Job Results:
Ligand
Structure
Job ID
9af75475a67248e060ab578f15db3d0b
Job name
NA
Time
2025-04-03 17:54:57
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Xanthine dehydrogenase/oxidase (XDH) | 2E1Q | 5.95 | |
Target general information Gen name XDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Xanthine oxidase; Xanthine dehydrogenase; XDHA Protein family Xanthine dehydrogenase family Biochemical class CH/CH(2) oxidoreductase Function Catalyzes the oxidation of hypoxanthine to xanthine. Catalyzes the oxidation of xanthine to uric acid. Contributes to the generation of reactive oxygen species. Has also low oxidase activity towards aldehydes (in vitro). Key enzyme in purine degradation. Related diseases Xanthinuria 1 (XAN1) [MIM:278300]: A disorder characterized by excretion of very large amounts of xanthine in the urine and a tendency to form xanthine stones. Uric acid is strikingly diminished in serum and urine. XAN1 is due to isolated xanthine dehydrogenase deficiency. Patients can metabolize allopurinol. {ECO:0000269|PubMed:10844591, ECO:0000269|PubMed:11379872, ECO:0000269|PubMed:14551354, ECO:0000269|PubMed:9153281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00640; DB00041; DB00437; DB00993; DB00958; DB01136; DB00856; DB00515; DB00746; DB03328; DB00997; DB03516; DB12466; DB04854; DB03147; DB04335; DB01020; DB00583; DB00170; DB01033; DB00157; DB03841; DB00336; DB01250; DB05262; DB06478; DB01168; DB00339; DB00127; DB01685; DB00831 Interacts with Q9Y3R0-3 EC number NA Uniprot keywords 2Fe-2S; 3D-structure; Cytoplasm; Disease variant; Disulfide bond; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase; Peroxisome; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 143697 Length 1307 Aromaticity 0.08 Instability index 37.9 Isoelectric point 8.01 Charge (pH=7) 7.07 2D Binding mode Binding energy (Kcal/mol) -8.12 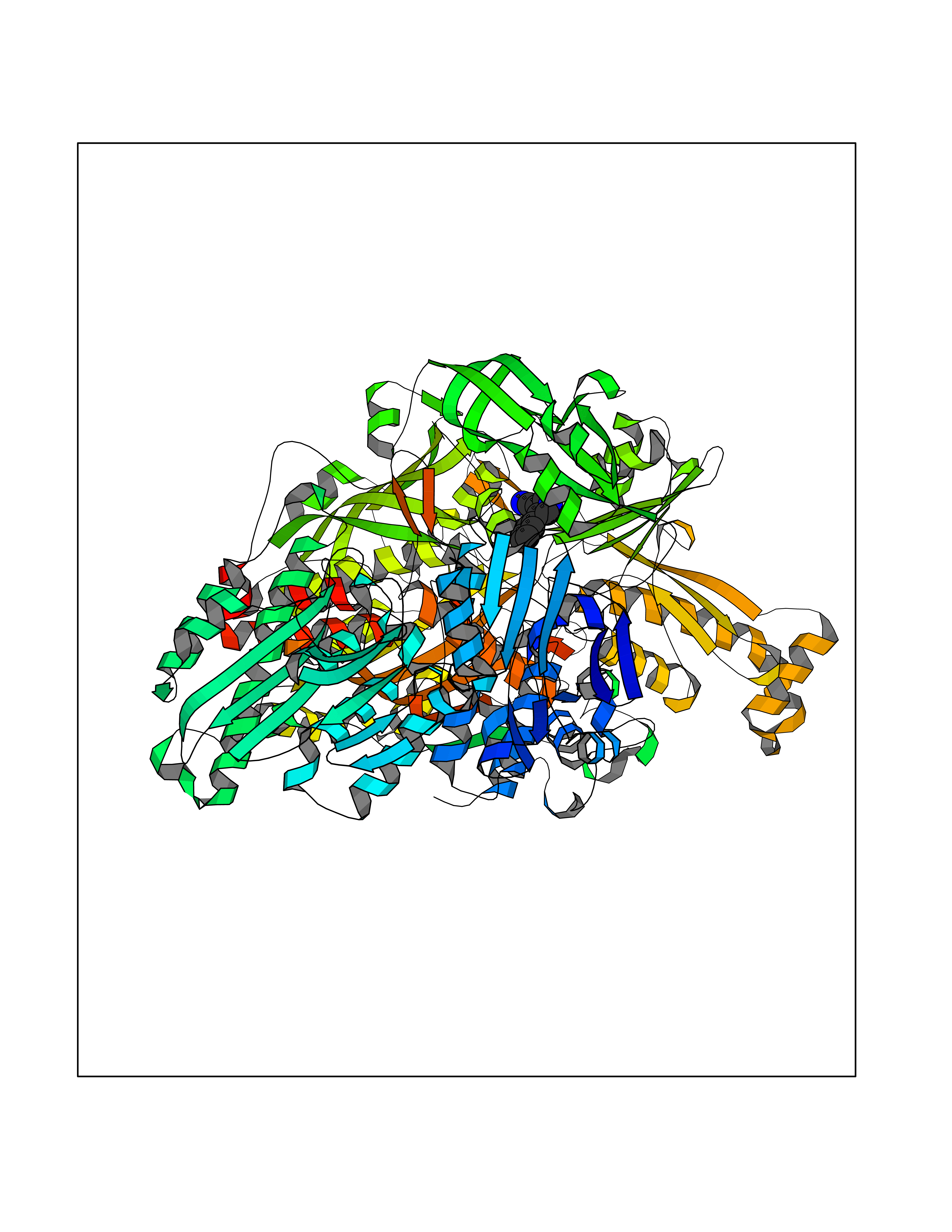 Molscript Map 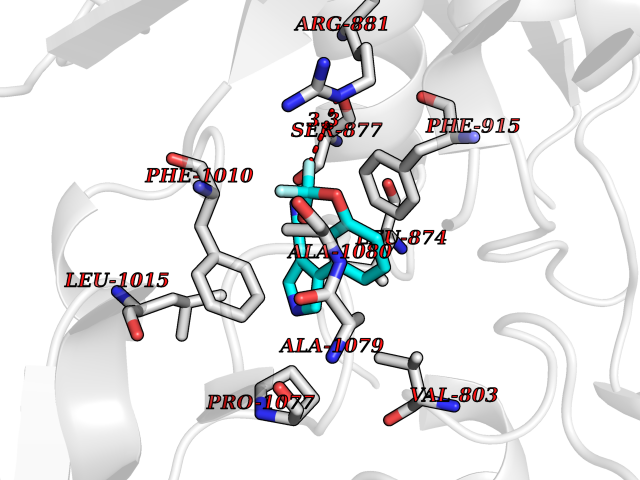 Pymol Map 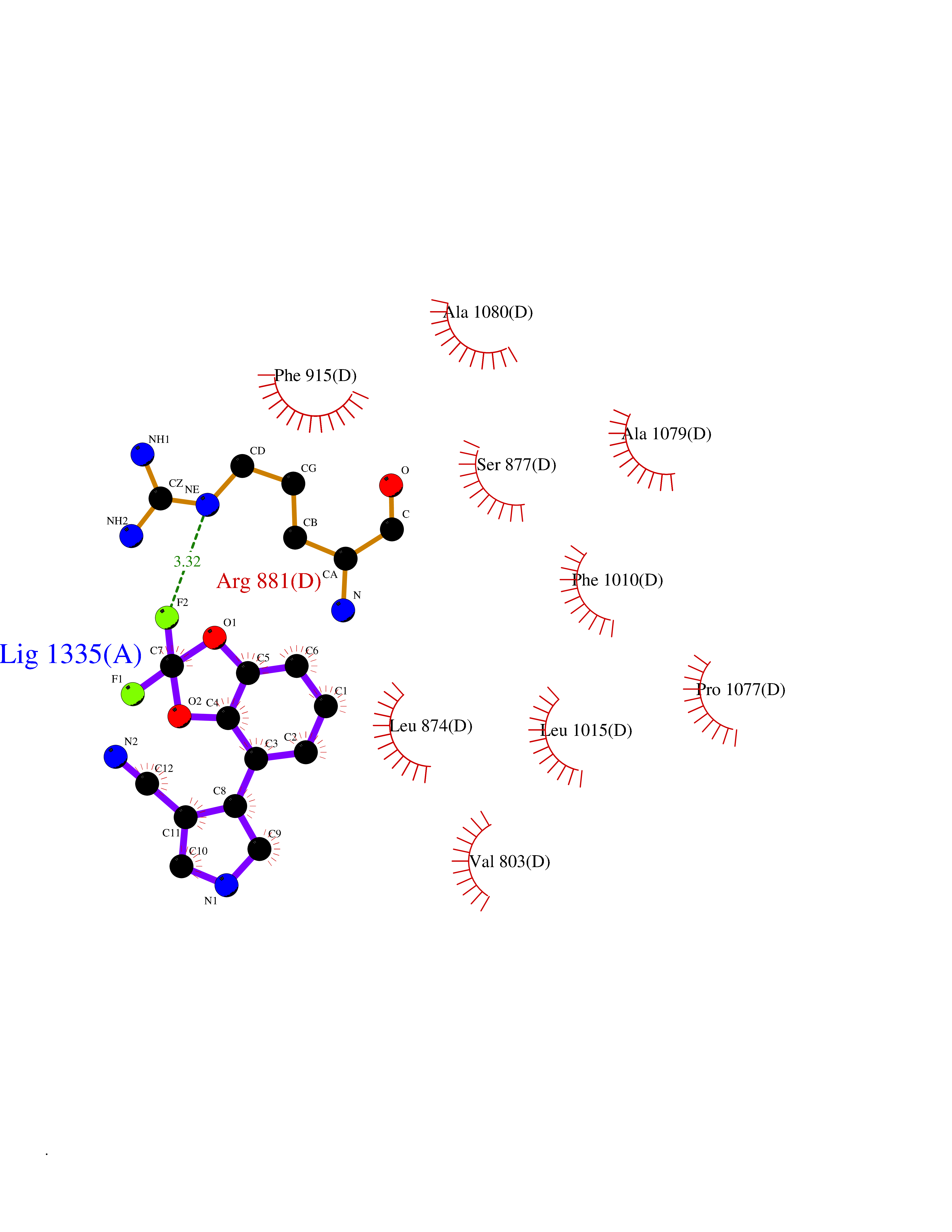 Ligplot Map 3D Binding mode Sequence ADKLVFFVNGRKVVEKNADPETTLLAYLRRKLGLSGTKLGCGEGGCGACTVMLSKYDRLQNKIVHFSANACLAPICSLHHVAVTTVEGIGSTKTRLHPVQERIAKSHGSQCGFCTPGIVMSMYTLLRNQPEPTMEEIENAFQGNLCRCTGYRPILQGFRTFARDGSPSLFKPEEFTPLDPTQEPIFPPELLRLKDTPRKQLRFEGERVTWIQASTLKELLDLKAQHPDAKLVVGNTEIGIEMKFKNMLFPMIVCPAWIPELNSVEHGPDGISFGAACPLSIVEKTLVDAVAKLPAQKTEVFRGVLEQLRWFAGKQVKSVASVGGNIITASPISDLNPVFMASGAKLTLVSRGTRRTVQMDHTFFPGYRKTLLSPEEILLSIEIPYSREGEYFSAFKQASRREDDIAKVTSGMRVLFKPGTTEVQELALCYGGMANRTISALKTTQRQLSKLWKEELLQDVCAGLAEELHLPPDAPGGMVDFRCTLTLSFFFKFYLTVLQKLGQENLEDKCGKLDPTFASATLLFQKDPPADVQLFQEVPKGQSEEDMVGRPLPHLAADMQASGEAVYCDDIPRYENELSLRLVTSTRAHAKIKSIDTSEAKKVPGFVCFISADDVPGSNITGICNDETVFAKDKVTCVGHIIGAVVADTPEHTQRAAQGVKITYEELPAIITIEDAIKNNSFYGPELKIEKGDLKKGFSEADNVVSGEIYIGGQEHFYLETHCTIAVPKGEAGEMELFVSTQNTMKTQSFVAKMLGVPANRIVVRVKRMGGGFGGKVTRSTVVSTAVALAAYKTGRPVRCMLDRDEDMLITGGRHPFLARYKVGFMKTGTVVALEVDHFSNVGNTQDLSQSIMERALFHMDNCYKIPNIRGTGRLCKTNLPSNTAFRGFGGPQGMLIAECWMSEVAVTCGMPAEEVRRKNLYKEGDLTHFNQKLEGFTLPRCWEECLASSQYHARKSEVDKFNKENCWKKRGLCIIPTKFGISFTVPFLNQAGALLHVYTDGSVLLTHGGTEMGQGLHTKMVQVASRALKIPTSKIYISETSTNTVPNTSPTAASVSADLNGQAVYAACQTILKRLEPYKKKNPSGSWEDWVTAAYMDTVSLSATGFYRTPNLGYSFETNSGNPFHYFSYGVACSEVEIDCLTGDHKNLRTDIVMDVGSSLNPAIDIGQVEGAFVQGLGLFTLEELHYSPEGSLHTRGPSTYKIPAFGSIPIEFRVSLLRDCPNKKAIYASKAVGEPPLFLAASIFFAIKDAIRAARAQHTGNNVKELFRLDSPATPEKIRNACVDKFTTLCVTGVPENCKPWSVRV Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Estrogen-related receptor-gamma (ESRRG) | 2E2R | 5.95 | |
Target general information Gen name ESRRG Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nuclear receptor subfamily 3 group B member 3; NR3B3; KIAA0832; Estrogen-related receptor gamma; Estrogen receptor-related protein 3; ERRG2; ERR3; ERR gamma-2 Protein family Nuclear hormone receptor family, NR3 subfamily Biochemical class Nuclear hormone receptor Function Binds specifically to an estrogen response element and activates reporter genes controlled by estrogen response elements. Induces the expression of PERM1 in the skeletal muscle. Orphan receptor that acts as transcription activator in the absence of bound ligand. Related diseases WHIM syndrome 1 (WHIMS1) [MIM:193670]: An autosomal dominant immunologic disease characterized by neutropenia, hypogammaglobulinemia and extensive human papillomavirus (HPV) infection. Despite the peripheral neutropenia, bone marrow aspirates from affected individuals contain abundant mature myeloid cells, a condition termed myelokathexis. {ECO:0000269|PubMed:12692554, ECO:0000269|PubMed:15536153}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: CXCR4 mutations play a role in the pathogenesis of Waldenstroem macroglobulinemia (WM) and influence disease presentation and outcome, as well as response to therapy. WM is a B-cell lymphoma characterized by accumulation of malignant lymphoplasmacytic cells in the bone marrow, lymph nodes and spleen, and hypersecretion of monoclonal immunoglobulin M (IgM). Excess IgM production results in serum hyperviscosity, tissue infiltration, and autoimmune-related pathology. {ECO:0000269|PubMed:24366360, ECO:0000269|PubMed:24553177}. Drugs (DrugBank ID) DB06884; DB04468; DB06973; DB07485; DB02659; DB00255; DB13952; DB13953; DB13954; DB13955; DB13956; DB06902; DB00675; DB00197 Interacts with Q05D60; Q9BVG8; P50222; P51843; Q12769; Q9UBK2; A0MZ66; G2XKQ0; Q8NFM4; Q13315; Q86WA6-2; Q9BZE7; Q13555-5; Q05D60; Q5JST6; P11474; O95718-2; P62508-3; Q15024; O95990-4; Q8IZU1; Q14296; P23508; Q6IN84; P51843; Q15466; P48552; P26367; Q9NPJ4; P01189; Q9UBK2; P62195; Q8N0T1-2; Q04864-2; Q6NUQ1; A0MZ66-4; Q8TAD8; P19237; P48788; Q96PN7; Q96S82; Q5SQQ9-2; Q7Z4V0 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 25755.7 Length 227 Aromaticity 0.07 Instability index 55.31 Isoelectric point 5.09 Charge (pH=7) -10.6 2D Binding mode Binding energy (Kcal/mol) -8.12 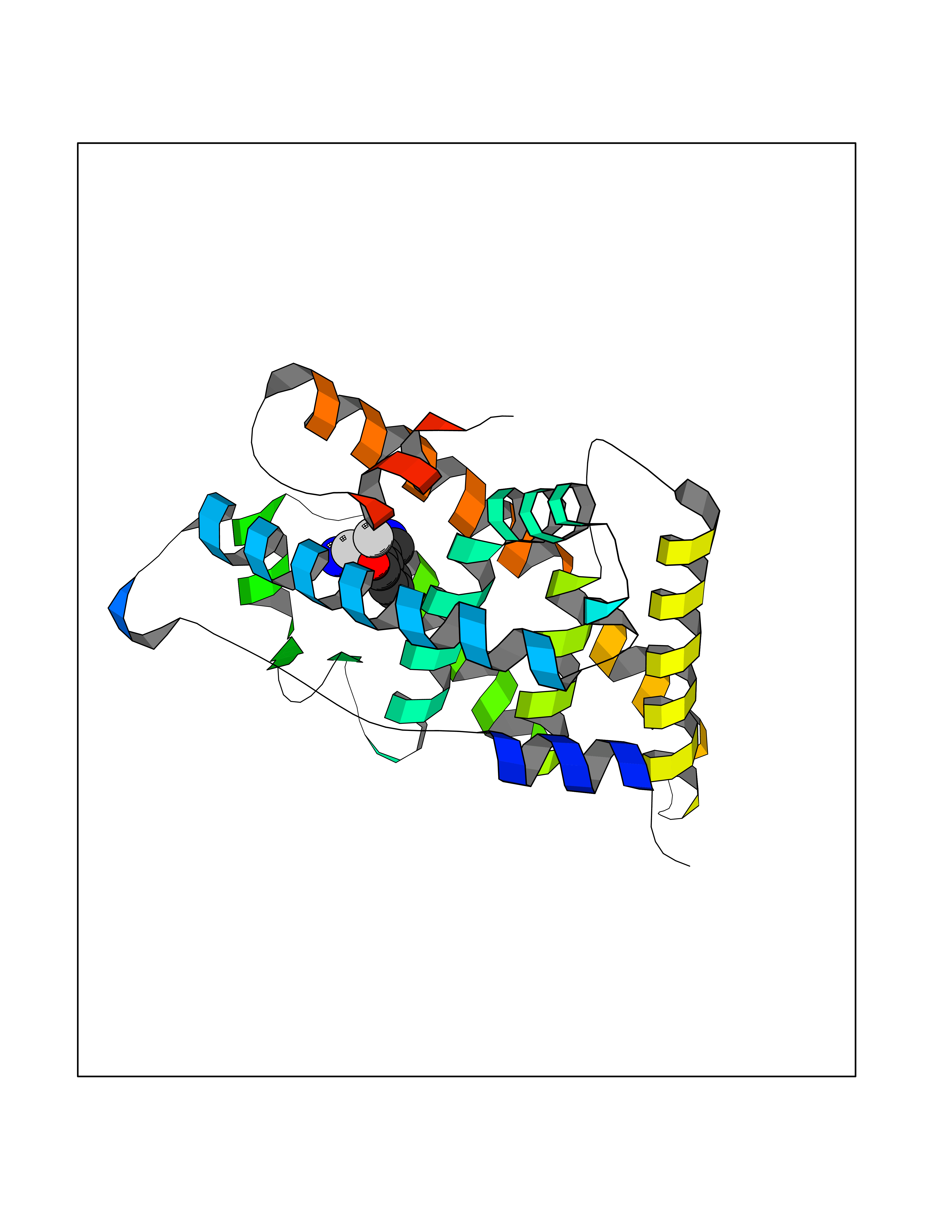 Molscript Map 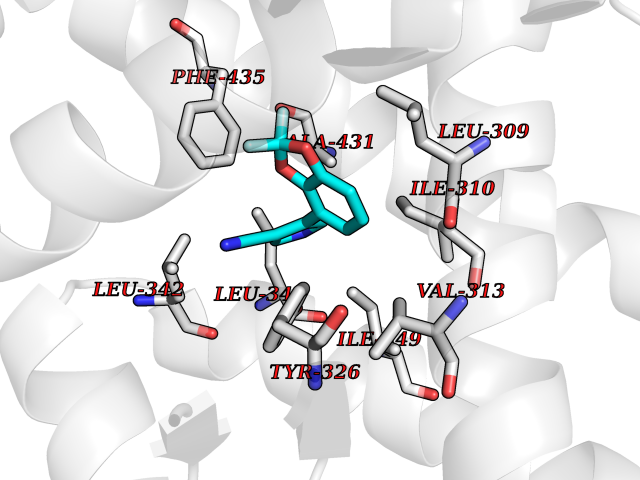 Pymol Map 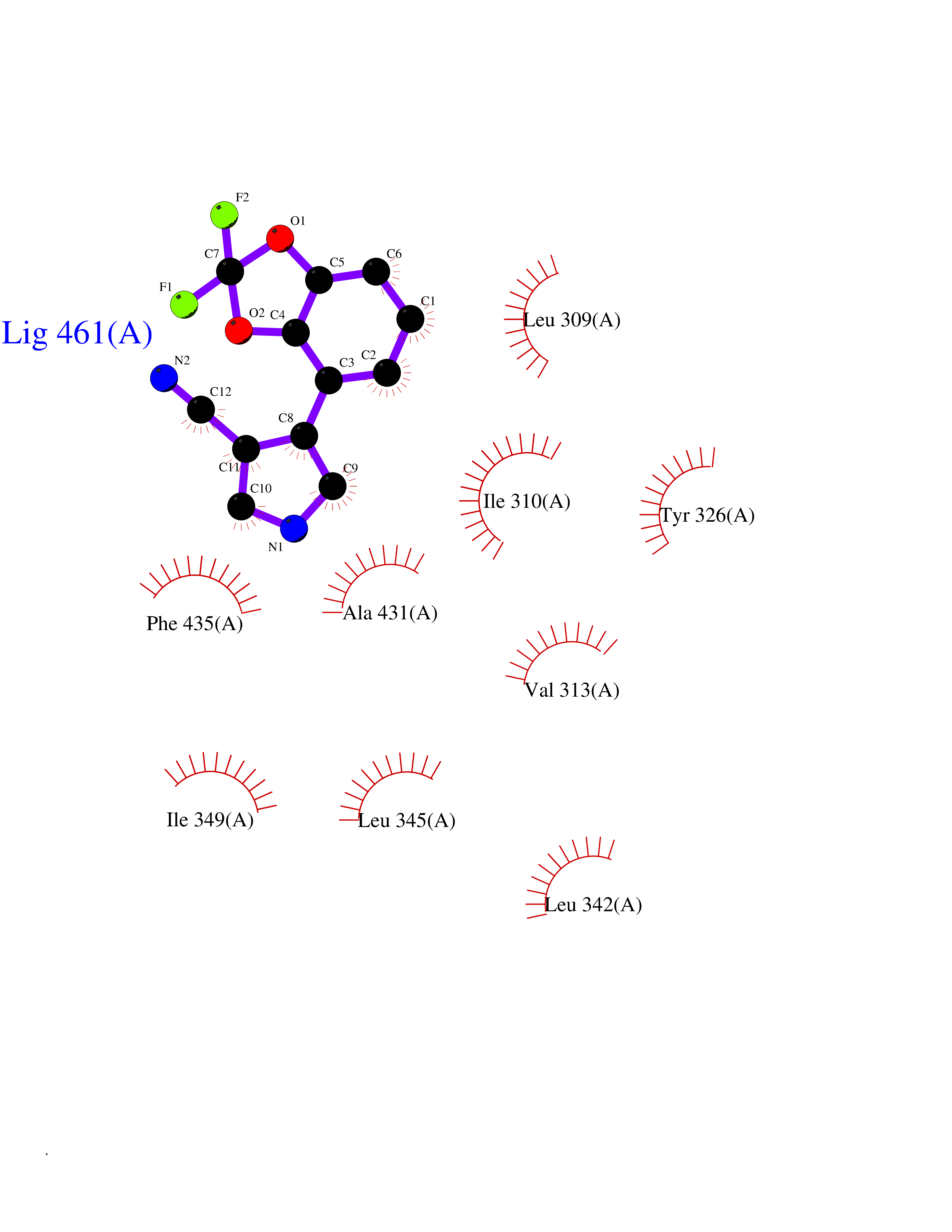 Ligplot Map 3D Binding mode Sequence KPYNKIVSHLLVAEPEKIYAMPDPTVPDSDIKALTTLCDLADRELVVIIGWAKHIPGFSTLSLADQMSLLQSAWMEILILGVVYRSLSFEDELVYADDYIMDEDQSKLAGLLDLNNAILQLVKKYKSMKLEKEEFVTLKAIALANSDSMHIEDVEAVQKLQDVLHEALQDYEAGQHMEDPRRAGKMLMTLPLLRQTSTKAVQHFYNIKLEGKVPMHKLFLEMLEAKV Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Glutamate receptor ionotropic NMDA 2A (NMDAR2A) | 5I2N | 5.95 | |
Target general information Gen name GRIN2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NR2A; NMDA receptor NR2A; N-methyl D-aspartate receptor subtype 2A; HNR2A; Glutamate receptor ionotropic, NMDA 2A; Glutamate [NMDA] receptor subunit epsilon-1; GluN2A Protein family Glutamate-gated ion channel (TC 1.A.10.1) family, NR1/GRIN1 subfamily Biochemical class Glutamate-gated ion channel Function Channel activation requires binding of the neurotransmitter glutamate to the epsilon subunit, glycine binding to the zeta subunit, plus membrane depolarization to eliminate channel inhibition by Mg(2+). Sensitivity to glutamate and channel kinetics depend on the subunit composition; channels containing GRIN1 and GRIN2A have higher sensitivity to glutamate and faster kinetics than channels formed by GRIN1 and GRIN2B. Contributes to the slow phase of excitatory postsynaptic current, long-term synaptic potentiation, and learning. Component of NMDA receptor complexes that function as heterotetrameric, ligand-gated ion channels with high calcium permeability and voltage-dependent sensitivity to magnesium. Related diseases Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal dominant (NDHMSD) [MIM:614254]: An autosomal dominant neurodevelopmental disorder characterized by severe intellectual disability and developmental delay, absent speech, muscular hypotonia, dyskinesia, and hyperkinetic movements. Cortical blindness, cerebral atrophy, and seizures are present in some patients. {ECO:0000269|PubMed:21376300, ECO:0000269|PubMed:25167861, ECO:0000269|PubMed:25864721, ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28095420, ECO:0000269|PubMed:28228639, ECO:0000269|PubMed:28389307, ECO:0000269|PubMed:38538865}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal recessive (NDHMSR) [MIM:617820]: An autosomal recessive neurodevelopmental disorder characterized by severe intellectual disability and psychomotor developmental delay, involuntary and stereotypic movements, spasticity, and inability to walk without support. Intractable seizures manifest in some patients. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28051072}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 101 (DEE101) [MIM:619814]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE101 is an autosomal recessive, severe form characterized by onset of seizures in early infancy. Death in infancy may occur. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:34611970}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01931; DB00659; DB06151; DB08838; DB01238; DB00289; DB05824; DB04620; DB03929; DB00647; DB00843; DB00228; DB11823; DB13146; DB06741; DB00142; DB00874; DB08954; DB06738; DB09409; DB09481; DB01043; DB00454; DB00333; DB04896; DB01173; DB00312; DB01174; DB01708; DB00418; DB00193 Interacts with P05067; P35637; Q12879-1; Q13224; Q62936 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Intellectual disability; Ion channel; Ion transport; Ligand-gated ion channel; Magnesium; Membrane; Metal-binding; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Zinc Protein physicochemical properties Chain ID B Molecular weight (Da) 63184.8 Length 559 Aromaticity 0.1 Instability index 29.67 Isoelectric point 8.51 Charge (pH=7) 6.66 2D Binding mode Binding energy (Kcal/mol) -8.11 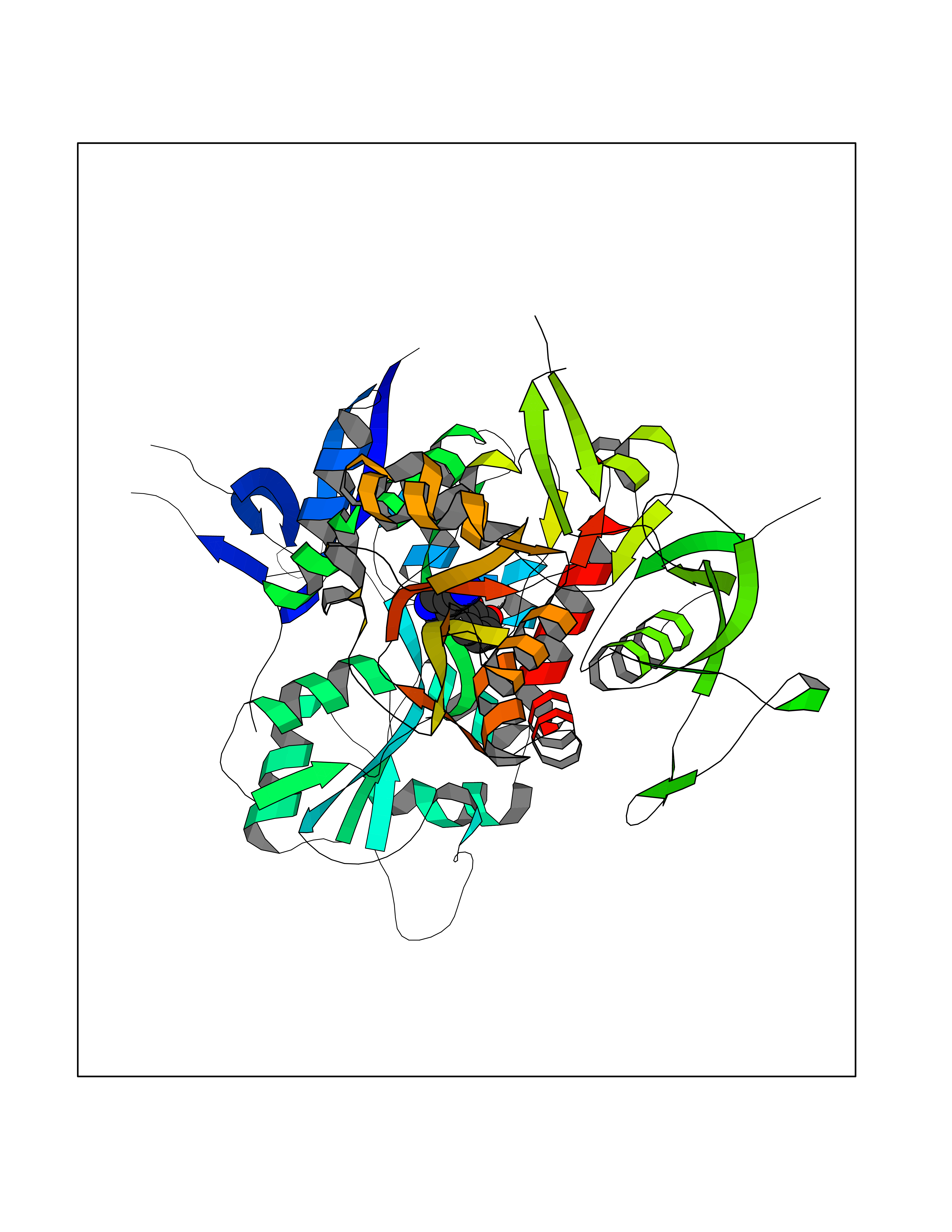 Molscript Map 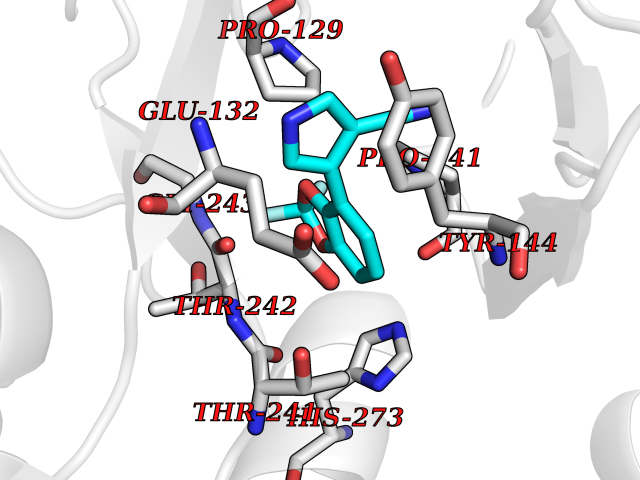 Pymol Map 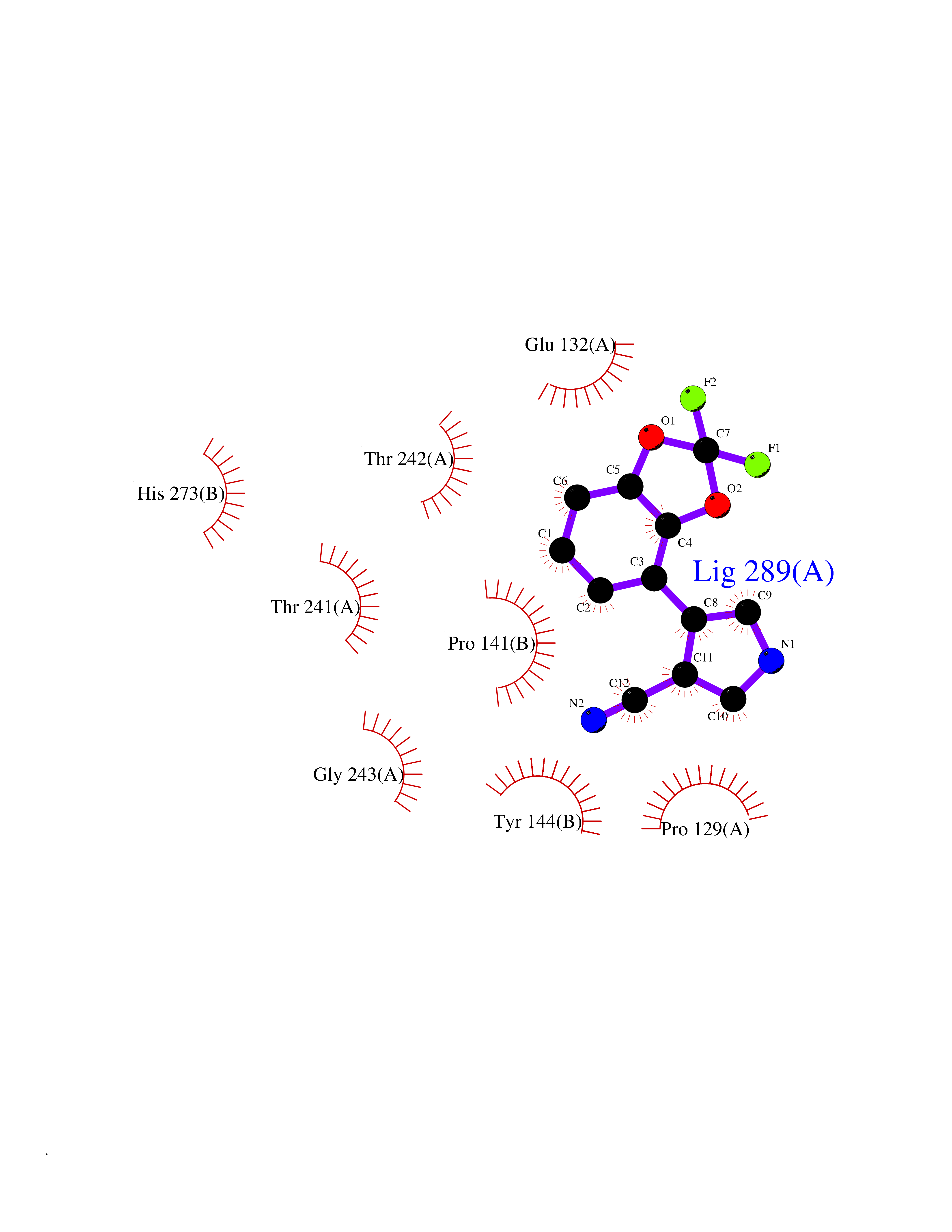 Ligplot Map 3D Binding mode Sequence DNHLSIVTLEEAPFVIVEDIDPETCVRNTVPCRKFVKINNSTNEGMNVKKCCKGFCIDILKKLSRTVKFTYDLYLVTNGKHGKKVNNVWNGMIGEVVYQRAVMAVGSLTINEERSEVVDFSVPFVETGISVMVSRGTQVTGLSDKKFQRPHDYSPPFRFGTVPNGSTERNIRNNYPYMHQYMTKFNQKGVEDALVSLKTGKLDAFIYDAAVLNYKAGRDEGCKLVTIGSGYIFATTGYGIALQKGSPWKRQIDLALLQFVGDGEMEELETLWLTGICMSTRLKIVTIHQEPFVYVKPTLSDGTCKEEFTVNGDPVKKVICTGPNDTSPGSPRHTVPQCCYGFCIDLLIKLARTMNFTYEVHLVADGKFGTQERVNKKEWNGMMGELLSGQADMIVAPLTINNERAQYIEFSKPFKYQGLTILVKKGTRITGINDPRLRNPSDKFIYATVKQSSVDIYFRRQVELSTMYRHMEKHNYESAAEAIQAVRDNKLHAFIWDSAVLEFEASQKCDLVTTGELFFRSGFGIGMRKDSPWKQNVSLSILKSHENGFMEDLDKTWVR Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Serine/threonine-protein kinase cot (COT) | 4Y85 | 5.95 | |
Target general information Gen name MAP3K8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tumor progression locus 2; TPL-2; Proto-oncogene c-Cot; Mitogen-activated protein kinase kinase kinase 8; ESTF; Cancer Osaka thyroid oncogene; COT Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, MAP kinase kinase kinase subfamily Biochemical class NA Function Required for lipopolysaccharide (LPS)-induced, TLR4-mediated activation of the MAPK/ERK pathway in macrophages, thus being critical for production of the proinflammatory cytokine TNF-alpha (TNF) during immune responses. Involved in the regulation of T-helper cell differentiation and IFNG expression in T-cells. Involved in mediating host resistance to bacterial infection through negative regulation of type I interferon (IFN) production. In vitro, activates MAPK/ERK pathway in response to IL1 in an IRAK1-independent manner, leading to up-regulation of IL8 and CCL4. Transduces CD40 and TNFRSF1A signals that activate ERK in B-cells and macrophages, and thus may play a role in the regulation of immunoglobulin production. May also play a role in the transduction of TNF signals that activate JNK and NF-kappa-B in some cell types. In adipocytes, activates MAPK/ERK pathway in an IKBKB-dependent manner in response to IL1B and TNF, but not insulin, leading to induction of lipolysis. Plays a role in the cell cycle. Isoform 1 shows some transforming activity, although it is much weaker than that of the activated oncogenic variant. Related diseases Hyperinsulinemic hypoglycemia, familial, 2 (HHF2) [MIM:601820]: A form of hyperinsulinemic hypoglycemia, a clinically and genetically heterogeneous disorder characterized by inappropriate insulin secretion from the pancreatic beta-cells in the presence of low blood glucose levels. HHF2 is a common cause of persistent hypoglycemia in infancy. Unless early and aggressive intervention is undertaken, brain damage from recurrent episodes of hypoglycemia may occur. HHF2 inheritance can be autosomal dominant or autosomal recessive. {ECO:0000269|PubMed:10204114, ECO:0000269|PubMed:12364426, ECO:0000269|PubMed:15562009, ECO:0000269|PubMed:15579781, ECO:0000269|PubMed:15807877, ECO:0000269|PubMed:15998776, ECO:0000269|PubMed:16332676, ECO:0000269|PubMed:16357843, ECO:0000269|PubMed:18596924, ECO:0000269|PubMed:19357197, ECO:0000269|PubMed:7847376, ECO:0000269|PubMed:8923010}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Diabetes mellitus, permanent neonatal, 2 (PNDM2) [MIM:618856]: A form of permanent neonatal diabetes mellitus, a type of diabetes characterized by onset of persistent hyperglycemia within the first six months of life. Initial clinical manifestations include intrauterine growth retardation, hyperglycemia, glycosuria, osmotic polyuria, severe dehydration, and failure to thrive. Some PNDM2 patients may also have developmental delay, muscle weakness, epilepsy and dysmorphic features. PNDM2 transmission pattern is consistent with autosomal dominant inheritance. {ECO:0000269|PubMed:15115830, ECO:0000269|PubMed:15292329, ECO:0000269|PubMed:15448106, ECO:0000269|PubMed:15448107, ECO:0000269|PubMed:15580558, ECO:0000269|PubMed:15583126, ECO:0000269|PubMed:16609879, ECO:0000269|PubMed:16731833, ECO:0000269|PubMed:17213273, ECO:0000269|PubMed:17652641, ECO:0000269|PubMed:17855752, ECO:0000269|PubMed:20022885, ECO:0000269|PubMed:28842488}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Transient neonatal diabetes mellitus 3 (TNDM3) [MIM:610582]: Neonatal diabetes mellitus, defined as insulin-requiring hyperglycemia within the first month of life, is a rare entity. In about half of the neonates, diabetes is transient and resolves at a median age of 3 months, whereas the rest have a permanent form of diabetes. In a significant number of patients with transient neonatal diabetes mellitus, diabetes type 2 appears later in life. The onset and severity of TNDM3 is variable with childhood-onset diabetes, gestational diabetes or adult-onset diabetes described. {ECO:0000269|PubMed:15718250, ECO:0000269|PubMed:15784703}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in KCNJ11 may contribute to non-insulin-dependent diabetes mellitus (NIDDM), also known as diabetes mellitus type 2.; DISEASE: Maturity-onset diabetes of the young 13 (MODY13) [MIM:616329]: A form of diabetes that is characterized by an autosomal dominant mode of inheritance, onset in childhood or early adulthood (usually before 25 years of age), a primary defect in insulin secretion and frequent insulin-independence at the beginning of the disease. {ECO:0000269|PubMed:22701567}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P08238; P19838; Q00653; Q13526; Q8NFZ5 EC number EC 2.7.11.25 Uniprot keywords 3D-structure; Alternative initiation; ATP-binding; Cell cycle; Cytoplasm; Immunity; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 34773.8 Length 307 Aromaticity 0.09 Instability index 39.24 Isoelectric point 6.68 Charge (pH=7) -1.2 2D Binding mode Binding energy (Kcal/mol) -8.11  Molscript Map 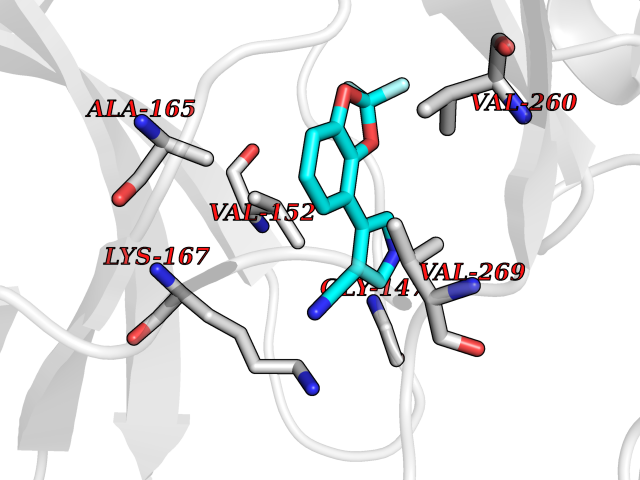 Pymol Map 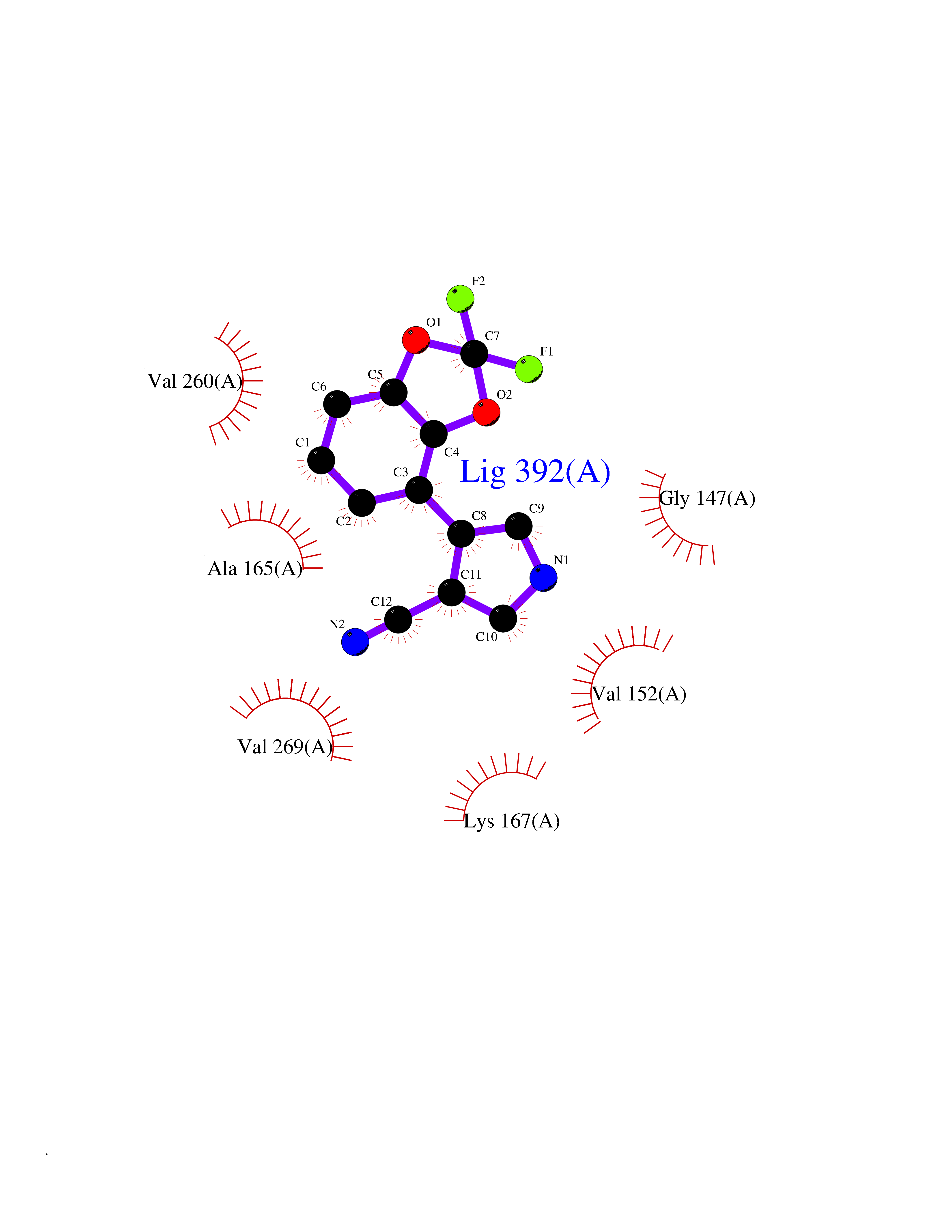 Ligplot Map 3D Binding mode Sequence LSSVRYGTVEDLLAFANHISNTPQESGILLNMVITPQNGRYQIDSDVLLIPWKLTYRNIFIPRGAFGKVYLAQDIKTKKRMACKLIPVDQFKPSDVEIQACFRHENIAELYGAVLWGETVHLFMEAGEGGSVLEKLESCGPMREFEIIWVTKHVLKGLDFLHSKKVIHHDIKPSNIVFMSTKAVLVDFGLSVQMTEDVYFPKDLRGTEIYMSPEVILCRGHSTKADIYSLGATLIHMQTGTPPWVKRYPRSAYPSYLYIIHKQAPPLEDIADDCSPGMRELIEASLERNPNHRPRAADLLKHEALNP Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Sphingosine-1-phosphate receptor 1 (S1PR1) | 7EW0 | 5.95 | |
