Job Results:
Ligand
Structure
Job ID
5217c115ac480b5185ad5781ec63f66c
Job name
NA
Time
2026-02-27 11:51:57
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Plasmodium Adenylosuccinate synthetase (Malaria Adss) | 1P9B | 4.55 | |
Target general information Gen name Malaria Adss Organism Plasmodium falciparum Uniprot ID TTD ID Synonyms IMP--aspartate ligase; Adenylosuccinate synthase; AdSS; AMPSase Protein family Adenylosuccinate synthetase family Biochemical class Carbon-nitrogen ligase Function Plays an important role in the salvage pathway for purine nucleotide biosynthesis. Catalyzes the first committed step in the biosynthesis of AMP from IMP. Related diseases Hypertension and brachydactyly syndrome (HTNB) [MIM:112410]: A syndrome characterized by brachydactyly type E, severe salt-independent but age-dependent hypertension, an increased fibroblast growth rate, neurovascular contact at the rostral-ventrolateral medulla, and altered baroreflex blood pressure regulation. It results in death from stroke before age 50 years when untreated. Brachydactyly type E is characterized by shortening of the fingers mainly in the metacarpals and metatarsals. {ECO:0000269|PubMed:25961942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03510; DB04315; DB02109 Interacts with NA EC number EC 6.3.4.4 Uniprot keywords 3D-structure; Cytoplasm; GTP-binding; Ligase; Magnesium; Metal-binding; Nucleotide-binding; Purine biosynthesis Protein physicochemical properties Chain ID A Molecular weight (Da) 47877.9 Length 424 Aromaticity 0.09 Instability index 31.72 Isoelectric point 7.63 Charge (pH=7) 1.58 2D Binding mode Binding energy (Kcal/mol) -6.2 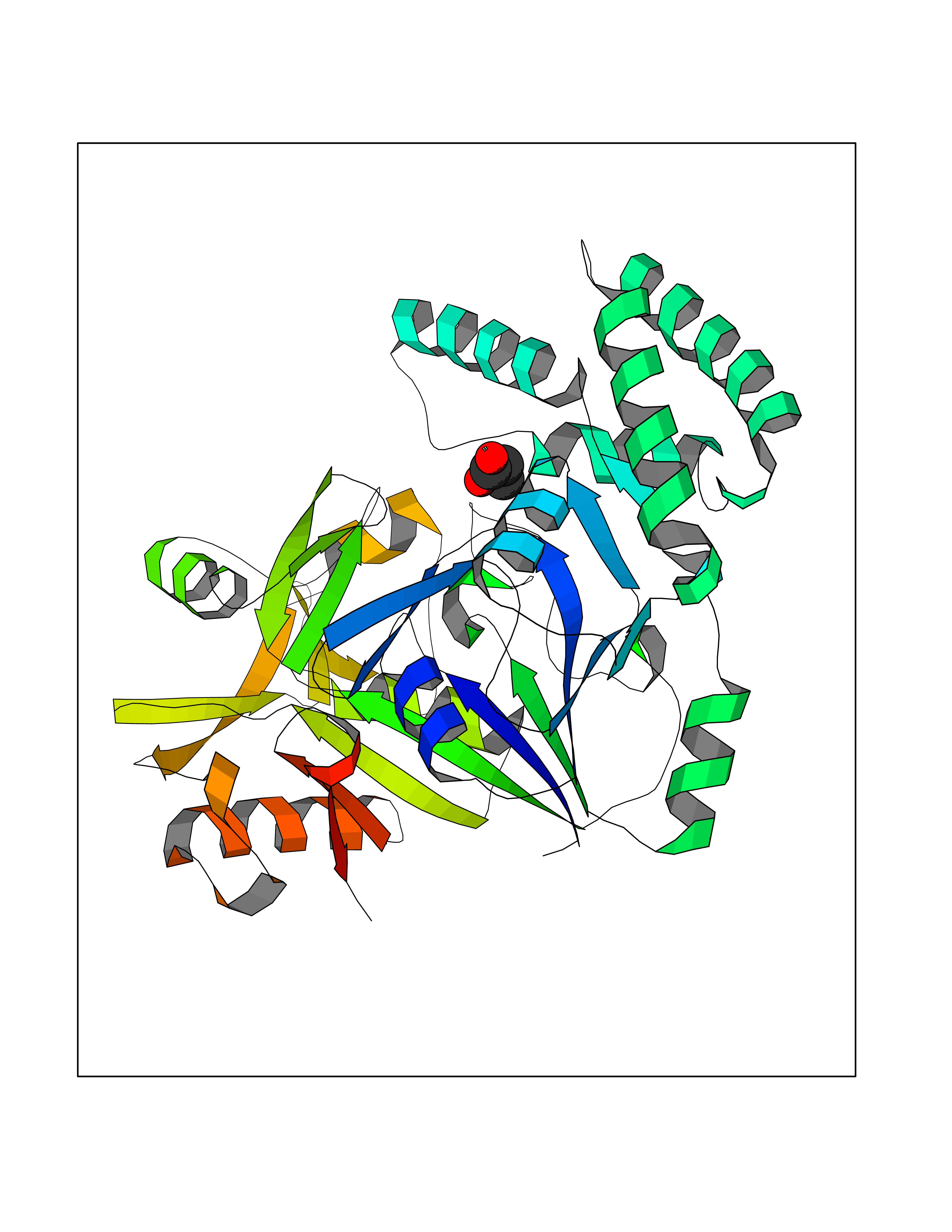 Molscript Map 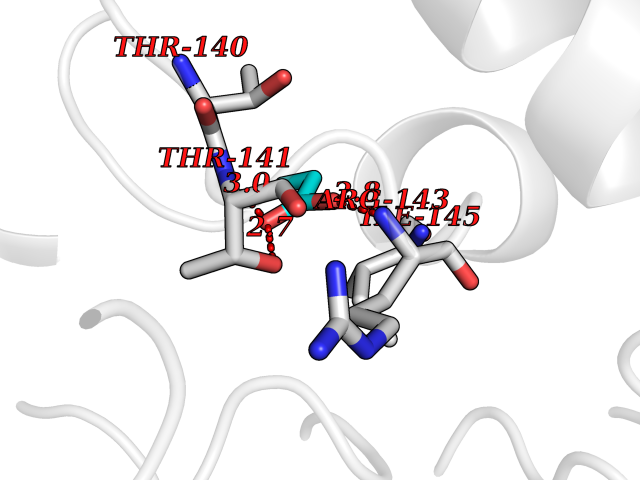 Pymol Map 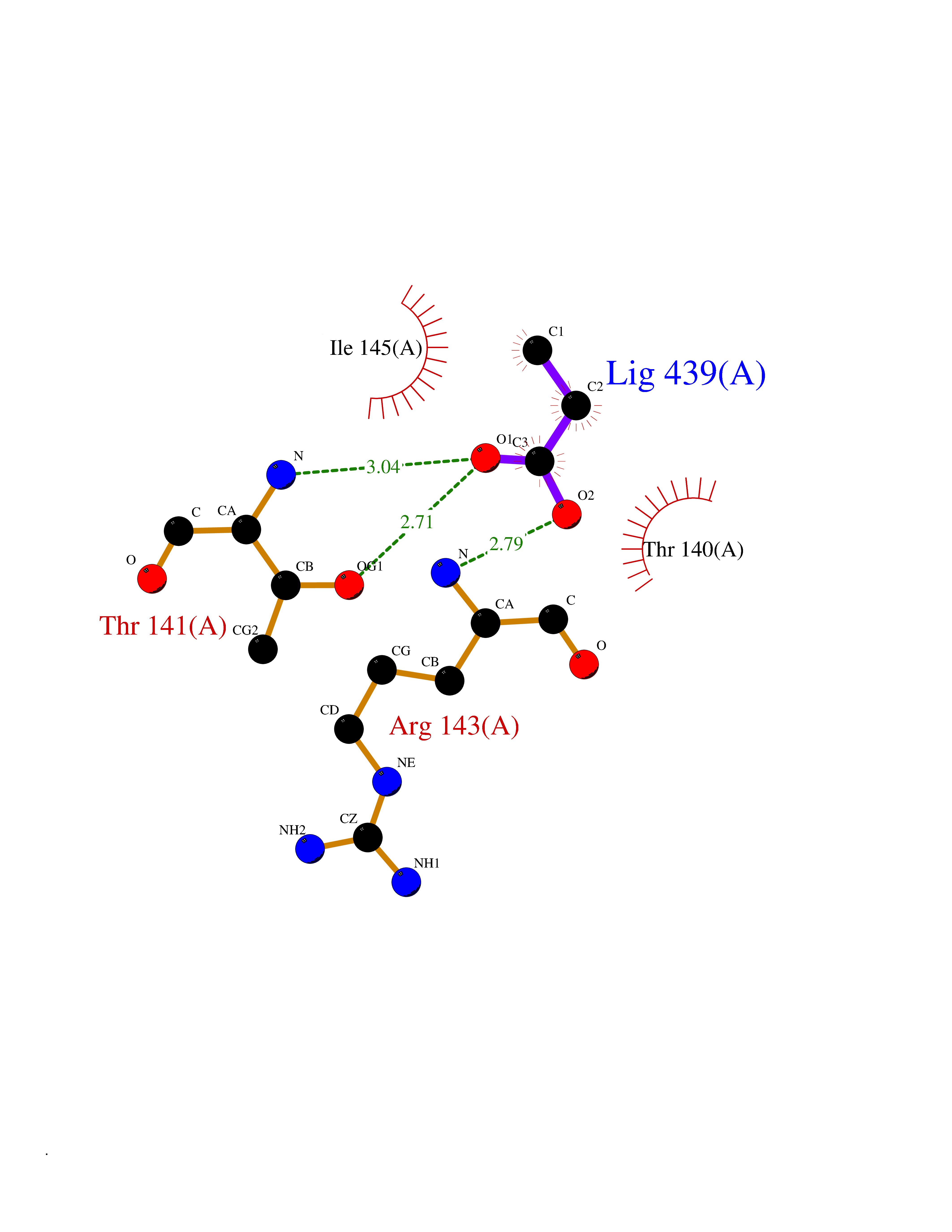 Ligplot Map 3D Binding mode Sequence GNVVAILGAQWGDEGKGKIIDMLSEYSDITCRFNGGANAGHTISVNDKKYALHLLPCGVLYDNNISVLGNGMVIHVKSLMEEIESVGGKLLDRLYLSNKAHILFDIHQIIDSIQETKKLKEGKQIGTTKRGIGPCYSTKASRIGIRLGTLKNFENFKNMYSKLIDHLMDLYNITEYDKEKELNLFYNYHIKLRDRIVDVISFMNTNLENNKKVLIEGANAAMLDIDFGTYPYVTSSCTTVGGVFSGLGIHHKKLNLVVGVVKSYLTRVGCGPFLTELNNDVGQYLREKGHEYGTTTKRPRRCGWLDIPMLLYVKCINSIDMINLTKLDVLSGLEEILLCVNFKNKKTGELLEKGCYPVEEEISEEYEPVYEKFSGWKEDISTCNEFDELPENAKKYILAIEKYLKTPIVWIGVGPNRKNMIVKK Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Branched-chain-amino-acid transaminase 2 (BCAT2) | 5CR5 | 4.54 | |
Target general information Gen name BCAT2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Placental protein 18; PP18; ECA39 protein; Branched-chain amino acid aminotransferase; BCAT2; BCAT(m); BCAT Protein family Class-IV pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transaminase Function Catalyzes the first reaction in the catabolism of the essential branched chain amino acids leucine, isoleucine, and valine. May also function as a transporter of branched chain alpha-keto acids. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB04074; DB00142; DB00167; DB00149; DB02635; DB00114; DB02142 Interacts with P10809 EC number EC 2.6.1.42 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Amino-acid biosynthesis; Aminotransferase; Branched-chain amino acid biosynthesis; Disease variant; Lipid metabolism; Mitochondrion; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase; Transit peptide Protein physicochemical properties Chain ID A,B Molecular weight (Da) 40725 Length 359 Aromaticity 0.09 Instability index 39.14 Isoelectric point 8.46 Charge (pH=7) 3.24 2D Binding mode Binding energy (Kcal/mol) -6.2 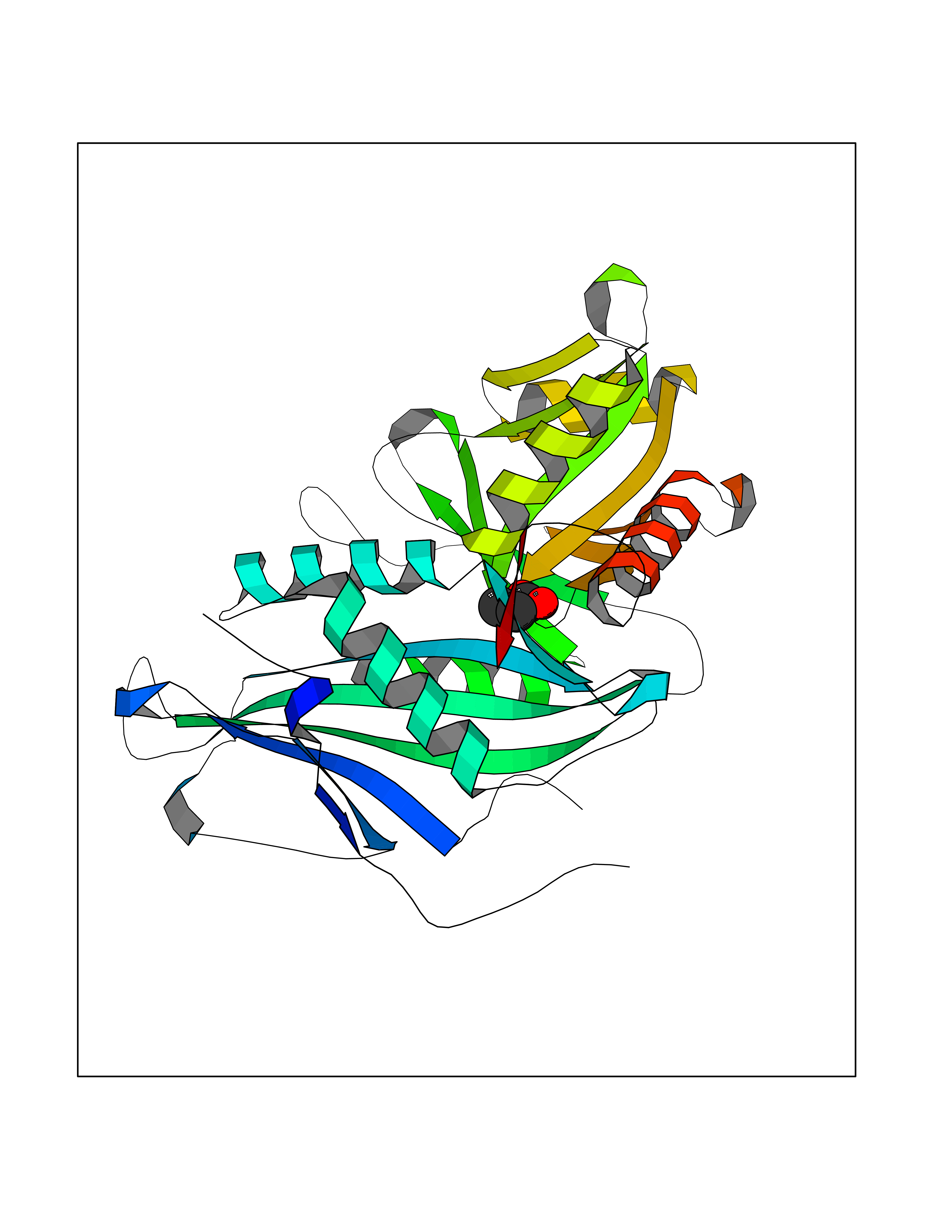 Molscript Map 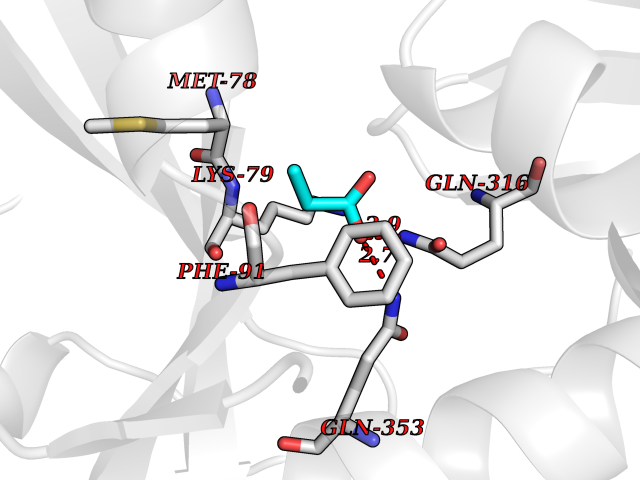 Pymol Map 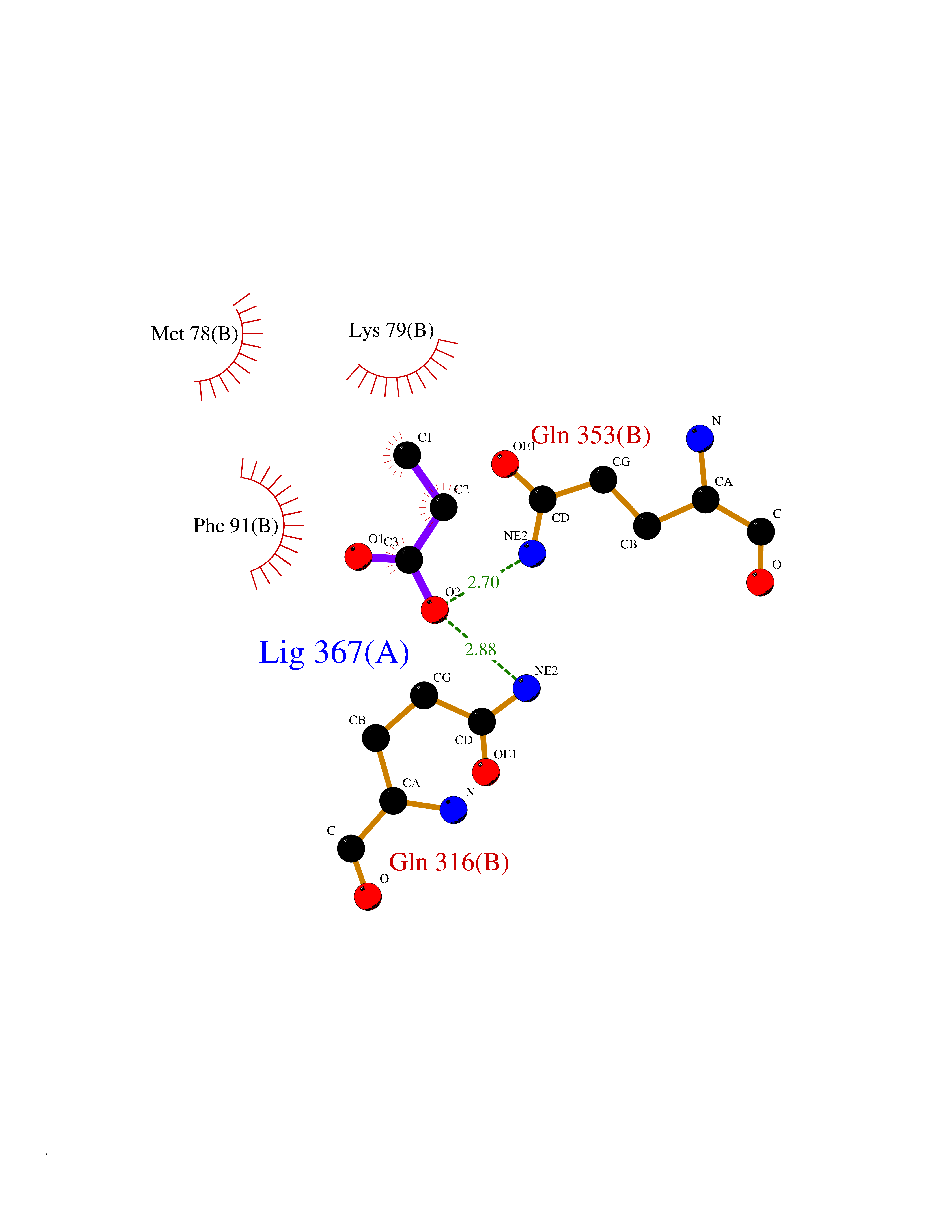 Ligplot Map 3D Binding mode Sequence SSFKAADLQLEMTQKPHKKPGLVFGKTFTDHMLMVEWNDKGWGQPRIQPFQNLTLHPASSSLHYSLQLFEGMKAFKGKDQQVRLFRPWLNMDRMLRSAMRLCLPSFDKLELLECIRRLIEVDKDWVPDAAGTSLYVRPVLIGNEPSLGVSQPTRALLFVILCPVGAYFPGGSVTPVSLLADPAFIRAWVGGVGNYKLGGNYGPTVLVQQEALKRGCEQVLWLYGPDHQLTEVGTMNIFVYWTHEDGVLELVTPPLNGVILPGVVRQSLLDMAQTWGEFRVVERTITMKQLLRALEEGRVREVFGSGTACQVCPVHRILYKDRNLHIPTMENGPELILRFQKELKEIQYGIRAHEWMFPV Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Tryptophan 5-hydroxylase 1 (TPH1) | 5TPG | 4.54 | |
Target general information Gen name TPH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tryptophan 5-monooxygenase 1; TRPH; TPRH Protein family Biopterin-dependent aromatic amino acid hydroxylase family Biochemical class Paired donor oxygen oxidoreductase Function Responsible for addition of the -HO group (hydroxylation) to the 5 position to form the amino acid 5-hydroxytryptophan (5-HTP), which is the initial and rate-limiting step in the synthesis of the neurotransmitter serotonin. Related diseases Tyrosinemia 2 (TYRSN2) [MIM:276600]: An inborn error of metabolism characterized by elevations of tyrosine in the blood and urine, and oculocutaneous manifestations. Typical features include palmoplantar keratosis, painful corneal ulcers, and intellectual