Job Results:
Ligand
Structure
Job ID
f5eac8ba4ac90e4335358bfb735bd782
Job name
NA
Time
2025-10-13 17:37:26
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Fatty acid synthase (FASN) | 3TJM | 5.66 | |
Target general information Gen name FASN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Yeast fatty acid synthase; Fatty-acyl-CoA synthase; Fatty acyl-CoA synthetase enzyme; FAS Protein family NA Biochemical class Acyltransferase Function Fatty acid synthetase catalyzes the formation of long-chain fatty acids from acetyl-CoA, malonyl-CoA and NADPH. This multifunctional protein has 7 catalytic activities as an acyl carrier protein. Related diseases Glycine encephalopathy 2 (GCE2) [MIM:620398]: A form of glycine encephalopathy, a metabolic disorder characterized by a high concentration of glycine in the body fluids. Affected individuals typically have severe neurological symptoms, including seizure, lethargy, and muscular hypotonia soon after birth. Most of them die within the neonatal period. Atypical cases have later disease onset and less severely affected psychomotor development. {ECO:0000269|PubMed:10873393, ECO:0000269|PubMed:11286506, ECO:0000269|PubMed:16051266, ECO:0000269|PubMed:26371980, ECO:0000269|PubMed:28244183, ECO:0000269|PubMed:8005589, ECO:0000269|PubMed:9600239, ECO:0000269|PubMed:9621520}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01034; DB01083 Interacts with Q15848; Q16665; P42858; Q8IV20; Q8TBB1; PRO_0000045603 [Q99IB8] EC number EC 2.3.1.85 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Isopeptide bond; Lipid biosynthesis; Lipid metabolism; Lyase; Multifunctional enzyme; NAD; NADP; Oxidoreductase; Phosphopantetheine; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; S-nitrosylation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30174.9 Length 275 Aromaticity 0.09 Instability index 43.28 Isoelectric point 5.92 Charge (pH=7) -5.4 2D Binding mode Binding energy (Kcal/mol) -7.72 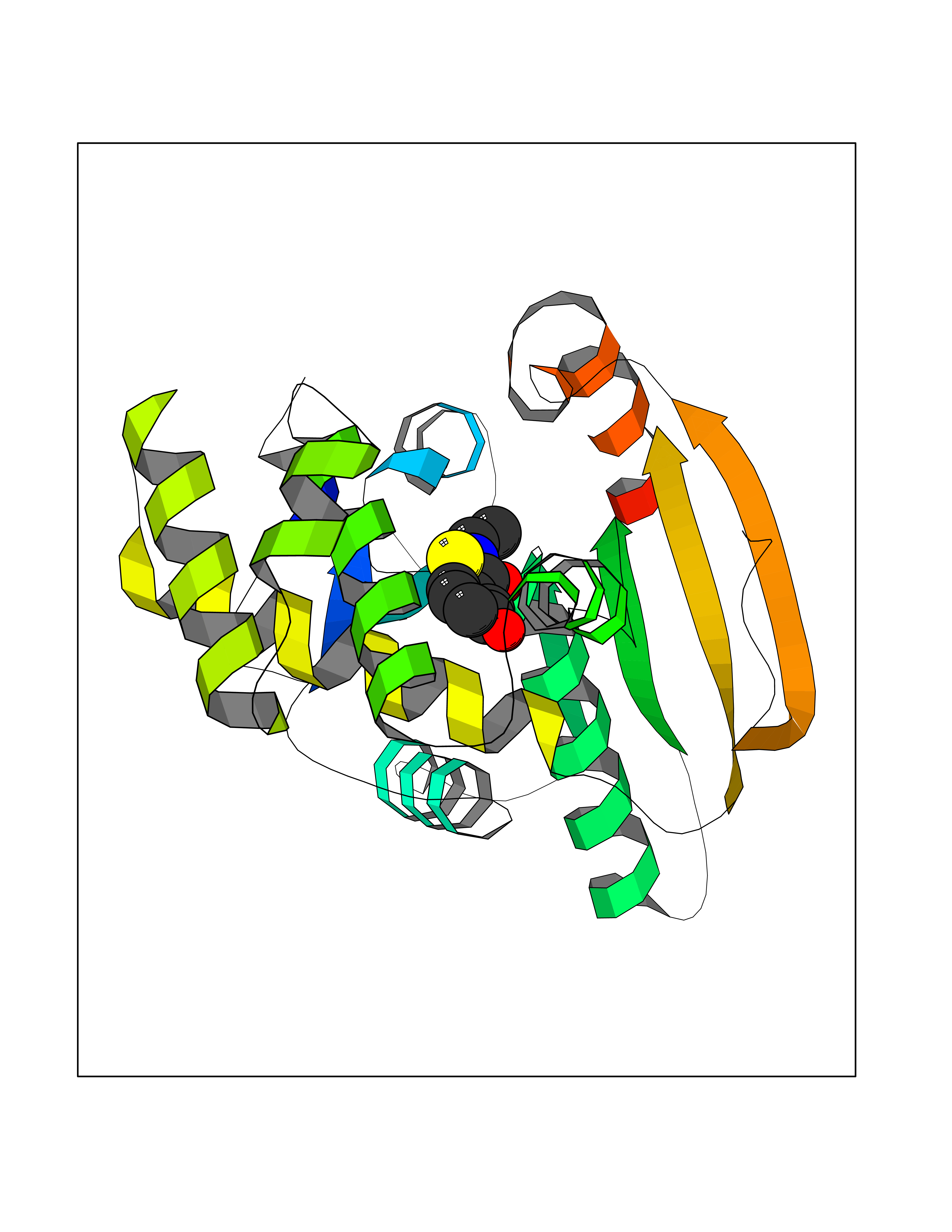 Molscript Map 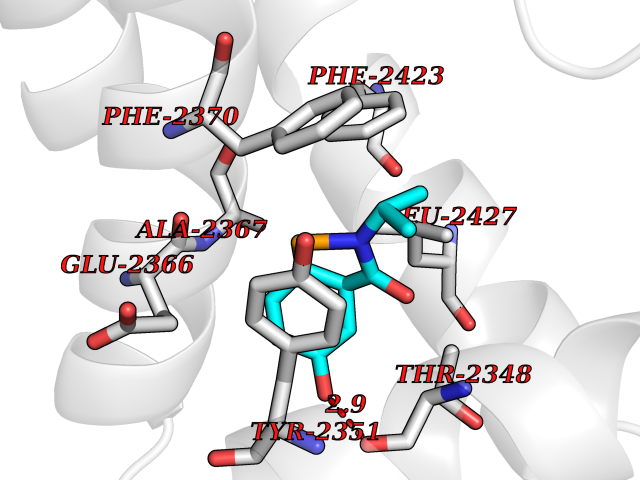 Pymol Map 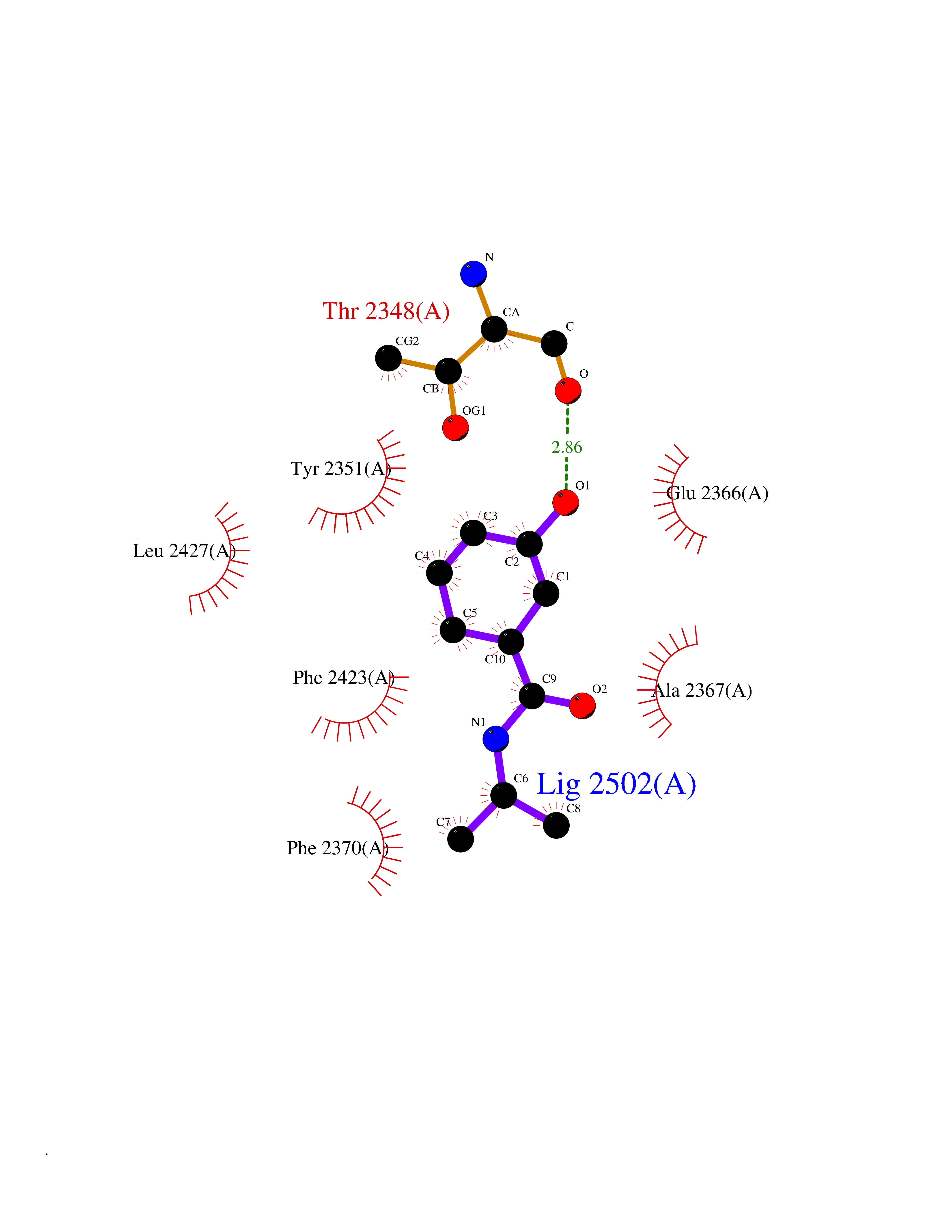 Ligplot Map 3D Binding mode Sequence NLRSLLVNPEGPTLMRLNSVQSSERPLFLVHPIEGSTTVFHSLASRLSIPTYGLQCTRAAPLDSIHSLAAYYIDCIRQVQPEGPYRVAGYSYGACVAFEMCSQLQAQQSPAPTHNSLFLFDGSPTYVLAYTGSYRAKLTPGCEAEAETEAICFFVQQFTDMEHNRVLEALLPLKGLEERVAAAVDLIIKSHQGLDRQELSFAARSFYYKLRAAEQYTPKAKYHGNVMLLRAAAGADYNLSQVCDGKVSVHVIEGDHATLLEGSGLESIISIIHSS Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Guanidinoacetate N-methyltransferase | 3ORH | 5.66 | |
Target general information Gen name GAMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, RMT2 methyltransferase family Biochemical class Transferase Function Guanidinoacetate N-methyltransferase activity.Methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 2 (CCDS2) [MIM:612736]: An autosomal recessive disorder characterized by developmental delay and regression, intellectual disability, severe disturbance of expressive and cognitive speech, intractable seizures, movement disturbances, severe depletion of creatine and phosphocreatine in the brain, and accumulation of guanidinoacetic acid in brain and body fluids. {ECO:0000269|PubMed:12468279, ECO:0000269|PubMed:15108290, ECO:0000269|PubMed:15651030, ECO:0000269|PubMed:16293431, ECO:0000269|PubMed:16855203, ECO:0000269|PubMed:17101918, ECO:0000269|PubMed:17466557, ECO:0000269|PubMed:19388150, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:24415674, ECO:0000269|PubMed:8651275}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00148; DB02751; DB00536; DB13191; DB01752 Interacts with O95363; Q969Q5; Q9HCM9-2 EC number 2.1.1.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Disease variant; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 24656 Length 219 Aromaticity 0.11 Instability index 46.5 Isoelectric point 5.91 Charge (pH=7) -4.34 2D Binding mode Binding energy (Kcal/mol) -7.72 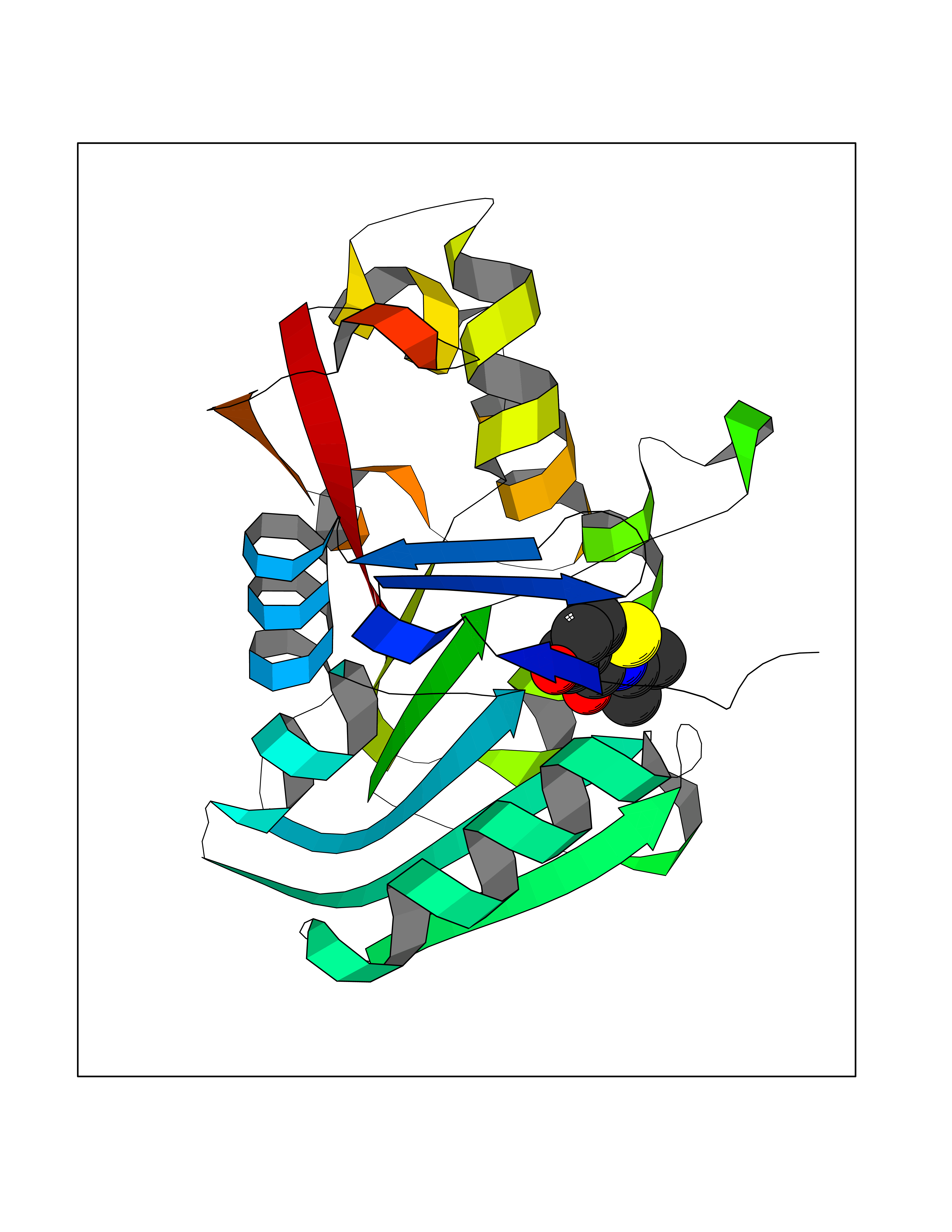 Molscript Map 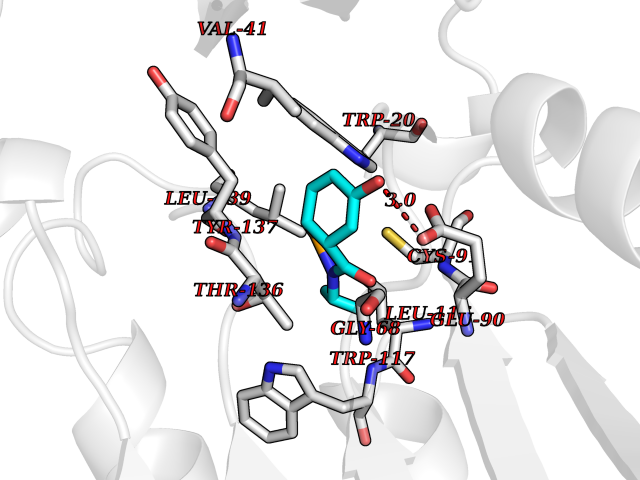 Pymol Map 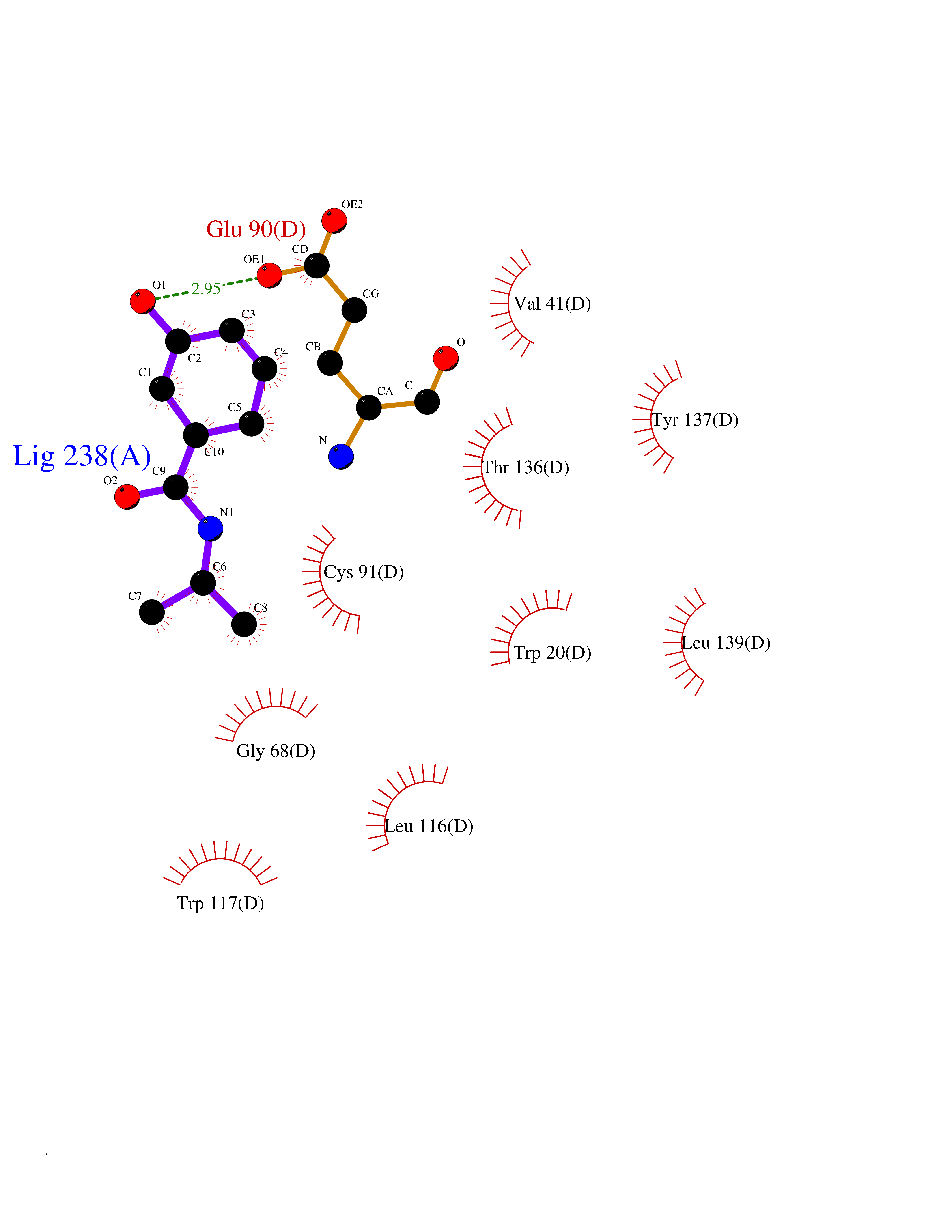 Ligplot Map 3D Binding mode Sequence PAWGAAPAAYDAADTHLRILGKPVMERWETPYMHALAAAASSKGGRVLEVGFGMAIAASKVQEAPIDEHWIIECNDGVFQRLRDWAPRQTHKVIPLKGLWEDVAPTLPDGHFDGILYDTYPLSEETWHTHQFNFIKNHAFRLLKPGGVLTYCNLTSWGELMKSKYSDITIMFEETQVPALLEAGFRRENIRTEVMALVPPADCRYYAFPQMITPLVTKG Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Opioid receptor sigma 1 (OPRS1) | 5HK1 | 5.66 | |
Target general information Gen name SIGMAR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hSigmaR1; Sigma1R; Sigma1-receptor; Sigma non-opioid intracellular receptor 1; Sigma 1-type opioid receptor; SRBP; SR31747-binding protein; SR31747 binding protein 1; SR-BP; SIG-1R; Opioid receptor, s Protein family ERG2 family Biochemical class GPCR rhodopsin Function Involved in the regulation of different receptors it plays a role in BDNF signaling and EGF signaling. Also regulates ion channels like the potassium channel and could modulate neurotransmitter release. Plays a role in calcium signaling through modulation together with ANK2 of the ITP3R-dependent calcium efflux at the endoplasmic reticulum. Plays a role in several other cell functions including proliferation, survival and death. Originally identified for its ability to bind various psychoactive drugs it is involved in learning processes, memory and mood alteration. Necessary for proper mitochondrial axonal transport in motor neurons, in particular the retrograde movement of mitochondria. Plays a role in protecting cells against oxidative stress-induced cell death via its interaction with RNF112. Functions in lipid transport from the endoplasmic reticulum and is involved in a wide array of cellular functions probably through regulation of the biogenesis of lipid microdomains at the plasma membrane. Related diseases Amyotrophic lateral sclerosis 16, juvenile (ALS16) [MIM:614373]: A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. {ECO:0000269|PubMed:21842496}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal recessive 2 (HMNR2) [MIM:605726]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. HMNR2 is characterized by onset of distal muscle weakness and wasting affecting the lower and upper limbs in the first decade. {ECO:0000269|PubMed:26078401, ECO:0000269|PubMed:27629094}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00321; DB09014; DB00907; DB00514; DB01488; DB00574; DB00502; DB00956; DB00704; DB00540; DB06174; DB00652; DB11186; DB03575; DB05316; DB01708; DB00409; DB01104 Interacts with Q92847-1; Q99720-1; O00213-2; P17612; P50454; P37173 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Amyotrophic lateral sclerosis; Cell junction; Cell membrane; Cell projection; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Lipid droplet; Lipid transport; Membrane; Neurodegeneration; Neuropathy; Nucleus; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 20805.3 Length 185 Aromaticity 0.14 Instability index 31.72 Isoelectric point 5.44 Charge (pH=7) -6.63 2D Binding mode Binding energy (Kcal/mol) -7.72 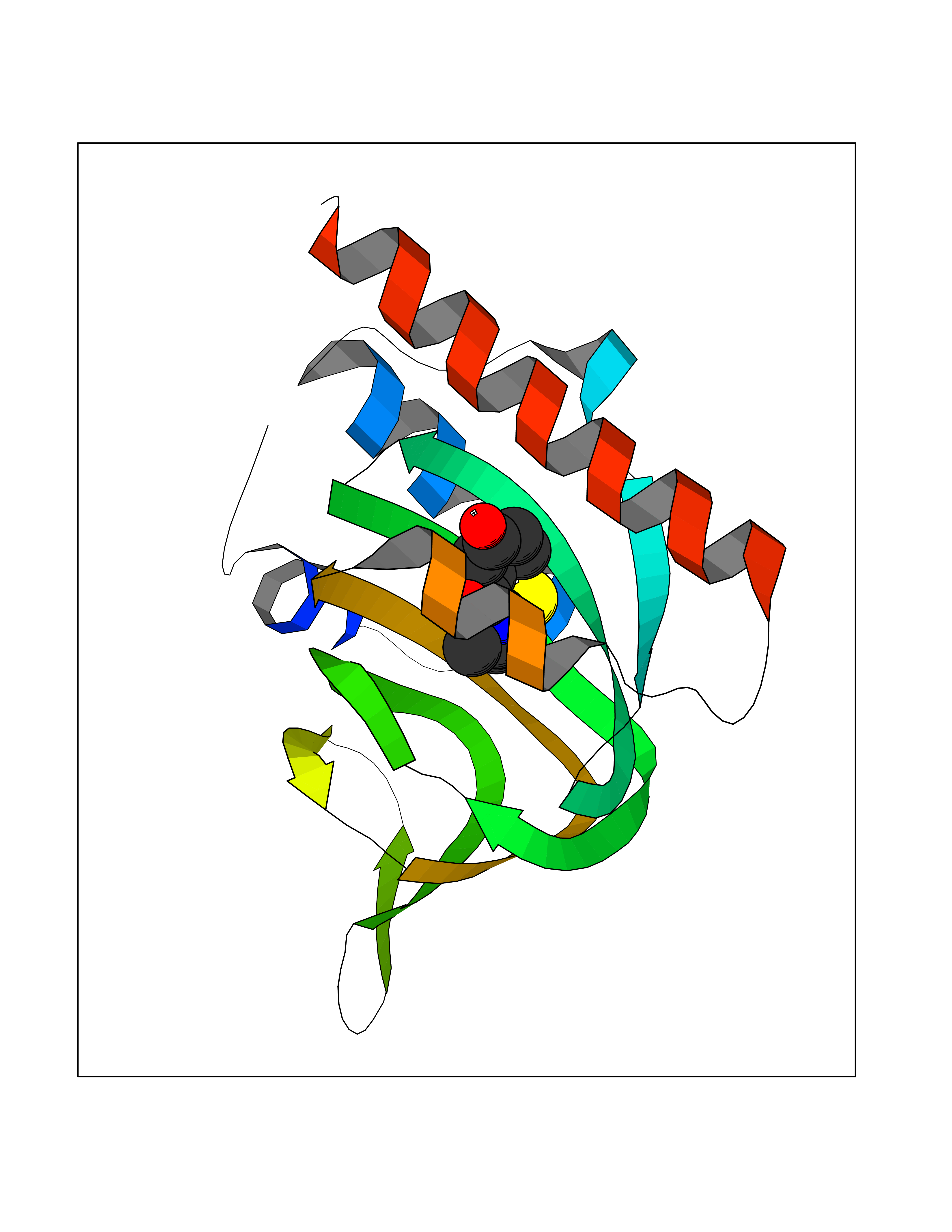 Molscript Map 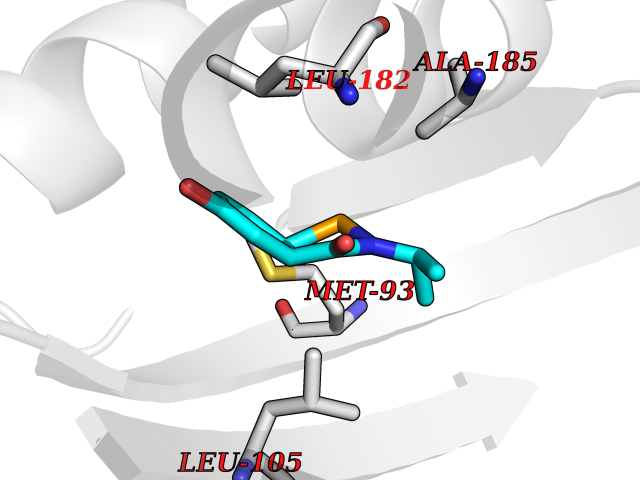 Pymol Map 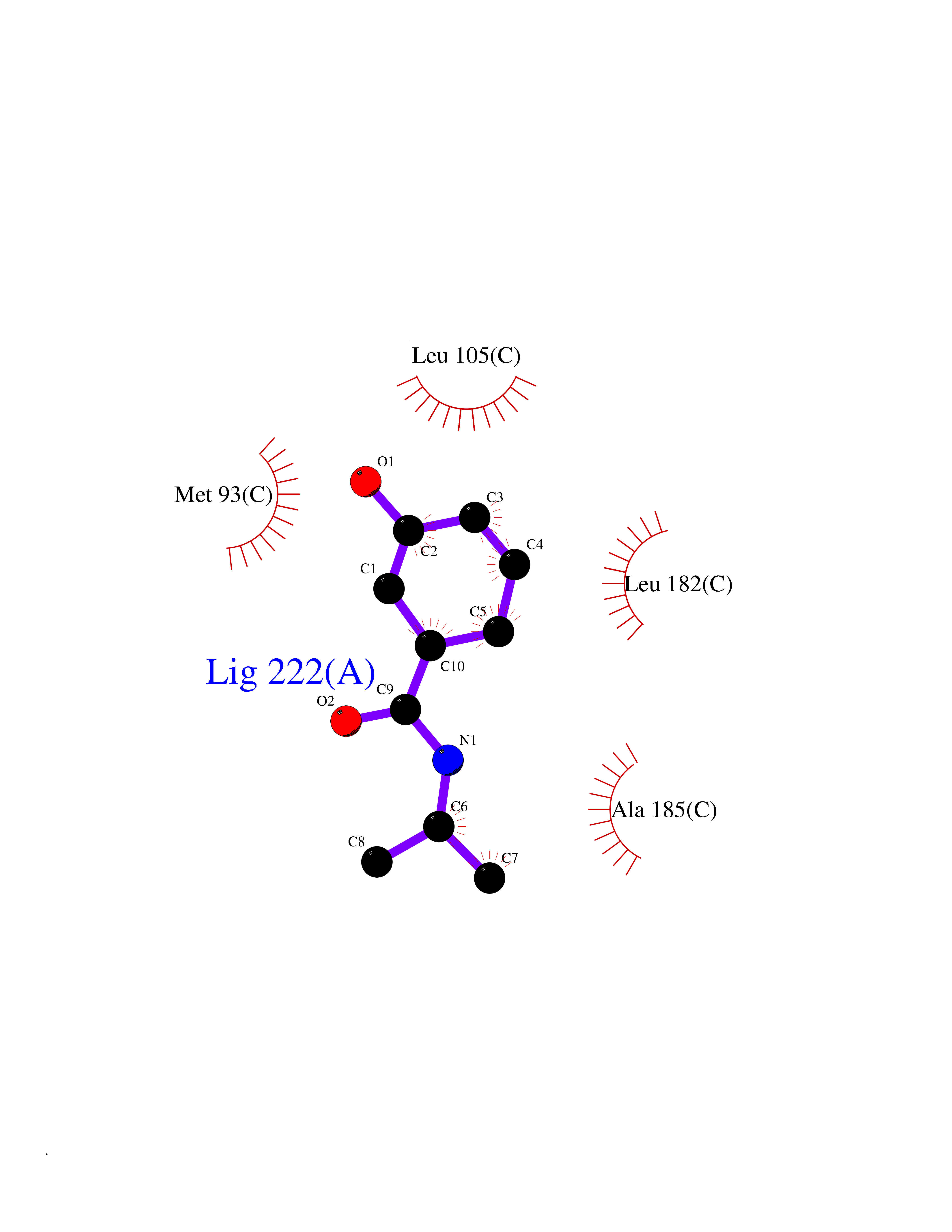 Ligplot Map 3D Binding mode Sequence VFQREEIAQLARQYAGLDHELAFSRLIVELRRLHPGHVLPDEELQWVFVNAGGWMGAMCLLHASLSEYVLLFGTALGSRGHSGRYWAEISDTIISGTFHQWREGTTKSEVFYPGETVVHGPGEATAVEWGPNTWMVEYGRGVIPSTLAFALADTVFSTQDFLTLFYTLRSYARGLRLELTTYLFG Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | MALT lymphoma-associated translocation (MALT1) | 7A41 | 5.66 | |
