Job Results:
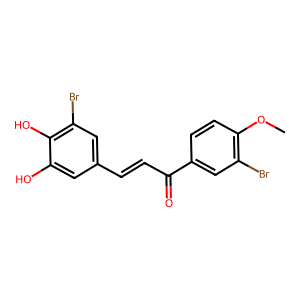
Ligand
Structure
Job ID
f5a9e34d4d6e0fad4e04e6dd0d3ae3aa
Job name
NA
Time
2026-01-10 22:50:50
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Bacterial Oxoacyl-[acyl-carrier-protein] synthase II (Bact fabF) | 2GFX | 6.99 | |
Target general information Gen name Bact fabF Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms KASB; KAS II; KAS 2; FabF; Condensing enzyme FabF; Beta-ketoacyl-acyl carrier protein synthase B; Beta-ketoacyl-ACP synthase II; Beta-ketoacyl-ACP synthase 2; 3-oxoacyl-[acyl-carrier-protein] synthase Protein family Thiolase-like superfamily, Beta-ketoacyl-ACP synthases family Biochemical class Acyltransferase Function Catalyzes the condensation reaction of fatty acid synthesis by the addition to an acyl acceptor of two carbons from malonyl-ACP. Has a preference for short chain acid substrates and may function to supply the octanoic substrates for lipoic acid biosynthesis. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08366; DB01034; DB03017; DB08407 Interacts with P0A6Y8 EC number EC 2.3.1.179 Uniprot keywords 3D-structure; Acyltransferase; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 42852 Length 411 Aromaticity 0.06 Instability index 31.41 Isoelectric point 5.72 Charge (pH=7) -7.34 2D Binding mode Binding energy (Kcal/mol) -9.54  Molscript Map 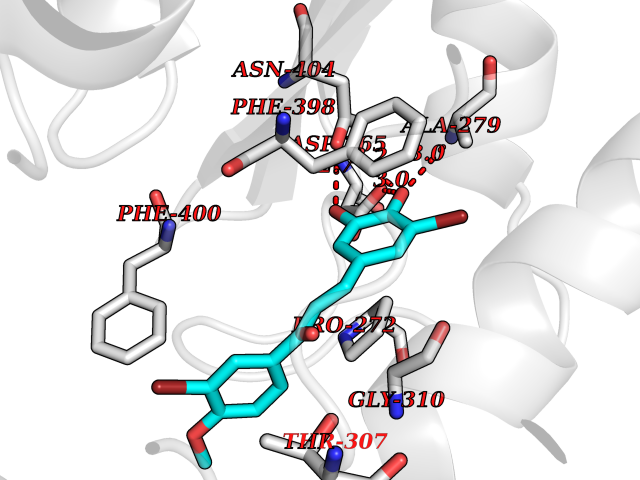 Pymol Map 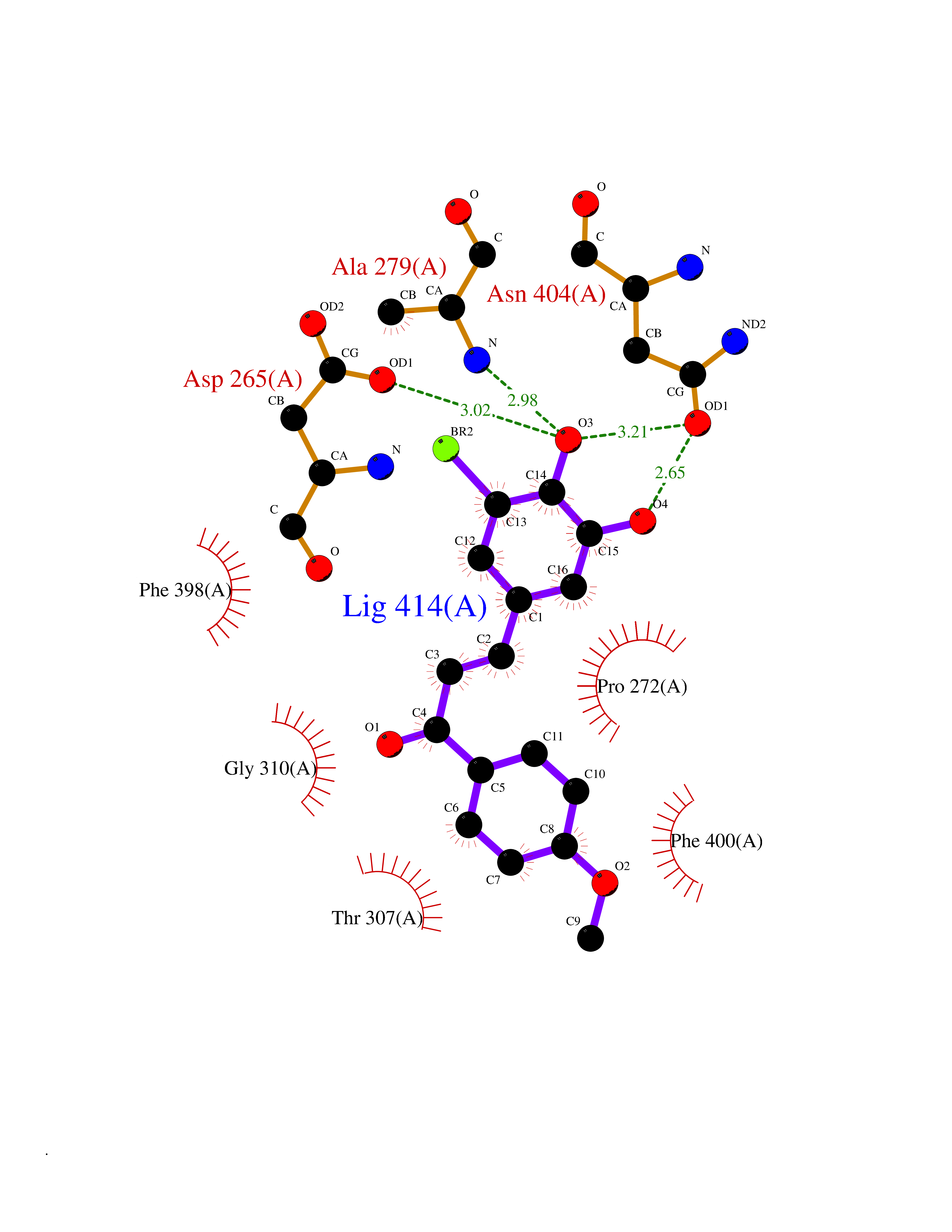 Ligplot Map 3D Binding mode Sequence KRRVVVTGLGMLSPVGNTVESTWKALLAGQSGISLIDHFDTSAYATKFAGLVKDFNCEDIISRKEQRKMDAFIQYGIVAGVQAMQDSGLEITEENATRIGAAIGSGIGGLGLIEENHTSLMNGGPRKISPFFVPSTIVNMVAGHLTIMYGLRGPSISIATAQTSGVHNIGHAARIIAYGDADVMVAGGAEKASTPLGVGGFGAARALSTRNDNPQAASRPWDKERDGFVLGDGAGMLVLEEYEHAKKRGAKIYAELVGFGMSSDAYHMTSPPENGAGAALAMANALRDAGIEASQIGYVNAHGTSTPAGDKAEAQAVKTIFGEAASRVLVSSTKSMTGHLLGAAGAVESIYSILALRDQAVPPTINLDNPDEGCDLDFVPHEARQVSGMEYTLCNSFGFGGTNGSLIFKKI Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Monocarboxylate transporter 1 (SLC16A1) | 6LZ0 | 6.99 | |
Target general information Gen name SLC16A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Solute carrier family 16 member 1; MCT1; MCT 1 Protein family Major facilitator superfamily, Monocarboxylate porter (TC 2.A.1.13) family Biochemical class Major facilitator Function Catalyzes the rapid transport across the plasma membrane of many monocarboxylates such as lactate, pyruvate, branched-chain oxo acids derived from leucine, valine and isoleucine, and the ketone bodies acetoacetate, beta-hydroxybutyrate and acetate. Depending on the tissue and on cicumstances, mediates the import or export of lactic acid and ketone bodies. Required for normal nutrient assimilation, increase of white adipose tissue and body weight gain when on a high-fat diet. Plays a role in cellular responses to a high-fat diet by modulating the cellular levels of lactate and pyruvate, small molecules that contribute to the regulation of central metabolic pathways and insulin secretion, with concomitant effects on plasma insulin levels and blood glucose homeostasis. Proton-coupled monocarboxylate transporter. Related diseases Symptomatic deficiency in lactate transport (SDLT) [MIM:245340]: Deficiency of lactate transporter may result in an acidic intracellular environment created by muscle activity with consequent degeneration of muscle and release of myoglobin and creatine kinase. This defect might compromise extreme performance in otherwise healthy individuals. {ECO:0000269|PubMed:10590411}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperinsulinemic hypoglycemia, familial, 7 (HHF7) [MIM:610021]: A form of hyperinsulinemic hypoglycemia, a clinically and genetically heterogeneous disorder characterized by inappropriate insulin secretion from the pancreatic beta-cells in the presence of low blood glucose levels. HHF7 features include exercise-induced hyperinsulinism, loss of consciousness due to hypoglycemia, and hypoglycemic seizures. HHF7 inheritance is autosomal dominant. {ECO:0000269|PubMed:17701893}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Monocarboxylate transporter 1 deficiency (MCT1D) [MIM:616095]: A metabolic disorder characterized by recurrent ketoacidosis, a pathologic state due to ketone formation exceeding ketone utilization. The clinical consequences of ketoacidosis are vomiting, osmotic diuresis, dehydration, and Kussmaul breathing. The condition may progress to decreased consciousness and, ultimately, death. {ECO:0000269|PubMed:25390740}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03166; DB01762; DB03773; DB04074; DB00345; DB00415; DB08892; DB03793; DB03066; DB07767; DB00529; DB01440; DB00142; DB04398; DB09338; DB00563; DB00731; DB00627; DB04552; DB00175; DB01032; DB00119; DB04216; DB00936; DB04348; DB00313 Interacts with Q66PJ3-4; Q92782-2; Q9UH65 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Membrane; Phosphoprotein; Proteomics identification; Reference proteome; Symport; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 40405.6 Length 375 Aromaticity 0.15 Instability index 32.61 Isoelectric point 9.12 Charge (pH=7) 10.98 2D Binding mode Binding energy (Kcal/mol) -9.53  Molscript Map 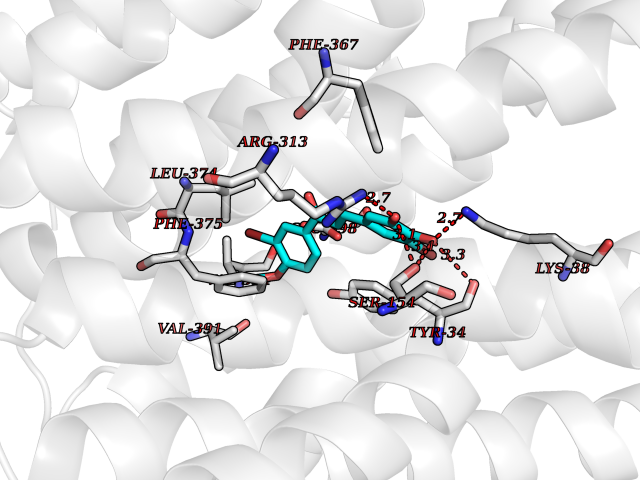 Pymol Map 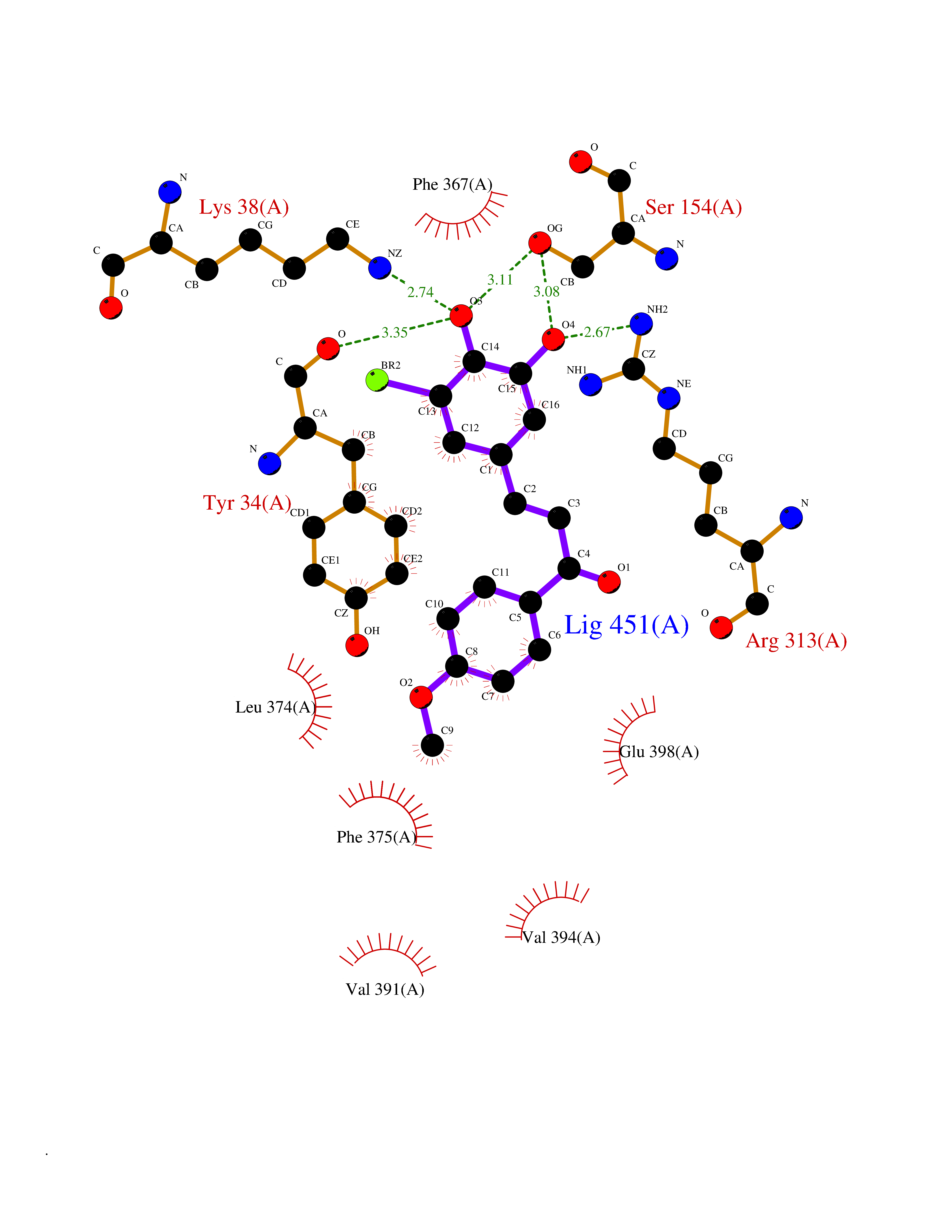 Ligplot Map 3D Binding mode Sequence GGWGWAVVIGAFISIGFSYAFPKSITVFFKEIEGIFHATTSEVSWISSIMLAVMYGGGPISSILVNKYGSRIVMIVGGCLSGCGLIAASFCNTVQQLYVCIGVIGGLGLAFNLNPALTMIGKYFYKRRPLANGLAMAGSPVFLCTLAPLNQVFFGIFGWRGSFLILGGLLLNCCVAGALMRPIGPHRGFLLYLSGNVIMFFGLFAPLVFLSSYGKSQHYSSEKSAFLLSILAFVDMVARPSMGLVANTKPIRPRIQYFFAASVVANGVCHMLAPLSTTYVGFCVYAGFFGFAFGWLSSVLFETLMDLVGPQRFSSAVGLVTIVECCPVLLGPPLLGRLNDMYGDYKYTYWACGVVLIISGIYLFIGMGINYRLLA Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Histone deacetylase 2 (HDAC2) | 4LY1 | 6.98 | |
Target general information Gen name HDAC2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HD2 Protein family Histone deacetylase family, HD type 1 subfamily Biochemical class Carbon-nitrogen hydrolase Function Gives a tag for epigenetic repression and plays an important role in transcriptional regulation, cell cycle progression and developmental events. Histone deacetylases act via the formation of large multiprotein complexes. Forms transcriptional repressor complexes by associating with MAD, SIN3, YY1 and N-COR. Interacts in the late S-phase of DNA-replication with DNMT1 in the other transcriptional repressor complex composed of DNMT1, DMAP1, PCNA, CAF1. Deacetylates TSHZ3 and regulates its transcriptional repressor activity. Component of a RCOR/GFI/KDM1A/HDAC complex that suppresses, via histone deacetylase (HDAC) recruitment, a number of genes implicated in multilineage blood cell development. May be involved in the transcriptional repression of circadian target genes, such as PER1, mediated by CRY1 through histone deacetylation. Involved in MTA1-mediated transcriptional corepression of TFF1 and CDKN1A. Responsible for the deacetylation of lysine residues on the N-terminal part of the core histones (H2A, H2B, H3 and H4). Related diseases Ventricular tachycardia, catecholaminergic polymorphic, 1, with or without atrial dysfunction and/or dilated cardiomyopathy (CPVT1) [MIM:604772]: An arrhythmogenic disorder characterized by stress-induced, bidirectional ventricular tachycardia that may degenerate into cardiac arrest and cause sudden death. Patients present with recurrent syncope, seizures, or sudden death after physical activity or emotional stress. CPVT1 inheritance is autosomal dominant. {ECO:0000269|PubMed:11157710, ECO:0000269|PubMed:11159936, ECO:0000269|PubMed:11208676, ECO:0000269|PubMed:12093772, ECO:0000269|PubMed:12106942, ECO:0000269|PubMed:14571276, ECO:0000269|PubMed:15046072, ECO:0000269|PubMed:15046073, ECO:0000269|PubMed:15466642, ECO:0000269|PubMed:15544015, ECO:0000269|PubMed:16188589, ECO:0000269|PubMed:24793461, ECO:0000269|PubMed:25372681, ECO:0000269|PubMed:27733687}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ventricular arrhythmias due to cardiac ryanodine receptor calcium release deficiency syndrome (VACRDS) [MIM:115000]: An autosomal dominant arrhythmogenic disorder characterized by syncope, cardiac arrest and/or sudden unexpected death, often in association with physical exertion or acute emotional stress. Patients who survive manifest polymorphic ventricular tachycardia and ventricular fibrillation. Unlike typical catecholaminergic ventricular tachycardia, arrhythmias are not reproducible on exercise stress testing or adrenaline challenge. {ECO:0000269|PubMed:12093772, ECO:0000269|PubMed:17984046, ECO:0000269|PubMed:33536282}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12565; DB01223; DB01076; DB05015; DB01262; DB11841; DB01095; DB12645; DB00227; DB11830; DB01303; DB06603; DB06819; DB05223; DB00175; DB03766; DB12847; DB06176; DB00641; DB00277; DB09091; DB00313; DB02546 Interacts with Q9C0K0; Q9HCU9; P68400; Q9UER7; P51610; Q13547; Q9UIS9; Q13330; P01106; P06748; P48382; Q96ST3; O95863; Q9HD15; O43463; Q9H3M7; Q92618; Q17R98; Q2HR82; PRO_0000449623 [P0DTD1] EC number EC 3.5.1.98 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Biological rhythms; Chromatin regulator; Cytoplasm; Hydrolase; Isopeptide bond; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-nitrosylation; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 42020.5 Length 366 Aromaticity 0.13 Instability index 29.52 Isoelectric point 6.52 Charge (pH=7) -2.16 2D Binding mode Binding energy (Kcal/mol) -9.53  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence KKKVCYYYDGDIGNYYYGQGHPMKPHRIRMTHNLLLNYGLYRKMEIYRPHKATAEEMTKYHSDEYIKFLRSIRPDNMSEYSKQMQRFNVGEDCPVFDGLFEFCQLSTGGSVAGAVKLNRQQTDMAVNWAGGLHHAKKSEASGFCYVNDIVLAILELLKYHQRVLYIDIDIHHGDGVEEAFYTTDRVMTVSFHKYGEYFPGTGDLRDIGAGKGKYYAVNFPMRDGIDDESYGQIFKPIISKVMEMYQPSAVVLQCGADSLSGDRLGCFNLTVKGHAKCVEVVKTFNLPLLMLGGGGYTIRNVARCWTYETAVALDCEIPNELPYNDYFEYFGPDFKLHISPSNMTNQNTPEYMEKIKQRLFENLRML Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Cyclin-dependent kinase 5 (CDK5) | 1UNL | 6.98 | |
