Job Results:
Ligand
Structure
Job ID
43b2a7cfabc12f2b0137afb38036989c
Job name
NA
Time
2025-03-05 09:37:27
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Glutathione S-transferase LANCL1 (LANCL1) | 3E73 | 4.00 | |
Target general information Gen name LANCL1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p40; LanC-like protein 1; GPR69A; 40 kDa erythrocyte membrane protein Protein family LanC-like protein family Biochemical class NA Function Functions as glutathione transferase. Catalyzes conjugation of the glutathione (GSH) to artificial substrates 1-chloro-2,4-dinitrobenzene (CDNB) and p-nitrophenyl acetate. Mitigates neuronal oxidative stress during normal postnatal development and in response to oxidative stresses probably through GSH antioxidant defense mechanism (By similarity). May play a role in EPS8 signaling. Binds glutathione. Related diseases Spermatogenic failure 5 (SPGF5) [MIM:243060]: An infertility disorder caused by spermatogenesis defects. Semen from affected men show close to 100% morphologically abnormal multiflagellar spermatozoa with low motility, oversized irregular heads, and abnormal midpiece and acrosome. {ECO:0000269|PubMed:17435757, ECO:0000269|PubMed:21733974}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9UHR4; P42858; Q08509 EC number EC 2.5.1.18 Uniprot keywords 3D-structure; Acetylation; Cell membrane; Cytoplasm; Direct protein sequencing; Membrane; Metal-binding; Proteomics identification; Reference proteome; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 46005.4 Length 405 Aromaticity 0.14 Instability index 33.7 Isoelectric point 7.13 Charge (pH=7) 0.4 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map 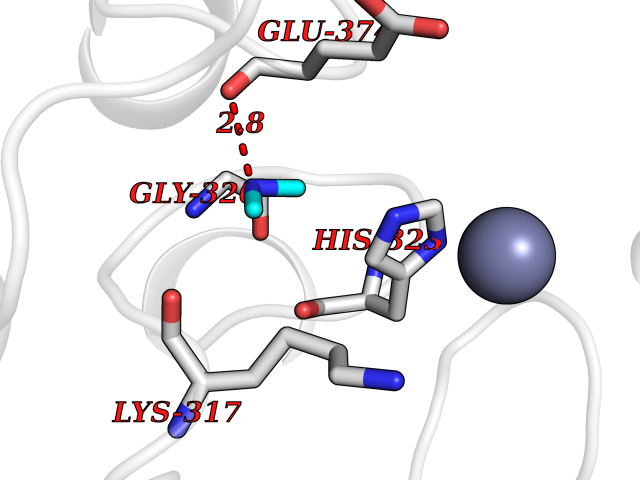 Pymol Map 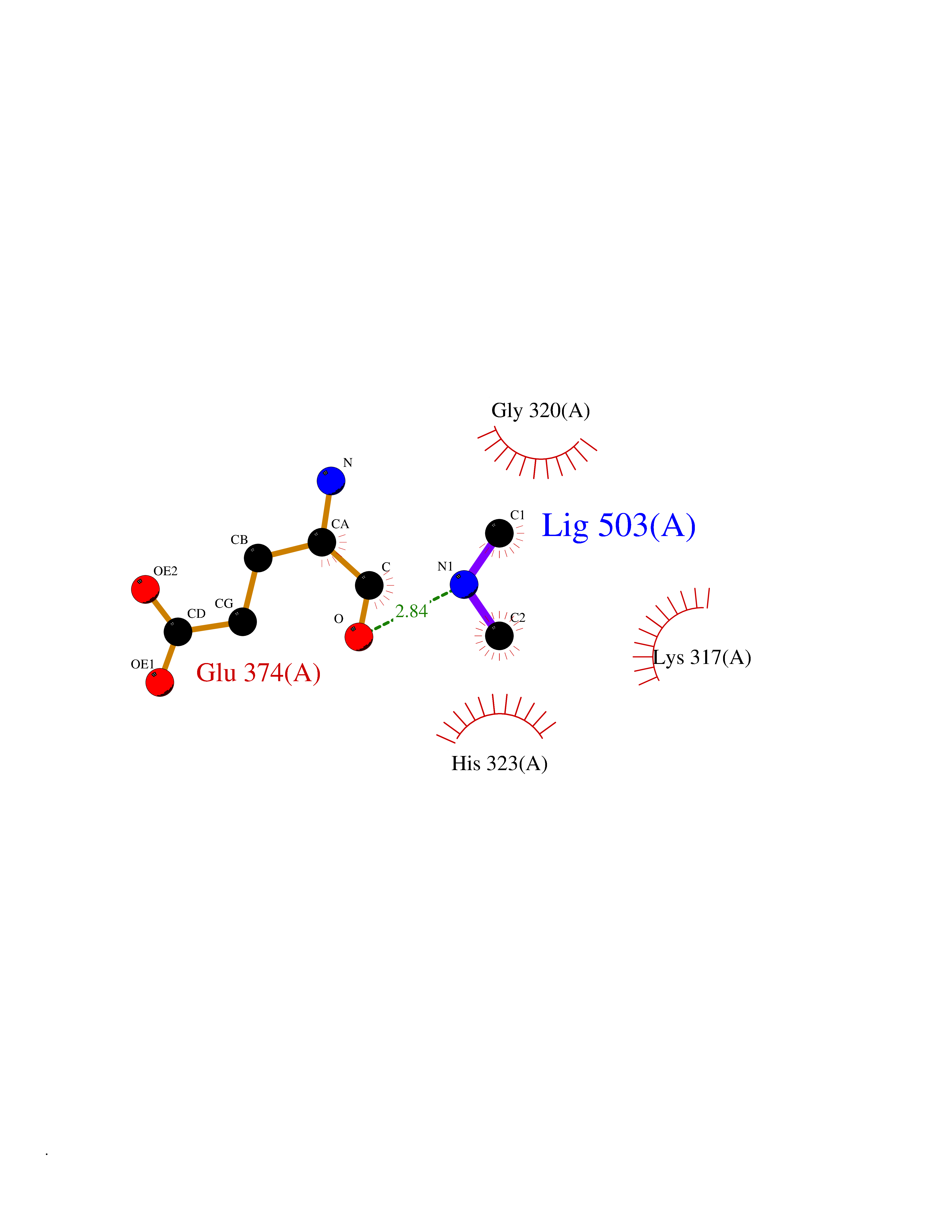 Ligplot Map 3D Binding mode Sequence SMDIEFMAQRAFPNPYADYNKSLAEGYFDAAGRLTPEFSQRLTNKIRELLQQMERGLKSADPRDGTGYTGWAGIAVLYLHLYDVFGDPAYLQLAHGYVKQSLNCLTKRSITFLCGDAGPLAVAAVLYHKMNNEKQAEDCITRLIHLNKIDPHAPNEMLYGRIGYIYALLFVNKNFGVEKIPQSHIQQICETILTSGENLARKRNFTAKSPLMYEWYQEYYVGAAHGLAGIYYYLMQPSLQVSQGKLHSLVKPSVDYVCQLKFPSGNYPPCIGDNRDLLVHWCHGAPGVIYMLIQAYKVFREEKYLCDAYQCADVIWQYGLLKKGYGLCHGSAGNAYAFLTLYNLTQDMKYLYRACKFAEWCLEYGEHGCRTPDTPFSLFEGMAGTIYFLADLLVPTKARFPAFEL Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Bifunctional protein PutA | 3E2Q | 3.99 | |
Target general information Gen name putA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b1014;poaA;JW0999 Protein family Proline dehydrogenase family; Aldehyde dehydrogenase family Biochemical class Oxidoreductase Function 1-pyrroline-5-carboxylate dehydrogenase activity.Bacterial-type RNA polymerase core promoter proximal region sequence-specific DNA binding.DNA binding.Flavin adenine dinucleotide binding.Identical protein binding.Proline dehydrogenase activity.Sequence-specific DNA binding.Transcriptional repressor activity, bacterial-type RNA polymerase core promoter proximal region sequence-specific binding. Related diseases Fructose-1,6-bisphosphatase deficiency (FBP1D) [MIM:229700]: An autosomal recessive metabolic disorder characterized by impaired gluconeogenesis, and episodes of hypoglycemia and metabolic acidosis that can be lethal in newborn infants or young children. {ECO:0000269|PubMed:12126934, ECO:0000269|PubMed:25601412, ECO:0000269|PubMed:9382095}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03051; DB03147; DB04398 Interacts with P09546 EC number 1.2.1.88; 1.5.5.2 Uniprot keywords 3D-structure; DNA-binding; FAD; Flavoprotein; Multifunctional enzyme; NAD; Oxidoreductase; Proline metabolism; Reference proteome; Repressor; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 45567.7 Length 407 Aromaticity 0.08 Instability index 33.2 Isoelectric point 7.22 Charge (pH=7) 0.47 2D Binding mode Binding energy (Kcal/mol) -5.44 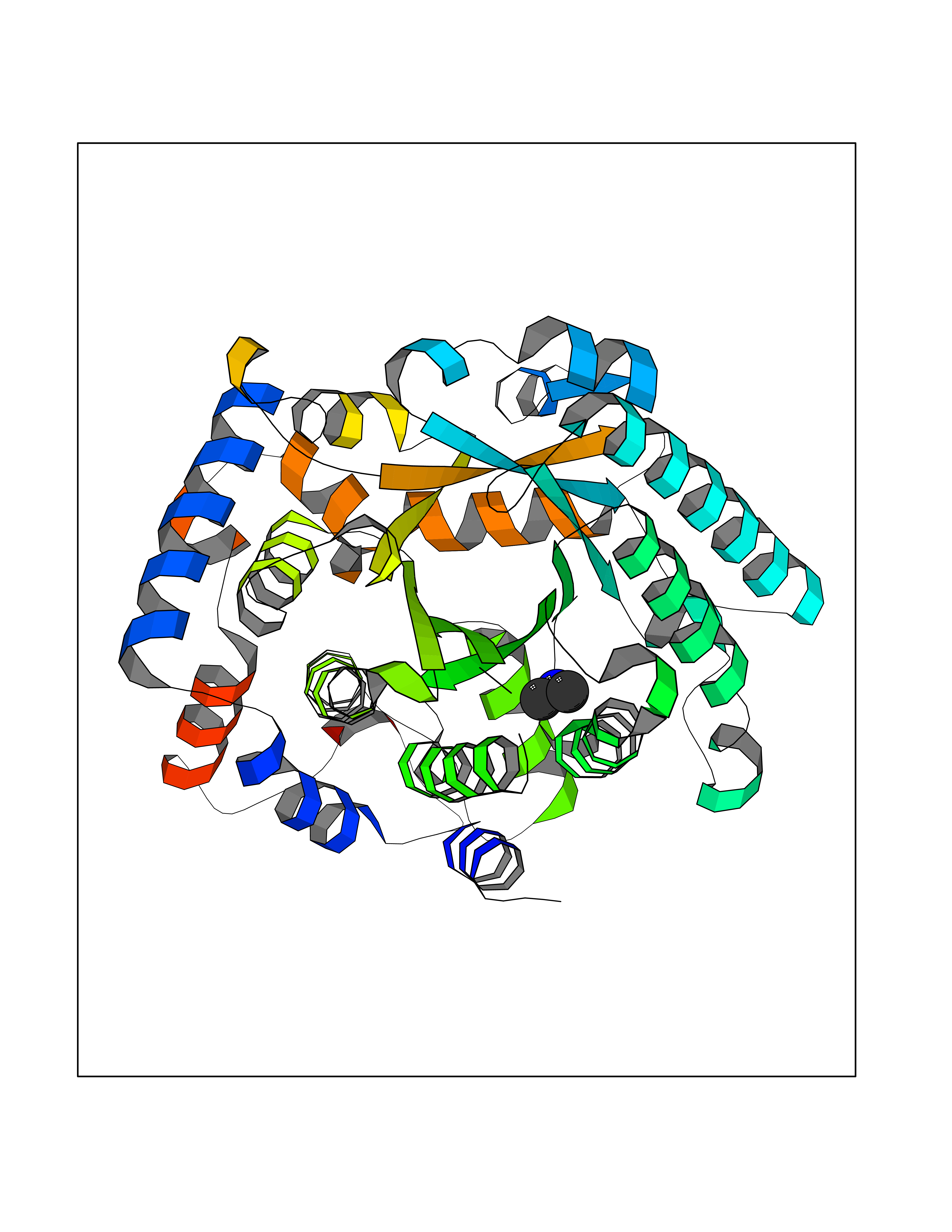 Molscript Map 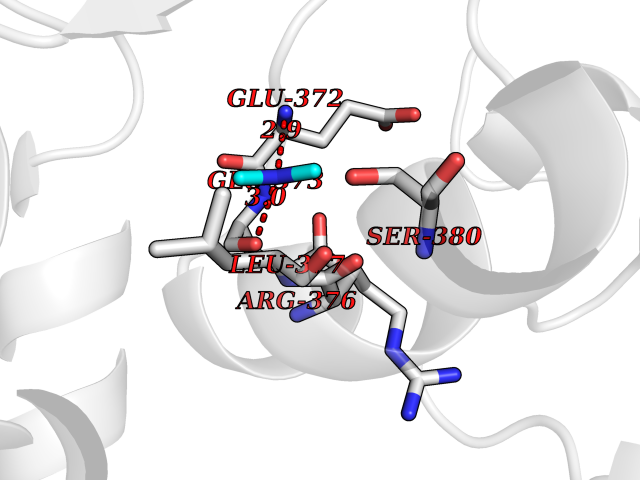 Pymol Map 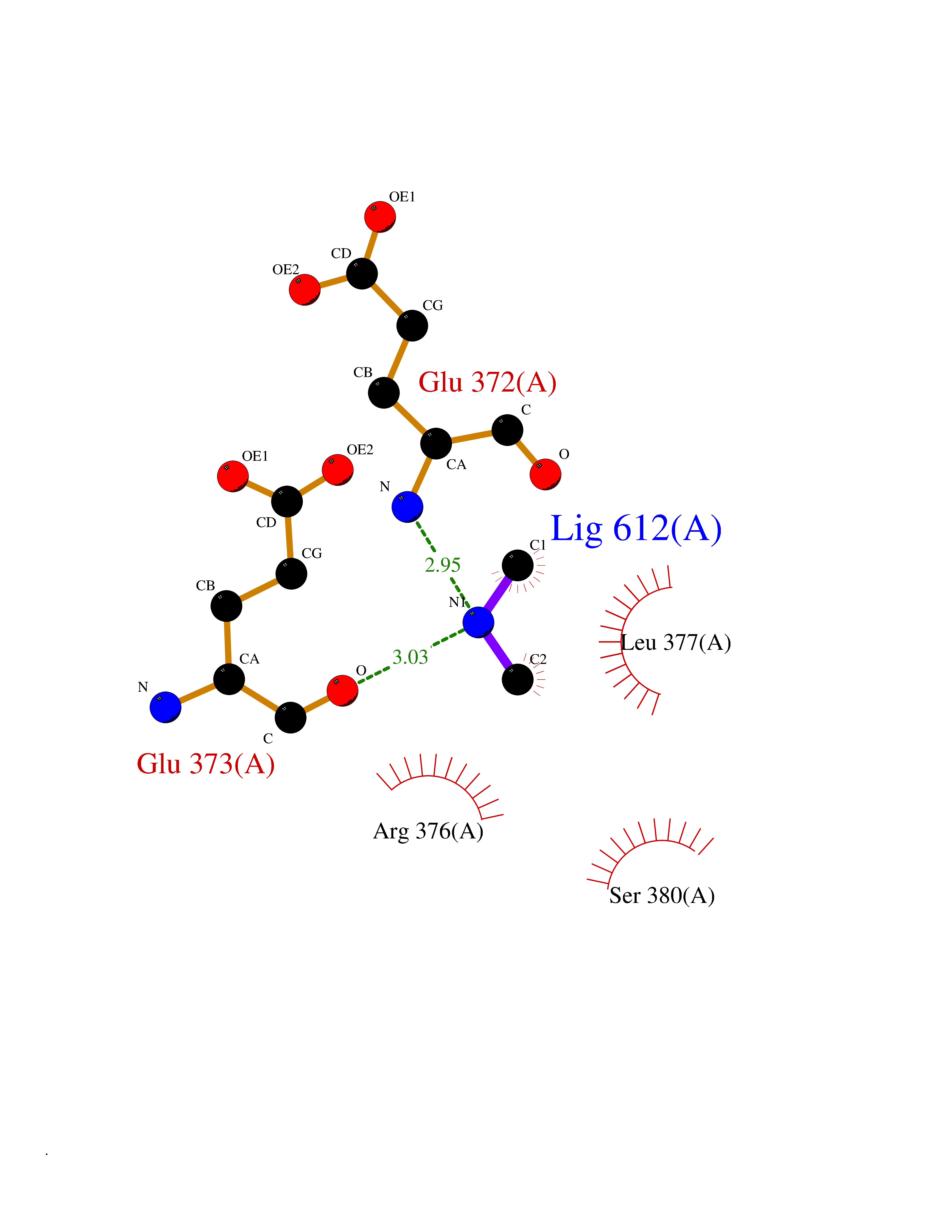 Ligplot Map 3D Binding mode Sequence QSVSRAAITAAYRRPETEAVSMLLEQARLPQPVAEQAHKLAYQLADKLRRLMGEQFVTGETIAEALANARKLEEKGFRYSYDMLGEAALTAADAQAYMVSYQQAIHAIGKASNGRGIYEGPGISIKLSALHPRYSRAQYDRVMEELYPRLKSLTLLARQYDIGINIDAEESDRLEISLDLLEKLCFEPELAGWNGIGFVIQAYQKRCPLVIDYLIDLATRSRRRLMIRLVKGAYWDSEIKRAQMDGLEGYPVYTRKVYTDVSYLACAKKLLAVPNLIYPQFATHNAHTLAAIYQLAGQNYYPGQYEFQCLHGMGEPLYEQVTGKVADGKLNRPCRISAPVGTHETLLAYLVRRLLENGANTSFVNRIADTSLPLDELVADPVTAVEKLAQQEGQTGLPHPKIPLPRD Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Poly [ADP-ribose] polymerase 1 (PARP1) | 5WS1 | 3.99 | |
Target general information Gen name PARP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein poly-ADP-ribosyltransferase PARP1; Poly[ADP-ribose] synthetase-1; Poly[ADP-ribose] synthase 1; Poly(ADP-ribose)polymerase-1; PPOL; PARP-1; NAD(+)Poly [ADP-ribose] polymerase-1 ADP-ribosyltrans Protein family ARTD/PARP family Biochemical class Glycosyltransferases Function Mainly mediates glutamate and aspartate ADP-ribosylation of target proteins: the ADP-D-ribosyl group of NAD(+) is transferred to the acceptor carboxyl group of glutamate and aspartate residues and further ADP-ribosyl groups are transferred to the 2'-position of the terminal adenosine moiety, building up a polymer with an average chain length of 20-30 units. Mediates the poly(ADP-ribosyl)ation of a number of proteins, including itself, APLF and CHFR. Also mediates serine ADP-ribosylation of target proteins following interaction with HPF1; HPF1 conferring serine specificity. Probably also catalyzes tyrosine ADP-ribosylation of target proteins following interaction with HPF1. Catalyzes the poly-ADP-ribosylation of histones in a HPF1-dependent manner. Involved in the base excision repair (BER) pathway by catalyzing the poly-ADP-ribosylation of a limited number of acceptor proteins involved in chromatin architecture and in DNA metabolism. ADP-ribosylation follows DNA damage and appears as an obligatory step in a detection/signaling pathway leading to the reparation of DNA strand breaks. In addition to base excision repair (BER) pathway, also involved in double-strand breaks (DSBs) repair: together with TIMELESS, accumulates at DNA damage sites and promotes homologous recombination repair by mediating poly-ADP-ribosylation. In addition to proteins, also able to ADP-ribosylate DNA: catalyzes ADP-ribosylation of DNA strand break termini containing terminal phosphates and a 2'-OH group in