Job Results:
Ligand
Structure
Job ID
0d7fe7ad13d85a9fa0b43f4c0711e72a
Job name
NA
Time
2026-02-27 11:51:24
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Estrogen sulfotransferase | 1G3M | 4.37 | |
Target general information Gen name SULT1E1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms STE Protein family Sulfotransferase 1 family Biochemical class Transferase Function Aryl sulfotransferase activity.Estrone sulfotransferase activity.Flavonol 3-sulfotransferase activity.Steroid binding.Steroid sulfotransferase activity.Sulfotransferase activity. Related diseases Neuromyotonia and axonal neuropathy, autosomal recessive (NMAN) [MIM:137200]: An autosomal recessive neurologic disorder characterized by onset in the first or second decade of a peripheral axonal neuropathy predominantly affecting motor more than sensory nerves. The axonal neuropathy is reminiscent of Charcot-Marie-Tooth disease type 2 and distal hereditary motor neuropathy. Individuals with NMAN also have delayed muscle relaxation and action myotonia associated with neuromyotonic discharges on needle EMG resulting from hyperexcitability of the peripheral nerves. {ECO:0000269|PubMed:16835243, ECO:0000269|PubMed:22961002, ECO:0000269|PubMed:28691797, ECO:0000269|PubMed:29787766, ECO:0000269|PubMed:31088288}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02902; DB03346; DB01812; DB00714; DB14635; DB01176; DB00977; DB09288; DB00675; DB09100 Interacts with O76083; O76083-2 EC number 2.8.2.4 Uniprot keywords 3D-structure; Cytoplasm; Lipid metabolism; Lipid-binding; Proteomics identification; Reference proteome; Steroid-binding; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34022.7 Length 285 Aromaticity 0.15 Instability index 32.76 Isoelectric point 6.33 Charge (pH=7) -2.75 2D Binding mode Binding energy (Kcal/mol) -5.97 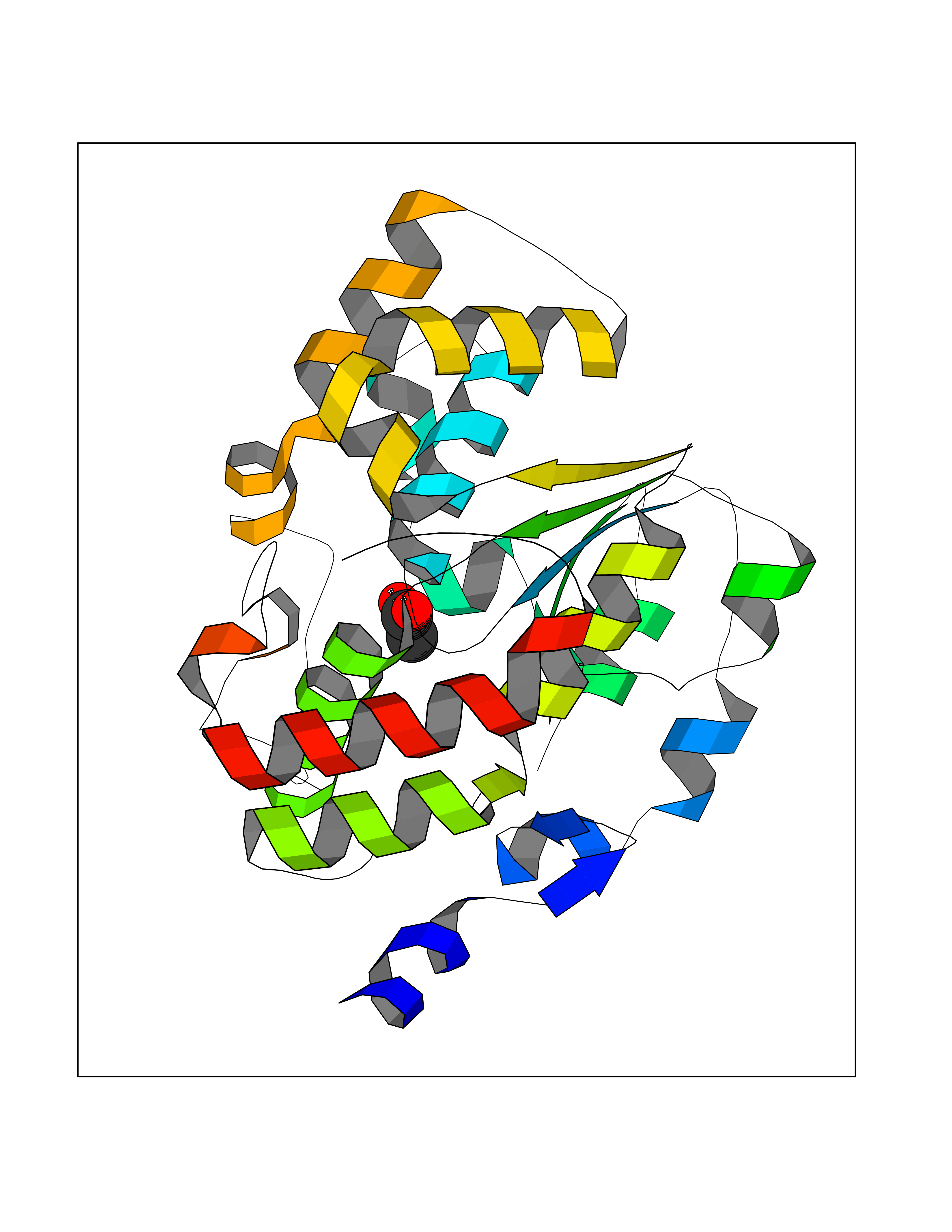 Molscript Map 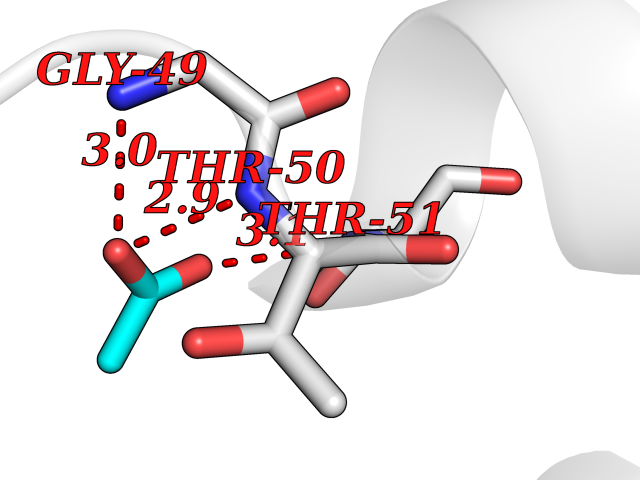 Pymol Map 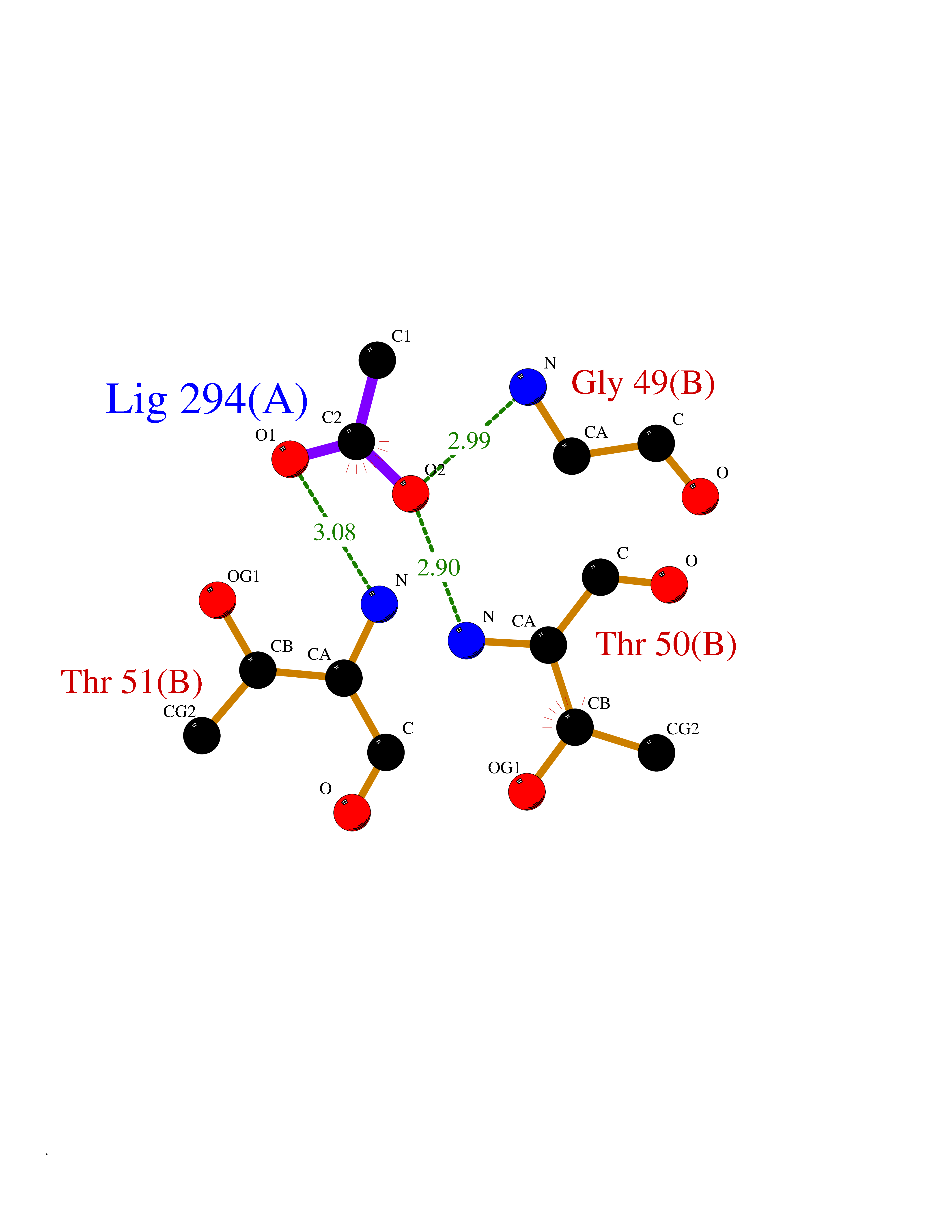 Ligplot Map 3D Binding mode Sequence SELDYYEKFEEVHGILMYKDFVKYWDNVEAFQARPDDLVIATYPKSGTTWVSEIVYMIYKEGDVEKCKEDVIFNRIPFLECRNGVKQLDEMNSPRIVKTHLPPELLPASFWEKDCKIIYLCRNAKDVAVSFYYFFLMVAGHPNPGSFPEFVEKFMQGQVPYGSWYKHVKSWWEKGKSPRVLFLFYEDLKEDIRKEVIKLIHFLERKPSEELVDRIIHHTSFQEMKNNPSTNYTTLPDEIMNQKLSPFMRKGITGDWKNHFTVALNEKFDKHYEQQMKESTLKFRT Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Folic acid synthesis protein FOL1 | 2BMB | 4.37 | |
Target general information Gen name FOL1 Organism Saccharomyces cerevisiae (strain ATCC 204508 / S288c) (Baker's yeast) Uniprot ID TTD ID NA Synonyms N0848;YNL256W Protein family DHNA family; HPPK family; DHPS family Biochemical class Transferase Function 2-amino-4-hydroxy-6-hydroxymethyldihydropteridine diphosphokinase activity.7,8-dihydromonapterin aldolase activity.ATP binding.Dihydroneopterin aldolase activity.Dihydropteroate synthase activity.Kinase activity.Metal ion binding. Related diseases LIMK1 is located in the Williams-Beuren syndrome (WBS) critical region. WBS results from a hemizygous deletion of several genes on chromosome 7q11.23, thought to arise as a consequence of unequal crossing over between highly homologous low-copy repeat sequences flanking the deleted region. Drugs (DrugBank ID) DB00634 Interacts with NA EC number 2.5.1.15; 2.7.6.3; 4.1.2.25 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Folate biosynthesis; Kinase; Lyase; Magnesium; Membrane; Metal-binding; Mitochondrion; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 58205.2 Length 513 Aromaticity 0.08 Instability index 42.41 Isoelectric point 5.92 Charge (pH=7) -8.31 2D Binding mode Binding energy (Kcal/mol) -5.96 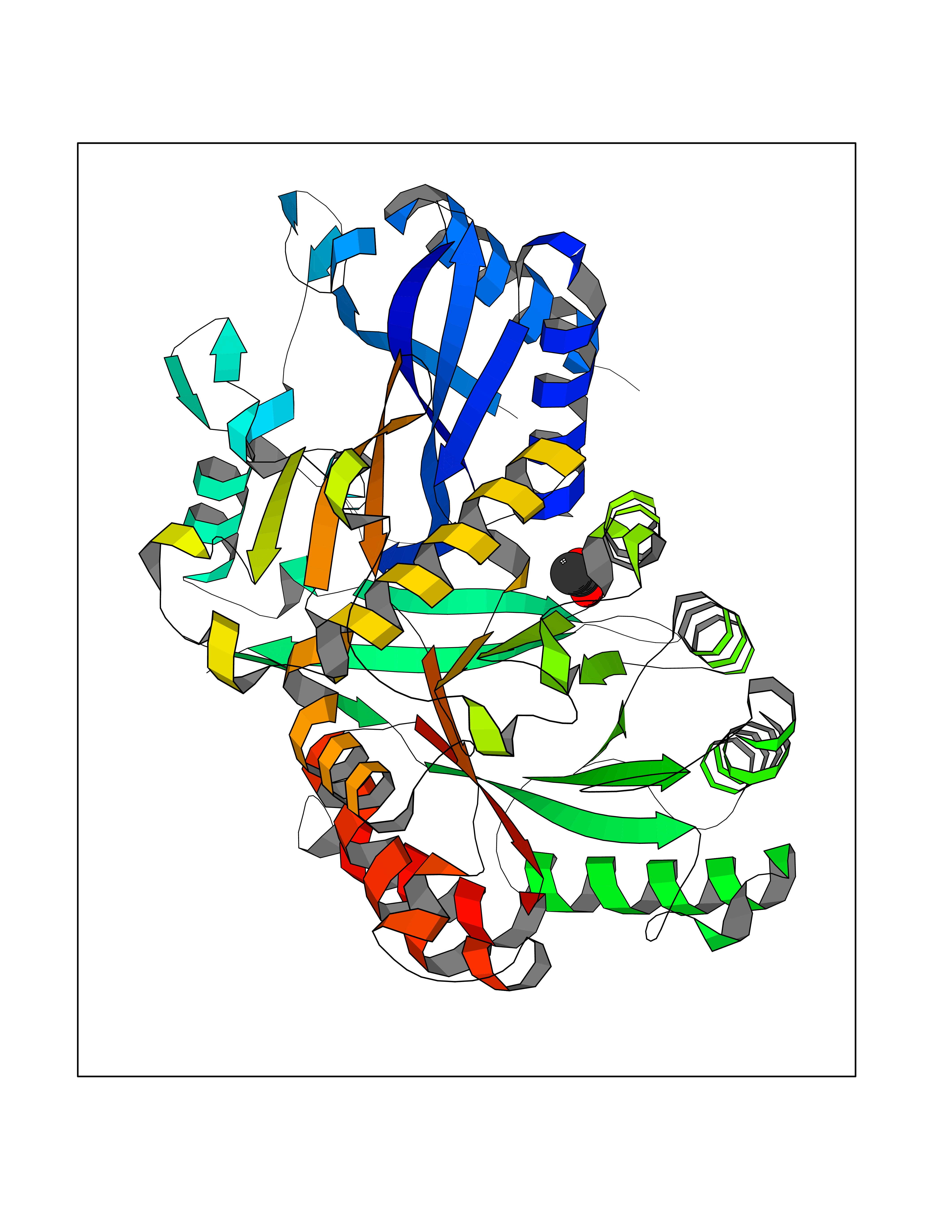 Molscript Map 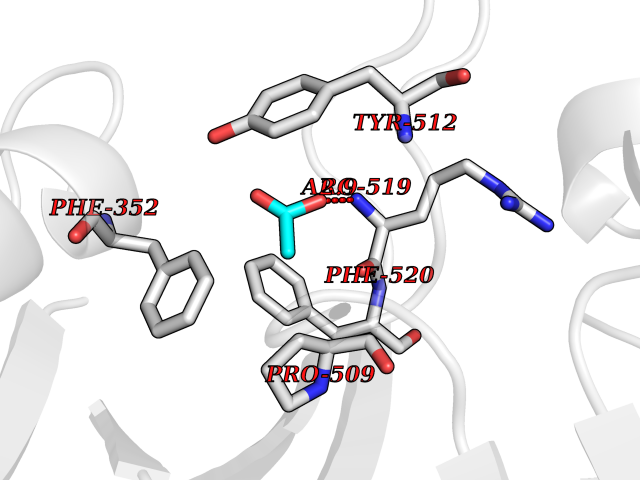 Pymol Map 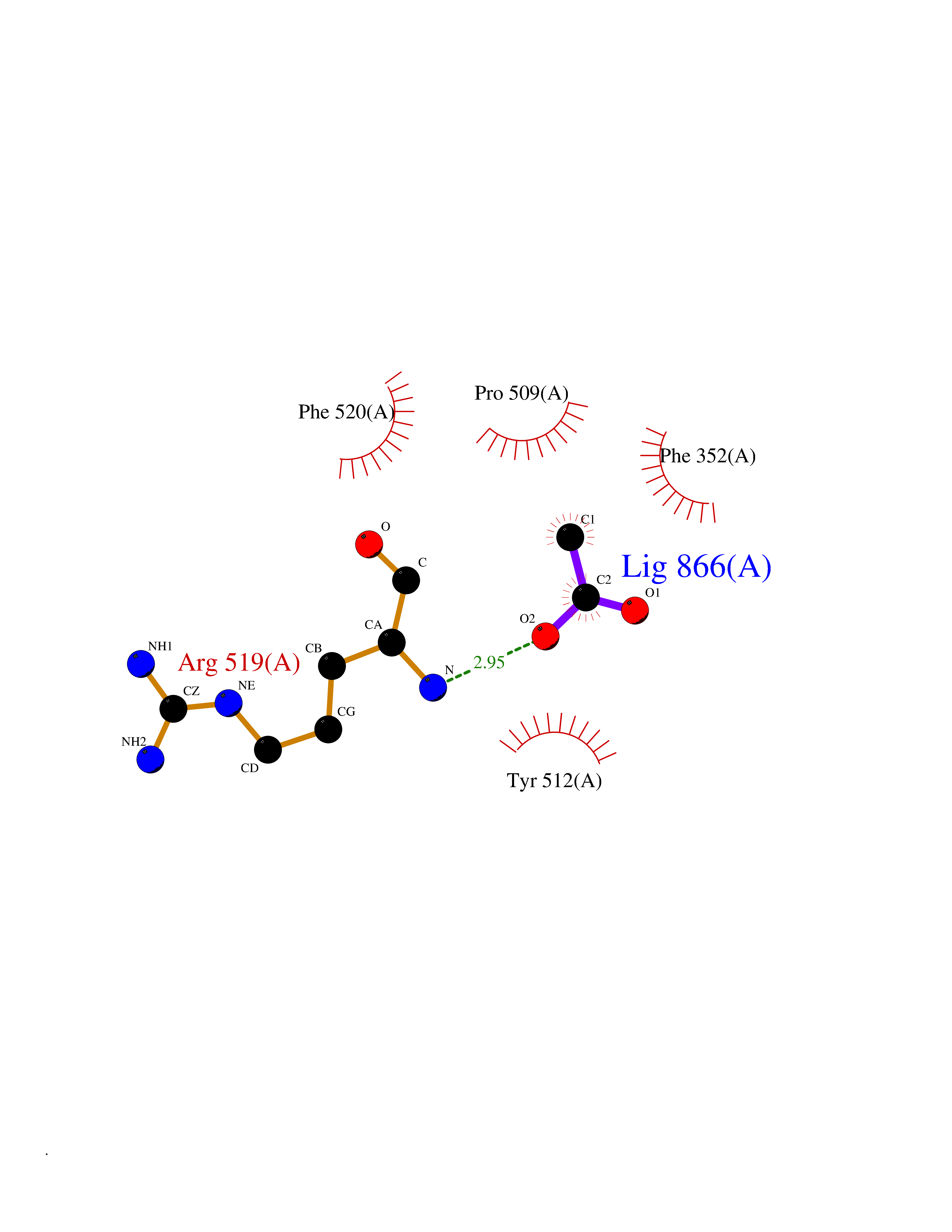 Ligplot Map 3D Binding mode Sequence SWKRAFLAFGSNIGDRFKHIQMALQLLSREKTVKLRNISSIFESEPMYFKDQTPFMNGCVEVETLLTPSELLKLCKKIEYEELQRTIDLDIVMFLNSAGEDIIVNEPDLNIPHPRMLERTFVLEPLCELISPVHLHPVTAEPIVDHLKQLYDKQHDEDTLWKLVPLPYRSGVEPRFLKFKTATKTNRITVSPTYIMAIFNATPDSFSDGGEHFADIESQLNDIIKLCKDALYLHESVIIDVGGCSTRPNSIQASEEEEIRRSIPLIKAIRESTELPQDKVILSIDTYRSNVAKEAIKVGVDIINDISGGLFDSNMFAVIAENPEICYILSHTRGDISTMNRLAHYENFALGDSIQQEFVHNTDIQQLDDLKDKTVLIRNVGQEIGERYIKAIDNGVKRWQILIDPGLGFAKTWKQNLQIIRHIPILKNYSFTMNSNNSQVYVNLRNMPVLLGPSRKKFIGHITKDVDAKQRDFATGAVVASCIGFGSDMVRVHDVKNCSKSIKLADAIYKGLE Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Acetyl-CoA carboxylase 1 | 2YL2 | 4.37 | |
Target general information Gen name ACACA Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ACAC;ACCA;ACC1 Protein family NA Biochemical class Ligase Function Acetyl-CoA carboxylase activity.ATP binding.Biotin carboxylase activity.Identical protein binding.Metal ion binding. Related diseases Acetyl-CoA carboxylase-alpha deficiency (ACACAD) [MIM:613933]: An autosomal recessive inborn error of de novo fatty acid synthesis associated with severe brain damage, persistent myopathy and poor growth. {ECO:0000269|PubMed:6114432}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00121 Interacts with Q13085; O60218; P38398; Q96EB6; Q9CQ20; P02654; Q92915-2; Q6NTF9-3 EC number 6.4.1.2 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative promoter usage; ATP-binding; Biotin; Cytoplasm; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Ligase; Lipid biosynthesis; Lipid metabolism; Magnesium; Manganese; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 54237.7 Length 486 Aromaticity 0.09 Instability index 39.18 Isoelectric point 6.37 Charge (pH=7) -2.46 2D Binding mode Binding energy (Kcal/mol) -5.96 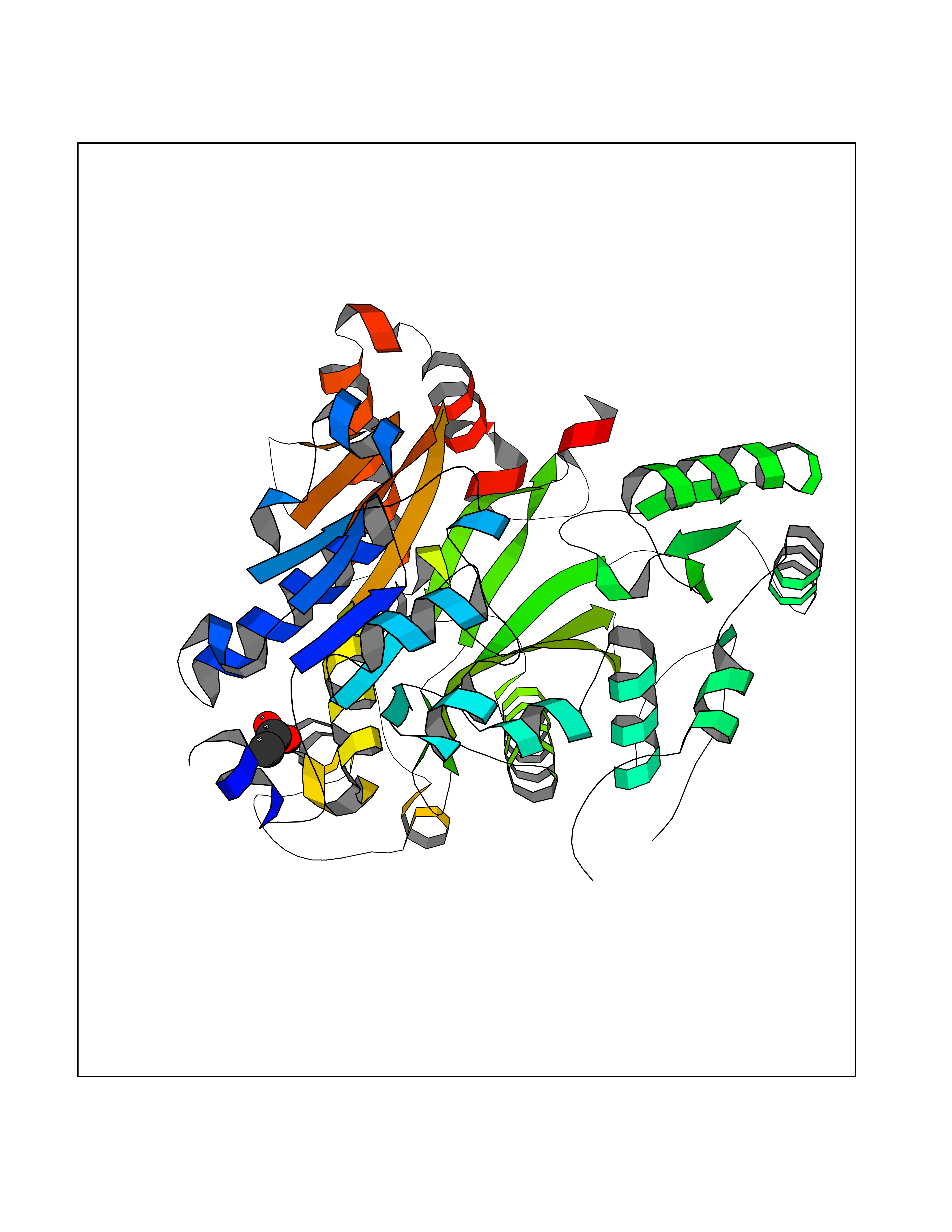 Molscript Map 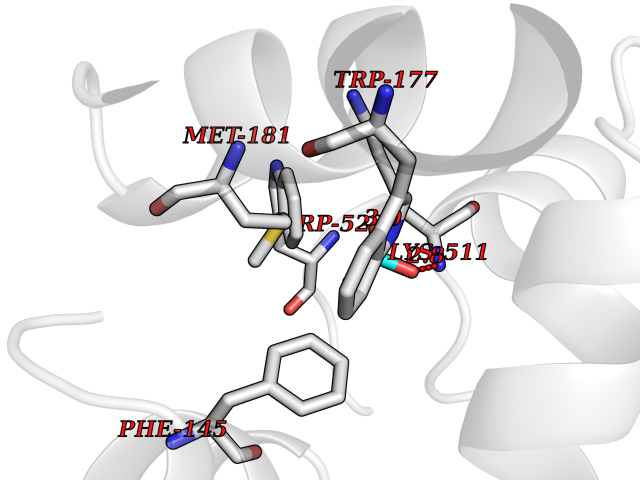 Pymol Map 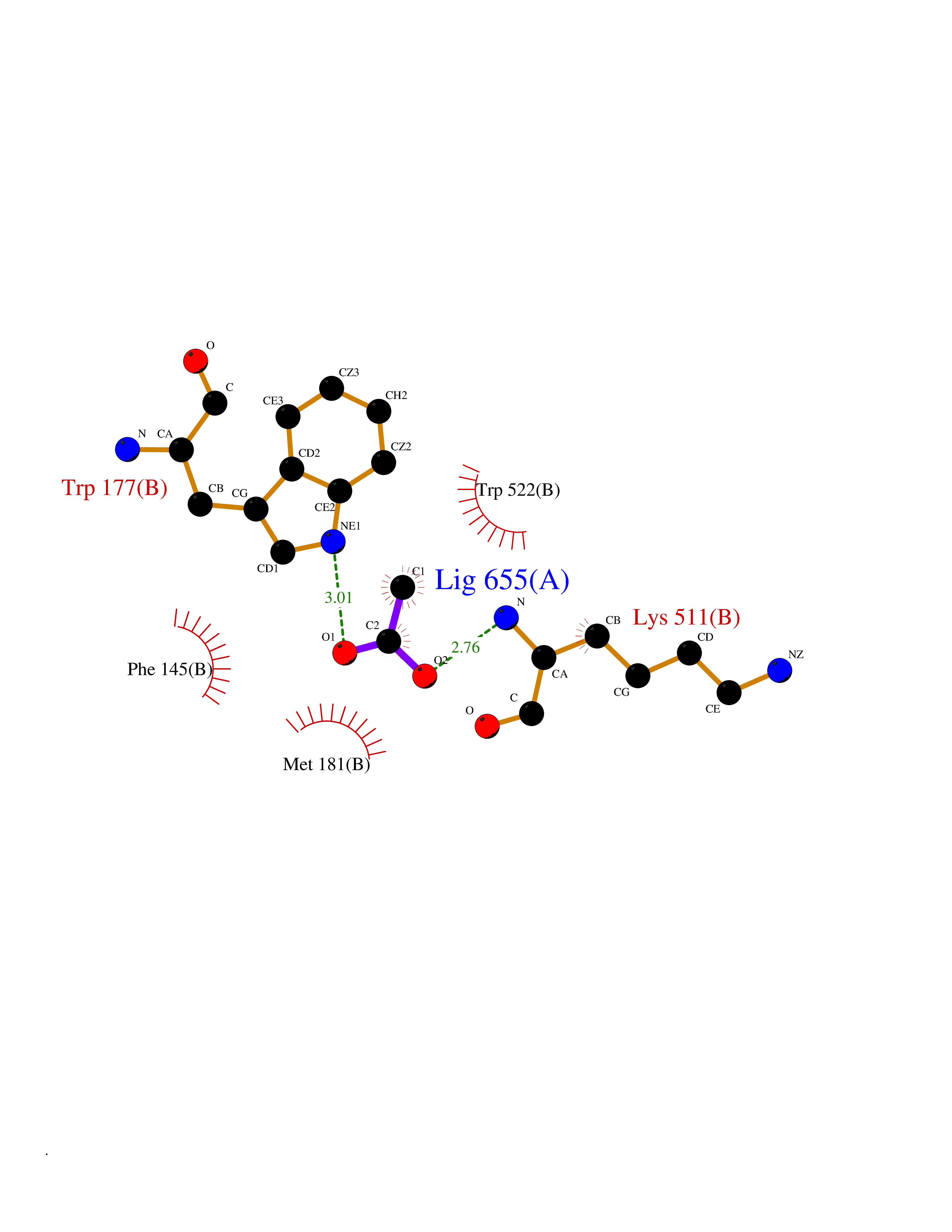 Ligplot Map 3D Binding mode Sequence VASPAEFVTRFGGNKVIEKVLIANNGIAAVKCMRSIRRWSYEMFRNERAIRFVVMVTPEDLKANAEYIKMADHYVPVPGGPNNNNYANVELILDIAKRIPVQAVWAGWGHASENPKLPELLLKNGIAFMGPPSQAMWALGDKIASSIVAQTAGIPTLPWSGSGLRVDWSKRILNVPQELYEKGYVKDVDDGLQAAEEVGYPVMIKASEGGGGKGIRKVNNADDFPNLFRQVQAEVPGSPIFVMRLAKQSRHLEVQILADQYGNAISLFGRDCSVQRRHQKIIEEAPATIATPAVFEHMEQCAVKLAKMVGYVSAGTVEYLYSQDGSFYFLELNPRLQVEHPCTEMVADVNLPAAQLQIAMGIPLYRIKDIRMMYGVSPWGDSPIDFEDSAHVPCPRGHVIAARITGTVQELNFRSNKNVWGYFSVQFGHCFSWGENREEAISNMVVALKELSIRGDFRTTVEYLIKLLETESFQMNRIDTGWLDRL Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Lanosterol 14-alpha-demethylase (EC 1.14.13.70) | 4G3J | 4.37 | |
Target general information Gen name Tb11.02.4080 Organism Trypanosoma brucei brucei (strain 927/4 GUTat10.1) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class NA Function NA Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) NA Interacts with NA EC number 1.14.13.70 Uniprot keywords 3D-structure; Heme; Iron; Metal-binding; Monooxygenase; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID D Molecular weight (Da) 50691.4 Length 448 Aromaticity 0.08 Instability index 54.09 Isoelectric point 6.99 Charge (pH=7) -0.03 2D Binding mode Binding energy (Kcal/mol) -5.96  Molscript Map  Pymol Map 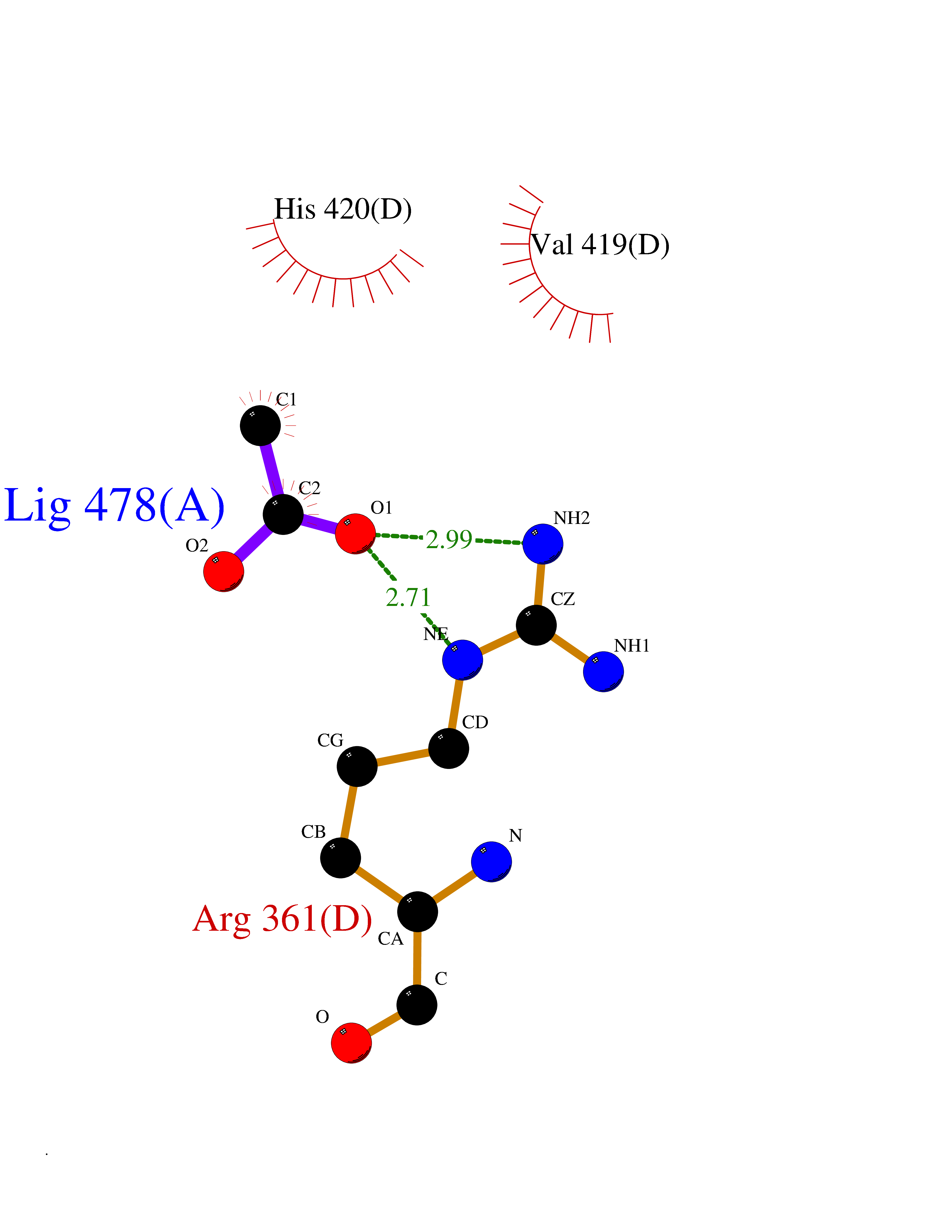 Ligplot Map 3D Binding mode Sequence GKLPPVYPVTVPILGHIIQFGKSPLGFMQECKRQLKSGIFTINIVGKRVTIVGDPHEHSRFFLPRNEVLSPREVYSFMVPVFGEGVAYAAPYPRMREQLNFLAEELTIAKFQNFVPAIQHEVRKFMAANWDKDEGEINLLEDCSTMIINTACQCLFGEDLRKRLDARRFAQLLAKMESSLIPAAVFLPILLKLPLPQSARCHEARTELQKILSEIIIARKEEEVNKDSSTSDLLSGLLSAVYRDGTPMSLHEVCGMIVAAMFAGQHTSSITTTWSMLHLMHPANVKHLEALRKEIEEFPAQLNYNNVMDEMPFAERCARESIRRDPPLLMLMRKVMADVKVGSYVVPKGDIIACSPLLSHHDEEAFPEPRRWDPERDEKVEGAFIGFGAGVHKCIGQKFGLLQVKTILATAFRSYDFQLLRDEVPDPDYHTMVVGPTASQCRVKYIRR Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Metabotropic glutamate receptor 3 (mGluR3) | 4XAR | 4.37 | |
