Job Results:
Ligand
Structure
Job ID
7eae9f0de32fdedcd28c1404448b5ea8
Job name
NA
Time
2025-04-07 15:33:07
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Tyrosine 3-monooxygenase (TH) | 2XSN | 4.26 | |
Target general information Gen name TH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine 3-hydroxylase; TH Protein family Biopterin-dependent aromatic amino acid hydroxylase family Biochemical class Paired donor oxygen oxidoreductase Function Plays an important role in the physiology of adrenergic neurones. Related diseases Segawa syndrome autosomal recessive (ARSEGS) [MIM:605407]: A form of DOPA-responsive dystonia presenting in infancy or early childhood. Dystonia is defined by the presence of sustained involuntary muscle contractions, often leading to abnormal postures. Some cases present with parkinsonian symptoms in infancy. Unlike all other forms of dystonia, it is an eminently treatable condition, due to a favorable response to L-DOPA. {ECO:0000269|PubMed:10585338, ECO:0000269|PubMed:11196107, ECO:0000269|PubMed:11246459, ECO:0000269|PubMed:15505183, ECO:0000269|PubMed:15747353, ECO:0000269|PubMed:16049992, ECO:0000269|PubMed:17696123, ECO:0000269|PubMed:18058633, ECO:0000269|PubMed:18554280, ECO:0000269|PubMed:19491146, ECO:0000269|PubMed:20056467, ECO:0000269|PubMed:20430833, ECO:0000269|PubMed:21940685, ECO:0000269|PubMed:22264700, ECO:0000269|PubMed:22815559, ECO:0000269|PubMed:23762320, ECO:0000269|PubMed:23939262, ECO:0000269|PubMed:24753243, ECO:0000269|PubMed:7814018, ECO:0000269|PubMed:8528210, ECO:0000269|PubMed:8817341, ECO:0000269|PubMed:9613851, ECO:0000269|PubMed:9703425}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: May play a role in the pathogenesis of Parkinson disease (PD). A genome-wide copy number variation analysis has identified a 34 kilobase deletion over the TH gene in a PD patient but not in any controls. {ECO:0000269|PubMed:20809526}. Drugs (DrugBank ID) DB03552; DB04400; DB00765; DB00120; DB00360; DB00135 Interacts with P29762; P61978-2; Q99750; P08651-5; O75928-2; Q9UHX1-2; P0DJD3-2; P07101-3; Q9UJ04; C9J7I0; Q5MCW4 EC number EC 1.14.16.2 Uniprot keywords 3D-structure; Alternative splicing; Catecholamine biosynthesis; Cell projection; Cytoplasm; Cytoplasmic vesicle; Disease variant; Dystonia; Iron; Metal-binding; Monooxygenase; Neurotransmitter biosynthesis; Nucleus; Oxidoreductase; Parkinson disease; Parkinsonism; Phosphoprotein; Proteomics identification; Reference proteome; Synapse Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 34997 Length 306 Aromaticity 0.12 Instability index 42.59 Isoelectric point 5.32 Charge (pH=7) -12.31 2D Binding mode Binding energy (Kcal/mol) -5.81 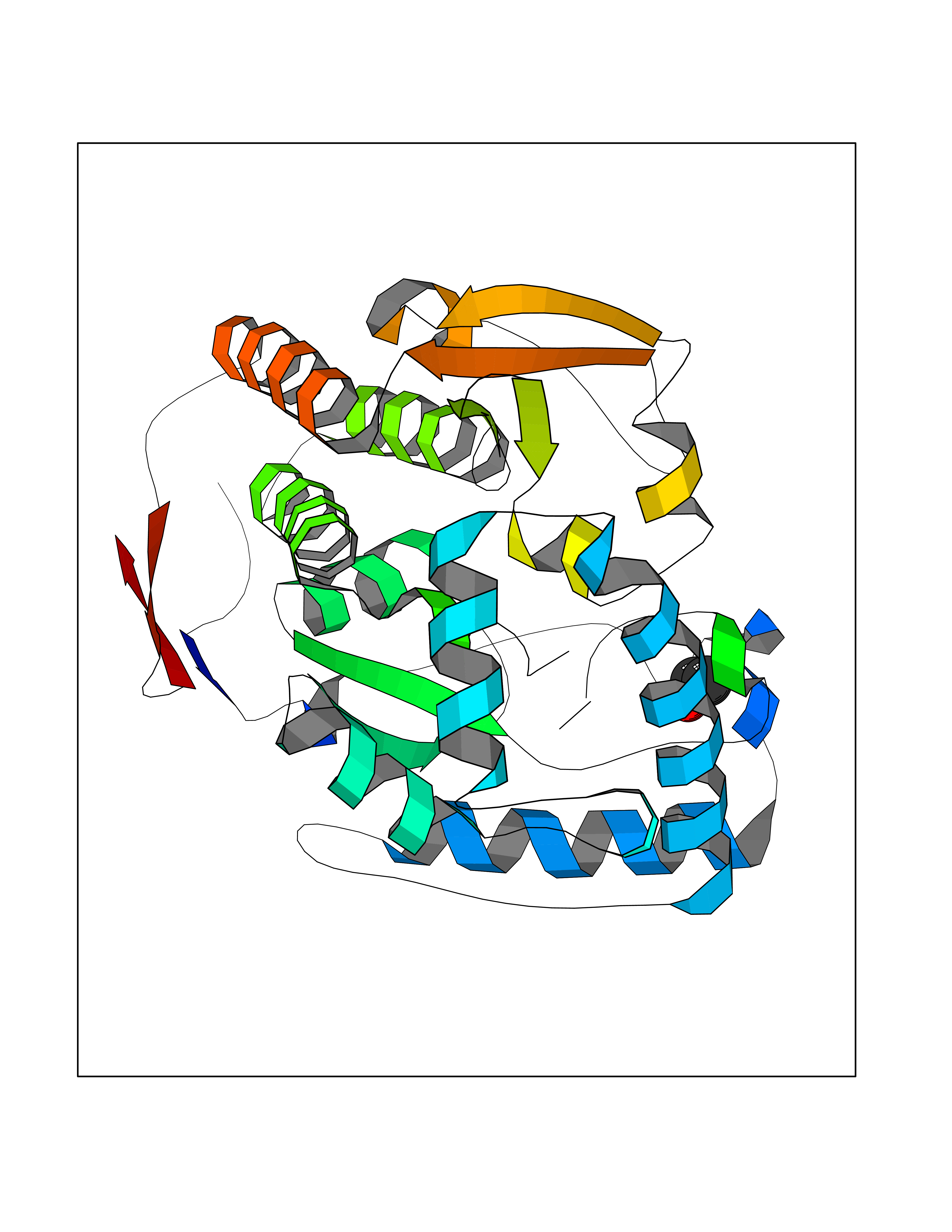 Molscript Map 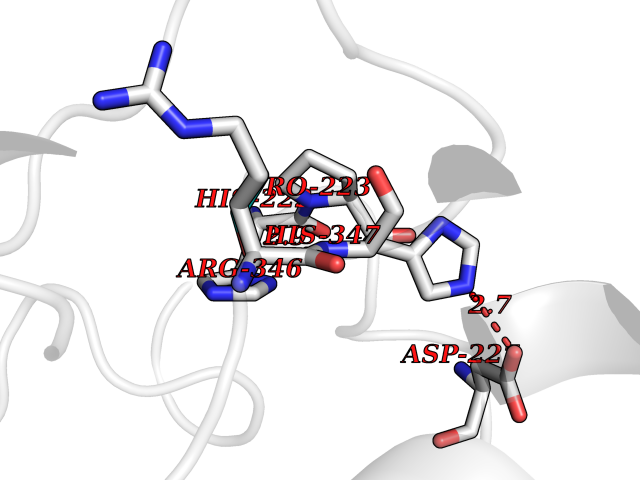 Pymol Map 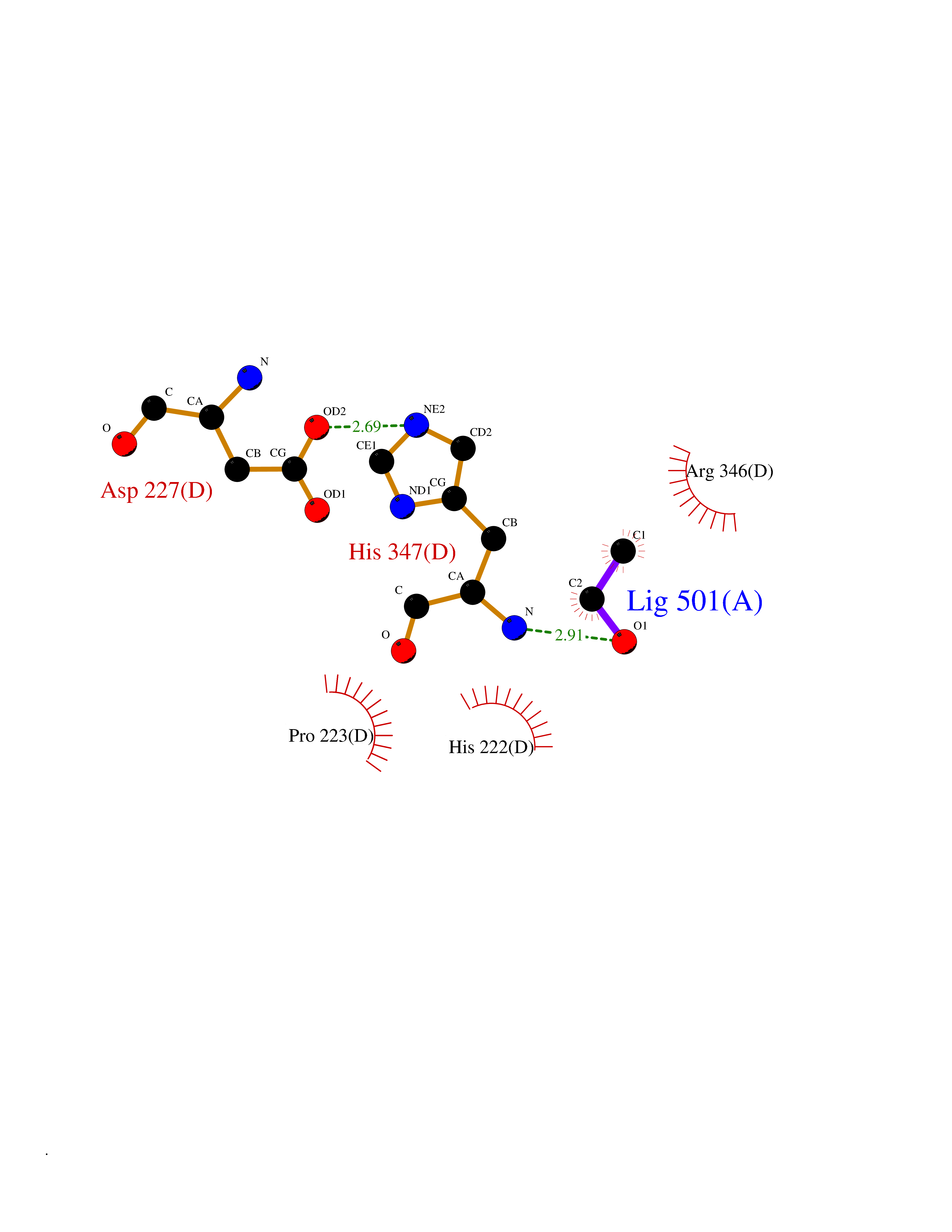 Ligplot Map 3D Binding mode Sequence VPWFPRKVSELDKCHHLVTKFDPDLDLDHPGFSDQVYRQRRKLIAEIAFQYRHGDPIPRVEYTAEEIATWKEVYTTLKGLYATHACGEHLEAFALLERFSGYREDNIPQLEDVSRFLKERTGFQLRPVAGLLSARDFLASLAFRVFQCTQYIRHASSPMHSPEPDCCHELLGHVPMLADRTFAQFSQDIGLASLGASDEEIEKLSTLYWFTVEFGLCKQNGEVKAYGAGLLSSYGELLHCLSEEPEIRAFDPEAAAVQPYQDQTYQSVYFVSESFSDAKDKLRSYASRIQRPFSVKFDPYTLAIDV Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | "Acetolactate synthase, chloroplastic (AtALS) (EC 2.2.1.6) (Acetohydroxy-acid synthase) (Protein CHLORSULFURON RESISTANT 1)" | 5K3S | 4.26 | |
Target general information Gen name ALS Organism Arabidopsis thaliana (Mouse-ear cress) Uniprot ID TTD ID NA Synonyms At3g48560;CSR1;AHAS;T8P19.70;TZP5 Protein family TPP enzyme family Biochemical class NA Function Catalyzes the formation of acetolactate from pyruvate, the first step in valine and isoleucine biosynthesis. {ECO:0000269|PubMed:10386618, ECO:0000269|PubMed:16665813, ECO:0000269|PubMed:16667374, ECO:0000269|PubMed:16668488, ECO:0000269|PubMed:2336405, ECO:0000269|PubMed:8913312, ECO:0000269|PubMed:9355748, ECO:0000269|PubMed:9677339, ECO:0000269|Ref.9}." Related diseases Niemann-Pick disease A (NPDA) [MIM:257200]: An early-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Niemann-Pick disease type A is a primarily neurodegenerative disorder characterized by onset within the first year of life, intellectual disability, digestive disorders, failure to thrive, major hepatosplenomegaly, and severe neurologic symptoms. The severe neurological disorders and pulmonary infections lead to an early death, often around the age of four. Clinical features are variable. A phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. {ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1391960, ECO:0000269|PubMed:15221801, ECO:0000269|PubMed:15877209, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:1718266, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:2023926, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430884, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:8680412, ECO:0000269|PubMed:8693491, ECO:0000269|PubMed:9266408, ECO:0000269|PubMed:9660788}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Niemann-Pick disease B (NPDB) [MIM:607616]: A late-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Clinical signs involve only visceral organs. The most constant sign is hepatosplenomegaly which can be associated with pulmonary symptoms. Patients remain free of neurologic manifestations. However, a phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. In Niemann-Pick disease type B, onset of the first symptoms occurs in early childhood and patients can survive into adulthood. {ECO:0000269|PubMed:12369017, ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1301192, ECO:0000269|PubMed:15241805, ECO:0000269|PubMed:16010684, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:16472269, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:1885770, ECO:0000269|PubMed:19050888, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:21098024, ECO:0000269|PubMed:21621718, ECO:0000269|PubMed:22613662, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430512, ECO:0000269|PubMed:25920558, ECO:0000269|PubMed:26084044, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:27659707, ECO:0000269|PubMed:8051942, ECO:0000269|PubMed:8664904}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 2.2.1.6 Uniprot keywords 3D-structure; Amino-acid biosynthesis; Branched-chain amino acid biosynthesis; Chloroplast; Coiled coil; FAD; Flavoprotein; Genetically modified food; Herbicide resistance; Magnesium; Metal-binding; Oxidation; Plastid; Reference proteome; Thiamine pyrophosphate; Transferase; Transit peptide Protein physicochemical properties Chain ID A Molecular weight (Da) 63431 Length 583 Aromaticity 0.07 Instability index 36.62 Isoelectric point 5.4 Charge (pH=7) -15.33 2D Binding mode Binding energy (Kcal/mol) -5.81 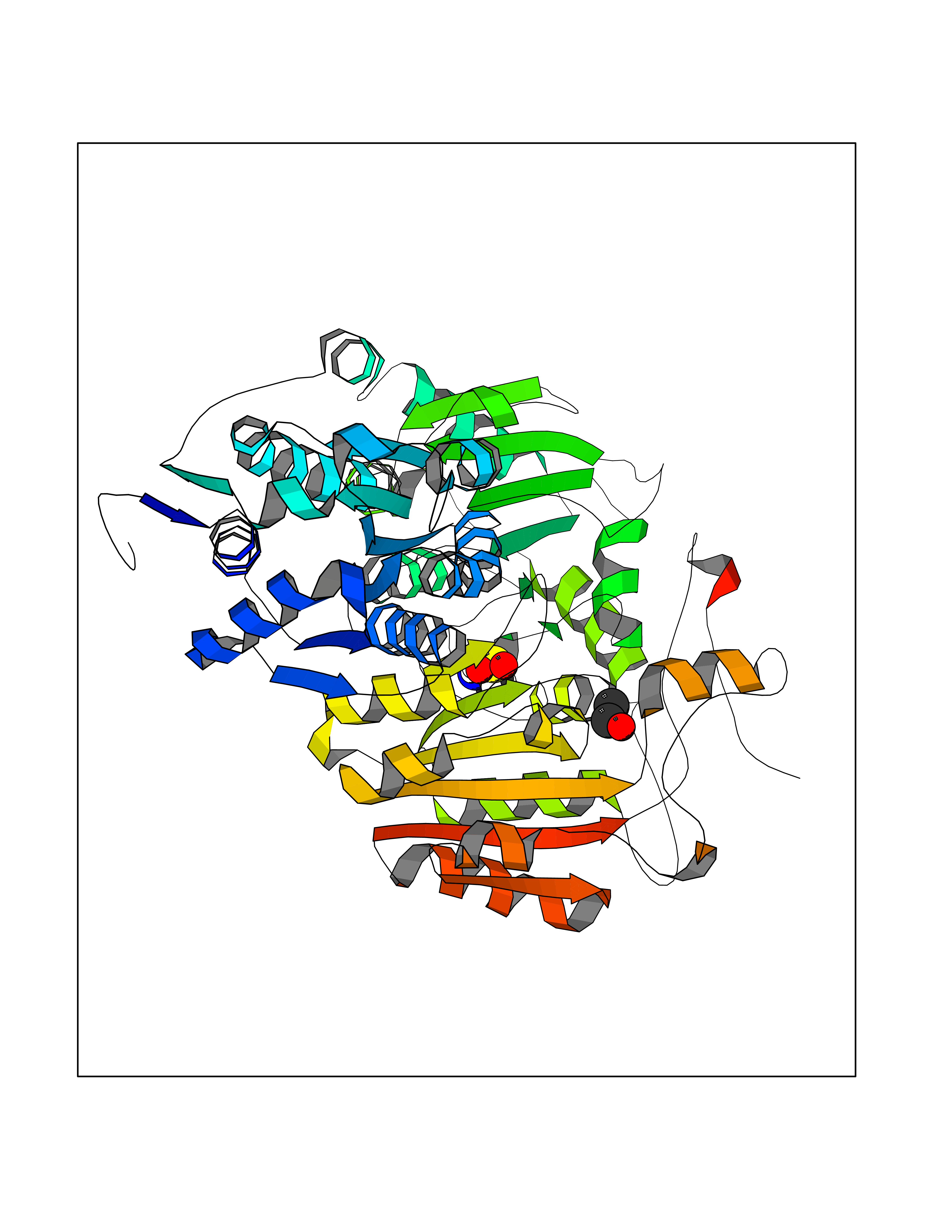 Molscript Map 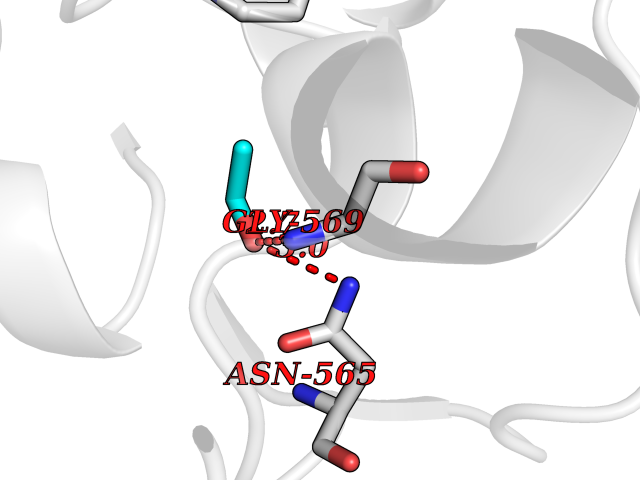 Pymol Map 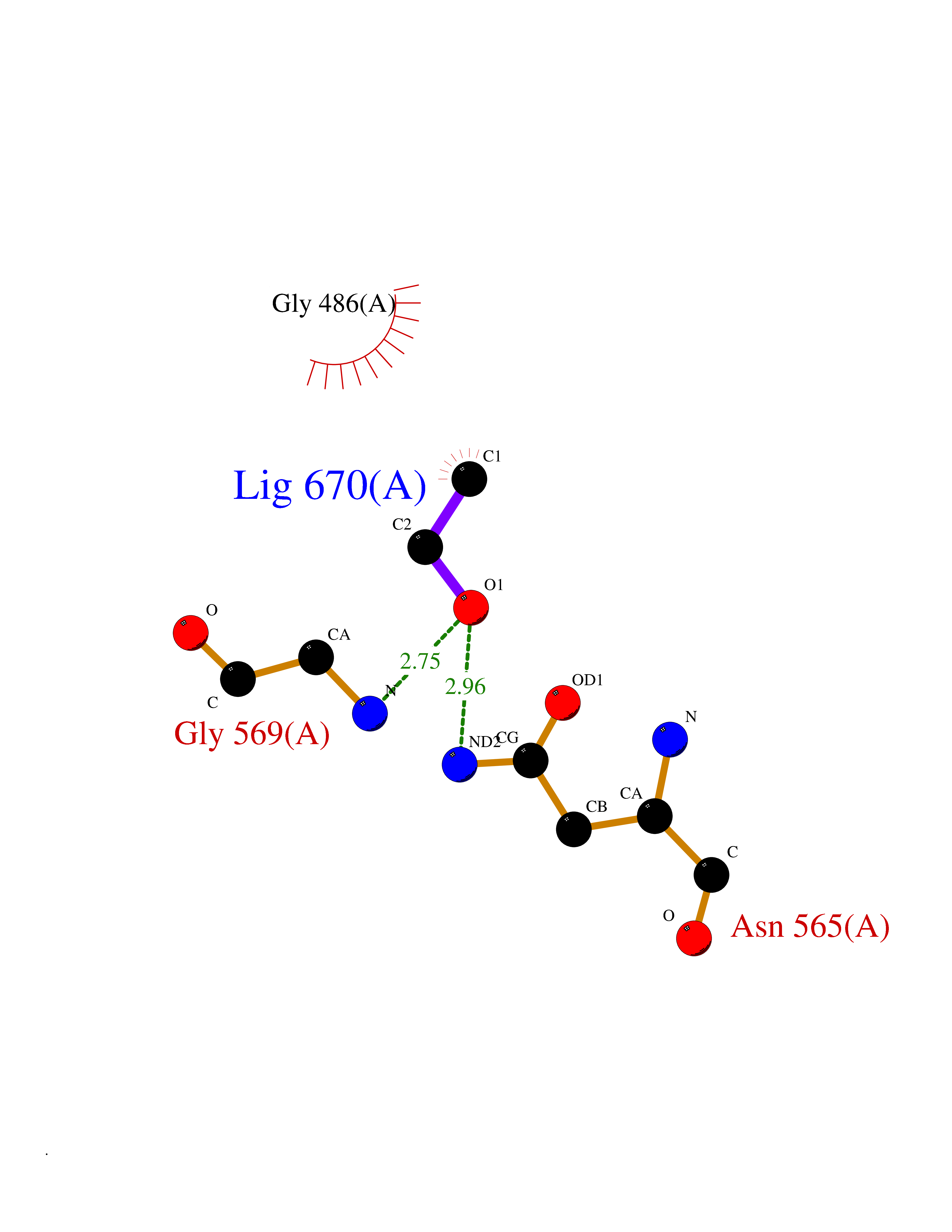 Ligplot Map 3D Binding mode Sequence TFISRFAPDQPRKGADILVEALERQGVETVFAYPGGASMEIHQALTRSSSIRNVLPRHEQGGVFAAEGYARSSGKPGICIATSGPGATNLVSGLADALLDSVPLVAITGQVPRRMIGTDAFQETPIVEVTRSITKHNYLVMDVEDIPRIIEEAFFLATSGRPGPVLVDVPKDIQQQLAIPNWEQAMRLPGYMSRMPKPPEDSHLEQIVRLISESKKPVLYVGGGCLNSSDELGRFVELTGIPVASTLMGLGSYPXDDELSLHMLGMHGTVYANYAVEHSDLLLAFGVRFDDRVTGKLEAFASRAKIVHIDIDSAEIGKNKTPHVSVCGDVKLALQGMNKVLENRAEELKLDFGVWRNELNVQKQKFPLSFKTFGEAIPPQYAIKVLDELTDGKAIISTGVGQHQMWAAQFYNYKKPRQWLSSGGLGAMGFGLPAAIGASVANPDAIVVDIDGDGSFIMNVQELATIRVENLPVKVLLLNNQHLGMVMQWEDRFYKANRAHTFLGDPAQEDEIFPNMLLFAAACGIPAARVTKKADLREAIQTMLDTPGPYLLDVICPHQEHVLPMIPSGGTFNDVITEGDGRL Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Cystathionine gamma-lyase (CTH) | 3COG | 4.26 | |