single- and double-stranded DNA, respectively. Required for PARP9 and DTX3L recruitment to DNA damage sites. PARP1-dependent PARP9-DTX3L-mediated ubiquitination promotes the rapid and specific recruitment of 53BP1/TP53BP1, UIMC1/RAP80, and BRCA1 to DNA damage sites. Acts as a regulator of transcription: positively regulates the transcription of MTUS1 and negatively regulates the transcription of MTUS2/TIP150. With EEF1A1 and TXK, forms a complex that acts as a T-helper 1 (Th1) cell-specific transcription factor and binds the promoter of IFN-gamma to directly regulate its transcription, and is thus involved importantly in Th1 cytokine production. Involved in the synthesis of ATP in the nucleus, together with NMNAT1, PARG and NUDT5. Nuclear ATP generation is required for extensive chromatin remodeling events that are energy-consuming. Poly-ADP-ribosyltransferase that mediates poly-ADP-ribosylation of proteins and plays a key role in DNA repair. Related diseases Dihydrolipoamide dehydrogenase deficiency (DLDD) [MIM:246900]: An autosomal recessive metabolic disorder characterized biochemically by a combined deficiency of the branched-chain alpha-keto acid dehydrogenase complex (BCKDC), pyruvate dehydrogenase complex (PDC), and alpha-ketoglutarate dehydrogenase complex (KGDC). Clinically, affected individuals have lactic acidosis and neurologic deterioration due to sensitivity of the central nervous system to defects in oxidative metabolism. {ECO:0000269|PubMed:10448086, ECO:0000269|PubMed:11687750, ECO:0000269|PubMed:12925875, ECO:0000269|PubMed:15712224, ECO:0000269|PubMed:16442803, ECO:0000269|PubMed:16770810, ECO:0000269|PubMed:17404228, ECO:0000269|PubMed:20160912, ECO:0000269|PubMed:8506365, ECO:0000269|PubMed:8968745, ECO:0000269|PubMed:9540846, ECO:0000269|PubMed:9934985}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04010; DB03509; DB03072; DB03722; DB03073; DB07787; DB07096; DB07330; DB02498; DB13877; DB02701; DB11793; DB02690; DB09074; DB12332; DB11760; DB00277; DB07232; DB01593; DB14487; DB14533; DB14548 Interacts with Q8IW19; Q7Z2E3; P42574; P49715; Q86WJ1-1; P26358; Q01094; Q96L91; P11308; O60741; P09429; Q13007; Q9BQ69; P08651; Q9Y530; P09874; Q8N2W9; P46063; Q9NTX7; Q14684-1; O95863; P63165; P04637; P0CG48; Q14191; P18887; P54577; Q2M1K9; Q02085 EC number EC 2.4.2.30 Uniprot keywords 3D-structure; Acetylation; ADP-ribosylation; Allosteric enzyme; Apoptosis; Chromosome; Cytoplasm; Direct protein sequencing; DNA damage; DNA repair; DNA-binding; Glycosyltransferase; Immunity; Innate immunity; Isopeptide bond; Metal-binding; NAD; Nucleotidyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 36904 Length 329 Aromaticity 0.08 Instability index 35.9 Isoelectric point 6.83 Charge (pH=7) -0.44 2D Binding mode Binding energy (Kcal/mol) -5.45 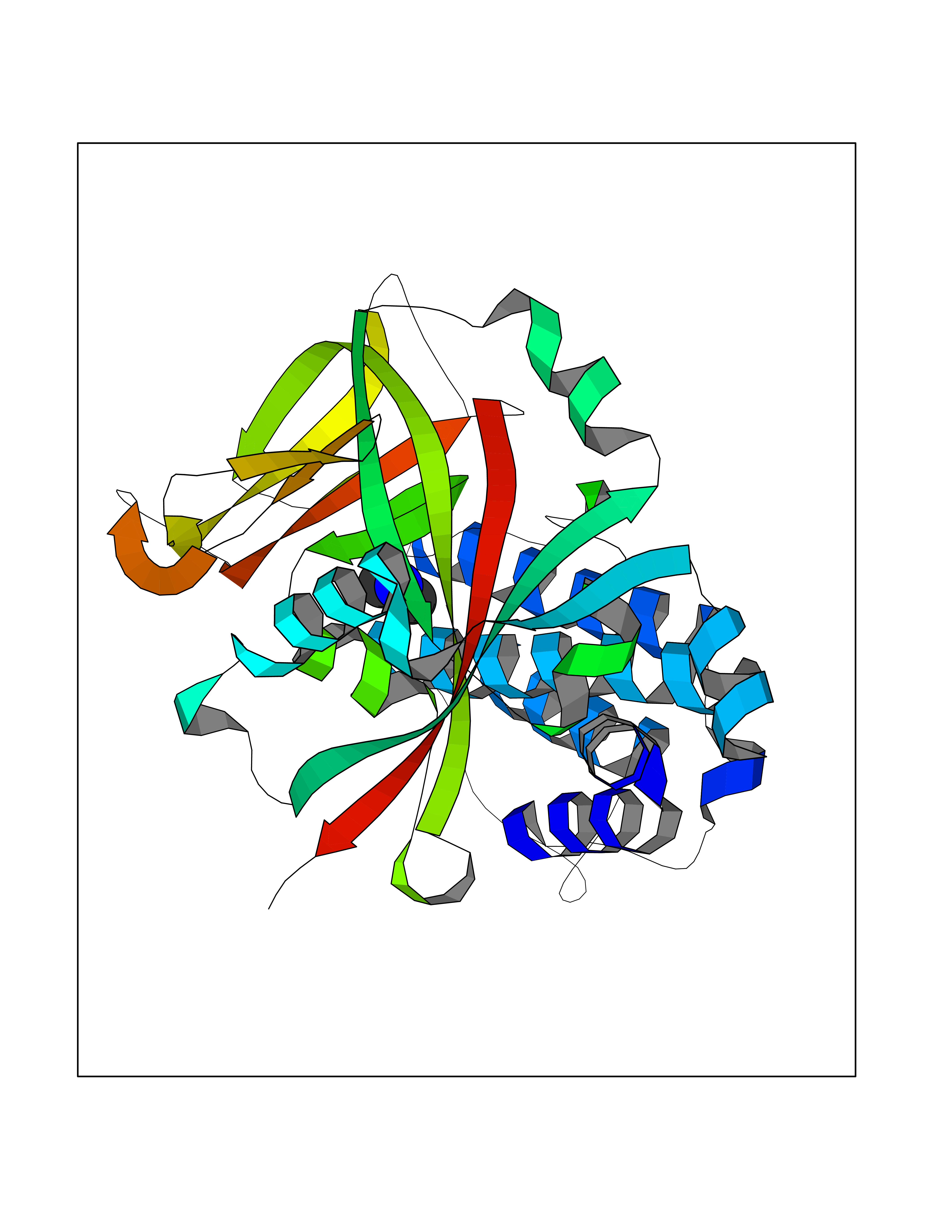 Molscript Map 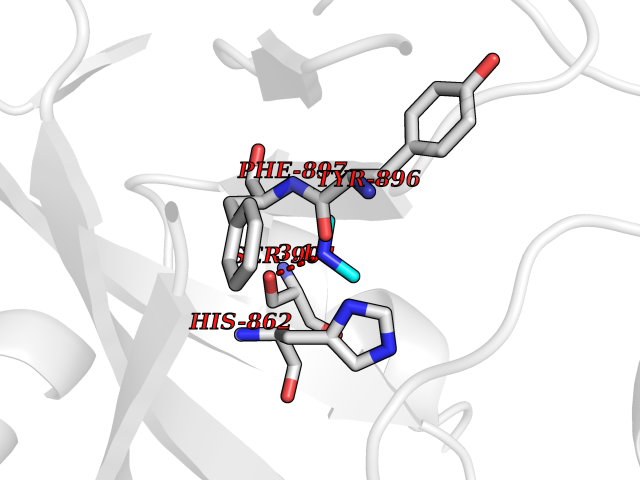 Pymol Map 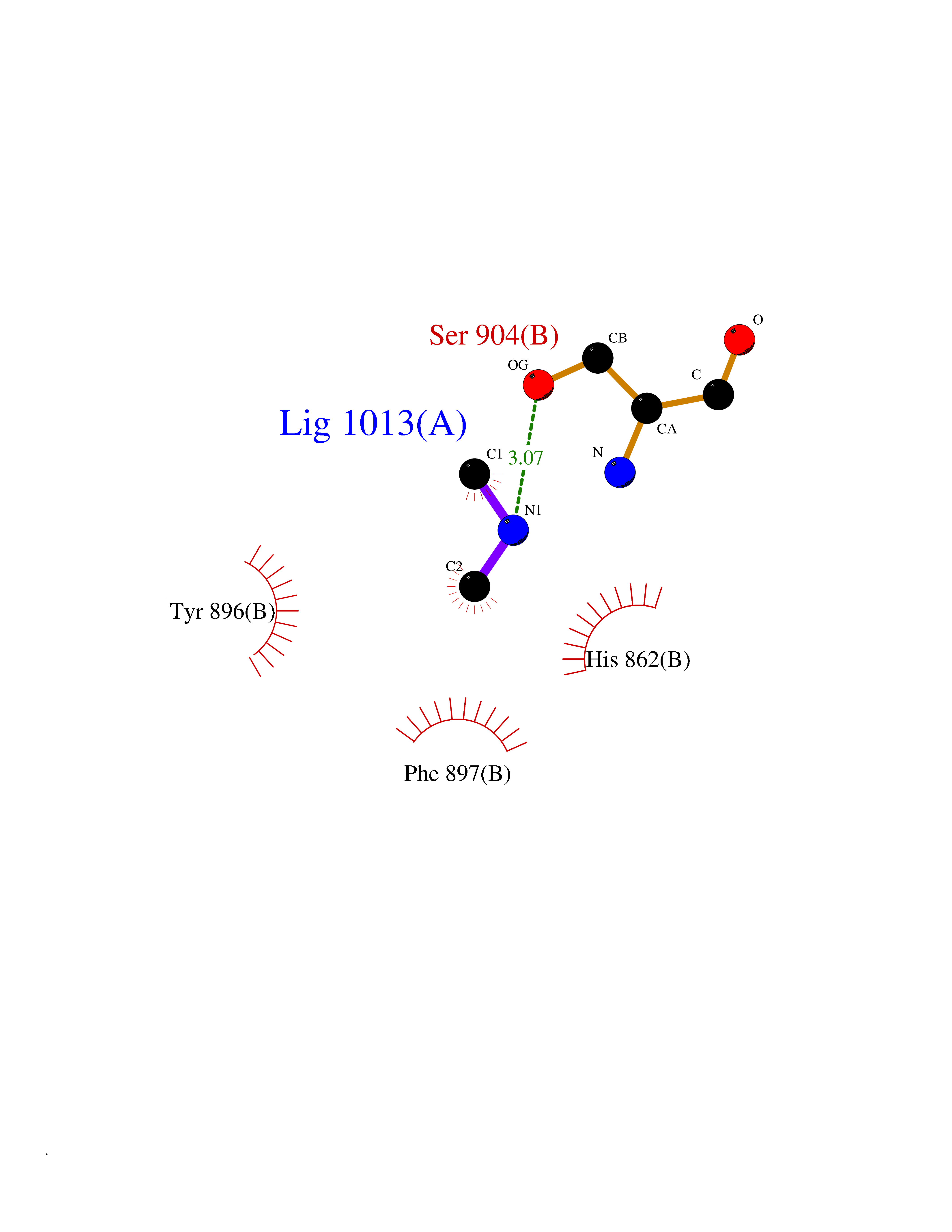 Ligplot Map 3D Binding mode Sequence DLIKMIFDVESMKKAMVEYEIDLQKMPLGKLSKRQIQAAYSILSEVQQAVSQGSDSQILDLSNRFYTLIPHDFGMKKPPLLNNADSVQAKAEMLDNLLDIEVAYSLPIDVNYEKLKTDIKVVDRDSEEAEIIRKYVKNTHATTHNAYDLEVIDIFKIEREGECQRYKPFKQLHNRRLLWHGSRTTNFAGILSQGLRIAPPEAPVTGYMFGKGIYFADMVSKSANYCHTSQGDPIGLILLGEVALGNMYELKHASHISKLPKGKHSVKGLGKTTPDPSANISLDGVDVPLGTGISSGVNDTSLLYNEYIVYDIAQVNLKYLLKLKFNFKT Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Endolysin | 1AM7 | 3.99 | |
Target general information Gen name R Organism Escherichia phage lambda (Bacteriophage lambda) Uniprot ID TTD ID NA Synonyms NA Protein family Glycosyl hydrolase 24 family Biochemical class Glycosidase Function Lyase activity.Lysozyme activity.Lytic transglycosylase activity. Related diseases Estrogen resistance (ESTRR) [MIM:615363]: A disorder characterized by partial or complete resistance to estrogens, in the presence of elevated estrogen serum levels. Clinical features include absence of the pubertal growth spurt, delayed bone maturation, unfused epiphyses, reduced bone mineral density, osteoporosis, continued growth into adulthood and very tall adult stature. Glucose intolerance, hyperinsulinemia and lipid abnormalities may also be present. {ECO:0000269|PubMed:23841731, ECO:0000269|PubMed:27754803}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04206 Interacts with NA EC number 4.2.2.n2 Uniprot keywords 3D-structure; Antimicrobial; Bacteriolytic enzyme; Cytolysis; Direct protein sequencing; Host cell lysis by virus; Host cytoplasm; Lyase; Reference proteome; Viral release from host cell Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 49834.9 Length 462 Aromaticity 0.07 Instability index 18.78 Isoelectric point 9.6 Charge (pH=7) 18.29 2D Binding mode Binding energy (Kcal/mol) -5.44 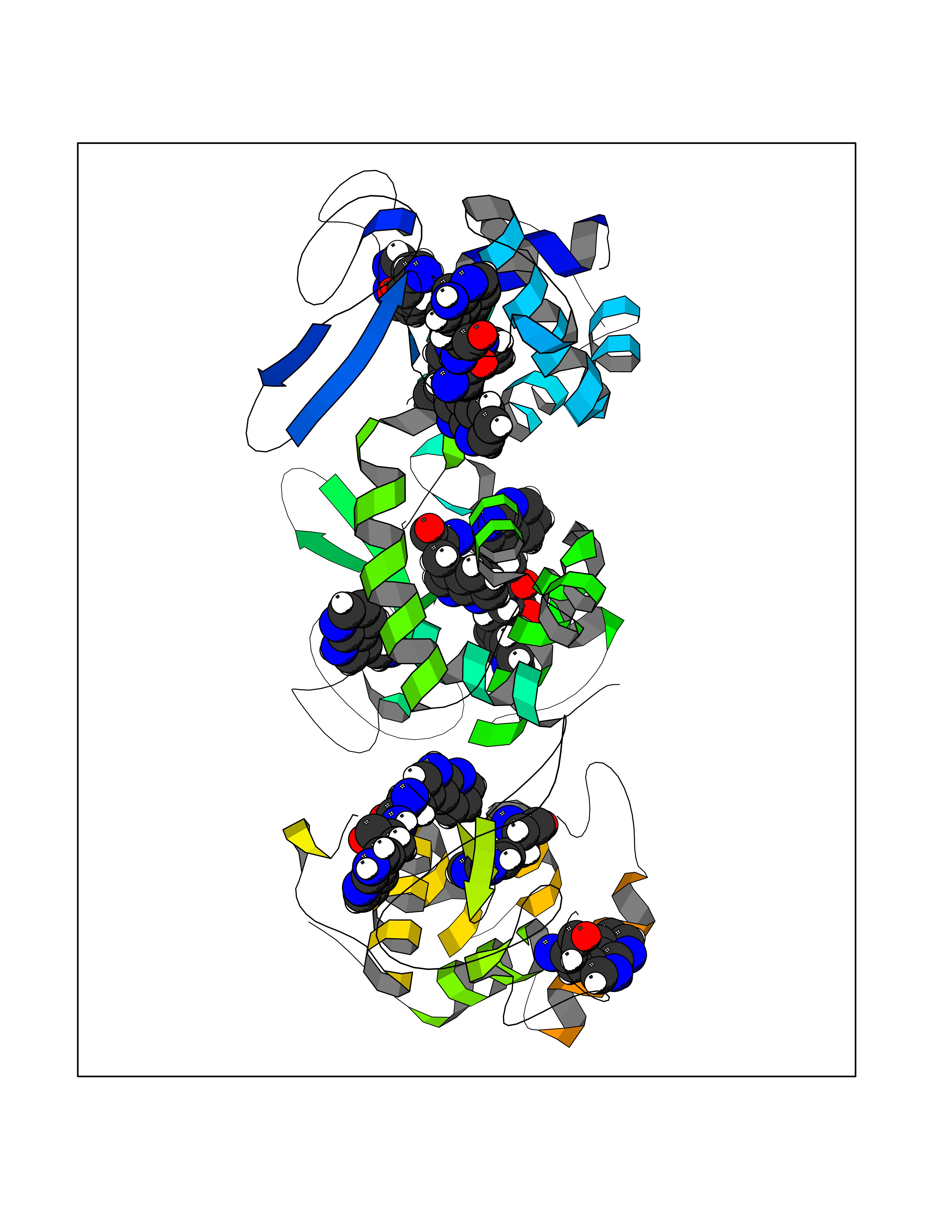 Molscript Map 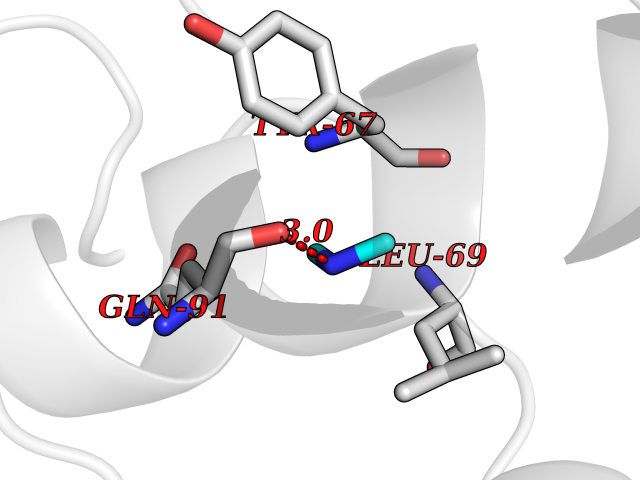 Pymol Map 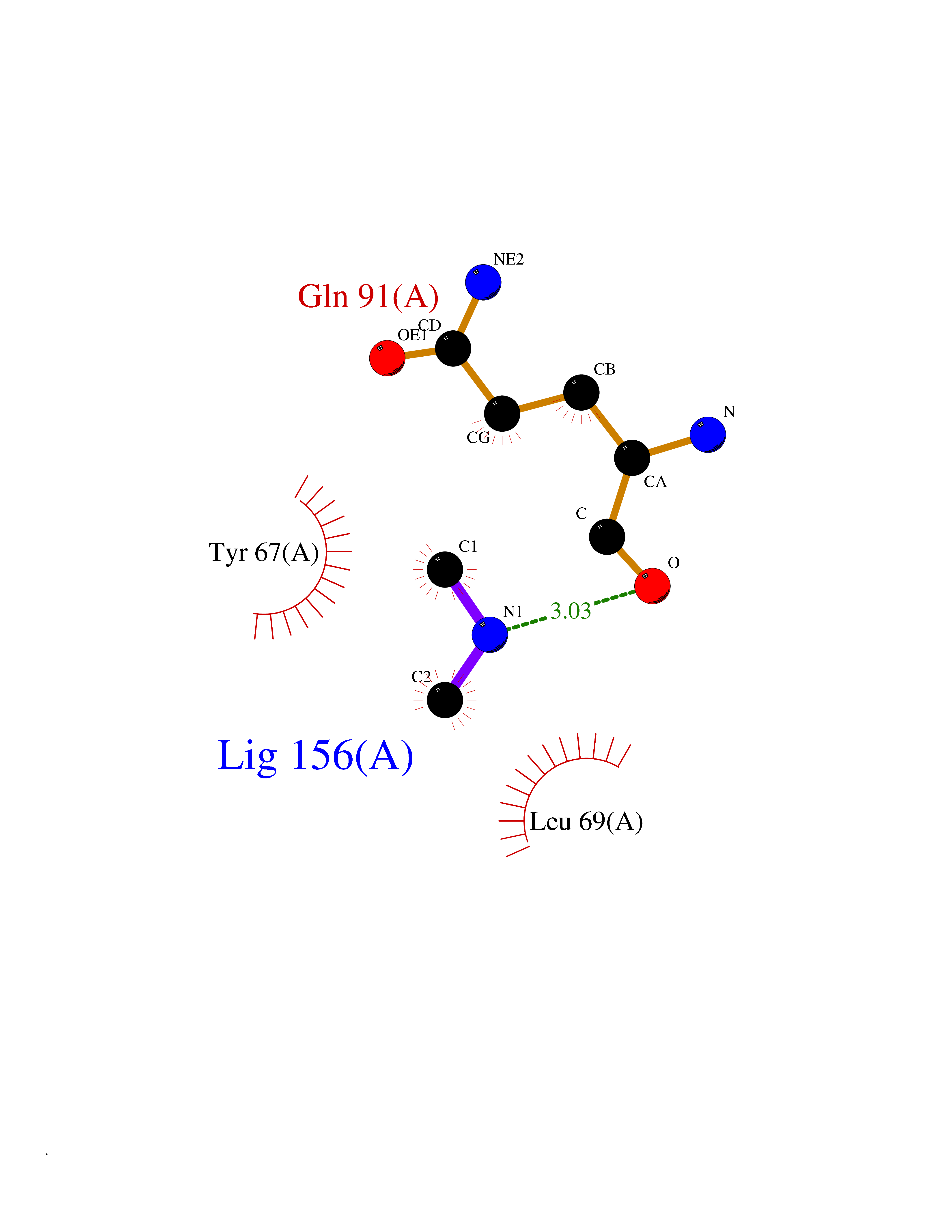 Ligplot Map 3D Binding mode Sequence MVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVRMVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVRMVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVR Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Coagulation factor VII (F7) | 4YLQ | 3.99 | |
Target general information Gen name F7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serum prothrombin conversion accelerator; SPCA; Proconvertin; Eptacog alfa Protein family Peptidase S1 family Biochemical class Peptidase Function Initiates the extrinsic pathway of blood coagulation. Serine protease that circulates in the blood in a zymogen form. Factor VII is converted to factor VIIa by factor Xa, factor XIIa, factor IXa, or thrombin by minor proteolysis. In the presence of tissue factor and calcium ions, factor VIIa then converts factor X to factor Xa by limited proteolysis. Factor VIIa will also convert factor IX to factor IXa in the presence of tissue factor and calcium. Related diseases Factor VII deficiency (FA7D) [MIM:227500]: A hemorrhagic disease with variable presentation. The clinical picture can be very severe, with the early occurrence of intracerebral hemorrhages or repeated hemarthroses, or, in contrast, moderate with cutaneous-mucosal hemorrhages (epistaxis, menorrhagia) or hemorrhages provoked by a surgical intervention. Finally, numerous subjects are completely asymptomatic despite very low factor VII