Target general information Gen name GRM3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms mGLUR3; Group III metabotropic glutamate receptor; GPRC1C Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling inhibits adenylate cyclase activity. G-protein coupled receptor for glutamate. Related diseases Paramyotonia congenita (PMC) [MIM:168300]: An autosomal dominant channelopathy characterized by myotonia, increased by exposure to cold, intermittent flaccid paresis, not necessarily dependent on cold or myotonia, lability of serum potassium, non-progressive nature and lack of atrophy or hypertrophy of muscles. In some patients, myotonia is not increased by cold exposure (paramyotonia without cold paralysis). Patients may have a combination phenotype of PMC and HYPP. {ECO:0000269|PubMed:10369308, ECO:0000269|PubMed:10727489, ECO:0000269|PubMed:1310898, ECO:0000269|PubMed:1316765, ECO:0000269|PubMed:1338909, ECO:0000269|PubMed:15318338, ECO:0000269|PubMed:15790667, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:18166706, ECO:0000269|PubMed:18690054, ECO:0000269|PubMed:19077043, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:8242056, ECO:0000269|PubMed:8308722, ECO:0000269|PubMed:8388676, ECO:0000269|PubMed:8580427}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hypokalemic 2 (HOKPP2) [MIM:613345]: An autosomal dominant disorder manifested by episodic flaccid generalized muscle weakness associated with falls of serum potassium levels. {ECO:0000269|PubMed:10599760, ECO:0000269|PubMed:10851391, ECO:0000269|PubMed:10944223, ECO:0000269|PubMed:11558801, ECO:0000269|PubMed:11591859, ECO:0000269|PubMed:16890191, ECO:0000269|PubMed:17898326, ECO:0000269|PubMed:18162704, ECO:0000269|PubMed:19118277, ECO:0000269|PubMed:20522878, ECO:0000269|PubMed:21043388, ECO:0000269|PubMed:24549961}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hyperkalemic (HYPP) [MIM:170500]: An autosomal dominant channelopathy characterized by episodic flaccid generalized muscle weakness associated with high levels of serum potassium. Concurrence of myotonia is found in HYPP patients. {ECO:0000269|PubMed:1659668, ECO:0000269|PubMed:1659948, ECO:0000269|PubMed:20076800}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis normokalemic (NKPP) [MIM:170500]: A disorder closely related to hyperkalemic periodic paralysis, but marked by a lack of alterations in potassium levels during attacks of muscle weakness. {ECO:0000269|PubMed:15596759, ECO:0000269|PubMed:18046642, ECO:0000269|PubMed:20522878}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myotonia SCN4A-related (MYOSCN4A) [MIM:608390]: A phenotypically highly variable myotonia aggravated by potassium loading, and sometimes by cold. Myotonia is characterized by sustained muscle tensing that prevents muscles from relaxing normally. It causes muscle stiffness that can interfere with movement. In some people the stiffness is very mild, while in other cases it may be severe enough to interfere with walking, running, and other activities of daily life. Myotonia SCN4A-related includes myotonia permanens and myotonia fluctuans. In myotonia permanens, the myotonia is generalized and there is a hypertrophy of the muscle, particularly in the neck and the shoulder. Attacks of severe muscle stiffness of the thoracic muscles may be life threatening due to impaired ventilation. In myotonia fluctuans, the muscle stiffness may fluctuate from day to day, provoked by exercise. {ECO:0000269|PubMed:10218481, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:16832098, ECO:0000269|PubMed:17212350, ECO:0000269|PubMed:17998485, ECO:0000269|PubMed:18203179, ECO:0000269|PubMed:18337100, ECO:0000269|PubMed:19015483, ECO:0000269|PubMed:19347921, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:27653901, ECO:0000269|PubMed:8058156, ECO:0000269|PubMed:9392583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 16 (CMS16) [MIM:614198]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS16 is characterized by fatigable generalized weakness and recurrent attacks of respiratory and bulbar paralysis since birth. The fatigable weakness involves lid-elevator, external ocular, facial, limb and truncal muscles and an decremental response of the compound muscle action potential on repetitive stimulation. {ECO:0000269|PubMed:12766226, ECO:0000269|PubMed:25707578, ECO:0000269|PubMed:26659129}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22A, classic (CMYO22A) [MIM:620351]: A form of congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22A is an autosomal recessive form characterized by fetal hypokinesia, polyhydramnios, and severe neonatal hypotonia associated with respiratory insufficiency. Affected individuals who survive the neonatal period have delayed motor development, difficulty walking, proximal muscle weakness of the upper and lower limbs, facial and neck muscle weakness, easy fatigability, and mild limb contractures or foot deformities. {ECO:0000269|PubMed:26700687, ECO:0000269|PubMed:28262468, ECO:0000269|PubMed:36090556}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22B, severe fetal (CMYO22B) [MIM:620369]: A severe congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22B is an autosomal recessive form characterized by onset in utero. Affected individuals show fetal akinesia, and develop fetal hydrops with pulmonary hypoplasia, severe joint contractures, and generalized muscle hypoplasia. Death occurs in utero or soon after birth. {ECO:0000269|PubMed:26700687}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05096 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50355.5 Length 445 Aromaticity 0.11 Instability index 38.26 Isoelectric point 6.52 Charge (pH=7) -1.53 2D Binding mode Binding energy (Kcal/mol) -5.97  Molscript Map 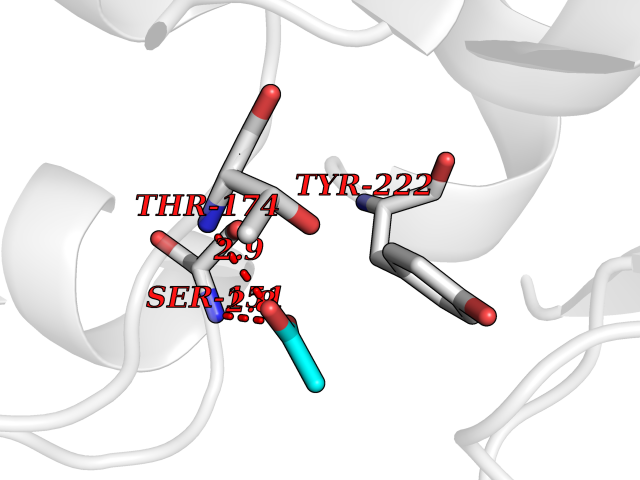 Pymol Map 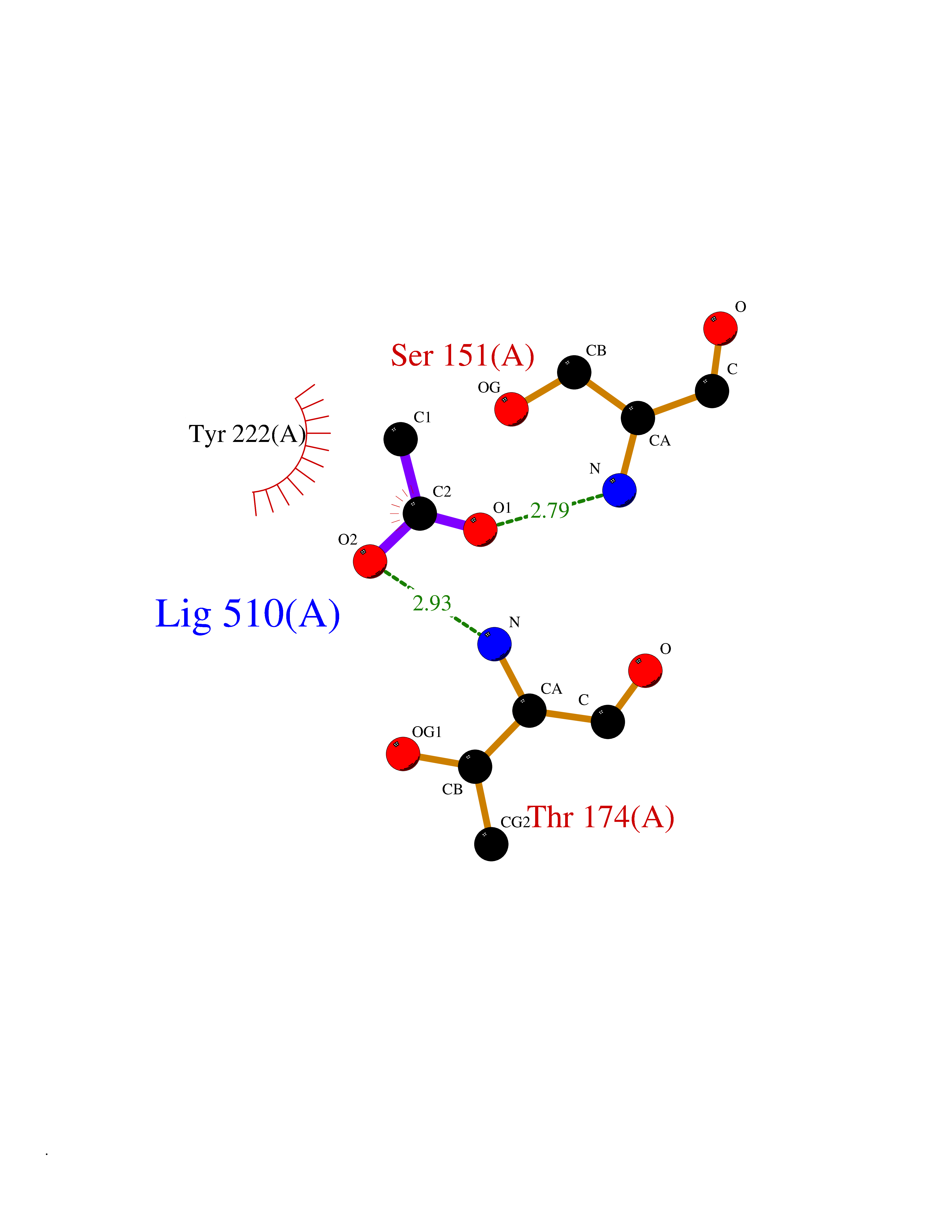 Ligplot Map 3D Binding mode Sequence RREIKIEGDLVLGGLFPINEKGTGTEECGRINEDRGIQRLEAMLFAIDEINKDDYLLPGVKLGVHILDTCSRDTYALEQSLEFVRASLLLIAGVIGGSYSSVSIQVANLLRLFQIPQISYASTSAKLSDKSRYDYFARTVPPDFYQAKAMAEILRFFNWTYVSTVASEGDYGETGIEAFEQEARLRNISIATAEKVGRSNIRKSYDSVIRELLQKPNARVVVLFMRSDDSRELIAAASRANASFTWVASDGWGAQESIIKGSEHVAYGAITLELASQPVRQFDRYFQSLNPYNNHRNPWFRDFWEQKFQCSLRVCDKHLAIDSSNYEQESKIMFVVNAVYAMAHALHKMQRTLCPNTTKLCDAMKILDGKKLYKDYLLKINFTAPDADSIVKFDTFGDGMGRYNVFNFQNVGGKYSYLKVGHWAETLSLDVNSIHWSRNSVPTSE Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Mycobacterium Membrane protein mmpL3 (MycB mmpL3) | 7NVH | 4.37 | |