Target general information Gen name CTH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Gamma-cystathionase; Cysteine-protein sulfhydrase Protein family Trans-sulfuration enzymes family Biochemical class NA Function Catalyzes the last step in the trans-sulfuration pathway from methionine to cysteine. Has broad substrate specificity. Converts cystathionine to cysteine, ammonia and 2-oxobutanoate. Converts two cysteine molecules to lanthionine and hydrogen sulfide. Can also accept homocysteine as substrate. Specificity depends on the levels of the endogenous substrates. Generates the endogenous signaling molecule hydrogen sulfide (H2S), and so contributes to the regulation of blood pressure. Acts as a cysteine-protein sulfhydrase by mediating sulfhydration of target proteins: sulfhydration consists of converting -SH groups into -SSH on specific cysteine residues of target proteins such as GAPDH, PTPN1 and NF-kappa-B subunit RELA, thereby regulating their function. Related diseases Cystathioninuria (CSTNU) [MIM:219500]: Autosomal recessive phenotype characterized by abnormal accumulation of plasma cystathionine, leading to increased urinary excretion. {ECO:0000269|PubMed:12574942, ECO:0000269|PubMed:18476726}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02328; DB03928; DB00151; DB04217; DB00114 Interacts with P32929; Q96NT3; Q96NT3-2; Q96HA8; Q6P9E2 EC number EC 4.4.1.1 Uniprot keywords 3D-structure; Alternative splicing; Amino-acid biosynthesis; Calmodulin-binding; Cysteine biosynthesis; Cytoplasm; Disease variant; Lipid metabolism; Lyase; Proteomics identification; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 86026 Length 782 Aromaticity 0.08 Instability index 32.4 Isoelectric point 6.27 Charge (pH=7) -9.46 2D Binding mode Binding energy (Kcal/mol) -5.81  Molscript Map 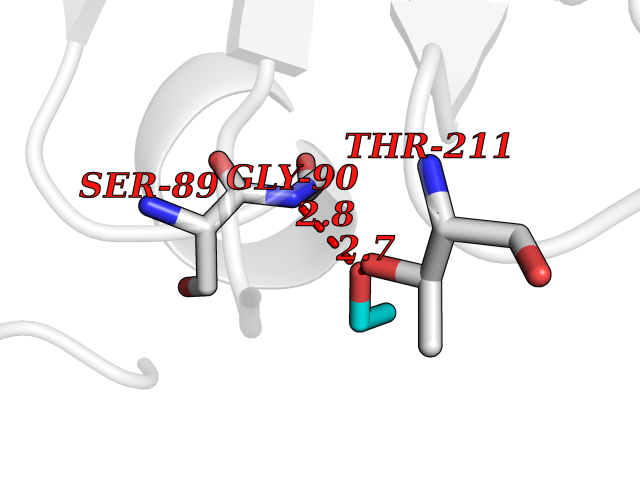 Pymol Map 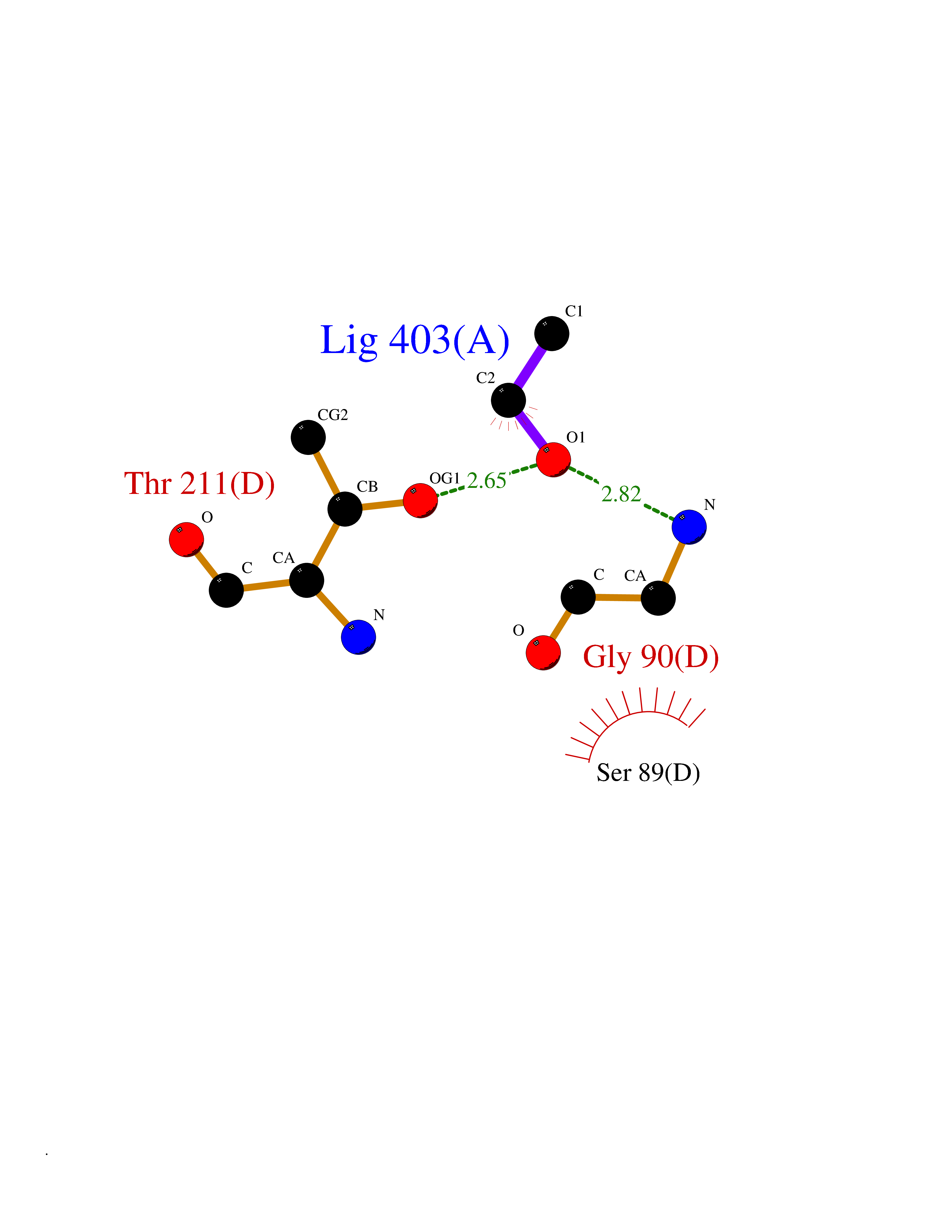 Ligplot Map 3D Binding mode Sequence GFLPHFQHFATQAIHVGQDPEQWTSRAVVPPISLSTTFKQGAPGQHSGFEYSRSGNPTRNCLEKAVAALDGAKYCLAFASGLAATVTITHLLKAGDQIICMDDVYGGTNRYFRQVASEFGLKISFVDCSKIKLLEAAITPETKLVWIETPTNPTQKVIDIEGCAHIVHKHGDIILVVDNTFMSPYFQRPLALGADISMYSATKYMNGHSDVVMGLVSVNCESLHNRLRFLQNSLGAVPSPIDCYLCNRGLKTLHVRMEKHFKNGMAVAQFLESNPWVEKVIYPGLPSHPQHELVKRQCTGCTGMVTFYIKGTLQHAEIFLKNLKLFTLAESLGGFESLAELPAIMTHASVLKNDRDVLGISDTLIRLSVGLEDEEDLLEDLDQALKAAHPPSGFLPHFQHFATQAIHVGQDPEQWTSRAVVPPISLSTTFKQGAPGQGFEYSRSGNPTRNCLEKAVAALDGAKYCLAFASGLAATVTITHLLKAGDQIICMDDVYGGTNRYFRQVASEFGLKISFVDCSKIKLLEAAITPETKLVWIETPTNPTQKVIDIEGCAHIVHKHGDIILVVDNTFMSPYFQRPLALGADISMYSATKYMNGHSDVVMGLVSVNCESLHNRLRFLQNSLGAVPSPIDCYLCNRGLKTLHVRMEKHFKNGMAVAQFLESNPWVEKVIYPGLPSHPQHELVKRQCTGCTGMVTFYIKGTLQHAEIFLKNLKLFTLAESLGGFESLAELPAIMTHASVLKNDRDVLGISDTLIRLSVGLEDEEDLLEDLDQALKAAHPPS Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Vasopressin V1a receptor | 1YTV | 4.26 | |
Target general information Gen name AVPR1A Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms AVPR1 Protein family G-protein coupled receptor 1 family, Vasopressin/oxytocin receptor subfamily Biochemical class Sugar binding protein Function Peptide binding.Peptide hormone binding.Protein kinase C binding.V1A vasopressin receptor binding.Vasopressin receptor activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB09059; DB00872; DB00035; DB00093; DB14642; DB16279; DB05452; DB13929; DB02638; DB06212; DB00067 Interacts with P25106 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID M,N Molecular weight (Da) 40697.6 Length 371 Aromaticity 0.1 Instability index 20.94 Isoelectric point 5.07 Charge (pH=7) -9.93 2D Binding mode Binding energy (Kcal/mol) -5.81  Molscript Map 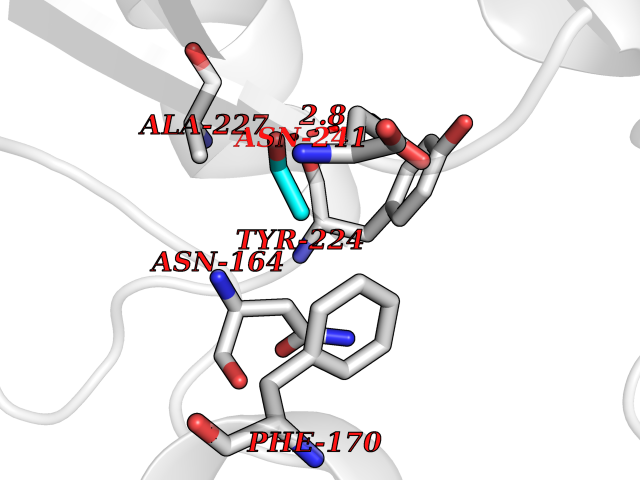 Pymol Map 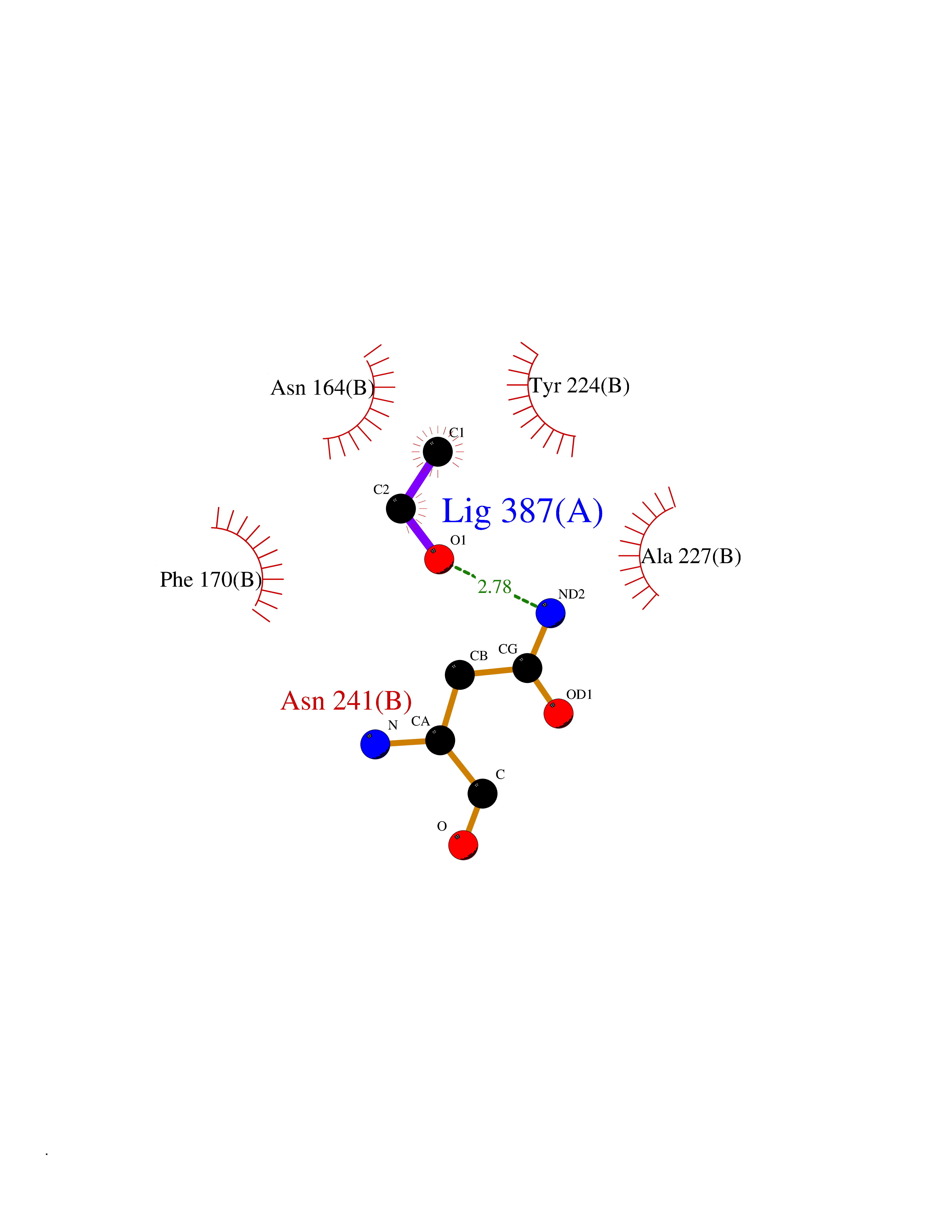 Ligplot Map 3D Binding mode Sequence NSSSNKIEEGKLVIWINGDKGYNGLAEVGKKFEKDTGIKVTVEHPDKLEEKFPQVAATGDGPDIIFWAHDRFGGYAQSGLLAEITPDKAFQDKLYPFTWDAVRYNGKLIAYPIAVEALSLIYNKDLLPNPPKTWEEIPALDKELKAKGKSALMFNLQEPYFTWPLIAADGGYAFKYENGKYDIKDVGVDNAGAKAGLTFLVDLIKNKHMNADTDYSIAEAAFNKGETAMTINGPWAWSNIDTSKVNYGVTVLPTFKGQPSKPFVGVLSAGINAASPNKELAKEFLENYLLTDEGLEAVNKDKPLGAVALKSYEEELAKDPRIAATMENAQKGEIMPNIPQMSAFWYAVRTAVINAASGRQTVDEALKDAQT Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Ribonucleoside-diphosphate reductase subunit M2 | 3OLJ | 4.26 | |
Target general information Gen name RRM2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms RR2 Protein family Ribonucleoside diphosphate reductase small chain family Biochemical class Oxidoreductase Function Metal ion binding.Ribonucleoside-diphosphate reductase activity, thioredoxin disulfide as acceptor. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00242; DB05260; DB05801; DB05003; DB05428 Interacts with P41002; Q9UM11; P23921; O00560 EC number 1.17.4.1 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Deoxyribonucleotide synthesis; Iron; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 33579.4 Length 286 Aromaticity 0.14 Instability index 43.7 Isoelectric point 5.12 Charge (pH=7) -12.86 2D Binding mode Binding energy (Kcal/mol) -5.81  Molscript Map 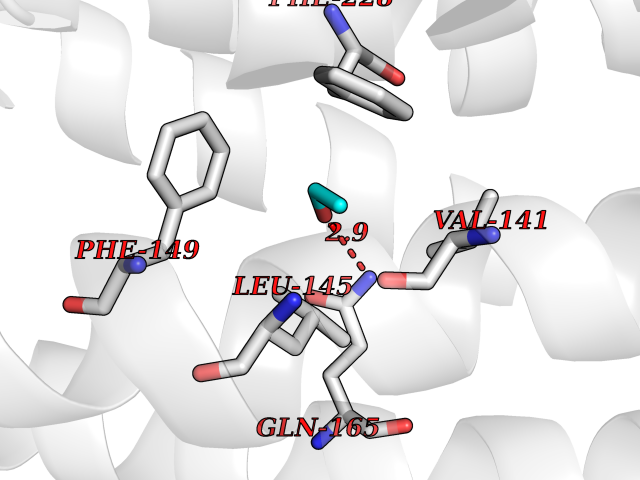 Pymol Map 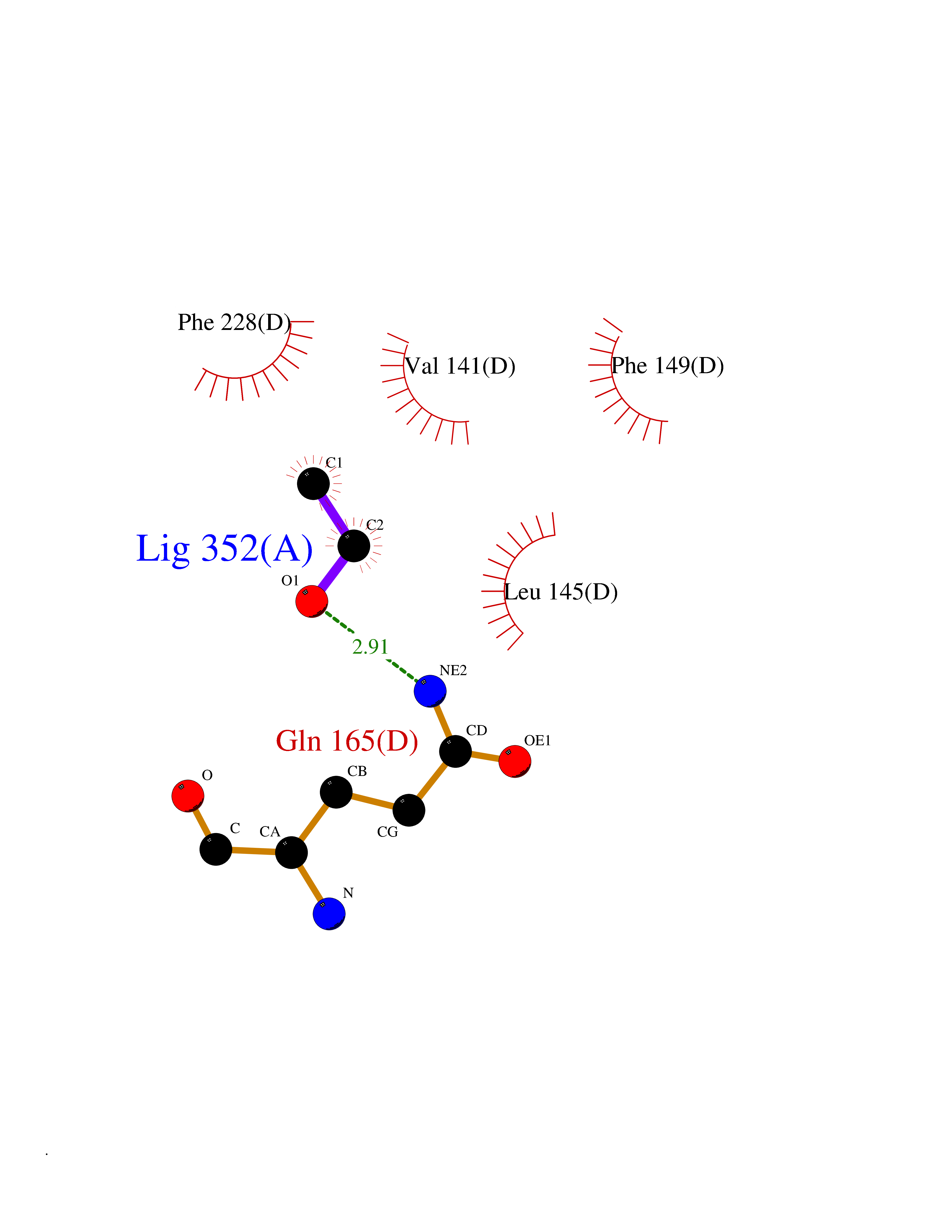 Ligplot Map 3D Binding mode Sequence MGVEDEPLLRENPRRFVIFPIEYHDIWQMYKKAEASFWTAEEVDLSKDIQHWESLKPEERYFISHVLAFFAASDGIVNENLVERFSQEVQITEARCFYGFQIAMENIHSEMYSLLIDTYIKDPKEREFLFNAIETMPCVKKKADWALRWIGDKEATYGERVVAFAAVEGIFFSGSFASIFWLKKRGLMPGLTFSNELISRDEGLHCDFACLMFKHLVHKPSEERVREIIINAVRIEQEFLTEALPVKLIGMNCTLMKQYIEFVADRLMLELGFSKVFRVENPFDFM Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Caspase-3 (CASP3) | 2XYG | 4.26 | |
Target general information Gen name CASP3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Yama protein; SREBP cleavage activity 1; SCA-1; Protein Yama; Cysteine protease CPP32; Caspase 3; CPP32; CPP-32; CASP-3; Apopain Protein family Peptidase C14A family Biochemical class Peptidase Function At the onset of apoptosis it proteolytically cleaves poly(ADP-ribose) polymerase (PARP) at a '216-Asp-|-Gly-217' bond. Cleaves and activates sterol regulatory element binding proteins (SREBPs) between the basic helix-loop-helix leucine zipper domain and the membrane attachment domain. Cleaves and activates caspase-6, -7 and -9. Involved in the cleavage of huntingtin. Triggers cell adhesion in sympathetic neurons through RET cleavage. Involved in the activation cascade of caspases responsible for apoptosis execution. Related diseases Smith-Kingsmore syndrome (SKS) [MIM:616638]: An autosomal dominant syndrome characterized by intellectual disability, macrocephaly, seizures, umbilical hernia, and facial dysmorphic features. {ECO:0000269|PubMed:25851998, ECO:0000269|PubMed:26542245, ECO:0000269|PubMed:27830187}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Focal cortical dysplasia 2 (FCORD2) [MIM:607341]: A form of focal cortical dysplasia, a malformation of cortical development that results in medically refractory epilepsy in the pediatric population and in adults. FCORD2 is a severe form, with onset usually in childhood, characterized by disrupted cortical lamination and specific cytological abnormalities. It is classified in 2 subtypes: type IIA characterized by dysmorphic neurons and lack of balloon cells; type IIB with dysmorphic neurons and balloon cells. {ECO:0000269|PubMed:25799227, ECO:0000269|PubMed:25878179, ECO:0000269|PubMed:26018084, ECO:0000269|PubMed:27830187}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08498; DB08497; DB08213; DB06862; DB08251; DB03124; DB08229; DB00945; DB05408; DB13751; DB06255; DB07696; DB01017; DB08499; DB12843; DB13048; DB00282; DB12709 Interacts with O43823; Q9Y243; P05067; P54252; P55212; P55211; Q14203-5; P42858; Q00987; O60551; P09874; Q5JUK2; P10599; Q9BYP7; P98170 EC number EC 3.4.22.56 Uniprot keywords 3D-structure; Acetylation; Apoptosis; Cytoplasm; Direct protein sequencing; Hydrolase; Phosphoprotein; Protease; Proteomics identification; Reference proteome; S-nitrosylation; Thiol protease; Ubl conjugation; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 27483.1 Length 239 Aromaticity 0.11 Instability index 38.09 Isoelectric point 8.39 Charge (pH=7) 3.03 2D Binding mode Binding energy (Kcal/mol) -5.81 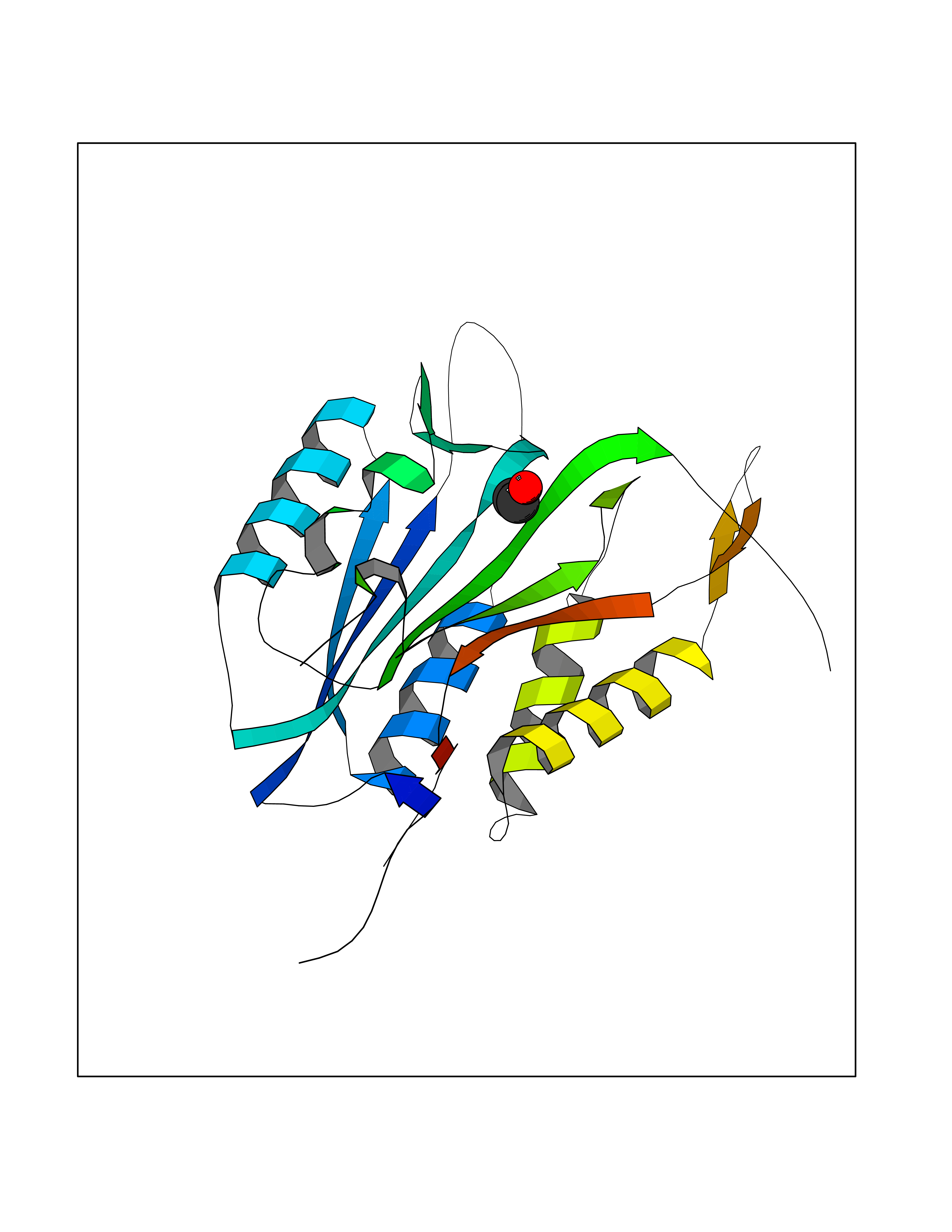 Molscript Map 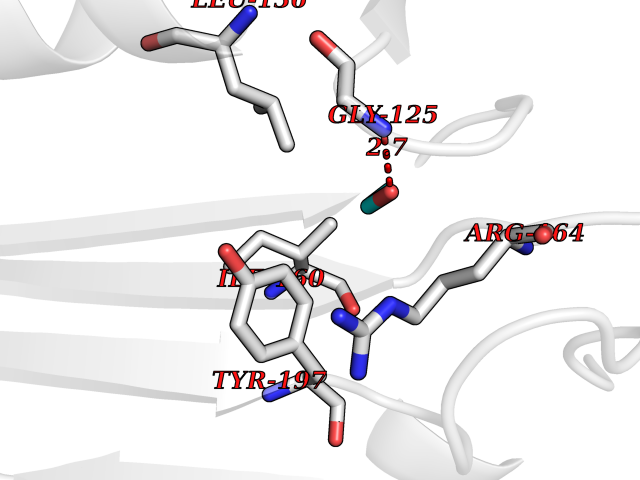 Pymol Map  Ligplot Map 3D Binding mode Sequence SGISLDNSYKMDYPEMGLCIIINNKNFHKSTGMTSRSGTDVDAANLRETFRNLKYEVRNKNDLTREEIVELMRDVSKEDHSKRSSFVCVLLSHGEEGIIFGTNGPVDLKKITNFFRGDRCRSLTGKPKLFIIQACRGTELDCGIETHKIPVEADFLYAYSTAPGYYSWRNSKDGSWFIQSLCAMLKQYADKLEFMHILTRVNRKVATEFESFSFDATFHAKKQIPCIVSMLTKELYFYH Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Peptidyl-prolyl cis-trans isomerase G | 2GW2 | 4.26 | |