Target general information Gen name MALT1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MALT lymphoma-associated translocation; Paracaspase Protein family Peptidase C14B family Biochemical class Peptidase Function Protease that enhances BCL10-induced activation of NF-kappa-B by mediating its cleavage. MALT1-dependent BCL10 cleavage plays an important role in T-cell antigen receptor-induced integrin adhesion. Involved in the induction of T helper 17 cells (Th17) differentiation. Cleaves RC3H1 and ZC3H12A in response to T-cell receptor (TCR) stimulation which releases their cooperatively repressed targets to promote Th17 cell differentiation (By similarity). Also mediates cleavage of N4BP1 in T-cells following TCR-mediated activation, leading to N4BP1 inactivation. Also has ubiquitin ligase activity: binds to TRAF6, inducing TRAF6 oligomerization and activation of its ligase activity. Related diseases Immunodeficiency 12 (IMD12) [MIM:615468]: A primary immunodeficiency characterized by onset in infancy of recurrent bacterial and candidal infections resulting in bronchiectasis and growth delay. Manifestations include mastoiditis, aphthous ulcers, cheilitis, gingivitis, esophagitis, gastritis, duodenitis, and meningitis. Levels of absolute lymphocytes and serum immunoglobulins are normal, but specific antibody titers are low despite immunization, and T-cells show impaired proliferative responses to mitogens. {ECO:0000269|PubMed:23727036}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: A chromosomal aberration involving MALT1 is recurrent in low-grade mucosa-associated lymphoid tissue (MALT lymphoma). Translocation t(11;18)(q21;q21) with BIRC2. This translocation is found in approximately 50% of cytogenetically abnormal low-grade MALT lymphoma. {ECO:0000269|PubMed:10339464, ECO:0000269|PubMed:10523859, ECO:0000269|PubMed:10702396, ECO:0000269|PubMed:11090634}. Drugs (DrugBank ID) NA Interacts with O95999; Q9BXL7; Q14790; P48729; Q9Y6K9; Q9UDY8; Q96PU8; Q9H0F6; Q13501; Q9Y4K3; P0CG48; P54252; P46379-2; G5E9A7; P50570-2; Q9BSK4; Q96JP0; P28799; P04792; O60333-2; O14901; O14832; P60891; Q7Z699; O76024 EC number EC 3.4.22.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Chromosomal rearrangement; Cytoplasm; Disease variant; Disulfide bond; Hydrolase; Immunity; Immunoglobulin domain; Nucleus; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Repeat; Ubl conjugation pathway Protein physicochemical properties Chain ID A Molecular weight (Da) 39995.8 Length 354 Aromaticity 0.09 Instability index 25.55 Isoelectric point 5.12 Charge (pH=7) -11.76 2D Binding mode Binding energy (Kcal/mol) -7.72 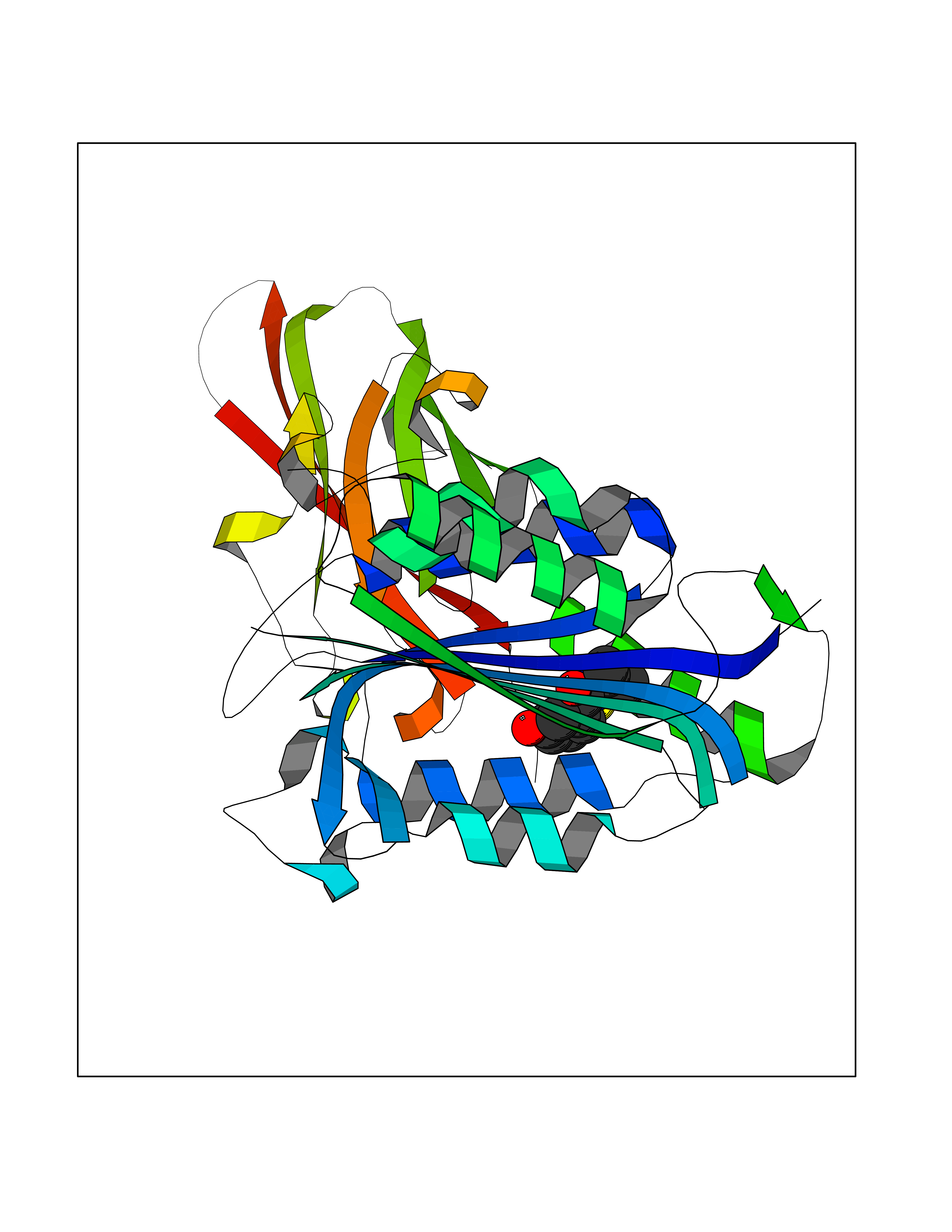 Molscript Map 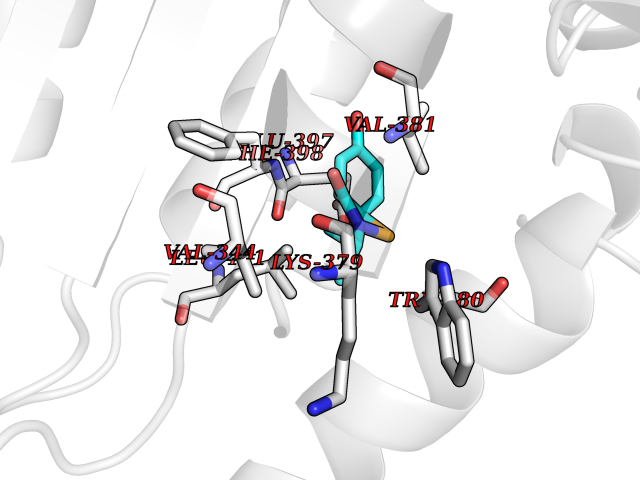 Pymol Map 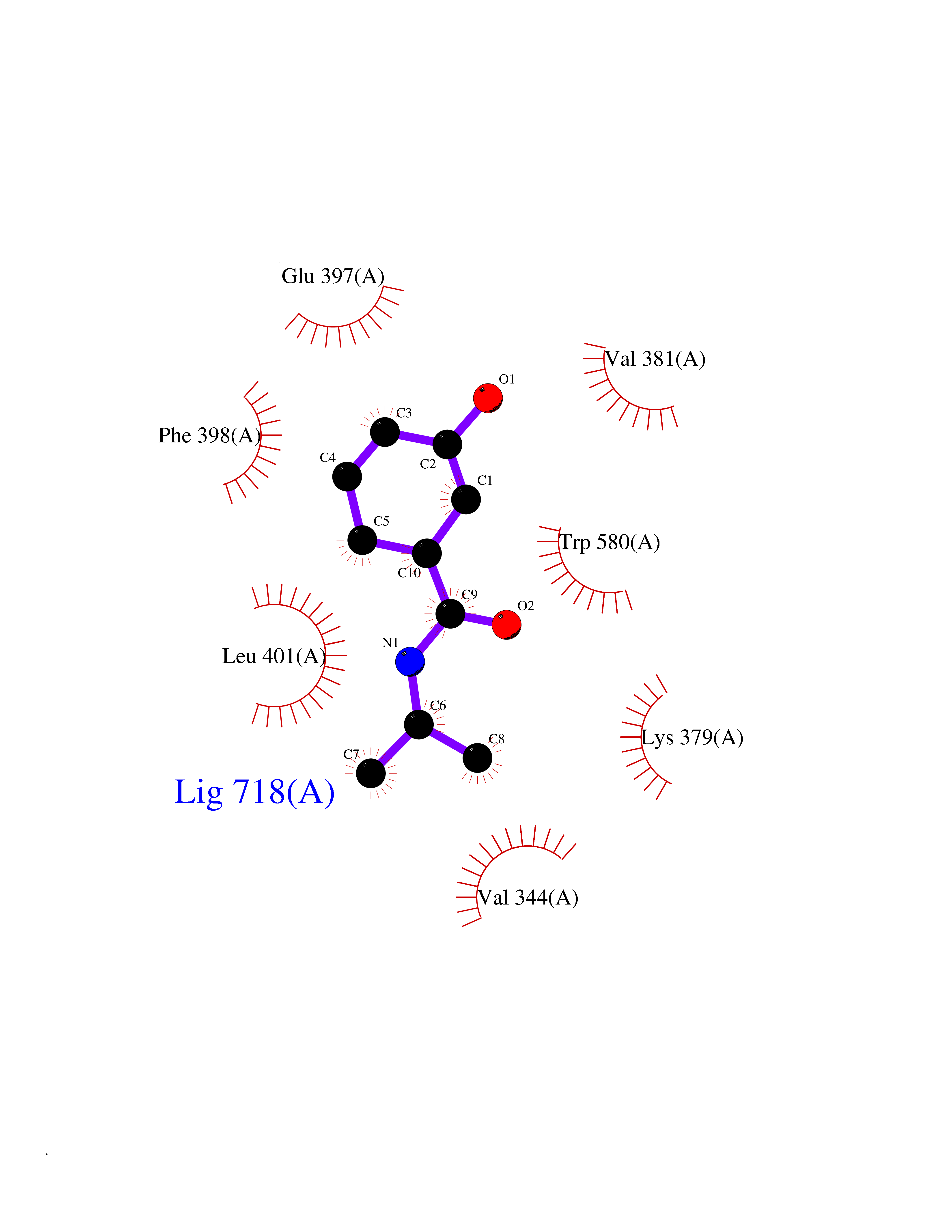 Ligplot Map 3D Binding mode Sequence QPLAKDKVALLIGNMNYREHPKLKAPLVDVYELTNLLRQLDFKVVSLLDLTEYEMRNAVDEFLLLLDKGVYGLLYYAGHGYENFGNSFMVPVDAPNPYRSENCLCVQNILKLMQEKETGLNVFLLDMCRTANIVFGYATCQGGLANGIFMKFLKDRLLEDKKITVLLDEVAEDMGKCHLTKGKQALEIRSSLSEKRALTDPIQGTEYSAESLVRNLQWAKAHELPESMCLKFDCGVQIQLGFAAEFSNVMIIYTSIVYKPPEIIMCDAYVTDFPLDLDIDPKDANKGTPEETGSYLVSKDLPKHCLYTRLSSLQKLKEHLVFTVCLSYQYSGLEDTVEDKQEVNVGKPLIAKLD Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Benzoate 1,2-dioxygenase electron transfer component | 1KRH | 5.65 | |
Target general information Gen name benC Organism Acinetobacter baylyi (strain ATCC 33305 / BD413 / ADP1) Uniprot ID TTD ID NA Synonyms ACIAD1438 Protein family Bacterial ring-hydroxylating dioxygenase ferredoxin reductase family Biochemical class Oxidoreductase Function 2 iron, 2 sulfur cluster binding.Electron carrier activity.Ferredoxin-NAD+ reductase activity.Metal ion binding. Related diseases Adenine phosphoribosyltransferase deficiency (APRTD) [MIM:614723]: An enzymatic deficiency that can lead to urolithiasis and renal failure. Patients have 2,8-dihydroxyadenine (DHA) urinary stones. {ECO:0000269|PubMed:11243733, ECO:0000269|PubMed:1353080, ECO:0000269|PubMed:15571218, ECO:0000269|PubMed:1746557, ECO:0000269|PubMed:21635362, ECO:0000269|PubMed:3343350, ECO:0000269|PubMed:3680503, ECO:0000269|PubMed:7915931}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.18.1.3 Uniprot keywords 2Fe-2S; 3D-structure; Aromatic hydrocarbons catabolism; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; NAD; Oxidoreductase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 37496.7 Length 337 Aromaticity 0.1 Instability index 47.02 Isoelectric point 4.75 Charge (pH=7) -18.87 2D Binding mode Binding energy (Kcal/mol) -7.71 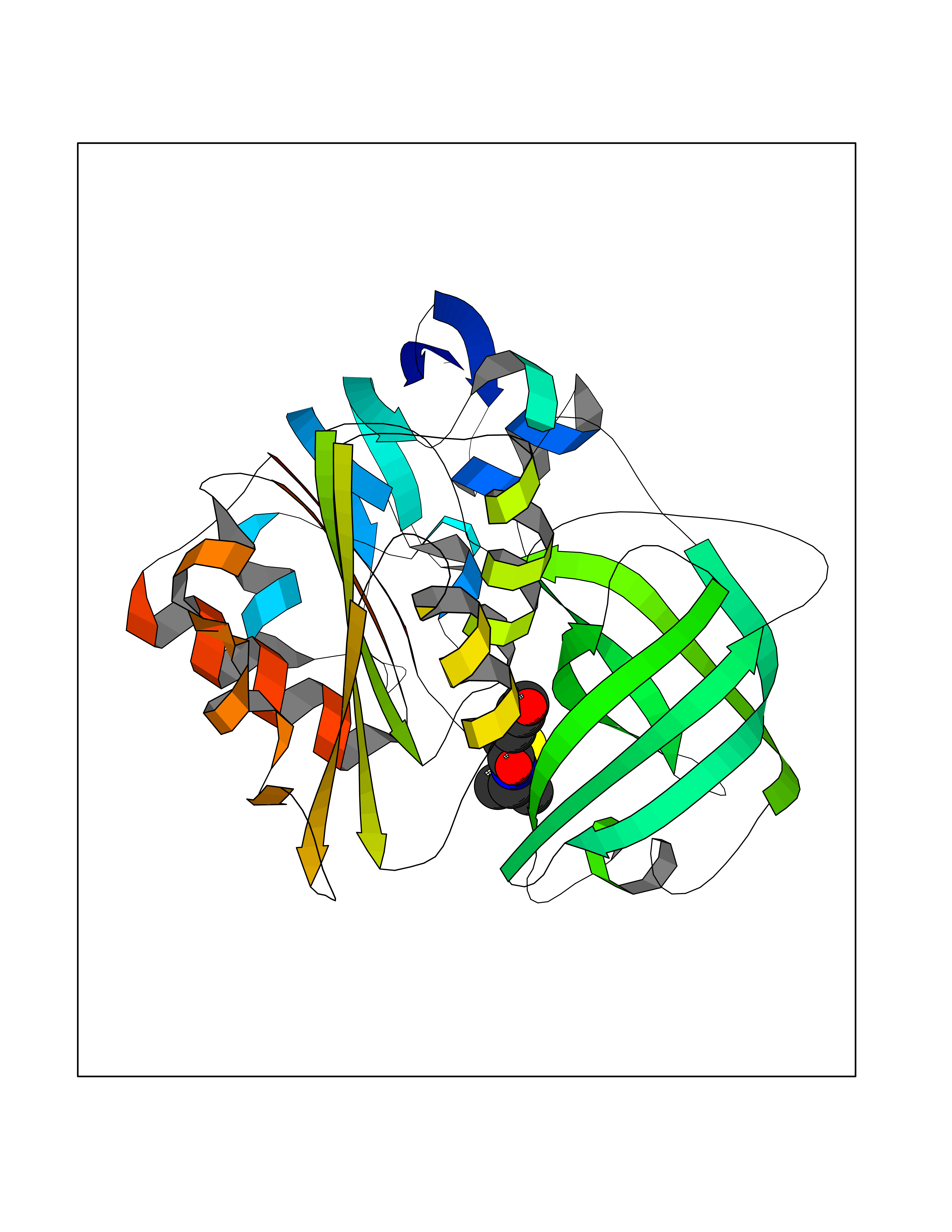 Molscript Map 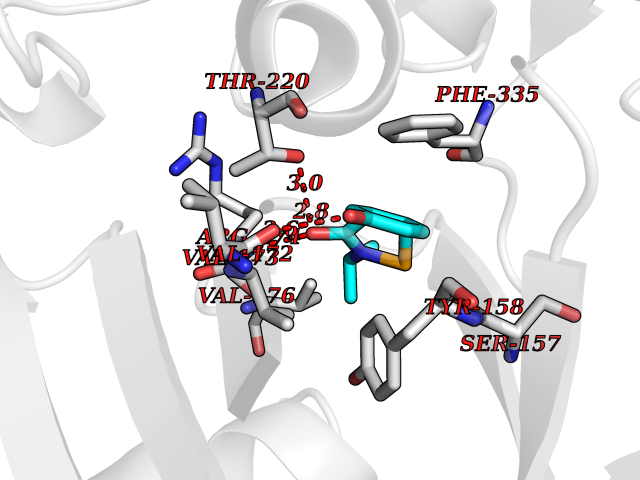 Pymol Map 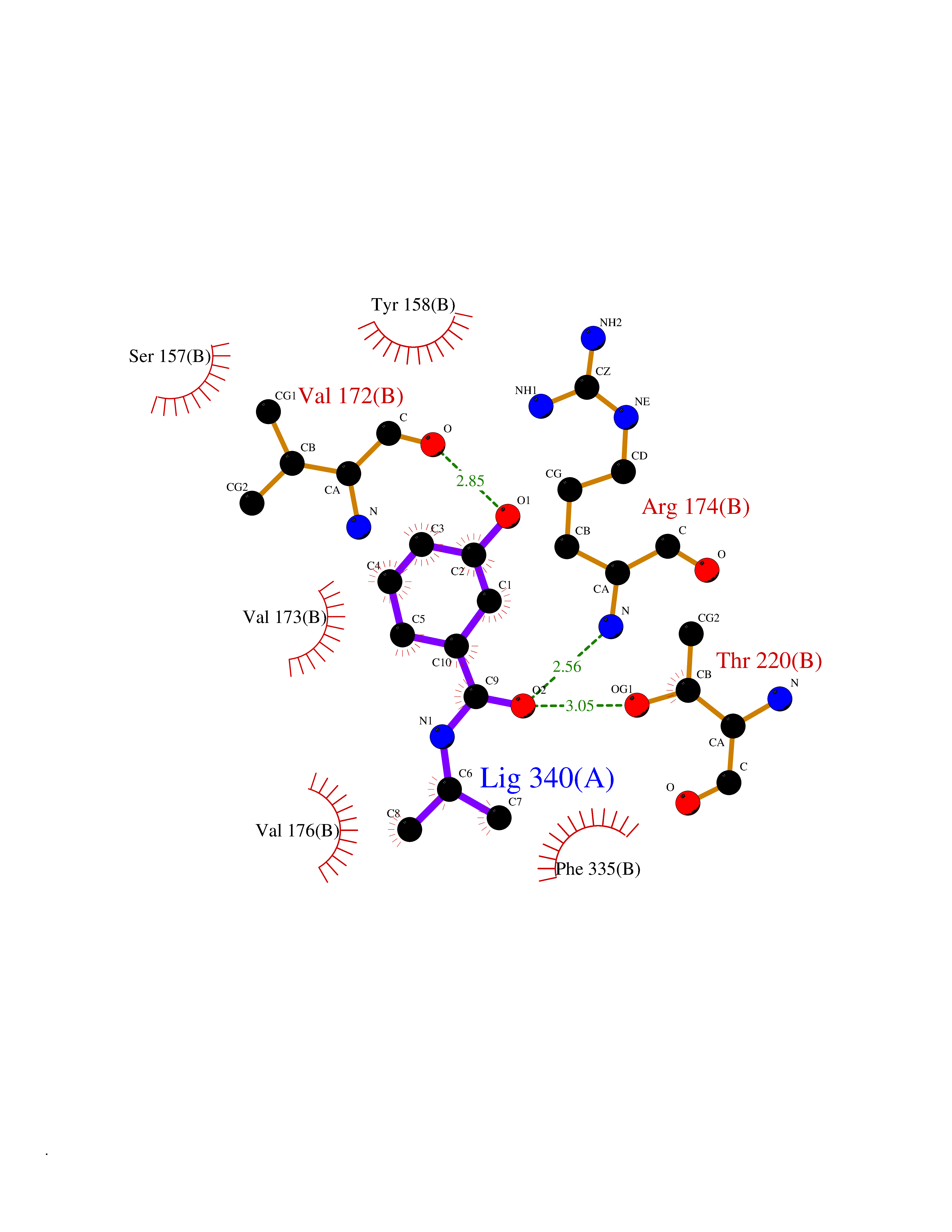 Ligplot Map 3D Binding mode Sequence SNHQVALQFEDGVTRFICIAQGETLSDAAYRQQINIPMDCREGECGTCRAFCESGNYDMPEDNYIEDALTPEEAQQGYVLACQCRPTSDAVFQIQASSEVCKTKIHHFEGTLARVENLSDSTITFDIQLDDGQPDIHFLAGQYVNVTLPGTTETRSYSFSSQPGNRLTGFVVRNVPQGKMSEYLSVQAKAGDKMSFTGPFGSFYLRDVKRPVLMLAGGTGIAPFLSMLQVLEQKGSEHPVRLVFGVTQDCDLVALEQLDALQQKLPWFEYRTVVAHAESQHERKGYVTGHIEYDWLNGGEVDVYLCGPVPMVEAVRSWLDTQGIQPANFLFEKFSAN Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Tyrosine aminotransferase | 3DYD | 5.65 | |
Target general information Gen name TAT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class-I pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transferase Function Amino acid binding.L-phenylalanine:2-oxoglutarate aminotransferase activity.L-tyrosine:2-oxoglutarate aminotransferase activity.Pyridoxal phosphate binding. Related diseases Tyrosinemia 2 (TYRSN2) [MIM:276600]: An inborn error of metabolism characterized by elevations of tyrosine in the blood and urine, and oculocutaneous manifestations. Typical features include palmoplantar keratosis, painful corneal ulcers, and intellectual disability. {ECO:0000269|PubMed:1357662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00142; DB00120; DB00114; DB00135 Interacts with P15104; P28799; P28799-2; P17735; Q05086; Q05086-3 EC number 2.6.1.5 Uniprot keywords 3D-structure; Acetylation; Aminotransferase; Disease variant; Intellectual disability; Palmoplantar keratoderma; Phenylalanine catabolism; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase; Tyrosine catabolism Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42209.5 Length 380 Aromaticity 0.08 Instability index 51.79 Isoelectric point 5.29 Charge (pH=7) -10.66 2D Binding mode Binding energy (Kcal/mol) -7.7 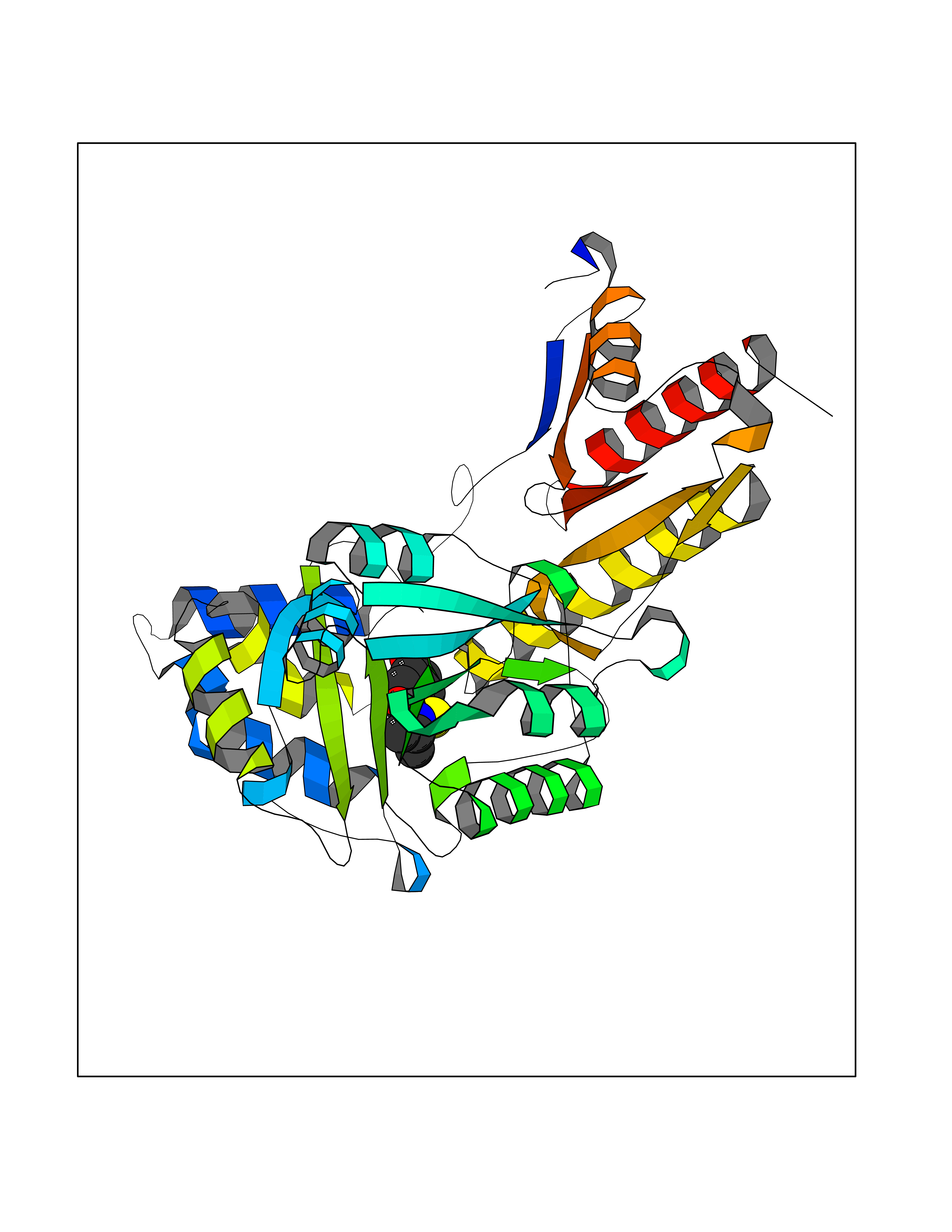 Molscript Map 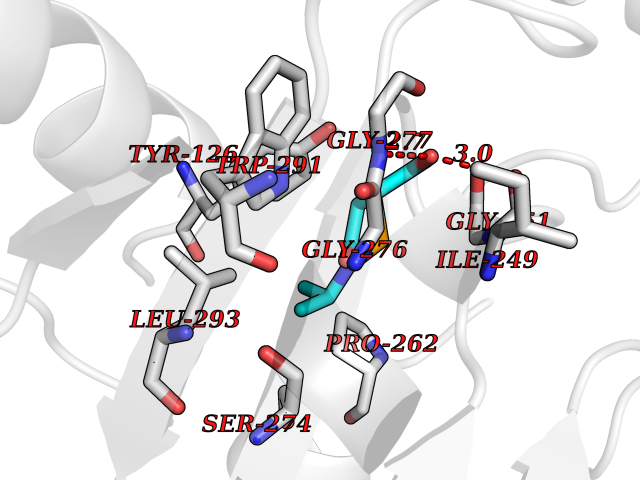 Pymol Map 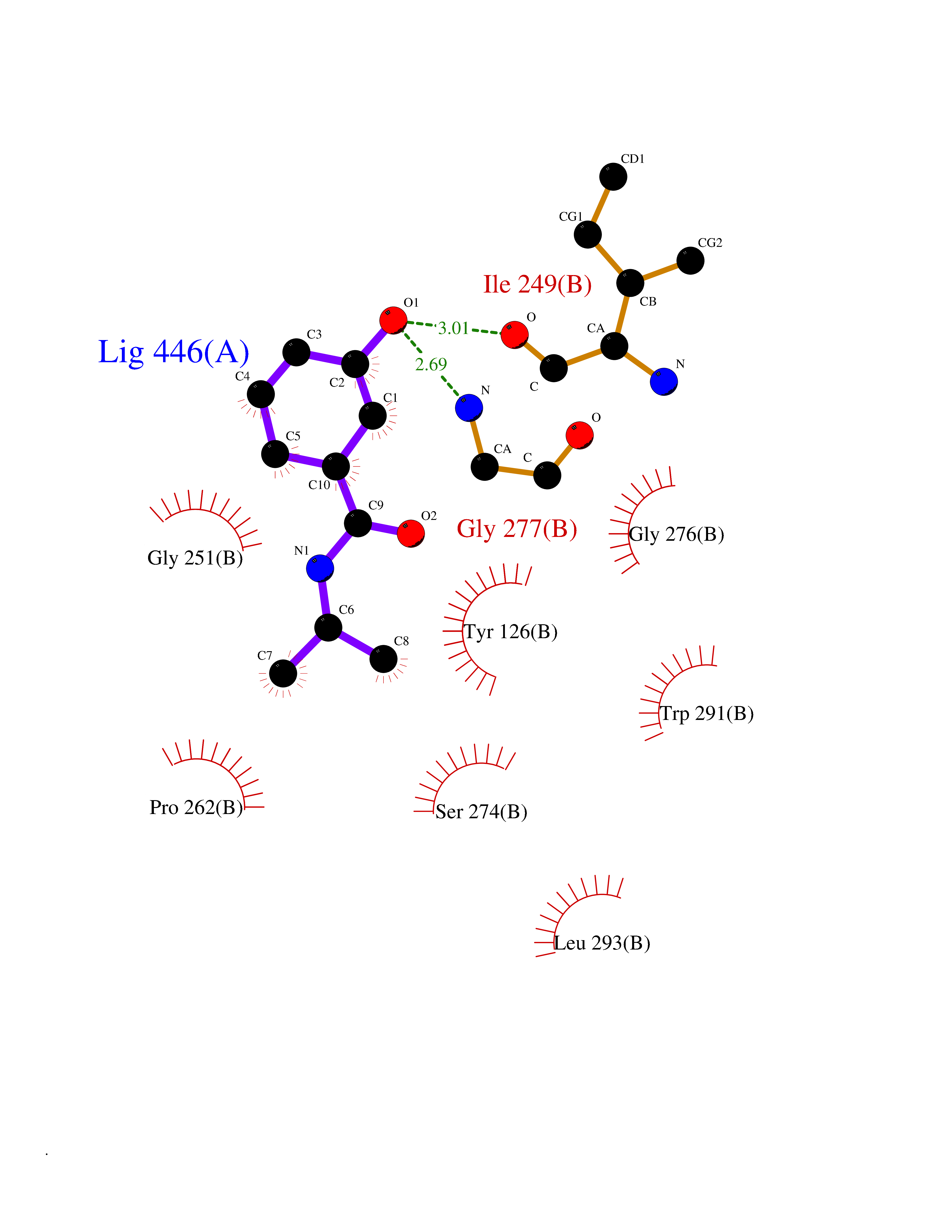 Ligplot Map 3D Binding mode Sequence VKPNPNKTMISLSIGDPTVFGNLPTDPEVTQAMKDALDSGKYNGYAPSIGFLSSREEIASYYHCPEAPLEAKDVILTSGCSQAIDLCLAVLANPGQNILVPRPGFSLYKTLAESMGIEVKLYNLLPEKSWEIDLKQLEYLIDEKTACLIVNNPSNPCGSVFSKRHLQKILAVAARQCVPILADEIYGDMVFSDCKYEPLATLSTDVPILSCGGLAKRWLVPGWRLGWILIHDRRDIFGNEIRDGLVKLSQRILGPCTIVQGALKSILCRTPGEFYHNTLSFLKSNADLCYGALAAIPGLRPVRPSGAMYLMVGIEMEHFPEFENDVEFTERLVAEQSVHCLPATCFEYPNFIRVVITVPEVMMLEACSRIQEFCEQHYHC Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Pseudomonas Methionine gamma-lyase (Pseudo mdeA) | 1PG8 | 5.65 | |
