Job Results:
Ligand
Structure
Job ID
4d96340bab84003cd843e60272b3611c
Job name
NA
Time
2025-02-13 15:23:33
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | 5,10-methylenetetrahydrofolate reductase | 3FST | 3.99 | |
Target general information Gen name metF Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b3941;JW3913 Protein family Methylenetetrahydrofolate reductase family Biochemical class Oxidoreductase Function FAD binding.Methylenetetrahydrofolate reductase (NAD(P)H) activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.5.1.54 Uniprot keywords 3D-structure; Amino-acid biosynthesis; FAD; Flavoprotein; Methionine biosynthesis; NAD; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A,C,E Molecular weight (Da) 30855.9 Length 274 Aromaticity 0.09 Instability index 27.54 Isoelectric point 5.84 Charge (pH=7) -4.61 2D Binding mode Binding energy (Kcal/mol) -5.44 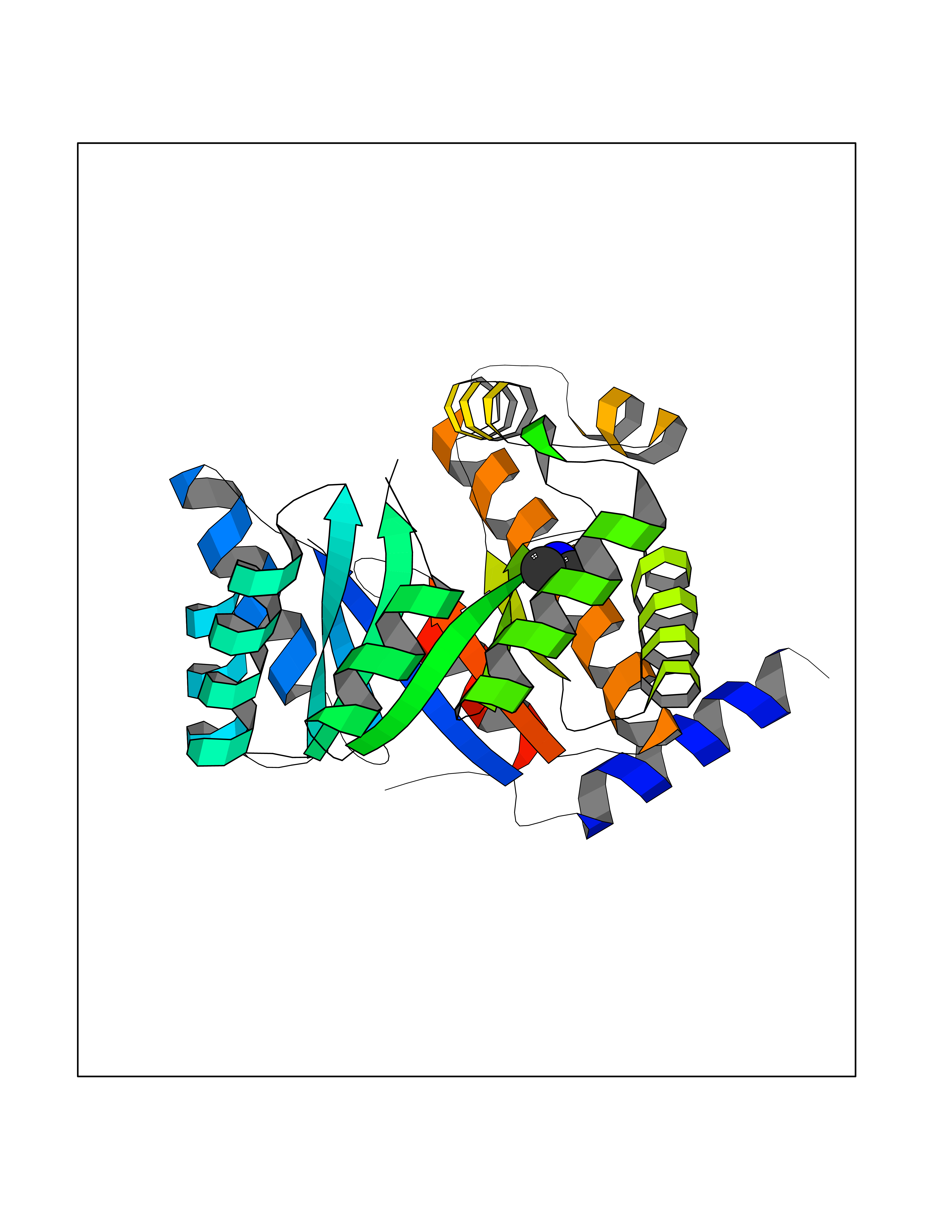 Molscript Map 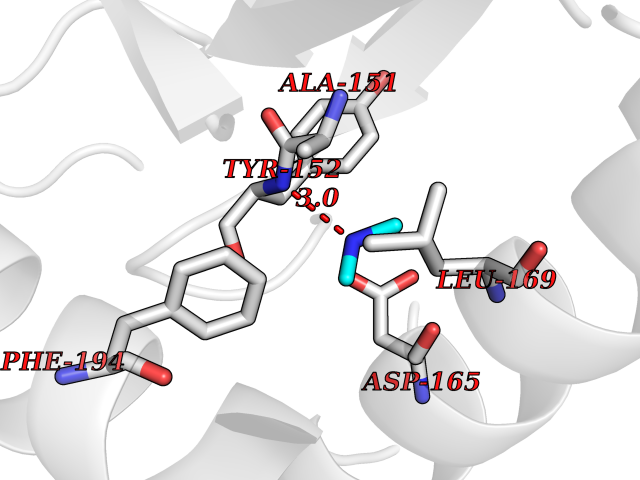 Pymol Map 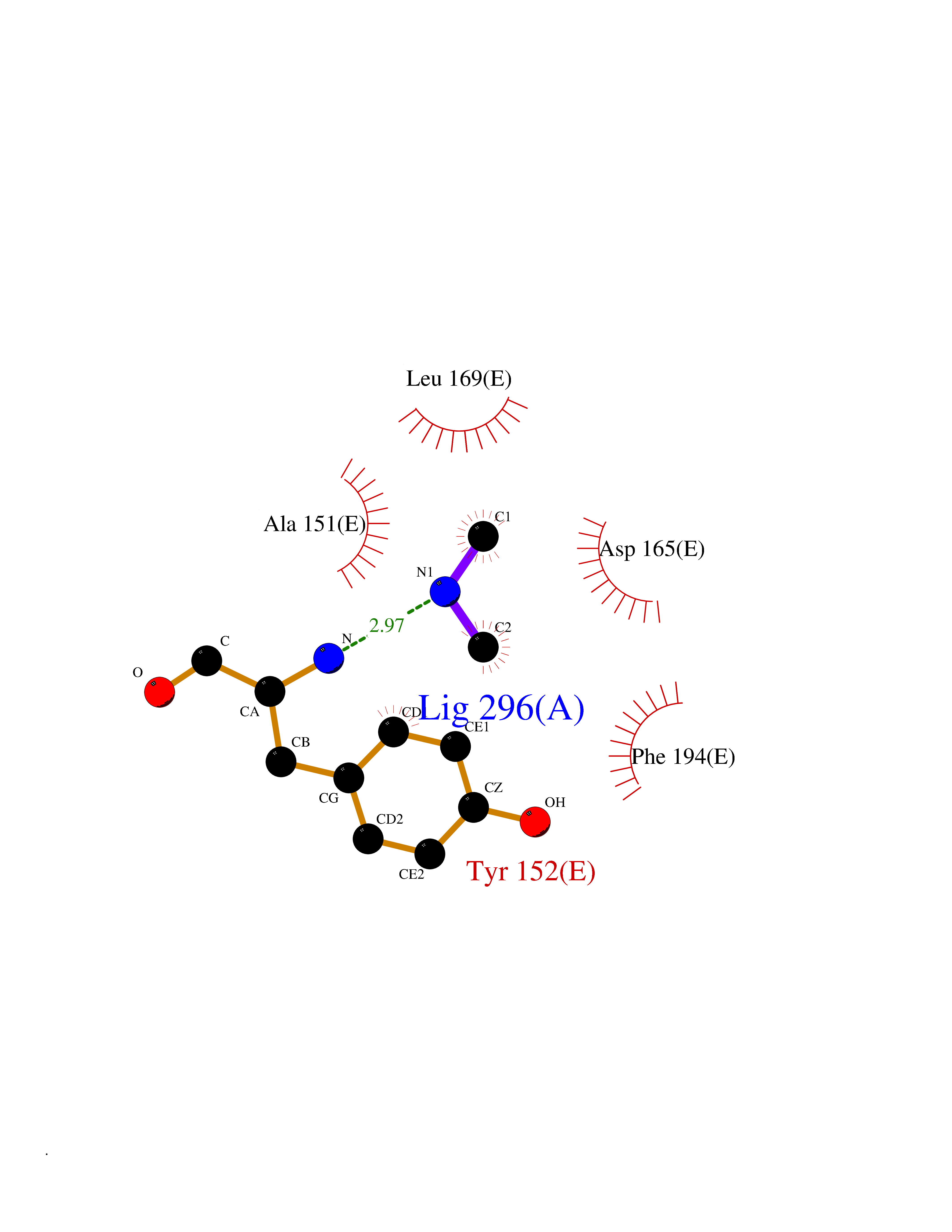 Ligplot Map 3D Binding mode Sequence FHASQRDALNQSLAEVQGQINVSFEFFPPRTSEMEQTLWNSIDRLSSLKPKFVSVTYTHSIIKGIKDRTGLEAAPHLTCIDATPDELRTIARDYWNNGIRHIVALRGDEMYASDLVTLLKEVADFDISVAAYPEVHPEAKSAQADLLNLKRKVDAGANRAITQFFFDVESYLRFRDRCVSAGIDVEIIPGILPVSNFKQAKKLADMTNVRIPAWMAQMFDGLDDDAETRKLVGANIAMDMVKILSREGVKDFHFYTLNRAEMSYAICHTLGVRP Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Bifunctional methylenetetrahydrofolate dehydrogenase/cyclohydrolase, mitochondrial | 5TC4 | 3.99 | |
Target general information Gen name MTHFD2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NMDMC Protein family Tetrahydrofolate dehydrogenase/cyclohydrolase family Biochemical class Oxidoreductase Function Magnesium ion binding.Methenyltetrahydrofolate cyclohydrolase activity.Methylenetetrahydrofolate dehydrogenase (NAD+) activity.Methylenetetrahydrofolate dehydrogenase (NADP+) activity.Phosphate ion binding. Related diseases Acute hepatic porphyria (AHEPP) [MIM:612740]: A form of porphyria. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. AHP is characterized by attacks of gastrointestinal disturbances, abdominal colic, paralyses and peripheral neuropathy. Most attacks are precipitated by drugs, alcohol, caloric deprivation, infections, or endocrine factors. {ECO:0000269|PubMed:10706561, ECO:0000269|PubMed:1309003, ECO:0000269|PubMed:1569184, ECO:0000269|PubMed:17236137, ECO:0000269|PubMed:2063868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00157; DB00116 Interacts with Q9UJ70-2 EC number 1.5.1.15; 3.5.4.9 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Hydrolase; Isopeptide bond; Magnesium; Mitochondrion; Multifunctional enzyme; NAD; NADP; One-carbon metabolism; Oxidoreductase; Proteomics identification; Reference proteome; Transit peptide; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 31600.3 Length 292 Aromaticity 0.03 Instability index 27.9 Isoelectric point 8 Charge (pH=7) 1.64 2D Binding mode Binding energy (Kcal/mol) -5.44 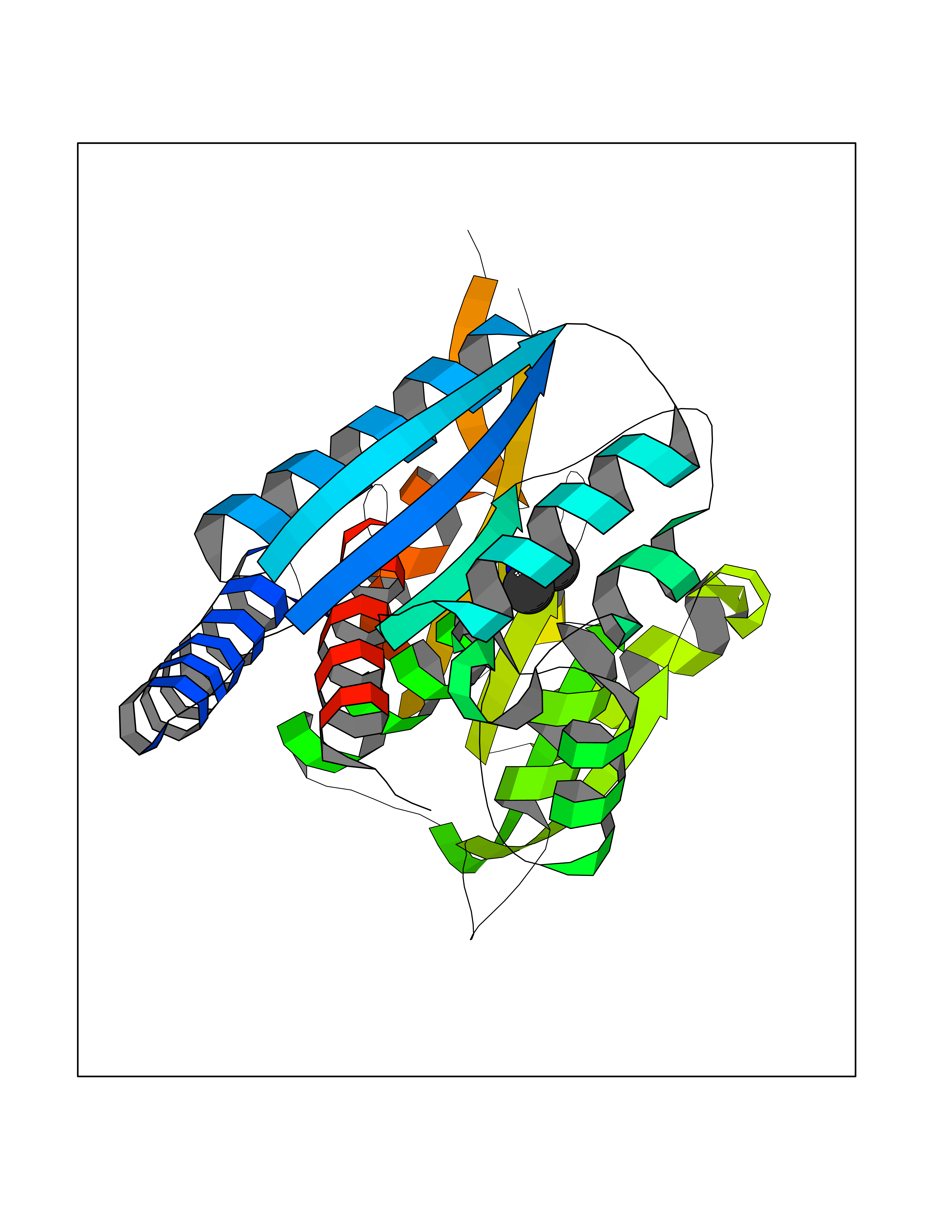 Molscript Map 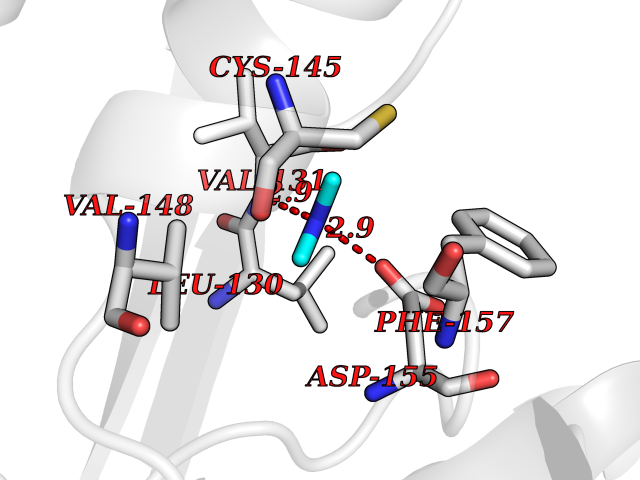 Pymol Map 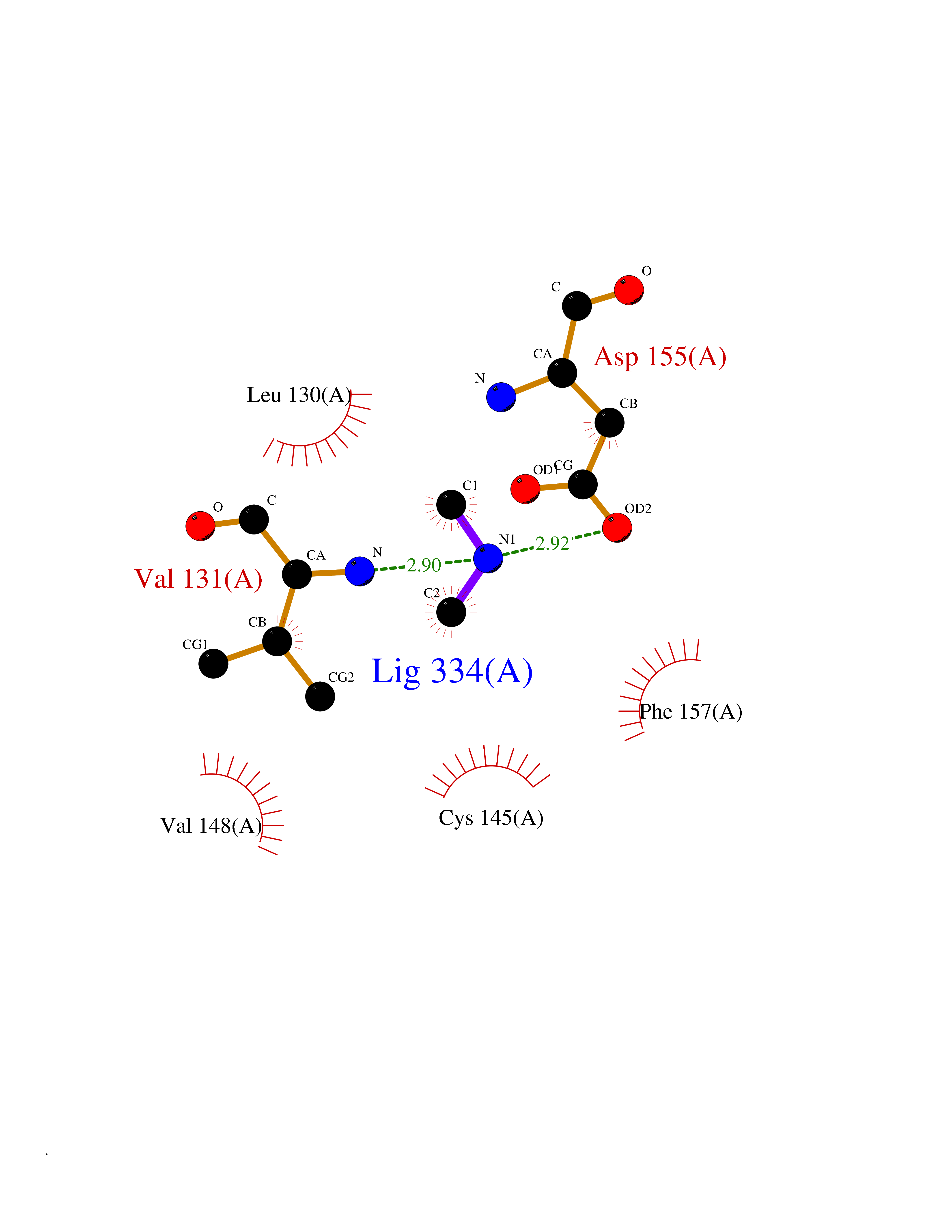 Ligplot Map 3D Binding mode Sequence EAVVISGRKLAQQIKQEVRQEVEEWVASGNKRPHLSVILVGENPASHSYVLNKTRAAAVVGINSETIMKPASISEEELLNLINKLNNDDNVDGLLVQLPLPEHIDERRICNAVSPDKDVDGFHVINVGRMCLDQYSMLPATPWGVWEIIKRTGIPTLGKNVVVAGRSKNVGMPIAMLLHTDGAHERPGGDATVTISHRYTPKEQLKKHTILADIVISAAGIPNLITADMIKEGAAVIDVGINRVHKPKLVGDVDFEGVRQKAGYITPVPGGVGPMTVAMLMKNTIIAAKKVL Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Endolysin | 1AM7 | 3.99 | |
Target general information Gen name R Organism Escherichia phage lambda (Bacteriophage lambda) Uniprot ID TTD ID NA Synonyms NA Protein family Glycosyl hydrolase 24 family Biochemical class Glycosidase Function Lyase activity.Lysozyme activity.Lytic transglycosylase activity. Related diseases Estrogen resistance (ESTRR) [MIM:615363]: A disorder characterized by partial or complete resistance to estrogens, in the presence of elevated estrogen serum levels. Clinical features include absence of the pubertal growth spurt, delayed bone maturation, unfused epiphyses, reduced bone mineral density, osteoporosis, continued growth into adulthood and very tall adult stature. Glucose intolerance, hyperinsulinemia and lipid abnormalities may also be present. {ECO:0000269|PubMed:23841731, ECO:0000269|PubMed:27754803}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04206 Interacts with NA EC number 4.2.2.n2 Uniprot keywords 3D-structure; Antimicrobial; Bacteriolytic enzyme; Cytolysis; Direct protein sequencing; Host cell lysis by virus; Host cytoplasm; Lyase; Reference proteome; Viral release from host cell Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 49834.9 Length 462 Aromaticity 0.07 Instability index 18.78 Isoelectric point 9.6 Charge (pH=7) 18.29 2D Binding mode Binding energy (Kcal/mol) -5.44 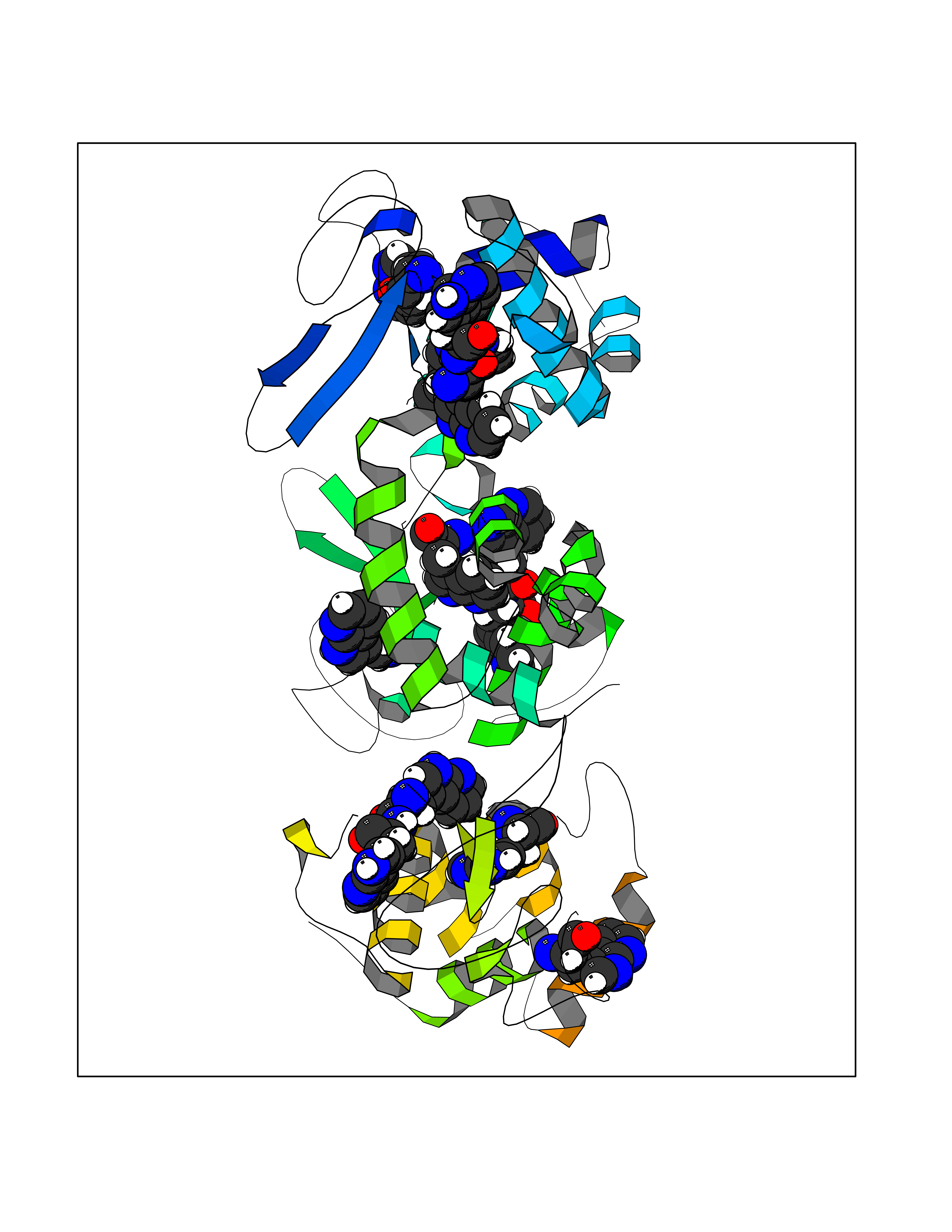 Molscript Map 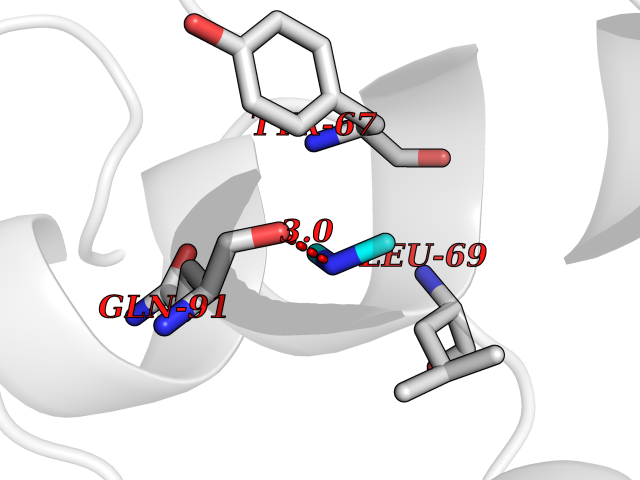 Pymol Map 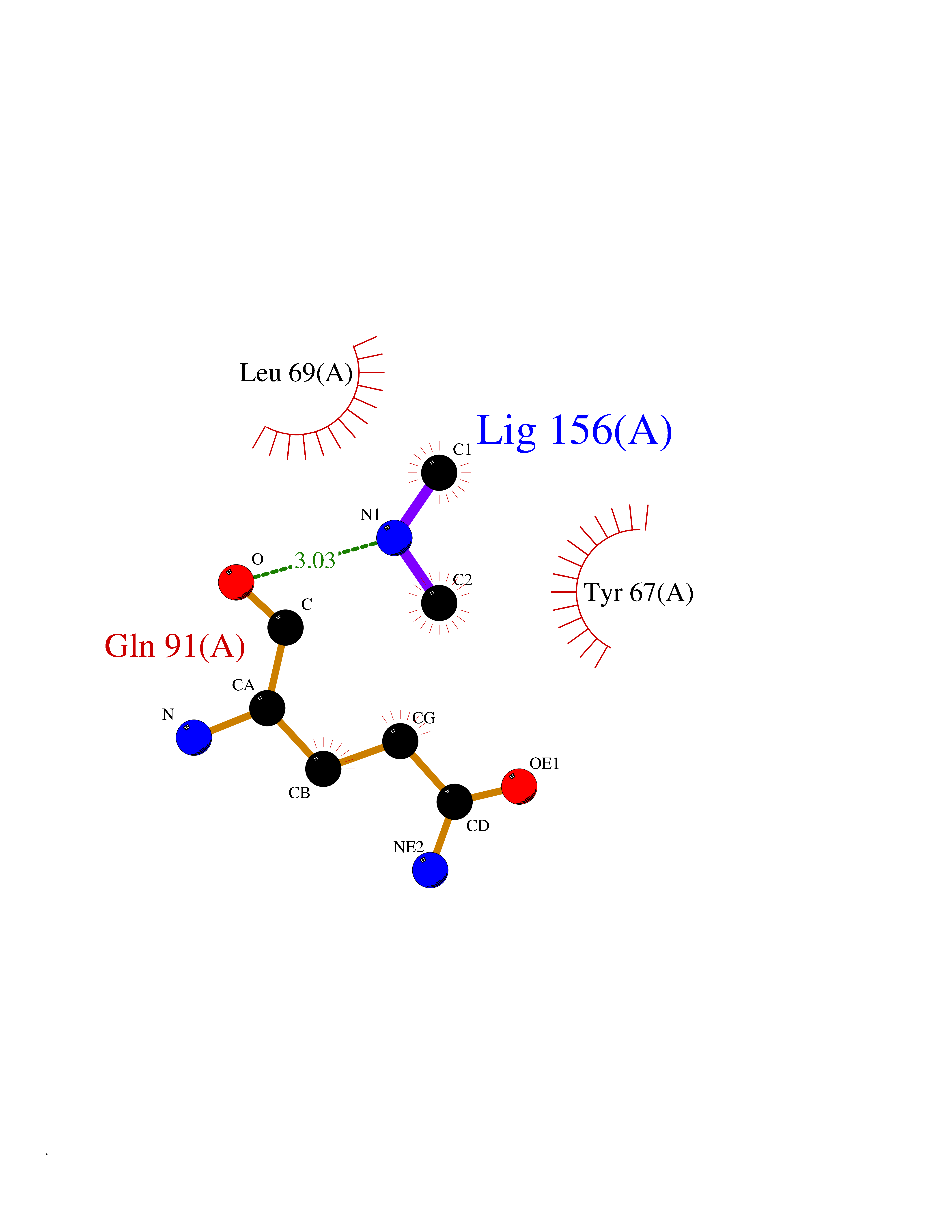 Ligplot Map 3D Binding mode Sequence MVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVRMVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVRMVEINNQRKAFLDMLAXSEGTDNGRQKTRNHGYDVIVGGELFTDYSDHPRKLVTLNPKLKSTGAGRYQLLSRXXDAYRKQLGLKDFSPKSQDAVALQQIKERGALPMIDRGDIRQAIDRCSNIXASLPGAGYGQFEHKADSLIAKFKEAGGTVR Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Alcohol dehydrogenase 1C | 1U3W | 3.99 | |
