Job Results:
Ligand
Structure
Job ID
500380c02188392c5ec501006b89ab5f
Job name
NA
Time
2025-02-18 14:12:25
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Cyclin-dependent kinase 6 (CDK6) | 5L2S | 4.00 | |
Target general information Gen name CDK6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine/threonine-protein kinase PLSTIRE; Serine/threonine protein kinase PLSTIRE; Cell division protein kinase 6; CDKN6 Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, CDC2/CDKX subfamily Biochemical class Kinase Function Phosphorylates pRB/RB1 and NPM1. Interacts with D-type G1 cyclins during interphase at G1 to form a pRB/RB1 kinase and controls the entrance into the cell cycle. Involved in initiation and maintenance of cell cycle exit during cell differentiation; prevents cell proliferation and regulates negatively cell differentiation, but is required for the proliferation of specific cell types (e. g. erythroid and hematopoietic cells). Essential for cell proliferation within the dentate gyrus of the hippocampus and the subventricular zone of the lateral ventricles. Required during thymocyte development. Promotes the production of newborn neurons, probably by modulating G1 length. Promotes, at least in astrocytes, changes in patterns of gene expression, changes in the actin cytoskeleton including loss of stress fibers, and enhanced motility during cell differentiation. Prevents myeloid differentiation by interfering with RUNX1 and reducing its transcription transactivation activity, but promotes proliferation of normal myeloid progenitors. Delays senescence. Promotes the proliferation of beta-cells in pancreatic islets of Langerhans. May play a role in the centrosome organization during the cell cycle phases. Serine/threonine-protein kinase involved in the control of the cell cycle and differentiation; promotes G1/S transition. Related diseases Microcephaly 12, primary, autosomal recessive (MCPH12) [MIM:616080]: A form of microcephaly, a disease defined as a head circumference more than 3 standard deviations below the age-related mean. Brain weight is markedly reduced and the cerebral cortex is disproportionately small. {ECO:0000269|PubMed:23918663}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07379; DB12001; DB03496; DB07795; DB09073; DB11730; DB15442 Interacts with P41238; Q8N5C1; P24385; P30281; P51946; Q14094; Q16543; P38936; P42771; P42772; P42773; P55273; Q08050-1; P08238; Q5XKR4; Q01196; Q9C019 EC number EC 2.7.11.22 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Cell cycle; Cell division; Cell projection; Cytoplasm; Cytoskeleton; Differentiation; Disease variant; Kinase; Nucleotide-binding; Nucleus; Phosphoprotein; Primary microcephaly; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 29850.2 Length 263 Aromaticity 0.1 Instability index 34.55 Isoelectric point 7.23 Charge (pH=7) 0.46 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence QQYECVAEIGEGAYGKVFKARDLKNGGRFVALKRVRVPLSTIREVAVLRHLETFEHPNVVRLFDVCTKLTLVFEHVDQDLTTYLDKVPEPGVPTETIKDMMFQLLRGLDFLHSHRVVHRDLKPQNILVTSSGQIKLADFGLAVTLWYRAPEVLLQSSYATPVDLWSVGCIFAEMFRRKPLFRGSSDVDQLGKILDVIGLPGEEDWPRDVALPRQAFHSKSAQPIEKFVTDIDELGKDLLLKCLTFNPAKRISAYSALSHPYFQ Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Alanine aminotransferase 2 | 3IHJ | 4.00 | |
Target general information Gen name GPT2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms AAT2;ALT2 Protein family Class-I pyridoxal-phosphate-dependent aminotransferase family, Alanine aminotransferase subfamily Biochemical class Transferase Function L-alanine:2-oxoglutarate aminotransferase activity.Pyridoxal phosphate binding. Related diseases Neurodevelopmental disorder with spastic paraplegia and microcephaly (NEDSPM) [MIM:616281]: An autosomal recessive syndrome characterized by severe psychomotor developmental delay, dysarthria, walking difficulties, moderately to severely impaired intellectual development, poor or absent speech, and progressive microcephaly. {ECO:0000269|PubMed:25758935}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00160; DB00142; DB00780; DB00114 Interacts with NA EC number 2.6.1.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminotransferase; Disease variant; Intellectual disability; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 48717.7 Length 436 Aromaticity 0.09 Instability index 47.17 Isoelectric point 6.07 Charge (pH=7) -4.32 2D Binding mode Binding energy (Kcal/mol) -5.45 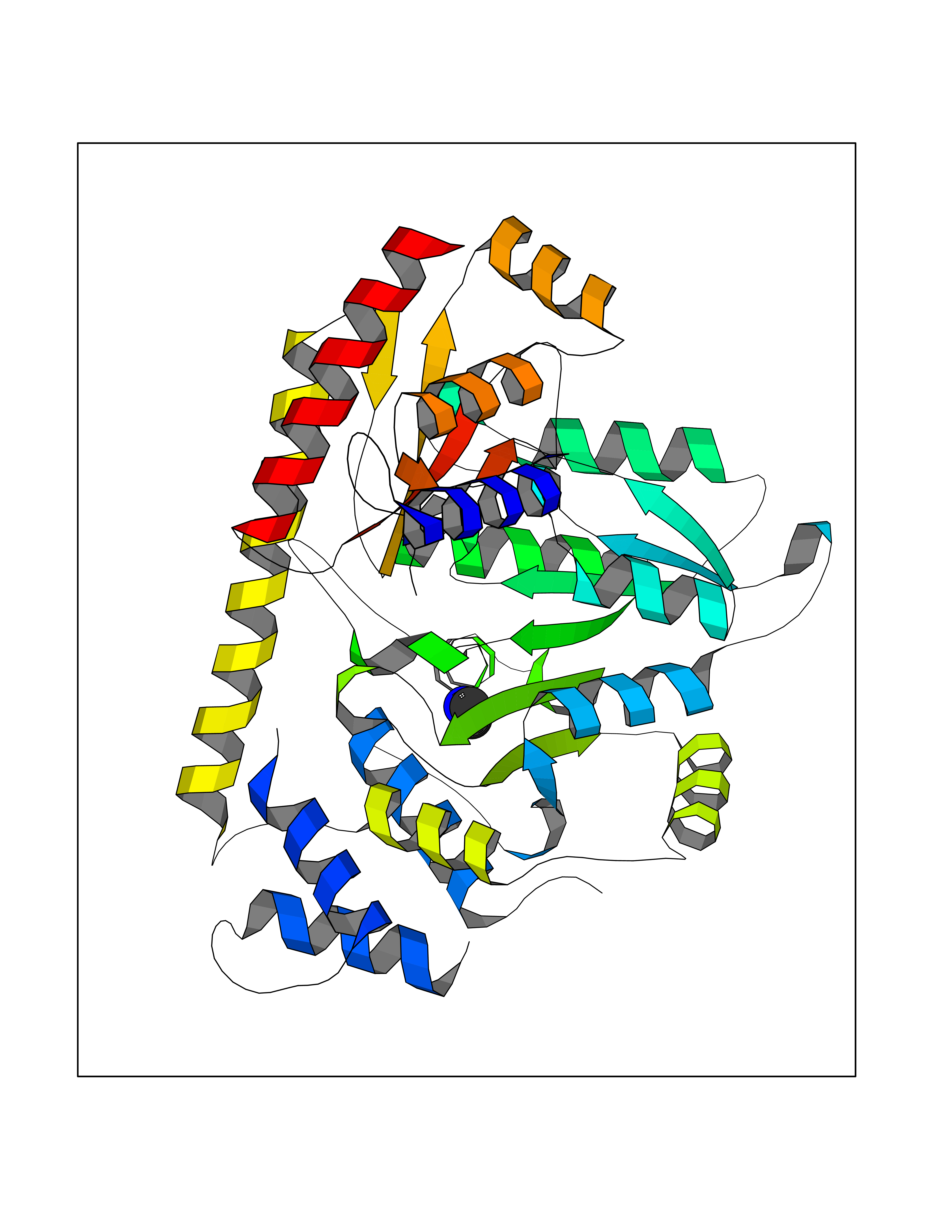 Molscript Map 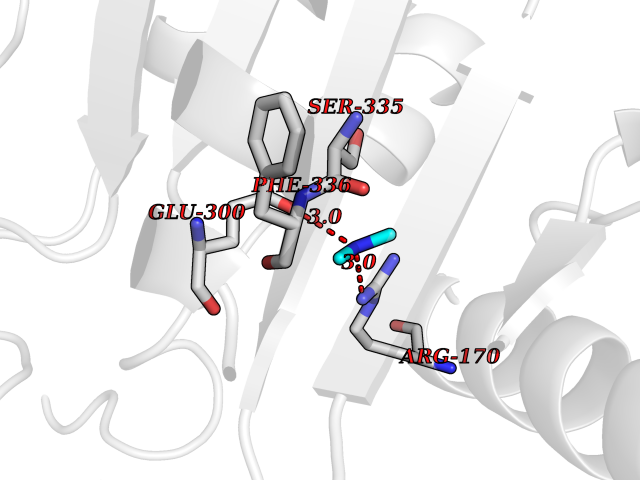 Pymol Map 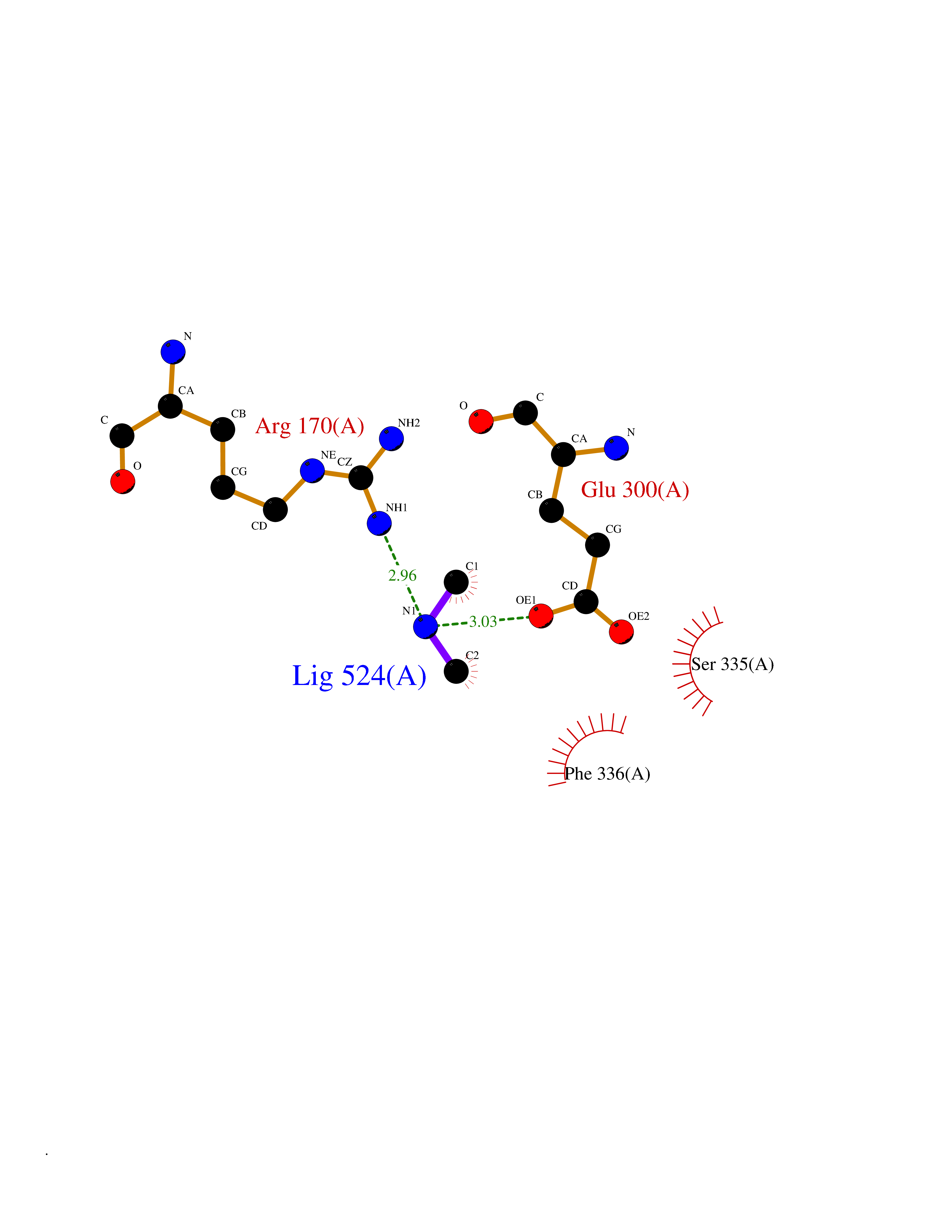 Ligplot Map 3D Binding mode Sequence PIVLKAGEIELELQRGIKKPFTEVIRANPITFLRQVMALCTYPNLLDSPSFPEDAKKRARRILQACSQGVNCIREDVAAYITRRDGGVPADPDNIYLTTGASDGISTILKILVSGGGKSRTGVMIPIPQYPLYSAVISELDAIQVNYYLDEENCWALNVNELRRAVQEAKDHCDPKVLCIINPGNPTGQVQSRKCIEDVIHFAWEEKLFLLADEVYQDNVYSPDCRFHSFKKVLYEMGPEYSSNVELASFHSTSKGYMGECGYRGGYMEVINLHPEIKGQLVKLLSVRLCPPVSGQAAMDIVVNPPVAGEESFEQFSREKESVLGNLAKKAKLTEDLFNQVPGIHCNPLQGAMYAFPRIFIPAKAVEAAQAHQMAPDMFYCMKLLEETGICVVPGSGFGQREGTYHFRMTILPPVEKLKTVLQKVKDFHINFLEKY Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Phospholipase D2 (PLD2) | 6OHP | 4.00 | |
Target general information Gen name PLD2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hPLD2; Phosphatidylcholine-hydrolyzing phospholipase D2; PLD1C; PLD 2; Choline phosphatase 2 Protein family Phospholipase D family Biochemical class Phosphoric diester hydrolase Function May have a role in signal-induced cytoskeletal regulation and/or endocytosis. Related diseases Intellectual developmental disorder, autosomal recessive 80, with variant lissencephaly (MRT80) [MIM:620653]: An autosomal recessive disorder characterized by global developmental delay, mildly to moderately impaired intellectual development, attention deficit-hyperactivity disorder, hypotonia, seizure, poor social skills, and autistic traits. Brain imaging shows fronto-temporal lissencephaly and pachygyria. {ECO:0000269|PubMed:37880421}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00122; DB14006 Interacts with P05067; P23528; P62993; P15153; P13051-2 EC number EC 3.1.4.4 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Hydrolase; Lipid degradation; Lipid metabolism; Lipoprotein; Membrane; Phosphoprotein; Proteomics identification; Reference proteome; Repeat Protein physicochemical properties Chain ID A Molecular weight (Da) 66788.7 Length 586 Aromaticity 0.1 Instability index 41.26 Isoelectric point 6.54 Charge (pH=7) -3.31 2D Binding mode Binding energy (Kcal/mol) -5.46 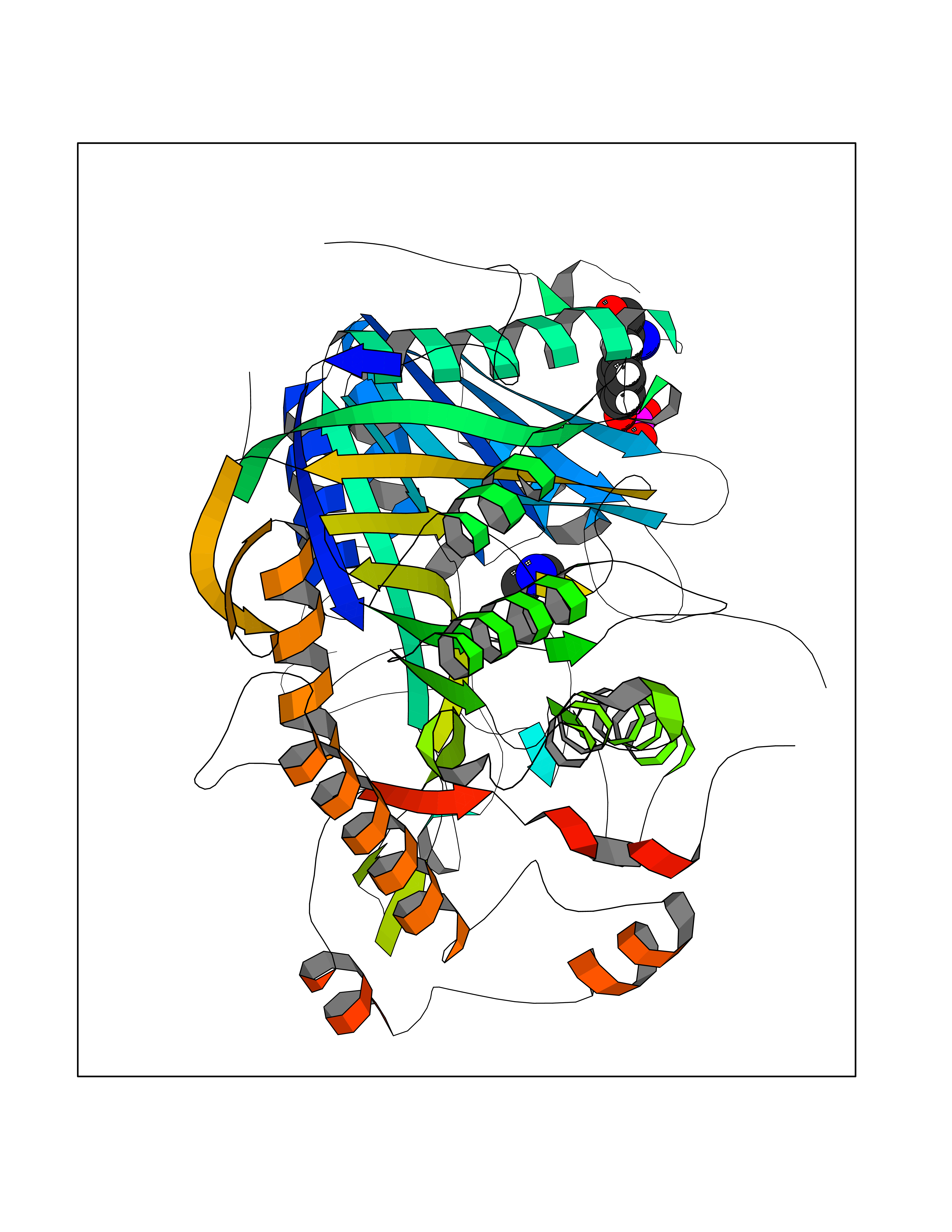 Molscript Map 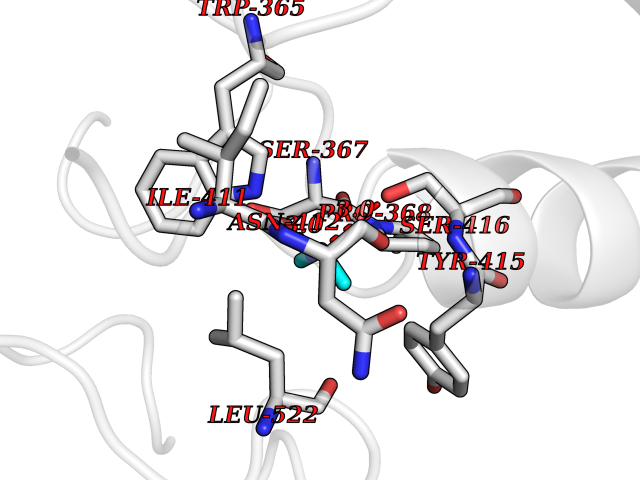 Pymol Map 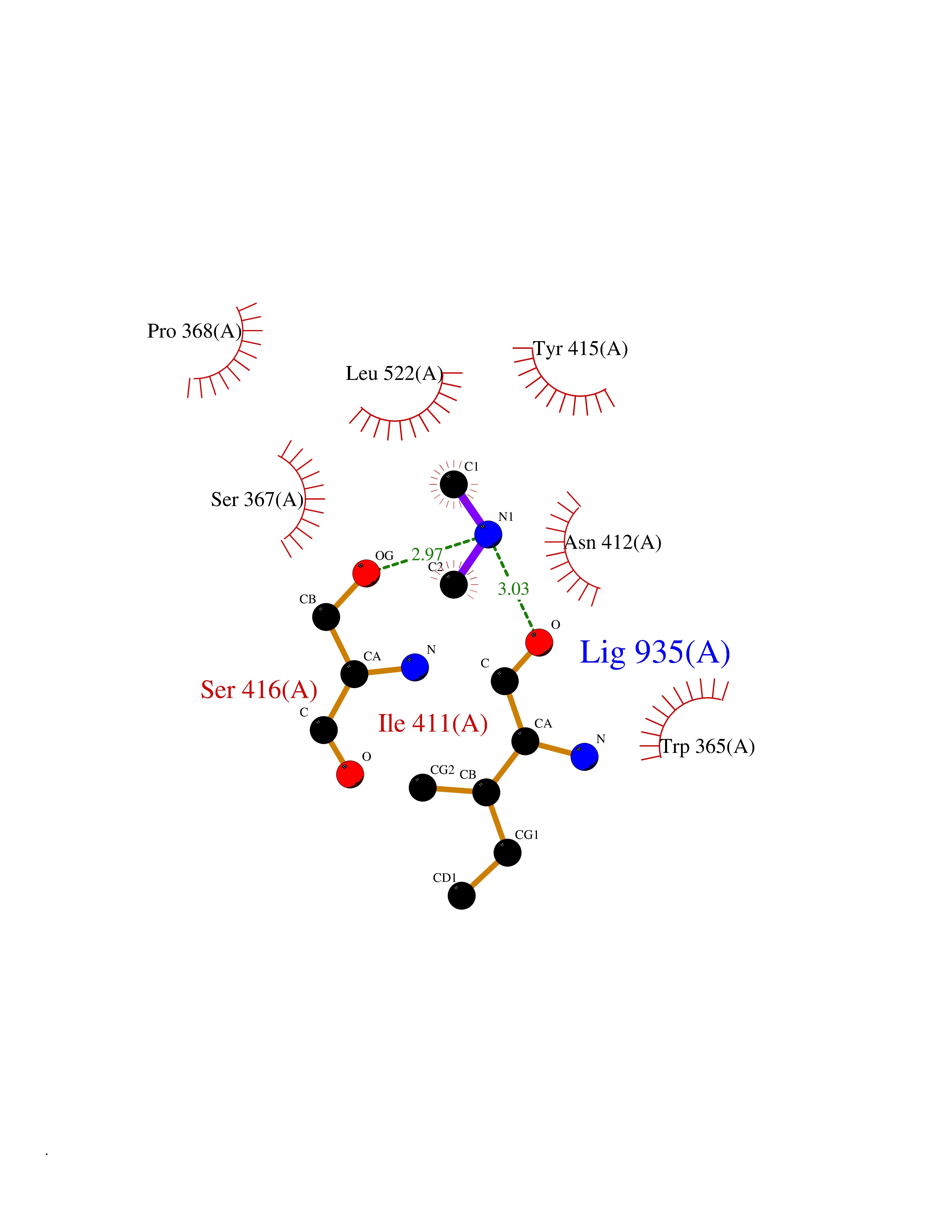 Ligplot Map 3D Binding mode Sequence FLQLHRHDSYAPPRPGTLARWFVNGAGYFAAVADAILRAQEEIFITDWWLSPEVYLKRPAHSDDWRLDIMLKRKAEEGVRVSILLFKEVELALGINSGYSKRALMLLHPNIKVMRHPDQVTLWAHHEKLLVVDQVVAFLGGLDLAYGRWDDLHYRLTDLGDLSHNQFFWLGKDYSNLITKDWVQLDRPFEDFIDRETTPRMPWRDVGVVVHGLPARDLARHFIQRWNFTKTTKAKXKTPTYPYLLPKSTSTFTLPGGQCTTVQVLRSVDRWSAGTLENSILNAYLHTIRESQHFLYIENQFFISCSDGRTVLNKVGDEIVDRILKAHKQGWCYRVYVLLPLLPGFEGDISTGGGNSIQAILHFTYRTLCRGEYSILHRLKAAMGTAWRDYISICGLRTHGELGGHPVSELIYIHSKVLIADDRTVIIGSANINDRSLLGKRDSELAVLIEDTETEPSLMNGAEYQAGRFALSLRKHCFGVILGANTRPDLDLRDPICDDFFQLWQDMAESNANIYEQIFRCLPSNATRSLRTLREYVAVEPLATVSPPLARSELTQVQGHLVHFPLKFLEDESLLPPGMIPLEVWT Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Thymidine kinase 1 (TK1) | 1W4R | 4.00 | |