Target general information Gen name MycB mmpL3 Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID Synonyms Trehalose monomycolate exporter MmpL3; TMM exporter MmpL3 Protein family Resistance-nodulation-cell division (RND) (TC 2.A.6) family, MmpL subfamily Biochemical class NA Function Transports trehalose monomycolate (TMM) across the inner membrane. Could also be part of a heme-iron acquisition system. Related diseases Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:8955068}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:17332249}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 3 (NS3) [MIM:609942]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:16474405, ECO:0000269|PubMed:16773572, ECO:0000269|PubMed:17056636, ECO:0000269|PubMed:17468812, ECO:0000269|PubMed:19396835, ECO:0000269|PubMed:20949621}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Gastric cancer (GASC) [MIM:613659]: A malignant disease which starts in the stomach, can spread to the esophagus or the small intestine, and can extend through the stomach wall to nearby lymph nodes and organs. It also can metastasize to other parts of the body. The term gastric cancer or gastric carcinoma refers to adenocarcinoma of the stomach that accounts for most of all gastric malignant tumors. Two main histologic types are recognized, diffuse type and intestinal type carcinomas. Diffuse tumors are poorly differentiated infiltrating lesions, resulting in thickening of the stomach. In contrast, intestinal tumors are usually exophytic, often ulcerating, and associated with intestinal metaplasia of the stomach, most often observed in sporadic disease. {ECO:0000269|PubMed:14534542, ECO:0000269|PubMed:3034404, ECO:0000269|PubMed:7773929}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in KRAS are a cause of pylocytic astrocytoma (PA). Pylocytic astrocytomas are neoplasms of the brain and spinal cord derived from glial cells which vary from histologically benign forms to highly anaplastic and malignant tumors. {ECO:0000269|PubMed:16247081}.; DISEASE: Cardiofaciocutaneous syndrome 2 (CFC2) [MIM:615278]: A form of cardiofaciocutaneous syndrome, a multiple congenital anomaly disorder characterized by a distinctive facial appearance, heart defects and intellectual disability. Heart defects include pulmonic stenosis, atrial septal defects and hypertrophic cardiomyopathy. Some affected individuals present with ectodermal abnormalities such as sparse, friable hair, hyperkeratotic skin lesions and a generalized ichthyosis-like condition. Typical facial features are similar to Noonan syndrome. They include high forehead with bitemporal constriction, hypoplastic supraorbital ridges, downslanting palpebral fissures, a depressed nasal bridge, and posteriorly angulated ears with prominent helices. CFC2 patients often do not have the skin abnormalities, such as ichthyosis, hyperkeratosis, and hemangioma observed in CFC1. {ECO:0000269|PubMed:16474404, ECO:0000269|PubMed:16474405, ECO:0000269|PubMed:17056636, ECO:0000269|PubMed:20949621, ECO:0000269|PubMed:21797849}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: KRAS mutations are involved in cancer development. {ECO:0000269|PubMed:14534542, ECO:0000269|PubMed:1553789, ECO:0000269|PubMed:16533793, ECO:0000269|PubMed:24623306, ECO:0000269|PubMed:3034404, ECO:0000269|PubMed:3627975, ECO:0000269|PubMed:6092920, ECO:0000269|PubMed:6695174, ECO:0000269|PubMed:7773929}.; DISEASE: Oculoectodermal syndrome (OES) [MIM:600268]: A syndrome characterized by the association of epibulbar dermoids and aplasia cutis congenita. Affected individuals show multiple, asymmetric, atrophic, non-scarring and hairless regions that may be associated with hamartomas. Ectodermal changes include linear hyperpigmentation that may follow the lines of Blaschko and rarely epidermal nevus-like lesions. Epibulbar dermoids may be uni-or bilateral. Additional ocular anomalies such as skin tags of the upper eyelid, rarely optic nerve or retinal changes, and microphthalmia can be present. The phenotypic expression is highly variable, and various other abnormalities have occasionally been reported including growth failure, lymphedema, cardiovascular defects, as well as neurodevelopmental symptoms like developmental delay, epilepsy, learning difficulties, and behavioral abnormalities. Benign tumor-like lesions such as nonossifying fibromas of the long bones and giant cell granulomas of the jaws have repeatedly been observed and appear to be age-dependent, becoming a common manifestation in individuals aged 5 years or older. {ECO:0000269|PubMed:25808193, ECO:0000269|PubMed:26970110, ECO:0000269|PubMed:30891959}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Schimmelpenning-Feuerstein-Mims syndrome (SFM) [MIM:163200]: A disease characterized by sebaceous nevi, often on the face, associated with variable ipsilateral abnormalities of the central nervous system, ocular anomalies, and skeletal defects. Many oral manifestations have been reported, not only including hypoplastic and malformed teeth, and mucosal papillomatosis, but also ankyloglossia, hemihyperplastic tongue, intraoral nevus, giant cell granuloma, ameloblastoma, bone cysts, follicular cysts, oligodontia, and odontodysplasia. Sebaceous nevi follow the lines of Blaschko and these can continue as linear intraoral lesions, as in mucosal papillomatosis. {ECO:0000269|PubMed:30891959}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Cell wall biogenesis/degradation; Lipid transport; Membrane; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 77237 Length 717 Aromaticity 0.09 Instability index 33.34 Isoelectric point 8.65 Charge (pH=7) 4.3 2D Binding mode Binding energy (Kcal/mol) -5.96  Molscript Map 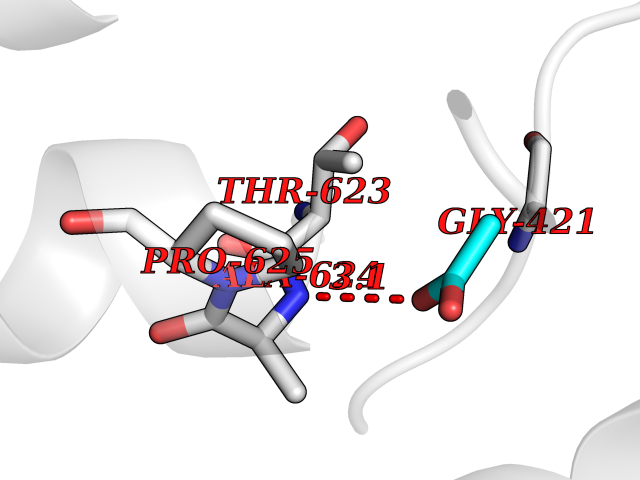 Pymol Map 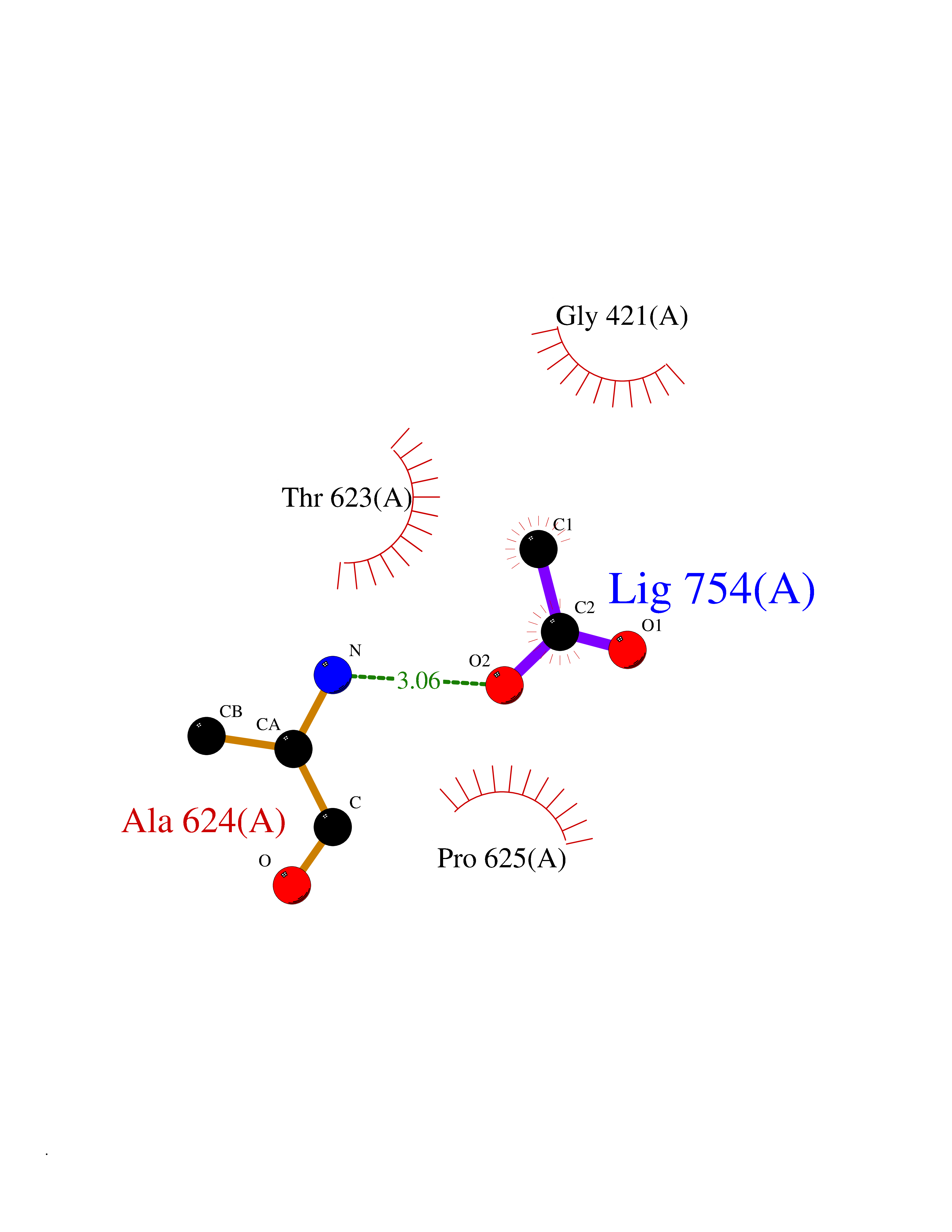 Ligplot Map 3D Binding mode Sequence MFAWWGRTVYRYRFIVIGVMVALCLGGGVFGLSLGKHVTQSGFYDDGSQSVQASVLGDQVYGRDRSGHIVAIFQAPAGKTVDDPAWSKKVVDELNRFQQDHPDQVLGWAGYLRASQATGMATADKKYTFVSIPLKGDDDDTILNNYKAIAPDLQRLDGGTVKLAGLQPVAEALTGTIATDQRRMEVLALPLVAVVLFFVFGGVIAAGLPVMVGGLCIAGALGIMRFLAIFGPVHYFAQPVVSLIGLGIAIDYGLFIVSRFREEIAEGYDTETAVRRTVITAGRTVTFSAVLIVASAIGLLLFPQGFLKSLTYATIASVMLSAILSITVLPACLGILGKHVDAEEVEAGFWGKLVNRVMKRPVLFAAPIVIIMILLIIPVGKLSLGGISEKYLPPTNSVRQAQEEFDKLFPGYRTNPLTLVIQTSNHQPVTDAQIADIRSKAMAIGGFIEPDNDPANMWQERAYAVGASKDPSVRVLQNGLINPADASKKLTELRAITPPKGITVLVGGTPALELDSIHGLFAKMPLMVVILLTTTIVLMFLAFGSVVLPIKATLMSALTLGSTMGILTWIFVDGHFSKWLNFTPTPLTAPVIGLIIALVFGLSTDYEVFLVSRMVEARERGMSTQEAIRIGTAATGRIITAAALIVAVVAGAFVFSDLVMMKYLAFGLMAALLLDATVVRMFLVPSVMKLLGDDCWWAPRWARRLQTRIGLGEIHLP Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Pseudomonas Methionine gamma-lyase (Pseudo mdeA) | 1PG8 | 4.37 | |
Target general information Gen name Pseudo mdeA Organism Pseudomonas putida (Arthrobacter siderocapsulatus) Uniprot ID TTD ID Synonyms Pseudo MGL; L-methionine gamma-lyase; L-methioninase; Homocysteine desulfhydrase Protein family Trans-sulfuration enzymes family, L-methionine gamma-lyase subfamily Biochemical class Carbon-sulfur lyases Function Catalyzes the alpha,gamma-elimination of L-methionine to produce methanethiol, 2-oxobutanoate and ammonia. Is involved in L-methionine catabolism. In fact, shows a multicatalytic function since it also catalyzes gamma-replacement of L-methionine with thiol compounds, alpha,gamma-elimination and gamma-replacement reactions of L-homocysteine and its S-substituted derivatives, O-substituted-L-homoserines and DL-selenomethionine, and, to a lesser extent, alpha,beta-elimination and beta-replacement reactions of L-cysteine, S-methyl-L-cysteine, and O-acetyl-L-serine. Also catalyzes deamination and gamma-addition reactions of L-vinylglycine. Thus, the enzyme is able to cleave C-S, C-Se, and C-O bonds of sulfur, selenium, and oxygen amino acids, respectively. Related diseases Lecithin-cholesterol acyltransferase deficiency (LCATD) [MIM:245900]: A disorder of lipoprotein metabolism characterized by inadequate esterification of plasmatic cholesterol. Two clinical forms are recognized: complete