Target general information Gen name ADH1C Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ADH3 Protein family Zinc-containing alcohol dehydrogenase family Biochemical class Oxidoreductase Function Alcohol dehydrogenase (NAD) activity.Alcohol dehydrogenase activity, zinc-dependent.Ethanol binding.Retinol dehydrogenase activity.Zinc ion binding. Related diseases Combined oxidative phosphorylation deficiency 6 (COXPD6) [MIM:300816]: A mitochondrial disease resulting in a neurodegenerative disorder characterized by psychomotor delay, hypotonia, areflexia, muscle weakness and wasting. Some patients manifest prenatal ventriculomegaly and severe postnatal encephalomyopathy. {ECO:0000269|PubMed:20362274, ECO:0000269|PubMed:22019070, ECO:0000269|PubMed:25583628, ECO:0000269|PubMed:26004228, ECO:0000269|PubMed:26173962, ECO:0000269|PubMed:27178839}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, X-linked recessive, 4, with or without cerebellar ataxia (CMTX4) [MIM:310490]: A neuromuscular disorder characterized by progressive sensorimotor axonal neuropathy, distal sensory impairment, difficulty walking due to peripheral neuropathy and/or cerebellar ataxia, and deafness due to auditory neuropathy. Additional features include cognitive impairment, cerebellar atrophy, dysarthria, abnormal extraocular movements, tremor, dysmetria and spasticity. The age at onset ranges from infancy to young adulthood. {ECO:0000269|PubMed:23217327, ECO:0000269|PubMed:26004228}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, X-linked, 5, with peripheral neuropathy (DFNX5) [MIM:300614]: A form of hearing loss characterized by absent or severely abnormal auditory brainstem response, abnormal middle ear reflexes, abnormal speech discrimination, loss of outer hair cell function, and cochlear nerve hypoplasia. DFNX5 patients manifest auditory neuropathy with childhood onset, associated with distal sensory impairment affecting the peripheral nervous system. {ECO:0000269|PubMed:25986071}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepimetaphyseal dysplasia, X-linked, with hypomyelinating leukodystrophy (SEMDHL) [MIM:300232]: An X-linked recessive developmental disorder characterized by slowly progressive skeletal and neurologic abnormalities, including short stature, large and deformed joints, significant motor impairment, visual defects, and sometimes cognitive deficits. Affected individuals typically have normal early development in the first year or so of life, followed by development regression and the development of symptoms. Brain imaging shows white matter abnormalities consistent with hypomyelinating leukodystrophy. {ECO:0000269|PubMed:28842795}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03061; DB01711; DB04312; DB04448; DB02249; DB02871; DB02721; DB03020; DB02659; DB04071; DB03559; DB00898; DB01213; DB02131; DB04113; DB00157; DB02822; DB02757; DB03226; DB14487; DB14533; DB14548 Interacts with P00325 EC number 1.1.1.1 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Metal-binding; NAD; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 39693.9 Length 374 Aromaticity 0.06 Instability index 23.06 Isoelectric point 8.53 Charge (pH=7) 5.87 2D Binding mode Binding energy (Kcal/mol) -5.45  Molscript Map 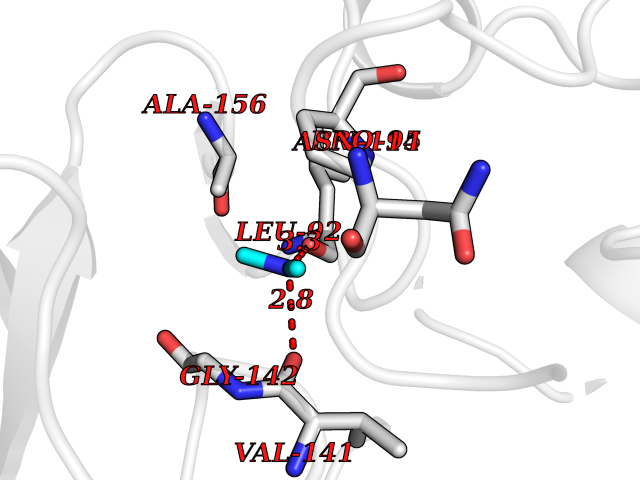 Pymol Map 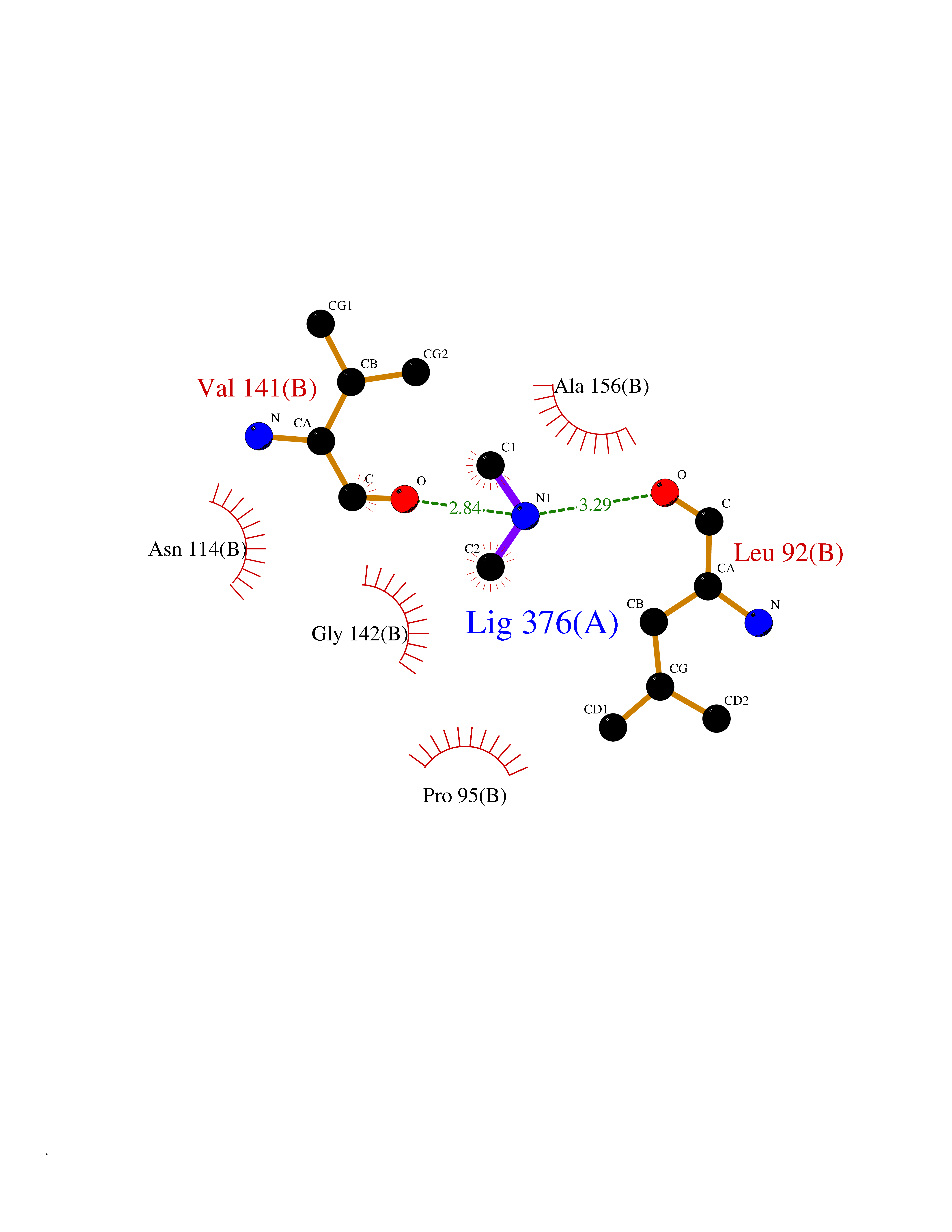 Ligplot Map 3D Binding mode Sequence STAGKVIKCKAAVLWELKKPFSIEEVEVAPPKAHEVRIKMVAAGICRSDEHVVSGNLVTPLPVILGHEAAGIVESVGEGVTTVKPGDKVIPLFTPQCGKCRICKNPESNYCLKNDLGNPRGTLQDGTRRFTCSGKPIHHFVGVSTFSQYTVVDENAVAKIDAASPLEKVCLIGCGFSTGYGSAVKVAKVTPGSTCAVFGLGGVGLSVVMGCKAAGAARIIAVDINKDKFAKAKELGATECINPQDYKKPIQEVLKEMTDGGVDFSFEVIGQLDTMMASLLCCHEACGTSVIVGVPPDSQNLSINPMLLLTGRTWKGAIFGGFKSKESVPKLVADFMAKKFSLDALITNVLPFEKINEGFDLLRSGKSIRTVLTF Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Transferrin (TF) | 1RYO | 3.99 | |
Target general information Gen name TF Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Siderophilin; Serotransferrin; PRO1400; Beta-1 metal-binding globulin Protein family Transferrin family Biochemical class Transferrin Function It is responsible for the transport of iron from sites of absorption and heme degradation to those of storage and utilization. Serum transferrin may also have a further role in stimulating cell proliferation. Transferrins are iron binding transport proteins which can bind two Fe(3+) ions in association with the binding of an anion, usually bicarbonate. Related diseases Atransferrinemia (ATRAF) [MIM:209300]: A rare autosomal recessive disorder characterized by abnormal synthesis of transferrin leading to iron overload and microcytic hypochromic anemia. {ECO:0000269|PubMed:11110675, ECO:0000269|PubMed:15466165}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01370; DB14517; DB14518; DB01294; DB14526; DB14527; DB11136; DB14528; DB14529; DB14530; DB00515; DB09130; DB11397; DB13949; DB14490; DB14491; DB14488; DB14501; DB14489; DB13257; DB06215; DB06784; DB05260; DB01592; DB00893; DB00677; DB06757; DB11182; DB14520; DB01593; DB14487; DB14533; DB14548 Interacts with O43315; O00501; Q7Z7G2; Q9GZR5; Q9Y282; Q96KR6; P01350; P08034; Q8NBJ4; O15529; Q8TED1; Q7Z5P4; A8MZ59; O15173; Q96TC7; Q3KNW5; Q9BXS9-3; Q99523; O43278-2; Q8N9I0; P02786; Q4KMG9; Q9K0U9; Q09057; Q9K0V0; P02786 EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Ion transport; Iron; Iron transport; Metal-binding; Methylation; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 35854.5 Length 324 Aromaticity 0.1 Instability index 34.17 Isoelectric point 7.58 Charge (pH=7) 1.42 2D Binding mode Binding energy (Kcal/mol) -5.44 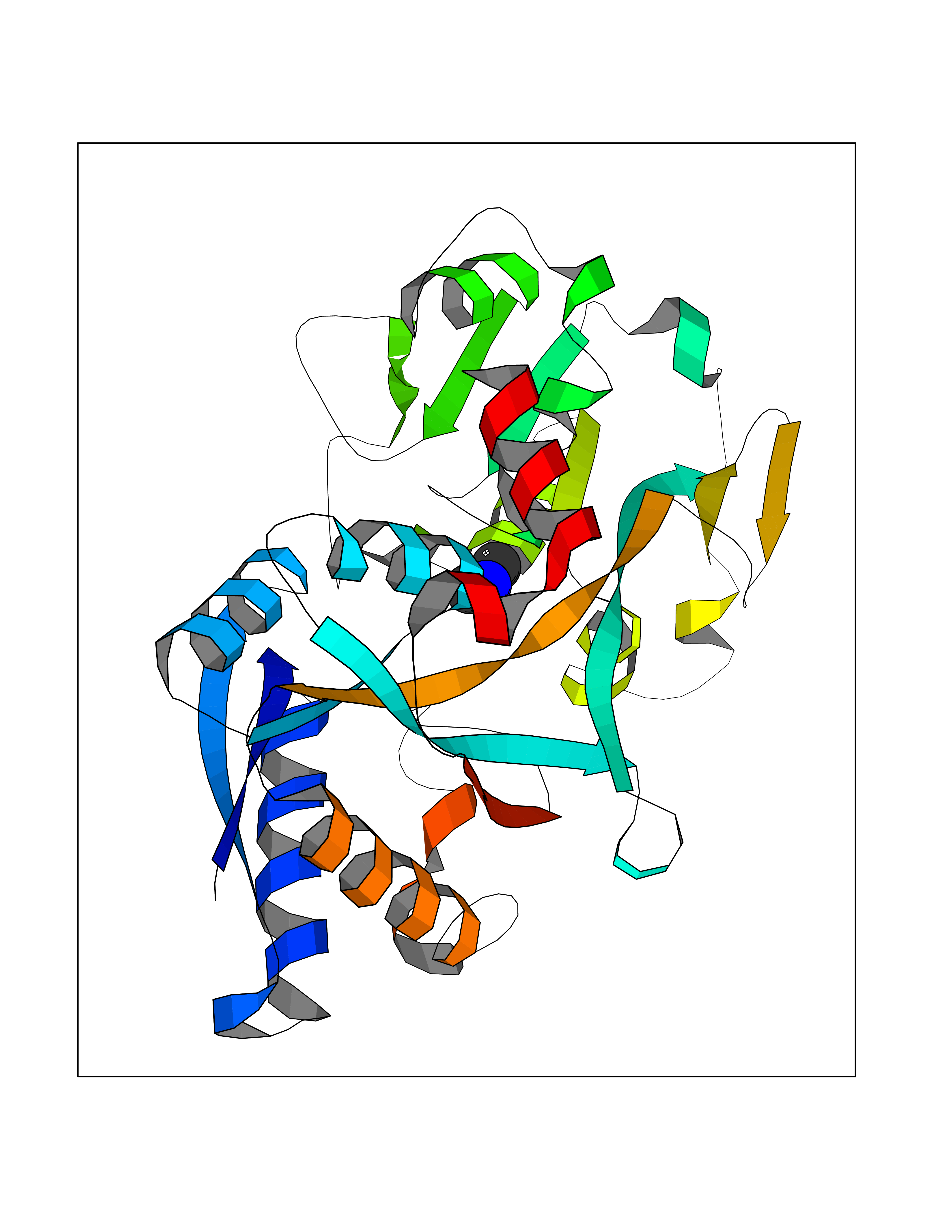 Molscript Map 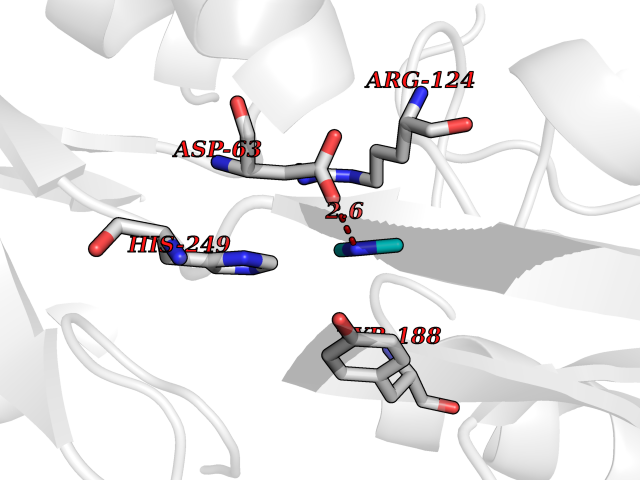 Pymol Map 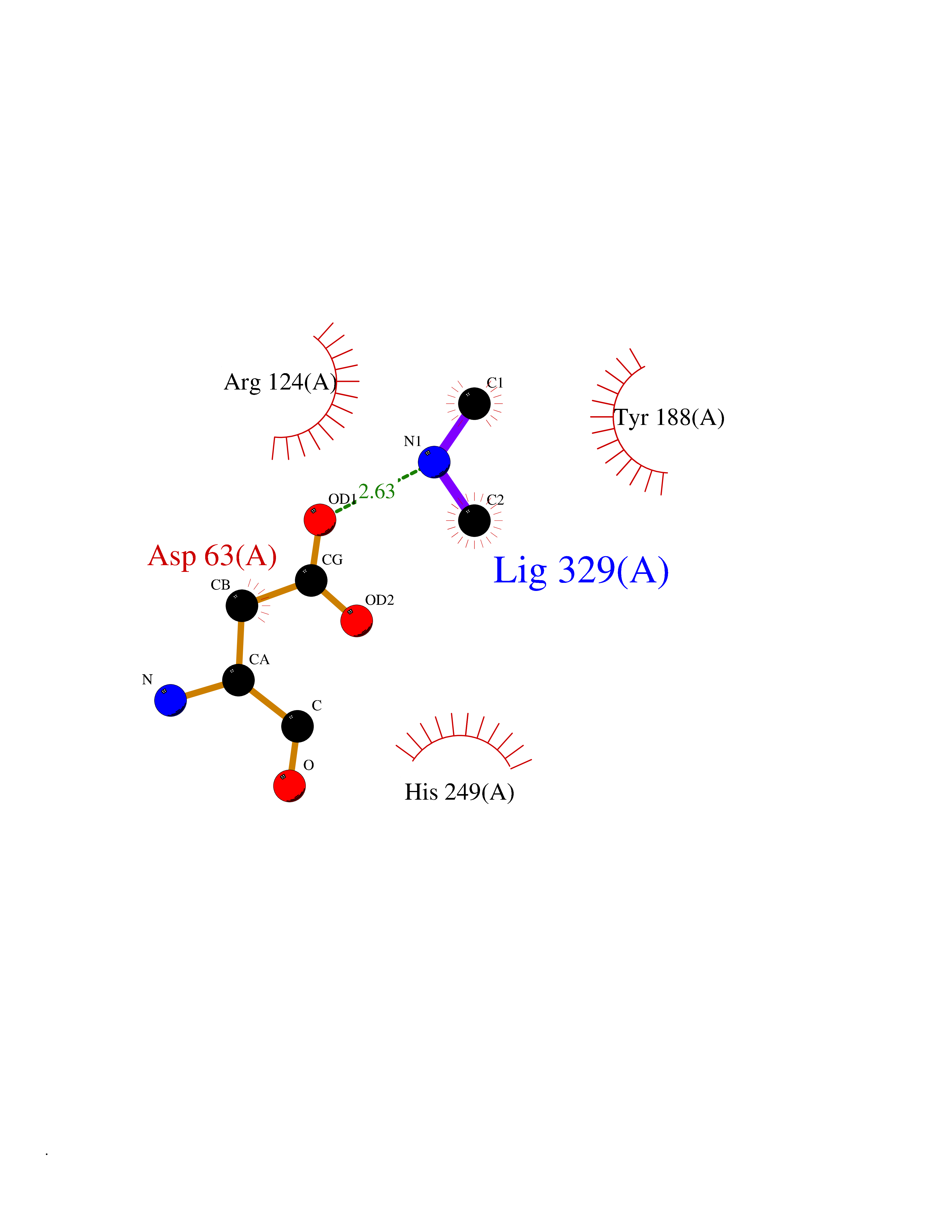 Ligplot Map 3D Binding mode Sequence KTVRWCAVSEHEATKCQSFRDHMKSVIPSDGPSVACVKKASYLDCIRAIAANEADAVTLDAGLVYDAYLAPNNLKPVVAEFYGSKEDPQTFYYAVAVVKKDSGFQMNQLRGKKSCHTGLGRSAGWNIPIGLLYCDLPEPRKPLEKAVANFFSGSCAPCADGTDFPQLCQLCPGCGCSTLNQYFGYSGAFKCLKDGAGDVAFVKHSTIFENLANKADRDQYELLCLDNTRKPVDEYKDCHLAQVPSHTVVARSMGGKEDLIWELLNQAQEHFGKDKSKEFQLFSSPHGKDLLFKDSAHGFLKVPPRMDAKMYLGYEYVTAIRNLR Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | 4-cresol dehydrogenase [hydroxylating] flavoprotein subunit | 1WVF | 3.99 | |