Target general information Gen name CDK5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tau protein kinase II catalytic subunit; TPKII catalytic subunit; Serine/threonine-protein kinase PSSALRE; Serine/threonine protein kinase PSSALRE; Proline-directed protein kinase F(A) (PDPK F(A)); Pr Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, CDC2/CDKX subfamily Biochemical class Kinase Function Interacts with D1 and D3-type G1 cyclins. Phosphorylates SRC, NOS3, VIM/vimentin, p35/CDK5R1, MEF2A, SIPA1L1, SH3GLB1, PXN, PAK1, MCAM/MUC18, SEPT5, SYN1, DNM1, AMPH, SYNJ1, CDK16, RAC1, RHOA, CDC42, TONEBP/NFAT5, MAPT/TAU, MAP1B, histone H1, p53/TP53, HDAC1, APEX1, PTK2/FAK1, huntingtin/HTT, ATM, MAP2, NEFH and NEFM. Regulates several neuronal development and physiological processes including neuronal survival, migration and differentiation, axonal and neurite growth, synaptogenesis, oligodendrocyte differentiation, synaptic plasticity and neurotransmission, by phosphorylating key proteins. Activated by interaction with CDK5R1 (p35) and CDK5R2 (p39), especially in post-mitotic neurons, and promotes CDK5R1 (p35) expression in an autostimulation loop. Phosphorylates many downstream substrates such as Rho and Ras family small GTPases (e. g. PAK1, RAC1, RHOA, CDC42) or microtubule-binding proteins (e. g. MAPT/TAU, MAP2, MAP1B), and modulates actin dynamics to regulate neurite growth and/or spine morphogenesis. Phosphorylates also exocytosis associated proteins such as MCAM/MUC18, SEPT5, SYN1, and CDK16/PCTAIRE1 as well as endocytosis associated proteins such as DNM1, AMPH and SYNJ1 at synaptic terminals. In the mature central nervous system (CNS), regulates neurotransmitter movements by phosphorylating substrates associated with neurotransmitter release and synapse plasticity; synaptic vesicle exocytosis, vesicles fusion with the presynaptic membrane, and endocytosis. Promotes cell survival by activating anti-apoptotic proteins BCL2 and STAT3, and negatively regulating of JNK3/MAPK10 activity. Phosphorylation of p53/TP53 in response to genotoxic and oxidative stresses enhances its stabilization by preventing ubiquitin ligase-mediated proteasomal degradation, and induces transactivation of p53/TP53 target genes, thus regulating apoptosis. Phosphorylation of p35/CDK5R1 enhances its stabilization by preventing calpain-mediated proteolysis producing p25/CDK5R1 and avoiding ubiquitin ligase-mediated proteasomal degradation. During aberrant cell-cycle activity and DNA damage, p25/CDK5 activity elicits cell-cycle activity and double-strand DNA breaks that precedes neuronal death by deregulating HDAC1. DNA damage triggered phosphorylation of huntingtin/HTT in nuclei of neurons protects neurons against polyglutamine expansion as well as DNA damage mediated toxicity. Phosphorylation of PXN reduces its interaction with PTK2/FAK1 in matrix-cell focal adhesions (MCFA) during oligodendrocytes (OLs) differentiation. Negative regulator of Wnt/beta-catenin signaling pathway. Activator of the GAIT (IFN-gamma-activated inhibitor of translation) pathway, which suppresses expression of a post-transcriptional regulon of proinflammatory genes in myeloid cells; phosphorylates the linker domain of glutamyl-prolyl tRNA synthetase (EPRS) in a IFN-gamma-dependent manner, the initial event in assembly of the GAIT complex. Phosphorylation of SH3GLB1 is required for autophagy induction in starved neurons. Phosphorylation of TONEBP/NFAT5 in response to osmotic stress mediates its rapid nuclear localization. MEF2 is inactivated by phosphorylation in nucleus in response to neurotoxin, thus leading to neuronal apoptosis. APEX1 AP-endodeoxyribonuclease is repressed by phosphorylation, resulting in accumulation of DNA damage and contributing to neuronal death. NOS3 phosphorylation down regulates NOS3-derived nitrite (NO) levels. SRC phosphorylation mediates its ubiquitin-dependent degradation and thus leads to cytoskeletal reorganization. May regulate endothelial cell migration and angiogenesis via the modulation of lamellipodia formation. Involved in dendritic spine morphogenesis by mediating the EFNA1-EPHA4 signaling. The complex p35/CDK5 participates in the regulation of the circadian clock by modulating the function of CLOCK protein: phosphorylates CLOCK at 'Thr-451' and 'Thr-461' and regulates the transcriptional activity of the CLOCK-ARNTL/BMAL1 heterodimer in association with altered stability and subcellular distribution. Proline-directed serine/threonine-protein kinase essential for neuronal cell cycle arrest and differentiation and may be involved in apoptotic cell death in neuronal diseases by triggering abortive cell cycle re-entry. Related diseases Lissencephaly 7, with cerebellar hypoplasia (LIS7) [MIM:616342]: A form of lissencephaly, a disorder of cortical development characterized by agyria or pachygyria and disorganization of the clear neuronal lamination of normal six-layered cortex. LIS7 patients manifest lack of psychomotor development, facial dysmorphism, arthrogryposis, and early-onset intractable seizures resulting in death in infancy. {ECO:0000269|PubMed:25560765}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07364; DB04014; DB03496; DB02950; DB02052; DB02116; DB03428; DB15442 Interacts with P61158; P05067; P23560-2; Q8TDN4; P14635; P24863; P30279; P30281; Q14094; Q15078; P38936; P46527; Q9UJC3; Q6FHY5; Q9Y6R0; P37231-2; P62937; O60260-5; Q5MJ70; A6NLX3; P20226; P09936 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Apoptosis; ATP-binding; Biological rhythms; Cell cycle; Cell division; Cell membrane; Cell projection; Cytoplasm; Kinase; Lissencephaly; Membrane; Neurodegeneration; Neurogenesis; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Synapse; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 33303.1 Length 292 Aromaticity 0.1 Instability index 28.4 Isoelectric point 8.04 Charge (pH=7) 2.07 2D Binding mode Binding energy (Kcal/mol) -9.52  Molscript Map  Pymol Map 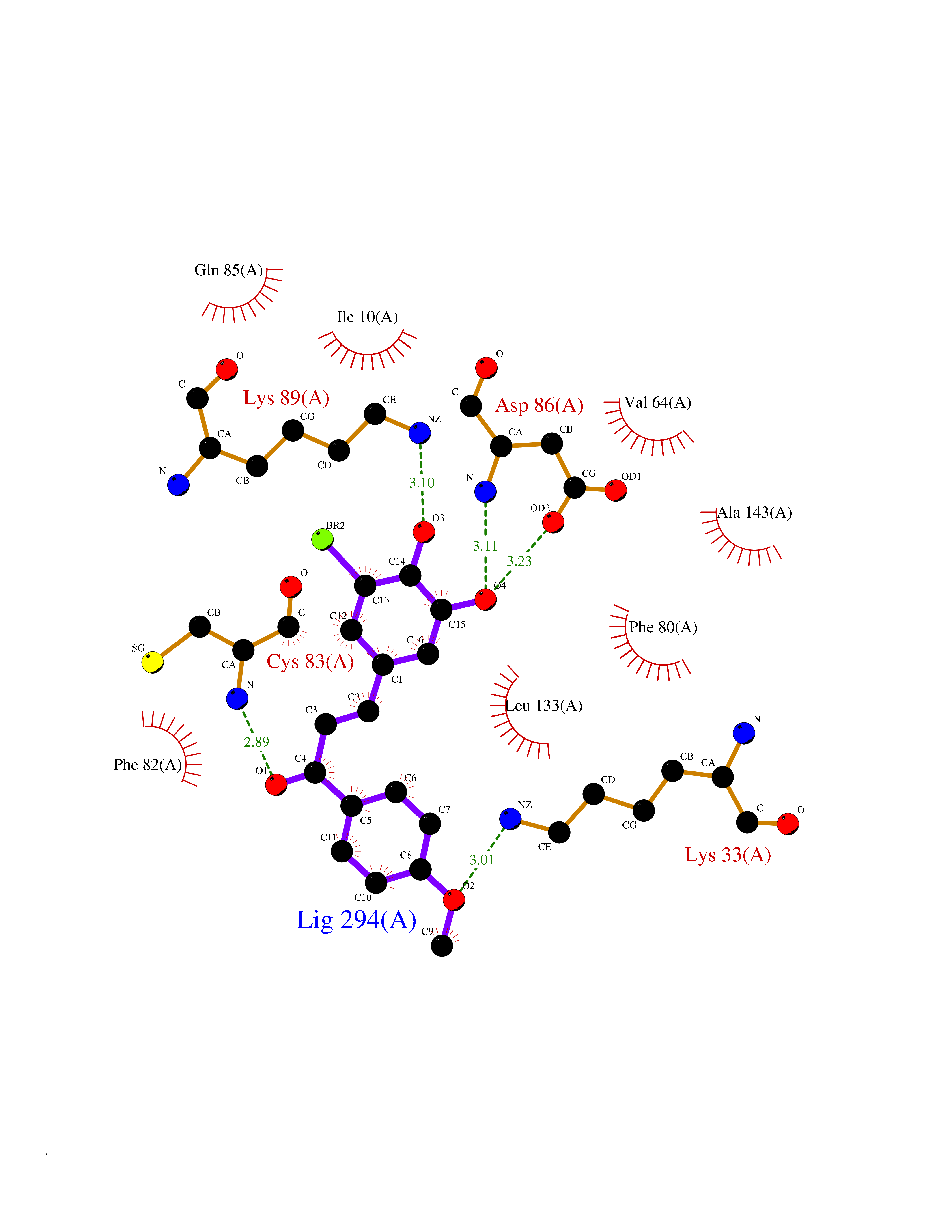 Ligplot Map 3D Binding mode Sequence MQKYEKLEKIGEGTYGTVFKAKNRETHEIVALKRVRLDDDDEGVPSSALREICLLKELKHKNIVRLHDVLHSDKKLTLVFEFCDQDLKKYFDSCNGDLDPEIVKSFLFQLLKGLGFCHSRNVLHRDLKPQNLLINRNGELKLANFGLARAFGIPVRCYSAEVVTLWYRPPDVLFGAKLYSTSIDMWSAGCIFAELANAGRPLFPGNDVDDQLKRIFRLLGTPTEEQWPSMTKLPDYKPYPMYPATTSLVNVVPKLNATGRDLLQNLLKCNPVQRISAEEALQHPYFSDFCPP Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Dual-specificity tyrosine-phosphorylation regulated kinase 3 (DYRK3) | 5Y86 | 6.98 | |
Target general information Gen name DYRK3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Regulatory erythroid kinase; REDK; Dual specificity tyrosine-phosphorylation-regulated kinase 3 Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, MNB/DYRK subfamily Biochemical class Kinase Function Dual-specificity tyrosine-regulated kinases (DYRKs) autophosphorylate a critical tyrosine residue in their activation loop and phosphorylate their substrate on serine and threonine residues. Acts as a central dissolvase of membraneless organelles during the G2-to-M transition, after the nuclear-envelope breakdown: acts by mediating phosphorylation of multiple serine and threonine residues in unstructured domains of proteins, such as SRRM1 and PCM1. Does not mediate disassembly of all membraneless organelles: disassembly of P-body and nucleolus is not regulated by DYRK3. Dissolution of membraneless organelles at the onset of mitosis is also required to release mitotic regulators, such as ZNF207, from liquid-unmixed organelles where they are sequestered and keep them dissolved during mitosis. Regulates mTORC1 by mediating the dissolution of stress granules: during stressful conditions, DYRK3 partitions from the cytosol to the stress granule, together with mTORC1 components, which prevents mTORC1 signaling. When stress signals are gone, the kinase activity of DYRK3 is required for the dissolution of stress granule and mTORC1 relocation to the cytosol: acts by mediating the phosphorylation of the mTORC1 inhibitor AKT1S1, allowing full reactivation of mTORC1 signaling. Also acts as a negative regulator of EPO-dependent erythropoiesis: may place an upper limit on red cell production during stress erythropoiesis. Inhibits cell death due to cytokine withdrawal in hematopoietic progenitor cells. Promotes cell survival upon genotoxic stress through phosphorylation of SIRT1: this in turn inhibits p53/TP53 activity and apoptosis. Dual-specificity protein kinase that promotes disassembly of several types of membraneless organelles during mitosis, such as stress granules, nuclear speckles and pericentriolar material. Related diseases Defects in MELK are associated with some cancers, such as brain or breast cancers. Expression is dramatically increased in aggressive undifferentiated tumors, correlating with poor patient outcome in breast and brain cancers, suggesting a role in tumor-initiating cells and proliferation via its function in cell proliferation regulation. Drugs (DrugBank ID) NA Interacts with Q9H8Y8 EC number EC 2.7.12.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell cycle; Cell division; Cytoplasm; Cytoskeleton; Kinase; Magnesium; Metal-binding; Mitosis; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 44821.5 Length 395 Aromaticity 0.1 Instability index 49.38 Isoelectric point 9.52 Charge (pH=7) 21.08 2D Binding mode Binding energy (Kcal/mol) -9.52 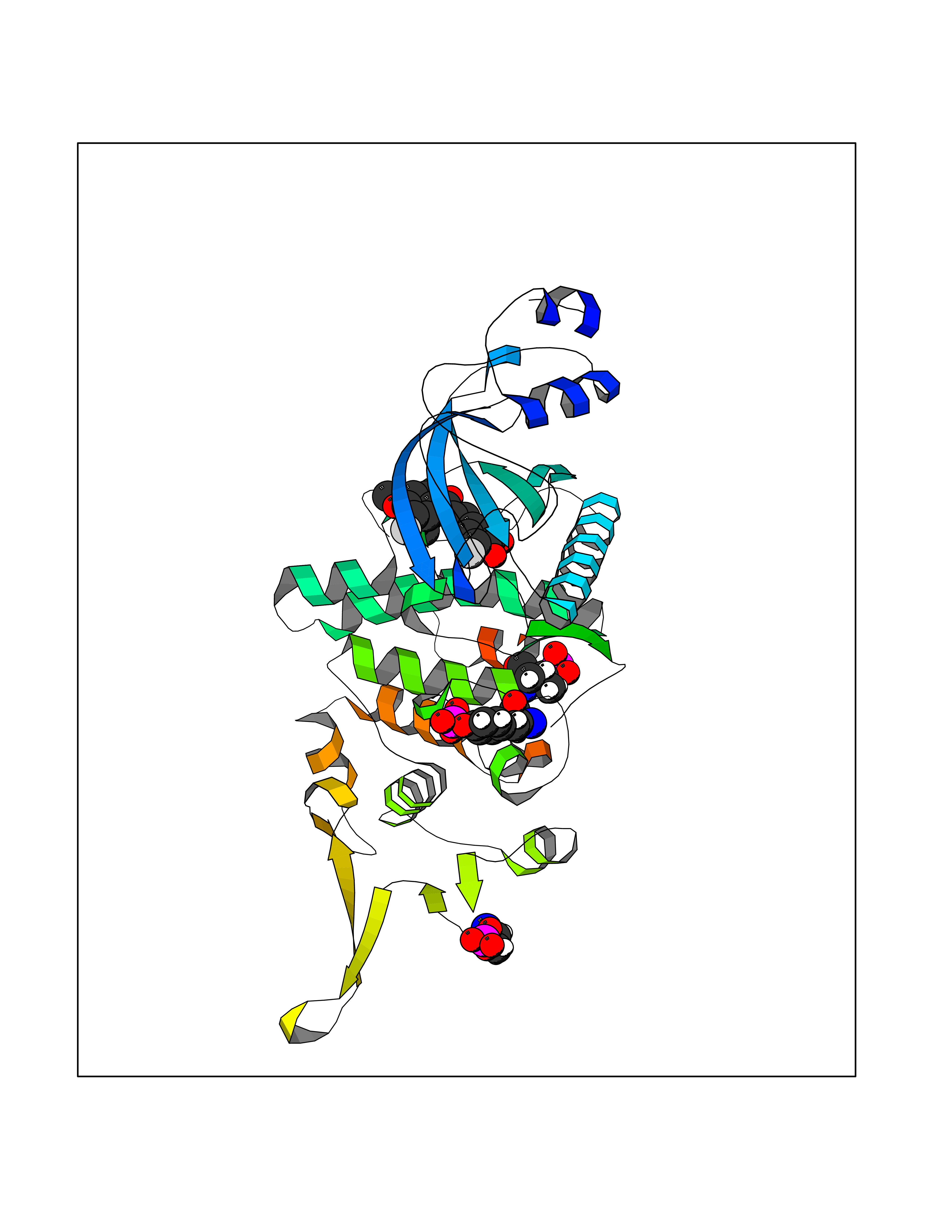 Molscript Map 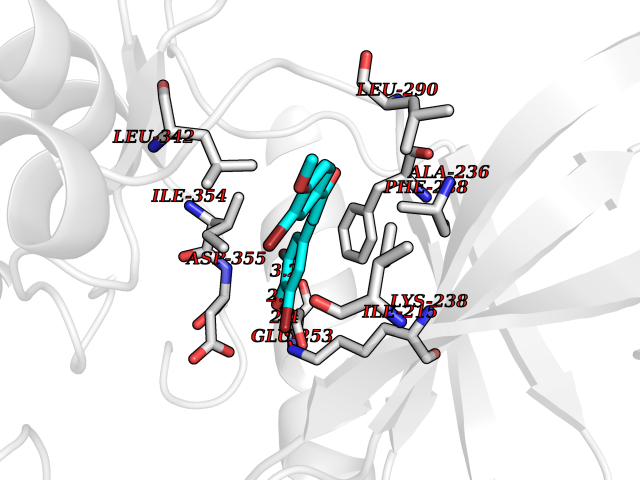 Pymol Map 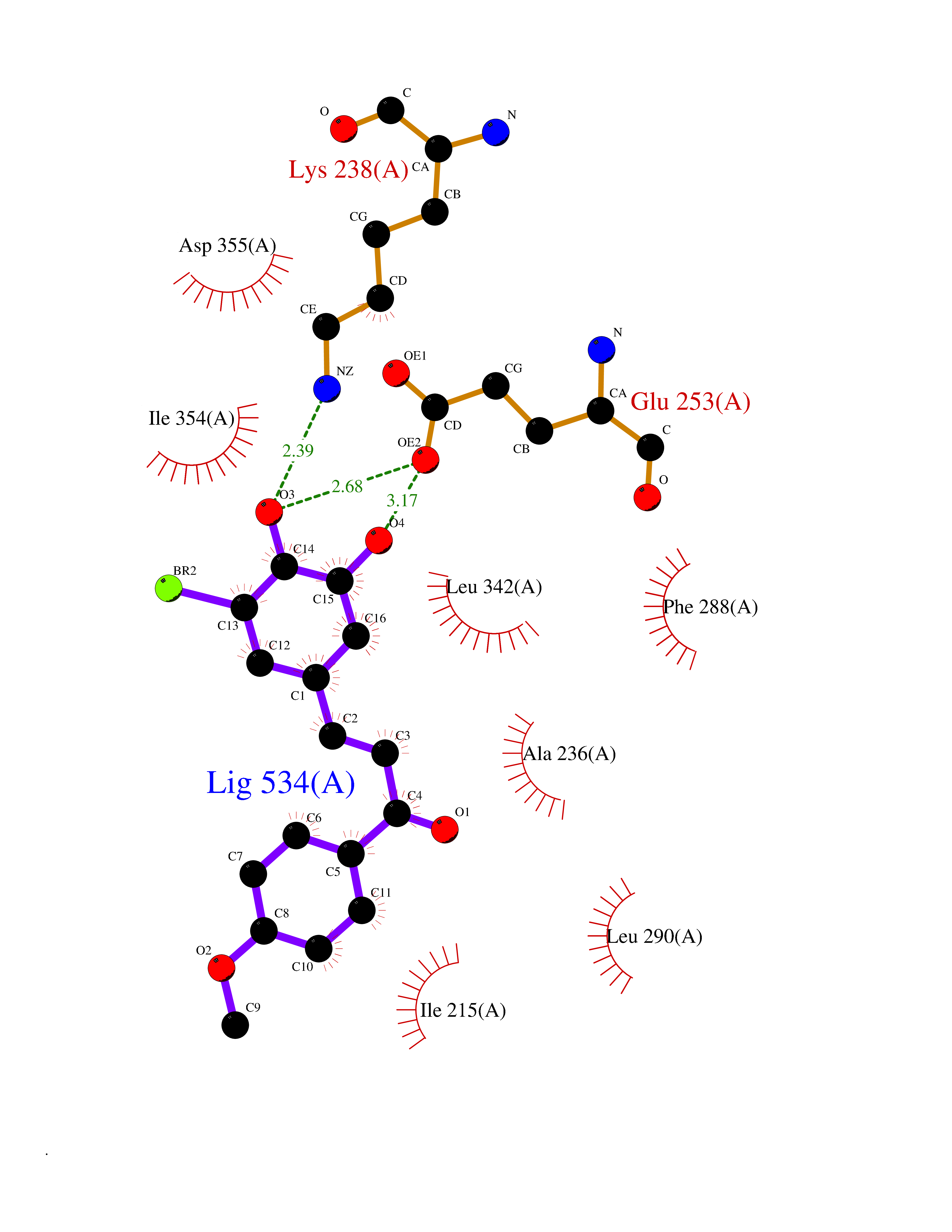 Ligplot Map 3D Binding mode Sequence VVPLTPEQALKQYKHHLTAYEKLEIINYPEIYFVGPNAKKRHGVIGGPNNGGYDDADGAYIHVPRDHLAYRYEVLKIIGKGSFGQVARVYDHKLRQYVALKMVRNEKRFHRQAAEEIRILEHLKKQDKTGSMNVIHMLESFTFRNHVCMAFELLSIDLYELIKKNKFQGFSVQLVRKFAQSILQSLDALHKNKIIHCDLKPENILLKHHGRSXTKVIDFGSSCFEYQKLYTXIQSRFYRAPEIILGSRYSTPIDIWSFGCILAELLTGQPLFPGEDEGDQLACMMELLGMPPPKLLEQSKRAKYFINXKGIPRYCSVTTQADGRVVLVGGRSRRGKKRGPPGSKDWGTALKGCDDYLFIEFLKRCLHWDPSARLXPAQALRHPWISKSVPRPLTT Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Cytosolic 10-formyltetrahydrofolate dehydrogenase | 2CFI | 6.97 | |
