Job Results:
Ligand
Structure
Job ID
3c17296c67deee94355bca3744afb8a3
Job name
NA
Time
2025-02-13 15:25:56
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Cathepsin F (CTSF) | 1M6D | 4.00 | |
Target general information Gen name CTSF Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CATSF Protein family Peptidase C1 family Biochemical class Peptidase Function Has also been implicated in tumor invasion and metastasis. Thiol protease which is believed to participate in intracellular degradation and turnover of proteins. Related diseases Ceroid lipofuscinosis, neuronal, 13 (Kufs type) (CLN13) [MIM:615362]: A form of neuronal ceroid lipofuscinosis characterized by adult onset of progressive cognitive decline and motor dysfunction leading to dementia and often early death. Some patients develop seizures. Neuronal ceroid lipofuscinoses are progressive neurodegenerative, lysosomal storage diseases characterized by intracellular accumulation of autofluorescent liposomal material. CLN13 inheritance is autosomal recessive. {ECO:0000269|PubMed:23297359}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02243; DB01871; DB01810; DB08775; DB03536; DB07913; DB03691; DB03573 Interacts with NA EC number EC 3.4.22.41 Uniprot keywords 3D-structure; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lysosome; Neurodegeneration; Neuronal ceroid lipofuscinosis; Protease; Proteomics identification; Reference proteome; Signal; Thiol protease; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 23634.4 Length 214 Aromaticity 0.11 Instability index 39.11 Isoelectric point 5.82 Charge (pH=7) -2.9 2D Binding mode Binding energy (Kcal/mol) -5.46 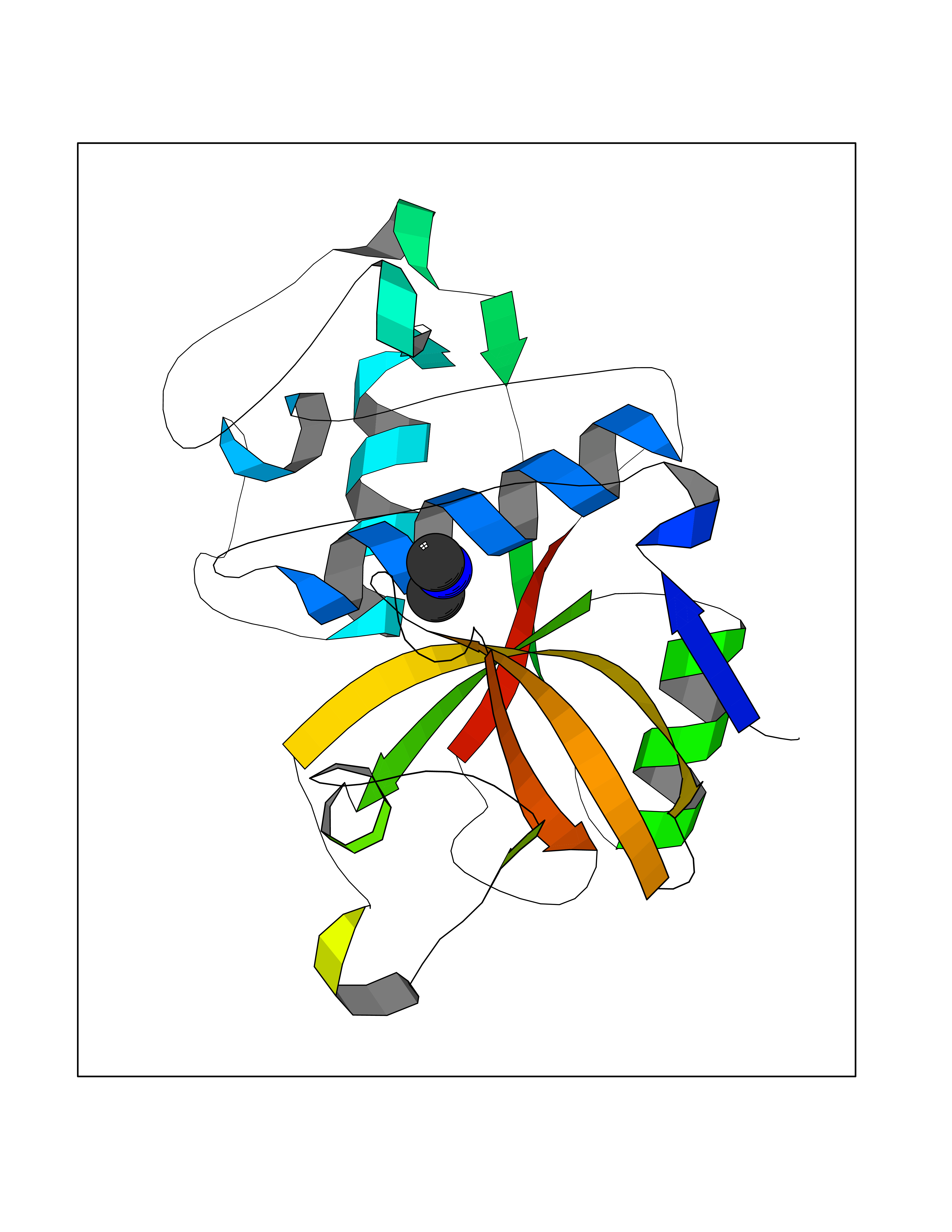 Molscript Map 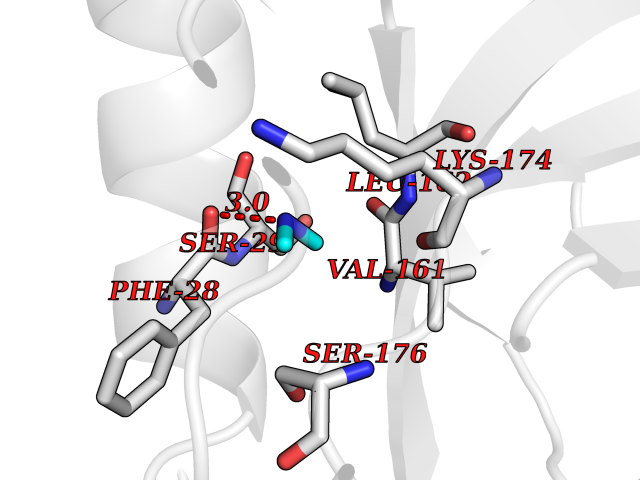 Pymol Map 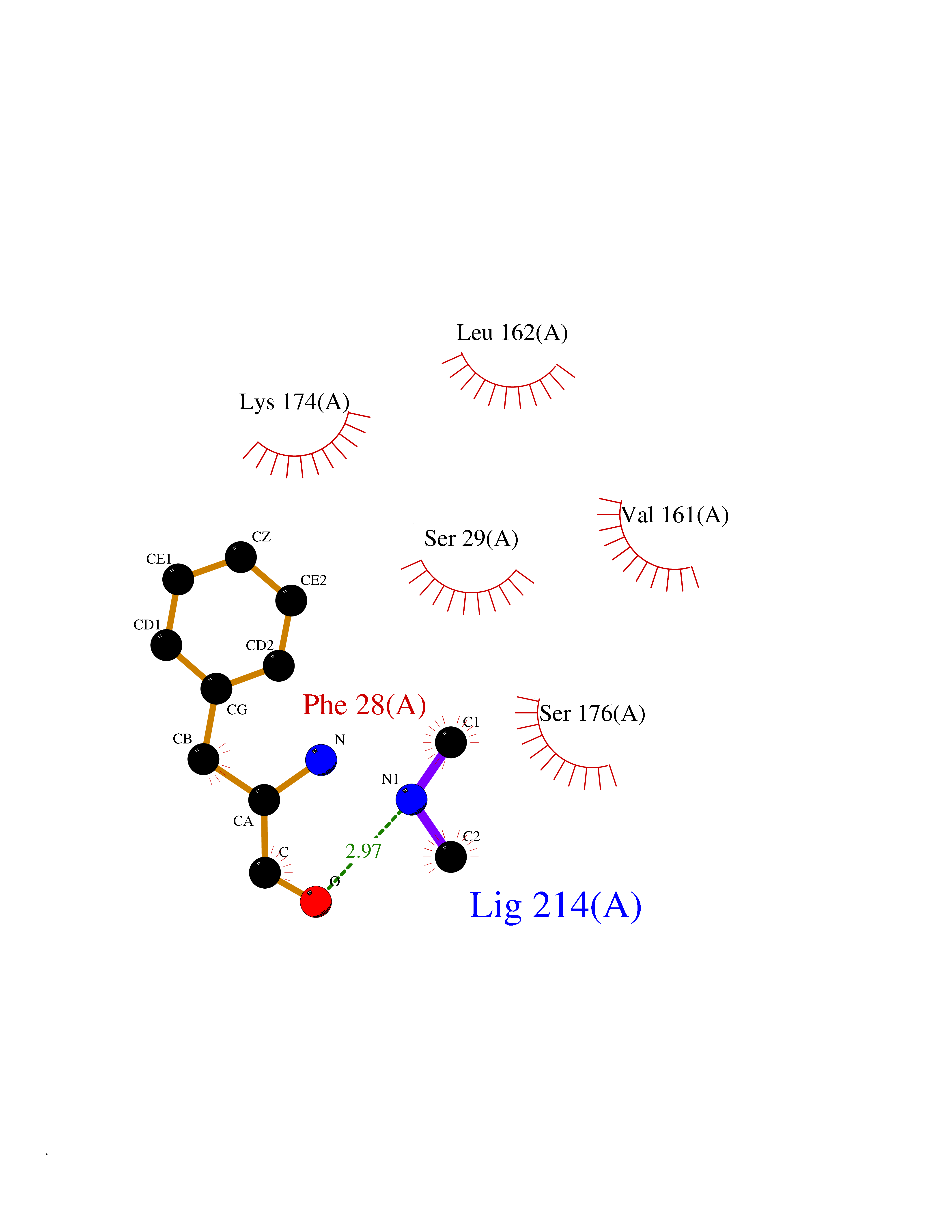 Ligplot Map 3D Binding mode Sequence APPEWDWRSKGAVTKVKDQGMCGSCWAFSVTGNVEGQWFLNQGTLLSLSEQELLDCDKMDKACMGGLPSNAYSAIKNLGGLETEDDYSYQGHMQSCQFSAEKAKVYIQDSVELSQNEQKLAAWLAKRGPISVAINAFGMQFYRHGISRPLRPLCSPWLIDHAVLLVGYGQRSDVPFWAIKNSWGTDWGEKGYYYLHRGSGACGVNTMASSAVVD Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Histone-lysine N-methyltransferase (HLNM) | 3QOW | 4.00 | |
Target general information Gen name DOT1L Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine N-methyltransferase 4; KMT4; KIAA1814; Histone-lysine N-methyltransferase, H3 lysine-79 specific; Histone H3-K79 methyltransferase; H3-K79-HMTase; DOT1-like protein Protein family Class I-like SAM-binding methyltransferase superfamily, DOT1 family Biochemical class Methyltransferase Function Histone methyltransferase. Methylates 'Lys-79' of histone H3. Nucleosomes are preferred as substrate compared to free histones. Binds to DNA. Related diseases Defects in DOTL1 are associated with an autosomal dominant form of global developmental delay and intellectual disability, with or without one or more major congenital anomalies (PubMed:37827158). The patient phenotypes are characterized by central nervous system (CNS) dysfunction, such as mild motor delay and significant speech and language delay, and a range of congenital anomalies, including brain structural anomalies, cardiac defects, varied urogenital features and growth restriction (PubMed:37827158). Variants may cause a gain-of-function effect leading to an increase in cellular H3K79 methylation levels (PubMed:37827158). {ECO:0000269|PubMed:37827158}. Drugs (DrugBank ID) NA Interacts with Q03111; P42568 EC number EC 2.1.1.43 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Disease variant; DNA-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 37367.1 Length 322 Aromaticity 0.11 Instability index 32.71 Isoelectric point 6.03 Charge (pH=7) -5.25 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map 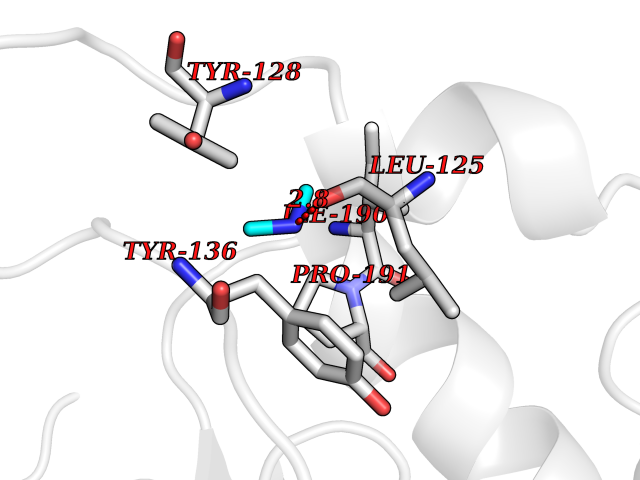 Pymol Map 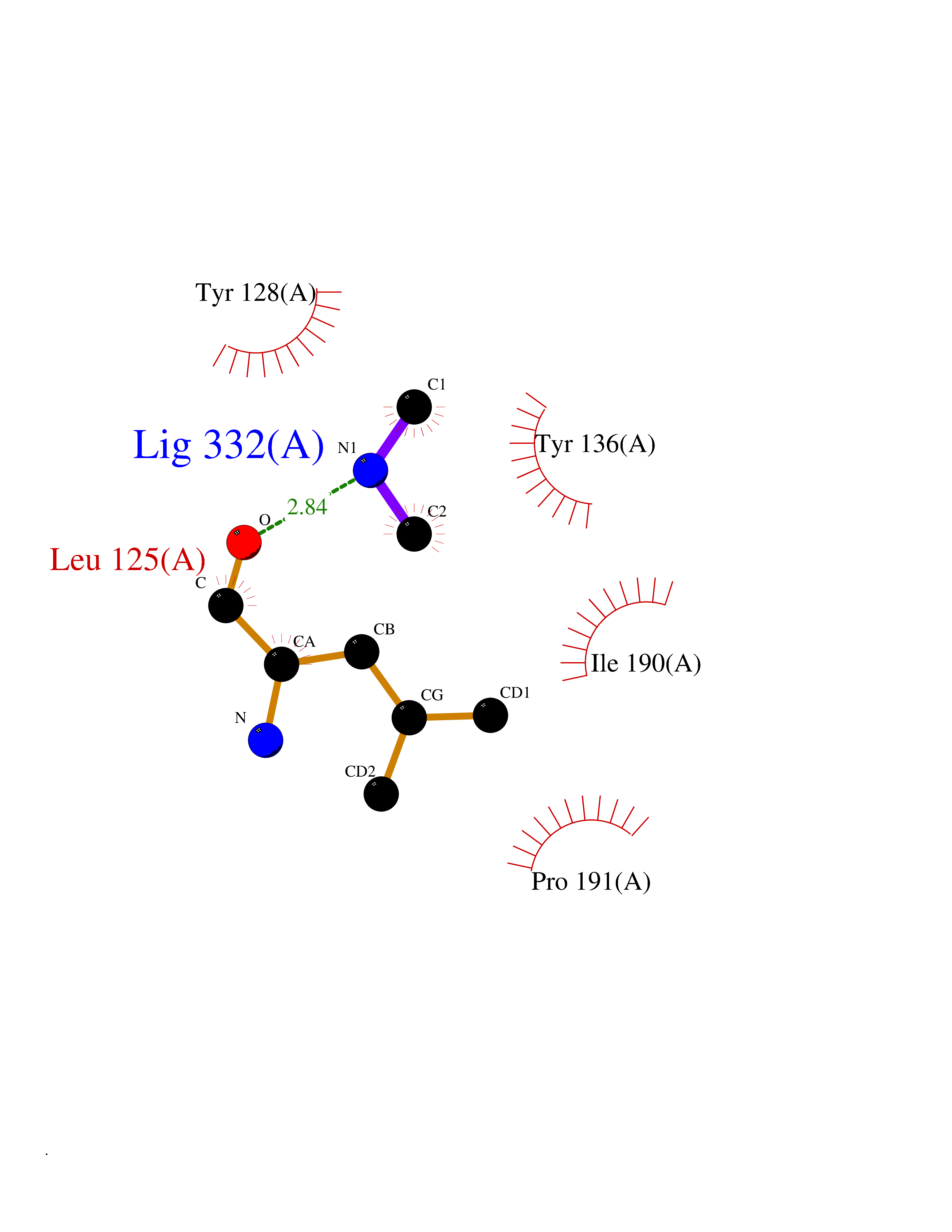 Ligplot Map 3D Binding mode Sequence KLELRLKSPVGAEPAVYPWPLPVYDKHHDAAHEIIETIRWVCEEIPDLKLAMENYLIDYDTKSFESMQRLCDKYNRAIDSIHQLWKGTTQPMKLNTRPSTGLLRHILQQVYNHSVTDPEKLNNYEPFSPEVYGETSFDLVAQMIDEIKMTDDDLFVDLGSGVGQVVLQVAAATNCKHHYGVEKADIPAKYAETMDREFRKWMKWYGKKHAEYTLERGDFLSEEWRERIANTSVIFVNNFAFGPEVDHQLKERFANMKEGGRIVSSKPFAPLNFRINSRNLSDIGTIMRVVELSPLKWTGKPVSYYLHTIDRTILENYFSSLK Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Geranyltranstransferase (FDPS) | 4KPJ | 4.00 | |
Target general information Gen name FDPS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms KIAA1293; Geranylgeranyl pyrophosphate synthase; Geranylgeranyl diphosphate synthase; GGPS1; GGPPSase; GGPP synthase; Farnesyltranstransferase; Farnesyl pyrophosphate synthase; Farnesyl diphosphate sy Protein family FPP/GGPP synthase family Biochemical class Alkyl aryl transferase Function FPP also serves as substrate for protein farnesylation and geranylgeranylation. Catalyzes the sequential condensation of isopentenyl pyrophosphate with the allylic pyrophosphates, dimethylallyl pyrophosphate, and then with the resultant geranylpyrophosphate to the ultimate product farnesyl pyrophosphate. Key enzyme in isoprenoid biosynthesis which catalyzes the formation of farnesyl diphosphate (FPP), a precursor for several classes of essential metabolites including sterols, dolichols, carotenoids, and ubiquinones. Related diseases Porokeratosis 9, multiple types (POROK9) [MIM:616631]: A form of porokeratosis, a disorder of faulty keratinization characterized by one or more atrophic patches surrounded by a distinctive hyperkeratotic ridgelike border called the cornoid lamella. The keratotic lesions can progress to overt cutaneous