Job Results:
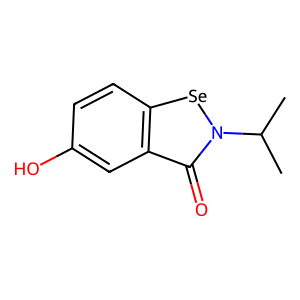
Ligand
Structure
Job ID
176b63e19640f4b1f79a8fdfdb32fb77
Job name
NA
Time
2025-10-13 17:39:01
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Receptor-type protein-tyrosine phosphatase zeta (PTPRZ1) | 5H08 | 5.75 | |
Target general information Gen name PTPRZ1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Receptor protein tyrosine phosphatase zeta; R-PTP-zeta; PTPRZ1 Protein family Protein-tyrosine phosphatase family, Receptor class 5 subfamily Biochemical class Phosphoric monoester hydrolase Function Protein tyrosine phosphatase that negatively regulates oligodendrocyte precursor proliferation in the embryonic spinal cord. Required for normal differentiation of the precursor cells into mature, fully myelinating oligodendrocytes. May play a role in protecting oligondendrocytes against apoptosis. May play a role in the establishment of contextual memory, probably via the dephosphorylation of proteins that are part of important signaling cascades. Related diseases Optic atrophy 1 (OPA1) [MIM:165500]: A condition that features progressive visual loss in association with optic atrophy. Atrophy of the optic disk indicates a deficiency in the number of nerve fibers which arise in the retina and converge to form the optic disk, optic nerve, optic chiasm and optic tracts. OPA1 is characterized by an insidious onset of visual impairment in early childhood with moderate to severe loss of visual acuity, temporal optic disk pallor, color vision deficits, and centrocecal scotoma of variable density. {ECO:0000269|PubMed:11017079, ECO:0000269|PubMed:11017080, ECO:0000269|PubMed:11440988, ECO:0000269|PubMed:11440989, ECO:0000269|PubMed:11810270, ECO:0000269|PubMed:12036970, ECO:0000269|PubMed:12566046, ECO:0000269|PubMed:14961560, ECO:0000269|PubMed:15948788, ECO:0000269|PubMed:16513463, ECO:0000269|PubMed:16617242, ECO:0000269|PubMed:18204809, ECO:0000269|PubMed:18360822, ECO:0000269|PubMed:19319978, ECO:0000269|PubMed:19325939, ECO:0000269|PubMed:19969356, ECO:0000269|PubMed:20185555, ECO:0000269|PubMed:22382025, ECO:0000269|PubMed:22857269, ECO:0000269|PubMed:23401657}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Dominant optic atrophy plus syndrome (DOA+) [MIM:125250]: A neurologic disorder characterized most commonly by an insidious onset of visual loss and sensorineural hearing loss in childhood with variable presentation of other clinical manifestations including progressive external ophthalmoplegia, muscle cramps, hyperreflexia, and ataxia. There appears to be a wide range of intermediate phenotypes. {ECO:0000269|PubMed:15531309, ECO:0000269|PubMed:16240368, ECO:0000269|PubMed:18065439, ECO:0000269|PubMed:18158317, ECO:0000269|PubMed:18195150, ECO:0000269|PubMed:20185555, ECO:0000269|PubMed:21112924, ECO:0000269|PubMed:23387428}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Behr syndrome (BEHRS) [MIM:210000]: An autosomal recessive syndrome characterized by optic atrophy beginning in early childhood associated with ataxia, pyramidal signs, spasticity, intellectual disability, and posterior column sensory loss. The ataxia, spasticity, and muscle contractures, mainly of the hip adductors, hamstrings, and soleus, are progressive and become more prominent in the second decade. {ECO:0000269|PubMed:21636302, ECO:0000269|PubMed:25012220, ECO:0000269|PubMed:25146916}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mitochondrial DNA depletion syndrome 14, cardioencephalomyopathic type (MTDPS14) [MIM:616896]: An autosomal recessive mitochondrial disorder characterized by lethal infantile encephalopathy, hypertrophic cardiomyopathy and optic atrophy. Skeletal muscle biopsies show significant mtDNA depletion and abnormal mitochondria. {ECO:0000269|PubMed:26561570}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9UM73; Q12860 EC number EC 3.1.3.48 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Hydrolase; Membrane; Phosphoprotein; Protein phosphatase; Proteoglycan; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 32208.1 Length 282 Aromaticity 0.11 Instability index 38.98 Isoelectric point 7.35 Charge (pH=7) 0.89 2D Binding mode Binding energy (Kcal/mol) -7.84 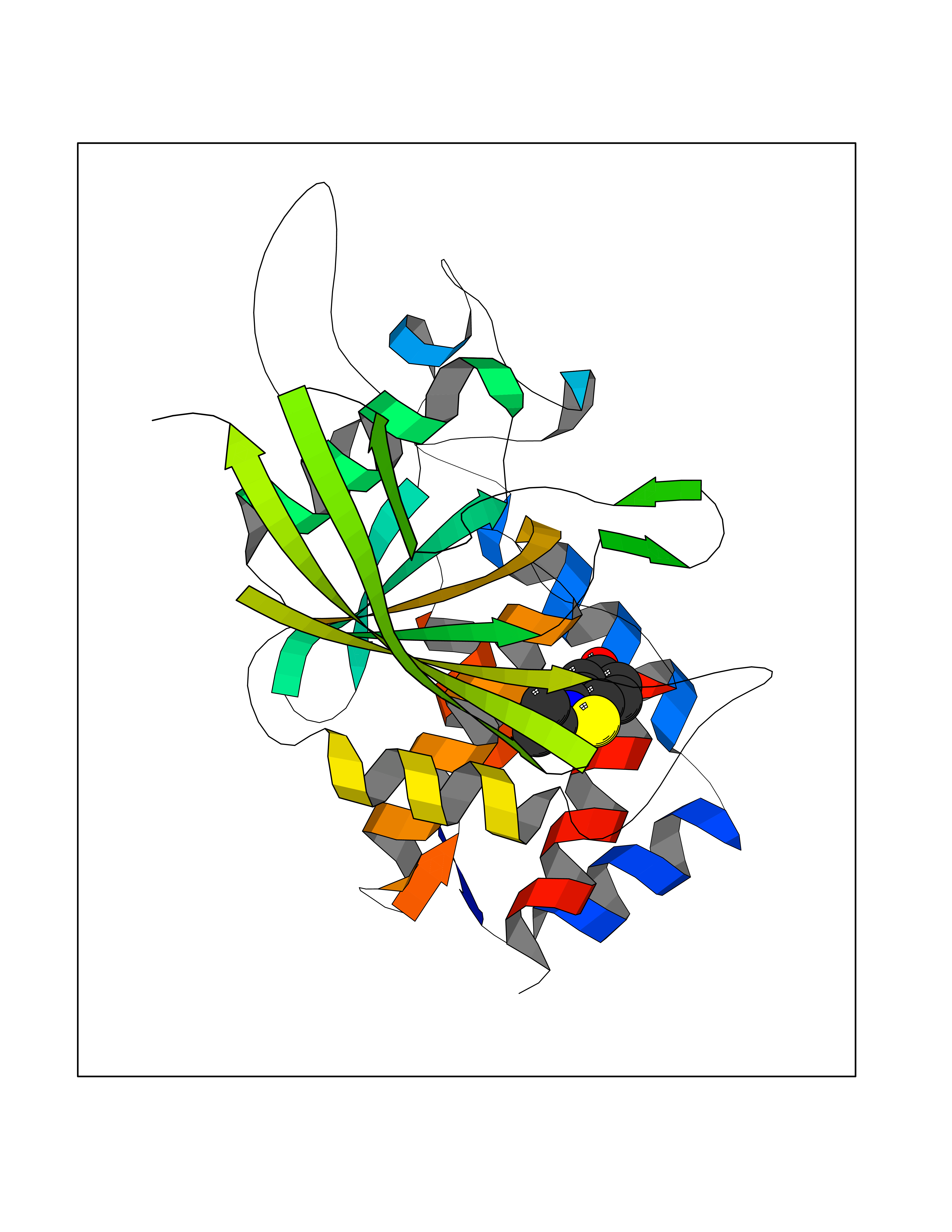 Molscript Map 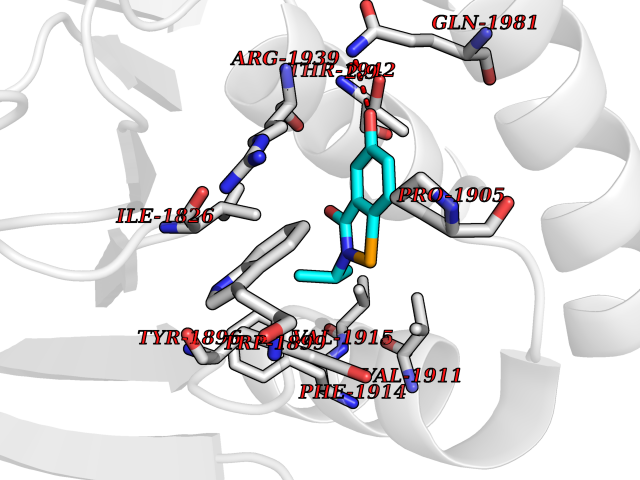 Pymol Map 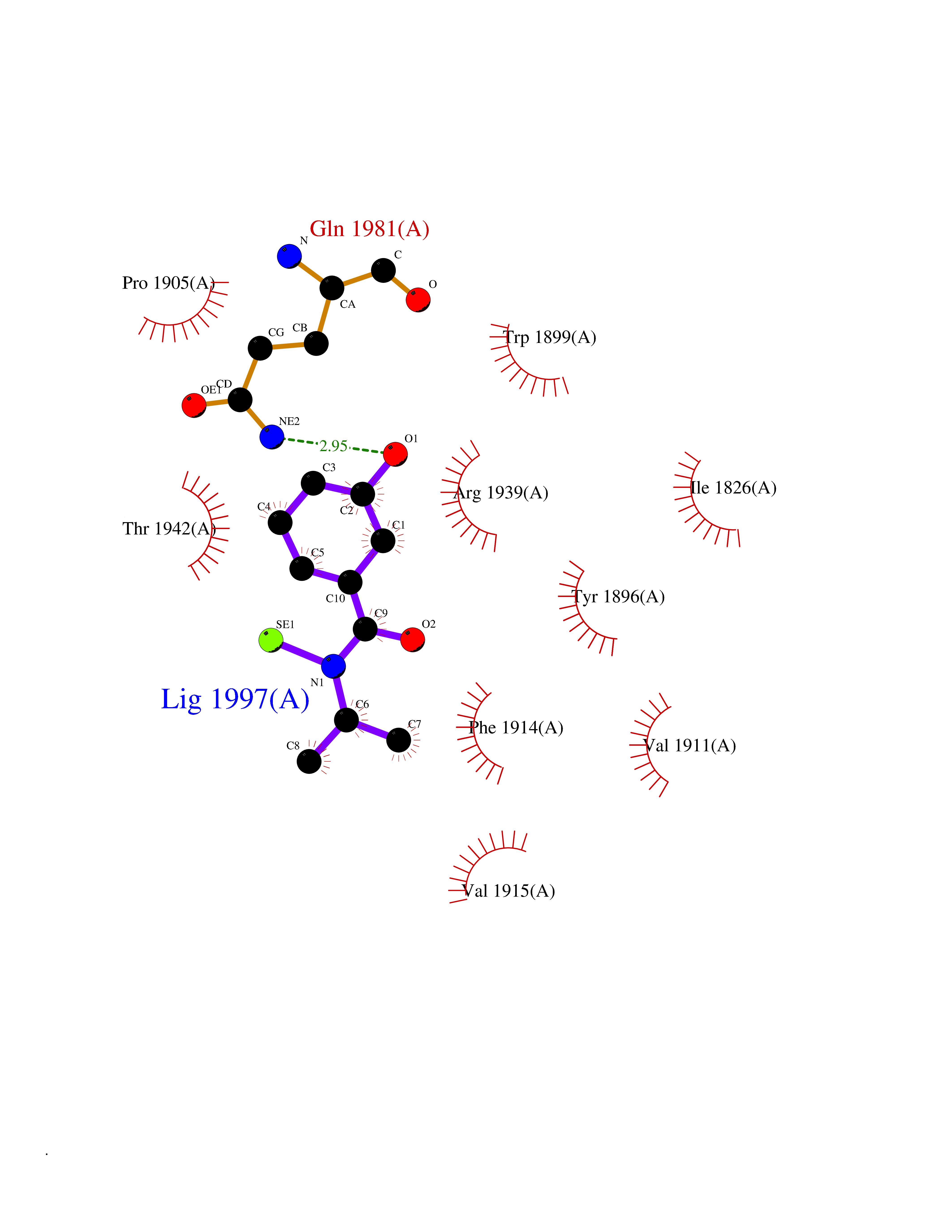 Ligplot Map 3D Binding mode Sequence GPAIPIKHFPKHVADLHASSGFTEEFEEVQSCTVDLGITADSSNHPDNKHKNRYINIVAYDHSRVKLAQLAEKDGKLTDYINANYVDGYNRPKAYIAAQGPLKSTAEDFWRMIWEHNVEVIVMITNLVEKGRRKCDQYWPADGSEEYGNFLVTQKSVQVLAYYTVRNFTLRNTKIRVVTQYHYTQWPDMGVPEYSLPVLTFVRKAAYAKRHAVGPVVVHCSAGVGRTGTYIVLDSMLQQIQHEGTVNIFGFLKHIRSQRNYLVQTEEQYVFIHDTLVEAILS Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Tyrosine 3-monooxygenase (TH) | 2XSN | 5.74 | |
Target general information Gen name TH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine 3-hydroxylase; TH Protein family Biopterin-dependent aromatic amino acid hydroxylase family Biochemical class Paired donor oxygen oxidoreductase Function Plays an important role in the physiology of adrenergic neurones. Related diseases Segawa syndrome autosomal recessive (ARSEGS) [MIM:605407]: A form of DOPA-responsive dystonia presenting in infancy or early childhood. Dystonia is defined by the presence of sustained involuntary muscle contractions, often leading to abnormal postures. Some cases present with parkinsonian symptoms in infancy. Unlike all other forms of dystonia, it is an eminently treatable condition, due to a favorable response to L-DOPA. {ECO:0000269|PubMed:10585338, ECO:0000269|PubMed:11196107, ECO:0000269|PubMed:11246459, ECO:0000269|PubMed:15505183, ECO:0000269|PubMed:15747353, ECO:0000269|PubMed:16049992, ECO:0000269|PubMed:17696123, ECO:0000269|PubMed:18058633, ECO:0000269|PubMed:18554280, ECO:0000269|PubMed:19491146, ECO:0000269|PubMed:20056467, ECO:0000269|PubMed:20430833, ECO:0000269|PubMed:21940685, ECO:0000269|PubMed:22264700, ECO:0000269|PubMed:22815559, ECO:0000269|PubMed:23762320, ECO:0000269|PubMed:23939262, ECO:0000269|PubMed:24753243, ECO:0000269|PubMed:7814018, ECO:0000269|PubMed:8528210, ECO:0000269|PubMed:8817341, ECO:0000269|PubMed:9613851, ECO:0000269|PubMed:9703425}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: May play a role in the pathogenesis of Parkinson disease (PD). A genome-wide copy number variation analysis has identified a 34 kilobase deletion over the TH gene in a PD patient but not in any controls. {ECO:0000269|PubMed:20809526}. Drugs (DrugBank ID) DB03552; DB04400; DB00765; DB00120; DB00360; DB00135 Interacts with P29762; P61978-2; Q99750; P08651-5; O75928-2; Q9UHX1-2; P0DJD3-2; P07101-3; Q9UJ04; C9J7I0; Q5MCW4 EC number EC 1.14.16.2 Uniprot keywords 3D-structure; Alternative splicing; Catecholamine biosynthesis; Cell projection; Cytoplasm; Cytoplasmic vesicle; Disease variant; Dystonia; Iron; Metal-binding; Monooxygenase; Neurotransmitter biosynthesis; Nucleus; Oxidoreductase; Parkinson disease; Parkinsonism; Phosphoprotein; Proteomics identification; Reference proteome; Synapse Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 34997 Length 306 Aromaticity 0.12 Instability index 42.59 Isoelectric point 5.32 Charge (pH=7) -12.31 2D Binding mode Binding energy (Kcal/mol) -7.83 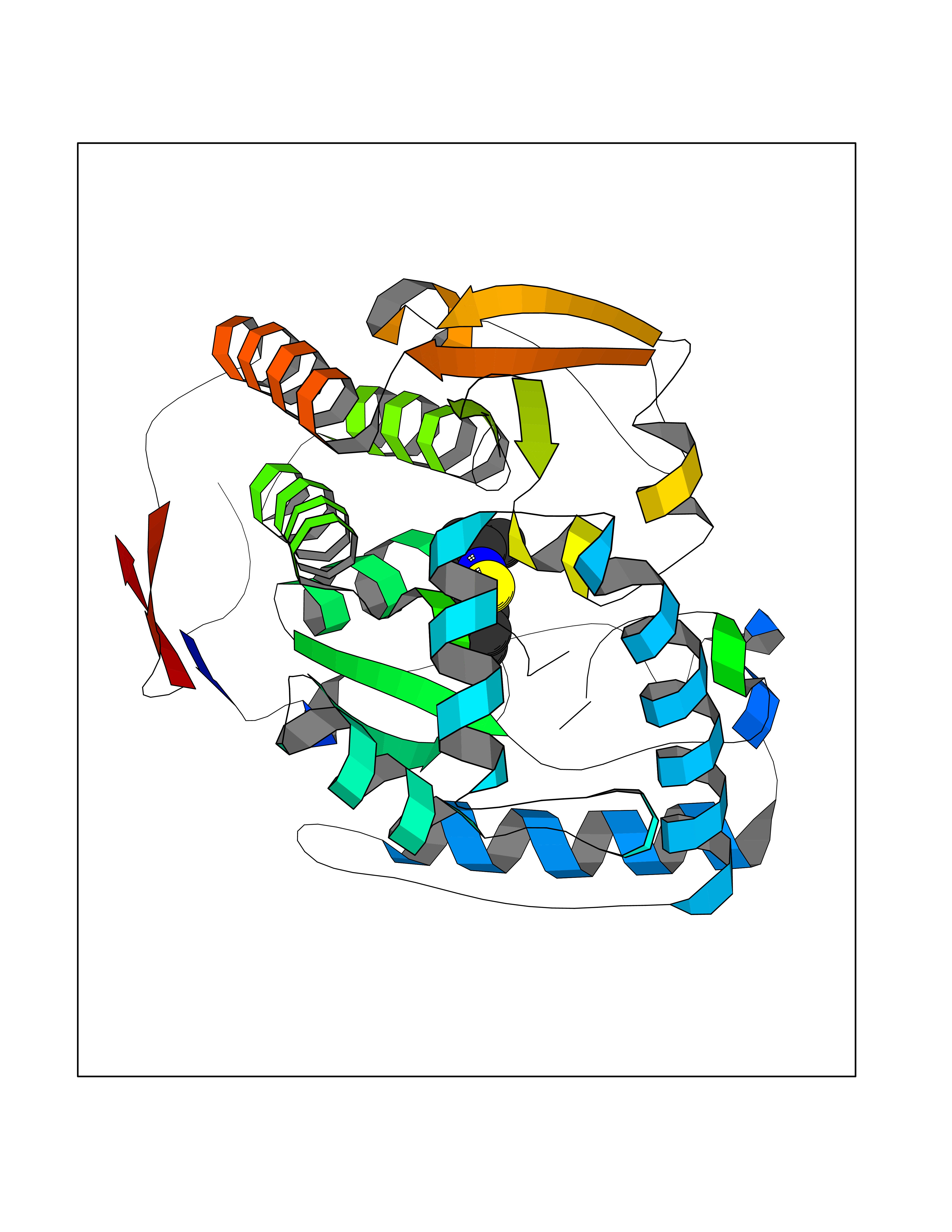 Molscript Map 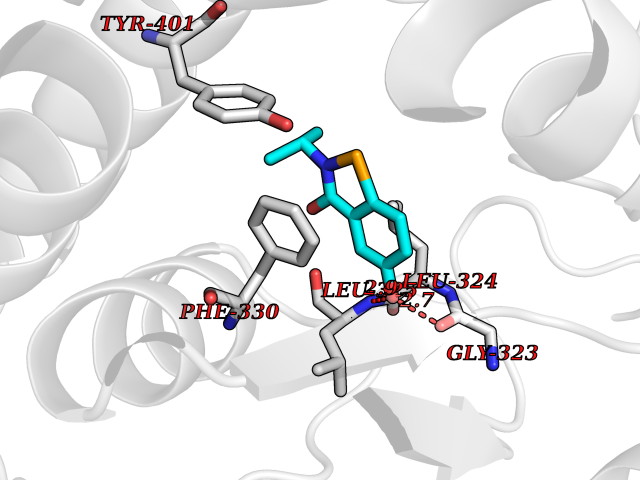 Pymol Map 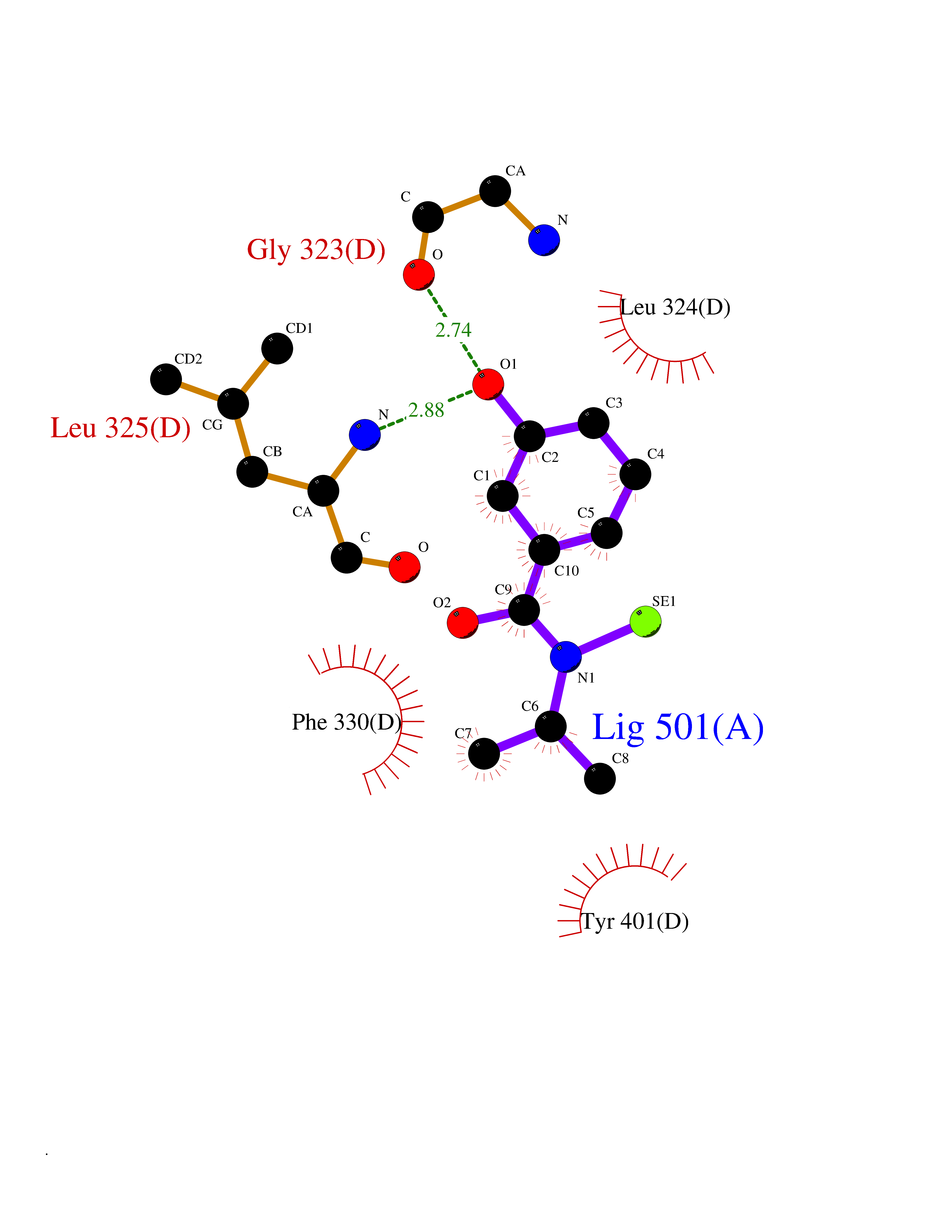 Ligplot Map 3D Binding mode Sequence VPWFPRKVSELDKCHHLVTKFDPDLDLDHPGFSDQVYRQRRKLIAEIAFQYRHGDPIPRVEYTAEEIATWKEVYTTLKGLYATHACGEHLEAFALLERFSGYREDNIPQLEDVSRFLKERTGFQLRPVAGLLSARDFLASLAFRVFQCTQYIRHASSPMHSPEPDCCHELLGHVPMLADRTFAQFSQDIGLASLGASDEEIEKLSTLYWFTVEFGLCKQNGEVKAYGAGLLSSYGELLHCLSEEPEIRAFDPEAAAVQPYQDQTYQSVYFVSESFSDAKDKLRSYASRIQRPFSVKFDPYTLAIDV Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Glutamate receptor ionotropic kainate 1 (GRIK1) | 3FV2 | 5.74 | |