disability. {ECO:0000269|PubMed:1357662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05199; DB00360; DB12095; DB00150 Interacts with Q14457; Q96IK1-2; Q9UKB3; Q9H8Y8; O43586; O95789-4 EC number EC 1.14.16.4 Uniprot keywords 3D-structure; Alternative splicing; Iron; Metal-binding; Monooxygenase; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Serotonin biosynthesis; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 31138.2 Length 271 Aromaticity 0.13 Instability index 43.43 Isoelectric point 6.73 Charge (pH=7) -0.86 2D Binding mode Binding energy (Kcal/mol) -6.2 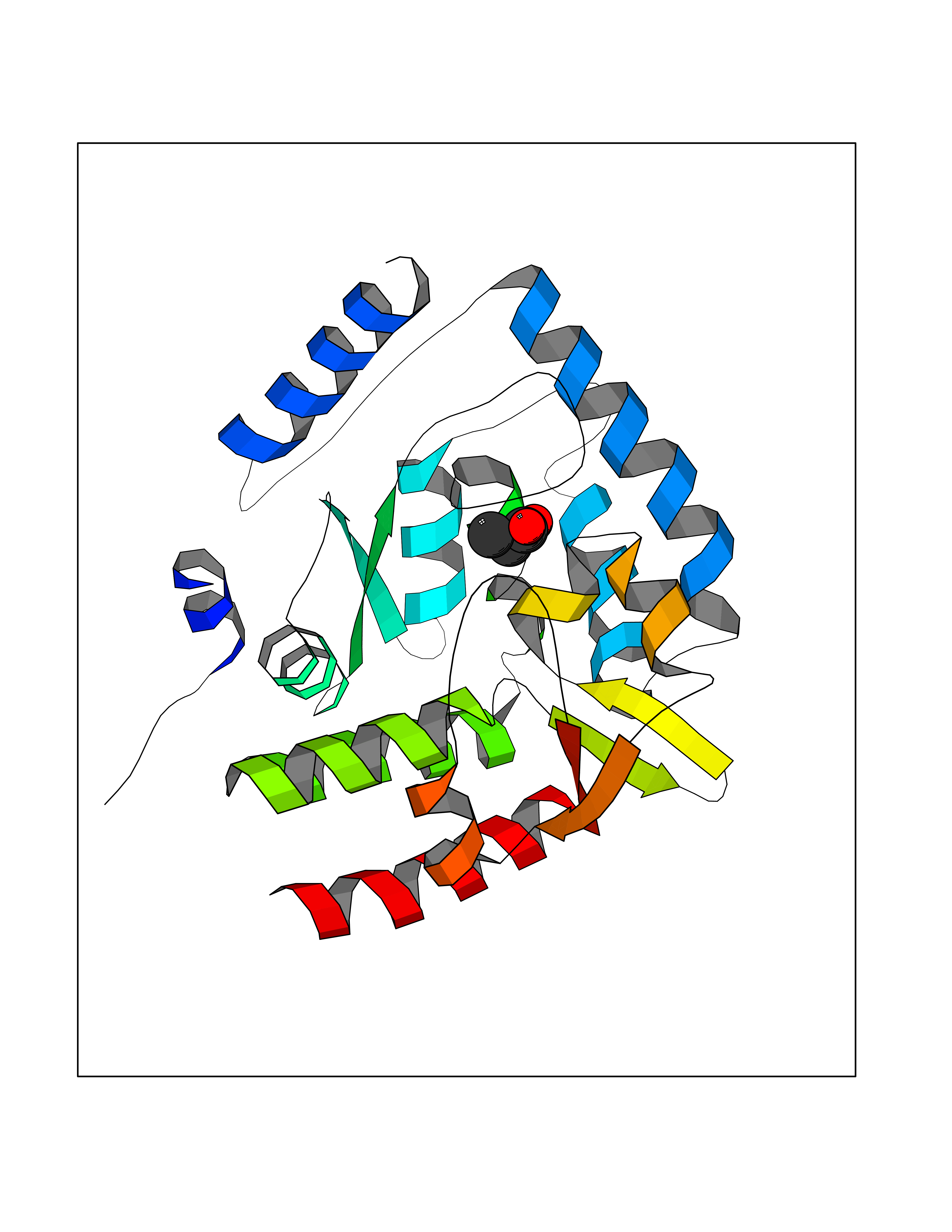 Molscript Map 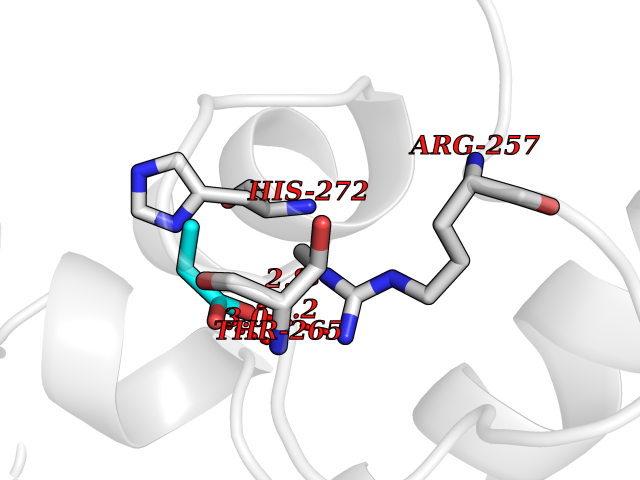 Pymol Map 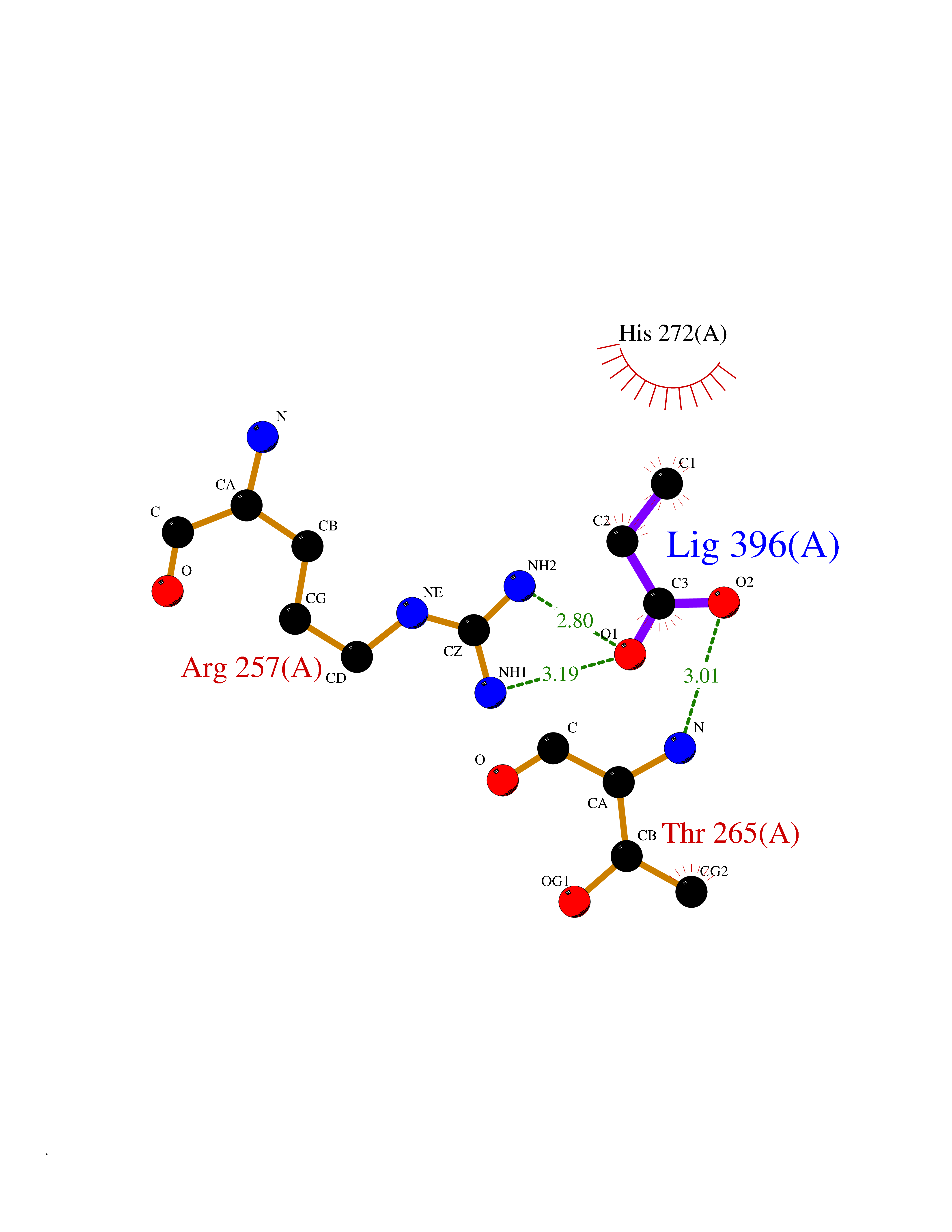 Ligplot Map 3D Binding mode Sequence TVPWFPKKISDLDHCNVYRKRRKYFADLAMNYKHGDPIPKVEFTEEEIKTWGTVFQELNKLYPTHACREYLKNLPLLSKYCGYREDNIPQLEDVSNFLKERTGFSIRPVAGYLSPRDFLSGLAFRVFHCTQYVRHSSDPFYTPEPDTCHELLGHVPLLAEPSFAQFSQEIGLASLGASEEAVQKLATCYFFTVEFGLCKQDGQLRVFGAGLLSSISELKHALSGHAKVKPFDPKITCKQECLITTFQDVYFVSESFEDAKEKMREFTKTIK Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Alkyl hydroperoxide reductase subunit F | 1HYU | 4.54 | |
Target general information Gen name ahpF Organism Salmonella typhimurium (strain LT2 / SGSC1412 / ATCC 700720) Uniprot ID TTD ID NA Synonyms STM0609 Protein family Class-II pyridine nucleotide-disulfide oxidoreductase family Biochemical class Oxidoreductase Function Alkyl hydroperoxide reductase activity.Electron carrier activity.Flavin adenine dinucleotide binding.NAD binding.Protein disulfide oxidoreductase activity. Related diseases Glutathionuria (GLUTH) [MIM:231950]: A very rare, autosomal recessive metabolic disorder characterized by the presence of glutathione in the urine, due to generalized gamma-glutamyl transpeptidase deficiency. Most patients manifest mild to moderate intellectual disability, and behavioral disturbance. Seizures, tremor, marfanoid features and strabismus are observed in some patients. {ECO:0000269|PubMed:29483667}. The disease is caused by variants affecting the gene represented in this entry. A large homozygous deletion that removes several exons of all isoforms of GGT1 has been found in one family affected by glutathionuria. {ECO:0000269|PubMed:29483667}. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.8.1.- Uniprot keywords 3D-structure; Direct protein sequencing; Disulfide bond; FAD; Flavoprotein; NAD; NADP; Oxidoreductase; Redox-active center; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 55949.2 Length 521 Aromaticity 0.06 Instability index 25.77 Isoelectric point 5.62 Charge (pH=7) -9.6 2D Binding mode Binding energy (Kcal/mol) -6.19 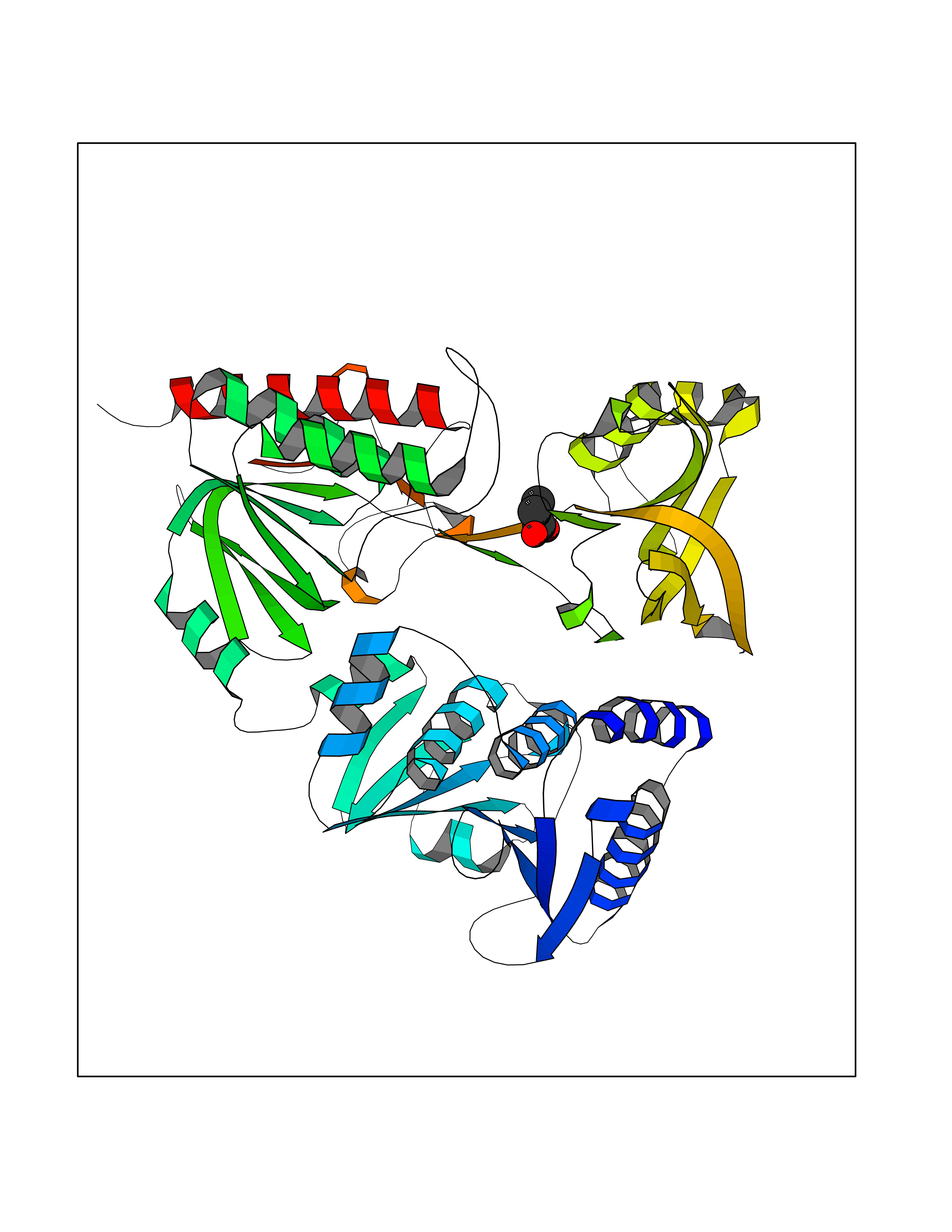 Molscript Map 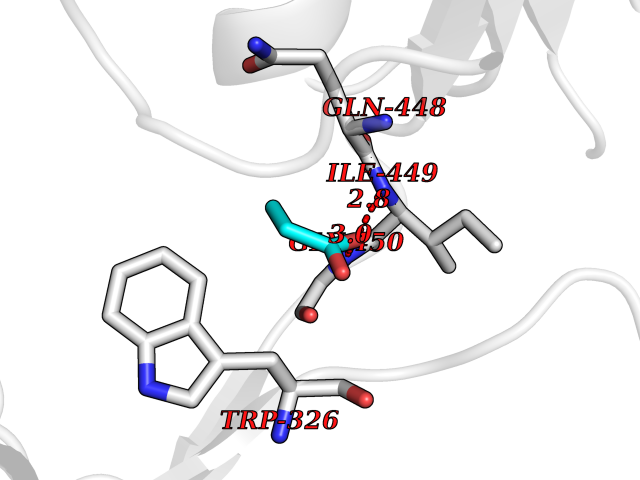 Pymol Map 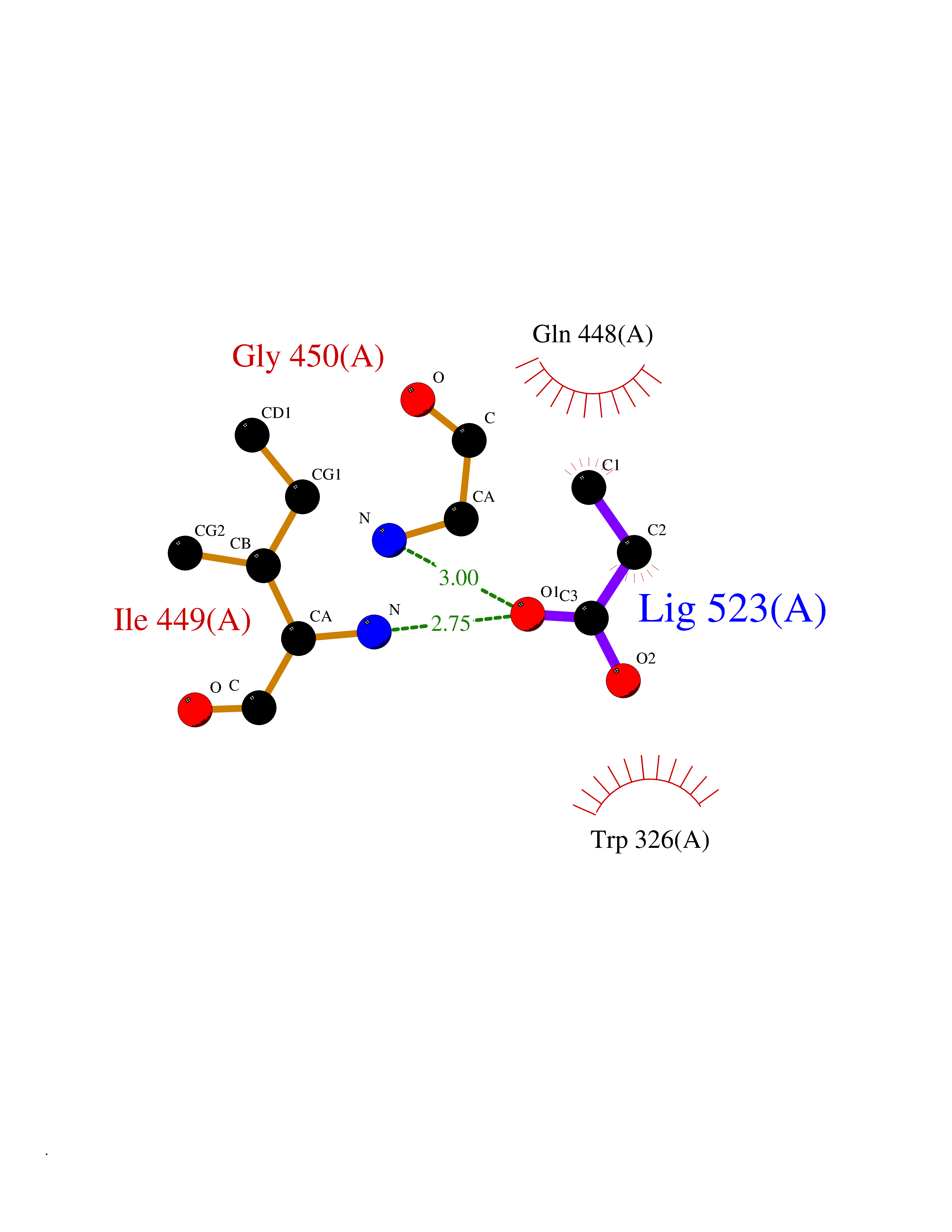 Ligplot Map 3D Binding mode Sequence MLDTNMKTQLRAYLEKLTKPVELIATLDDSAKSAEIKELLAEIAELSDKVTFKEDNTLPVRKPSFLITNPGSQQGPRFAGSPLGHEFTSLVLALLWTGGHPSKEAQSLLEQIRDIDGDFEFETYYSLSCHNCPDVVQALNLMAVLNPRIKHTAIDGGTFQNEITERNVMGVPAVFVNGKEFGQGRMTLTEIVAKVDTGAEKRAAEALNKRDAYDVLIVGSGPAGAAAAVYSARKGIRTGLMGERFGGQVLDTVDIENYISVPKTEGQKLAGALKAHVSDYDVDVIDSQSASKLVPAATEGGLHQIETASGAVLKARSIIIATGAKWRNMNVPGEDQYRTKGVTYCPHCDGPLFKGKRVAVIGGGNSGVEAAIDLAGIVEHVTLLEFAPEMKADQVLQDKVRSLKNVDIILNAQTTEVKGDGSKVVGLEYRDRVSGDIHSVALAGIFVQIGLLPNTHWLEGALERNRMGEIIIDAKCETSVKGVFAAGDCTTVPYKQIIIATGEGAKASLSAFDYLIRTKIA Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Ribonucleoside-diphosphate reductase subunit M2 | 3OLJ | 4.54 | |
Target general information Gen name RRM2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms RR2 Protein family Ribonucleoside diphosphate reductase small chain family Biochemical class Oxidoreductase Function Metal ion binding.Ribonucleoside-diphosphate reductase activity, thioredoxin disulfide as acceptor. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00242; DB05260; DB05801; DB05003; DB05428 Interacts with P41002; Q9UM11; P23921; O00560 EC number 1.17.4.1 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Deoxyribonucleotide synthesis; Iron; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 33579.4 Length 286 Aromaticity 0.14 Instability index 43.7 Isoelectric point 5.12 Charge (pH=7) -12.86 2D Binding mode Binding energy (Kcal/mol) -6.19  Molscript Map 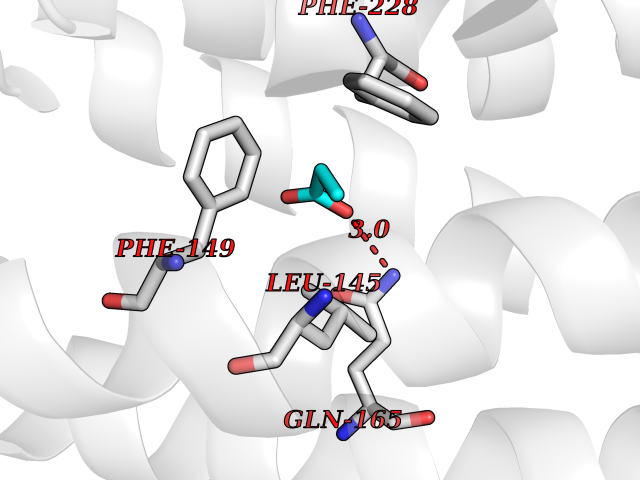 Pymol Map 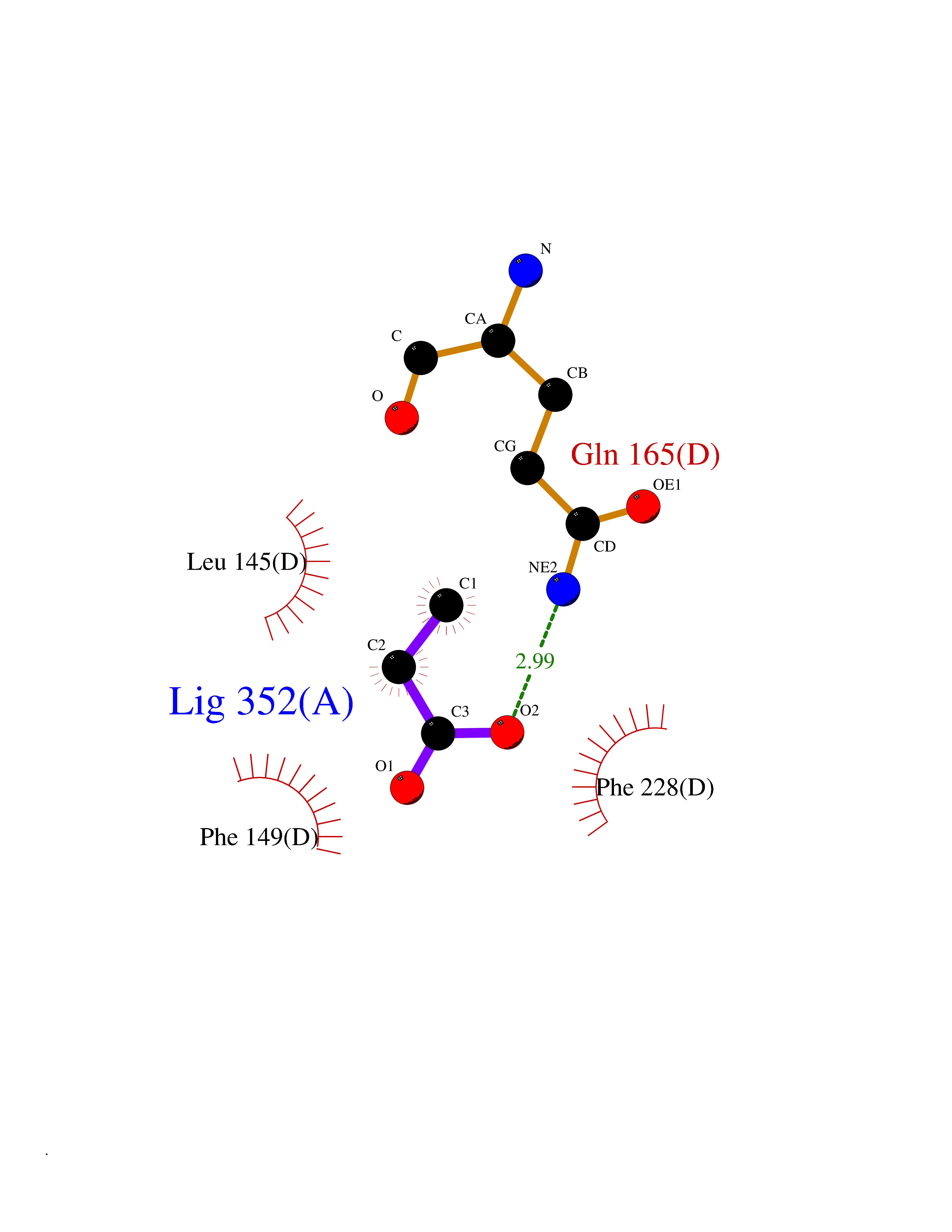 Ligplot Map 3D Binding mode Sequence MGVEDEPLLRENPRRFVIFPIEYHDIWQMYKKAEASFWTAEEVDLSKDIQHWESLKPEERYFISHVLAFFAASDGIVNENLVERFSQEVQITEARCFYGFQIAMENIHSEMYSLLIDTYIKDPKEREFLFNAIETMPCVKKKADWALRWIGDKEATYGERVVAFAAVEGIFFSGSFASIFWLKKRGLMPGLTFSNELISRDEGLHCDFACLMFKHLVHKPSEERVREIIINAVRIEQEFLTEALPVKLIGMNCTLMKQYIEFVADRLMLELGFSKVFRVENPFDFM Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Cytosolic purine 5'-nucleotidase | 2JC9 | 4.54 | |