Target general information Gen name PPIG Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Isomerase Function Cyclosporin A binding.Peptidyl-prolyl cis-trans isomerase activity.RNA binding. Related diseases Intellectual developmental disorder, autosomal dominant 6, with or without seizures (MRD6) [MIM:613970]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD6 additional features may include seizures, hypotonia, abnormal movements, such as dystonia, and autistic features. {ECO:0000269|PubMed:20890276, ECO:0000269|PubMed:23033978, ECO:0000269|PubMed:23160955, ECO:0000269|PubMed:24863970, ECO:0000269|PubMed:25356899, ECO:0000269|PubMed:27839871, ECO:0000269|PubMed:28095420, ECO:0000269|PubMed:38538865}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 27 (DEE27) [MIM:616139]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. {ECO:0000269|PubMed:24272827, ECO:0000269|PubMed:27839871, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:38538865}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: A chromosomal aberrations involving GRIN2B has been found in patients with intellectual disability. Translocations t(9;12)(p23;p13.1) and t(10;12)(q21.1;p13.1) with a common breakpoint in 12p13.1. Drugs (DrugBank ID) DB00172 Interacts with Q8N7W2-2; Q8NHQ1; O75553; Q9UI36-2; Q96C98; Q8NC69; P17931; Q6NVH9; Q15365; Q9UL42; Q96CD2; Q14498; Q16637; Q12800; Q9NVV9; PRO_0000037309 [P0C6X7] EC number 5.2.1.8 Uniprot keywords 3D-structure; Alternative splicing; Isomerase; Isopeptide bond; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Rotamase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 19125.4 Length 173 Aromaticity 0.1 Instability index 26.46 Isoelectric point 7.14 Charge (pH=7) 0.24 2D Binding mode Binding energy (Kcal/mol) -5.81  Molscript Map 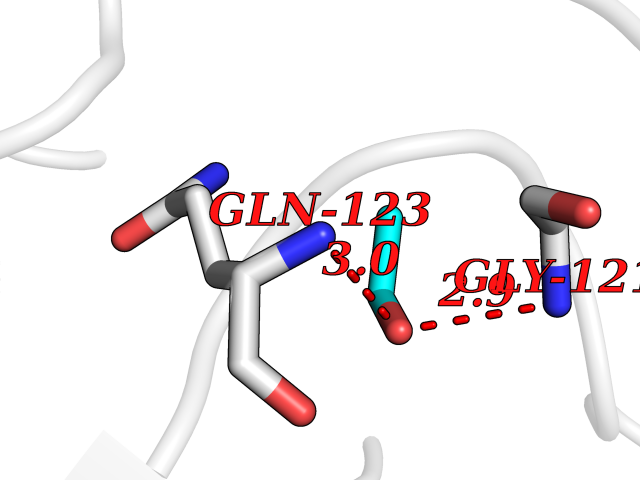 Pymol Map 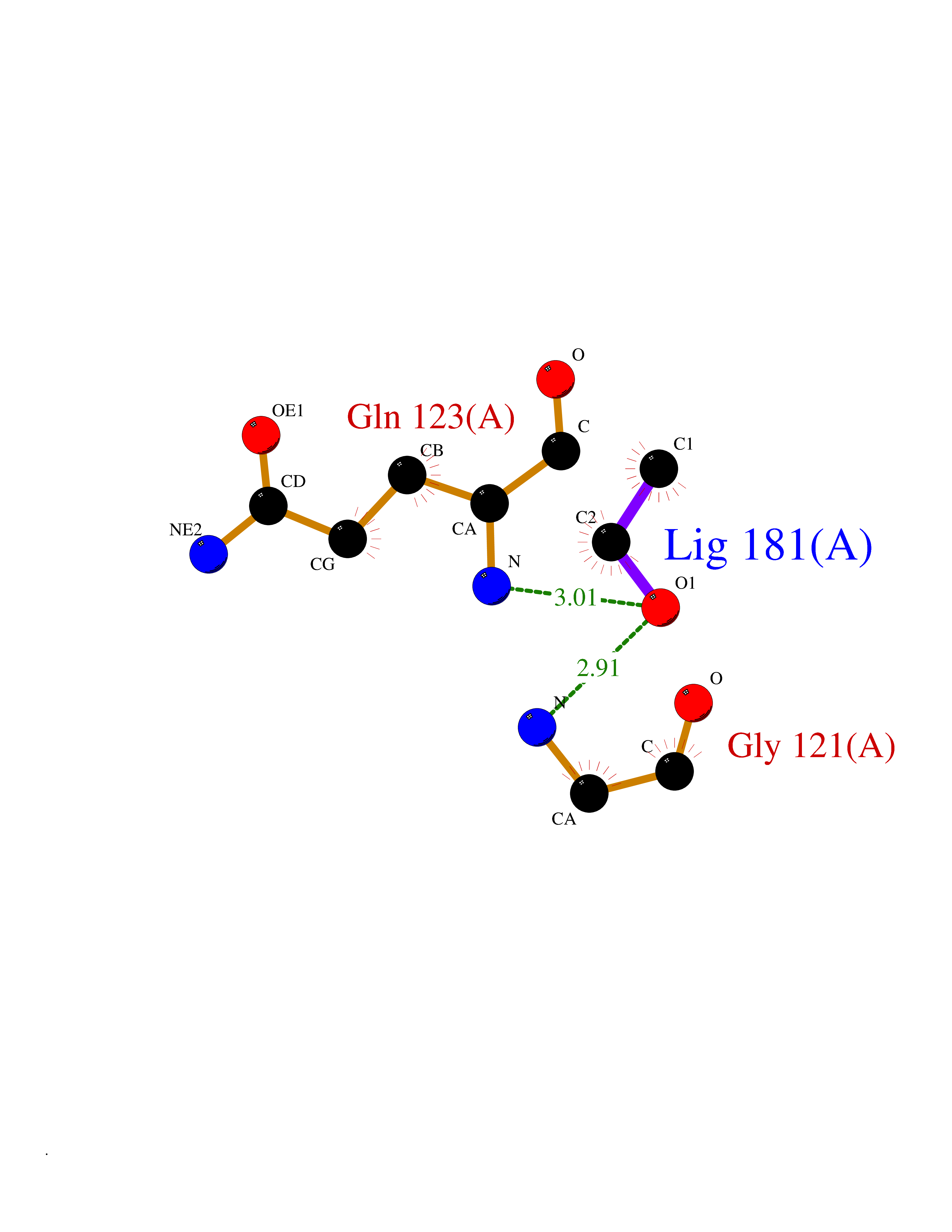 Ligplot Map 3D Binding mode Sequence RPRCFFDIAINNQPAGRVVFELFSDVCPKTCENFRCLCTGEKGTGKSTQKPLHYKSCLFHRVVKDFMVQGGDFSEGNGRGGESIYGGFFEDESFAVKHNAAFLLSMANRGKDTNGSQFFITTKPTPHLDGHHVVFGQVISGQEVVREIENQKTDAASKPFAEVRILSCGELIP Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Sphingosine-1-phosphate lyase 1 (SGPL1) | 4Q6R | 4.26 | |
Target general information Gen name SGPL1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hSPL; Sphingosine-1-phosphate aldolase; SPL 1; SP-lyase 1; S1PL; KIAA1252 Protein family Group II decarboxylase family, Sphingosine-1-phosphate lyase subfamily Biochemical class Carbon-carbon lyase Function Elevates stress-induced ceramide production and apoptosis. Required for global lipid homeostasis in liver and cholesterol homeostasis in fibroblasts. Involved in the regulation of pro-inflammatory response and neutrophil trafficking. Modulates neuronal autophagy via phosphoethanolamine production which regulates accumulation of aggregate-prone proteins such as APP. Seems to play a role in establishing neuronal contact sites and axonal maintenance. Cleaves phosphorylated sphingoid bases (PSBs), such as sphingosine-1-phosphate, into fatty aldehydes and phosphoethanolamine. Related diseases RENI syndrome (RENI) [MIM:617575]: An autosomal recessive, steroid-resistant nephrotic syndrome that manifests in infancy or early childhood, and progresses to end-stage renal failure within a few years. Additional clinical features include ichthyosis, adrenal insufficiency, immunodeficiency, and neurological defects. In rare cases, patients present with isolated primary adrenal insufficiency. Some patients present in utero with fetal hydrops and fetal demise. {ECO:0000269|PubMed:28165339, ECO:0000269|PubMed:28165343, ECO:0000269|PubMed:28181337, ECO:0000269|PubMed:30090628}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00114 Interacts with Q92482; O43315; Q92843; Q6PL45-2; Q8NHW4; Q8N6F1-2; P52803; Q9UKR5; Q7L5A8; Q8N128-2; Q14802-3; P30519; Q01628; Q9NX47; Q6ZSS7; P30301; Q8IY49-2; Q9NXK6; Q04941; Q5VZY2; Q5GAN6; Q5QGT7; Q8TAC9; Q99726; P02808; Q86WV6; Q12846; Q9UNK0; Q96HP8; Q9NWH2; Q5BJF2; O14817; A5PKU2; Q53HI1 EC number EC 4.1.2.27 Uniprot keywords 3D-structure; Acetylation; Apoptosis; Disease variant; Endoplasmic reticulum; Lipid metabolism; Lyase; Membrane; Nitration; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Signal-anchor; Sphingolipid metabolism; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 97521.7 Length 887 Aromaticity 0.1 Instability index 32.99 Isoelectric point 8.97 Charge (pH=7) 18.32 2D Binding mode Binding energy (Kcal/mol) -5.81  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence EYVKALPSQGLSSSAVLEKLKEYSSMDAFWQEGRASGTVYSGEEKLTELLVKAYGDFAWSNPLHPDIFPGLRKIEAEIVRIACSLFNGGPDSCGCVTSGGTESILMACKAYRDLAFEKGIKTPEIVAPQSAHAAFNKAASYFGMKIVRVPLTKMMEVDVRAMRRAISRNTAMLVCSTPQFPHGVIDPVPEVAKLAVKYKIPLHVDACLGGFLIVFMEKAGYPLEHPFDFRVKGVTSISADTHXYGYAPKGSSLVLYSDKKYRNYQFFVDTDWQGGIYASPTIAGSRPGGISAACWAALMHFGENGYVEATKQIIKTARFLKSELENIKGIFVFGNPQLSVIALGSRDFDIYRLSNLMTAKGWNLNQLQFPPSIHFCITLLHARKRVAIQFLKDIRESVTQIMKNPKAKTTGMGAIYGMAQTTVDRNMVAELSSVFLDSLYSTDKEYVKALPSQGLSSSAVLEKLKEYSSMDAFWQEGRASGTVYSGEEKLTELLVKAYGDFAWSNPLHPDIFPGLRKIEAEIVRIACSLFNGGPDSCGCVTSGGTESILMACKAYRDLAFEKGIKTPEIVAPQSAHAAFNKAASYFGMKIVRVPLTKMMEVDVRAMRRAISRNTAMLVCSTPQFPHGVIDPVPEVAKLAVKYKIPLHVDACLGGFLIVFMEKAGYPLEHPFDFRVKGVTSISADTHXYGYAPKGSSLVLYSDKKYRNYQFFVDTDWQGGIYASPTIAGSRPGGISAACWAALMHFGENGYVEATKQIIKTARFLKSELENIKGIFVFGNPQLSVIALGSRDFDIYRLSNLMTAKGWNLNQLQFPPSIHFCITLLHARKRVAIQFLKDIRESVTQIMKNPKAKTTGMGAIYGMAQTTVDRNMVAELSSVFLDSLYSTD Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Tryptophan--tRNA ligase, mitochondrial | 5EKD | 4.26 | |
Target general information Gen name WARS2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class-I aminoacyl-tRNA synthetase family Biochemical class Ligase / antibiotic Function ATP binding.Tryptophan-tRNA ligase activity. Related diseases Neurodevelopmental disorder, mitochondrial, with abnormal movements and lactic acidosis, with or without seizures (NEMMLAS) [MIM:617710]: An autosomal recessive, mitochondrial disorder with a broad phenotypic spectrum ranging from severe neonatal lactic acidosis, encephalomyopathy and early death to an attenuated course with milder manifestations. Clinical features include delayed psychomotor development, intellectual disability, hypotonia, dystonia, ataxia, and spasticity. Severe combined respiratory chain deficiency may be found in severely affected individuals. {ECO:0000269|PubMed:28236339, ECO:0000269|PubMed:28650581, ECO:0000269|PubMed:28905505, ECO:0000269|PubMed:30920170, ECO:0000269|PubMed:35074316}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Parkinsonism-dystonia 3, childhood-onset (PKDYS3) [MIM:619738]: An autosomal recessive neurodegenerative disorder with onset in infancy or early childhood. Affected individuals present with progressive movement abnormalities, including parkinsonism with tremor, dystonia, myoclonus ataxia, and hyperkinetic movements such as ballismus. The parkinsonism features may be responsive to treatment with levodopa, although many patients develop levodopa-induced dyskinesia. Some patients may have mild cognitive impairment or psychiatric disturbances. {ECO:0000269|PubMed:29120065, ECO:0000269|PubMed:31970218, ECO:0000269|PubMed:34890876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00150 Interacts with NA EC number 6.1.1.2 Uniprot keywords 3D-structure; Alternative splicing; Aminoacyl-tRNA synthetase; ATP-binding; Disease variant; Dystonia; Ligase; Mitochondrion; Nucleotide-binding; Parkinsonism; Primary mitochondrial disease; Protein biosynthesis; Proteomics identification; Reference proteome; Transit peptide Protein physicochemical properties Chain ID A,B Molecular weight (Da) 36376.7 Length 327 Aromaticity 0.06 Instability index 51.57 Isoelectric point 8.75 Charge (pH=7) 4.55 2D Binding mode Binding energy (Kcal/mol) -5.81  Molscript Map  Pymol Map 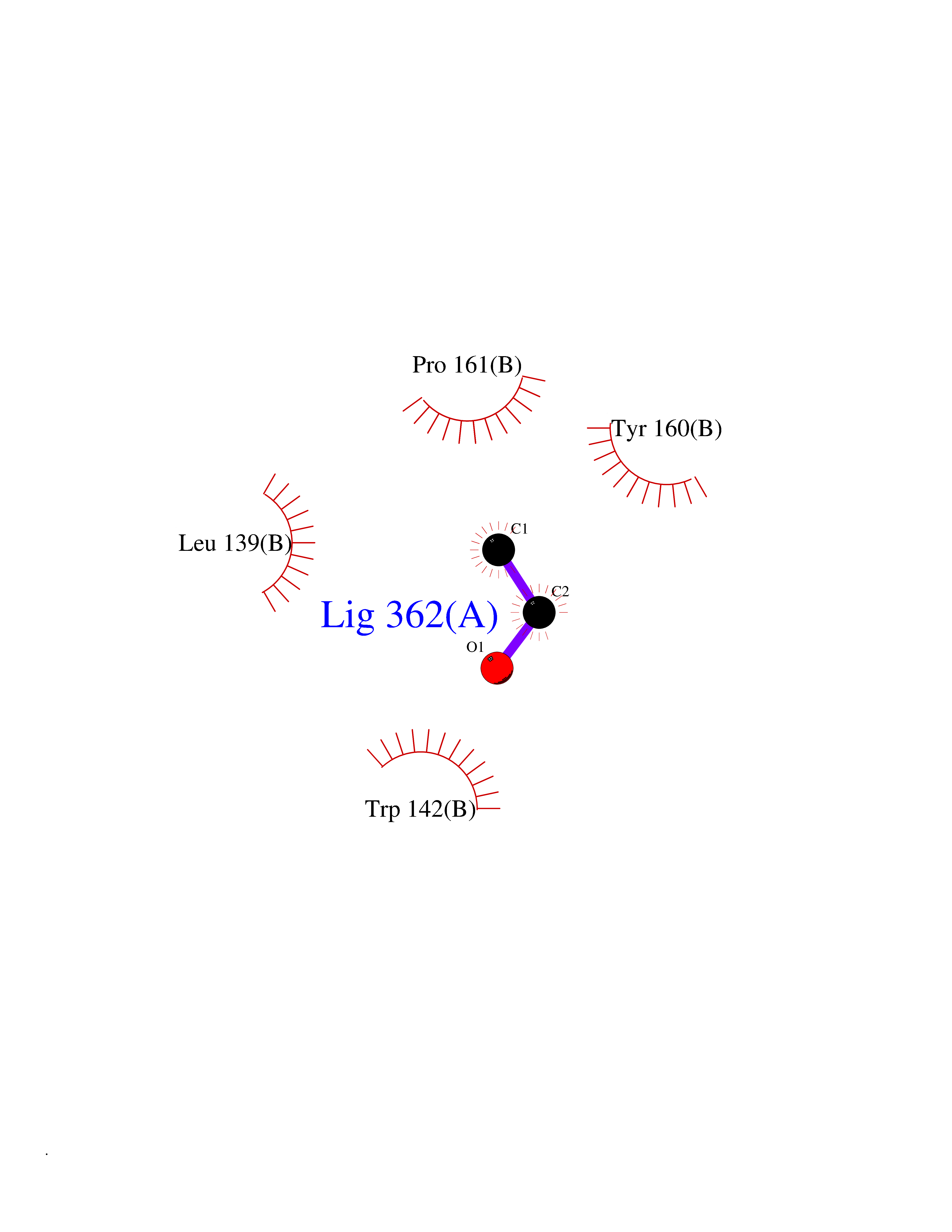 Ligplot Map 3D Binding mode Sequence LQKDSKKRVFSGIQPTGILHLGNYLGAIESWVRLQDEYDSVLYSIVDLHSITVPQDPAVLRQSILDMTAVLLACGINPEKSILFQQSQVSEHTQLSWILSCMVRLPRLQHLHQWKAKTTGTVGLLTYPVLQAADILLYKSTHVPVGEDQVQHMELVQDLAQGFNKKYGEFFPVPESILTSMKKVKSLRDPSAKMSKSDPDKLATVRITDSPEEIVQKFRKAVTDFTSEVTYDPAGRAGVSNIVAVHAAVTGLSVEEVVRRSAGMNTARYKLAVADAVIEKFAPIKREIEKLKLDKDHLEKVLQIGSAKAKELAYTVCQEVKKLVGFL Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Chymase (CYM) | 4K69 | 4.26 | |