Target general information Gen name pchF Organism Pseudomonas putida (Arthrobacter siderocapsulatus) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Oxidoreductase Function 4-cresol dehydrogenase (hydroxylating) activity.Flavin adenine dinucleotide binding.Oxidoreductase activity, acting on CH-OH group of donors. Related diseases Dihydrolipoamide dehydrogenase deficiency (DLDD) [MIM:246900]: An autosomal recessive metabolic disorder characterized biochemically by a combined deficiency of the branched-chain alpha-keto acid dehydrogenase complex (BCKDC), pyruvate dehydrogenase complex (PDC), and alpha-ketoglutarate dehydrogenase complex (KGDC). Clinically, affected individuals have lactic acidosis and neurologic deterioration due to sensitivity of the central nervous system to defects in oxidative metabolism. {ECO:0000269|PubMed:10448086, ECO:0000269|PubMed:11687750, ECO:0000269|PubMed:12925875, ECO:0000269|PubMed:15712224, ECO:0000269|PubMed:16442803, ECO:0000269|PubMed:16770810, ECO:0000269|PubMed:17404228, ECO:0000269|PubMed:20160912, ECO:0000269|PubMed:8506365, ECO:0000269|PubMed:8968745, ECO:0000269|PubMed:9540846, ECO:0000269|PubMed:9934985}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.17.9.1 Uniprot keywords 3D-structure; Direct protein sequencing; FAD; Flavoprotein; Oxidoreductase; Plasmid Protein physicochemical properties Chain ID A Molecular weight (Da) 57240.8 Length 515 Aromaticity 0.1 Instability index 30.94 Isoelectric point 6.06 Charge (pH=7) -4.42 2D Binding mode Binding energy (Kcal/mol) -5.44 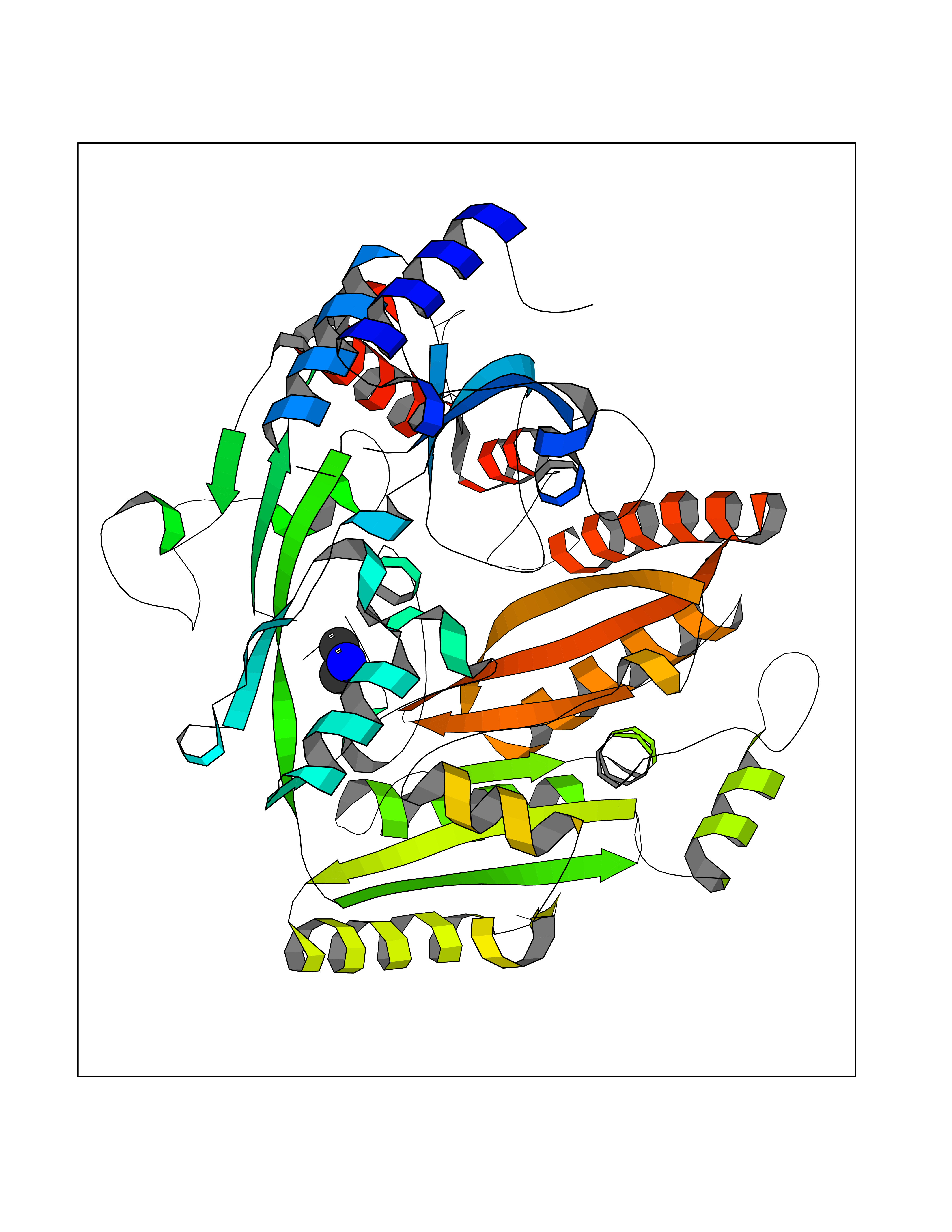 Molscript Map 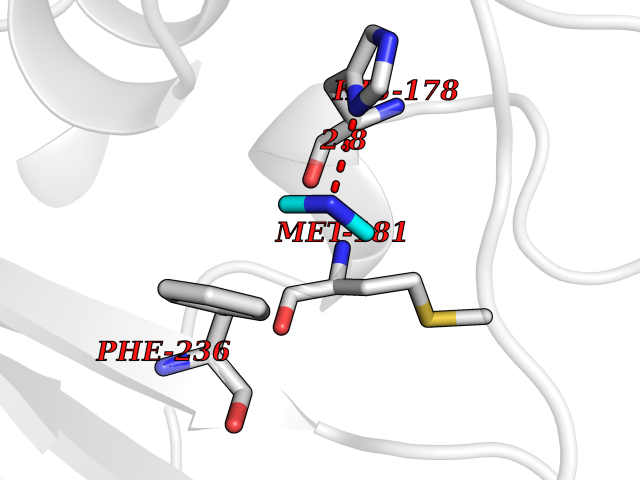 Pymol Map 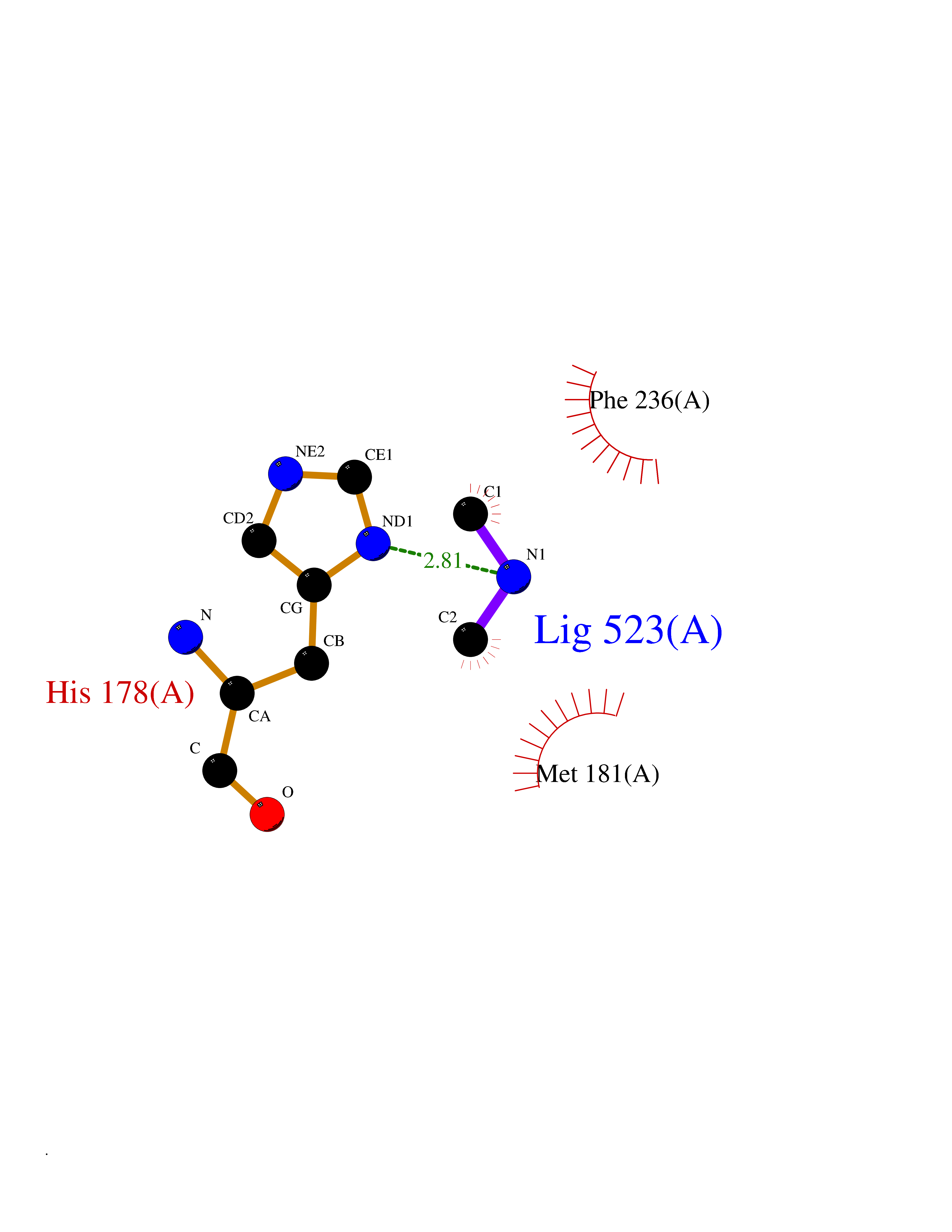 Ligplot Map 3D Binding mode Sequence AVLPKGVTQGEFNKAVQKFRALLGDDNVLVESDQLVPYNKIMMPVENAAHAPSAAVTATTVEQVQGVVKICNEHKIPIWTISTGRNFGYGSAAPVQRGQVILDLKKMNKIIKIDPEMCYALVEPGVTFGQMYDYIQENNLPVMLSFSAPSAIAGPVGNTMDRGVGYTPYGEHFMMQCGMEVVLANGDVYRTGMGGVPGSNTWQIFKWGYGPTLDGMFTQANYGICTKMGFWLMPKPPVFKPFEVIFEDEADIVEIVDALRPLRMSNTIPNSVVIASTLWEAGSAHLTRAQYTTEPGHTPDSVIKQMQKDTGMGAWNLYAALYGTQEQVDVNWKIVTDVFKKLGKGRIVTQEEAGDTQPFKYRAQLMSGVPNLQEFGLYNWRGGGGSMWFAPVSEARGSECKKQAAMAKRVLHKYGLDYVAEFIVAPRDMHHVIDVLYDRTNPEETKRADACFNELLDEFEKEGYAVYRVNTRFQDRVAQSYGPVKRKLEHAIKRAVDPNNILAPGRSGIDLNNDF Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Poly [ADP-ribose] polymerase 1 (PARP1) | 5WS1 | 3.99 | |
Target general information Gen name PARP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein poly-ADP-ribosyltransferase PARP1; Poly[ADP-ribose] synthetase-1; Poly[ADP-ribose] synthase 1; Poly(ADP-ribose)polymerase-1; PPOL; PARP-1; NAD(+)Poly [ADP-ribose] polymerase-1 ADP-ribosyltrans Protein family ARTD/PARP family Biochemical class Glycosyltransferases Function Mainly mediates glutamate and aspartate ADP-ribosylation of target proteins: the ADP-D-ribosyl group of NAD(+) is transferred to the acceptor carboxyl group of glutamate and aspartate residues and further ADP-ribosyl groups are transferred to the 2'-position of the terminal adenosine moiety, building up a polymer with an average chain length of 20-30 units. Mediates the poly(ADP-ribosyl)ation of a number of proteins, including itself, APLF and CHFR. Also mediates serine ADP-ribosylation of target proteins following interaction with HPF1; HPF1 conferring serine specificity. Probably also catalyzes tyrosine ADP-ribosylation of target proteins following interaction with HPF1. Catalyzes the poly-ADP-ribosylation of histones in a HPF1-dependent manner. Involved in the base excision repair (BER) pathway by catalyzing the poly-ADP-ribosylation of a limited number of acceptor proteins involved in chromatin architecture and in DNA metabolism. ADP-ribosylation follows DNA damage and appears as an obligatory step in a detection/signaling pathway leading to the reparation of DNA strand breaks. In addition to base excision repair (BER) pathway, also involved in double-strand breaks (DSBs) repair: together with TIMELESS, accumulates at DNA damage sites and promotes homologous recombination repair by mediating poly-ADP-ribosylation. In addition to proteins, also able to ADP-ribosylate DNA: catalyzes ADP-ribosylation of DNA strand break termini containing terminal phosphates and a 2'-OH group in single- and double-stranded DNA, respectively. Required for PARP9 and DTX3L recruitment to DNA damage sites. PARP1-dependent PARP9-DTX3L-mediated ubiquitination promotes the rapid and specific recruitment of 53BP1/TP53BP1, UIMC1/RAP80, and BRCA1 to DNA damage sites. Acts as a regulator of transcription: positively regulates the transcription of MTUS1 and negatively regulates the transcription of MTUS2/TIP150. With EEF1A1 and TXK, forms a complex that acts as a T-helper 1 (Th1) cell-specific transcription factor and binds the promoter of IFN-gamma to directly regulate its transcription, and is thus involved importantly in Th1 cytokine production. Involved in the synthesis of ATP in the nucleus, together with NMNAT1, PARG and NUDT5. Nuclear ATP generation is required for extensive chromatin remodeling events that are energy-consuming. Poly-ADP-ribosyltransferase that mediates poly-ADP-ribosylation of proteins and plays a key role in DNA repair. Related diseases Dihydrolipoamide dehydrogenase deficiency (DLDD) [MIM:246900]: An autosomal recessive metabolic disorder characterized biochemically by a combined deficiency of the branched-chain alpha-keto acid dehydrogenase complex (BCKDC), pyruvate dehydrogenase complex (PDC), and alpha-ketoglutarate dehydrogenase complex (KGDC). Clinically, affected individuals have lactic acidosis and neurologic deterioration due to sensitivity of the central nervous system to defects in oxidative