Target general information Gen name NT5C2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PNT5;NT5B;NT5CP Protein family 5'(3')-deoxyribonucleotidase family Biochemical class Hydrolase Function 5'-nucleotidase activity.Metal ion binding.Nucleoside phosphotransferase activity.Nucleotide binding. Related diseases Spastic paraplegia 45, autosomal recessive (SPG45) [MIM:613162]: A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. Some SPG45 patients manifest intellectual disability, contractures and learning disability. {ECO:0000269|PubMed:24482476, ECO:0000269|PubMed:28884889}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00171; DB00811; DB06408 Interacts with P48047; P51116; Q8IVS8; Q7L9L4; Q86TA1; Q70IA8; Q9Y5B8; Q6ZVK8; O00560; Q9NRS6 EC number 2.7.1.77; 3.1.3.5; 3.1.3.99 Uniprot keywords 3D-structure; Allosteric enzyme; Alternative splicing; ATP-binding; Cytoplasm; Disease variant; Hereditary spastic paraplegia; Hydrolase; Magnesium; Metal-binding; Neurodegeneration; Nucleotide metabolism; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 53978.4 Length 467 Aromaticity 0.14 Instability index 30.87 Isoelectric point 8.25 Charge (pH=7) 3.41 2D Binding mode Binding energy (Kcal/mol) -6.19 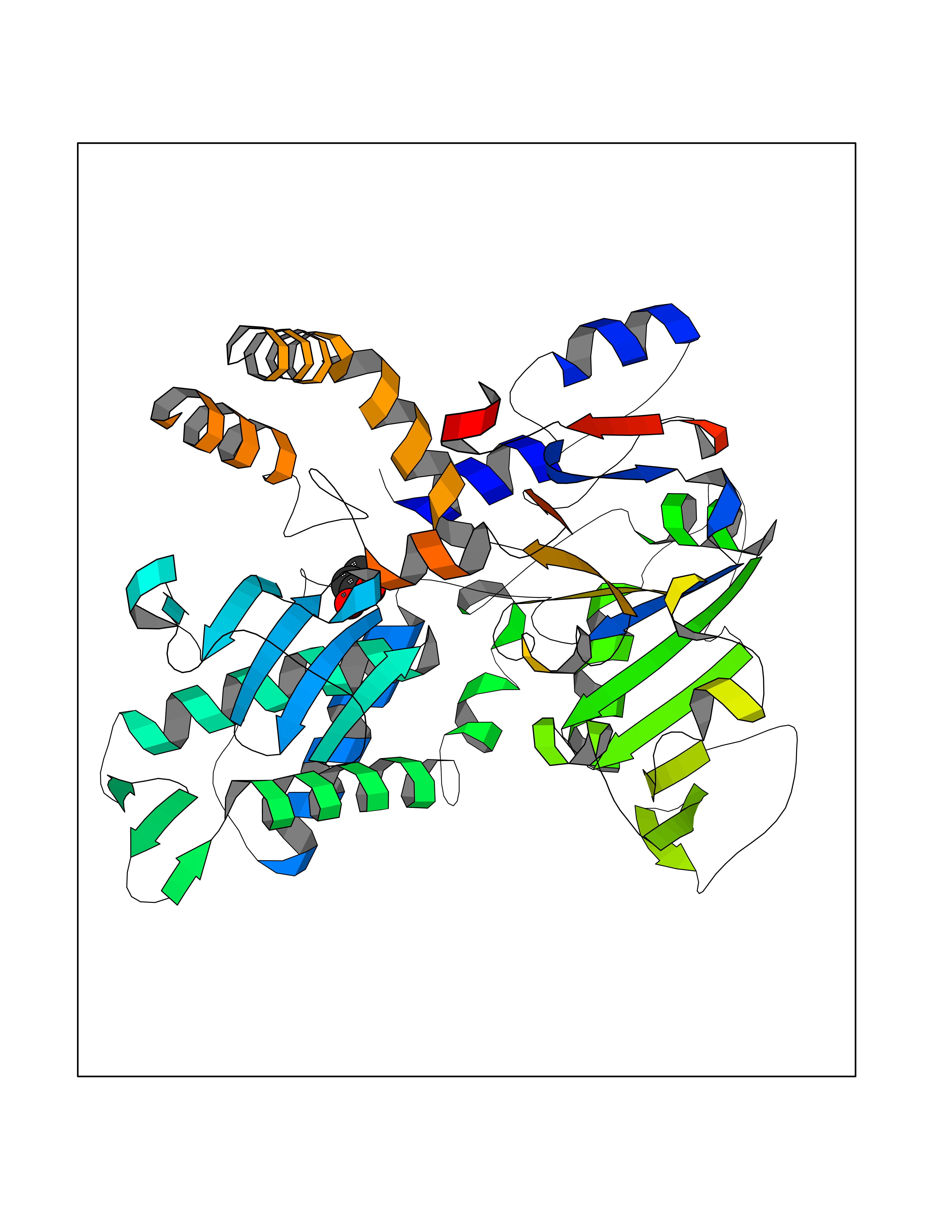 Molscript Map 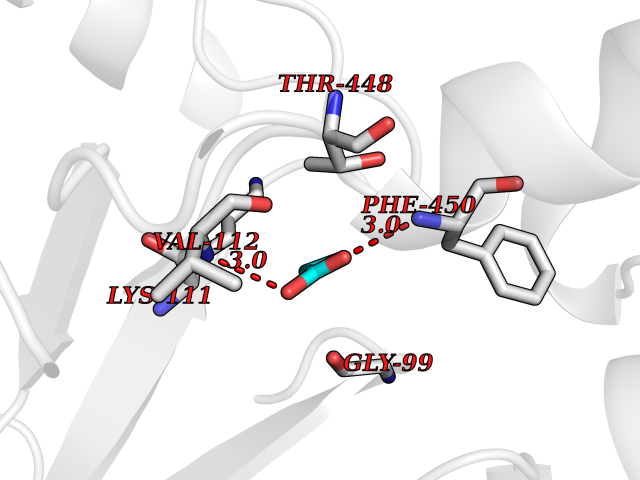 Pymol Map 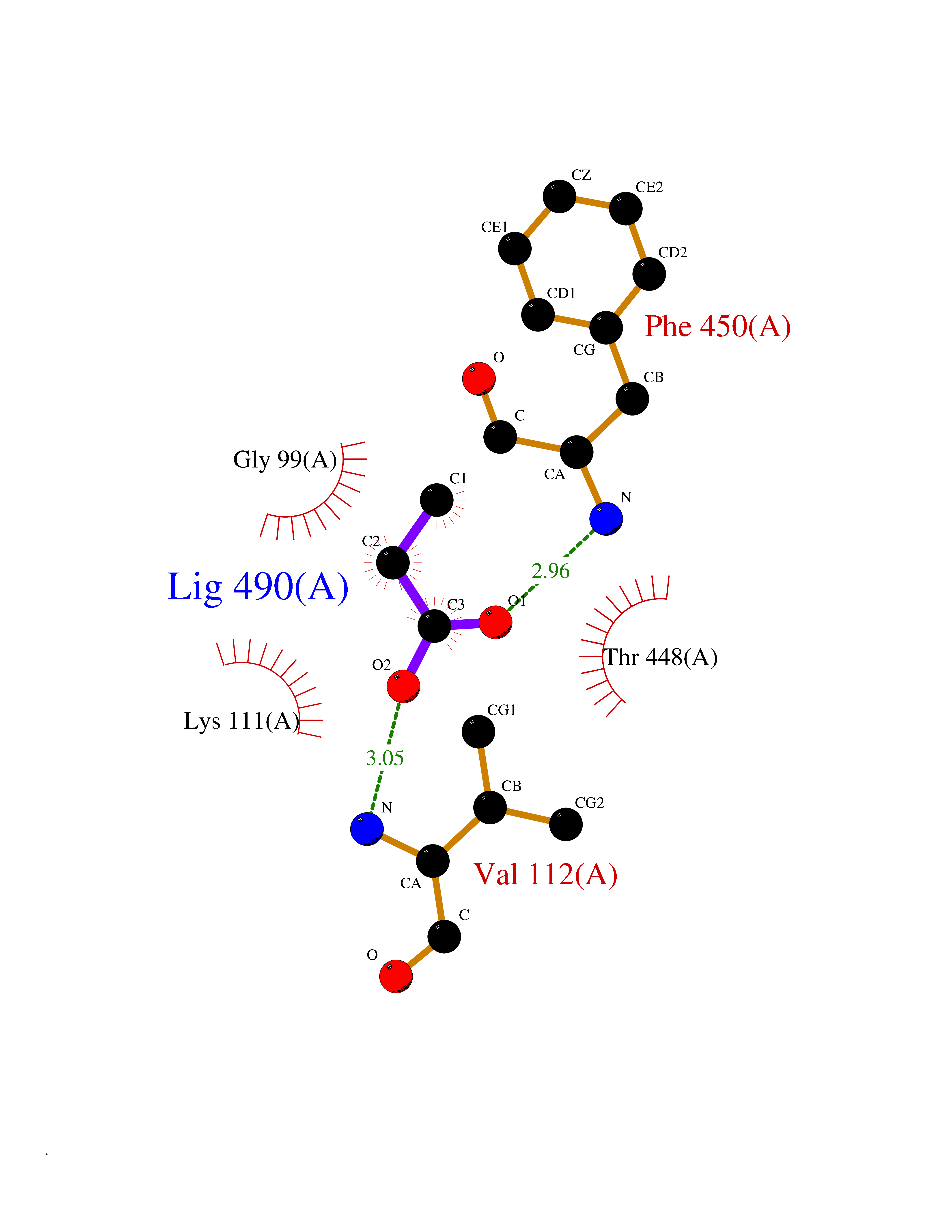 Ligplot Map 3D Binding mode Sequence TSWSDRLQNAADMPANMDKHALKKYRREAYHRVFVNRSLAMEKIKCFGFDMDYTLAVYKSPEYESLGFELTVERLVSIGYPQELLSFAYDSTFPTRGLVFDTLYGNLLKVDAYGNLLVCAHGFNFIRGPETREQYPNKFIQRDDTERFYILNTLFNLPETYLLACLVDFFTNCPRYTSCETGFKDGDLFMSYRSMFQDVRDAVDWVHYKGSLKEKTVENLEKYVVKDGKLPLLLSRMKEVGKVFLATNSDYKYTDKIMTYLFDFPHGPKPGSSHRPWQSYFDLILVDARKPLFFGEGTVLRQVDTKTGKLKIGTYTGPLQHGIVYSGGSSDTICDLLGAKGKDILYIGDHIFGDILKSKKRQGWRTFLVIPELAQELHVWTDKSSLFEELQSLDIFLAQRRIKKVTHDMDMCYGMMGSLFRSGSRQTLFASQVMRYADLYAASFINLLYYPFSYLFRAAHVLMPHES Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Acetyl-CoA carboxylase 1 | 2YL2 | 4.54 | |
Target general information Gen name ACACA Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ACAC;ACCA;ACC1 Protein family NA Biochemical class Ligase Function Acetyl-CoA carboxylase activity.ATP binding.Biotin carboxylase activity.Identical protein binding.Metal ion binding. Related diseases Acetyl-CoA carboxylase-alpha deficiency (ACACAD) [MIM:613933]: An autosomal recessive inborn error of de novo fatty acid synthesis associated with severe brain damage, persistent myopathy and poor growth. {ECO:0000269|PubMed:6114432}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00121 Interacts with Q13085; O60218; P38398; Q96EB6; Q9CQ20; P02654; Q92915-2; Q6NTF9-3 EC number 6.4.1.2 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative promoter usage; ATP-binding; Biotin; Cytoplasm; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Ligase; Lipid biosynthesis; Lipid metabolism; Magnesium; Manganese; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 54237.7 Length 486 Aromaticity 0.09 Instability index 39.18 Isoelectric point 6.37 Charge (pH=7) -2.46 2D Binding mode Binding energy (Kcal/mol) -6.19 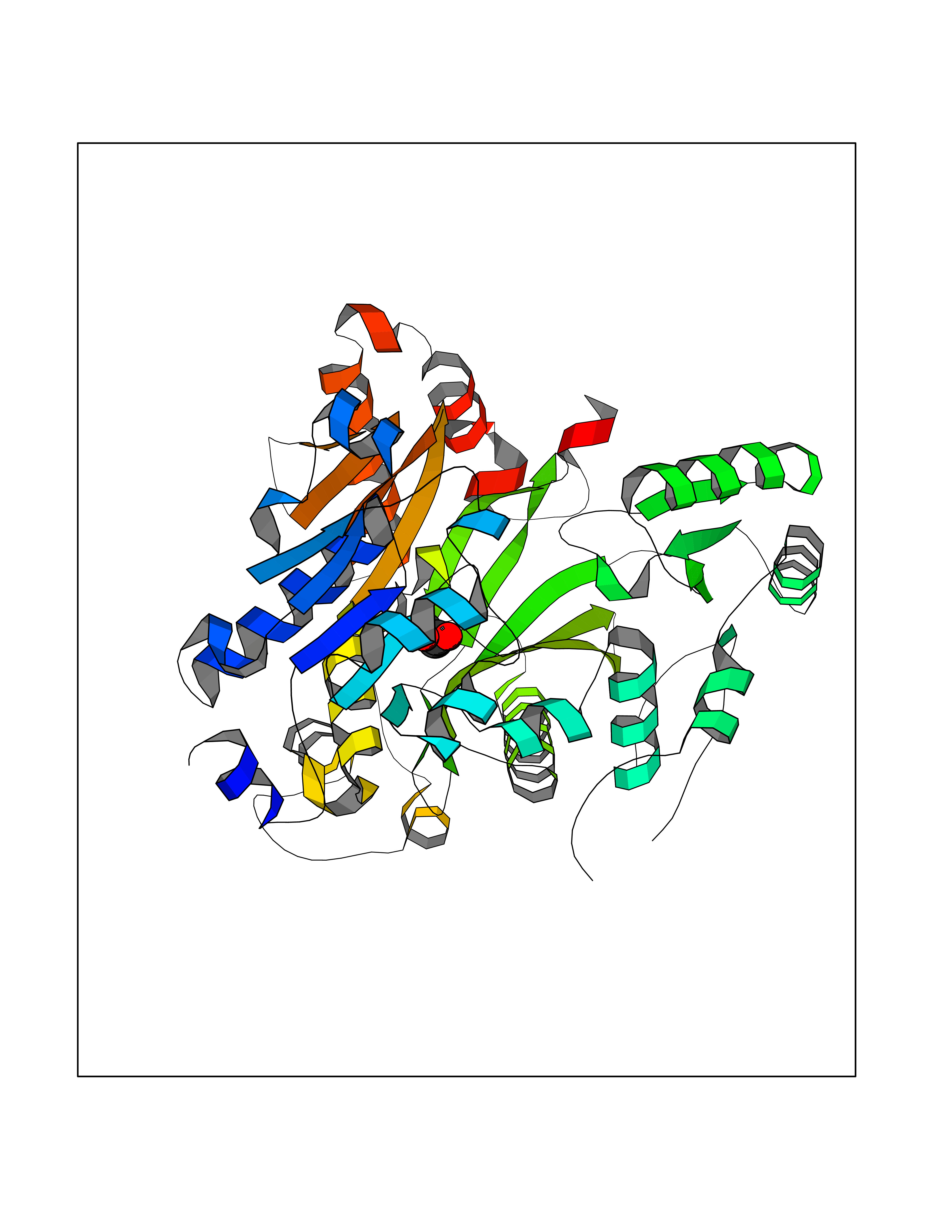 Molscript Map 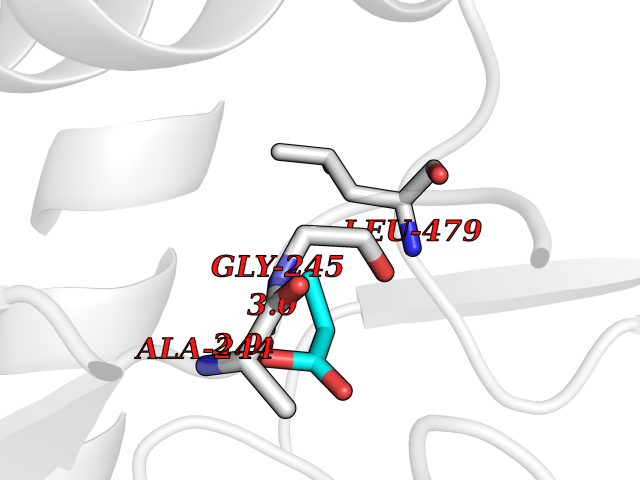 Pymol Map 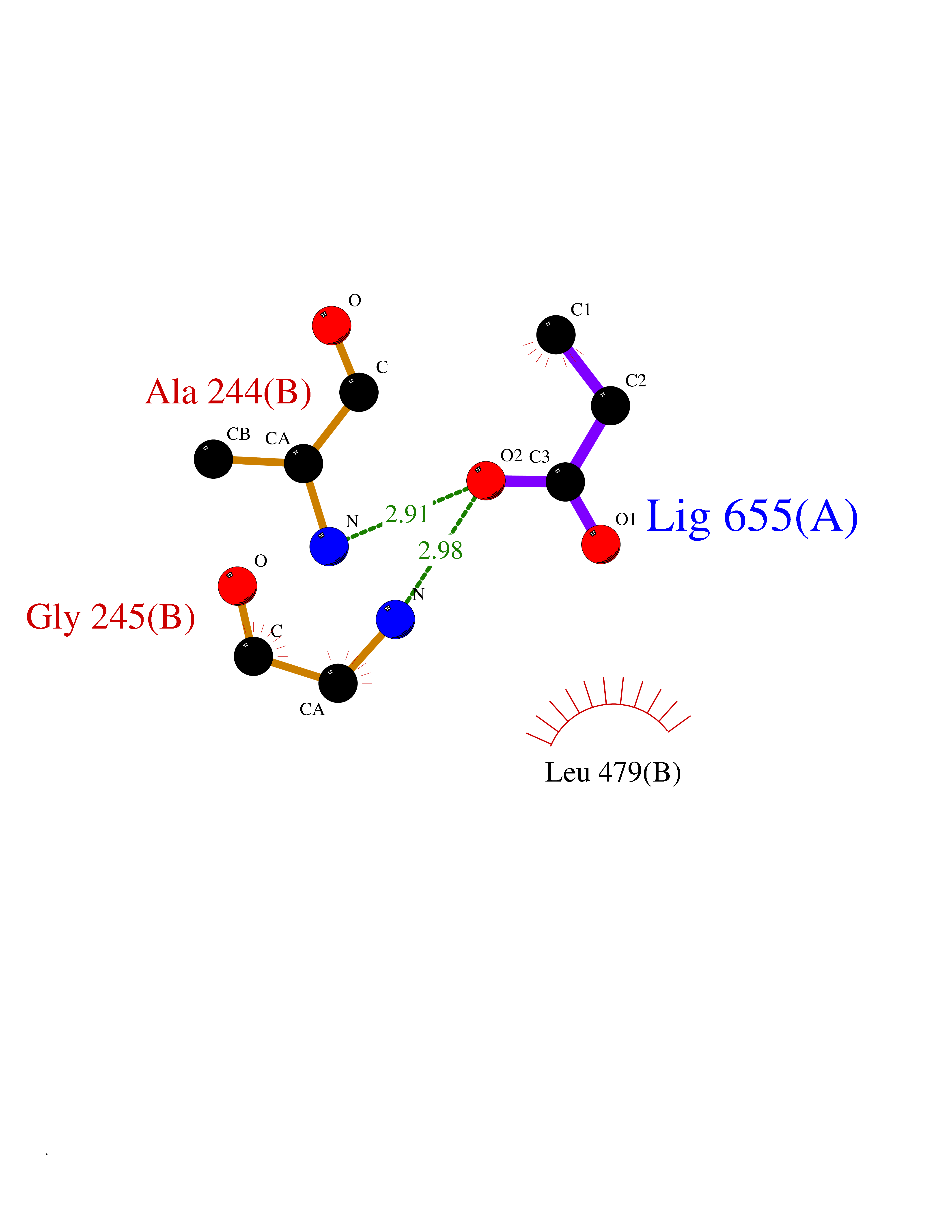 Ligplot Map 3D Binding mode Sequence VASPAEFVTRFGGNKVIEKVLIANNGIAAVKCMRSIRRWSYEMFRNERAIRFVVMVTPEDLKANAEYIKMADHYVPVPGGPNNNNYANVELILDIAKRIPVQAVWAGWGHASENPKLPELLLKNGIAFMGPPSQAMWALGDKIASSIVAQTAGIPTLPWSGSGLRVDWSKRILNVPQELYEKGYVKDVDDGLQAAEEVGYPVMIKASEGGGGKGIRKVNNADDFPNLFRQVQAEVPGSPIFVMRLAKQSRHLEVQILADQYGNAISLFGRDCSVQRRHQKIIEEAPATIATPAVFEHMEQCAVKLAKMVGYVSAGTVEYLYSQDGSFYFLELNPRLQVEHPCTEMVADVNLPAAQLQIAMGIPLYRIKDIRMMYGVSPWGDSPIDFEDSAHVPCPRGHVIAARITGTVQELNFRSNKNVWGYFSVQFGHCFSWGENREEAISNMVVALKELSIRGDFRTTVEYLIKLLETESFQMNRIDTGWLDRL Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Phenylacetone monooxygenase | 1W4X | 4.54 | |
Target general information Gen name pamO Organism Thermobifida fusca (strain YX) Uniprot ID TTD ID NA Synonyms Tfu_1490 Protein family FAD-binding monooxygenase family Biochemical class Oxygenase Function Phenylacetone monooxygenase activity. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.14.13.92 Uniprot keywords 3D-structure; FAD; Flavoprotein; Monooxygenase; NADP; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 60232 Length 533 Aromaticity 0.12 Instability index 32.05 Isoelectric point 5.25 Charge (pH=7) -17.1 2D Binding mode Binding energy (Kcal/mol) -6.19 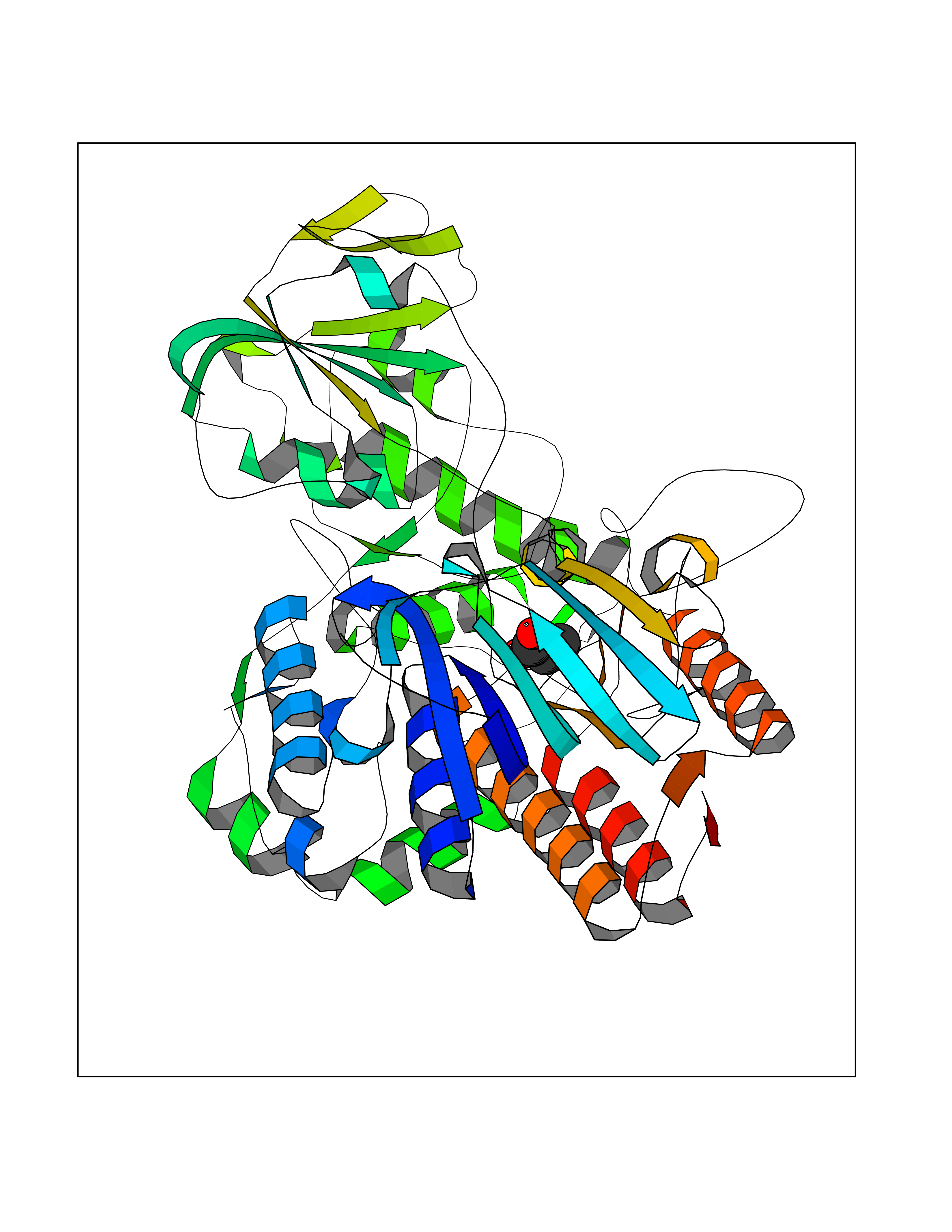 Molscript Map 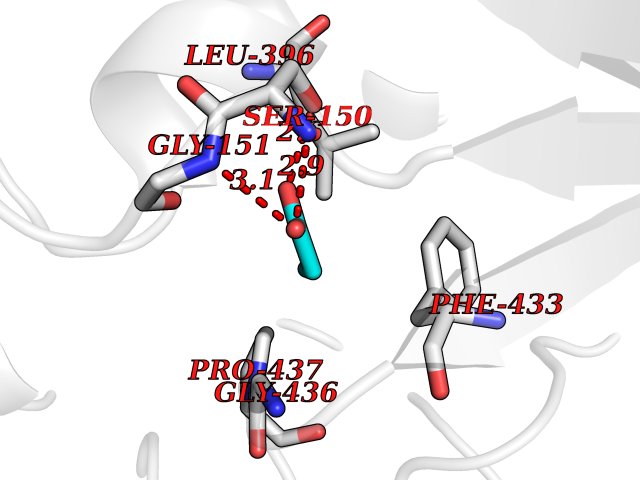 Pymol Map 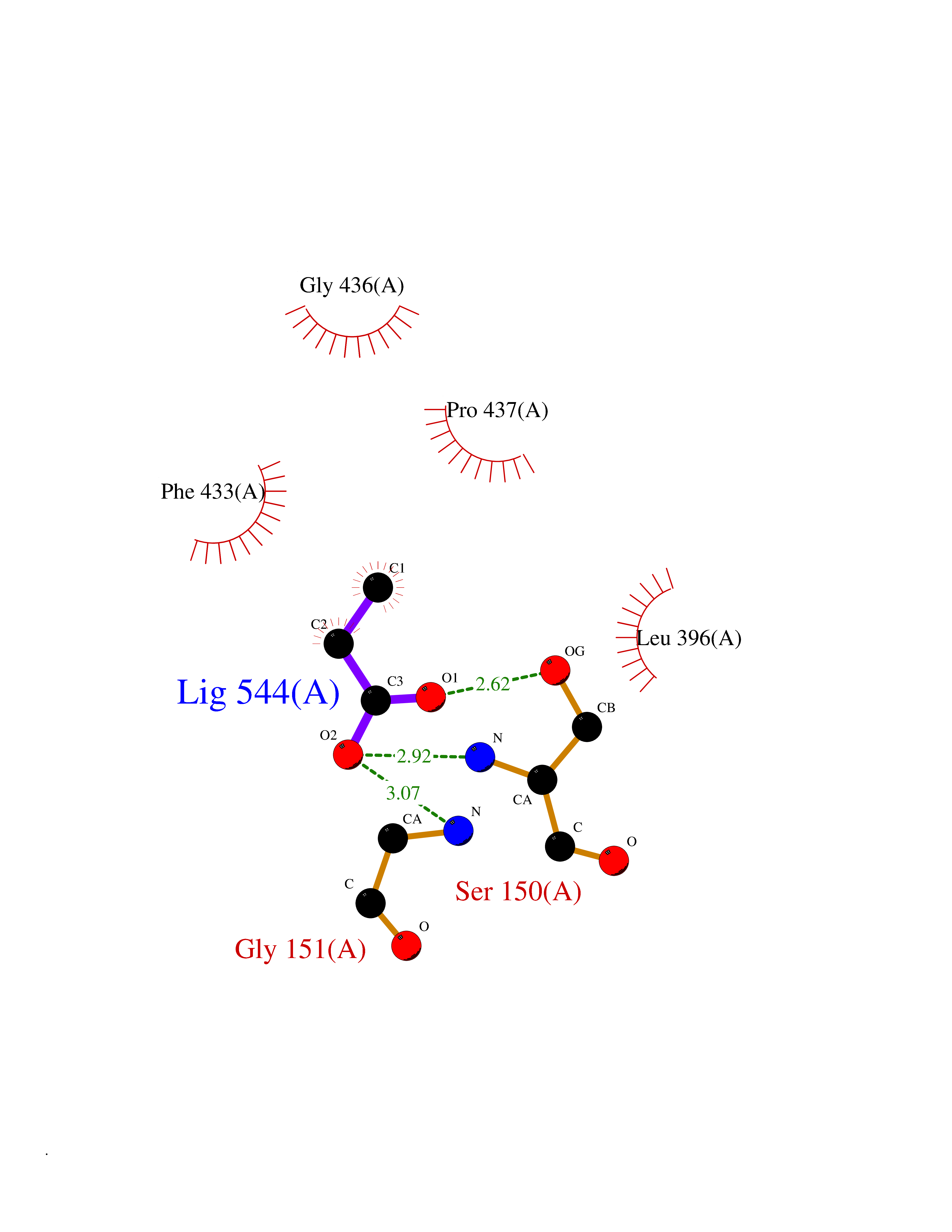 Ligplot Map 3D Binding mode Sequence RRQPPEEVDVLVVGAGFSGLYALYRLRELGRSVHVIETAGDVGGVWYWNRYPGARCDIESIEYCYSFSEEVLQEWNWTERYASQPEILRYINFVADKFDLRSGITFHTTVTAAAFDEATNTWTVDTNHGDRIRARYLIMASGQLSVPQLPNFPGLKDFAGNLYHTGNWPHEPVDFSGQRVGVIGTGSSGIQVSPQIAKQAAELFVFQRTPHFAVPARNAPLDPEFLADLKKRYAEFREESRNTPGGTHRYQGPKSALEVSDEELVETLERYWQEGGPDILAAYRDILRDRDANERVAEFIRNKIRNTVRDPEVAERLVPKGYPFGTKRLILEIDYYEMFNRDNVHLVDTLSAPIETITPRGVRTSEREYELDSLVLATGFDALTGALFKIDIRGVGNVALKEKWAAGPRTYLGLSTAGFPNLFFIAGPGSPSALSNMLVSIEQHVEWVTDHIAYMFKNGLTRSEAVLEKEDEWVEHVNEIADETLYPMTASWYTGANVPGKPRVFMLYVGGFHRYRQICDEVAAKGYEGFVLT Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | 2-oxopropyl-CoM reductase, carboxylating | 1MO9 | 4.54 | |
