Job Results:
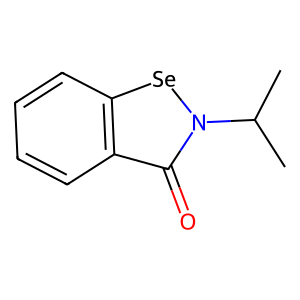
Ligand
Structure
Job ID
0e55eb5f8c726e55360091fb91d44f8e
Job name
NA
Time
2025-12-22 14:49:06
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Optic atrophy protein 1 (OPA1) | 6JTG | 5.74 | |
Target general information Gen name OPA1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms OPA1; Dynaminlike 120 kDa protein, mitochondrial; Dynaminlike 120 kDa protein, form S1 Protein family TRAFAC class dynamin-like GTPase superfamily, Dynamin/Fzo/YdjA family Biochemical class Carbon-carbon hydrolase Function Dynamin-like 120 kDa protein, form S1: Inactive form produced by cleavage at S1 position by OMA1 following stress conditions that induce loss of mitochondrial membrane potential, leading to negative regulation of mitochondrial fusion. Related diseases Optic atrophy 1 (OPA1) [MIM:165500]: A condition that features progressive visual loss in association with optic atrophy. Atrophy of the optic disk indicates a deficiency in the number of nerve fibers which arise in the retina and converge to form the optic disk, optic nerve, optic chiasm and optic tracts. OPA1 is characterized by an insidious onset of visual impairment in early childhood with moderate to severe loss of visual acuity, temporal optic disk pallor, color vision deficits, and centrocecal scotoma of variable density. {ECO:0000269|PubMed:11017079, ECO:0000269|PubMed:11017080, ECO:0000269|PubMed:11440988, ECO:0000269|PubMed:11440989, ECO:0000269|PubMed:11810270, ECO:0000269|PubMed:12036970, ECO:0000269|PubMed:12566046, ECO:0000269|PubMed:14961560, ECO:0000269|PubMed:15948788, ECO:0000269|PubMed:16513463, ECO:0000269|PubMed:16617242, ECO:0000269|PubMed:18204809, ECO:0000269|PubMed:18360822, ECO:0000269|PubMed:19319978, ECO:0000269|PubMed:19325939, ECO:0000269|PubMed:19969356, ECO:0000269|PubMed:20185555, ECO:0000269|PubMed:22382025, ECO:0000269|PubMed:22857269, ECO:0000269|PubMed:23401657}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Dominant optic atrophy plus syndrome (DOA+) [MIM:125250]: A neurologic disorder characterized most commonly by an insidious onset of visual loss and sensorineural hearing loss in childhood with variable presentation of other clinical manifestations including progressive external ophthalmoplegia, muscle cramps, hyperreflexia, and ataxia. There appears to be a wide range of intermediate phenotypes. {ECO:0000269|PubMed:15531309, ECO:0000269|PubMed:16240368, ECO:0000269|PubMed:18065439, ECO:0000269|PubMed:18158317, ECO:0000269|PubMed:18195150, ECO:0000269|PubMed:20185555, ECO:0000269|PubMed:21112924, ECO:0000269|PubMed:23387428}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Behr syndrome (BEHRS) [MIM:210000]: An autosomal recessive syndrome characterized by optic atrophy beginning in early childhood associated with ataxia, pyramidal signs, spasticity, intellectual disability, and posterior column sensory loss. The ataxia, spasticity, and muscle contractures, mainly of the hip adductors, hamstrings, and soleus, are progressive and become more prominent in the second decade. {ECO:0000269|PubMed:21636302, ECO:0000269|PubMed:25012220, ECO:0000269|PubMed:25146916}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mitochondrial DNA depletion syndrome 14, cardioencephalomyopathic type (MTDPS14) [MIM:616896]: An autosomal recessive mitochondrial disorder characterized by lethal infantile encephalopathy, hypertrophic cardiomyopathy and optic atrophy. Skeletal muscle biopsies show significant mtDNA depletion and abnormal mitochondria. {ECO:0000269|PubMed:26561570}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q12983; Q5S007; Q9NTG7 EC number EC 3.6.5.5 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Apoptosis; Cardiomyopathy; Coiled coil; Deafness; Disease variant; Disulfide bond; GTP-binding; Hydrolase; Lipid-binding; Membrane; Mitochondrion; Mitochondrion inner membrane; Neurodegeneration; Nucleotide-binding; Primary mitochondrial disease; Proteomics identification; Reference proteome; Sensory transduction; Transit peptide; Transmembrane; Transmembrane helix; Vision Protein physicochemical properties Chain ID A Molecular weight (Da) 36858.7 Length 330 Aromaticity 0.06 Instability index 41.24 Isoelectric point 5.51 Charge (pH=7) -7.89 2D Binding mode Binding energy (Kcal/mol) -7.83 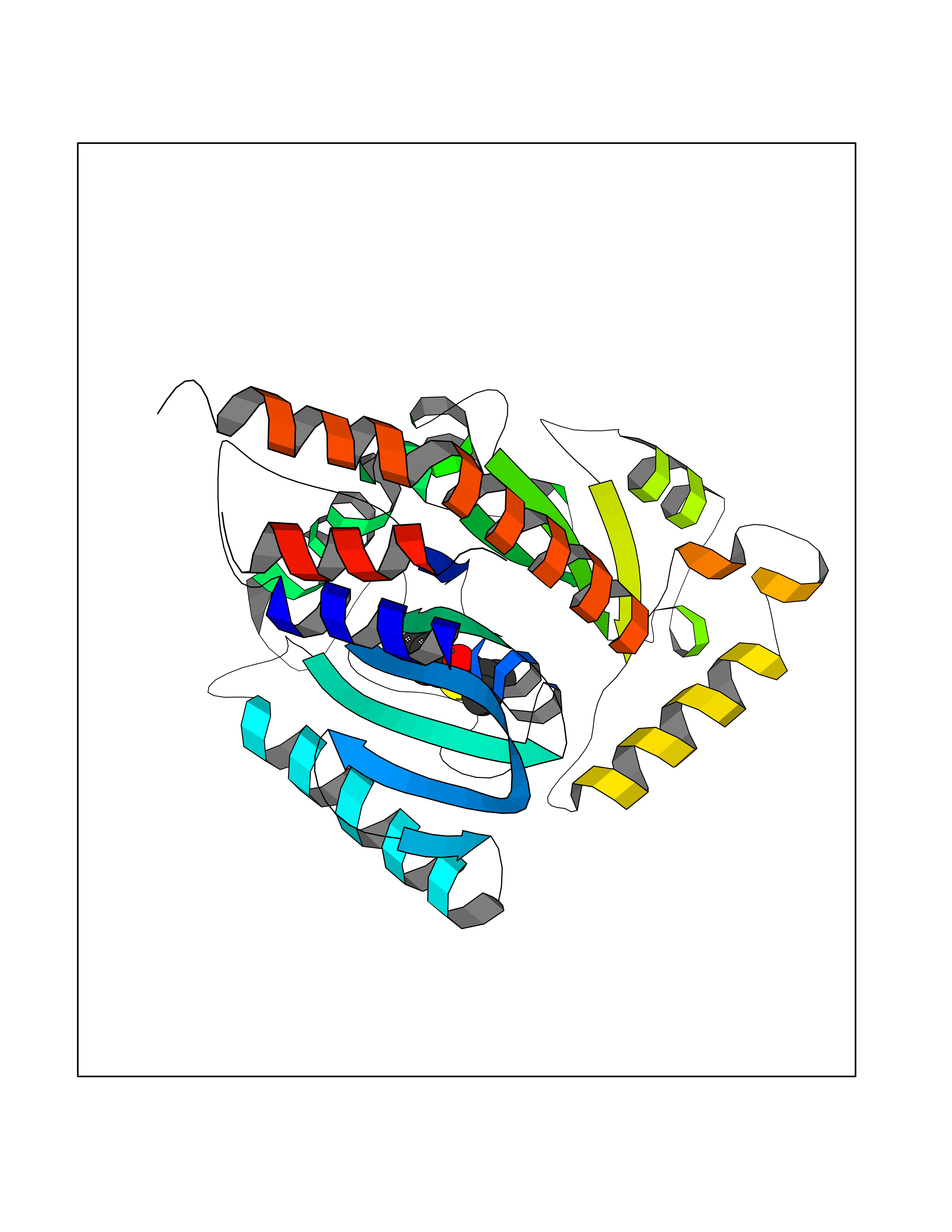 Molscript Map 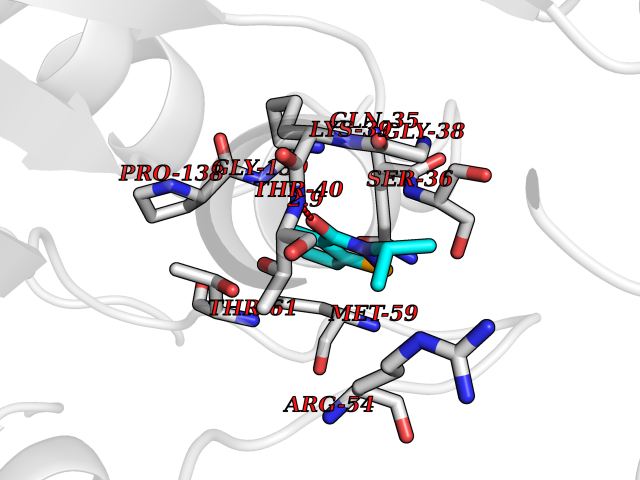 Pymol Map 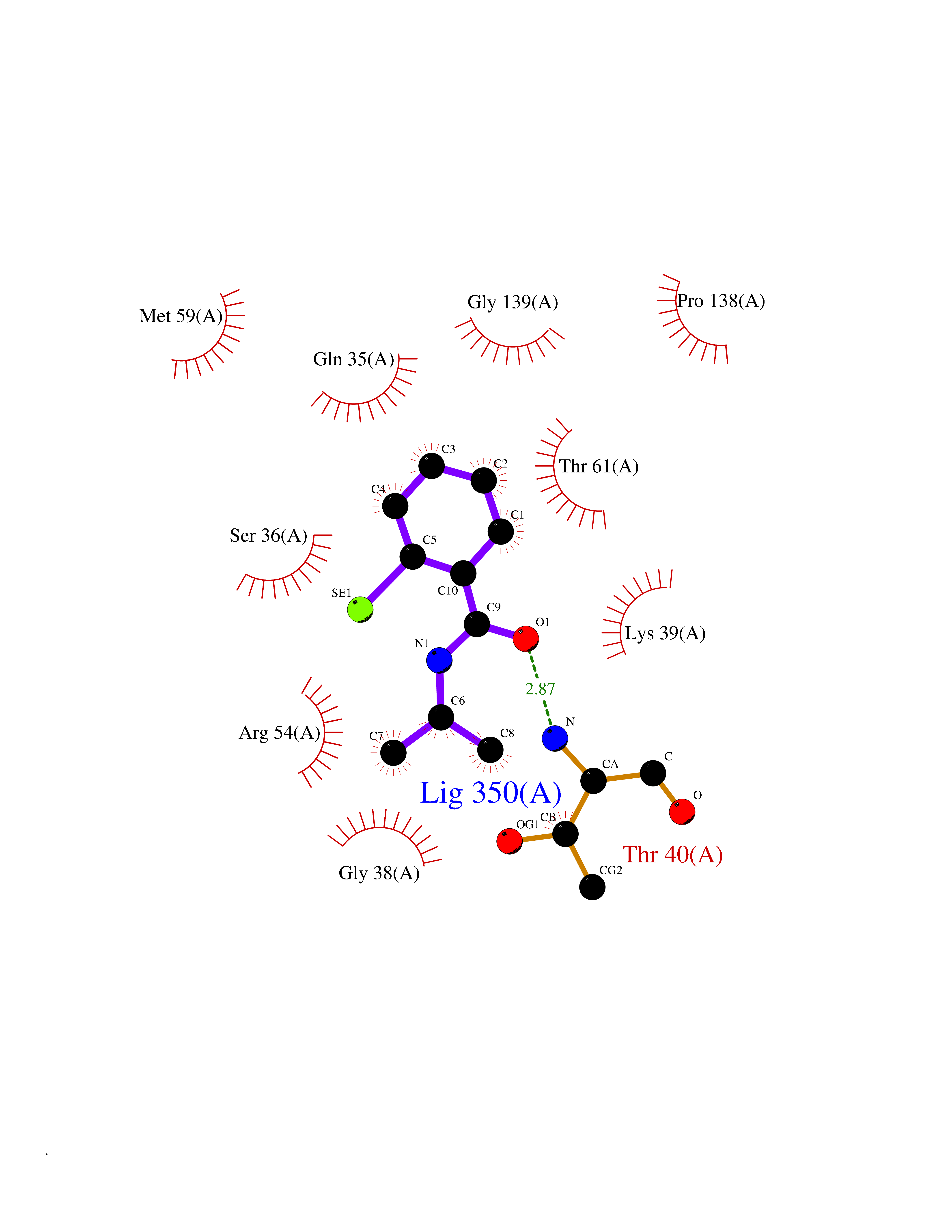 Ligplot Map 3D Binding mode Sequence SLIDMYSEVLDVLSDYDASYNTQDHLPRVVVVGDQSAGKTSVLEMIAQARIFPRGSGEMMTRSPVKVTLSEGPHHVALFKDSSREFDLTKEEDLAALRHEIELRMRKNVKEGCTVSPETISLNVKGPGLQRMVLVDLPGVINTVTSGMAPDTKETIFSISKAYMQNPNAIILCIQDGSVDAERSIVTDLVSQMDPHGRRTIFVLTKVDLAEKNVASPSRIQQIIEGKLFPMKALGYFAVVTGKGNSSESIEAIREYEEEFFQNSKLLKTSMLKAHQVTTRNLSLAVSDCFWKMVRESVEQQADSFKATRFNLEVREIQEKLDAFIEALHQ Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Potassium channel subfamily K member 2 | 4TWK | 5.73 | |
Target general information Gen name KCNK2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms TREK;TREK1 Protein family Two pore domain potassium channel (TC 1.A.1.8) family Biochemical class Transport protein Function Outward rectifier potassium channel activity.Potassium channel inhibitor activity.Potassium ion leak channel activity. Related diseases Diaphragmatic hernia 4, with cardiovascular defects (DIH4) [MIM:620025]: An autosomal recessive form of congenital diaphragmatic hernia, a posterolateral defect of the diaphragm, generally located on the left side, that permits the herniation of abdominal viscera into the thorax. The lungs are hypoplastic and have abnormal vessels that cause respiratory insufficiency and persistent pulmonary hypertension with high mortality. About one third of cases have cardiovascular malformations and lesser proportions have skeletal, neural, genitourinary, gastrointestinal or other defects. {ECO:0000269|PubMed:33565183, ECO:0000269|PubMed:36263470}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00204; DB04855 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Cell projection; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Ion channel; Ion transport; Membrane; Metal-binding; Phosphoprotein; Postsynaptic cell membrane; Potassium; Potassium channel; Potassium transport; Proteomics identification; Reference proteome; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 18512.8 Length 171 Aromaticity 0.08 Instability index 33.48 Isoelectric point 5.34 Charge (pH=7) -5.71 2D Binding mode Binding energy (Kcal/mol) -7.81 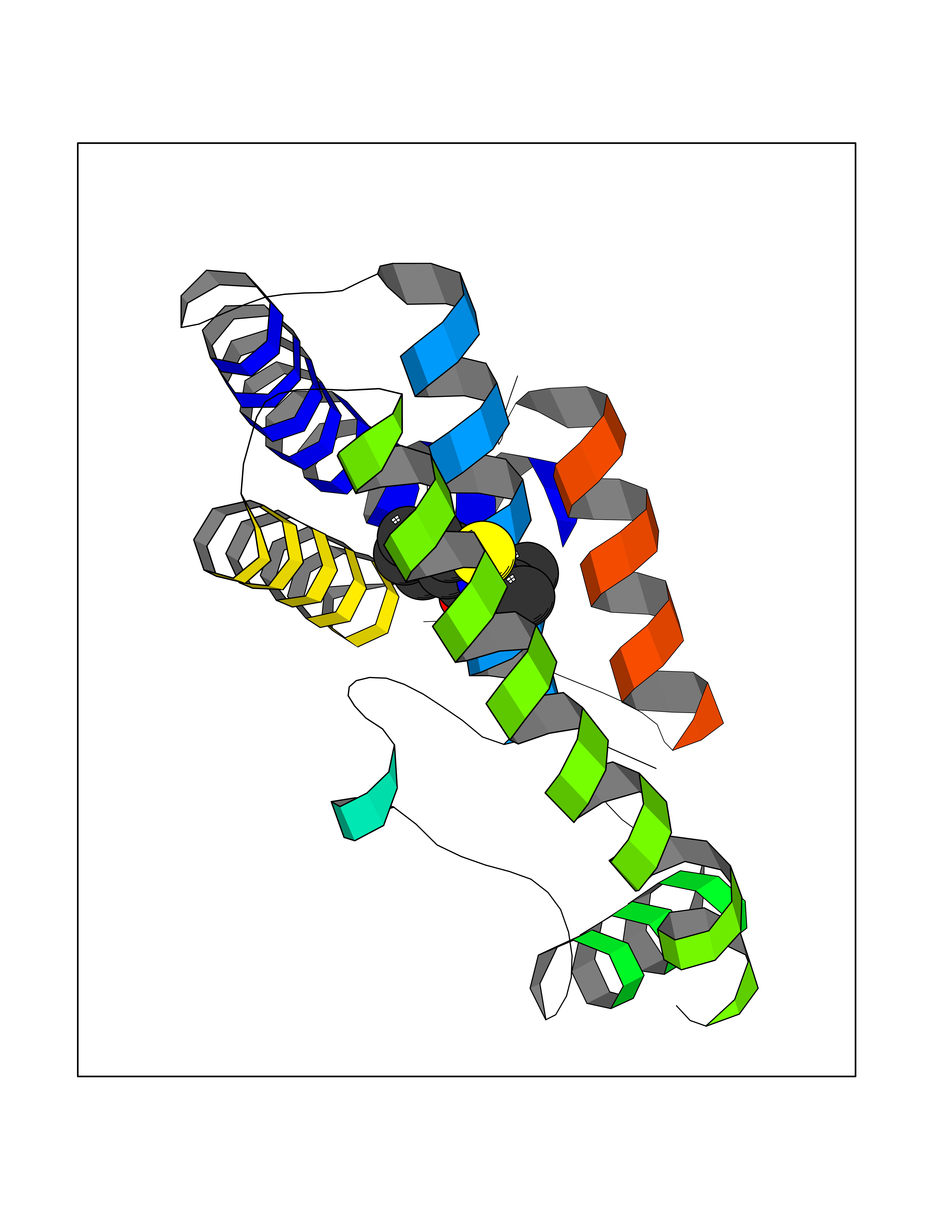 Molscript Map 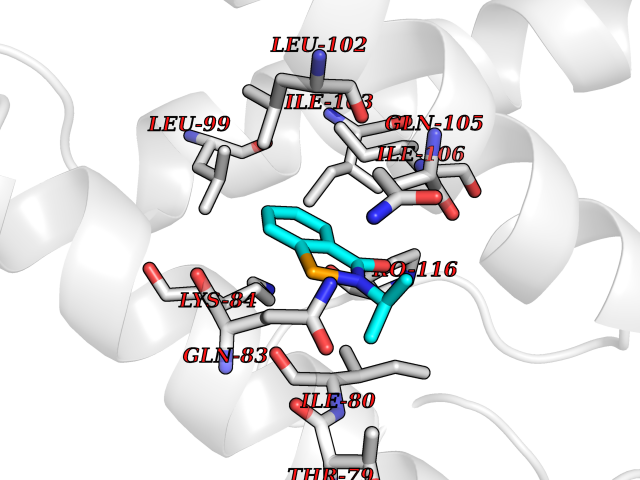 Pymol Map 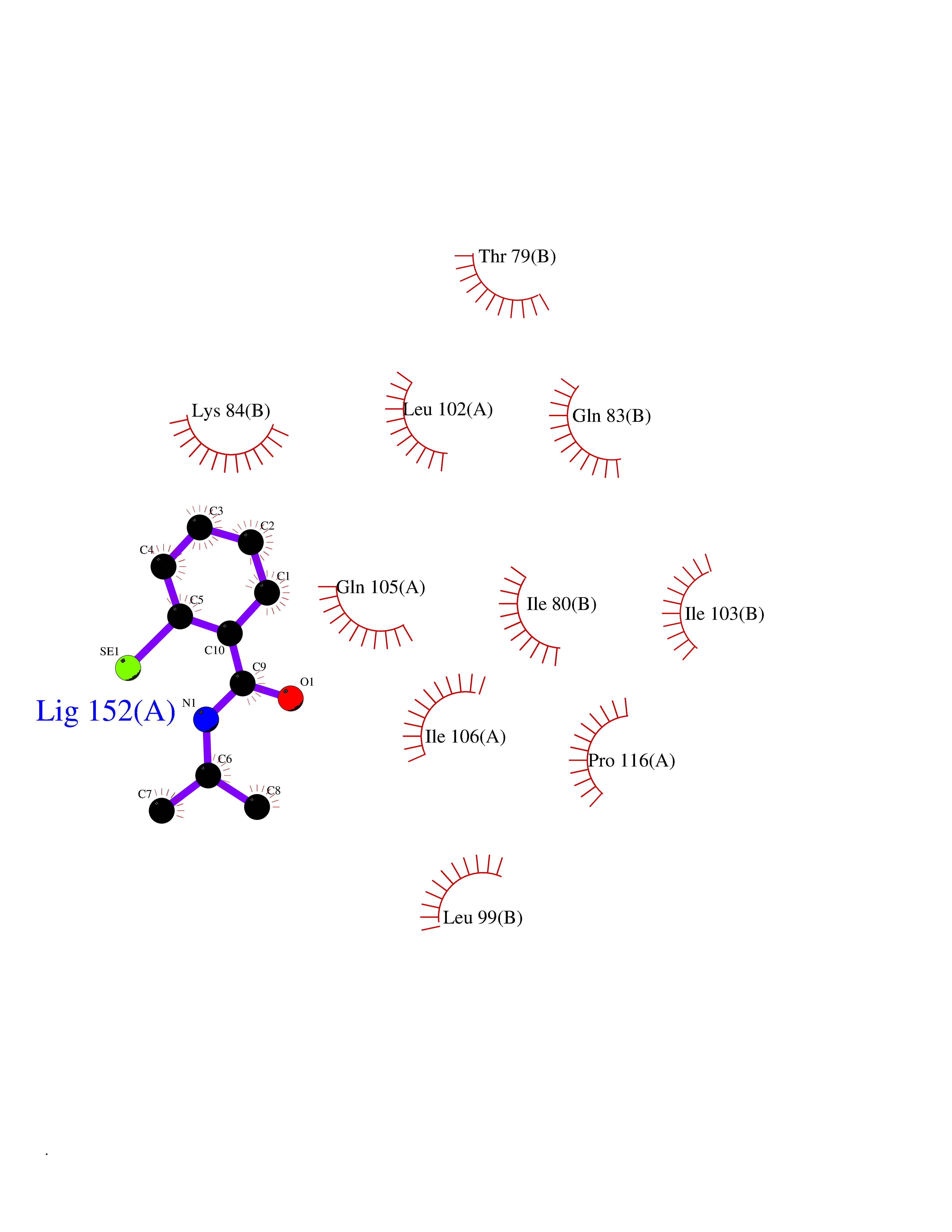 Ligplot Map 3D Binding mode Sequence GATVFKALEQPHEISQRTTIVIQKQTFISQHSCVNSTELDELIQQIVAAINAGIIPLGNTSNQISHWDLGSSFFFAGTVITTIGFGNISATVFKALEQPHEISQRTTIVIQKQTFISQHSCVNSTELDELIQQIVAAINAGIIPISHWDLGSSFFFAGTVITTIGFGNISP Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Pyruvate synthase | 2C42 | 5.73 | |