LCAT deficiency and fish-eye disease. LCATD is generally referred to the complete form which is associated with absence of both alpha and beta LCAT activities resulting in esterification anomalies involving both HDL (alpha-LCAT activity) and LDL (beta-LCAT activity). It causes a typical triad of diffuse corneal opacities, target cell hemolytic anemia, and proteinuria with renal failure. {ECO:0000269|PubMed:11423760, ECO:0000269|PubMed:12957688, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:16051254, ECO:0000269|PubMed:16216249, ECO:0000269|PubMed:1681161, ECO:0000269|PubMed:1859405, ECO:0000269|PubMed:2370048, ECO:0000269|PubMed:7607641, ECO:0000269|PubMed:7711728, ECO:0000269|PubMed:8318557, ECO:0000269|PubMed:8432868, ECO:0000269|PubMed:8807342, ECO:0000269|PubMed:9007616, ECO:0000269|PubMed:9741700}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fish-eye disease (FED) [MIM:136120]: A disorder of lipoprotein metabolism due to partial lecithin-cholesterol acyltransferase deficiency that affects only alpha-LCAT activity. FED is characterized by low plasma HDL and corneal opacities due to accumulation of cholesterol deposits in the cornea ('fish-eye'). {ECO:0000269|PubMed:1516702, ECO:0000269|PubMed:1571050, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:1737840, ECO:0000269|PubMed:21901787, ECO:0000269|PubMed:8620346, ECO:0000269|PubMed:9261271}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04083 Interacts with NA EC number EC 4.4.1.11 Uniprot keywords 3D-structure; Direct protein sequencing; Lyase; Pyridoxal phosphate Protein physicochemical properties Chain ID A,C Molecular weight (Da) 85234.4 Length 796 Aromaticity 0.07 Instability index 37.1 Isoelectric point 6.21 Charge (pH=7) -11.34 2D Binding mode Binding energy (Kcal/mol) -5.96  Molscript Map 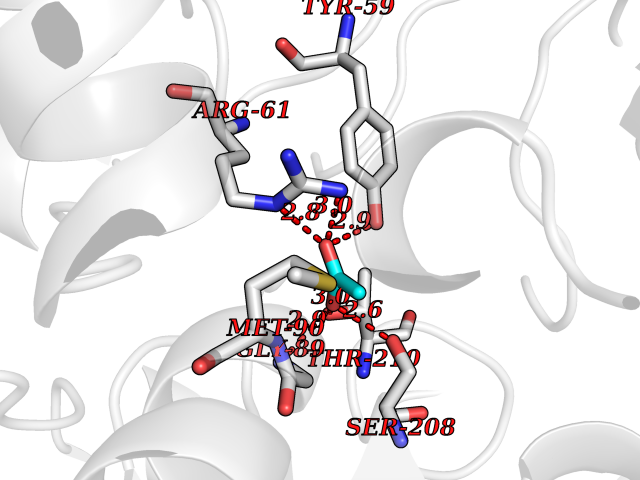 Pymol Map 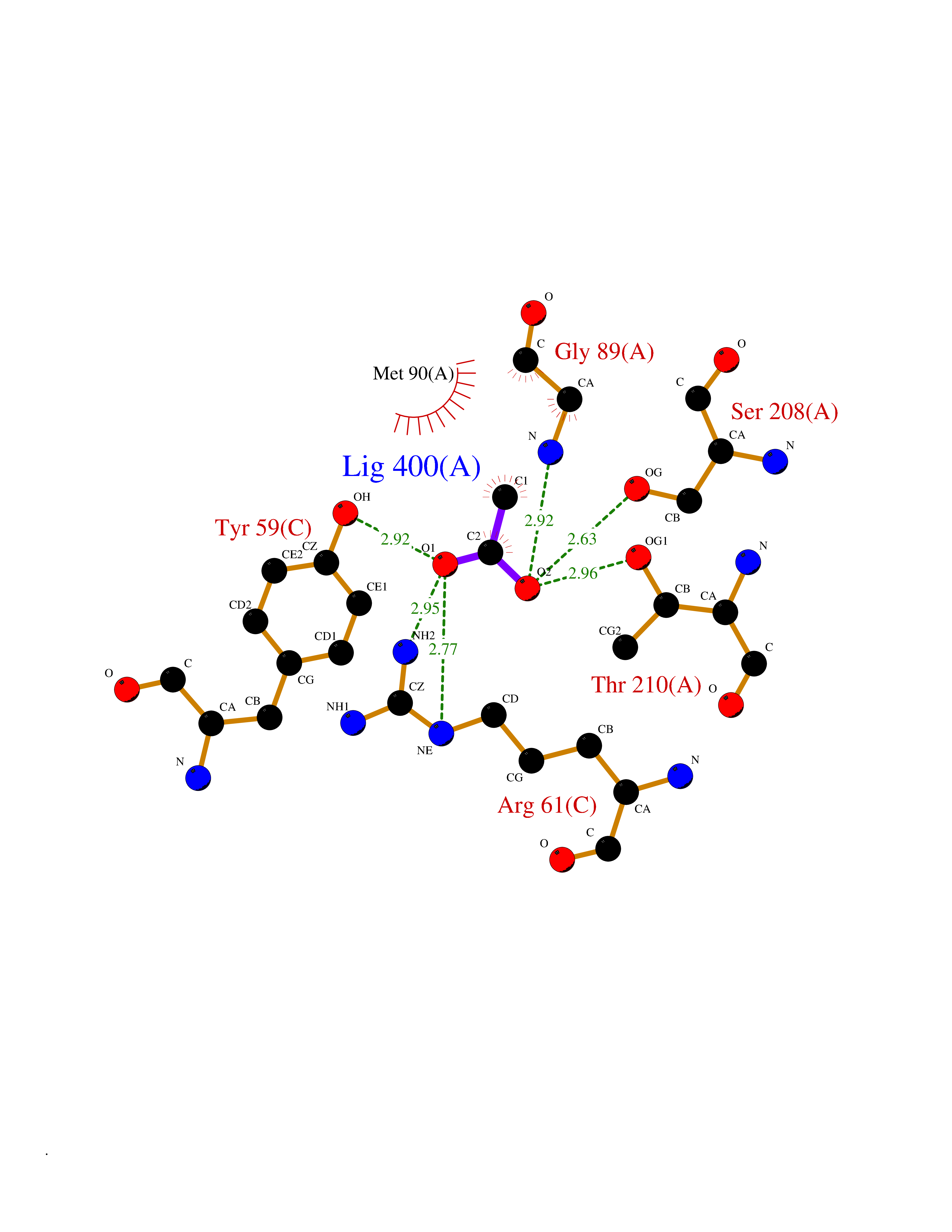 Ligplot Map 3D Binding mode Sequence MHGSNKLPGFATRAIHHGYDPQDHGGALVPPVYQTATFTFPTVEYGAACFAGEQAGHFYSRISNPTLNLLEARMASLEGGEAGLALASGMGAITSTLWTLLRPGDEVLLGNTLYGCTFAFLHHGIGEFGVKLRHVDMADLQALEAAMTPATRVIYFESPANPNMHMADIAGVAKIARKHGATVVVDNTYCTPYLQRPLELGADLVVHSATKYLSGHGDITAGIVVGSQALVDRIRLQGLKDMTGAVLSPHDAALLMRGIKTLNLRMDRHCANAQVLAEFLARQPQVELIHYPGLASFPQYTLARQQMSQPGGMIAFELKGGIGAGRRFMNALQLFSRAVSLGDAESLAQHPASMTHSSYTPEERAHYGISEGLVRLSVGLEDIDDLLADVQQALKASAMHGSNKLPGFATRAIHHGYDPQDHGGALVPPVYQTATFTFPTVEYGAACFAGEQAGHFYSRISNPTLNLLEARMASLEGGEAGLALASGMGAITSTLWTLLRPGDEVLLGNTLYGCTFAFLHHGIGEFGVKLRHVDMADLQALEAAMTPATRVIYFESPANPNMHMADIAGVAKIARKHGATVVVDNTYCTPYLQRPLELGADLVVHSATKYLSGHGDITAGIVVGSQALVDRIRLQGLKDMTGAVLSPHDAALLMRGIKTLNLRMDRHCANAQVLAEFLARQPQVELIHYPGLASFPQYTLARQQMSQPGGMIAFELKGGIGAGRRFMNALQLFSRAVSLGDAESLAQHPASMTHSSYTPEERAHYGISEGLVRLSVGLEDIDDLLADVQQALKASA Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Cerebron E3 ubiquitin ligase complex (CRL4-CRBN E3 ubiquitin ligase) | 4CI1 | 4.37 | |
Target general information Gen name CUL4A/CUL4B-DDB1-CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NA Protein family Cullin family Biochemical class NA Function NA Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P54253; Q86VP6; Q16531; Q92466; P08238; O94888; P55072 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Biological rhythms; DNA damage; DNA repair; Host-virus interaction; Isopeptide bond; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID B Molecular weight (Da) 42669.7 Length 368 Aromaticity 0.1 Instability index 44.94 Isoelectric point 8.72 Charge (pH=7) 6.58 2D Binding mode Binding energy (Kcal/mol) -5.96  Molscript Map  Pymol Map 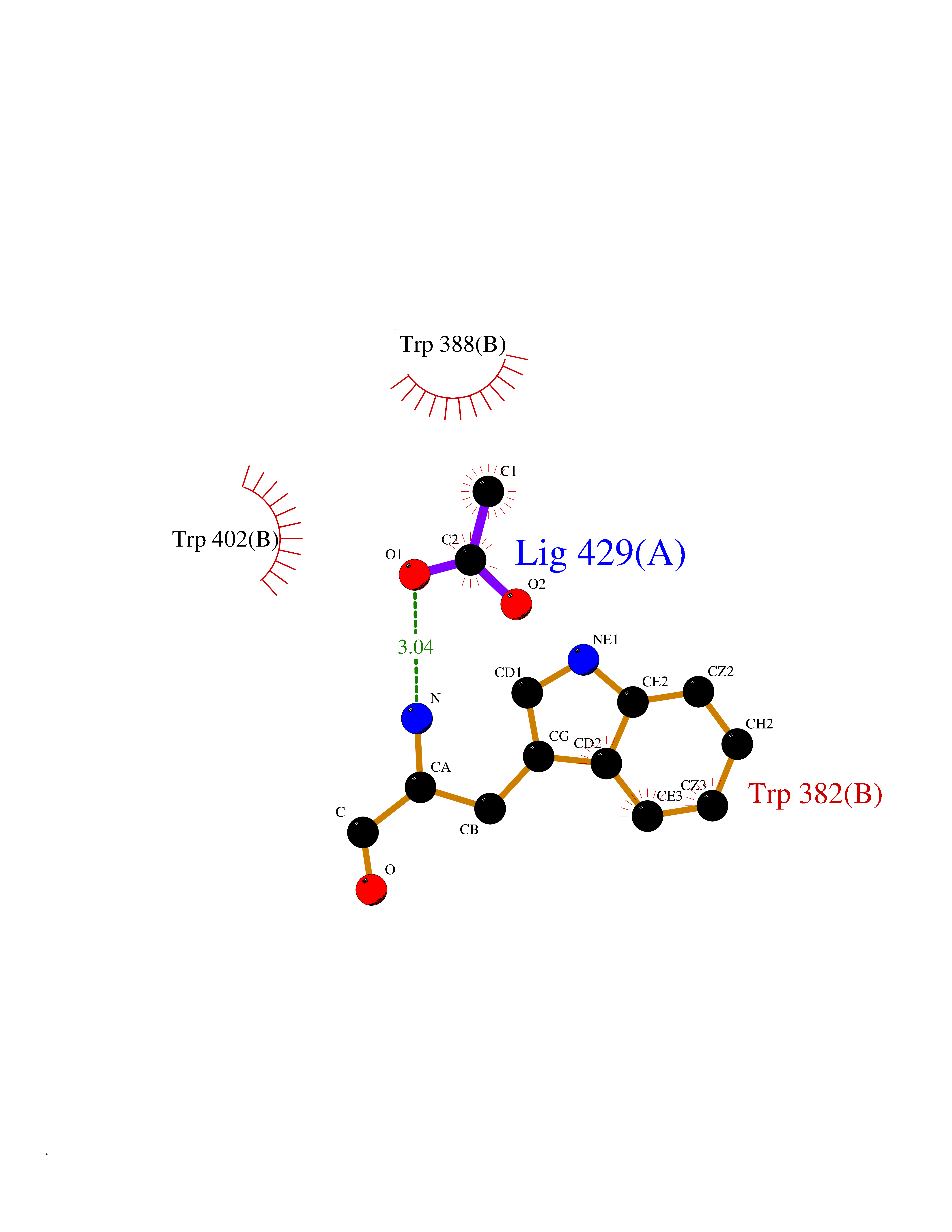 Ligplot Map 3D Binding mode Sequence MINFDTSLPTSHMYLGSDMEEFHGRTLHDDDSCQVIPVLPHVMVMLIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVREREAHFGTTAEIYAYREEQEYGIETVKVKAIGRQRFKVLEIRTQSDGIQQAKVQILPERVLPSTMSAVQLQSLSRRHIRAFRQWWQKYQKRKFHCASLTSWPPWLYSLYDAETLMERVKRQLHEWDENLKDESLPTNPIDFSYRVAACLPIDDALRIQLLKIGSAIQRLRELDIMNKTSLCCKQCQDTEITTKNEIFSLSLCGPMAAYVNPHGYIHETLTVYKACNLNLSGRPSTEHSWFPGYAWTIAQCRICGNHMGWKFTATKKDMSPQKFWGLTRSALLPR Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | SET domain containing 8 (KMT5A) | 5TEG | 4.37 | |
Target general information Gen name KMT5A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SETD8; SET8; SET07; SET domain-containing protein 8; PRSET7; PR/SET07; PR/SET domain-containing protein 07; PR-Set7; N-lysine methyltransferase KMT5A; Lysine-specific methylase 5A; Lysine N-methyltran Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, PR/SET subfamily Biochemical class Methyltransferase Function Specifically monomethylates 'Lys-20' of histone H4 (H4K20me1). H4K20me1 is enriched during mitosis and represents a specific tag for epigenetic transcriptional repression. Mainly functions in euchromatin regions, thereby playing a central role in the silencing of euchromatic genes. Required for cell proliferation, probably by contributing to the maintenance of proper higher-order structure of DNA during mitosis. Involved in chromosome condensation and proper cytokinesis. Nucleosomes are preferred as substrate compared to free histones. Mediates monomethylation of p53/TP53 at 'Lys-382', leading to repress p53/TP53-target genes. Plays a negative role in TGF-beta response regulation and a positive role in cell migration. Protein-lysine N-methyltransferase that monomethylates both histones and non-histone proteins. Related diseases Sick sinus syndrome 2 (SSS2) [MIM:163800]: The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS2 onset is in utero or at birth. {ECO:0000269|PubMed:15123648, ECO:0000269|PubMed:16407510, ECO:0000269|PubMed:20662977, ECO:0000269|PubMed:23103389}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 8 (BRGDA8) [MIM:613123]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:19165230}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Epilepsy, idiopathic generalized 18 (EIG18) [MIM:619521]: An autosomal dominant form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. EIG18 is characterized by onset of myoclonic seizures in infancy. Although the seizures remit, some patients may have later speech or cognitive impairment. {ECO:0000269|PubMed:30127718}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P62805; P07910; Q15672 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell cycle; Cell division; Chromatin regulator; Chromosome; Coiled coil; Direct protein sequencing; Methyltransferase; Mitosis; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A,D Molecular weight (Da) 19129.4 Length 167 Aromaticity 0.08 Instability index 49.18 Isoelectric point 7.88 Charge (pH=7) 1.37 2D Binding mode Binding energy (Kcal/mol) -5.96 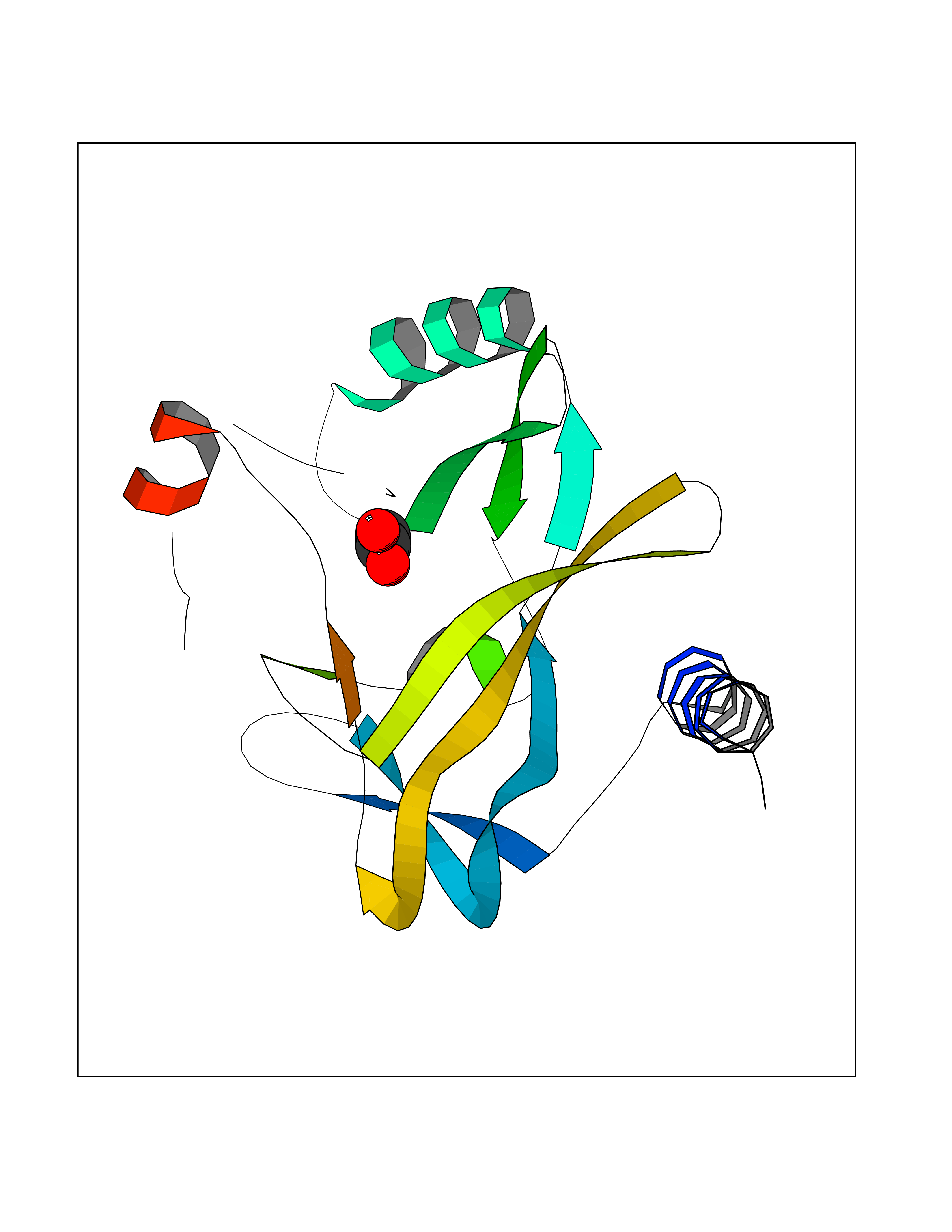 Molscript Map 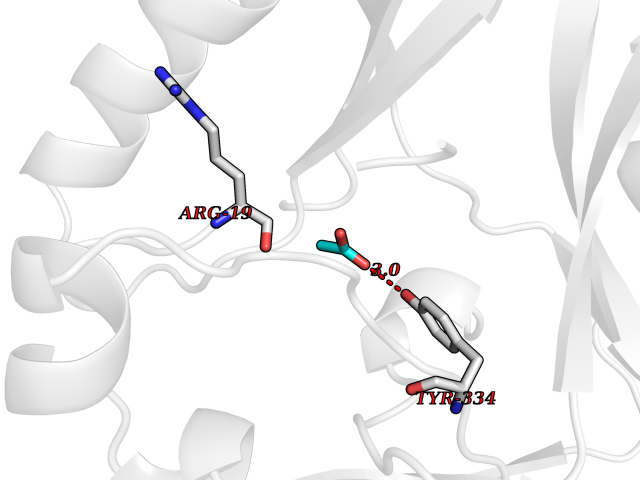 Pymol Map 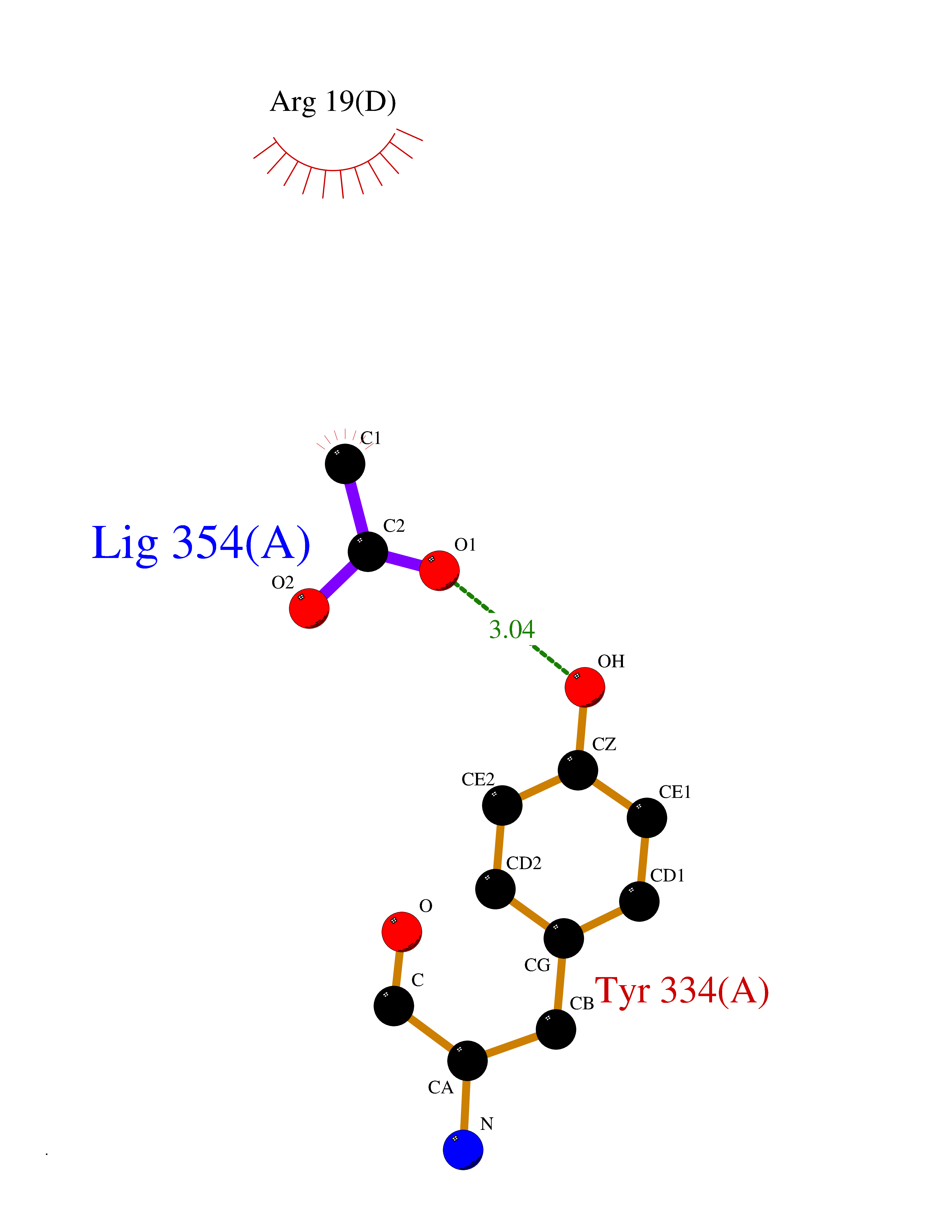 Ligplot Map 3D Binding mode Sequence KSKAELQSEERKRIDELIESGKEEGMKIDLIDGKGRGVIATKQFSRGDFVVEYHGDLIEITDAKKREALYAQDPSTGCYMYYFQYLSKTYCVDATRETNRLGRLINHSKSGNCQTKLHDIDGVPHLILIASRDIAAGEELLYDYGDRSKASIEAHPWLKHKRHRVLR Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Cholesterol 24-hydroxylase (CYP46A1) | 3MDR | 4.37 | |
