Job Results:
Ligand
Structure
Job ID
369cc27f15dbe7b3a0e0dfe22b2eb6d9
Job name
NA
Time
2025-03-05 09:37:27
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Human immunodeficiency virus Negative factor (HIV nef) | 6B72 | 4.00 | |
Target general information Gen name HIV nef Organism Human immunodeficiency virus type 1 group M subtype B (isolate ARV2/SF2) (HIV-1) Uniprot ID TTD ID Synonyms nef; Nef protein; F-protein; 3'ORF; 27 kDa protein Protein family Lentivirus primate group Nef protein family Biochemical class Lentivirus primate group Nef Function Extracellular Nef protein targets CD4(+) T-lymphocytes for apoptosis by interacting with CXCR4 surface receptors. Related diseases Neurodegeneration due to cerebral folate transport deficiency (NCFTD) [MIM:613068]: An autosomal recessive neurodegenerative disorder resulting from brain-specific folate deficiency early in life. Onset is apparent in late infancy with severe developmental regression, movement disturbances, epilepsy and leukodystrophy. {ECO:0000269|PubMed:19732866}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q14457; P01730 EC number NA Uniprot keywords 3D-structure; AIDS; Apoptosis; Early protein; Host cell membrane; Host Golgi apparatus; Host membrane; Host-virus interaction; Inhibition of host adaptive immune response by virus; Inhibition of host autophagy by virus; Inhibition of host MHC class I molecule presentation by virus; Inhibition of host MHC class II molecule presentation by virus; Lipoprotein; Membrane; Myristate; Phosphoprotein; Reference proteome; Secreted; SH3-binding; Viral immunoevasion; Virion; Virulence Protein physicochemical properties Chain ID A,B Molecular weight (Da) 29232.1 Length 245 Aromaticity 0.16 Instability index 50.97 Isoelectric point 5.71 Charge (pH=7) -7.32 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map 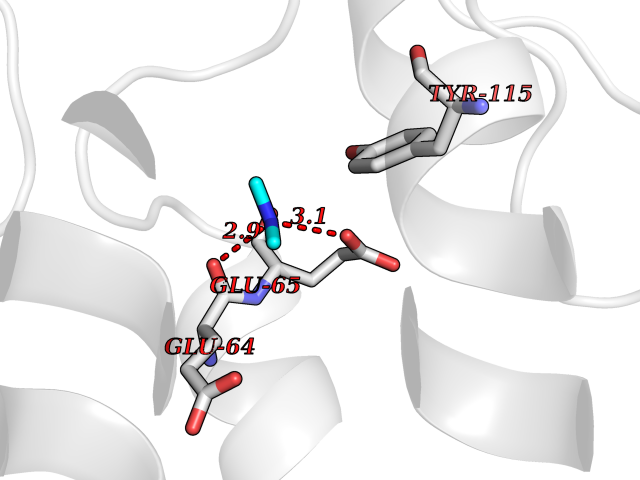 Pymol Map 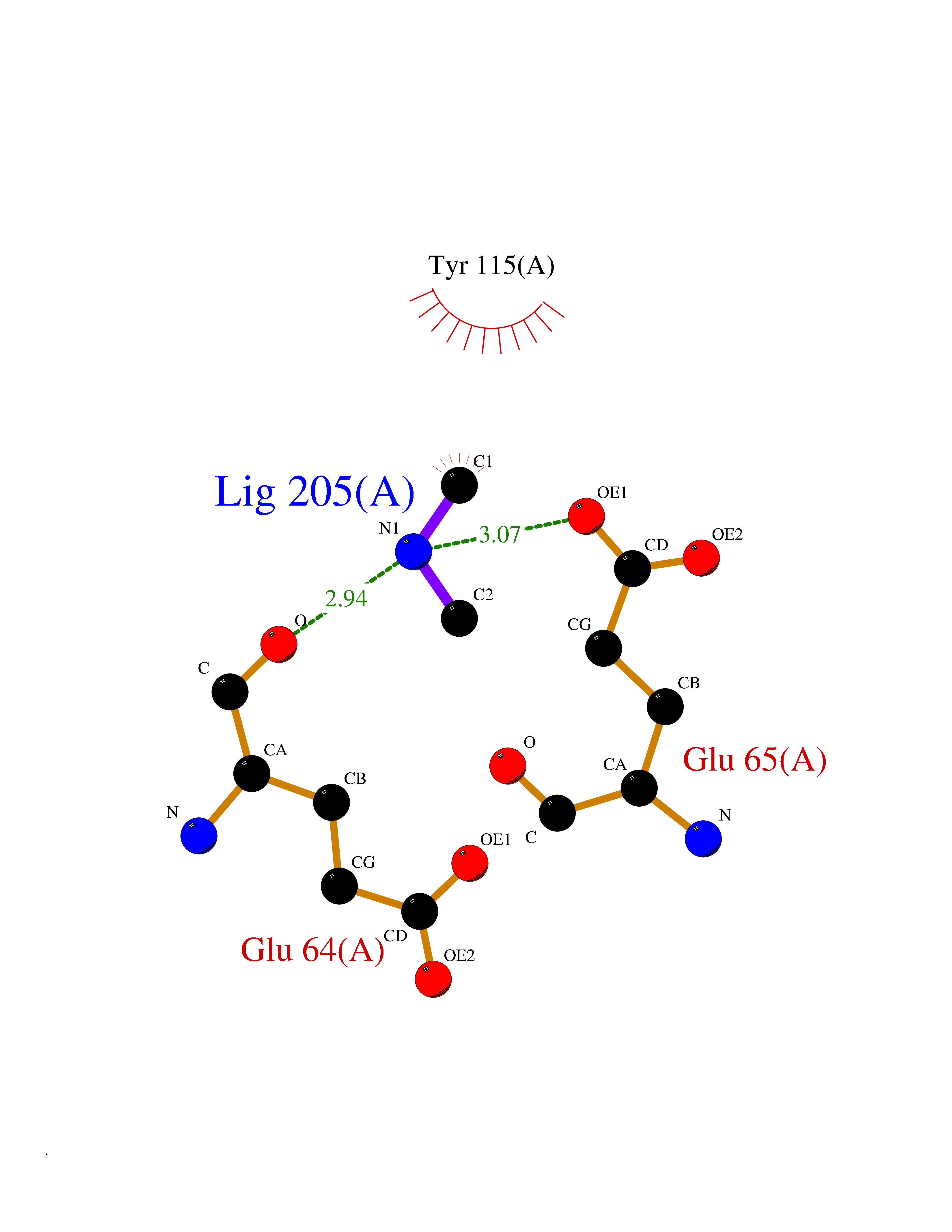 Ligplot Map 3D Binding mode Sequence ADCAWLEAQEEEEVGFPVRPQVPLRPMTYKAALDISHFLKEKGGLEGLIWSQRRQEILDLWIYHTQGYFPDWQNYTPGPGIRYPLTFGWCFKLVPVEEKEVLVWRFDSKLAFHHMARELHPEYYCAWLEAQEEEEVGFPVRPQVPLRPMTYKAALDISHFLKEKGGLEGLIWSQRRQEILDLWIYHTQGYFPDWQNYTPGPGIRYPLTFGWCFKLVPVEKEVLVWRFDSKLAFHHMARELHPEYY Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Kynurenine oxoglutarate transaminase II (AADAT) | 2XH1 | 4.00 | |
Target general information Gen name AADAT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Kynurenineoxoglutarate transaminase II; Kynurenineoxoglutarate transaminase 2; Kynurenineoxoglutarate aminotransferase II; Kynurenine/alphaaminoadipate aminotransferase, mitochondrial; Kynurenine amin Protein family Class-I pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transaminase Function Transaminase with broad substrate specificity. Has transaminase activity towards aminoadipate, kynurenine, methionine and glutamate. Shows activity also towards tryptophan, aspartate and hydroxykynurenine. Accepts a variety of oxo-acids as amino- group acceptors, with a preference for 2-oxoglutarate, 2- oxocaproic acid, phenylpyruvate and alpha-oxo-gamma-methiol butyric acid. Can also use glyoxylate as amino-group acceptor (in vitro). Related diseases Epilepsy, nocturnal frontal lobe, 3 (ENFL3) [MIM:605375]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:11062464, ECO:0000269|PubMed:11104662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00142; DB00114 Interacts with NA EC number EC 2.6.1.39 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminotransferase; Mitochondrion; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase; Transit peptide Protein physicochemical properties Chain ID A,B Molecular weight (Da) 91792.6 Length 824 Aromaticity 0.09 Instability index 47.71 Isoelectric point 5.96 Charge (pH=7) -8.73 2D Binding mode Binding energy (Kcal/mol) -5.45  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ENYARFITAASAARNPSPIMISLAGGLPNPNMFPFKTAVITVENGKTIQFGEEMMKRALQYSPSAGIPELLSWLKQLQIKLHNPPTIHYPPSQGQMDLCVTSGSQQGLCKVFEMIINPGDNVLLDEPAYSGTLQSLHPLGCNIINVASDESGIVPDSLRDILSRWKPEDAKNPQKNTPKFLYTVPNGNNPTGNSLTSERKKEIYELARKYDFLIIEDDPYYFLQFNKFRVPTFLSMDVDGRVIRADSFSKIISSGLRIGFLTGPKPLIERVILHIQVSTLHPSTFNQLMISQLLHEWGEEGFMAHVDRVIDFYSNQKDAILAAADKWLTGLAEWHVPAAGMFLWIKVKGINDVKELIEEKAVKMGVLMLPGNAFYVDSSAPSPYLRASFSSASPEQMDVAFQVLAQLIKESLENYARFITAASAARNPSPIMISLAGGLPNPNMFPFKTAVITVENGKTIQFGEEMMKRALQYSPSAGIPELLSWLKQLQIKLHNPPTIHYPPSQGQMDLCVTSGSQQGLCKVFEMIINPGDNVLLDEPAYSGTLQSLHPLGCNIINVASDESGIVPDSLRDILSRWKPEDAKNPQKNTPKFLYTVPNGNNPTGNSLTSERKKEIYELARKYDFLIIEDDPYYFLQFNKFRVPTFLSMDVDGRVIRADSFSKIISSGLRIGFLTGPKPLIERVILHIQVSTLHPSTFNQLMISQLLHEWGEEGFMAHVDRVIDFYSNQKDAILAAADKWLTGLAEWHVPAAGMFLWIKVKGINDVKELIEEKAVKMGVLMLPGNAFYVDSSAPSPYLRASFSSASPEQMDVAFQVLAQLIKESL Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Cathepsin V (CTSV) | 1FH0 | 4.00 | |
Target general information Gen name CTSV Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UNQ268/PRO305; Cathepsin U; Cathepsin L2; CTSU; CTSL2; CATL2 Protein family Peptidase C1 family Biochemical class Peptidase Function May have an important role in corneal physiology. Cysteine protease. Related diseases Developmental and epileptic encephalopathy 24 (DEE24) [MIM:615871]: A disease characterized by early-onset seizures, intellectual disability of varying degrees, and behavioral disturbances or autistic features in most individuals. {ECO:0000269|PubMed:24747641, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Generalized epilepsy with febrile seizures plus 10 (GEFSP10) [MIM:618482]: An autosomal dominant neurologic disorder with incomplete penetrance, characterized by variable types of seizures including absence, tonic-clonic, febrile, focal, and eyelid myoclonia. Some patients have normal neurologic development. Others have mild-to-moderate intellectual disability or autism spectrum disorder. {ECO:0000269|PubMed:29936235, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02869; DB04451 Interacts with P07711 EC number EC 3.4.22.43 Uniprot keywords 3D-structure; Disulfide bond; Glycoprotein; Hydrolase; Lysosome; Protease; Proteomics identification; Reference proteome; Signal; Thiol protease; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 24013.7 Length 221 Aromaticity 0.11 Instability index 16.86 Isoelectric point 8.4 Charge (pH=7) 2.96 2D Binding mode Binding energy (Kcal/mol) -5.45  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LPKSVDWRKKGYVTPVKNQKQCGSCWAFSATGALEGQMFRKTGKLVSLSEQNLVDCSRPQGNQGCNGGFMARAFQYVKENGGLDSEESYPYVAVDEICKYRPENSVAQDTGFTVVAPGKEKALMKAVATVGPISVAMDAGHSSFQFYKSGIYFEPDCSSKNLDHGVLVVGYGFEGANSDNSKYWLVKNSWGPEWGSNGYVKIAKDKNNHCGIATAASYPNV Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Aldehyde oxidase (AOX1) | 7OPN | 4.00 | |
