Job Results:
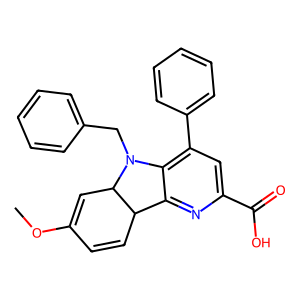
Ligand
Structure
Job ID
b9ed182cddc54993fbd50fd04d8e2354
Job name
NA
Time
2025-09-26 08:49:45
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Lysine-specific demethylase 5B (KDM5B) | 5FY9 | 7.65 | |
Target general information Gen name KDM5B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Retinoblastomabinding protein 2 homolog 1; Retinoblastoma-binding protein 2 homolog 1; RBP2H1; RBP2-H1; RBBP2H1; PLU1; PLU-1; Lysinespecific demethylase 5B; Jumonji/ARID domaincontaining protein 1B; J Protein family JARID1 histone demethylase family Biochemical class Paired donor oxygen oxidoreductase Function Does not demethylate histone H3 'Lys-9' or H3 'Lys-27'. Demethylates trimethylated, dimethylated and monomethylated H3 'Lys-4'. Acts as a transcriptional corepressor for FOXG1B and PAX9. Favors the proliferation of breast cancer cells by repressing tumor suppressor genes such as BRCA1 and HOXA5. In contrast, may act as a tumor suppressor for melanoma. Represses the CLOCK-ARNTL/BMAL1 heterodimer-mediated transcriptional activation of the core clock component PER2. Histone demethylase that demethylates 'Lys-4' of histone H3, thereby playing a central role in histone code. Related diseases Intellectual developmental disorder, autosomal recessive 65 (MRT65) [MIM:618109]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT65 patients have moderate to severe intellectual disability, developmental delay, and facial dysmorphism. Camptodactyly is present in some patients. {ECO:0000269|PubMed:29276005, ECO:0000269|PubMed:30409806}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P49711 EC number EC 1.14.11.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Biological rhythms; Chromatin regulator; Dioxygenase; Disease variant; Intellectual disability; Iron; Isopeptide bond; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Repressor; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 53020.6 Length 460 Aromaticity 0.12 Instability index 44.23 Isoelectric point 5.28 Charge (pH=7) -18.32 2D Binding mode Binding energy (Kcal/mol) -10.44  Molscript Map 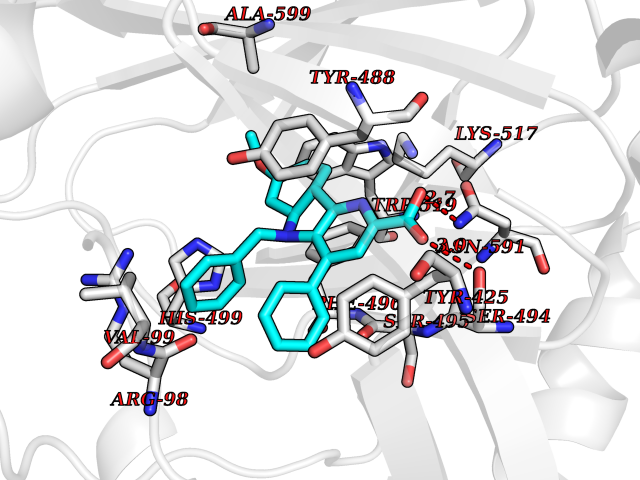 Pymol Map 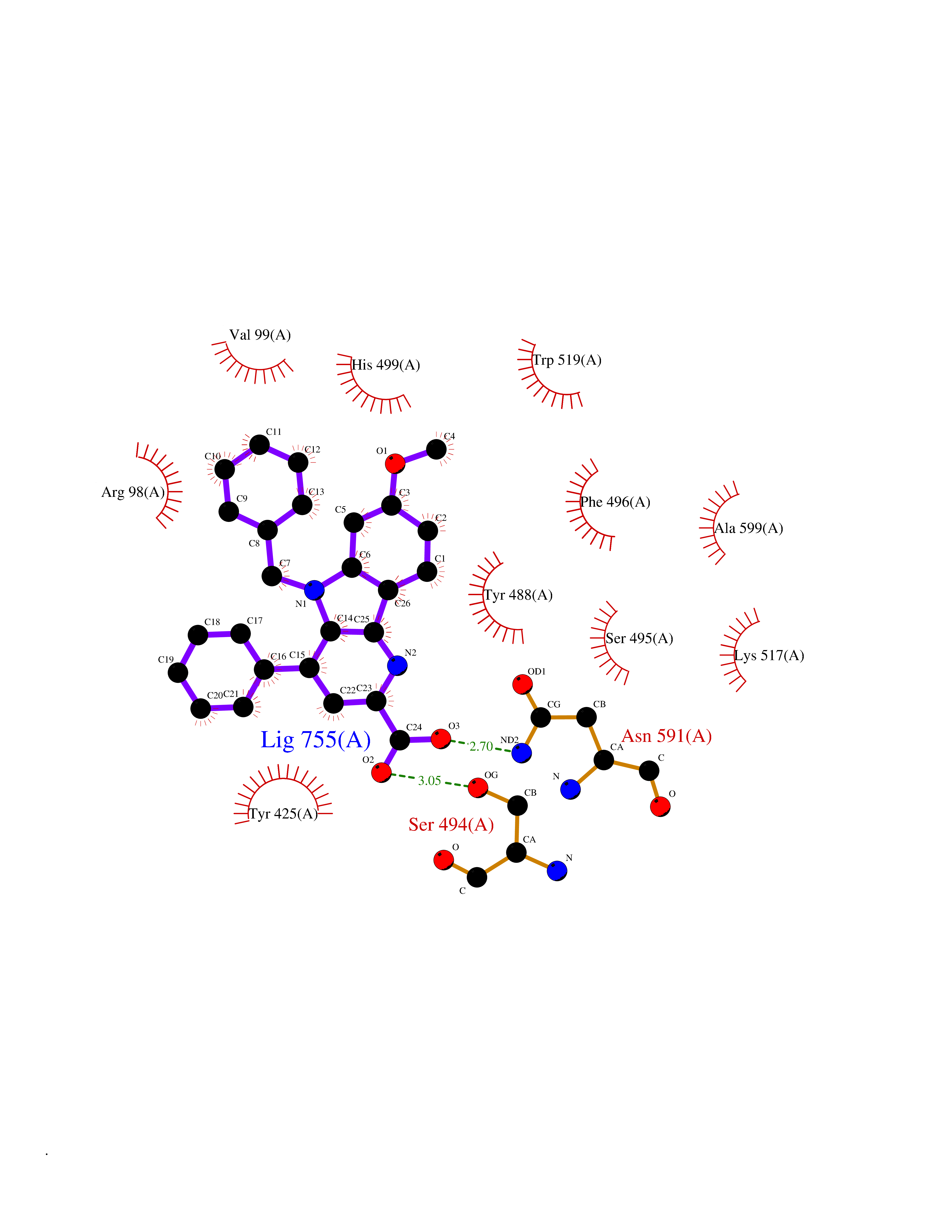 Ligplot Map 3D Binding mode Sequence SMFLPPPECPVFEPSWEEFADPFAFIHKIRPIAEQTGICKVRPPPDWQPPFACDVDKLHFTPRIQRLNELEAQTRVKLGGGGARDYTLRTFGEMADAFKSDYFNMPVHMVPTELVEKEFWRLVSTIEEDVTVEYGADIASKEFGSGFPVRDIKLSPEEEEYLDSGWNLNNMPVMEQSVLAHITADICGMKLPWLYVGMCFSSFCWHIEDHWSYSINYLHWGEPKTWYGVPGYAAEQLENVMKKLAPELFVSQPDLLHQLVTIMNPNTLMTHEVPVYRTNQCAGEFVITFPRAYHSGFNQGFNFAEAVNFCTVDWLPLGRQCVEHYRLLHRYCVFSHDEMICKMASKADVLDVVVASTVQKDMAIMIEDEKALRETVRKLGVIDSERMDFELLPDDERQCVKCKTTCFMSAISCSCKPGLLVCLHHVKELCSCPPYKYKLRYRYTLDDLYPMMNALKLRAE Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Metabotropic glutamate receptor 2 (mGluR2) | 7E9G | 7.65 | |
Target general information Gen name GRM2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms mGLUR2; Group II metabotropic glutamate receptor; Glutamate receptor mGLU2; GPRC1B Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors, such as adenylate cyclase. Signaling inhibits adenylate cyclase activity. May mediate suppression of neurotransmission or may be involved in synaptogenesis or synaptic stabilization. G-protein coupled receptor for glutamate. Related diseases Oocyte/zygote/embryo maturation arrest 21 (OZEMA21) [MIM:620610]: An autosomal dominant, female infertility disorder characterized by zygote development arrest due to failure of pronuclei fusion. {ECO:0000269|PubMed:33948904, ECO:0000269|PubMed:33953335}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05096 Interacts with Q5T8D3-2; Q9BYF1; Q13520; Q13323; Q8WV48; P57739; O95484; Q7Z7G2; P00387; P27487; P28223-1; Q5SR56; O14880; Q8N4V1; Q58DX5; Q13113; Q9NR31; Q8IWU4; Q9H2H9; P27105; Q8N3G9; Q96Q45-2; Q9NWD8; Q8WUV1; Q9UMX0-2; P0DTC2 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cell projection; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 85146.2 Length 769 Aromaticity 0.11 Instability index 36.84 Isoelectric point 8.48 Charge (pH=7) 9.7 2D Binding mode Binding energy (Kcal/mol) -10.43 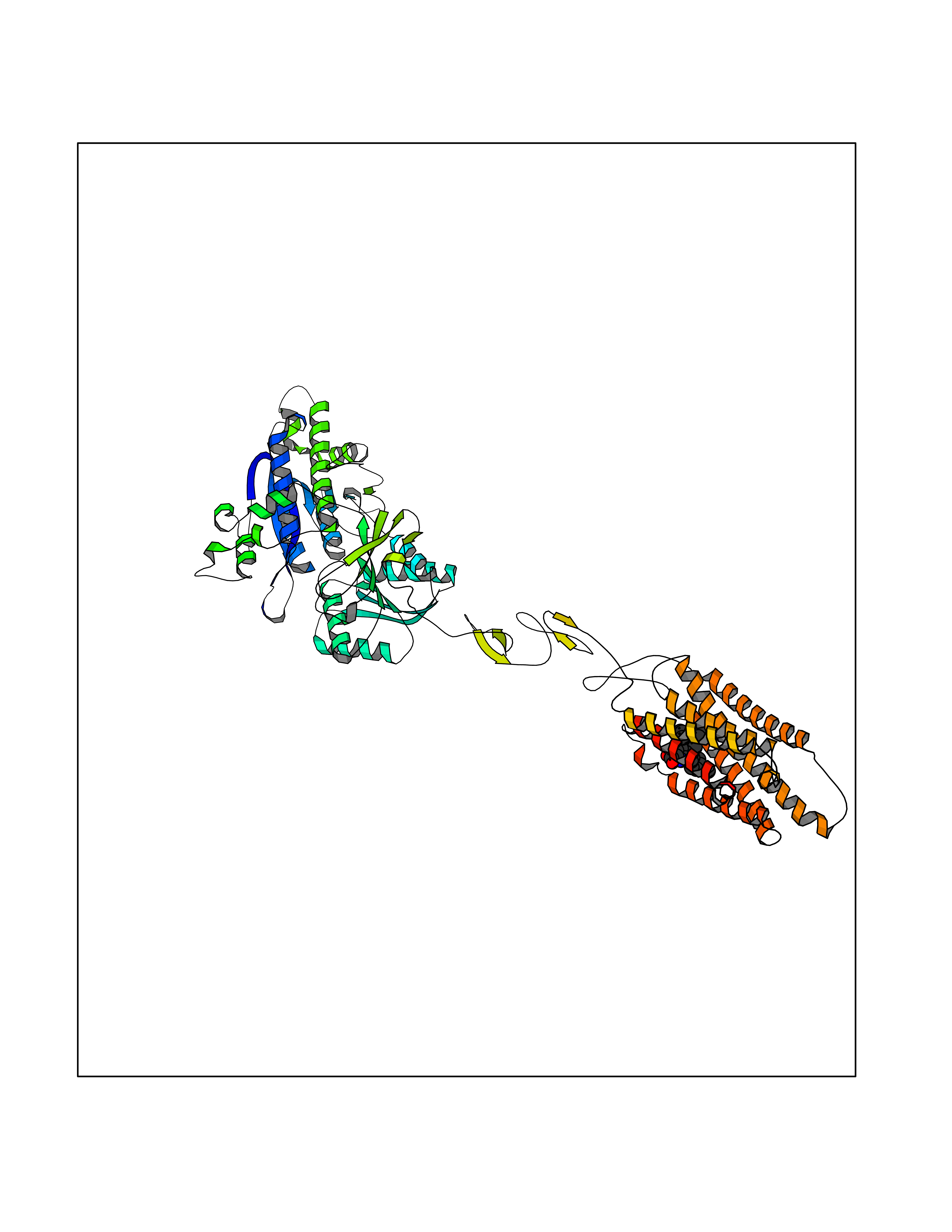 Molscript Map 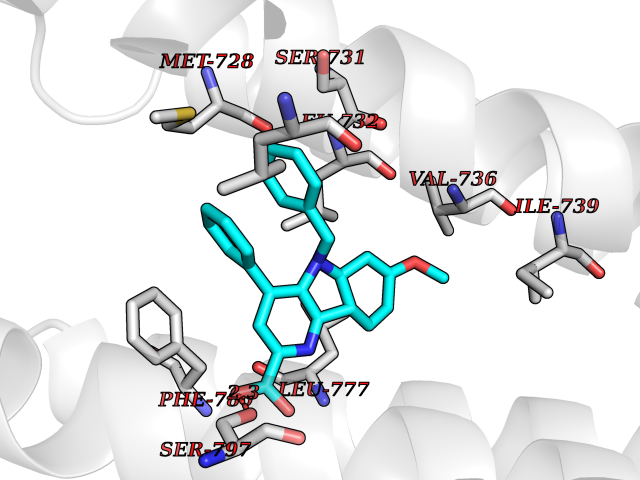 Pymol Map 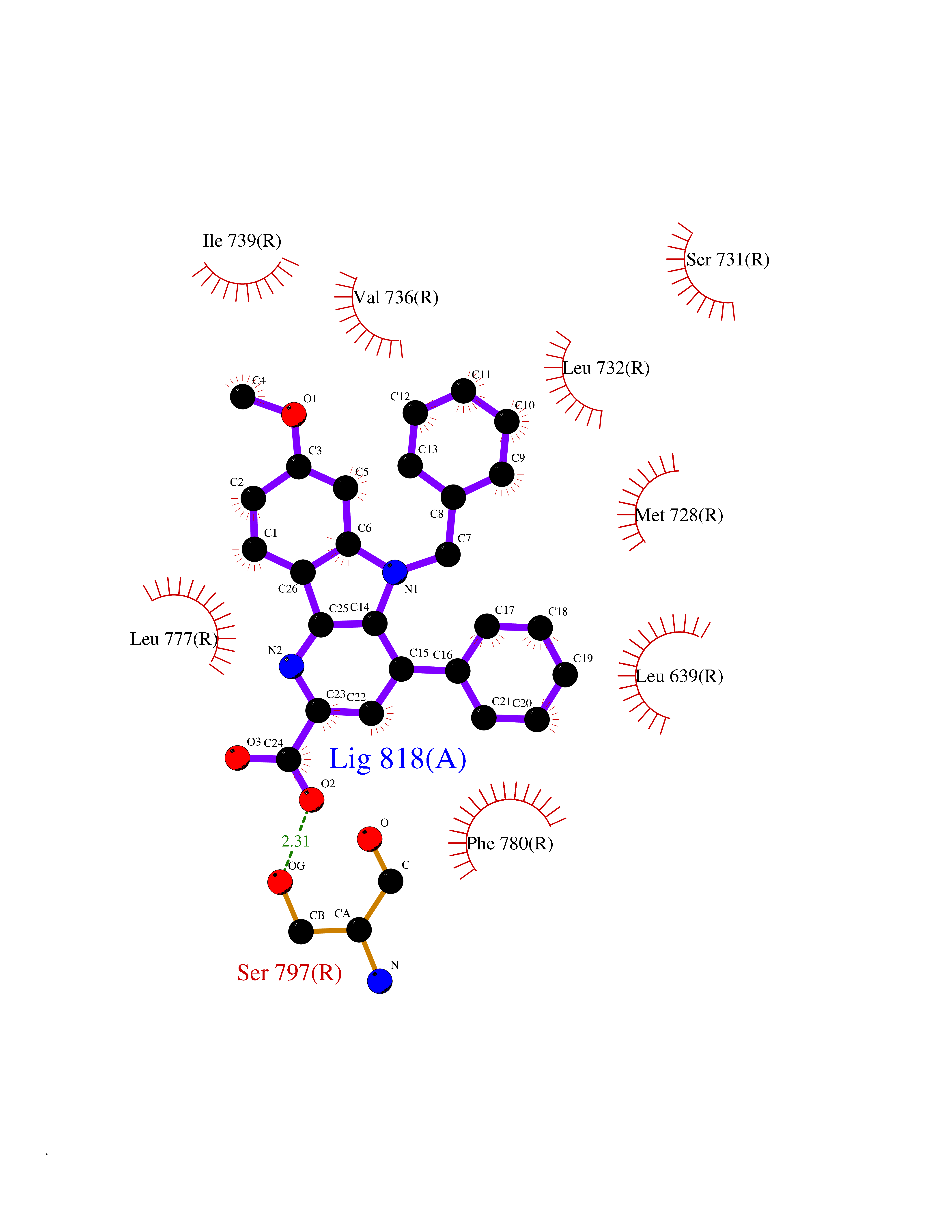 Ligplot Map 3D Binding mode Sequence KKVLTLEGDLVLGGLFPVHQKGGPAEDCGPVNEHRGIQRLEAMLFALDRINRDPHLLPGVRLGAHILDSCSKDTHALEQALDFVRASLITGVIGGSYSDVSIQVANLLRLFQIPQISYASTSAKLSDKSRYDYFARTVPPDFFQAKAMAEILRFFNWTYVSTVASEGDYGETGIEAFELEARARNICVATSEKVGRAMSRAAFEGVVRALLQKPSARVAVLFTRSEDARELLAASQRLNASFTWVASDGWGALESVVAGSEGAAEGAITIELASYPISDFASYFQSLDPWNNSRNPWFREFWEQRFRCSFRQRDCAAHSLRAVPFEQESKIMFVVNAVYAMAHALHNMHRALCPNTTRLCDAMRPVNGRRLYKDFVLNVKFDAPFRPADTHNEVRFDRFGDGIGRYNIFTYLRAGSGRYRYQKVGYWAEGLTLDTSLIPWASPSAGPLPASRCSEPCLQNEVKSVQPGEVCCWLCIPCQPYEYRLDEFTCADCGLGYWPNASLTGCFELPQEYIRWGDAWAVGPVTIACLGALATLFVLGVFVRHNATPVVKAAGRELCYILLGGVFLCYCMTFIFIAKPSTAVCTLRRLGLGTAFSVCYSALLTKTNRIARIFGGAREGAQRPRFISPASQVAICLALISGQLLIVVAWLVVEAPGTGKETAPERREVVTLRCNHRDASMLGSLAYNVLLIALCTLYAFKTRKCPENFNEAKFIGFTMYTTCIIWLAFLPIFYVTSSDYRVQTTTMCVSVSLSGSVVLGCLFAPKLHI Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Hemoglobin subunit alpha | 1IRD | 7.64 | |
Target general information Gen name HBA1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms HBA2 Protein family Globin family Biochemical class Oxygen storage / transport Function Heme binding.Iron ion binding.Oxygen binding.Oxygen transporter activity. Related diseases Heinz body anemias (HEIBAN) [MIM:140700]: Form of non-spherocytic hemolytic anemia of Dacie type 1. After splenectomy, which has little benefit, basophilic inclusions called Heinz bodies are demonstrable in the erythrocytes. Before splenectomy, diffuse or punctate basophilia may be evident. Most of these cases are probably instances of hemoglobinopathy. The hemoglobin demonstrates heat lability. Heinz bodies are observed also with the Ivemark syndrome (asplenia with cardiovascular anomalies) and with glutathione peroxidase deficiency. {ECO:0000269|PubMed:2833478}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Alpha-thalassemia (A-THAL) [MIM:604131]: A form of thalassemia. Thalassemias are common monogenic diseases occurring mostly in Mediterranean and Southeast Asian populations. The hallmark of alpha-thalassemia is an imbalance in globin-chain production in the adult HbA molecule. The level of alpha chain production can range from none to very nearly normal levels. Deletion of both copies of each of the two alpha-globin genes causes alpha(0)-thalassemia, also known as homozygous alpha thalassemia. Due to the complete absence of alpha chains, the predominant fetal hemoglobin is a tetramer of gamma-chains (Bart hemoglobin) that has essentially no oxygen carrying capacity. This causes oxygen starvation in the fetal tissues leading to prenatal lethality or early neonatal death. The loss of two alpha genes results in mild alpha-thalassemia, also known as heterozygous alpha-thalassemia. Affected individuals have small red cells and a mild anemia (microcytosis). If three of the four alpha-globin genes are functional, individuals are completely asymptomatic. Some rare forms of alpha-thalassemia are due to point mutations (non-deletional alpha-thalassemia). The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Alpha(0)-thalassemia is associated with non-immune hydrops fetalis, a generalized edema of the fetus with fluid accumulation in the body cavities due to non-immune causes. Non-immune hydrops fetalis is not a diagnosis in itself but a symptom, a feature of many genetic disorders, and the end-stage of a wide variety of disorders.; DISEASE: Hemoglobin H disease (HBH) [MIM:613978]: A form of alpha-thalassemia due to the loss of three alpha genes. This results in high levels of a tetramer of four beta chains (hemoglobin H), causing a severe and life-threatening anemia. Untreated, most patients die in childhood or early adolescence. {ECO:0000269|PubMed:10569720}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08262; DB07427; DB08077; DB07428; DB02126; DB09130; DB08486; DB15617; DB09147; DB13995; DB14490; DB14491; DB14488; DB14501; DB14489; DB13257; DB01592; DB00893; DB09112; DB09140; DB06154; DB07645; DB09517; DB08632; DB14975; DB01593; DB14487; DB14533; DB14548 Interacts with Q9NZD4; Q2TAC2; P00387; P68871; P02042; P02100; P69892; P09105; Q15323; O76011; Q6A162; P29474; P0DPK4 EC number NA Uniprot keywords 3D-structure; Acetylation; Direct protein sequencing; Disease variant; Glycation; Glycoprotein; Heme; Hereditary hemolytic anemia; Iron; Metal-binding; Oxygen transport; Phosphoprotein; Proteomics identification; Reference proteome; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 30975.2 Length 287 Aromaticity 0.08 Instability index 6.57 Isoelectric point 7.92 Charge (pH=7) 2.38 2D Binding mode Binding energy (Kcal/mol) -10.42 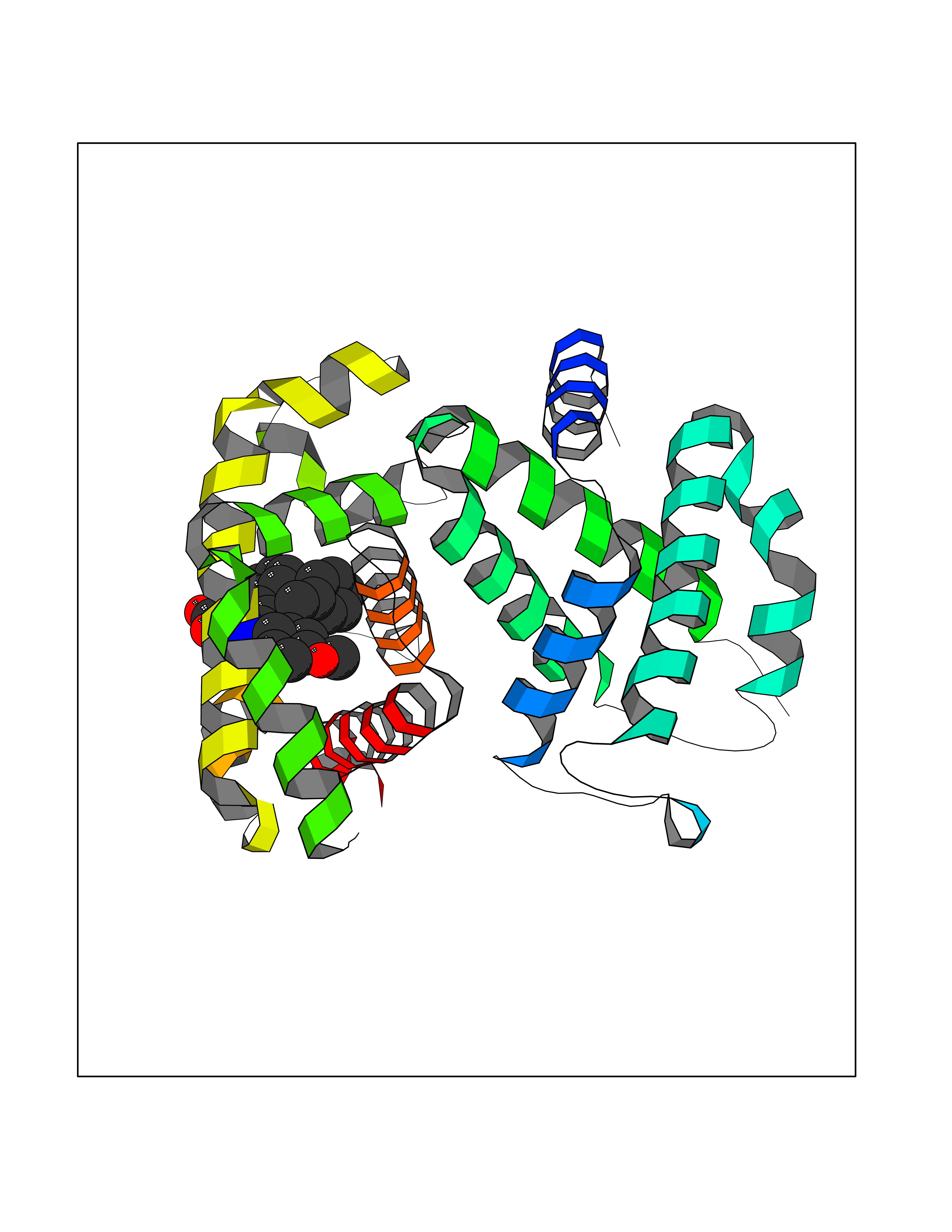 Molscript Map 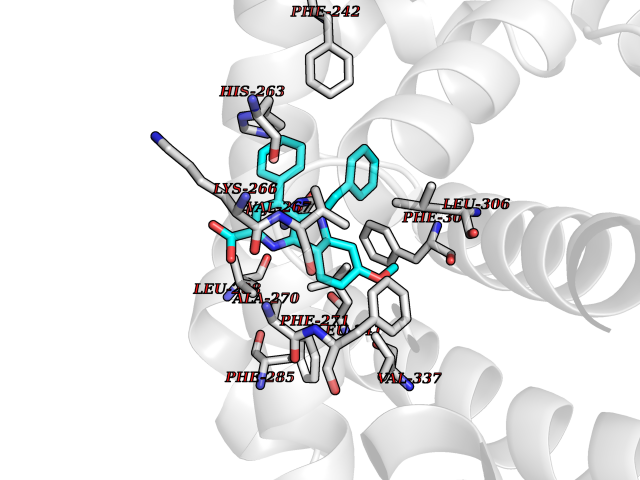 Pymol Map 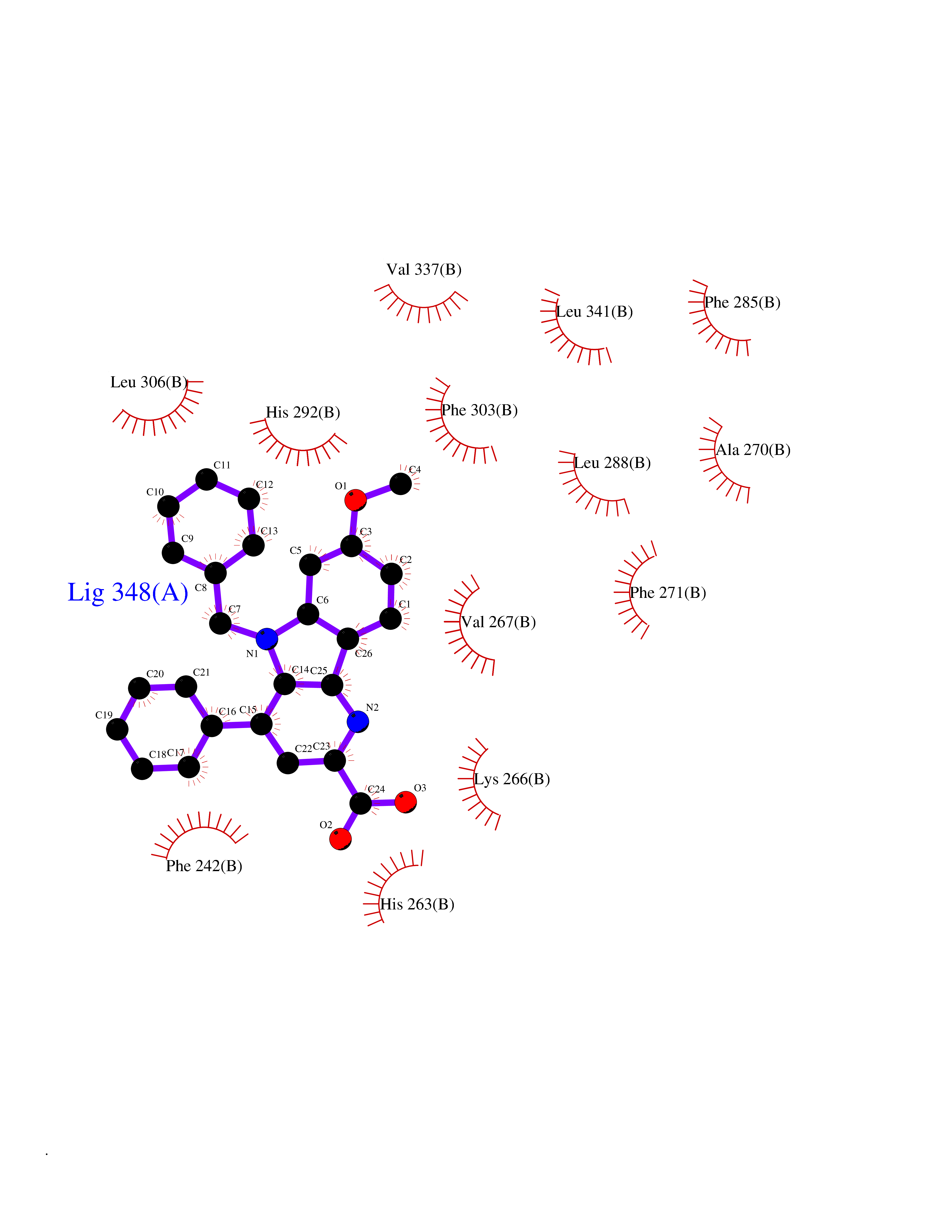 Ligplot Map 3D Binding mode Sequence VLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDLSHGSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTSKYRVHLTPEEKSAVTALWGKVNVDEVGGEALGRLLVVYPWTQRFFESFGDLSTPDAVMGNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATLSELHCDKLHVDPENFRLLGNVLVCVLAHHFGKEFTPPVQAAYQKVVAGVANALAHKYH Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Thyrotropin receptor (TSHR) | 7XW6 | 7.64 | |
