Job Results:
Ligand
Structure
Job ID
a9ea1a8efdae070d2ad25867c2a2c1f3
Job name
NA
Time
2026-02-24 16:12:06
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | p-hydroxybenzoate hydroxylase | 1K0I | 5.57 | |
Target general information Gen name pobA Organism Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Uniprot ID TTD ID NA Synonyms PA0247 Protein family Aromatic-ring hydroxylase family Biochemical class Hydrolase Function 4-hydroxybenzoate 3-monooxygenase activity.FAD binding.Flavin adenine dinucleotide binding. Related diseases Immunodeficiency, common variable, 12, with autoimmunity (CVID12) [MIM:616576]: A primary immunodeficiency characterized by hypogammaglobulinemia and recurrent bacterial infections. About half of patients develop autoimmune features, including cytopenia, as well as generalized inflammation and lymphoproliferation manifest as lymphadenopathy or hepatosplenomegaly. {ECO:0000269|PubMed:26279205}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02839; DB04242; DB03482; DB02362; DB03147 Interacts with NA EC number 1.14.13.2 Uniprot keywords 3D-structure; Aromatic hydrocarbons catabolism; FAD; Flavoprotein; Monooxygenase; NADP; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 44295 Length 394 Aromaticity 0.09 Instability index 37.66 Isoelectric point 6.2 Charge (pH=7) -3.67 2D Binding mode Binding energy (Kcal/mol) -7.6  Molscript Map 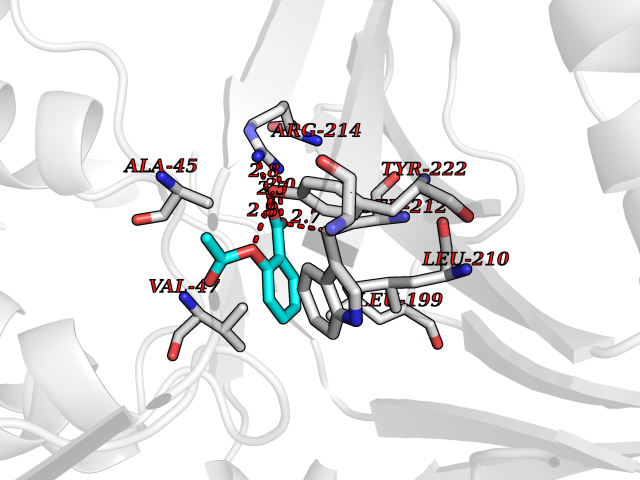 Pymol Map 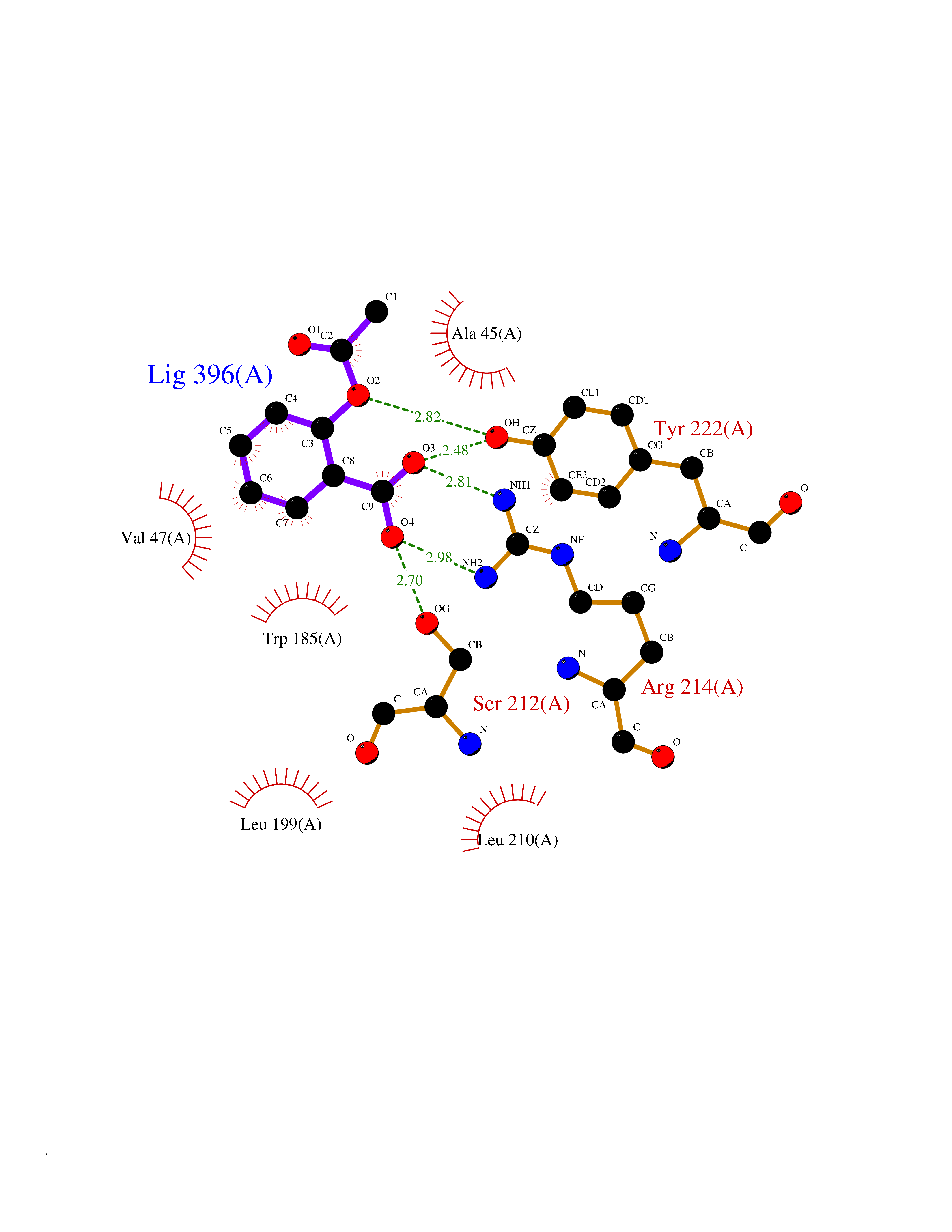 Ligplot Map 3D Binding mode Sequence MKTQVAIIGAGPSGLLLGQLLHKAGIDNVILERQTPDYVLGRIRAGVLEQGMVDLLREAGVDRRMARDGLVHEGVEIAFAGQRRRIDLKRLSGGKTVTVYGQTEVTRDLMEAREACGATTVYQAAEVRLHDLQGERPYVTFERDGERLRLDCDYIAGCDGFHGISRQSIPAERLKVFERVYPFGWLGLLADTPPVSHELIYANHPRGFALCSQRSATRSQYYVQVPLSEKVEDWSDERFWTELKARLPSEVAEKLVTGPSLEKSIAPLRSFVVEPMQHGRLFLAGDAAHIVPPTGAKGLNLAASDVSTLYRLLLKAYREGRGELLERYSAICLRRIWKAERFSWWMTSVLHRFPDTDAFSQRIQQTELEYYLGSEAGLATIAENYVGLPYEEIE Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Fatty acid synthase (FASN) | 3TJM | 5.57 | |
Target general information Gen name FASN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Yeast fatty acid synthase; Fatty-acyl-CoA synthase; Fatty acyl-CoA synthetase enzyme; FAS Protein family NA Biochemical class Acyltransferase Function Fatty acid synthetase catalyzes the formation of long-chain fatty acids from acetyl-CoA, malonyl-CoA and NADPH. This multifunctional protein has 7 catalytic activities as an acyl carrier protein. Related diseases Glycine encephalopathy 2 (GCE2) [MIM:620398]: A form of glycine encephalopathy, a metabolic disorder characterized by a high concentration of glycine in the body fluids. Affected individuals typically have severe neurological symptoms, including seizure, lethargy, and muscular hypotonia soon after birth. Most of them die within the neonatal period. Atypical cases have later disease onset and less severely affected psychomotor development. {ECO:0000269|PubMed:10873393, ECO:0000269|PubMed:11286506, ECO:0000269|PubMed:16051266, ECO:0000269|PubMed:26371980, ECO:0000269|PubMed:28244183, ECO:0000269|PubMed:8005589, ECO:0000269|PubMed:9600239, ECO:0000269|PubMed:9621520}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01034; DB01083 Interacts with Q15848; Q16665; P42858; Q8IV20; Q8TBB1; PRO_0000045603 [Q99IB8] EC number EC 2.3.1.85 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Isopeptide bond; Lipid biosynthesis; Lipid metabolism; Lyase; Multifunctional enzyme; NAD; NADP; Oxidoreductase; Phosphopantetheine; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; S-nitrosylation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30174.9 Length 275 Aromaticity 0.09 Instability index 43.28 Isoelectric point 5.92 Charge (pH=7) -5.4 2D Binding mode Binding energy (Kcal/mol) -7.59  Molscript Map 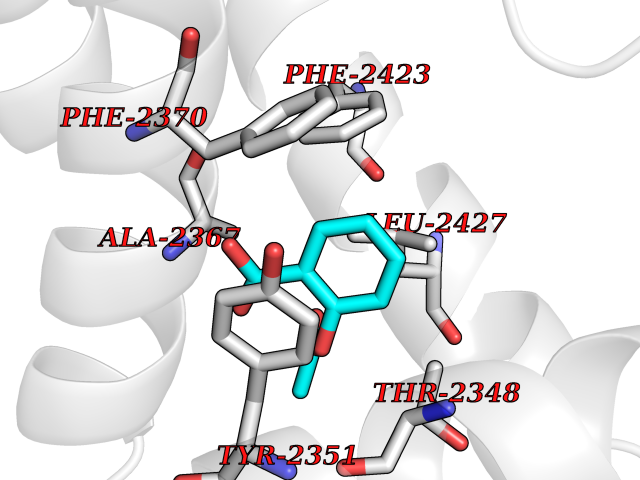 Pymol Map 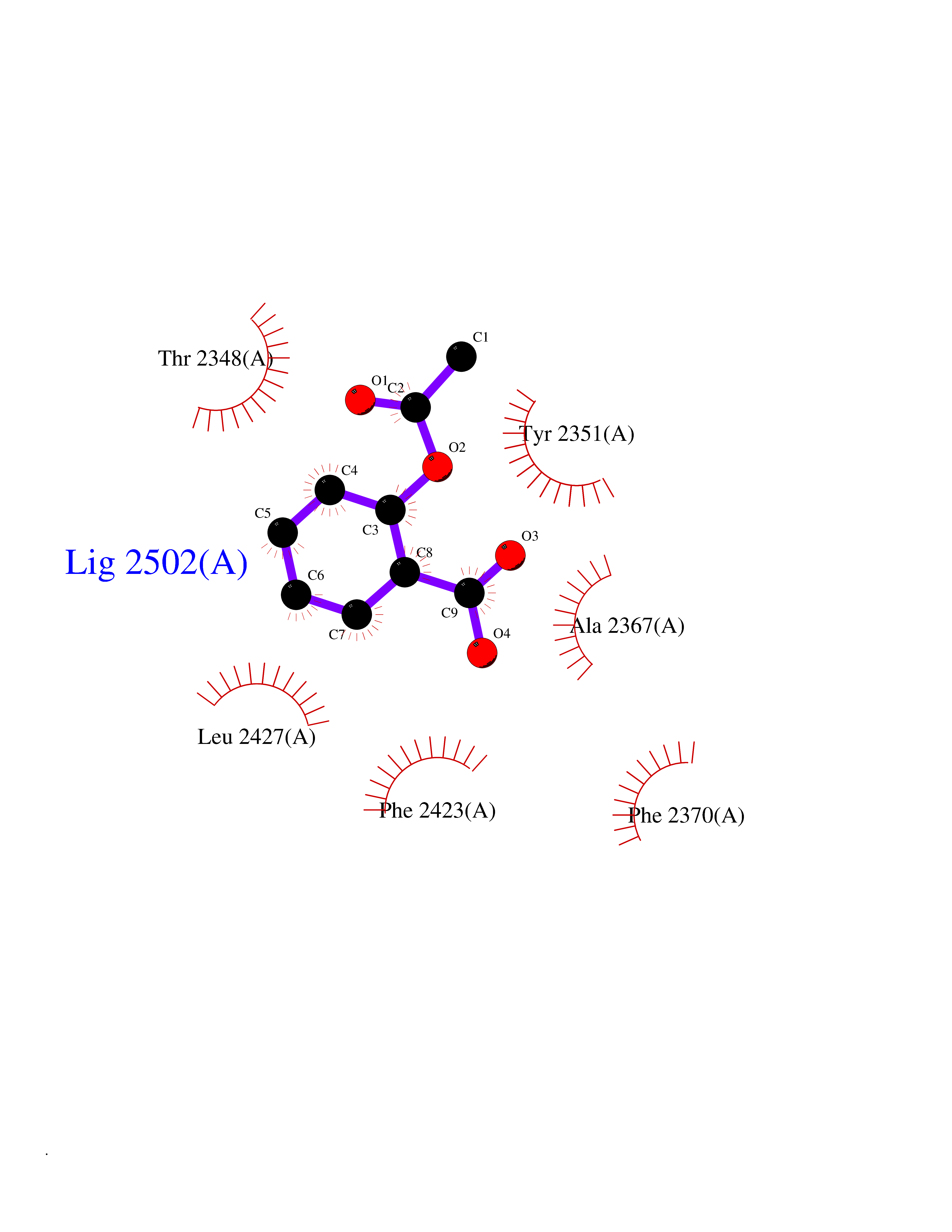 Ligplot Map 3D Binding mode Sequence NLRSLLVNPEGPTLMRLNSVQSSERPLFLVHPIEGSTTVFHSLASRLSIPTYGLQCTRAAPLDSIHSLAAYYIDCIRQVQPEGPYRVAGYSYGACVAFEMCSQLQAQQSPAPTHNSLFLFDGSPTYVLAYTGSYRAKLTPGCEAEAETEAICFFVQQFTDMEHNRVLEALLPLKGLEERVAAAVDLIIKSHQGLDRQELSFAARSFYYKLRAAEQYTPKAKYHGNVMLLRAAAGADYNLSQVCDGKVSVHVIEGDHATLLEGSGLESIISIIHSS Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Estrogen sulfotransferase | 1G3M | 5.57 | |
Target general information Gen name SULT1E1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms STE Protein family Sulfotransferase 1 family Biochemical class Transferase Function Aryl sulfotransferase activity.Estrone sulfotransferase activity.Flavonol 3-sulfotransferase activity.Steroid binding.Steroid sulfotransferase activity.Sulfotransferase activity. Related diseases Neuromyotonia and axonal neuropathy, autosomal recessive (NMAN) [MIM:137200]: An autosomal recessive neurologic disorder characterized by onset in the first or second decade of a peripheral axonal neuropathy predominantly affecting motor more than sensory nerves. The axonal neuropathy is reminiscent of Charcot-Marie-Tooth disease type 2 and distal hereditary motor neuropathy. Individuals with NMAN also have delayed muscle relaxation and action myotonia associated with neuromyotonic discharges on needle EMG resulting from hyperexcitability of the peripheral nerves. {ECO:0000269|PubMed:16835243, ECO:0000269|PubMed:22961002, ECO:0000269|PubMed:28691797, ECO:0000269|PubMed:29787766, ECO:0000269|PubMed:31088288}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02902; DB03346; DB01812; DB00714; DB14635; DB01176; DB00977; DB09288; DB00675; DB09100 Interacts with O76083; O76083-2 EC number 2.8.2.4 Uniprot keywords 3D-structure; Cytoplasm; Lipid metabolism; Lipid-binding; Proteomics identification; Reference proteome; Steroid-binding; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34022.7 Length 285 Aromaticity 0.15 Instability index 32.76 Isoelectric point 6.33 Charge (pH=7) -2.75 2D Binding mode Binding energy (Kcal/mol) -7.6 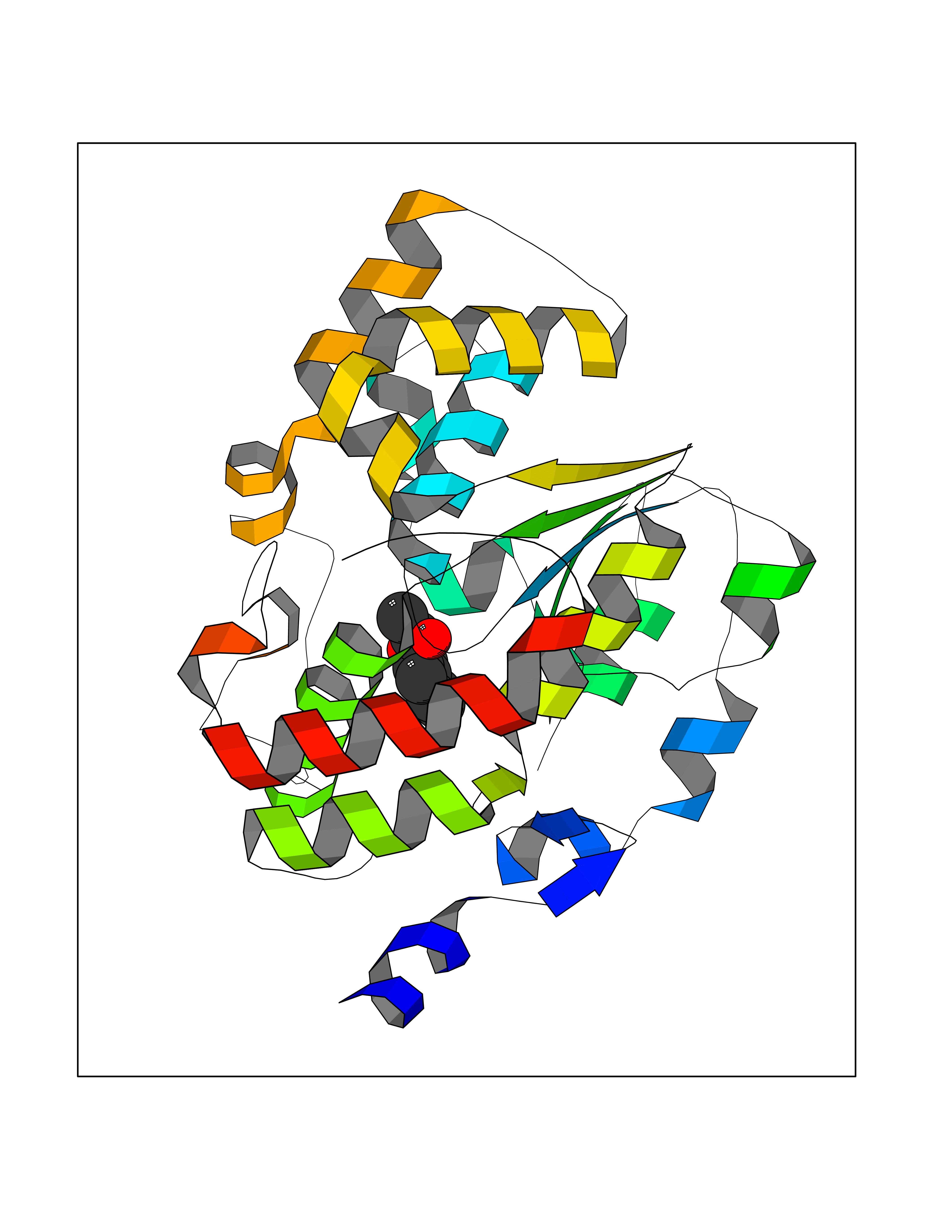 Molscript Map 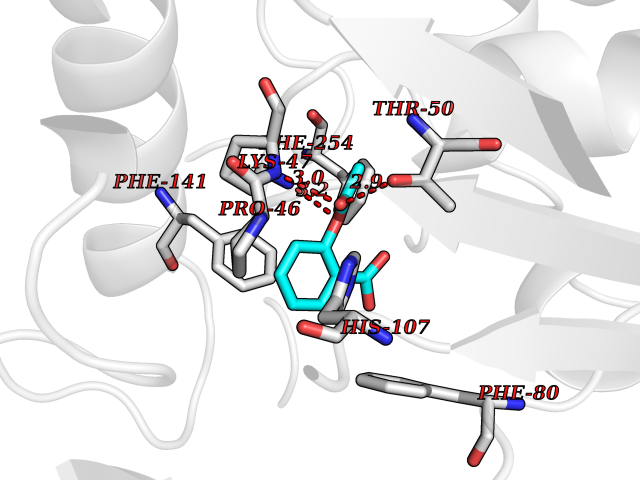 Pymol Map 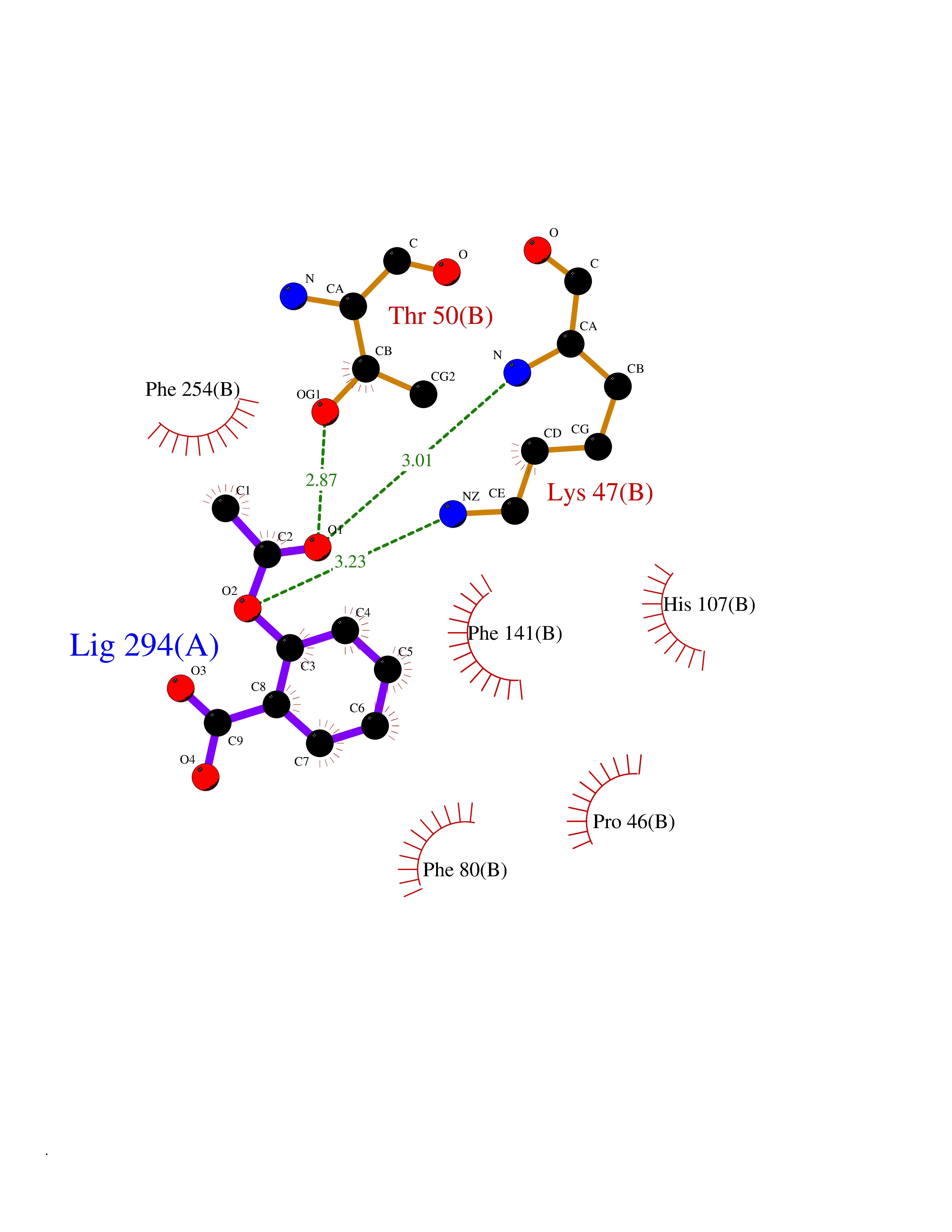 Ligplot Map 3D Binding mode Sequence SELDYYEKFEEVHGILMYKDFVKYWDNVEAFQARPDDLVIATYPKSGTTWVSEIVYMIYKEGDVEKCKEDVIFNRIPFLECRNGVKQLDEMNSPRIVKTHLPPELLPASFWEKDCKIIYLCRNAKDVAVSFYYFFLMVAGHPNPGSFPEFVEKFMQGQVPYGSWYKHVKSWWEKGKSPRVLFLFYEDLKEDIRKEVIKLIHFLERKPSEELVDRIIHHTSFQEMKNNPSTNYTTLPDEIMNQKLSPFMRKGITGDWKNHFTVALNEKFDKHYEQQMKESTLKFRT Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Cyclin-dependent kinase 8 (CDK8) | 3RGF | 5.57 | |
Target general information Gen name CDK8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein kinase K35; Mediator of RNA polymerase II transcription subunit CDK8; Mediator complex subunit CDK8; Cell division protein kinase 8 Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, CDC2/CDKX subfamily Biochemical class Kinase Function Mediator functions as a bridge to convey information from gene-specific regulatory proteins to the basal RNA polymerase II transcription machinery. Mediator is recruited to promoters by direct interactions with regulatory proteins and serves as a scaffold for the assembly of a functional preinitiation complex with RNA polymerase II and the general transcription factors. Phosphorylates the CTD (C-terminal domain) of the large subunit of RNA polymerase II (RNAp II), which may inhibit the formation of a transcription initiation complex. Phosphorylates CCNH leading to down-regulation of the TFIIH complex and transcriptional repression. Recruited through interaction with MAML1 to hyperphosphorylate the intracellular domain of NOTCH, leading to its degradation. Component of the Mediator complex, a coactivator involved in regulated gene