Target general information Gen name xecC Organism Xanthobacter autotrophicus (strain ATCC BAA-1158 / Py2) Uniprot ID TTD ID NA Synonyms Xaut_4867 Protein family Class-I pyridine nucleotide-disulfide oxidoreductase family Biochemical class Oxidoreductase Function 2-oxopropyl-CoM reductase (carboxylating) activity.Flavin adenine dinucleotide binding. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB03163; DB03147 Interacts with NA EC number 1.8.1.5 Uniprot keywords 3D-structure; Disulfide bond; FAD; Flavoprotein; NADP; Oxidoreductase; Plasmid; Redox-active center; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 114413 Length 1044 Aromaticity 0.08 Instability index 25.66 Isoelectric point 5.68 Charge (pH=7) -21.74 2D Binding mode Binding energy (Kcal/mol) -6.2 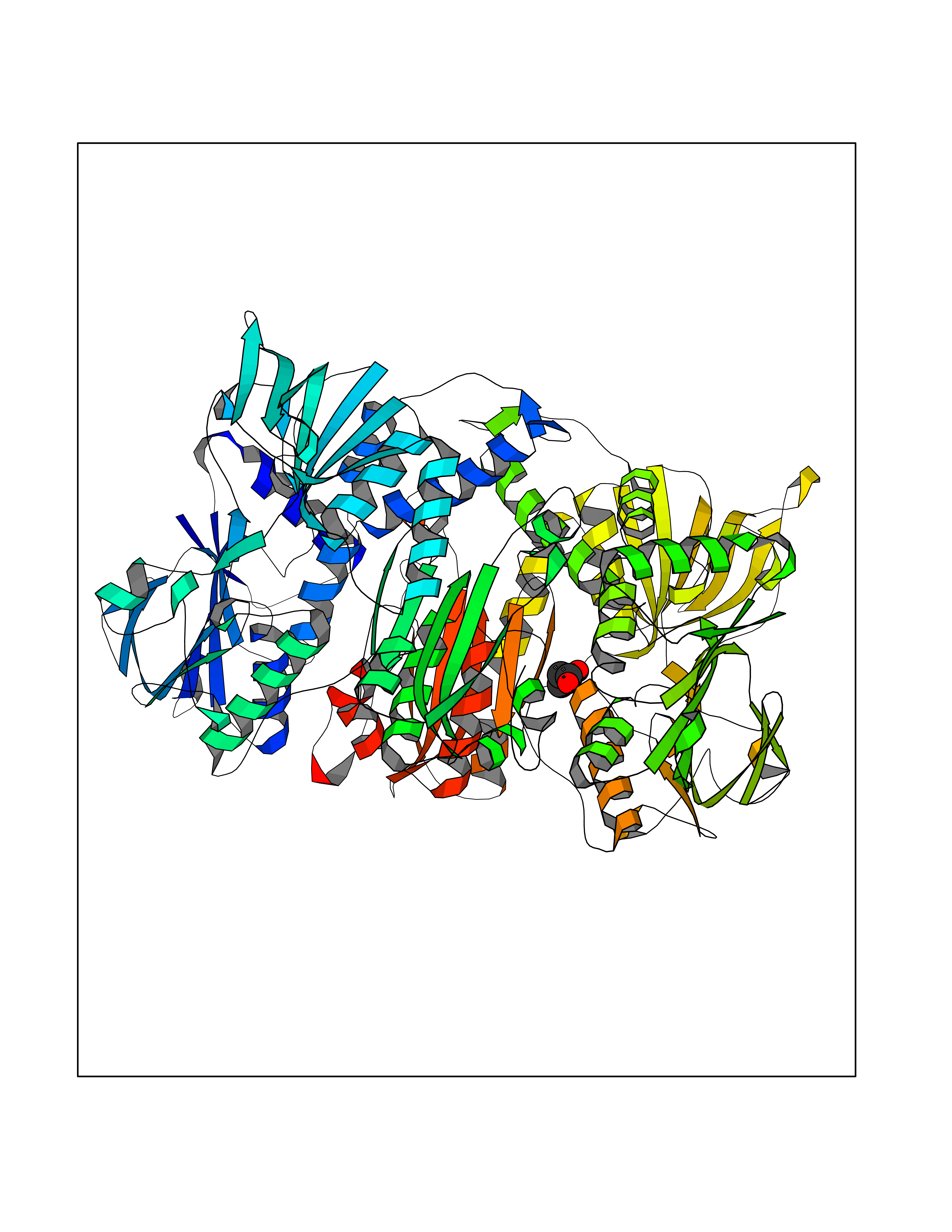 Molscript Map 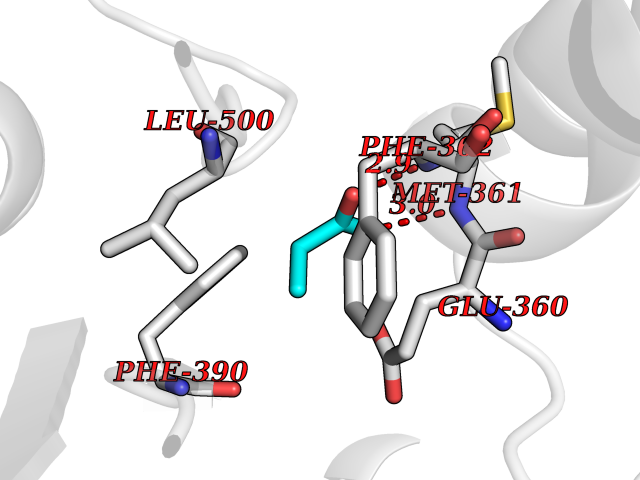 Pymol Map 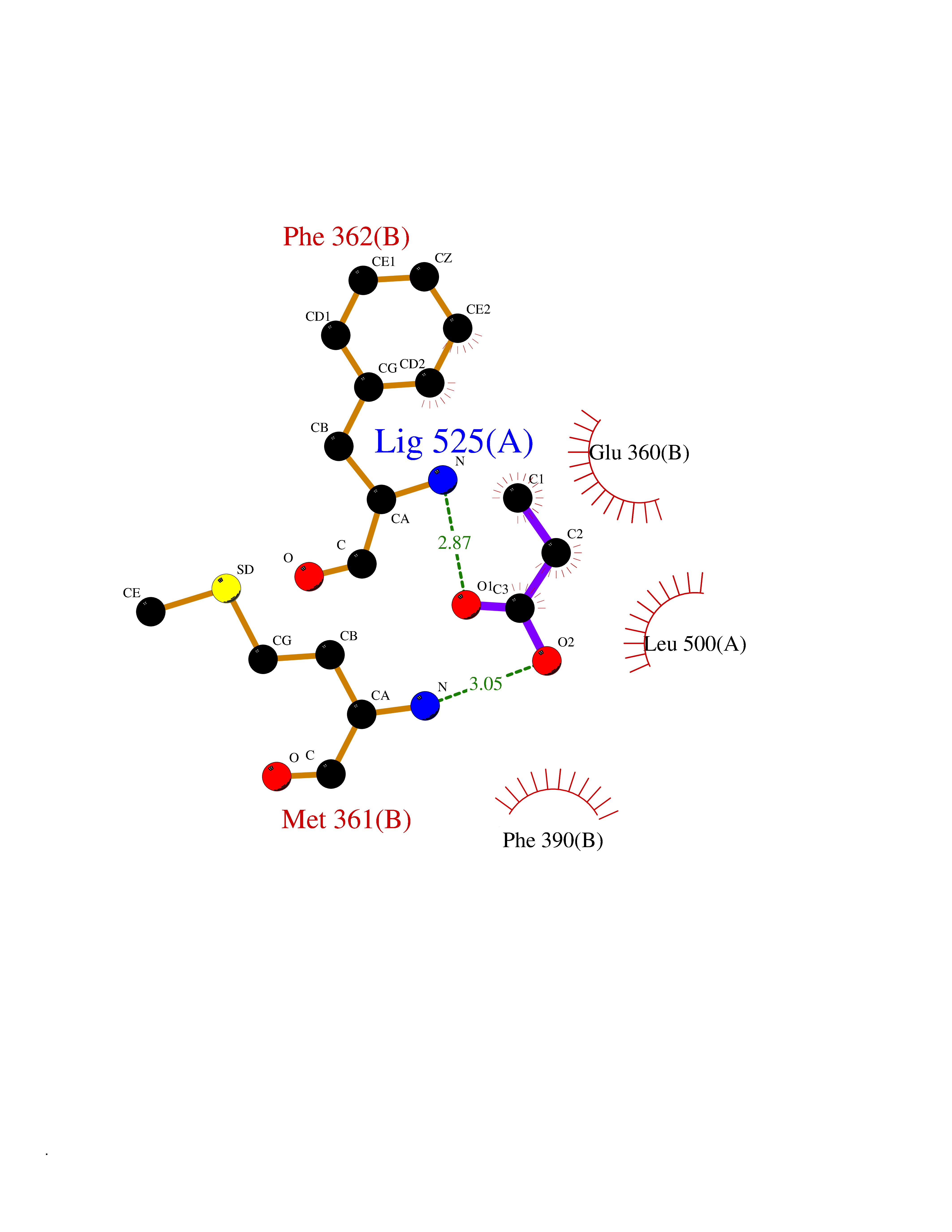 Ligplot Map 3D Binding mode Sequence KVWNARNDHLTINQWATRIDEILEAPDGGEVIYNVDENDPREYDAIFIGGGAAGRFGSAYLRAMGGRQLIVDRWPFLGGSCPHNACVPHHLFSDCAAELMLARTFSGQYWFPDMTEKVVGIKEVVDLFRAGRNGPHGIMNFQSKEQLNLEYILNCPAKVIDNHTVEAAGKVFKAKNLILAVGAGPGTLDVPGVNAKGVFDHATLVEELDYEPGSTVVVVGGSKTAVEYGCFFNATGRRTVMLVRTEPLKLIKDNETRAYVLDRMKEQGMEIISGSNVTRIEEDANGRVQAVVAMTPNGEMRIETDFVFLGLGEQPRSAELAKILGLDLGPKGEVLVNEYLQTSVPNVYAVGDLIGGPMEMFKARKSGCYAARNVMGEKISYTPKNYPDFLHTHYEVSFLGMGEEEARAAGHEIVTIKMPPDTENGLNVALPASDRTMLYAFGKGTAHMSGFQKIVIDAKTRKVLGAHHVGYGAKDAFQYLNVLIKQGLTVDELGDMDELFLNPTHFIQLSRLRAGSKNLVSLKVWNARNDHLTINQWATRIDEILEAPDGGEVIYNVDENDPREYDAIFIGGGAAGRFGSAYLRAMGGRQLIVDRWPFLGGSCPHNACVPHHLFSDCAAELMLARTFSGQYWFPDMTEKVVGIKEVVDLFRAGRNGPHGIMNFQSKEQLNLEYILNCPAKVIDNHTVEAAGKVFKAKNLILAVGAGPGTLDVPGVNAKGVFDHATLVEELDYEPGSTVVVVGGSKTAVEYGCFFNATGRRTVMLVRTEPLKLIKDNETRAYVLDRMKEQGMEIISGSNVTRIEEDANGRVQAVVAMTPNGEMRIETDFVFLGLGEQPRSAELAKILGLDLGPKGEVLVNEYLQTSVPNVYAVGDLIGGPMEMFKARKSGCYAARNVMGEKISYTPKNYPDFLHTHYEVSFLGMGEEEARAAGHEIVTIKMPPDTENGLNVALPASDRTMLYAFGKGTAHMSGFQKIVIDAKTRKVLGAHHVGYGAKDAFQYLNVLIKQGLTVDELGDMDELFLNPTHFIQLSRLRAGSKNLVSL Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Chymase (CYM) | 4K69 | 4.54 | |
Target general information Gen name CMA1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mast cell protease I; CYH; Alpha-chymase Protein family Peptidase S1 family, Granzyme subfamily Biochemical class Peptidase Function Major secreted protease of mast cells with suspected roles in vasoactive peptide generation, extracellular matrix degradation, and regulation of gland secretion. Related diseases Weaver syndrome (WVS) [MIM:277590]: A syndrome of accelerated growth and osseous maturation, unusual craniofacial appearance, hoarse and low-pitched cry, and hypertonia with camptodactyly. Distinguishing features of Weaver syndrome include broad forehead and face, ocular hypertelorism, prominent wide philtrum, micrognathia, deep horizontal chin groove, and deep-set nails. In addition, carpal bone development is advanced over the rest of the hand. {ECO:0000269|PubMed:22177091, ECO:0000269|PubMed:22190405, ECO:0000269|PubMed:23239504, ECO:0000269|PubMed:26694085, ECO:0000269|PubMed:28229514}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03814; DB04016; DB07680; DB03297 Interacts with NA EC number EC 3.4.21.39 Uniprot keywords 3D-structure; Alternative splicing; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 23715.1 Length 215 Aromaticity 0.07 Instability index 37.79 Isoelectric point 9.51 Charge (pH=7) 11.49 2D Binding mode Binding energy (Kcal/mol) -6.19 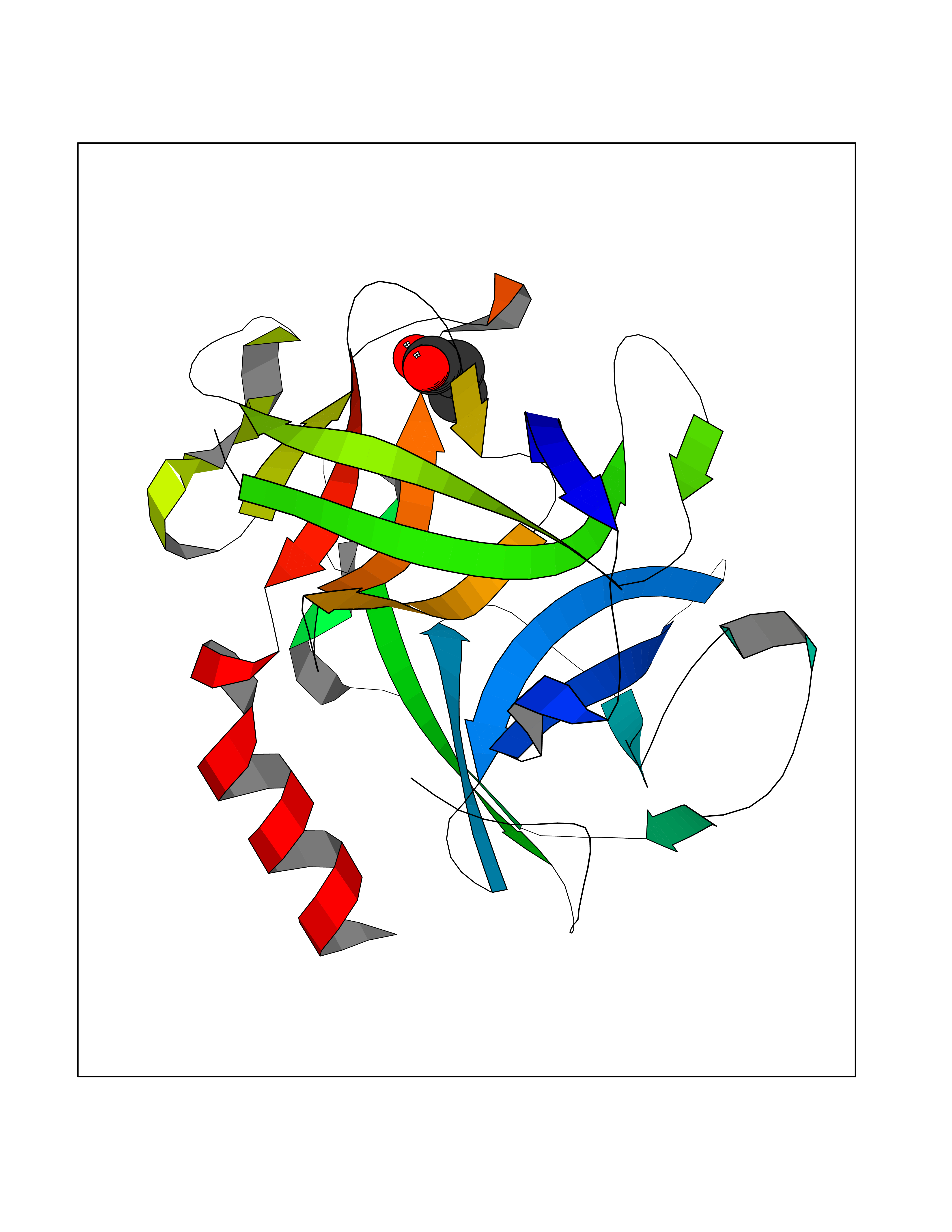 Molscript Map 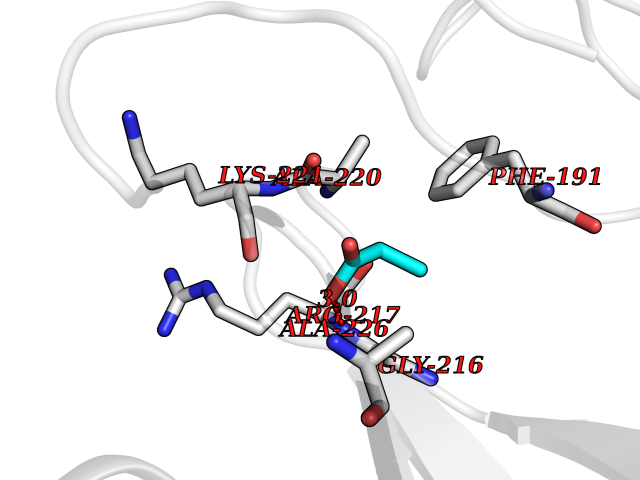 Pymol Map 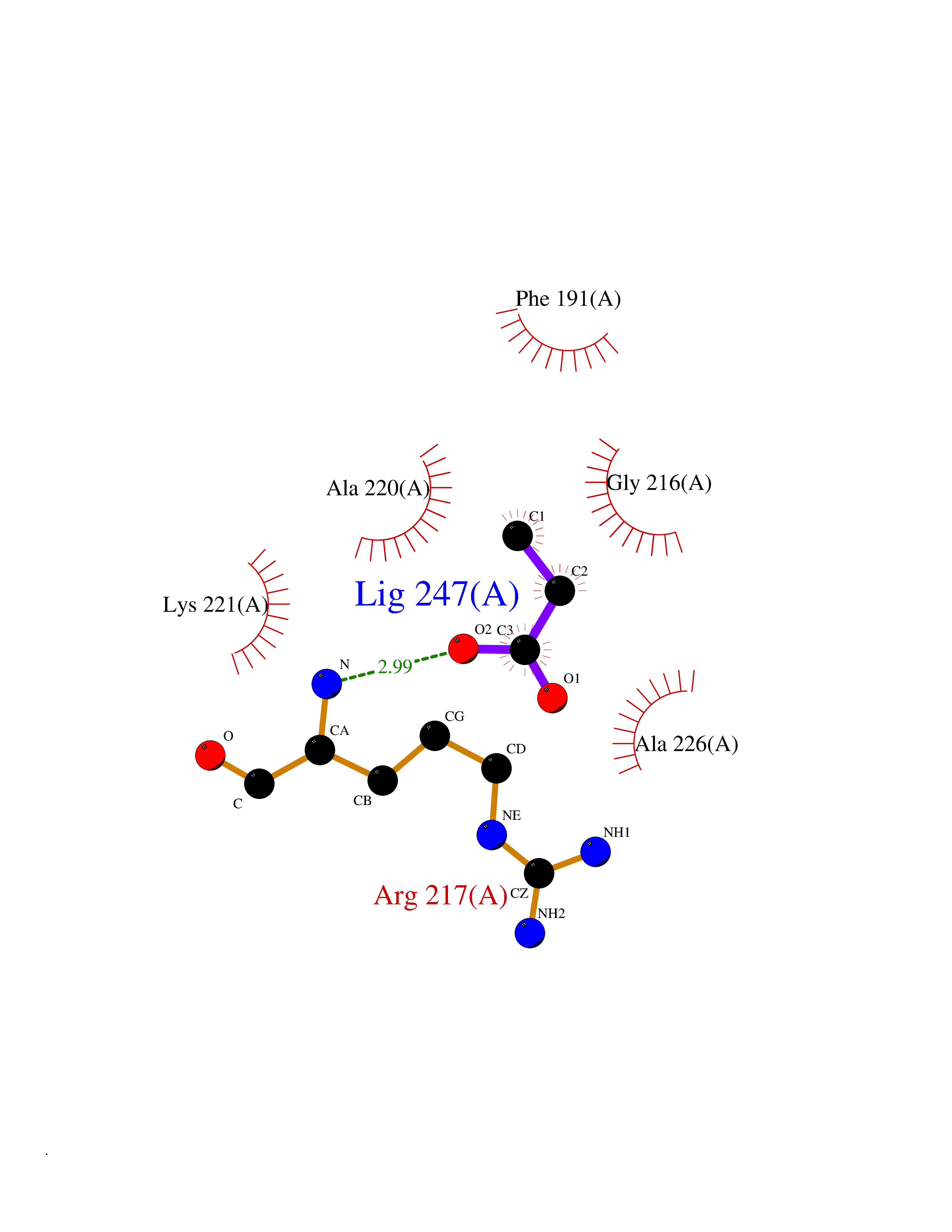 Ligplot Map 3D Binding mode Sequence IIGGTECKPHSRPYMAYLEIVTSNGPSKFCGGFLIRRNFVLTAAHCAGRSITVTLGAHNITEEEDTWQKLEVIKQFRHPKYNTSTLHHDIMLLKLKEKASLTLAVGTLGRMCRVAGWGRTGVLKPGSDTLQEVKLRLMDPQACSHFRDFDHNLQLCVGNPRKTKSAFKGDSGGPLLCAGAAQGIVSYGRSDAKPPAVFTRISHYQPWINQILQAN Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Alpha-1-antitrypsin (SERPINA1) | 5NBU | 4.54 | |
Target general information Gen name SERPINA1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SERPINA1; PRO0684/PRO2209; Alpha1-proteinase; Alpha-1-antiproteinase; Alpha-1 protease inhibitor Protein family Serpin family Biochemical class Serpin protein Function Inhibitor of serine proteases. Its primary target is elastase, but it also has a moderate affinity for plasmin and thrombin. Related diseases Alpha-1-antitrypsin deficiency (A1ATD) [MIM:613490]: A disorder whose most common manifestation is emphysema, which becomes evident by the third to fourth decade. A less common manifestation of the deficiency is liver disease, which occurs in children and adults, and may result in cirrhosis and liver failure. Environmental factors, particularly cigarette