Target general information Gen name S1PR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Sphingosine 1-phosphate receptor Edg-1; S1P1; S1P receptor Edg-1; S1P receptor 1; Endothelial differentiation G-protein coupled receptor 1; CHEDG1; CD363 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Signaling leads to the activation of RAC1, SRC, PTK2/FAK1 and MAP kinases. Plays an important role in cell migration, probably via its role in the reorganization of the actin cytoskeleton and the formation of lamellipodia in response to stimuli that increase the activity of the sphingosine kinase SPHK1. Required for normal chemotaxis toward sphingosine 1-phosphate. Required for normal embryonic heart development and normal cardiac morphogenesis. Plays an important role in the regulation of sprouting angiogenesis and vascular maturation. Inhibits sprouting angiogenesis to prevent excessive sprouting during blood vessel development. Required for normal egress of mature T-cells from the thymus into the blood stream and into peripheral lymphoid organs. Plays a role in the migration of osteoclast precursor cells, the regulation of bone mineralization and bone homeostasis. Plays a role in responses to oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine by pulmonary endothelial cells and in the protection against ventilator-induced lung injury. G-protein coupled receptor for the bioactive lysosphingolipid sphingosine 1-phosphate (S1P) that seems to be coupled to the G(i) subclass of heteromeric G proteins. Related diseases Wolcott-Rallison syndrome (WRS) [MIM:226980]: A rare autosomal recessive disorder, characterized by permanent neonatal or early infancy insulin-dependent diabetes and, at a later age, epiphyseal dysplasia, osteoporosis, growth retardation and other multisystem manifestations, such as hepatic and renal dysfunctions, intellectual disability and cardiovascular abnormalities. {ECO:0000269|PubMed:10932183, ECO:0000269|PubMed:12086964, ECO:0000269|PubMed:12960215, ECO:0000269|PubMed:16813601, ECO:0000269|PubMed:24168455, ECO:0000269|PubMed:24194294, ECO:0000269|PubMed:27145240, ECO:0000269|PubMed:28220546, ECO:0000269|PubMed:30906465, ECO:0000269|PubMed:30922274, ECO:0000269|PubMed:32216767, ECO:0000269|PubMed:34123975}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14766; DB08868; DB12612; DB12016; DB12371 Interacts with Q07108 EC number NA Uniprot keywords 3D-structure; Acetylation; Angiogenesis; Cell membrane; Chemotaxis; Disulfide bond; Endosome; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID D Molecular weight (Da) 32418.9 Length 284 Aromaticity 0.13 Instability index 39.5 Isoelectric point 9.71 Charge (pH=7) 19.09 2D Binding mode Binding energy (Kcal/mol) -8.11 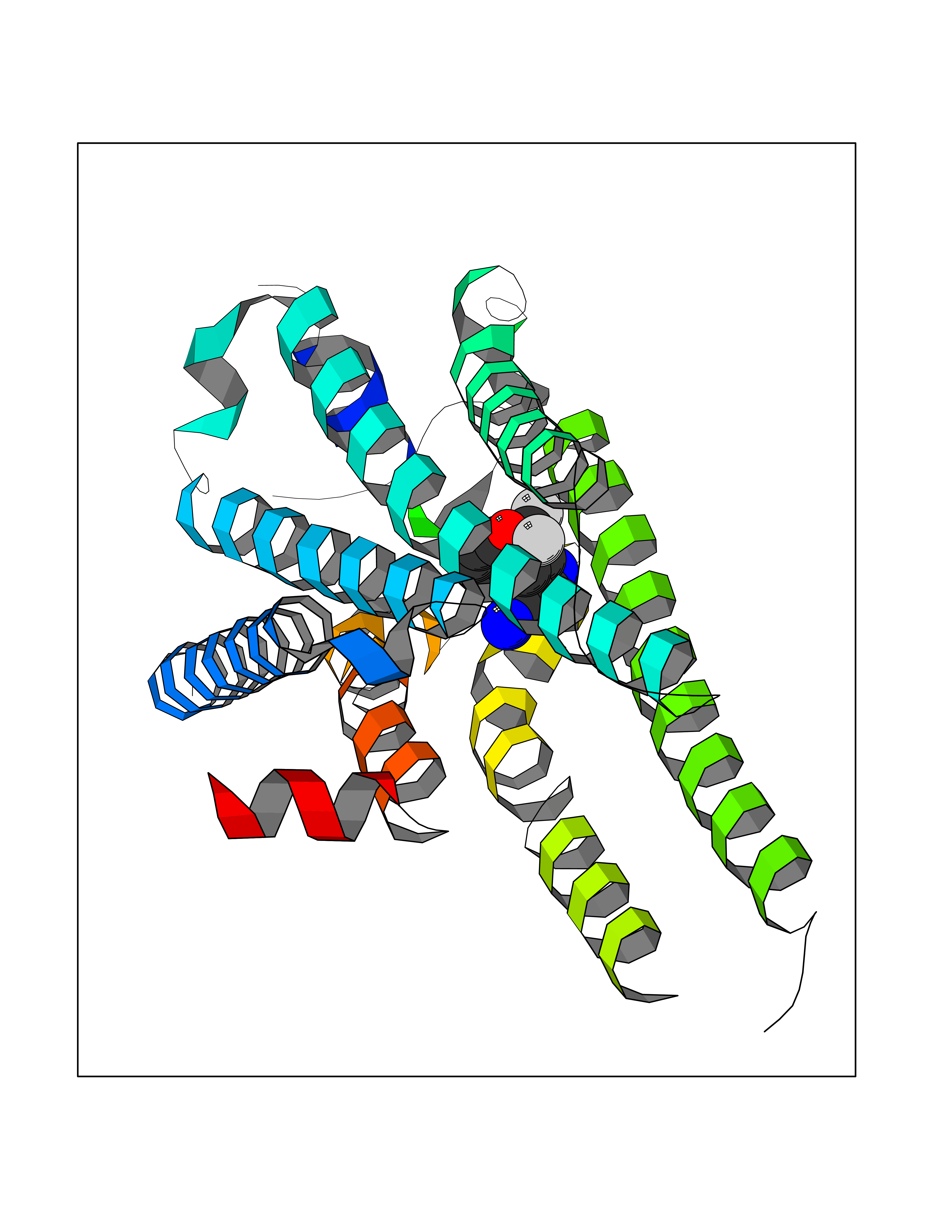 Molscript Map 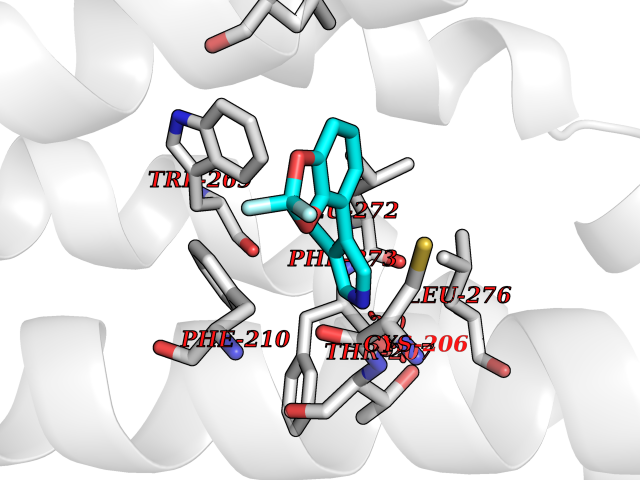 Pymol Map  Ligplot Map 3D Binding mode Sequence YDIIVRHYNYTGKLTSVVFILICCFIILENIFVLLTIWKTKKFHRPMYYFIGNLALSDLLAGVAYTANLLLSGATTYKLTPAQWFLREGSMFVALSASVFSLLAIAIERYITMLKMKLHNGSNNFRLFLLISACWVISLILGGLPIMGWNCISALSSCSTVLPLYHKHYILFCTTVFTLLLLSIVILYCRIYSLVRTRSRRLTFRKSEKSLALLKTVIIVLSVFIACWAPLFILLLLDVGCKVKTCDILFRAEYFLVLAVLNSGTNPIIYTLTNKEMRRAFIRI Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Caspase-6 (CASP6) | 4NBL | 5.95 | |
Target general information Gen name CASP6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MCH2; Caspase-6 subunit p18; Caspase-6 subunit p11; CASP-6; Apoptotic protease Mch-2 Protein family Peptidase C14A family Biochemical class Peptidase Function Cleaves poly(ADP-ribose) polymerase in vitro, as well as lamins. Overexpression promotes programmed cell death. Involved in the activation cascade of caspases responsible for apoptosis execution. Related diseases Growth hormone deficiency, isolated, 1A (IGHD1A) [MIM:262400]: An autosomal recessive, severe deficiency of growth hormone leading to dwarfism. Patients often develop antibodies to administered growth hormone. {ECO:0000269|PubMed:8364549}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 1B (IGHD1B) [MIM:612781]: An autosomal recessive deficiency of growth hormone leading to short stature. Patients have low but detectable levels of growth hormone, significantly retarded bone age, and a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:12655557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Kowarski syndrome (KWKS) [MIM:262650]: A syndrome clinically characterized by short stature associated with bioinactive growth hormone, normal or slightly increased growth hormone secretion, pathologically low insulin-like growth factor 1 levels, and normal catch-up growth on growth hormone replacement therapy. {ECO:0000269|PubMed:17519310, ECO:0000269|PubMed:8552145, ECO:0000269|PubMed:9276733}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 2 (IGHD2) [MIM:173100]: An autosomal dominant deficiency of growth hormone leading to short stature. Clinical severity is variable. Patients have a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:11502836, ECO:0000269|PubMed:9152628}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9Y614; Q6DHV7-2; Q6UY14-3; Q96MA6; Q5T2L2; Q96Q83-2; Q9Y303-2; Q9NU02; P09525; P06727; Q8WW27; Q66PJ3-4; Q6XD76; P18848; Q9H0Y0; Q14032; P54687-4; P06276; Q9NSI6-4; Q96Q07-2; Q9H0W9-3; Q9NQ89; Q13901; Q3SXR2; Q8N1A6; P17655; P20807-4; P42574; P55212; O00257-3; P24863; Q9NNX6-10; Q9UJX2; P42773; O95674; Q494V2-2; Q8WUX9; Q9Y3D0; Q8N365; Q3SX64; Q99966; P09496-2; Q6PJW8-3; Q96BR5; P02458-1; Q9UGL9; Q9UKG9-2; P26998; P35222; Q53TN4; P61962; O60479; Q96EY1-3; Q92782-2; Q9BPU6; A0AVK6; Q658K8; O00303; Q13347; O00472; O00423; Q6NXG1-3; Q49AJ0-4; Q8N128-2; Q8IZU1; Q6ZNL6; Q9NSA1; Q06547-3; Q49A26-4; Q9HAV0; Q6NXT2; Q9BT25; Q9NRZ9-6; Q96EW2-2; P42858; Q8N6M8-2; Q92613; P0C870; Q9UK76; Q8N5Z5; Q8TBB5-2; Q9UH77; Q8N4N3-2; Q5JUW0-3; Q8N1A0; P13473-2; Q6DKI2; Q9H2C1; Q8N0U6; Q9Y234; Q8TBB1; Q1L5Z9; Q96JB6; Q16609; Q8IYG6; P0DP58-2; Q969L2; P27338; A6NJ78-4; Q96C03-3; Q8N5J2-3; A0A0A0MR05; P34949-2; Q9BV20; Q6IN84-2; A2RUH7; P01106; Q9H7X0; Q15742-2; Q9UJ70-2; Q8NDH3-5; Q96HA8; P36639-4; Q8NFH4; Q8NFH3; Q7Z3B4; Q6N063-2; Q6GQQ9-2; Q9H8K7; Q99447; P27815-4; O15534; Q9BUL5; Q00169; P48739; P61925; Q58EX7-2; O60664; Q14181; P0DPB6; P36954; Q07869; O60927; Q6ZMI0-5; P54619; Q8NCQ7-2; P41222; P29074; Q8WUD1-2; Q5R372-9; Q9HD47-3; Q09028; Q04206; P47804-3; Q15382; Q06587; Q8N5U6; P62701; Q66K80; Q01826; O15126; P22307-3; Q9BRK5; Q9NTN9-3; P01011; Q15393; Q9NR46; Q9BZQ2; O60902-3; Q86US8; P37840; Q96H20; Q13573; Q7Z6I5; Q496A3; Q9C004; Q5W111-2; Q96BD6; Q92797-2; O60506-4; O15273; Q86WV5; Q96A09; P54274-2; P22735; O43548; Q9NQ88; Q9UIK5-2; Q53NU3; P04637; Q12888; P36406; Q86WT6-2; Q13885; P49459; Q9P1Q0-4; Q9NX94; Q8NA23-2; Q9BQA1; O00755; O95070; O43829; Q8IWT0-2; Q53FD0-2; Q05CR2; Q96JL9-2; Q96LX8; Q3KNS6-3; Q5JTY5; A0A384MDV8; B7Z3E8; Q86V28 EC number EC 3.4.22.59 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; Autocatalytic cleavage; Cytoplasm; Hydrolase; Lipoprotein; Nucleus; Palmitate; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Thiol protease; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 57170 Length 500 Aromaticity 0.12 Instability index 31.33 Isoelectric point 8.05 Charge (pH=7) 4.52 2D Binding mode Binding energy (Kcal/mol) -8.12  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence AFYKREMFDPAEKYKMDHRRRGIALIFNHERFFWHLTLPERRGTCADRDNLTRRFSDLGFEVKCFNDLKAEELLLKIHEVSTVSHADADCFVCVFLSHGEGNHIYAYDAKIEIQTLTGLFKGDKCHSLVGKPKIFIIQAARGNQHDVPVIPDTNITEVDAASVYTLPAGADFLMCYSVAEGYYSHRETVNGSWYIQDLCEMLGKYGSSLEFTELLTLVNRKVSQRRVDFCKDPSAIGKKQVPCFASMLTKKLHFFPKSMFDPAEKYKMDHRRRGIALIFNHERFFWHLTLPERRGTCADRDNLTRRFSDLGFEVKCFNDLKAEELLLKIHEVSTVSHADADCFVCVFLSHGEGNHIYAYDAKIEIQTLTGLFKGDKCHSLVGKPKIFIIQAARGNTNITEVDAASVYTLPAGADFLMCYSVAEGYYSHRETVNGSWYIQDLCEMLGKYGSSLEFTELLTLVNRKVSQRRVDFCKDPSAIGKKQVPCFASMLTKKLHFFPK Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | GABA(A) receptor alpha-1 (GABRA1) | 6X3T | 5.95 | |
