Job Results:
Ligand
Structure
Job ID
7f371ea108ffadeed56066fb8d4cae63
Job name
NA
Time
2024-12-09 11:06:55
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Histone-lysine N-methyltransferase EHMT2 (EHMT2) | 5VSC | 4.73 | |
Target general information Gen name EHMT2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein G9a; NG36; Lysine N-methyltransferase 1C; KMT1C; Histone H3-K9 methyltransferase 3; HLA-B-associated transcript 8; H3-K9-HMTase 3; G9A; Euchromatic histone-lysine N-methyltransferase 2; C6orf3 Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, Suvar3-9 subfamily Biochemical class Methyltransferase Function H3K9me represents a specific tag for epigenetic transcriptional repression by recruiting HP1 proteins to methylated histones. Also mediates monomethylation of 'Lys-56' of histone H3 (H3K56me1) in G1 phase, leading to promote interaction between histone H3 and PCNA and regulating DNA replication. Also weakly methylates 'Lys-27' of histone H3 (H3K27me). Also required for DNA methylation, the histone methyltransferase activity is not required for DNA methylation, suggesting that these 2 activities function independently. Probably targeted to histone H3 by different DNA-binding proteins like E2F6, MGA, MAX and/or DP1. May also methylate histone H1. In addition to the histone methyltransferase activity, also methylates non-histone proteins: mediates dimethylation of 'Lys-373' of p53/TP53. Also methylates CDYL, WIZ, ACIN1, DNMT1, HDAC1, ERCC6, KLF12 and itself. Histone methyltransferase that specifically mono- and dimethylates 'Lys-9' of histone H3 (H3K9me1 and H3K9me2, respectively) in euchromatin. Related diseases Pseudohypoaldosteronism 2C (PHA2C) [MIM:614492]: An autosomal dominant disorder characterized by severe hypertension, hyperkalemia, hyperchloremia, mild hyperchloremic metabolic acidosis in some cases, and correction of physiologic abnormalities by thiazide diuretics. {ECO:0000269|PubMed:11498583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuropathy, hereditary