metabolism. {ECO:0000269|PubMed:10448086, ECO:0000269|PubMed:11687750, ECO:0000269|PubMed:12925875, ECO:0000269|PubMed:15712224, ECO:0000269|PubMed:16442803, ECO:0000269|PubMed:16770810, ECO:0000269|PubMed:17404228, ECO:0000269|PubMed:20160912, ECO:0000269|PubMed:8506365, ECO:0000269|PubMed:8968745, ECO:0000269|PubMed:9540846, ECO:0000269|PubMed:9934985}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04010; DB03509; DB03072; DB03722; DB03073; DB07787; DB07096; DB07330; DB02498; DB13877; DB02701; DB11793; DB02690; DB09074; DB12332; DB11760; DB00277; DB07232; DB01593; DB14487; DB14533; DB14548 Interacts with Q8IW19; Q7Z2E3; P42574; P49715; Q86WJ1-1; P26358; Q01094; Q96L91; P11308; O60741; P09429; Q13007; Q9BQ69; P08651; Q9Y530; P09874; Q8N2W9; P46063; Q9NTX7; Q14684-1; O95863; P63165; P04637; P0CG48; Q14191; P18887; P54577; Q2M1K9; Q02085 EC number EC 2.4.2.30 Uniprot keywords 3D-structure; Acetylation; ADP-ribosylation; Allosteric enzyme; Apoptosis; Chromosome; Cytoplasm; Direct protein sequencing; DNA damage; DNA repair; DNA-binding; Glycosyltransferase; Immunity; Innate immunity; Isopeptide bond; Metal-binding; NAD; Nucleotidyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 36904 Length 329 Aromaticity 0.08 Instability index 35.9 Isoelectric point 6.83 Charge (pH=7) -0.44 2D Binding mode Binding energy (Kcal/mol) -5.44 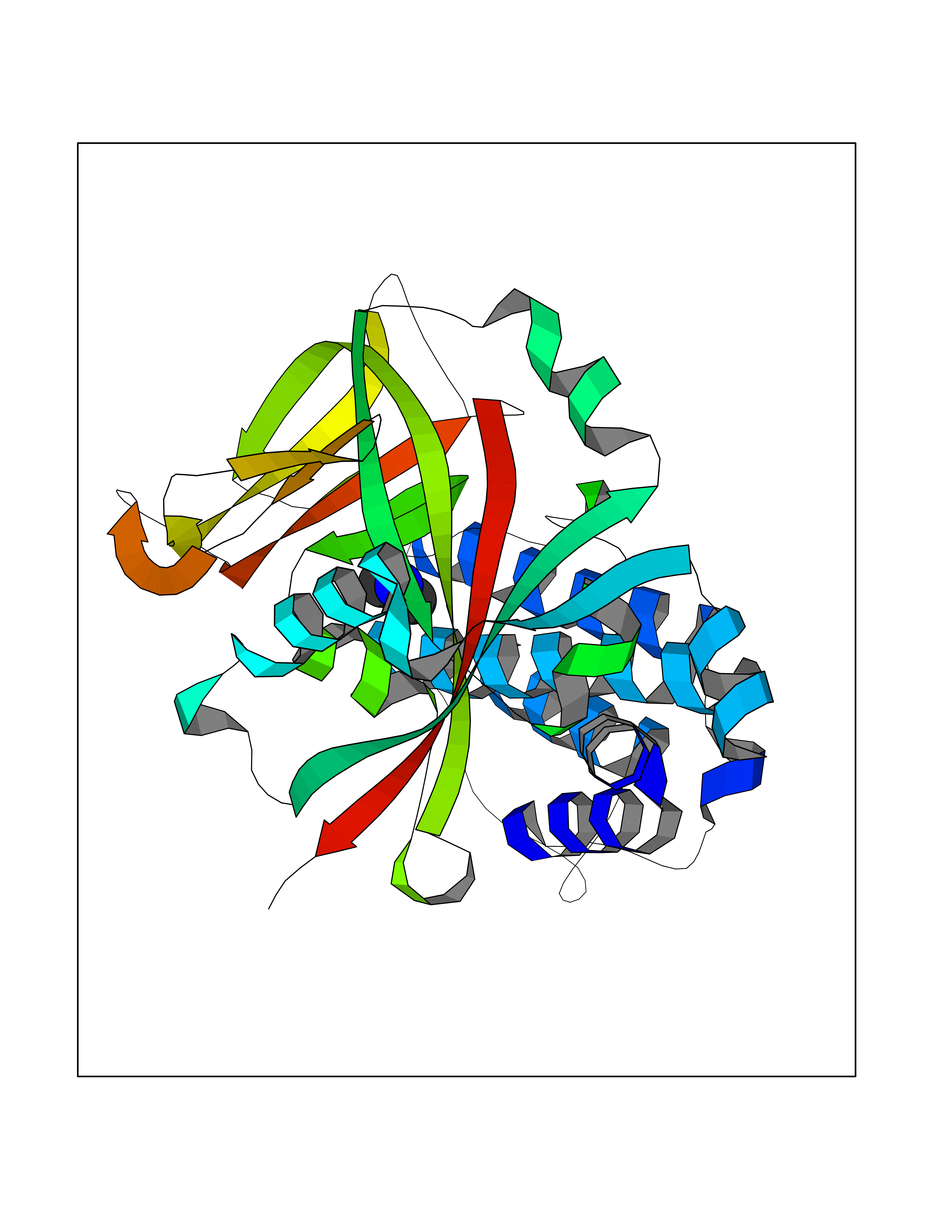 Molscript Map 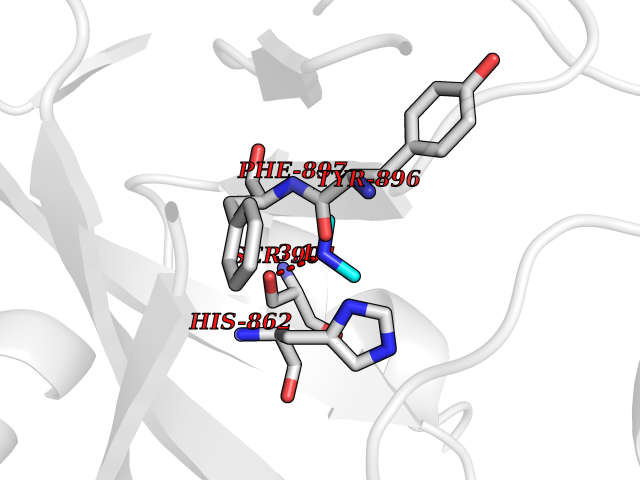 Pymol Map 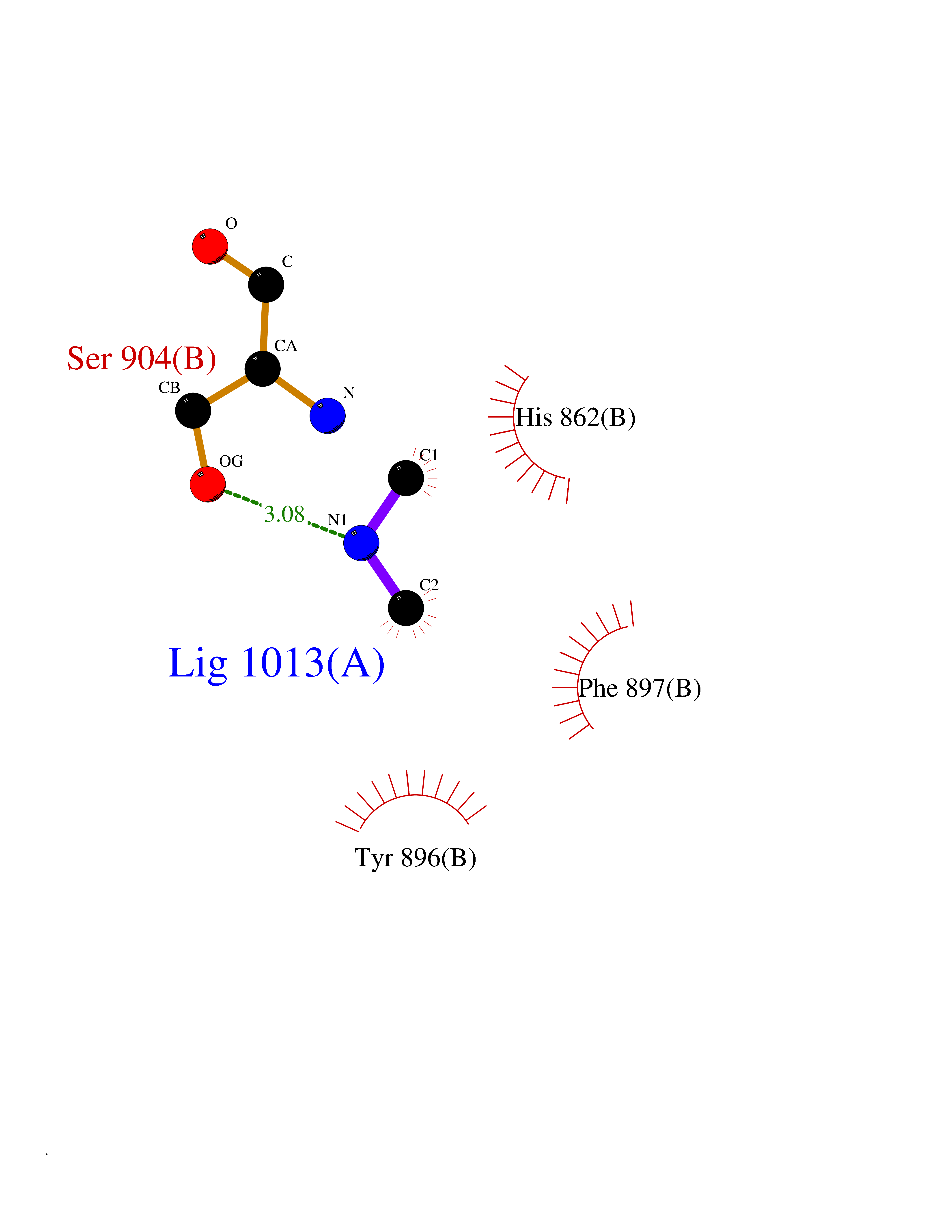 Ligplot Map 3D Binding mode Sequence DLIKMIFDVESMKKAMVEYEIDLQKMPLGKLSKRQIQAAYSILSEVQQAVSQGSDSQILDLSNRFYTLIPHDFGMKKPPLLNNADSVQAKAEMLDNLLDIEVAYSLPIDVNYEKLKTDIKVVDRDSEEAEIIRKYVKNTHATTHNAYDLEVIDIFKIEREGECQRYKPFKQLHNRRLLWHGSRTTNFAGILSQGLRIAPPEAPVTGYMFGKGIYFADMVSKSANYCHTSQGDPIGLILLGEVALGNMYELKHASHISKLPKGKHSVKGLGKTTPDPSANISLDGVDVPLGTGISSGVNDTSLLYNEYIVYDIAQVNLKYLLKLKFNFKT Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Penicillin-binding protein 2B | 2WAD | 3.99 | |
Target general information Gen name penA Organism Streptococcus pneumoniae (strain ATCC BAA-255 / R6) Uniprot ID TTD ID NA Synonyms spr1517;pbp2b Protein family Transpeptidase family Biochemical class Peptide binding protein Function Penicillin binding. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01163; DB00415; DB08795; DB01140; DB00456; DB01066; DB00493; DB01331; DB01212; DB00567; DB03313; DB00485; DB00739; DB01603; DB00607; DB00713; DB00319 Interacts with NA EC number NA Uniprot keywords 3D-structure; Antibiotic resistance; Cell membrane; Cell shape; Cell wall biogenesis/degradation; Membrane; Peptidoglycan synthesis; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 65444.4 Length 607 Aromaticity 0.08 Instability index 30.15 Isoelectric point 4.95 Charge (pH=7) -20.68 2D Binding mode Binding energy (Kcal/mol) -5.44 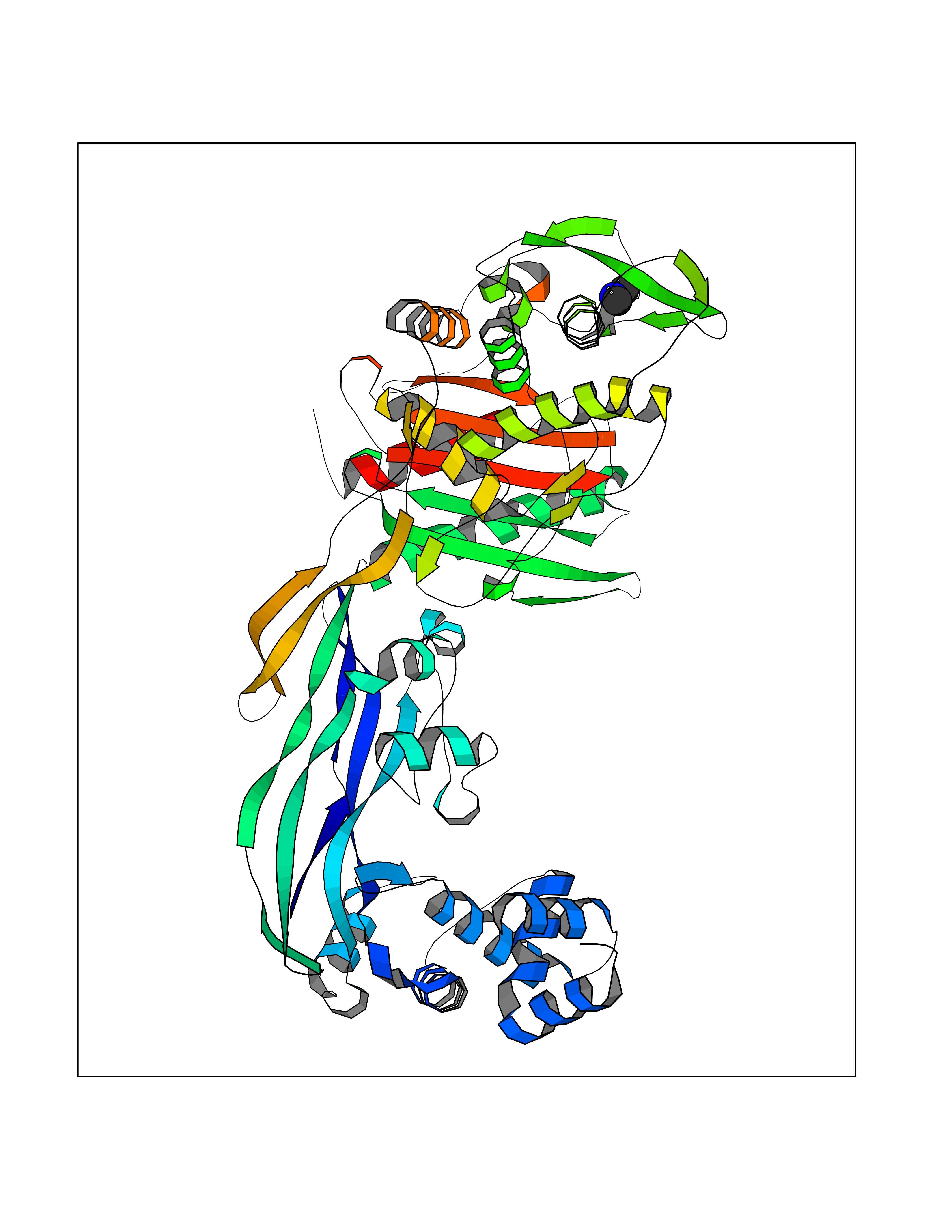 Molscript Map 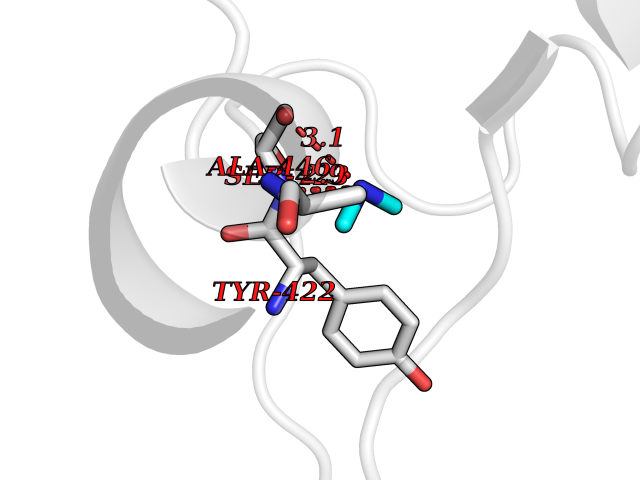 Pymol Map 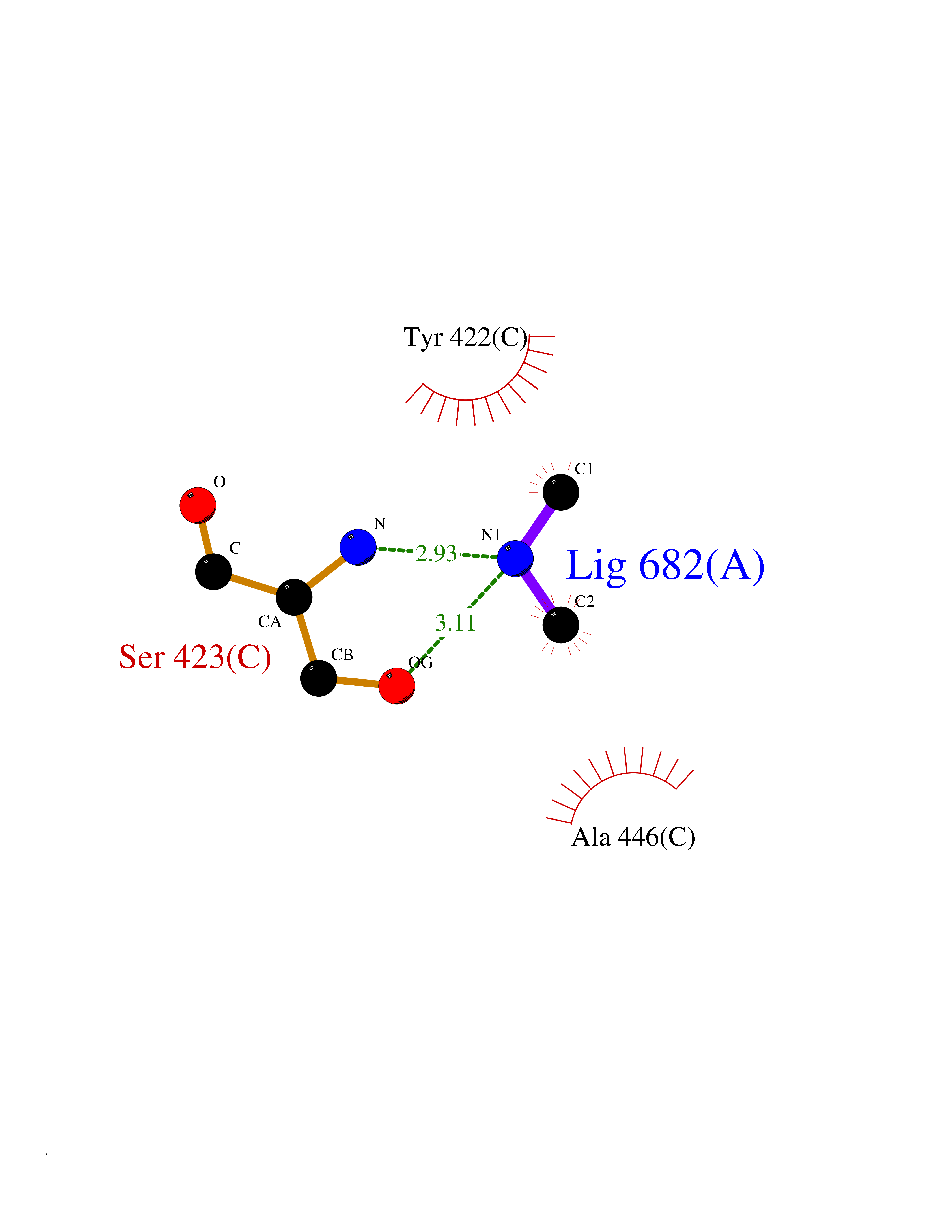 Ligplot Map 3D Binding mode Sequence SQTKVTTSSARGEIYDASGKPLVENTLKQVVSFTRSNKMTATDLKEIAKKLLTYVSISSPNLTERQLADYYLADPEIYKKTVEALPSESELYNNAVDSVPTSQLNYTEDEKKEIYLFSQLNAVGNFATGTIATDPLNDSQVAVIASISKEMPGISISTSWDRKILETSLSSIVGSVSSEKAGLPAEEAESYLKKGYSLNDRVGTSYLEKQYEEVLQGKRPVKEIHLDKHGDMESVENIEEGSKGKNIKLTIDLAFQDSVDALLKSYFNSELGNGGAKYSEGVYAVALNPQTGAVLSMSGLKHDLKTGELTPDSLGTVTNVFVPGSVVKAATISSGWENGVLSGNQTLTDQPIVFQGSAPIYSWYKLAYGSFPITAVEALEYSSNAYVVQTALGIMGQTYQPNMFVGTSNLESAMGKLRSTFGEYGLGSATGIDLPDESTGLVPKEYNFANFITNAFGQFDNYTPMQLAQYVATIANNGVRLAPHIVEGIYDNNDKGGLGELIQAIDTKEINKVNISESDMAILHQGFYQVSHGTSPLTTGRAFSDGATVSISGKTGTNTNAVAYAPTENPQIAVAVVFPHNTNLTKNVGPAIARDIINLYNQHHPMN Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Riboflavin synthase | 1PKV | 3.99 | |
Target general information Gen name ribC Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1654;ribE;b1662 Protein family NA Biochemical class Transferase Function Riboflavin synthase activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00140 Interacts with NA EC number 2.5.1.9 Uniprot keywords 3D-structure; Reference proteome; Repeat; Riboflavin biosynthesis; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 19023.5 Length 174 Aromaticity 0.05 Instability index 2.85 Isoelectric point 5.13 Charge (pH=7) -9.8 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map 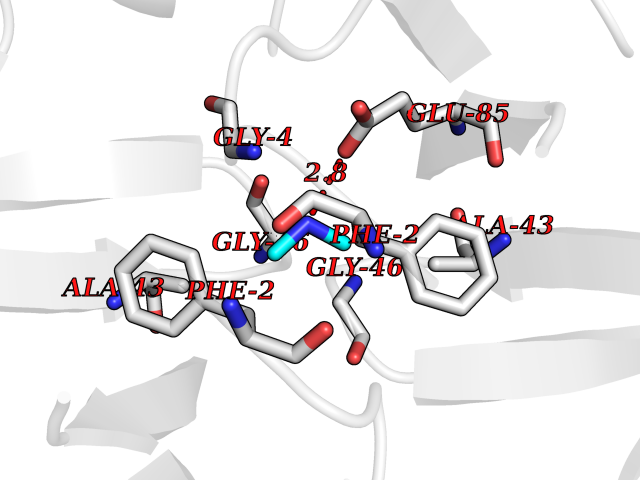 Pymol Map 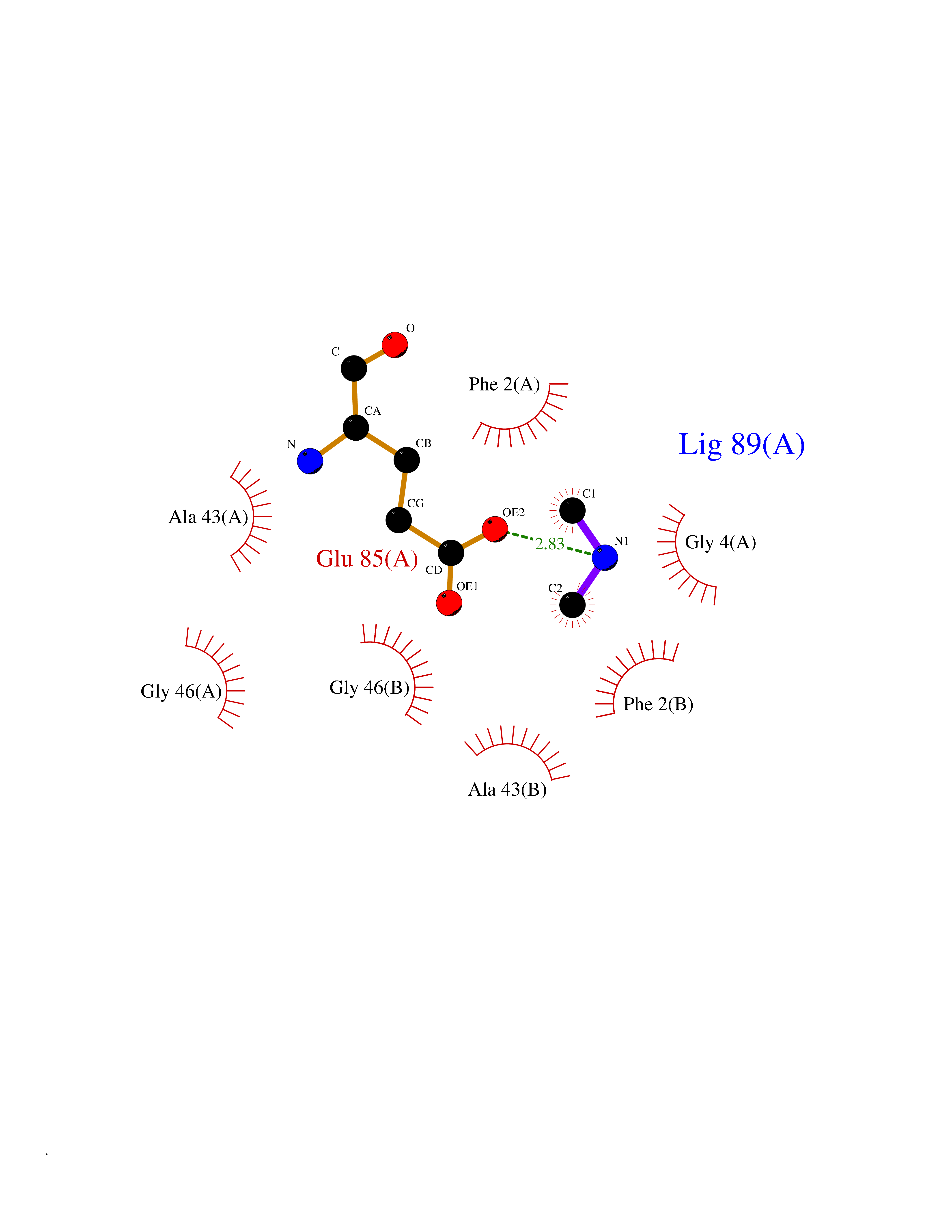 Ligplot Map 3D Binding mode Sequence MFTGIVQGTAKLVSIDEKPNFRTHVVELPDHMLDGLETGASVAHNGCCLTVTEINGNHVSFDLMKETLRITNLGDLKVGDWVNVERAMFTGIVQGTAKLVSIDEKPNFRTHVVELPDHMLDGLETGASVAHNGCCLTVTEINGNHVSFDLMKETLRITNLGDLKVGDWVNVERA Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Sodium channel subunit beta-2 | 5FEB | 3.99 | |
Target general information Gen name SCN2B Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms UNQ326/PRO386 Protein family Sodium channel auxiliary subunit SCN2B (TC 8.A.17) family Biochemical class Membrane protein Function Sodium channel regulator activity.Voltage-gated sodium channel activity involved in cardiac muscle cell action potential. Related diseases Atrial fibrillation, familial, 14 (ATFB14) [MIM:615378]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:19808477}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Genetic variations in SCN2B may be involved in Brugada syndrome (PubMed:23559163). This tachyarrhythmia is characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:23559163}. Drugs (DrugBank ID) DB05541; DB00907; DB13269; DB13961; DB00776; DB00243; DB00313; DB00909 Interacts with NA EC number NA Uniprot keywords 3D-structure; Atrial fibrillation; Brugada syndrome; Cell membrane; Cell projection; Disease variant; Disulfide bond; Glycoprotein; Immunoglobulin domain; Ion transport; Membrane; Phosphoprotein; Proteomics identification; Reference proteome; Signal; Sodium; Sodium transport; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 14190 Length 122 Aromaticity 0.1 Instability index 37.04 Isoelectric point 6.39 Charge (pH=7) -1.21 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SNAMEVTVPATLNVLNGSDARLPCTFNSAYTVNHKQFSLNWTYQECNNCSEEMFLQFRMKIINLKLERFQDRVEFSGNPSKYDVSVMLRNVQPEDEGIYNCYIMNPPDRHRGHGKIHLQVLM Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Sphingomyelin phosphodiesterase | 5I85 | 3.99 | |