Target general information Gen name GRIK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Glutamate receptor 5; GluR5 kainate receptor; GluR5; GluR-5; GRIK1; Excitatory amino acid receptor 3; EAA3 Protein family Glutamate-gated ion channel (TC 1.A.10.1) family, GRIK1 subfamily Biochemical class Glutamate-gated ion channel Function Ionotropic glutamate receptor. L-glutamate acts as an excitatory neurotransmitter at many synapses in the central nervous system. Binding of the excitatory neurotransmitter L- glutamate induces a conformation change, leading tothe opening of the cation channel, and thereby converts the chemical signal to an electrical impulse. The receptor then desensitizes rapidly and enters a transient inactive state, characterized by the presence of bound agonist. May be involved in the transmission of light information from the retina to the hypothalamus. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB00237; DB00142; DB06354; DB00273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; RNA editing; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 29057.1 Length 256 Aromaticity 0.1 Instability index 24.38 Isoelectric point 8.3 Charge (pH=7) 1.97 2D Binding mode Binding energy (Kcal/mol) -7.84 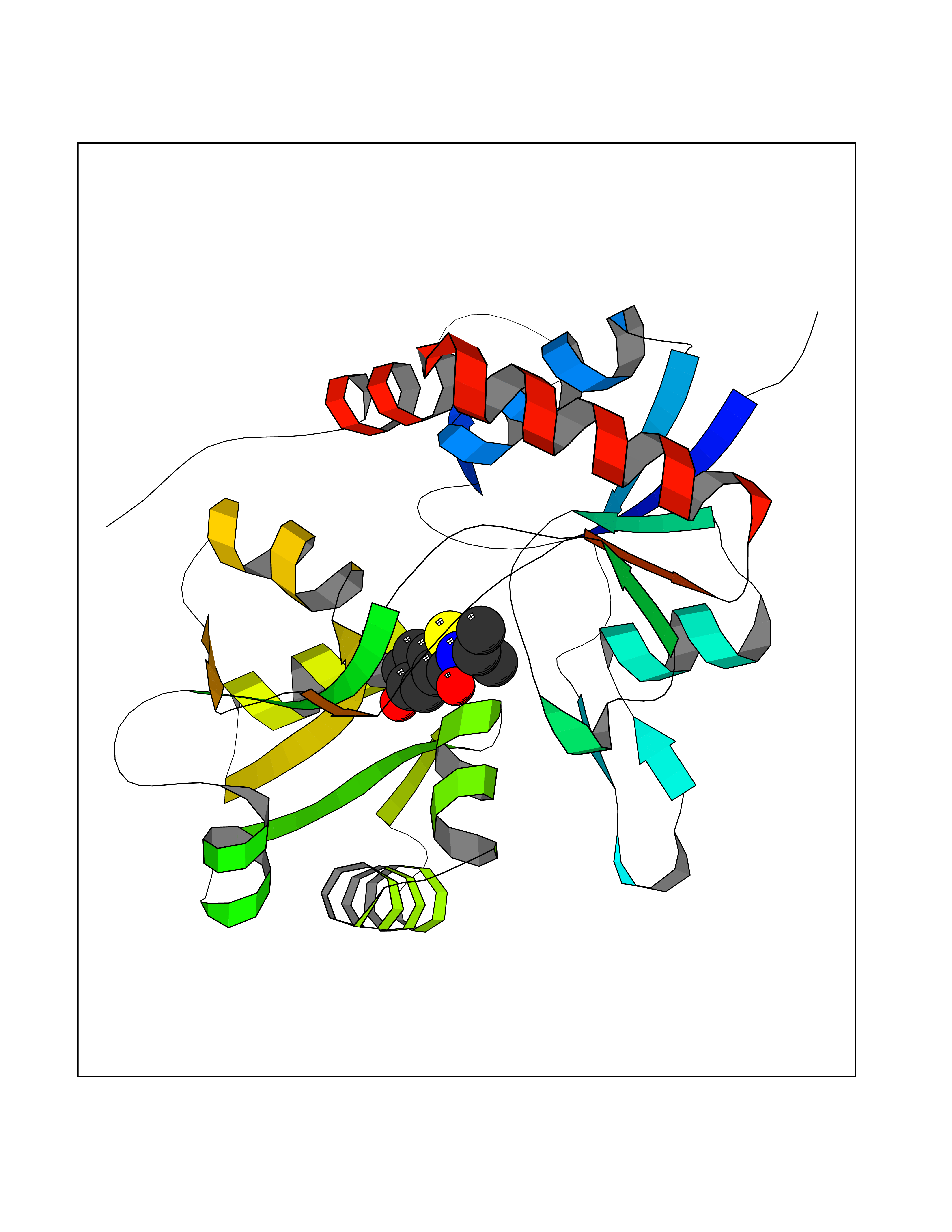 Molscript Map 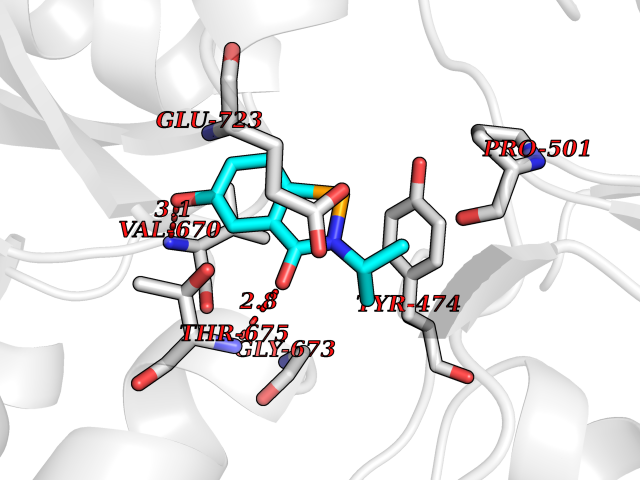 Pymol Map 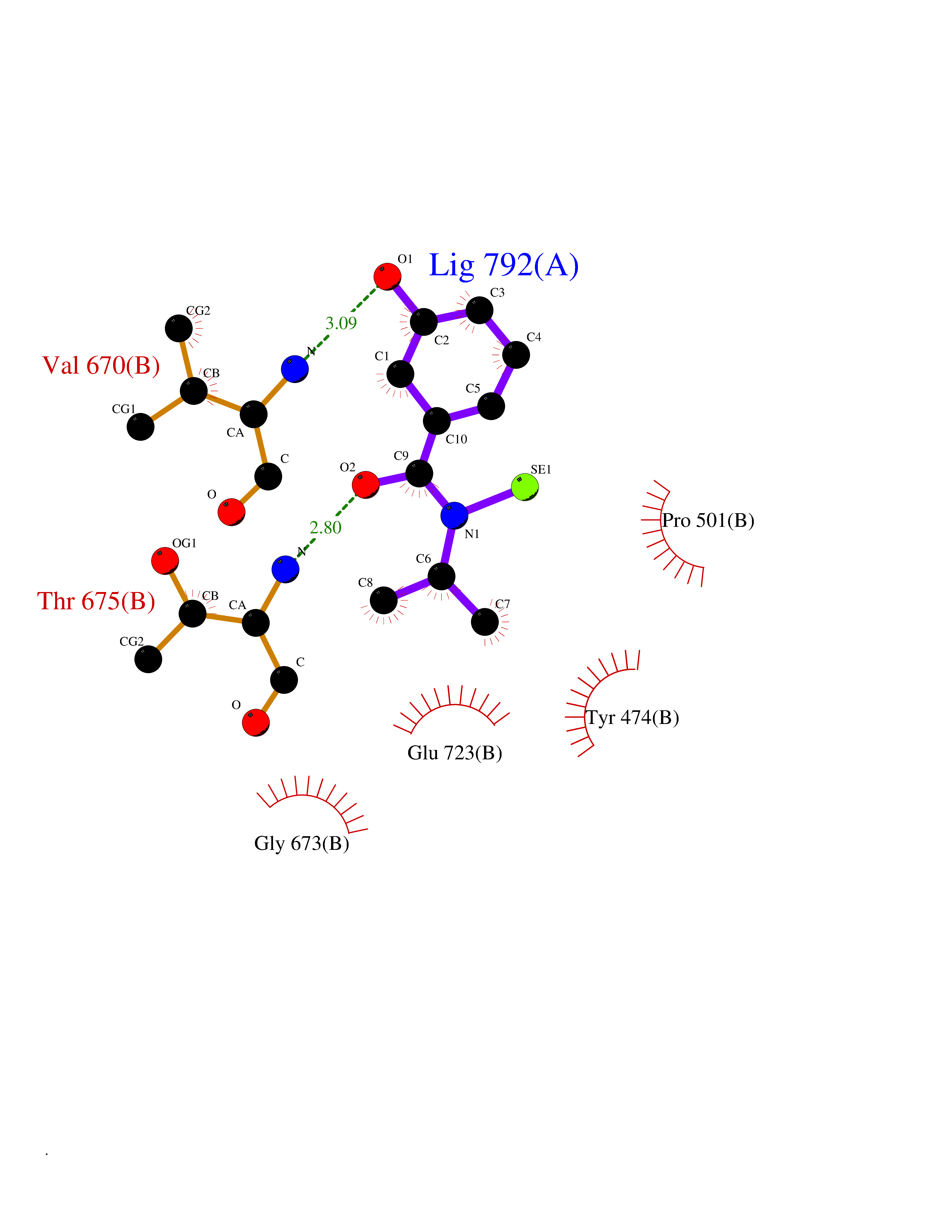 Ligplot Map 3D Binding mode Sequence ANRTLIVTTILEEPYVMYRKSDKPLYGNDRFEGYCLDLLKELSNILGFIYDVKLVPDGKYGAQNDKGEWNGMVKELIDHRADLAVAPLTITYVREKVIDFSKPFMTLGISILYRKGTPIDSADDLAKQTKIEYGAVRDGSTMTFFKKSKISTYEKMWAFMSSRQQTALVRNSDEGIQRVLTTDYALLMESTSIEYVTQRNCNLTQIGGLIDSKGYGVGTPIGSPYRDKITIAILQLQEEGKLHMMKEKWWRGNGCP Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Methionine aminopeptidase 2 (METAP2) | 5D6E | 5.74 | |
Target general information Gen name METAP2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Peptidase M 2; P67eIF2; P67; MetAP 2; METAP2; Initiation factor 2 associated 67 kDa glycoprotein; (MetAP)-2 Protein family Peptidase M24A family, Methionine aminopeptidase eukaryotic type 2 subfamily Biochemical class Peptidase Function Cotranslationally removes the N-terminal methionine from nascent proteins. The N-terminal methionine is often cleaved when the second residue in the primary sequence is small and uncharged (Met-Ala-, Cys, Gly, Pro, Ser, Thr, or Val). Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03396; DB04108; DB07322; DB07323; DB02310; DB07746; DB07309; DB07313; DB02893; DB02640; DB00134; DB07377; DB03086; DB01422; DB04324; DB05864; DB03900; DB08633 Interacts with P21917; Q96LA5; Q9HBH5 EC number EC 3.4.11.18 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminopeptidase; Cytoplasm; Direct protein sequencing; Glycoprotein; Hydrolase; Metal-binding; Phosphoprotein; Protease; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 41399.7 Length 370 Aromaticity 0.08 Instability index 32.42 Isoelectric point 5.65 Charge (pH=7) -8.25 2D Binding mode Binding energy (Kcal/mol) -7.83  Molscript Map 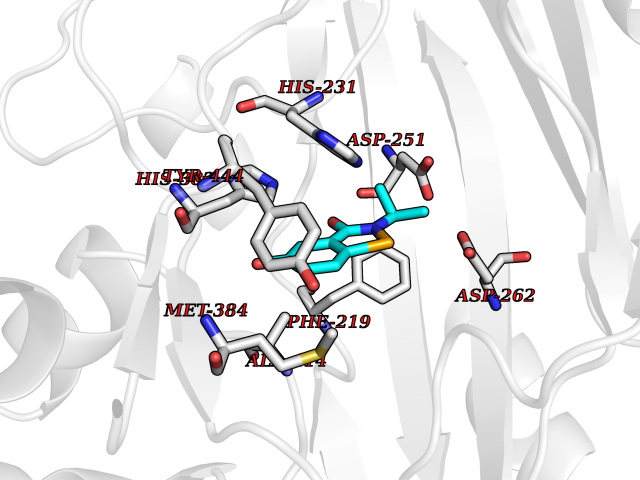 Pymol Map 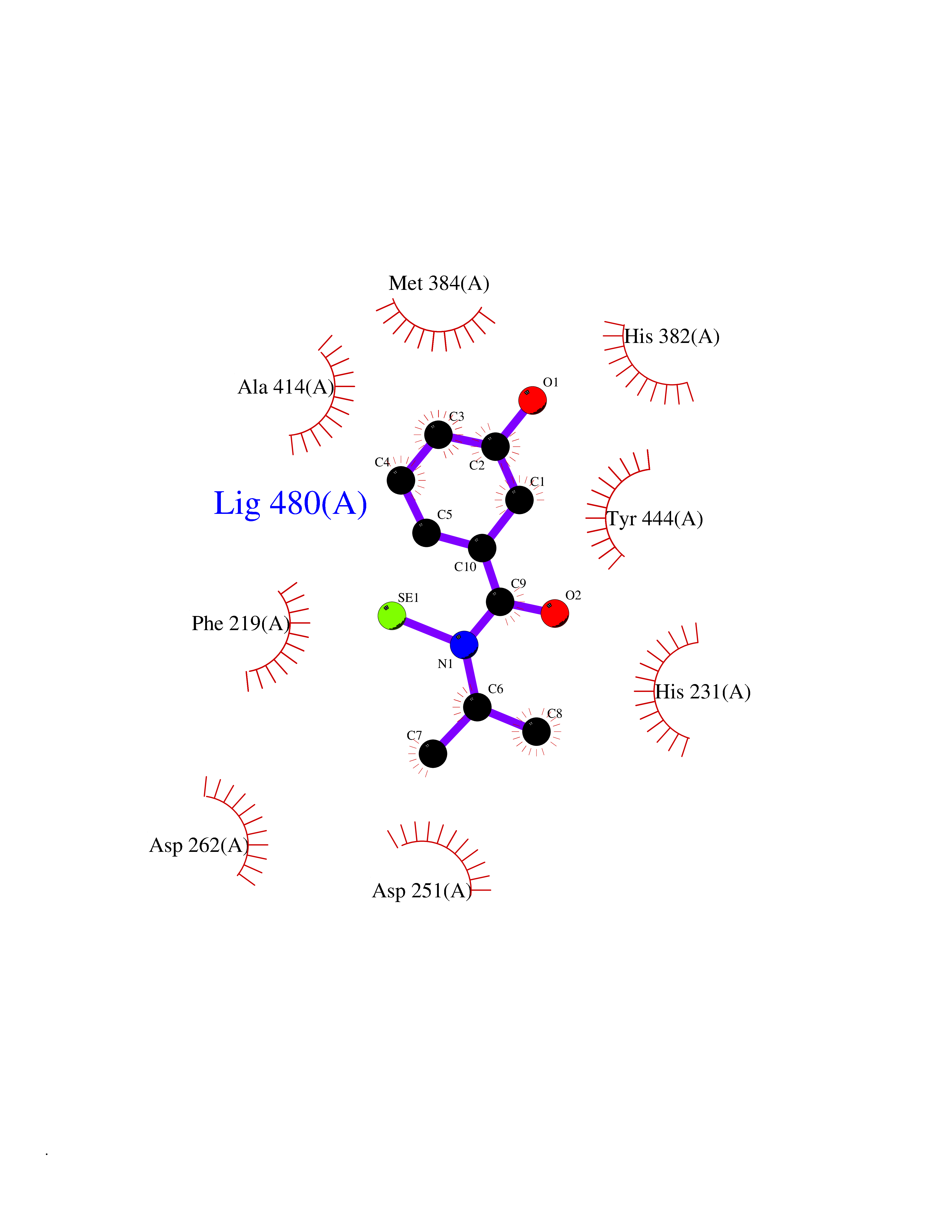 Ligplot Map 3D Binding mode Sequence PKVQTDPPSVPICDLYPNGVFPKGQECEYPPTQDGRTAAWRTTSEEKKALDQASEEIWNDFREAAEAHRQVRKYVMSWIKPGMTMIEICEKLEDCSRKLIKENGLNAGLAFPTGCSLNNCAAHYTPNAGDTTVLQYDDICKIDFGTHISGRIIDCAFTVTFNPKYDTLLKAVKDATNTGIKCAGIDVRLCDVGEAIQEVMESYEVEIDGKTYQVKPIRNLNGHSIGQYRIHAGKTVPIVKGGEATRMEEGEVYAIETFGSTGKGVVHDDMECSHYMKNFDVGHVPIRLPRTKHLLNVINENFGTLAFCRRWLDRLGESKYLMALKNLCDLGIVDPYPPLCDIKGSYTAQFEHTILLRPTCKEVVSRGDDY Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Pyruvate dehydrogenase kinase 1 (PDHK1) | 2Q8G | 5.74 | |
Target general information Gen name PDK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Pyruvate dehydrogenase kinase isoform 1; Pyruvate dehydrogenase (acetyl-transferring) kinase isozyme 1, mitochondrial; PDHK1; PDH kinase 1 Protein family PDK/BCKDK protein kinase family Biochemical class Kinase Function Kinase that plays a key role in regulation of glucose and fatty acid metabolism and homeostasis via phosphorylation of the pyruvate dehydrogenase subunits PDHA1 and PDHA2. This inhibits pyruvate dehydrogenase activity, and thereby regulates metabolite flux through the tricarboxylic acid cycle, down-regulates aerobic respiration and inhibits the formation of acetyl-coenzyme A from pyruvate. Plays an important role in cellular responses to hypoxia and is important for cell proliferation under hypoxia. Protects cells against apoptosis in response to hypoxia and oxidative stress. Related diseases TP53 is found in increased amounts in a wide variety of transformed cells. TP53 is frequently mutated or inactivated in about 60% of cancers. TP53 defects are found in Barrett metaplasia a condition in which the normally stratified squamous epithelium of the lower esophagus is replaced by a metaplastic columnar epithelium. The condition develops as a complication in approximately 10% of patients with chronic gastroesophageal reflux disease and predisposes to the development of esophageal adenocarcinoma.; DISEASE: Esophageal cancer (ESCR) [MIM:133239]: A malignancy of the esophagus. The most common types are esophageal squamous cell carcinoma and adenocarcinoma. Cancer of the esophagus remains a devastating disease because it is usually not detected until it has progressed to an advanced incurable stage. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Li-Fraumeni syndrome (LFS) [MIM:151623]: An autosomal dominant familial cancer syndrome that in its classic form is defined by the existence of a proband affected by a sarcoma before 45 years with a first degree relative affected by any tumor before 45 years and another first degree relative with any tumor before 45 years or a sarcoma at any age. Other clinical definitions for LFS have been proposed and called Li-Fraumeni like syndrome (LFL). In these families affected relatives develop a diverse set of malignancies at unusually early ages. Four types of cancers account for 80% of tumors occurring in TP53 germline mutation carriers: breast cancers, soft tissue and bone sarcomas, brain tumors (astrocytomas) and adrenocortical carcinomas. Less frequent tumors include choroid plexus carcinoma or papilloma before the age of 15, rhabdomyosarcoma before the age of 5, leukemia, Wilms tumor, malignant phyllodes tumor, colorectal and gastric cancers. {ECO:0000269|PubMed:10484981, ECO:0000269|PubMed:1565144, ECO:0000269|PubMed:1737852, ECO:0000269|PubMed:1933902, ECO:0000269|PubMed:1978757, ECO:0000269|PubMed:2259385, ECO:0000269|PubMed:36108750, ECO:0000269|PubMed:7887414, ECO:0000269|PubMed:8825920, ECO:0000269|PubMed:9452042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Squamous cell carcinoma of the head and neck (HNSCC) [MIM:275355]: A non-melanoma skin cancer affecting the head and neck. The hallmark of cutaneous SCC is malignant transformation of normal epidermal keratinocytes. The gene represented in this entry is involved in disease pathogenesis.; DISEASE: Lung cancer (LNCR) [MIM:211980]: A common malignancy affecting tissues of the lung. The most common form of lung cancer is non-small cell lung cancer (NSCLC) that can be divided into 3 major histologic subtypes: squamous cell carcinoma, adenocarcinoma, and large cell lung cancer. NSCLC is often diagnosed at an advanced stage and has a poor prognosis. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Papilloma of choroid plexus (CPP) [MIM:260500]: A benign tumor of neuroectodermal origin that generally occurs in childhood, but has also been reported in adults. Although generally found within the ventricular system, choroid plexus papillomas can arise ectopically in the brain parenchyma or disseminate throughout the neuraxis. Patients present with signs and symptoms of increased intracranial pressure including headache, hydrocephalus, papilledema, nausea, vomiting, cranial nerve deficits, gait impairment, and seizures. {ECO:0000269|PubMed:12085209}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Adrenocortical carcinoma (ADCC) [MIM:202300]: A malignant neoplasm of the adrenal cortex and a rare childhood tumor. It occurs with increased frequency in patients with Beckwith-Wiedemann syndrome and Li-Fraumeni syndrome. {ECO:0000269|PubMed:11481490}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Basal cell carcinoma 7 (BCC7) [MIM:614740]: A common malignant skin neoplasm that typically appears on hair-bearing skin, most commonly on sun-exposed areas. It is slow growing and rarely metastasizes, but has potentialities for local invasion and destruction. It usually develops as a flat, firm, pale area that is small, raised, pink or red, translucent, shiny, and waxy, and the area may bleed following minor injury. Tumor size can vary from a few millimeters to several centimeters in diameter. {ECO:0000269|PubMed:21946351}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Bone marrow failure syndrome 5 (BMFS5) [MIM:618165]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFS5 is an autosomal dominant form characterized by infantile onset of severe red cell anemia requiring transfusion. Additional features include hypogammaglobulinemia, poor growth with microcephaly, developmental delay, and seizures. {ECO:0000269|PubMed:30146126}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07403; DB08809 Interacts with P05067; P08559; Q16513; P31749-1; P31751-1 EC number EC 2.7.11.2 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Kinase; Mitochondrion; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Transit peptide Protein physicochemical properties Chain ID A Molecular weight (Da) 42249.9 Length 368 Aromaticity 0.11 Instability index 49.91 Isoelectric point 6.83 Charge (pH=7) -0.46 2D Binding mode Binding energy (Kcal/mol) -7.83 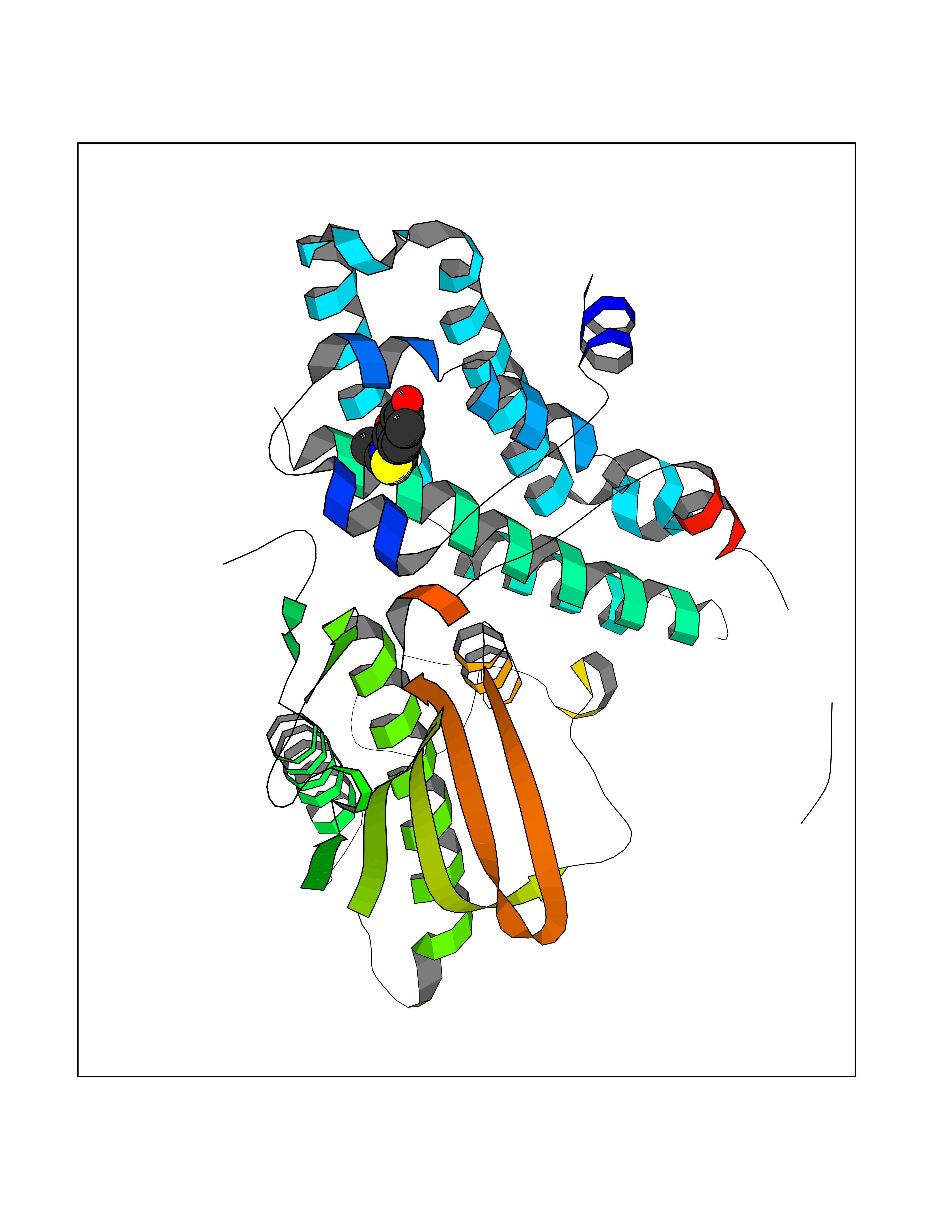 Molscript Map 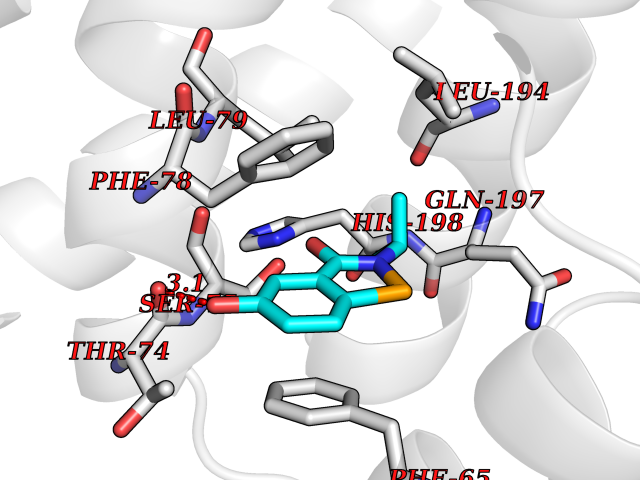 Pymol Map 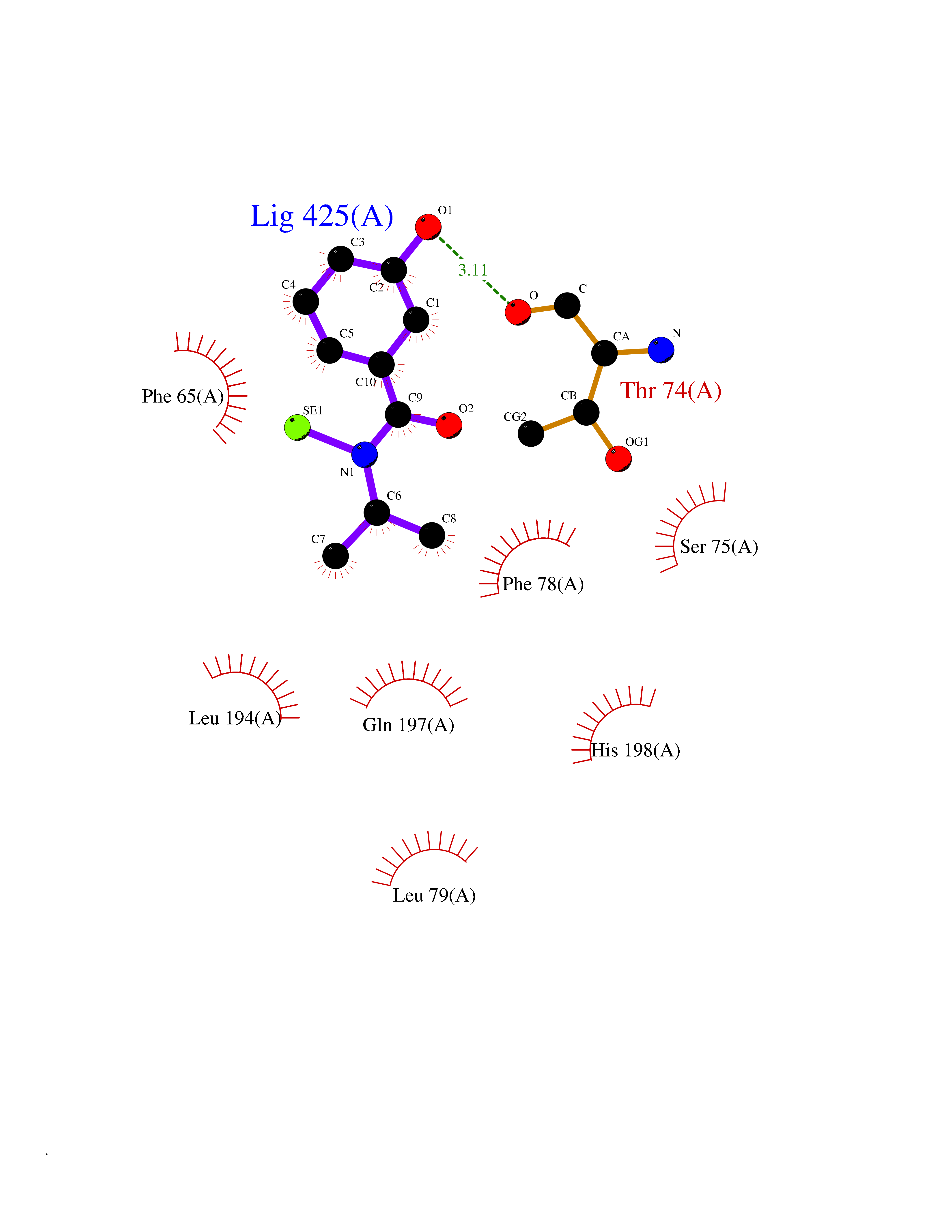 Ligplot Map 3D Binding mode Sequence GVPGQVDFYARFSPSPLSMKQFLDFGSVNACEKTSFMFLRQELPVRLANIMKEISLLPDNLLRTPSVQLVQSWYIQSLQELLDFKDKSAEDAKAIYDFTDTVIRIRNRHNDVIPTMAQGVIEYKESFDPVTSQNVQYFLDRFYMSRISIRMLLNQHSLLFGKHIGSINPNCNVLEVIKDGYENARRLCDLYYINSPELELEELNAKSPGQPIQVVYVPSHLYHMVFELFKNAMRATMEHHANRGVYPPIQVHVTLGNEDLTVKMSDRGGGVPLRKIDRLFNYMYSTAPRPRVETSRAVPLAGFGYGLPISRLYAQYFQGDLKLYSLEGYGTDAVIYIKALSTDSIERLPVYNKAAWKHYNTNDDWCVP Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Histone-lysine N-methyltransferase SMYD3 (SMYD3) | 6P7Z | 5.74 | |
Target general information Gen name SMYD3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SET and MYND domain-containing protein 3; Zinc finger MYND domain-containing protein 1 Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family Biochemical class NA Function Histone methyltransferase. Specifically methylates 'Lys-4' of histone H3, inducing di- and tri-methylation, but not monomethylation . Also methylates 'Lys-5' of histone H4. Plays an important role in transcriptional activation as a member of an RNA polymerase complex. Binds DNA containing 5'-CCCTCC-3' or 5'-GAGGGG-3' sequences. Related diseases Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:17332249}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 6 (NS6) [MIM:613224]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:19966803}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: RAS-associated autoimmune leukoproliferative disorder (RALD) [MIM:614470]: A disorder of apoptosis, characterized by chronic accumulation of non-malignant lymphocytes, defective lymphocyte apoptosis, and an increased risk for the development of hematologic malignancies. {ECO:0000269|PubMed:17517660}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanocytic nevus syndrome, congenital (CMNS) [MIM:137550]: A syndrome characterized by congenital pigmentary skin lesions which can occur at any site and can cover most of the body surface. These lesions may or may not be hairy. Congenital melanocytic nevi are associated with neuromelanosis (the presence of melanin-producing cells within the brain parenchyma or leptomeninges). Less commonly they are associated with malignant melanoma in childhood, both in the skin and the central nervous system. CMNS patients also tend to have a characteristic facial appearance, including wide or prominent forehead, periorbital fullness, small short nose with narrow nasal bridge, round face, full cheeks, prominent premaxilla, and everted lower lip. {ECO:0000269|PubMed:18633438, ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanosis, neurocutaneous (NCMS) [MIM:249400]: A rare congenital disease characterized by the presence of giant or multiple melanocytic nevi on the skin, foci of melanin-producing cells within the brain parenchyma, and infiltration of leptomeninges by abnormal melanin deposits. Neurologic abnormalities include seizures, hydrocephalus, arachnoid cysts, tumors, and syringomyelia. Some patients may develop malignant melanoma. {ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Keratinocytic non-epidermolytic nevus (KNEN) [MIM:162900]: Epidermal nevi of the common, non-organoid and non-epidermolytic type are benign skin lesions and may vary in their extent from a single (usually linear) lesion to widespread and systematized involvement. They may be present at birth or develop early during childhood. {ECO:0000269|PubMed:22499344}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid cancer, non-medullary, 2 (NMTC2) [MIM:188470]: A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms. {ECO:0000269|PubMed:12727991}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9H0L4; Q9H0I2; Q13064; Q7Z3B4; Q16512; Q92529; Q15915; Q9Y2U5 EC number EC 2.1.1.354 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Chromatin regulator; Cytoplasm; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 48694.1 Length 425 Aromaticity 0.08 Instability index 44.91 Isoelectric point 6.86 Charge (pH=7) -0.4 2D Binding mode Binding energy (Kcal/mol) -7.83 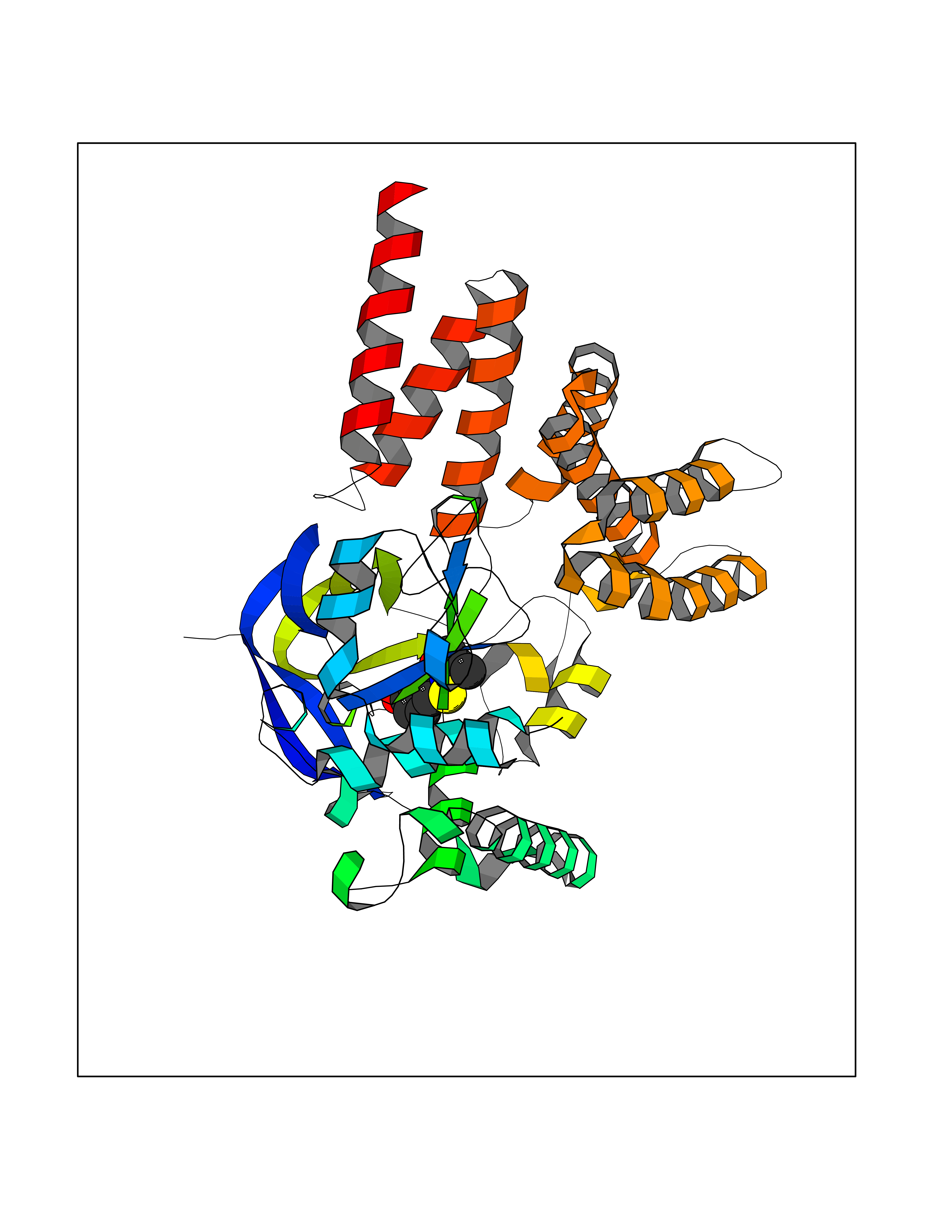 Molscript Map 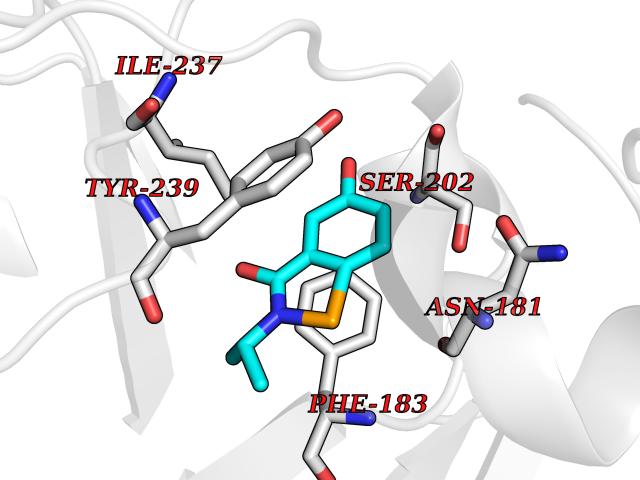 Pymol Map 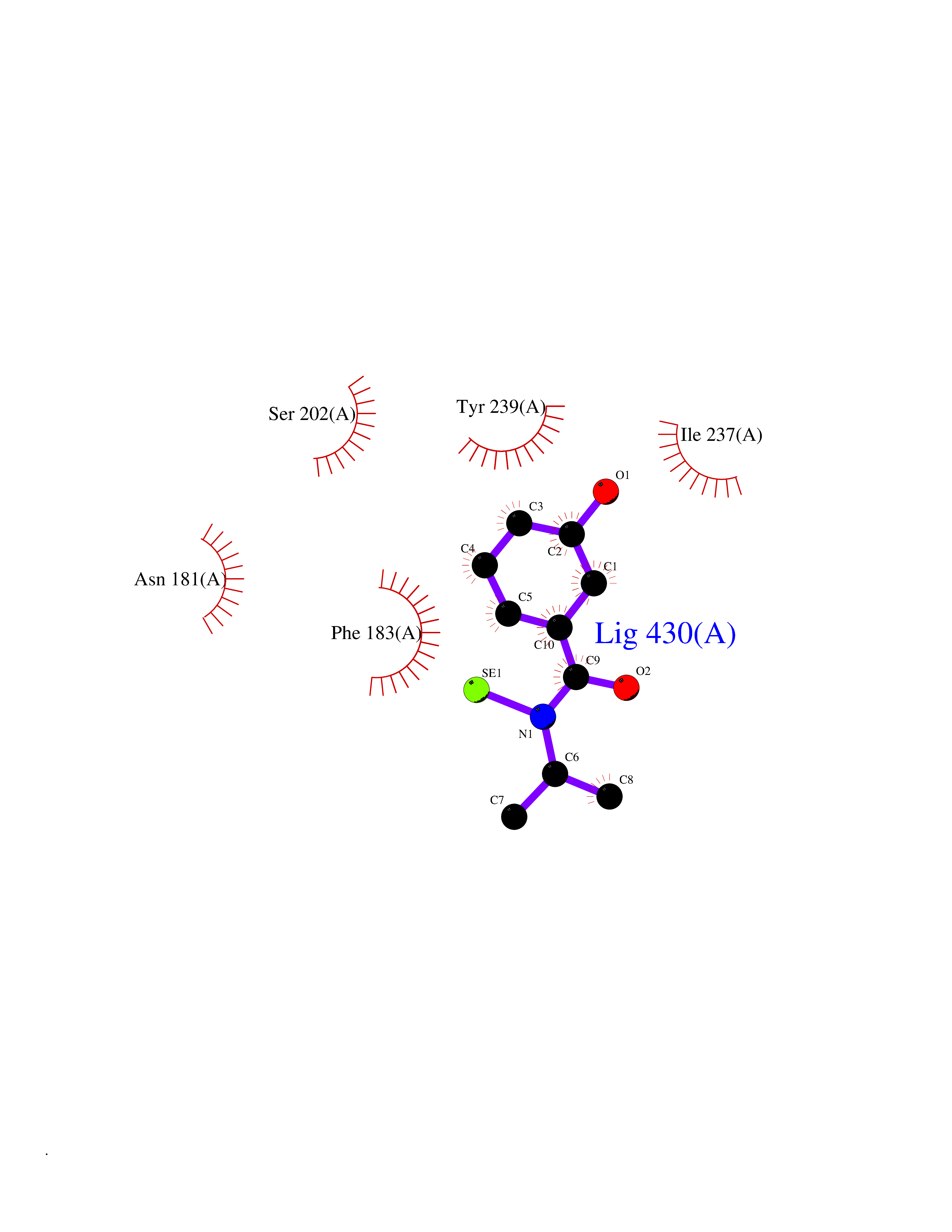 Ligplot Map 3D Binding mode Sequence PLKVEKFATANRGNGLRAVTPLRPGELLFRSDPLAYTVCKGSRGVVCDRCLLGKEKLMRCSQCRVAKYCSAKCQKKAWPDHKRECKCLKSCPRYPPDSVRLLGRVVFKLMDGAPSESEKLYSFYDLESNINKLTEDKKEGLRQLVMTFQHFMREEIQDASQLPPAFDLFEAFAKVICNSFTICNAEMQEVGVGLYPSISLLNHSCDPNCSIVFNGPHLLLRAVRDIEVGEELTICYLDMLMTSEERRKQLRDQYCFECDCFRCQTQDKDADMLTGDEQVWKEVQESLKKIEELKAHWKWEQVLAMCQAIISSNSERLPDINIYQLKVLDCAMDACINLGLLEEALFYGTRTMEPYRIFFPGSHPVRGVQVMKVGKLQLHQGMFPQAMKNLRLAFDIMRVTHGREHSLIEDLILLLEECDANIRAS Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Dual-specificity tyrosine-phosphorylation regulated kinase 2 (DYRK2) | 6HDR | 5.74 | |
Target general information Gen name DYRK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dual specificity tyrosine-phosphorylation-regulated kinase 2 Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, MNB/DYRK subfamily Biochemical class Kinase Function Functions in part via its role in ubiquitin-dependent proteasomal protein degradation. Functions downstream of ATM and phosphorylates p53/TP53 at 'Ser-46', and thereby contributes to the induction of apoptosis in response to DNA damage. Phosphorylates NFATC1, and thereby inhibits its accumulation in the nucleus and its transcription factor activity. Phosphorylates EIF2B5 at 'Ser-544', enabling its subsequent phosphorylation and inhibition by GSK3B. Likewise, phosphorylation of NFATC1, CRMP2/DPYSL2 and CRMP4/DPYSL3 promotes their subsequent phosphorylation by GSK3B. May play a general role in the priming of GSK3 substrates. Inactivates GYS1 by phosphorylation at 'Ser-641', and potentially also a second phosphorylation site, thus regulating glycogen synthesis. Mediates EDVP E3 ligase complex formation and is required for the phosphorylation and subsequent degradation of KATNA1. Phosphorylates TERT at 'Ser-457', promoting TERT ubiquitination by the EDVP complex. Phosphorylates SIAH2, and thereby increases its ubiquitin ligase activity. Promotes the proteasomal degradation of MYC and JUN, and thereby regulates progress through the mitotic cell cycle and cell proliferation. Promotes proteasomal degradation of GLI2 and GLI3, and thereby plays a role in smoothened