00145; DB04083; DB00114; DB00133 Interacts with Q5BKX5-3; P21549; P0C7T5; Q9NR55; Q9H5F2; Q9UKJ5; A8MQ03; O43281-2; A1KXE4-2; Q5TD97; P49356; P53539; P49639; Q15323; O76011; O76014; Q6A162; Q07627; P60328; Q9BYR8; Q3LI67; Q8IUC2; Q9BYQ4; O60711; Q99750; Q5VZ52; P0DPK4; O43482; P50542-1; O15496; Q6ZR37; Q9NQX0; Q8HWS3; Q5W111-2; Q8WVR3; Q6DKK2 EC number 2.6.1.44; 2.6.1.51 Uniprot keywords 3D-structure; Acetylation; Aminotransferase; Disease variant; Peroxisome; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84537 Length 771 Aromaticity 0.07 Instability index 36.81 Isoelectric point 8.42 Charge (pH=7) 6.81 2D Binding mode Binding energy (Kcal/mol) -6.19 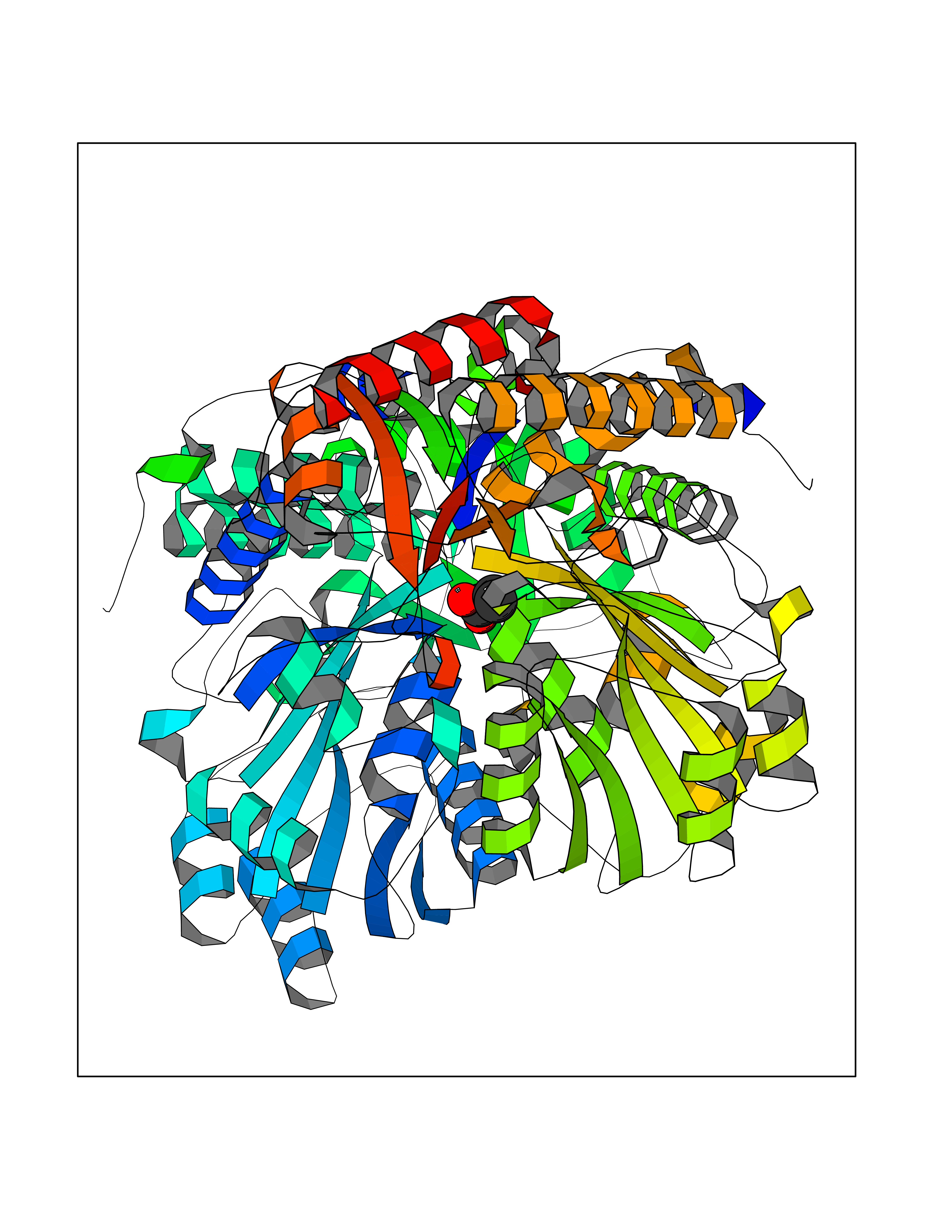 Molscript Map 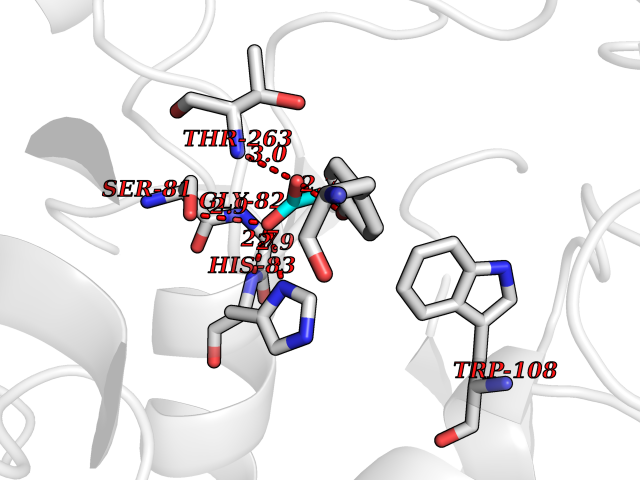 Pymol Map 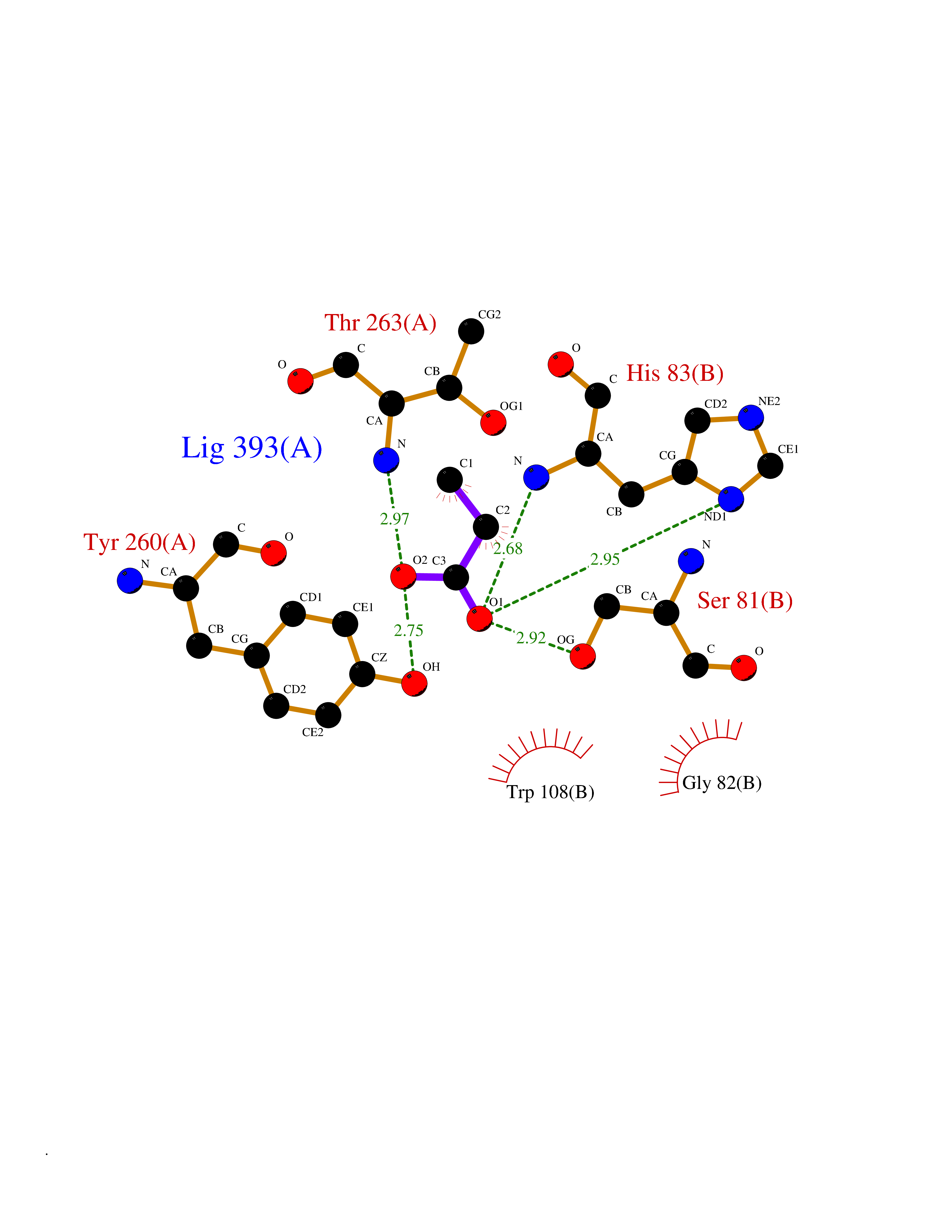 Ligplot Map 3D Binding mode Sequence LLVTPPKALLKPLSIPNQLLLGPGPSNLPPRIMAAGGLQMIGSMSKDMYQIMDEIKEGIQYVFQTRNPLTLVISGSGHCALEAALVNVLEPGDSFLVGANGIWGQRAVDIGERIGARVHPMTKDPGGHYTLQEVEEGLAQHKPVLLFLTHGESSTGVLQPLDGFGELCHRYKCLLLVDSVASLGGTPLYMDRQGIDILYSGSQKALNAPPGTSLISFSDKAKKKMYSRKTKPFSFYLDIKWLANFWGCDDQPRMYHHTIPVISLYSLRESLALIAEQGLENSWRQHREAAAYLHGRLQALGLQLFVKDPALRLPTVTTVAVPAGYDWRDIVSYVIDHFDIEIMGGLGPSTGKVLRIGLLGCNATRENVDRVTEALRAALQHCPKKLLVTPPKALLKPLSIPNQLLLGPGPSNLPPRIMAAGGLQMIGSMSKDMYQIMDEIKEGIQYVFQTRNPLTLVISGSGHCALEAALVNVLEPGDSFLVGANGIWGQRAVDIGERIGARVHPMTKDPGGHYTLQEVEEGLAQHKPVLLFLTHGESSTGVLQPLDGFGELCHRYKCLLLVDSVASLGGTPLYMDRQGIDILYSGSQKALNAPPGTSLISFSDKAKKKMYSRKTKPFSFYLDIKWLANFWGCDDQPRMYHHTIPVISLYSLRESLALIAEQGLENSWRQHREAAAYLHGRLQALGLQLFVKDPALRLPTVTTVAVPAGYDWRDIVSYVIDHFDIEIMGGLGPSTGKVLRIGLLGCNATRENVDRVTEALRAALQHCPKKK Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Ribonucleoside-diphosphate reductase subunit M2 | 3OLJ | 4.54 | |
Target general information Gen name RRM2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms RR2 Protein family Ribonucleoside diphosphate reductase small chain family Biochemical class Oxidoreductase Function Metal ion binding.Ribonucleoside-diphosphate reductase activity, thioredoxin disulfide as acceptor. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00242; DB05260; DB05801; DB05003; DB05428 Interacts with P41002; Q9UM11; P23921; O00560 EC number 1.17.4.1 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Deoxyribonucleotide synthesis; Iron; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 33579.4 Length 286 Aromaticity 0.14 Instability index 43.7 Isoelectric point 5.12 Charge (pH=7) -12.86 2D Binding mode Binding energy (Kcal/mol) -6.2  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MGVEDEPLLRENPRRFVIFPIEYHDIWQMYKKAEASFWTAEEVDLSKDIQHWESLKPEERYFISHVLAFFAASDGIVNENLVERFSQEVQITEARCFYGFQIAMENIHSEMYSLLIDTYIKDPKEREFLFNAIETMPCVKKKADWALRWIGDKEATYGERVVAFAAVEGIFFSGSFASIFWLKKRGLMPGLTFSNELISRDEGLHCDFACLMFKHLVHKPSEERVREIIINAVRIEQEFLTEALPVKLIGMNCTLMKQYIEFVADRLMLELGFSKVFRVENPFDFM Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Pyruvate kinase PKLR | 4IP7 | 4.54 | |
Target general information Gen name PKLR Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PKL;PK1 Protein family Pyruvate kinase family Biochemical class Transferase Function ATP binding.Kinase activity.Magnesium ion binding.Potassium ion binding.Pyruvate kinase activity. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02726; DB00787; DB04551; DB16236; DB00119 Interacts with Q9UBL6-2 EC number 2.7.1.40 Uniprot keywords 3D-structure; Allosteric enzyme; Alternative splicing; ATP-binding; Disease variant; Glycolysis; Hereditary hemolytic anemia; Kinase; Magnesium; Manganese; Metal-binding; Nucleotide-binding; Phosphoprotein; Potassium; Proteomics identification; Pyruvate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 45695.1 Length 421 Aromaticity 0.06 Instability index 34.44 Isoelectric point 6.88 Charge (pH=7) -0.35 2D Binding mode Binding energy (Kcal/mol) -6.19  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GTAFFQQQQLPAAMADTFLEHLCLLDIDSEPVAARSTSIIATIGPASRSVERLKEMIKAGMNIARLNFSHGSHEYHAESIANVREAVESFSPLSYRPVAIALDTKGPEIGLSEQDVRDLRFGVEHGVDIVFASFVRKASDVAAVRAALGPEGHGIKIISKIENHEGVKRFDEILEVSDGIMVARGDLGIEIPAEKVFLAQKMMIGRCNLAGKPVVCATQMLESMITKPRPTRAETSDVANAVLDGADCIMLSGETAKGNFPVEAVKMQHAIAREAEAAVYHRQLFEELRRAAPLSRDPTEVTAIGAVEAAFKCCAAAIIVLTTTGRSAQLLSRYRPRAAVIAVTRSAQAARQVHLCRGVFPLLYREPPEAIWADDVDRRVQFGIESGKLRGFLRVGDLVIVVTGWRPGSGYTNIMRVLSIS Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Acetyl-CoA carboxylase 1 | 2YL2 | 4.54 | |
Target general information Gen name ACACA Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ACAC;ACCA;ACC1 Protein family NA Biochemical class Ligase Function Acetyl-CoA carboxylase activity.ATP binding.Biotin carboxylase activity.Identical protein binding.Metal ion binding. Related diseases Acetyl-CoA carboxylase-alpha deficiency (ACACAD) [MIM:613933]: An autosomal recessive inborn error of de novo fatty acid synthesis associated with severe brain damage, persistent myopathy and poor growth. {ECO:0000269|PubMed:6114432}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00121 Interacts with Q13085; O60218; P38398; Q96EB6; Q9CQ20; P02654; Q92915-2; Q6NTF9-3 EC number 6.4.1.2 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative promoter usage; ATP-binding; Biotin; Cytoplasm; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Ligase; Lipid biosynthesis; Lipid metabolism; Magnesium; Manganese; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 54237.7 Length 486 Aromaticity 0.09 Instability index 39.18 Isoelectric point 6.37 Charge (pH=7) -2.46 2D Binding mode Binding energy (Kcal/mol) -6.19 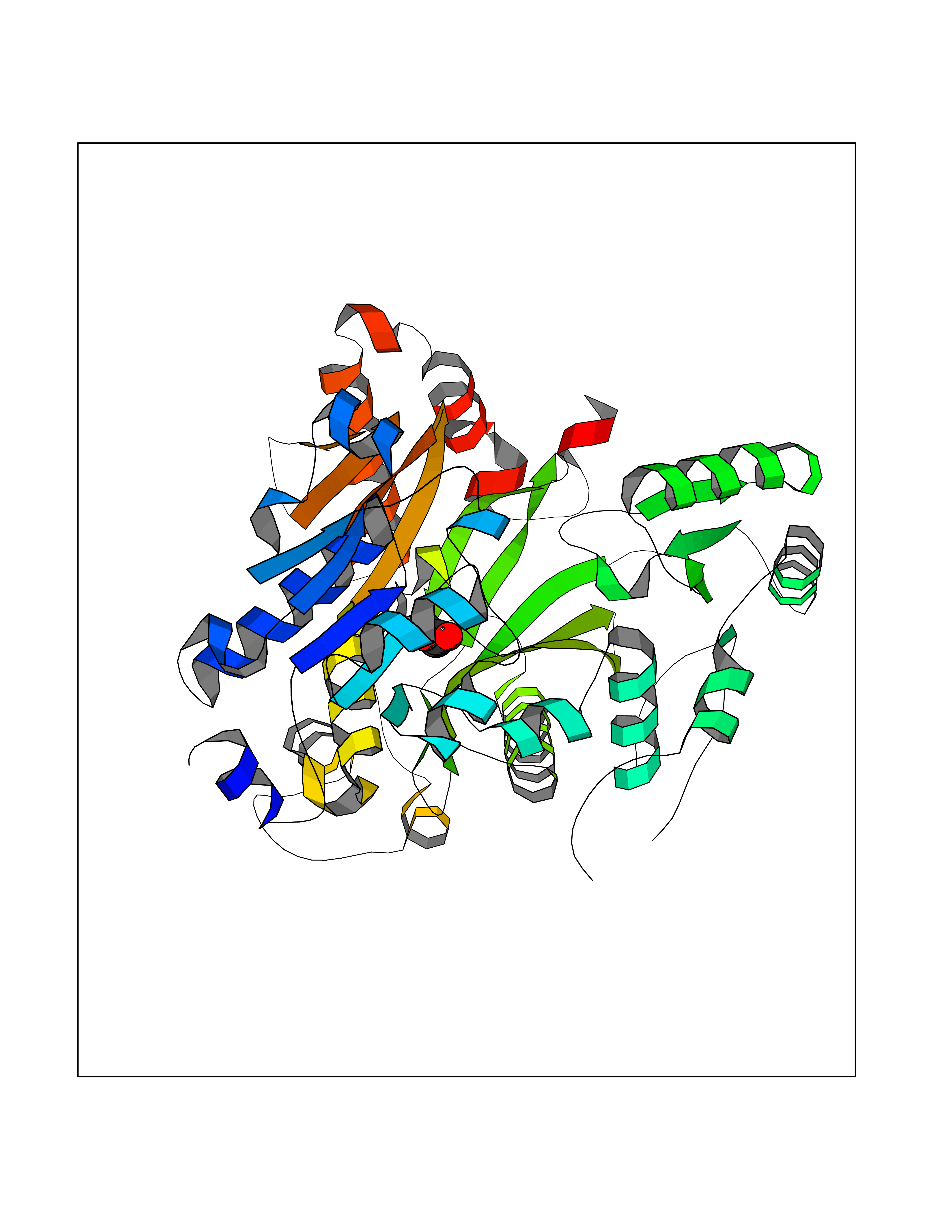 Molscript Map 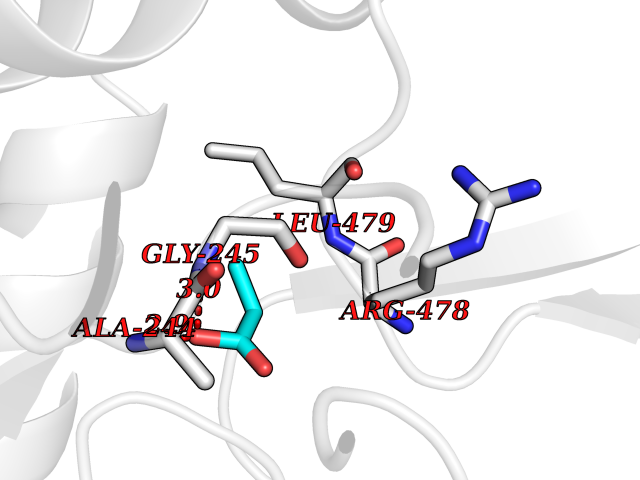 Pymol Map 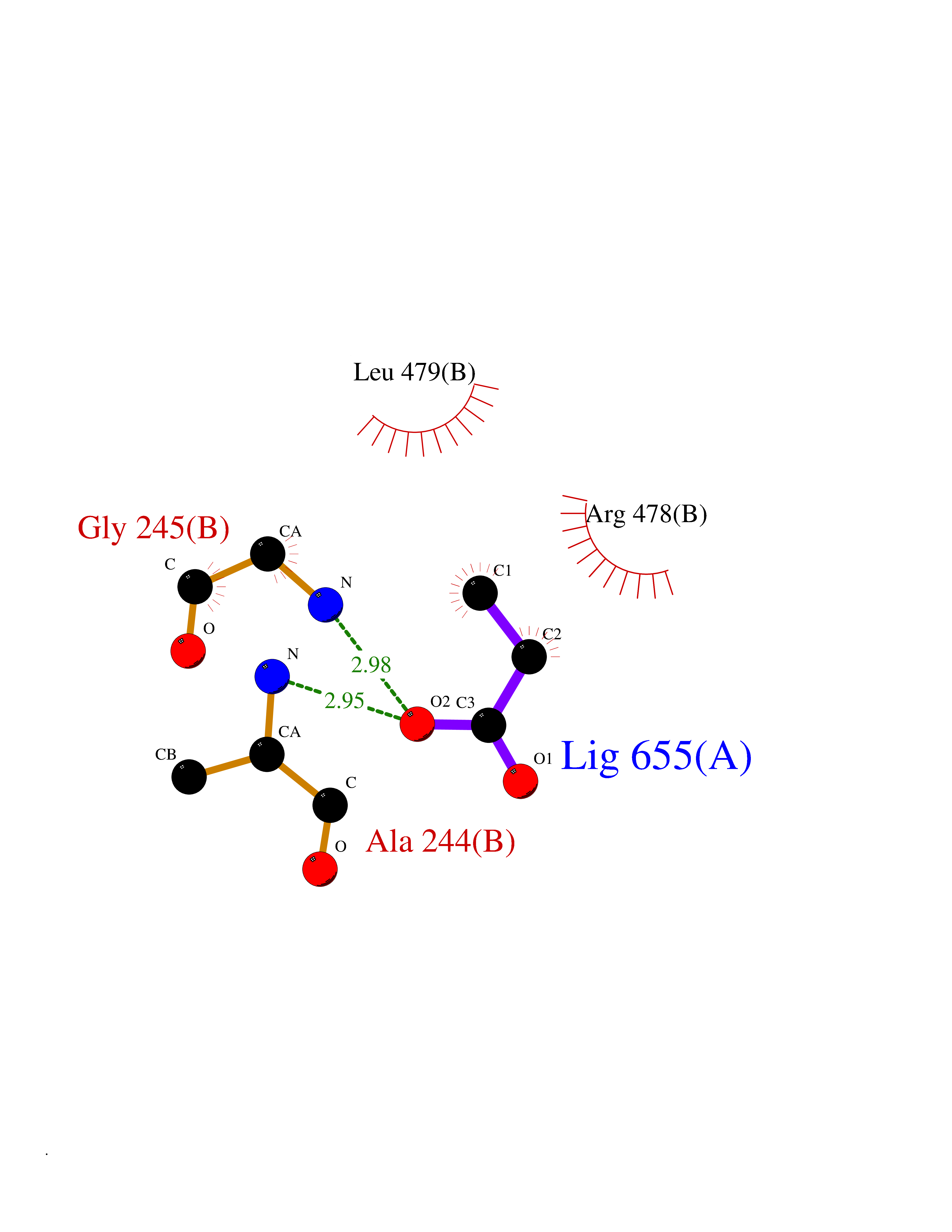 Ligplot Map 3D Binding mode Sequence VASPAEFVTRFGGNKVIEKVLIANNGIAAVKCMRSIRRWSYEMFRNERAIRFVVMVTPEDLKANAEYIKMADHYVPVPGGPNNNNYANVELILDIAKRIPVQAVWAGWGHASENPKLPELLLKNGIAFMGPPSQAMWALGDKIASSIVAQTAGIPTLPWSGSGLRVDWSKRILNVPQELYEKGYVKDVDDGLQAAEEVGYPVMIKASEGGGGKGIRKVNNADDFPNLFRQVQAEVPGSPIFVMRLAKQSRHLEVQILADQYGNAISLFGRDCSVQRRHQKIIEEAPATIATPAVFEHMEQCAVKLAKMVGYVSAGTVEYLYSQDGSFYFLELNPRLQVEHPCTEMVADVNLPAAQLQIAMGIPLYRIKDIRMMYGVSPWGDSPIDFEDSAHVPCPRGHVIAARITGTVQELNFRSNKNVWGYFSVQFGHCFSWGENREEAISNMVVALKELSIRGDFRTTVEYLIKLLETESFQMNRIDTGWLDRL Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Phenylacetone monooxygenase | 1W4X | 4.54 | |