Target general information Gen name por Organism Desulfocurvibacter africanus (Desulfovibrio africanus) Uniprot ID TTD ID NA Synonyms NA Protein family Pyruvate:ferredoxin/flavodoxin oxidoreductase family Biochemical class Oxidoreductase Function 4 iron, 4 sulfur cluster binding.Iron ion binding.Pyruvate synthase activity.Thiamine pyrophosphate binding. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02410; DB01987; DB00507 Interacts with NA EC number 1.2.7.1 Uniprot keywords 3D-structure; 4Fe-4S; Calcium; Cytoplasm; Direct protein sequencing; Disulfide bond; Electron transport; Iron; Iron-sulfur; Magnesium; Metal-binding; Oxidoreductase; Pyruvate; Thiamine pyrophosphate; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 115569 Length 1065 Aromaticity 0.09 Instability index 31.51 Isoelectric point 6.32 Charge (pH=7) -5.62 2D Binding mode Binding energy (Kcal/mol) -7.81 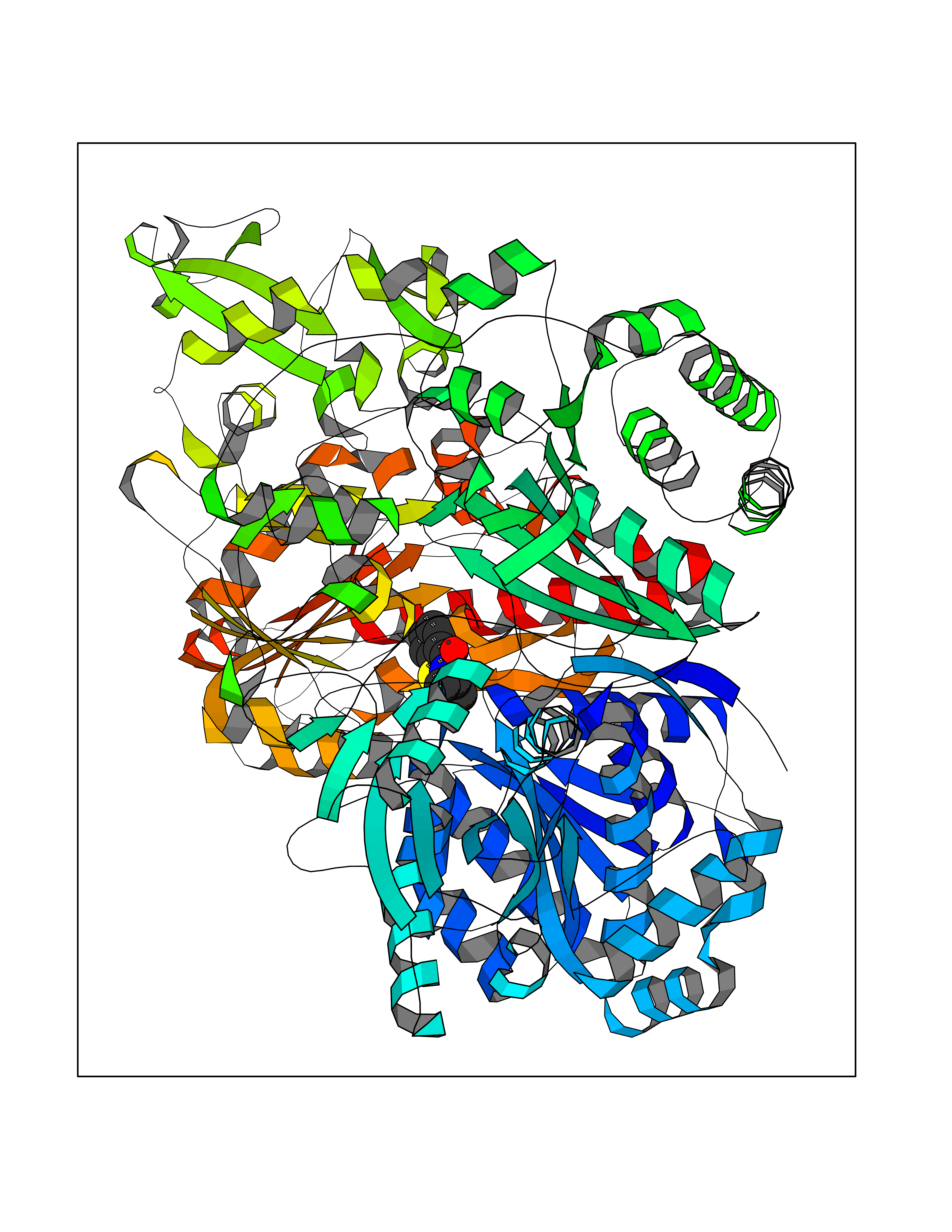 Molscript Map 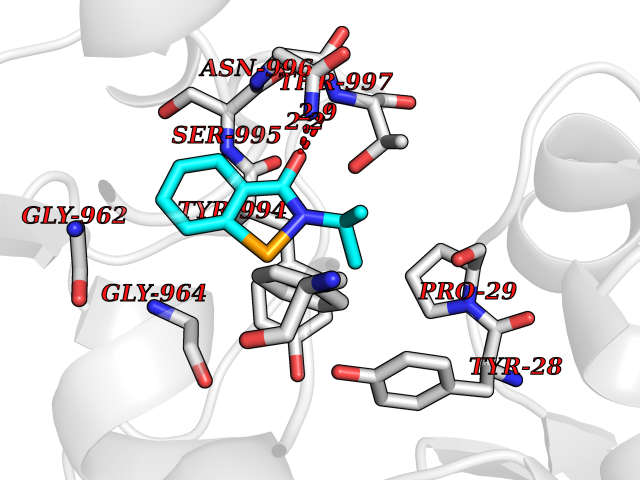 Pymol Map 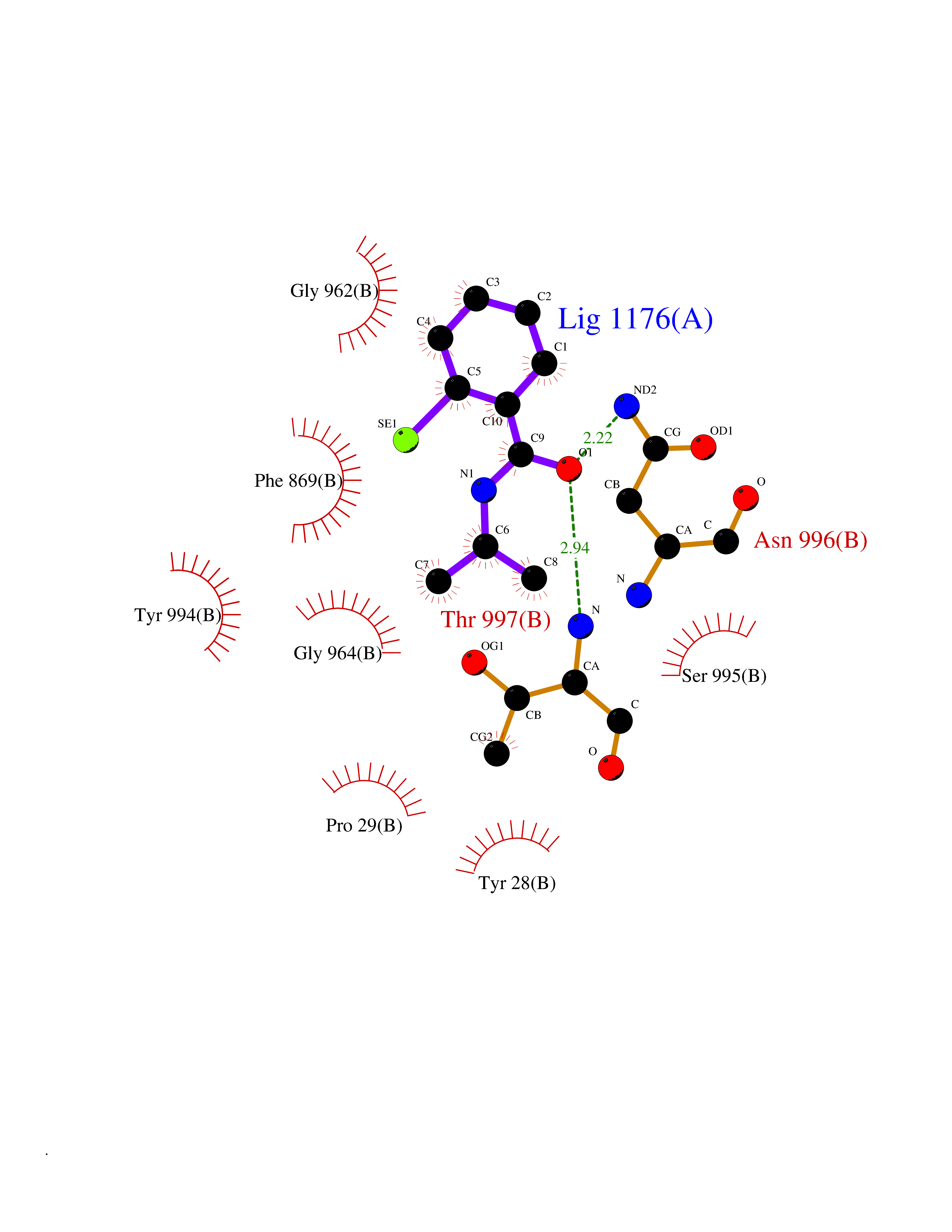 Ligplot Map 3D Binding mode Sequence GKKMMTTDGNTATAHVAYAMSEVAAIYPITPSSTMGEEADDWAAQGRKNIFGQTLTIREMQSEAGAAGAVHGALAAGALTTTFTASQGLLLMIPNMYKISGELLPGVFHVTARAIAAHALSIFGDHQDIYAARQTGFAMLASSSVQEAHDMALVAHLAAIESNVPFMHFFDGFRTSHEIQKIEVLDYADMASLVNQKALAEFRAKSPGIVAEYMQKVASLTGRSYKLFDYVGAPDAERVIVSMGSSCETIEEVINHLAAKGEKIGLIKVRLYRPFVSEAFFAALPASAKVITVLDRTKEPGAPGDPLYLDVCSAFVERGEAMPKILAGRYGLGSKEFSPAMVKSVYDNMSGAKKNHFTVGIEDDVTGTSLPVDNAFADTTPKGTIQCQFWGLGADGTVGANKQAIKIIGDNTDLFAQGYFSYDSKKSGGITISHLRFGEKPIQSTYLVNRADYVACHNPAYVGIYDILEGIKDGGTFVLNSPWSSLEDMDKHLPSGIKRTIANKKLKFYNIDAVKIATDVGLGGRINMIMQTAFFKLAGVLPFEKAVDLLKKSIHKAYGKKGEKIVKMNTDAVDQAVTSLQEFKYPDSWKDAPAETKAEPMTNEFFKNVVKPILTQQGDKLPVSAFEADGRFPLGTSQFEKRGVAINVPQWVPENCIQCNQCAFVCPHSAILPVLAKEEELVGAPANFTALEAKGKELKGYKFRIQINTLDCMGCGNCADICPPKEKALVMQPLDTQRDAQVPNLEYAARIPVKSEVLPRDSLKGSQFQEPLMEFSGACSGCGETPYVRVITQLFGERMFIANATGCSSIWGASAPSMPYKTNRLGQGPAWGNSLFEDAAEYGFGMSVWIFGGDGWAYDIGYGGLDHVLASGEDVNVFVMDTEVYSNTGGQSSKATPTGAVAKFAAAGKRTGKKDLARMVMTYGYVYVATVSMGYSKQQFLKVLKEAESFPGPSLVIAYATCINQGLRKGMGKSQDVMNTAVKSGYWPLFRYDPRLAAQGKNPFQLDSKAPDGSVEEFLMAQNRFAVLDRSFPEDAKRLRAQVAHELDVRFKELEHMAATNIFES Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Riboflavin kinase | 1NB0 | 5.73 | |
Target general information Gen name RFK Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Transferase Function ATP binding.Metal ion binding.Riboflavin kinase activity. Related diseases Glutaric aciduria 1 (GA1) [MIM:231670]: An autosomal recessive metabolic disorder characterized by progressive dystonia and athetosis due to gliosis and neuronal loss in the basal ganglia. {ECO:0000269|PubMed:14707522, ECO:0000269|PubMed:18775954, ECO:0000269|PubMed:24973495, ECO:0000269|PubMed:8541831, ECO:0000269|PubMed:8900227, ECO:0000269|PubMed:8900228, ECO:0000269|PubMed:9600243, ECO:0000269|PubMed:9711871}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03247; DB00140 Interacts with Q9NXG0-2; P19438; P19438-1 EC number 2.7.1.26 Uniprot keywords 3D-structure; ATP-binding; Cytoplasm; Flavoprotein; FMN; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Proteomics identification; Reference proteome; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 16749.9 Length 147 Aromaticity 0.12 Instability index 41.55 Isoelectric point 7.09 Charge (pH=7) 0.12 2D Binding mode Binding energy (Kcal/mol) -7.81  Molscript Map 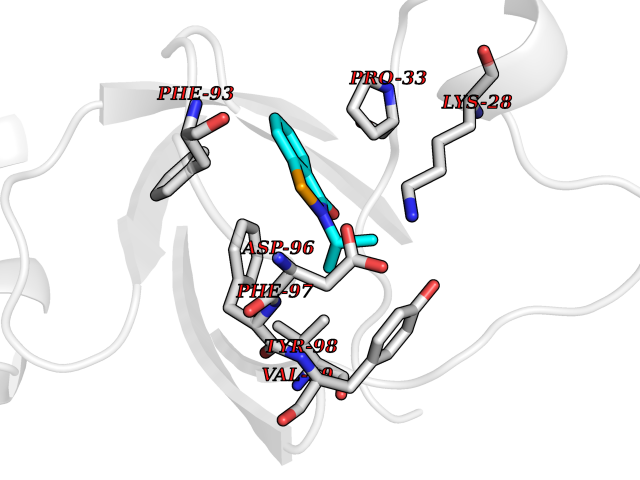 Pymol Map 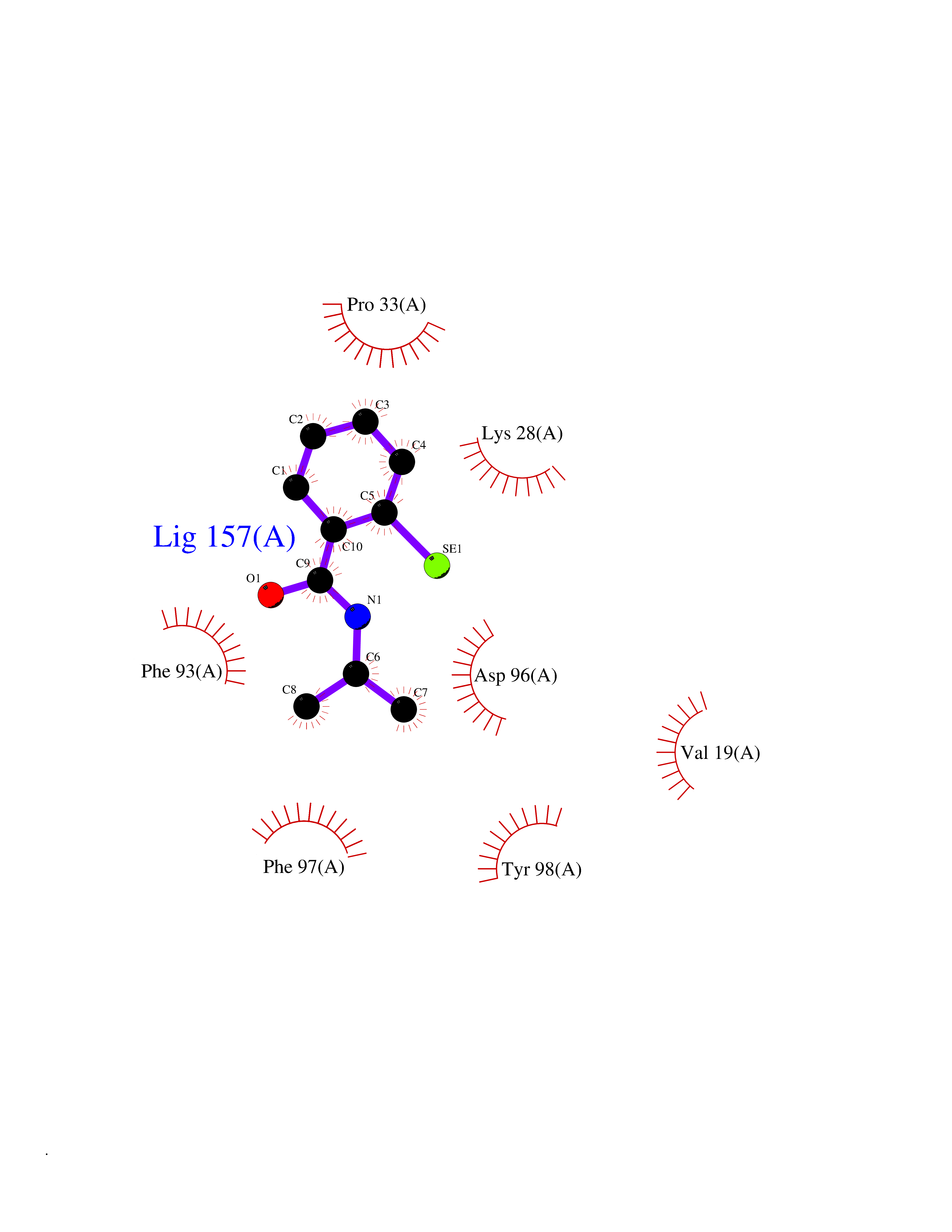 Ligplot Map 3D Binding mode Sequence RHLPYFCRGQVVRGFGRGSKQLGIPTANFPEQVVDNLPADISTGIYYGWASVGSGDVHKMVVSIGWNPYYKNTKKSMETHIMHTFKEDFYGEILNVAIVGYLRPEKNFDSLESLISAIQGDIEEAKKRLELPEYLKIKEDNFFQVSK Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | SEC14-like protein 4 | 4TLG | 5.73 | |
Target general information Gen name SEC14L4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms TAP3 Protein family NA Biochemical class Transport protein Function Lipid binding.Transporter activity. Related diseases Chondrodysplasia with platyspondyly, distinctive brachydactyly, hydrocephaly, and microphthalmia (CDP-PBHM) [MIM:300863]: A disease characterized by chondrodysplasia, severe platyspondyly, hydrocephaly, and facial features with microphthalmia. Bone abnormalities include a distinctive metaphyseal cupping of the metacarpals, metatarsals, and phalanges. Affected females show a milder phenotype with small stature, sometimes associated with body asymmetry and mild intellectual disability. {ECO:0000269|PubMed:20181727}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB11635; DB11251; DB00163 Interacts with Q96LC9; O43186; P78358; Q9NYQ3; Q0VD86; Q15323; O76011; P50221; Q6FHY5; Q02548; P26367; Q9H8W4; Q04864; Q04864-2; Q9UHV2; P15884; P15884-3; Q96N21; Q9BYV2; Q8N6Y0; Q9H0C1 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Lipid-binding; Proteomics identification; Reference proteome; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 23947.6 Length 210 Aromaticity 0.1 Instability index 50.84 Isoelectric point 5.55 Charge (pH=7) -3.11 2D Binding mode Binding energy (Kcal/mol) -7.81  Molscript Map 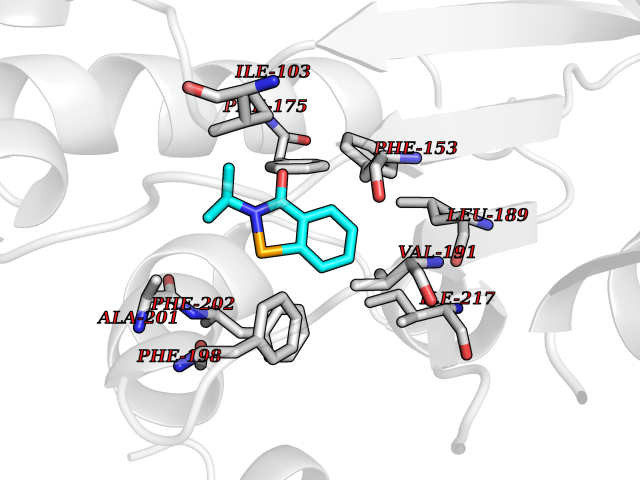 Pymol Map 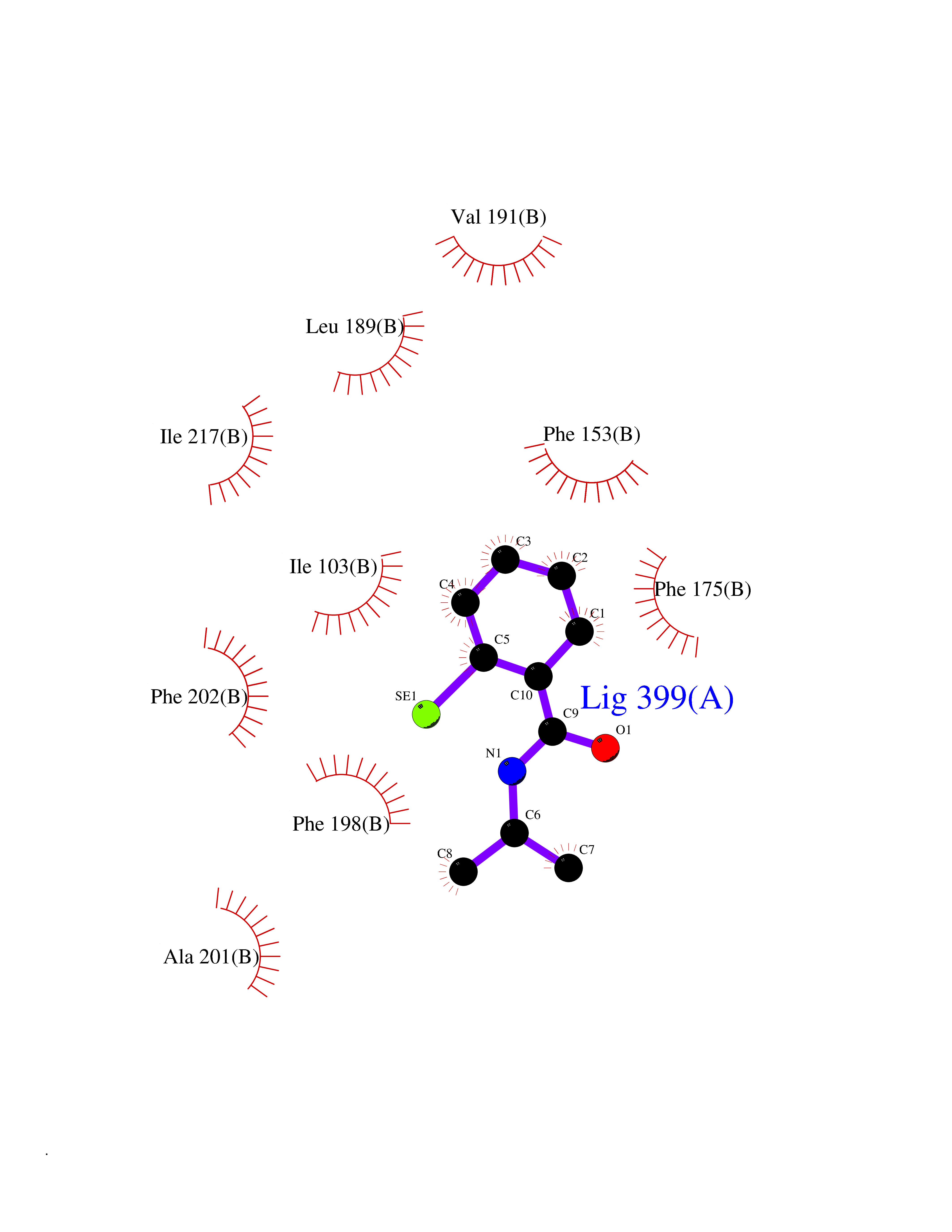 Ligplot Map 3D Binding mode Sequence VTWQPPEVIQLYDSGGLCGYDYEGCPVYFNIIGSLDPKGLLLSASKQDMIRKRIKVCELLLHECELQTQKLGRKIEMALMVFDMEGLSLKHLWKPAVEVYQQFFSILEANYPETLKNLIVIRAPKLFPVAFNLVKSFMSEETRRKIVILGDNWKQELTKFISPDQLPVEFGGTMTDPDGNPKCLTKINYGGEVPKSYYPDKASEETLQSM Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Leucine carboxyl methyltransferase 1 | 3IEI | 5.73 | |
Target general information Gen name LCMT1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CGI-68;LCMT Protein family Methyltransferase superfamily, LCMT family Biochemical class Transferase Function Protein C-terminal carboxyl O-methyltransferase activity.Protein C-terminal leucine carboxyl O-methyltransferase activity.S-adenosylmethionine-dependent methyltransferase activity. Related diseases Neurodevelopmental disorder, mitochondrial, with abnormal movements and lactic acidosis, with or without seizures (NEMMLAS) [MIM:617710]: An autosomal recessive, mitochondrial disorder with a broad phenotypic spectrum ranging from severe neonatal lactic acidosis, encephalomyopathy and early death to an attenuated course with milder manifestations. Clinical features include delayed psychomotor development, intellectual disability, hypotonia, dystonia, ataxia, and spasticity. Severe combined respiratory chain deficiency may be found in severely affected individuals. {ECO:0000269|PubMed:28236339, ECO:0000269|PubMed:28650581, ECO:0000269|PubMed:28905505, ECO:0000269|PubMed:30920170, ECO:0000269|PubMed:35074316}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Parkinsonism-dystonia 3, childhood-onset (PKDYS3) [MIM:619738]: An autosomal recessive neurodegenerative disorder with onset in infancy or early childhood. Affected individuals present with progressive movement abnormalities, including parkinsonism with tremor, dystonia, myoclonus ataxia, and hyperkinetic movements