transcription of nearly all RNA polymerase II-dependent genes. Related diseases Intellectual developmental disorder with hypotonia and behavioral abnormalities (IDDHBA) [MIM:618748]: An autosomal dominant neurodevelopmental disorder with onset in infancy. IDDHBA is characterized by hypotonia, global developmental delay, learning disability, and behavioral abnormalities, such as autistic features and attention deficit-hyperactivity disorder. Additional variable features may include non-specific facial dysmorphism, congenital heart defects, ocular anomalies, and poor feeding. {ECO:0000269|PubMed:30905399}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03496 Interacts with P24863; Q01094; P02489; Q14204; Q92876 EC number EC 2.7.11.22 Uniprot keywords 3D-structure; Activator; Alternative splicing; ATP-binding; Autism spectrum disorder; Disease variant; Intellectual disability; Kinase; Nucleotide-binding; Nucleus; Proteomics identification; Reference proteome; Repressor; Serine/threonine-protein kinase; Transcription; Transcription regulation; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 37655.2 Length 321 Aromaticity 0.11 Instability index 37.04 Isoelectric point 8.56 Charge (pH=7) 5.13 2D Binding mode Binding energy (Kcal/mol) -7.6 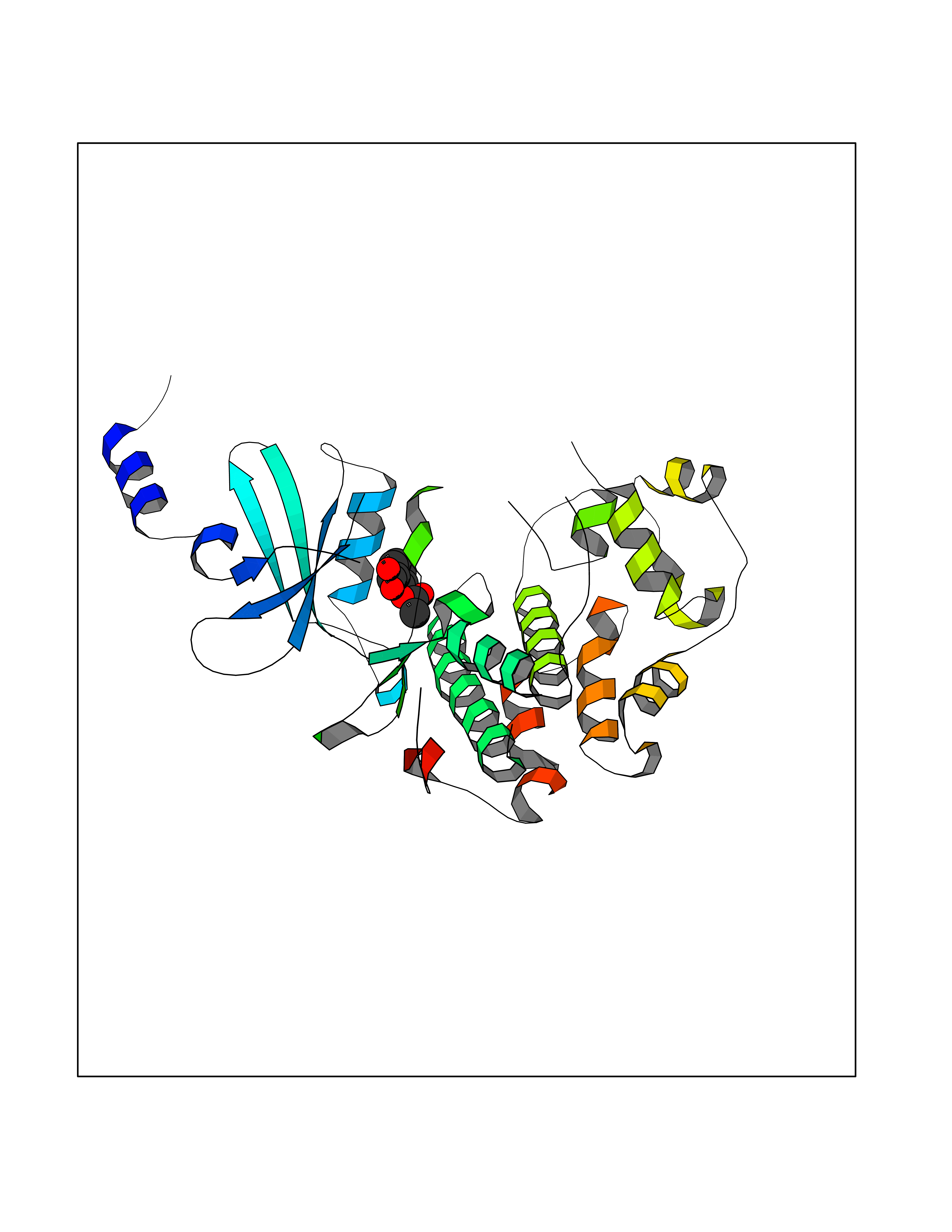 Molscript Map 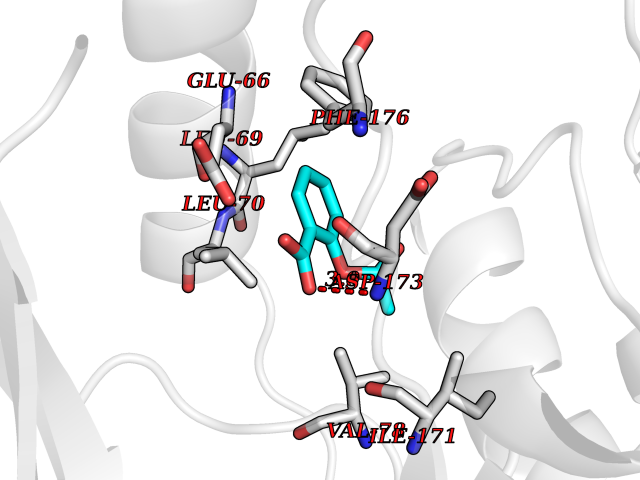 Pymol Map 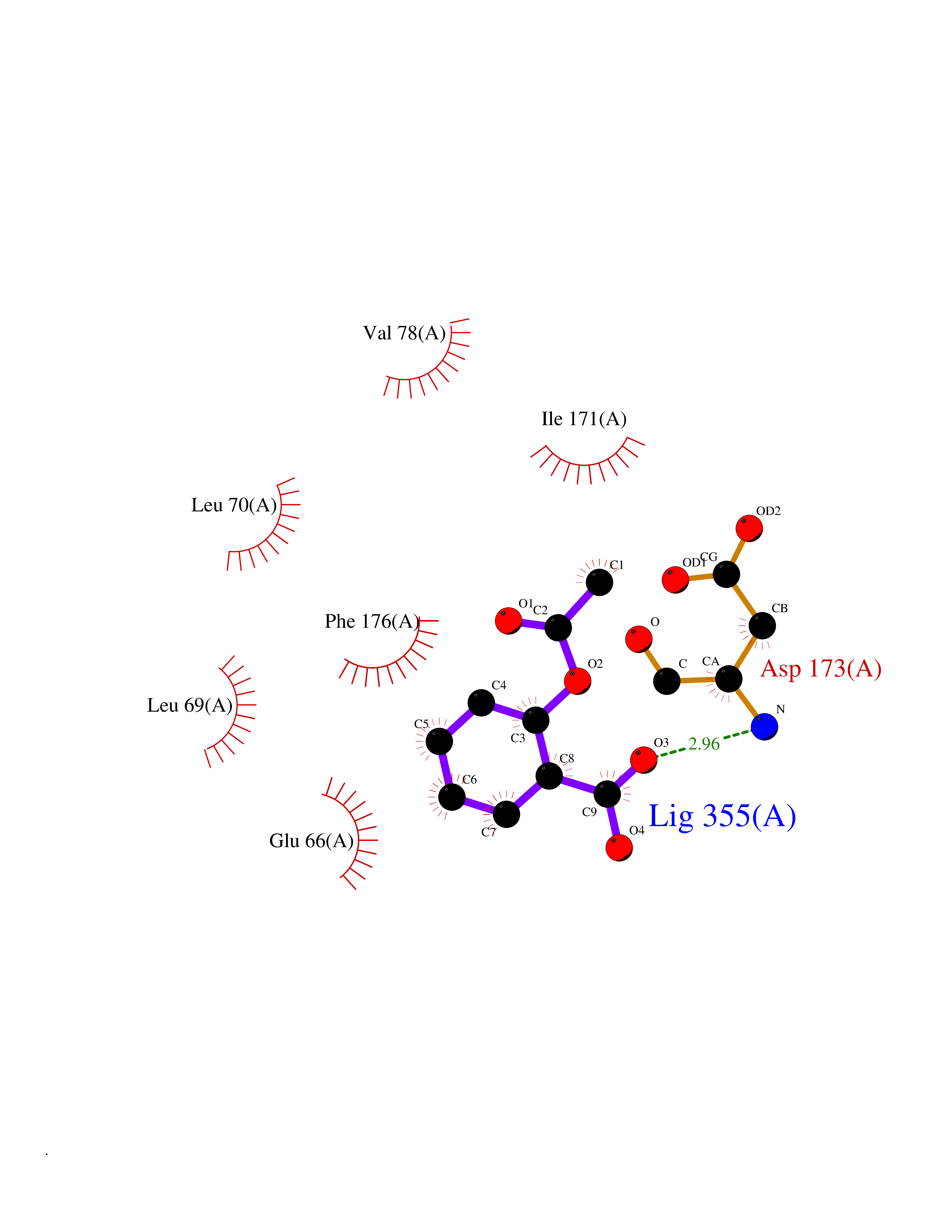 Ligplot Map 3D Binding mode Sequence DKMDYDFKVKLSSERERVEDLFEYEGCKVGHVYKAKRKDGKDDKDYALKQIEGTGISMSACREIALLRELKHPNVISLQKVFLSHADRKVWLLFDYAEHDLWHIIKFHRASKLPRGMVKSLLYQILDGIHYLHANWVLHRDLKPANILVMGEGPERGRVKIADMGFARVTFWYRAPELLLGARHYTKAIDIWAIGCIFAELLTSEPIFHCRQNPYHHDQLDRIFNVMGFPADKDWEDIKKMPEHSTLMKDFRRNTYTNCSLIKYMEKHKVKPDSKAFHLLQKLLTMDPIKRITSEQAMQDPYFLEDPLPTSDVFAGCQIPY Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Glucagon-like peptide 1 receptor (GLP1R) | 6X1A | 5.57 | |
Target general information Gen name GLP1R Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GLP-1R; GLP-1-R; GLP-1 receptor Protein family G-protein coupled receptor 2 family Biochemical class GPCR secretin Function Ligand binding triggers activation of a signaling cascade that leads to the activation of adenylyl cyclase and increased intracellular cAMP levels. Plays a role in regulating insulin secretion in response to GLP-1. G-protein coupled receptor for glucagon-like peptide 1 (GLP-1). Related diseases Lynch syndrome 2 (LYNCH2) [MIM:609310]: A form of Lynch syndrome, an autosomal dominant disease associated with marked increase in cancer susceptibility. It is characterized by a familial predisposition to early-onset colorectal carcinoma (CRC) and extra-colonic tumors of the gastrointestinal, urological and female reproductive tracts. Lynch syndrome is reported to be the most common form of inherited colorectal cancer in the Western world. Clinically, it is often divided into two subgroups. Type I is characterized by hereditary predisposition to colorectal cancer, a young age of onset, and carcinoma observed in the proximal colon. Type II is characterized by increased risk for cancers in certain tissues such as the uterus, ovary, breast, stomach, small intestine, skin, and larynx in addition to the colon. Diagnosis of classical Lynch syndrome is based on the Amsterdam criteria: 3 or more relatives affected by colorectal cancer, one a first degree relative of the other two; 2 or more generation affected; 1 or more colorectal cancers presenting before 50 years of age; exclusion of hereditary polyposis syndromes. The term 'suspected Lynch syndrome' or 'incomplete Lynch syndrome' can be used to describe families who do not or only partially fulfill the Amsterdam criteria, but in whom a genetic basis for colon cancer is strongly suspected. {ECO:0000269|PubMed:10323887, ECO:0000269|PubMed:10375096, ECO:0000269|PubMed:10386556, ECO:0000269|PubMed:10413423, ECO:0000269|PubMed:10480359, ECO:0000269|PubMed:10598809, ECO:0000269|PubMed:10627141, ECO:0000269|PubMed:10660333, ECO:0000269|PubMed:10671064, ECO:0000269|PubMed:10713887, ECO:0000269|PubMed:10777691, ECO:0000269|PubMed:10882759, ECO:0000269|PubMed:11139242, ECO:0000269|PubMed:11427529, ECO:0000269|PubMed:11726306, ECO:0000269|PubMed:11748856, ECO:0000269|PubMed:11754112, ECO:0000269|PubMed:11781295, ECO:0000269|PubMed:11793442, ECO:0000269|PubMed:11839723, ECO:0000269|PubMed:11870161, ECO:0000269|PubMed:12095971, ECO:0000269|PubMed:12132870, ECO:0000269|PubMed:12200596, ECO:0000269|PubMed:12362047, ECO:0000269|PubMed:12373605, ECO:0000269|PubMed:12655562, ECO:0000269|PubMed:12658575, ECO:0000269|PubMed:14635101, ECO:0000269|PubMed:14961575, ECO:0000269|PubMed:15064764, ECO:0000269|PubMed:15139004, ECO:0000269|PubMed:15365995, ECO:0000269|PubMed:15365996, ECO:0000269|PubMed:16083711, ECO:0000269|PubMed:16451135, ECO:0000269|PubMed:17301300, ECO:0000269|PubMed:17510385, ECO:0000269|PubMed:18561205, ECO:0000269|PubMed:20020535, ECO:0000269|PubMed:21120944, ECO:0000269|PubMed:22753075, ECO:0000269|PubMed:7757073, ECO:0000269|PubMed:8566964, ECO:0000269|PubMed:8571956, ECO:0000269|PubMed:8574961, ECO:0000269|PubMed:8797773, ECO:0000269|PubMed:8872463, ECO:0000269|PubMed:8993976, ECO:0000269|PubMed:9048925, ECO:0000269|PubMed:9067757, ECO:0000269|PubMed:9218993, ECO:0000269|PubMed:9272156, ECO:0000269|PubMed:9298827, ECO:0000269|PubMed:9311737, ECO:0000269|PubMed:9326924, ECO:0000269|PubMed:9399661, ECO:0000269|PubMed:9559627, ECO:0000269|PubMed:9718327, ECO:0000269|PubMed:9833759, ECO:0000269|PubMed:9927034, ECO:0000269|Ref.5}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mismatch repair cancer syndrome 1 (MMRCS1) [MIM:276300]: An autosomal recessive form of mismatch repair cancer syndrome, a childhood cancer predisposition syndrome encompassing a broad tumor spectrum. This includes hematological malignancies, central nervous system tumors, Lynch syndrome-associated malignancies such as colorectal tumors as well as multiple intestinal polyps, embryonic tumors and rhabdomyosarcoma. Multiple cafe-au-lait macules, a feature reminiscent of neurofibromatosis type 1, are often found as first manifestation of the underlying cancer. {ECO:0000269|PubMed:11427529, ECO:0000269|PubMed:17440981, ECO:0000269|PubMed:7661930}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Muir-Torre syndrome (MRTES) [MIM:158320]: Rare autosomal dominant disorder characterized by sebaceous neoplasms and visceral malignancy. {ECO:0000269|PubMed:8751876}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in MLH1 may contribute to lobular carcinoma in situ (LCIS), a non-invasive neoplastic disease of the breast.; DISEASE: Endometrial cancer (ENDMC) [MIM:608089]: A malignancy of endometrium, the mucous lining of the uterus. Most endometrial cancers are adenocarcinomas, cancers that begin in cells that make and release mucus and other fluids. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Some epigenetic changes can be transmitted unchanged through the germline (termed 'epigenetic inheritance'). Evidence that this mechanism occurs in humans is provided by the identification of individuals in whom 1 allele of the MLH1 gene is epigenetically silenced throughout the soma (implying a germline event). These individuals are affected by Lynch syndrome but does not have identifiable mutations in MLH1, even though it is silenced, which demonstrates that an epimutation can phenocopy a genetic disease.; DISEASE: Colorectal cancer (CRC) [MIM:114500]: A complex disease characterized by malignant lesions arising from the inner wall of the large intestine (the colon) and the rectum. Genetic alterations are often associated with progression from premalignant lesion (adenoma) to invasive adenocarcinoma. Risk factors for cancer of the colon and rectum include colon polyps, long-standing ulcerative colitis, and genetic family history. {ECO:0000269|PubMed:10598809, ECO:0000269|PubMed:10882759, ECO:0000269|PubMed:12132870, ECO:0000269|PubMed:12655564, ECO:0000269|PubMed:14504054, ECO:0000269|PubMed:15184898, ECO:0000269|PubMed:18033691, ECO:0000269|PubMed:8872463, ECO:0000269|PubMed:9032648, ECO:0000269|PubMed:9087566, ECO:0000269|PubMed:9611074}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09043; DB09045; DB15650; DB01276; DB00040; DB16697; DB06655; DB09265; DB13928; DB14027; DB15171 Interacts with A8MQ03; Q07627; Q8IUG1; P60409; P60410; P60411; Q9BYP8; P26371; Q7Z3S9; P0DPK4 EC number NA Uniprot keywords 3D-structure; ADP-ribosylation; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 45579.6 Length 390 Aromaticity 0.16 Instability index 39.66 Isoelectric point 6.73 Charge (pH=7) -0.68 2D Binding mode Binding energy (Kcal/mol) -7.59 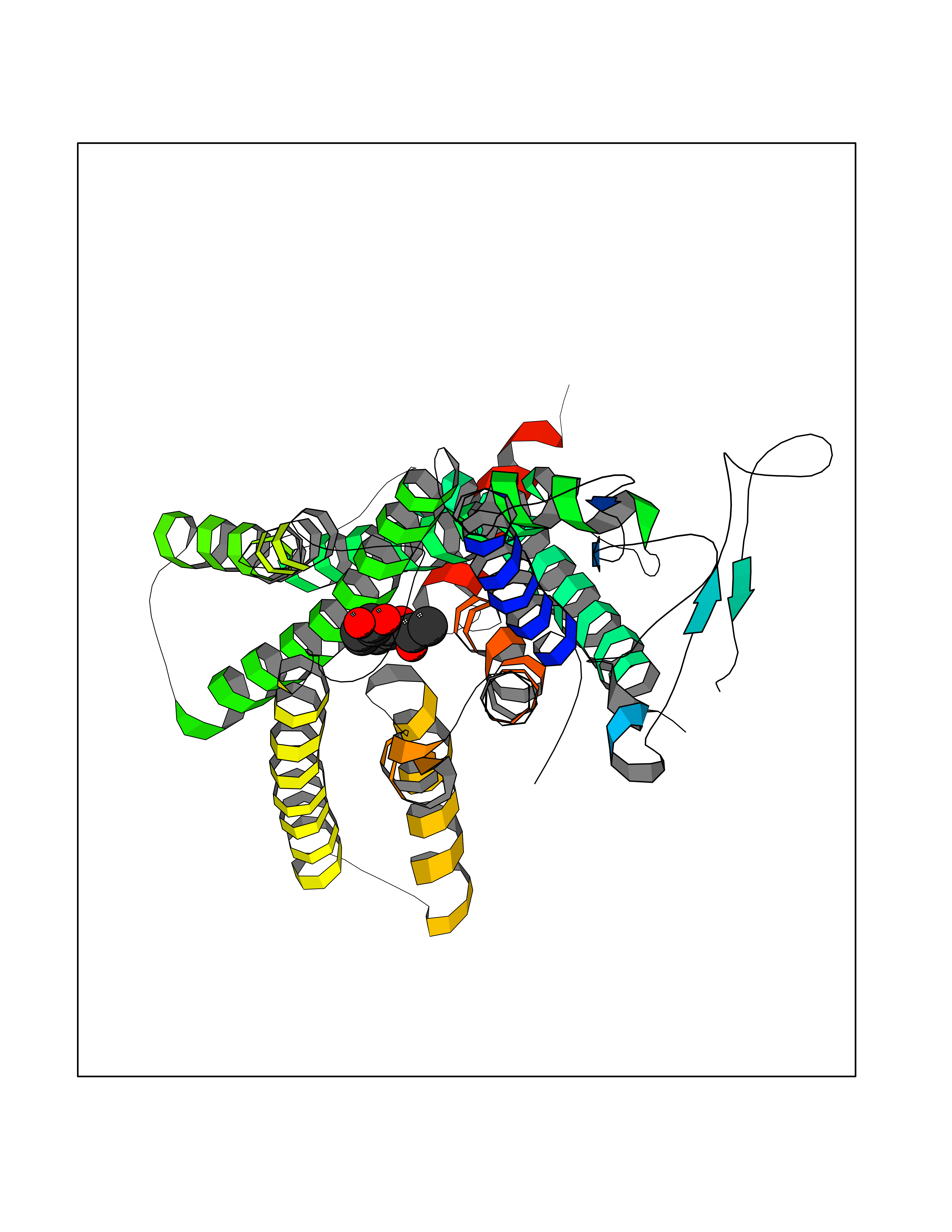 Molscript Map 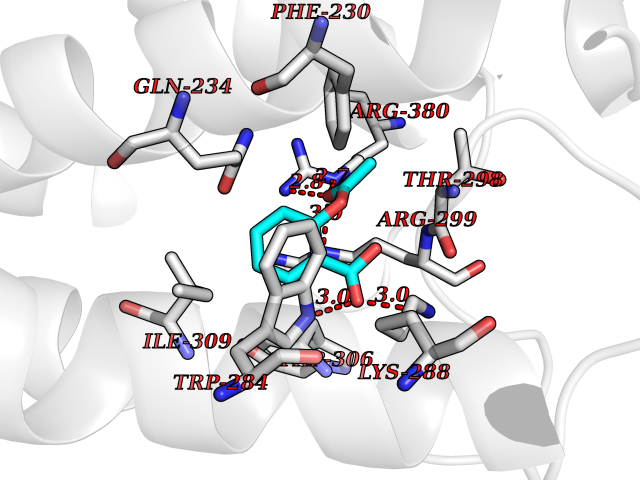 Pymol Map 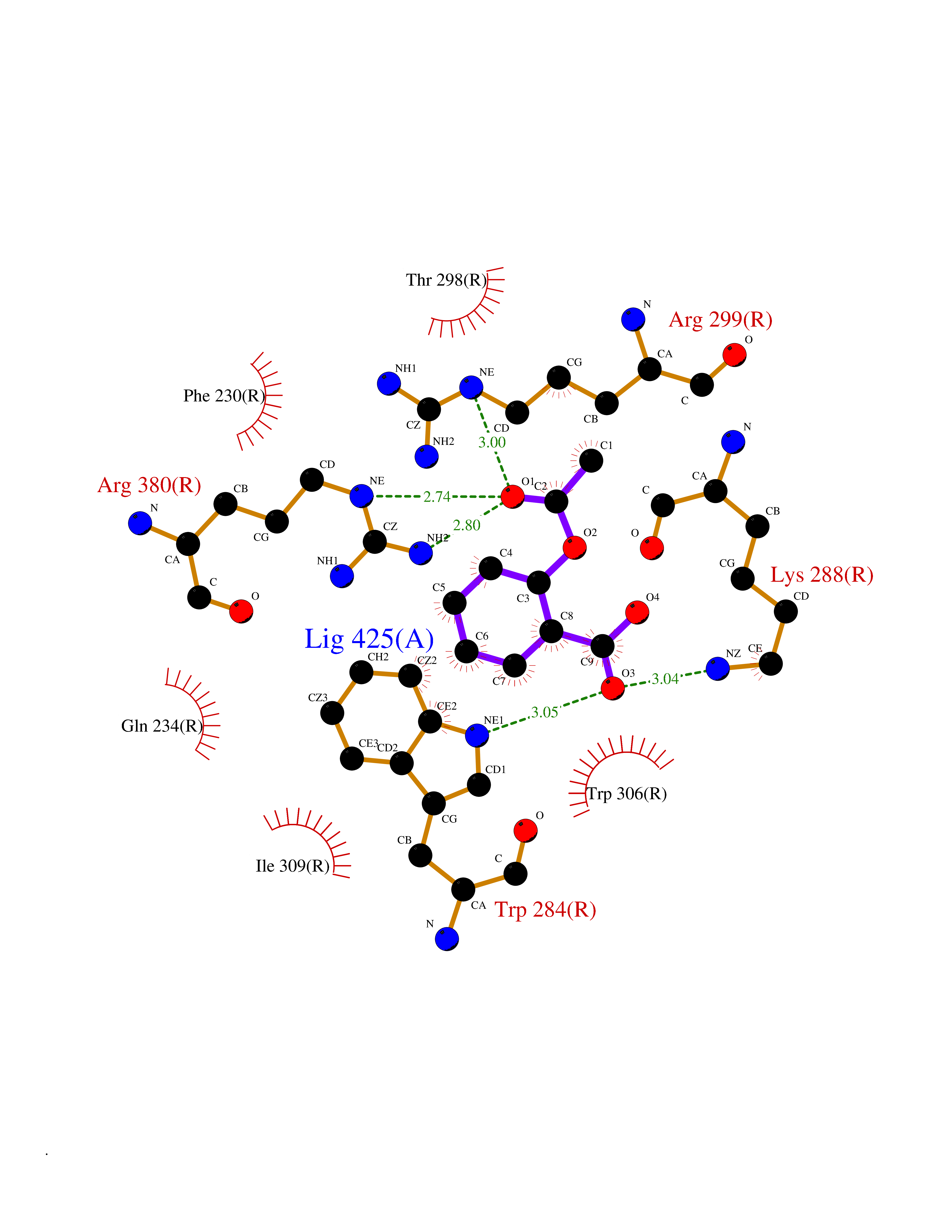 Ligplot Map 3D Binding mode Sequence ATVSLWETVQKWREYRRQCQRSLTEDPPPATDLFCNRTFDEYACWPDGEPGSFVNVSCPWYLPWASSVPQGHVYRFCTAEGLWLQKDNSSLPWRDLSECEESSPEEQLLFLYIIYTVGYALSFSALVIASAILLGFRHLHCTRNYIHLNLFASFILRALSVFIKDAALKWMYSTAAQQHQWDGLLSYQDSLSCRLVFLLMQYCVAANYYWLLVEGVYLYTLLAFSVFSEQWIFRLYVSIGWGVPLLFVVPWGIVKYLYEDEGCWTRNSNMNYWLIIRLPILFAIGVNFLIFVRVICIVVSKLKANLMCKTDIKCRLAKSTLTLIPLLGTHEVIFAFVMDEHARGTLRFIKLFTELSFTSFQGLMVAILYCFVNNEVQLEFRKSWERWRLE Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Proteinase-activated receptor 1 | 3VW7 | 5.56 | |
Target general information Gen name F2R Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms TR;PAR1;CF2R Protein family G-protein coupled receptor 1 family Biochemical class Signaling protein / antagonist Function G-protein alpha-subunit binding.G-protein beta-subunit binding.G-protein coupled receptor activity.Receptor binding.Thrombin-activated receptor activity. Related diseases 3-ketothiolase deficiency (3KTD) [MIM:203750]: An autosomal recessive inborn error of isoleucine catabolism characterized by intermittent ketoacidotic attacks associated with unconsciousness. Some patients die during an attack or are mentally retarded. Urinary excretion of 2-methyl-3-hydroxybutyric acid, 2-methylacetoacetic acid, triglylglycine, butanone is increased. It seems likely that the severity of this disease correlates better with the environmental or acquired factors than with the ACAT1 genotype. {ECO:0000269|PubMed:1346617, ECO:0000269|PubMed:1715688, ECO:0000269|PubMed:7728148, ECO:0000269|PubMed:9744475}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05361; DB00086; DB11300; DB09030 Interacts with Q03135; Q9UNN8 EC number NA Uniprot keywords 3D-structure; Blood coagulation; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Hemostasis; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 31193.7 Length 282 Aromaticity 0.16 Instability index 36.7 Isoelectric point 8.2 Charge (pH=7) 2.98 2D Binding mode Binding energy (Kcal/mol) -7.58 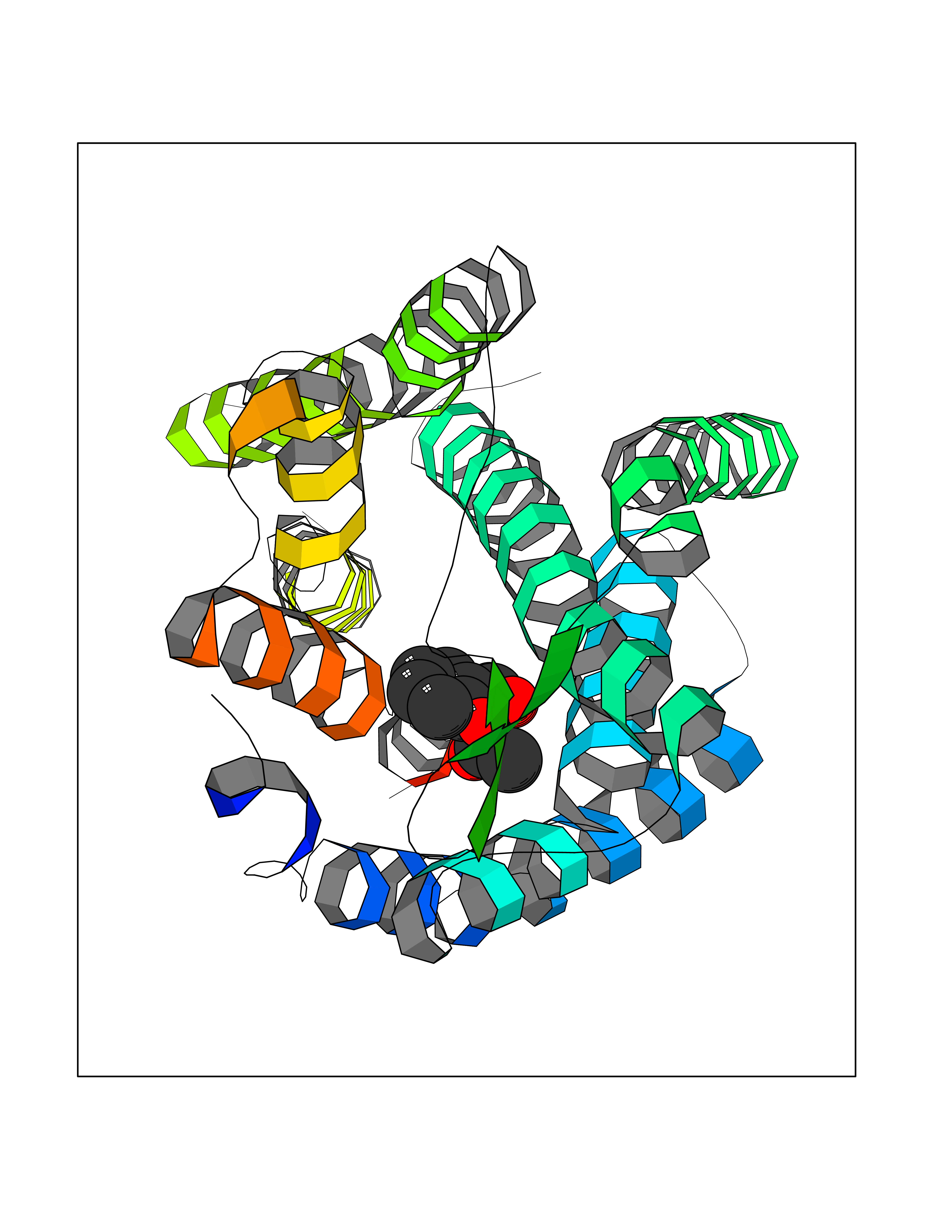 Molscript Map 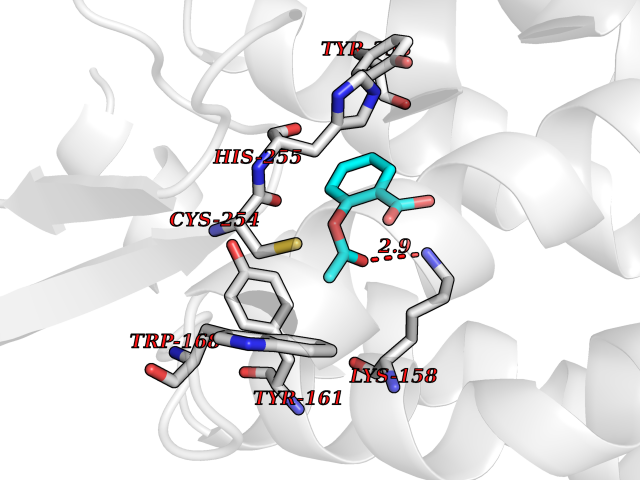 Pymol Map 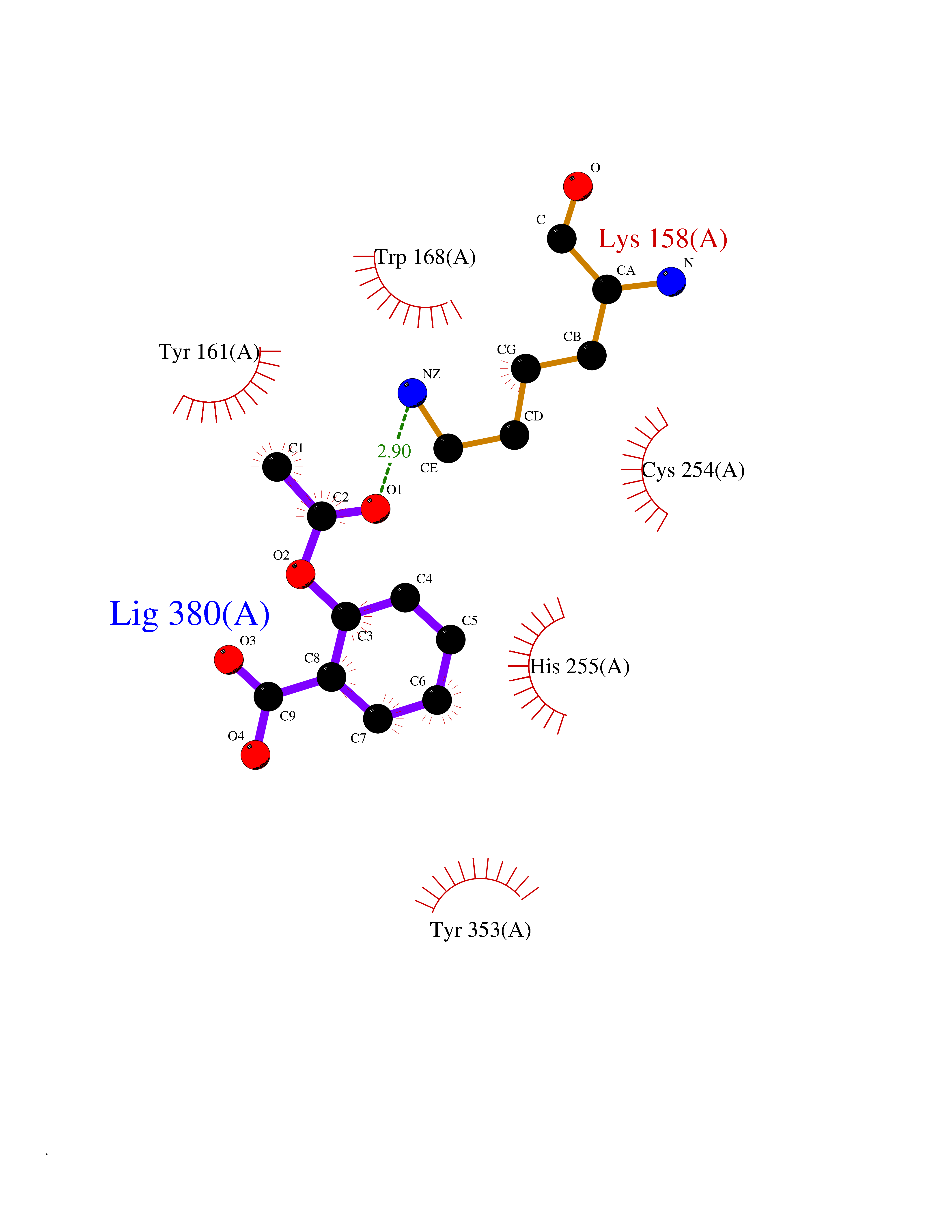 Ligplot Map 3D Binding mode Sequence DASGYLTSSWLTLFVPSVYTGVFVVSLPLNIMAIVVFILKMKVKKPAVVYMLHLATADVLFVSVLPFKISYYFSGSDWQFGSELCRFVTAAFYCNMYASILLMTVISIDRFLAVVYPMRTLGRASFTCLAIWALAIAGVVPLLLKEQTIQVPGLGITTCHDVLSETLLEGYYAYYFSAFSAVFFFVPLIISTVCYVSIIRCLSSSAANRSKKSRALFLSAAVFCIFIICFGPTNVLLIAHYSFLSHTSTTEAAYFAYLLCVCVSSISCCIDPLIYYYASSEC Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | TRANSPORT INHIBITOR RESPONSE 1 protein | 2P1Q | 5.56 | |
Target general information Gen name IAA7 Organism Arabidopsis thaliana (Mouse-ear cress) Uniprot ID TTD ID NA Synonyms AXR2;At3g23050;MXC7.8 Protein family Aux/IAA family Biochemical class Signaling protein Function DNA binding transcription factor activity. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) NA Interacts with Q9LW29; Q9C5W9; Q8RYC8; Q94AH6; Q9ZR12; P49677; Q38828; Q38829; Q38830; Q38831; O24407; O24408; O24409; P49678; O24410; Q8LAL2; Q9XFM0; Q38822; Q9M1R4; Q9C5X0; Q9C8Y3; Q39255; Q570C0 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Auxin signaling pathway; Nucleus; Reference proteome; Repressor; Transcription; Transcription regulation Protein physicochemical properties Chain ID B,C Molecular weight (Da) 65385.2 Length 581 Aromaticity 0.09 Instability index 47.83 Isoelectric point 7.46 Charge (pH=7) 1.23 2D Binding mode Binding energy (Kcal/mol) -7.58 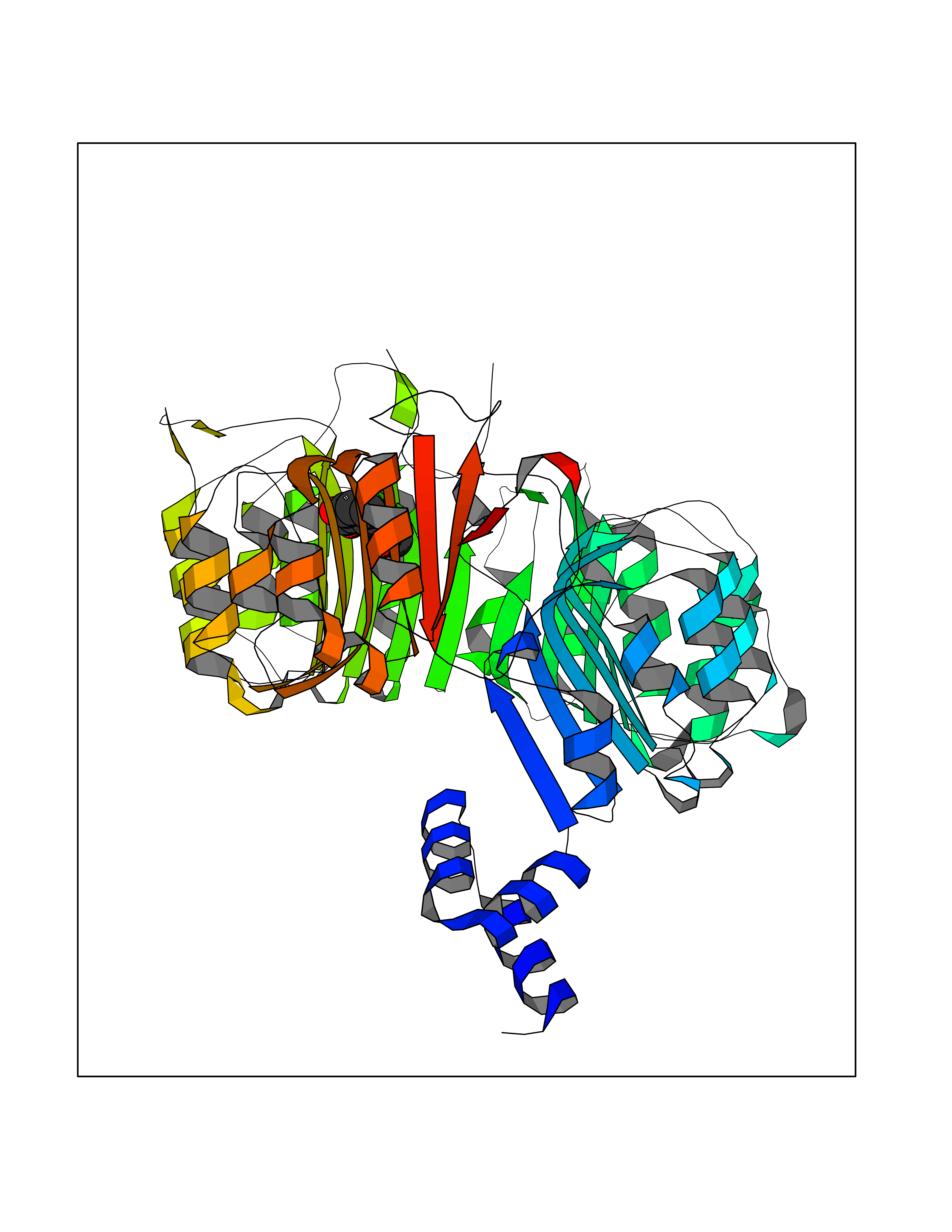 Molscript Map 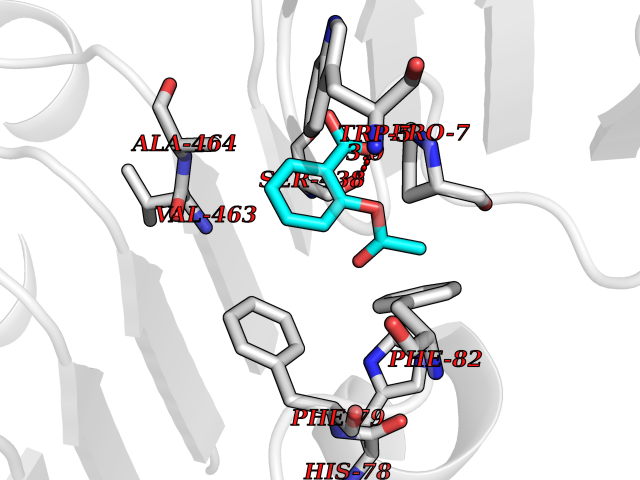 Pymol Map 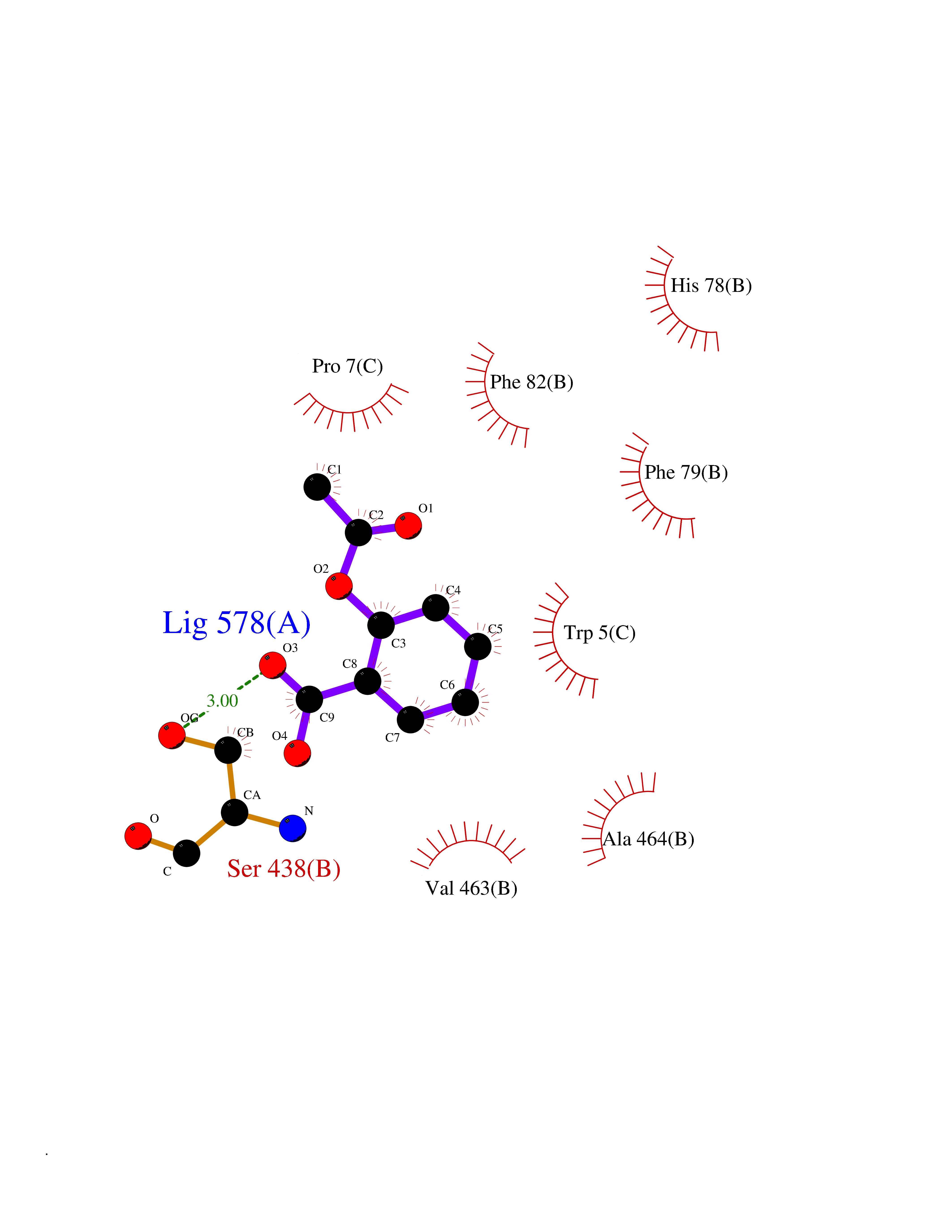 Ligplot Map 3D Binding mode Sequence QVVGWPPVRNYRKFPEEVLEHVFSFIQLDKDRNSVSLVCKSWYEIERWCRRKVFIGNCYAVSPATVIRRFPKVRSVELKGKPHFADFNLVPDGWGGYVYPWIEAMSSSYTWLEEIRLKRMVVTDDCLELIAKSFKNFKVLVLSSCEGFSTDGLAAIAATCRNLKELDLRESDVDDVSGHWLSHFPDTYTSLVSLNISCLASEVSFSALERLVTRCPNLKSLKLNRAVPLEKLATLLQRAPQLEELGTGGYTAEVRPDVYSGLSVALSGCKELRCLSGFWDAVPAYLPAVYSVCSRLTTLNLSYATVQSYDLVKLLCQCPKLQRLWVLDYIEDAGLEVLASTCKDLRELRVFPSEPFVMEPNVALTEQGLVSVSMGCPKLESVLYFCRQMTNAALITIARNRPNMTRFRLCIIEPKAPDYLTLEPLDIGFGAIVEHCKDLRRLSLSGLLTDKVFEYIGTYAKKMEMLSVAFAGDSDLGMHHVLSGCDSLRKLEIRDCPFGDKALLANASKLETMRSLWMSSCSVSFGACKLLGQKMPKLNVEVIDERGAPDSRPESCPVERVFIYRTVAGPRFDMPGFVWNM Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Riboflavin kinase | 1NB0 | 5.56 | |
Target general information Gen name RFK Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Transferase Function ATP binding.Metal ion binding.Riboflavin kinase activity. Related diseases Glutaric aciduria 1 (GA1) [MIM:231670]: An autosomal recessive metabolic disorder characterized by progressive dystonia and athetosis due to gliosis and neuronal loss in the basal ganglia. {ECO:0000269|PubMed:14707522, ECO:0000269|PubMed:18775954, ECO:0000269|PubMed:24973495, ECO:0000269|PubMed:8541831, ECO:0000269|PubMed:8900227, ECO:0000269|PubMed:8900228, ECO:0000269|PubMed:9600243, ECO:0000269|PubMed:9711871}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03247; DB00140 Interacts with Q9NXG0-2; P19438; P19438-1 EC number 2.7.1.26 Uniprot keywords 3D-structure; ATP-binding; Cytoplasm; Flavoprotein; FMN; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Proteomics identification; Reference proteome; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 16749.9 Length 147 Aromaticity 0.12 Instability index 41.55 Isoelectric point 7.09 Charge (pH=7) 0.12 2D Binding mode Binding energy (Kcal/mol) -7.58 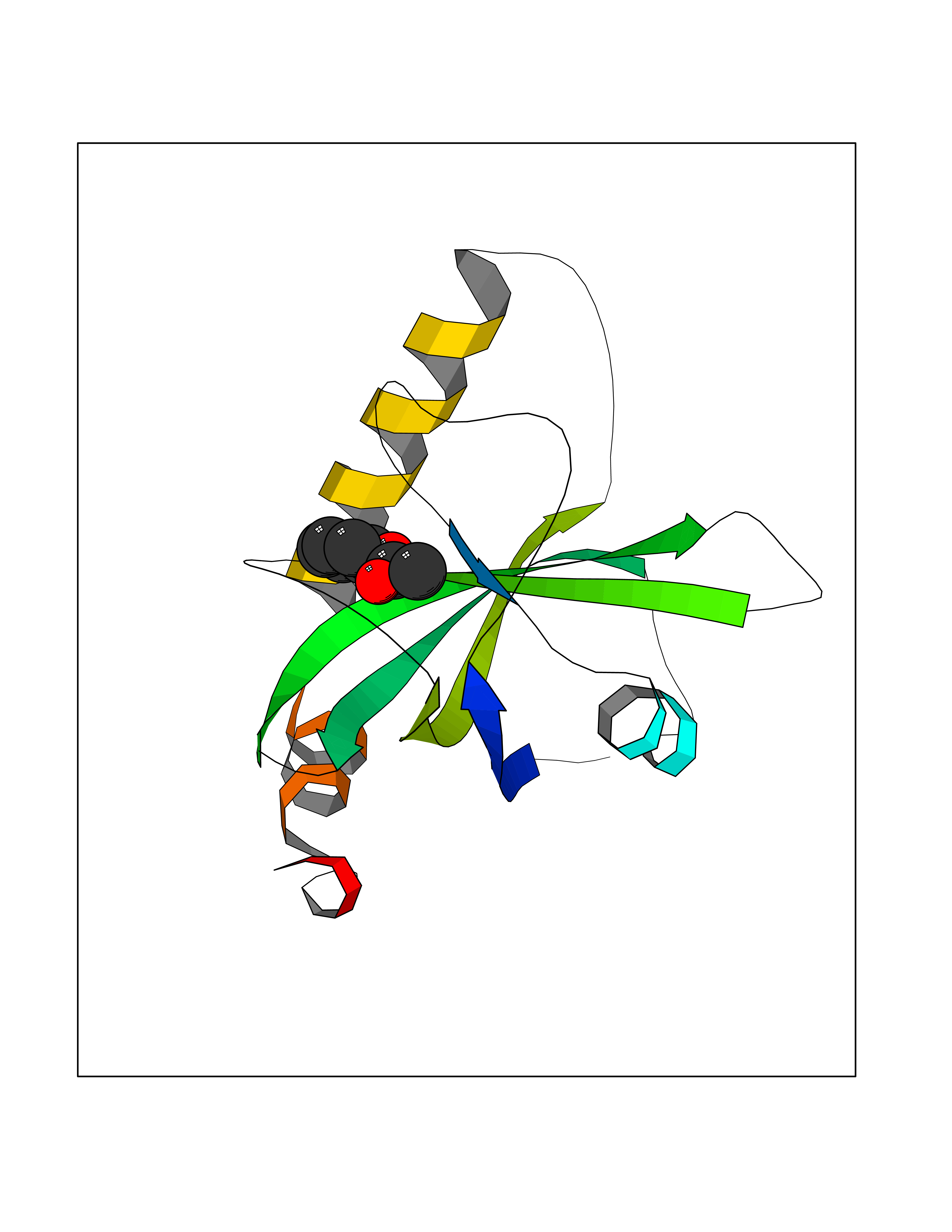 Molscript Map 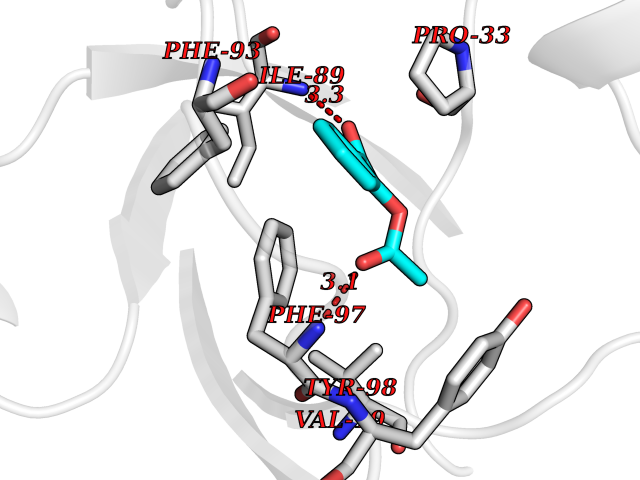 Pymol Map 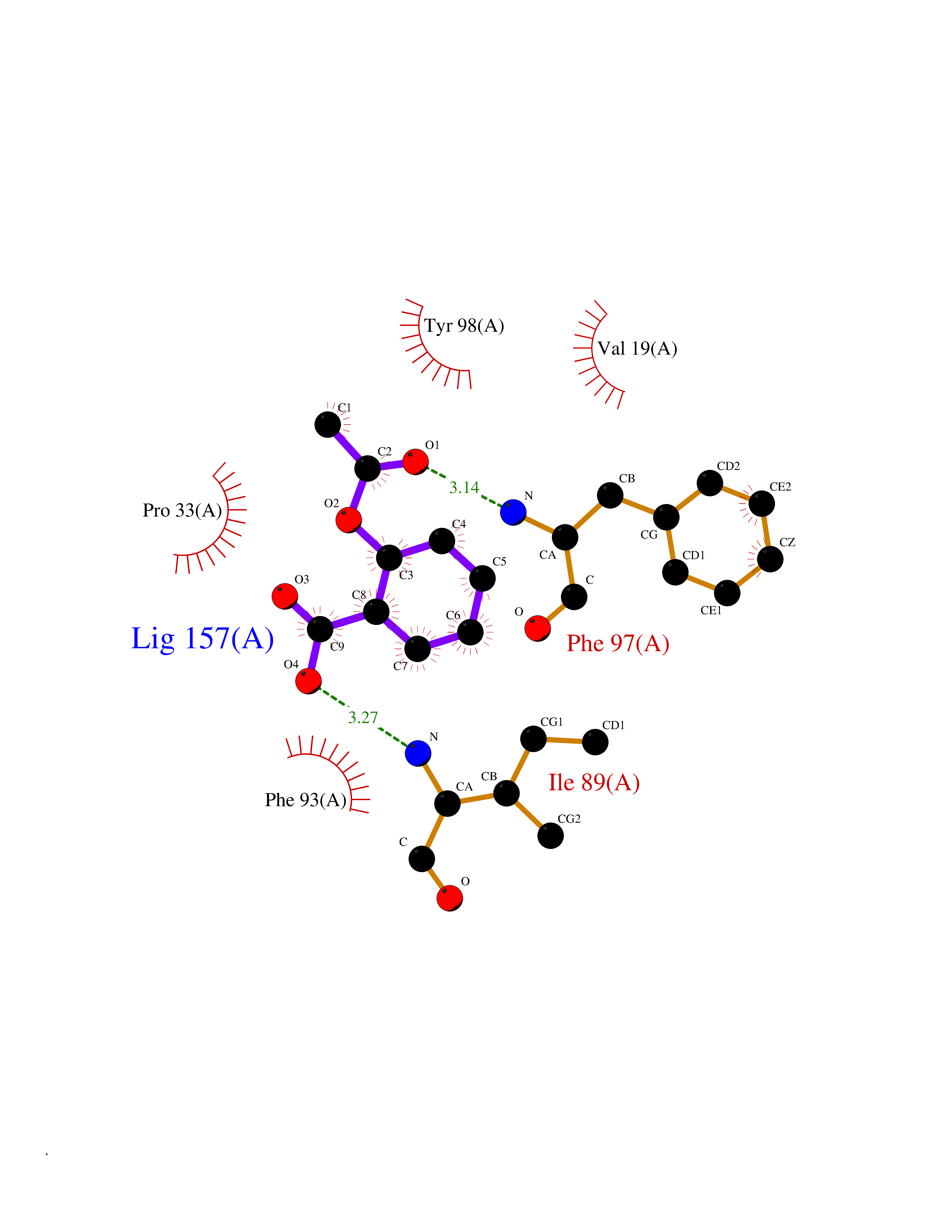 Ligplot Map 3D Binding mode Sequence RHLPYFCRGQVVRGFGRGSKQLGIPTANFPEQVVDNLPADISTGIYYGWASVGSGDVHKMVVSIGWNPYYKNTKKSMETHIMHTFKEDFYGEILNVAIVGYLRPEKNFDSLESLISAIQGDIEEAKKRLELPEYLKIKEDNFFQVSK Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Histone-lysine N-methyltransferase KMT5B (KMT5B) | 3S8P | 5.56 | |