Target general information Gen name CYP46A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cytochrome P450 46A1; CYP46; Cholesterol 24S-hydroxylase; Cholesterol 24-monooxygenase; CH24H Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function Primarily catalyzes the hydroxylation (with S stereochemistry) at C-24 of cholesterol side chain, triggering cholesterol diffusion out of neurons and its further degradation. By promoting constant cholesterol elimination in neurons, may activate the mevalonate pathway and coordinate the synthesis of new cholesterol and nonsterol isoprenoids involved in synaptic activity and learning. Further hydroxylates cholesterol derivatives and hormone steroids on both the ring and side chain of these molecules, converting them into active oxysterols involved in lipid signaling and biosynthesis. Acts as an epoxidase converting cholesta-5,24-dien-3beta-ol/desmosterol into (24S),25-epoxycholesterol, an abundant lipid ligand of nuclear NR1H2 and NR1H3 receptors shown to promote neurogenesis in developing brain. May also catalyze the oxidative metabolism of xenobiotics, such as clotrimazole. P450 monooxygenase that plays a major role in cholesterol homeostasis in the brain. Related diseases Spinocerebellar ataxia, autosomal recessive, with axonal neuropathy 1 (SCAN1) [MIM:607250]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCAN1 is an autosomal recessive cerebellar ataxia (ARCA) associated with peripheral axonal motor and sensory neuropathy, distal muscular atrophy, pes cavus and steppage gait as seen in Charcot-Marie-Tooth neuropathy. All affected individuals have normal intelligence. {ECO:0000269|PubMed:12244316, ECO:0000269|PubMed:15647511, ECO:0000269|PubMed:15920477, ECO:0000269|PubMed:16141202, ECO:0000269|PubMed:17948061}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.14.14.25 Uniprot keywords 3D-structure; Alternative splicing; Cell projection; Cholesterol metabolism; Endoplasmic reticulum; Heme; Iron; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism; Synapse; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 48977 Length 427 Aromaticity 0.1 Instability index 49.59 Isoelectric point 9.04 Charge (pH=7) 6.25 2D Binding mode Binding energy (Kcal/mol) -5.96  Molscript Map 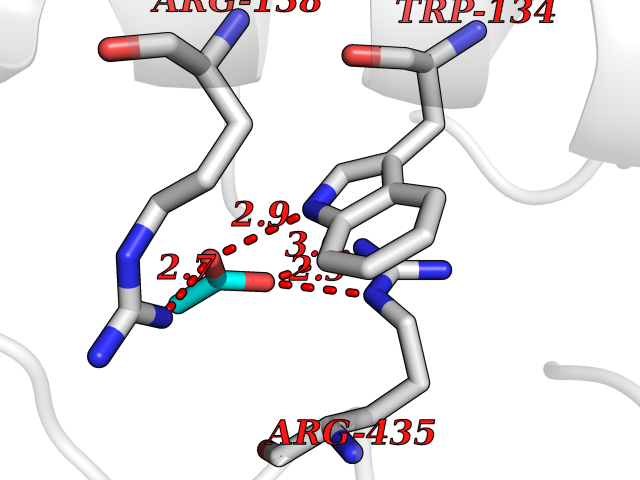 Pymol Map 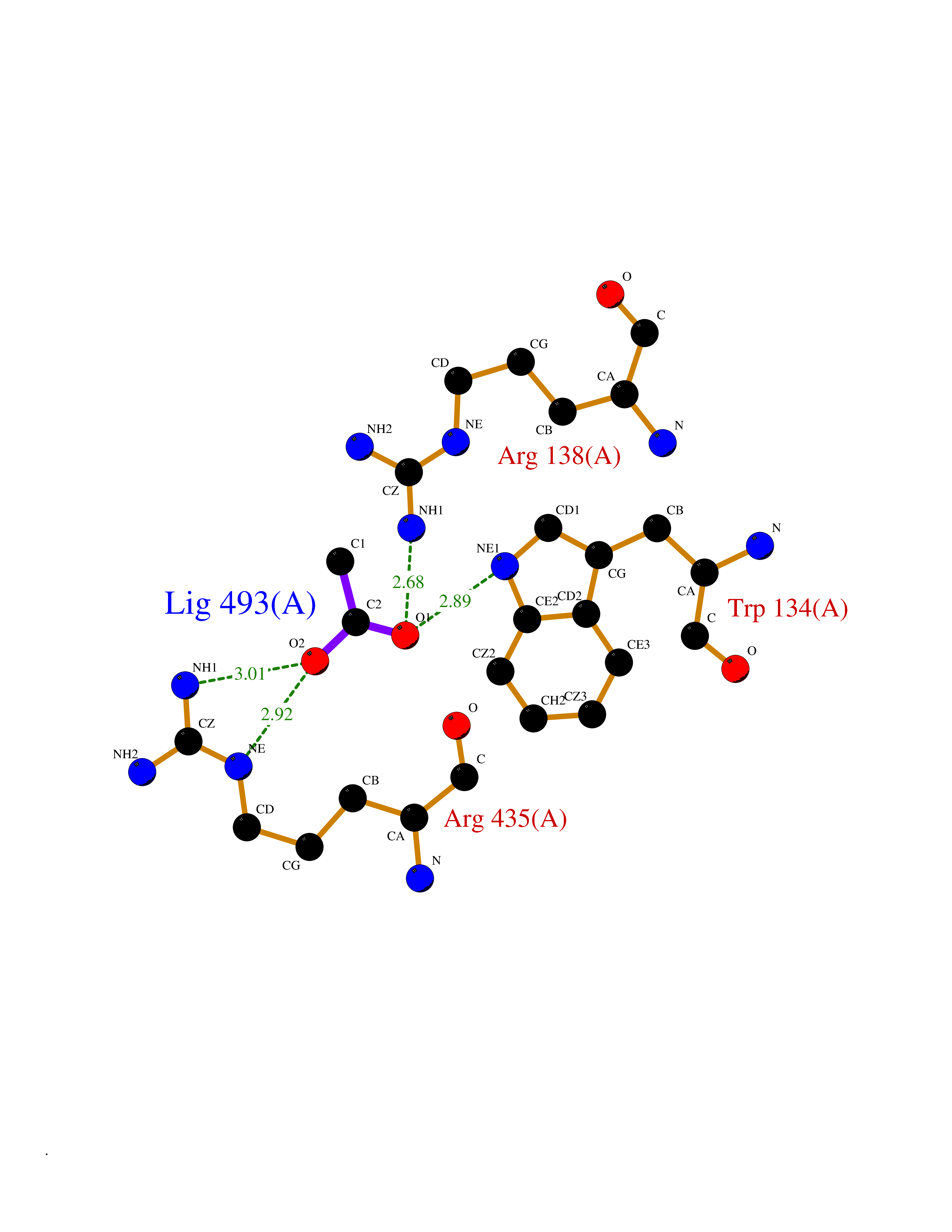 Ligplot Map 3D Binding mode Sequence RVLQDVFLDWAKKYGPVVRVNVFHKTSVIVTSPESVKKFLMSTKYNKDSKMYRALQTVFGERLFGQGLVSECNYERWHKQRRVIDLAFSRSSLVSLMETFNEKAEQLVEILEAKADGQTPVSMQDMLTYTAMDILAKAAFGMETSMLLGAQKPLSQAVKLMLEGITASRNTKRKQLREVRESIRFLRQVGRDWVQRRREALKRGEEVPADILTQILKAEEGAQDDEGLLDNFVTFFIAGHETSANHLAFTVMELSRQPEIVARLQAEVDEVIGSKRYLDFEDLGRLQYLSQVLKESLRLYPPAWGTFRLLEEETLIDGVRVPGNTPLLFSTYVMGRMDTYFEDPLTFNPDRFGPGAPKPRFTYFPFSLGHRSCIGQQFAQMEVKVVMAKLLQRLEFRLVPGQRFGLQEQATLKPLDPVLCTLRPRGW Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Cathepsin L (CTSL) | 3BC3 | 4.37 | |
Target general information Gen name CTSL Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Major excreted protein; MEP; Cathepsin L1; CTSL1 Protein family Peptidase C1 family Biochemical class Peptidase Function Important for the overall degradation of proteins in lysosomes. Related diseases Charcot-Marie-Tooth disease, axonal, 2DD (CMT2DD) [MIM:618036]: A dominant axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:29499166}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hypomagnesemia, seizures, and impaired intellectual development 2 (HOMGSMR2) [MIM:618314]: An autosomal dominant disease characterized by generalized seizures in infancy, severe hypomagnesemia, and renal magnesium wasting. Seizures persist despite magnesium supplementation and are associated with significant intellectual disability. {ECO:0000269|PubMed:30388404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07477; DB12010; DB03661; DB14962 Interacts with O60911; O43765; P0DTC2; P59594; G5EFH4 EC number EC 3.4.22.15 Uniprot keywords 3D-structure; Alternative initiation; Cell membrane; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Glycoprotein; Host-virus interaction; Hydrolase; Lysosome; Membrane; Nucleus; Protease; Proteomics identification; Reference proteome; Secreted; Signal; Thiol protease; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 23519.9 Length 214 Aromaticity 0.12 Instability index 30.95 Isoelectric point 4.79 Charge (pH=7) -9.97 2D Binding mode Binding energy (Kcal/mol) -5.97  Molscript Map 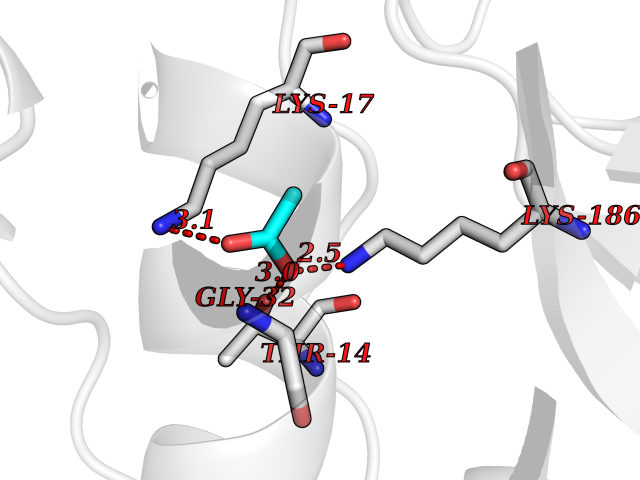 Pymol Map 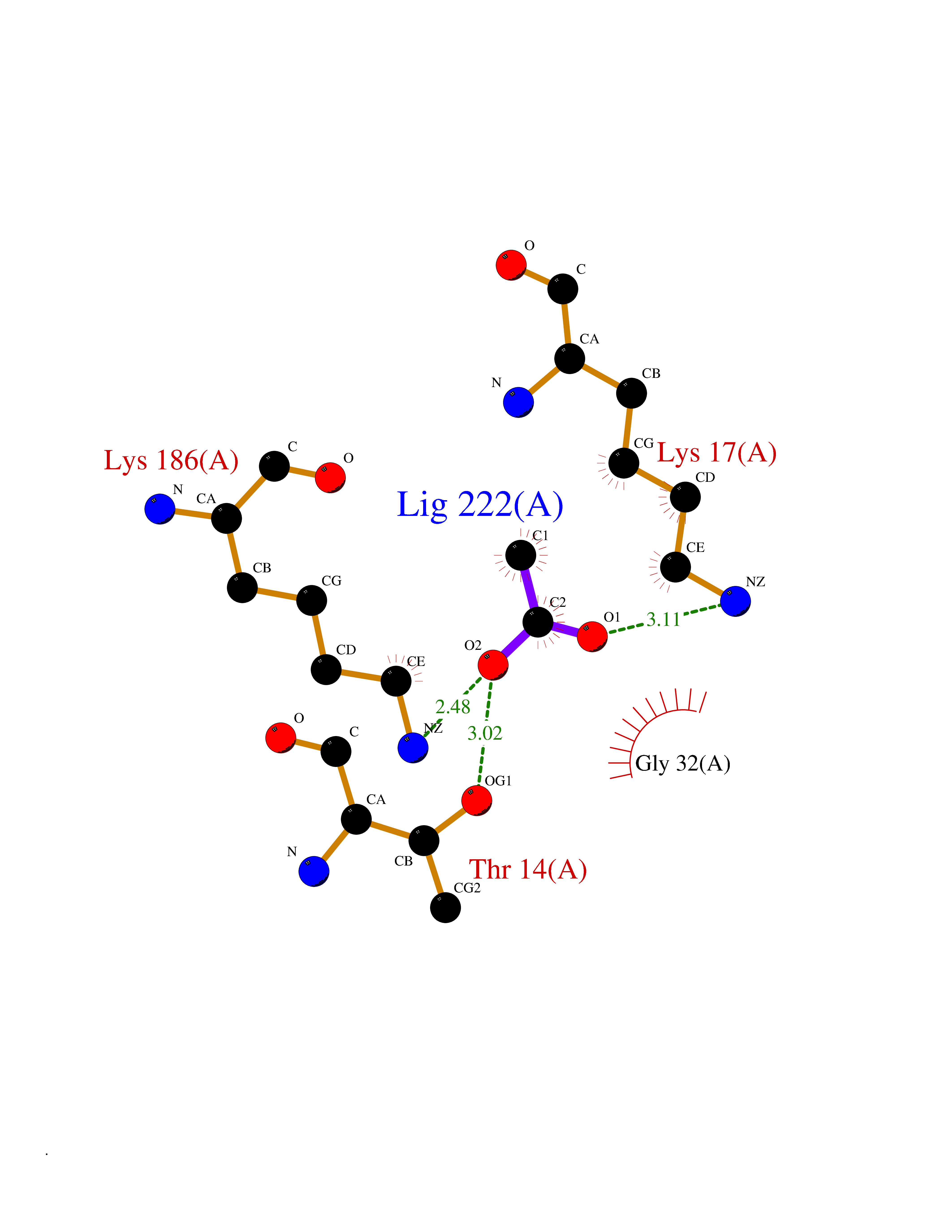 Ligplot Map 3D Binding mode Sequence APRSVDWREKGYVTPVKNQGQCGSWAFSATGALEGQMFRKTGRLISLSEQNLVDCSGPQGNEGCNGGLMDYAFQYVQDNGGLDSEESYPYEATEESCKYNPKYSVANDTGFVDIPKQEKALMKAVATVGPISVAIDAGHESFLFYKEGIYFEPDCSSEDMDHGVLVVGYGFESNKYWLVKNSWGEEWGMGGYVKMAKDRRNHCGIASAASYPTV Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Phenylethanolamine N-methyltransferase (PNMT) | 2G72 | 4.37 | |
Target general information Gen name PNMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNMTase; PENT; Noradrenaline N-methyltransferase Protein family Class I-like SAM-binding methyltransferase superfamily, NNMT/PNMT/TEMT family Biochemical class NA Function Converts noradrenaline to adrenaline. Related diseases A chromosomal aberration involving TRIM24/TIF1 is found in papillary thyroid carcinomas (PTCs). Translocation t(7;10)(q32;q11) with RET. The translocation generates the TRIM24/RET (PTC6) oncogene. {ECO:0000269|PubMed:10439047}. Drugs (DrugBank ID) DB08129; DB08128; DB07739; DB07798; DB07747; DB03468; DB08550; DB03824; DB04273; DB07906; DB07597; DB09571; DB00968; DB08631; DB01752; DB08654 Interacts with Q9P2G9-2; Q8TBB1 EC number EC 2.1.1.28 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 29198.9 Length 264 Aromaticity 0.09 Instability index 54.33 Isoelectric point 5.91 Charge (pH=7) -3.69 2D Binding mode Binding energy (Kcal/mol) -5.96  Molscript Map 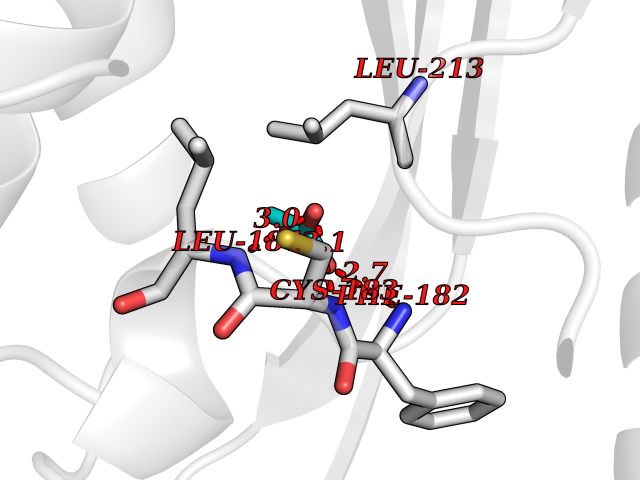 Pymol Map 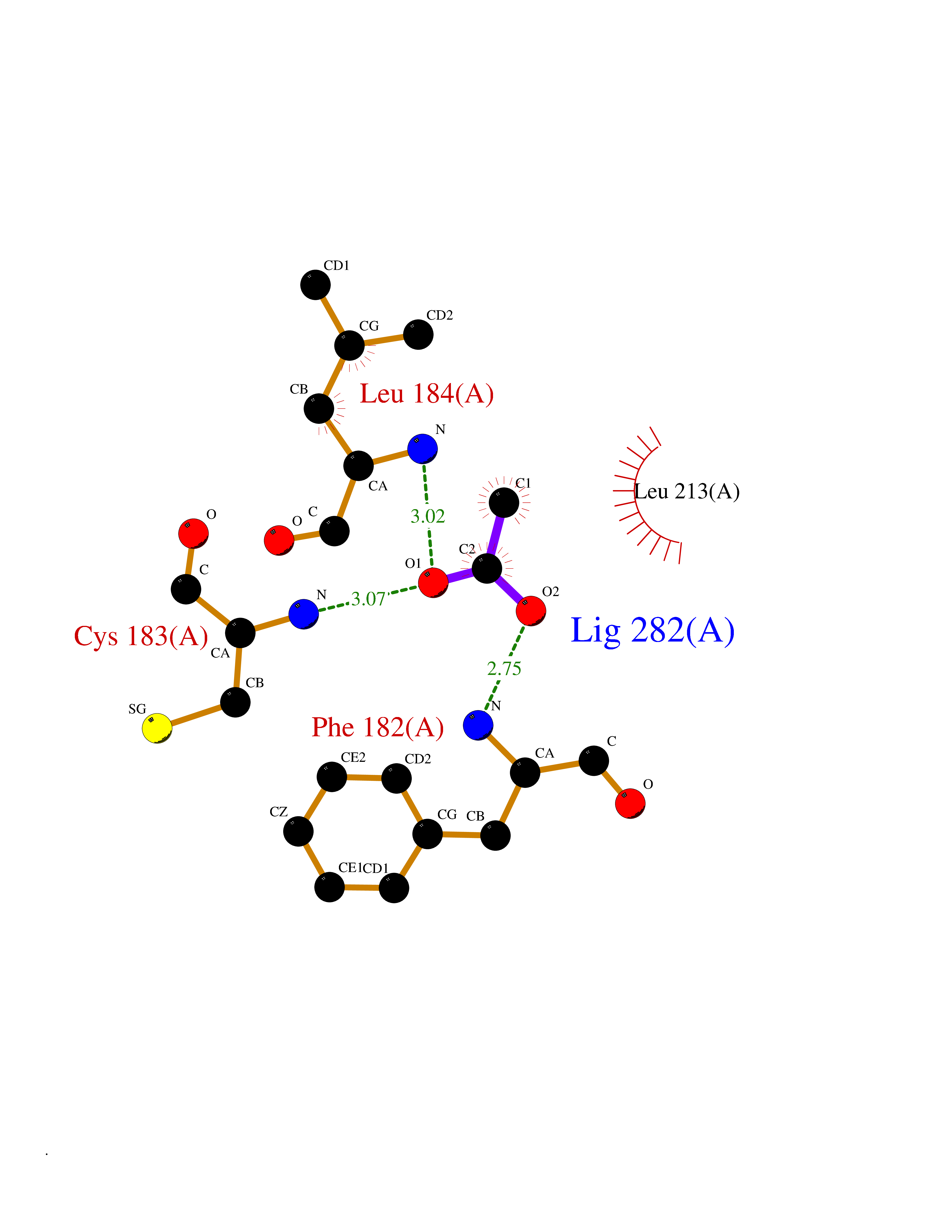 Ligplot Map 3D Binding mode Sequence APGQAAVASAYQRFEPRAYLRNNYAPPRGDLCNPNGVGPWKLRCLAQTFATGEVSGRTLIDIGSGPTVYQLLSACSHFEDITMTDFLEVNRQELGRWLQEEPGAFNWSMYSQHACLIEGKGECWQDKERQLRARVKRVLPIDVHQPQPLGAGSPAPLPADALVSAFCLEAVSPDLASFQRALDHITTLLRPGGHLLLIGALEESWYLAGEARLTVVPVSEEEVREALVRSGYKVRDLRTYIMPAHLQTGVDDVKGVFFAWAQKV Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Hepatitis A virus cellular receptor 2 (TIM3) | 7M3Z | 4.37 | |
Target general information Gen name Hepatitis A virus HAVCR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms TIMD3; TIMD-3; TIM3; TIM-3; T-cell membrane protein 3; T-cell immunoglobulin mucin receptor 3; T-cell immunoglobulin and mucin domain-containing protein 3; HAVcr-2; HAVCR2; CD366 Protein family Immunoglobulin superfamily, TIM family Biochemical class Immunoglobulin Function Generally accepted to have an inhibiting function. Reports on stimulating functions suggest that the activity may be influenced by the cellular context and/or the respective ligand. Regulates macrophage activation. Inhibits T-helper type 1 lymphocyte (Th1)-mediated auto- and alloimmune responses and promotes immunological tolerance. In CD8+ cells attenuates TCR-induced signaling, specifically by blocking NF-kappaB and NFAT promoter activities resulting in the loss of IL-2 secretion. The function may implicate its association with LCK proposed to impair phosphorylation of TCR subunits, and/or LGALS9-dependent recruitment of PTPRC to the immunological synapse. In contrast, shown to activate TCR-induced signaling in T-cells probably implicating ZAP70, LCP2, LCK and FYN. Expressed on Treg cells can inhibit Th17 cell responses. Receptor for LGALS9. Binding to LGALS9 is believed