Target general information Gen name pamO Organism Thermobifida fusca (strain YX) Uniprot ID TTD ID NA Synonyms Tfu_1490 Protein family FAD-binding monooxygenase family Biochemical class Oxygenase Function Phenylacetone monooxygenase activity. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.14.13.92 Uniprot keywords 3D-structure; FAD; Flavoprotein; Monooxygenase; NADP; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 60232 Length 533 Aromaticity 0.12 Instability index 32.05 Isoelectric point 5.25 Charge (pH=7) -17.1 2D Binding mode Binding energy (Kcal/mol) -6.19 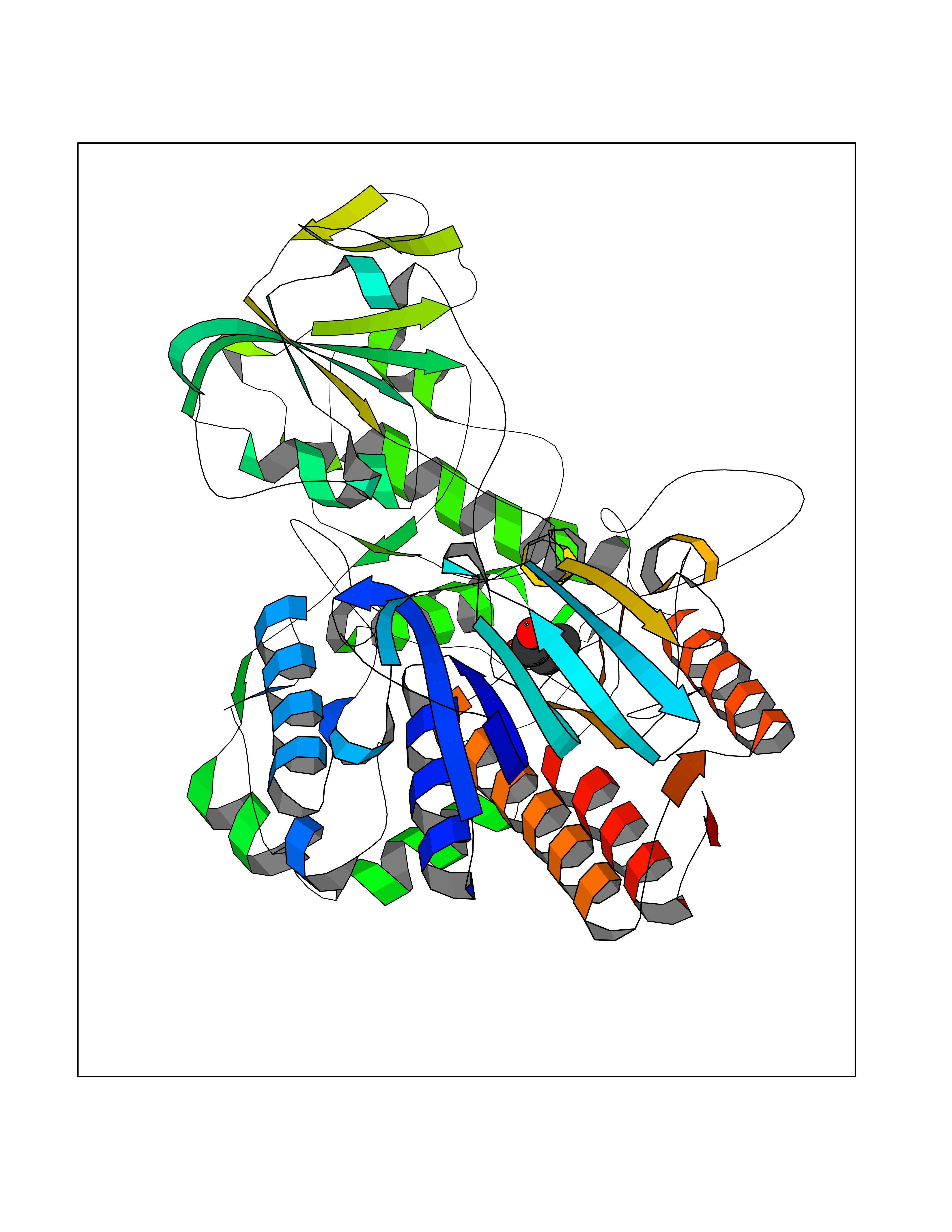 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence RRQPPEEVDVLVVGAGFSGLYALYRLRELGRSVHVIETAGDVGGVWYWNRYPGARCDIESIEYCYSFSEEVLQEWNWTERYASQPEILRYINFVADKFDLRSGITFHTTVTAAAFDEATNTWTVDTNHGDRIRARYLIMASGQLSVPQLPNFPGLKDFAGNLYHTGNWPHEPVDFSGQRVGVIGTGSSGIQVSPQIAKQAAELFVFQRTPHFAVPARNAPLDPEFLADLKKRYAEFREESRNTPGGTHRYQGPKSALEVSDEELVETLERYWQEGGPDILAAYRDILRDRDANERVAEFIRNKIRNTVRDPEVAERLVPKGYPFGTKRLILEIDYYEMFNRDNVHLVDTLSAPIETITPRGVRTSEREYELDSLVLATGFDALTGALFKIDIRGVGNVALKEKWAAGPRTYLGLSTAGFPNLFFIAGPGSPSALSNMLVSIEQHVEWVTDHIAYMFKNGLTRSEAVLEKEDEWVEHVNEIADETLYPMTASWYTGANVPGKPRVFMLYVGGFHRYRQICDEVAAKGYEGFVLT Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Pyridoxal phosphate phosphatase (PDXP) | 2CFT | 4.54 | |
Target general information Gen name PDXP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PLPP; PLP phosphatase; PDXP Protein family HAD-like hydrolase superfamily Biochemical class Phosphoric monoester hydrolase Function Protein serine phosphatase that dephosphorylates 'Ser-3' in cofilin and probably also dephosphorylates phospho-serine residues in DSTN. Regulates cofilin-dependent actin cytoskeleton reorganization. Required for normal progress through mitosis and normal cytokinesis. Does not dephosphorylate phospho-threonines in LIMK1. Does not dephosphorylate peptides containing phospho- tyrosine. Pyridoxal phosphate phosphatase. Has some activity towards pyridoxal 5'-phosphate (PLP), pyridoxine 5'-phosphate (PMP) and pyridoxine 5'-phosphate (PNP), with a highest activity with PLP followed by PNP. Related diseases Paramyotonia congenita (PMC) [MIM:168300]: An autosomal dominant channelopathy characterized by myotonia, increased by exposure to cold, intermittent flaccid paresis, not necessarily dependent on cold or myotonia, lability of serum potassium, non-progressive nature and lack of atrophy or hypertrophy of muscles. In some patients, myotonia is not increased by cold exposure (paramyotonia without cold paralysis). Patients may have a combination phenotype of PMC and HYPP. {ECO:0000269|PubMed:10369308, ECO:0000269|PubMed:10727489, ECO:0000269|PubMed:1310898, ECO:0000269|PubMed:1316765, ECO:0000269|PubMed:1338909, ECO:0000269|PubMed:15318338, ECO:0000269|PubMed:15790667, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:18166706, ECO:0000269|PubMed:18690054, ECO:0000269|PubMed:19077043, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:8242056, ECO:0000269|PubMed:8308722, ECO:0000269|PubMed:8388676, ECO:0000269|PubMed:8580427}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hypokalemic 2 (HOKPP2) [MIM:613345]: An autosomal dominant disorder manifested by episodic flaccid generalized muscle weakness associated with falls of serum potassium levels. {ECO:0000269|PubMed:10599760, ECO:0000269|PubMed:10851391, ECO:0000269|PubMed:10944223, ECO:0000269|PubMed:11558801, ECO:0000269|PubMed:11591859, ECO:0000269|PubMed:16890191, ECO:0000269|PubMed:17898326, ECO:0000269|PubMed:18162704, ECO:0000269|PubMed:19118277, ECO:0000269|PubMed:20522878, ECO:0000269|PubMed:21043388, ECO:0000269|PubMed:24549961}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hyperkalemic (HYPP) [MIM:170500]: An autosomal dominant channelopathy characterized by episodic flaccid generalized muscle weakness associated with high levels of serum potassium. Concurrence of myotonia is found in HYPP patients. {ECO:0000269|PubMed:1659668, ECO:0000269|PubMed:1659948, ECO:0000269|PubMed:20076800}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis normokalemic (NKPP) [MIM:170500]: A disorder closely related to hyperkalemic periodic paralysis, but marked by a lack of alterations in potassium levels during attacks of muscle weakness. {ECO:0000269|PubMed:15596759, ECO:0000269|PubMed:18046642, ECO:0000269|PubMed:20522878}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myotonia SCN4A-related (MYOSCN4A) [MIM:608390]: A phenotypically highly variable myotonia aggravated by potassium loading, and sometimes by cold. Myotonia is characterized by sustained muscle tensing that prevents muscles from relaxing normally. It causes muscle stiffness that can interfere with movement. In some people the stiffness is very mild, while in other cases it may be severe enough to interfere with walking, running, and other activities of daily life. Myotonia SCN4A-related includes myotonia permanens and myotonia fluctuans. In myotonia permanens, the myotonia is generalized and there is a hypertrophy of the muscle, particularly in the neck and the shoulder. Attacks of severe muscle stiffness of the thoracic muscles may be life threatening due to impaired ventilation. In myotonia fluctuans, the muscle stiffness may fluctuate from day to day, provoked by exercise. {ECO:0000269|PubMed:10218481, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:16832098, ECO:0000269|PubMed:17212350, ECO:0000269|PubMed:17998485, ECO:0000269|PubMed:18203179, ECO:0000269|PubMed:18337100, ECO:0000269|PubMed:19015483, ECO:0000269|PubMed:19347921, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:27653901, ECO:0000269|PubMed:8058156, ECO:0000269|PubMed:9392583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 16 (CMS16) [MIM:614198]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS16 is characterized by fatigable generalized weakness and recurrent attacks of respiratory and bulbar paralysis since birth. The fatigable weakness involves lid-elevator, external ocular, facial, limb and truncal muscles and an decremental response of the compound muscle action potential on repetitive stimulation. {ECO:0000269|PubMed:12766226, ECO:0000269|PubMed:25707578, ECO:0000269|PubMed:26659129}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22A, classic (CMYO22A) [MIM:620351]: A form of congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22A is an autosomal recessive form characterized by fetal hypokinesia, polyhydramnios, and severe neonatal hypotonia associated with respiratory insufficiency. Affected individuals who survive the neonatal period have delayed motor development, difficulty