Target general information Gen name GABRA1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Gamma-aminobutyric acid receptor subunit alpha-1; GABA(A) receptor subunit alpha-1 Protein family Ligand-gated ion channel (TC 1.A.9) family, Gamma-aminobutyric acid receptor (TC 1.A.9.5) subfamily, GABRA1 sub-subfamily Biochemical class Ligand-gated ion channel Function Ligand-gated chloride channel which is a component of the heteropentameric receptor for GABA, the major inhibitory neurotransmitter in the brain. Plays an important role in the formation of functional inhibitory GABAergic synapses in addition to mediating synaptic inhibition as a GABA-gated ion channel. The gamma2 subunit is necessary but not sufficient for a rapid formation of active synaptic contacts and the synaptogenic effect of this subunit is influenced by the type of alpha and beta subunits present in the receptor pentamer (By similarity). The alpha1/beta2/gamma2 receptor and the alpha1/beta3/gamma2 receptor exhibit synaptogenic activity. GABRA1-mediated plasticity in the orbitofrontal cortex regulates context-dependent action selection (By similarity). Functions also as histamine receptor and mediates cellular responses to histamine (By similarity). Related diseases Epilepsy, childhood absence 4 (ECA4) [MIM:611136]: A subtype of idiopathic generalized epilepsy characterized by an onset at age 6-7 years, frequent absence seizures (several per day) and bilateral, synchronous, symmetric 3-Hz spike waves on EEG. Tonic-clonic seizures often develop in adolescence. Absence seizures may either remit or persist into adulthood. {ECO:0000269|PubMed:16718694}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Epilepsy, idiopathic generalized 13 (EIG13) [MIM:611136]: A disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. {ECO:0000269|PubMed:21714819}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Juvenile myoclonic epilepsy 5 (EJM5) [MIM:611136]: A subtype of idiopathic generalized epilepsy. Patients have afebrile seizures only, with onset in adolescence (rather than in childhood) and myoclonic jerks which usually occur after awakening and are triggered by sleep deprivation and fatigue. {ECO:0000269|PubMed:11992121}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 19 (DEE19) [MIM:615744]: A severe neurologic disorder characterized by onset of seizures in the first months of life and usually associated with EEG abnormalities. Affected infants have convulsive seizures (hemiclonic or generalized) that are often prolonged and triggered by fever. Other seizure types include focal, myoclonic, absence seizures, and drop attacks. Development is normal in the first year of life with later slowing and intellectual disability. {ECO:0000269|PubMed:24623842, ECO:0000269|PubMed:27864847}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12537; DB00546; DB06579; DB00404; DB01351; DB00543; DB11901; DB01352; DB01483; DB14719; DB11859; DB01558; DB09017; DB00237; DB00241; DB01353; DB01489; DB00395; DB00475; DB14715; DB01594; DB00349; DB06470; DB01068; DB00628; DB01559; DB01553; DB00363; DB01511; DB01189; DB00829; DB01496; DB01341; DB13837; DB00228; DB01215; DB00402; DB00898; DB00189; DB01545; DB09166; DB00292; DB01567; DB01205; DB01544; DB00690; DB05087; DB01381; DB01437; DB00801; DB01159; DB01354; DB01355; DB00753; DB01587; DB00555; DB13643; DB00186; DB13872; DB13437; DB00603; DB01043; DB00371; DB00463; DB00474; DB01028; DB00849; DB01107; DB15489; DB00683; DB12458; DB01595; DB14028; DB00334; DB00842; DB14672; DB00312; DB01174; DB00252; DB00466; DB13335; DB01708; DB01588; DB00794; DB00837; DB00818; DB01589; DB01346; DB12404; DB00418; DB01236; DB09118; DB00306; DB01956; DB00231; DB01154; DB11582; DB00599; DB00273; DB00897; DB00962; DB00425; DB00909; DB01198; DB15490 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Chloride; Chloride channel; Cytoplasmic vesicle; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 77460 Length 672 Aromaticity 0.14 Instability index 35.95 Isoelectric point 8.75 Charge (pH=7) 7.31 2D Binding mode Binding energy (Kcal/mol) -8.12  Molscript Map 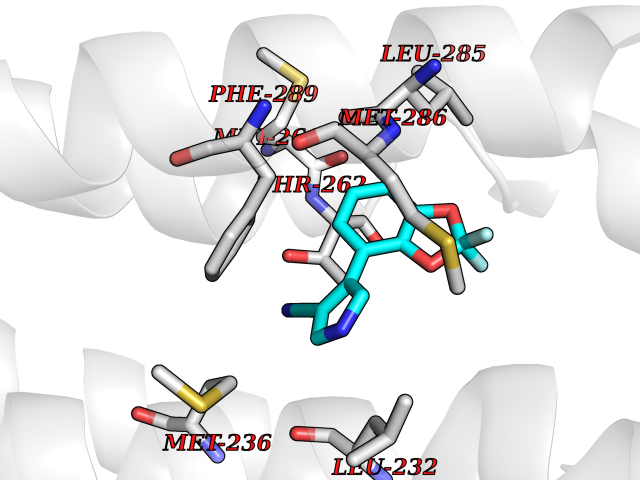 Pymol Map 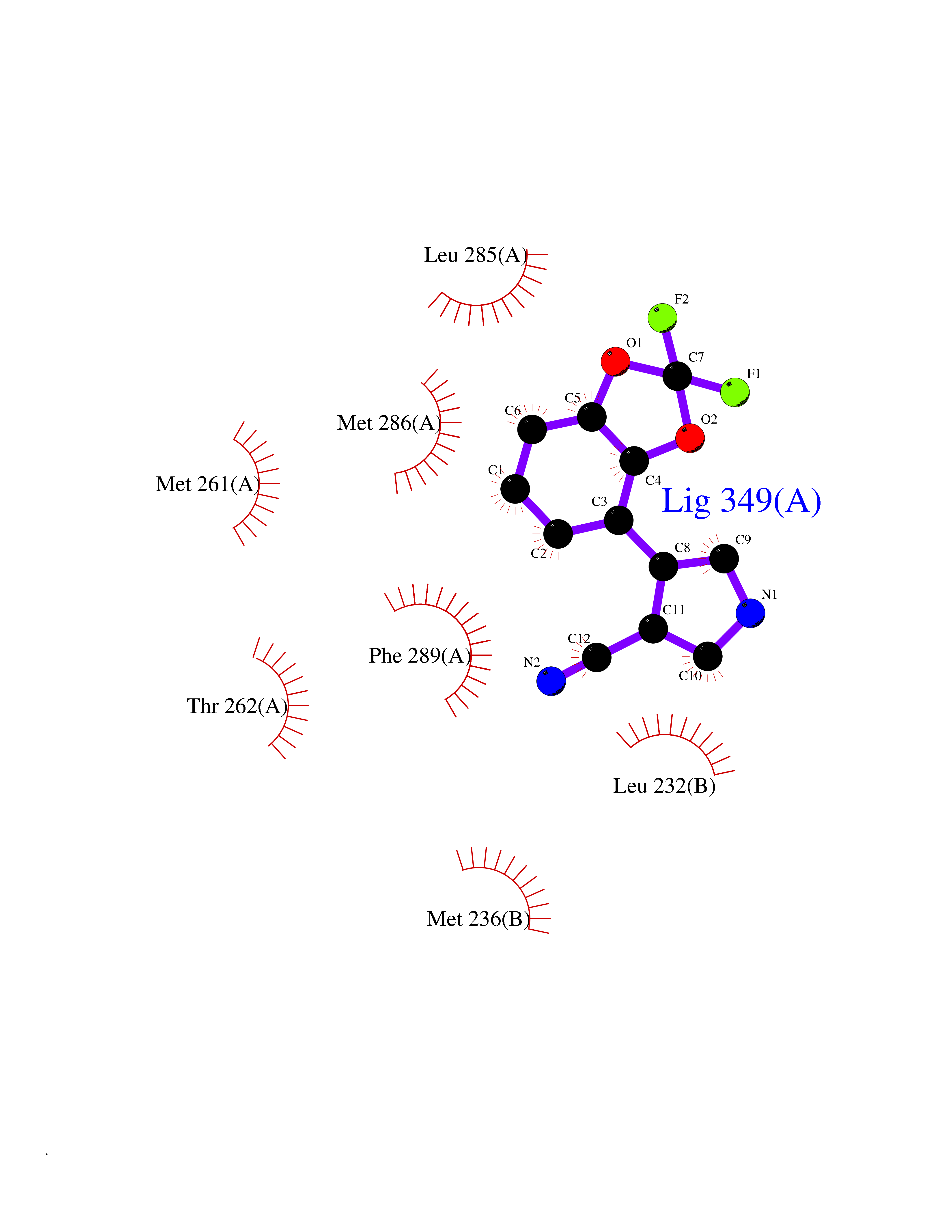 Ligplot Map 3D Binding mode Sequence SNMSLVKETVDRLLKGYDIRLRPDFGGPPVAVGMNIDIASIDMVSEVNMDYTLTMYFQQAWRDKRLSYNVIPLNLTLDNRVADQLWVPDTYFLNDKKSFVHGVTVKNRMIRLHPDGTVLYGLRITTTAACMMDLRRYPLDEQNCTLEIESYGYTTDDIEFYWRGDDNAVTGVTKIELPQFSIVDYKLITKKVVFSTGSYPRLSLSFKLKRNIGYFILQTYMPSILITILSWVSFWINYDASAARVALGITTVLTMTTINTHLRETLPKIPYVKAIDMYLMGCFVFVFMALLEYALVNYIFFSQPARAAAIDRWSRIFFPVVFSFFNIVYWLYYVDNTTVFTRILDRLLDGYDNRLRPGLGERVTEVKTDIFVTSFGPVSDHDMEYTIDVFFRQSWKDERLKFKGPMTVLRLNNLMASKIWTPDTFFHNGKKSVAHNMTMPNKLLRITEDGTLLYTMRLTVRAECPMHLEDFPMDAHACPLKFGSYAYTRAEVVYEWTREPARSVVVAEDGSRLNQYDLLGQTVDSGIVQSSTGEYVVMTTHFHLKRKIGYFVIQTYLPCIMTVILSQVSFWLNRESVPARTVFGVTTVLTMTTLSISARNSLPKVAYATAMDWFIAVCYAFVFSALIEFATVNYFTKSQPARAAKIDRLSRIAFPLLFGIFNLVYWATYLNR Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Renal carcinoma antigen NY-REN-64 (IRAK-4) | 6EG9 | 5.95 | |
Target general information Gen name IRAK4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Interleukin-1 receptor-associated kinase 4; IRAK-4 Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family, Pelle subfamily Biochemical class Kinase Function Serine/threonine-protein kinase that plays a critical role in initiating innate immune response against foreign pathogens. Involved in Toll-like receptor (TLR) and IL-1R signaling pathways. Is rapidly recruited by MYD88 to the receptor-signaling complex upon TLR activation to form the Myddosome together with IRAK2. Phosphorylates initially IRAK1, thus stimulating the kinase activity and intensive autophosphorylation of IRAK1. Phosphorylates E3 ubiquitin ligases Pellino proteins (PELI1, PELI2 and PELI3) to promote pellino-mediated polyubiquitination of IRAK1. Then, the ubiquitin-binding domain of IKBKG/NEMO binds to polyubiquitinated IRAK1 bringing together the IRAK1-MAP3K7/TAK1-TRAF6 complex and the NEMO-IKKA-IKKB complex. In turn, MAP3K7/TAK1 activates IKKs (CHUK/IKKA and IKBKB/IKKB) leading to NF-kappa-B nuclear translocation and activation. Alternatively, phosphorylates TIRAP to promote its ubiquitination and subsequent degradation. Phosphorylates NCF1 and regulates NADPH oxidase activation after LPS stimulation suggesting a similar mechanism during microbial infections. Related diseases Immunodeficiency 67 (IMD67) [MIM:607676]: An autosomal recessive primary immunodeficiency characterized by recurrent, life-threatening systemic and invasive bacterial infections beginning in infancy or early childhood. {ECO:0000269|PubMed:12637671, ECO:0000269|PubMed:12925671, ECO:0000269|PubMed:16950813, ECO:0000269|PubMed:17878374, ECO:0000269|PubMed:19663824, ECO:0000269|PubMed:21057262, ECO:0000269|PubMed:24316379}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08590; DB12010 Interacts with Q9UBH0; O43187; Q99836; Q99836-1; Q96FA3; Q9HAT8; P58753; Q9C029; P0DPA2; Q96LX8; Q8K4B2 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cytoplasm; Disease variant; Immunity; Innate immunity; Kinase; Magnesium; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 30948.8 Length 276 Aromaticity 0.08 Instability index 43.33 Isoelectric point 5.09 Charge (pH=7) -13.45 2D Binding mode Binding energy (Kcal/mol) -8.11 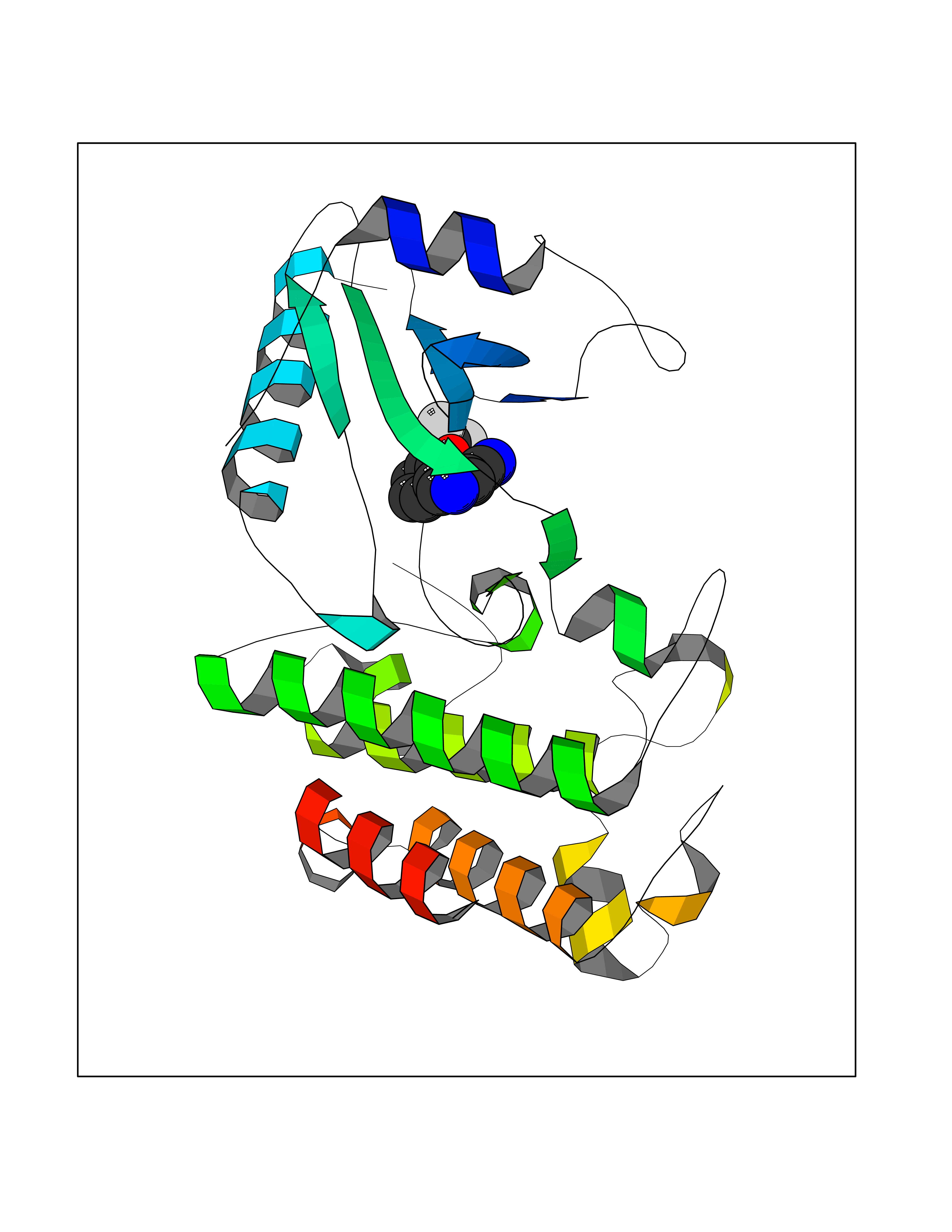 Molscript Map 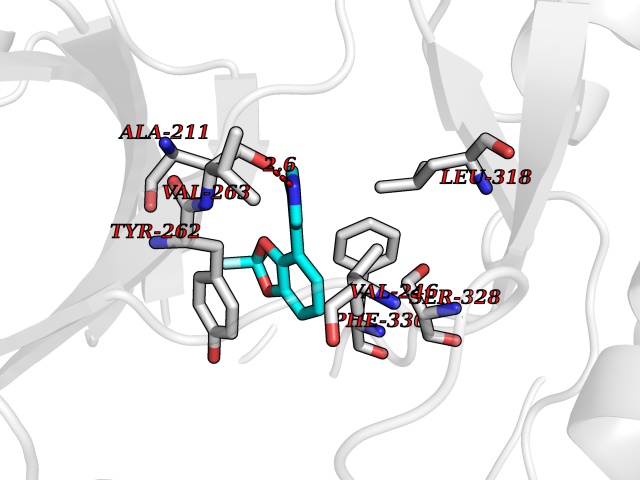 Pymol Map 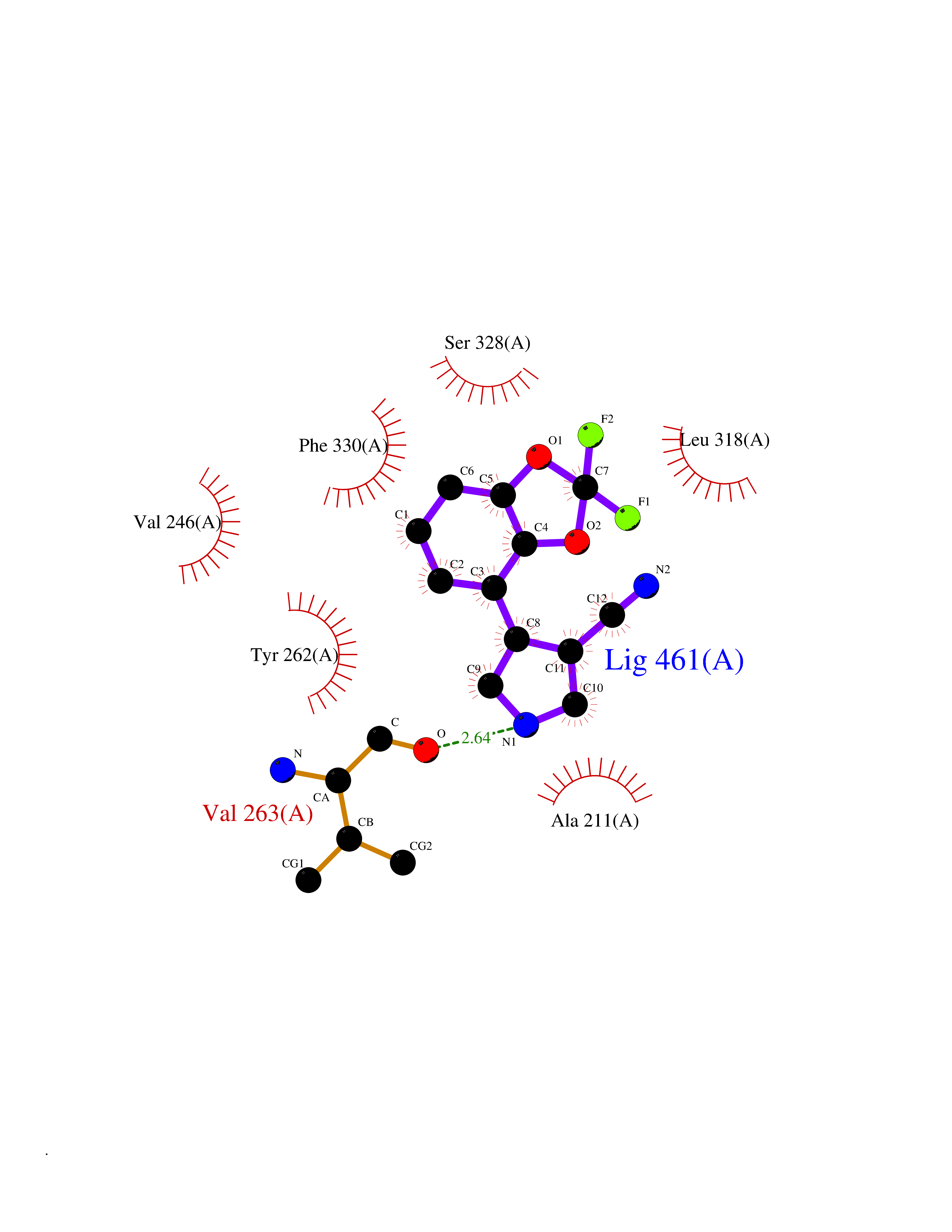 Ligplot Map 3D Binding mode Sequence RFHSFSFYELKNVTNNFDERPISVGGNKMGEGGFGVVYKGYVNNTTVAVKKLAAITTEELKQQFDQEIKVMAKCQHENLVELLGFSSDGDDLCLVYVYMPNGSLLDRLSCLDGTPPLSWHMRCKIAQGAANGINFLHENHHIHRDIKSANILLDEAFTAKISDFGVGTTAYMAPEALRGEITPKSDIYSFGVVLLEIITGLPAVDEHREPQLLLDIKEEIEDEEKTIEDYIDKKMNDADSTSVEAMYSVASQCLHEKKNKRPDIKKVQQLLQEMTA Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | GABA(A) receptor beta-2 (GABRB2) | 6X3T | 5.95 | |