and sonic hedgehog signaling. Plays a role in cytoskeleton organization and neurite outgrowth via its phosphorylation of DCX and DPYSL2. Phosphorylates CRMP2/DPYSL2, CRMP4/DPYSL3, DCX, EIF2B5, EIF4EBP1, GLI2, GLI3, GYS1, JUN, MDM2, MYC, NFATC1, p53/TP53, TAU/MAPT and KATNA1. Can phosphorylate histone H1, histone H3 and histone H2B (in vitro). Can phosphorylate CARHSP1 (in vitro). Serine/threonine-protein kinase involved in the regulation of the mitotic cell cycle, cell proliferation, apoptosis, organization of the cytoskeleton and neurite outgrowth. Related diseases Bone marrow failure and diabetes mellitus syndrome (BMFDMS) [MIM:620044]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFDMS is an autosomal recessive form characterized by various degrees of bone marrow failure, ranging from dyserythropoiesis to bone marrow aplasia, with onset in infancy or early childhood, and non-autoimmune insulin-dependent diabetes mellitus appearing in the first or second decades. Many patients show pigmentary skin abnormalities and short stature. {ECO:0000269|PubMed:28073829, ECO:0000269|PubMed:35611808, ECO:0000269|PubMed:35931051}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NR20; Q13422; Q9BQD3; Q9BRK4; P23497; O43379; P62258; Q96C00 EC number EC 2.7.12.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cytoplasm; Kinase; Magnesium; Manganese; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID A Molecular weight (Da) 46422.1 Length 407 Aromaticity 0.09 Instability index 44.91 Isoelectric point 9.09 Charge (pH=7) 12.37 2D Binding mode Binding energy (Kcal/mol) -7.83 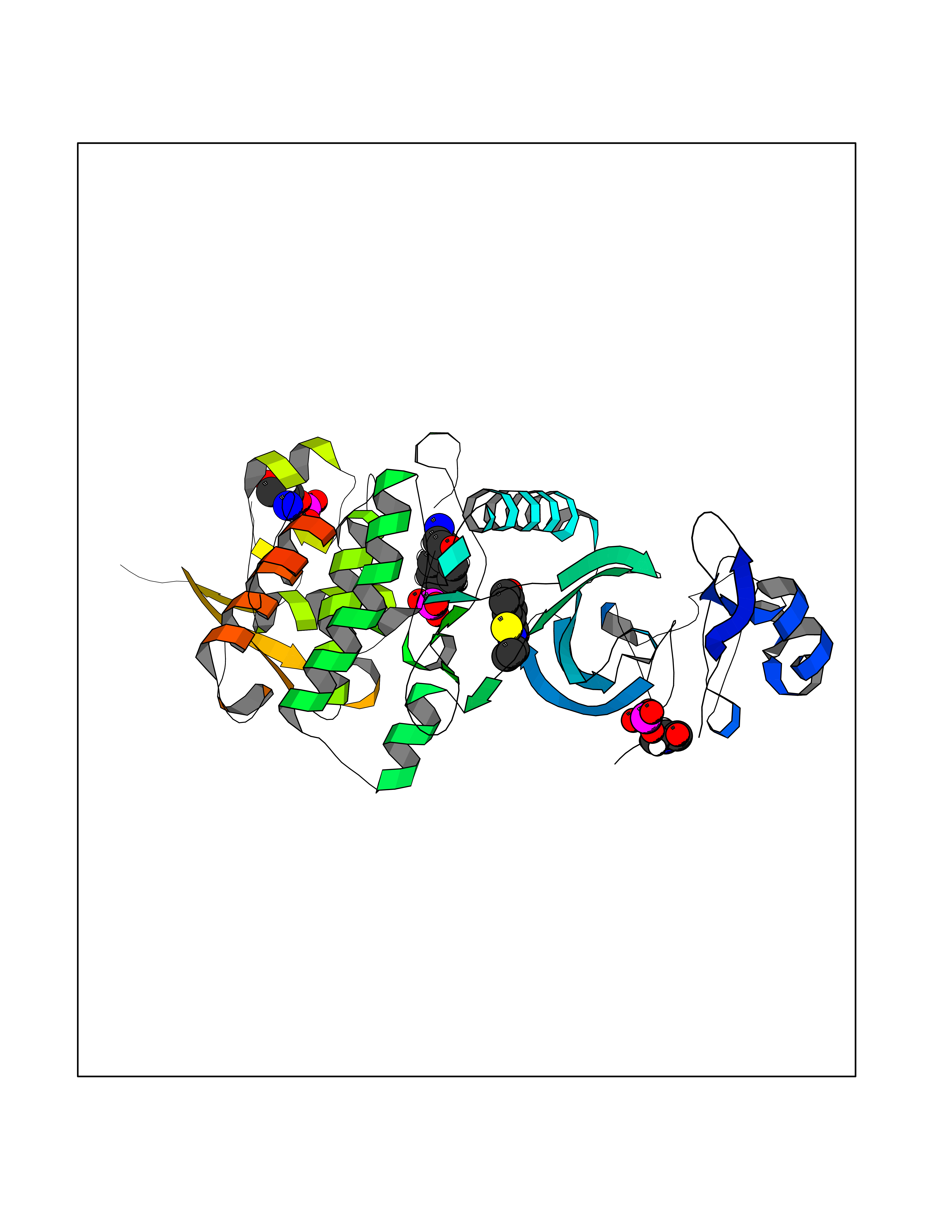 Molscript Map 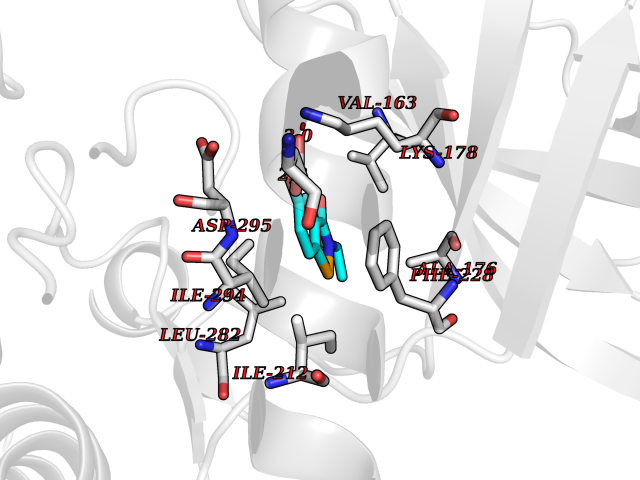 Pymol Map 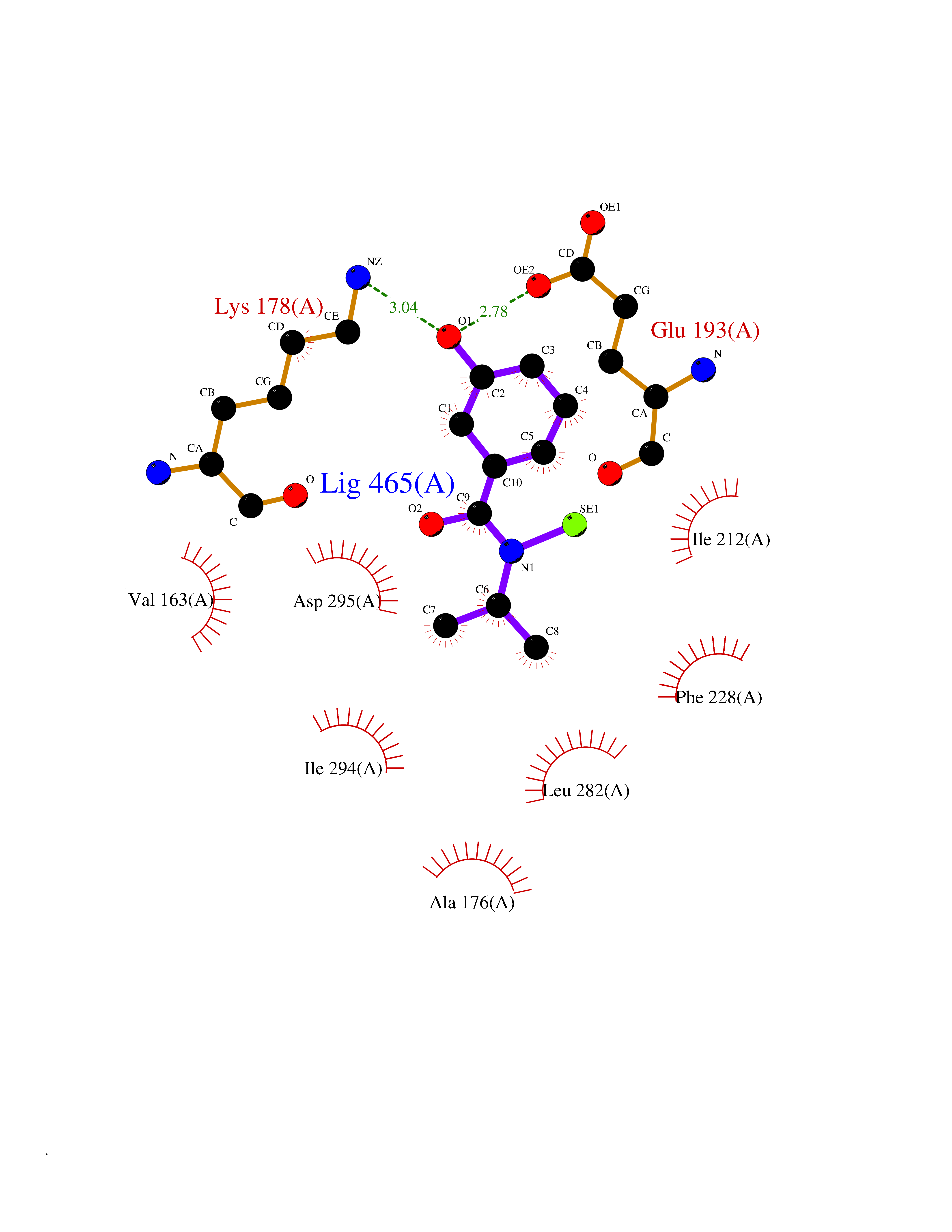 Ligplot Map 3D Binding mode Sequence HHHSXGVDLGTENLYFQSMGKVKATPMTPEQAMKQYMQKLTAFEHHEIFSYPEIYFLGLNAKKRQGMTGGPNNGGYDDDQGSYVQVPHDHVAYRYEVLKVIGKGSFGQVVKAYDHKVHQHVALKMVRNEKRFHRQAAEEIRILEHLRKQDKDNTMNVIHMLENFTFRNHICMTFELLSMNLYELIKKNKFQGFSLPLVRKFAHSILQCLDALHKNRIIHCDLKPENILLKQQGRSGIKVIDFGSSCYEHQRVYTXIQSRFYRAPEVILGARYGMPIDMWSLGCILAELLTGYPLLPGEDEGDQLACMIELLGMPSQKLLDASKRAKNFVSXKGYPRYCTVTTLSDVVLNGGRSRRGKLRGPPESREWGNALKGCDDPLFLDFLKQCLEWDPAVRMTPGQALRHPWLR Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Estrogen-related receptor-beta (ESRRB) | 6LIT | 5.74 | |
Target general information Gen name ESRRB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Steroid hormone receptor ERR2; Nuclear receptor subfamily 3 group B member 2; NR3B2; Estrogen-related receptor beta; Estrogen receptor-like 2; ESRL2; ERRB2; ERR-beta; ERR beta-2 Protein family Nuclear hormone receptor family, NR3 subfamily Biochemical class Nuclear hormone receptor Function Isoform 3: Transcription factor that binds a canonical ESRRB recognition (ERRE) sequence 5'TCAAGGTCA-3' localized on promoter and enhancer of targets genes regulating their expression or their transcription activity. Plays a role, in a LIF-independent manner, in maintainance of self-renewal and pluripotency of embryonic and trophoblast stem cells through different signaling pathways including FGF signaling pathway and Wnt signaling pathways. Upon FGF signaling pathway activation, interacts with KDM1A by directly binding to enhancer site of ELF5 and EOMES and activating their transcription leading to self-renewal of trophoblast stem cells. Also regulates expression of multiple rod-specific genes and is required for survival of this cell type (By similarity). Plays a role as transcription factor activator of GATA6, NR0B1, POU5F1 and PERM1. Plays a role as transcription factor repressor of NFE2L2 transcriptional activity and ESR1 transcriptional activity. During mitosis remains bound to a subset of interphase target genes, including pluripotency regulators, through the canonical ESRRB recognition (ERRE) sequence, leading to their transcriptional activation in early G1 phase. Can coassemble on structured DNA elements with other transcription factors like SOX2, POU5F1, KDM1A and NCOA3 to trigger ESRRB-dependent gene activation. This mechanism, in the case of SOX2 corecruitment prevents the embryonic stem cells (ESCs) to epiblast stem cells (EpiSC) transition through positive regulation of NR0B1 that inhibits the EpiSC transcriptional program. Also plays a role inner ear development by controlling expression of ion channels and transporters and in early placentation (By similarity). Related diseases Deafness, autosomal recessive, 35 (DFNB35) [MIM:608565]: A form of non-syndromic deafness characterized by non-progressive, prelingual hearing loss. {ECO:0000269|PubMed:18179891}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00255; DB07776; DB01645 Interacts with P62508-3 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Chromosome; Cytoplasm; Deafness; Disease variant; DNA-binding; Metal-binding; Non-syndromic deafness; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 22767.3 Length 198 Aromaticity 0.08 Instability index 56.41 Isoelectric point 5.76 Charge (pH=7) -5.51 2D Binding mode Binding energy (Kcal/mol) -7.83 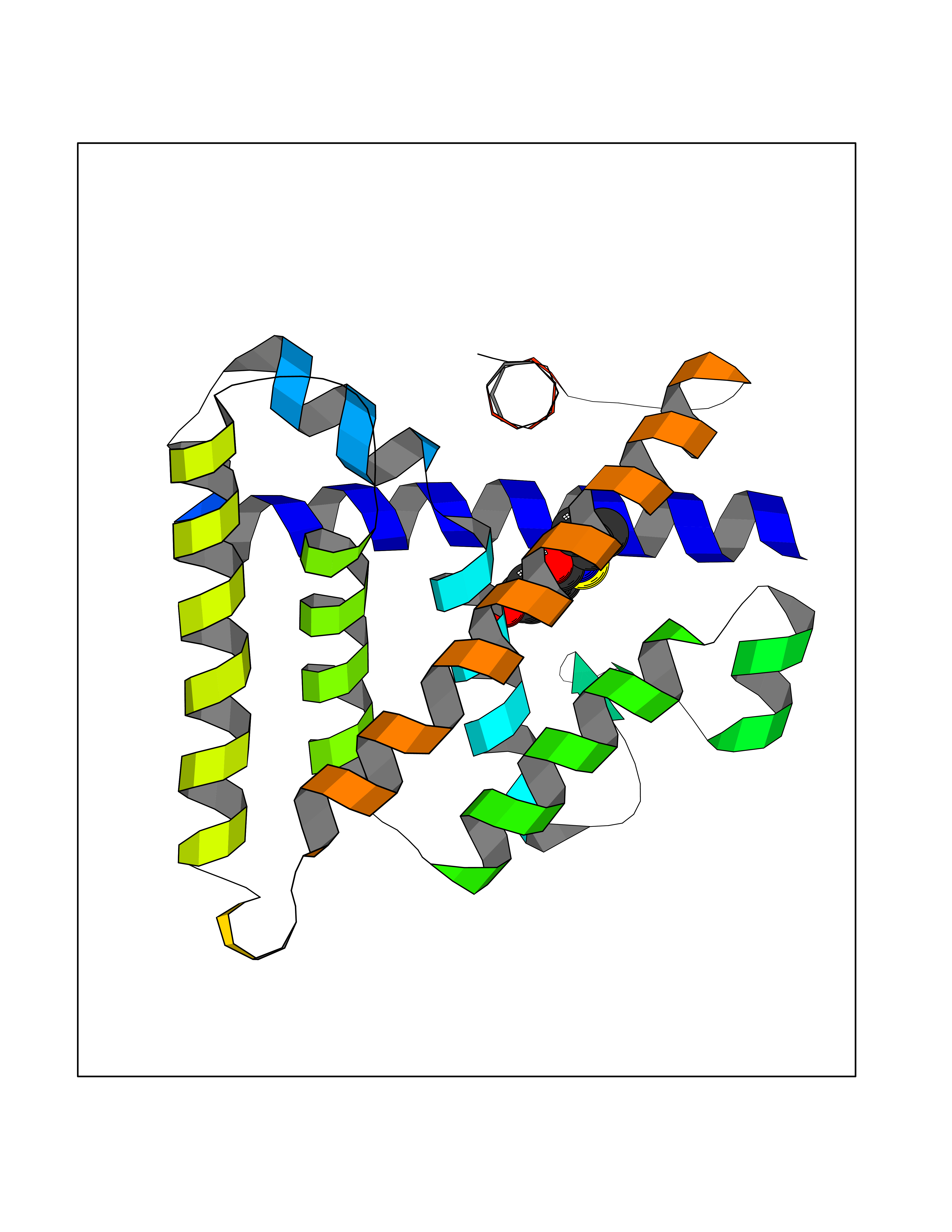 Molscript Map  Pymol Map 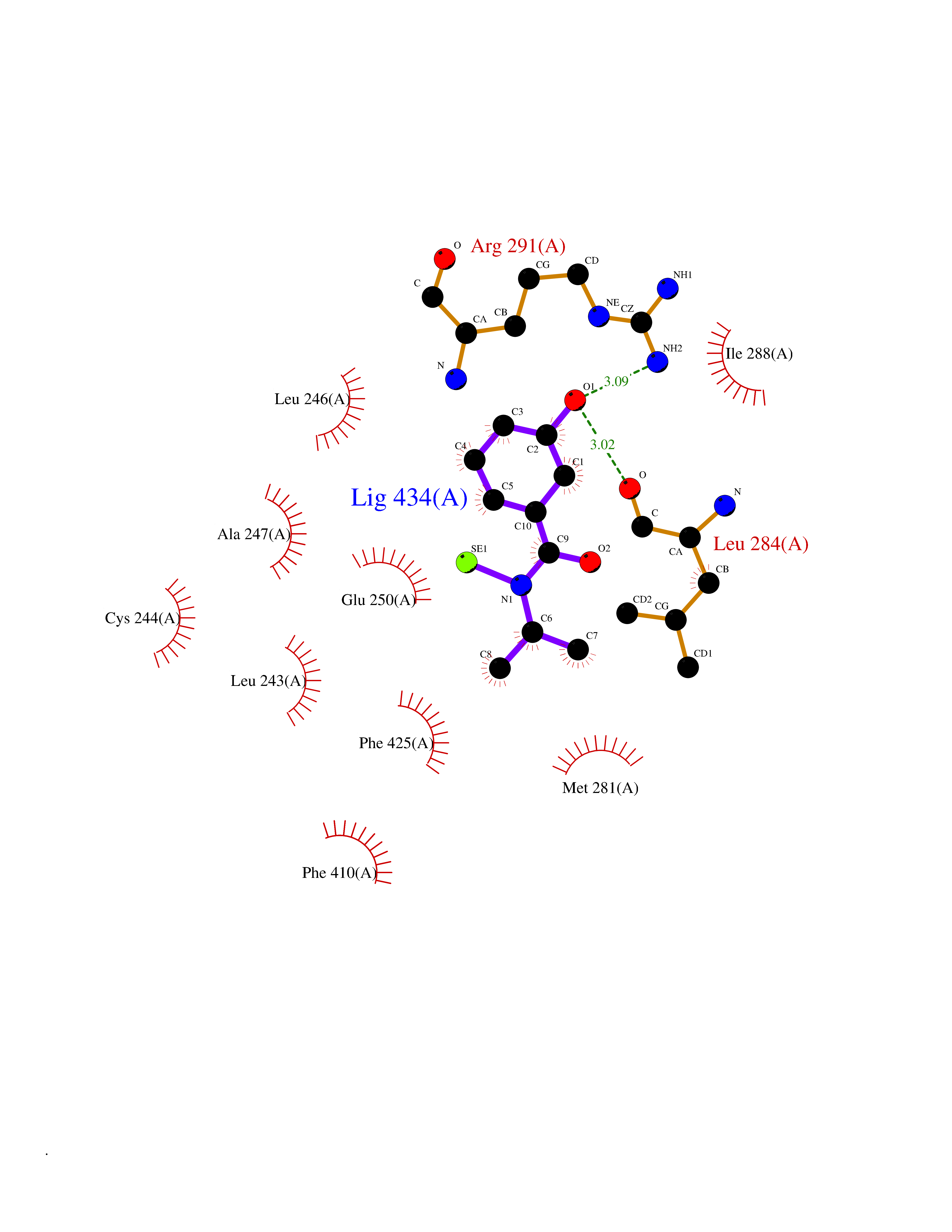 Ligplot Map 3D Binding mode Sequence GDIKALTTLCDLADRELVVIIGWAKHIPGFSSLSLGDQMSLLQSAWMEILILGIVYRSLPYDDKLVYAEDYIMDEEHSRLAGLLELYRAILQLVRRYKKLKVEKEEFVTLKALALANSDSMHIEDLEAVQKLQDLLHEALQDYELSQHHEEPWRTGKLLLTLPLLRQTAAKAVQHFYSVKLQGKVPMHKLFLEMLEAK Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Bacterial DNA ligase (Bact ligA) | 2OWO | 5.74 | |
Target general information Gen name Bact ligA Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms ligA; Polydeoxyribonucleotide synthase [NAD+]; NAD+-dependent DNA ligase Protein family NAD-dependent DNA ligase family, LigA subfamily Biochemical class Phosphoric ester ligase Function DNA ligase that catalyzes the formation of phosphodiester linkages between 5'-phosphoryl and 3'-hydroxyl groups in double-stranded DNA using NAD as a coenzyme and as the energy source for the reaction. It is essential for DNA replication and repair of damaged DNA. Related diseases Dystonia 1, torsion, autosomal dominant (DYT1) [MIM:128100]: A primary torsion dystonia, and the most common and severe form. Dystonia is defined by the presence of sustained involuntary muscle contractions, often leading to abnormal postures. Dystonia type 1 is characterized by involuntary, repetitive, sustained muscle contractions or postures involving one or more sites of the body, in the absence of other neurological symptoms. Typically, symptoms develop first in an arm or leg in middle to late childhood and progress in approximately 30% of patients to other body regions (generalized dystonia) within about five years. 'Torsion' refers to the twisting nature of body movements observed in DYT1, often affecting the trunk. Distribution and severity of symptoms vary widely between affected individuals, ranging from mild focal dystonia to severe generalized dystonia, even within families. {ECO:0000269|PubMed:14970196, ECO:0000269|PubMed:15505207, ECO:0000269|PubMed:16361107, ECO:0000269|PubMed:17428918, ECO:0000269|PubMed:18167355, ECO:0000269|PubMed:18477710, ECO:0000269|PubMed:18827015, ECO:0000269|PubMed:19955557, ECO:0000269|PubMed:20169475, ECO:0000269|PubMed:21102408, ECO:0000269|PubMed:24930953, ECO:0000269|PubMed:27490483, ECO:0000269|PubMed:9288096}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Arthrogryposis multiplex congenita 5 (AMC5) [MIM:618947]: A form of arthrogryposis multiplex congenita, a developmental condition characterized by multiple joint contractures resulting from reduced or absent fetal movements. AMC5 is an autosomal recessive form characterized by severe congenital contractures, developmental delay, strabismus and tremor. {ECO:0000269|PubMed:28516161, ECO:0000269|PubMed:29053766, ECO:0000269|PubMed:30244176}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07813 EC number EC 6.5.1.2 Uniprot keywords 3D-structure; Direct protein sequencing; DNA damage; DNA repair; DNA replication; Ligase; Magnesium; Metal-binding; NAD; Reference proteome; Zinc Protein physicochemical properties Chain ID D,A,C Molecular weight (Da) 66922.6 Length 612 Aromaticity 0.06 Instability index 37.23 Isoelectric point 5.48 Charge (pH=7) -16.8 2D Binding mode Binding energy (Kcal/mol) -7.83 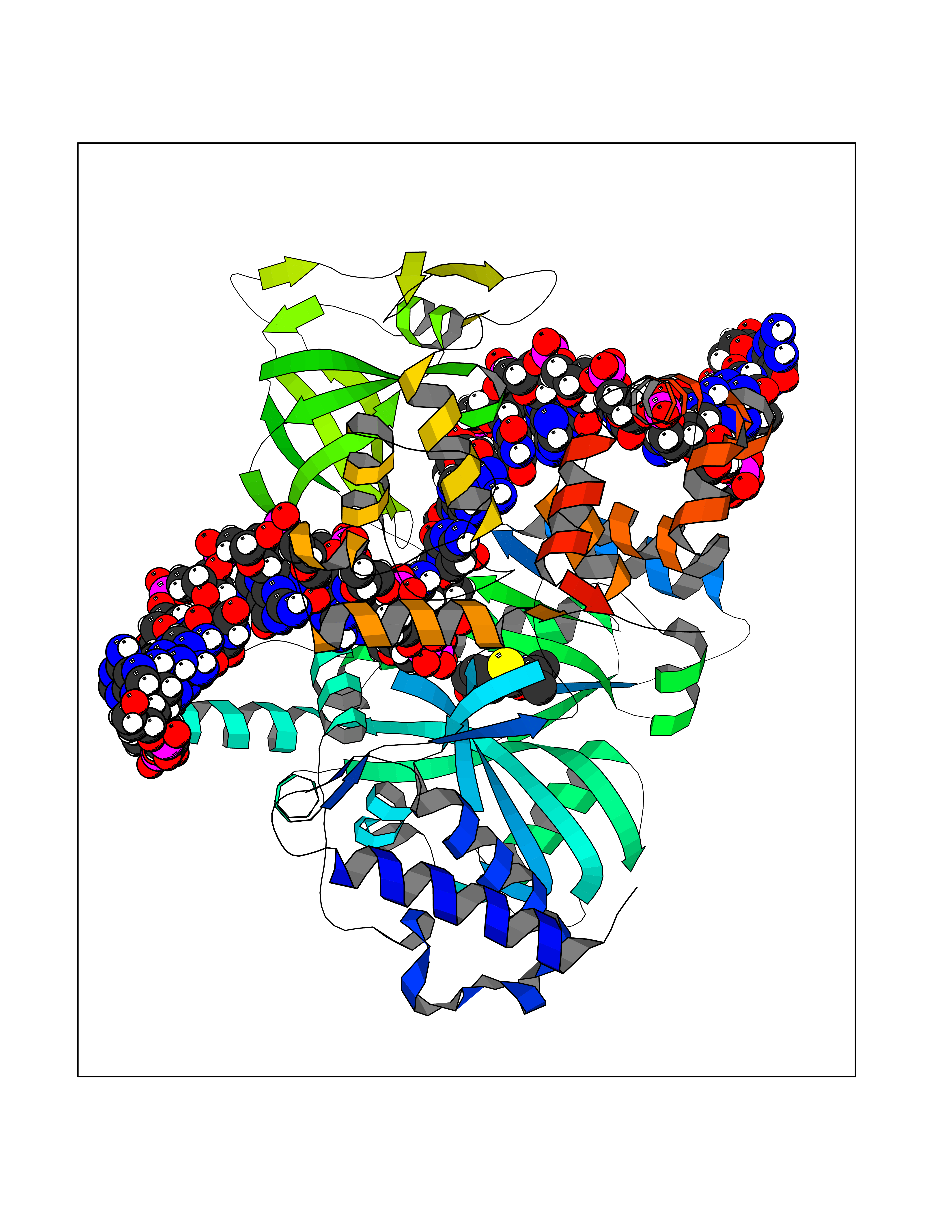 Molscript Map 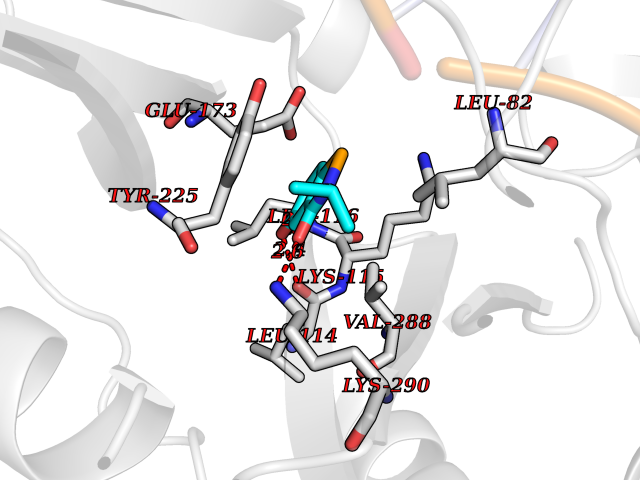 Pymol Map 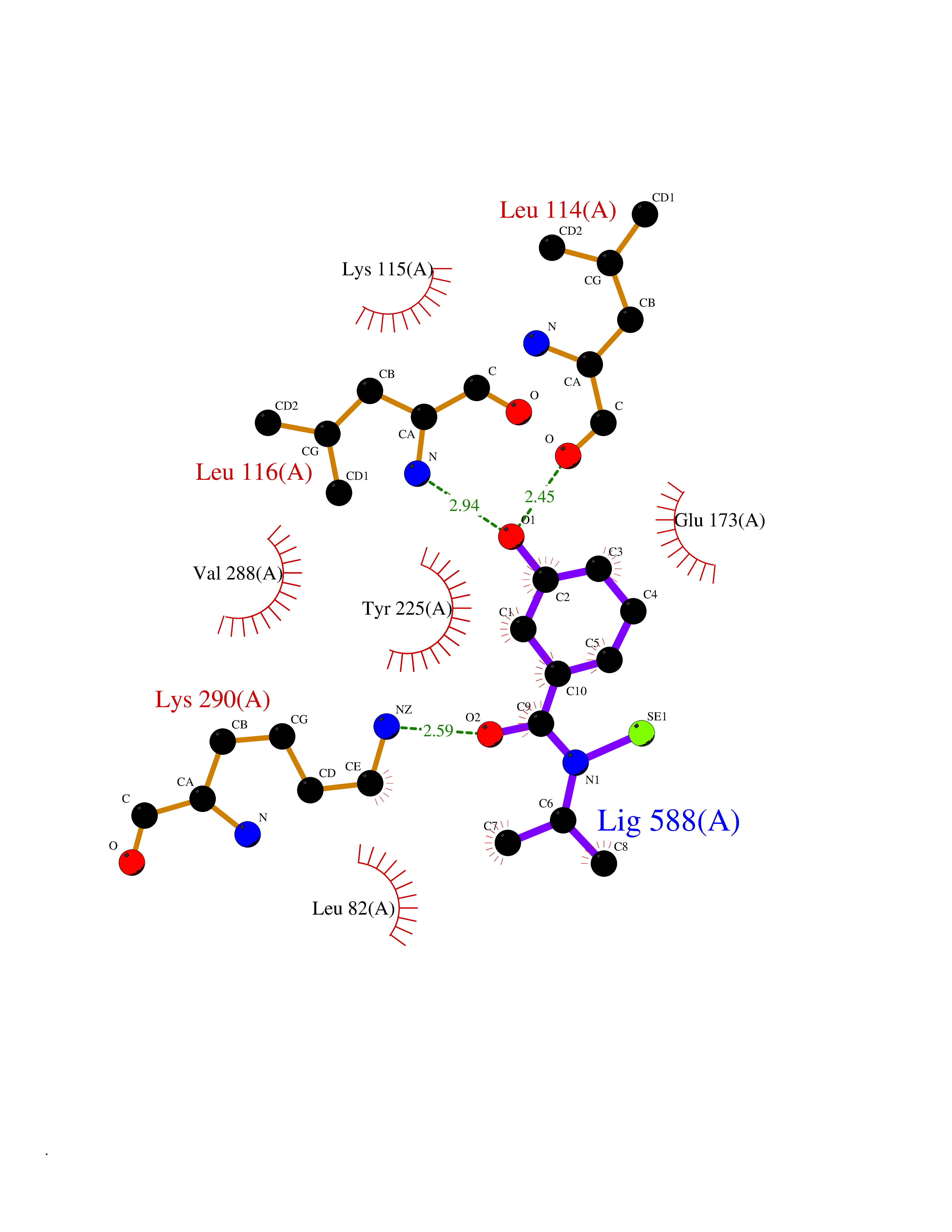 Ligplot Map 3D Binding mode Sequence ACAATTGCGACXCCACTATCGGAATGMESIEQQLTELRTTLRHHEYLYHVMDAPEIPDAEYDRLMRELRELETKHPELITPDSPTQRVGAAPLAAFSQIRHEVPMLSLDNVFDEESFLAFNKRVQDRLKNNEKVTWCCELKLDGLAVSILYENGVLVSAATRGDGTTGEDITSNVRTIRAIPLKLHGENIPARLEVRGEVFLPQAGFEKINEDARRTGGKVFANPRNAAAGSLRQLDPRITAKRPLTFFCYGVGVLEGGELPDTHLGRLLQFKKWGLPVSDRVTLCESAEEVLAFYHKVEEDRPTLGFDIDGVVIKVNSLAQQEQLGFVARAPRWAVAFKFPAQEQMTFVRDVEFQVGRTGAITPVARLEPVHVAGVLVSNATLHNADEIERLGLRIGDKVVIRRAGDVIPQVVNVVLSERPEDTREVVFPTHCPVCGSDVERVEGEAVARCTGGLICGAQRKESLKHFVSRRAMDVDGMGDKIIDQLVEKEYVHTPADLFKLTAGKLTGLERMGPKSAQNVVNALEKAKETTFARFLYALGIREVGEATAAGLAAYFGTLEALEAASIEELQKVPDVGIVVASHVHNFFAEESNRNVISELLAEGVHWPAP Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Neuronal acetylcholine receptor alpha-3/beta-4 (CHRNA3/B4) | 6PV7 | 5.74 | |
