Job Results:
Ligand
Structure
Job ID
e8ee1117e51baec8034629c7ce0a1a1e
Job name
NA
Time
2025-12-29 08:34:20
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Bacterial Fatty acid synthetase I (Bact inhA) | 4DTI | 5.89 | |
Target general information Gen name Bact inhA Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID Synonyms Enoyl-[acyl-carrier-protein] reductase [NADH]; Bacterial InhA Protein family Short-chain dehydrogenases/reductases (SDR) family, FabI subfamily Biochemical class CH-CH donor oxidoreductase Function Involved in the resistance against the antituberculosis drugs isoniazid and ethionamide. Related diseases Immunodeficiency 35 (IMD35) [MIM:611521]: A primary immunodeficiency characterized by recurrent skin abscesses, pneumonia, and highly elevated serum IgE. {ECO:0000269|PubMed:17088085}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07155; DB07188; DB07192; DB07090; DB07222; DB07287; DB07178; DB00609; DB04289; DB00951; DB07123; DB05154; DB02990; DB08604 Interacts with NA EC number NA Uniprot keywords 3D-structure; Antibiotic resistance; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; NAD; Oxidoreductase; Phosphoprotein; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 28279.2 Length 267 Aromaticity 0.06 Instability index 39.93 Isoelectric point 5.74 Charge (pH=7) -3.78 2D Binding mode Binding energy (Kcal/mol) -8.04 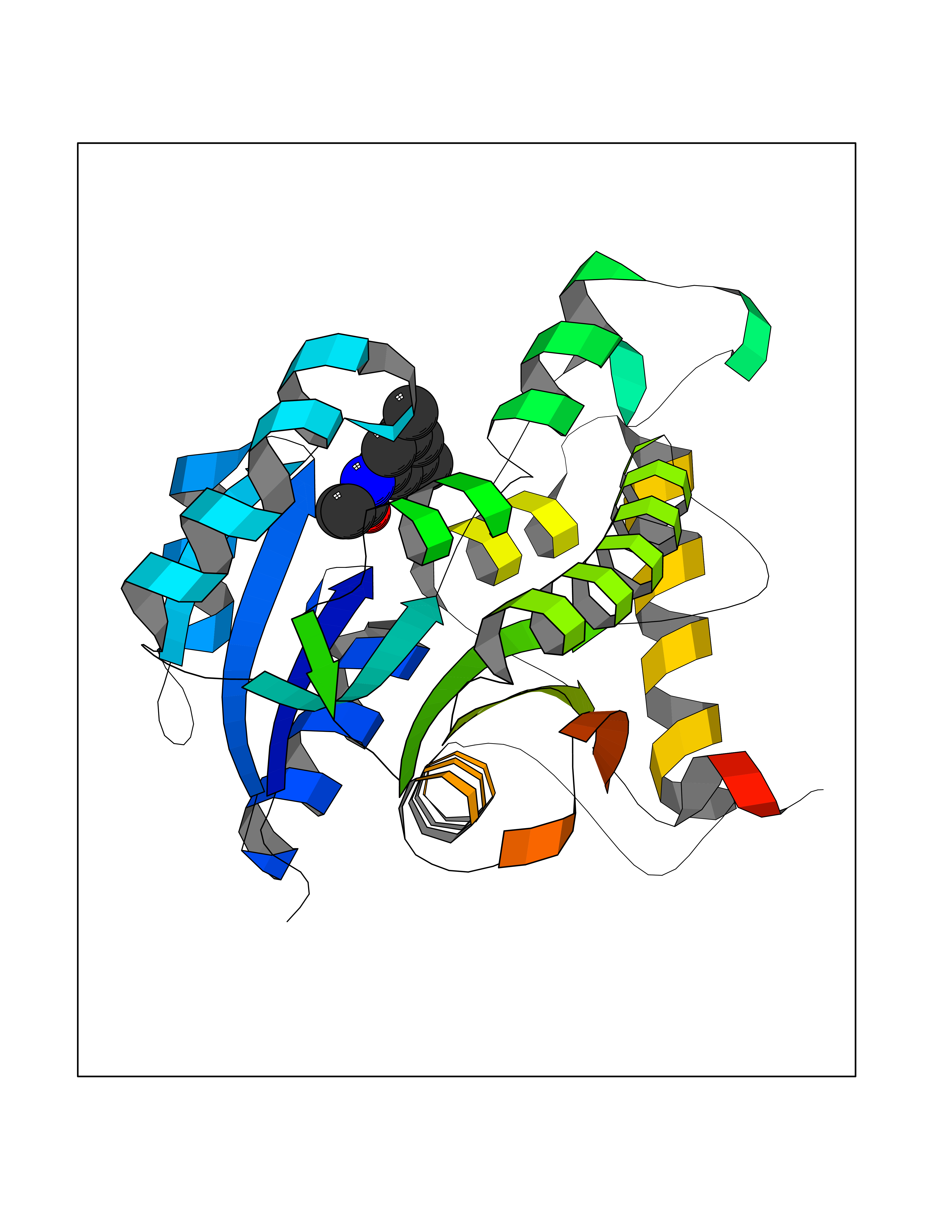 Molscript Map 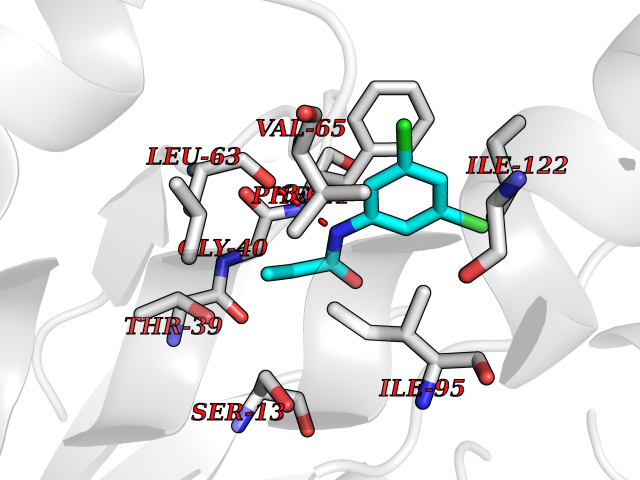 Pymol Map 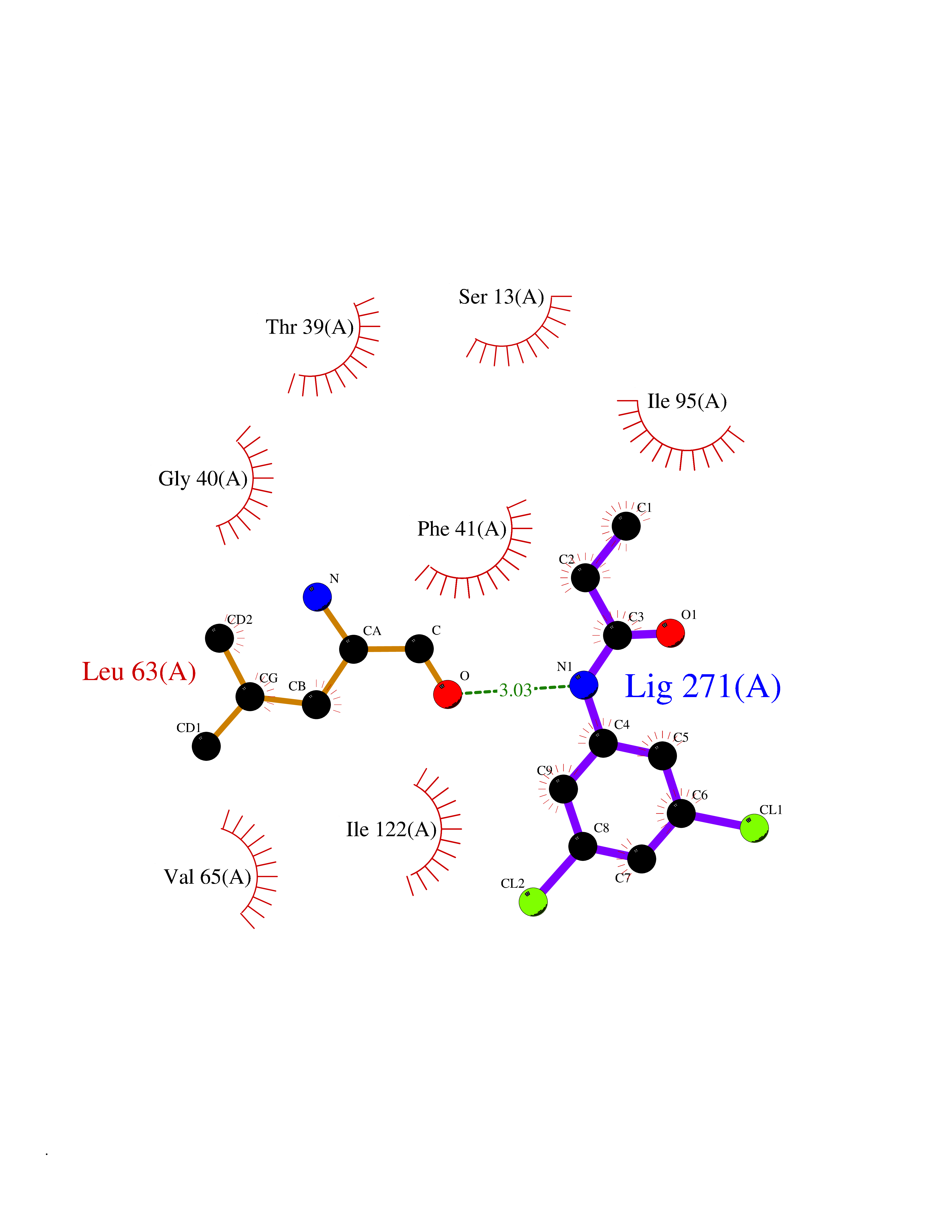 Ligplot Map 3D Binding mode Sequence GLLDGKRILVSGIITDSSIAFHIARVAQEQGAQLVLTGFDRLRLIQRITDRLPAKAPLLELDVQNEEHLASLAGRVTEAIGAGNKLDGVVHAIGFMPQTGMGINPFFDAPYADVSKGIHISAYSYASMAKALLPIMNPGGSIVGMDFDPSRAMPAYNWMTVAKSALESVNRFVAREAGKYGVRSNLVAAGPIRTLAMSAIVGGALGEEAGAQIQLLEEGWDQRAPIGWNMKDATPVAKTVCALLSDWLPATTGDIIYADGGAHTQLL Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Histamine H1 receptor (H1R) | 7DFL | 5.89 | |
Target general information Gen name HRH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HH1R; H1R Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function In peripheral tissues, the H1 subclass of histamine receptors mediates the contraction of smooth muscles, increase in capillary permeability due to contraction of terminal venules, and catecholamine release from adrenal medulla, as well as mediating neurotransmission in the central nervous system. Related diseases Hypertriglyceridemia, transient infantile (HTGTI) [MIM:614480]: An autosomal recessive disorder characterized by onset of moderate to severe transient hypertriglyceridemia in infancy that normalizes with age. The hypertriglyceridemia is associated with hepatomegaly, moderately elevated transaminases, persistent fatty liver, and the development of hepatic fibrosis. {ECO:0000269|PubMed:22226083, ECO:0000269|PubMed:24549054}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01615; DB09488; DB06766; DB01246; DB00321; DB00543; DB08799; DB01238; DB14185; DB06216; DB00637; DB00719; DB00972; DB00245; DB00767; DB04890; DB06698; DB11591; DB09128; DB01237; DB00835; DB00354; DB09016; DB00748; DB06016; DB00341; DB08936; DB08800; DB01114; DB00477; DB01239; DB00568; DB00215; DB00283; DB04837; DB00363; DB01176; DB00434; DB01151; DB00967; DB00405; DB09555; DB00985; DB08801; DB01075; DB01146; DB09167; DB01142; DB00366; DB01084; DB05492; DB00751; DB01175; DB06678; DB00950; DB04841; DB00502; DB05381; DB05079; DB00557; DB04946; DB00458; DB08802; DB00920; DB00555; DB01106; DB06282; DB00455; DB09195; DB00408; DB00934; DB00737; DB06691; DB01071; DB00902; DB01403; DB06148; DB00370; DB00540; DB05080; DB06229; DB00334; DB00768; DB01173; DB01267; DB00715; DB08922; DB01619; DB01620; DB06153; DB00433; DB00420; DB01069; DB00777; DB01224; DB00912; DB00734; DB11614; DB05345; DB00342; DB04905; DB11235; DB00797; DB00656; DB00726; DB00792; DB00427; DB09185; DB00246; DB01624 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32298.5 Length 275 Aromaticity 0.15 Instability index 36.59 Isoelectric point 9.54 Charge (pH=7) 16.01 2D Binding mode Binding energy (Kcal/mol) -8.04 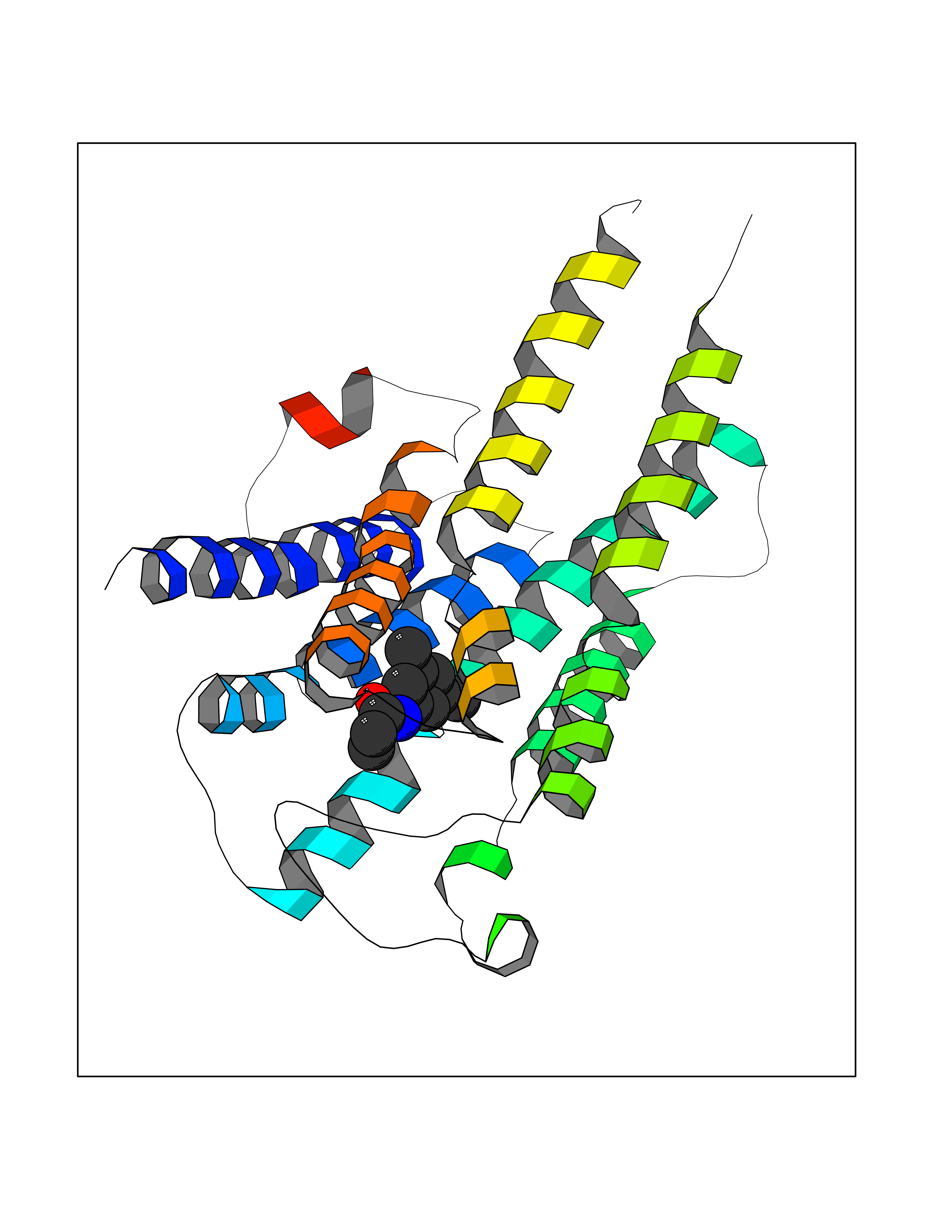 Molscript Map 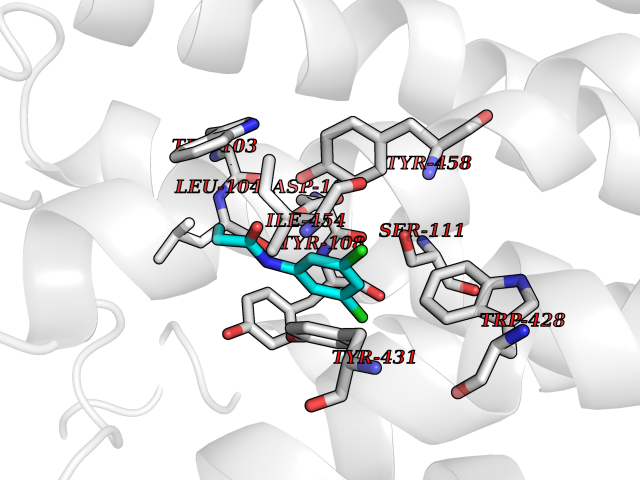 Pymol Map 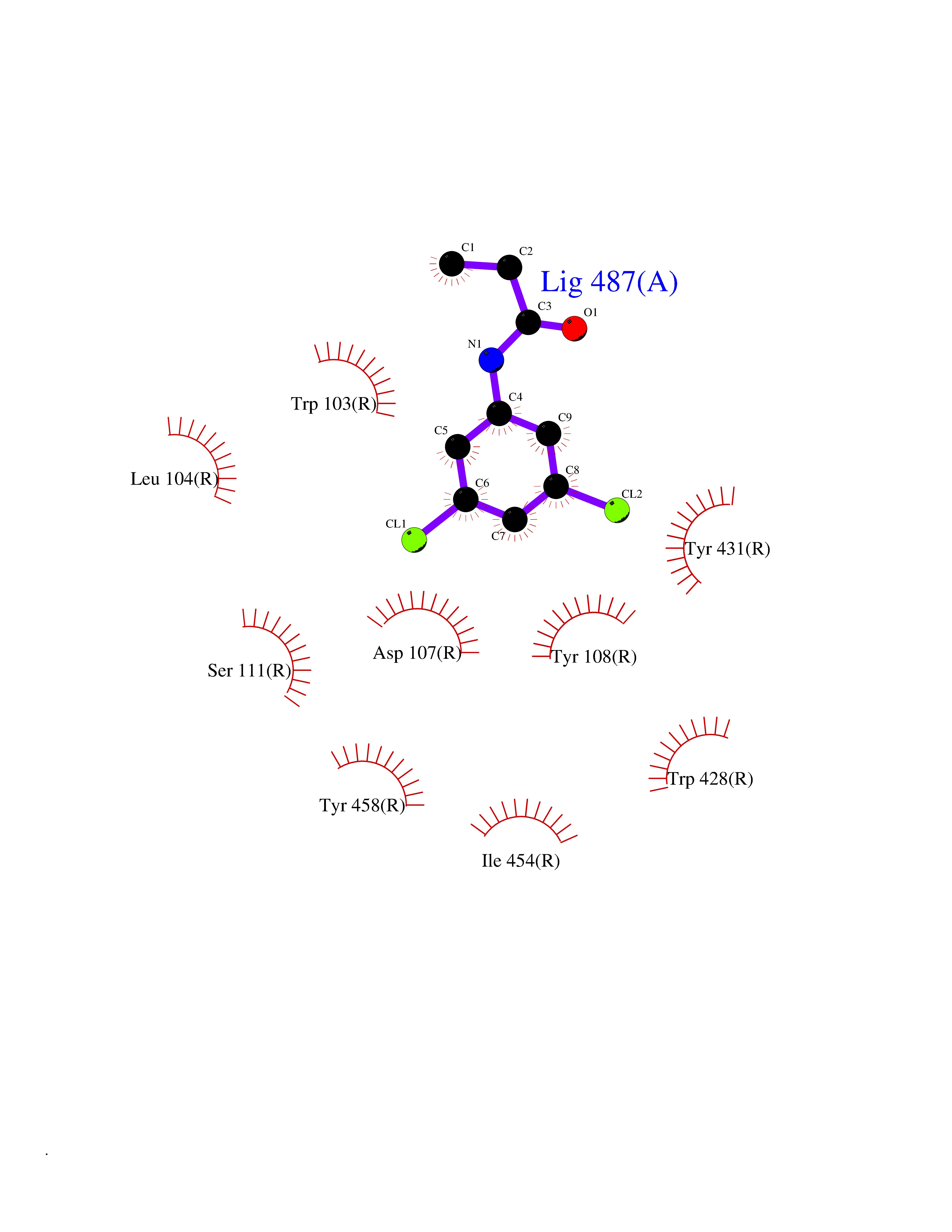 Ligplot Map 3D Binding mode Sequence MPLVVVLSTICLVTVGLNLLVLYAVRSERKLHTVGNLYIVSLSVADLIVGAVVMPMNILYLLMSKWSLGRPLCLFWLSMDYVASTASIFSVFILCIDRYRSVQQPLRYLKYRTKTRASATILGAWFLSFLWVIPILGWNHFMQQTSVRREDKCETDFYDVTWFKVMTAIINFYLPTLLMLWFYAKIYKAVRQHCLHMNRERKAAKQLGFIMAAFILCWIPYFIFFMVIAFCKNCCNEHLHMFTIWLGYINSTLNPLIYPLCNENFKKTFKRILHI Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Aggrecanase (ADAMTS5) | 3HY7 | 5.89 | |
Target general information Gen name ADAMTS5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aggrecanase-2; ADMP-2; ADAMTS5; ADAMTS-5; ADAM-TS5; ADAM-TS 5; ADAM-TS 11; A disintegrin and metalloproteinase with thrombospondinmotifs 5 Protein family NA Biochemical class Peptidase Function Cleaves aggrecan, a cartilage proteoglycan, and may be involved in its turnover. May play an important role in the destruction of aggrecan in arthritic diseases. May play a role in proteolytic processing mostly during the peri-implantation period. Related diseases Mucopolysaccharidosis 1H (MPS1H) [MIM:607014]: A severe form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1H usually present, within the first year of life, a combination of hepatosplenomegaly, skeletal deformities, corneal clouding and severe intellectual disability. Obstructive airways disease, respiratory infection and cardiac complications usually result in death before 10 years of age. {ECO:0000269|PubMed:10466419, ECO:0000269|PubMed:10735634, ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:1301941, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:19396826, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:24036510, ECO:0000269|PubMed:31194252, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:7951228, ECO:0000269|PubMed:8019563, ECO:0000269|PubMed:8328452, ECO:0000269|PubMed:8401515, ECO:0000269|Ref.20}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 1H/S (MPS1H/S) [MIM:607015]: A form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. MPS1H/S represents an intermediate phenotype of the MPS1 clinical spectrum. It is characterized by relatively little neurological involvement, but most of the somatic symptoms described for severe MPS1 develop in the early to mid-teens, causing considerable loss of mobility. {ECO:0000269|PubMed:10466419, ECO:0000269|PubMed:10735634, ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:8401515}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 1S (MPS1S) [MIM:607016]: A mild form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1S may have little or no neurological involvement, normal stature and life span, but present development of joints stiffness, mild hepatosplenomegaly, aortic valve disease and corneal clouding. {ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:19396826, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:25256405, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:8213840}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06837; DB03880; DB06945 Interacts with P13608 EC number EC 3.4.24.- Uniprot keywords 3D-structure; Cleavage on pair of basic residues; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 23997.8 Length 217 Aromaticity 0.06 Instability index 46.68 Isoelectric point 5.82 Charge (pH=7) -8.36 2D Binding mode Binding energy (Kcal/mol) -8.04 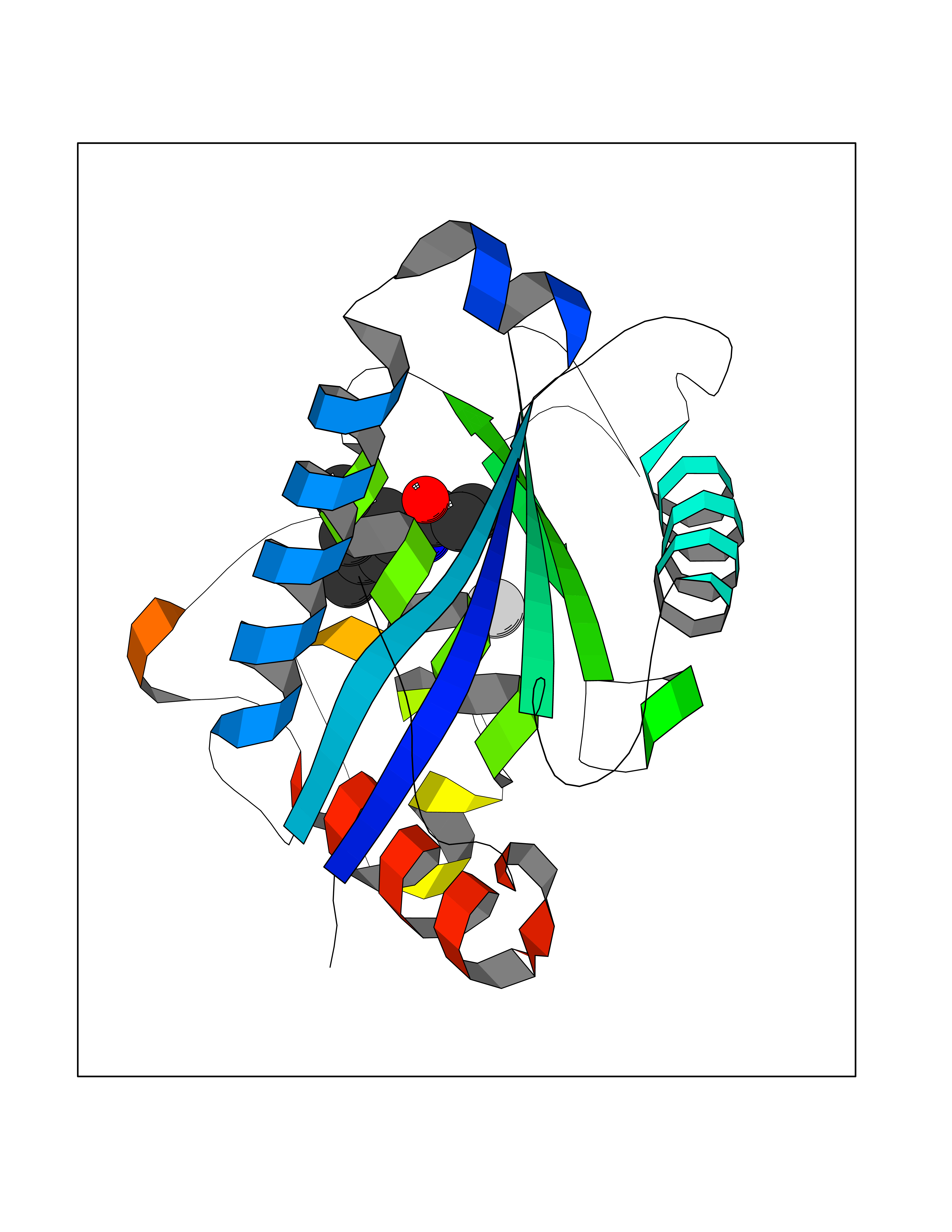 Molscript Map 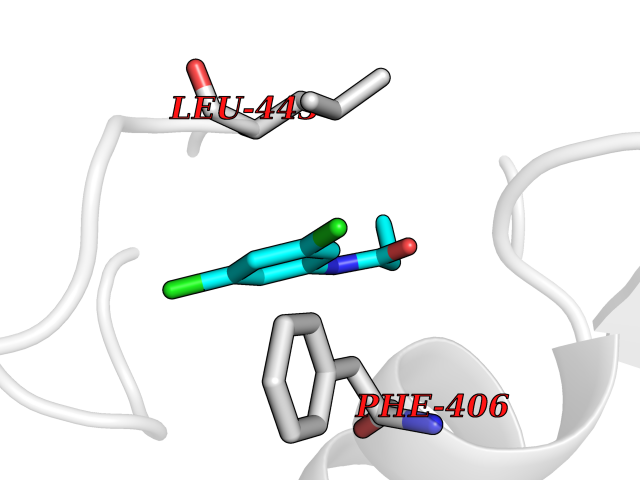 Pymol Map 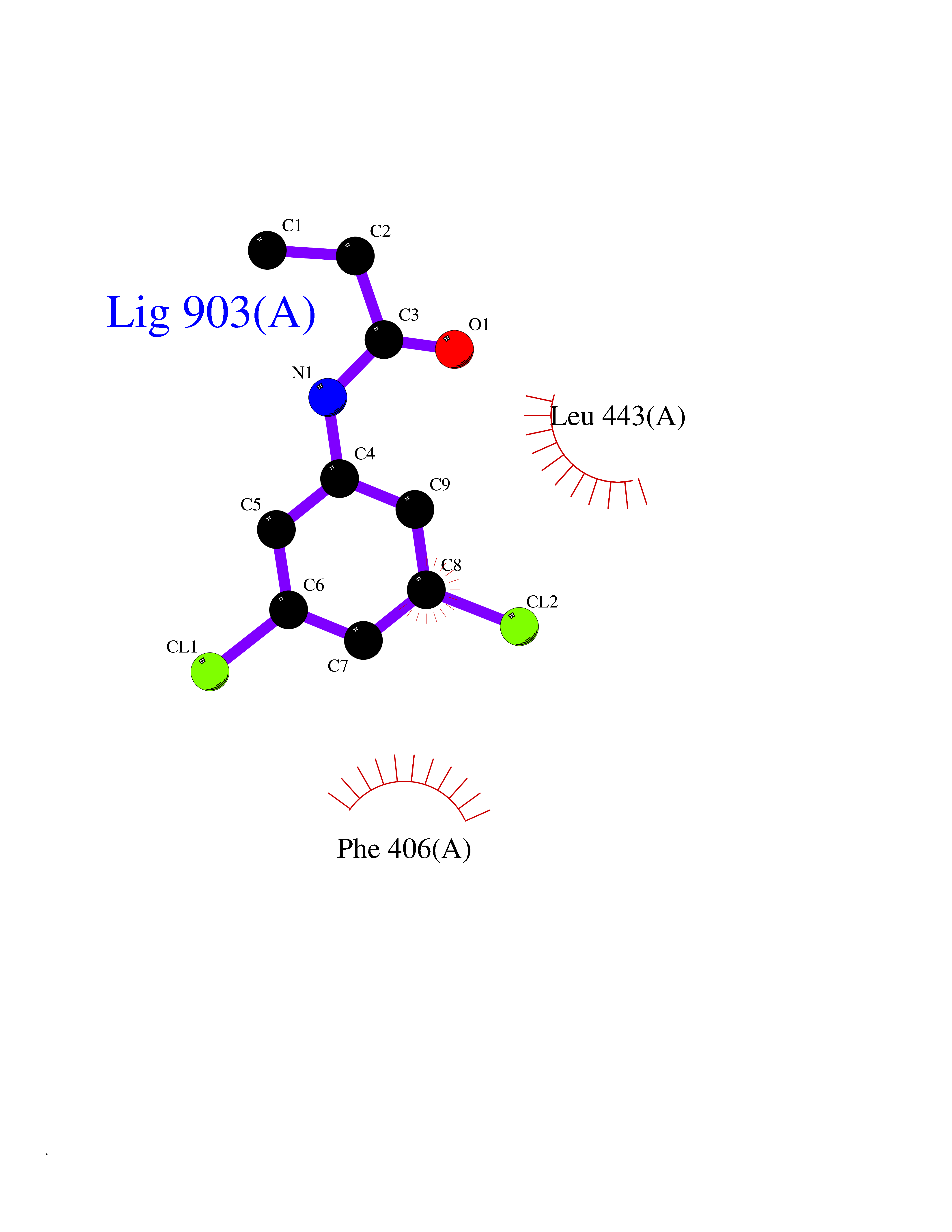 Ligplot Map 3D Binding mode Sequence SRARQVELLLVADASMARKYGRGLQHYLLTLASIANRLYSHASIENHIRLAVVKVVVLGDKDKSLEVSKNAATTLKNFCKWQHQHNQLGDDHEEHYDAAILFTREDLCGHHSCDTLGMADVGTICSPERSCAVIEDDGLHAAFTVAHEIGHLLGLSHDDSKFCEETFGSTEDKRLMSSILTSIDASKPWSKCTSATITEFLDDGHGNCLLDLPRKQI Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | SEC14-like protein 2 | 4OMJ | 5.88 | |