smoking, greatly increase the risk of emphysema at an earlier age. {ECO:0000269|PubMed:1905728, ECO:0000269|PubMed:2227940, ECO:0000269|PubMed:2390072}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01998; DB09130; DB00080; DB03345; DB14007; DB05961; DB05481; DB01593; DB14487; DB14533; DB14548 Interacts with Q9Y282; Q8N7X4; P01009; P43307; O15393; P00772; P71213; P00760 EC number NA Uniprot keywords 3D-structure; Acute phase; Alternative splicing; Blood coagulation; Direct protein sequencing; Endoplasmic reticulum; Extracellular matrix; Glycoprotein; Hemostasis; Phosphoprotein; Protease inhibitor; Proteomics identification; Reference proteome; Secreted; Serine protease inhibitor; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 41542.2 Length 370 Aromaticity 0.09 Instability index 30.24 Isoelectric point 5.56 Charge (pH=7) -9.66 2D Binding mode Binding energy (Kcal/mol) -6.2 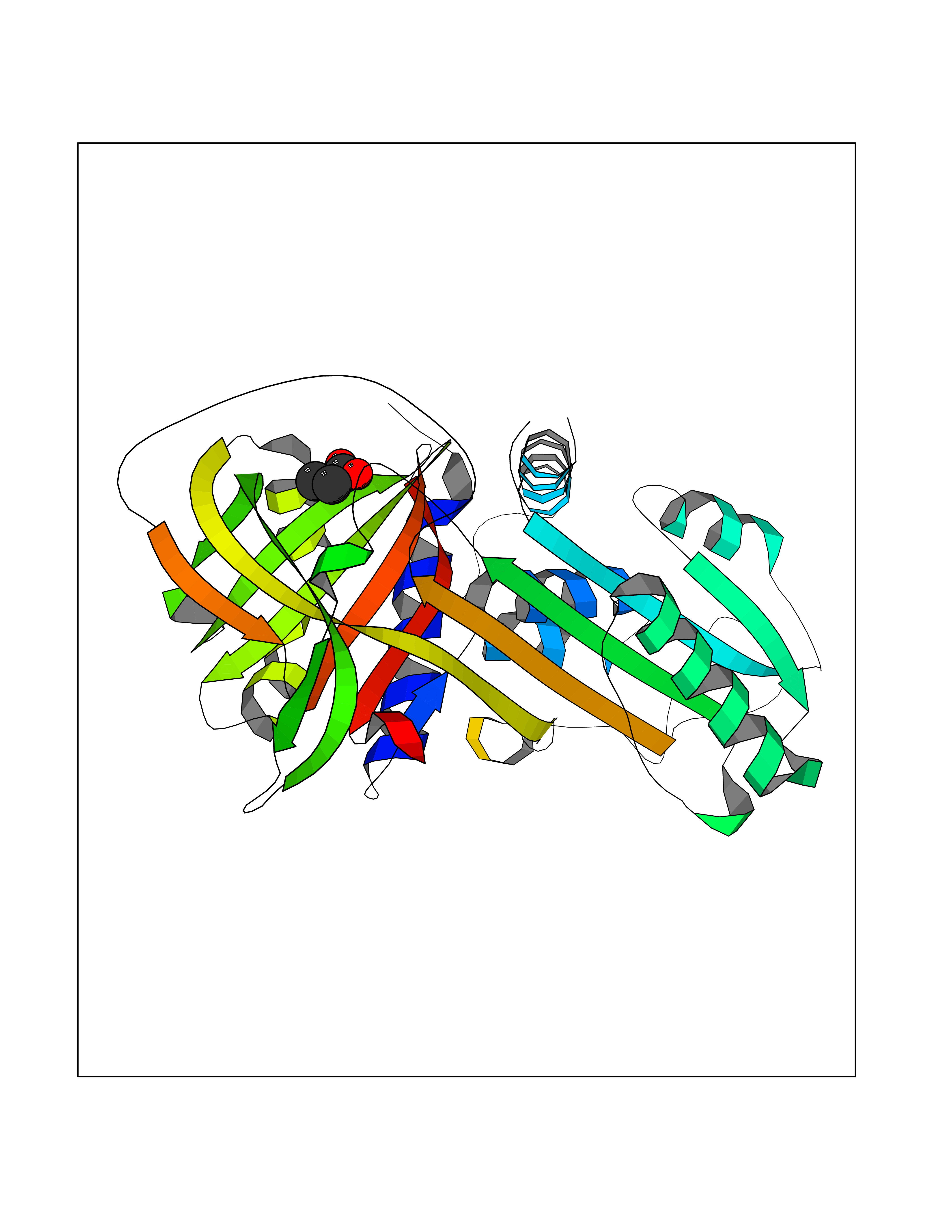 Molscript Map 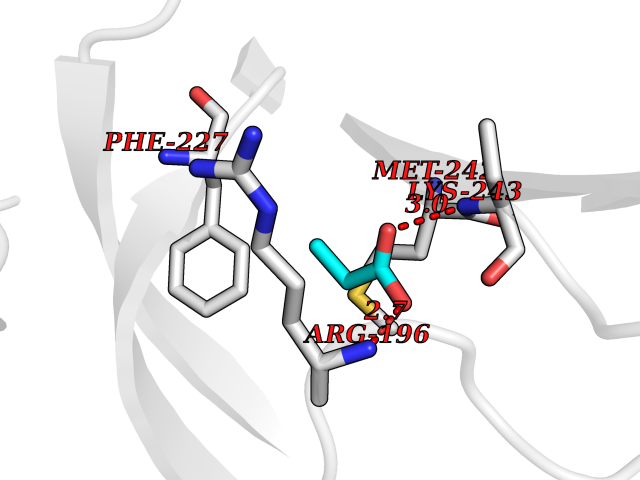 Pymol Map 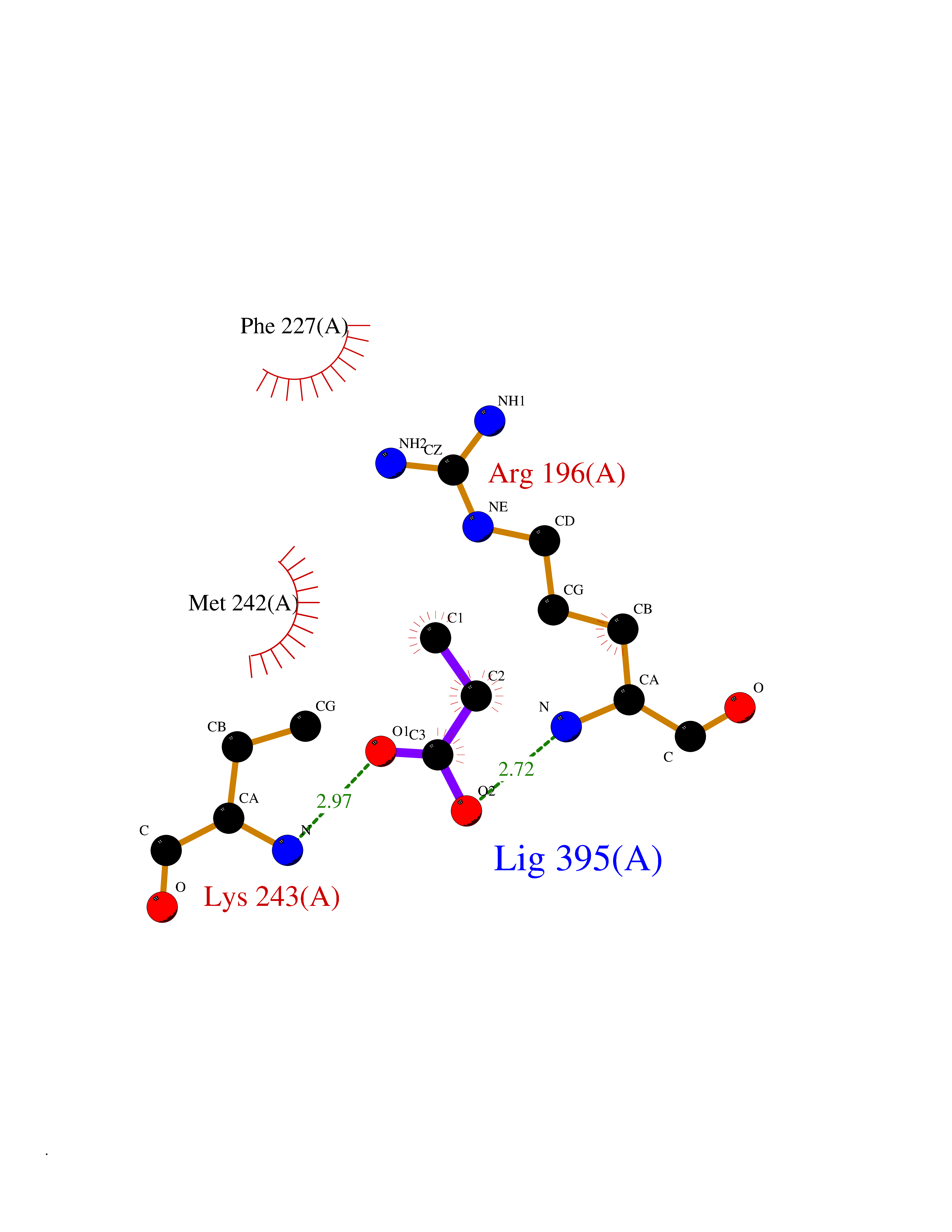 Ligplot Map 3D Binding mode Sequence TFNKITPNLAEFAFSLYRQLAHQSNSTNILFSPVSIAAAFAMLSLGAKGDTHDEILEGLNFNLTEIPEAQIHEGFQELLRTLNQSQLQLTTGNGLFLSEGLKLVDKFLEDVKKLYHSEAFTVNFGDTEEAKKQINDYVEKGTQGKIVDLVKELDRDTVFALVNYIFFKGKWERPFEVKDTEEEDFHVDQVTTVKVPMMKRLGMFNIQHSKKLSSWVLLMKYLGNATAIFFLPDEGKLQHLENELTHDIITKFLENEDRRSASLHLPKLSITGTYDLKSVLGQLGITKVFSNGADLSGVTEEAPLKLSKAVHKAVLTIDEKGTEAAGAMFLEAIPMSIPPEVKFNKPFVFLIIEQNTKAPLFMGRVVNPTQ Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Chloride channel protein 7 (ClC-7) | 7CQ5 | 4.54 | |
Target general information Gen name CLCN7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms H(+)/Cl(-) exchange transporter 7; ClC-7; Chloride channel 7 alpha subunit Protein family Chloride channel (TC 2.A.49) family, ClC-7/CLCN7 subfamily Biochemical class Chloride channel Function Slowly voltage-gated channel mediating the exchange of chloride ions against protons. Functions as antiporter and contributes to the acidification of the lysosome lumen. Related diseases Osteopetrosis, autosomal recessive 4 (OPTB4) [MIM:611490]: A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves. {ECO:0000269|PubMed:11207362, ECO:0000269|PubMed:11741829, ECO:0000269|PubMed:14584882, ECO:0000269|PubMed:17033731, ECO:0000269|PubMed:19953639, ECO:0000269|PubMed:26395888, ECO:0000269|PubMed:26477479}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Osteopetrosis, autosomal dominant 2 (OPTA2) [MIM:166600]: A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. OPTA2 is the most common form of osteopetrosis, occurring in adolescence or adulthood. It is characterized by sclerosis, predominantly involving the spine, the pelvis and the skull base. {ECO:0000269|PubMed:11741829, ECO:0000269|PubMed:14584882, ECO:0000269|PubMed:19288050, ECO:0000269|PubMed:19953639, ECO:0000269|PubMed:26395888}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hypopigmentation, organomegaly, and delayed myelination and development (HOD) [MIM:618541]: An autosomal dominant pleiotropic syndrome characterized by skin and hair hypopigmentation, growth and developmental delay, organomegaly including enlarged liver, spleen and kidneys, delayed brain myelination and developmental deficit in motor skills. Skin and liver biopsies show cellular accumulation of large intracellular vacuoles. {ECO:0000269|PubMed:31155284}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q6P5T0; P19397; Q8TB36; O00258; Q53GS7; Q9UGI6-2; Q8TAF8; Q9Y5Y7; P35372-10; Q86WC4; Q9NVC3; Q8N2U9; Q7Z699; Q96MV1; Q7Z7N9 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Antiport; ATP-binding; CBS domain; Chloride; Disease variant; Ion transport; Lysosome; Membrane; Nucleotide-binding; Osteopetrosis; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID C Molecular weight (Da) 75175 Length 681 Aromaticity 0.11 Instability index 38.7 Isoelectric point 8.88 Charge (pH=7) 8.95 2D Binding mode Binding energy (Kcal/mol) -6.2  Molscript Map 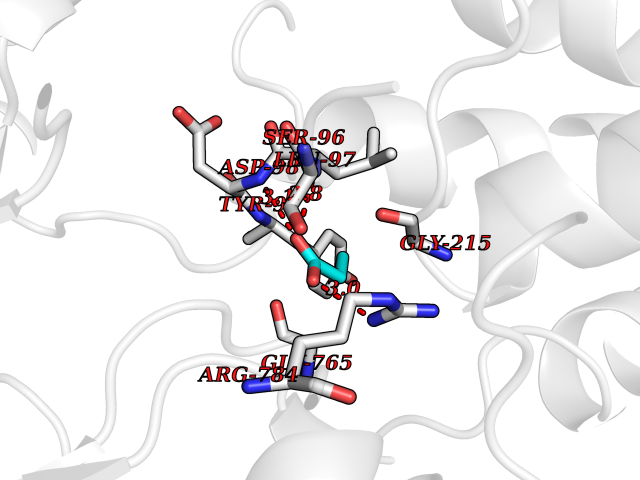 Pymol Map 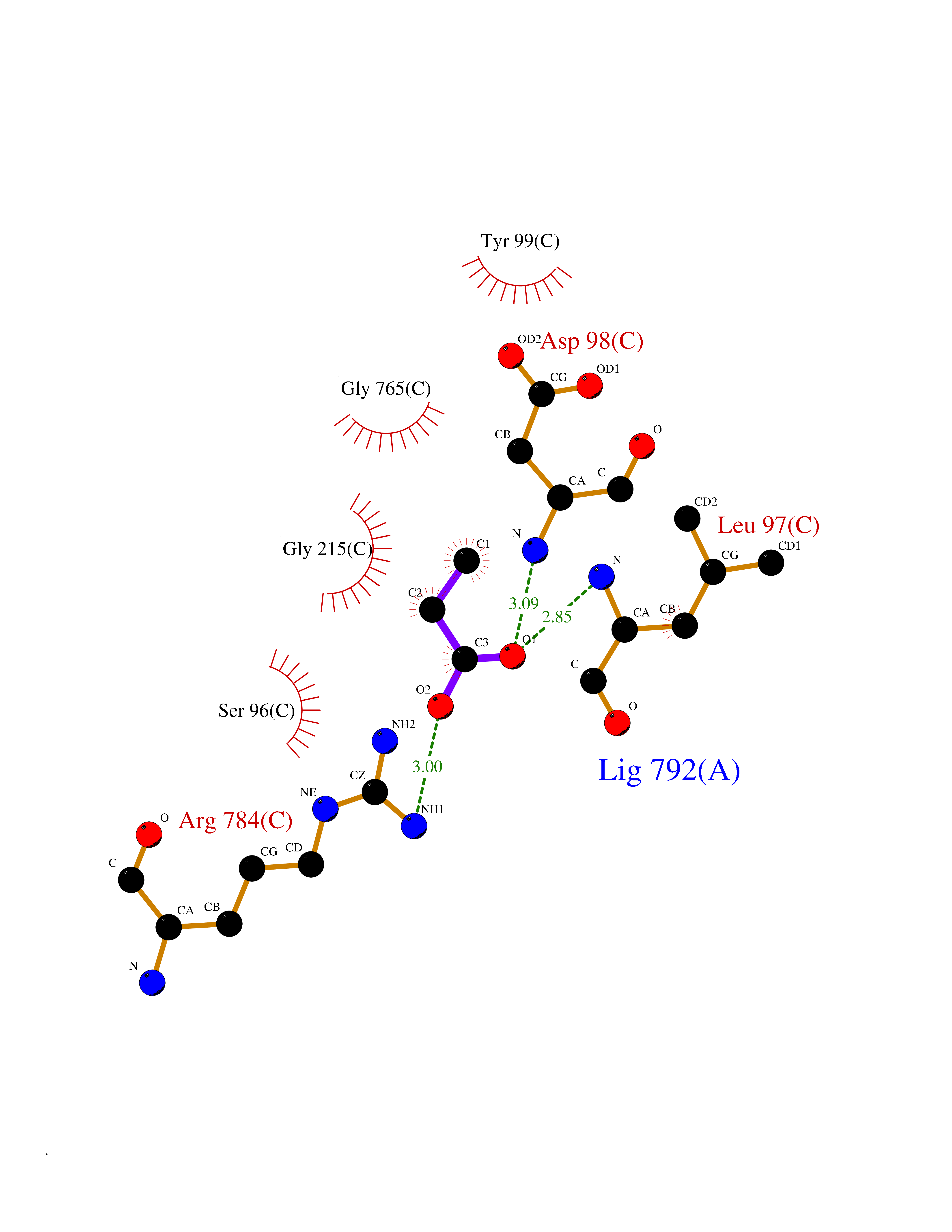 Ligplot Map 3D Binding mode Sequence YESLDYDNSENQLFLEEERRINHTAFRTVEIKRWVICALIGILTGLVACFIDIVVENLAGLKYRVIKGNIDKFTEKGGLSFSLLLWATLNAAFVLVGSVIVAFIEPVAAGSGIPQIKCFLNGVKIPHVVRLKTLVIKVSGVILSVVGGLAVGKEGPMIHSGSVIAAGISQGRSTSLKRDFKIFEYFRRDTEKRDFVSAGAAAGVSAAFGAPVGGVLFSLEEGASFWNQFLTWRIFFASMISTFTLNFVLSIYHGNMWDLSSPGLINFGRFDSEKMAYTIHEIPVFIAMGVVGGVLGAVFNALNYWLTMFRIRYIHRPCLQVIEAVLVAAVTATVAFVLIYSSRDCQPLQGGSMSYPLQLFCADGEYNSMAAAFFNTPEKSVVSLFHDPPGSYNPLTLGLFTLVYFFLACWTYGLTVSAGVFIPSLLIGAAWGRLFGISLSYLTGAAIWADPGKYALMGAAAQLGGIVRMTLSLTVIMMEATSNVTYGFPIMLVLMTAKIVGDVFIEGLYDMHIQLQSVPFLHWEAPVTSHSLTAREVMSTPVTCLRRREKVGVIVDVLSDTASNHNGFPVVEARLQGLILRSQLIVLLKHKVFVRRLRLKDFRDAYPRFPPIQSIHVSQDERECTMDLSEFMNPSPYTVPQEASLPRVFKLFRALGLRHLVVVDNRNQVVGLVTRKDLARY Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Deoxyhypusine synthase (DHPS) | 6XXJ | 4.54 | |