Target general information Gen name CHRNA3-CHRNB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Neuronal acetylcholine receptor Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Neurotransmitter receptor Function A type of nicotinic acetylcholine receptor, consisting of 3 and 4 subunits. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89661.2 Length 775 Aromaticity 0.12 Instability index 35.11 Isoelectric point 6.07 Charge (pH=7) -5.67 2D Binding mode Binding energy (Kcal/mol) -7.83  Molscript Map 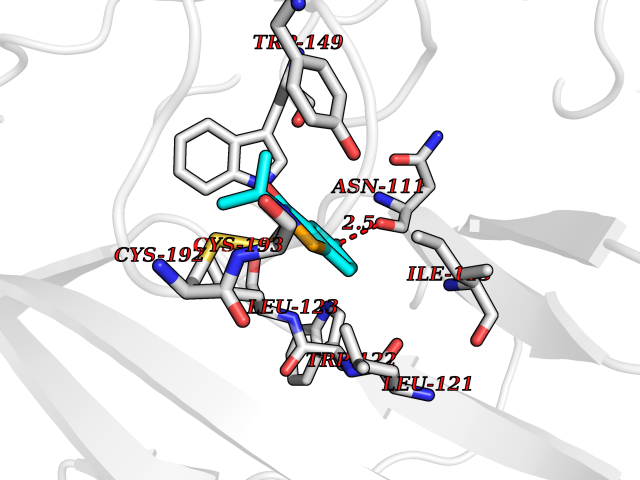 Pymol Map 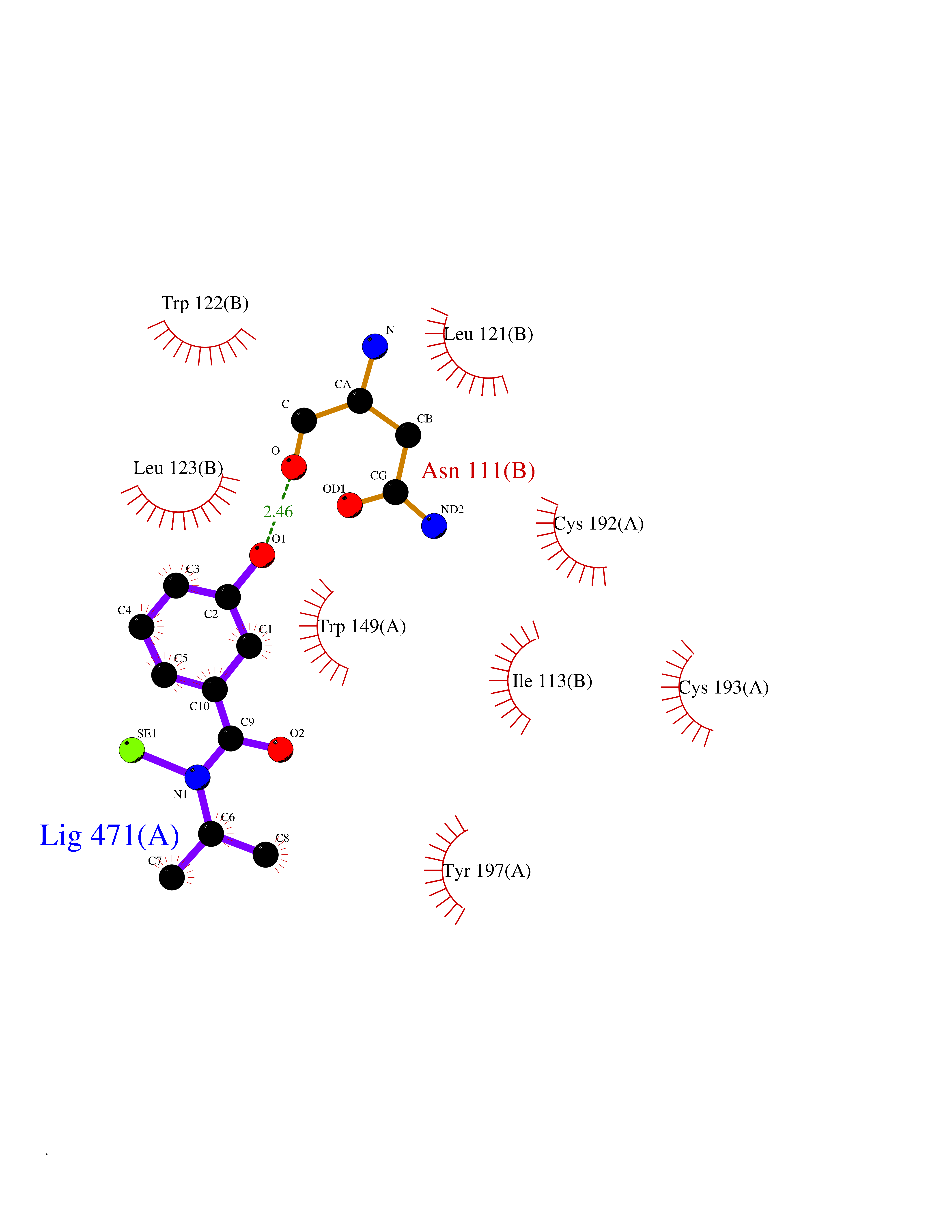 Ligplot Map 3D Binding mode Sequence SEAEHRLFERLFEDYNEIIRPVANVSDPVIIHFEVSMSQLVKVDEVNQIMETNLWLKQIWNDYKLKWNPSDYGGAEFMRVPAQKIWKPDIVLYNNAVGDFQVDDKTKALLKYTGEVTWIPPAIFKSSCKIDVTYFPFDYQNCTMKFGSWSYDKAKIDLVLIGSSMNLKDYWESGEWAIIKAPGYKHDIKYNCCEEIYPDITYSLYIRRLPLFYTINLIIPCLLISFLTVLVFYLPSDCGEKVTLCISVLLSLTVFLLVITETIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHYRTPTTHTMPSWVKTVFLNLLPRVMFMTRIKEAIQSVKYIAENMKAQNEAKEIQDDWKYVAMVIDRIFLWVFTLVCILGTAGLFLQPLMRVANAEEKLMDDLLNKTRYNNLIRPATSSSQLISIKLQLSLAQLISVNEREQIMTTNVWLKQEWTDYRLTWNSSRYEGVNILRIPAKRIWLPDIVLYNNADGTYEVSVYTNLIVRSNGSVLWLPPAIYKSACKIEVKYFPFDQQNCTLKFRSWTYDHTEIDMVLMTPTASMDDFTPSGEWDIVALPGRRTVNPQDPSYVDVTYDFIIKRKPLFYTINLIIPCVLTTLLAILVFYLPSDCGEKMTLCISVLLALTFFLLLISKIVPPTSLDVPLIGKYLMFTMVLVTFSIVTSVCVLNVHHRSPSTHTMAPWVKRCFLHKLPTFLFMKRRQDVQEALEGVSFIAQHMKNDDEDQSVVEDWKYVAMVVDRLFLWVFMFVCVLGTVGLFLP Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Cardiac phospholamban (PLN) | 7E0Z | 5.74 | |
Target general information Gen name PLN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Phospholamban; PLN; PLB Protein family Phospholamban family Biochemical class NA Function Reversibly inhibits the activity of ATP2A2 in cardiac sarcoplasmic reticulum by decreasing the apparent affinity of the ATPase for Ca(2+). Modulates the contractility of the heart muscle in response to physiological stimuli via its effects on ATP2A2. Modulatescalcium re-uptake during muscle relaxation and plays an important role in calcium homeostasis in the heart muscle. The degree of ATP2A2 inhibition depends on the oligomeric state of PLN. ATP2A2 inhibition is alleviated by PLN phosphorylation. Related diseases Cardiomyopathy, dilated, 1P (CMD1P) [MIM:609909]: A disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death. {ECO:0000269|PubMed:12610310, ECO:0000269|PubMed:16432188, ECO:0000269|PubMed:22137083, ECO:0000269|PubMed:22427649, ECO:0000269|PubMed:22707725}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Cardiomyopathy, familial hypertrophic, 18 (CMH18) [MIM:613874]: A hereditary heart disorder characterized by ventricular hypertrophy, which is usually asymmetric and often involves the interventricular septum. The symptoms include dyspnea, syncope, collapse, palpitations, and chest pain. They can be readily provoked by exercise. The disorder has inter- and intrafamilial variability ranging from benign to malignant forms with high risk of cardiac failure and sudden cardiac death. {ECO:0000269|PubMed:12705874}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q3SXY8; P07307-3; O15342; Q9BXK5; Q13323; P19397; O95471; Q9UHP7-3; Q7Z7G2; O43889-2; Q96BA8; Q09013; Q92838; Q9GZR5; Q5JX71; Q14318; Q8TBE3; P48165; Q8TDT2; O60883; Q8TED1; P31937; Q7Z5P4; P43628; Q5T700; Q8N112; Q9GZY8-5; Q6N075; Q99735; O14880; Q9GZW8; Q9H2K0; P15941-11; Q8TBJ4; O95197-3; Q9NR31; A0A0S2Z4U3; Q9Y3P8; Q15849; Q8IWU4; O95436-2; Q9NQQ7-3; Q9NP94; Q9HBV2; Q9NPE6; Q16623; P32856-2; Q9BVX2; Q7Z7N9; Q6UW68; Q9NWC5; Q96B21; Q4KMG9; Q8N661; O15393-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Cardiomyopathy; Disease variant; Endoplasmic reticulum; Lipoprotein; Membrane; Mitochondrion; Palmitate; Phosphoprotein; Proteomics identification; Reference proteome; Sarcoplasmic reticulum; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 40393 Length 349 Aromaticity 0.13 Instability index 35.1 Isoelectric point 8.98 Charge (pH=7) 6.56 2D Binding mode Binding energy (Kcal/mol) -7.83  Molscript Map 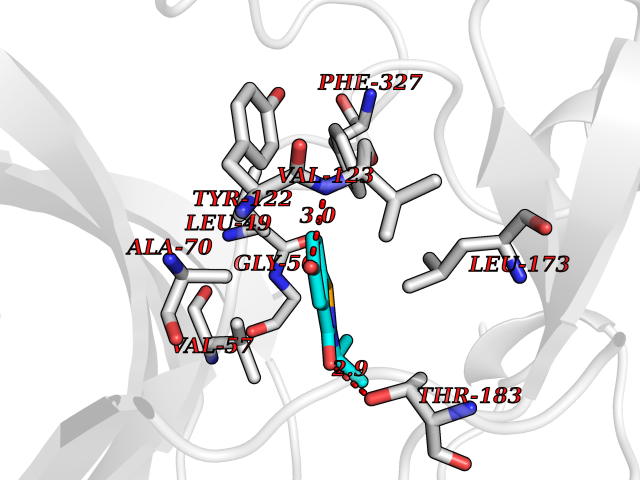 Pymol Map 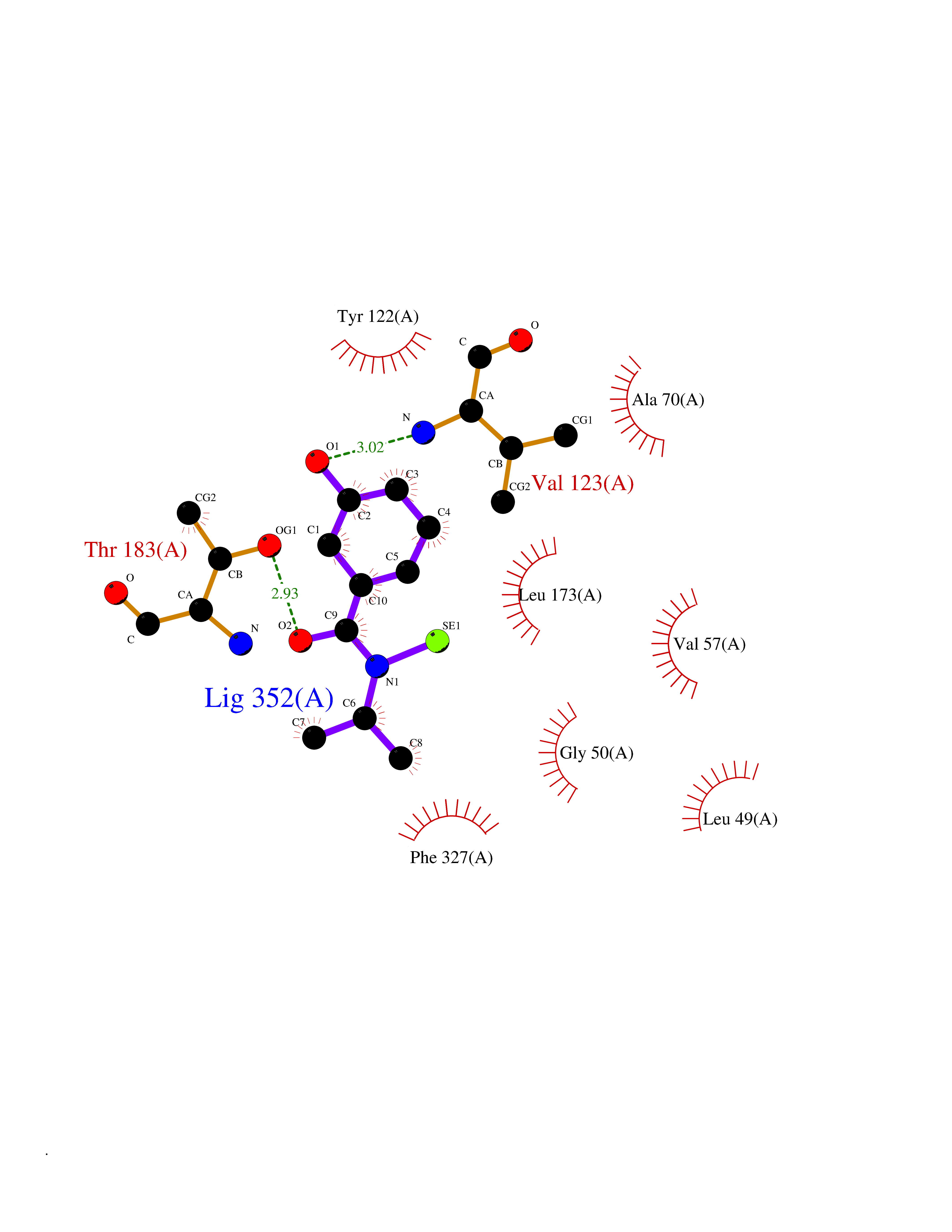 Ligplot Map 3D Binding mode Sequence ESVKEFLAKAKEDFLKKWETPQNTAQLDQFDRIKTLGTGSFGRVMLVKHKESGNHYAMKILDKQKVVKLKQIEHTLNEKRILQAVNFPFLVKLEFSFKDNSNLYMVMEYVAGGEMFSHLRRIGRFSEPHARFYAAQIVLTFEYLHSLDLIYRDLKPENLLIDQQGYIQVTDFGFAKRVKGRTWXLCGTPEYLAPEIILSKGYNKAVDWWALGVLIYEMAAGYPPFFADQPIQIYEKIVSGKVRFPSHFSSDLKDLLRNLLQVDLTKRFGNLKNGVNDIKNHKWFATTDWIAIYQRKVEAPFIPKFKGPGDTSNFDDYEEEEIRVXINEKCGKEFTEFTRSAIRRASTIE Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Muscarinic acetylcholine receptor M1 (CHRM1) | 5CXV | 5.73 | |
Target general information Gen name CHRM1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms M1 receptor Protein family G-protein coupled receptor 1 family, Muscarinic acetylcholine receptor subfamily, CHRM1 sub-subfamily Biochemical class GPCR rhodopsin Function Primary transducing effect is Pi turnover. The muscarinic acetylcholine receptor mediates various cellular responses, including inhibition of adenylate cyclase, breakdown of phosphoinositides and modulation of potassium channels through the action of G proteins. Related diseases Pyruvate dehydrogenase E1-beta deficiency (PDHBD) [MIM:614111]: An enzymatic defect causing primary lactic acidosis in children. It is associated with a broad clinical spectrum ranging from fatal lactic acidosis in the newborn to chronic neurologic dysfunction with structural abnormalities in the central nervous system without systemic acidosis. {ECO:0000269|PubMed:15138885}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03128; DB08897; DB05752; DB00321; DB00543; DB00517; DB04365; DB01238; DB14185; DB00572; DB00245; DB00767; DB01019; DB00810; DB09128; DB00835; DB00354; DB00411; DB00185; DB00477; DB01239; DB00568; DB00771; DB00363; DB00907; DB00979; DB00942; DB00434; DB00496; DB01151; DB00804; DB01231; DB00280; DB09167; DB01142; DB00366; DB01175; DB09194; DB06702; DB01148; DB00875; DB00483; DB00986; DB06787; DB11181; DB00725; DB00424; DB09262; DB00458; DB00332; DB01221; DB00408; DB00934; DB04843; DB00454; DB06709; DB00940; DB01403; DB00462; DB00340; DB01233; DB00805; DB01618; DB05152; DB00622; DB05766; DB00540; DB00334; DB01062; DB00383; DB00219; DB00715; DB01085; DB00670; DB06153; DB00387; DB00392; DB00420; DB01069; DB00782; DB00777; DB12278; DB11156; DB01224; DB11855; DB13581; DB00747; DB01591; DB02010; DB00342; DB11235; DB01409; DB01036; DB00193; DB00505; DB00508; DB00376; DB09089; DB00726; DB00809; DB00209; DB09076; DB09185; DB00246 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 33337 Length 291 Aromaticity 0.15 Instability index 26.7 Isoelectric point 8.75 Charge (pH=7) 5.76 2D Binding mode Binding energy (Kcal/mol) -7.82  Molscript Map 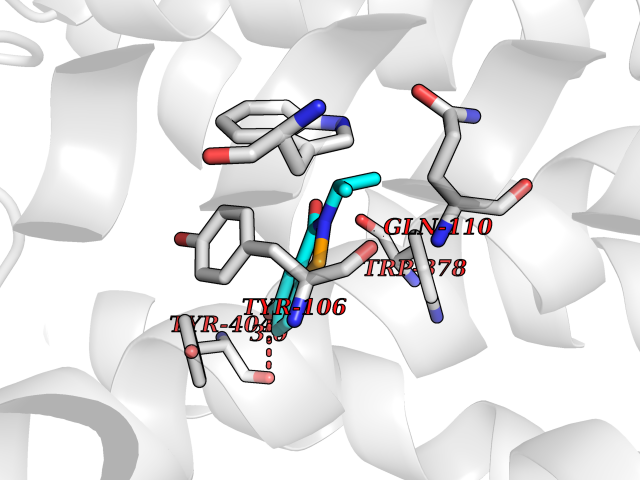 Pymol Map  Ligplot Map 3D Binding mode Sequence KGPWQVAFIGITTGLLSLATVTGNLLVLISFKVNTELKTVNNYFLLSLACADLIIGTFSMNLYTTYLLMGHWALGTLACDLWLALDYVASQASVMNLLLISFDRYFSVTRPLSYRAKRTPRRAALMIGLAWLVSFVLWAPAILFWQYLVGERTVLAGQCYIQFLSQPIITFGTAMAAFYLPVTVMCTLYWRIYRETENRFSLVKEKKAARTLSAILLAFILTWTPYNIMVLVSTFCKDCVPETLWELGYWLCYVNSTINPMCYALCNKAFRDTFRLLLLCRWDKDYKDDDD Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Adenosine deaminase (ADA) | 3IAR | 5.73 | |