sensory and autonomic, 2A (HSAN2A) [MIM:201300]: A form of hereditary sensory and autonomic neuropathy, a genetically and clinically heterogeneous group of disorders characterized by degeneration of dorsal root and autonomic ganglion cells, and by sensory and/or autonomic abnormalities. HSAN2A is an autosomal recessive disorder characterized by impairment of pain, temperature and touch sensation, onset of symptoms in infancy or early childhood, occurrence of distal extremity pathologies (paronychia, whitlows, ulcers, and Charcot joints), frequent amputations, sensory loss that affects all modalities of sensation (lower and upper limbs and perhaps the trunk as well), absence or diminution of tendon reflexes (usually in all limbs), minimal autonomic dysfunction, absence of sensory nerve action potentials, and virtual absence of myelinated fibers with decreased numbers of unmyelinated fibers in sural nerves. {ECO:0000269|PubMed:15060842, ECO:0000269|PubMed:15911806, ECO:0000269|PubMed:18521183}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q6VMQ6-2; Q6P1J9; Q9UBC3; P38919; Q9UM22; P23771; Q99684; Q13547; Q96JB3; Q92831; O60341-1; Q9Y4X4; P57682; Q13330; O94776; Q9BTC8; P20592; Q9BSU3; Q99801-1; O60568; Q9NQX1; Q5JSZ5; Q7Z3Z2; Q9P2R6; Q14119; Q96GT9; O60315; Q9NWS9-2; Q96JM2; A0A0S2Z5X4; Q96BV0; Q96EG3; Q07120; O60341-1 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ANK repeat; Chromatin regulator; Chromosome; Isopeptide bond; Metal-binding; Methylation; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; S-adenosyl-L-methionine; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 31010.9 Length 269 Aromaticity 0.1 Instability index 47.49 Isoelectric point 5.16 Charge (pH=7) -9.31 2D Binding mode Binding energy (Kcal/mol) -6.45 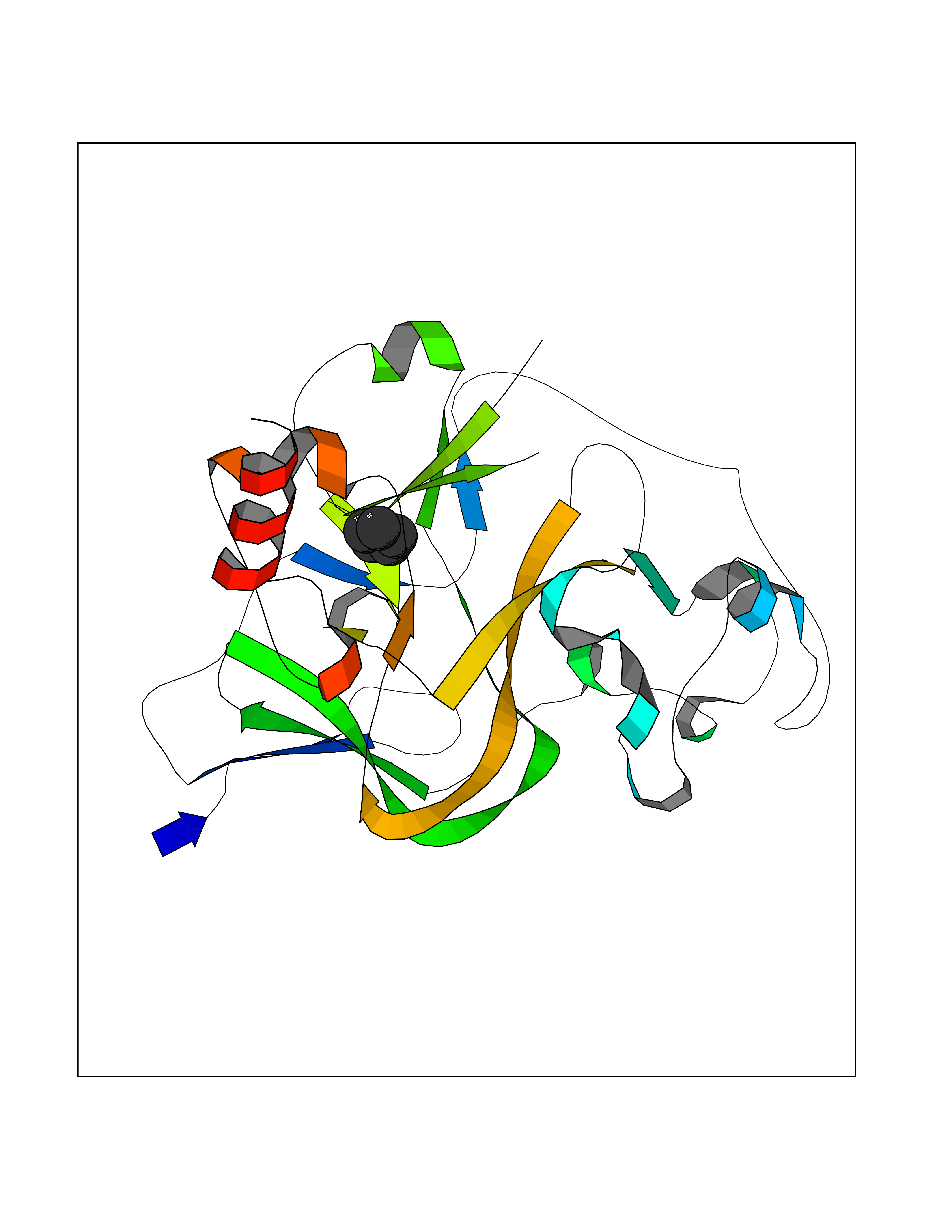 Molscript Map 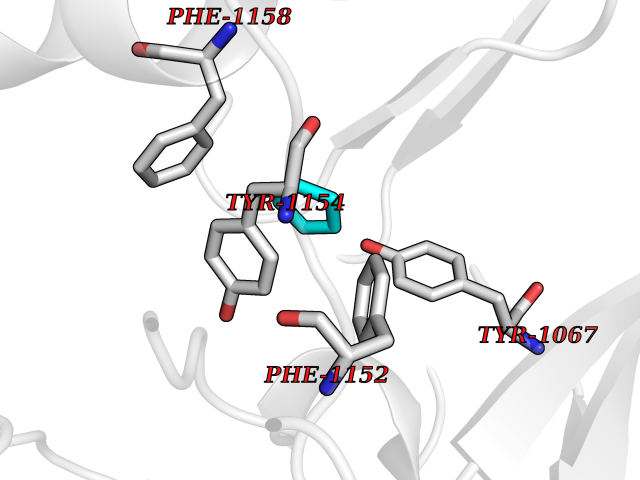 Pymol Map 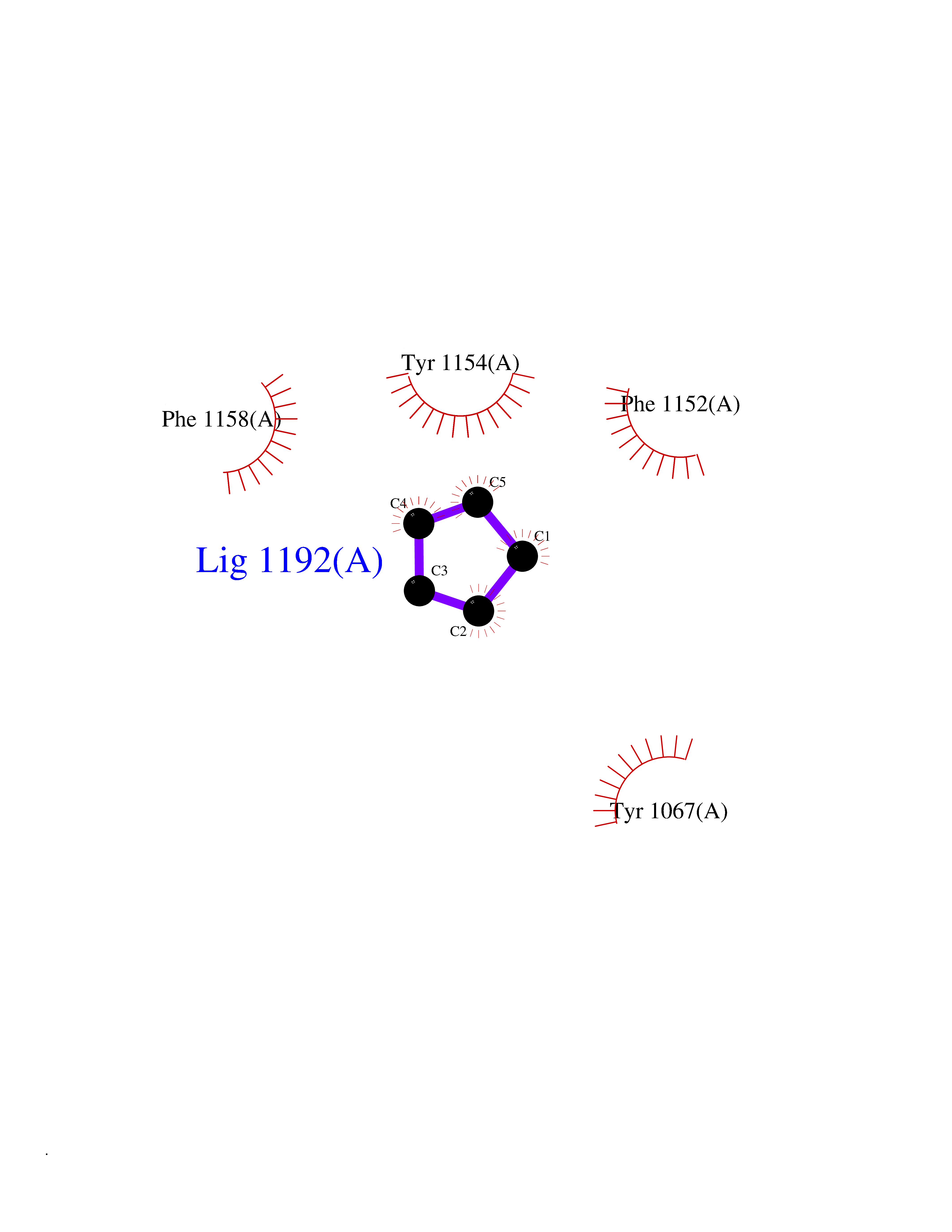 Ligplot Map 3D Binding mode Sequence