Target general information Gen name TSHR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Thyroid stimulating hormone receptor; TSHR; TSH-R; TSH receptor Protein family G-protein coupled receptor 1 family, FSH/LSH/TSH subfamily Biochemical class GPCR rhodopsin Function Receptor for thyrothropin. Plays a central role in controlling thyroid cell metabolism. The activity of this receptor is mediated by G proteins which activate adenylate cyclase. Also acts as a receptor for thyrostimulin (gpa2+gpb5). Related diseases Defects in TSHR are found in patients affected by hyperthyroidism with different etiologies. Somatic, constitutively activating TSHR mutations and/or constitutively activating G(s)alpha mutations have been identified in toxic thyroid nodules (TTNs) that are the predominant cause of hyperthyroidism in iodine deficient areas. These mutations lead to TSH independent activation of the cAMP cascade resulting in thyroid growth and hormone production. TSHR mutations are found in autonomously functioning thyroid nodules (AFTN), toxic multinodular goiter (TMNG) and hyperfunctioning thyroid adenomas (HTA). TMNG encompasses a spectrum of different clinical entities, ranging from a single hyperfunctioning nodule within an enlarged thyroid, to multiple hyperfunctioning areas scattered throughout the gland. HTA are discrete encapsulated neoplasms characterized by TSH-independent autonomous growth, hypersecretion of thyroid hormones, and TSH suppression. Defects in TSHR are also a cause of thyroid neoplasms (papillary and follicular cancers).; DISEASE: Autoantibodies against TSHR are directly responsible for the pathogenesis and hyperthyroidism of Graves disease. Antibody interaction with TSHR results in an uncontrolled receptor stimulation.; DISEASE: Hypothyroidism, congenital, non-goitrous, 1 (CHNG1) [MIM:275200]: A non-autoimmune condition characterized by resistance to thyroid-stimulating hormone (TSH) leading to increased levels of plasma TSH and low levels of thyroid hormone. It presents variable severity depending on the completeness of the defect. Most patients are euthyroid and asymptomatic, with a normal sized thyroid gland. Only a subset of patients develop hypothyroidism and present a hypoplastic thyroid gland. {ECO:0000269|PubMed:10720030, ECO:0000269|PubMed:11095460, ECO:0000269|PubMed:11442002, ECO:0000269|PubMed:12050212, ECO:0000269|PubMed:14725684, ECO:0000269|PubMed:15531543, ECO:0000269|PubMed:25978107, ECO:0000269|PubMed:7528344, ECO:0000269|PubMed:8954020, ECO:0000269|PubMed:9100579, ECO:0000269|PubMed:9185526, ECO:0000269|PubMed:9329388}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Familial gestational hyperthyroidism (HTFG) [MIM:603373]: A condition characterized by abnormally high levels of serum thyroid hormones occurring during early pregnancy. {ECO:0000269|PubMed:9854118}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperthyroidism, non-autoimmune (HTNA) [MIM:609152]: A condition characterized by abnormally high levels of serum thyroid hormones, thyroid hyperplasia, goiter and lack of anti-thyroid antibodies. Typical features of Graves disease such as exophthalmia, myxedema, antibodies anti-TSH receptor and lymphocytic infiltration of the thyroid gland are absent. {ECO:0000269|PubMed:10199795, ECO:0000269|PubMed:10852462, ECO:0000269|PubMed:11081252, ECO:0000269|PubMed:11127522, ECO:0000269|PubMed:11201847, ECO:0000269|PubMed:11517004, ECO:0000269|PubMed:11549687, ECO:0000269|PubMed:15163335, ECO:0000269|PubMed:7800007, ECO:0000269|PubMed:7920658, ECO:0000269|PubMed:8636266, ECO:0000269|PubMed:8964822, ECO:0000269|PubMed:9349581, ECO:0000269|PubMed:9360555, ECO:0000269|PubMed:9398746, ECO:0000269|PubMed:9589634}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00024 Interacts with P30542; Q9NPA3; Q14160; P21579 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Congenital hypothyroidism; Direct protein sequencing; Disease variant; Disulfide bond; G-protein coupled receptor; Glycoprotein; Leucine-rich repeat; Membrane; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Sulfation; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 63643.2 Length 562 Aromaticity 0.12 Instability index 33.57 Isoelectric point 8.41 Charge (pH=7) 6.83 2D Binding mode Binding energy (Kcal/mol) -10.43 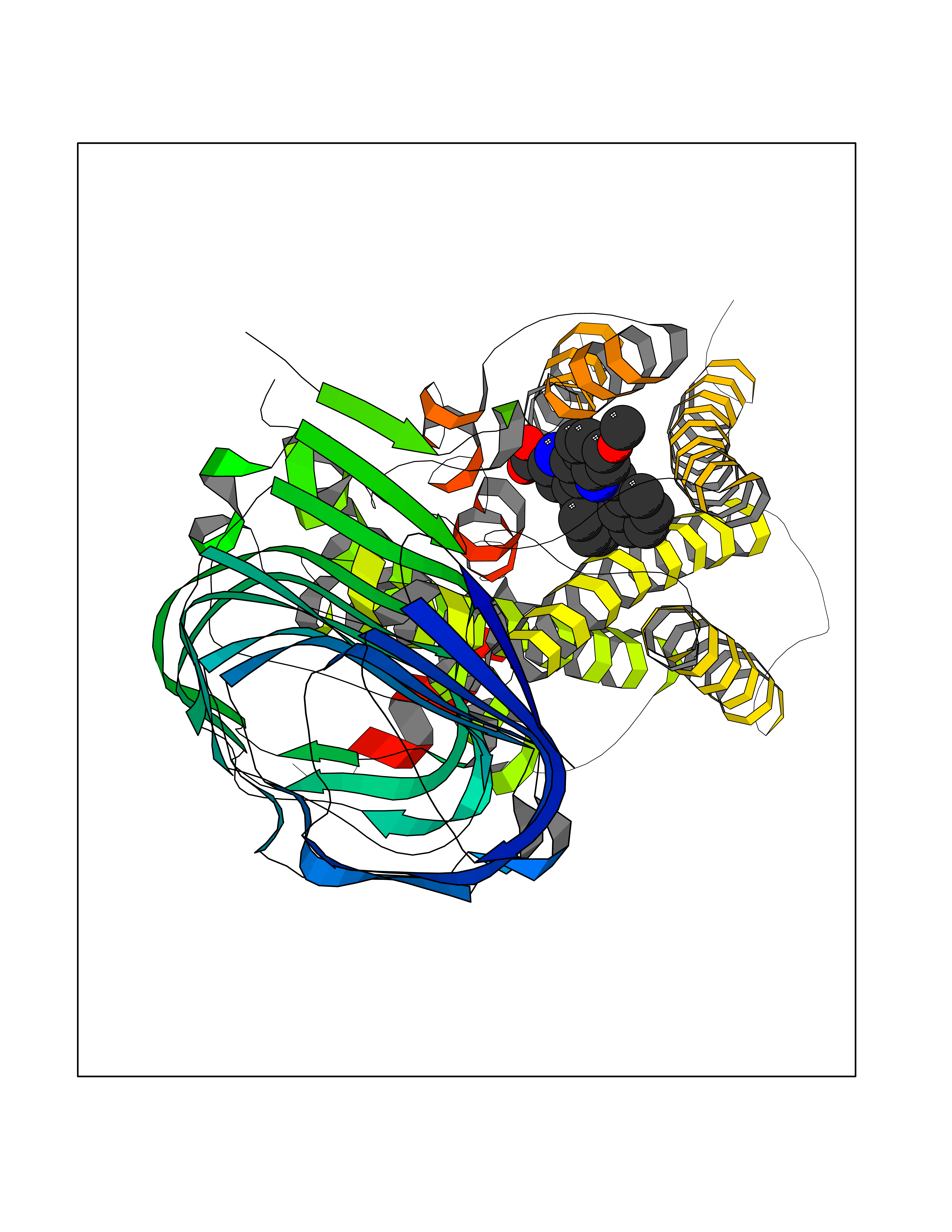 Molscript Map 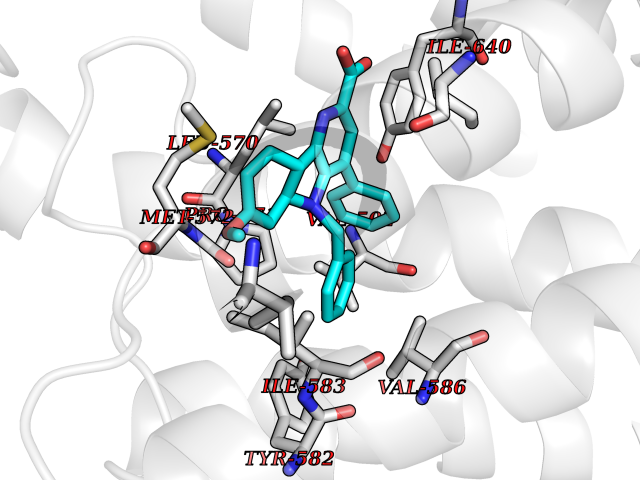 Pymol Map 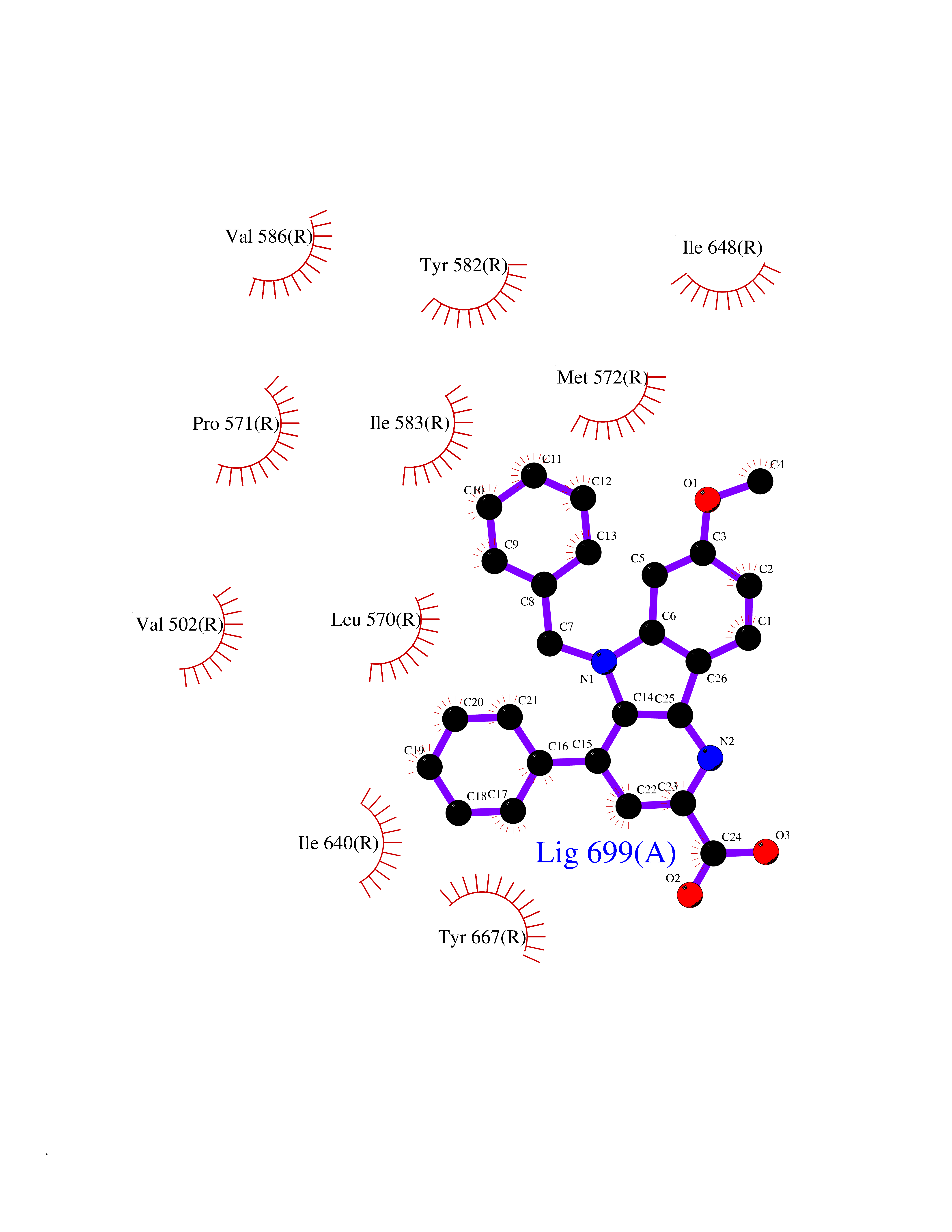 Ligplot Map 3D Binding mode Sequence PCECHQEEDFRVTCKDIQRIPSLPPSTQTLKLIETHLRTIPSHAFSNLPNISRIYVSIDVTLQQLESHSFYNLSKVTHIEIRNTRNLTYIDPDALKELPLLKFLGIFNTGLKMFPDLTKVYSTDIFFILEITDNPYMTSIPVNAFQGLCNETLTLKLYNNGFTSVQGYAFNGTKLDAVYLNKNKYLTVIDKDAFGGVYSGPSLLDVSQTSVTALPSKGLEHLKELIARNTWTLKKLPLSLSFLHLTRADLSYPIHCCAFKNQKEDMVCTPKSDEFNPCEDIMGYKFLRIVVWFVSLLALLGNVFVLLILLTSHYKLNVPRFLMCNLAFADFCMGMYLLLIASVDLYTHSEYYNHAIDWQTGPGCNTAGFFTVFASELSVYTLTVITLERWYAITFAMRLDRKIRLRHACAIMVGGWVCCFLLALLPLVGISSYAKVSICLPMDTETPLALAYIVFVLTLNIVAFVIVCCCYVKIYITVRNPDKDTKIAKRMAVLIFTDFICMAPISFYALSAILNKPLITVSNSKILLVLFYPLNSCANPFLYAIFTKAFQRDVFILLSKFG Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Matrix metalloproteinase-10 (MMP-10) | 1Q3A | 7.63 | |
Target general information Gen name MMP10 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Transin-2; Stromelysin-2; STMY2; SL-2 Protein family Peptidase M10A family Biochemical class Peptidase Function Activates procollagenase. Can degrade fibronectin, gelatins of type I, III, IV, and V; weakly collagens III, IV, and V. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00786; DB08271 Interacts with NA EC number EC 3.4.24.22 Uniprot keywords 3D-structure; Calcium; Collagen degradation; Disulfide bond; Extracellular matrix; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 52822 Length 471 Aromaticity 0.12 Instability index 21.13 Isoelectric point 4.83 Charge (pH=7) -35.32 2D Binding mode Binding energy (Kcal/mol) -10.41 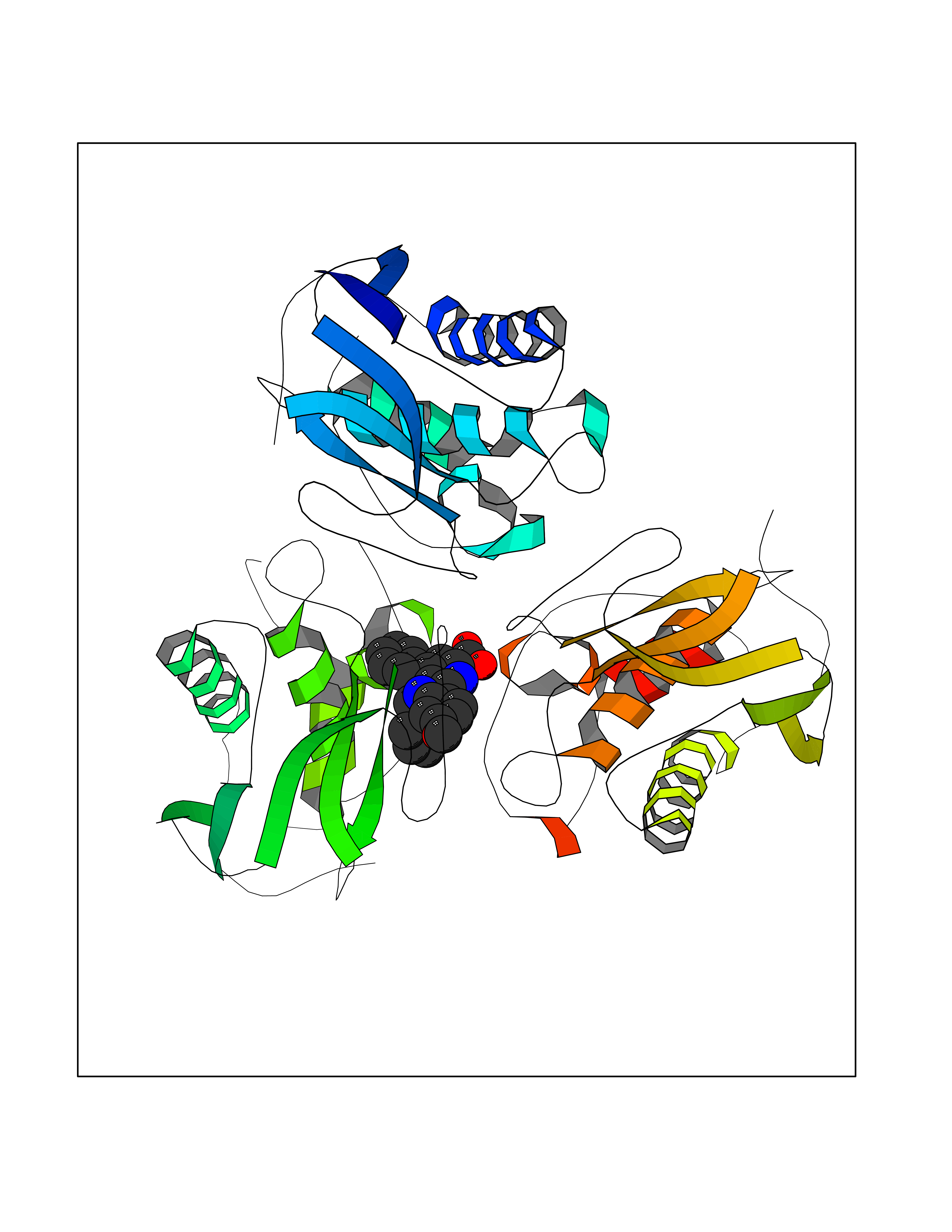 Molscript Map  Pymol Map 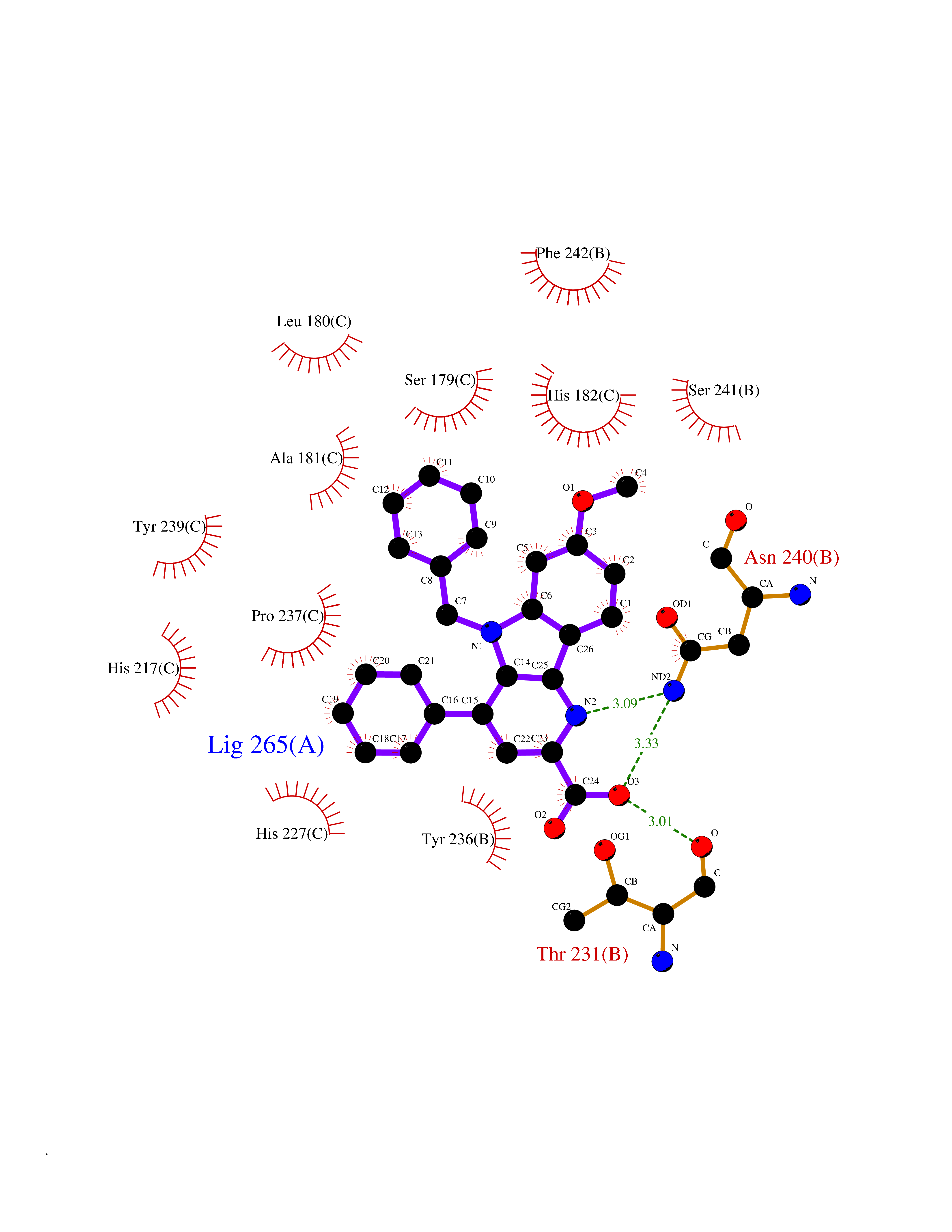 Ligplot Map 3D Binding mode Sequence MPKWRKTHLTYRIVNYTPDLPRDAVDSAIEKALKVWEEVTPLTFSRLYEGEADIMISFAVKEHGDNYSFDGPGHSLAHAYPPGPGLYGDIHFDDDEKWTEDASGTNLFLVAAHELGHSLGLFHSANTEALMYPLYNSLAQFRLSQDDVNGIQSLYGPKWRKTHLTYRIVNYTPDLPRDAVDSAIEKALKVWEEVTPLTFSRLYEGEADIMISFAVKEHGDNYSFDGPGHSLAHAYPPGPGLYGDIHFDDDEKWTEDASGTNLFLVAAHELGHSLGLFHSANTEALMYPLYNSLAQFRLSQDDVNGIQSLYGGMPKWRKTHLTYRIVNYTPDLPRDAVDSAIEKALKVWEEVTPLTFSRLYEGEADIMISFAVKEHGDNYSFDGPGHSLAHAYPPGPGLYGDIHFDDDEKWTEDASGTNLFLVAAHELGHSLGLFHSANTEALMYPLYNSFTELAQFRLSQDDVNGIQSLYG Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Pyruvate kinase M2 (PKM) | 3GR4 | 7.63 | |
Target general information Gen name PKM Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p58; Tumor M2-PK; Thyroid hormone-binding protein 1; THBP1; Pyruvate kinase muscle isozyme; Pyruvate kinase isozymes M1/M2; Pyruvate kinase PKM; Pyruvate kinase 2/3; PKM2; PK3; PK2; Opa-interacting pr Protein family Pyruvate kinase family Biochemical class Kinase Function Stimulates POU5F1-mediated transcriptional activation. Plays a general role in caspase independent cell death of tumor cells. The ratio between the highly active tetrameric form and nearly inactive dimeric form determines whether glucose carbons are channeled to biosynthetic processes or used for glycolytic ATP production. The transition between the 2 forms contributes to the control of glycolysis and is important for tumor cell proliferation and survival. Promotes in a STAT1-dependent manner, the expression of the immune checkpoint protein CD274 in ARNTL/BMAL1-deficient macrophages. Glycolytic enzyme that catalyzes the transfer of a phosphoryl group from phosphoenolpyruvate (PEP) to ADP, generating ATP. Related diseases Congenital sucrase-isomaltase deficiency (CSID) [MIM:222900]: Autosomal recessive intestinal disorder that is clinically characterized by fermentative diarrhea, abdominal pain, and cramps upon ingestion of sugar. The symptoms are the consequence of absent or drastically reduced enzymatic activities of sucrase and isomaltase. The prevalence of CSID is 0.02 % in individuals of European descent and appears to be much higher in Greenland, Alaskan, and Canadian native people. CSID arises due to post-translational perturbations in the intracellular transport, polarized sorting, aberrant processing, and defective function of SI. {ECO:0000269|PubMed:10903344, ECO:0000269|PubMed:11340066, ECO:0000269|PubMed:14724820, ECO:0000269|PubMed:16329100, ECO:0000269|PubMed:8609217}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07697; DB07692; DB02726; DB07628; DB00787; DB11638; DB09130; DB08951; DB01733; DB11263; DB00119 Interacts with P49407; P32121; Q96IK1-2; P35222; P53355; P22607; P42858; P04049; Q8N488; Q7Z699; Q9BSI4; Q9UMX0; Q9Y649; Q9WMX2; P35222; P53355; Q9H6Z9; P68431; Q16665; P27361 