Target general information Gen name Pseudo mdeA Organism Pseudomonas putida (Arthrobacter siderocapsulatus) Uniprot ID TTD ID Synonyms Pseudo MGL; L-methionine gamma-lyase; L-methioninase; Homocysteine desulfhydrase Protein family Trans-sulfuration enzymes family, L-methionine gamma-lyase subfamily Biochemical class Carbon-sulfur lyases Function Catalyzes the alpha,gamma-elimination of L-methionine to produce methanethiol, 2-oxobutanoate and ammonia. Is involved in L-methionine catabolism. In fact, shows a multicatalytic function since it also catalyzes gamma-replacement of L-methionine with thiol compounds, alpha,gamma-elimination and gamma-replacement reactions of L-homocysteine and its S-substituted derivatives, O-substituted-L-homoserines and DL-selenomethionine, and, to a lesser extent, alpha,beta-elimination and beta-replacement reactions of L-cysteine, S-methyl-L-cysteine, and O-acetyl-L-serine. Also catalyzes deamination and gamma-addition reactions of L-vinylglycine. Thus, the enzyme is able to cleave C-S, C-Se, and C-O bonds of sulfur, selenium, and oxygen amino acids, respectively. Related diseases Lecithin-cholesterol acyltransferase deficiency (LCATD) [MIM:245900]: A disorder of lipoprotein metabolism characterized by inadequate esterification of plasmatic cholesterol. Two clinical forms are recognized: complete LCAT deficiency and fish-eye disease. LCATD is generally referred to the complete form which is associated with absence of both alpha and beta LCAT activities resulting in esterification anomalies involving both HDL (alpha-LCAT activity) and LDL (beta-LCAT activity). It causes a typical triad of diffuse corneal opacities, target cell hemolytic anemia, and proteinuria with renal failure. {ECO:0000269|PubMed:11423760, ECO:0000269|PubMed:12957688, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:16051254, ECO:0000269|PubMed:16216249, ECO:0000269|PubMed:1681161, ECO:0000269|PubMed:1859405, ECO:0000269|PubMed:2370048, ECO:0000269|PubMed:7607641, ECO:0000269|PubMed:7711728, ECO:0000269|PubMed:8318557, ECO:0000269|PubMed:8432868, ECO:0000269|PubMed:8807342, ECO:0000269|PubMed:9007616, ECO:0000269|PubMed:9741700}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fish-eye disease (FED) [MIM:136120]: A disorder of lipoprotein metabolism due to partial lecithin-cholesterol acyltransferase deficiency that affects only alpha-LCAT activity. FED is characterized by low plasma HDL and corneal opacities due to accumulation of cholesterol deposits in the cornea ('fish-eye'). {ECO:0000269|PubMed:1516702, ECO:0000269|PubMed:1571050, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:1737840, ECO:0000269|PubMed:21901787, ECO:0000269|PubMed:8620346, ECO:0000269|PubMed:9261271}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04083 Interacts with NA EC number EC 4.4.1.11 Uniprot keywords 3D-structure; Direct protein sequencing; Lyase; Pyridoxal phosphate Protein physicochemical properties Chain ID A,C Molecular weight (Da) 85234.4 Length 796 Aromaticity 0.07 Instability index 37.1 Isoelectric point 6.21 Charge (pH=7) -11.34 2D Binding mode Binding energy (Kcal/mol) -7.7 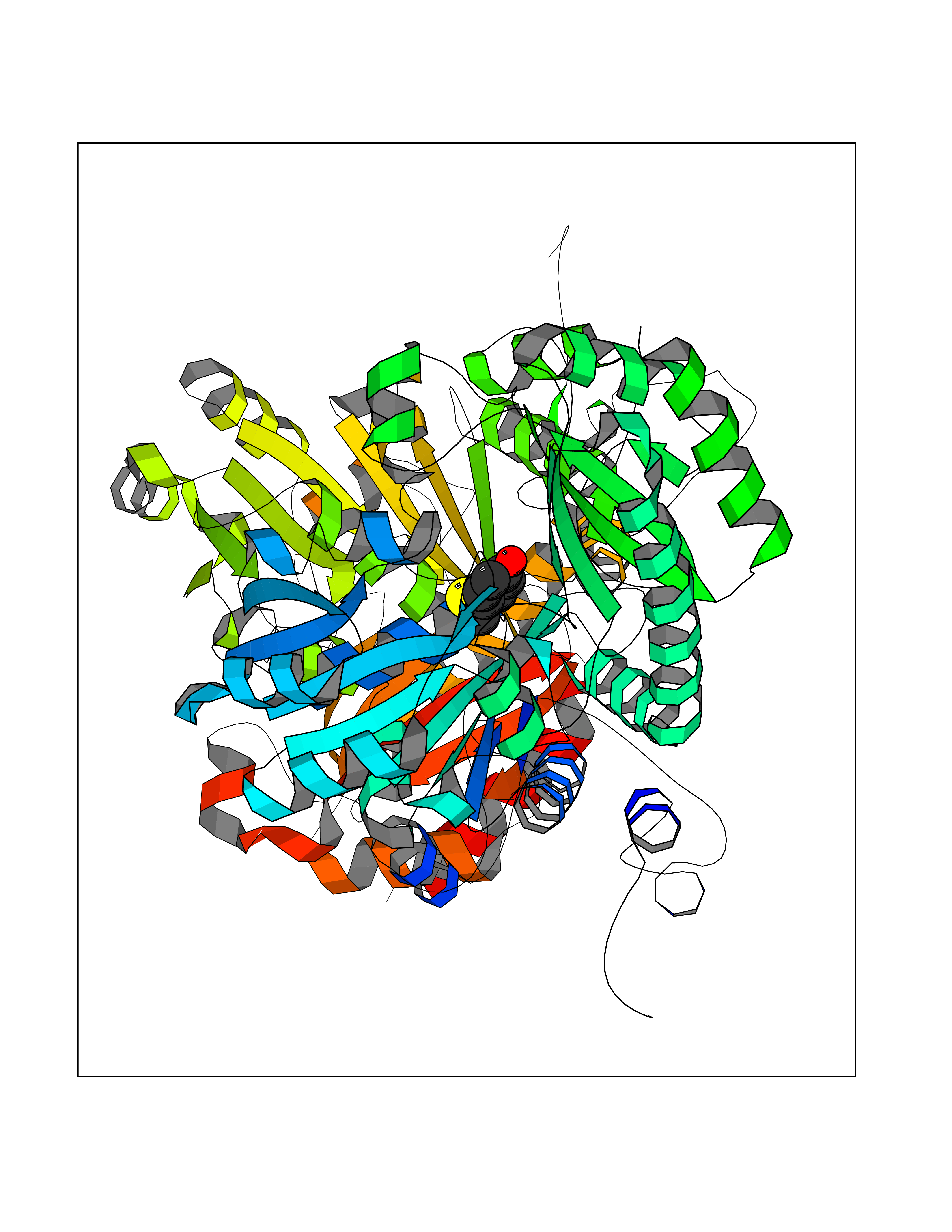 Molscript Map 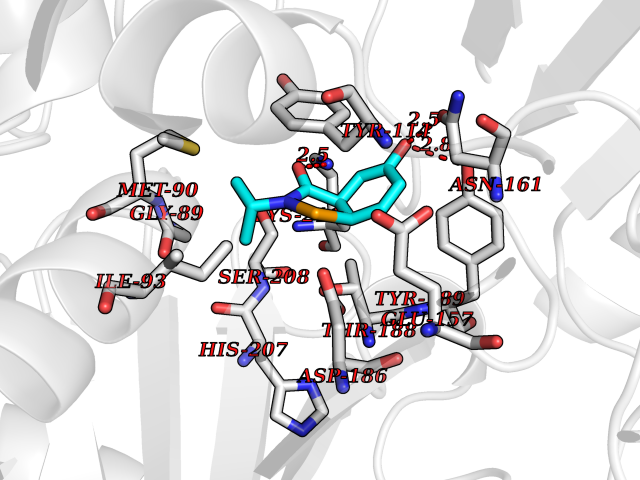 Pymol Map 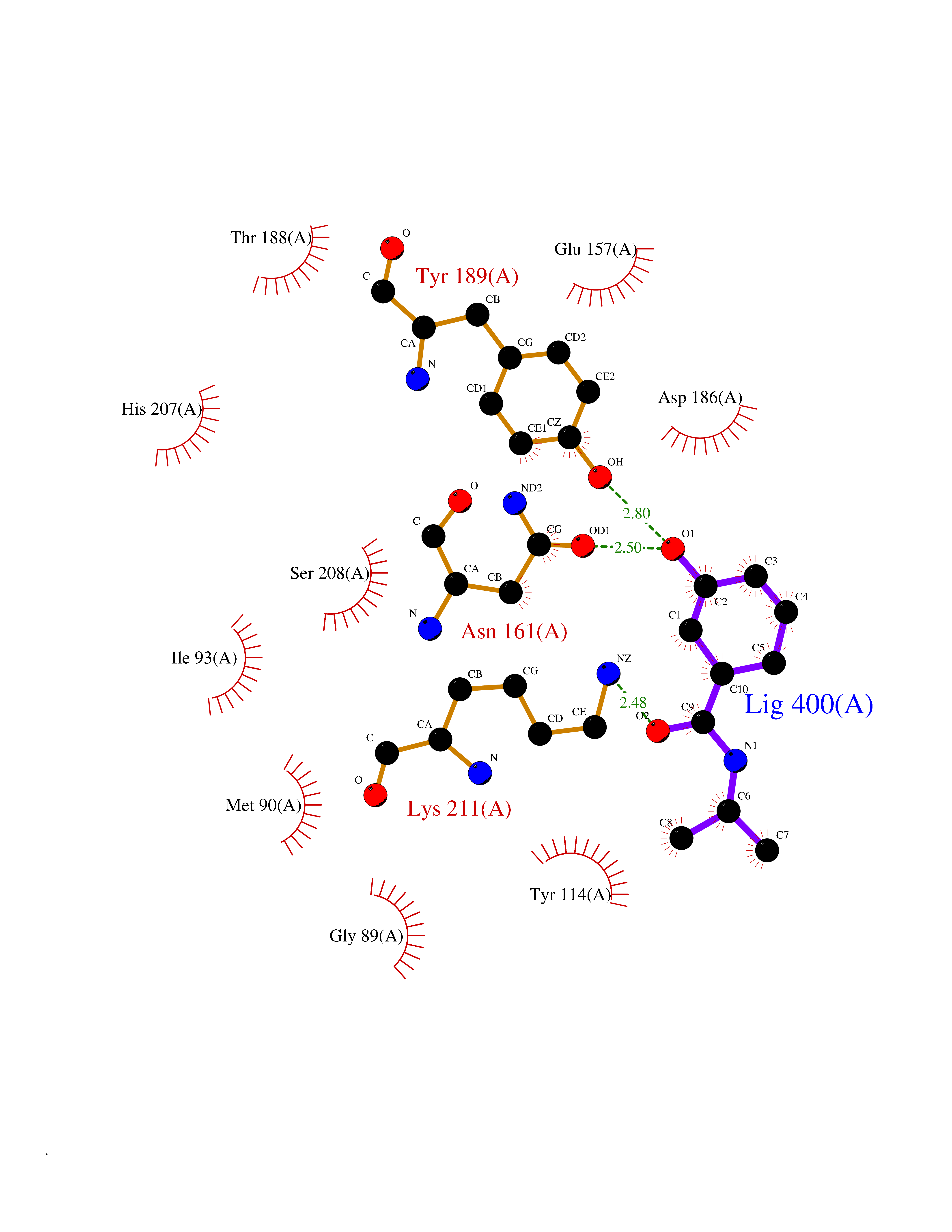 Ligplot Map 3D Binding mode Sequence MHGSNKLPGFATRAIHHGYDPQDHGGALVPPVYQTATFTFPTVEYGAACFAGEQAGHFYSRISNPTLNLLEARMASLEGGEAGLALASGMGAITSTLWTLLRPGDEVLLGNTLYGCTFAFLHHGIGEFGVKLRHVDMADLQALEAAMTPATRVIYFESPANPNMHMADIAGVAKIARKHGATVVVDNTYCTPYLQRPLELGADLVVHSATKYLSGHGDITAGIVVGSQALVDRIRLQGLKDMTGAVLSPHDAALLMRGIKTLNLRMDRHCANAQVLAEFLARQPQVELIHYPGLASFPQYTLARQQMSQPGGMIAFELKGGIGAGRRFMNALQLFSRAVSLGDAESLAQHPASMTHSSYTPEERAHYGISEGLVRLSVGLEDIDDLLADVQQALKASAMHGSNKLPGFATRAIHHGYDPQDHGGALVPPVYQTATFTFPTVEYGAACFAGEQAGHFYSRISNPTLNLLEARMASLEGGEAGLALASGMGAITSTLWTLLRPGDEVLLGNTLYGCTFAFLHHGIGEFGVKLRHVDMADLQALEAAMTPATRVIYFESPANPNMHMADIAGVAKIARKHGATVVVDNTYCTPYLQRPLELGADLVVHSATKYLSGHGDITAGIVVGSQALVDRIRLQGLKDMTGAVLSPHDAALLMRGIKTLNLRMDRHCANAQVLAEFLARQPQVELIHYPGLASFPQYTLARQQMSQPGGMIAFELKGGIGAGRRFMNALQLFSRAVSLGDAESLAQHPASMTHSSYTPEERAHYGISEGLVRLSVGLEDIDDLLADVQQALKASA Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Vitamin K epoxide reductase complex 1 (VKORC1) | 6WV3 | 5.65 | |
Target general information Gen name VKORC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin K1 2,3-epoxide reductase subunit 1; VKORC1; VKOR; UNQ308/PRO351; MSTP576; MSTP134 Protein family VKOR family Biochemical class Short-chain dehydrogenases reductase Function Involved invitamin K metabolism. Catalytic subunit of the vitamin K epoxide reductase (VKOR) complex which reduces inactive vitamin K 2,3-epoxide to active vitamin K. Vitamin K is required for the gamma-carboxylation of various proteins, including clotting factors, and is required for normal blood coagulation, but also for normal bone development. Related diseases Combined deficiency of vitamin K-dependent clotting factors 2 (VKCFD2) [MIM:607473]: VKCFD leads to a bleeding tendency that is usually reversed by oral administration of vitamin K. {ECO:0000269|PubMed:14765194, ECO:0000269|PubMed:16270630}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Coumarin resistance (CMRES) [MIM:122700]: A condition characterized by partial or complete resistance to warfarin or other 4-hydroxycoumarin derivatives. These drugs are used as anti-coagulants for the prevention of thromboembolic diseases in subjects with deep vein thrombosis, atrial fibrillation, or mechanical heart valve replacement. {ECO:0000269|PubMed:14765194, ECO:0000269|PubMed:20946155}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01418; DB00266; DB09332; DB00170; DB00498; DB00946; DB01022; DB00682 Interacts with Q13323; Q7Z7G2; Q96BA8; Q9Y282; Q5JX71; Q96KR6; Q5T7V8; Q8TDT2; Q9NQG1; P15941-11; Q96TC7; Q9NR31; A0A0S2Z4U3; Q8TBB6; O15393-2; Q19QW4 EC number EC 1.17.4.4 Uniprot keywords 3D-structure; Alternative splicing; Disease variant; Disulfide bond; Endoplasmic reticulum; Membrane; Oxidoreductase; Proteomics identification; Quinone; Redox-active center; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 42656.4 Length 381 Aromaticity 0.1 Instability index 32.12 Isoelectric point 7.73 Charge (pH=7) 1.93 2D Binding mode Binding energy (Kcal/mol) -7.7 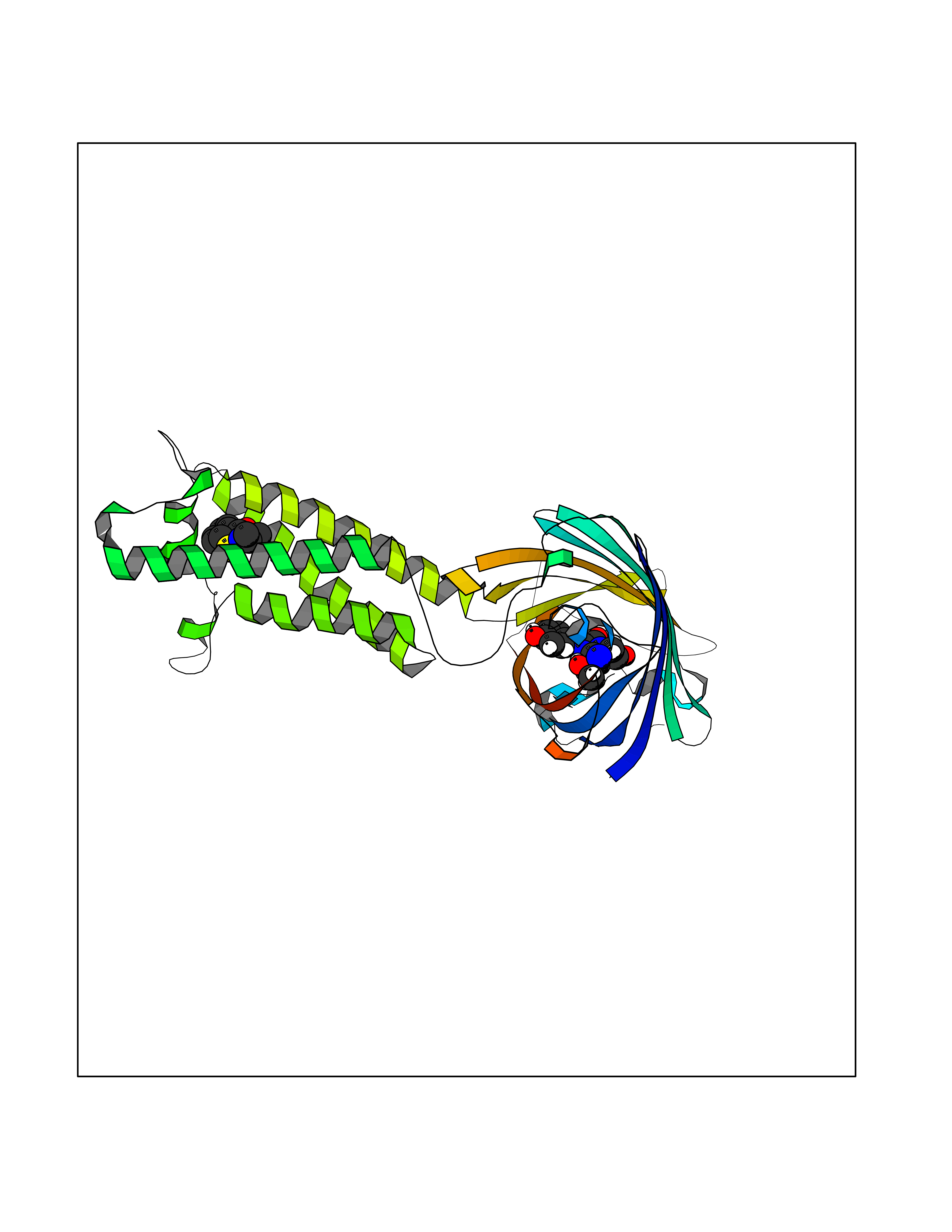 Molscript Map  Pymol Map 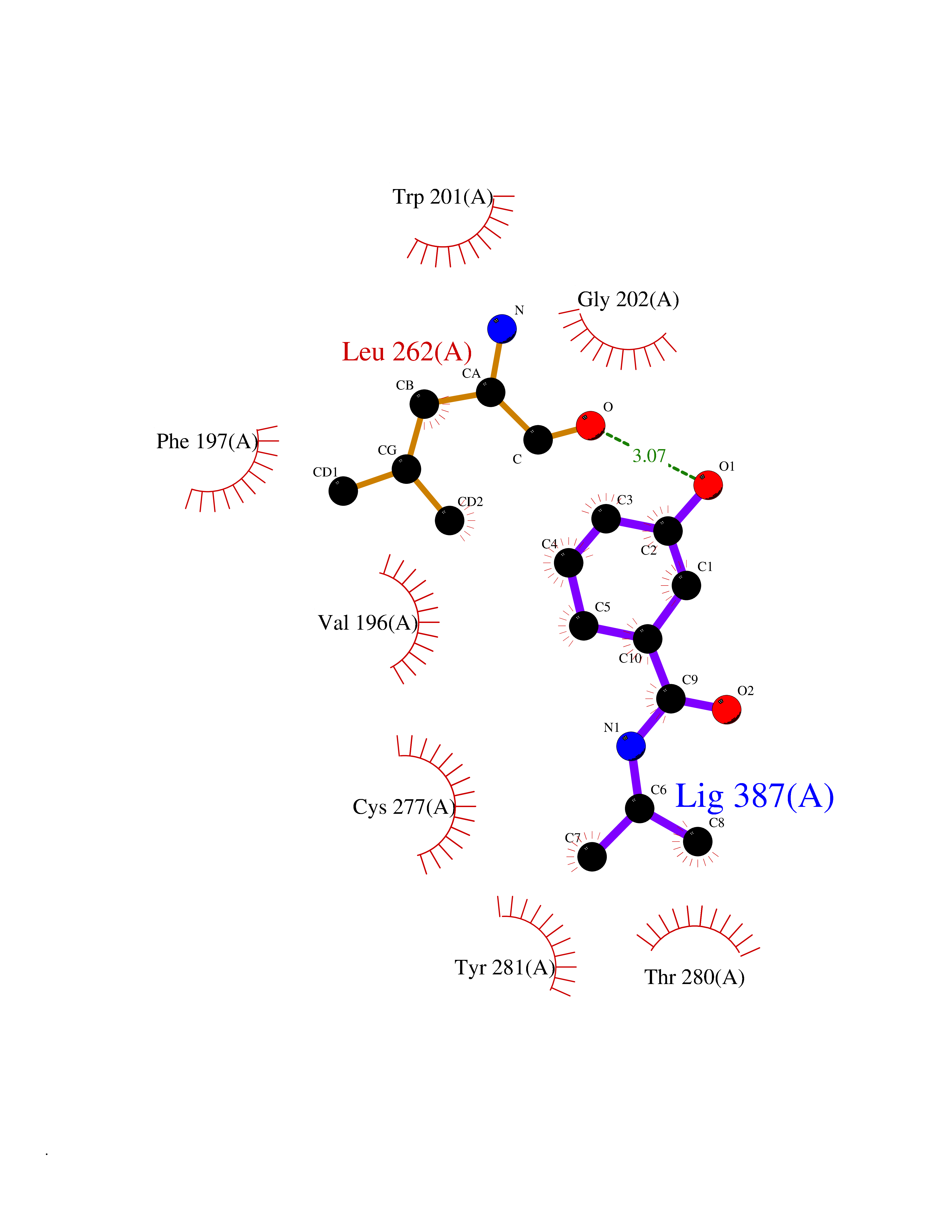 Ligplot Map 3D Binding mode Sequence KGEELFTGVVPILVELDGDVNGHKFSVRGEGEGDATNGKLTLKFICTTGKLPVPWPTLVTTLXVQCFSRYPDHMKRHDFFKSAMPEGYVQERTISFKDDGTYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNSTWGSPGWVRLALCLTGLVLSLYALHVKAARARDRDYRALCDVGTAISCSRVFSSRWGRGFGLVEHVLGQDSILNQSNSIFGCIFYTLQLLLGCLRTRWASVLMLLSSLVSLAGSVYLAWILFFVLYDFCIVCITTYAINVSLMWLSFRKVQENSHNVYITADKQKNGIKANFKIRHNVEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSVLSKDPNEKRDHMVLLEFVTAAGITHHH Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Pyruvate dehydrogenase kinase 1 (PDHK1) | 2Q8G | 5.65 | |