levels. {ECO:0000269|PubMed:10862079, ECO:0000269|PubMed:11091194, ECO:0000269|PubMed:11129332, ECO:0000269|PubMed:12472587, ECO:0000269|PubMed:14717781, ECO:0000269|PubMed:1634227, ECO:0000269|PubMed:18976247, ECO:0000269|PubMed:19432927, ECO:0000269|PubMed:19751712, ECO:0000269|PubMed:2070047, ECO:0000269|PubMed:21206266, ECO:0000269|PubMed:21372693, ECO:0000269|PubMed:26761581, ECO:0000269|PubMed:7974346, ECO:0000269|PubMed:7981691, ECO:0000269|PubMed:8043443, ECO:0000269|PubMed:8204879, ECO:0000269|PubMed:8242057, ECO:0000269|PubMed:8364544, ECO:0000269|PubMed:8652821, ECO:0000269|PubMed:8844208, ECO:0000269|PubMed:8883260, ECO:0000269|PubMed:8940045, ECO:0000269|PubMed:9414278, ECO:0000269|PubMed:9452082, ECO:0000269|PubMed:9576180}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04590; DB07207; DB04758; DB04606; DB04593; DB07376; DB07247; DB08232; DB06552; DB13151; DB00100; DB13152; DB00036; DB09332; DB04767; DB13933 Interacts with P13726 EC number EC 3.4.21.21 Uniprot keywords 3D-structure; Alternative splicing; Blood coagulation; Calcium; Cleavage on pair of basic residues; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Gamma-carboxyglutamic acid; Glycoprotein; Hemostasis; Hydrolase; Hydroxylation; Pharmaceutical; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID H Molecular weight (Da) 26492.2 Length 240 Aromaticity 0.09 Instability index 34.61 Isoelectric point 6.81 Charge (pH=7) -0.52 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map 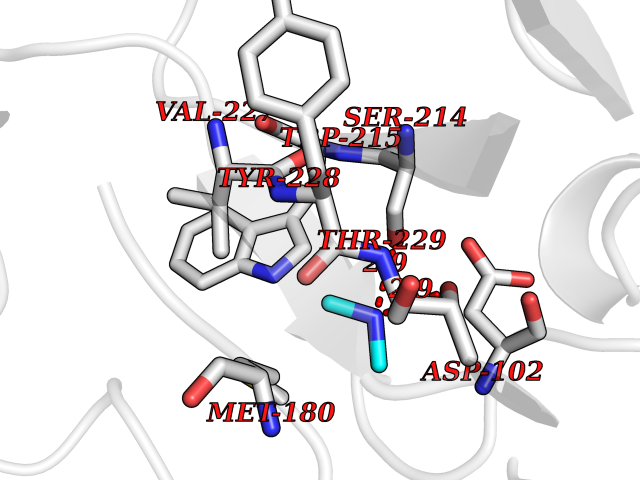 Pymol Map 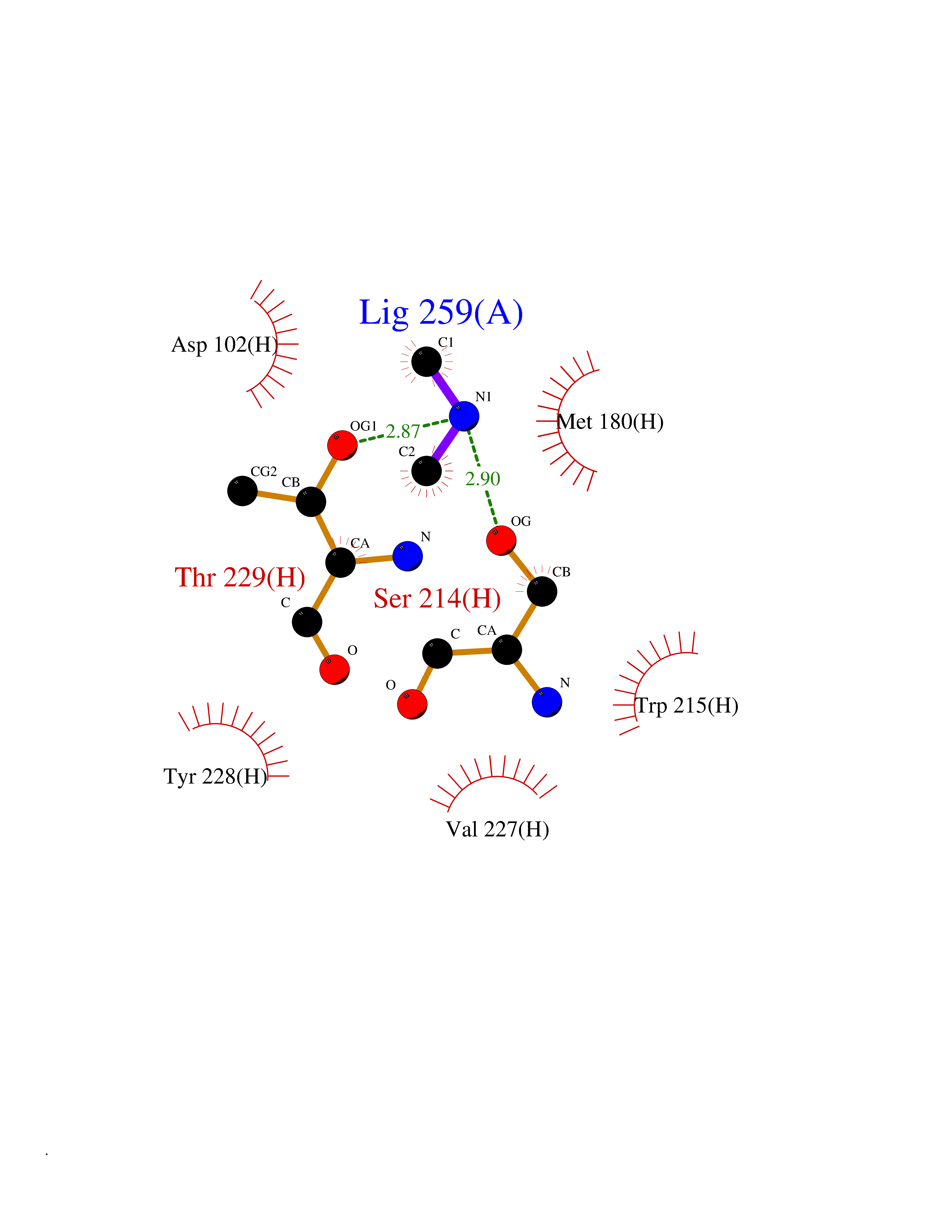 Ligplot Map 3D Binding mode Sequence IVGGKVCPKGECPWQVLLLVNGAQLCGGTLINTIWVVSAAHCFNWRNLIAVLGEHDLSEHDGDEQSRRVAQVIIPSTYVPGTTNHDIALLRLHQPVVLTDHVVPLCLPERTFTLAFVRFSLVSGWGQLLDRGATALELMVLNVPRLMTQDCEASFPGKITEYMFCGYSDSKDSCKGDSGGPHATHYRGTWYLTGIVSWGQGCATVGHFGVYTRVSQYIEWLQKLMRSEPRPGVLLRAPFP Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Riboflavin synthase | 1PKV | 3.99 | |
Target general information Gen name ribC Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1654;ribE;b1662 Protein family NA Biochemical class Transferase Function Riboflavin synthase activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00140 Interacts with NA EC number 2.5.1.9 Uniprot keywords 3D-structure; Reference proteome; Repeat; Riboflavin biosynthesis; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 19023.5 Length 174 Aromaticity 0.05 Instability index 2.85 Isoelectric point 5.13 Charge (pH=7) -9.8 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map 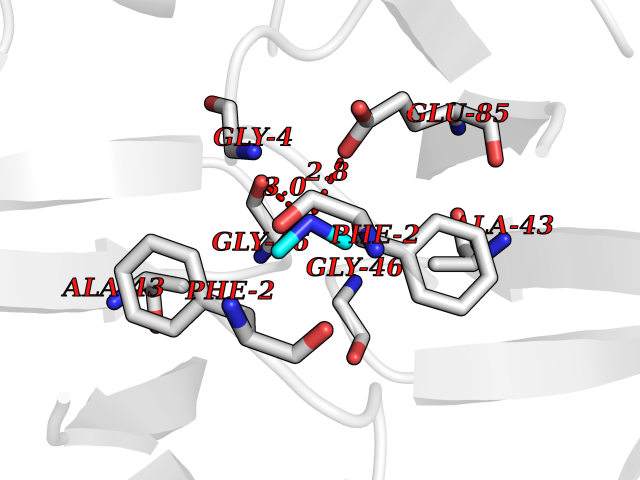 Pymol Map 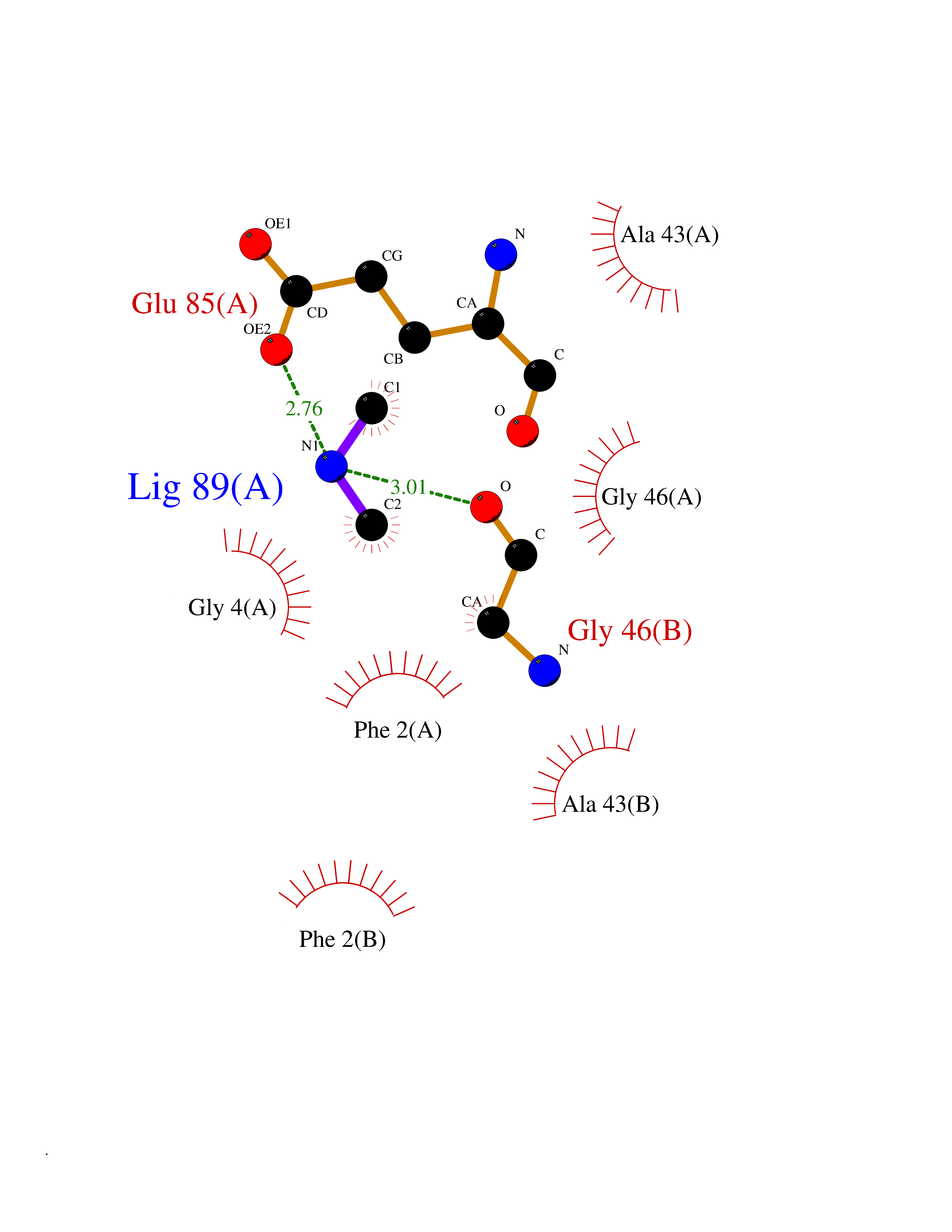 Ligplot Map 3D Binding mode Sequence MFTGIVQGTAKLVSIDEKPNFRTHVVELPDHMLDGLETGASVAHNGCCLTVTEINGNHVSFDLMKETLRITNLGDLKVGDWVNVERAMFTGIVQGTAKLVSIDEKPNFRTHVVELPDHMLDGLETGASVAHNGCCLTVTEINGNHVSFDLMKETLRITNLGDLKVGDWVNVERA Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Geranylgeranyl pyrophosphate synthase | 2Q80 | 3.99 | |
Target general information Gen name GGPS1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family FPP/GGPP synthase family Biochemical class Transferase Function Dimethylallyltranstransferase activity.Farnesyltranstransferase activity.Geranyltranstransferase activity.Identical protein binding.Metal ion binding. Related diseases Muscular dystrophy, congenital hearing loss, and ovarian insufficiency syndrome (MDHLO) [MIM:619518]: An autosomal recessive disorder characterized by early-onset progressive muscle weakness, sensorineural hearing loss, and primary amenorrhea due to ovarian insufficiency. Some patients become wheelchair-bound by the second decade, whereas others have a milder phenotype and maintain independent ambulation into adulthood. Most patients have respiratory insufficiency. {ECO:0000269|PubMed:32403198}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06830; DB06931; DB07221; DB08529; DB07410; DB07780; DB04695; DB02552; DB07841; DB00710; DB04714; DB07873; DB06548; DB00282; DB00399 Interacts with O00244; O95749; O00560 EC number 2.5.1.-; 2.5.1.1; 2.5.1.10; 2.5.1.29 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Congenital muscular dystrophy; Cytoplasm; Deafness; Disease variant; Isoprene biosynthesis; Lipid metabolism; Magnesium; Metal-binding; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D,E,F Molecular weight (Da) 32872.4 Length 284 Aromaticity 0.11 Instability index 41.09 Isoelectric point 6.17 Charge (pH=7) -3.37 2D Binding mode Binding energy (Kcal/mol) -5.45 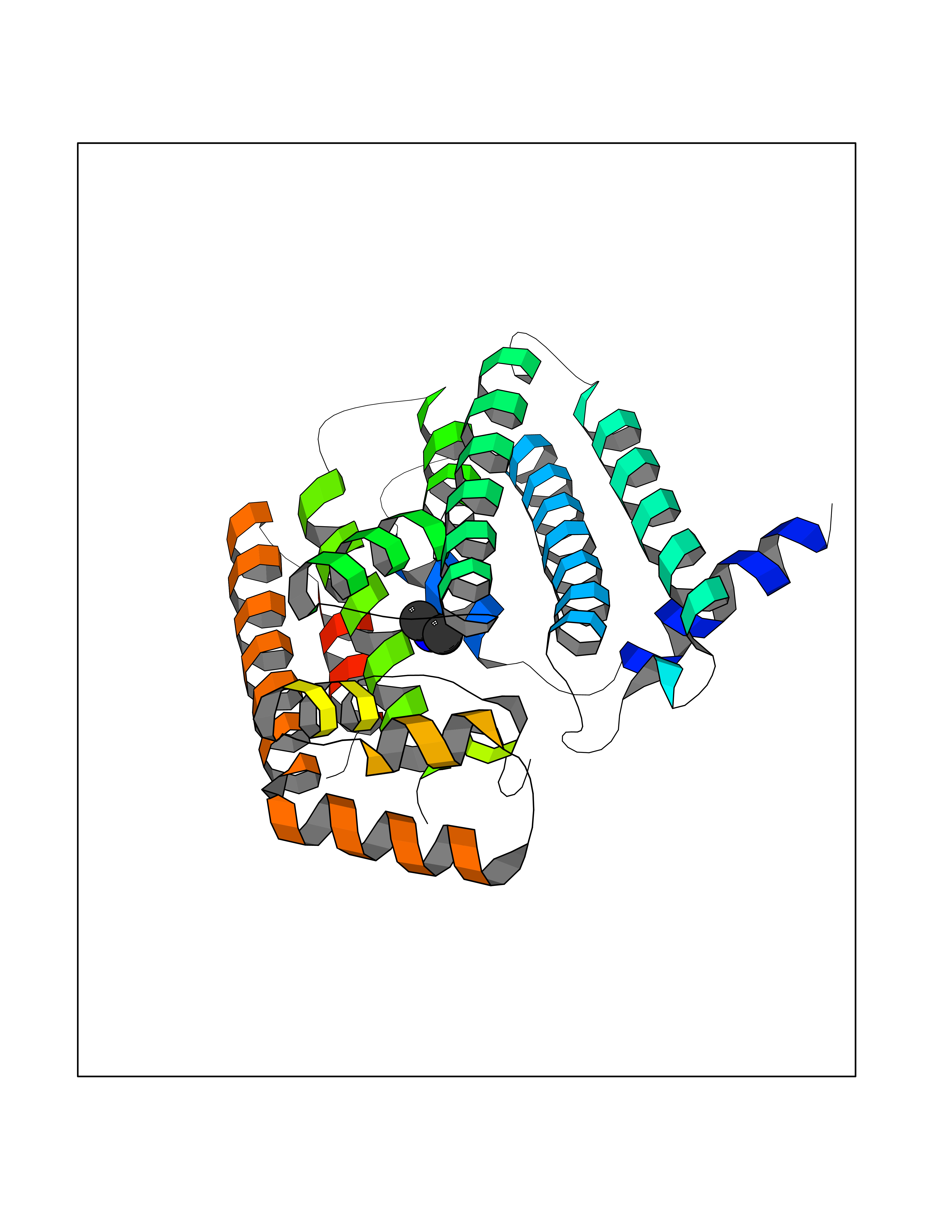 Molscript Map 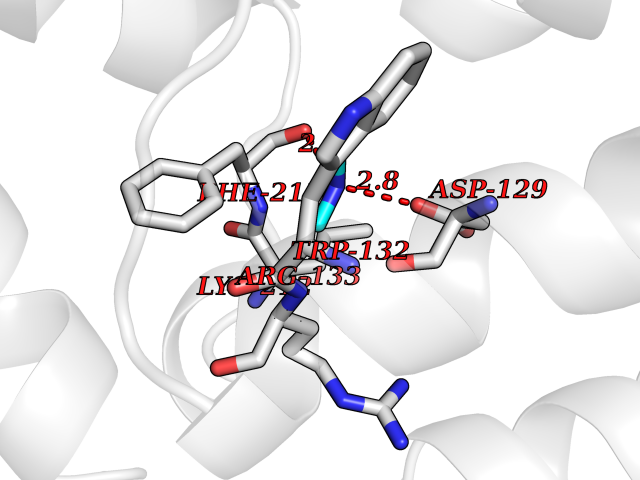 Pymol Map 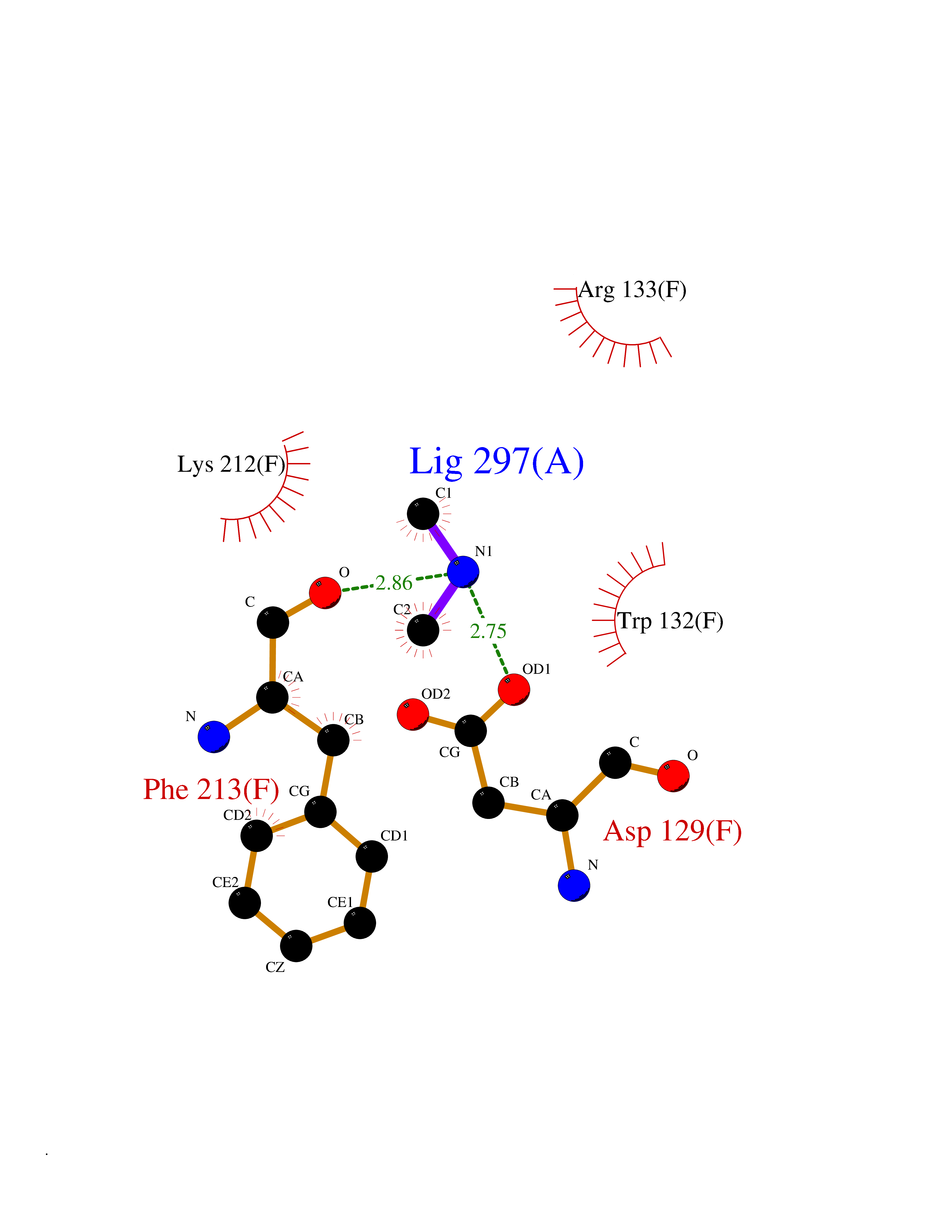 Ligplot Map 3D Binding mode Sequence ETVQRILLEPYKYLLQLPGKQVRTKLSQAFNHWLKVPEDKLQIIIEVTEMLHNASLLIDDIEDNSKLRRGFPVAHSIYGIPSVINSANYVYFLGLEKVLTLDHPDAVKLFTRQLLELHQGQGLDIYWRDNYTCPTEEEYKAMVLQKTGGLFGLAVGLMQLFSDYKEDLKPLLNTLGLFFQIRDDYANLHSKSFCEDLTEGKFSFPTIHAIWSRPESTQVQNILRQRTENIDIKKYCVHYLEDVGSFEYTRNTLKELEAKAYKQIDARGGNPELVALVKHLSKMF Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Neprilysin | 1R1H | 3.99 | |
Target general information Gen name MME Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms EPN Protein family Peptidase M13 family Biochemical class Hydrolase Function Endopeptidase activity.Exopeptidase activity.Metalloendopeptidase activity.Metallopeptidase activity.Peptide binding.Zinc ion binding. Related diseases Charcot-Marie-Tooth disease, axonal, 2T (CMT2T) [MIM:617017]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:26991897, ECO:0000269|PubMed:27588448}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinocerebellar ataxia 43 (SCA43) [MIM:617018]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA43 is a slowly progressive, autosomal dominant form. {ECO:0000269|PubMed:27583304}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08575; DB02597; DB00616; DB11623; DB05796; DB06655; DB02558; DB02062; DB00886; DB02557; DB09292; DB13928; DB08626 Interacts with P05067; P21926; Q06787-7; P08107; P04792 EC number 3.4.24.11 Uniprot keywords 3D-structure; Cell membrane; Charcot-Marie-Tooth disease; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lipoprotein; Membrane; Metal-binding; Metalloprotease; Myristate; Neurodegeneration; Neuropathy; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Spinocerebellar ataxia; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 79435.8 Length 696 Aromaticity 0.11 Instability index 37.5 Isoelectric point 5.53 Charge (pH=7) -11.46 2D Binding