such as ballismus. The parkinsonism features may be responsive to treatment with levodopa, although many patients develop levodopa-induced dyskinesia. Some patients may have mild cognitive impairment or psychiatric disturbances. {ECO:0000269|PubMed:29120065, ECO:0000269|PubMed:31970218, ECO:0000269|PubMed:34890876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00149 Interacts with P51116 EC number 2.1.1.233 Uniprot keywords 3D-structure; Alternative splicing; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H Molecular weight (Da) 35803 Length 310 Aromaticity 0.08 Instability index 42.77 Isoelectric point 6.13 Charge (pH=7) -3.58 2D Binding mode Binding energy (Kcal/mol) -7.81  Molscript Map 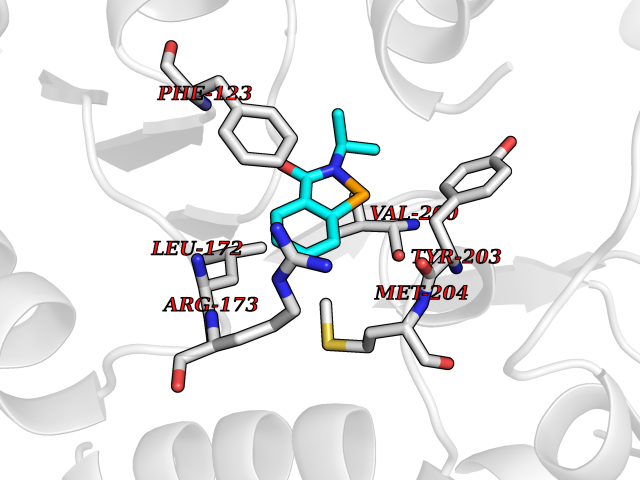 Pymol Map 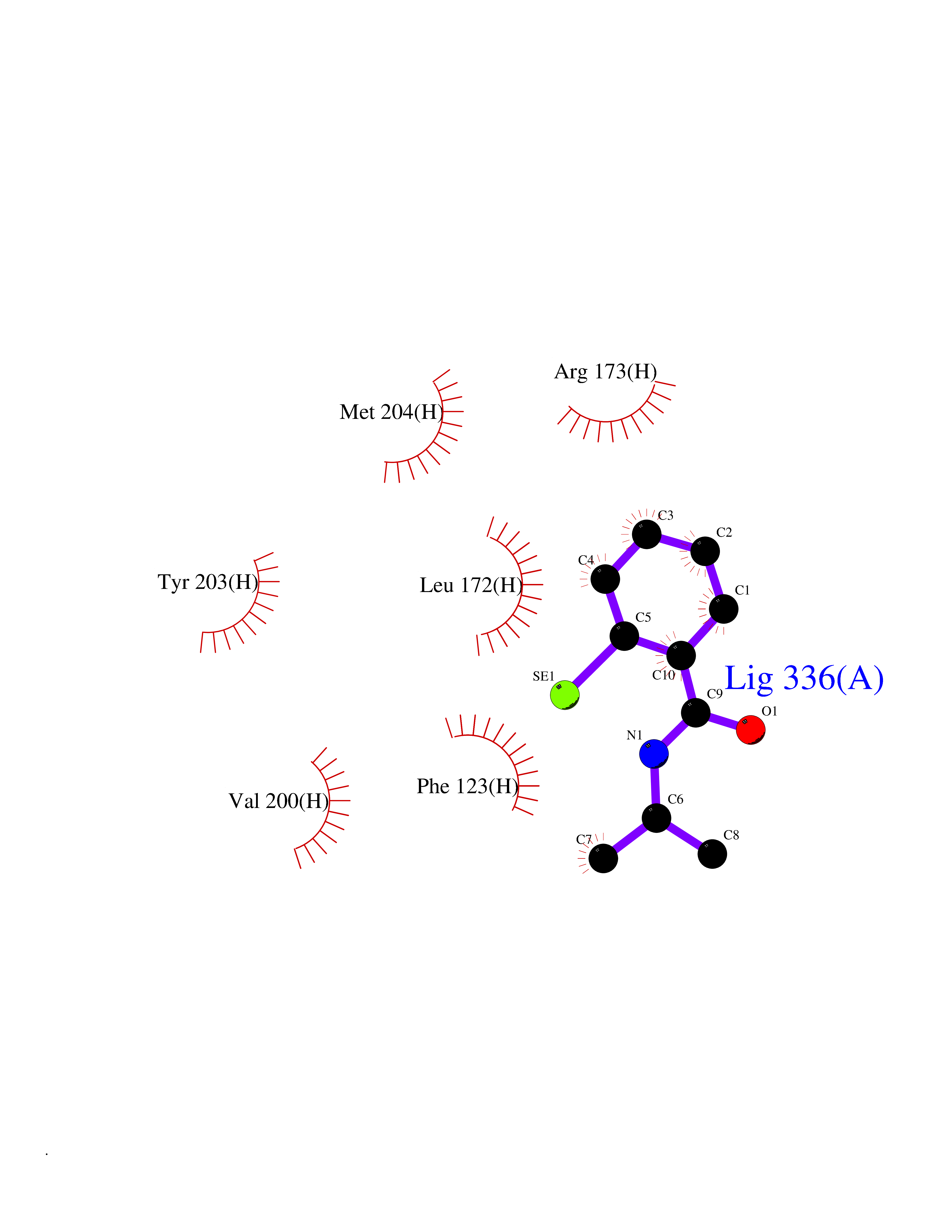 Ligplot Map 3D Binding mode Sequence GVRGTCEDASLCKRFAVSIGYWHDPYIQHFVRLSKERKAPEINRGYFARVHGVSQLIKAFLRKTECHCQIVNLGAGMDTTFWRLKDEDLLSSKYFEVDFPMIVTRKLHSIKCKPPLSSPILELHSEDTLQMDGHILDSKRYAVIGADLRDLSELEEKLKKCNMNTQLPTLLIAECVLVYMTPEQSANLLKWAANSFERAMFINYEQVNMGDRFGQIMIENLRRRQCDLAGVETCKSLESQKERLLSNGWETASAVDMMELYNRLPRAEVSRIESLEFLDEMELLEQLMRHYCLCWATKGGNELGLKEITY Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Thymidine kinase 1 (TK1) | 1W4R | 5.73 | |
Target general information Gen name TK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Thymidine kinase, cytosolic Protein family Thymidine kinase family Biochemical class Kinase Function cytosol, identical protein binding, thymidine kinase activity, zinc ion binding, DNA metabolic process, nucleobase-containing compound metabolic process, protein homotetramerization, pyrimidine nucleoside salvage, thymidine metabolic process Related diseases Seizures, benign familial infantile, 3 (BFIS3) [MIM:607745]: A form of benign familial infantile epilepsy, a neurologic disorder characterized by afebrile seizures occurring in clusters during the first year of life, without neurologic sequelae. BFIS3 inheritance is autosomal dominant. {ECO:0000269|PubMed:11371648, ECO:0000269|PubMed:12243921, ECO:0000269|PubMed:15048894, ECO:0000269|PubMed:16417554, ECO:0000269|PubMed:17021166, ECO:0000269|PubMed:17386050, ECO:0000269|PubMed:18479388, ECO:0000269|PubMed:20371507, ECO:0000269|PubMed:22612257, ECO:0000269|PubMed:23360469, ECO:0000269|PubMed:23758435, ECO:0000269|PubMed:25982755, ECO:0000269|PubMed:26291284, ECO:0000269|PubMed:29844171, ECO:0000269|PubMed:30144217}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 11 (DEE11) [MIM:613721]: An autosomal dominant seizure disorder characterized by neonatal or infantile onset of refractory seizures with resultant delayed neurologic development and persistent neurologic abnormalities. Patients may progress to West syndrome, which is characterized by tonic spasms with clustering, arrest of psychomotor development, and hypsarrhythmia on EEG. {ECO:0000269|PubMed:19783390, ECO:0000269|PubMed:19786696, ECO:0000269|PubMed:20956790, ECO:0000269|PubMed:22677033, ECO:0000269|PubMed:23033978, ECO:0000269|PubMed:23195492, ECO:0000269|PubMed:23550958, ECO:0000269|PubMed:23662938, ECO:0000269|PubMed:23708187, ECO:0000269|PubMed:23935176, ECO:0000269|PubMed:23988467, ECO:0000269|PubMed:24463883, ECO:0000269|PubMed:24579881, ECO:0000269|PubMed:24659627, ECO:0000269|PubMed:24710820, ECO:0000269|PubMed:25457084, ECO:0000269|PubMed:25459969, ECO:0000269|PubMed:25772804, ECO:0000269|PubMed:25818041, ECO:0000269|PubMed:26138355, ECO:0000269|PubMed:26291284, ECO:0000269|PubMed:26993267, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:29625812, ECO:0000269|PubMed:29844171, ECO:0000269|PubMed:30144217, ECO:0000269|PubMed:30415926}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in SCN2A are associated with genetic epilepsy with febrile seizures plus (GEFS+), a familial autosomal dominant epilepsy syndrome, a clinical subset of febrile seizures, characterized by frequent episodes after 6 years of age and various types of subsequent epilepsy. {ECO:0000269|PubMed:29635106}.; DISEASE: Defects in SCN2A are associated with autism spectrum disorders (ASD). It seems that mutations resulting in sodium channel gain of function and increased neuron excitability lead to infantile seizures, whereas variants resulting in sodium channel loss of function and decrease neuron excitability are associated with ASD. {ECO:0000269|PubMed:28256214}.; DISEASE: Episodic ataxia 9 (EA9) [MIM:618924]: An autosomal dominant neurologic disorder characterized by episodic ataxia manifesting in the first years of life, early-onset seizures, difficulty walking, dizziness, slurred speech, headache, vomiting, and pain. The duration of ataxic episodes is heterogeneous. Most patients show episodes lasting minutes to maximum several hours, but periods lasting days up to weeks have been reported. Some patients have mildly delayed development with speech delay and/or autistic features or mildly impaired intellectual development. {ECO:0000269|PubMed:26645390, ECO:0000269|PubMed:27159988, ECO:0000269|PubMed:27328862, ECO:0000269|PubMed:28065826}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01692; DB04485; DB02452; DB00432; DB00495 Interacts with P05067; A0A087WZT3; Q92993; Q1RN33; P04183 EC number EC 2.7.1.21 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Cytoplasm; DNA synthesis; Kinase; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 19373.5 Length 174 Aromaticity 0.09 Instability index 36.21 Isoelectric point 8.63 Charge (pH=7) 3.88 2D Binding mode Binding energy (Kcal/mol) -7.81 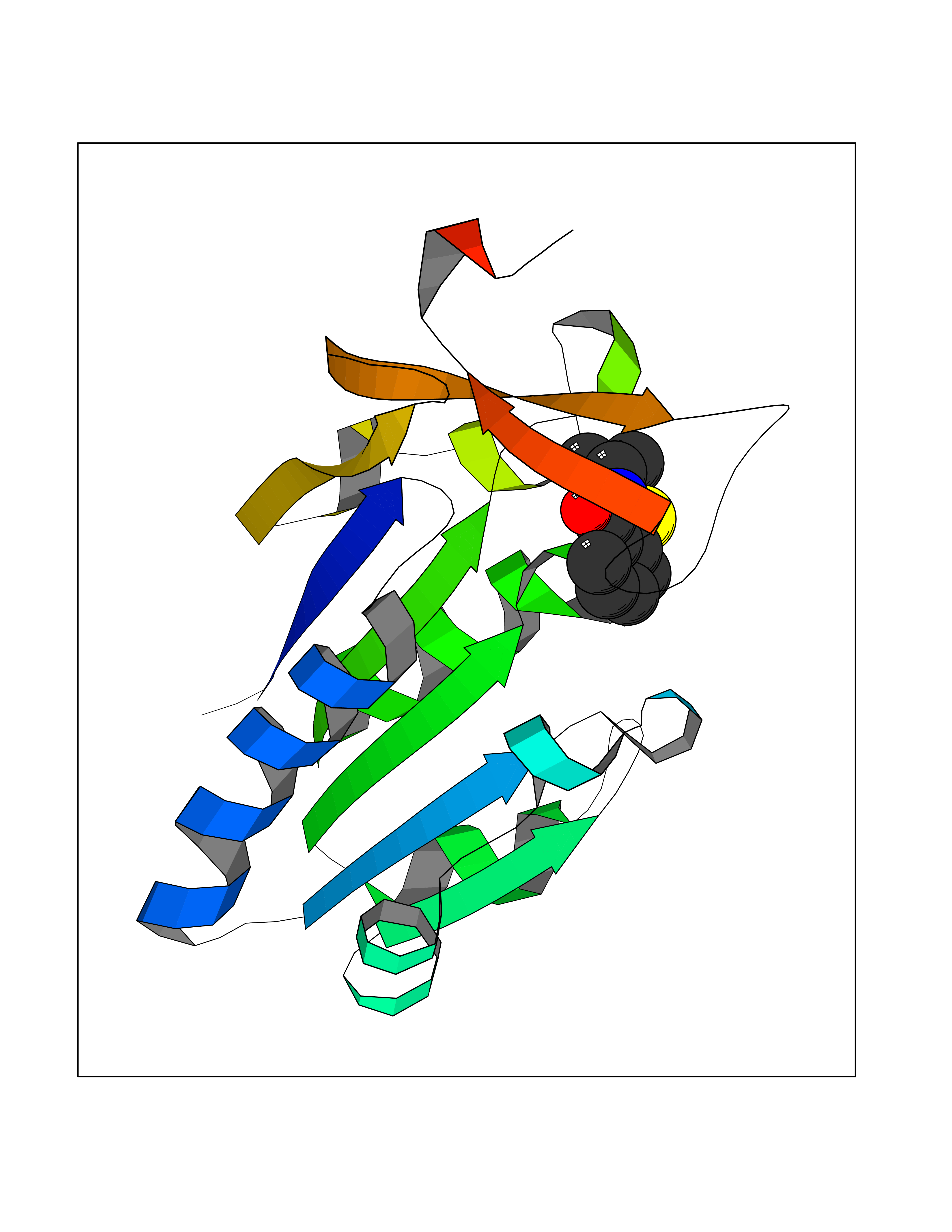 Molscript Map 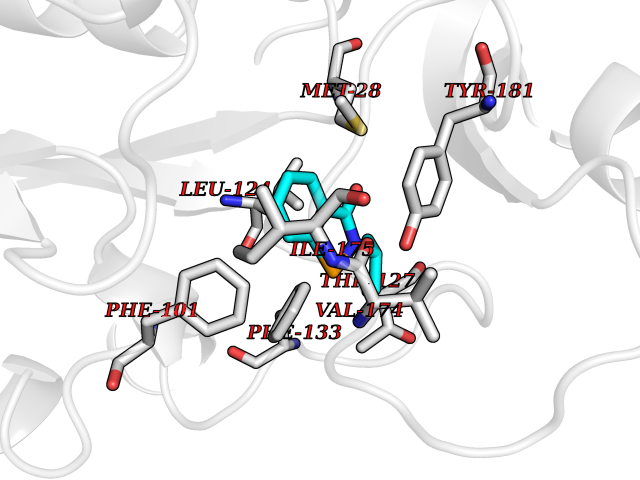 Pymol Map 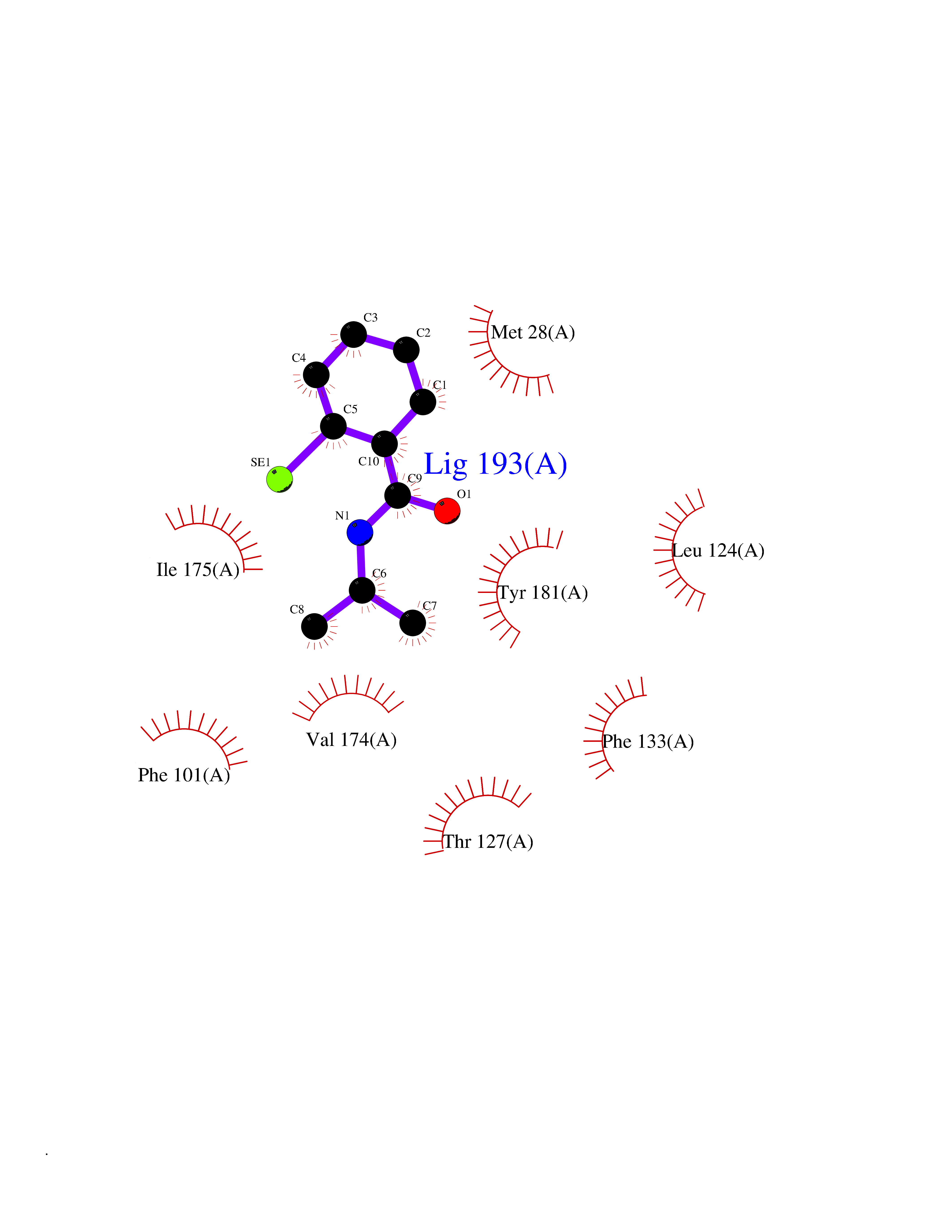 Ligplot Map 3D Binding mode Sequence RGQIQVILGPMFSGKSTELMRRVRRFQIAQYKCLVIKYAKDTRYSSSFCTHDRNTMEALPACLLRDVAQEALGVAVIGIDEGQFFPDIVEFCEAMANAGKTVIVAALDGTFQRKPFGAILNLVPLAESVVKLTAVCMECFREAAYTKRLGTEKEVEVIGGADKYHSVCRLCYFK Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Lethal(3)malignant brain tumor-like 3 (L3MBTL3) | 4FL6 | 5.73 | |