walking, proximal muscle weakness of the upper and lower limbs, facial and neck muscle weakness, easy fatigability, and mild limb contractures or foot deformities. {ECO:0000269|PubMed:26700687, ECO:0000269|PubMed:28262468, ECO:0000269|PubMed:36090556}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22B, severe fetal (CMYO22B) [MIM:620369]: A severe congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22B is an autosomal recessive form characterized by onset in utero. Affected individuals show fetal akinesia, and develop fetal hydrops with pulmonary hypoplasia, severe joint contractures, and generalized muscle hypoplasia. Death occurs in utero or soon after birth. {ECO:0000269|PubMed:26700687}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00114; DB00165 Interacts with P29066 EC number EC 3.1.3.3 Uniprot keywords 3D-structure; Alternative initiation; Alternative splicing; Cell membrane; Cell projection; Cytoplasm; Cytoskeleton; Direct protein sequencing; Hydrolase; Magnesium; Membrane; Metal-binding; Proteomics identification; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 31285.4 Length 292 Aromaticity 0.06 Instability index 41.9 Isoelectric point 6.61 Charge (pH=7) -0.98 2D Binding mode Binding energy (Kcal/mol) -6.19  Molscript Map  Pymol Map 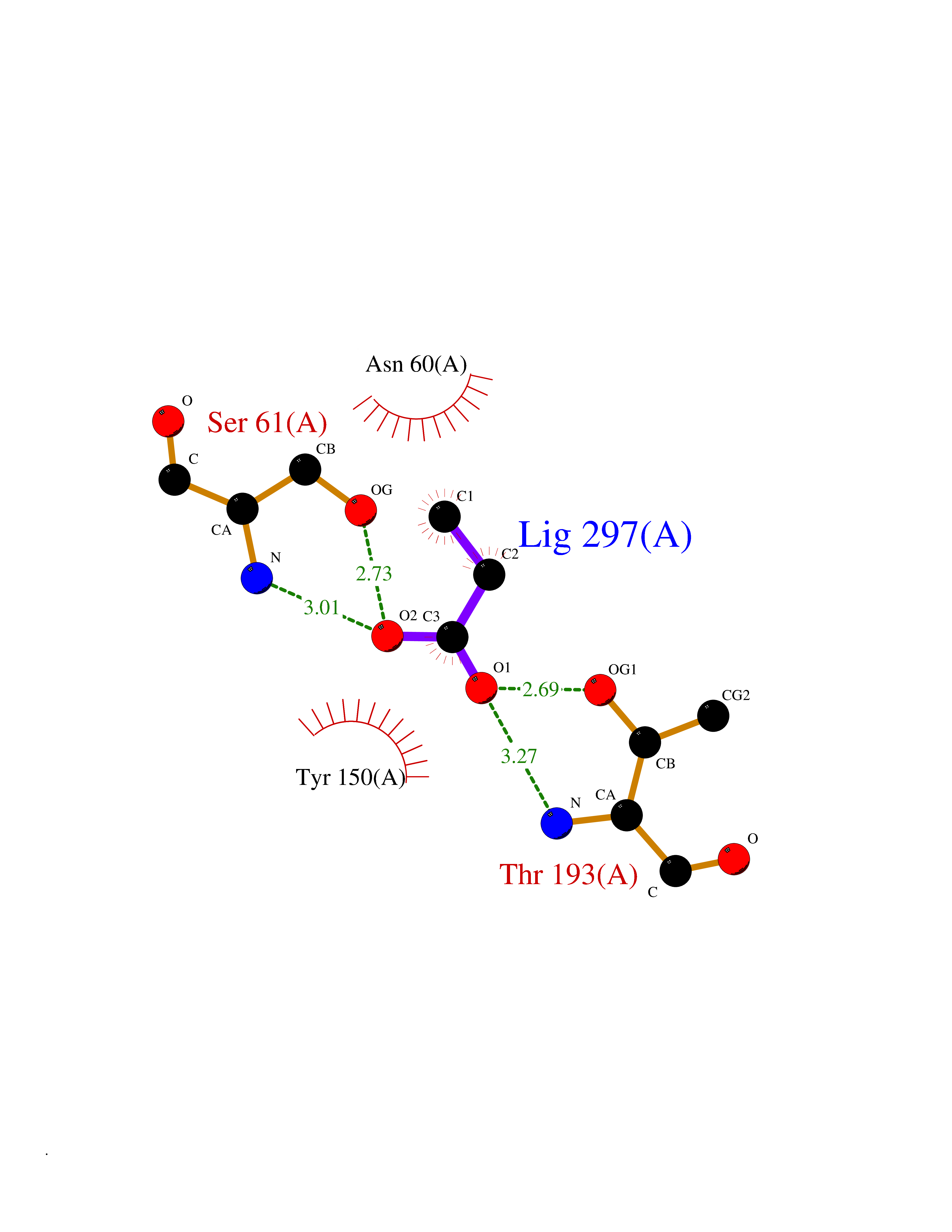 Ligplot Map 3D Binding mode Sequence MARCERLRGAALRDVLGRAQGVLFDCDGVLWNGERAVPGAPELLERLARAGKAALFVSNNSRRARPELALRFARLGFGGLRAEQLFSSALCAARLLRQRLPGPPDGAVFVLGGEGLRAELRAAGLRLAGDPSAGDGAAPRVRAVLVGYDEHFSFAKLREACAHLRDPECLLVATDRDPWHPLSDGSRTPGTGSLAAAVETASGRQALVVGKPSPYMFECITENFSIDPARTLMVGDRLETDILFGHRCGMTTVLTLTGVSRLEEAQAYLAAGQHDLVPHYYVESIADLTEGL Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Chymase (CYM) | 4K69 | 4.54 | |
Target general information Gen name CMA1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mast cell protease I; CYH; Alpha-chymase Protein family Peptidase S1 family, Granzyme subfamily Biochemical class Peptidase Function Major secreted protease of mast cells with suspected roles in vasoactive peptide generation, extracellular matrix degradation, and regulation of gland secretion. Related diseases Weaver syndrome (WVS) [MIM:277590]: A syndrome of accelerated growth and osseous maturation, unusual craniofacial appearance, hoarse and low-pitched cry, and hypertonia with camptodactyly. Distinguishing features of Weaver syndrome include broad forehead and face, ocular hypertelorism, prominent wide philtrum, micrognathia, deep horizontal chin groove, and deep-set nails. In addition, carpal bone development is advanced over the rest of the hand. {ECO:0000269|PubMed:22177091, ECO:0000269|PubMed:22190405, ECO:0000269|PubMed:23239504, ECO:0000269|PubMed:26694085, ECO:0000269|PubMed:28229514}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03814; DB04016; DB07680; DB03297 Interacts with NA EC number EC 3.4.21.39 Uniprot keywords 3D-structure; Alternative splicing; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 23715.1 Length 215 Aromaticity 0.07 Instability index 37.79 Isoelectric point 9.51 Charge (pH=7) 11.49 2D Binding mode Binding energy (Kcal/mol) -6.19  Molscript Map  Pymol Map 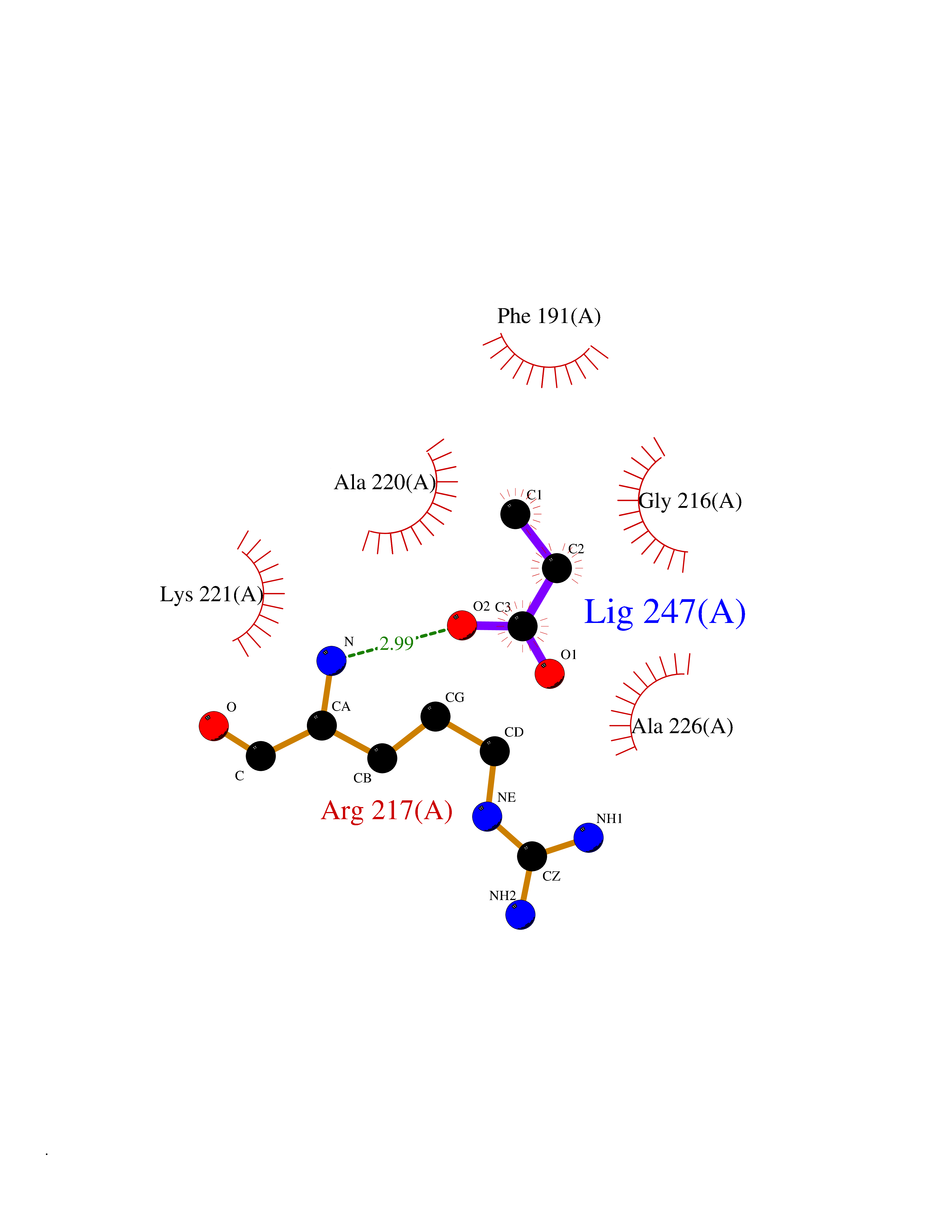 Ligplot Map 3D Binding mode Sequence IIGGTECKPHSRPYMAYLEIVTSNGPSKFCGGFLIRRNFVLTAAHCAGRSITVTLGAHNITEEEDTWQKLEVIKQFRHPKYNTSTLHHDIMLLKLKEKASLTLAVGTLGRMCRVAGWGRTGVLKPGSDTLQEVKLRLMDPQACSHFRDFDHNLQLCVGNPRKTKSAFKGDSGGPLLCAGAAQGIVSYGRSDAKPPAVFTRISHYQPWINQILQAN Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Mycobacterium Isocitrate lyase (MycB icl) | 1F8M | 4.54 | |
Target general information Gen name MycB icl Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID Synonyms Isocitratase; Isocitrase; ICL Protein family Isocitrate lyase/PEP mutase superfamily, Isocitrate lyase family Biochemical class Carbon-carbon lyase Function Catalyzes the formation of succinate and glyoxylate from isocitrate, a key step of the glyoxylate cycle. May be involved in the assimilation of one-carbon compounds via the isocitrate lyase- positive serine pathway. Related diseases Intellectual developmental disorder, autosomal recessive 80, with variant lissencephaly (MRT80) [MIM:620653]: An autosomal recessive disorder characterized by global developmental delay, mildly to moderately impaired intellectual development, attention deficit-hyperactivity disorder, hypotonia, seizure, poor social skills, and autistic traits. Brain imaging shows fronto-temporal lissencephaly and pachygyria. {ECO:0000269|PubMed:37880421}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04343 Interacts with NA EC number EC 4.1.3.1 Uniprot keywords 3D-structure; Glyoxylate bypass; Isopeptide bond; Lyase; Magnesium; Manganese; Metal-binding; Reference proteome; Tricarboxylic acid cycle; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 93759.5 Length 854 Aromaticity 0.09 Instability index 28.02 Isoelectric point 4.98 Charge (pH=7) -34.53 2D Binding mode Binding energy (Kcal/mol) -6.19  Molscript Map 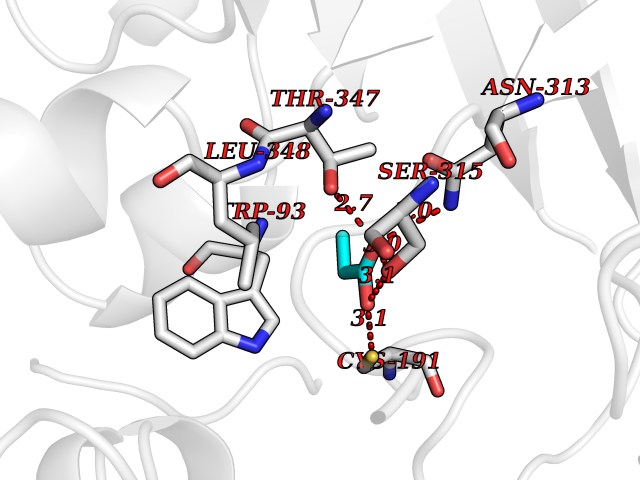 Pymol Map 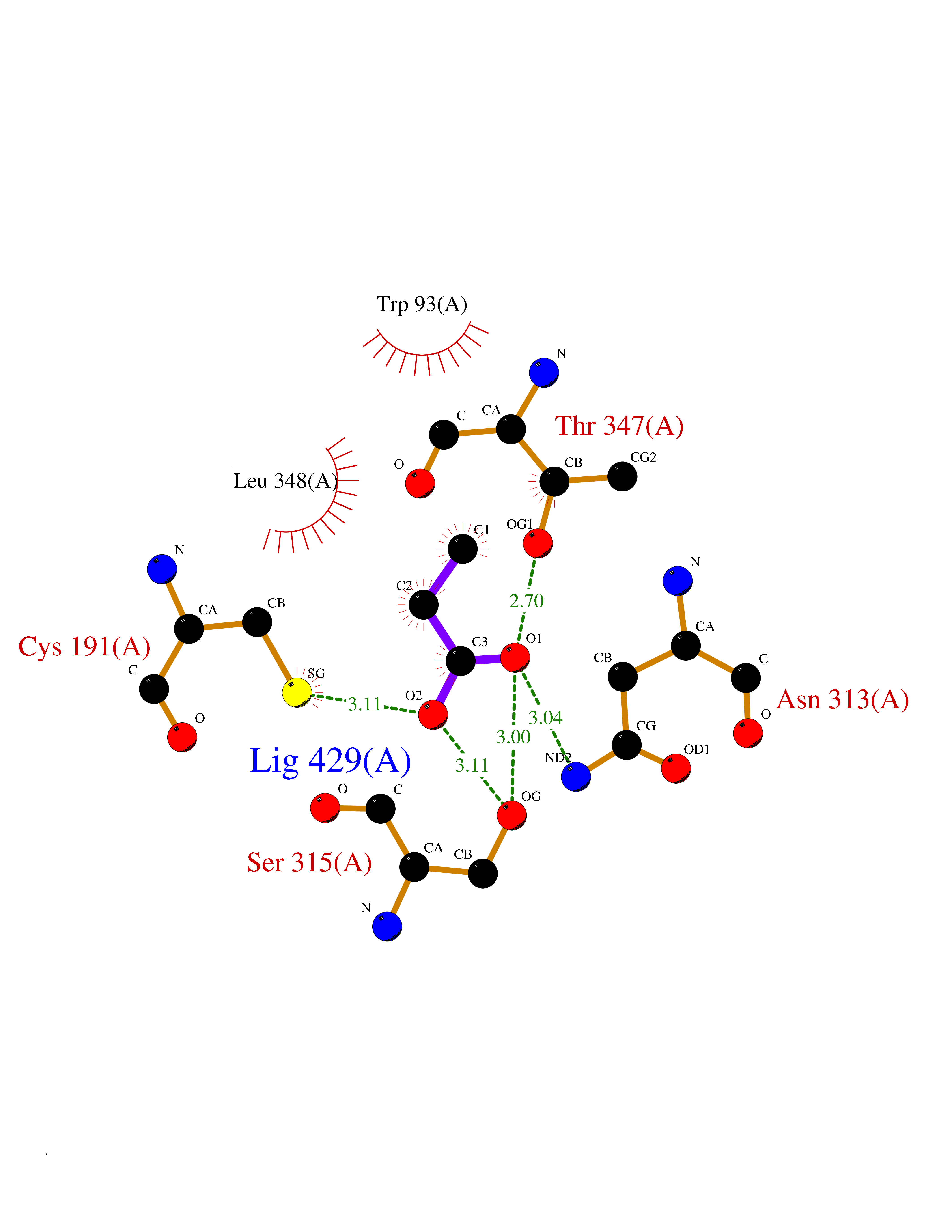 Ligplot Map 3D Binding mode Sequence ASVVGTPKSAEQIQQEWDTNPRWKDVTRTYSAEDVVALQGSVVEEHTLARRGAEVLWEQLHDLEWVNALGALTGNMAVQQVRAGLKAIYLSGWQVAGDANLSGHTYPDQSLYPANSVPQVVRRINNALQRADQIAKIEGDTSVENWLAPIVADGEAGFGGALNVYELQKALIAAGVAGSHWEDQLASEKKCGHLGGKVLIPTQQHIRTLTSARLAADVADVPTVVIARTDAEAATLITSDVDERDQPFITGERTREGFYRTKNGIEPCIARAKAYAPFADLIWMETGTPDLEAARQFSEAVKAEYPDQMLAYNCSPSFNWKKHLDDATIAKFQKELAAMGFKFQFITLAGFHALNYSMFDLAYGYAQNQMSAYVELQEREFAAEERGYTATKHQREVGAGYFDRIATTVDPNSSTTALTGSTEEGQFASVVGTPKSAEQIQQEWDTNPRWKDVTRTYSAEDVVALQGSVVEEHTLARRGAEVLWEQLHDLEWVNALGALTGNMAVQQVRAGLKAIYLSGWQVAGDANLSGHTYPDQSLYPANSVPQVVRRINNALQRADQIAKIEGDTSVENWLAPIVADGEAGFGGALNVYELQKALIAAGVAGSHWEDQLASEKKCGHLGGKVLIPTQQHIRTLTSARLAADVADVPTVVIARTDAEAATLITSDVDERDQPFITGERTREGFYRTKNGIEPCIARAKAYAPFADLIWMETGTPDLEAARQFSEAVKAEYPDQMLAYNCSPSFNWKKHLDDATIAKFQKELAAMGFKFQFITLAGFHALNYSMFDLAYGYAQNQMSAYVELQEREFAAEERGYTATKHQREVGAGYFDRIATTVDPNSSTTALTGSTEEGQF Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Gastrin (GAST) | 5WRJ | 4.54 | |