mode Binding energy (Kcal/mol) -5.45  Molscript Map 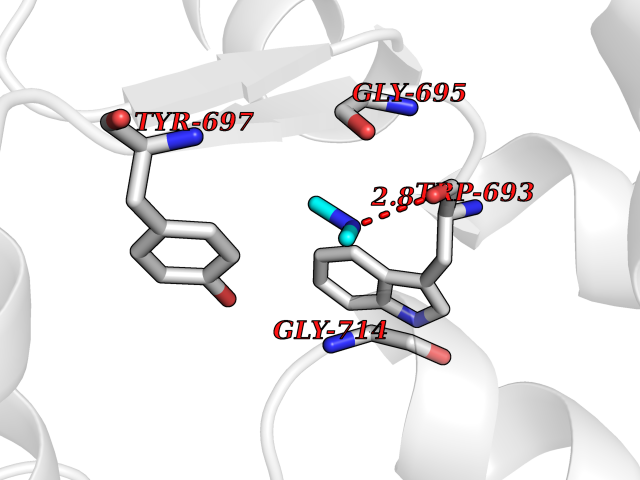 Pymol Map 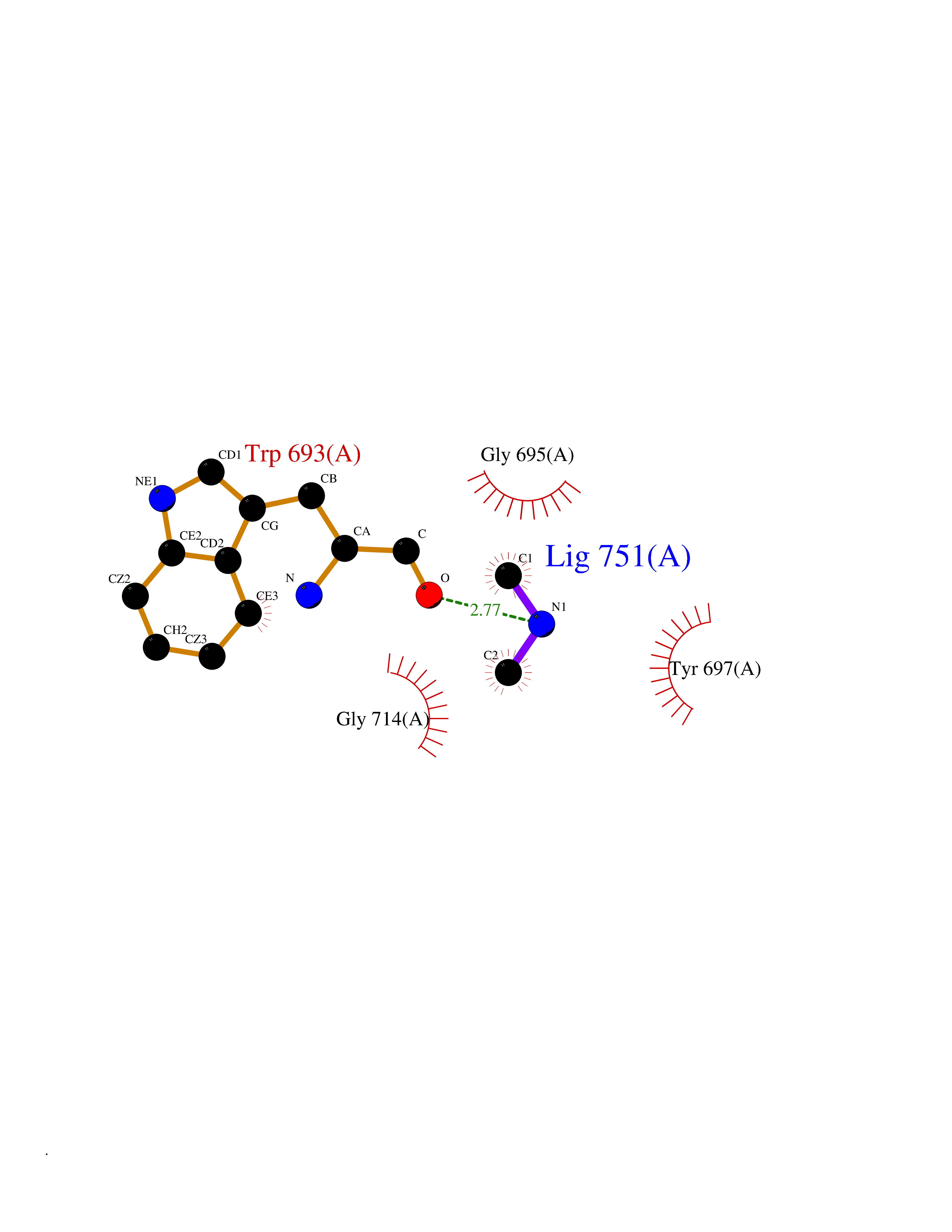 Ligplot Map 3D Binding mode Sequence GICKSSDCIKSAARLIQNMDATTEPCTDFFKYACGGWLKRNVIPETSSRYGNFDILRDELEVVLKDVLQEPKTEDIVAVQKAKALYRSCINESAIDSRGGEPLLKLLPDIYGWPVATENWEQKYGASWTAEKAIAQLNSKYGKKVLINLFVGTDDKNSVNHVIHIDQPRLGLPSRDYYECTGIYKEACTAYVDFMISVARLIRQEERLPIDENQLALEMNKVMELEKEIANATAKPEDRNDPMLLYNKMTLAQIQNNFSLEINGKPFSWLNFTNEIMSTVNISITNEEDVVVYAPEYLTKLKPILTKYSARDLQNLMSWRFIMDLVSSLSRTYKESRNAFRKALYGTTSETATWRRCANYVNGNMENAVGRLYVEAAFAGESKHVVEDLIAQIREVFIQTLDDLTWMDAETKKRAEEKALAIKERIGYPDDIVSNDNKLNNEYLELNYKEDEYFENIIQNLKFSQSKQLKKLREKVDKDEWISGAAVVNAFYSSGRNQIVFPAGILQPPFFSAQQSNSLNYGGIGMVIGHEITHGFDDNGRNFNKDGDLVDWWTQQSASNFKEQSQCMVYQYGNFSWDLAGGQHLNGINTLGENIADNGGLGQAYRAYQNYIKKNGEEKLLPGLDLNHKQLFFLNFAQVWCGTYRPEYAVNSIKTDVHSPGNFRIIGTLQNSAEFSEAFHCRKNSYMNPEKKCRVW Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Transferrin (TF) | 1RYO | 3.99 | |
Target general information Gen name TF Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Siderophilin; Serotransferrin; PRO1400; Beta-1 metal-binding globulin Protein family Transferrin family Biochemical class Transferrin Function It is responsible for the transport of iron from sites of absorption and heme degradation to those of storage and utilization. Serum transferrin may also have a further role in stimulating cell proliferation. Transferrins are iron binding transport proteins which can bind two Fe(3+) ions in association with the binding of an anion, usually bicarbonate. Related diseases Atransferrinemia (ATRAF) [MIM:209300]: A rare autosomal recessive disorder characterized by abnormal synthesis of transferrin leading to iron overload and microcytic hypochromic anemia. {ECO:0000269|PubMed:11110675, ECO:0000269|PubMed:15466165}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01370; DB14517; DB14518; DB01294; DB14526; DB14527; DB11136; DB14528; DB14529; DB14530; DB00515; DB09130; DB11397; DB13949; DB14490; DB14491; DB14488; DB14501; DB14489; DB13257; DB06215; DB06784; DB05260; DB01592; DB00893; DB00677; DB06757; DB11182; DB14520; DB01593; DB14487; DB14533; DB14548 Interacts with O43315; O00501; Q7Z7G2; Q9GZR5; Q9Y282; Q96KR6; P01350; P08034; Q8NBJ4; O15529; Q8TED1; Q7Z5P4; A8MZ59; O15173; Q96TC7; Q3KNW5; Q9BXS9-3; Q99523; O43278-2; Q8N9I0; P02786; Q4KMG9; Q9K0U9; Q09057; Q9K0V0; P02786 EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Ion transport; Iron; Iron transport; Metal-binding; Methylation; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 35854.5 Length 324 Aromaticity 0.1 Instability index 34.17 Isoelectric point 7.58 Charge (pH=7) 1.42 2D Binding mode Binding energy (Kcal/mol) -5.44 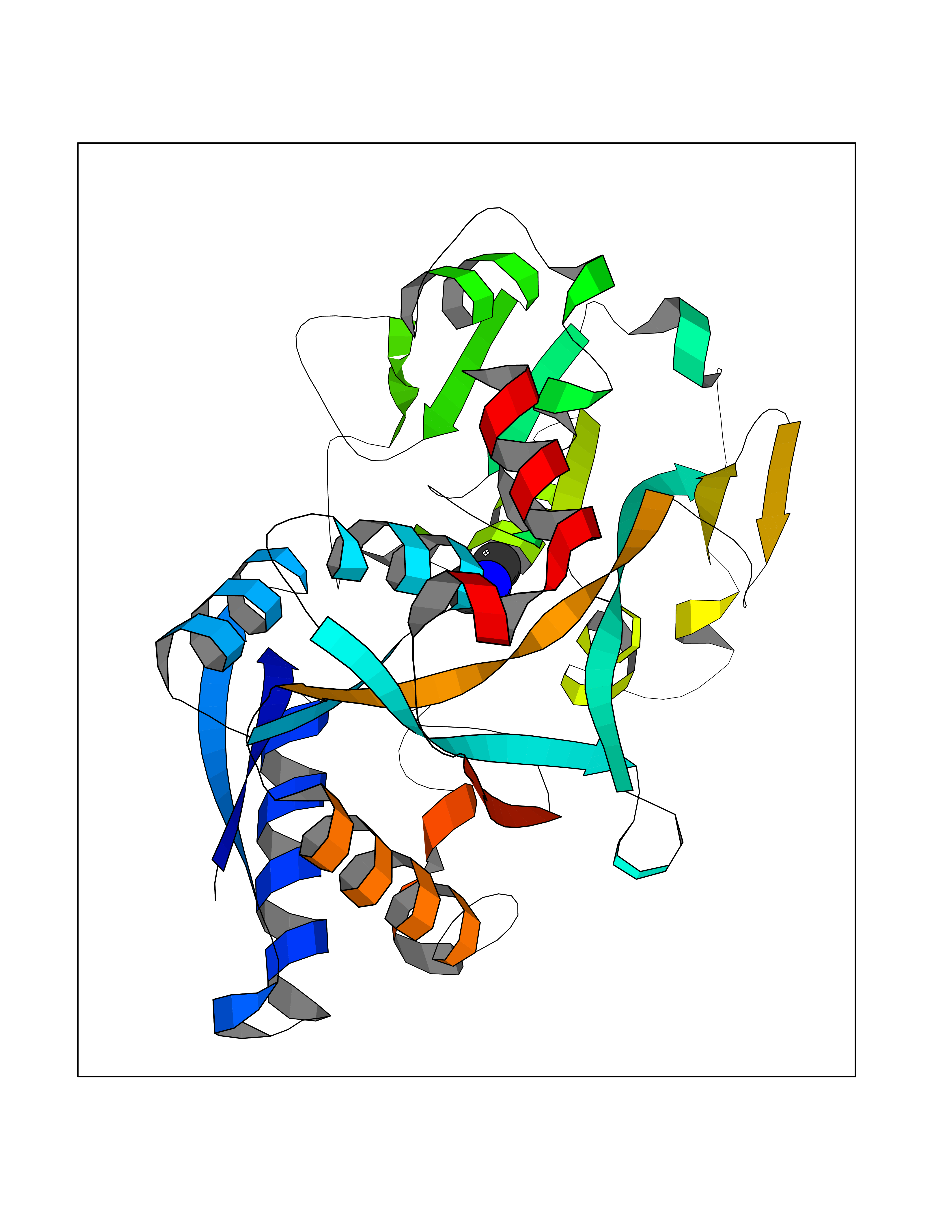 Molscript Map 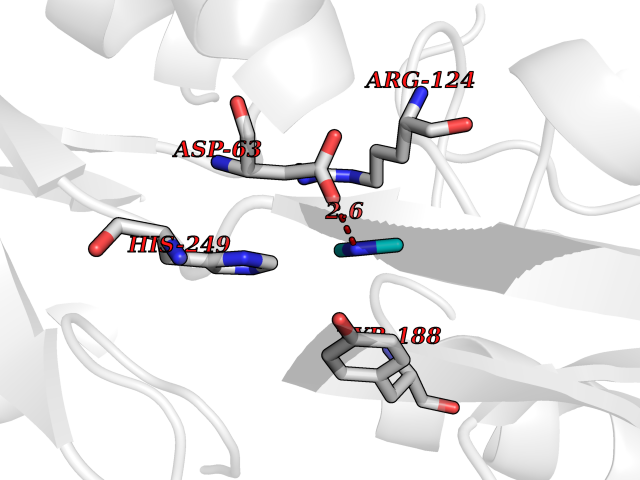 Pymol Map 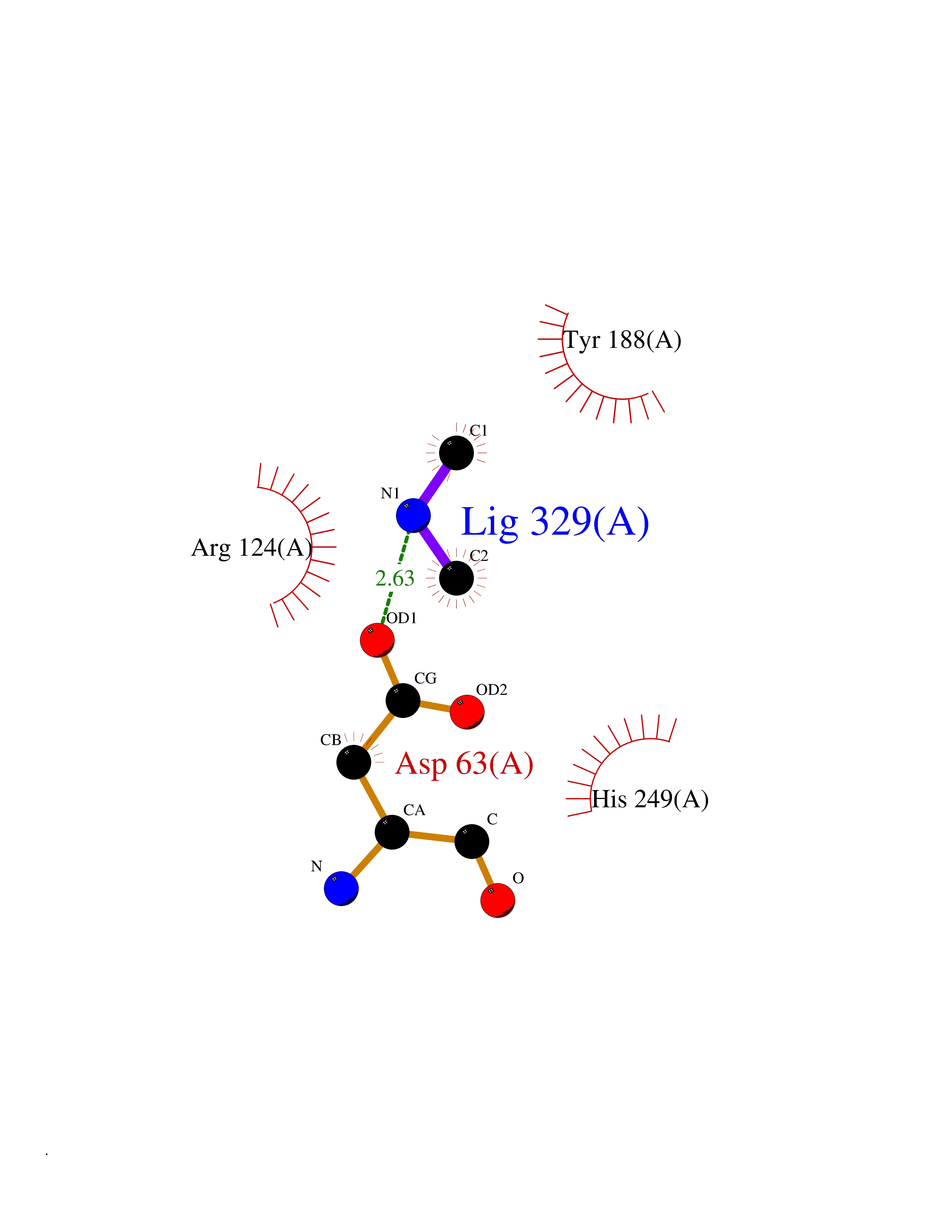 Ligplot Map 3D Binding mode Sequence KTVRWCAVSEHEATKCQSFRDHMKSVIPSDGPSVACVKKASYLDCIRAIAANEADAVTLDAGLVYDAYLAPNNLKPVVAEFYGSKEDPQTFYYAVAVVKKDSGFQMNQLRGKKSCHTGLGRSAGWNIPIGLLYCDLPEPRKPLEKAVANFFSGSCAPCADGTDFPQLCQLCPGCGCSTLNQYFGYSGAFKCLKDGAGDVAFVKHSTIFENLANKADRDQYELLCLDNTRKPVDEYKDCHLAQVPSHTVVARSMGGKEDLIWELLNQAQEHFGKDKSKEFQLFSSPHGKDLLFKDSAHGFLKVPPRMDAKMYLGYEYVTAIRNLR Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Steroid 17-alpha-monooxygenase (S17AH) | 3SWZ | 3.99 | |
Target general information Gen name CYP17A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Steroid 17-alpha-hydroxylase/17,20 lyase; P450c17; P450-C17; P450 17; CYPXVII; CYP17A1; CYP 17; 17 alpha-Hydroxylase/C17-20-lyase Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function Conversion of pregnenolone and progesterone to their 17- alpha-hydroxylated products and subsequently to dehydroepiandrosterone (DHEA) and androstenedione. Catalyzes both the 17-alpha-hydroxylation and the 17,20-lyase reaction. Involved in sexual development during fetal life and at puberty. Related diseases Adrenal hyperplasia 5 (AH5) [MIM:202110]: A form of congenital adrenal hyperplasia, a common recessive disease due to defective synthesis of cortisol. Congenital adrenal hyperplasia is characterized by androgen excess leading to ambiguous genitalia in affected females, rapid somatic growth during childhood in both sexes with premature closure of the epiphyses and short adult stature. Four clinical types: 'salt wasting' (SW, the most severe type), 'simple virilizing' (SV, less severely affected patients), with normal aldosterone biosynthesis, 'non-classic form' or late-onset (NC or LOAH) and 'cryptic' (asymptomatic). {ECO:0000269|PubMed:10720067, ECO:0000269|PubMed:11549685, ECO:0000269|PubMed:11836339, ECO:0000269|PubMed:12466376, ECO:0000269|PubMed:14671162, ECO:0000269|PubMed:1515452, ECO:0000269|PubMed:1714904, ECO:0000269|PubMed:1740503, ECO:0000269|PubMed:19793597, ECO:0000269|PubMed:24140098, ECO:0000269|PubMed:24498484, ECO:0000269|PubMed:25650406, ECO:0000269|PubMed:2808364, ECO:0000269|PubMed:8027220, ECO:0000269|PubMed:8245018, ECO:0000269|PubMed:8345056, ECO:0000269|PubMed:8396144, ECO:0000269|PubMed:8550762, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05812; DB04630; DB01424; DB09061; DB00882; DB01234; DB14649; DB01026; DB05667; DB14009; DB14011; DB00157; DB01708; DB00396; DB02901 Interacts with NA EC number EC 1.14.14.19 Uniprot keywords 3D-structure; Congenital adrenal hyperplasia; Disease variant; Endoplasmic reticulum; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Steroidogenesis Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 51385.8 Length 453 Aromaticity 0.08 Instability index 32.67 Isoelectric point 8.48 Charge (pH=7) 4.08 2D Binding mode Binding energy (Kcal/mol) -5.45  Molscript Map 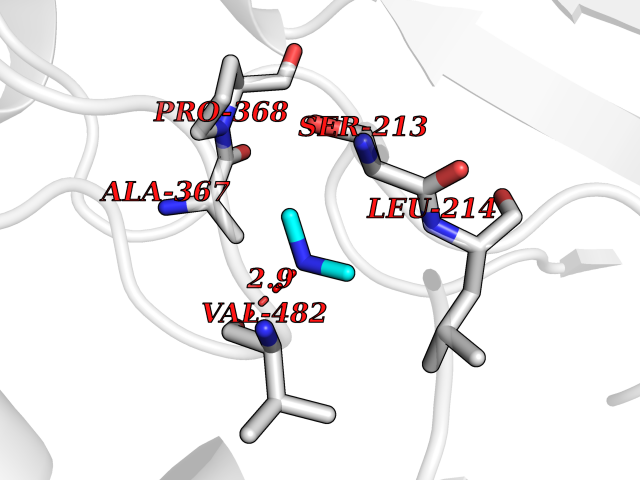 Pymol Map 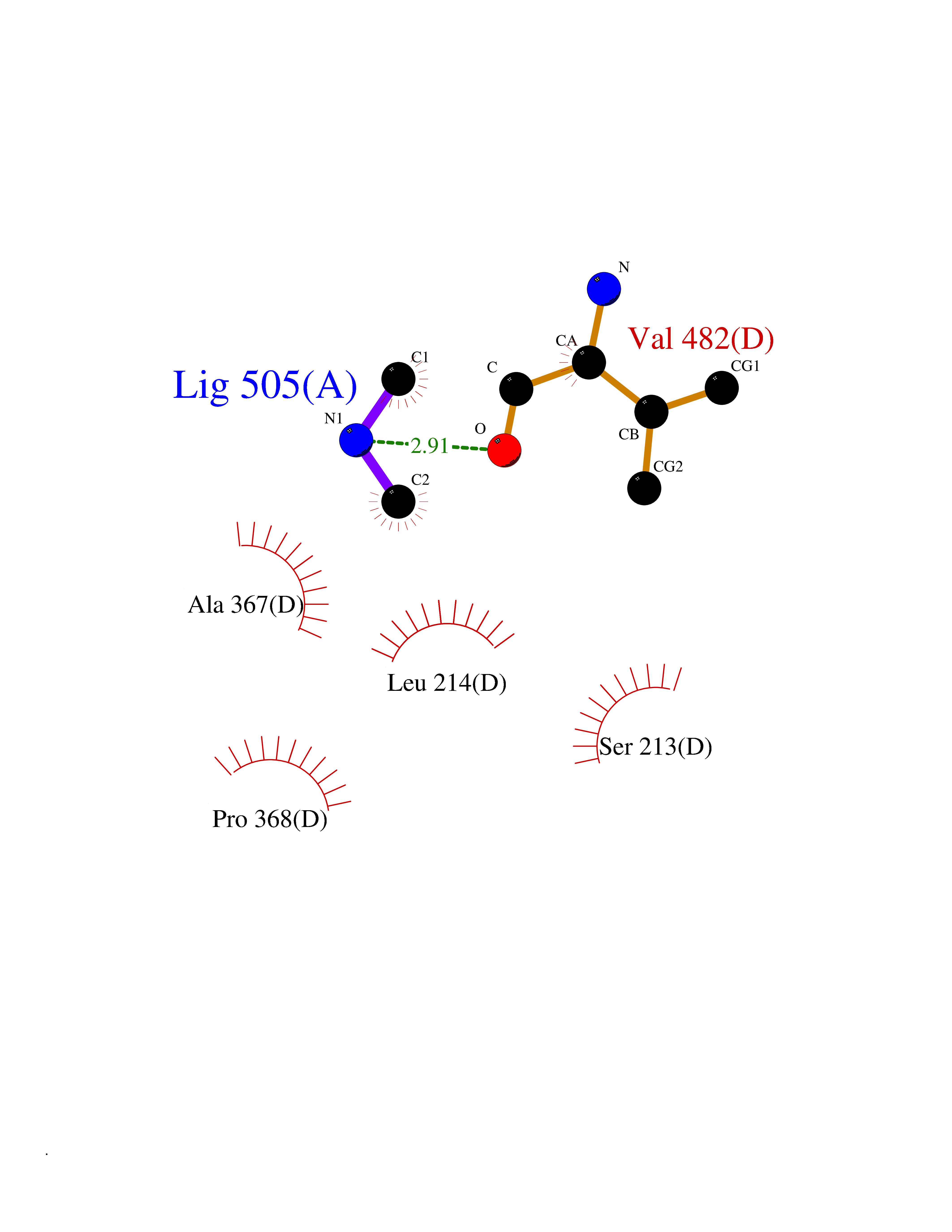 Ligplot Map 3D Binding mode Sequence LPRHGHMHNNFFKLQKKYGPIYSVRMGTKTTVIVGHHQLAKEVLIKKGKDFSGRPQMATLDIASNNRKGIAFADSGAHWQLHRRLAMATFALFKDGDQKLEKIICQEISTLCDMLATHNGQSIDISFPVFVAVTNVISLICFNTSYKNGDPELNVIQNYNEGIIDNLSKDSLVDLVPWLKIFPNKTLEKLKSHVKIRNDLLNKILENYKEKFRSDSITNMLDTLMQAKMNSDDSELLSDNHILTTIGDIFGAGVETTTSVVKWTLAFLLHNPQVKKKLYEEIDQNVGFSRTPTISDRNRLLLLEATIREVLRLRPVAPMLIPHKANVDSSIGEFAVDKGTEVIINLWALHHNEKEWHQPDQFMPERFLNPAGTQLISPSVSYLPFGAGPRSCIGEILARQELFLIMAWLLQRFDLEVPDDGQLPSLEGIPKVVFLIDSFKVKIKVRQAWREAQ Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Hydrogen peroxide-inducible genes activator | 1I69 | 3.99 | |