EC number EC 2.7.1.40 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; ATP-binding; Cytoplasm; Direct protein sequencing; Glycolysis; Hydroxylation; Isopeptide bond; Kinase; Magnesium; Metal-binding; Methylation; Nucleotide-binding; Nucleus; Phosphoprotein; Potassium; Proteomics identification; Pyruvate; Reference proteome; S-nitrosylation; Transferase; Translation regulation; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 112053 Length 1024 Aromaticity 0.05 Instability index 27.06 Isoelectric point 7.34 Charge (pH=7) 1.66 2D Binding mode Binding energy (Kcal/mol) -10.41 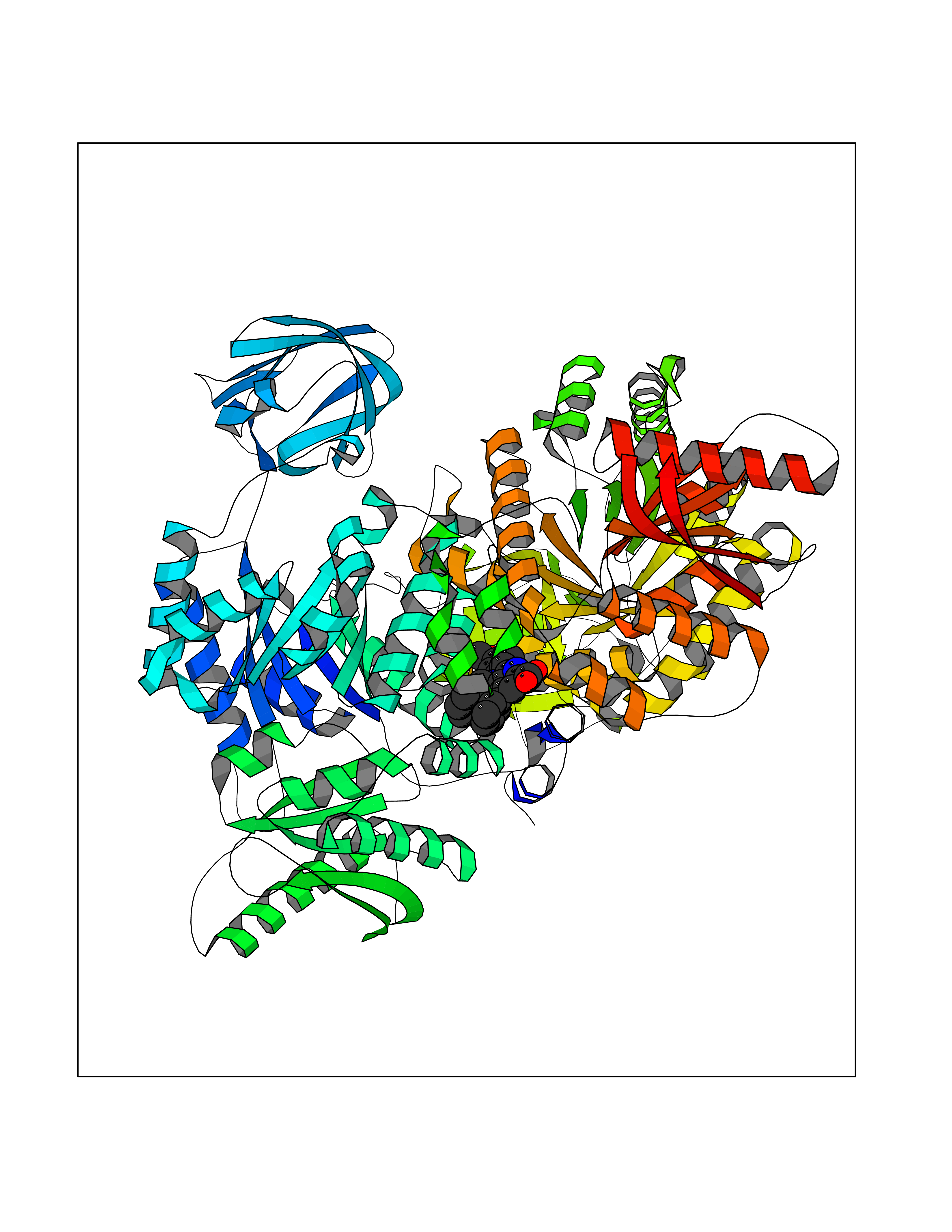 Molscript Map 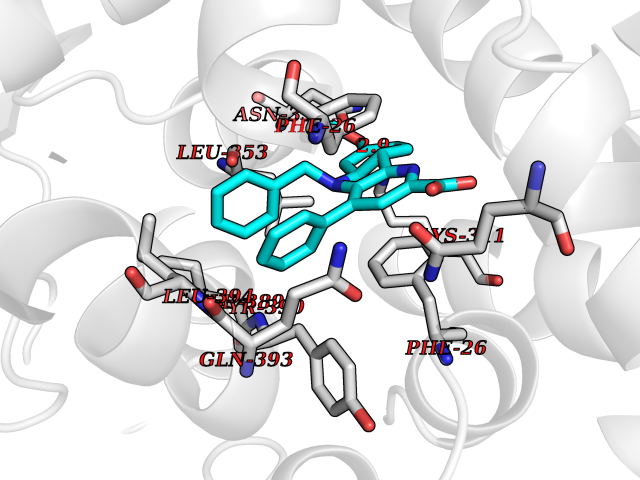 Pymol Map 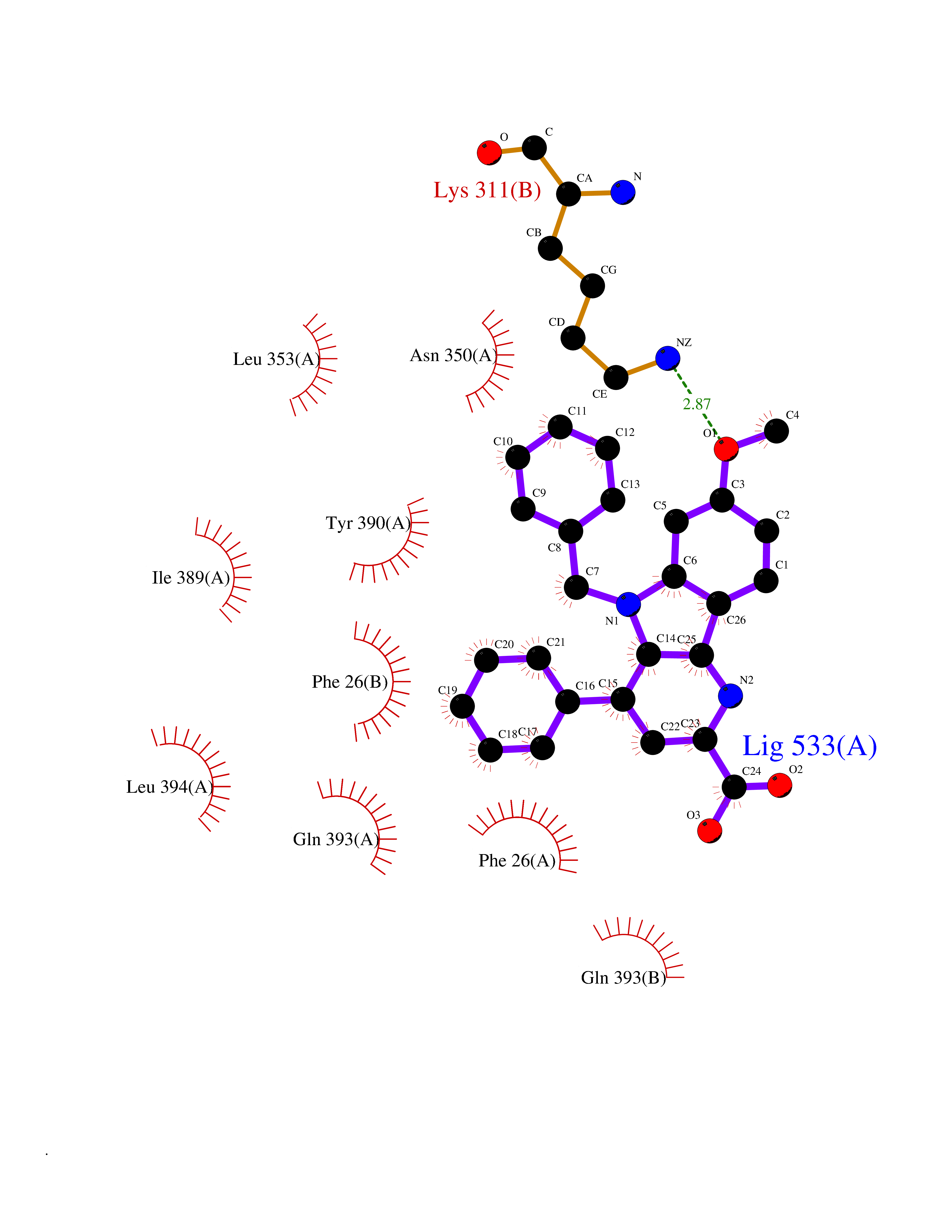 Ligplot Map 3D Binding mode Sequence IQTQQLHAAMADTFLEHMCRLDIDSPPITARNTGIICTIGPASRSVETLKEMIKSGMNVARLNFSHGTHEYHAETIKNVRTATESFASDPILYRPVAVALDTKGPEIRTGLIKGSGTAEVELKKGATLKITLDNAYMEKCDENILWLDYKNICKVVEVGSKIYVDDGLISLQVKQKGADFLVTEVENGGSLGSKKGVNLPGAAVDLPAVSEKDIQDLKFGVEQDVDMVFASFIRKASDVHEVRKVLGEKGKNIKIISKIENHEGVRRFDEILEASDGIMVARGDLGIEIPAEKVFLAQKMMIGRCNRAGKPVICATQMLESMIKKPRPTRAEGSDVANAVLDGADCIMLSGETAKGDYPLEAVRMQHLIAREAEAAIYHLQLFEELRRLAPITSDPTEATAVGAVEASFKCCSGAIIVLTKSGRSAHQVARYRPRAPIIAVTRNPQTARQAHLYRGIFPVLCKDPVQEAWAEDVDLRVNFAMNVGKARGFFKKGDVVIVLTGWRPGSGFTNTMRVVPVPIQTQQLHAAMADTFLEHMCRLDIDSPPITARNTGIICTIGPASRSVETLKEMIKSGMNVARLNFSHGTHEYHAETIKNVRTATESFASDPILYRPVAVALDTKGPEIRTGLIKEVEATLKITLDNAYMEKCDENILWLDYKNICKVVEVGSKIYVDDGLISLQVDFLVTEVENGGSLGSKKGVNLPGAAVDLPAVSEKDIQDLKFGVEQDVDMVFASFIRKASDVHEVRKVLGEKGKNIKIISKIENHEGVRRFDEILEASDGIMVARGDLGIEIPAEKVFLAQKMMIGRCNRAGKPVICATQMLESMIKKPRPTRAEGSDVANAVLDGADCIMLSGETAKGDYPLEAVRMQHLIAREAEAAIYHLQLFEELRRLAPITSDPTEATAVGAVEASFKCCSGAIIVLTKSGRSAHQVARYRPRAPIIAVTRNPQTARQAHLYRGIFPVLCKDPVQEAWAEDVDLRVNFAMNVGKARGFFKKGDVVIVLTGWRPGSGFTNTMRVVPVP Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Cholesterol oxidase | 1COY | 7.63 | |
Target general information Gen name choB Organism Brevibacterium sterolicum Uniprot ID TTD ID NA Synonyms NA Protein family GMC oxidoreductase family Biochemical class Oxidoreductase(oxygen receptor) Function Cholesterol oxidase activity.Flavin adenine dinucleotide binding.Steroid delta-isomerase activity. Related diseases Achondroplasia (ACH) [MIM:100800]: A frequent form of short-limb dwarfism. It is characterized by a long, narrow trunk, short extremities, particularly in the proximal (rhizomelic) segments, a large head with frontal bossing, hypoplasia of the midface and a trident configuration of the hands. ACH is an autosomal dominant disease. {ECO:0000269|PubMed:10611230, ECO:0000269|PubMed:12297284, ECO:0000269|PubMed:7758520, ECO:0000269|PubMed:7847369, ECO:0000269|PubMed:8078586, ECO:0000269|PubMed:8599935}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Crouzon syndrome with acanthosis nigricans (CAN) [MIM:612247]: Classic Crouzon disease which is caused by mutations in the FGFR2 gene is characterized by craniosynostosis (premature fusion of the skull sutures), and facial hypoplasia. Crouzon syndrome with acanthosis nigricans (a skin disorder characterized by pigmentation anomalies), CAN, is considered to be an independent disorder from classic Crouzon syndrome. CAN is characterized by additional more severe physical manifestation, such as Chiari malformation, hydrocephalus, and atresia or stenosis of the choanas, and is caused by a specific mutation (Ala-391 to Glu) in the transmembrane domain of FGFR3. It is proposed to have an autosomal dominant mode of inheritance. {ECO:0000269|PubMed:17935505, ECO:0000269|PubMed:7493034}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thanatophoric dysplasia 1 (TD1) [MIM:187600]: A neonatal lethal skeletal dysplasia. Affected individuals manifest severe shortening of the limbs with macrocephaly, narrow thorax, short ribs, and curved femurs. {ECO:0000269|PubMed:10360402, ECO:0000269|PubMed:10671061, ECO:0000269|PubMed:7773297, ECO:0000269|PubMed:8589699, ECO:0000269|PubMed:8845844, ECO:0000269|PubMed:9790257}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thanatophoric dysplasia 2 (TD2) [MIM:187601]: A neonatal lethal skeletal dysplasia causing severe shortening of the limbs, narrow thorax and short ribs. Patients with thanatophoric dysplasia type 2 have straight femurs and cloverleaf skull. {ECO:0000269|PubMed:12297284, ECO:0000269|PubMed:7773297, ECO:0000269|PubMed:8754806}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hypochondroplasia (HCH) [MIM:146000]: Autosomal dominant disease and is characterized by disproportionate short stature. It resembles achondroplasia, but with a less severe phenotype. {ECO:0000269|PubMed:10215410, ECO:0000269|PubMed:10777366, ECO:0000269|PubMed:11055896, ECO:0000269|PubMed:12707965, ECO:0000269|PubMed:7670477, ECO:0000269|PubMed:9452043}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Bladder cancer (BLC) [MIM:109800]: A malignancy originating in tissues of the urinary bladder. It often presents with multiple tumors appearing at different times and at different sites in the bladder. Most bladder cancers are transitional cell carcinomas that begin in cells that normally make up the inner lining of the bladder. Other types of bladder cancer include squamous cell carcinoma (cancer that begins in thin, flat cells) and adenocarcinoma (cancer that begins in cells that make and release mucus and other fluids). Bladder cancer is a complex disorder with both genetic and environmental influences. {ECO:0000269|PubMed:10471491, ECO:0000269|PubMed:11314002}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Somatic mutations can constitutively activate FGFR3.; DISEASE: Cervical cancer (CERCA) [MIM:603956]: A malignant neoplasm of the cervix, typically originating from a dysplastic or premalignant lesion previously present at the active squamocolumnar junction. The transformation from mild dysplastic to invasive carcinoma generally occurs slowly within several years, although the rate of this process varies widely. Carcinoma in situ is particularly known to precede invasive cervical cancer in most cases. Cervical cancer is strongly associated with infection by oncogenic types of human papillomavirus. {ECO:0000269|PubMed:10471491}. The gene represented in this entry is involved in disease pathogenesis.; DISEASE: Camptodactyly, tall stature, and hearing loss syndrome (CATSHLS) [MIM:610474]: An autosomal dominant syndrome characterized by permanent and irreducible flexion of one or more fingers of the hand and/or feet, tall stature, scoliosis and/or a pectus excavatum, and hearing loss. Affected individuals have developmental delay and/or intellectual disability, and several of these have microcephaly. Radiographic findings included tall vertebral bodies with irregular borders and broad femoral metaphyses with long tubular shafts. On audiological exam, each tested member have bilateral sensorineural hearing loss and absent otoacoustic emissions. The hearing loss was congenital or developed in early infancy, progressed variably in early childhood, and range from mild to severe. Computed tomography and magnetic resonance imaging reveal that the brain, middle ear, and inner ear are structurally normal. {ECO:0000269|PubMed:17033969}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Multiple myeloma (MM) [MIM:254500]: A malignant tumor of plasma cells usually arising in the bone marrow and characterized by diffuse involvement of the skeletal system, hyperglobulinemia, Bence-Jones proteinuria and anemia. Complications of multiple myeloma are bone pain, hypercalcemia, renal failure and spinal cord compression. The aberrant antibodies that are produced lead to impaired humoral immunity and patients have a high prevalence of infection. Amyloidosis may develop in some patients. Multiple myeloma is part of a spectrum of diseases ranging from monoclonal gammopathy of unknown significance (MGUS) to plasma cell leukemia. {ECO:0000269|PubMed:11529856, ECO:0000269|PubMed:9207791}. The gene represented in this entry may be involved in disease pathogenesis. A chromosomal aberration involving FGFR3 is found in multiple myeloma. Translocation t(4;14)(p16.3;q32.3) with the IgH locus.; DISEASE: Lacrimo-auriculo-dento-digital syndrome 2 (LADD2) [MIM:620192]: A form of lacrimo-auriculo-dento-digital syndrome, an autosomal dominant disease characterized by aplastic/hypoplastic lacrimal and salivary glands and ducts, cup-shaped ears, hearing loss, hypodontia and enamel hypoplasia, and distal limb segments anomalies. In addition to these cardinal features, facial dysmorphism, malformations of the kidney and respiratory system and abnormal genitalia have been reported. Craniosynostosis and severe syndactyly are not observed. {ECO:0000269|PubMed:16501574}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Keratinocytic non-epidermolytic nevus (KNEN) [MIM:162900]: Epidermal nevi of the common, non-organoid and non-epidermolytic type are benign skin lesions and may vary in their extent from a single (usually linear) lesion to widespread and systematized involvement. They