Target general information Gen name PDK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Pyruvate dehydrogenase kinase isoform 1; Pyruvate dehydrogenase (acetyl-transferring) kinase isozyme 1, mitochondrial; PDHK1; PDH kinase 1 Protein family PDK/BCKDK protein kinase family Biochemical class Kinase Function Kinase that plays a key role in regulation of glucose and fatty acid metabolism and homeostasis via phosphorylation of the pyruvate dehydrogenase subunits PDHA1 and PDHA2. This inhibits pyruvate dehydrogenase activity, and thereby regulates metabolite flux through the tricarboxylic acid cycle, down-regulates aerobic respiration and inhibits the formation of acetyl-coenzyme A from pyruvate. Plays an important role in cellular responses to hypoxia and is important for cell proliferation under hypoxia. Protects cells against apoptosis in response to hypoxia and oxidative stress. Related diseases TP53 is found in increased amounts in a wide variety of transformed cells. TP53 is frequently mutated or inactivated in about 60% of cancers. TP53 defects are found in Barrett metaplasia a condition in which the normally stratified squamous epithelium of the lower esophagus is replaced by a metaplastic columnar epithelium. The condition develops as a complication in approximately 10% of patients with chronic gastroesophageal reflux disease and predisposes to the development of esophageal adenocarcinoma.; DISEASE: Esophageal cancer (ESCR) [MIM:133239]: A malignancy of the esophagus. The most common types are esophageal squamous cell carcinoma and adenocarcinoma. Cancer of the esophagus remains a devastating disease because it is usually not detected until it has progressed to an advanced incurable stage. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Li-Fraumeni syndrome (LFS) [MIM:151623]: An autosomal dominant familial cancer syndrome that in its classic form is defined by the existence of a proband affected by a sarcoma before 45 years with a first degree relative affected by any tumor before 45 years and another first degree relative with any tumor before 45 years or a sarcoma at any age. Other clinical definitions for LFS have been proposed and called Li-Fraumeni like syndrome (LFL). In these families affected relatives develop a diverse set of malignancies at unusually early ages. Four types of cancers account for 80% of tumors occurring in TP53 germline mutation carriers: breast cancers, soft tissue and bone sarcomas, brain tumors (astrocytomas) and adrenocortical carcinomas. Less frequent tumors include choroid plexus carcinoma or papilloma before the age of 15, rhabdomyosarcoma before the age of 5, leukemia, Wilms tumor, malignant phyllodes tumor, colorectal and gastric cancers. {ECO:0000269|PubMed:10484981, ECO:0000269|PubMed:1565144, ECO:0000269|PubMed:1737852, ECO:0000269|PubMed:1933902, ECO:0000269|PubMed:1978757, ECO:0000269|PubMed:2259385, ECO:0000269|PubMed:36108750, ECO:0000269|PubMed:7887414, ECO:0000269|PubMed:8825920, ECO:0000269|PubMed:9452042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Squamous cell carcinoma of the head and neck (HNSCC) [MIM:275355]: A non-melanoma skin cancer affecting the head and neck. The hallmark of cutaneous SCC is malignant transformation of normal epidermal keratinocytes. The gene represented in this entry is involved in disease pathogenesis.; DISEASE: Lung cancer (LNCR) [MIM:211980]: A common malignancy affecting tissues of the lung. The most common form of lung cancer is non-small cell lung cancer (NSCLC) that can be divided into 3 major histologic subtypes: squamous cell carcinoma, adenocarcinoma, and large cell lung cancer. NSCLC is often diagnosed at an advanced stage and has a poor prognosis. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Papilloma of choroid plexus (CPP) [MIM:260500]: A benign tumor of neuroectodermal origin that generally occurs in childhood, but has also been reported in adults. Although generally found within the ventricular system, choroid plexus papillomas can arise ectopically in the brain parenchyma or disseminate throughout the neuraxis. Patients present with signs and symptoms of increased intracranial pressure including headache, hydrocephalus, papilledema, nausea, vomiting, cranial nerve deficits, gait impairment, and seizures. {ECO:0000269|PubMed:12085209}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Adrenocortical carcinoma (ADCC) [MIM:202300]: A malignant neoplasm of the adrenal cortex and a rare childhood tumor. It occurs with increased frequency in patients with Beckwith-Wiedemann syndrome and Li-Fraumeni syndrome. {ECO:0000269|PubMed:11481490}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Basal cell carcinoma 7 (BCC7) [MIM:614740]: A common malignant skin neoplasm that typically appears on hair-bearing skin, most commonly on sun-exposed areas. It is slow growing and rarely metastasizes, but has potentialities for local invasion and destruction. It usually develops as a flat, firm, pale area that is small, raised, pink or red, translucent, shiny, and waxy, and the area may bleed following minor injury. Tumor size can vary from a few millimeters to several centimeters in diameter. {ECO:0000269|PubMed:21946351}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Bone marrow failure syndrome 5 (BMFS5) [MIM:618165]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFS5 is an autosomal dominant form characterized by infantile onset of severe red cell anemia requiring transfusion. Additional features include hypogammaglobulinemia, poor growth with microcephaly, developmental delay, and seizures. {ECO:0000269|PubMed:30146126}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07403; DB08809 Interacts with P05067; P08559; Q16513; P31749-1; P31751-1 EC number EC 2.7.11.2 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Kinase; Mitochondrion; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Transit peptide Protein physicochemical properties Chain ID A Molecular weight (Da) 42249.9 Length 368 Aromaticity 0.11 Instability index 49.91 Isoelectric point 6.83 Charge (pH=7) -0.46 2D Binding mode Binding energy (Kcal/mol) -7.7  Molscript Map 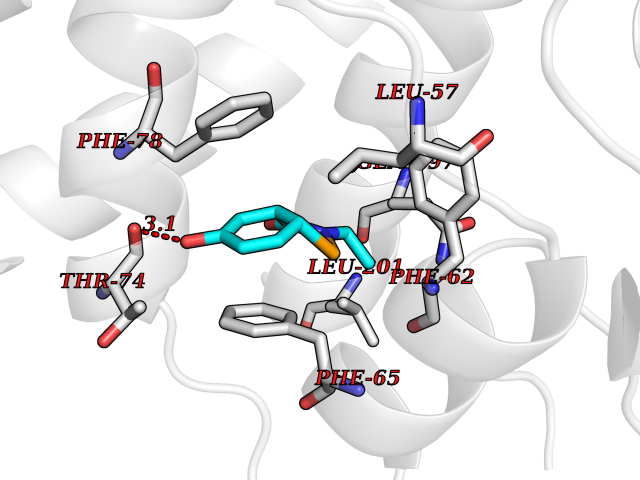 Pymol Map 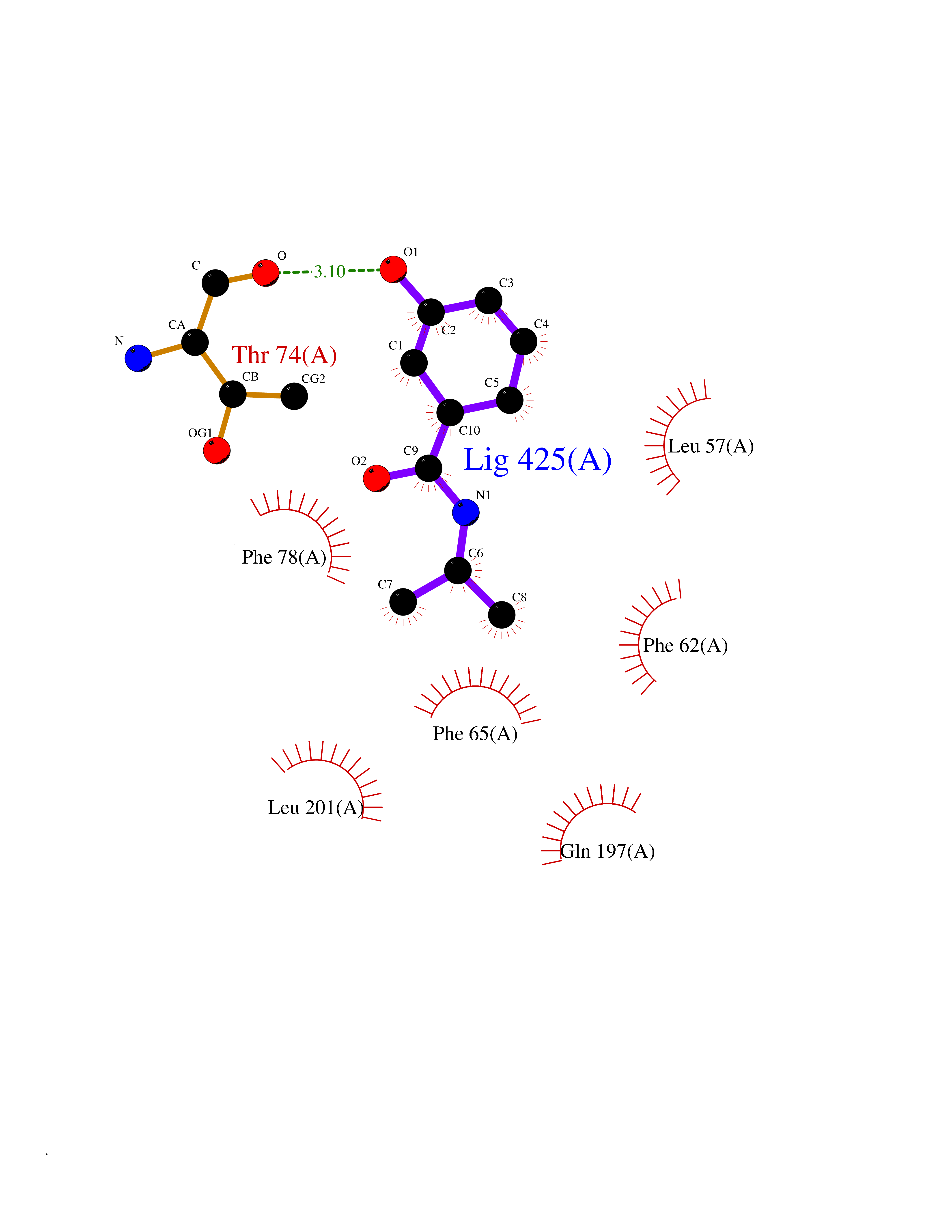 Ligplot Map 3D Binding mode Sequence GVPGQVDFYARFSPSPLSMKQFLDFGSVNACEKTSFMFLRQELPVRLANIMKEISLLPDNLLRTPSVQLVQSWYIQSLQELLDFKDKSAEDAKAIYDFTDTVIRIRNRHNDVIPTMAQGVIEYKESFDPVTSQNVQYFLDRFYMSRISIRMLLNQHSLLFGKHIGSINPNCNVLEVIKDGYENARRLCDLYYINSPELELEELNAKSPGQPIQVVYVPSHLYHMVFELFKNAMRATMEHHANRGVYPPIQVHVTLGNEDLTVKMSDRGGGVPLRKIDRLFNYMYSTAPRPRVETSRAVPLAGFGYGLPISRLYAQYFQGDLKLYSLEGYGTDAVIYIKALSTDSIERLPVYNKAAWKHYNTNDDWCVP Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Lysine-specific histone demethylase 1B (KDM1B) | 4HSU | 5.65 | |