Target general information Gen name TK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Thymidine kinase, cytosolic Protein family Thymidine kinase family Biochemical class Kinase Function cytosol, identical protein binding, thymidine kinase activity, zinc ion binding, DNA metabolic process, nucleobase-containing compound metabolic process, protein homotetramerization, pyrimidine nucleoside salvage, thymidine metabolic process Related diseases Seizures, benign familial infantile, 3 (BFIS3) [MIM:607745]: A form of benign familial infantile epilepsy, a neurologic disorder characterized by afebrile seizures occurring in clusters during the first year of life, without neurologic sequelae. BFIS3 inheritance is autosomal dominant. {ECO:0000269|PubMed:11371648, ECO:0000269|PubMed:12243921, ECO:0000269|PubMed:15048894, ECO:0000269|PubMed:16417554, ECO:0000269|PubMed:17021166, ECO:0000269|PubMed:17386050, ECO:0000269|PubMed:18479388, ECO:0000269|PubMed:20371507, ECO:0000269|PubMed:22612257, ECO:0000269|PubMed:23360469, ECO:0000269|PubMed:23758435, ECO:0000269|PubMed:25982755, ECO:0000269|PubMed:26291284, ECO:0000269|PubMed:29844171, ECO:0000269|PubMed:30144217}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 11 (DEE11) [MIM:613721]: An autosomal dominant seizure disorder characterized by neonatal or infantile onset of refractory seizures with resultant delayed neurologic development and persistent neurologic abnormalities. Patients may progress to West syndrome, which is characterized by tonic spasms with clustering, arrest of psychomotor development, and hypsarrhythmia on EEG. {ECO:0000269|PubMed:19783390, ECO:0000269|PubMed:19786696, ECO:0000269|PubMed:20956790, ECO:0000269|PubMed:22677033, ECO:0000269|PubMed:23033978, ECO:0000269|PubMed:23195492, ECO:0000269|PubMed:23550958, ECO:0000269|PubMed:23662938, ECO:0000269|PubMed:23708187, ECO:0000269|PubMed:23935176, ECO:0000269|PubMed:23988467, ECO:0000269|PubMed:24463883, ECO:0000269|PubMed:24579881, ECO:0000269|PubMed:24659627, ECO:0000269|PubMed:24710820, ECO:0000269|PubMed:25457084, ECO:0000269|PubMed:25459969, ECO:0000269|PubMed:25772804, ECO:0000269|PubMed:25818041, ECO:0000269|PubMed:26138355, ECO:0000269|PubMed:26291284, ECO:0000269|PubMed:26993267, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:29625812, ECO:0000269|PubMed:29844171, ECO:0000269|PubMed:30144217, ECO:0000269|PubMed:30415926}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in SCN2A are associated with genetic epilepsy with febrile seizures plus (GEFS+), a familial autosomal dominant epilepsy syndrome, a clinical subset of febrile seizures, characterized by frequent episodes after 6 years of age and various types of subsequent epilepsy. {ECO:0000269|PubMed:29635106}.; DISEASE: Defects in SCN2A are associated with autism spectrum disorders (ASD). It seems that mutations resulting in sodium channel gain of function and increased neuron excitability lead to infantile seizures, whereas variants resulting in sodium channel loss of function and decrease neuron excitability are associated with ASD. {ECO:0000269|PubMed:28256214}.; DISEASE: Episodic ataxia 9 (EA9) [MIM:618924]: An autosomal dominant neurologic disorder characterized by episodic ataxia manifesting in the first years of life, early-onset seizures, difficulty walking, dizziness, slurred speech, headache, vomiting, and pain. The duration of ataxic episodes is heterogeneous. Most patients show episodes lasting minutes to maximum several hours, but periods lasting days up to weeks have been reported. Some patients have mildly delayed development with speech delay and/or autistic features or mildly impaired intellectual development. {ECO:0000269|PubMed:26645390, ECO:0000269|PubMed:27159988, ECO:0000269|PubMed:27328862, ECO:0000269|PubMed:28065826}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01692; DB04485; DB02452; DB00432; DB00495 Interacts with P05067; A0A087WZT3; Q92993; Q1RN33; P04183 EC number EC 2.7.1.21 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Cytoplasm; DNA synthesis; Kinase; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 19373.5 Length 174 Aromaticity 0.09 Instability index 36.21 Isoelectric point 8.63 Charge (pH=7) 3.88 2D Binding mode Binding energy (Kcal/mol) -5.46 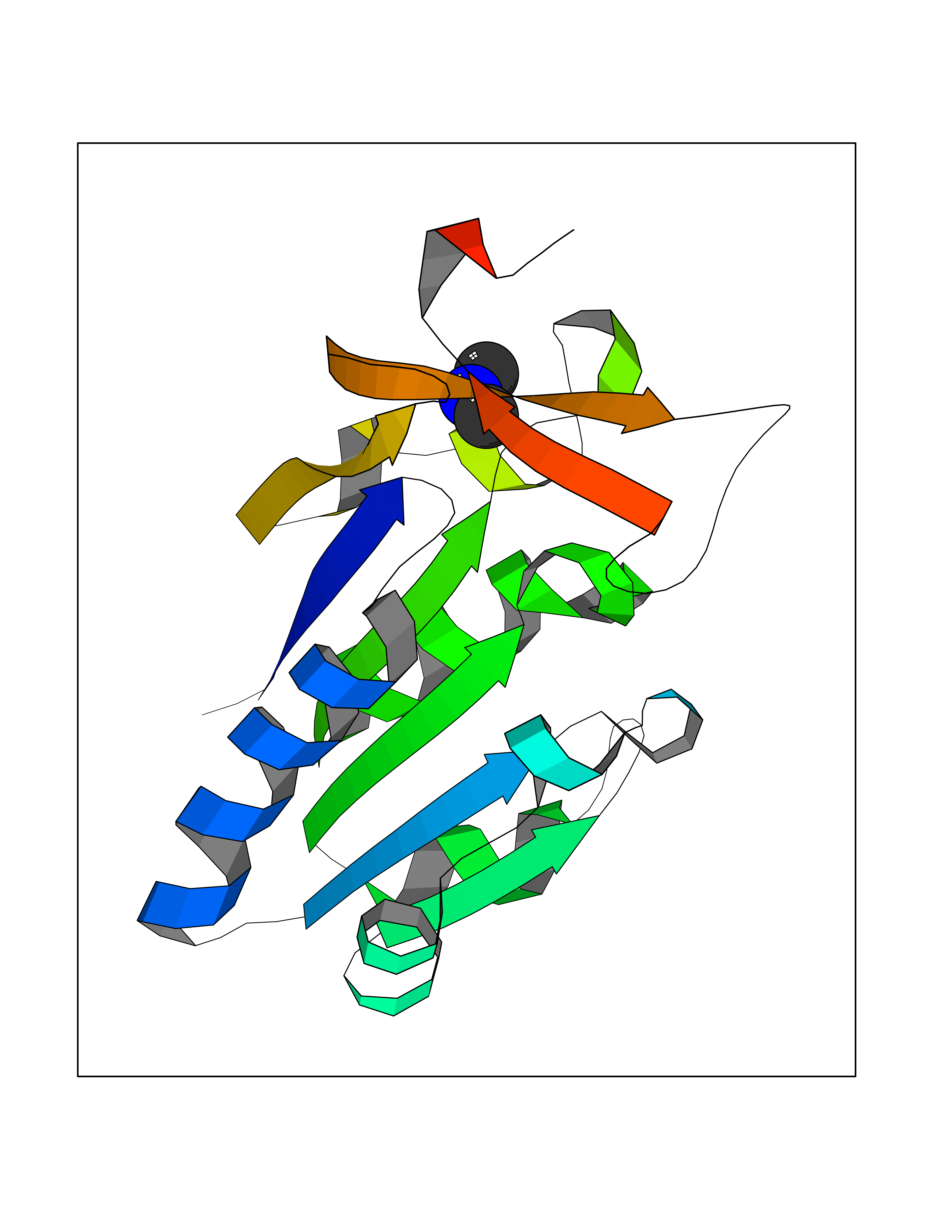 Molscript Map 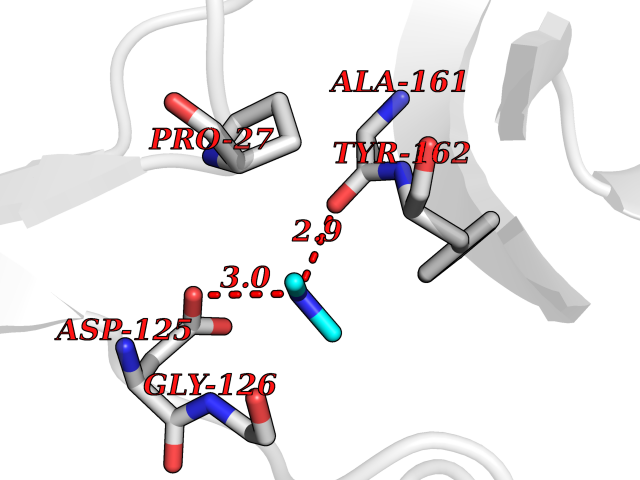 Pymol Map 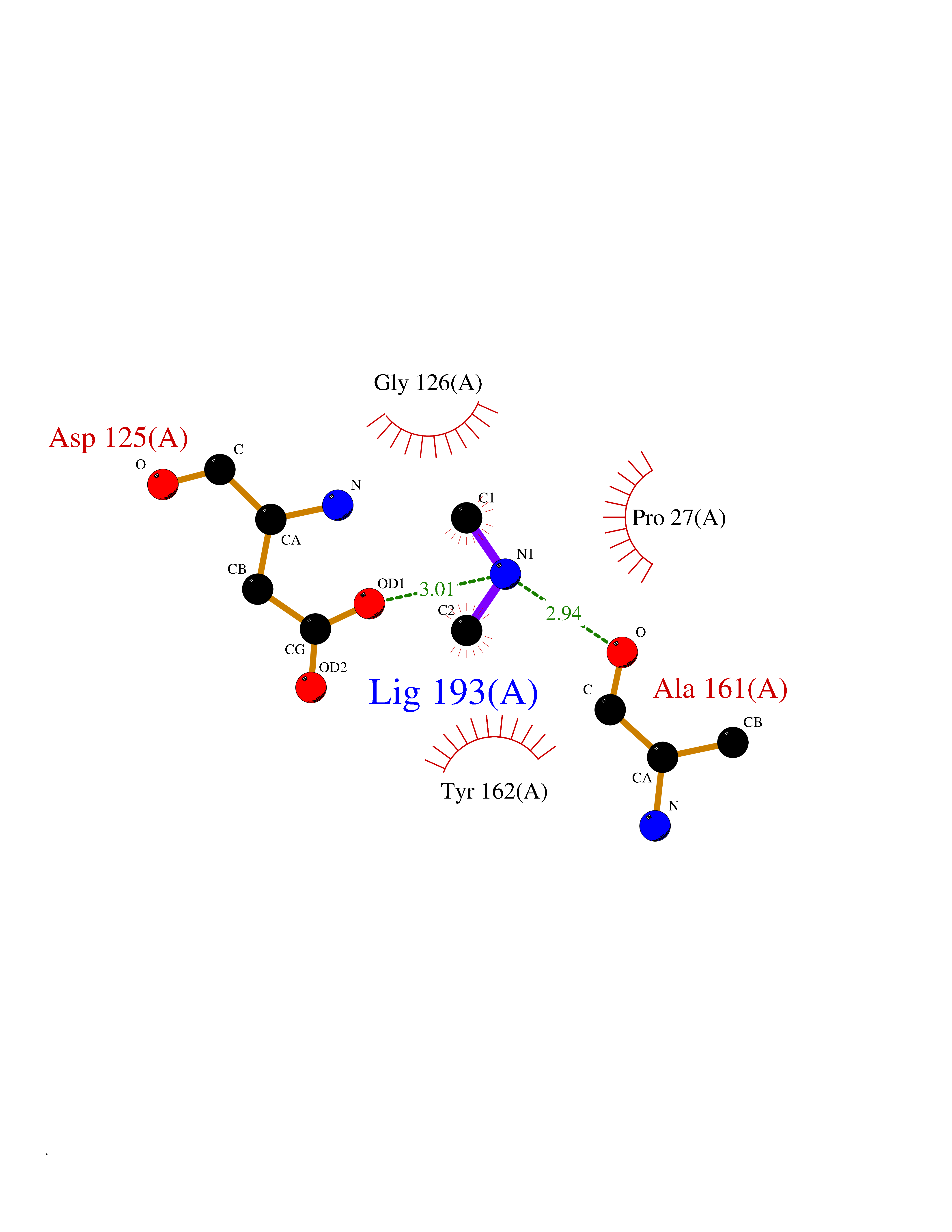 Ligplot Map 3D Binding mode Sequence RGQIQVILGPMFSGKSTELMRRVRRFQIAQYKCLVIKYAKDTRYSSSFCTHDRNTMEALPACLLRDVAQEALGVAVIGIDEGQFFPDIVEFCEAMANAGKTVIVAALDGTFQRKPFGAILNLVPLAESVVKLTAVCMECFREAAYTKRLGTEKEVEVIGGADKYHSVCRLCYFK Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Scavenger decapping enzyme DcpS (DCPS) | 1ST4 | 4.00 | |
Target general information Gen name DCPS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Scavenger mRNA-decapping enzyme DcpS; Histidine triad protein member5; Hint-related 7meGMP-directed hydrolase; HINT-5; DCS-1; DCPS Protein family HIT family Biochemical class Acid anhydrides hydrolase Function Decapping scavenger enzyme that catalyzes the cleavage of a residual cap structure following the degradation of mRNAs by the 3'->5' exosome-mediated mRNA decay pathway. Hydrolyzes cap analog structures like 7-methylguanosine nucleoside triphosphate (m7GpppG) with up to 10 nucleotide substrates (small capped oligoribonucleotides) and specifically releases 5'-phosphorylated RNA fragments and 7-methylguanosine monophosphate (m7GMP). Cleaves cap analog structures like tri-methyl guanosine nucleoside triphosphate (m3(2,2,7)GpppG) with very poor efficiency. Does not hydrolyze unmethylated cap analog (GpppG) and shows no decapping activity on intact m7GpppG-capped mRNA molecules longer than 25 nucleotides. Does not hydrolyze 7-methylguanosine diphosphate (m7GDP) to m7GMP (PubMed:22985415). May also play a role in the 5'->3 mRNA decay pathway; m7GDP, the downstream product released by the 5'->3' mRNA mediated decapping activity, may be also converted by DCPS to m7GMP (PubMed:14523240). Binds to m7GpppG and strongly to m7GDP. Plays a role in first intron splicing of pre- mRNAs. Inhibits activation-induced cell death. Related diseases Al-Raqad syndrome (ARS) [MIM:616459]: A syndrome characterized by delayed psychomotor development, moderate to severe intellectual disability, poor or absent speech, microcephaly, congenital hypotonia, and severe growth delay. {ECO:0000269|PubMed:25701870, ECO:0000269|PubMed:25712129}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07644; DB07643; DB07642; DB03593; DB01960; DB01649; DB03958 Interacts with Q96C86; P52292; O15131; O60684 EC number EC 3.6.1.59 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Disease variant; Hydrolase; Intellectual disability; mRNA processing; mRNA splicing; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID B,A Molecular weight (Da) 69192.9 Length 597 Aromaticity 0.09 Instability index 54.62 Isoelectric point 6.12 Charge (pH=7) -9.94 2D Binding mode Binding energy (Kcal/mol) -5.45 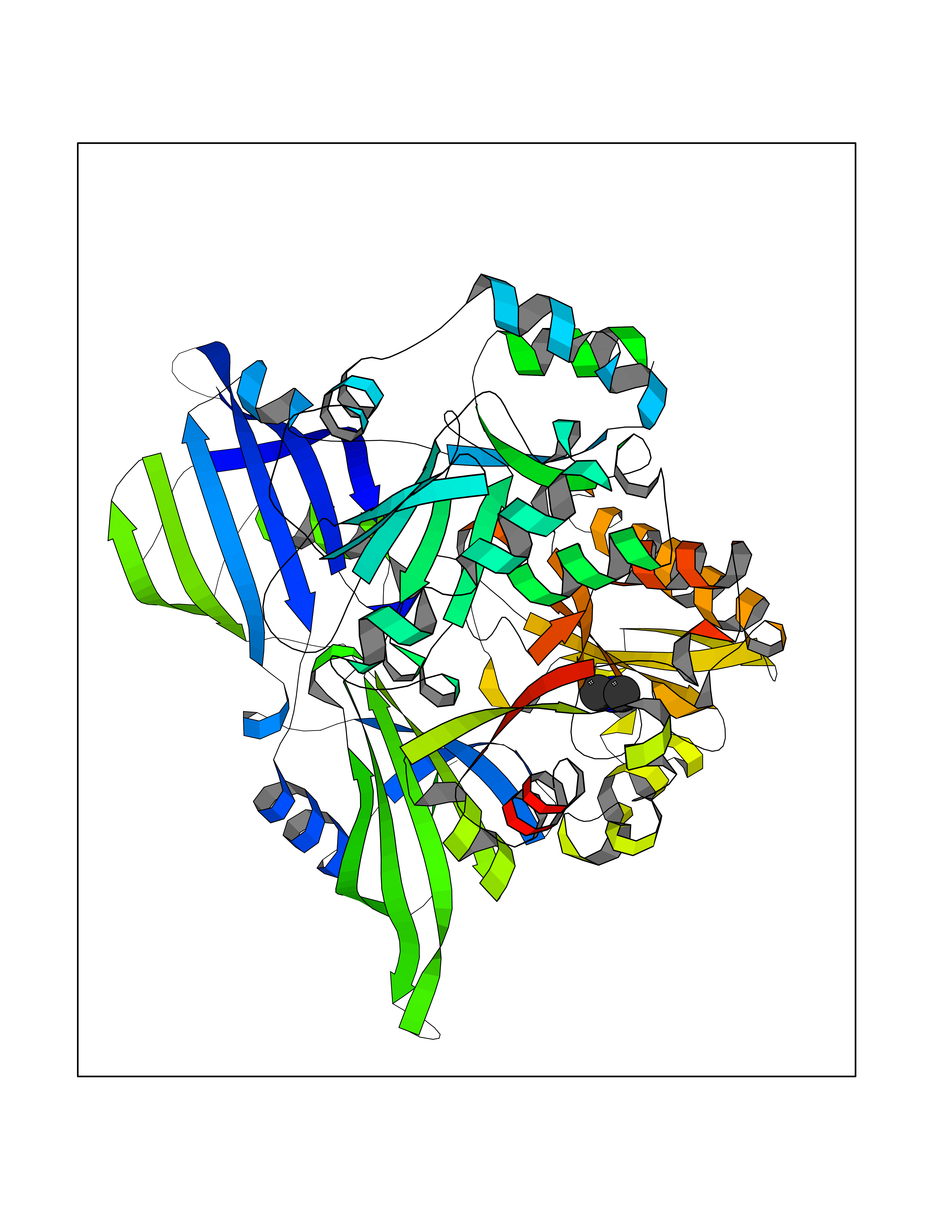 Molscript Map 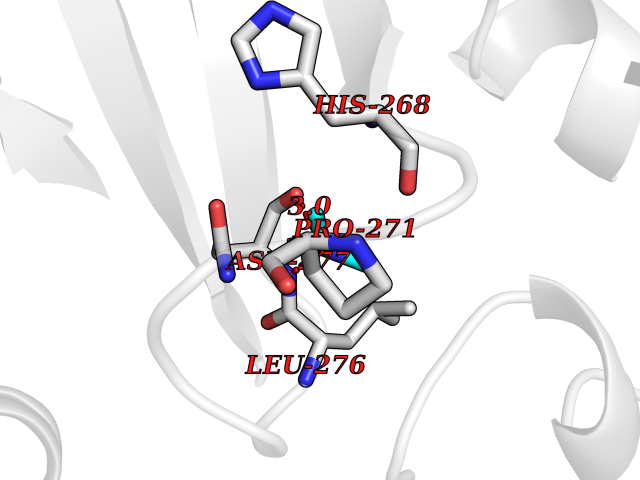 Pymol Map 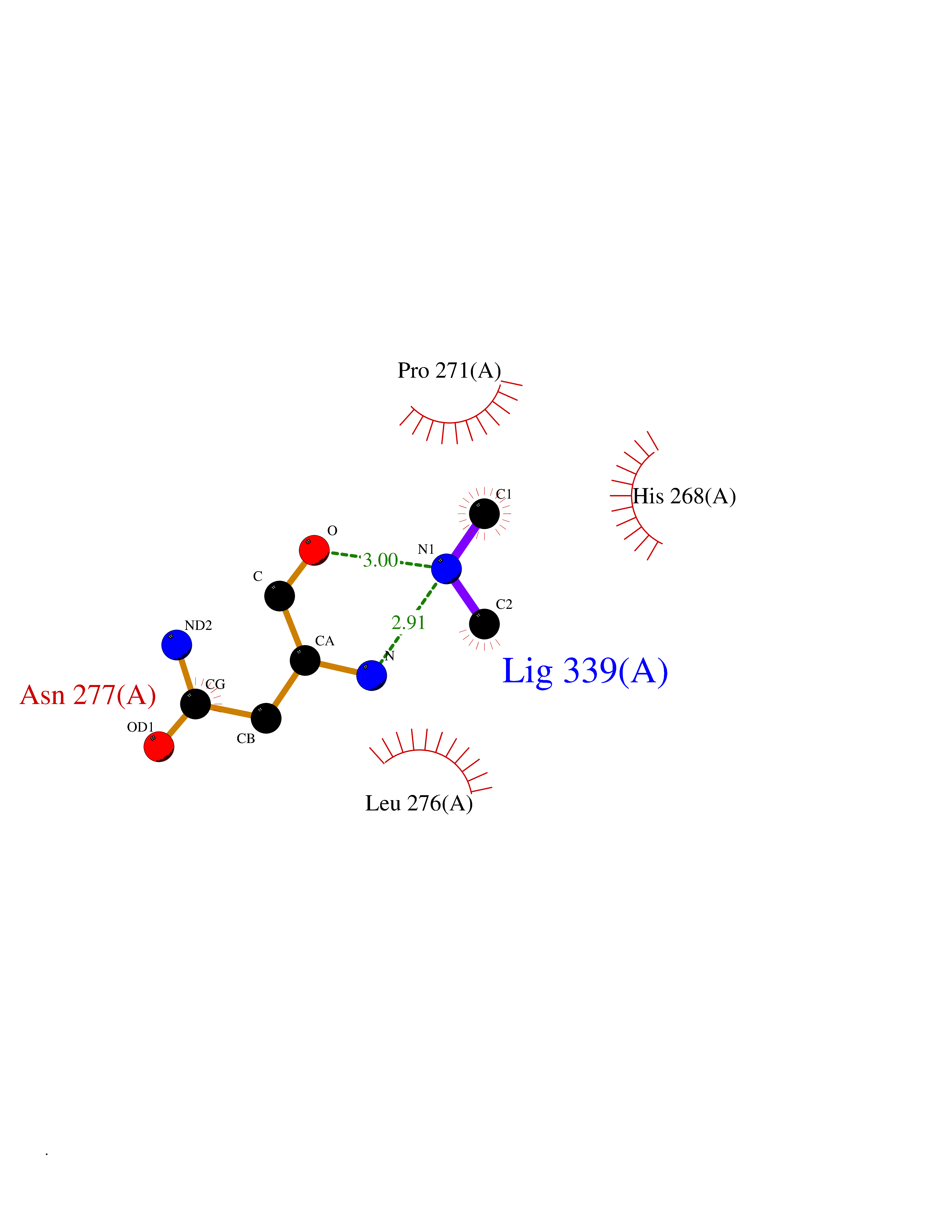 Ligplot Map 3D Binding mode Sequence VRLPFSGFRLQKVLRESARDKIIFLHGKVNEASGDGDGEDAVVILEKTPFQVEQVAQLLTGSPELQLQFSNDIYSTYHLFPPRQLNDVKTTVVYPATEKHLQKYLRQDLRLIRETGDDYRNITLPHLESQSLSIQWVYNILDKKAEADRIVFENPDPSDGFVLIPDLKWNQQQLDDLYLIAICHRRGIRSLRDLTPEHLPLLRNILHQGQEAILQRYRMKGDHLRVYLHYLPSYYHLNVHFTALGFEAPGSGVERAHLLAEVIENLECDPRHYQQRTLTFALRADDPLLKLLQEAQQAPVRLPFSGFRLQKVLRESARDKIIFLHGKVNEASGDGDGEDAVVILEKTPFQVEQVAQLLTGSPELQLQFSNDIYSTYHLFPPRQLNDVKTTVVYPATEKHLQKYLRQDLRLIRETGDDYRNITLPHLESQSLSIQWVYNILDKKAEADRIVFENPDPSDGFVLIPDLKWNQQQLDDLYLIAICHRRGIRSLRDLTPEHLPLLRNILHQGQEAILQRYRMKGDHLRVYLHYLPSYYHLNVHFTALGFEAPGSGVERAHLLAEVIENLECDPRHYQQRTLTFALRADDPLLKLLQEAQQS Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Bromodomain adjacent to zinc finger 2A (BAZ2A) | 7BL9 | 4.00 | |