may be present at birth or develop early during childhood. {ECO:0000269|PubMed:16841094}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Muenke syndrome (MNKS) [MIM:602849]: A condition characterized by premature closure of coronal suture of skull during development (coronal craniosynostosis), which affects the shape of the head and face. It may be uni- or bilateral. When bilateral, it is characterized by a skull with a small antero-posterior diameter (brachycephaly), often with a decrease in the depth of the orbits and hypoplasia of the maxillae. Unilateral closure of the coronal sutures leads to flattening of the orbit on the involved side (plagiocephaly). The intellect is normal. In addition to coronal craniosynostosis some affected individuals show skeletal abnormalities of hands and feet, sensorineural hearing loss, intellectual disability and respiratory insufficiency. {ECO:0000269|PubMed:11746040, ECO:0000269|PubMed:9042914, ECO:0000269|PubMed:9950359}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Keratosis, seborrheic (KERSEB) [MIM:182000]: A common benign skin tumor. Seborrheic keratoses usually begin with the appearance of one or more sharply defined, light brown, flat macules. The lesions may be sparse or numerous. As they initially grow, they develop a velvety to finely verrucous surface, followed by an uneven warty surface with multiple plugged follicles and a dull or lackluster appearance. {ECO:0000269|PubMed:15772091}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Testicular germ cell tumor (TGCT) [MIM:273300]: A common malignancy in males representing 95% of all testicular neoplasms. TGCTs have various pathologic subtypes including: unclassified intratubular germ cell neoplasia, seminoma (including cases with syncytiotrophoblastic cells), spermatocytic seminoma, embryonal carcinoma, yolk sac tumor, choriocarcinoma, and teratoma. {ECO:0000269|PubMed:19855393}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Achondroplasia, severe, with developmental delay and acanthosis nigricans (SADDAN) [MIM:616482]: A severe form of achondroplasia associated with developmental delay and acanthosis nigricans. Patients manifest short-limb dwarfism, with a long, narrow trunk, short extremities, particularly in the proximal (rhizomelic) segments, a large head with frontal bossing, hypoplasia of the midface and a trident configuration of the hands. Acanthosis nigricans is a skin condition characterized by brown-pigmented, velvety verrucosities in body folds and creases. {ECO:0000269|PubMed:10053006}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147; DB01708 Interacts with NA EC number 1.1.3.6; 5.3.3.1 Uniprot keywords 3D-structure; Cholesterol metabolism; Direct protein sequencing; FAD; Flavoprotein; Isomerase; Lipid metabolism; Oxidoreductase; Secreted; Signal; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54295.8 Length 501 Aromaticity 0.1 Instability index 19.97 Isoelectric point 8.88 Charge (pH=7) 5.13 2D Binding mode Binding energy (Kcal/mol) -10.4 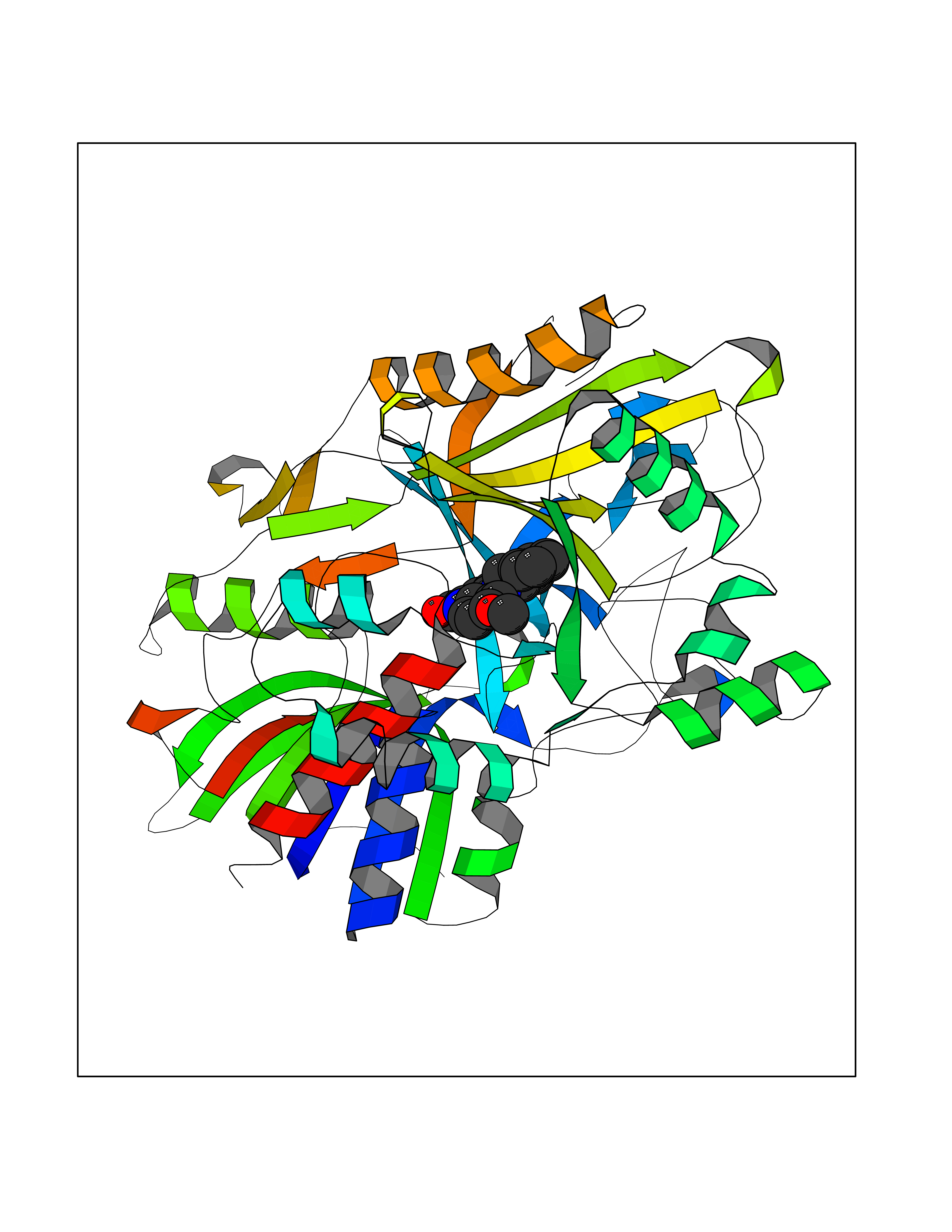 Molscript Map 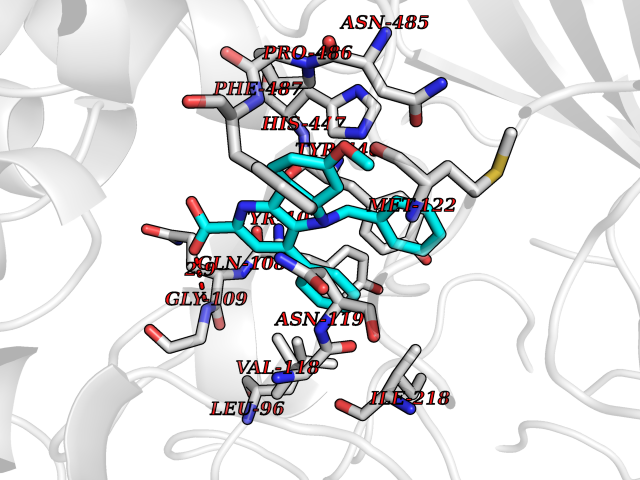 Pymol Map 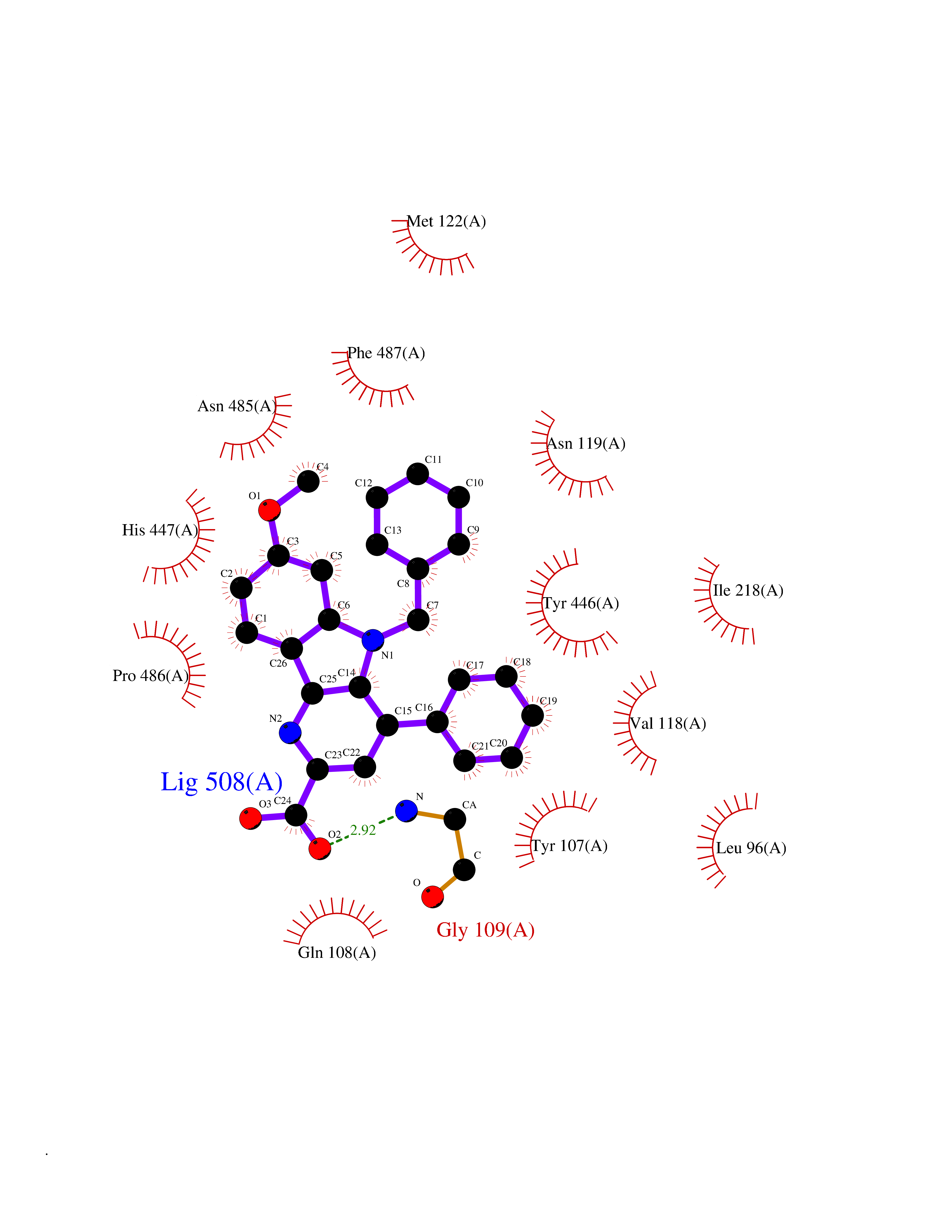 Ligplot Map 3D Binding mode Sequence RTLADGDRVPALVIGSGYGGAVAALRLTQAGIPTQIVEMGRSWDTPGSDGKIFCGMLNPDKRSMWLADKTDQPVSNFMGFGINKSIDRYVGVLDSERFSGIKVYQGRGVGGGSLVNGGMAVTPKRNYFEEILPSVDSNEMYNKYFPRANTGLGVNNIDQAWFESTEWYKFARTGRKTAQRSGFTTAFVPNVYDFEYMKKEAAGQVTKSGLGGEVIYGNNAGKKSLDKTYLAQAAATGKLTITTLHRVTKVAPATGSGYSVTMEQIDEQGNVVATKVVTADRVFFAAGSVGTSKLLVSMKAQGHLPNLSSQVGEGWGNNGNIMVGRANHMWDATGSKQATIPTMGIDNWADPTAPIFAEIAPLPAGLETYVSLYLAITKNPERARFQFNSGTGKVDLTWAQSQNQKGIDMAKKVFDKINQKEGTIYRTDLFYYKTWGDDFTYHPLGGVLLNKATDNFGRLPEYPGLYVVDGSLVPGNVGVNPFVTITALAERNMDKIISSDI Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Nitric-oxide synthase inducible (NOS2) | 3E7G | 7.63 | |
Target general information Gen name NOS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms iNOS; Peptidyl-cysteine S-nitrosylase NOS2; Nitric oxide synthase, inducible; NOS2A; NOS type II; Inducible NOS; Inducible NO synthase; Hepatocyte NOS; HEP-NOS Protein family NOS family Biochemical class Paired donor oxygen oxidoreductase Function Produces nitric oxide (NO) which is a messenger molecule with diverse functions throughout the body. In macrophages, NO mediates tumoricidal and bactericidal actions. Also has nitrosylase activity and mediates cysteine S-nitrosylation of cytoplasmic target proteins such PTGS2/COX2 (By similarity). As component of the iNOS-S100A8/9 transnitrosylase complex involved in the selective inflammatory stimulus-dependent S-nitrosylation of GAPDH on 'Cys-247' implicated in regulation of the GAIT complex activity and probably multiple targets including ANXA5, EZR, MSN and VIM. Involved in inflammation, enhances the synthesis of proinflammatory mediators such as IL6 and IL8. Related diseases Cerebellar ataxia, impaired intellectual development, and dysequilibrium syndrome 3 (CAMRQ3) [MIM:613227]: An autosomal recessive, congenital cerebellar ataxia associated with dysarthia, quadrupedal gait and intellectual disability. {ECO:0000269|PubMed:19461874}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07003; DB07007; DB07011; DB07405; DB08750; DB01997; DB07029; DB07008; DB08214; DB07002; DB01835; DB06879; DB04534; DB03100; DB02207; DB00125; DB00155; DB01234; DB14649; DB11327; DB00997; DB07306; DB07388; DB05252; DB01381; DB03366; DB05214; DB04400; DB09237; DB00244; DB01110; DB01017; DB03144; DB01686; DB03449; DB06916; DB07318; DB07389; DB02044; DB02644; DB05383; DB02234; DB03953; DB02462; DB08814 Interacts with P04406 EC number EC 1.14.13.39 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Calmodulin-binding; Cytoplasm; FAD; Flavoprotein; FMN; Heme; Iron; Metal-binding; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 48633 Length 421 Aromaticity 0.12 Instability index 46.5 Isoelectric point 6.75 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -10.4 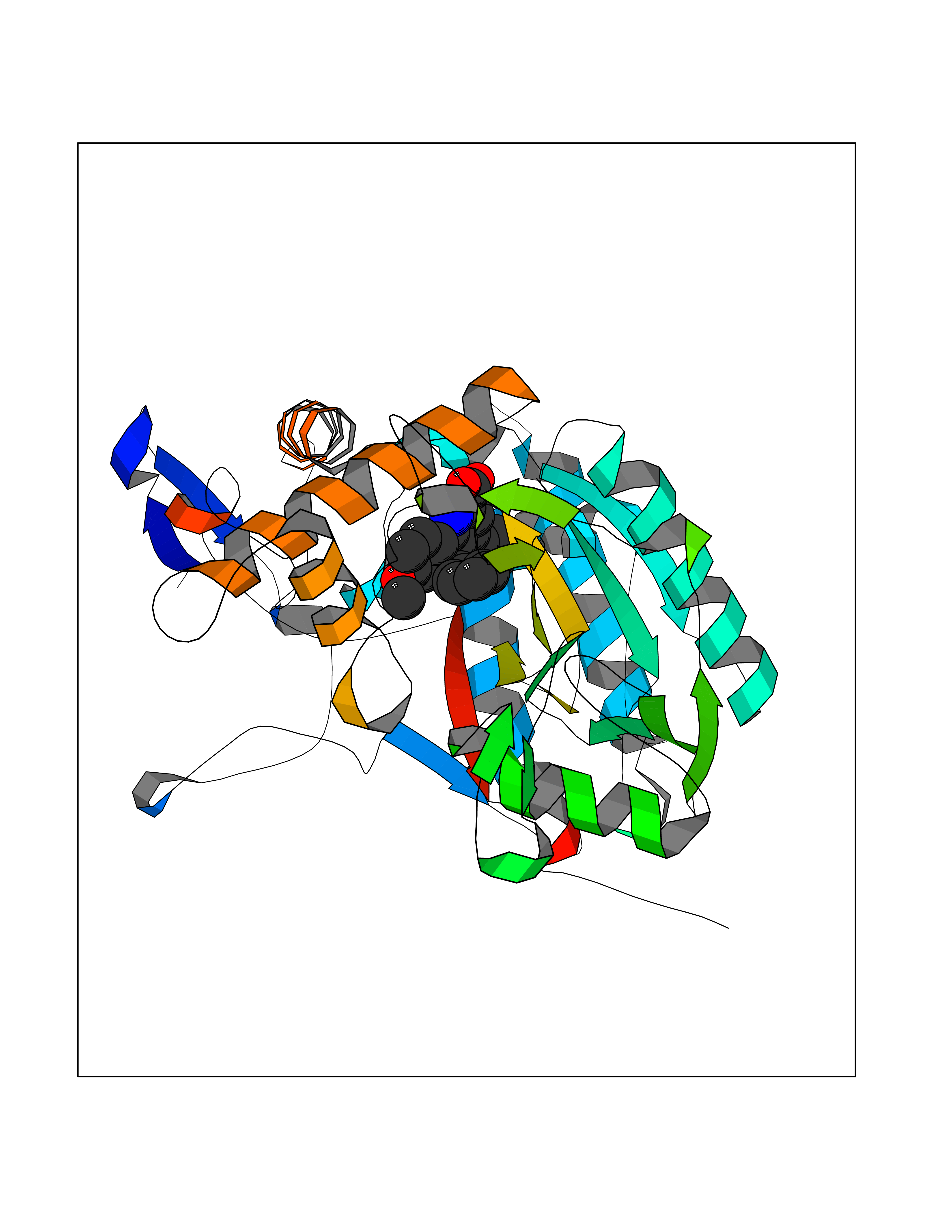 Molscript Map 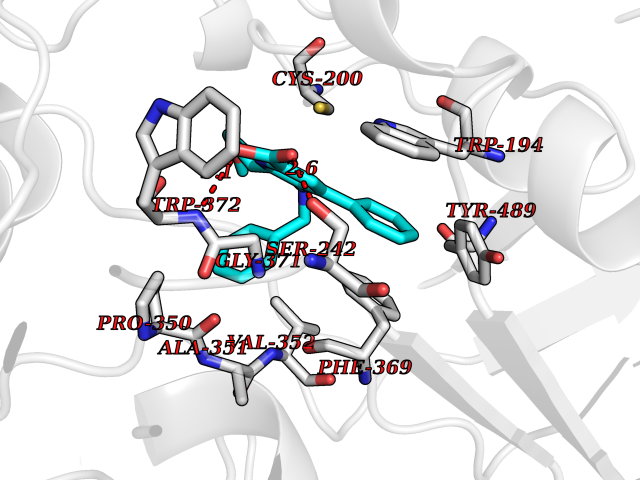 Pymol Map  Ligplot Map 3D Binding mode Sequence RHVRIKNWGSGMTFQDTLHHKAKGILTCRSKSCLGSIMTPKSLTRGPRDKPTPPDELLPQAIEFVNQYYGSFKEAKIEEHLARVEAVTKEIETTGTYQLTGDELIFATKQAWRNAPRCIGRIQWSNLQVFDARSCSTAREMFEHICRHVRYSTNNGNIRSAITVFPQRSDGKHDFRVWNAQLIRYAGYQMPDGSIRGDPANVEFTQLCIDLGWKPKYGRFDVVPLVLQANGRDPELFEIPPDLVLEVAMEHPKYEWFRELELKWYALPAVANMLLEVGGLEFPGCPFNGWYMGTEIGVRDFCDVQRYNILEEVGRRMGLETHKLASLWKDQAVVEINIAVLHSFQKQNVTIMDHHSAAESFMKYMQNEYRSRGGCPADWIWLVPPMSGSITPVFHQEMLNYVLSPFYYYQVEAWKTHVWQD Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | GABA(A) receptor gamma-2 (GABRG2) | 6X3X | 7.63 | |
Target general information Gen name GABRG2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Gamma-aminobutyric acid receptor subunit gamma-2; GABA(A) receptor subunit gamma-2 Protein family Ligand-gated ion channel (TC 1.A.9) family, Gamma-aminobutyric acid receptor (TC 1.A.9.5) subfamily, GABRG2 sub-subfamily Biochemical class Neurotransmitter receptor Function Plays an important role in the formation of functional inhibitory GABAergic synapses in addition to mediating synaptic inhibition as a GABA-gated ion channel. The gamma2 subunit is necessary but not sufficient for a rapid formation of active synaptic contacts and the synaptogenic effect of this subunit is influenced by the type of alpha and beta subunits present in the receptor pentamer. The alpha1/beta2/gamma2 receptor and the alpha1/beta3/gamma2 receptor exhibit synaptogenic activity. The alpha2/beta2/gamma2 receptor exhibits synatogenic activity whereas the alpha2/beta3/gamma2 receptor shows very little or no synaptogenic activity. Functions also as histamine receptor and mediates cellular responses to histamine. Ligand-gated chloride channel which is a component of the heteropentameric receptor for GABA, the major inhibitory neurotransmitter in the brain. Related diseases Developmental and epileptic encephalopathy 74 (DEE74) [MIM:618396]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE74 is an autosomal dominant form with onset in the first year of life. {ECO:0000269|PubMed:27864268}. The gene represented in this entry is involved in disease pathogenesis.; DISEASE: Epilepsy, childhood absence 2 (ECA2) [MIM:607681]: A subtype of idiopathic generalized epilepsy characterized by an onset at age 6-7 years, frequent absence seizures (several per day) and bilateral, synchronous, symmetric 3-Hz spike waves on EEG. Tonic-clonic seizures often develop in adolescence. Some individuals manifest febrile seizures. Absence seizures may either remit or persist into adulthood. {ECO:0000269|PubMed:11326275}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Febrile seizures, familial, 8 (FEB8) [MIM:607681]: Seizures associated with febrile episodes in childhood without any evidence of intracranial infection or defined pathologic or traumatic cause. It is a common condition, affecting 2-5% of children aged 3 months to 5 years. The majority are simple febrile seizures (generally defined as generalized onset, single seizures with a duration of less than 30 minutes). Complex