Target general information Gen name KDM1B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine-specific histone demethylase 2; LSD2; Flavin-containing amine oxidase domain-containing protein 1; C6orf193; AOF1 Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Required for de novo DNA methylation of a subset of imprinted genes during oogenesis. Acts by oxidizing the substrate by FAD to generate the corresponding imine that is subsequently hydrolyzed. Demethylates both mono- and di-methylated 'Lys-4' of histone H3. Has no effect on tri-methylated 'Lys-4', mono-, di- or tri-methylated 'Lys-9', mono-, di- or tri-methylated 'Lys-27', mono-, di- or tri-methylated 'Lys-36' of histone H3, or on mono-, di- or tri-methylated 'Lys-20' of histone H4. Histone demethylase that demethylates 'Lys-4' of histone H3, a specific tag for epigenetic transcriptional activation, thereby acting as a corepressor. Related diseases Angioedema, hereditary, 1 (HAE1) [MIM:106100]: An autosomal dominant disorder characterized by episodic local swelling involving subcutaneous or submucous tissue of the upper respiratory and gastrointestinal tracts, face, extremities, and genitalia. Hereditary angioedema due to C1 esterase inhibitor deficiency is comprised of two clinically indistinguishable forms. In hereditary angioedema type 1, serum levels of C1 esterase inhibitor are decreased, while in type 2, the levels are normal or elevated, but the protein is non-functional. {ECO:0000269|PubMed:12773530, ECO:0000269|PubMed:1363816, ECO:0000269|PubMed:1451784, ECO:0000269|PubMed:14635117, ECO:0000269|PubMed:16409206, ECO:0000269|PubMed:2118657, ECO:0000269|PubMed:2296585, ECO:0000269|PubMed:22994404, ECO:0000269|PubMed:2365061, ECO:0000269|PubMed:24456027, ECO:0000269|PubMed:3178731, ECO:0000269|PubMed:7814636, ECO:0000269|PubMed:7883978, ECO:0000269|PubMed:8172583, ECO:0000269|PubMed:8529136, ECO:0000269|PubMed:8755917, ECO:0000269|Ref.41}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q96L03 EC number EC 1.-.-.- Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Developmental protein; FAD; Flavoprotein; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,C Molecular weight (Da) 85795.5 Length 763 Aromaticity 0.1 Instability index 37.87 Isoelectric point 8.41 Charge (pH=7) 9.16 2D Binding mode Binding energy (Kcal/mol) -7.71 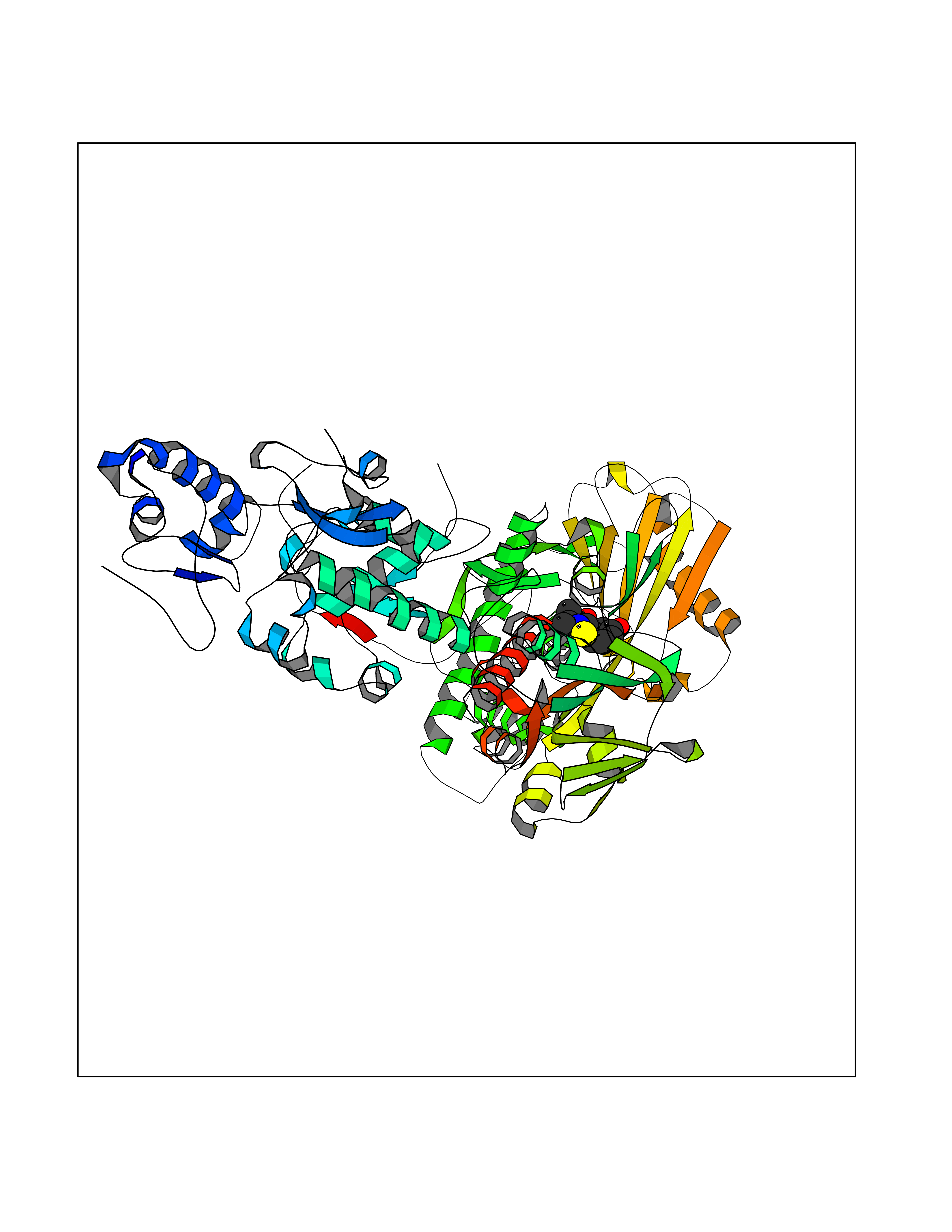 Molscript Map 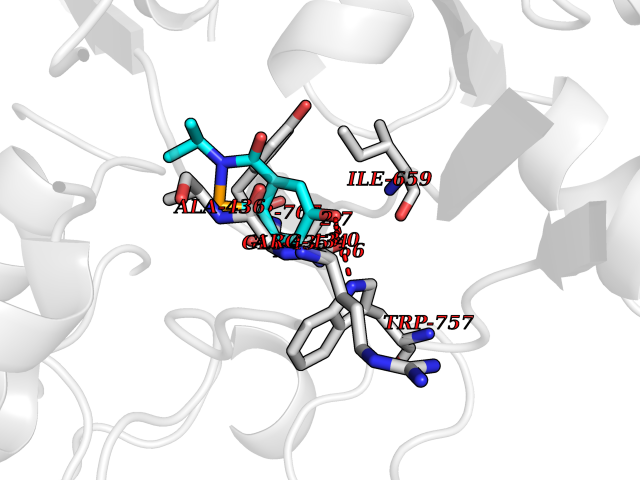 Pymol Map 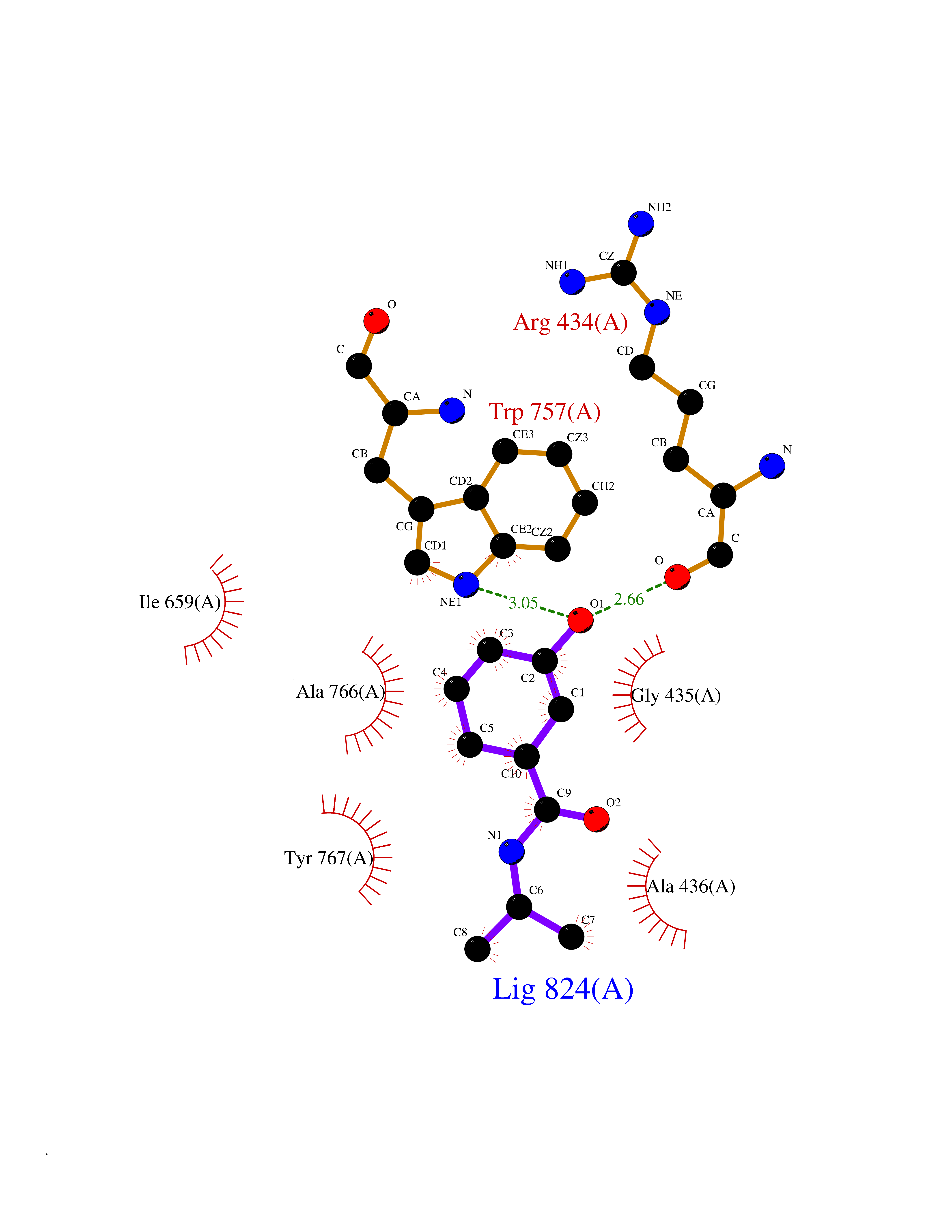 Ligplot Map 3D Binding mode Sequence GSRKCEKAGCTATCPVCFASASERCAKNGYTSRWYHLSCGEHFCNECFDHYYRSHKDGYDKYTTWKKIWTSNGKTEPSPKAFMADQQLPYWVQCTKPECRKWRQLTKEIQLTPQIAKTYRCGMKSDHCSLPEDLRVLEVSNHWWYSMLILPPLLKDSVAAPLLSAYYPDCVGMSPSCTGMNRYFQPFYQPNECGKALCVRPDVMELDELYEFPEYSRDPTMYLALRNLILALWYTNCKEALTPQKCIPHIIVRGLVRIRCVQEVERILYFMTRKGLINTGVLSVGADQYLLPKDYHNKSVIIIGAGPAGLAAARQLHNFGIKVTVLEAKDRIGGRVWDDKSFKGVTVGRGAQIVNGCINNPVALMCEQLGISMHKFGERCDLIQEGGRITDPTIDKRMDFHFNALLDVVSEWRKDKTQLQDVPLGEKIEEIYKAFIKESGIQFSELEGQVLQFHLSNLEYACGSNLHQVSARSWDHNEFFAQFAGDHTLLTPGYSVIIEKLAEGLDIQLKSPVQCIDYSGDEVQVTTTDGTGYSAQKVLVTVPLALLQKGAIQFNPPLSEKKMKAINSLGAGIIEKIALQFPYRFWDSKVQGADFFGHVPPSASKRGLFAVFYDMDPQKKHSVLMSVIAGEAVASVRTLDDKQVLQQCMATLRELFKEQEVPDPTKYFVTRWSTDPWIQMAYSFVKTGGSGEAYDIIAEDIQGTVFFAGEATNRHFPQTVTGAYLSGVREASKIAAFARTMQTARKSTGGKAPRKQLATKAAR Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Estrogen-related receptor-beta (ESRRB) | 6LIT | 5.65 | |
Target general information Gen name ESRRB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Steroid hormone receptor ERR2; Nuclear receptor subfamily 3 group B member 2; NR3B2; Estrogen-related receptor beta; Estrogen receptor-like 2; ESRL2; ERRB2; ERR-beta; ERR beta-2 Protein family Nuclear hormone receptor family, NR3 subfamily Biochemical class Nuclear hormone receptor Function Isoform 3: Transcription factor that binds a canonical ESRRB recognition (ERRE) sequence 5'TCAAGGTCA-3' localized on promoter and enhancer of targets genes regulating their expression or their transcription activity. Plays a role, in a LIF-independent manner, in maintainance of self-renewal and pluripotency of embryonic and trophoblast stem cells through different signaling pathways including FGF signaling pathway and Wnt signaling pathways. Upon FGF signaling pathway activation, interacts with KDM1A by directly binding to enhancer site of ELF5 and EOMES and activating their transcription leading to self-renewal of trophoblast stem cells. Also regulates expression of multiple rod-specific genes and is required for survival of this cell type (By similarity). Plays a role as transcription factor activator of GATA6, NR0B1, POU5F1 and PERM1. Plays a role as transcription factor repressor of NFE2L2 transcriptional activity and ESR1 transcriptional activity. During mitosis remains bound to a subset of interphase target genes, including pluripotency regulators, through the canonical ESRRB recognition (ERRE) sequence, leading to their transcriptional activation in early G1 phase. Can coassemble on structured DNA elements with other transcription factors like SOX2, POU5F1, KDM1A and NCOA3 to trigger ESRRB-dependent gene activation. This mechanism, in the case of SOX2 corecruitment prevents the embryonic stem cells (ESCs) to epiblast stem cells (EpiSC) transition through positive regulation of NR0B1 that inhibits the EpiSC transcriptional program. Also plays a role inner ear development by controlling expression of ion channels and transporters and in early placentation (By similarity). Related diseases Deafness, autosomal recessive, 35 (DFNB35) [MIM:608565]: A form of non-syndromic deafness characterized by non-progressive, prelingual hearing loss. {ECO:0000269|PubMed:18179891}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00255; DB07776; DB01645 Interacts with P62508-3 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Chromosome; Cytoplasm; Deafness; Disease variant; DNA-binding; Metal-binding; Non-syndromic deafness; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 22767.3 Length 198 Aromaticity 0.08 Instability index 56.41 Isoelectric point 5.76 Charge (pH=7) -5.51 2D Binding mode Binding energy (Kcal/mol) -7.7 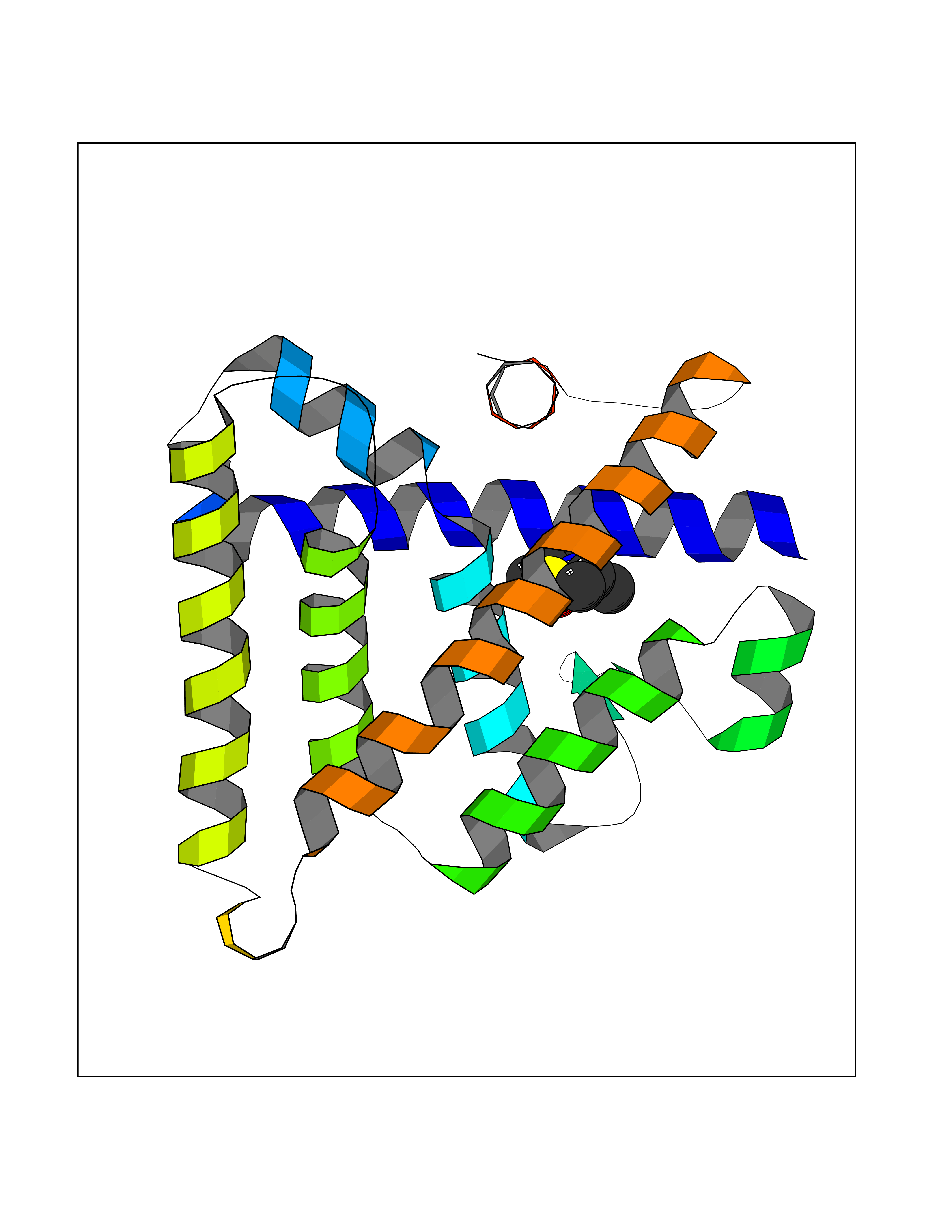 Molscript Map 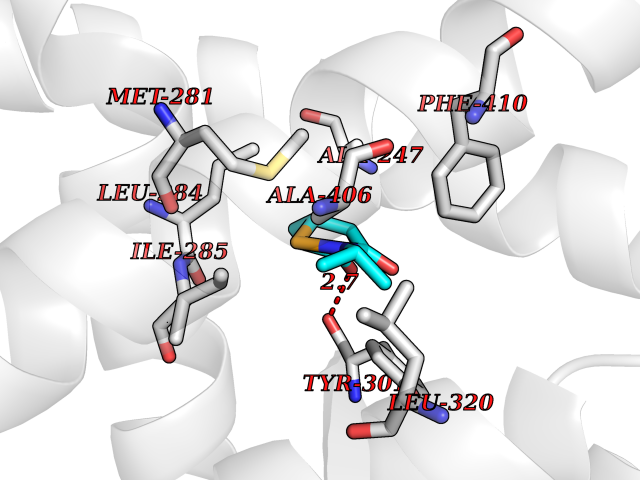 Pymol Map 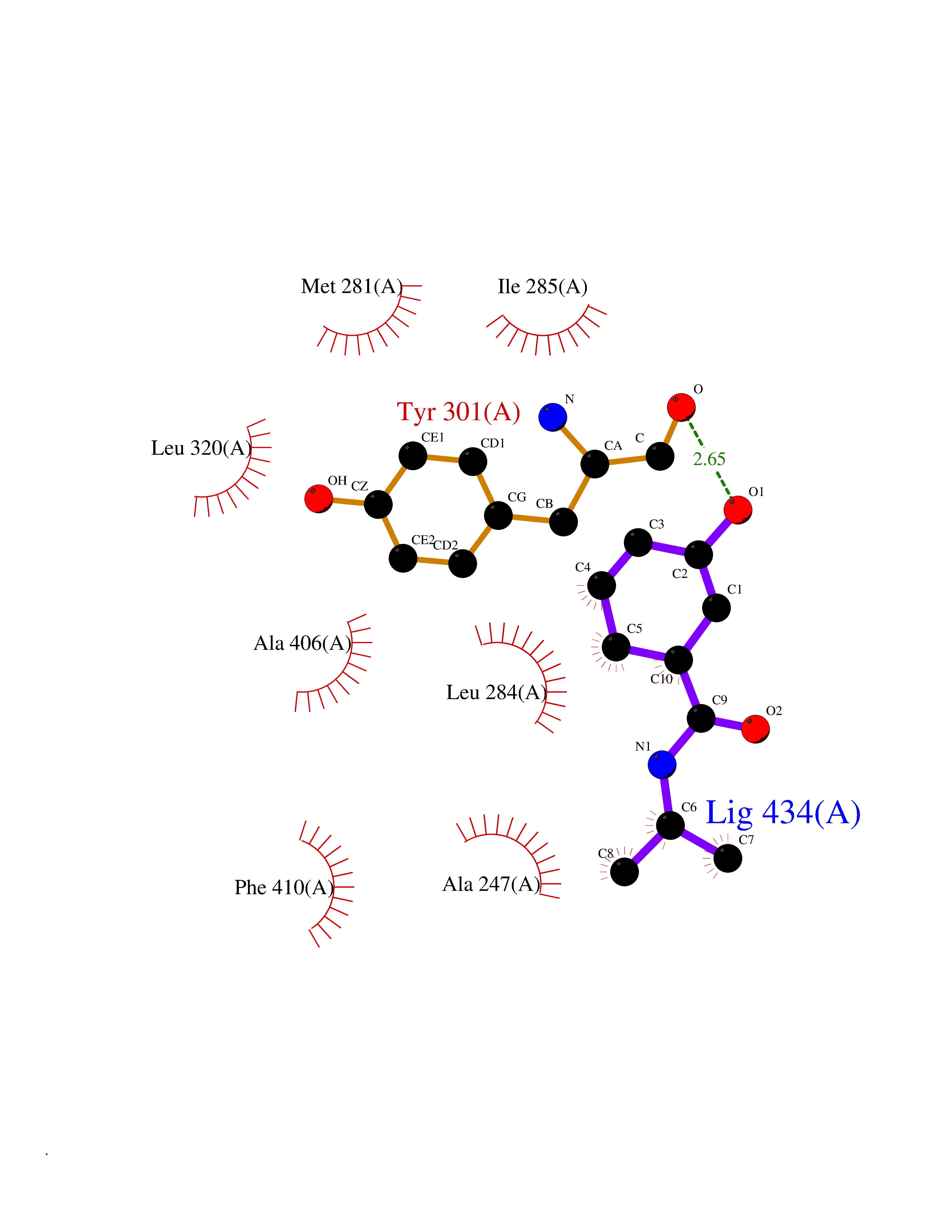 Ligplot Map 3D Binding mode Sequence GDIKALTTLCDLADRELVVIIGWAKHIPGFSSLSLGDQMSLLQSAWMEILILGIVYRSLPYDDKLVYAEDYIMDEEHSRLAGLLELYRAILQLVRRYKKLKVEKEEFVTLKALALANSDSMHIEDLEAVQKLQDLLHEALQDYELSQHHEEPWRTGKLLLTLPLLRQTAAKAVQHFYSVKLQGKVPMHKLFLEMLEAK Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Lysine-specific demethylase 5B (KDM5B) | 5FY9 | 5.65 | |