Target general information Gen name SMPD1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ASM Protein family Acid sphingomyelinase family Biochemical class Hydrolase Function Acid sphingomyelin phosphodiesterase activity.Hydrolase activity, acting on glycosyl bonds.Sphingomyelin phosphodiesterase activity.Zinc ion binding. Related diseases Niemann-Pick disease A (NPDA) [MIM:257200]: An early-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Niemann-Pick disease type A is a primarily neurodegenerative disorder characterized by onset within the first year of life, intellectual disability, digestive disorders, failure to thrive, major hepatosplenomegaly, and severe neurologic symptoms. The severe neurological disorders and pulmonary infections lead to an early death, often around the age of four. Clinical features are variable. A phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. {ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1391960, ECO:0000269|PubMed:15221801, ECO:0000269|PubMed:15877209, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:1718266, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:2023926, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430884, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:8680412, ECO:0000269|PubMed:8693491, ECO:0000269|PubMed:9266408, ECO:0000269|PubMed:9660788}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Niemann-Pick disease B (NPDB) [MIM:607616]: A late-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Clinical signs involve only visceral organs. The most constant sign is hepatosplenomegaly which can be associated with pulmonary symptoms. Patients remain free of neurologic manifestations. However, a phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. In Niemann-Pick disease type B, onset of the first symptoms occurs in early childhood and patients can survive into adulthood. {ECO:0000269|PubMed:12369017, ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1301192, ECO:0000269|PubMed:15241805, ECO:0000269|PubMed:16010684, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:16472269, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:1885770, ECO:0000269|PubMed:19050888, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:21098024, ECO:0000269|PubMed:21621718, ECO:0000269|PubMed:22613662, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430512, ECO:0000269|PubMed:25920558, ECO:0000269|PubMed:26084044, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:27659707, ECO:0000269|PubMed:8051942, ECO:0000269|PubMed:8664904}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00381; DB12151; DB00477; DB01151; DB14009 Interacts with P55210 EC number 3.1.4.12; 3.1.4.3 Uniprot keywords 3D-structure; Alternative splicing; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Glycosidase; Host-virus interaction; Hydrolase; Lipid droplet; Lipid metabolism; Lysosome; Metal-binding; Neurodegeneration; Niemann-Pick disease; Phosphoprotein; Proteomics identification; Reference proteome; Secreted; Signal; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 58913.8 Length 528 Aromaticity 0.11 Instability index 46.93 Isoelectric point 6.48 Charge (pH=7) -3.6 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map  Pymol Map 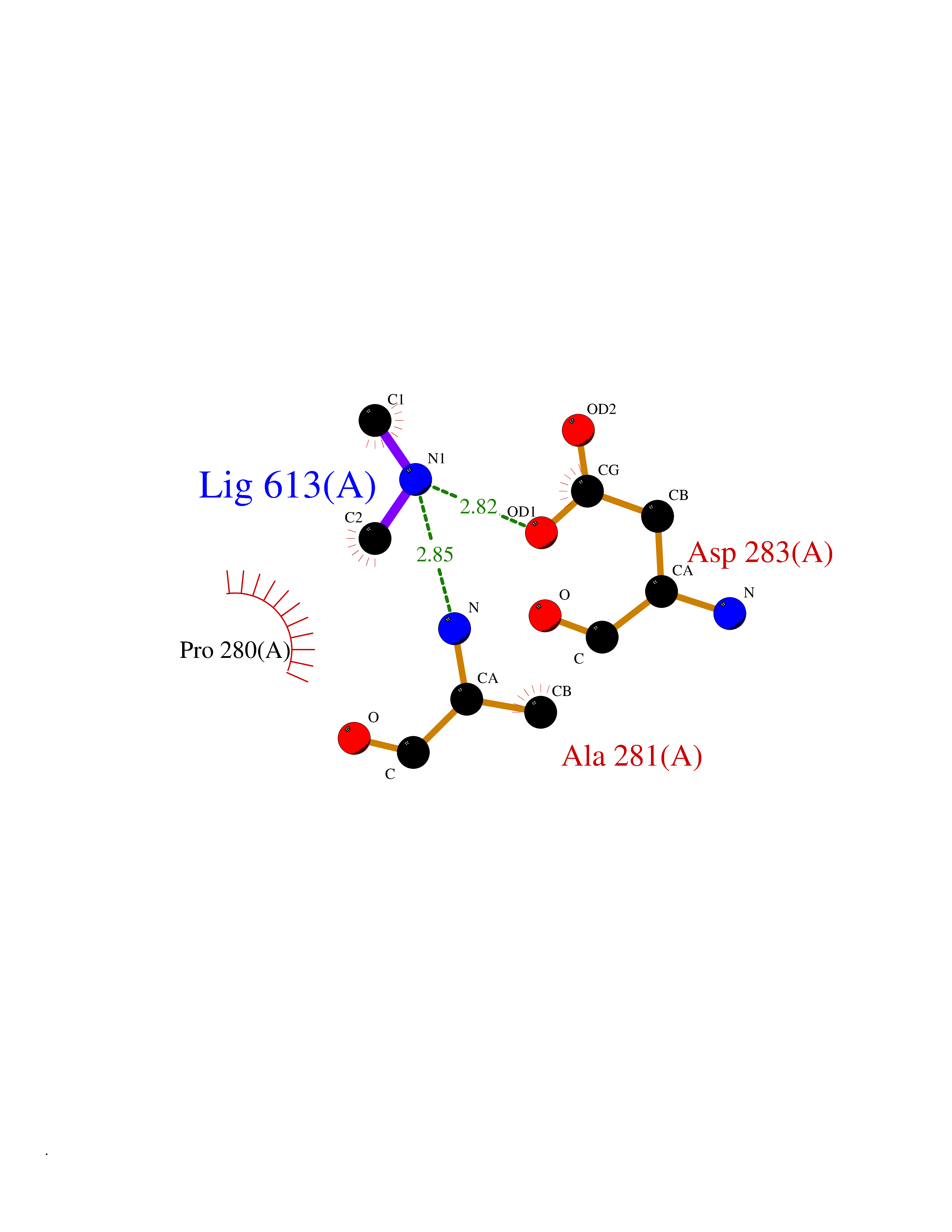 Ligplot Map 3D Binding mode Sequence WGNLTCPICKGLFTAINLGLKKEPNVARVGSVAIKLCNLLKIAPPAVCQSIVHLFEDDMVEVWRRSVLSPSEACGLLLGSTCGHWDIFSSWNISLPTVPKPPPKPPSPPAPGAPVSRILFLTDLHWDHDYLEGTDPDCADPLCCRRGSGLPPASRPGAGYWGEYSKCDLPLRTLESLLSGLGPAGPFDMVYWTGDIPAHDVWHQTRQDQLRALTTVTALVRKFLGPVPVYPAVGNHESTPVNSFPPPFIEGNHSSRWLYEAMAKAWEPWLPAEALRTLRIGGFYALSPYPGLRLISLNMNFCSRENFWLLINSTDPAGQLQWLVGELQAAEDRGDKVHIIGHIPPGHCLKSWSWNYYRIVARYENTLAAQFFGHTHVDEFEVFYDEETLSRPLAVAFLAPSATTYIGLNPGYRVYQIDGNYSGSSHVVLDHETYILNLTQANIPGAIPHWQLLYRARETYGLPNTLPTAWHNLVYRMRGDMQLFQTFWFLYHKGHPPSEPCGTPCRLATLCAQLSARADSPALCRHLM Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Catechol-O-methyl-transferase (COMT) | 3BWY | 3.99 | |
Target general information Gen name COMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms S-COMT; MB-COMT; Catechol-O-methyltransferase; COMT Protein family Class I-like SAM-binding methyltransferase superfamily, Cation-dependent O-methyltransferase family Biochemical class Methyltransferase Function Catalyzes the O-methylation, and thereby the inactivation, of catecholamine neurotransmitters and catechol hormones. Also shortens the biological half-lives of certain neuroactive drugs, like L-DOPA, alpha-methyl DOPA and isoproterenol. Related diseases Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:15645182}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07462; DB02342; DB02105; DB08049; DB00118; DB00714; DB03336; DB00286; DB00255; DB00841; DB00988; DB15488; DB00494; DB00668; DB00783; DB00977; DB01064; DB00968; DB01141; DB03907; DB04820; DB06152; DB11632; DB00252; DB01420; DB00323 Interacts with Q6P5T0; P30518; Q8NFU1; Q8NHW4; P34972; Q96BA8; P50402; Q5JX71; O14843; O00258; P08034; O75712; Q9NTQ9; O95377; Q8TDT2; Q8N6U8; O15529; P31937; Q9H2F3; O95279; Q5SR56; A6NDP7; Q0D2K0; Q7RTS5; Q9UHJ9-5; Q8IY26; Q9H6H4; Q6NTF9-3; O75783; Q99500; Q9Y6D0; Q3KNW5; O60669; P22732; Q96G79; Q5T1Q4; Q9NY26; Q9NP94; Q6P1K1; P30825; Q9UHI5; B2RUZ4; Q9UPZ6; Q96MV1; Q9NV29; A0PK00; Q9NUH8; Q9P0S9; Q14656; Q6UW68; Q9H0R3; O95807; P34981; Q15645; Q15836; O95183; O76024; P30260; Q9H816; Q92997; P29323-3; P22607; P06396; Q15323; Q6A162; P26371; O15116; P20645; O14744; Q5T160; Q9UJD0; Q2MKA7; Q8N488; O75880; Q14141; Q9UNE7; Q15645; Q9NYH9; Q8NA23-2 