neoplasms, typically squamous cell carcinomas. Multiple clinical variants of porokeratosis are recognized, including porokeratosis of Mibelli, linear porokeratosis, disseminated superficial actinic porokeratosis, palmoplantar porokeratosis, and punctate porokeratosis. Different clinical presentations can be observed among members of the same family. Individuals expressing more than one variant have also been reported. {ECO:0000269|PubMed:26202976}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00630; DB01785; DB07780; DB02552; DB07841; DB00710; DB06255; DB04714; DB02508; DB06548; DB00282; DB00884; DB00399 Interacts with O95870; P54253; Q6ZMZ0; Q9BRI3; Q8WWF3 EC number EC 2.5.1.10 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Cytoplasm; Disease variant; Host-virus interaction; Hydroxylation; Isoprene biosynthesis; Lipid biosynthesis; Lipid metabolism; Magnesium; Metal-binding; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 38705 Length 338 Aromaticity 0.12 Instability index 44.66 Isoelectric point 5.1 Charge (pH=7) -9.92 2D Binding mode Binding energy (Kcal/mol) -5.45  Molscript Map 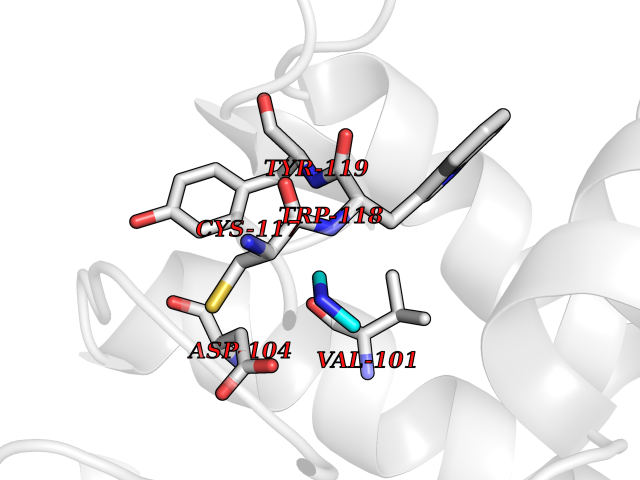 Pymol Map 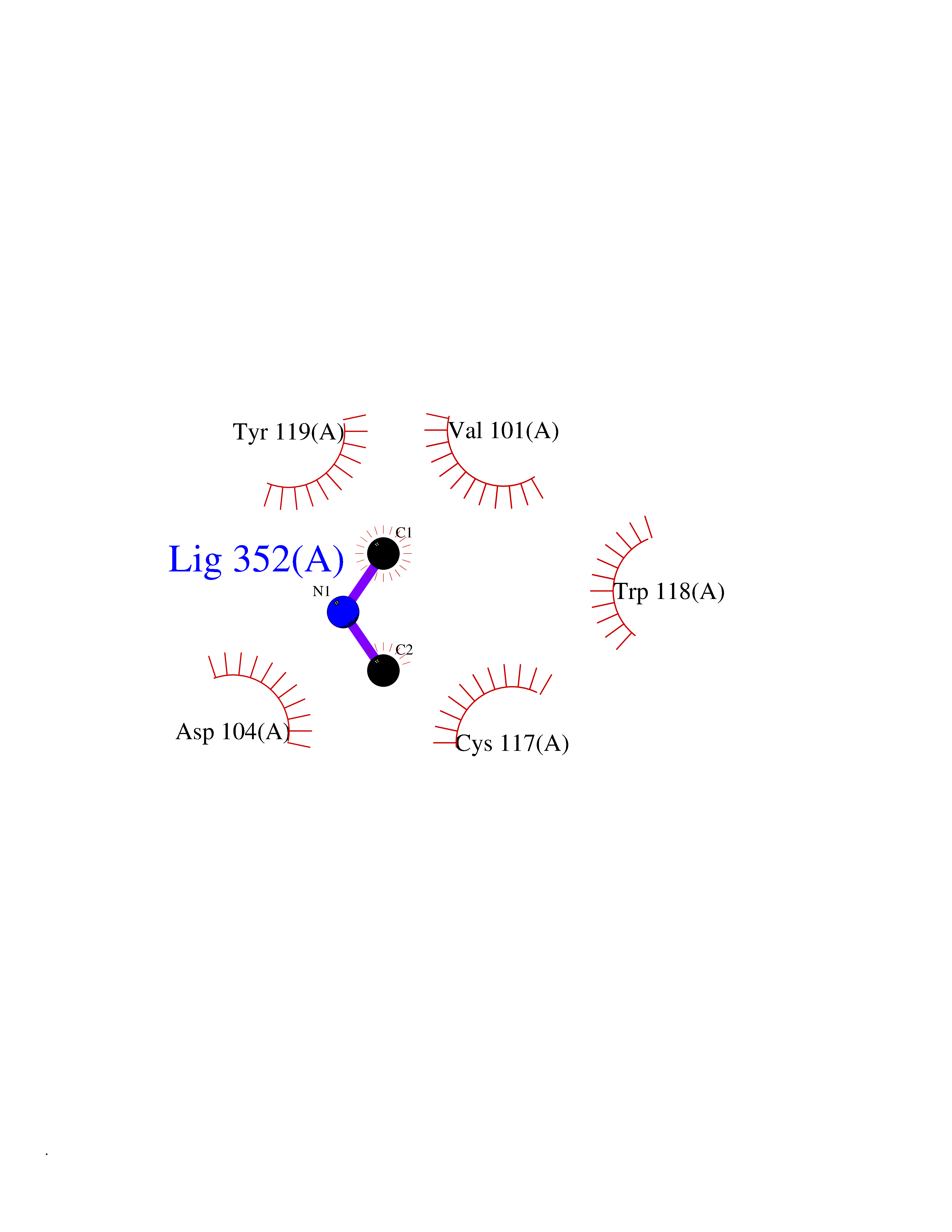 Ligplot Map 3D Binding mode Sequence VYAQEKQDFVQHFSQIVRVLTEHPEIGDAIARLKEVLEYNAIGGKYNRGLTVVVAFRELVEPRKQDADSLQRAWTVGWCVELLQAFFLVADDIMDSSLTRRGQICWYQKPGVGLDAINDANLLEACIYRLLKLYCREQPYYLNLIELFLQSSYQTEIGQTLDLLTAPQGNVDLVRFTEKRYKSIVKYKTAFASFYLPIAAAMYMAGIDGEKEHANAKKILLEMGEFFQIQDDYLDLFGDPSVTGKIGTDIQDNKCSWLVVQCLQRATPEQYQILKENYGQKEAEKVARVKALYEELDLPAVFLQYEEDSYSHIMALIEQYAAPLPPAVFLGLARKIYK Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Glutathione S-transferase LANCL1 (LANCL1) | 3E73 | 4.00 | |
Target general information Gen name LANCL1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p40; LanC-like protein 1; GPR69A; 40 kDa erythrocyte membrane protein Protein family LanC-like protein family Biochemical class NA Function Functions as glutathione transferase. Catalyzes conjugation of the glutathione (GSH) to artificial substrates 1-chloro-2,4-dinitrobenzene (CDNB) and p-nitrophenyl acetate. Mitigates neuronal oxidative stress during normal postnatal development and in response to oxidative stresses probably through GSH antioxidant defense mechanism (By similarity). May play a role in EPS8 signaling. Binds glutathione. Related diseases Spermatogenic failure 5 (SPGF5) [MIM:243060]: An infertility disorder caused by spermatogenesis defects. Semen from affected men show close to 100% morphologically abnormal multiflagellar spermatozoa with low motility, oversized irregular heads, and abnormal midpiece and acrosome. {ECO:0000269|PubMed:17435757, ECO:0000269|PubMed:21733974}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9UHR4; P42858; Q08509 EC number EC 2.5.1.18 Uniprot keywords 3D-structure; Acetylation; Cell membrane; Cytoplasm; Direct protein sequencing; Membrane; Metal-binding; Proteomics identification; Reference proteome; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 46005.4 Length 405 Aromaticity 0.14 Instability index 33.7 Isoelectric point 7.13 Charge (pH=7) 0.4 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map 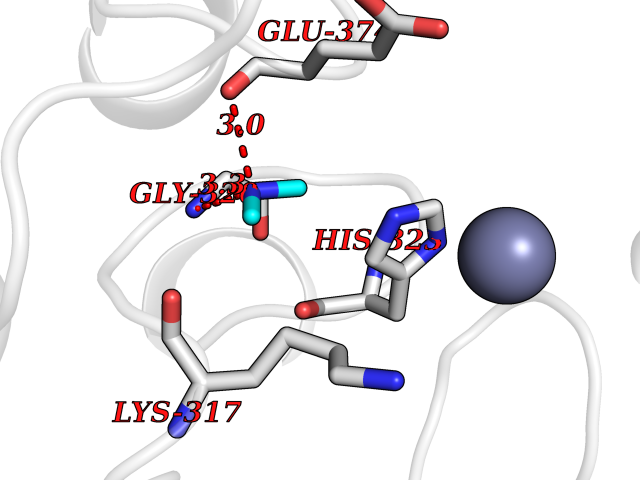 Pymol Map 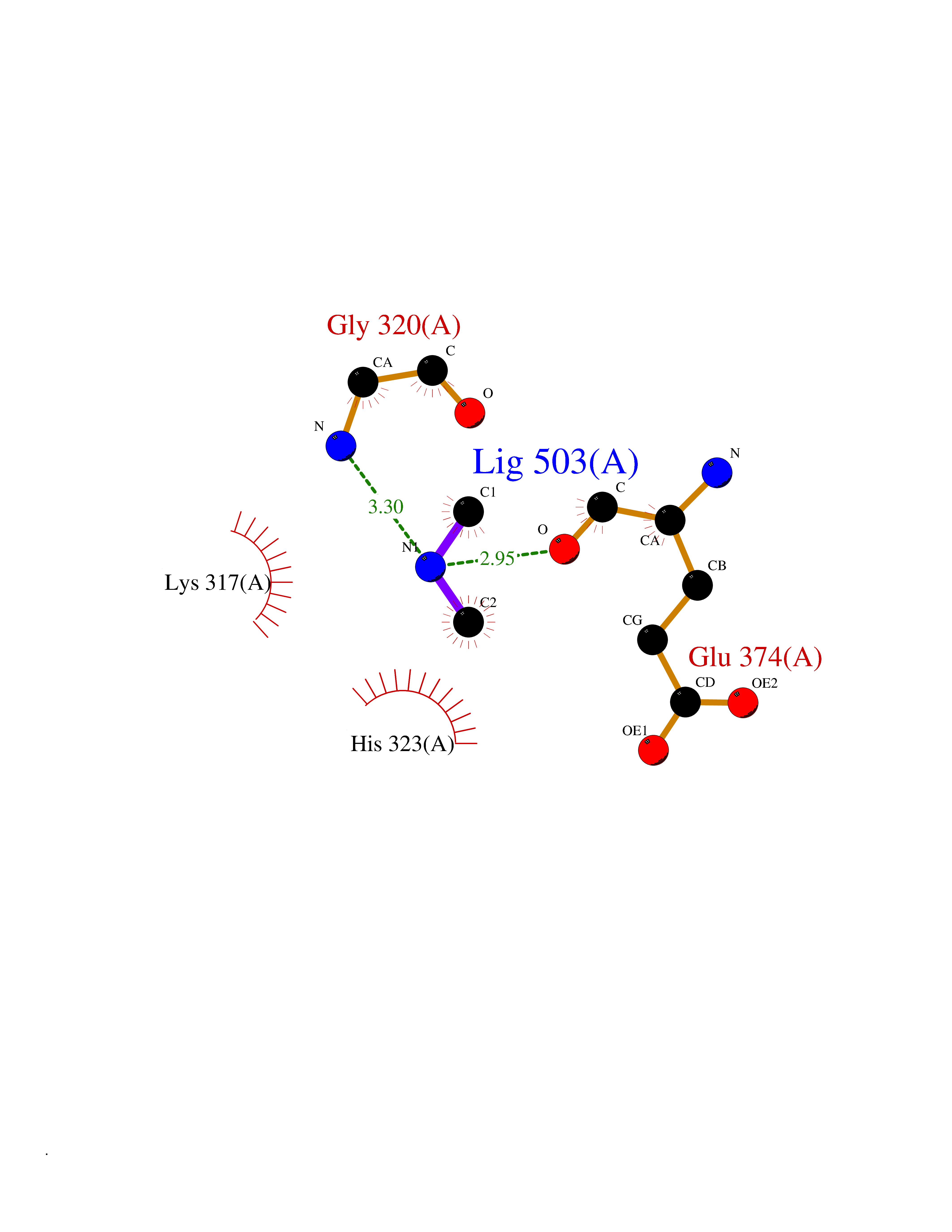 Ligplot Map 3D Binding mode Sequence SMDIEFMAQRAFPNPYADYNKSLAEGYFDAAGRLTPEFSQRLTNKIRELLQQMERGLKSADPRDGTGYTGWAGIAVLYLHLYDVFGDPAYLQLAHGYVKQSLNCLTKRSITFLCGDAGPLAVAAVLYHKMNNEKQAEDCITRLIHLNKIDPHAPNEMLYGRIGYIYALLFVNKNFGVEKIPQSHIQQICETILTSGENLARKRNFTAKSPLMYEWYQEYYVGAAHGLAGIYYYLMQPSLQVSQGKLHSLVKPSVDYVCQLKFPSGNYPPCIGDNRDLLVHWCHGAPGVIYMLIQAYKVFREEKYLCDAYQCADVIWQYGLLKKGYGLCHGSAGNAYAFLTLYNLTQDMKYLYRACKFAEWCLEYGEHGCRTPDTPFSLFEGMAGTIYFLADLLVPTKARFPAFEL Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Bacterial Dihydropteroate synthetase (Bact folP) | 1AJ0 | 3.99 | |
Target general information Gen name Bact folP Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms folP; H2Pte synthase; Dihydropteroate synthase; Dihydropteroate pyrophosphorylase; DHPS Protein family DHPS family Biochemical class Alkyl aryl transferase Function Dhps catalyzes the formation of the immediate precursor of folic acid. It is implicated in resistance to sulfonamide. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14033; DB00634; DB01298; DB01581; DB06821; DB01582; DB00576; DB01015; DB00259; DB06729; DB00263 Interacts with NA EC number EC 2.5.1.15 Uniprot keywords 3D-structure; Direct protein sequencing; Folate biosynthesis; Magnesium; Metal-binding; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 30614.8 Length 282 Aromaticity 0.05 Instability index 32.55 Isoelectric point 5.68 Charge (pH=7) -8.5 2D Binding mode Binding energy (Kcal/mol) -5.45 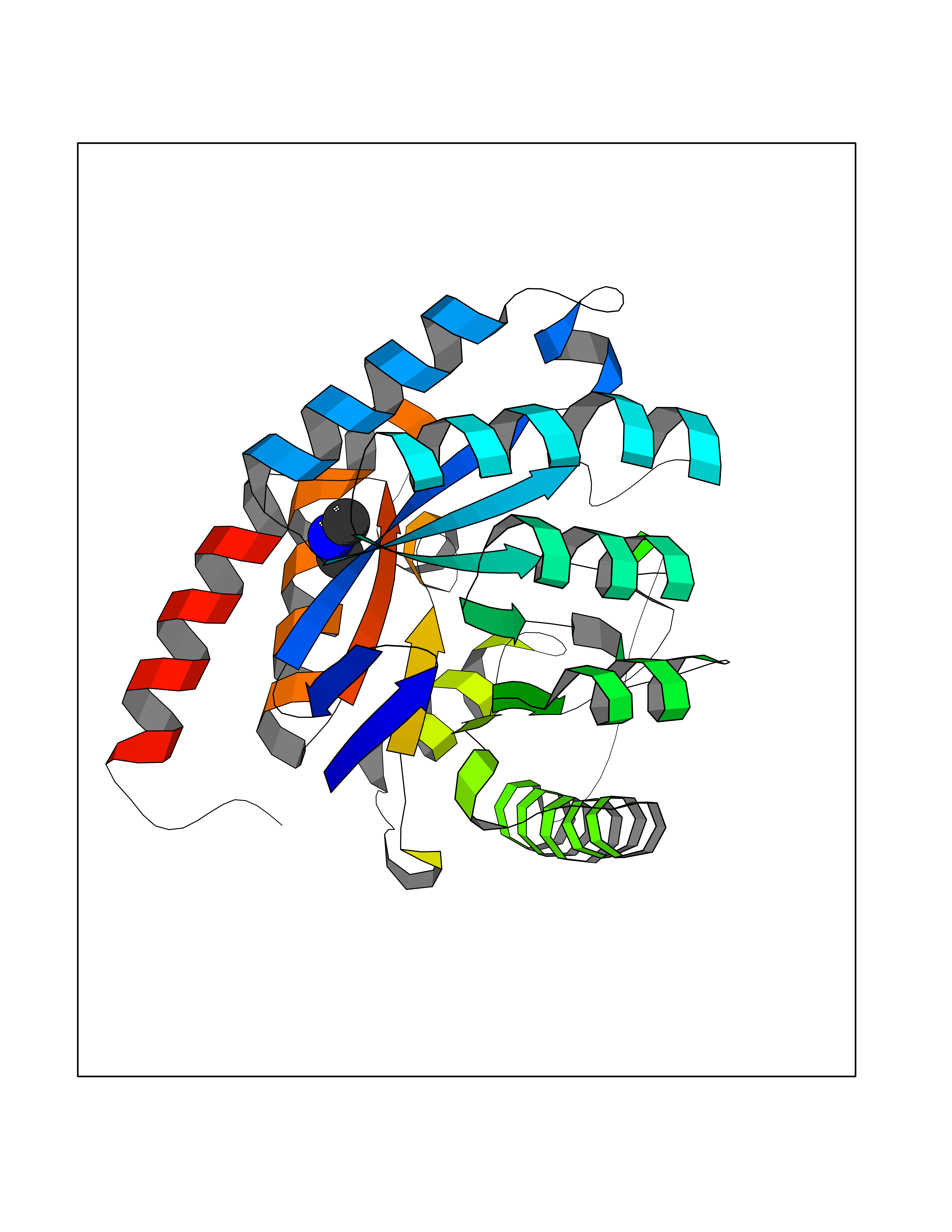 Molscript Map 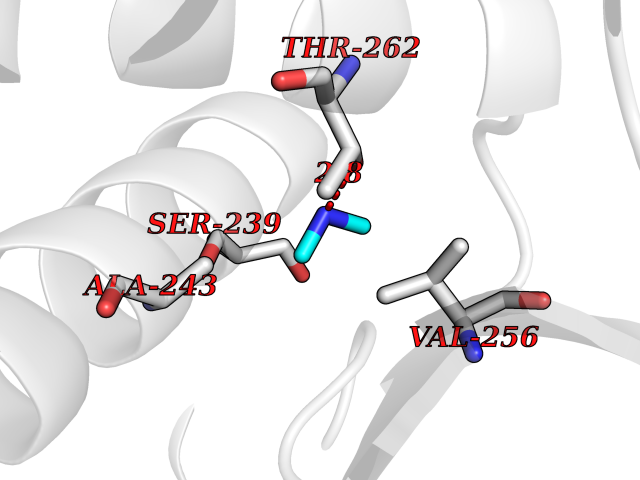 Pymol Map 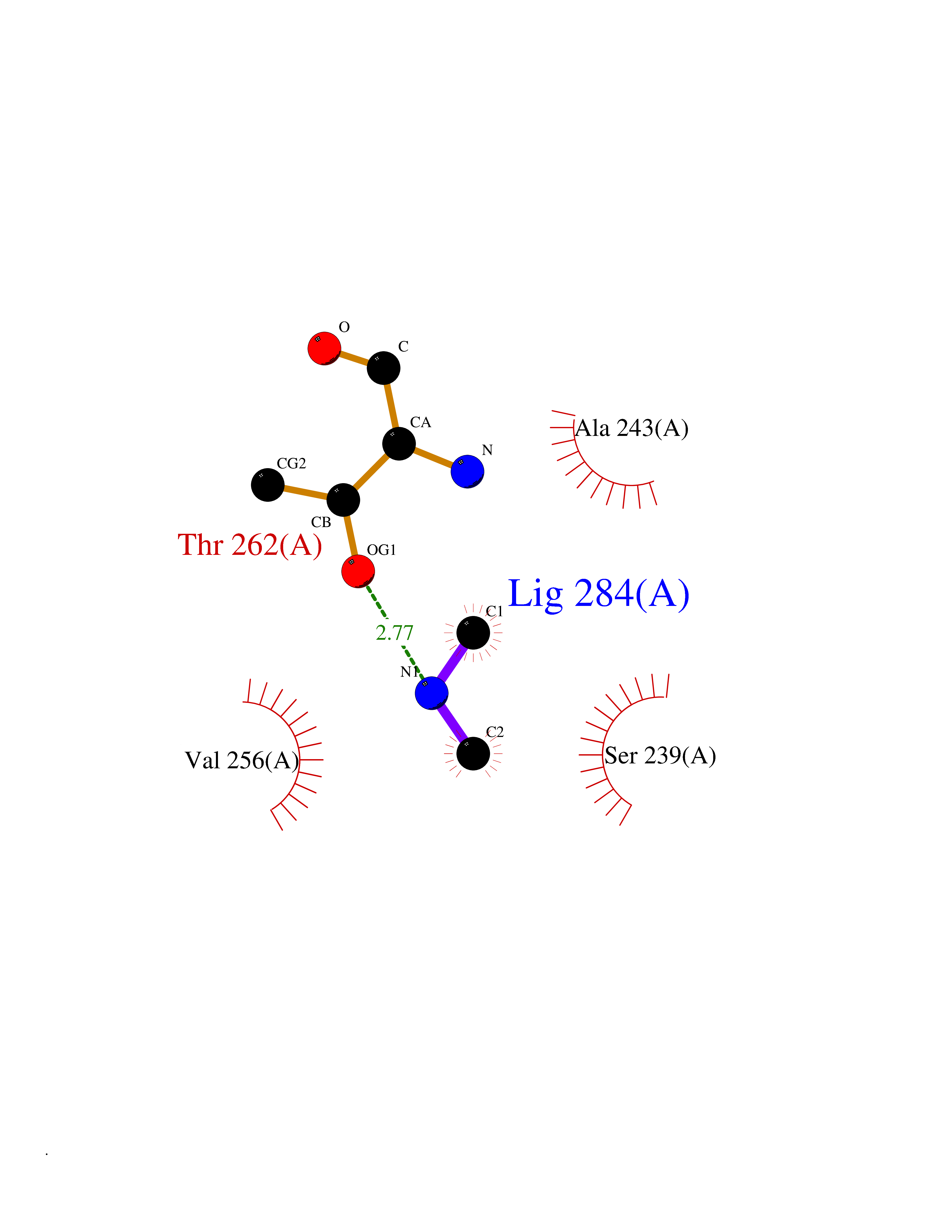 Ligplot Map 3D Binding mode Sequence MKLFAQGTSLDLSHPHVMGILNVTPDSFSDGGTHNSLIDAVKHANLMINAGATIIDVGGESTRPGAAEVSVEEELQRVIPVVEAIAQRFEVWISVDTSKPEVIRESAKVGAHIINDIRSLSEPGALEAAAETGLPVCLMHMQGNPKTMQEAPKYDDVFAEVNRYFIEQIARCEQAGIAKEKLLLDPGFGFGKNLSHNYSLLARLAEFHHFNLPLLVGMSRKSMIGQLLNVGPSERLSGSLACAVIAAMQGAHIIRVHDVKETVEAMRVVEATLSAKENKRYE Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Endolysin | 1AM7 | 3.99 | |