Target general information Gen name DHPS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms DHS Protein family Deoxyhypusine synthase family Biochemical class Alkyl aryl transferase Function Catalyzes the NAD-dependent oxidativecleavage of spermidine and the subsequent transfer of the butylamine moiety of spermidine to the epsilon-amino group of a specific lysine residue of the eIF-5A precursor protein to form the intermediate deoxyhypusine residue. Related diseases Neurodevelopmental disorder with seizures and speech and walking impairment (NEDSSWI) [MIM:618480]: An autosomal recessive disorder characterized by global developmental delay with intellectual disability and poor speech acquisition, and walking difficulties due to hypotonia, hypertonia, spasticity, or poor coordination. Additional features include seizures, mild dysmorphic features, and variable short stature. {ECO:0000269|PubMed:30661771}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03639 Interacts with Q53QZ3; P49366; P63241; Q9GZV4; Q8TBB1; P27361; Q6FHY5; Q9GZT8; Q96HA8; Q6ZVK8; P26367; Q9HD43; O00194; P09455; Q04864; Q04864-2; P32969; Q7L8A9; P52744; P52744-2 EC number EC 2.5.1.46 Uniprot keywords 3D-structure; Alternative splicing; Direct protein sequencing; Disease variant; Hypusine biosynthesis; Intellectual disability; NAD; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 76663.5 Length 692 Aromaticity 0.09 Instability index 36.19 Isoelectric point 5.35 Charge (pH=7) -15.99 2D Binding mode Binding energy (Kcal/mol) -6.19  Molscript Map 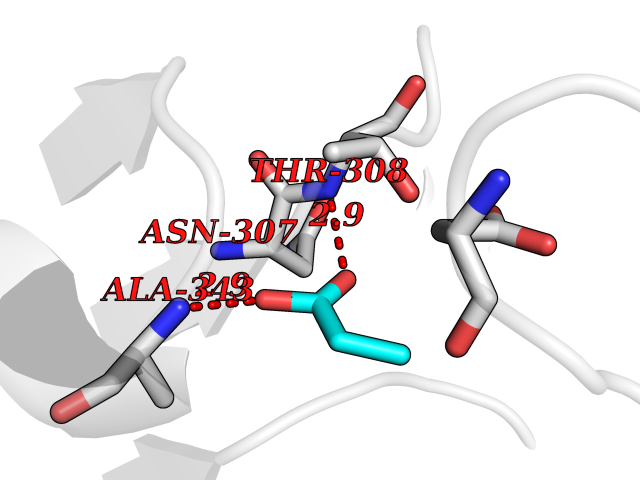 Pymol Map 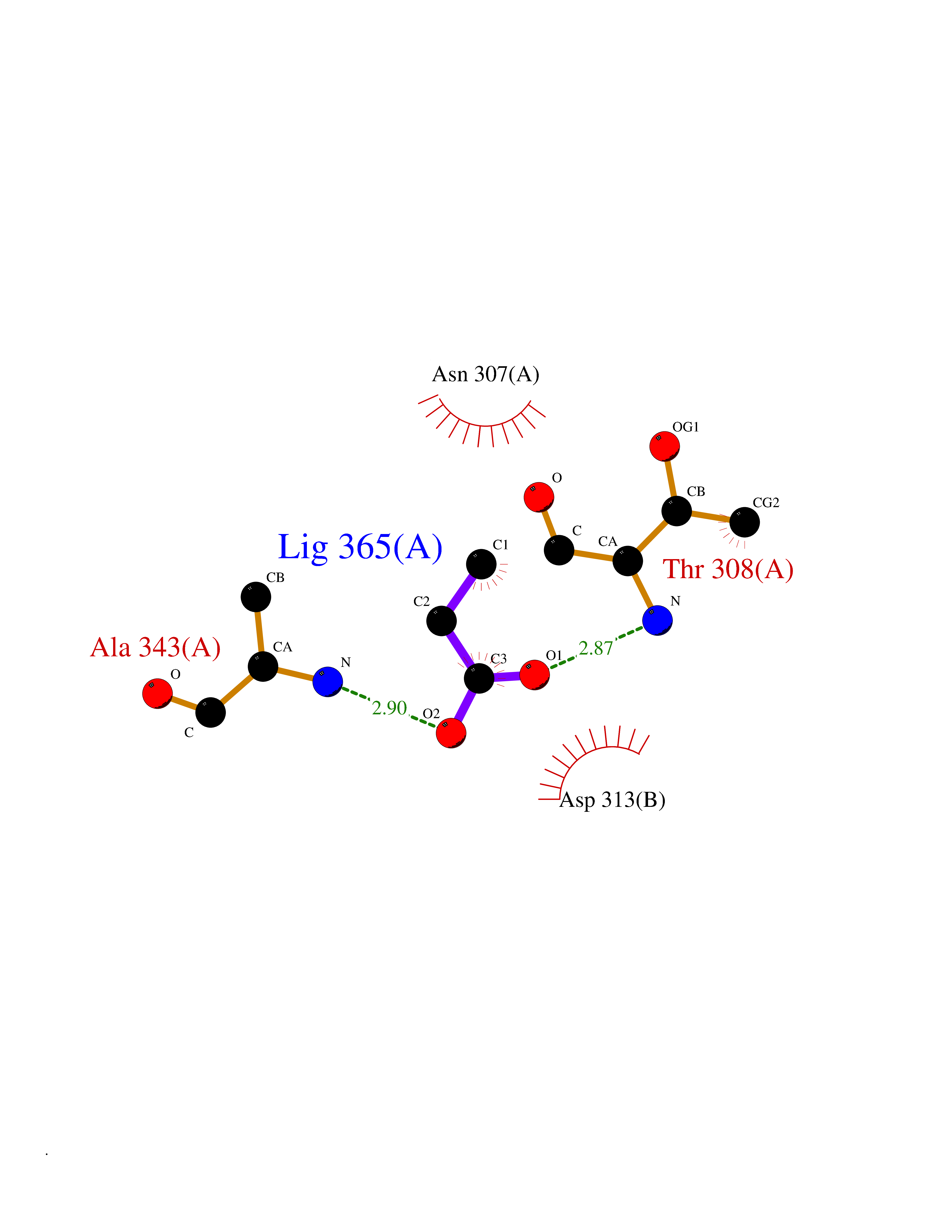 Ligplot Map 3D Binding mode Sequence EAPAGALAAVLKHSSTLPPESTQVRGYDFNRGVNYRALLEAFGTTGFQATNFGRAVQQVNAMIEKKLEPLSQDEDQHADLTQSRRPLTSCTIFLGYTSNLISSGIRETIRYLVQHNMVDVLVTTAGGVEEDLIKCLAPTYLGEFSLRGKELRENGINRIGNLLVPNENYXKFEDWLMPILDQMVMEQNTEGVKWTPSKMIARLGKEINNPESVYYWAQKNHIPVFSPALTDGSLGDMIFFHSYKNPGLVLDIVEDLRLINTQAIFAKCTGMIILGGGVVKHHIANANLMRNGADYAVYINTAQEFDGSDSGARPDEAVSWGKIRVDAQPVKVYADASLVFPLLVAETFAQKMDAFMSTQVRGYDFNRGVNYRALLEAFGTTGFQATNFGRAVQQVNAMIEKKLEPLSQDEDQHADLTQSRRPLTSCTIFLGYTSNLISSGIRETIRYLVQHNMVDVLVTTAGGVEEDLIKCLAPTYLGEFSLRGKELRENGINRIGNLLVPNENYXKFEDWLMPILDQMVMEQNTEGVKWTPSKMIARLGKEINNPESVYYWAQKNHIPVFSPALTDGSLGDMIFFHSYKNPGLVLDIVEDLRLINTQAIFAKCTGMIILGGGVVKHHIANANLMRNGADYAVYINTAQEFDGSDSGARPDEAVSWGKIRVDAQPVKVYADASLVFPLLVAETFAQKMDAFM Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Geranyltranstransferase (FDPS) | 4KPJ | 4.54 | |
Target general information Gen name FDPS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms KIAA1293; Geranylgeranyl pyrophosphate synthase; Geranylgeranyl diphosphate synthase; GGPS1; GGPPSase; GGPP synthase; Farnesyltranstransferase; Farnesyl pyrophosphate synthase; Farnesyl diphosphate sy Protein family FPP/GGPP synthase family Biochemical class Alkyl aryl transferase Function FPP also serves as substrate for protein farnesylation and geranylgeranylation. Catalyzes the sequential condensation of isopentenyl pyrophosphate with the allylic pyrophosphates, dimethylallyl pyrophosphate, and then with the resultant geranylpyrophosphate to the ultimate product farnesyl pyrophosphate. Key enzyme in isoprenoid biosynthesis which catalyzes the formation of farnesyl diphosphate (FPP), a precursor for several classes of essential metabolites including sterols, dolichols, carotenoids, and ubiquinones. Related diseases Porokeratosis 9, multiple types (POROK9) [MIM:616631]: A form of porokeratosis, a disorder of faulty keratinization characterized by one or more atrophic patches surrounded by a distinctive hyperkeratotic ridgelike border called the cornoid lamella. The keratotic lesions can progress to overt cutaneous neoplasms, typically squamous cell carcinomas. Multiple clinical variants of porokeratosis are recognized, including porokeratosis of Mibelli, linear porokeratosis, disseminated superficial actinic porokeratosis, palmoplantar porokeratosis, and punctate porokeratosis. Different clinical presentations can be observed among members of the same family. Individuals expressing more than one variant have also been reported. {ECO:0000269|PubMed:26202976}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00630; DB01785; DB07780; DB02552; DB07841; DB00710; DB06255; DB04714; DB02508; DB06548; DB00282; DB00884; DB00399 Interacts with O95870; P54253; Q6ZMZ0; Q9BRI3; Q8WWF3 EC number EC 2.5.1.10 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Cytoplasm; Disease variant; Host-virus interaction; Hydroxylation; Isoprene biosynthesis; Lipid biosynthesis; Lipid metabolism; Magnesium; Metal-binding; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 38705 Length 338 Aromaticity 0.12 Instability index 44.66 Isoelectric point 5.1 Charge (pH=7) -9.92 2D Binding mode Binding energy (Kcal/mol) -6.19 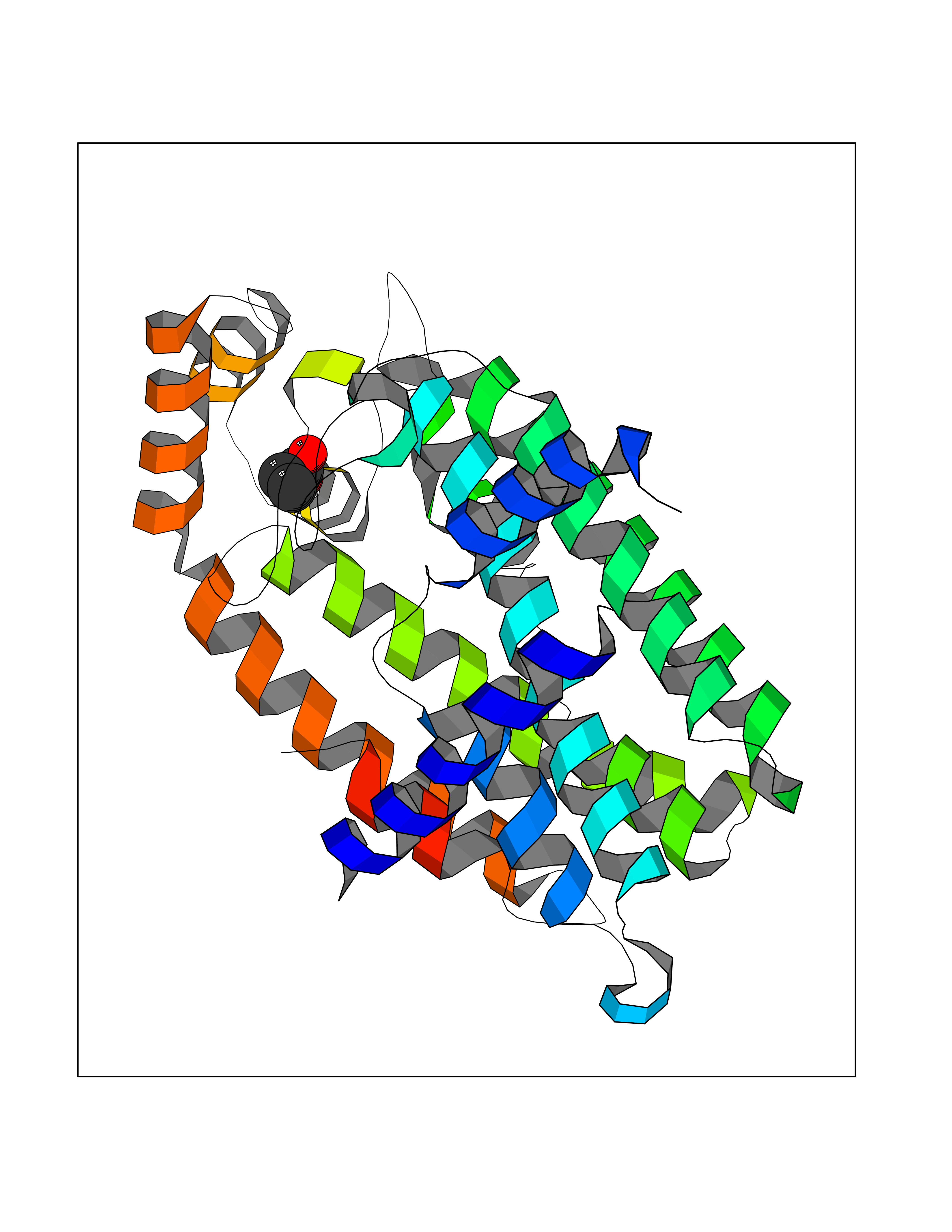 Molscript Map 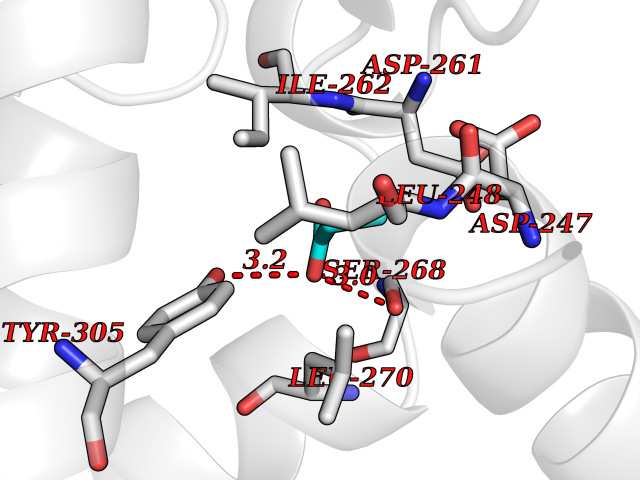 Pymol Map 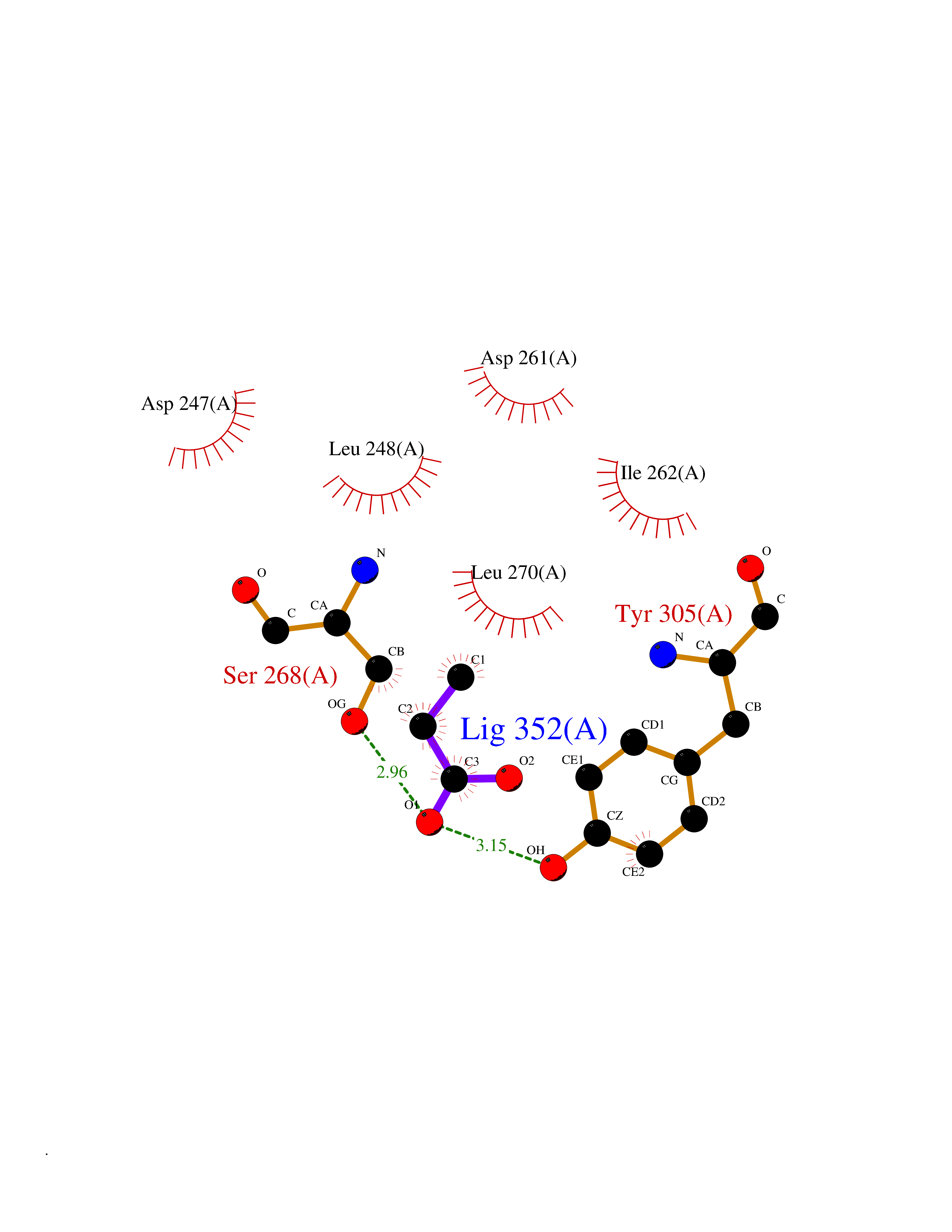 Ligplot Map 3D Binding mode Sequence VYAQEKQDFVQHFSQIVRVLTEHPEIGDAIARLKEVLEYNAIGGKYNRGLTVVVAFRELVEPRKQDADSLQRAWTVGWCVELLQAFFLVADDIMDSSLTRRGQICWYQKPGVGLDAINDANLLEACIYRLLKLYCREQPYYLNLIELFLQSSYQTEIGQTLDLLTAPQGNVDLVRFTEKRYKSIVKYKTAFASFYLPIAAAMYMAGIDGEKEHANAKKILLEMGEFFQIQDDYLDLFGDPSVTGKIGTDIQDNKCSWLVVQCLQRATPEQYQILKENYGQKEAEKVARVKALYEELDLPAVFLQYEEDSYSHIMALIEQYAAPLPPAVFLGLARKIYK Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Aldose reductase (AKR1B1) | 1US0 | 4.54 | |