Target general information Gen name L3MBTL3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MBT1; MBT-1; Lethal(3)malignant brain tumor-like protein 3; L(3)mbt-like protein 3; KIAA1798; H-l(3)mbt-like protein 3 Protein family NA Biochemical class NA Function Putative Polycomb group (PcG) protein. PcG proteins maintain the transcriptionally repressive state of genes, probably via a modification of chromatin, rendering it heritably changed in its expressibility. Required for normal maturation of myeloid progenitor cells (By similarity). Related diseases Spinocerebellar ataxia, autosomal recessive, with axonal neuropathy 1 (SCAN1) [MIM:607250]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCAN1 is an autosomal recessive cerebellar ataxia (ARCA) associated with peripheral axonal motor and sensory neuropathy, distal muscular atrophy, pes cavus and steppage gait as seen in Charcot-Marie-Tooth neuropathy. All affected individuals have normal intelligence. {ECO:0000269|PubMed:12244316, ECO:0000269|PubMed:15647511, ECO:0000269|PubMed:15920477, ECO:0000269|PubMed:16141202, ECO:0000269|PubMed:17948061}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NWX5; Q8N9N5; Q13895; Q96JK2; Q16531; P26358; Q01094; O00716; Q96JM7; Q9Y4Z0; P45984; Q9UBU8; Q15014; Q9NPG2; Q9UMX2; P18545; Q8IXK0; Q8N381; Q96S99; Q9H8W4; P62875; Q13131; P54646; P14678-2; P23497; G2XKQ0; P10827; Q6DKK2; P54253; A0A0S2Z5G4; Q13895; Q9BXJ3; Q8IUI8; Q14203-5; Q9H4E7; Q8NFF5-2; Q53EP0-3; O95995; O75031; P42858; Q14005-2; Q63ZY3; Q96JM7-2; P61968; P45984; P55081; Q9UBU8-2; Q15014; Q9NPG2; Q16656-4; Q96HA8; Q96BD5; Q92569; Q96S99; Q9H8W4; O60568; A0A6Q8PF08; P67775; P54646; P63000; O94955; Q5VUG0; P37840; P00441; Q5MJ10; Q9UMX1; Q13148; Q86TI0; Q8N8B7-2; Q9Y228; Q5T7W7; Q6DKK2; P09936; P31930; P61758; A0A0S2Z6A9; Q9H0M4-4; P36508 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 71079 Length 615 Aromaticity 0.14 Instability index 29.71 Isoelectric point 6.42 Charge (pH=7) -6.56 2D Binding mode Binding energy (Kcal/mol) -7.81 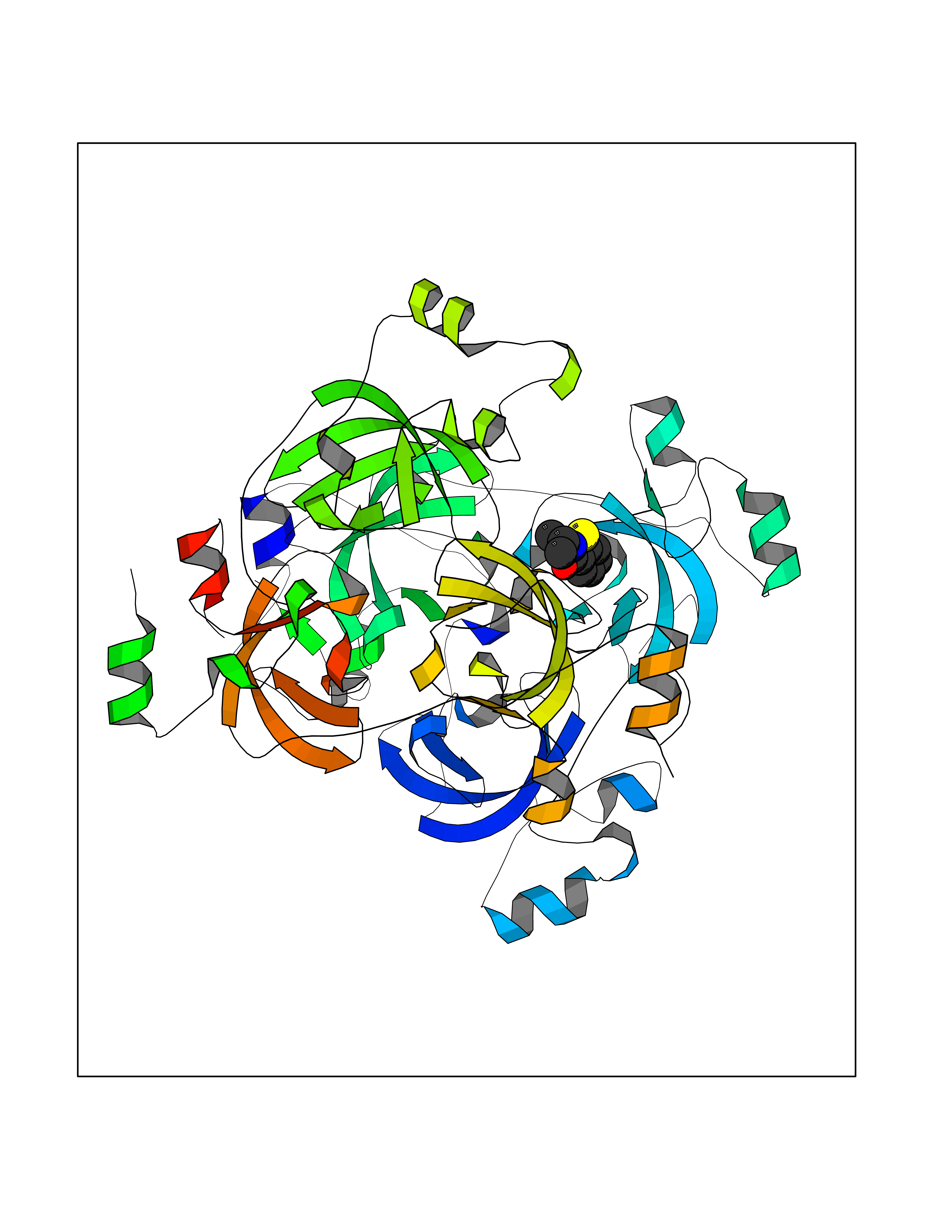 Molscript Map 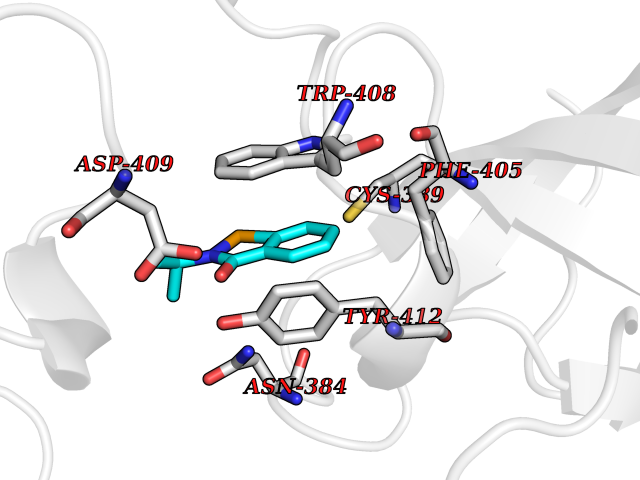 Pymol Map 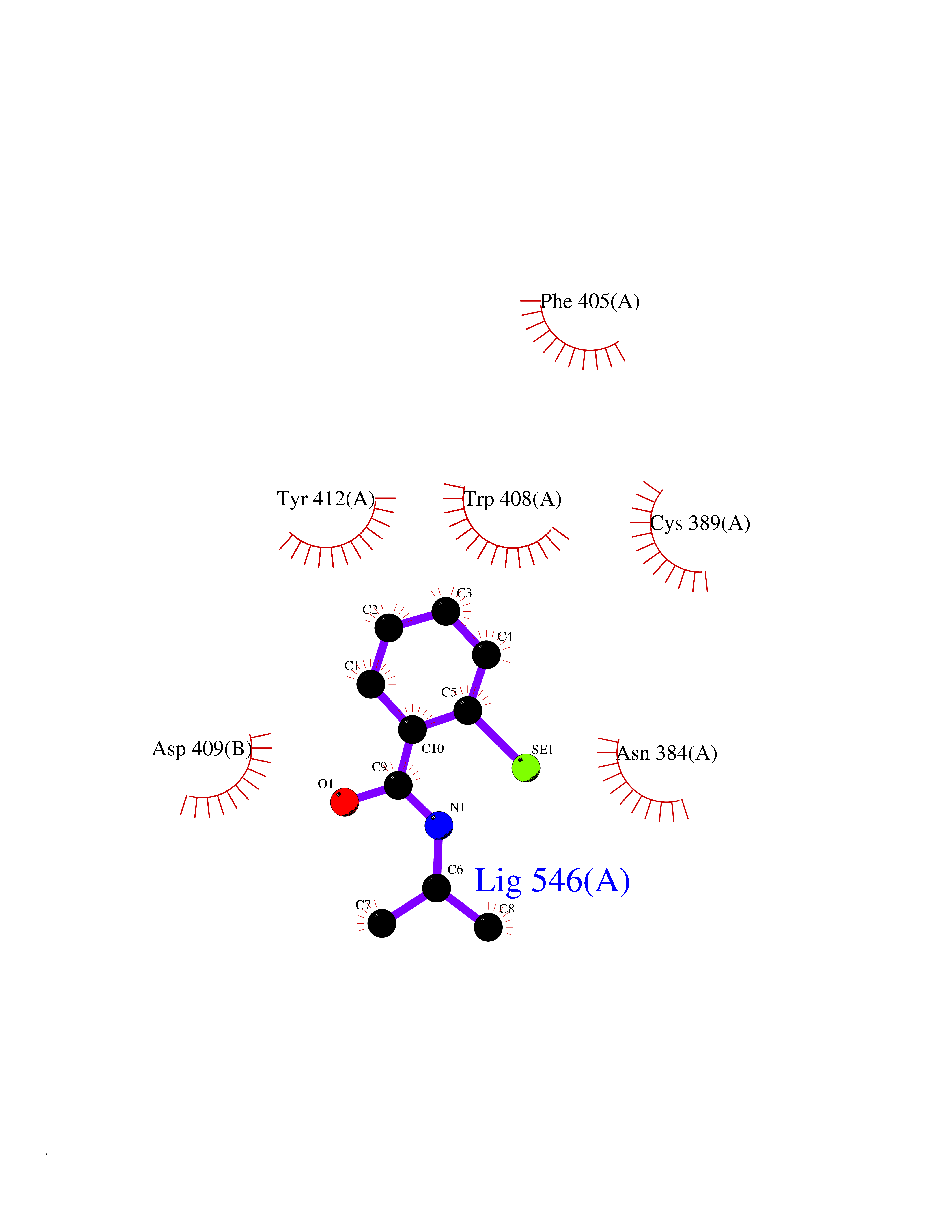 Ligplot Map 3D Binding mode Sequence AWCWASYLEEEKAVAVPAKLFKEHQSFPYNKNGFKVGMKLEGVDPEHQSVYCVLTVAEVCGYRIKLHFDGYSDCYDFWVNADALDIHPVGWCEKTGHKLHPPKGYKEEEFNWQTYLKTCKAQAAPKSLFEVIPSGFRVGMKLEAVDKKNPSFICVATVTDMVDNRFLVHFDNWDESYDYWCEASSPHIHPVGWCKEHRRTLITPPGYPNVKHFSWDKYLEETNSLPAPARAFKVKPPHGFQKKMKLEVVDKRNPMFIRVATVADTDDHRVKVHFDGWNNCYDYWIDADSPDIHPVGWCSKTGHPLQPPLAWCWASYLEEEKAVAVPAKLFKEHQSFPYNKNGFKVGMKLEGVDPEHQSVYCVLTVAEVCGYRIKLHFDGYSDCYDFWVNADALDIHPVGWCEKTGHKLHPPKGYKEEEFNWQTYLKTCKAQAAPKSLFENSGFRVGMKLEAVDKKNPSFICVATVTDMVDNRFLVHFDNWDESYDYWCEASSPHIHPVGWCKEHRRTLITPPGYPNVHFSWDKYLEETNSLPAPARAFKVKPPHGFQKKMKLEVVDKRNPMFIRVATVADTDDHRVKVHFDGWNNCYDYWIDADSPDIHPVGWCSKTGHPLQPPL Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Oxysterols receptor LXR-alpha (NR1H3) | 3IPQ | 5.73 | |
Target general information Gen name NR1H3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nuclear receptor subfamily 1 group H member 3; Nuclear receptor LXRalpha; Nuclear orphan receptor LXR-alpha; Liver X receptor alpha; LXRalpha; LXRA Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Interaction with retinoic acid receptor (RXR) shifts RXR from its role as a silent DNA-binding partner to an active ligand-binding subunit in mediating retinoid responses through target genes defined by LXRES. LXRES are DR4-type response elements characterized by direct repeats of two similar hexanuclotide half-sites spaced by four nucleotides. Plays an important role in the regulation of cholesterol homeostasis, regulating cholesterol uptake through MYLIP-dependent ubiquitination of LDLR, VLDLR and LRP8. Interplays functionally with RORA for the regulation of genes involved in liver metabolism. Nuclear receptor that exhibits a ligand-dependent transcriptional activation activity. Related diseases Okur-Chung neurodevelopmental syndrome (OCNDS) [MIM:617062]: An autosomal dominant neurodevelopmental disorder characterized by developmental delay, intellectual disability, behavioral problems, hypotonia, speech problems, microcephaly, pachygyria and variable dysmorphic features. {ECO:0000269|PubMed:27048600}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08175; DB08063; DB11994; DB07929; DB13174; DB07080 Interacts with O60869; O60341; Q99750; Q15788; O75376; Q07869; Q07869-1; Q03181; P37231; P19793; P28702; P48443; O43463; P42858; Q99750; O95817; G5E9A7; O95872; P02545; Q99750; P28702; P28702-3; P48443; Q7Z699 EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Cytoplasm; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 25389.9 Length 220 Aromaticity 0.09 Instability index 46.42 Isoelectric point 5.51 Charge (pH=7) -6.58 2D Binding mode Binding energy (Kcal/mol) -7.82 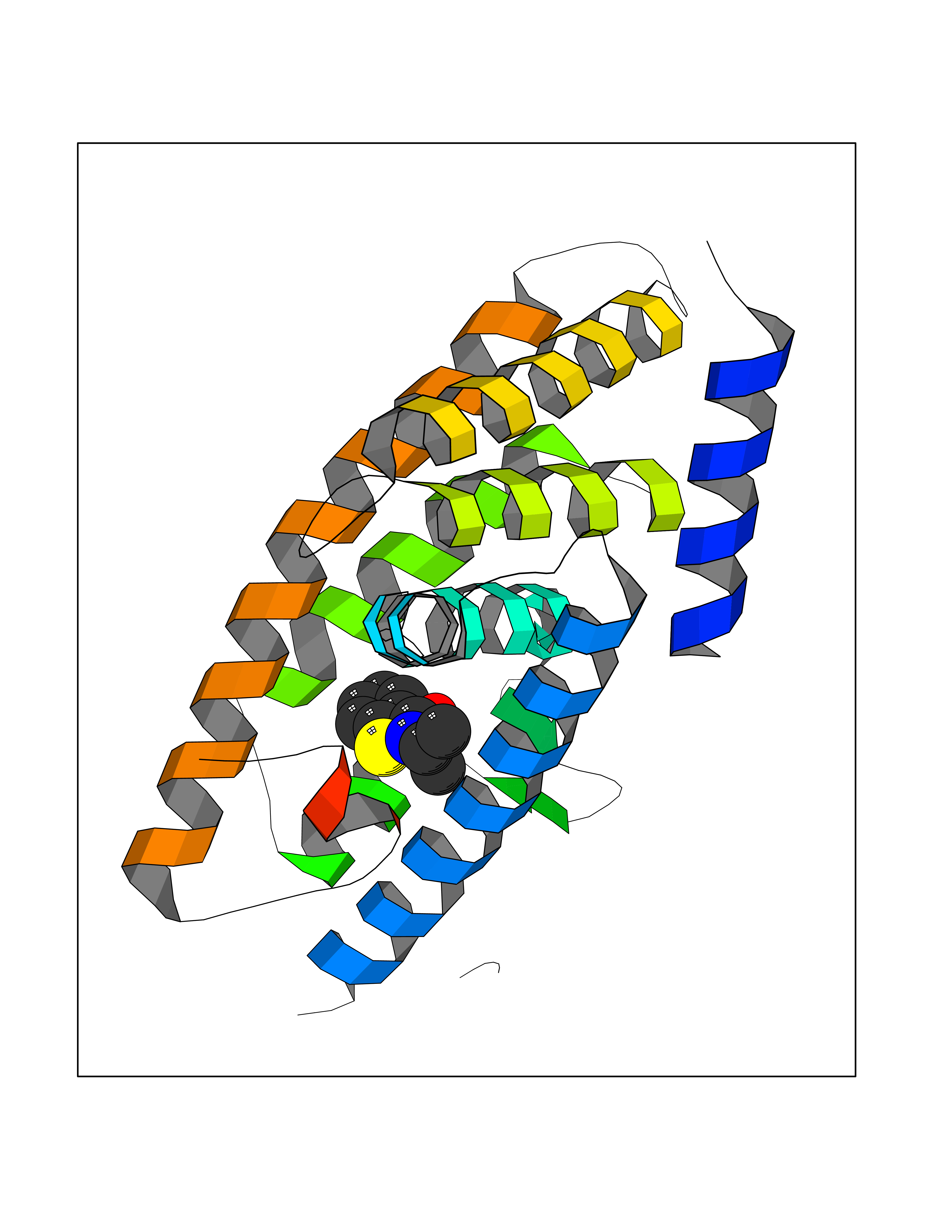 Molscript Map 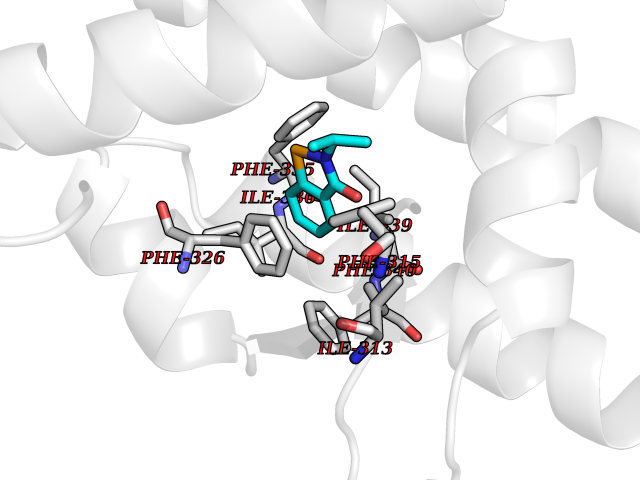 Pymol Map 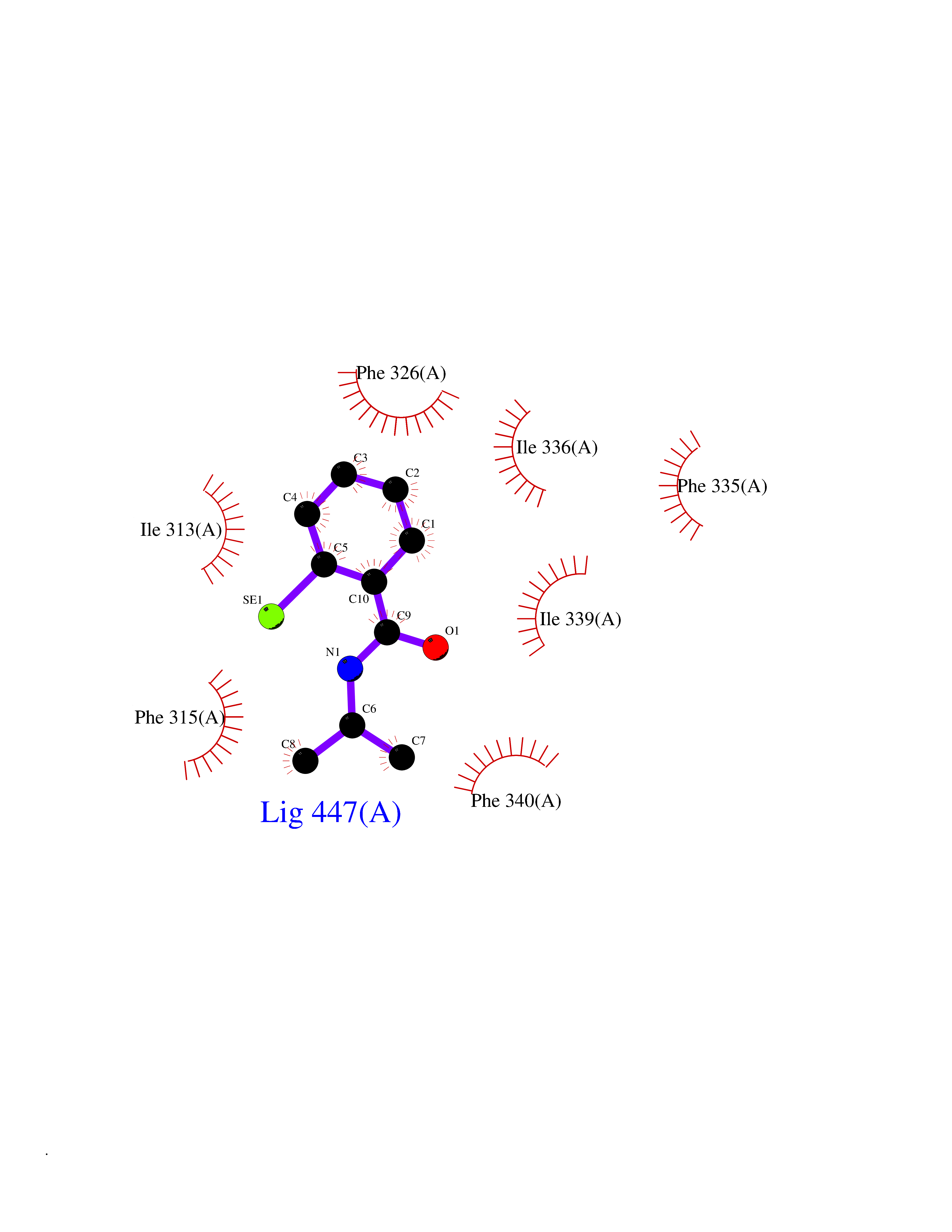 Ligplot Map 3D Binding mode Sequence QLSPEQLGMIEKLVAAQQTPWPEARQQRFAHFTELAIVSVQEIVDFAKQLPGFLQLSREDQIALLKTSAIEVMLLETSRRYNPGSESITFLKDFSYNREDFAKAGLQVEFINPIFEFSRAMNELQLNDAEFALLIAISIFSADRPNVQDQLQVERLQHTYVEALHAYVSIHHPHDRLMFPRMLMKLVSLRTLSSVHSEQVFALRLQDKKLPPLLSEIWDV Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Neuronal acetylcholine receptor alpha-4 (CHRNA4) | 6CNJ | 5.73 | |
Target general information Gen name CHRNA4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nicotinic acetylcholine receptor alpha4; CHRNA4; Alpha-4 nAChR Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-4/CHRNA4 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasmamembrane permeable to sodium ions. Related diseases Epilepsy, nocturnal frontal lobe, 1 (ENFL1) [MIM:600513]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:10563623, ECO:0000269|PubMed:14623738, ECO:0000269|PubMed:7550350}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01351; DB01352; DB00572; DB01483; DB00237; DB00241; DB01353; DB00564; DB00565; DB09028; DB01245; DB00514; DB01496; DB07720; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00898; DB01354; DB01355; DB00753; DB00657; DB00333; DB00463; DB00849; DB00184; DB00312; DB01174; DB00981; DB05458; DB00794; DB05740; DB00747; DB00418; DB00202; DB00306; DB00599; DB01273 Interacts with Q6UY14-3; P05067; P83916; Q6UXH1-1; Q6UXH1-3; P20042; Q9NZR2; Q92673; P17787 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84601.2 Length 728 Aromaticity 0.13 Instability index 39.72 Isoelectric point 5.86 Charge (pH=7) -9.84 2D Binding mode Binding energy (Kcal/mol) -7.81 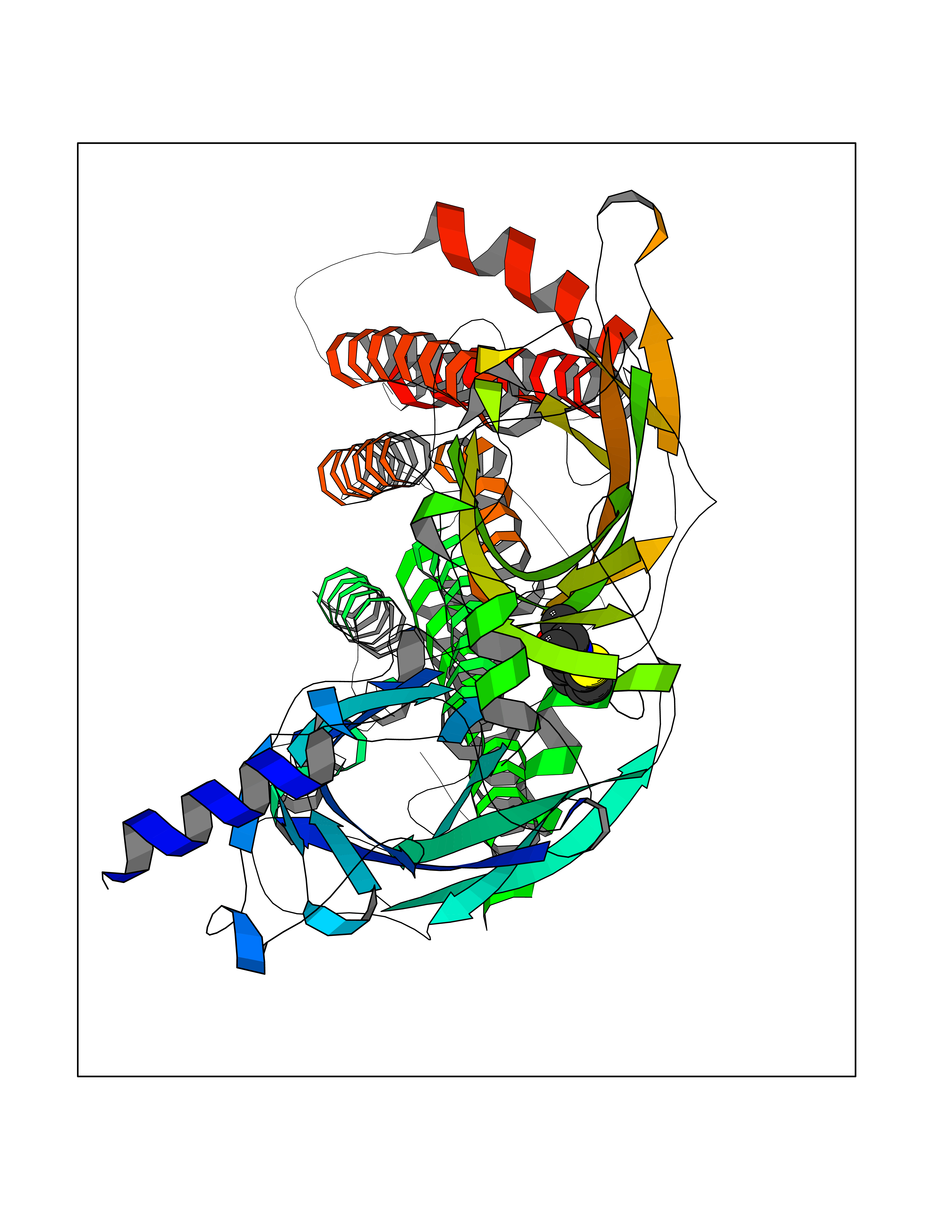 Molscript Map 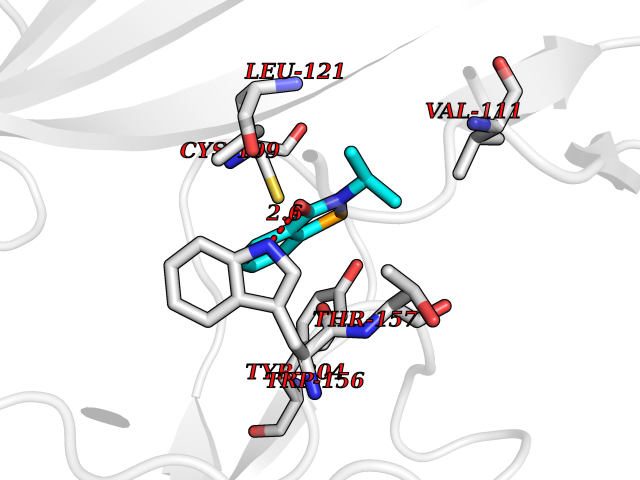 Pymol Map 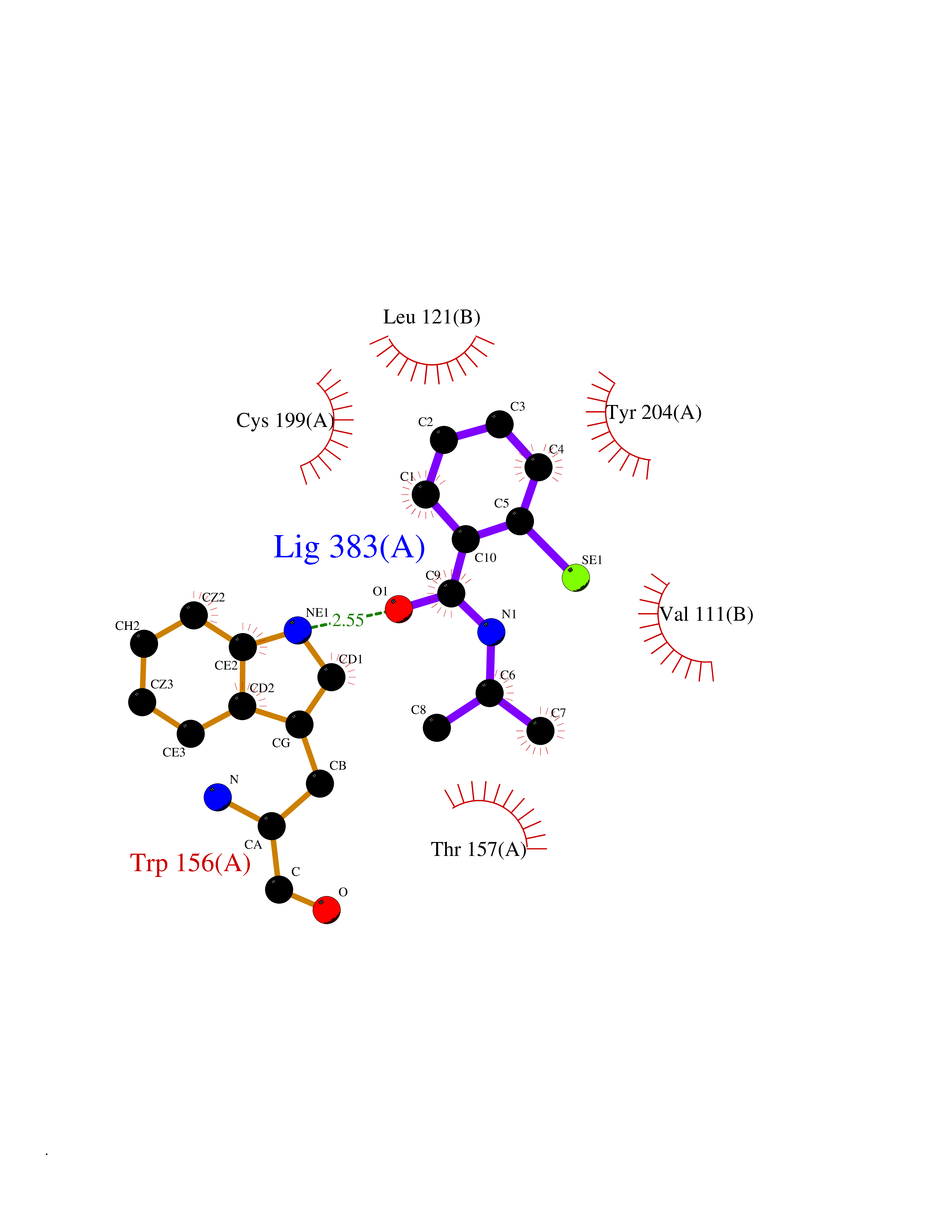 Ligplot Map 3D Binding mode Sequence ETRAHAEERLLKKLFSGYNKWSRPVANISDVVLVRFGLSIAQLIDVDEKNQMMTTNVWVKQEWHDYKLRWDPADYENVTSIRIPSELIWRPDIVLYNNADGDFAVTHLTKAHLFHDGRVQWTPPAIYKSSCSIDVTFFPFDQQNCTMKFGSWTYDKAKIDLVNMHSRVDQLDFWESGEWVIVDAVGTYNTRKYECCAEIYPDITYAFVIRRLPLFYTINLIIPCLLISCLTVLVFYLPSECGEKITLCISVLLSLTVFLLLITEIIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHHRSPRTHTMPTWVRRVFLDIVPRLLLMKRFERSVKEDWKYVAMVIDRIFLWMFIIVCLLGTVGLFLPPWDTEERLVEHLLDPSRYNKLIRPATNGSELVTVQLMVSLAQLISVHEREQIMTTNVWLTQEWEDYRLTWKPEEFDNMKKVRLPSKHIWLPDVVLYNNADGMYEVSFYSNAVVSYDGSIFWLPPAIYKSACKIEVKHFPFDQQNCTMKFRSWTYDRTEIDLVLKSEVASLDDFTPSGEWDIVALPGRRNENPDDSTYVDITYDFIIRRKPLFYTINLIIPCVLITSLAILVFYLPSDCGEKMTLCISVLLALTVFLLLISKIVPPTSLDVPLVGKYLMFTMVLVTFSIVTSVCVLNVHHRSPTTHTMAPWVKVVFLEKLPALLFMQQSVSEDWKYVAMVIDRLFLWIFVFVCVFGTIGMF Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Neuronal acetylcholine receptor beta-2 (CHRNB2) | 6CNJ | 5.73 | |