Target general information Gen name GABRB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GABRB2; GABA(A) receptor subunit beta-2 Protein family Ligand-gated ion channel (TC 1.A.9) family, Gamma-aminobutyric acid receptor (TC 1.A.9.5) subfamily, GABRB2 sub-subfamily Biochemical class Neurotransmitter receptor Function Component of the heteropentameric receptor for GABA, the major inhibitory neurotransmitter in the vertebrate brain. Functions also as histamine receptor and mediates cellular responses to histamine. Functions as receptor for diazepines and various anesthetics, such as pentobarbital; these are bound at a separate allosteric effector binding site. Functions as ligand- gated chloride channel. Related diseases Epileptic encephalopathy, infantile or early childhood, 2 (IECEE2) [MIM:617829]: A form of epileptic encephalopathy, a heterogeneous group of severe childhood onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. IECEE2 is an autosomal dominant condition with variable age at seizure onset, ranging from early infancy to 6 years. {ECO:0000269|PubMed:25124326, ECO:0000269|PubMed:27789573, ECO:0000269|PubMed:29100083}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12537; DB00546; DB00404; DB00543; DB11901; DB14719; DB11859; DB01558; DB09017; DB00237; DB00241; DB01489; DB00395; DB00475; DB14715; DB01594; DB00349; DB01068; DB00628; DB01559; DB01553; DB01511; DB01189; DB00829; DB13837; DB00228; DB01215; DB00402; DB00898; DB00189; DB01545; DB09166; DB00292; DB01567; DB01205; DB01544; DB00690; DB06716; DB05087; DB01381; DB01437; DB00801; DB01159; DB00753; DB01587; DB00555; DB13643; DB00186; DB13872; DB13437; DB00603; DB01043; DB00371; DB00463; DB01028; DB01107; DB15489; DB00683; DB01595; DB14028; DB00842; DB14672; DB00312; DB00252; DB13335; DB01708; DB01588; DB00794; DB00818; DB01589; DB12404; DB01236; DB09118; DB00306; DB01956; DB00231; DB11582; DB00897; DB15490 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Chloride; Chloride channel; Cytoplasmic vesicle; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 77460 Length 672 Aromaticity 0.14 Instability index 35.95 Isoelectric point 8.75 Charge (pH=7) 7.31 2D Binding mode Binding energy (Kcal/mol) -8.12  Molscript Map 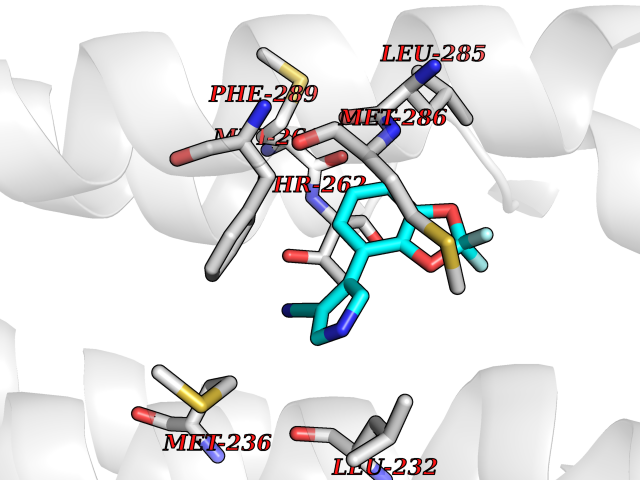 Pymol Map 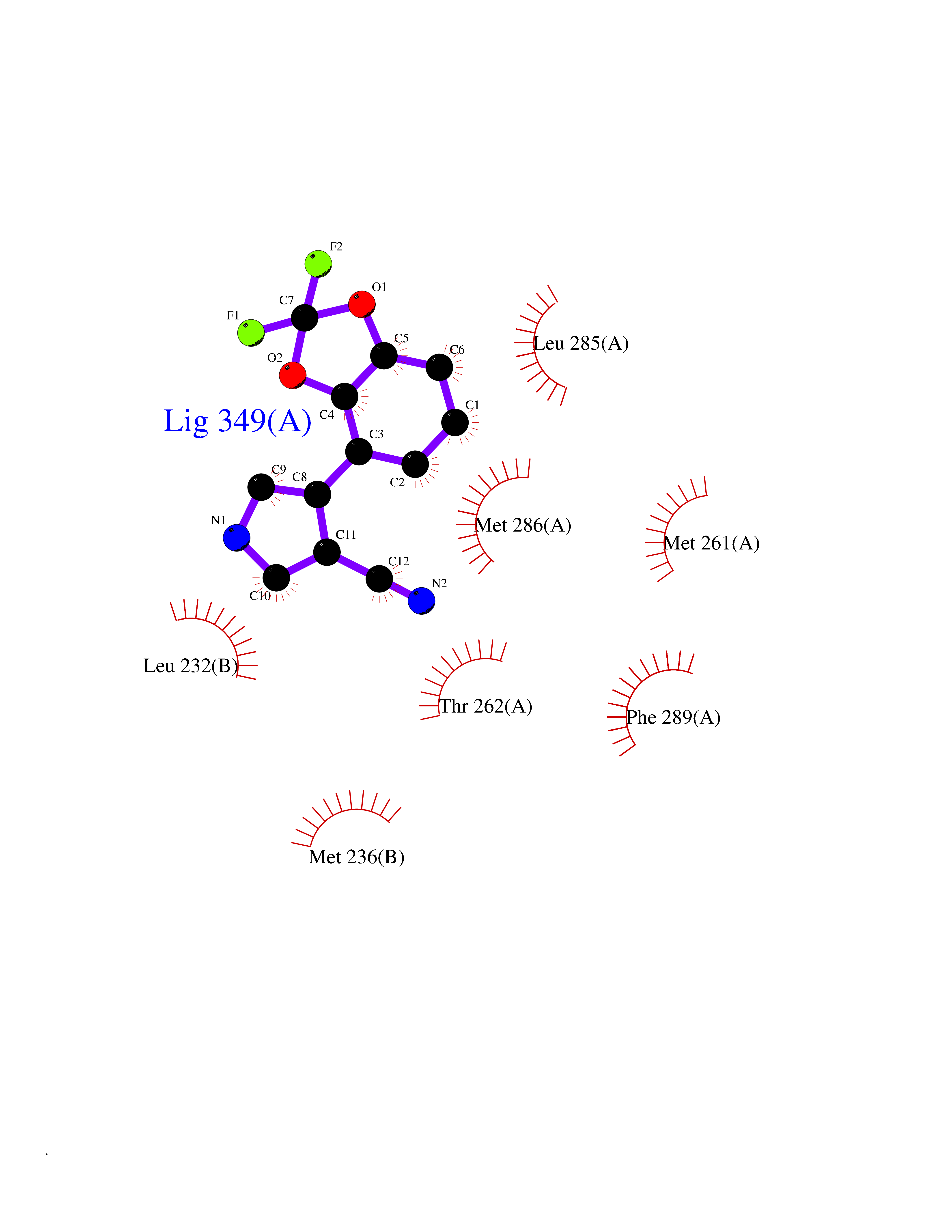 Ligplot Map 3D Binding mode Sequence SNMSLVKETVDRLLKGYDIRLRPDFGGPPVAVGMNIDIASIDMVSEVNMDYTLTMYFQQAWRDKRLSYNVIPLNLTLDNRVADQLWVPDTYFLNDKKSFVHGVTVKNRMIRLHPDGTVLYGLRITTTAACMMDLRRYPLDEQNCTLEIESYGYTTDDIEFYWRGDDNAVTGVTKIELPQFSIVDYKLITKKVVFSTGSYPRLSLSFKLKRNIGYFILQTYMPSILITILSWVSFWINYDASAARVALGITTVLTMTTINTHLRETLPKIPYVKAIDMYLMGCFVFVFMALLEYALVNYIFFSQPARAAAIDRWSRIFFPVVFSFFNIVYWLYYVDNTTVFTRILDRLLDGYDNRLRPGLGERVTEVKTDIFVTSFGPVSDHDMEYTIDVFFRQSWKDERLKFKGPMTVLRLNNLMASKIWTPDTFFHNGKKSVAHNMTMPNKLLRITEDGTLLYTMRLTVRAECPMHLEDFPMDAHACPLKFGSYAYTRAEVVYEWTREPARSVVVAEDGSRLNQYDLLGQTVDSGIVQSSTGEYVVMTTHFHLKRKIGYFVIQTYLPCIMTVILSQVSFWLNRESVPARTVFGVTTVLTMTTLSISARNSLPKVAYATAMDWFIAVCYAFVFSALIEFATVNYFTKSQPARAAKIDRLSRIAFPLLFGIFNLVYWATYLNR Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | MALT lymphoma-associated translocation (MALT1) | 7A41 | 5.95 | |
Target general information Gen name MALT1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MALT lymphoma-associated translocation; Paracaspase Protein family Peptidase C14B family Biochemical class Peptidase Function Protease that enhances BCL10-induced activation of NF-kappa-B by mediating its cleavage. MALT1-dependent BCL10 cleavage plays an important role in T-cell antigen receptor-induced integrin adhesion. Involved in the induction of T helper 17 cells (Th17) differentiation. Cleaves RC3H1 and ZC3H12A in response to T-cell receptor (TCR) stimulation which releases their cooperatively repressed targets to promote Th17 cell differentiation (By similarity). Also mediates cleavage of N4BP1 in T-cells following TCR-mediated activation, leading to N4BP1 inactivation. Also has ubiquitin ligase activity: binds to TRAF6, inducing TRAF6 oligomerization and activation of its ligase activity. Related diseases Immunodeficiency 12 (IMD12) [MIM:615468]: A primary immunodeficiency characterized by onset in infancy of recurrent bacterial and candidal infections resulting in bronchiectasis and growth delay. Manifestations include mastoiditis, aphthous ulcers, cheilitis, gingivitis, esophagitis, gastritis, duodenitis, and meningitis. Levels of absolute lymphocytes and serum immunoglobulins are normal, but specific antibody titers are low despite immunization, and T-cells show impaired proliferative responses to mitogens. {ECO:0000269|PubMed:23727036}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: A chromosomal aberration involving MALT1 is recurrent in low-grade mucosa-associated lymphoid tissue (MALT lymphoma). Translocation t(11;18)(q21;q21) with BIRC2. This translocation is found in approximately 50% of cytogenetically abnormal low-grade MALT lymphoma. {ECO:0000269|PubMed:10339464, ECO:0000269|PubMed:10523859, ECO:0000269|PubMed:10702396, ECO:0000269|PubMed:11090634}. Drugs (DrugBank ID) NA Interacts with O95999; Q9BXL7; Q14790; P48729; Q9Y6K9; Q9UDY8; Q96PU8; Q9H0F6; Q13501; Q9Y4K3; P0CG48; P54252; P46379-2; G5E9A7; P50570-2; Q9BSK4; Q96JP0; P28799; P04792; O60333-2; O14901; O14832; P60891; Q7Z699; O76024 EC number EC 3.4.22.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Chromosomal rearrangement; Cytoplasm; Disease variant; Disulfide bond; Hydrolase; Immunity; Immunoglobulin domain; Nucleus; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Repeat; Ubl conjugation pathway Protein physicochemical properties Chain ID A Molecular weight (Da) 39995.8 Length 354 Aromaticity 0.09 Instability index 25.55 Isoelectric point 5.12 Charge (pH=7) -11.76 2D Binding mode Binding energy (Kcal/mol) -8.11 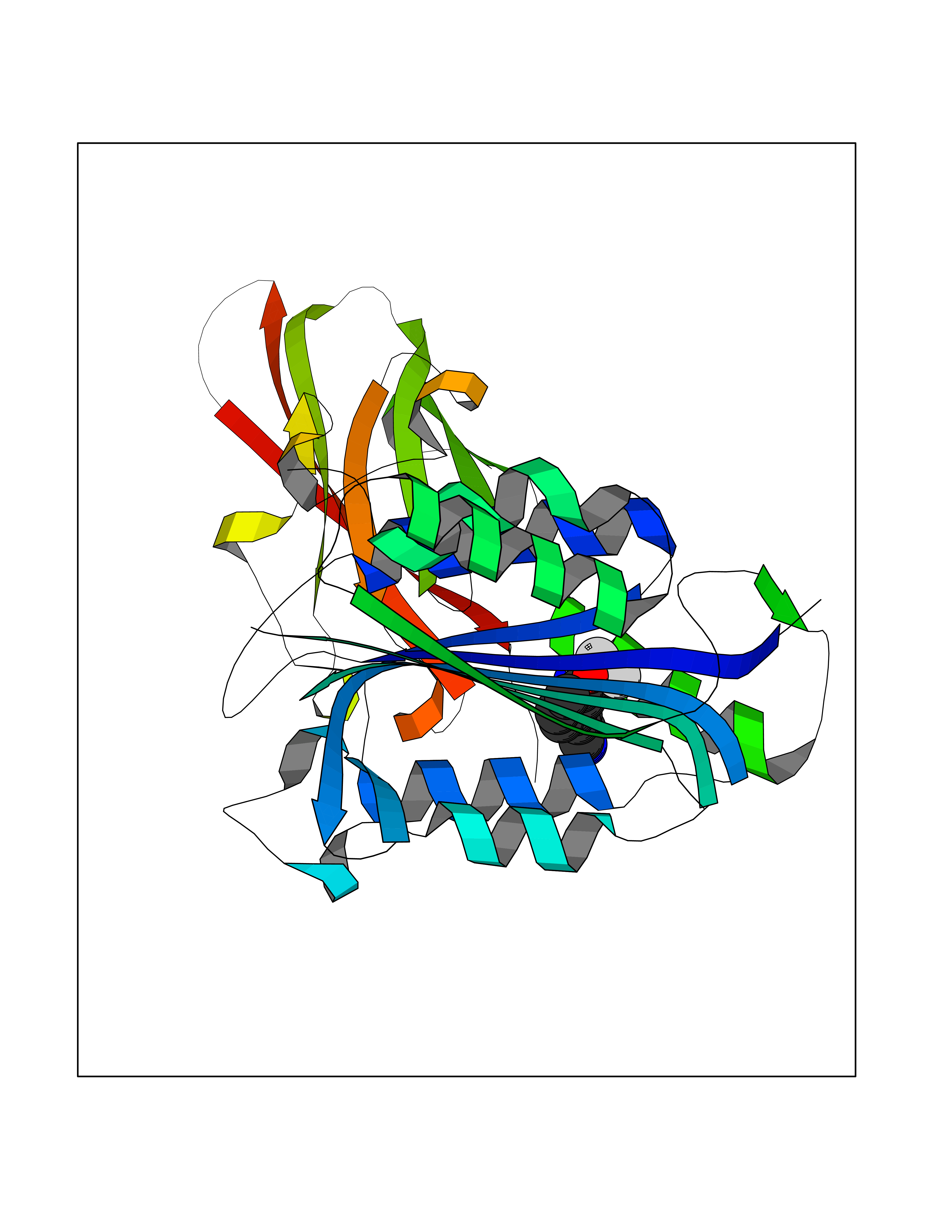 Molscript Map 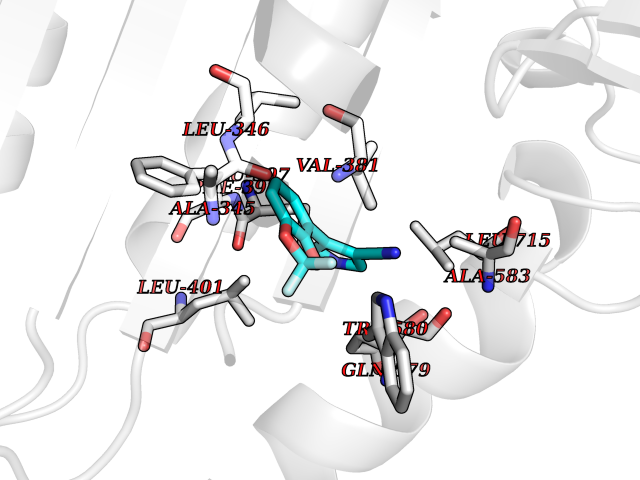 Pymol Map 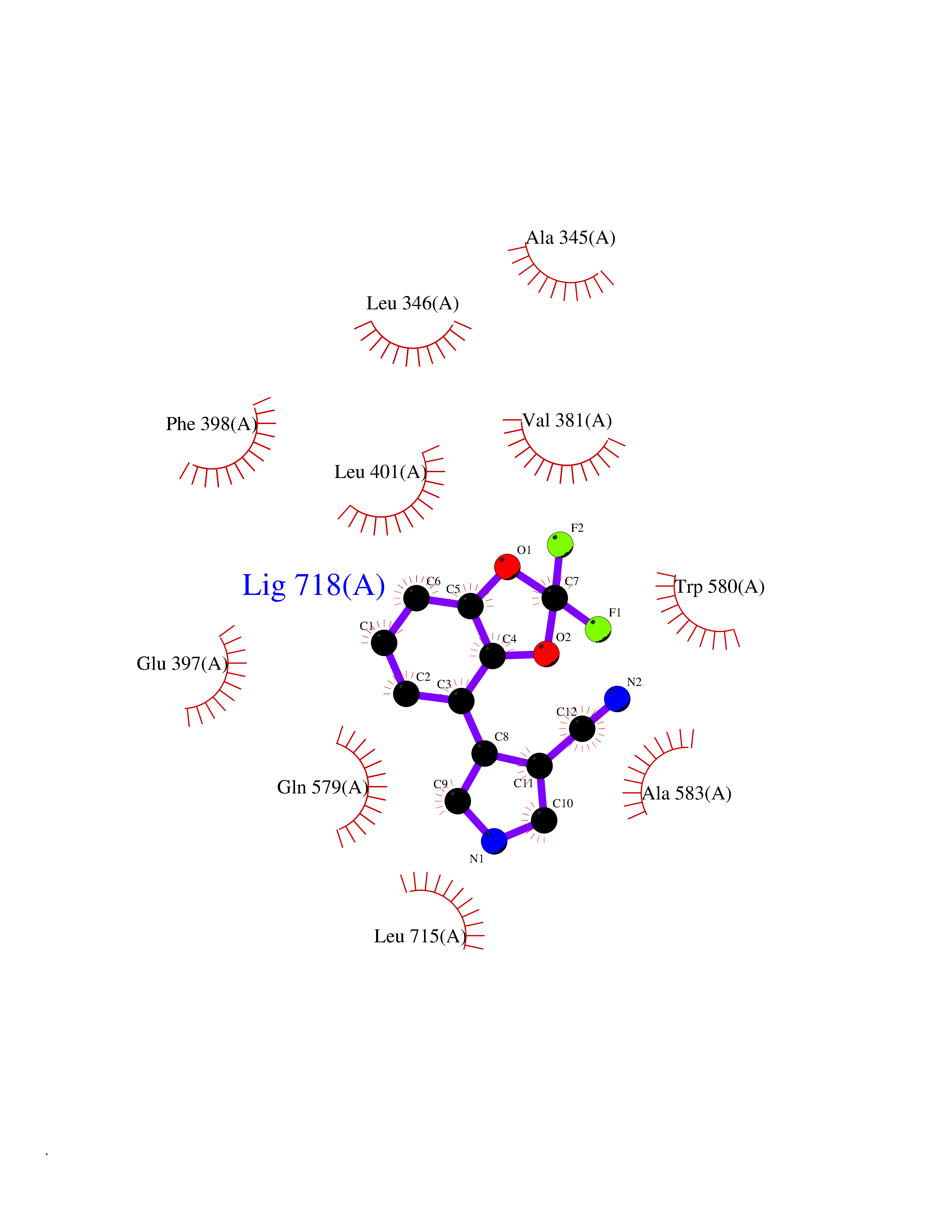 Ligplot Map 3D Binding mode Sequence QPLAKDKVALLIGNMNYREHPKLKAPLVDVYELTNLLRQLDFKVVSLLDLTEYEMRNAVDEFLLLLDKGVYGLLYYAGHGYENFGNSFMVPVDAPNPYRSENCLCVQNILKLMQEKETGLNVFLLDMCRTANIVFGYATCQGGLANGIFMKFLKDRLLEDKKITVLLDEVAEDMGKCHLTKGKQALEIRSSLSEKRALTDPIQGTEYSAESLVRNLQWAKAHELPESMCLKFDCGVQIQLGFAAEFSNVMIIYTSIVYKPPEIIMCDAYVTDFPLDLDIDPKDANKGTPEETGSYLVSKDLPKHCLYTRLSSLQKLKEHLVFTVCLSYQYSGLEDTVEDKQEVNVGKPLIAKLD Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | "Periplasmic trehalase (EC 3.2.1.28) (Alpha,alpha-trehalase) (Alpha,alpha-trehalose glucohydrolase) (Tre37A)" | 2JG0 | 5.94 | |
Target general information Gen name treA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1186;osmA;b1197 Protein family Glycosyl hydrolase 37 family Biochemical class NA Function Provides the cells with the ability to utilize trehalose at high osmolarity by splitting it into glucose molecules that can subsequently be taken up by the phosphotransferase-mediated uptake system. Related diseases SRC kinase activity has been shown to be increased in several tumor tissues and tumor cell lines such as colon carcinoma cells. {ECO:0000269|PubMed:2498394, ECO:0000269|PubMed:3093483}.; DISEASE: Thrombocytopenia 6 (THC6) [MIM:616937]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC6 is an autosomal dominant form. Affected individuals may also have bone abnormalities and an increased risk for myelofibrosis. {ECO:0000269|PubMed:26936507}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.2.1.28 Uniprot keywords 3D-structure; Direct protein sequencing; Glycosidase; Hydrolase; Periplasm; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 57508.9 Length 507 Aromaticity 0.11 Instability index 48.32 Isoelectric point 5.48 Charge (pH=7) -10.13 2D Binding mode Binding energy (Kcal/mol) -8.1 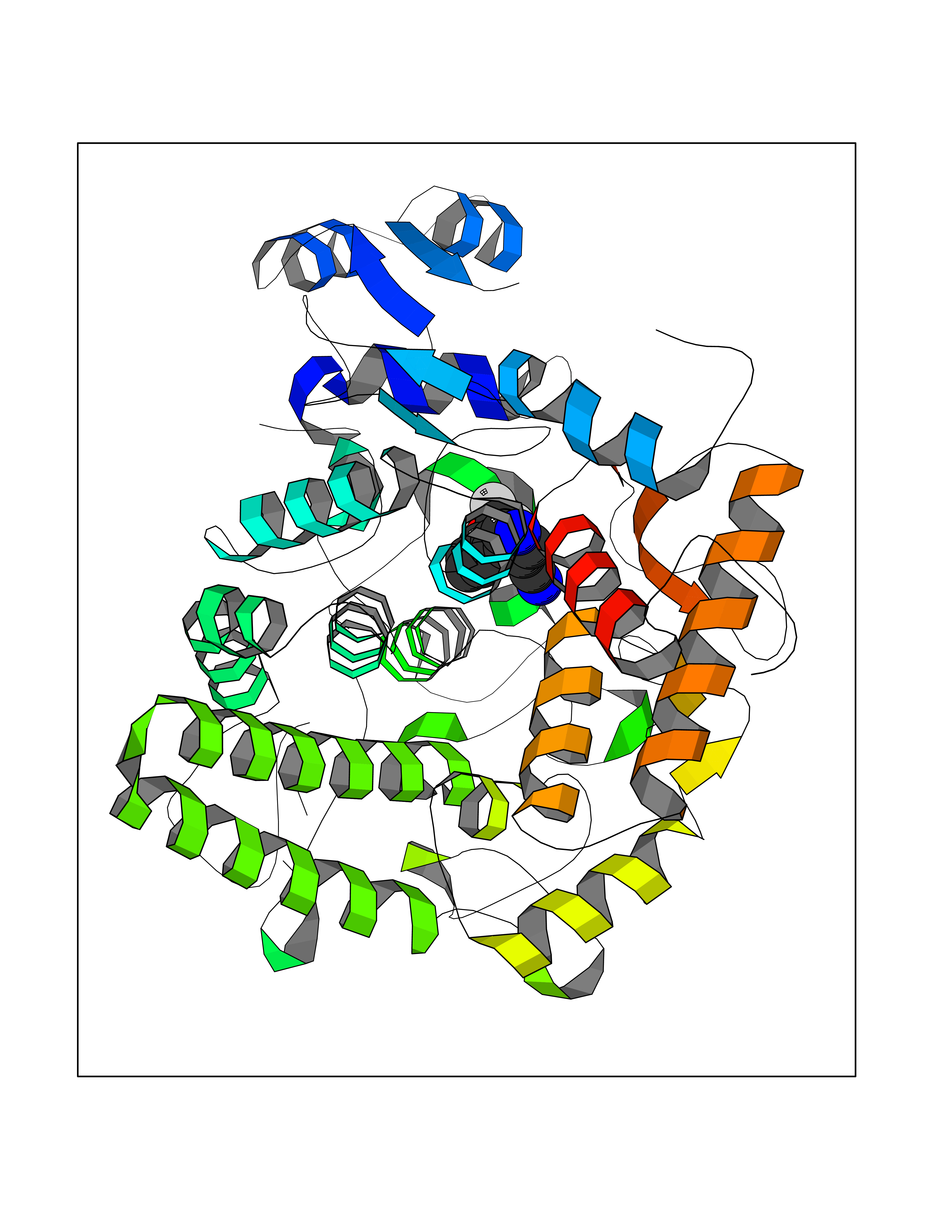 Molscript Map 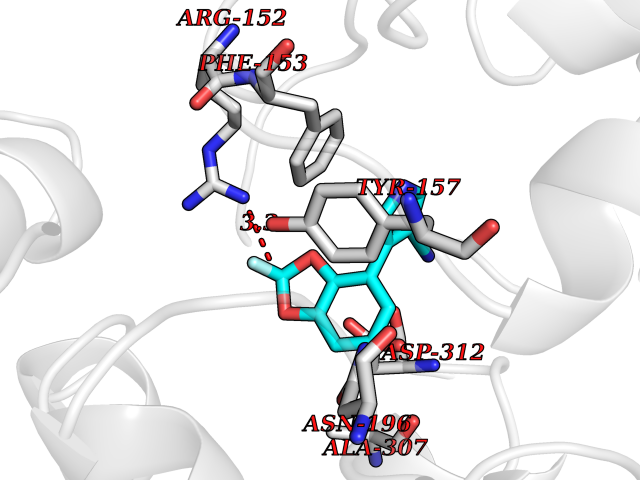 Pymol Map 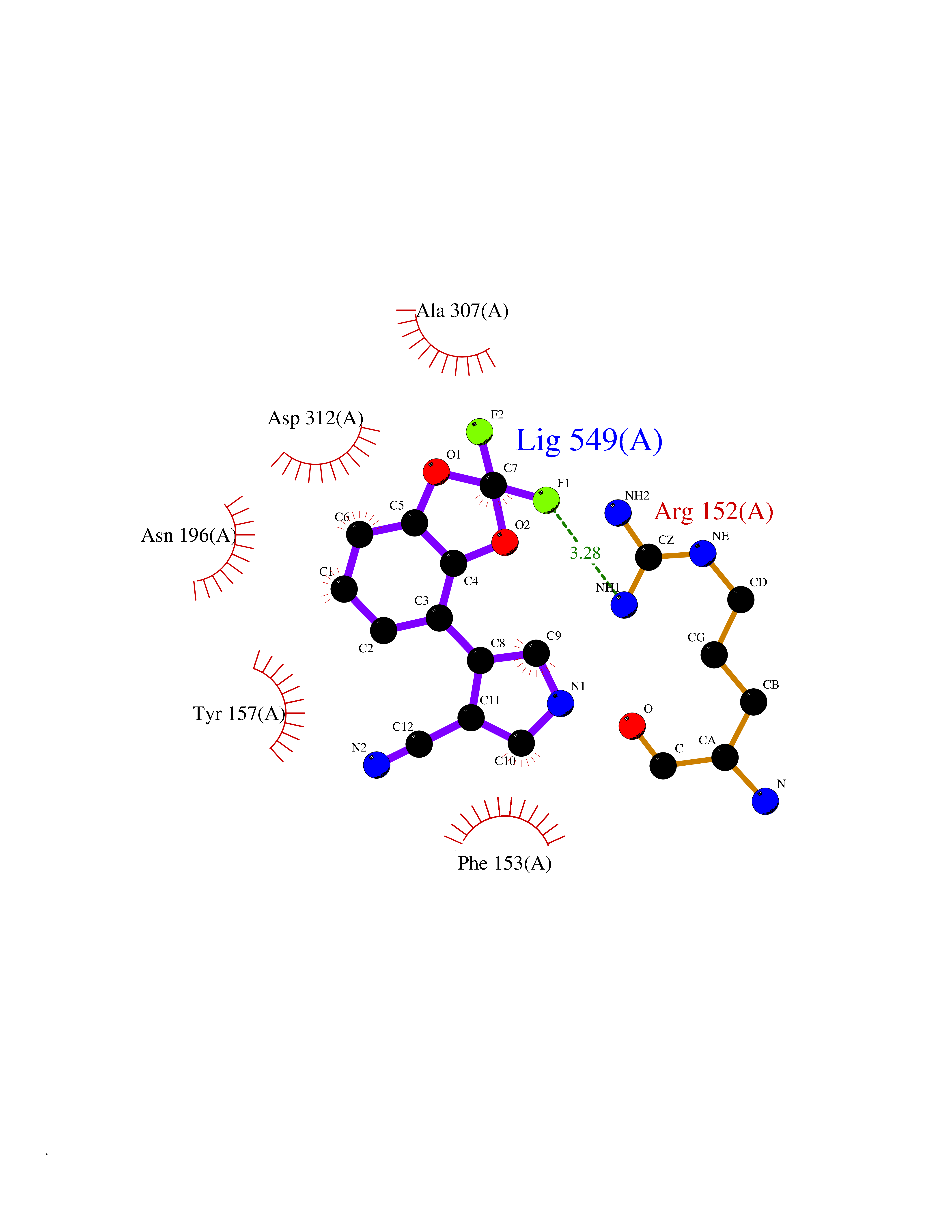 Ligplot Map 3D Binding mode Sequence PQPPDILLGPLFNDVQNAKLFPDQKTFADAVPNSDPLMILADYRMQQNQSGFDLRHFVNVNFTLPKYVPPEGQSLREHIDGLWPVLTRSTENTEKWDSLLPLPEPYVVPGGRFREVYYWDSYFTMLGLAESGHWDKVADMVANFAHEIDTYGHIPNGNRSYYLSRSQPPFFALMVELLAQHEGDAALKQYLPQMQKEYAYWMDGVENLQAGQQEKRVVKLQDGTLLNRYWDDRDTPRPESWVEDIATAKSNPNRPATEIYRDLRSAAASGWDFSSRWMDNPQQLNTLRTTSIVPVDLNSLMFKMEKILARASKAAGDNAMANQYETLANARQKGIEKYLWNDQQGWYADYDLKSHKVRNQLTAAALFPLYVNAAAKDRANKMATATKTHLLQPGGLNTTSVKSGQQWDAPNGWAPLQWVATEGLQNYGQKEVAMDISWHFLTNVQHTYDREKKLVEKYDVSTTGTGGGGGEYPLQDGFGWTNGVTLKMLDLICPKEQPCDNVPATRP Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Adenosine deaminase (EC 3.5.4.4) (S-methyl-5'-thioadenosine deaminase) (EC 3.5.4.31) | 3EWD | 5.94 | |