Target general information Gen name SEC14L2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms KIAA1658;C22orf6;KIAA1186 Protein family NA Biochemical class Transport protein Function Phospholipid binding.Transporter activity.Vitamin E binding. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB14001; DB14002; DB11635; DB11251; DB00163 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Cytoplasm; Lipid-binding; Nucleus; Proteomics identification; Reference proteome; Transcription; Transcription regulation; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 31533.3 Length 274 Aromaticity 0.1 Instability index 49.26 Isoelectric point 8.34 Charge (pH=7) 2.81 2D Binding mode Binding energy (Kcal/mol) -8.03 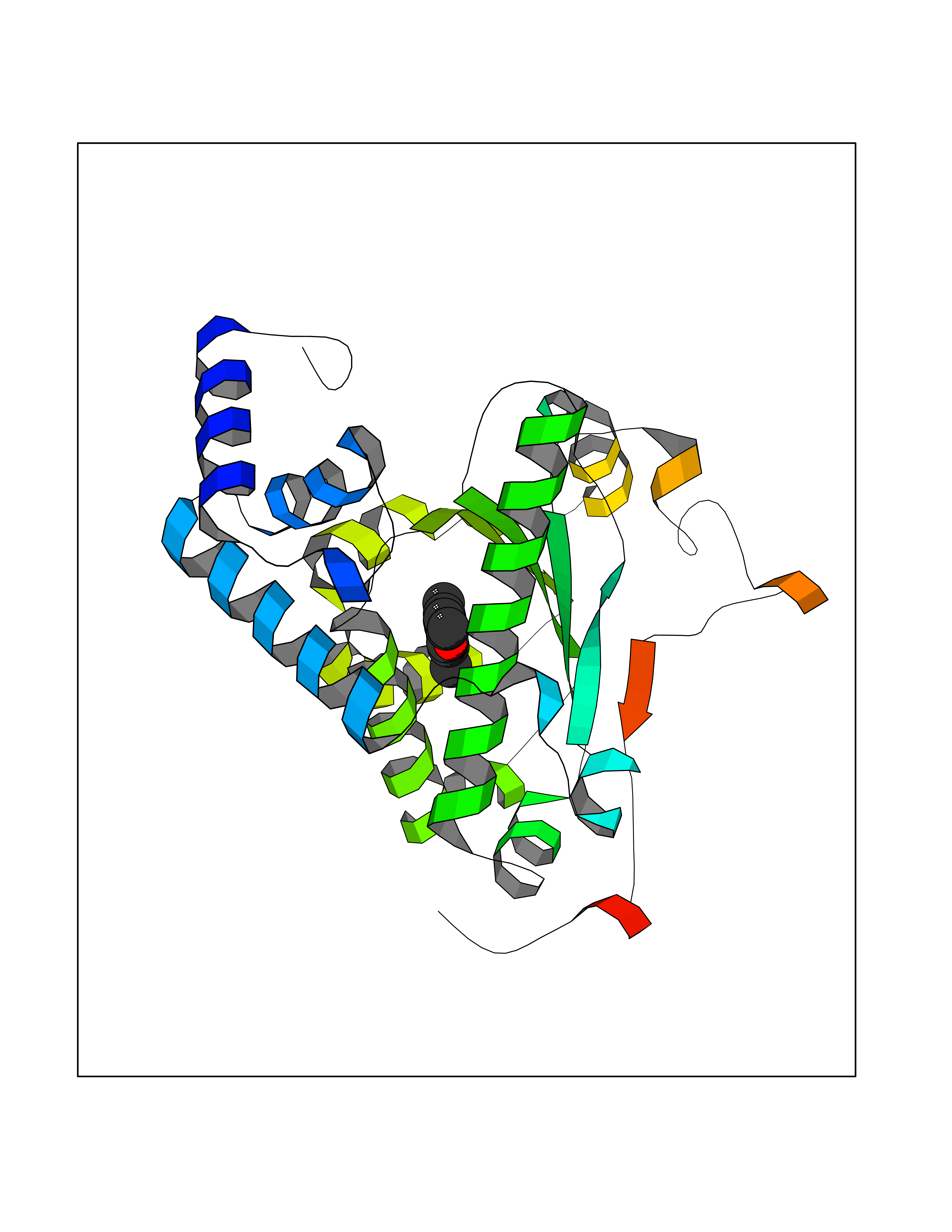 Molscript Map 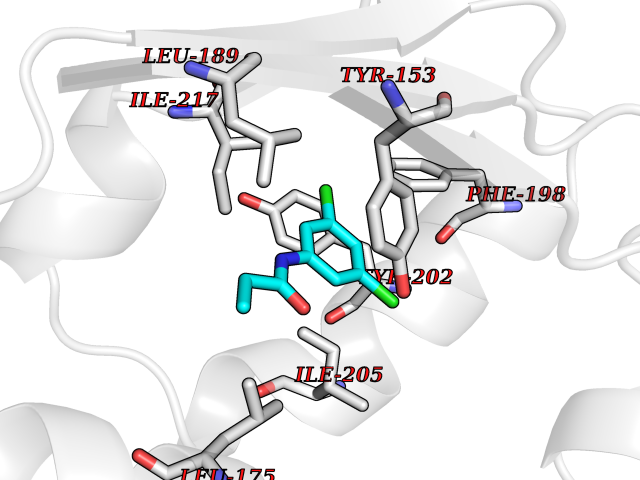 Pymol Map 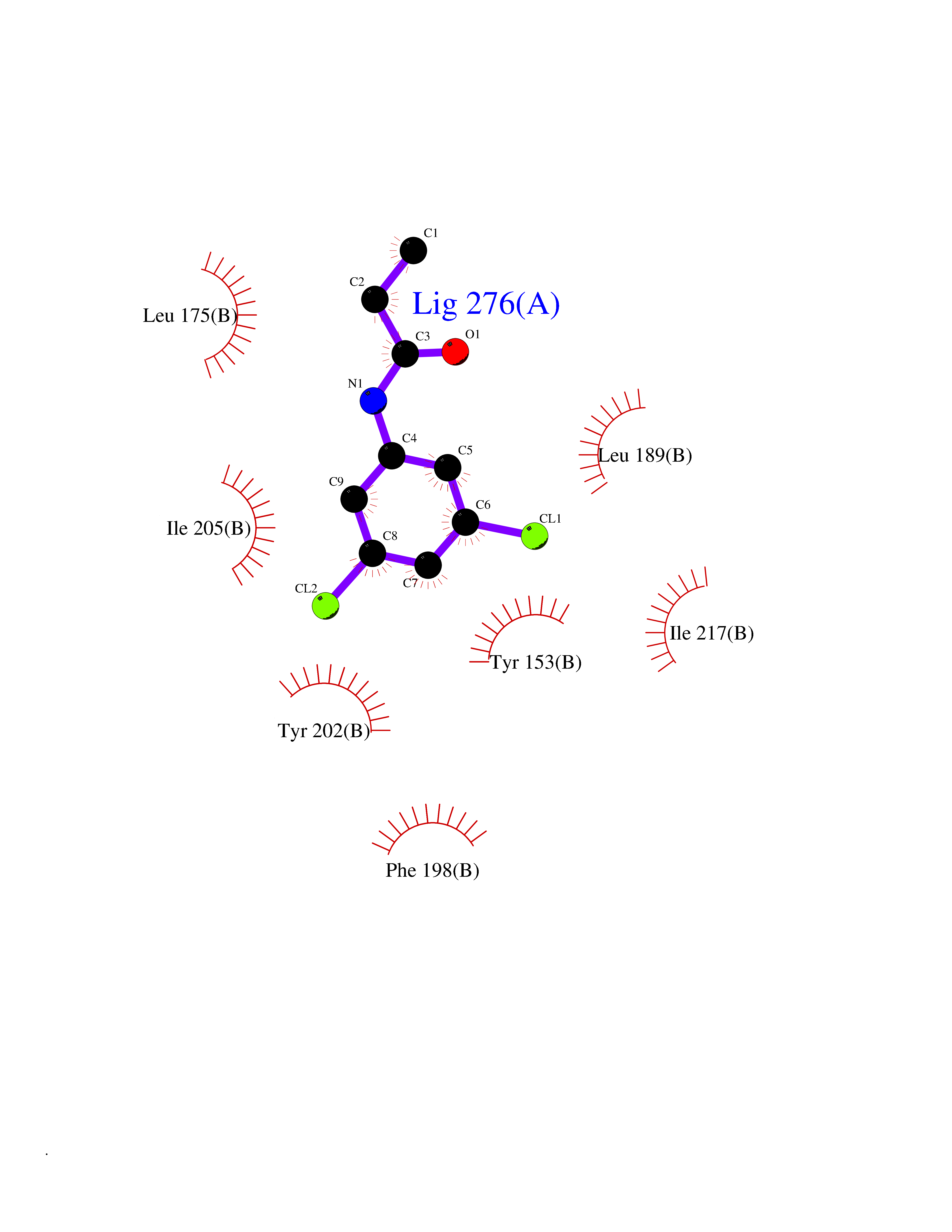 Ligplot Map 3D Binding mode Sequence MSGRVGDLSPRQKEALAKFRENVQDVLPALPNPDDYFLLRWLRARSFDLQKSEAMLRKHVEFRKQKDIDNIISWQPPEVIQQYLSGGMCGYDLDGCPVWYDIIGPLDAKGLLFSASKQDLLRTKMRECELLLQECAHQTTKLGRKVETITIIYDCEGLGLKHLWKPAVEAYGEFLCMFEENYPETLKRLFVVKAPKLFPVAYNLIKPFLSEDTRKKIMVLGANWKEVLLKHISPDQVPVEYGGTMTDPDGNPKCKSKINYGGDIPRKYYVRDQV Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Dual-specificity tyrosine-phosphorylation regulated kinase 2 (DYRK2) | 6HDR | 5.88 | |
Target general information Gen name DYRK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dual specificity tyrosine-phosphorylation-regulated kinase 2 Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, MNB/DYRK subfamily Biochemical class Kinase Function Functions in part via its role in ubiquitin-dependent proteasomal protein degradation. Functions downstream of ATM and phosphorylates p53/TP53 at 'Ser-46', and thereby contributes to the induction of apoptosis in response to DNA damage. Phosphorylates NFATC1, and thereby inhibits its accumulation in the nucleus and its transcription factor activity. Phosphorylates EIF2B5 at 'Ser-544', enabling its subsequent phosphorylation and inhibition by GSK3B. Likewise, phosphorylation of NFATC1, CRMP2/DPYSL2 and CRMP4/DPYSL3 promotes their subsequent phosphorylation by GSK3B. May play a general role in the priming of GSK3 substrates. Inactivates GYS1 by phosphorylation at 'Ser-641', and potentially also a second phosphorylation site, thus regulating glycogen synthesis. Mediates EDVP E3 ligase complex formation and is required for the phosphorylation and subsequent degradation of KATNA1. Phosphorylates TERT at 'Ser-457', promoting TERT ubiquitination by the EDVP complex. Phosphorylates SIAH2, and thereby increases its ubiquitin ligase activity. Promotes the proteasomal degradation of MYC and JUN, and thereby regulates progress through the mitotic cell cycle and cell proliferation. Promotes proteasomal degradation of GLI2 and GLI3, and thereby plays a role in smoothened and sonic hedgehog signaling. Plays a role in cytoskeleton organization and neurite outgrowth via its phosphorylation of DCX and DPYSL2. Phosphorylates CRMP2/DPYSL2, CRMP4/DPYSL3, DCX, EIF2B5, EIF4EBP1, GLI2, GLI3, GYS1, JUN, MDM2, MYC, NFATC1, p53/TP53, TAU/MAPT and KATNA1. Can phosphorylate histone H1, histone H3 and histone H2B (in vitro). Can phosphorylate CARHSP1 (in vitro). Serine/threonine-protein kinase involved in the regulation of the mitotic cell cycle, cell proliferation, apoptosis, organization of the cytoskeleton and neurite outgrowth. Related diseases Bone marrow failure and diabetes mellitus syndrome (BMFDMS) [MIM:620044]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFDMS is an autosomal recessive form characterized by various degrees of bone marrow failure, ranging from dyserythropoiesis to bone marrow aplasia, with onset in infancy or early childhood, and non-autoimmune insulin-dependent diabetes mellitus appearing in the first or second decades. Many patients show pigmentary skin abnormalities and short stature. {ECO:0000269|PubMed:28073829, ECO:0000269|PubMed:35611808, ECO:0000269|PubMed:35931051}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NR20; Q13422; Q9BQD3; Q9BRK4; P23497; O43379; P62258; Q96C00 EC number EC 2.7.12.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cytoplasm; Kinase; Magnesium; Manganese; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID A Molecular weight (Da) 46422.1 Length 407 Aromaticity 0.09 Instability index 44.91 Isoelectric point 9.09 Charge (pH=7) 12.37 2D Binding mode Binding energy (Kcal/mol) -8.02 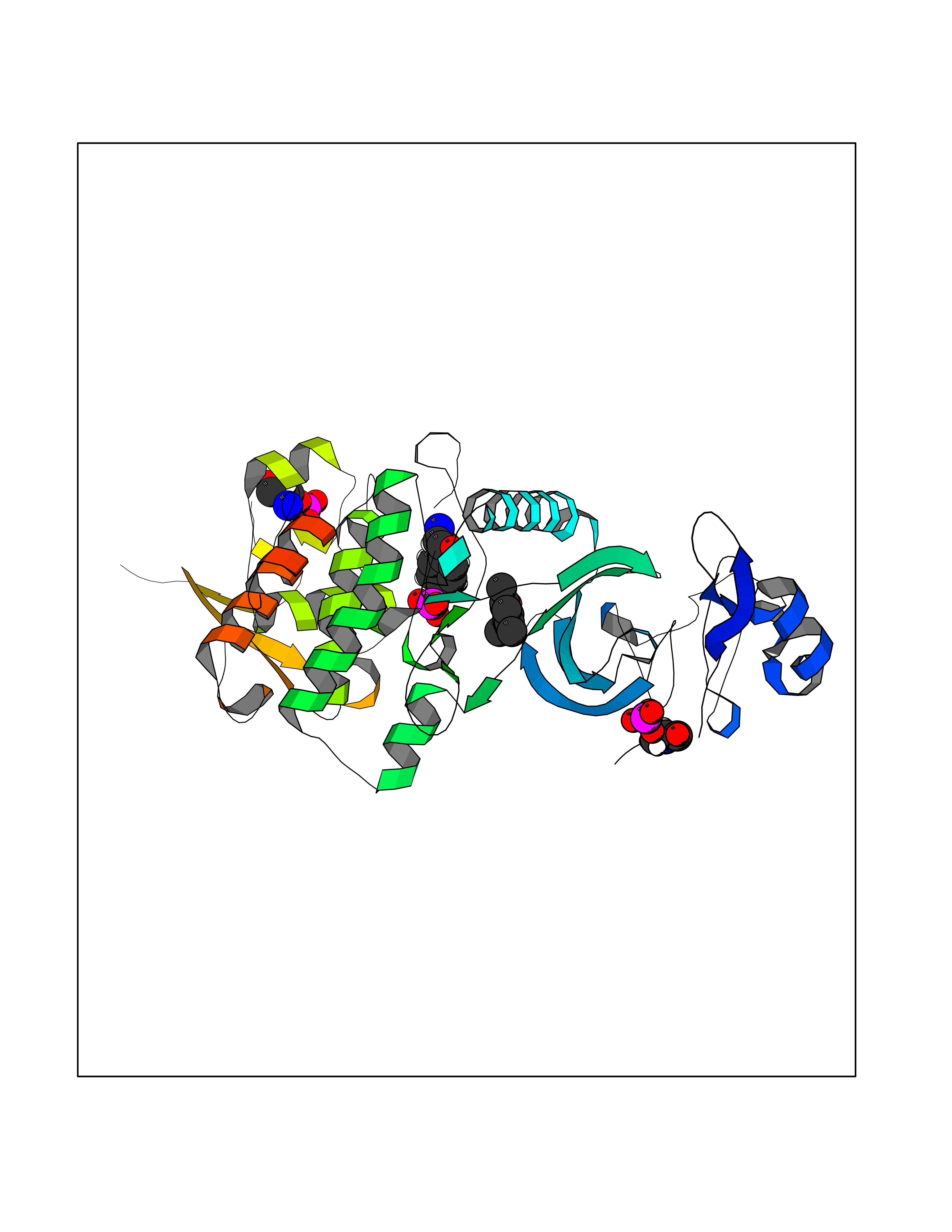 Molscript Map 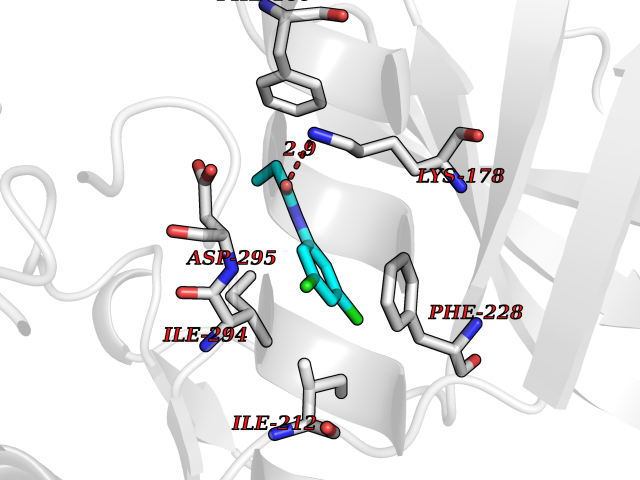 Pymol Map 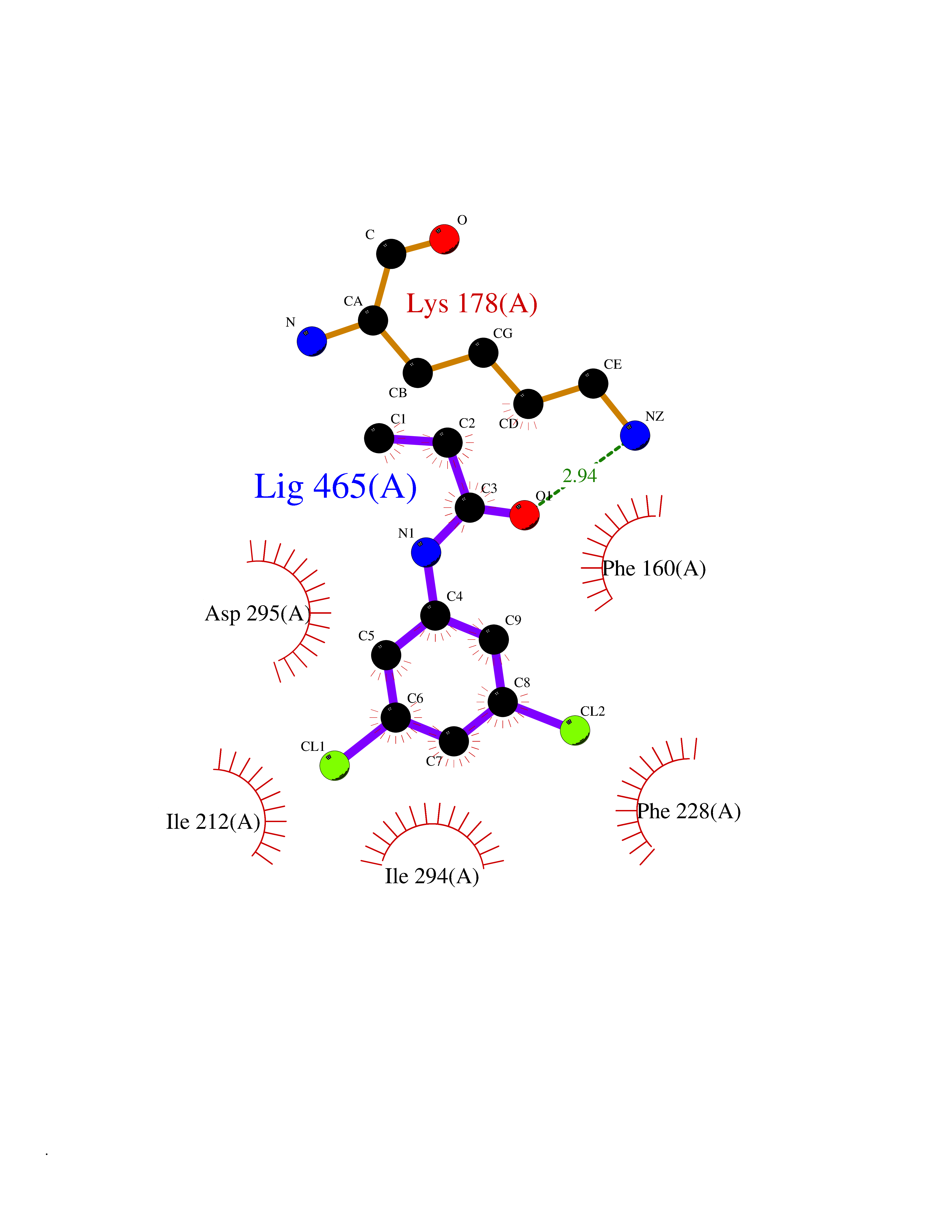 Ligplot Map 3D Binding mode Sequence HHHSXGVDLGTENLYFQSMGKVKATPMTPEQAMKQYMQKLTAFEHHEIFSYPEIYFLGLNAKKRQGMTGGPNNGGYDDDQGSYVQVPHDHVAYRYEVLKVIGKGSFGQVVKAYDHKVHQHVALKMVRNEKRFHRQAAEEIRILEHLRKQDKDNTMNVIHMLENFTFRNHICMTFELLSMNLYELIKKNKFQGFSLPLVRKFAHSILQCLDALHKNRIIHCDLKPENILLKQQGRSGIKVIDFGSSCYEHQRVYTXIQSRFYRAPEVILGARYGMPIDMWSLGCILAELLTGYPLLPGEDEGDQLACMIELLGMPSQKLLDASKRAKNFVSXKGYPRYCTVTTLSDVVLNGGRSRRGKLRGPPESREWGNALKGCDDPLFLDFLKQCLEWDPAVRMTPGQALRHPWLR Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Sphingosine-1-phosphate receptor 2 (S1PR2) | 7T6B | 5.88 | |
Target general information Gen name S1PR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Sphingosine 1-phosphate receptor Edg-5; S1PR2; S1P2; S1P receptor Edg-5; S1P receptor 2; Endothelial differentiation G-protein coupled receptor 5 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for the lysosphingolipid sphingosine 1- phosphate (S1P). S1P is a bioactive lysophospholipid that elicits diverse physiological effect on most types of cells and tissues. When expressed in rat HTC4 hepatoma cells, is capable of mediating S1P-induced cell proliferation and suppression of apoptosis. Related diseases Deafness, autosomal recessive, 68 (DFNB68) [MIM:610419]: A form of non-syndromic sensorineural hearing loss. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. {ECO:0000269|PubMed:26805784}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P16144; Q9JK11-1 EC number NA Uniprot keywords 3D-structure; Cell membrane; Deafness; Disease variant; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Non-syndromic deafness; Palmitate; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 28917.3 Length 264 Aromaticity 0.11 Instability index 38.95 Isoelectric point 9.11 Charge (pH=7) 9.27 2D Binding mode Binding energy (Kcal/mol) -8.02 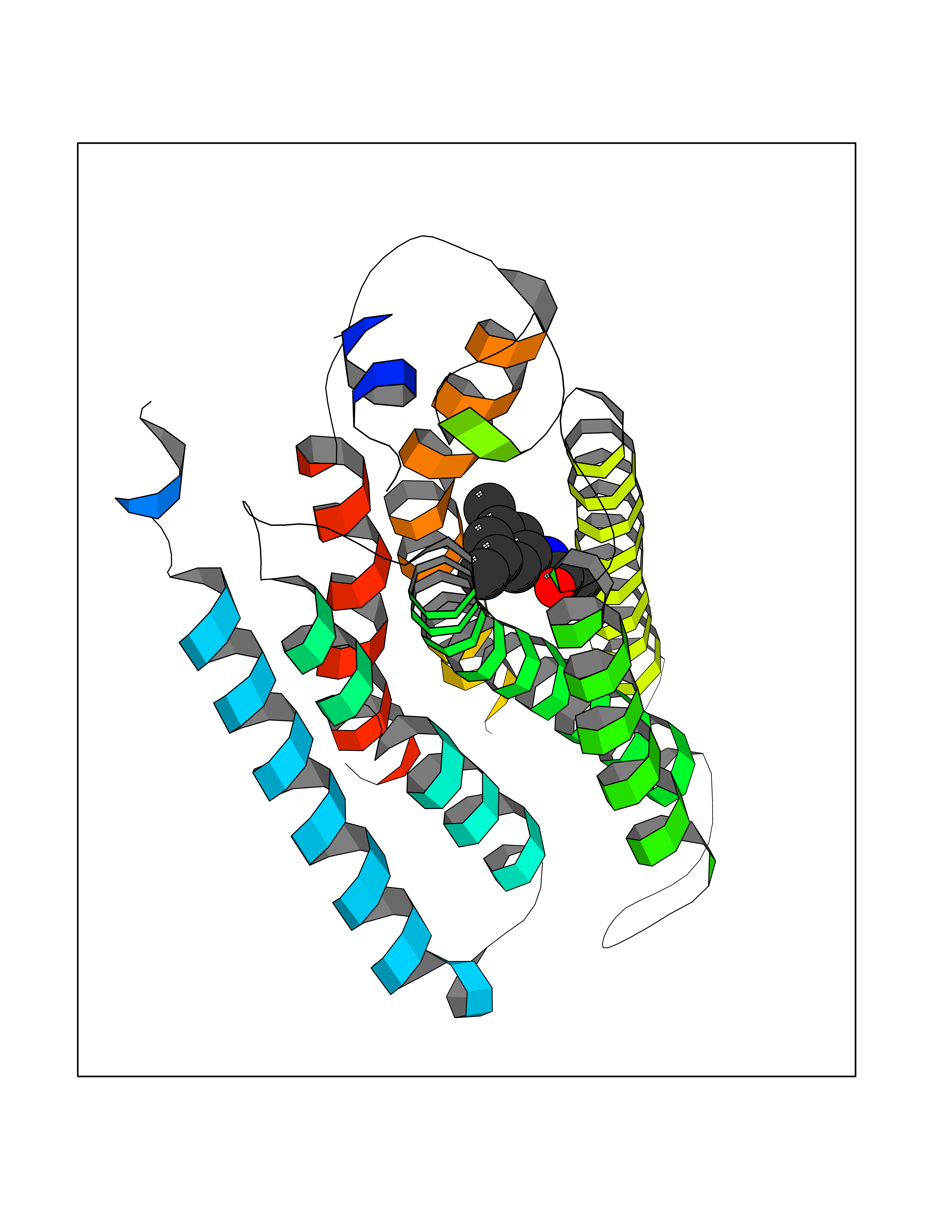 Molscript Map 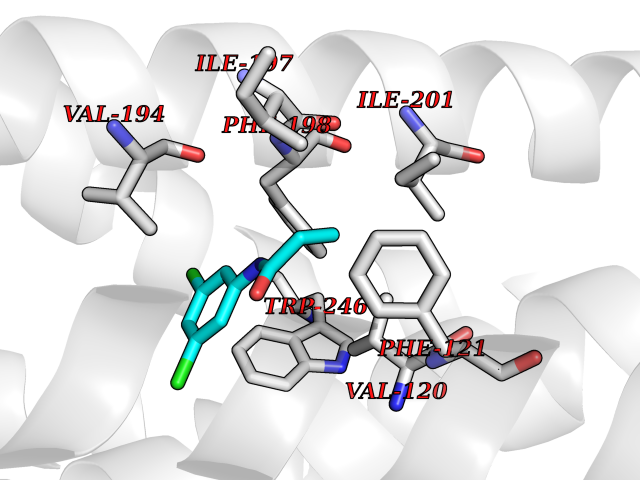 Pymol Map 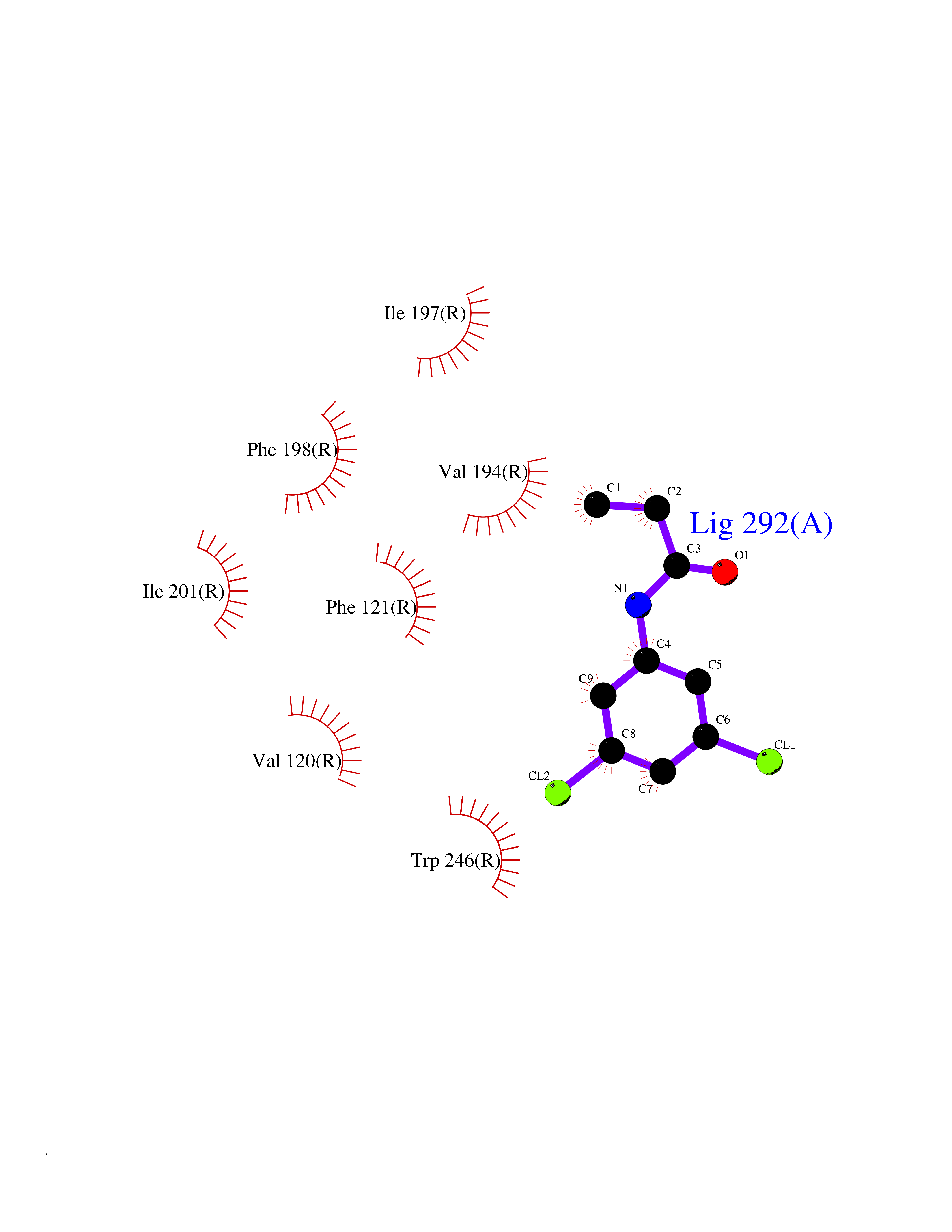 Ligplot Map 3D Binding mode Sequence NKVQEHYNYTKTSRQVASAFIVILCCAIVVENLLVLIAVARNSKFHSAMYLFLGNLAASDLLAGVAFVANTLLSGSVTLRLTPVQWFAREGSAFITLSASVFSLLAIAIERHVAIAKVKLYGSDKSCRMLLLIGASWLISLVLGGLPILGWNCLGHLEACSTVLPLYAKHYVLCVVTIFSIILLAIVALYVRIYCVVRSSQTLALLKTVTIVLGVFIVCWLPAFSILLLDYACPVHSCPILYKAHYFFAVSTLNSLLNPVIYTW Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Glutamate carboxypeptidase III (NAALAD2) | 3FED | 5.88 | |