Target general information Gen name ALDH1L1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms FTHFD Protein family GART family; Aldehyde dehydrogenase family, ALDH1L subfamily Biochemical class Oxidoreductase Function Aldehyde dehydrogenase (NAD) activity.Catalytic activity.Formyltetrahydrofolate dehydrogenase activity.Hydroxymethyl-, formyl- and related transferase activity. Related diseases Developmental and epileptic encephalopathy 39 with leukodystrophy (DEE39) [MIM:612949]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE39 is characterized by global hypomyelination of the central nervous system, with the gray matter appearing relatively unaffected. Inheritance is autosomal recessive. {ECO:0000269|PubMed:19641205, ECO:0000269|PubMed:24515575}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00116 Interacts with Q3SY69; Q92624 EC number 1.5.1.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; NADP; One-carbon metabolism; Oxidoreductase; Phosphopantetheine; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 33869.5 Length 308 Aromaticity 0.08 Instability index 30.42 Isoelectric point 6.09 Charge (pH=7) -3.83 2D Binding mode Binding energy (Kcal/mol) -9.5 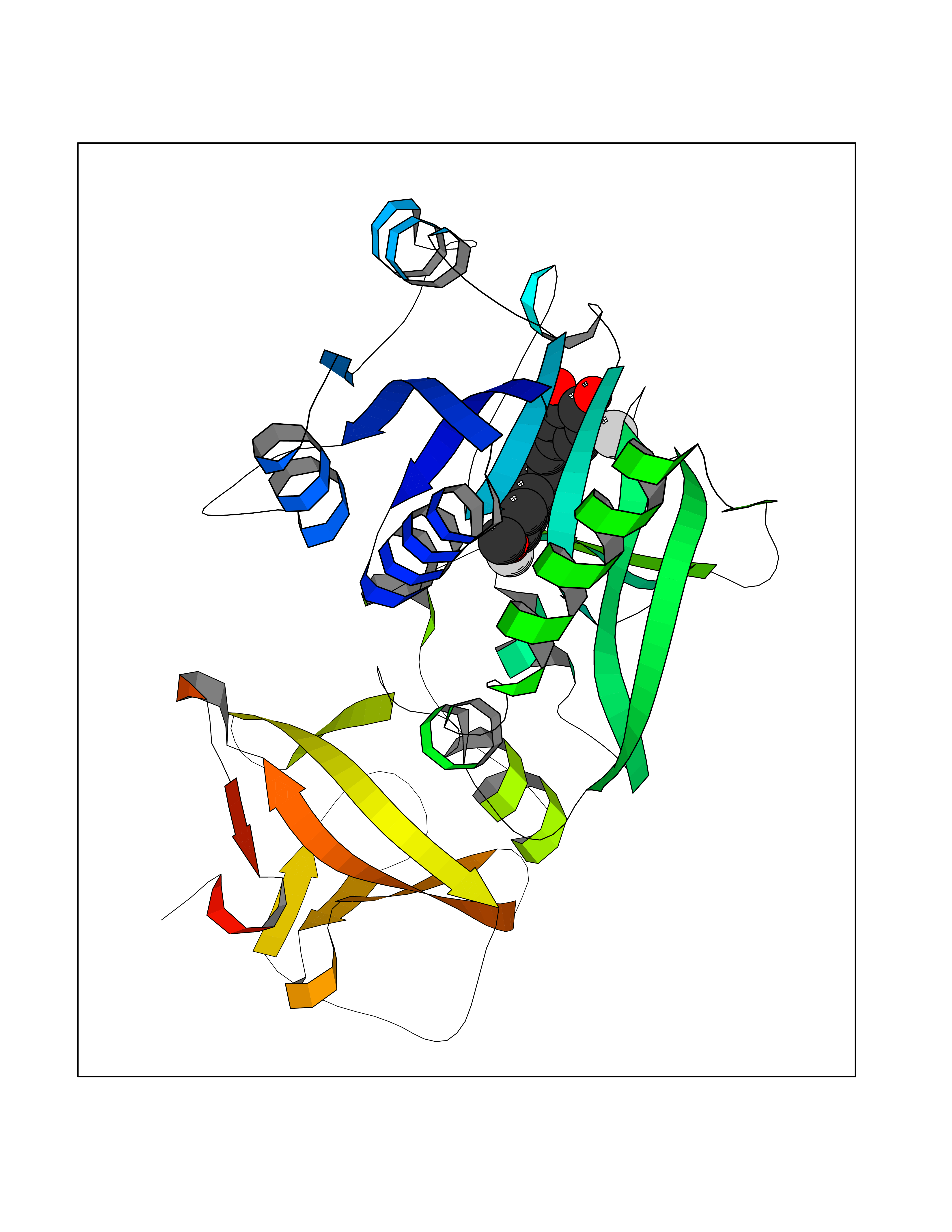 Molscript Map 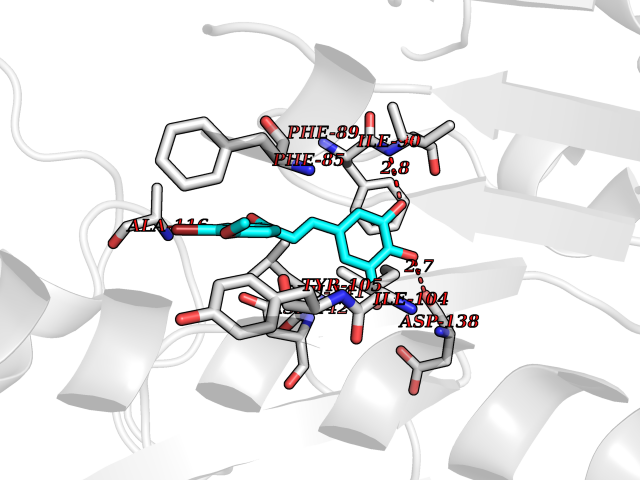 Pymol Map 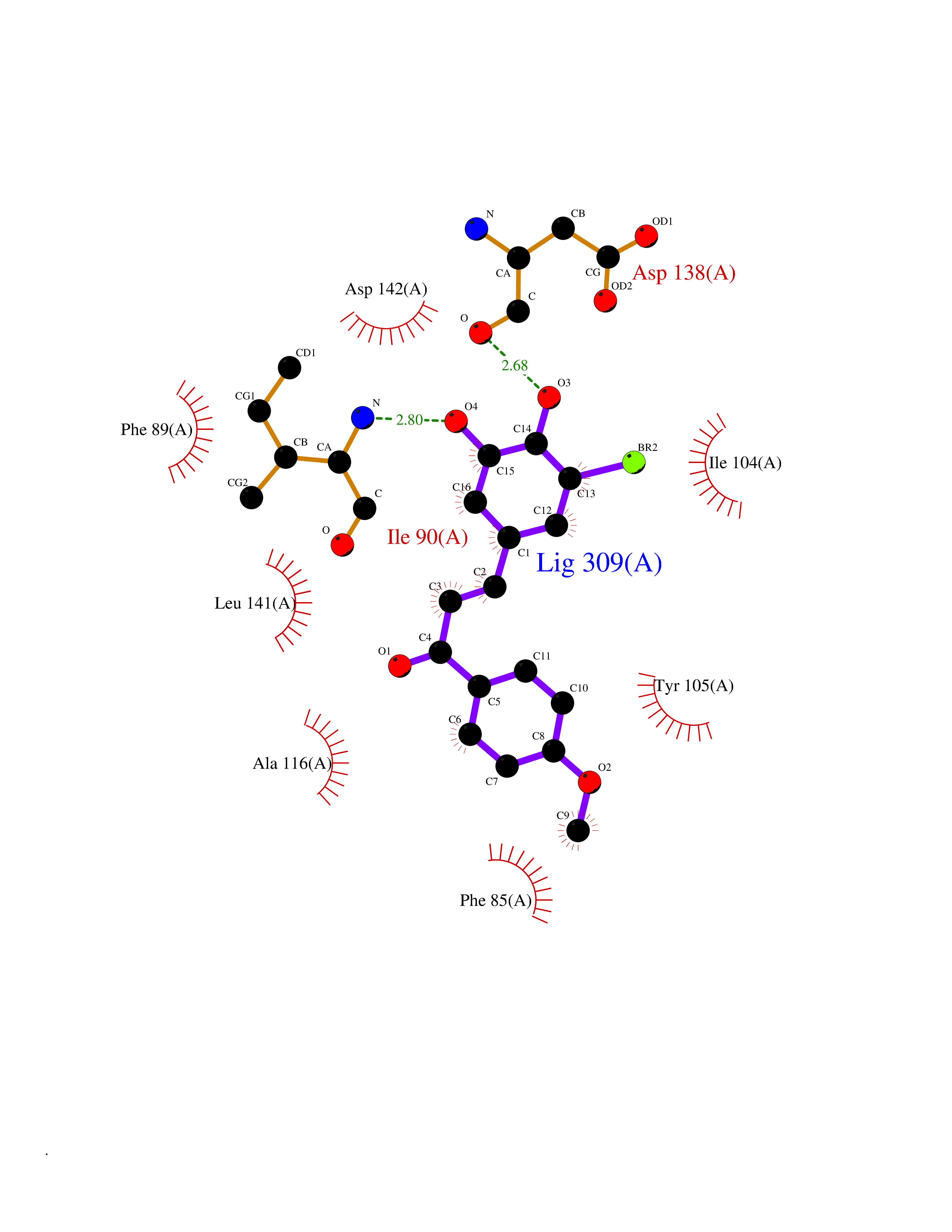 Ligplot Map 3D Binding mode Sequence SMKIAVIGQSLFGQEVYCHLRKEGHEVVGVFTVPDKDGKADPLGLEAEKDGVPVFKYSRWRAKGQALPDVVAKYQALGAELNVLPFCSQFIPMEIISAPRHGSIIYHPSLLPRHRGASAINWTLIHGDKKGGFSIFWADDGLDTGDLLLQKECEVLPDDTVSTLYNRFLFPEGIKGMVQAVRLIAEGKAPRLPQPEEGATYEGIQKKETAKINWDQPAEAIHNWIRGNDKVPGAWTEACEQKLTFFNSTLNTSGLVPEGDALPIPGAHRPGVVTKAGLILFGNDDKMLLVKNIQLEDGKMILASNFFK Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Matrix metalloproteinase-9 (MMP-9) | 4XCT | 6.97 | |
Target general information Gen name MMP9 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Matrix metalloproteinase 9; GELB; CLG4B; 92 kDa type IV collagenase; 92 kDa gelatinase Protein family Peptidase M10A family Biochemical class Peptidase Function Could play a role in bone osteoclastic resorption. Cleaves KiSS1 at a Gly-|-Leu bond. Cleaves type IV and type V collagen into large C-terminal three quarter fragments and shorter N-terminal one quarter fragments. Degrades fibronectin but not laminin or Pz-peptide. May play an essential role in local proteolysis of the extracellular matrix and in leukocyte migration. Related diseases Intervertebral disc disease (IDD) [MIM:603932]: A common musculo-skeletal disorder caused by degeneration of intervertebral disks of the lumbar spine. It results in low-back pain and unilateral leg pain. {ECO:0000269|PubMed:18455130}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Metaphyseal anadysplasia 2 (MANDP2) [MIM:613073]: A bone development disorder characterized by skeletal anomalies that resolve spontaneously with age. Clinical characteristics are evident from the first months of life and include slight shortness of stature and a mild varus deformity of the legs. Patients attain a normal stature in adolescence and show improvement or complete resolution of varus deformity of the legs and rhizomelic micromelia. {ECO:0000269|PubMed:19615667}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07246; DB07285; DB01949; DB03683; DB07117; DB01197; DB06423; DB00143; DB00786; DB01017; DB05387; DB12843; DB05495; DB01593; DB14487; DB14533; DB14548 Interacts with Q16819; Q16820; P14780; Q8IX30; P13611; Q9ZFS6 EC number EC 3.4.24.35 Uniprot keywords 3D-structure; Calcium; Collagen degradation; Direct protein sequencing; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 17550.3 Length 157 Aromaticity 0.14 Instability index 27.98 Isoelectric point 5.12 Charge (pH=7) -11.26 2D Binding mode Binding energy (Kcal/mol) -9.5 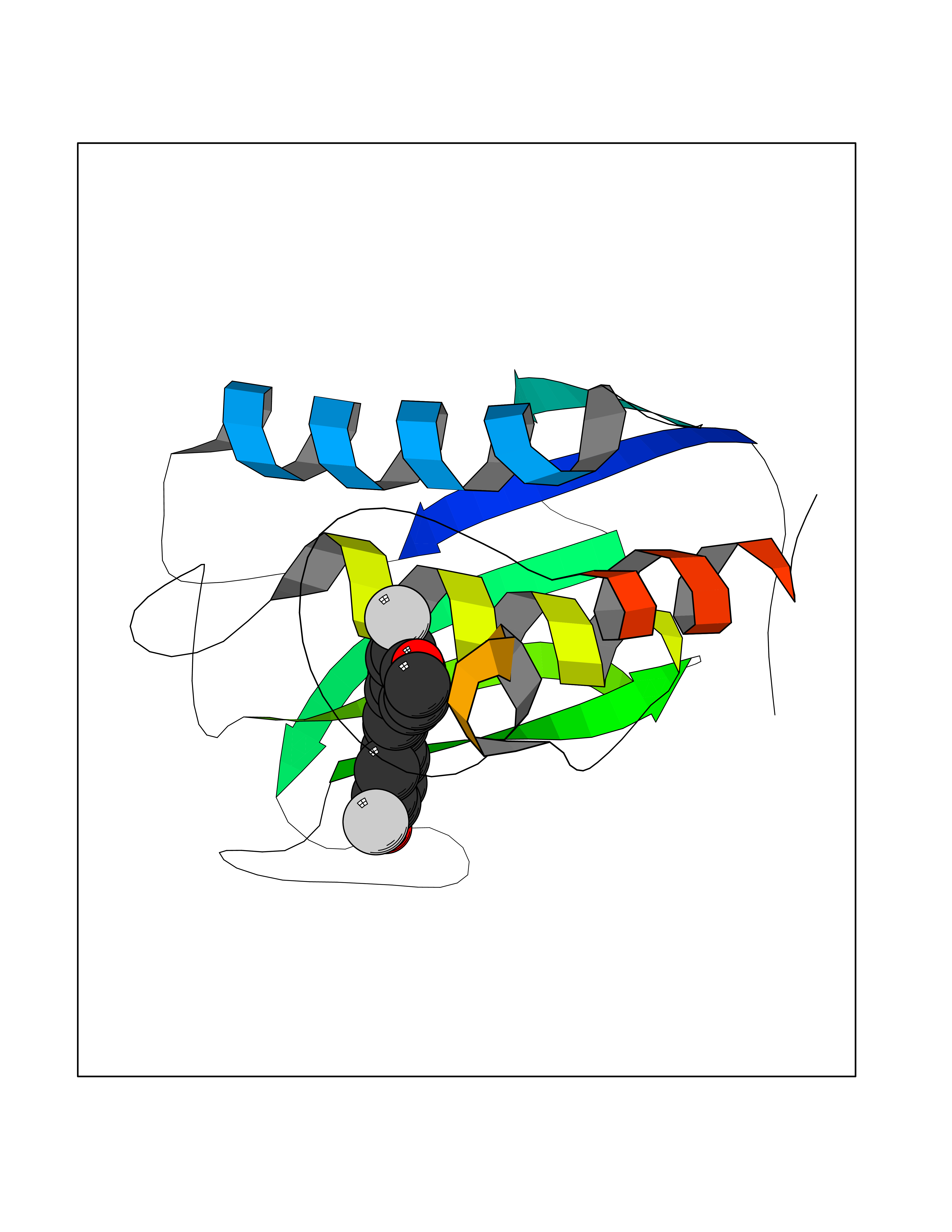 Molscript Map 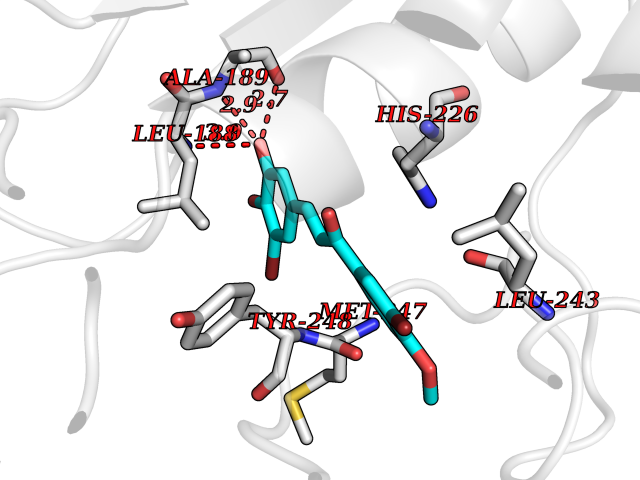 Pymol Map 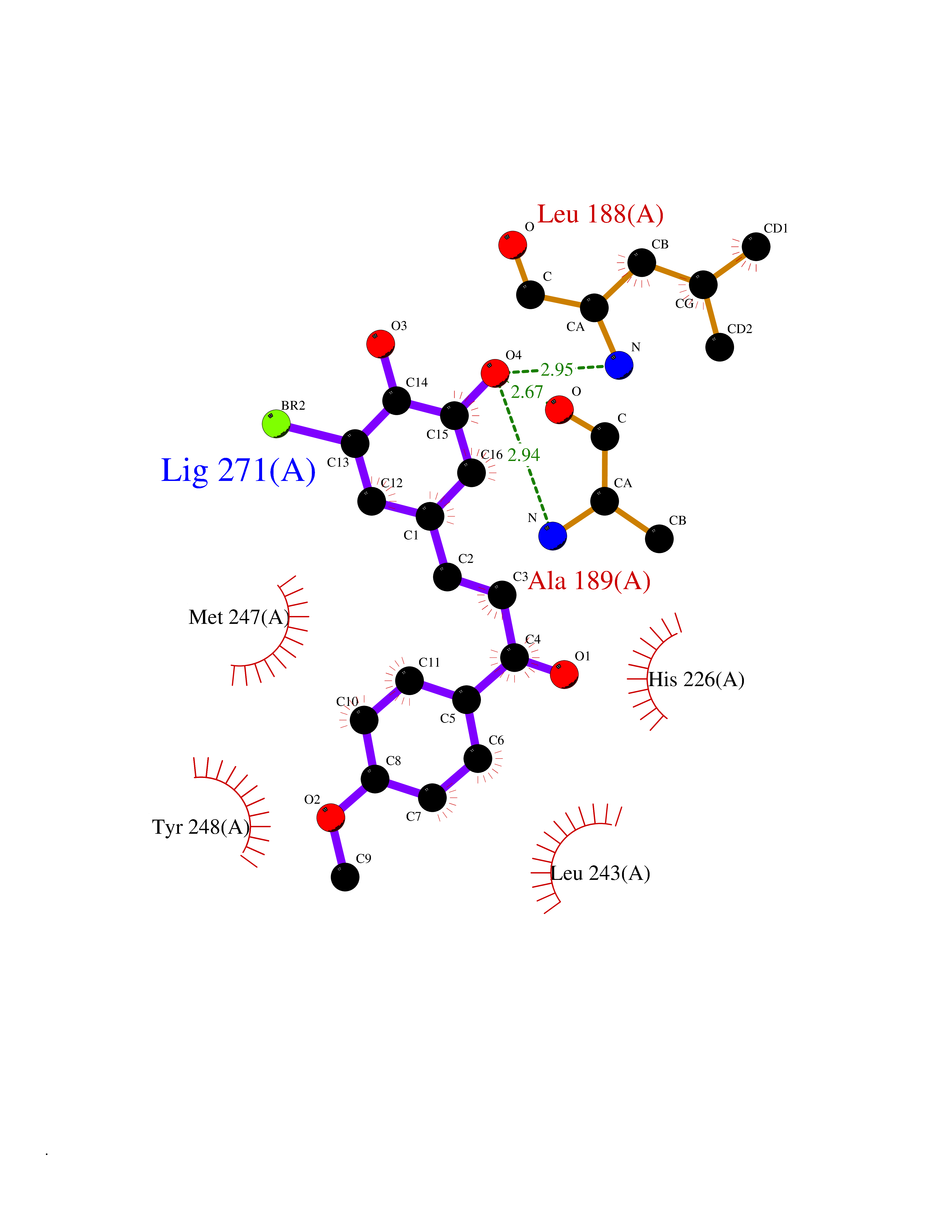 Ligplot Map 3D Binding mode Sequence DLKWHHHNITYWIQNYSEDLPRAVIDDAFARAFALWSAVTPLTFTRVYSRDADIVIQFGVAEHGDGYPFDGKDGLLAHAFPPGPGIQGDAHFDDDELWSLGKGVGYSLFLVAAHEFGHALGLDHSSVPEALMYPMYRFTEGPPLHKDDVNGIRHLYG Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Retinaldehyde-binding protein 1 | 3HX3 | 6.97 | |
Target general information Gen name RLBP1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CRALBP Protein family NA Biochemical class Transport protein Function 11-cis retinal binding.Retinol binding.Transporter activity. Related diseases Bothnia retinal dystrophy (BRD) [MIM:607475]: A type of retinitis punctata albescens. Affected individuals show night blindness from early childhood with features consistent with retinitis punctata albescens and macular degeneration. {ECO:0000269|PubMed:10102298}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rod-cone dystrophy Newfoundland (NFRCD) [MIM:607476]: A rod-cone dystrophy reminiscent of retinitis punctata albescens but with a substantially lower age at onset and more-rapid and distinctive progression. Rod-cone dystrophies results from initial loss of rod photoreceptors, later followed by cone photoreceptors loss. {ECO:0000269|PubMed:11868161}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Retinitis punctata albescens (RPA) [MIM:136880]: A form of fleck retina disease characterized by aggregation of white flecks posteriorly in the retina, causing night blindness and delayed dark adaptation. It differs from fundus albipunctatus in being progressive and evolving to generalized atrophy of the retina. {ECO:0000269|PubMed:10102299, ECO:0000269|PubMed:11453974, ECO:0000269|PubMed:9326942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00162 Interacts with Q9P2G9-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Disease variant; Proteomics identification; Reference proteome; Retinol-binding; Sensory transduction; Transport; Vision Protein physicochemical properties Chain ID A Molecular weight (Da) 28328.6 Length 250 Aromaticity 0.14 Instability index 52.64 Isoelectric point 4.96 Charge (pH=7) -9.87 2D Binding mode Binding energy (Kcal/mol) -9.5 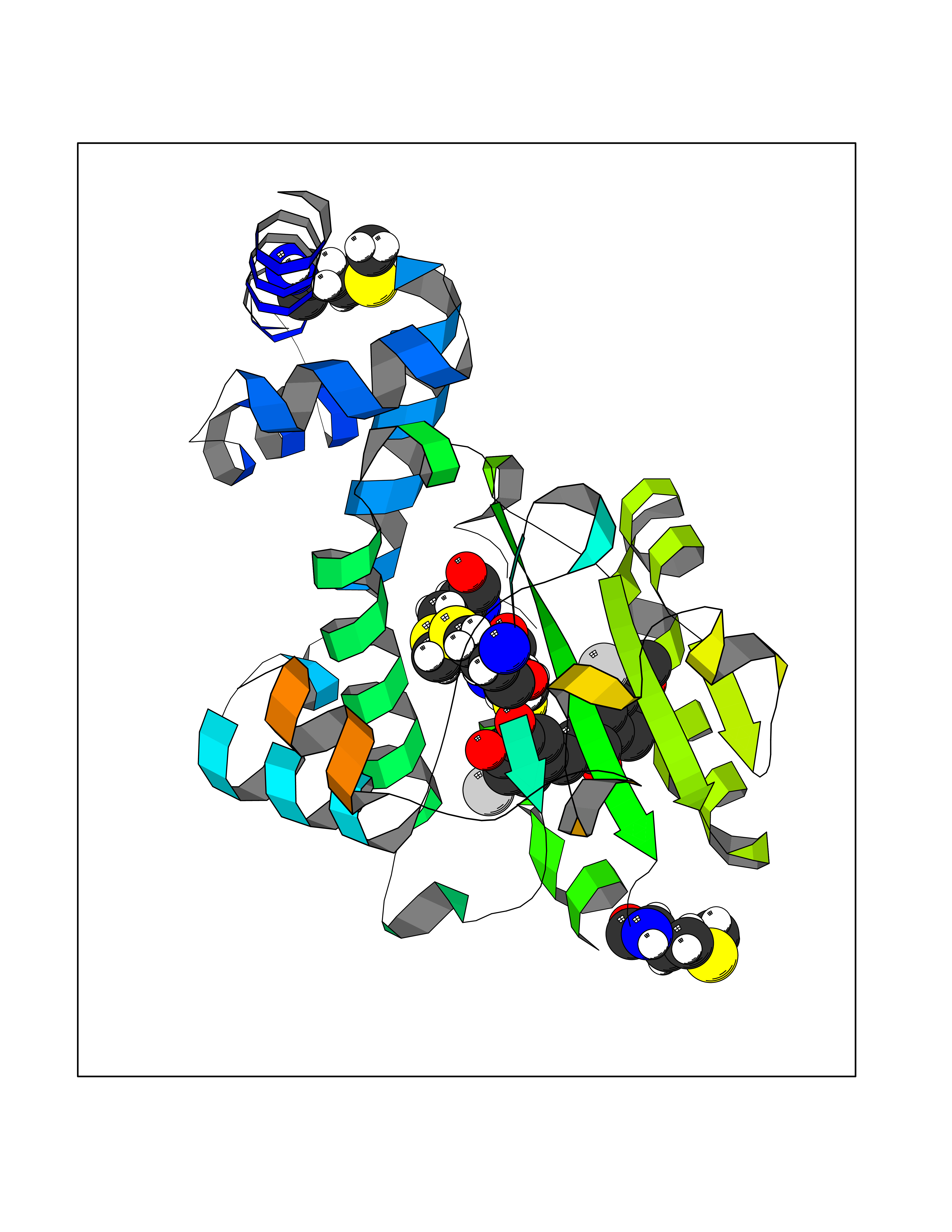 Molscript Map 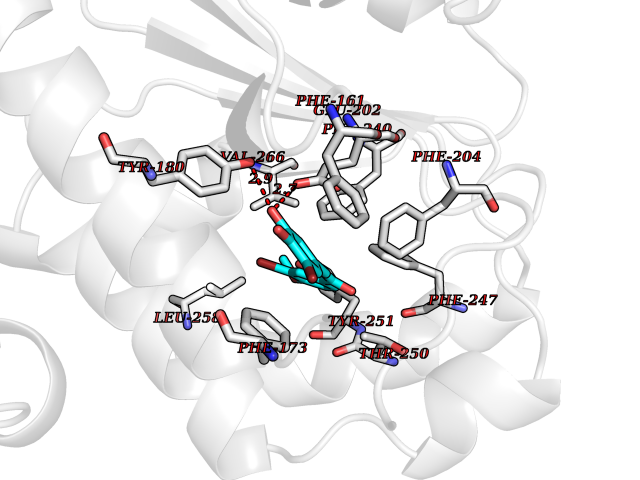 Pymol Map 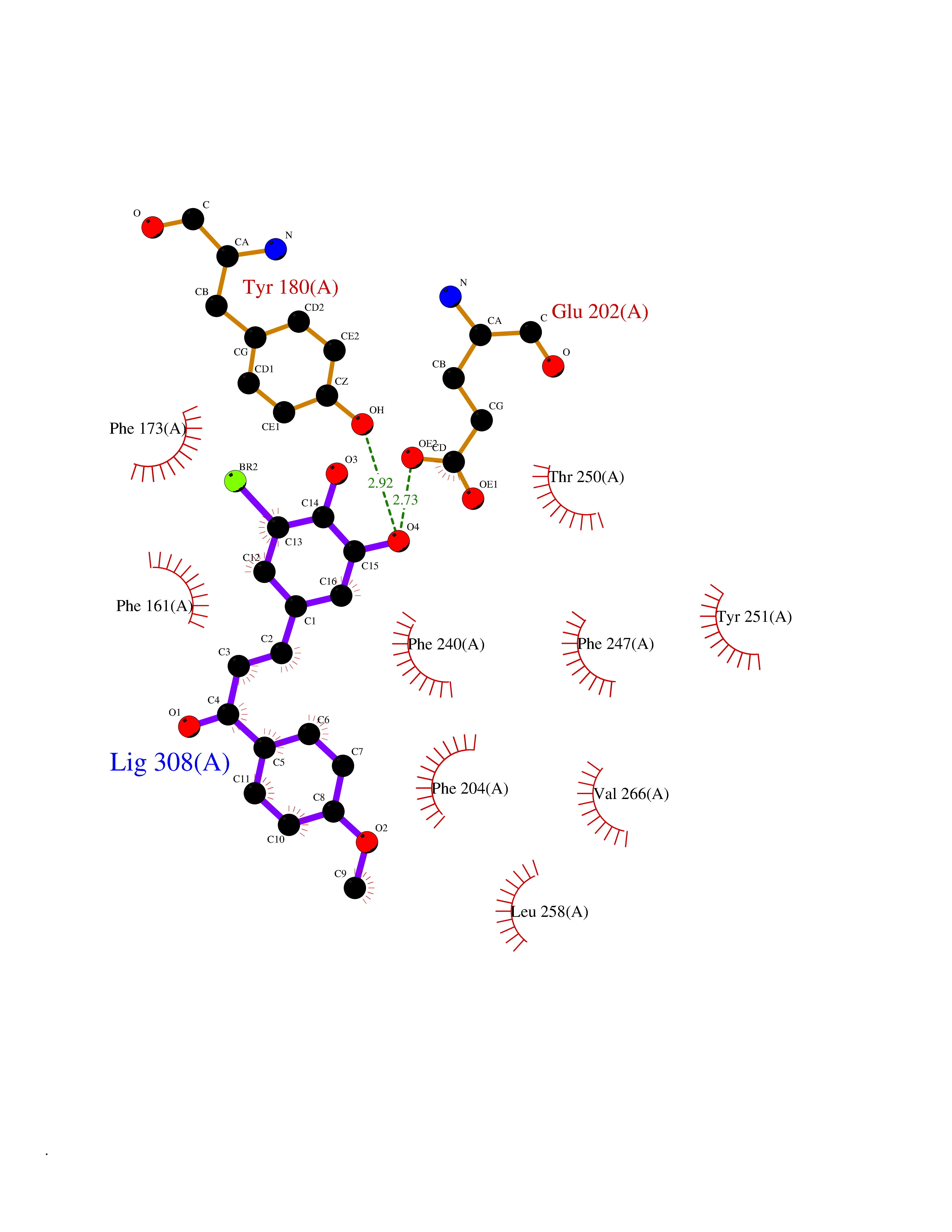 Ligplot Map 3D Binding mode Sequence ETREEAVRELQEXVQAQAASGEELAVAVAERVQEKDSGFFLRFIRARKFNVGRAYELLRGYVNFRLQYPELFDSLSPEAVRCTIEAGYPGVLSSRDKYGRVVXLFNIENWQSQEITFDEILQAYCFILEKLLENEETQINGFCIIENFKGFTXQQAASLRTSDLRKXVDXLQDSFPAWFKAIHFIHQPWYFTTTYNVVKPFLKSKLLERVFVHGDDLSGFYQEIDENILPSDFGGTLPKYDGKAVAEQLF Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Bacterial Flavohemoglobin (Bact hmp) | 1GVH | 6.97 | |
Target general information Gen name Bact hmp Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms Nitric oxide dioxygenase; NOD; NO oxygenase; Hemoglobin-like protein; HMP; Ferrisiderophore reductase B; Dihydropteridine reductase Protein family Globin family, Two-domain flavohemoproteins subfamily; Flavoprotein pyridine nucleotide cytochrome reductase family Biochemical class Paired donor oxygen oxidoreductase Function Various electron acceptors arealso reduced by HMP in vitro, including dihydropterine, ferrisiderophores, ferric citrate, cytochrome c, nitrite, S-nitrosoglutathione, and alkylhydroperoxides. However, it is unknown if these reactions are of any biological significance in vivo. Related diseases Ovarian dysgenesis 1 (ODG1) [MIM:233300]: An autosomal recessive disease characterized by primary amenorrhea, variable development of secondary sex characteristics, poorly developed streak ovaries, and high serum levels of follicle-stimulating hormone (FSH) and luteinizing hormone (LH). {ECO:0000269|PubMed:10551778, ECO:0000269|PubMed:11889179, ECO:0000269|PubMed:12571157, ECO:0000269|PubMed:12915623, ECO:0000269|PubMed:7553856, ECO:0000269|PubMed:9769327, ECO:0000269|PubMed:9851774}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ovarian hyperstimulation syndrome (OHSS) [MIM:608115]: Disorder which occurs either spontaneously or most often as an iatrogenic complication of ovarian stimulation treatments for in vitro fertilization. The clinical manifestations vary from abdominal distention and discomfort to potentially life-threatening, massive ovarian enlargement and capillary leak