TEKIICRDVARGYENVPIPCVNGVDGEPCPEDYKYISENCETSTMNIDRNITHLQHCTCVDDCSSSNCLCGQLSIRCWYDKDGRLLQEFNKIEPPLIFECNQACSCWRNCKNRVVQSGIKVRLQLYRTAKMGWGVRALQTIPQGTFICEYVGELISDAEADVREDDSYLFDLDEVYCIDARYYGNISRFINHLCDPNIIPVRVFMLHQDLRFPRIAFFSSRDIRTGEELGFDYGDRFWDIKSKYFTCQCGSEKCKHSAEAIALEQSRLA Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | T-cell surface glycoprotein CD1a (CD1A) | 6NUX | 4.73 | |
Target general information Gen name CD1A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hTa1 thymocyteantigen; hTa1 thymocyte antigen; T-cell surfaceantigen T6/Leu-6; T-cell surface antigen T6/Leu-6; CD1a Protein family NA Biochemical class Immunoglobulin Function Antigen-presenting protein that binds self and non-self lipid and glycolipid antigens and presents them to T-cell receptors on natural killer T-cells. Related diseases Pulmonary hypertension, primary, 1 (PPH1) [MIM:178600]: A rare disorder characterized by plexiform lesions of proliferating endothelial cells in pulmonary arterioles. The lesions lead to elevated pulmonary arterial pression, right ventricular failure, and death. The disease can occur from infancy throughout life and it has a mean age at onset of 36 years. Penetrance is reduced. Although familial pulmonary hypertension is rare, cases secondary to known etiologies are more common and include those associated with the appetite-suppressant drugs. {ECO:0000269|PubMed:10903931, ECO:0000269|PubMed:10973254, ECO:0000269|PubMed:11015450, ECO:0000269|PubMed:11115378, ECO:0000269|PubMed:12045205, ECO:0000269|PubMed:12358323, ECO:0000269|PubMed:15965979, ECO:0000269|PubMed:24936649, ECO:0000269|PubMed:25187962, ECO:0000269|PubMed:28507310}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pulmonary venoocclusive disease 1, autosomal dominant (PVOD1) [MIM:265450]: A disease characterized by widespread fibrous obstruction and intimal thickening of septal veins and preseptal venules, a low diffusing capacity for carbon monoxide, occult alveolar hemorrhage, and