febrile seizures are characterized by focal onset, duration greater than 30 minutes, and/or more than one seizure in a 24 hour period. The likelihood of developing epilepsy following simple febrile seizures is low. Complex febrile seizures are associated with a moderately increased incidence of epilepsy. {ECO:0000269|PubMed:16924025}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Generalized epilepsy with febrile seizures plus 3 (GEFSP3) [MIM:607681]: A rare autosomal dominant, familial condition with incomplete penetrance and large intrafamilial variability. Patients display febrile seizures persisting sometimes beyond the age of 6 years and/or a variety of afebrile seizure types. This disease combines febrile seizures, generalized seizures often precipitated by fever at age 6 years or more, and partial seizures, with a variable degree of severity. {ECO:0000269|PubMed:11326274, ECO:0000269|PubMed:23708187}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12537; DB00546; DB06579; DB00404; DB00543; DB11901; DB14719; DB11859; DB01558; DB09017; DB00237; DB00241; DB01489; DB00395; DB00475; DB14715; DB01594; DB00349; DB01068; DB00628; DB01559; DB01553; DB01511; DB01189; DB00829; DB13837; DB00228; DB01215; DB00402; DB00189; DB01545; DB09166; DB00292; DB01567; DB01205; DB01544; DB00690; DB05087; DB01381; DB01437; DB00801; DB01159; DB00753; DB01587; DB00555; DB13643; DB00186; DB13872; DB13437; DB00603; DB01043; DB00371; DB00463; DB01028; DB01107; DB15489; DB00683; DB12458; DB01595; DB14028; DB00334; DB00842; DB14672; DB00312; DB00252; DB13335; DB01708; DB01588; DB00794; DB00818; DB01589; DB12404; DB01236; DB09118; DB00306; DB01956; DB00231; DB11582; DB00897; DB00425; DB00909; DB15490 Interacts with P51513; O95166 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Cell projection; Chloride; Chloride channel; Cytoplasmic vesicle; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Lipoprotein; Membrane; Palmitate; Postsynaptic cell membrane; Proteomics identification; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 77460 Length 672 Aromaticity 0.14 Instability index 35.95 Isoelectric point 8.75 Charge (pH=7) 7.31 2D Binding mode Binding energy (Kcal/mol) -10.41  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SNMSLVKETVDRLLKGYDIRLRPDFGGPPVAVGMNIDIASIDMVSEVNMDYTLTMYFQQAWRDKRLSYNVIPLNLTLDNRVADQLWVPDTYFLNDKKSFVHGVTVKNRMIRLHPDGTVLYGLRITTTAACMMDLRRYPLDEQNCTLEIESYGYTTDDIEFYWRGDDNAVTGVTKIELPQFSIVDYKLITKKVVFSTGSYPRLSLSFKLKRNIGYFILQTYMPSILITILSWVSFWINYDASAARVALGITTVLTMTTINTHLRETLPKIPYVKAIDMYLMGCFVFVFMALLEYALVNYIFFSQPARAAAIDRWSRIFFPVVFSFFNIVYWLYYVDNTTVFTRILDRLLDGYDNRLRPGLGERVTEVKTDIFVTSFGPVSDHDMEYTIDVFFRQSWKDERLKFKGPMTVLRLNNLMASKIWTPDTFFHNGKKSVAHNMTMPNKLLRITEDGTLLYTMRLTVRAECPMHLEDFPMDAHACPLKFGSYAYTRAEVVYEWTREPARSVVVAEDGSRLNQYDLLGQTVDSGIVQSSTGEYVVMTTHFHLKRKIGYFVIQTYLPCIMTVILSQVSFWLNRESVPARTVFGVTTVLTMTTLSISARNSLPKVAYATAMDWFIAVCYAFVFSALIEFATVNYFTKSQPARAAKIDRLSRIAFPLLFGIFNLVYWATYLNR Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Cannabinoid receptor 2 (CB2) | 6PT0 | 7.63 | |
Target general information Gen name CNR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hCB2; Cannabinoid CB2 receptor; CX5; CB2B; CB2A; CB-2 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function May function in inflammatory response, nociceptive transmission and bone homeostasis. Heterotrimeric G protein-coupled receptor for endocannabinoid 2-arachidonoylglycerol mediating inhibition of adenylate cyclase. Related diseases Factor V deficiency (FA5D) [MIM:227400]: A blood coagulation disorder leading to a hemorrhagic diathesis known as parahemophilia. {ECO:0000269|PubMed:10942390, ECO:0000269|PubMed:12393490}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thrombophilia due to activated protein C resistance (THPH2) [MIM:188055]: A hemostatic disorder due to defective degradation of factor V by activated protein C. It is characterized by a poor anticoagulant response to activated protein C resulting in tendency to thrombosis. {ECO:0000269|PubMed:10391209, ECO:0000269|PubMed:10942390, ECO:0000269|PubMed:11435304, ECO:0000269|PubMed:11858490, ECO:0000269|PubMed:14617013, ECO:0000269|PubMed:14695241, ECO:0000269|PubMed:16710414, ECO:0000269|PubMed:8164741, ECO:0000269|PubMed:9454742}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Budd-Chiari syndrome (BDCHS) [MIM:600880]: A syndrome caused by obstruction of hepatic venous outflow involving either the hepatic veins or the terminal segment of the inferior vena cava. Obstructions are generally caused by thrombosis and lead to hepatic congestion and ischemic necrosis. Clinical manifestations observed in the majority of patients include hepatomegaly, right upper quadrant pain and abdominal ascites. Budd-Chiari syndrome is associated with a combination of disease states including primary myeloproliferative syndromes and thrombophilia due to factor V Leiden, protein C deficiency and antithrombin III deficiency. Budd-Chiari syndrome is a rare but typical complication in patients with polycythemia vera. {ECO:0000269|PubMed:9245936}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Ischemic stroke (ISCHSTR) [MIM:601367]: A stroke is an acute neurologic event leading to death of neural tissue of the brain and resulting in loss of motor, sensory and/or cognitive function. Ischemic strokes, resulting from vascular occlusion, is considered to be a highly complex disease consisting of a group of heterogeneous disorders with multiple genetic and environmental risk factors. {ECO:0000269|PubMed:15534175}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Pregnancy loss, recurrent, 1 (RPRGL1) [MIM:614389]: A common complication of pregnancy, resulting in spontaneous abortion before the fetus has reached viability. The term includes all miscarriages from the time of conception until 24 weeks of gestation. Recurrent pregnancy loss is defined as 3 or more consecutive spontaneous abortions. {ECO:0000269|PubMed:11018168}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09061; DB00470; DB06202; DB14009; DB00486; DB14011; DB02955; DB16321; DB11755 Interacts with Q9UKJ8; Q15848; Q9NRZ5; P13236; P21964; Q14802-3; Q8N387; Q8IXM6; I3L0A0; Q96AA3; Q9Y6D0; Q6ICL7; Q9NP94; Q13501; Q96HH6; Q969S6; Q9NWH2; Q9H2L4; Q8N2M4; Q6ZT21; Q5TGU0; Q9Y548; Q9BSR8; Q96EC8 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cell projection; G-protein coupled receptor; Glycoprotein; Inflammatory response; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32999.2 Length 298 Aromaticity 0.11 Instability index 30.98 Isoelectric point 9.49 Charge (pH=7) 14.35 2D Binding mode Binding energy (Kcal/mol) -10.4 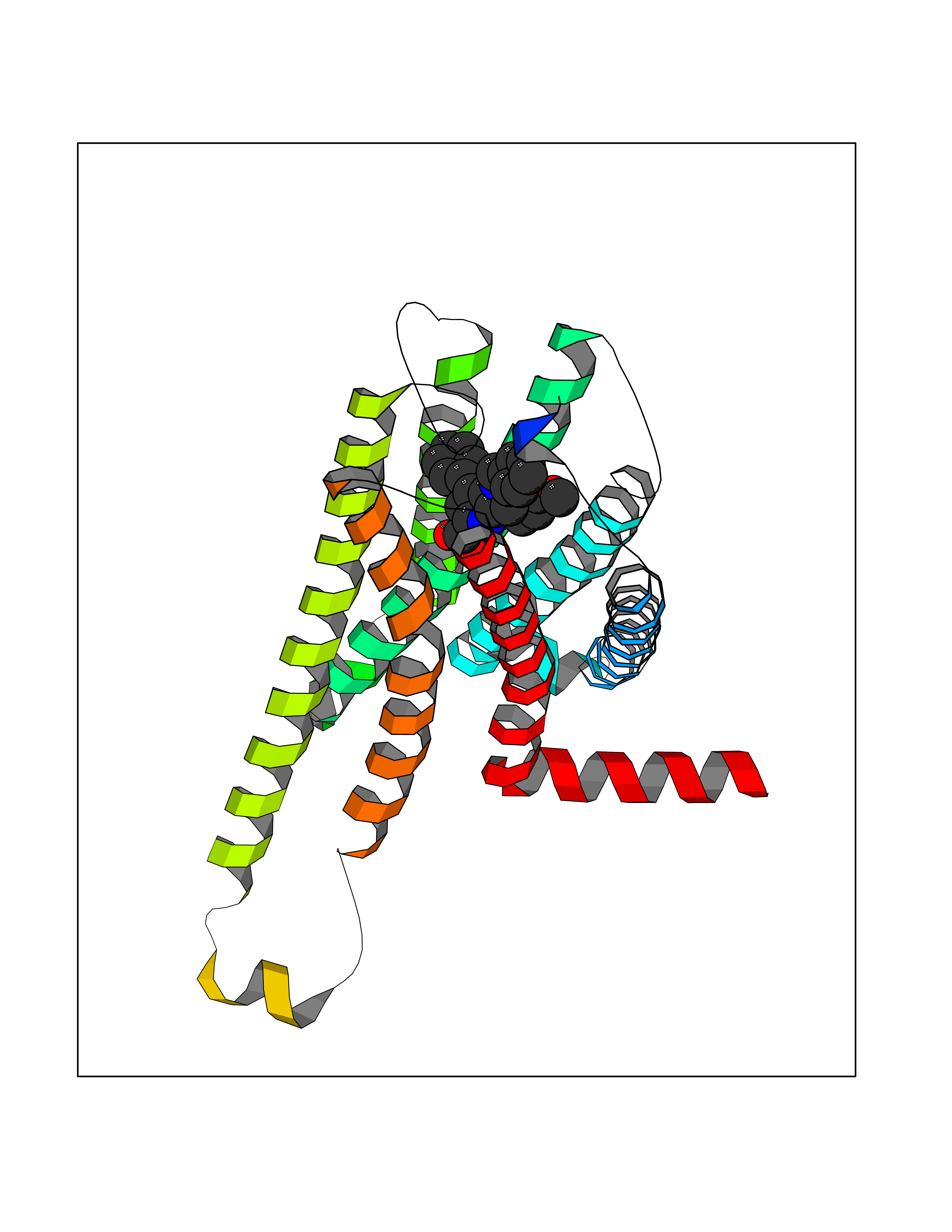 Molscript Map 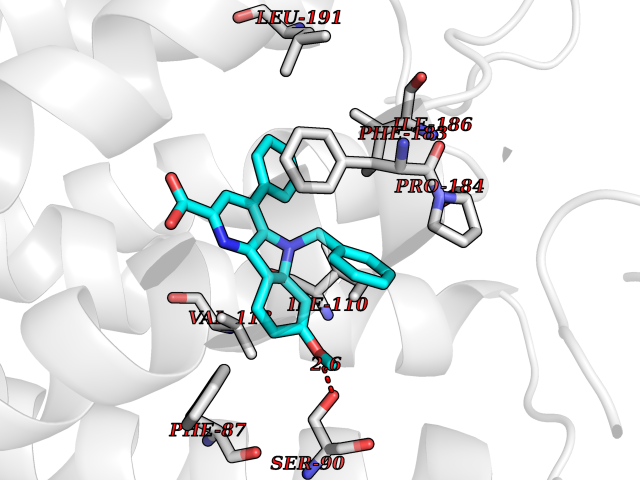 Pymol Map 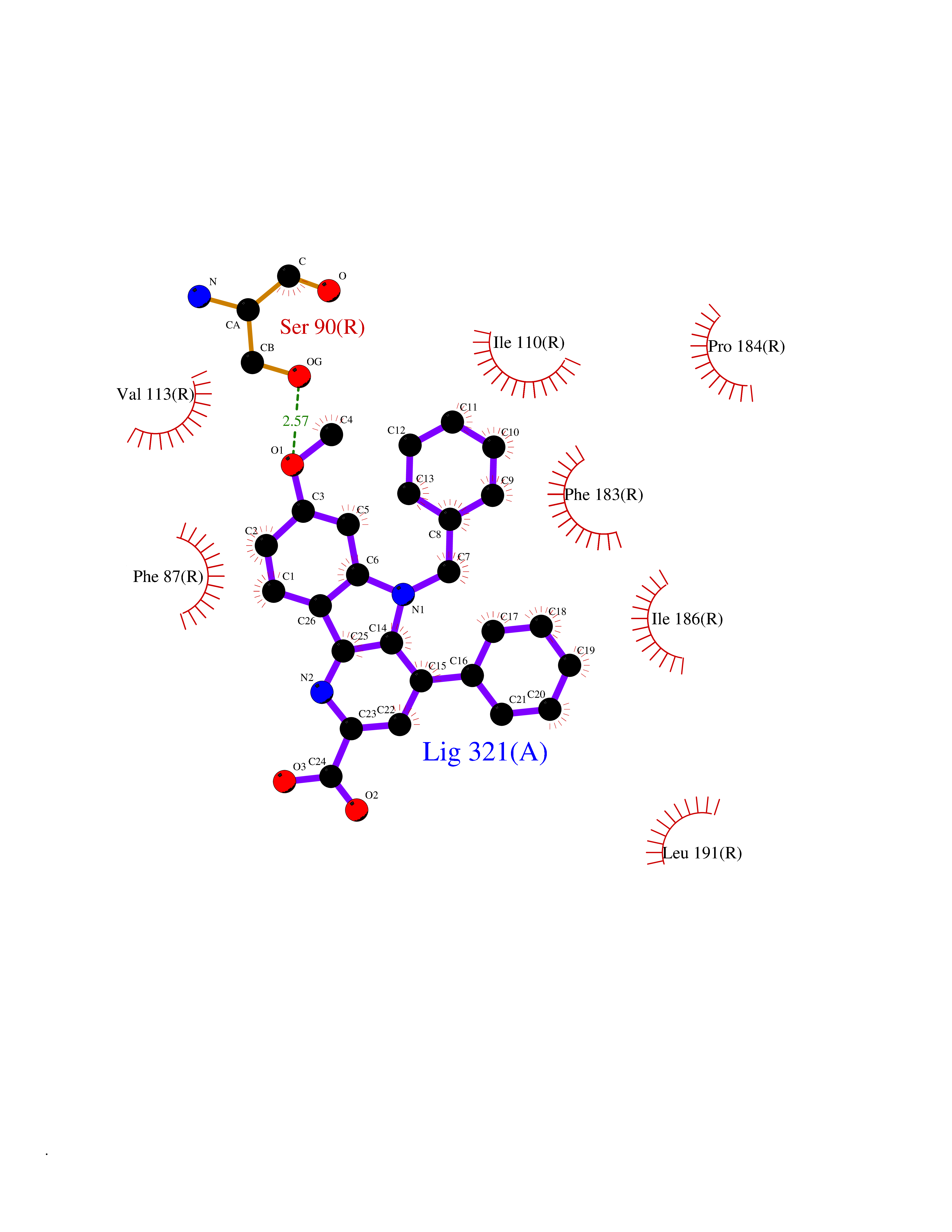 Ligplot Map 3D Binding mode Sequence MKDYMILSGPQKTAVAVLCTLLGLLSALENVAVLYLILSSHQLRRKPSYLFIGSLAGADFLASVVFACSFVNFHVFHGVDSKAVFLLKIGSVTMTFTASVGSLLLTAIDRYLCLRYPPSYKALLTRGRALVTLGIMWVLSALVSYLPLMGWTCCPRPCSELFPLIPNDYLLSWLLFIAFLFSGIIYTYGHVLWKAHQHVASLSGHQDRQVPGMARMRLDVRLAKTLGLVLAVLLICWFPVLALMAHSLATTLSDQVKKAFAFCSMLCLINSMVNPVIYALRSGEIRSSAHHCLAHWKK Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Dual-specificity tyrosine-phosphorylation regulated kinase 2 (DYRK2) | 6HDR | 7.63 | |
Target general information Gen name DYRK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dual specificity tyrosine-phosphorylation-regulated kinase 2 Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, MNB/DYRK subfamily Biochemical class Kinase Function Functions in part via its role in ubiquitin-dependent proteasomal protein degradation. Functions downstream of ATM and phosphorylates p53/TP53 at 'Ser-46', and thereby contributes to the induction of apoptosis in response to DNA damage. Phosphorylates NFATC1, and thereby inhibits its accumulation in the nucleus and its transcription factor activity. Phosphorylates EIF2B5 at 'Ser-544', enabling its subsequent phosphorylation and inhibition by GSK3B. Likewise, phosphorylation of NFATC1, CRMP2/DPYSL2 and CRMP4/DPYSL3 promotes their subsequent phosphorylation by GSK3B. May play a general role in the priming of GSK3 substrates. Inactivates GYS1 by phosphorylation at 'Ser-641', and potentially also a second phosphorylation site, thus regulating glycogen synthesis. Mediates EDVP E3 ligase complex formation and is required for the phosphorylation and subsequent degradation of KATNA1. Phosphorylates TERT at 'Ser-457', promoting TERT ubiquitination by the EDVP complex. Phosphorylates SIAH2, and thereby increases its ubiquitin ligase activity. Promotes the proteasomal degradation of MYC and JUN, and thereby regulates progress through the mitotic cell cycle and cell proliferation. Promotes proteasomal degradation of GLI2 and GLI3, and thereby plays a role in smoothened and sonic hedgehog signaling. Plays a role in cytoskeleton organization and neurite outgrowth via its phosphorylation of DCX and DPYSL2. Phosphorylates CRMP2/DPYSL2, CRMP4/DPYSL3, DCX, EIF2B5, EIF4EBP1, GLI2, GLI3, GYS1, JUN, MDM2, MYC, NFATC1, p53/TP53, TAU/MAPT and KATNA1. Can phosphorylate histone H1, histone H3 and histone H2B (in vitro). Can phosphorylate CARHSP1 (in vitro). Serine/threonine-protein kinase involved in the regulation of the mitotic cell cycle, cell proliferation, apoptosis, organization of the cytoskeleton and neurite outgrowth. Related diseases Bone marrow failure and diabetes mellitus syndrome (BMFDMS) [MIM:620044]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFDMS is an autosomal recessive form characterized by various degrees of bone marrow failure, ranging from dyserythropoiesis to bone marrow aplasia, with onset in infancy or early childhood, and non-autoimmune insulin-dependent diabetes mellitus appearing in the first or second decades. Many patients show pigmentary skin abnormalities and short stature. {ECO:0000269|PubMed:28073829, ECO:0000269|PubMed:35611808, ECO:0000269|PubMed:35931051}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NR20; Q13422; Q9BQD3; Q9BRK4; P23497; O43379; P62258; Q96C00 EC number EC 2.7.12.