Target general information Gen name R Organism Escherichia phage lambda (Bacteriophage lambda) Uniprot ID TTD ID NA Synonyms NA Protein family Glycosyl hydrolase 24 family Biochemical class Glycosidase Function Lyase activity.Lysozyme activity.Lytic transglycosylase activity. Related diseases Estrogen resistance (ESTRR) [MIM:615363]: A disorder characterized by partial or complete resistance to estrogens, in the presence of elevated estrogen serum levels. Clinical features include absence of the pubertal growth spurt, delayed bone maturation, unfused epiphyses, reduced bone mineral density, osteoporosis, continued growth into adulthood and very tall adult stature. Glucose intolerance, hyperinsulinemia and lipid abnormalities may also be present. {ECO:0000269|PubMed:23841731, ECO:0000269|PubMed:27754803}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04206 Interacts with NA EC number 4.2.2.n2 Uniprot keywords 3D-structure; Antimicrobial; Bacteriolytic enzyme; Cytolysis; Direct protein sequencing; Host cell lysis by virus; Host cytoplasm; Lyase; Reference proteome; Viral release from host cell Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 49834.9 Length 462 Aromaticity 0.07 Instability index 18.78 Isoelectric point 9.6 Charge (pH=7) 18.29 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map 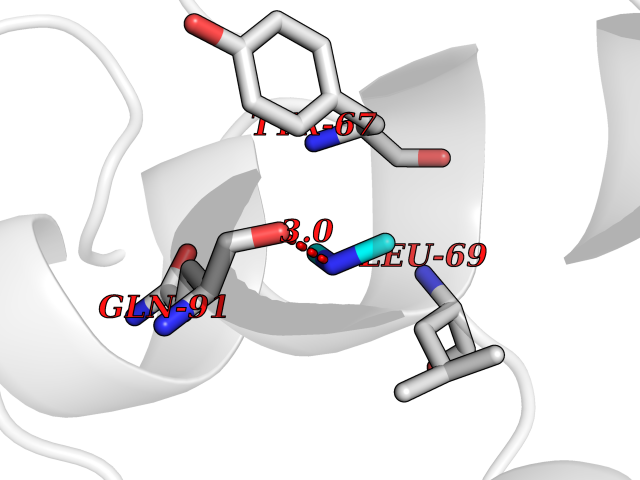 Pymol Map 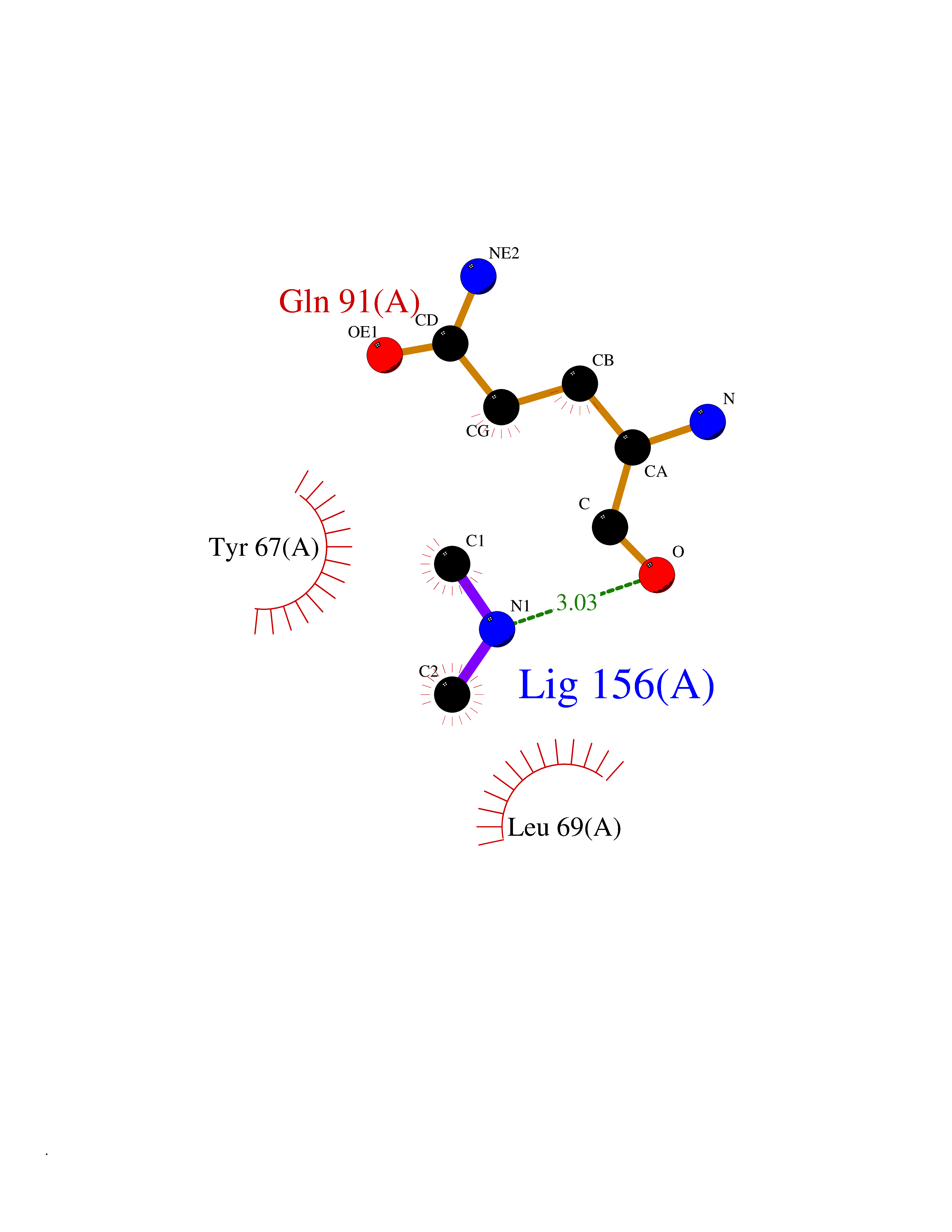 Ligplot Map 3D Binding mode Sequence MVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVRMVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVRMVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVR Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Interleukin 5 receptor alpha (IL5RA) | 3QT2 | 3.99 | |
Target general information Gen name IL5RA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Interleukin-5 receptor subunit alpha; IL5R; IL-5RA; IL-5R-alpha; IL-5R subunit alpha; IL-5 receptor subunit alpha; CDw125; CD125 Protein family IL-5 family Biochemical class Cytokine receptor Function The alpha chain binds to IL5. This is the receptor for interleukin-5. Related diseases Leukocyte adhesion deficiency 1 (LAD1) [MIM:116920]: LAD1 patients have recurrent bacterial infections and their leukocytes are deficient in a wide range of adhesion-dependent functions. {ECO:0000269|PubMed:1346613, ECO:0000269|PubMed:1347532, ECO:0000269|PubMed:1352501, ECO:0000269|PubMed:1590804, ECO:0000269|PubMed:1694220, ECO:0000269|PubMed:1968911, ECO:0000269|PubMed:20529581, ECO:0000269|PubMed:20549317, ECO:0000269|PubMed:7509236, ECO:0000269|PubMed:7686755, ECO:0000269|PubMed:9884339}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06612; DB01411; DB06602 Interacts with P32927; Q01344; Q01344-2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytokine; Direct protein sequencing; Disulfide bond; Glycoprotein; Growth factor; Reference proteome; Secreted; Signal Protein physicochemical properties Chain ID C,D,E,F Molecular weight (Da) 23806.5 Length 210 Aromaticity 0.1 Instability index 43.54 Isoelectric point 5.66 Charge (pH=7) -4.73 2D Binding mode Binding energy (Kcal/mol) -5.45  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ISLLPPVNFTIKVTGLAQVLLQWKPNPDQEQRNVNLEYQVKINAPKEDDYETRITESKAVTILHMGFSASVRTILQNDHSLLASSWASAELHAPPGSPGTSIVNLTCTTNTTEDNYSRLRSYQVSLHCTWMVGTDAPEDTQYFLYYRYGSWTEECQEYSMDTLGRNIACWFPRTFILSKGRDWLAVLVNGSSKHSAIRPFDQLFALHAID Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Cholinesterase (BCHE) | 1P0I | 3.99 | |
Target general information Gen name BCHE Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Pseudocholinesterase; Choline esterase II; CHE1; Butyrylcholine esterase; Acylcholine acylhydrolase Protein family Type-B carboxylesterase/lipase family Biochemical class Type-B carboxylesterase/lipase Function Esterase with broad substrate specificity. Contributes to the inactivation of the neurotransmitter acetylcholine. Can degrade neurotoxic organophosphate esters. Related diseases Butyrylcholinesterase deficiency (BCHED) [MIM:617936]: An autosomal recessive metabolic condition characterized by increased sensitivity to certain anesthetic drugs, including the muscle relaxants succinylcholine or mivacurium. BCHED results in slower hydrolysis of these drugs and, consequently, a prolonged neuromuscular block, leading to apnea. The duration of the prolonged apnea varies significantly depending on the extent of the enzyme deficiency. {ECO:0000269|PubMed:10404729, ECO:0000269|PubMed:11928765, ECO:0000269|PubMed:12881446, ECO:0000269|PubMed:1306123, ECO:0000269|PubMed:1349196, ECO:0000269|PubMed:1415224, ECO:0000269|PubMed:15563885, ECO:0000269|PubMed:15781196, ECO:0000269|PubMed:1611188, ECO:0000269|PubMed:16788378, ECO:0000269|PubMed:17700357, ECO:0000269|PubMed:18075469, ECO:0000269|PubMed:18300943, ECO:0000269|PubMed:25054547, ECO:0000269|PubMed:25264279, ECO:0000269|PubMed:2915989, ECO:0000269|PubMed:7634491, ECO:0000269|PubMed:8554068, ECO:0000269|PubMed:8680411, ECO:0000269|PubMed:9110359, ECO:0000269|PubMed:9191541, ECO:0000269|PubMed:9388484, ECO:0000269|PubMed:9543549, ECO:0000269|PubMed:9694584}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08200; DB08201; DB03814; DB07940; DB03672; DB03128; DB08897; DB01122; DB06692; DB01408; DB00868; DB06756; DB11148; DB03568; DB04250; DB06774; DB01161; DB00856; DB00477; DB00122; DB14006; DB00527; DB00515; DB04920; DB00907; DB00979; DB01245; DB00944; DB11397; DB02811; DB00711; DB00449; DB07681; DB00843; DB01135; DB01057; DB01010; DB01364; DB03822; DB08658; DB00674; DB00941; DB00762; DB06636; DB00677; DB01064; DB01221; DB00772; DB00888; DB00358; DB02845; DB08893; DB01226; DB09205; DB01400; DB00585; DB00892; DB01337; DB00082; DB00183; DB00790; DB04892; DB03976; DB01338; DB00733; DB01035; DB00721; DB00392; DB09288; DB00545; DB00178; DB05386; DB00989; DB05875; DB00202; DB00391; DB00382; DB00871; DB04572; DB14031; DB00620; DB00508; DB01116; DB01199 