nodular ground-glass opacities, septal lines and lymph node enlargement showed by high-resolution computed tomography of the chest. It is frequently associated with pulmonary capillary dilatation and proliferation, and is a rare and devastating cause of pulmonary hypertension. {ECO:0000269|PubMed:12446270, ECO:0000269|PubMed:16429395}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00098 Interacts with P61769 EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Cell membrane; Disulfide bond; Endosome; Glycoprotein; Immunity; Immunoglobulin domain; Membrane; Proteomics identification; Reference proteome; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 30867.3 Length 270 Aromaticity 0.13 Instability index 37.45 Isoelectric point 6.16 Charge (pH=7) -4.5 2D Binding mode Binding energy (Kcal/mol) -6.46 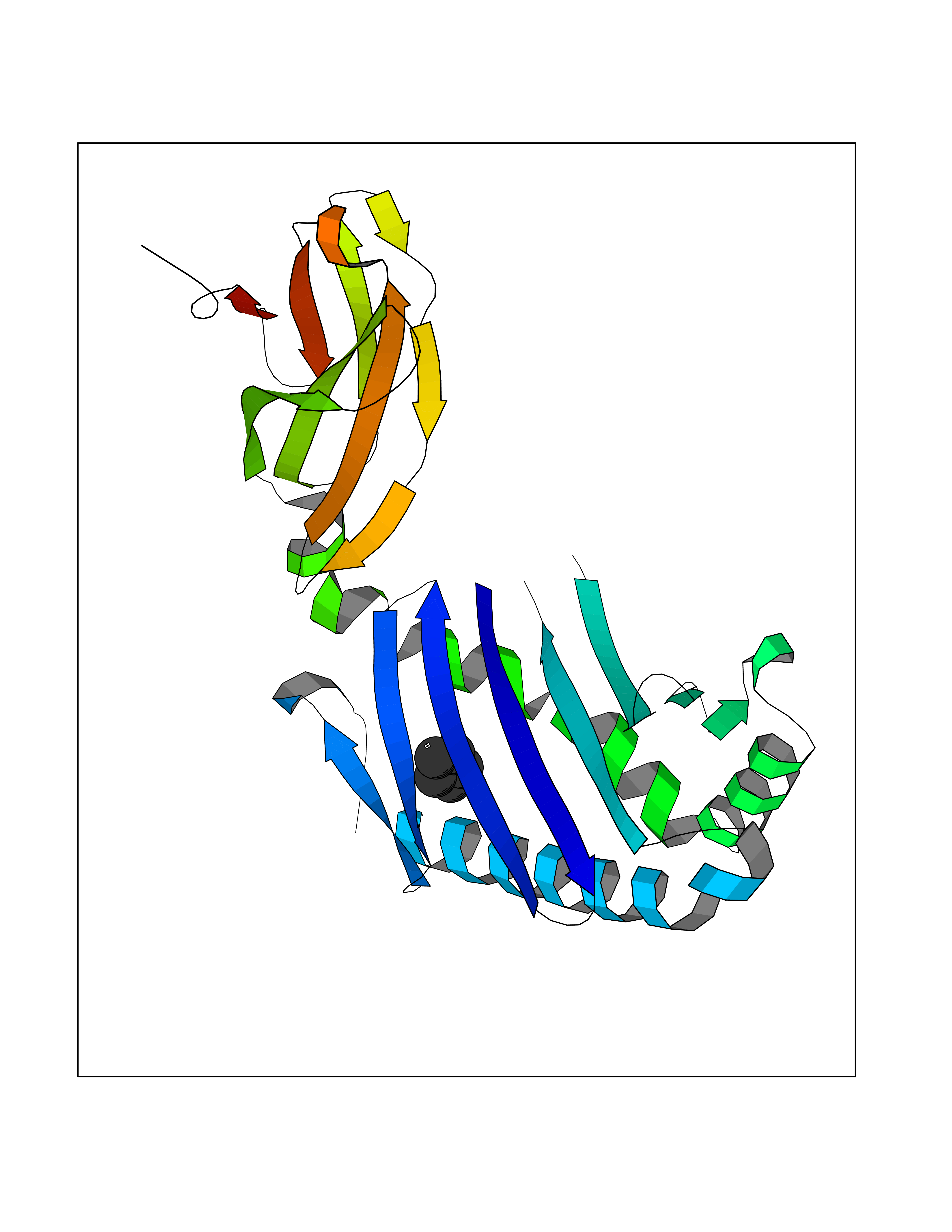 Molscript Map 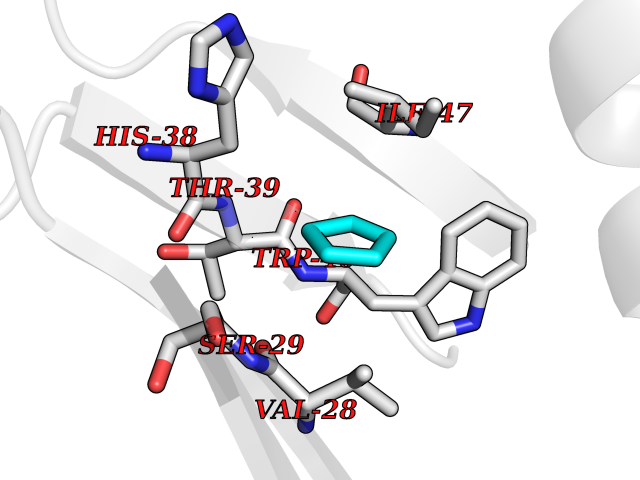 Pymol Map 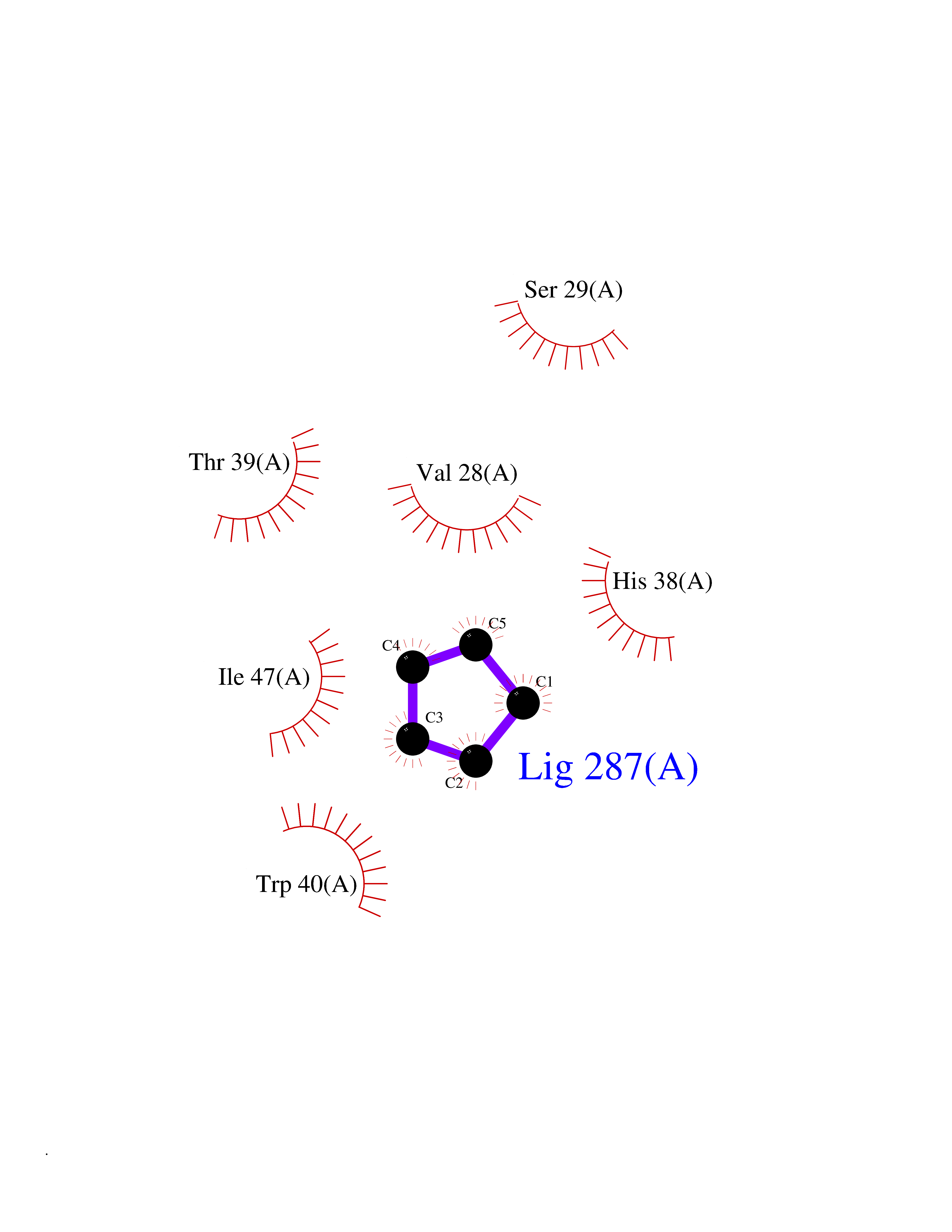 Ligplot Map 3D Binding mode Sequence SFHVIWIASFYNHSWKQNLVSGWLSDLQTHTWDSNSSTIVFLWPWSRGNFSNEWKELETLFRIRTIRSFEGIRRYAHELQFEYPFEIQVTGGCESGSFLQLAYQGSDFVSFQNNSWLPYPVAGNMAKHFCKVLNQNQHENDITHNLLSDTCPRFILGLLDAGKAHLQRQVKPEAWLSHGPSPGPGHLQLVCHVSGFYPKPVWVMWMRGEQEQQGTQRGDILPSADGTWYLRATLEVAAGEAADLSCRVKHSSLEGQDIVLYWEGSLVPRG Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Histone-lysine N-methyltransferase KMT5C (KMT5C) | 3RQ4 | 4.73 | |
Target general information Gen name KMT5C Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine N-methyltransferase 5C; Lysine-specific methyltransferase 5C; Suppressor of variegation 4-20 homolog 2; Su(var)4-20 homolog 2; Suv4-20h2; [histone H4]-N-methyl-L-lysine20 N-methyltransferase KM Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, Suvar4-20 subfamily Biochemical class NA Function Histone methyltransferase that specifically methylates monomethylated 'Lys-20' (H4K20me1) and dimethylated 'Lys-20' (H4K20me2) of histone H4 to produce respectively dimethylated 'Lys-20' (H4K20me2) and