Target general information Gen name NAALAD2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NAALADase II; NAALAD2 Protein family Peptidase M28 family, M28B subfamily Biochemical class Peptidase Function Has N-acetylated-alpha-linked-acidic dipeptidase (NAALADase) activity. Also exhibits a dipeptidyl-peptidase IV type activity. Inactivate the peptide neurotransmitter N- acetylaspartylglutamate. Related diseases Dystonia 1, torsion, autosomal dominant (DYT1) [MIM:128100]: A primary torsion dystonia, and the most common and severe form. Dystonia is defined by the presence of sustained involuntary muscle contractions, often leading to abnormal postures. Dystonia type 1 is characterized by involuntary, repetitive, sustained muscle contractions or postures involving one or more sites of the body, in the absence of other neurological symptoms. Typically, symptoms develop first in an arm or leg in middle to late childhood and progress in approximately 30% of patients to other body regions (generalized dystonia) within about five years. 'Torsion' refers to the twisting nature of body movements observed in DYT1, often affecting the trunk. Distribution and severity of symptoms vary widely between affected individuals, ranging from mild focal dystonia to severe generalized dystonia, even within families. {ECO:0000269|PubMed:14970196, ECO:0000269|PubMed:15505207, ECO:0000269|PubMed:16361107, ECO:0000269|PubMed:17428918, ECO:0000269|PubMed:18167355, ECO:0000269|PubMed:18477710, ECO:0000269|PubMed:18827015, ECO:0000269|PubMed:19955557, ECO:0000269|PubMed:20169475, ECO:0000269|PubMed:21102408, ECO:0000269|PubMed:24930953, ECO:0000269|PubMed:27490483, ECO:0000269|PubMed:9288096}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Arthrogryposis multiplex congenita 5 (AMC5) [MIM:618947]: A form of arthrogryposis multiplex congenita, a developmental condition characterized by multiple joint contractures resulting from reduced or absent fetal movements. AMC5 is an autosomal recessive form characterized by severe congenital contractures, developmental delay, strabismus and tremor. {ECO:0000269|PubMed:28516161, ECO:0000269|PubMed:29053766, ECO:0000269|PubMed:30244176}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9UHD4; Q6NTF9-3; B2RUZ4; O76024 EC number EC 3.4.17.21 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Carboxypeptidase; Cell membrane; Dipeptidase; Glycoprotein; Hydrolase; Membrane; Metal-binding; Metalloprotease; Multifunctional enzyme; Protease; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 77761.6 Length 690 Aromaticity 0.12 Instability index 39.42 Isoelectric point 8.48 Charge (pH=7) 4.65 2D Binding mode Binding energy (Kcal/mol) -8.02 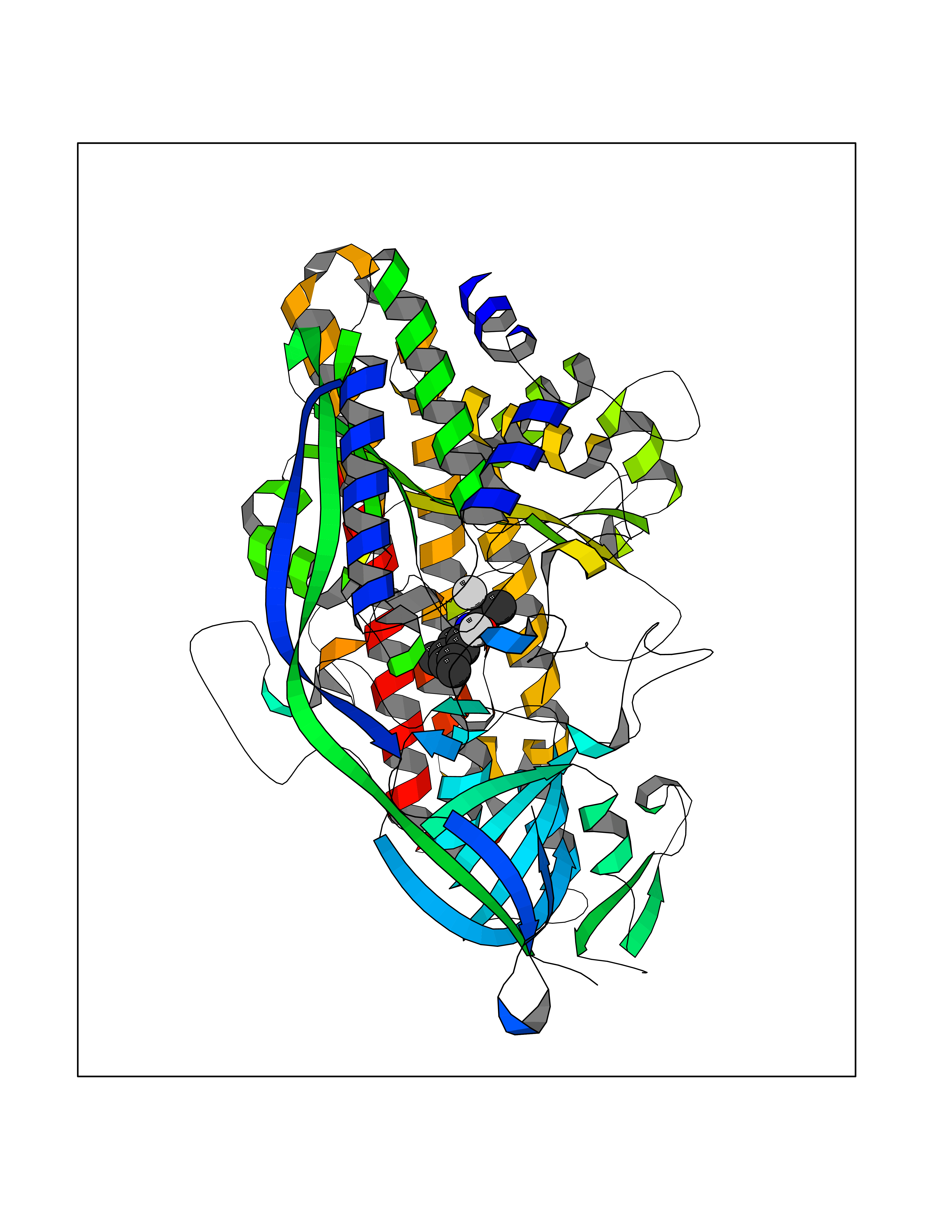 Molscript Map 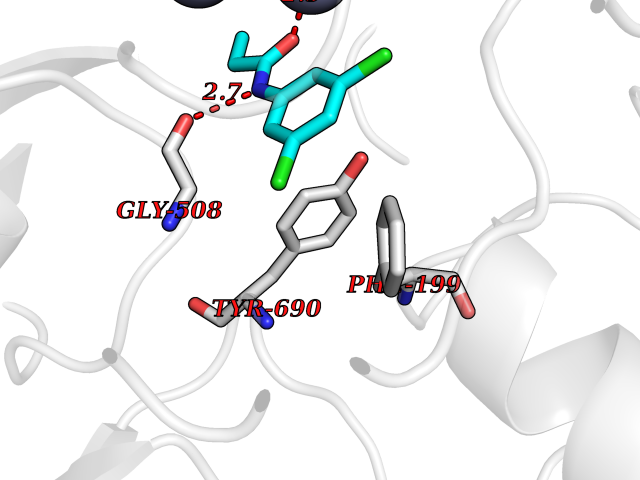 Pymol Map 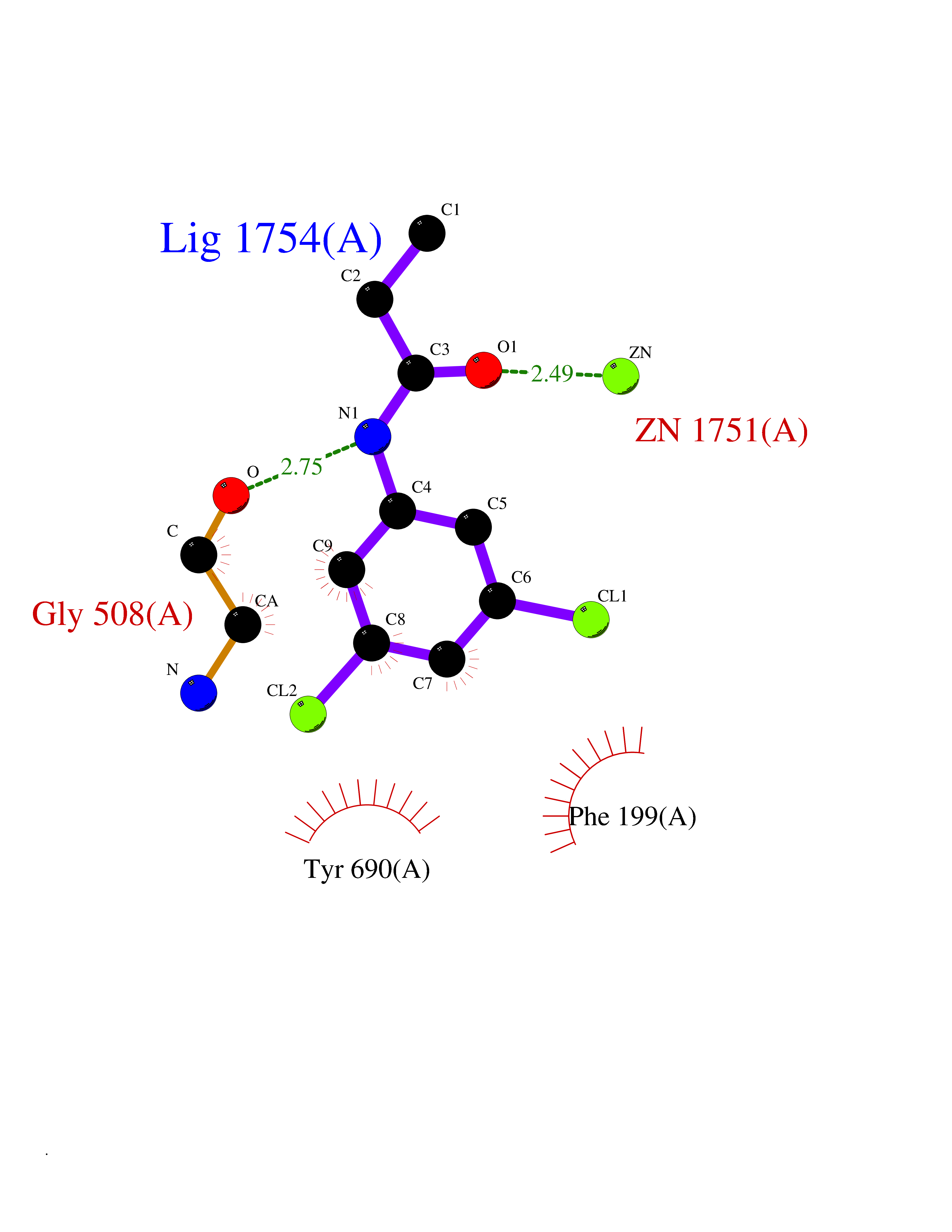 Ligplot Map 3D Binding mode Sequence SIRWKLVSEMKAENIKSFLRSFTKLPHLAGTEQNFLLAKKIQTQWKKFGLDSAKLVHYDVLLSYPNETNANYISIVDEHETEIFKTSPPPDGYENVTNIVPPYNAFSAQGMPEGDLVYVNYARTEDFFKLEREMGINCTGKIVIARYGKIFRGNKVKNAMLAGAIGIILYSDPADYFAPEVQPYPKGWNLPGTAAQRGNVLNLNGAGDPLTPGYPAKEYTFRLDVEEGVGIPRIPVHPIGYNDAEILLRYLGGIAPPDKSWKGALNVSYSIGPGFTGSSFRKVRMHVYNINKITRIYNVVGTIRGSVEPDRYVILGGHRDSWVFGAIDPTSGVAVLQEIARSFGKLMSKGWRPRRTIIFASWDAEEFGLLGSTEWAEENVKILQERSIAYINSDSSIEGNYTLRVDCTPLLYQLVYKLTKEIPSPDDGFESKSLYESWLEKDPSPENKNLPRINKLGSGSDFEAYFQRLGIASGRARYTKNKKTDKYSSYPVYHTIYETFELVEKFYDPTFKKQLSVAQLRGALVYELVDSKIIPFNIQDYAEALKNYAASIYNLSKKHDQQLTDHGVSFDSLFSAVKNFSEAASDFHKRLIQVDLNNPIAVRMMNDQLMLLERAFIDPLGLPGKLFYRHIIFAPSSHNKYAGESFPGIYDAIFDIENKANSRLAWKEVKKHISIAAFTIQAAAGTLKEV Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Zinc finger protein Helios (IKZF2) | 7LPS | 5.88 | |
Target general information Gen name IKZF2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ikaros family zinc finger protein 2 Protein family Ikaros C2H2-type zinc-finger protein family Biochemical class NA Function Associates with Ikaros at centromeric heterochromatin. Related diseases Developmental and epileptic encephalopathy 25, with amelogenesis imperfecta (DEE25) [MIM:615905]: An autosomal recessive disease characterized by subclinical seizures appearing in the first days of life, evolving to severe epileptic disease. Affected individuals have profound or severe delayed development with lack of speech, and most patients do not acquire the ability to sit. Additional variable features include axial hypotonia, peripheral hypertonia, and abnormal involuntary movements such as dystonia and choreoathetosis. Dental abnormalities, including delayed eruption, hypodontia, tooth hypoplasia, yellow discoloration, thin enamel, and enamel chipping are observed in most patients. {ECO:0000269|PubMed:24995870, ECO:0000269|PubMed:26384929, ECO:0000269|PubMed:30054523}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P29972; P56545; P56545-3; Q17RB8; P09022; Q8N8B7-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID B,C Molecular weight (Da) 47006.6 Length 410 Aromaticity 0.09 Instability index 44.28 Isoelectric point 7.23 Charge (pH=7) 0.69 2D Binding mode Binding energy (Kcal/mol) -8.02 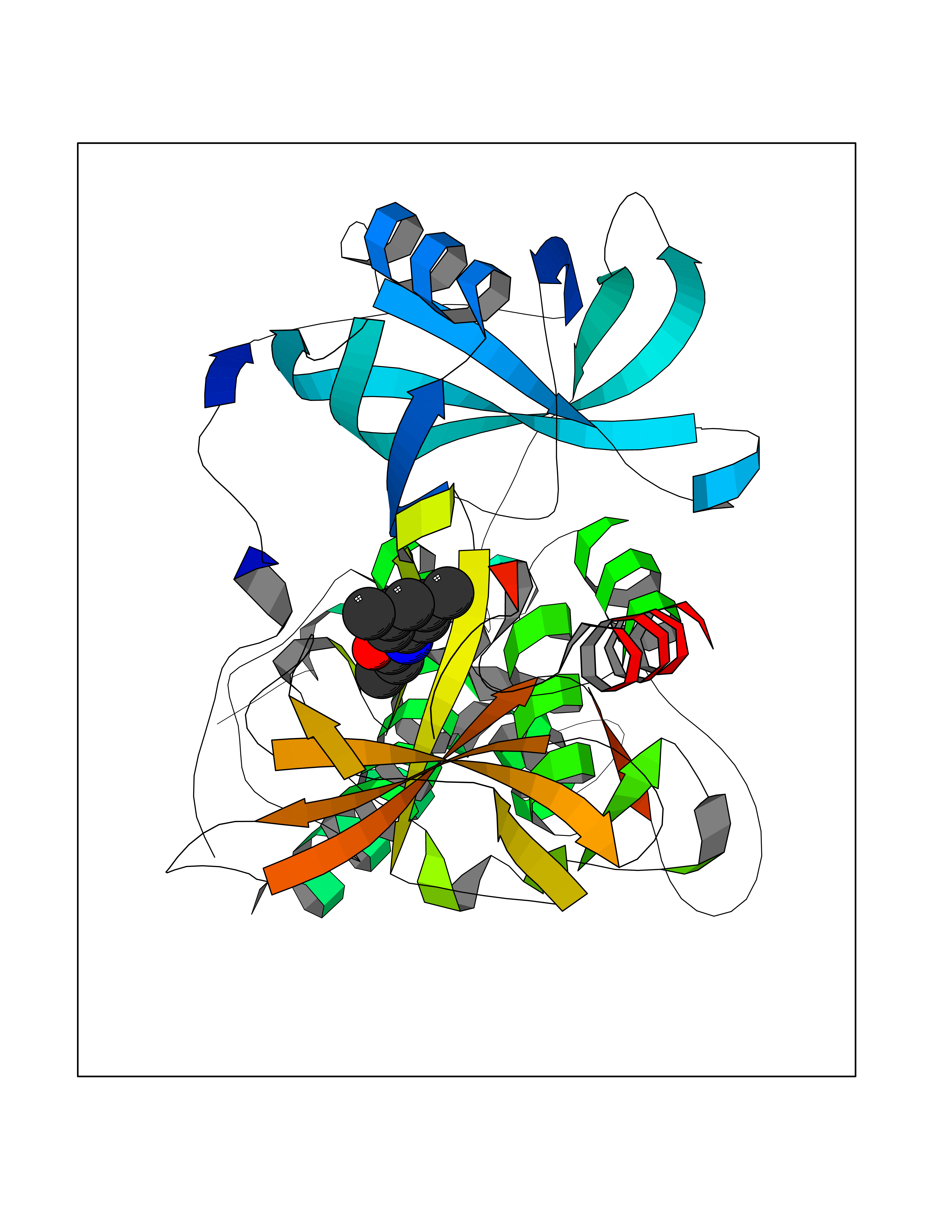 Molscript Map 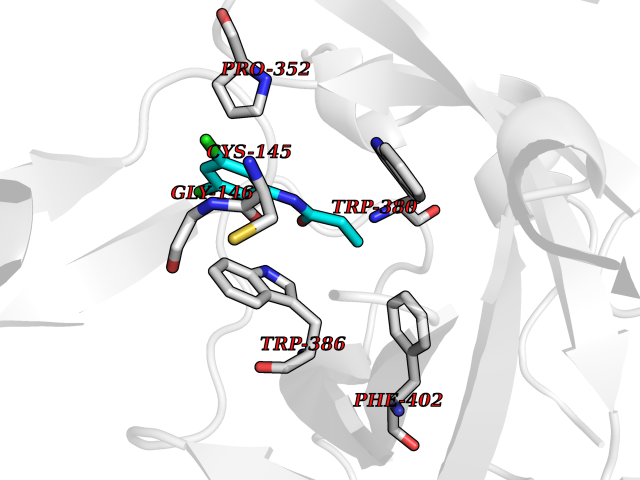 Pymol Map 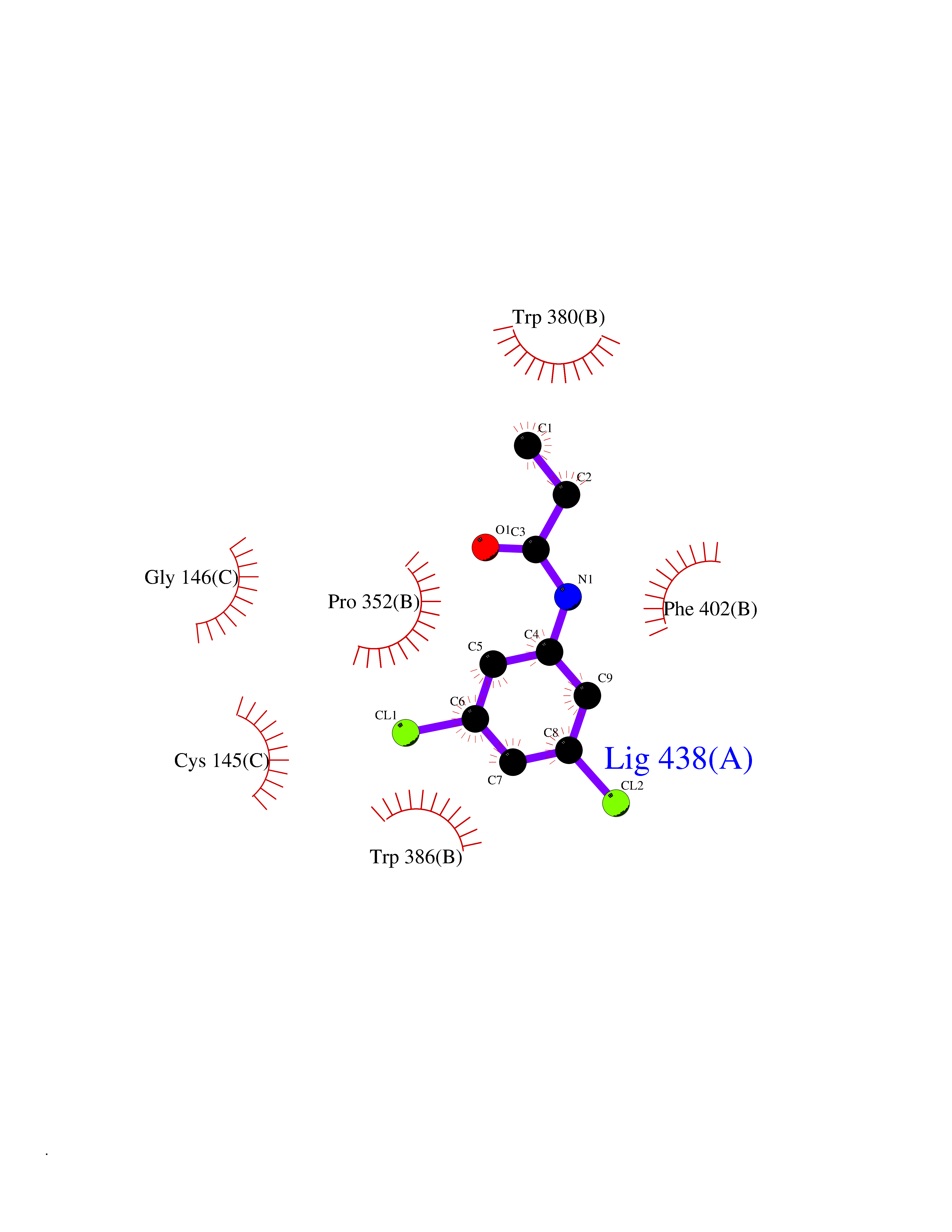 Ligplot Map 3D Binding mode Sequence INFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEQDFGIEIVKVKAIGRQRFKVLELRTQSDGIQQAKVQILPECVLPSTMSAVQLESLNKCQIFPCSYKWWQKYQKRKFHCANLTSWPRWLYSLYDAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPDGERPFHCNQCGASFTQKGNLLRHIKLHSG Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Angiopoietin-related protein 4 (ANGPTL4) | 6U73 | 5.88 | |
Target general information Gen name ANGPTL4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UNQ171/PRO197; PSEC0166; PP1158; PGAR; Hepatic fibrinogen/angiopoietin-related protein; HFARP; Angiopoietin-like protein PP1158; Angiopoietin-like protein 4; Angiopoietin-like 4; ARP4 Protein family NA Biochemical class Fibrinogen protein Function May also play a role in regulating glucose homeostasis and insulin sensitivity. Inhibits proliferation, migration, and tubule formation of endothelial cells and reduces vascular leakage. Upon heterologous expression, inhibits the adhesion of endothelial cell to the extracellular matrix (ECM), and inhibits the reorganization of the actin cytoskeleton, formation of actin stress fibers and focal adhesions in endothelial cells that have adhered to ANGPTL4-containing ECM (in vitro). Depending on context, may modulate tumor-related angiogenesis. Mediates inactivation of the lipoprotein lipase LPL, and thereby plays a role in the regulation of triglyceride clearance from the blood serum and in lipid metabolism. Related diseases Mucopolysaccharidosis 1H (MPS1H) [MIM:607014]: A severe form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1H usually present, within the