Interacts with P54252; P46379-2; P06276; P55212; O75190-2; O14901; P13473-2; O75400-2; P62826; P67812; P02814 EC number EC 3.1.1.8 Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Phosphoprotein; Proteomics identification; Reference proteome; Secreted; Serine esterase; Sialic acid; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 58935.5 Length 523 Aromaticity 0.13 Instability index 38.71 Isoelectric point 8.63 Charge (pH=7) 5.36 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map  Pymol Map 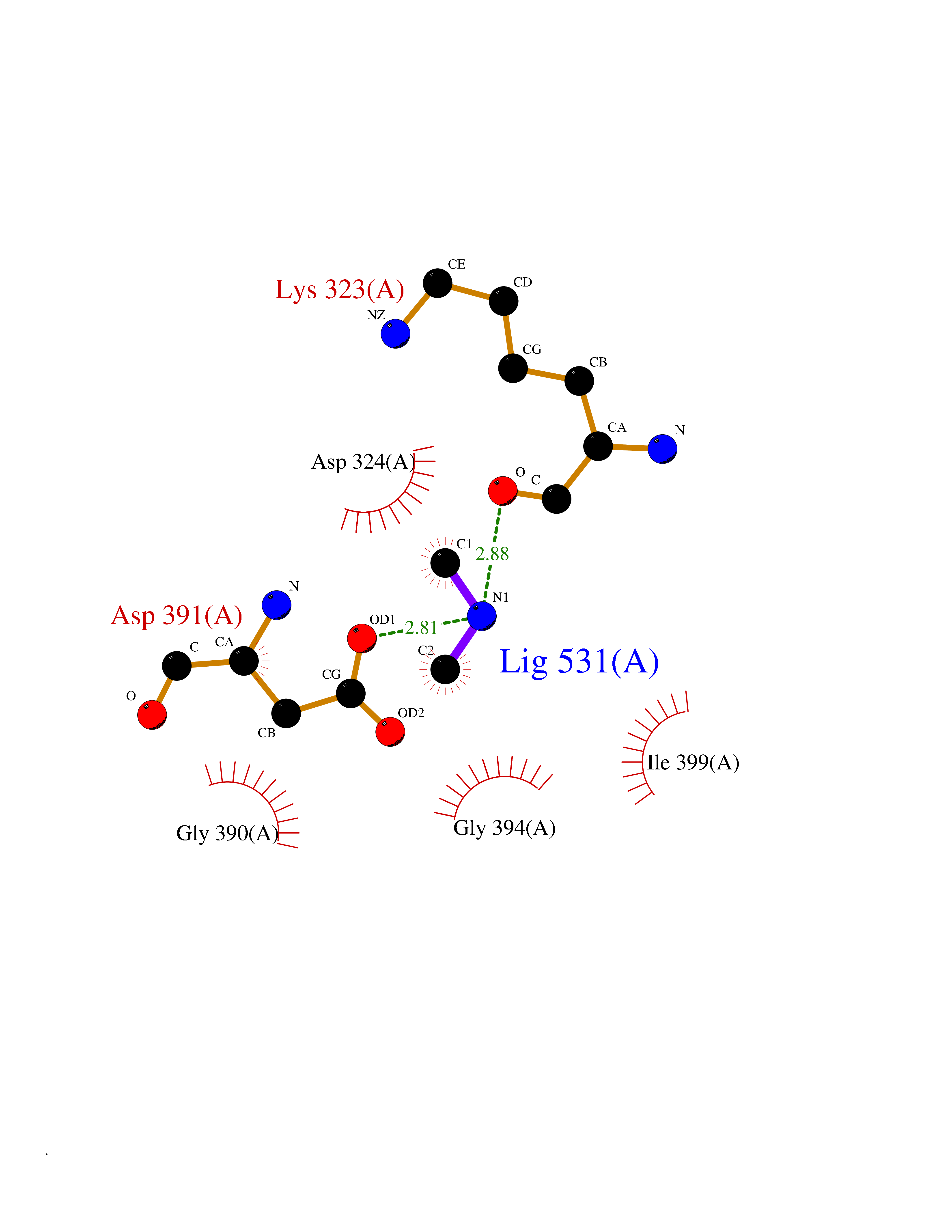 Ligplot Map 3D Binding mode Sequence IIIATKNGKVRGMQLTVFGGTVTAFLGIPYAQPPLGRLRFKKPQSLTKWSDIWNATKYANSCCQNIDQSFPGFHGSEMWNPNTDLSEDCLYLNVWIPAPKPKNATVLIWIYGGGFQTGTSSLHVYDGKFLARVERVIVVSMNYRVGALGFLALPGNPEAPGNMGLFDQQLALQWVQKNIAAFGGNPKSVTLFGESAGAASVSLHLLSPGSHSLFTRAILQSGSFNAPWAVTSLYEARNRTLNLAKLTGCSRENETEIIKCLRNKDPQEILLNEAFVVPYGTPLSVNFGPTVDGDFLTDMPDILLELGQFKKTQILVGVNKDEGTAFLVYGAPGFSKDNNSIITRKEFQEGLKIFFPGVSEFGKESILFHYTDWVQRPENYREALGDVVGDYNFICPALEFTKKFSEWGNNAFFYYFEHRSSKLPWPEWMGVMHGYEIEFVFGLPLERRDYTKAEEILSRSIVKRWANFAKYGNPQETQNQSTSWPVFKSTEQKYLTLNTESTRIMTKLRAQQCRFWTSFFPKV Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Riboflavin synthase | 1PKV | 3.99 | |
Target general information Gen name ribC Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1654;ribE;b1662 Protein family NA Biochemical class Transferase Function Riboflavin synthase activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00140 Interacts with NA EC number 2.5.1.9 Uniprot keywords 3D-structure; Reference proteome; Repeat; Riboflavin biosynthesis; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 19023.5 Length 174 Aromaticity 0.05 Instability index 2.85 Isoelectric point 5.13 Charge (pH=7) -9.8 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map 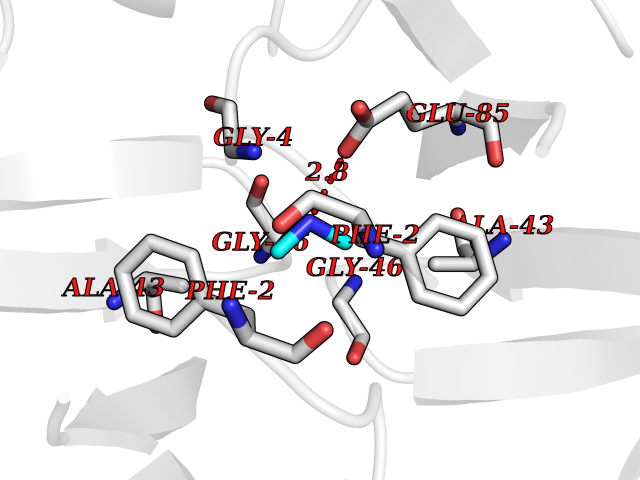 Pymol Map 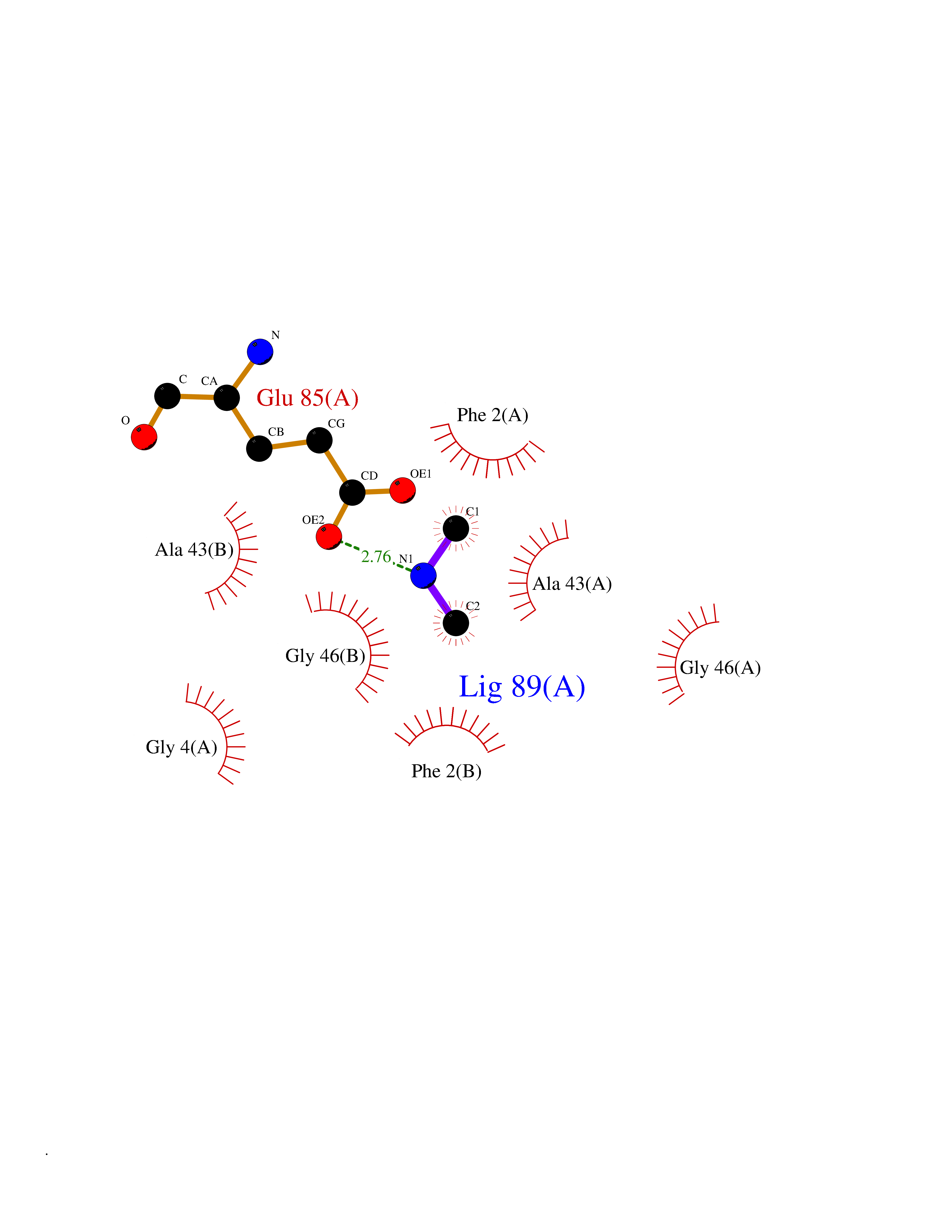 Ligplot Map 3D Binding mode Sequence MFTGIVQGTAKLVSIDEKPNFRTHVVELPDHMLDGLETGASVAHNGCCLTVTEINGNHVSFDLMKETLRITNLGDLKVGDWVNVERAMFTGIVQGTAKLVSIDEKPNFRTHVVELPDHMLDGLETGASVAHNGCCLTVTEINGNHVSFDLMKETLRITNLGDLKVGDWVNVERA Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Gamma-aminobutyric acid type B receptor subunit 1 | 4MS4 | 3.99 | |
Target general information Gen name GABBR1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms GPRC3B;GPR51 Protein family G-protein coupled receptor 3 family, GABA-B receptor subfamily Biochemical class Signaling protein / antagonist Function G-protein coupled GABA receptor activity. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08891; DB08892; DB00181; DB00363; DB02530; DB05010; DB09072 Interacts with Q9UBS5; Q9UBS5-2; P46459; Q86UR5 EC number NA Uniprot keywords 3D-structure; Cell membrane; Coiled coil; Direct protein sequencing; Disease variant; Disulfide bond; Epilepsy; G-protein coupled receptor; Glycoprotein; Intellectual disability; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 46502.1 Length 408 Aromaticity 0.12 Instability index 50.05 Isoelectric point 5.78 Charge (pH=7) -5.62 2D Binding mode Binding energy (Kcal/mol) -5.45  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence RRAVYIGALFPMSGGWPGGQACQPAVEMALEDVNSRRDILPDYELKLIHHDSKCDPGQATKYLYELLYNDPIKIILMPGCSSVSTLVAEAARMWNLIVLSYGSSSPALSNRQRFPTFFRTHPSATLHNPTRVKLFEKWGWKKIATIQQTTEVFTSTLDDLEERVKEAGIEITFRQSFFSDPAVPVKNLKRQDARIIVGLFYETEARKVFCEVYKERLFGKKYVWFLIGWYADNWFKIYDPSINCTVDEMTEAVEGHITTEIVMLNPANTRSISNMTSQEFVEKLTKRLKRHPEETGGFQEAPLAYDAIWALALALNKTSRLEDFNYNNQTITDQIYRAMNSSSFEGVSGHVVFDASGSRMAWTLIEQLQGGSYKKIGYYDSTKDDLSWSKTDKWIGGSPPADDYKDDD Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Poly [ADP-ribose] polymerase 1 (PARP1) | 5WS1 | 3.99 | |
Target general information Gen name PARP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein poly-ADP-ribosyltransferase PARP1; Poly[ADP-ribose] synthetase-1; Poly[ADP-ribose] synthase 1; Poly(ADP-ribose)polymerase-1; PPOL; PARP-1; NAD(+)Poly [ADP-ribose] polymerase-1 ADP-ribosyltrans Protein family ARTD/PARP family Biochemical class Glycosyltransferases Function Mainly mediates glutamate and aspartate ADP-ribosylation of target proteins: the ADP-D-ribosyl group of NAD(+) is transferred to the acceptor carboxyl group of glutamate and aspartate residues and further ADP-ribosyl groups are transferred to the 2'-position of the terminal adenosine moiety, building up a polymer with an average chain length of 20-30 units. Mediates the poly(ADP-ribosyl)ation of a number of proteins, including itself, APLF and CHFR. Also mediates serine ADP-ribosylation of target proteins following interaction with HPF1; HPF1 conferring serine specificity. Probably also catalyzes tyrosine ADP-ribosylation of target proteins following interaction with HPF1. Catalyzes the poly-ADP-ribosylation of histones in a HPF1-dependent manner. Involved in the base excision repair (BER) pathway by catalyzing the poly-ADP-ribosylation of a limited number of acceptor proteins involved in chromatin architecture and in DNA metabolism. ADP-ribosylation follows DNA damage and appears as an obligatory step in a detection/signaling pathway leading to the reparation of DNA strand breaks. In addition to base excision repair (BER) pathway, also involved in double-strand breaks (DSBs) repair: together with TIMELESS, accumulates at DNA damage sites and promotes homologous recombination repair by mediating poly-ADP-ribosylation. In addition to proteins, also able to ADP-ribosylate DNA: catalyzes ADP-ribosylation