trimethylated 'Lys-20' (H4K20me3) and thus regulates transcription and maintenance of genome integrity. In vitro also methylates unmodified 'Lys-20' (H4K20me0) of histone H4 and nucleosomes. H4 'Lys-20' trimethylation represents a specific tag for epigenetic transcriptional repression. Mainly functions in pericentric heterochromatin regions, thereby playing a central role in the establishment of constitutive heterochromatin in these regions. KMT5C is targeted to histone H3 via its interaction with RB1 family proteins (RB1, RBL1 and RBL2) (By similarity). Facilitates TP53BP1 foci formation upon DNA damage and proficient non-homologous end-joining (NHEJ)-directed DNA repair by catalyzing the di- and trimethylation of 'Lys-20' of histone H4. May play a role in class switch reconbination by catalyzing the di- and trimethylation of 'Lys-20' of histone H4 (By similarity). Related diseases Brachydactyly A2 (BDA2) [MIM:112600]: A form of brachydactyly. Brachydactyly defines a group of inherited malformations characterized by shortening of the digits due to abnormal development of the phalanges and/or the metacarpals. In brachydactyly type A2 shortening of the middle phalanges is confined to the index finger and the second toe, all other digits being more or less normal. Because of a rhomboid or triangular shape of the affected middle phalanx, the end of the second finger usually deviates radially. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}. The gene represented in this entry is involved in disease pathogenesis. Duplications of a cis-regulatory element located approximately 110 kb downstream of BMP2 have been found in BDA2 families. They likely cause altered BMP2 expression with pathological consequences. {ECO:0000269|PubMed:19327734, ECO:0000269|PubMed:21357617}.; DISEASE: Short stature, facial dysmorphism, and skeletal anomalies with or without cardiac anomalies 1 (SSFSC1) [MIM:617877]: An autosomal dominant disorder characterized by short stature, facial