with fluid sequestration. Pathologic features of this syndrome include the presence of multiple serous and hemorrhagic follicular cysts lined by luteinized cells, a condition called hyperreactio luteinalis. {ECO:0000269|PubMed:12930927, ECO:0000269|PubMed:12930928, ECO:0000269|PubMed:15080154, ECO:0000269|PubMed:16278261, ECO:0000269|PubMed:17721928, ECO:0000269|PubMed:24058690, ECO:0000269|PubMed:25581598}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number EC 1.14.12.17 Uniprot keywords 3D-structure; Cytoplasm; Detoxification; Direct protein sequencing; FAD; Flavoprotein; Heme; Iron; Metal-binding; NAD; NADP; Oxidoreductase; Oxygen transport; Reference proteome; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 43867.1 Length 396 Aromaticity 0.1 Instability index 28.85 Isoelectric point 5.48 Charge (pH=7) -12.32 2D Binding mode Binding energy (Kcal/mol) -9.51 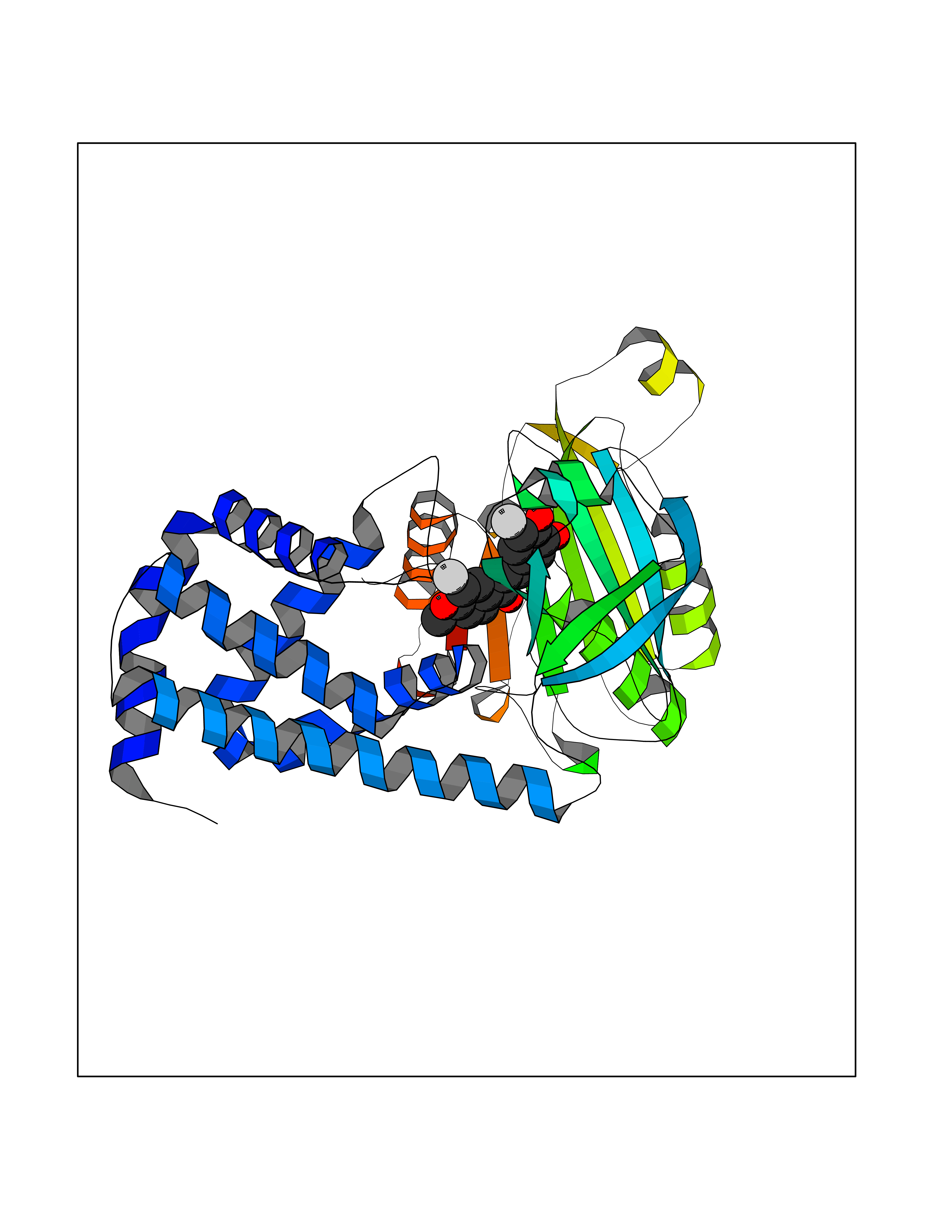 Molscript Map 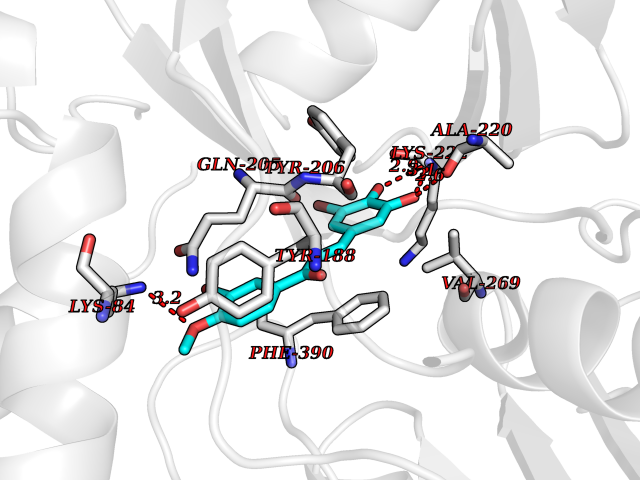 Pymol Map 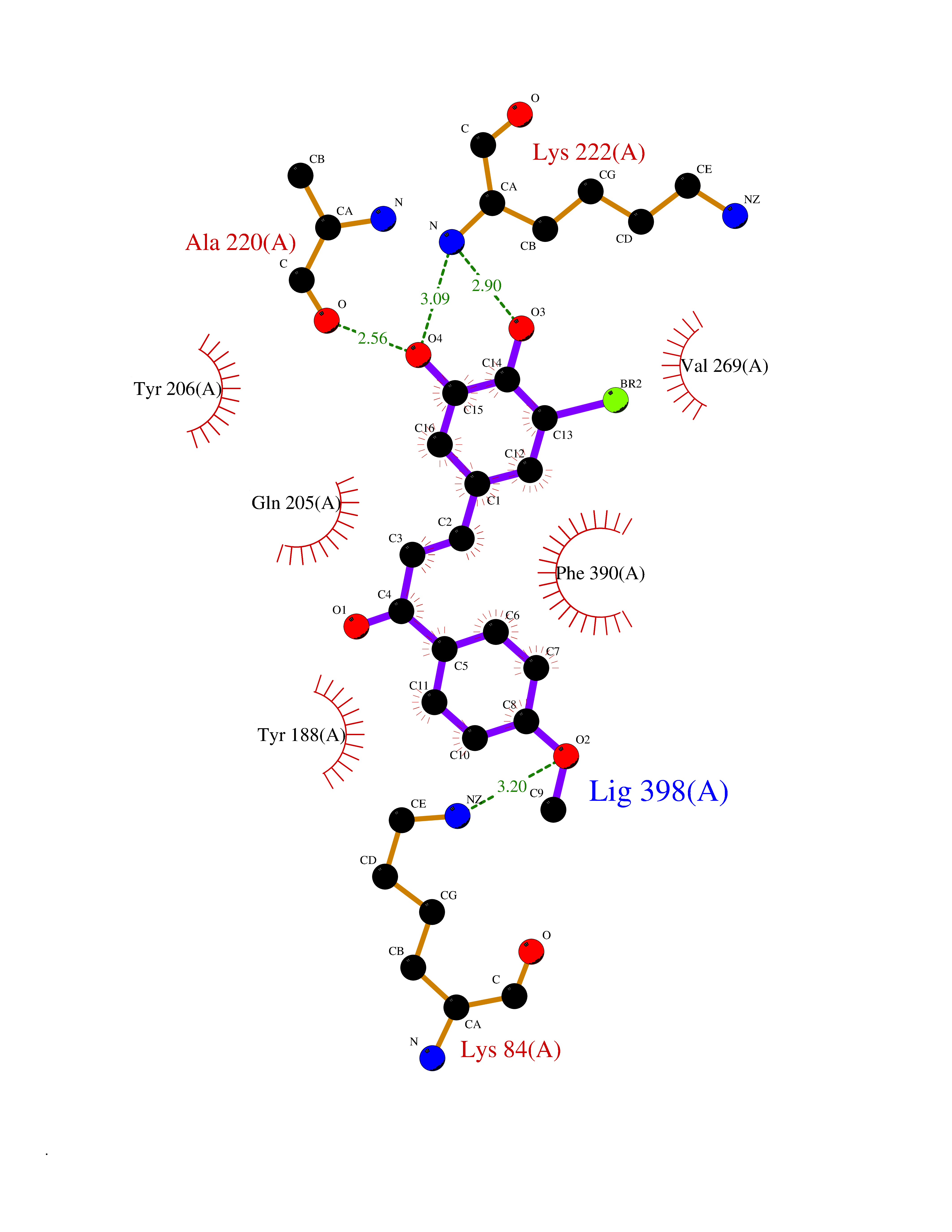 Ligplot Map 3D Binding mode Sequence MLDAQTIATVKATIPLLVETGPKLTAHFYDRMFTHNPELKEIFNMSNQRNGDQREALFNAIAAYASNIENLPALLPAVEKIAQKHTSFQIKPEQYNIVGEHLLATLDEMFSPGQEVLDAWGKAYGVLANVFINREAEIYNENASKAGGWEGTRDFRIVAKTPRSALITSFELEPVDGGAVAEYRPGQYLGVWLKPEGFPHQEIRQYSLTRKPDGKGYRIAVKREEGGQVSNWLHNHANVGDVVKLVAPAGDFFMAVADDTPVTLISAGVGQTPMLAMLDTLAKAGHTAQVNWFHAAENGDVHAFADEVKELGQSLPRFTAHTWYRQPSEADRAKGQFDSEGLMDLSKLEGAFSDPTMQFYLCGPVGFMQFTAKQLVDLGVKQENIHYECFGPHKVL Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Androgen receptor (AR) | 2AM9 | 6.97 | |
Target general information Gen name AR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Testosterone receptor; Nuclear receptor subfamily 3 group C member 4; NR3C4; Dihydrotestosterone receptor; DHTR Protein family Nuclear hormone receptor family, NR3 subfamily Biochemical class Nuclear hormone receptor Function Transcription factor activity is modulated by bound coactivator and corepressor proteins like ZBTB7A that recruits NCOR1 and NCOR2 to the androgen response elements/ARE on target genes, negatively regulating androgen receptor signaling and androgen-induced cell proliferation. Transcription activation is also down-regulated by NR0B2. Activated, but not phosphorylated, by HIPK3 and ZIPK/DAPK3. Steroid hormone receptors are ligand-activated transcription factors that regulate eukaryotic gene expression and affect cellular proliferation and differentiation in target tissues. Related diseases Androgen insensitivity syndrome (AIS) [MIM:300068]: An X-linked recessive form of pseudohermaphroditism due end-organ resistance to androgen. Affected males have female external genitalia, female breast development, blind vagina, absent uterus and female adnexa, and abdominal or inguinal testes, despite a normal 46,XY karyotype. {ECO:0000269|PubMed:10022458, ECO:0000269|PubMed:10221692, ECO:0000269|PubMed:10221770, ECO:0000269|PubMed:10404311, ECO:0000269|PubMed:10458483, ECO:0000269|PubMed:10571951, ECO:0000269|PubMed:10590024, ECO:0000269|PubMed:10690872, ECO:0000269|PubMed:11587068, ECO:0000269|PubMed:11744994, ECO:0000269|PubMed:1307250, ECO:0000269|PubMed:1316540, ECO:0000269|PubMed:1426313, ECO:0000269|PubMed:1430233, ECO:0000269|PubMed:1464650, ECO:0000269|PubMed:14756668, ECO:0000269|PubMed:1480178, ECO:0000269|PubMed:1487249, ECO:0000269|PubMed:1569163, ECO:0000269|PubMed:1609793, ECO:0000269|PubMed:16129672, ECO:0000269|PubMed:16595706, ECO:0000269|PubMed:1775137, ECO:0000269|PubMed:1999491, ECO:0000269|PubMed:2082179, ECO:0000269|PubMed:2594783, ECO:0000269|PubMed:7537149, ECO:0000269|PubMed:7581399, ECO:0000269|PubMed:7633398, ECO:0000269|PubMed:7641413, ECO:0000269|PubMed:7671849, ECO:0000269|PubMed:7929841, ECO:0000269|PubMed:7962294, ECO:0000269|PubMed:7970939, ECO:0000269|PubMed:7981687, ECO:0000269|PubMed:7981689, ECO:0000269|PubMed:7993455, ECO:0000269|PubMed:8040309, ECO:0000269|PubMed:8096390, ECO:0000269|PubMed:8103398, ECO:0000269|PubMed:8162033, ECO:0000269|PubMed:8224266, ECO:0000269|PubMed:8281140, ECO:0000269|PubMed:8325950, ECO:0000269|PubMed:8339746, ECO:0000269|PubMed:8413310, ECO:0000269|PubMed:8446106, ECO:0000269|PubMed:8626869, ECO:0000269|PubMed:8647313, ECO:0000269|PubMed:8683794, ECO:0000269|PubMed:8723113, ECO:0000269|PubMed:8768864, ECO:0000269|PubMed:8809734, ECO:0000269|PubMed:8830623, ECO:0000269|PubMed:8918984, ECO:0000269|PubMed:8990010, ECO:0000269|PubMed:9001799, ECO:0000269|PubMed:9007482, ECO:0000269|PubMed:9039340, ECO:0000269|PubMed:9106550, ECO:0000269|PubMed:9160185, ECO:0000269|PubMed:9252933, ECO:0000269|PubMed:9255042, ECO:0000269|PubMed:9302173, ECO:0000269|PubMed:9328206, ECO:0000269|PubMed:9544375, ECO:0000269|PubMed:9554754, ECO:0000269|PubMed:9610419, ECO:0000269|PubMed:9627582, ECO:0000269|PubMed:9698822, ECO:0000269|PubMed:9851768, ECO:0000269|PubMed:9856504, ECO:0000269|Ref.116, ECO:0000269|Ref.182}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinal and bulbar muscular atrophy X-linked 1 (SMAX1) [MIM:313200]: An X-linked recessive form of spinal muscular atrophy. Spinal muscular atrophy refers to a group of neuromuscular disorders characterized by degeneration of the anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. SMAX1 occurs only in men. Age at onset is usually in the third to fifth decade of life, but earlier involvement has been reported. It is characterized by slowly progressive limb and bulbar muscle weakness with fasciculations, muscle atrophy, and gynecomastia. The disorder is clinically similar to classic forms of autosomal spinal muscular atrophy. {ECO:0000269|PubMed:15851746}. The disease is caused by variants affecting the gene represented in this entry. Caused by trinucleotide CAG repeat expansion. In SMAX1 patients the number of Gln ranges from 38 to 62. Longer expansions result in earlier onset and more severe clinical manifestations of the disease.; DISEASE: Prostate cancer, hereditary, X-linked 3 (HPCX3) [MIM:301120]: A condition associated with familial predisposition to cancer of the prostate. Most prostate cancers are adenocarcinomas that develop in the acini of the prostatic ducts. Other rare histopathologic types of prostate cancer that occur in approximately 5% of patients include small cell carcinoma, mucinous carcinoma, prostatic ductal carcinoma, transitional cell carcinoma, squamous cell carcinoma, basal cell carcinoma, adenoid cystic carcinoma (basaloid), signet-ring cell carcinoma and neuroendocrine carcinoma. {ECO:0000269|PubMed:8530589}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Defects in AR may play a role in metastatic prostate cancer. The mutated receptor stimulates prostate growth and metastases development despite of androgen ablation. This treatment can reduce primary and metastatic lesions probably by inducing apoptosis of tumor cells when they express the wild-type receptor. {ECO:0000269|PubMed:10363963, ECO:0000269|PubMed:10569618, ECO:0000269|PubMed:1562539, ECO:0000269|PubMed:16129672, ECO:0000269|PubMed:17311914, ECO:0000269|PubMed:2260966, ECO:0000269|PubMed:25091737, ECO:0000269|PubMed:8187068, ECO:0000269|PubMed:8274409, ECO:0000269|PubMed:8827083}.; DISEASE: Androgen insensitivity, partial (PAIS) [MIM:312300]: A disorder that is characterized by hypospadias, hypogonadism, gynecomastia, genital ambiguity, normal XY karyotype, and a pedigree pattern consistent with X-linked recessive inheritance. Some patients present azoospermia or severe oligospermia without other clinical manifestations. {ECO:0000269|PubMed:10022458, ECO:0000269|PubMed:10221692, ECO:0000269|PubMed:10470409, ECO:0000269|PubMed:10502786, ECO:0000269|PubMed:10543676, ECO:0000269|PubMed:11587068, ECO:0000269|PubMed:1303262, ECO:0000269|PubMed:1307250, ECO:0000269|PubMed:1316540, ECO:0000269|PubMed:1424203, ECO:0000269|PubMed:1430233, ECO:0000269|PubMed:14756668, ECO:0000269|PubMed:2010552, ECO:0000269|PubMed:7581399, ECO:0000269|PubMed:7649358, ECO:0000269|PubMed:7671849, ECO:0000269|PubMed:7909256, ECO:0000269|PubMed:7910529, ECO:0000269|PubMed:7929841, ECO:0000269|PubMed:7970939, ECO:0000269|PubMed:7981687, ECO:0000269|PubMed:8033918, ECO:0000269|PubMed:8097257, ECO:0000269|PubMed:8126121, ECO:0000269|PubMed:8205256, ECO:0000269|PubMed:8281139, ECO:0000269|PubMed:8325932, ECO:0000269|PubMed:8325950, ECO:0000269|PubMed:8446106, ECO:0000269|PubMed:8550758, ECO:0000269|PubMed:8809734, ECO:0000269|PubMed:8823308, ECO:0000269|PubMed:8824883, ECO:0000269|PubMed:9039340, ECO:0000269|PubMed:9196614, ECO:0000269|PubMed:9302173, ECO:0000269|PubMed:9329414, ECO:0000269|PubMed:9543136, ECO:0000269|PubMed:9607727, ECO:0000269|PubMed:9768671, ECO:0000269|PubMed:9856504, ECO:0000269|Ref.124}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hypospadias 1, X-linked (HYSP1) [MIM:300633]: A common malformation in which the urethra opens on the ventral side of the penis, due to developmental arrest of urethral fusion. The opening can be located glandular, penile, or even more posterior in the scrotum or perineum. Hypospadias is a feature of several syndromic disorders, including the androgen insensitivity syndrome and Opitz