Target general information Gen name BAZ2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hWALp3; Transcription termination factor I-interacting protein 5; Tip5; TTF-I-interacting protein 5; KIAA0314; Bromodomain adjacent to zinc finger domain protein 2A Protein family WAL family Biochemical class NA Function In the complex, it plays a central role by being recruited to rDNA and by targeting chromatin modifying enzymes such as HDAC1, leading to repress RNA polymerase I transcription. Recruited to rDNA via its interaction with TTF1 and its ability to recognize and bind histone H4 acetylated on 'Lys-16' (H4K16ac), leading to deacetylation of H4K5ac, H4K8ac, H4K12ac but not H4K16ac. Specifically binds pRNAs, 150-250 nucleotide RNAs that are complementary in sequence to the rDNA promoter; pRNA-binding is required for heterochromatin formation and rDNA silencing. Essential component of the NoRC (nucleolar remodeling complex) complex, a complex that mediates silencing of a fraction of rDNA by recruiting histone-modifying enzymes and DNA methyltransferases, leading to heterochromatin formation and transcriptional silencing. Related diseases Systemic lupus erythematosus 6 (SLEB6) [MIM:609939]: A chronic, relapsing, inflammatory, and often febrile multisystemic disorder of connective tissue, characterized principally by involvement of the skin, joints, kidneys and serosal membranes. It is of unknown etiology, but is thought to represent a failure of the regulatory mechanisms of the autoimmune system. The disease is marked by a wide range of system dysfunctions, an elevated erythrocyte sedimentation rate, and the formation of LE cells in the blood or bone marrow. {ECO:0000269|PubMed:18204446, ECO:0000269|PubMed:18204448}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P16333; Q62187 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Bromodomain; Chromatin regulator; Coiled coil; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Repressor; RNA-binding; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 12366.8 Length 105 Aromaticity 0.13 Instability index 62.4 Isoelectric point 4.76 Charge (pH=7) -8.26 2D Binding mode Binding energy (Kcal/mol) -5.45 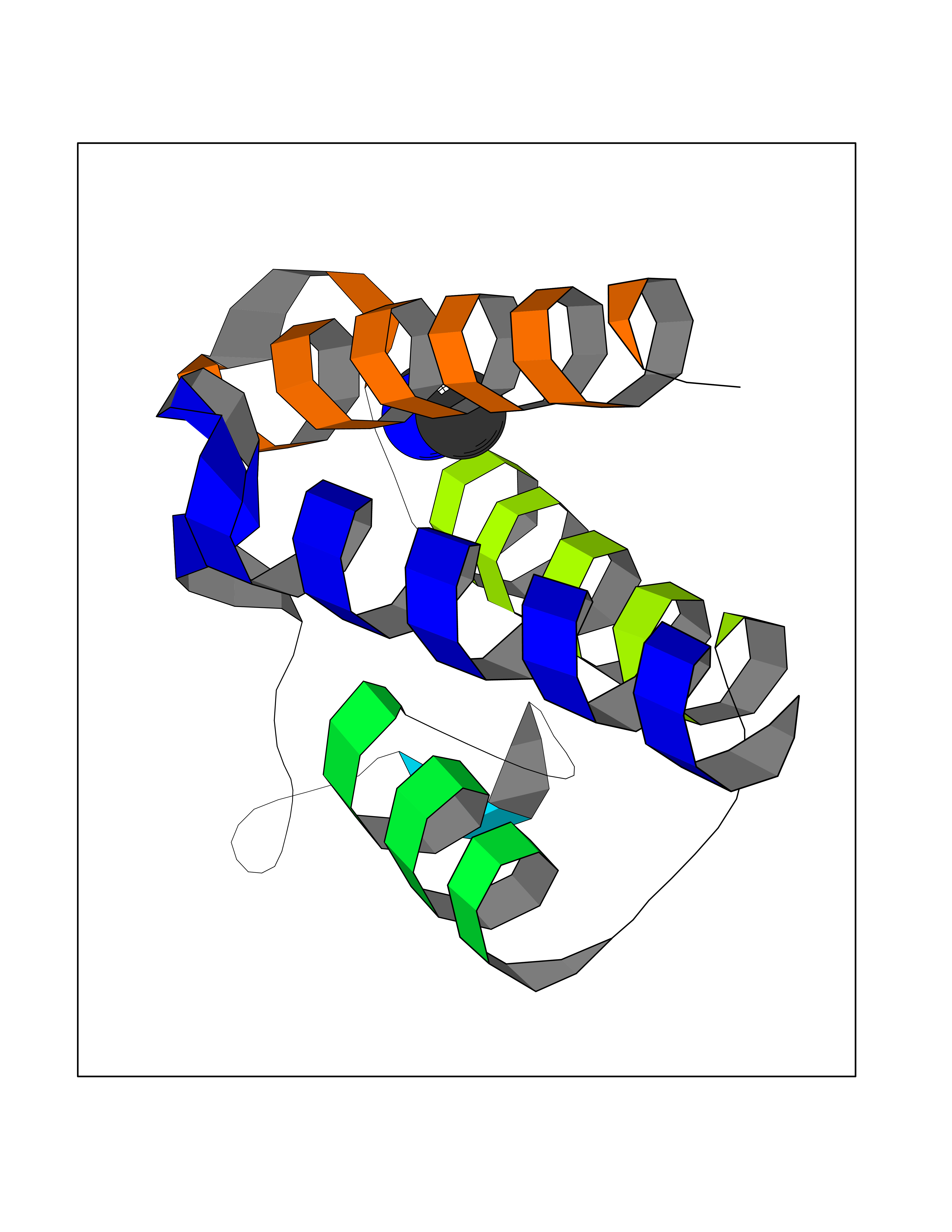 Molscript Map 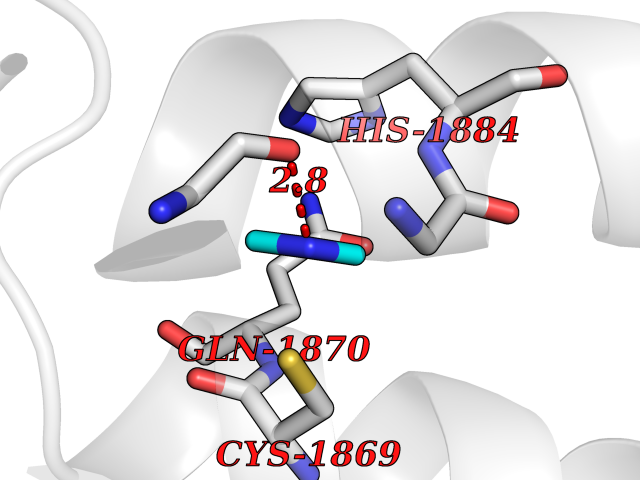 Pymol Map 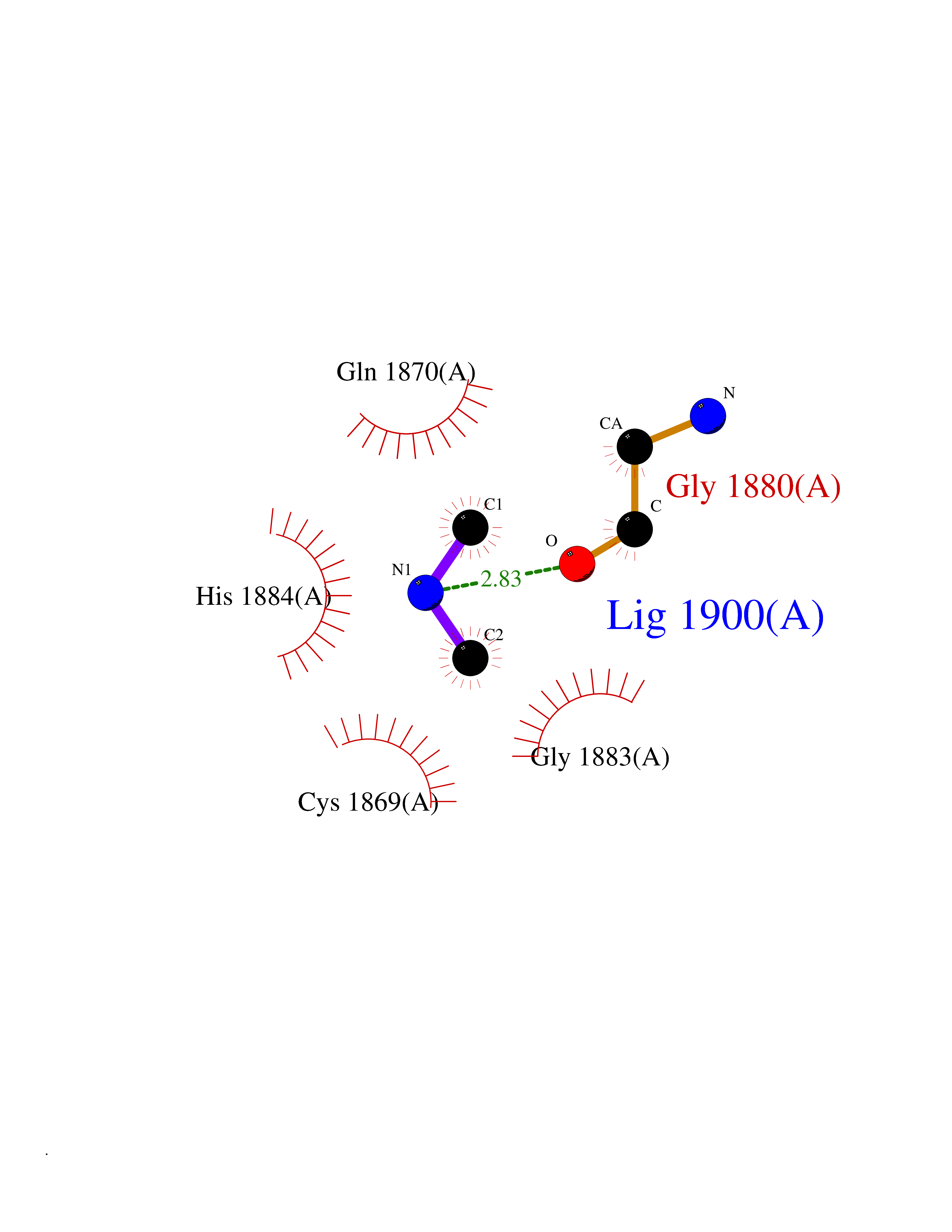 Ligplot Map 3D Binding mode Sequence SMHSDLTFCEIILMEMESHDAAWPFLEPVNPRLVSGYRRIIKNPMDFSTMRERLLRGGYTSSEEFAADALLVFDNCQTFNEDDSEVGKAGHIMRRFFESRWEEFY Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Glutathione S-transferase LANCL1 (LANCL1) | 3E73 | 4.00 | |
Target general information Gen name LANCL1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p40; LanC-like protein 1; GPR69A; 40 kDa erythrocyte membrane protein Protein family LanC-like protein family Biochemical class NA Function Functions as glutathione transferase. Catalyzes conjugation of the glutathione (GSH) to artificial substrates 1-chloro-2,4-dinitrobenzene (CDNB) and p-nitrophenyl acetate. Mitigates neuronal oxidative stress during normal postnatal development and in response to oxidative stresses probably through GSH antioxidant defense mechanism (By similarity). May play a role in EPS8 signaling. Binds glutathione. Related diseases Spermatogenic failure 5 (SPGF5) [MIM:243060]: An infertility disorder caused by spermatogenesis defects. Semen from affected men show close to 100% morphologically abnormal multiflagellar spermatozoa with low motility, oversized irregular heads, and abnormal midpiece and acrosome. {ECO:0000269|PubMed:17435757, ECO:0000269|PubMed:21733974}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9UHR4; P42858; Q08509 EC number EC 2.5.1.18 Uniprot keywords 3D-structure; Acetylation; Cell membrane; Cytoplasm; Direct protein sequencing; Membrane; Metal-binding; Proteomics identification; Reference proteome; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 46005.4 Length 405 Aromaticity 0.14 Instability index 33.7 Isoelectric point 7.13 Charge (pH=7) 0.4 2D Binding mode Binding energy (Kcal/mol) -5.46 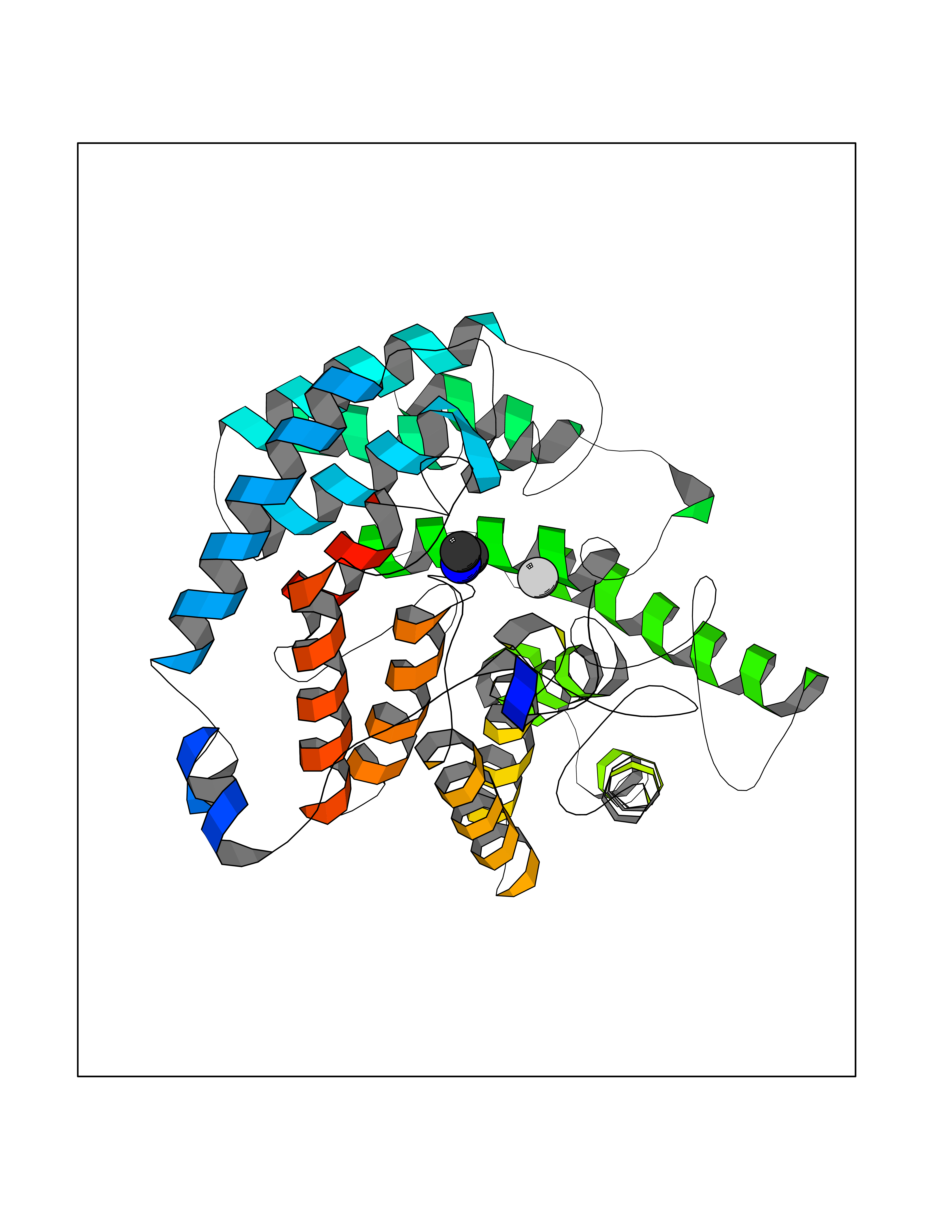 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SMDIEFMAQRAFPNPYADYNKSLAEGYFDAAGRLTPEFSQRLTNKIRELLQQMERGLKSADPRDGTGYTGWAGIAVLYLHLYDVFGDPAYLQLAHGYVKQSLNCLTKRSITFLCGDAGPLAVAAVLYHKMNNEKQAEDCITRLIHLNKIDPHAPNEMLYGRIGYIYALLFVNKNFGVEKIPQSHIQQICETILTSGENLARKRNFTAKSPLMYEWYQEYYVGAAHGLAGIYYYLMQPSLQVSQGKLHSLVKPSVDYVCQLKFPSGNYPPCIGDNRDLLVHWCHGAPGVIYMLIQAYKVFREEKYLCDAYQCADVIWQYGLLKKGYGLCHGSAGNAYAFLTLYNLTQDMKYLYRACKFAEWCLEYGEHGCRTPDTPFSLFEGMAGTIYFLADLLVPTKARFPAFEL Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Cholinesterase (BCHE) | 1P0I | 3.99 | |
Target general information Gen name BCHE Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Pseudocholinesterase; Choline esterase II; CHE1; Butyrylcholine esterase; Acylcholine acylhydrolase Protein family Type-B carboxylesterase/lipase family Biochemical class Type-B carboxylesterase/lipase Function Esterase with broad substrate specificity. Contributes to the inactivation of the neurotransmitter acetylcholine. Can degrade neurotoxic organophosphate esters. Related diseases Butyrylcholinesterase deficiency (BCHED) [MIM:617936]: An autosomal recessive metabolic condition characterized by increased sensitivity to certain anesthetic drugs, including the muscle relaxants succinylcholine or mivacurium. BCHED results in slower hydrolysis of these drugs and, consequently, a prolonged neuromuscular block, leading to apnea. The duration of the prolonged apnea varies significantly depending on the extent of the enzyme deficiency. {ECO:0000269|PubMed:10404729, ECO:0000269|PubMed:11928765, ECO:0000269|PubMed:12881446, ECO:0000269|PubMed:1306123, ECO:0000269|PubMed:1349196, ECO:0000269|PubMed:1415224, ECO:0000269|PubMed:15563885, ECO:0000269|PubMed:15781196, ECO:0000269|PubMed:1611188, ECO:0000269|PubMed:16788378, ECO:0000269|PubMed:17700357, ECO:0000269|PubMed:18075469, ECO:0000269|PubMed:18300943, ECO:0000269|PubMed:25054547, ECO:0000269|PubMed:25264279, ECO:0000269|PubMed:2915989, ECO:0000269|PubMed:7634491, ECO:0000269|PubMed:8554068, ECO:0000269|PubMed:8680411, ECO:0000269|PubMed:9110359, ECO:0000269|PubMed:9191541, ECO:0000269|PubMed:9388484, ECO:0000269|PubMed:9543549, ECO:0000269|PubMed:9694584}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08200; DB08201; DB03814; DB07940; DB03672; DB03128; DB08897; DB01122; DB06692; DB01408; DB00868; DB06756; DB11148; DB03568; DB04250; DB06774; DB01161; DB00856; DB00477; DB00122; DB14006; DB00527; DB00515; DB04920; DB00907; DB00979; DB01245; DB00944; DB11397; DB02811; DB00711; DB00449; DB07681; DB00843; DB01135; DB01057; DB01010; DB01364; DB03822; DB08658; DB00674; DB00941; DB00762; DB06636; DB00677; DB01064; DB01221; DB00772; DB00888; DB00358; DB02845; DB08893; DB01226; DB09205; DB01400; DB00585; DB00892; DB01337; DB00082; DB00183; DB00790; DB04892; DB03976; DB01338; DB00733; DB01035; DB00721; DB00392; DB09288; DB00545; DB00178; DB05386; DB00989; DB05875; DB00202; DB00391; DB00382; DB00871; DB04572; DB14031; DB00620; DB00508; DB01116; DB01199 Interacts with P54252; P46379-2; P06276; P55212; O75190-2; O14901; P13473-2; O75400-2; P62826; P67812; P02814 EC number EC 3.1.1.8 Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Phosphoprotein; Proteomics identification; Reference proteome; Secreted; Serine esterase; Sialic acid; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 58935.5 Length 523 Aromaticity 0.13 Instability index 38.71 Isoelectric point 8.63 Charge (pH=7) 5.36 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map  Pymol Map 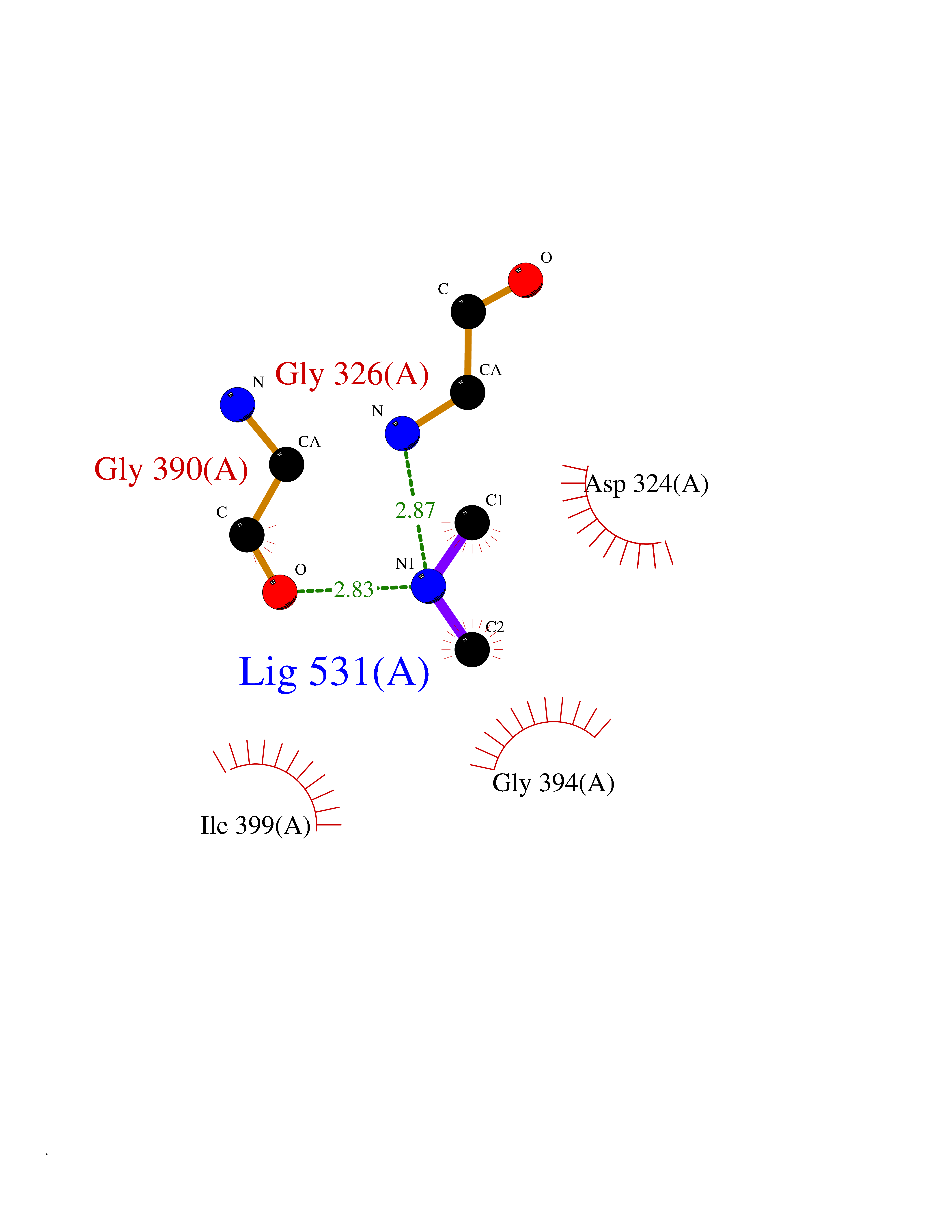 Ligplot Map 3D Binding mode Sequence IIIATKNGKVRGMQLTVFGGTVTAFLGIPYAQPPLGRLRFKKPQSLTKWSDIWNATKYANSCCQNIDQSFPGFHGSEMWNPNTDLSEDCLYLNVWIPAPKPKNATVLIWIYGGGFQTGTSSLHVYDGKFLARVERVIVVSMNYRVGALGFLALPGNPEAPGNMGLFDQQLALQWVQKNIAAFGGNPKSVTLFGESAGAASVSLHLLSPGSHSLFTRAILQSGSFNAPWAVTSLYEARNRTLNLAKLTGCSRENETEIIKCLRNKDPQEILLNEAFVVPYGTPLSVNFGPTVDGDFLTDMPDILLELGQFKKTQILVGVNKDEGTAFLVYGAPGFSKDNNSIITRKEFQEGLKIFFPGVSEFGKESILFHYTDWVQRPENYREALGDVVGDYNFICPALEFTKKFSEWGNNAFFYYFEHRSSKLPWPEWMGVMHGYEIEFVFGLPLERRDYTKAEEILSRSIVKRWANFAKYGNPQETQNQSTSWPVFKSTEQKYLTLNTESTRIMTKLRAQQCRFWTSFFPKV Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Tyrosine-protein kinase ABL1 (ABL) | 5HU9 | 3.99 | |