Target general information Gen name oxyR Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b3961;mor;momR;JW3933 Protein family LysR transcriptional regulatory family Biochemical class Transcription Function Bacterial-type RNA polymerase core promoter proximal region sequence-specific DNA binding.Transcription factor activity, bacterial-type RNA polymerase core promoter proximal region sequence-specific binding. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; Direct protein sequencing; Disulfide bond; DNA-binding; Glutathionylation; Oxidation; Reference proteome; Repressor; S-nitrosylation; Stress response; Transcription; Transcription regulation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 46013 Length 412 Aromaticity 0.07 Instability index 44.76 Isoelectric point 5.57 Charge (pH=7) -14.52 2D Binding mode Binding energy (Kcal/mol) -5.45 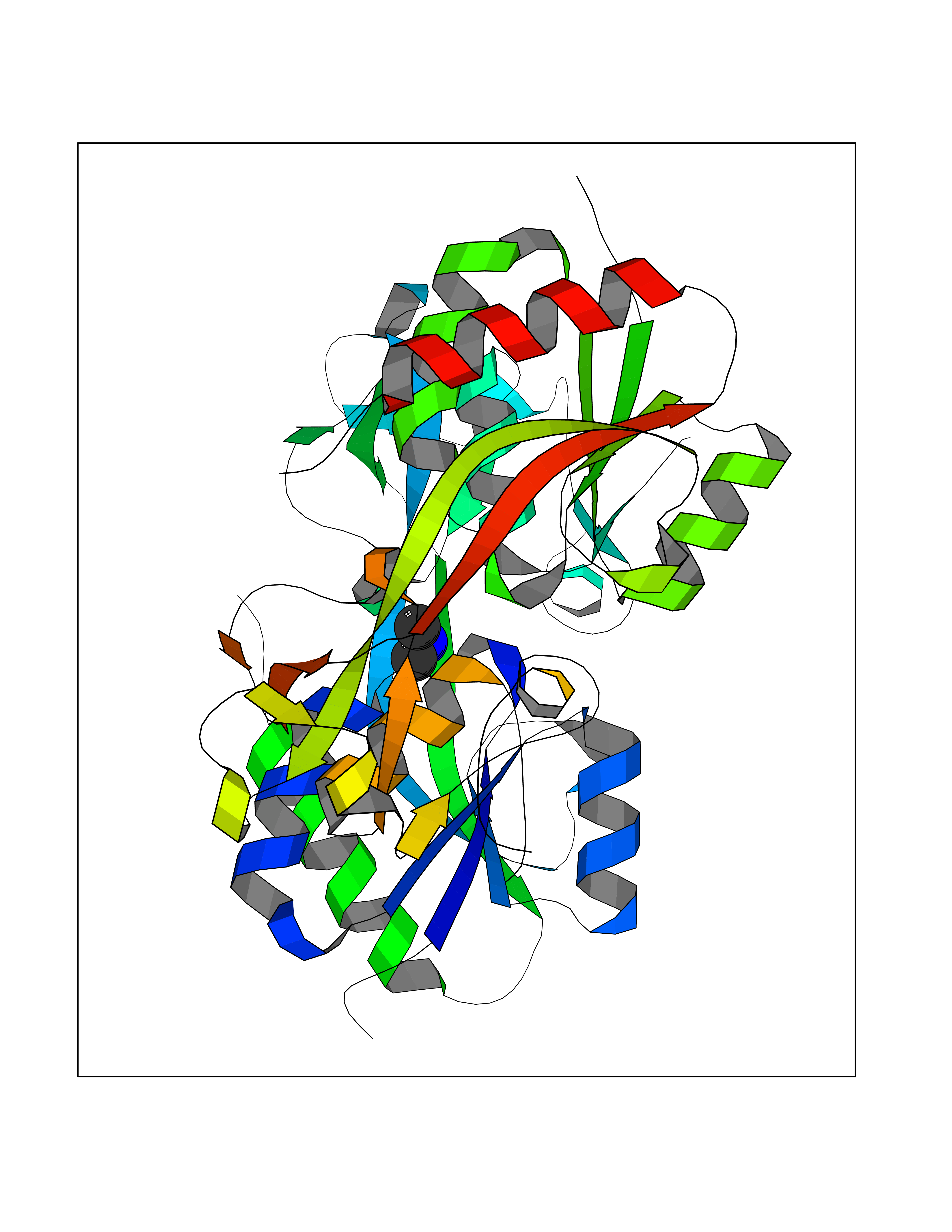 Molscript Map 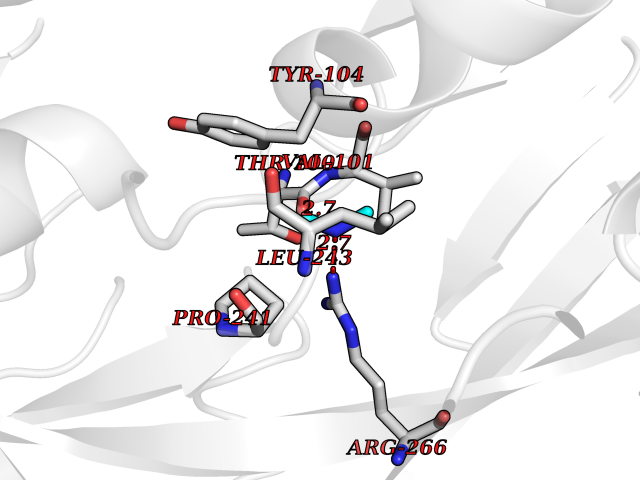 Pymol Map 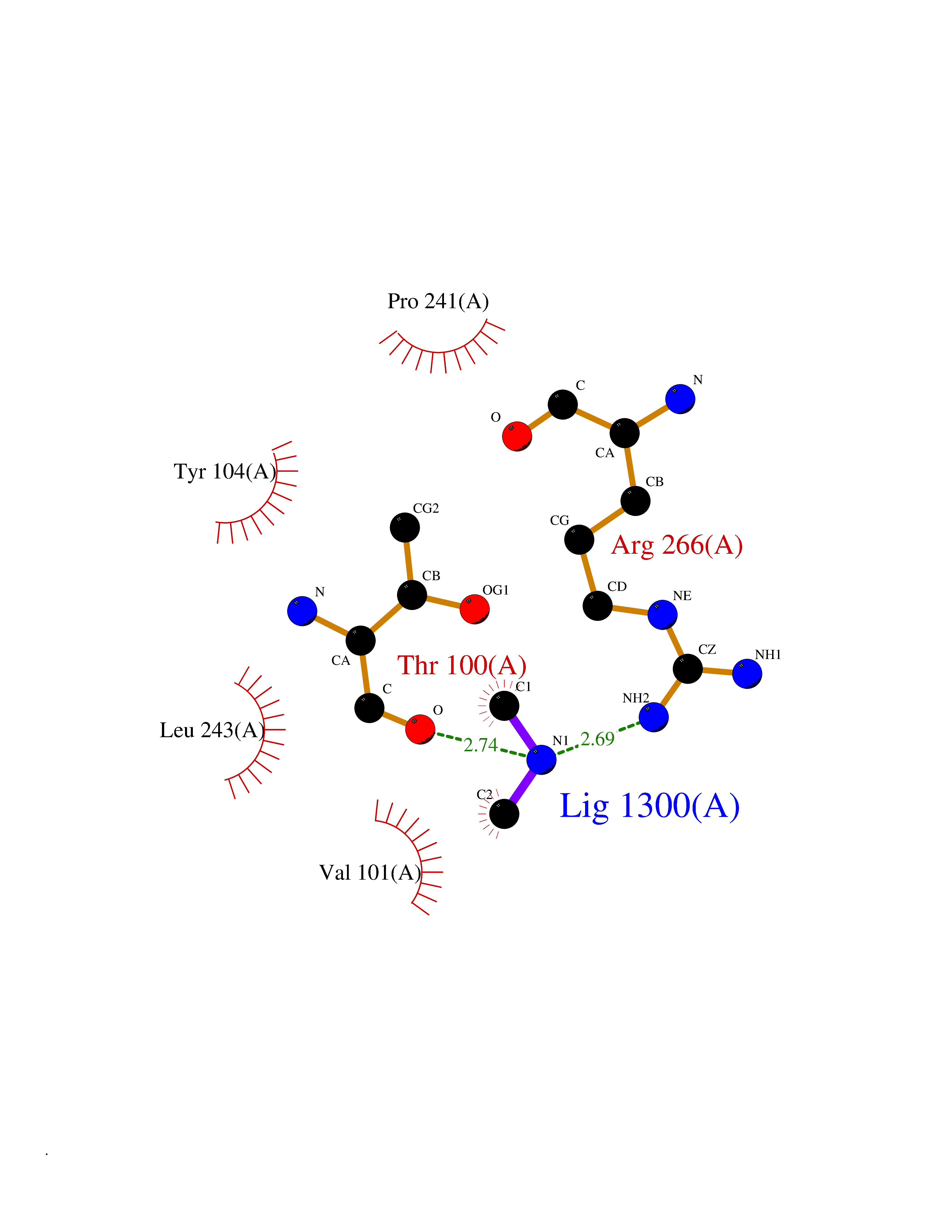 Ligplot Map 3D Binding mode Sequence ETMSGPLHIGLIPTVGPYLLPHIIPMLHQTFPKLEMYLHEAQTHQLLAQLDSGKLDAVILALVKESEAFIEVPLFDEPMLLAIYEDHPWANREAVPMADLAGEKLLMLEDGHSLRDQAMGFCFDTHFRATSLETLRNMVAAGSGITLLPALAVPPERKRDGVVYLPAIKPEPRRTIGLVYRPGSPLRSRYEQLAEAIRARMDGHFDETMSGPLHIGLIPTVGPYLLPHIIPMLHQTFPKLEMYLHEAQTHQLLAQLDSGKLDAVILALVKESEAFIEVPLFDEPMLLAIYEDHPWANREAVPMADLAGEKLLMLEDGHSLRDQAMGFCFETHFRATSLETLRNMVAAGSGITLLPALAVPPERKRDGVVYLPAIKPEPRRTIGLVYRPGSPLRSRYEQLAEAIRARMDGHFD Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Tartrate-resistant acid phosphatase type 5 | 2BQ8 | 3.99 | |
Target general information Gen name ACP5 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Metallophosphoesterase superfamily, Purple acid phosphatase family Biochemical class Hydrolase Function Acid phosphatase activity.Ferric iron binding.Ferrous iron binding. Related diseases Spondyloenchondrodysplasia with immune dysregulation (SPENCDI) [MIM:607944]: A disease characterized by vertebral and metaphyseal dysplasia, spasticity with cerebral calcifications, and strong predisposition to autoimmune diseases. The skeletal dysplasia is characterized by radiolucent and irregular spondylar and metaphyseal lesions that represent islands of chondroid tissue within bone. {ECO:0000269|PubMed:21217752, ECO:0000269|PubMed:21217755}. The disease is caused by variants affecting the gene represented in this entry. ACP5 inactivating mutations result in a functional excess of phosphorylated osteopontin causing deregulation of osteopontin signaling and consequential autoimmune disease. Drugs (DrugBank ID) NA Interacts with NA EC number 3.1.3.2 Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Iron; Lysosome; Metal-binding; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID X Molecular weight (Da) 34330.6 Length 304 Aromaticity 0.12 Instability index 42.3 Isoelectric point 9.11 Charge (pH=7) 6.75 2D Binding mode Binding energy (Kcal/mol) -5.44 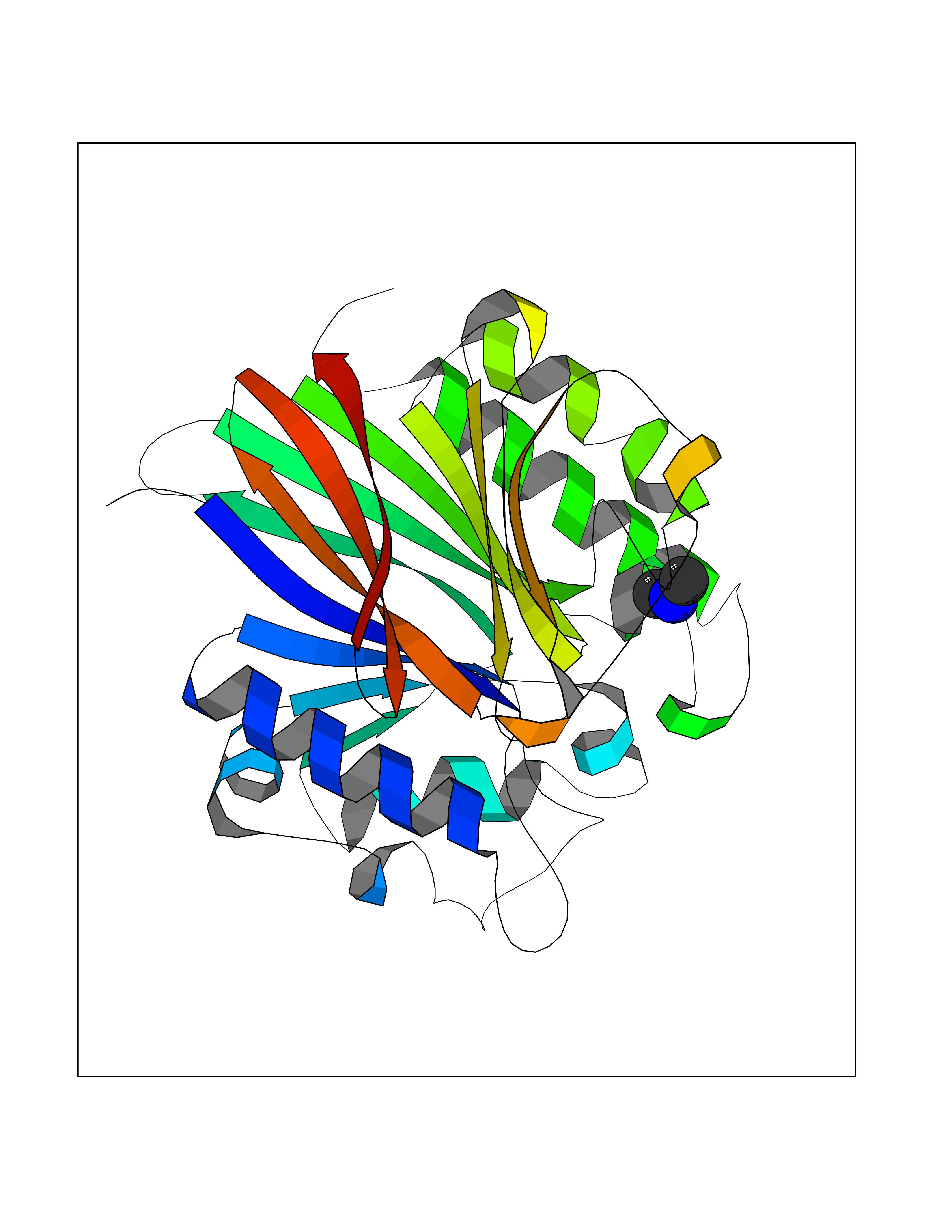 Molscript Map 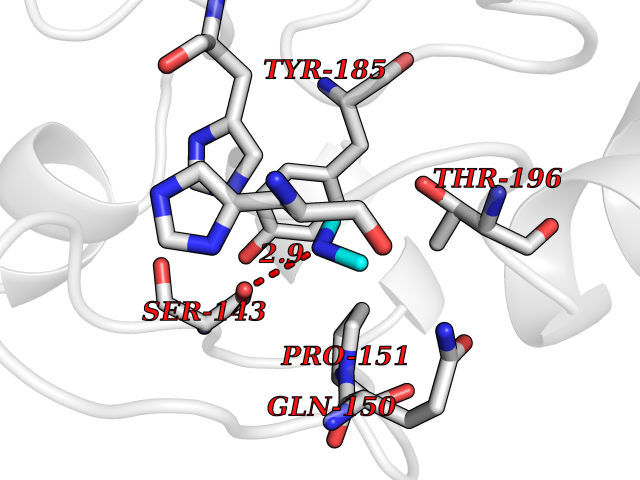 Pymol Map 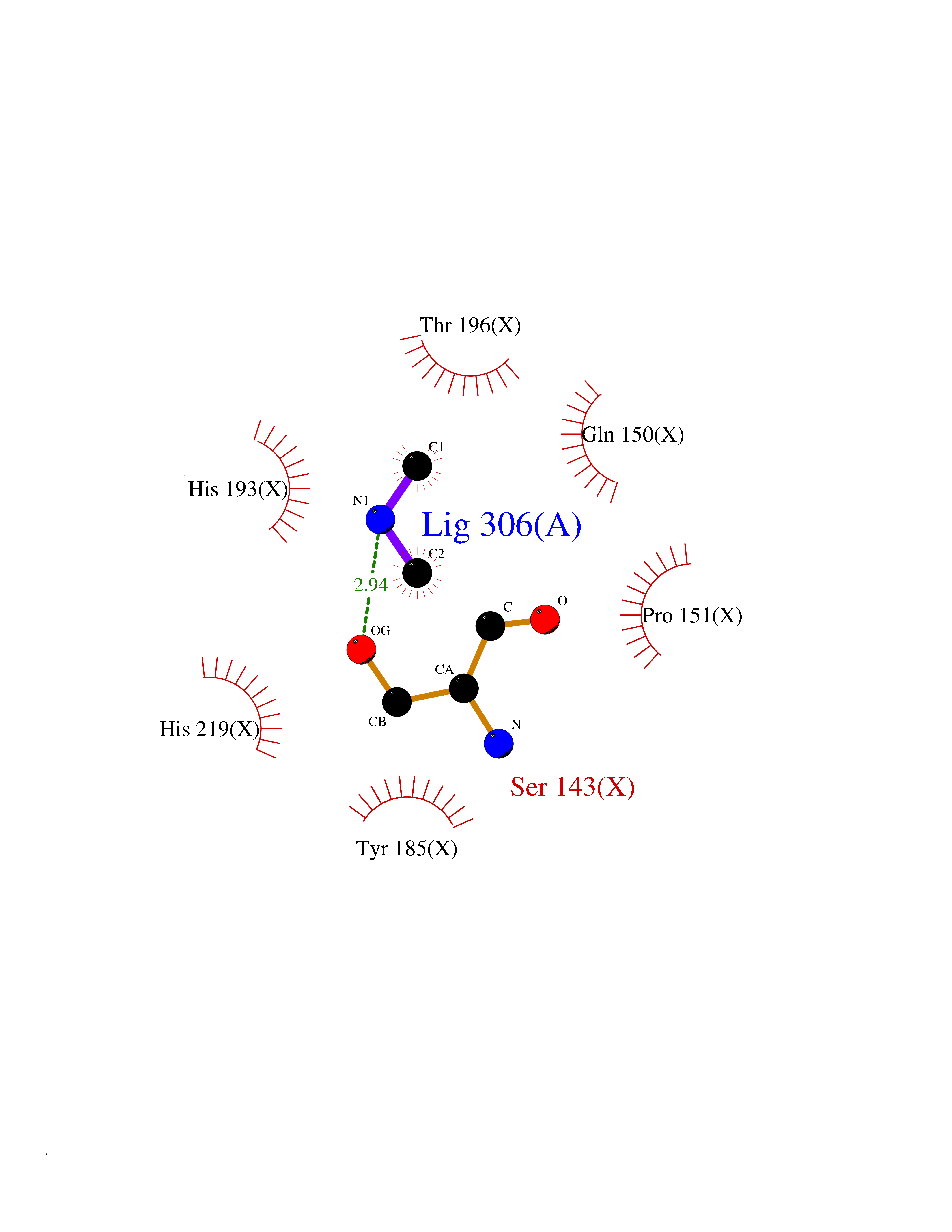 Ligplot Map 3D Binding mode Sequence ATPALRFVAVGDWGGVPNAPFHTAREMANAKEIARTVQILGADFILSLGDNFYFTGVQDINDKRFQETFEDVFSDRSLRKVPWYVLAGNHDHLGNVSAQIAYSKISKRWNFPSPFYRLHFKIPQTNVSVAIFMLDTVTLCGNSDDFLSQQPERPRDVKLARTQLSWLKKQLAAAREDYVLVAGHYPVWSIAEHGPTHCLVKQLRPLLATYGVTAYLCGHDHNLQYLQDENGVGYVLSGAGNFMDPSKRHQRKVPNGYLRFHYGTEDSLGGFAYVEISSKEMTVTYIEASGKSLFKTRLPRRARP Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Penicillin-binding protein 2B | 2WAD | 3.99 | |
Target general information Gen name penA Organism Streptococcus pneumoniae (strain ATCC BAA-255 / R6) Uniprot ID TTD ID NA Synonyms spr1517;pbp2b Protein family Transpeptidase family Biochemical class Peptide binding protein Function Penicillin binding. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01163; DB00415; DB08795; DB01140; DB00456; DB01066; DB00493; DB01331; DB01212; DB00567; DB03313; DB00485; DB00739; DB01603; DB00607; DB00713; DB00319 Interacts with NA EC number NA Uniprot keywords 3D-structure; Antibiotic resistance; Cell membrane; Cell shape; Cell wall biogenesis/degradation; Membrane; Peptidoglycan synthesis; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 65444.4 Length 607 Aromaticity 0.08 Instability index 30.15 Isoelectric point 4.95 Charge (pH=7) -20.68 2D Binding mode Binding energy (Kcal/mol) -5.44 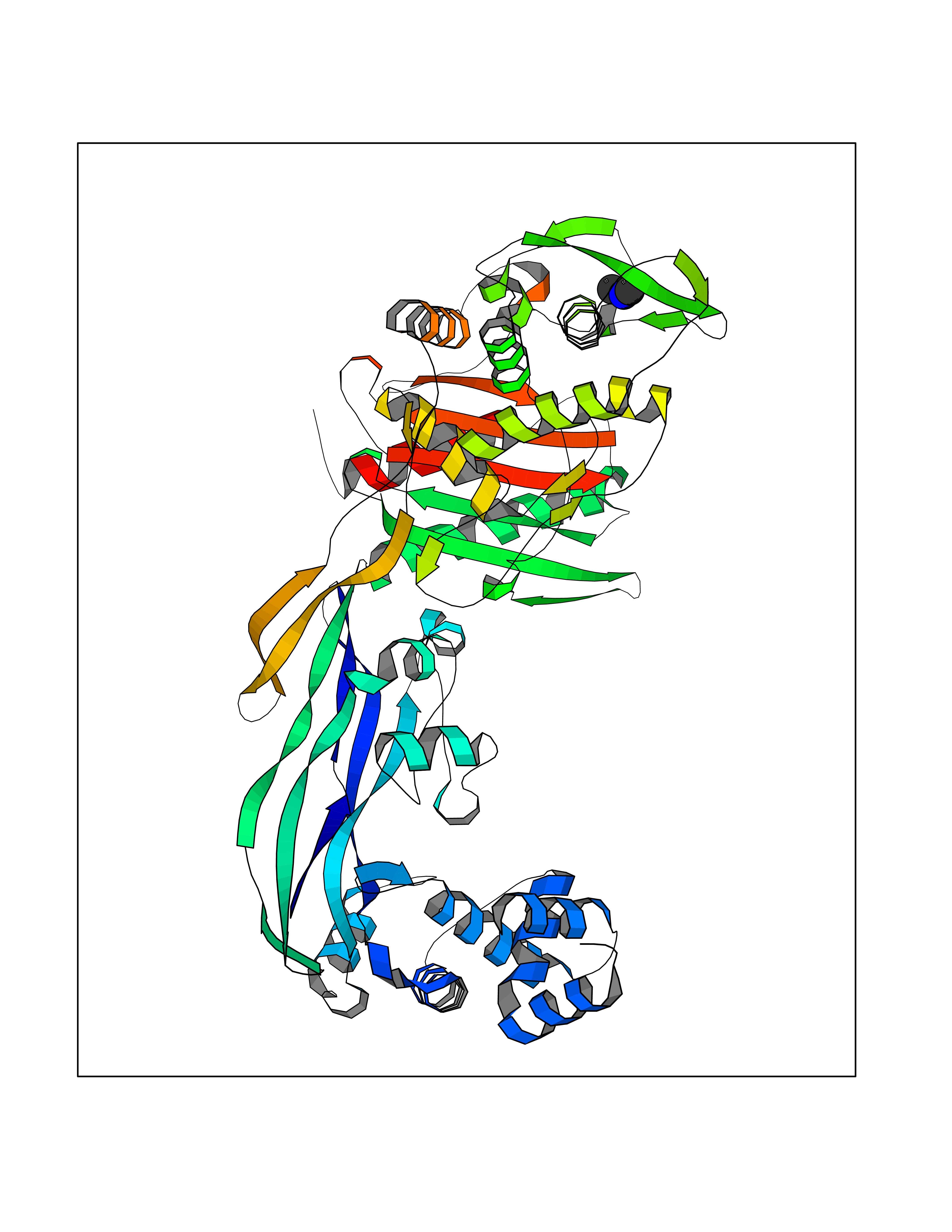 Molscript Map 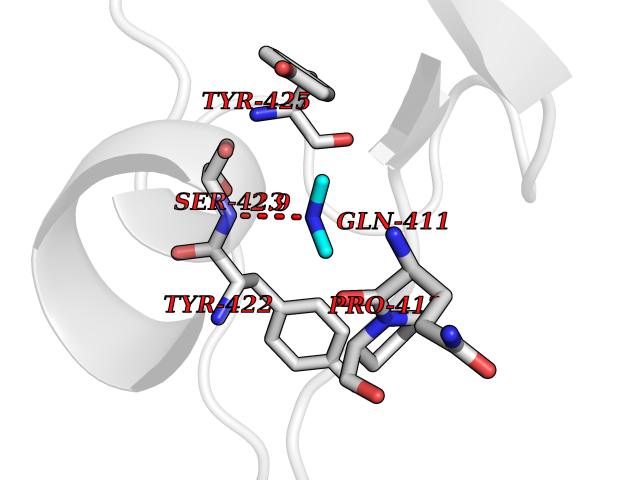 Pymol Map 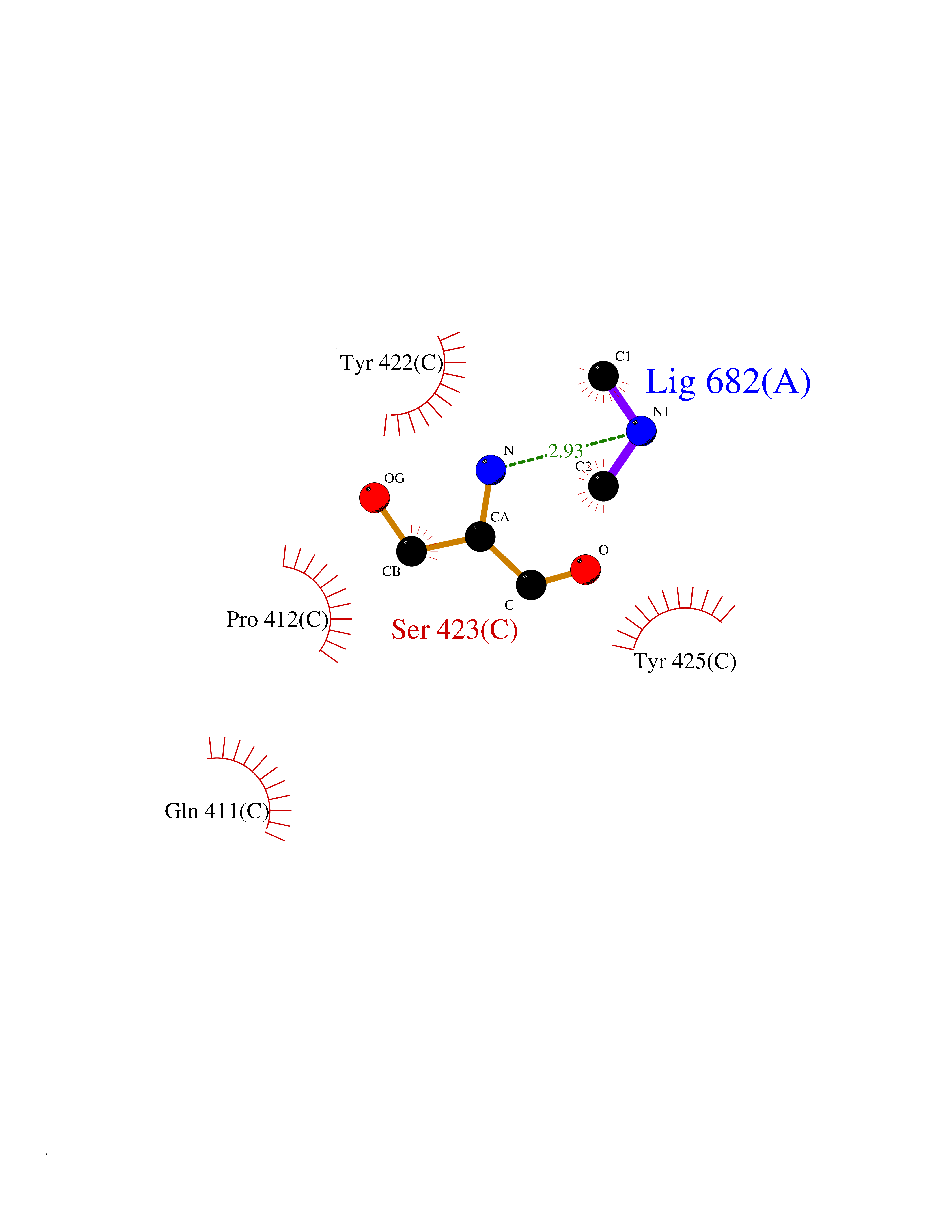 Ligplot Map 3D Binding mode Sequence SQTKVTTSSARGEIYDASGKPLVENTLKQVVSFTRSNKMTATDLKEIAKKLLTYVSISSPNLTERQLADYYLADPEIYKKTVEALPSESELYNNAVDSVPTSQLNYTEDEKKEIYLFSQLNAVGNFATGTIATDPLNDSQVAVIASISKEMPGISISTSWDRKILETSLSSIVGSVSSEKAGLPAEEAESYLKKGYSLNDRVGTSYLEKQYEEVLQGKRPVKEIHLDKHGDMESVENIEEGSKGKNIKLTIDLAFQDSVDALLKSYFNSELGNGGAKYSEGVYAVALNPQTGAVLSMSGLKHDLKTGELTPDSLGTVTNVFVPGSVVKAATISSGWENGVLSGNQTLTDQPIVFQGSAPIYSWYKLAYGSFPITAVEALEYSSNAYVVQTALGIMGQTYQPNMFVGTSNLESAMGKLRSTFGEYGLGSATGIDLPDESTGLVPKEYNFANFITNAFGQFDNYTPMQLAQYVATIANNGVRLAPHIVEGIYDNNDKGGLGELIQAIDTKEINKVNISESDMAILHQGFYQVSHGTSPLTTGRAFSDGATVSISGKTGTNTNAVAYAPTENPQIAVAVVFPHNTNLTKNVGPAIARDIINLYNQHHPMN Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Cholinesterase (BCHE) | 1P0I | 3.99 | |
Target general information Gen name BCHE Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Pseudocholinesterase; Choline esterase II; CHE1; Butyrylcholine esterase; Acylcholine acylhydrolase Protein family Type-B carboxylesterase/lipase family Biochemical class Type-B carboxylesterase/lipase Function Esterase with broad substrate specificity. Contributes to the inactivation of the neurotransmitter acetylcholine. Can degrade neurotoxic organophosphate esters. Related diseases Butyrylcholinesterase deficiency (BCHED) [MIM:617936]: An autosomal recessive metabolic condition characterized by increased sensitivity to certain anesthetic drugs, including the muscle relaxants succinylcholine or mivacurium. BCHED results in slower hydrolysis of these drugs and, consequently, a prolonged neuromuscular block, leading to apnea. The duration of the prolonged apnea varies significantly depending on the extent of the enzyme