Target general information Gen name AOX1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms AOX1 Protein family Xanthine dehydrogenase family Biochemical class Aldehyde/oxo donor oxidoreductase Function Oxidase with broad substrate specificity, oxidizing aromatic azaheterocycles, such as N1-methylnicotinamide and N- methylphthalazinium, as well as aldehydes, such as benzaldehyde, retinal, pyridoxal, and vanillin. Plays a key role in the metabolism of xenobiotics and drugs containing aromatic azaheterocyclic substituents. Participates in the bioactivation of prodrugs such as famciclovir, catalyzing the oxidation step from 6-deoxypenciclovir topenciclovir, which is a potent antiviral agent. Is probably involved in the regulation of reactive oxygen species homeostasis. May be a prominent source of superoxide generation via the one-electron reduction of molecular oxygen. Also may catalyze nitric oxide (NO) production via the reduction of nitrite to NO with NADH or aldehyde as electron donor. May play a role in adipogenesis. Related diseases Progressive familial heart block 1B (PFHB1B) [MIM:604559]: A cardiac bundle branch disorder characterized by progressive alteration of cardiac conduction through the His-Purkinje system, with a pattern of a right bundle-branch block and/or left anterior hemiblock occurring individually or together. It leads to complete atrio-ventricular block causing syncope and sudden death. {ECO:0000269|PubMed:19726882, ECO:0000269|PubMed:20562447, ECO:0000269|PubMed:21887725}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythrokeratodermia variabilis et progressiva 6 (EKVP6) [MIM:618531]: A form of erythrokeratodermia variabilis et progressiva, a genodermatosis characterized by the coexistence of two independent skin lesions: transient erythema and hyperkeratosis that is usually localized but occasionally occurs in its generalized form. Clinical presentation varies significantly within a family and from one family to another. Palmoplantar keratoderma is present in around 50% of cases. EKVP6 inheritance is autosomal dominant. {ECO:0000269|PubMed:30528822}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00437; DB00513; DB00484; DB11791; DB00215; DB00924; DB03516; DB01175; DB00426; DB12466; DB09054; DB09078; DB00170; DB01033; DB00563; DB08840; DB00157; DB00339; DB00481; DB04827; DB00962; DB00246; DB00909 Interacts with Q06278 EC number EC 1.2.3.1 Uniprot keywords 2Fe-2S; 3D-structure; Cytoplasm; FAD; Flavoprotein; Iron; Iron-sulfur; Lipid metabolism; Metal-binding; Molybdenum; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 286845 Length 2590 Aromaticity 0.08 Instability index 42.7 Isoelectric point 6.92 Charge (pH=7) -1.62 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ASELLFYVNGRKVIEKNVDPETMLLPYLRKKLRLTGTKYGCGGGGCGACTVMISRYNPITKRIRHHPANACLIPICSLYGAAVTTVEGIGSTHTRIHPVQERIAKCHGTQCGFCTPGMVMSIYTLLRNHPEPTLDQLTDALGGNLCRCTGYRPIIDACKTFCKPKLFAEEEFLPLDPTQELIFPPELMIMAEKQSQRTRVFGSERMMWFSPVTLKELLEFKFKYPQAPVIMGNTSVGPEVKFKGVFHPVIISPDRIEELSVVNHAYNGLTLGAGLSLAQVKDILADVVQKLPEEKTQMYHALLKHLGTLAGSQIRNMASLGGHIISRHPDSDLNPILAVGNCTLNLLSKEGKRQIPLNEQFLSKCPNADLKPQEILVSVNIPYSRKWEFVSAFRQAQRQENALAIVNSGMRVFFGEGDGIIRELCISYGGVGPATICAKNSCQKLIGRHWNEQMLDIACRLILNEVSLLGSAPGGKVEFKRTLIISFLFKFYLEVSQILKKMDPVHYPSLADKYESALEDLHSKHHCSTLKYQNQHPEDPIGHPIMHLSGVKHATGEAIYCDDMPLVDQELFLTFVTSSRAHAKIVSIDLSEALSMPGVVDIMTAEHLSDVNSFCFFTEAEKFLATDKVFCVGQLVCAVLADSEVQAKRAAKRVKIVYQDLEPLILTIEESIQHNSSFKPERKLEYGNVDEAFKVVDQILEGEIHMGGQEHFYMETQSMLVVPKGEDQEMDVYVSTQFPKYIQDIVASTLKLPANKVMCHVRRVGGAFGGKVLKTGIIAAVTAFAANKHGRAVRCVLERGEDMLITGGRHPYLGKYKAGFMNDGRILALDMEHYSNAGASLDESLFVIEMGLLKMDNAYKFPNLRCRGWACRTNLPSNTAFRGFGFPQAALITESCITEVAAKCGLSPEKVRIINMYKEIDQTPYKQEINAKNLIQCWRECMAMSSYSLRKVAVEKFNAENYWKKKGLAMVPLKFPVGLGSRAAGQAAALVHIYLDGSVLVTHGGIEMGQGVHTKMIQVVSRELRMPMSNVHLRGTSTETVPNANISGGSVVADLNGLAVKDACQTLLKRLEPIISKNPKGTWKDWAQTAFDESINLSAVGYFRGYESDMNWEKGEGQPFEYFVYGAACSEVEIDCLTGDHKNIRTDIVMDVGCSINPAIDIGQIEGAFIQGMGLYTIEELNYSPQGILHTHGPDQYKIPAICDMPTELHIALLPPSQNSNTLYSSKGLGESGVFLGCSVFFAIHDAVSAARQERGLHLTLNSPLTPEKIRMACEDKFTKMIPRDEPGSYVPWNVASELLFYVNGRKVIEKNVDPETMLLPYLRKKLRLTGTKYGCGGGGCGACTVMISRYNPITKRIRHHPANACLIPICSLYGAAVTTVEGIGSTHTRIHPVQERIAKCHGTQCGFCTPGMVMSIYTLLRNHPEPTLDQLTDALGGNLCRCTGYRPIIDACKTFCKPKLFAEEEFLPLDPTQELIFPPELMIMAEKQSQRTRVFGSERMMWFSPVTLKELLEFKFKYPQAPVIMGNTSVGPEVKFKGVFHPVIISPDRIEELSVVNHAYNGLTLGAGLSLAQVKDILADVVQKLPEEKTQMYHALLKHLGTLAGSQIRNMASLGGHIISRHPDSDLNPILAVGNCTLNLLSKEGKRQIPLNEQFLSKCPNADLKPQEILVSVNIPYSRKWEFVSAFRQAQRQENALAIVNSGMRVFFGEGDGIIRELCISYGGVGPATICAKNSCQKLIGRHWNEQMLDIACRLILNEVSLLGSAPGGKVEFKRTLIISFLFKFYLEVSQILKKMDPVHYPSLADKYESALEDLHSKHHCSTLKYQNQHPEDPIGHPIMHLSGVKHATGEAIYCDDMPLVDQELFLTFVTSSRAHAKIVSIDLSEALSMPGVVDIMTAEHLSDVNSFCFFTEAEKFLATDKVFCVGQLVCAVLADSEVQAKRAAKRVKIVYQDLEPLILTIEESIQHNSSFKPERKLEYGNVDEAFKVVDQILEGEIHMGGQEHFYMETQSMLVVPKGEDQEMDVYVSTQFPKYIQDIVASTLKLPANKVMCHVRRVGGAFGGKVLKTGIIAAVTAFAANKHGRAVRCVLERGEDMLITGGRHPYLGKYKAGFMNDGRILALDMEHYSNAGASLDESLFVIEMGLLKMDNAYKFPNLRCRGWACRTNLPSNTAFRGFGFPQAALITESCITEVAAKCGLSPEKVRIINMYKEIDQTPYKQEINAKNLIQCWRECMAMSSYSLRKVAVEKFNAENYWKKKGLAMVPLKFPVGLGSRAAGQAAALVHIYLDGSVLVTHGGIEMGQGVHTKMIQVVSRELRMPMSNVHLRGTSTETVPNANISGGSVVADLNGLAVKDACQTLLKRLEPIISKNPKGTWKDWAQTAFDESINLSAVGYFRGYESDMNWEKGEGQPFEYFVYGAACSEVEIDCLTGDHKNIRTDIVMDVGCSINPAIDIGQIEGAFIQGMGLYTIEELNYSPQGILHTHGPDQYKIPAICDMPTELHIALLPPSQNSNTLYSSKGLGESGVFLGCSVFFAIHDAVSAARQERGLHLTLNSPLTPEKIRMACEDKFTKMIPRDEPGSYVPWNV Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Glutathione S-transferase LANCL1 (LANCL1) | 3E73 | 4.00 | |
Target general information Gen name LANCL1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p40; LanC-like protein 1; GPR69A; 40 kDa erythrocyte membrane protein Protein family LanC-like protein family Biochemical class NA Function Functions as glutathione transferase. Catalyzes conjugation of the glutathione (GSH) to artificial substrates 1-chloro-2,4-dinitrobenzene (CDNB) and p-nitrophenyl acetate. Mitigates neuronal oxidative stress during normal postnatal development and in response to oxidative stresses probably through GSH antioxidant defense mechanism (By similarity). May play a role in EPS8 signaling. Binds glutathione. Related diseases Spermatogenic failure 5 (SPGF5) [MIM:243060]: An infertility disorder caused by spermatogenesis defects. Semen from affected men show close to 100% morphologically abnormal multiflagellar spermatozoa with low motility, oversized irregular heads, and abnormal midpiece and acrosome. {ECO:0000269|PubMed:17435757, ECO:0000269|PubMed:21733974}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9UHR4; P42858; Q08509 EC number EC 2.5.1.18 Uniprot keywords 3D-structure; Acetylation; Cell membrane; Cytoplasm; Direct protein sequencing; Membrane; Metal-binding; Proteomics identification; Reference proteome; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 46005.4 Length 405 Aromaticity 0.14 Instability index 33.7 Isoelectric point 7.13 Charge (pH=7) 0.4 2D Binding mode Binding energy (Kcal/mol) -5.46 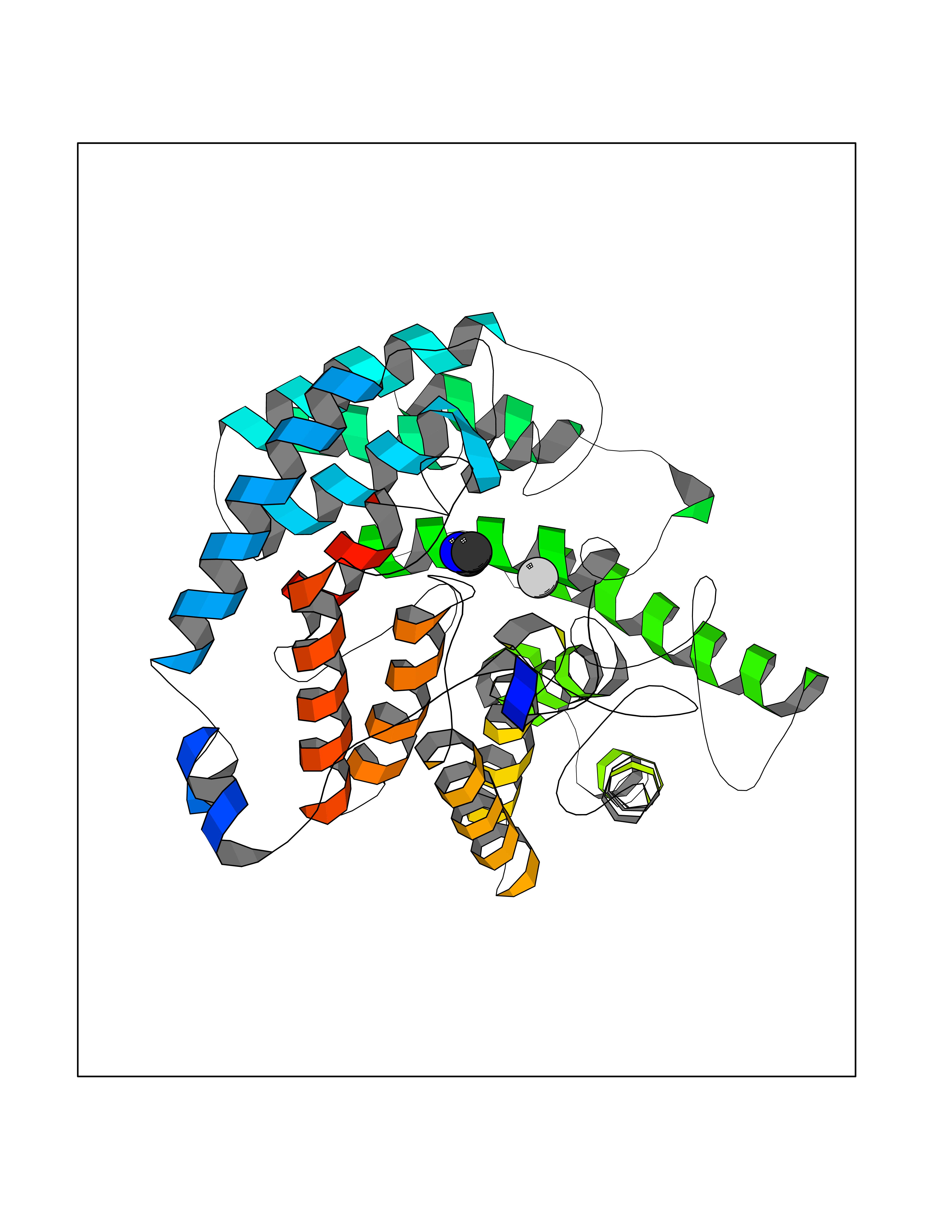 Molscript Map 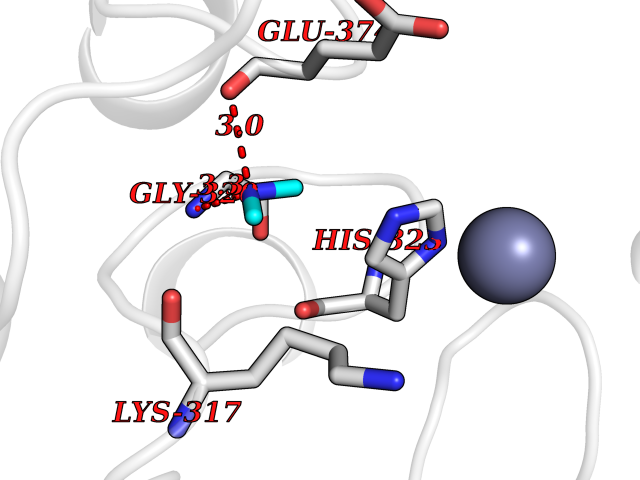 Pymol Map 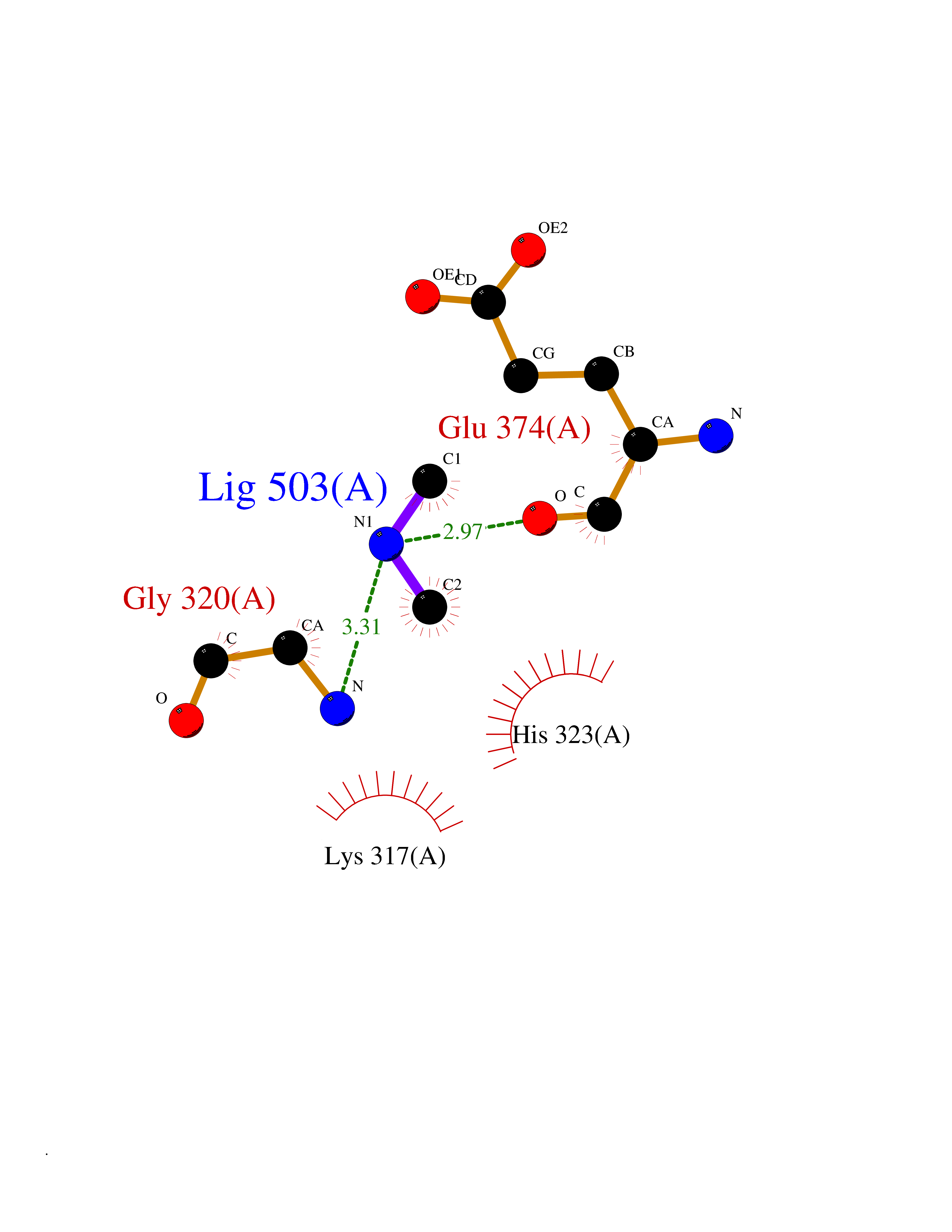 Ligplot Map 3D Binding mode Sequence SMDIEFMAQRAFPNPYADYNKSLAEGYFDAAGRLTPEFSQRLTNKIRELLQQMERGLKSADPRDGTGYTGWAGIAVLYLHLYDVFGDPAYLQLAHGYVKQSLNCLTKRSITFLCGDAGPLAVAAVLYHKMNNEKQAEDCITRLIHLNKIDPHAPNEMLYGRIGYIYALLFVNKNFGVEKIPQSHIQQICETILTSGENLARKRNFTAKSPLMYEWYQEYYVGAAHGLAGIYYYLMQPSLQVSQGKLHSLVKPSVDYVCQLKFPSGNYPPCIGDNRDLLVHWCHGAPGVIYMLIQAYKVFREEKYLCDAYQCADVIWQYGLLKKGYGLCHGSAGNAYAFLTLYNLTQDMKYLYRACKFAEWCLEYGEHGCRTPDTPFSLFEGMAGTIYFLADLLVPTKARFPAFEL Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | 5,10-methylenetetrahydrofolate reductase | 3FST | 3.99 | |