Target general information Gen name ADA Organism Plasmodium vivax (strain Salvador I) Uniprot ID TTD ID NA Synonyms PVX_111245 Protein family Metallo-dependent hydrolases superfamily, Adenosine and AMP deaminases family Biochemical class NA Function Catalyzes the hydrolytic deamination of adenosine to produce inosine (PubMed:19728741). Unlike mammalian adenosine deaminases, also catalyzes the deamination of 5'-methylthioadenosine (MTA), a by-product of polyamine biosynthesis, to produce 5'-methylthioinosine (MTI) (PubMed:19728741). Plays an essential role in the purine salvage pathway which allows the parasite to use host cell purines for the synthesis of nucleic acids (PubMed:19728741). {ECO:0000269|PubMed:19728741}." Related diseases NA Drugs (DrugBank ID) NA Interacts with NA EC number 3.5.4.31; 3.5.4.4 Uniprot keywords 3D-structure; Hydrolase; Metal-binding; Purine salvage; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 41950.8 Length 364 Aromaticity 0.11 Instability index 27.19 Isoelectric point 5.49 Charge (pH=7) -13.73 2D Binding mode Binding energy (Kcal/mol) -8.1 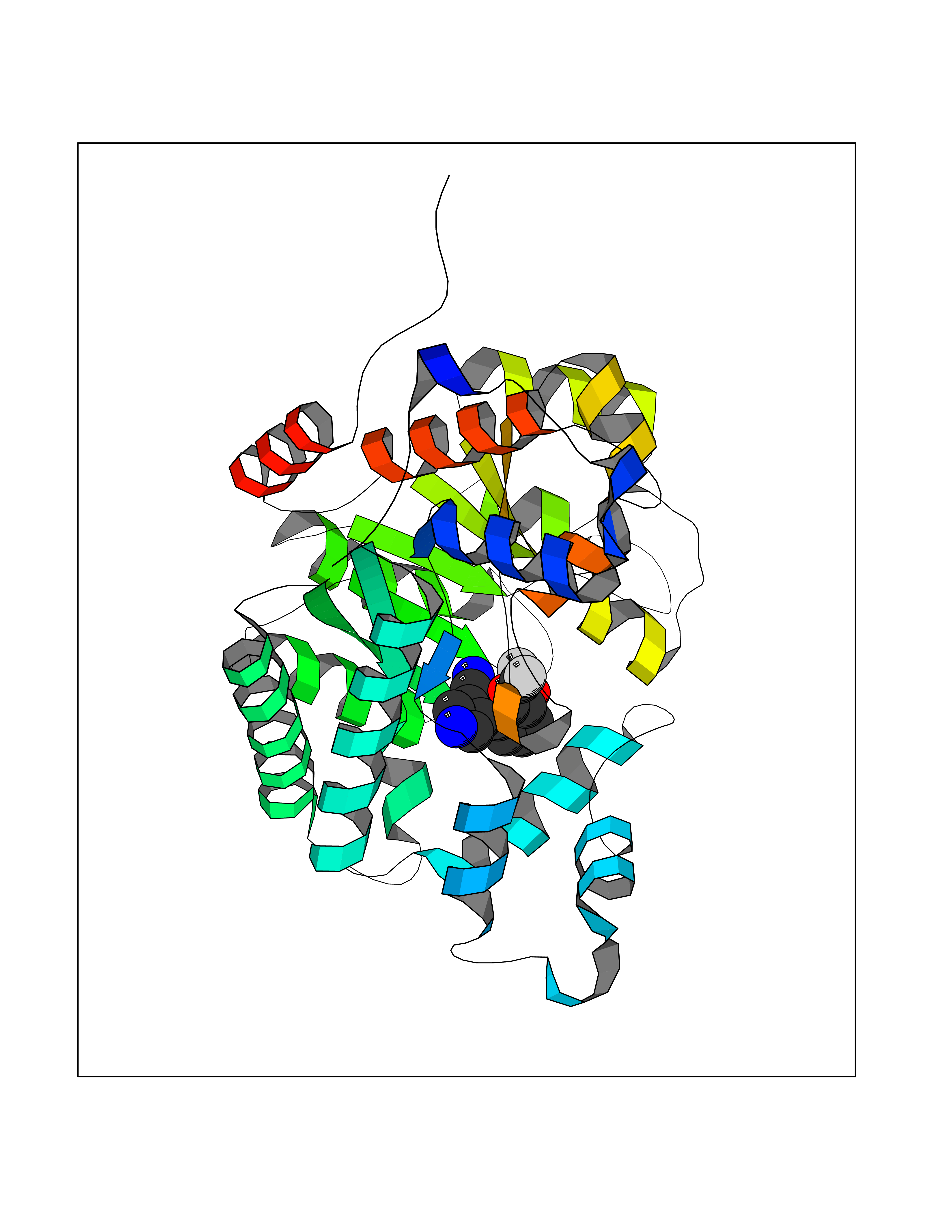 Molscript Map 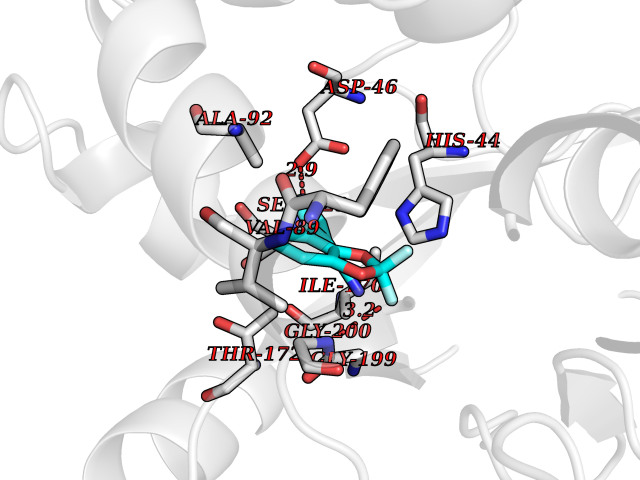 Pymol Map 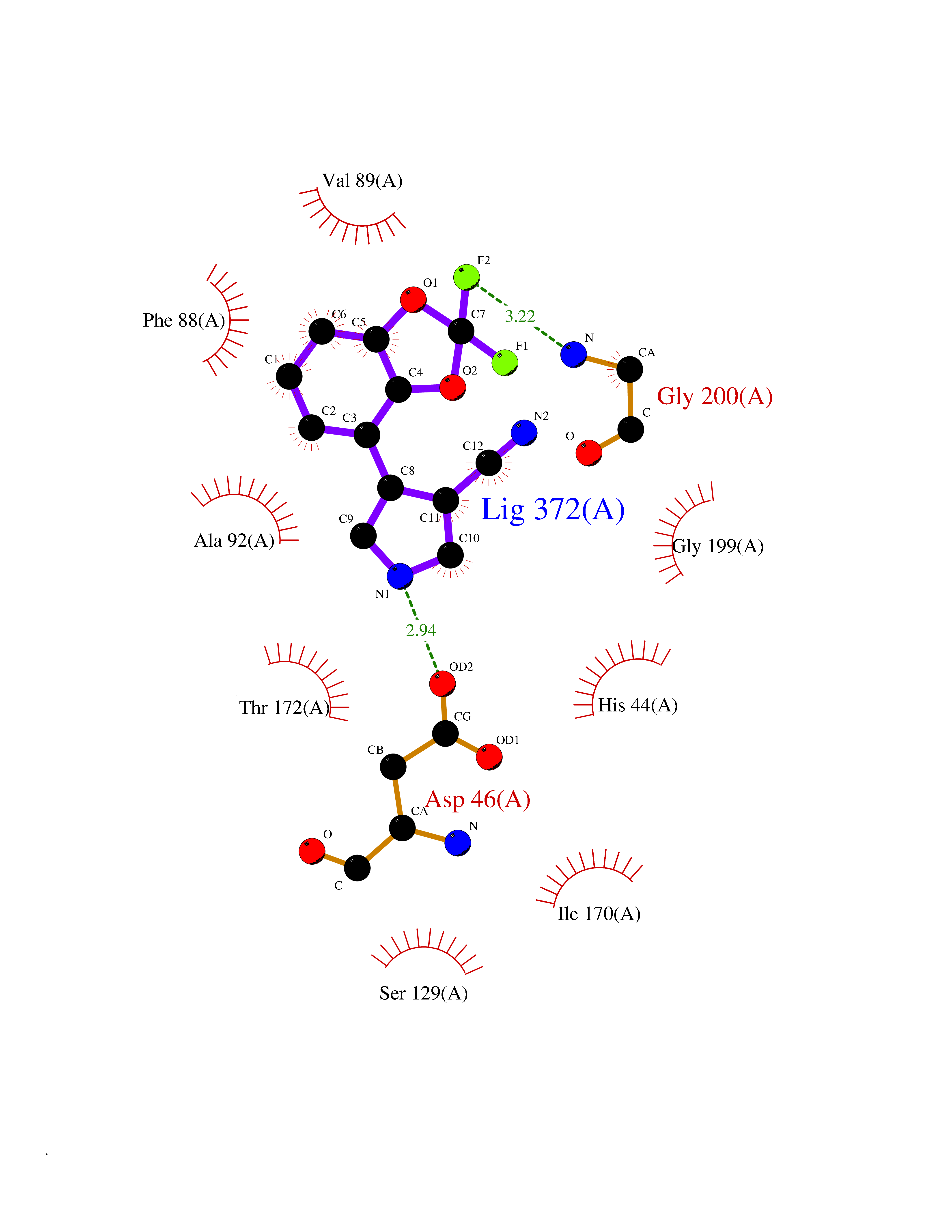 Ligplot Map 3D Binding mode Sequence PIDFLKKEELKNIDLSQMSKKERYKIWKRIPKCELHCHLDLCFSADFFVSCIRKYNLQPNLSDEEVLDYYLFAKGGKSLGEFVEKAIKVADIFHDYEVIEDLAKHAVFNKYKEGVVLMEFRYSPTFVAFKYNLDIELIHQAIVKGIKEVVELLDHKIHVALMCIGTGHEAANIKASADFCLKHKADFVGFDHGGHEVDLKEYKEIFDYVRESGVPLSVHAGEDVTLPNLNTLYSAIQVLKVERIGHGIRVAESQELIDMVKEKNILLEVCPISNVLLKNAKSMDTHPIRQLYDAGVKVSVNSDDPGMFLTNINDDYEELYTHLNFTLEDFMKMNEWALEKSFMDSNIKDKIKNLYFKGEFEAYV Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Serine--pyruvate aminotransferase | 5HHY | 5.94 | |
Target general information Gen name AGXT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms AGT1;SPAT Protein family Class-V pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transferase Function Alanine-glyoxylate transaminase activity.Amino acid binding.Identical protein binding.Protein homodimerization activity.Protein self-association.Pyridoxal phosphate binding.Receptor binding.Serine-pyruvate transaminase activity.Transaminase activity. Related diseases Hyperoxaluria primary 1 (HP1) [MIM:259900]: An inborn error of glyoxylate metabolism characterized by increased excretion of oxalate and glycolate, and progressive tissue accumulation of insoluble calcium oxalate. Affected individuals are at risk for nephrolithiasis, nephrocalcinosis and early onset end-stage renal disease. {ECO:0000269|PubMed:10394939, ECO:0000269|PubMed:10453743, ECO:0000269|PubMed:10541294, ECO:0000269|PubMed:10862087, ECO:0000269|PubMed:10960483, ECO:0000269|PubMed:12559847, ECO:0000269|PubMed:12777626, ECO:0000269|PubMed:1301173, ECO:0000269|PubMed:1349575, ECO:0000269|PubMed:15253729, ECO:0000269|PubMed:15849466, ECO:0000269|PubMed:15961946, ECO:0000269|PubMed:15963748, ECO:0000269|PubMed:16971151, ECO:0000269|PubMed:1703535, ECO:0000269|PubMed:17495019, ECO:0000269|PubMed:2039493, ECO:0000269|PubMed:23229545, ECO:0000269|PubMed:24055001, ECO:0000269|PubMed:24934730, ECO:0000269|PubMed:26149463, ECO:0000269|PubMed:8101040, ECO:0000269|PubMed:9192270, ECO:0000269|PubMed:9604803}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08060; DB00160; DB02079; DB00145; DB04083; DB00114; DB00133 Interacts with Q5BKX5-3; P21549; P0C7T5; Q9NR55; Q9H5F2; Q9UKJ5; A8MQ03; O43281-2; A1KXE4-2; Q5TD97; P49356; P53539; P49639; Q15323; O76011; O76014; Q6A162; Q07627; P60328; Q9BYR8; Q3LI67; Q8IUC2; Q9BYQ4; O60711; Q99750; Q5VZ52; P0DPK4; O43482; P50542-1; O15496; Q6ZR37; Q9NQX0; Q8HWS3; Q5W111-2; Q8WVR3; Q6DKK2 EC number 2.6.1.44; 2.6.1.51 Uniprot keywords 3D-structure; Acetylation; Aminotransferase; Disease variant; Peroxisome; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84537 Length 771 Aromaticity 0.07 Instability index 36.81 Isoelectric point 8.42 Charge (pH=7) 6.81 2D Binding mode Binding energy (Kcal/mol) -8.1 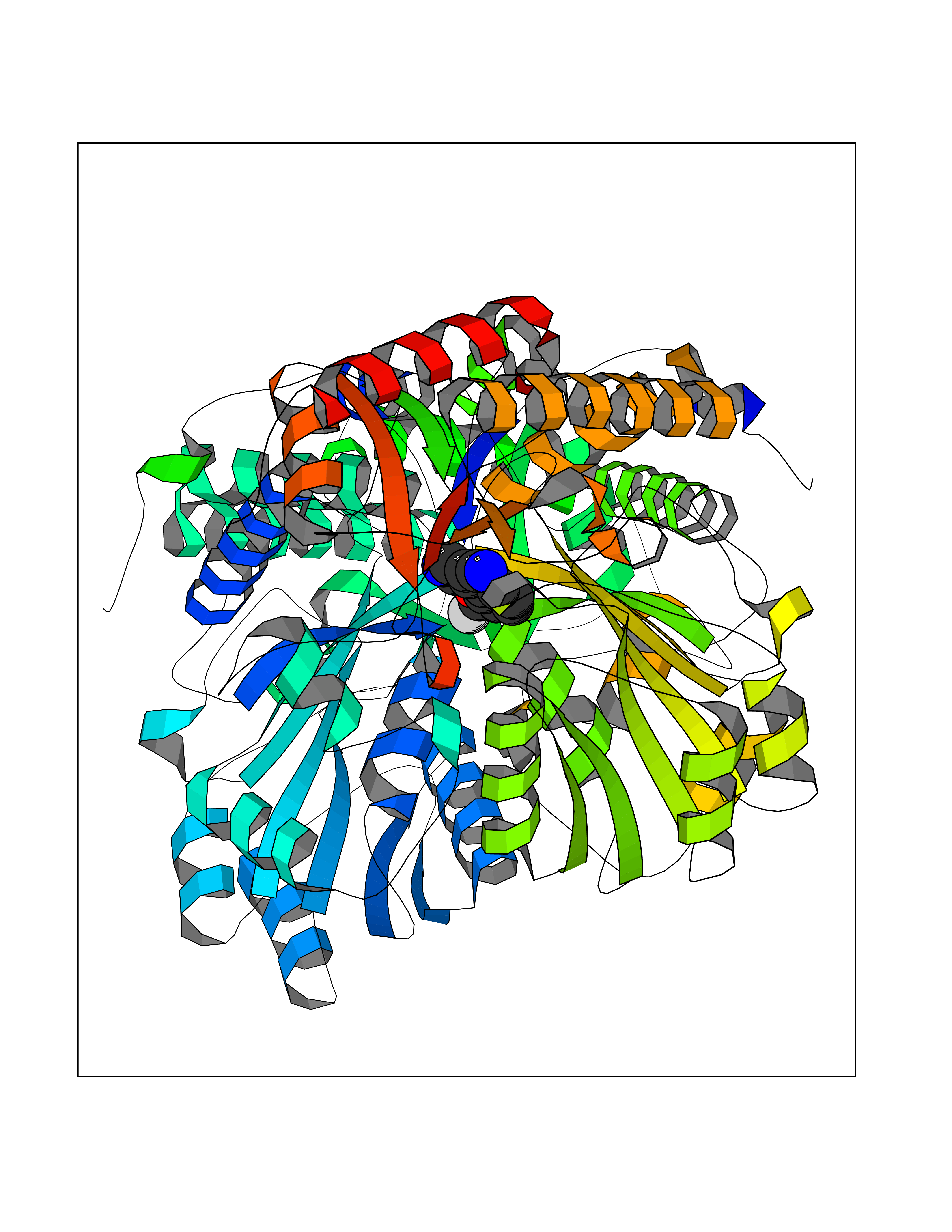 Molscript Map 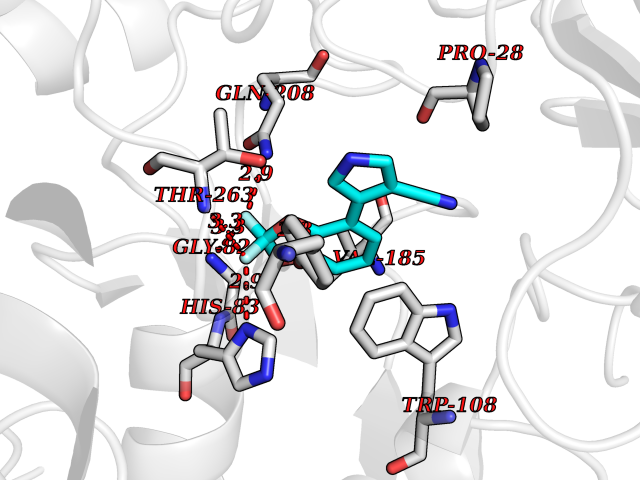 Pymol Map 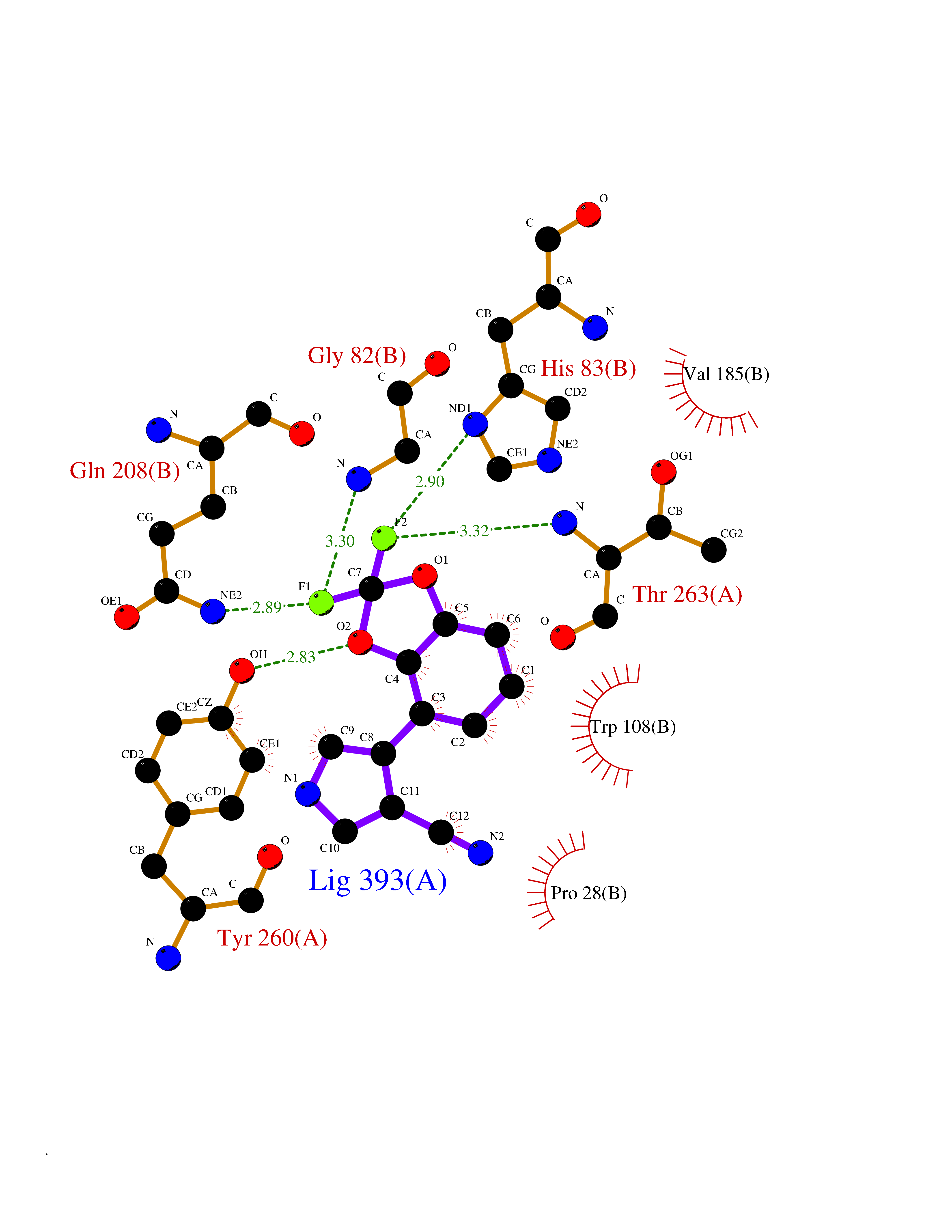 Ligplot Map 3D Binding mode Sequence LLVTPPKALLKPLSIPNQLLLGPGPSNLPPRIMAAGGLQMIGSMSKDMYQIMDEIKEGIQYVFQTRNPLTLVISGSGHCALEAALVNVLEPGDSFLVGANGIWGQRAVDIGERIGARVHPMTKDPGGHYTLQEVEEGLAQHKPVLLFLTHGESSTGVLQPLDGFGELCHRYKCLLLVDSVASLGGTPLYMDRQGIDILYSGSQKALNAPPGTSLISFSDKAKKKMYSRKTKPFSFYLDIKWLANFWGCDDQPRMYHHTIPVISLYSLRESLALIAEQGLENSWRQHREAAAYLHGRLQALGLQLFVKDPALRLPTVTTVAVPAGYDWRDIVSYVIDHFDIEIMGGLGPSTGKVLRIGLLGCNATRENVDRVTEALRAALQHCPKKLLVTPPKALLKPLSIPNQLLLGPGPSNLPPRIMAAGGLQMIGSMSKDMYQIMDEIKEGIQYVFQTRNPLTLVISGSGHCALEAALVNVLEPGDSFLVGANGIWGQRAVDIGERIGARVHPMTKDPGGHYTLQEVEEGLAQHKPVLLFLTHGESSTGVLQPLDGFGELCHRYKCLLLVDSVASLGGTPLYMDRQGIDILYSGSQKALNAPPGTSLISFSDKAKKKMYSRKTKPFSFYLDIKWLANFWGCDDQPRMYHHTIPVISLYSLRESLALIAEQGLENSWRQHREAAAYLHGRLQALGLQLFVKDPALRLPTVTTVAVPAGYDWRDIVSYVIDHFDIEIMGGLGPSTGKVLRIGLLGCNATRENVDRVTEALRAALQHCPKKK Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | 2-iminobutanoate/2-iminopropanoate deaminase | 1ONI | 5.94 | |
Target general information Gen name RIDA Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms HRSP12 Protein family RutC family Biochemical class Translation Function Deaminase activity.Endoribonuclease activity, producing 3'-phosphomonoesters.Long-chain fatty acid binding.Platinum binding.Protein homodimerization activity.RNA binding.Transition metal ion binding.Xenon atom binding. Related diseases Congenital bile acid synthesis defect 2 (CBAS2) [MIM:235555]: A condition characterized by jaundice, intrahepatic cholestasis and hepatic failure. Patients with this liver disease show absence or low levels of chenodeoxycholic acid and cholic acid in plasma and urine. {ECO:0000269|PubMed:12970144, ECO:0000269|PubMed:15030995, ECO:0000269|PubMed:19175828, ECO:0000269|PubMed:20522910}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q8N9N5-2 EC number 3.5.99.10 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Hydrolase; Lipid metabolism; Mitochondrion; Nucleus; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; RNA-binding Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H,I Molecular weight (Da) 42624.3 Length 404 Aromaticity 0.07 Instability index 36.76 Isoelectric point 8.99 Charge (pH=7) 5.46 2D Binding mode Binding energy (Kcal/mol) -8.11 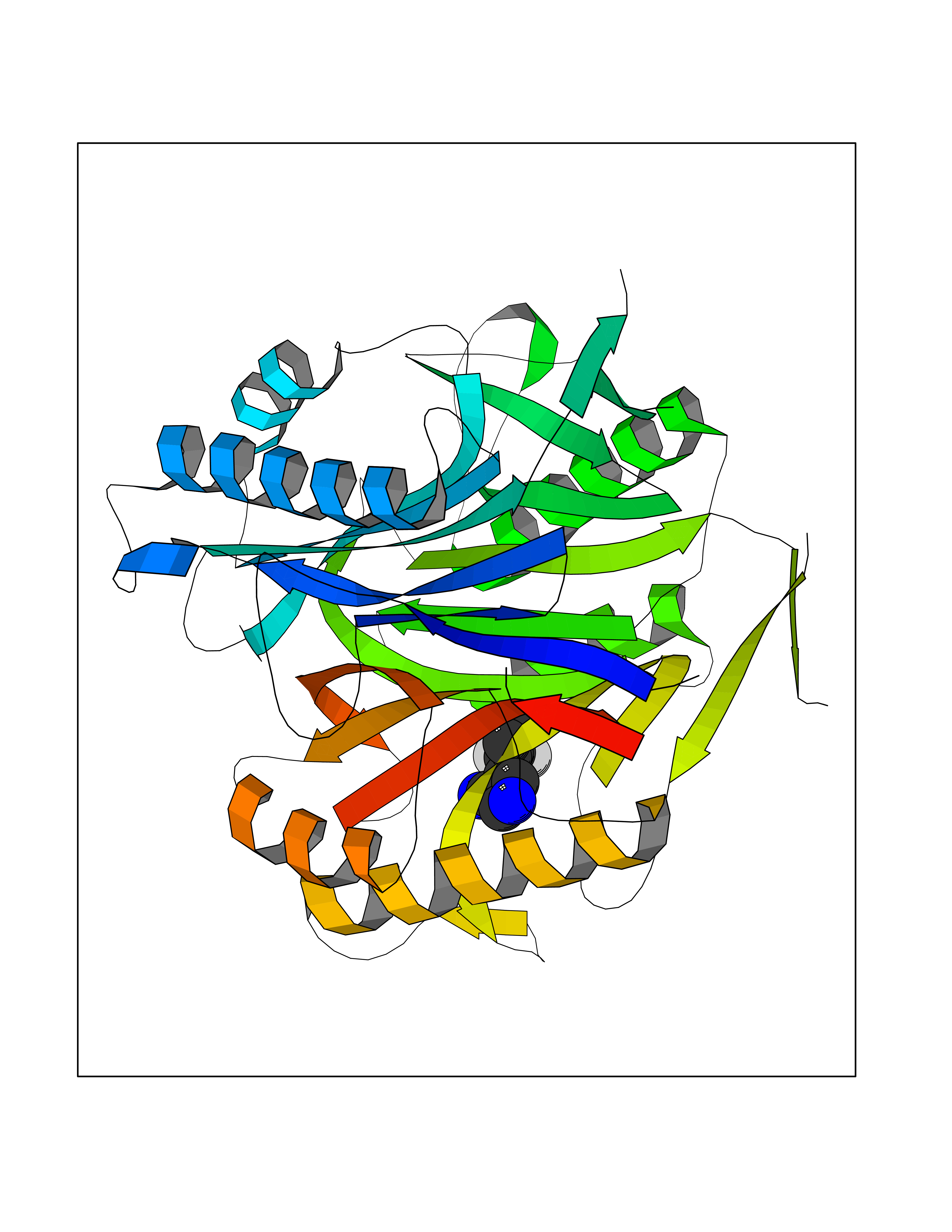 Molscript Map 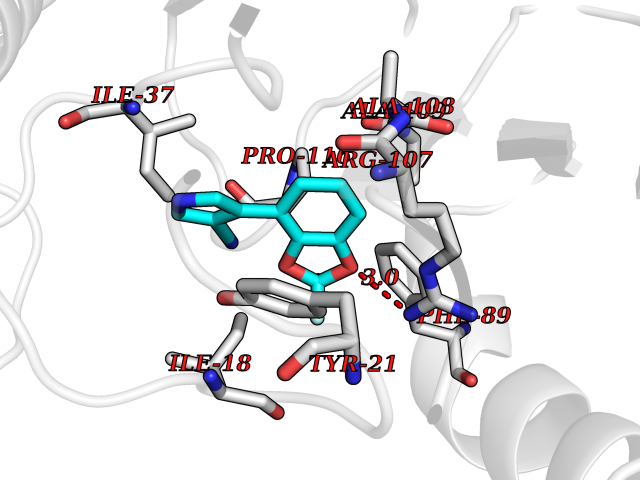 Pymol Map 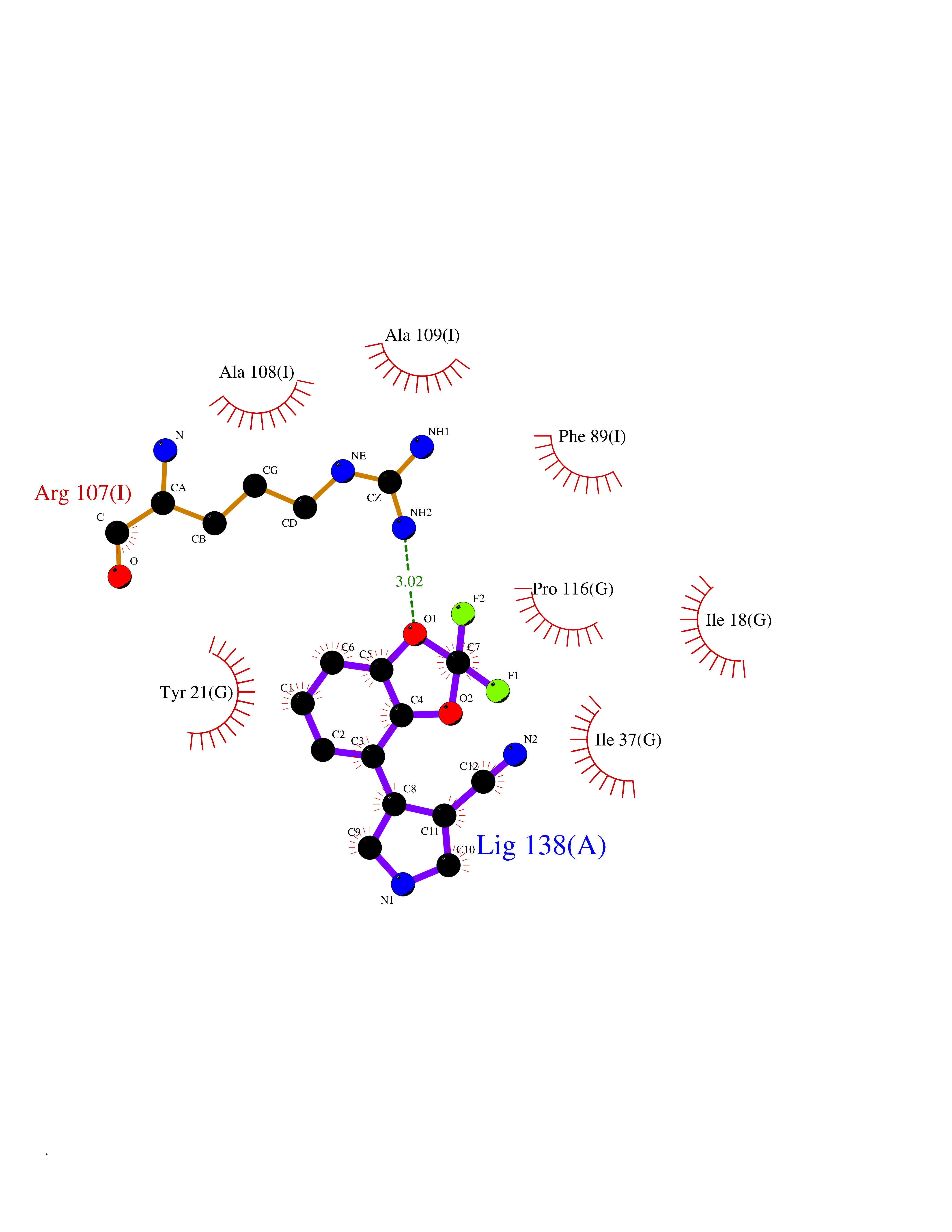 Ligplot Map 3D Binding mode Sequence SSLIRRVISTAKAPGAIGPYSQAVLVDRTIYISGQIGMDPSSGQLVSGGVAEEAKQALKNMGEILKAAGCDFTNVVKTTVLLADINDFNTVNEIYKQYFKSNFPARAAYQVAALPKGSRIEIEAVAIQGPLTTASSSLIRRVISTAKAPGAIGPYSQAVLVDRTIYISGQIGMDPSSGQLVSGGVAEEAKQALKNMGEILKAAGCDFTNVVKTTVLLADINDFNTVNEIYKQYFKSNFPARAAYQVAALPKGSRIEIEAVAIQGPLTTASSSLIRRVISTAKAPGAIGPYSQAVLVDRTIYISGQIGMDPSSGQLVSGGVAEEAKQALKNMGEILKAAGCDFTNVVKTTVLLADINDFNTVNEIYKQYFKSNFPARAAYQVAALPKGSRIEIEAVAIQGPLTTA Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Epithelial discoidin domain receptor 1 (DDR1) | 4BKJ | 5.94 | |
Target general information Gen name DDR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine-protein kinase CAK; Tyrosine kinase DDR; TRKE; TRK E; RTK6; Protein-tyrosine kinase RTK-6; Protein-tyrosine kinase 3A; PTK3A; NTRK4; NEP; Mammary carcinoma kinase 10; MCK-10; HGK2; Epithelial Protein family Protein kinase superfamily, Tyr protein kinase family, Insulin receptor subfamily Biochemical class Kinase Function Collagen binding triggers a signaling pathway that involves SRC and leads to the activation of MAP kinases. Regulates remodeling of the extracellular matrix by up-regulation of the matrix metalloproteinases MMP2, MMP7 and MMP9, and thereby facilitates cell migration and wound healing. Required for normal blastocyst implantation during pregnancy, for normal mammary gland differentiation and normal lactation. Required for normal ear morphology and normal hearing. Promotes smooth muscle cell migration, and thereby contributes to arterial wound healing. Also plays a role in tumor cell invasion. Phosphorylates PTPN11. Tyrosine kinase that functions as cell surface receptor for fibrillar collagen and regulates cell attachment to the extracellular matrix, remodeling of the extracellular matrix, cell migration, differentiation, survival and cell proliferation. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010; DB00619; DB15822 Interacts with Q16832; O43639; Q06124; Q9UHD9 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Calcium; Cell membrane; Direct protein sequencing; Disulfide bond; Glycoprotein; Kinase; Lactation; Membrane; Metal-binding; Nucleotide-binding; Phosphoprotein; Pregnancy; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34061.1 Length 297 Aromaticity 0.1 Instability index 42.8 Isoelectric point 6.32 Charge (pH=7) -2.01 2D Binding mode Binding energy (Kcal/mol) -8.1 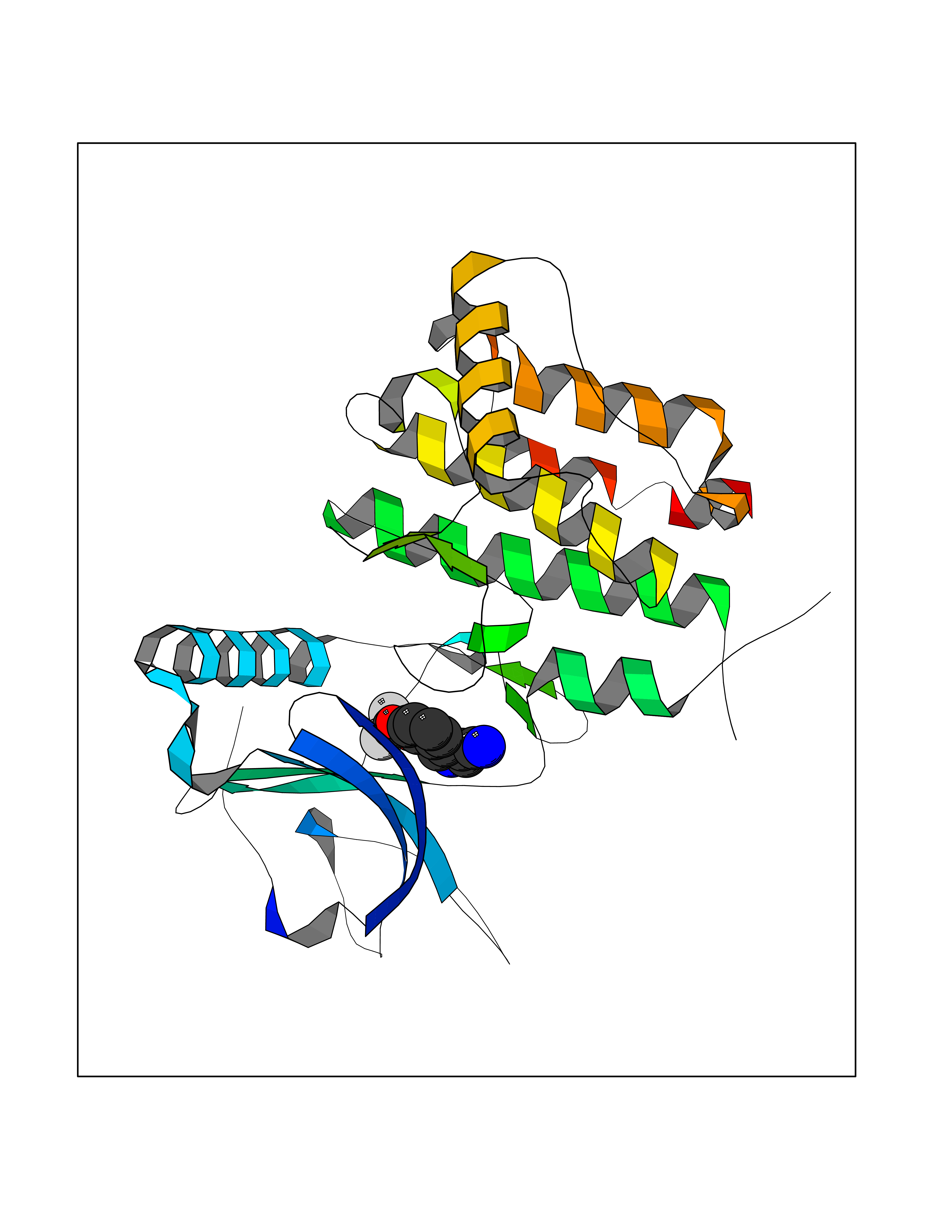 Molscript Map 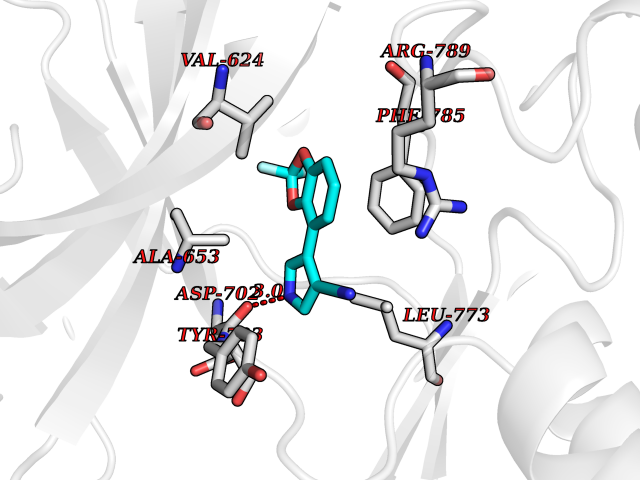 Pymol Map 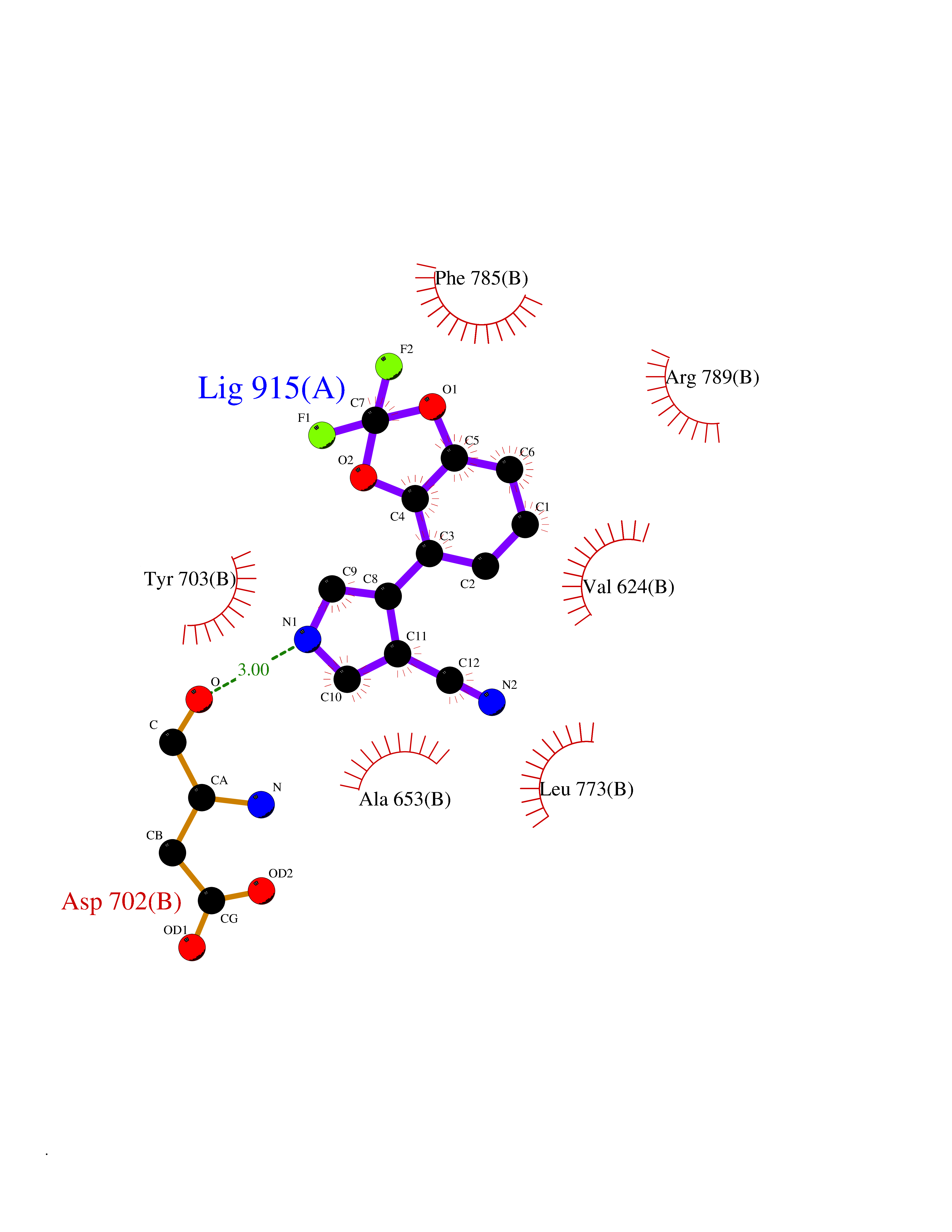 Ligplot Map 3D Binding mode Sequence MPRVDFPRSRLRFKEKLGEGQFGEVHLCEVDSPQDLVSLDFPLNVRKGHPLLVAVKILRPDATKNARNDFLKEVKIMSRLKDPNIIRLLGVCVQDDPLCMITDYMENGDLNQFLSAHQLEDKGPTISYPMLLHVAAQIASGMRYLATLNFVHRDLATRNCLVGENFTIKIADFGMSRNLYAGDYYRAVLPIRWMAWECILMGKFTTASDVWAFGVTLWEVLMLCRAQPFGQLTDEQVIENAGEFFRDQGRQVYLSRPPACPQGLYELMLRCWSRESEQRPPFSQLHRFLAEDALNTV Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Dimethylglycine oxidase | 1PJ5 | 5.94 | |