Target general information Gen name ADA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Adenosine aminohydrolase; ADA1 Protein family Metallo-dependent hydrolases superfamily, Adenosine and AMP deaminases family Biochemical class Carbon-nitrogen hydrolase Function Plays an important role in purine metabolism and in adenosine homeostasis. Modulates signaling by extracellular adenosine, and so contributes indirectly to cellular signaling events. Acts as a positive regulator of T-cell coactivation, by binding DPP4. Its interaction with DPP4 regulates lymphocyte-epithelial cell adhesion. Enhances dendritic cell immunogenicity by affecting dendritic cell costimulatory molecule expression and cytokines and chemokines secretion. Enhances CD4+ T-cell differentiation and proliferation. Acts as a positive modulator of adenosine receptors ADORA1 and ADORA2A, by enhancing their ligand affinity via conformational change. Stimulates plasminogen activation. Plays a role in male fertility. Plays a protective role in early postimplantation embryonic development. Catalyzes the hydrolytic deamination of adenosine and 2-deoxyadenosine. Related diseases Severe combined immunodeficiency autosomal recessive T-cell-negative/B-cell-negative/NK-cell-negative due to adenosine deaminase deficiency (ADASCID) [MIM:102700]: An autosomal recessive disorder accounting for about 50% of non-X-linked SCIDs. SCID refers to a genetically and clinically heterogeneous group of rare congenital disorders characterized by impairment of both humoral and cell-mediated immunity, leukopenia, and low or absent antibody levels. Patients with SCID present in infancy with recurrent, persistent infections by opportunistic organisms. The common characteristic of all types of SCID is absence of T-cell-mediated cellular immunity due to a defect in T-cell development. ADA deficiency has been diagnosed in chronically ill teenagers and adults (late or adult onset). Population and newborn screening programs have also identified several healthy individuals with normal immunity who have partial ADA deficiency. {ECO:0000269|PubMed:10200056, ECO:0000269|PubMed:1284479, ECO:0000269|PubMed:2166947, ECO:0000269|PubMed:2783588, ECO:0000269|PubMed:3182793, ECO:0000269|PubMed:3839802, ECO:0000269|PubMed:6208479, ECO:0000269|PubMed:7599635, ECO:0000269|PubMed:8227344, ECO:0000269|PubMed:8299233, ECO:0000269|PubMed:9361033}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07711; DB07783; DB07786; DB04218; DB07785; DB03015; DB02472; DB00640; DB00975; DB14598; DB14600; DB00974; DB05057; DB03220; DB02616; DB02096; DB03572; DB02830; DB03370; DB04440; DB01280; DB00552; DB03000; DB00277; DB00194 Interacts with A5A3E0 EC number EC 3.5.4.4 Uniprot keywords 3D-structure; Acetylation; Cell adhesion; Cell junction; Cell membrane; Cytoplasm; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Hereditary hemolytic anemia; Hydrolase; Lysosome; Membrane; Metal-binding; Nucleotide metabolism; Proteomics identification; Reference proteome; SCID; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 40403.5 Length 360 Aromaticity 0.08 Instability index 38.7 Isoelectric point 5.63 Charge (pH=7) -9.06 2D Binding mode Binding energy (Kcal/mol) -7.81  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence PAFDKPKVELHVHLDGSIKPETILYYGRRRGIALPANTAEGLLNVIGMDKPLTLPDFLAKFDYYMPAIAGCREAIKRIAYEFVEMKAKEGVVYVEVRYSPHLLANSKVEPIPWNQAEGDLTPDEVVALVGQGLQEGERDFGVKARSILCCMRHQPNWSPKVVELCKKYQQQTVVAIDLAGDETIPGSSLLPGHVQAYQEAVKSGIHRTVHAGEVGSAEVVKEAVDILKTERLGHGYHTLEDQALYNRLRQENMHFEICPWSSYLTGAWKPDTEHAVIRLKNDQANYSLNTDDPLIFKSTLDTDYQMTKRDMGFTEEEFKRLNINAAKSSFLPEDEKRELLDLLYKAYGMPPSASAGQNLA Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Non-heme chloroperoxidase | 1A8U | 5.73 | |
Target general information Gen name cpo Organism Kitasatospora aureofaciens (Streptomyces aureofaciens) Uniprot ID TTD ID NA Synonyms cpoT Protein family AB hydrolase superfamily, Bacterial non-heme haloperoxidase / perhydrolase family Biochemical class Haloperoxidase Function Chloride peroxidase activity. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB03793 Interacts with NA EC number 1.11.1.- Uniprot keywords 3D-structure; Chloride; Oxidoreductase; Peroxidase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 60428.4 Length 554 Aromaticity 0.13 Instability index 31.26 Isoelectric point 4.65 Charge (pH=7) -32.72 2D Binding mode Binding energy (Kcal/mol) -7.82 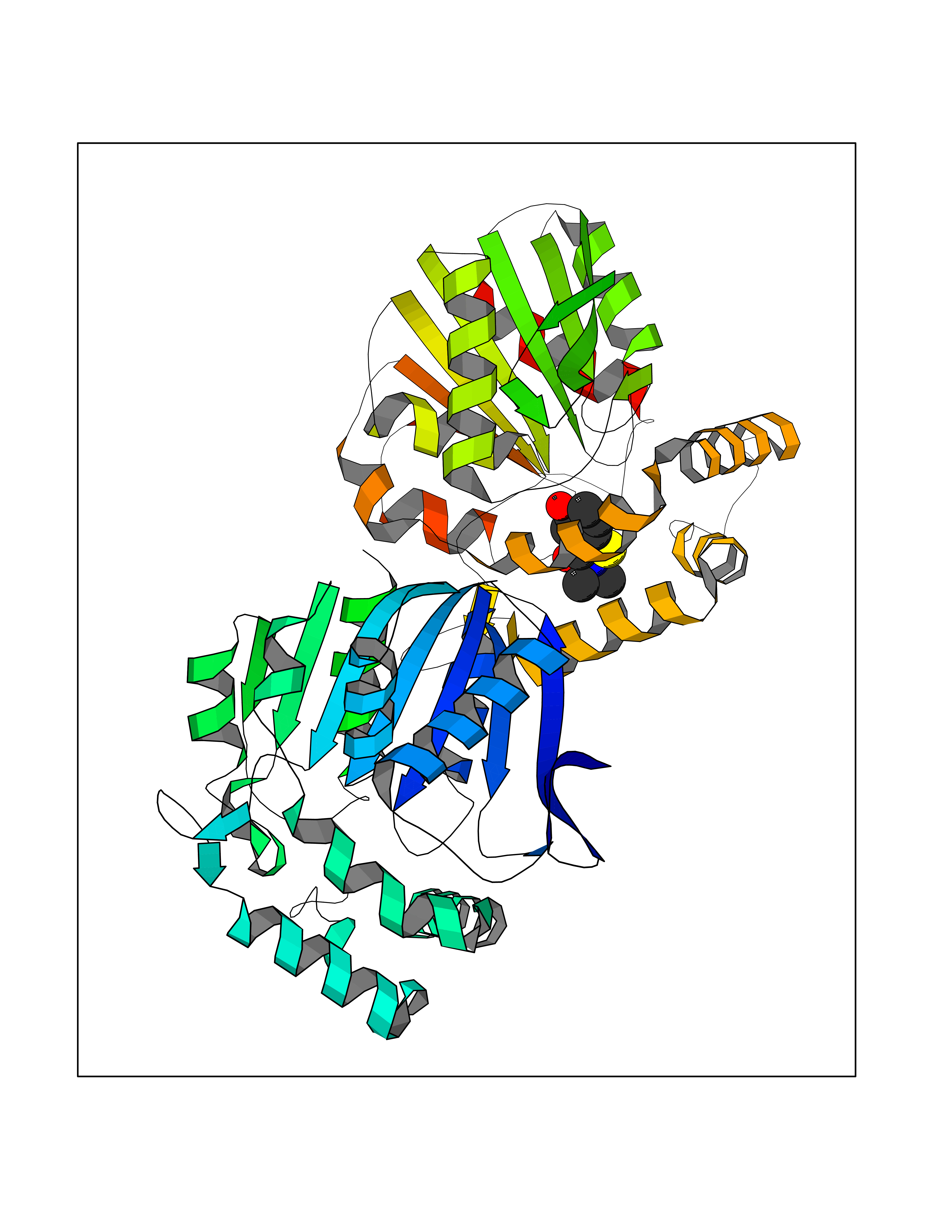 Molscript Map 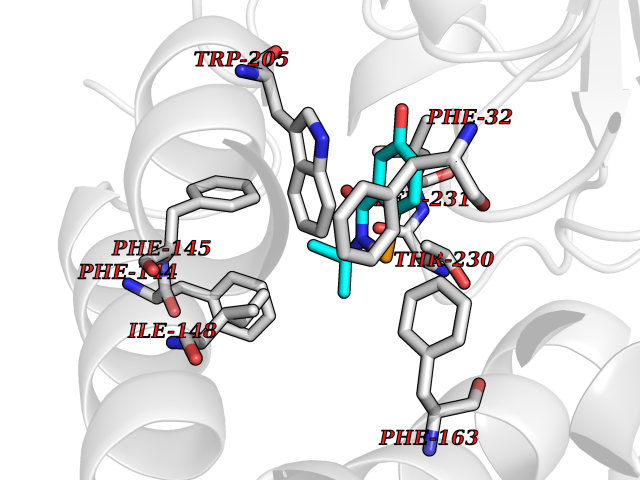 Pymol Map 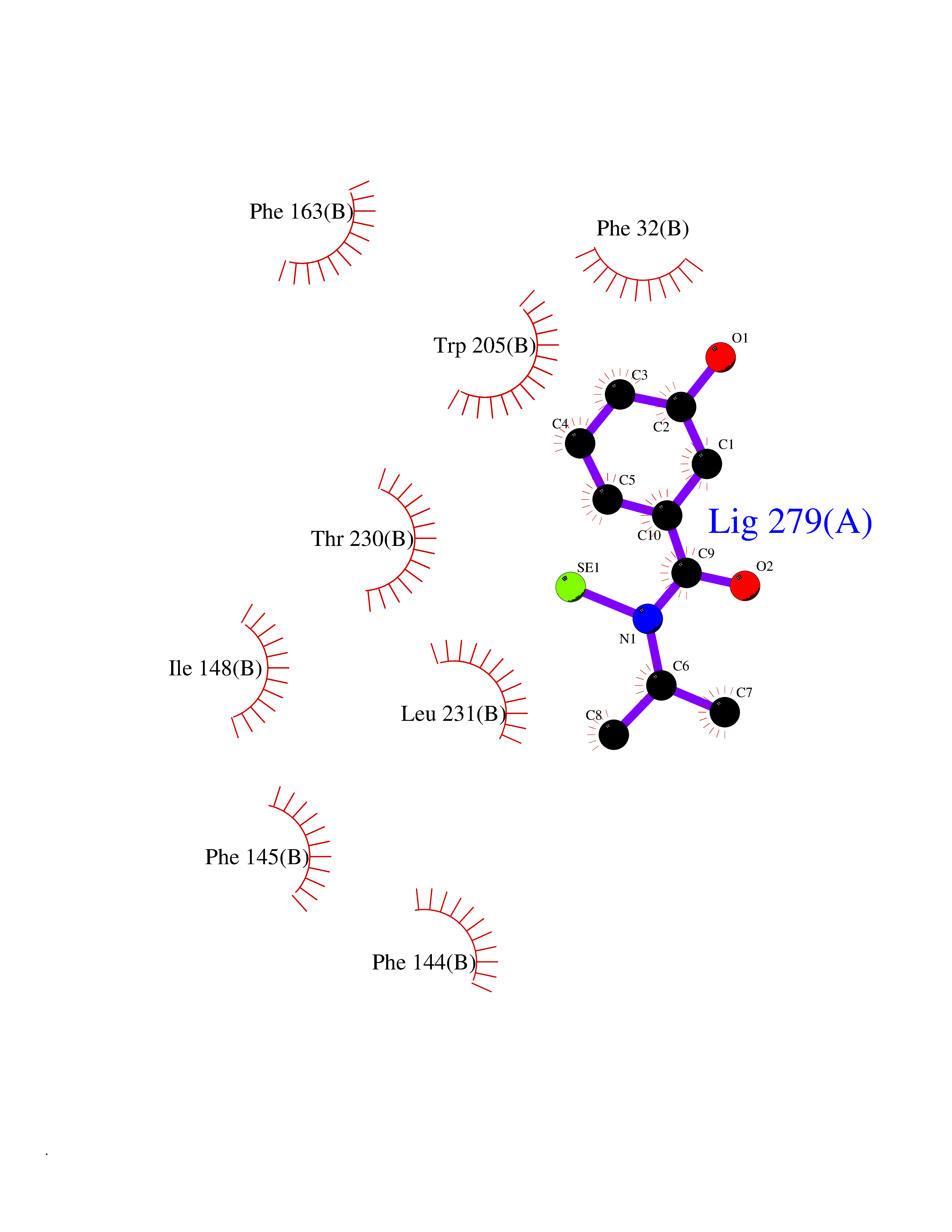 Ligplot Map 3D Binding mode Sequence PFITVGQENSTSIDLYYEDHGAGQPVVLIHGFPLSGHSWERQSAALLDAGYRVITYDRRGFGQSSQPTTGYDYDTFAADLNTVLETLDLQDAVLVGFSMGTGEVARYVSSYGTARIAKVAFLASLEPFLLKTDDNPDGAAPKEFFDGIVAAVKADRYAFYTGFFNDFYNLDENLGTRISEEAVRNSWNTAASGGFFAAAAAPTTWYTDFRADIPRIDVPALILHGTGDRTLPIENTARVFHKALPSAEYVEVEGAPHGLLWTHAEEVNTALLAFLAKPFITVGQENSTSIDLYYEDHGAGQPVVLIHGFPLSGHSWERQSAALLDAGYRVITYDRRGFGQSSQPTTGYDYDTFAADLNTVLETLDLQDAVLVGFSMGTGEVARYVSSYGTARIAKVAFLASLEPFLLKTDDNPDGAAPKEFFDGIVAAVKADRYAFYTGFFNDFYNLDENLGTRISEEAVRNSWNTAASGGFFAAAAAPTTWYTDFRADIPRIDVPALILHGTGDRTLPIENTARVFHKALPSAEYVEVEGAPHGLLWTHAEEVNTALLAFLAK Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Cannabinoid receptor 1 (CB1) | 5U09 | 5.73 | |
Target general information Gen name CNR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cannabinoid CB1 receptor; CNR; CB-R; CANN6 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Mediates many cannabinoid-induced effects, acting, among others, on food intake, memory loss, gastrointestinal motility, catalepsy, ambulatory activity, anxiety, chronic pain. Signaling typically involves reduction in cyclic AMP. In the hypothalamus, may have a dual effect on mitochondrial respiration depending upon the agonist dose and possibly upon the cell type. Increases respiration at low doses, while decreases respiration at high doses. At high doses, CNR1 signal transduction involves G-protein alpha-i protein activation and subsequent inhibition of mitochondrial soluble adenylate cyclase, decrease in cyclic AMP concentration, inhibition of protein kinase A (PKA)-dependent phosphorylation of specific subunits of the mitochondrial electron transport system, including NDUFS2. In the hypothalamus, inhibits leptin-induced reactive oxygen species (ROS) formation and mediates cannabinoid-induced increase in SREBF1 and FASN gene expression. In response to cannabinoids, drives the release of orexigenic beta-endorphin, but not that of melanocyte-stimulating hormone alpha/alpha-MSH, from hypothalamic POMC neurons, hence promoting food intake. In the hippocampus, regulates cellular respiration and energy production in response to cannabinoids. Involved in cannabinoid-dependent depolarization-induced suppression of inhibition (DSI), a process in which depolarization of CA1 postsynaptic pyramidal neurons mobilizes eCBs, which retrogradely activate presynaptic CB1 receptors, transiently decreasing GABAergic inhibitory neurotransmission. Also reduces excitatory synaptic transmission. In superior cervical ganglions and cerebral vascular smooth muscle cells, inhibits voltage-gated Ca(2+) channels in a constitutive, as well as agonist-dependent manner. In cerebral vascular smooth muscle cells, cannabinoid-induced inhibition of voltage-gated Ca(2+) channels leads to vasodilation and decreased vascular tone. Induces leptin production in adipocytes and reduces LRP2-mediated leptin clearance in the kidney, hence participating in hyperleptinemia. In adipose tissue, CNR1 signaling leads to increased expression of SREBF1, ACACA and FASN genes. In the liver, activation by