Target general information Gen name ABL1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p150; Proto-oncogene tyrosine-protein kinase ABL1; Proto-oncogene c-Abl; JTK7; C-ABL; Abl; Abelson tyrosine-protein kinase 1; Abelson murine leukemia viral oncogene homolog 1 Protein family Protein kinase superfamily, Tyr protein kinase family, ABL subfamily Biochemical class Kinase Function Coordinates actin remodeling through tyrosine phosphorylation of proteins controlling cytoskeleton dynamics like WASF3 (involved in branch formation); ANXA1 (involved in membrane anchoring); DBN1, DBNL, CTTN, RAPH1 and ENAH (involved in signaling); or MAPT and PXN (microtubule-binding proteins). Phosphorylation of WASF3 is critical for the stimulation of lamellipodia formation and cell migration. Involved in the regulation of cell adhesion and motility through phosphorylation of key regulators of these processes such as BCAR1, CRK, CRKL, DOK1, EFS or NEDD9. Phosphorylates multiple receptor tyrosine kinases and more particularly promotes endocytosis of EGFR, facilitates the formation of neuromuscular synapses through MUSK, inhibits PDGFRB-mediated chemotaxis and modulates the endocytosis of activated B-cell receptor complexes. Other substrates which are involved in endocytosis regulation are the caveolin (CAV1) and RIN1. Moreover, ABL1 regulates the CBL family of ubiquitin ligases that drive receptor down-regulation and actin remodeling. Phosphorylation of CBL leads to increased EGFR stability. Involved in late-stage autophagy by regulating positively the trafficking and function of lysosomal components. ABL1 targets to mitochondria in response to oxidative stress and thereby mediates mitochondrial dysfunction and cell death. In response to oxidative stress, phosphorylates serine/threonine kinase PRKD2 at 'Tyr-717'. ABL1 is also translocated in the nucleus where it has DNA-binding activity and is involved in DNA-damage response and apoptosis. Many substrates are known mediators of DNA repair: DDB1, DDB2, ERCC3, ERCC6, RAD9A, RAD51, RAD52 or WRN. Activates the proapoptotic pathway when the DNA damage is too severe to be repaired. Phosphorylates TP73, a primary regulator for this type of damage-induced apoptosis. Phosphorylates the caspase CASP9 on 'Tyr-153' and regulates its processing in the apoptotic response to DNA damage. Phosphorylates PSMA7 that leads to an inhibition of proteasomal activity and cell cycle transition blocks. ABL1 acts also as a regulator of multiple pathological signaling cascades during infection. Several known tyrosine-phosphorylated microbial proteins have been identified as ABL1 substrates. This is the case of A36R of Vaccinia virus, Tir (translocated intimin receptor) of pathogenic E. coli and possibly Citrobacter, CagA (cytotoxin-associated gene A) of H. pylori, or AnkA (ankyrin repeat-containing protein A) of A. phagocytophilum. Pathogens can highjack ABL1 kinase signaling to reorganize the host actin cytoskeleton for multiple purposes, like facilitating intracellular movement and host cell exit. Finally, functions as its own regulator through autocatalytic activity as well as through phosphorylation of its inhibitor, ABI1. Regulates T-cell differentiation in a TBX21-dependent manner. Phosphorylates TBX21 on tyrosine residues leading to an enhancement of its transcriptional activator activity. Non-receptor tyrosine-protein kinase that plays a role in many key processes linked to cell growth and survival such as cytoskeleton remodeling in response to extracellular stimuli, cell motility and adhesion, receptor endocytosis, autophagy, DNA damage response and apoptosis. Related diseases Leukemia, chronic myeloid (CML) [MIM:608232]: A clonal myeloproliferative disorder of a pluripotent stem cell with a specific cytogenetic abnormality, the Philadelphia chromosome (Ph), involving myeloid, erythroid, megakaryocytic, B-lymphoid, and sometimes T-lymphoid cells, but not marrow fibroblasts. The gene represented in this entry is involved in disease pathogenesis.; DISEASE: A chromosomal aberration involving ABL1 has been found in patients with chronic myeloid leukemia. Translocation t(9;22)(q34;q11) with BCR. The translocation produces a BCR-ABL found also in acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL). {ECO:0000269|PubMed:3021337}.; DISEASE: A chromosomal aberration involving ABL1 is found in a form of acute lymphoblastic leukemia (PubMed:15361874). Translocation t(9;9)(q34;q34) with NUP214 (PubMed:15361874). {ECO:0000269|PubMed:15361874}.; DISEASE: Congenital heart defects and skeletal malformations syndrome (CHDSKM) [MIM:617602]: An autosomal dominant disorder characterized by congenital heart disease with atrial and ventricular septal defects, variable skeletal abnormalities, and failure to thrive. Skeletal defects include pectus excavatum, scoliosis, and finger contractures. Some patient exhibit joint laxity. {ECO:0000269|PubMed:28288113}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08043; DB08583; DB07831; DB08350; DB12597; DB00171; DB06616; DB12267; DB01254; DB12010; DB00619; DB13749; DB08231; DB03878; DB04868; DB08339; DB08901; DB12323; DB08896; DB14989; DB05184 Interacts with Q8IZP0; Q9NYB9; O14672; P10275; Q13315; Q4KMG0; P46108; P46109; P35222; P00533; P04626; Q03468; Q14315; P36888; P05107; P10721; Q38SD2; Q92918; Q7Z434; O43196; P15941; P15941-12; P16333; O43900; Q13905; Q86UR5; Q13671; P31947; Q15464; O75751; P37840; Q9BX66; O60504-2; Q07890; P12931; P51692; Q9Y4G6; P11387; P04637; P15498; Q9Y6W5; P62258; P61981; P63104; O35158; P37840; P48165; Q15323; P37840 EC number EC 2.7.10.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Apoptosis; ATP-binding; Autophagy; Cell adhesion; Chromosomal rearrangement; Cytoplasm; Cytoskeleton; Disease variant; DNA damage; DNA repair; DNA-binding; Endocytosis; Kinase; Lipoprotein; Magnesium; Manganese; Membrane; Metal-binding; Mitochondrion; Myristate; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Reference proteome; SH2 domain; SH3 domain; Transferase; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 31264.6 Length 270 Aromaticity 0.12 Instability index 37.99 Isoelectric point 5.42 Charge (pH=7) -7.67 2D Binding mode Binding energy (Kcal/mol) -5.44 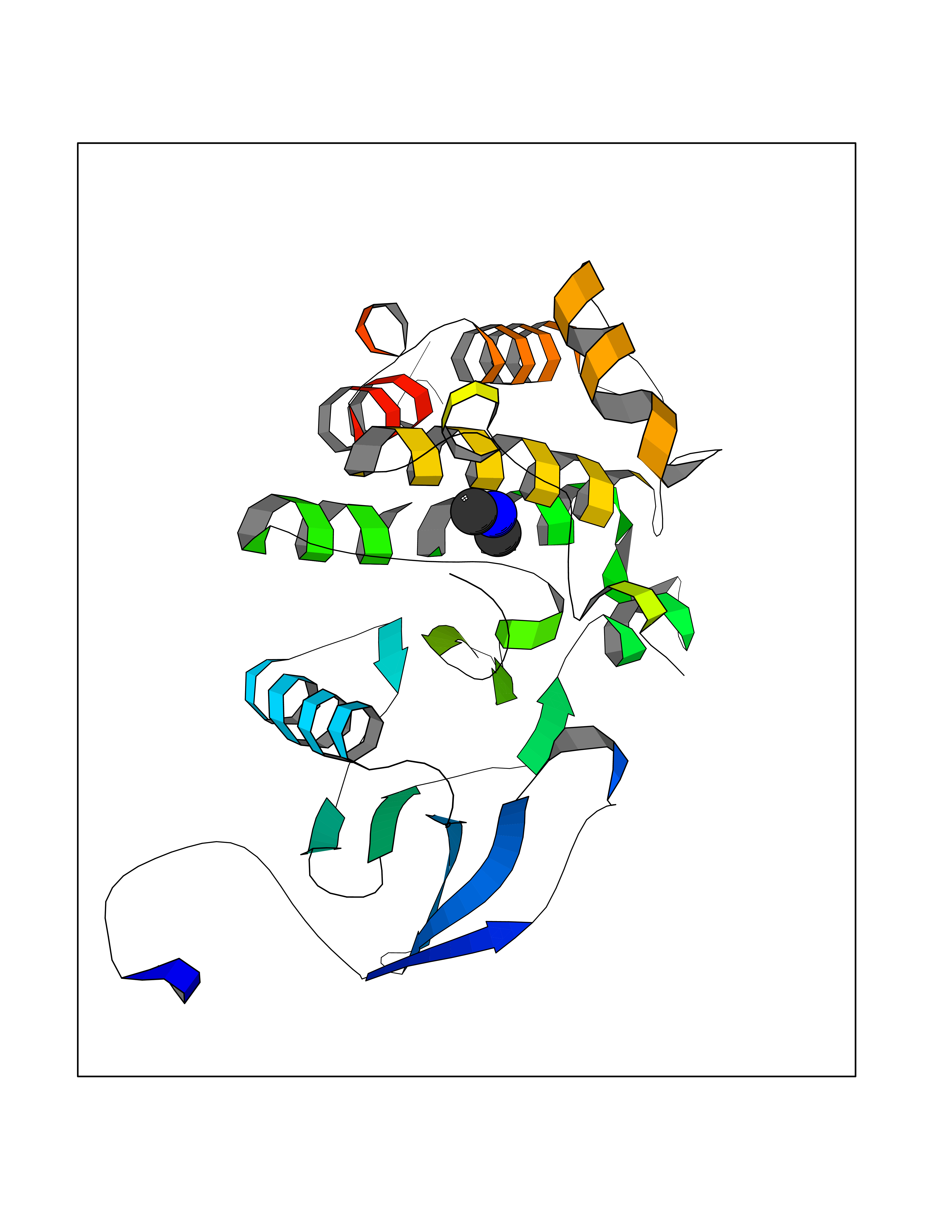 Molscript Map 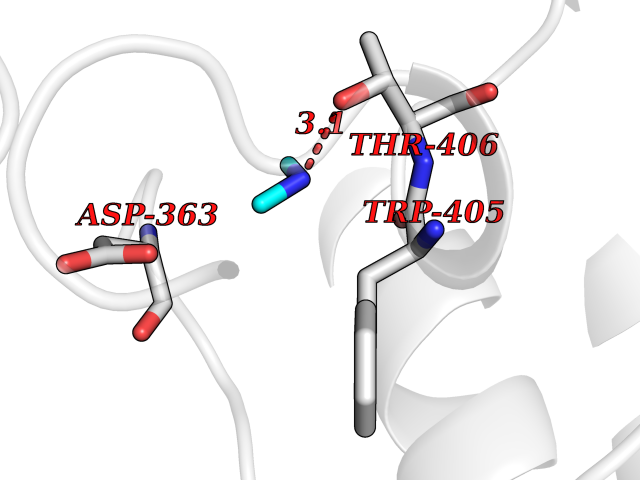 Pymol Map 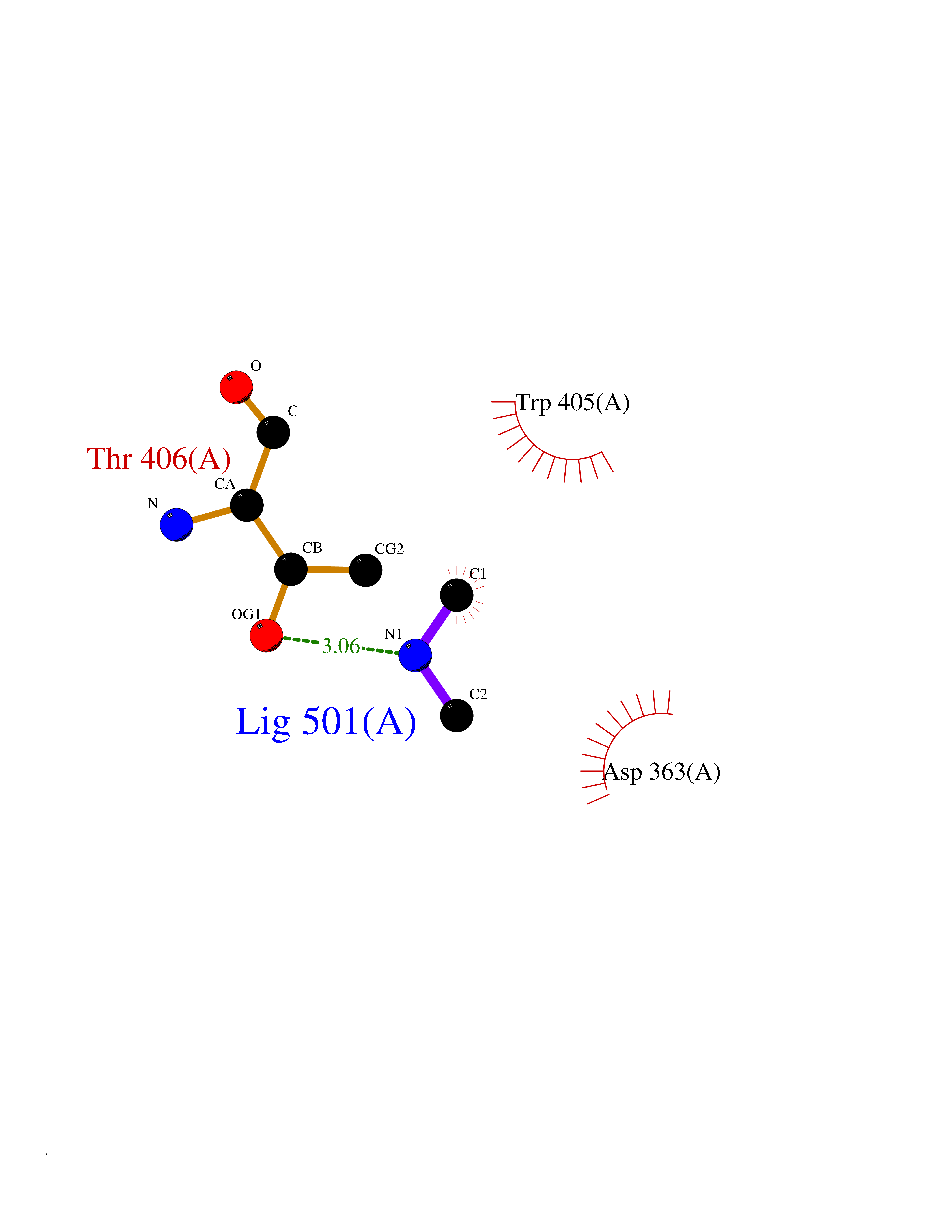 Ligplot Map 3D Binding mode Sequence AMGSSPNYDKWEMERTDITMKHKLGGGQYGEVYEGVWKKYSLTVAVKTLKEDTMEVEEFLKEAAVMKEIKHPNLVQLLGVCTREPPFYIITEFMTYGNLLDYLRECNRQEVNAVVLLYMATQISSAMEYLEKKNFIHRDLAARNCLVGENHLVKVADFGLSRLMTAHAGAKFPIKWTAPESLAYNKFSIKSDVWAFGVLLWEIATYGMSPYPGIDLSQVYELLEKDYRMERPEGCPEKVYELMRACWQWNPSDRPSFAEIHQAFETMFQE Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Endolysin | 1AM7 | 3.99 | |