of DNA strand break termini containing terminal phosphates and a 2'-OH group in single- and double-stranded DNA, respectively. Required for PARP9 and DTX3L recruitment to DNA damage sites. PARP1-dependent PARP9-DTX3L-mediated ubiquitination promotes the rapid and specific recruitment of 53BP1/TP53BP1, UIMC1/RAP80, and BRCA1 to DNA damage sites. Acts as a regulator of transcription: positively regulates the transcription of MTUS1 and negatively regulates the transcription of MTUS2/TIP150. With EEF1A1 and TXK, forms a complex that acts as a T-helper 1 (Th1) cell-specific transcription factor and binds the promoter of IFN-gamma to directly regulate its transcription, and is thus involved importantly in Th1 cytokine production. Involved in the synthesis of ATP in the nucleus, together with NMNAT1, PARG and NUDT5. Nuclear ATP generation is required for extensive chromatin remodeling events that are energy-consuming. Poly-ADP-ribosyltransferase that mediates poly-ADP-ribosylation of proteins and plays a key role in DNA repair. Related diseases Dihydrolipoamide dehydrogenase deficiency (DLDD) [MIM:246900]: An autosomal recessive metabolic disorder characterized biochemically by a combined deficiency of the branched-chain alpha-keto acid dehydrogenase complex (BCKDC), pyruvate dehydrogenase complex (PDC), and alpha-ketoglutarate dehydrogenase complex (KGDC). Clinically, affected individuals have lactic acidosis and neurologic deterioration due to sensitivity of the central nervous system to defects in oxidative metabolism. {ECO:0000269|PubMed:10448086, ECO:0000269|PubMed:11687750, ECO:0000269|PubMed:12925875, ECO:0000269|PubMed:15712224, ECO:0000269|PubMed:16442803, ECO:0000269|PubMed:16770810, ECO:0000269|PubMed:17404228, ECO:0000269|PubMed:20160912, ECO:0000269|PubMed:8506365, ECO:0000269|PubMed:8968745, ECO:0000269|PubMed:9540846, ECO:0000269|PubMed:9934985}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04010; DB03509; DB03072; DB03722; DB03073; DB07787; DB07096; DB07330; DB02498; DB13877; DB02701; DB11793; DB02690; DB09074; DB12332; DB11760; DB00277; DB07232; DB01593; DB14487; DB14533; DB14548 Interacts with Q8IW19; Q7Z2E3; P42574; P49715; Q86WJ1-1; P26358; Q01094; Q96L91; P11308; O60741; P09429; Q13007; Q9BQ69; P08651; Q9Y530; P09874; Q8N2W9; P46063; Q9NTX7; Q14684-1; O95863; P63165; P04637; P0CG48; Q14191; P18887; P54577; Q2M1K9; Q02085 EC number EC 2.4.2.30 Uniprot keywords 3D-structure; Acetylation; ADP-ribosylation; Allosteric enzyme; Apoptosis; Chromosome; Cytoplasm; Direct protein sequencing; DNA damage; DNA repair; DNA-binding; Glycosyltransferase; Immunity; Innate immunity; Isopeptide bond; Metal-binding; NAD; Nucleotidyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 36904 Length 329 Aromaticity 0.08 Instability index 35.9 Isoelectric point 6.83 Charge (pH=7) -0.44 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map  Pymol Map 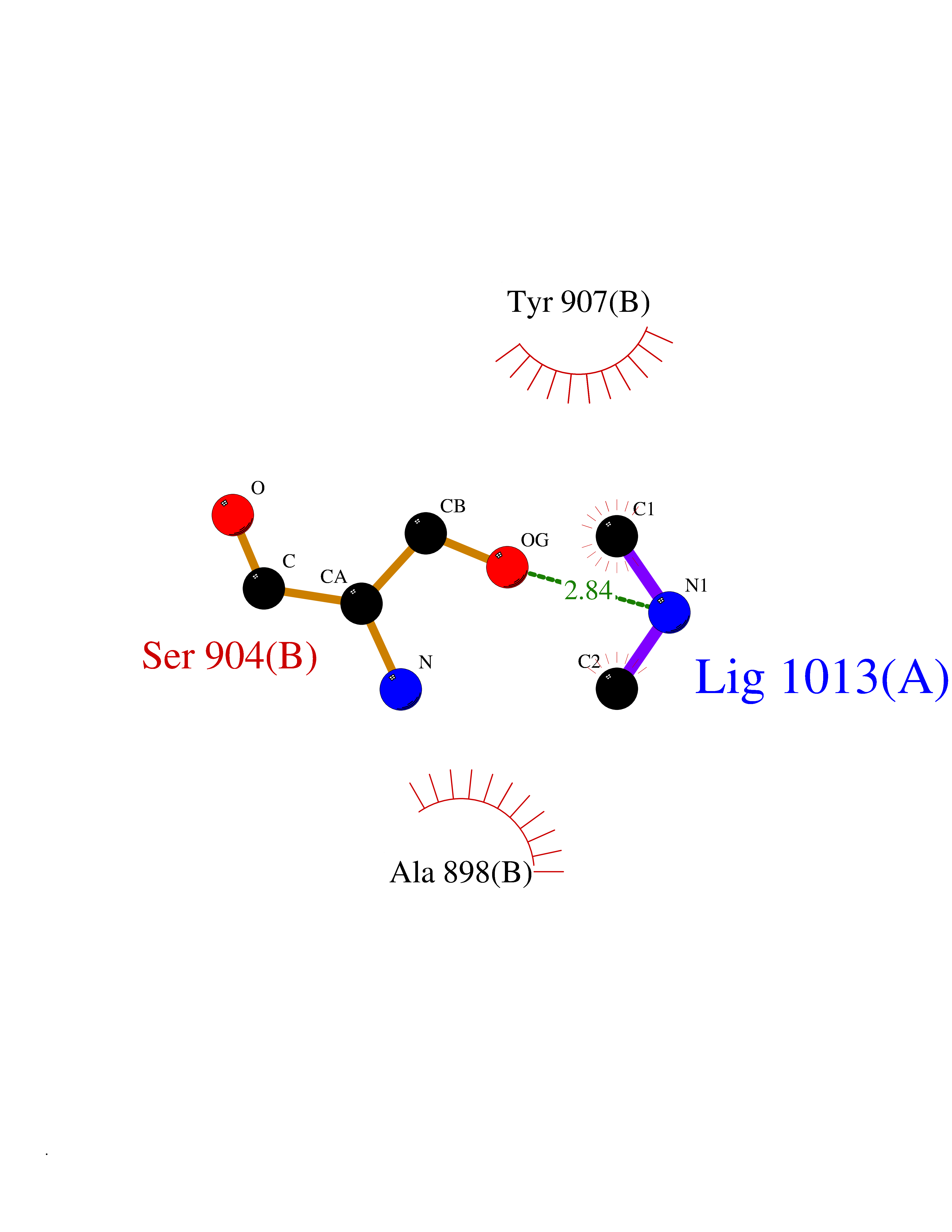 Ligplot Map 3D Binding mode Sequence DLIKMIFDVESMKKAMVEYEIDLQKMPLGKLSKRQIQAAYSILSEVQQAVSQGSDSQILDLSNRFYTLIPHDFGMKKPPLLNNADSVQAKAEMLDNLLDIEVAYSLPIDVNYEKLKTDIKVVDRDSEEAEIIRKYVKNTHATTHNAYDLEVIDIFKIEREGECQRYKPFKQLHNRRLLWHGSRTTNFAGILSQGLRIAPPEAPVTGYMFGKGIYFADMVSKSANYCHTSQGDPIGLILLGEVALGNMYELKHASHISKLPKGKHSVKGLGKTTPDPSANISLDGVDVPLGTGISSGVNDTSLLYNEYIVYDIAQVNLKYLLKLKFNFKT Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Penicillin-binding protein 2B | 2WAD | 3.99 | |
Target general information Gen name penA Organism Streptococcus pneumoniae (strain ATCC BAA-255 / R6) Uniprot ID TTD ID NA Synonyms spr1517;pbp2b Protein family Transpeptidase family Biochemical class Peptide binding protein Function Penicillin binding. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01163; DB00415; DB08795; DB01140; DB00456; DB01066; DB00493; DB01331; DB01212; DB00567; DB03313; DB00485; DB00739; DB01603; DB00607; DB00713; DB00319 Interacts with NA EC number NA Uniprot keywords 3D-structure; Antibiotic resistance; Cell membrane; Cell shape; Cell wall biogenesis/degradation; Membrane; Peptidoglycan synthesis; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 65444.4 Length 607 Aromaticity 0.08 Instability index 30.15 Isoelectric point 4.95 Charge (pH=7) -20.68 2D Binding mode Binding energy (Kcal/mol) -5.45  Molscript Map 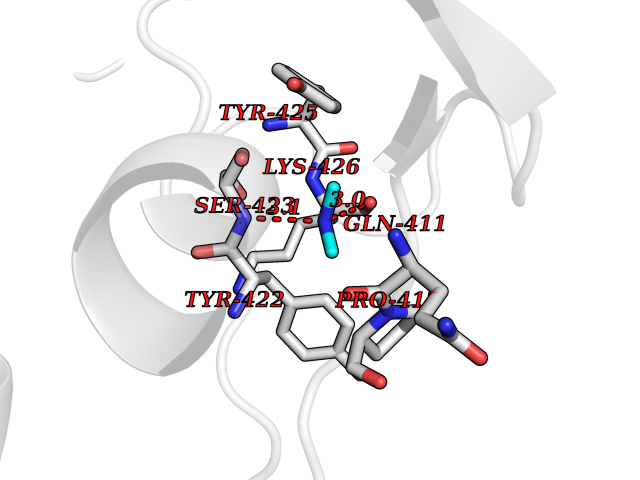 Pymol Map 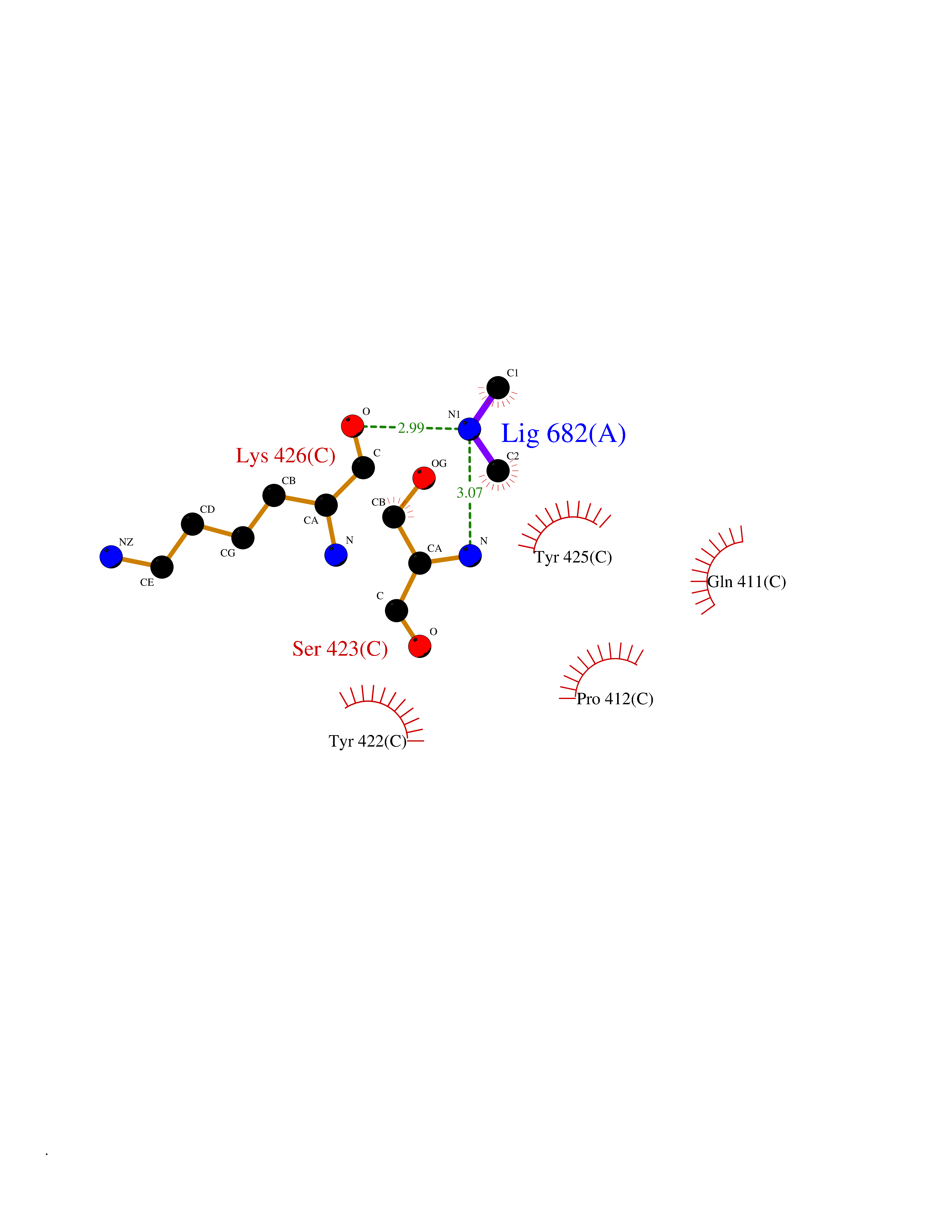 Ligplot Map 3D Binding mode Sequence SQTKVTTSSARGEIYDASGKPLVENTLKQVVSFTRSNKMTATDLKEIAKKLLTYVSISSPNLTERQLADYYLADPEIYKKTVEALPSESELYNNAVDSVPTSQLNYTEDEKKEIYLFSQLNAVGNFATGTIATDPLNDSQVAVIASISKEMPGISISTSWDRKILETSLSSIVGSVSSEKAGLPAEEAESYLKKGYSLNDRVGTSYLEKQYEEVLQGKRPVKEIHLDKHGDMESVENIEEGSKGKNIKLTIDLAFQDSVDALLKSYFNSELGNGGAKYSEGVYAVALNPQTGAVLSMSGLKHDLKTGELTPDSLGTVTNVFVPGSVVKAATISSGWENGVLSGNQTLTDQPIVFQGSAPIYSWYKLAYGSFPITAVEALEYSSNAYVVQTALGIMGQTYQPNMFVGTSNLESAMGKLRSTFGEYGLGSATGIDLPDESTGLVPKEYNFANFITNAFGQFDNYTPMQLAQYVATIANNGVRLAPHIVEGIYDNNDKGGLGELIQAIDTKEINKVNISESDMAILHQGFYQVSHGTSPLTTGRAFSDGATVSISGKTGTNTNAVAYAPTENPQIAVAVVFPHNTNLTKNVGPAIARDIINLYNQHHPMN Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Transferrin (TF) | 1RYO | 3.99 | |