endocannabinoids leads to increased de novo lipogenesis and reduced fatty acid catabolism, associated with increased expression of SREBF1/SREBP-1, GCK, ACACA, ACACB and FASN genes. May also affect de novo cholesterol synthesis and HDL-cholesteryl ether uptake. Peripherally modulates energy metabolism. In high carbohydrate diet-induced obesity, may decrease the expression of mitochondrial dihydrolipoyl dehydrogenase/DLD in striated muscles, as well as that of selected glucose/ pyruvate metabolic enzymes, hence affecting energy expenditure through mitochondrial metabolism. In response to cannabinoid anandamide, elicits a proinflammatory response in macrophages, which involves NLRP3 inflammasome activation and IL1B and IL18 secretion. In macrophages infiltrating pancreatic islets, this process may participate in the progression of type-2 diabetes and associated loss of pancreatic beta-cells. G-protein coupled receptor for endogenous cannabinoids (eCBs), including N-arachidonoylethanolamide (also called anandamide or AEA) and 2-arachidonoylglycerol (2-AG), as well as phytocannabinoids, such as delta(9)-tetrahydrocannabinol (THC). Related diseases Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:18177726}. The protein represented in this entry may be involved in disease pathogenesis. May contribute to the development of diet-induced obesity and several obesity-associated features, such as dyslipidemia and liver steatosis, regulating peripheral lipogenesis, energy expenditure and feeding behavior. CNR1 inverse agonists have been shown to reduce body weight and improve metabolic abnormalities in obese subjects, although adverse neuropsychiatric effects, including anxiety, irritability, and depressed mood, halted their therapeutic development (PubMed:18177726). In obese mice, peripherally restricted CNR1 inverse agonists have been shown to normalize metabolic abnormalities, including insulin resistance and fatty liver, and to reverse leptin resistance. {ECO:0000269|PubMed:18177726}.; DISEASE: Dysfunction of the endogenous cannabinoid system including CNR1 has been implicated in the pathogenesis of a number of central nervous system disorders, including Huntington disease, Parkinson disease, and Alzheimer disease (PubMed:32549916). In post-mortem brains from Huntington disease patients, a progressive CNR1 loss has been observed in the caudate nucleus, putamen, and substantia nigra pars reticulata, and altered expression and abnormal endocannabinoid levels precede motor symptoms in a disease mouse model (PubMed:10828533, PubMed:19524019, PubMed:8255419). In Parkinson disease, low CNR1 expression in mid-superior frontal gyrus and mid-cingulate cortex has been associated with poor mind, poor executive functioning and poor episode memory, while patients with more severe visuospatial dysfunction showed decreased receptor availability in the precuneus, mid-cingulate, supplementary motor cortex, inferior orbitofrontal gyrus and thalamus (PubMed:31342135). In an animal model for Alzheimer disease, CNR1 heterozygous deletion has been associated with decreased levels of postsynaptic density protein 95 (DLG4/PSD95) and accelerated memory impairment, suggesting synaptic dysfunction and a crucial role for CNR1 in the progression of disease symptoms (PubMed:10828533, PubMed:19524019, PubMed:30096288, PubMed:31342135, PubMed:8255419). {ECO:0000269|PubMed:10828533, ECO:0000269|PubMed:19524019, ECO:0000269|PubMed:30096288, ECO:0000269|PubMed:31342135, ECO:0000269|PubMed:32549916, ECO:0000269|PubMed:8255419}. Drugs (DrugBank ID) DB05750; DB09061; DB00470; DB14009; DB00486; DB14011; DB11745; DB09288; DB02955; DB06155; DB05077; DB11755; DB05201 Interacts with P29274; P21554 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Cell projection; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Neurodegeneration; Obesity; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 32070.3 Length 282 Aromaticity 0.13 Instability index 40.15 Isoelectric point 9.16 Charge (pH=7) 9.36 2D Binding mode Binding energy (Kcal/mol) -7.81 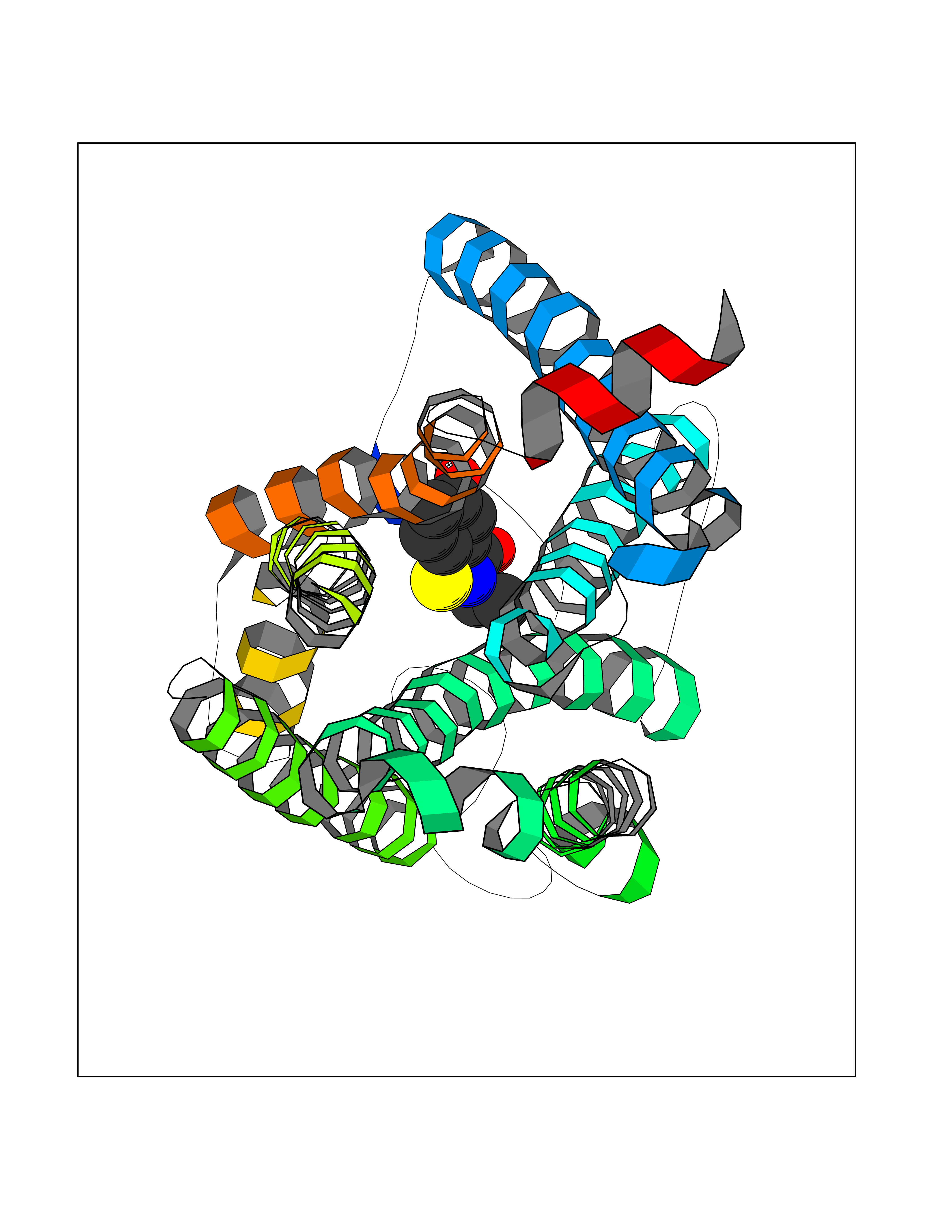 Molscript Map 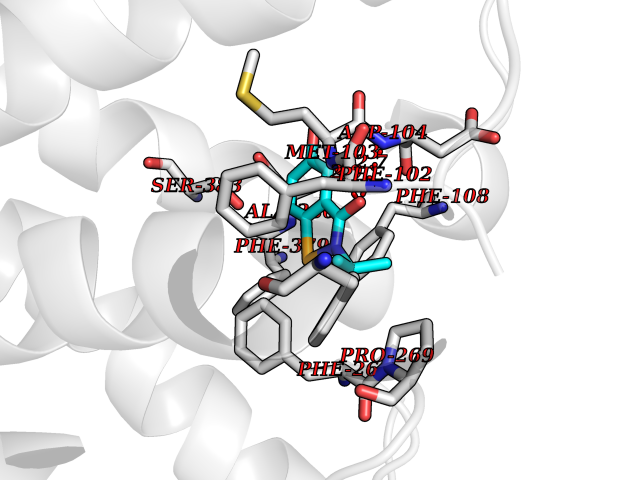 Pymol Map 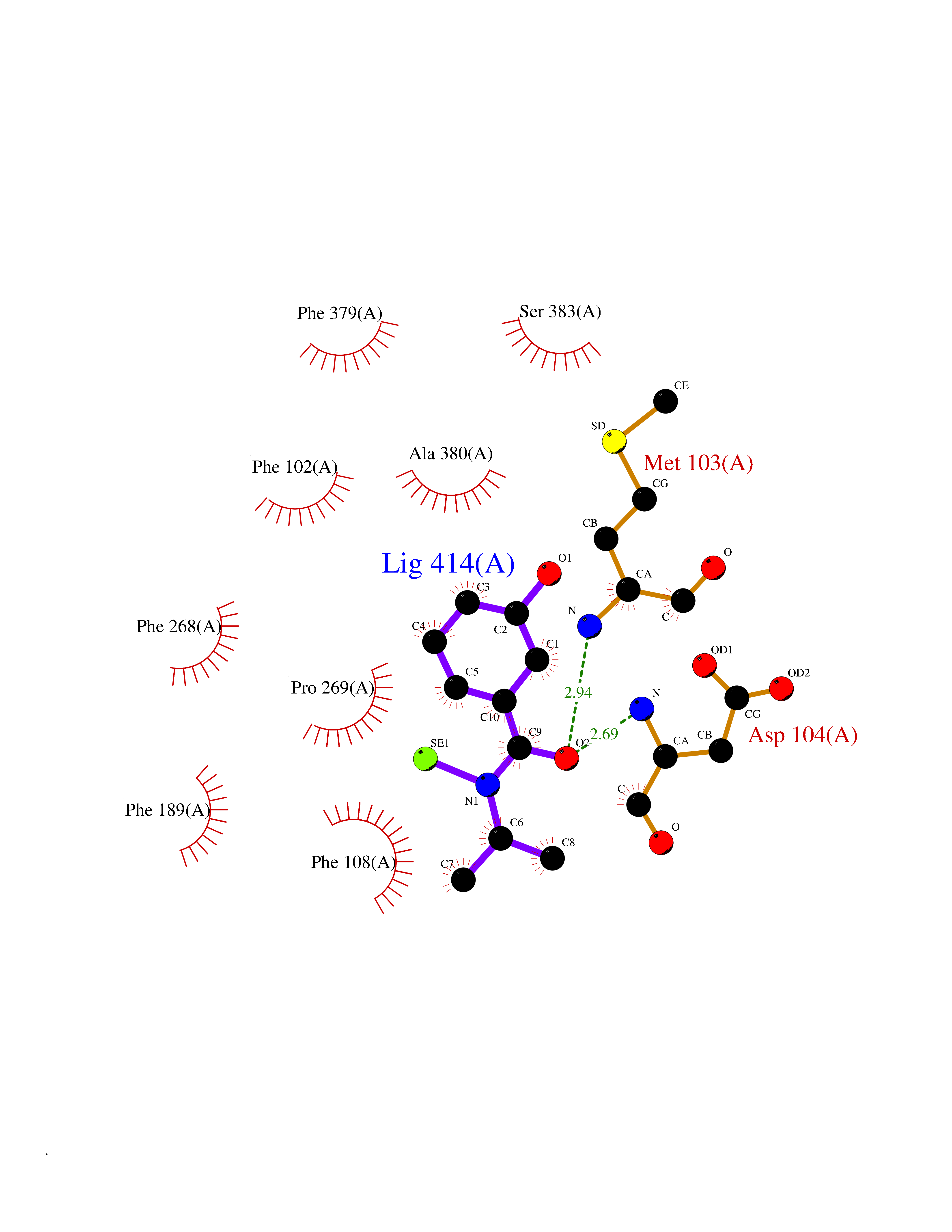 Ligplot Map 3D Binding mode Sequence ENFMDIECFMVLNPSQQLAIAVLSLTLGTFTVLENLLVLCVILHSRSLRCRPSYHFIGSLAVADLLGSVIFVYSFIDFHVFHRKDSRNVFLFKLGGVTASFTASVGSLFLAAIDRYISIHRPLAYKRIVTRPKAVVAFCLMWTIAIVIAVLPLLGWNCEKLQSVCSDIFPHIDETYLMFWIGVTSVLLLFIVYAYMYILWKADQARMDIRLAKTLVLILVVLIICWGPLLAIMVYDVFGKMNKLIKTVFAFCSMLCLLNSTVNPIIYALRSKDLRHAFRSMF Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Xanthine dehydrogenase/oxidase (XDH) | 2E1Q | 5.73 | |
Target general information Gen name XDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Xanthine oxidase; Xanthine dehydrogenase; XDHA Protein family Xanthine dehydrogenase family Biochemical class CH/CH(2) oxidoreductase Function Catalyzes the oxidation of hypoxanthine to xanthine. Catalyzes the oxidation of xanthine to uric acid. Contributes to the generation of reactive oxygen species. Has also low oxidase activity towards aldehydes (in vitro). Key enzyme in purine degradation. Related diseases Xanthinuria 1 (XAN1) [MIM:278300]: A disorder characterized by excretion of very large amounts of xanthine in the urine and a tendency to form xanthine stones. Uric acid is strikingly diminished in serum and urine. XAN1 is due to isolated xanthine dehydrogenase deficiency. Patients can metabolize allopurinol. {ECO:0000269|PubMed:10844591, ECO:0000269|PubMed:11379872, ECO:0000269|PubMed:14551354, ECO:0000269|PubMed:9153281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00640; DB00041; DB00437; DB00993; DB00958; DB01136; DB00856; DB00515; DB00746; DB03328; DB00997; DB03516; DB12466; DB04854; DB03147; DB04335; DB01020; DB00583; DB00170; DB01033; DB00157; DB03841; DB00336; DB01250; DB05262; DB06478; DB01168; DB00339; DB00127; DB01685; DB00831 Interacts with Q9Y3R0-3 EC number NA Uniprot keywords 2Fe-2S; 3D-structure; Cytoplasm; Disease variant; Disulfide bond; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase; Peroxisome; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 143697 Length 1307 Aromaticity 0.08 Instability index 37.9 Isoelectric point 8.01 Charge (pH=7) 7.07 2D Binding mode Binding energy (Kcal/mol) -7.81  Molscript Map 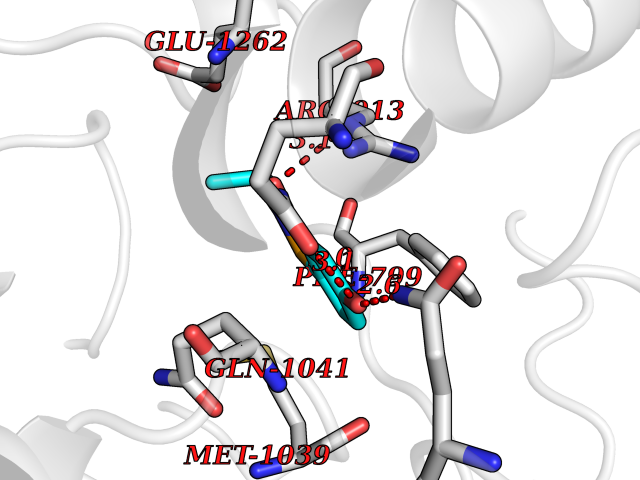 Pymol Map 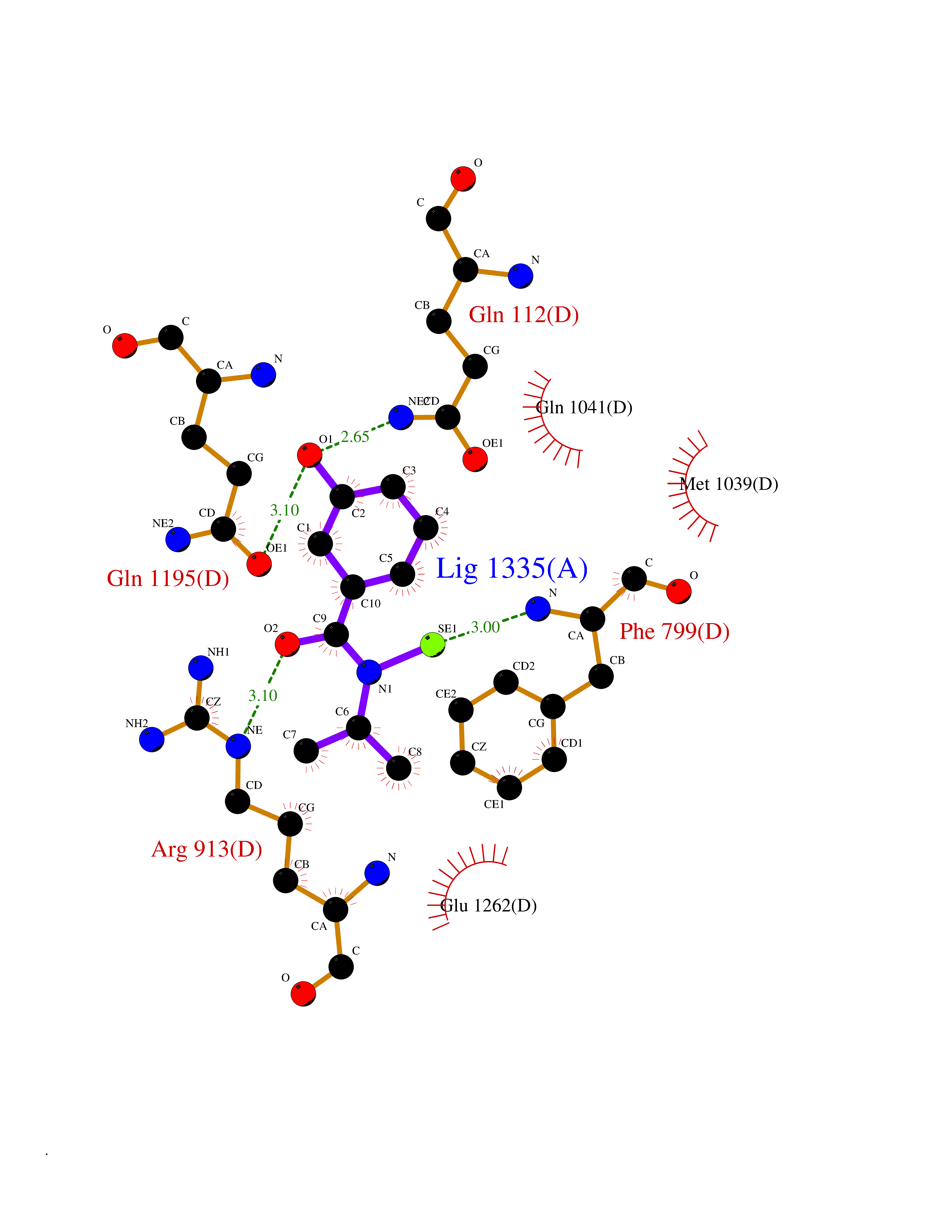 Ligplot Map 3D Binding mode Sequence ADKLVFFVNGRKVVEKNADPETTLLAYLRRKLGLSGTKLGCGEGGCGACTVMLSKYDRLQNKIVHFSANACLAPICSLHHVAVTTVEGIGSTKTRLHPVQERIAKSHGSQCGFCTPGIVMSMYTLLRNQPEPTMEEIENAFQGNLCRCTGYRPILQGFRTFARDGSPSLFKPEEFTPLDPTQEPIFPPELLRLKDTPRKQLRFEGERVTWIQASTLKELLDLKAQHPDAKLVVGNTEIGIEMKFKNMLFPMIVCPAWIPELNSVEHGPDGISFGAACPLSIVEKTLVDAVAKLPAQKTEVFRGVLEQLRWFAGKQVKSVASVGGNIITASPISDLNPVFMASGAKLTLVSRGTRRTVQMDHTFFPGYRKTLLSPEEILLSIEIPYSREGEYFSAFKQASRREDDIAKVTSGMRVLFKPGTTEVQELALCYGGMANRTISALKTTQRQLSKLWKEELLQDVCAGLAEELHLPPDAPGGMVDFRCTLTLSFFFKFYLTVLQKLGQENLEDKCGKLDPTFASATLLFQKDPPADVQLFQEVPKGQSEEDMVGRPLPHLAADMQASGEAVYCDDIPRYENELSLRLVTSTRAHAKIKSIDTSEAKKVPGFVCFISADDVPGSNITGICNDETVFAKDKVTCVGHIIGAVVADTPEHTQRAAQGVKITYEELPAIITIEDAIKNNSFYGPELKIEKGDLKKGFSEADNVVSGEIYIGGQEHFYLETHCTIAVPKGEAGEMELFVSTQNTMKTQSFVAKMLGVPANRIVVRVKRMGGGFGGKVTRSTVVSTAVALAAYKTGRPVRCMLDRDEDMLITGGRHPFLARYKVGFMKTGTVVALEVDHFSNVGNTQDLSQSIMERALFHMDNCYKIPNIRGTGRLCKTNLPSNTAFRGFGGPQGMLIAECWMSEVAVTCGMPAEEVRRKNLYKEGDLTHFNQKLEGFTLPRCWEECLASSQYHARKSEVDKFNKENCWKKRGLCIIPTKFGISFTVPFLNQAGALLHVYTDGSVLLTHGGTEMGQGLHTKMVQVASRALKIPTSKIYISETSTNTVPNTSPTAASVSADLNGQAVYAACQTILKRLEPYKKKNPSGSWEDWVTAAYMDTVSLSATGFYRTPNLGYSFETNSGNPFHYFSYGVACSEVEIDCLTGDHKNLRTDIVMDVGSSLNPAIDIGQVEGAFVQGLGLFTLEELHYSPEGSLHTRGPSTYKIPAFGSIPIEFRVSLLRDCPNKKAIYASKAVGEPPLFLAASIFFAIKDAIRAARAQHTGNNVKELFRLDSPATPEKIRNACVDKFTTLCVTGVPENCKPWSVRV Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Fatty acid synthase (FASN) | 3TJM | 5.73 | |