Target general information Gen name R Organism Escherichia phage lambda (Bacteriophage lambda) Uniprot ID TTD ID NA Synonyms NA Protein family Glycosyl hydrolase 24 family Biochemical class Glycosidase Function Lyase activity.Lysozyme activity.Lytic transglycosylase activity. Related diseases Estrogen resistance (ESTRR) [MIM:615363]: A disorder characterized by partial or complete resistance to estrogens, in the presence of elevated estrogen serum levels. Clinical features include absence of the pubertal growth spurt, delayed bone maturation, unfused epiphyses, reduced bone mineral density, osteoporosis, continued growth into adulthood and very tall adult stature. Glucose intolerance, hyperinsulinemia and lipid abnormalities may also be present. {ECO:0000269|PubMed:23841731, ECO:0000269|PubMed:27754803}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04206 Interacts with NA EC number 4.2.2.n2 Uniprot keywords 3D-structure; Antimicrobial; Bacteriolytic enzyme; Cytolysis; Direct protein sequencing; Host cell lysis by virus; Host cytoplasm; Lyase; Reference proteome; Viral release from host cell Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 49834.9 Length 462 Aromaticity 0.07 Instability index 18.78 Isoelectric point 9.6 Charge (pH=7) 18.29 2D Binding mode Binding energy (Kcal/mol) -5.44 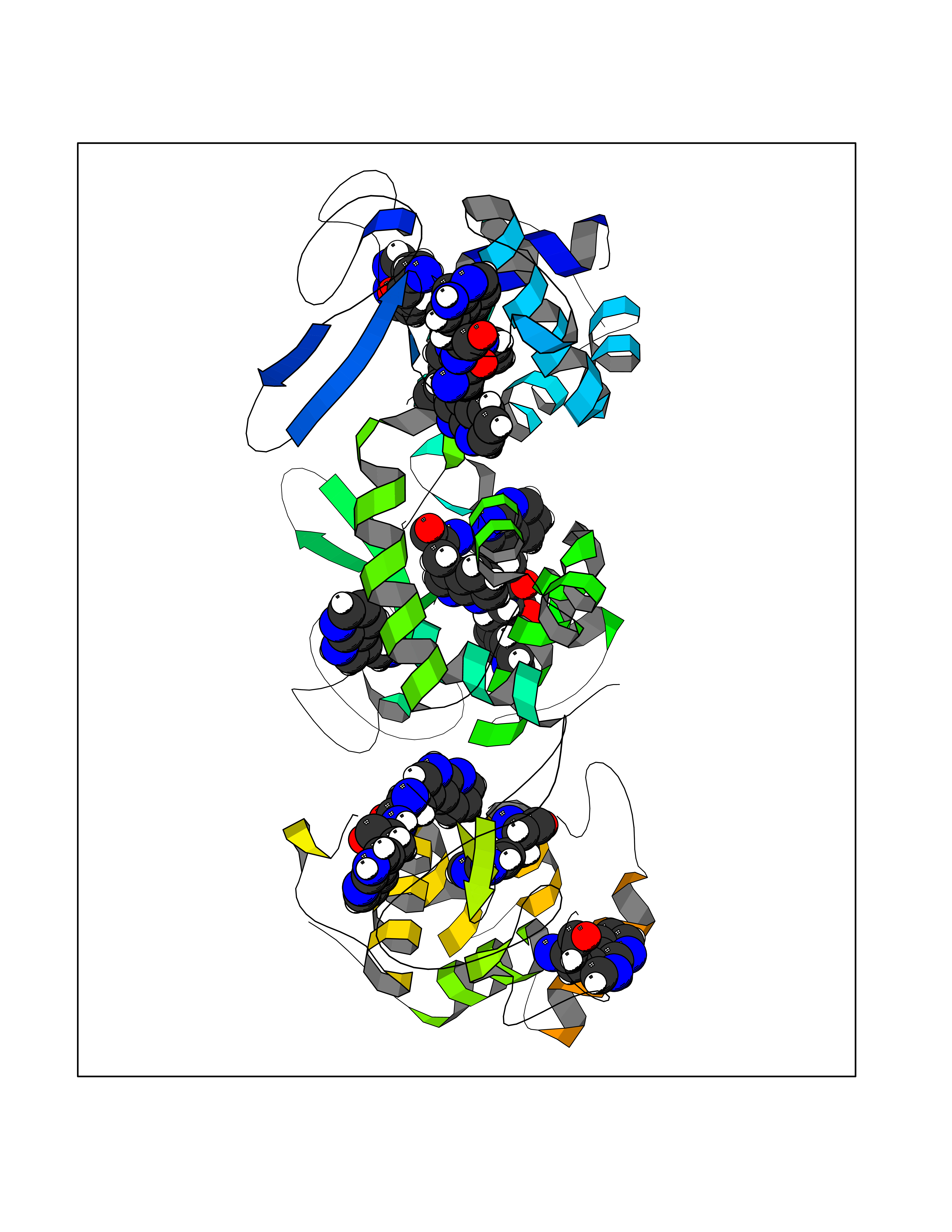 Molscript Map 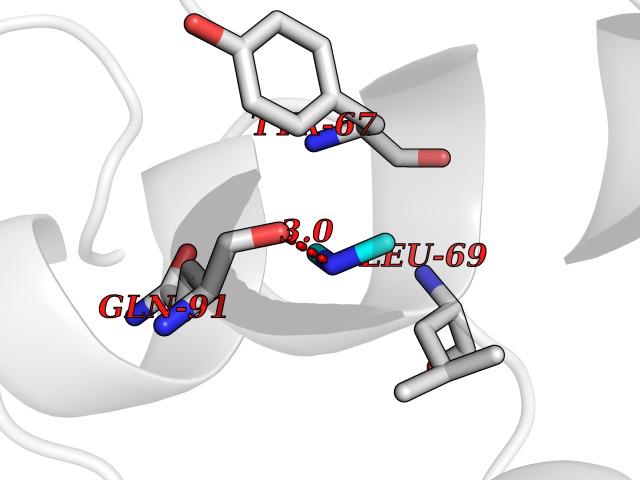 Pymol Map 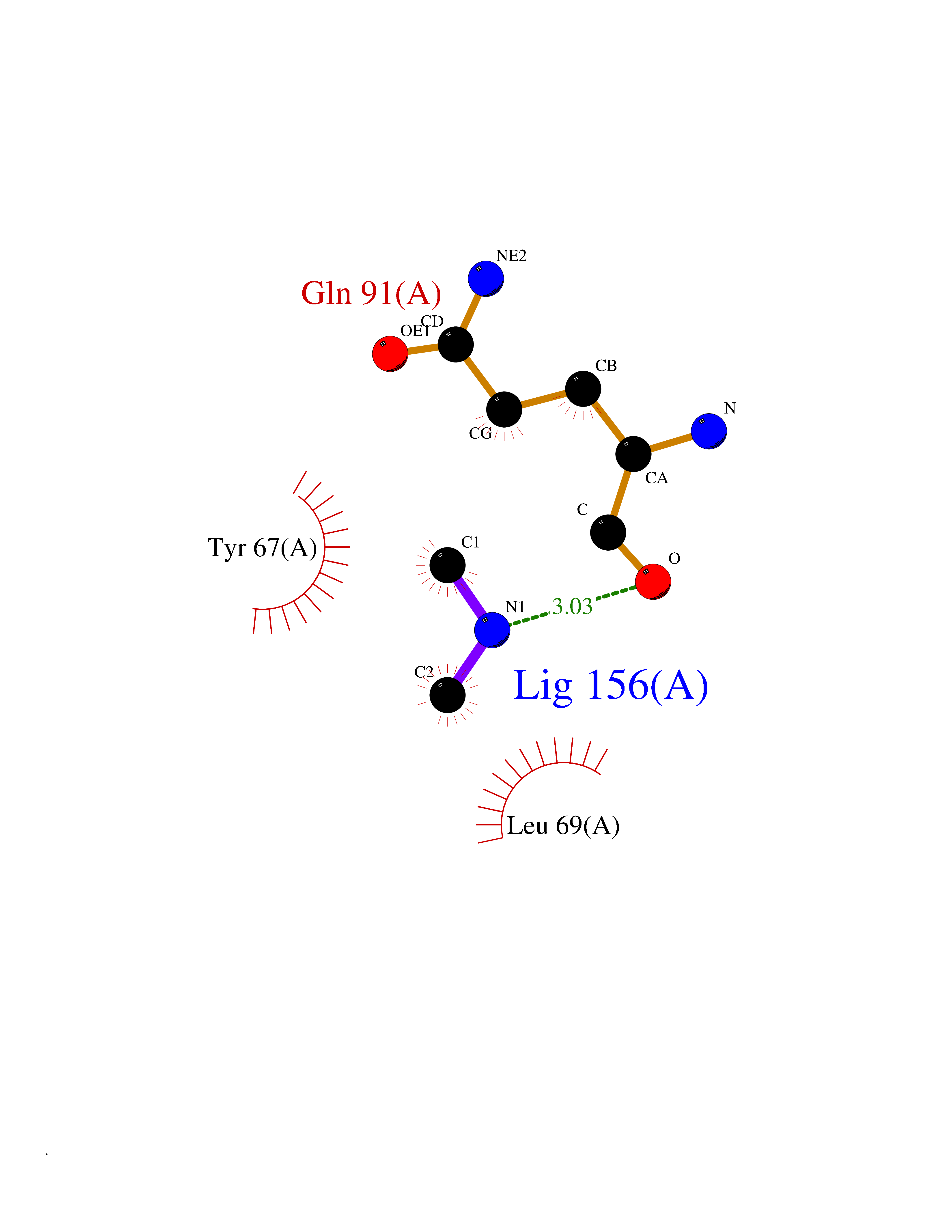 Ligplot Map 3D Binding mode Sequence MVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVRMVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVRMVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVR Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Transferrin (TF) | 1RYO | 3.99 | |
Target general information Gen name TF Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Siderophilin; Serotransferrin; PRO1400; Beta-1 metal-binding globulin Protein family Transferrin family Biochemical class Transferrin Function It is responsible for the transport of iron from sites of absorption and heme degradation to those of storage and utilization. Serum transferrin may also have a further role in stimulating cell proliferation. Transferrins are iron binding transport proteins which can bind two Fe(3+) ions in association with the binding of an anion, usually bicarbonate. Related diseases Atransferrinemia (ATRAF) [MIM:209300]: A rare autosomal recessive disorder characterized by abnormal synthesis of transferrin leading to iron overload and microcytic hypochromic anemia. {ECO:0000269|PubMed:11110675, ECO:0000269|PubMed:15466165}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01370; DB14517; DB14518; DB01294; DB14526; DB14527; DB11136; DB14528; DB14529; DB14530; DB00515; DB09130; DB11397; DB13949; DB14490; DB14491; DB14488; DB14501; DB14489; DB13257; DB06215; DB06784; DB05260; DB01592; DB00893; DB00677; DB06757; DB11182; DB14520; DB01593; DB14487; DB14533; DB14548 Interacts with O43315; O00501; Q7Z7G2; Q9GZR5; Q9Y282; Q96KR6; P01350; P08034; Q8NBJ4; O15529; Q8TED1; Q7Z5P4; A8MZ59; O15173; Q96TC7; Q3KNW5; Q9BXS9-3; Q99523; O43278-2; Q8N9I0; P02786; Q4KMG9; Q9K0U9; Q09057; Q9K0V0; P02786 EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Ion transport; Iron; Iron transport; Metal-binding; Methylation; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 35854.5 Length 324 Aromaticity 0.1 Instability index 34.17 Isoelectric point 7.58 Charge (pH=7) 1.42 2D Binding mode Binding energy (Kcal/mol) -5.44 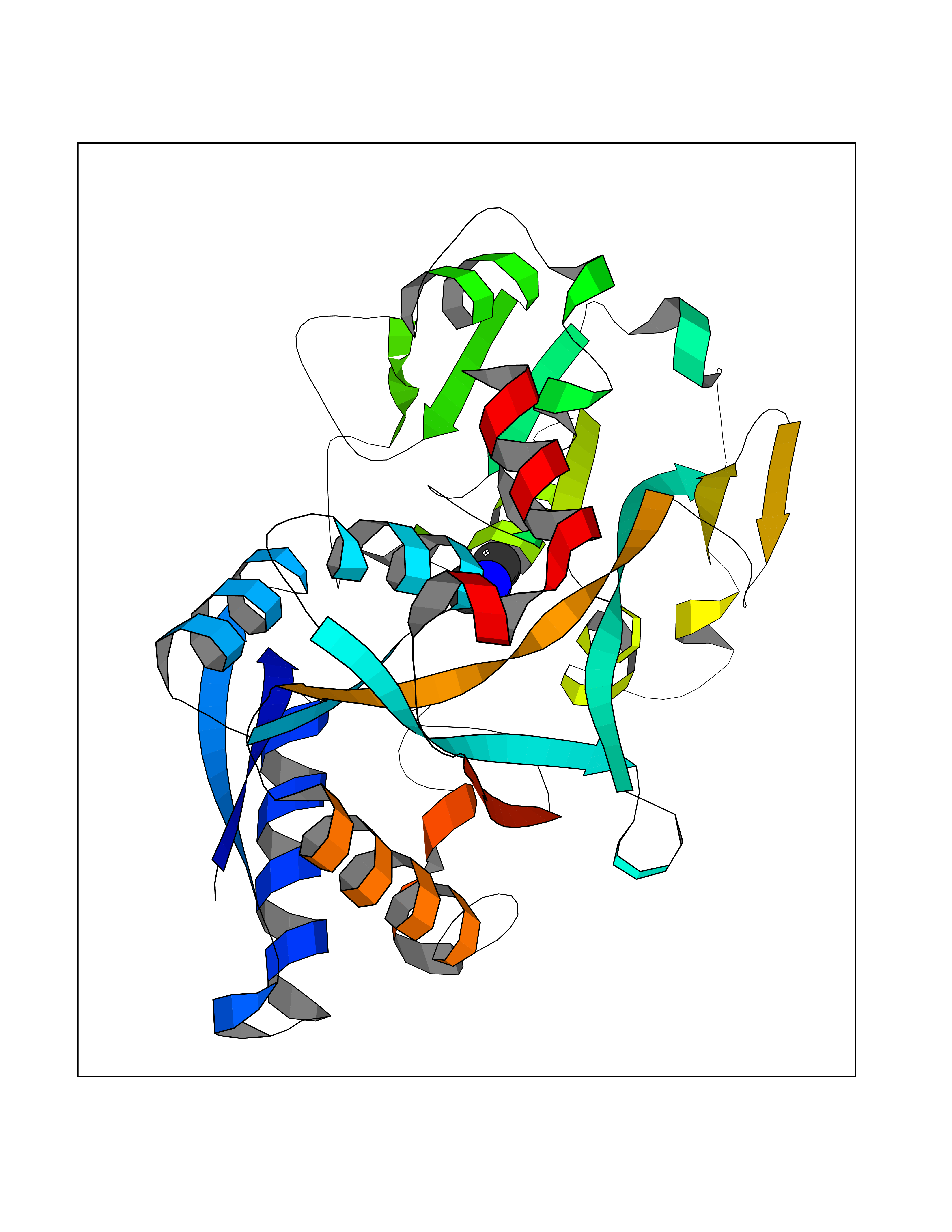 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence KTVRWCAVSEHEATKCQSFRDHMKSVIPSDGPSVACVKKASYLDCIRAIAANEADAVTLDAGLVYDAYLAPNNLKPVVAEFYGSKEDPQTFYYAVAVVKKDSGFQMNQLRGKKSCHTGLGRSAGWNIPIGLLYCDLPEPRKPLEKAVANFFSGSCAPCADGTDFPQLCQLCPGCGCSTLNQYFGYSGAFKCLKDGAGDVAFVKHSTIFENLANKADRDQYELLCLDNTRKPVDEYKDCHLAQVPSHTVVARSMGGKEDLIWELLNQAQEHFGKDKSKEFQLFSSPHGKDLLFKDSAHGFLKVPPRMDAKMYLGYEYVTAIRNLR Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Aromatase (CYP19A1) | 3S79 | 3.99 | |
Target general information Gen name CYP19A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms P-450AROM; Estrogen synthetase; Estrogen synthase; Cytochrome P450 19A1; Cytochrome P-450AROM; CYPXIX; CYP19; CYAR; ARO1 Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function Catalyzes the formation of aromatic C18 estrogens from C19 androgens. Related diseases Aromatase excess syndrome (AEXS) [MIM:139300]: An autosomal dominant disorder characterized by increased extraglandular aromatization of steroids that presents with heterosexual precocity in males and isosexual precocity in females. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Aromatase deficiency (AROD) [MIM:613546]: A rare disease in which fetal androgens are not converted into estrogens due to placental aromatase deficiency. Thus, pregnant women exhibit a hirsutism, which spontaneously resolves after post-partum. At birth, female babies present with pseudohermaphroditism due to virilization of extern genital organs. In adult females, manifestations include delay of puberty, breast hypoplasia and primary amenorrhoea with multicystic ovaries. {ECO:0000269|PubMed:24705274, ECO:0000269|PubMed:8265607, ECO:0000269|PubMed:8530621, ECO:0000269|PubMed:9211678}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02342; DB00357; DB01217; DB00443; DB04794; DB06719; DB13009; DB00389; DB00269; DB00856; DB04839; DB01406; DB00255; DB00858; DB01127; DB14598; DB14600; DB00974; DB06423; DB00783; DB00655; DB00926; DB00990; DB04539; DB01026; DB01006; DB05667; DB00358; DB01065; DB00333; DB06710; DB01110; DB16236; DB05749; DB08804; DB03467; DB00184; DB09389; DB01229; DB05804; DB00481; DB05875; DB02901; DB06147; DB00675; DB00894; DB00624; DB13943; DB13944; DB13946; DB01007; DB00197 Interacts with NA EC number EC 1.14.14.14 Uniprot keywords 3D-structure; Alternative splicing; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Heme; Iron; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Pseudohermaphroditism; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 52141.6 Length 452 Aromaticity 0.1 Instability index 36.02 Isoelectric point 8.45 Charge (pH=7) 4.41 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map  Pymol Map 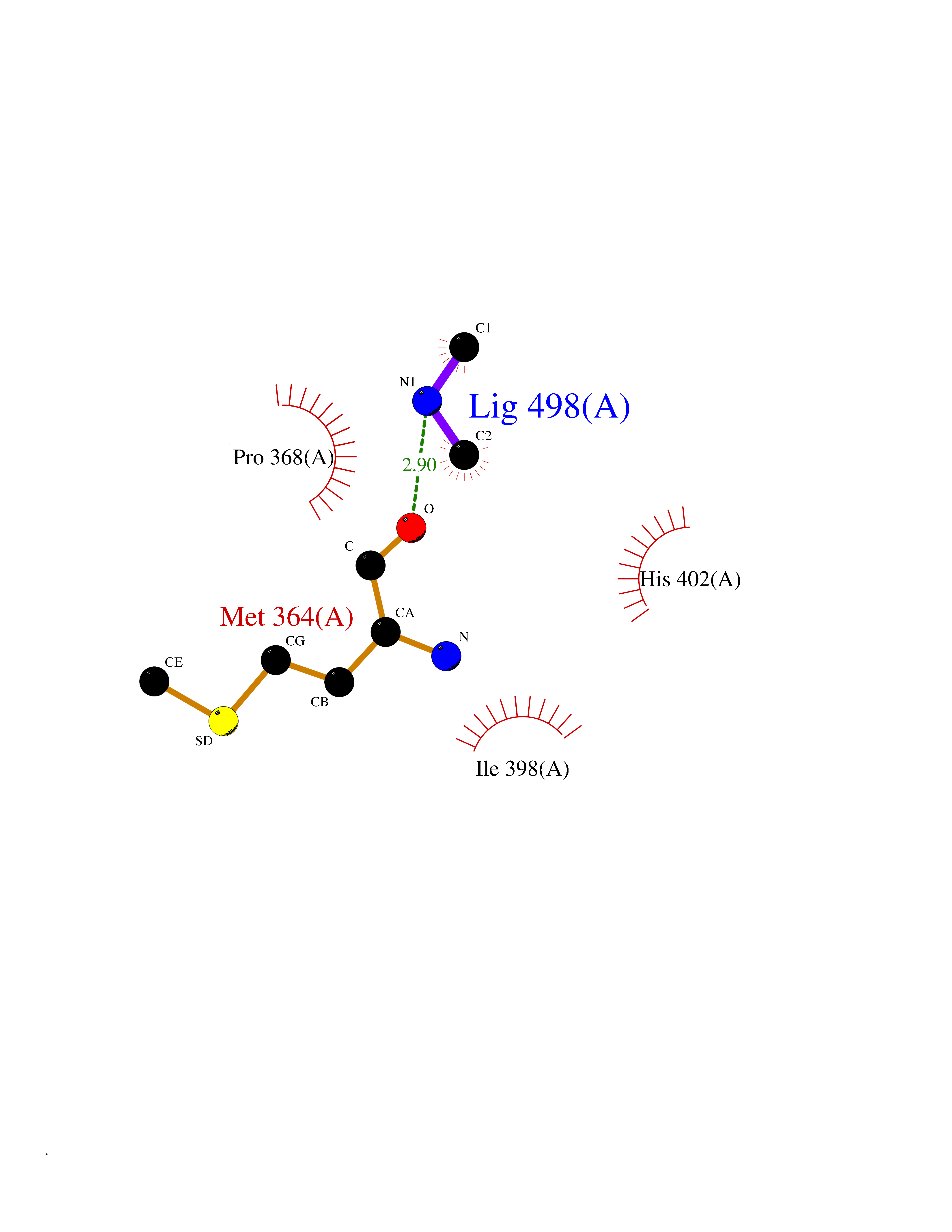 Ligplot Map 3D Binding mode Sequence SSIPGPGYCMGIGPLISHGRFLWMGIGSACNYYNRVYGEFMRVWISGEETLIISKSSSMFHIMKHNHYSSRFGSKLGLQCIGMHEKGIIFNNNPELWKTTRPFFMKALSGPGLVRMVTVCAESLKTHLDRLEEVTNESGYVDVLTLLRRVMLDTSNTLFLRIPLDESAIVVKIQGYFDAWQALLIKPDIFFKISWLYKKYEKSVKDLKDAIEVLIAEKRRRISTEEKLEECMDFATELILAEKRGDLTRENVNQCILEMLIAAPDTMSVSLFFMLFLIAKHPNVEEAIIKEIQTVIGERDIKIDDIQKLKVMENFIYESMRYQPVVDLVMRKALEDDVIDGYPVKKGTNIILNIGRMHRLEFFPKPNEFTLENFAKNVPYRYFQPFGFGPRGCAGKYIAMVMMKAILVTLLRRFHVKTLQGQCVESIQKIHDLSLHPDETKNMLEMIFTPRN Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | NH(3)-dependent NAD(+) synthetase | 1KQP | 3.99 | |