deficiency. {ECO:0000269|PubMed:10404729, ECO:0000269|PubMed:11928765, ECO:0000269|PubMed:12881446, ECO:0000269|PubMed:1306123, ECO:0000269|PubMed:1349196, ECO:0000269|PubMed:1415224, ECO:0000269|PubMed:15563885, ECO:0000269|PubMed:15781196, ECO:0000269|PubMed:1611188, ECO:0000269|PubMed:16788378, ECO:0000269|PubMed:17700357, ECO:0000269|PubMed:18075469, ECO:0000269|PubMed:18300943, ECO:0000269|PubMed:25054547, ECO:0000269|PubMed:25264279, ECO:0000269|PubMed:2915989, ECO:0000269|PubMed:7634491, ECO:0000269|PubMed:8554068, ECO:0000269|PubMed:8680411, ECO:0000269|PubMed:9110359, ECO:0000269|PubMed:9191541, ECO:0000269|PubMed:9388484, ECO:0000269|PubMed:9543549, ECO:0000269|PubMed:9694584}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08200; DB08201; DB03814; DB07940; DB03672; DB03128; DB08897; DB01122; DB06692; DB01408; DB00868; DB06756; DB11148; DB03568; DB04250; DB06774; DB01161; DB00856; DB00477; DB00122; DB14006; DB00527; DB00515; DB04920; DB00907; DB00979; DB01245; DB00944; DB11397; DB02811; DB00711; DB00449; DB07681; DB00843; DB01135; DB01057; DB01010; DB01364; DB03822; DB08658; DB00674; DB00941; DB00762; DB06636; DB00677; DB01064; DB01221; DB00772; DB00888; DB00358; DB02845; DB08893; DB01226; DB09205; DB01400; DB00585; DB00892; DB01337; DB00082; DB00183; DB00790; DB04892; DB03976; DB01338; DB00733; DB01035; DB00721; DB00392; DB09288; DB00545; DB00178; DB05386; DB00989; DB05875; DB00202; DB00391; DB00382; DB00871; DB04572; DB14031; DB00620; DB00508; DB01116; DB01199 Interacts with P54252; P46379-2; P06276; P55212; O75190-2; O14901; P13473-2; O75400-2; P62826; P67812; P02814 EC number EC 3.1.1.8 Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Phosphoprotein; Proteomics identification; Reference proteome; Secreted; Serine esterase; Sialic acid; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 58935.5 Length 523 Aromaticity 0.13 Instability index 38.71 Isoelectric point 8.63 Charge (pH=7) 5.36 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map 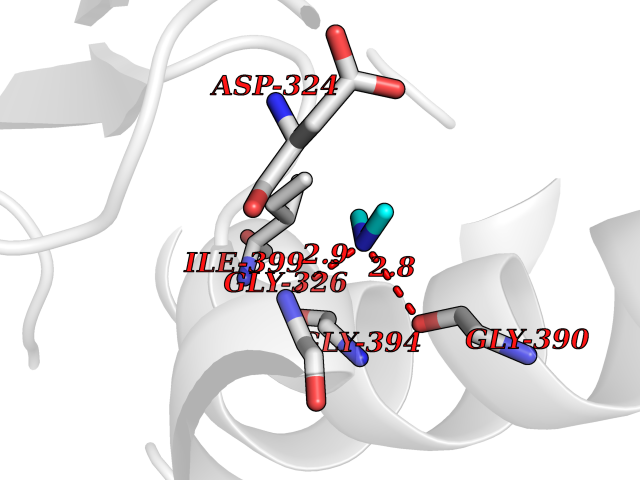 Pymol Map 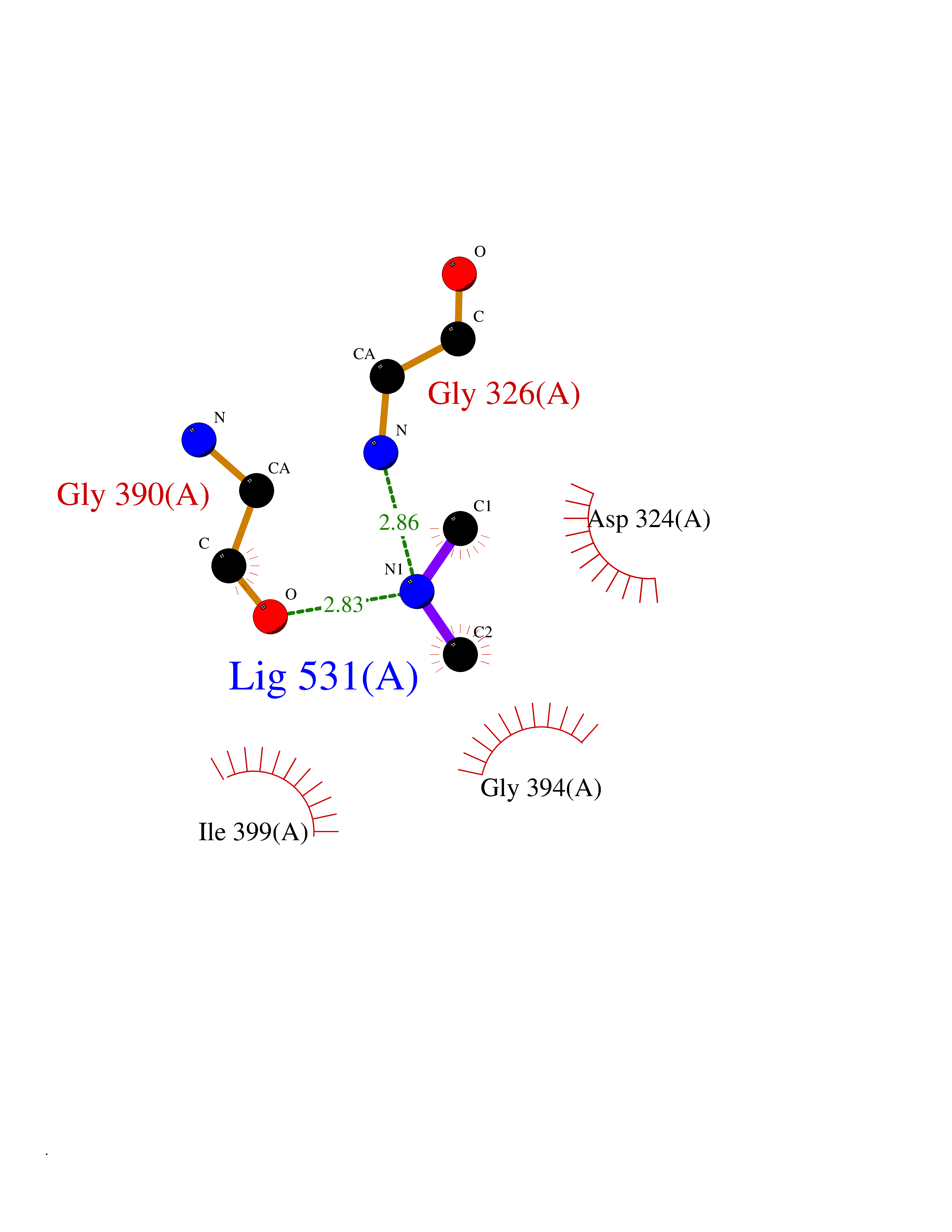 Ligplot Map 3D Binding mode Sequence IIIATKNGKVRGMQLTVFGGTVTAFLGIPYAQPPLGRLRFKKPQSLTKWSDIWNATKYANSCCQNIDQSFPGFHGSEMWNPNTDLSEDCLYLNVWIPAPKPKNATVLIWIYGGGFQTGTSSLHVYDGKFLARVERVIVVSMNYRVGALGFLALPGNPEAPGNMGLFDQQLALQWVQKNIAAFGGNPKSVTLFGESAGAASVSLHLLSPGSHSLFTRAILQSGSFNAPWAVTSLYEARNRTLNLAKLTGCSRENETEIIKCLRNKDPQEILLNEAFVVPYGTPLSVNFGPTVDGDFLTDMPDILLELGQFKKTQILVGVNKDEGTAFLVYGAPGFSKDNNSIITRKEFQEGLKIFFPGVSEFGKESILFHYTDWVQRPENYREALGDVVGDYNFICPALEFTKKFSEWGNNAFFYYFEHRSSKLPWPEWMGVMHGYEIEFVFGLPLERRDYTKAEEILSRSIVKRWANFAKYGNPQETQNQSTSWPVFKSTEQKYLTLNTESTRIMTKLRAQQCRFWTSFFPKV Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Sphingomyelin phosphodiesterase | 5I85 | 3.99 | |
Target general information Gen name SMPD1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ASM Protein family Acid sphingomyelinase family Biochemical class Hydrolase Function Acid sphingomyelin phosphodiesterase activity.Hydrolase activity, acting on glycosyl bonds.Sphingomyelin phosphodiesterase activity.Zinc ion binding. Related diseases Niemann-Pick disease A (NPDA) [MIM:257200]: An early-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Niemann-Pick disease type A is a primarily neurodegenerative disorder characterized by onset within the first year of life, intellectual disability, digestive disorders, failure to thrive, major hepatosplenomegaly, and severe neurologic symptoms. The severe neurological disorders and pulmonary infections lead to an early death, often around the age of four. Clinical features are variable. A phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. {ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1391960, ECO:0000269|PubMed:15221801, ECO:0000269|PubMed:15877209, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:1718266, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:2023926, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430884, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:8680412, ECO:0000269|PubMed:8693491, ECO:0000269|PubMed:9266408, ECO:0000269|PubMed:9660788}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Niemann-Pick disease B (NPDB) [MIM:607616]: A late-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Clinical signs involve only visceral organs. The most constant sign is hepatosplenomegaly which can be associated with pulmonary symptoms. Patients remain free of neurologic manifestations. However, a phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. In Niemann-Pick disease type B, onset of the first symptoms occurs in early childhood and patients can survive into adulthood. {ECO:0000269|PubMed:12369017, ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1301192, ECO:0000269|PubMed:15241805, ECO:0000269|PubMed:16010684, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:16472269, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:1885770, ECO:0000269|PubMed:19050888, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:21098024, ECO:0000269|PubMed:21621718, ECO:0000269|PubMed:22613662, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430512, ECO:0000269|PubMed:25920558, ECO:0000269|PubMed:26084044, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:27659707, ECO:0000269|PubMed:8051942, ECO:0000269|PubMed:8664904}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00381; DB12151; DB00477; DB01151; DB14009 Interacts with P55210 EC number 3.1.4.12; 3.1.4.3 Uniprot keywords 3D-structure; Alternative splicing; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Glycosidase; Host-virus interaction; Hydrolase; Lipid droplet; Lipid metabolism; Lysosome; Metal-binding; Neurodegeneration; Niemann-Pick disease; Phosphoprotein; Proteomics identification; Reference proteome; Secreted; Signal; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 58913.8 Length 528 Aromaticity 0.11 Instability index 46.93 Isoelectric point 6.48 Charge (pH=7) -3.6 2D Binding mode Binding energy (Kcal/mol) -5.44 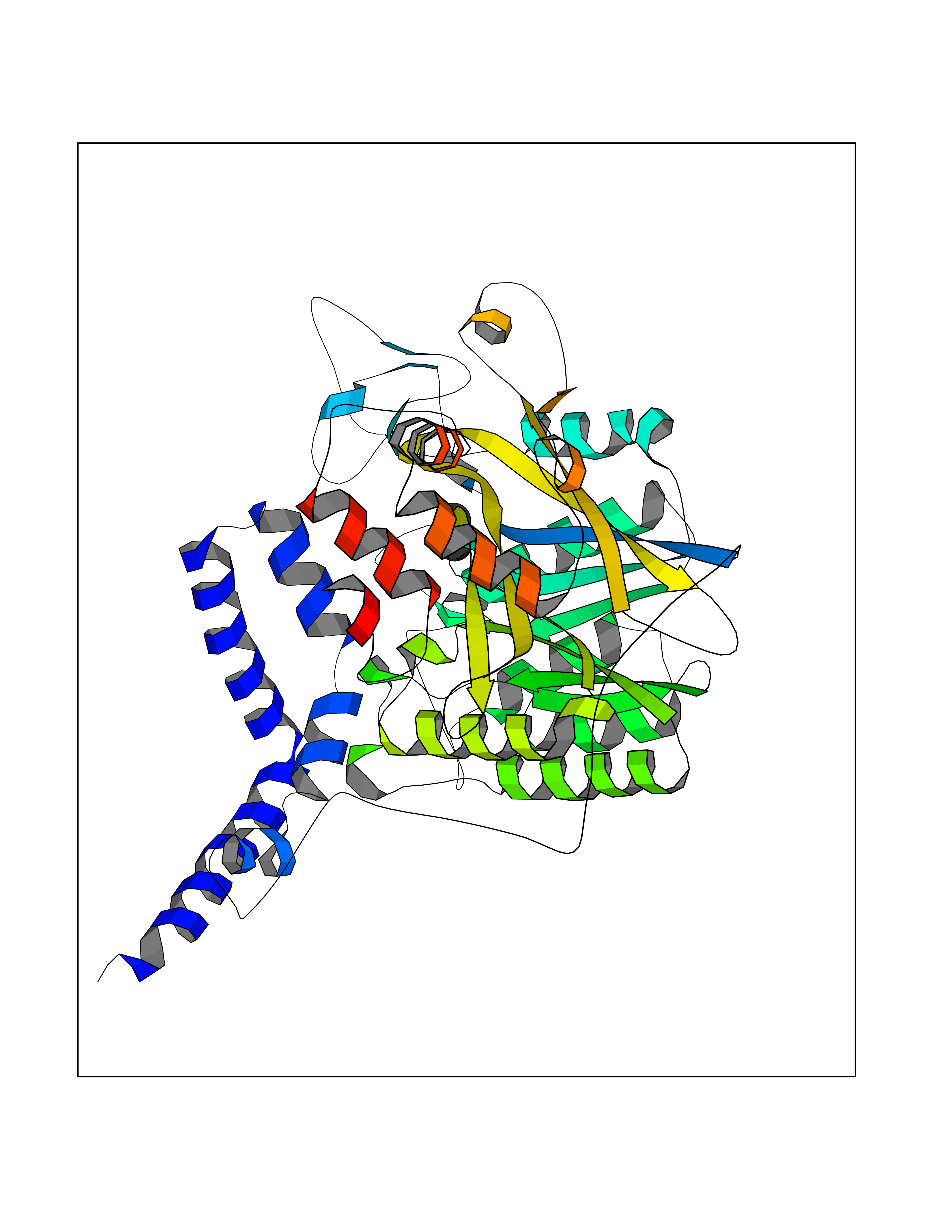 Molscript Map 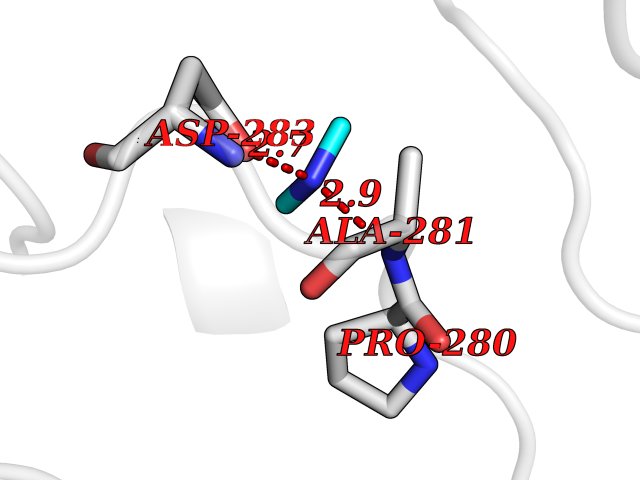 Pymol Map 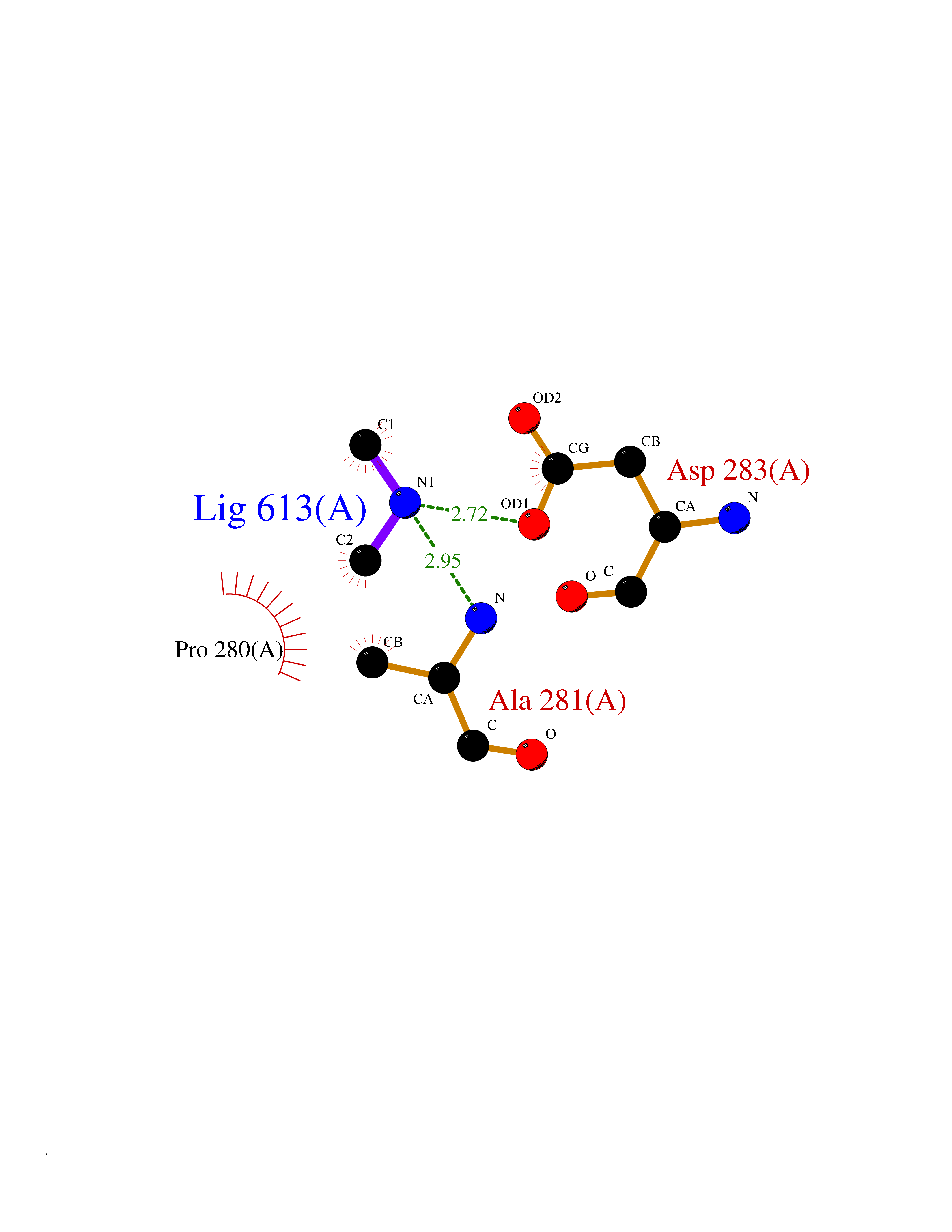 Ligplot Map 3D Binding mode Sequence WGNLTCPICKGLFTAINLGLKKEPNVARVGSVAIKLCNLLKIAPPAVCQSIVHLFEDDMVEVWRRSVLSPSEACGLLLGSTCGHWDIFSSWNISLPTVPKPPPKPPSPPAPGAPVSRILFLTDLHWDHDYLEGTDPDCADPLCCRRGSGLPPASRPGAGYWGEYSKCDLPLRTLESLLSGLGPAGPFDMVYWTGDIPAHDVWHQTRQDQLRALTTVTALVRKFLGPVPVYPAVGNHESTPVNSFPPPFIEGNHSSRWLYEAMAKAWEPWLPAEALRTLRIGGFYALSPYPGLRLISLNMNFCSRENFWLLINSTDPAGQLQWLVGELQAAEDRGDKVHIIGHIPPGHCLKSWSWNYYRIVARYENTLAAQFFGHTHVDEFEVFYDEETLSRPLAVAFLAPSATTYIGLNPGYRVYQIDGNYSGSSHVVLDHETYILNLTQANIPGAIPHWQLLYRARETYGLPNTLPTAWHNLVYRMRGDMQLFQTFWFLYHKGHPPSEPCGTPCRLATLCAQLSARADSPALCRHLM Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Aromatic-L-amino-acid decarboxylase (DDC) | 3RCH | 3.99 | |