Target general information Gen name AKR1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aldehyde reductase; AKR1B1 Protein family Aldo/keto reductase family Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the NADPH-dependent reduction of a wide variety of carbonyl-containing compounds to their corresponding alcohols with a broad range of catalytic efficiencies. Related diseases Glutamine deficiency, congenital (GLND) [MIM:610015]: An autosomal recessive disorder characterized by variable brain malformations, encephalopathy, severe developmental delay, seizures, and decreased glutamine levels in bodily fluids. Death in early infancy may occur. {ECO:0000269|PubMed:16267323, ECO:0000269|PubMed:26711351, ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 116 (DEE116) [MIM:620806]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE116 is autosomal dominant form characterized by severe developmental delay, seizures, and white matter abnormalities. {ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry. DEE116 is caused by variants that disrupt the canonical translation start codon in GLUL resulting in initiation of translation at Met-18 (PubMed:38579670). The resulting protein is enzymatically competent but insensitive to negative feedback regulation via glutamine-induced degradation. {ECO:0000269|PubMed:38579670}. Drugs (DrugBank ID) DB07028; DB07030; DB07450; DB02101; DB08449; DB08000; DB07139; DB07498; DB02007; DB02020; DB11859; DB02994; DB04272; DB07187; DB00694; DB00997; DB06246; DB01039; DB02021; DB16707; DB00143; DB02834; DB08084; DB01689; DB07063; DB06077; DB02518; DB00157; DB03461; DB05383; DB05533; DB05327; DB02712; DB00605; DB02383; DB02132; DB08772; DB07093; DB07999; DB08098 Interacts with Q9BUY7 EC number EC 1.1.1.300 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Lipid metabolism; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 35447.6 Length 313 Aromaticity 0.09 Instability index 36.41 Isoelectric point 7.1 Charge (pH=7) 0.26 2D Binding mode Binding energy (Kcal/mol) -6.19 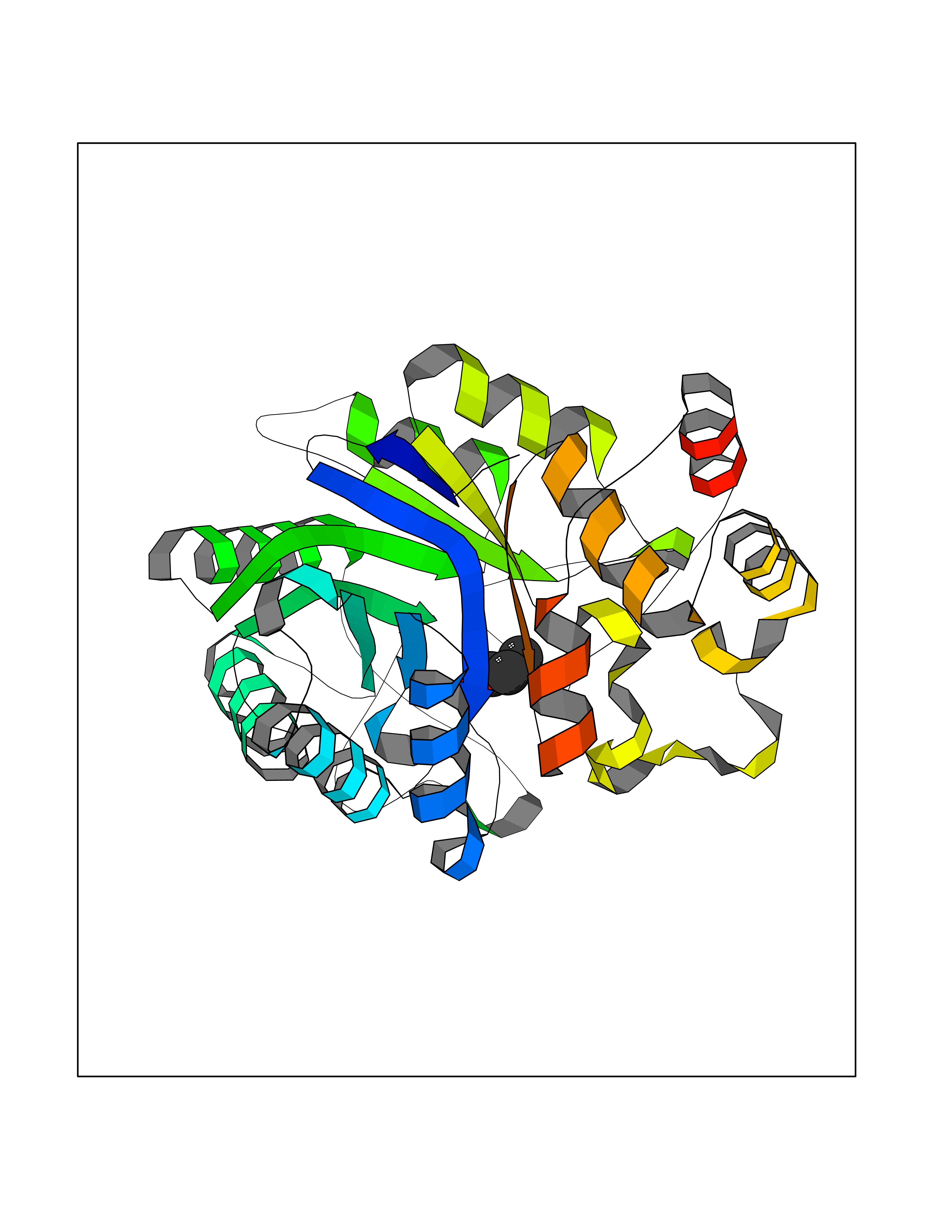 Molscript Map 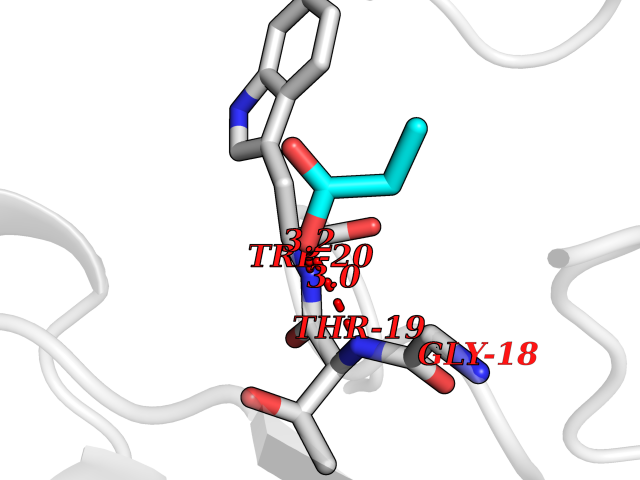 Pymol Map 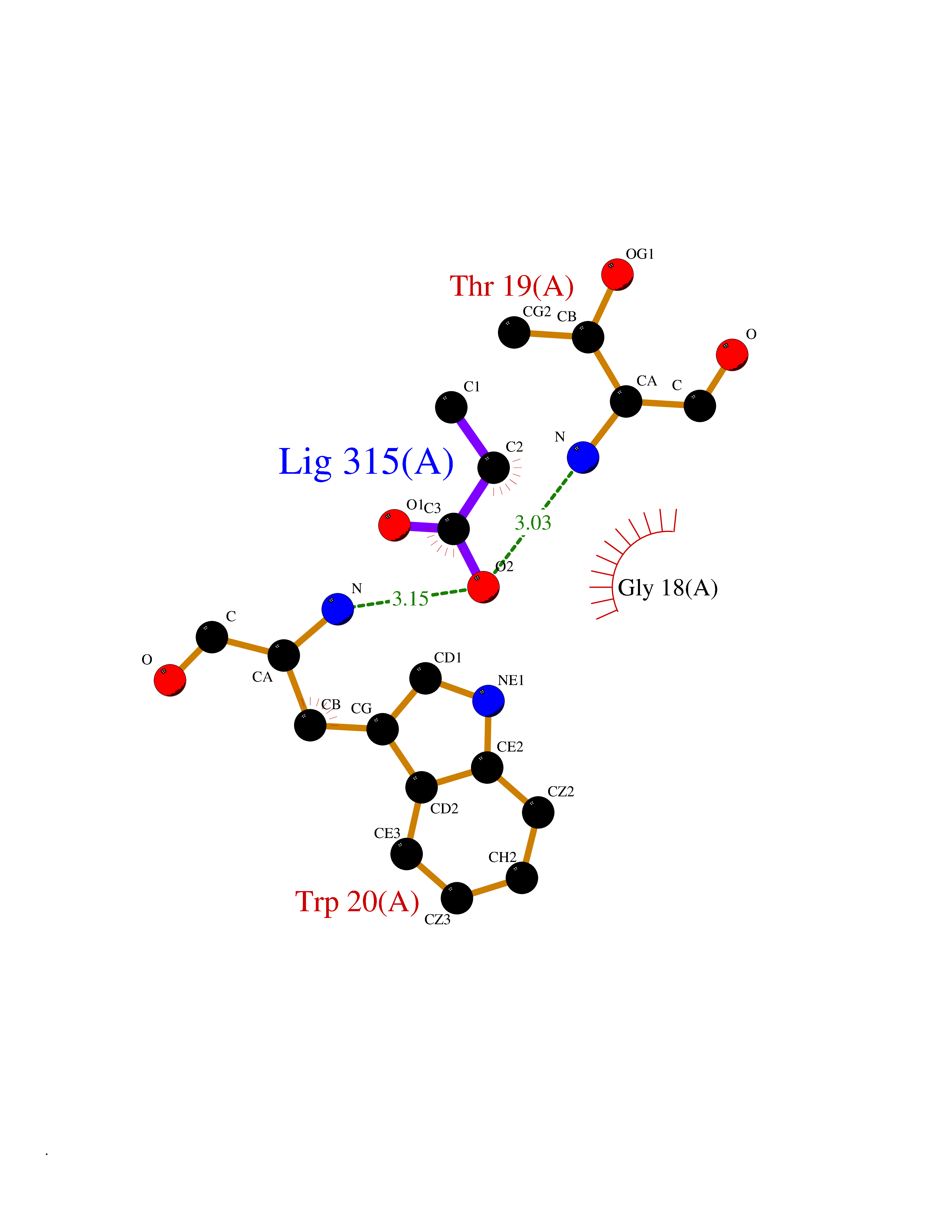 Ligplot Map 3D Binding mode Sequence MASRILLNNGAKMPILGLGTWKSPPGQVTEAVKVAIDVGYRHIDCAHVYQNENEVGVAIQEKLREQVVKREELFIVSKLWCTYHEKGLVKGACQKTLSDLKLDYLDLYLIHWPTGFKPGKEFFPLDESGNVVPSDTNILDTWAAMEELVDEGLVKAIGISNFNHLQVEMILNKPGLKYKPAVNQIECHPYLTQEKLIQYCQSKGIVVTAYSPLGSPDRPWAKPEDPSLLEDPRIKAIAAKHNKTTAQVLIRFPMQRNLVVIPKSVTPERIAENFKVFDFELSSQDMTTLLSYNRNWRVCALLSCTSHKDYPFH Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | "Periplasmic trehalase (EC 3.2.1.28) (Alpha,alpha-trehalase) (Alpha,alpha-trehalose glucohydrolase) (Tre37A)" | 2JG0 | 4.54 | |
Target general information Gen name treA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1186;osmA;b1197 Protein family Glycosyl hydrolase 37 family Biochemical class NA Function Provides the cells with the ability to utilize trehalose at high osmolarity by splitting it into glucose molecules that can subsequently be taken up by the phosphotransferase-mediated uptake system. Related diseases SRC kinase activity has been shown to be increased in several tumor tissues and tumor cell lines such as colon carcinoma cells. {ECO:0000269|PubMed:2498394, ECO:0000269|PubMed:3093483}.; DISEASE: Thrombocytopenia 6 (THC6) [MIM:616937]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC6 is an autosomal dominant form. Affected individuals may also have bone abnormalities and an increased risk for myelofibrosis. {ECO:0000269|PubMed:26936507}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.2.1.28 Uniprot keywords 3D-structure; Direct protein sequencing; Glycosidase; Hydrolase; Periplasm; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 57508.9 Length 507 Aromaticity 0.11 Instability index 48.32 Isoelectric point 5.48 Charge (pH=7) -10.13 2D Binding mode Binding energy (Kcal/mol) -6.2 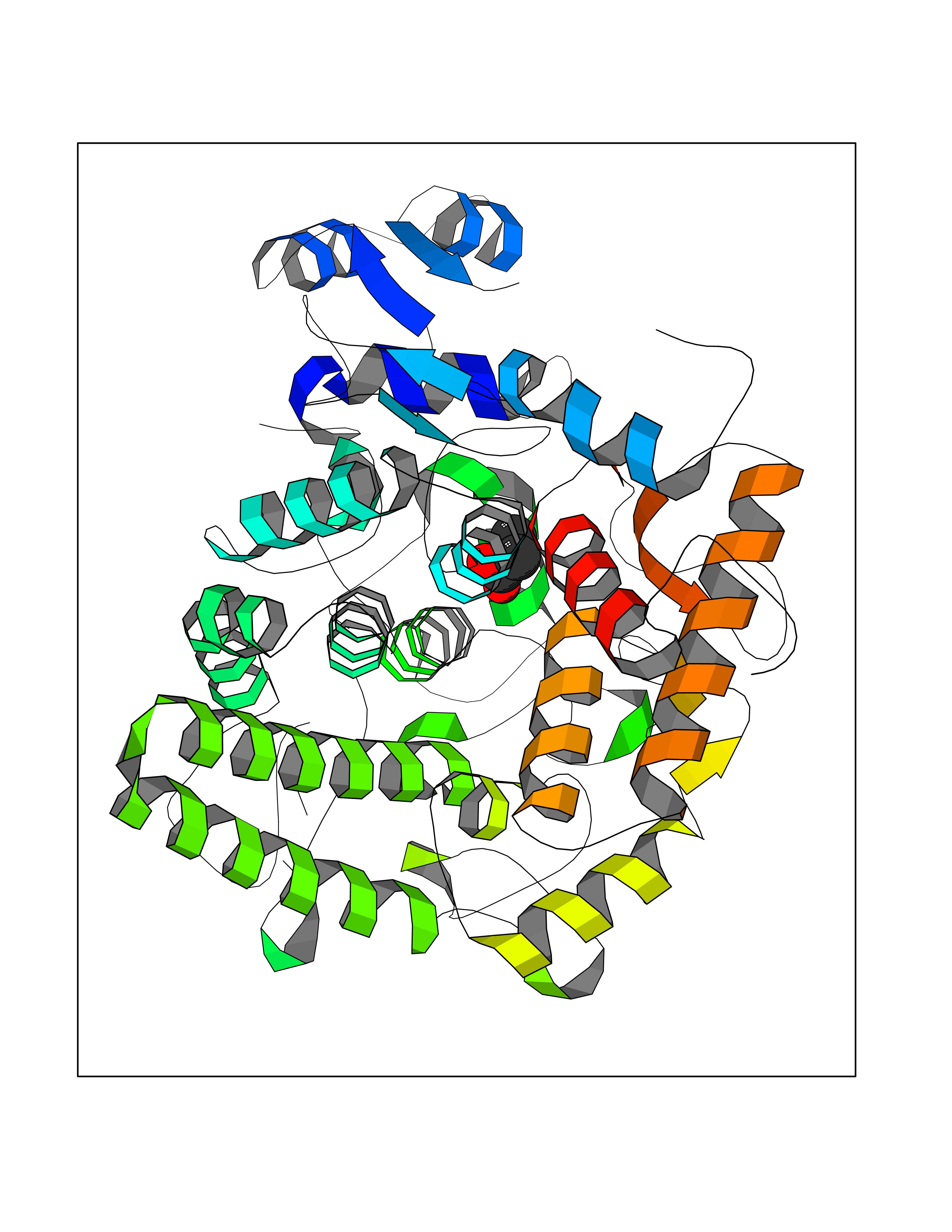 Molscript Map 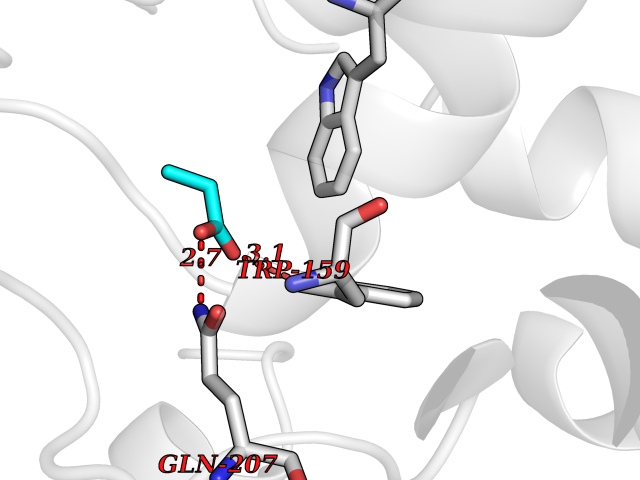 Pymol Map 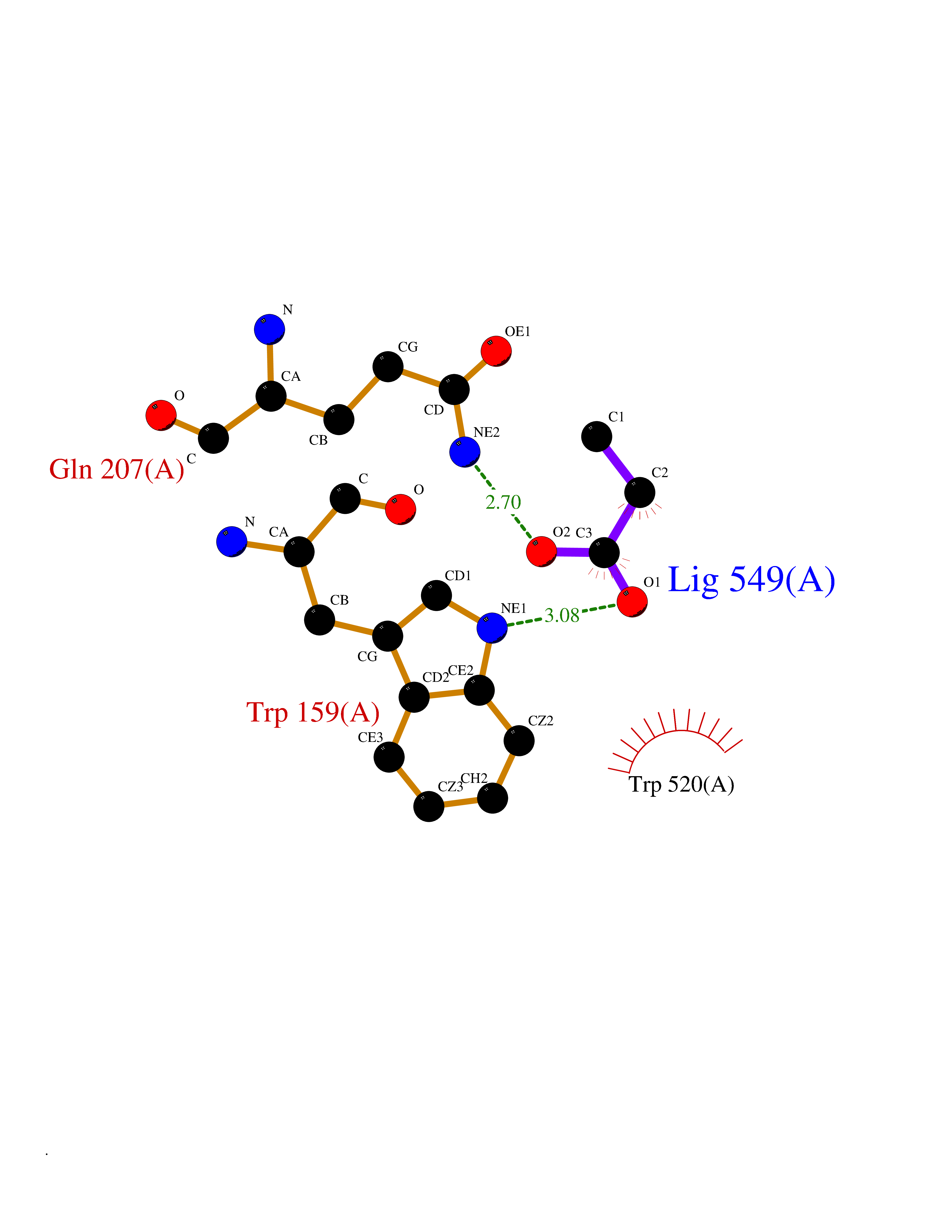 Ligplot Map 3D Binding mode Sequence PQPPDILLGPLFNDVQNAKLFPDQKTFADAVPNSDPLMILADYRMQQNQSGFDLRHFVNVNFTLPKYVPPEGQSLREHIDGLWPVLTRSTENTEKWDSLLPLPEPYVVPGGRFREVYYWDSYFTMLGLAESGHWDKVADMVANFAHEIDTYGHIPNGNRSYYLSRSQPPFFALMVELLAQHEGDAALKQYLPQMQKEYAYWMDGVENLQAGQQEKRVVKLQDGTLLNRYWDDRDTPRPESWVEDIATAKSNPNRPATEIYRDLRSAAASGWDFSSRWMDNPQQLNTLRTTSIVPVDLNSLMFKMEKILARASKAAGDNAMANQYETLANARQKGIEKYLWNDQQGWYADYDLKSHKVRNQLTAAALFPLYVNAAAKDRANKMATATKTHLLQPGGLNTTSVKSGQQWDAPNGWAPLQWVATEGLQNYGQKEVAMDISWHFLTNVQHTYDREKKLVEKYDVSTTGTGGGGGEYPLQDGFGWTNGVTLKMLDLICPKEQPCDNVPATRP Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Carbonic anhydrase 3 | 3UYQ | 4.54 | |