Target general information Gen name GAST Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Gastrin6; GAST; G52; G34; G14 Protein family Gastrin/cholecystokinin family Biochemical class Gastrin cholecystokinin Function Gastrin stimulates the stomach mucosa to produce and secrete hydrochloric acid and the pancreas to secrete its digestive enzymes. It also stimulates smooth muscle contraction and increases blood circulation and water secretion in the stomach and intestine. Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12532 Interacts with Q13520; O43315; Q12797-6; Q9BXK5; Q8N5K1; Q96BA8; P00387; Q9Y282; Q5JX71; Q8NBJ4; Q8TDT2; P43628; O76011; Q6ZUX7; Q15546; P15941-11; Q13113; P60201-2; Q14973; P02787; Q4KMG9 EC number NA Uniprot keywords 3D-structure; Amidation; Cleavage on pair of basic residues; Direct protein sequencing; Hormone; Phosphoprotein; Proteomics identification; Pyrrolidone carboxylic acid; Reference proteome; Secreted; Signal; Sulfation Protein physicochemical properties Chain ID A,F Molecular weight (Da) 31827.9 Length 280 Aromaticity 0.08 Instability index 41.06 Isoelectric point 8.84 Charge (pH=7) 5.56 2D Binding mode Binding energy (Kcal/mol) -6.19  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence AYHKDMPLIFIGGVPRSGTTLMRAMLDAHPDIRCGEETRVIPRILALKQMWSRSSKEKIRLDEAGVTDEVLDSAMQAFLLEIIVKHGEPAPYLCNKDPFALKSLTYLSRLFPNAKFLLMVRDGRASVHSMISRKVTIAGFDLNSYRDCLTKWNRAIETMYNQCMEVGYKKCMLVHYEQLVLHPERWMRTLLKFLQIPWNHSVLHHEEMIGKAGGVSLSKVERSTDQVIKPVNVGALSKWVGKIPPDVLQDMAVIAPMLAKLGYDPYANPPNYGKPEEEAY Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Torsin-1A (TOR1A) | 5J1S | 4.54 | |
Target general information Gen name TOR1A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Torsin family 1 member A; Torsin ATPase-1A; TORA; TA; Dystonia 1 protein; DYT1; DQ2 Protein family ClpA/ClpB family, Torsin subfamily Biochemical class Acid anhydride hydrolase Function Involved in the regulation of synaptic vesicle recycling, controls STON2 protein stability in collaboration with the COP9 signalosome complex (CSN). In the nucleus, may link the cytoskeleton with the nuclear envelope, this mechanism seems to be crucial for the control of nuclear polarity, cell movement and, specifically in neurons, nuclear envelope integrity. Participates in the cellular trafficking and may regulate the subcellular location of multipass membrane proteins such as the dopamine transporter SLC6A3, leading to the modulation of dopamine neurotransmission. In the endoplasmic reticulum, plays a role in the quality control of protein folding by increasing clearance of misfolded proteins such as SGCE variants or holding them in an intermediate state for proper refolding. May have a redundant function with TOR1B in non-neural tissues. Protein with chaperone functions important for the control of protein folding, processing, stability and localization as well as for the reduction of misfolded protein aggregates. Related diseases Dystonia 1, torsion, autosomal dominant (DYT1) [MIM:128100]: A primary torsion dystonia, and the most common and severe form. Dystonia is defined by the presence of sustained involuntary muscle contractions, often leading to abnormal postures. Dystonia type 1 is characterized by involuntary, repetitive, sustained muscle contractions or postures involving one or more sites of the body, in the absence of other neurological symptoms. Typically, symptoms develop first in an arm or leg in middle to late childhood and progress in approximately 30% of patients to other body regions (generalized dystonia) within about five years. 'Torsion' refers to the twisting nature of body movements observed in DYT1, often affecting the trunk. Distribution and severity of symptoms vary widely between affected individuals, ranging from mild focal dystonia to severe generalized dystonia, even within families. {ECO:0000269|PubMed:14970196, ECO:0000269|PubMed:15505207, ECO:0000269|PubMed:16361107, ECO:0000269|PubMed:17428918, ECO:0000269|PubMed:18167355, ECO:0000269|PubMed:18477710, ECO:0000269|PubMed:18827015, ECO:0000269|PubMed:19955557, ECO:0000269|PubMed:20169475, ECO:0000269|PubMed:21102408, ECO:0000269|PubMed:24930953, ECO:0000269|PubMed:27490483, ECO:0000269|PubMed:9288096}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Arthrogryposis multiplex congenita 5 (AMC5) [MIM:618947]: A form of arthrogryposis multiplex congenita, a developmental condition characterized by multiple joint contractures resulting from reduced or absent fetal movements. AMC5 is an autosomal recessive form characterized by severe congenital contractures, developmental delay, strabismus and tremor. {ECO:0000269|PubMed:28516161, ECO:0000269|PubMed:29053766, ECO:0000269|PubMed:30244176}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q6PCB6; P05067; Q96FT7-4; Q9UNS2; P22692; P60409; Q99750; Q5JTV8; Q8NFQ8; Q6PCB6; P63010-2; P05067; Q9UII2; Q9UJX2; Q53EZ4; Q96FZ7; Q92478; P35222; Q9NR90-2; Q9UHI6; Q14154; Q9H147; O75616; Q9UI08-2; Q6PIV2; Q9H4A5; P0C0S5; Q6DN90-2; Q8NA54; Q92993; Q8IUB9; A2RU56; Q8TDB4; P41218; Q9Y605; O43196-2; O00746; Q96CV9-2; Q99497; Q13113; P27986-2; P12273; O75626-3; P57729; Q96QF0-7; Q9BY12-3; Q15019-3; Q9NQ40; Q12824; Q05C28; Q7Z6I5; O75558; P21980-2; Q9BUZ4; Q9NX07-2; Q5JTY5 EC number EC 3.6.4.- Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell projection; Chaperone; Cytoplasm; Cytoplasmic vesicle; Cytoskeleton; Disease variant; Dystonia; Endoplasmic reticulum; Glycoprotein; Hydrolase; Membrane; Nucleotide-binding; Nucleus; Proteomics identification; Reference proteome; Signal; Synapse Protein physicochemical properties Chain ID A,B Molecular weight (Da) 58238.4 Length 512 Aromaticity 0.12 Instability index 39.75 Isoelectric point 5.81 Charge (pH=7) -11.04 2D Binding mode Binding energy (Kcal/mol) -6.2  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SLSREALQKDLDDNLFGQHLAKKIILNAVFGFINNPKPKKPLTLSLHGWTGTGKNFVSKIIAENIYEGGLNSDYVHLFVATLHFPHASNITLYKDQLQLWIRGNVSACARSIFIFDQMDKMHAGLIDAIKPFLDYYDLVDGVSYQKAMFIFLSNAGAERITDVALDFWRSGKQREDIKLKDIEHALSVSVFNNKNSGFWHSSLIDRNLIDYFVPFLPLEYKHLKMCIRVEMQSRGYEIDEDIVSRVAEEMTFFPKEERVFSDKGCKTVFTKLDYYYDNSYYSSPAQQVPKNPALEAFLAQFSQLEDKFPGQSSFLWQRGRKFLQKHLNASNPTEPATIIFTAAREGRETLKCLSHHVADAYTSSQKVSPIQIDGAGRTWQDSDTVKLLVDLELSYGFENGQKAAVVHHFESFPAGSTLIFYKYCDHENAAFKDVALVLTVLLEEETLEASVGPRETEEKVRDLLWAKFTNSDTPTSFNHMDSDKLSGLWSRISHLVLPVQPVSSIEEQGCLF Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Transketolase (TK) | 4KXV | 4.54 | |