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cytoplasm; Kinase; Magnesium; Manganese; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID A Molecular weight (Da) 46422.1 Length 407 Aromaticity 0.09 Instability index 44.91 Isoelectric point 9.09 Charge (pH=7) 12.37 2D Binding mode Binding energy (Kcal/mol) -10.4 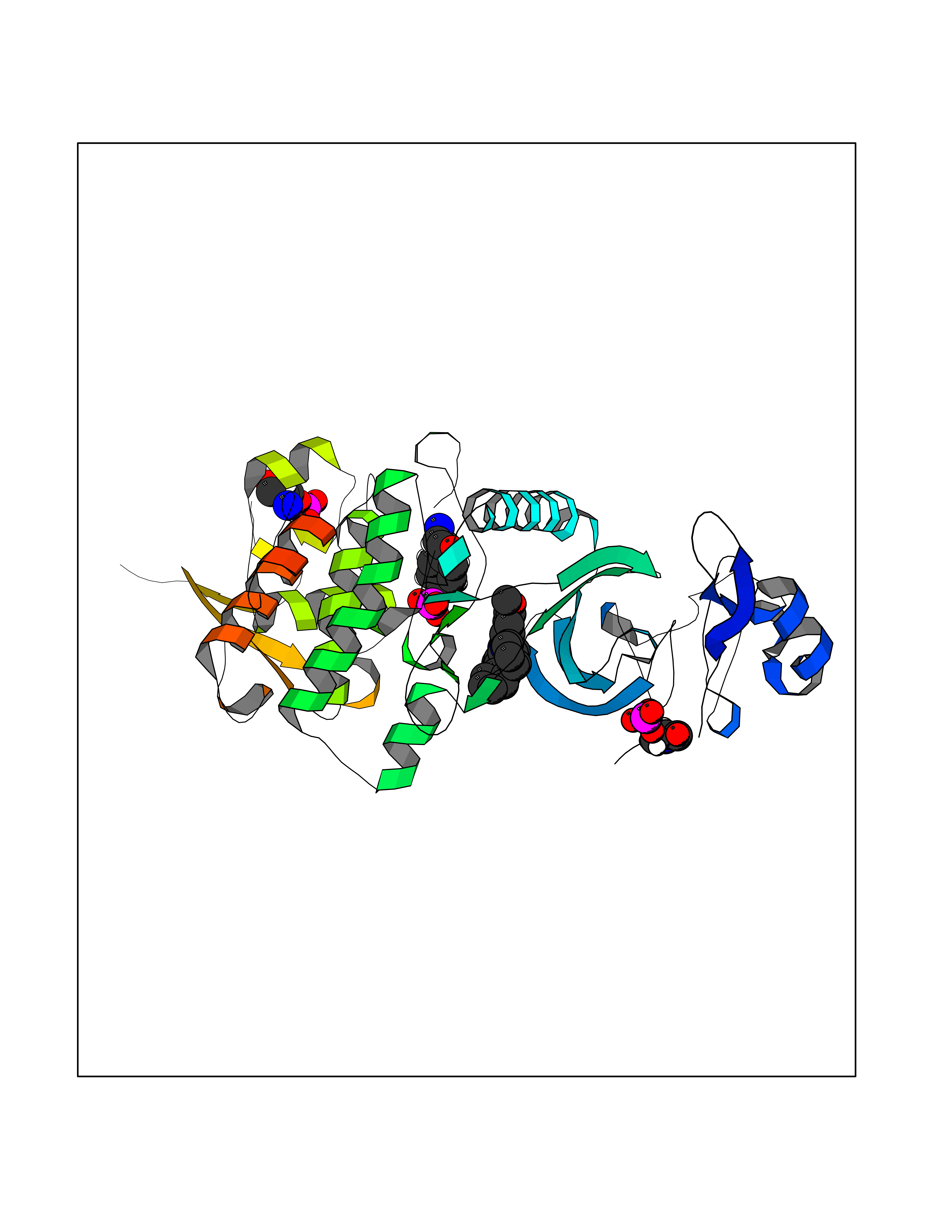 Molscript Map 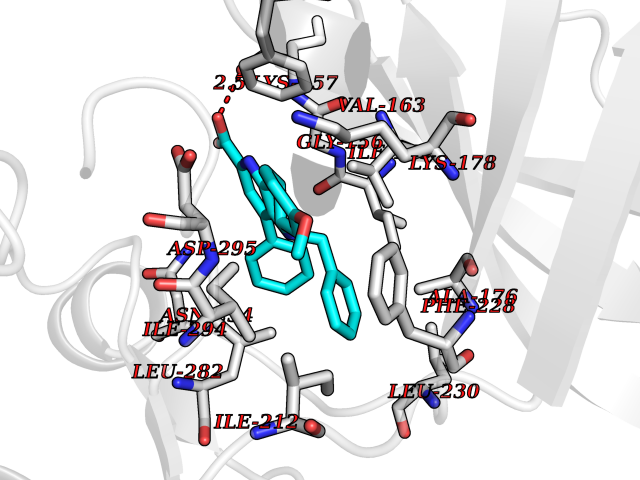 Pymol Map 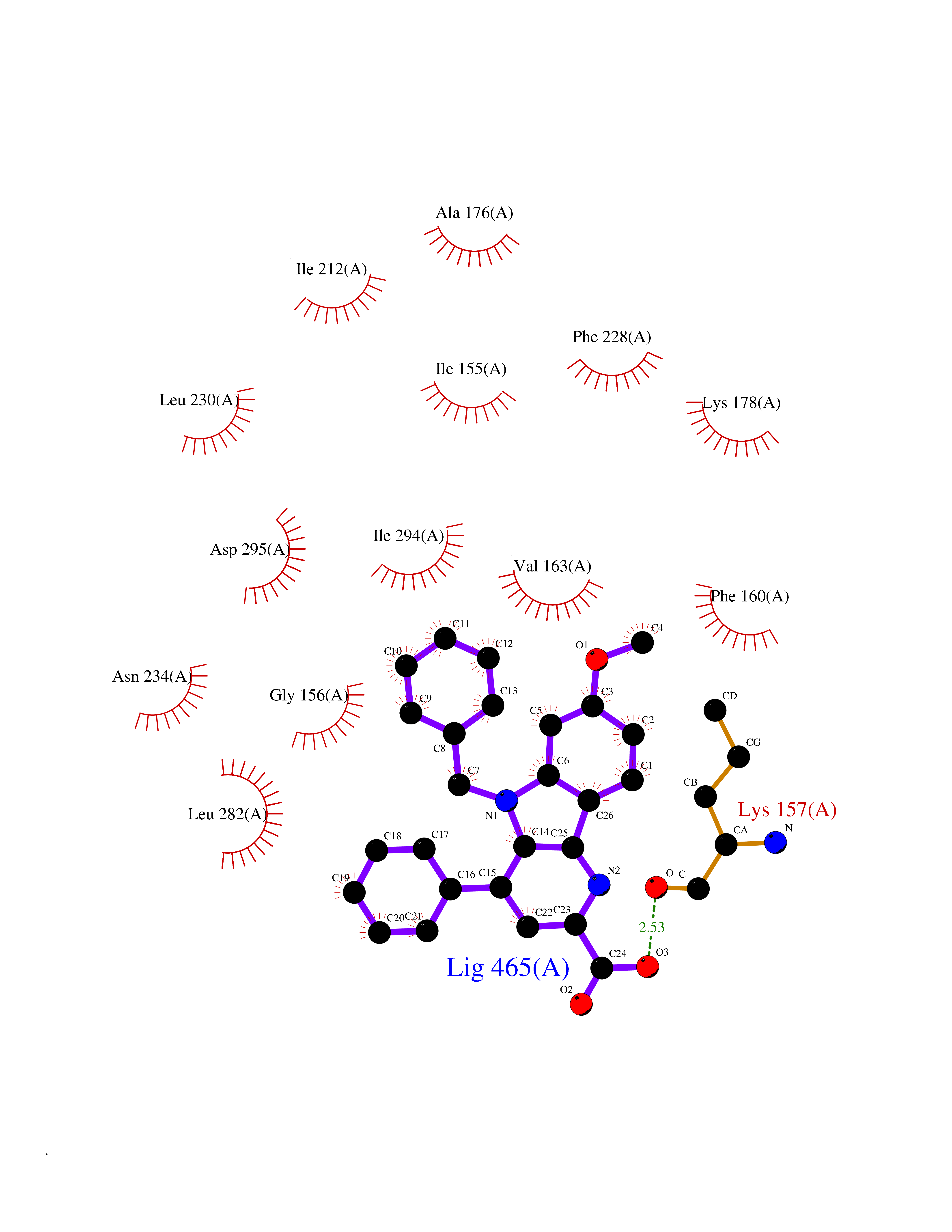 Ligplot Map 3D Binding mode Sequence HHHSXGVDLGTENLYFQSMGKVKATPMTPEQAMKQYMQKLTAFEHHEIFSYPEIYFLGLNAKKRQGMTGGPNNGGYDDDQGSYVQVPHDHVAYRYEVLKVIGKGSFGQVVKAYDHKVHQHVALKMVRNEKRFHRQAAEEIRILEHLRKQDKDNTMNVIHMLENFTFRNHICMTFELLSMNLYELIKKNKFQGFSLPLVRKFAHSILQCLDALHKNRIIHCDLKPENILLKQQGRSGIKVIDFGSSCYEHQRVYTXIQSRFYRAPEVILGARYGMPIDMWSLGCILAELLTGYPLLPGEDEGDQLACMIELLGMPSQKLLDASKRAKNFVSXKGYPRYCTVTTLSDVVLNGGRSRRGKLRGPPESREWGNALKGCDDPLFLDFLKQCLEWDPAVRMTPGQALRHPWLR Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Sphingosine kinase 1 (SPHK1) | 3VZB | 7.63 | |
Target general information Gen name SPHK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SPK 1; SPK; SPHK1; SK 1; Acetyltransferase SPHK1 Protein family NA Biochemical class Kinase Function Acts on D-erythro-sphingosine and to a lesser extent sphinganine, but not other lipids, such as D,L-threo-dihydrosphingosine, N,N-dimethylsphingosine, diacylglycerol, ceramide, or phosphatidylinositol. In contrast to proapoptotic SPHK2, has a negative effect on intracellular ceramide levels, enhances cell growth and inhibits apoptosis. Involved in the regulation of inflammatory response and neuroinflammation. Via the product sphingosine 1-phosphate, stimulates TRAF2 E3 ubiquitin ligase activity, and promotes activation of NF-kappa-B in response to TNF signaling leading to IL17 secretion. In response to TNF and in parallel to NF-kappa-B activation, negatively regulates RANTES inducion through p38 MAPK signaling pathway. Involved in endocytic membrane trafficking induced by sphingosine, recruited to dilate endosomes, also plays a role on later stages of endosomal maturation and membrane fusion independently of its kinase activity. In Purkinje cells, seems to be also involved in the regulation of autophagosome-lysosome fusion upon VEGFA. Catalyzes the phosphorylation of sphingosine to form sphingosine 1-phosphate (SPP), a lipid mediator with both intra- and extracellular functions. Related diseases Intellectual developmental disorder, X-linked, syndromic, Claes-Jensen type (MRXSCJ) [MIM:300534]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRXSCJ patients manifest intellectual disability associated with variable features such as slowly progressive spastic paraplegia, seizures, facial dysmorphism. {ECO:0000269|PubMed:15586325, ECO:0000269|PubMed:16538222, ECO:0000269|PubMed:16541399, ECO:0000269|PubMed:17320160, ECO:0000269|PubMed:17468742, ECO:0000269|PubMed:23356856, ECO:0000269|PubMed:25666439}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08868 Interacts with P07858; P68104; Q14192; Q2M3C7; Q9Y4K3; P13473-2; Q9Y371 EC number EC 2.7.1.91 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Calmodulin-binding; Cell membrane; Coated pit; Cytoplasm; Endosome; Kinase; Lipid metabolism; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Synapse; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 39813 Length 360 Aromaticity 0.08 Instability index 43.79 Isoelectric point 7.34 Charge (pH=7) 0.84 2D Binding mode Binding energy (Kcal/mol) -10.41 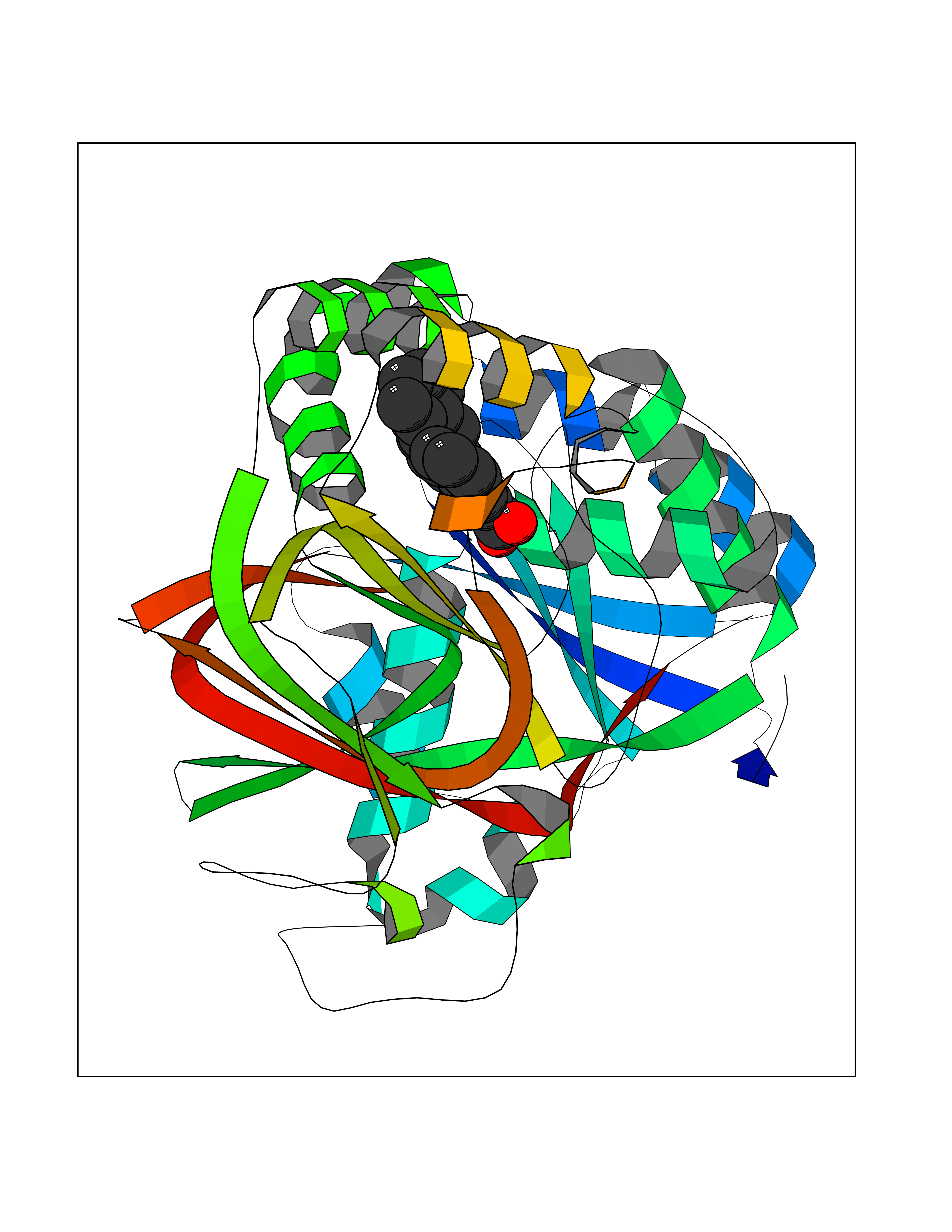 Molscript Map 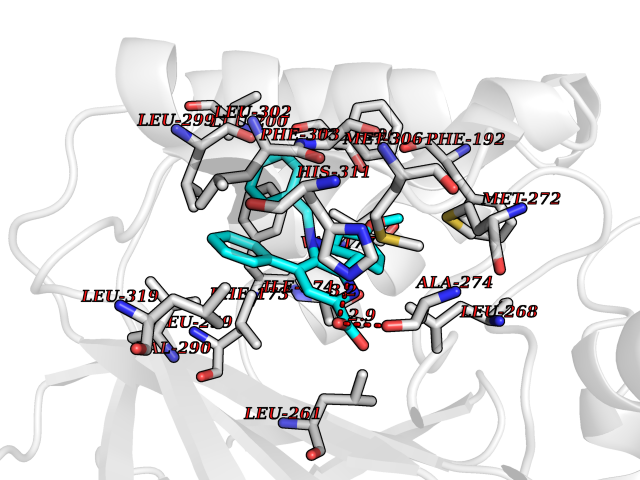 Pymol Map 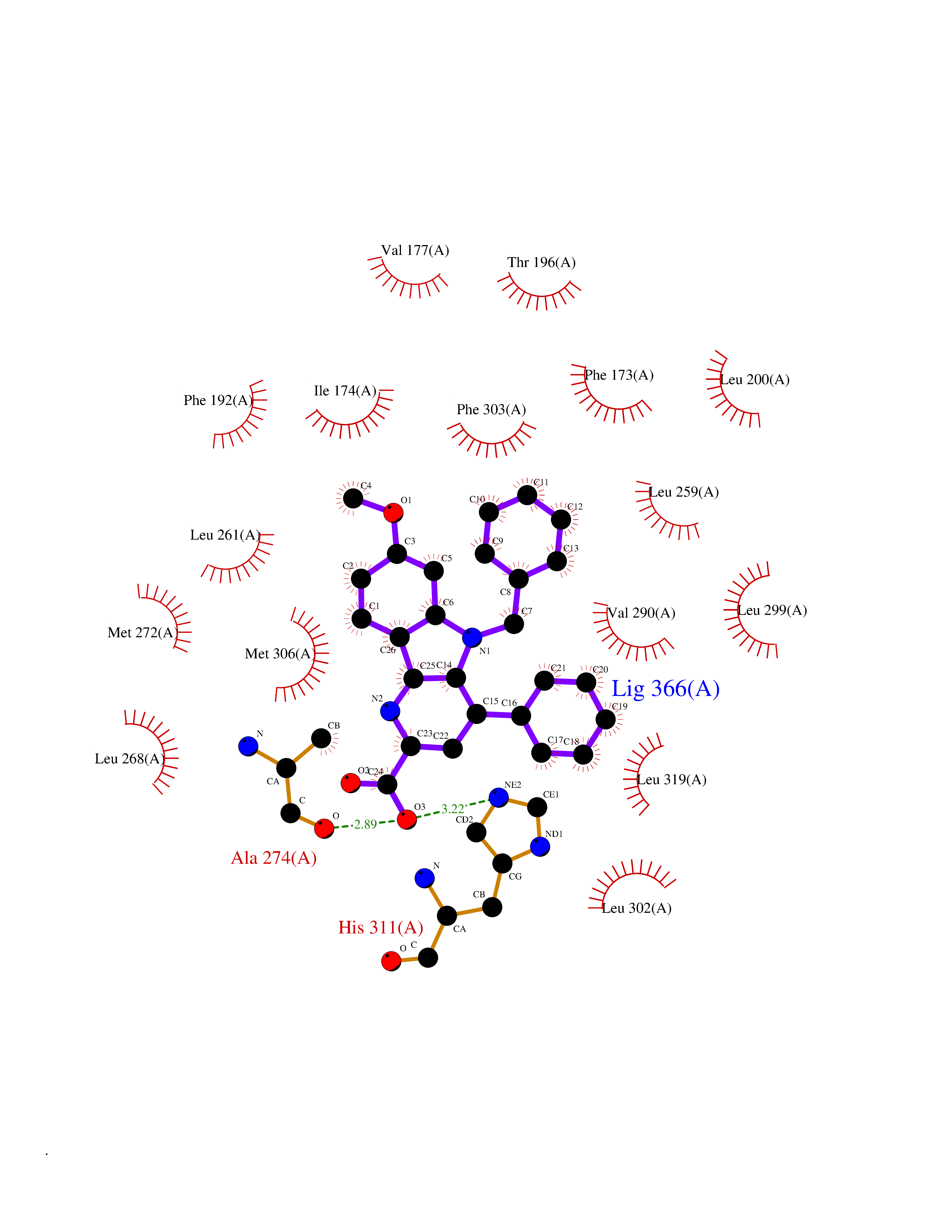 Ligplot Map 3D Binding mode Sequence AMGSGVLPRPCRVLVLLNPRGGKGKALQLFRSHVQPLLAEAEISFTLMLTERRNHARELVRSEELGRWDALVVMSGDGLMHEVVNGLMERPDWETAIQKPLCSLPAGSGNALAASLNHYAGYEQVTNEDLLTNCTLLLCRRLLSPMNLLSLHTASGLRLFSVLSLAWGFIADVDLESEKYRRLGEMRFTLGTFLRLAALRTYRGRLAYLPVGRVGSKTPASPVVVQQGPVDAHLVPLEEPVPSHWTVVPDEDFVLVLALLHSHLGSEMFAAPMGRCAAGVMHLFYVRAGVSRAMLLRLFLAMEKGRHMEYECPYLVYVPVVAFRLEPKDGKGVFAVDGELMVSEAVQGQVHPNYFWMVSG Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Trypanosoma Trypanothione reductase (Trypano TPR) | 2WBA | 7.63 | |
Target general information Gen name Trypano TPR Organism Trypanosoma brucei brucei Uniprot ID TTD ID Synonyms TRYR; TPR; Parasite-specific trypanothione reductase; N(1),N(8)-bis(glutathionyl)spermidine reductase Protein family Class-I pyridine nucleotide-disulfide oxidoreductase family Biochemical class Sulfur donor oxidoreductase Function Trypanothione is the parasite analog of glutathione; this enzyme is the equivalent of glutathione reductase. Related diseases Immunodeficiency 57 with autoinflammation (IMD57) [MIM:618108]: An autosomal recessive primary immunodeficiency characterized by lymphopenia and recurrent viral, bacterial, and fungal infections. Patients exhibit early-onset inflammatory bowel disease involving the upper and lower gastrointestinal tract, and develop progressive polyarthritis. {ECO:0000269|PubMed:30026316}. The disease is caused by variants affecting the gene represented in this entry. RIPK1-deficient immune cells from IMD57 patients have impaired proinflammatory signaling leading to dysregulated cytokine secretion and are prone to necroptosis. {ECO:0000269|PubMed:30026316}.; DISEASE: Autoinflammation with episodic fever and lymphadenopathy (AIEFL) [MIM:618852]: An autosomal dominant immunologic disorder characterized by early onset of recurrent episodes of unexplained fever, lymphadenopathy, hepatosplenomegaly, and increased levels of inflammatory cytokines and chemokines in patient serum. {ECO:0000269|PubMed:31827280, ECO:0000269|PubMed:31827281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.8.1.12 Uniprot keywords 3D-structure; Cytoplasm; Disulfide bond; FAD; Flavoprotein; NADP; Oxidoreductase; Redox-active center Protein physicochemical properties Chain ID A,B Molecular weight (Da) 105578 Length 978 Aromaticity 0.08 Instability index 33.76 Isoelectric point 6.25 Charge (pH=7) -6.81 2D Binding mode Binding energy (Kcal/mol) -10.41 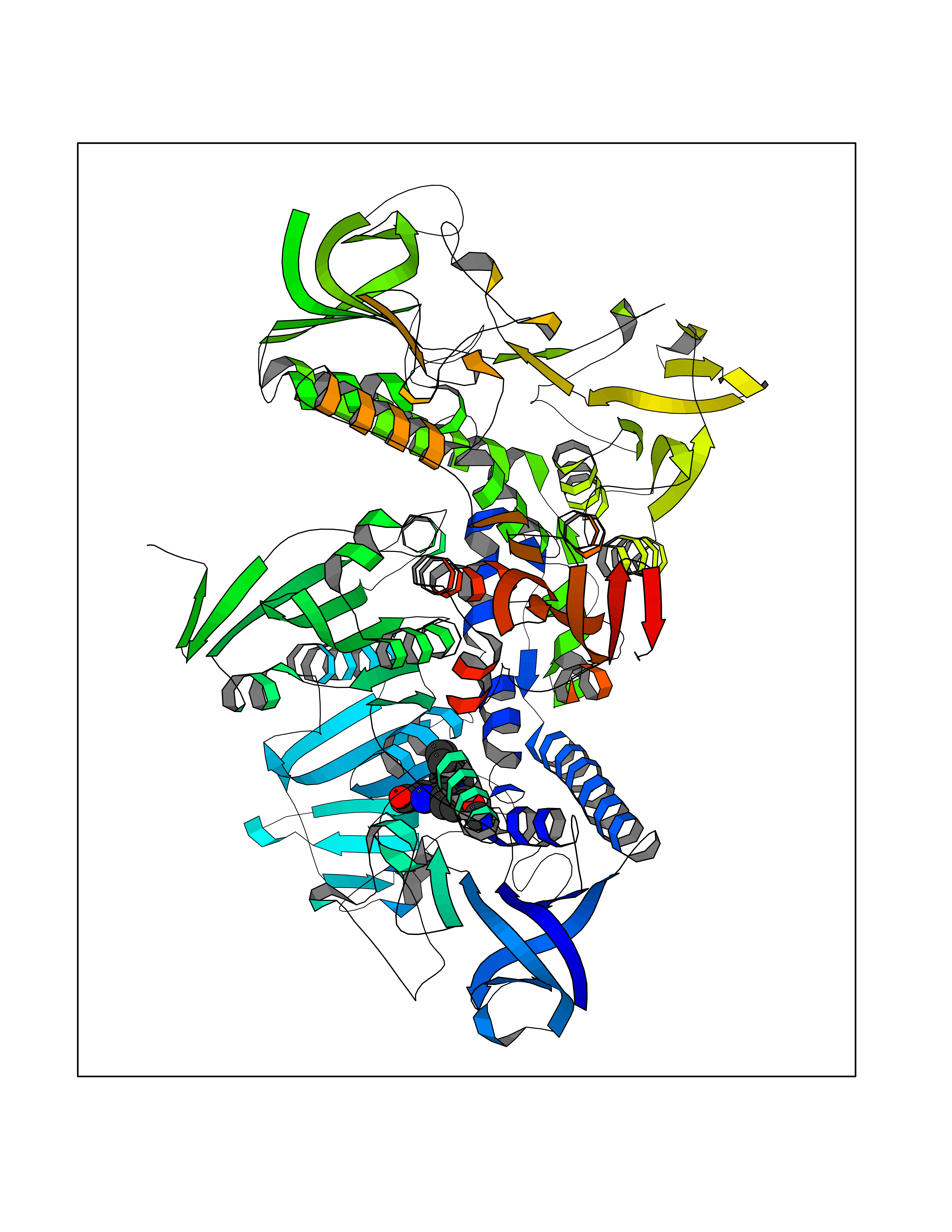 Molscript Map 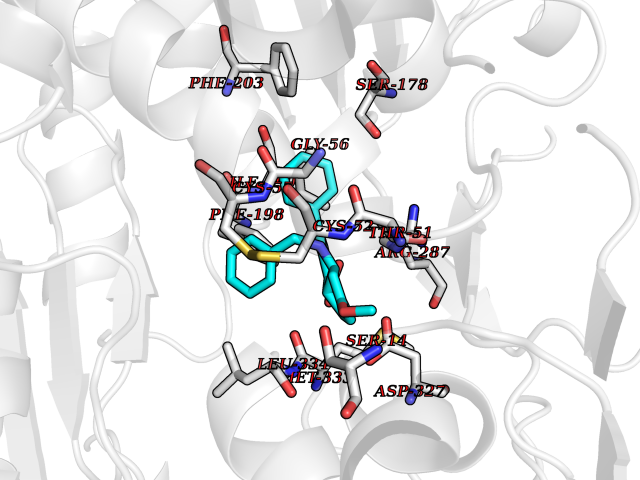 Pymol Map 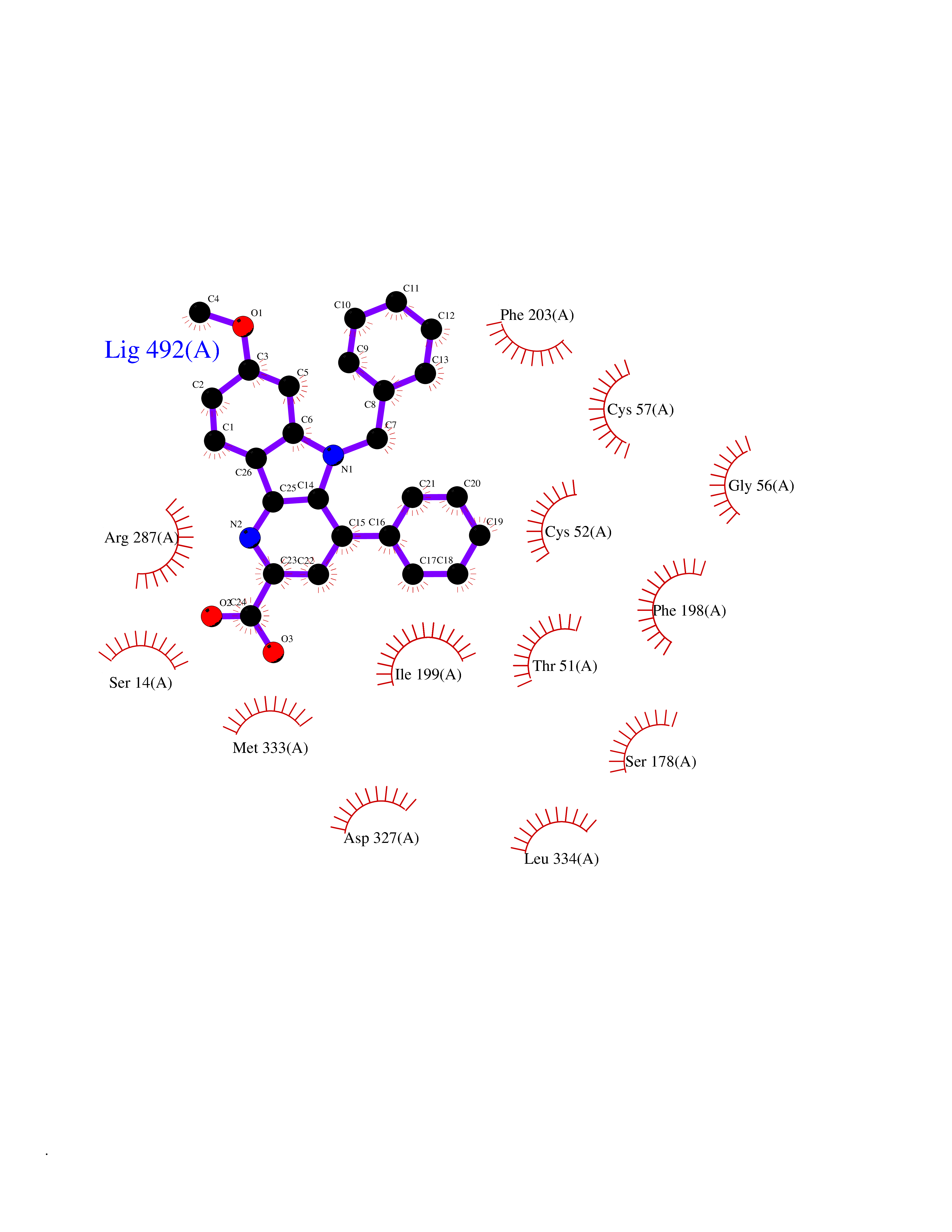 Ligplot Map 3D Binding mode Sequence SKAFDLVVIGAGSGGLEAGWNAATLYGKRVAVVDVQTSHGPPFYAALGGTCVNVGCVPKKLMVTGAQYMDHLRESAGFGWEFDGSSVKANWKKLIAAKNEAVLDINKSYEGMFNDTEGLDFFLGWGSLESKNVVVVRETADPKSAVKERLQADHILLATGSWPQMPAIPGIEHCISSNEAFYLPEPPRRVLTVGGGFISVEFAGIFNAYKPPGGKVTLCYRNNLILRGFDETIREEVTKQLTANGIEIMTNENPAKVSLNTDGSKHVTFESGKTLDVDVVMMAIGRIPRTNDLQLGNVGVKLTPKGGVQVDEFSRTNVPNIYAIGDITDRLMLTPVAINEGAALVDTVFGNKPRKTDHTRVASAVFSIPPIGTCGLIEEVAAKEFEKVAVYMSSFTPLMHNISGSKYKKFVAKIVTNHSDGTVLGVHLLGDGAPEIIQAVGVCLRLNAKISDFYNTIGVHPTSAEELCSMRTPSYYYVKGEKMEKLPDSSKAFDLVVIGAGSGGLEAGWNAATLYGKRVAVVDVQTSHGPPFYAALGGTCVNVGCVPKKLMVTGAQYMDHLRESAGFGWEFDGSSVKANWKKLIAAKNEAVLDINKSYEGMFNDTEGLDFFLGWGSLESKNVVVVRETADPKSAVKERLQADHILLATGSWPQMPAIPGIEHCISSNEAFYLPEPPRRVLTVGGGFISVEFAGIFNAYKPPGGKVTLCYRNNLILRGFDETIREEVTKQLTANGIEIMTNENPAKVSLNTDGSKHVTFESGKTLDVDVVMMAIGRIPRTNDLQLGNVGVKLTPKGGVQVDEFSRTNVPNIYAIGDITDRLMLTPVAINEGAALVDTVFGNKPRKTDHTRVASAVFSIPPIGTCGLIEEVAAKEFEKVAVYMSSFTPLMHNISGSKYKKFVAKIVTNHSDGTVLGVHLLGDGAPEIIQAVGVCLRLNAKISDFYNTIGVHPTSAEELCSMRTPSYYYVKGEKMEKLPDS Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | TWIK-related acid-sensitive potassium channel 1 (TASK1) | 6RV3 | 7.62 | |