first year of life, a combination of hepatosplenomegaly, skeletal deformities, corneal clouding and severe intellectual disability. Obstructive airways disease, respiratory infection and cardiac complications usually result in death before 10 years of age. {ECO:0000269|PubMed:10466419, ECO:0000269|PubMed:10735634, ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:1301941, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:19396826, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:24036510, ECO:0000269|PubMed:31194252, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:7951228, ECO:0000269|PubMed:8019563, ECO:0000269|PubMed:8328452, ECO:0000269|PubMed:8401515, ECO:0000269|Ref.20}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 1H/S (MPS1H/S) [MIM:607015]: A form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. MPS1H/S represents an intermediate phenotype of the MPS1 clinical spectrum. It is characterized by relatively little neurological involvement, but most of the somatic symptoms described for severe MPS1 develop in the early to mid-teens, causing considerable loss of mobility. {ECO:0000269|PubMed:10466419, ECO:0000269|PubMed:10735634, ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:8401515}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 1S (MPS1S) [MIM:607016]: A mild form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1S may have little or no neurological involvement, normal stature and life span, but present development of joints stiffness, mild hepatosplenomegaly, aortic valve disease and corneal clouding. {ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:19396826, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:25256405, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:8213840}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9BY76; P05556; P18084 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Angiogenesis; Coiled coil; Direct protein sequencing; Disulfide bond; Extracellular matrix; Glycoprotein; Lipid metabolism; Proteomics identification; Reference proteome; Secreted; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 24429.1 Length 216 Aromaticity 0.12 Instability index 40.6 Isoelectric point 8.51 Charge (pH=7) 2.46 2D Binding mode Binding energy (Kcal/mol) -8.02 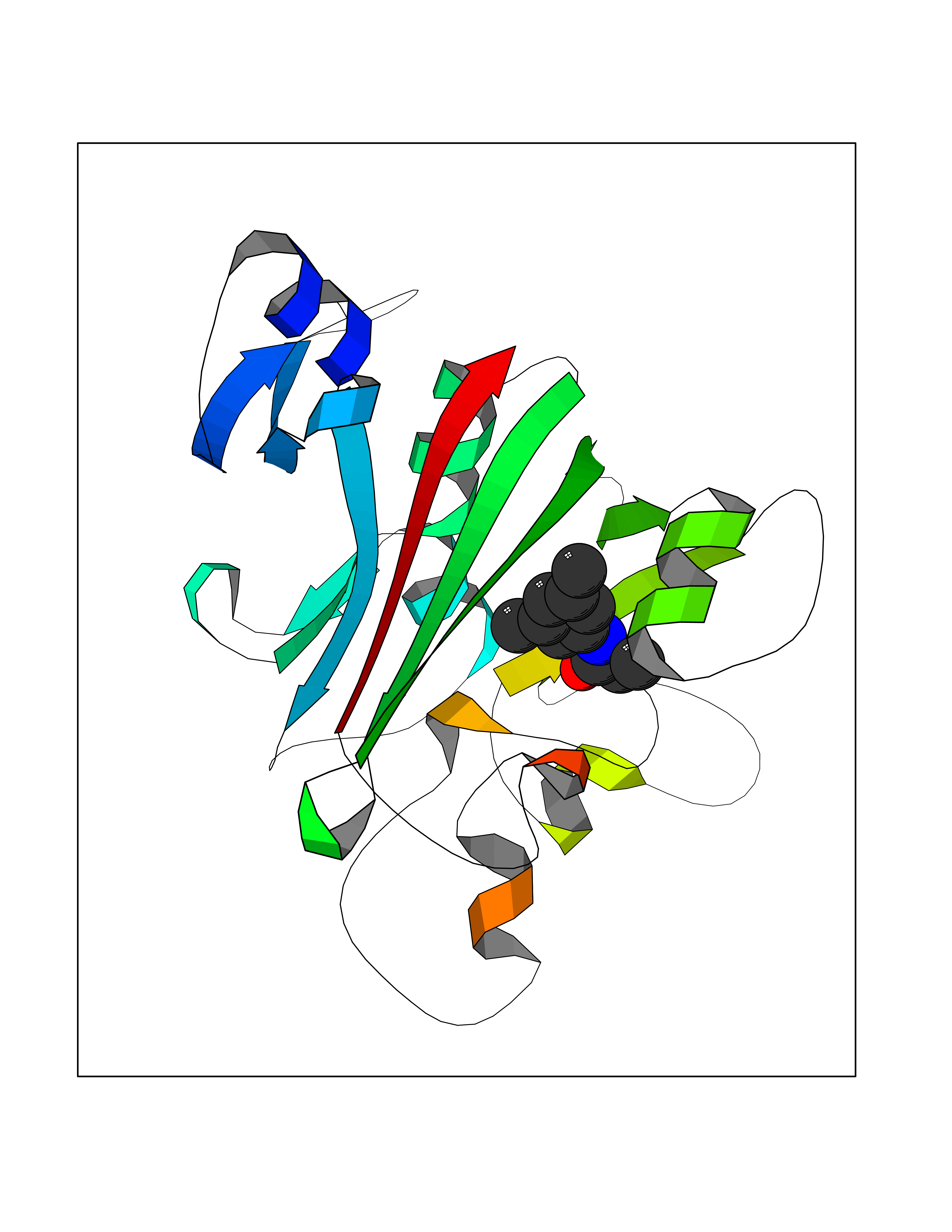 Molscript Map  Pymol Map 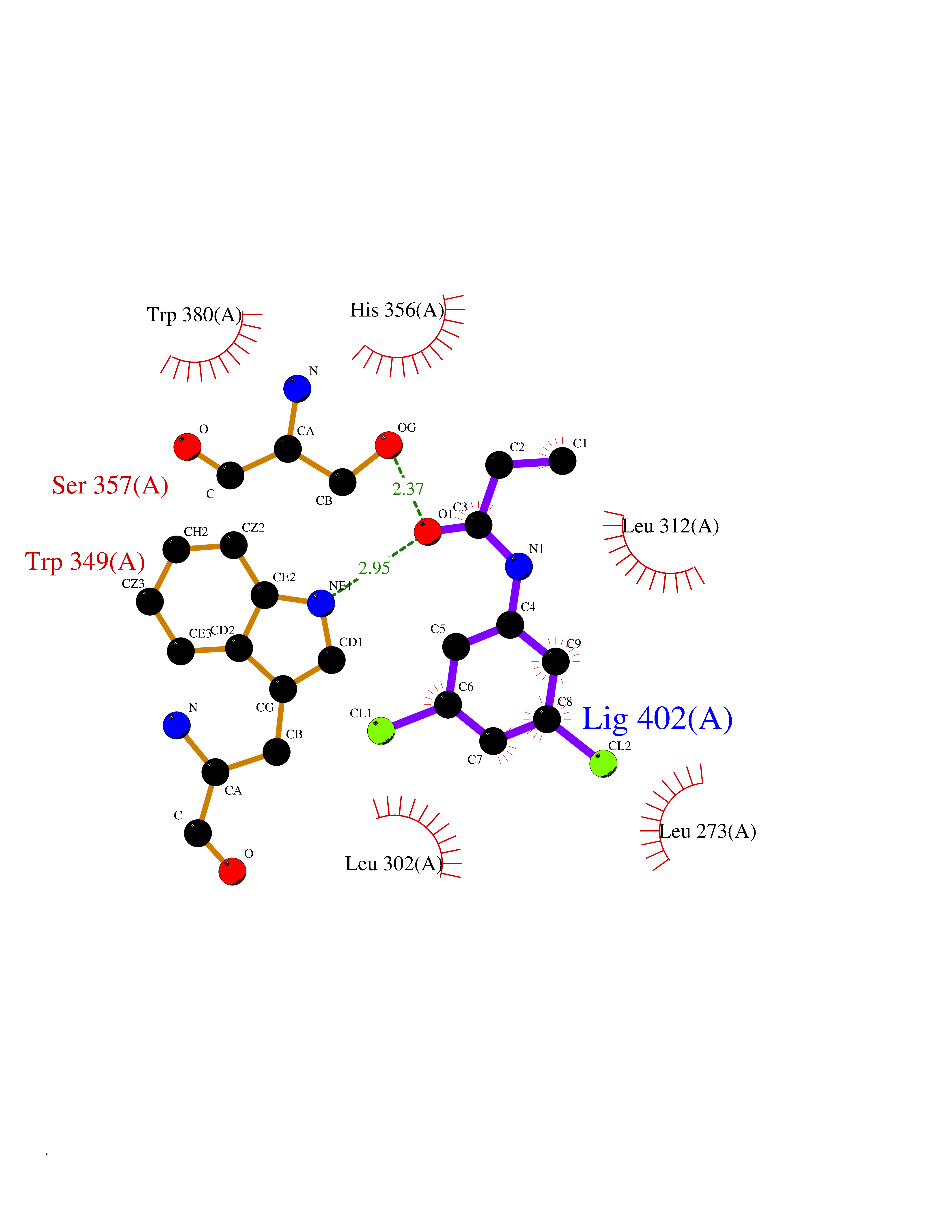 Ligplot Map 3D Binding mode Sequence PRDCQELFQVGERQSGLFEIQPQGSPPFLVNCKMTSDGGWTVIQRRHDGSVDFNRPWEAYKAGFGDPHGEFWLGLEKVHSITGDRNSRLAVQLRDWDGNAELLQFSVHLGGEDTAYSLQLTAPVAGQLGATTVPPSGLSVPFSTWDQDHDLRRDKNCAKSLSGGWWFGTCSHSNLNGQYFRSIPQQRQKLKKGIFWKTWRGRYYPLQATTMLIQPM Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | UDP-N-acetylglucosamine 1-carboxyvinyltransferase | 1C3R | 5.87 | |
Target general information Gen name murA Organism Aquifex aeolicus (strain VF5) Uniprot ID TTD ID NA Synonyms aq_1023 Protein family Histone deacetylase family Biochemical class Lyase Function UDP-N-acetylglucosamine 1-carboxyvinyltransferase activity. Related diseases Atrial fibrillation, familial, 14 (ATFB14) [MIM:615378]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:19808477}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Genetic variations in SCN2B may be involved in Brugada syndrome (PubMed:23559163). This tachyarrhythmia is characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:23559163}. Drugs (DrugBank ID) DB04297; DB02546 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetoin catabolism; Metal-binding; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42338.9 Length 372 Aromaticity 0.12 Instability index 35.74 Isoelectric point 5.77 Charge (pH=7) -6.5 2D Binding mode Binding energy (Kcal/mol) -8.01  Molscript Map 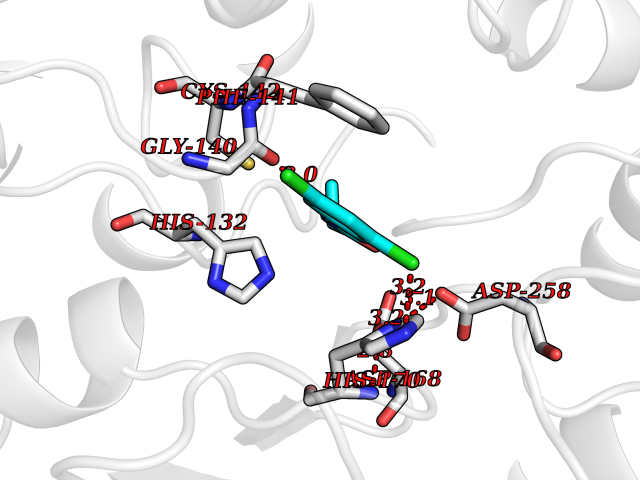 Pymol Map 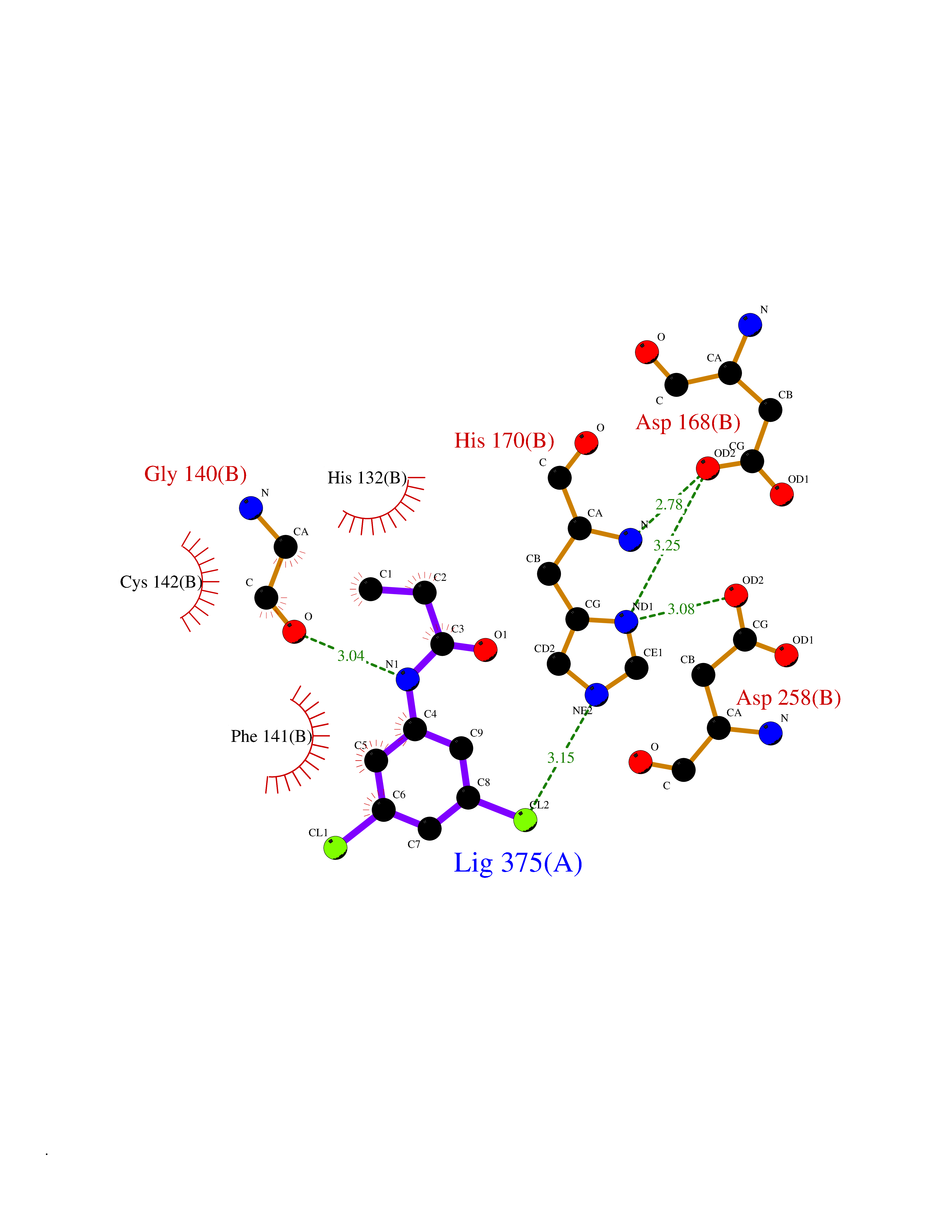 Ligplot Map 3D Binding mode Sequence KKVKLIGTLDYGKYRYPKNHPLKIPRVSLLLRFKDAMNLIDEKELIKSRPATKEELLLFHTEDYINTLMEAERSQSVPKGAREKYNIGGYENPVSYAMFTGSSLATGSTVQAIEEFLKGNVAFNPAGGMHHAFKSRANGFCYINNPAVGIEYLRKKGFKRILYIDLDAHHCDGVQEAFYDTDQVFVLSLHQSPEYAFPFEKGFLEEIGEGKGKGYNLNIPLPKGLNDNEFLFALEKSLEIVKEVFEPEVYLLQLGTDPLLEDYLSKFNLSNVAFLKAFNIVREVFGEGVYLGGGGYHPYALARAWTLIWCELSGREVPEKLNNKAKELLKSIDFEEFDDEVDRSYMLETLKDPWRGGEVRKEVKDTLEKAKA Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Serum albumin (ALB) | 4L8U | 5.87 | |
Target general information Gen name ALB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serum albumin Protein family ALB/AFP/VDB family Biochemical class NA Function Serum albumin, the main protein of plasma, has a good binding capacity for water, Ca(2+), Na(+), K(+), fatty acids, hormones, bilirubin and drugs. Its main function is the regulation of the colloidal osmotic pressure of blood. Major zinc transporter in plasma, typically binds about 80% of all plasma zinc. Related diseases Hyperthyroxinemia, familial dysalbuminemic (FDAH) [MIM:615999]: A disorder characterized by abnormally elevated levels of total serum thyroxine (T4) in euthyroid patients. It is due to abnormal serum albumin that binds T4 with enhanced affinity. {ECO:0000269|PubMed:7852505, ECO:0000269|PubMed:8048949, ECO:0000269|PubMed:9329347, ECO:0000269|PubMed:9589637}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Analbuminemia (ANALBA) [MIM:616000]: A rare autosomal recessive disorder manifested by the presence of a very low amount of circulating serum albumin. Affected individuals manifest mild edema, hypotension, fatigue, and, occasionally, lower body lipodystrophy (mainly in adult females). The most common biochemical finding is hyperlipidemia, with a significant increase in the total and LDL cholesterol concentrations, but normal concentrations of HDL cholesterol and triglycerides. {ECO:0000269|PubMed:8134387}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08496; DB07517; DB12001; DB05812; DB14973; DB11703; DB01418; DB01614; DB00316; DB00414; DB09347; DB06151; DB00459; DB00787; DB00640; DB00802; DB00346; DB00404; DB00770; DB01370; DB14517; DB14518; DB01118; DB00321; DB01060; DB00415; DB00276; DB06728; DB11901; DB00714; DB04557; DB09229; DB11217; DB00278; DB01238; DB14185; DB09204; DB01169; DB11638; DB09274; DB00126; DB06216; DB01072; DB00335; DB00289; DB01076; DB00995; DB06237; DB07402; DB00993; DB08822; DB08903; DB16703; DB00245; DB01086; DB01053; DB00443; DB14669; DB11967; DB13909; DB01294; DB09223; DB00083; DB09128; DB01222; DB15248; DB00490; DB00237; DB11148; DB06772; DB11751; DB11093; DB11348; DB14481; DB04690; DB01101; DB03600; DB01197; DB01136; DB00456; DB01327; DB14879; DB00274; DB01328; DB01329; DB00493; DB01330; DB00430; DB00438; DB01212; DB06119; DB00567; DB07565; DB08936; DB00878; DB00608; DB00477; DB09093; DB00310; DB00501; DB00568; DB00537; DB00515; DB00349; DB01013; DB00845; DB01242; DB01068; DB00575; DB00758; DB01147; DB00363; DB15534; DB01394; DB00286; DB12483; DB09130; DB01380; DB08865; DB11134; DB06778; DB01176; DB00924; DB00434; DB00847; DB01914; DB06695; DB08912; DB11963; DB04816; DB00080; DB12941; DB01264; DB11943; DB11637; DB01189; DB00304; DB01234; DB14649; DB09213; DB00829; DB01119; DB11397; DB00586; DB00485; DB00266; DB00900; DB00861; DB01396; DB00343; DB08995; DB08930; DB01142; DB00997; DB00254; DB00366; DB04855; DB00476; DB01126; DB01057; DB12243; DB13421; DB00625; DB15444; DB00879; DB00