EC number EC 2.1.1.6 Uniprot keywords 3D-structure; Alternative initiation; Catecholamine metabolism; Cell membrane; Cytoplasm; Direct protein sequencing; Lipid metabolism; Magnesium; Membrane; Metal-binding; Methyltransferase; Neurotransmitter degradation; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Schizophrenia; Signal-anchor; Transferase; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 23851.2 Length 214 Aromaticity 0.07 Instability index 25.99 Isoelectric point 5.25 Charge (pH=7) -7.75 2D Binding mode Binding energy (Kcal/mol) -5.45 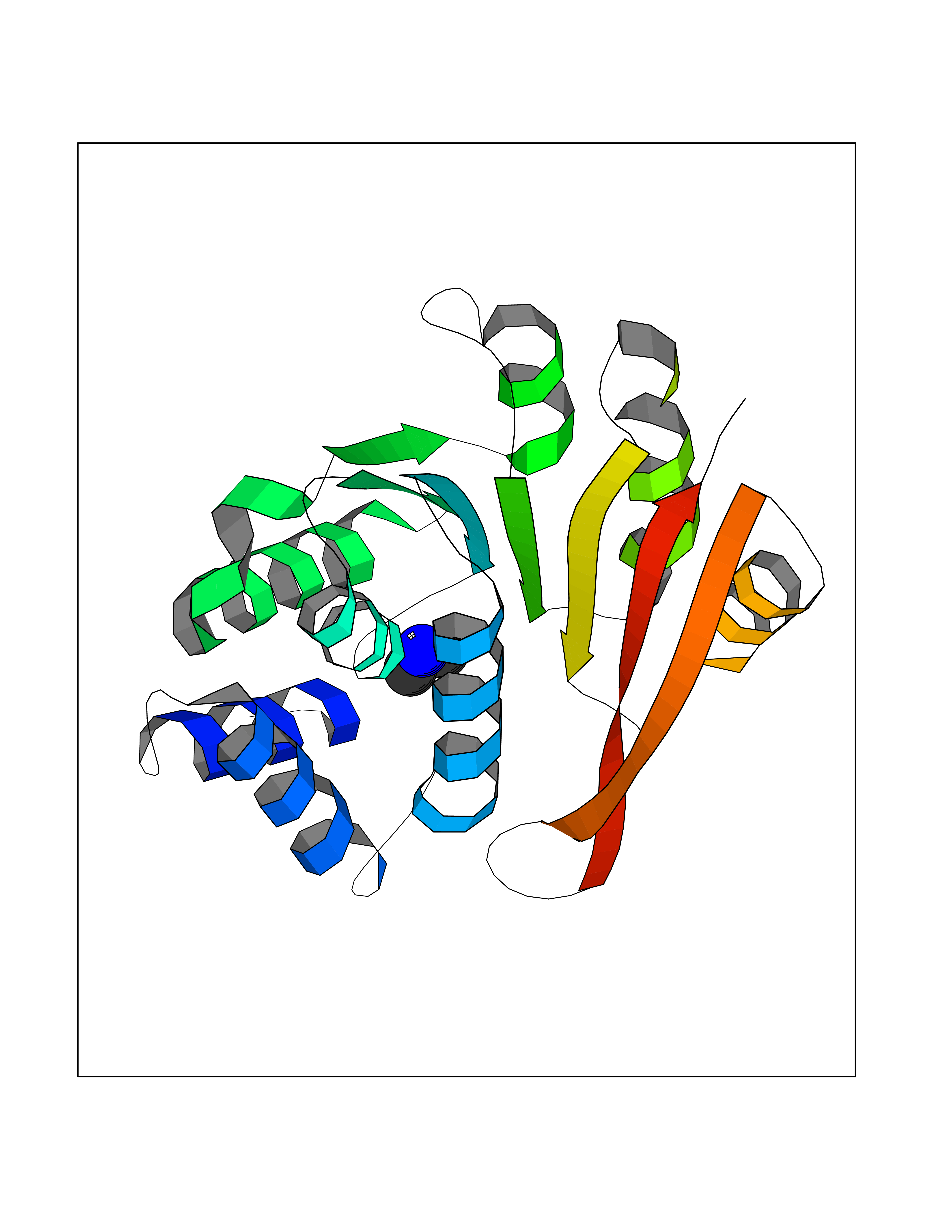 Molscript Map 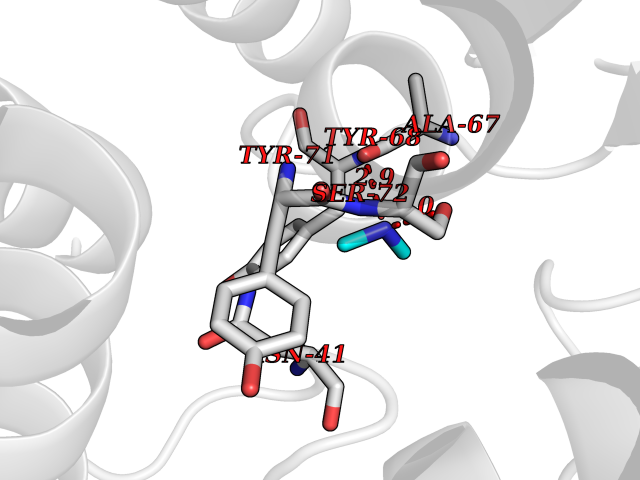 Pymol Map 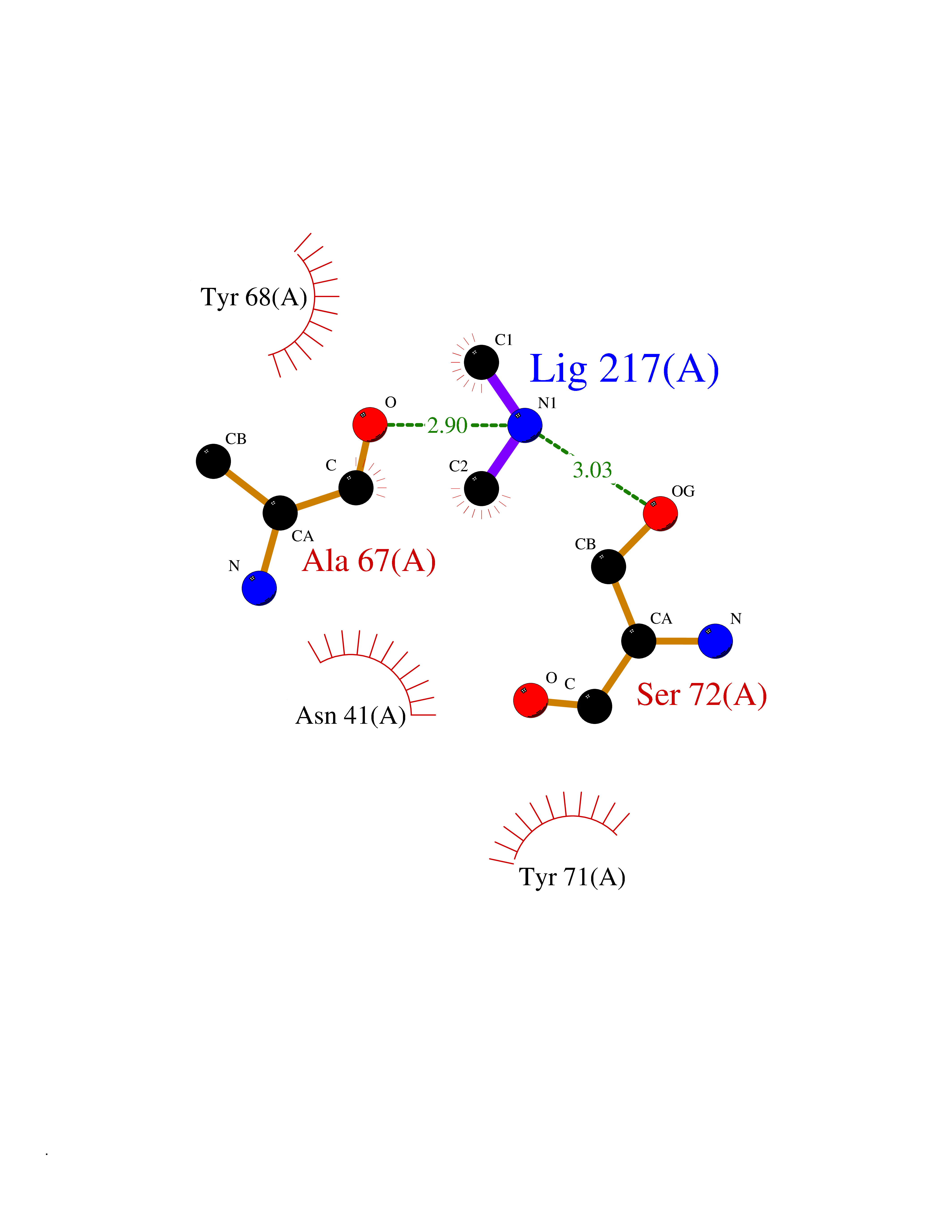 Ligplot Map 3D Binding mode Sequence GDTKEQRILNHVLQHAEPGNAQSVLEAIDTYCEQKEWAMNVGDKKGKIVDAVIQEHQPSVLLELGAYCGYSAVRMARLLSPGARLITIEINPDCAAITQRMVDFAGMKDKVTLVVGASQDIIPQLKKKYDVDTLDMVFLDHWKDRYLPDTLLLEECGLLRKGTVLLADNVICPGAPDFLAHVRGSSCFECTHYQSFLEYREVVDGLEKAIYKGP Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | 4-hydroxyphenylpyruvate dioxygenase | 3ISQ | 3.99 | |
Target general information Gen name HPD Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PPD Protein family 4HPPD family Biochemical class Oxidoreductase Function 4-hydroxyphenylpyruvate dioxygenase activity.Metal ion binding. Related diseases Tyrosinemia 3 (TYRSN3) [MIM:276710]: An inborn error of metabolism characterized by elevations of tyrosine in the blood and urine, seizures and mild intellectual disability. {ECO:0000269|PubMed:10942115, ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hawkinsinuria (HWKS) [MIM:140350]: An inborn error of tyrosine metabolism characterized by failure to thrive, persistent metabolic acidosis, fine and sparse hair, and excretion of the unusual cyclic amino acid metabolite, hawkinsin, in the urine. {ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02850; DB00348 Interacts with NA EC number 1.13.11.27 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Dioxygenase; Disease variant; Endoplasmic reticulum; Golgi apparatus; Intellectual disability; Iron; Membrane; Metal-binding; Oxidoreductase; Phenylalanine catabolism; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Tyrosine catabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 43164.8 Length 376 Aromaticity 0.11 Instability index 32.38 Isoelectric point 6.73 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -5.44 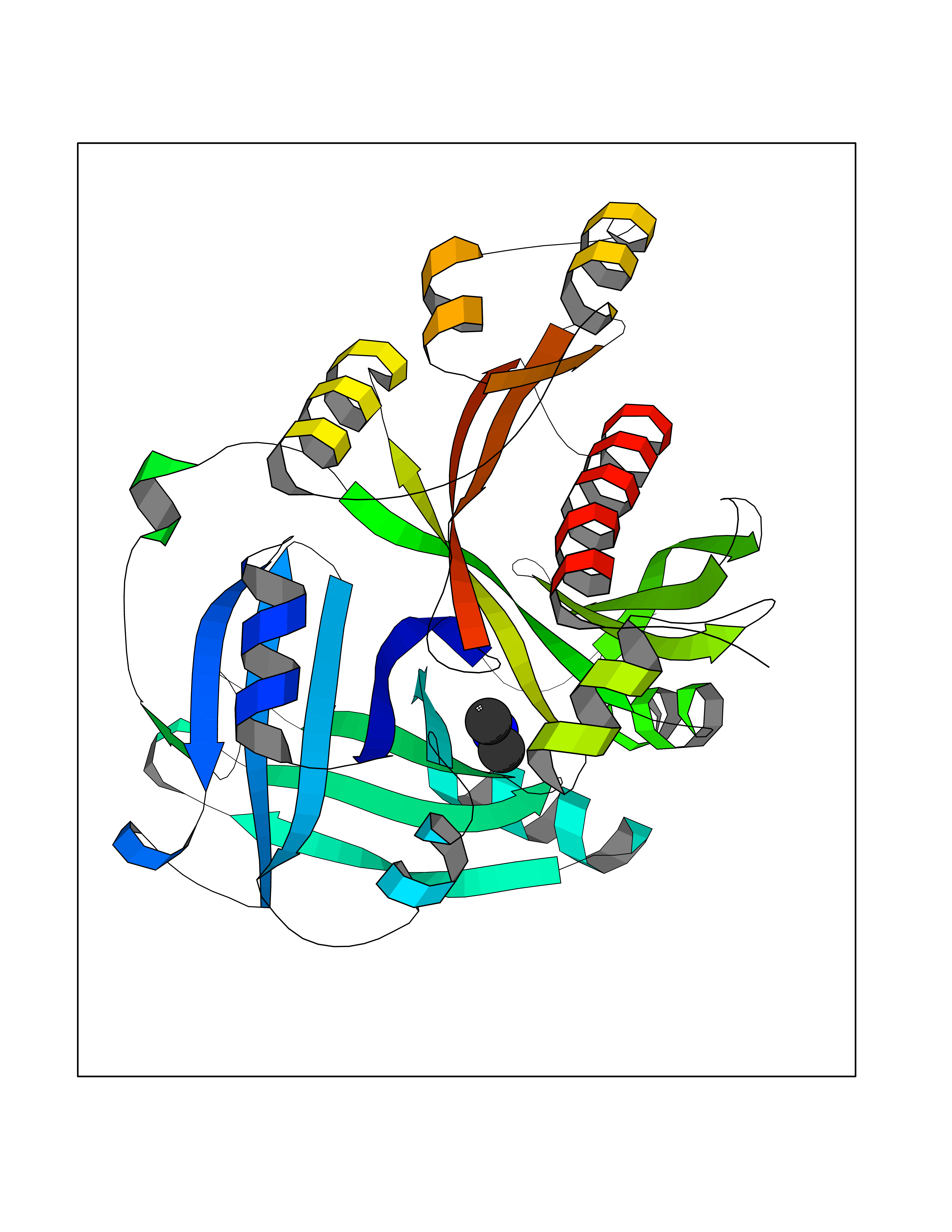 Molscript Map 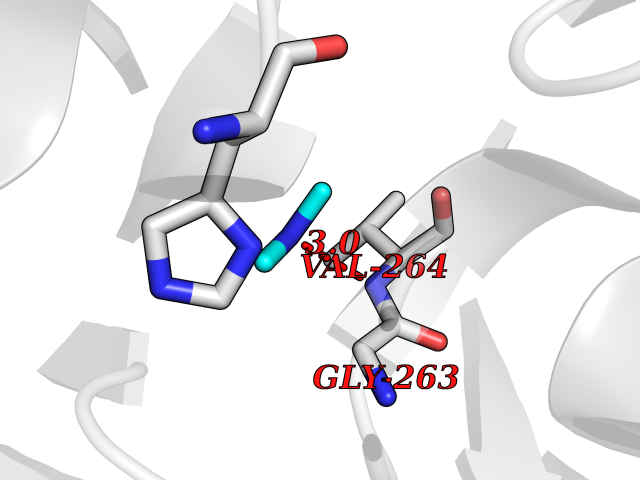 Pymol Map 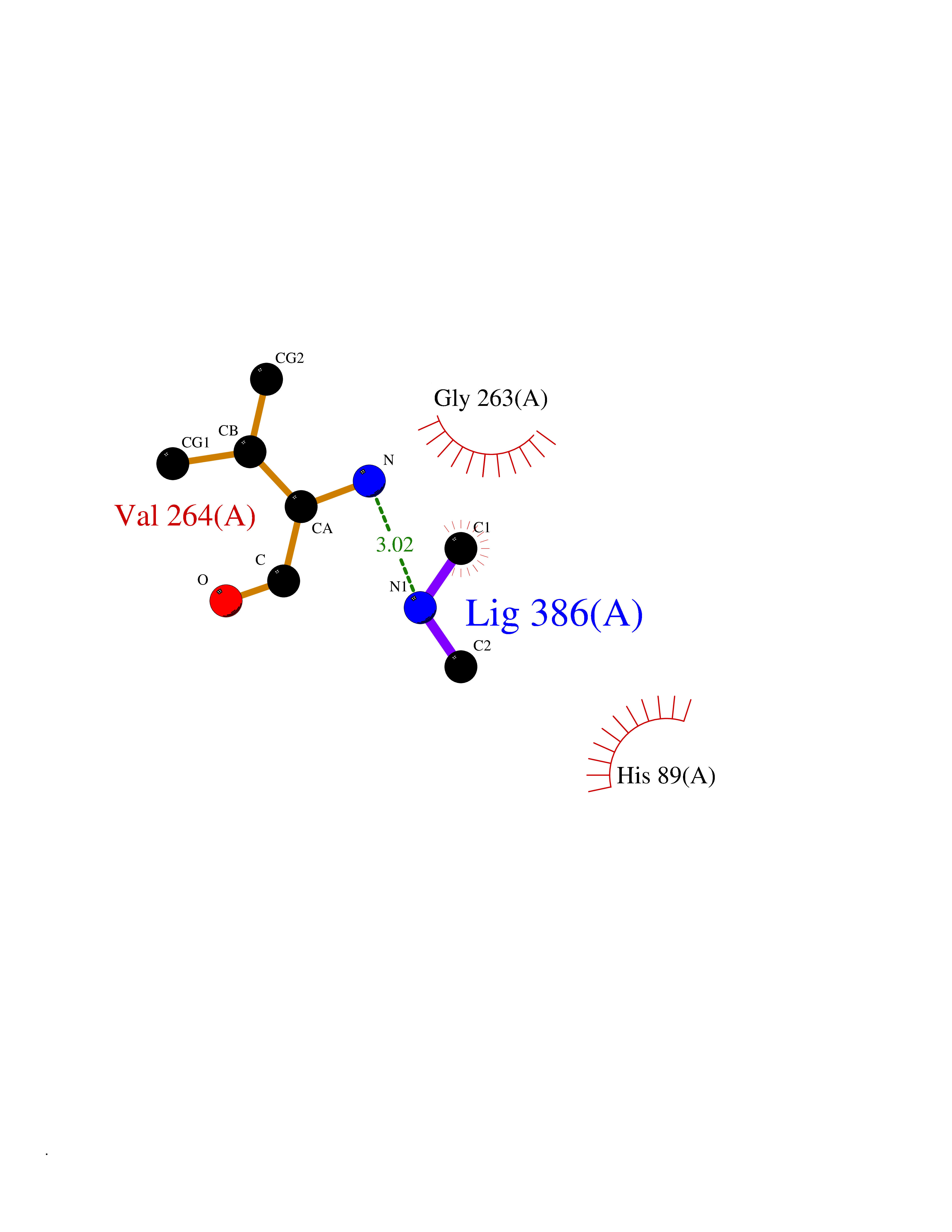 Ligplot Map 3D Binding mode Sequence AKPERGRFLHFHSVTFWVGNAKQAASFYCSKMGFEPLAYRGLETGSREVVSHVIKQGKIVFVLSSALNPWNKEMGDHLVKHGDGVKDIAFEVEDCDYIVQKARERGAKIMREPWVEQDKFGKVKFAVLQTYGDTTHTLVEKMNYIGQFLPGYEAPAFMDPLLPKLPKCSLEMIDHIVGNQPDQEMVSASEWYLKNLQFHRFWSVDDTQVHTEYSSLRSIVVANYEESIKMPINEPAPGKKKSQIQEYVDYNGGAGVQHIALKTEDIITAIRHLRERGLEFLSVPSTYYKQLREKLKTAKIKVKENIDALEELKILVDYDEKGYLLQIFTKPVQDRPTLFLEVIQRHNHQGFGAGNFNSLFKAFEEEQNLRGNLTNM Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Aspartoacylase | 4MXU | 3.99 | |
Target general information Gen name ASPA Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ASP;ACY2 Protein family AspA/AstE family, Aspartoacylase subfamily Biochemical class Hydrolase Function Aminoacylase activity.Aspartoacylase activity.Hydrolase activity, acting on ester bonds.Identical protein binding.Metal ion binding. Related diseases Canavan disease (CAND) [MIM:271900]: A rare neurodegenerative condition of infancy or childhood characterized by white matter vacuolization and demyelination that gives rise to a spongy appearance. The clinical features are onset in early infancy, atonia of neck muscles, hypotonia, hyperextension of legs and flexion of arms, blindness, severe mental defect, megalocephaly, and death by 18 months on the average. {ECO:0000269|PubMed:10407784, ECO:0000269|PubMed:10564886, ECO:0000269|PubMed:10909858, ECO:0000269|PubMed:12205125, ECO:0000269|PubMed:12638939, ECO:0000269|PubMed:12706335, ECO:0000269|PubMed:24036223, ECO:0000269|PubMed:28101991, ECO:0000269|PubMed:7599639, ECO:0000269|PubMed:7668285, ECO:0000269|PubMed:8023850, ECO:0000269|PubMed:8252036, ECO:0000269|PubMed:8659549, ECO:0000269|PubMed:9452117}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00128; DB01593; DB14487; DB14533; DB14548 Interacts with Q96HD9; P45381; Q9UNS2; Q68J44; Q14145; O75925; Q9UHD9; Q9H347; Q8IYU4 EC number 3.5.1.15 Uniprot keywords 3D-structure; Cytoplasm; Disease variant; Hydrolase; Leukodystrophy; Metal-binding; Nucleus; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34380.1 Length 301 Aromaticity 0.1 Instability index 44.33 Isoelectric point 5.96 Charge (pH=7) -6.22 2D Binding mode Binding energy (Kcal/mol) -5.44 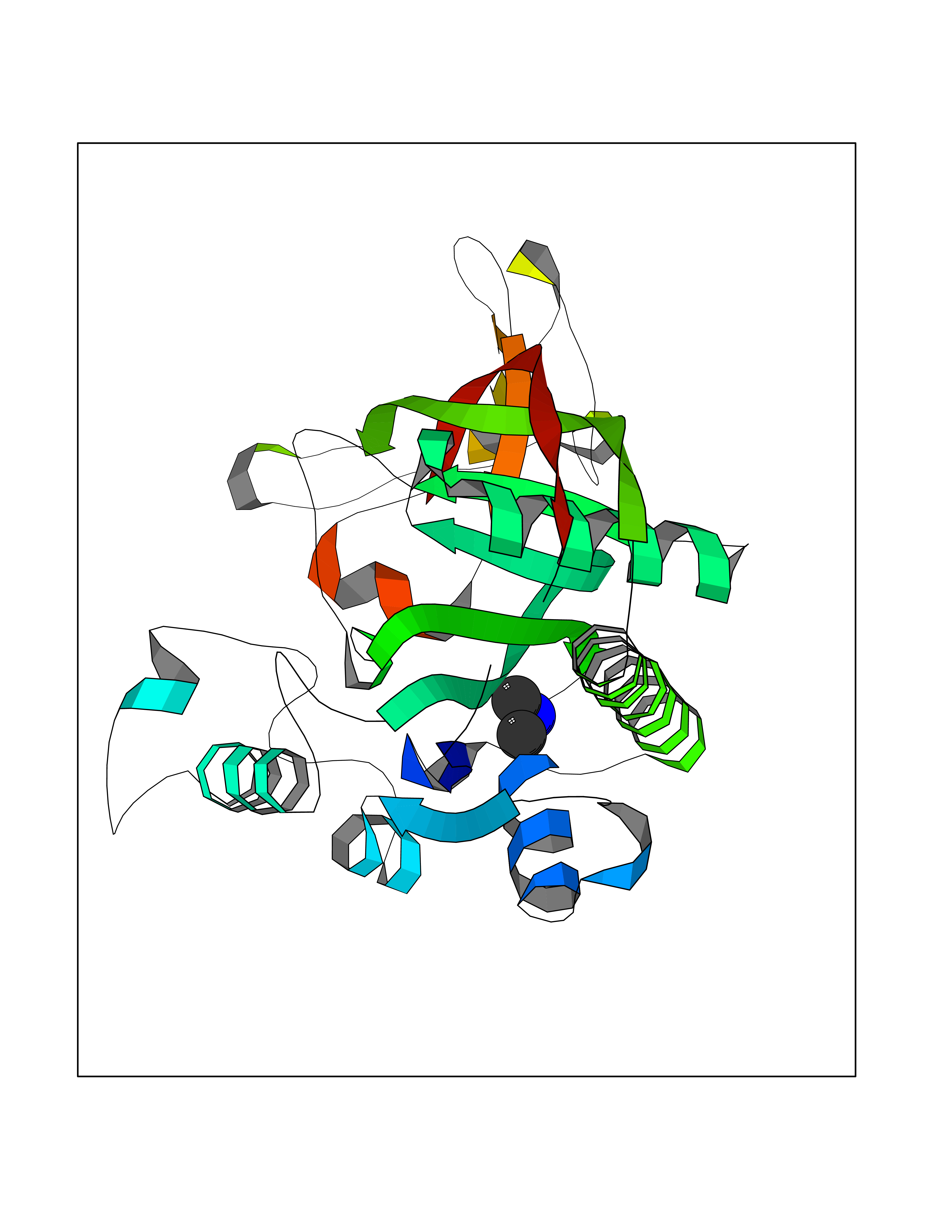 Molscript Map 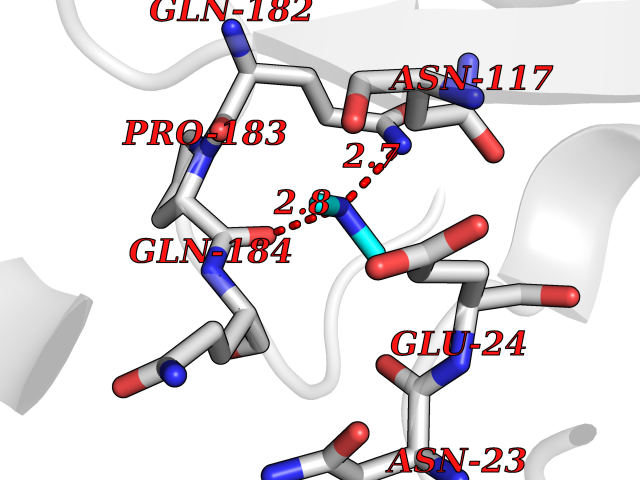 Pymol Map 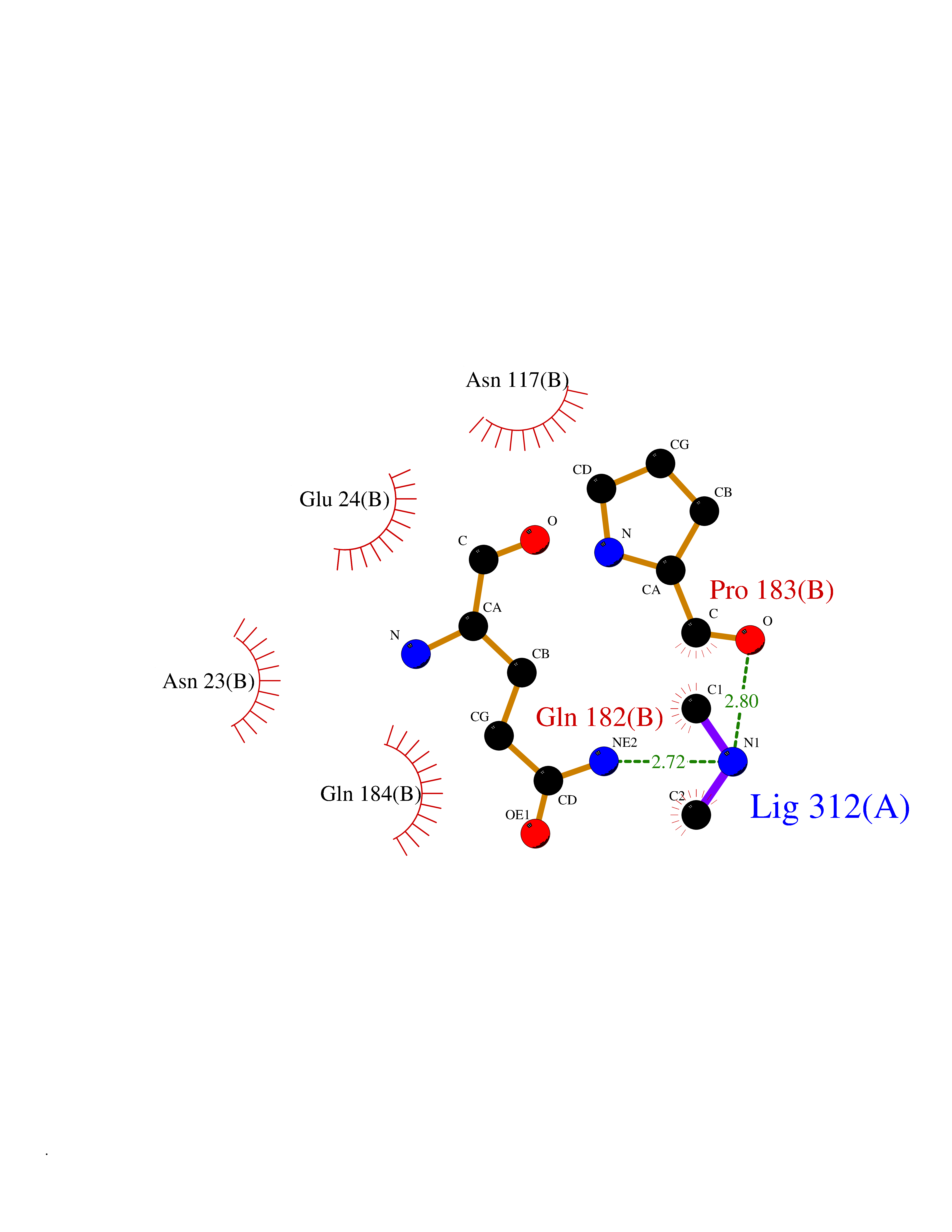 Ligplot Map 3D Binding mode Sequence HIQKVAIFGGTHGNELTGVFLVKHWLENGAEIQRTGLEVKPFITNPRAVKKCTRYIDCDLNRIFDLENLGKKMSEDLPYEVRRAQEINHLFGPKDSEDSYDIIFDLHNTTSNMGCTLILEDSRNNFLIQMFHYIKTSLAPLPCYVYLIEHPSLKYATTRSIAKYPVGIEVGPQPQGVLRADILDQMRKMIKHALDFIHHFNEGEEFPPCAIEVYKIIEKVDYPRDENGEIAAIIHPNLQDQDWKPLHPGDPMFLTLDGKTIPLGGDCTVYPVFVNEAAYYEKKEAFAKTTKLTLNAKSIRC Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Beta-lactamase TEM | 1M40 | 3.99 | |