to result in suppression of T-cell responses; the resulting apoptosis of antigen-specific cells may implicate HAVCR2 phosphorylation and disruption of its association with BAG6. Binding to LGALS9 is proposed to be involved in innate immune response to intracellular pathogens. Expressed on Th1 cells interacts with LGALS9 expressed on Mycobacterium tuberculosis-infected macrophages to stimulate antibactericidal activity including IL-1 beta secretion and to restrict intracellular bacterial growth. However, the function as receptor for LGALS9 has been challenged. Also reported to enhance CD8+ T-cell responses to an acute infection such as by Listeria monocytogenes. Receptor for phosphatidylserine (PtSer); PtSer-binding is calcium-dependent. May recognize PtSer on apoptotic cells leading to their phagocytosis. Mediates the engulfment of apoptotic cells by dendritic cells. Expressed on T-cells, promotes conjugation but not engulfment of apoptotic cells. Expressed on dendritic cells (DCs) positively regulates innate immune response and in synergy with Toll-like receptors promotes secretion of TNF-alpha. In tumor-imfiltrating DCs suppresses nucleic acid-mediated innate immune repsonse by interaction with HMGB1 and interfering with nucleic acid-sensing and trafficking of nucleid acids to endosomes. Expressed on natural killer (NK) cells acts as a coreceptor to enhance IFN-gamma production in response to LGALS9. In contrast, shown to suppress NK cell-mediated cytotoxicity. Negatively regulates NK cell function in LPS-induced endotoxic shock. Cell surface receptor implicated in modulating innate and adaptive immune responses. Related diseases May be involved in T-cell exhaustion associated with chronic viral infections such as with human immunodeficiency virus (HIV) and hepatitic C virus (HCV). {ECO:0000269|PubMed:19001139, ECO:0000269|PubMed:19587053}.; DISEASE: T-cell lymphoma, subcutaneous panniculitis-like (SPTCL) [MIM:618398]: An uncommon form of T-cell non-Hodgkin lymphoma, in which cytotoxic CD8+ T-cells infiltrate subcutaneous adipose tissue, and rimming adipocytes in a lace-like pattern. Affected individuals typically present with multiple subcutaneous nodules, systemic B-cell symptoms, and, in a subset of cases, autoimmune disorders, most commonly systemic lupus erythematosus. A subset of patients develop hemophagocytic lymphohistiocytosis. SPTCL transmission pattern is consistent with autosomal recessive inheritance with incomplete penetrance. {ECO:0000269|PubMed:30374066, ECO:0000269|PubMed:30792187, ECO:0000269|Ref.2}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P13688; Q96IW7; Q8N2M4 EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Alternative splicing; Cell junction; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Immunity; Immunoglobulin domain; Inflammatory response; Innate immunity; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 12286.9 Length 109 Aromaticity 0.12 Instability index 26.55 Isoelectric point 5.04 Charge (pH=7) -2.58 2D Binding mode Binding energy (Kcal/mol) -5.96 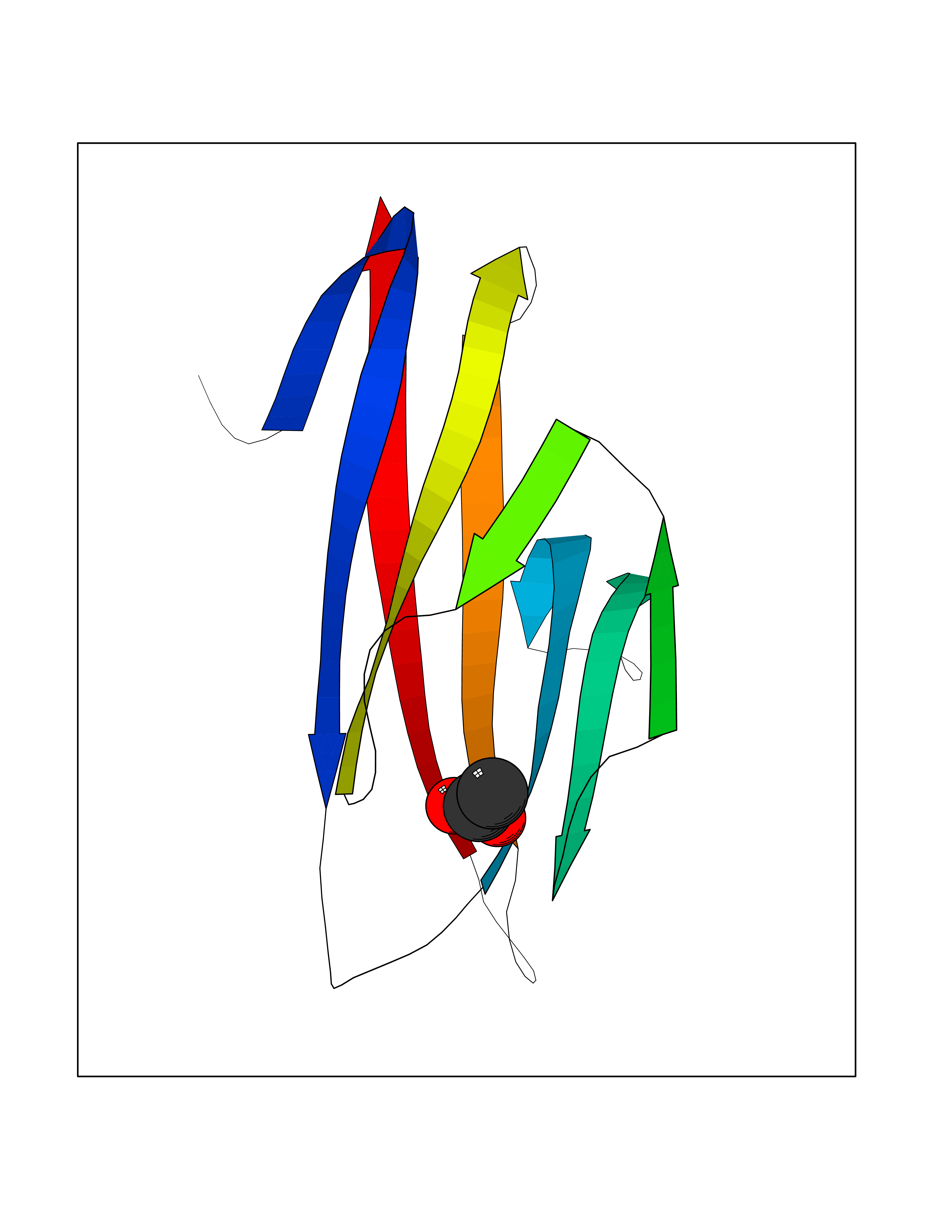 Molscript Map 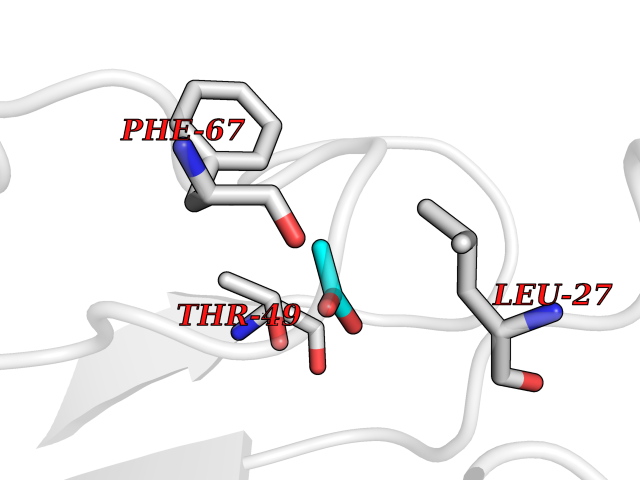 Pymol Map 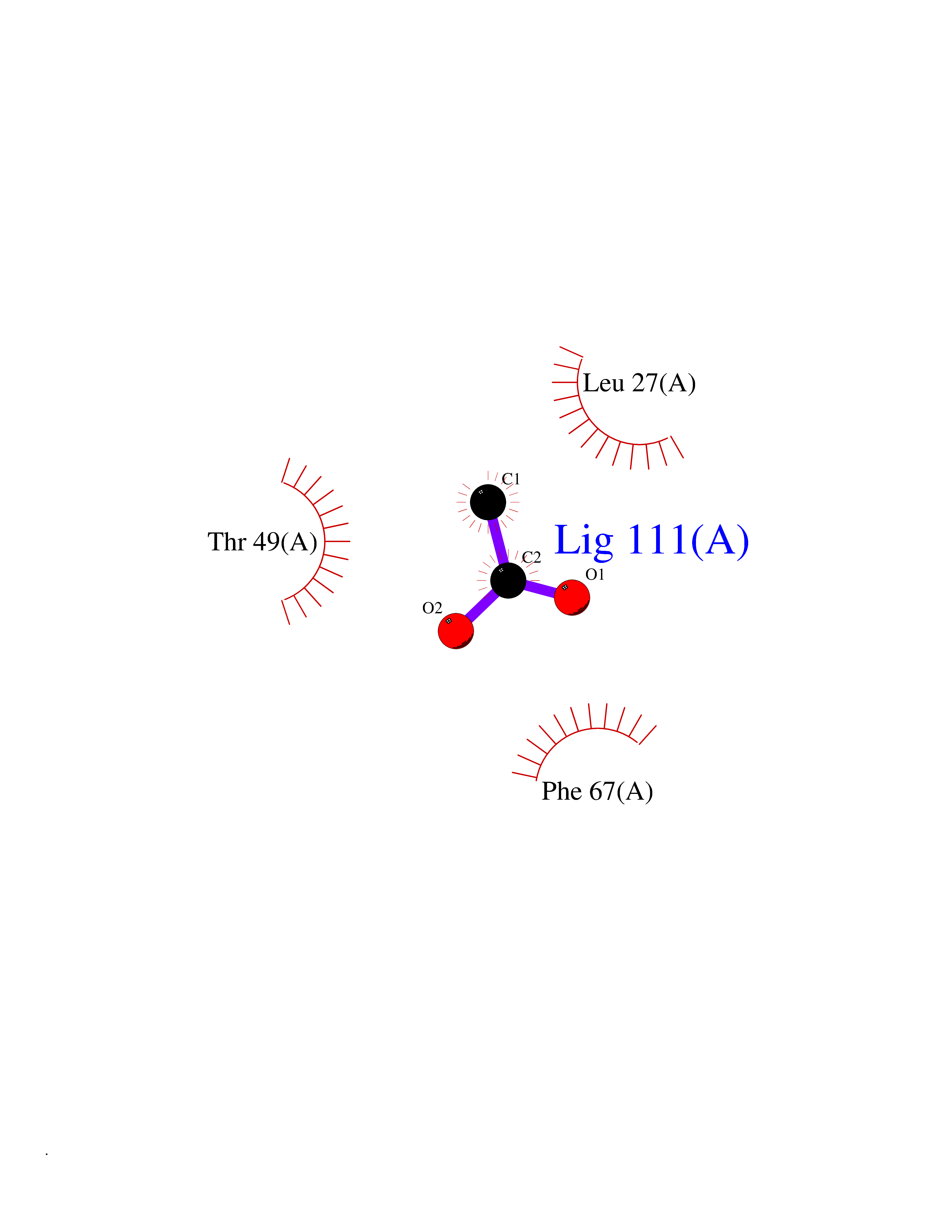 Ligplot Map 3D Binding mode Sequence SEVEYRAEVGQNAYLPCFYTPAAPGNLVPVCWGKGACPVFECGNVVLRTDERDVNYWTSRYWLNGDFRKGDVSLTIENVTLADSGIYCCRIQIPGIMNDEKFNLKLVIK Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Purine nucleoside phosphorylase (PNP) | 4EAR | 4.36 | |
Target general information Gen name PNP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNP; Inosine phosphorylase Protein family PNP/MTAP phosphorylase family Biochemical class Pentosyltransferase Function The purine nucleoside phosphorylases catalyze the phosphorolytic breakdown of the N-glycosidic bond in the beta- (deoxy)ribonucleoside molecules, with the formation of the corresponding free purine bases and pentose-1-phosphate. Related diseases Purine nucleoside phosphorylase deficiency (PNPD) [MIM:613179]: A disorder that interrupts both the catabolism of inosine into hypoxanthine and guanosine into guanine, and leads to the accumulation of guanosine, inosine, and their deoxified by-products. The main clinical presentation is recurrent infections due to severe T-cell immunodeficiency. Some patients also have neurologic impairment. {ECO:0000269|PubMed:1384322, ECO:0000269|PubMed:3029074, ECO:0000269|PubMed:8931706}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03881; DB03551; DB02222; DB02391; DB03609; DB01667; DB04260; DB02796; DB04753; DB00640; DB00242; DB00900; DB06185; DB02377; DB02857; DB04754; DB04757; DB04076; DB02230; DB04335; DB02568; DB03101 Interacts with P05067; Q9UQM7; O14576-2; P06241; P14136; Q92993-2; Q9BXM7; P00491; P17612; P63000; Q92673; Q15583 EC number EC 2.4.2.1 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Disease variant; Glycosyltransferase; Phosphoprotein; Proteomics identification; Purine salvage; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 31849.2 Length 288 Aromaticity 0.1 Instability index 34.77 Isoelectric point 6.42 Charge (pH=7) -1.63 2D Binding mode Binding energy (Kcal/mol) -5.94 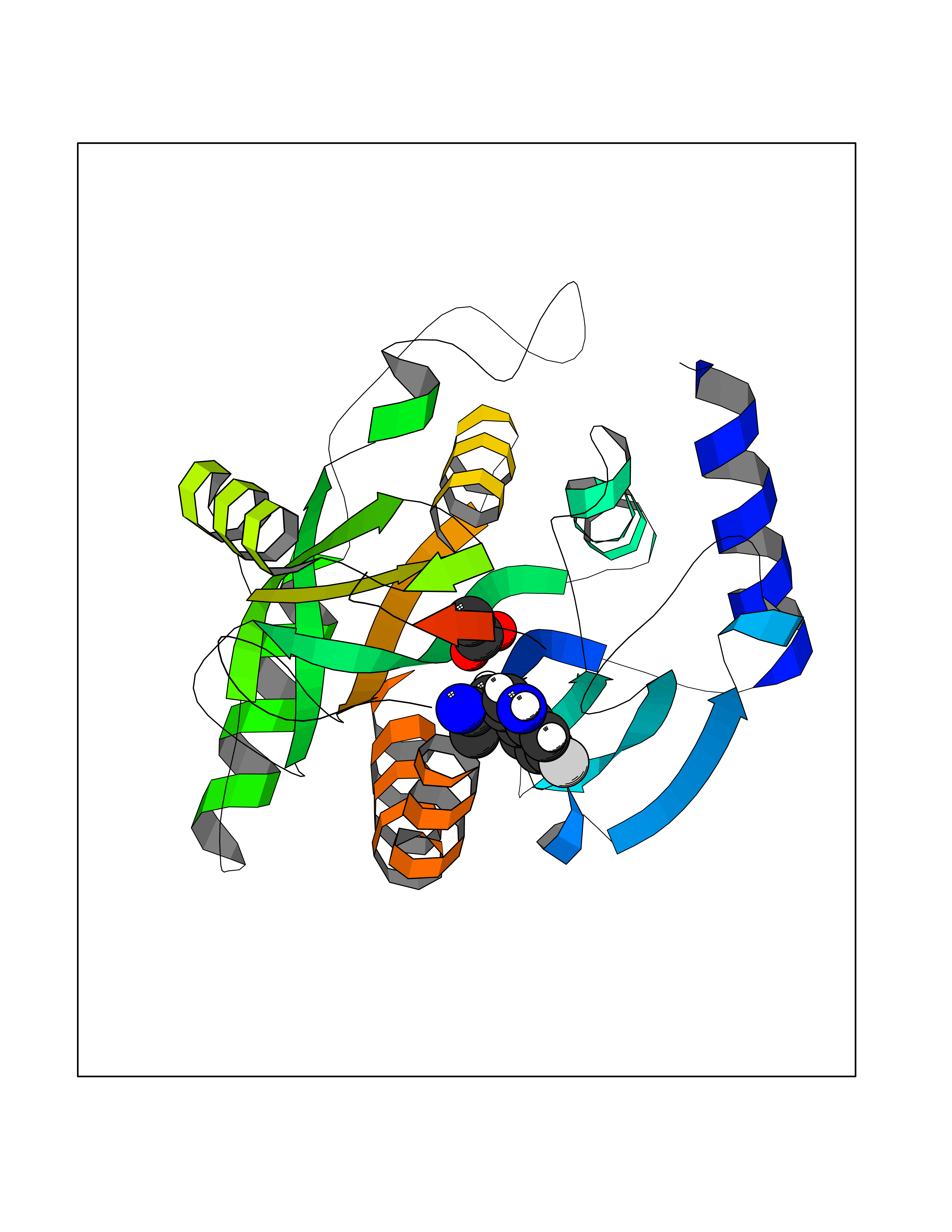 Molscript Map 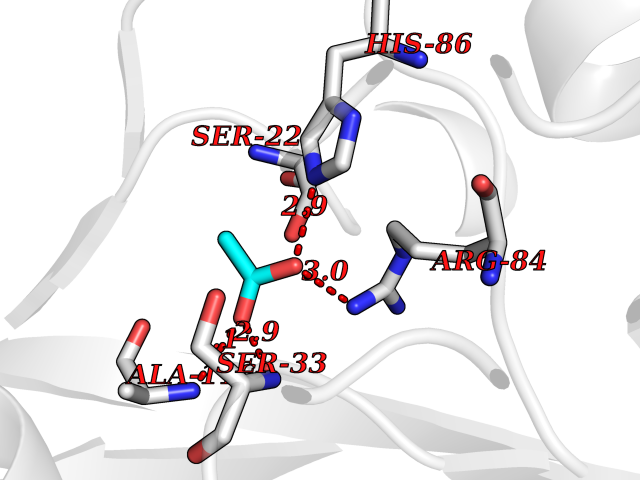 Pymol Map 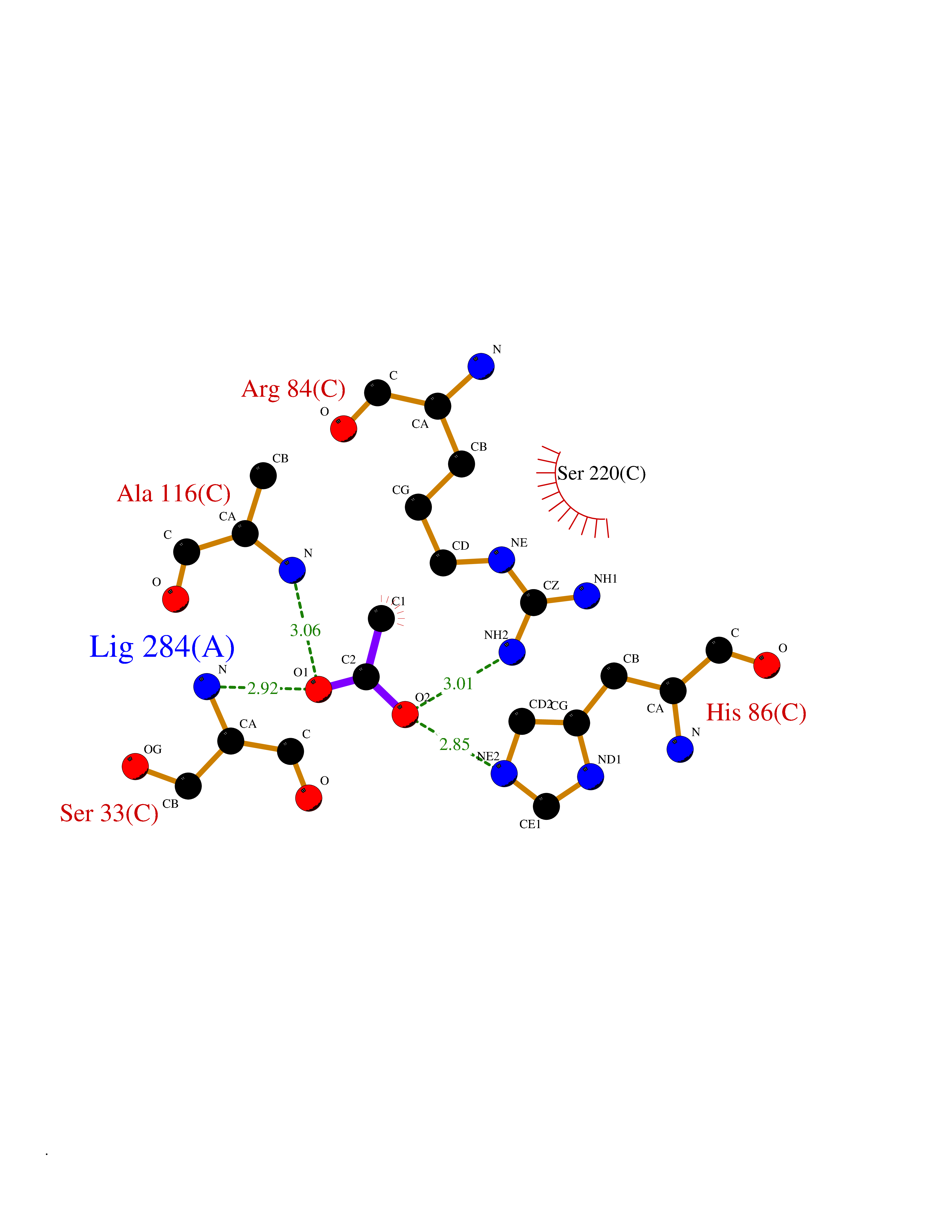 Ligplot Map 3D Binding mode Sequence GYTYEDYKNTAEYLLSHTKHRPQVAIICGSGLGGLTDKLTQAQIFDYSEIPNFPRSTVPGHAGRLVFGFLNGRACVMMQGRFHMYEGYPLYKVTFPVRVFHLLGVDTLVVTNAAGGLNPKFEVGDIMLIRDHINLPGFSGQNPLRGPNDERFGDRFPAMSDAYDRTMRQRALSTYKQMGEQRELQEGTYVMVAGPSFETVAECRVLQKLGADAVGMSTVPEVIVARHCGLRVFGFSLITNKVIMDYESLEKANXEEVLAAGKQAAQKLEQFVSILMASIDRFPAMSDA Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | NH(3)-dependent NAD(+) synthetase | 1KQP | 4.36 | |
Target general information Gen name nadE Organism Bacillus subtilis (strain 168) Uniprot ID TTD ID NA Synonyms outB;BSU03130 Protein family NAD synthetase family Biochemical class Ligase Function ATP binding.Metal ion binding.NAD+ synthase (glutamine-hydrolyzing) activity.NAD+ synthase activity. Related diseases Leukodystrophy, hypomyelinating, 15 (HLD15) [MIM:617951]: An autosomal recessive disorder characterized by hypomyelinating leukodystrophy with thinning of the corpus callosum. Clinical features include motor and cognitive impairment appearing in the first or second decade of life, dystonia, ataxia, spasticity, and dysphagia. Most patients develop severe optic atrophy, and some have hearing loss. {ECO:0000269|PubMed:29576217}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02596; DB04099; DB00798 Interacts with NA EC number 6.3.1.5 Uniprot keywords 3D-structure; ATP-binding; Direct protein sequencing; Ligase; Magnesium; Metal-binding; NAD; Nucleotide-binding; Phosphoprotein; Reference proteome; Sporulation; Stress response Protein physicochemical properties Chain ID A,B Molecular weight (Da) 60509.3 Length 542 Aromaticity 0.08 Instability index 31.96 Isoelectric point 5.07 Charge (pH=7) -19.73 2D Binding mode Binding energy (Kcal/mol) -5.95  Molscript Map 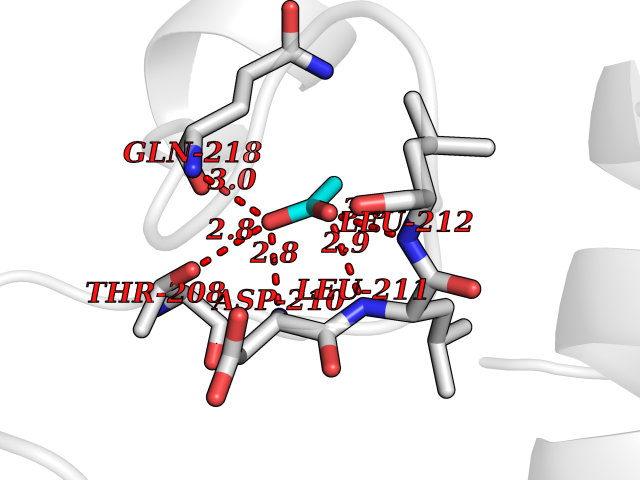 Pymol Map 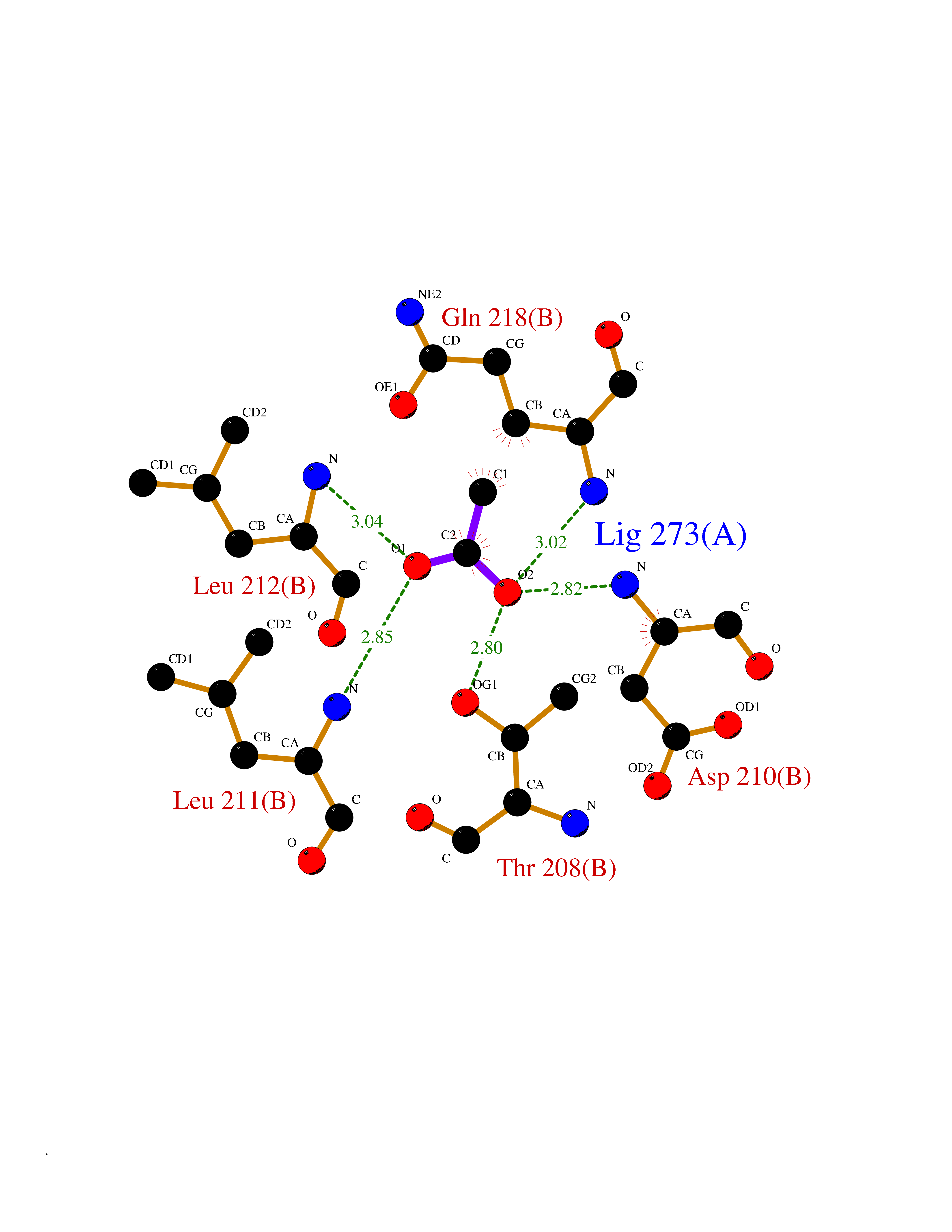 Ligplot Map 3D Binding mode Sequence SMQEKIMRELHVKPSIDPKQEIEDRVNFLKQYVKKTGAKGFVLGISGGQDSTLAGRLAQLAVESIREEGGDAQFIAVRLPHGTQQDEDDAQLALKFIKPDKSWKFDIKSTVSAFSDQYQQETGDQLTDFNKGNVKARTRMIAQYAIGGQEGLLVLGTDHAAEAVTGFFTKYGDGGADLLPLTGLTKRQGRTLLKELGAPERLYLKEPTADLLDEKPQQSDETELGISYDEIDDYLEGKEVSAKVSEALEKRYSMTEHKRQVPASMFDDWWKSMQEKIMRELHVKPSIDPKQEIEDRVNFLKQYVKKTGAKGFVLGISGGQDSTLAGRLAQLAVESIREEGGDAQFIAVRLPHGTQQDEDDAQLALKFIKPDKSWKFDIKSTVSAFSDQYQQETGDQLTDFNKGNVKARTRMIAQYAIGGQEGLLVLGTDHAAEAVTGFFTKYGDGGADLLPLTGLTKRQGRTLLKELGAPERLYLKEPTADLLDEKPQQSDETELGISYDEIDDYLEGKEVSAKVSEALEKRYSMTEHKRQVPASMFDDWWK Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Cholesterol desmolase (CYP11A1) | 3N9Z | 4.36 | |