dysmorphism, skeletal anomalies, and variable cardiac defects. Distinctive facial features include midface retrusion, short upturned nose, long philtrum, high-arched or cleft palate, and variable degrees of micrognathia and dental crowding. Skeletal anomalies include patterning defects of the axial skeleton, characterized by 11 pairs of ribs and brachydactyly of the fifth ray. Congenital heart defects are variably observed and appear to involve primarily the cardiac outflow tract. {ECO:0000269|PubMed:29198724}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q13185 EC number EC 2.1.1.361 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 27285.8 Length 240 Aromaticity 0.1 Instability index 42.74 Isoelectric point 8.32 Charge (pH=7) 3.24 2D Binding mode Binding energy (Kcal/mol) -6.45 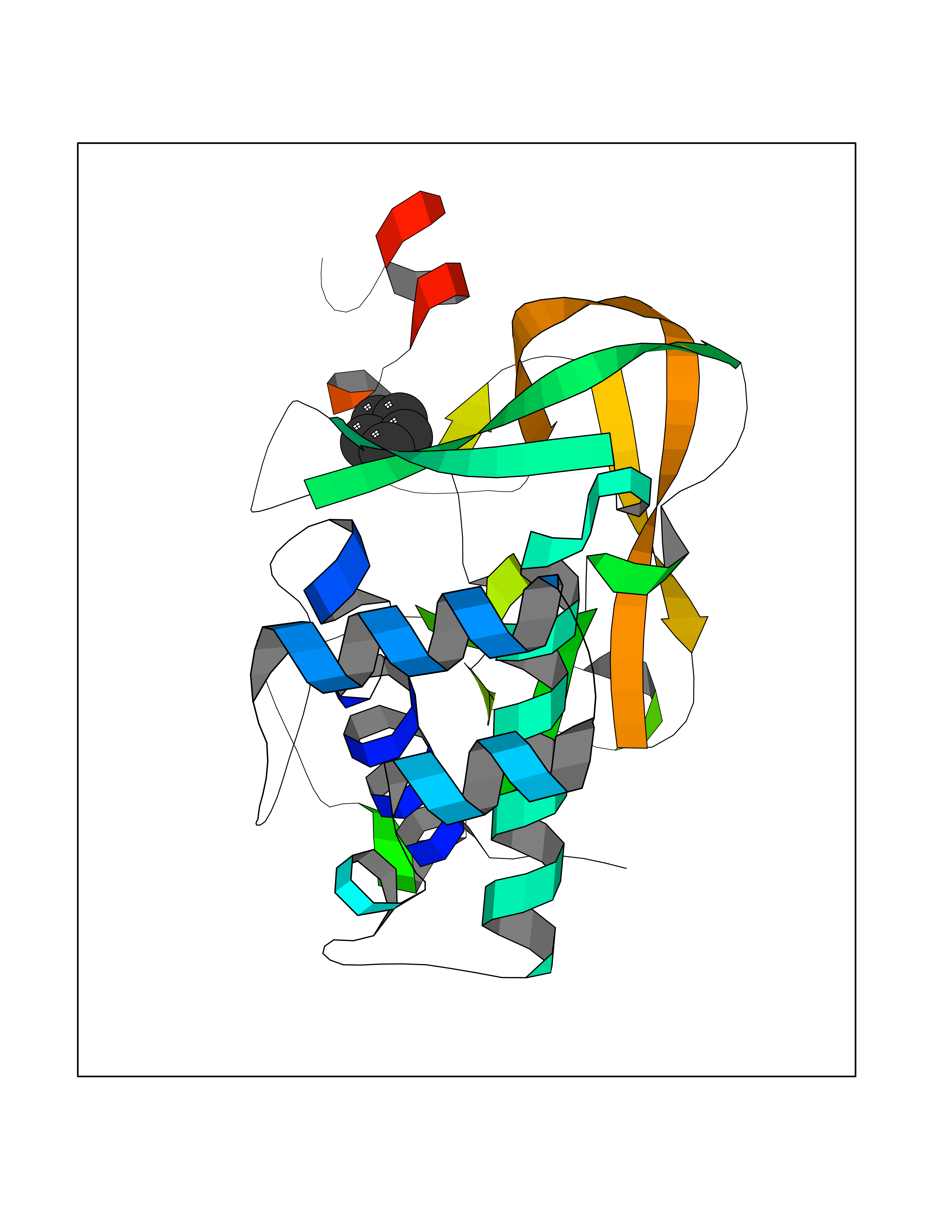 Molscript Map 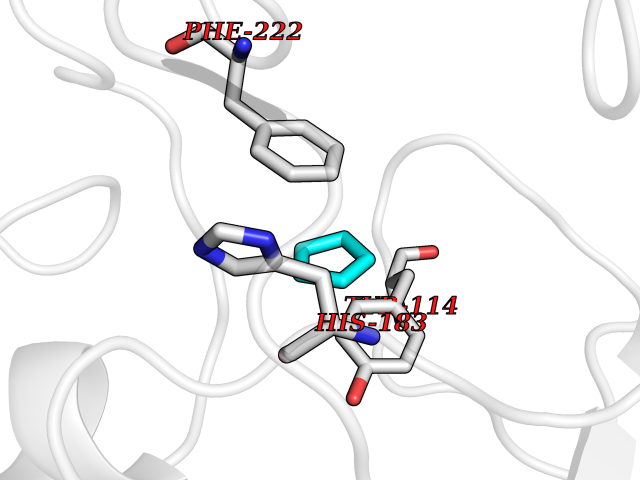 Pymol Map 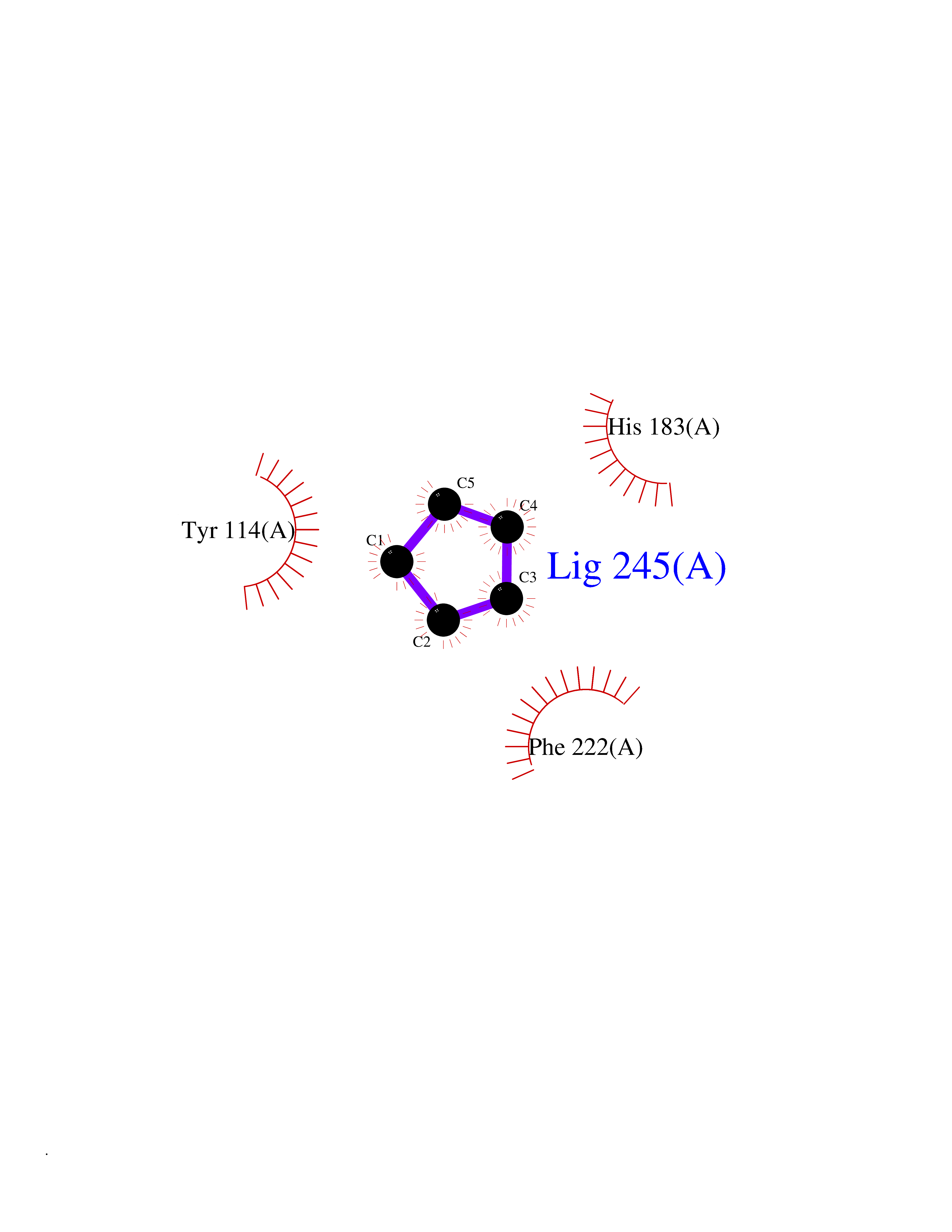 Ligplot Map 3D Binding mode Sequence DRVTARELCENDDLATSLVLDPYLGFRTHKMNVSPVPPLRRQQHLRSALETFLRQRDLEAAYRALTLGGWTARYFQSRGPRQEAALKTHVYRYLRAFLPESGFTILPCTRYSMETNGAKIVSTRAWKKNEKLELLVGCIAELREADEGLLRAGENDFSIMYSTRKRSAQLWLGPAAFINHDCKPNCKFVPADGNAACVKVLRDIEPGDEVTCFYGEGFFGEKNEHCECHTCERKGEGAFR Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Neuronal acetylcholine receptor beta-4 (CHRNB4) | 6PV7 | 4.73 | |
Target general information Gen name CHRNB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CHRNB4; Beta-4 nAChR Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Beta-4/CHRNB4 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane. Related diseases Renal cell carcinoma (RCC) [MIM:144700]: Renal cell carcinoma is a heterogeneous group of sporadic or hereditary carcinoma derived from cells of the proximal renal tubular epithelium. It is subclassified into clear cell renal carcinoma (non-papillary carcinoma), papillary renal cell carcinoma, chromophobe renal cell carcinoma, collecting duct carcinoma with medullary carcinoma of the