Target general information Gen name nadE Organism Bacillus subtilis (strain 168) Uniprot ID TTD ID NA Synonyms outB;BSU03130 Protein family NAD synthetase family Biochemical class Ligase Function ATP binding.Metal ion binding.NAD+ synthase (glutamine-hydrolyzing) activity.NAD+ synthase activity. Related diseases Leukodystrophy, hypomyelinating, 15 (HLD15) [MIM:617951]: An autosomal recessive disorder characterized by hypomyelinating leukodystrophy with thinning of the corpus callosum. Clinical features include motor and cognitive impairment appearing in the first or second decade of life, dystonia, ataxia, spasticity, and dysphagia. Most patients develop severe optic atrophy, and some have hearing loss. {ECO:0000269|PubMed:29576217}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02596; DB04099; DB00798 Interacts with NA EC number 6.3.1.5 Uniprot keywords 3D-structure; ATP-binding; Direct protein sequencing; Ligase; Magnesium; Metal-binding; NAD; Nucleotide-binding; Phosphoprotein; Reference proteome; Sporulation; Stress response Protein physicochemical properties Chain ID A,B Molecular weight (Da) 60509.3 Length 542 Aromaticity 0.08 Instability index 31.96 Isoelectric point 5.07 Charge (pH=7) -19.73 2D Binding mode Binding energy (Kcal/mol) -5.44 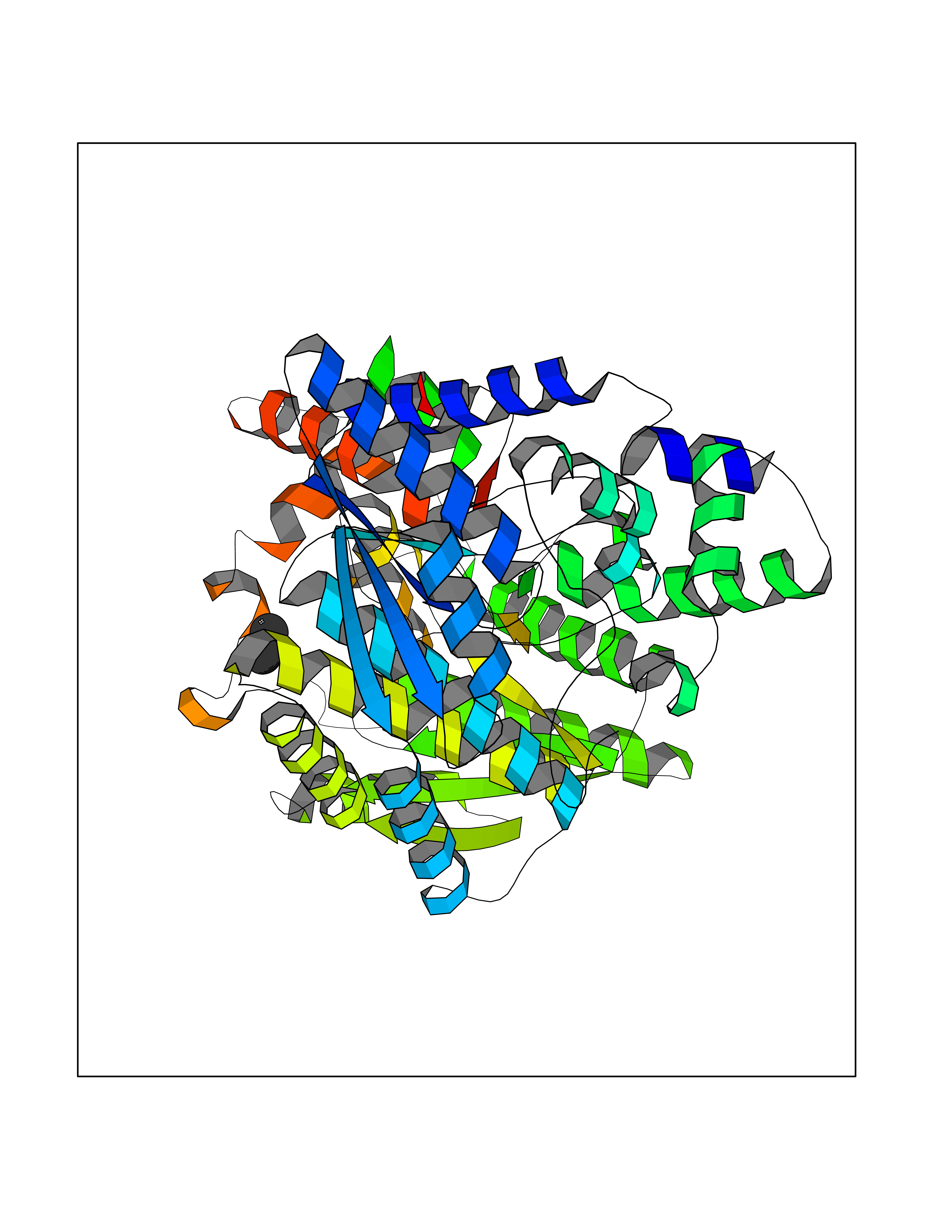 Molscript Map 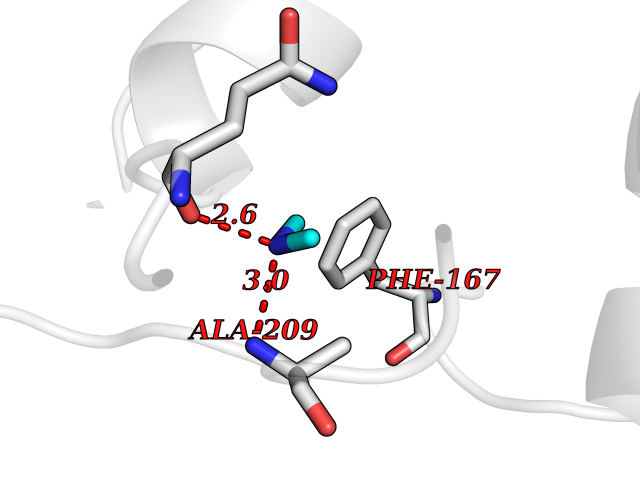 Pymol Map 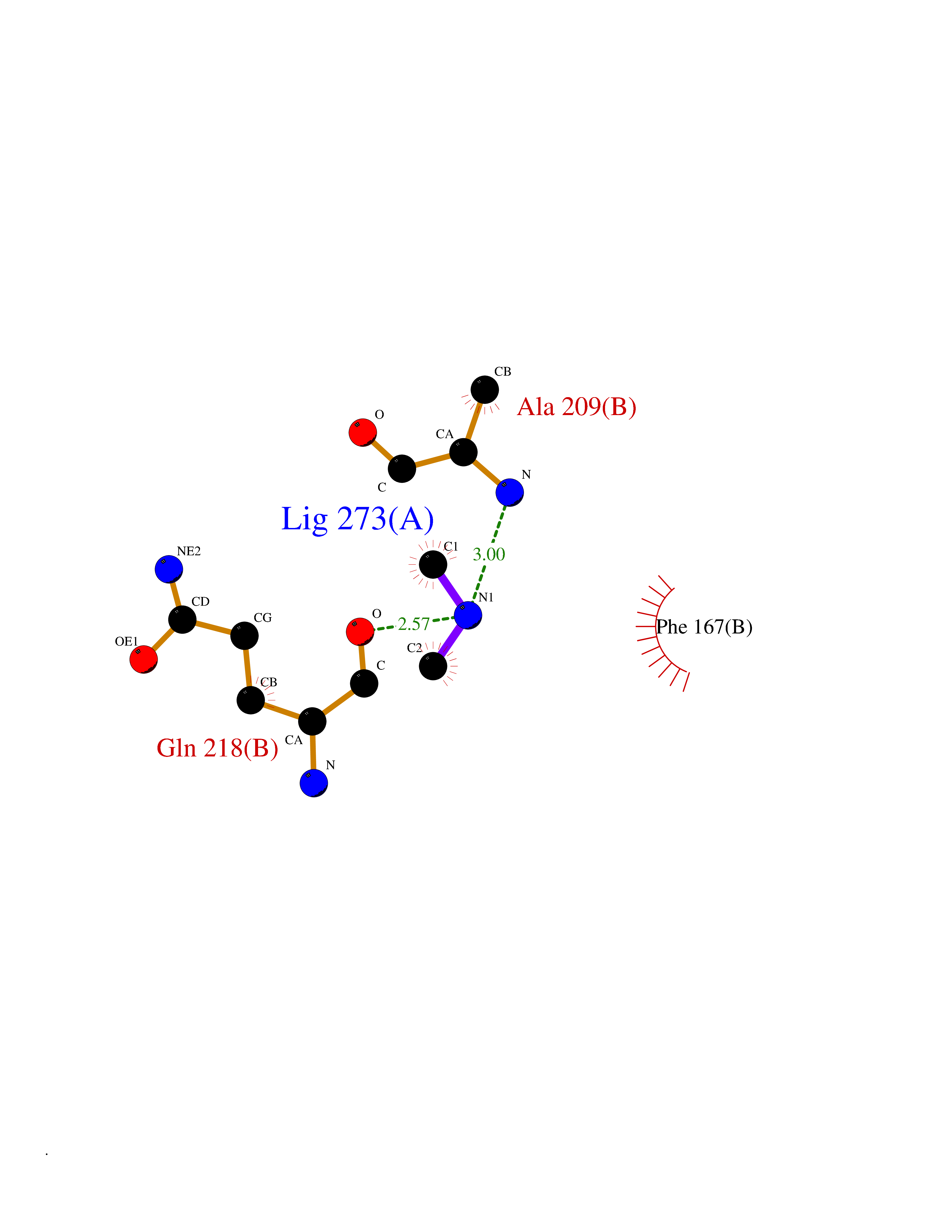 Ligplot Map 3D Binding mode Sequence SMQEKIMRELHVKPSIDPKQEIEDRVNFLKQYVKKTGAKGFVLGISGGQDSTLAGRLAQLAVESIREEGGDAQFIAVRLPHGTQQDEDDAQLALKFIKPDKSWKFDIKSTVSAFSDQYQQETGDQLTDFNKGNVKARTRMIAQYAIGGQEGLLVLGTDHAAEAVTGFFTKYGDGGADLLPLTGLTKRQGRTLLKELGAPERLYLKEPTADLLDEKPQQSDETELGISYDEIDDYLEGKEVSAKVSEALEKRYSMTEHKRQVPASMFDDWWKSMQEKIMRELHVKPSIDPKQEIEDRVNFLKQYVKKTGAKGFVLGISGGQDSTLAGRLAQLAVESIREEGGDAQFIAVRLPHGTQQDEDDAQLALKFIKPDKSWKFDIKSTVSAFSDQYQQETGDQLTDFNKGNVKARTRMIAQYAIGGQEGLLVLGTDHAAEAVTGFFTKYGDGGADLLPLTGLTKRQGRTLLKELGAPERLYLKEPTADLLDEKPQQSDETELGISYDEIDDYLEGKEVSAKVSEALEKRYSMTEHKRQVPASMFDDWWK Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Alcohol dehydrogenase 1C | 1U3W | 3.99 | |
Target general information Gen name ADH1C Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ADH3 Protein family Zinc-containing alcohol dehydrogenase family Biochemical class Oxidoreductase Function Alcohol dehydrogenase (NAD) activity.Alcohol dehydrogenase activity, zinc-dependent.Ethanol binding.Retinol dehydrogenase activity.Zinc ion binding. Related diseases Combined oxidative phosphorylation deficiency 6 (COXPD6) [MIM:300816]: A mitochondrial disease resulting in a neurodegenerative disorder characterized by psychomotor delay, hypotonia, areflexia, muscle weakness and wasting. Some patients manifest prenatal ventriculomegaly and severe postnatal encephalomyopathy. {ECO:0000269|PubMed:20362274, ECO:0000269|PubMed:22019070, ECO:0000269|PubMed:25583628, ECO:0000269|PubMed:26004228, ECO:0000269|PubMed:26173962, ECO:0000269|PubMed:27178839}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, X-linked recessive, 4, with or without cerebellar ataxia (CMTX4) [MIM:310490]: A neuromuscular disorder characterized by progressive sensorimotor axonal neuropathy, distal sensory impairment, difficulty walking due to peripheral neuropathy and/or cerebellar ataxia, and deafness due to auditory neuropathy. Additional features include cognitive impairment, cerebellar atrophy, dysarthria, abnormal extraocular movements, tremor, dysmetria and spasticity. The age at onset ranges from infancy to young adulthood. {ECO:0000269|PubMed:23217327, ECO:0000269|PubMed:26004228}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, X-linked, 5, with peripheral neuropathy (DFNX5) [MIM:300614]: A form of hearing loss characterized by absent or severely abnormal auditory brainstem response, abnormal middle ear reflexes, abnormal speech discrimination, loss of outer hair cell function, and cochlear nerve hypoplasia. DFNX5 patients manifest auditory neuropathy with childhood onset, associated with distal sensory impairment affecting the peripheral nervous system. {ECO:0000269|PubMed:25986071}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepimetaphyseal dysplasia, X-linked, with hypomyelinating leukodystrophy (SEMDHL) [MIM:300232]: An X-linked recessive developmental disorder characterized by slowly progressive skeletal and neurologic abnormalities, including short stature, large and deformed joints, significant motor impairment, visual defects, and sometimes cognitive deficits. Affected individuals typically have normal early development in the first year or so of life, followed by development regression and the development of symptoms. Brain imaging shows white matter abnormalities consistent with hypomyelinating leukodystrophy. {ECO:0000269|PubMed:28842795}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03061; DB01711; DB04312; DB04448; DB02249; DB02871; DB02721; DB03020; DB02659; DB04071; DB03559; DB00898; DB01213; DB02131; DB04113; DB00157; DB02822; DB02757; DB03226; DB14487; DB14533; DB14548 Interacts with P00325 EC number 1.1.1.1 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Metal-binding; NAD; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 39693.9 Length 374 Aromaticity 0.06 Instability index 23.06 Isoelectric point 8.53 Charge (pH=7) 5.87 2D Binding mode Binding energy (Kcal/mol) -5.45 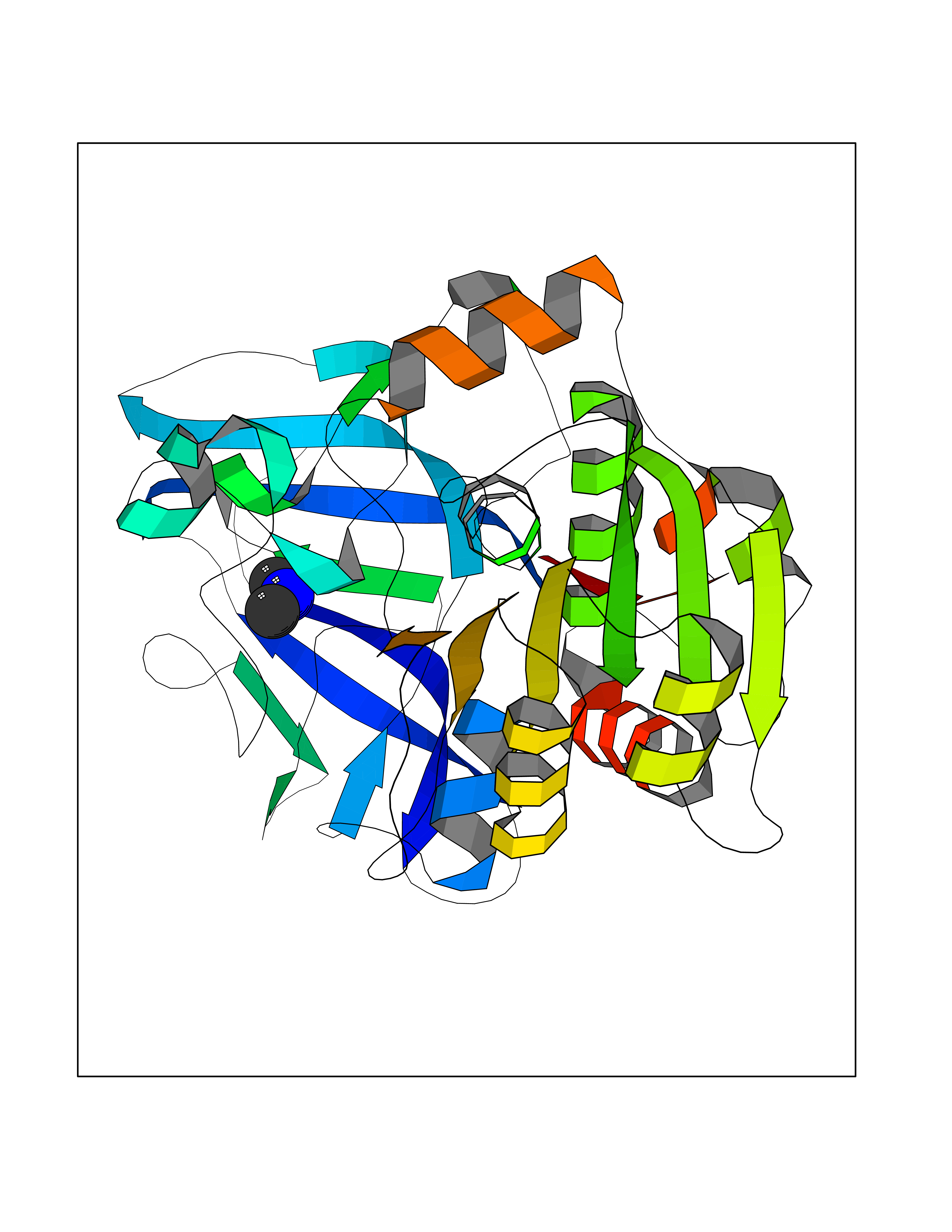 Molscript Map 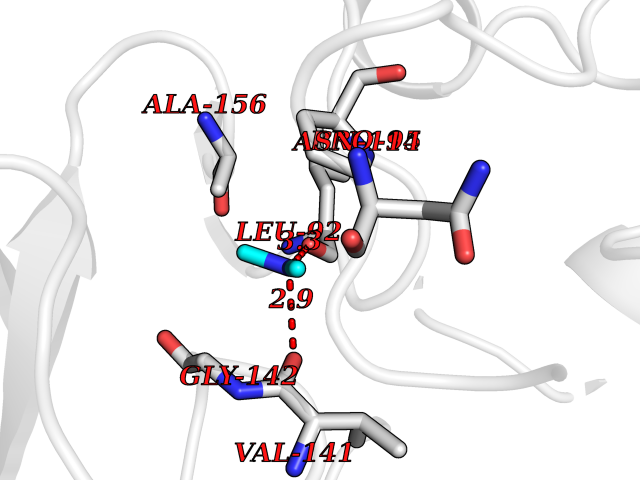 Pymol Map 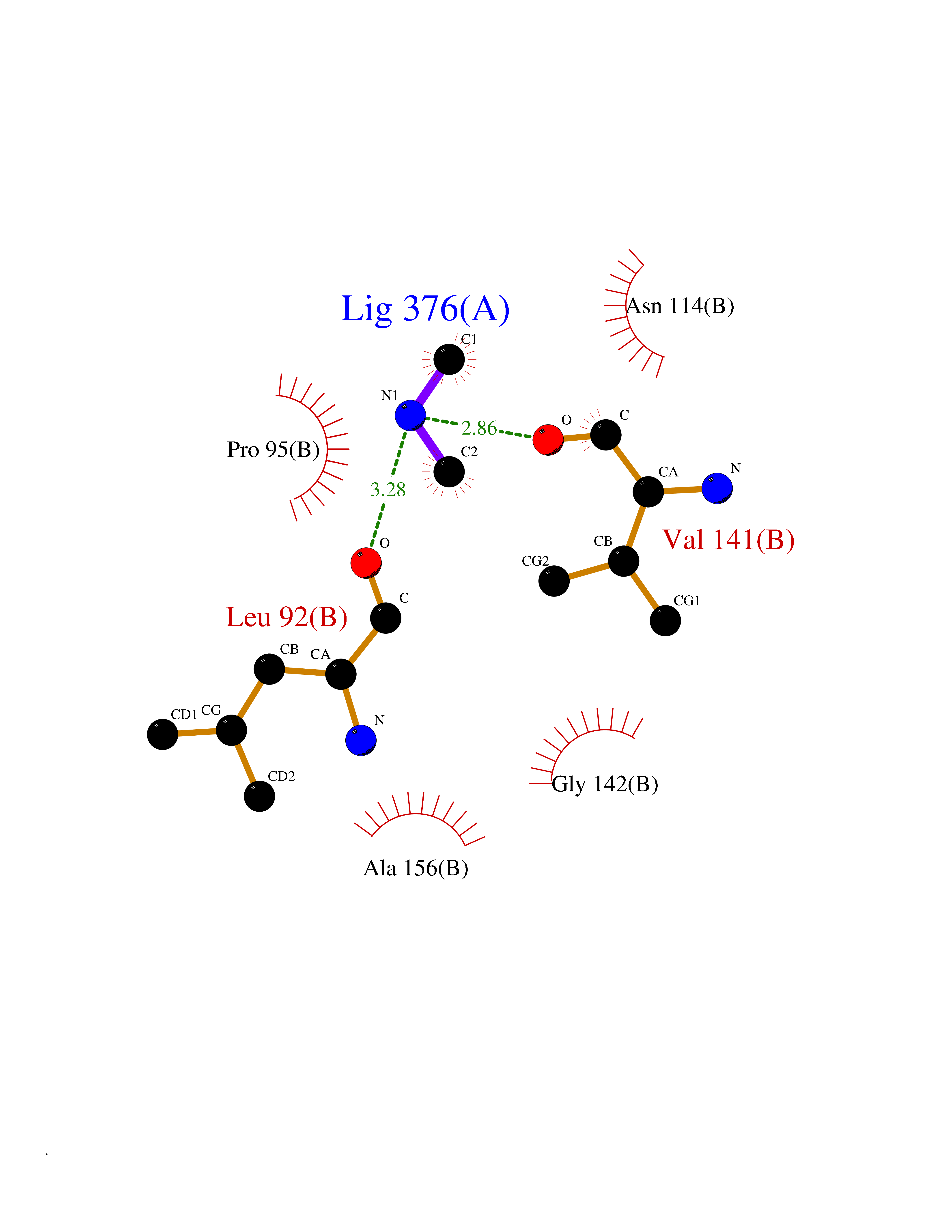 Ligplot Map 3D Binding mode Sequence STAGKVIKCKAAVLWELKKPFSIEEVEVAPPKAHEVRIKMVAAGICRSDEHVVSGNLVTPLPVILGHEAAGIVESVGEGVTTVKPGDKVIPLFTPQCGKCRICKNPESNYCLKNDLGNPRGTLQDGTRRFTCSGKPIHHFVGVSTFSQYTVVDENAVAKIDAASPLEKVCLIGCGFSTGYGSAVKVAKVTPGSTCAVFGLGGVGLSVVMGCKAAGAARIIAVDINKDKFAKAKELGATECINPQDYKKPIQEVLKEMTDGGVDFSFEVIGQLDTMMASLLCCHEACGTSVIVGVPPDSQNLSINPMLLLTGRTWKGAIFGGFKSKESVPKLVADFMAKKFSLDALITNVLPFEKINEGFDLLRSGKSIRTVLTF Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Steroid 17-alpha-monooxygenase (S17AH) | 3SWZ | 3.99 | |
Target general information Gen name CYP17A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Steroid 17-alpha-hydroxylase/17,20 lyase; P450c17; P450-C17; P450 17; CYPXVII; CYP17A1; CYP 17; 17 alpha-Hydroxylase/C17-20-lyase Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function Conversion of pregnenolone and progesterone to their 17- alpha-hydroxylated products and subsequently to dehydroepiandrosterone (DHEA) and androstenedione. Catalyzes both the 17-alpha-hydroxylation and the 17,20-lyase reaction. Involved in sexual development during fetal life and at puberty. Related diseases Adrenal hyperplasia 5 (AH5) [MIM:202110]: A form of congenital adrenal hyperplasia, a common recessive disease due to defective synthesis of cortisol. Congenital adrenal hyperplasia is characterized by androgen excess leading to ambiguous genitalia in affected females, rapid somatic growth during childhood in both sexes with premature closure of the epiphyses and short adult stature. Four clinical types: 'salt wasting' (SW, the most severe type), 'simple virilizing' (SV, less severely affected patients), with normal aldosterone biosynthesis, 'non-classic form' or late-onset (NC or LOAH) and 'cryptic' (asymptomatic). {ECO:0000269|PubMed:10720067, ECO:0000269|PubMed:11549685, ECO:0000269|PubMed:11836339, ECO:0000269|PubMed:12466376, ECO:0000269|PubMed:14671162, ECO:0000269|PubMed:1515452, ECO:0000269|PubMed:1714904, ECO:0000269|PubMed:1740503, ECO:0000269|PubMed:19793597, ECO:0000269|PubMed:24140098, ECO:0000269|PubMed:24498484, ECO:0000269|PubMed:25650406, ECO:0000269|PubMed:2808364, ECO:0000269|PubMed:8027220, ECO:0000269|PubMed:8245018, ECO:0000269|PubMed:8345056, ECO:0000269|PubMed:8396144, ECO:0000269|PubMed:8550762, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05812; DB04630; DB01424; DB09061; DB00882; DB01234; DB14649; DB01026; DB05667; DB14009; DB14011; DB00157; DB01708; DB00396; DB02901 Interacts with NA EC number EC 1.14.14.19 Uniprot keywords 3D-structure; Congenital adrenal hyperplasia; Disease variant; Endoplasmic reticulum; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Steroidogenesis Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 51385.8 Length 453 Aromaticity 0.08 Instability index 32.67 Isoelectric point 8.48 Charge (pH=7) 4.08 2D Binding mode Binding energy (Kcal/mol) -5.45 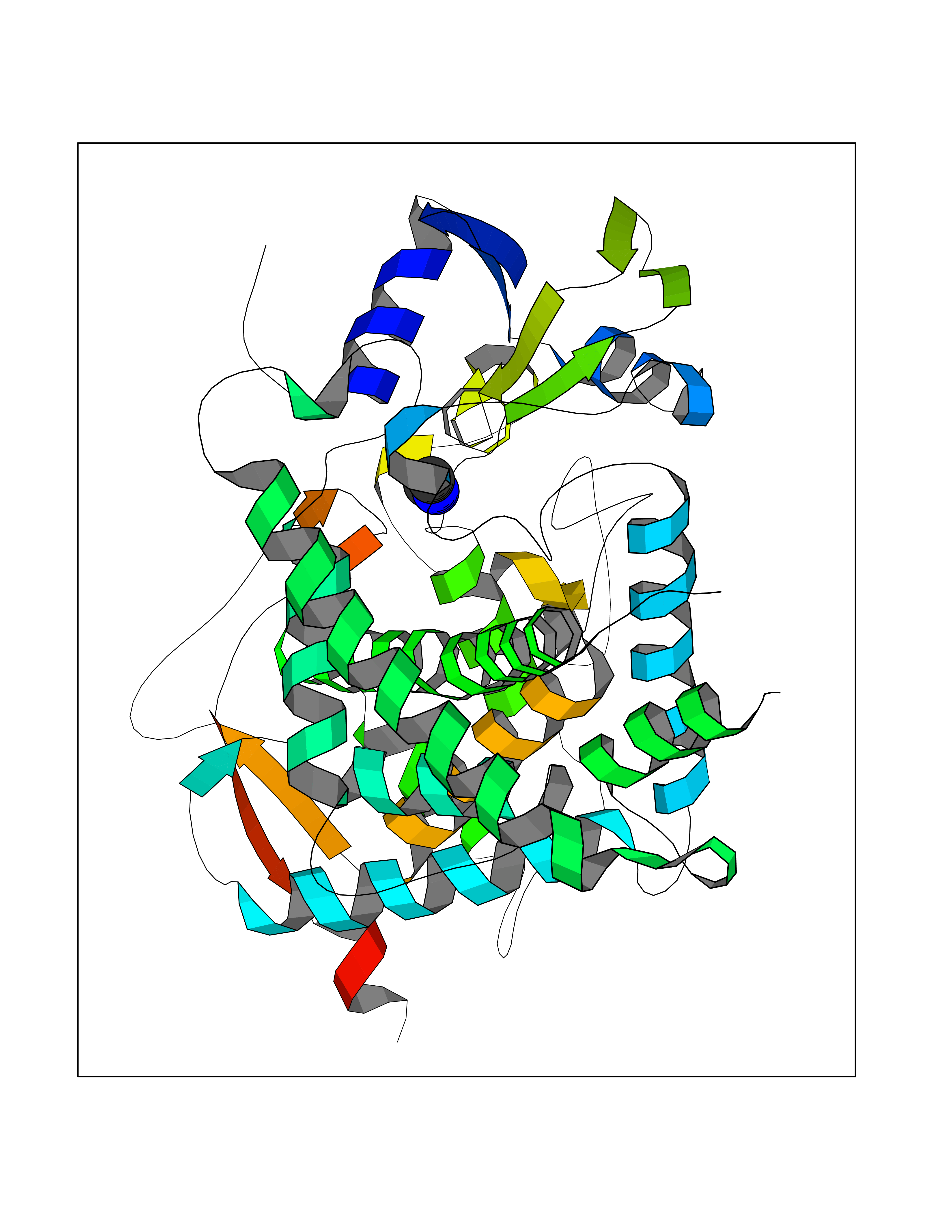 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LPRHGHMHNNFFKLQKKYGPIYSVRMGTKTTVIVGHHQLAKEVLIKKGKDFSGRPQMATLDIASNNRKGIAFADSGAHWQLHRRLAMATFALFKDGDQKLEKIICQEISTLCDMLATHNGQSIDISFPVFVAVTNVISLICFNTSYKNGDPELNVIQNYNEGIIDNLSKDSLVDLVPWLKIFPNKTLEKLKSHVKIRNDLLNKILENYKEKFRSDSITNMLDTLMQAKMNSDDSELLSDNHILTTIGDIFGAGVETTTSVVKWTLAFLLHNPQVKKKLYEEIDQNVGFSRTPTISDRNRLLLLEATIREVLRLRPVAPMLIPHKANVDSSIGEFAVDKGTEVIINLWALHHNEKEWHQPDQFMPERFLNPAGTQLISPSVSYLPFGAGPRSCIGEILARQELFLIMAWLLQRFDLEVPDDGQLPSLEGIPKVVFLIDSFKVKIKVRQAWREAQ Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Poly [ADP-ribose] polymerase 1 (PARP1) | 5WS1 | 3.99 | |