584; DB13874; DB11718; DB00228; DB08899; DB01364; DB00530; DB00303; DB11827; DB12235; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB04574; DB00903; DB00977; DB00749; DB00294; DB01276; DB12466; DB04854; DB01039; DB00573; DB00813; DB00950; DB16165; DB01195; DB00687; DB15690; DB00544; DB00472; DB00712; DB08906; DB00983; DB01320; DB06716; DB11796; DB00695; DB15149; DB00743; DB06705; DB01044; DB00317; DB01241; DB12141; DB11978; DB01120; DB01067; DB01016; DB00986; DB13751; DB04539; DB12836; DB11575; DB11359; DB01159; DB14999; DB00070; DB01275; DB00999; DB00774; DB00327; DB09526; DB01611; DB01005; DB00557; DB13014; DB12471; DB09053; DB01050; DB00159; DB01088; DB00619; DB09262; DB00458; DB00808; DB00328; DB07992; DB09564; DB01307; DB05382; DB04711; DB09333; DB00332; DB16200; DB01029; DB00762; DB06636; DB00753; DB00677; DB00951; DB01064; DB00982; DB11757; DB01167; DB08820; DB01587; DB01026; DB01009; DB00598; DB09236; DB00709; DB00555; DB03017; DB01006; DB09237; DB06282; DB01235; DB01137; DB00451; DB00601; DB17083; DB00279; DB01583; DB06655; DB01601; DB09195; DB00678; DB00227; DB09280; DB15935; DB12674; DB00137; DB08932; DB14513; DB01397; DB06796; DB06234; DB00737; DB13959; DB09124; DB00603; DB00784; DB00814; DB01042; DB00454; DB09383; DB00931; DB00333; DB00563; DB00968; DB09241; DB00959; DB06710; DB00264; DB01110; DB00683; DB08893; DB00295; DB01024; DB08231; DB00461; DB00607; DB01183; DB00788; DB00731; DB04861; DB00220; DB11828; DB00238; DB01115; DB11820; DB09079; DB11793; DB12005; DB06713; DB00717; DB00957; DB00540; DB00104; DB00334; DB09074; DB04224; DB00768; DB12455; DB11130; DB04911; DB01083; DB13310; DB01173; DB00526; DB00842; DB00776; DB01062; DB00497; DB06412; DB03585; DB00595; DB15575; DB09073; DB03796; DB13967; DB14582; DB00642; DB00850; DB12978; DB01619; DB03255; DB00946; DB00252; DB01132; DB01621; DB04951; DB00554; DB08860; DB11642; DB01324; DB09087; DB09418; DB06813; DB13514; DB06209; DB01058; DB00860; DB15566; DB14631; DB00635; DB01032; DB01069; DB09348; DB00818; DB00571; DB06480; DB00852; DB00165; DB04216; DB00881; DB00908; DB12874; DB08735; DB00481; DB11853; DB12404; DB00912; DB02709; DB11855; DB01045; DB11753; DB08864; DB08931; DB14840; DB15305; DB00734; DB00503; DB11182; DB00412; DB01098; DB04847; DB06201; DB08877; DB08736; DB00936; DB00938; DB01232; DB11689; DB13928; DB01104; DB01236; DB12965; DB06290; DB00877; DB00815; DB15093; DB00421; DB00649; DB03193; DB06150; DB01581; DB01582; DB00576; DB01015; DB00795; DB00605; DB00391; DB00870; DB00864; DB00675; DB05134; DB09139; DB05521; DB00853; DB14126; DB09299; DB15133; DB00857; DB00342; DB00624; DB13943; DB13944; DB01420; DB13946; DB00759; DB00152; DB11590; DB01622; DB01623; DB09100; DB09070; DB08816; DB01133; DB15171; DB11800; DB01056; DB08895; DB01124; DB00500; DB00273; DB01685; DB00214; DB00755; DB00620; DB00432; DB08814; DB11677; DB00376; DB09069; DB00792; DB00427; DB08867; DB09076; DB12255; DB00313; DB00512; DB05294; DB08881; DB00661; DB15456; DB11641; DB08828; DB00162; DB11739; DB16699; DB00682; DB00943; DB00495; DB00744; DB14533; DB14548; DB00246; DB04828 Interacts with P02768; P02786; Q8N5Z5; Q6GQQ9-2; Q07869; Q09028; Q86WT6-2; O76024 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cleavage on pair of basic residues; Copper; Direct protein sequencing; Disease variant; Disulfide bond; Glycation; Glycoprotein; Lipid-binding; Metal-binding; Methylation; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 34028.4 Length 298 Aromaticity 0.09 Instability index 43.45 Isoelectric point 5.49 Charge (pH=7) -10.44 2D Binding mode Binding energy (Kcal/mol) -8.01  Molscript Map 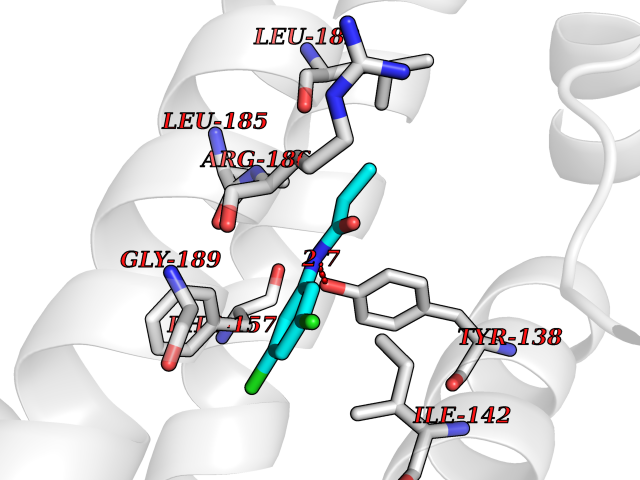 Pymol Map 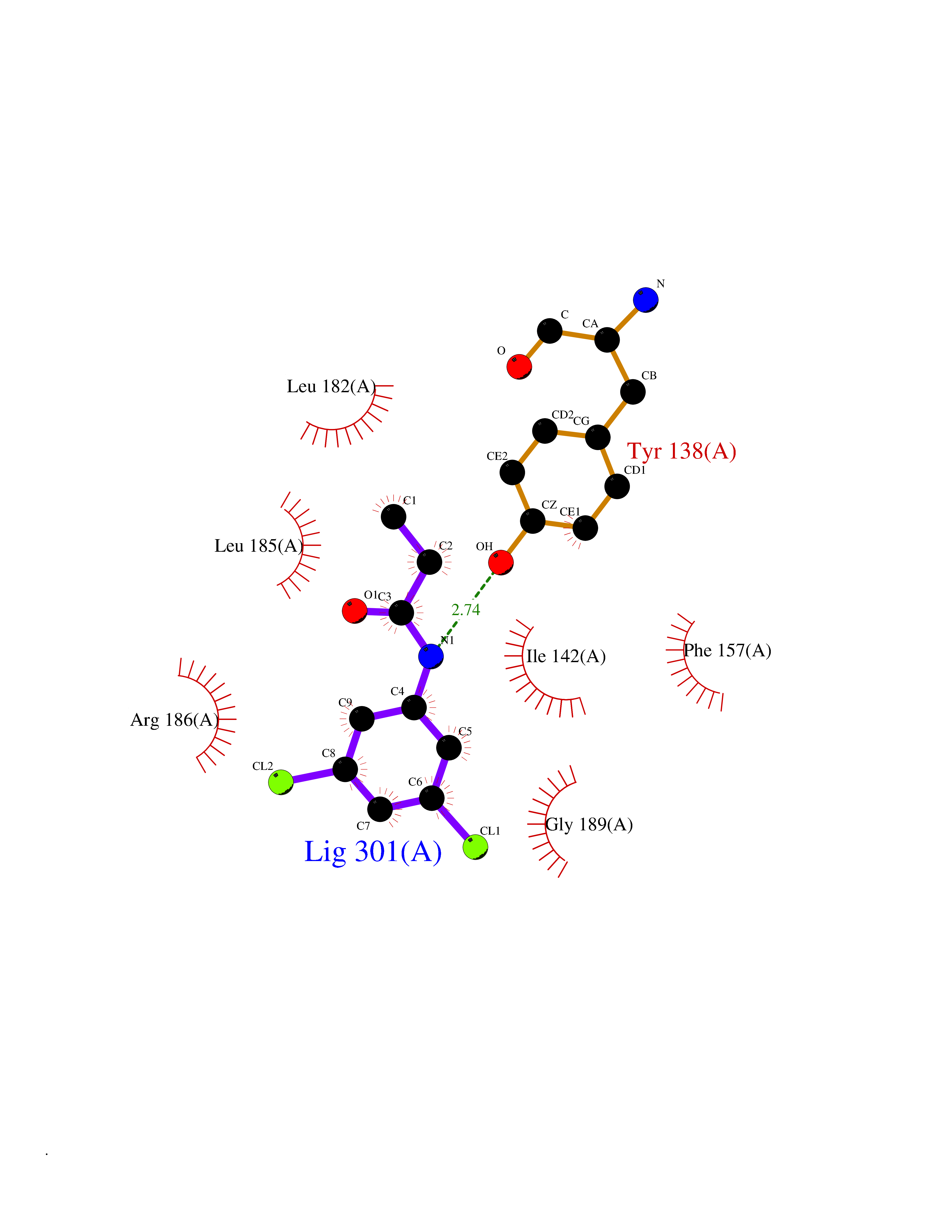 Ligplot Map 3D Binding mode Sequence AHKSEVAHRFKDLGEENFKALVLIAFAQYLQQCPFEDHVKLVNEVTEFAKTCVADESAENCDKSLHTLFGDKLCTVATLRETYGEMADCCAKQEPERNECFLQHKDDNPNLPRLVRPEVDVMCTAFHDNEETFLKKYLYEIARRHPYFYAPELLFFAKRYKAAFTECCQAADKAACLLPKLDELRDEGKASSAKQRLKCASLQKFGERAFKAWAVARLSQRFPKAEFAEVSKLVTDLTKVHTECCHGDLLECADDRADLAKYICENQDSISSKLKECCEKPLLEKSHCIAEVENDEMP Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Fatty acid-binding protein, intestinal | 3AKM | 5.87 | |
Target general information Gen name FABP2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms FABPI Protein family Calycin superfamily, Fatty-acid binding protein (FABP) family Biochemical class Transport protein Function Fatty acid binding.Transporter activity. Related diseases Usher syndrome 3B (USH3B) [MIM:614504]: A syndrome characterized by progressive vision and hearing loss during early childhood. Some patients have the so-called 'Charles Bonnet syndrome,' involving decreased visual acuity and vivid visual hallucinations. USH is a genetically heterogeneous condition characterized by the association of retinitis pigmentosa with sensorineural deafness. Age at onset and differences in auditory and vestibular function distinguish Usher syndrome type 1 (USH1), Usher syndrome type 2 (USH2) and Usher syndrome type 3 (USH3). USH3 is characterized by postlingual, progressive hearing loss, variable vestibular dysfunction, and onset of retinitis pigmentosa symptoms, including nyctalopia, constriction of the visual fields, and loss of central visual acuity, usually by the second decade of life. {ECO:0000269|PubMed:22279524}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, axonal, 2W (CMT2W) [MIM:616625]: An autosomal dominant, axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. CMT2W patients manifest a peripheral neuropathy mainly affecting the lower limbs and resulting in gait difficulties and distal sensory impairment. Most patients also have upper limb involvement. {ECO:0000269|PubMed:22930593, ECO:0000269|PubMed:26072516, ECO:0000269|PubMed:29235198}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04557; DB09213; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB01050; DB08231; DB03796; DB01138 Interacts with O95994; Q9NYB0 EC number NA Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Lipid-binding; Proteomics identification; Reference proteome; Transport Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 15075.9 Length 131 Aromaticity 0.11 Instability index 32.01 Isoelectric point 6.88 Charge (pH=7) -0.09 2D Binding mode Binding energy (Kcal/mol) -8.01 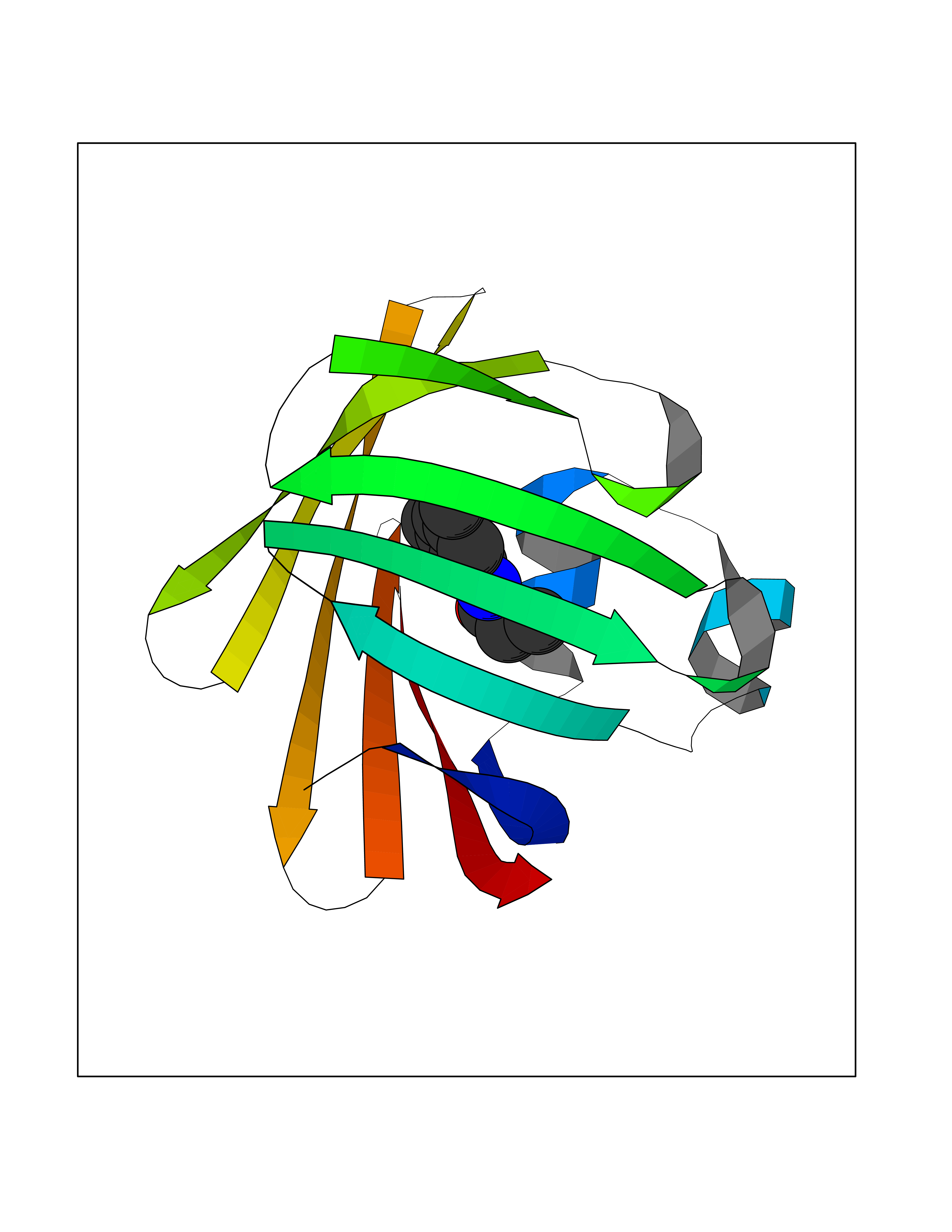 Molscript Map 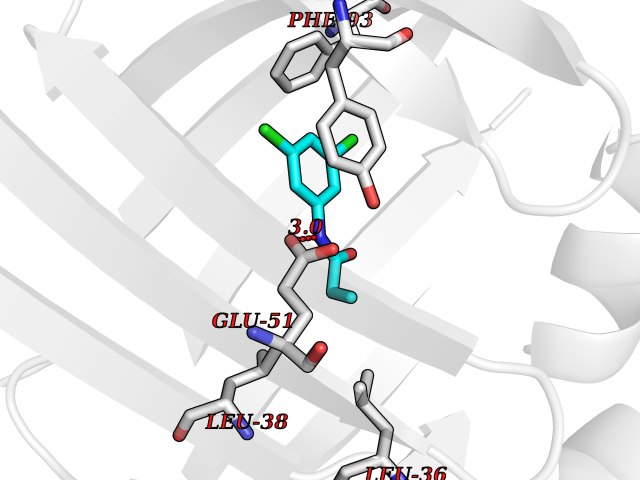 Pymol Map 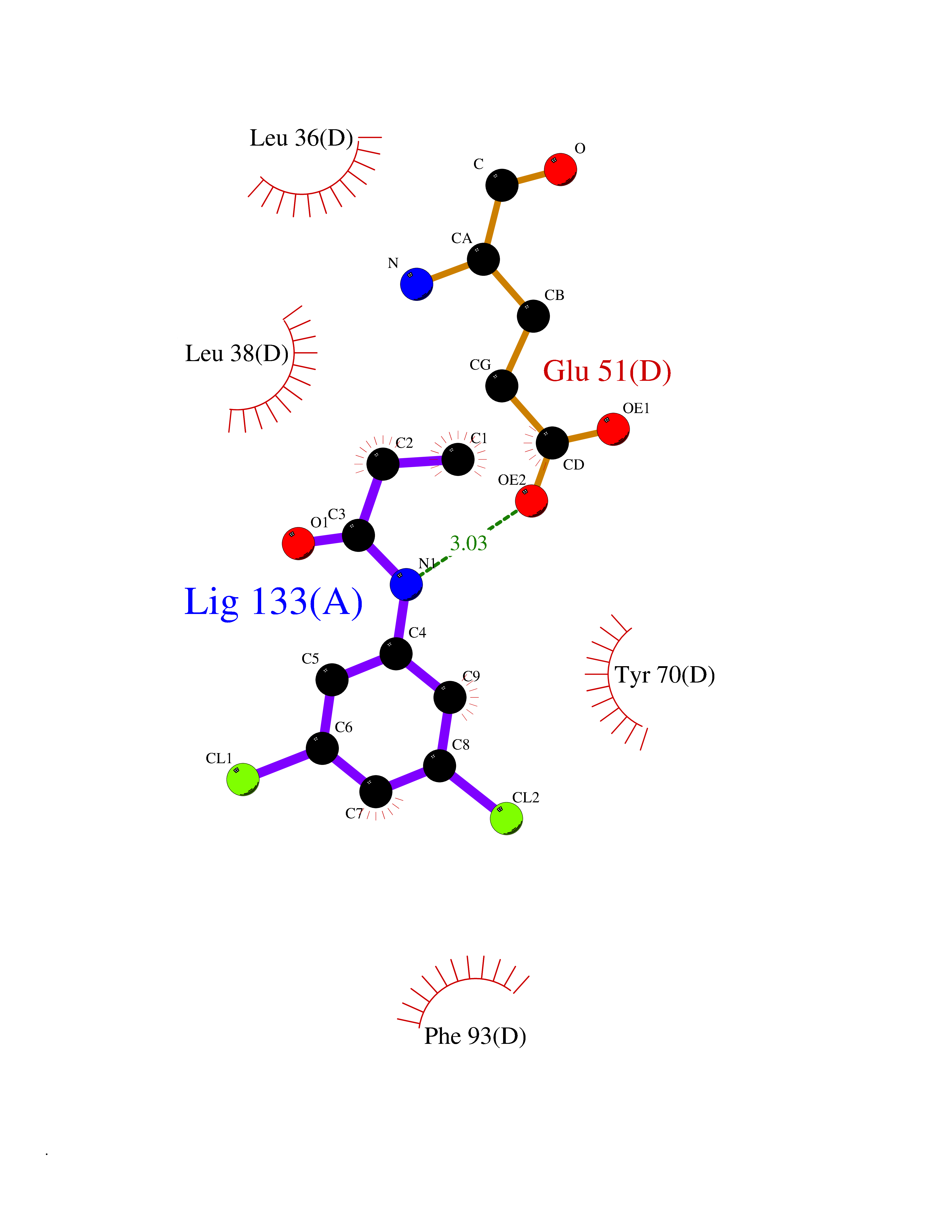 Ligplot Map 3D Binding mode Sequence AFDSTWKVDRSENYDKFMEKMGVNIVKRKLAAHDNLKLTITQEGNKFTVKESSAFRNIEVVFELGVTFNYNLADGTELRGTWSLEGNKLIGKFKRTDNGNELNTVREIIGDELVQTYVYEGVEAKRIFKKD Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Cytochrome P450 1A2 | 2HI4 | 5.87 | |