Target general information Gen name CYP11A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms P450(scc); Cytochrome P450(scc); Cytochrome P450 11A1; Cholesterol side-chain cleavage enzyme, mitochondrial; CYPXIA1; CYP11A Protein family Adrenodoxin/putidaredoxin family Biochemical class Paired donor oxygen oxidoreductase Function Catalyzes the side-chain cleavage reaction of cholesterol to pregnenolone, the precursor of most steroid hormones. Related diseases Leukocyte adhesion deficiency 1 (LAD1) [MIM:116920]: LAD1 patients have recurrent bacterial infections and their leukocytes are deficient in a wide range of adhesion-dependent functions. {ECO:0000269|PubMed:1346613, ECO:0000269|PubMed:1347532, ECO:0000269|PubMed:1352501, ECO:0000269|PubMed:1590804, ECO:0000269|PubMed:1694220, ECO:0000269|PubMed:1968911, ECO:0000269|PubMed:20529581, ECO:0000269|PubMed:20549317, ECO:0000269|PubMed:7509236, ECO:0000269|PubMed:7686755, ECO:0000269|PubMed:9884339}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00648 Interacts with NA EC number EC 1.14.15.6 Uniprot keywords 2Fe-2S; 3D-structure; Acetylation; Cholesterol metabolism; Electron transport; Iron; Iron-sulfur; Lipid metabolism; Metal-binding; Mitochondrion; Phosphoprotein; Proteomics identification; Reference proteome; Steroid metabolism; Steroidogenesis; Sterol metabolism; Transit peptide; Transport Protein physicochemical properties Chain ID C,D Molecular weight (Da) 54754.5 Length 470 Aromaticity 0.13 Instability index 33.2 Isoelectric point 8.16 Charge (pH=7) 2.32 2D Binding mode Binding energy (Kcal/mol) -5.94 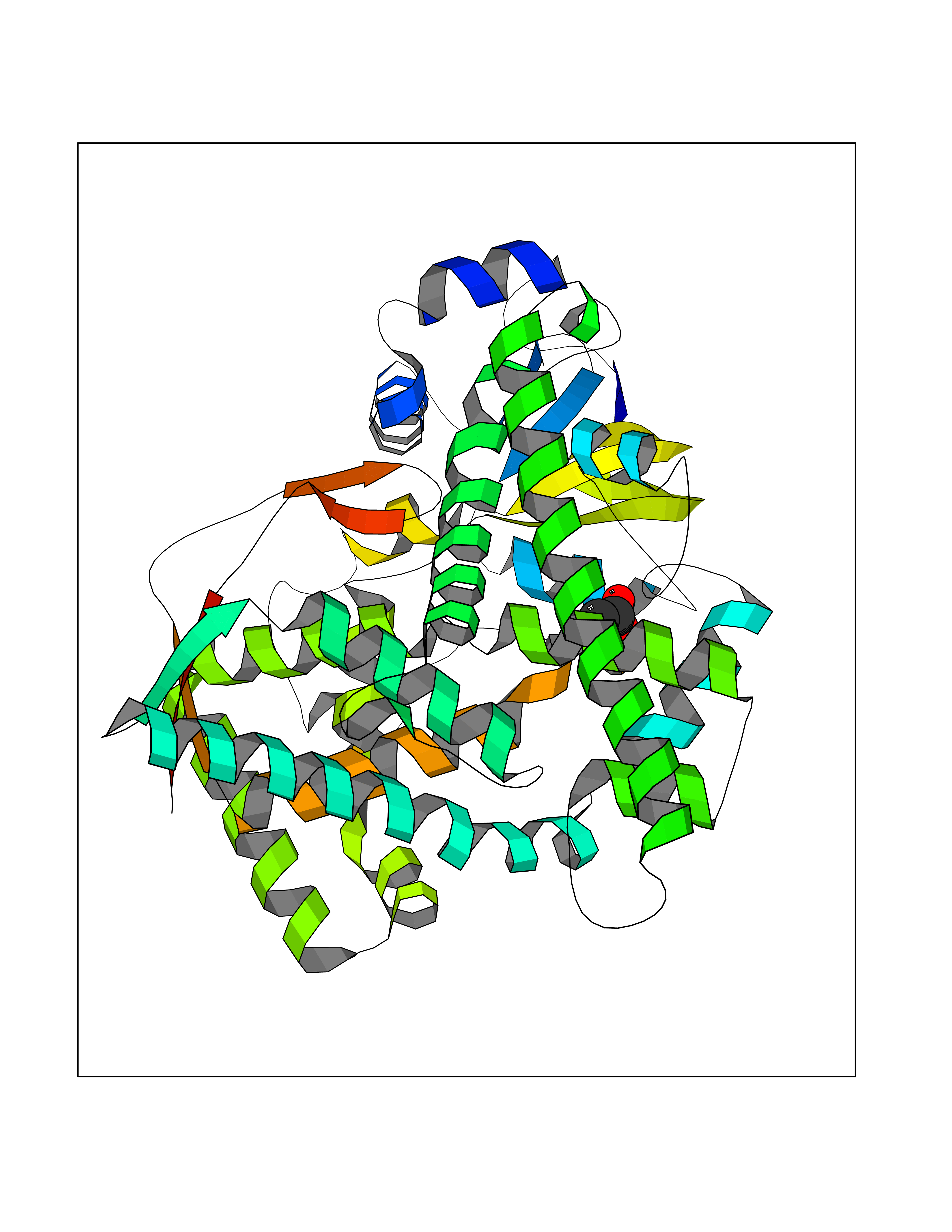 Molscript Map 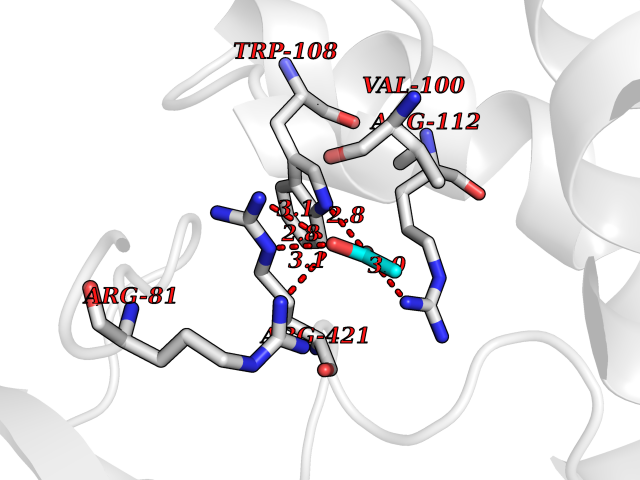 Pymol Map 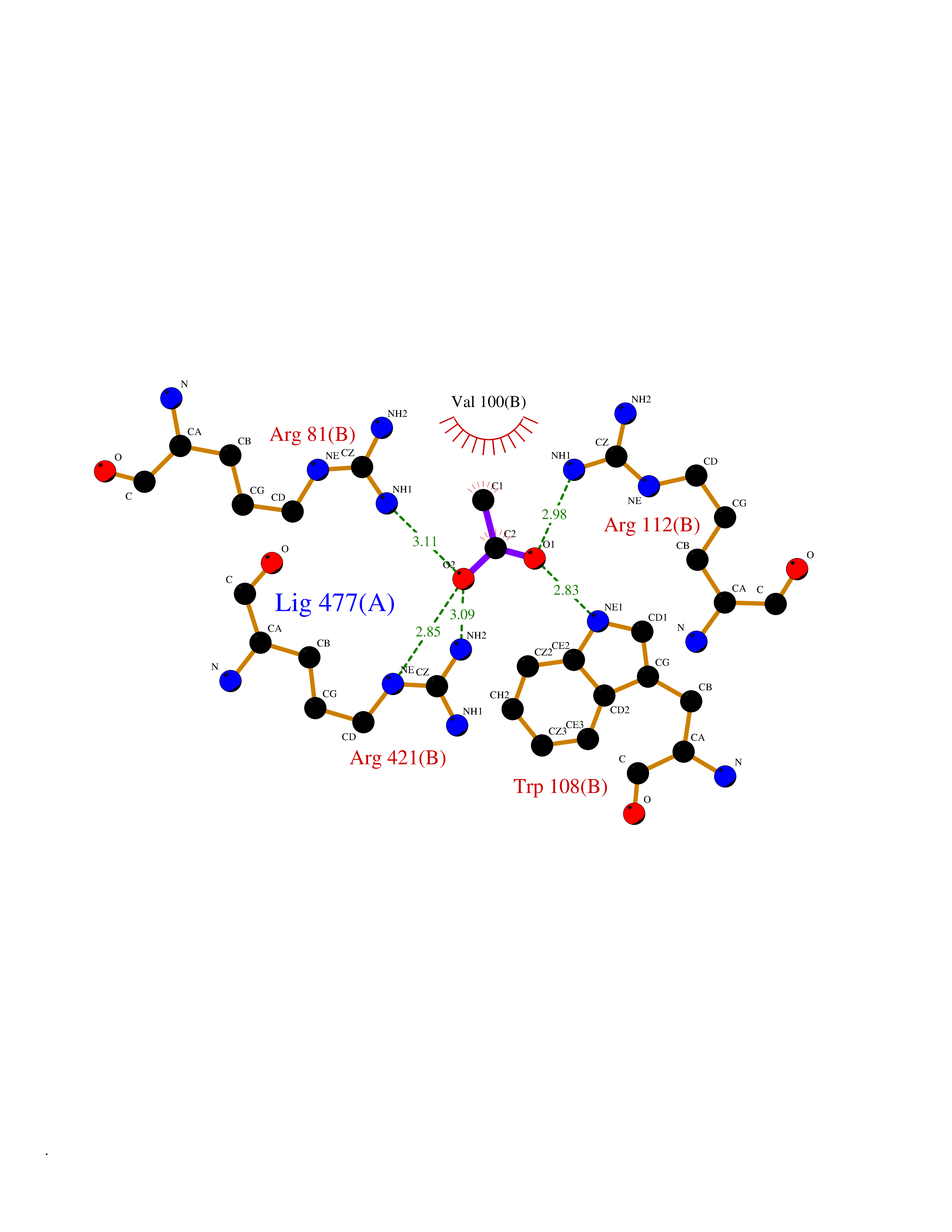 Ligplot Map 3D Binding mode Sequence PRPFNEIPSPGDNGWLNLYHFWRETGTHKVHLHHVQNFQKYGPIYREKLGNVESVYVIDPEDVALLFKSEGPNPERFLIPPWVAYHQYYQRPIGVLLKKSAAWKKDRVALNQEVMAPEATKNFLPLLDAVSRDFVSVLHRRIKKAGSGNYSGDISDDLFRFAFESITNVIFGERQGMLEEVVNPEAQRFIDAIYQMFHTSVPMLNLPPDLFRLFRTKTWKDHVAAWDVIFSKADIYTQNFYWELRQKGSVHHDYRGILYRLLGDSKMSFEDIKANVTEMLAGGVDTTSMTLQWHLYEMARNLKVQDMLRAEVLAARHQAQGDMATMLQLVPLLKASIKETLRLHPISVTLQRYLVNDLVLRDYMIPAKTLVQVAIYALGREPTFFFDPENFDPTRWLSKDKNITYFRNLGFGWGVRQCLGRRIAELEMTIFLINMLENFRVEIQHLSDVGTTFNLILMPEKPISFTFWPF Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Non-heme chloroperoxidase | 1A8U | 4.36 | |
Target general information Gen name cpo Organism Kitasatospora aureofaciens (Streptomyces aureofaciens) Uniprot ID TTD ID NA Synonyms cpoT Protein family AB hydrolase superfamily, Bacterial non-heme haloperoxidase / perhydrolase family Biochemical class Haloperoxidase Function Chloride peroxidase activity. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB03793 Interacts with NA EC number 1.11.1.- Uniprot keywords 3D-structure; Chloride; Oxidoreductase; Peroxidase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 60428.4 Length 554 Aromaticity 0.13 Instability index 31.26 Isoelectric point 4.65 Charge (pH=7) -32.72 2D Binding mode Binding energy (Kcal/mol) -5.95 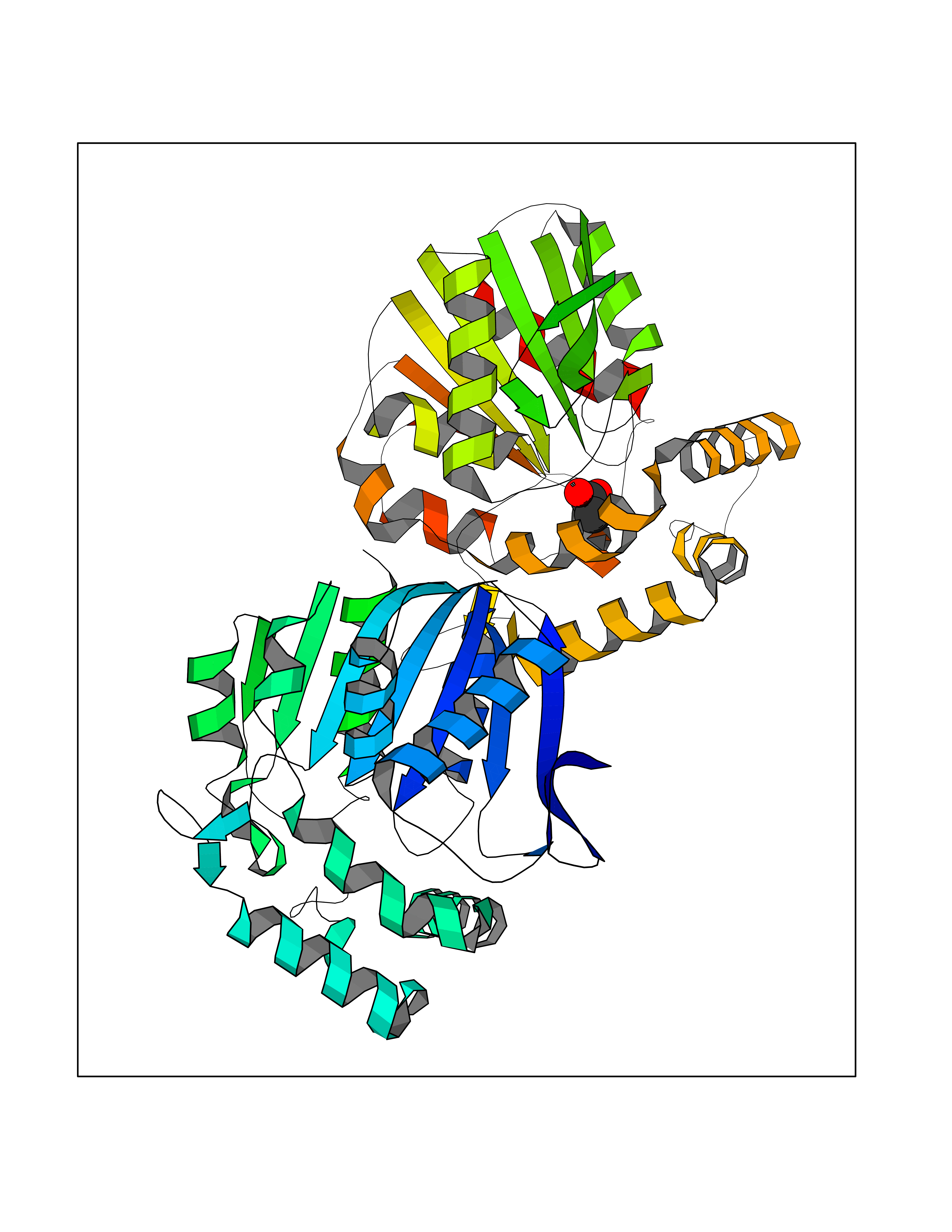 Molscript Map 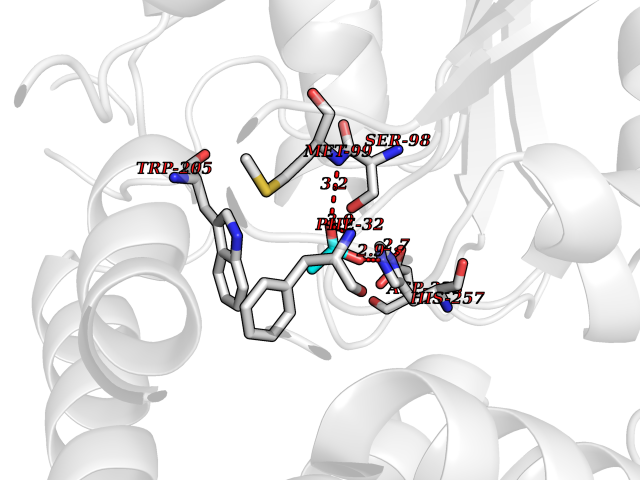 Pymol Map 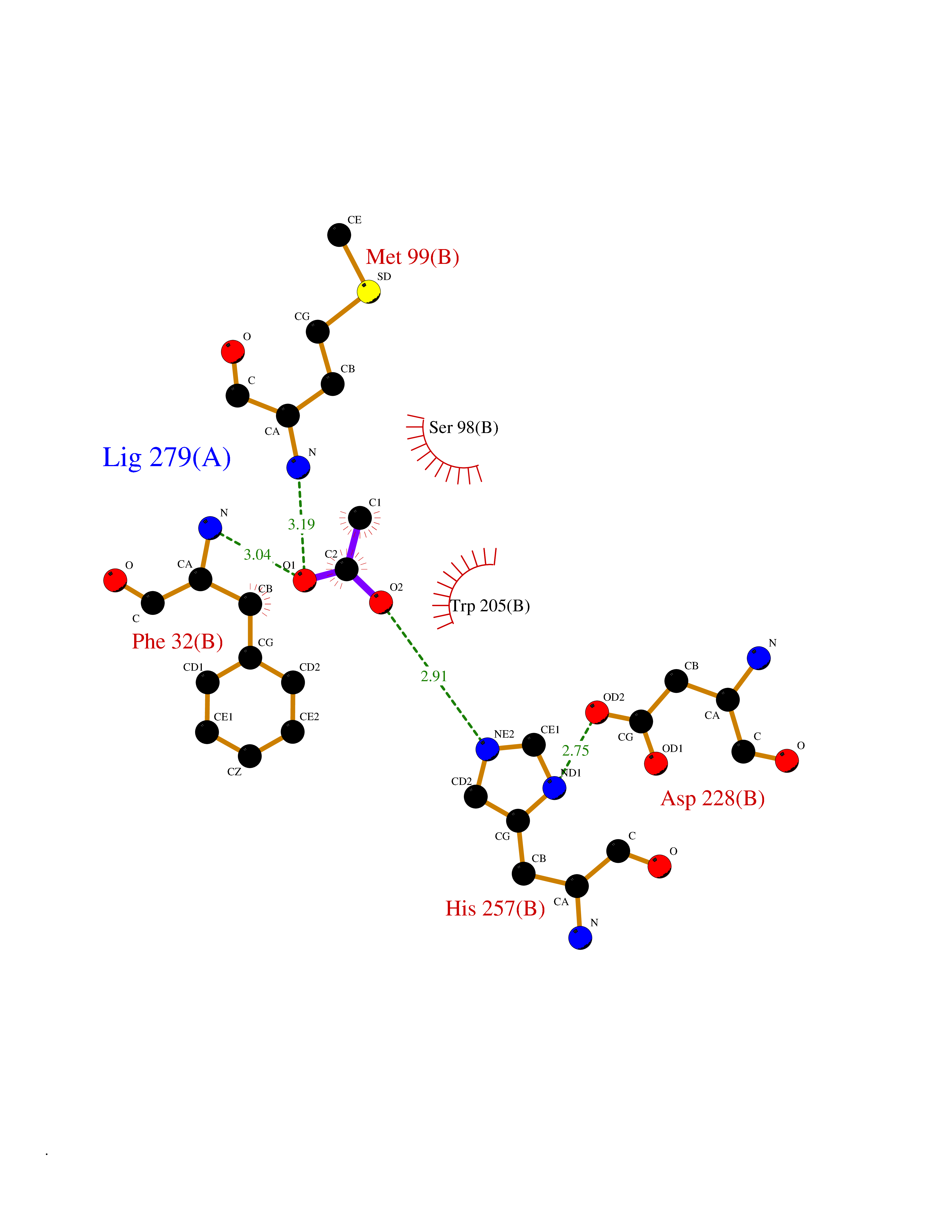 Ligplot Map 3D Binding mode Sequence PFITVGQENSTSIDLYYEDHGAGQPVVLIHGFPLSGHSWERQSAALLDAGYRVITYDRRGFGQSSQPTTGYDYDTFAADLNTVLETLDLQDAVLVGFSMGTGEVARYVSSYGTARIAKVAFLASLEPFLLKTDDNPDGAAPKEFFDGIVAAVKADRYAFYTGFFNDFYNLDENLGTRISEEAVRNSWNTAASGGFFAAAAAPTTWYTDFRADIPRIDVPALILHGTGDRTLPIENTARVFHKALPSAEYVEVEGAPHGLLWTHAEEVNTALLAFLAKPFITVGQENSTSIDLYYEDHGAGQPVVLIHGFPLSGHSWERQSAALLDAGYRVITYDRRGFGQSSQPTTGYDYDTFAADLNTVLETLDLQDAVLVGFSMGTGEVARYVSSYGTARIAKVAFLASLEPFLLKTDDNPDGAAPKEFFDGIVAAVKADRYAFYTGFFNDFYNLDENLGTRISEEAVRNSWNTAASGGFFAAAAAPTTWYTDFRADIPRIDVPALILHGTGDRTLPIENTARVFHKALPSAEYVEVEGAPHGLLWTHAEEVNTALLAFLAK Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Integrin beta-3 | 3NIF | 4.36 | |
Target general information Gen name ITGB3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms GP3A Protein family Integrin beta chain family Biochemical class Cell adhesion Function Cell adhesion molecule binding.Coreceptor activity.Enzyme binding.Extracellular matrix binding.Fibronectin binding.Identical protein binding.Platelet-derived growth factor receptor binding.Protease binding.Protein disulfide isomerase activity.Vascular endothelial growth factor receptor 2 binding.Virus receptor activity. Related diseases Glanzmann thrombasthenia 2 (GT2) [MIM:619267]: A form of Glanzmann thrombasthenia, a disorder characterized by failure of platelet aggregation, absent or diminished clot retraction, and mucocutaneous bleeding of mild-to-moderate severity. Glanzmann thrombasthenia has been classified into clinical types I and II. In type I, platelets show absence of glycoprotein IIb-IIIa complexes at their surface and lack fibrinogen and clot retraction capability. In type II, the platelets express glycoprotein IIb-IIIa complexes at reduced levels, have detectable amounts of fibrinogen, and have low or moderate clot retraction capability. {ECO:0000269|PubMed:10233432, ECO:0000269|PubMed:11588040, ECO:0000269|PubMed:11897046, ECO:0000269|PubMed:12083483, ECO:0000269|PubMed:12353082, ECO:0000269|PubMed:1371279, ECO:0000269|PubMed:1438206, ECO:0000269|PubMed:15583747, ECO:0000269|PubMed:15634267, ECO:0000269|PubMed:15748237, ECO:0000269|PubMed:1602006, ECO:0000269|PubMed:20020534, ECO:0000269|PubMed:2392682, ECO:0000269|PubMed:29084015, ECO:0000269|PubMed:8781422, ECO:0000269|PubMed:9215749, ECO:0000269|PubMed:9376589, ECO:0000269|PubMed:9684783, ECO:0000269|PubMed:9790984}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Bleeding disorder, platelet-type, 24 (BDPLT24) [MIM:619271]: An autosomal dominant disorder of platelet production characterized by congenital macrothrombocytopenia and platelet anisocytosis. Affected individuals may have no or only mildly increased bleeding tendency. {ECO:0000269|PubMed:18065693, ECO:0000269|PubMed:29380037}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00054; DB00098; DB00063; DB13949; DB15598; DB06472; DB04863; DB00451; DB05787; DB02709; DB14520; DB00775 Interacts with P78423; P21333; P08514; P08514-1; P06756; P05106; P18031; Q9Y490; P05094; F5HB81; Q62101; P05480-2; P26039; P54939; P06935 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell adhesion; Cell junction; Cell membrane; Cell projection; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Glycoprotein; Host cell receptor for virus entry; Host-virus interaction; Integrin; Magnesium; Membrane; Metal-binding; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Synapse; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID B,D Molecular weight (Da) 70443.1 Length 646 Aromaticity 0.11 Instability index 41.21 Isoelectric point 5.02 Charge (pH=7) -21.12 2D Binding mode Binding energy (Kcal/mol) -5.95  Molscript Map 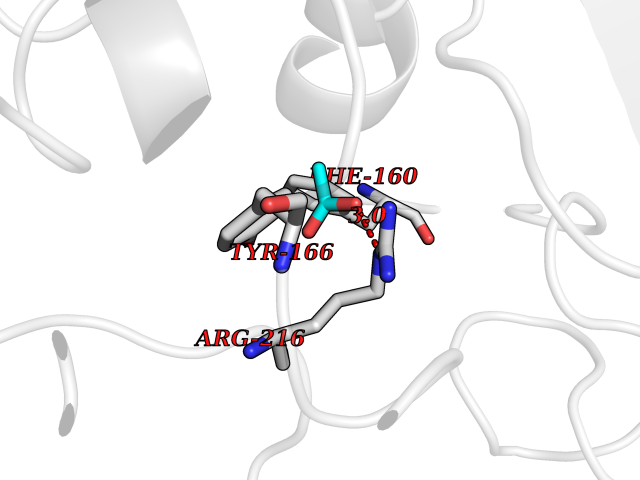 Pymol Map 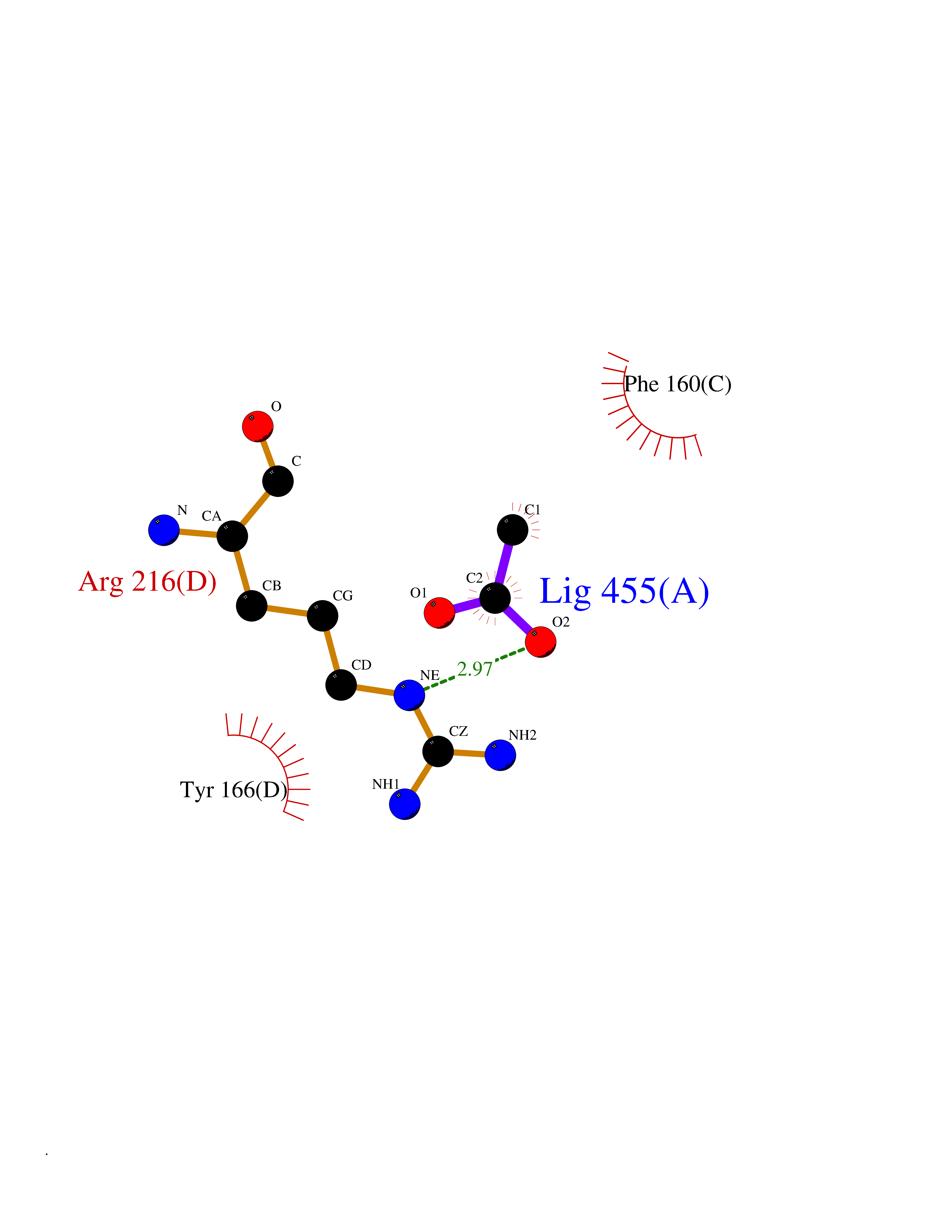 Ligplot Map 3D Binding mode Sequence LNLDPVQLTFYAGPNGSQFGFSLDFHKDSHGRVAIVVGAPRTLGPSQEETGGVFLCPWRAEGGQCPSLLFDLRDETRNVGSQTLQTFKARQGLGASVVSWSDVIVACAPWQHWNVLEKTEEAEKTPVGSCFLAQPESGRRAEYSPCRGNTLSRIYVENDFSWDKRYCEAGFSSVVTQAGELVLGAPGGYYFLGLLAQAPVADIFSSYRPGILLWHVSSQSLSFDSSNPEYFDGYWGYSVAVGEFDGDLNTTEYVVGAPTWSWTLGAVEILDSYYQRLHRLRGEQMASYFGHSVAVTDVNGDGRHDLLVGAPLYMESRADRKLAEVGRVYLFLQPRGPHALGAPSLLLTGTQLYGRFGSAIAPLGDLDRDGYNDIAVAAPYGGPSGRGQVLVFLGQSEGLRSRPSQVLDSPFPTGSAFGFSLRGAVDIDDNGYPDLIVGAYGANQVAVYRAQPVVSPYMYISPPEALENPCYDMKTTCLPMFGYKHVLTLTDQVTRFNEEVKKQSVSRNRDAPEGGFDAIMQATVCDEKIGWRNDASHLLVFTTDAKTHIALDGRLAGIVQPNDGQCHVGSDNHYSASTTMDYPSLGLMTEKLSQKNINLIFAVTENVVNLYQNYSELIPGTTVGVLSMDSSNVLQLIVDAYGKIRS Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Dihydrofolate synthase/folylpolyglutamate synthase | 1W78 | 4.36 | |