Target general information Gen name KMT5B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine N-methyltransferase 5B; Lysine-specific methyltransferase 5B; Suppressor of variegation 4-20 homolog 1; Su(var)4-20 homolog 1; Suv4-20h1; [histone H4]-N-methyl-L-lysine20 N-methyltransferase KM Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, Suvar4-20 subfamily Biochemical class NA Function Histone methyltransferase that specifically methylates monomethylated 'Lys-20' (H4K20me1) and dimethylated 'Lys-20' (H4K20me2) of histone H4 to produce respectively dimethylated 'Lys-20' (H4K20me2) and trimethylated 'Lys-20' (H4K20me3) and thus regulates transcription and maintenance of genome integrity. In vitro also methylates unmodified 'Lys-20' (H4K20me0) of histone H4 and nucleosomes. H4 'Lys-20' trimethylation represents a specific tag for epigenetic transcriptional repression. Mainly functions in pericentric heterochromatin regions, thereby playing a central role in the establishment of constitutive heterochromatin in these regions. KMT5B is targeted to histone H3 via its interaction with RB1 family proteins (RB1, RBL1 and RBL2) (By similarity). Plays a role in myogenesis by regulating the expression of target genes, such as EID3. Facilitates TP53BP1 foci formation upon DNA damage and proficient non-homologous end-joining (NHEJ)-directed DNA repair by catalyzing the di- and trimethylation of 'Lys-20' of histone H4. May play a role in class switch reconbination by catalyzing the di- and trimethylation of 'Lys-20' of histone H4 (By similarity). Related diseases Intellectual developmental disorder, autosomal dominant 51 (MRD51) [MIM:617788]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. {ECO:0000269|PubMed:28191889, ECO:0000269|PubMed:29276005}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9H2G4; Q61026 EC number EC 2.1.1.361 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Disease variant; Intellectual disability; Isopeptide bond; Metal-binding; Methyltransferase; Myogenesis; Nucleus; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 26230.2 Length 233 Aromaticity 0.11 Instability index 42.29 Isoelectric point 5.64 Charge (pH=7) -6.07 2D Binding mode Binding energy (Kcal/mol) -7.58  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence XSAKELCENDDLATSLVLDPYLGFQTHKXNTRQEELKEVIERFKKDEHLEKAFKCLTSGEWARHYFLNKNKXQEKLFKEHVFIYLRXFATDSGFEILPCNRYSSEQNGAKIVATKEWKRNDKIELLVGCIAELSEIEENXLLRHGENDFSVXYSTRKNCAQLWLGPAAFINHDCRPNCKFVSTGRDTACVKALRDIEPGEEISCYYGDGFFGENNEFCECYTCERRGTGAFKS Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Histone-lysine N-methyltransferase KMT5C (KMT5C) | 3RQ4 | 5.56 | |
Target general information Gen name KMT5C Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine N-methyltransferase 5C; Lysine-specific methyltransferase 5C; Suppressor of variegation 4-20 homolog 2; Su(var)4-20 homolog 2; Suv4-20h2; [histone H4]-N-methyl-L-lysine20 N-methyltransferase KM Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, Suvar4-20 subfamily Biochemical class NA Function Histone methyltransferase that specifically methylates monomethylated 'Lys-20' (H4K20me1) and dimethylated 'Lys-20' (H4K20me2) of histone H4 to produce respectively dimethylated 'Lys-20' (H4K20me2) and trimethylated 'Lys-20' (H4K20me3) and thus regulates transcription and maintenance of genome integrity. In vitro also methylates unmodified 'Lys-20' (H4K20me0) of histone H4 and nucleosomes. H4 'Lys-20' trimethylation represents a specific tag for epigenetic transcriptional repression. Mainly functions in pericentric heterochromatin regions, thereby playing a central role in the establishment of constitutive heterochromatin in these regions. KMT5C is targeted to histone H3 via its interaction with RB1 family proteins (RB1, RBL1 and RBL2) (By similarity). Facilitates TP53BP1 foci formation upon DNA damage and proficient non-homologous end-joining (NHEJ)-directed DNA repair by catalyzing the di- and trimethylation of 'Lys-20' of histone H4. May play a role in class switch reconbination by catalyzing the di- and trimethylation of 'Lys-20' of histone H4 (By similarity). Related diseases Brachydactyly A2 (BDA2) [MIM:112600]: A form of brachydactyly. Brachydactyly defines a group of inherited malformations characterized by shortening of the digits due to abnormal development of the phalanges and/or the metacarpals. In brachydactyly type A2 shortening of the middle phalanges is confined to the index finger and the second toe, all other digits being more or less normal. Because of a rhomboid or triangular shape of the affected middle phalanx, the end of the second finger usually deviates radially. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}. The gene represented in this entry is involved in disease pathogenesis. Duplications of a cis-regulatory element located approximately 110 kb downstream of BMP2 have been found in BDA2 families. They likely cause altered BMP2 expression with pathological consequences. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}.; DISEASE: Short stature, facial dysmorphism, and skeletal anomalies with or without cardiac anomalies 1 (SSFSC1) [MIM:617877]: An autosomal dominant disorder characterized by short stature, facial dysmorphism, skeletal anomalies, and variable cardiac defects. Distinctive facial features include midface retrusion, short upturned nose, long philtrum, high-arched or cleft palate, and variable degrees of micrognathia and dental crowding. Skeletal anomalies include patterning defects of the axial skeleton, characterized by 11 pairs of ribs and brachydactyly of the fifth ray. Congenital heart defects are variably observed and appear to involve primarily the cardiac outflow tract. {ECO:0000269|PubMed:29198724}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q13185 EC number EC 2.1.1.361 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 27285.8 Length 240 Aromaticity 0.1 Instability index 42.74 Isoelectric point 8.32 Charge (pH=7) 3.24 2D Binding mode Binding energy (Kcal/mol) -7.59  Molscript Map 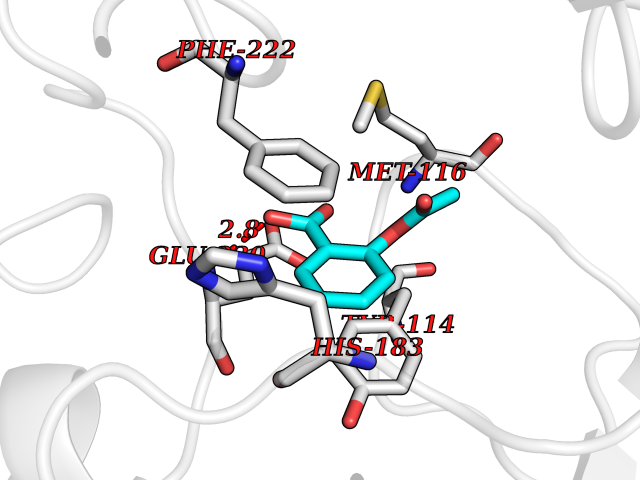 Pymol Map 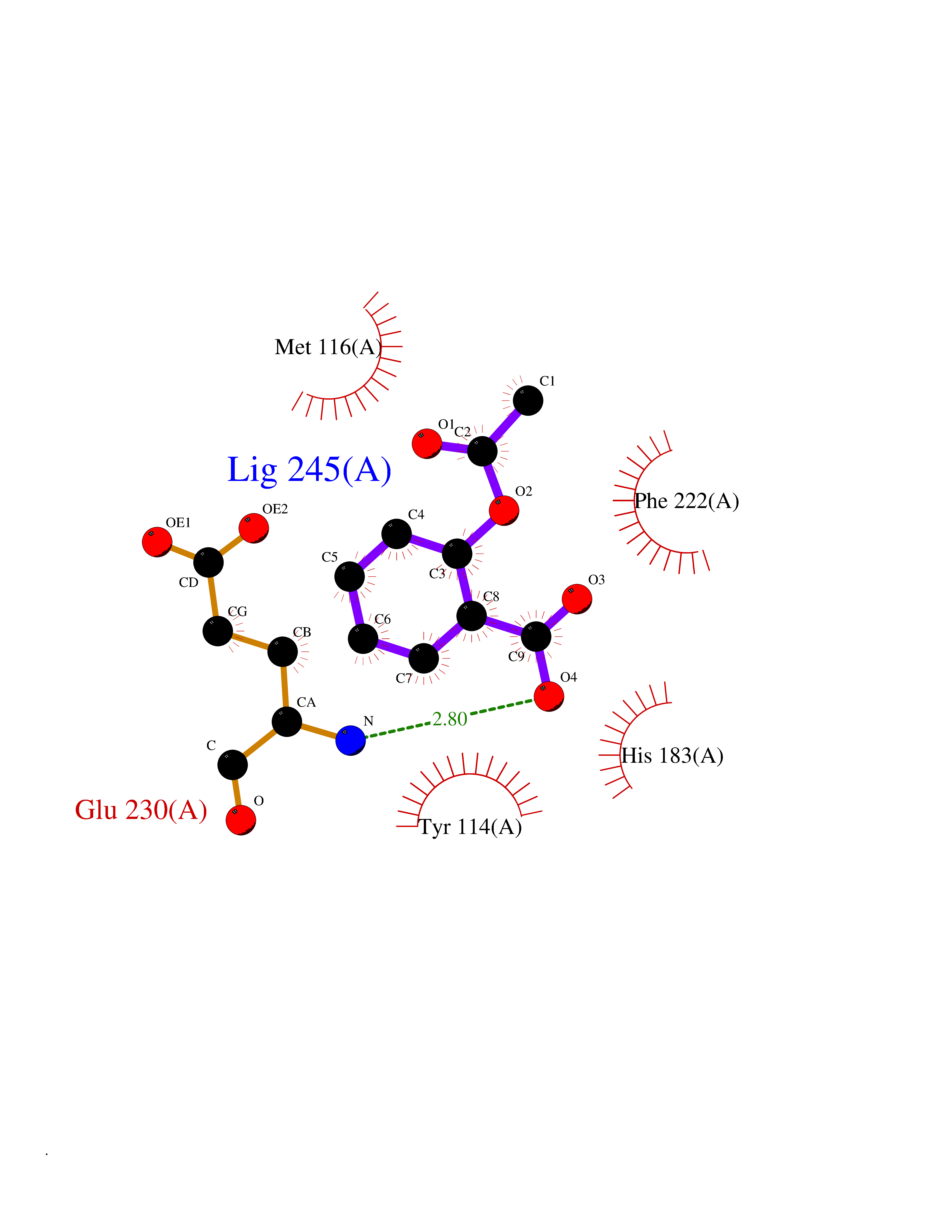 Ligplot Map 3D Binding mode Sequence DRVTARELCENDDLATSLVLDPYLGFRTHKMNVSPVPPLRRQQHLRSALETFLRQRDLEAAYRALTLGGWTARYFQSRGPRQEAALKTHVYRYLRAFLPESGFTILPCTRYSMETNGAKIVSTRAWKKNEKLELLVGCIAELREADEGLLRAGENDFSIMYSTRKRSAQLWLGPAAFINHDCKPNCKFVPADGNAACVKVLRDIEPGDEVTCFYGEGFFGEKNEHCECHTCERKGEGAFR Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Caspase-7 (CASP7) | 1SHJ | 5.56 | |
Target general information Gen name CASP7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MCH3; ICE-like apoptotic protease 3; ICE-LAP3; CMH-1; CASP-7; Apoptotic protease Mch-3 Protein family Peptidase C14A family Biochemical class Peptidase Function Cleaves and activates sterol regulatory element binding proteins (SREBPs). Proteolytically cleaves poly(ADP-ribose) polymerase (PARP) at a '216-Asp-|-Gly-217' bond. Overexpression promotes programmed cell death. Involved in the activation cascade of caspases responsible for apoptosis execution. Related diseases Pregnancy loss, recurrent, 3 (RPRGL3) [MIM:614391]: A common complication of pregnancy, resulting in spontaneous abortion before the fetus has reached viability. The term includes all miscarriages from the time of conception until 24 weeks of gestation. Recurrent pregnancy loss is defined as 3 or more consecutive spontaneous abortions. {ECO:0000269|PubMed:17339269}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05408; DB03384; DB06255 Interacts with Q13490; P83105; P42858; Q8N4N3-2; P43364; Q16236; Q9GZT8; Q13177; P27986-2; P21673; Q86WV1-2; P17405; P98170 EC number EC 3.4.22.60 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; Apoptosis; Cytoplasm; Hydrolase; Nucleus; Phosphoprotein; Protease; Proteomics identification; Reference proteome; RNA-binding; Secreted; Thiol protease; Ubl conjugation; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 47441.5 Length 417 Aromaticity 0.11 Instability index 20.98 Isoelectric point 8.38 Charge (pH=7) 6.12 2D Binding mode Binding energy (Kcal/mol) -7.58 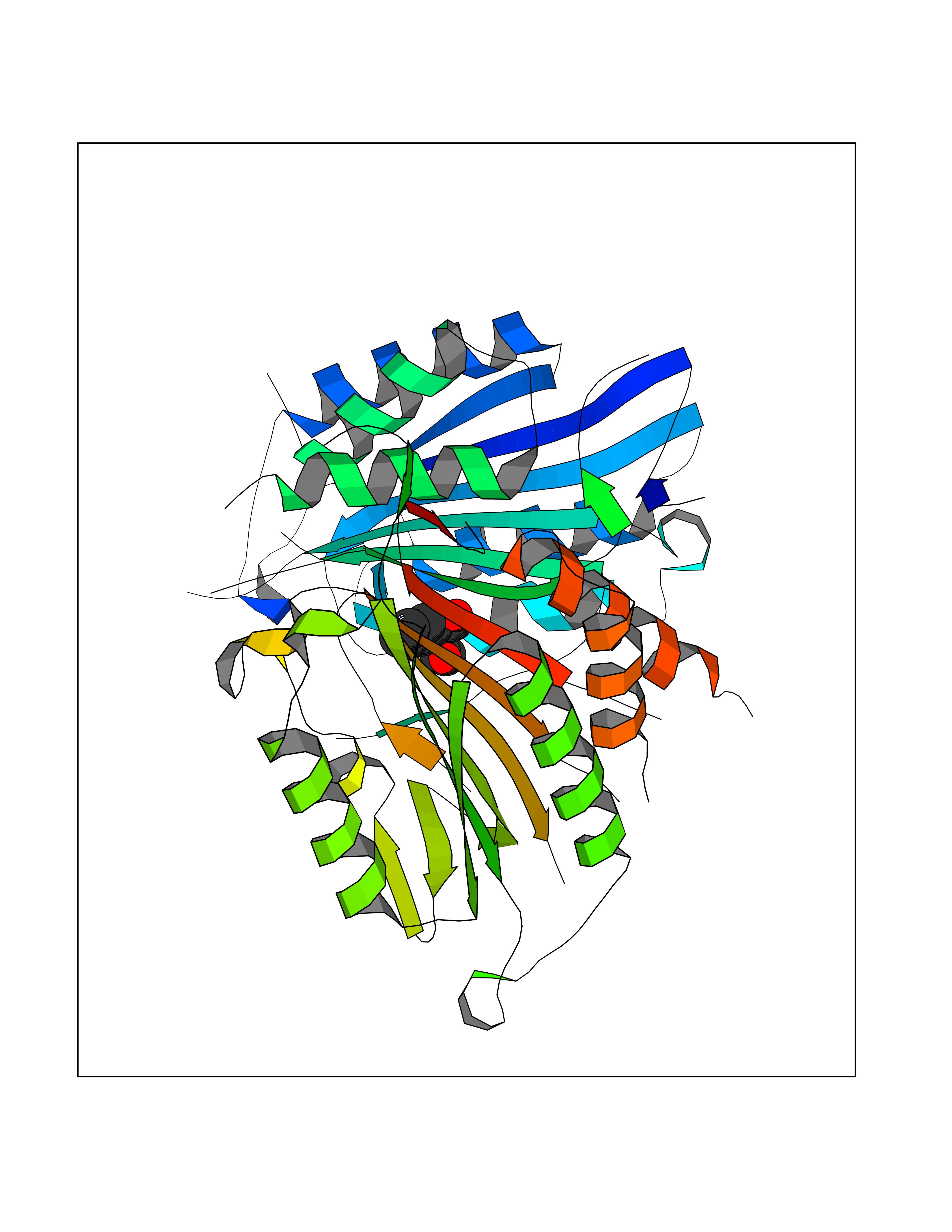 Molscript Map 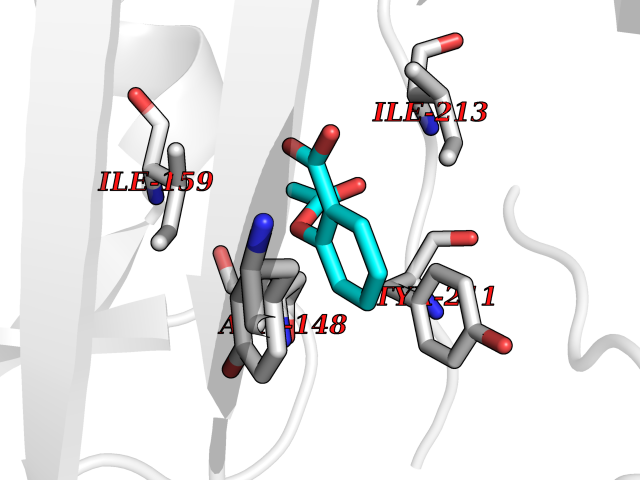 Pymol Map 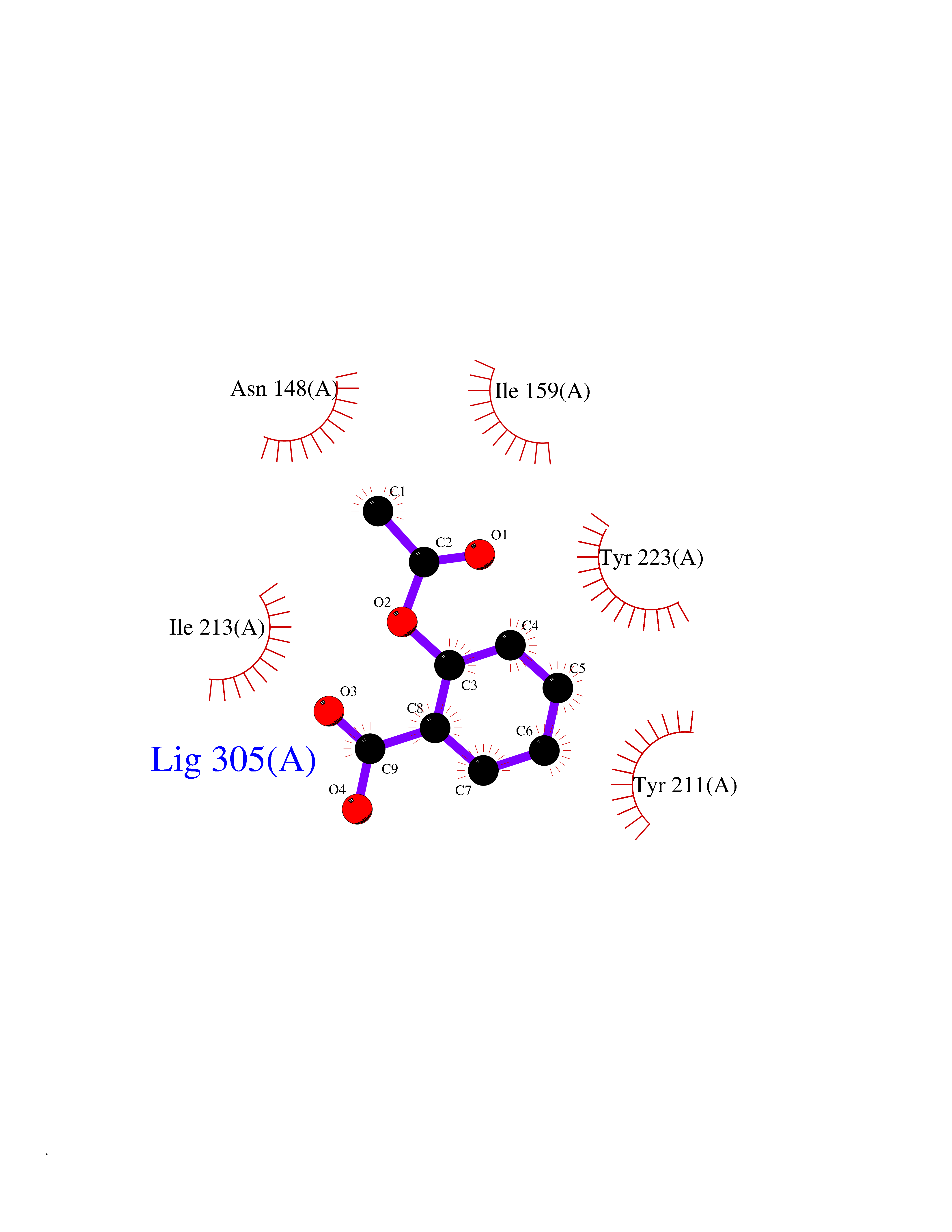 Ligplot Map 3D Binding mode Sequence TYQYNMNFEKLGKCIIINNKNFDKVTGMGVRNGTDKDAEALFKCFRSLGFDVIVYNDCSCAKMQDLLKKASEEDHTNAACFACILLSHGEENVIYGKDGVTPIKDLTAHFRGARCKTLLEKPKLFFIQACRGTEPRYKIPVEADFLFAYSTVRGSWFVQALCSILEEHGKDLEIMQILTRVNDRVARHFKKQIPCVVSMLTKELYFSQVPTYQYNMNFEKLGKCIIINNKNFDKVTGMGVRNGTDKDAEALFKCFRSLGFDVIVYNDCSCAKMQDLLKKASEEDHTNAACFACILLSHGEENVIYGKDGVTPIKDLTAHFRGARCKTLLEKPKLFFIQACRGPRYKIPVEADFLFAYSTVPGSWFVQALCSILEEHGKDLEIMQILTRVNDRVARHFESKQIPCVVSMLTKELYFSQ Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Cytochrome P450 1B1 (CYP1B1) | 3PM0 | 5.56 | |