Target general information Gen name metF Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b3941;JW3913 Protein family Methylenetetrahydrofolate reductase family Biochemical class Oxidoreductase Function FAD binding.Methylenetetrahydrofolate reductase (NAD(P)H) activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.5.1.54 Uniprot keywords 3D-structure; Amino-acid biosynthesis; FAD; Flavoprotein; Methionine biosynthesis; NAD; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A,C,E Molecular weight (Da) 30855.9 Length 274 Aromaticity 0.09 Instability index 27.54 Isoelectric point 5.84 Charge (pH=7) -4.61 2D Binding mode Binding energy (Kcal/mol) -5.45 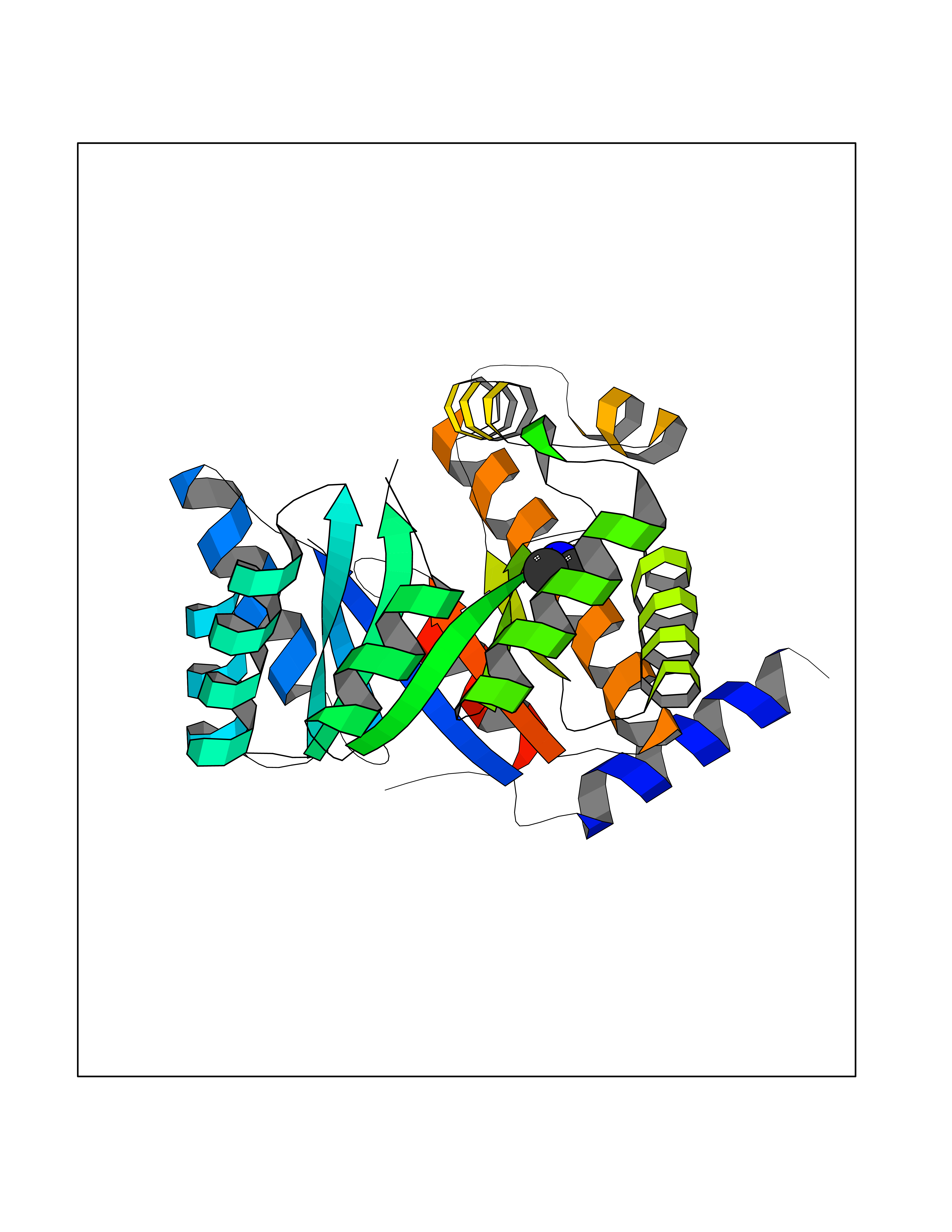 Molscript Map 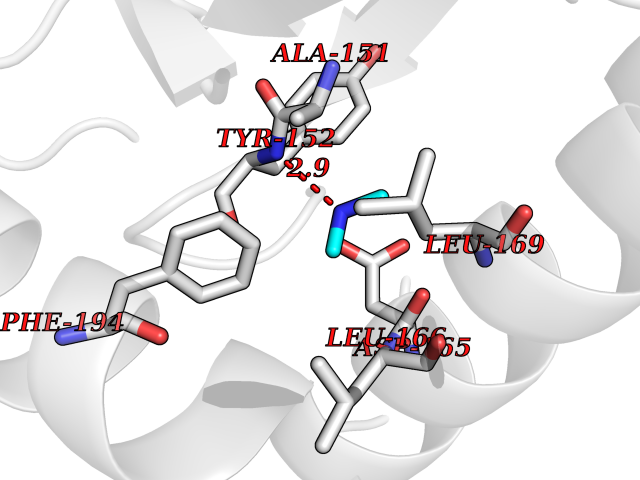 Pymol Map 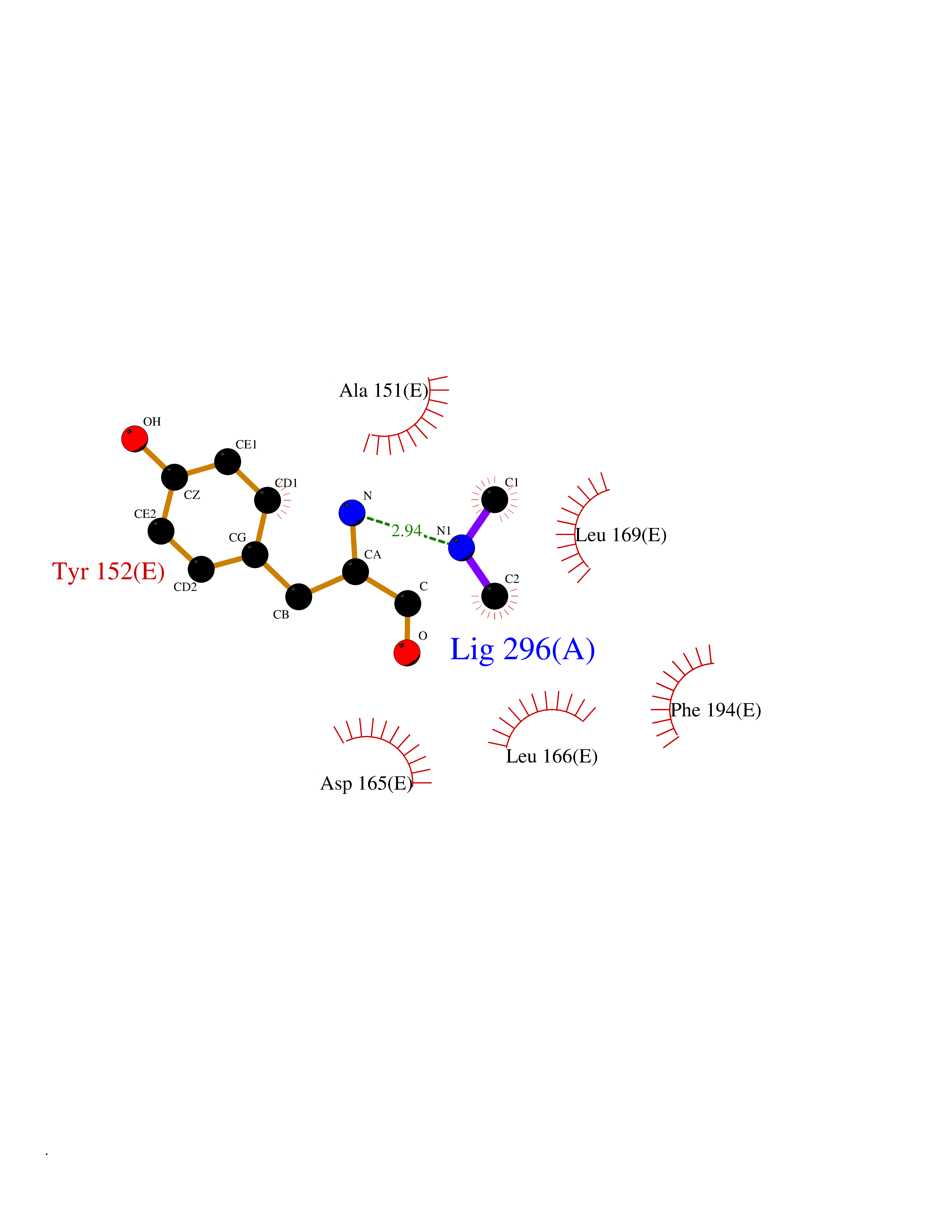 Ligplot Map 3D Binding mode Sequence FHASQRDALNQSLAEVQGQINVSFEFFPPRTSEMEQTLWNSIDRLSSLKPKFVSVTYTHSIIKGIKDRTGLEAAPHLTCIDATPDELRTIARDYWNNGIRHIVALRGDEMYASDLVTLLKEVADFDISVAAYPEVHPEAKSAQADLLNLKRKVDAGANRAITQFFFDVESYLRFRDRCVSAGIDVEIIPGILPVSNFKQAKKLADMTNVRIPAWMAQMFDGLDDDAETRKLVGANIAMDMVKILSREGVKDFHFYTLNRAEMSYAICHTLGVRP Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Steroid 17-alpha-monooxygenase (S17AH) | 3SWZ | 3.99 | |
Target general information Gen name CYP17A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Steroid 17-alpha-hydroxylase/17,20 lyase; P450c17; P450-C17; P450 17; CYPXVII; CYP17A1; CYP 17; 17 alpha-Hydroxylase/C17-20-lyase Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function Conversion of pregnenolone and progesterone to their 17- alpha-hydroxylated products and subsequently to dehydroepiandrosterone (DHEA) and androstenedione. Catalyzes both the 17-alpha-hydroxylation and the 17,20-lyase reaction. Involved in sexual development during fetal life and at puberty. Related diseases Adrenal hyperplasia 5 (AH5) [MIM:202110]: A form of congenital adrenal hyperplasia, a common recessive disease due to defective synthesis of cortisol. Congenital adrenal hyperplasia is characterized by androgen excess leading to ambiguous genitalia in affected females, rapid somatic growth during childhood in both sexes with premature closure of the epiphyses and short adult stature. Four clinical types: 'salt wasting' (SW, the most severe type), 'simple virilizing' (SV, less severely affected patients), with normal aldosterone biosynthesis, 'non-classic form' or late-onset (NC or LOAH) and 'cryptic' (asymptomatic). {ECO:0000269|PubMed:10720067, ECO:0000269|PubMed:11549685, ECO:0000269|PubMed:11836339, ECO:0000269|PubMed:12466376, ECO:0000269|PubMed:14671162, ECO:0000269|PubMed:1515452, ECO:0000269|PubMed:1714904, ECO:0000269|PubMed:1740503, ECO:0000269|PubMed:19793597, ECO:0000269|PubMed:24140098, ECO:0000269|PubMed:24498484, ECO:0000269|PubMed:25650406, ECO:0000269|PubMed:2808364, ECO:0000269|PubMed:8027220, ECO:0000269|PubMed:8245018, ECO:0000269|PubMed:8345056, ECO:0000269|PubMed:8396144, ECO:0000269|PubMed:8550762, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05812; DB04630; DB01424; DB09061; DB00882; DB01234; DB14649; DB01026; DB05667; DB14009; DB14011; DB00157; DB01708; DB00396; DB02901 Interacts with NA EC number EC 1.14.14.19 Uniprot keywords 3D-structure; Congenital adrenal hyperplasia; Disease variant; Endoplasmic reticulum; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Steroidogenesis Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 51385.8 Length 453 Aromaticity 0.08 Instability index 32.67 Isoelectric point 8.48 Charge (pH=7) 4.08 2D Binding mode Binding energy (Kcal/mol) -5.45  Molscript Map 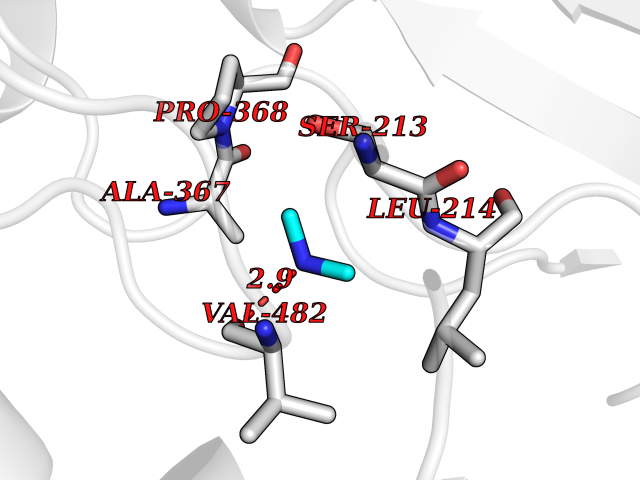 Pymol Map 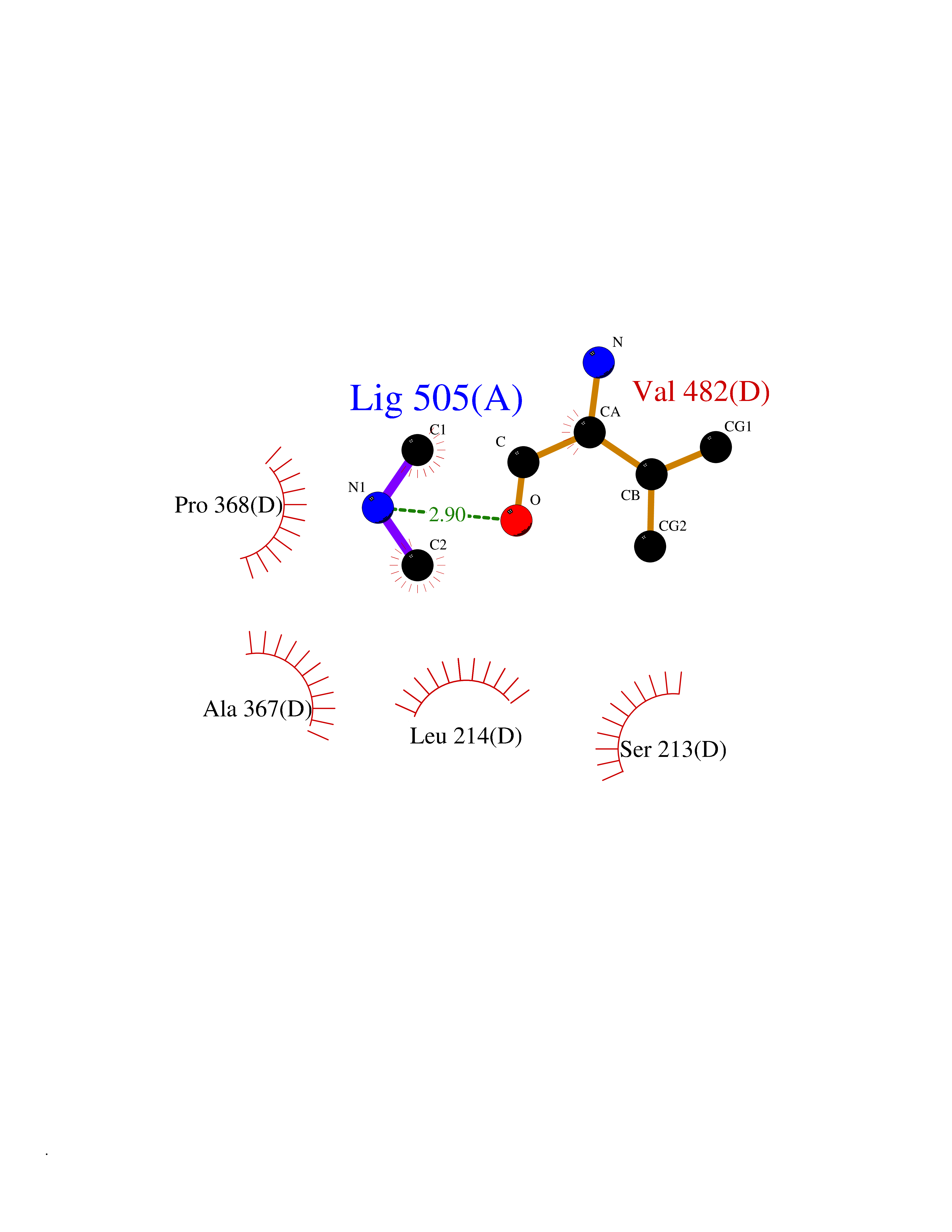 Ligplot Map 3D Binding mode Sequence LPRHGHMHNNFFKLQKKYGPIYSVRMGTKTTVIVGHHQLAKEVLIKKGKDFSGRPQMATLDIASNNRKGIAFADSGAHWQLHRRLAMATFALFKDGDQKLEKIICQEISTLCDMLATHNGQSIDISFPVFVAVTNVISLICFNTSYKNGDPELNVIQNYNEGIIDNLSKDSLVDLVPWLKIFPNKTLEKLKSHVKIRNDLLNKILENYKEKFRSDSITNMLDTLMQAKMNSDDSELLSDNHILTTIGDIFGAGVETTTSVVKWTLAFLLHNPQVKKKLYEEIDQNVGFSRTPTISDRNRLLLLEATIREVLRLRPVAPMLIPHKANVDSSIGEFAVDKGTEVIINLWALHHNEKEWHQPDQFMPERFLNPAGTQLISPSVSYLPFGAGPRSCIGEILARQELFLIMAWLLQRFDLEVPDDGQLPSLEGIPKVVFLIDSFKVKIKVRQAWREAQ Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Endolysin | 1AM7 | 3.99 | |
Target general information Gen name R Organism Escherichia phage lambda (Bacteriophage lambda) Uniprot ID TTD ID NA Synonyms NA Protein family Glycosyl hydrolase 24 family Biochemical class Glycosidase Function Lyase activity.Lysozyme activity.Lytic transglycosylase activity. Related diseases Estrogen resistance (ESTRR) [MIM:615363]: A disorder characterized by partial or complete resistance to estrogens, in the presence of elevated estrogen serum levels. Clinical features include absence of the pubertal growth spurt, delayed bone maturation, unfused epiphyses, reduced bone mineral density, osteoporosis, continued growth into adulthood and very tall adult stature. Glucose intolerance, hyperinsulinemia and lipid abnormalities may also be present. {ECO:0000269|PubMed:23841731, ECO:0000269|PubMed:27754803}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04206 Interacts with NA EC number 4.2.2.n2 Uniprot keywords 3D-structure; Antimicrobial; Bacteriolytic enzyme; Cytolysis; Direct protein sequencing; Host cell lysis by virus; Host cytoplasm; Lyase; Reference proteome; Viral release from host cell Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 49834.9 Length 462 Aromaticity 0.07 Instability index 18.78 Isoelectric point 9.6 Charge (pH=7) 18.29 2D Binding mode Binding energy (Kcal/mol) -5.44 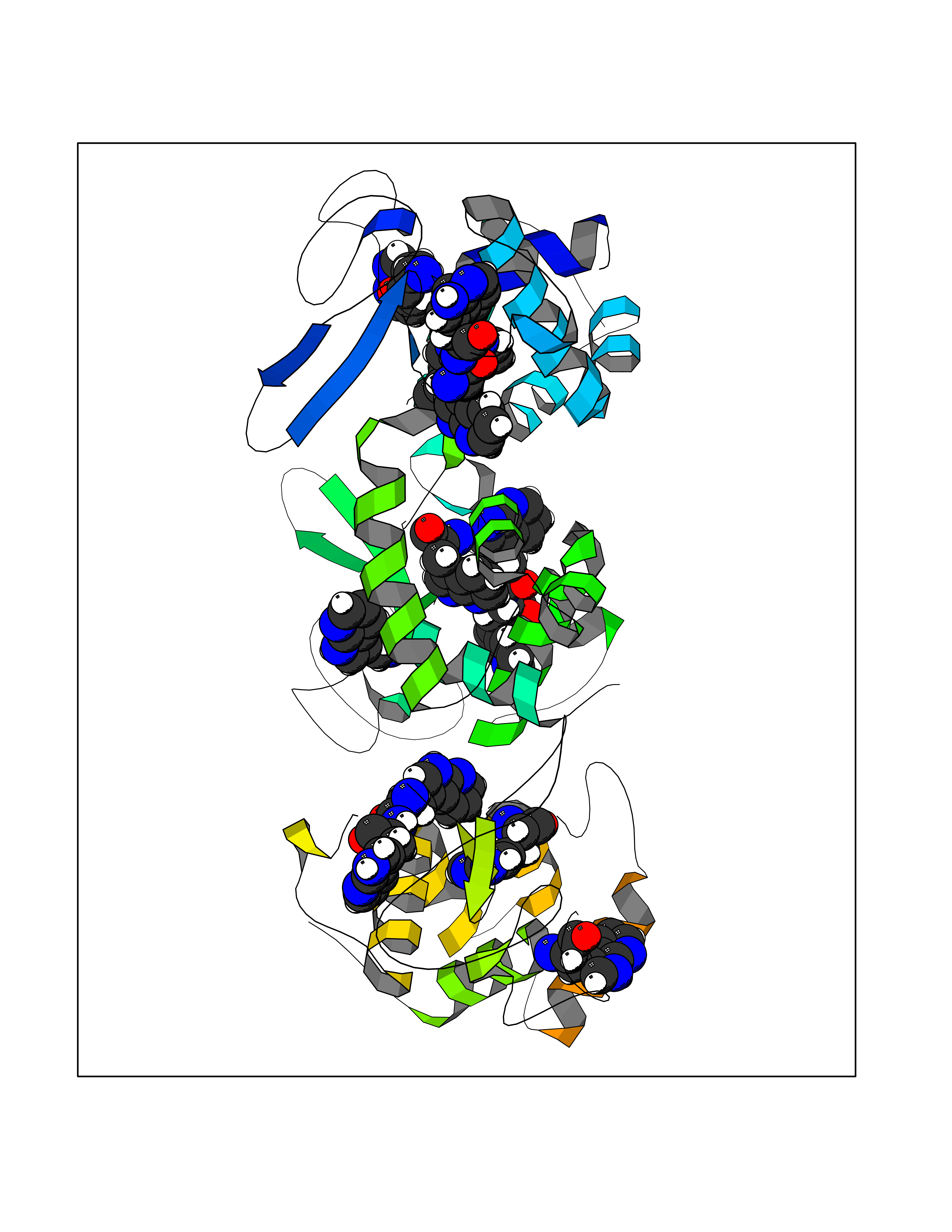 Molscript Map  Pymol Map 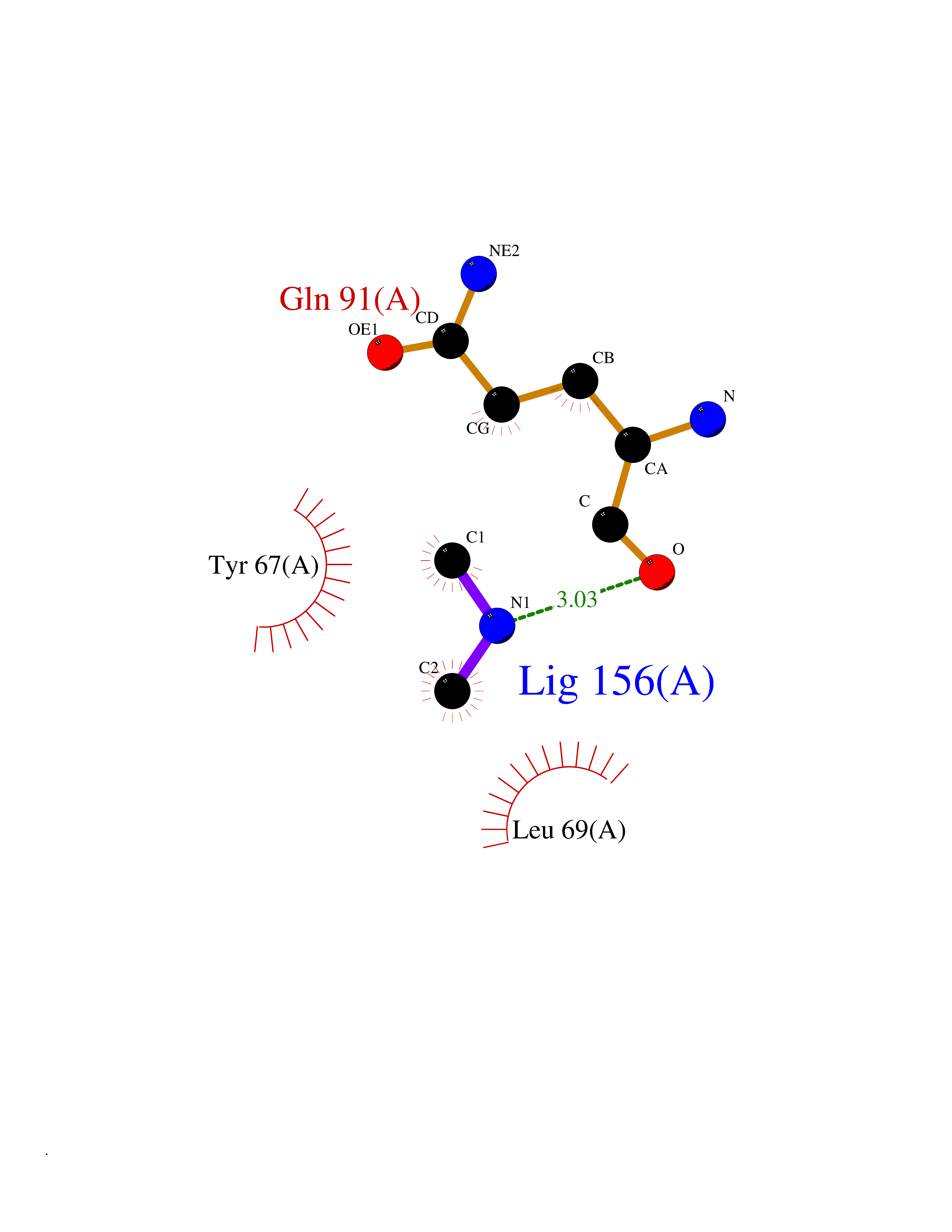 Ligplot Map 3D Binding mode Sequence MVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVRMVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVRMVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVR Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Transferrin (TF) | 1RYO | 3.99 | |