Target general information Gen name DDC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms DOPA decarboxylase; AADC Protein family Group II decarboxylase family Biochemical class Carbon-carbon lyase Function Catalyzes the decarboxylation of L-3,4-dihydroxyphenylalanine (DOPA) to dopamine, L-5-hydroxytryptophan to serotonin and L-tryptophan to tryptamine. Related diseases Aromatic L-amino-acid decarboxylase deficiency (AADCD) [MIM:608643]: An inborn error in neurotransmitter metabolism that leads to combined serotonin and catecholamine deficiency. It causes developmental and psychomotor delay, poor feeding, lethargy, ptosis, intermittent hypothermia, gastrointestinal disturbances. The onset is early in infancy and inheritance is autosomal recessive. {ECO:0000269|PubMed:14991824, ECO:0000269|PubMed:15079002, ECO:0000269|Ref.12}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB12783; DB00190; DB00260; DB06262; DB13848; DB00875; DB01235; DB00968; DB00114; DB00150 Interacts with P10275 EC number EC 4.1.1.28 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Catecholamine biosynthesis; Decarboxylase; Disease variant; Lyase; Proteomics identification; Pyridoxal phosphate; Reference proteome; Repeat Protein physicochemical properties Chain ID A,B Molecular weight (Da) 41117.4 Length 369 Aromaticity 0.09 Instability index 37.52 Isoelectric point 8.26 Charge (pH=7) 3.76 2D Binding mode Binding energy (Kcal/mol) -5.44 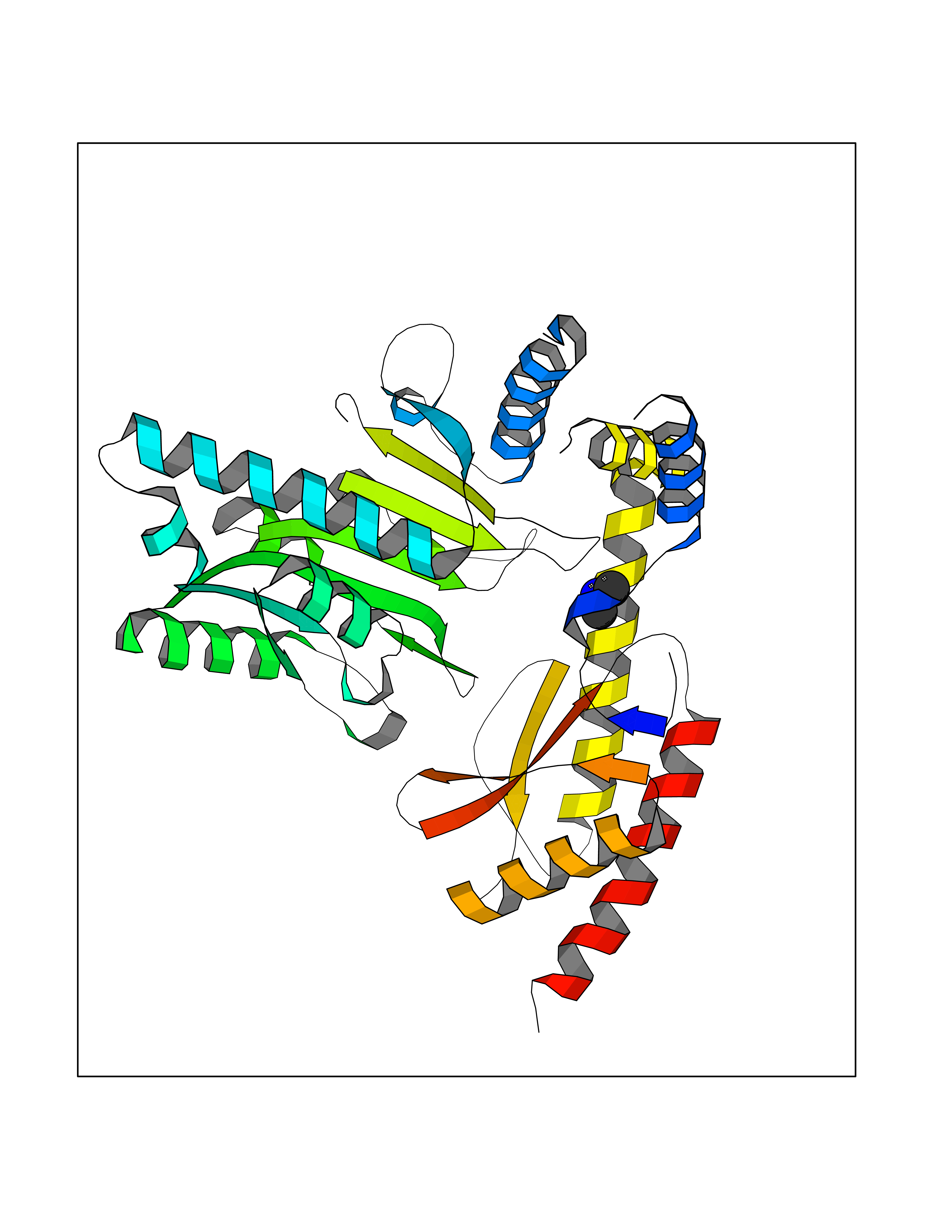 Molscript Map 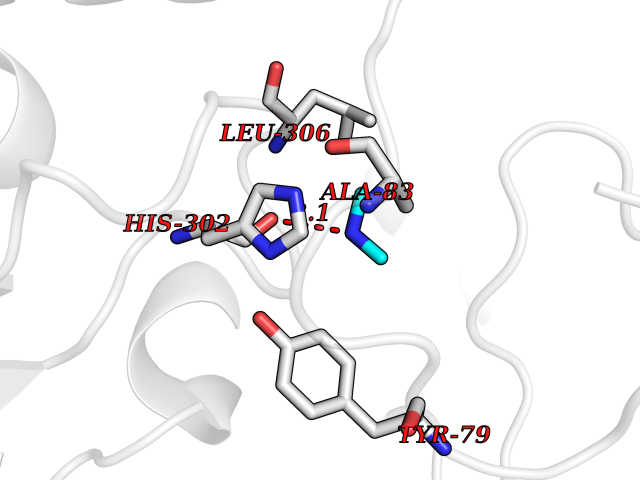 Pymol Map 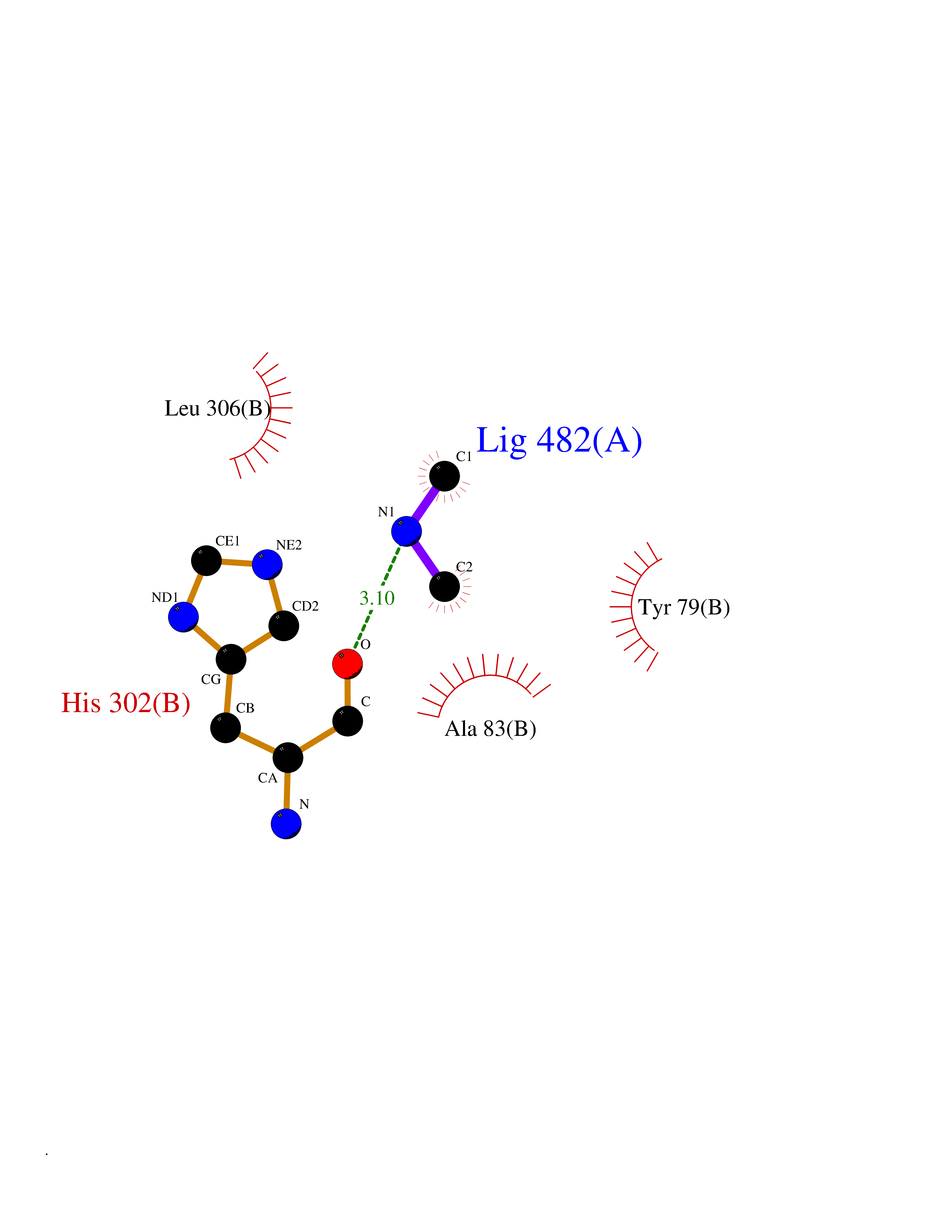 Ligplot Map 3D Binding mode Sequence HSPYFFAYFPTASSYPAMLADMLCGAIGASPACTELETVMMDWLGKMLELPKAFLNEKAGEGGGVIQGSASEATLVALLAARTKVIHRLQAASPELTQAAIMEKLVAYSSDQAHSSVERAGLIGGVKLKAIPSDGNFAMRASALQEALERDKAAGLIPFFMVATLGTTTCCSFDNLLEVGPICNKEDIWLHVDAAYAGSAFICPEFRHLLNGVEFADSFNFNPHKWLLVNFDCSAMWVKKRTDLRFRSLKMWFVFRMYGVKGLQAYIRKHVQLSHEFESLVRQDPRFEICVEVILGLVCFRLKGSNKVNEALLQRINSAKKIHLVPCHLRDKFVLRFAICSRTVESAHVQRAWEHIKELAADVLRAERE Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Caspase-3 (CASP3) | 2XYG | 3.99 | |
Target general information Gen name CASP3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Yama protein; SREBP cleavage activity 1; SCA-1; Protein Yama; Cysteine protease CPP32; Caspase 3; CPP32; CPP-32; CASP-3; Apopain Protein family Peptidase C14A family Biochemical class Peptidase Function At the onset of apoptosis it proteolytically cleaves poly(ADP-ribose) polymerase (PARP) at a '216-Asp-|-Gly-217' bond. Cleaves and activates sterol regulatory element binding proteins (SREBPs) between the basic helix-loop-helix leucine zipper domain and the membrane attachment domain. Cleaves and activates caspase-6, -7 and -9. Involved in the cleavage of huntingtin. Triggers cell adhesion in sympathetic neurons through RET cleavage. Involved in the activation cascade of caspases responsible for apoptosis execution. Related diseases Smith-Kingsmore syndrome (SKS) [MIM:616638]: An autosomal dominant syndrome characterized by intellectual disability, macrocephaly, seizures, umbilical hernia, and facial dysmorphic features. {ECO:0000269|PubMed:25851998, ECO:0000269|PubMed:26542245, ECO:0000269|PubMed:27830187}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Focal cortical dysplasia 2 (FCORD2) [MIM:607341]: A form of focal cortical dysplasia, a malformation of cortical development that results in medically refractory epilepsy in the pediatric population and in adults. FCORD2 is a severe form, with onset usually in childhood, characterized by disrupted cortical lamination and specific cytological abnormalities. It is classified in 2 subtypes: type IIA characterized by dysmorphic neurons and lack of balloon cells; type IIB with dysmorphic neurons and balloon cells. {ECO:0000269|PubMed:25799227, ECO:0000269|PubMed:25878179, ECO:0000269|PubMed:26018084, ECO:0000269|PubMed:27830187}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08498; DB08497; DB08213; DB06862; DB08251; DB03124; DB08229; DB00945; DB05408; DB13751; DB06255; DB07696; DB01017; DB08499; DB12843; DB13048; DB00282; DB12709 Interacts with O43823; Q9Y243; P05067; P54252; P55212; P55211; Q14203-5; P42858; Q00987; O60551; P09874; Q5JUK2; P10599; Q9BYP7; P98170 EC number EC 3.4.22.56 Uniprot keywords 3D-structure; Acetylation; Apoptosis; Cytoplasm; Direct protein sequencing; Hydrolase; Phosphoprotein; Protease; Proteomics identification; Reference proteome; S-nitrosylation; Thiol protease; Ubl conjugation; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 27483.1 Length 239 Aromaticity 0.11 Instability index 38.09 Isoelectric point 8.39 Charge (pH=7) 3.03 2D Binding mode Binding energy (Kcal/mol) -5.44 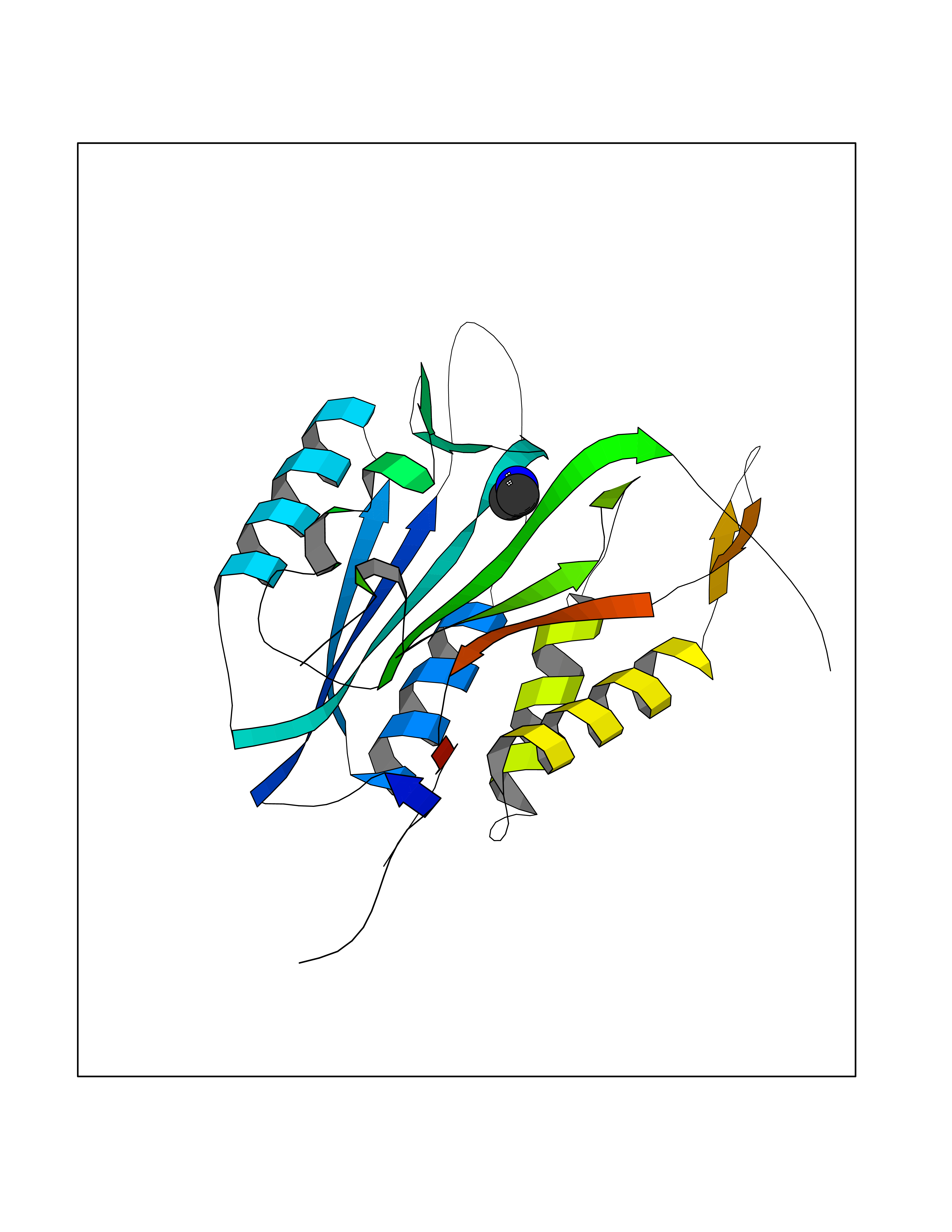 Molscript Map 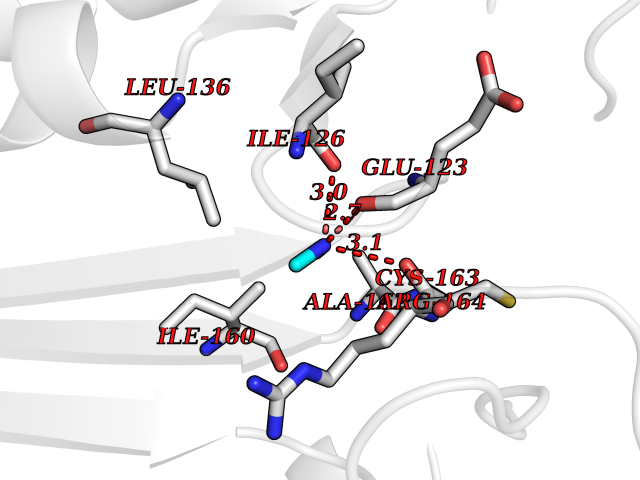 Pymol Map 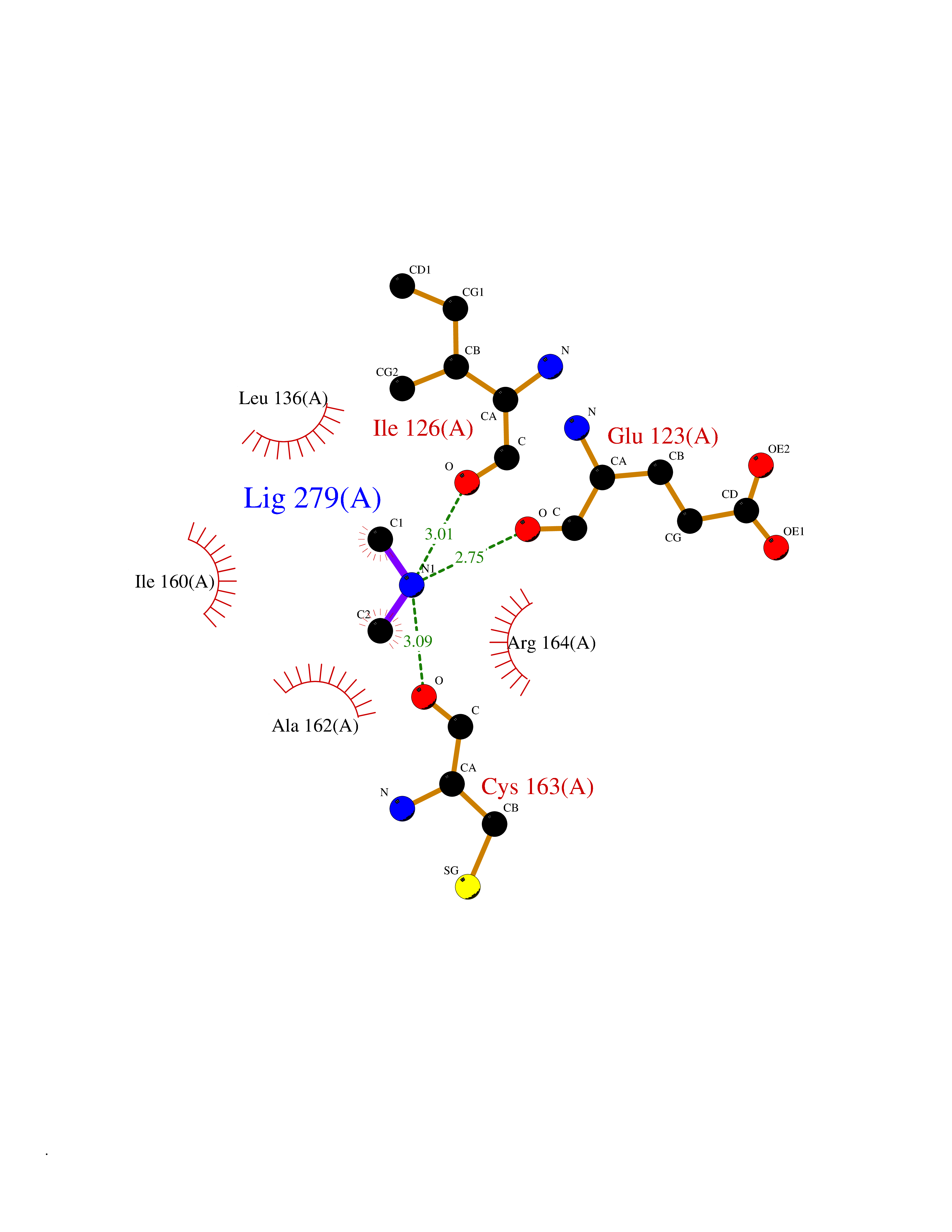 Ligplot Map 3D Binding mode Sequence SGISLDNSYKMDYPEMGLCIIINNKNFHKSTGMTSRSGTDVDAANLRETFRNLKYEVRNKNDLTREEIVELMRDVSKEDHSKRSSFVCVLLSHGEEGIIFGTNGPVDLKKITNFFRGDRCRSLTGKPKLFIIQACRGTELDCGIETHKIPVEADFLYAYSTAPGYYSWRNSKDGSWFIQSLCAMLKQYADKLEFMHILTRVNRKVATEFESFSFDATFHAKKQIPCIVSMLTKELYFYH Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | 4-hydroxyphenylpyruvate dioxygenase | 3ISQ | 3.99 | |