Target general information Gen name CMA1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mast cell protease I; CYH; Alpha-chymase Protein family Peptidase S1 family, Granzyme subfamily Biochemical class Peptidase Function Major secreted protease of mast cells with suspected roles in vasoactive peptide generation, extracellular matrix degradation, and regulation of gland secretion. Related diseases Weaver syndrome (WVS) [MIM:277590]: A syndrome of accelerated growth and osseous maturation, unusual craniofacial appearance, hoarse and low-pitched cry, and hypertonia with camptodactyly. Distinguishing features of Weaver syndrome include broad forehead and face, ocular hypertelorism, prominent wide philtrum, micrognathia, deep horizontal chin groove, and deep-set nails. In addition, carpal bone development is advanced over the rest of the hand. {ECO:0000269|PubMed:22177091, ECO:0000269|PubMed:22190405, ECO:0000269|PubMed:23239504, ECO:0000269|PubMed:26694085, ECO:0000269|PubMed:28229514}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03814; DB04016; DB07680; DB03297 Interacts with NA EC number EC 3.4.21.39 Uniprot keywords 3D-structure; Alternative splicing; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 23715.1 Length 215 Aromaticity 0.07 Instability index 37.79 Isoelectric point 9.51 Charge (pH=7) 11.49 2D Binding mode Binding energy (Kcal/mol) -5.81  Molscript Map 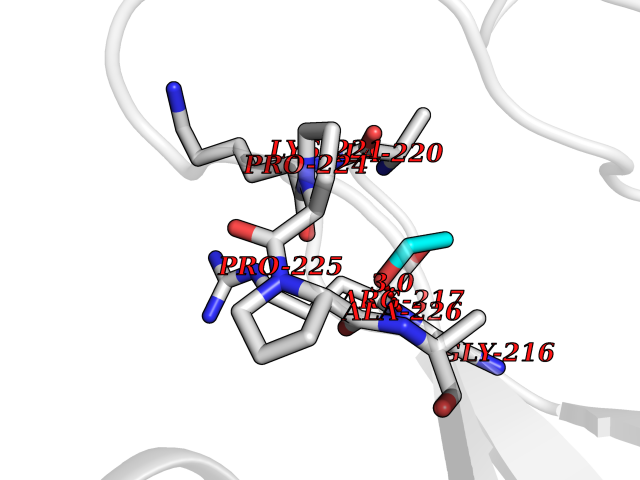 Pymol Map 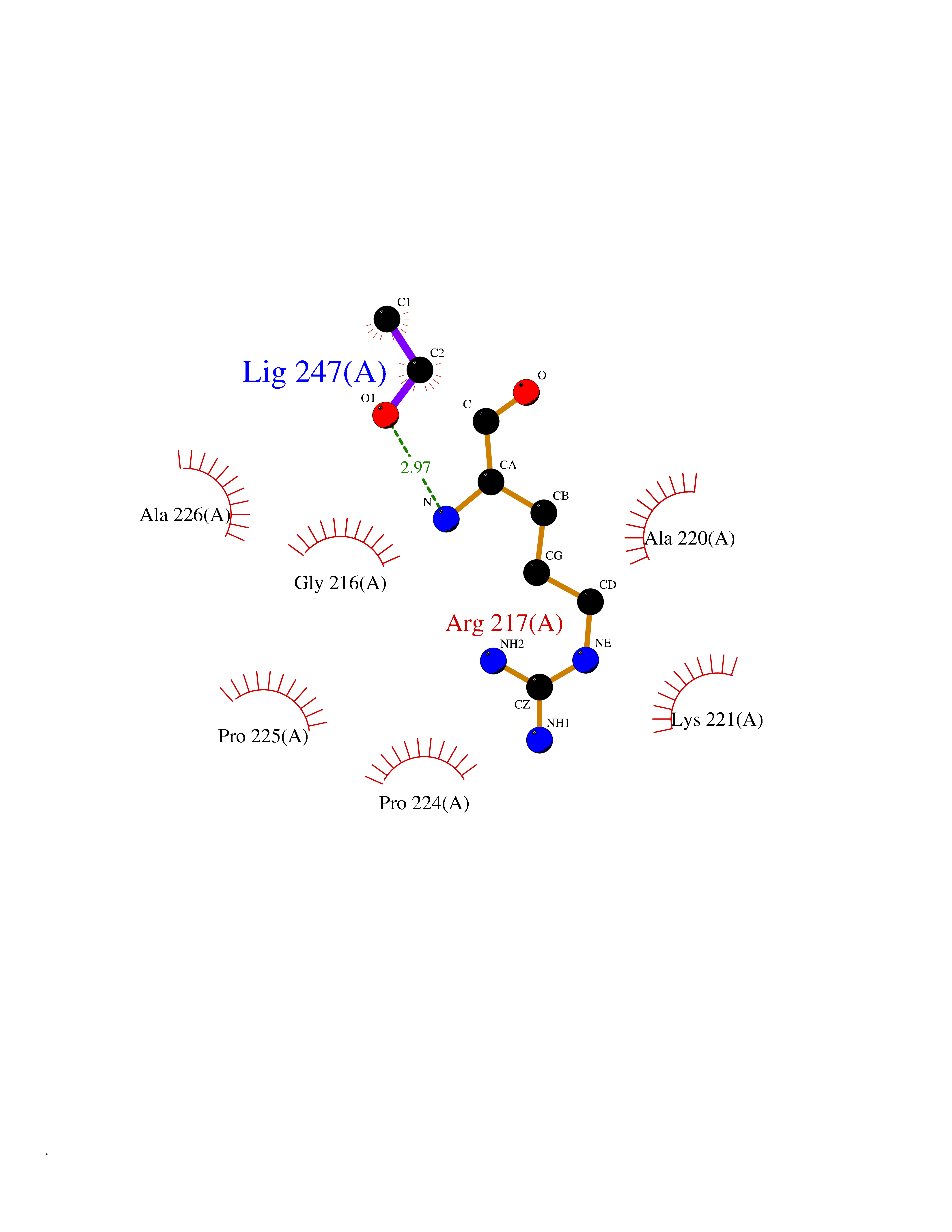 Ligplot Map 3D Binding mode Sequence IIGGTECKPHSRPYMAYLEIVTSNGPSKFCGGFLIRRNFVLTAAHCAGRSITVTLGAHNITEEEDTWQKLEVIKQFRHPKYNTSTLHHDIMLLKLKEKASLTLAVGTLGRMCRVAGWGRTGVLKPGSDTLQEVKLRLMDPQACSHFRDFDHNLQLCVGNPRKTKSAFKGDSGGPLLCAGAAQGIVSYGRSDAKPPAVFTRISHYQPWINQILQAN Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Phosphodiesterase 3B (PDE3B) | 1SOJ | 4.26 | |
Target general information Gen name PDE3B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cGMPinhibited 3',5'cyclic phosphodiesterase B; cGMP-inhibited 3',5'-cyclic phosphodiesterase B; Cyclic GMPinhibited phosphodiesterase B; Cyclic GMP-inhibited phosphodiesterase B; CGIPDE1; CGIPDE B; CG Protein family Cyclic nucleotide phosphodiesterase family, PDE3 subfamily Biochemical class Phosphoric diester hydrolase Function May play a role in fat metabolism. Regulates cAMP binding of RAPGEF3. Through simultaneous binding to RAPGEF3 and PIK3R6 assembles a signaling complex in which the PI3K gamma complex is activated by RAPGEF3 and which is involved in angiogenesis. Cyclic nucleotide phosphodiesterase with a dual-specificity for the second messengers cAMP and cGMP, which are key regulators of many important physiological processes. Related diseases Erythrocytosis, familial, 4 (ECYT4) [MIM:611783]: An autosomal dominant disorder characterized by elevated serum hemoglobin and hematocrit, and normal platelet and leukocyte counts. {ECO:0000269|PubMed:18184961, ECO:0000269|PubMed:18378852, ECO:0000269|PubMed:19208626, ECO:0000269|PubMed:22367913}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07954; DB01640; DB01427; DB00201; DB01970; DB01113; DB09283 Interacts with O60760; Q8TBB1; P48736; Q5UE93; O95398 EC number EC 3.1.4.17 Uniprot keywords 3D-structure; Alternative splicing; Angiogenesis; cAMP; cGMP; Hydrolase; Magnesium; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 41872.2 Length 364 Aromaticity 0.11 Instability index 48.97 Isoelectric point 5.64 Charge (pH=7) -11.84 2D Binding mode Binding energy (Kcal/mol) -5.8  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LDLILVEEYDSLIEKMSNWNFPIFELVEKMGEKSGRILSQVMYTLFQDTGLLEIFKIPTQQFMNYFRALENGYRDIPYHNRIHATDVLHAVWYLTTRPVPGLQQIHNGRIAYISSKSCSNPDESYGCLSSNIPALELMALYVAAAMHDYDHPGRTNAFLVATNAPQAVLYNDRSVLENHHAASAWNLYLSRPEYNFLLHLDHVEFKRFRFLVIEAILATDLKKHFDFLAEFNAKANDVNSNGIEWSNENDRLLVCQVCIKLADINGPAKVRDLHLKWTEGIVNEFYEQGDEEANLGLPISPFMDRSSPQLAKLQESFITHIVGPLCNSYDAAGLLPGQWLEAESRRRIFCQLMHHLTENHKIWK Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Albendazole monooxygenase (CYP3A4) | 3UA1 | 4.26 | |
Target general information Gen name CYP3A4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Taurochenodeoxycholate 6-alpha-hydroxylase; Quinine 3-monooxygenase; P450-PCN1; Nifedipine oxidase; NF-25; HLp; Cytochrome P450-PCN1; Cytochrome P450 NF-25; Cytochrome P450 HLp; Cytochrome P450 3A4; C Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function In liver microsomes, this enzyme is involved in an NADPH-dependent electron transport pathway. It performs a variety of oxidation reactions (e. g. caffeine 8-oxidation, omeprazole sulphoxidation, midazolam 1'-hydroxylation and midazolam 4-hydroxylation) of structurally unrelated compounds, including steroids, fatty acids, and xenobiotics. Acts as a 1,8-cineole 2-exo-monooxygenase. The enzyme also hydroxylates etoposide. Catalyzes 4-beta-hydroxylation of cholesterol. May catalyze 25-hydroxylation of cholesterol in vitro. Catalyzes sulfoxidation of the anthelmintics albendazole and fenbendazole. Cytochromes P450 are a group of heme-thiolate monooxygenases. Related diseases Vitamin D-dependent rickets 3 (VDDR3) [MIM:619073]: An autosomal dominant disorder of vitamin D metabolism resulting in early-onset rickets, reduced serum levels of the vitamin D metabolites 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D, and deficient responsiveness to parent and activated forms of vitamin D. {ECO:0000269|PubMed:29461981}. The gene represented in this entry is involved in disease pathogenesis. Drugs (DrugBank ID) DB08496; DB14055; DB12537; DB12629; DB01456; DB04070; DB11919; DB12515; DB11932; DB12001; DB05812; DB14973; DB11703; DB01418; DB00316; DB00819; DB15568; DB00546; DB08838; DB00518; DB00240; DB00041; DB04630; DB00802; DB00346; DB09026; DB00918; DB06203; DB00969; DB12015; DB14003; DB00404; DB06403; DB06742; DB13141; DB00288; DB00357; DB01424; DB01223; DB01118; DB00321; DB00381; DB00701; DB01217; DB01536; DB01435; DB11901; DB06605; DB00714; DB05676; DB00673; DB01352; DB09229; DB00278; DB01238; DB14185; DB06413; DB01169; DB06697; DB12597; DB06216; DB00637; DB11586; DB01072; DB16098; DB01076; DB01117; DB15011; DB06237; DB15233; DB06442; DB11995; DB06318; DB06626; DB00972; DB09230; DB04957; DB00207; DB12781; DB13997; DB04975; DB01483; DB11817; DB09227; DB00394; DB08903; DB05015; DB16703; DB15463; DB13488; DB09231; DB00865; DB01244; DB15982; DB00443; DB14669; DB12236; DB00307; DB01393; DB01128; DB11799; DB04794; DB00905; DB13746; DB16536; DB00612; DB13975; DB09223; DB08873; DB00188; DB00559; DB06616; DB07348; DB08870; DB09128; DB12267; DB01194; DB05541; DB01200; DB09017; DB11752; DB01222; DB00297; DB00921; DB00490; DB01008; DB09173; DB06772; DB00248; DB08875; DB00201; DB04886; DB00136; DB08907; DB01152; DB09061; DB14737; DB12218; DB11791; DB08502; DB06774; DB00564; DB11383; DB11960; DB06016; DB13835; DB01136; DB14984; DB06634; DB00520; DB01333; DB00482; DB06119; DB09063; DB00439; DB06419; DB00185; DB06777; DB00446; DB00475; DB13528; DB00608; DB00856; DB01114; DB00477; DB00356; DB00169; DB01410; DB09201; DB09232; DB01166; DB00501; DB01012; DB00568; DB00537; DB00604; DB00215; DB01211; DB12499; DB04920; DB01190; DB00349; DB11750; DB01013; DB13158; DB14652; DB00845; DB00636; DB06470; DB01242; DB01068; DB00575; DB00758; DB13843; DB00628; DB01559; DB00257; DB00363; DB09065; DB05239; DB00907; DB00318; DB01394; DB06342; DB00872; DB00286; DB12483; DB04652; DB01285; DB14681; DB01380; DB13003; DB08865; DB11672; DB14635; DB04838; DB00924; DB00531; DB00091; DB04839; DB00987; DB08912; DB09102; DB11963; DB01764; DB01406; DB11779; DB06292; DB04884; DB11682; DB00250; DB15031; DB00496; DB09234; DB12941; DB01264; DB09183; DB01254; DB00694; DB01609; DB11921; DB11943; DB11637; DB00705; DB13857; DB01151; DB00304; DB01260; DB06780; DB01134; DB06700; DB12161; DB01234; DB14649; DB11487; DB09555; DB05351; DB04856; DB14068; DB00514; DB00647; DB14063; DB11994; DB00829; DB00586; DB00485; DB09123; DB00255; DB09095; DB06781; DB01396; DB11274; DB01551; DB11273; DB13345; DB13385; DB00320; DB00343; DB01093; DB08995; DB13347; DB00954; DB00280; DB00822; DB02520; DB01248; DB00204; DB00757; DB08930; DB01184; DB00843; DB11400; DB12301; DB06446; DB05928; DB00590; DB01142; DB00997; DB00254; DB00470; DB04855; DB01395; DB00476; DB11952; DB00378; DB11742; DB14240; DB01127; DB14598; DB14600; DB00625; DB09235; DB06374; DB11979; DB11574; DB00216; DB15444; DB09039; DB09101; DB14064; DB13874; DB11718; DB13007; DB11986; DB08899; DB08992; DB00751; DB00668; DB00700; DB12266; DB01873; DB11405; DB03515; DB02187; DB12329; DB12147; DB01049; DB01253; DB00696; DB00530; DB00199; DB01175; DB11823; DB14575; DB09119; DB00736; DB01215; DB09381; DB12235; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB01196; DB00655; DB04574; DB00402; DB00330; DB00898; DB00977; DB00593; DB08794; DB01466; DB00823; DB09166; DB00294; DB00773; DB01628; DB14766; DB06414; DB13866; DB01590; DB00990; DB00973; DB12500; DB00949; DB01023; DB08980; DB00574; DB00813; DB06702; DB12265; DB08874; DB01216; DB16165; DB13961; DB04908; DB00301; DB00196; DB00687; DB00663; DB04841; DB00180; DB01544; DB00591; DB01047; DB08971; DB00324; DB00472; DB08970; DB14634; DB09378; DB14637; DB00846; DB00690; DB13338; DB04842; DB00499; DB13867; DB08906; DB00588; DB01095; DB00176; DB12307; DB08905; DB01319; DB06717; DB14019; DB01320; DB12010; DB11796; DB11679; DB00947; DB02703; DB15149; DB00674; DB12923; DB05087; DB00317; DB01241; DB01645; DB12184; DB06730; DB11619; DB12141; DB01381; DB11978; DB13879; DB00143; DB01016; DB08909; DB00986; DB05814; DB00889; DB10534; DB11575; DB00365; DB00400; DB01018; DB06786; DB01218; DB13728; DB00502; DB01159; DB05212; DB01275; DB00956; DB00769; DB00741; DB14538; DB14539; DB14540; DB14541; DB14542; DB14543; DB14545; DB14544; DB01611; DB14570; DB06789; DB00557; DB12471; DB09053; DB01050; DB11737; DB09054; DB01181; DB04946; DB00619; DB09262; DB00458; DB00724; DB05039; DB08953; DB00808; DB00224; DB06370; DB11886; DB13293; DB01029; DB00762; DB11633; DB06636; DB00951; DB00982; DB00270; DB11757; DB01167; DB09083; DB08820; DB00602; DB14568; DB04845; DB09570; DB01221; DB01587; DB06738; DB01026; DB09309; DB05903; DB09236; DB06218; DB06791; DB00448; DB01259; DB06685; DB14723; DB12825; DB11951; DB15673; DB16217; DB09078; DB00528; DB11560; DB06469; DB12070; DB01006; DB01227; DB09237; DB01002; DB06282; DB05667; DB00825; DB08918; DB00367; DB00281; DB13766; DB08882; DB17083; DB01583; DB00589; DB09198; DB14065; DB08827; DB01206; DB06448; DB16222; DB00836; DB01601; DB00455; DB00186; DB04871; DB12130; DB09195; DB12089; DB00678; DB14596; DB00227; DB09212; DB08933; DB09280; DB06077; DB06708; DB08815; DB12674; DB12474; DB04829; DB13074; DB08932; DB09238; DB16226; DB04835; DB06234; DB14921; DB00643; DB14009; DB09124; DB00603; DB00253; DB00358; DB00351; DB11529; DB14659; DB00814; DB00170; DB00454; DB09383; DB01071; DB01357; DB04817; DB00333; DB04833; DB00763; DB00563; DB01028; DB09241; DB00353; DB00959; DB14644; DB12952; DB06710; DB00247; DB01233; DB00264; DB00916; DB01011; DB15489; DB00379; DB06148; DB01388; DB01110; DB00683; DB13456; DB06595; DB00834; DB04896; DB13287; DB08893; DB11792; DB00370; DB12489; DB16236; DB06587; DB00648; DB01204; DB16390; DB00745; DB11763; DB00764; DB14512; DB00471; DB00295; DB09205; DB00688; DB01024; DB11605; DB00486; DB14011; DB00607; DB12092; DB11691; DB06230; DB09049; DB01183; DB00731; DB04861; DB01149; DB00220; DB11828; DB09199; DB09048; DB00238; DB00627; DB00622; DB02701; DB00184; DB01115; DB09239; DB04868; DB09240; DB06712; DB04743; DB00393; DB09079; DB16691; DB12005; DB00401; DB01595; DB01054; DB00435; DB11636; DB13981; DB06713; DB14678; DB00717; DB09371; DB01059; DB00957; DB09389; DB00540; DB06174; DB06152; DB00104; DB06670; DB00334; DB09074; DB11442; DB14881; DB00768; DB16267; DB12513; DB09568; DB00338; DB00904; DB11130; DB04911; DB01083; DB01173; DB11837; DB09330; DB04938; DB13500; DB00776; DB12532; DB00239; DB01062; DB00497; DB06412; DB01192; DB12612; DB01229; DB11697; DB09073; DB01267; DB00377; DB05467; DB06603; DB00213; DB00617; DB01384; DB08439; DB00910; DB09297; DB00715; DB06663; DB03010; DB06589; DB00082; DB15102; DB13791; DB00312; DB11198; DB08883; DB01186; DB01074; DB08922; DB00850; DB12978; DB03783; DB00780; DB01174; DB00946; DB00191; DB00812; DB00252; DB13878; DB01085; DB05316; DB00337; DB01100; DB06762; DB09090; DB01132; DB13941; DB12582; DB01621; DB04951; DB17472; DB11642; DB04977; DB12240; DB08910; DB08901; DB12016; DB01263; DB05478; DB15822; DB01411; DB06209; DB01588; DB01058; DB01130; DB00860; DB15566; DB14633; DB14631; DB00635; DB14646; DB13208; DB02789; DB04825; DB05154; DB01087; DB00794; DB01032; DB00396; DB00420; DB13602; DB09288; DB01182; DB12278; DB00571; DB06480; DB00545; DB01589; DB04216; DB01224; DB01103; DB13685; DB00908; DB00468; DB01369; DB12874; DB01129; DB00481; DB00980; DB00863; DB00243; DB00234; DB08896; DB11853; DB06458; DB14761; DB00409; DB00912; DB16826; DB02709; DB01256; DB13174; DB11730; DB06233; DB00615; DB04934; DB01045; DB11753; DB01201; DB01220; DB08864; DB12457; DB00896; DB06155; DB08931; DB14840; DB15305; DB00734; DB14924; DB00503; DB06228; DB09200; DB00533; DB01656; DB13409; DB09291; DB06176; DB00296; DB00412; DB05271; DB00778; DB12332; DB06201; DB11614; DB01698; DB08877; DB06654; DB12391; DB01001; DB00938; DB12543; DB01232; DB11805; DB11767; DB06335; DB00747; DB12834; DB14583; DB11459; DB01037; DB05885; DB11362; DB11942; DB15685; DB11689; DB06731; DB06739; DB06144; DB01104; DB01236; DB01105; DB00203; DB06207; DB09036; DB06290; DB00641; DB12371; DB00877; DB01261; DB06268; DB05482; DB01591; DB09308; DB09099; DB09143; DB00398; DB12713; DB15569; DB12548; DB01323; DB09118; DB00708; DB00359; DB01015; DB01138; DB01268; DB09034; DB09317; DB09318; DB00864; DB00820; DB00675; DB00706; DB06083; DB09071; DB01349; DB08833; DB12887; DB12020; DB05521; DB00976; DB12095; DB00231; DB06287; DB11761; DB00444; DB09299; DB15133; DB00857; DB00342; DB13399; DB13725; DB04905; DB00624; DB13943; DB13944; DB01420; DB13946; DB00759; DB12093; DB14066; DB11712; DB01041; DB00277; DB01154; DB00599; DB04572; DB00906; DB09289; DB08816; DB11470; DB00911; DB01007; DB01409; DB00932; DB06137; DB16732; DB11800; DB06273; DB11635; DB11251; DB08895; DB08811; DB09216; DB01036; DB06212; DB00273; DB01685; DB00539; DB05109; DB00193; DB08911; DB07615; DB00752; DB14962; DB05773; DB00656; DB00755; DB00620; DB00897; DB12245; DB12808; DB09089; DB00347; DB00440; DB06045; DB00197; DB13179; DB11652; DB15328; DB06267; DB08867; DB14989; DB13609; DB15091; DB01586; DB12255; DB11915; DB00580; DB00313; DB15114; DB05294; DB03701; DB04894; DB00862; DB11613; DB08881; DB11581; DB00285; DB00661; DB14895; DB06652; DB09082; DB06684; DB09185; DB00570; DB00541; DB00309; DB11641; DB00361; DB12131; DB08828; DB11094; DB00163; DB11693; DB11739; DB09030; DB00582; DB09068; DB14975; DB12026; DB00682; DB13950; DB01392; DB00549; DB00962; DB15035; DB15688; DB00495; DB00744; DB04832; DB00246; DB00425; DB04828; DB00909; DB01198; DB09225; DB01624; DB15490 Interacts with O15287; Q6ZQX7-4 EC number EC 1.14.14.- Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Fatty acid metabolism; Heme; Iron; Lipid biosynthesis; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol metabolism; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 52195.6 Length 456 Aromaticity 0.11 Instability index 44.02 Isoelectric point 8.48 Charge (pH=7) 4.36 2D Binding mode Binding energy (Kcal/mol) -5.8  Molscript Map 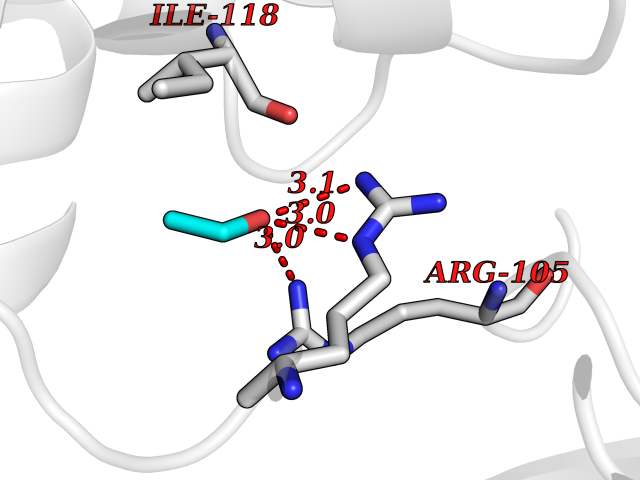 Pymol Map 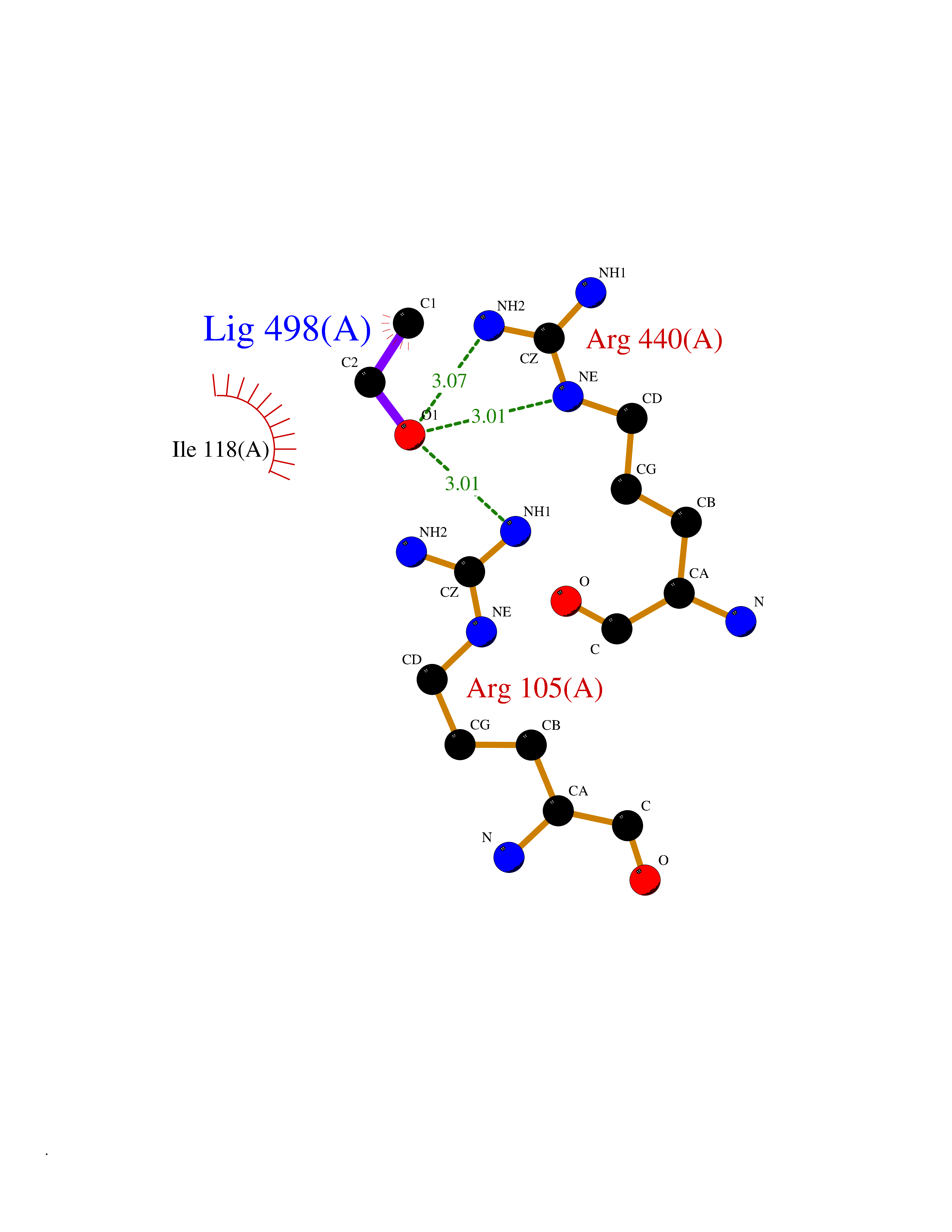 Ligplot Map 3D Binding mode Sequence HSHGLFKKLGIPGPTPLPFLGNILSYHKGFCMFDMECHKKYGKVWGFYDGQQPVLAITDPDMIKTVLVKECYSVFTNRRPFGPVGFMKSAISIAEDEEWKRLRSLLSPTFTSGKLKEMVPIIAQYGDVLVRNLRREAETGKPVTLKDVFGAYSMDVITSTSFGVNIDSLNNPQDPFVENTKKLLRFDFLDPFFLSITVFPFLIPILEVLNICVFPREVTNFLRKSVKRMKEEDTQVDFLQLMIDSQHKALSDLELVAQSIIFIFAGYETTSSVLSFIMYELATHPDVQQKLQEEIDAVLPNKAPPTYDTVLQMEYLDMVVNETLRLFPIAMRLERVCKKDVEINGMFIPKGVVVMIPSYALHRDPKYWTEPEKFLPERFSKKNKDNIDPYIYTPFGSGPRNCIGMRFALMNMKLALIRVLQNFSFKPCKETQIPLKLSLGGLLQPEKPVVLKVESR Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Tankyrase-2 (TNKS-2) | 3U9H | 4.26 | |
Target general information Gen name TNKS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tankyrase-related protein; Tankyrase-like protein; Tankyrase II; TRF1-interacting ankyrin-related ADP-ribose polymerase 2; TNKL; TANK2; Protein poly-ADP-ribosyltransferase tankyrase-2; Poly [ADP-ribos Protein family ARTD/PARP family Biochemical class Glycosyltransferases Function Acts as an activator of the Wnt signaling pathway by mediating poly-ADP-ribosylation of AXIN1 and AXIN2, 2 key components of the beta-catenin destruction complex: poly-ADP-ribosylated target proteins are recognized by RNF146, which mediates their ubiquitination and subsequent degradation. Also mediates poly-ADP-ribosylation of BLZF1 and CASC3, followed by recruitment of RNF146 and subsequent ubiquitination. Mediates poly-ADP-ribosylation of TERF1, thereby contributing to the regulation of telomere length. Stimulates 26S proteasome activity. Poly-ADP-ribosyltransferase involved in various processes such as Wnt signaling pathway, telomere length and vesicle trafficking. Related diseases Intellectual developmental disorder with macrocephaly, seizures, and speech delay (IDDMSSD) [MIM:618158]: An autosomal dominant neurodevelopmental disorder characterized by impaired intellectual development, poor speech, postnatal macrocephaly, and seizures. {ECO:0000269|PubMed:30290153}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O15084; Q7Z6K5-1; O15169; Q9NWV8; P11274; Q13698; Q9NRI5; Q6V0I7; Q9NWT6; P14652; Q9UIQ6; Q14980; Q9BZL4; Q92698; P78314; O43815; P54274; Q9C0C2; Q9UHP3; Q06649 EC number EC 2.4.2.30 Uniprot keywords 3D-structure; ADP-ribosylation; ANK repeat; Chromosome; Cytoplasm; Glycosyltransferase; Golgi apparatus; Hydroxylation; Membrane; Metal-binding; NAD; Nucleotidyltransferase; Nucleus; Proteomics identification; Reference proteome; Repeat; Telomere; Transferase; Ubl conjugation; Wnt signaling pathway; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 23695.5 Length 208 Aromaticity 0.11 Instability index 47.61 Isoelectric point 8.28 Charge (pH=7) 2.88 2D Binding mode Binding energy (Kcal/mol) -5.8 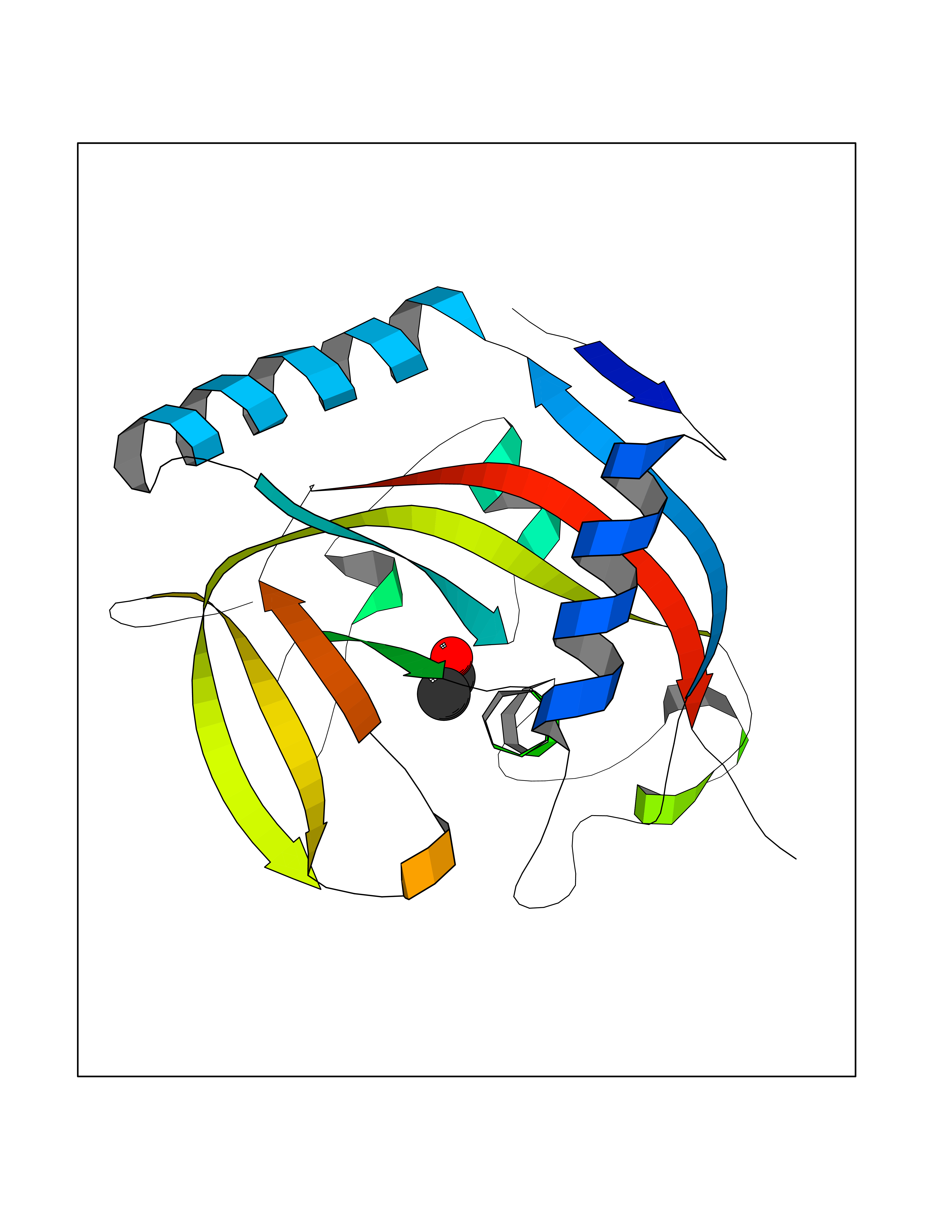 Molscript Map 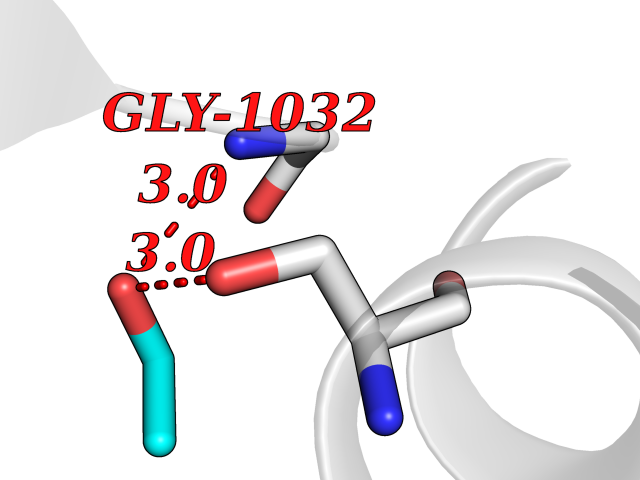 Pymol Map 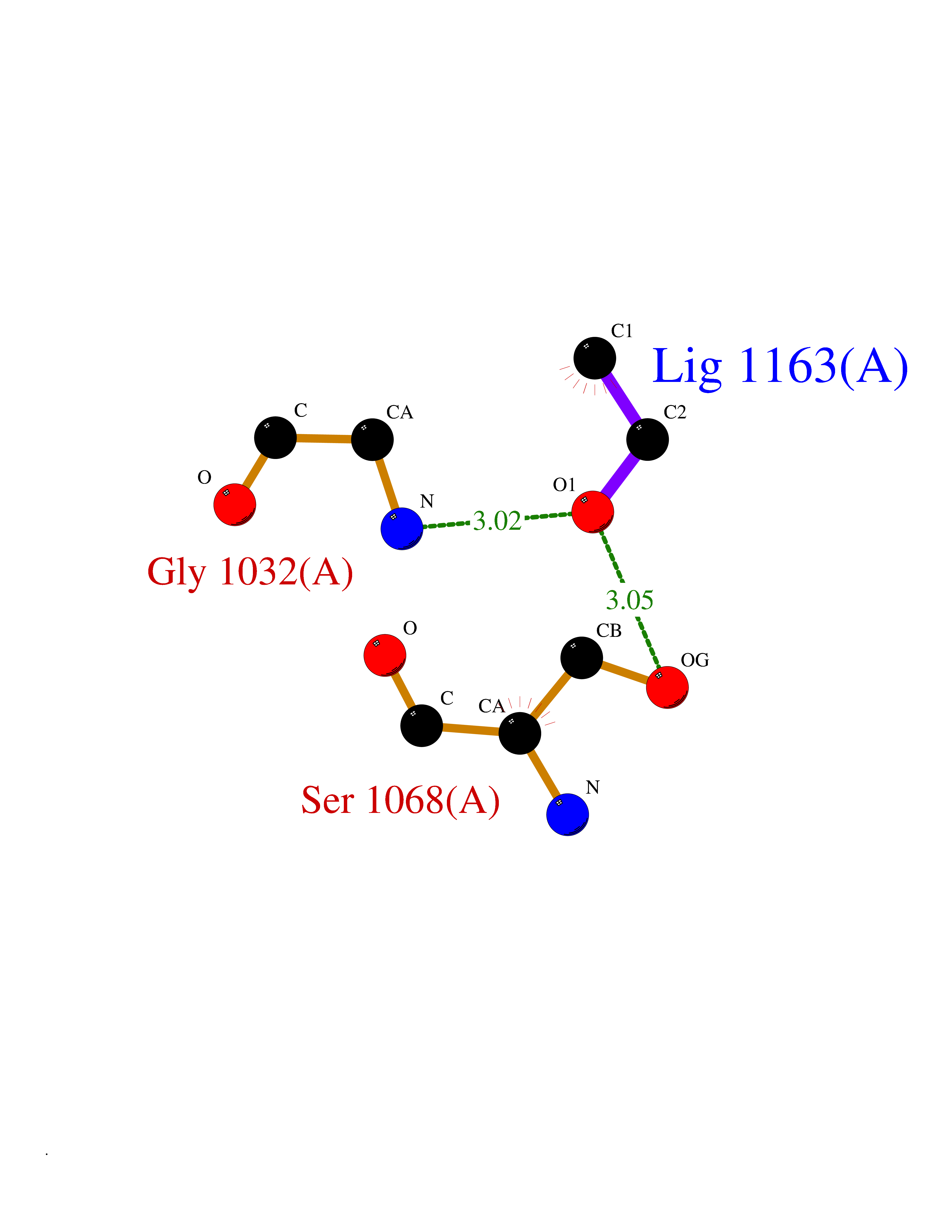 Ligplot Map 3D Binding mode Sequence GTILIDLSPDDKEFQSVEEEMQSTVREHRDGGHAGGIFNRYNILKIQKVCNKKLWERYTHRRKEVSEENHNHANERMLFHGSPFVNAIIHKGFDERHAYIGGMFGAGIYFAENSSKSNQYVYGIGGGTGCPVHKDRSCYICHRQLLFCRVTLGKSFLQFSAMAHSPPGHHSVTGRPSVNGLALAEYVIYRGEQAYPEYLITYQIMRPE Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Fungal Isoleucyl t-RNA synthetase (Fung ILS1) | 7D5C | 4.26 | |
Target general information Gen name Fung ILS1 Organism Saccharomyces cerevisiae (strain ATCC 204508 / S288c) (Baker's yeast) Uniprot ID TTD ID Synonyms Fung Isoleucyl-tRNA synthetase; Fung Isoleucine--tRNA ligase, cytoplasmic; Fung IleRS Protein family Class-I aminoacyl-tRNA synthetase family Biochemical class Carbon-oxygen ligase Function Has aminoacyl-tRNA editing and isoleucine-tRNA ligase activity. Related diseases Intellectual developmental disorder with dysmorphic facies and ptosis (IDDDFP) [MIM:617333]: An autosomal dominant neurodevelopmental disorder characterized by delayed psychomotor development, intellectual disability, delayed language, and facial dysmorphisms, most notably ptosis. Additional features may include poor growth, hypotonia, and seizures. {ECO:0000269|PubMed:27939639, ECO:0000269|PubMed:27939640}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 6.1.1.5 Uniprot keywords 3D-structure; Aminoacyl-tRNA synthetase; ATP-binding; Cytoplasm; Direct protein sequencing; Isopeptide bond; Ligase; Nucleotide-binding; Phosphoprotein; Protein biosynthesis; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 105860 Length 919 Aromaticity 0.12 Instability index 37.01 Isoelectric point 5.73 Charge (pH=7) -14.6 2D Binding mode Binding energy (Kcal/mol) -5.81  Molscript Map 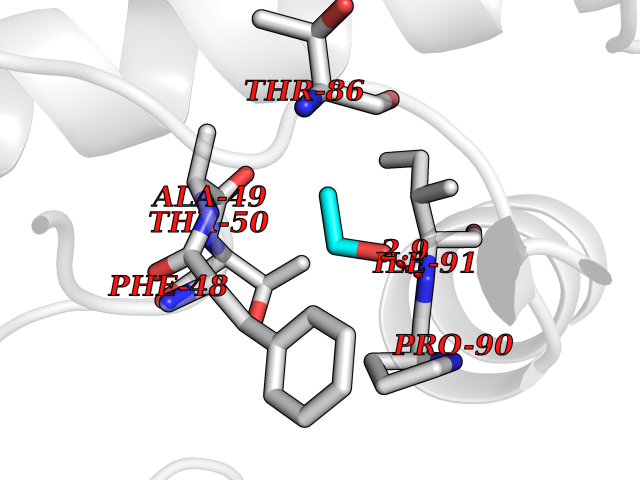 Pymol Map 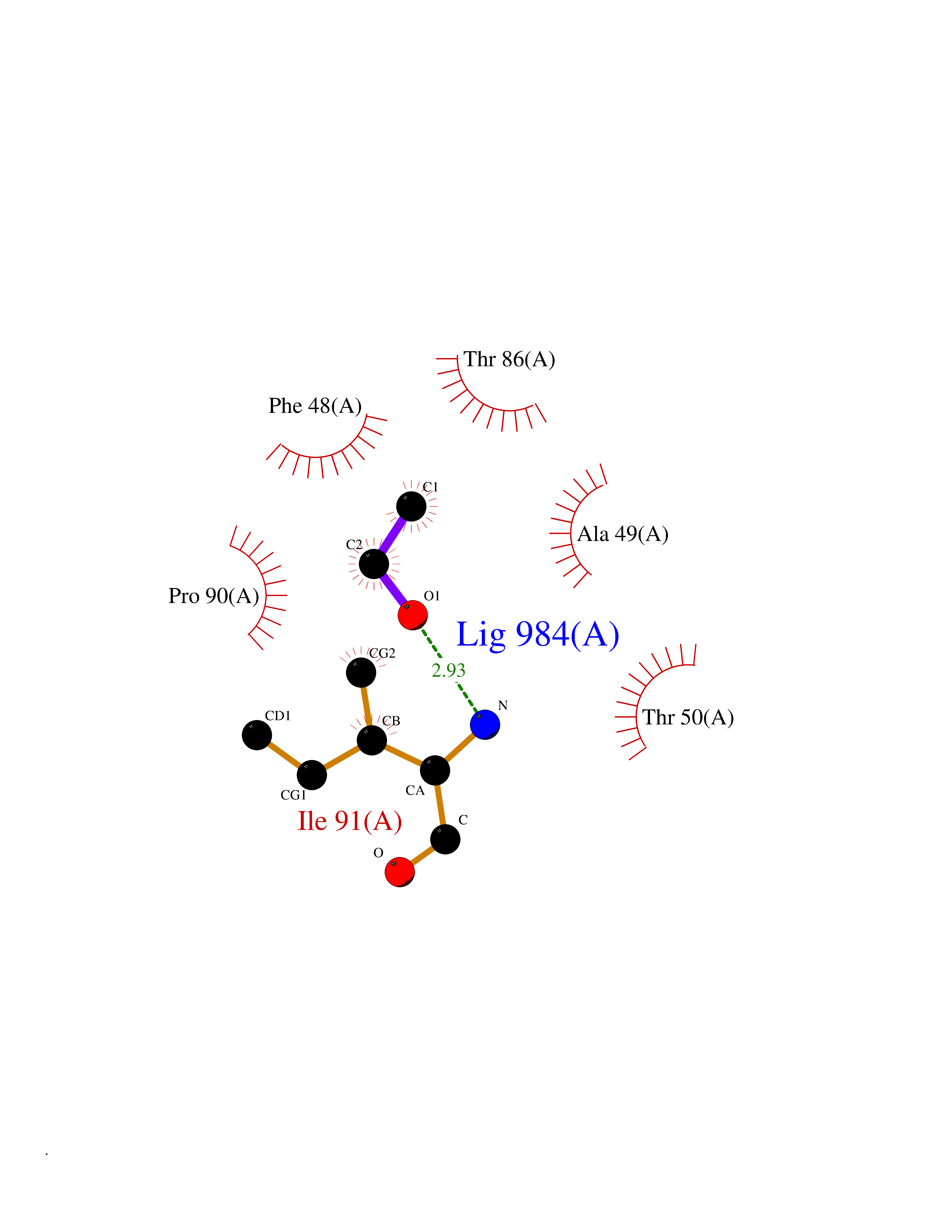 Ligplot Map 3D Binding mode Sequence FSFPKEEEKVLSLWDEIDAFHTSLELTKDKPEFSFFDGPPFATGTPHYGHILASTIKDIVPRYATMTGHHVERRFGWDTHGVPIEHIIDKKLGITGKDDVFKYGLENYNNECRSIVMTYASDWRKTIGRLGRWIDFDNDYKTMYPSFMESTWWAFKQLHEKGQVYRGFKVMPYSTGLTTPLSNFEAQQNYKDVNDPAVTIGFNVIGQEKTQLVAWTTTPWTLPSNLSLCVNADFEYVKIYDETRDRYFILLESLIKTLYKKPKNEKYKIVEKIKGSDLVGLKYEPLFPYFAEQFHETAFRVISDDYVTSDSGTGIVHNAPAFGEEDNAACLKNGVISEDSVLPNAIDDLGRFTKDVPDFEGVYVKDADKLIIKYLTNTGNLLLASQIRHSYPFCWRSDTPLLYRSVPAWFVRVKNIVPQMLDSVMKSHWVPNTIKEKRFANWIANARDWNVSRNRYWGTPIPLWVSDDFEEVVCVGSIKELEELTGVRNITDLHRDVIDKLTIPSKQGKGDLKRIEEVFDCWFESGSMPYASQHYPFENTEKFDERVPANFISEGLDQTRGWFYTLAVLGTHLFGSVPYKNVIVSGIVLAADGRKMSKSLKNYPDPSIVLNKYGADALRLYLINSPVLKAESLKFKEEGVKEVVSKVLLPWWNSFKFLDGQIALLKKMSNIDFQYDDSVKSDNVMDRWILASMQSLVQFIHEEMGQYKLYTVVPKLLNFIDELTNWYIRFNRRRLKGENGVEDCLKALNSLFDALFTFVRAMAPFTPFLSESIYLRLKEYIPEAVLAKYGKDGRSVHFLSYPVVKKEYFDEAIETAVSRMQSVIDLGRNIREKKTISLKTPLKTLVILHSDESYLKDVEALKNYIIEELNVRDVVITSDEAKYGVEYRGLPESAVQAGQETRTDQDVLIIMDTNIYSEL Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Bromodomain-containing protein 9 (BRD9) | 6V0X | 4.26 | |
Target general information Gen name BRD9 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Rhabdomyosarcoma antigen MU-RMS-40.8 Protein family NA Biochemical class Bromodomain Function Plays a role in chromatin remodeling and regulation of transcription. Acts as a chromatin reader that recognizes and binds acylated histones: binds histones that are acetylated and/or butyrylated. Component of SWI/SNF chromatin remodeling subcomplex GBAF that carries out key enzymatic activities, changing chromatin structure by altering DNA-histone contacts within a nucleosome in an ATP-dependent manner. Related diseases Major depressive disorder (MDD) [MIM:608516]: A common psychiatric disorder. It is a complex trait characterized by one or more major depressive episodes without a history of manic, mixed, or hypomanic episodes. A major depressive episode is characterized by at least 2 weeks during which there is a new onset or clear worsening of either depressed mood or loss of interest or pleasure in nearly all activities. Four additional symptoms must also be present including changes in appetite, weight, sleep, and psychomotor activity; decreased energy; feelings of worthlessness or guilt; difficulty thinking, concentrating, or making decisions; or recurrent thoughts of death or suicidal ideation, plans, or attempts. The episode must be accompanied by distress or impairment in social, occupational, or other important areas of functioning. {ECO:0000269|PubMed:15229186}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q7Z7H3; Q7Z7H3 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Bromodomain; Chromatin regulator; Isopeptide bond; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 11491.3 Length 99 Aromaticity 0.13 Instability index 13.58 Isoelectric point 9.24 Charge (pH=7) 3.8 2D Binding mode Binding energy (Kcal/mol) -5.81  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence STPIQQLLEHFLRQLQRKDPHGFFAFPVTDAIAPGYSMIIKHPMDFGTMKDKIVANEYKSVTEFKADFKLMCDNAMTYNRPDTVYYKLAKKILHAGFKM Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Ubiquitin carboxyl-terminal hydrolase 30 (USP30) | 5OHK | 4.26 | |