syndrome. {ECO:0000269|PubMed:8097257}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07422; DB07039; DB04709; DB07717; DB07454; DB02932; DB08035; DB01481; DB08088; DB08461; DB08087; DB07421; DB01063; DB07423; DB11901; DB01128; DB07286; DB01541; DB14639; DB01564; DB12499; DB04839; DB01406; DB12941; DB09123; DB00255; DB06133; DB01395; DB00858; DB15488; DB11219; DB08899; DB13155; DB00655; DB09086; DB02266; DB01185; DB00623; DB00499; DB11619; DB11064; DB01026; DB15647; DB00367; DB08089; DB05234; DB13934; DB11425; DB06710; DB02998; DB11429; DB00648; DB08804; DB00984; DB00665; DB06713; DB00717; DB09371; DB00957; DB09389; DB00621; DB01428; DB06412; DB01608; DB11447; DB01708; DB00396; DB07419; DB07769; DB14583; DB00421; DB02901; DB13951; DB06718; DB00675; DB00624; DB13943; DB13944; DB01420; DB13946; DB06870; DB08604; DB08867 Interacts with P00519; Q9UBL3; P51451; Q8WV28; O60885-1; P78543; Q14790; P24385; Q92793; O14595; P35222; Q9UER7; P20711; P11308; P07332; P09769; Q02790; P55317; O75593; Q14451; P06396; P56524; Q16665; Q16666; O15357; Q15652; O95251; Q9BY66; Q9BY66-3; Q03164; O14686; P06239; P07948; P20794; P42679; Q00987; Q15596; Q14686; O96028; Q99497; P27986; O00459; Q92569; P19174; P16885; Q06830; P78527; Q06124; P20936; Q9UBS8; Q9Y252; O14796; Q9NP31; P29353; Q6S5L8; Q5VZ18; Q15797; O14544; P12931; Q9ULZ2; P63165; Q9HBL0; P07947; Q9R1E0; Q06986 EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Isopeptide bond; Lipid-binding; Lipoprotein; Metal-binding; Neurodegeneration; Nucleus; Palmitate; Phosphoprotein; Proteomics identification; Pseudohermaphroditism; Receptor; Reference proteome; Steroid-binding; Transcription; Transcription regulation; Triplet repeat expansion; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 29137.9 Length 250 Aromaticity 0.11 Instability index 42.11 Isoelectric point 8.94 Charge (pH=7) 5.43 2D Binding mode Binding energy (Kcal/mol) -9.51 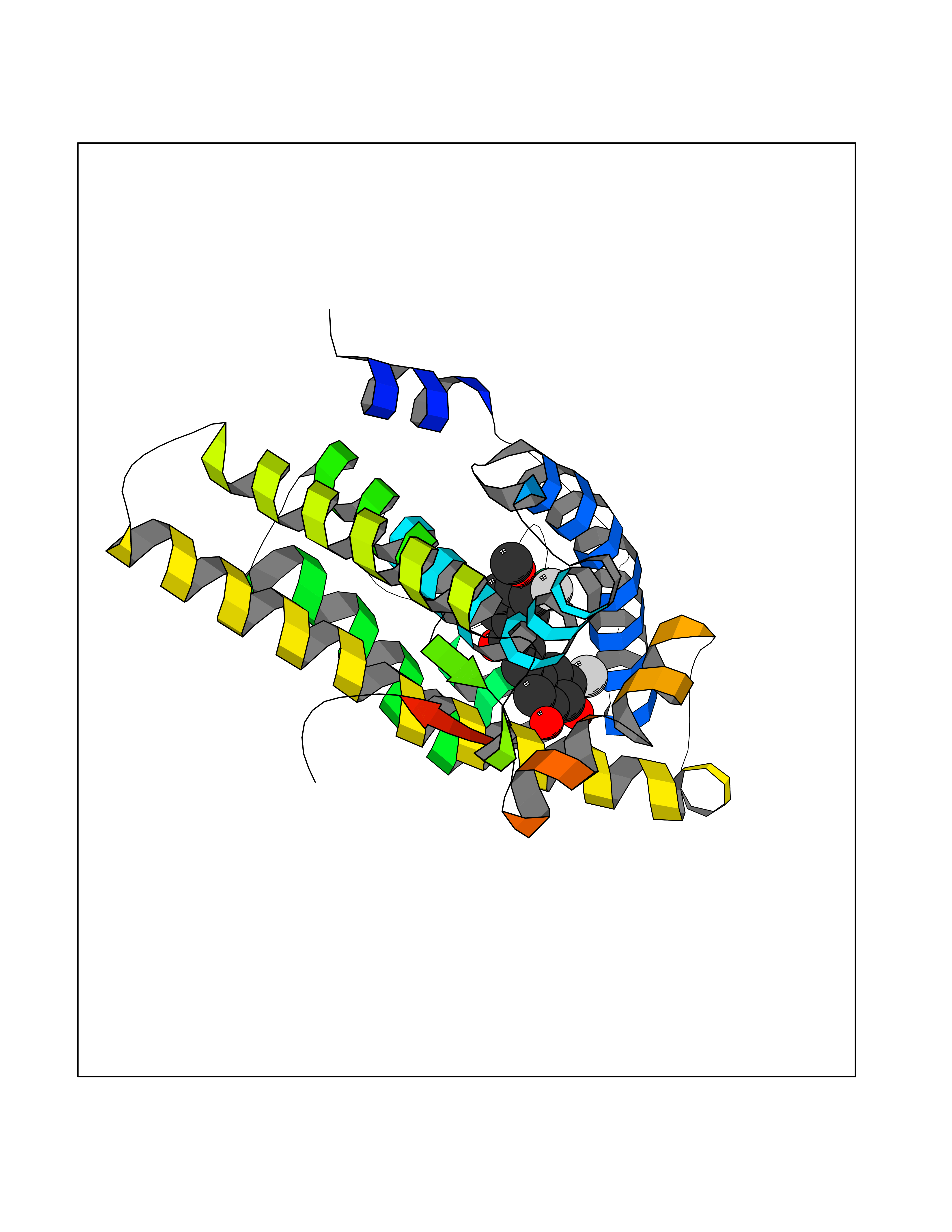 Molscript Map 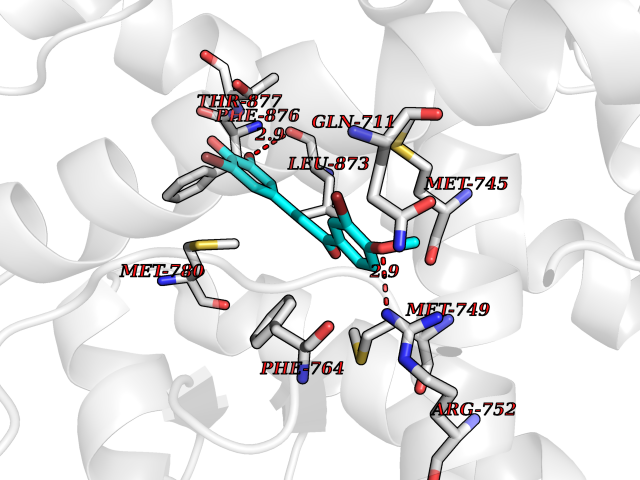 Pymol Map 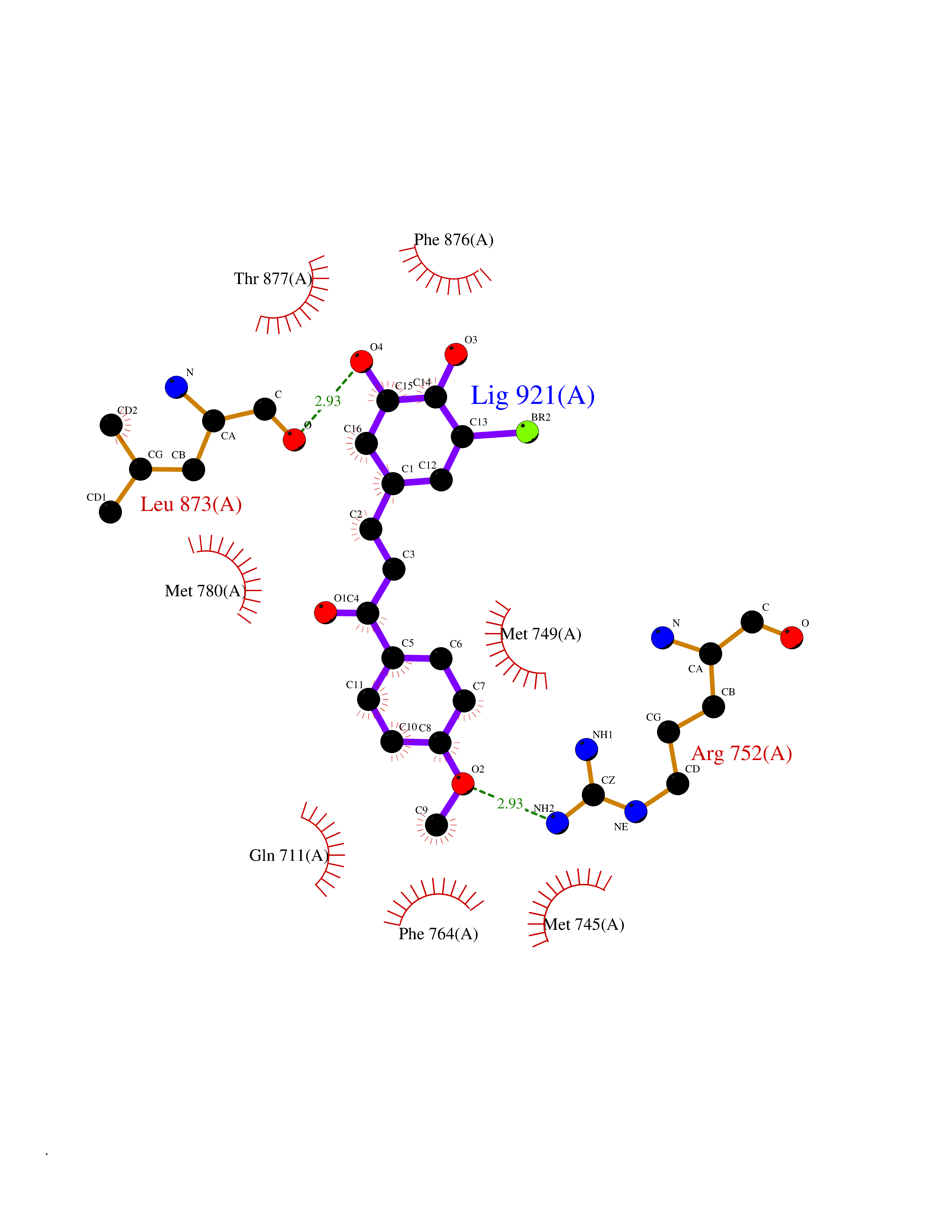 Ligplot Map 3D Binding mode Sequence QPIFLNVLEAIEPGVVCAGHDNNQPDSFAALLSSLNELGERQLVHVVKWAKALPGFRNLHVDDQMAVIQYSWMGLMVFAMGWRSFTNVNSRMLYFAPDLVFNEYRMHKSRMYSQCVRMRHLSQEFGWLQITPQEFLCMKALLLFSIIPVDGLKNQKFFDELRMNYIKELDRIIACKRKNPTSCSRRFYQLTKLLDSVQPIARELHQFTFDLLIKSHMVSVDFPEMMAEIISVQVPKILSGKVKPIYFHTQ Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Plasmodium DOXP reductoisomerase (Malaria DXR) | 3AU9 | 6.97 | |
Target general information Gen name Malaria DXR Organism Plasmodium falciparum (isolate HB3) Uniprot ID TTD ID Synonyms IspC; DXR; DXP reductoisomerase; DOXP reductoisomerase; 2-C-Methyl-d-erythritol 4-phosphate synthase; 1-deoxyxylulose-5-phosphate reductoisomerase Protein family DXR family Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the NADP-dependent rearrangement and reduction of 1-deoxy-D-xylulose-5-phosphate (DXP) to 2-C-methyl-D-erythritol 4-phosphate (MEP). Related diseases Ichthyosis, congenital, autosomal recessive 11 (ARCI11) [MIM:602400]: A form of autosomal recessive congenital ichthyosis, a disorder of keratinization with abnormal differentiation and desquamation of the epidermis, resulting in abnormal skin scaling over the whole body. The main skin phenotypes are lamellar ichthyosis (LI) and non-bullous congenital ichthyosiform erythroderma (NCIE), although phenotypic overlap within the same patient or among patients from the same family can occur. Lamellar ichthyosis is a condition often associated with an embedment in a collodion-like membrane at birth; skin scales later develop, covering the entire body surface. Non-bullous congenital ichthyosiform erythroderma characterized by fine whitish scaling on an erythrodermal background; larger brownish scales are present on the buttocks, neck and legs. {ECO:0000269|PubMed:17273967, ECO:0000269|PubMed:18843291}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.1.1.267 Uniprot keywords 3D-structure; Apicoplast; Isoprene biosynthesis; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Plastid; Transit peptide Protein physicochemical properties Chain ID A Molecular weight (Da) 46644.4 Length 410 Aromaticity 0.09 Instability index 36.77 Isoelectric point 6.95 Charge (pH=7) -0.14 2D Binding mode Binding energy (Kcal/mol) -9.51 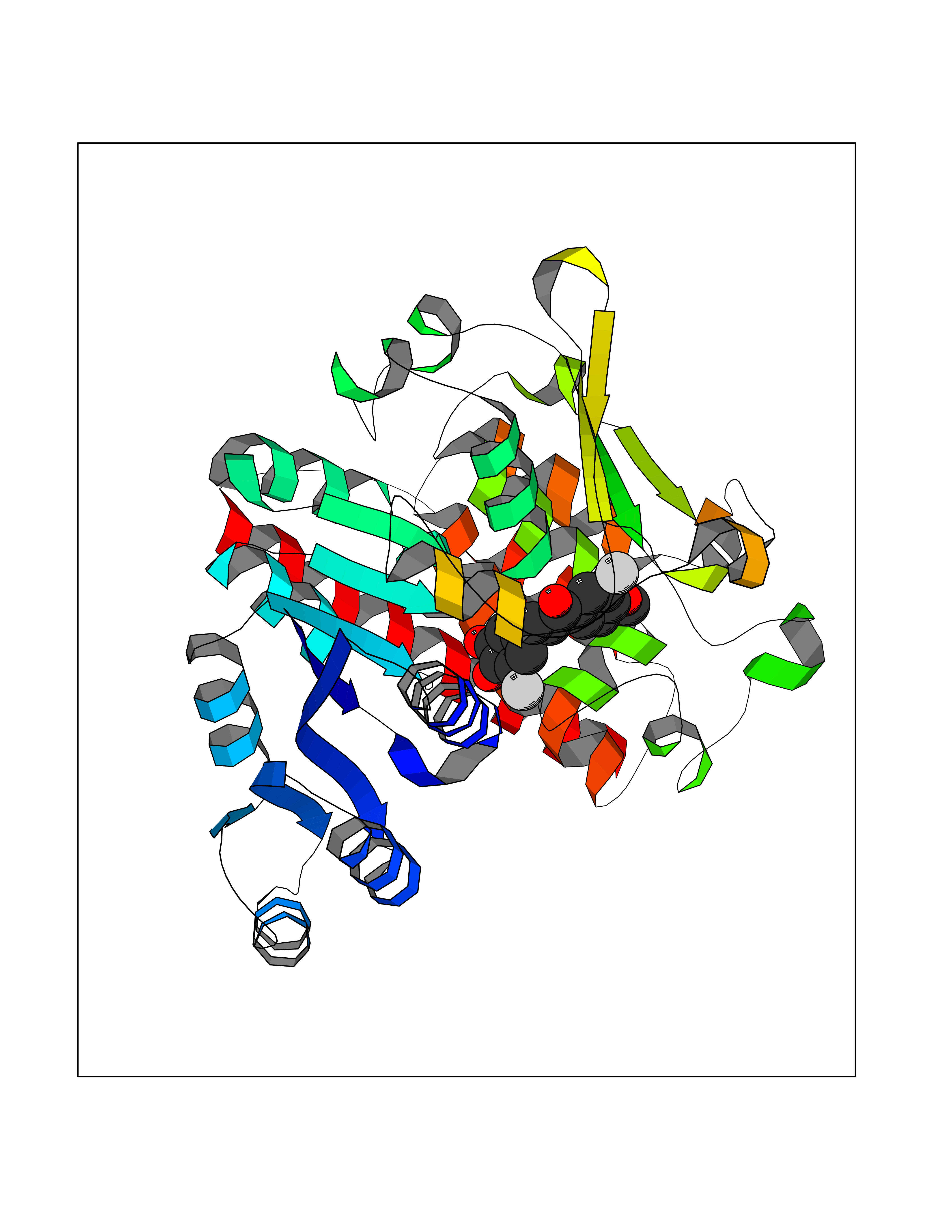 Molscript Map 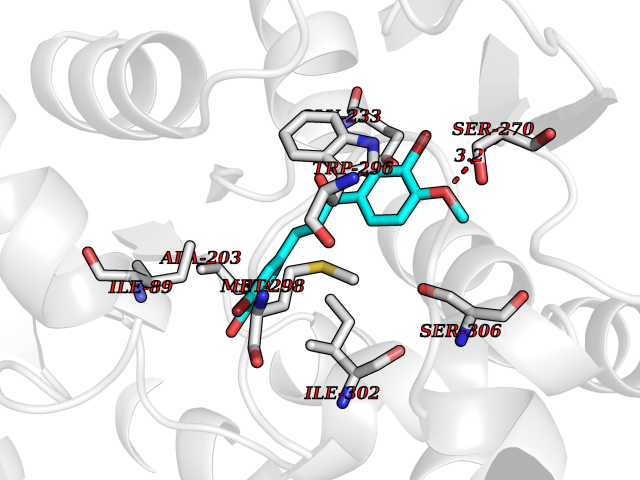 Pymol Map 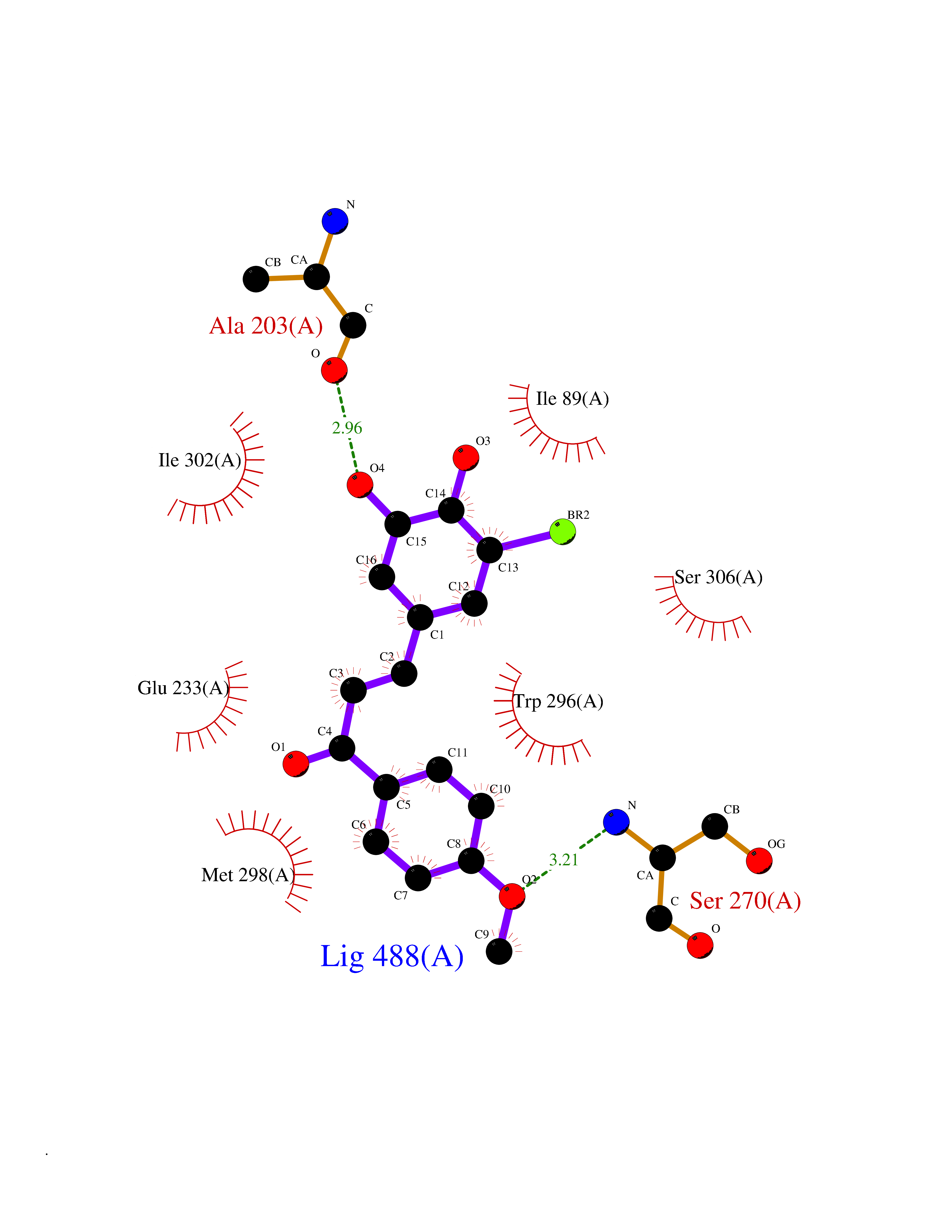 Ligplot Map 3D Binding mode Sequence PINVAIFGSTGSIGTNALNIIRECNKIENVFNVKALYVNKSVNELYEQAREFLPEYLCIHDKSVYEELKELVKNIKDYKPIILCGDEGMKEICSSNSIDKIVIGIDSFQGLYSTMYAIMNNKIVALANKESIVSAGFFLKKLLNIHKNAKIIPVDSEHSAIFQCLDNNKVLKTKCLQDNFSKINNINKIFLCSSGGPFQNLTMDELKNVTSENALKHPKWKMGKKITIDSATMMNKGLEVIETHFLFDVDYNDIEVIVHKECIIHSCVEFIDKSVISQMYYPDMQIPILYSLTWPDRIKTNLKPLDLAQVSTLTFHKPSLEHFPCIKLAYQAGIKGNFYPTVLNASNEIANNLFLNNKIKYFDISSIISQVLESFNSQKVSENSEDLMKQILQIHSWAKDKATDIYNKHN Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | N-acylethanolamine-hydrolyzing acidamidase (NAAA) | 6DXX | 6.97 | |
Target general information Gen name NAAA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nacylsphingosine amidohydrolaselike; Nacylethanolaminehydrolyzing acid amidase subunit beta; NAAA; Acid ceramidaselike protein; ASAHlike protein Protein family Acid ceramidase family Biochemical class Carbon-nitrogen hydrolase Function Degrades bioactive fatty acid amides to their corresponding acids, with the following preference: N- palmitoylethanolamine > N-myristoylethanolamine > N- lauroylethanolamine = N-stearoylethanolamine > N- arachidonoylethanolamine > N-oleoylethanolamine. Also exhibits weak hydrolytic activity against the ceramides N- lauroylsphingosine and N-palmitoylsphingosine. Related diseases Hypertriglyceridemia, transient infantile (HTGTI) [MIM:614480]: An autosomal recessive disorder characterized by onset of moderate to severe transient hypertriglyceridemia in infancy that normalizes with age. The hypertriglyceridemia is associated with hepatomegaly, moderately elevated transaminases, persistent fatty liver, and the development of hepatic fibrosis. {ECO:0000269|PubMed:22226083, ECO:0000269|PubMed:24549054}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09061; DB14009; DB14011 Interacts with NA EC number EC 3.5.1.- Uniprot keywords 3D-structure; Alternative splicing; Autocatalytic cleavage; Direct protein sequencing; Disulfide bond; Fatty acid metabolism; Glycoprotein; Hydrolase; Lipid degradation; Lipid metabolism; Lysosome; Membrane; Proteomics identification; Reference proteome; Signal; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 36877.8 Length 328 Aromaticity 0.11 Instability index 44.37 Isoelectric point 7.72 Charge (pH=7) 1.08 2D Binding mode Binding energy (Kcal/mol) -9.51  Molscript Map 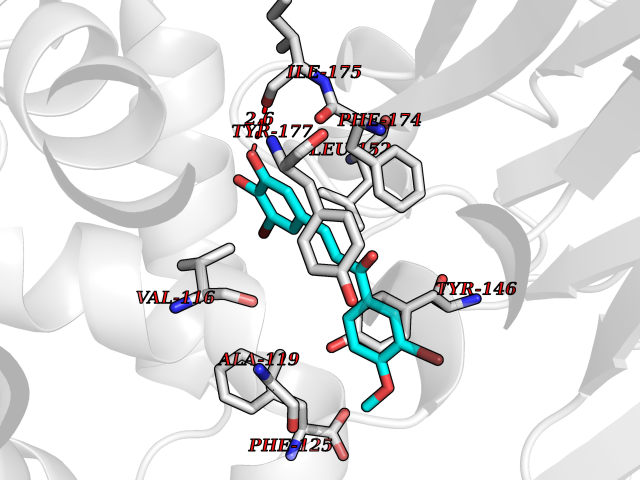 Pymol Map 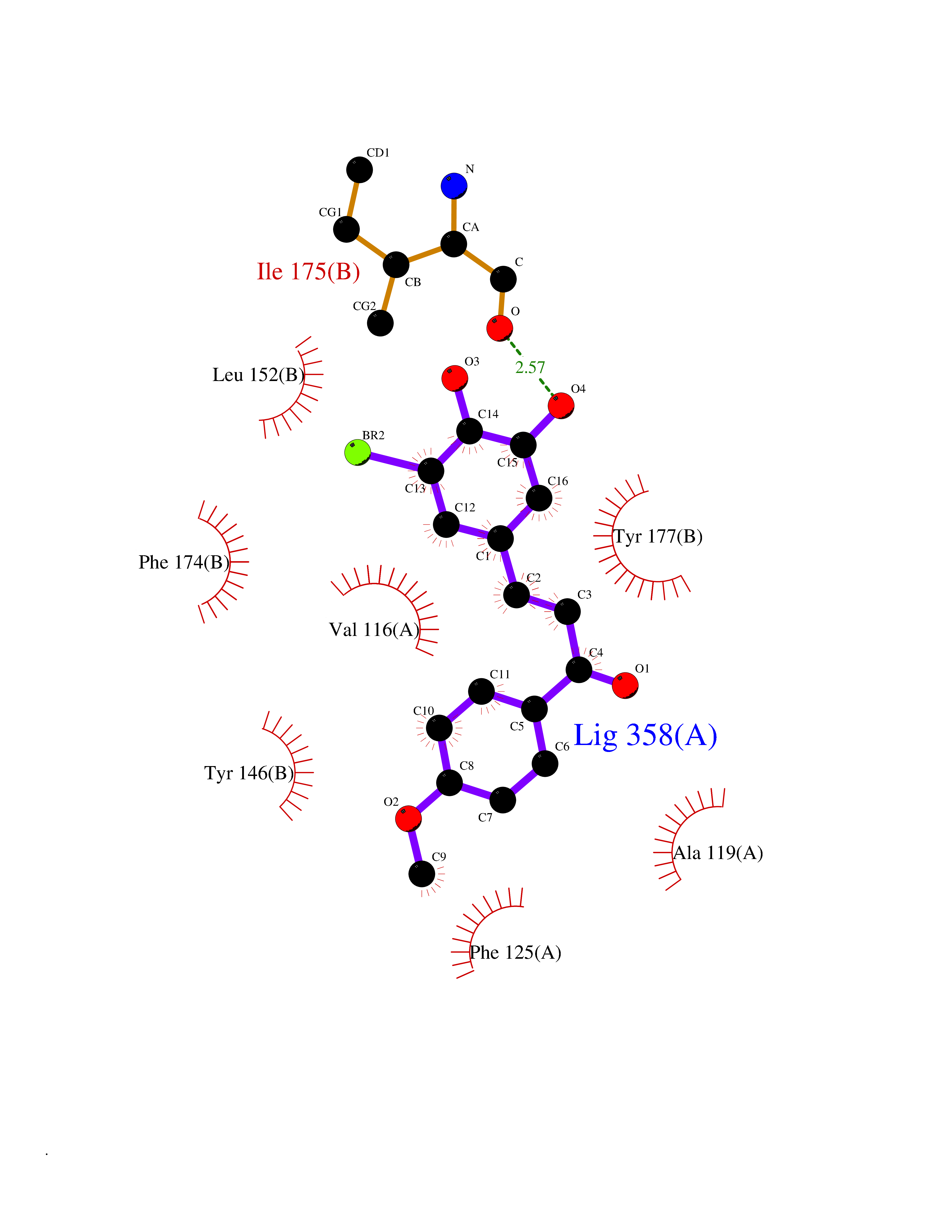 Ligplot Map 3D Binding mode Sequence SPPAAPRFNVSLDSVPELRWLPVLRHYDLDLVRAAMAQVIGDRVPKWVHVLIGKVVLELERFLPQPFTGEIRGMCDFMNLSLADCLLVNLAYESSVFCTSIVAQDSRGHIYHGRNLDYPFGNVLRKLTVDVQFLKNGQIAFTGTTFIGYVGLWTGQSPHKFTVSGDERDKGWWWENAIAALFRRHIPVSWLIRATLSESENFEAAVGKLAKTPLIADVYYIVGGTSPREGVVITRNRDGPADIWPLDPLNGAWFRVETNYDHWKPAPKEDDRRTSAIKALNATGQANLSLEALFQILSVVPVYNNFTIYTTVMSAGSPDKYMTRIRNP Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Purine nucleoside phosphorylase (PNP) | 4EAR | 6.96 | |
Target general information Gen name PNP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNP; Inosine phosphorylase Protein family PNP/MTAP phosphorylase family Biochemical class Pentosyltransferase Function The purine nucleoside phosphorylases catalyze the phosphorolytic breakdown of the N-glycosidic bond in the beta- (deoxy)ribonucleoside molecules, with the formation of the corresponding free purine bases and pentose-1-phosphate. Related diseases Purine nucleoside phosphorylase deficiency (PNPD) [MIM:613179]: A disorder that interrupts both the catabolism of inosine into hypoxanthine and guanosine into guanine, and leads to the accumulation of guanosine, inosine, and their deoxified by-products. The main clinical presentation is recurrent infections due to severe T-cell immunodeficiency. Some patients also have neurologic impairment. {ECO:0000269|PubMed:1384322, ECO:0000269|PubMed:3029074, ECO:0000269|PubMed:8931706}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03881; DB03551; DB02222; DB02391; DB03609; DB01667; DB04260; DB02796; DB04753; DB00640; DB00242; DB00900; DB06185; DB02377; DB02857; DB04754; DB04757; DB04076; DB02230; DB04335; DB02568; DB03101 Interacts with P05067; Q9UQM7; O14576-2; P06241; P14136; Q92993-2; Q9BXM7; P00491; P17612; P63000; Q92673; Q15583 EC number EC 2.4.2.1 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Disease variant; Glycosyltransferase; Phosphoprotein; Proteomics identification; Purine salvage; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 31849.2 Length 288 Aromaticity 0.1 Instability index 34.77 Isoelectric point 6.42 Charge (pH=7) -1.63 2D Binding mode Binding energy (Kcal/mol) -9.49 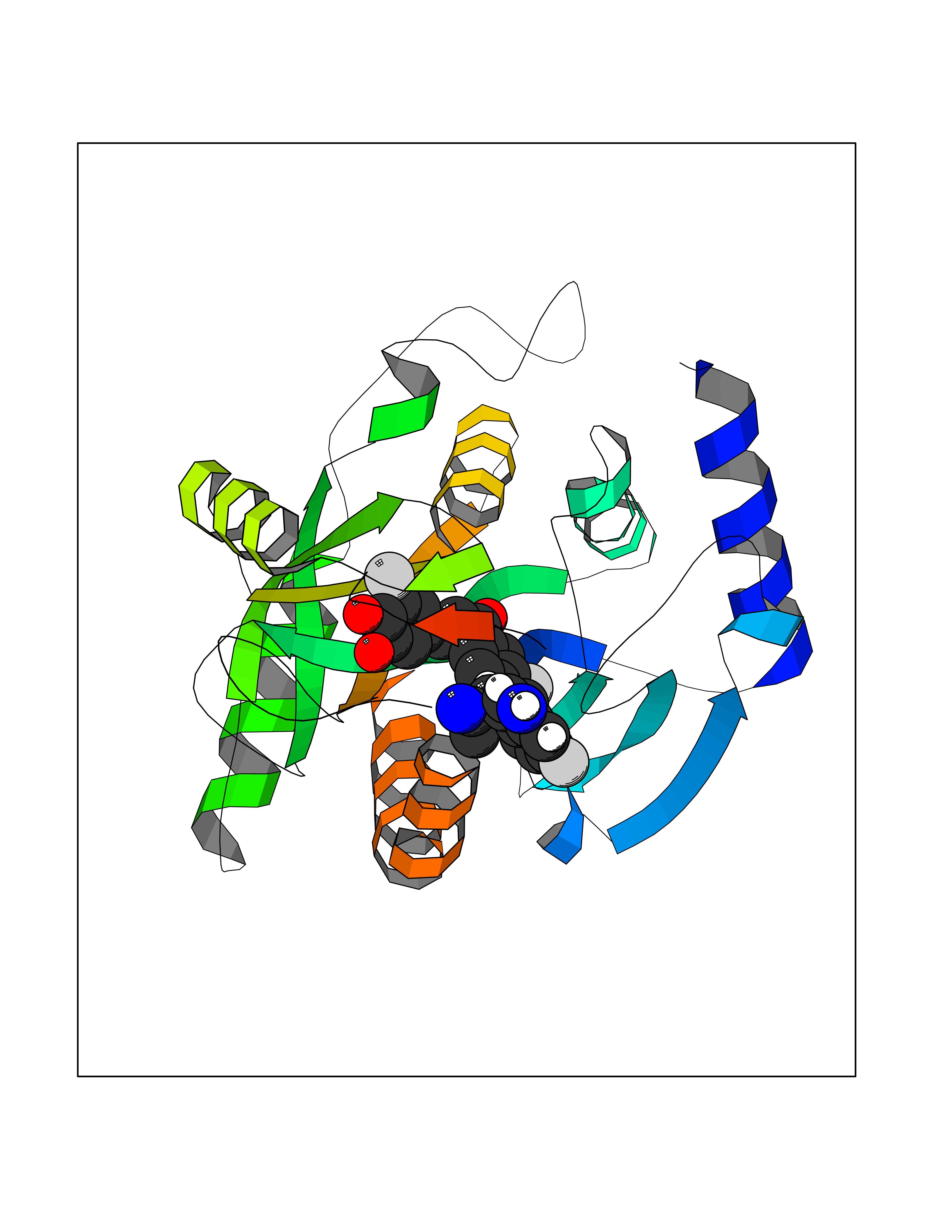 Molscript Map 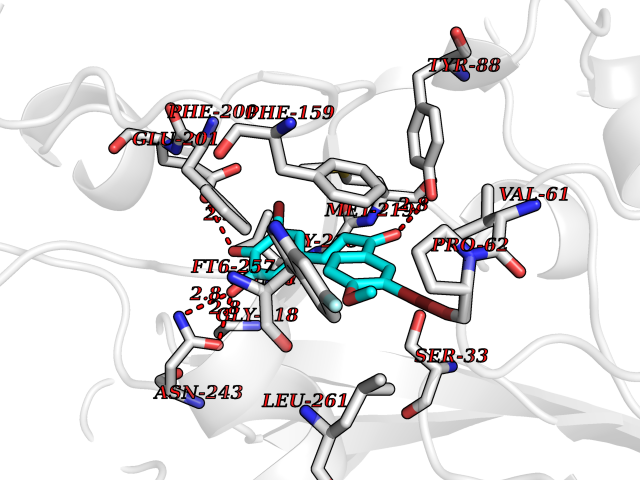 Pymol Map 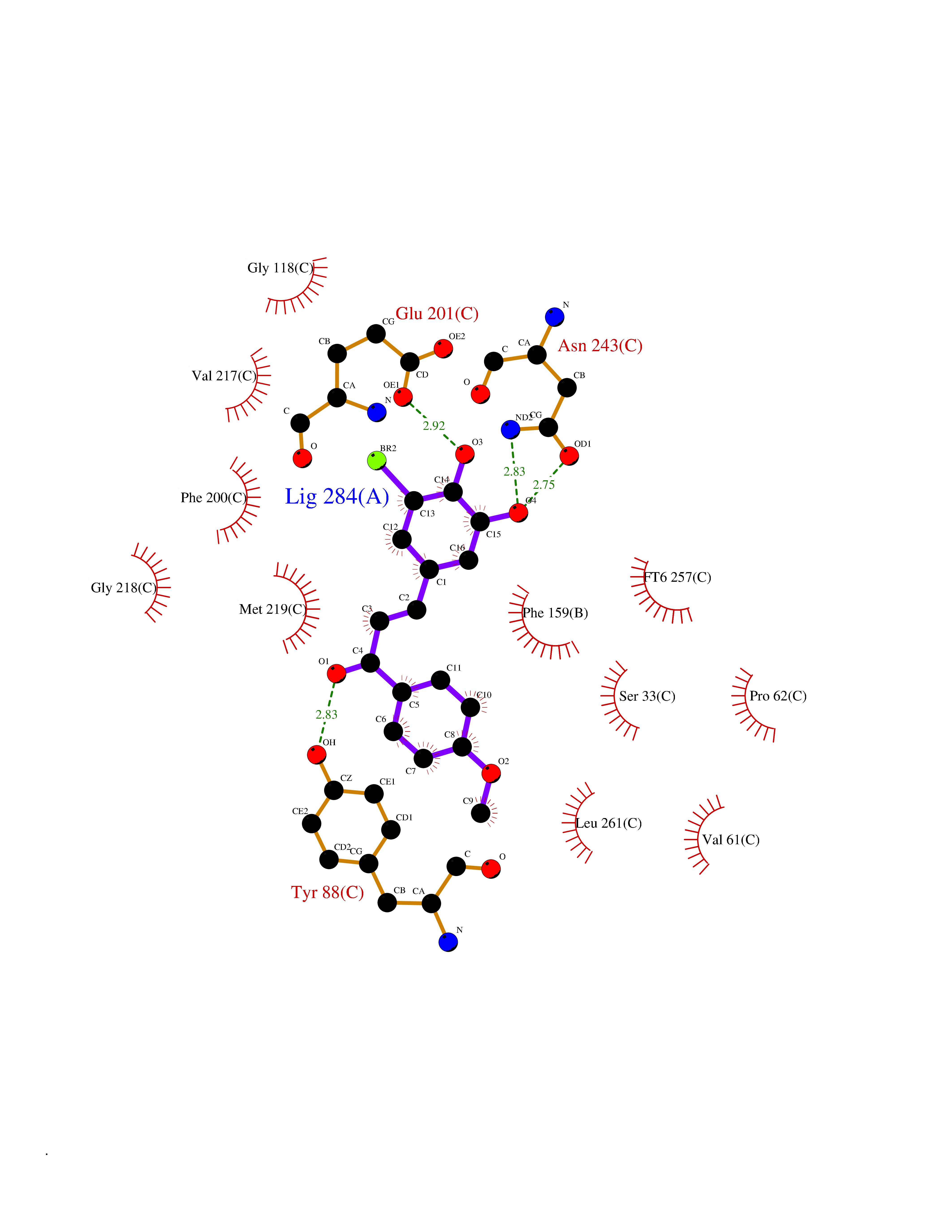 Ligplot Map 3D Binding mode Sequence GYTYEDYKNTAEYLLSHTKHRPQVAIICGSGLGGLTDKLTQAQIFDYSEIPNFPRSTVPGHAGRLVFGFLNGRACVMMQGRFHMYEGYPLYKVTFPVRVFHLLGVDTLVVTNAAGGLNPKFEVGDIMLIRDHINLPGFSGQNPLRGPNDERFGDRFPAMSDAYDRTMRQRALSTYKQMGEQRELQEGTYVMVAGPSFETVAECRVLQKLGADAVGMSTVPEVIVARHCGLRVFGFSLITNKVIMDYESLEKANXEEVLAAGKQAAQKLEQFVSILMASIDRFPAMSDA Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Glycine oxidase | 1RYI | 6.96 | |