Target general information Gen name CHRNB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nicotinic acetylcholine receptor beta2; Nicotinic acetylcholine receptor beta 2-subunit protein; CHRNB2; Beta-2 nAChR; Alpha-4/beta-2 nicotinic receptor Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Beta-2/CHRNB2 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane permeable to sodiun ions. Related diseases Epilepsy, nocturnal frontal lobe, 3 (ENFL3) [MIM:605375]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:11062464, ECO:0000269|PubMed:11104662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00572; DB00237; DB00565; DB09028; DB01245; DB00514; DB07720; DB00898; DB00753; DB00657; DB00333; DB00184; DB00981; DB05458; DB05855; DB05740; DB00747; DB00202; DB01273 Interacts with P43681-1; P30532 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84601.2 Length 728 Aromaticity 0.13 Instability index 39.72 Isoelectric point 5.86 Charge (pH=7) -9.84 2D Binding mode Binding energy (Kcal/mol) -7.81 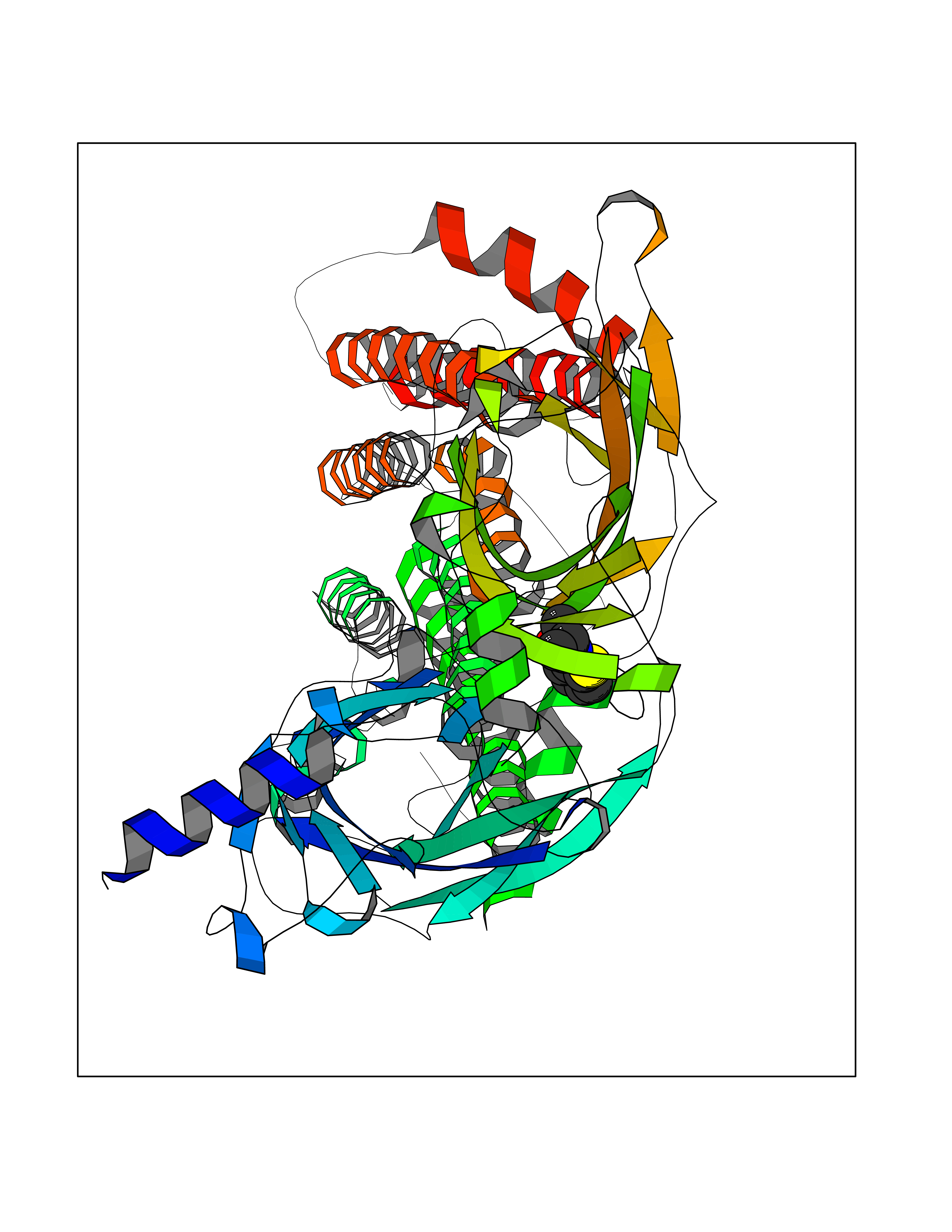 Molscript Map 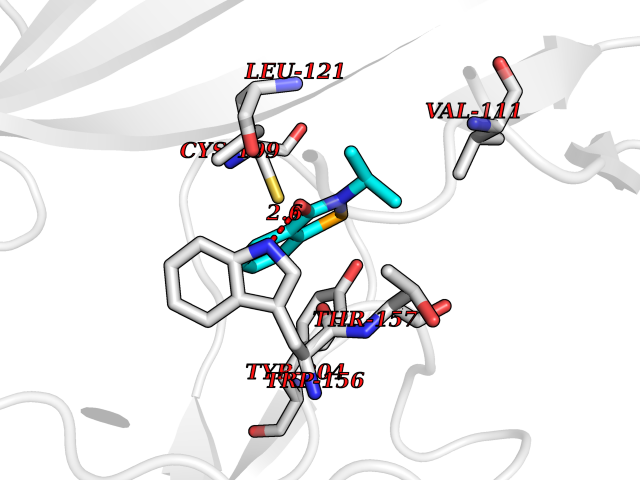 Pymol Map 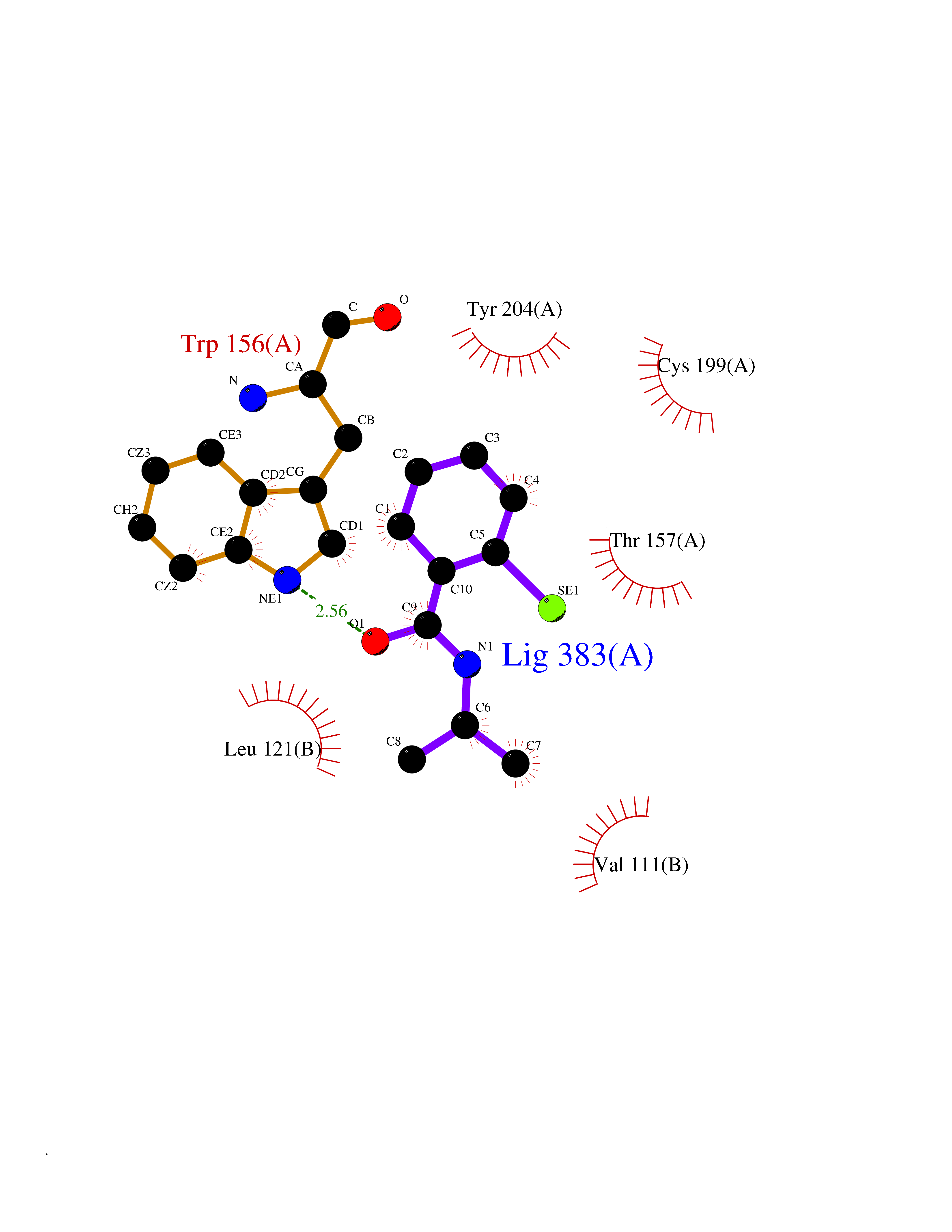 Ligplot Map 3D Binding mode Sequence ETRAHAEERLLKKLFSGYNKWSRPVANISDVVLVRFGLSIAQLIDVDEKNQMMTTNVWVKQEWHDYKLRWDPADYENVTSIRIPSELIWRPDIVLYNNADGDFAVTHLTKAHLFHDGRVQWTPPAIYKSSCSIDVTFFPFDQQNCTMKFGSWTYDKAKIDLVNMHSRVDQLDFWESGEWVIVDAVGTYNTRKYECCAEIYPDITYAFVIRRLPLFYTINLIIPCLLISCLTVLVFYLPSECGEKITLCISVLLSLTVFLLLITEIIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHHRSPRTHTMPTWVRRVFLDIVPRLLLMKRFERSVKEDWKYVAMVIDRIFLWMFIIVCLLGTVGLFLPPWDTEERLVEHLLDPSRYNKLIRPATNGSELVTVQLMVSLAQLISVHEREQIMTTNVWLTQEWEDYRLTWKPEEFDNMKKVRLPSKHIWLPDVVLYNNADGMYEVSFYSNAVVSYDGSIFWLPPAIYKSACKIEVKHFPFDQQNCTMKFRSWTYDRTEIDLVLKSEVASLDDFTPSGEWDIVALPGRRNENPDDSTYVDITYDFIIRRKPLFYTINLIIPCVLITSLAILVFYLPSDCGEKMTLCISVLLALTVFLLLISKIVPPTSLDVPLVGKYLMFTMVLVTFSIVTSVCVLNVHHRSPTTHTMAPWVKVVFLEKLPALLFMQQSVSEDWKYVAMVIDRLFLWIFVFVCVFGTIGMF Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Folate receptor alpha (FOLR1) | 4LRH | 5.73 | |
Target general information Gen name FOLR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ovarian tumorassociated antigen MOv18; KB cells FBP; Folate receptor, adult; Folate receptor 1; FRalpha; FOLR1; Adult folatebinding protein Protein family Folate receptor family Biochemical class Folate receptor Function Binds to folate and reduced folic acid derivatives and mediates delivery of 5-methyltetrahydrofolate and folate analogs into the interior of cells. Has high affinity for folate and folic acid analogs at neutral pH. Exposure to slightly acidic pHafter receptor endocytosis triggers a conformation change that strongly reduces its affinity for folates and mediates their release. Required for normal embryonic development and normal cell proliferation. Related diseases Neurodegeneration due to cerebral folate transport deficiency (NCFTD) [MIM:613068]: An autosomal recessive neurodegenerative disorder resulting from brain-specific folate deficiency early in life. Onset is apparent in late infancy with severe developmental regression, movement disturbances, epilepsy and leukodystrophy. {ECO:0000269|PubMed:19732866}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05595; DB00158; DB00563; DB12489; DB15413; DB05168 Interacts with Q8N357 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Endosome; Folate-binding; Glycoprotein; GPI-anchor; Lipoprotein; Membrane; Neurodegeneration; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 24216 Length 207 Aromaticity 0.13 Instability index 49.36 Isoelectric point 8.14 Charge (pH=7) 3.41 2D Binding mode Binding energy (Kcal/mol) -7.82  Molscript Map 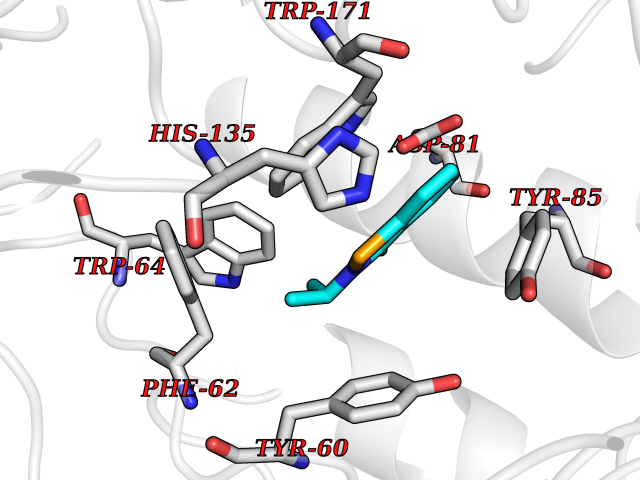 Pymol Map 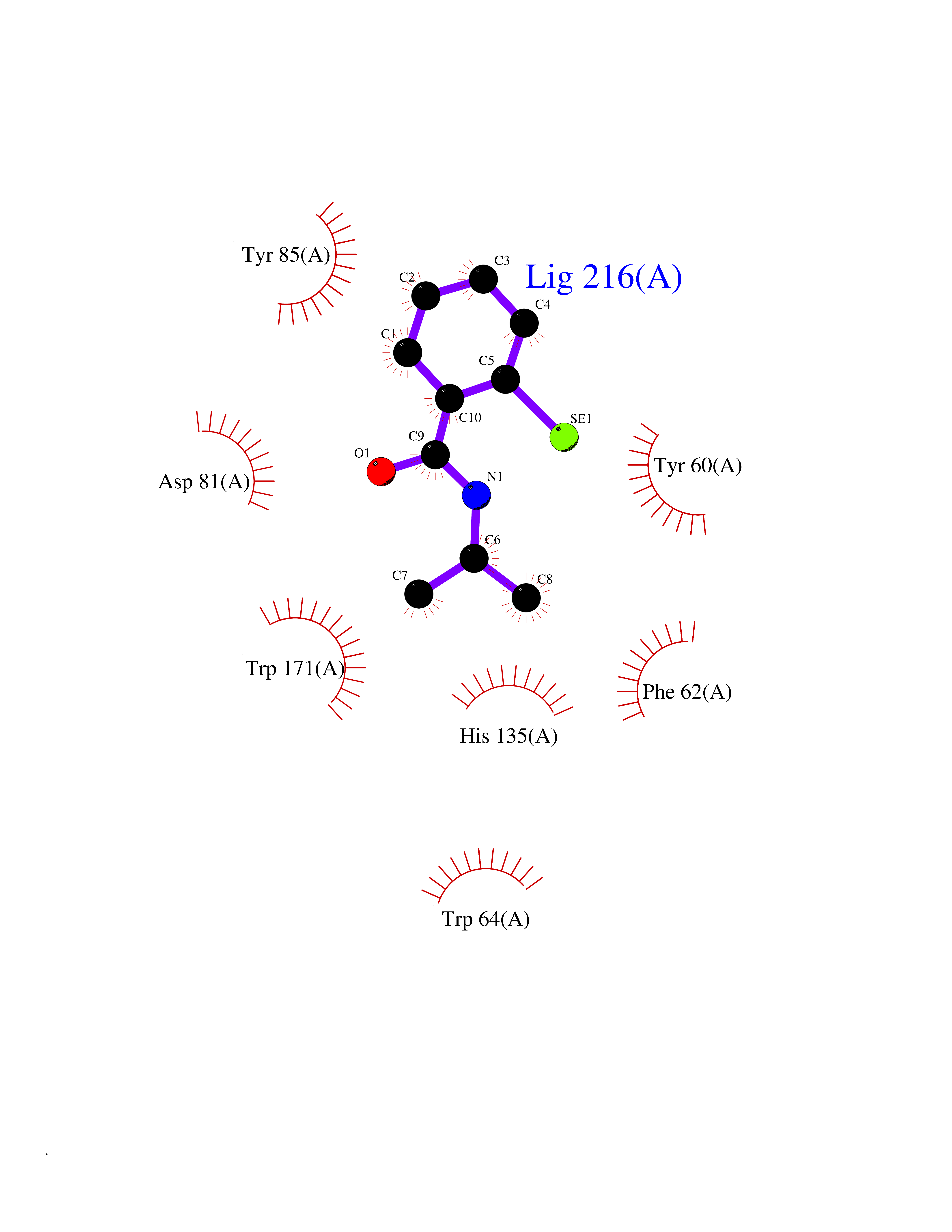 Ligplot Map 3D Binding mode Sequence RTELLNVCMNAKHHKEKPGPEDKLHEQCRPWRKNACCSTNTSQEAHKDVSYLYRFNWNHCGEMAPACKRHFIQDTCLYECSPNLGPWIQQVDQSWRKERVLNVPLCKEDCEQWWEDCRTSYTCKSNWHKGWNWTSGFNKCAVGAACQPFHFYFPTPTVLCNEIWTHSYKVSNYSRGSGRCIQMWFDPAQGNPNEEVARFYAAAMSGT Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Hydroxymethylglutaryl-CoA synthase 2 (HMGCS2) | 2WYA | 5.73 | |
Target general information Gen name HMGCS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HMGCS2; HMG-CoAsynthase; 3-hydroxy-3-methylglutaryl coenzyme A synthase 2 Protein family Thiolase-like superfamily, HMG-CoA synthase family Biochemical class Acyltransferase Function This enzyme condenses acetyl-CoA with acetoacetyl-CoA to form HMG-CoA, which is the substrate for HMG-CoA reductase. Related diseases 3-hydroxy-3-methylglutaryl-CoA synthase-2 deficiency (HMGCS2D) [MIM:605911]: A metabolic disorder characterized by severe hypoketotic hypoglycemia, encephalopathy, and hepatomegaly. {ECO:0000269|PubMed:11228257, ECO:0000269|PubMed:11479731, ECO:0000269|PubMed:12647205, ECO:0000269|PubMed:16601895, ECO:0000269|PubMed:23751782, ECO:0000269|PubMed:25511235, ECO:0000269|PubMed:29597274}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 2.3.3.10 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Disease variant; Lipid biosynthesis; Lipid metabolism; Mitochondrion; Phosphoprotein; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transferase; Transit peptide Protein physicochemical properties Chain ID A,D Molecular weight (Da) 102499 Length 920 Aromaticity 0.1 Instability index 33.48 Isoelectric point 6.72 Charge (pH=7) -1.41 2D Binding mode Binding energy (Kcal/mol) -7.82 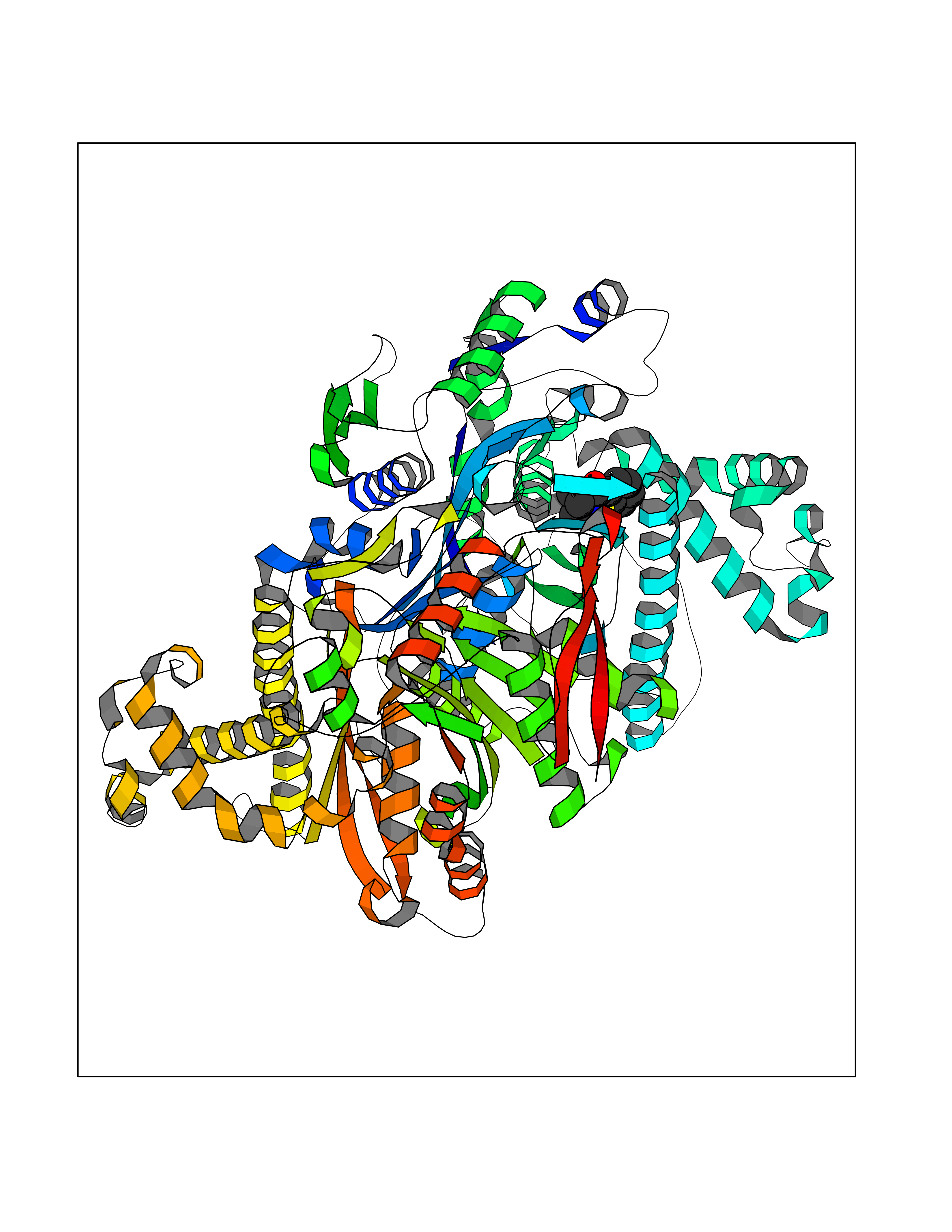 Molscript Map 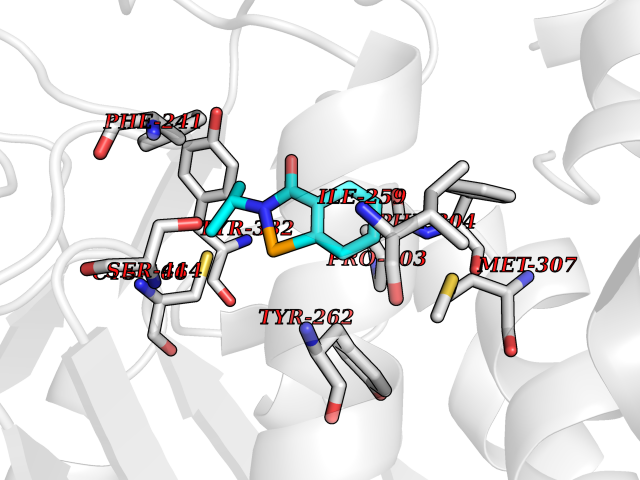 Pymol Map 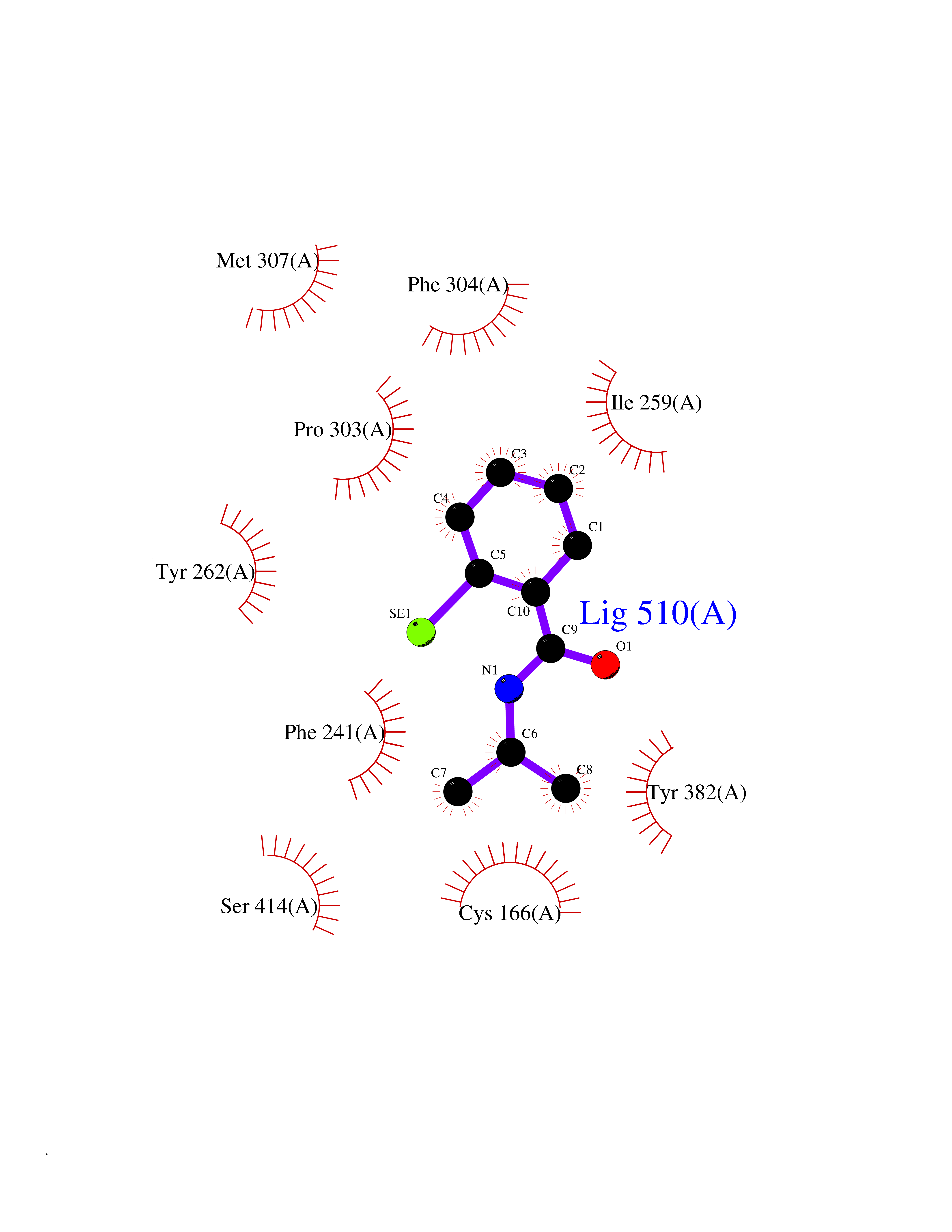 Ligplot Map 3D Binding mode Sequence SMPKDVGILALEVYFPAQYVDQTDLEKYNNVEAGKYTVGLGQTRMGFCSVQEDINSLCLTVVQRLMERIQLPWDSVGRLEVGTETIIDKSKAVKTVLMELFQDSGNTDIEGIDTTNACYGGTASLFNAANWMESSSWDGRYAMVVCGDIAVYPSGNARPTGGAGAVAMLIGPKAPLALERGLRGTHMENVYDFYKPNLASEYPIVDGKLSIQCYLRALDRCYTSYRKKIQNQWKQAGSDRPFTLDDLQYMIFHTPFCKMVQKSLARLMFNDFLSASSDTQTSLYKGLEAFGGLKLEDTYTNKDLDKALLKASQDMFDKKTKASLYLSTHNGNMYTSSLYGCLASLLSHHSAQELAGSRIGAFSYGSGLAASFFSFRVSQDAAPGSPLDKLVSSTSDLPKRLASRKCVSPEEFTEIMNQREQFYHKVNFSPPGDTNSLFPGTWYLERVDEQHRRKYARRPVSMPKDVGILALEVYFPAQYVDQTDLEKYNNVEAGKYTVGLGQTRMGFCSVQEDINSLCLTVVQRLMERIQLPWDSVGRLEVGTETIIDKSKAVKTVLMELFQDSGNTDIEGIDTTNACYGGTASLFNAANWMESSSWDGRYAMVVCGDIAVYPSGNARPTGGAGAVAMLIGPKAPLALERGLRGTHMENVYDFYKPNLASEYPIVDGKLSIQCYLRALDRCYTSYRKKIQNQWKQAGSDRPFTLDDLQYMIFHTPFCKMVQKSLARLMFNDFLSASSDTQTSLYKGLEAFGGLKLEDTYTNKDLDKALLKASQDMFDKKTKASLYLSTHNGNMYTSSLYGCLASLLSHHSAQELAGSRIGAFSYGSGLAASFFSFRVSQDAAPGSPLDKLVSSTSDLPKRLASRKCVSPEEFTEIMNQREQFYHKVNFSPPGDTNSLFPGTWYLERVDEQHRRKYARRPV Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Serum albumin (ALB) | 4L8U | 5.72 | |