Target general information Gen name KDM5B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Retinoblastomabinding protein 2 homolog 1; Retinoblastoma-binding protein 2 homolog 1; RBP2H1; RBP2-H1; RBBP2H1; PLU1; PLU-1; Lysinespecific demethylase 5B; Jumonji/ARID domaincontaining protein 1B; J Protein family JARID1 histone demethylase family Biochemical class Paired donor oxygen oxidoreductase Function Does not demethylate histone H3 'Lys-9' or H3 'Lys-27'. Demethylates trimethylated, dimethylated and monomethylated H3 'Lys-4'. Acts as a transcriptional corepressor for FOXG1B and PAX9. Favors the proliferation of breast cancer cells by repressing tumor suppressor genes such as BRCA1 and HOXA5. In contrast, may act as a tumor suppressor for melanoma. Represses the CLOCK-ARNTL/BMAL1 heterodimer-mediated transcriptional activation of the core clock component PER2. Histone demethylase that demethylates 'Lys-4' of histone H3, thereby playing a central role in histone code. Related diseases Intellectual developmental disorder, autosomal recessive 65 (MRT65) [MIM:618109]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT65 patients have moderate to severe intellectual disability, developmental delay, and facial dysmorphism. Camptodactyly is present in some patients. {ECO:0000269|PubMed:29276005, ECO:0000269|PubMed:30409806}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P49711 EC number EC 1.14.11.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Biological rhythms; Chromatin regulator; Dioxygenase; Disease variant; Intellectual disability; Iron; Isopeptide bond; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Repressor; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 53020.6 Length 460 Aromaticity 0.12 Instability index 44.23 Isoelectric point 5.28 Charge (pH=7) -18.32 2D Binding mode Binding energy (Kcal/mol) -7.71  Molscript Map 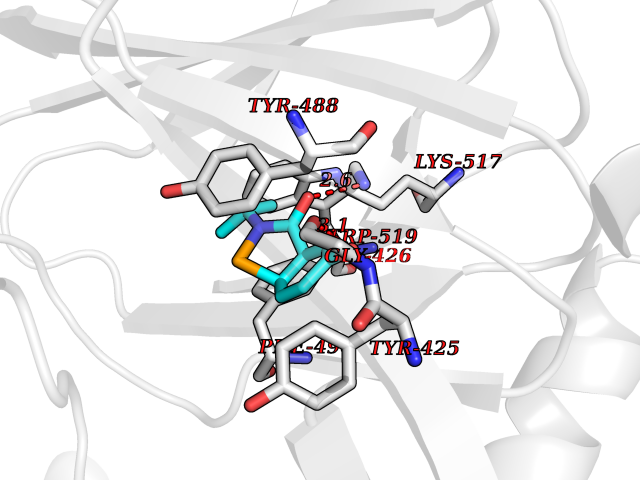 Pymol Map 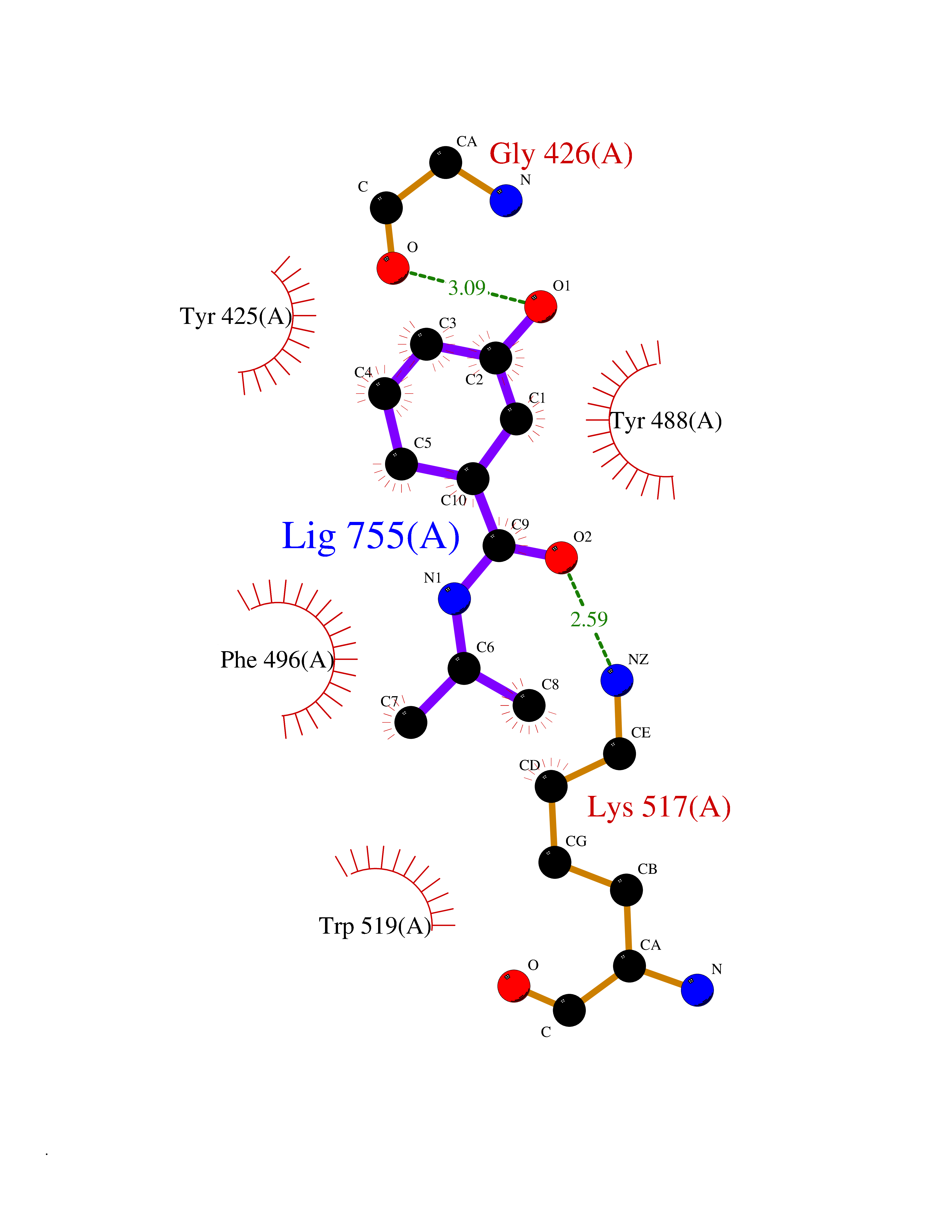 Ligplot Map 3D Binding mode Sequence SMFLPPPECPVFEPSWEEFADPFAFIHKIRPIAEQTGICKVRPPPDWQPPFACDVDKLHFTPRIQRLNELEAQTRVKLGGGGARDYTLRTFGEMADAFKSDYFNMPVHMVPTELVEKEFWRLVSTIEEDVTVEYGADIASKEFGSGFPVRDIKLSPEEEEYLDSGWNLNNMPVMEQSVLAHITADICGMKLPWLYVGMCFSSFCWHIEDHWSYSINYLHWGEPKTWYGVPGYAAEQLENVMKKLAPELFVSQPDLLHQLVTIMNPNTLMTHEVPVYRTNQCAGEFVITFPRAYHSGFNQGFNFAEAVNFCTVDWLPLGRQCVEHYRLLHRYCVFSHDEMICKMASKADVLDVVVASTVQKDMAIMIEDEKALRETVRKLGVIDSERMDFELLPDDERQCVKCKTTCFMSAISCSCKPGLLVCLHHVKELCSCPPYKYKLRYRYTLDDLYPMMNALKLRAE Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Insulin-degrading enzyme (IDE) | 4PES | 5.64 | |
Target general information Gen name IDE Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Insulysin; Insulinase; Insulin protease; Abeta-degrading protease Protein family Peptidase M16 family Biochemical class Peptidase Function Substrate binding induces important conformation changes, making it possible to bind and degrade larger substrates, such as insulin. Contributes to the regulation of peptide hormone signaling cascades and regulation of blood glucose homeostasis via its role in the degradation of insulin, glucagon and IAPP. Plays a role in the degradation and clearance of APP-derived amyloidogenic peptides that are secreted by neurons and microglia. Involved in antigen processing. Produces both the N terminus and the C terminus of MAGEA3-derived antigenic peptide (EVDPIGHLY) that is presented to cytotoxic T lymphocytes by MHC class I. Plays a role in the cellular breakdown of insulin, APP peptides, IAPP peptides, glucagon, bradykinin, kallidin and other peptides, and thereby plays a role in intercellular peptide signaling. Related diseases Congenital sucrase-isomaltase deficiency (CSID) [MIM:222900]: Autosomal recessive intestinal disorder that is clinically characterized by fermentative diarrhea, abdominal pain, and cramps upon ingestion of sugar. The symptoms are the consequence of absent or drastically reduced enzymatic activities of sucrase and isomaltase. The prevalence of CSID is 0.02 % in individuals of European descent and appears to be much higher in Greenland, Alaskan, and Canadian native people. CSID arises due to post-translational perturbations in the intracellular transport, polarized sorting, aberrant processing, and defective function of SI. {ECO:0000269|PubMed:10903344, ECO:0000269|PubMed:11340066, ECO:0000269|PubMed:14724820, ECO:0000269|PubMed:16329100, ECO:0000269|PubMed:8609217}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00626; DB09456; DB09564; DB01307; DB00030; DB00046; DB00071; DB14487; DB14533; DB14548 Interacts with P05067; P55773; P10147; P02686; PRO_0000000093 [P05067]; P01275; P10997; P14735-1; P01308; Q9J3M8 EC number EC 3.4.24.56 Uniprot keywords 3D-structure; Allosteric enzyme; Alternative splicing; ATP-binding; Cell membrane; Cytoplasm; Direct protein sequencing; Host cell receptor for virus entry; Host-virus interaction; Hydrolase; Membrane; Metal-binding; Metalloprotease; Nucleotide-binding; Protease; Proteomics identification; Receptor; Reference proteome; Secreted; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 55426 Length 483 Aromaticity 0.11 Instability index 30.18 Isoelectric point 5.79 Charge (pH=7) -12.48 2D Binding mode Binding energy (Kcal/mol) -7.7 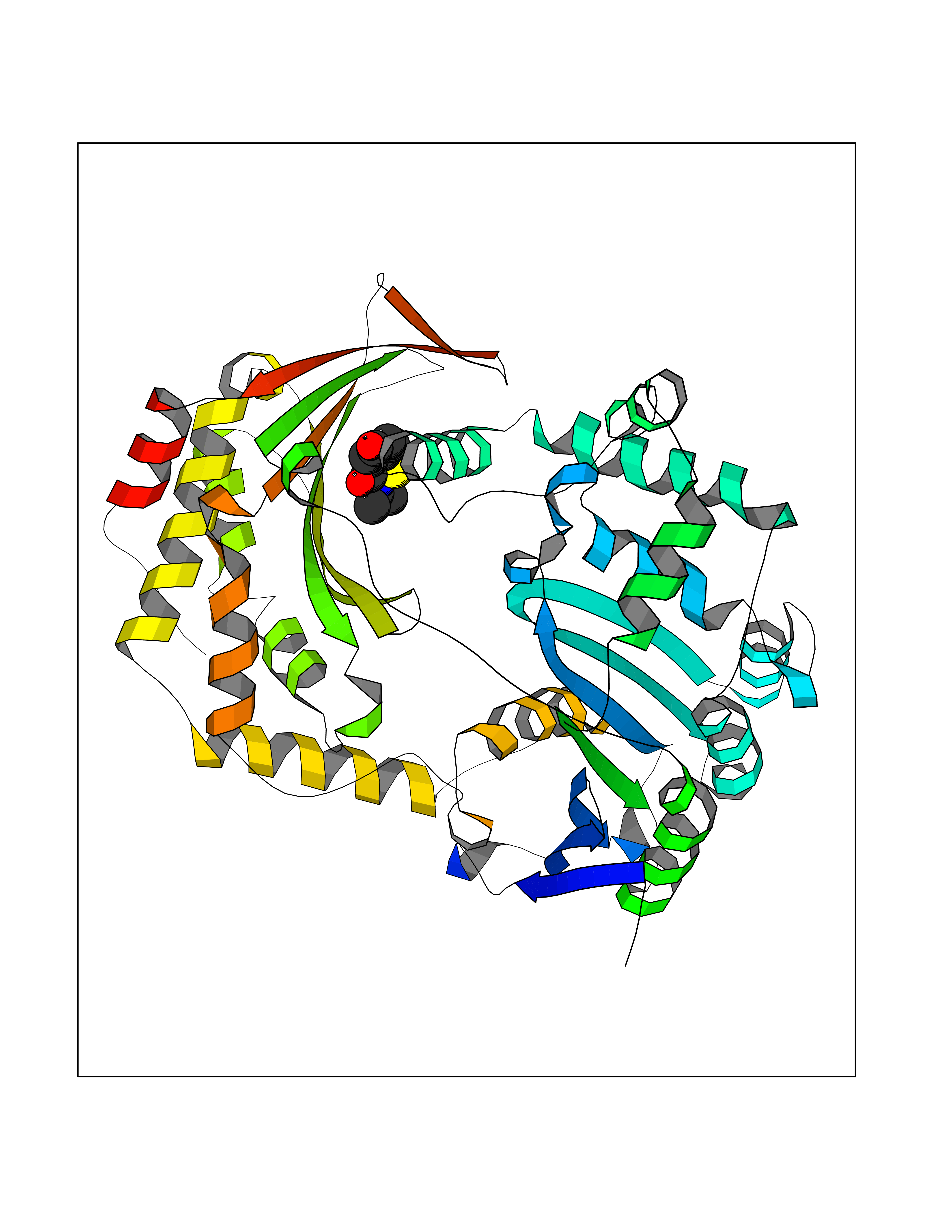 Molscript Map 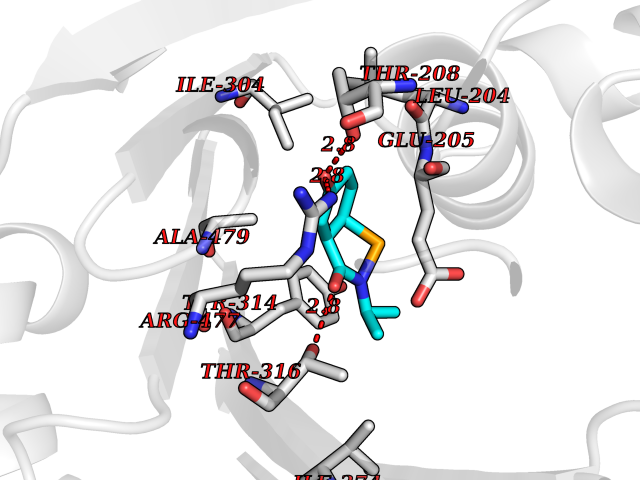 Pymol Map 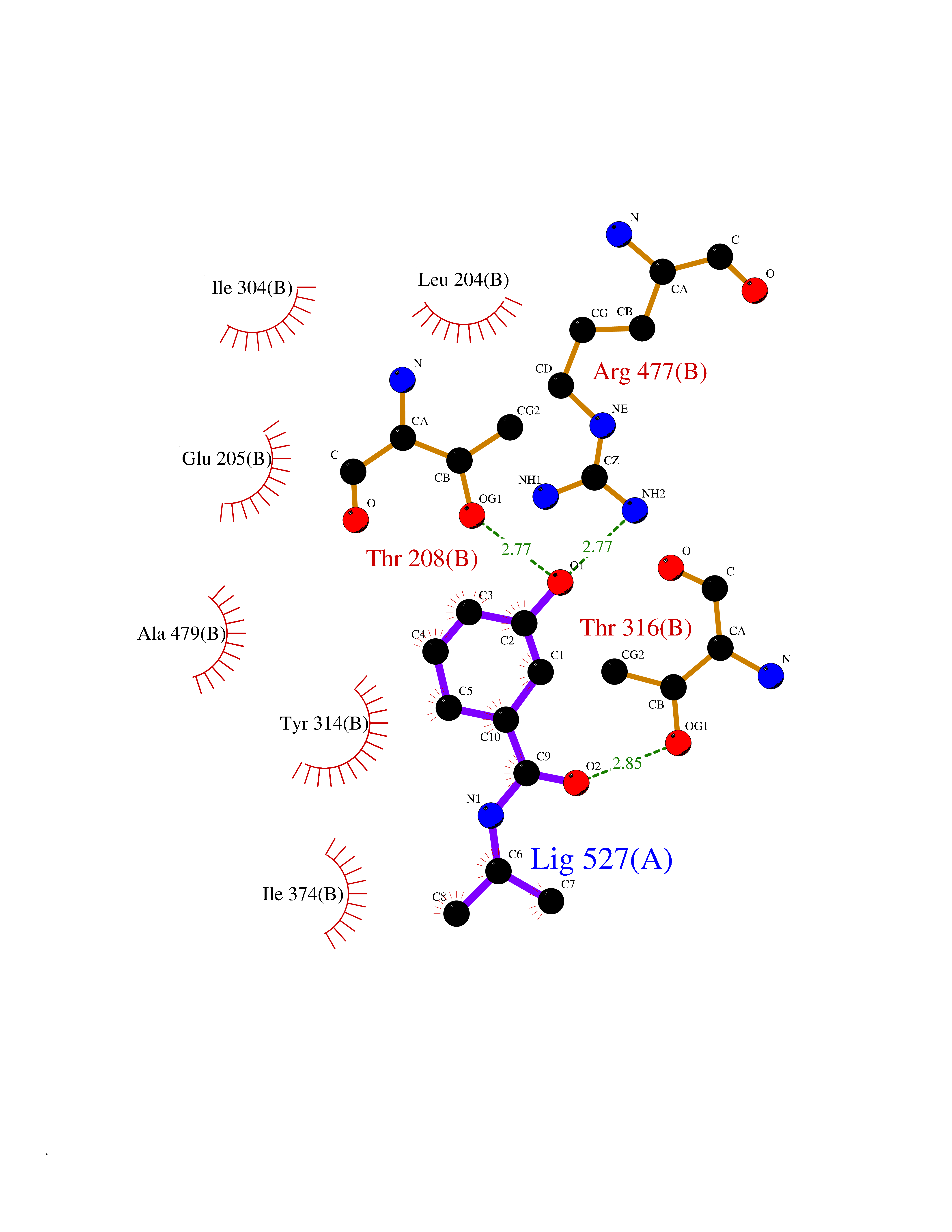 Ligplot Map 3D Binding mode Sequence NNPAIKRIGNHITKSPEDKREYRGLELANGIKVLLISDPTTDKSSAALDVHIGSLSDPPNIAGLSHFLQHMLFLGTKKYPKENEYSQFLSEHAGSSNAFTSGEHTNYYFDVSHEHLEGALDRFAQFFLSPLFDESAKDREVNAVDSEHEKNVMNDAWRLFQLEKATGNPKHPFSKFGTGNKYTLETRPNQEGIDVRQELLKFHSAYYSSNLMAVVVLGRESLDDLTNLVVKLFSEVENKNVPLPEFPEHPFQEEHLKQLYKIVPIKDIRNLYVTFPIPDLQKYYKSNPGHYLGHLIGHEGPGSLLSELKSKGWVNTLVGGQKEGARGFMFFIINVDLTEEGLLHVEDIILHMFQYIQKLRAEGPQEWVFQELKDLNAVAFRFKDKERPRGYTSKIAGILHYYPLEEVLTAEYLLEEFRPDLIEMVLDKLRPENVRVAIVSKSFEGKTDRTEEWYGTQYKQEAIPDEVIKKWQNADLNGKFKLP Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | 3-oxoacyl-[acyl-carrier-protein] synthase 2 | 3I8P | 5.64 | |