Target general information Gen name dmg Organism Arthrobacter globiformis Uniprot ID TTD ID NA Synonyms NA Protein family GcvT family Biochemical class Oxidoreductase Function Dimethylglycine oxidase activity.Nucleotide binding. Related diseases Curry-Jones syndrome (CRJS) [MIM:601707]: A multisystem disorder characterized by patchy skin lesions, polysyndactyly, diverse cerebral malformations, unicoronal craniosynostosis, iris colobomas, microphthalmia, and intestinal malrotation with myofibromas or hamartomas. {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. The disease is caused by variants affecting the gene represented in this entry. 8 individuals have been identified with the disease-causing mutation Phe-412 and all were mosaic. The mutation could not be reliably detected in blood, greatest success rates were obtained with affected tissues obtained by invasive procedures. It is thought that the mutation has arisen postzygotically early during embryonic development (PubMed:27236920). This mutation has also been identified in ameloblastoma, medulloblastoma, meningioma, and basal cell carcinoma, and has been reported as the oncogenic driver in some of these tumors (PubMed:24859340). {ECO:0000269|PubMed:24859340, ECO:0000269|PubMed:27236920}. Drugs (DrugBank ID) DB03256; DB03147 Interacts with NA EC number 1.5.3.10 Uniprot keywords 3D-structure; Direct protein sequencing; FAD; Flavoprotein; Nucleotide-binding; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 45912.2 Length 427 Aromaticity 0.07 Instability index 43.46 Isoelectric point 4.83 Charge (pH=7) -20.69 2D Binding mode Binding energy (Kcal/mol) -8.1 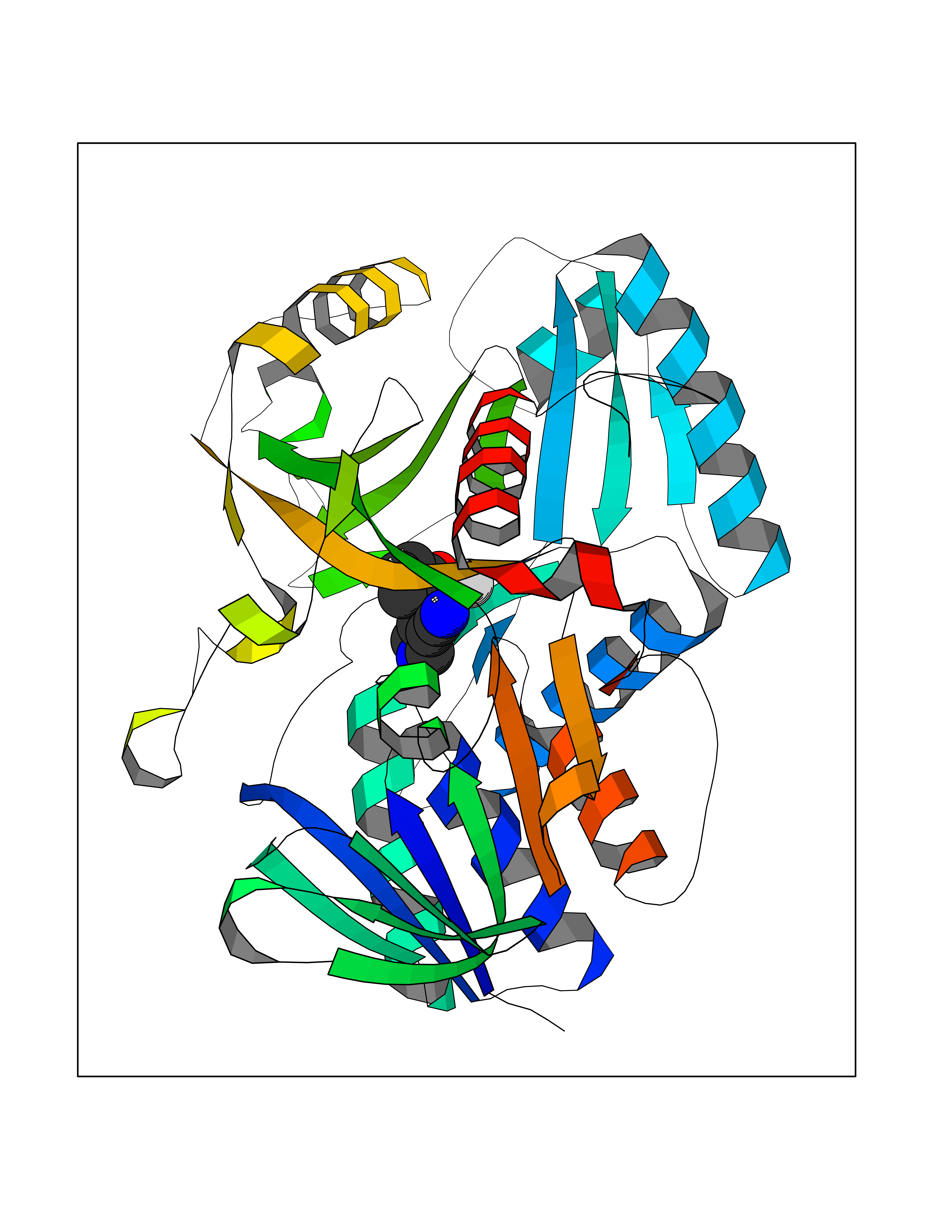 Molscript Map 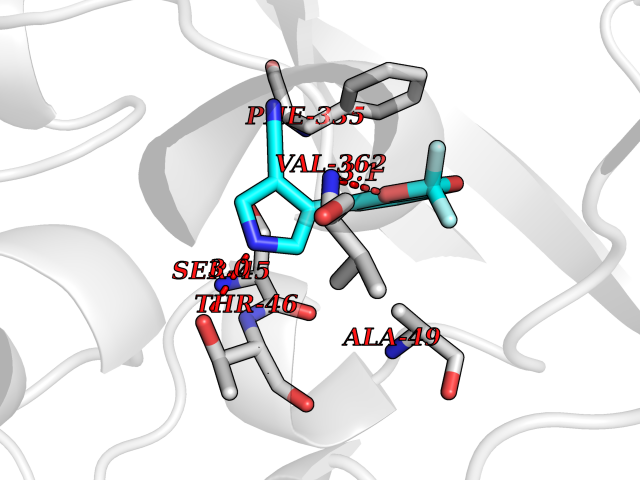 Pymol Map 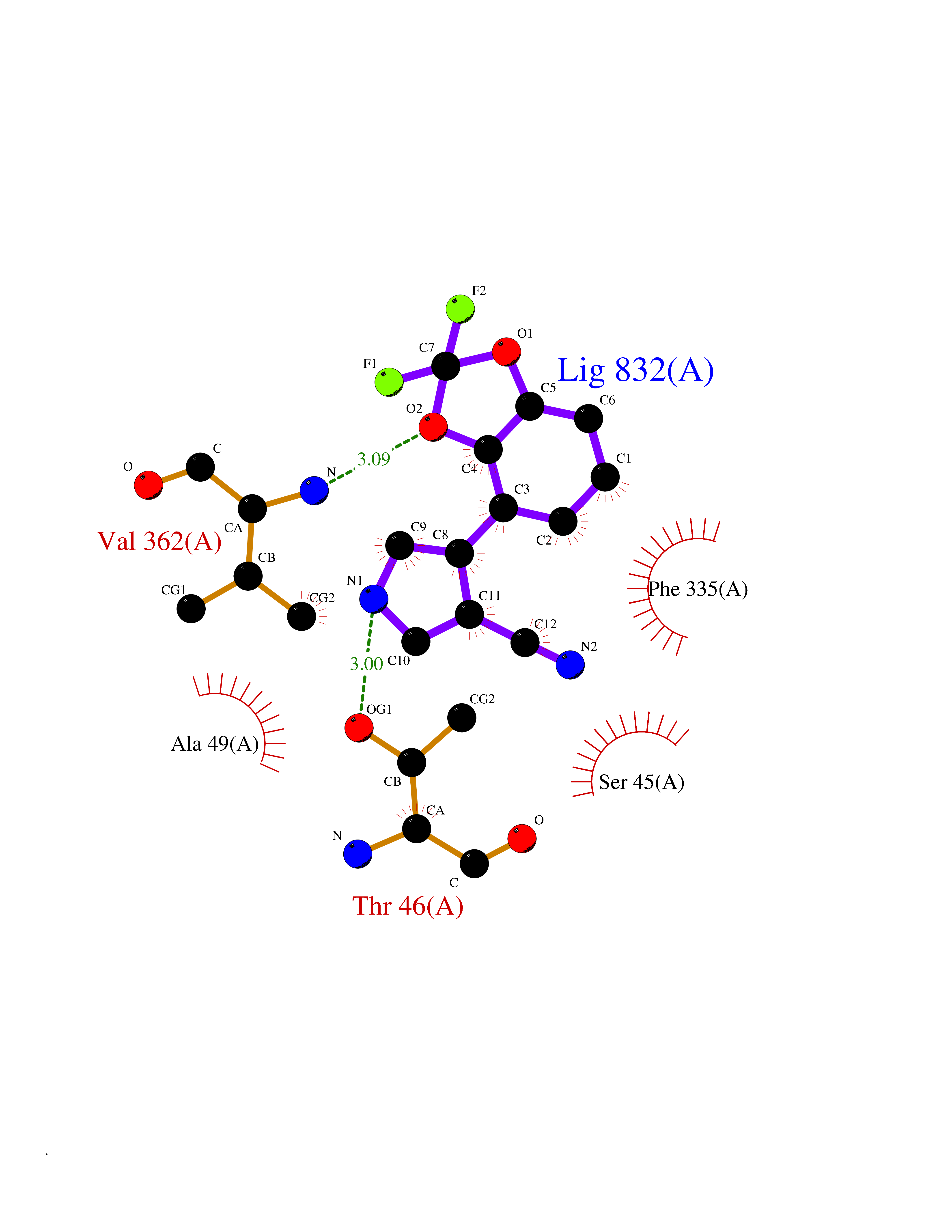 Ligplot Map 3D Binding mode Sequence TPRIVIIGAGIVGTNLADELVTRGWNNITVLDQGPLNMPGGSTSHAPGLVFQTNPSKTMASFAKYTVEKLLSLTEDGVSCFNQVGGLEVATTETRLADLKRKLGYAAAWGIEGRLLSPAECQELYPLLDGENILGGLHVPSDGLASAARAVQLLIKRTESAGVTYRGSTTVTGIEQSGGRVTGVQTADGVIPADIVVSCAGFWGAKIGAMIGMAVPLLPLAHQYVKTTPVPAQQGRNDQPNGARLPILRHQDQDLYYREHGDRYGIGSYAHRPMPVDVDTLGAYAPETVSEHHMPSRLDFTLEDFLPAWEATKQLLPALADSEIEDGFNGIFSFTPDGGPLLGESKELDGFYVAEAVWVTHSAGVAKAMAELLTTGRSETDLGECDITRFEDVQLTPEYVSETSQQNFVEIYDVLHPLQPRLSPRNL Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Influenza Polymerase acidic endonuclease (Influ PA) | 4ZI0 | 5.94 | |
Target general information Gen name Influ PA Organism Influenza A virus (strain A/Puerto Rico/8/1934 H1N1) Uniprot ID TTD ID Synonyms RNA-directed RNA polymerase subunit P2; Polymerase acidic protein Protein family Influenza viruses PA family Biochemical class NA Function Plays an essential role in viral RNA transcription and replication by forming the heterotrimeric polymerase complex together with PB1 and PB2 subunits. The complex transcribes viral mRNAs by using a unique mechanism called cap-snatching. It consists in the hijacking and cleavage of host capped pre-mRNAs. These short capped RNAs are then used as primers for viral mRNAs. The PB2 subunit is responsible for the binding of the 5' cap of cellular pre-mRNAs which are subsequently cleaved after 10-13 nucleotides by the PA subunit that carries the endonuclease activity. Related diseases Lymphatic malformation 8 (LMPHM8) [MIM:618773]: A form of primary lymphedema, a disease characterized by swelling of body parts due to developmental anomalies and functional defects of the lymphatic system. Adult patients with lymphedema may suffer from recurrent local infections. Impaired lymphatic drainage in the fetus can develop into hydrops fetalis, a severe condition characterized by excessive fluid accumulation in more than two fetal extra-vascular compartments and body cavities, placental enlargement and edema, pericardial or pleural effusion, or ascites. LMPHM8 is an autosomal recessive form characterized by onset in utero and fetal death due to non-immune hydrops fetalis. {ECO:0000269|PubMed:30115739}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB13997 Interacts with P03485; P03466; P03431 EC number EC 3.1.-.- Uniprot keywords 3D-structure; Cap snatching; Endonuclease; Eukaryotic host gene expression shutoff by virus; Eukaryotic host transcription shutoff by virus; Host cytoplasm; Host gene expression shutoff by virus; Host nucleus; Host-virus interaction; Hydrolase; Inhibition of host RNA polymerase II by virus; Manganese; Metal-binding; Nuclease; Phosphoprotein; Reference proteome; Ribosomal frameshifting Protein physicochemical properties Chain ID A Molecular weight (Da) 21288.1 Length 181 Aromaticity 0.11 Instability index 49.23 Isoelectric point 6.27 Charge (pH=7) -1.8 2D Binding mode Binding energy (Kcal/mol) -8.11 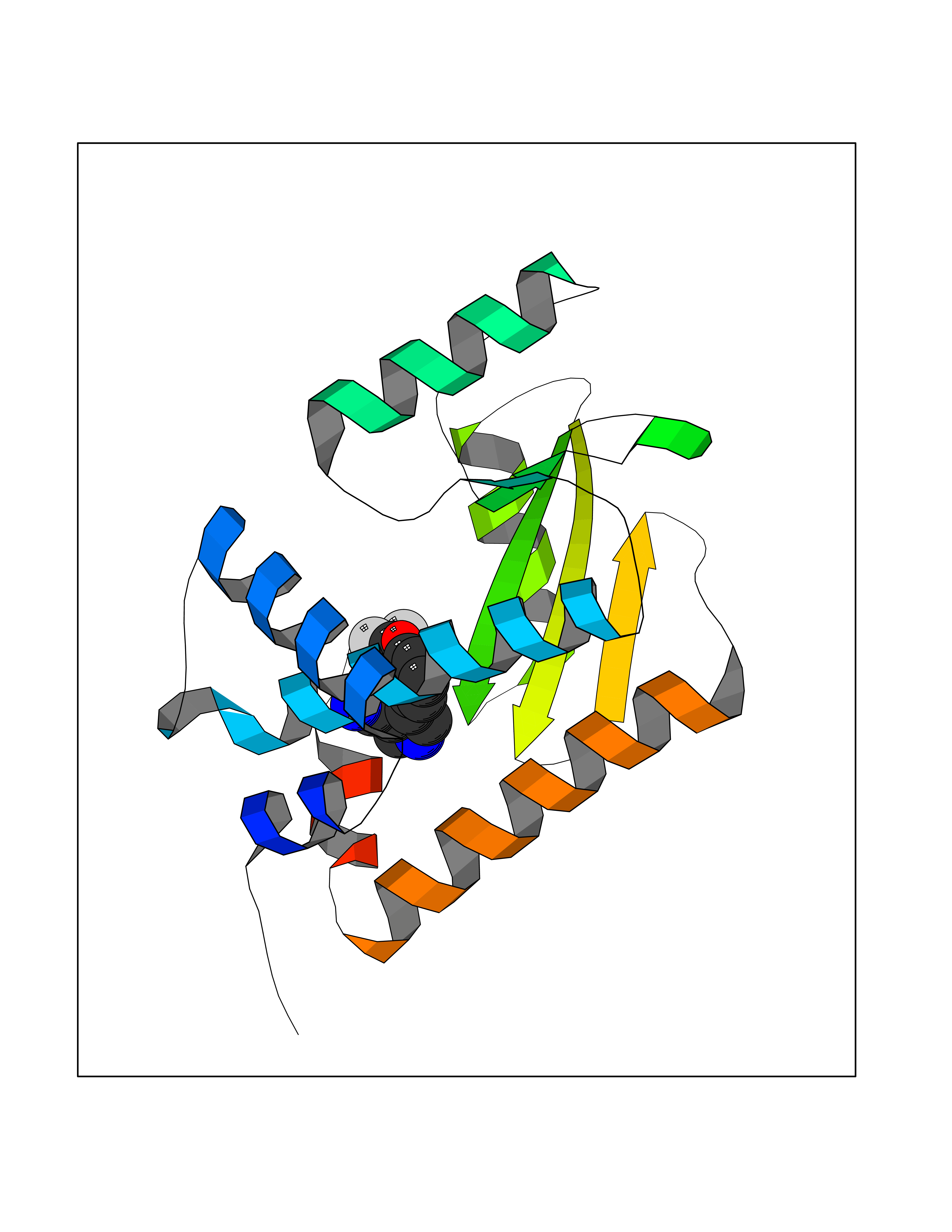 Molscript Map 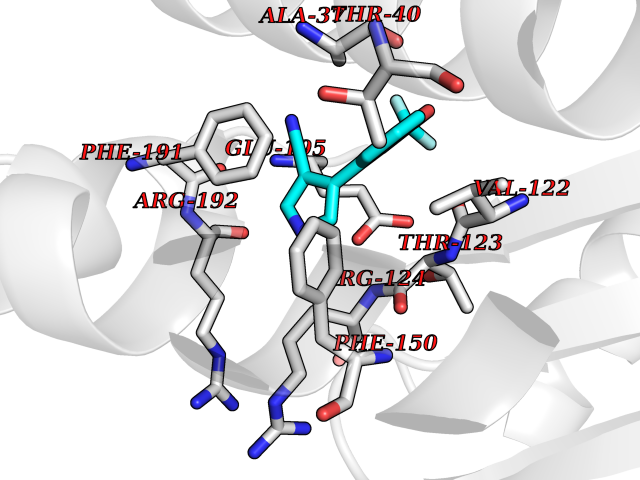 Pymol Map 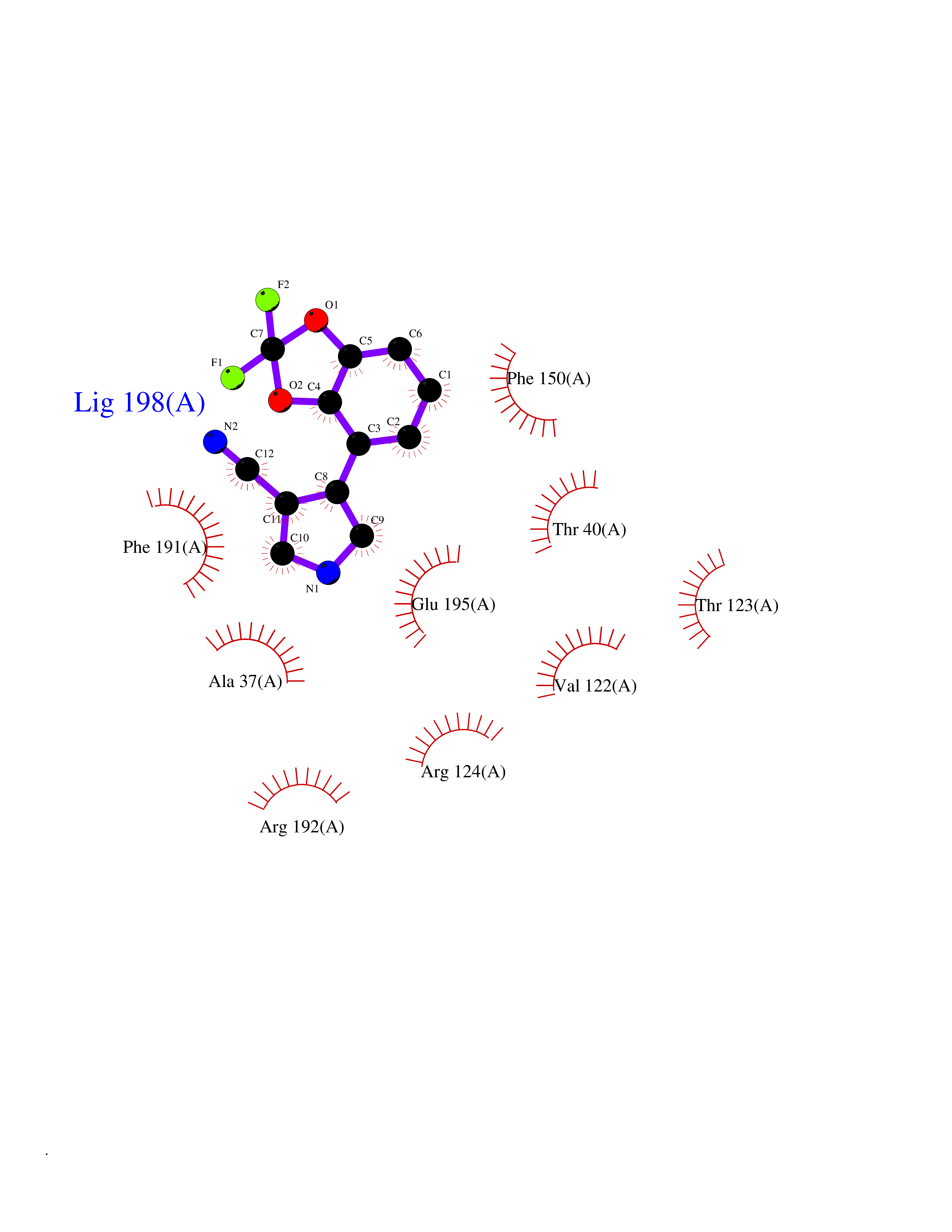 Ligplot Map 3D Binding mode Sequence GPLGSMEDFVRQCFNPMIVELAEKTMKEYGEDLKIETNKFAAICTHLEVCFMYSDASKHRFEIIEGRDRTMAWTVVNSICNTTGAEKPKFLPDLYDYKENRFIEIGVTRREVHIYYLEKANKIKSEKTHIHIFSFTGEEMATKADYTLDEESRARIKTRLFTIRQEMASRGLWDSFRQSER Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Diamine oxidase (AOC1) | 3HII | 5.94 | |
Target general information Gen name AOC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Kidney amine oxidase; KAO; Histaminase; Amiloride-binding protein; AOC1; ABP Protein family Copper/topaquinone oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the degradation of compounds such as putrescine, histamine, spermine, and spermidine, substances involved in allergic and immune responses, cell proliferation, tissue differentiation, tumor formation, and possibly apoptosis. Placental DAO is thought to play a role in the regulation of the female reproductive function. Related diseases Lichtenstein-Knorr syndrome (LIKNS) [MIM:616291]: An autosomal recessive neurologic disorder characterized by progressive cerebellar ataxia and severe progressive sensorineural hearing loss. {ECO:0000269|PubMed:25205112, ECO:0000269|PubMed:30237576}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00594; DB01373; DB09130; DB03608; DB05383 Interacts with Q15038; O75593; Q8IUC2; Q96HA8; Q7Z3K3; Q6ZRY4; Q01085-2; O43711; Q96K80 EC number EC 1.4.3.22 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Copper; Direct protein sequencing; Disulfide bond; Glycoprotein; Heparin-binding; Membrane; Metal-binding; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal; TPQ Protein physicochemical properties Chain ID A,B Molecular weight (Da) 162607 Length 1425 Aromaticity 0.13 Instability index 45.72 Isoelectric point 6.76 Charge (pH=7) -4.07 2D Binding mode Binding energy (Kcal/mol) -8.11  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence PRKAGVFSDLSNQELKAVHSFLWSKKELRLQPSSTTTMAKNTVFLIEMLLPKKYHVLRFLDKGERHPVREARAVIFFGDQEHPNVTEFAVGPLPGPCYMRALSPRPGYQSSWASRPISTAEYALLYHTLQEATKPLHQFFLNTTGFSFQDCHDRCLAFTDVAPRGVASGQRRSWLIIQRYVEGYFLHPTGLELLVDHGSTDAGHWAVEQVWYNGKFYGSPEELARKYADGEVDVVVLEDPLEPPLFSSHKPRGDFPSPIHVSGPRLVQPHGPRFRLEGNAVLYGGWSFAFRLRSSSGLQVLNVHFGGERIAYEVSVQEAVALYGGHTPAGMQTKYLDVGWGLGSVTHELAPGIDCPETATFLDTFHYYDADDPVHYPRALCLFEMPTGVPLRRHFNSNFKGGFNFYAGLKGQVLVLRTTSTVYNXDYIWDFIFYPNGVMEAKMHATGYVHATFYTPEGLRHGTRLHTHLIGNIHTHLVHYRVDLDVAGTKNSFQTLQMKLENITNPWSPRHRVVQPTLEQTQYSWERQAAFRFKRKLPKYLLFTSPQENPWGHKRSYRLQIHSMADQVLPPGWQEEQAITWARYPLAVTKYRESELCSSSIYHQNDPWDPPVVFEQFLHNNENIENEDLVAWVTVGFLHIPHSEDIPNTATPGNSVGFLLRPFNFFPEDPSLASRDTVIVWPRDNGPNYVQRWIPEDRDCSMPPPFSYNGTYRPVRKAGVFSDLSNQELKAVHSFLWSKKELRLQPSSTTTMAKNTVFLIEMLLPKKYHVLRFLDKGERHPVREARAVIFFGDQEHPNVTEFAVGPLPGPCYMRALSPRPGYQSSWASRPISTAEYALLYHTLQEATKPLHQFFLNTTGFSFQDCHDRCLAFTDVAPRGVASGQRRSWLIIQRYVEGYFLHPTGLELLVDHGSTDAGHWAVEQVWYNGKFYGSPEELARKYADGEVDVVVLEPPLFSSHKPRGDFPSPIHVSGPRLVQPHGPRFRLEGNAVLYGGWSFAFRLRSSSGLQVLNVHFGGERIAYEVSVQEAVALYGGHTPAGMQTKYLDVGWGLGSVTHELAPGIDCPETATFLDTFHYYDADDPVHYPRALCLFEMPTGVPLRRHFNSNFKGGFNFYAGLKGQVLVLRTTSTVYNXDYIWDFIFYPNGVMEAKMHATGYVHATFYTPEGLRHGTRLHTHLIGNIHTHLVHYRVDLDVAGTKNSFQTLQMKLENITNPWSPRHRVVQPTLEQTQYSWERQAAFRFKRKLPKYLLFTSPQENPWGHKRSYRLQIHSMADQVLPPGWQEEQAITWARYPLAVTKYRESELCSSSIYHQNDPWDPPVVFEQFLHNNENIENEDLVAWVTVGFLHIPHSEDIPNTATPGNSVGFLLRPFNFFPEDPSLASRDTVIVWPRDNGPNYVQRWIPEDRDCSMPPPFSYNGTYRPV Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Phosphodiesterase 3A (PDE3A) | 7EG0 | 5.94 | |