Target general information Gen name CYP1A2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Aromatase activity.Caffeine oxidase activity.Demethylase activity.Electron carrier activity.Enzyme binding.Heme binding.Iron ion binding.Monooxygenase activity.Oxidoreductase activity.Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, reduced flavin or flavoprotein as one donor, and incorporation of one atom of oxygen.Oxygen binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08496; DB01667; DB14132; DB04356; DB02489; DB11932; DB12001; DB05812; DB13573; DB01418; DB00316; DB15568; DB06594; DB00518; DB05396; DB00969; DB07453; DB01424; DB01223; DB01118; DB00321; DB00261; DB01217; DB01435; DB06605; DB05676; DB06413; DB06216; DB01072; DB15011; DB06442; DB06626; DB00993; DB00972; DB13203; DB05015; DB16703; DB06769; DB01086; DB06770; DB06771; DB06732; DB00195; DB04889; DB11967; DB13975; DB00188; DB12151; DB01558; DB14018; DB13812; DB00201; DB09061; DB14737; DB11791; DB06774; DB00564; DB06016; DB01136; DB12814; DB00477; DB00356; DB01166; DB00501; DB01012; DB00568; DB00827; DB00537; DB00215; DB12499; DB14025; DB00349; DB01242; DB00575; DB00758; DB00363; DB00286; DB11672; DB14635; DB00924; DB08912; DB00851; DB06292; DB01254; DB01609; DB01151; DB16650; DB12161; DB01191; DB00633; DB11994; DB00586; DB11511; DB12945; DB00280; DB01184; DB09167; DB05928; DB01142; DB09273; DB00470; DB00476; DB00625; DB15444; DB06210; DB13874; DB11718; DB00467; DB11404; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB04574; DB13592; DB00330; DB00898; DB00977; DB00773; DB01628; DB00927; DB04854; DB01482; DB00574; DB12265; DB15669; DB01195; DB08972; DB04841; DB00544; DB00472; DB00499; DB00176; DB01320; DB00998; DB14029; DB06160; DB01044; DB01241; DB01155; DB01645; DB01381; DB00986; DB00365; DB00400; DB05708; DB00629; DB00502; DB01094; DB14999; DB04076; DB11737; DB00619; DB00458; DB11564; DB01306; DB09456; DB09564; DB01307; DB00047; DB01309; DB00030; DB00046; DB11567; DB00071; DB11568; DB05258; DB00034; DB00105; DB15131; DB00011; DB00018; DB00069; DB00060; DB00068; DB00033; DB00951; DB11757; DB09570; DB01026; DB01097; DB16217; DB09078; DB01002; DB05667; DB00281; DB12406; DB09198; DB04948; DB00978; DB06448; DB16220; DB01601; DB00455; DB04871; DB06077; DB01283; DB00772; DB00934; DB06234; DB14009; DB00784; DB01065; DB00170; DB00454; DB00532; DB00333; DB00763; DB00553; DB01028; DB09241; DB01233; DB00379; DB06148; DB01388; DB06595; DB00370; DB16236; DB00745; DB11763; DB00218; DB06510; DB14011; DB00461; DB00607; DB00779; DB00788; DB06600; DB00238; DB06803; DB00184; DB01115; DB11793; DB00435; DB05115; DB00717; DB01059; DB00540; DB05990; DB01165; DB00334; DB16267; DB00338; DB00904; DB11632; DB11443; DB01173; DB11837; DB09330; DB01303; DB11697; DB00377; DB00715; DB06589; DB11774; DB00487; DB00008; DB00022; DB09122; DB13634; DB00806; DB11198; DB08883; DB00850; DB03783; DB01174; DB00388; DB00252; DB11450; DB01100; DB13823; DB04951; DB17472; DB11642; DB08910; DB15822; DB01058; DB01087; DB00794; DB00420; DB09288; DB01182; DB06479; DB00818; DB00571; DB13449; DB11892; DB04216; DB00908; DB00468; DB01129; DB00980; DB09290; DB00863; DB01367; DB00409; DB02709; DB13174; DB01045; DB11753; DB00740; DB14924; DB00503; DB00533; DB01656; DB15119; DB00268; DB00296; DB00412; DB00817; DB12332; DB13772; DB06654; DB11491; DB00418; DB01037; DB11689; DB06290; DB13261; DB15093; DB00052; DB00398; DB01208; DB09118; DB00428; DB06820; DB00382; DB00675; DB06083; DB09071; DB05488; DB09256; DB01079; DB01405; DB00857; DB08880; DB11712; DB01412; DB00277; DB00730; DB01623; DB00208; DB06137; DB00697; DB01056; DB06264; DB00752; DB00384; DB12245; DB00831; DB15442; DB00440; DB00685; DB08867; DB14989; DB13609; DB06235; DB00313; DB08881; DB00661; DB09185; DB12026; DB00682; DB02134; DB00549; DB00744; DB00315; DB00425; DB09225; DB09120 Interacts with O95870 EC number 1.14.14.1; 4.2.1.152 Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Fatty acid metabolism; Glycoprotein; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54475 Length 480 Aromaticity 0.1 Instability index 40.43 Isoelectric point 9.16 Charge (pH=7) 9.89 2D Binding mode Binding energy (Kcal/mol) -8.01  Molscript Map 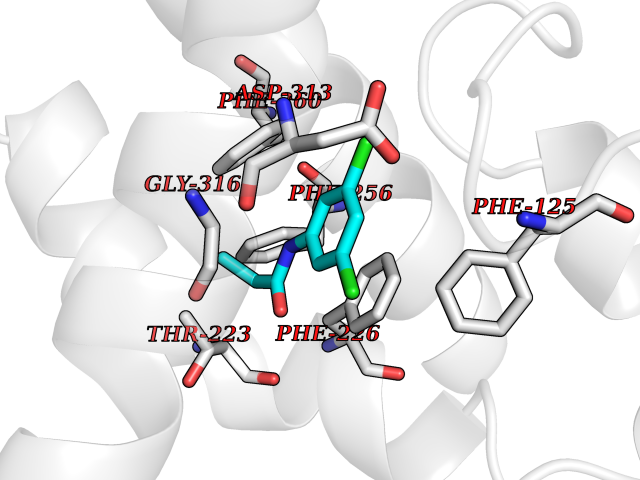 Pymol Map 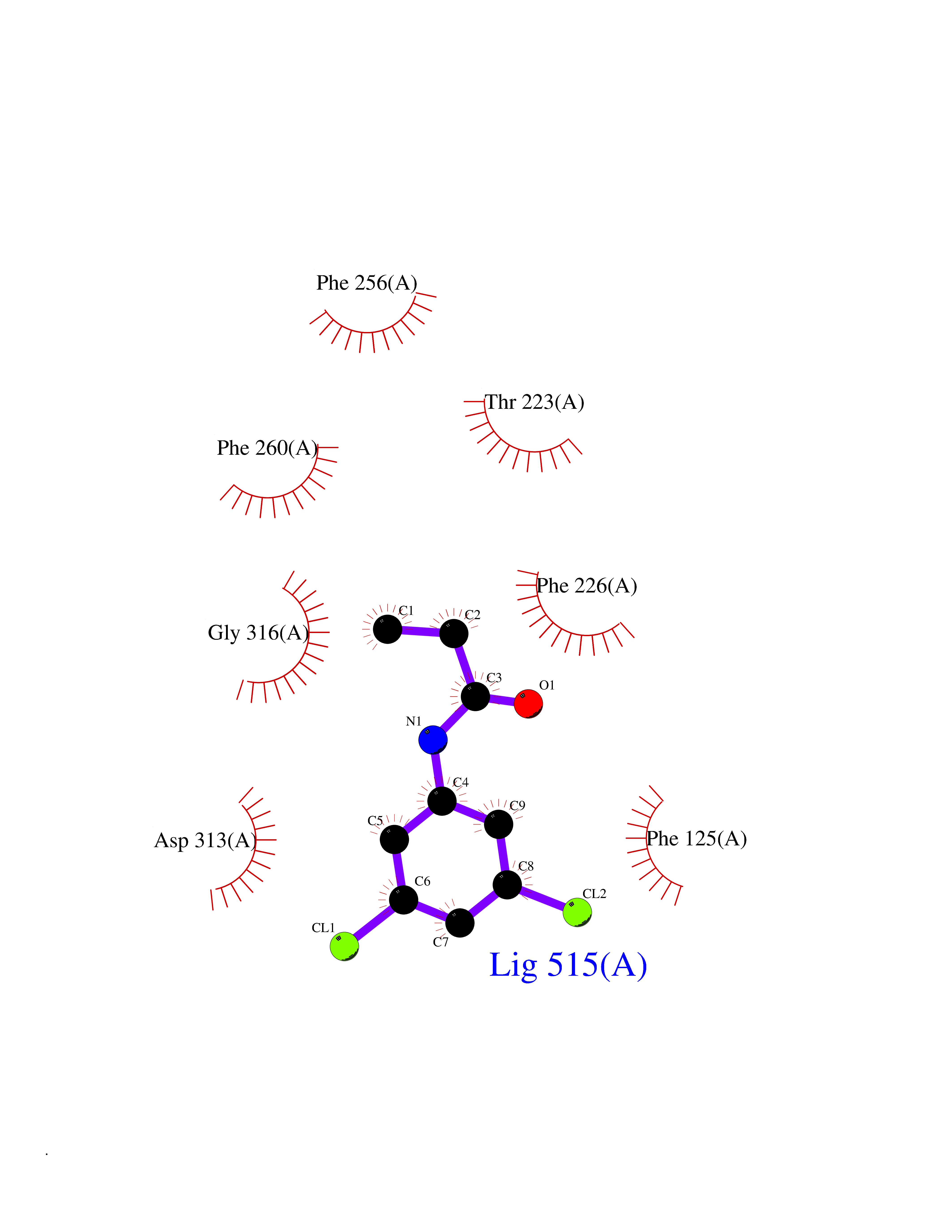 Ligplot Map 3D Binding mode Sequence RVPKGLKSPPEPWGWPLLGHVLTLGKNPHLALSRMSQRYGDVLQIRIGSTPVLVLSRLDTIRQALVRQGDDFKGRPDLYTSTLITDGQSLTFSTDSGPVWAARRRLAQNALNTFSIASDPASSSSCYLEEHVSKEAKALISRLQELMAGPGHFDPYNQVVVSVANVIGAMCFGQHFPESSDEMLSLVKNTHEFVETASSGNPLDFFPILRYLPNPALQRFKAFNQRFLWFLQKTVQEHYQDFDKNSVRDITGALFKHSKKGPRASGNLIPQEKIVNLVNDIFGAGFDTVTTAISWSLMYLVTKPEIQRKIQKELDTVIGRERRPRLSDRPQLPYLEAFILETFRHSSFLPFTIPHSTTRDTTLNGFYIPKKCCVFVNQWQVNHDPELWEDPSEFRPERFLTADGTAINKPLSEKMMLFGMGKRRCIGEVLAKWEIFLFLAILLQQLEFSVPPGVKVDLTPIYGLTMKHARCEHVQARRFS Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Fumarate reductase flavoprotein subunit | 1KF6 | 5.87 | |
Target general information Gen name frdA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW4115;b4154 Protein family FAD-dependent oxidoreductase 2 family, FRD/SDH subfamily Biochemical class Oxidoreductase Function Electron carrier activity.FAD binding.Fumarate reductase (menaquinone).Succinate dehydrogenase activity. Related diseases Glycogen storage disease 11 (GSD11) [MIM:612933]: A metabolic disorder that results in exertional myoglobinuria, pain, cramps and easy fatigue. {ECO:0000269|PubMed:2334430}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07490; DB07918; DB00730 Interacts with P0AC47; P0ACB4; P76111 EC number 1.3.5.1 Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Direct protein sequencing; Electron transport; FAD; Flavoprotein; Membrane; Nucleotide-binding; Oxidoreductase; Reference proteome; Transport Protein physicochemical properties Chain ID A,M Molecular weight (Da) 90370.7 Length 820 Aromaticity 0.08 Instability index 28.88 Isoelectric point 5.86 Charge (pH=7) -16.21 2D Binding mode Binding energy (Kcal/mol) -8  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MQTFQADLAIVGAGGAGLRAAIAAAQANPNAKIALISKVYPMRSHTVAAEGGSAAVAQDHDSFEYHFHDTVAGGDWLCEQDVVDYFVHHCPTEMTQLELWGCPWSRRPDGSVNVRRFGGMKIERTWFAADKTGFHMLHTLFQTSLQFPQIQRFDEHFVLDILVDDGHVRGLVAMNMMEGTLVQIRANAVVMATGGAGRVYRYNTNGGIVTGDGMGMALSHGVPLRDMEFVQYHPTGLPGSGILMTEGCRGEGGILVNKNGYRYLQDYGMGPETPLGEPKNKYMELGPRDKVSQAFWHEWRKGNTISTPRGDVVYLDLRHLGEKKLHERLPFICELAKAYVGVDPVKEPIPVRPTAHYTMGGIETDQNCETRIKGLFAVGECSSVGLHGANRLGSNSLAELVVFGRLAGEQATERAATAGNGNEAAIEAQAAGVEQRLKDLVNQDGGENWAKIRDEMGLAMEEGCGIYRTPELMQKTIDKLAELQERFKRVRITDTSSVFNTDLLYTIELGHGLNVAECMAHSAMARKESRGAHQRLDEGCTERDDVNFLKHTLAFRDADGTTRLEYSDVKITTLPPAAEMKNLKIEVVRYNPEVDTAPHSAFYEVPYDATTSLLDALGYIKDNLAPDLSYRWSCRMAICGSCGMMVNNVPKLACKTFLRDYTDGMKVEALANFPIERDLVVDMTHFIESLEAIKPYIIGNSRTADQGTNIQTPAQMAKYHQFSGCINCGLCYAACPQFGLNPEFIGPAAITLAHRYNEDSRDHGKKERMAQLNSQNGVWSCTFVGYCSEVCPKHVDPAAAIQQGKVESSKDFLIATLKPR Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Thyroid hormone receptor beta (THRB) | 1N46 | 5.87 | |