Target general information Gen name thiO Organism Bacillus subtilis (strain 168) Uniprot ID TTD ID NA Synonyms goxB;yjbR;BSU11670 Protein family DAO family, ThiO subfamily Biochemical class Oxidoreductase Function FAD binding.Glycine oxidase activity.Oxidoreductase activity. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB02713; DB03147 Interacts with NA EC number 1.4.3.19 Uniprot keywords 3D-structure; Cytoplasm; FAD; Flavoprotein; Herbicide resistance; Oxidoreductase; Reference proteome; Thiamine biosynthesis Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 40395.8 Length 364 Aromaticity 0.1 Instability index 36.07 Isoelectric point 6.02 Charge (pH=7) -6.35 2D Binding mode Binding energy (Kcal/mol) -9.5  Molscript Map 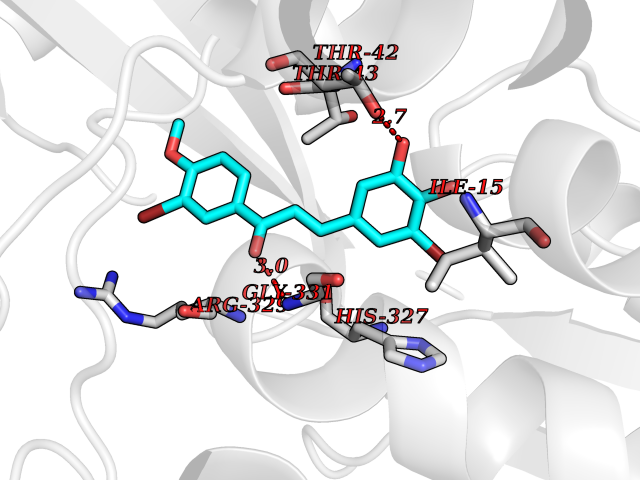 Pymol Map 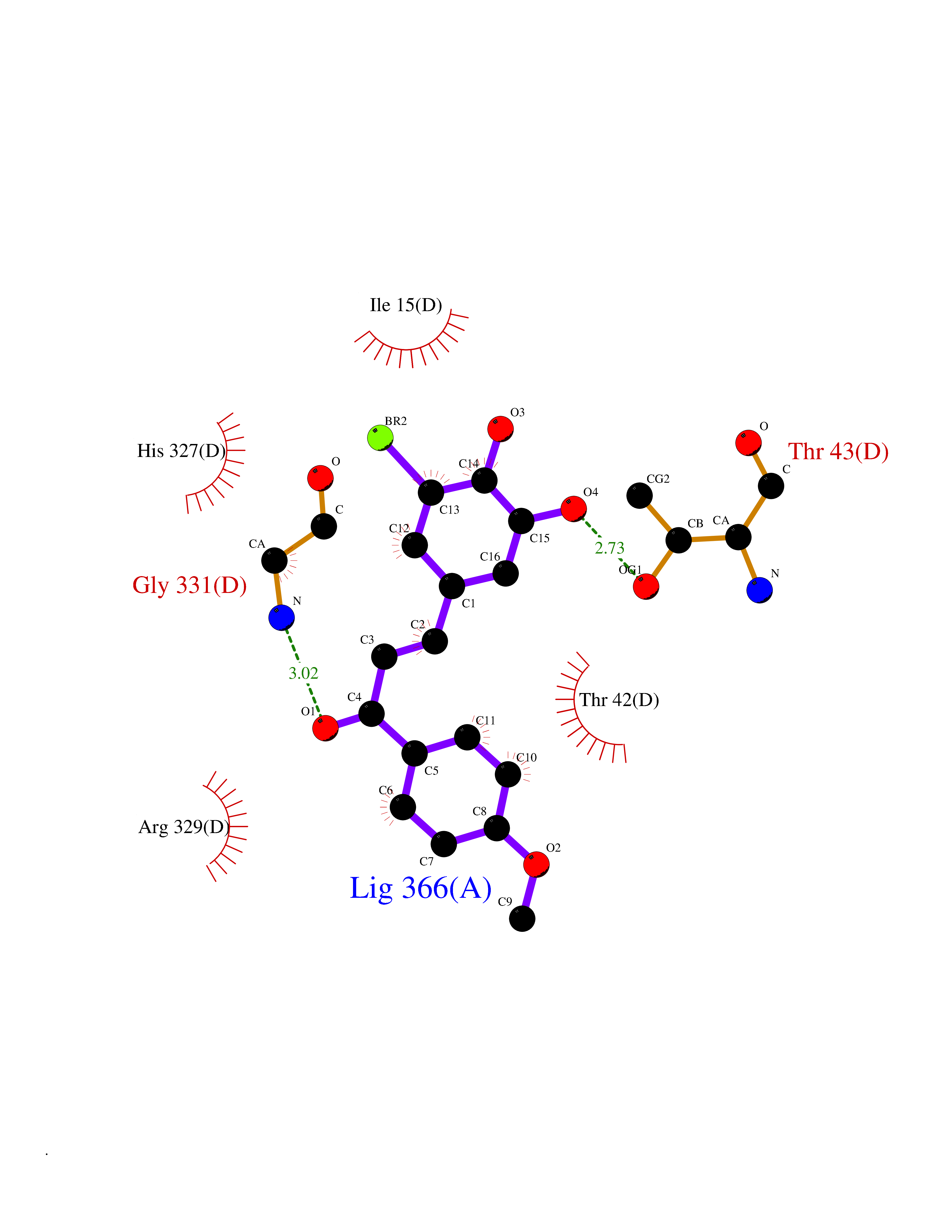 Ligplot Map 3D Binding mode Sequence MKRHYEAVVIGGGIIGSAIAYYLAKENKNTALFESGTMGGRTTSAAAGMLGAHAECEERDAFFDFAMHSQRLYKGLGEELYALSGVDIRQHNGGMFKLAFSEEDVLQLRQMDDLDSVSWYSKEEVLEKEPYASGDIFGASFIQDDVHVEPYFVCKAYVKAAKMLGAEIFEHTPVLHVERDGEALFIKTPSGDVWANHVVVASGVWSGMFFKQLGLNNAFLPVKGECLSVWNDDIPLTKTLYHDHCYIVPRKSGRLVVGATMKPGDWSETPDLGGLESVMKKAKTMLPAIQNMKVDRFWAGLRPGTKDGKPYIGRHPEDSRILFAAGHFRNGILLAPATGALISDLIMNKEVNQDWLHAFRIDRK Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Janus kinase 2 (JAK-2) | 3UGC | 6.96 | |
Target general information Gen name JAK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine-protein kinase JAK2 Protein family Protein kinase superfamily, Tyr protein kinase family, JAK subfamily Biochemical class Kinase Function Mediates essential signaling events in both innate and adaptive immunity. In the cytoplasm, plays a pivotal role in signal transduction via its association with type I receptors such as growth hormone (GHR), prolactin (PRLR), leptin (LEPR), erythropoietin (EPOR), thrombopoietin (THPO); or type II receptors including IFN-alpha, IFN-beta, IFN-gamma and multiple interleukins. Following ligand-binding to cell surface receptors, phosphorylates specific tyrosine residues on the cytoplasmic tails of the receptor, creating docking sites for STATs proteins. Subsequently, phosphorylates the STATs proteins once they are recruited to the receptor. Phosphorylated STATs then form homodimer or heterodimers and translocate to the nucleus to activate gene transcription. For example, cell stimulation with erythropoietin (EPO) during erythropoiesis leads to JAK2 autophosphorylation, activation, and its association with erythropoietin receptor (EPOR) that becomes phosphorylated in its cytoplasmic domain. Then, STAT5 (STAT5A or STAT5B) is recruited, phosphorylated and activated by JAK2. Once activated, dimerized STAT5 translocates into the nucleus and promotes the transcription of several essential genes involved in the modulation of erythropoiesis. Part of a signaling cascade that is activated by increased cellular retinol and that leads to the activation of STAT5 (STAT5A or STAT5B). In addition, JAK2 mediates angiotensin-2-induced ARHGEF1 phosphorylation. Plays a role in cell cycle by phosphorylating CDKN1B. Cooperates with TEC through reciprocal phosphorylation to mediate cytokine-driven activation of FOS transcription. In the nucleus, plays a key role in chromatin by specifically mediating phosphorylation of 'Tyr-41' of histone H3 (H3Y41ph), a specific tag that promotes exclusion of CBX5 (HP1 alpha) from chromatin. Non-receptor tyrosine kinase involved in various processes such as cell growth, development, differentiation or histone modifications. Related diseases Chromosomal aberrations involving JAK2 are found in both chronic and acute forms of eosinophilic, lymphoblastic and myeloid leukemia. Translocation t(8;9)(p22;p24) with PCM1 links the protein kinase domain of JAK2 to the major portion of PCM1. Translocation t(9;12)(p24;p13) with ETV6.; DISEASE: Budd-Chiari syndrome (BDCHS) [MIM:600880]: A syndrome caused by obstruction of hepatic venous outflow involving either the hepatic veins or the terminal segment of the inferior vena cava. Obstructions are generally caused by thrombosis and lead to hepatic congestion and ischemic necrosis. Clinical manifestations observed in the majority of patients include hepatomegaly, right upper quadrant pain and abdominal ascites. Budd-Chiari syndrome is associated with a combination of disease states including primary myeloproliferative syndromes and thrombophilia due to factor V Leiden, protein C deficiency and antithrombin III deficiency. Budd-Chiari syndrome is a rare but typical complication in patients with polycythemia vera. {ECO:0000269|PubMed:16707754}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Polycythemia vera (PV) [MIM:263300]: A myeloproliferative disorder characterized by abnormal proliferation of all hematopoietic bone marrow elements, erythroid hyperplasia, an absolute increase in total blood volume, but also by myeloid leukocytosis, thrombocytosis and splenomegaly. {ECO:0000269|PubMed:15781101, ECO:0000269|PubMed:15793561, ECO:0000269|PubMed:15858187, ECO:0000269|PubMed:16603627, ECO:0000269|PubMed:25644777}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thrombocythemia 3 (THCYT3) [MIM:614521]: A myeloproliferative disorder characterized by excessive platelet production, resulting in increased numbers of circulating platelets. It can be associated with spontaneous hemorrhages and thrombotic episodes. {ECO:0000269|PubMed:16325696, ECO:0000269|PubMed:22397670}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Myelofibrosis (MYELOF) [MIM:254450]: A disorder characterized by replacement of the bone marrow by fibrous tissue, occurring in association with a myeloproliferative disorder. Clinical manifestations may include anemia, pallor, splenomegaly, hypermetabolic state, petechiae, ecchymosis, bleeding, lymphadenopathy, hepatomegaly, portal hypertension. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:16247455}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04716; DB07162; DB08067; DB07161; DB14973; DB11817; DB11986; DB12500; DB12010; DB11763; DB11697; DB15822; DB08877; DB08895; DB05243; DB15035 Interacts with P32927; Q01344; P23458; O60674; P40238; P16333; P18031; O75116; P29597; Q9JHI9 EC number EC 2.7.10.2 Uniprot keywords 3D-structure; Adaptive immunity; ATP-binding; Chromatin regulator; Chromosomal rearrangement; Cytoplasm; Disease variant; Immunity; Innate immunity; Kinase; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Reference proteome; Repeat; SH2 domain; Transferase; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32174.5 Length 274 Aromaticity 0.1 Instability index 50.94 Isoelectric point 7.78 Charge (pH=7) 1.46 2D Binding mode Binding energy (Kcal/mol) -9.49  Molscript Map 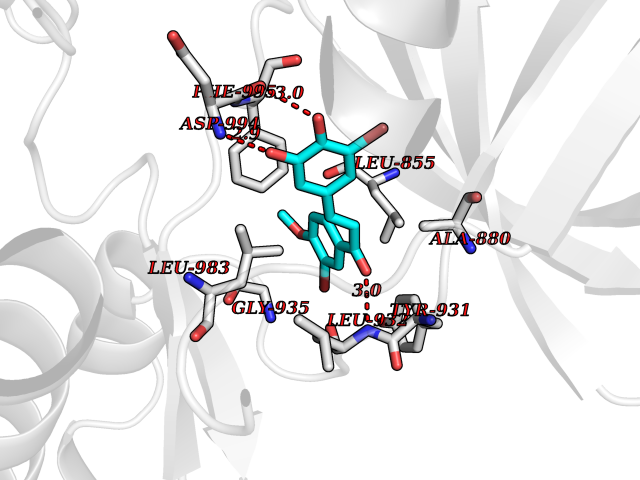 Pymol Map 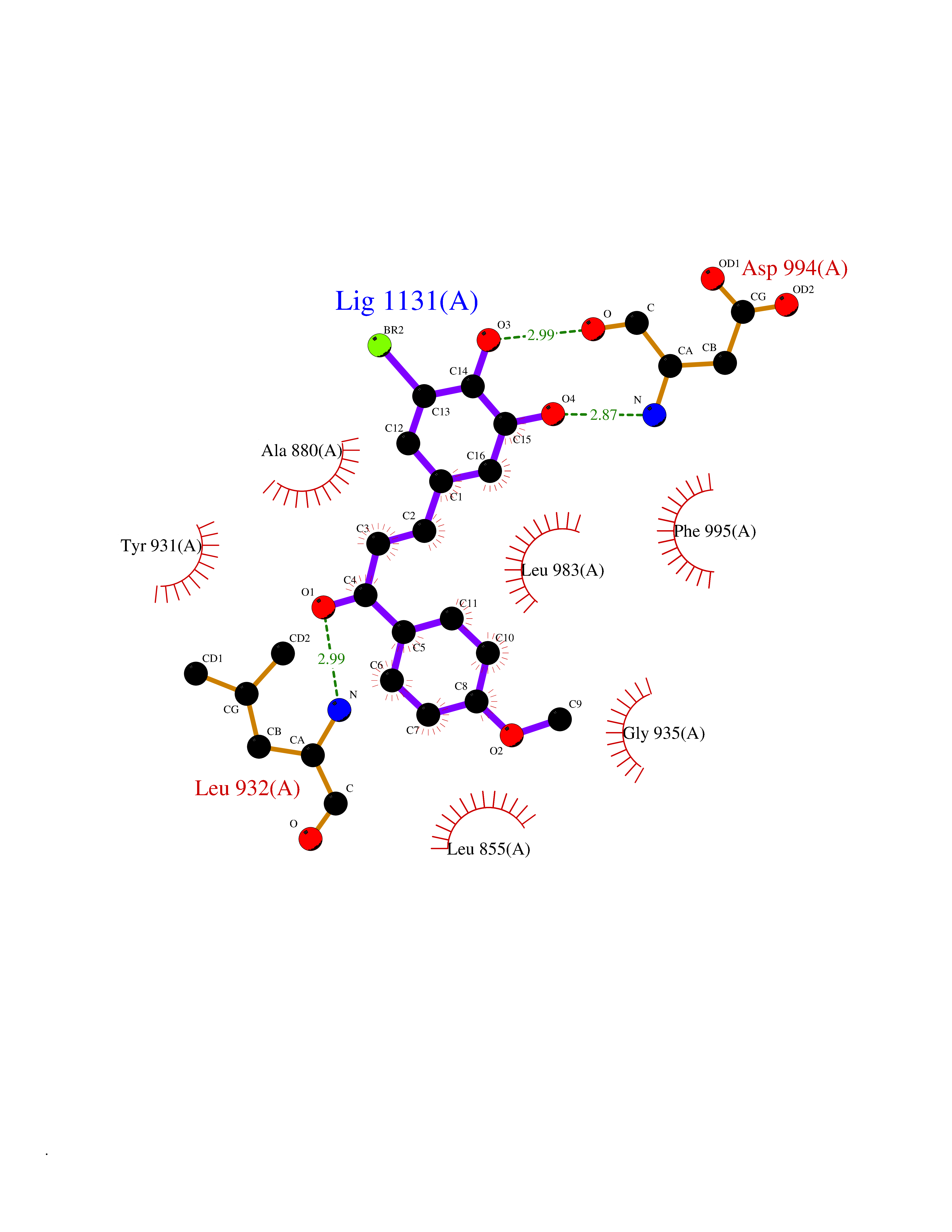 Ligplot Map 3D Binding mode Sequence QFEERHLKFLQQLGKGNFGSVEMCRYDPLQDNTGEVVAVKKLQHSTEEHLRDFEREIEILKSLQHDNIVKYKGVCYSAGRRNLKLIMEYLPYGSLRDYLQKHKERIDHIKLLQYTSQICKGMEYLGTKRYIHRDLATRNILVENENRVKIGDFGLTKPGESPIFWYAPESLTESKFSVASDVWSFGVVLYELFTYIEKSKSPPAEFMRMIGNDKQGQMIVFHLIELLKNNGRLPRPDGCPDEIYMIMTECWNNNVNQRPSFRDLALRVDQIRDN Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Xanthine dehydrogenase/oxidase (XDH) | 2E1Q | 6.96 | |
Target general information Gen name XDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Xanthine oxidase; Xanthine dehydrogenase; XDHA Protein family Xanthine dehydrogenase family Biochemical class CH/CH(2) oxidoreductase Function Catalyzes the oxidation of hypoxanthine to xanthine. Catalyzes the oxidation of xanthine to uric acid. Contributes to the generation of reactive oxygen species. Has also low oxidase activity towards aldehydes (in vitro). Key enzyme in purine degradation. Related diseases Xanthinuria 1 (XAN1) [MIM:278300]: A disorder characterized by excretion of very large amounts of xanthine in the urine and a tendency to form xanthine stones. Uric acid is strikingly diminished in serum and urine. XAN1 is due to isolated xanthine dehydrogenase deficiency. Patients can metabolize allopurinol. {ECO:0000269|PubMed:10844591, ECO:0000269|PubMed:11379872, ECO:0000269|PubMed:14551354, ECO:0000269|PubMed:9153281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00640; DB00041; DB00437; DB00993; DB00958; DB01136; DB00856; DB00515; DB00746; DB03328; DB00997; DB03516; DB12466; DB04854; DB03147; DB04335; DB01020; DB00583; DB00170; DB01033; DB00157; DB03841; DB00336; DB01250; DB05262; DB06478; DB01168; DB00339; DB00127; DB01685; DB00831 Interacts with Q9Y3R0-3 EC number NA Uniprot keywords 2Fe-2S; 3D-structure; Cytoplasm; Disease variant; Disulfide bond; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase; Peroxisome; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 143697 Length 1307 Aromaticity 0.08 Instability index 37.9 Isoelectric point 8.01 Charge (pH=7) 7.07 2D Binding mode Binding energy (Kcal/mol) -9.49  Molscript Map 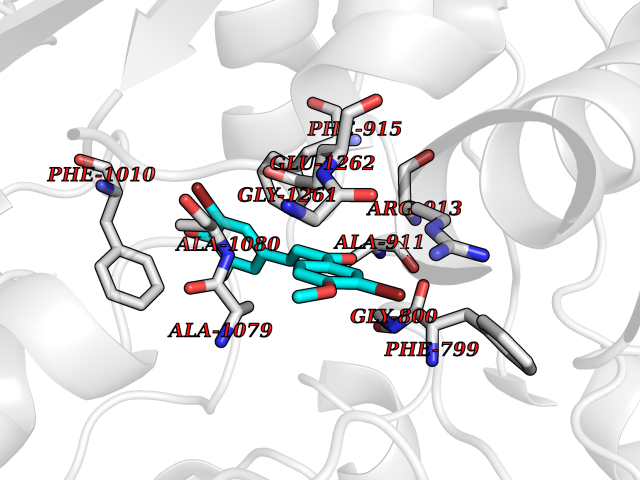 Pymol Map 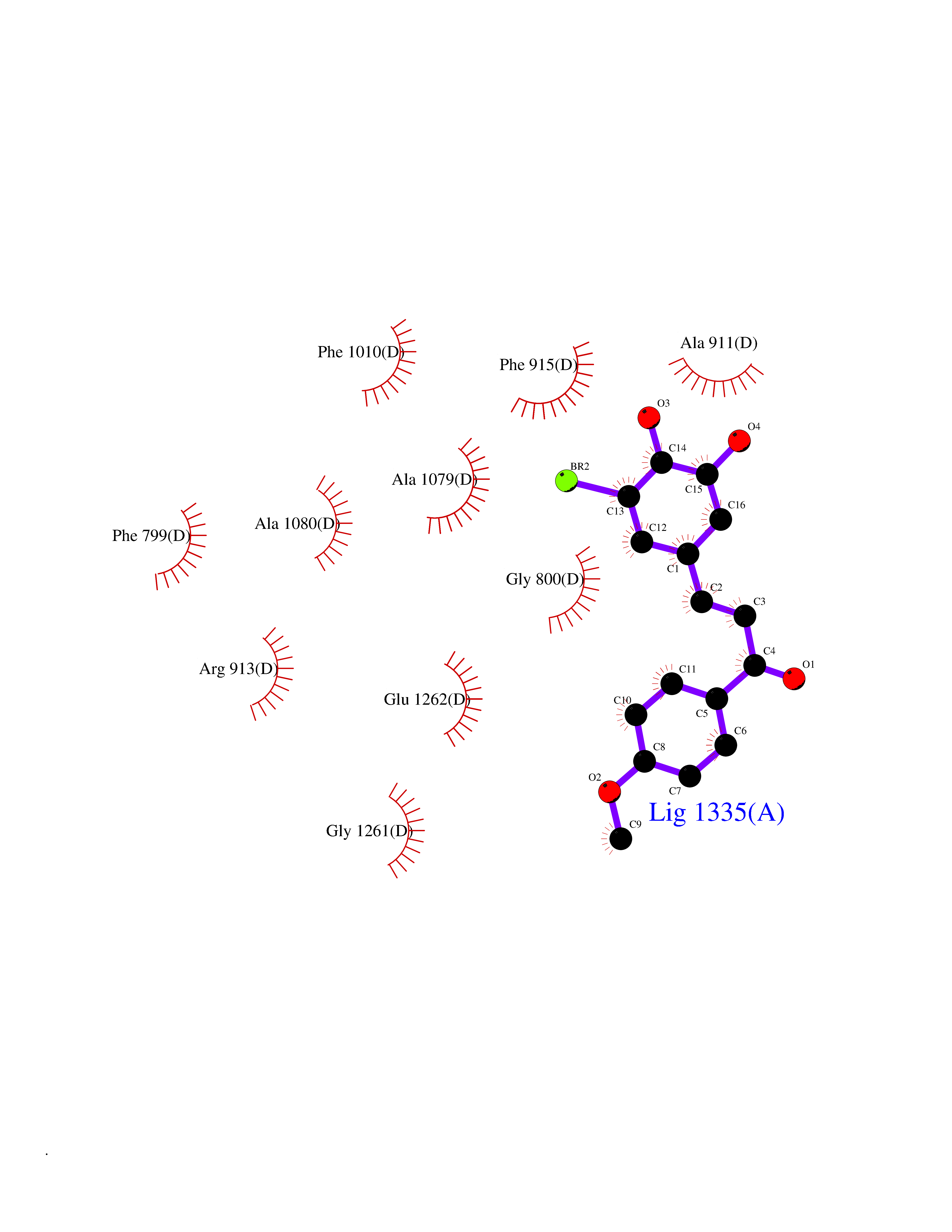 Ligplot Map 3D Binding mode Sequence ADKLVFFVNGRKVVEKNADPETTLLAYLRRKLGLSGTKLGCGEGGCGACTVMLSKYDRLQNKIVHFSANACLAPICSLHHVAVTTVEGIGSTKTRLHPVQERIAKSHGSQCGFCTPGIVMSMYTLLRNQPEPTMEEIENAFQGNLCRCTGYRPILQGFRTFARDGSPSLFKPEEFTPLDPTQEPIFPPELLRLKDTPRKQLRFEGERVTWIQASTLKELLDLKAQHPDAKLVVGNTEIGIEMKFKNMLFPMIVCPAWIPELNSVEHGPDGISFGAACPLSIVEKTLVDAVAKLPAQKTEVFRGVLEQLRWFAGKQVKSVASVGGNIITASPISDLNPVFMASGAKLTLVSRGTRRTVQMDHTFFPGYRKTLLSPEEILLSIEIPYSREGEYFSAFKQASRREDDIAKVTSGMRVLFKPGTTEVQELALCYGGMANRTISALKTTQRQLSKLWKEELLQDVCAGLAEELHLPPDAPGGMVDFRCTLTLSFFFKFYLTVLQKLGQENLEDKCGKLDPTFASATLLFQKDPPADVQLFQEVPKGQSEEDMVGRPLPHLAADMQASGEAVYCDDIPRYENELSLRLVTSTRAHAKIKSIDTSEAKKVPGFVCFISADDVPGSNITGICNDETVFAKDKVTCVGHIIGAVVADTPEHTQRAAQGVKITYEELPAIITIEDAIKNNSFYGPELKIEKGDLKKGFSEADNVVSGEIYIGGQEHFYLETHCTIAVPKGEAGEMELFVSTQNTMKTQSFVAKMLGVPANRIVVRVKRMGGGFGGKVTRSTVVSTAVALAAYKTGRPVRCMLDRDEDMLITGGRHPFLARYKVGFMKTGTVVALEVDHFSNVGNTQDLSQSIMERALFHMDNCYKIPNIRGTGRLCKTNLPSNTAFRGFGGPQGMLIAECWMSEVAVTCGMPAEEVRRKNLYKEGDLTHFNQKLEGFTLPRCWEECLASSQYHARKSEVDKFNKENCWKKRGLCIIPTKFGISFTVPFLNQAGALLHVYTDGSVLLTHGGTEMGQGLHTKMVQVASRALKIPTSKIYISETSTNTVPNTSPTAASVSADLNGQAVYAACQTILKRLEPYKKKNPSGSWEDWVTAAYMDTVSLSATGFYRTPNLGYSFETNSGNPFHYFSYGVACSEVEIDCLTGDHKNLRTDIVMDVGSSLNPAIDIGQVEGAFVQGLGLFTLEELHYSPEGSLHTRGPSTYKIPAFGSIPIEFRVSLLRDCPNKKAIYASKAVGEPPLFLAASIFFAIKDAIRAARAQHTGNNVKELFRLDSPATPEKIRNACVDKFTTLCVTGVPENCKPWSVRV Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Folate receptor alpha (FOLR1) | 4LRH | 6.96 | |