Target general information Gen name PARP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein poly-ADP-ribosyltransferase PARP1; Poly[ADP-ribose] synthetase-1; Poly[ADP-ribose] synthase 1; Poly(ADP-ribose)polymerase-1; PPOL; PARP-1; NAD(+)Poly [ADP-ribose] polymerase-1 ADP-ribosyltrans Protein family ARTD/PARP family Biochemical class Glycosyltransferases Function Mainly mediates glutamate and aspartate ADP-ribosylation of target proteins: the ADP-D-ribosyl group of NAD(+) is transferred to the acceptor carboxyl group of glutamate and aspartate residues and further ADP-ribosyl groups are transferred to the 2'-position of the terminal adenosine moiety, building up a polymer with an average chain length of 20-30 units. Mediates the poly(ADP-ribosyl)ation of a number of proteins, including itself, APLF and CHFR. Also mediates serine ADP-ribosylation of target proteins following interaction with HPF1; HPF1 conferring serine specificity. Probably also catalyzes tyrosine ADP-ribosylation of target proteins following interaction with HPF1. Catalyzes the poly-ADP-ribosylation of histones in a HPF1-dependent manner. Involved in the base excision repair (BER) pathway by catalyzing the poly-ADP-ribosylation of a limited number of acceptor proteins involved in chromatin architecture and in DNA metabolism. ADP-ribosylation follows DNA damage and appears as an obligatory step in a detection/signaling pathway leading to the reparation of DNA strand breaks. In addition to base excision repair (BER) pathway, also involved in double-strand breaks (DSBs) repair: together with TIMELESS, accumulates at DNA damage sites and promotes homologous recombination repair by mediating poly-ADP-ribosylation. In addition to proteins, also able to ADP-ribosylate DNA: catalyzes ADP-ribosylation of DNA strand break termini containing terminal phosphates and a 2'-OH group in single- and double-stranded DNA, respectively. Required for PARP9 and DTX3L recruitment to DNA damage sites. PARP1-dependent PARP9-DTX3L-mediated ubiquitination promotes the rapid and specific recruitment of 53BP1/TP53BP1, UIMC1/RAP80, and BRCA1 to DNA damage sites. Acts as a regulator of transcription: positively regulates the transcription of MTUS1 and negatively regulates the transcription of MTUS2/TIP150. With EEF1A1 and TXK, forms a complex that acts as a T-helper 1 (Th1) cell-specific transcription factor and binds the promoter of IFN-gamma to directly regulate its transcription, and is thus involved importantly in Th1 cytokine production. Involved in the synthesis of ATP in the nucleus, together with NMNAT1, PARG and NUDT5. Nuclear ATP generation is required for extensive chromatin remodeling events that are energy-consuming. Poly-ADP-ribosyltransferase that mediates poly-ADP-ribosylation of proteins and plays a key role in DNA repair. Related diseases Dihydrolipoamide dehydrogenase deficiency (DLDD) [MIM:246900]: An autosomal recessive metabolic disorder characterized biochemically by a combined deficiency of the branched-chain alpha-keto acid dehydrogenase complex (BCKDC), pyruvate dehydrogenase complex (PDC), and alpha-ketoglutarate dehydrogenase complex (KGDC). Clinically, affected individuals have lactic acidosis and neurologic deterioration due to sensitivity of the central nervous system to defects in oxidative metabolism. {ECO:0000269|PubMed:10448086, ECO:0000269|PubMed:11687750, ECO:0000269|PubMed:12925875, ECO:0000269|PubMed:15712224, ECO:0000269|PubMed:16442803, ECO:0000269|PubMed:16770810, ECO:0000269|PubMed:17404228, ECO:0000269|PubMed:20160912, ECO:0000269|PubMed:8506365, ECO:0000269|PubMed:8968745, ECO:0000269|PubMed:9540846, ECO:0000269|PubMed:9934985}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04010; DB03509; DB03072; DB03722; DB03073; DB07787; DB07096; DB07330; DB02498; DB13877; DB02701; DB11793; DB02690; DB09074; DB12332; DB11760; DB00277; DB07232; DB01593; DB14487; DB14533; DB14548 Interacts with Q8IW19; Q7Z2E3; P42574; P49715; Q86WJ1-1; P26358; Q01094; Q96L91; P11308; O60741; P09429; Q13007; Q9BQ69; P08651; Q9Y530; P09874; Q8N2W9; P46063; Q9NTX7; Q14684-1; O95863; P63165; P04637; P0CG48; Q14191; P18887; P54577; Q2M1K9; Q02085 EC number EC 2.4.2.30 Uniprot keywords 3D-structure; Acetylation; ADP-ribosylation; Allosteric enzyme; Apoptosis; Chromosome; Cytoplasm; Direct protein sequencing; DNA damage; DNA repair; DNA-binding; Glycosyltransferase; Immunity; Innate immunity; Isopeptide bond; Metal-binding; NAD; Nucleotidyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 36904 Length 329 Aromaticity 0.08 Instability index 35.9 Isoelectric point 6.83 Charge (pH=7) -0.44 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map  Pymol Map 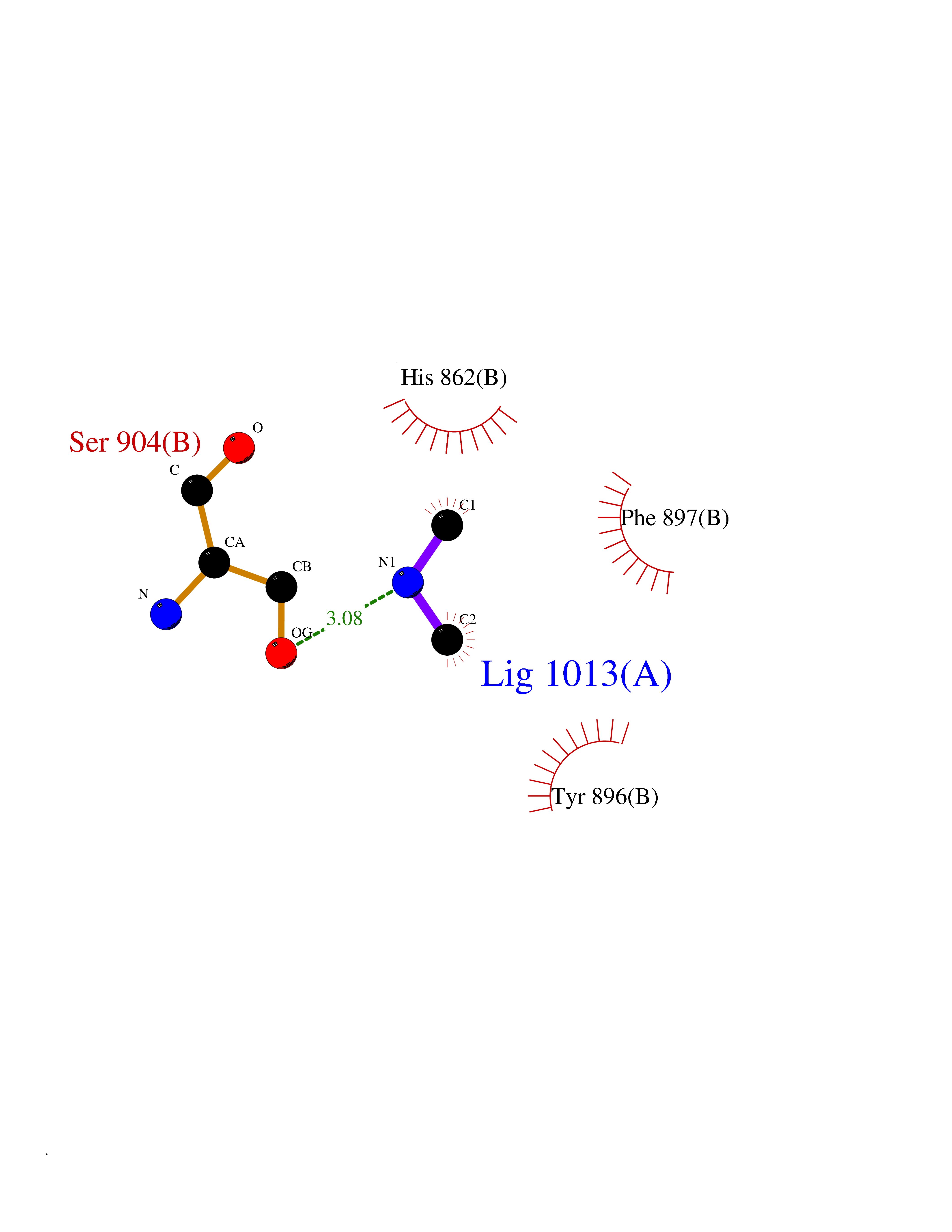 Ligplot Map 3D Binding mode Sequence DLIKMIFDVESMKKAMVEYEIDLQKMPLGKLSKRQIQAAYSILSEVQQAVSQGSDSQILDLSNRFYTLIPHDFGMKKPPLLNNADSVQAKAEMLDNLLDIEVAYSLPIDVNYEKLKTDIKVVDRDSEEAEIIRKYVKNTHATTHNAYDLEVIDIFKIEREGECQRYKPFKQLHNRRLLWHGSRTTNFAGILSQGLRIAPPEAPVTGYMFGKGIYFADMVSKSANYCHTSQGDPIGLILLGEVALGNMYELKHASHISKLPKGKHSVKGLGKTTPDPSANISLDGVDVPLGTGISSGVNDTSLLYNEYIVYDIAQVNLKYLLKLKFNFKT Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Bifunctional methylenetetrahydrofolate dehydrogenase/cyclohydrolase, mitochondrial | 5TC4 | 3.99 | |
Target general information Gen name MTHFD2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NMDMC Protein family Tetrahydrofolate dehydrogenase/cyclohydrolase family Biochemical class Oxidoreductase Function Magnesium ion binding.Methenyltetrahydrofolate cyclohydrolase activity.Methylenetetrahydrofolate dehydrogenase (NAD+) activity.Methylenetetrahydrofolate dehydrogenase (NADP+) activity.Phosphate ion binding. Related diseases Acute hepatic porphyria (AHEPP) [MIM:612740]: A form of porphyria. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. AHP is characterized by attacks of gastrointestinal disturbances, abdominal colic, paralyses and peripheral neuropathy. Most attacks are precipitated by drugs, alcohol, caloric deprivation, infections, or endocrine factors. {ECO:0000269|PubMed:10706561, ECO:0000269|PubMed:1309003, ECO:0000269|PubMed:1569184, ECO:0000269|PubMed:17236137, ECO:0000269|PubMed:2063868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00157; DB00116 Interacts with Q9UJ70-2 EC number 1.5.1.15; 3.5.4.9 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Hydrolase; Isopeptide bond; Magnesium; Mitochondrion; Multifunctional enzyme; NAD; NADP; One-carbon metabolism; Oxidoreductase; Proteomics identification; Reference proteome; Transit peptide; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 31600.3 Length 292 Aromaticity 0.03 Instability index 27.9 Isoelectric point 8 Charge (pH=7) 1.64 2D Binding mode Binding energy (Kcal/mol) -5.44 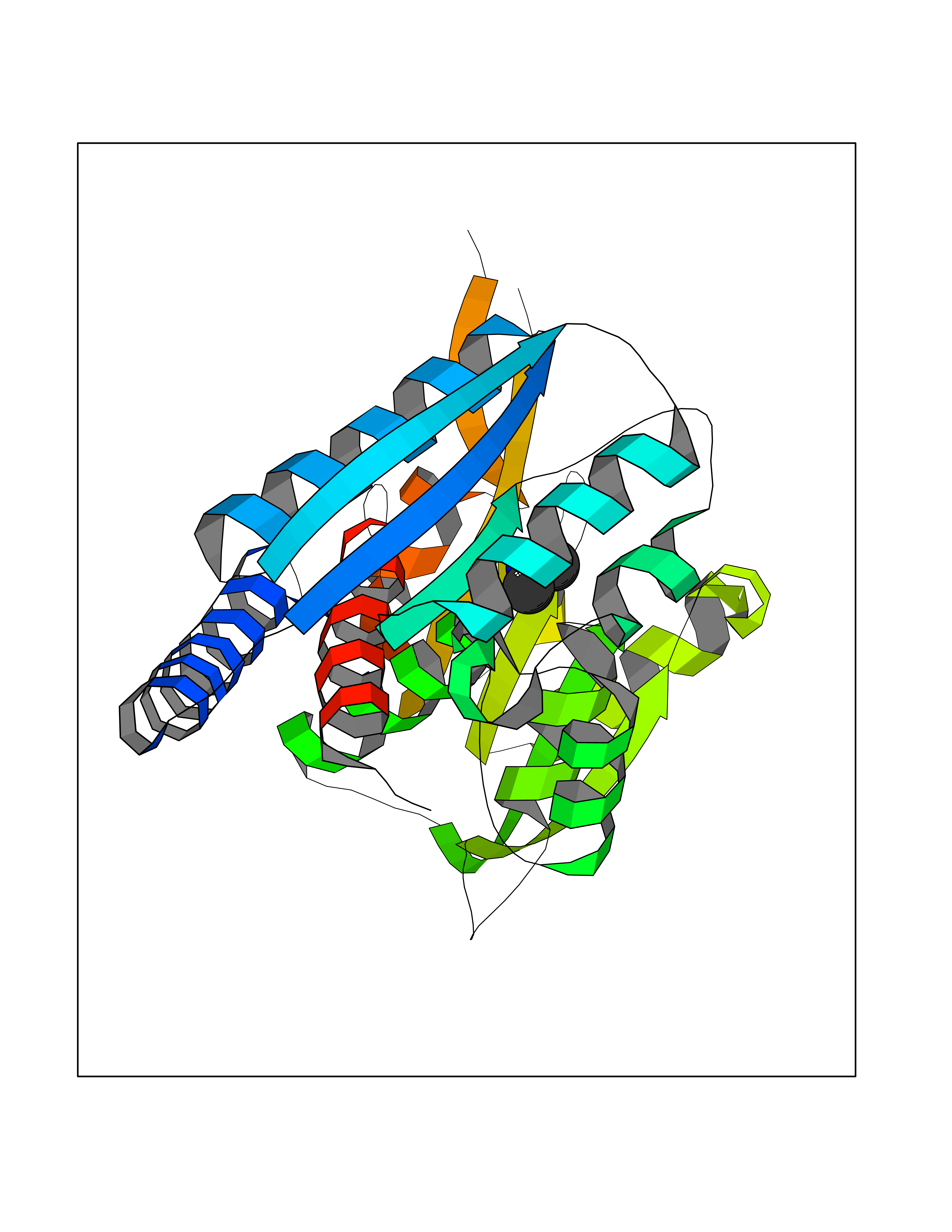 Molscript Map 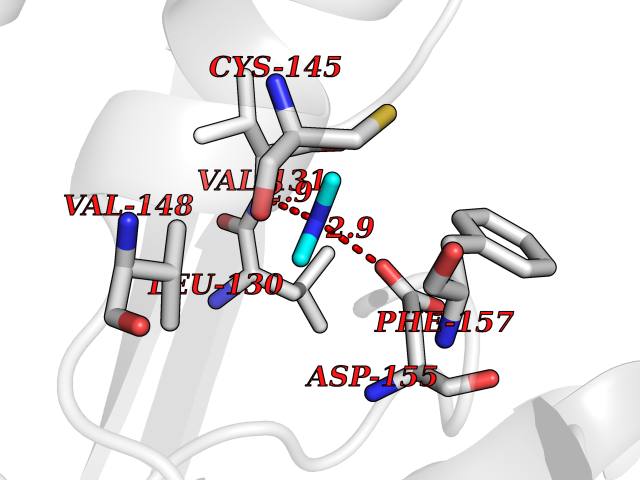 Pymol Map 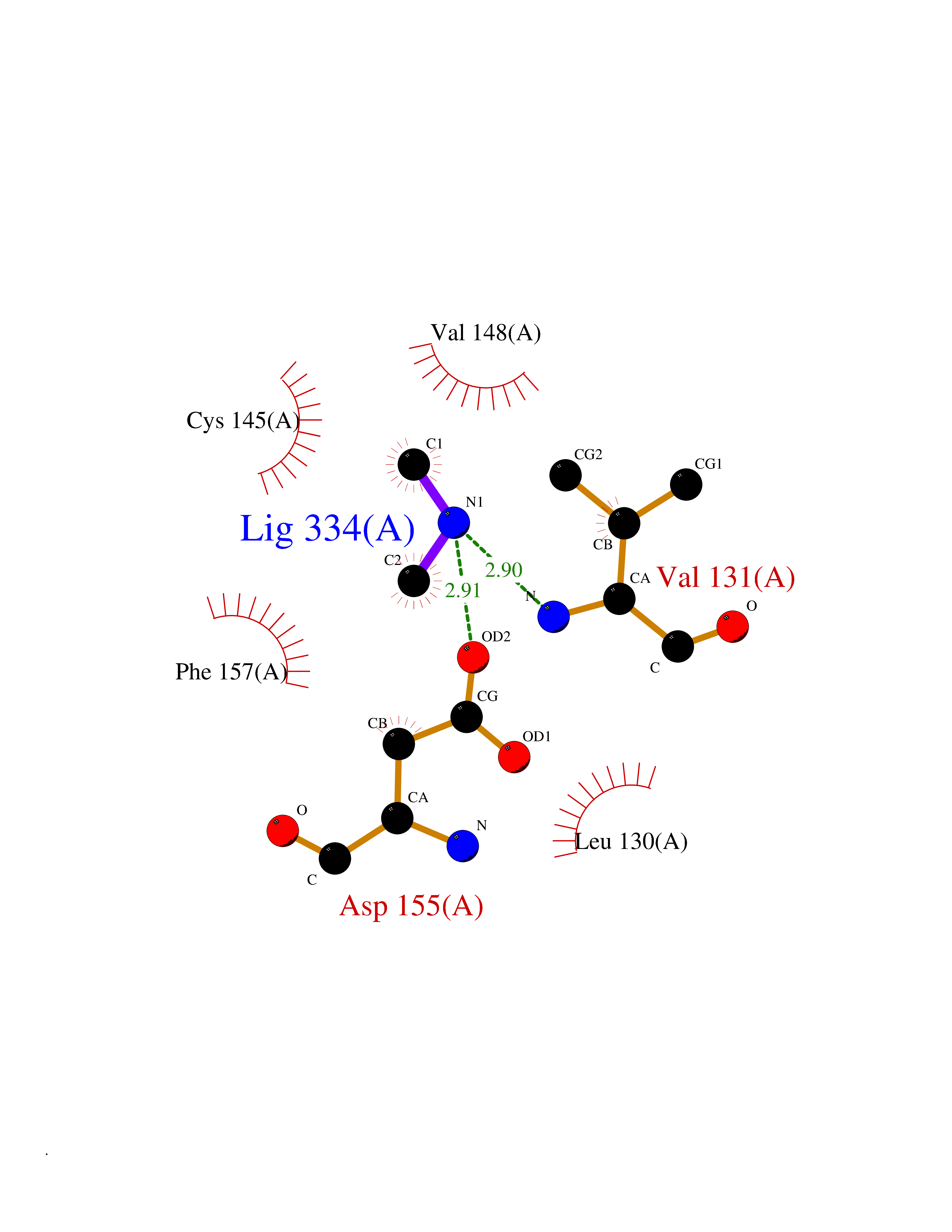 Ligplot Map 3D Binding mode Sequence EAVVISGRKLAQQIKQEVRQEVEEWVASGNKRPHLSVILVGENPASHSYVLNKTRAAAVVGINSETIMKPASISEEELLNLINKLNNDDNVDGLLVQLPLPEHIDERRICNAVSPDKDVDGFHVINVGRMCLDQYSMLPATPWGVWEIIKRTGIPTLGKNVVVAGRSKNVGMPIAMLLHTDGAHERPGGDATVTISHRYTPKEQLKKHTILADIVISAAGIPNLITADMIKEGAAVIDVGINRVHKPKLVGDVDFEGVRQKAGYITPVPGGVGPMTVAMLMKNTIIAAKKVL Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Chloramphenicol acetyltransferase 3 | 3CLA | 3.99 | |