Target general information Gen name TF Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Siderophilin; Serotransferrin; PRO1400; Beta-1 metal-binding globulin Protein family Transferrin family Biochemical class Transferrin Function It is responsible for the transport of iron from sites of absorption and heme degradation to those of storage and utilization. Serum transferrin may also have a further role in stimulating cell proliferation. Transferrins are iron binding transport proteins which can bind two Fe(3+) ions in association with the binding of an anion, usually bicarbonate. Related diseases Atransferrinemia (ATRAF) [MIM:209300]: A rare autosomal recessive disorder characterized by abnormal synthesis of transferrin leading to iron overload and microcytic hypochromic anemia. {ECO:0000269|PubMed:11110675, ECO:0000269|PubMed:15466165}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01370; DB14517; DB14518; DB01294; DB14526; DB14527; DB11136; DB14528; DB14529; DB14530; DB00515; DB09130; DB11397; DB13949; DB14490; DB14491; DB14488; DB14501; DB14489; DB13257; DB06215; DB06784; DB05260; DB01592; DB00893; DB00677; DB06757; DB11182; DB14520; DB01593; DB14487; DB14533; DB14548 Interacts with O43315; O00501; Q7Z7G2; Q9GZR5; Q9Y282; Q96KR6; P01350; P08034; Q8NBJ4; O15529; Q8TED1; Q7Z5P4; A8MZ59; O15173; Q96TC7; Q3KNW5; Q9BXS9-3; Q99523; O43278-2; Q8N9I0; P02786; Q4KMG9; Q9K0U9; Q09057; Q9K0V0; P02786 EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Ion transport; Iron; Iron transport; Metal-binding; Methylation; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 35854.5 Length 324 Aromaticity 0.1 Instability index 34.17 Isoelectric point 7.58 Charge (pH=7) 1.42 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence KTVRWCAVSEHEATKCQSFRDHMKSVIPSDGPSVACVKKASYLDCIRAIAANEADAVTLDAGLVYDAYLAPNNLKPVVAEFYGSKEDPQTFYYAVAVVKKDSGFQMNQLRGKKSCHTGLGRSAGWNIPIGLLYCDLPEPRKPLEKAVANFFSGSCAPCADGTDFPQLCQLCPGCGCSTLNQYFGYSGAFKCLKDGAGDVAFVKHSTIFENLANKADRDQYELLCLDNTRKPVDEYKDCHLAQVPSHTVVARSMGGKEDLIWELLNQAQEHFGKDKSKEFQLFSSPHGKDLLFKDSAHGFLKVPPRMDAKMYLGYEYVTAIRNLR Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Geranylgeranyl pyrophosphate synthase | 2Q80 | 3.99 | |
Target general information Gen name GGPS1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family FPP/GGPP synthase family Biochemical class Transferase Function Dimethylallyltranstransferase activity.Farnesyltranstransferase activity.Geranyltranstransferase activity.Identical protein binding.Metal ion binding. Related diseases Muscular dystrophy, congenital hearing loss, and ovarian insufficiency syndrome (MDHLO) [MIM:619518]: An autosomal recessive disorder characterized by early-onset progressive muscle weakness, sensorineural hearing loss, and primary amenorrhea due to ovarian insufficiency. Some patients become wheelchair-bound by the second decade, whereas others have a milder phenotype and maintain independent ambulation into adulthood. Most patients have respiratory insufficiency. {ECO:0000269|PubMed:32403198}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06830; DB06931; DB07221; DB08529; DB07410; DB07780; DB04695; DB02552; DB07841; DB00710; DB04714; DB07873; DB06548; DB00282; DB00399 Interacts with O00244; O95749; O00560 EC number 2.5.1.-; 2.5.1.1; 2.5.1.10; 2.5.1.29 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Congenital muscular dystrophy; Cytoplasm; Deafness; Disease variant; Isoprene biosynthesis; Lipid metabolism; Magnesium; Metal-binding; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D,E,F Molecular weight (Da) 32872.4 Length 284 Aromaticity 0.11 Instability index 41.09 Isoelectric point 6.17 Charge (pH=7) -3.37 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ETVQRILLEPYKYLLQLPGKQVRTKLSQAFNHWLKVPEDKLQIIIEVTEMLHNASLLIDDIEDNSKLRRGFPVAHSIYGIPSVINSANYVYFLGLEKVLTLDHPDAVKLFTRQLLELHQGQGLDIYWRDNYTCPTEEEYKAMVLQKTGGLFGLAVGLMQLFSDYKEDLKPLLNTLGLFFQIRDDYANLHSKSFCEDLTEGKFSFPTIHAIWSRPESTQVQNILRQRTENIDIKKYCVHYLEDVGSFEYTRNTLKELEAKAYKQIDARGGNPELVALVKHLSKMF Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Poly [ADP-ribose] polymerase 1 (PARP1) | 5WS1 | 3.99 | |
Target general information Gen name PARP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein poly-ADP-ribosyltransferase PARP1; Poly[ADP-ribose] synthetase-1; Poly[ADP-ribose] synthase 1; Poly(ADP-ribose)polymerase-1; PPOL; PARP-1; NAD(+)Poly [ADP-ribose] polymerase-1 ADP-ribosyltrans Protein family ARTD/PARP family Biochemical class Glycosyltransferases Function Mainly mediates glutamate and aspartate ADP-ribosylation of target proteins: the ADP-D-ribosyl group of NAD(+) is transferred to the acceptor carboxyl group of glutamate and aspartate residues and further ADP-ribosyl groups are transferred to the 2'-position of the terminal adenosine moiety, building up a polymer with an average chain length of 20-30 units. Mediates the poly(ADP-ribosyl)ation of a number of proteins, including itself, APLF and CHFR. Also mediates serine ADP-ribosylation of target proteins following interaction with HPF1; HPF1 conferring serine specificity. Probably also catalyzes tyrosine ADP-ribosylation of target proteins following interaction with HPF1. Catalyzes the poly-ADP-ribosylation of histones in a HPF1-dependent manner. Involved in the base excision repair (BER) pathway by catalyzing the poly-ADP-ribosylation of a limited number of acceptor proteins involved in chromatin architecture and in DNA metabolism. ADP-ribosylation follows DNA damage and appears as an obligatory step in a detection/signaling pathway leading to the reparation of DNA strand breaks. In addition to base excision repair (BER) pathway, also involved in double-strand breaks (DSBs) repair: together with TIMELESS, accumulates at DNA damage sites and promotes homologous recombination repair by mediating poly-ADP-ribosylation. In addition to proteins, also able to ADP-ribosylate DNA: catalyzes ADP-ribosylation of DNA strand break termini containing terminal phosphates and a 2'-OH group in single- and double-stranded DNA, respectively. Required for PARP9 and DTX3L recruitment to DNA damage sites. PARP1-dependent PARP9-DTX3L-mediated ubiquitination promotes the rapid and specific recruitment of 53BP1/TP53BP1, UIMC1/RAP80, and BRCA1 to DNA damage sites. Acts as a regulator of transcription: positively regulates the transcription of MTUS1 and negatively regulates the transcription of MTUS2/TIP150. With EEF1A1 and TXK, forms a complex that acts as a T-helper 1 (Th1) cell-specific transcription factor and binds the promoter of IFN-gamma to directly