Target general information Gen name ALB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serum albumin Protein family ALB/AFP/VDB family Biochemical class NA Function Serum albumin, the main protein of plasma, has a good binding capacity for water, Ca(2+), Na(+), K(+), fatty acids, hormones, bilirubin and drugs. Its main function is the regulation of the colloidal osmotic pressure of blood. Major zinc transporter in plasma, typically binds about 80% of all plasma zinc. Related diseases Hyperthyroxinemia, familial dysalbuminemic (FDAH) [MIM:615999]: A disorder characterized by abnormally elevated levels of total serum thyroxine (T4) in euthyroid patients. It is due to abnormal serum albumin that binds T4 with enhanced affinity. {ECO:0000269|PubMed:7852505, ECO:0000269|PubMed:8048949, ECO:0000269|PubMed:9329347, ECO:0000269|PubMed:9589637}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Analbuminemia (ANALBA) [MIM:616000]: A rare autosomal recessive disorder manifested by the presence of a very low amount of circulating serum albumin. Affected individuals manifest mild edema, hypotension, fatigue, and, occasionally, lower body lipodystrophy (mainly in adult females). The most common biochemical finding is hyperlipidemia, with a significant increase in the total and LDL cholesterol concentrations, but normal concentrations of HDL cholesterol and triglycerides. {ECO:0000269|PubMed:8134387}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08496; DB07517; DB12001; DB05812; DB14973; DB11703; DB01418; DB01614; DB00316; DB00414; DB09347; DB06151; DB00459; DB00787; DB00640; DB00802; DB00346; DB00404; DB00770; DB01370; DB14517; DB14518; DB01118; DB00321; DB01060; DB00415; DB00276; DB06728; DB11901; DB00714; DB04557; DB09229; DB11217; DB00278; DB01238; DB14185; DB09204; DB01169; DB11638; DB09274; DB00126; DB06216; DB01072; DB00335; DB00289; DB01076; DB00995; DB06237; DB07402; DB00993; DB08822; DB08903; DB16703; DB00245; DB01086; DB01053; DB00443; DB14669; DB11967; DB13909; DB01294; DB09223; DB00083; DB09128; DB01222; DB15248; DB00490; DB00237; DB11148; DB06772; DB11751; DB11093; DB11348; DB14481; DB04690; DB01101; DB03600; DB01197; DB01136; DB00456; DB01327; DB14879; DB00274; DB01328; DB01329; DB00493; DB01330; DB00430; DB00438; DB01212; DB06119; DB00567; DB07565; DB08936; DB00878; DB00608; DB00477; DB09093; DB00310; DB00501; DB00568; DB00537; DB00515; DB00349; DB01013; DB00845; DB01242; DB01068; DB00575; DB00758; DB01147; DB00363; DB15534; DB01394; DB00286; DB12483; DB09130; DB01380; DB08865; DB11134; DB06778; DB01176; DB00924; DB00434; DB00847; DB01914; DB06695; DB08912; DB11963; DB04816; DB00080; DB12941; DB01264; DB11943; DB11637; DB01189; DB00304; DB01234; DB14649; DB09213; DB00829; DB01119; DB11397; DB00586; DB00485; DB00266; DB00900; DB00861; DB01396; DB00343; DB08995; DB08930; DB01142; DB00997; DB00254; DB00366; DB04855; DB00476; DB01126; DB01057; DB12243; DB13421; DB00625; DB15444; DB00879; DB00584; DB13874; DB11718; DB00228; DB08899; DB01364; DB00530; DB00303; DB11827; DB12235; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB04574; DB00903; DB00977; DB00749; DB00294; DB01276; DB12466; DB04854; DB01039; DB00573; DB00813; DB00950; DB16165; DB01195; DB00687; DB15690; DB00544; DB00472; DB00712; DB08906; DB00983; DB01320; DB06716; DB11796; DB00695; DB15149; DB00743; DB06705; DB01044; DB00317; DB01241; DB12141; DB11978; DB01120; DB01067; DB01016; DB00986; DB13751; DB04539; DB12836; DB11575; DB11359; DB01159; DB14999; DB00070; DB01275; DB00999; DB00774; DB00327; DB09526; DB01611; DB01005; DB00557; DB13014; DB12471; DB09053; DB01050; DB00159; DB01088; DB00619; DB09262; DB00458; DB00808; DB00328; DB07992; DB09564; DB01307; DB05382; DB04711; DB09333; DB00332; DB16200; DB01029; DB00762; DB06636; DB00753; DB00677; DB00951; DB01064; DB00982; DB11757; DB01167; DB08820; DB01587; DB01026; DB01009; DB00598; DB09236; DB00709; DB00555; DB03017; DB01006; DB09237; DB06282; DB01235; DB01137; DB00451; DB00601; DB17083; DB00279; DB01583; DB06655; DB01601; DB09195; DB00678; DB00227; DB09280; DB15935; DB12674; DB00137; DB08932; DB14513; DB01397; DB06796; DB06234; DB00737; DB13959; DB09124; DB00603; DB00784; DB00814; DB01042; DB00454; DB09383; DB00931; DB00333; DB00563; DB00968; DB09241; DB00959; DB06710; DB00264; DB01110; DB00683; DB08893; DB00295; DB01024; DB08231; DB00461; DB00607; DB01183; DB00788; DB00731; DB04861; DB00220; DB11828; DB00238; DB01115; DB11820; DB09079; DB11793; DB12005; DB06713; DB00717; DB00957; DB00540; DB00104; DB00334; DB09074; DB04224; DB00768; DB12455; DB11130; DB04911; DB01083; DB13310; DB01173; DB00526; DB00842; DB00776; DB01062; DB00497; DB06412; DB03585; DB00595; DB15575; DB09073; DB03796; DB13967; DB14582; DB00642; DB00850; DB12978; DB01619; DB03255; DB00946; DB00252; DB01132; DB01621; DB04951; DB00554; DB08860; DB11642; DB01324; DB09087; DB09418; DB06813; DB13514; DB06209; DB01058; DB00860; DB15566; DB14631; DB00635; DB01032; DB01069; DB09348; DB00818; DB00571; DB06480; DB00852; DB00165; DB04216; DB00881; DB00908; DB12874; DB08735; DB00481; DB11853; DB12404; DB00912; DB02709; DB11855; DB01045; DB11753; DB08864; DB08931; DB14840; DB15305; DB00734; DB00503; DB11182; DB00412; DB01098; DB04847; DB06201; DB08877; DB08736; DB00936; DB00938; DB01232; DB11689; DB13928; DB01104; DB01236; DB12965; DB06290; DB00877; DB00815; DB15093; DB00421; DB00649; DB03193; DB06150; DB01581; DB01582; DB00576; DB01015; DB00795; DB00605; DB00391; DB00870; DB00864; DB00675; DB05134; DB09139; DB05521; DB00853; DB14126; DB09299; DB15133; DB00857; DB00342; DB00624; DB13943; DB13944; DB01420; DB13946; DB00759; DB00152; DB11590; DB01622; DB01623; DB09100; DB09070; DB08816; DB01133; DB15171; DB11800; DB01056; DB08895; DB01124; DB00500; DB00273; DB01685; DB00214; DB00755; DB00620; DB00432; DB08814; DB11677; DB00376; DB09069; DB00792; DB00427; DB08867; DB09076; DB12255; DB00313; DB00512; DB05294; DB08881; DB00661; DB15456; DB11641; DB08828; DB00162; DB11739; DB16699; DB00682; DB00943; DB00495; DB00744; DB14533; DB14548; DB00246; DB04828 Interacts with P02768; P02786; Q8N5Z5; Q6GQQ9-2; Q07869; Q09028; Q86WT6-2; O76024 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cleavage on pair of basic residues; Copper; Direct protein sequencing; Disease variant; Disulfide bond; Glycation; Glycoprotein; Lipid-binding; Metal-binding; Methylation; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 34028.4 Length 298 Aromaticity 0.09 Instability index 43.45 Isoelectric point 5.49 Charge (pH=7) -10.44 2D Binding mode Binding energy (Kcal/mol) -7.81 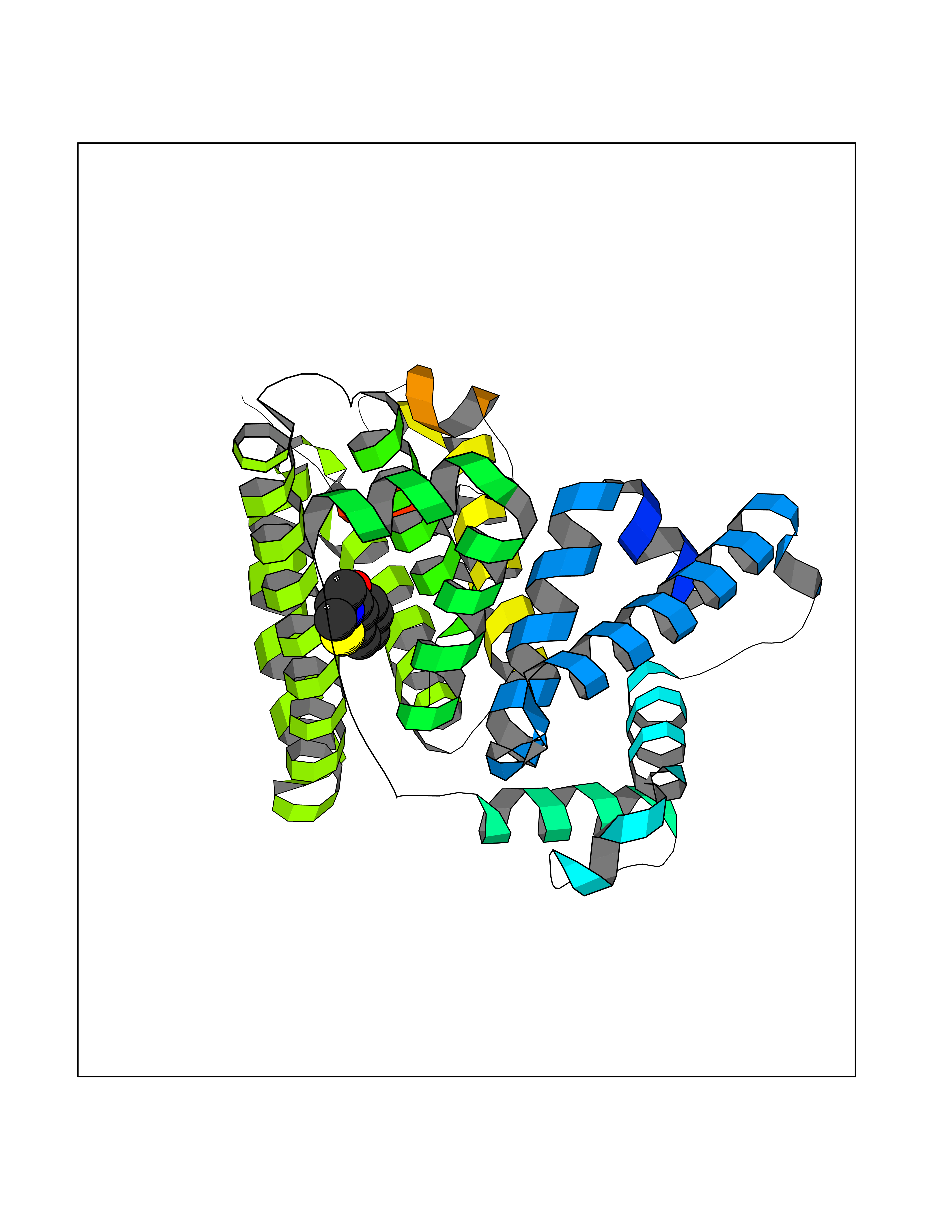 Molscript Map 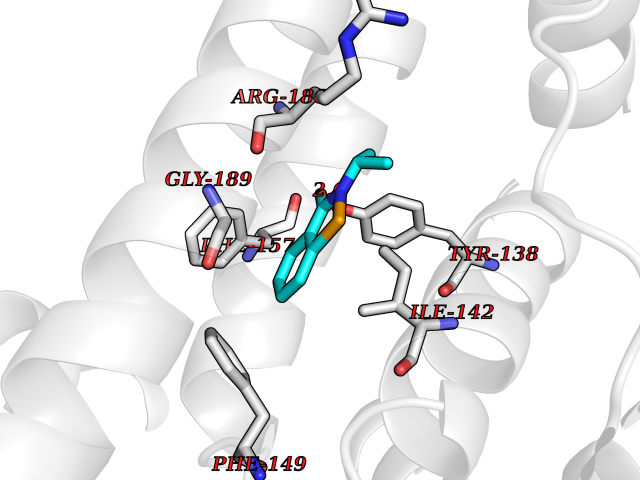 Pymol Map 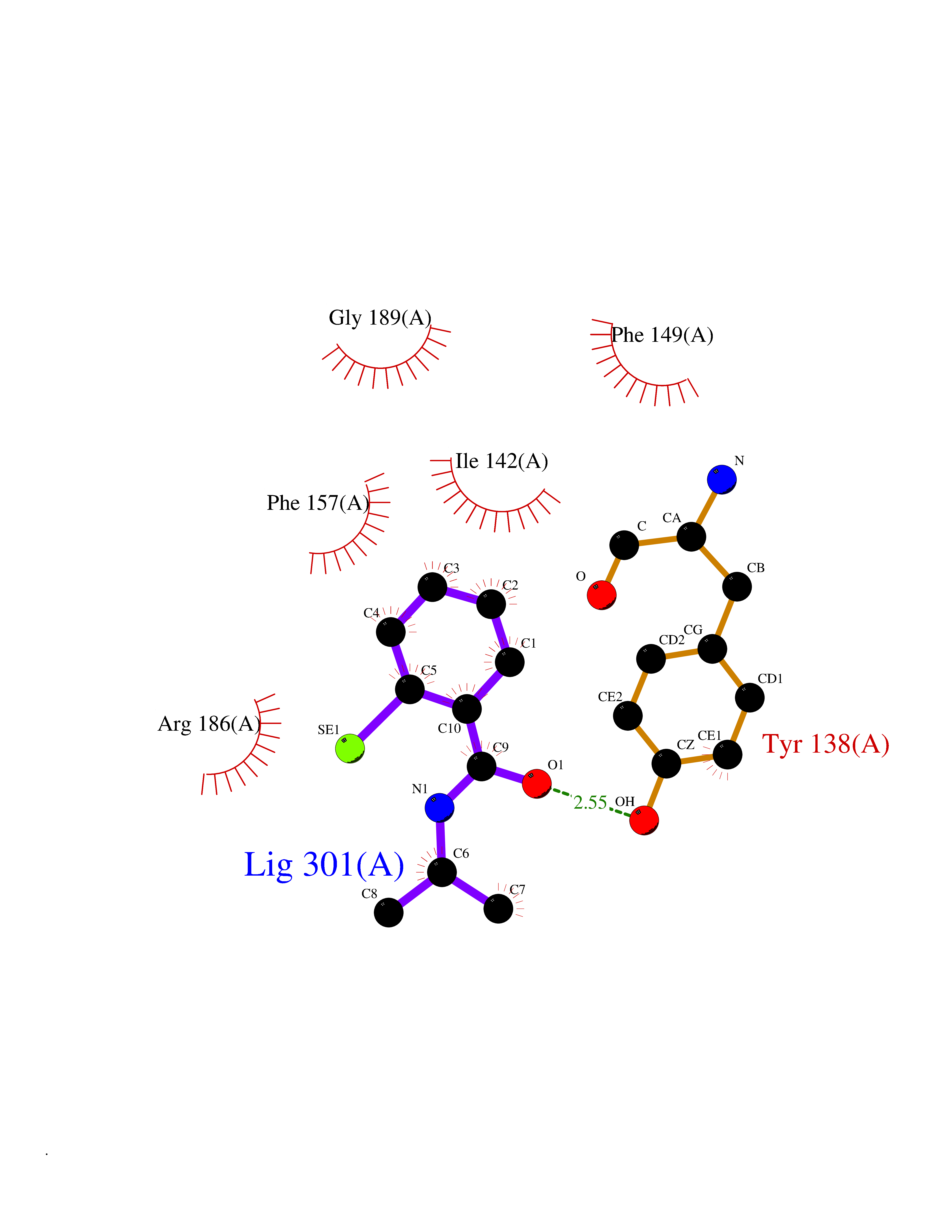 Ligplot Map 3D Binding mode Sequence AHKSEVAHRFKDLGEENFKALVLIAFAQYLQQCPFEDHVKLVNEVTEFAKTCVADESAENCDKSLHTLFGDKLCTVATLRETYGEMADCCAKQEPERNECFLQHKDDNPNLPRLVRPEVDVMCTAFHDNEETFLKKYLYEIARRHPYFYAPELLFFAKRYKAAFTECCQAADKAACLLPKLDELRDEGKASSAKQRLKCASLQKFGERAFKAWAVARLSQRFPKAEFAEVSKLVTDLTKVHTECCHGDLLECADDRADLAKYICENQDSISSKLKECCEKPLLEKSHCIAEVENDEMP Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Retinoic acid receptor alpha (RARA) | 3KMR | 5.72 | |