Target general information Gen name CYP1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CYPIB1 Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function In liver microsomes, this enzyme is involved in an NADPH-dependent electron transport pathway. It oxidizes a variety of structurally unrelated compounds, including steroids, fatty acids, retinoid and xenobiotics. Preferentially oxidizes 17beta-estradiol to the carcinogenic 4-hydroxy derivative, and a variety of procarcinogenic compounds to their activated forms, including polycyclic aromatic hydrocarbons. Promotes angiogenesis by removing cellular oxygenation products, thereby decreasing oxidative stress, release of antiangiogenic factor THBS2, then allowing endothelial cells migration, cell adhesion and capillary morphogenesis. These changes are concommitant with the endothelial nitric oxide synthase activity and nitric oxide synthesis. Plays an important role in the regulation of perivascular cell proliferation, migration, and survival through modulation of the intracellular oxidative state and NF-kappa-B expression and/or activity, during angiogenesis. Contributes to oxidative homeostasis and ultrastructural organization and function of trabecular meshwork tissue through modulation of POSTN expression. Cytochromes P450 are a group of heme-thiolate monooxygenases. Related diseases Anterior segment dysgenesis 6 (ASGD6) [MIM:617315]: A form of anterior segment dysgenesis, a group of defects affecting anterior structures of the eye including cornea, iris, lens, trabecular meshwork, and Schlemm canal. Anterior segment dysgeneses result from abnormal migration or differentiation of the neural crest derived mesenchymal cells that give rise to components of the anterior chamber during eye development. Different anterior segment anomalies may exist alone or in combination, including iris hypoplasia, enlarged or reduced corneal diameter, corneal vascularization and opacity, posterior embryotoxon, corectopia, polycoria, abnormal iridocorneal angle, ectopia lentis, and anterior synechiae between the iris and posterior corneal surface. Clinical conditions falling within the phenotypic spectrum of anterior segment dysgeneses include aniridia, Axenfeld anomaly, Reiger anomaly/syndrome, Peters anomaly, and iridogoniodysgenesis. ASGD6 patients predominantly manifest Peters anomaly. Peters anomaly consists of corneal leukoma, defects in the posterior structures of the cornea such as absence of the posterior corneal stroma and Descemet membrane, and a variable degree of iridocorneal and/or keratolenticular adhesions. Over 50% of patients develop glaucoma in childhood. {ECO:0000269|PubMed:11403040}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glaucoma 3, primary congenital, A (GLC3A) [MIM:231300]: An autosomal recessive form of primary congenital glaucoma (PCG). PCG is characterized by marked increase of intraocular pressure at birth or early childhood, large ocular globes (buphthalmos) and corneal edema. It results from developmental defects of the trabecular meshwork and anterior chamber angle of the eye that prevent adequate drainage of aqueous humor. {ECO:0000269|PubMed:10227395, ECO:0000269|PubMed:10655546, ECO:0000269|PubMed:11184479, ECO:0000269|PubMed:11527932, ECO:0000269|PubMed:11774072, ECO:0000269|PubMed:11980847, ECO:0000269|PubMed:12036985, ECO:0000269|PubMed:12525557, ECO:0000269|PubMed:14635112, ECO:0000269|PubMed:14640114, ECO:0000269|PubMed:15255109, ECO:0000269|PubMed:15342693, ECO:0000269|PubMed:15475877, ECO:0000269|PubMed:16490498, ECO:0000269|PubMed:16688110, ECO:0000269|PubMed:16735994, ECO:0000269|PubMed:16862072, ECO:0000269|PubMed:18470941, ECO:0000269|PubMed:9463332, ECO:0000269|PubMed:9497261}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Glaucoma 1, open angle, A (GLC1A) [MIM:137750]: A form of primary open angle glaucoma (POAG). POAG is characterized by a specific pattern of optic nerve and visual field defects. The angle of the anterior chamber of the eye is open, and usually the intraocular pressure is increased. However, glaucoma can occur at any intraocular pressure. The disease is generally asymptomatic until the late stages, by which time significant and irreversible optic nerve damage has already taken place. {ECO:0000269|PubMed:11774072}. The gene represented in this entry acts as a disease modifier. Digenic mutations in CYP1B1 and MYOC have been found in a family segregating both primary adult-onset and juvenile forms of open angle glaucoma (PubMed:11774072). All affected family members with mutations in both MYOC and CYP1B1 had juvenile glaucoma, whereas those with only the MYOC mutation had the adult-onset form (PubMed:11774072). {ECO:0000269|PubMed:11774072}. Drugs (DrugBank ID) DB02342; DB00613; DB06732; DB00443; DB00121; DB01222; DB00201; DB09061; DB14737; DB01254; DB00694; DB01248; DB00997; DB00470; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB07776; DB00499; DB01645; DB01381; DB00741; DB01064; DB01026; DB00448; DB14009; DB01065; DB00170; DB00959; DB01204; DB14011; DB03467; DB00338; DB01229; DB14631; DB00635; DB01087; DB00396; DB00818; DB04216; DB02709; DB00675; DB00624; DB13946; DB00277; DB12245; DB11155 Interacts with Q02763 EC number EC 1.14.14.- Uniprot keywords 3D-structure; Disease variant; Endoplasmic reticulum; Fatty acid metabolism; Glaucoma; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Mitochondrion; Monooxygenase; Oxidoreductase; Peters anomaly; Proteomics identification; Reference proteome; Steroid metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 51875.9 Length 459 Aromaticity 0.1 Instability index 34.16 Isoelectric point 8.64 Charge (pH=7) 4.89 2D Binding mode Binding energy (Kcal/mol) -7.59 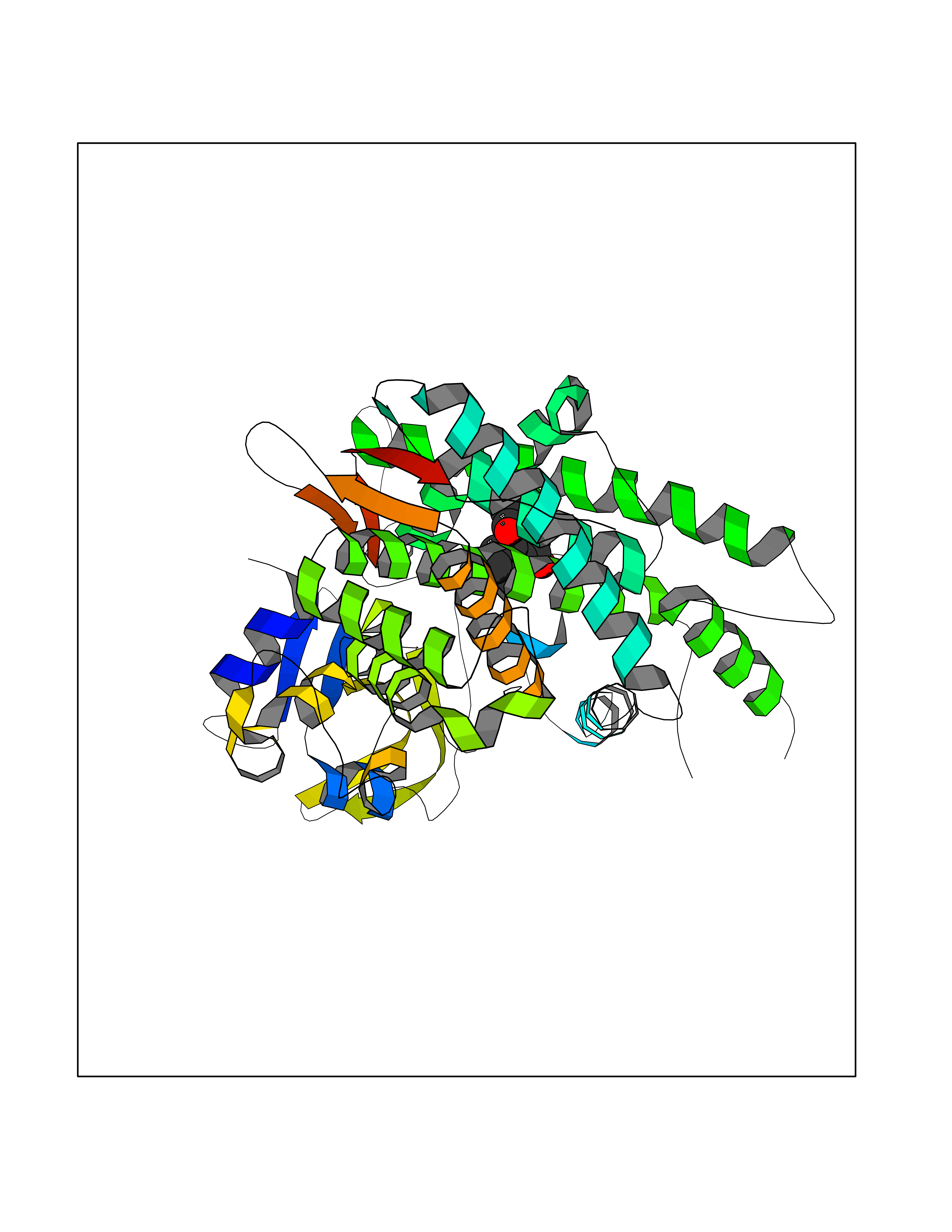 Molscript Map 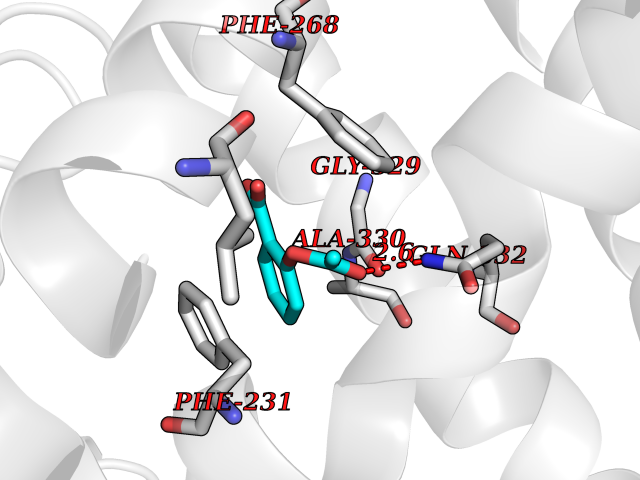 Pymol Map 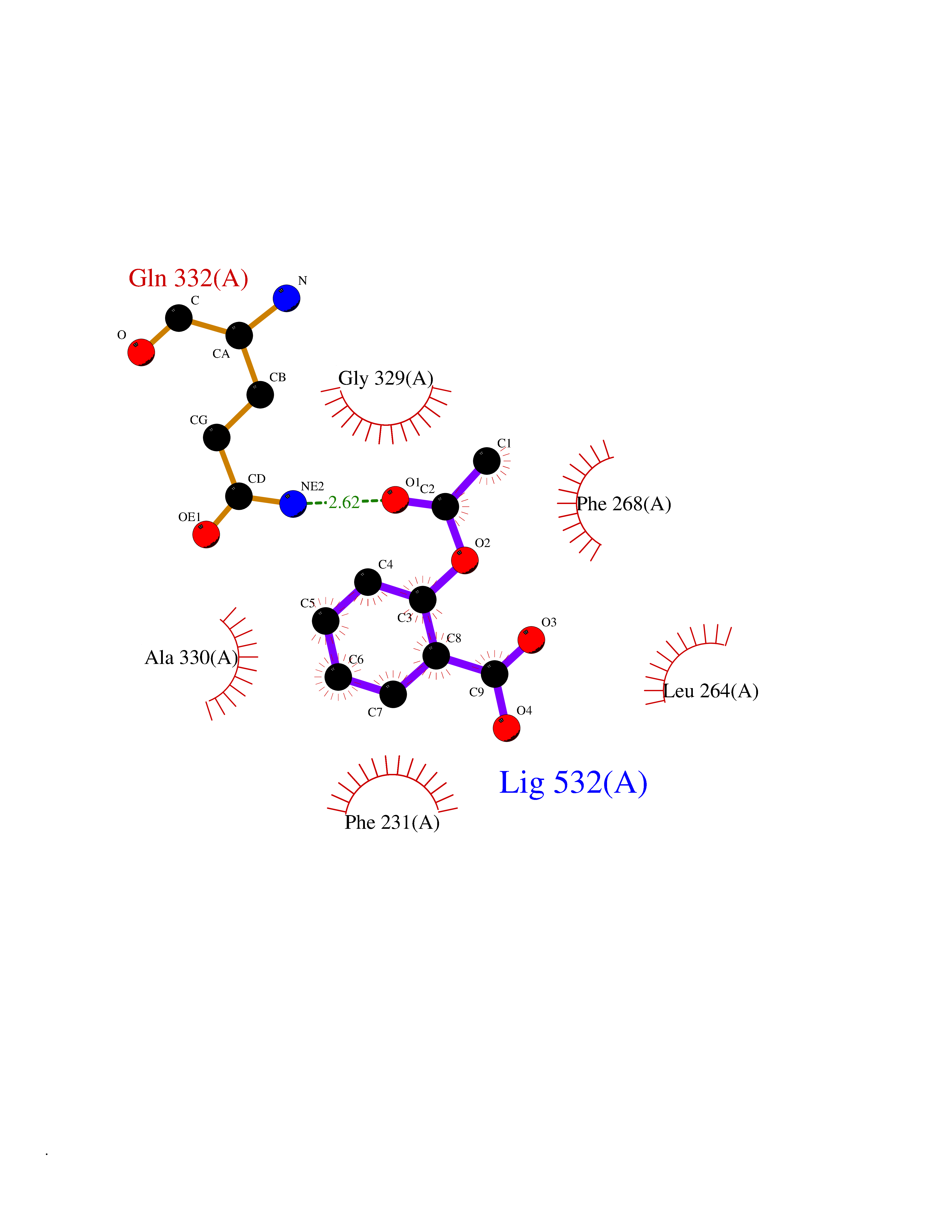 Ligplot Map 3D Binding mode Sequence QAAHLSFARLARRYGDVFQIRLGSCPIVVLNGERAIHQALVQQGSAFADRPSFASFRVVSGGRSMAFGHYSEHWKVQRRAAHSMMRNFFTRQPRSRQVLEGHVLSEARELVALLVRGSADGAFLDPRPLTVVAVANVMSAVCFGCRYSHDDPEFRELLSHNEEFGRTVGAGSLVDVMPWLQYFPNPVRTVFREFEQLNRNFSNFILDKFLRHCESLRPGAAPRDMMDAFILSAEKKAAGDGARLDLENVPATITDIFGASQDTLSTALQWLLLLFTRYPDVQTRVQAELDQVVGRDRLPCMGDQPNLPYVLAFLYEAMRFSSFVPVTIPHATTANTSVLGYHIPKDTVVFVNQWSVNHDPLKWPNPENFDPARFLDKDGLINKDLTSRVMIFSVGKRRCIGEELSKMQLFLFISILAHQCDFRANPNEPAKMNFSYGLTIKPKSFKVNVTLRESMELLD Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Orexin receptor type 2 (HCRTR2) | 4S0V | 5.55 | |