Target general information Gen name KCNK3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Two pore potassium channel KT3.1; Two pore K(+) channel KT3.1; TWIK-related acid-sensitive K(+) channel 1; Acid-sensitive potassium channel protein TASK-1 Protein family Two pore domain potassium channel (TC 1.A.1.8) family Biochemical class NA Function pH-dependent, voltage-insensitive, background potassium channel protein. Rectification direction results from potassium ion concentration on either side of the membrane. Acts as an outward rectifier when external potassium concentration is low. When external potassium concentration is high, current is inward. Related diseases Pulmonary hypertension, primary, 4 (PPH4) [MIM:615344]: A rare disorder characterized by plexiform lesions of proliferating endothelial cells in pulmonary arterioles. The lesions lead to elevated pulmonary arterial pression, right ventricular failure, and death. The disease can occur from infancy throughout life and it has a mean age at onset of 36 years. Penetrance is reduced. Although familial pulmonary hypertension is rare, cases secondary to known etiologies are more common and include those associated with the appetite-suppressant drugs. {ECO:0000269|PubMed:23883380}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in this gene may cause developmental delay with sleep apnea (DDSA). A disorder characterized by developmental neurologic, skeletal and respiratory anomalies including microcephaly, arthrogryposis, scoliosis, cleft palate, facial dysmorphology, bilateral talipes, feeding difficulties and central and/or obstructive sleep apnea. Malformations are detected as early as 21 weeks post gestation. Severely affected patients require ongoing treatment with nocturnal O2 or pressure-controlled ventilation. The disease is associated with recurrent de novo gain of function variants. {ECO:0000269|PubMed:36195757}. Drugs (DrugBank ID) DB00561; DB01159 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Disease variant; Glycoprotein; Ion channel; Ion transport; Membrane; Metal-binding; Potassium; Potassium channel; Potassium transport; Proteomics identification; Reference proteome; Sodium; Sodium channel; Sodium transport; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 58522.5 Length 517 Aromaticity 0.14 Instability index 39.16 Isoelectric point 8.4 Charge (pH=7) 4.32 2D Binding mode Binding energy (Kcal/mol) -10.39  Molscript Map 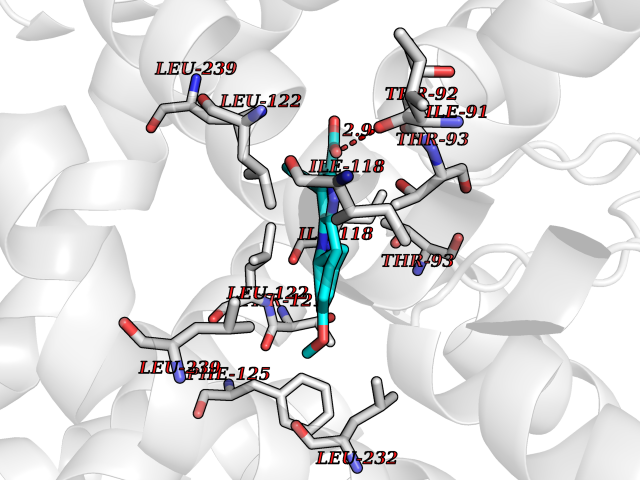 Pymol Map 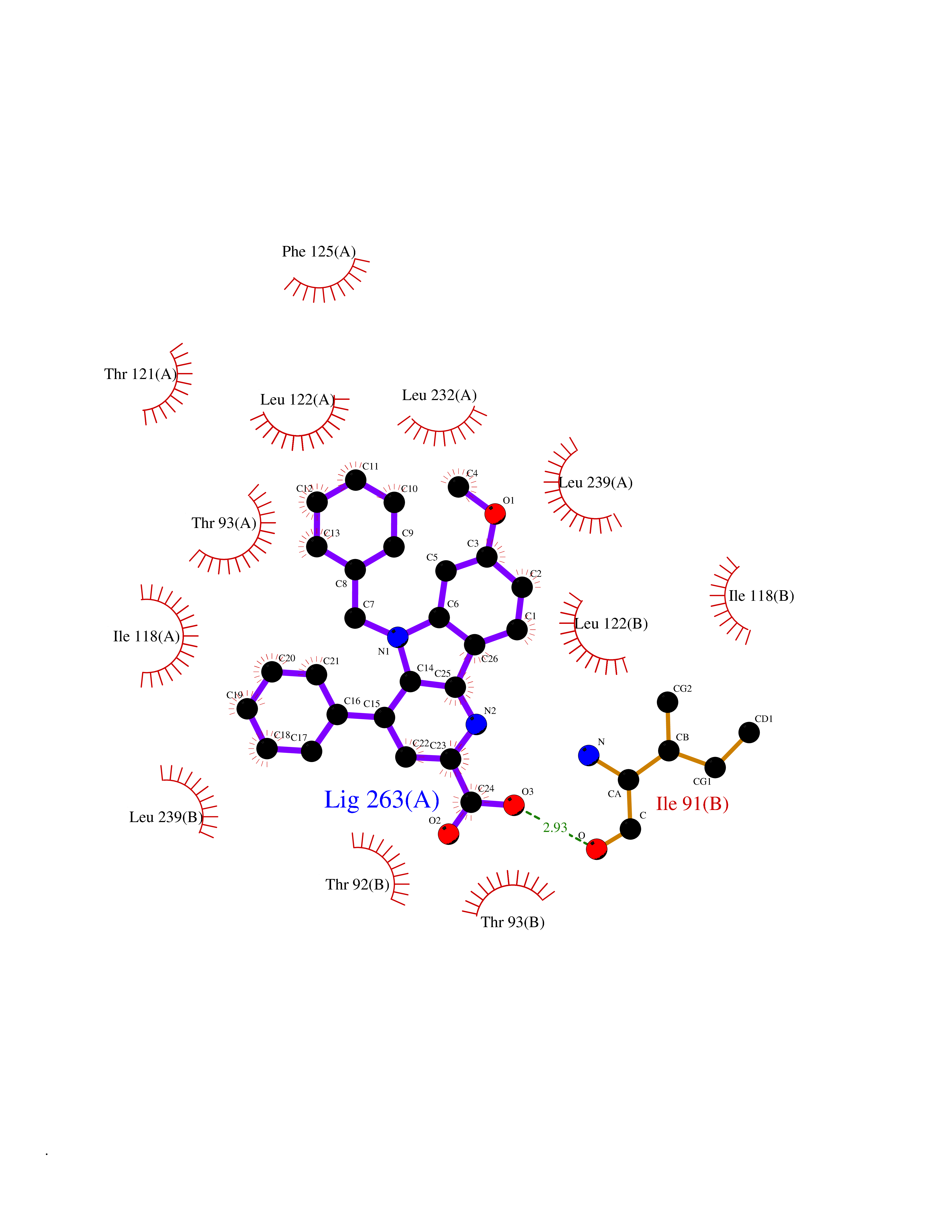 Ligplot Map 3D Binding mode Sequence MKRQNVRTLALIVCTFTYLLVGAAVFDALESEPELIERQRLELRQQELRARYNLSQGGYEELERVVLRLKPHKAGVQWRFAGSFYFAITVITTIGYGHAAPSTDGGKVFCMFYALLGIPLTLVMFQSLGERINTLVRYLLHRAKKGLGADVSMANMVLIGFFSCISTLCIGAAAFSHYEHWTFFQAYYYCFITLTTIGFGDYVALQKDQALQTQPQYVAFSFVYILTGLTVIGAFLNLVVLRFMTMNAEDEKRDAENLMKRQNVRTLALIVCTFTYLLVGAAVFDALESEPELIERQRLELRQQELRARYNLSQGGYEELERVVLRLKPHKAGVQWRFAGSFYFAITVITTIGYGHAAPSTDGGKVFCMFYALLGIPLTLVMFQSLGERINTLVRYLLHRAKKGLGMADVSMANMVLIGFFSCISTLCIGAAAFSHYEHWTFFQAYYYCFITLTTIGFGDYVALQKDQALQTQPQYVAFSFVYILTGLTVIGAFLNLVVLRFMTMNAEDEKRDAENL Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Glycogen phosphorylase, liver form | 3DDS | 7.61 | |
Target general information Gen name PYGL Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Glycogen phosphorylase family Biochemical class Transferase Function AMP binding.ATP binding.Bile acid binding.Drug binding.Glucose binding.Glycogen phosphorylase activity.Linear malto-oligosaccharide phosphorylase activity.Protein homodimerization activity.Purine nucleobase binding.Pyridoxal phosphate binding.SHG alpha-glucan phosphorylase activity.Vitamin binding. Related diseases Glycogen storage disease 6 (GSD6) [MIM:232700]: A metabolic disorder characterized by mild to moderate hypoglycemia, mild ketosis, growth retardation, and prominent hepatomegaly. Heart and skeletal muscle are not affected. {ECO:0000269|PubMed:9529348}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P11216; P11217 EC number 2.4.1.1 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; Carbohydrate metabolism; Cytoplasm; Disease variant; Glycogen metabolism; Glycogen storage disease; Glycosyltransferase; Nucleotide-binding; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89481.9 Length 778 Aromaticity 0.11 Instability index 34.96 Isoelectric point 6.49 Charge (pH=7) -3.54 2D Binding mode Binding energy (Kcal/mol) -10.37  Molscript Map 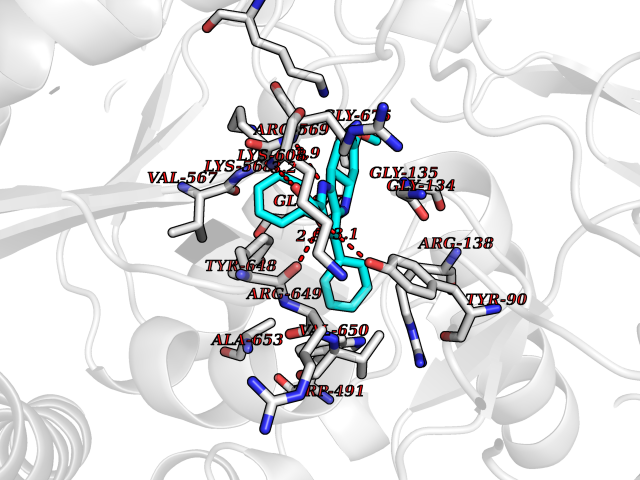 Pymol Map 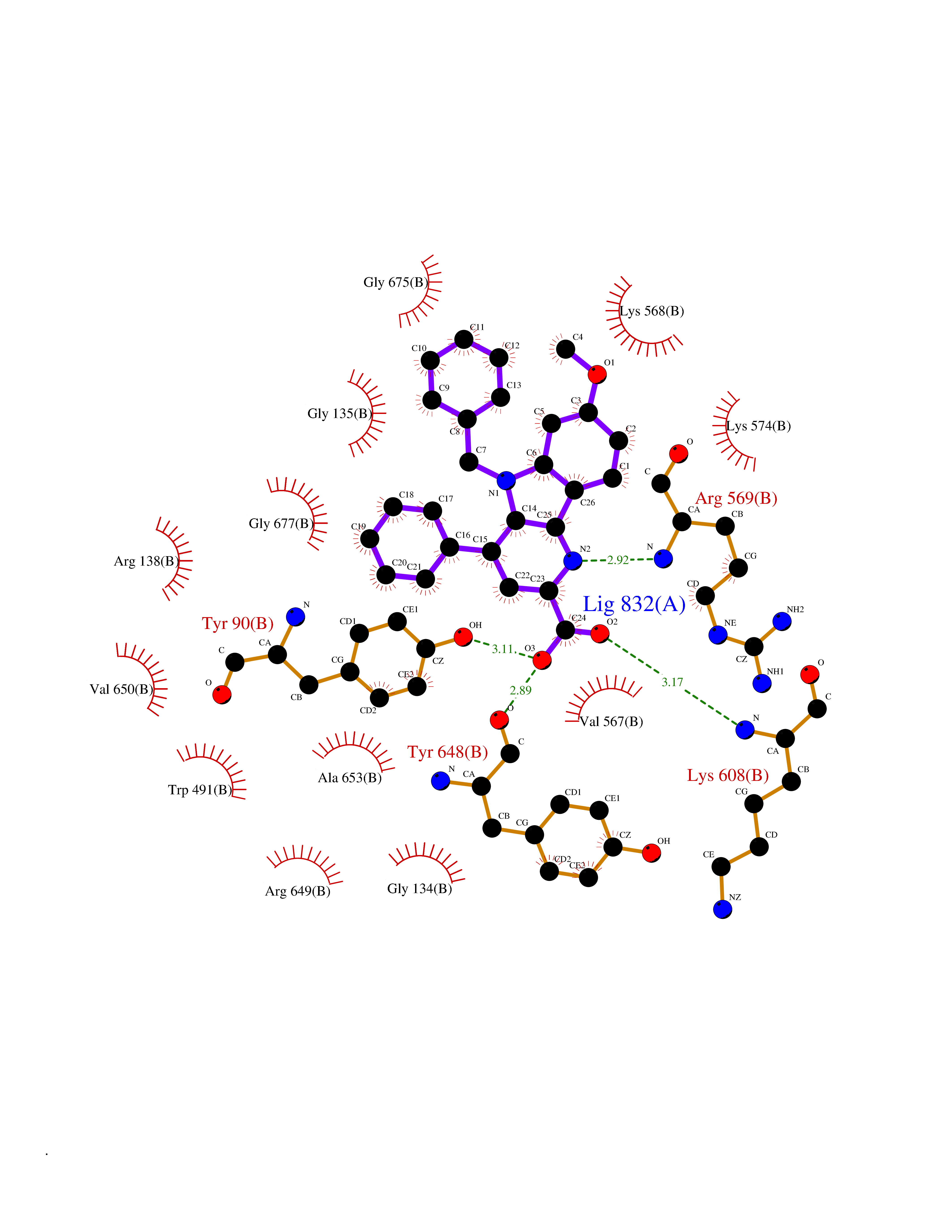 Ligplot Map 3D Binding mode Sequence NVAELKKSFNRHLHFTLVKDRNVATTRDYYFALAHTVRDHLVGRWIRTQQHYYDKCPKRVYYLSLEFYMGRTLQNTMINLGLQNACDEAIYQLGLDIEELEEIEEDAGLGNGGLGRLAACFLDSMATLGLAAYGYGIRYEYGIFNQKIRDGWQVEEADDWLRYGNPWEKSRPEFMLPVHFYGKVEHTNTGTKWIDTQVVLALPYDTPVPGYMNNTVNTMRLWSENISRVLYPNDNFFEGKELRLKQEYFVVAATLQDIIRRFKASKFGSTGTVFDAFPDQVAIQLNDTHPALAIPELMRIFVDIEKLPWSKAWELTQKTFAYTNHTVLPEALERWPVDLVEKLLPRHLEIIYEINQKHLDRIVALFPKDVDRLRRMSLIEEEGSKRINMAHLCIVGSHAVNGVAKIHSDIVKTKVFKDFSELEPDKFQNKTNGITPRRWLLLCNPGLAELIAEKIGEDYVKDLSQLTKLHSFLGDDVFLRELAKVKQENKLKFSQFLETEYKVKINPSSMFDVQVKRIHEYKRQLLNCLHVITMYNRIKKDPKKLFVPRTVIIGGKAAPGYHMAKMIIKLITSVADVVNNDPMVGSKLKVIFLENYRVSLAEKVIPATDLSEQISTAGTEASGTGNMKFMLNGALTIGTMDGANVEMAEEAGEENLFIFGMRIDDVAALDKKGYEAKEYYEALPELKLVIDQIDNGFFSPKQPDLFKDIINMLFYHDRFKVFADYEAYVKCQDKVSQLYMNPKAWNTMVLKNIAASGKFSSDRTIKEYAQNIWNVEPS Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Bifunctional protein PutA | 3E2Q | 7.61 | |
Target general information Gen name putA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b1014;poaA;JW0999 Protein family Proline dehydrogenase family; Aldehyde dehydrogenase family Biochemical class Oxidoreductase Function 1-pyrroline-5-carboxylate dehydrogenase activity.Bacterial-type RNA polymerase core promoter proximal region sequence-specific DNA binding.DNA binding.Flavin adenine dinucleotide binding.Identical protein binding.Proline dehydrogenase activity.Sequence-specific DNA binding.Transcriptional repressor activity, bacterial-type RNA polymerase core promoter proximal region sequence-specific binding. Related diseases Fructose-1,6-bisphosphatase deficiency (FBP1D) [MIM:229700]: An autosomal recessive metabolic disorder characterized by impaired gluconeogenesis, and episodes of hypoglycemia and metabolic acidosis that can be lethal in newborn infants or young children. {ECO:0000269|PubMed:12126934, ECO:0000269|PubMed:25601412, ECO:0000269|PubMed:9382095}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03051; DB03147; DB04398 Interacts with P09546 EC number 1.2.1.88; 1.5.5.2 Uniprot keywords 3D-structure; DNA-binding; FAD; Flavoprotein; Multifunctional enzyme; NAD; Oxidoreductase; Proline metabolism; Reference proteome; Repressor; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 45567.7 Length 407 Aromaticity 0.08 Instability index 33.2 Isoelectric point 7.22 Charge (pH=7) 0.47 2D Binding mode Binding energy (Kcal/mol) -10.38 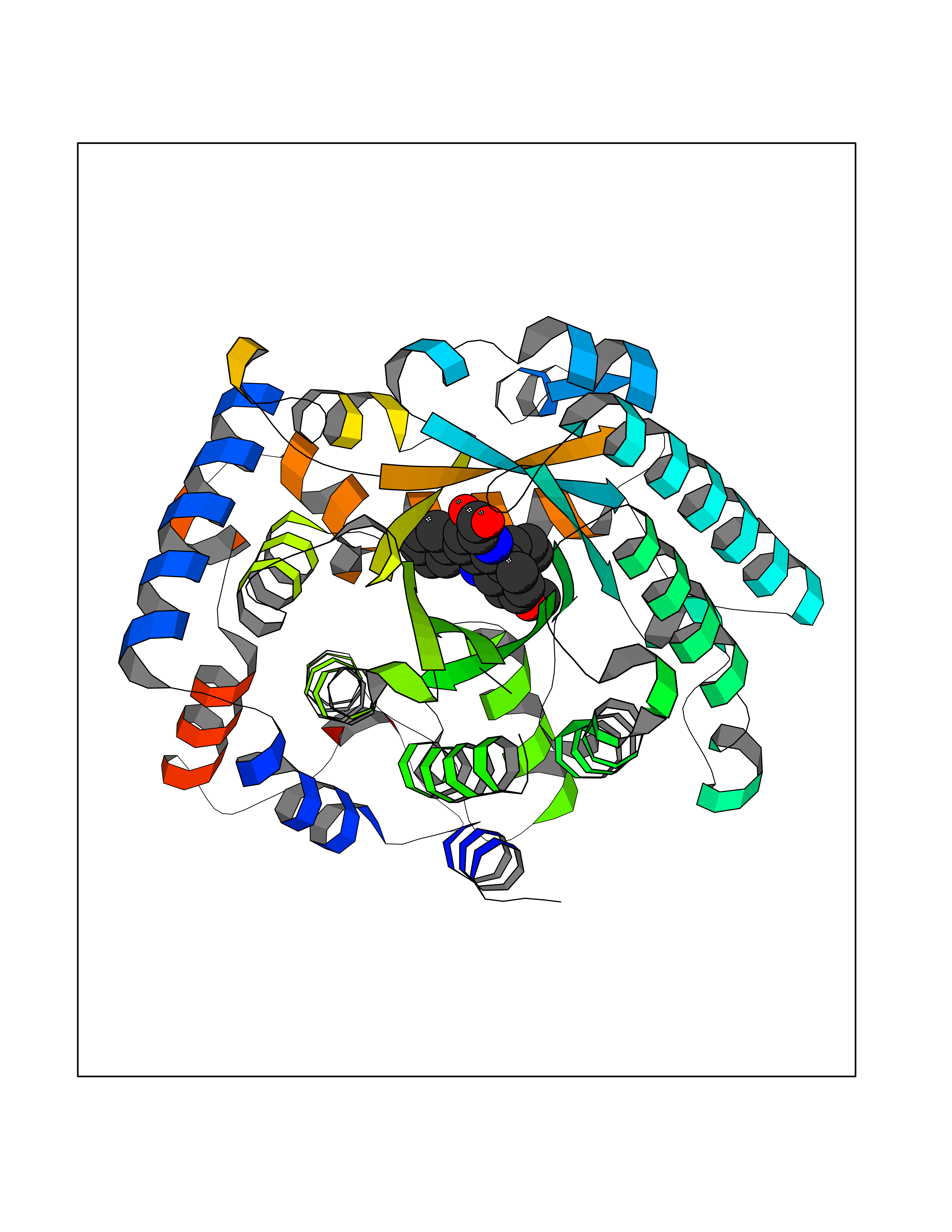 Molscript Map 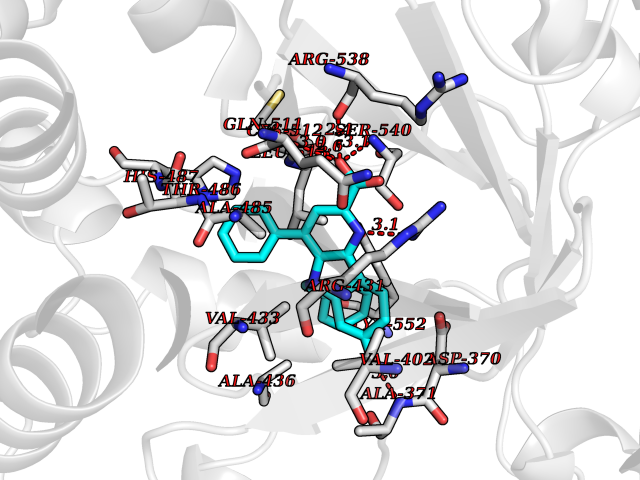 Pymol Map 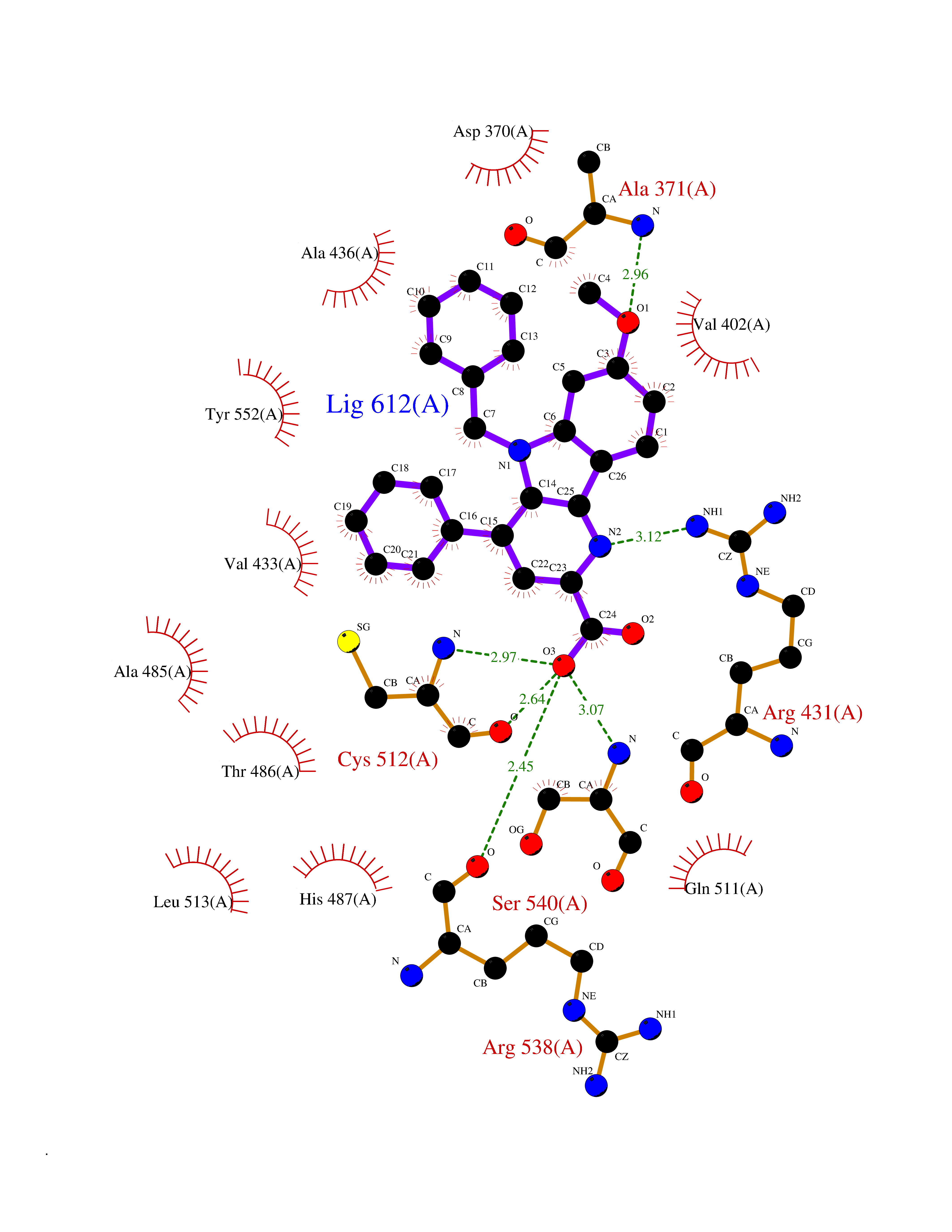 Ligplot Map 3D Binding mode Sequence QSVSRAAITAAYRRPETEAVSMLLEQARLPQPVAEQAHKLAYQLADKLRRLMGEQFVTGETIAEALANARKLEEKGFRYSYDMLGEAALTAADAQAYMVSYQQAIHAIGKASNGRGIYEGPGISIKLSALHPRYSRAQYDRVMEELYPRLKSLTLLARQYDIGINIDAEESDRLEISLDLLEKLCFEPELAGWNGIGFVIQAYQKRCPLVIDYLIDLATRSRRRLMIRLVKGAYWDSEIKRAQMDGLEGYPVYTRKVYTDVSYLACAKKLLAVPNLIYPQFATHNAHTLAAIYQLAGQNYYPGQYEFQCLHGMGEPLYEQVTGKVADGKLNRPCRISAPVGTHETLLAYLVRRLLENGANTSFVNRIADTSLPLDELVADPVTAVEKLAQQEGQTGLPHPKIPLPRD Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Aldose reductase (AKR1B1) | 1US0 | 7.61 | |
Target general information Gen name AKR1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aldehyde reductase; AKR1B1 Protein family Aldo/keto reductase family Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the NADPH-dependent reduction of a wide variety of carbonyl-containing compounds to their corresponding alcohols with a broad range of catalytic efficiencies. Related diseases Glutamine deficiency, congenital (GLND) [MIM:610015]: An autosomal recessive disorder characterized by variable brain malformations, encephalopathy, severe developmental delay, seizures, and decreased glutamine levels in bodily fluids. Death in early infancy may occur. {ECO:0000269|PubMed:16267323, ECO:0000269|PubMed:26711351, ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 116 (DEE116) [MIM:620806]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE116 is autosomal dominant form characterized by severe developmental delay, seizures, and white matter abnormalities. {ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry. DEE116 is caused by variants that disrupt the canonical translation start codon in GLUL resulting in initiation of translation at Met-18 (PubMed:38579670). The resulting protein is enzymatically competent but insensitive to negative feedback regulation via glutamine-induced degradation. {ECO:0000269|PubMed:38579670}. Drugs (DrugBank ID) DB07028; DB07030; DB07450; DB02101; DB08449; DB08000; DB07139; DB07498; DB02007; DB02020; DB11859; DB02994; DB04272; DB07187; DB00694; DB00997; DB06246; DB01039; DB02021; DB16707; DB00143; DB02834; DB08084; DB01689; DB07063; DB06077; DB02518; DB00157; DB03461; DB05383; DB05533; DB05327; DB02712; DB00605; DB02383; DB02132; DB08772; DB07093; DB07999; DB08098 Interacts with Q9BUY7 EC number EC 1.1.1.300 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Lipid metabolism; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 35447.6 Length 313 Aromaticity 0.09 Instability index 36.41 Isoelectric point 7.1 Charge (pH=7) 0.26 2D Binding mode Binding energy (Kcal/mol) -10.38  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MASRILLNNGAKMPILGLGTWKSPPGQVTEAVKVAIDVGYRHIDCAHVYQNENEVGVAIQEKLREQVVKREELFIVSKLWCTYHEKGLVKGACQKTLSDLKLDYLDLYLIHWPTGFKPGKEFFPLDESGNVVPSDTNILDTWAAMEELVDEGLVKAIGISNFNHLQVEMILNKPGLKYKPAVNQIECHPYLTQEKLIQYCQSKGIVVTAYSPLGSPDRPWAKPEDPSLLEDPRIKAIAAKHNKTTAQVLIRFPMQRNLVVIPKSVTPERIAENFKVFDFELSSQDMTTLLSYNRNWRVCALLSCTSHKDYPFH Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Guanylate cyclase soluble beta-1 (GUCY1B1) | 7D9R | 7.61 | |