Target general information Gen name FOLR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ovarian tumorassociated antigen MOv18; KB cells FBP; Folate receptor, adult; Folate receptor 1; FRalpha; FOLR1; Adult folatebinding protein Protein family Folate receptor family Biochemical class Folate receptor Function Binds to folate and reduced folic acid derivatives and mediates delivery of 5-methyltetrahydrofolate and folate analogs into the interior of cells. Has high affinity for folate and folic acid analogs at neutral pH. Exposure to slightly acidic pHafter receptor endocytosis triggers a conformation change that strongly reduces its affinity for folates and mediates their release. Required for normal embryonic development and normal cell proliferation. Related diseases Neurodegeneration due to cerebral folate transport deficiency (NCFTD) [MIM:613068]: An autosomal recessive neurodegenerative disorder resulting from brain-specific folate deficiency early in life. Onset is apparent in late infancy with severe developmental regression, movement disturbances, epilepsy and leukodystrophy. {ECO:0000269|PubMed:19732866}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05595; DB00158; DB00563; DB12489; DB15413; DB05168 Interacts with Q8N357 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Endosome; Folate-binding; Glycoprotein; GPI-anchor; Lipoprotein; Membrane; Neurodegeneration; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 24216 Length 207 Aromaticity 0.13 Instability index 49.36 Isoelectric point 8.14 Charge (pH=7) 3.41 2D Binding mode Binding energy (Kcal/mol) -9.5 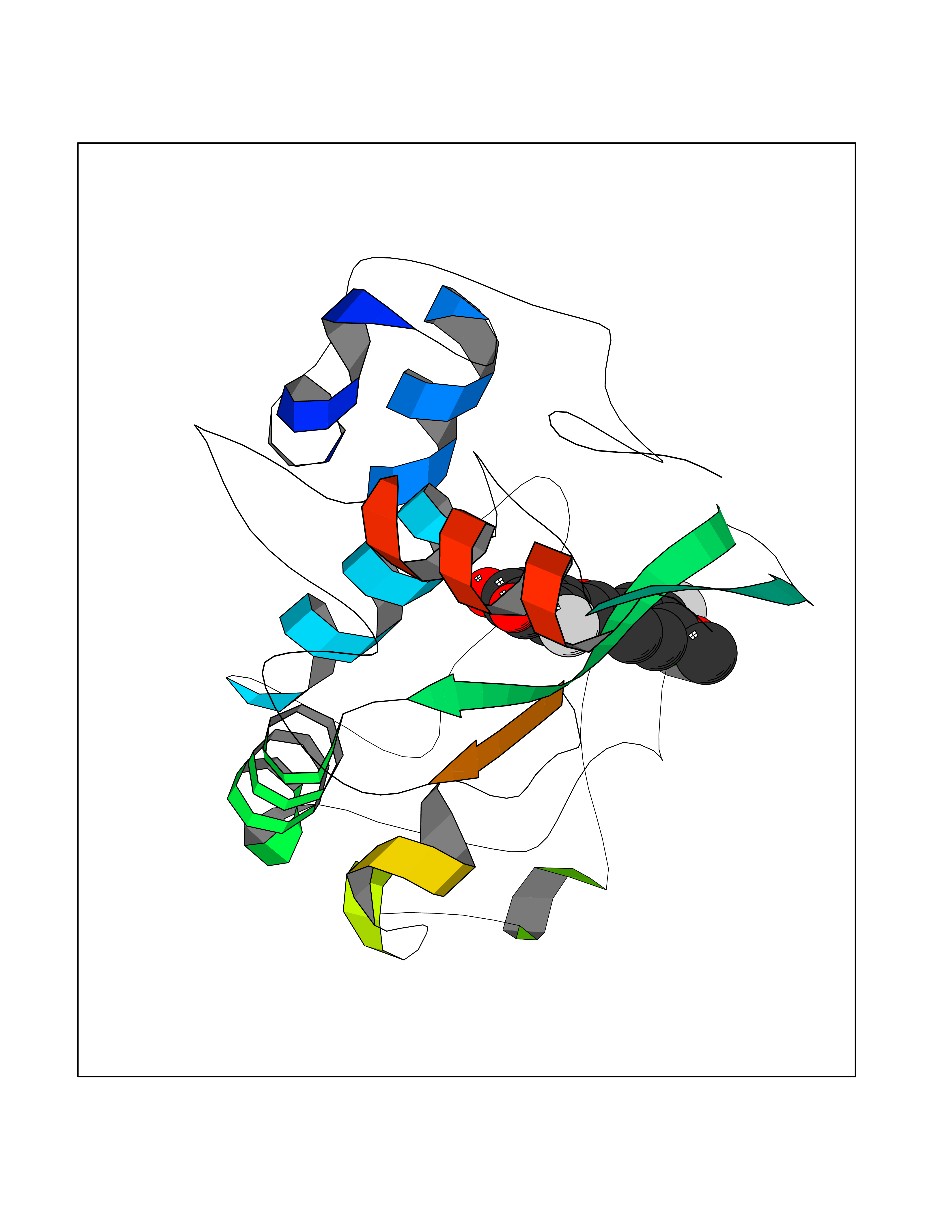 Molscript Map 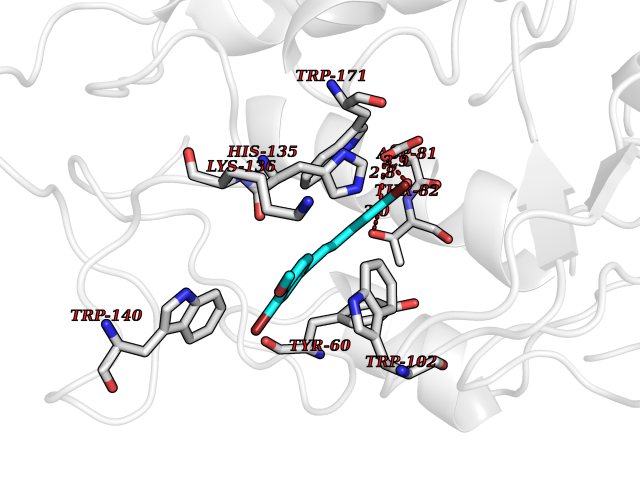 Pymol Map 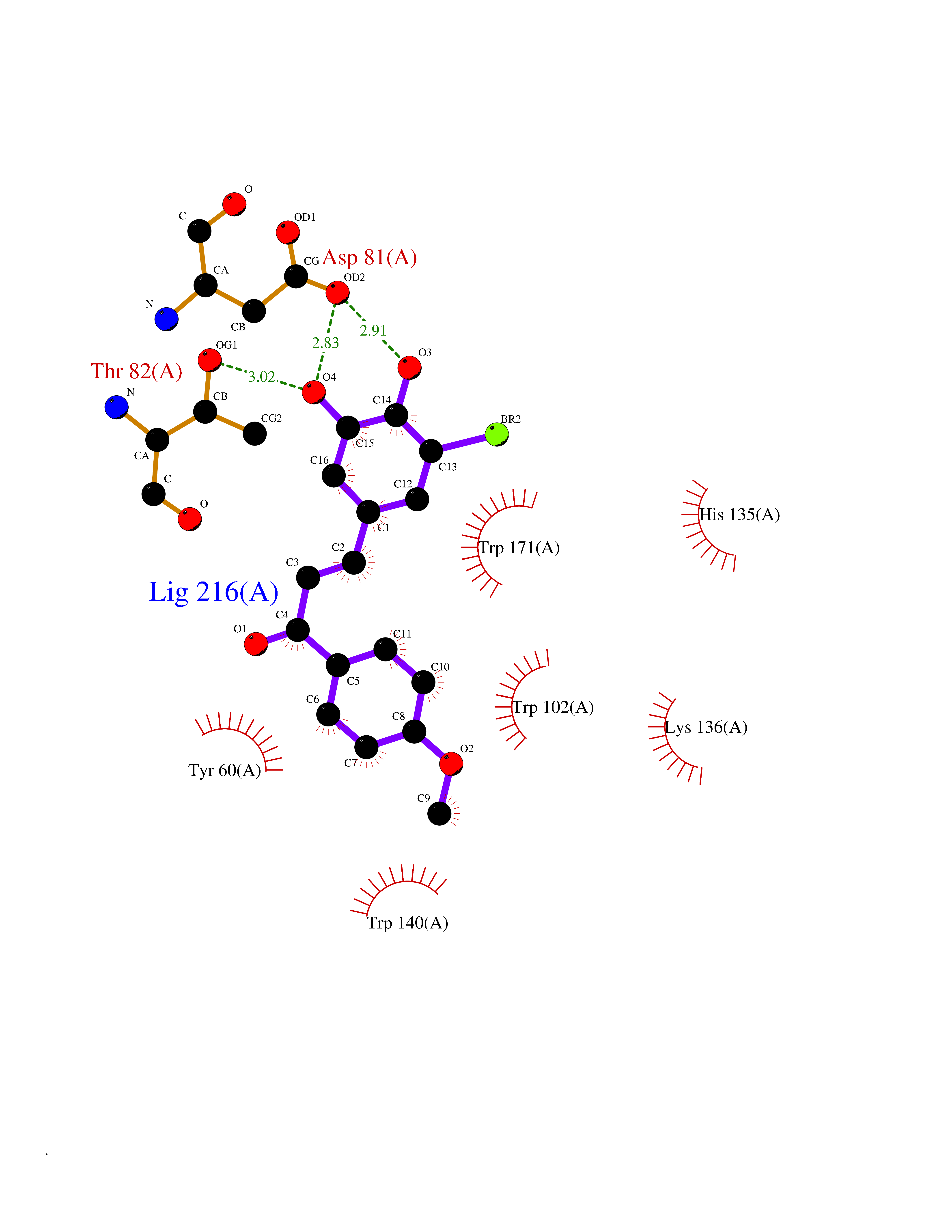 Ligplot Map 3D Binding mode Sequence RTELLNVCMNAKHHKEKPGPEDKLHEQCRPWRKNACCSTNTSQEAHKDVSYLYRFNWNHCGEMAPACKRHFIQDTCLYECSPNLGPWIQQVDQSWRKERVLNVPLCKEDCEQWWEDCRTSYTCKSNWHKGWNWTSGFNKCAVGAACQPFHFYFPTPTVLCNEIWTHSYKVSNYSRGSGRCIQMWFDPAQGNPNEEVARFYAAAMSGT Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | NAD-dependent deacetylase sirtuin-2 (SIRT2) | 4RMJ | 6.96 | |
Target general information Gen name SIRT2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SIR2like protein 2; SIR2L2; SIR2L; SIR2-like protein 2; Regulatory protein SIR2 homolog 2; NADdependent protein deacetylase sirtuin2; NAD-dependent protein deacetylase sirtuin-2 Protein family Sirtuin family, Class I subfamily Biochemical class Carbon-nitrogen hydrolase Function Participates in the modulation of multiple and diverse biological processes such as cell cycle control, genomic integrity, microtubule dynamics, cell differentiation, metabolic networks, and autophagy. Plays a major role in the control of cell cycle progression and genomic stability. Functions in the antephase checkpoint preventing precocious mitotic entry in response to microtubule stress agents, and hence allowing proper inheritance of chromosomes. Positively regulates the anaphase promoting complex/cyclosome (APC/C) ubiquitin ligase complex activity by deacetylating CDC20 and FZR1, then allowing progression through mitosis. Associates both with chromatin at transcriptional start sites (TSSs) and enhancers of active genes. Plays a role in cell cycle and chromatin compaction through epigenetic modulation of the regulation of histone H4 'Lys-20' methylation (H4K20me1) during early mitosis. Specifically deacetylates histone H4 at 'Lys-16' (H4K16ac) between the G2/M transition and metaphase enabling H4K20me1 deposition by KMT5A leading to ulterior levels of H4K20me2 and H4K20me3 deposition throughout cell cycle, and mitotic S-phase progression. Deacetylates KMT5A modulating KMT5A chromatin localization during the mitotic stress response. Deacetylates also histone H3 at 'Lys-57' (H3K56ac) during the mitotic G2/M transition. Upon bacterium Listeria monocytogenes infection, deacetylates 'Lys-18' of histone H3 in a receptor tyrosine kinase MET- and PI3K/Akt-dependent manner, thereby inhibiting transcriptional activity and promoting late stages of listeria infection. During oocyte meiosis progression, may deacetylate histone H4 at 'Lys-16' (H4K16ac) and alpha-tubulin, regulating spindle assembly and chromosome alignment by influencing microtubule dynamics and kinetochore function. Deacetylates histone H4 at 'Lys-16' (H4K16ac) at the VEGFA promoter and thereby contributes to regulate expression of VEGFA, a key regulator of angiogenesis. Deacetylates alpha-tubulin at 'Lys-40' and hence controls neuronal motility, oligodendroglial cell arbor projection processes and proliferation of non-neuronal cells. Phosphorylation at Ser-368 by a G1/S-specific cyclin E-CDK2 complex inactivates SIRT2-mediated alpha-tubulin deacetylation, negatively regulating cell adhesion, cell migration and neurite outgrowth during neuronal differentiation. Deacetylates PARD3 and participates in the regulation of Schwann cell peripheral myelination formation during early postnatal development and during postinjury remyelination. Involved in several cellular metabolic pathways. Plays a role in the regulation of blood glucose homeostasis by deacetylating and stabilizing phosphoenolpyruvate carboxykinase PCK1 activity in response to low nutrient availability. Acts as a key regulator in the pentose phosphate pathway (PPP) by deacetylating and activating the glucose-6-phosphate G6PD enzyme, and therefore, stimulates the production of cytosolic NADPH to counteract oxidative damage. Maintains energy homeostasis in response to nutrient deprivation as well as energy expenditure by inhibiting adipogenesis and promoting lipolysis. Attenuates adipocyte differentiation by deacetylating and promoting FOXO1 interaction to PPARG and subsequent repression of PPARG-dependent transcriptional activity. Plays a role in the regulation of lysosome-mediated degradation of protein aggregates by autophagy in neuronal cells. Deacetylates FOXO1 in response to oxidative stress or serum deprivation, thereby negatively regulating FOXO1-mediated autophagy. Deacetylates a broad range of transcription factors and co-regulators regulating target gene expression. Deacetylates transcriptional factor FOXO3 stimulating the ubiquitin ligase SCF(SKP2)-mediated FOXO3 ubiquitination and degradation. Deacetylates HIF1A and therefore promotes HIF1A degradation and inhibition of HIF1A transcriptional activity in tumor cells in response to hypoxia. Deacetylates RELA in the cytoplasm inhibiting NF-kappaB-dependent transcription activation upon TNF-alpha stimulation. Inhibits transcriptional activation by deacetylating p53/TP53 and EP300. Deacetylates also EIF5A. Functions as a negative regulator on oxidative stress-tolerance in response to anoxia-reoxygenation conditions. Plays a role as tumor suppressor. NAD-dependent protein deacetylase, which deacetylates internal lysines on histone and alpha-tubulin as well as many other proteins such as key transcription factors. Related diseases Deafness, autosomal recessive, 39 (DFNB39) [MIM:608265]: A form of profound prelingual sensorineural hearing loss. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. {ECO:0000269|PubMed:19576567}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15493 Interacts with O60566; O60729; P11413; Q92831; Q04206; Q9BYB0; Q12834; Q9UM11 EC number EC 3.5.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Autophagy; Cell cycle; Cell division; Cell membrane; Cell projection; Chromosome; Cytoplasm; Cytoskeleton; Differentiation; Immunity; Innate immunity; Meiosis; Membrane; Metal-binding; Microtubule; Mitosis; NAD; Neurodegeneration; Neurogenesis; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 34093.1 Length 301 Aromaticity 0.1 Instability index 45.2 Isoelectric point 5.59 Charge (pH=7) -6.94 2D Binding mode Binding energy (Kcal/mol) -9.49 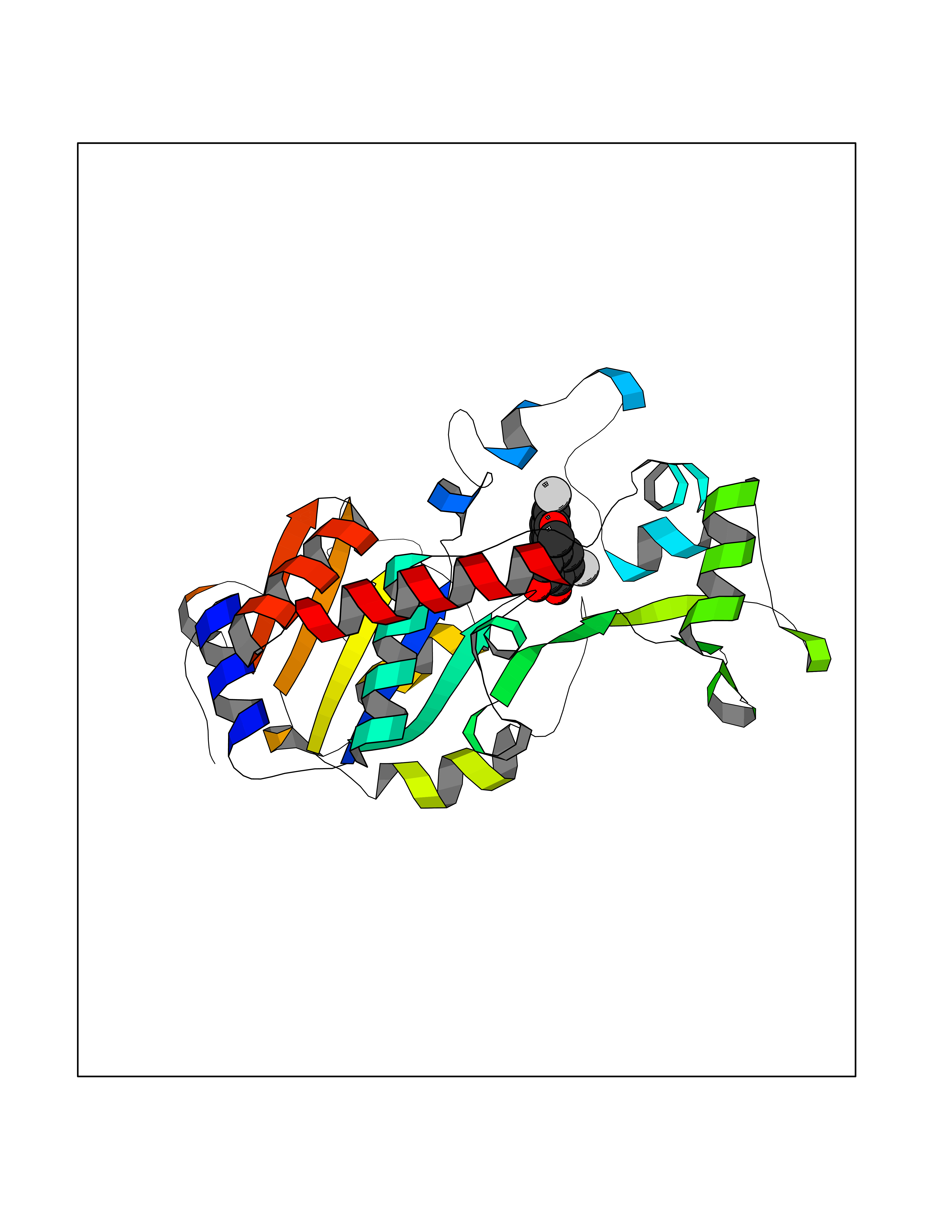 Molscript Map 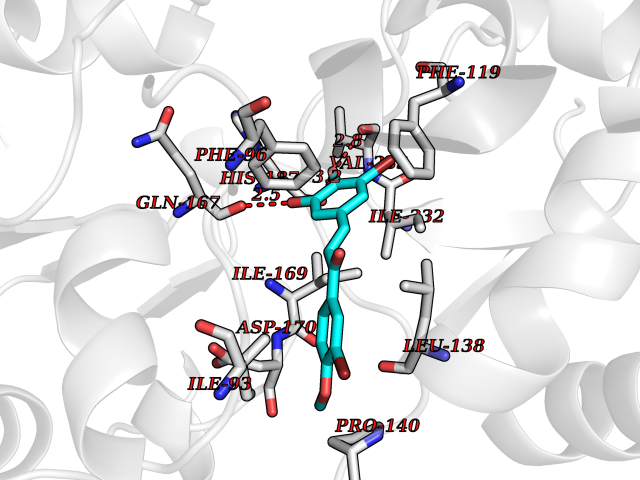 Pymol Map 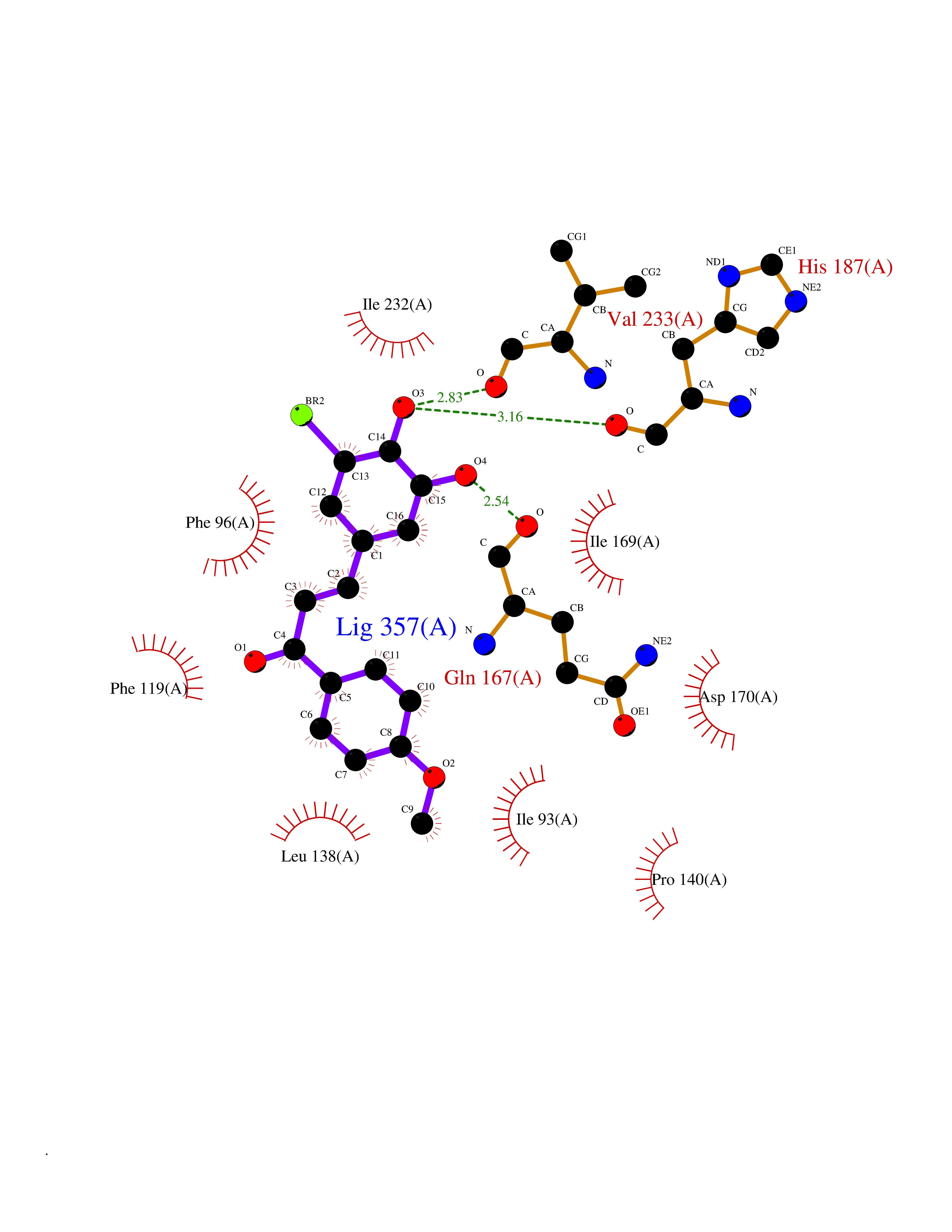 Ligplot Map 3D Binding mode Sequence MERLLDELTLEGVARYMQSERCRRVICLVGAGISTSAGIPDFRSPSTGLYDNLEKYHLPYPEAIFEISYFKKHPEPFFALAKELYPGQFKPTICHYFMRLLKDKGLLLRCYTQNIDTLERIAGLEQEDLVEAHGTFYTSHCVSASCRHEYPLSWMKEKIFSEVTPKCEDCQSLVKPDIVFFGESLPARFFSCMQSDFLKVDLLLVMGTSLQVQPFASLISKAPLSTPRLLINKEKAGQSDPFLGMIMGLGGGMDFDSKKAYRDVAWLGECDQGCLALAELLGWKKELEDLVRREHASIDAQ Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Dihydrolipoyl dehydrogenase, mitochondrial | 1ZMD | 6.95 | |