Target general information Gen name folC Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms dedC;JW2312;b2315 Protein family Folylpolyglutamate synthase family Biochemical class Synthase Function ATP binding.Dihydrofolate synthase activity.Metal ion binding.Tetrahydrofolylpolyglutamate synthase activity. Related diseases Complement hyperactivation, angiopathic thrombosis, and protein-losing enteropathy (CHAPLE) [MIM:226300]: An autosomal recessive disease characterized by abdominal pain and diarrhea, primary intestinal lymphangiectasia, edema due to hypoproteinemia, malabsorption, and less frequently, bowel inflammation, recurrent infections, and angiopathic thromboembolic disease. Patients' T lymphocytes show increased complement activation causing surface deposition of complement and the generation of soluble C5a. {ECO:0000269|PubMed:28657829, ECO:0000269|PubMed:28657861}. The disease is caused by variants affecting the gene represented in this entry. CHAPLE is caused by biallelic mutations in the CD55 gene. Drugs (DrugBank ID) DB02437; DB03830 Interacts with NA EC number 6.3.2.12; 6.3.2.17 Uniprot keywords 3D-structure; ATP-binding; Folate biosynthesis; Ligase; Magnesium; Metal-binding; Nucleotide-binding; One-carbon metabolism; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 44392.8 Length 414 Aromaticity 0.07 Instability index 29.47 Isoelectric point 5.2 Charge (pH=7) -15.5 2D Binding mode Binding energy (Kcal/mol) -5.94  Molscript Map  Pymol Map 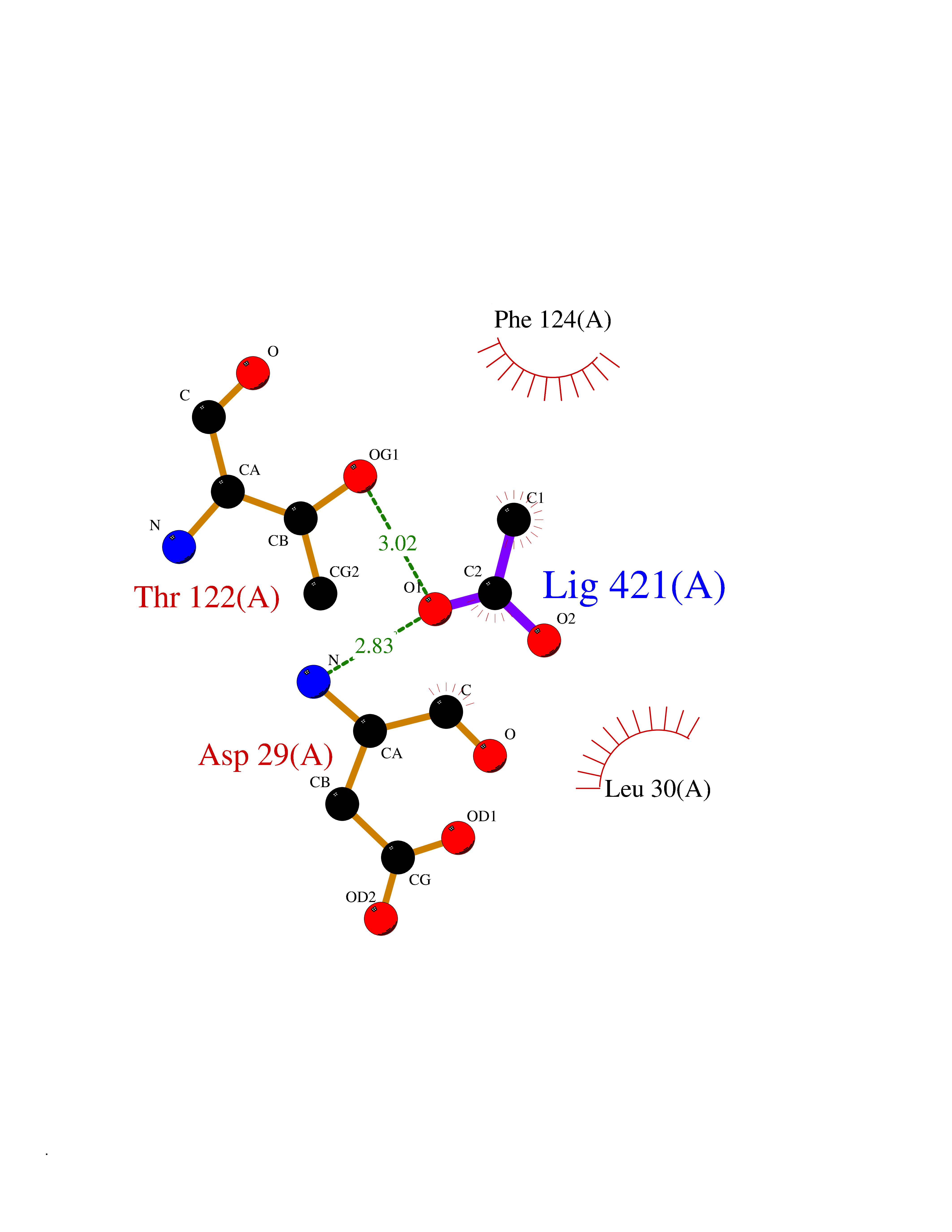 Ligplot Map 3D Binding mode Sequence TPQAASPLASWLSYLENLHSKTIDLGLERVSLVAARLGVLKPAPFVFTVAGTNGKGTTCRTLESILMAAGYKVGVYSSPHLVRYTERVRVQGQELPESAHTASFAEIESARGDISLTYFEYGTLSALWLFKQAQLDVVILEVGLGGRLDATNIVDADVAVVTSIALDHTDWLGPDRESIGREXAGIFRSEKPAIVGEPEMPSTIADVAQEKGALLQRRGVEWNYSVTDHDWAFSDAHGTLENLPLPLVPQPNAATALAALRASGLEVSENAIRDGIASAILPGRFQIVSESPRVIFDVAHNPHAAEYLTGRMKALPKNGRVLAVIGMLHDKDIAGTLAWLKSVVDDWYCAPLEGPRGATAEQLLEHLGNGKSFDSVAQAWDAAMADAKAEDTVLVCGSFHTVAHVMEVIDARRS Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Cytochrome P450 2C8 | 2NNJ | 4.36 | |
Target general information Gen name CYP2C8 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Arachidonic acid epoxygenase activity.Aromatase activity.Caffeine oxidase activity.Estrogen 16-alpha-hydroxylase activity.Heme binding.Iron ion binding.Monooxygenase activity.Oxygen binding. Related diseases A chromosomal aberration involving BCL2 has been found in chronic lymphatic leukemia. Translocation t(14;18)(q32;q21) with immunoglobulin gene regions. BCL2 mutations found in non-Hodgkin lymphomas carrying the chromosomal translocation could be attributed to the Ig somatic hypermutation mechanism resulting in nucleotide transitions. {ECO:0000269|PubMed:2875799, ECO:0000269|PubMed:3285301}. Drugs (DrugBank ID) DB08607; DB08496; DB14055; DB12001; DB05812; DB15568; DB00918; DB12015; DB01424; DB01118; DB00321; DB00381; DB00613; DB01060; DB17449; DB01217; DB01435; DB11901; DB06605; DB00714; DB01072; DB01076; DB11995; DB00972; DB08822; DB12781; DB13997; DB05015; DB16703; DB06770; DB05229; DB00443; DB12236; DB00307; DB01393; DB13746; DB16536; DB06616; DB12267; DB12151; DB01194; DB01222; DB00921; DB06772; DB08875; DB00201; DB13919; DB00796; DB09061; DB08502; DB00564; DB00482; DB06119; DB00439; DB00608; DB00169; DB09201; DB00501; DB00604; DB12499; DB00349; DB00845; DB00758; DB00257; DB00363; DB00907; DB01394; DB05219; DB00531; DB08912; DB11682; DB00250; DB09183; DB01609; DB01234; DB14649; DB09213; DB00829; DB00586; DB00255; DB00343; DB01184; DB00625; DB11979; DB15444; DB06210; DB13874; DB11718; DB08899; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00402; DB00977; DB14766; DB00973; DB12466; DB04854; DB01023; DB01039; DB16165; DB00544; DB13867; DB08906; DB00588; DB01095; DB11679; DB01241; DB01645; DB11978; DB01218; DB00741; DB14538; DB14539; DB14540; DB14541; DB14542; DB14543; DB14545; DB14544; DB01611; DB12471; DB01050; DB09054; DB01181; DB00619; DB16200; DB01029; DB11633; DB06636; DB00951; DB11757; DB14568; DB09570; DB01221; DB06738; DB01026; DB01009; DB00465; DB00448; DB01259; DB09078; DB12070; DB05667; DB08918; DB00451; DB04725; DB00281; DB17083; DB01583; DB09198; DB06448; DB00836; DB00455; DB12130; DB00678; DB00227; DB09280; DB15935; DB06077; DB08932; DB14921; DB14009; DB00603; DB00784; DB00814; DB00170; DB00532; DB01357; DB00333; DB09241; DB00959; DB00916; DB01110; DB06595; DB00834; DB16236; DB11763; DB00764; DB14512; DB00471; DB00295; DB06510; DB00688; DB01024; DB00486; DB00788; DB09199; DB00622; DB00184; DB01115; DB04868; DB06712; DB12005; DB06670; DB09080; DB16267; DB12513; DB09296; DB00338; DB11632; DB01062; DB12612; DB01229; DB03796; DB05467; DB00617; DB06589; DB08922; DB00850; DB00780; DB01174; DB00946; DB00252; DB01132; DB00554; DB17472; DB08860; DB08901; DB15822; DB14631; DB00635; DB01032; DB00818; DB00205; DB04216; DB00908; DB00468; DB01129; DB00481; DB08896; DB11853; DB14761; DB00912; DB16826; DB00615; DB01045; DB11753; DB01201; DB01220; DB08864; DB08931; DB14840; DB14924; DB00503; DB09200; DB00533; DB00412; DB04847; DB12332; DB00938; DB12543; DB01232; DB00418; DB01037; DB11362; DB15685; DB11689; DB06739; DB00641; DB01261; DB00398; DB15569; DB00421; DB09118; DB00359; DB06729; DB01138; DB00675; DB00799; DB12020; DB09256; DB01079; DB12095; DB15133; DB00857; DB00342; DB08880; DB00624; DB13943; DB13944; DB13946; DB11712; DB00208; DB06137; DB01124; DB01685; DB00214; DB08911; DB00374; DB00755; DB00897; DB12245; DB12808; DB00347; DB00440; DB00197; DB13179; DB11652; DB15328; DB12255; DB15114; DB00862; DB11613; DB08881; DB00661; DB08828; DB09068; DB12026; DB00682; DB00549; DB01198 Interacts with P13473-2; O75400-2; Q9Y371 EC number 1.14.14.1 Uniprot keywords 3D-structure; Alternative splicing; Direct protein sequencing; Endoplasmic reticulum; Heme; Iron; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Steroid metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 52511 Length 463 Aromaticity 0.1 Instability index 37.03 Isoelectric point 8.6 Charge (pH=7) 6.74 2D Binding mode Binding energy (Kcal/mol) -5.94  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence KLPPGPTPLPIIGNMLQIDVKDICKSFTNFSKVYGPVFTVYFGMNPIVVFHGYEAVKEALIDNGEEFSGRGNSPISQRITKGLGIISSNGKRWKEIRRFSLTTLRNFGMGKRSIEDRVQEEAHCLVEELRKTKASPCDPTFILGCAPCNVICSVVFQKRFDYKDQNFLTLMKRFNENFRILNSPWIQVCNNFPLLIDCFPGTHNKVLKNVALTRSYIREKVKEHQASLDVNNPRDFIDCFLIKMEQEKDNQKSEFNIENLVGTVADLFVAGTETTSTTLRYGLLLLLKHPEVTAKVQEEIDHVIGRHRSPCMQDRSHMPYTDAVVHEIQRYSDLVPTGVPHAVTTDTKFRNYLIPKGTTIMALLTSVLHDDKEFPNPNIFDPGHFLDKNGNFKKSDYFMPFSAGKRICAGEGLARMELFLFLTTILQNFNLKSVDDLKNLNTTAVTKGIVSLPPSYQICFIPV Hydrogen bonds contact Hydrophobic contact | ||||