Target general information Gen name THRB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms c-erbA-beta; c-erbA-2; THR1; Nuclear receptor subfamily 1 group A member 2; NR1A2; ERBA2 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function High affinity receptor for thyroid hormones, including triiodothyronine and thyroxine. Nuclear hormone receptor that can act as a repressor or activator of transcription. Related diseases Thyroid hormone resistance, generalized, autosomal dominant (GRTHD) [MIM:188570]: An autosomal dominant disease characterized by high levels of circulating thyroid hormones (T3-T4), goiter, abnormal mental functions, increased susceptibility to infections, abnormal growth and bone maturation, tachycardia and deafness. Affected individuals may also have attention deficit-hyperactivity disorders (ADHD) and language difficulties. Patients have normal or slightly elevated thyroid stimulating hormone (TSH). {ECO:0000269|PubMed:10660344, ECO:0000269|PubMed:12511610, ECO:0000269|PubMed:12554782, ECO:0000269|PubMed:1314846, ECO:0000269|PubMed:1324420, ECO:0000269|PubMed:1563081, ECO:0000269|PubMed:1587388, ECO:0000269|PubMed:1619012, ECO:0000269|PubMed:1661299, ECO:0000269|PubMed:16804041, ECO:0000269|PubMed:1846005, ECO:0000269|PubMed:19268523, ECO:0000269|PubMed:2153155, ECO:0000269|PubMed:2510172, ECO:0000269|PubMed:7833659, ECO:0000269|PubMed:8175986, ECO:0000269|PubMed:8514853, ECO:0000269|PubMed:8664910, ECO:0000269|PubMed:8889584}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid hormone resistance, generalized, autosomal recessive (GRTHR) [MIM:274300]: An autosomal recessive disorder characterized by goiter, clinical euthyroidism, end-organ unresponsiveness to thyroid hormone, abnormal growth and bone maturation, and deafness. Patients also have high levels of circulating thyroid hormones, with elevated thyroid stimulating hormone. {ECO:0000269|PubMed:1653889}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Selective pituitary thyroid hormone resistance (PRTH) [MIM:145650]: Variant form of thyroid hormone resistance and is characterized by clinical hyperthyroidism, with elevated free thyroid hormones, but inappropriately normal serum TSH. Unlike GRTH, where the syndrome usually segregates with a dominant allele, the mode of inheritance in PRTH has not been established. {ECO:0000269|PubMed:7528740, ECO:0000269|PubMed:8381821}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08085; DB03181; DB02106; DB01118; DB00509; DB05035; DB03788; DB03176; DB00451; DB00279; DB01583; DB05192; DB07425; DB09100; DB03604 Interacts with Q60974; Q9Y618 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Deafness; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 27235.4 Length 239 Aromaticity 0.09 Instability index 43.29 Isoelectric point 5.42 Charge (pH=7) -8.55 2D Binding mode Binding energy (Kcal/mol) -8.01  Molscript Map 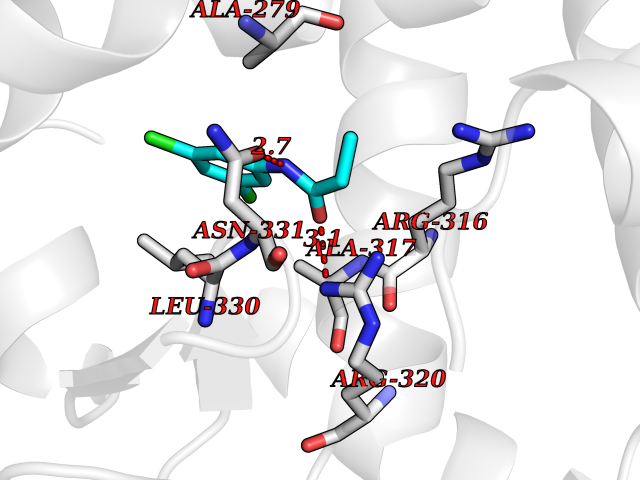 Pymol Map 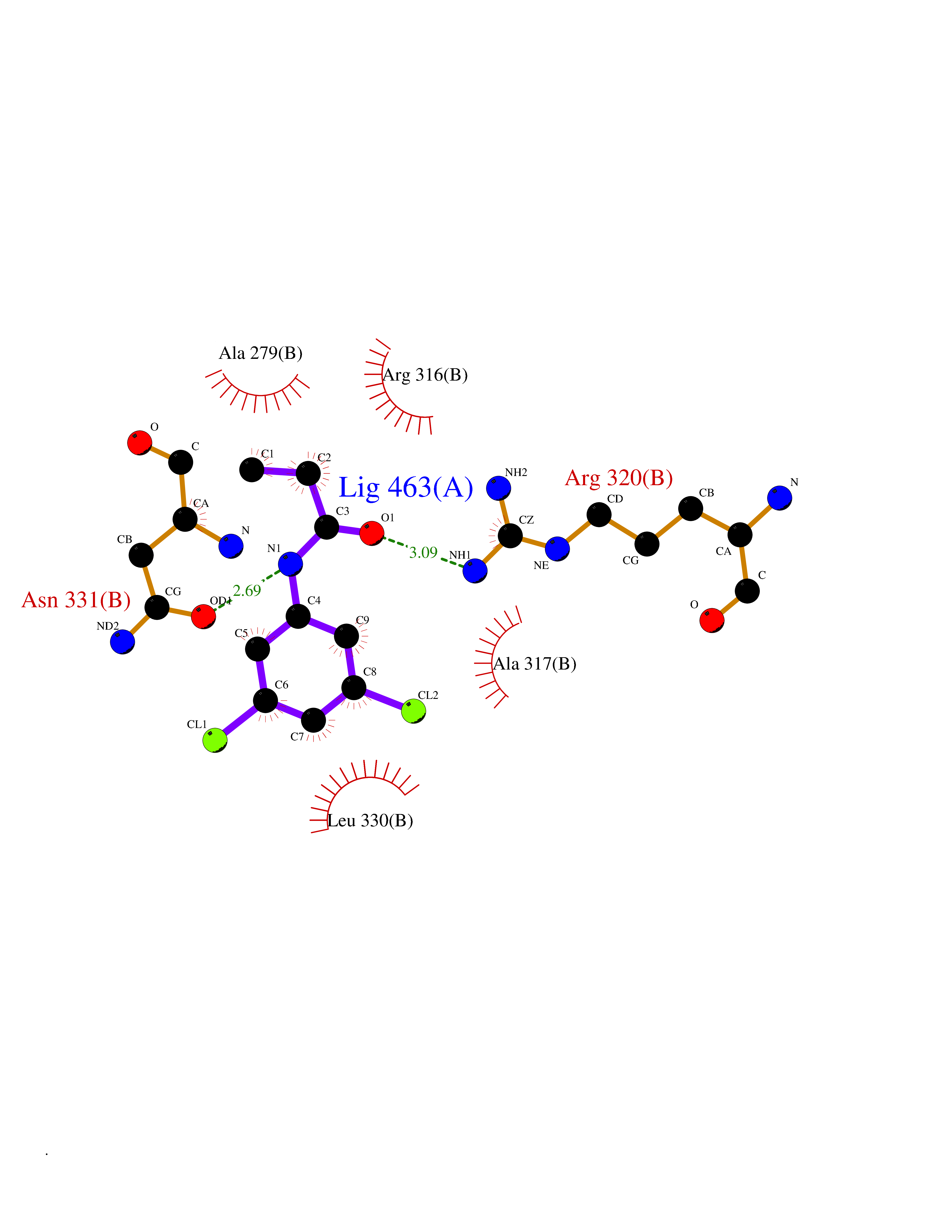 Ligplot Map 3D Binding mode Sequence KPEPTDEEWELIKTVTEAHVATNAQWKQKRKFLPEDIGQAKVDLEAFSHFTKIITPAITRVVDFAKKLPMFCELPCEDQIILLKGCCMEIMSLRAAVRYDPESETLTLNGEMAVTRGQLKNGGLGVVSDAIFDLGMSLSSFNLDDTEVALLQAVLLMSSDRPGLACVERIEKYQDSFLLAFEHYINYRKHHVTHFWPKLLMKVTDLRMIGACHASRFLHMKVECPTELFPPLFLEVFED Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Acetylcholinesterase (AChE) | 4M0E | 5.87 | |
Target general information Gen name ACHE Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms YT; N-ACHE; ARACHE Protein family Type-B carboxylesterase/lipase family Biochemical class Carboxylic ester hydrolase Function Role in neuronal apoptosis. Terminates signal transduction at the neuromuscular junction by rapid hydrolysis of the acetylcholine released into the synaptic cleft. Related diseases Phosphoribosylaminoimidazole carboxylase deficiency (PAICSD) [MIM:619859]: An autosomal recessive inborn error of purine metabolism, clinically characterized by multiple congenital anomalies and early neonatal death. {ECO:0000269|PubMed:31600779}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07846; DB02673; DB04617; DB04614; DB04615; DB07756; DB07701; DB02404; DB03814; DB08615; DB02343; DB02226; DB03005; DB04114; DB03128; DB01122; DB03283; DB00411; DB00122; DB14006; DB01245; DB00944; DB08357; DB08996; DB00449; DB00843; DB01010; DB01364; DB00898; DB00674; DB00483; DB06525; DB04864; DB03348; DB07555; DB00677; DB04924; DB03359; DB00358; DB00940; DB02825; DB02845; DB08167; DB04021; DB00805; DB01805; DB03740; DB04556; DB01400; DB04892; DB00981; DB00733; DB02166; DB00545; DB00863; DB00989; DB00382; DB04616; DB12816; DB01199; DB13503; DB04859 Interacts with Q9Y215; P06733; P63244 EC number EC 3.1.1.7 Uniprot keywords 3D-structure; Alternative splicing; Blood group antigen; Cell membrane; Direct protein sequencing; Disulfide bond; Glycoprotein; GPI-anchor; Hydrolase; Lipoprotein; Membrane; Neurotransmitter degradation; Nucleus; Proteomics identification; Reference proteome; Secreted; Serine esterase; Signal; Synapse Protein physicochemical properties Chain ID A,B Molecular weight (Da) 58804.1 Length 537 Aromaticity 0.11 Instability index 40.85 Isoelectric point 5.73 Charge (pH=7) -8.18 2D Binding mode Binding energy (Kcal/mol) -8 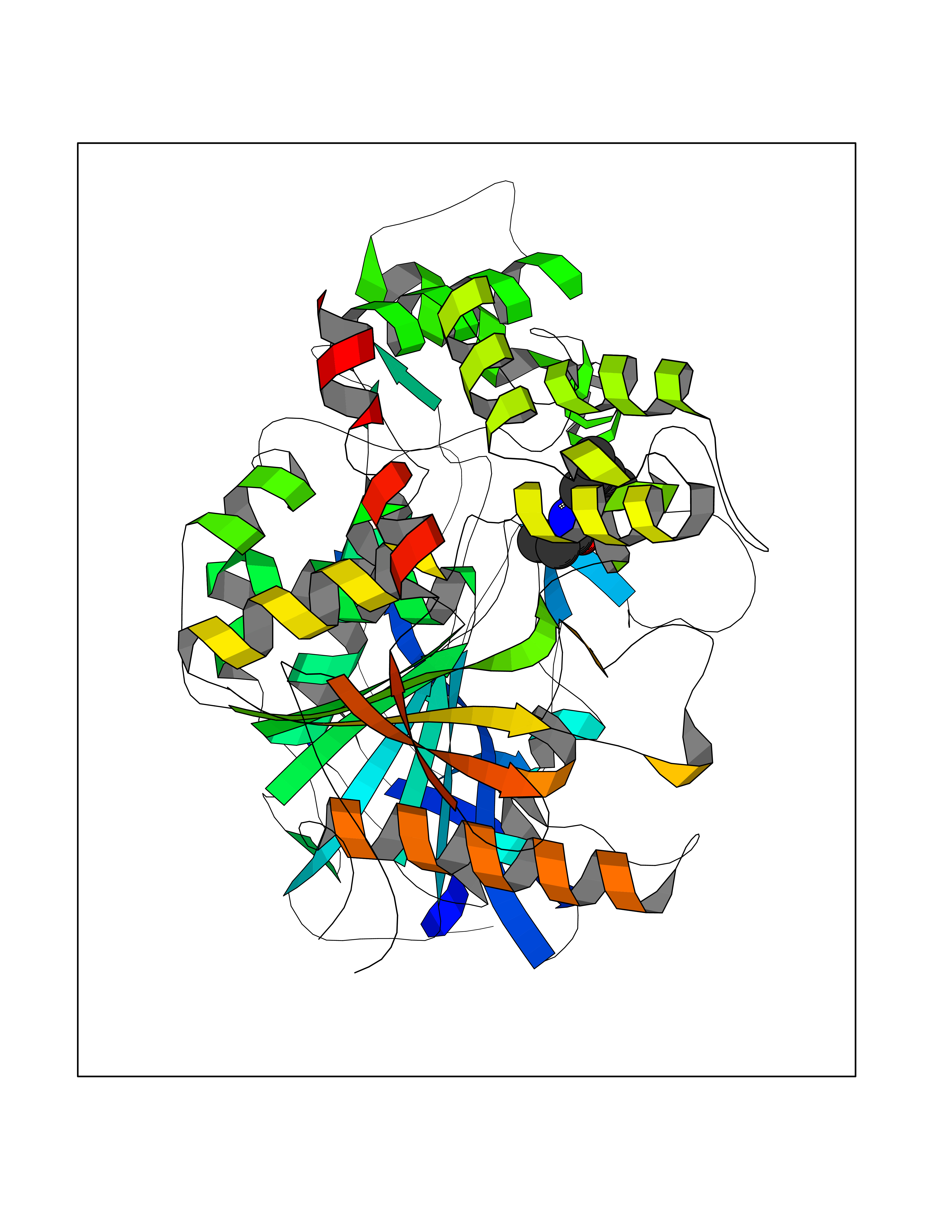 Molscript Map 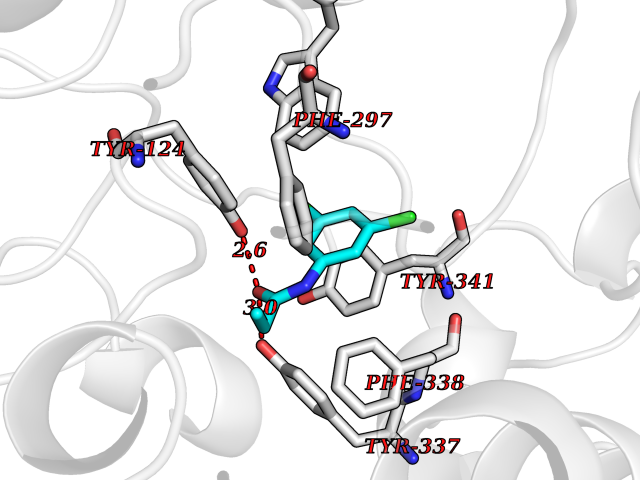 Pymol Map 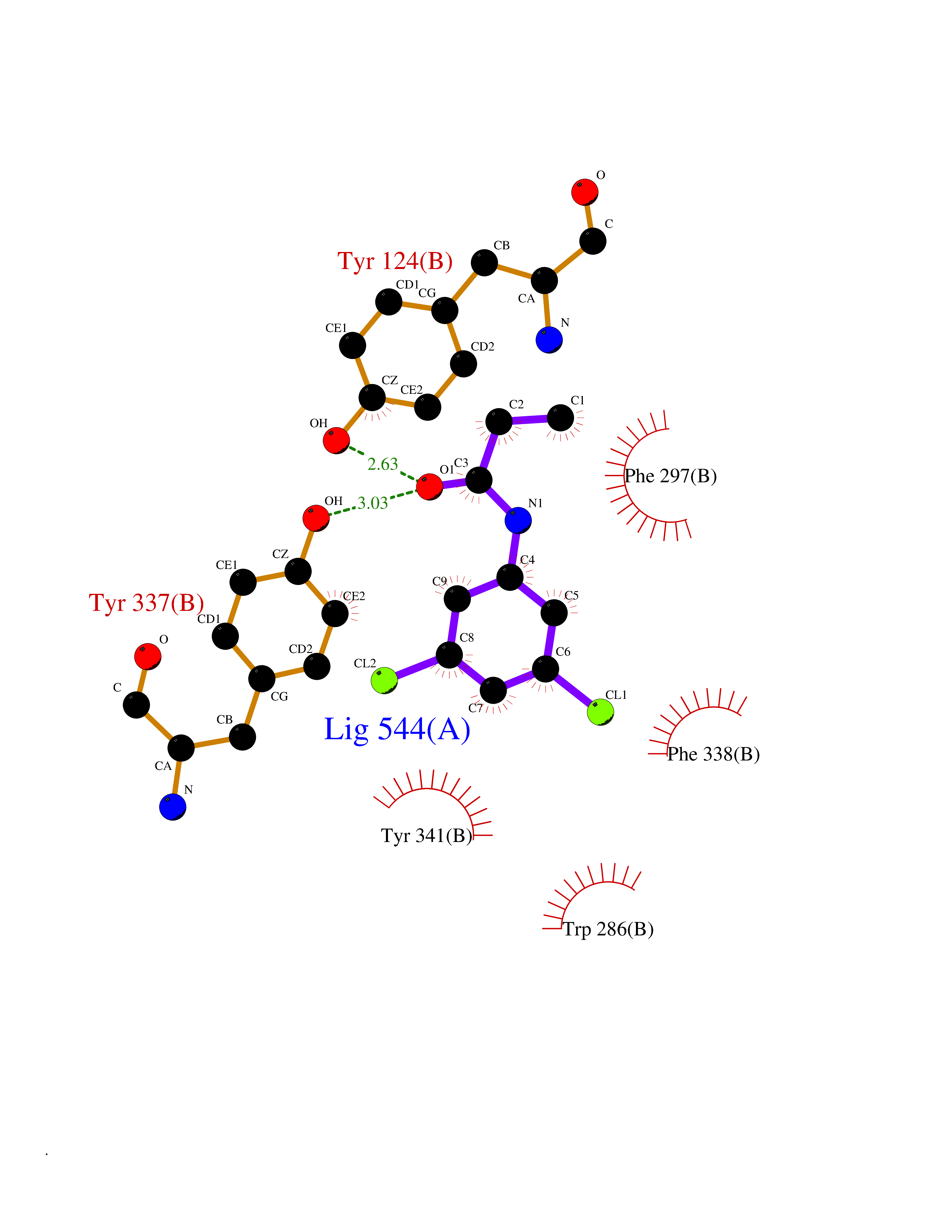 Ligplot Map 3D Binding mode Sequence EDAELLVTVRGGRLRGIRLKTPGGPVSAFLGIPFAEPPMGPRRFLPPEPKQPWSGVVDATTFQSVCYQYVDTLYPGFEGTEMWNPNRELSEDCLYLNVWTPYPRPTSPTPVLVWIYGGGFYSGASSLDVYDGRFLVQAERTVLVSMNYRVGAFGFLALPGSREAPGNVGLLDQRLALQWVQENVAAFGGDPTSVTLFGESAGAASVGMHLLSPPSRGLFHRAVLQSGAPNGPWATVGMGEARRRATQLAHLVGCPPGGTGGNDTELVACLRTRPAQVLVNHEWHVLPQESVFRFSFVPVVDGDFLSDTPEALINAGDFHGLQVLVGVVKDEGSYFLVYGAPGFSKDNESLISRAEFLAGVRVGVPQVSDLAAEAVVLHYTDWLHPEDPARLREALSDVVGDHNVVCPVAQLAGRLAAQGARVYAYVFEHRASTLSWPLWMGVPHGYEIEFIFGIPLDPSRNYTAEEKIFAQRLMRYWANFARTGDPNEPPKAPQWPPYTAGAQQYVSLDLRPLEVRRGLRAQACAFWNRFLPKLLSA Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | L-serine dehydratase/L-threonine deaminase | 1P5J | 5.87 | |
Target general information Gen name SDS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms SDH Protein family Serine/threonine dehydratase family Biochemical class Lyase Function L-serine ammonia-lyase activity.L-threonine ammonia-lyase activity.Protein homodimerization activity.Pyridoxal phosphate binding. Related diseases Immunodeficiency, common variable, 12, with autoimmunity (CVID12) [MIM:616576]: A primary immunodeficiency characterized by hypogammaglobulinemia and recurrent bacterial infections. About half of patients develop autoimmune features, including cytopenia, as well as generalized inflammation and lymphoproliferation manifest as lymphadenopathy or hepatosplenomegaly. {ECO:0000269|PubMed:26279205}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00114; DB00133 Interacts with Q8WTU0; O14964; Q96PF1 EC number 4.3.1.17; 4.3.1.19 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Gluconeogenesis; Lipid metabolism; Lyase; Proteomics identification; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 33559.7 Length 319 Aromaticity 0.06 Instability index 34.65 Isoelectric point 7.2 Charge (pH=7) 0.42 2D Binding mode Binding energy (Kcal/mol) -8 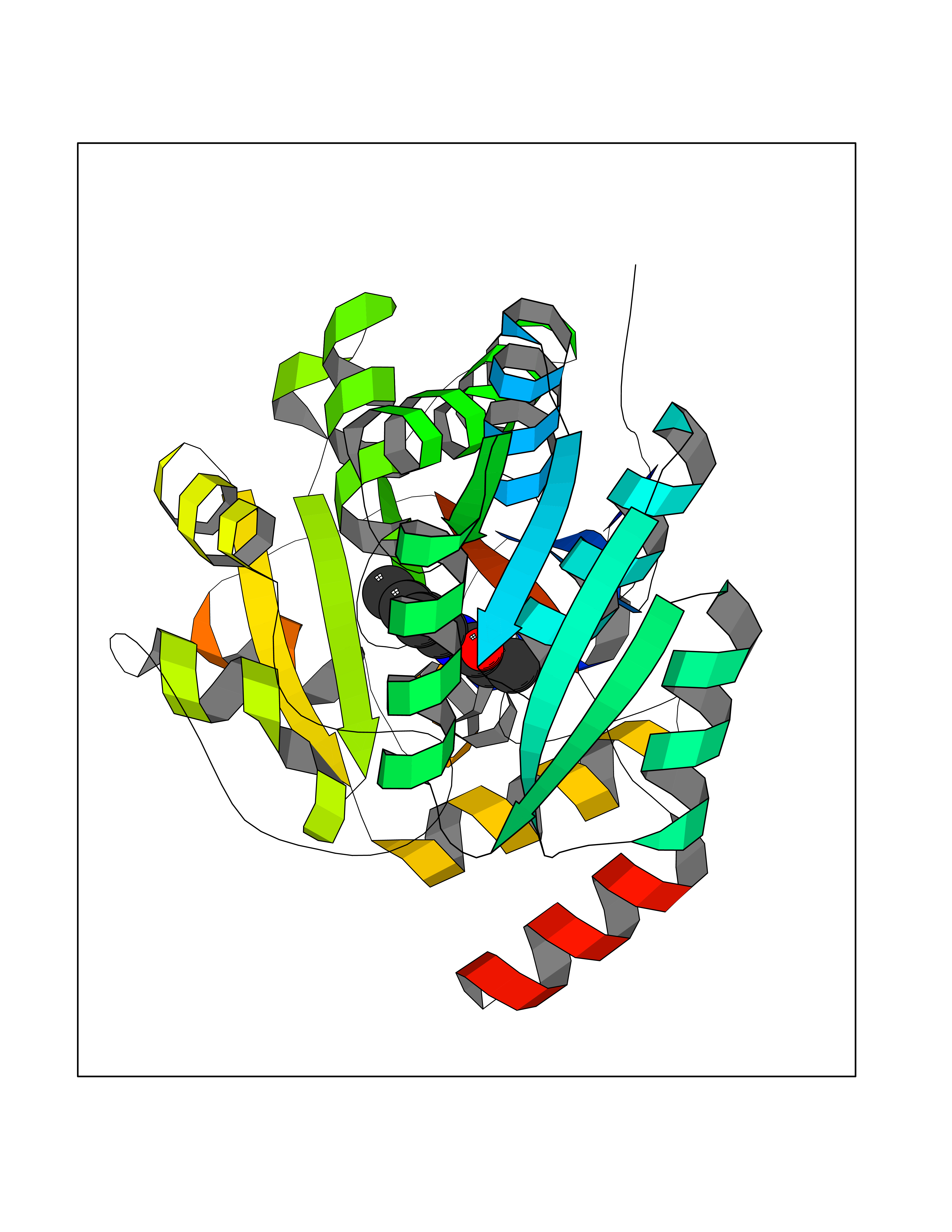 Molscript Map 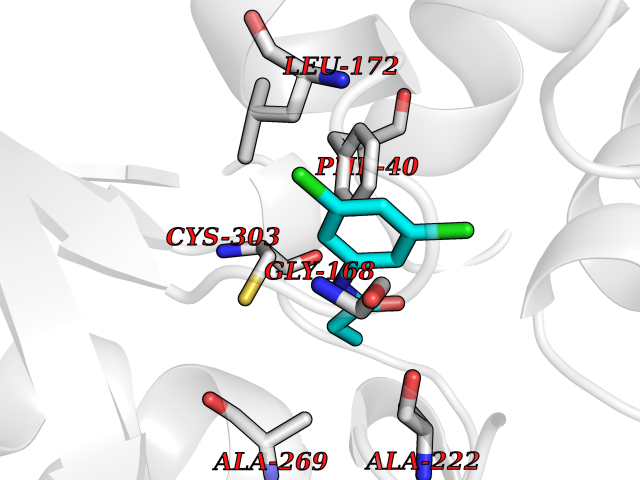 Pymol Map 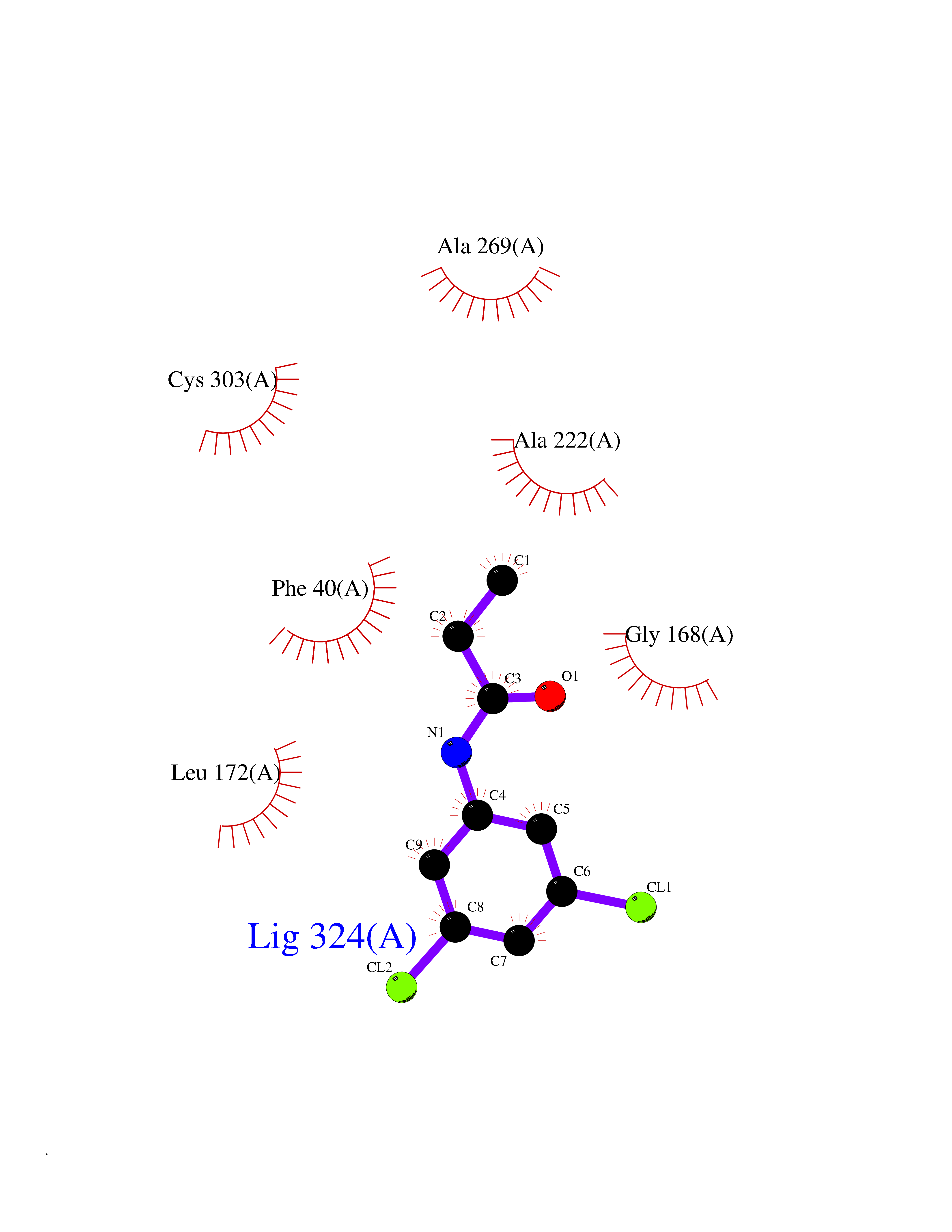 Ligplot Map 3D Binding mode Sequence GEPLHVKTPIRDSMALSKMAGTSVYLKMDSAQPSGSFKIRGIGHFCKRWAKQGCAHFVCSSAGNAGMAAAYAARQLGVPATIVVPGTTPALTIERLKNEGATCKVVGELLDEAFELAKALAKNNPGWVYIPPFDDPLIWEGHASIVKELKETLWEKPGAIALSVGGGGLLCGVVQGLQECGWGDVPVIAMETFGAHSFHAATTAGKLVSLPKITSVAKALGVKTVGSQALKLFQEHPIFSEVISDQEAVAAIEKFVDDEKILVEPACGAALAAVYSHVIQKLQLEGNLRTPLPSLVVIVCGGSNISLAQLRALKEQLGM Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Metabotropic glutamate receptor 5 (mGluR5) | 4OO9 | 5.87 | |