regulate its transcription, and is thus involved importantly in Th1 cytokine production. Involved in the synthesis of ATP in the nucleus, together with NMNAT1, PARG and NUDT5. Nuclear ATP generation is required for extensive chromatin remodeling events that are energy-consuming. Poly-ADP-ribosyltransferase that mediates poly-ADP-ribosylation of proteins and plays a key role in DNA repair. Related diseases Dihydrolipoamide dehydrogenase deficiency (DLDD) [MIM:246900]: An autosomal recessive metabolic disorder characterized biochemically by a combined deficiency of the branched-chain alpha-keto acid dehydrogenase complex (BCKDC), pyruvate dehydrogenase complex (PDC), and alpha-ketoglutarate dehydrogenase complex (KGDC). Clinically, affected individuals have lactic acidosis and neurologic deterioration due to sensitivity of the central nervous system to defects in oxidative metabolism. {ECO:0000269|PubMed:10448086, ECO:0000269|PubMed:11687750, ECO:0000269|PubMed:12925875, ECO:0000269|PubMed:15712224, ECO:0000269|PubMed:16442803, ECO:0000269|PubMed:16770810, ECO:0000269|PubMed:17404228, ECO:0000269|PubMed:20160912, ECO:0000269|PubMed:8506365, ECO:0000269|PubMed:8968745, ECO:0000269|PubMed:9540846, ECO:0000269|PubMed:9934985}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04010; DB03509; DB03072; DB03722; DB03073; DB07787; DB07096; DB07330; DB02498; DB13877; DB02701; DB11793; DB02690; DB09074; DB12332; DB11760; DB00277; DB07232; DB01593; DB14487; DB14533; DB14548 Interacts with Q8IW19; Q7Z2E3; P42574; P49715; Q86WJ1-1; P26358; Q01094; Q96L91; P11308; O60741; P09429; Q13007; Q9BQ69; P08651; Q9Y530; P09874; Q8N2W9; P46063; Q9NTX7; Q14684-1; O95863; P63165; P04637; P0CG48; Q14191; P18887; P54577; Q2M1K9; Q02085 EC number EC 2.4.2.30 Uniprot keywords 3D-structure; Acetylation; ADP-ribosylation; Allosteric enzyme; Apoptosis; Chromosome; Cytoplasm; Direct protein sequencing; DNA damage; DNA repair; DNA-binding; Glycosyltransferase; Immunity; Innate immunity; Isopeptide bond; Metal-binding; NAD; Nucleotidyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 36904 Length 329 Aromaticity 0.08 Instability index 35.9 Isoelectric point 6.83 Charge (pH=7) -0.44 2D Binding mode Binding energy (Kcal/mol) -5.45  Molscript Map  Pymol Map 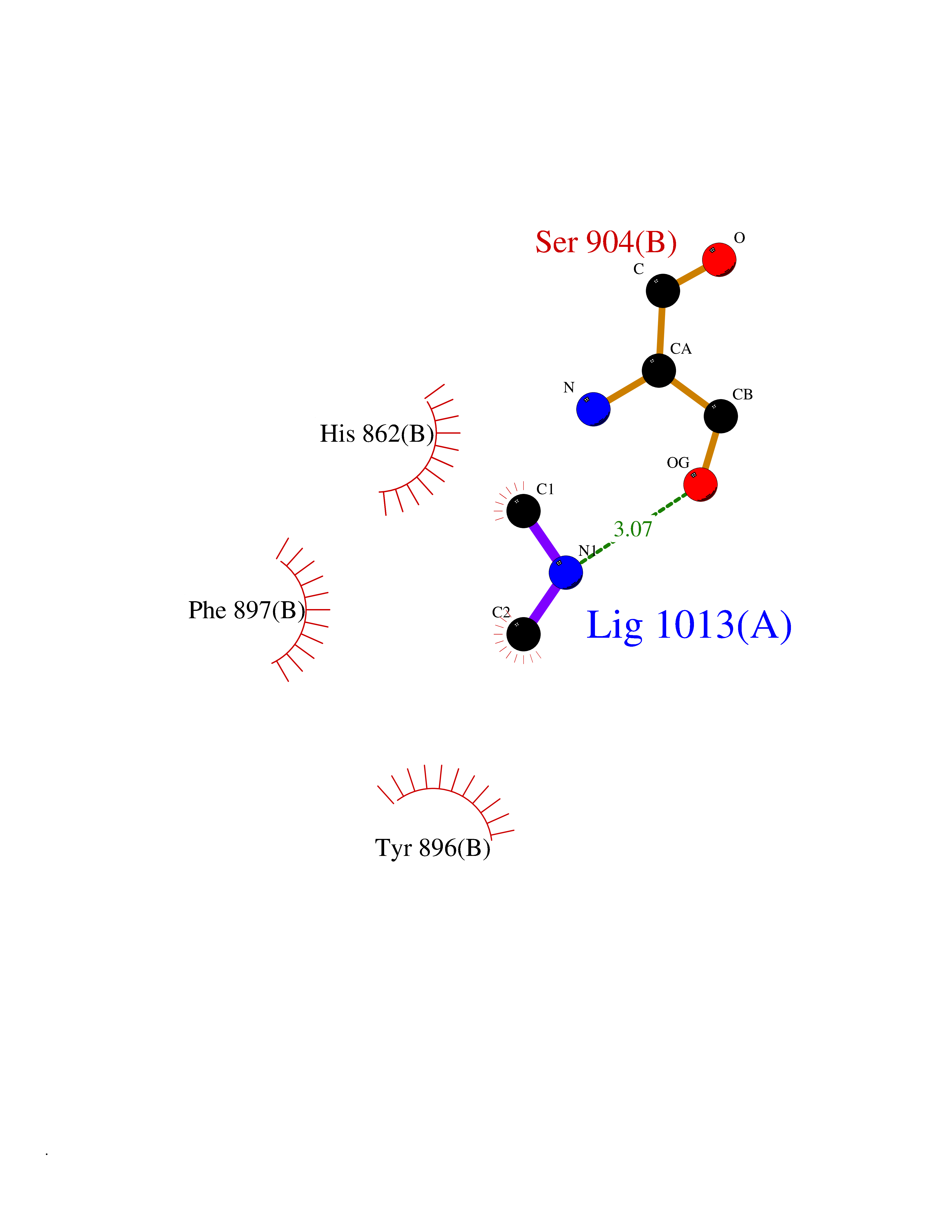 Ligplot Map 3D Binding mode Sequence DLIKMIFDVESMKKAMVEYEIDLQKMPLGKLSKRQIQAAYSILSEVQQAVSQGSDSQILDLSNRFYTLIPHDFGMKKPPLLNNADSVQAKAEMLDNLLDIEVAYSLPIDVNYEKLKTDIKVVDRDSEEAEIIRKYVKNTHATTHNAYDLEVIDIFKIEREGECQRYKPFKQLHNRRLLWHGSRTTNFAGILSQGLRIAPPEAPVTGYMFGKGIYFADMVSKSANYCHTSQGDPIGLILLGEVALGNMYELKHASHISKLPKGKHSVKGLGKTTPDPSANISLDGVDVPLGTGISSGVNDTSLLYNEYIVYDIAQVNLKYLLKLKFNFKT Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Cyclin-dependent kinase 4 | 2W96 | 3.99 | |
Target general information Gen name CDK4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, CDC2/CDKX subfamily Biochemical class Cell cycle Function ATP binding.Cyclin binding.Cyclin-dependent protein serine/threonine kinase activity.Cyclin-dependent protein serine/threonine kinase regulator activity.Protein complex binding. Related diseases Melanoma, cutaneous malignant 3 (CMM3) [MIM:609048]: A malignant neoplasm of melanocytes, arising de novo or from a pre-existing benign nevus, which occurs most often in the skin but may also involve other sites. {ECO:0000269|PubMed:7652577, ECO:0000269|PubMed:8528263, ECO:0000269|PubMed:9311594, ECO:0000269|PubMed:9425228}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12001; DB03496; DB12010; DB09073; DB02733; DB11730; DB15442 Interacts with Q9UH17; P24385; P30279; P30281; Q16543; P50613; P38936; P46527; P49918; P42771; P42772; P42773; P55273; Q9UJC3; P08238; Q9UKT9; Q0VD86; P01106; Q9ULD0; P28749; Q08999; P09936; Q8N720 EC number 2.7.11.22 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cell cycle; Cell division; Cytoplasm; Disease variant; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID B Molecular weight (Da) 30138.4 Length 267 Aromaticity 0.09 Instability index 36.2 Isoelectric point 5.78 Charge (pH=7) -5.83 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map 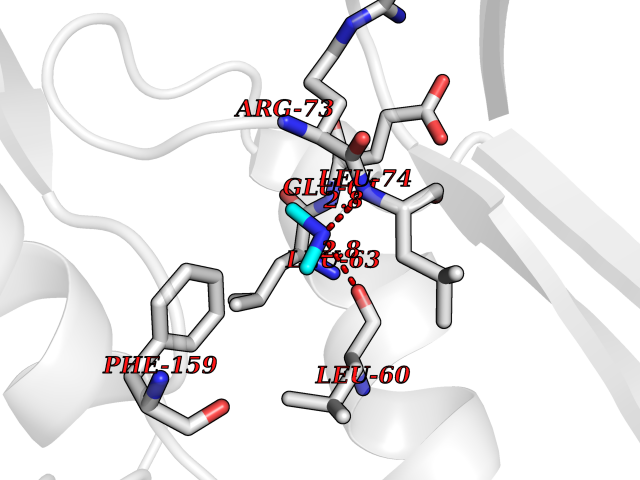 Pymol Map 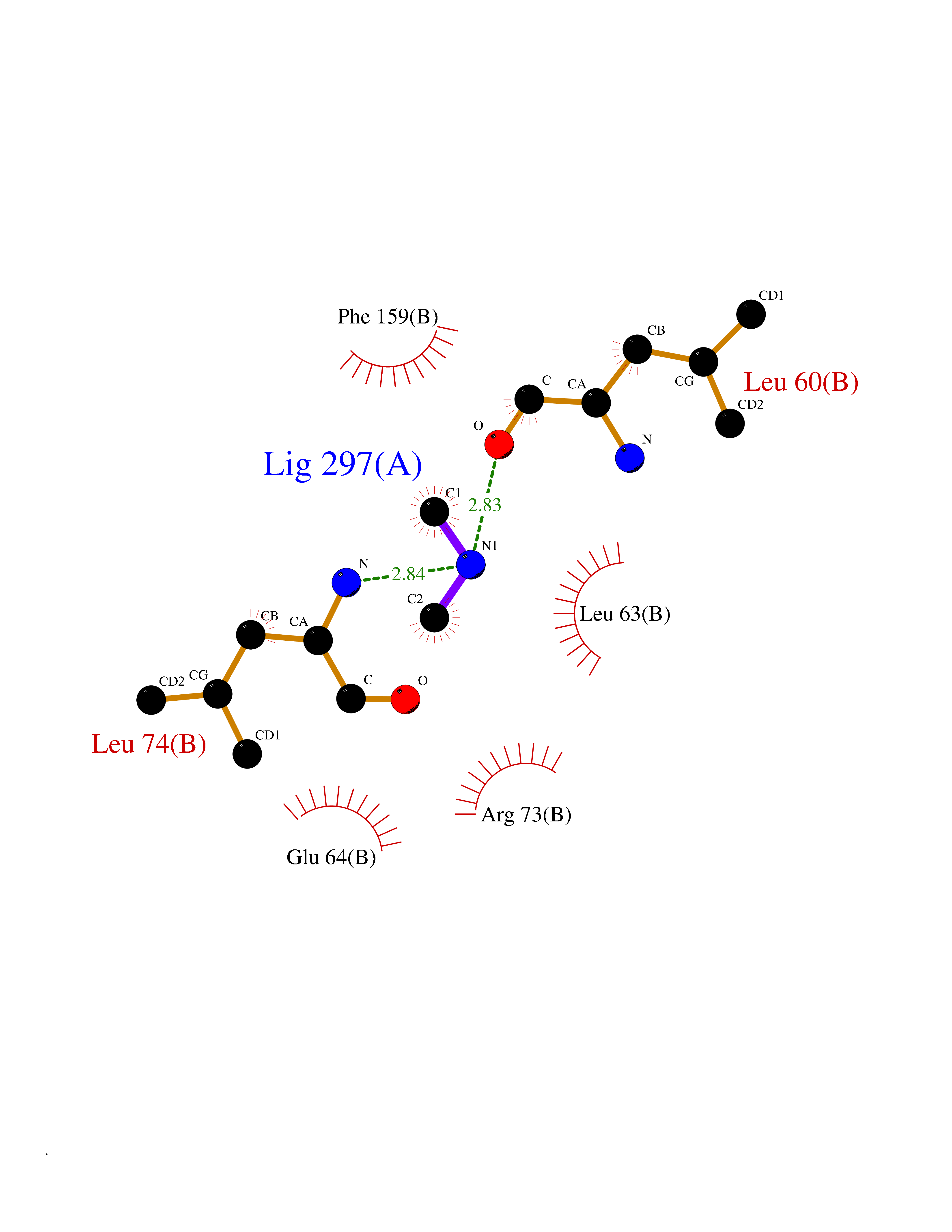 Ligplot Map 3D Binding mode Sequence SRYEPVAEIGVGAYGTVYKARDPHSGHFVALKSVRVPNGEEGLPISTVREVALLRRLEAFEHPNVVRLMDVCATSRTDREIKVTLVFEHVDQDLRTYLDKAPPPGLPAETIKDLMRQFLRGLDFLHANCIVHRDLKPENILVTSGGTVKLADFGLARIYSYQMALDPVVVTLWYRAPEVLLQSTYATPVDMWSVGCIFAEMFRRKPLFCGNSEADQLGKIFDLIGLPPEDDWVPEMEESGAQLLLEMLTFNPHKRISAFRALQHSYL Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Camphor 5-monooxygenase | 4L4E | 3.99 | |
Target general information Gen name camC Organism Pseudomonas putida (Arthrobacter siderocapsulatus) Uniprot ID TTD ID NA Synonyms cyp101 Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Camphor 5-monooxygenase activity.Heme binding.Iron ion binding. Related diseases Combined oxidative phosphorylation deficiency 6 (COXPD6) [MIM:300816]: A mitochondrial disease resulting in a neurodegenerative disorder characterized by psychomotor delay, hypotonia, areflexia, muscle weakness and wasting. Some patients manifest prenatal ventriculomegaly and severe postnatal encephalomyopathy. {ECO:0000269|PubMed:20362274, ECO:0000269|PubMed:22019070, ECO:0000269|PubMed:25583628, ECO:0000269|PubMed:26004228, ECO:0000269|PubMed:26173962, ECO:0000269|PubMed:27178839}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, X-linked recessive, 4, with or without cerebellar ataxia (CMTX4) [MIM:310490]: A neuromuscular disorder characterized by progressive sensorimotor axonal neuropathy, distal sensory impairment, difficulty walking due to peripheral neuropathy and/or cerebellar ataxia, and deafness due to auditory neuropathy. Additional features include cognitive impairment, cerebellar atrophy, dysarthria, abnormal extraocular movements, tremor, dysmetria and spasticity. The age at onset ranges from infancy to young adulthood. {ECO:0000269|PubMed:23217327, ECO:0000269|PubMed:26004228}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, X-linked, 5, with peripheral neuropathy (DFNX5) [MIM:300614]: A form of hearing loss characterized by absent or severely abnormal auditory brainstem response, abnormal middle ear reflexes, abnormal speech discrimination, loss of outer hair cell function, and cochlear nerve hypoplasia. DFNX5 patients manifest auditory neuropathy with childhood onset, associated with distal sensory impairment affecting the peripheral nervous system. {ECO:0000269|PubMed:25986071}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepimetaphyseal dysplasia, X-linked, with hypomyelinating leukodystrophy (SEMDHL) [MIM:300232]: An X-linked recessive developmental disorder