Target general information Gen name TKT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms TKT1; SDDHD; HEL107; HEL-S-48 Protein family Transketolase family Biochemical class Transketolase Function Catalyzes the transfer of a two-carbon ketol group from a ketose donor to an aldose acceptor, via a covalent intermediate with the cofactor thiamine pyrophosphate. Related diseases Short stature, developmental delay, and congenital heart defects (SDDHD) [MIM:617044]: An autosomal recessive syndrome characterized by short stature, developmental delay, intellectual disability and congenital heart defects including ventricular septal defect, atrial septal defect and patent foramen ovale. Cataract and uveitis are observed in some patients. {ECO:0000269|PubMed:27259054}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09130 Interacts with P54274 EC number EC 2.2.1.1 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Calcium; Direct protein sequencing; Disease variant; Dwarfism; Isopeptide bond; Magnesium; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Thiamine pyrophosphate; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 67546.4 Length 620 Aromaticity 0.07 Instability index 35.53 Isoelectric point 7.37 Charge (pH=7) 1.28 2D Binding mode Binding energy (Kcal/mol) -6.19  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ESYHKPDQQKLQALKDTANRLRISSIQATTAAGSGHPTSCCSAAEIMAVLFFHTMRYKSQDPRNPHNDRFVLSKGHAAPILYAVWAEAGFLAEAELLNLRKISSDLDGHPVPKQAFTDVATGSLGQGLGAACGMAYTGKYFDKASYRVYCLLGDGELSEGSVWEAMAFASIYKLDNLVAILDINRLGQSDPAPLQHQMDIYQKRCEAFGWHAIIVDGHSVEELCKAFGQAKHQPTAIIAKTFKGRGITGVEDKESWHGKPLPKNMAEQIIQEIYSQIQSKKKILATPPQEDAPSVDIANIRMPSLPSYKVGDKIATRKAYGQALAKLGHASDRIIALDGDTKNSTFSEIFKKEHPDRFIECYIAEQNMVSIAVGCATRNRTVPFCSTFAAFFTRAFDQIRMAAISESNINLCGSHCGVSIGEDGPSQMALEDLAMFRSVPTSTVFYPSDGVATEKAVELAANTKGICFIRTSRPENAIIYNNNEDFQVGQAKVVLKSKDDQVTVIGAGVTLHEALAAAELLKKEKINIRVLDPFTIKPLDRKLILDSARATKGRILTVEDHYYEGGIGEAVSSAVVGEPGITVTHLAVNRVPRSGKPAELLKMFGIDRDAIAQAVRGLIT Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | PAK-4 protein kinase (PAK4) | 2X4Z | 4.54 | |
Target general information Gen name PAK4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p21-activated kinase 4; Serine/threonine-protein kinase PAK 4; PAK-4; KIAA1142 Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, STE20 subfamily Biochemical class Kinase Function Activation by various effectors including growth factor receptors or active CDC42 and RAC1 results in a conformational change and a subsequent autophosphorylation on several serine and/or threonine residues. Phosphorylates and inactivates the protein phosphatase SSH1, leading to increased inhibitory phosphorylation of the actin binding/depolymerizing factor cofilin. Decreased cofilin activity may lead to stabilization of actin filaments. Phosphorylates LIMK1, a kinase that also inhibits the activity of cofilin. Phosphorylates integrin beta5/ITGB5 and thus regulates cell motility. Phosphorylates ARHGEF2 and activates the downstream target RHOA that plays a role in the regulation of assembly of focal adhesions and actin stress fibers. Stimulates cell survival by phosphorylating the BCL2 antagonist of cell death BAD. Alternatively, inhibits apoptosis by preventing caspase-8 binding to death domain receptors in a kinase independent manner. Plays a role in cell-cycle progression by controlling levels of the cell-cycle regulatory protein CDKN1A and by phosphorylating RAN. Serine/threonine protein kinase that plays a role in a variety of different signaling pathways including cytoskeleton regulation, cell migration, growth, proliferation or cell survival. Related diseases Metachromatic leukodystrophy (MLD) [MIM:250100]: An autosomal recessive disease caused by abnormal intralysosomal accumulation of cerebroside-3-sulfate in central and peripheral nervous systems, as well as other organs. MLD is clinically characterized by leukodystrophy, progressive demyelination and a variety of neurological symptoms, including gait disturbances, ataxias, optical atrophy, dementia, seizures, and spastic tetraparesis. Decreased arylsulfatase A activity is detected in urine, leukocytes, and fibroblasts of affected individuals. Several forms of the disease can be distinguished according to the age at onset and disease severity: late infantile, juvenile and adult forms, partial cerebroside sulfate deficiency, and pseudoarylsulfatase A deficiency. Individuals with pseudoarylsulfatase A deficiency have low arylsulfatase A activity but lack neurological manifestations and are apparently healthy. {ECO:0000269|PubMed:10220151, ECO:0000269|PubMed:10381328, ECO:0000269|PubMed:10477432, ECO:0000269|PubMed:10533072, ECO:0000269|PubMed:10751093, ECO:0000269|PubMed:11020646, ECO:0000269|PubMed:11061266, ECO:0000269|PubMed:11456299, ECO:0000269|PubMed:11941485, ECO:0000269|PubMed:12503099, ECO:0000269|PubMed:12788103, ECO:0000269|PubMed:1353340, ECO:0000269|PubMed:14517960, ECO:0000269|PubMed:14680985, ECO:0000269|PubMed:15026521, ECO:0000269|PubMed:15326627, ECO:0000269|PubMed:15710861, ECO:0000269|PubMed:1670590, ECO:0000269|PubMed:1673291, ECO:0000269|PubMed:1678251, ECO:0000269|PubMed:18693274, ECO:0000269|PubMed:19606494, ECO:0000269|PubMed:20339381, ECO:0000269|PubMed:21265945, ECO:0000269|PubMed:2574462, ECO:0000269|PubMed:7581401, ECO:0000269|PubMed:7825603, ECO:0000269|PubMed:7860068, ECO:0000269|PubMed:7902317, ECO:0000269|PubMed:7906588, ECO:0000269|PubMed:7909527, ECO:0000269|PubMed:8095918, ECO:0000269|PubMed:8101038, ECO:0000269|PubMed:8101083, ECO:0000269|PubMed:8104633, ECO:0000269|PubMed:8891236, ECO:0000269|PubMed:9090526, ECO:0000269|PubMed:9272717, ECO:0000269|PubMed:9452102, ECO:0000269|PubMed:9490297, ECO:0000269|PubMed:9600244, ECO:0000269|PubMed:9819708}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Multiple sulfatase deficiency (MSD) [MIM:272200]: A clinically and biochemically heterogeneous disorder caused by the simultaneous impairment of all sulfatases, due to defective post-translational modification and activation. It combines features of individual sulfatase deficiencies such as metachromatic leukodystrophy, mucopolysaccharidosis, chondrodysplasia punctata, hydrocephalus, ichthyosis, neurologic deterioration and developmental delay. {ECO:0000269|PubMed:15146462}. The protein represented in this entry is involved in disease pathogenesis. Arylsulfatase A activity is impaired in multiple sulfatase deficiency due to mutations in SUMF1 (PubMed:15146462). SUMF1 mutations result in defective post-translational modification of ARSA at residue Cys-69 that is not converted to 3-oxoalanine (PubMed:7628016). {ECO:0000269|PubMed:15146462, ECO:0000269|PubMed:7628016}. Drugs (DrugBank ID) DB12010 Interacts with P60953; Q96EL1; P31947; P54274; O00401; P62258; P63104; P05067; P37840 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cell cycle; Cytoplasm; Kinase; Methylation; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32672.8 Length 291 Aromaticity 0.07 Instability index 36.81 Isoelectric point 8.36 Charge (pH=7) 2.21 2D Binding mode Binding energy (Kcal/mol) -6.2  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MSHEQFRAALQLVVDPGDPRSYLDNFIKIGEGSTGIVCIATVRSSGKLVAVKKMDLRKQQRRELLFNEVVIMRDYQHENVVEMYNSYLVGDELWVVMEFLEGGALTDIVTHTRMNEEQIAAVCLAVLQALSVLHAQGVIHRDIKSDSILLTHDGRVKLSDFGFCAQVSKEVPRRKXLVGTPYWMAPELISRLPYGPEVDIWSLGIMVIEMVDGEPPYFNEPPLKAMKMIRDNLPPRLKNLHKVSPSLKGFLDRLLVRDPAQRATAAELLKHPFLAKAGPPASIVPLMRQNR Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Ornithine decarboxylase (ODC1) | 2OO0 | 4.53 | |