Target general information Gen name GUCY1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Soluble guanylate cyclase small subunit; Guanylate cyclase soluble subunit beta-3; Guanylate cyclase soluble subunit beta-1; GUCY1B3; GUCSB3; GUC1B3; GCS-beta-3; GCS-beta-1 Protein family Adenylyl cyclase class-4/guanylyl cyclase family Biochemical class Phosphorus-oxygen lyase Function Mediates responses to nitric oxide (NO) by catalyzing the biosynthesis of the signaling molecule cGMP. Related diseases Neurodevelopmental disorder with seizures, hypotonia, and brain imaging abnormalities (NEDSHBA) [MIM:618922]: An autosomal recessive neurodevelopmental disorder characterized by global developmental delay, hypotonia, severe to profound intellectual disability, early-onset epilepsy, and microcephaly. Neuroimaging shows cerebral atrophy, thin corpus callosum and hypomyelination in a majority of cases. Death in childhood may occur. {ECO:0000269|PubMed:27435318, ECO:0000269|PubMed:28097321, ECO:0000269|PubMed:32286009, ECO:0000269|PubMed:33476302, ECO:0000269|PubMed:33500274}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09401; DB15456 Interacts with Q02108; Q02108-1 EC number EC 4.6.1.2 Uniprot keywords 3D-structure; Alternative splicing; cGMP biosynthesis; Cytoplasm; Direct protein sequencing; GTP-binding; Heme; Iron; Lyase; Metal-binding; Nucleotide-binding; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 121177 Length 1071 Aromaticity 0.09 Instability index 42.35 Isoelectric point 6.18 Charge (pH=7) -11.86 2D Binding mode Binding energy (Kcal/mol) -10.38  Molscript Map 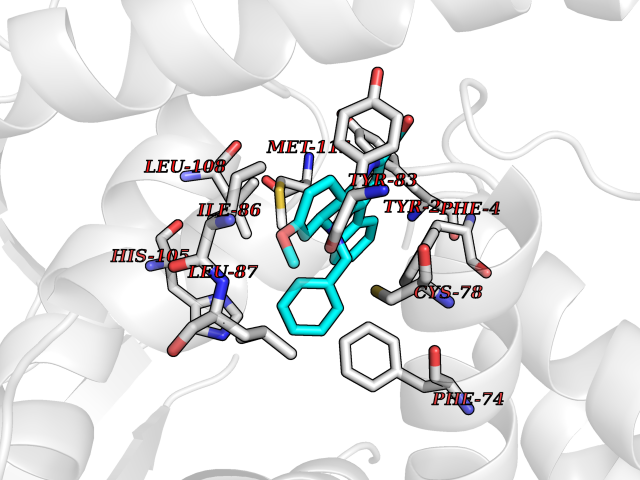 Pymol Map 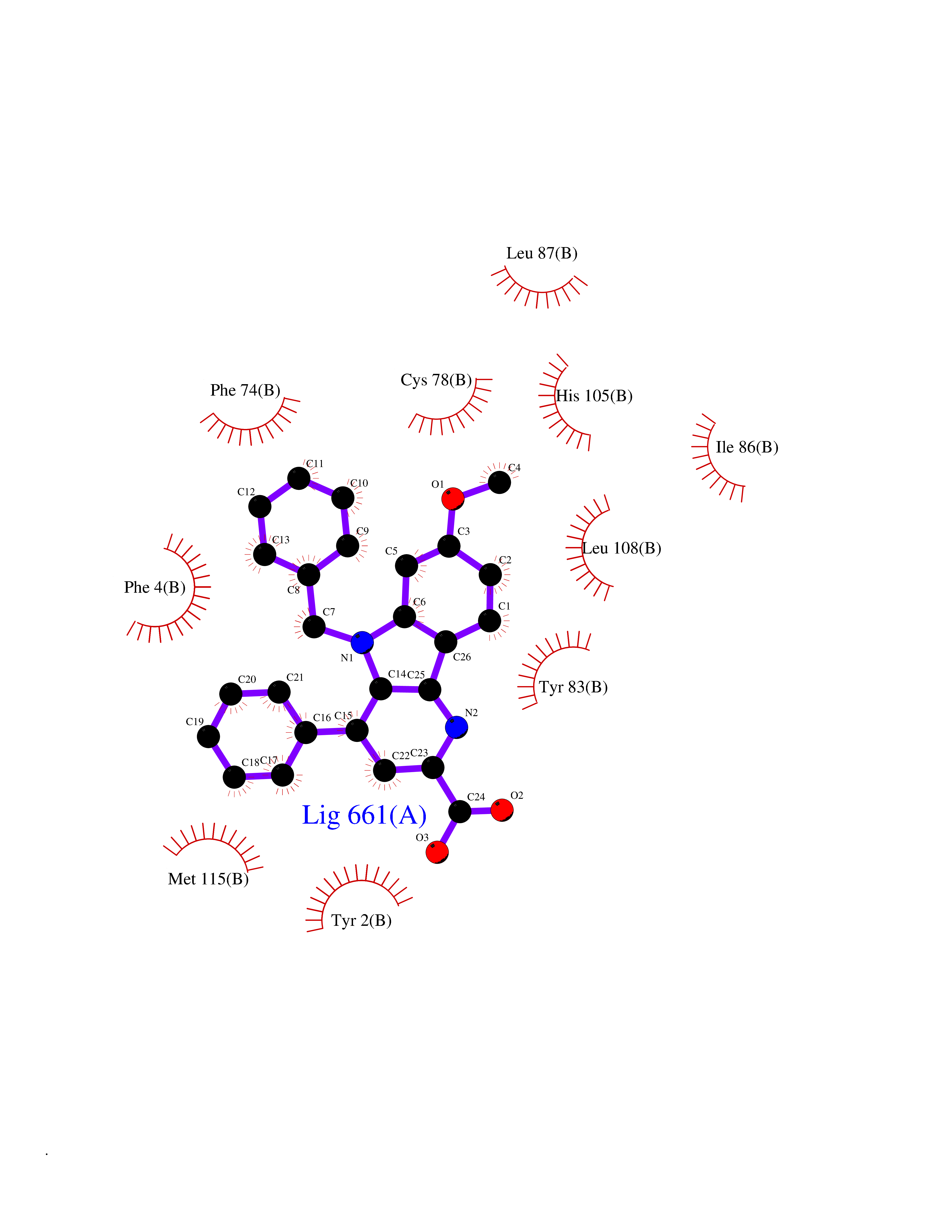 Ligplot Map 3D Binding mode Sequence SRVYLHTLAESICKLIFPEFERLNVALQRTLAKHKFEKTIAEQAVAAGVPVEVIKESLGEEVFKICYEEDENILGVVGGTLKDFLNSFSTLLKQASILCLLHVYYFTTSLILPGIIKAAAHVLYETEVEVSLYLLYSVHSLVIPTSLFCKTFPFHFMFDKDMTILQFGNGIRRLMNRRDKPNFEEYFEILTPKINQTFSGIMTMLNMQFVVRVRRVMDLKGQMIYIVESSAILFLGSPCVLYLSDIPIHNALRDVVLIGEQARAQDGLKKRLGKLKATLEQAHQALEEEKKKTVDLLCSIFPCEVAQQLWQGQVVQAKKFSNVTMLFSDIVGFTAICSQCSPLQVITMLNALYTRFDQQCGELDVYKVETIGDAYCVAGGLHKESDTHAVQIALMAVKMMELSDEVMSPHGEPIKMRIGLHSGSVFAGVVGVKMPRYCLFGNNVTLANKFESCSVPRKINVSPTTYRLLKDCPGFVFTPRSREELPPNFPSEIPGICHFLDAMYGFVNHALELLVIRNYGPEVWEDIKKEAQLDEEGQFLVRIIYDDSKTYDLVAAASKVLNLNAGEILQMFGKMFFVFCQESGYDTILRVLGSNVREFLQNLDALHDHLATIYPGMRAPSFRCTDAEKGKGLILHYYSEREGLQDIVIGIIKTVAQQIHGTEIDMKVIQQRNEECDHTQFLIEEKEESRISPYTFCKAFPFHIIFDRDLVVTQCGNAIYRVLPQLQPGNCSLLSVFSLVRPHIDISFHGILSHINTVFVLRSKEGLLDSCLRLKGQMIYLPEADSILFLCSPSVMNLDDLTRRGLYLSDIPLHDATRDLVLLGEQFREEYKLTQELEILTDRLQLTLRALEDEKKKTDTLLYSVLPPSVANELRHKRPVPAKRYDNVTILFSGIVGFNAFCSKHAGAMKIVNLLNDLYTRFDTLTDSRKNPFVYKVETVGDKYMTVSGLPEPCIHHARSICHLALDMMEIAGQVQVDGESVQITIGIHTGEVVTGVIGQRMPRYCLFGNTVNLTSRTETTGEKGKINVSEYTYRCLMSPENSDPQFHLEHRGPVSMKGKKEPMQVWFLSR Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Bacterial DNA ligase (Bact ligA) | 2OWO | 7.61 | |
Target general information Gen name Bact ligA Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms ligA; Polydeoxyribonucleotide synthase [NAD+]; NAD+-dependent DNA ligase Protein family NAD-dependent DNA ligase family, LigA subfamily Biochemical class Phosphoric ester ligase Function DNA ligase that catalyzes the formation of phosphodiester linkages between 5'-phosphoryl and 3'-hydroxyl groups in double-stranded DNA using NAD as a coenzyme and as the energy source for the reaction. It is essential for DNA replication and repair of damaged DNA. Related diseases Dystonia 1, torsion, autosomal dominant (DYT1) [MIM:128100]: A primary torsion dystonia, and the most common and severe form. Dystonia is defined by the presence of sustained involuntary muscle contractions, often leading to abnormal postures. Dystonia type 1 is characterized by involuntary, repetitive, sustained muscle contractions or postures involving one or more sites of the body, in the absence of other neurological symptoms. Typically, symptoms develop first in an arm or leg in middle to late childhood and progress in approximately 30% of patients to other body regions (generalized dystonia) within about five years. 'Torsion' refers to the twisting nature of body movements observed in DYT1, often affecting the trunk. Distribution and severity of symptoms vary widely between affected individuals, ranging from mild focal dystonia to severe generalized dystonia, even within families. {ECO:0000269|PubMed:14970196, ECO:0000269|PubMed:15505207, ECO:0000269|PubMed:16361107, ECO:0000269|PubMed:17428918, ECO:0000269|PubMed:18167355, ECO:0000269|PubMed:18477710, ECO:0000269|PubMed:18827015, ECO:0000269|PubMed:19955557, ECO:0000269|PubMed:20169475, ECO:0000269|PubMed:21102408, ECO:0000269|PubMed:24930953, ECO:0000269|PubMed:27490483, ECO:0000269|PubMed:9288096}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Arthrogryposis multiplex congenita 5 (AMC5) [MIM:618947]: A form of arthrogryposis multiplex congenita, a developmental condition characterized by multiple joint contractures resulting from reduced or absent fetal movements. AMC5 is an autosomal recessive form characterized by severe congenital contractures, developmental delay, strabismus and tremor. {ECO:0000269|PubMed:28516161, ECO:0000269|PubMed:29053766, ECO:0000269|PubMed:30244176}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07813 EC number EC 6.5.1.2 Uniprot keywords 3D-structure; Direct protein sequencing; DNA damage; DNA repair; DNA replication; Ligase; Magnesium; Metal-binding; NAD; Reference proteome; Zinc Protein physicochemical properties Chain ID D,A,C Molecular weight (Da) 66922.6 Length 612 Aromaticity 0.06 Instability index 37.23 Isoelectric point 5.48 Charge (pH=7) -16.8 2D Binding mode Binding energy (Kcal/mol) -10.38 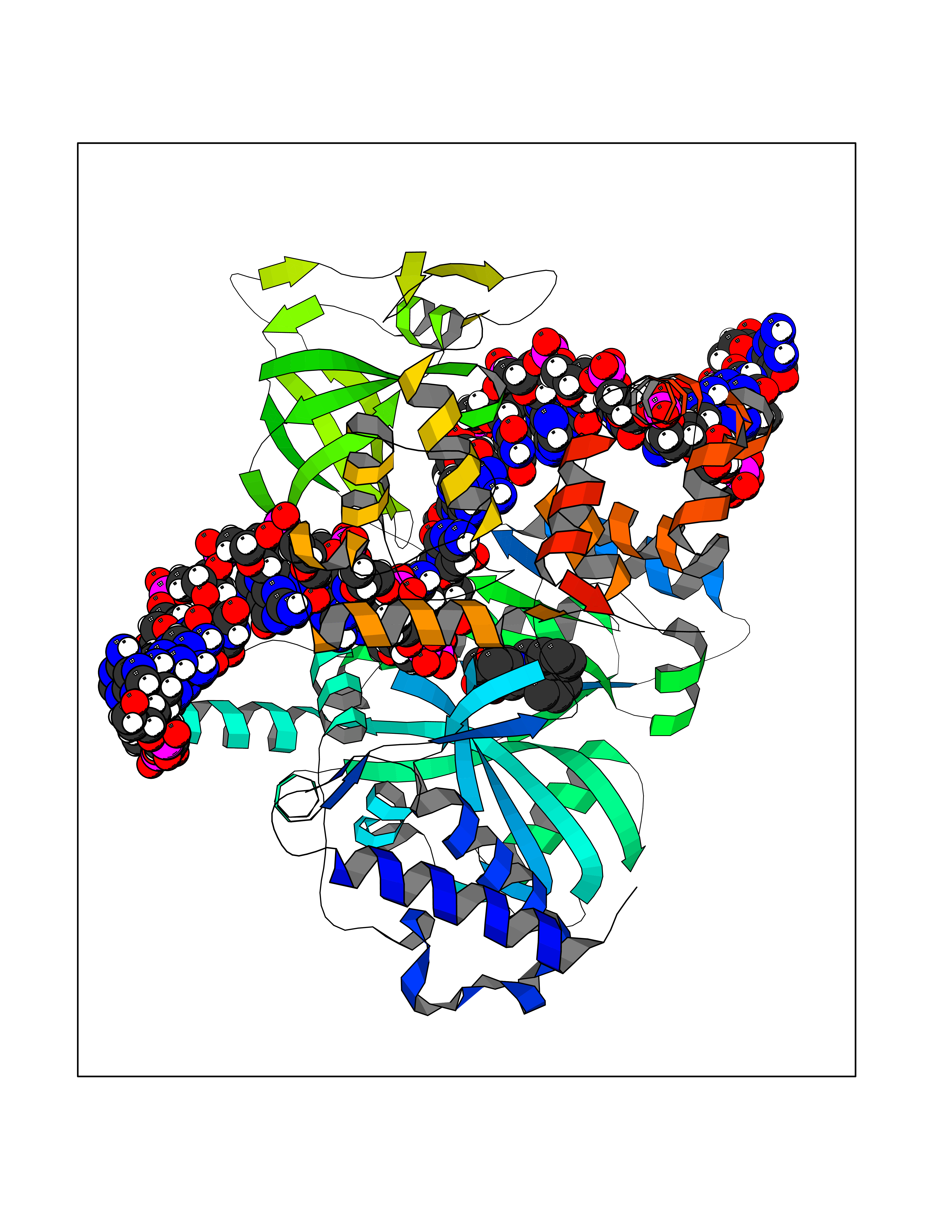 Molscript Map 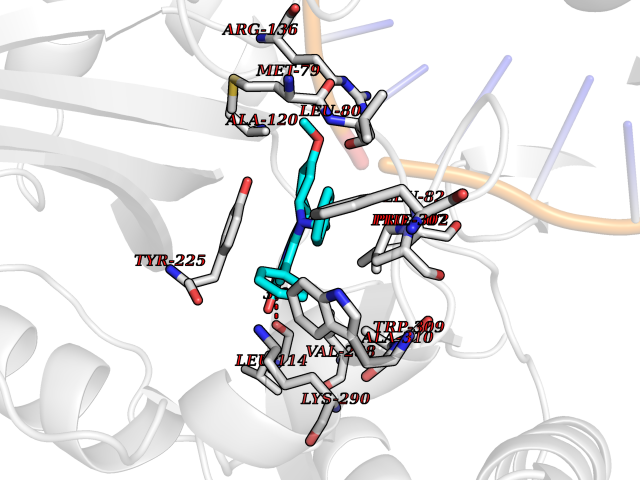 Pymol Map 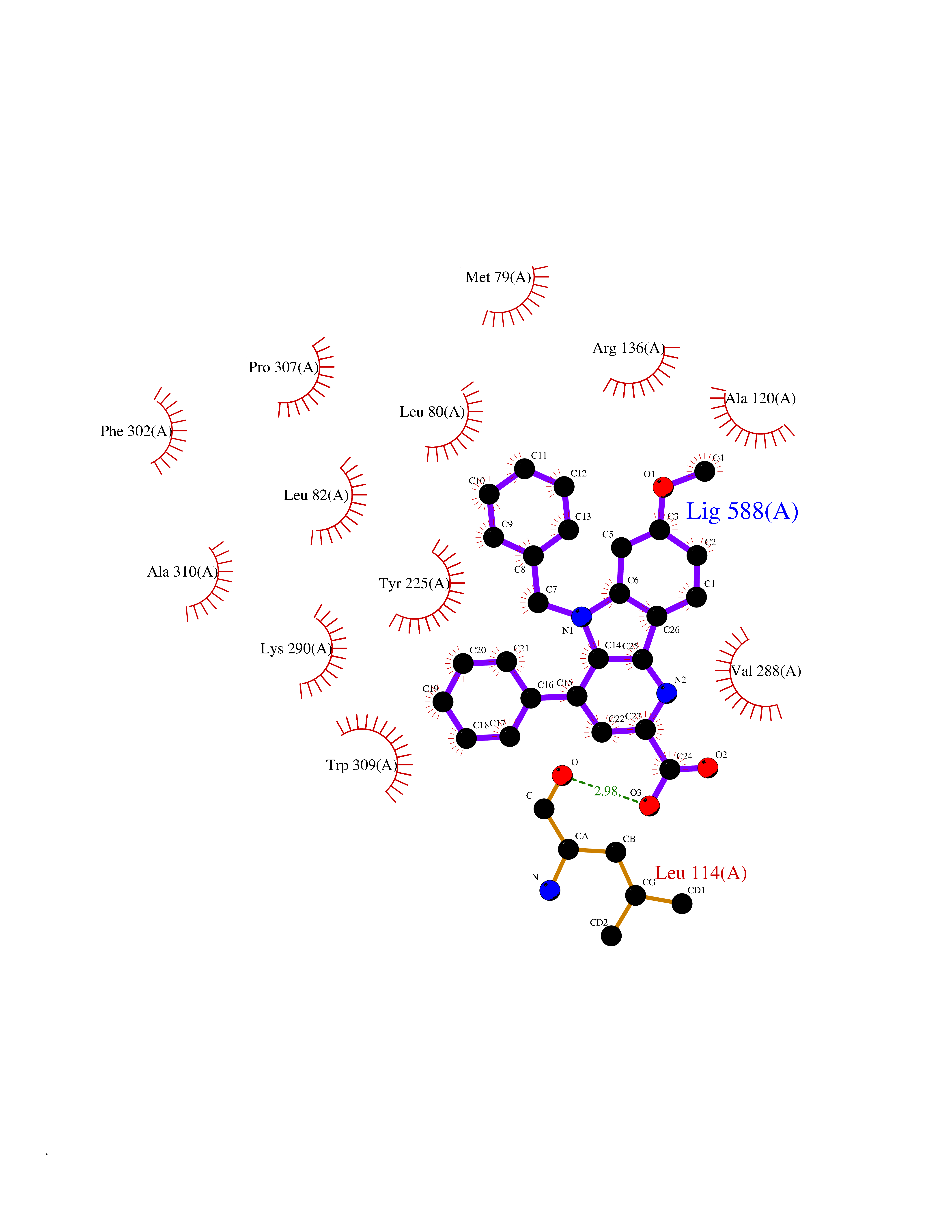 Ligplot Map 3D Binding mode Sequence ACAATTGCGACXCCACTATCGGAATGMESIEQQLTELRTTLRHHEYLYHVMDAPEIPDAEYDRLMRELRELETKHPELITPDSPTQRVGAAPLAAFSQIRHEVPMLSLDNVFDEESFLAFNKRVQDRLKNNEKVTWCCELKLDGLAVSILYENGVLVSAATRGDGTTGEDITSNVRTIRAIPLKLHGENIPARLEVRGEVFLPQAGFEKINEDARRTGGKVFANPRNAAAGSLRQLDPRITAKRPLTFFCYGVGVLEGGELPDTHLGRLLQFKKWGLPVSDRVTLCESAEEVLAFYHKVEEDRPTLGFDIDGVVIKVNSLAQQEQLGFVARAPRWAVAFKFPAQEQMTFVRDVEFQVGRTGAITPVARLEPVHVAGVLVSNATLHNADEIERLGLRIGDKVVIRRAGDVIPQVVNVVLSERPEDTREVVFPTHCPVCGSDVERVEGEAVARCTGGLICGAQRKESLKHFVSRRAMDVDGMGDKIIDQLVEKEYVHTPADLFKLTAGKLTGLERMGPKSAQNVVNALEKAKETTFARFLYALGIREVGEATAAGLAAYFGTLEALEAASIEELQKVPDVGIVVASHVHNFFAEESNRNVISELLAEGVHWPAP Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | G-protein coupled bile acid receptor 1 (GPBAR1) | 7CFM | 7.61 | |
Target general information Gen name GPBAR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hGPCR19; hBG37; TGR5; Membrane-type receptor for bile acids; M-BAR; G-protein coupled receptor GPCR19; BG37 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Bile acid-binding induces its internalization, activation of extracellular signal-regulated kinase and intracellular cAMP production. May be involved in the suppression of macrophage functions by bile acids. Receptor for bile acid. Related diseases Dystonia 34, myoclonic (DYT34) [MIM:619724]: A form of dystonia, a disorder defined by the presence of sustained involuntary muscle contraction, often leading to abnormal postures. DYT34 is an autosomal dominant form characterized by childhood-onset dystonia predominantly affecting hands and neck, with a fast tremor with superimposed myoclonus and, in some individuals, subtle cerebellar signs. {ECO:0000269|PubMed:32212350}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Neurodevelopmental disorder with or without variable movement or behavioral abnormalities (NEDMAB) [MIM:619725]: An autosomal dominant disorder characterized by motor and language developmental delay, intellectual disability often associated with early-onset movement disorders comprising cerebellar ataxia and/or extrapyramidal symptoms. Other variable features include autism spectrum disorder or autistic features and epilepsy. {ECO:0000269|PubMed:33242881}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06777; DB02659; DB03619 Interacts with P38646 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 29418.5 Length 273 Aromaticity 0.1 Instability index 40.15 Isoelectric point 9.83 Charge (pH=7) 9.97 2D Binding mode Binding energy (Kcal/mol) -10.38 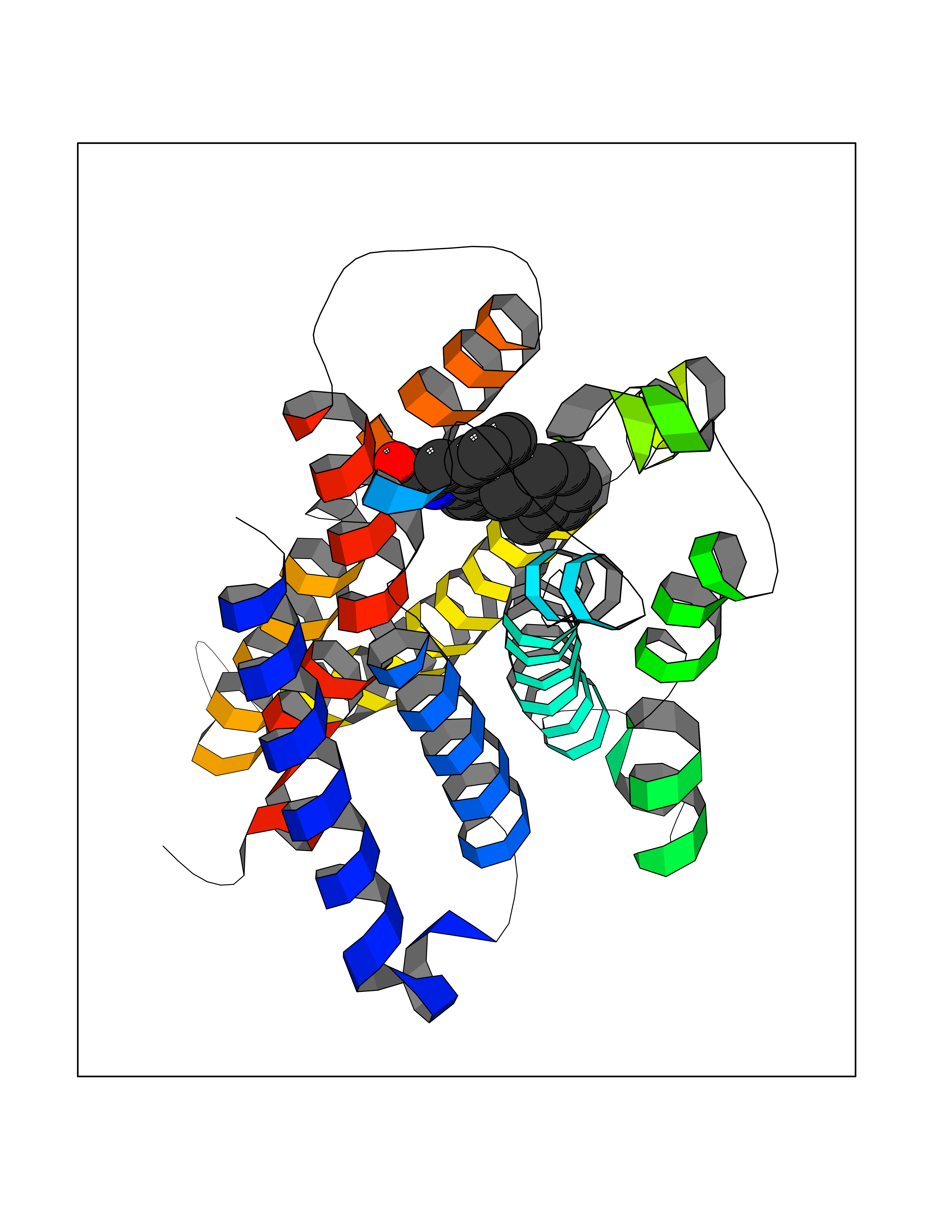 Molscript Map  Pymol Map 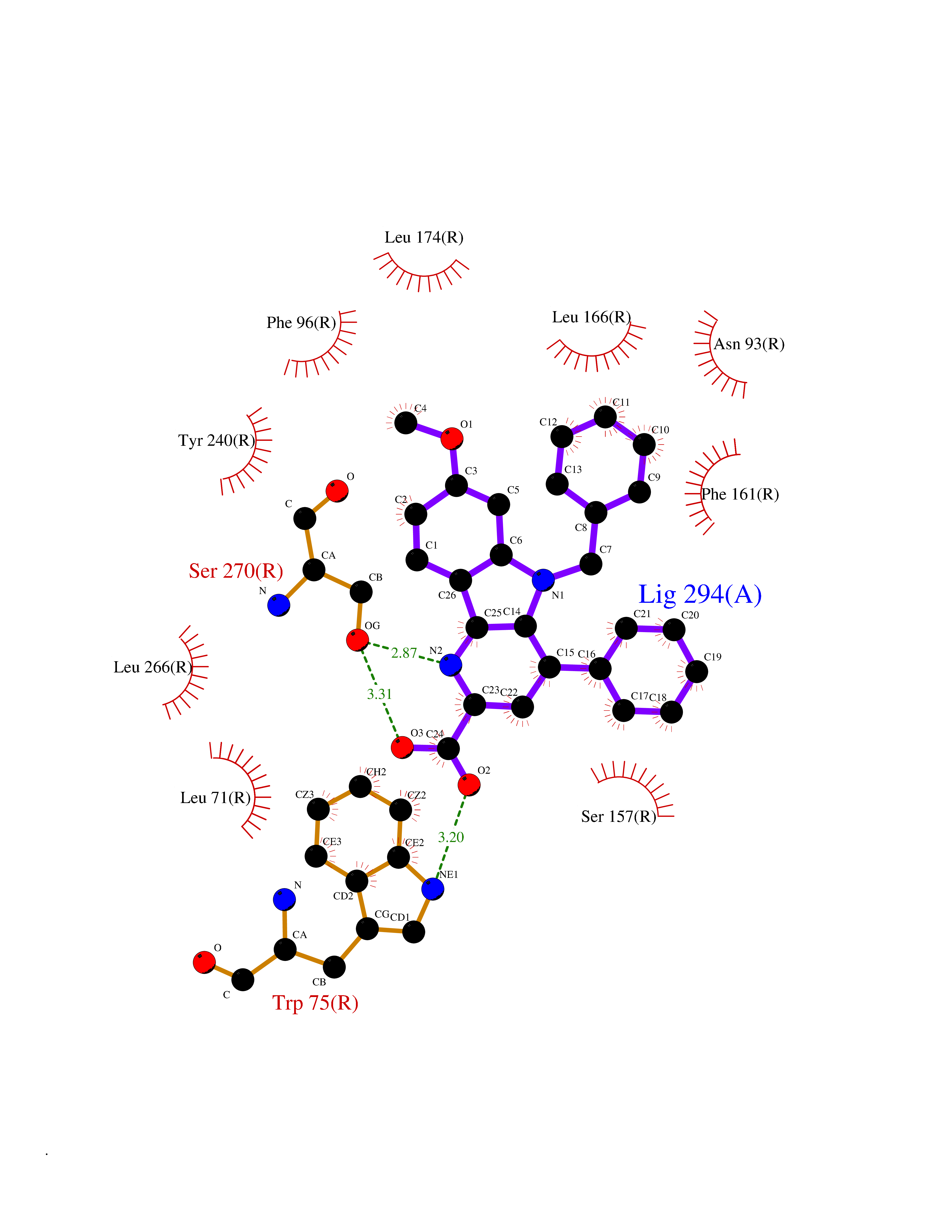 Ligplot Map 3D Binding mode Sequence LSLALASLIITANLLLALGIAWDRRLRSPPAGCFFLSLLLAGLLTGLALPTLPGLWNQSRRGYWSCLLVYLAPNFSFLSLLANLLLVHGERYMAVLRPLQPPGSIRLALLLTWAGPLLFASLPALGWNHWTPGANCSSQAIFPAPYLYLEVYGLLLPAVGAAAFLSVRVLATAHRQLQDICRLERAVCRDEPSALARALTWRQARAQAGAMLLFGLCWGPYVATLLLSVLAYEQRPPLGPGTLLSLLSLGSASAAAVPVAMGLGDQRYTAPWR Hydrogen bonds contact Hydrophobic contact | ||||