Target general information Gen name PDE3A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cGMP-inhibited 3',5'-cyclic phosphodiesterase A; Phosphodiesterase 3A; Cyclic GMP-inhibited phosphodiesterase A; Cyclic GMP inhibited phosphodiesterase A; CGI-PDE A Protein family Cyclic nucleotide phosphodiesterase family, PDE3 subfamily Biochemical class Phosphoric diester hydrolase Function Cyclic nucleotide phosphodiesterase with a dual-specificity for the second messengers cAMP and cGMP, which are key regulators of many important physiological processes. Related diseases Hypertension and brachydactyly syndrome (HTNB) [MIM:112410]: A syndrome characterized by brachydactyly type E, severe salt-independent but age-dependent hypertension, an increased fibroblast growth rate, neurovascular contact at the rostral-ventrolateral medulla, and altered baroreflex blood pressure regulation. It results in death from stroke before age 50 years when untreated. Brachydactyly type E is characterized by shortening of the fingers mainly in the metacarpals and metatarsals. {ECO:0000269|PubMed:25961942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01223; DB01427; DB00261; DB00201; DB01166; DB04880; DB05266; DB00922; DB00235; DB01303; DB00277; DB08811; DB09283 Interacts with Q9Y6D6; Q9Y6D5 EC number EC 3.1.4.17 Uniprot keywords 3D-structure; cAMP; cGMP; Cytoplasm; Direct protein sequencing; Disease variant; Hydrolase; Isopeptide bond; Manganese; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 108171 Length 939 Aromaticity 0.11 Instability index 39.75 Isoelectric point 6.53 Charge (pH=7) -4.15 2D Binding mode Binding energy (Kcal/mol) -8.1  Molscript Map  Pymol Map 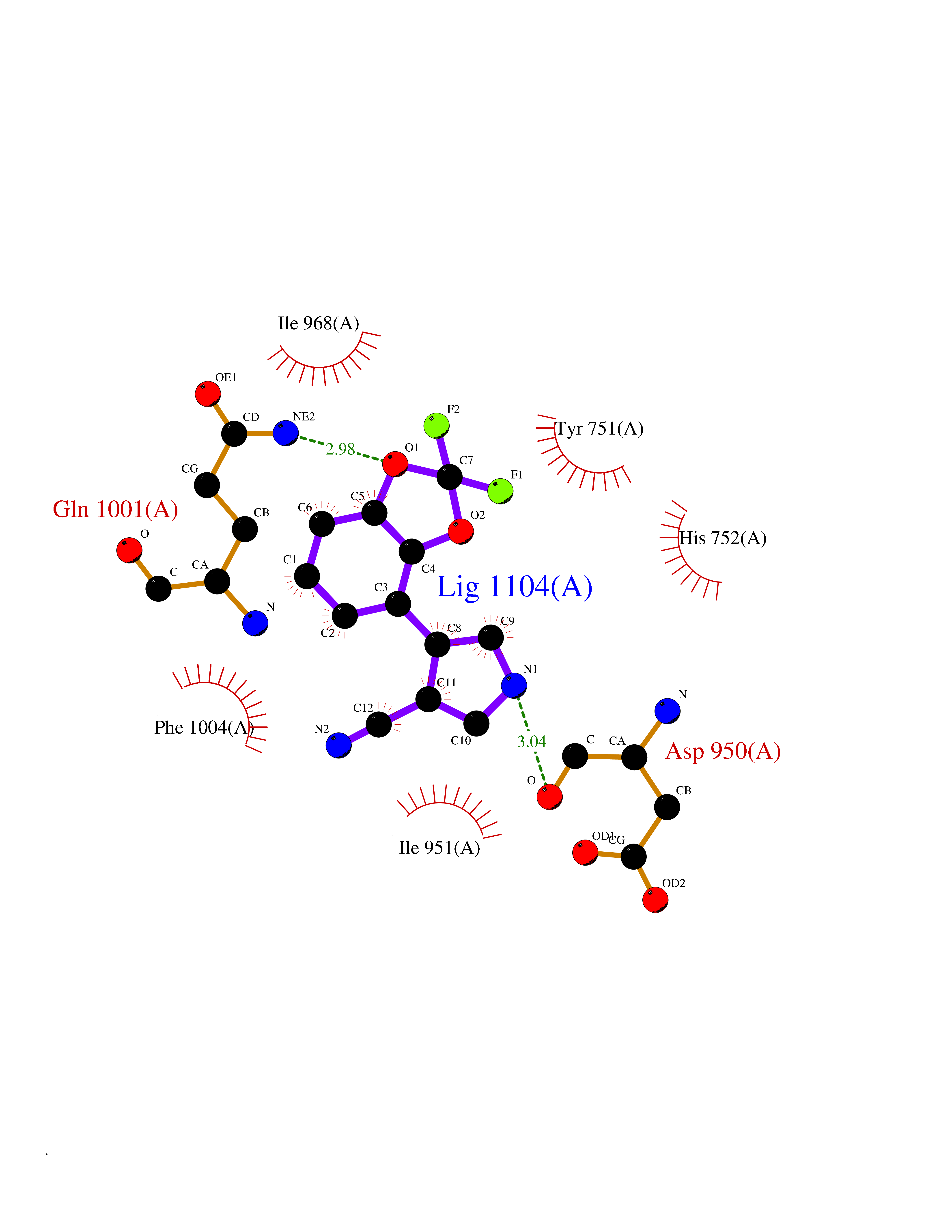 Ligplot Map 3D Binding mode Sequence KPILAPEPLVMDNLDSIMEQLNTWNFPIFDLVENIGRKCGRILSQVSYRLFEDMGLFEAFKIPIREFMNYFHALEIGYRDIPYHNRIHATDVLHAVWYLTTQPIPGLSTVGYVFSKTYNVTDDKYGCLSGNIPALELMALYVAAAMHDYDHPGRTNAFLVATSAPQAVLYNDRSVLENHHAAAAWNLFMSRPEYNFLINLDHVEFKHFRFLVIEAILATDLKKHFDFVAKFNGKVNDDVGIDWTNENDRLLVCQMCIKLADINGPAKCKELHLQWTDGIVNEFYEQGDEEASLGLPISPFMDRSAPQLANLQESFISHIVGPLCNSYDSAGLMPGKWVEKIYCQITQHLLQNHKMWKKVIEEEQRLAGIENQNISVDLETNYAELVLDVGRVTLGENSRKKMKDCKLRKKQNESVSRAMCALLNSGGGVIKAEIENEDYSYTKDGIGLDLENSFSNILLFVPEYLDFMQNGNYFLIFVKSWSLNTSGLRITTLSSNLYKRDITSAKVMNATAALEFLKDMKKTRGRLYLRPELLAKRPCVDIQEENNMKALAGVFFDRTELDRKEKLTFTESTHVEIKNFSTERLLQRIKEILPQYVSAFANTDGGYLFIGLNEDKEIIGFKAEMSDLDDLEREIEKSIRKMPVHHFCMEKKKINYSCKFLGVYDKGSLCGYVCALRVERFCCAVFAKEPDSWHVKDNRVMQLTRKEWIQFMVEAEPKFSSAYEEVISQINTSLPAPHSWPLLEWQRQRHHCPGLSGRITYTPENLCRKLFLQHEGLKQLICEEMSSVRKGSLIFSRSWSVDLGLQENHKVLCDALLISQDSPPVLYTFHMVQDEEFKGYSTQTALTLKQKLAKIGGYTKKVCVMTKIFYLSPEGMTSCQYDLRSQVIYPESYYFTRRKYLLKALFKALKRLKSLRDQFSFAENLYQIIGIDCFQKNDK Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Mutated Histone H3.3 (H3F3A) | 4GUS | 5.94 | |
Target general information Gen name H3F3A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PP781; Histone H3.3; H3F3; H3.3B; H3.3A Protein family Histone H3 family Biochemical class NA Function Variant histone H3 which replaces conventional H3 in a wide range of nucleosomes in active genes. Constitutes the predominant form of histone H3 in non-dividing cells and is incorporated into chromatin independently of DNA synthesis. Deposited at sites of nucleosomal displacement throughout transcribed genes, suggesting that it represents an epigenetic imprint of transcriptionally active chromatin. Nucleosomes wrap and compact DNA into chromatin, limiting DNA accessibility to the cellular machineries which require DNA as a template. Histones thereby play a central role in transcription regulation, DNA repair, DNA replication and chromosomal stability. DNA accessibility is regulated via a complex set of post-translational modifications of histones, also called histone code, and nucleosome remodeling. Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539269}. The gene represented in this entry is involved in disease pathogenesis. H3F3A mutations affecting residues involved in post-translational modifications of histone H3.3 are recurrent in malignant, aggressive gliomas including glioblastoma multiforme (GBM) and diffuse intrinsic pontine glioma (DIPG) (PubMed:22286061, PubMed:22286216). The mechanism through which mutations lead to tumorigenesis involves altered histones methylation, impaired regulation of Polycomb repressive complex 2 (PRC2) activity, and aberrant epigenetic regulation of gene expression (PubMed:23539183, PubMed:23539269, PubMed:23603901). {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539183, ECO:0000269|PubMed:23539269, ECO:0000269|PubMed:23603901}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 1 (BRYLIB1) [MIM:619720]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB1 is caused by variants in H3-3A. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 2 (BRYLIB2) [MIM:619721]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB2 is caused by variants in H3-3B. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: H3F3A and H3F3B mutations affecting residues involved in post-translational modifications of histone H3.3 are implicated in the pathogenesis of some bone and cartilage neoplasms. Mutations have been found with high prevalence in chondroblastoma and giant cell tumors of bone, and with low frequency in osteosarcoma, conventional chondrosarcoma and clear cell chondrosarcoma. Chondroblastoma samples frequently carry a H3F3B mutation affecting residue Lys-37 (H3K36), although H3F3A is mutated in some cases. Most giant cell tumors of bone harbor H3F3A mutations affecting residue Gly-35 (H3G34). {ECO:0000269|PubMed:24162739}. Drugs (DrugBank ID) NA Interacts with Q9NVP2; P45973; Q13111; Q9UER7; Q9UER7-1; Q9Y6K1; P62805; P49321-2; Q8IZL8; Q5VWG9; Q9VK33; Q8R5C8 EC number NA Uniprot keywords 3D-structure; Acetylation; ADP-ribosylation; Chromosome; Citrullination; Direct protein sequencing; Disease variant; DNA-binding; Hydroxylation; Intellectual disability; Lipoprotein; Methylation; Nucleosome core; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A,C Molecular weight (Da) 86148.9 Length 766 Aromaticity 0.1 Instability index 37.57 Isoelectric point 8.25 Charge (pH=7) 7.16 2D Binding mode Binding energy (Kcal/mol) -8.1  Molscript Map 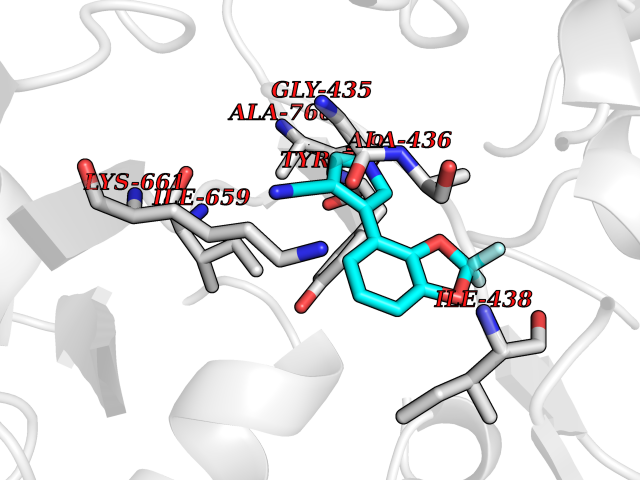 Pymol Map 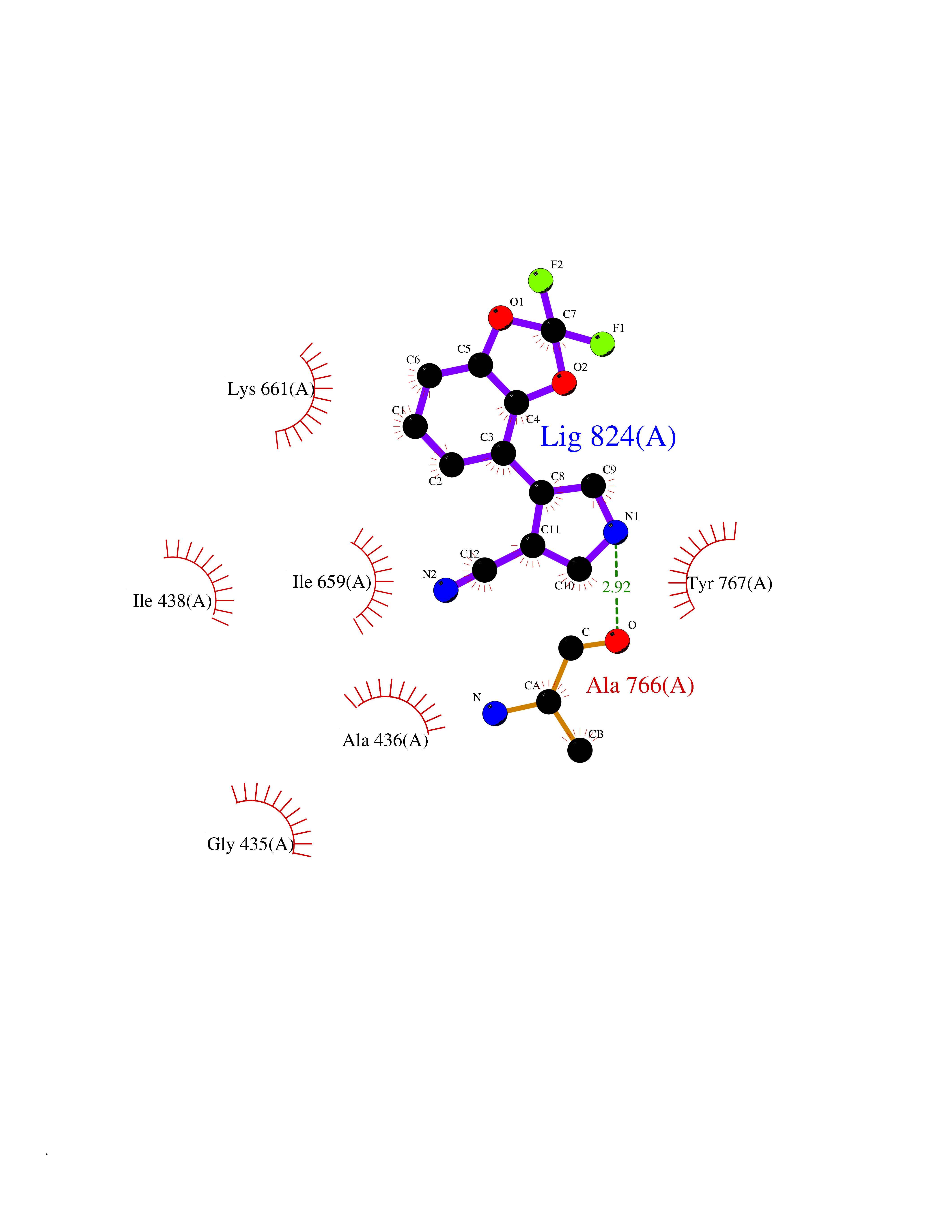 Ligplot Map 3D Binding mode Sequence GSRKCEKAGCTATCPVCFASASERCAKNGYTSRWYHLSCGEHFCNECFDHYYRSHKDGYDKYTTWKKIWTSNGKTEPSPKAFMADQQLPYWVQCTKPECRKWRQLTKEIQLTPQIAKTYRCGMKPNTAIKPETSDHCSLPEDLRVLEVSNHWWYSMLILPPLLKDSVAAPLLSAYYPDCVGMSPSCTGMNRYFQPFYQPNECGKALCVRPDVMELDELYEFPEYSRDPTMYLALRNLILALWYTNCKEALTPQKCIPHIIVRGLVRIRCVQEVERILYFMTRKGLINTGVLSVGADQYLLPKDYHNKSVIIIGAGPAGLAAARQLHNFGIKVTVLEAKDRIGGRVWDDKSFKGVTVGRGAQIVNGCINNPVALMCEQLGISMHKFGERCDLIQEGGRITDPTIDKRMDFHFNALLDVVSEWRKDKTQLQDVPLGEKIEEIYKAFIKESGIQFSELEGQVLQFHLSNLEYACGSNLHQVSARSWDHNEFFAQFAGDHTLLTPGYSVIIEKLAEGLDIQLKSPVQCIDYSGDEVQVTTTDGTGYSAQKVLVTVPLALLQKGAIQFNPPLSEKKMKAINSLGAGIIEKIALQFPYRFWDSKVQGADFFGHVPPSASKRGLFAVFYDMDPQKKHSVLMSVIAGEAVASVRTLDDKQVLQQCMATLRELFKEQEVPDPTKYFVTRWSTDPWIQMAYSFVKTGGSGEAYDIIAEDIQGTVFFAGEATNRHFPQTVTGAYLSGVREASKIAAFARTMQTARKSTGGKAPRKQL Hydrogen bonds contact Hydrophobic contact | ||||