Target general information Gen name ODC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ODC Protein family Orn/Lys/Arg decarboxylase class-II family Biochemical class Carbon-carbon lyase Function Polyamines are essential for cell proliferation and are implicated in cellular processes, ranging from DNA replication to apoptosis. Catalyzes the first and rate-limiting step of polyamine biosynthesis that converts ornithine into putrescine, which is the precursor for the polyamines, spermidine and spermine. Related diseases Bachmann-Bupp syndrome (BABS) [MIM:619075]: An autosomal dominant disorder characterized by global developmental delay, alopecia, absolute or relative macrocephaly, and facial dysmorphism. Neuroimaging shows white matter abnormalities, prominent Virchow-Robin spaces, periventricular cysts, and abnormalities of the corpus callosum. {ECO:0000269|PubMed:30239107, ECO:0000269|PubMed:30475435}. The disease is caused by variants affecting the gene represented in this entry. BABS is due to truncating variants that lead to a gain of function. This phenomenon apparently results from truncation proximal to or involving the C-terminal region of ODC1 protein, distal enough to allow escape from nonsense-mediated decay. A gain of function is corroborated by elevated plasma levels of N-acetylputrescine, with otherwise normal polyamine levels, in affected individuals. {ECO:0000269|PubMed:30475435}. Drugs (DrugBank ID) DB06243; DB04263; DB03856; DB04083; DB02824; DB01917; DB00114; DB02209; DB00203; DB00127; DB00313 Interacts with Q9H8Y8; Q92993; Q9UMX2; Q9UMX2-2 EC number EC 4.1.1.17 Uniprot keywords 3D-structure; Decarboxylase; Disease variant; Hypotrichosis; Lyase; Phosphoprotein; Polyamine biosynthesis; Proteomics identification; Pyridoxal phosphate; Reference proteome; S-nitrosylation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 45682.9 Length 410 Aromaticity 0.11 Instability index 40.93 Isoelectric point 5.61 Charge (pH=7) -6.68 2D Binding mode Binding energy (Kcal/mol) -6.18  Molscript Map 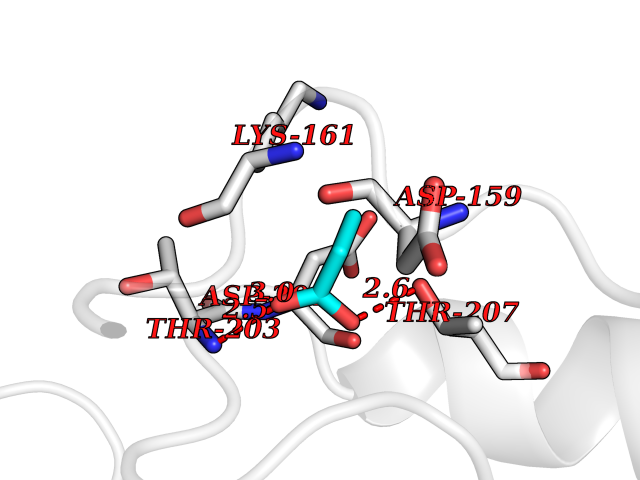 Pymol Map 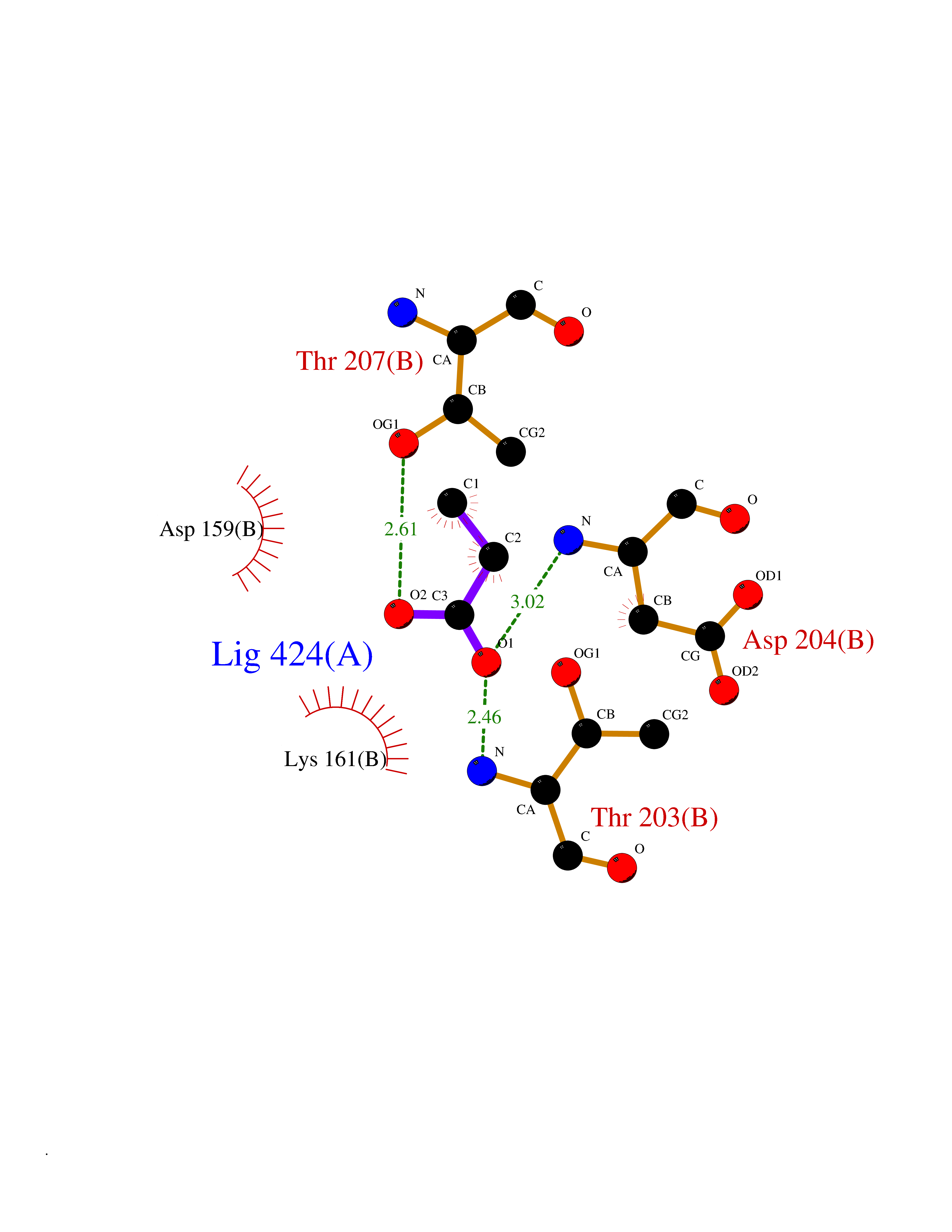 Ligplot Map 3D Binding mode Sequence LMNNFGNEEFDCHFLDEGFTAKDILDQKINEVSSSDDKDAFYVADLGDILKKHLRWLKALPRVTPFYAVKCNDSKAIVKTLAATGTGFDCASKTEIQLVQSLGVPPERIIYANPCKQVSQIKYAANNGVQMMTFDSEVELMKVARAHPKAKLVLRIATDDSKAVCRLSVKFGATLRTSRLLLERAKELNIDVVGVSFHVGSGCTDPETFVQAISDARCVFDMGAEVGFSMYLLDIGGGFPGSEDVKLKFEEITGVINPALDKYFPSDSGVRIIAEPGRYYVASAFTLAVNIIAKKIVLEQTFMYYVNDGVYGSFNCILYDHAHVKPLLQKRPKPDEKYYSSSIWGPTCDGLDRIVERCDLPEMHVGDWMLFENMGAYTVAAASTFNGFQRPTIYYVMSGPAWQLMQQFQN Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Bifunctional methylenetetrahydrofolate dehydrogenase/cyclohydrolase, mitochondrial | 5TC4 | 4.53 | |
Target general information Gen name MTHFD2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NMDMC Protein family Tetrahydrofolate dehydrogenase/cyclohydrolase family Biochemical class Oxidoreductase Function Magnesium ion binding.Methenyltetrahydrofolate cyclohydrolase activity.Methylenetetrahydrofolate dehydrogenase (NAD+) activity.Methylenetetrahydrofolate dehydrogenase (NADP+) activity.Phosphate ion binding. Related diseases Acute hepatic porphyria (AHEPP) [MIM:612740]: A form of porphyria. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. AHP is characterized by attacks of gastrointestinal disturbances, abdominal colic, paralyses and peripheral neuropathy. Most attacks are precipitated by drugs, alcohol, caloric deprivation, infections, or endocrine factors. {ECO:0000269|PubMed:10706561, ECO:0000269|PubMed:1309003, ECO:0000269|PubMed:1569184, ECO:0000269|PubMed:17236137, ECO:0000269|PubMed:2063868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00157; DB00116 Interacts with Q9UJ70-2 EC number 1.5.1.15; 3.5.4.9 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Hydrolase; Isopeptide bond; Magnesium; Mitochondrion; Multifunctional enzyme; NAD; NADP; One-carbon metabolism; Oxidoreductase; Proteomics identification; Reference proteome; Transit peptide; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 31600.3 Length 292 Aromaticity 0.03 Instability index 27.9 Isoelectric point 8 Charge (pH=7) 1.64 2D Binding mode Binding energy (Kcal/mol) -6.17 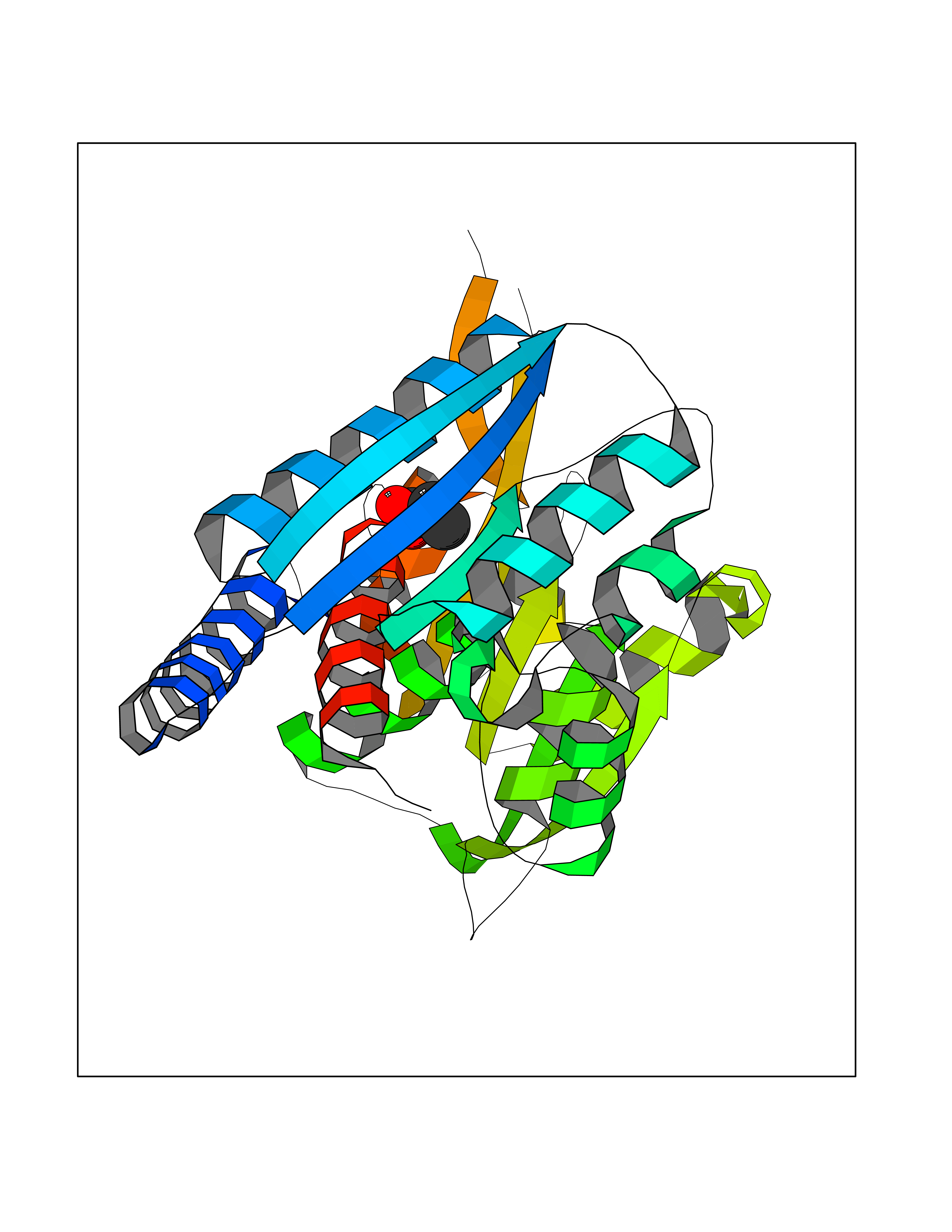 Molscript Map 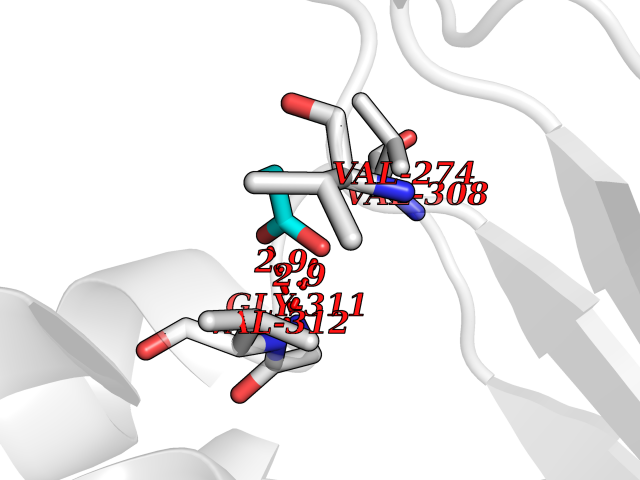 Pymol Map 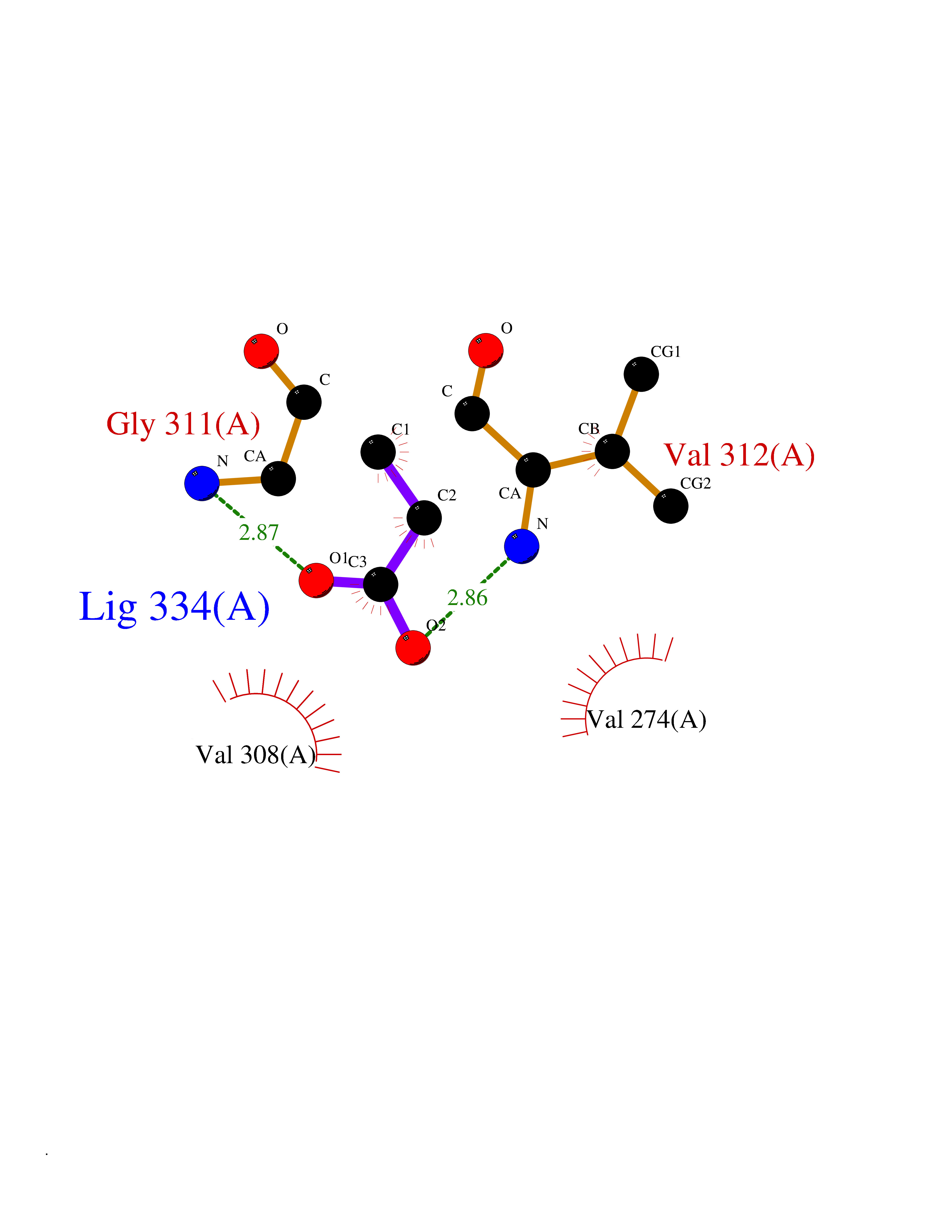 Ligplot Map 3D Binding mode Sequence EAVVISGRKLAQQIKQEVRQEVEEWVASGNKRPHLSVILVGENPASHSYVLNKTRAAAVVGINSETIMKPASISEEELLNLINKLNNDDNVDGLLVQLPLPEHIDERRICNAVSPDKDVDGFHVINVGRMCLDQYSMLPATPWGVWEIIKRTGIPTLGKNVVVAGRSKNVGMPIAMLLHTDGAHERPGGDATVTISHRYTPKEQLKKHTILADIVISAAGIPNLITADMIKEGAAVIDVGINRVHKPKLVGDVDFEGVRQKAGYITPVPGGVGPMTVAMLMKNTIIAAKKVL Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Alkyl hydroperoxide reductase subunit F | 1HYU | 4.53 | |
Target general information Gen name ahpF Organism Salmonella typhimurium (strain LT2 / SGSC1412 / ATCC 700720) Uniprot ID TTD ID NA Synonyms STM0609 Protein family Class-II pyridine nucleotide-disulfide oxidoreductase family Biochemical class Oxidoreductase Function Alkyl hydroperoxide reductase activity.Electron carrier activity.Flavin adenine dinucleotide binding.NAD binding.Protein disulfide oxidoreductase activity. Related diseases Glutathionuria (GLUTH) [MIM:231950]: A very rare, autosomal recessive metabolic disorder characterized by the presence of glutathione in the urine, due to generalized gamma-glutamyl transpeptidase deficiency. Most patients manifest mild to moderate intellectual disability, and behavioral disturbance. Seizures, tremor, marfanoid features and strabismus are observed in some patients. {ECO:0000269|PubMed:29483667}. The disease is caused by variants affecting the gene represented in this entry. A large homozygous deletion that removes several exons of all isoforms of GGT1 has been found in one family affected by glutathionuria. {ECO:0000269|PubMed:29483667}. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.8.1.- Uniprot keywords 3D-structure; Direct protein sequencing; Disulfide bond; FAD; Flavoprotein; NAD; NADP; Oxidoreductase; Redox-active center; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 55949.2 Length 521 Aromaticity 0.06 Instability index 25.77 Isoelectric point 5.62 Charge (pH=7) -9.6 2D Binding mode Binding energy (Kcal/mol) -6.18 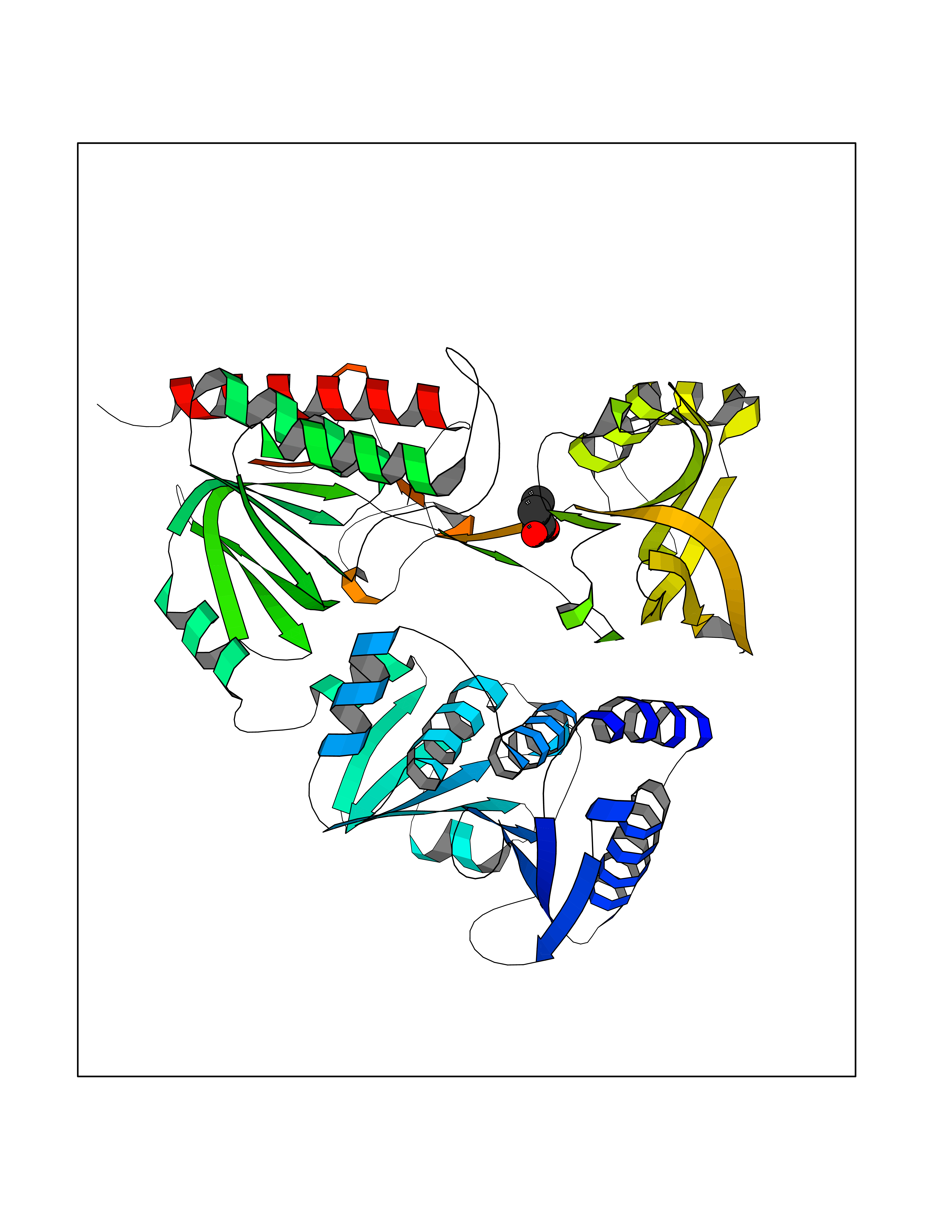 Molscript Map 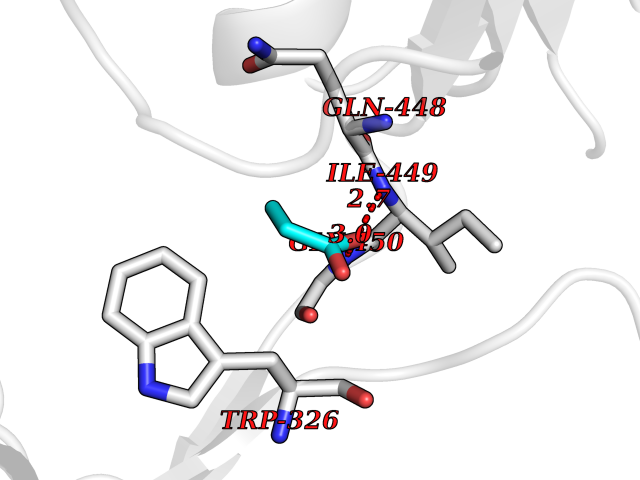 Pymol Map 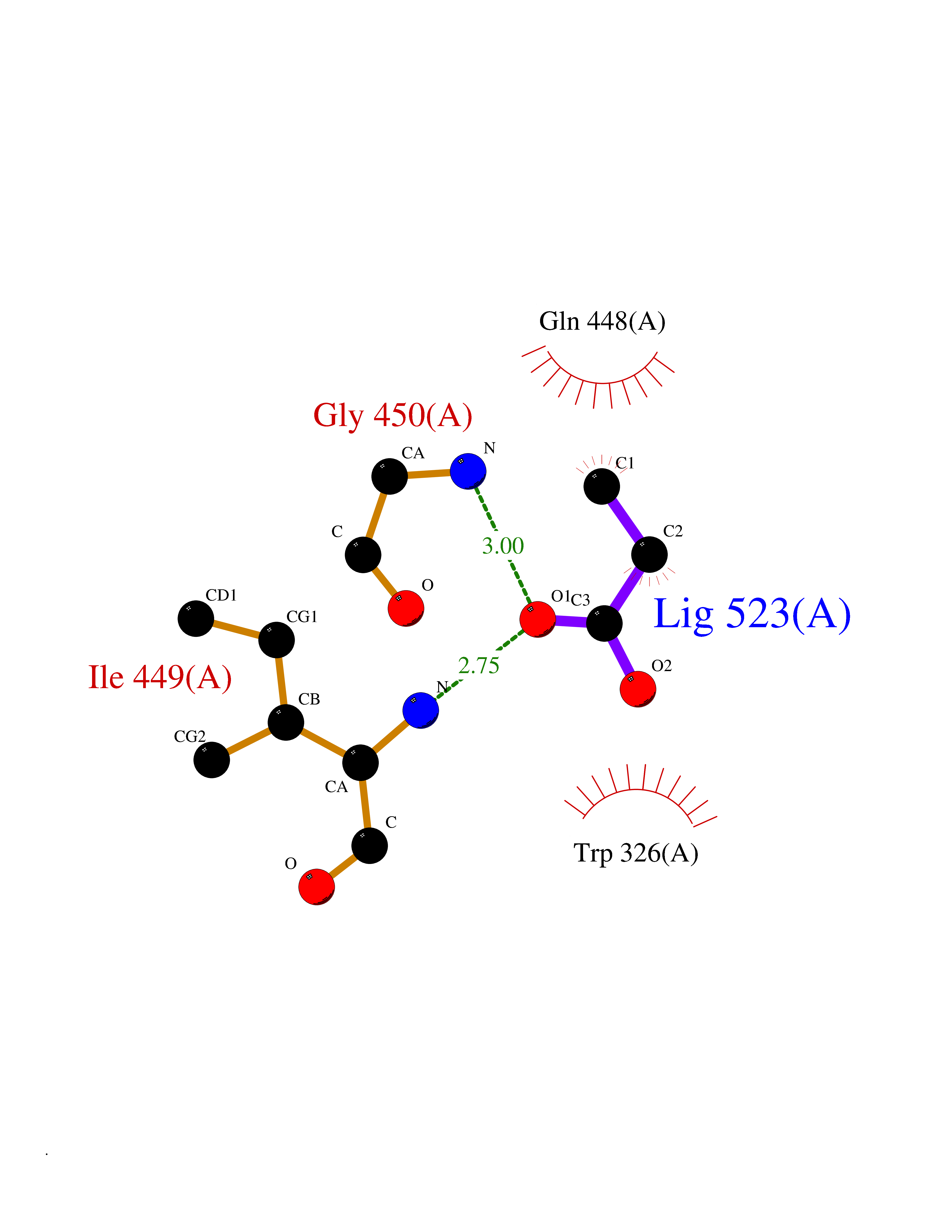 Ligplot Map 3D Binding mode Sequence MLDTNMKTQLRAYLEKLTKPVELIATLDDSAKSAEIKELLAEIAELSDKVTFKEDNTLPVRKPSFLITNPGSQQGPRFAGSPLGHEFTSLVLALLWTGGHPSKEAQSLLEQIRDIDGDFEFETYYSLSCHNCPDVVQALNLMAVLNPRIKHTAIDGGTFQNEITERNVMGVPAVFVNGKEFGQGRMTLTEIVAKVDTGAEKRAAEALNKRDAYDVLIVGSGPAGAAAAVYSARKGIRTGLMGERFGGQVLDTVDIENYISVPKTEGQKLAGALKAHVSDYDVDVIDSQSASKLVPAATEGGLHQIETASGAVLKARSIIIATGAKWRNMNVPGEDQYRTKGVTYCPHCDGPLFKGKRVAVIGGGNSGVEAAIDLAGIVEHVTLLEFAPEMKADQVLQDKVRSLKNVDIILNAQTTEVKGDGSKVVGLEYRDRVSGDIHSVALAGIFVQIGLLPNTHWLEGALERNRMGEIIIDAKCETSVKGVFAAGDCTTVPYKQIIIATGEGAKASLSAFDYLIRTKIA Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Caspase-1 (CASP1) | 1SC3 | 4.53 | |
Target general information Gen name CASP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms P45; Interleukin-1 beta-converting enzyme; Interleukin-1 beta converting enzyme; Interleukin-1 beta convertase; IL1BCE; IL1BC; IL-1BC; IL-1 beta-converting enzyme; IL-1 beta converting enzyme; ICE; CA Protein family Peptidase C14A family Biochemical class Peptidase Function Important for defense against pathogens. Cleaves and activates sterol regulatory element binding proteins (SREBPs). Can also promote apoptosis. Upon inflammasome activation, during DNA virus infection but not RNA virus challenge, controls antiviral immunity through the cleavage of CGAS, rendering it inactive. Thiol protease that cleaves IL-1 beta between an Asp and an Ala, releasing the mature cytokine which is involved in a variety of inflammatory processes. Related diseases Cataract 6, multiple types (CTRCT6) [MIM:116600]: An opacification of the crystalline lens of the eye that frequently results in visual impairment or blindness. Opacities vary in morphology, are often confined to a portion of the lens, and may be static or progressive. CTRCT6 includes posterior polar and age-related cortical cataracts, among others. Posterior polar cataract is a subcapsular opacity, usually disk-shaped, located at the back of the lens. Age-related cortical cataract is a developmental punctate opacity restricted to the cortex. The cataract is white or cerulean, increases in number with age, but rarely affects vision. {ECO:0000269|PubMed:19005574, ECO:0000269|PubMed:19306328, ECO:0000269|PubMed:19649315, ECO:0000269|PubMed:22570727}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Overexpressed in several cancer types and promotes malignancy. {ECO:0000269|PubMed:19573808}. Drugs (DrugBank ID) DB07733; DB07916; DB00945; DB05408; DB05301; DB01017; DB04875; DB05507; DB07744 Interacts with Q5XLA6; P57730; P29466; P09038; P57764; P01583; O15553; O15553-2; Q9NPP4; Q9C000; Q9HC29; Q9ULZ3; P58753-2; Q08AM6; P05067; P54252; Q13867; P14136; P42858; Q13387-4; P27986-2; O14494; O60260-5; P49810; P84022; Q9BX74; O60784-2; Q08AM6; Q9UBQ0-2; O76024; P01583 EC number EC 3.4.22.36 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; Cell membrane; Cytoplasm; Direct protein sequencing; Hydrolase; Isopeptide bond; Membrane; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Thiol protease; Ubl conjugation; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 29526.6 Length 261 Aromaticity 0.08 Instability index 43.65 Isoelectric point 7.03 Charge (pH=7) 0.06 2D Binding mode Binding energy (Kcal/mol) -6.18  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence TSSGSEGNVKLCSLEEAQRIWKQKSAEIYPIMDKSSRTRLALIICNEEFDSIPRRTGAEVDITGMTMLLQNLGYSVDVKKNLTASDMTTELEAFAHRPEHKTSDSTFLVFMSHGIREGICGKKHSEQVPDILQLNAIFNMLNTKNCPSLKDKPKVIIIQAARGDSPGVVWFKDAIKKAHIEKDFIAFCSSTPDNVSWRHPTMGSVFIGRLIEHMQEYACSCDVEEIFRKVRFSFEQPDGRAQMPTTERVTLTRCFYLFPGH Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Cytosolic purine 5'-nucleotidase | 2JC9 | 4.53 | |
Target general information Gen name NT5C2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PNT5;NT5B;NT5CP Protein family 5'(3')-deoxyribonucleotidase family Biochemical class Hydrolase Function 5'-nucleotidase activity.Metal ion binding.Nucleoside phosphotransferase activity.Nucleotide binding. Related diseases Spastic paraplegia 45, autosomal recessive (SPG45) [MIM:613162]: A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. Some SPG45 patients manifest intellectual disability, contractures and learning disability. {ECO:0000269|PubMed:24482476, ECO:0000269|PubMed:28884889}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00171; DB00811; DB06408 Interacts with P48047; P51116; Q8IVS8; Q7L9L4; Q86TA1; Q70IA8; Q9Y5B8; Q6ZVK8; O00560; Q9NRS6 EC number 2.7.1.77; 3.1.3.5; 3.1.3.99 Uniprot keywords 3D-structure; Allosteric enzyme; Alternative splicing; ATP-binding; Cytoplasm; Disease variant; Hereditary spastic paraplegia; Hydrolase; Magnesium; Metal-binding; Neurodegeneration; Nucleotide metabolism; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 53978.4 Length 467 Aromaticity 0.14 Instability index 30.87 Isoelectric point 8.25 Charge (pH=7) 3.41 2D Binding mode Binding energy (Kcal/mol) -6.18  Molscript Map 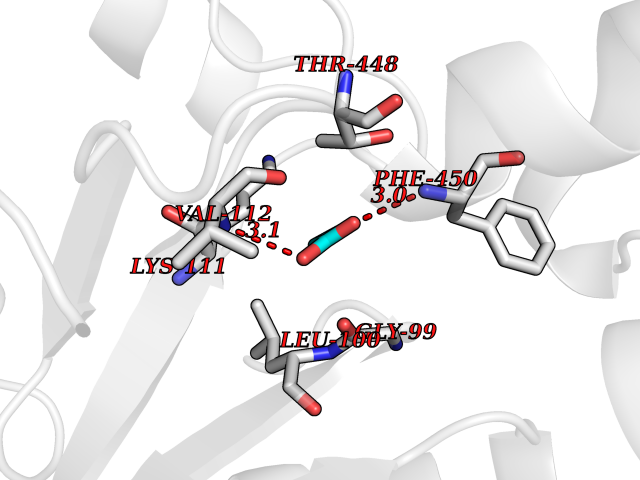 Pymol Map 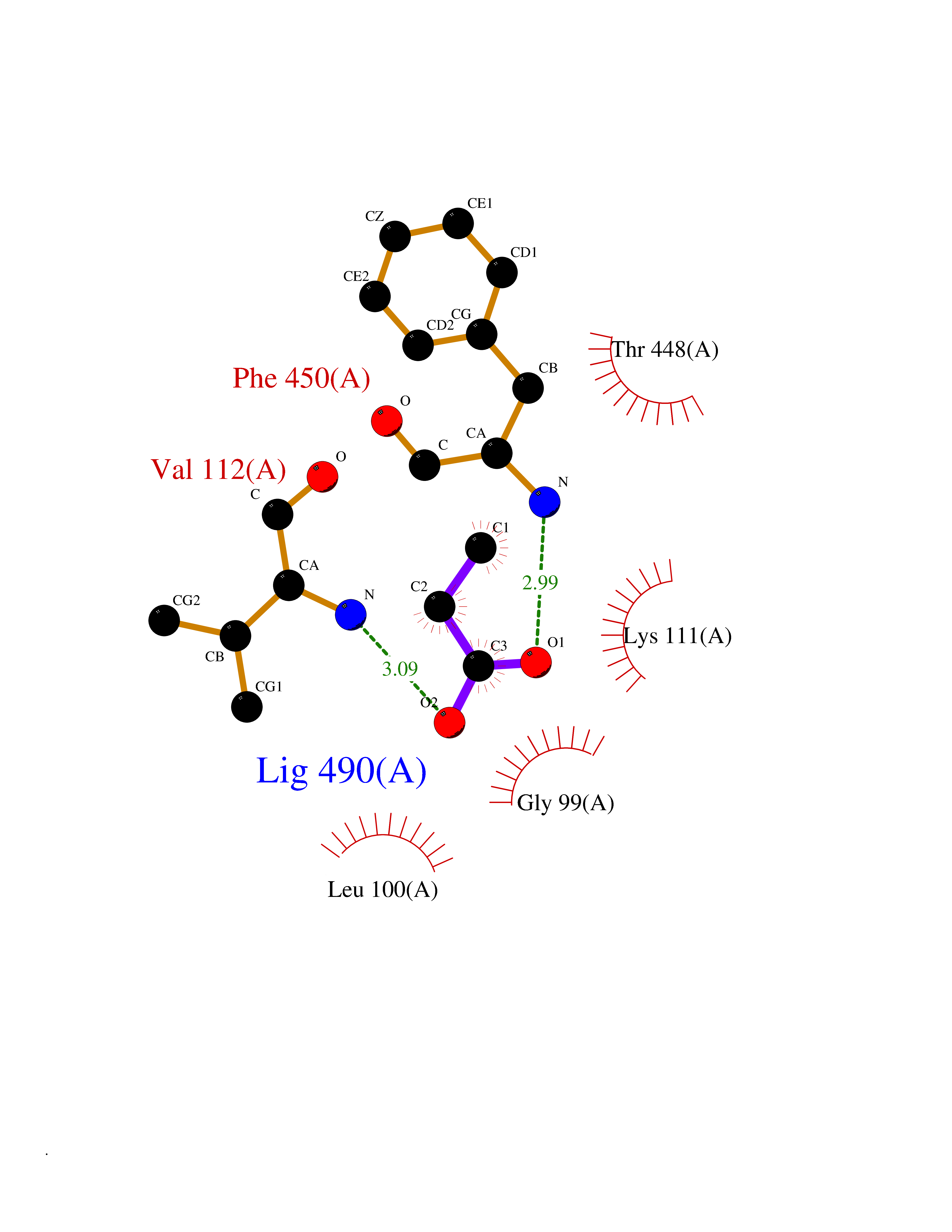 Ligplot Map 3D Binding mode Sequence TSWSDRLQNAADMPANMDKHALKKYRREAYHRVFVNRSLAMEKIKCFGFDMDYTLAVYKSPEYESLGFELTVERLVSIGYPQELLSFAYDSTFPTRGLVFDTLYGNLLKVDAYGNLLVCAHGFNFIRGPETREQYPNKFIQRDDTERFYILNTLFNLPETYLLACLVDFFTNCPRYTSCETGFKDGDLFMSYRSMFQDVRDAVDWVHYKGSLKEKTVENLEKYVVKDGKLPLLLSRMKEVGKVFLATNSDYKYTDKIMTYLFDFPHGPKPGSSHRPWQSYFDLILVDARKPLFFGEGTVLRQVDTKTGKLKIGTYTGPLQHGIVYSGGSSDTICDLLGAKGKDILYIGDHIFGDILKSKKRQGWRTFLVIPELAQELHVWTDKSSLFEELQSLDIFLAQRRIKKVTHDMDMCYGMMGSLFRSGSRQTLFASQVMRYADLYAASFINLLYYPFSYLFRAAHVLMPHES Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Mycobacterium Membrane protein mmpL3 (MycB mmpL3) | 7NVH | 4.53 | |