Target general information Gen name CA3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Alpha-carbonic anhydrase family Biochemical class Lyase Function Carbonate dehydratase activity.Nickel cation binding.Phosphatase activity.Zinc ion binding. Related diseases Cortical dysplasia, complex, with other brain malformations 6 (CDCBM6) [MIM:615771]: A disorder of aberrant neuronal migration and disturbed axonal guidance. Affected individuals have microcephaly, ataxia, and severe delayed psychomotor development. Brain imaging shows variable malformations of cortical development, including white matter streaks, dysmorphic basal ganglia, corpus callosum abnormalities, brainstem and cerebellar hypoplasia, cortical dysplasia, polymicrogyria. {ECO:0000269|PubMed:23246003}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Skin creases, congenital symmetric circumferential, 1 (CSCSC1) [MIM:156610]: An autosomal dominant disease characterized by multiple, symmetric, circumferential rings of folded skin, affecting primarily the limbs. Affected individuals also exhibit intellectual disability, cleft palate, and dysmorphic features. {ECO:0000269|PubMed:26637975}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00819; DB00562; DB01194; DB00482; DB00606; DB01144; DB00869; DB08846; DB00311; DB00703; DB12418; DB00391; DB00273; DB00580; DB00909 Interacts with P37235; Q9BS40 EC number 4.2.1.1 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Glutathionylation; Lyase; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 29416.7 Length 259 Aromaticity 0.11 Instability index 38.2 Isoelectric point 6.71 Charge (pH=7) -1.07 2D Binding mode Binding energy (Kcal/mol) -6.19  Molscript Map 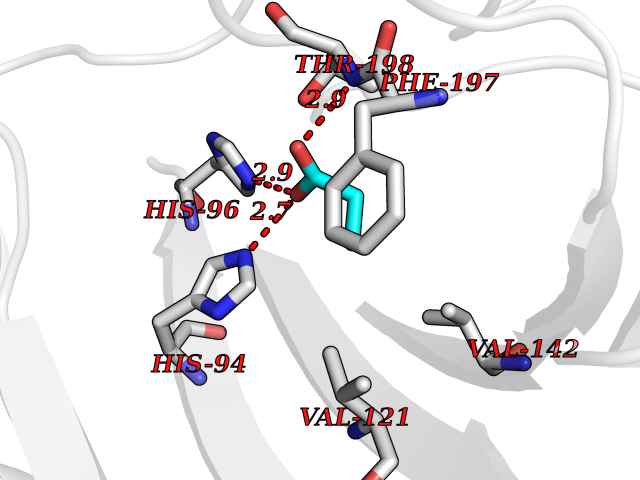 Pymol Map 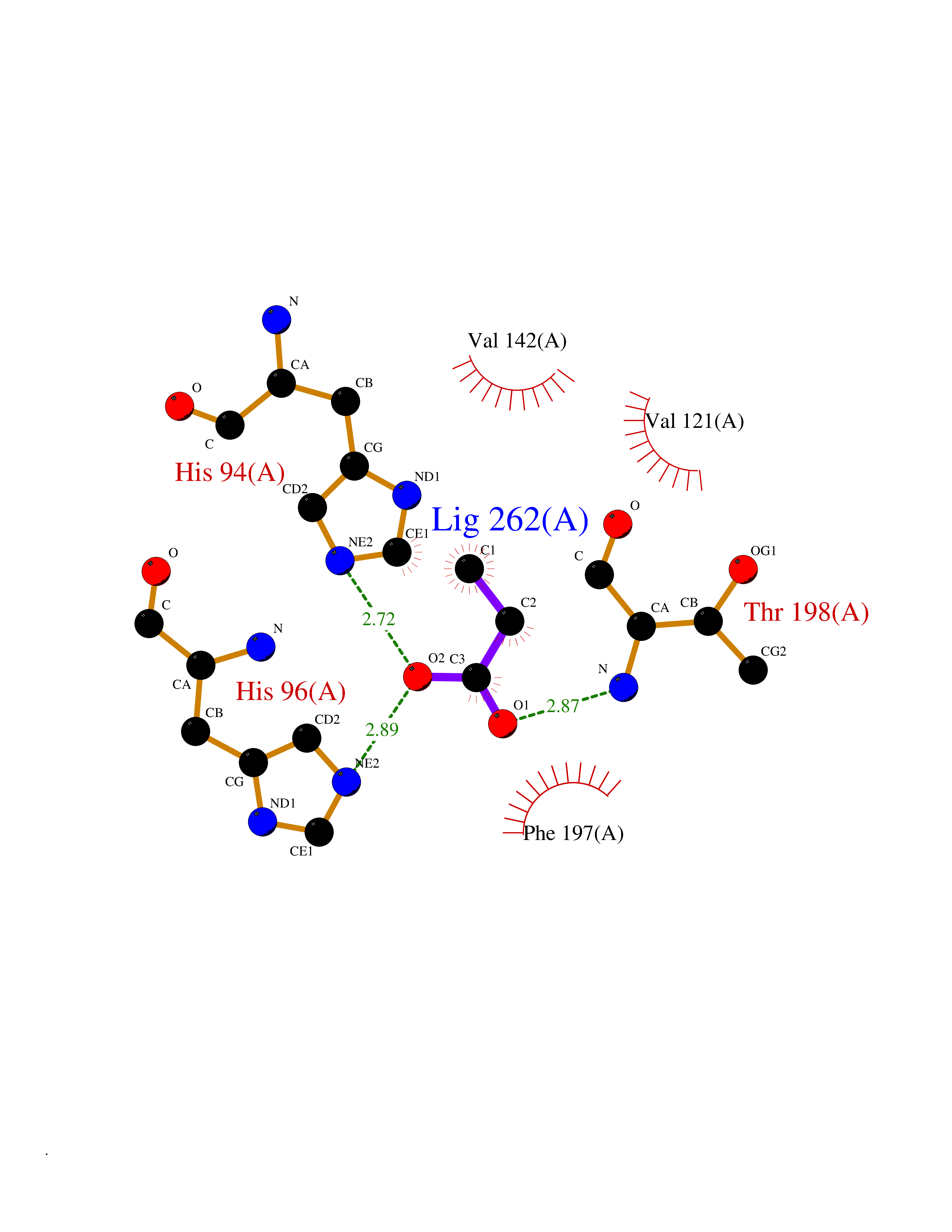 Ligplot Map 3D Binding mode Sequence AKEWGYASHNGPDHWHELFPNAKGENQSPIELHTKDIRHDPSLQPWSVSYDGGSAKTILNNGHTCRVVFDDTYDRSMLRGGPLPGPYRLRQFHLHWGSSDDHGSEHTVDGVKYAAELHLVHWNPKYNTFKEALKQRDGIAVIGIFLKIGHENGEFQIFLDALDKIKTKGKEAPFTKFDPSSLFPASRDYWTYQGSFTTPPCEECIVWLLLKEPMTVSSDQMAKLRSLLSSAENEPPVPLVSNWRPPQPINNRVVRASFK Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Dopamine beta-hydroxylase | 4ZEL | 4.54 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -6.2  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Argininosuccinate lyase | 1K62 | 4.54 | |
Target general information Gen name ASL Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Lyase 1 family, Argininosuccinate lyase subfamily Biochemical class Lyase Function Argininosuccinate lyase activity.Identical protein binding. Related diseases Argininosuccinic aciduria (ARGINSA) [MIM:207900]: An autosomal recessive disorder of the urea cycle. The disease is characterized by mental and physical retardation, liver enlargement, skin lesions, dry and brittle hair showing trichorrhexis nodosa microscopically and fluorescing red, convulsions, and episodic unconsciousness. {ECO:0000269|PubMed:11747432, ECO:0000269|PubMed:11747433, ECO:0000269|PubMed:12408190, ECO:0000269|PubMed:1705937, ECO:0000269|PubMed:17326097, ECO:0000269|PubMed:19703900, ECO:0000269|PubMed:22081021, ECO:0000269|PubMed:2263616, ECO:0000269|PubMed:24166829, ECO:0000269|PubMed:9045711}. The disease is caused by variants affecting the gene represented in this entry. The phenotype heterogeneity among patients is associated with interallelic complementation resulting in either complete loss of activity or partial regeneration of functional active sites in the heterotetrameric mutant protein. {ECO:0000269|PubMed:11747433}. Drugs (DrugBank ID) DB03814; DB00125; DB02267 Interacts with P04424; Q9BTE3-2; Q96HA8; O75382 EC number 4.3.2.1 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Amino-acid biosynthesis; Arginine biosynthesis; Disease variant; Lyase; Proteomics identification; Reference proteome; Urea cycle Protein physicochemical properties Chain ID A,B Molecular weight (Da) 51364.1 Length 459 Aromaticity 0.08 Instability index 35.82 Isoelectric point 6.66 Charge (pH=7) -1.25 2D Binding mode Binding energy (Kcal/mol) -6.19  Molscript Map 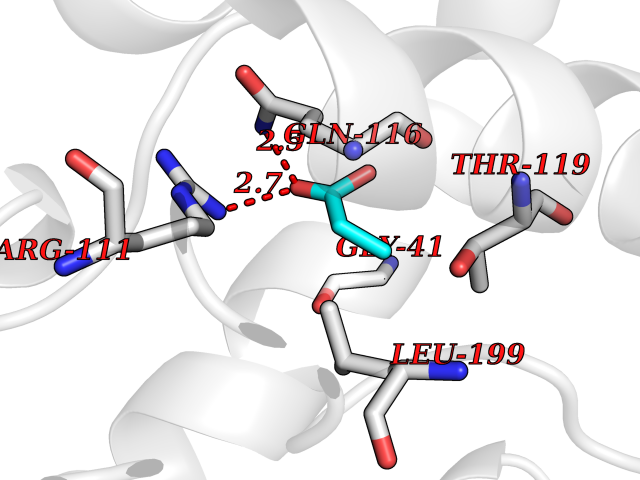 Pymol Map 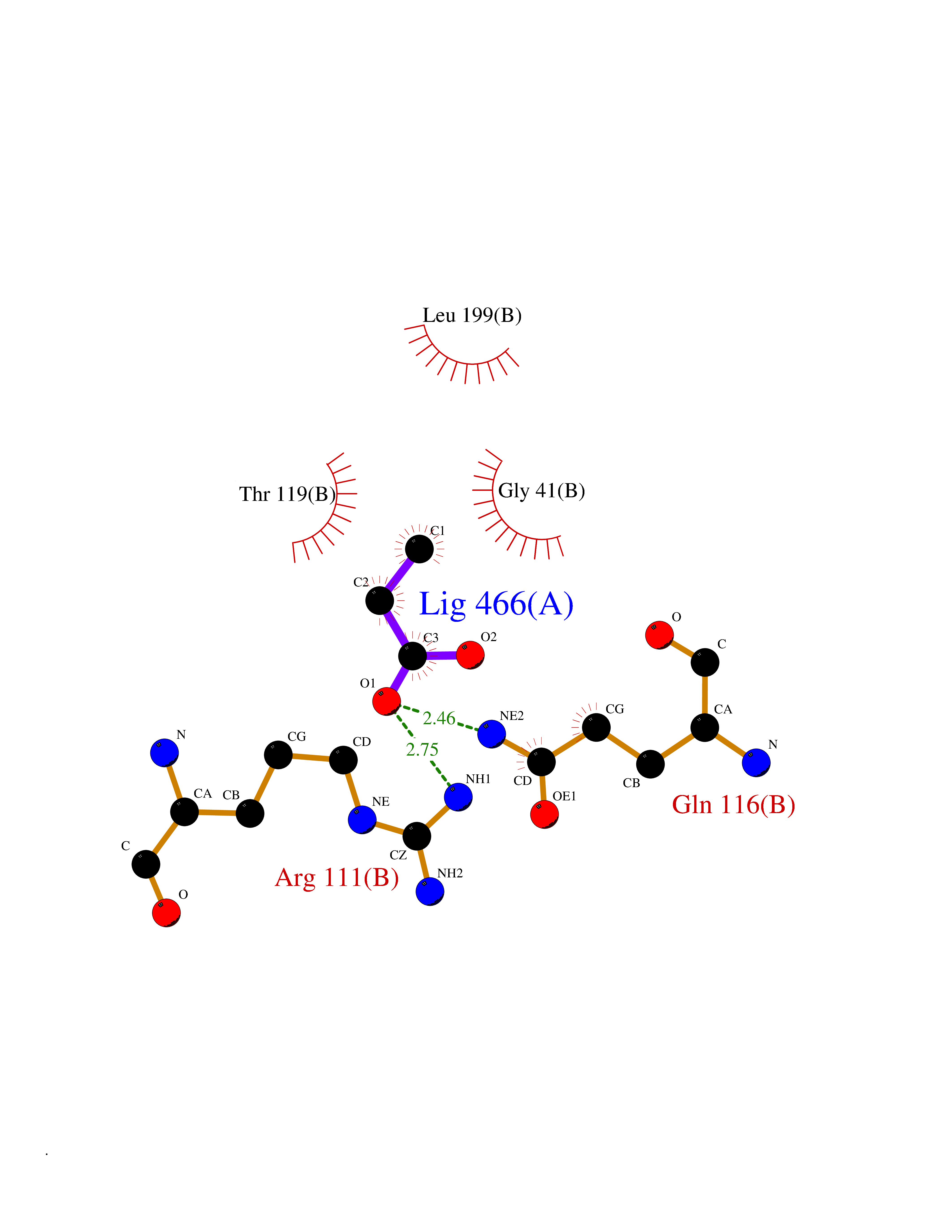 Ligplot Map 3D Binding mode Sequence GKLWGGRFVGAVDPIMEKFNASIAYDRHLWEVDVQGSKAYSRGLEKAGLLTKAEMDQILHGLDKVAEEWAQGTFKLNSNDEDIHTANERRLKELIGATAGKLHTGRSRNDQVVTDLRLWMRQTCSTLSGLLWELIRTMVDRAEAERDVLFPGYTHLQRAQPIRWSHWILSHAVALTRDSERLLEVRKRINVLPLGSGAIAGNPLGVDRELLRAELNFGAITLNSMDATSERDFVAEFLFWRSLCMTHLSRMAEDLILYCTKEFSFVQLSDAYSTGSSLMPRKKNPDSLELIRSKAGRVFGRCAGLLMTLKGLPSTYNKDLQEDKEAVFEVSDTMSAVLQVATGVISTLQIHQENMGQALSPDMLATDLAYYLVRKGMPFRQAHEASGKAVFMAETKGVALNQLSLQELQTISPLFSGDVICVWDYRHSVEQYGALGGTARSSVDWQIRQVRALLQAQQA Hydrogen bonds contact Hydrophobic contact | ||||