Target general information Gen name GRM5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MGLUR5; GPRC1E Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function G-protein coupled receptor for glutamate. Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling activates a phosphatidylinositol-calcium second messenger system and generates a calcium-activated chloride current. Plays an important role in the regulation of synaptic plasticity and the modulation of the neural network activity. Related diseases Charcot-Marie-Tooth disease, axonal, 2D (CMT2D) [MIM:601472]: A dominant axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:12690580, ECO:0000269|PubMed:17035524, ECO:0000269|PubMed:17101916, ECO:0000269|PubMed:17663003, ECO:0000269|PubMed:20169446, ECO:0000269|PubMed:24604904, ECO:0000269|PubMed:25168514, ECO:0000269|PubMed:26244500, ECO:0000269|PubMed:26503042, ECO:0000269|PubMed:31173493}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal dominant 5 (HMND5) [MIM:600794]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. {ECO:0000269|PubMed:12690580, ECO:0000269|PubMed:17035524, ECO:0000269|PubMed:23279345, ECO:0000269|PubMed:24627108, ECO:0000269|PubMed:26503042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinal muscular atrophy, infantile, James type (SMAJI) [MIM:619042]: An autosomal dominant form of spinal muscular atrophy, a group of neuromuscular disorders characterized by degeneration of the anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. SMAJI is a severe disease characterized by hypotonia manifesting in the first weeks or months of life, delayed motor development, motor regression, and muscle weakness and atrophy primarily affecting distal muscles. Additional variable features include feeding difficulties, poor overall growth, foot deformities, kyphosis, hyperlordosis, scoliosis, vocal cord dysfunction, and respiratory insufficiency. {ECO:0000269|PubMed:32181591}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00659; DB05070; DB12733; DB06201 Interacts with P41594; Q7Z6G3 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Methylation; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 27065.4 Length 247 Aromaticity 0.13 Instability index 42.92 Isoelectric point 9.24 Charge (pH=7) 11.34 2D Binding mode Binding energy (Kcal/mol) -8 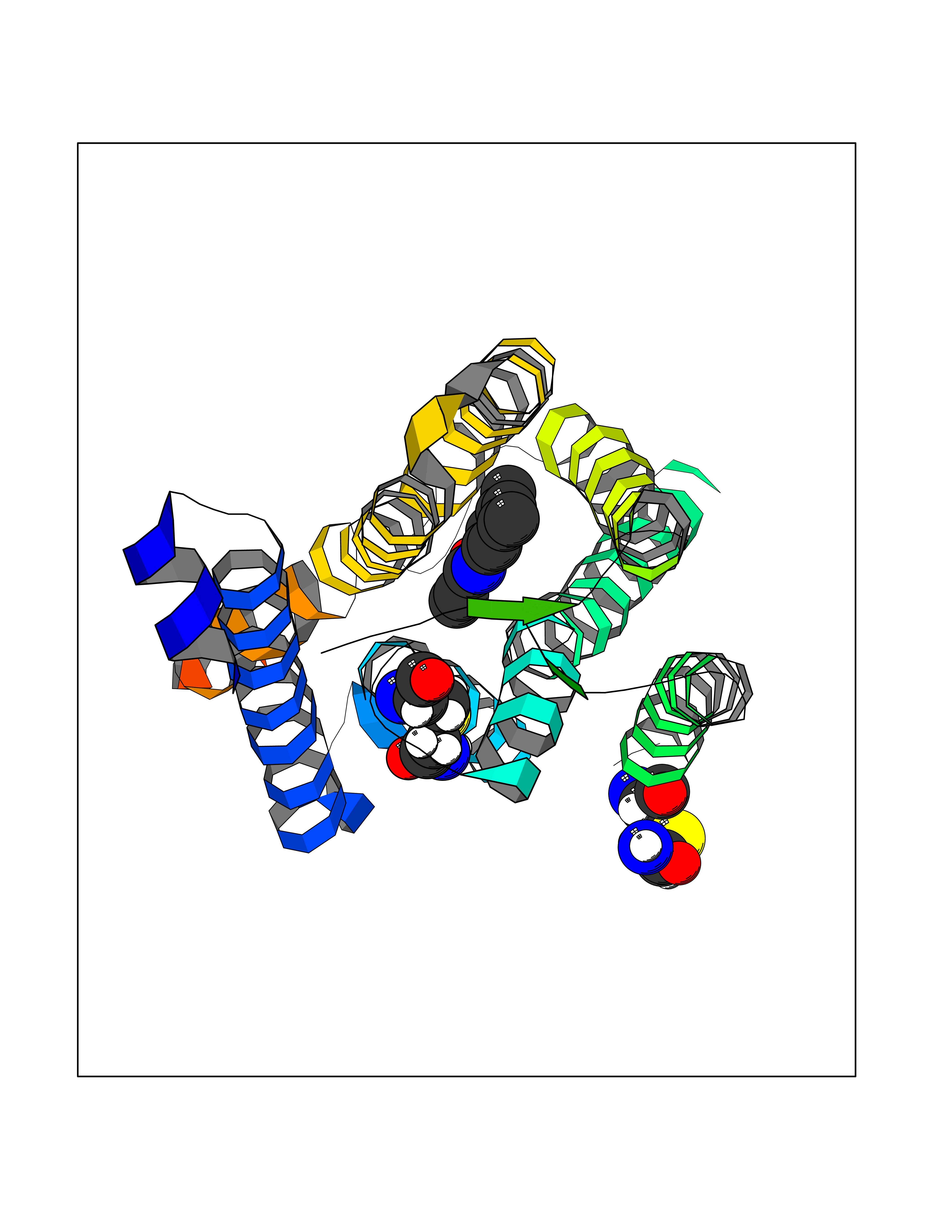 Molscript Map 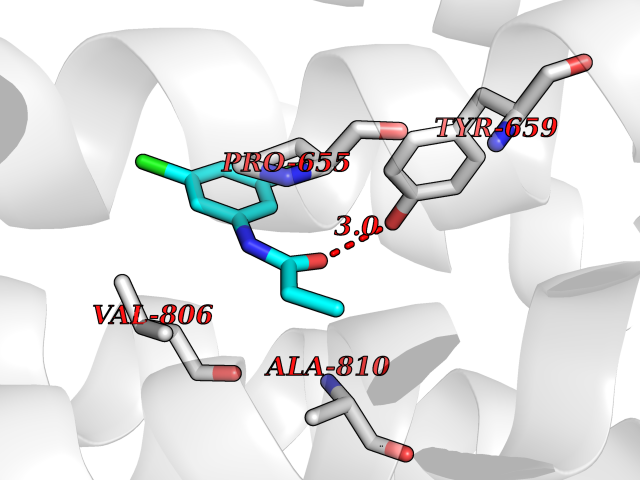 Pymol Map 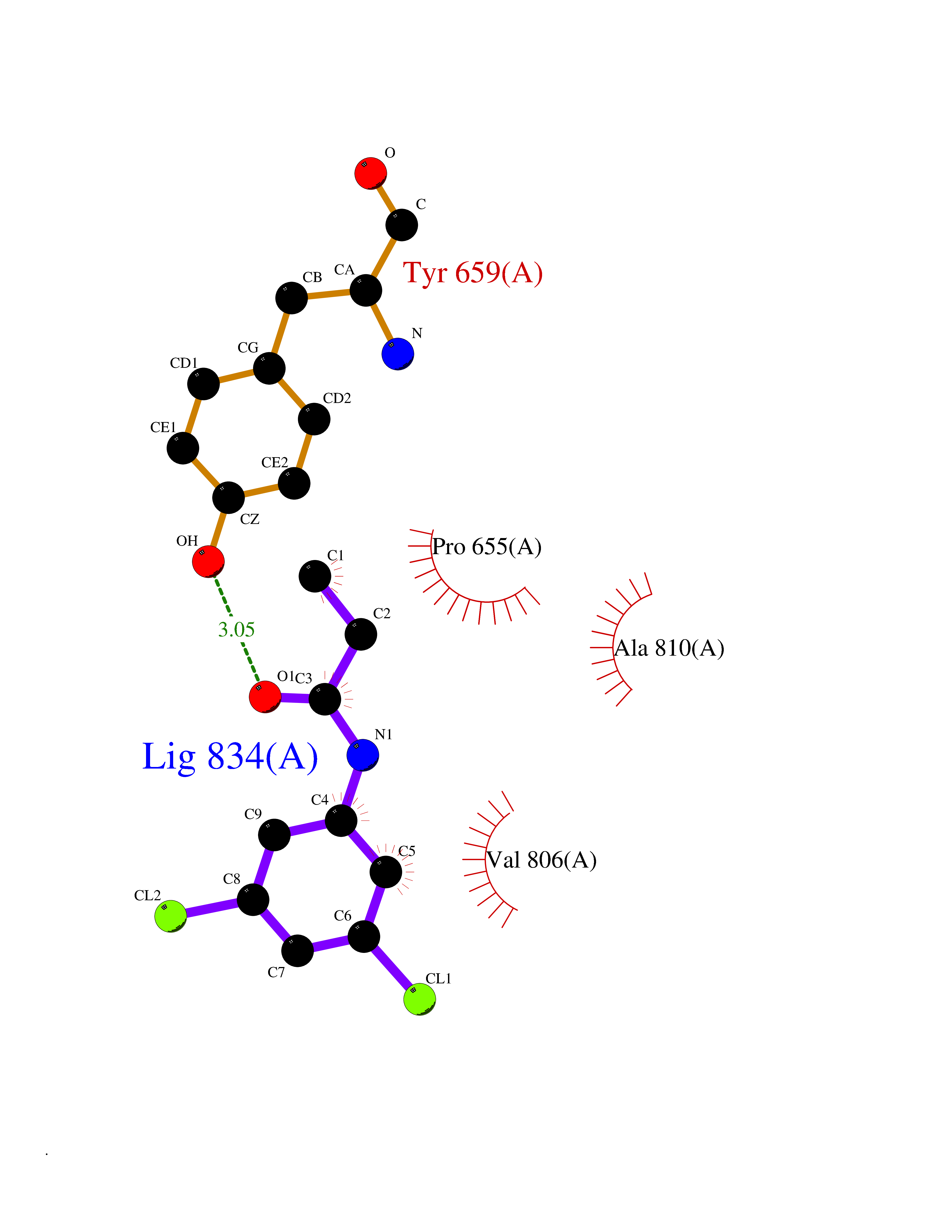 Ligplot Map 3D Binding mode Sequence SPVQYLRWGDPAPIAAVVFACLGLLATLFVTVVFIIYRDTPVVKSSSRELCYIILAGICLGYLCTFXLIAKPKQIYCYLQRIGIGLSPAMSYSALVTKTYRAARILAMSKKSAXAQLVIAFILICIQLGIIVALFIMEPPDIMVYLICNTTNLGVVAPLGYNGLLILACTFYAFKTRNVPANFNEAKYIAFTMYTTCIIWLAFVPIYFGSNYKIITMCFSVSLSATVALGCMFVPKVYIILAKPERN Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Succinyl-CoA:3-ketoacid coenzyme A transferase 1, mitochondrial | 3DLX | 5.87 | |
Target general information Gen name OXCT1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms OXCT;SCOT Protein family 3-oxoacid CoA-transferase family Biochemical class Transferase Function 3-oxoacid CoA-transferase activity.Protein homodimerization activity. Related diseases Succinyl-CoA:3-oxoacid CoA transferase deficiency (SCOTD) [MIM:245050]: A disorder of ketone body metabolism, characterized by episodic ketoacidosis. Patients are usually asymptomatic between episodes. {ECO:0000269|PubMed:10964512, ECO:0000269|PubMed:21296660, ECO:0000269|PubMed:31073471, ECO:0000269|PubMed:33596448, ECO:0000269|PubMed:9671268}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02731; DB00139 Interacts with NA EC number 2.8.3.5 Uniprot keywords 3D-structure; Alternative splicing; Direct protein sequencing; Disease variant; Lipid metabolism; Mitochondrion; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Transit peptide Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 49904.1 Length 459 Aromaticity 0.07 Instability index 28.89 Isoelectric point 7.05 Charge (pH=7) 0.12 2D Binding mode Binding energy (Kcal/mol) -8.01 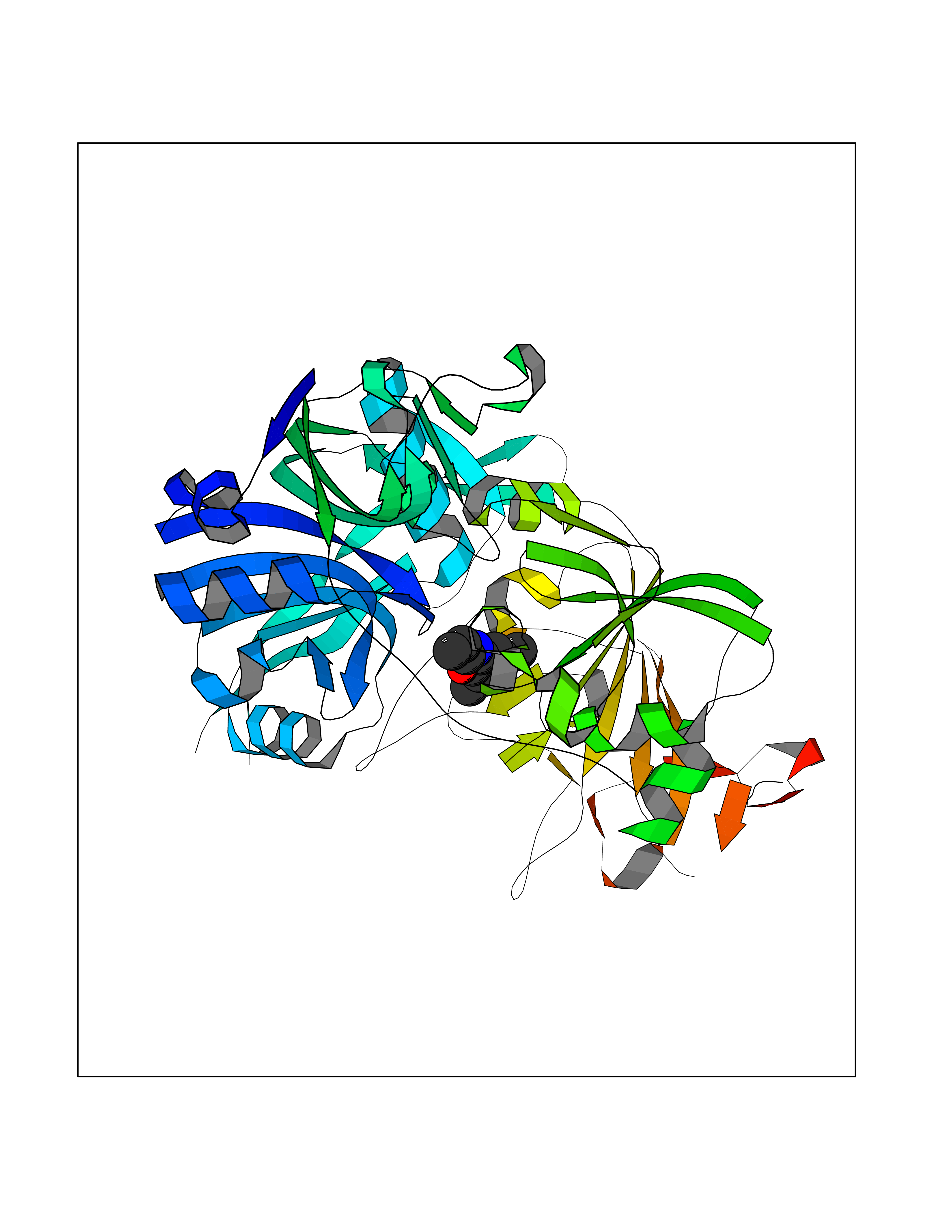 Molscript Map 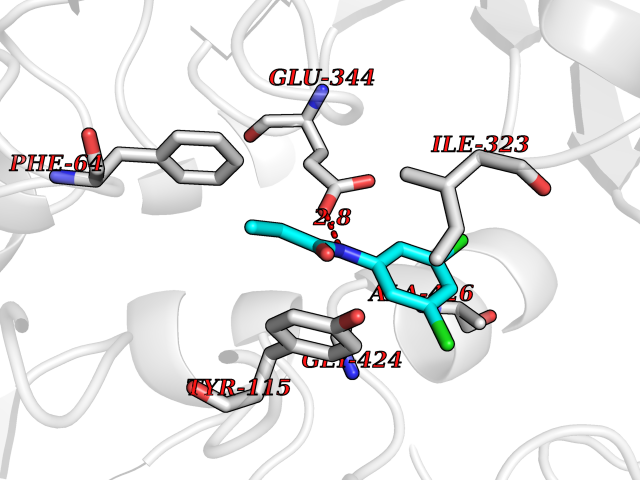 Pymol Map 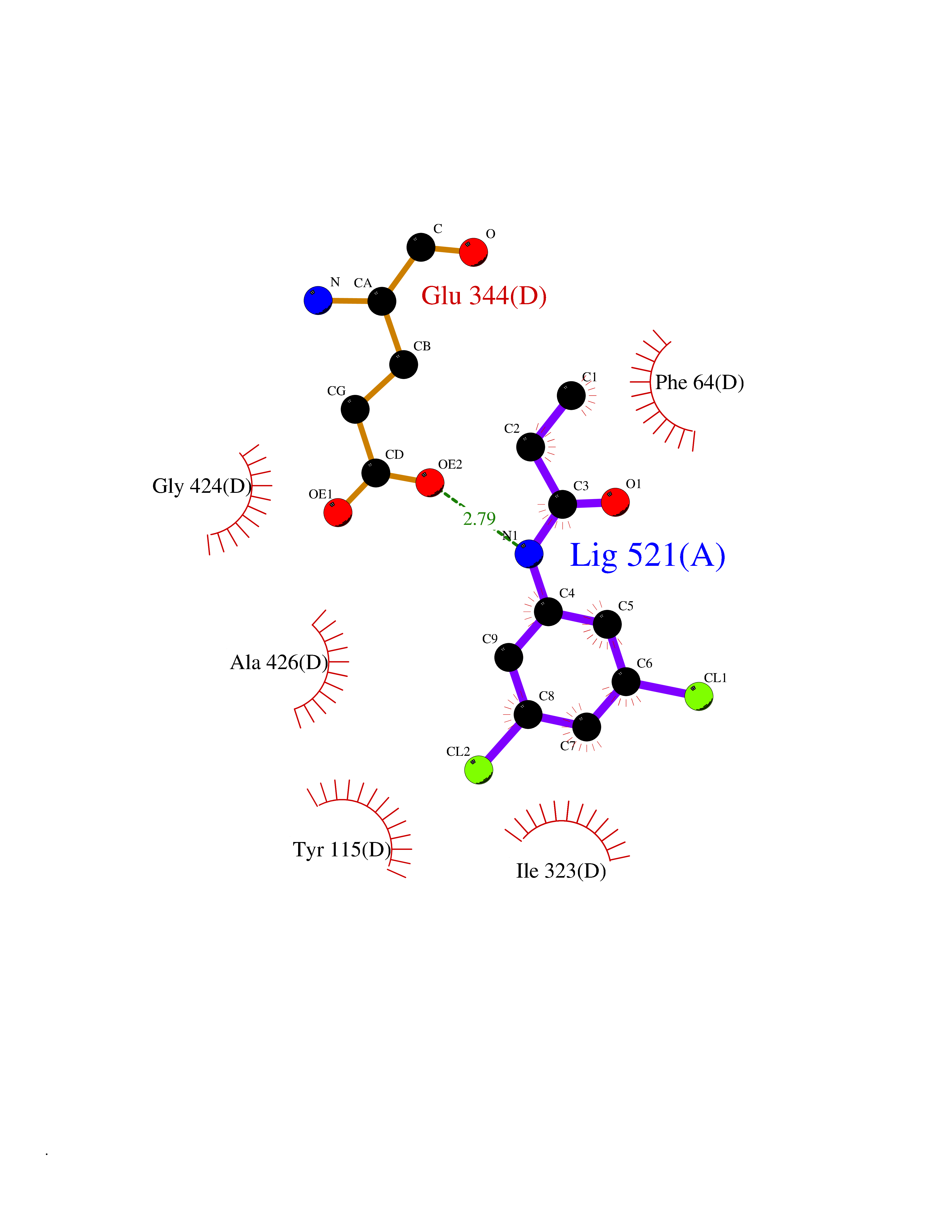 Ligplot Map 3D Binding mode Sequence TKFYTDPVEAVKDIPDGATVLVGGFGLCGIPENLIDALLKTGVKGLTAVSNNAGVDNFGLGLLLRSKQIKRMVSSYVGENAEFERQYLSGELEVELTPQGTLAERIRAGGAGVPAFYTPTGYGTLVQEGGSPIKYNKGSVAIASKPREVREFNGQHFILEEAITGDFALVKAWKADRAGNVIFRKSARNFNLPMCKAAETTVVEVEEIVDIGAFAPEDIHIPQIYVHRLIKGEKYEKRIERLSIRKRERIIKRAALEFEDGMYANLGIGIPLLASNFISPNITVHLQSENGVLGLGPYPRQHEADADLINAGKETVTILPGASFFSSDESFAMIRGGHVDLTMLGAMQVSKYGDLANWMIPGKMVKGMGGAMDLVSSAKTKVVVTMEHSAKGNAHKIMEKCTLPLTGKQCVNRIITEKAVFDVDKKKGLTLIELWDDVQKSTGCDFAVSPKLMPMQQIA Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Sodium channel subunit beta-3 | 4L1D | 5.87 | |
Target general information Gen name SCN3B Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms KIAA1158 Protein family Sodium channel auxiliary subunit SCN3B (TC 8.A.17) family Biochemical class Membrane protein Function Ion channel binding.Sodium channel regulator activity.Voltage-gated sodium channel activity.Voltage-gated sodium channel activity involved in cardiac muscle cell action potential. Related diseases Brugada syndrome 7 (BRGDA7) [MIM:613120]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:20031595}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Atrial fibrillation, familial, 16 (ATFB16) [MIM:613120]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:20558140, ECO:0000269|PubMed:21051419}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05541; DB00907; DB13269; DB13961; DB00776; DB00243; DB00313; DB00909 Interacts with Q15848; Q9NRZ5; Q9NUQ2; Q86W74-2; Q96PS8; Q9H2C2; Q9HD20-3; P27449; Q12981; Q12983; Q6PL45-2; Q4LDR2; P78329; P81534; Q9BUN8; Q6ZPD8; Q9UKR5; Q96D05-2; Q92520; Q9Y3D6; Q14318; Q14802-3; Q8WWP7; Q96F15; Q8TDV0; Q9BZJ8; Q9Y5U9; Q9Y5U4; Q6H9L7; P11215; Q68G75; O95214; Q9UBY5; Q969L2; Q9P0N8; Q6N075; O14880; Q5J8X5; Q9NZG7; Q9P0S3; Q53FV1; Q8NHP8; Q04941; Q96GM1; Q01453; Q02161-2; O76064; Q96GQ5; Q9NTJ5; O00767; Q96IW7; Q8N6R1; Q8WV19; Q9H9B4; Q9BWM7; P22732; Q99726; Q8IWU4; Q96G79; Q6ICL7; Q9H2H9; Q9NVC3; P30825; Q9NRQ5; B2RUZ4; P0DN84; Q13277; O43752; Q9UNK0; Q9UPZ6; O14925; Q9BZW4; P55061; Q9BXJ8; A0PK00; Q5BJH2-2; Q9NV12; Q9Y6G1; Q9NUH8; Q9BTX3; Q8WW34-2; Q9BU79; Q69YG0; P56557; Q9H2L4; Q6PI78; Q8N2M4; Q6ZT21; Q6ZUI0; O60636; A5PKU2; Q9NYZ1; Q9Y5Z9; Q9NZ43; P23763-3; P63027; Q15836; Q9P0L0; O95292; O95070; Q9BSR8; O95159 EC number NA Uniprot keywords 3D-structure; Atrial fibrillation; Brugada syndrome; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Immunoglobulin domain; Ion channel; Ion transport; Membrane; Proteomics identification; Reference proteome; Signal; Sodium; Sodium channel; Sodium transport; Transmembrane; Transmembrane helix; Transport; Voltage-gated channel Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 38121 Length 332 Aromaticity 0.09 Instability index 42.72 Isoelectric point 6.59 Charge (pH=7) -1.45 2D Binding mode Binding energy (Kcal/mol) -8.01 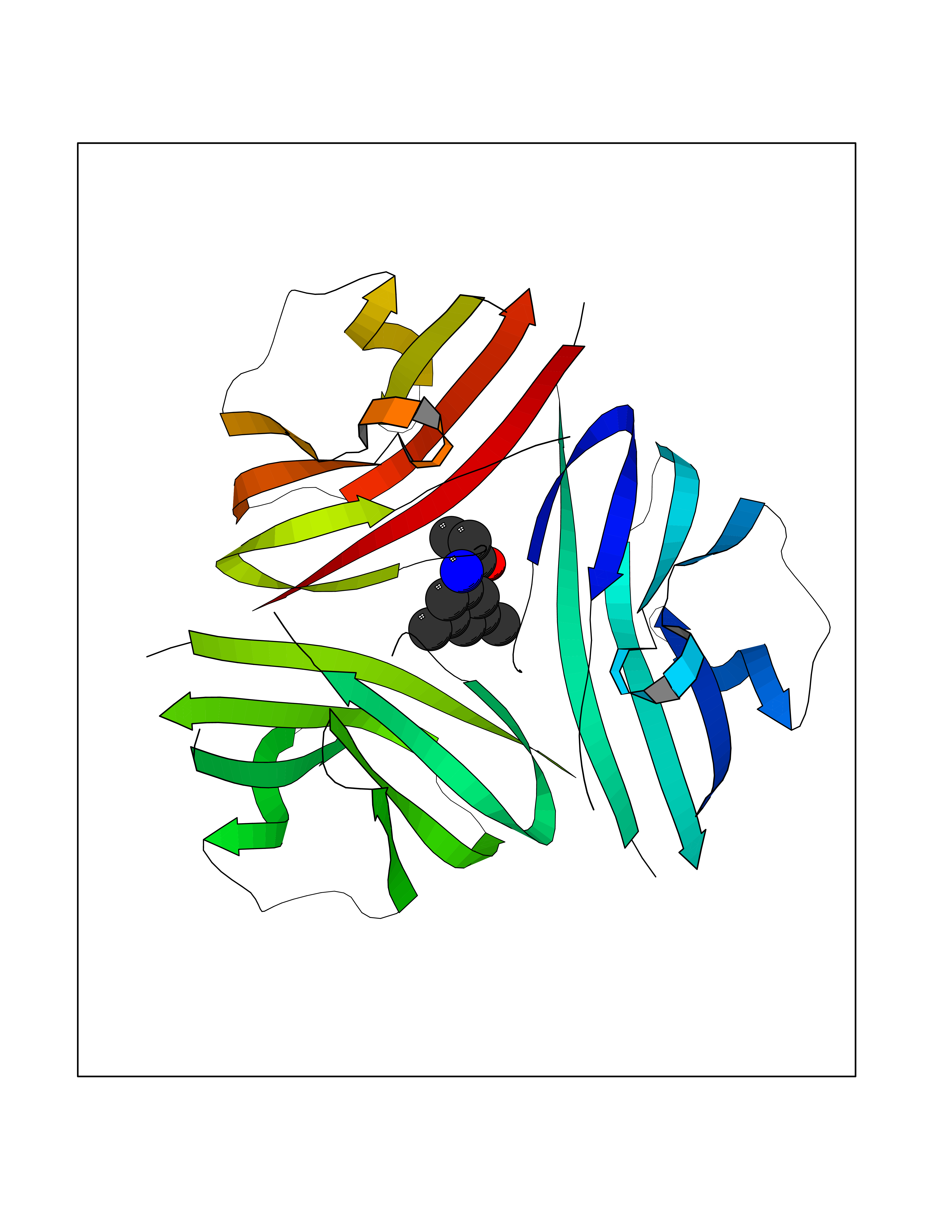 Molscript Map 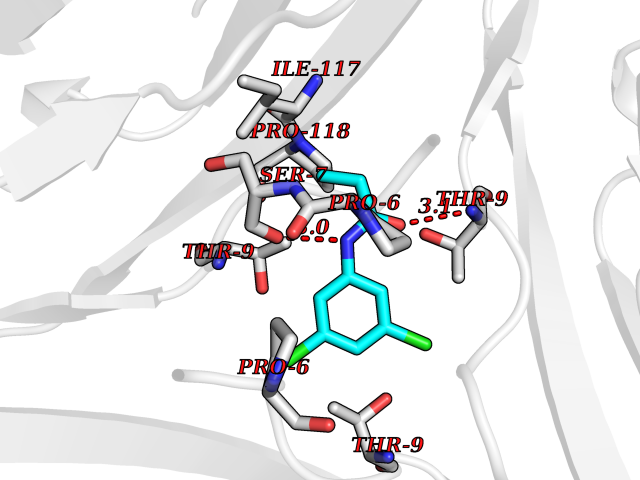 Pymol Map 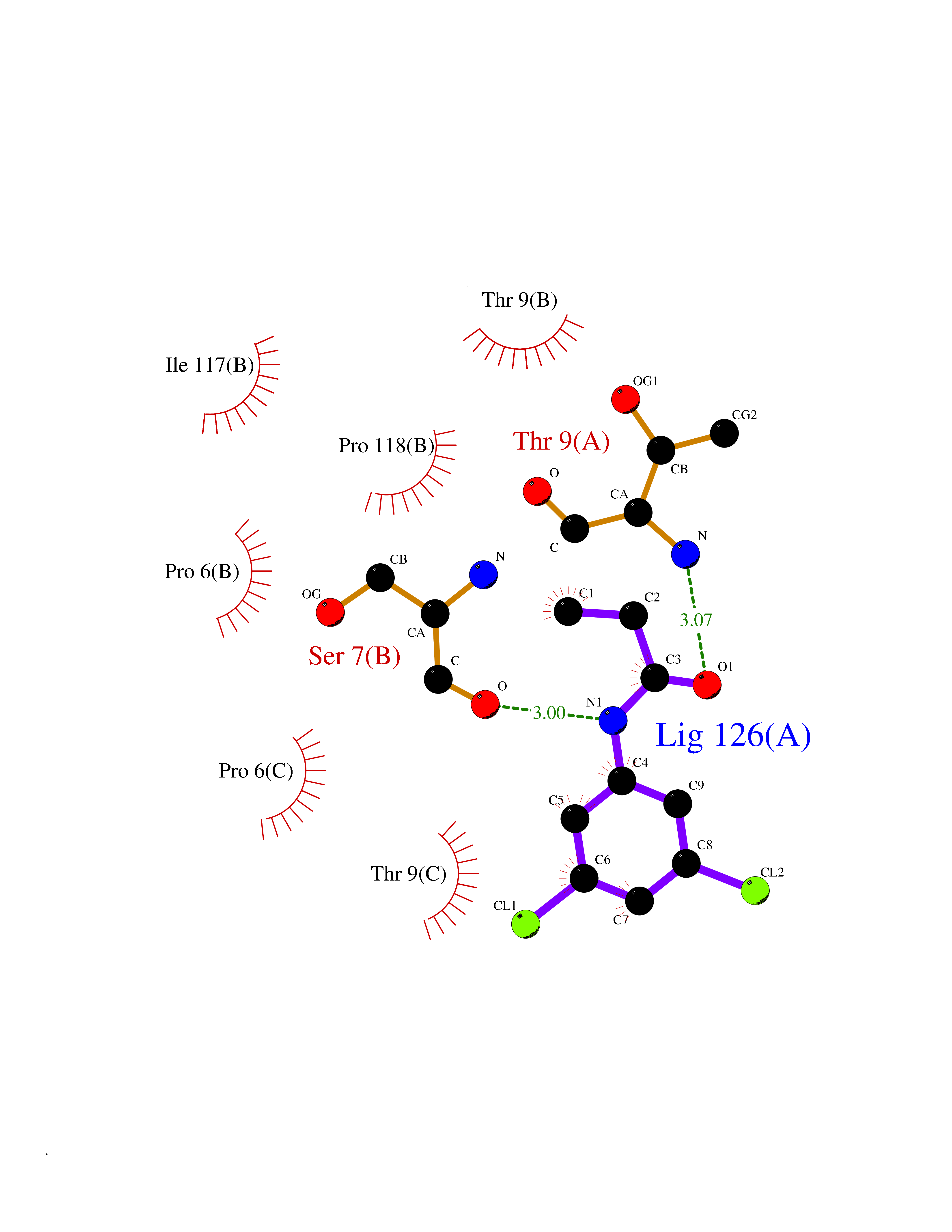 Ligplot Map 3D Binding mode Sequence VCVEVPSETEAVQGNPMKLRCISCMKRATTVVEWFYRPEGGKDFLIYEYRNGHQEVESPFQGRLQWNGSKDLQDVSITVLNVTLNDSGLYTCNVSREFFVKTTRLIPLRVHHHCVEVPSETEAVQGNPMKLRCISCMKATTVVEWFYRPEGGKDFLIYEYRNGHQEVESPFQGRLQWNGSKDLQDVSITVLNVTLNDSGLYTCNVSREFVKTTRLIPLRVHHVCVEVPSETEAVQGNPMKLRCISCMKATTVVEWFYRPEGGKDFLIYEYRNGHQEVESPFQGRLQWNGSKDLQDVSITVLNVTLNDSGLYTCNVSREFVKTTRLIPLRVHH Hydrogen bonds contact Hydrophobic contact | ||||