Target general information Gen name cat3 Organism Escherichia coli Uniprot ID TTD ID NA Synonyms NA Protein family Chloramphenicol acetyltransferase family Biochemical class Transferase (acyltransferase) Function Chloramphenicol O-acetyltransferase activity. Related diseases Ornithine carbamoyltransferase deficiency (OTCD) [MIM:311250]: An X-linked disorder of the urea cycle which causes a form of hyperammonemia. Mutations with no residual enzyme activity are always expressed in hemizygote males by a very severe neonatal hyperammonemic coma that generally proves to be fatal. Heterozygous females are either asymptomatic or express orotic aciduria spontaneously or after protein intake. The disorder is treatable with supplemental dietary arginine and low protein diet. The arbitrary classification of patients into the 'neonatal' group (clinical hyperammonemia in the first few days of life) and 'late' onset (clinical presentation after the neonatal period) has been used to differentiate severe from mild forms. {ECO:0000269|PubMed:10070627, ECO:0000269|PubMed:10502831, ECO:0000269|PubMed:10737985, ECO:0000269|PubMed:11793483, ECO:0000269|PubMed:1480464, ECO:0000269|PubMed:1671317, ECO:0000269|PubMed:1721894, ECO:0000269|PubMed:2347583, ECO:0000269|PubMed:2474822, ECO:0000269|PubMed:2556444, ECO:0000269|PubMed:3170748, ECO:0000269|PubMed:7474905, ECO:0000269|PubMed:7951259, ECO:0000269|PubMed:8019569, ECO:0000269|PubMed:8081373, ECO:0000269|PubMed:8081398, ECO:0000269|PubMed:8099056, ECO:0000269|PubMed:8112735, ECO:0000269|PubMed:8530002, ECO:0000269|PubMed:8807340, ECO:0000269|PubMed:8830175, ECO:0000269|PubMed:8956038, ECO:0000269|PubMed:8956045, ECO:0000269|PubMed:9065786, ECO:0000269|PubMed:9143919, ECO:0000269|PubMed:9266388, ECO:0000269|PubMed:9286441, ECO:0000269|PubMed:9452024, ECO:0000269|PubMed:9452049, ECO:0000269|PubMed:9452065, ECO:0000269|Ref.32, ECO:0000269|Ref.43}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00446; DB07565; DB02703 Interacts with NA EC number 2.3.1.28 Uniprot keywords 3D-structure; Acyltransferase; Antibiotic resistance; Direct protein sequencing; Plasmid; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 24993.2 Length 213 Aromaticity 0.14 Instability index 32.26 Isoelectric point 5.78 Charge (pH=7) -4.91 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MNYTKFDVKNWVRREHFEFYRHRLPCGFSLTSKIDITTLKKSLDDSAYKFYPVMIYLIAQAVNQFDELRMAIKDDELIVWDSVDPQFTVFHQETETFSALSCPYSSDIDQFMVNYLSVMERYKSDTKLFPQGVTPENHLNISALPWVNFDSFNLNVANFTDYFAPIITMAKYQQEGDRLLLPLSVQVHHAVCDGFHVARFINRLQELCNSKLK Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Urease subunit alpha | 1FWE | 3.99 | |
Target general information Gen name ureC Organism Klebsiella aerogenes (Enterobacter aerogenes) Uniprot ID TTD ID NA Synonyms NA Protein family Metallo-dependent hydrolases superfamily, Urease alpha subunit family Biochemical class Hydrolase Function Nickel cation binding.Urease activity. Related diseases Can contribute to cancer cell survival, proliferation, migration, and invasion, and tumor angiogenesis and metastasis. May contribute to cancer pathogenesis by promoting inflammatory responses and recruitment of tumor-infiltrating macrophages.; DISEASE: Abnormally high expression of soluble isoforms (isoform 2, isoform 3 or isoform 4) may be a cause of preeclampsia. Drugs (DrugBank ID) DB00551; DB05265 Interacts with P18316 EC number 3.5.1.5 Uniprot keywords 3D-structure; Cytoplasm; Hydrolase; Metal-binding; Nickel Protein physicochemical properties Chain ID C Molecular weight (Da) 80688.3 Length 753 Aromaticity 0.06 Instability index 28.4 Isoelectric point 5.58 Charge (pH=7) -21.17 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map 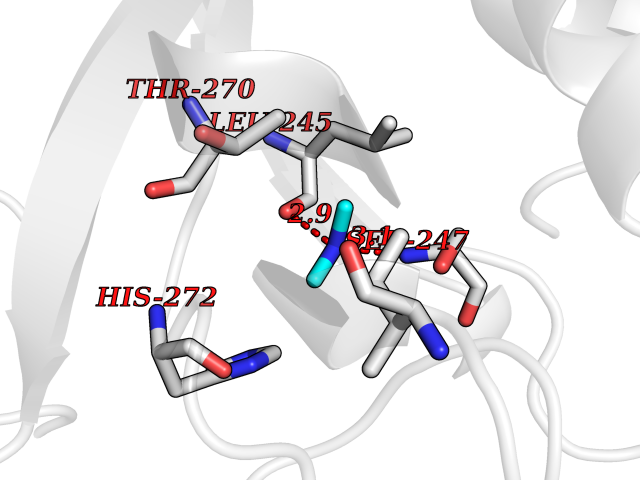 Pymol Map 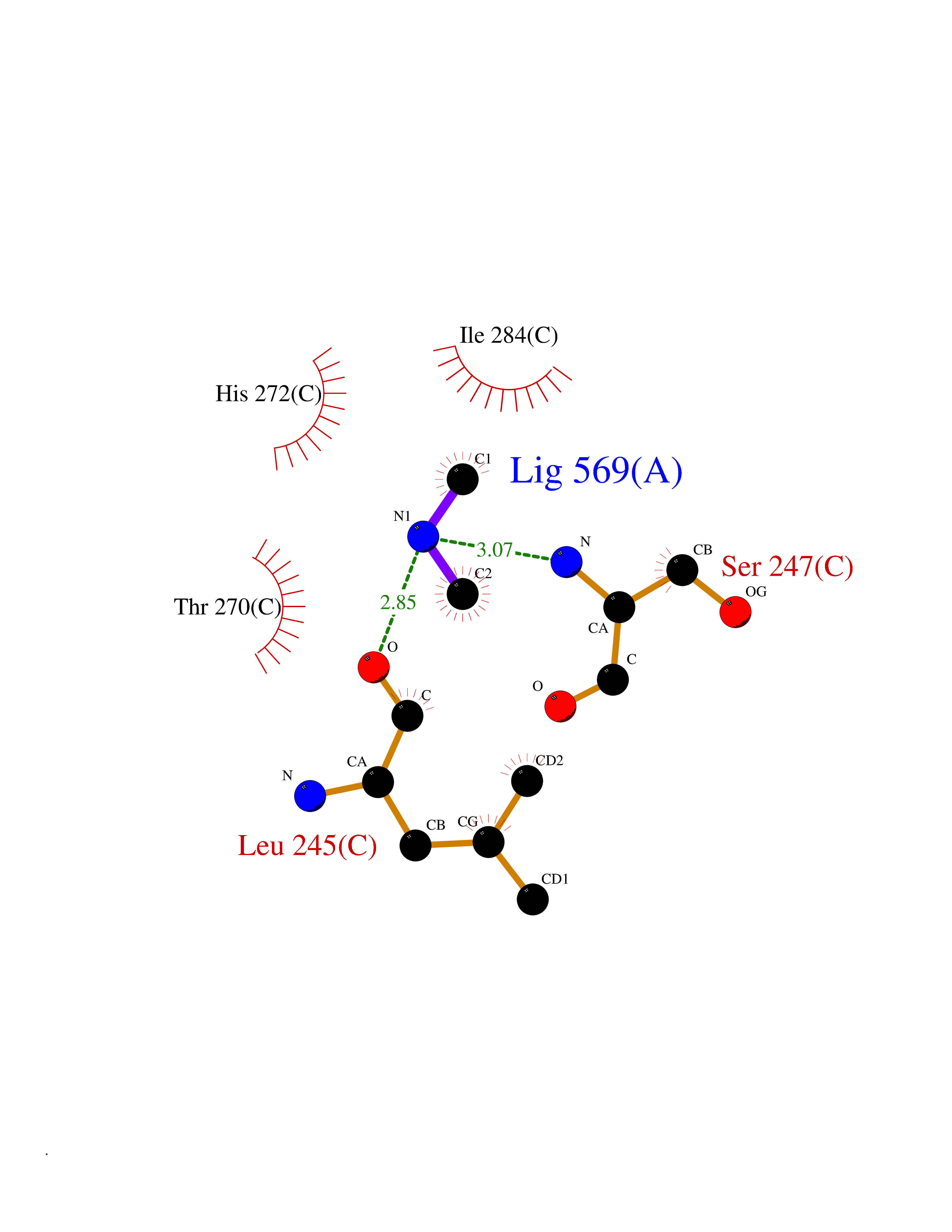 Ligplot Map 3D Binding mode Sequence MELTPREKDKLLLFTAALVAERRLARGLKLNYPESVALISAFIMEGARDGKSVASLMEEGRHVLTREQVMEGVPEMIPDIQVEATFPDGSKLVTVHNPIISNISRQAYADMFGPTVGDKVRLADTELWIEVEDDLTTYGEEVKFGGGKVIRDGMGQGQMLAADCVDLVLTNALIVDHWGIVKADIGVKDGRIFAIGKAGNPDIQPNVTIPIGAATEVIAAEGKIVTAGGIDTHIHWICPQQAEEALVSGVTTMVGGGTGPAAGTHATTCTPGPWYISRMLQAADSLPVNIGLLGKGNVSQPDALREQVAAGVIGLXIHEDWGATPAAIDCALTVADEMDIQVALHSDTLNESGFVEDTLAAIGGRTIHTFHTEGAGGGHAPDIITACAHPNILPSSTNPTLPYTLNTIDEHLDMLMFAESRIRRETIAAEDVLHDLGAFSLTSSDSQAMGRVGEVILRTWQVAHRMKVQRGALAEETGDNDNFRVKRYIAKYTINPALTHGIAHEVGSIEVGKLADLVVWSPAFFGVKPATVIKGGMIAIAPMGDINASIPTPQPVHYRPMFGALGSARHHCRLTFLSQAAAANGVAERLNLRSAIAVVKGCRTVQKADMVHNSLQPNITVDAQTYEVRVDGELITSEPADVLPMAQRYFLFMIPGEYHVKPGQIALNTGRATCRVVVENHGDRPIQVGSHYHFAEVNPALKFDRQQAAGYRLNIPAGTAVRFEPGQKREVELVAFAGHRAVFGFRGEVMGPL Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Monoamine oxidase type B (MAO-B) | 2V5Z | 3.99 | |
Target general information Gen name MAOB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MAO-B; Amine oxidase [flavin-containing] B Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. MAOB preferentially degrades benzylamine and phenylethylamine. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB08176; DB02211; DB08516; DB08480; DB01472; DB04307; DB07512; DB07513; DB00915; DB00182; DB06698; DB04889; DB00215; DB09130; DB04147; DB00988; DB01363; DB00668; DB01175; DB02509; DB03147; DB14914; DB00614; DB04818; DB02095; DB01247; DB00601; DB01577; DB01442; DB01171; DB08082; DB02643; DB04677; DB03894; DB08804; DB04820; DB00184; DB04821; DB12612; DB01626; DB00780; DB00191; DB00388; DB01132; DB00721; DB01168; DB01367; DB09363; DB06654; DB01037; DB01104; DB14569; DB09042; DB00752; DB16446; DB09185; DB04832; DB00909 Interacts with P55212; P28329-3; Q8NI60; Q5RI15; Q92915-2; P22607; Q53GS7; P06396; P01112; O14901; P13473-2; P21397; Q9BVL2; O75400-2; P62826; Q6NTF9-3; Q9Y371; Q7Z699; Q9UMX0; Q9Y649 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; FAD; Flavoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Oxidoreductase; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56019.9 Length 494 Aromaticity 0.09 Instability index 34.81 Isoelectric point 6.51 Charge (pH=7) -2.2 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map 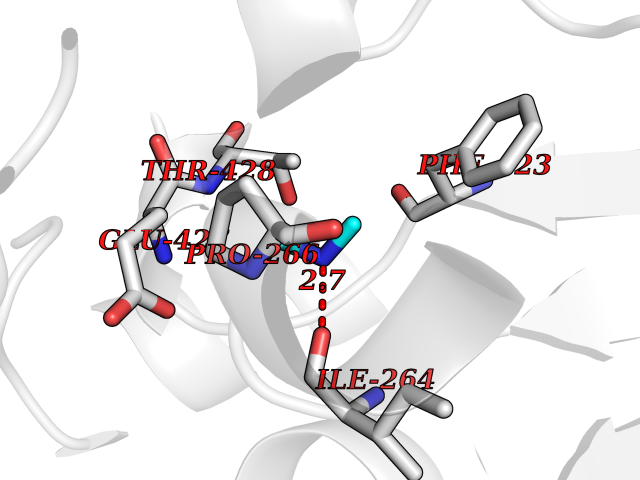 Pymol Map 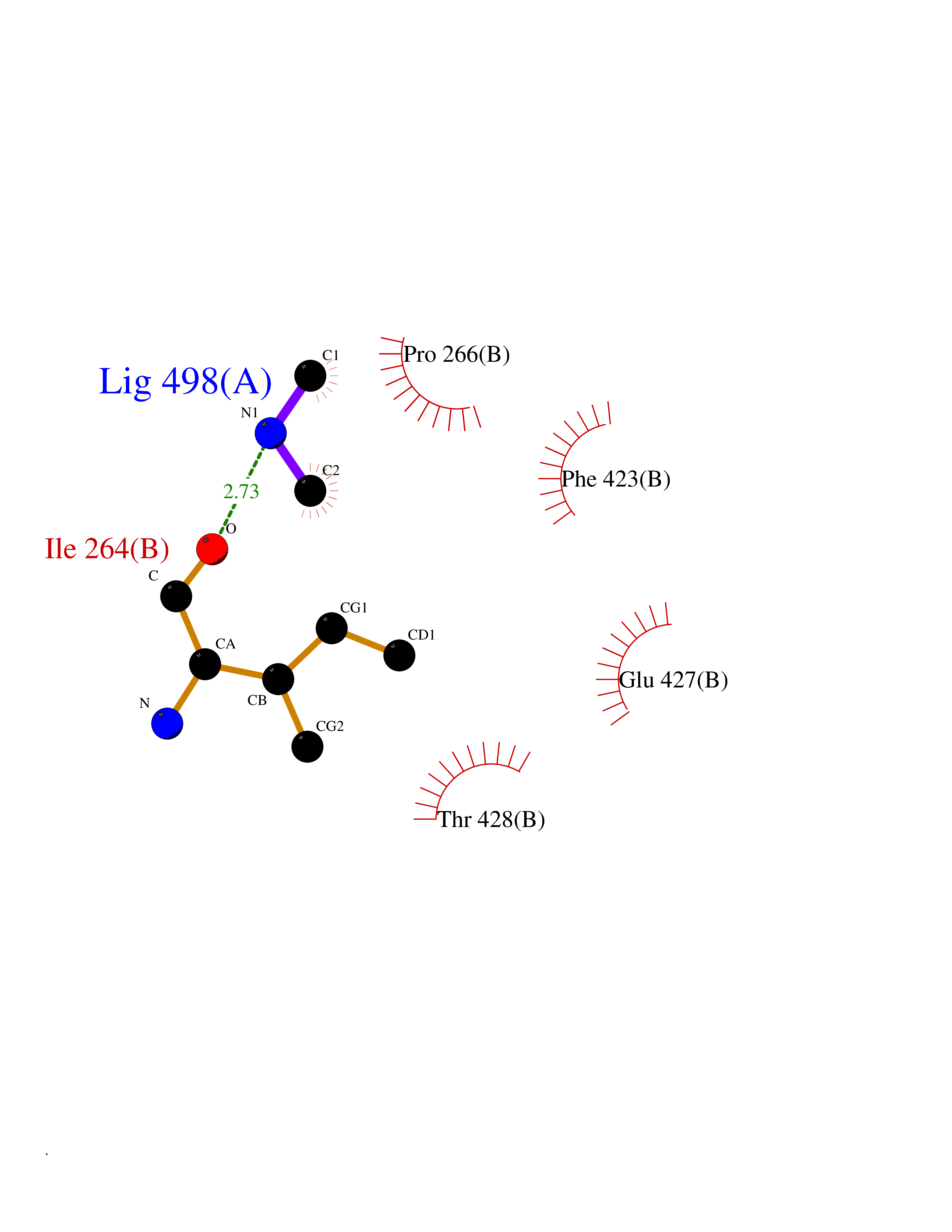 Ligplot Map 3D Binding mode Sequence NKCDVVVVGGGISGMAAAKLLHDSGLNVVVLEARDRVGGRTYTLRNQKVKYVDLGGSYVGPTQNRILRLAKELGLETYKVNEVERLIHHVKGKSYPFRGPFPPVWNPITYLDHNNFWRTMDDMGREIPSDAPWKAPLAEEWDNMTMKELLDKLCWTESAKQLATLFVNLCVTAETHEVSALWFLWYVKQCGGTTRIISTTNGGQERKFVGGSGQVSERIMDLLGDRVKLERPVIYIDQTRENVLVETLNHEMYEAKYVISAIPPTLGMKIHFNPPLPMMRNQMITRVPLGSVIKCIVYYKEPFWRKKDYCGTMIIDGEEAPVAYTLDDTKPEGNYAAIMGFILAHKARKLARLTKEERLKKLCELYAKVLGSLEALEPVHYEEKNWCEEQYSGGCYTTYFPPGILTQYGRVLRQPVDRIYFAGTETATHWSGYMEGAVEAGERAAREILHAMGKIPEDEIWQSEPESVDVPAQPITTTFLERHLPSVPGLLRLI Hydrogen bonds contact Hydrophobic contact | ||||