Target general information Gen name RARA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-alpha; RAR alpha; Nuclear receptor subfamily 1 group B member 1; NR1B1 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RXR/RAR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence of ligand, the RXR-RAR heterodimers associate with a multiprotein complex containing transcription corepressors that induce histone acetylation, chromatin condensation and transcriptional suppression. On ligand binding, the corepressors dissociate from the receptors and associate with the coactivators leading to transcriptional activation. RARA plays an essential role in the regulation of retinoic acid-induced germ cell development during spermatogenesis. Has a role in the survival of early spermatocytes at the beginning prophase of meiosis. In Sertoli cells, may promote the survival and development of early meiotic prophase spermatocytes. In concert with RARG, required for skeletal growth, matrix homeostasis and growth plate function. Receptor for retinoic acid. Related diseases Chromosomal aberrations involving RARA are commonly found in acute promyelocytic leukemia. Translocation t(11;17)(q32;q21) with ZBTB16/PLZF; translocation t(15;17)(q21;q21) with PML; translocation t(5;17)(q32;q11) with NPM. The PML-RARA oncoprotein requires both the PML ring structure and coiled-coil domain for both interaction with UBE2I, nuclear microspeckle location and sumoylation. In addition, the coiled-coil domain functions in blocking RA-mediated transactivation and cell differentiation. {ECO:0000269|PubMed:12691149, ECO:0000269|PubMed:8302850, ECO:0000269|PubMed:8562957}. Drugs (DrugBank ID) DB00459; DB00210; DB00523; DB00926; DB00982; DB05785; DB04942; DB00799; DB00755; DB12808 Interacts with O43707-1; O15296; Q15699; Q96RK4; O95273; P51946; Q15910; P50148; Q9UKP3; Q96EZ8; Q15648; Q71SY5; Q15788; Q9Y6Q9; O75376; Q9Y618; Q16236; P13056-2; P48552; Q9UPP1-2; Q9H8W4; P37231; P78527; P19793; P28702; P28702-3; P48443; Q96EB6; P63165; Q8WW24; Q2M1K9; Q91XC0; P59598; Q14457; P48552; Q96CV9; P28702; P48443; Q8WW24 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Chromosomal rearrangement; Cytoplasm; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 27724.1 Length 244 Aromaticity 0.05 Instability index 50.8 Isoelectric point 5.82 Charge (pH=7) -3.61 2D Binding mode Binding energy (Kcal/mol) -7.8  Molscript Map 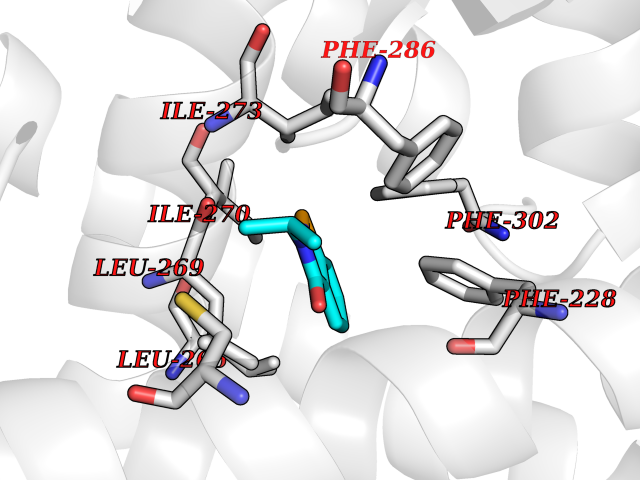 Pymol Map 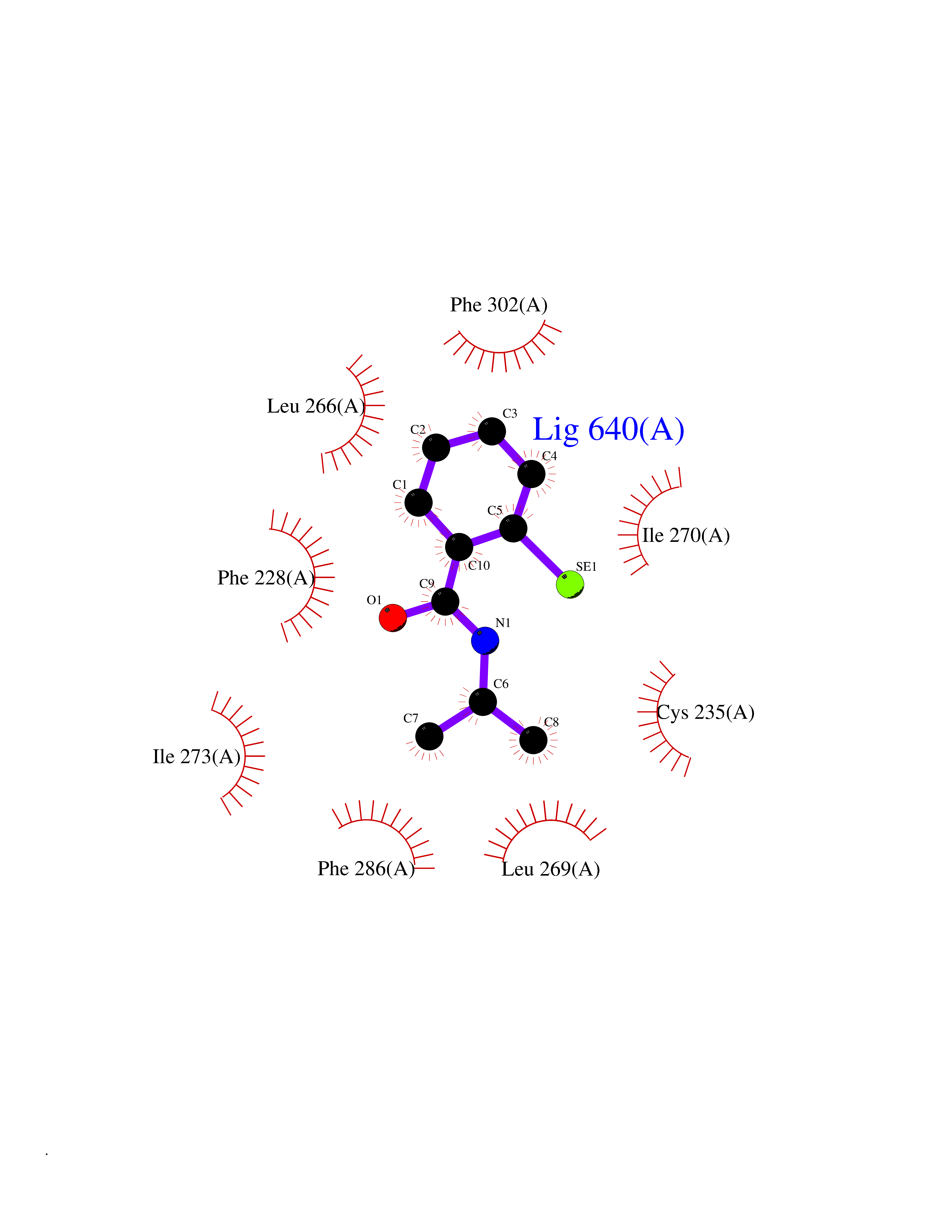 Ligplot Map 3D Binding mode Sequence PEVGELIEKVRKAHQETFPALCQLGKYTTNNSSEQRVSLDIDLWDKFSELSTKCIIKTVEFAKQLPGFTTLTIADQITLLKAACLDILILRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFAFANQLLPLEMDDAETGLLSAICLICGDRQDLEQPDRVDMLQEPLLEALKVYVRKRRPSRPHMFPKMLMKITDLRSISAKGAERVITLKMEIPGSMPPLIQEMLEHKILHRLLQE Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Monoamine oxidase type B (MAO-B) | 2V5Z | 5.72 | |
Target general information Gen name MAOB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MAO-B; Amine oxidase [flavin-containing] B Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. MAOB preferentially degrades benzylamine and phenylethylamine. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB08176; DB02211; DB08516; DB08480; DB01472; DB04307; DB07512; DB07513; DB00915; DB00182; DB06698; DB04889; DB00215; DB09130; DB04147; DB00988; DB01363; DB00668; DB01175; DB02509; DB03147; DB14914; DB00614; DB04818; DB02095; DB01247; DB00601; DB01577; DB01442; DB01171; DB08082; DB02643; DB04677; DB03894; DB08804; DB04820; DB00184; DB04821; DB12612; DB01626; DB00780; DB00191; DB00388; DB01132; DB00721; DB01168; DB01367; DB09363; DB06654; DB01037; DB01104; DB14569; DB09042; DB00752; DB16446; DB09185; DB04832; DB00909 Interacts with P55212; P28329-3; Q8NI60; Q5RI15; Q92915-2; P22607; Q53GS7; P06396; P01112; O14901; P13473-2; P21397; Q9BVL2; O75400-2; P62826; Q6NTF9-3; Q9Y371; Q7Z699; Q9UMX0; Q9Y649 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; FAD; Flavoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Oxidoreductase; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56019.9 Length 494 Aromaticity 0.09 Instability index 34.81 Isoelectric point 6.51 Charge (pH=7) -2.2 2D Binding mode Binding energy (Kcal/mol) -7.8 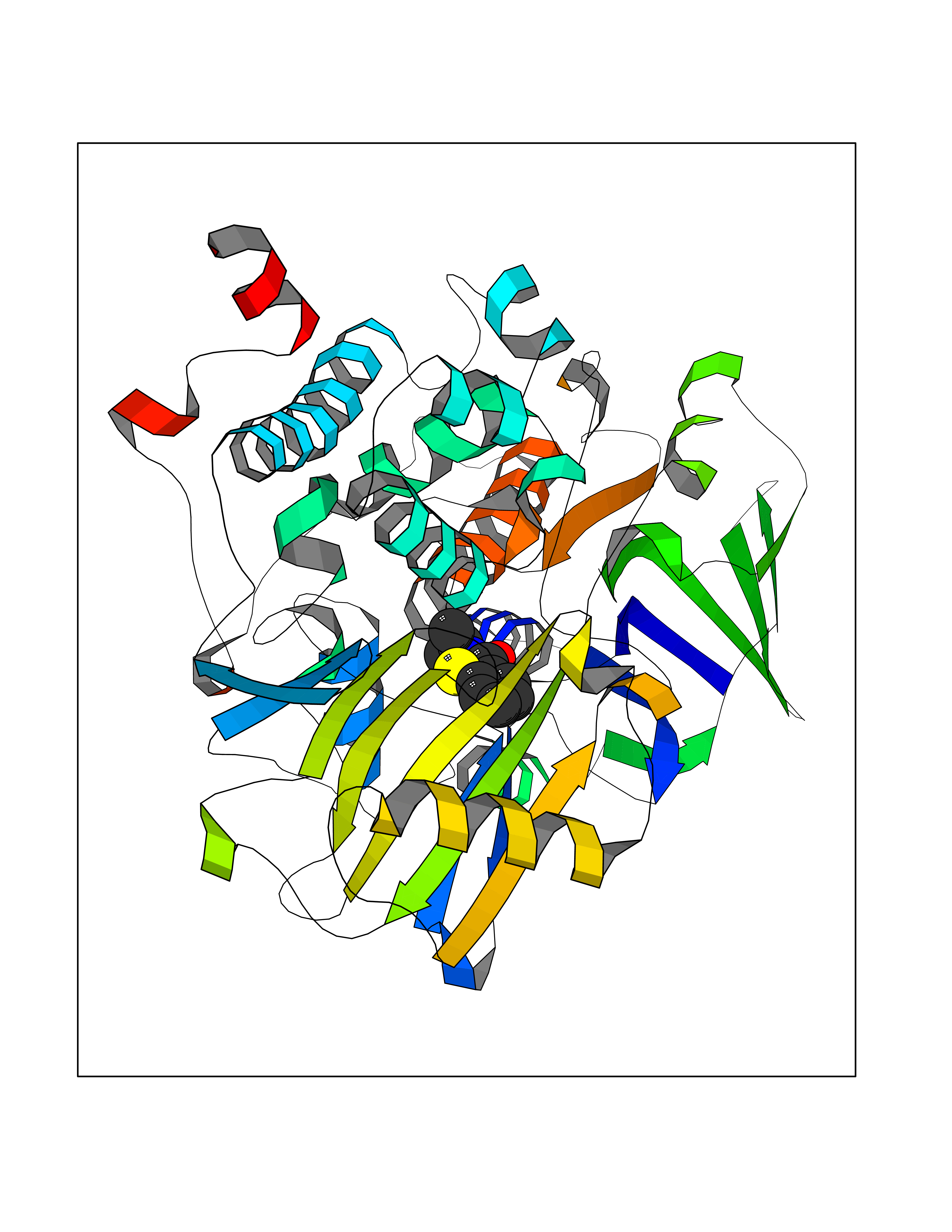 Molscript Map 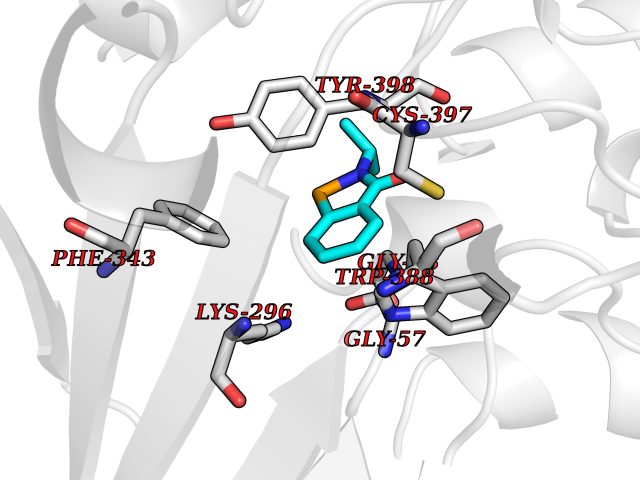 Pymol Map 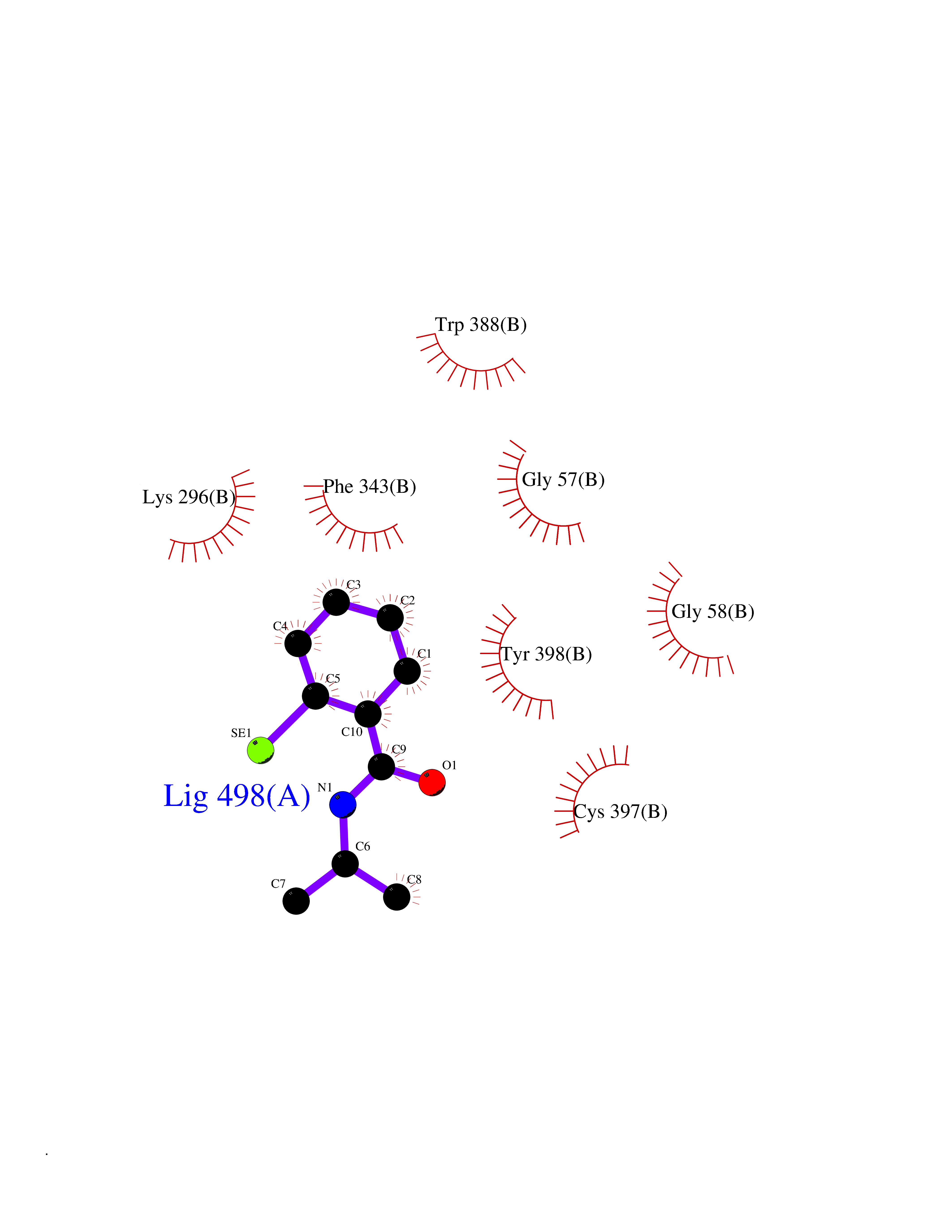 Ligplot Map 3D Binding mode Sequence NKCDVVVVGGGISGMAAAKLLHDSGLNVVVLEARDRVGGRTYTLRNQKVKYVDLGGSYVGPTQNRILRLAKELGLETYKVNEVERLIHHVKGKSYPFRGPFPPVWNPITYLDHNNFWRTMDDMGREIPSDAPWKAPLAEEWDNMTMKELLDKLCWTESAKQLATLFVNLCVTAETHEVSALWFLWYVKQCGGTTRIISTTNGGQERKFVGGSGQVSERIMDLLGDRVKLERPVIYIDQTRENVLVETLNHEMYEAKYVISAIPPTLGMKIHFNPPLPMMRNQMITRVPLGSVIKCIVYYKEPFWRKKDYCGTMIIDGEEAPVAYTLDDTKPEGNYAAIMGFILAHKARKLARLTKEERLKKLCELYAKVLGSLEALEPVHYEEKNWCEEQYSGGCYTTYFPPGILTQYGRVLRQPVDRIYFAGTETATHWSGYMEGAVEAGERAAREILHAMGKIPEDEIWQSEPESVDVPAQPITTTFLERHLPSVPGLLRLI Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Nitric-oxide synthase inducible (NOS2) | 3E7G | 5.72 | |
Target general information Gen name NOS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms iNOS; Peptidyl-cysteine S-nitrosylase NOS2; Nitric oxide synthase, inducible; NOS2A; NOS type II; Inducible NOS; Inducible NO synthase; Hepatocyte NOS; HEP-NOS Protein family NOS family Biochemical class Paired donor oxygen oxidoreductase Function Produces nitric oxide (NO) which is a messenger molecule with diverse functions throughout the body. In macrophages, NO mediates tumoricidal and bactericidal actions. Also has nitrosylase activity and mediates cysteine S-nitrosylation of cytoplasmic target proteins such PTGS2/COX2 (By similarity). As component of the iNOS-S100A8/9 transnitrosylase complex involved in the selective inflammatory stimulus-dependent S-nitrosylation of GAPDH on 'Cys-247' implicated in regulation of the GAIT complex activity and probably multiple targets including ANXA5, EZR, MSN and VIM. Involved in inflammation, enhances the synthesis of proinflammatory mediators such as IL6 and IL8. Related diseases Cerebellar ataxia, impaired intellectual development, and dysequilibrium syndrome 3 (CAMRQ3) [MIM:613227]: An autosomal recessive, congenital cerebellar ataxia associated with dysarthia, quadrupedal gait and intellectual disability. {ECO:0000269|PubMed:19461874}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07003; DB07007; DB07011; DB07405; DB08750; DB01997; DB07029; DB07008; DB08214; DB07002; DB01835; DB06879; DB04534; DB03100; DB02207; DB00125; DB00155; DB01234; DB14649; DB11327; DB00997; DB07306; DB07388; DB05252; DB01381; DB03366; DB05214; DB04400; DB09237; DB00244; DB01110; DB01017; DB03144; DB01686; DB03449; DB06916; DB07318; DB07389; DB02044; DB02644; DB05383; DB02234; DB03953; DB02462; DB08814 Interacts with P04406 EC number EC 1.14.13.39 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Calmodulin-binding; Cytoplasm; FAD; Flavoprotein; FMN; Heme; Iron; Metal-binding; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 48633 Length 421 Aromaticity 0.12 Instability index 46.5 Isoelectric point 6.75 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -7.8 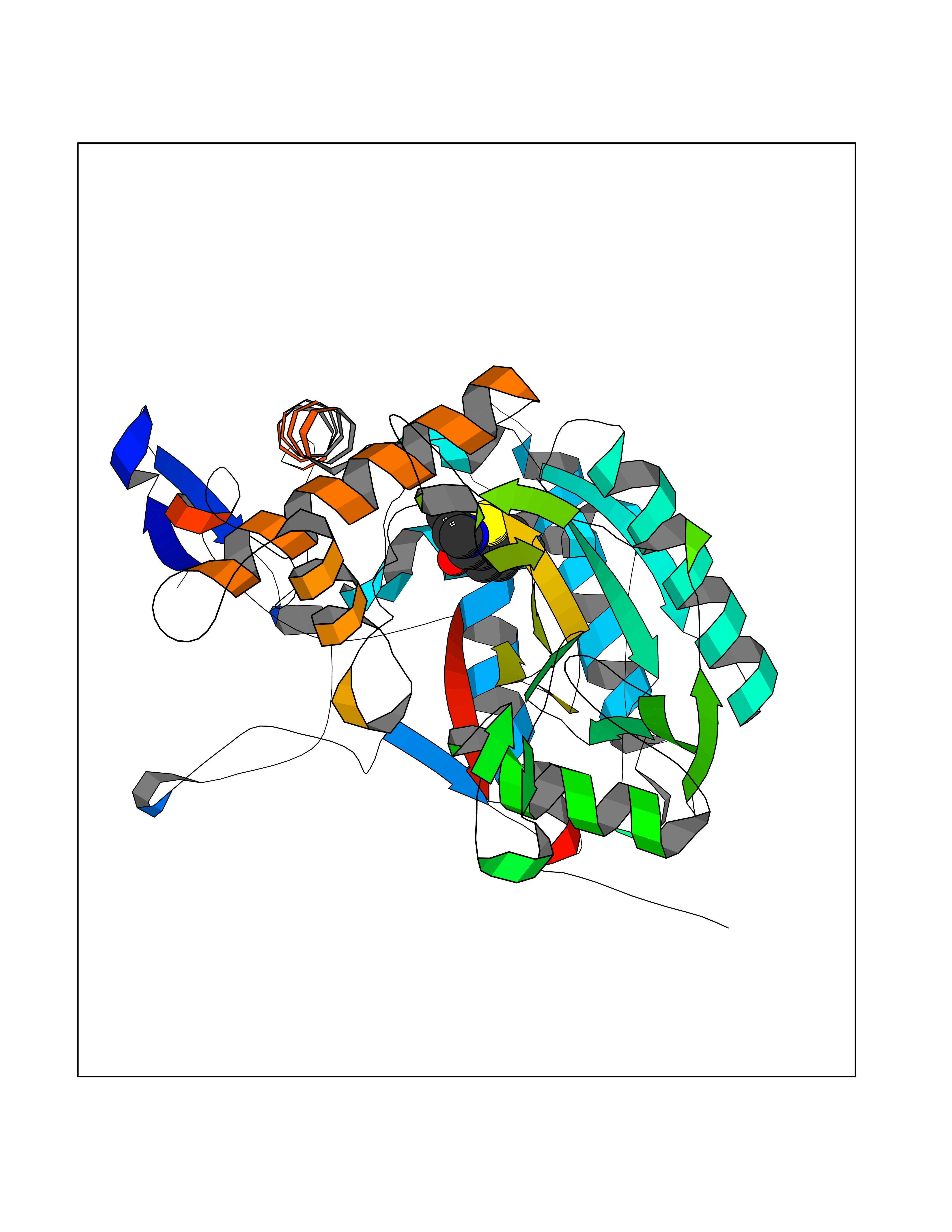 Molscript Map 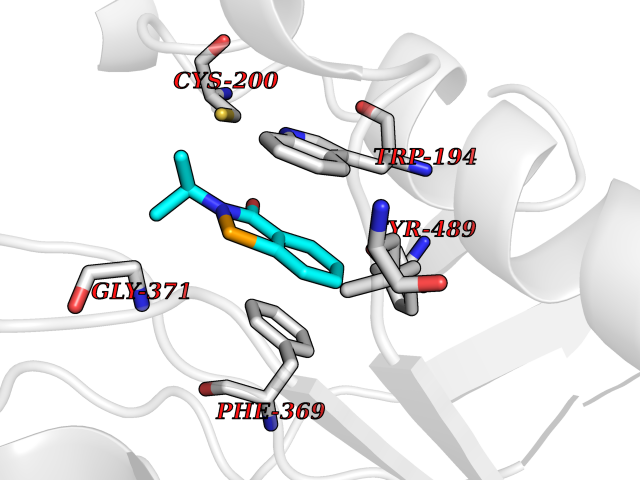 Pymol Map 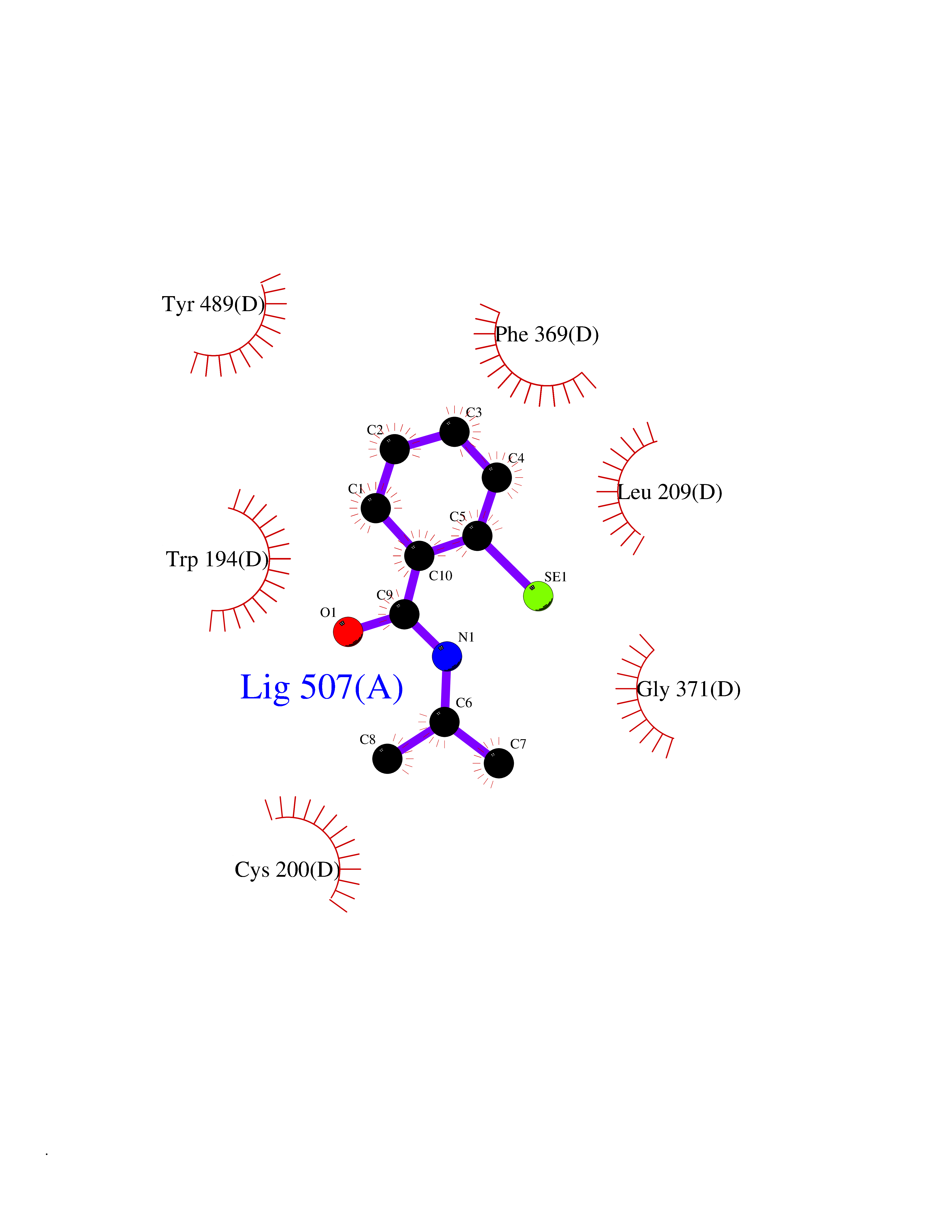 Ligplot Map 3D Binding mode Sequence RHVRIKNWGSGMTFQDTLHHKAKGILTCRSKSCLGSIMTPKSLTRGPRDKPTPPDELLPQAIEFVNQYYGSFKEAKIEEHLARVEAVTKEIETTGTYQLTGDELIFATKQAWRNAPRCIGRIQWSNLQVFDARSCSTAREMFEHICRHVRYSTNNGNIRSAITVFPQRSDGKHDFRVWNAQLIRYAGYQMPDGSIRGDPANVEFTQLCIDLGWKPKYGRFDVVPLVLQANGRDPELFEIPPDLVLEVAMEHPKYEWFRELELKWYALPAVANMLLEVGGLEFPGCPFNGWYMGTEIGVRDFCDVQRYNILEEVGRRMGLETHKLASLWKDQAVVEINIAVLHSFQKQNVTIMDHHSAAESFMKYMQNEYRSRGGCPADWIWLVPPMSGSITPVFHQEMLNYVLSPFYYYQVEAWKTHVWQD Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Guanidinoacetate N-methyltransferase | 3ORH | 5.72 | |