Target general information Gen name fabF Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1081;b1095;fabJ Protein family Thiolase-like superfamily, Beta-ketoacyl-ACP synthases family Biochemical class Transferase Function 3-oxoacyl-[acyl-carrier-protein] synthase activity.Beta-ketoacyl-acyl-carrier-protein synthase II activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08366; DB01034; DB03017; DB08407 Interacts with P0A6Y8 EC number 2.3.1.179 Uniprot keywords 3D-structure; Acyltransferase; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 42794.9 Length 411 Aromaticity 0.06 Instability index 31.41 Isoelectric point 5.72 Charge (pH=7) -7.34 2D Binding mode Binding energy (Kcal/mol) -7.69 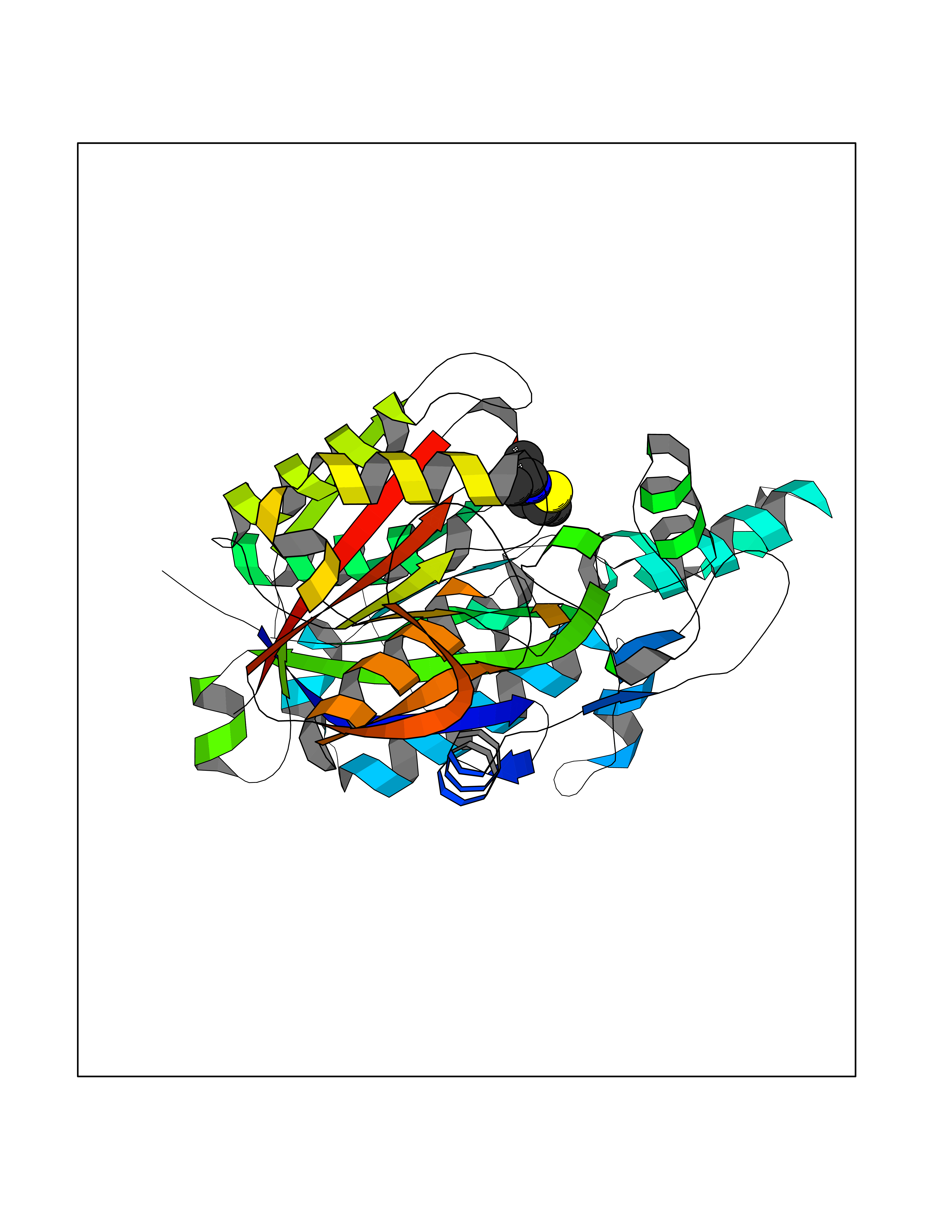 Molscript Map 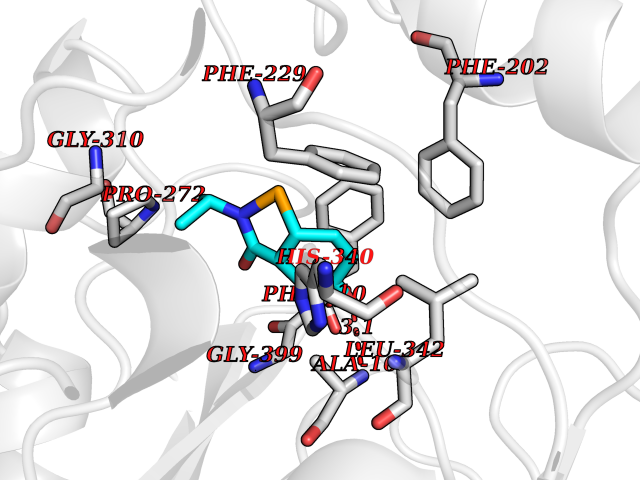 Pymol Map 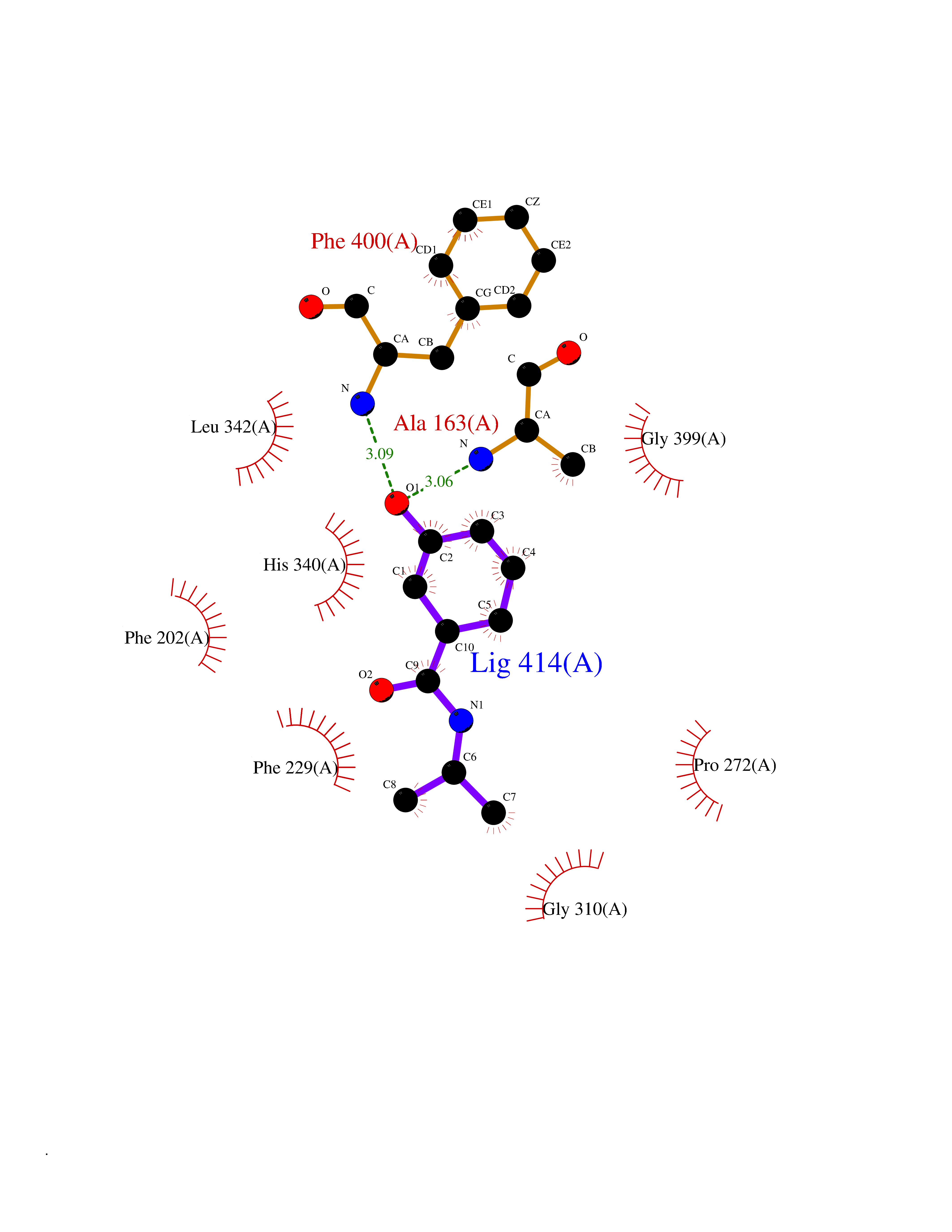 Ligplot Map 3D Binding mode Sequence KRRVVVTGLGMLSPVGNTVESTWKALLAGQSGISLIDHFDTSAYATKFAGLVKDFNCEDIISRKEQRKMDAFIQYGIVAGVQAMQDSGLEITEENATRIGAAIGSGIGGLGLIEENHTSLMNGGPRKISPFFVPSTIVNMVAGHLTIMYGLRGPSISIATAATSGVHNIGHAARIIAYGDADVMVAGGAEKASTPLGVGGFGAARALSTRNDNPQAASRPWDKERDGFVLGDGAGMLVLEEYEHAKKRGAKIYAELVGFGMSSDAYHMTSPPENGAGAALAMANALRDAGIEASQIGYVNAHGTSTPAGDKAEAQAVKTIFGEAASRVLVSSTKSMTGHLLGAAGAVESIYSILALRDQAVPPTINLDNPDEGCDLDFVPHEARQVSGMEYTLCNSFGFGGTNGSLIFKKI Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Dopamine beta-hydroxylase | 4ZEL | 5.64 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -7.69 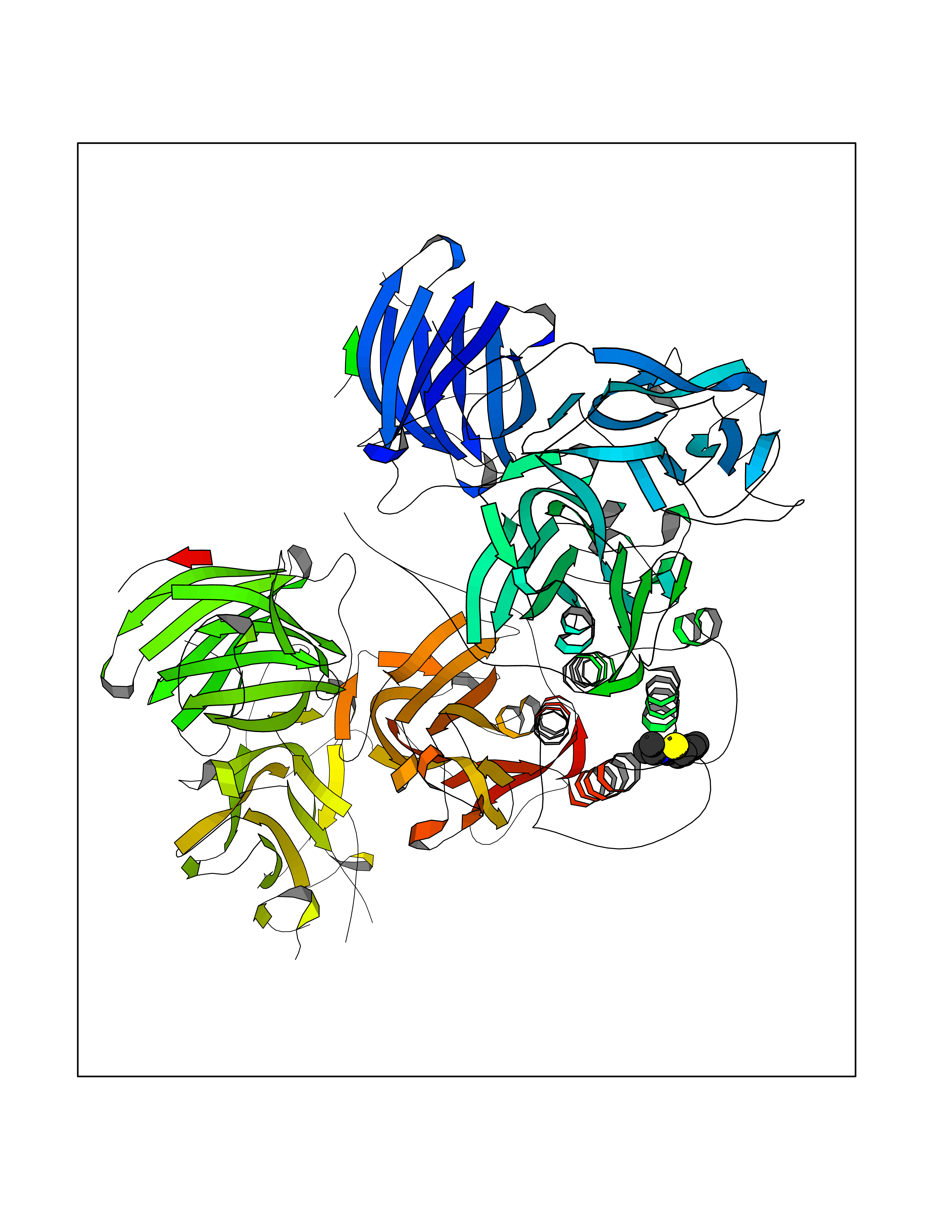 Molscript Map 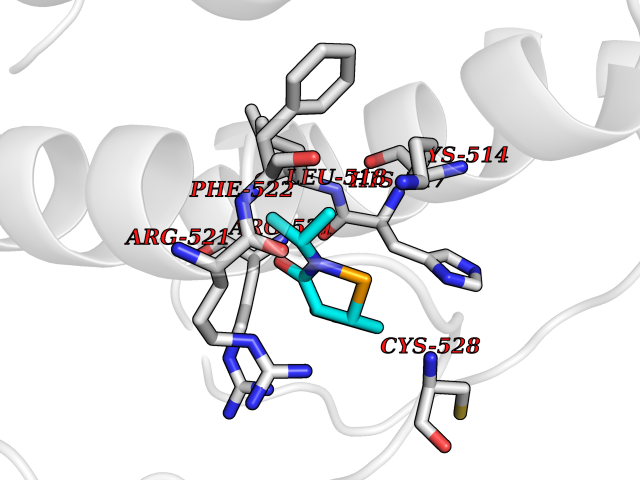 Pymol Map  Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Muscarinic acetylcholine receptor M4 (CHRM4) | 5DSG | 5.64 | |
Target general information Gen name CHRM4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms M4 receptor; CHRM4 Protein family G-protein coupled receptor 1 family, Muscarinic acetylcholine receptor subfamily, CHRM4 sub-subfamily Biochemical class GPCR rhodopsin Function The muscarinic acetylcholine receptor mediates various cellular responses, including inhibition of adenylate cyclase, breakdown of phosphoinositides and modulation of potassium channels through the action of G proteins. Primary transducing effect is inhibition of adenylate cyclase. Related diseases Leukodystrophy, hypomyelinating, 15 (HLD15) [MIM:617951]: An autosomal recessive disorder characterized by hypomyelinating leukodystrophy with thinning of the corpus callosum. Clinical features include motor and cognitive impairment appearing in the first or second decade of life, dystonia, ataxia, spasticity, and dysphagia. Most patients develop severe optic atrophy, and some have hearing loss. {ECO:0000269|PubMed:29576217}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03128; DB08897; DB05752; DB00321; DB00543; DB01238; DB14185; DB00572; DB00767; DB01019; DB00835; DB00411; DB01239; DB00568; DB00363; DB00496; DB01151; DB09167; DB01142; DB00366; DB09194; DB06702; DB00986; DB06787; DB11181; DB00725; DB00424; DB00458; DB01625; DB01221; DB00408; DB00934; DB00454; DB06709; DB00940; DB01403; DB00340; DB00622; DB00540; DB00334; DB00715; DB01085; DB00387; DB01069; DB00777; DB12278; DB01224; DB11855; DB13581; DB00747; DB01591; DB00342; DB11235; DB01409; DB01036; DB00376; DB09089; DB00726; DB00809; DB09076; DB09185; DB00246 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 32545.4 Length 287 Aromaticity 0.14 Instability index 30.63 Isoelectric point 9.34 Charge (pH=7) 11.91 2D Binding mode Binding energy (Kcal/mol) -7.69  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence HNRYETVEMVFIATVTGSLSLVTVVGNILVMLSIKVNRQLQTVNNYFLFSLACADLIIGAFSMNLYTVYIIKGYWPLGAVVCDLWLALDYVVSNASVMNLLIISFDRYFCVTKPLTYPARRTTKMAGLMIAAAWVLSFVLWAPAILFWQFVVGKRTVPDNQCFIQFLSNPAVTFGTAIAAFYLPVVIMTVLYIHISLASRSRVQMAARERKVTRTIFAILLAFILTWTPYNVMVLVNTFCQSCIPDTVWSIGYWLCYVNSTINPACYALCNATFKKTFRHLLLCQYR Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Estrogen-related receptor-gamma (ESRRG) | 2E2R | 5.64 | |
Target general information Gen name ESRRG Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nuclear receptor subfamily 3 group B member 3; NR3B3; KIAA0832; Estrogen-related receptor gamma; Estrogen receptor-related protein 3; ERRG2; ERR3; ERR gamma-2 Protein family Nuclear hormone receptor family, NR3 subfamily Biochemical class Nuclear hormone receptor Function Binds specifically to an estrogen response element and activates reporter genes controlled by estrogen response elements. Induces the expression of PERM1 in the skeletal muscle. Orphan receptor that acts as transcription activator in the absence of bound ligand. Related diseases WHIM syndrome 1 (WHIMS1) [MIM:193670]: An autosomal dominant immunologic disease characterized by neutropenia, hypogammaglobulinemia and extensive human papillomavirus (HPV) infection. Despite the peripheral neutropenia, bone marrow aspirates from affected individuals contain abundant mature myeloid cells, a condition termed myelokathexis. {ECO:0000269|PubMed:12692554, ECO:0000269|PubMed:15536153}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: CXCR4 mutations play a role in the pathogenesis of Waldenstroem macroglobulinemia (WM) and influence disease presentation and outcome, as well as response to therapy. WM is a B-cell lymphoma characterized by accumulation of malignant lymphoplasmacytic cells in the bone marrow, lymph nodes and spleen, and hypersecretion of monoclonal immunoglobulin M (IgM). Excess IgM production results in serum hyperviscosity, tissue infiltration, and autoimmune-related pathology. {ECO:0000269|PubMed:24366360, ECO:0000269|PubMed:24553177}. Drugs (DrugBank ID) DB06884; DB04468; DB06973; DB07485; DB02659; DB00255; DB13952; DB13953; DB13954; DB13955; DB13956; DB06902; DB00675; DB00197 Interacts with Q05D60; Q9BVG8; P50222; P51843; Q12769; Q9UBK2; A0MZ66; G2XKQ0; Q8NFM4; Q13315; Q86WA6-2; Q9BZE7; Q13555-5; Q05D60; Q5JST6; P11474; O95718-2; P62508-3; Q15024; O95990-4; Q8IZU1; Q14296; P23508; Q6IN84; P51843; Q15466; P48552; P26367; Q9NPJ4; P01189; Q9UBK2; P62195; Q8N0T1-2; Q04864-2; Q6NUQ1; A0MZ66-4; Q8TAD8; P19237; P48788; Q96PN7; Q96S82; Q5SQQ9-2; Q7Z4V0 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 25755.7 Length 227 Aromaticity 0.07 Instability index 55.31 Isoelectric point 5.09 Charge (pH=7) -10.6 2D Binding mode Binding energy (Kcal/mol) -7.69  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence KPYNKIVSHLLVAEPEKIYAMPDPTVPDSDIKALTTLCDLADRELVVIIGWAKHIPGFSTLSLADQMSLLQSAWMEILILGVVYRSLSFEDELVYADDYIMDEDQSKLAGLLDLNNAILQLVKKYKSMKLEKEEFVTLKAIALANSDSMHIEDVEAVQKLQDVLHEALQDYEAGQHMEDPRRAGKMLMTLPLLRQTSTKAVQHFYNIKLEGKVPMHKLFLEMLEAKV Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Cocaine esterase | 3I2K | 5.64 | |