kidney, and unclassified renal cell carcinoma. Clear cell renal cell carcinoma is the most common subtype. {ECO:0000269|PubMed:20054297, ECO:0000269|PubMed:23622243, ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}. The disease may be caused by variants affecting the gene represented in this entry. Defects of SETD2 are associated with loss of DNA methylation at non-promoter regions (PubMed:23792563). SETD2 defects lead to aberrant and reduced nucleosome compaction and chromatin association of key replication proteins, such as MCM7 and DNA polymerase delta, leading to hinder replication fork progression and prevent loading of RAD51 homologous recombination repair factor at DNA breaks (PubMed:25728682). {ECO:0000269|PubMed:23792563, ECO:0000269|PubMed:25728682}.; DISEASE: Luscan-Lumish syndrome (LLS) [MIM:616831]: An autosomal dominant syndrome with a variable phenotype. Clinical features include macrocephaly, distinctive facial appearance, postnatal overgrowth, various degrees of learning difficulties, autism spectrum disorder, and intellectual disability. {ECO:0000269|PubMed:23160955, ECO:0000269|PubMed:24852293, ECO:0000269|PubMed:26084711, ECO:0000269|PubMed:27317772}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, acute lymphoblastic (ALL) [MIM:613065]: A subtype of acute leukemia, a cancer of the white blood cells. ALL is a malignant disease of bone marrow and the most common malignancy diagnosed in children. The malignant cells are lymphoid precursor cells (lymphoblasts) that are arrested in an early stage of development. The lymphoblasts replace the normal marrow elements, resulting in a marked decrease in the production of normal blood cells. Consequently, anemia, thrombocytopenia, and neutropenia occur to varying degrees. The lymphoblasts also proliferate in organs other than the marrow, particularly the liver, spleen, and lymphnodes. {ECO:0000269|PubMed:24509477, ECO:0000269|PubMed:24662245}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:16314571, ECO:0000269|PubMed:24509477}. The