Target general information Gen name HCRTR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ox2r; Ox2-R; Ox-2-R; Orexin-2 receptor; Hypocretin receptor type 2; HFGANP Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Triggers an increase in cytoplasmic Ca(2+) levels in response to orexin-A binding. Nonselective, high-affinity receptor for both orexin-A and orexin-B neuropeptides. Related diseases Hyperchlorhidrosis, isolated (HYCHL) [MIM:143860]: An autosomal recessive disorder characterized by excessive sweating and increased sweat chloride levels. Affected individuals suffer from episodes of hyponatremic dehydration and report increased amounts of visible salt precipitates in sweat. {ECO:0000269|PubMed:21035102, ECO:0000269|PubMed:21184099, ECO:0000269|PubMed:26911677}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15031; DB11951; DB09034 Interacts with P62937 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 32524.7 Length 282 Aromaticity 0.14 Instability index 38.96 Isoelectric point 9.18 Charge (pH=7) 12.36 2D Binding mode Binding energy (Kcal/mol) -7.57  Molscript Map 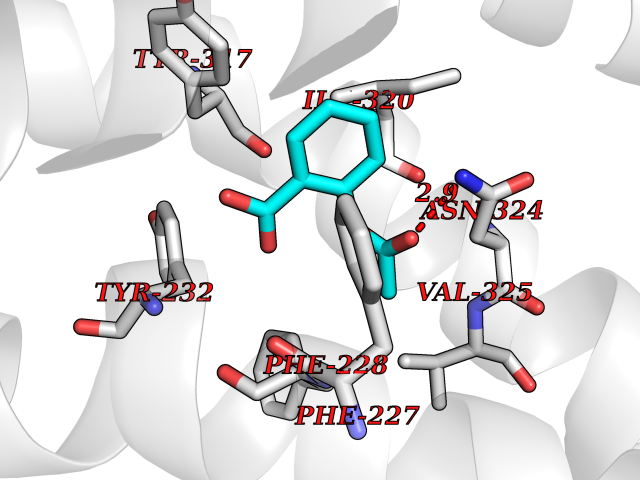 Pymol Map 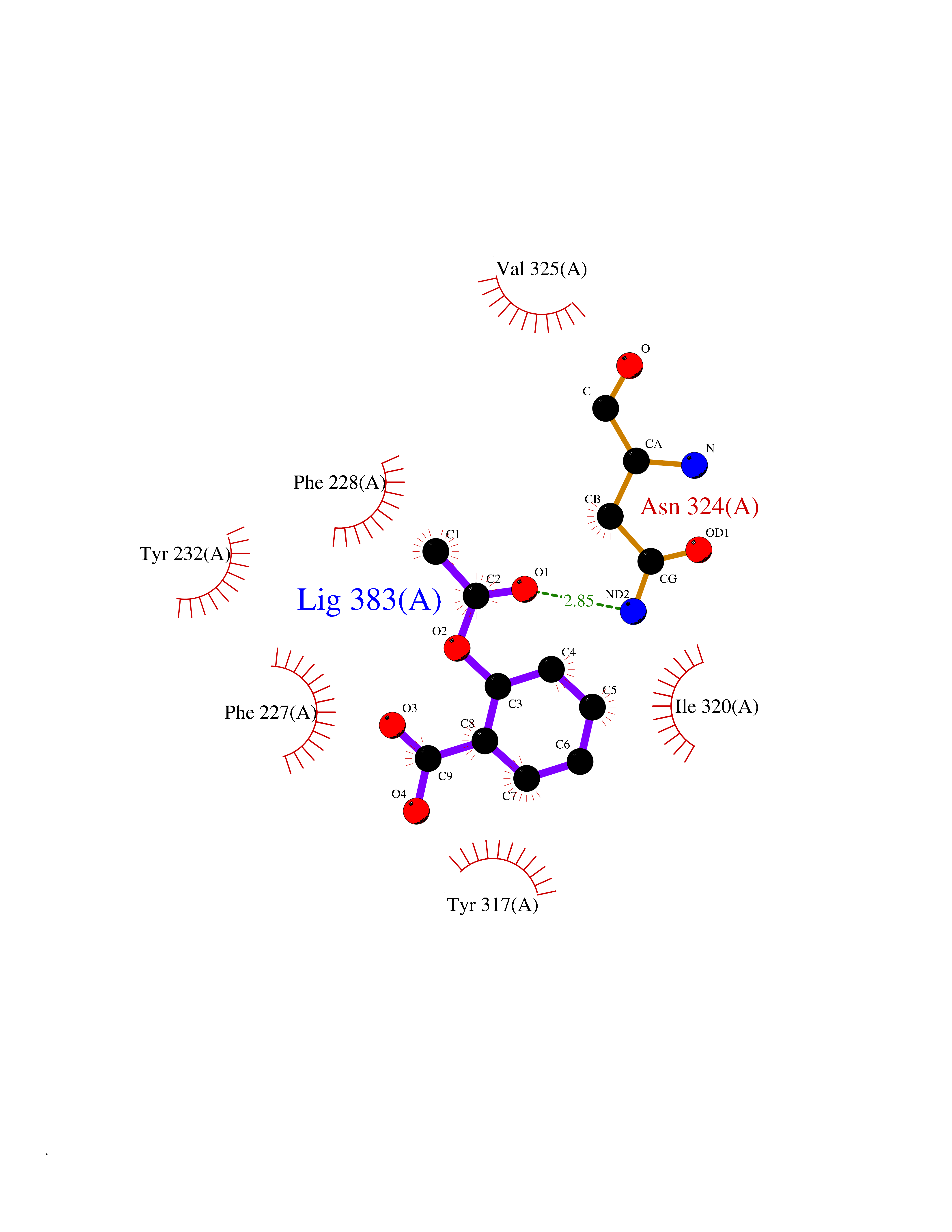 Ligplot Map 3D Binding mode Sequence PKEYEWVLIAGYIIVFVVALIGNVLVCVAVWKNHHMRTVTNYFIVNLSLADVLVTITCLPATLVVDITETWFFGQSLCKVIPYLQTVSVSVSVLTLSCIALDRWYAICHPSTAKRARNSIVIIWIVSCIIMIPQAIVMECSTVFKTTLFTVCDERWGGEIYPKMYHICFFLVTYMAPLCLMVLAYLQIFRKLWCRQKQIRARRKTARMLMVVLLVFAICYLPISILNVLKRVFGMFAHDRETVYAWFTFSHWLVYANSAANPIIYNFLSGKFREEFKAAFSC Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | L-serine dehydratase/L-threonine deaminase | 1P5J | 5.55 | |
Target general information Gen name SDS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms SDH Protein family Serine/threonine dehydratase family Biochemical class Lyase Function L-serine ammonia-lyase activity.L-threonine ammonia-lyase activity.Protein homodimerization activity.Pyridoxal phosphate binding. Related diseases Immunodeficiency, common variable, 12, with autoimmunity (CVID12) [MIM:616576]: A primary immunodeficiency characterized by hypogammaglobulinemia and recurrent bacterial infections. About half of patients develop autoimmune features, including cytopenia, as well as generalized inflammation and lymphoproliferation manifest as lymphadenopathy or hepatosplenomegaly. {ECO:0000269|PubMed:26279205}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00114; DB00133 Interacts with Q8WTU0; O14964; Q96PF1 EC number 4.3.1.17; 4.3.1.19 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Gluconeogenesis; Lipid metabolism; Lyase; Proteomics identification; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 33559.7 Length 319 Aromaticity 0.06 Instability index 34.65 Isoelectric point 7.2 Charge (pH=7) 0.42 2D Binding mode Binding energy (Kcal/mol) -7.57  Molscript Map 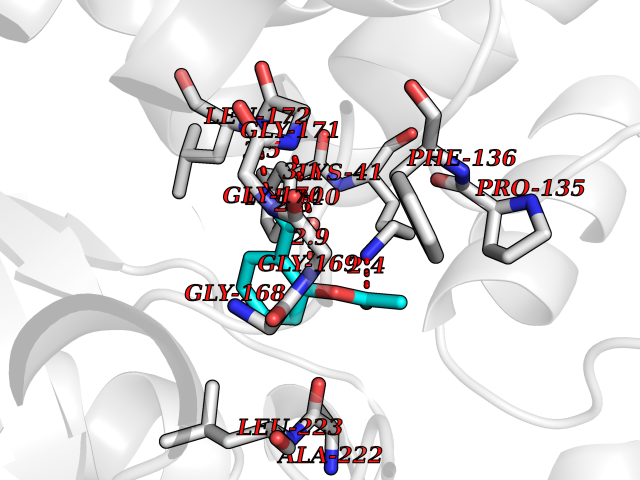 Pymol Map 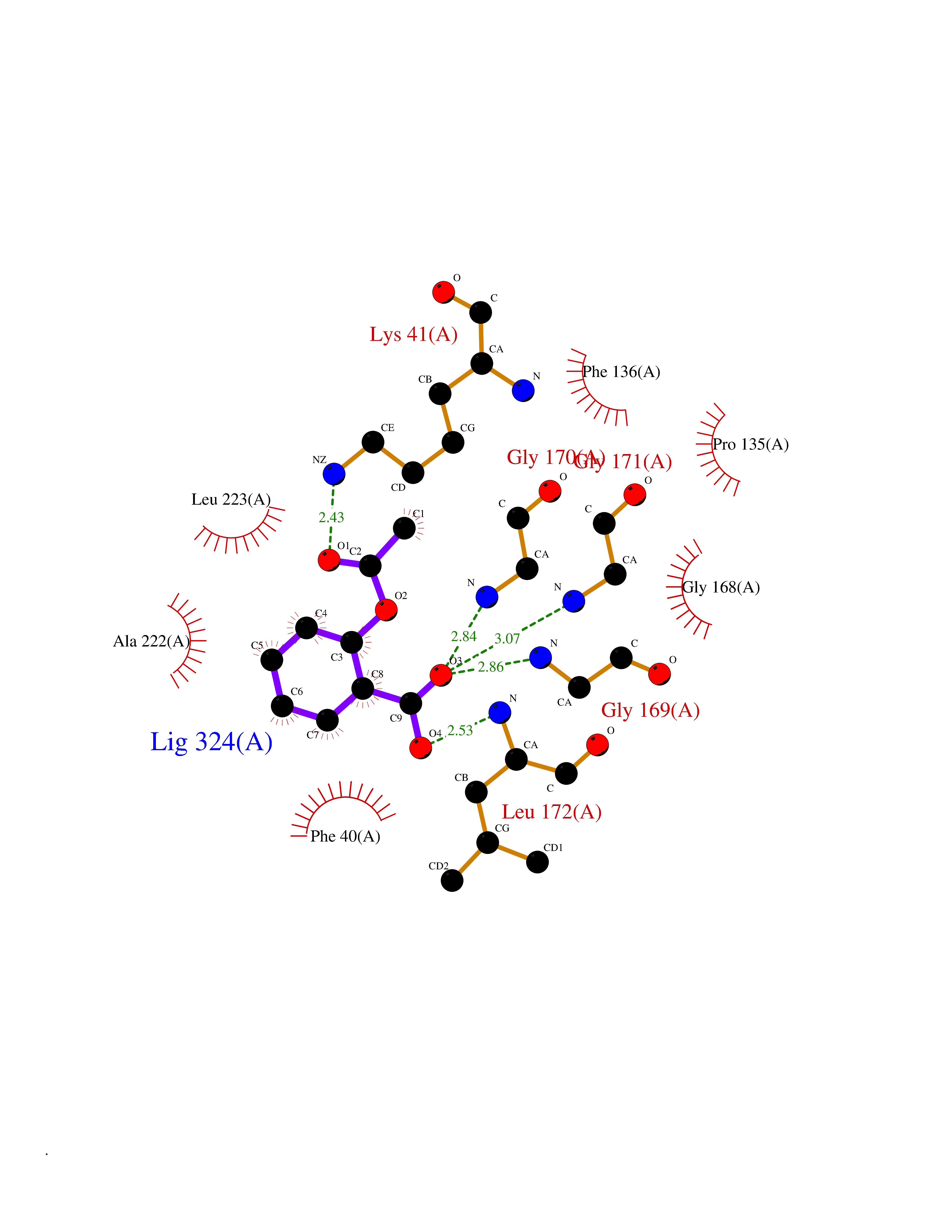 Ligplot Map 3D Binding mode Sequence GEPLHVKTPIRDSMALSKMAGTSVYLKMDSAQPSGSFKIRGIGHFCKRWAKQGCAHFVCSSAGNAGMAAAYAARQLGVPATIVVPGTTPALTIERLKNEGATCKVVGELLDEAFELAKALAKNNPGWVYIPPFDDPLIWEGHASIVKELKETLWEKPGAIALSVGGGGLLCGVVQGLQECGWGDVPVIAMETFGAHSFHAATTAGKLVSLPKITSVAKALGVKTVGSQALKLFQEHPIFSEVISDQEAVAAIEKFVDDEKILVEPACGAALAAVYSHVIQKLQLEGNLRTPLPSLVVIVCGGSNISLAQLRALKEQLGM Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Low molecular weight phosphotyrosine protein phosphatase | 5KQL | 5.55 | |
Target general information Gen name ACP1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Low molecular weight phosphotyrosine protein phosphatase family Biochemical class hydrolase / hydrolase inhibitor Function Acid phosphatase activity.Non-membrane spanning protein tyrosine phosphatase activity. Related diseases Waardenburg syndrome 4A (WS4A) [MIM:277580]: A disorder characterized by the association of Waardenburg features (depigmentation and deafness) with the absence of enteric ganglia in the distal part of the intestine (Hirschsprung disease). {ECO:0000269|PubMed:12189494, ECO:0000269|PubMed:8634719}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hirschsprung disease 2 (HSCR2) [MIM:600155]: A disorder of neural crest development characterized by absence of enteric ganglia along a variable length of the intestine. It is the most common cause of congenital intestinal obstruction. Early symptoms range from complete acute neonatal obstruction, characterized by vomiting, abdominal distention and failure to pass stool, to chronic constipation in the older child. {ECO:0000269|PubMed:11471546, ECO:0000269|PubMed:28236341, ECO:0000269|PubMed:8001158, ECO:0000269|PubMed:8630503, ECO:0000269|PubMed:8852660}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: ABCD syndrome (ABCDS) [MIM:600501]: An autosomal recessive syndrome characterized by albinism, black lock at temporal occipital region, bilateral deafness, aganglionosis of the large intestine and total absence of neurocytes and nerve fibers in the small intestine. {ECO:0000269|PubMed:11891690}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Heterozygous mutations in EDNRB may be responsible for Waardenburg syndrome 2, an autosomal dominant disorder characterized by sensorineural deafness and pigmentary disturbances. {ECO:0000269|PubMed:28236341}. Drugs (DrugBank ID) DB04214; DB00173 Interacts with Q96CV9 EC number 3.1.3.2; 3.1.3.48 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Hydrolase; Phosphoprotein; Protein phosphatase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 17582.8 Length 154 Aromaticity 0.09 Instability index 50.52 Isoelectric point 7 Charge (pH=7) -0 2D Binding mode Binding energy (Kcal/mol) -7.57 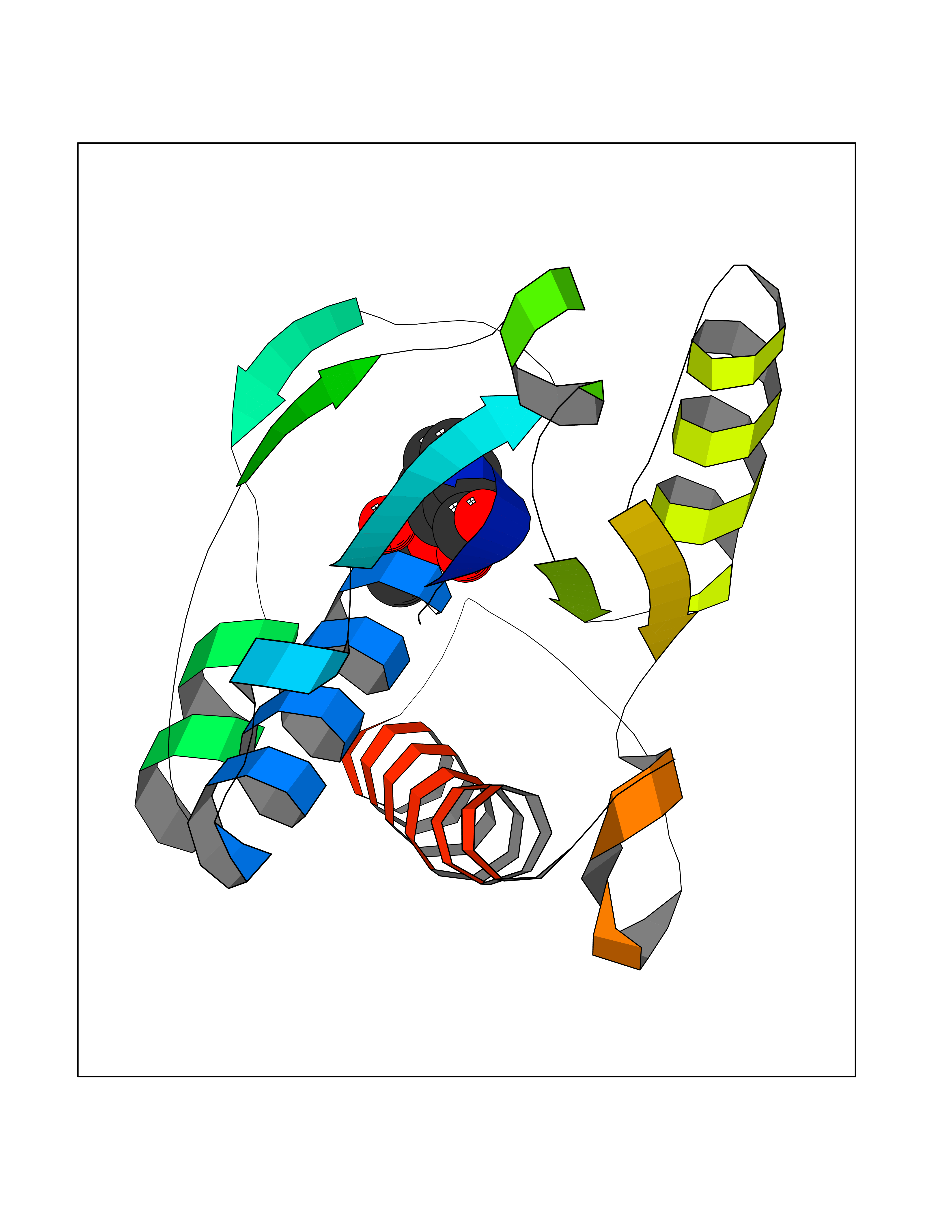 Molscript Map 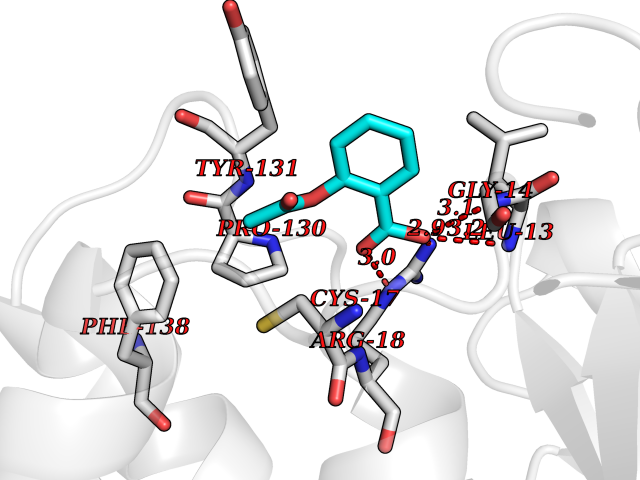 Pymol Map 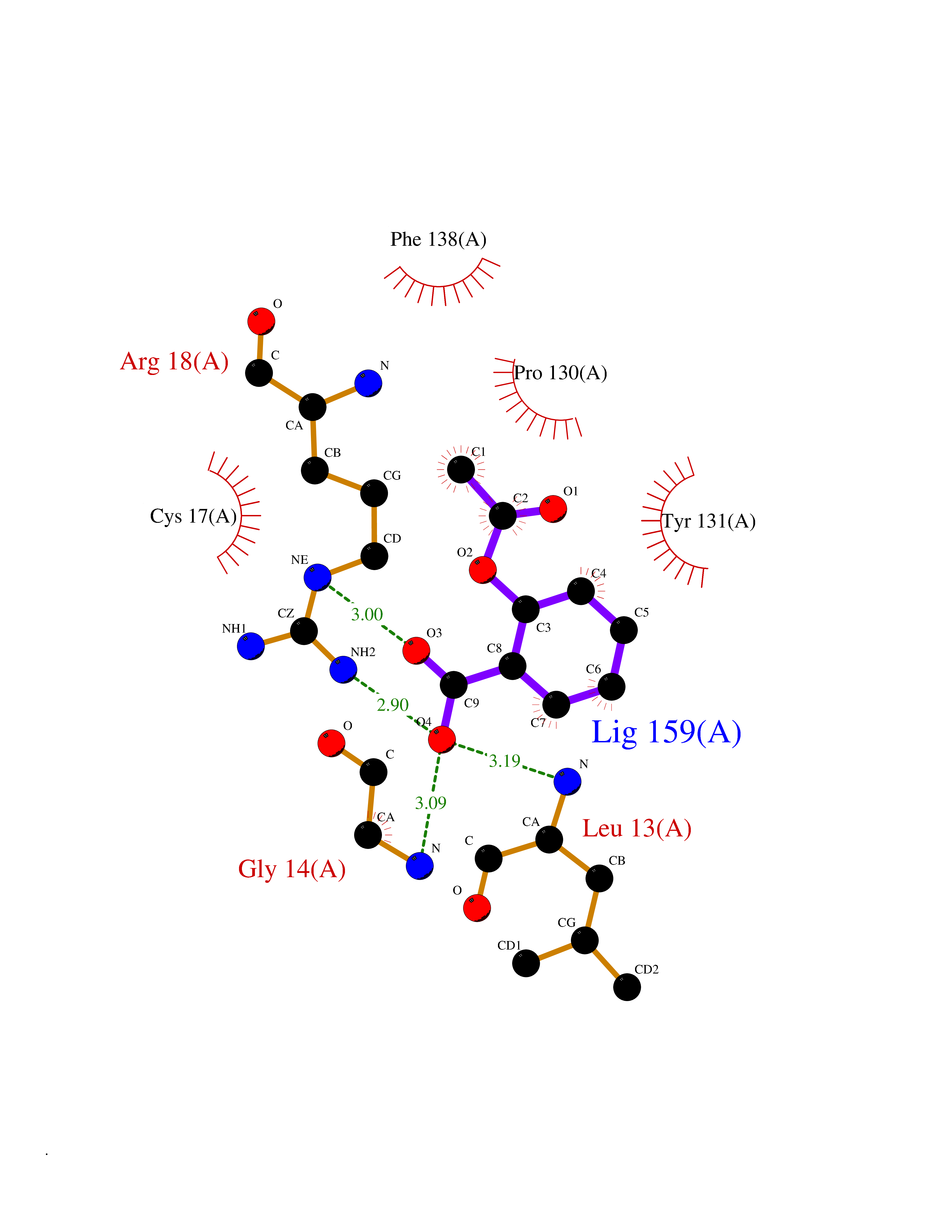 Ligplot Map 3D Binding mode Sequence ATKSVLFVCLGNICRSPIAEAVFRKLVTDQNISENWRVDSAATSGYEIGNPPDYRGQSCMKRHGIPMSHVARQITKEDFATFDYILCMDESNLRDLNRKSNQVKTCKAKIELLGSYDPQKQLIIEDPYYGNDSDFETVYQQCVRCCRAFLEKAH Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Cytochrome P450 2B6 (CYP2B6) | 3IBD | 5.55 | |
Target general information Gen name CYP2B6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cytochrome P450 IIB1; CYPIIB6; 1,4-cineole 2-exo-monooxygenase Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function In liver microsomes, this enzyme is involved in an NADPH-dependent electron transport pathway. It oxidizes a variety of structurally unrelated compounds, including steroids, fatty acids, and xenobiotics. Acts as a 1,4-cineole 2-exo-monooxygenase. Cytochromes P450 are a group of heme-thiolate monooxygenases. Related diseases Aromatic L-amino-acid decarboxylase deficiency (AADCD) [MIM:608643]: An inborn error in neurotransmitter metabolism that leads to combined serotonin and catecholamine deficiency. It causes developmental and psychomotor delay, poor feeding, lethargy, ptosis, intermittent hypothermia, gastrointestinal disturbances. The onset is early in infancy and inheritance is autosomal recessive. {ECO:0000269|PubMed:14991824, ECO:0000269|PubMed:15079002, ECO:0000269|Ref.12}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08369; DB02974; DB11932; DB12001; DB14973; DB15568; DB00321; DB00381; DB00701; DB01435; DB00714; DB06413; DB06697; DB13132; DB11586; DB01076; DB15011; DB00972; DB08822; DB13997; DB04975; DB01086; DB00865; DB00443; DB04794; DB12151; DB01194; DB05541; DB01222; DB01156; DB09061; DB14737; DB00564; DB06119; DB00439; DB00568; DB00604; DB00515; DB12499; DB00349; DB06470; DB00758; DB01559; DB00257; DB01394; DB05219; DB08865; DB11672; DB14635; DB04664; DB00531; DB08912; DB01151; DB16650; DB01234; DB14649; DB04856; DB00514; DB00829; DB00586; DB01184; DB00997; DB00470; DB00476; DB00625; DB15444; DB13874; DB11718; DB08899; DB00751; DB11823; DB00655; DB00898; DB01466; DB00574; DB12265; DB01544; DB00472; DB01095; DB00176; DB01320; DB05087; DB00986; DB01159; DB00956; DB00741; DB09054; DB01181; DB00458; DB00762; DB11633; DB06636; DB00753; DB11757; DB01167; DB14568; DB09570; DB01221; DB06738; DB01026; DB11951; DB09078; DB12070; DB05667; DB00281; DB00836; DB01601; DB00455; DB04871; DB12130; DB09280; DB00772; DB09238; DB14921; DB14009; DB01043; DB00170; DB00454; DB00532; DB04817; DB00333; DB00763; DB01028; DB09241; DB00849; DB00959; DB06710; DB00379; DB06148; DB01110; DB06595; DB16236; DB00745; DB11763; DB00220; DB00238; DB00622; DB00184; DB01115; DB04868; DB12005; DB00435; DB00957; DB09074; DB16267; DB11632; DB01173; DB11837; DB04938; DB05467; DB00715; DB08883; DB01074; DB04930; DB12978; DB03575; DB01174; DB00252; DB13941; DB17472; DB11642; DB06209; DB14631; DB00635; DB01069; DB00818; DB00908; DB00481; DB08896; DB11853; DB16826; DB02709; DB00615; DB01045; DB11753; DB01201; DB08864; DB00503; DB06176; DB00296; DB00412; DB00778; DB01037; DB06739; DB01104; DB01236; DB00641; DB00398; DB15569; DB12548; DB09118; DB06729; DB01138; DB00675; DB12020; DB12095; DB00231; DB00624; DB13943; DB13944; DB13946; DB11712; DB01041; DB09499; DB04572; DB08816; DB00208; DB06137; DB00193; DB00755; DB12245; DB12808; DB00197; DB12255; DB00313; DB11613; DB08881; DB00661; DB09185; DB11739; DB00582; DB09068; DB14975; DB15035 Interacts with NA EC number EC 1.14.13.