Target general information Gen name TF Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Siderophilin; Serotransferrin; PRO1400; Beta-1 metal-binding globulin Protein family Transferrin family Biochemical class Transferrin Function It is responsible for the transport of iron from sites of absorption and heme degradation to those of storage and utilization. Serum transferrin may also have a further role in stimulating cell proliferation. Transferrins are iron binding transport proteins which can bind two Fe(3+) ions in association with the binding of an anion, usually bicarbonate. Related diseases Atransferrinemia (ATRAF) [MIM:209300]: A rare autosomal recessive disorder characterized by abnormal synthesis of transferrin leading to iron overload and microcytic hypochromic anemia. {ECO:0000269|PubMed:11110675, ECO:0000269|PubMed:15466165}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01370; DB14517; DB14518; DB01294; DB14526; DB14527; DB11136; DB14528; DB14529; DB14530; DB00515; DB09130; DB11397; DB13949; DB14490; DB14491; DB14488; DB14501; DB14489; DB13257; DB06215; DB06784; DB05260; DB01592; DB00893; DB00677; DB06757; DB11182; DB14520; DB01593; DB14487; DB14533; DB14548 Interacts with O43315; O00501; Q7Z7G2; Q9GZR5; Q9Y282; Q96KR6; P01350; P08034; Q8NBJ4; O15529; Q8TED1; Q7Z5P4; A8MZ59; O15173; Q96TC7; Q3KNW5; Q9BXS9-3; Q99523; O43278-2; Q8N9I0; P02786; Q4KMG9; Q9K0U9; Q09057; Q9K0V0; P02786 EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Ion transport; Iron; Iron transport; Metal-binding; Methylation; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 35854.5 Length 324 Aromaticity 0.1 Instability index 34.17 Isoelectric point 7.58 Charge (pH=7) 1.42 2D Binding mode Binding energy (Kcal/mol) -5.44 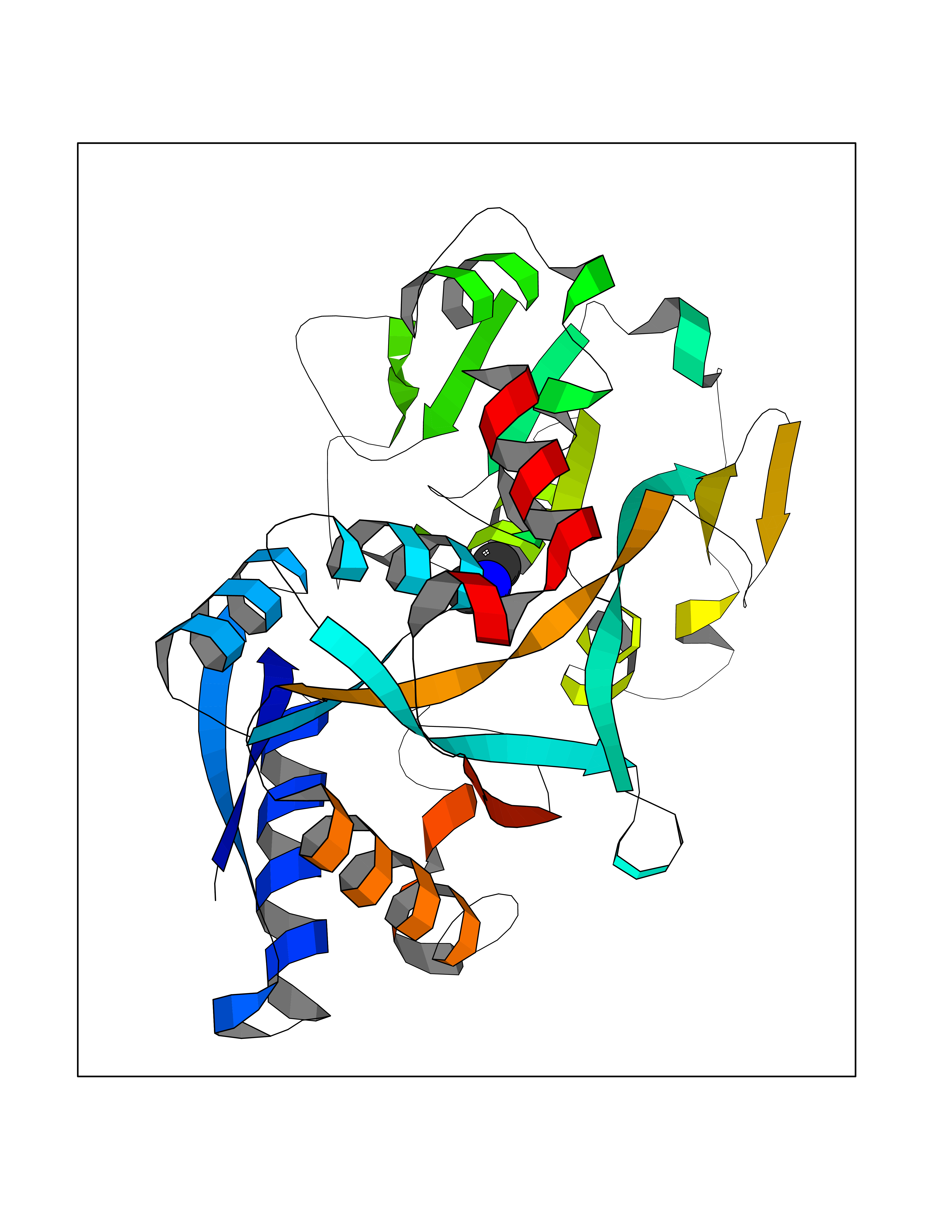 Molscript Map 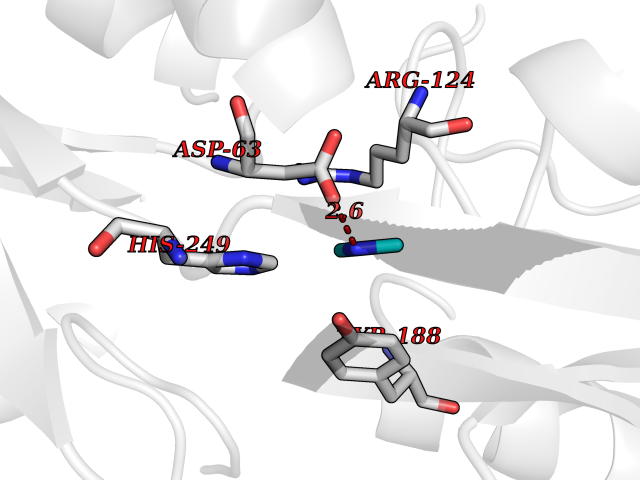 Pymol Map 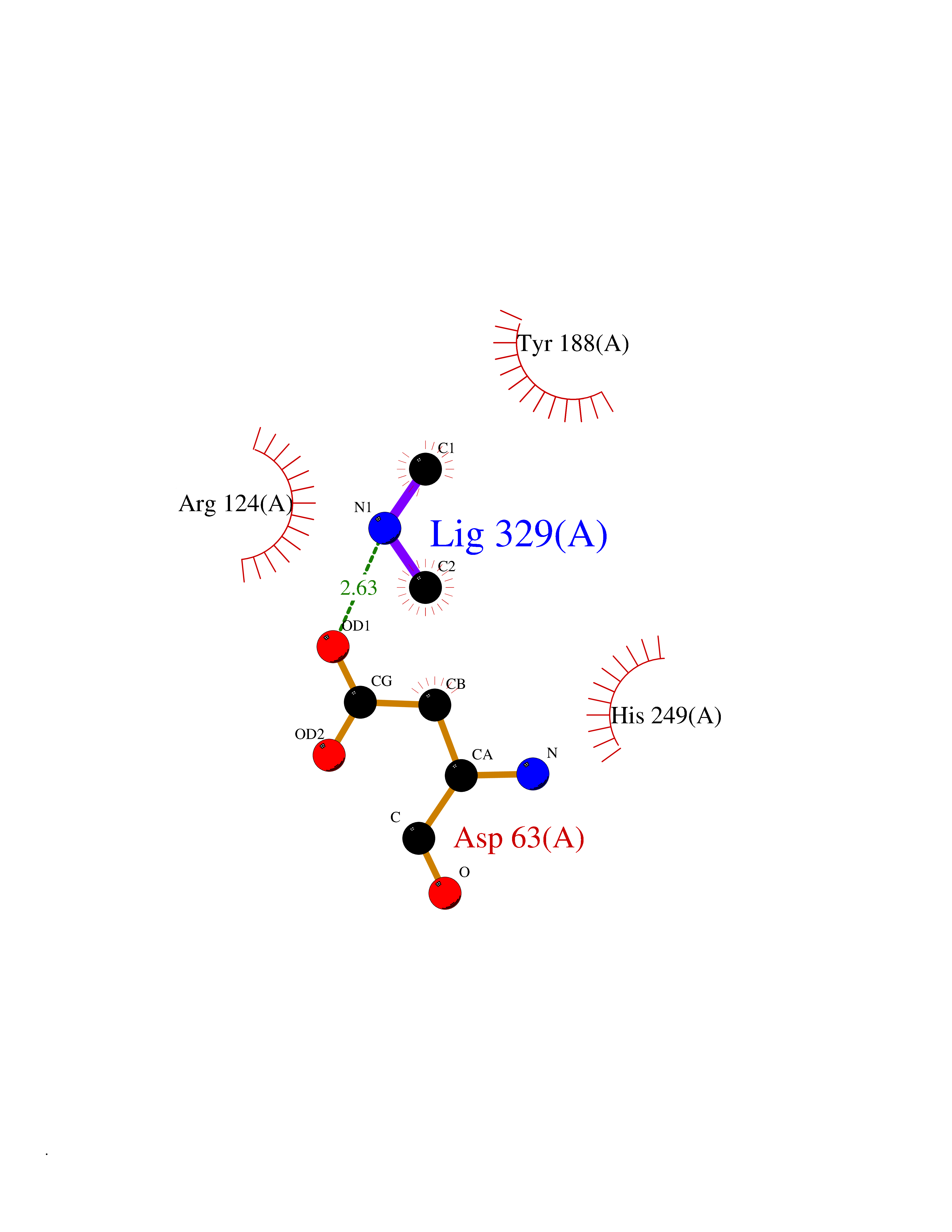 Ligplot Map 3D Binding mode Sequence KTVRWCAVSEHEATKCQSFRDHMKSVIPSDGPSVACVKKASYLDCIRAIAANEADAVTLDAGLVYDAYLAPNNLKPVVAEFYGSKEDPQTFYYAVAVVKKDSGFQMNQLRGKKSCHTGLGRSAGWNIPIGLLYCDLPEPRKPLEKAVANFFSGSCAPCADGTDFPQLCQLCPGCGCSTLNQYFGYSGAFKCLKDGAGDVAFVKHSTIFENLANKADRDQYELLCLDNTRKPVDEYKDCHLAQVPSHTVVARSMGGKEDLIWELLNQAQEHFGKDKSKEFQLFSSPHGKDLLFKDSAHGFLKVPPRMDAKMYLGYEYVTAIRNLR Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Beta-glucosidase A | 1E4I | 3.99 | |
Target general information Gen name bglA Organism Paenibacillus polymyxa (Bacillus polymyxa) Uniprot ID TTD ID NA Synonyms NA Protein family Glycosyl hydrolase 1 family Biochemical class Hydrolase Function Beta-glucosidase activity.Scopolin beta-glucosidase activity. Related diseases Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:15645182}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02658; DB04282; DB04304 Interacts with NA EC number 3.2.1.21 Uniprot keywords 3D-structure; Carbohydrate metabolism; Cellulose degradation; Glycosidase; Hydrolase; Polysaccharide degradation Protein physicochemical properties Chain ID A Molecular weight (Da) 51515.2 Length 447 Aromaticity 0.14 Instability index 38.44 Isoelectric point 5.28 Charge (pH=7) -18.1 2D Binding mode Binding energy (Kcal/mol) -5.45 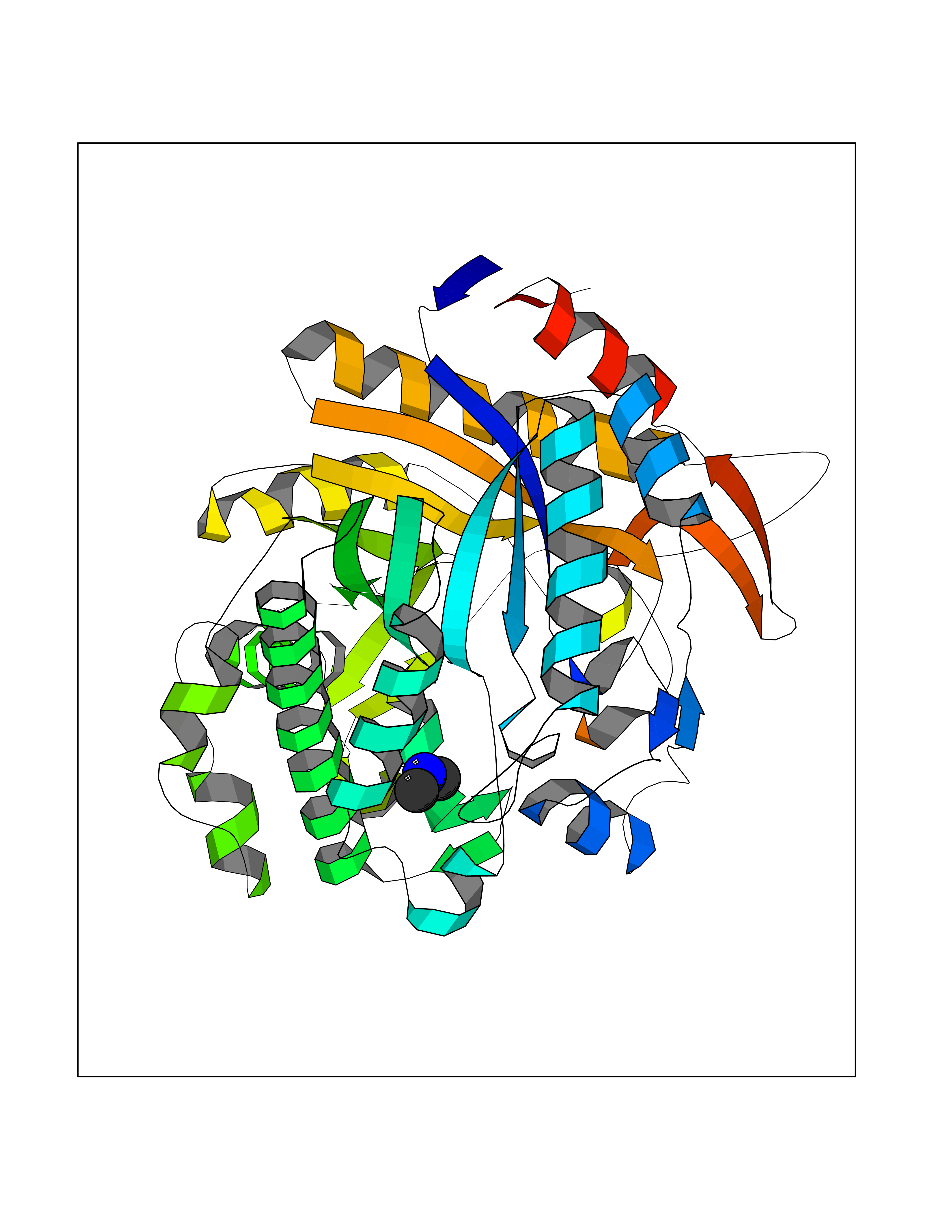 Molscript Map 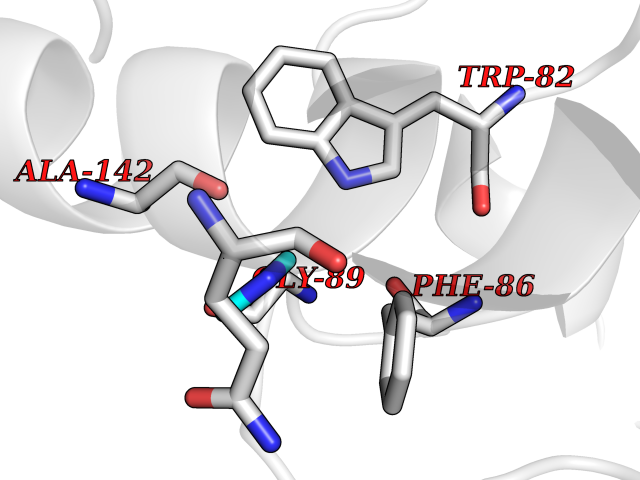 Pymol Map 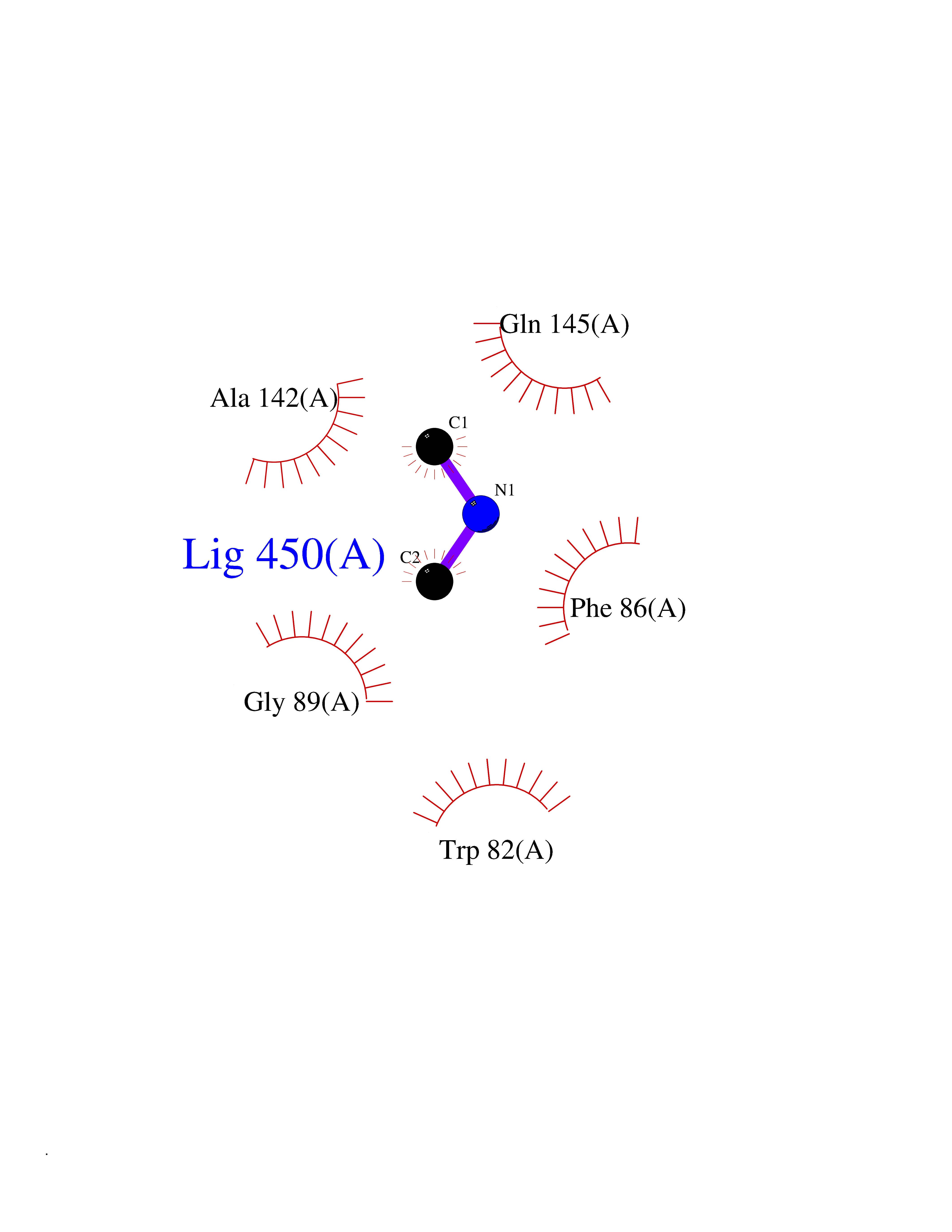 Ligplot Map 3D Binding mode Sequence TIFQFPQDFMWGTATAAYQIEGAYQEDGRGLSIWDTFAHTPGKVFNGDNGNVACDSYHRYEEDIRLMKELGIRTYRFSVSWPRIFPNGDGEVNQKGLDYYHRVVDLLNDNGIEPFCTLYHWDLPQALQDAGGWGNRRTIQAFVQFAETMFREFHGKIQHWLTFNEPWCIAFLSNMLGVHAPGLTNLQTAIDVGHHLLVAHGLSVRRFRELGTSGQIGIAPNVSWAVPYSTSEEDKAACARTISLHSDWFLQPIYQGSYPQFLVDWFAEQGATVPIQDGDMDIIGEPIDMIGINYYSMSVNRFNPEAGFLQSEEINMGLPVTDIGWPVESRGLYEVLHYLQKYGNIDIYITENGACINDEVVNGKVQDDRRISYMQQHLVQVHRTIHDGLHVKGYMAWSLLDNFEWAEGYNMRFGMIHVDFRTQVRTPKQSYYWYRNVVSNNWLETRR Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Neutrophil collagenase | 4QKZ | 3.99 | |
Target general information Gen name MMP8 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CLG1 Protein family Peptidase M10A family Biochemical class Hydrolase / hydrolase inhibitor Function Metalloendopeptidase activity.Serine-type endopeptidase activity.Zinc ion binding. Related diseases Protoporphyria, erythropoietic, 1 (EPP1) [MIM:177000]: An autosomal recessive form of porphyria with onset usually before age 10 years. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. Erythropoietic protoporphyria is marked by excessive protoporphyrin in erythrocytes, plasma, liver and feces, and by widely varying photosensitive skin changes ranging from a burning or pruritic sensation to erythema, edema and wheals. {ECO:0000269|PubMed:10942404, ECO:0000269|PubMed:11375302, ECO:0000269|PubMed:12063482, ECO:0000269|PubMed:12601550, ECO:0000269|PubMed:1376018, ECO:0000269|PubMed:15286165, ECO:0000269|PubMed:17196862, ECO:0000269|PubMed:1755842, ECO:0000269|PubMed:7910885, ECO:0000269|PubMed:8757534, ECO:0000269|PubMed:9211198, ECO:0000269|PubMed:9585598, ECO:0000269|PubMed:9740232}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07772; DB07713; DB07397; DB02326; DB03207; DB02953; DB08476; DB07900; DB03622; DB03880; DB08028; DB03636; DB00786; DB08403; DB06971 Interacts with NA EC number 3.4.24.34 Uniprot keywords 3D-structure; Calcium; Collagen degradation; Direct protein sequencing; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 18096.6 Length 163 Aromaticity 0.12 Instability index 27.13 Isoelectric point 4.64 Charge (pH=7) -11.95 2D Binding mode Binding energy (Kcal/mol) -5.45 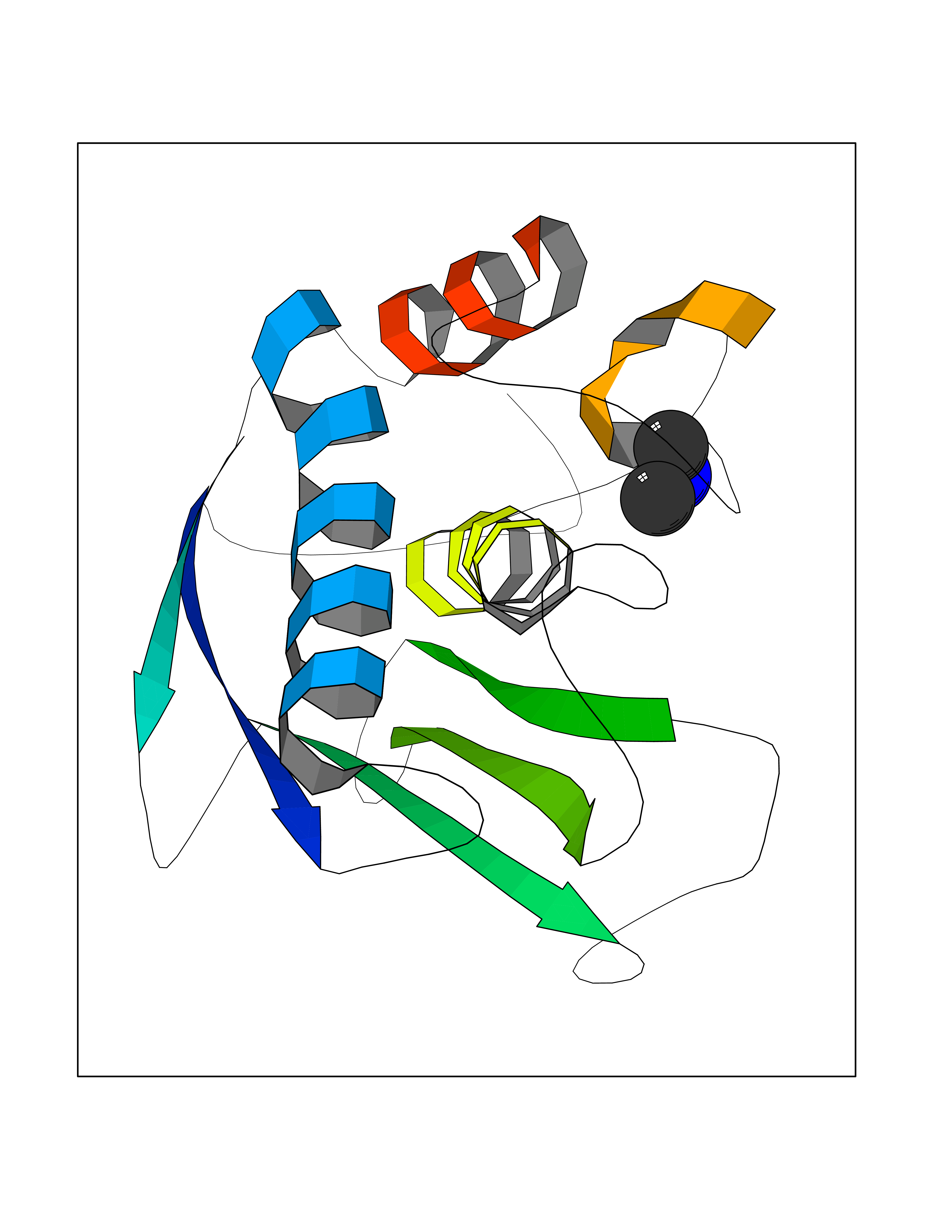 Molscript Map  Pymol Map 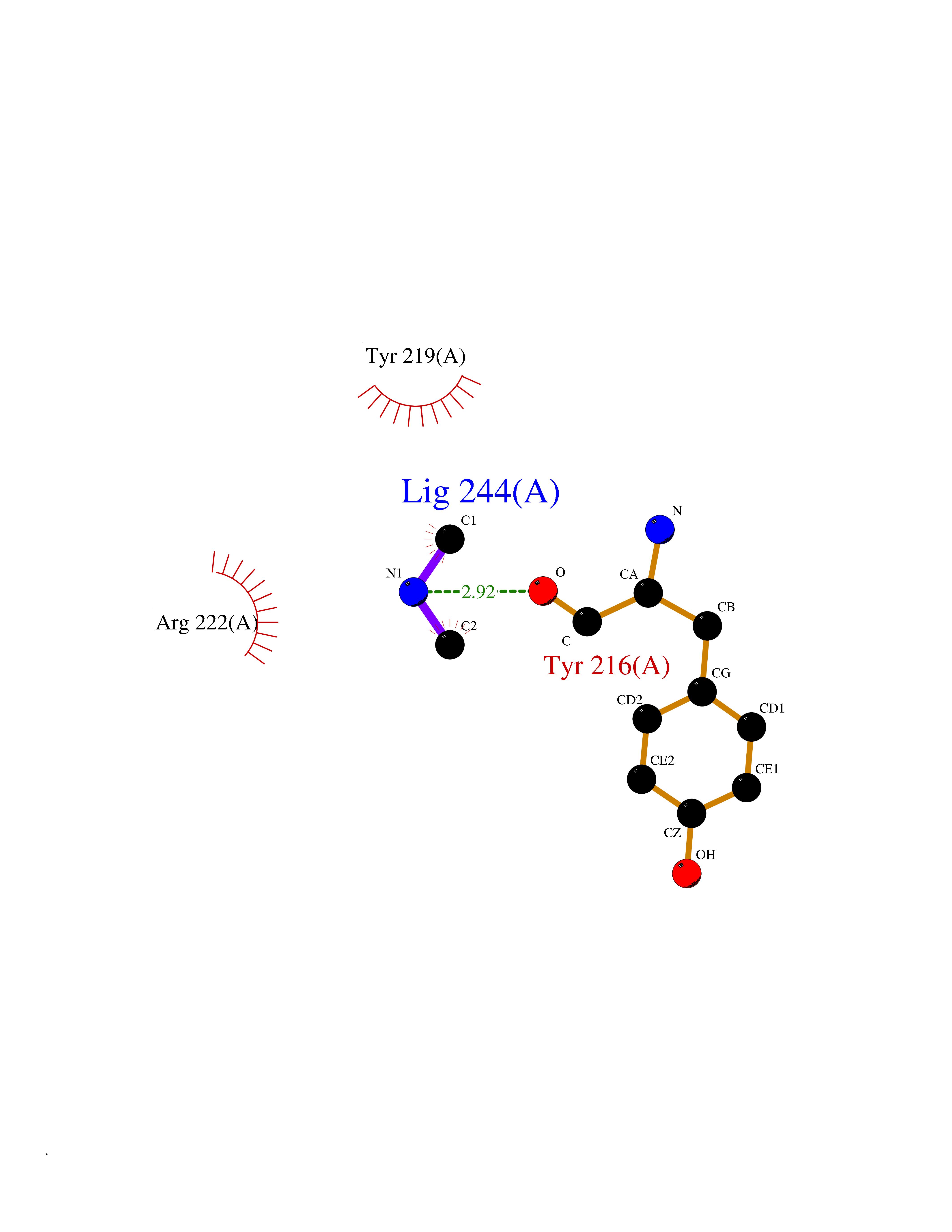 Ligplot Map 3D Binding mode Sequence MLTPGNPKWERTNLTYRIRNYTPQLSEAEVERAIKDAFELWSVASPLIFTRISQGEADINIAFYQRDHGDNSPFDGPNGILAHAFQPGQGIGGDAHFDAEETWTNTSANYNLFLVAAHEFGHSLGLAHSSDPGALMYPNYAFRETSNYSLPQDDIDGIQAIYG Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Aromatic-L-amino-acid decarboxylase (DDC) | 3RCH | 3.99 | |