Target general information Gen name GAMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, RMT2 methyltransferase family Biochemical class Transferase Function Guanidinoacetate N-methyltransferase activity.Methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 2 (CCDS2) [MIM:612736]: An autosomal recessive disorder characterized by developmental delay and regression, intellectual disability, severe disturbance of expressive and cognitive speech, intractable seizures, movement disturbances, severe depletion of creatine and phosphocreatine in the brain, and accumulation of guanidinoacetic acid in brain and body fluids. {ECO:0000269|PubMed:12468279, ECO:0000269|PubMed:15108290, ECO:0000269|PubMed:15651030, ECO:0000269|PubMed:16293431, ECO:0000269|PubMed:16855203, ECO:0000269|PubMed:17101918, ECO:0000269|PubMed:17466557, ECO:0000269|PubMed:19388150, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:24415674, ECO:0000269|PubMed:8651275}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00148; DB02751; DB00536; DB13191; DB01752 Interacts with O95363; Q969Q5; Q9HCM9-2 EC number 2.1.1.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Disease variant; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 24656 Length 219 Aromaticity 0.11 Instability index 46.5 Isoelectric point 5.91 Charge (pH=7) -4.34 2D Binding mode Binding energy (Kcal/mol) -7.8 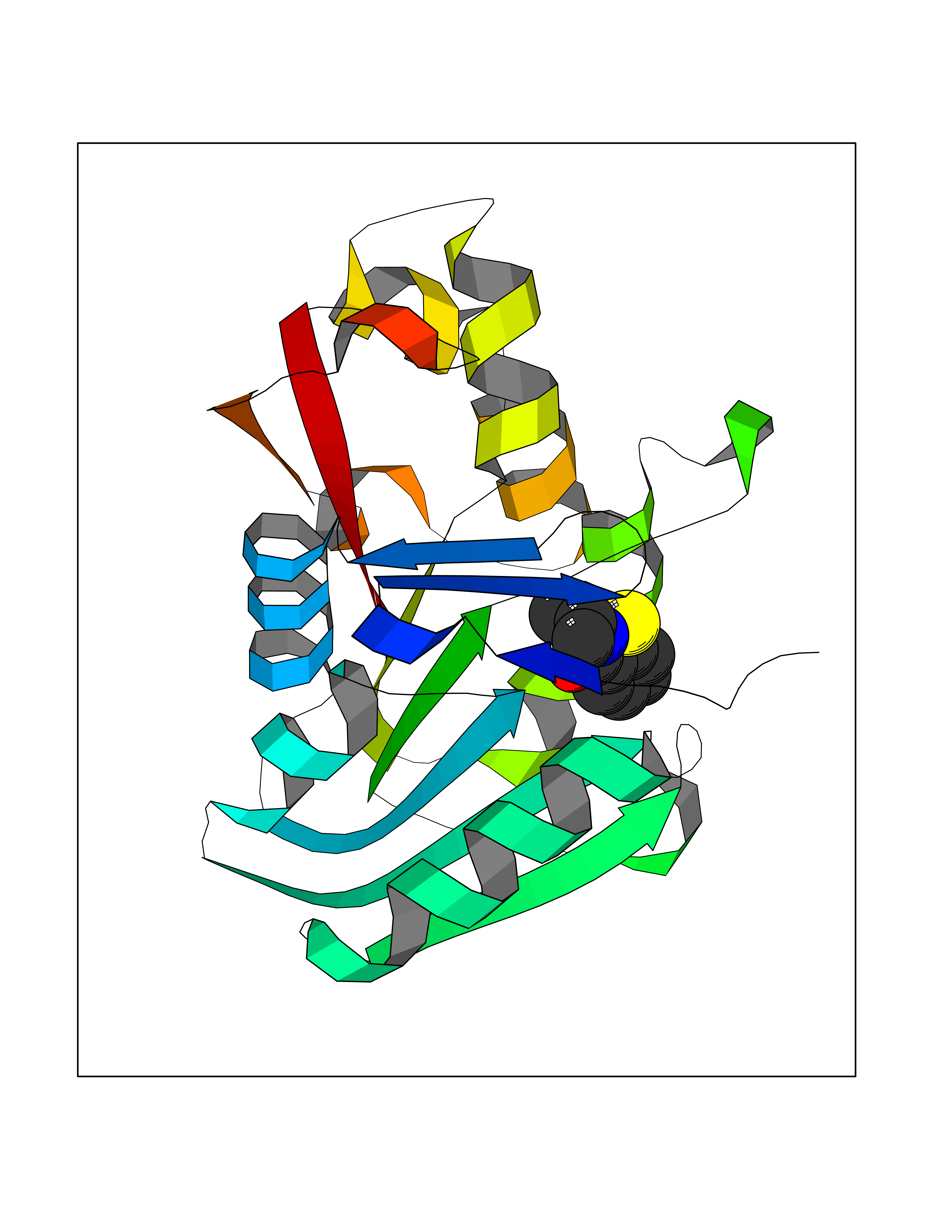 Molscript Map 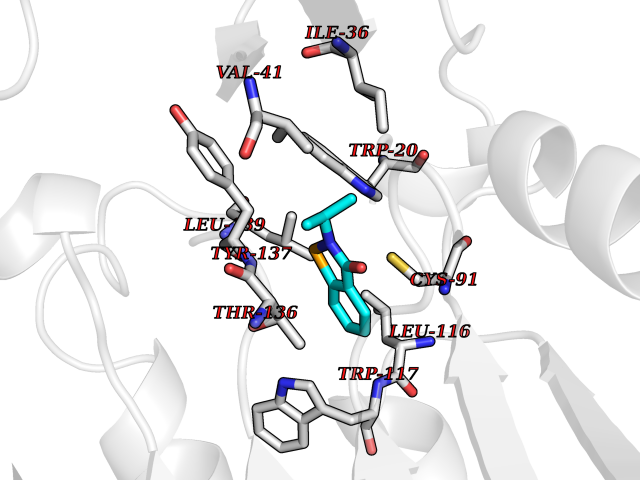 Pymol Map 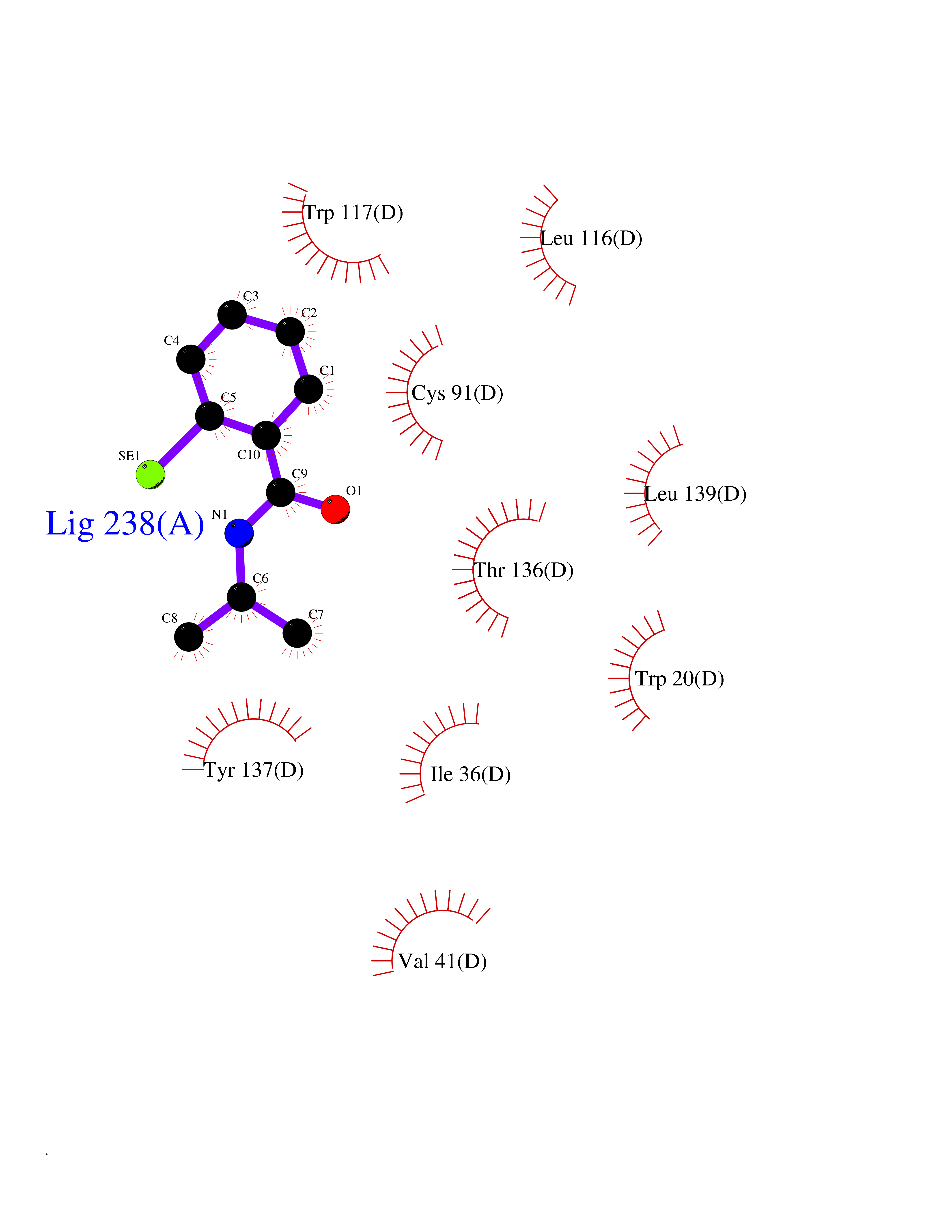 Ligplot Map 3D Binding mode Sequence PAWGAAPAAYDAADTHLRILGKPVMERWETPYMHALAAAASSKGGRVLEVGFGMAIAASKVQEAPIDEHWIIECNDGVFQRLRDWAPRQTHKVIPLKGLWEDVAPTLPDGHFDGILYDTYPLSEETWHTHQFNFIKNHAFRLLKPGGVLTYCNLTSWGELMKSKYSDITIMFEETQVPALLEAGFRRENIRTEVMALVPPADCRYYAFPQMITPLVTKG Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Neuronal acetylcholine receptor alpha-2 (CHRNA2) | 5FJV | 5.72 | |
Target general information Gen name CHRNA2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CHRNA2 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-2/CHRNA2 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane. Related diseases Epilepsy, nocturnal frontal lobe, 4 (ENFL4) [MIM:610353]: An autosomal dominant focal epilepsy characterized by nocturnal seizures associated with fear sensation, tongue movements, and nocturnal wandering, closely resembling nightmares and sleep walking. {ECO:0000269|PubMed:16826524}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Seizures, benign familial infantile, 6 (BFIS6) [MIM:610353]: A form of benign familial infantile epilepsy, a neurologic disorder characterized by afebrile seizures occurring in clusters during the first year of life, without neurologic sequelae. BFIS6 inheritance is autosomal dominant. {ECO:0000269|PubMed:25847220}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00732; DB00237; DB00411; DB00565; DB01245; DB00514; DB01135; DB07720; DB00898; DB00472; DB00483; DB08960; DB00657; DB01336; DB00416; DB01226; DB00184; DB01337; DB01338; DB00721; DB00728; DB05740; DB00202; DB01199; DB01339 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 48323.4 Length 413 Aromaticity 0.15 Instability index 32 Isoelectric point 5.69 Charge (pH=7) -6.58 2D Binding mode Binding energy (Kcal/mol) -7.8 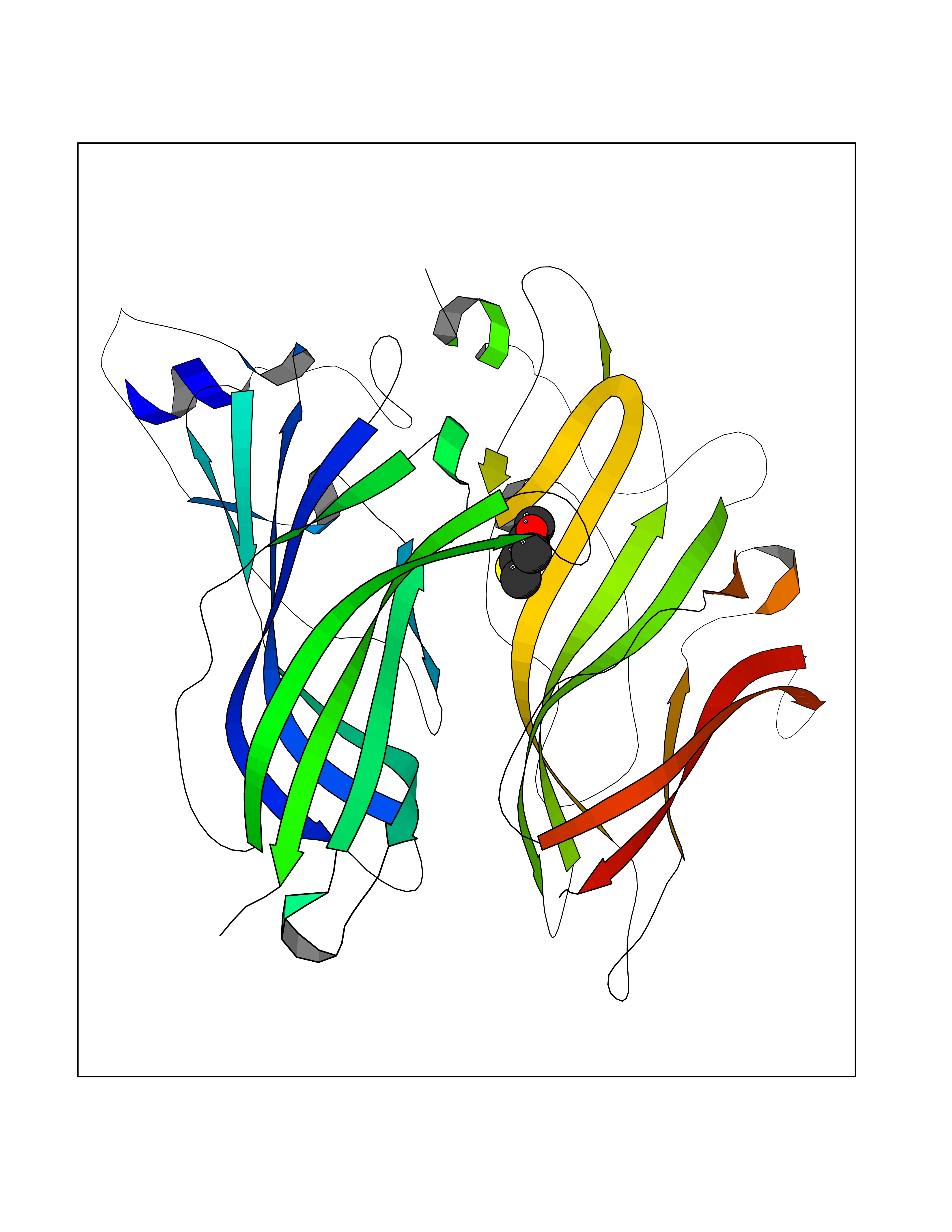 Molscript Map 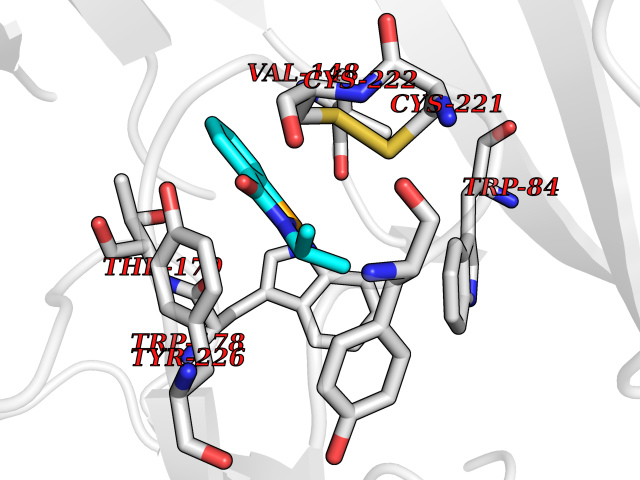 Pymol Map 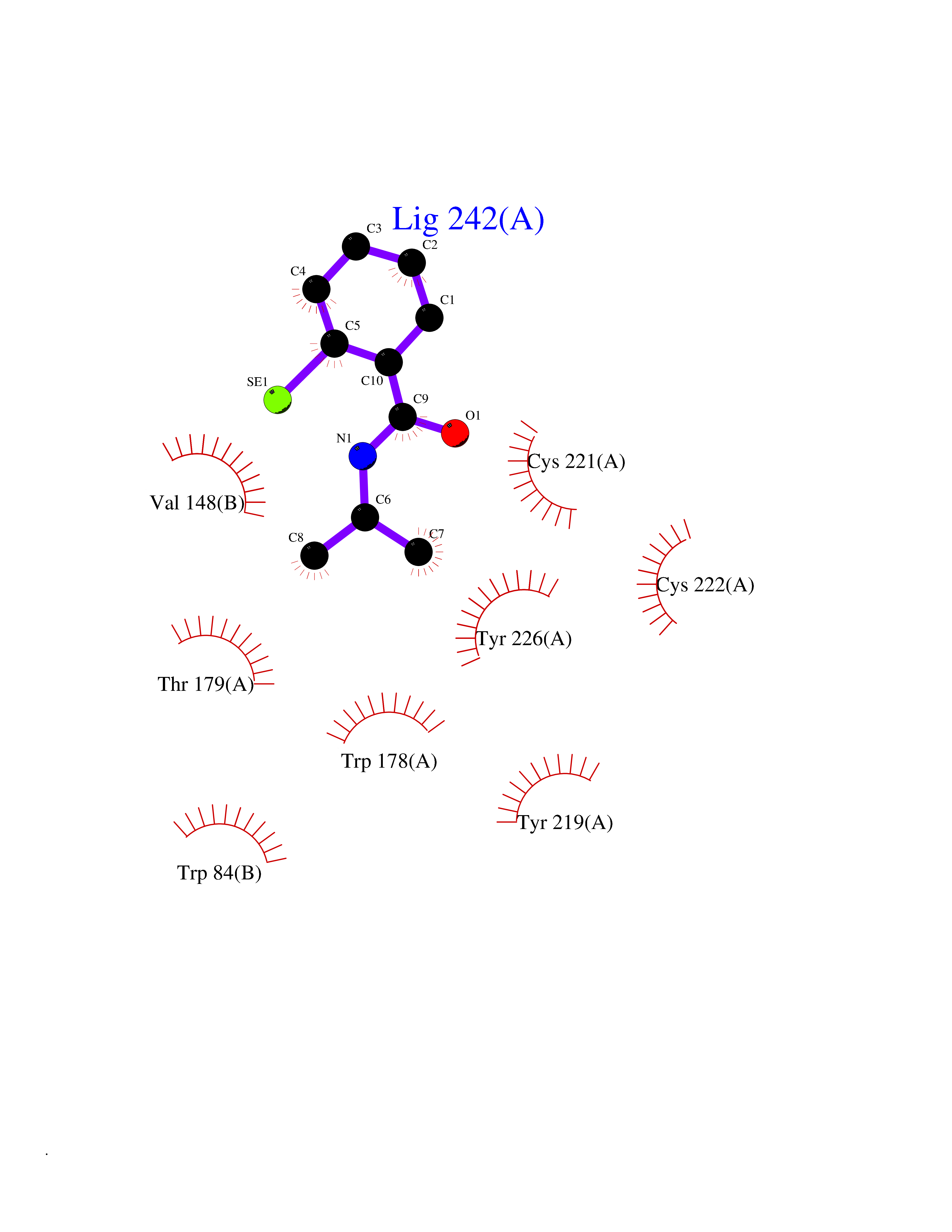 Ligplot Map 3D Binding mode Sequence DRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLP Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Hyperpolarization cyclic nucleotide-gated channel 2 (HCN2) | 3U10 | 5.72 | |
Target general information Gen name HCN2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channel 2; Brain cyclic nucleotide-gated channel 2; BCNG2; BCNG-2 Protein family Potassium channel HCN family Biochemical class Voltage-gated ion channel Function Contributes to the native pacemaker currents in heart (If) and in neurons (Ih). Can also transport ammonium in the distal nephron. Produces a large instantaneous current. Modulated by intracellular chloride ions and pH; acidic pH shifts the activation to more negative voltages. Hyperpolarization-activated ion channel exhibiting weak selectivity for potassium over sodium ions. Related diseases Epilepsy, idiopathic generalized 17 (EIG17) [MIM:602477]: A form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. Both autosomal dominant and autosomal recessive EIG17 inheritance have been reported. {ECO:0000269|PubMed:22131395, ECO:0000269|PubMed:29064616}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Febrile seizures, familial, 2 (FEB2) [MIM:602477]: Seizures associated with febrile episodes in childhood without any evidence of intracranial infection or defined pathologic or traumatic cause. It is a common condition, affecting 2-5% of children aged 3 months to 5 years. The majority are simple febrile seizures (generally defined as generalized onset, single seizures with a duration of less than 30 minutes). Complex febrile seizures are characterized by focal onset, duration greater than 30 minutes, and/or more than one seizure in a 24 hour period. The likelihood of developing epilepsy following simple febrile seizures is low. Complex febrile seizures are associated with a moderately increased incidence of epilepsy. FEB2 transmission pattern is consistent with autosomal dominant inheritance. {ECO:0000269|PubMed:24324597}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02527; DB02315; DB09083 Interacts with Q9UL51; Q4ACU6-1 EC number NA Uniprot keywords 3D-structure; Ammonia transport; cAMP; cAMP-binding; Cell membrane; Disease variant; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Lipoprotein; Membrane; Methylation; Nucleotide-binding; Palmitate; Phosphoprotein; Potassium; Potassium channel; Potassium transport; Proteomics identification; Reference proteome; Sodium; Sodium channel; Sodium transport; Transmembrane; Transmembrane helix; Transport; Voltage-gated channel Protein physicochemical properties Chain ID A Molecular weight (Da) 23672.9 Length 202 Aromaticity 0.12 Instability index 38.05 Isoelectric point 8.85 Charge (pH=7) 4.11 2D Binding mode Binding energy (Kcal/mol) -7.8 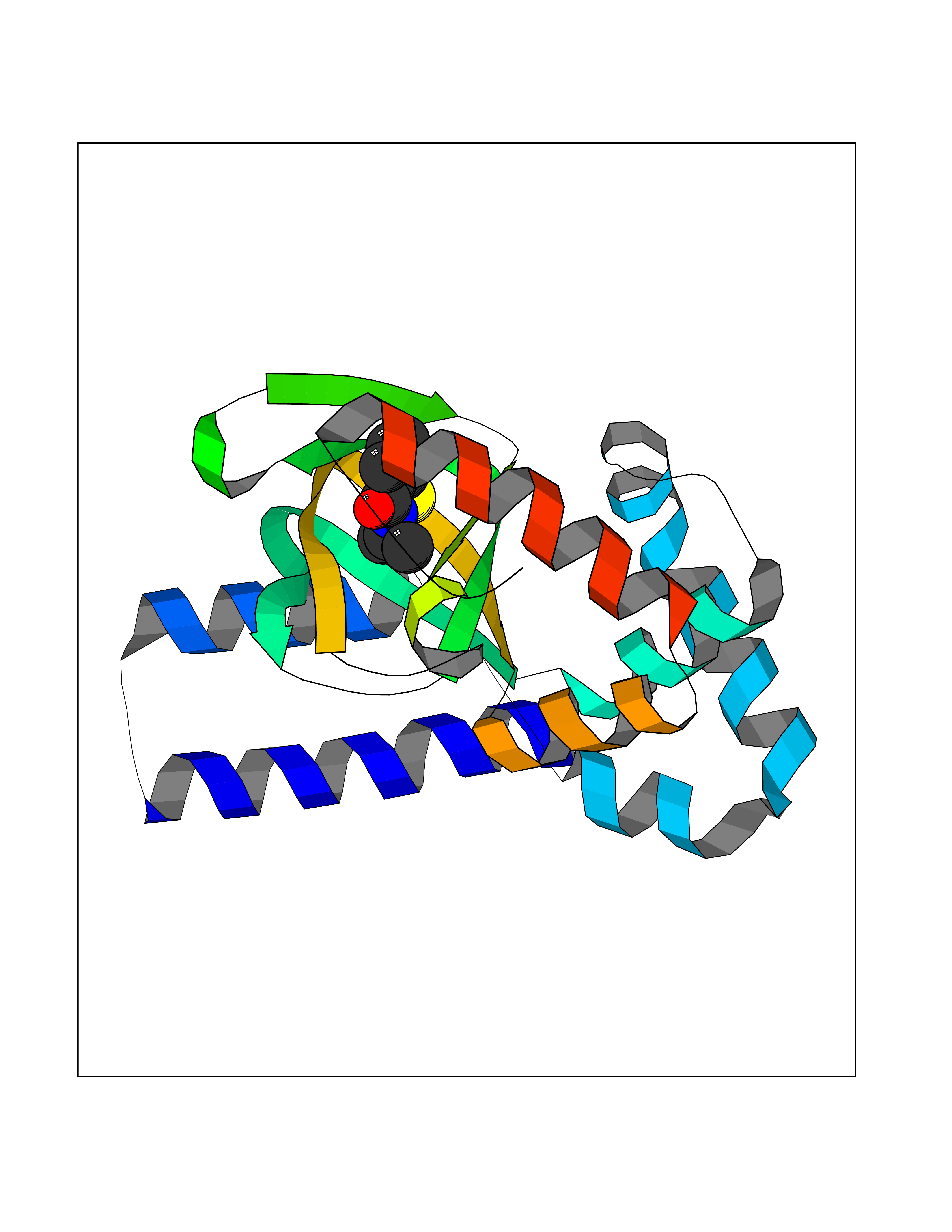 Molscript Map 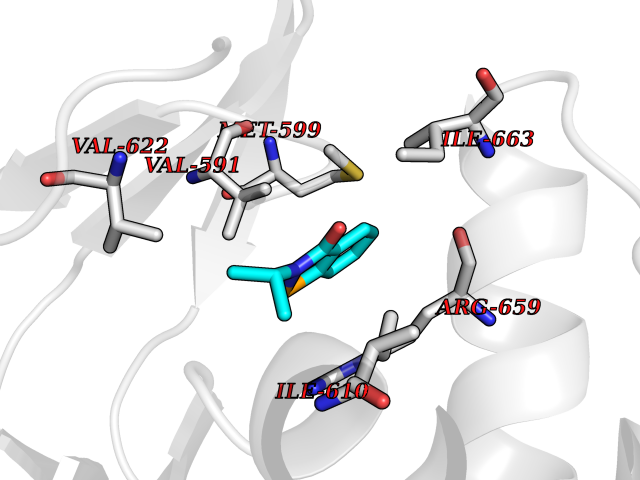 Pymol Map 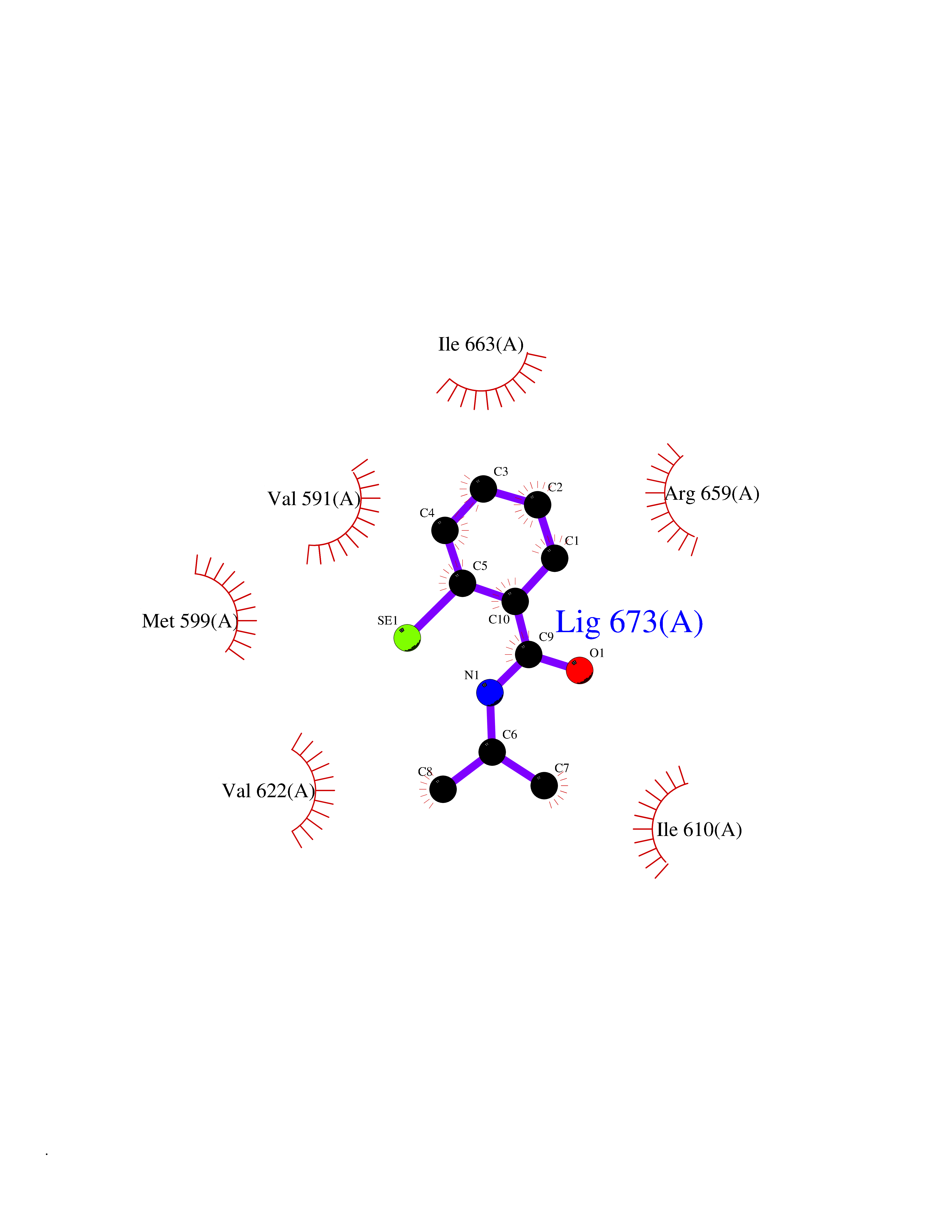 Ligplot Map 3D Binding mode Sequence DSSRRQYQEKYKQVEQYMSFHKLPADFRQKIHDYYEHRYQGKMFDEDSILGELNGPLREEIVNFNCRKLVASMPLFANADPNFVTAMLTKLKFEVFQPGDYIIREGTIGKKMYFIQHGVVSVLTKGNKEMKLSDGSYFGEICLLTRGRRTASVRADTYCRLYSLSVDNFNEVLEEYPMMRRAFETVAIDRLDRIGKKNSILL Hydrogen bonds contact Hydrophobic contact | ||||