- Uniprot keywords 3D-structure; Alternative splicing; Endoplasmic reticulum; Heme; Iron; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 53293 Length 465 Aromaticity 0.12 Instability index 38.79 Isoelectric point 8.63 Charge (pH=7) 5.35 2D Binding mode Binding energy (Kcal/mol) -7.57 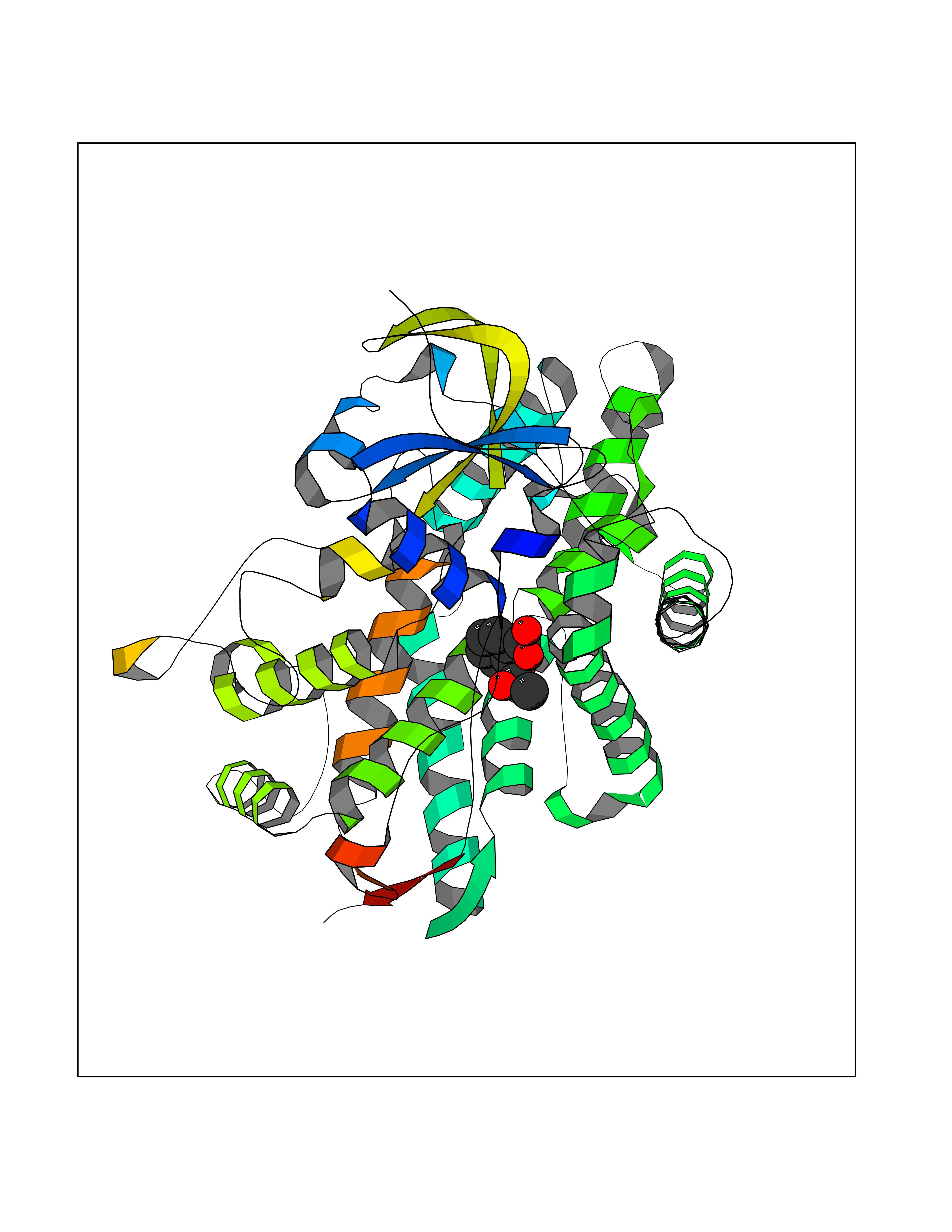 Molscript Map 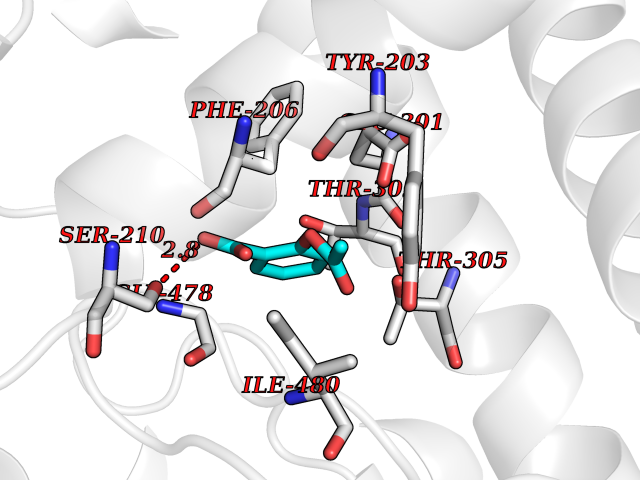 Pymol Map 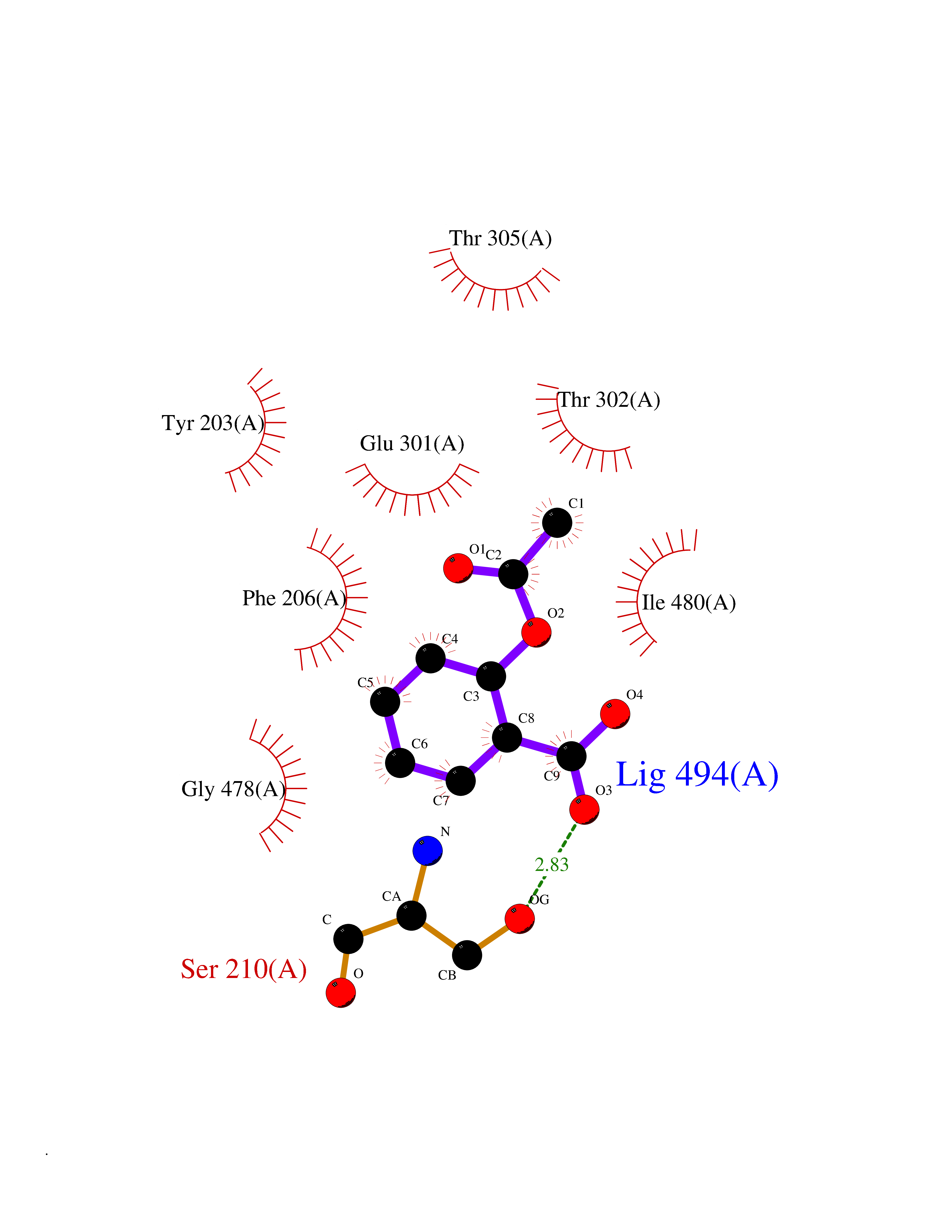 Ligplot Map 3D Binding mode Sequence GKLPPGPRPLPLLGNLLQMDRRGLLKSFLRFREKYGDVFTVHLGPRPVVMLCGVEAIREALVDKAEAFSGRGKIAMVDPFFRGYGVIFANGNRWKVLRRFSVTTMRDFGMGKRSVEERIQEEAQCLIEELRKSKGALMDPTFLFQSITANIICSIVFGKRFHYQDQEFLKMLNLFYQTFSLISSVFGQLFELFSGFLKHFPGAHRQVYKNLQEINAYIGHSVEKHRETLDPSAPRDLIDTYLLHMEKEKSNAHSEFSHQNLNLNTLSLFFAGTETTSTTLRYGFLLMLKYPHVAERVYREIEQVIGPHRPPELHDRAKMPYTEAVIYEIQRFSDLLPMGVPHIVTQHTSFRGYIIPKDTEVFLILSTALHDPHYFEKPDAFNPDHFLDANGALKKTEAFIPFSLGKRICLGEGIARAELFLFFTTILQNFSMASPVAPEDIDLTPQECGVGKIPPTYQIRFLPRH Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Nicotinamide N-methyltransferase | 2IIP | 5.55 | |
Target general information Gen name NNMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, NNMT/PNMT/TEMT family Biochemical class Transferase Function Nicotinamide N-methyltransferase activity.Pyridine N-methyltransferase activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB00627 Interacts with NA EC number 2.1.1.1 Uniprot keywords 3D-structure; Acetylation; Citrullination; Cytoplasm; Direct protein sequencing; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27886.8 Length 251 Aromaticity 0.1 Instability index 40.66 Isoelectric point 5.23 Charge (pH=7) -5.11 2D Binding mode Binding energy (Kcal/mol) -7.57  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GFTSKDTYLSHFNPRDYLEKYYSAESQILKHLLKNLFKIFCLDGVKGDLLIDIGSGPTIYQLLSACESFKEIVVTDYSDQNLQELEKWLKAAPAAFDWSPVVTYVCDLEGNRVKGPEKEEKLRQAVKQVLKCDVTQSQPLGAVPLPPADCVLSTLCLDAACPDLPTYCRALRNLGSLLKPGGFLVIMDALKSSYYMIGEQKFSSLPLGREAVEAAVKEAGYTIEWFEVISQSYSSTMANNEGLFSLVARKL Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Carboxypeptidase A1 (CPA1) | 5OM9 | 5.55 | |
Target general information Gen name CPA1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CPA Protein family Peptidase M14 family Biochemical class Peptidase Function Carboxypeptidase that catalyzes the release of a C-terminal amino acid, but has little or no action with -Asp, -Glu, -Arg, -Lys or -Pro. Related diseases Spinocerebellar ataxia, autosomal recessive, with axonal neuropathy 1 (SCAN1) [MIM:607250]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCAN1 is an autosomal recessive cerebellar ataxia (ARCA) associated with peripheral axonal motor and sensory neuropathy, distal muscular atrophy, pes cavus and steppage gait as seen in Charcot-Marie-Tooth neuropathy. All affected individuals have normal intelligence. {ECO:0000269|PubMed:12244316, ECO:0000269|PubMed:15647511, ECO:0000269|PubMed:15920477, ECO:0000269|PubMed:16141202, ECO:0000269|PubMed:17948061}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06924; DB07484; DB02494; DB03441; DB04316; DB03201; DB11095; DB02652; DB07506; DB04058; DB07351; DB08762; DB08368; DB03012 Interacts with P19399 EC number EC 3.4.17.1 Uniprot keywords 3D-structure; Carboxypeptidase; Direct protein sequencing; Disulfide bond; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 44660.7 Length 396 Aromaticity 0.12 Instability index 25.06 Isoelectric point 5.23 Charge (pH=7) -15.98 2D Binding mode Binding energy (Kcal/mol) -7.58  Molscript Map 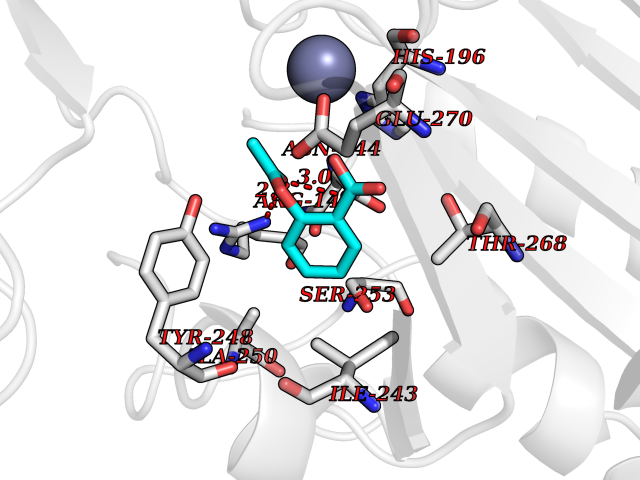 Pymol Map 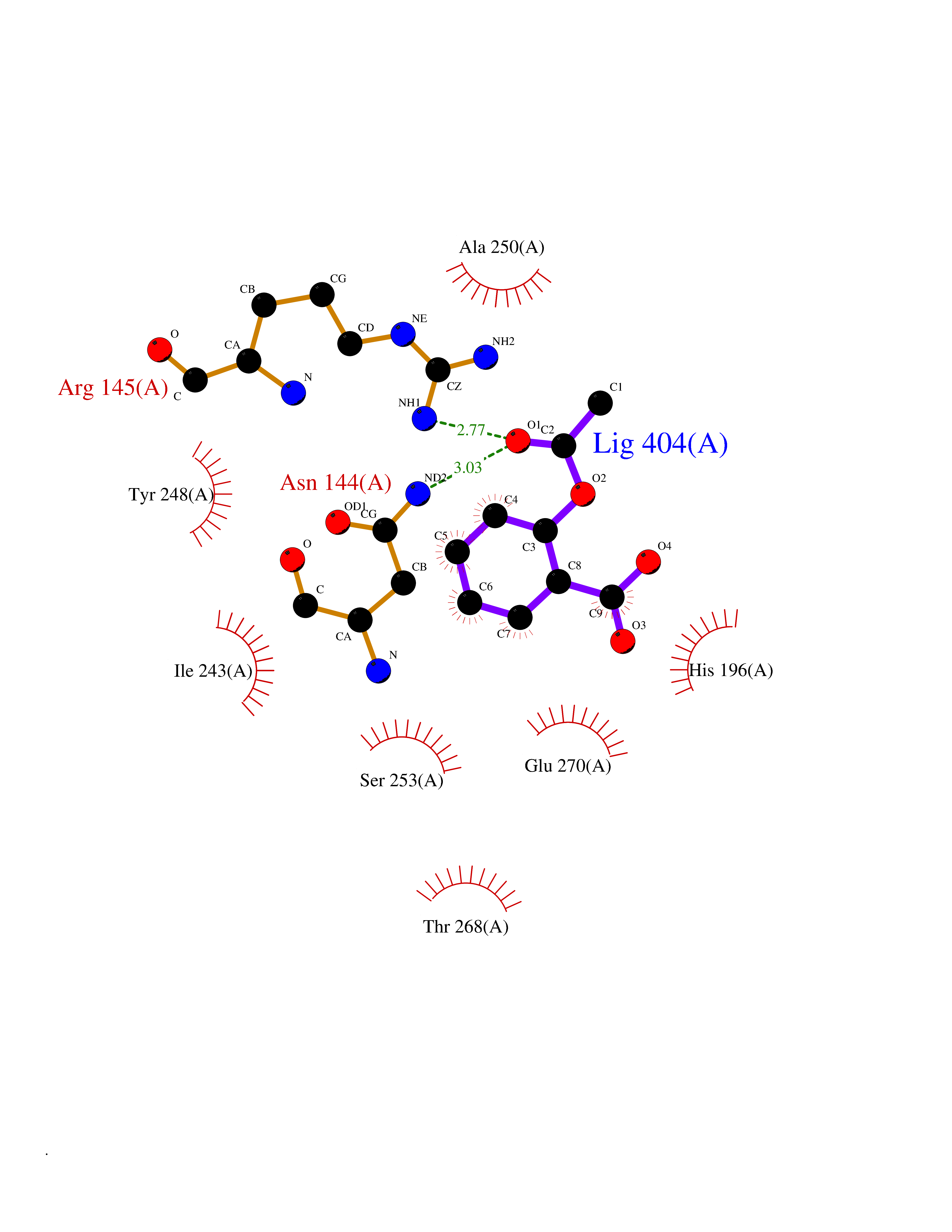 Ligplot Map 3D Binding mode Sequence KEDFVGHQVLRISVADEAQVQKVKELEDLEHLQLDFWRGPAHPGSPIDVRVPFPSIQAVKIFLESHGISYETMIEDVQSLLDEEQEQMFRSTDTFNYATYHTLEEIYDFLDLLVAENPHLVSKIQIGNTYEGRPIYVLKFSTGGSKRPAIWIDTGIHSREWVTQASGVWFAKKITQDYGQDAAFTAILDTLDIFLEIVTNPDGFAFTHSTNRMWRKTRSHTAGSLCIGVDPNRNWDAGFGLSGASSNPCSETYHGKFANSEVEVKSIVDFVKDHGNIKAFISIHSYSQLLMYPYGYKTEPVPDQDELDQLSKAAVTALASLYGTKFNYGSIIKAIYQASGSTIDWTYSQGIKYSFTFELRDTGRYGFLLPASQIIPTAKETWLALLTIMEHTLNHP Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Beta-arrestin-1 (ARRB1) | 6TKO | 5.55 | |
Target general information Gen name ARRB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Non-visual arrestin-2; Betaarrestin1; Arrestin beta1; Arrestin beta-1; ARR1 Protein family Arrestin family Biochemical class Arrestin protein Function During homologous desensitization, beta-arrestins bind to the GPRK-phosphorylated receptor and sterically preclude its coupling to the cognate G-protein; the binding appears to require additional receptor determinants exposed only in the active receptor conformation. The beta-arrestins target many receptors for internalization by acting as endocytic adapters (CLASPs, clathrin-associated sorting proteins) and recruiting the GPRCs to the adapter protein 2 complex 2 (AP-2) in clathrin-coated pits (CCPs). However, the extent of beta-arrestin involvement appears to vary significantly depending on the receptor, agonist and cell type. Internalized arrestin-receptor complexes traffic to intracellular endosomes, where they remain uncoupled from G-proteins. Two different modes of arrestin-mediated internalization occur. Class A receptors, like ADRB2, OPRM1, ENDRA, D1AR and ADRA1B dissociate from beta-arrestin at or near the plasma membrane and undergo rapid recycling. Class B receptors, like AVPR2, AGTR1, NTSR1, TRHR and TACR1 internalize as a complex with arrestin and traffic with it to endosomal vesicles, presumably as desensitized receptors, for extended periods of time. Receptor resensitization then requires that receptor-bound arrestin is removed so that the receptor can be dephosphorylated and returned to the plasma membrane. Involved in internalization of P2RY4 and UTP-stimulated internalization of P2RY2. Involved in phosphorylation-dependent internalization of OPRD1 ands subsequent recycling. Involved in the degradation of cAMP by recruiting cAMP phosphodiesterases to ligand-activated receptors. Beta-arrestins function as multivalent adapter proteins that can switch the GPCR from a G-protein signaling mode that transmits short-lived signals from the plasma membrane via small molecule second messengers and ion channels to a beta-arrestin signaling mode that transmits a distinct set of signals that are initiated as the receptor internalizes and transits the intracellular compartment. Acts as signaling scaffold for MAPK pathways such as MAPK1/3 (ERK1/2). ERK1/2 activated by the beta-arrestin scaffold is largely excluded from the nucleus and confined to cytoplasmic locations such as endocytic vesicles, also called beta-arrestin signalosomes. Recruits c-Src/SRC to ADRB2 resulting in ERK activation. GPCRs for which the beta-arrestin-mediated signaling relies on both ARRB1 and ARRB2 (codependent regulation) include ADRB2, F2RL1 and PTH1R. For some GPCRs the