Target general information Gen name DDC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms DOPA decarboxylase; AADC Protein family Group II decarboxylase family Biochemical class Carbon-carbon lyase Function Catalyzes the decarboxylation of L-3,4-dihydroxyphenylalanine (DOPA) to dopamine, L-5-hydroxytryptophan to serotonin and L-tryptophan to tryptamine. Related diseases Aromatic L-amino-acid decarboxylase deficiency (AADCD) [MIM:608643]: An inborn error in neurotransmitter metabolism that leads to combined serotonin and catecholamine deficiency. It causes developmental and psychomotor delay, poor feeding, lethargy, ptosis, intermittent hypothermia, gastrointestinal disturbances. The onset is early in infancy and inheritance is autosomal recessive. {ECO:0000269|PubMed:14991824, ECO:0000269|PubMed:15079002, ECO:0000269|Ref.12}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB12783; DB00190; DB00260; DB06262; DB13848; DB00875; DB01235; DB00968; DB00114; DB00150 Interacts with P10275 EC number EC 4.1.1.28 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Catecholamine biosynthesis; Decarboxylase; Disease variant; Lyase; Proteomics identification; Pyridoxal phosphate; Reference proteome; Repeat Protein physicochemical properties Chain ID A,B Molecular weight (Da) 41117.4 Length 369 Aromaticity 0.09 Instability index 37.52 Isoelectric point 8.26 Charge (pH=7) 3.76 2D Binding mode Binding energy (Kcal/mol) -5.44 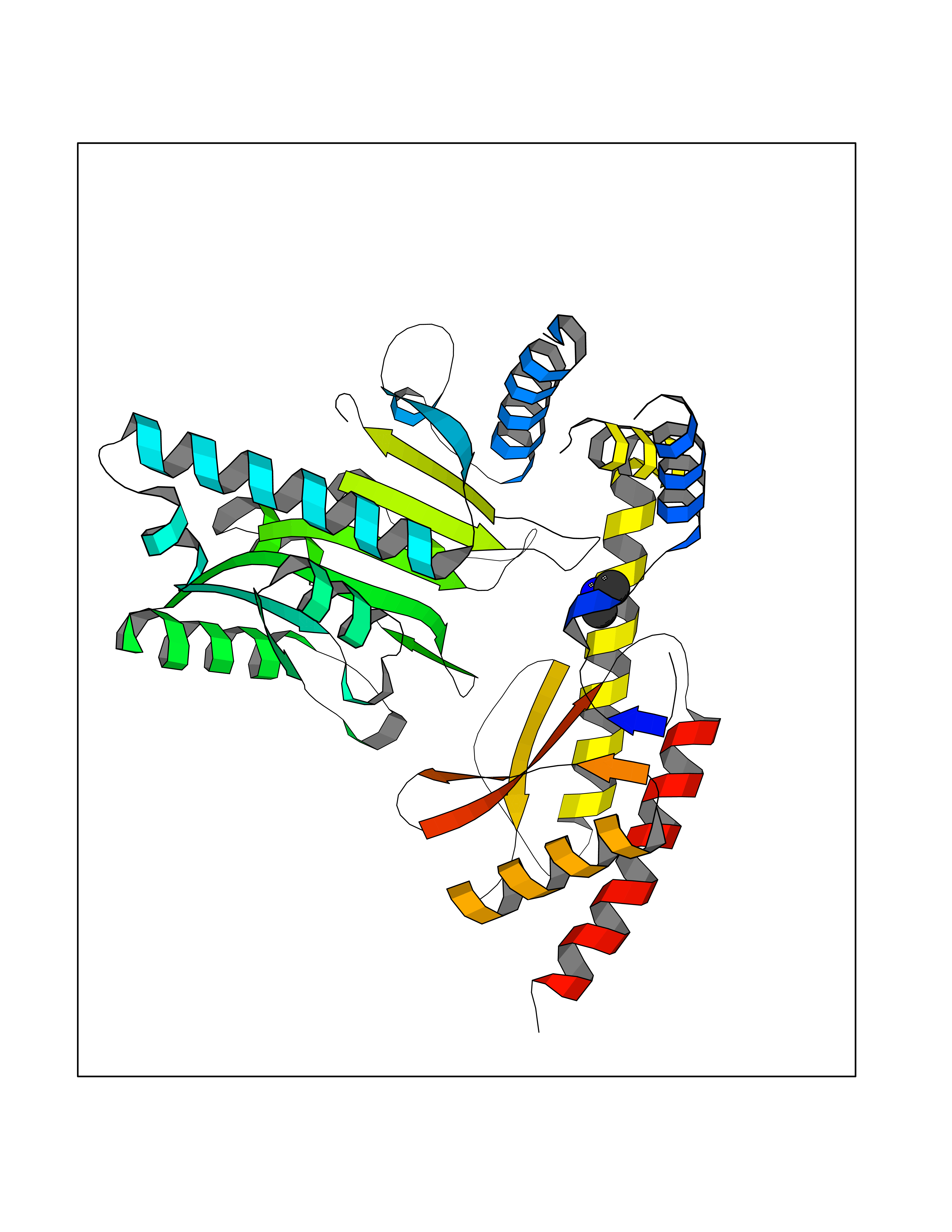 Molscript Map  Pymol Map 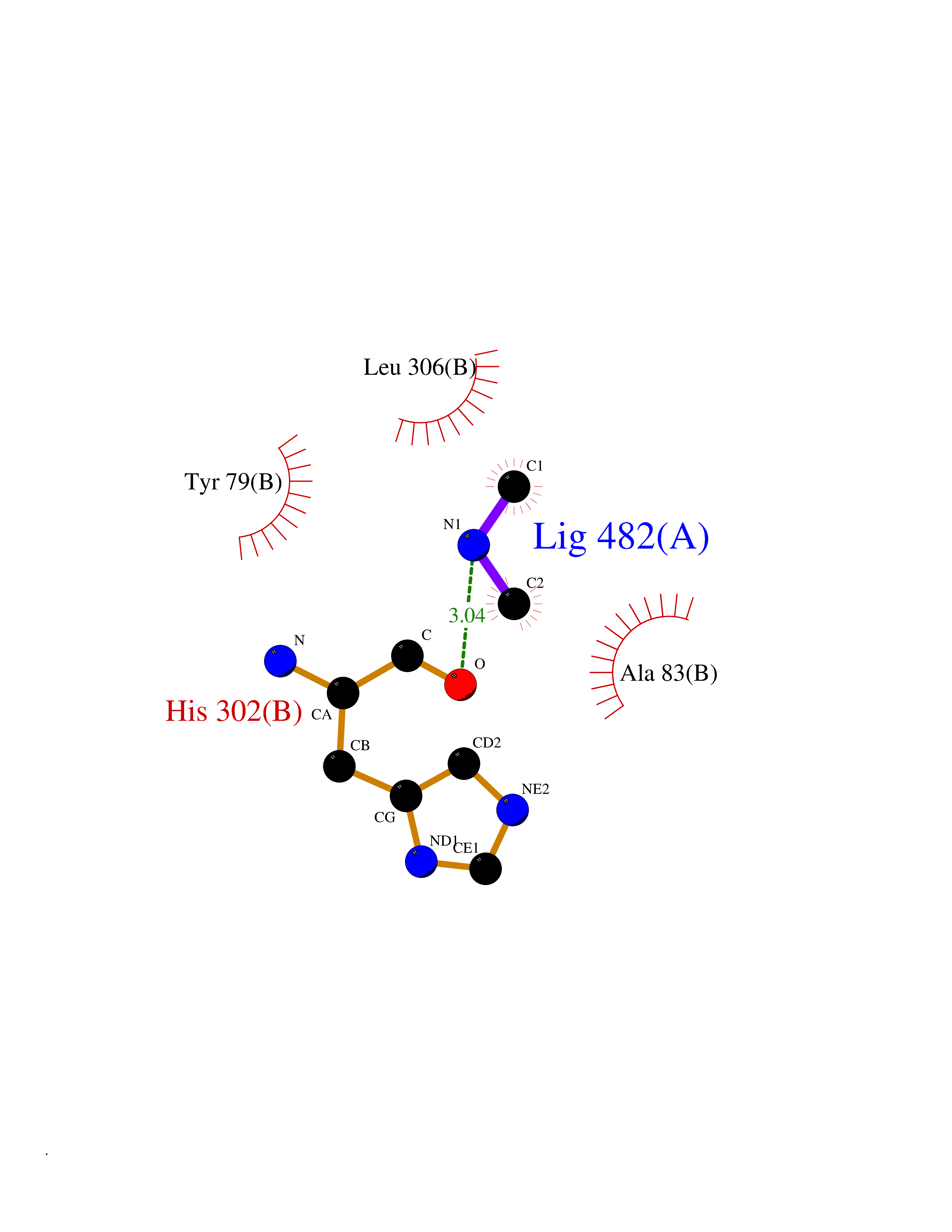 Ligplot Map 3D Binding mode Sequence HSPYFFAYFPTASSYPAMLADMLCGAIGASPACTELETVMMDWLGKMLELPKAFLNEKAGEGGGVIQGSASEATLVALLAARTKVIHRLQAASPELTQAAIMEKLVAYSSDQAHSSVERAGLIGGVKLKAIPSDGNFAMRASALQEALERDKAAGLIPFFMVATLGTTTCCSFDNLLEVGPICNKEDIWLHVDAAYAGSAFICPEFRHLLNGVEFADSFNFNPHKWLLVNFDCSAMWVKKRTDLRFRSLKMWFVFRMYGVKGLQAYIRKHVQLSHEFESLVRQDPRFEICVEVILGLVCFRLKGSNKVNEALLQRINSAKKIHLVPCHLRDKFVLRFAICSRTVESAHVQRAWEHIKELAADVLRAERE Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Catechol-O-methyl-transferase (COMT) | 3BWY | 3.99 | |
Target general information Gen name COMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms S-COMT; MB-COMT; Catechol-O-methyltransferase; COMT Protein family Class I-like SAM-binding methyltransferase superfamily, Cation-dependent O-methyltransferase family Biochemical class Methyltransferase Function Catalyzes the O-methylation, and thereby the inactivation, of catecholamine neurotransmitters and catechol hormones. Also shortens the biological half-lives of certain neuroactive drugs, like L-DOPA, alpha-methyl DOPA and isoproterenol. Related diseases Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:15645182}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07462; DB02342; DB02105; DB08049; DB00118; DB00714; DB03336; DB00286; DB00255; DB00841; DB00988; DB15488; DB00494; DB00668; DB00783; DB00977; DB01064; DB00968; DB01141; DB03907; DB04820; DB06152; DB11632; DB00252; DB01420; DB00323 Interacts with Q6P5T0; P30518; Q8NFU1; Q8NHW4; P34972; Q96BA8; P50402; Q5JX71; O14843; O00258; P08034; O75712; Q9NTQ9; O95377; Q8TDT2; Q8N6U8; O15529; P31937; Q9H2F3; O95279; Q5SR56; A6NDP7; Q0D2K0; Q7RTS5; Q9UHJ9-5; Q8IY26; Q9H6H4; Q6NTF9-3; O75783; Q99500; Q9Y6D0; Q3KNW5; O60669; P22732; Q96G79; Q5T1Q4; Q9NY26; Q9NP94; Q6P1K1; P30825; Q9UHI5; B2RUZ4; Q9UPZ6; Q96MV1; Q9NV29; A0PK00; Q9NUH8; Q9P0S9; Q14656; Q6UW68; Q9H0R3; O95807; P34981; Q15645; Q15836; O95183; O76024; P30260; Q9H816; Q92997; P29323-3; P22607; P06396; Q15323; Q6A162; P26371; O15116; P20645; O14744; Q5T160; Q9UJD0; Q2MKA7; Q8N488; O75880; Q14141; Q9UNE7; Q15645; Q9NYH9; Q8NA23-2 EC number EC 2.1.1.6 Uniprot keywords 3D-structure; Alternative initiation; Catecholamine metabolism; Cell membrane; Cytoplasm; Direct protein sequencing; Lipid metabolism; Magnesium; Membrane; Metal-binding; Methyltransferase; Neurotransmitter degradation; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Schizophrenia; Signal-anchor; Transferase; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 23851.2 Length 214 Aromaticity 0.07 Instability index 25.99 Isoelectric point 5.25 Charge (pH=7) -7.75 2D Binding mode Binding energy (Kcal/mol) -5.45  Molscript Map 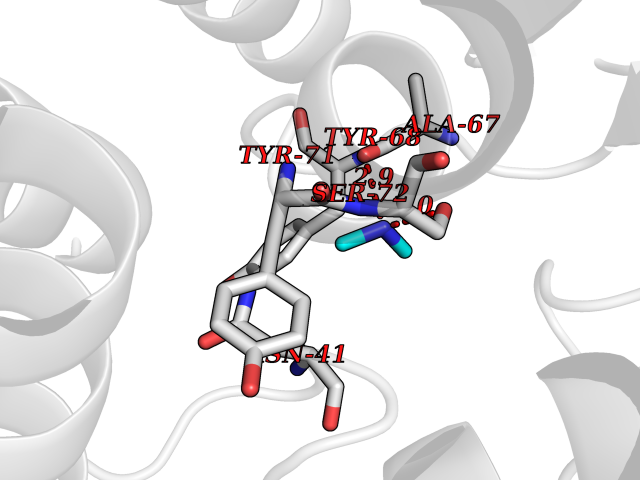 Pymol Map 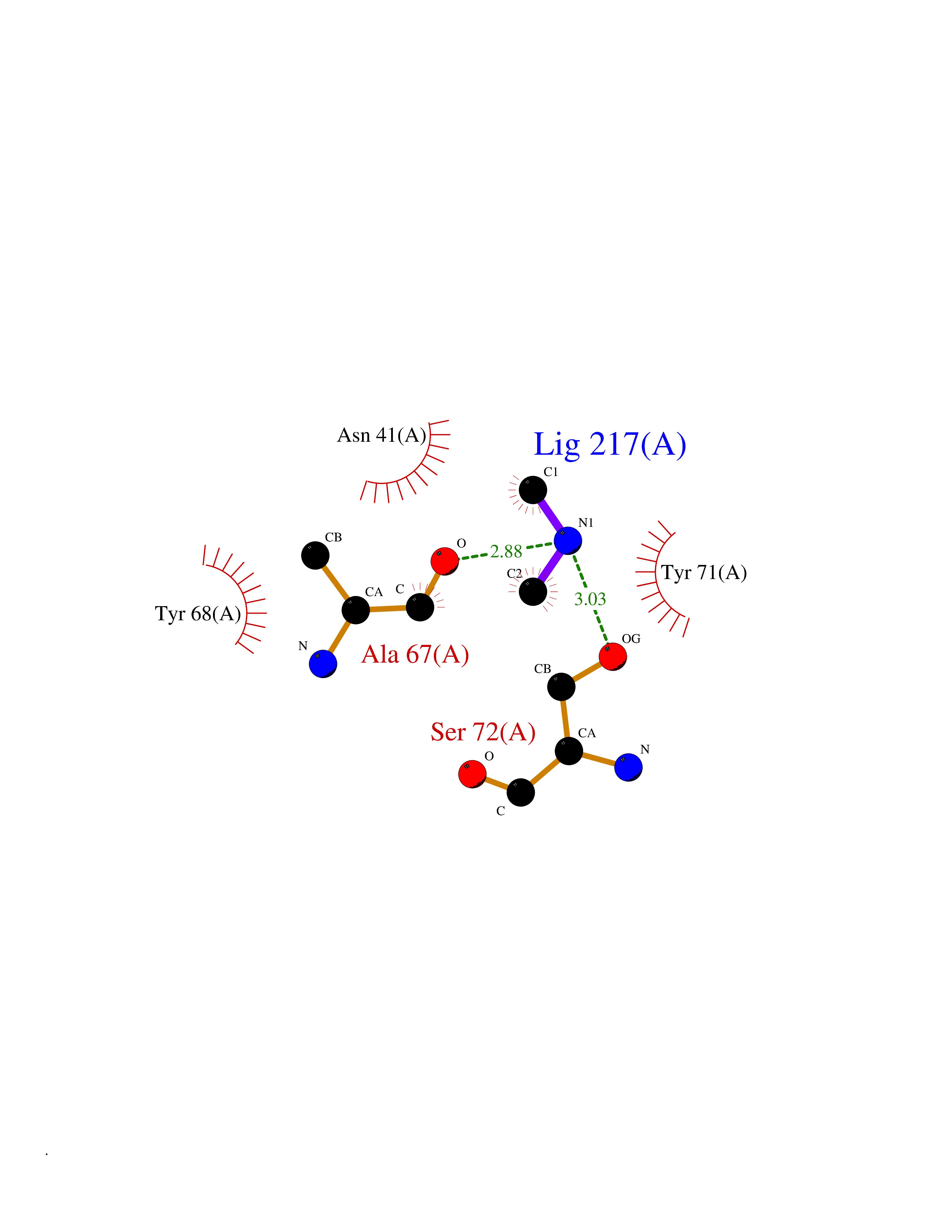 Ligplot Map 3D Binding mode Sequence GDTKEQRILNHVLQHAEPGNAQSVLEAIDTYCEQKEWAMNVGDKKGKIVDAVIQEHQPSVLLELGAYCGYSAVRMARLLSPGARLITIEINPDCAAITQRMVDFAGMKDKVTLVVGASQDIIPQLKKKYDVDTLDMVFLDHWKDRYLPDTLLLEECGLLRKGTVLLADNVICPGAPDFLAHVRGSSCFECTHYQSFLEYREVVDGLEKAIYKGP Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Vasopressin V2 receptor | 4JQI | 3.99 | |
Target general information Gen name AVPR2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ADHR;DIR3;V2R;DIR Protein family G-protein coupled receptor 1 family, Vasopressin/oxytocin receptor subfamily Biochemical class Signaling protein Function Peptide binding.Vasopressin receptor activity. Related diseases Nephrogenic syndrome of inappropriate antidiuresis (NSIAD) [MIM:300539]: Characterized by an inability to excrete a free water load, with inappropriately concentrated urine and resultant hyponatremia, hypoosmolarity, and natriuresis. {ECO:0000269|PubMed:15872203}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Diabetes insipidus, nephrogenic, 1, X-linked (NDI1) [MIM:304800]: A disorder caused by the inability of the renal collecting ducts to absorb water in response to arginine vasopressin. Characterized by excessive water drinking (polydipsia), excessive urine excretion (polyuria), persistent hypotonic urine, and