Target general information Gen name cocE Organism Rhodococcus sp. (strain MB1 Bresler) Uniprot ID TTD ID NA Synonyms NA Protein family CocE/NonD hydrolase family Biochemical class Hydrolase Function Carboxylic ester hydrolase activity.Dipeptidyl-peptidase activity. Related diseases Thiamine metabolism dysfunction syndrome 5, episodic encephalopathy type (THMD5) [MIM:614458]: An autosomal recessive metabolic disorder due to an inborn error of thiamine metabolism. The phenotype is highly variable, but in general, affected individuals have onset in early childhood of acute encephalopathic episodes associated with increased serum and CSF lactate. These episodes result in progressive neurologic dysfunction manifest as gait disturbances, ataxia, dystonia, and spasticity, which in some cases may result in loss of ability to walk. Cognitive function is usually preserved, although mildly delayed development has been reported. These episodes are usually associated with infection and metabolic decompensation. Some patients may have recovery of some neurologic deficits. {ECO:0000269|PubMed:22152682}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03793; DB01795 Interacts with NA EC number 3.1.1.84 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Hydrolase; Serine esterase Protein physicochemical properties Chain ID A Molecular weight (Da) 62127.9 Length 574 Aromaticity 0.09 Instability index 26.62 Isoelectric point 4.56 Charge (pH=7) -33.24 2D Binding mode Binding energy (Kcal/mol) -7.69  Molscript Map 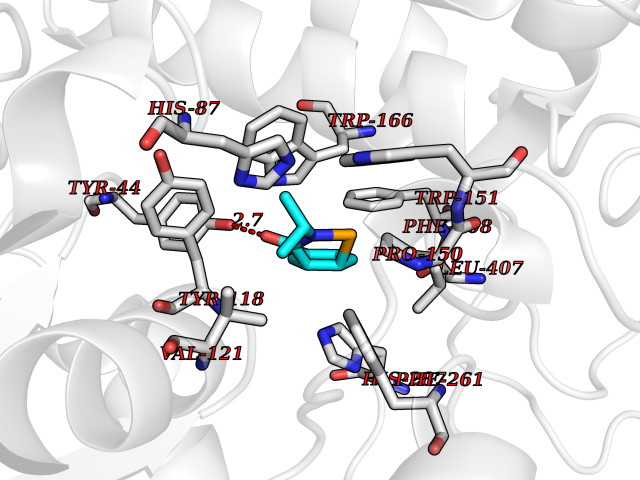 Pymol Map 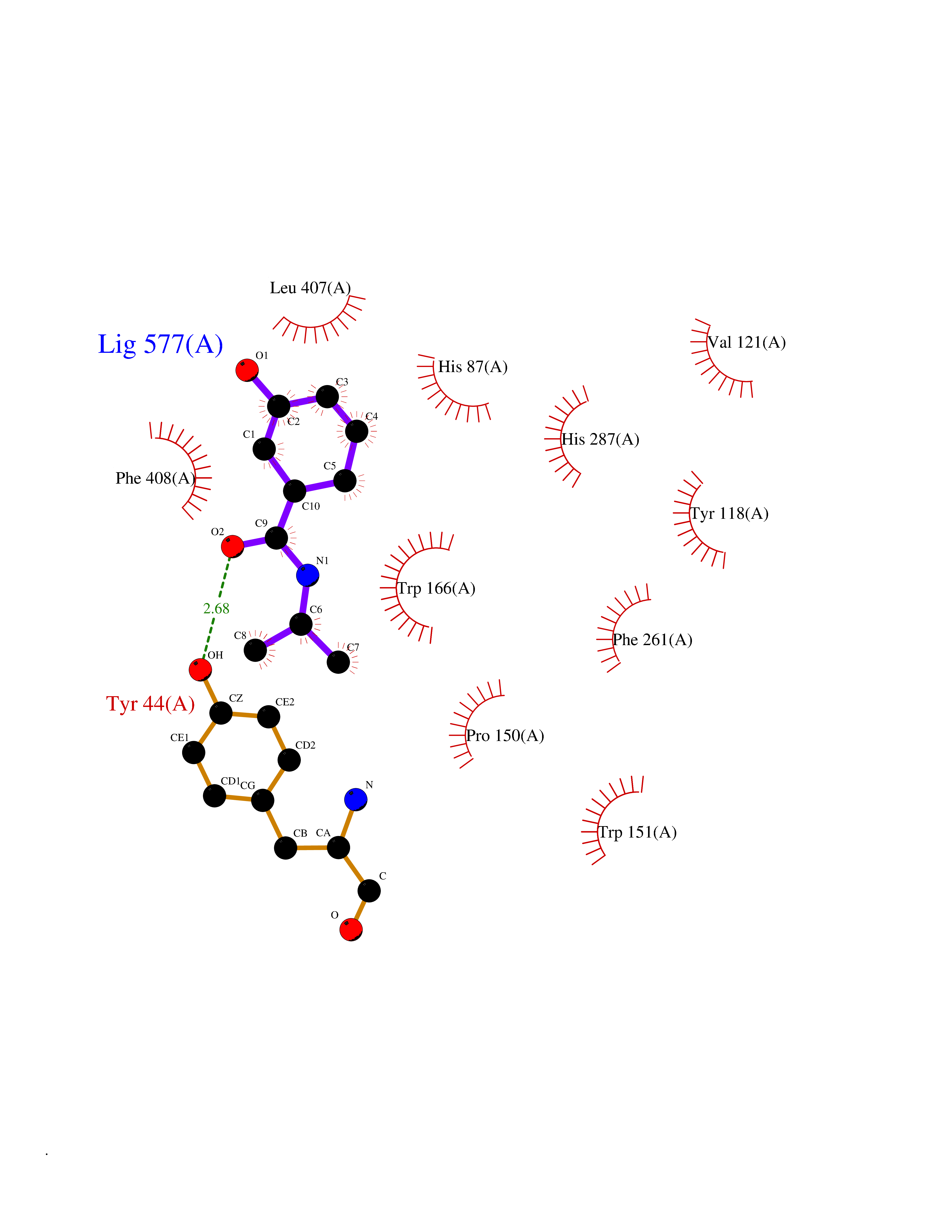 Ligplot Map 3D Binding mode Sequence VDGNYSVASNVMVPMRDGVRLAVDLYRPDADGPVPVLLVRNPYDKFDVFAWSTQSTNWLEFVRDGYAVVIQDTRGLFASEGEFVPHVDDEADAEDTLSWILEQAWCDGNVGMFGVSYLGVTQWQAAVSGVGGLKAIAPSMASADLYRAPWYGPGGALSVEALLGWSALIGTGLITSRSDARPEDAADFVQLAAILNDVAGAASVTPLAEQPLLGRLIPWVIDQVVDHPDNDESWQSISLFERLGGLATPALITAGWYDGFVGESLRTFVAVKDNADARLVVGPWSHSNLTGRNADRKFGIAATYPIQEATTMHKAFFDRHLRGETDALAGVPKVRLFVMGIDEWRDETDWPLPDTAYTPFYLGGSGAANTSTGGGTLSTSISGTESADTYLYDPADPVPSLGGTLLFHNGDNGPADQRPIHDRDDVLCYSTEVLTDPVEVTGTVSARLFVSSSAVDTDFTAKLVDVFPDGRAIALCDGIVRMRYRETLVNPTLIEAGEIYEVAIDMLATSNVFLPGHRIMVQVSSSNFPKYDRNSNTGGVIAREQLEEMCTAVNRIHRGPEHPSHIVLPIIKRK Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Hyperpolarization cyclic nucleotide-gated channel 4 (HCN4) | 3OTF | 5.64 | |
Target general information Gen name HCN4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channel 4 Protein family Potassium channel HCN family Biochemical class Voltage-gated ion channel Function Contributes to the native pacemaker currents in heart (If) that regulate the rhythm of heart beat. May contribute to the native pacemaker currents in neurons (Ih). May mediate responses to sour stimuli. Hyperpolarization-activated ion channel with very slow activation and inactivation exhibiting weak selectivity for potassium over sodium ions. Related diseases Sick sinus syndrome 2 (SSS2) [MIM:163800]: The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS2 onset is in utero or at birth. {ECO:0000269|PubMed:15123648, ECO:0000269|PubMed:16407510, ECO:0000269|PubMed:20662977, ECO:0000269|PubMed:23103389}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 8 (BRGDA8) [MIM:613123]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:19165230}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Epilepsy, idiopathic generalized 18 (EIG18) [MIM:619521]: An autosomal dominant form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. EIG18 is characterized by onset of myoclonic seizures in infancy. Although the seizures remit, some patients may have later speech or cognitive impairment. {ECO:0000269|PubMed:30127718}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O60741; Q9Y3Q4 EC number NA Uniprot keywords 3D-structure; Brugada syndrome; cAMP; cAMP-binding; Cell membrane; Disease variant; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Nucleotide-binding; Phosphoprotein; Potassium; Potassium channel; Potassium transport; Proteomics identification; Reference proteome; Sodium; Sodium channel; Sodium transport; Transmembrane; Transmembrane helix; Transport; Voltage-gated channel Protein physicochemical properties Chain ID A Molecular weight (Da) 23211.2 Length 197 Aromaticity 0.12 Instability index 42.69 Isoelectric point 8.67 Charge (pH=7) 3.11 2D Binding mode Binding energy (Kcal/mol) -7.69 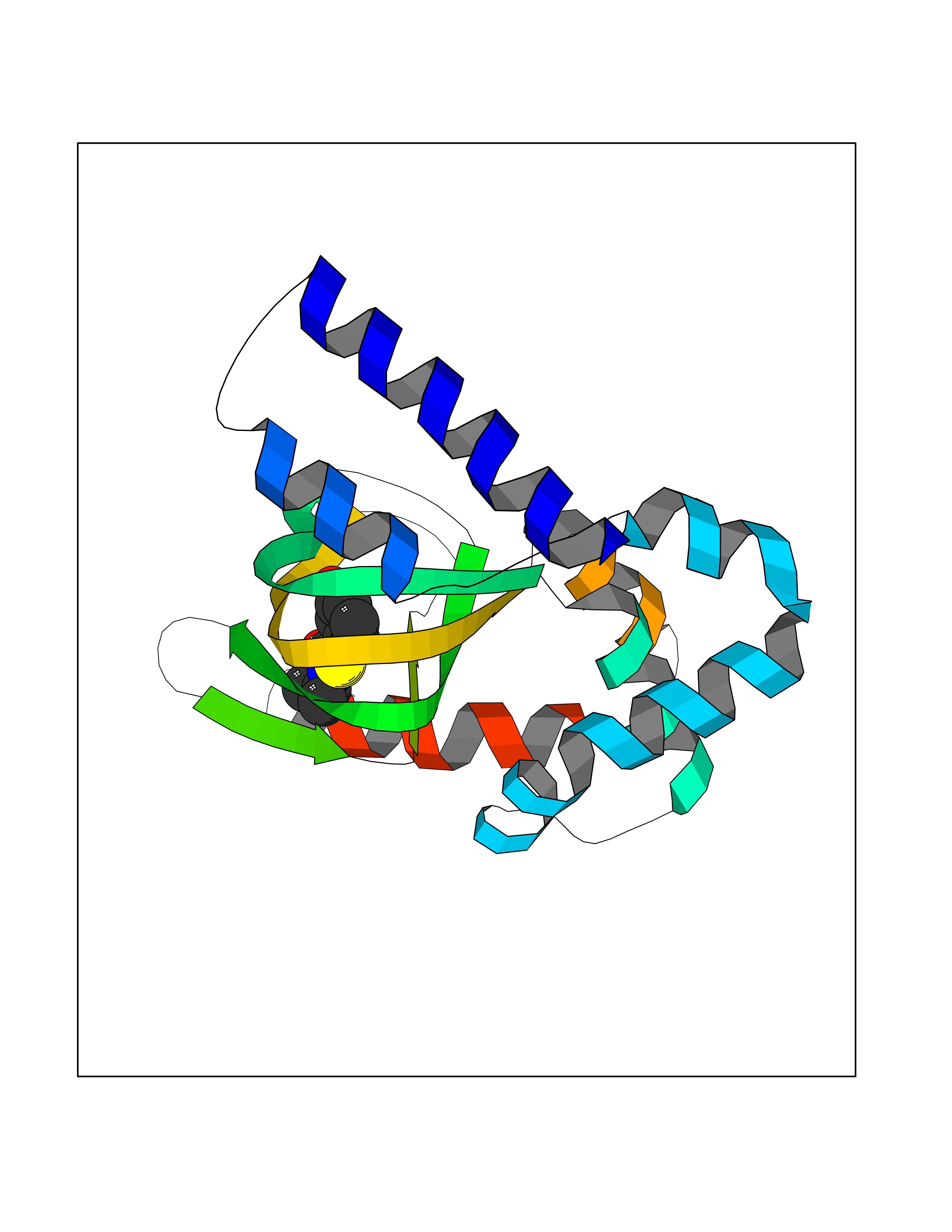 Molscript Map 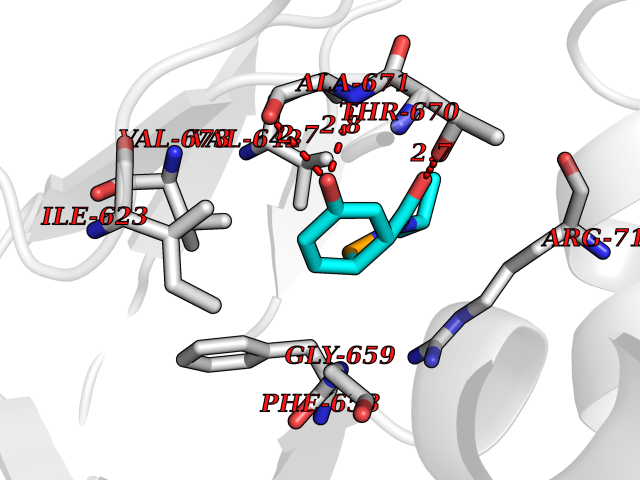 Pymol Map 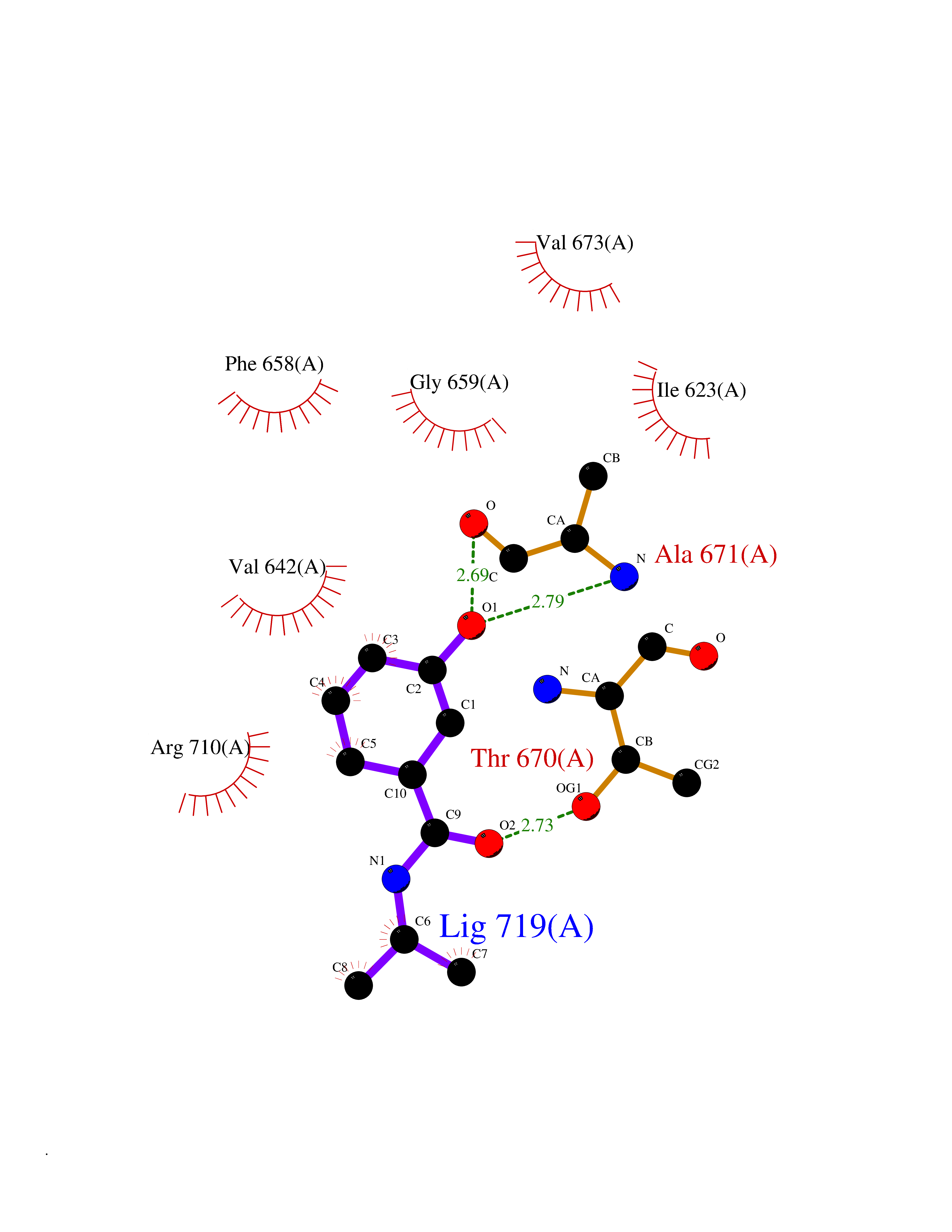 Ligplot Map 3D Binding mode Sequence DSSRRQYQEKYKQVEQYMSFHKLPPDTRQRIHDYYEHRYQGKMFDEESILGELSEPLREEIINFNCRKLVASMPLFANADPNFVTSMLTKLRFEVFQPGDYIIREGTIGKKMYFIQHGVVSVLTKGNKETKLADGSYFGEICLLTRGRRTASVRADTYCRLYSLSVDNFNEVLEEYPMMRRAFETVALDRLDRIGKK Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | N-acylethanolamine-hydrolyzing acidamidase (NAAA) | 6DXX | 5.64 | |
Target general information Gen name NAAA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nacylsphingosine amidohydrolaselike; Nacylethanolaminehydrolyzing acid amidase subunit beta; NAAA; Acid ceramidaselike protein; ASAHlike protein Protein family Acid ceramidase family Biochemical class Carbon-nitrogen hydrolase Function Degrades bioactive fatty acid amides to their corresponding acids, with the following preference: N- palmitoylethanolamine > N-myristoylethanolamine > N- lauroylethanolamine = N-stearoylethanolamine > N- arachidonoylethanolamine > N-oleoylethanolamine. Also exhibits weak hydrolytic activity against the ceramides N- lauroylsphingosine and N-palmitoylsphingosine. Related diseases Hypertriglyceridemia, transient infantile (HTGTI) [MIM:614480]: An autosomal recessive disorder characterized by onset of moderate to severe transient hypertriglyceridemia in infancy that normalizes with age. The hypertriglyceridemia is associated with hepatomegaly, moderately elevated transaminases, persistent fatty liver, and the development of hepatic fibrosis. {ECO:0000269|PubMed:22226083, ECO:0000269|PubMed:24549054}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09061; DB14009; DB14011 Interacts with NA EC number EC 3.5.1.- Uniprot keywords 3D-structure; Alternative splicing; Autocatalytic cleavage; Direct protein sequencing; Disulfide bond; Fatty acid metabolism; Glycoprotein; Hydrolase; Lipid degradation; Lipid metabolism; Lysosome; Membrane; Proteomics identification; Reference proteome; Signal; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 36877.8 Length 328 Aromaticity 0.11 Instability index 44.37 Isoelectric point 7.72 Charge (pH=7) 1.08 2D Binding mode Binding energy (Kcal/mol) -7.7  Molscript Map 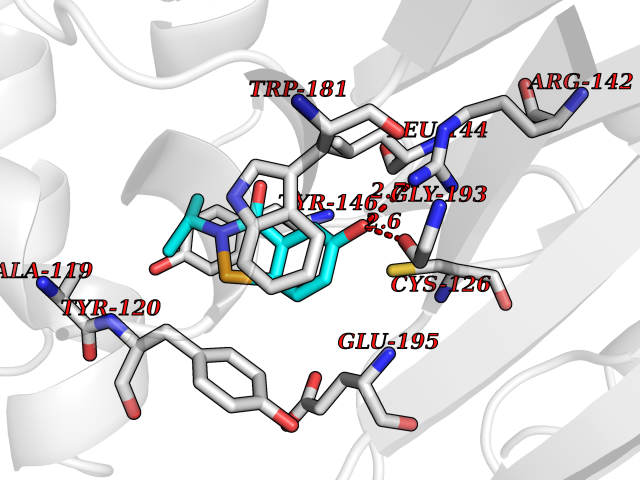 Pymol Map 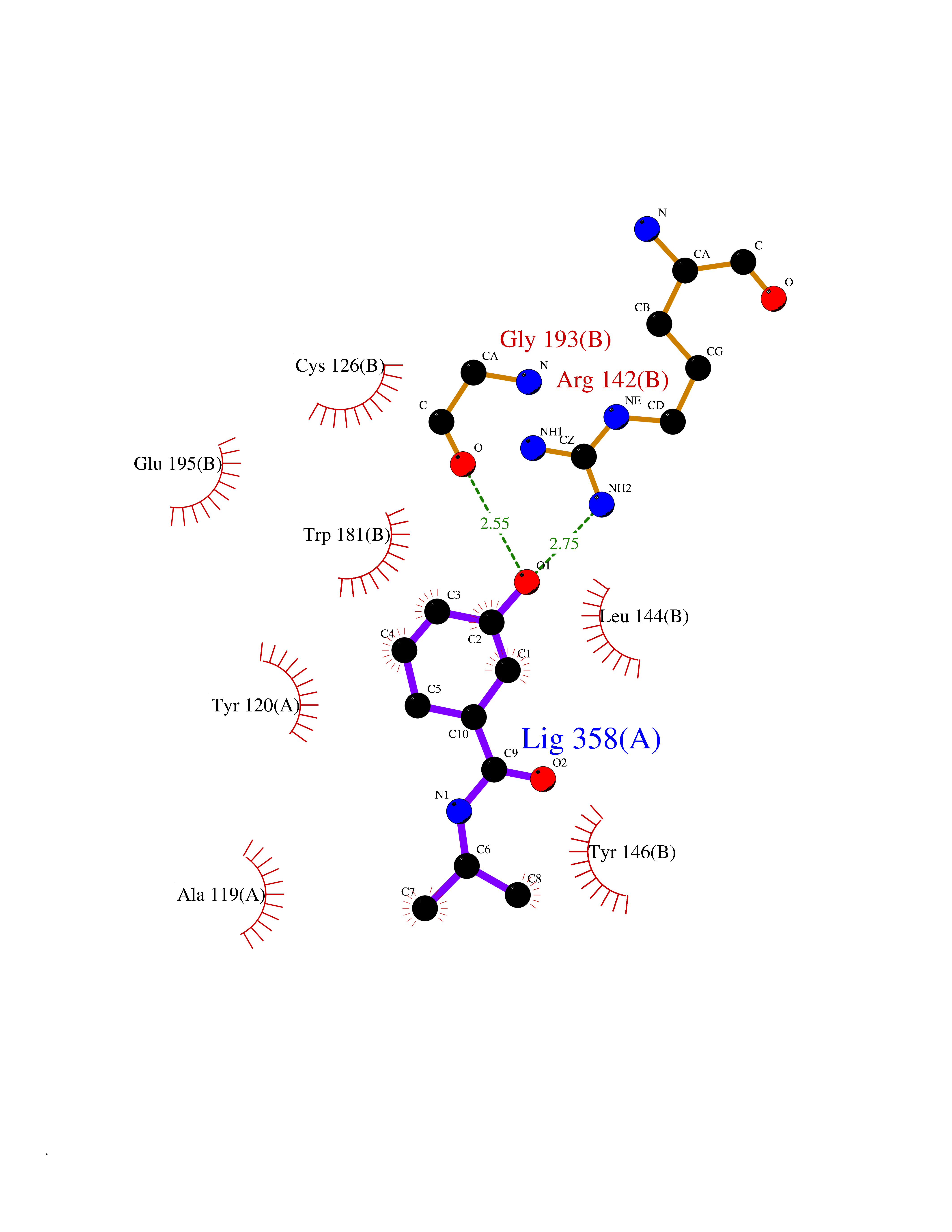 Ligplot Map 3D Binding mode Sequence SPPAAPRFNVSLDSVPELRWLPVLRHYDLDLVRAAMAQVIGDRVPKWVHVLIGKVVLELERFLPQPFTGEIRGMCDFMNLSLADCLLVNLAYESSVFCTSIVAQDSRGHIYHGRNLDYPFGNVLRKLTVDVQFLKNGQIAFTGTTFIGYVGLWTGQSPHKFTVSGDERDKGWWWENAIAALFRRHIPVSWLIRATLSESENFEAAVGKLAKTPLIADVYYIVGGTSPREGVVITRNRDGPADIWPLDPLNGAWFRVETNYDHWKPAPKEDDRRTSAIKALNATGQANLSLEALFQILSVVPVYNNFTIYTTVMSAGSPDKYMTRIRNP Hydrogen bonds contact Hydrophobic contact | ||||