characterized by slowly progressive skeletal and neurologic abnormalities, including short stature, large and deformed joints, significant motor impairment, visual defects, and sometimes cognitive deficits. Affected individuals typically have normal early development in the first year or so of life, followed by development regression and the development of symptoms. Brain imaging shows white matter abnormalities consistent with hypomyelinating leukodystrophy. {ECO:0000269|PubMed:28842795}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03836; DB02617; DB02817; DB03627; DB04032; DB03031; DB02125; DB04501; DB01744; DB01663; DB01011; DB01703; DB01826; DB03540; DB02851 Interacts with P00259 EC number 1.14.15.1 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Heme; Iron; Metal-binding; Monooxygenase; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 45446.3 Length 405 Aromaticity 0.08 Instability index 45.33 Isoelectric point 5.23 Charge (pH=7) -16.08 2D Binding mode Binding energy (Kcal/mol) -5.44 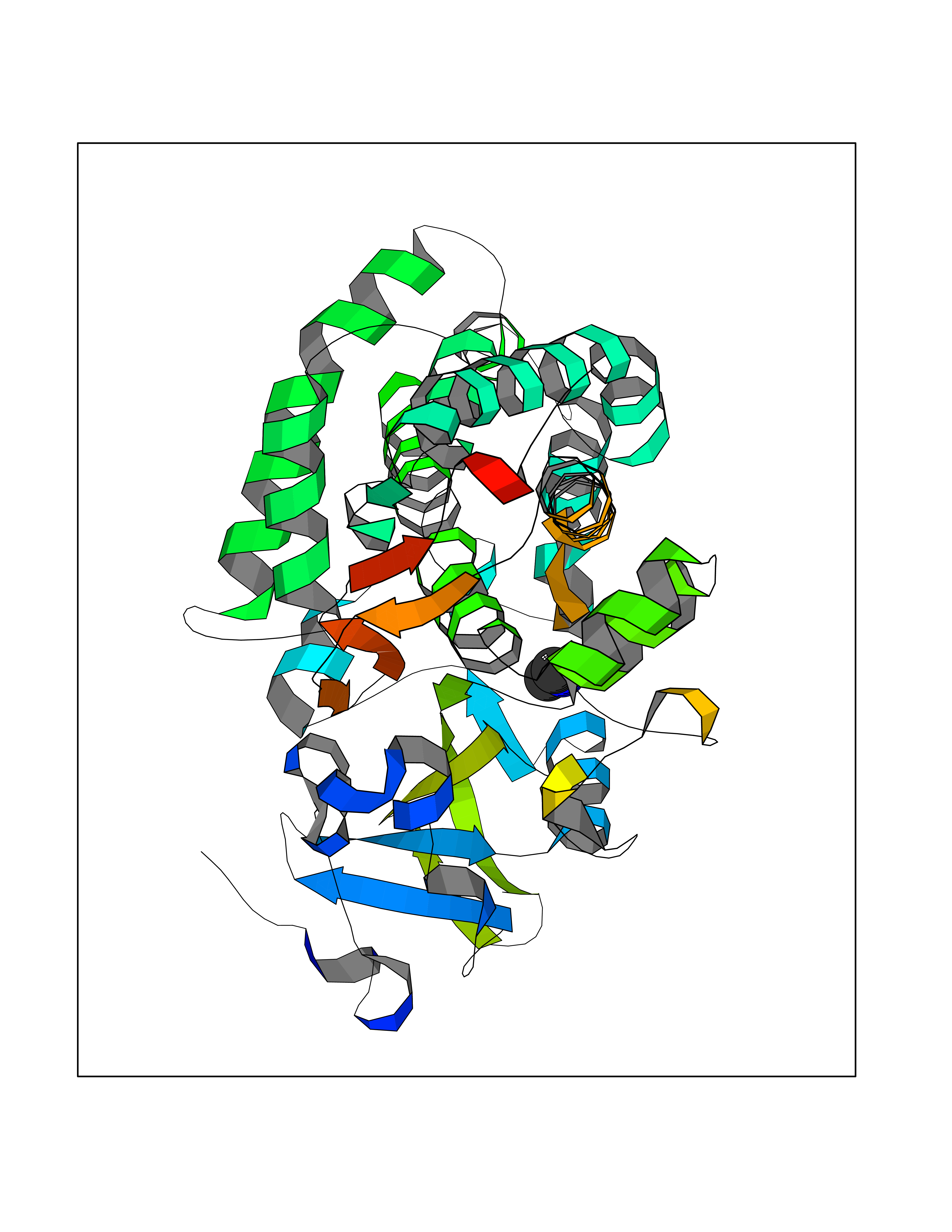 Molscript Map 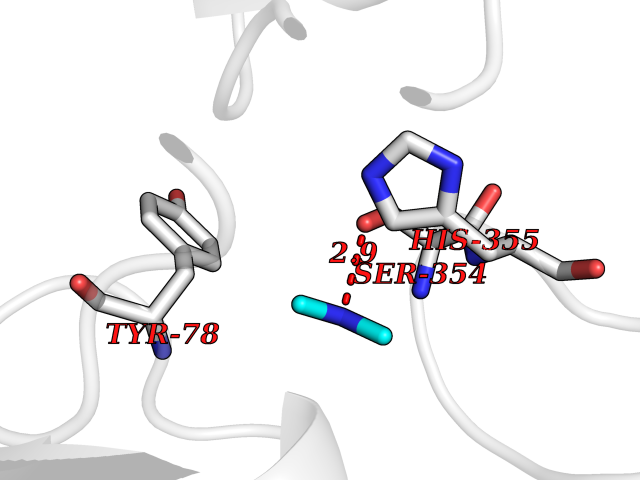 Pymol Map 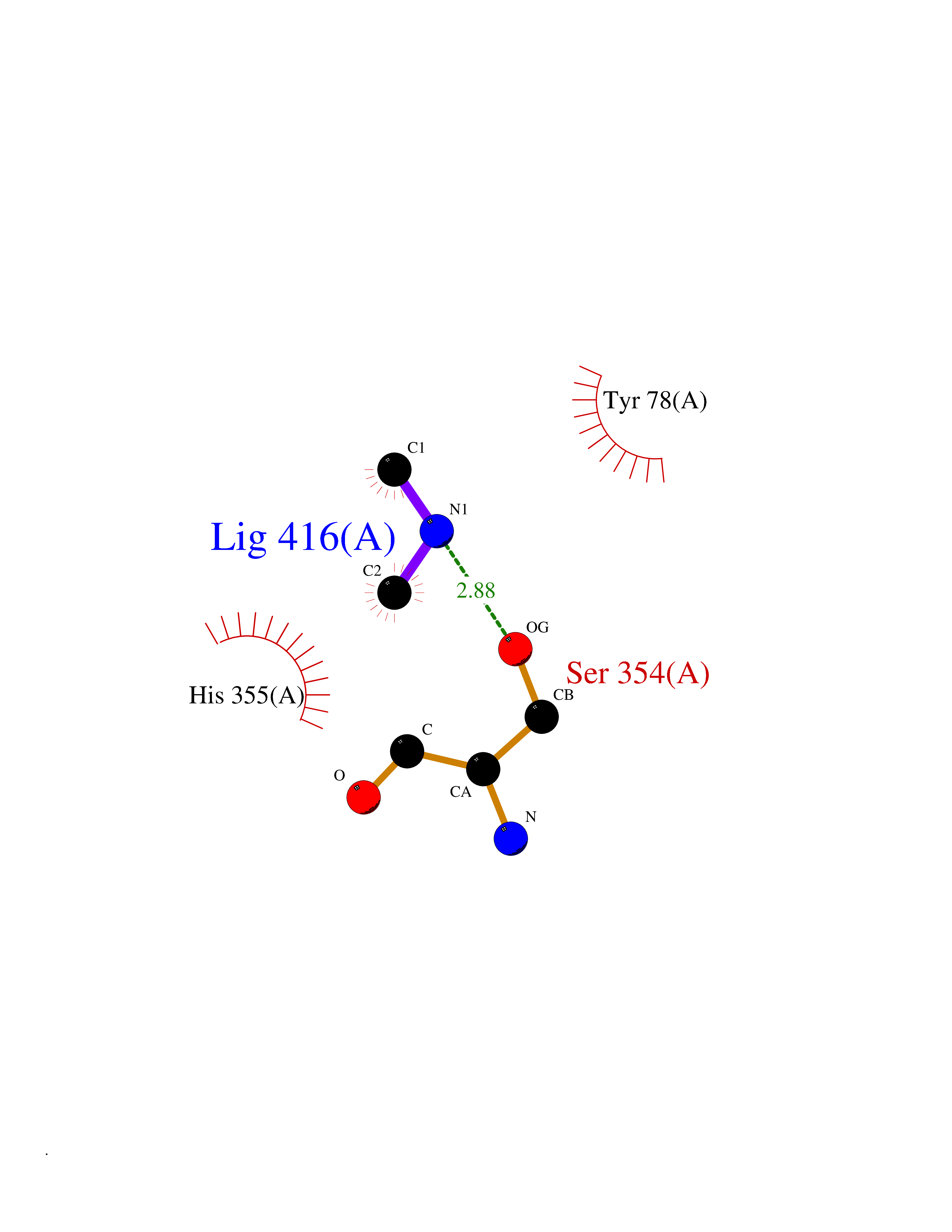 Ligplot Map 3D Binding mode Sequence NLAPLPPHVPEHLVFDFDMYNPSNLSAGVQEAWAVLQESNVPDLVWTRCNGGHWIATRGQLIREAYEDYRHFSSECPFIPREAGEAYDFIPTSMDPPEQRQFRALANQVVGMPVVDKLENRIQELACSLIESLRPQGQCNFTEDYAEPFPIRIFMLLAGLPEEDIPHLGYLTDQMTRPDGSMTFAEAKEALYDYLIPIIEQRRQKPGTDAISIVANGQVNGRPITSDEAKRMCGLLLVGGLDTVVNFLSFSMEFLAKSPEHRQELIERPERIPAACEELLRRFSLVADGRILTSDYEFHGVQLKKGDQILLPQMLSGLDERENAAPMHVDFSRQKVSHTTFGHGSHLCAGQHLARREIIVTLKEWLTRIPDFSIAPGAQIQHKSGIVSGVQALPLVWDPATTKAV Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Glutamate dehydrogenase 1, mitochondrial | 1L1F | 3.99 | |
Target general information Gen name GLUD1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms GLUD Protein family Glu/Leu/Phe/Val dehydrogenases family Biochemical class Oxidoreductase Function ADP binding.ATP binding.Glutamate dehydrogenase (NAD+) activity.Glutamate dehydrogenase [NAD(P)+] activity.GTP binding.Identical protein binding.Leucine binding.NAD+ binding. Related diseases Hyperinsulinemic hypoglycemia, familial, 6 (HHF6) [MIM:606762]: A form of hyperinsulinemic hypoglycemia, a clinically and genetically heterogeneous disorder characterized by inappropriate insulin secretion from the pancreatic beta-cells in the presence of low blood glucose levels. HHF6 is an autosomal dominant form characterized by hypoglycemia due to congenital hyperinsulinism combined with persistent hyperammonemia. Clinical features include loss of consciousness due to hypoglycemia, hypoglycemic seizures, and mental retardation. {ECO:0000269|PubMed:10636977, ECO:0000269|PubMed:11214910, ECO:0000269|PubMed:11297618, ECO:0000269|PubMed:9571255}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11081; DB00142; DB04137; DB00756; DB00157 Interacts with P49448 EC number 1.4.1.3 Uniprot keywords 3D-structure; Acetylation; ADP-ribosylation; Alternative splicing; ATP-binding; Direct protein sequencing; Disease variant; Endoplasmic reticulum; GTP-binding; Hydroxylation; Mitochondrion; NADP; Nucleotide-binding; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Transit peptide Protein physicochemical properties Chain ID A,B,C,D,E,F Molecular weight (Da) 49471.9 Length 447 Aromaticity 0.09 Instability index 27.17 Isoelectric point 7.3 Charge (pH=7) 0.81 2D Binding mode Binding energy (Kcal/mol) -5.44 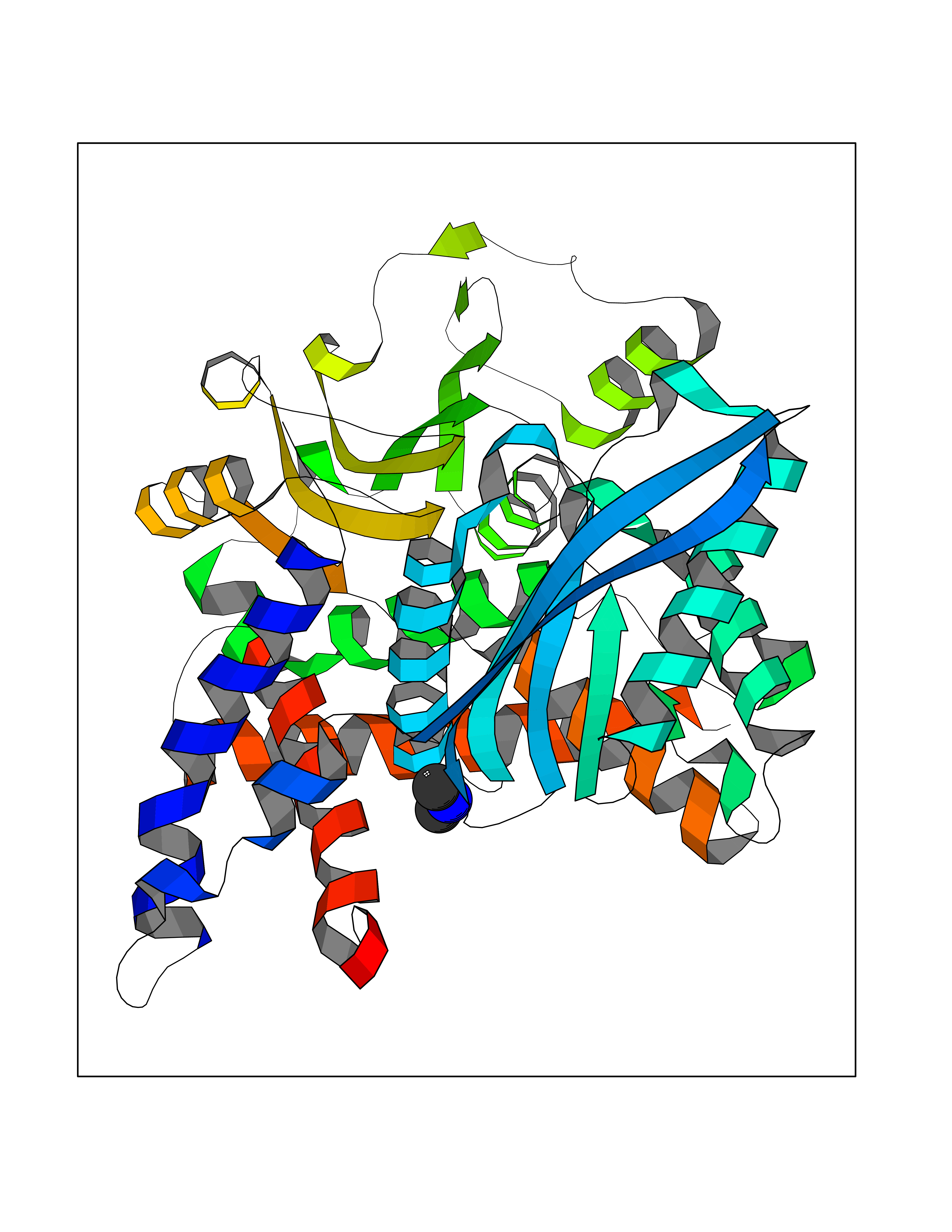 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence DPNFFKMVEGFFDRGASIVEDKLVEDLRTRESEEQKRNRVRGILRIIKPCNHVLSLSFPIRRDDGSWEVIEGYRAQHSQHRTPCKGGIRYSTDVSVDEVKALASLMTYKCAVVDVPFGGAKAGVKINPKNYTDNELEKITRRFTMELAKKGFIGPGIDVPAPDMSTGEREMSWIADTYASTIGHYDINAHACVTGKPISQGGIHGRISATGRGVFHGIENFINEASYMSILGMTPGFGDKTFVVQGFGNVGLHSMRYLHRFGAKCIAVGESDGSIWNPDGIDPKELEDFKLQHGSILGFPKAKPYEGSILEADCDILIPAASEKQLTKSNAPRVKAKIIAEGANGPTTPEADKIFLERNIMVIPDLYLNAGGVTVSYFEWLKNLNHVSYSEKDIVHSGLAYTMERSARQIMRTAMKYNLGLDLRTAAYVNAIEKVFKVYNEAGVTFT Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Phenylalanine hydroxylase (PAH) | 1J8U | 3.99 | |
Target general information Gen name PAH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Phenylalanine4hydroxylase; Phenylalanine-4-hydroxylase; Phe4monooxygenase; Phe-4-monooxygenase Protein family Biopterin-dependent aromatic amino acid hydroxylase family Biochemical class Paired donor oxygen oxidoreductase Function Catalyzes the hydroxylation of L-phenylalanine to L-tyrosine. Related diseases Phenylalanine hydroxylase deficiency (PAH deficiency) [MIM:261600]: An autosomal recessive inborn error of phenylalanine metabolism characterized by intolerance to dietary intake of the essential amino acid phenylalanine. The disease spectrum depends on the degree of PAH deficiency and the phenylalanine levels in plasma. Severe deficiency causes classic phenylketonuria (PKU) that is characterized by plasma concentrations of phenylalanine persistently above 1200 umol/L. PKU patients develop profound and irreversible intellectual disability, unless low phenylalanine diet is introduced early in life. They tend to have light pigmentation, rashes similar to eczema, epilepsy, extreme hyperactivity, psychotic states and an unpleasant 'mousy' odor. Less severe forms of PAH deficiency are characterized by phenylalanine levels above normal (120 umol/L) but below 1200 umol/L and include moderate PKU, mild PKU, non-PKU hyperphenylalaninemia (non-PKU HPA) and mild hyperphenylalaninemia. Individuals with PAH deficiency who have plasma phenylalanine concentrations consistently below 600 umol/L on an unrestricted diet are not at higher risk of developing intellectual, neurologic, and neuropsychological impairment than are individuals without PAH deficiency. {ECO:0000269|PubMed:10200057, ECO:0000269|PubMed:10679941, ECO:0000269|PubMed:11180595, ECO:0000269|PubMed:11326337, ECO:0000269|PubMed:11385716, ECO:0000269|PubMed:11461196, ECO:0000269|PubMed:11935335, ECO:0000269|PubMed:12501224, ECO:0000269|PubMed:1301187, ECO:0000269|PubMed:1355066, ECO:0000269|PubMed:1358789, ECO:0000269|PubMed:1363837, ECO:0000269|PubMed:1363838, ECO:0000269|PubMed:1671810, ECO:0000269|PubMed:1672290, ECO:0000269|PubMed:1672294, ECO:0000269|PubMed:1679030, ECO:0000269|PubMed:1709636, ECO:0000269|PubMed:18538294, ECO:0000269|PubMed:1975559, ECO:0000269|PubMed:2014802, ECO:0000269|PubMed:22513348, ECO:0000269|PubMed:22526846, ECO:0000269|PubMed:23792259, ECO:0000269|PubMed:2564729, ECO:0000269|PubMed:2615649, ECO:0000269|PubMed:2840952, ECO:0000269|PubMed:32668217, ECO:0000269|PubMed:7833954, ECO:0000269|PubMed:8068076, ECO:0000269|PubMed:8088845, ECO:0000269|PubMed:8098245, ECO:0000269|PubMed:8406445, ECO:0000269|PubMed:8889583, ECO:0000269|PubMed:8889590, ECO:0000269|PubMed:9048935, ECO:0000269|PubMed:9101291, ECO:0000269|PubMed:9450897, ECO:0000269|PubMed:9452061, ECO:0000269|PubMed:9452062, ECO:0000269|PubMed:9521426, ECO:0000269|PubMed:9600453, ECO:0000269|PubMed:9634518, ECO:0000269|PubMed:9792407, ECO:0000269|PubMed:9792411, ECO:0000269|PubMed:9852673, ECO:0000269|PubMed:9950317}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03673; DB04339; DB06778; DB04419; DB06262; DB04400; DB00368; DB00120; DB02562; DB00360 