Target general information Gen name bla Organism Escherichia coli Uniprot ID TTD ID NA Synonyms blaT-4;blaT-6;blaT-5;blaT-3 Protein family Class-A beta-lactamase family Biochemical class Hydrolase Function Beta-lactamase activity. Related diseases WHIM syndrome 1 (WHIMS1) [MIM:193670]: An autosomal dominant immunologic disease characterized by neutropenia, hypogammaglobulinemia and extensive human papillomavirus (HPV) infection. Despite the peripheral neutropenia, bone marrow aspirates from affected individuals contain abundant mature myeloid cells, a condition termed myelokathexis. {ECO:0000269|PubMed:12692554, ECO:0000269|PubMed:15536153}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: CXCR4 mutations play a role in the pathogenesis of Waldenstroem macroglobulinemia (WM) and influence disease presentation and outcome, as well as response to therapy. WM is a B-cell lymphoma characterized by accumulation of malignant lymphoplasmacytic cells in the bone marrow, lymph nodes and spleen, and hypersecretion of monoclonal immunoglobulin M (IgM). Excess IgM production results in serum hyperviscosity, tissue infiltration, and autoimmune-related pathology. {ECO:0000269|PubMed:24366360, ECO:0000269|PubMed:24553177}. Drugs (DrugBank ID) DB07466; DB07464; DB02614; DB04430; DB08551; DB07599; DB02841; DB02642; DB09060; DB01053; DB04035; DB01598; DB04037; DB12377; DB12107 Interacts with P35804 EC number 3.5.2.6 Uniprot keywords 3D-structure; Antibiotic resistance; Direct protein sequencing; Disulfide bond; Hydrolase; Plasmid; Signal; Transposable element Protein physicochemical properties Chain ID A Molecular weight (Da) 28876.6 Length 263 Aromaticity 0.05 Instability index 37.98 Isoelectric point 5.46 Charge (pH=7) -6.68 2D Binding mode Binding energy (Kcal/mol) -5.45 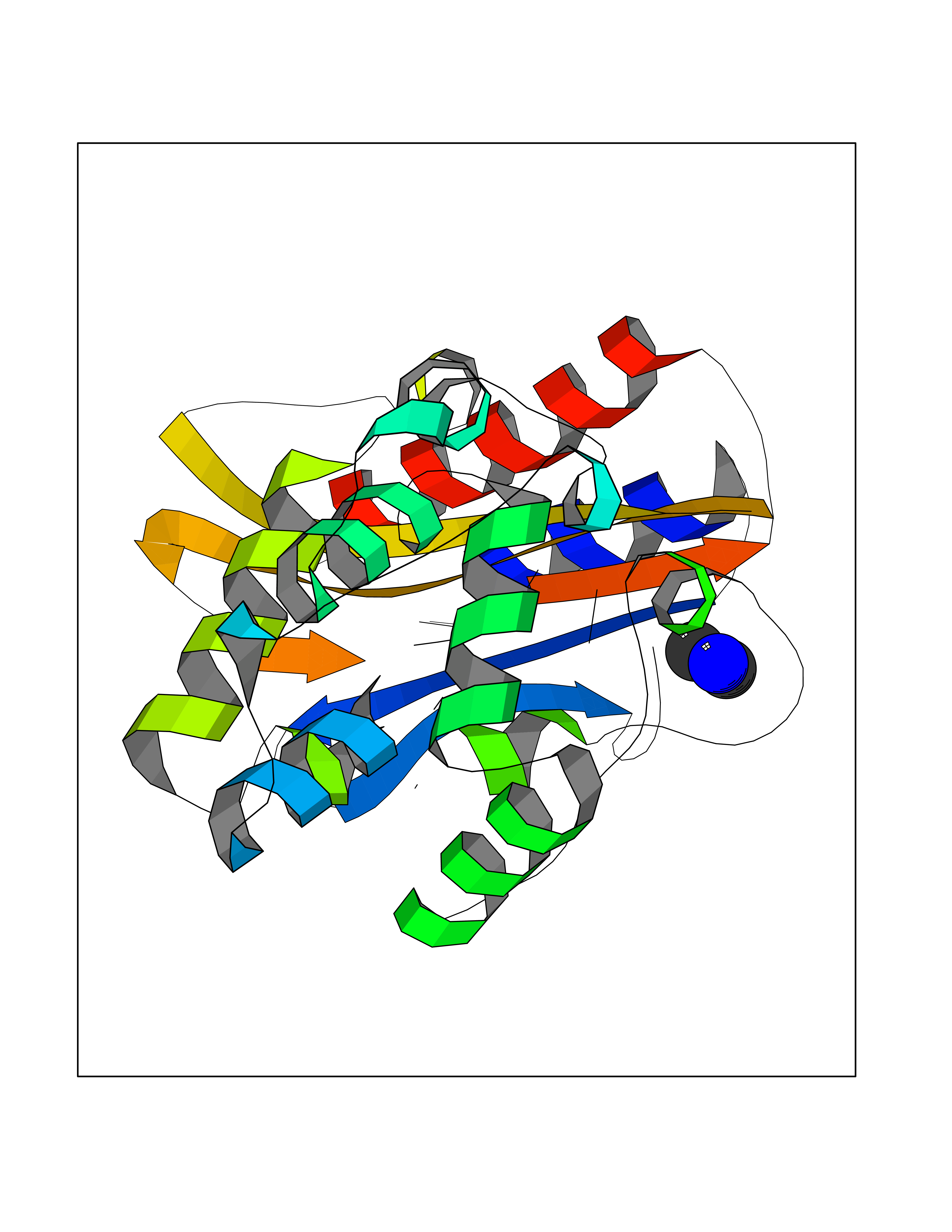 Molscript Map 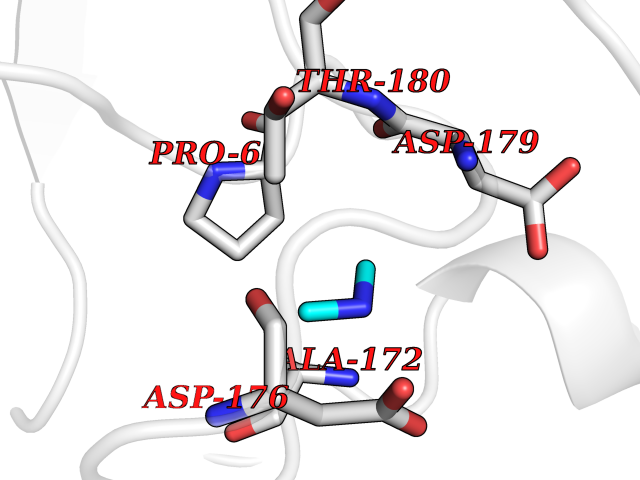 Pymol Map 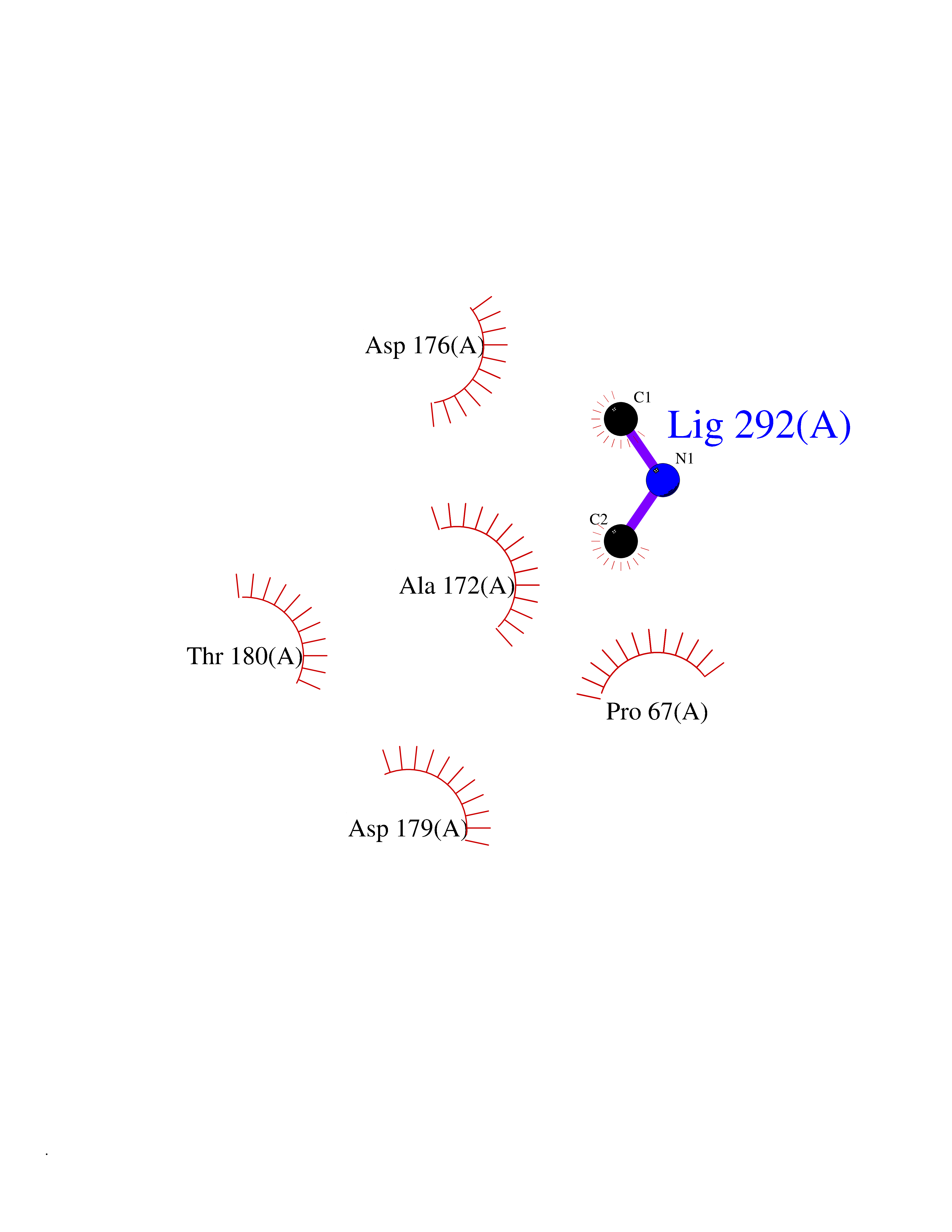 Ligplot Map 3D Binding mode Sequence HPETLVKVKDAEDQLGARVGYIELDLNSGKILESFRPEERFPMMSTFKVLLCGAVLSRVDAGQEQLGRRIHYSQNDLVEYSPVTEKHLTDGMTVRELCSAAITMSDNTAANLLLTTIGGPKELTAFLHNMGDHVTRLDRWEPELNEAIPNDERDTTTPAAMATTLRKLLTGELLTLASRQQLIDWMEADKVAGPLLRSALPAGWFIADKSGAGERGSRGIIAALGPDGKPSRIVVIYTTGSQATMDERNRQIAEIGASLIKHW Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Estrogen-related receptor-gamma (ESRRG) | 2E2R | 3.99 | |
Target general information Gen name ESRRG Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nuclear receptor subfamily 3 group B member 3; NR3B3; KIAA0832; Estrogen-related receptor gamma; Estrogen receptor-related protein 3; ERRG2; ERR3; ERR gamma-2 Protein family Nuclear hormone receptor family, NR3 subfamily Biochemical class Nuclear hormone receptor Function Binds specifically to an estrogen response element and activates reporter genes controlled by estrogen response elements. Induces the expression of PERM1 in the skeletal muscle. Orphan receptor that acts as transcription activator in the absence of bound ligand. Related diseases WHIM syndrome 1 (WHIMS1) [MIM:193670]: An autosomal dominant immunologic disease characterized by neutropenia, hypogammaglobulinemia and extensive human papillomavirus (HPV) infection. Despite the peripheral neutropenia, bone marrow aspirates from affected individuals contain abundant mature myeloid cells, a condition termed myelokathexis. {ECO:0000269|PubMed:12692554, ECO:0000269|PubMed:15536153}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: CXCR4 mutations play a role in the pathogenesis of Waldenstroem macroglobulinemia (WM) and influence disease presentation and outcome, as well as response to therapy. WM is a B-cell lymphoma characterized by accumulation of malignant lymphoplasmacytic cells in the bone marrow, lymph nodes and spleen, and hypersecretion of monoclonal immunoglobulin M (IgM). Excess IgM production results in serum hyperviscosity, tissue infiltration, and autoimmune-related pathology. {ECO:0000269|PubMed:24366360, ECO:0000269|PubMed:24553177}. Drugs (DrugBank ID) DB06884; DB04468; DB06973; DB07485; DB02659; DB00255; DB13952; DB13953; DB13954; DB13955; DB13956; DB06902; DB00675; DB00197 Interacts with Q05D60; Q9BVG8; P50222; P51843; Q12769; Q9UBK2; A0MZ66; G2XKQ0; Q8NFM4; Q13315; Q86WA6-2; Q9BZE7; Q13555-5; Q05D60; Q5JST6; P11474; O95718-2; P62508-3; Q15024; O95990-4; Q8IZU1; Q14296; P23508; Q6IN84; P51843; Q15466; P48552; P26367; Q9NPJ4; P01189; Q9UBK2; P62195; Q8N0T1-2; Q04864-2; Q6NUQ1; A0MZ66-4; Q8TAD8; P19237; P48788; Q96PN7; Q96S82; Q5SQQ9-2; Q7Z4V0 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 25755.7 Length 227 Aromaticity 0.07 Instability index 55.31 Isoelectric point 5.09 Charge (pH=7) -10.6 2D Binding mode Binding energy (Kcal/mol) -5.45 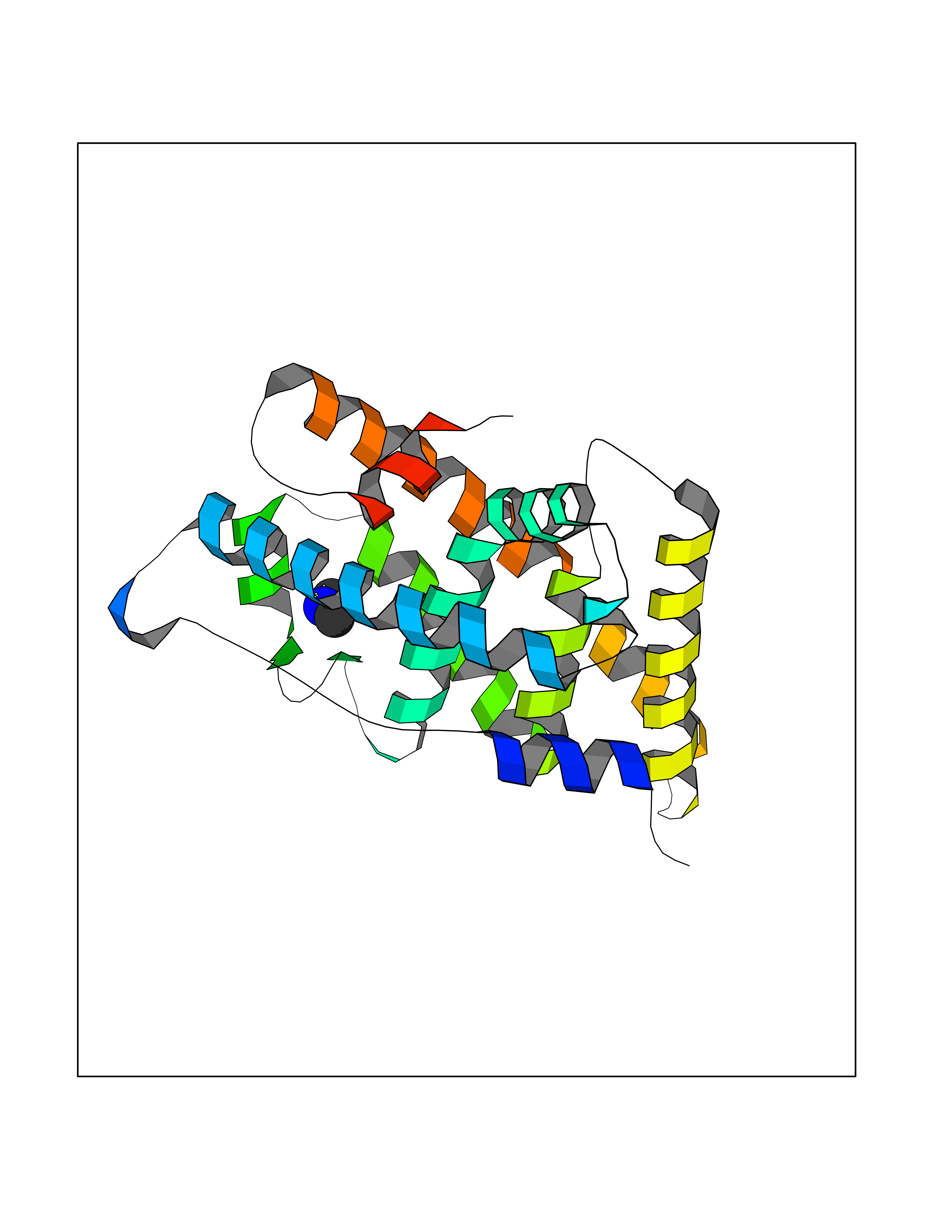 Molscript Map  Pymol Map 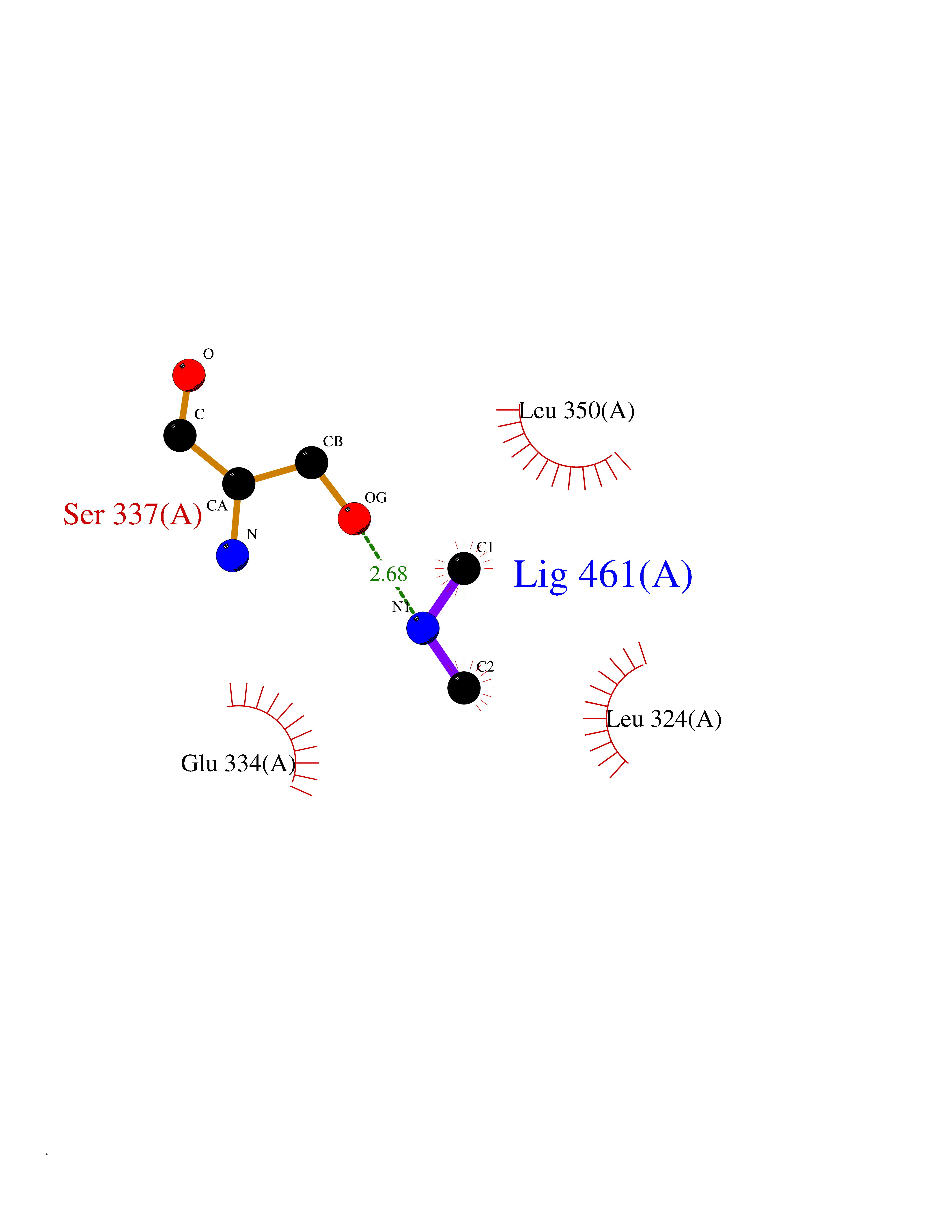 Ligplot Map 3D Binding mode Sequence KPYNKIVSHLLVAEPEKIYAMPDPTVPDSDIKALTTLCDLADRELVVIIGWAKHIPGFSTLSLADQMSLLQSAWMEILILGVVYRSLSFEDELVYADDYIMDEDQSKLAGLLDLNNAILQLVKKYKSMKLEKEEFVTLKAIALANSDSMHIEDVEAVQKLQDVLHEALQDYEAGQHMEDPRRAGKMLMTLPLLRQTSTKAVQHFYNIKLEGKVPMHKLFLEMLEAKV Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Aldehyde oxidoreductase | 4USA | 3.99 | |
Target general information Gen name mop Organism Megalodesulfovibrio gigas (Desulfovibrio gigas) Uniprot ID TTD ID NA Synonyms NA Protein family Xanthine dehydrogenase family Biochemical class Oxidoreductase Function 2 iron, 2 sulfur cluster binding.Aldehyde dehydrogenase (FAD-independent) activity.Electron carrier activity.Metal ion binding. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB02137 Interacts with NA EC number 1.2.99.7 Uniprot keywords 2Fe-2S; 3D-structure; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 96930.4 Length 907 Aromaticity 0.07 Instability index 29.17 Isoelectric point 5.69 Charge (pH=7) -17.56 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map 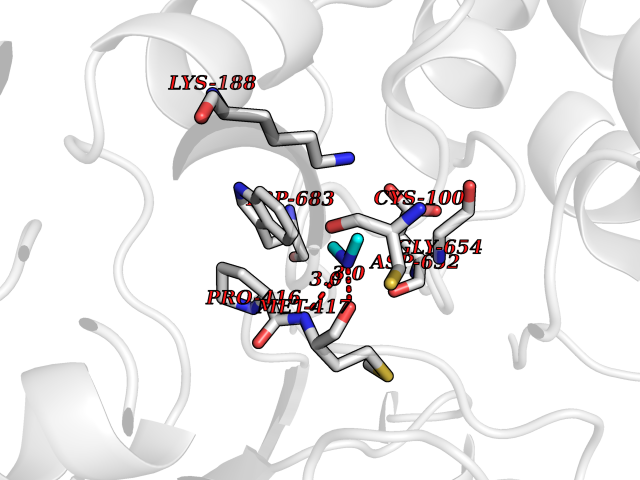 Pymol Map 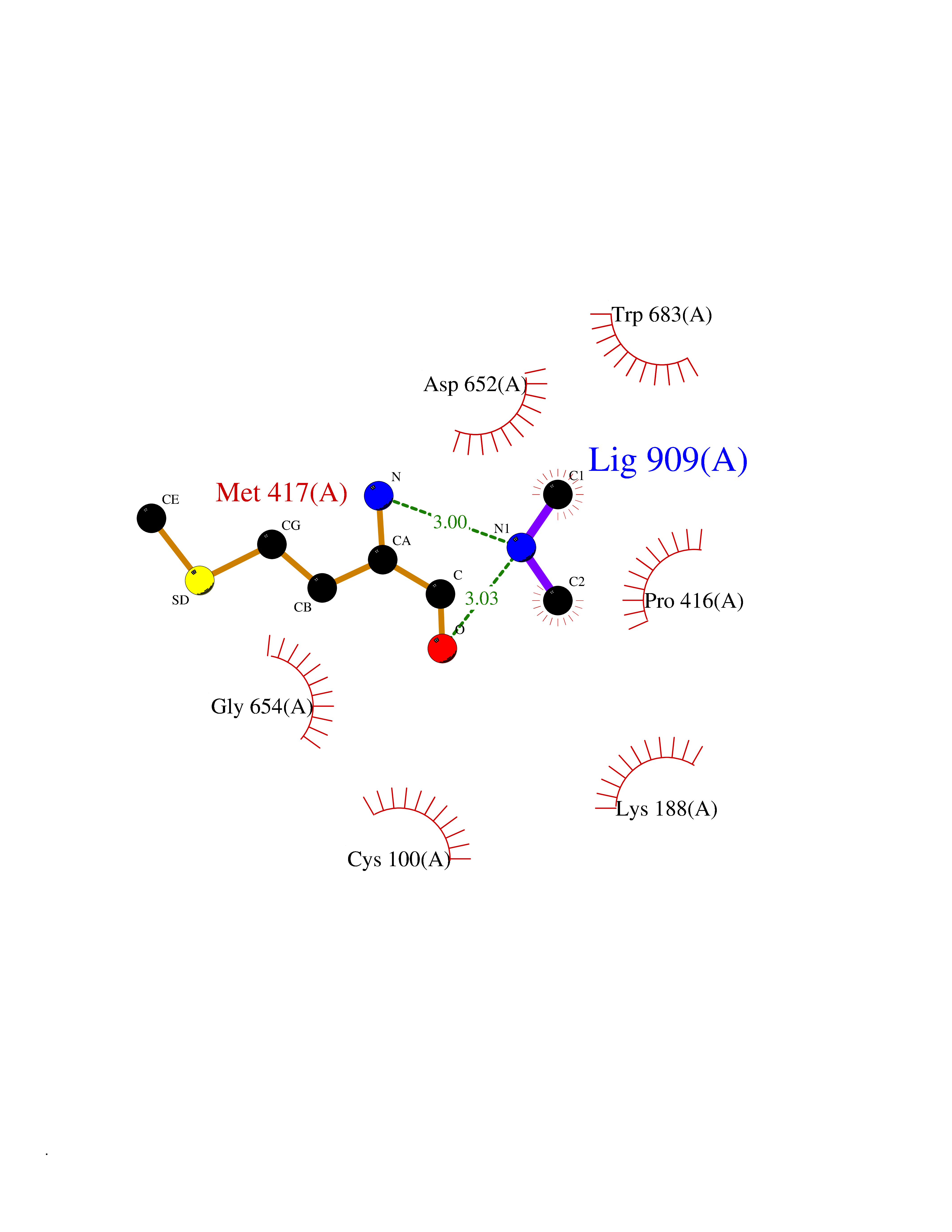 Ligplot Map 3D Binding mode Sequence MIQKVITVNGIEQNLFVDAEALLSDVLRQQLGLTGVKVGCEQGQCGACSVILDGKVVRACVTKMKRVADGAQITTIEGVGQPENLHPLQKAWVLHGGAQCGFCSPGFIVSAKGLLDTNADPSREDVRDWFQKHRNACRCTGYKPLVDAVMDAAAVINGKKPETDLEFKMPADGRIWGSKYPRPTAVAKVTGTLDYGADLGLKMPAGTLHLAMVQAKVSHANIKGIDTSEALTMPGVHSVITHKDVKGKNRITGLITFPTNKGDGWDRPILXDEKVFQYGDCIALVCADSEANARAAAEKVKVDLEELPAYMSGPAAAAEDAIEIHPGTPNVYFEQPIVKGEDTGPIFASADVTVEGDFYVGRQPHMPIEPDVAFAYMGDDGKCYIHSKSIGVHLHLYMIAPGVGLEPDQLVLVANPMGGTFGYKFSPTSEALVAVAAMATGRPVHLRYNYQQQQQYTGKRSPWEMNVKFAAKKDGTLLAMESDWLVDHGPYSEFGDLLTLRGAQFIGAGYNIPNIRGLGRTVATNHVWGSAFRGYGAPQSMFASECLMDMLAEKLGMDPLELRYKNAYRPGDTNPTGQEPEVFSLPDMIDQLRPKYQAALEKAQKESTATHKKGVGISIGVYGSGLDGPDASEAWAELNADGTITVHTAWEDHGQGADIGCVGTAHEALRPMGVAPEKIKFTWPNTATTPNSGPSGGSRQQVMTGNAIRVACENLLKACEKPGGGYYTYDELKAADKPTKITGNWTASGATHCDAVTGLGKPFVVYMYGVFMAEVTVDVATGQTTVDGMTLMADLGSLCNQLATDGQIYGGLAQGIGLALSEDFEDIKKHATLVGAGFPFIKQIPDKLDIVYVNHPRPDGPFGASGVGELPLTSPHAAIINAIKSATGVRIYRLPAYPEKVLEALKA Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Tissue kallikrein (KLK2) | 4NFE | 3.99 | |
Target general information Gen name KLK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hGK-1; Tissue kallikrein-2; Kallikrein-2; Glandular kallikrein-1 Protein family Peptidase S1 family, Kallikrein subfamily Biochemical class Peptidase Function Glandular kallikreins cleave Met-Lys and Arg-Ser bonds in kininogen to release Lys-bradykinin. Related diseases Nivelon-Nivelon-Mabille syndrome (NNMS) [MIM:600092]: An autosomal recessive syndrome characterized by progressive microcephaly, cerebellar vermis hypoplasia, and skeletal dysplasia. Additional variable features include early infantile-onset seizures, intrauterine and postnatal growth retardation, generalized chondrodysplasia, and micromelia. 46,XY gonadal dysgenesis may be present. {ECO:0000269|PubMed:24784881, ECO:0000269|PubMed:30912300}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.21.35 Uniprot keywords 3D-structure; Alternative splicing; Disulfide bond; Glycoprotein; Hydrolase; Protease; Proteomics identification; Reference proteome; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 24954.3 Length 227 Aromaticity 0.08 Instability index 43.7 Isoelectric point 6.42 Charge (pH=7) -2.35 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map 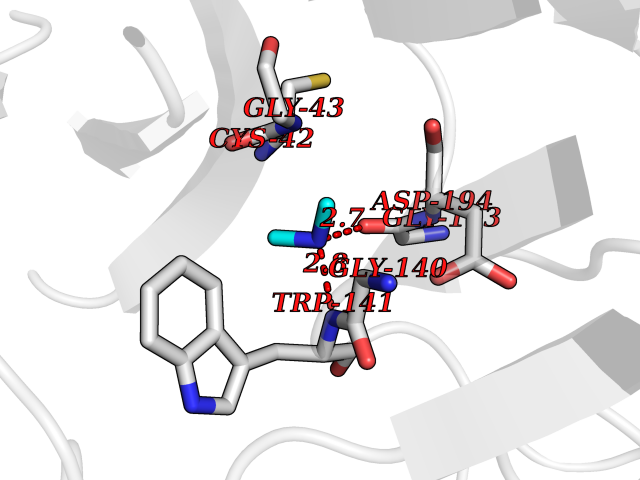 Pymol Map 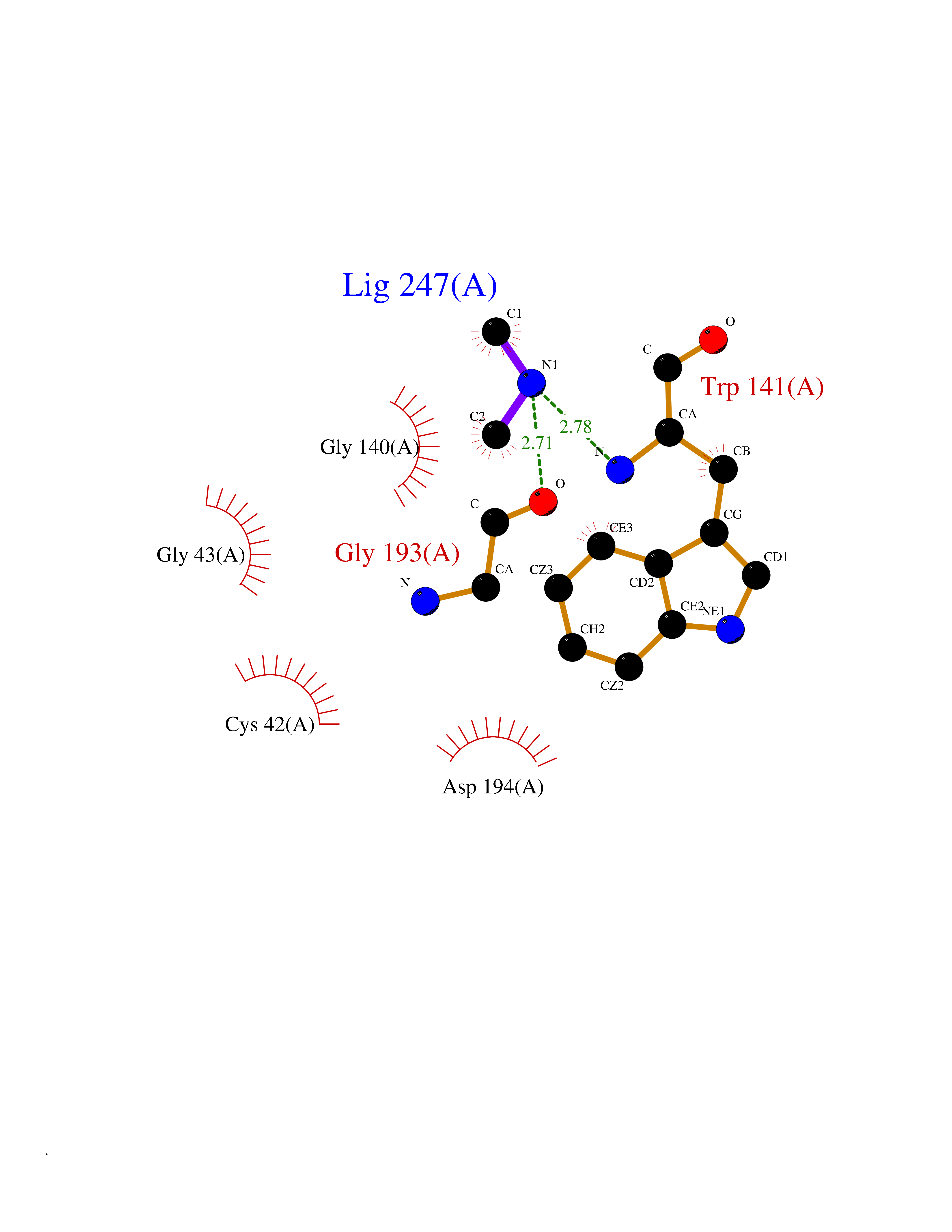 Ligplot Map 3D Binding mode Sequence IVGGWECEKHSQPWQVAVYSHGWAHCGGVLVHPQWVLTAAHCLKKNSQVWLGRHNLFEPEDTGQRVPVSHSFPHPLYNMSLDSSHDLMLLRLSEPAKITDVVKVLGLPTQEPALGTTCYASGWGSIEPEEFLRPRSLQCVSLHLLSNDMCARAYSEKVTEFMLCAGLWTGGKDTCGGDSGGPLVCNGVLQGITSWGPEPCALPEKPAVYTKVVHYRKWIKDTIAANP Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Lysozyme (LYZ) | 207L | 3.99 | |
Target general information Gen name LYZ Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysozyme C; LYZ; 1,4betaNacetylmuramidase C Protein family Glycosyl hydrolase 22 family Biochemical class Glycosylase Function Lysozymes have primarily a bacteriolytic function; those intissues and body fluids are associated with the monocyte- macrophage system and enhance the activity of immunoagents. Related diseases Amyloidosis, hereditary systemic 5 (AMYLD5) [MIM:620658]: A form of hereditary systemic amyloidosis, a disorder characterized by amyloid deposition in multiple tissues resulting in a wide clinical spectrum. AMYLD5 primarily affects the viscera, and the predominant clinical features are renal dysfunction of varying severity, and intra-abdominal bleeding. Inheritance is autosomal dominant. {ECO:0000269|PubMed:8464497}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02159; DB03487; DB02759; DB03006; DB00128; DB03189; DB03967; DB04268; DB03013; DB03120; DB03175; DB11182; DB02772; DB04194; DB06912 Interacts with P61626 EC number EC 3.2.1.17 Uniprot keywords 3D-structure; Amyloid; Amyloidosis; Antimicrobial; Bacteriolytic enzyme; Direct protein sequencing; Disease variant; Disulfide bond; Glycosidase; Hydrolase; Proteomics identification; Reference proteome; Secreted; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 14668.5 Length 130 Aromaticity 0.1 Instability index 25.59 Isoelectric point 9.37 Charge (pH=7) 7.78 2D Binding mode Binding energy (Kcal/mol) -5.44 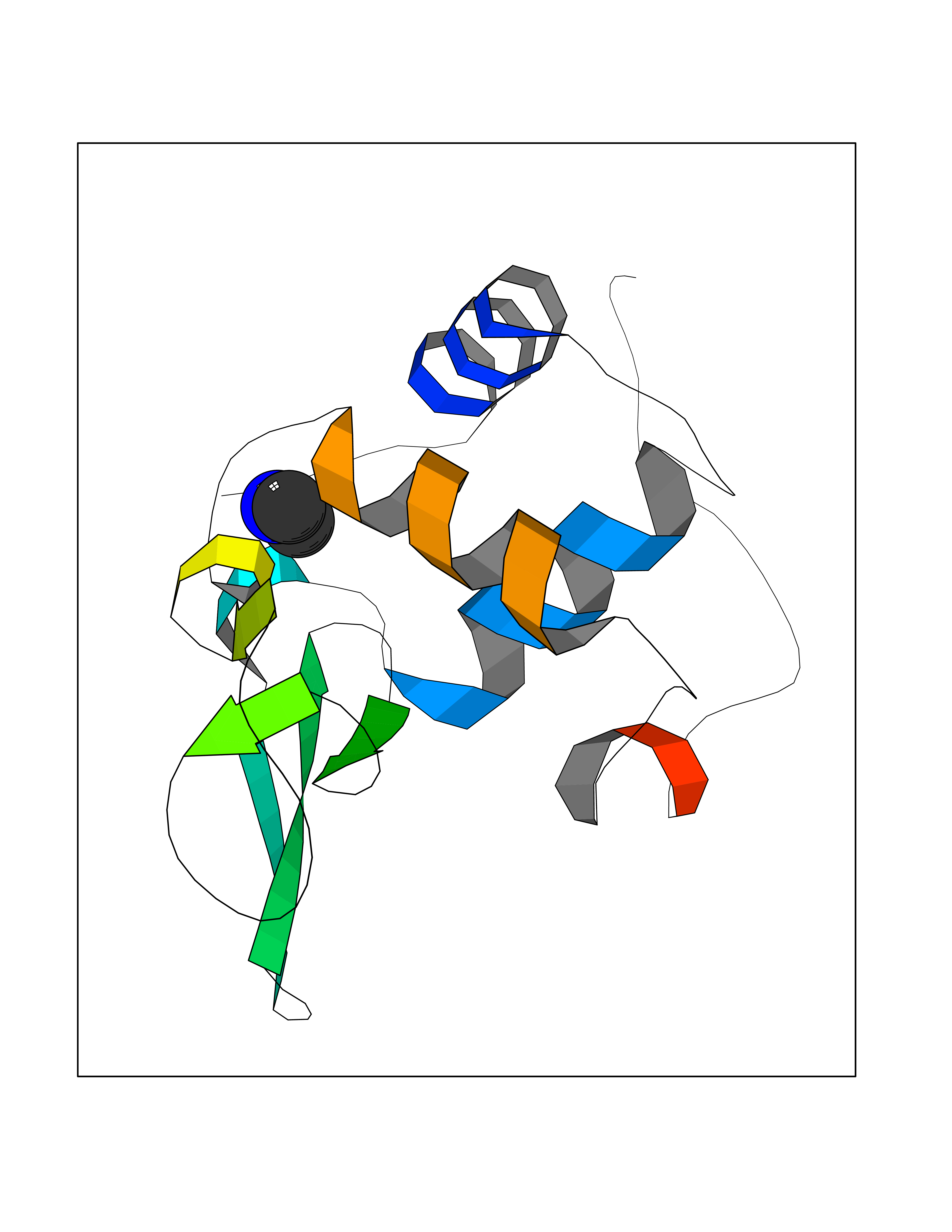 Molscript Map 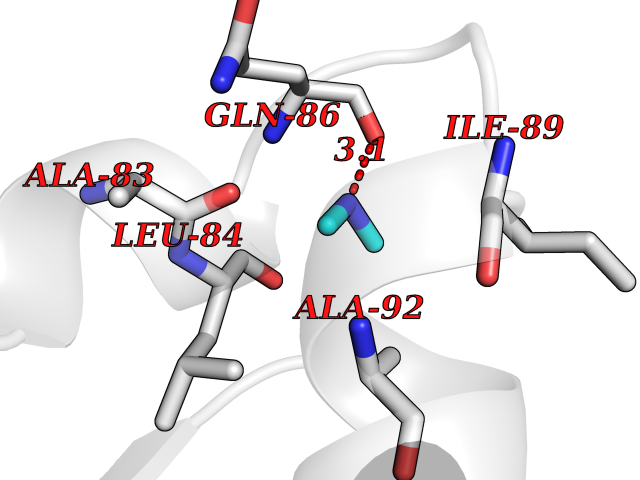 Pymol Map 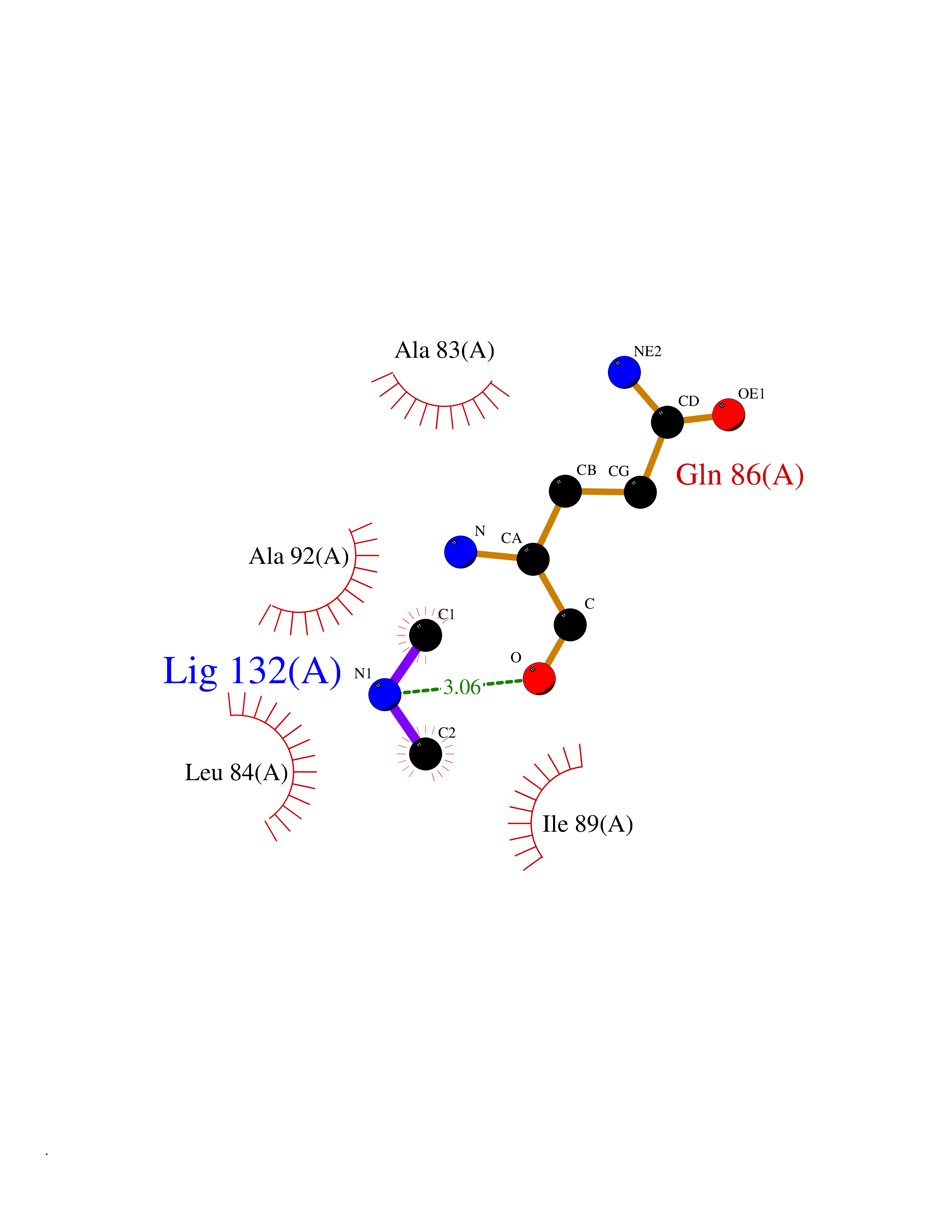 Ligplot Map 3D Binding mode Sequence KVFERCELARTLKRLGMDGYRGISLANWMCLAKWESGYNTRATNYNAGDRSTDYGIFQINSRYWCNDGKTPGAVNAAHLSCSALLQDNIADAVACAKRVVRDPQGIRAWVAWRNRCQNRDVRQYVQGCGV Hydrogen bonds contact Hydrophobic contact | ||||
| 100 | Cerebron E3 ubiquitin ligase complex (CRL4-CRBN E3 ubiquitin ligase) | 4CI1 | 3.99 | |
Target general information Gen name CUL4A/CUL4B-DDB1-CRBN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NA Protein family Cullin family Biochemical class NA Function NA Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P54253; Q86VP6; Q16531; Q92466; P08238; O94888; P55072 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Biological rhythms; DNA damage; DNA repair; Host-virus interaction; Isopeptide bond; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID B Molecular weight (Da) 42669.7 Length 368 Aromaticity 0.1 Instability index 44.94 Isoelectric point 8.72 Charge (pH=7) 6.58 2D Binding mode Binding energy (Kcal/mol) -5.44  Molscript Map 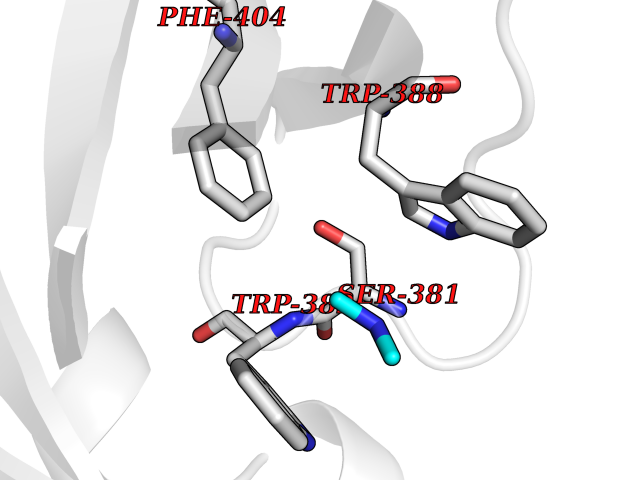 Pymol Map 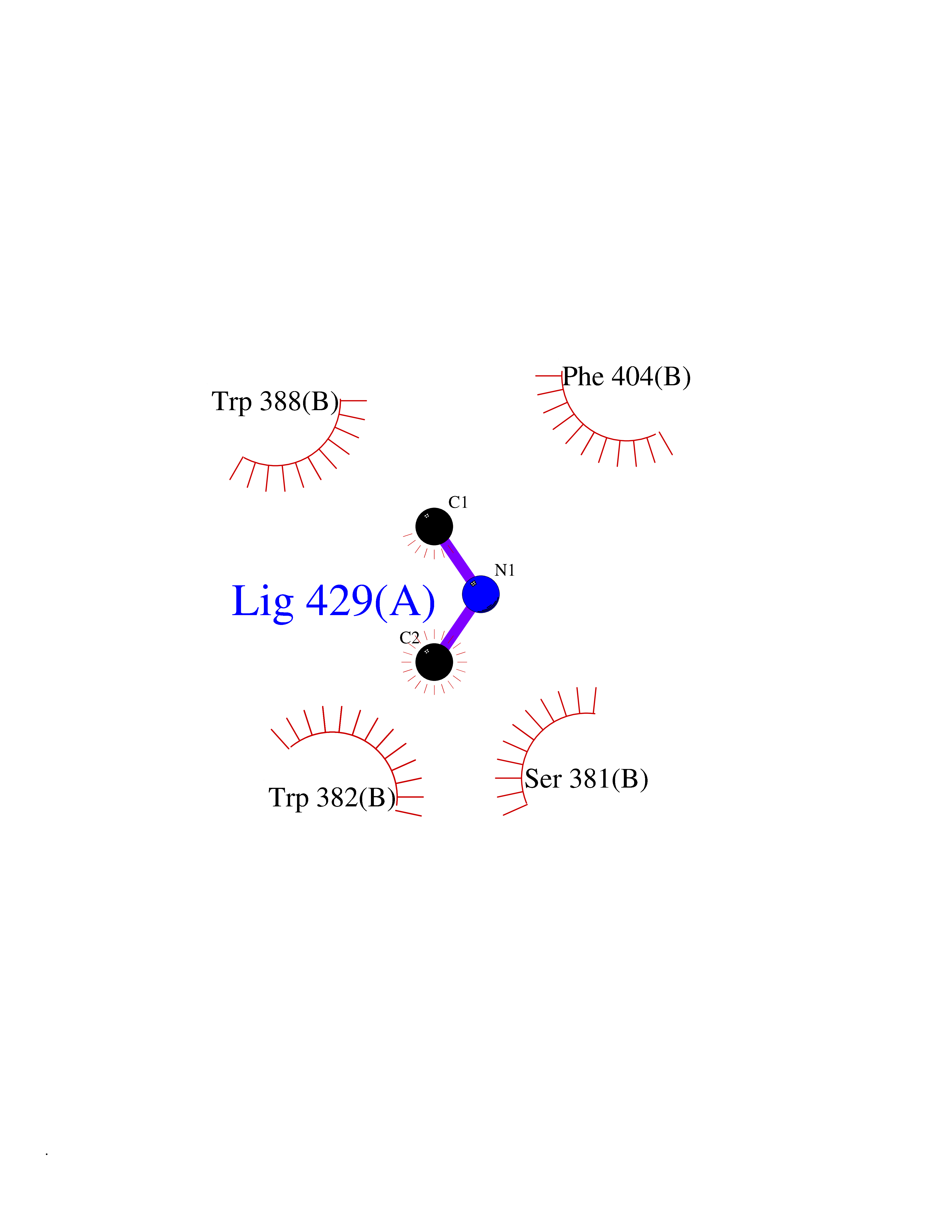 Ligplot Map 3D Binding mode Sequence MINFDTSLPTSHMYLGSDMEEFHGRTLHDDDSCQVIPVLPHVMVMLIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVREREAHFGTTAEIYAYREEQEYGIETVKVKAIGRQRFKVLEIRTQSDGIQQAKVQILPERVLPSTMSAVQLQSLSRRHIRAFRQWWQKYQKRKFHCASLTSWPPWLYSLYDAETLMERVKRQLHEWDENLKDESLPTNPIDFSYRVAACLPIDDALRIQLLKIGSAIQRLRELDIMNKTSLCCKQCQDTEITTKNEIFSLSLCGPMAAYVNPHGYIHETLTVYKACNLNLSGRPSTEHSWFPGYAWTIAQCRICGNHMGWKFTATKKDMSPQKFWGLTRSALLPR Hydrogen bonds contact Hydrophobic contact | ||||