Target general information Gen name FASN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Yeast fatty acid synthase; Fatty-acyl-CoA synthase; Fatty acyl-CoA synthetase enzyme; FAS Protein family NA Biochemical class Acyltransferase Function Fatty acid synthetase catalyzes the formation of long-chain fatty acids from acetyl-CoA, malonyl-CoA and NADPH. This multifunctional protein has 7 catalytic activities as an acyl carrier protein. Related diseases Glycine encephalopathy 2 (GCE2) [MIM:620398]: A form of glycine encephalopathy, a metabolic disorder characterized by a high concentration of glycine in the body fluids. Affected individuals typically have severe neurological symptoms, including seizure, lethargy, and muscular hypotonia soon after birth. Most of them die within the neonatal period. Atypical cases have later disease onset and less severely affected psychomotor development. {ECO:0000269|PubMed:10873393, ECO:0000269|PubMed:11286506, ECO:0000269|PubMed:16051266, ECO:0000269|PubMed:26371980, ECO:0000269|PubMed:28244183, ECO:0000269|PubMed:8005589, ECO:0000269|PubMed:9600239, ECO:0000269|PubMed:9621520}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01034; DB01083 Interacts with Q15848; Q16665; P42858; Q8IV20; Q8TBB1; PRO_0000045603 [Q99IB8] EC number EC 2.3.1.85 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Isopeptide bond; Lipid biosynthesis; Lipid metabolism; Lyase; Multifunctional enzyme; NAD; NADP; Oxidoreductase; Phosphopantetheine; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; S-nitrosylation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30174.9 Length 275 Aromaticity 0.09 Instability index 43.28 Isoelectric point 5.92 Charge (pH=7) -5.4 2D Binding mode Binding energy (Kcal/mol) -7.82 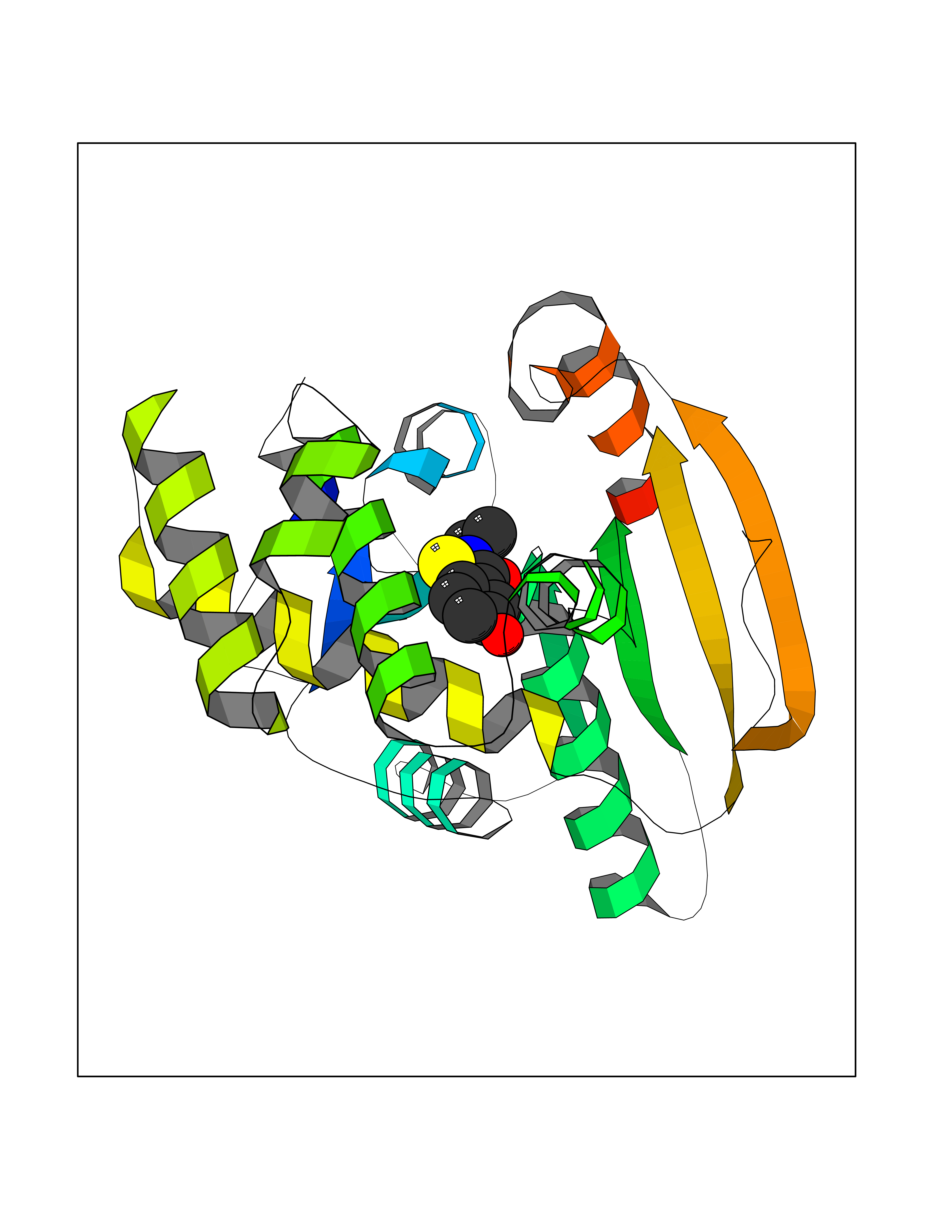 Molscript Map 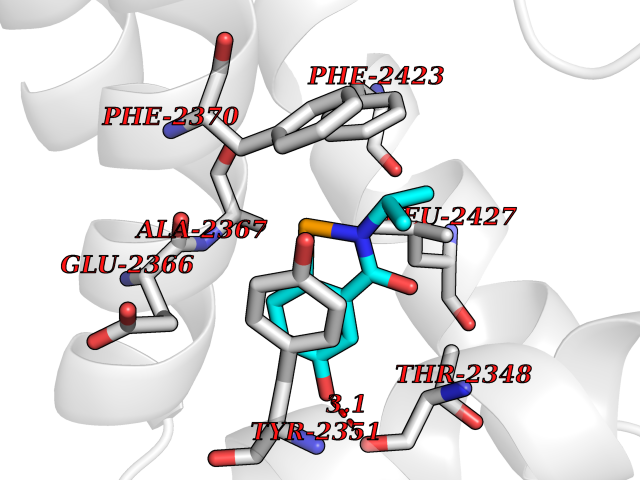 Pymol Map 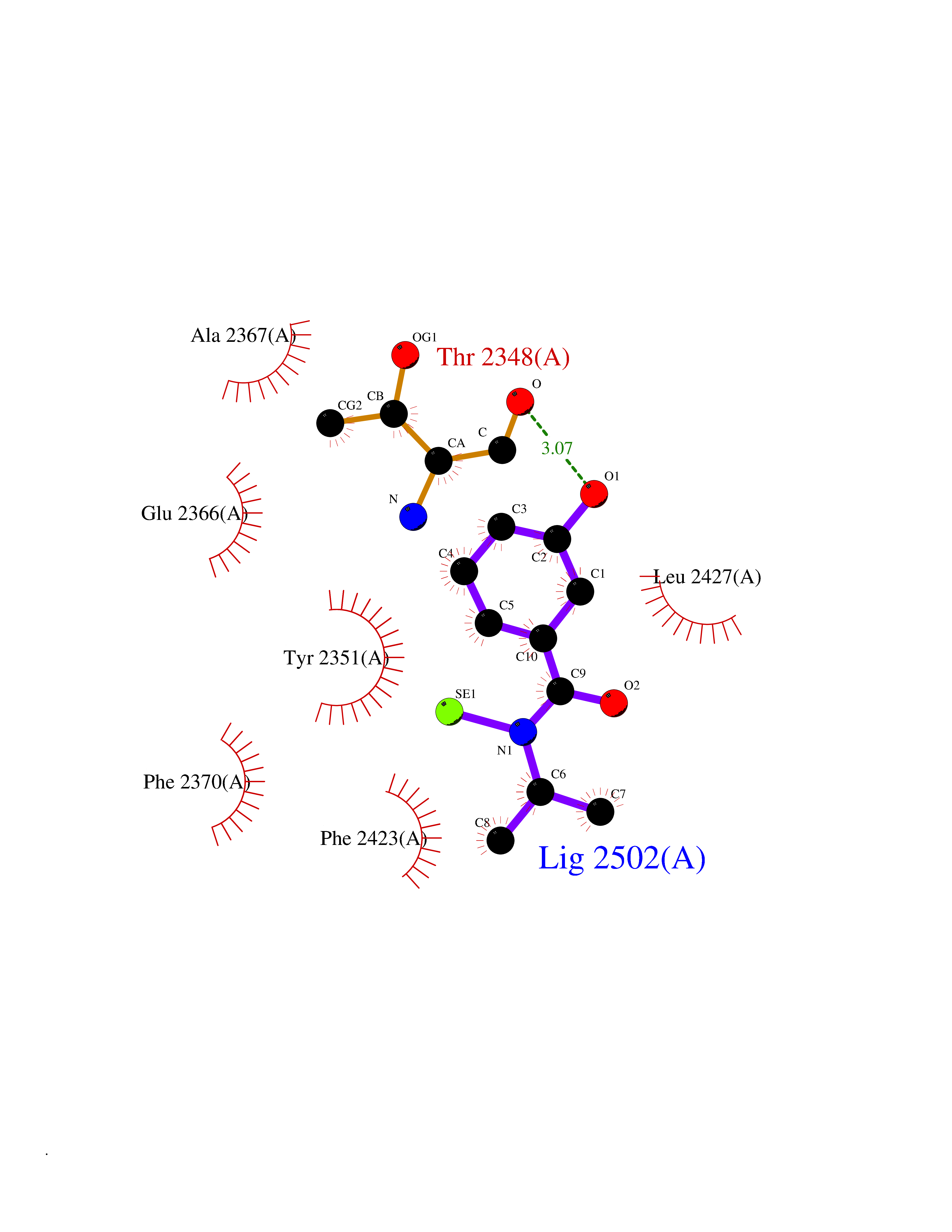 Ligplot Map 3D Binding mode Sequence NLRSLLVNPEGPTLMRLNSVQSSERPLFLVHPIEGSTTVFHSLASRLSIPTYGLQCTRAAPLDSIHSLAAYYIDCIRQVQPEGPYRVAGYSYGACVAFEMCSQLQAQQSPAPTHNSLFLFDGSPTYVLAYTGSYRAKLTPGCEAEAETEAICFFVQQFTDMEHNRVLEALLPLKGLEERVAAAVDLIIKSHQGLDRQELSFAARSFYYKLRAAEQYTPKAKYHGNVMLLRAAAGADYNLSQVCDGKVSVHVIEGDHATLLEGSGLESIISIIHSS Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Pyruvate synthase | 2C42 | 5.73 | |
Target general information Gen name por Organism Desulfocurvibacter africanus (Desulfovibrio africanus) Uniprot ID TTD ID NA Synonyms NA Protein family Pyruvate:ferredoxin/flavodoxin oxidoreductase family Biochemical class Oxidoreductase Function 4 iron, 4 sulfur cluster binding.Iron ion binding.Pyruvate synthase activity.Thiamine pyrophosphate binding. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02410; DB01987; DB00507 Interacts with NA EC number 1.2.7.1 Uniprot keywords 3D-structure; 4Fe-4S; Calcium; Cytoplasm; Direct protein sequencing; Disulfide bond; Electron transport; Iron; Iron-sulfur; Magnesium; Metal-binding; Oxidoreductase; Pyruvate; Thiamine pyrophosphate; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 115569 Length 1065 Aromaticity 0.09 Instability index 31.51 Isoelectric point 6.32 Charge (pH=7) -5.62 2D Binding mode Binding energy (Kcal/mol) -7.82  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GKKMMTTDGNTATAHVAYAMSEVAAIYPITPSSTMGEEADDWAAQGRKNIFGQTLTIREMQSEAGAAGAVHGALAAGALTTTFTASQGLLLMIPNMYKISGELLPGVFHVTARAIAAHALSIFGDHQDIYAARQTGFAMLASSSVQEAHDMALVAHLAAIESNVPFMHFFDGFRTSHEIQKIEVLDYADMASLVNQKALAEFRAKSPGIVAEYMQKVASLTGRSYKLFDYVGAPDAERVIVSMGSSCETIEEVINHLAAKGEKIGLIKVRLYRPFVSEAFFAALPASAKVITVLDRTKEPGAPGDPLYLDVCSAFVERGEAMPKILAGRYGLGSKEFSPAMVKSVYDNMSGAKKNHFTVGIEDDVTGTSLPVDNAFADTTPKGTIQCQFWGLGADGTVGANKQAIKIIGDNTDLFAQGYFSYDSKKSGGITISHLRFGEKPIQSTYLVNRADYVACHNPAYVGIYDILEGIKDGGTFVLNSPWSSLEDMDKHLPSGIKRTIANKKLKFYNIDAVKIATDVGLGGRINMIMQTAFFKLAGVLPFEKAVDLLKKSIHKAYGKKGEKIVKMNTDAVDQAVTSLQEFKYPDSWKDAPAETKAEPMTNEFFKNVVKPILTQQGDKLPVSAFEADGRFPLGTSQFEKRGVAINVPQWVPENCIQCNQCAFVCPHSAILPVLAKEEELVGAPANFTALEAKGKELKGYKFRIQINTLDCMGCGNCADICPPKEKALVMQPLDTQRDAQVPNLEYAARIPVKSEVLPRDSLKGSQFQEPLMEFSGACSGCGETPYVRVITQLFGERMFIANATGCSSIWGASAPSMPYKTNRLGQGPAWGNSLFEDAAEYGFGMSVWIFGGDGWAYDIGYGGLDHVLASGEDVNVFVMDTEVYSNTGGQSSKATPTGAVAKFAAAGKRTGKKDLARMVMTYGYVYVATVSMGYSKQQFLKVLKEAESFPGPSLVIAYATCINQGLRKGMGKSQDVMNTAVKSGYWPLFRYDPRLAAQGKNPFQLDSKAPDGSVEEFLMAQNRFAVLDRSFPEDAKRLRAQVAHELDVRFKELEHMAATNIFES Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Dimethylglycine oxidase | 1PJ5 | 5.73 | |
Target general information Gen name dmg Organism Arthrobacter globiformis Uniprot ID TTD ID NA Synonyms NA Protein family GcvT family Biochemical class Oxidoreductase Function Dimethylglycine oxidase activity.Nucleotide binding. Related diseases Curry-Jones syndrome (CRJS) [MIM:601707]: A multisystem disorder characterized by patchy skin lesions, polysyndactyly, diverse cerebral malformations, unicoronal craniosynostosis, iris colobomas, microphthalmia, and intestinal malrotation with myofibromas or hamartomas. {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. The disease is caused by variants affecting the gene represented in this entry. 8 individuals have been identified with the disease-causing mutation Phe-412 and all were mosaic. The mutation could not be reliably detected in blood, greatest success rates were obtained with affected tissues obtained by invasive procedures. It is thought that the mutation has arisen postzygotically early during embryonic development (PubMed:27236920). This mutation has also been identified in ameloblastoma, medulloblastoma, meningioma, and basal cell carcinoma, and has been reported as the oncogenic driver in some of these tumors (PubMed:24859340). {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. Drugs (DrugBank ID) DB03256; DB03147 Interacts with NA EC number 1.5.3.10 Uniprot keywords 3D-structure; Direct protein sequencing; FAD; Flavoprotein; Nucleotide-binding; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 45912.2 Length 427 Aromaticity 0.07 Instability index 43.46 Isoelectric point 4.83 Charge (pH=7) -20.69 2D Binding mode Binding energy (Kcal/mol) -7.82  Molscript Map 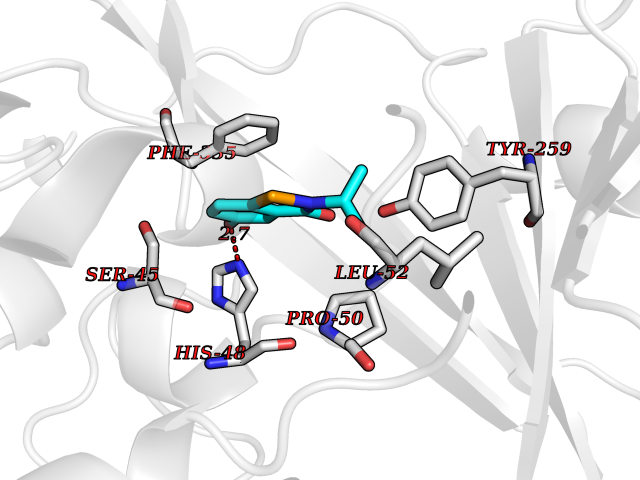 Pymol Map 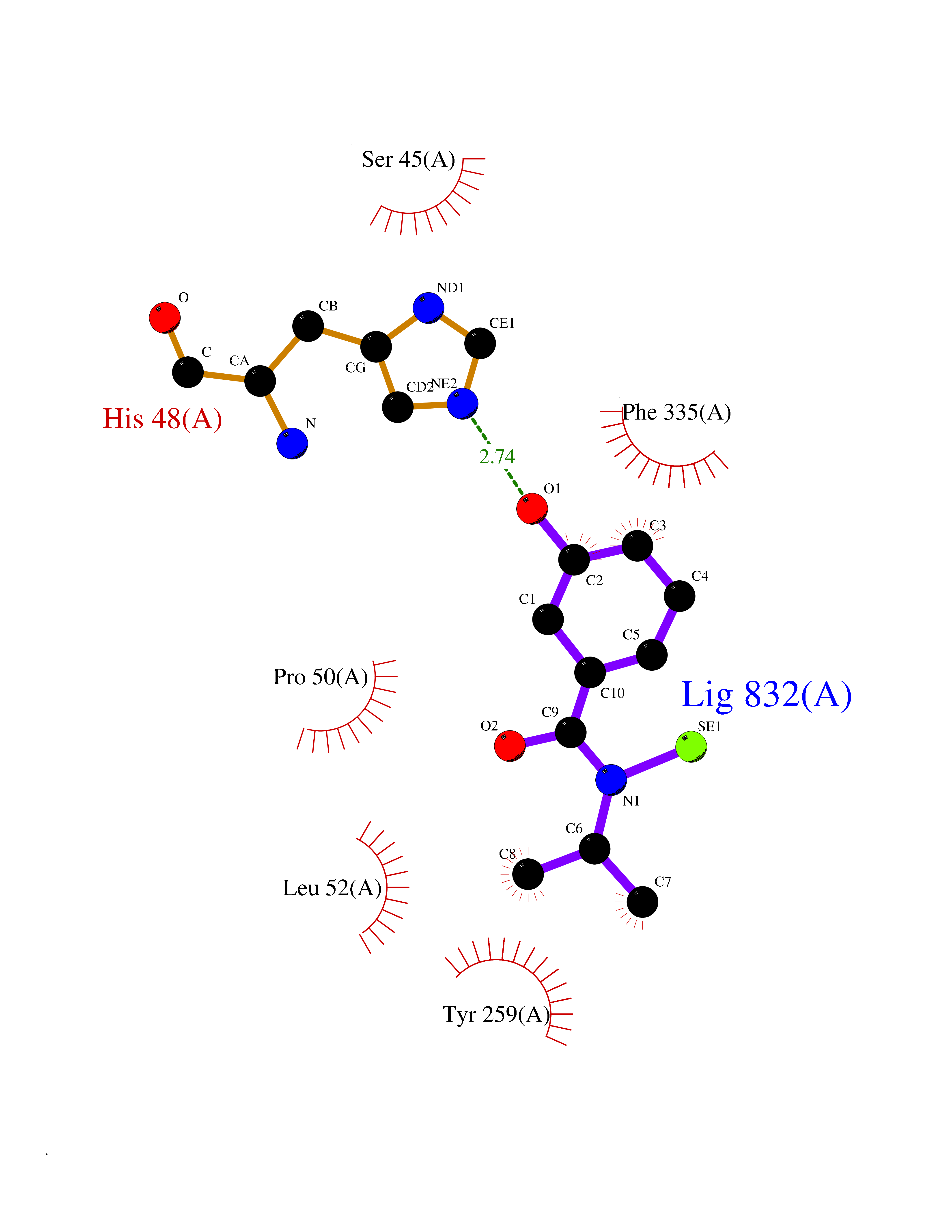 Ligplot Map 3D Binding mode Sequence TPRIVIIGAGIVGTNLADELVTRGWNNITVLDQGPLNMPGGSTSHAPGLVFQTNPSKTMASFAKYTVEKLLSLTEDGVSCFNQVGGLEVATTETRLADLKRKLGYAAAWGIEGRLLSPAECQELYPLLDGENILGGLHVPSDGLASAARAVQLLIKRTESAGVTYRGSTTVTGIEQSGGRVTGVQTADGVIPADIVVSCAGFWGAKIGAMIGMAVPLLPLAHQYVKTTPVPAQQGRNDQPNGARLPILRHQDQDLYYREHGDRYGIGSYAHRPMPVDVDTLGAYAPETVSEHHMPSRLDFTLEDFLPAWEATKQLLPALADSEIEDGFNGIFSFTPDGGPLLGESKELDGFYVAEAVWVTHSAGVAKAMAELLTTGRSETDLGECDITRFEDVQLTPEYVSETSQQNFVEIYDVLHPLQPRLSPRNL Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Glycolipid transfer protein | 3RZN | 5.73 | |
Target general information Gen name GLTP Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family GLTP family Biochemical class Lipid transport Function Glycolipid binding.Glycolipid transporter activity.Identical protein binding.Intermembrane lipid transfer activity.Lipid binding. Related diseases Brugada syndrome 7 (BRGDA7) [MIM:613120]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:20031595}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Atrial fibrillation, familial, 16 (ATFB16) [MIM:613120]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:20558140, ECO:0000269|PubMed:21051419}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03600; DB04465; DB03017; DB03203 Interacts with Q96DZ9; Q9NZD2 EC number NA Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Lipid transport; Proteomics identification; Reference proteome; Repeat; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23534.1 Length 206 Aromaticity 0.11 Instability index 36.45 Isoelectric point 7.08 Charge (pH=7) 0.1 2D Binding mode Binding energy (Kcal/mol) -7.81 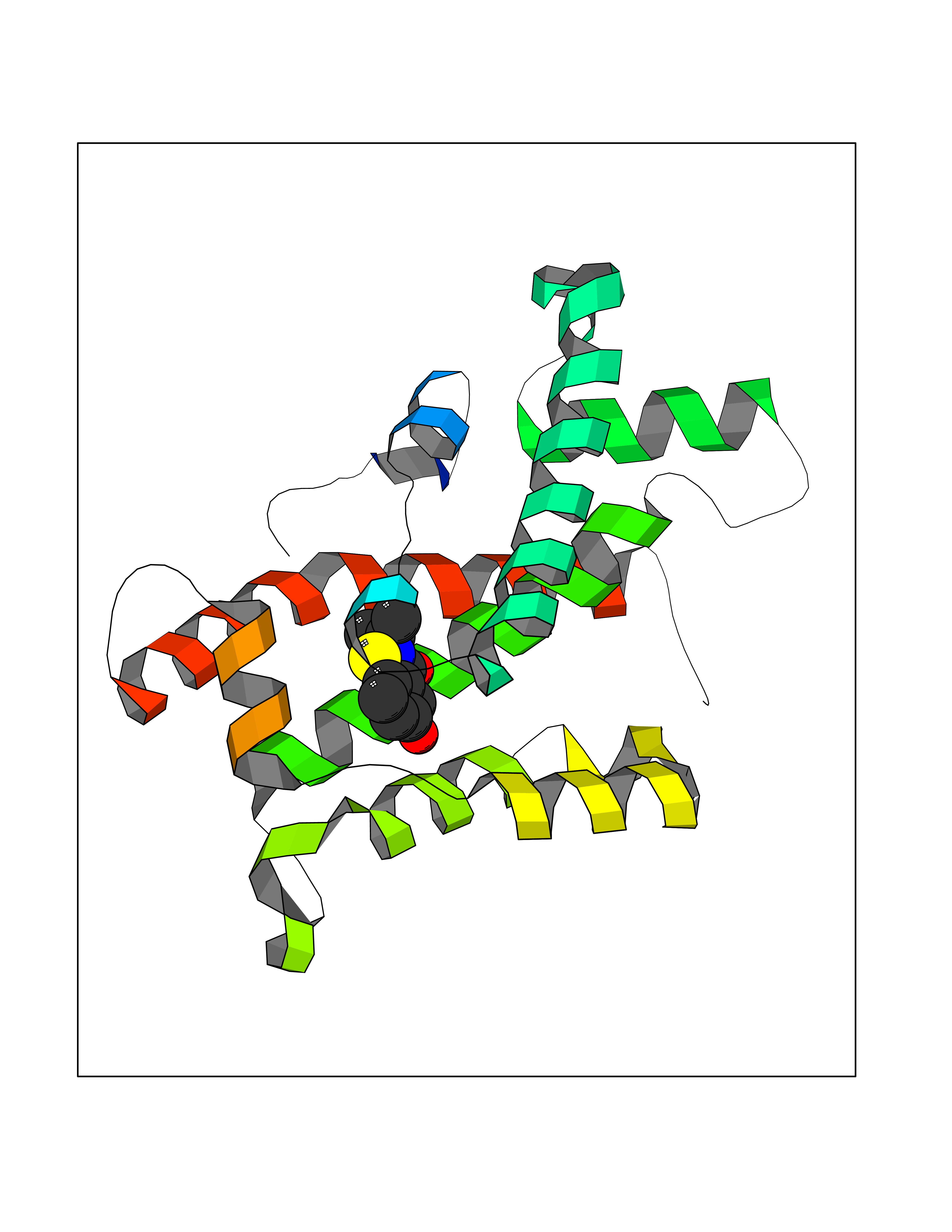 Molscript Map 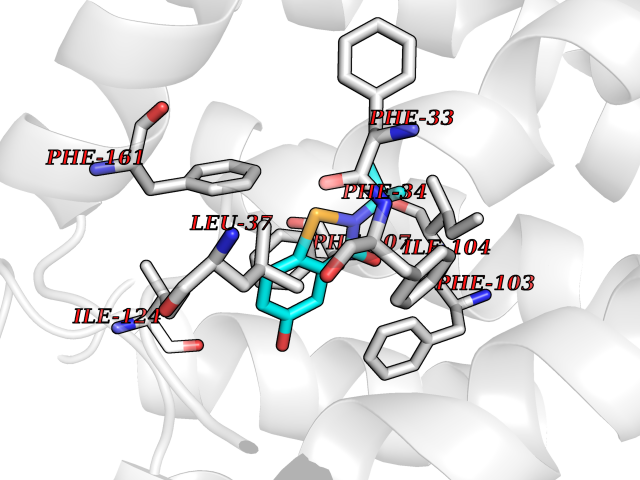 Pymol Map 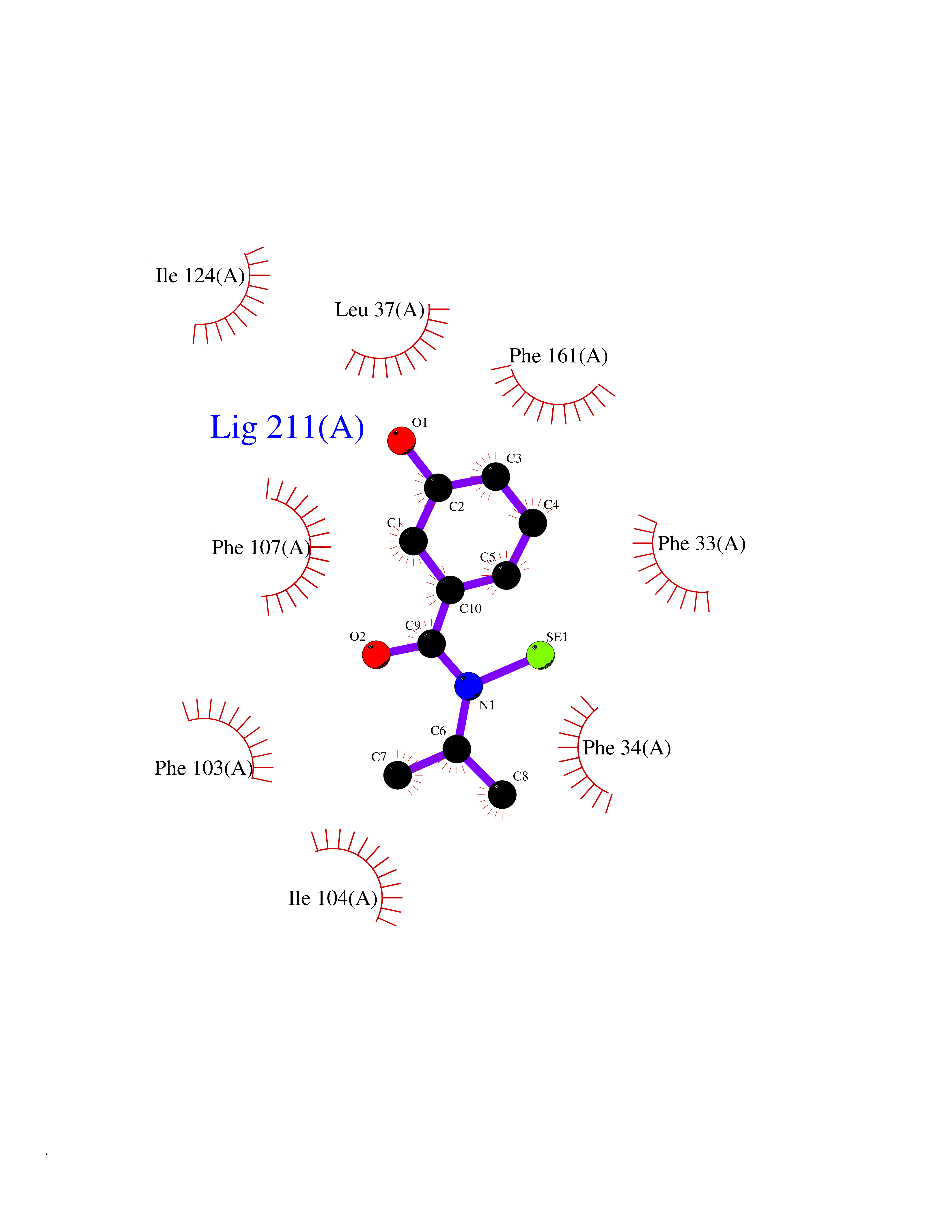 Ligplot Map 3D Binding mode Sequence LAEHLLKPLPADKQIETGPFLEAVSHLPPFFDCLGSPVFTPIKADISGNITKIKAVYDTNPAKFRTLQNILEVEKEMYGAEWPKVGATLALMWLKRGLRFIQVFLQSICDGERDENHPNLIRVNATKAYEMALKKYHGWIVQKIFQAALYAAPYKSDFLKALSKGQNVTEEECLEKIRLFLVNYTATIDVIYEMYTQMNAELNYKV Hydrogen bonds contact Hydrophobic contact | ||||