Interacts with NA EC number EC 1.14.16.1 Uniprot keywords 3D-structure; Allosteric enzyme; Direct protein sequencing; Disease variant; Iron; Metal-binding; Monooxygenase; Oxidoreductase; Phenylalanine catabolism; Phenylketonuria; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 35556.1 Length 307 Aromaticity 0.15 Instability index 39.96 Isoelectric point 6.17 Charge (pH=7) -3.5 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map  Pymol Map 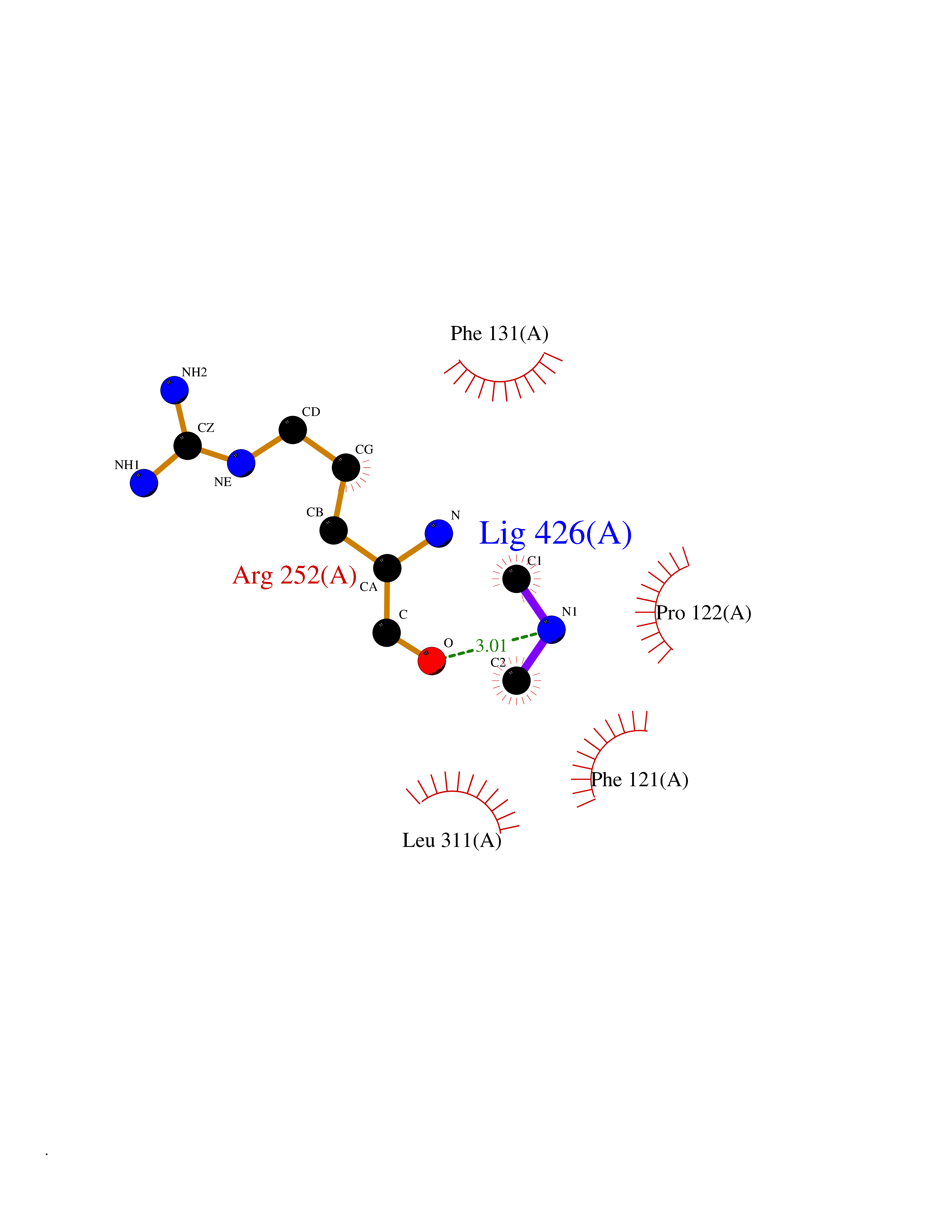 Ligplot Map 3D Binding mode Sequence VPWFPRTIQELDRFANQILSYGAELDADHPGFKDPVYRARRKQFADIAYNYRHGQPIPRVEYMEEEKKTWGTVFKTLKSLYKTHACYEYNHIFPLLEKYCGFHEDNIPQLEDVSQFLQTCTGFRLRPVAGLLSSRDFLGGLAFRVFHCTQYIRHGSKPMYTPEPDICHELLGHVPLFSDRSFAQFSQEIGLASLGAPDEYIEKLATIYWFTVEFGLCKQGDSIKAYGAGLLSSFGELQYCLSEKPKLLPLELEKTAIQNYTVTEFQPLYYVAESFNDAKEKVRNFAATIPRPFSVRYDPYTQRIEVL Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Bifunctional methylenetetrahydrofolate dehydrogenase/cyclohydrolase, mitochondrial | 5TC4 | 3.99 | |
Target general information Gen name MTHFD2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NMDMC Protein family Tetrahydrofolate dehydrogenase/cyclohydrolase family Biochemical class Oxidoreductase Function Magnesium ion binding.Methenyltetrahydrofolate cyclohydrolase activity.Methylenetetrahydrofolate dehydrogenase (NAD+) activity.Methylenetetrahydrofolate dehydrogenase (NADP+) activity.Phosphate ion binding. Related diseases Acute hepatic porphyria (AHEPP) [MIM:612740]: A form of porphyria. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. AHP is characterized by attacks of gastrointestinal disturbances, abdominal colic, paralyses and peripheral neuropathy. Most attacks are precipitated by drugs, alcohol, caloric deprivation, infections, or endocrine factors. {ECO:0000269|PubMed:10706561, ECO:0000269|PubMed:1309003, ECO:0000269|PubMed:1569184, ECO:0000269|PubMed:17236137, ECO:0000269|PubMed:2063868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00157; DB00116 Interacts with Q9UJ70-2 EC number 1.5.1.15; 3.5.4.9 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Hydrolase; Isopeptide bond; Magnesium; Mitochondrion; Multifunctional enzyme; NAD; NADP; One-carbon metabolism; Oxidoreductase; Proteomics identification; Reference proteome; Transit peptide; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 31600.3 Length 292 Aromaticity 0.03 Instability index 27.9 Isoelectric point 8 Charge (pH=7) 1.64 2D Binding mode Binding energy (Kcal/mol) -5.44 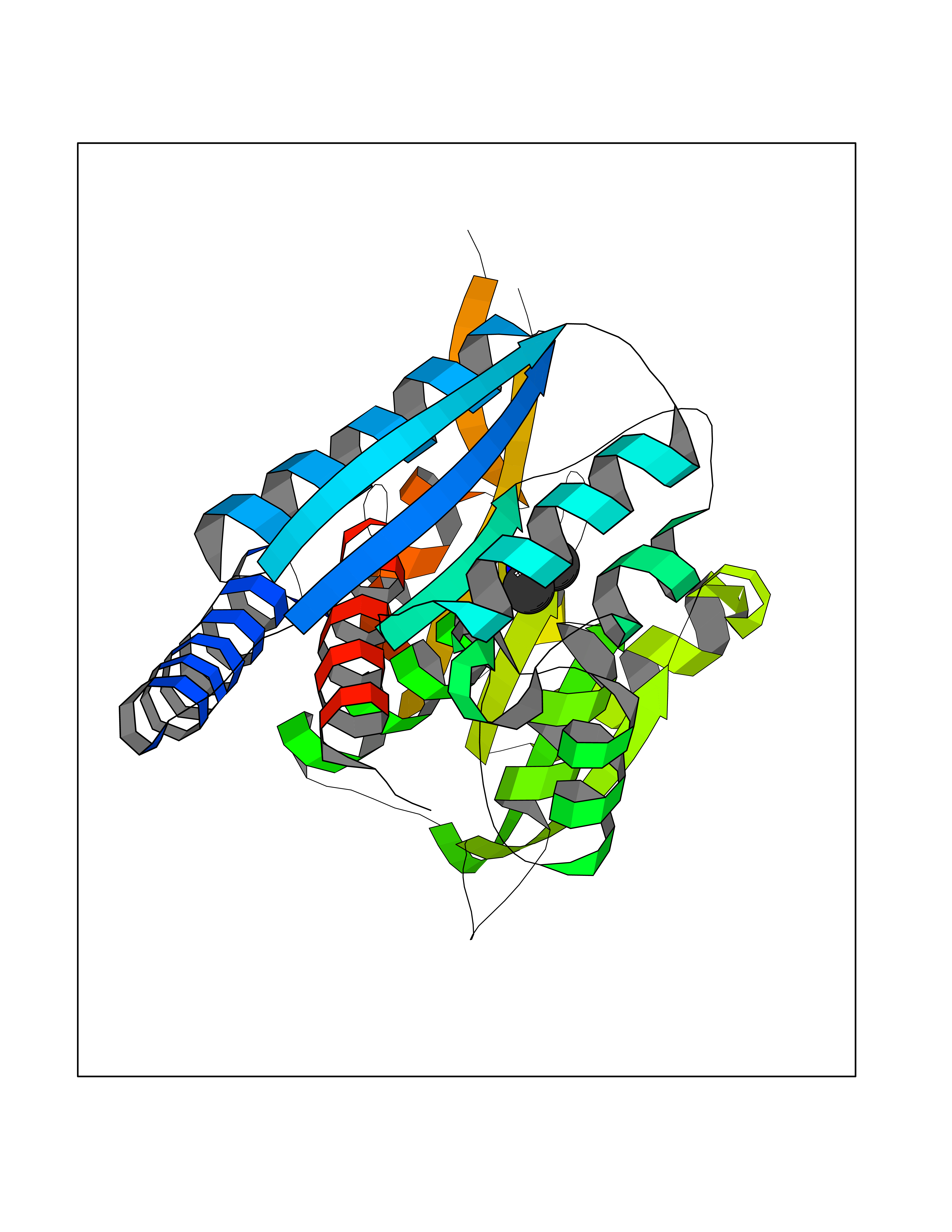 Molscript Map 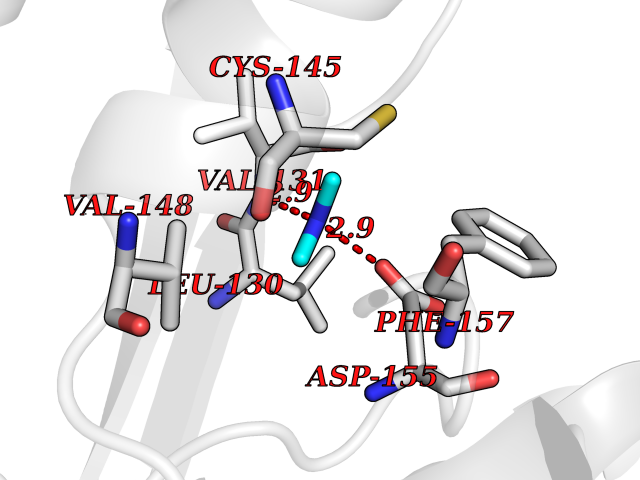 Pymol Map 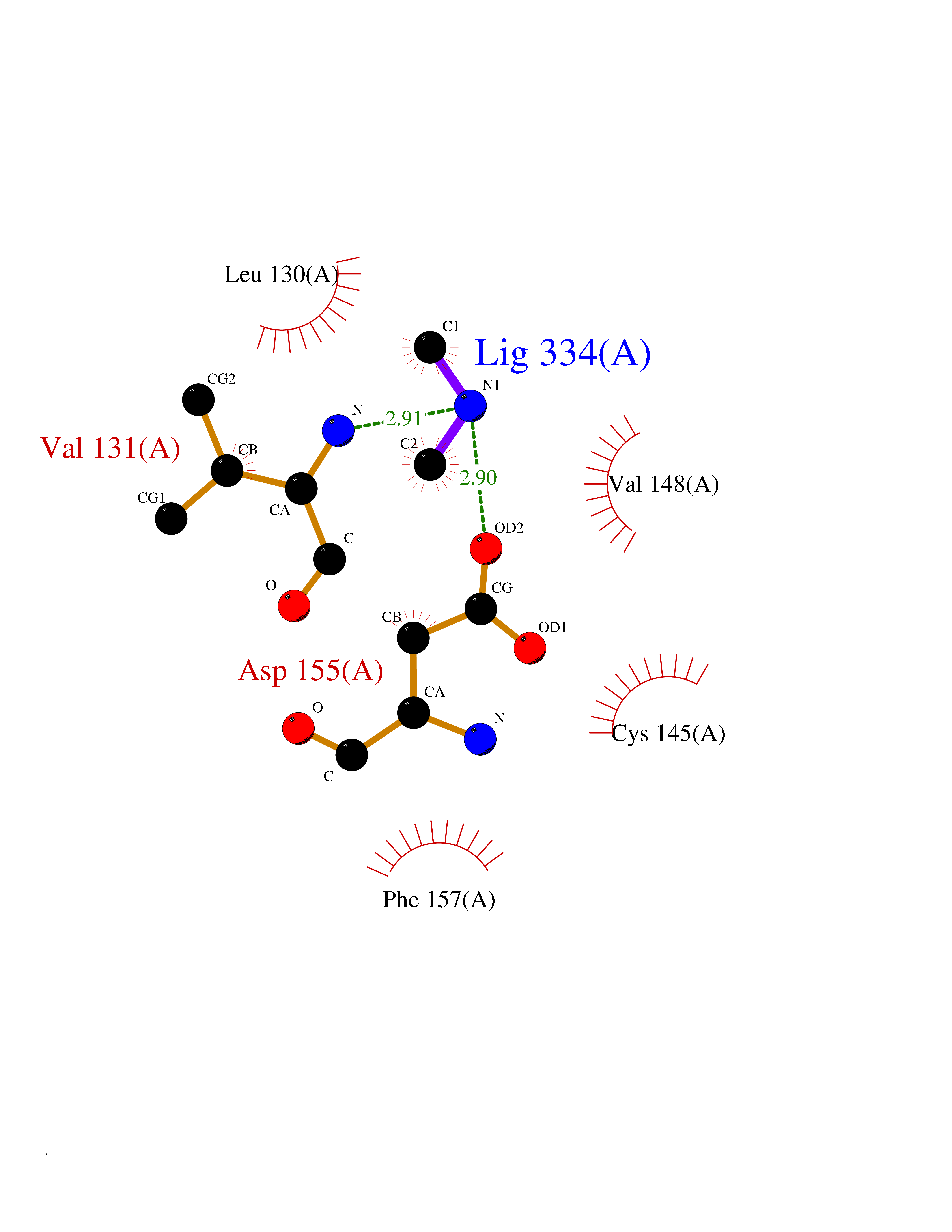 Ligplot Map 3D Binding mode Sequence EAVVISGRKLAQQIKQEVRQEVEEWVASGNKRPHLSVILVGENPASHSYVLNKTRAAAVVGINSETIMKPASISEEELLNLINKLNNDDNVDGLLVQLPLPEHIDERRICNAVSPDKDVDGFHVINVGRMCLDQYSMLPATPWGVWEIIKRTGIPTLGKNVVVAGRSKNVGMPIAMLLHTDGAHERPGGDATVTISHRYTPKEQLKKHTILADIVISAAGIPNLITADMIKEGAAVIDVGINRVHKPKLVGDVDFEGVRQKAGYITPVPGGVGPMTVAMLMKNTIIAAKKVL Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Catechol-O-methyl-transferase (COMT) | 3BWY | 3.99 | |
Target general information Gen name COMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms S-COMT; MB-COMT; Catechol-O-methyltransferase; COMT Protein family Class I-like SAM-binding methyltransferase superfamily, Cation-dependent O-methyltransferase family Biochemical class Methyltransferase Function Catalyzes the O-methylation, and thereby the inactivation, of catecholamine neurotransmitters and catechol hormones. Also shortens the biological half-lives of certain neuroactive drugs, like L-DOPA, alpha-methyl DOPA and isoproterenol. Related diseases Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:15645182}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07462; DB02342; DB02105; DB08049; DB00118; DB00714; DB03336; DB00286; DB00255; DB00841; DB00988; DB15488; DB00494; DB00668; DB00783; DB00977; DB01064; DB00968; DB01141; DB03907; DB04820; DB06152; DB11632; DB00252; DB01420; DB00323 Interacts with Q6P5T0; P30518; Q8NFU1; Q8NHW4; P34972; Q96BA8; P50402; Q5JX71; O14843; O00258; P08034; O75712; Q9NTQ9; O95377; Q8TDT2; Q8N6U8; O15529; P31937; Q9H2F3; O95279; Q5SR56; A6NDP7; Q0D2K0; Q7RTS5; Q9UHJ9-5; Q8IY26; Q9H6H4; Q6NTF9-3; O75783; Q99500; Q9Y6D0; Q3KNW5; O60669; P22732; Q96G79; Q5T1Q4; Q9NY26; Q9NP94; Q6P1K1; P30825; Q9UHI5; B2RUZ4; Q9UPZ6; Q96MV1; Q9NV29; A0PK00; Q9NUH8; Q9P0S9; Q14656; Q6UW68; Q9H0R3; O95807; P34981; Q15645; Q15836; O95183; O76024; P30260; Q9H816; Q92997; P29323-3; P22607; P06396; Q15323; Q6A162; P26371; O15116; P20645; O14744; Q5T160; Q9UJD0; Q2MKA7; Q8N488; O75880; Q14141; Q9UNE7; Q15645; Q9NYH9; Q8NA23-2 EC number EC 2.1.1.6 Uniprot keywords 3D-structure; Alternative initiation; Catecholamine metabolism; Cell membrane; Cytoplasm; Direct protein sequencing; Lipid metabolism; Magnesium; Membrane; Metal-binding; Methyltransferase; Neurotransmitter degradation; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Schizophrenia; Signal-anchor; Transferase; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 23851.2 Length 214 Aromaticity 0.07 Instability index 25.99 Isoelectric point 5.25 Charge (pH=7) -7.75 2D Binding mode Binding energy (Kcal/mol) -5.45 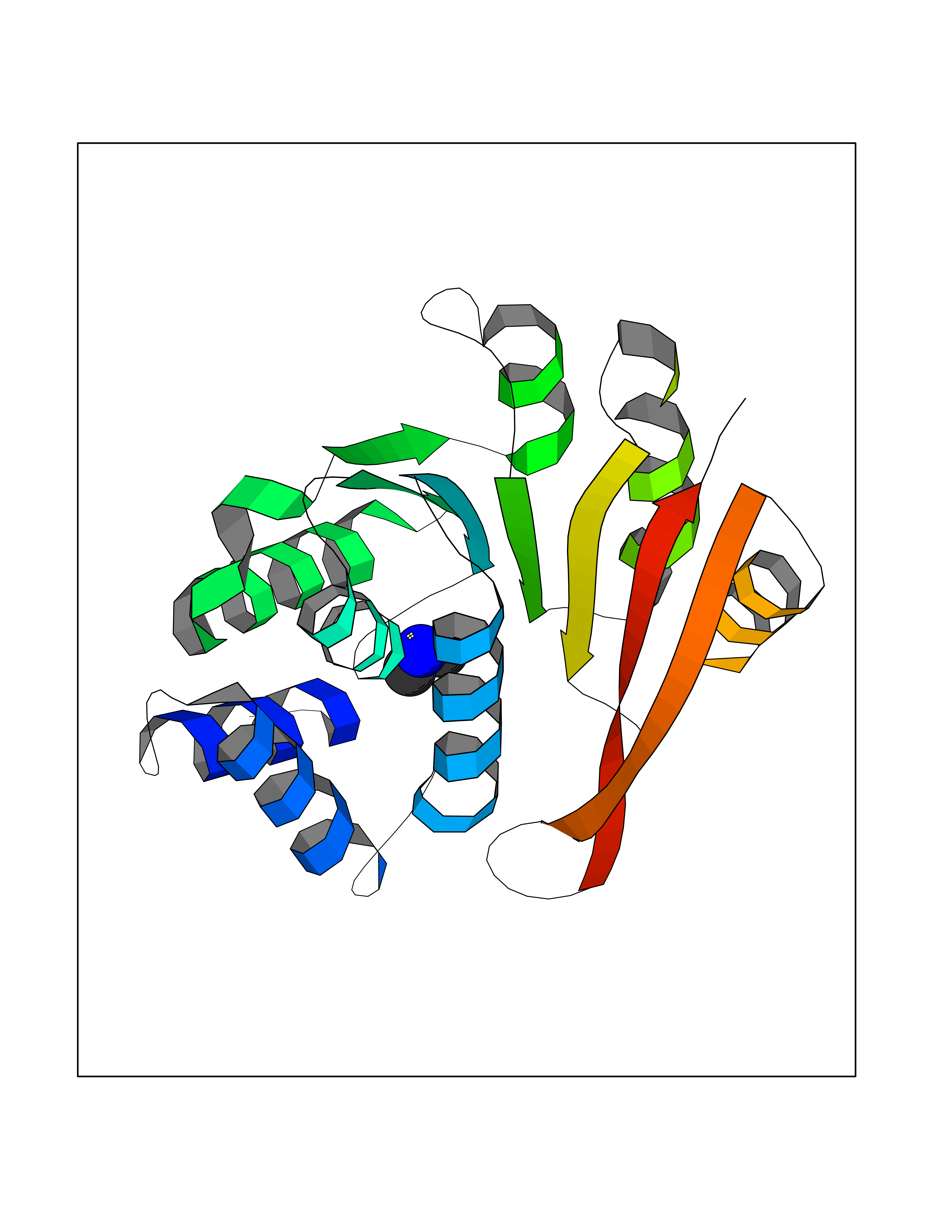 Molscript Map 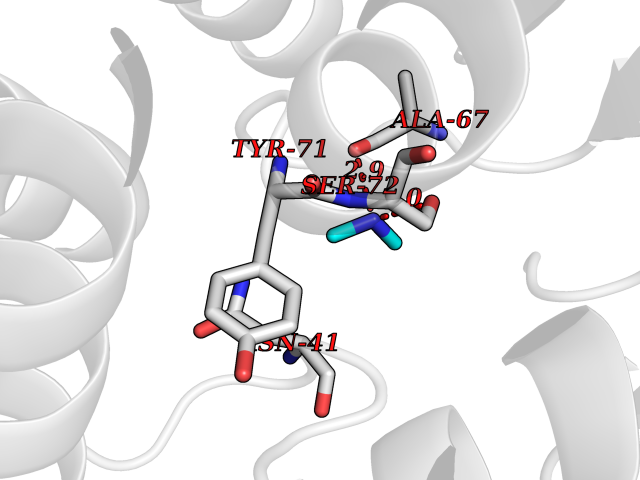 Pymol Map 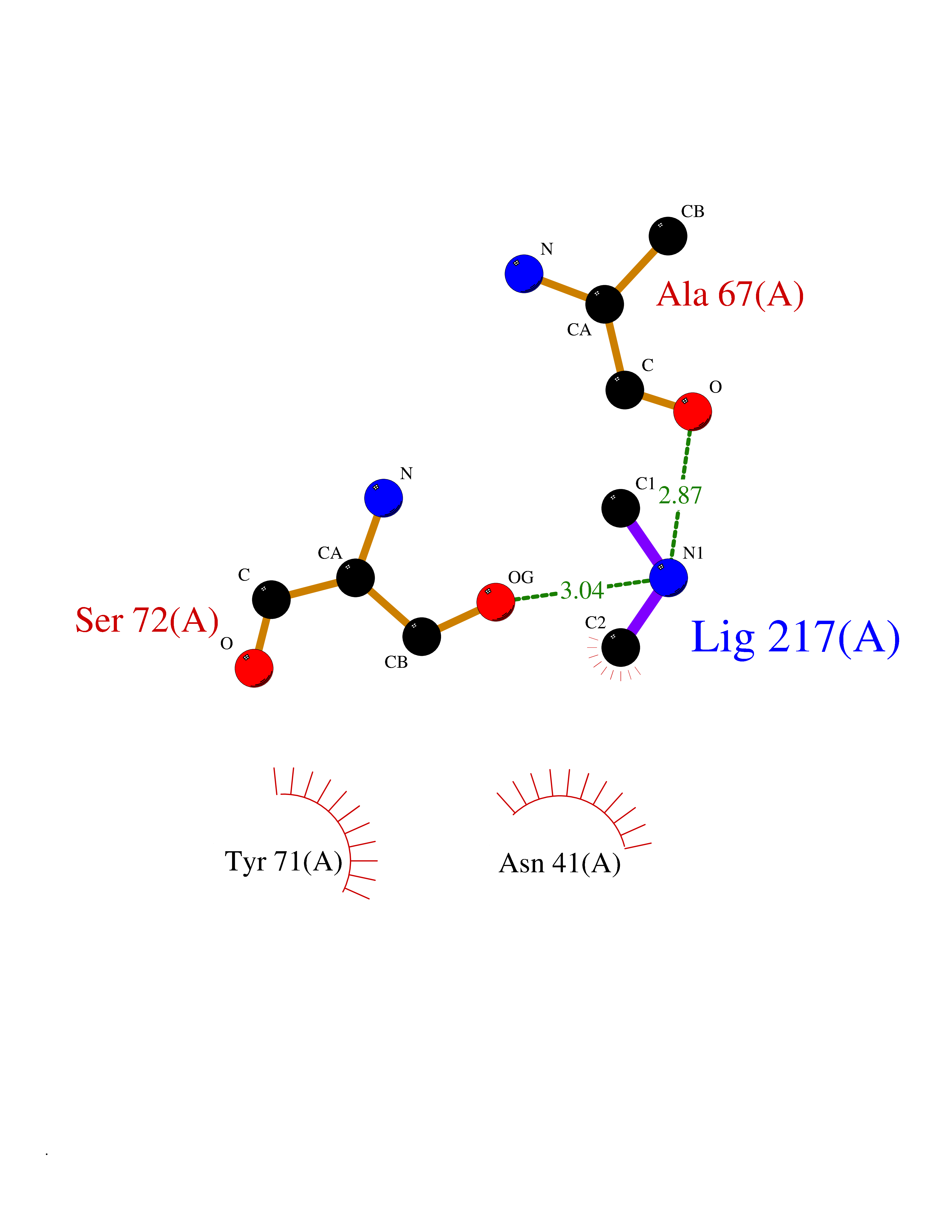 Ligplot Map 3D Binding mode Sequence GDTKEQRILNHVLQHAEPGNAQSVLEAIDTYCEQKEWAMNVGDKKGKIVDAVIQEHQPSVLLELGAYCGYSAVRMARLLSPGARLITIEINPDCAAITQRMVDFAGMKDKVTLVVGASQDIIPQLKKKYDVDTLDMVFLDHWKDRYLPDTLLLEECGLLRKGTVLLADNVICPGAPDFLAHVRGSSCFECTHYQSFLEYREVVDGLEKAIYKGP Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Aspartate carbamoyltransferase (CAD) | 4C6E | 3.99 | |