Target general information Gen name DLD Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PHE3;LAD;GCSL Protein family Class-I pyridine nucleotide-disulfide oxidoreductase family Biochemical class Oxidoreductase Function Dihydrolipoyl dehydrogenase activity.Electron carrier activity.Flavin adenine dinucleotide binding.Lipoamide binding.NAD binding. Related diseases Dihydrolipoamide dehydrogenase deficiency (DLDD) [MIM:246900]: An autosomal recessive metabolic disorder characterized biochemically by a combined deficiency of the branched-chain alpha-keto acid dehydrogenase complex (BCKDC), pyruvate dehydrogenase complex (PDC), and alpha-ketoglutarate dehydrogenase complex (KGDC). Clinically, affected individuals have lactic acidosis and neurologic deterioration due to sensitivity of the central nervous system to defects in oxidative metabolism. {ECO:0000269|PubMed:10448086, ECO:0000269|PubMed:11687750, ECO:0000269|PubMed:12925875, ECO:0000269|PubMed:15712224, ECO:0000269|PubMed:16442803, ECO:0000269|PubMed:16770810, ECO:0000269|PubMed:17404228, ECO:0000269|PubMed:20160912, ECO:0000269|PubMed:8506365, ECO:0000269|PubMed:8968745, ECO:0000269|PubMed:9540846, ECO:0000269|PubMed:9934985}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147; DB00145; DB00157 Interacts with P42858; O14713; O00330; P30041; P62258 EC number 1.8.1.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell projection; Cilium; Cytoplasmic vesicle; Disease variant; Disulfide bond; FAD; Flagellum; Flavoprotein; Mitochondrion; NAD; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Redox-active center; Reference proteome; Transit peptide Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H Molecular weight (Da) 49832.7 Length 471 Aromaticity 0.06 Instability index 26.19 Isoelectric point 6.51 Charge (pH=7) -2.02 2D Binding mode Binding energy (Kcal/mol) -9.48 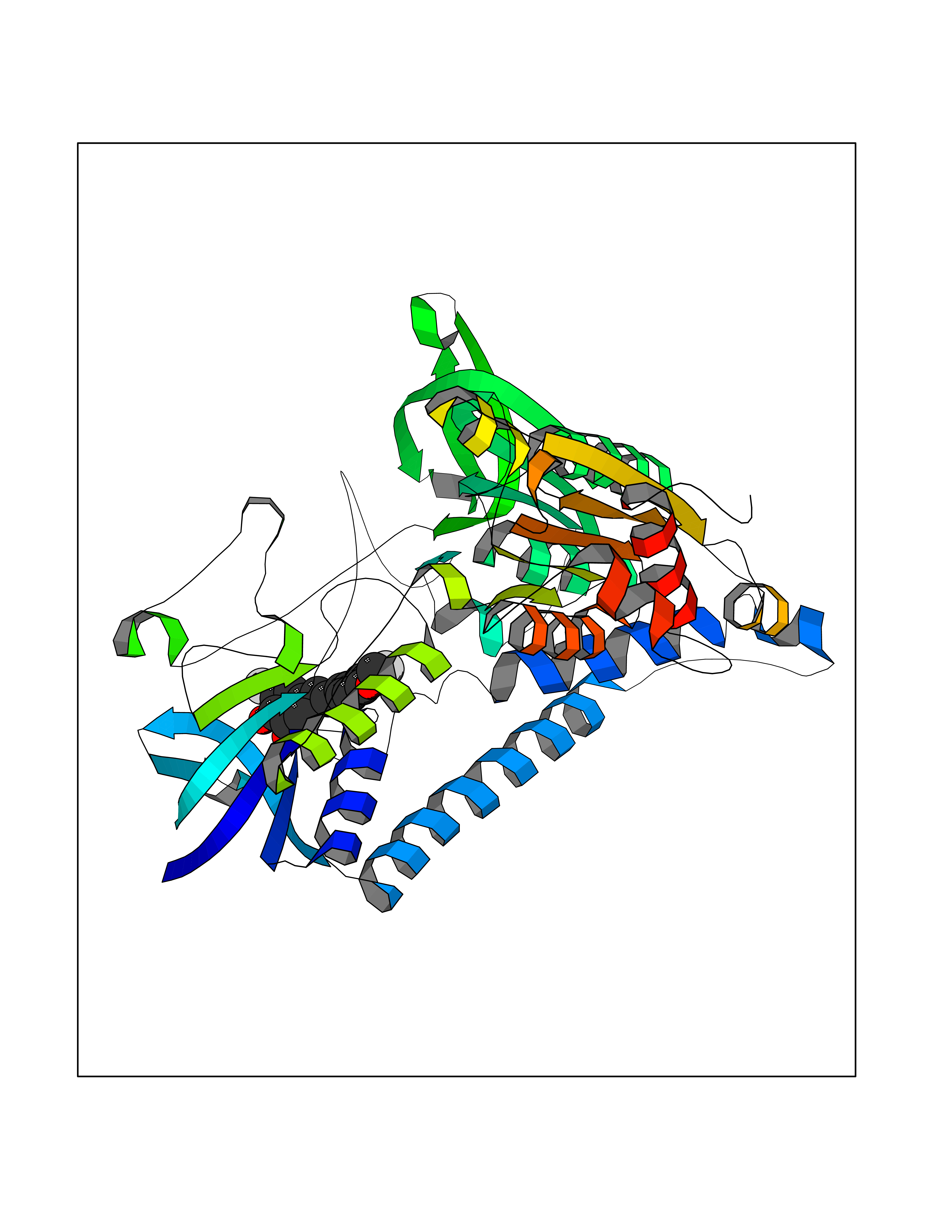 Molscript Map 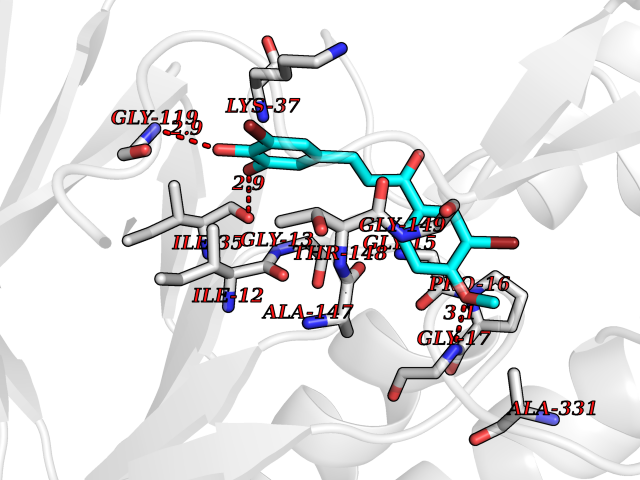 Pymol Map 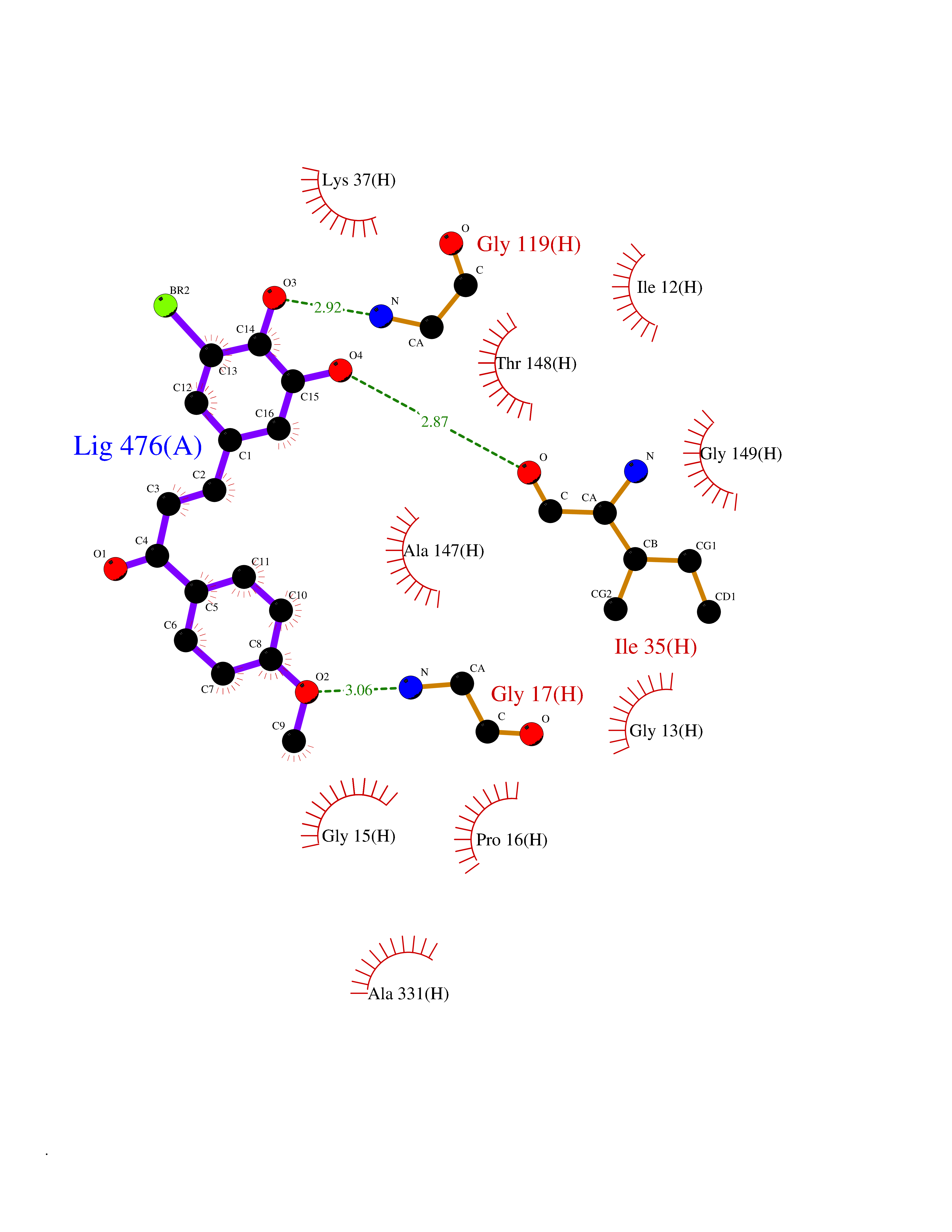 Ligplot Map 3D Binding mode Sequence PIDADVTVIGSGPGGYVAAIKAAQLGFKTVCIEKNETLGGTCLNVGCIPSKALLNNSHYYHMAHGTDFASRGIEMSEVRLNLDKMMEQKSTAVKALTGGIAHLFKQNKVVHVNGYGKITGKNQVTATKADGGTQVIDTKNILIATGSEVTPFPGITIDEDTIVSSTGALSLKKVPEKMVVIGAGVIGVELGSVWQRLGADVTAVEFLGHVGGVGIDMEISKNFQRILQKQGFKFKLNTKVTGATKKSDGKIDVSIEAASGGKAEVITCDVLLVCIGRRPFTKNLGLEELGIELDPRGRIPVNTRFQTKIPNIYAIGDVVAGPMLAHKAEDEGIICVEGMAGGAVHIDYNCVPSVIYTHPEVAWVGKSEEQLKEEGIEYKVGKFPFAANSRAKTNADTDGMVKILGQKSTDRVLGAHILGPGAGEMVNEAALALEYGASCEDIARVCHAHPTLSEAFREANLAASFGKSINF Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Serum albumin (ALB) | 4L8U | 6.95 | |
Target general information Gen name ALB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serum albumin Protein family ALB/AFP/VDB family Biochemical class NA Function Serum albumin, the main protein of plasma, has a good binding capacity for water, Ca(2+), Na(+), K(+), fatty acids, hormones, bilirubin and drugs. Its main function is the regulation of the colloidal osmotic pressure of blood. Major zinc transporter in plasma, typically binds about 80% of all plasma zinc. Related diseases Hyperthyroxinemia, familial dysalbuminemic (FDAH) [MIM:615999]: A disorder characterized by abnormally elevated levels of total serum thyroxine (T4) in euthyroid patients. It is due to abnormal serum albumin that binds T4 with enhanced affinity. {ECO:0000269|PubMed:7852505, ECO:0000269|PubMed:8048949, ECO:0000269|PubMed:9329347, ECO:0000269|PubMed:9589637}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Analbuminemia (ANALBA) [MIM:616000]: A rare autosomal recessive disorder manifested by the presence of a very low amount of circulating serum albumin. Affected individuals manifest mild edema, hypotension, fatigue, and, occasionally, lower body lipodystrophy (mainly in adult females). The most common biochemical finding is hyperlipidemia, with a significant increase in the total and LDL cholesterol concentrations, but normal concentrations of HDL cholesterol and triglycerides. {ECO:0000269|PubMed:8134387}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08496; DB07517; DB12001; DB05812; DB14973; DB11703; DB01418; DB01614; DB00316; DB00414; DB09347; DB06151; DB00459; DB00787; DB00640; DB00802; DB00346; DB00404; DB00770; DB01370; DB14517; DB14518; DB01118; DB00321; DB01060; DB00415; DB00276; DB06728; DB11901; DB00714; DB04557; DB09229; DB11217; DB00278; DB01238; DB14185; DB09204; DB01169; DB11638; DB09274; DB00126; DB06216; DB01072; DB00335; DB00289; DB01076; DB00995; DB06237; DB07402; DB00993; DB08822; DB08903; DB16703; DB00245; DB01086; DB01053; DB00443; DB14669; DB11967; DB13909; DB01294; DB09223; DB00083; DB09128; DB01222; DB15248; DB00490; DB00237; DB11148; DB06772; DB11751; DB11093; DB11348; DB14481; DB04690; DB01101; DB03600; DB01197; DB01136; DB00456; DB01327; DB14879; DB00274; DB01328; DB01329; DB00493; DB01330; DB00430; DB00438; DB01212; DB06119; DB00567; DB07565; DB08936; DB00878; DB00608; DB00477; DB09093; DB00310; DB00501; DB00568; DB00537; DB00515; DB00349; DB01013; DB00845; DB01242; DB01068; DB00575; DB00758; DB01147; DB00363; DB15534; DB01394; DB00286; DB12483; DB09130; DB01380; DB08865; DB11134; DB06778; DB01176; DB00924; DB00434; DB00847; DB01914; DB06695; DB08912; DB11963; DB04816; DB00080; DB12941; DB01264; DB11943; DB11637; DB01189; DB00304; DB01234; DB14649; DB09213; DB00829; DB01119; DB11397; DB00586; DB00485; DB00266; DB00900; DB00861; DB01396; DB00343; DB08995; DB08930; DB01142; DB00997; DB00254; DB00366; DB04855; DB00476; DB01126; DB01057; DB12243; DB13421; DB00625; DB15444; DB00879; DB00584; DB13874; DB11718; DB00228; DB08899; DB01364; DB00530; DB00303; DB11827; DB12235; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB04574; DB00903; DB00977; DB00749; DB00294; DB01276; DB12466; DB04854; DB01039; DB00573; DB00813; DB00950; DB16165; DB01195; DB00687; DB15690; DB00544; DB00472; DB00712; DB08906; DB00983; DB01320; DB06716; DB11796; DB00695; DB15149; DB00743; DB06705; DB01044; DB00317; DB01241; DB12141; DB11978; DB01120; DB01067; DB01016; DB00986; DB13751; DB04539; DB12836; DB11575; DB11359; DB01159; DB14999; DB00070; DB01275; DB00999; DB00774; DB00327; DB09526; DB01611; DB01005; DB00557; DB13014; DB12471; DB09053; DB01050; DB00159; DB01088; DB00619; DB09262; DB00458; DB00808; DB00328; DB07992; DB09564; DB01307; DB05382; DB04711; DB09333; DB00332; DB16200; DB01029; DB00762; DB06636; DB00753; DB00677; DB00951; DB01064; DB00982; DB11757; DB01167; DB08820; DB01587; DB01026; DB01009; DB00598; DB09236; DB00709; DB00555; DB03017; DB01006; DB09237; DB06282; DB01235; DB01137; DB00451; DB00601; DB17083; DB00279; DB01583; DB06655; DB01601; DB09195; DB00678; DB00227; DB09280; DB15935; DB12674; DB00137; DB08932; DB14513; DB01397; DB06796; DB06234; DB00737; DB13959; DB09124; DB00603; DB00784; DB00814; DB01042; DB00454; DB09383; DB00931; DB00333; DB00563; DB00968; DB09241; DB00959; DB06710; DB00264; DB01110; DB00683; DB08893; DB00295; DB01024; DB08231; DB00461; DB00607; DB01183; DB00788; DB00731; DB04861; DB00220; DB11828; DB00238; DB01115; DB11820; DB09079; DB11793; DB12005; DB06713; DB00717; DB00957; DB00540; DB00104; DB00334; DB09074; DB04224; DB00768; DB12455; DB11130; DB04911; DB01083; DB13310; DB01173; DB00526; DB00842; DB00776; DB01062; DB00497; DB06412; DB03585; DB00595; DB15575; DB09073; DB03796; DB13967; DB14582; DB00642; DB00850; DB12978; DB01619; DB03255; DB00946; DB00252; DB01132; DB01621; DB04951; DB00554; DB08860; DB11642; DB01324; DB09087; DB09418; DB06813; DB13514; DB06209; DB01058; DB00860; DB15566; DB14631; DB00635; DB01032; DB01069; DB09348; DB00818; DB00571; DB06480; DB00852; DB00165; DB04216; DB00881; DB00908; DB12874; DB08735; DB00481; DB11853; DB12404; DB00912; DB02709; DB11855; DB01045; DB11753; DB08864; DB08931; DB14840; DB15305; DB00734; DB00503; DB11182; DB00412; DB01098; DB04847; DB06201; DB08877; DB08736; DB00936; DB00938; DB01232; DB11689; DB13928; DB01104; DB01236; DB12965; DB06290; DB00877; DB00815; DB15093; DB00421; DB00649; DB03193; DB06150; DB01581; DB01582; DB00576; DB01015; DB00795; DB00605; DB00391; DB00870; DB00864; DB00675; DB05134; DB09139; DB05521; DB00853; DB14126; DB09299; DB15133; DB00857; DB00342; DB00624; DB13943; DB13944; DB01420; DB13946; DB00759; DB00152; DB11590; DB01622; DB01623; DB09100; DB09070; DB08816; DB01133; DB15171; DB11800; DB01056; DB08895; DB01124; DB00500; DB00273; DB01685; DB00214; DB00755; DB00620; DB00432; DB08814; DB11677; DB00376; DB09069; DB00792; DB00427; DB08867; DB09076; DB12255; DB00313; DB00512; DB05294; DB08881; DB00661; DB15456; DB11641; DB08828; DB00162; DB11739; DB16699; DB00682; DB00943; DB00495; DB00744; DB14533; DB14548; DB00246; DB04828 Interacts with P02768; P02786; Q8N5Z5; Q6GQQ9-2; Q07869; Q09028; Q86WT6-2; O76024 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cleavage on pair of basic residues; Copper; Direct protein sequencing; Disease variant; Disulfide bond; Glycation; Glycoprotein; Lipid-binding; Metal-binding; Methylation; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 34028.4 Length 298 Aromaticity 0.09 Instability index 43.45 Isoelectric point 5.49 Charge (pH=7) -10.44 2D Binding mode Binding energy (Kcal/mol) -9.48 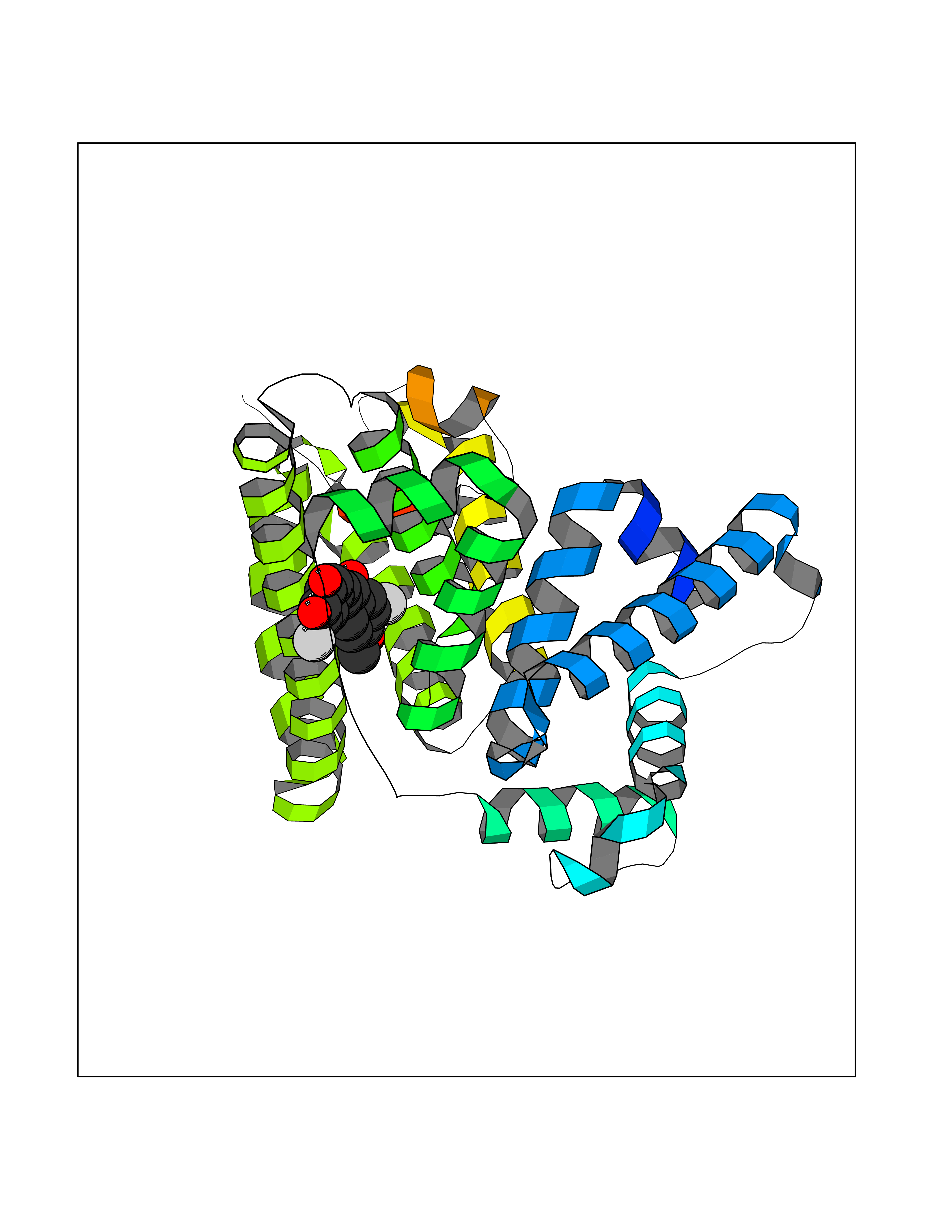 Molscript Map 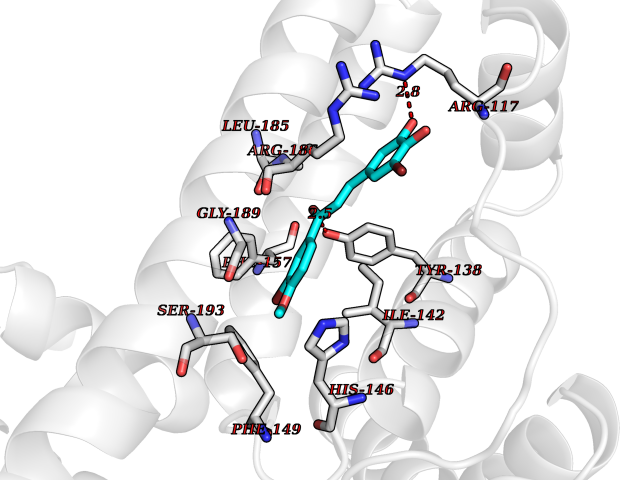 Pymol Map 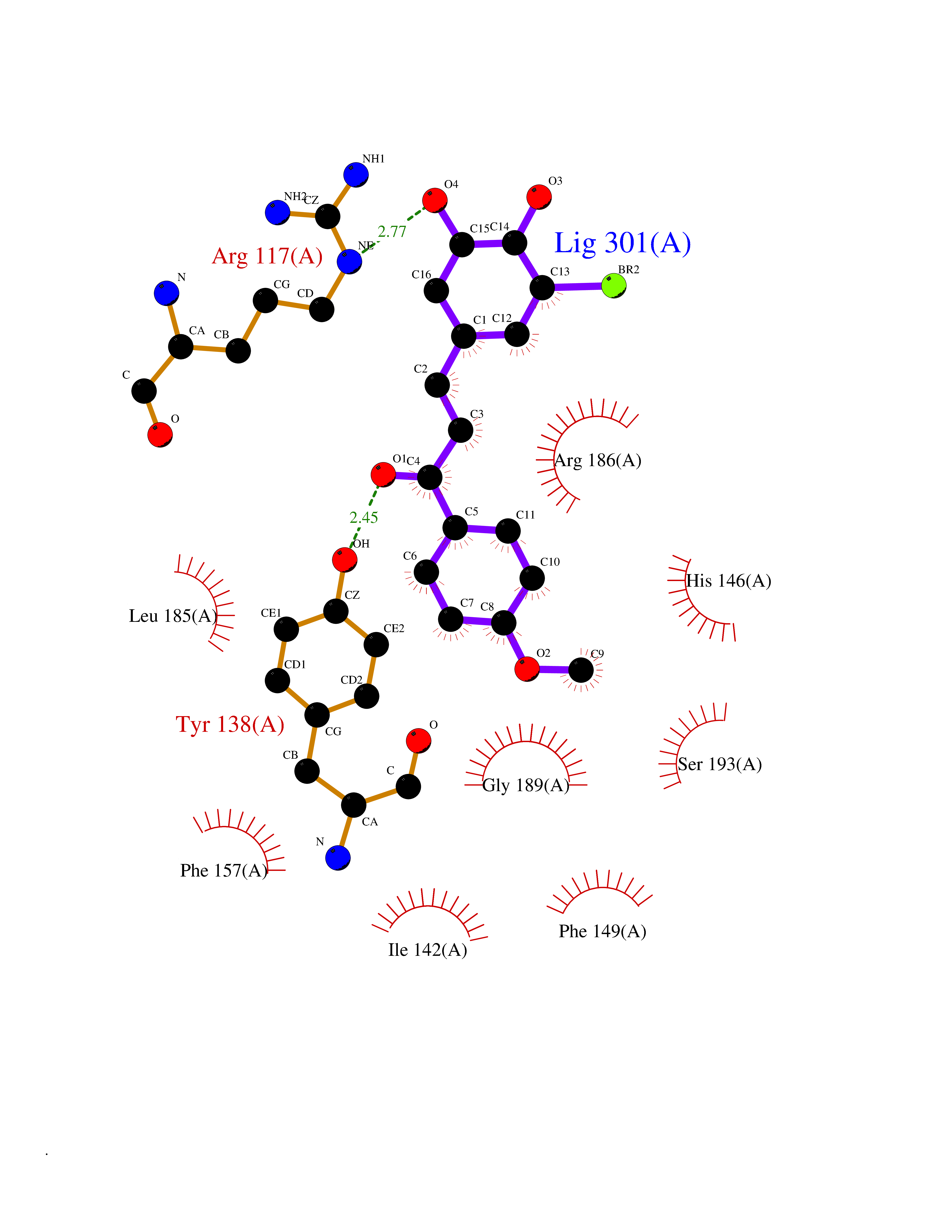 Ligplot Map 3D Binding mode Sequence AHKSEVAHRFKDLGEENFKALVLIAFAQYLQQCPFEDHVKLVNEVTEFAKTCVADESAENCDKSLHTLFGDKLCTVATLRETYGEMADCCAKQEPERNECFLQHKDDNPNLPRLVRPEVDVMCTAFHDNEETFLKKYLYEIARRHPYFYAPELLFFAKRYKAAFTECCQAADKAACLLPKLDELRDEGKASSAKQRLKCASLQKFGERAFKAWAVARLSQRFPKAEFAEVSKLVTDLTKVHTECCHGDLLECADDRADLAKYICENQDSISSKLKECCEKPLLEKSHCIAEVENDEMP Hydrogen bonds contact Hydrophobic contact | ||||