Target general information Gen name USP30 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Deubiquitinating enzyme 30; Ubiquitin thioesterase 30; Ubiquitin-specific-processing protease 30; Ub-specific protease 30 Protein family Peptidase C19 family Biochemical class Peptidase Function Deubiquitinating enzyme tethered to the mitochondrial outer membrane that acts as a key inhibitor of mitophagy by counteracting the action of parkin (PRKN): hydrolyzes ubiquitin attached by parkin on target proteins, such as RHOT1/MIRO1 and TOMM20, thereby blocking parkin's ability to drive mitophagy. Preferentially cleaves 'Lys-6'- and 'Lys-11'-linked polyubiquitin chains, 2 types of linkage that participate in mitophagic signaling. Does not cleave efficiently polyubiquitin phosphorylated at 'Ser-65'. Acts as negative regulator of mitochondrial fusion by mediating deubiquitination of MFN1 and MFN2 (By similarity). Related diseases Pseudohypoaldosteronism 2C (PHA2C) [MIM:614492]: An autosomal dominant disorder characterized by severe hypertension, hyperkalemia, hyperchloremia, mild hyperchloremic metabolic acidosis in some cases, and correction of physiologic abnormalities by thiazide diuretics. {ECO:0000269|PubMed:11498583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuropathy, hereditary sensory and autonomic, 2A (HSAN2A) [MIM:201300]: A form of hereditary sensory and autonomic neuropathy, a genetically and clinically heterogeneous group of disorders characterized by degeneration of dorsal root and autonomic ganglion cells, and by sensory and/or autonomic abnormalities. HSAN2A is an autosomal recessive disorder characterized by impairment of pain, temperature and touch sensation, onset of symptoms in infancy or early childhood, occurrence of distal extremity pathologies (paronychia, whitlows, ulcers, and Charcot joints), frequent amputations, sensory loss that affects all modalities of sensation (lower and upper limbs and perhaps the trunk as well), absence or diminution of tendon reflexes (usually in all limbs), minimal autonomic dysfunction, absence of sensory nerve action potentials, and virtual absence of myelinated fibers with decreased numbers of unmyelinated fibers in sural nerves. {ECO:0000269|PubMed:15060842, ECO:0000269|PubMed:15911806, ECO:0000269|PubMed:18521183}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P54253; Q6NTF9-3 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Hydrolase; Isopeptide bond; Membrane; Mitochondrion; Mitochondrion outer membrane; Protease; Proteomics identification; Reference proteome; Thiol protease; Transmembrane; Transmembrane helix; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 43456.9 Length 379 Aromaticity 0.09 Instability index 50.2 Isoelectric point 6.43 Charge (pH=7) -4.37 2D Binding mode Binding energy (Kcal/mol) -5.81  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence KGLVPGLVNLGNTCFMNSLLQGLSACPAFIRWLEEFTSQYSRQYLSLTLLHLLKALSCQEVTDDEVLDASCLLDVLRMYRWQISSFEEQDAHELFHVITSSLEDERDGSGSHWKSQHPFHGRLTSNMVCKHCEHQSPVRFDTFDSLSLSIPAATWGHPLTLDHCLHHFISSESVRDVVCDNCTKRTTFVKQLKLGKLPQCLCIHLQRLSWSSHGTPLKRHEHVQFNEDLSMDEYKYHSNASTYLFRLMAVVVHHGDMHSGHFVTYRRSPPSSNQWLWVSDDTVRKASLQEVLSSSAYLLFYERVMQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRG Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Folate receptor beta (FOLR2) | 4KN0 | 4.26 | |
Target general information Gen name FOLR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Placental folate-binding protein; Folate receptor, fetal/placental; Folate receptor type-beta; Folate receptor 2; FR-beta; FOLR2 Protein family Folate receptor family Biochemical class Folate receptor Function Binds to folate and reduced folic acid derivatives and mediates delivery of 5-methyltetrahydrofolate and folate analogs into the interior of cells. Has high affinity for folate and folic acid analogs at neutral pH. Exposure to slightly acidic pH after receptor endocytosis triggers a conformation change that strongly reduces its affinity for folates and mediates their release. Related diseases Acute hepatic porphyria (AHEPP) [MIM:612740]: A form of porphyria. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. AHP is characterized by attacks of gastrointestinal disturbances, abdominal colic, paralyses and peripheral neuropathy. Most attacks are precipitated by drugs, alcohol, caloric deprivation, infections, or endocrine factors. {ECO:0000269|PubMed:10706561, ECO:0000269|PubMed:1309003, ECO:0000269|PubMed:1569184, ECO:0000269|PubMed:17236137, ECO:0000269|PubMed:2063868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00158; DB00563; DB05168 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Direct protein sequencing; Disulfide bond; Folate-binding; Glycoprotein; GPI-anchor; Lipoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23841.6 Length 205 Aromaticity 0.12 Instability index 56.78 Isoelectric point 7.92 Charge (pH=7) 2.58 2D Binding mode Binding energy (Kcal/mol) -5.81  Molscript Map  Pymol Map 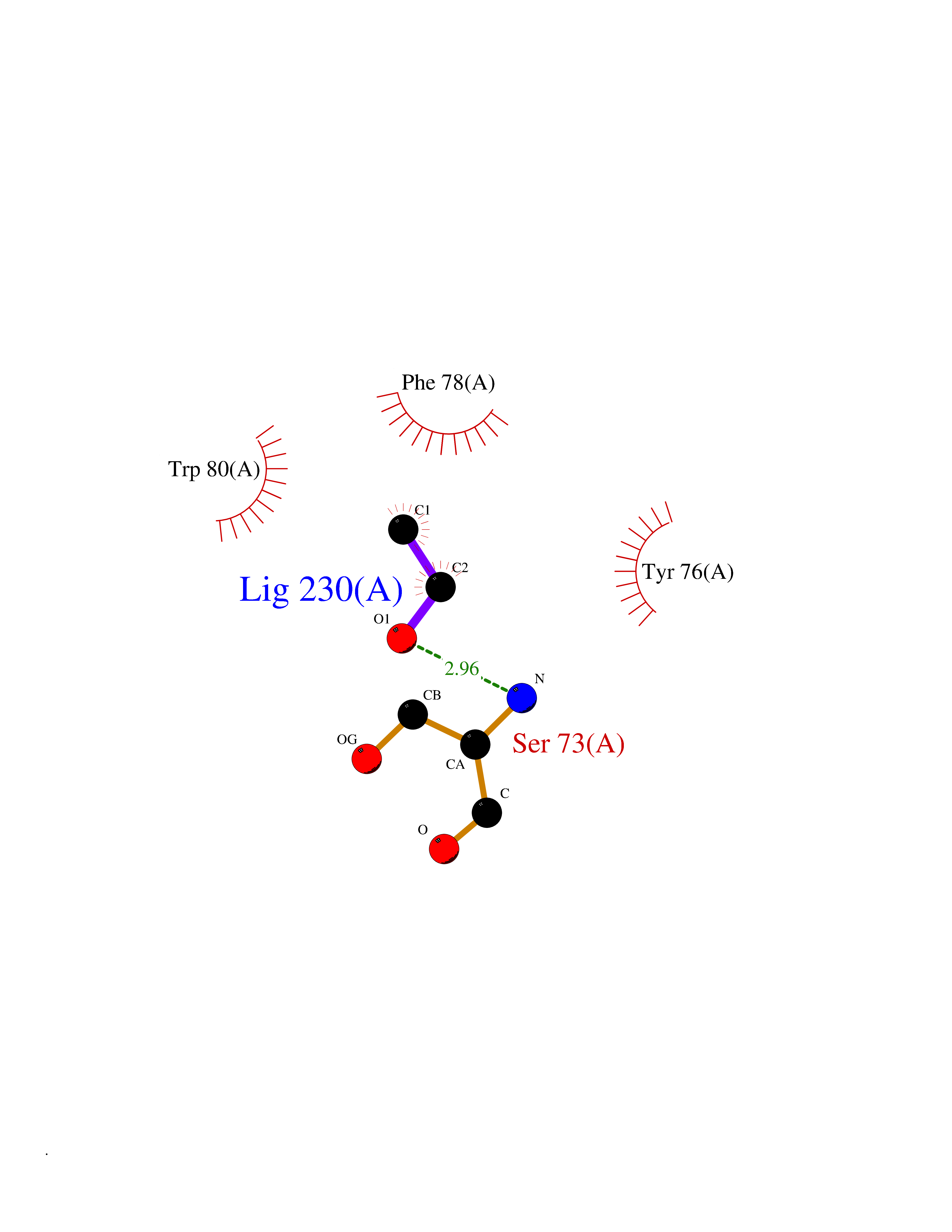 Ligplot Map 3D Binding mode Sequence RTDLLNVCMDAKHHKTKPGPEDKLHDQCSPWKKNACCTASTSQELHKDTSRLYNFNWDHCGKMEPACKRHFIQDTCLYECSPNLGPWIQQVNQSWRKERFLDVPLCKEDCQRWWEDCHTSHTCKSNWHRGWDWTSGVNKCPAGALCRTFESYFPTPAALCEGLWSHSYKVSNYSRGSGRCIQMWFDSAQGNPNEEVARFYAAAMH Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Hepatocyte growth factor receptor | 4R1V | 4.26 | |
Target general information Gen name MET Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Protein kinase superfamily, Tyr protein kinase family Biochemical class Transferase / transferase inhibitor Function ATP binding.Hepatocyte growth factor-activated receptor activity.Identical protein binding.Phosphatidylinositol-4,5-bisphosphate 3-kinase activity.Protein phosphatase binding.Protein tyrosine kinase activity.Ras guanyl-nucleotide exchange factor activity. Related diseases Activation of MET after rearrangement with the TPR gene produces an oncogenic protein.; DISEASE: Defects in MET may be associated with gastric cancer.; DISEASE: Hepatocellular carcinoma (HCC) [MIM:114550]: A primary malignant neoplasm of epithelial liver cells. The major risk factors for HCC are chronic hepatitis B virus (HBV) infection, chronic hepatitis C virus (HCV) infection, prolonged dietary aflatoxin exposure, alcoholic cirrhosis, and cirrhosis due to other causes. {ECO:0000269|PubMed:9927037}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Renal cell carcinoma papillary (RCCP) [MIM:605074]: A subtype of renal cell carcinoma tending to show a tubulo-papillary architecture formed by numerous, irregular, finger-like projections of connective tissue. Renal cell carcinoma is a heterogeneous group of sporadic or hereditary carcinoma derived from cells of the proximal renal tubular epithelium. {ECO:0000269|PubMed:10327054, ECO:0000269|PubMed:10417759, ECO:0000269|PubMed:10433944, ECO:0000269|PubMed:9140397, ECO:0000269|PubMed:9563489}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: A common allele in the promoter region of the MET shows genetic association with susceptibility to autism in some families. Functional assays indicate a decrease in MET promoter activity and altered binding of specific transcription factor complexes.; DISEASE: MET activating mutations may be involved in the development of a highly malignant, metastatic syndrome known as cancer of unknown primary origin (CUP) or primary occult malignancy. Systemic neoplastic spread is generally a late event in cancer progression. However, in some instances, distant dissemination arises at a very early stage, so that metastases reach clinical relevance before primary lesions. Sometimes, the primary lesions cannot be identified in spite of the progresses in the diagnosis of malignancies.; DISEASE: Deafness, autosomal recessive, 97 (DFNB97) [MIM:616705]: A form of non-syndromic sensorineural hearing loss with prelingual onset. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. {ECO:0000269|PubMed:25941349}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Osteofibrous dysplasia (OSFD) [MIM:607278]: A congenital disorder of osteogenesis characterized by non-neoplastic, radiolucent lesions that affect the cortical bone immediately under the periosteum. It usually manifests as a painless swelling or anterior bowing of the long bones, most commonly the tibia and fibula. {ECO:0000269|PubMed:26637977}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Disease-associated variants identified in 4 families cause the deletion of exon 14. This results in the exclusion of an ubiquitination target site within the cytoplasmic domain, hence in protein stabilization. The persistent presence of MET at the cell surface in conditions of ligand-dependent activation retards osteoblastic differentiation. {ECO:0000269|PubMed:26637977}.; DISEASE: Arthrogryposis, distal, 11 (DA11) [MIM:620019]: A form of distal arthrogryposis, a disease characterized by congenital joint contractures that mainly involve two or more distal parts of the limbs, in the absence of a primary neurological or muscle disease. DA11 is an autosomal dominant form characterized mainly by camptodactyly. Other features include absent flexion creases and limited forearm supination. {ECO:0000269|PubMed:30777867}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06896; DB08791; DB06997; DB07969; DB08079; DB16695; DB12742; DB12267; DB08875; DB11791; DB08865; DB12010; DB02152; DB07369; DB06995; DB06314; DB01268; DB15133; DB12200; DB11800 Interacts with P22681; Q96EY1; Q96EY1-2; P00533; P09769; P14210; P14210-6; O15357; P35968; P06239; P07948; P08581; P41218; P15941; P16333; O43639; Q16288; P27986; O00459; Q92569; P19174; O43157; O15031; Q9ULL4; Q8TCU6; P18031; Q06124; P23467; Q12913; Q16827; P20936; Q9UQQ2; O60880; O14796; Q9NP31; Q8N5H7; Q15464; P29353; P98077; Q6S5L8; Q96IW2; Q9H6Q3; O75159; O14544; P12931; Q9ULZ2; P43405; P42680; Q9HBL0; Q63HR2; Q68CZ2; Q9UKW4; P07947; P43403; Q08048; P0DQD2; P35918; Q00944 EC number 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Chromosomal rearrangement; Deafness; Disease variant; Disulfide bond; Glycoprotein; Kinase; Membrane; Non-syndromic deafness; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Secreted; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32760.8 Length 289 Aromaticity 0.1 Instability index 37.12 Isoelectric point 8.79 Charge (pH=7) 5.68 2D Binding mode Binding energy (Kcal/mol) -5.8  Molscript Map 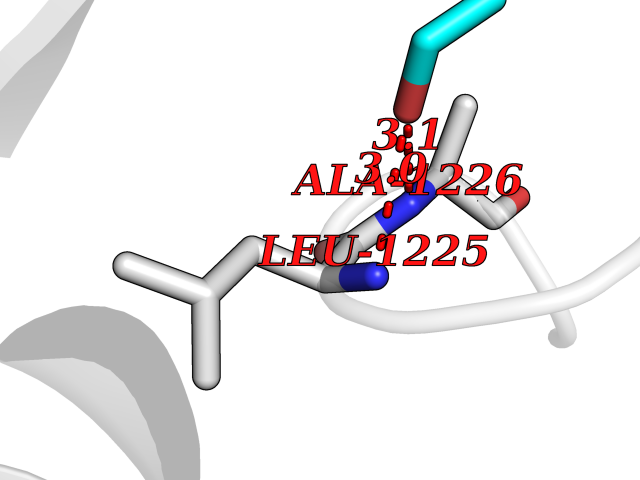 Pymol Map 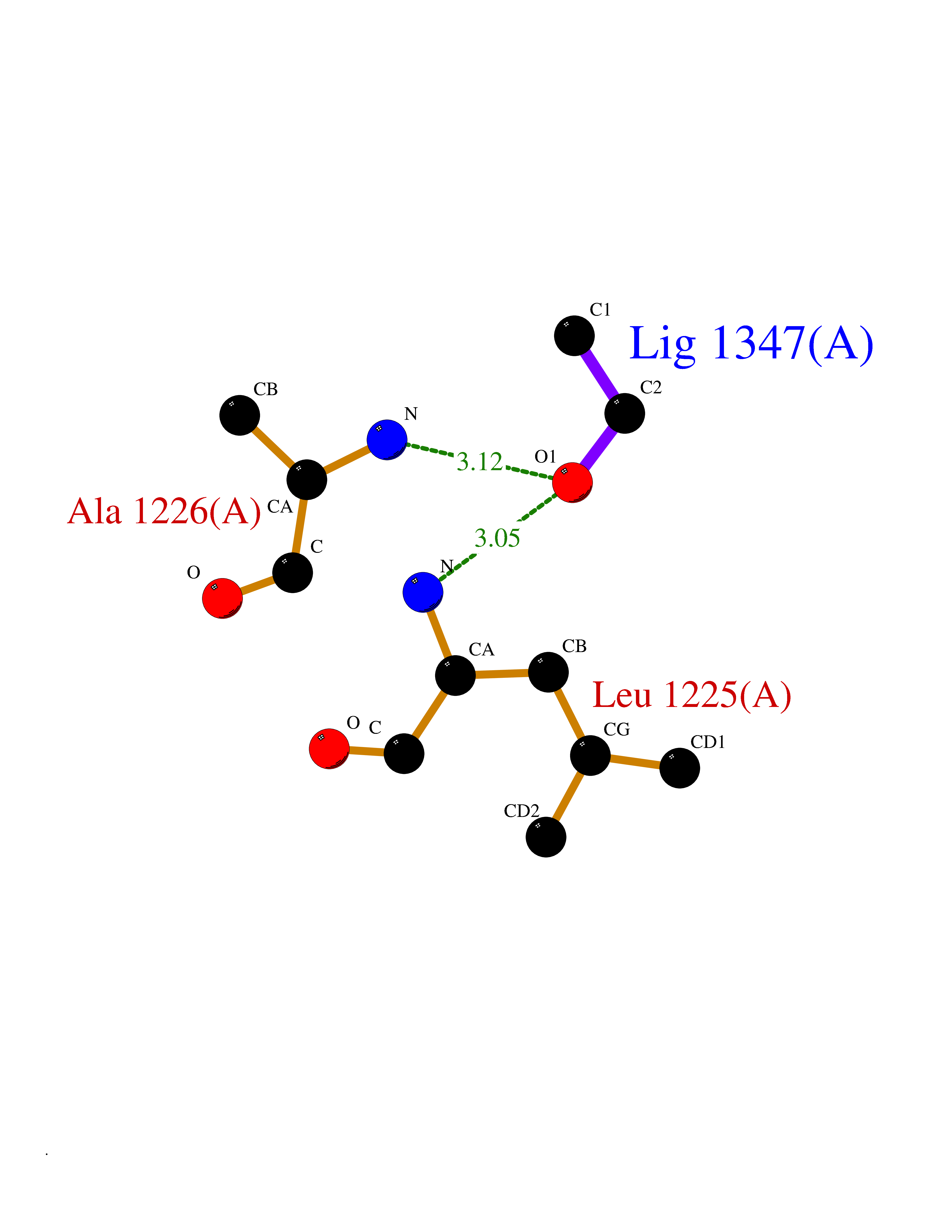 Ligplot Map 3D Binding mode Sequence LSALNPELVQAVQHVVIGPSSLIVHFNEVIGRGHFGCVYHGTLLDNDGKKIHCAVKSLNRITDIGEVSQFLTEGIIMKDFSHPNVLSLLGICLRSSPLVVLPYMKHGDLRNFIRNETHNPTVKDLIGFGLQVAKGMKYLASKKFVHRDLAARNCMLDEKFTVKVADFGLARDMYDKEYYSVHNKTGAKLPVKWMALESLQTQKFTTKSDVWSFGVLLWELMTRGAPPYPDVNTFDITVYLLQGRRLLQPEYCPDPLYEVMLKCWHPKAEMRPSFSELVSRISAIFSTFI Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Pyridoxal kinase (PDXK) | 3KEU | 4.26 | |
Target general information Gen name PDXK Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Pyridoxine kinase; PDXK Protein family Pyridoxine kinase family Biochemical class Kinase Function Required for synthesisof pyridoxal-5-phosphate from vitamin B6. Related diseases Neuropathy, hereditary motor and sensory, 6C, with optic atrophy (HMSN6C) [MIM:618511]: An autosomal recessive neurologic disorder characterized by childhood onset of axonal, sensorimotor polyneuropathy affecting mainly the lower limbs, and adult-onset optic atrophy. Clinical features include progressive distal muscle weakness and atrophy, significant standing and walking difficulties, areflexia, neurogenic pain and progressive visual impairment. {ECO:0000269|PubMed:31187503}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04776; DB03909; DB04770; DB00147; DB00165 Interacts with NA EC number EC 2.7.1.35 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Charcot-Marie-Tooth disease; Cobalt; Cytoplasm; Disease variant; Kinase; Magnesium; Manganese; Metal-binding; Neurodegeneration; Neuropathy; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Sodium; Transferase; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34370.1 Length 305 Aromaticity 0.07 Instability index 39.92 Isoelectric point 6.16 Charge (pH=7) -3.37 2D Binding mode Binding energy (Kcal/mol) -5.81 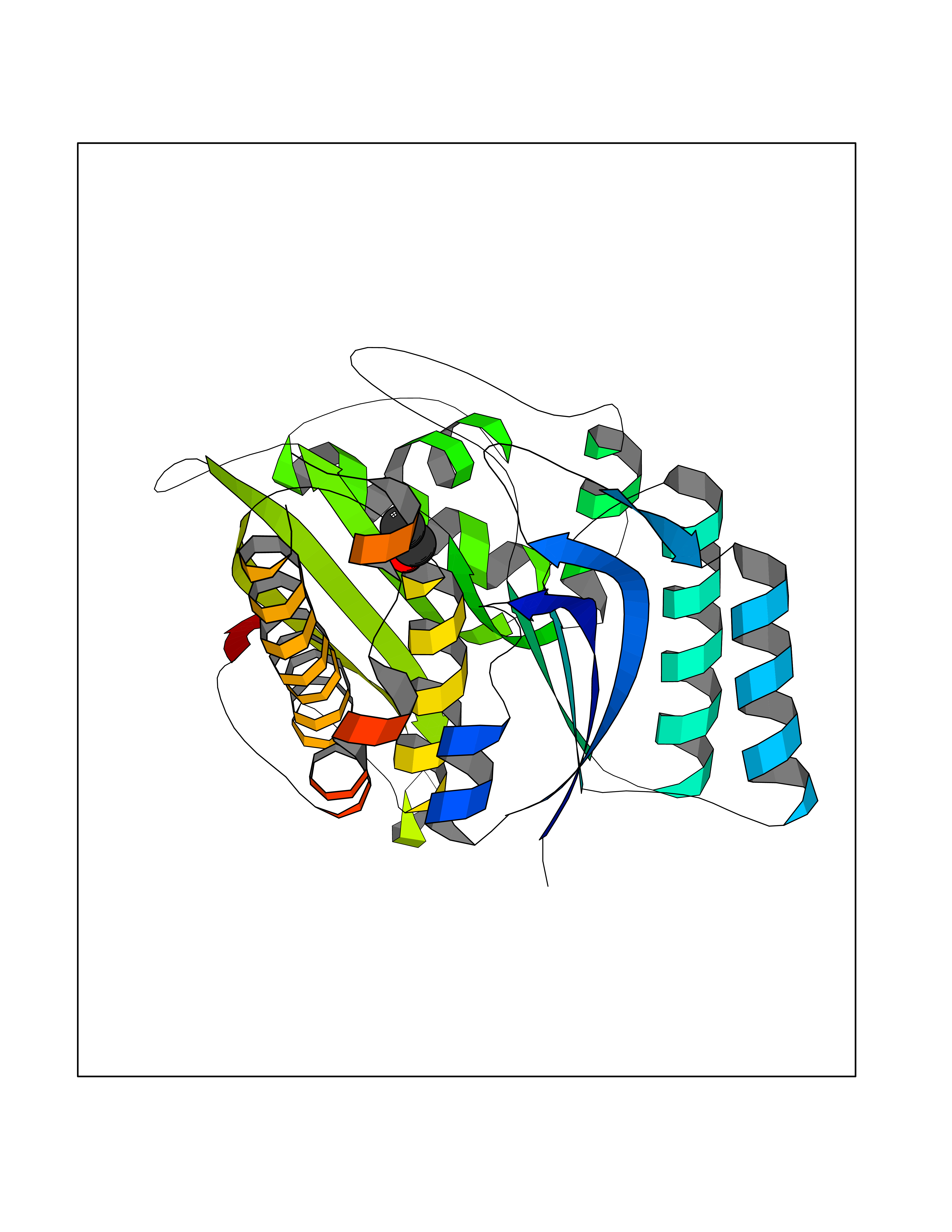 Molscript Map 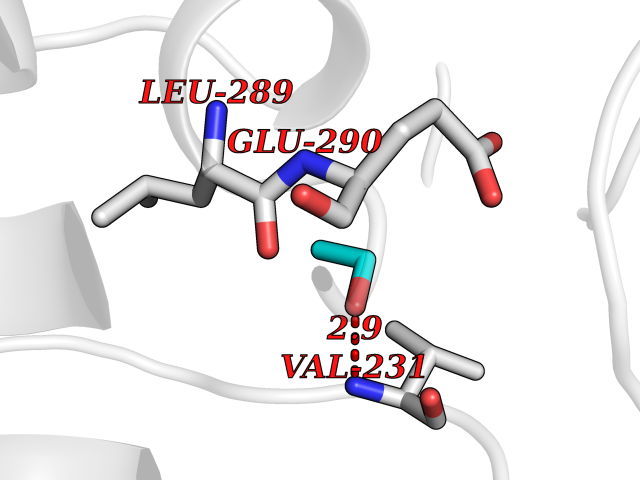 Pymol Map 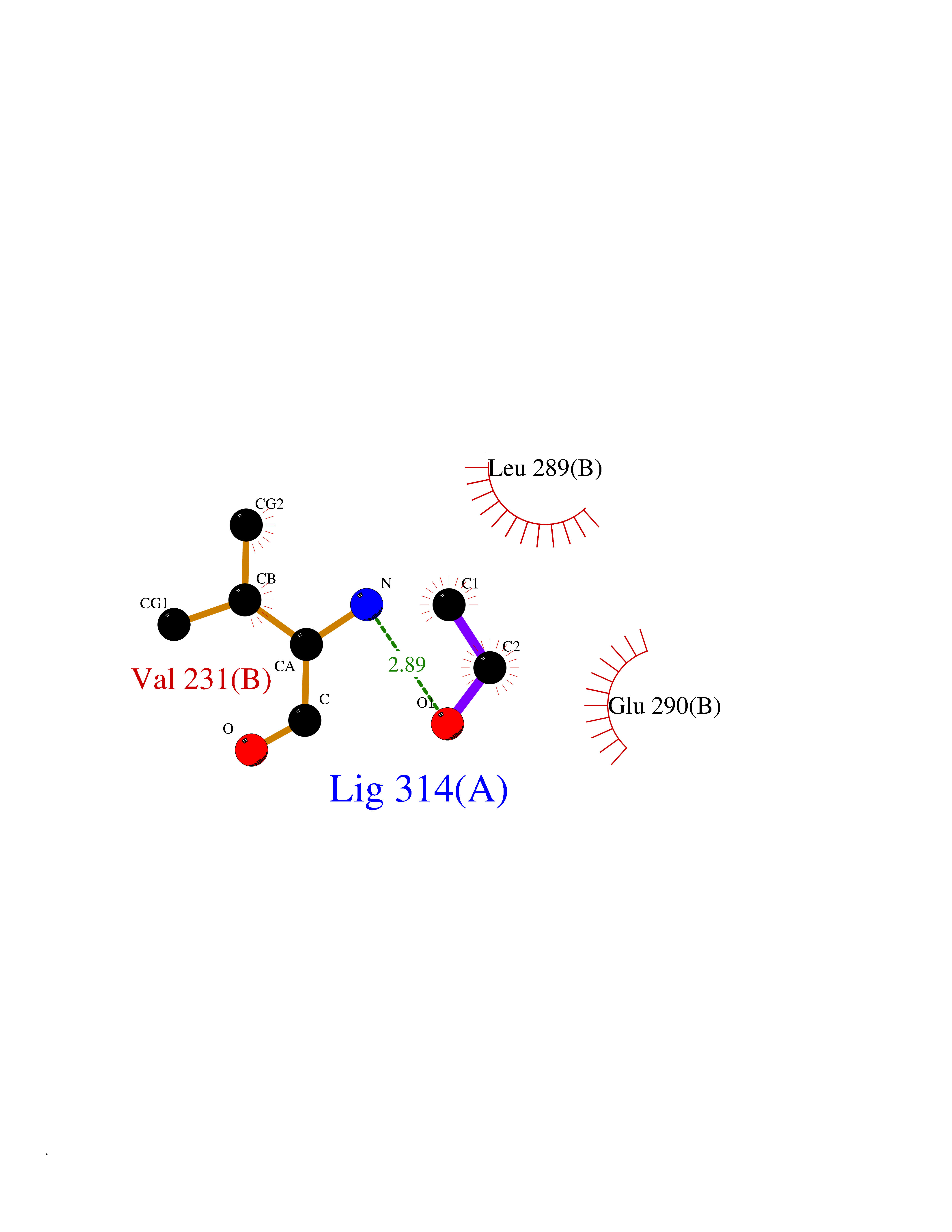 Ligplot Map 3D Binding mode Sequence ECRVLSIQSHVIRGYVGNRAATFPLQVLGFEIDAVNSVQFSNHTGYAHWKGQVLNSDELQELYEGLRLNNMNKYDYVLTGYTRDKSFLAMVVDIVQELKQQNPRLVYVCDPVLGDKWDGEGSMYVPEDLLPVYKEKVVPLADIITPNQFEAELLSGRKIHSQEEALRVMDMLHSMGPDTVVITSSDLPSPQGSNYLIVLGSQRRRNPAGSVVMERIRMDIRKVDAVFVGTGDLFAAMLLAWTHKHPNNLKVACEKTVSTLHHVLQRTIQCAKAQARPSPMQLELRMVQSKRDIEDPEIVVQATVL Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Glutathione S-transferase P (GSTP1) | 5J41 | 4.26 | |
Target general information Gen name GSTP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GSTP11; GSTP1-1; GST3; GST classpi; GST class-pi; FAEES3 Protein family GST superfamily, Pi family Biochemical class Alkyl aryl transferase Function Regulates negatively CDK5 activity via p25/p35 translocation to prevent neurodegeneration. Conjugation of reduced glutathione to a wide number of exogenous and endogenous hydrophobic electrophiles. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01834; DB03814; DB00316; DB14001; DB00321; DB01008; DB04972; DB00958; DB00291; DB02633; DB00515; DB01242; DB00363; DB11672; DB14635; DB14002; DB03619; DB11831; DB00903; DB00773; DB06246; DB05460; DB00143; DB03310; DB03003; DB13014; DB00526; DB14924; DB08370; DB03686; DB04132; DB01915; DB07849; DB00197; DB00163 Interacts with Q6UY14-3; Q92624; Q9BWT7; A8MQ03; Q9NRD0; Q5TD97; P49639; Q15323; Q14525; O76011; P78385; Q07627; Q8IUG1; P60409; P60411; Q9BYR8; Q9BYR6; Q3LI66; Q9P2M1; Q5JR59-3; Q7Z3S9; P0DPK4; P22735; Q12933; Q8N720 EC number EC 2.5.1.18 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Lipid metabolism; Mitochondrion; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 46333.6 Length 417 Aromaticity 0.1 Instability index 30.67 Isoelectric point 5.44 Charge (pH=7) -5.73 2D Binding mode Binding energy (Kcal/mol) -5.81  Molscript Map 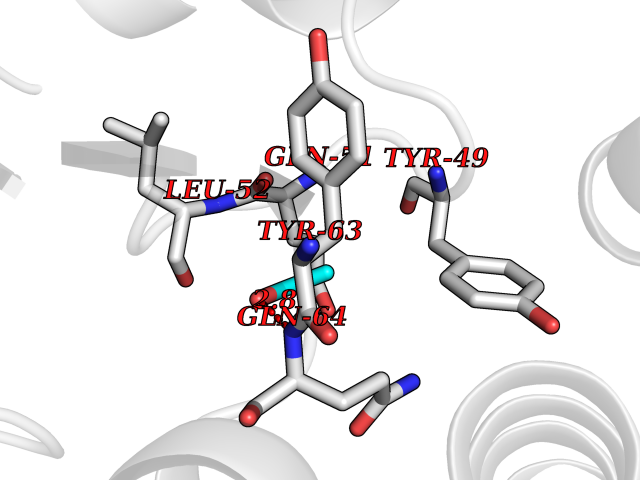 Pymol Map 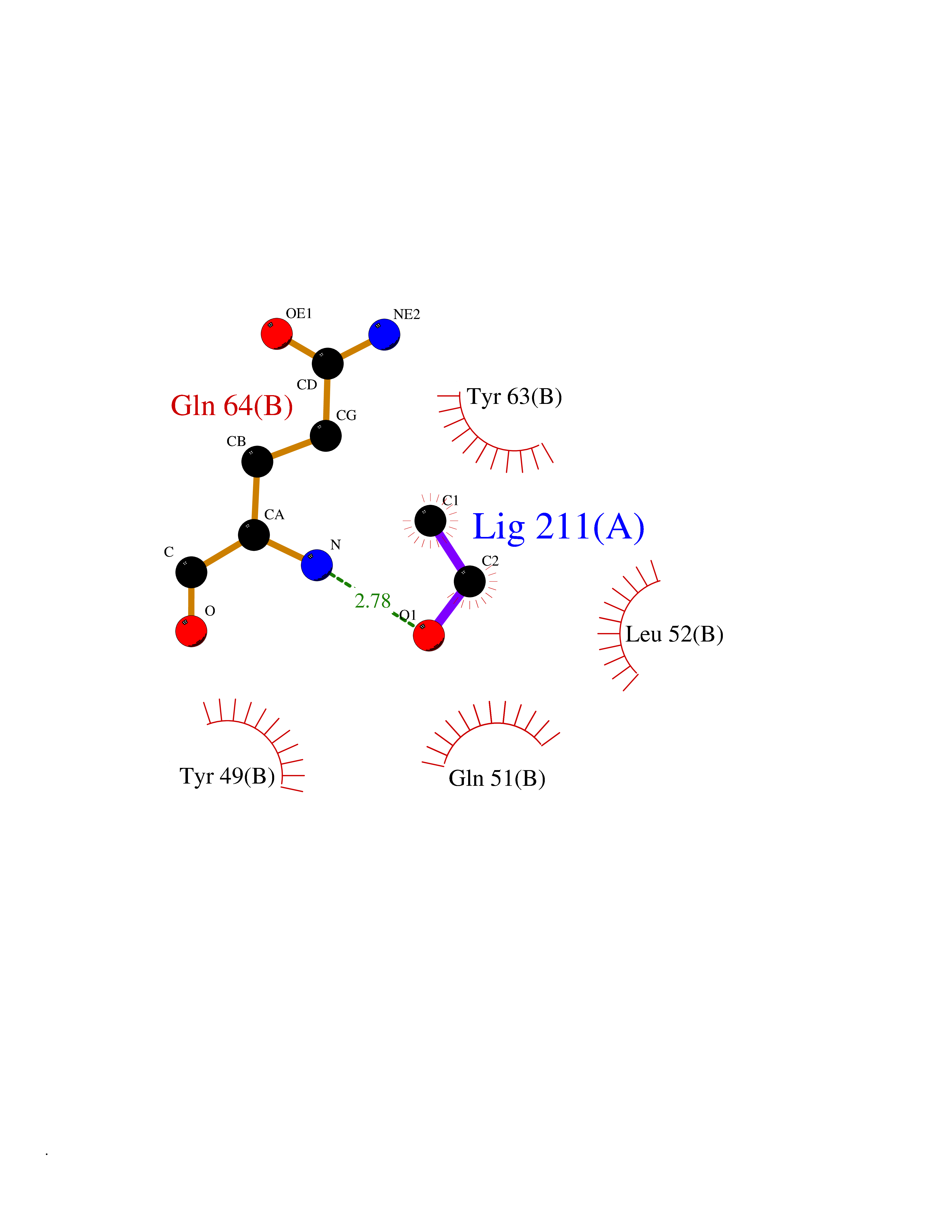 Ligplot Map 3D Binding mode Sequence PPYTVVYFPVRGRCAALRMLLADQGQSWKEEVVTVETWQEGSLKASCLYGQLPKFQDGDLTLYQSNTILRHLGRTLGLYGKDQQEAALVDMVNDGVEDLRCKYISLIYTNYEAGKDDYVKALPGQLKPFETLLSQNQGGKTFIVGDQISFADYNLLDLLLIHEVLAPGCLDAFPLLSAYVGRLSARPKLKAFLASPEYVNLPINGNGKQPYTVVYFPVRGRCAALRMLLADQGQSWKEEVVTVETWQEGSLKASCLYGQLPKFQDGDLTLYQSNTILRHLGRTLGLYGKDQQEAALVDMVNDGVEDLRCKYISLIYTNYEAGKDDYVKALPGQLKPFETLLSQNQGGKTFIVGDQISFADYNLLDLLLIHEVLAPGCLDAFPLLSAYVGRLSARPKLKAFLASPEYVNLPINGNGKQ Hydrogen bonds contact Hydrophobic contact | ||||