Target general information Gen name HPD Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PPD Protein family 4HPPD family Biochemical class Oxidoreductase Function 4-hydroxyphenylpyruvate dioxygenase activity.Metal ion binding. Related diseases Tyrosinemia 3 (TYRSN3) [MIM:276710]: An inborn error of metabolism characterized by elevations of tyrosine in the blood and urine, seizures and mild intellectual disability. {ECO:0000269|PubMed:10942115, ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hawkinsinuria (HWKS) [MIM:140350]: An inborn error of tyrosine metabolism characterized by failure to thrive, persistent metabolic acidosis, fine and sparse hair, and excretion of the unusual cyclic amino acid metabolite, hawkinsin, in the urine. {ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02850; DB00348 Interacts with NA EC number 1.13.11.27 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Dioxygenase; Disease variant; Endoplasmic reticulum; Golgi apparatus; Intellectual disability; Iron; Membrane; Metal-binding; Oxidoreductase; Phenylalanine catabolism; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Tyrosine catabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 43164.8 Length 376 Aromaticity 0.11 Instability index 32.38 Isoelectric point 6.73 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -5.44 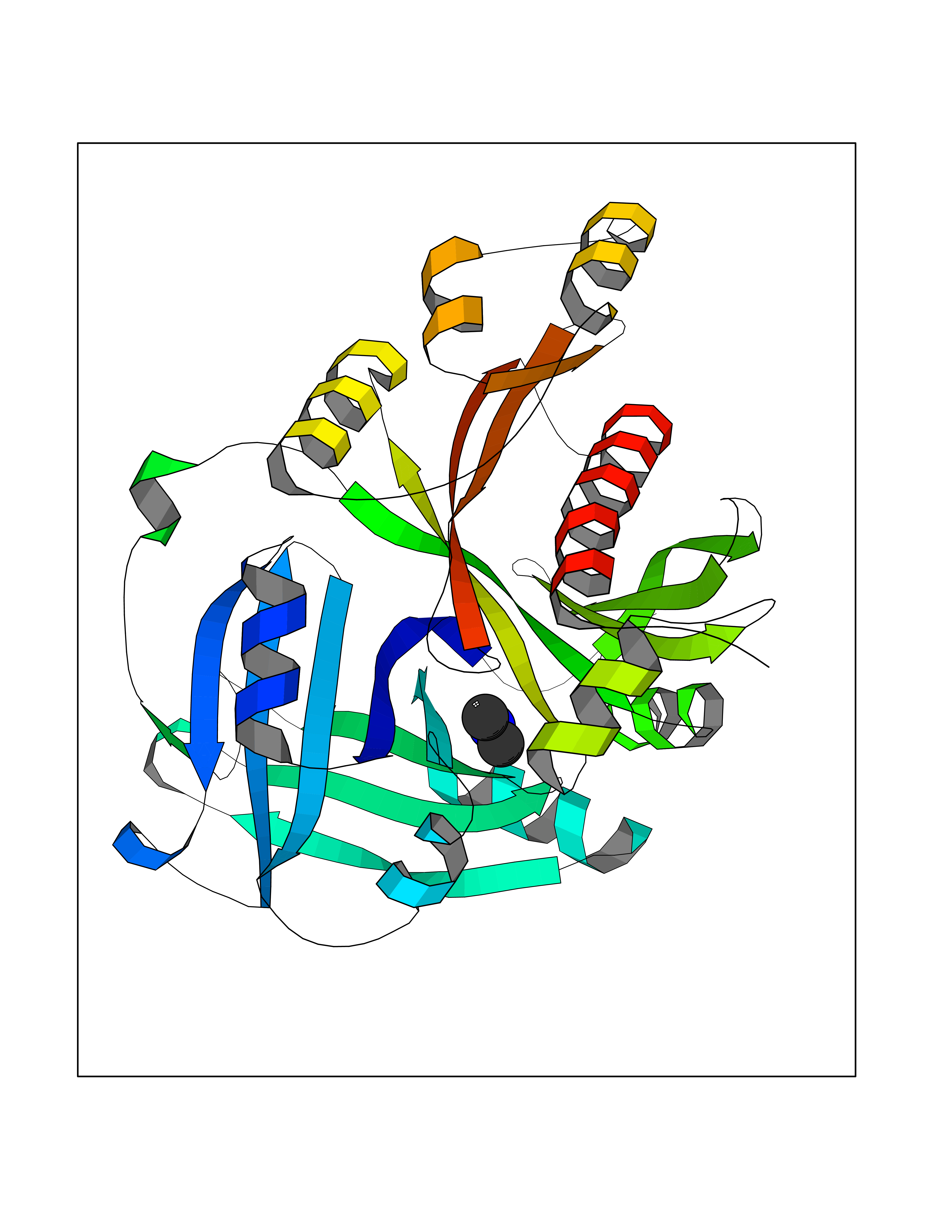 Molscript Map  Pymol Map 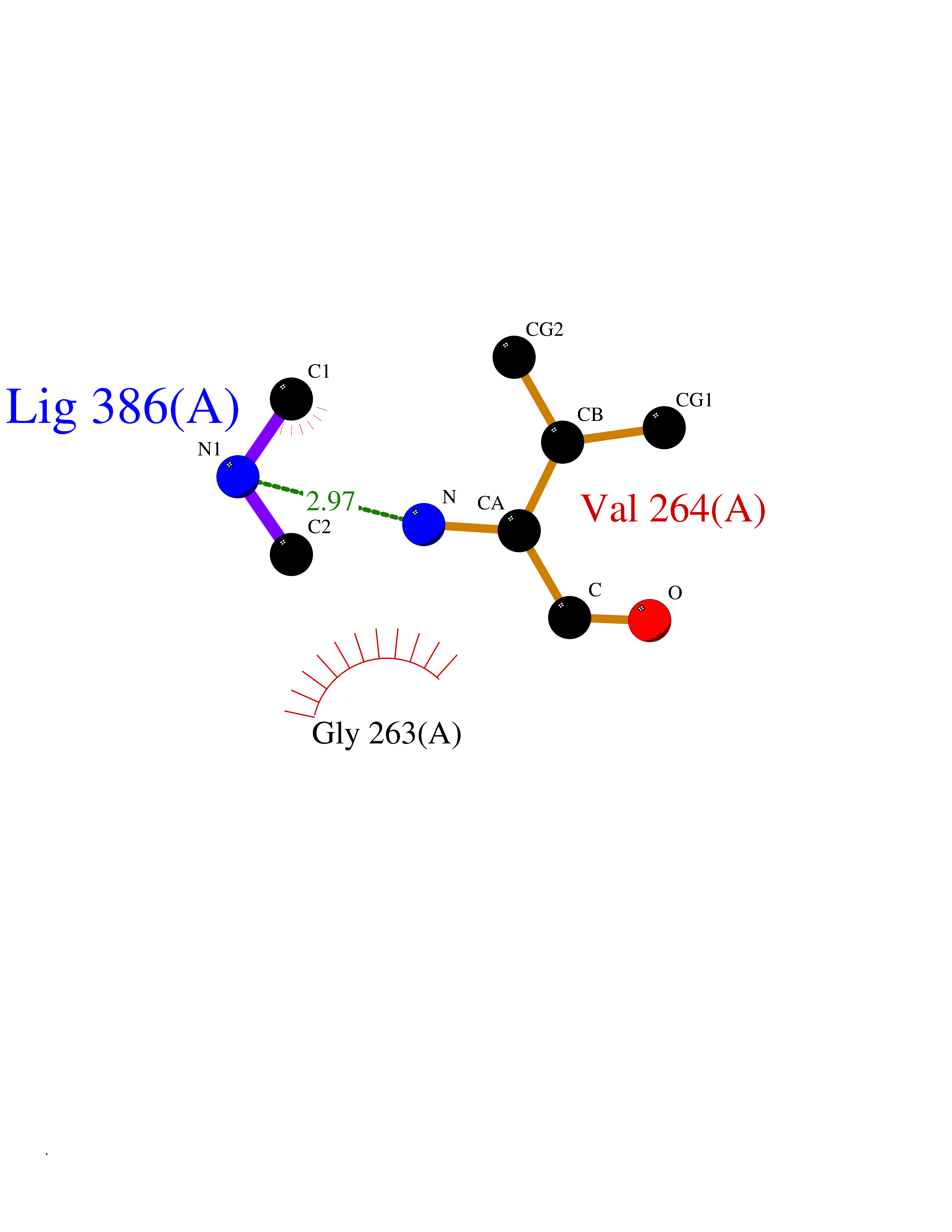 Ligplot Map 3D Binding mode Sequence AKPERGRFLHFHSVTFWVGNAKQAASFYCSKMGFEPLAYRGLETGSREVVSHVIKQGKIVFVLSSALNPWNKEMGDHLVKHGDGVKDIAFEVEDCDYIVQKARERGAKIMREPWVEQDKFGKVKFAVLQTYGDTTHTLVEKMNYIGQFLPGYEAPAFMDPLLPKLPKCSLEMIDHIVGNQPDQEMVSASEWYLKNLQFHRFWSVDDTQVHTEYSSLRSIVVANYEESIKMPINEPAPGKKKSQIQEYVDYNGGAGVQHIALKTEDIITAIRHLRERGLEFLSVPSTYYKQLREKLKTAKIKVKENIDALEELKILVDYDEKGYLLQIFTKPVQDRPTLFLEVIQRHNHQGFGAGNFNSLFKAFEEEQNLRGNLTNM Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Branched-chain-amino-acid transaminase 1 (BCAT1) | 2COI | 3.99 | |
Target general information Gen name BCAT1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein ECA39; ECA39; Branched-chain-amino-acid aminotransferase, cytosolic; BCT1; BCAT(c) Protein family Class-IV pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transaminase Function Catalyzes the first reaction in the catabolism of the essential branched chain amino acids leucine, isoleucine, and valine. Related diseases Charcot-Marie-Tooth disease, dominant intermediate C (CMTDIC) [MIM:608323]: A form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. The dominant intermediate type C is characterized by clinical and pathologic features intermediate between demyelinating and axonal peripheral neuropathies, and motor median nerve conduction velocities ranging from 25 to 45 m/sec. {ECO:0000269|PubMed:16429158}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurologic, endocrine, and pancreatic disease, multisystem, infantile-onset 2 (IMNEPD2) [MIM:619418]: An autosomal recessive disorder with variable clinical manifestations and severity. Main features include cholestatic hepatitis, poor feeding, poor overall growth, and hypoglycemia apparent from infancy. Most patients have variable global developmental delay, sensorineural deafness, retinal abnormalities with visual defects, and hypotonia. Some patients have endocrine abnormalities. Brain imaging often shows dysmyelination, thin corpus callosum, cerebral atrophy, and white matter abnormalities. Death in early childhood may occur. {ECO:0000269|PubMed:27633801, ECO:0000269|PubMed:29232904, ECO:0000269|PubMed:30304524}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in YARS1 may be the cause of proximal-predominant motor neuropathy. Affected individuals may develop tremors, cramping of hands, asymmetric weakness in the upper and lower extremities, and present with elevated creatine kinase levels. {ECO:0000269|PubMed:36307205}. Drugs (DrugBank ID) DB00996; DB00142; DB00167; DB00149; DB07544; DB00114; DB00161 Interacts with P55212; O75190-2; O14645; P22607; P06396; O14901; P13473-2; O75400-2 EC number EC 2.6.1.42 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Amino-acid biosynthesis; Aminotransferase; Branched-chain amino acid biosynthesis; Cytoplasm; Lipid metabolism; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 40123.7 Length 356 Aromaticity 0.1 Instability index 32.34 Isoelectric point 5.39 Charge (pH=7) -9 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map 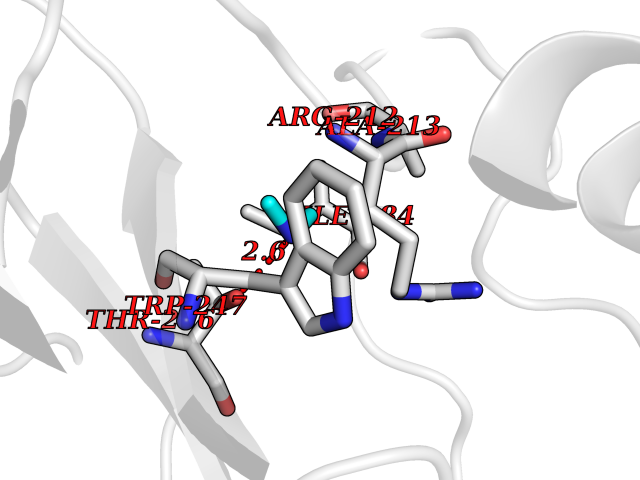 Pymol Map 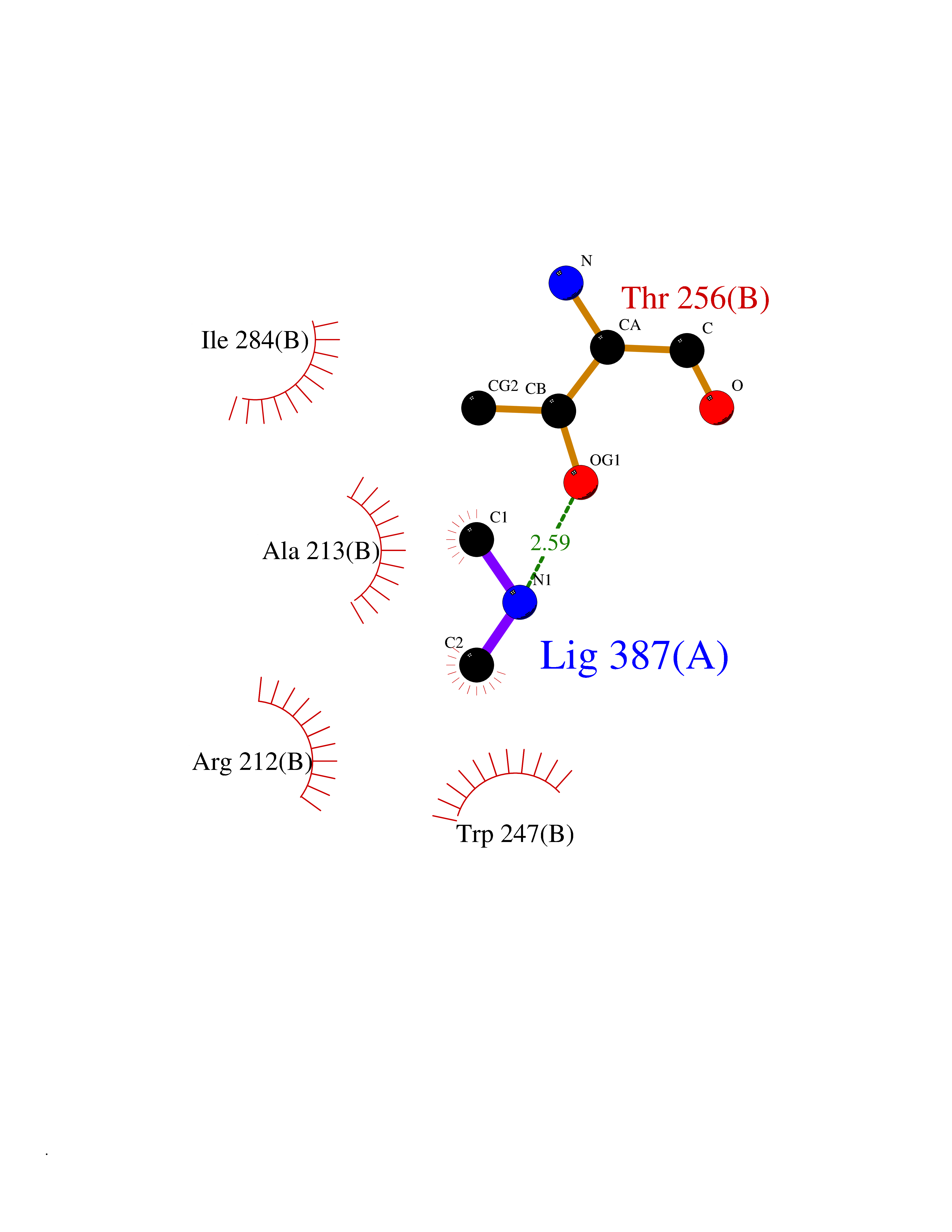 Ligplot Map 3D Binding mode Sequence TFKAKDLIVTPATILKEKPDPNLVFGTVFTDHMLTVEWSSEFGWEKPHIKPLQNLSLHPGSSALHYAVELFEGLKAFRGVDNKIRLFQPNLNMDRMYRSAVRATLPVFDKEELLECIQQLVKLDQEWVPYSTSASLYIRPTFIGTEPSLGVKKPTKALLFVLLSPVGPYFNPVSLWANPKYVRAWKGGTGDCKMGGNYGSSLFAQCEAVDNGCQQVLWLYGEDHQITEVGTMNLFLYWINEDGEEELATPPLDGIILPGVTRRCILDLAHQWGEFKVSERYLTMDDLTTALEGNRVREMFGSGTACVVCPVSDILYKGETIHIPTMENGPKLASRILSKLTDIQYGREERDWTIVL Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Oxygen-insensitive NAD(P)H nitroreductase | 1KQB | 3.99 | |
Target general information Gen name nfsB Organism Enterobacter cloacae Uniprot ID TTD ID NA Synonyms nfsI;nfnB Protein family Nitroreductase family Biochemical class Oxidoreductase Function Oxidoreductase activity. Related diseases A chromosomal aberration involving NFKB2 is found in a case of B-cell non Hodgkin lymphoma (B-NHL). Translocation t(10;14)(q24;q32) with IGHA1. The resulting oncogene is also called Lyt-10C alpha variant.; DISEASE: A chromosomal aberration involving NFKB2 is found in a cutaneous T-cell leukemia (C-TCL) cell line. This rearrangement produces the p80HT gene which codes for a truncated 80 kDa protein (p80HT).; DISEASE: In B-cell leukemia (B-CLL) cell line, LB40 and EB308, can be found after heterogeneous chromosomal aberrations, such as internal deletions.; DISEASE: Immunodeficiency, common variable, 10 (CVID10) [MIM:615577]: A primary immunodeficiency characterized by childhood-onset of recurrent infections, hypogammaglobulinemia, and decreased numbers of memory and marginal zone B-cells. Some patients may develop autoimmune features and have circulating autoantibodies. An unusual feature is central adrenal insufficiency. {ECO:0000269|PubMed:24140114, ECO:0000269|PubMed:25524009}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03793; DB03247 Interacts with NA EC number 1.-.-.- Uniprot keywords 3D-structure; Flavoprotein; FMN; NAD; NADP; Oxidoreductase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 47619.4 Length 432 Aromaticity 0.08 Instability index 38.43 Isoelectric point 5.52 Charge (pH=7) -12.98 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map 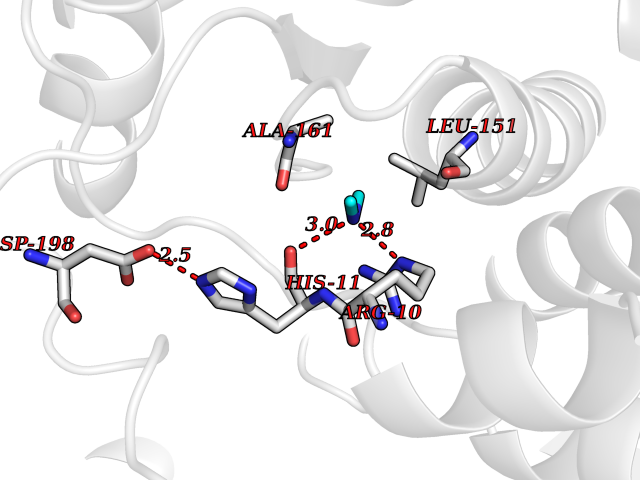 Pymol Map 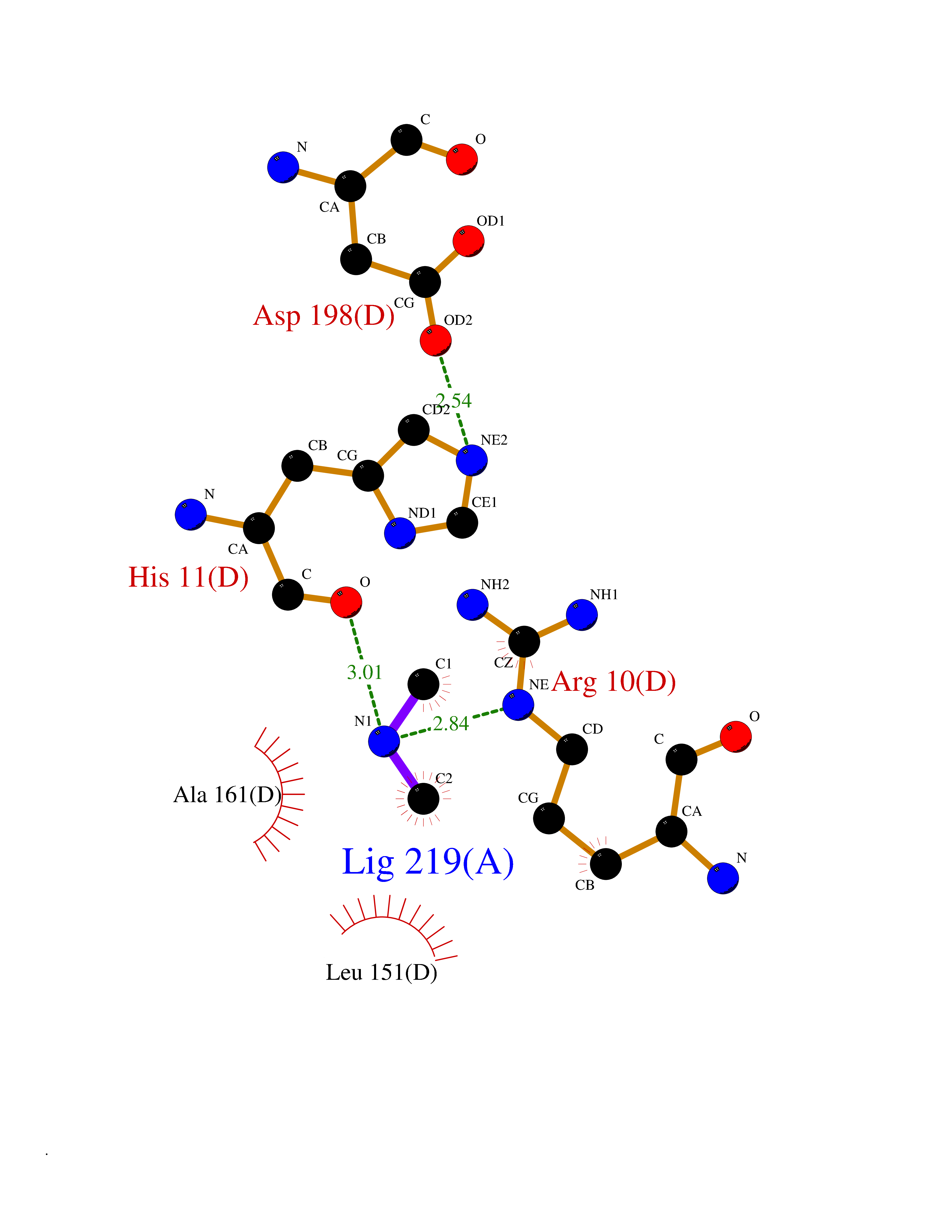 Ligplot Map 3D Binding mode Sequence DIISVALKRHSTKAFDASKKLTAEEAEKIKTLLQYSPSSTNSQPWHFIVASTEEGKARVAKSAAGTYVFNERKMLDASHVVVFCAKTAMDDAWLERVVDQEEADGRFNTPEAKAANHKGRTYFADMHRVDLKDDDQWMAKQVYLNVGNFLLGVGAMGLDAVPIEGFDAAILDEEFGLKEKGFTSLVVVPVGHHSVEDFNATLPKSRLPLSTIVTECDIISVALKRHSTKAFDASKKLTAEEAEKIKTLLQYSPSSTNSQPWHFIVASTEEGKARVAKSAAGTYVFNERKMLDASHVVVFCAKTAMDDAWLERVVDQEEADGRFNTPEAKAANHKGRTYFADMHRVDLKDDDQWMAKQVYLNVGNFLLGVGAMGLDAVPIEGFDAAILDEEFGLKEKGFTSLVVVPVGHHSVEDFNATLPKSRLPLSTIVTEC Hydrogen bonds contact Hydrophobic contact | ||||