hypokalemia. {ECO:0000269|PubMed:10477431, ECO:0000269|PubMed:10694923, ECO:0000269|PubMed:10770218, ECO:0000269|PubMed:10820167, ECO:0000269|PubMed:10820168, ECO:0000269|PubMed:11026555, ECO:0000269|PubMed:11232028, ECO:0000269|PubMed:11793119, ECO:0000269|PubMed:11916004, ECO:0000269|PubMed:1303257, ECO:0000269|PubMed:1303271, ECO:0000269|PubMed:1356229, ECO:0000269|PubMed:16845277, ECO:0000269|PubMed:7560098, ECO:0000269|PubMed:7607658, ECO:0000269|PubMed:7833930, ECO:0000269|PubMed:7913579, ECO:0000269|PubMed:7933835, ECO:0000269|PubMed:7984150, ECO:0000269|PubMed:7987330, ECO:0000269|PubMed:7999078, ECO:0000269|PubMed:8037205, ECO:0000269|PubMed:8045948, ECO:0000269|PubMed:8078903, ECO:0000269|PubMed:8267567, ECO:0000269|PubMed:8479490, ECO:0000269|PubMed:8514744, ECO:0000269|PubMed:8929875, ECO:0000269|PubMed:9127330, ECO:0000269|PubMed:9369448, ECO:0000269|PubMed:9402087, ECO:0000269|PubMed:9452109, ECO:0000269|PubMed:9711877, ECO:0000269|PubMed:9773787, ECO:0000269|PubMed:9853256}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09059; DB00872; DB00618; DB00035; DB14642; DB05091; DB05838; DB16279; DB02638; DB06212; DB00067 Interacts with Q8NCT1; P21964; Q8IVJ1 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Diabetes insipidus; Disease variant; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Palmitate; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID V Molecular weight (Da) 41084.9 Length 374 Aromaticity 0.07 Instability index 39.38 Isoelectric point 8.46 Charge (pH=7) 4.07 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map 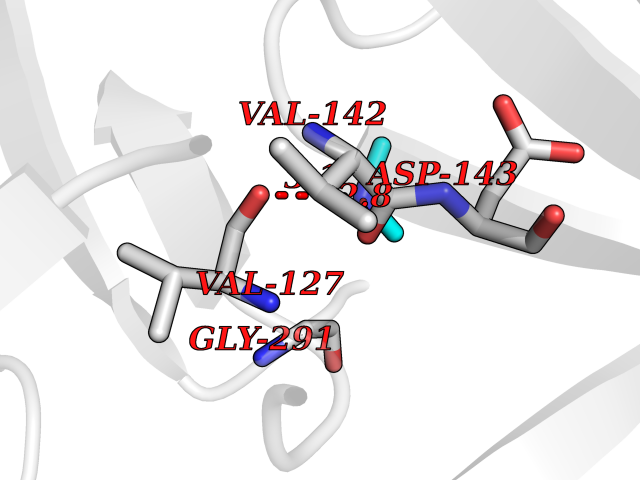 Pymol Map 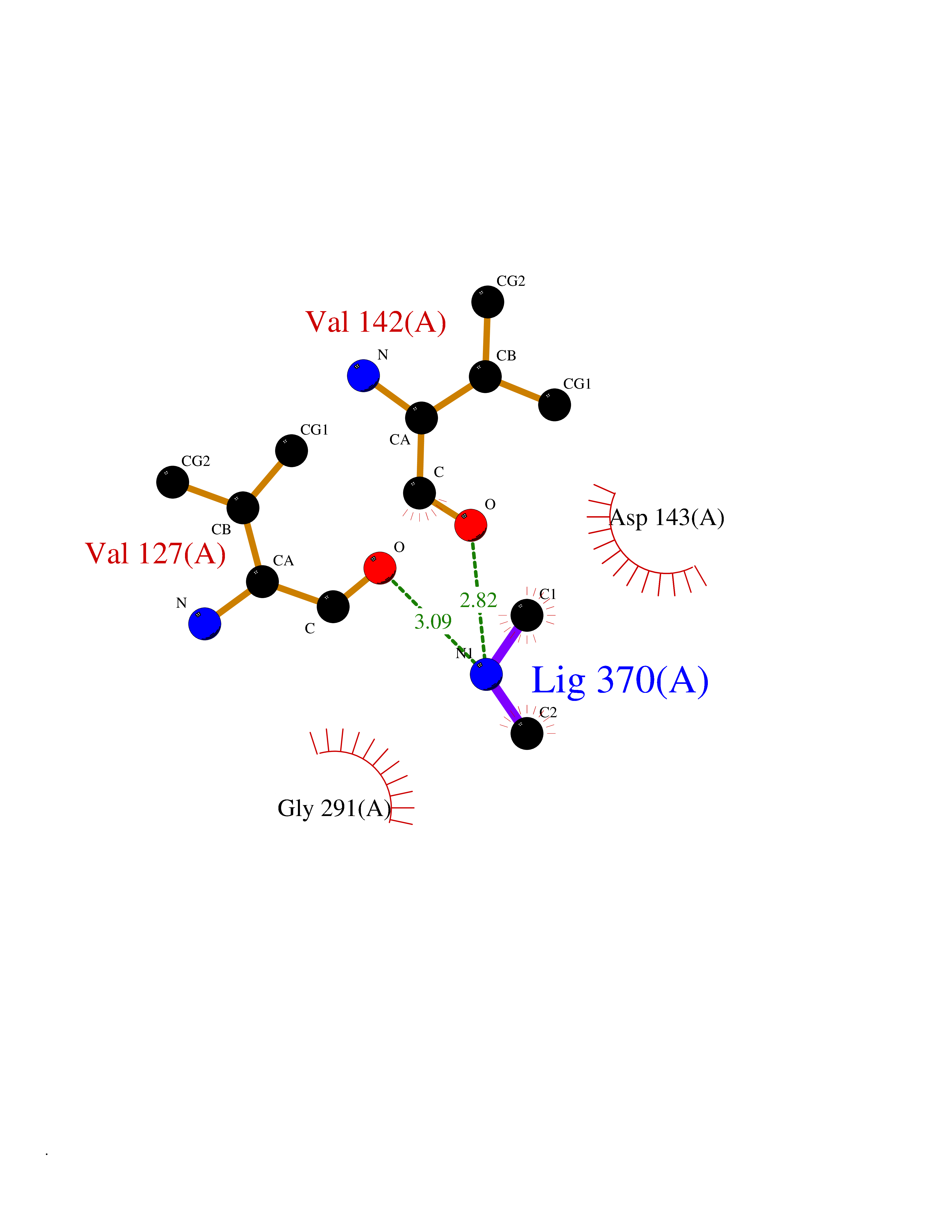 Ligplot Map 3D Binding mode Sequence TRVFKKASPNGKLTVYLGKRDFVDHIDLVDPVDGVVLVDPEYLKERRVYVTLTCAFRYGREDLDVLGLTFRKDLFVANVQSFPPAPEDKKPLTRLQERLIKKLGEHAYPFTFEIPPNLPCSVTLQPGPEDTGKACGVDYEVKAFCAENLEEKIHKRNSVRLVIRKVQYAPERPGPQPTAETTRQFLMSDKPLHLEASLDKEIYYHGEPISVNVHVTNNTNKTVKKIKISVRQYADICLFNTAQYKCPVAMEEADDTVAPSSTFCKVYTLTPFLANNREKRGLALDGKLKHEDTNLASSTLLREREILGIIVSYKVKVKLVVSRGGLLGDLASSDVAVELPFTLMHPKPKEEPPRXPPXLGPEXCXXAXXXLAKD Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | UDP-galactopyranose mutase | 1I8T | 3.99 | |
Target general information Gen name glf Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b2036;JW2021;yefE Protein family UDP-galactopyranose/dTDP-fucopyranose mutase family Biochemical class Isomerase Function Flavin adenine dinucleotide binding.UDP-galactopyranose mutase activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB03147 Interacts with P11868 EC number 5.4.99.9 Uniprot keywords 3D-structure; Direct protein sequencing; FAD; Flavoprotein; Isomerase; Lipopolysaccharide biosynthesis; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42965.3 Length 367 Aromaticity 0.14 Instability index 32.48 Isoelectric point 6.62 Charge (pH=7) -1.52 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map 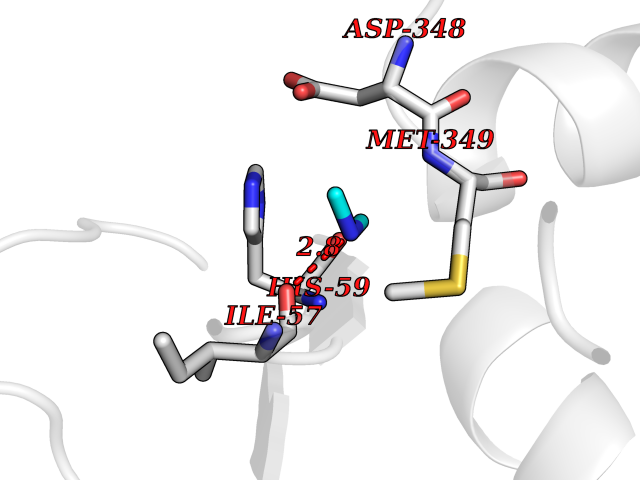 Pymol Map 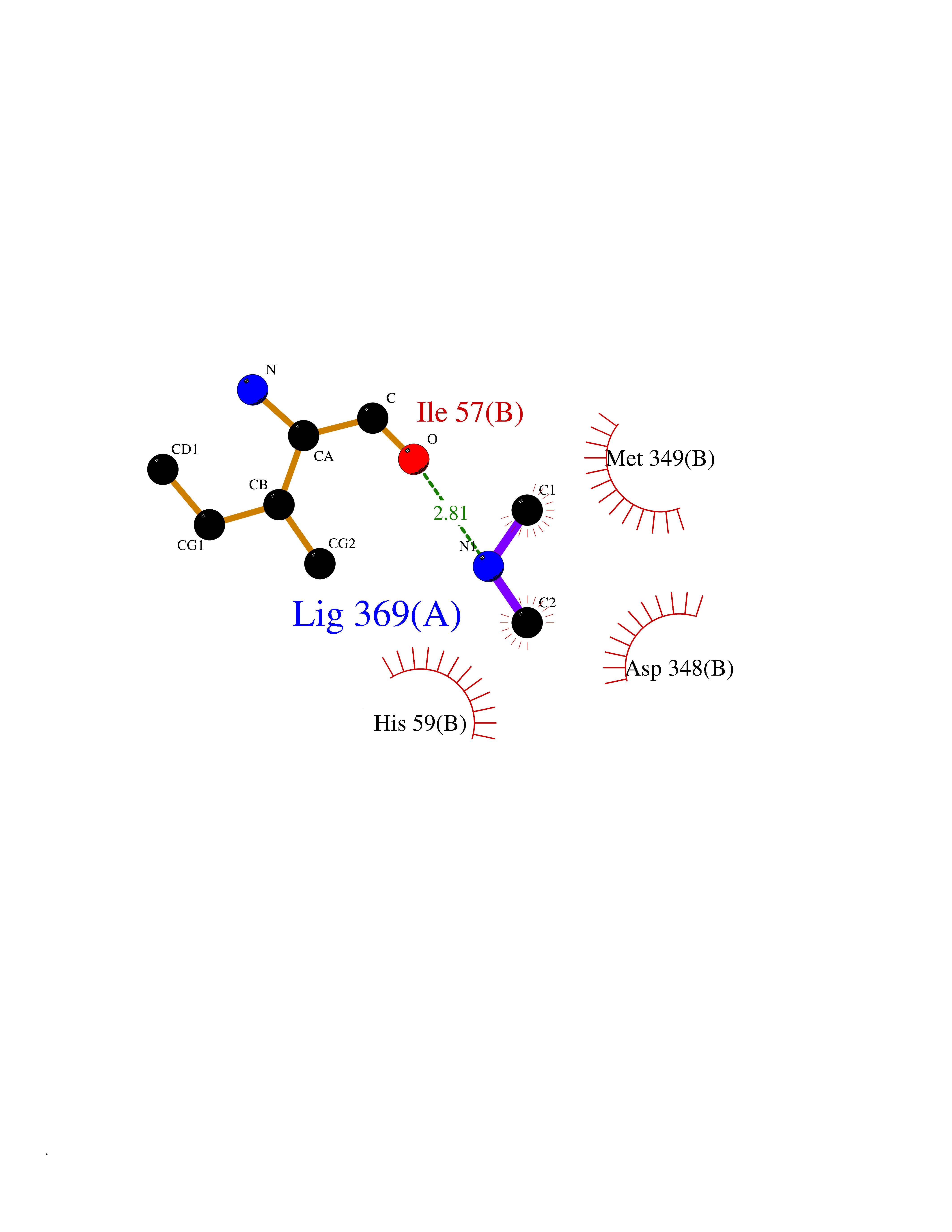 Ligplot Map 3D Binding mode Sequence MYDYIIVGSGLFGAVCANELKKLNKKVLVIEKRNHIGGNAYTEDCEGIQIHKYGAHIFHTNDKYIWDYVNDLVEFNRFTNSPLAIYKDKLFNLPFNMNTFHQMWGVKDPQEAQNIINAQKKKYGDKVPENLEEQAISLVGEDLYQALIKGYTEKQWGRSAKELPAFIIKRIPVRFTFDNNYFSDRYQGIPVGGYTKLIEKMLEGVDVKLGIDFLKDKDSLASKAHRIIYTGPIDQYFDYRFGALEYRSLKFETERHEFPNFQGNAVINFTDANVPYTRIIEHKHFDYVETKHTVVTKEYPLEWKVGDEPYYPVNDNKNMELFKKYRELASREDKVIFGGRLAEYKYYDMHQVISAALYQVKNIMSTD Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Nitric-oxide synthase brain (NOS1) | 5ADF | 3.99 | |
Target general information Gen name NOS1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Peptidyl-cysteine S-nitrosylase NOS1; Nitric oxide synthase, brain; Neuronal NOS; NOS, type I; NOS type I; NNOS; NC-NOS; N-NOS; BNOS Protein family NOS family Biochemical class Paired donor oxygen oxidoreductase Function In the brain and peripheral nervous system, NO displays many properties of a neurotransmitter. Probably has nitrosylase activity and mediates cysteine S-nitrosylation of cytoplasmic target proteins such SRR. Produces nitric oxide (NO) which is a messenger molecule with diverse functions throughout the body. Related diseases Variation Asp-298 in NOS3 may be associated with susceptibility to coronary spasm. {ECO:0000269|PubMed:11740345, ECO:0000269|PubMed:9737779}. Drugs (DrugBank ID) DB02143; DB02727; DB01997; DB03892; DB02207; DB03710; DB00155; DB00843; DB00997; DB03147; DB03247; DB01942; DB01221; DB02077; DB01821; DB09241; DB03144; DB03449; DB02044; DB02644; DB08019; DB08018; DB02027; DB03461; DB04223; DB06096; DB02991; DB03707 Interacts with Q08AM6 EC number EC 1.14.13.39 Uniprot keywords 3D-structure; Alternative splicing; Calmodulin-binding; Cell membrane; Cell projection; FAD; Flavoprotein; FMN; Heme; Iron; Membrane; Metal-binding; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Synapse; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34875.7 Length 299 Aromaticity 0.1 Instability index 42.94 Isoelectric point 5.96 Charge (pH=7) -6.25 2D Binding mode Binding energy (Kcal/mol) -5.45 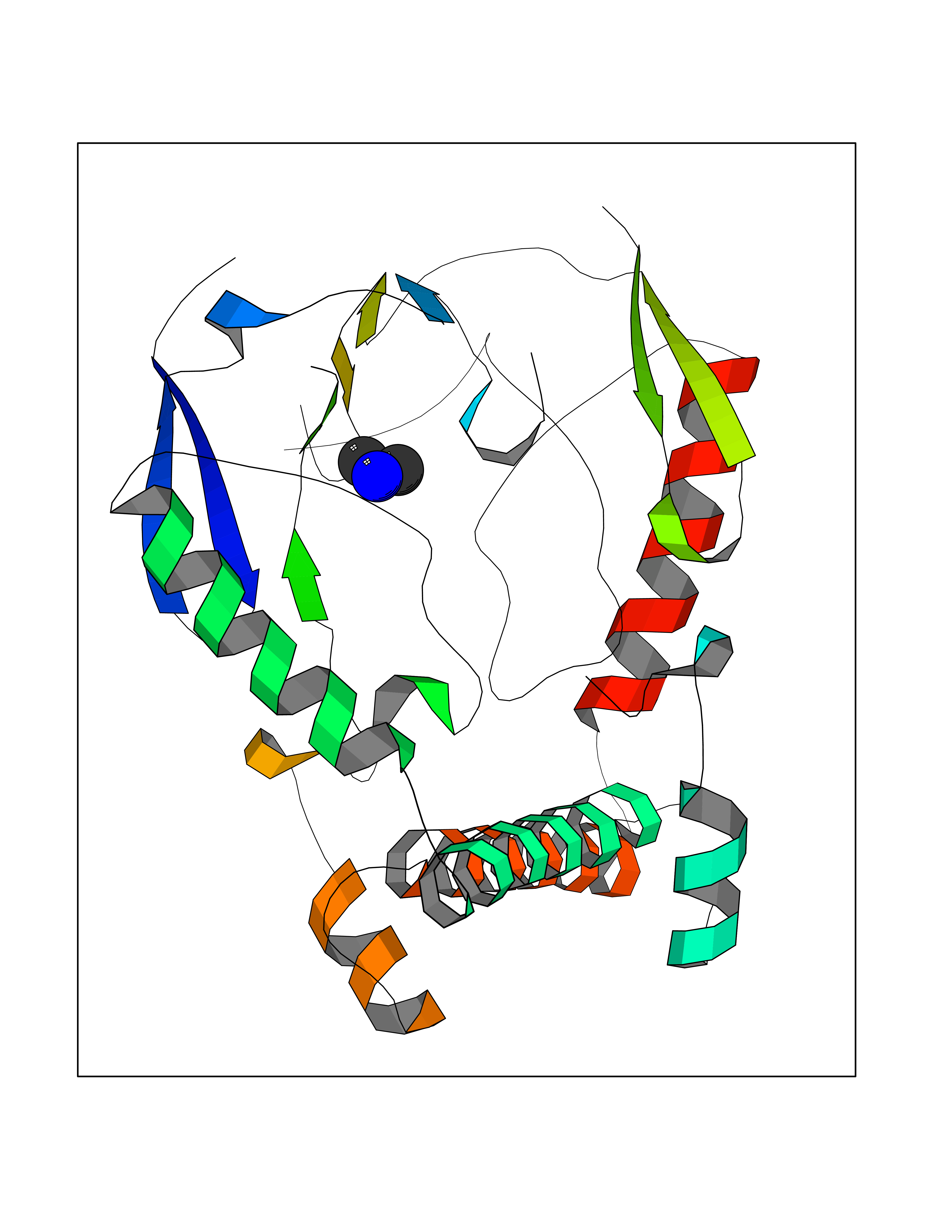 Molscript Map 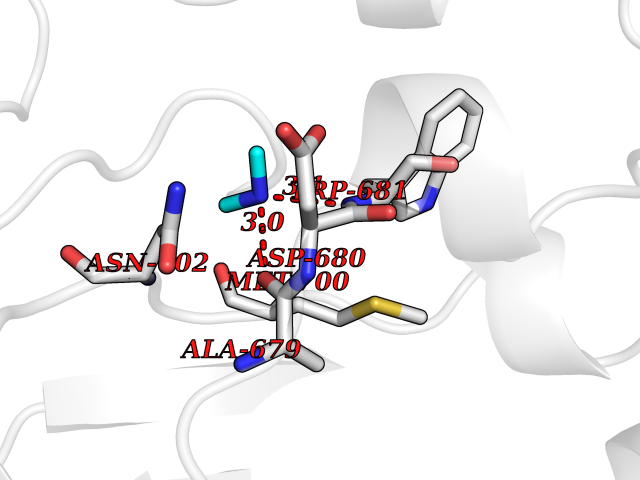 Pymol Map 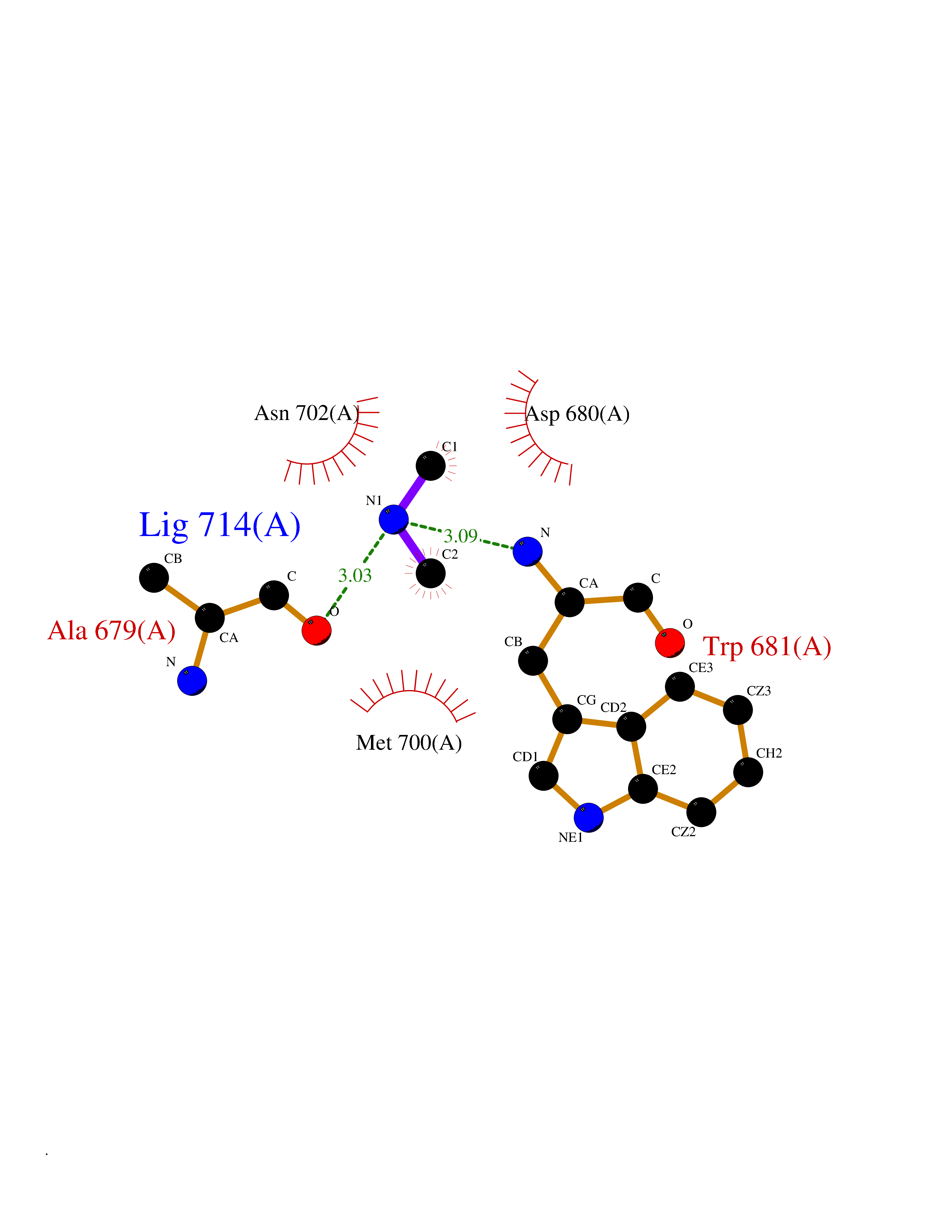 Ligplot Map 3D Binding mode Sequence CPRFLKVKNWETEVVLTDTLHLKSTLETGCTEYICMGSIMHPRDYCDNSRYNILEEVAKKMNLDMRKTSSLWKDQALVEINIAVLYSFQSDKVTIVDHHSATESFIKHMENEYRCRGGCPADWVWIVPPMSGSITPVFHQEMLNYRLRFLKVKNWETEVVLTDTLHLKSTLETGCTEYICMGSIMHPRDYCDNSRYNILEEVAKKMNLDMRKTSSLWKDQALVEINIAVLYSFQSDKVTIVDHHSATESFIKHMENEYRCRGGCPADWVWIVPPMSGSITPVFHQEMLNYRLTPSFEYQ Hydrogen bonds contact Hydrophobic contact | ||||