Target general information Gen name CAD Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CAD Protein family CarA family; CarB family; Metallo-dependent hydrolases superfamily, DHOase family, CAD subfamily; Aspartate/ornithine carbamoyltransferase superfamily, ATCase family Biochemical class Carbon-nitrogen ligase Function This protein is a "fusion" protein encoding four enzymatic activities of the pyrimidine pathway (GATase, CPSase, ATCase and DHOase). Related diseases Developmental and epileptic encephalopathy 50 (DEE50) [MIM:616457]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE50 is an autosomal recessive, progressive disease with onset in infancy and favorable response to treatment with oral uridine. {ECO:0000269|PubMed:25678555, ECO:0000269|PubMed:28087732}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00128; DB00130; DB03459 Interacts with P27708; Q8N137; P63104 EC number NA Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; ATP-binding; Congenital disorder of glycosylation; Cytoplasm; Direct protein sequencing; Disease variant; Epilepsy; Hydrolase; Ligase; Magnesium; Manganese; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Pyrimidine biosynthesis; Reference proteome; Repeat; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 38268.4 Length 351 Aromaticity 0.06 Instability index 41.29 Isoelectric point 5.86 Charge (pH=7) -10.56 2D Binding mode Binding energy (Kcal/mol) -5.45 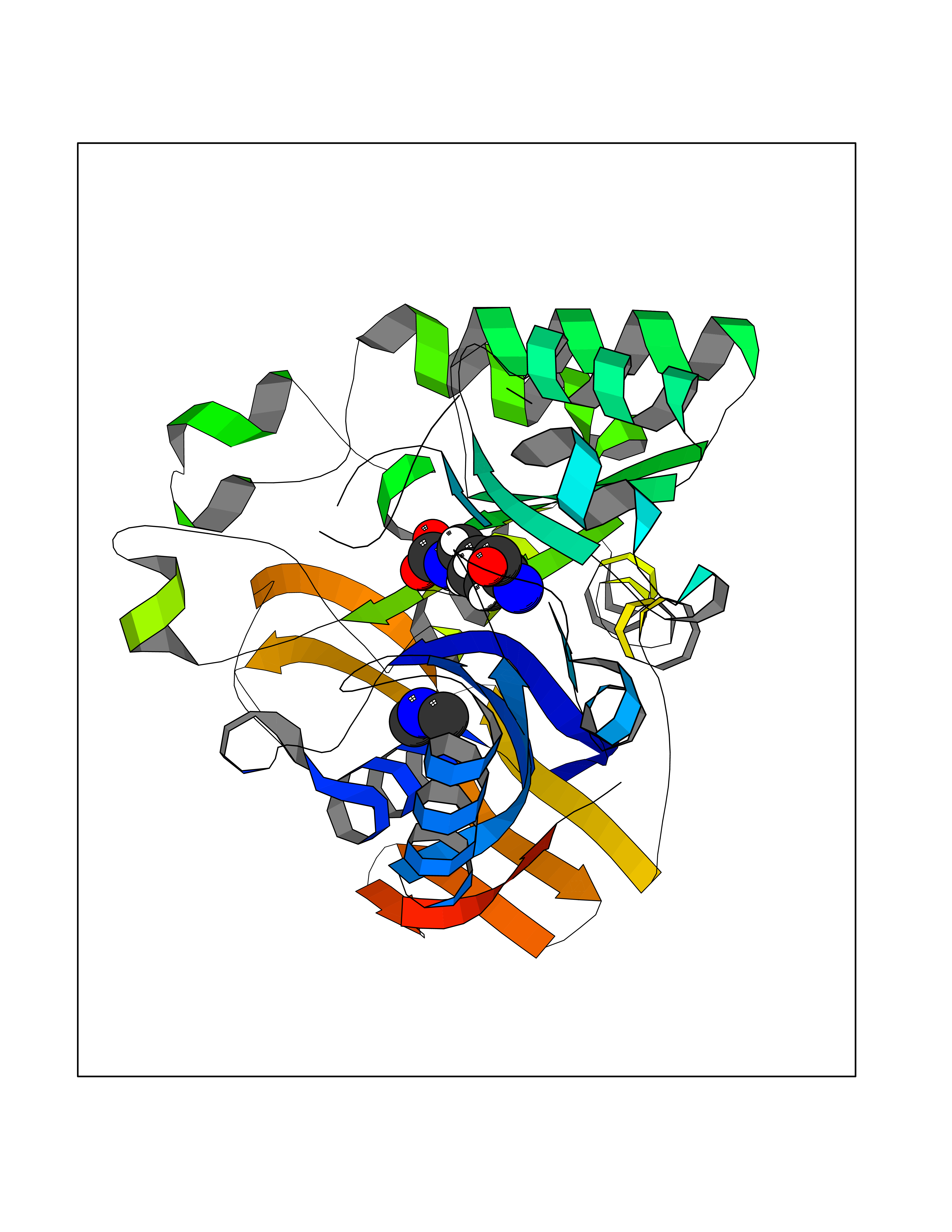 Molscript Map 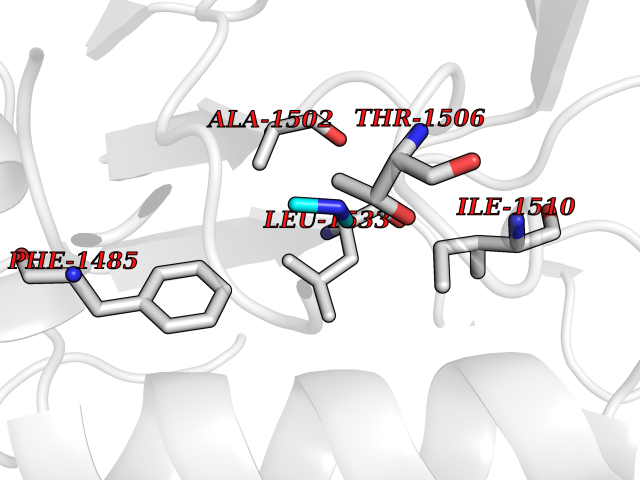 Pymol Map 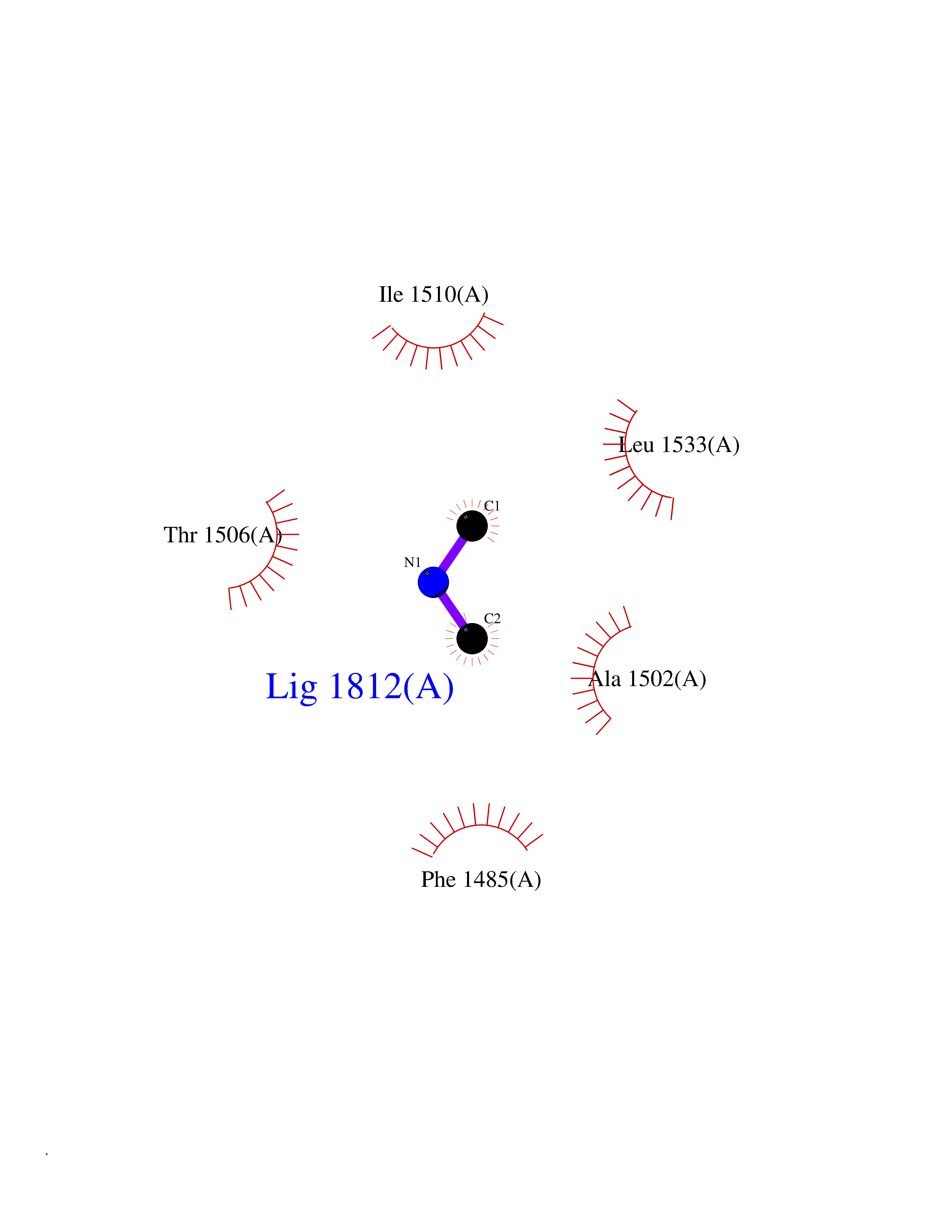 Ligplot Map 3D Binding mode Sequence KLVRLPGLIDVHVHLREPGGTHKEDFASGTAAALAGGITMVCAMPNTRPPIIDAPALALAQKLAEAGARCDFALFLGASSENAGTLGTVAGSAAGLXLYLNETFSELRLDSVVQWMEHFETWPSHLPIVAHAEQQTVAAVLMVAQLTQRSVHICHVARKEEILLIKAAKARGLPVTCEVAPHHLFLSHDDLERLGPGKGEVRPELGSRQDVEALWENMAVIDCFASDHAPHTLEEKCGSRPPPGFPGLETMLPLLLTAVSEGRLSLDDLLQRLHHNPRRIFHLPPQEDTYVEVDLEHEWTIPSHMPFSKAHWTPFEGQKVKGTVRRVVLRGEVAYIDGQVLVPPGYGQDVR Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | 4-hydroxyphenylpyruvate dioxygenase | 3ISQ | 3.99 | |
Target general information Gen name HPD Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PPD Protein family 4HPPD family Biochemical class Oxidoreductase Function 4-hydroxyphenylpyruvate dioxygenase activity.Metal ion binding. Related diseases Tyrosinemia 3 (TYRSN3) [MIM:276710]: An inborn error of metabolism characterized by elevations of tyrosine in the blood and urine, seizures and mild intellectual disability. {ECO:0000269|PubMed:10942115, ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hawkinsinuria (HWKS) [MIM:140350]: An inborn error of tyrosine metabolism characterized by failure to thrive, persistent metabolic acidosis, fine and sparse hair, and excretion of the unusual cyclic amino acid metabolite, hawkinsin, in the urine. {ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02850; DB00348 Interacts with NA EC number 1.13.11.27 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Dioxygenase; Disease variant; Endoplasmic reticulum; Golgi apparatus; Intellectual disability; Iron; Membrane; Metal-binding; Oxidoreductase; Phenylalanine catabolism; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Tyrosine catabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 43164.8 Length 376 Aromaticity 0.11 Instability index 32.38 Isoelectric point 6.73 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map 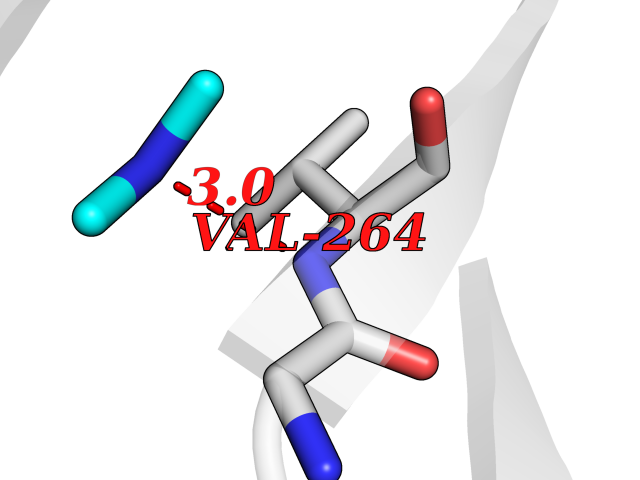 Pymol Map 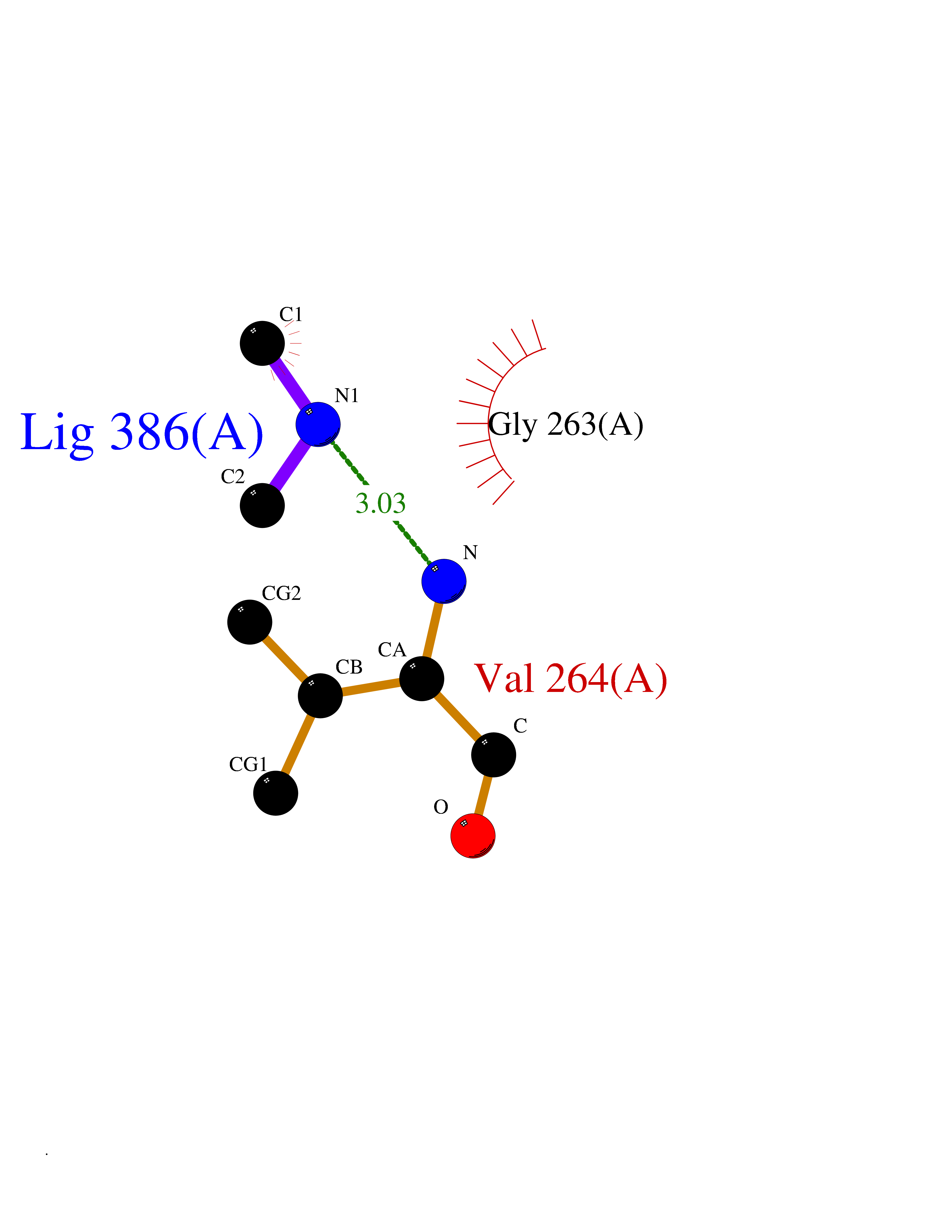 Ligplot Map 3D Binding mode Sequence AKPERGRFLHFHSVTFWVGNAKQAASFYCSKMGFEPLAYRGLETGSREVVSHVIKQGKIVFVLSSALNPWNKEMGDHLVKHGDGVKDIAFEVEDCDYIVQKARERGAKIMREPWVEQDKFGKVKFAVLQTYGDTTHTLVEKMNYIGQFLPGYEAPAFMDPLLPKLPKCSLEMIDHIVGNQPDQEMVSASEWYLKNLQFHRFWSVDDTQVHTEYSSLRSIVVANYEESIKMPINEPAPGKKKSQIQEYVDYNGGAGVQHIALKTEDIITAIRHLRERGLEFLSVPSTYYKQLREKLKTAKIKVKENIDALEELKILVDYDEKGYLLQIFTKPVQDRPTLFLEVIQRHNHQGFGAGNFNSLFKAFEEEQNLRGNLTNM Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Cyclin-dependent kinase 6 (CDK6) | 5L2S | 3.99 | |
Target general information Gen name CDK6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine/threonine-protein kinase PLSTIRE; Serine/threonine protein kinase PLSTIRE; Cell division protein kinase 6; CDKN6 Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, CDC2/CDKX subfamily Biochemical class Kinase Function Phosphorylates pRB/RB1 and NPM1. Interacts with D-type G1 cyclins during interphase at G1 to form a pRB/RB1 kinase and controls the entrance into the cell cycle. Involved in initiation and maintenance of cell cycle exit during cell differentiation; prevents cell proliferation and regulates negatively cell differentiation, but is required for the proliferation of specific cell types (e. g. erythroid and hematopoietic cells). Essential for cell proliferation within the dentate gyrus of the hippocampus and the subventricular zone of the lateral ventricles. Required during thymocyte development. Promotes the production of newborn neurons, probably by modulating G1 length. Promotes, at least in astrocytes, changes in patterns of gene expression, changes in the actin cytoskeleton including loss of stress fibers, and enhanced motility during cell differentiation. Prevents myeloid differentiation by interfering with RUNX1 and reducing its transcription transactivation activity, but promotes proliferation of normal myeloid progenitors. Delays senescence. Promotes the proliferation of beta-cells in pancreatic islets of Langerhans. May play a role in the centrosome organization during the cell cycle phases. Serine/threonine-protein kinase involved in the control of the cell cycle and differentiation; promotes G1/S transition. Related diseases Microcephaly 12, primary, autosomal recessive (MCPH12) [MIM:616080]: A form of microcephaly, a disease defined as a head circumference more than 3 standard deviations below the age-related mean. Brain weight is markedly reduced and the cerebral cortex is disproportionately small. {ECO:0000269|PubMed:23918663}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07379; DB12001; DB03496; DB07795; DB09073; DB11730; DB15442 Interacts with P41238; Q8N5C1; P24385; P30281; P51946; Q14094; Q16543; P38936; P42771; P42772; P42773; P55273; Q08050-1; P08238; Q5XKR4; Q01196; Q9C019 EC number EC 2.7.11.22 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Cell cycle; Cell division; Cell projection; Cytoplasm; Cytoskeleton; Differentiation; Disease variant; Kinase; Nucleotide-binding; Nucleus; Phosphoprotein; Primary microcephaly; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 29850.2 Length 263 Aromaticity 0.1 Instability index 34.55 Isoelectric point 7.23 Charge (pH=7) 0.46 2D Binding mode Binding energy (Kcal/mol) -5.45 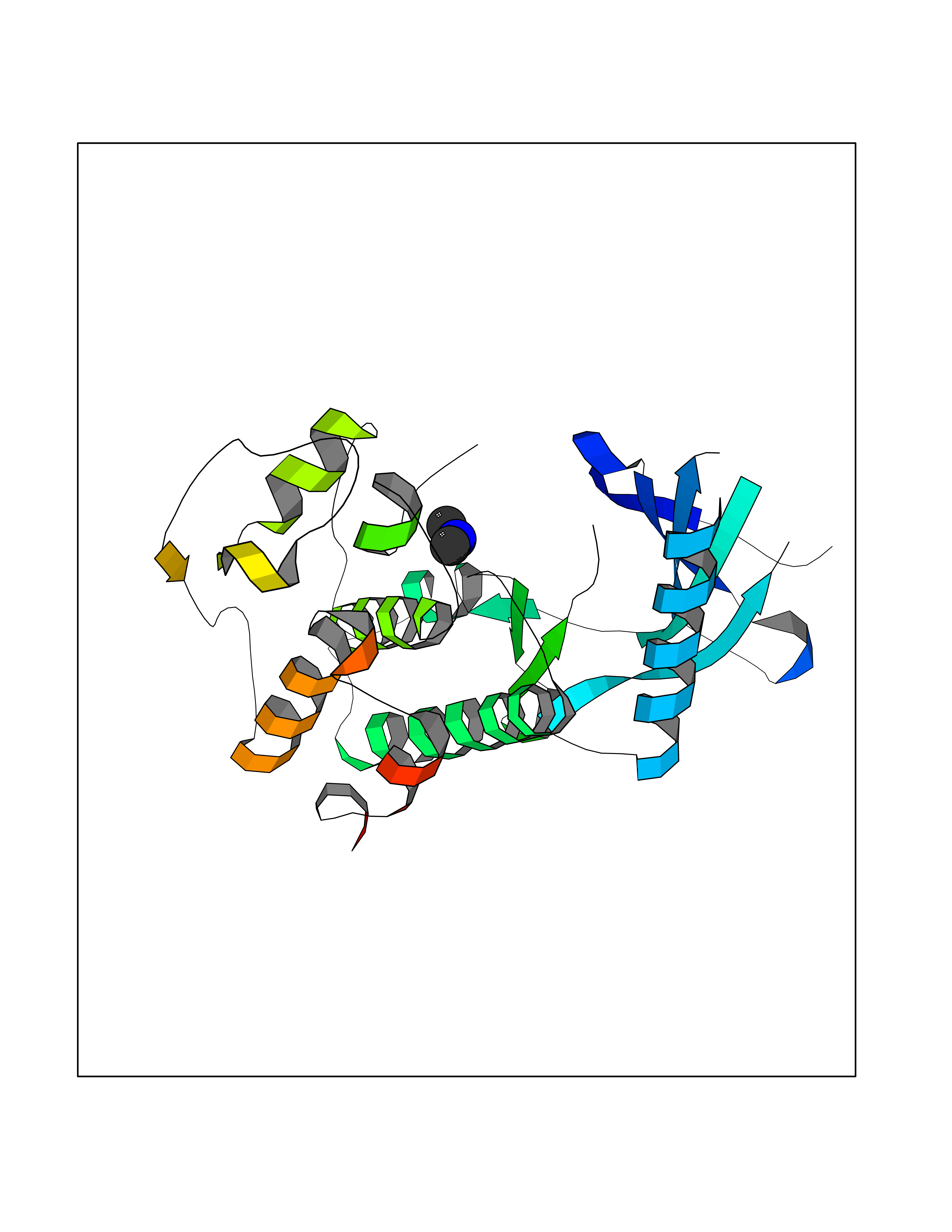 Molscript Map 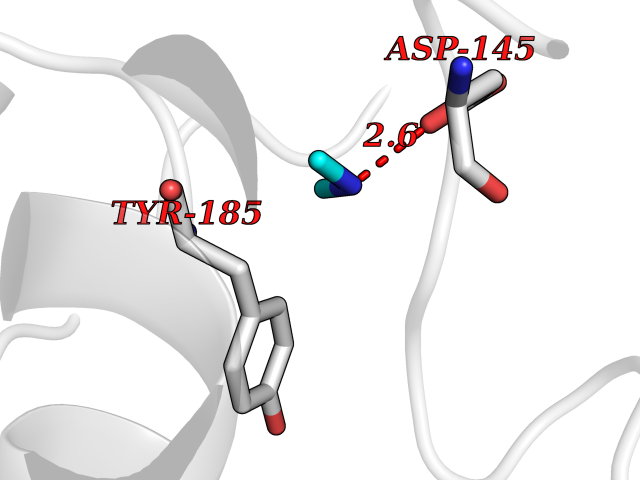 Pymol Map 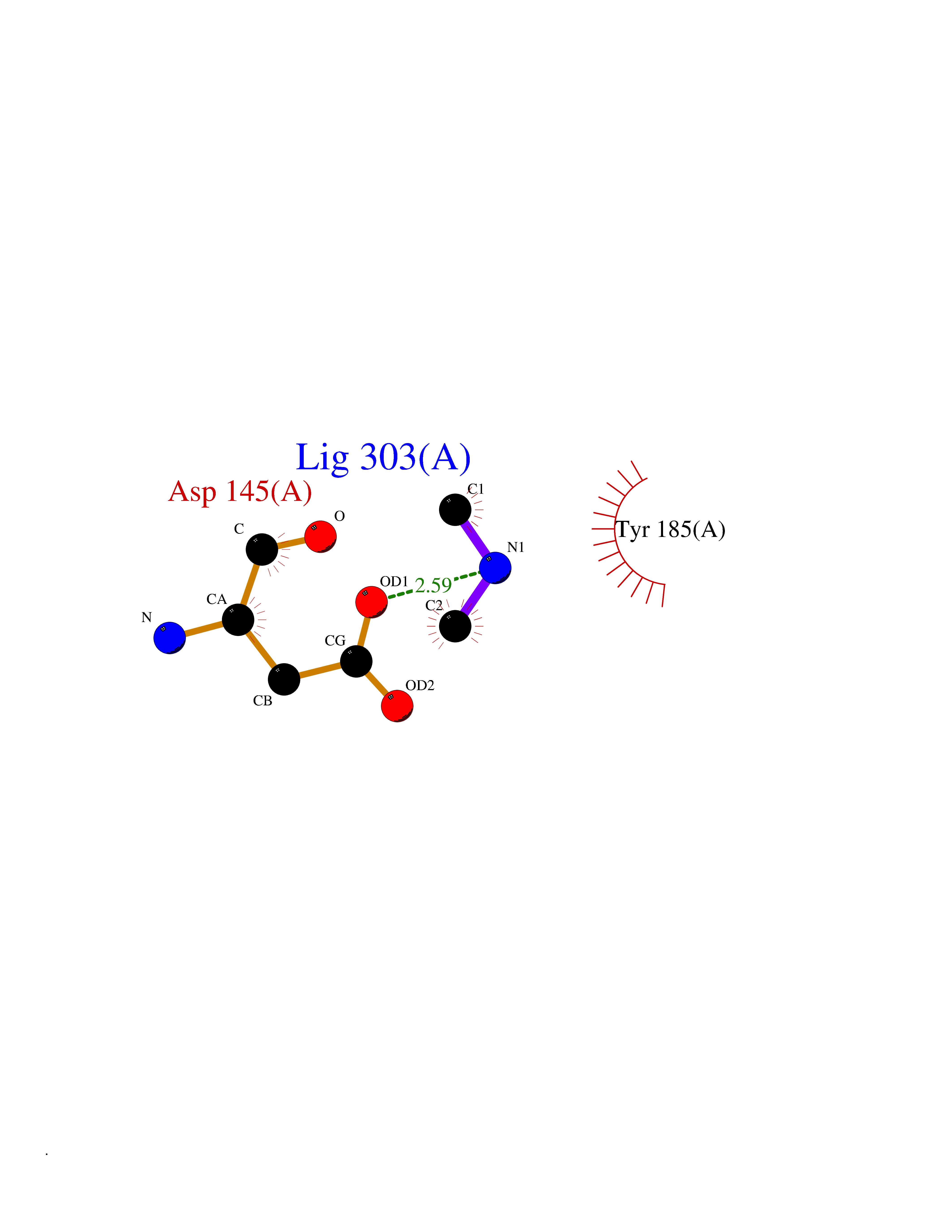 Ligplot Map 3D Binding mode Sequence QQYECVAEIGEGAYGKVFKARDLKNGGRFVALKRVRVPLSTIREVAVLRHLETFEHPNVVRLFDVCTKLTLVFEHVDQDLTTYLDKVPEPGVPTETIKDMMFQLLRGLDFLHSHRVVHRDLKPQNILVTSSGQIKLADFGLAVTLWYRAPEVLLQSSYATPVDLWSVGCIFAEMFRRKPLFRGSSDVDQLGKILDVIGLPGEEDWPRDVALPRQAFHSKSAQPIEKFVTDIDELGKDLLLKCLTFNPAKRISAYSALSHPYFQ Hydrogen bonds contact Hydrophobic contact | ||||