beta-arrestin-mediated signaling relies on either ARRB1 or ARRB2 and is inhibited by the other respective beta-arrestin form (reciprocal regulation). Inhibits ERK1/2 signaling in AGTR1- and AVPR2-mediated activation (reciprocal regulation). Is required for SP-stimulated endocytosis of NK1R and recruits c-Src/SRC to internalized NK1R resulting in ERK1/2 activation, which is required for the antiapoptotic effects of SP. Is involved in proteinase-activated F2RL1-mediated ERK activity. Acts as signaling scaffold for the AKT1 pathway. Is involved in alpha-thrombin-stimulated AKT1 signaling. Is involved in IGF1-stimulated AKT1 signaling leading to increased protection from apoptosis. Involved in activation of the p38 MAPK signaling pathway and in actin bundle formation. Involved in F2RL1-mediated cytoskeletal rearrangement and chemotaxis. Involved in AGTR1-mediated stress fiber formation by acting together with GNAQ to activate RHOA. Appears to function as signaling scaffold involved in regulation of MIP-1-beta-stimulated CCR5-dependent chemotaxis. Involved in attenuation of NF-kappa-B-dependent transcription in response to GPCR or cytokine stimulation by interacting with and stabilizing CHUK. May serve as nuclear messenger for GPCRs. Involved in OPRD1-stimulated transcriptional regulation by translocating to CDKN1B and FOS promoter regions and recruiting EP300 resulting in acetylation of histone H4. Involved in regulation of LEF1 transcriptional activity via interaction with DVL1 and/or DVL2 Also involved in regulation of receptors other than GPCRs. Involved in Toll-like receptor and IL-1 receptor signaling through the interaction with TRAF6 which prevents TRAF6 autoubiquitination and oligomerization required for activation of NF-kappa-B and JUN. Binds phosphoinositides. Binds inositolhexakisphosphate (InsP6). Involved in IL8-mediated granule release in neutrophils. Required for atypical chemokine receptor ACKR2-induced RAC1-LIMK1-PAK1-dependent phosphorylation of cofilin (CFL1) and for the up-regulation of ACKR2 from endosomal compartment to cell membrane, increasing its efficiency in chemokine uptake and degradation. Involved in the internalization of the atypical chemokine receptor ACKR3. Negatively regulates the NOTCH signaling pathway by mediating the ubiquitination and degradation of NOTCH1 by ITCH. Participates to the recruitment of the ubiquitin-protein ligase to the receptor. Functions in regulating agonist-mediated G-protein coupled receptor (GPCR) signaling by mediating both receptor desensitization and resensitization processes. Related diseases Intellectual developmental disorder with dysmorphic facies and ptosis (IDDDFP) [MIM:617333]: An autosomal dominant neurodevelopmental disorder characterized by delayed psychomotor development, intellectual disability, delayed language, and facial dysmorphisms, most notably ptosis. Additional features may include poor growth, hypotonia, and seizures. {ECO:0000269|PubMed:27939639, ECO:0000269|PubMed:27939640}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P63010-2; O15169; P0DP25; P20963; P25101; P50148; Q5JWF2; Q14749; P06396; Q16665; P11142; Q99683; P53779; P45984; Q00987; P19338; Q14978; P14618; P14859-6; P35813; O75688; Q13523; P06702; P12931; Q15208; Q13428; P04637; P27348; P25490; O43298; O95218; Q7DB77 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Cell projection; Coated pit; Cytoplasm; Cytoplasmic vesicle; Membrane; Nucleus; Phosphoprotein; Protein transport; Proteomics identification; Reference proteome; Signal transduction inhibitor; Transcription; Transcription regulation; Transport; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32455.6 Length 293 Aromaticity 0.12 Instability index 28.99 Isoelectric point 9.12 Charge (pH=7) 9.86 2D Binding mode Binding energy (Kcal/mol) -7.57  Molscript Map 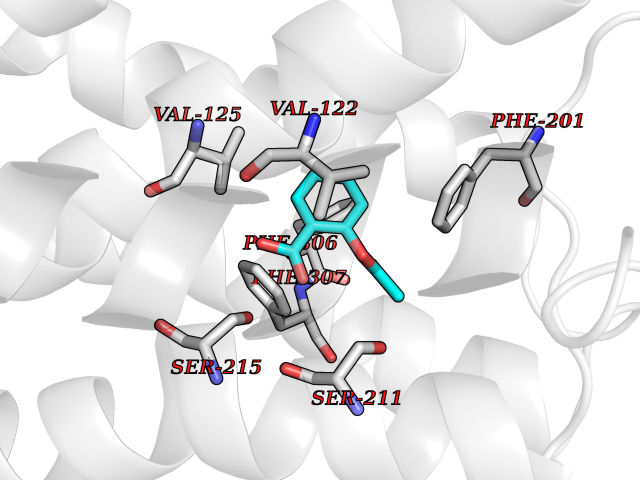 Pymol Map 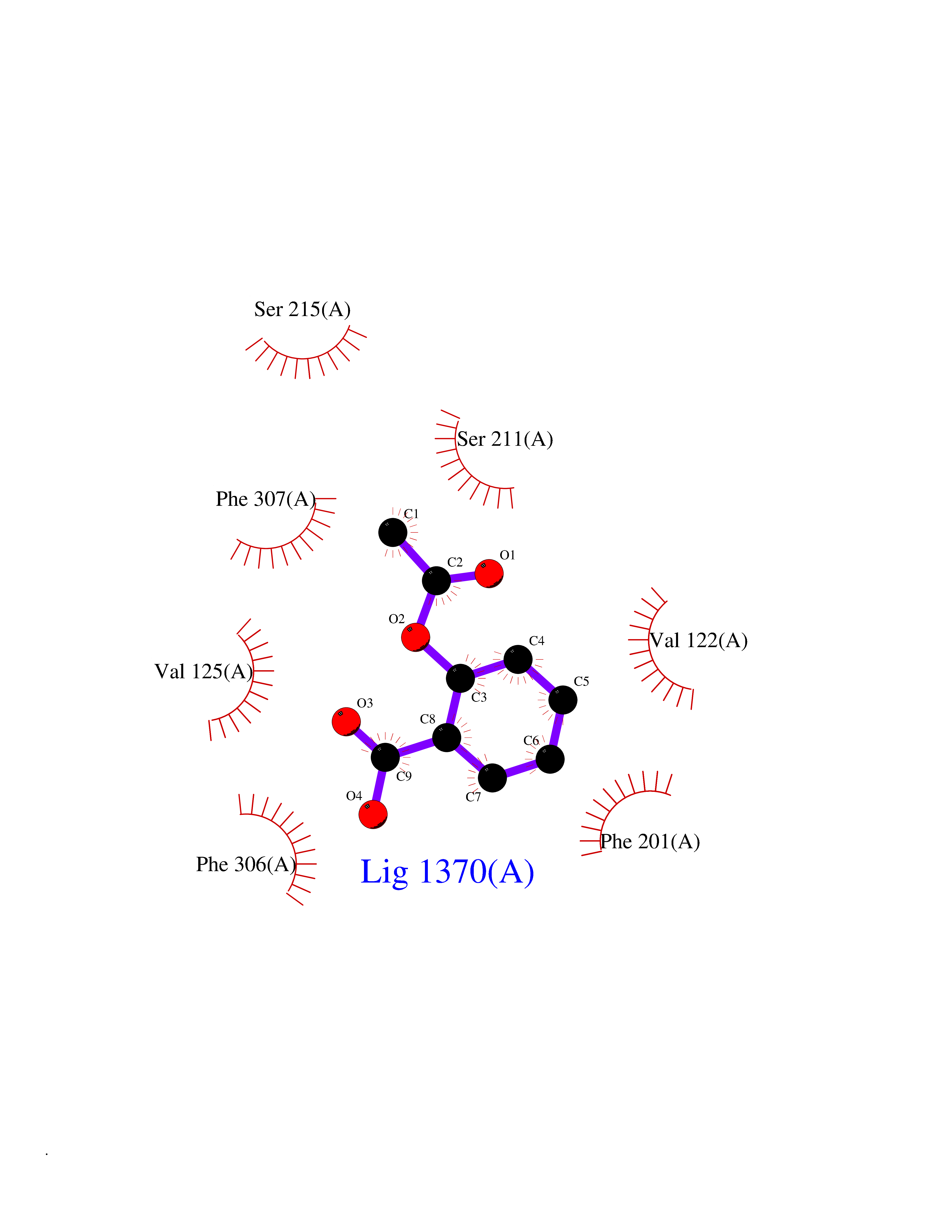 Ligplot Map 3D Binding mode Sequence AGCSLLMALVVLLIVAGNVLVIAAIGRTQRLQTLTNLFITSLACADLVVGLLVVPFGATLVCRGTWLWGSFLCELWTSLDVLCVTASIWTLCVIAIDRYLAITSPFRYQSLMTRARAKVIICTVWAISALVSFLPIMMHWWRDEDPQALKCYQDPGCCDFVTNRAYAIASSIISFYIPLLIMIFVYLRVYREAKEQIRKIDVMAMREHKALKTLGIIMGVFTLCWLPFFLVNIVNVFNRDLVPKWLFVAFNWLGYANSAMNPIIYCRSPDFRKAFKRLLAEXAXXAXXXLAKD Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Nicotinamide phosphoribosyltransferase (NAMPT) | 2E5D | 5.55 | |
Target general information Gen name NAMPT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Visfatin; PreBcell colonyenhancing factor 1; PreB cellenhancing factor; Pre-B-cell colony-enhancing factor 1; Pre-B cell-enhancing factor; PBEF1; PBEF; Nampt; NAmPRTase Protein family NAPRTase family Biochemical class Glycosyltransferases Function It is the rate limiting component in the mammalian NAD biosynthesis pathway. The secreted form behaves both as a cytokine with immunomodulating properties and an adipokine with anti-diabetic properties, it has no enzymatic activity, partly because of lack of activation by ATP, which has a low level in extracellular space and plasma. Plays a role in the modulation of circadian clock function. NAMPT-dependent oscillatory production of NAD regulates oscillation of clock target gene expression by releasing the core clock component: CLOCK-ARNTL/BMAL1 heterodimer from NAD-dependent SIRT1-mediated suppression. Catalyzes the condensation of nicotinamide with 5-phosphoribosyl-1-pyrophosphate to yield nicotinamide mononucleotide, an intermediate in the biosynthesis of NAD. Related diseases Hemolytic anemia, non-spherocytic, due to glucose phosphate isomerase deficiency (HA-GPID) [MIM:613470]: A form of anemia in which there is no abnormal hemoglobin or spherocytosis. It is caused by glucose phosphate isomerase deficiency. {ECO:0000269|PubMed:28803808, ECO:0000269|PubMed:7989588, ECO:0000269|PubMed:8499925, ECO:0000269|PubMed:8822952, ECO:0000269|PubMed:8822954, ECO:0000269|PubMed:9446754, ECO:0000269|PubMed:9856489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12980; DB12731; DB05217 Interacts with P02792; Q01628; P03886; P43490; Q70CQ1-2 EC number EC 2.4.2.12 Uniprot keywords 3D-structure; Acetylation; Biological rhythms; Cytokine; Cytoplasm; Glycosyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Pyridine nucleotide biosynthesis; Reference proteome; Secreted; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 105483 Length 932 Aromaticity 0.11 Instability index 34.4 Isoelectric point 6.68 Charge (pH=7) -2.24 2D Binding mode Binding energy (Kcal/mol) -7.57 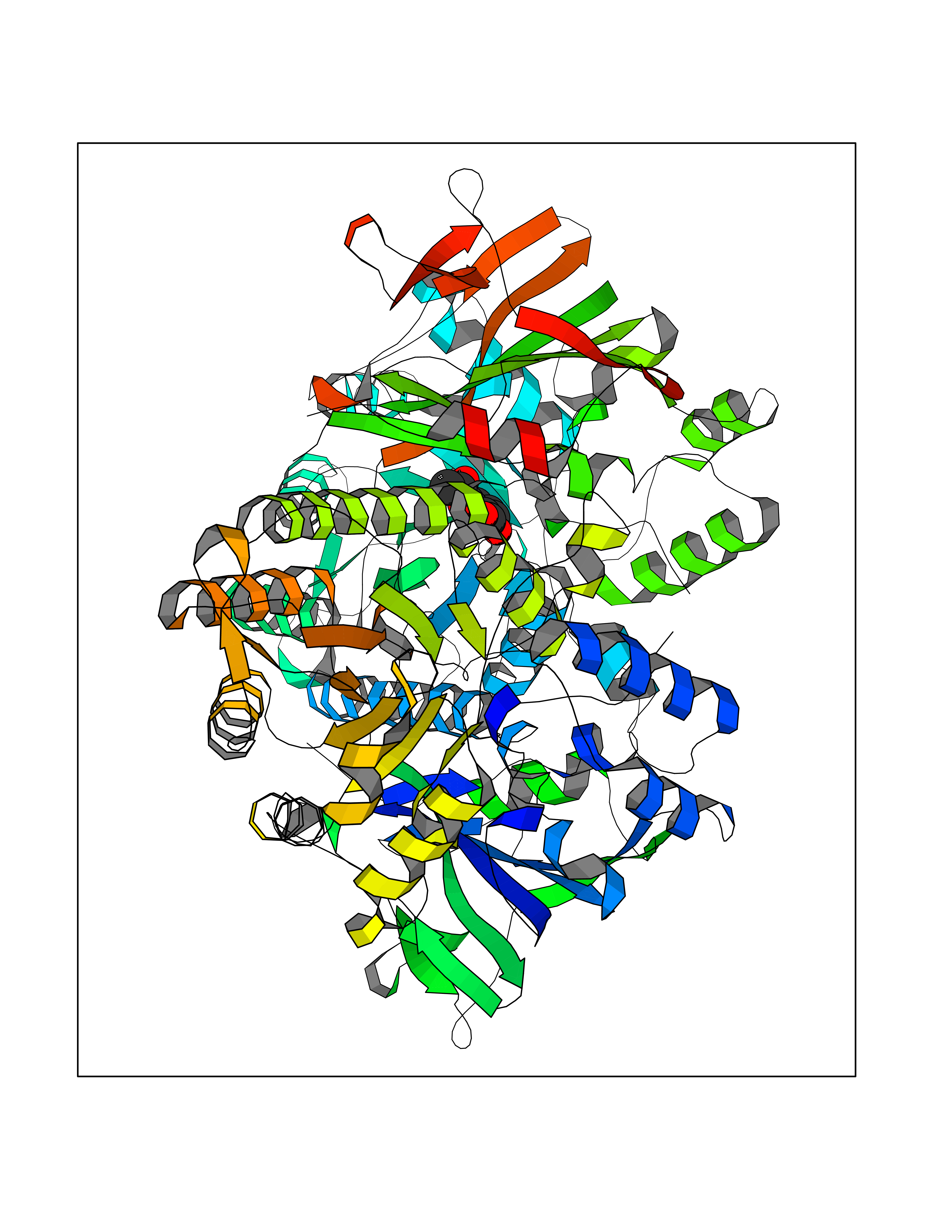 Molscript Map 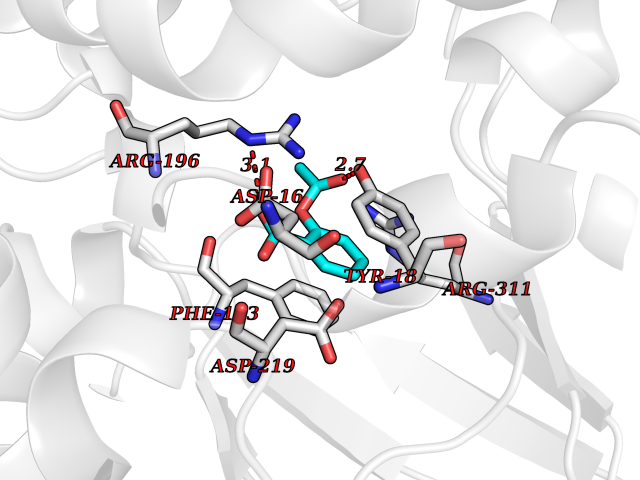 Pymol Map 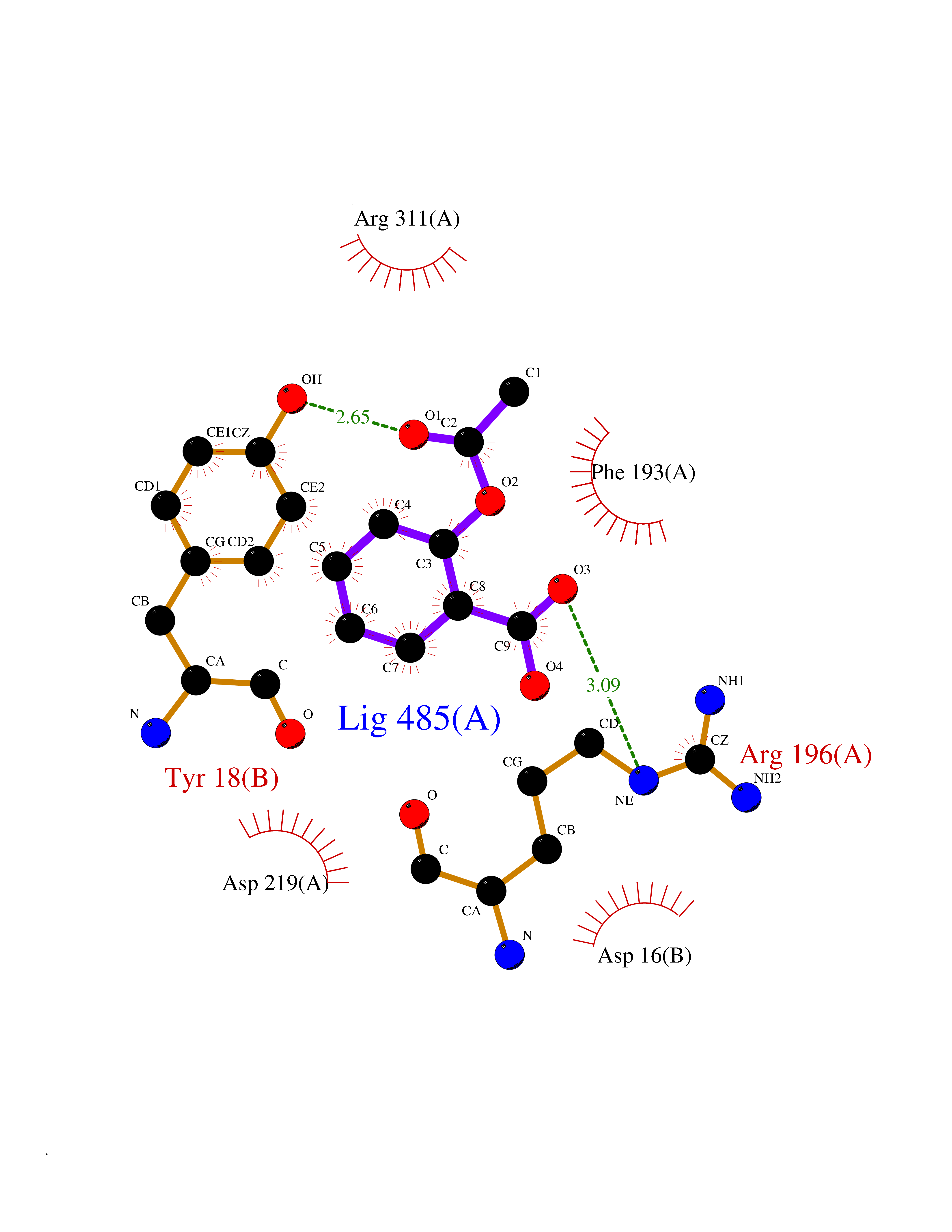 Ligplot Map 3D Binding mode Sequence EFNILLATDSYKVTHYKQYPPNTSKVYSYFECREKKYEETVFYGLQYILNKYLKGKVVTKEKIQEAKDVYKEHFQDDVFNEKGWNYILEKYDGHLPIEIKAVPEGFVIPRGNVLFTVENTDPECYWLTNWIETILVQSWYPITVATNSREQKKILAKYLLETSGNLDGLEYKLHDFGYRGVSSQETAGIGASAHLVNFKGTDTVAGLALIKKYYGTKDPVPGYSVPAAEHSTITAWGKDHEKDAFEHIVTQFSSVPVSVVSDSYDIYNACEKIWGEDLRHLIVSRSTQAPLIIRPDSGNPLDTVLKVLEILGKKFPVTENSKGYKLLPPYLRVIQGDGVDINTLQEIVEGMKQKMWSIENIAFGSGGGLLQKLTRDLLNCSFKCSYVVTNGLGINVFKDPVADPNKRSKKGRLSLHRTPAGNFVTLEEGKGDLEEYGQDLLHTVFKNGKVTKSYSFDEIRKNAQLNEFNILLATDSYKVTHYKQYPPNTSKVYSYFECREKKYEETVFYGLQYILNKYLKGKVVTKEKIQEAKDVYKEHFQDDVFNEKGWNYILEKYDGHLPIEIKAVPEGFVIPRGNVLFTVENTDPECYWLTNWIETILVQSWYPITVATNSREQKKILAKYLLETSGNLDGLEYKLHDFGYRGVSSQETAGIGASAHLVNFKGTDTVAGLALIKKYYGTKDPVPGYSVPAAEHSTITAWGKDHEKDAFEHIVTQFSSVPVSVVSDSYDIYNACEKIWGEDLRHLIVSRSTQAPLIIRPDSGNPLDTVLKVLEILGKKFPVTENSKGYKLLPPYLRVIQGDGVDINTLQEIVEGMKQKMWSIENIAFGSGGGLLQKLTRDLLNCSFKCSYVVTNGLGINVFKDPVADPNKRSKKGRLSLHRTPAGNFVTLEEGKGDLEEYGQDLLHTVFKNGKVTKSYSFDEIRKNAQLN Hydrogen bonds contact Hydrophobic contact | ||||