Target general information Gen name MycB mmpL3 Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID Synonyms Trehalose monomycolate exporter MmpL3; TMM exporter MmpL3 Protein family Resistance-nodulation-cell division (RND) (TC 2.A.6) family, MmpL subfamily Biochemical class NA Function Transports trehalose monomycolate (TMM) across the inner membrane. Could also be part of a heme-iron acquisition system. Related diseases Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:8955068}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:17332249}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 3 (NS3) [MIM:609942]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:16474405, ECO:0000269|PubMed:16773572, ECO:0000269|PubMed:17056636, ECO:0000269|PubMed:17468812, ECO:0000269|PubMed:19396835, ECO:0000269|PubMed:20949621}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Gastric cancer (GASC) [MIM:613659]: A malignant disease which starts in the stomach, can spread to the esophagus or the small intestine, and can extend through the stomach wall to nearby lymph nodes and organs. It also can metastasize to other parts of the body. The term gastric cancer or gastric carcinoma refers to adenocarcinoma of the stomach that accounts for most of all gastric malignant tumors. Two main histologic types are recognized, diffuse type and intestinal type carcinomas. Diffuse tumors are poorly differentiated infiltrating lesions, resulting in thickening of the stomach. In contrast, intestinal tumors are usually exophytic, often ulcerating, and associated with intestinal metaplasia of the stomach, most often observed in sporadic disease. {ECO:0000269|PubMed:14534542, ECO:0000269|PubMed:3034404, ECO:0000269|PubMed:7773929}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in KRAS are a cause of pylocytic astrocytoma (PA). Pylocytic astrocytomas are neoplasms of the brain and spinal cord derived from glial cells which vary from histologically benign forms to highly anaplastic and malignant tumors. {ECO:0000269|PubMed:16247081}.; DISEASE: Cardiofaciocutaneous syndrome 2 (CFC2) [MIM:615278]: A form of cardiofaciocutaneous syndrome, a multiple congenital anomaly disorder characterized by a distinctive facial appearance, heart defects and intellectual disability. Heart defects include pulmonic stenosis, atrial septal defects and hypertrophic cardiomyopathy. Some affected individuals present with ectodermal abnormalities such as sparse, friable hair, hyperkeratotic skin lesions and a generalized ichthyosis-like condition. Typical facial features are similar to Noonan syndrome. They include high forehead with bitemporal constriction, hypoplastic supraorbital ridges, downslanting palpebral fissures, a depressed nasal bridge, and posteriorly angulated ears with prominent helices. CFC2 patients often do not have the skin abnormalities, such as ichthyosis, hyperkeratosis, and hemangioma observed in CFC1. {ECO:0000269|PubMed:16474404, ECO:0000269|PubMed:16474405, ECO:0000269|PubMed:17056636, ECO:0000269|PubMed:20949621, ECO:0000269|PubMed:21797849}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: KRAS mutations are involved in cancer development. {ECO:0000269|PubMed:14534542, ECO:0000269|PubMed:1553789, ECO:0000269|PubMed:16533793, ECO:0000269|PubMed:24623306, ECO:0000269|PubMed:3034404, ECO:0000269|PubMed:3627975, ECO:0000269|PubMed:6092920, ECO:0000269|PubMed:6695174, ECO:0000269|PubMed:7773929}.; DISEASE: Oculoectodermal syndrome (OES) [MIM:600268]: A syndrome characterized by the association of epibulbar dermoids and aplasia cutis congenita. Affected individuals show multiple, asymmetric, atrophic, non-scarring and hairless regions that may be associated with hamartomas. Ectodermal changes include linear hyperpigmentation that may follow the lines of Blaschko and rarely epidermal nevus-like lesions. Epibulbar dermoids may be uni-or bilateral. Additional ocular anomalies such as skin tags of the upper eyelid, rarely optic nerve or retinal changes, and microphthalmia can be present. The phenotypic expression is highly variable, and various other abnormalities have occasionally been reported including growth failure, lymphedema, cardiovascular defects, as well as neurodevelopmental symptoms like developmental delay, epilepsy, learning difficulties, and behavioral abnormalities. Benign tumor-like lesions such as nonossifying fibromas of the long bones and giant cell granulomas of the jaws have repeatedly been observed and appear to be age-dependent, becoming a common manifestation in individuals aged 5 years or older. {ECO:0000269|PubMed:25808193, ECO:0000269|PubMed:26970110, ECO:0000269|PubMed:30891959}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Schimmelpenning-Feuerstein-Mims syndrome (SFM) [MIM:163200]: A disease characterized by sebaceous nevi, often on the face, associated with variable ipsilateral abnormalities of the central nervous system, ocular anomalies, and skeletal defects. Many oral manifestations have been reported, not only including hypoplastic and malformed teeth, and mucosal papillomatosis, but also ankyloglossia, hemihyperplastic tongue, intraoral nevus, giant cell granuloma, ameloblastoma, bone cysts, follicular cysts, oligodontia, and odontodysplasia. Sebaceous nevi follow the lines of Blaschko and these can continue as linear intraoral lesions, as in mucosal papillomatosis. {ECO:0000269|PubMed:30891959}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Cell wall biogenesis/degradation; Lipid transport; Membrane; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 77237 Length 717 Aromaticity 0.09 Instability index 33.34 Isoelectric point 8.65 Charge (pH=7) 4.3 2D Binding mode Binding energy (Kcal/mol) -6.18  Molscript Map 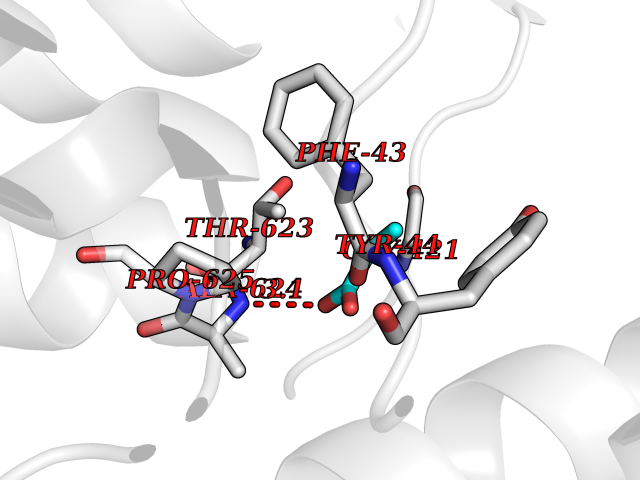 Pymol Map 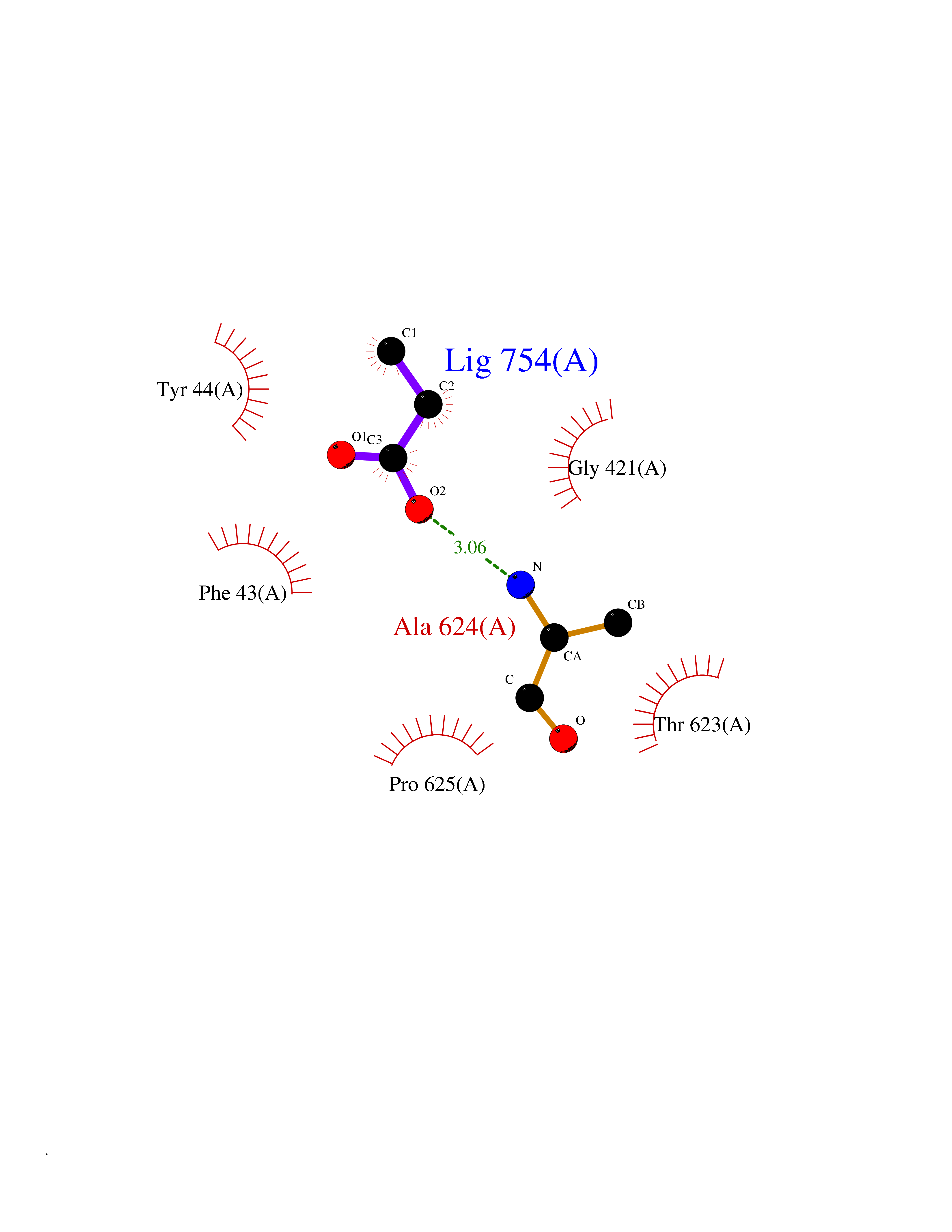 Ligplot Map 3D Binding mode Sequence MFAWWGRTVYRYRFIVIGVMVALCLGGGVFGLSLGKHVTQSGFYDDGSQSVQASVLGDQVYGRDRSGHIVAIFQAPAGKTVDDPAWSKKVVDELNRFQQDHPDQVLGWAGYLRASQATGMATADKKYTFVSIPLKGDDDDTILNNYKAIAPDLQRLDGGTVKLAGLQPVAEALTGTIATDQRRMEVLALPLVAVVLFFVFGGVIAAGLPVMVGGLCIAGALGIMRFLAIFGPVHYFAQPVVSLIGLGIAIDYGLFIVSRFREEIAEGYDTETAVRRTVITAGRTVTFSAVLIVASAIGLLLFPQGFLKSLTYATIASVMLSAILSITVLPACLGILGKHVDAEEVEAGFWGKLVNRVMKRPVLFAAPIVIIMILLIIPVGKLSLGGISEKYLPPTNSVRQAQEEFDKLFPGYRTNPLTLVIQTSNHQPVTDAQIADIRSKAMAIGGFIEPDNDPANMWQERAYAVGASKDPSVRVLQNGLINPADASKKLTELRAITPPKGITVLVGGTPALELDSIHGLFAKMPLMVVILLTTTIVLMFLAFGSVVLPIKATLMSALTLGSTMGILTWIFVDGHFSKWLNFTPTPLTAPVIGLIIALVFGLSTDYEVFLVSRMVEARERGMSTQEAIRIGTAATGRIITAAALIVAVVAGAFVFSDLVMMKYLAFGLMAALLLDATVVRMFLVPSVMKLLGDDCWWAPRWARRLQTRIGLGEIHLP Hydrogen bonds contact Hydrophobic contact | ||||