disease may be caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Intellectual developmental disorder, autosomal dominant 70 (MRD70) [MIM:620157]: An autosomal dominant disorder characterized by mild global developmental delay, moderately impaired intellectual disability with speech difficulties, and behavioral abnormalities. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rabin-Pappas syndrome (RAPAS) [MIM:620155]: An autosomal dominant neurodevelopmental disorder characterized by severely impaired global development, intellectual disability, microcephaly, facial dysmorphism, and variable congenital anomalies affecting the skeletal, genitourinary, cardiac, and other organ systems. {ECO:0000269|PubMed:32710489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00237; DB00565; DB00514; DB07720; DB00898; DB00472; DB01227; DB00184; DB01090; DB00202 Interacts with Q6FHY5 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89661.2 Length 775 Aromaticity 0.12 Instability index 35.11 Isoelectric point 6.07 Charge (pH=7) -5.67 2D Binding mode Binding energy (Kcal/mol) -6.45 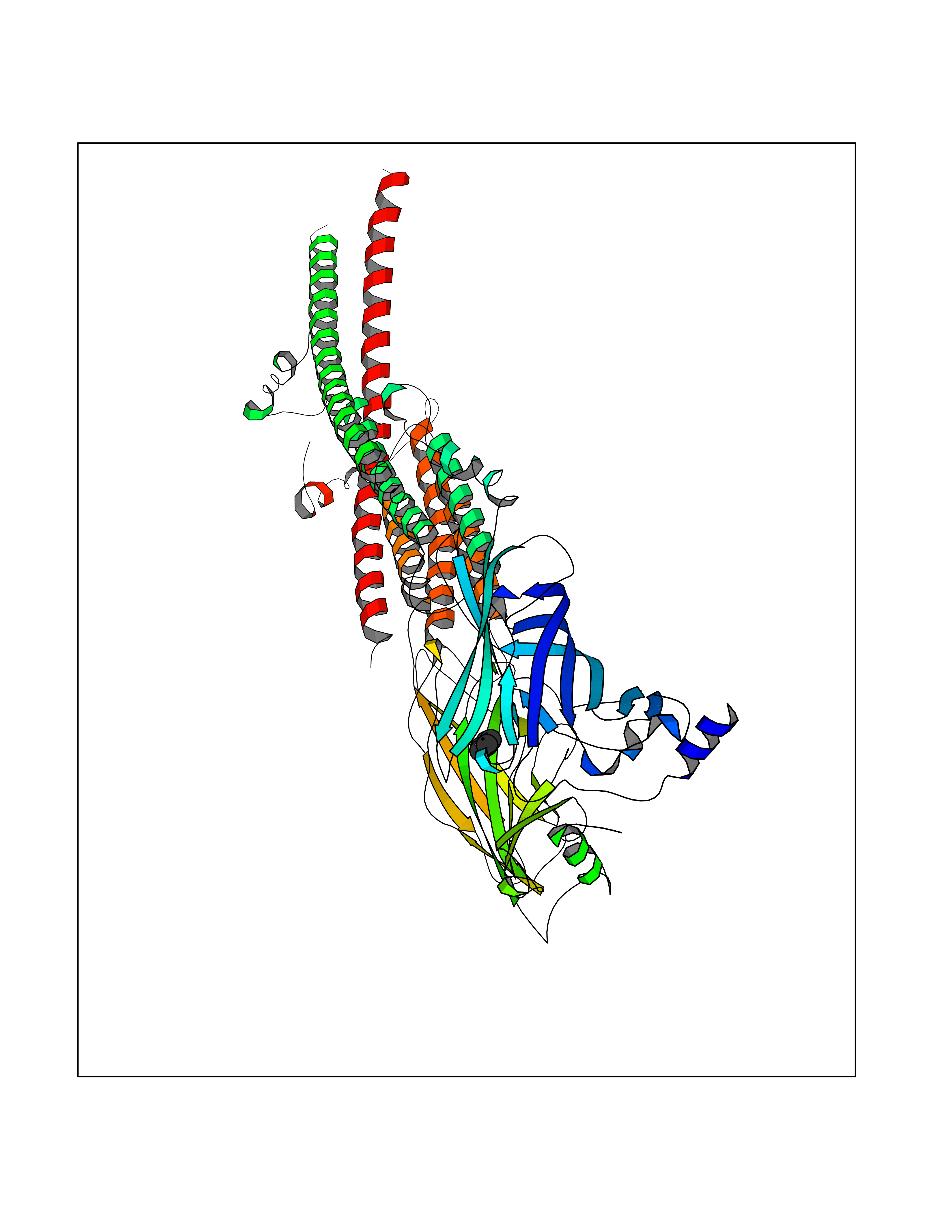 Molscript Map  Pymol Map 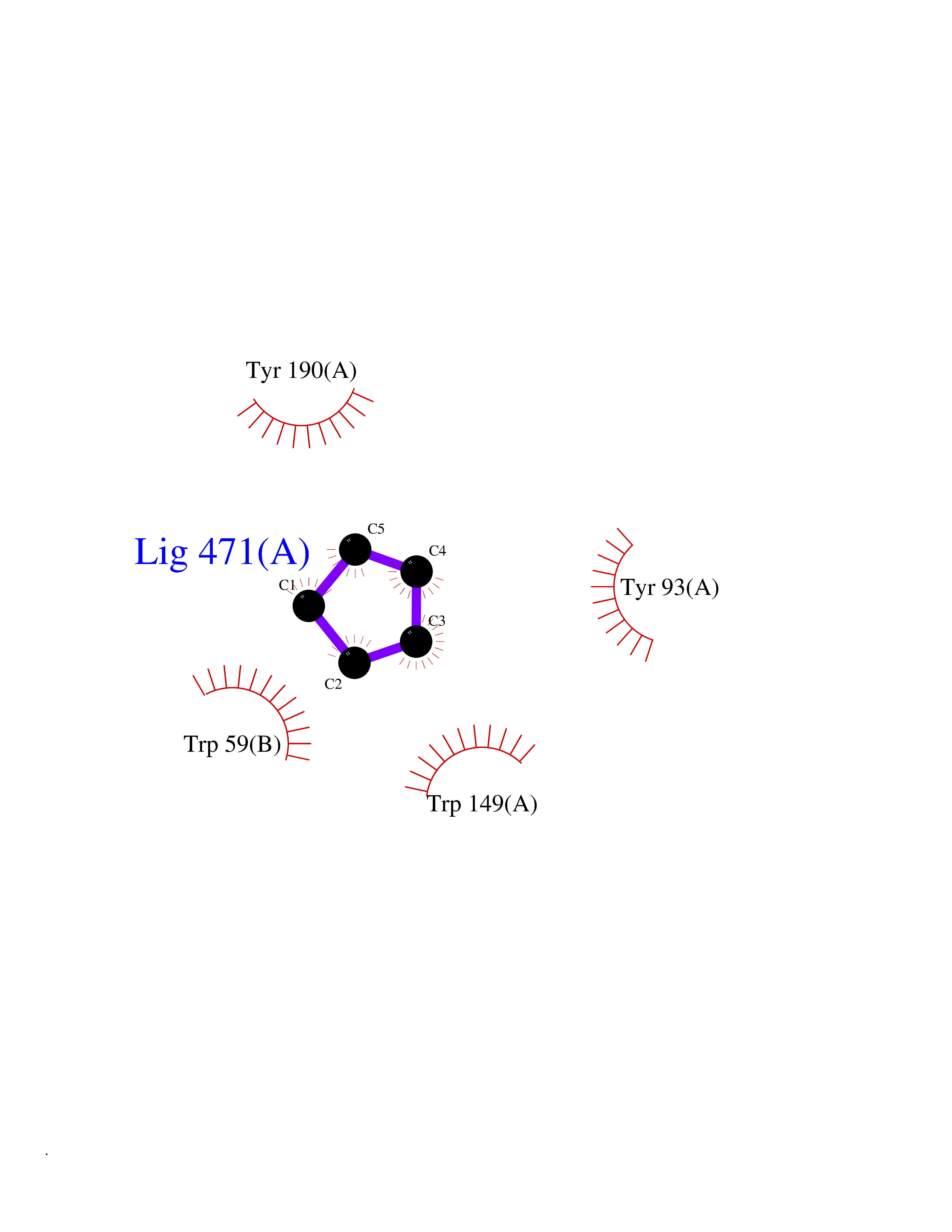 Ligplot Map 3D Binding mode Sequence SEAEHRLFERLFEDYNEIIRPVANVSDPVIIHFEVSMSQLVKVDEVNQIMETNLWLKQIWNDYKLKWNPSDYGGAEFMRVPAQKIWKPDIVLYNNAVGDFQVDDKTKALLKYTGEVTWIPPAIFKSSCKIDVTYFPFDYQNCTMKFGSWSYDKAKIDLVLIGSSMNLKDYWESGEWAIIKAPGYKHDIKYNCCEEIYPDITYSLYIRRLPLFYTINLIIPCLLISFLTVLVFYLPSDCGEKVTLCISVLLSLTVFLLVITETIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHYRTPTTHTMPSWVKTVFLNLLPRVMFMTRIKEAIQSVKYIAENMKAQNEAKEIQDDWKYVAMVIDRIFLWVFTLVCILGTAGLFLQPLMRVANAEEKLMDDLLNKTRYNNLIRPATSSSQLISIKLQLSLAQLISVNEREQIMTTNVWLKQEWTDYRLTWNSSRYEGVNILRIPAKRIWLPDIVLYNNADGTYEVSVYTNLIVRSNGSVLWLPPAIYKSACKIEVKYFPFDQQNCTLKFRSWTYDHTEIDMVLMTPTASMDDFTPSGEWDIVALPGRRTVNPQDPSYVDVTYDFIIKRKPLFYTINLIIPCVLTTLLAILVFYLPSDCGEKMTLCISVLLALTFFLLLISKIVPPTSLDVPLIGKYLMFTMVLVTFSIVTSVCVLNVHHRSPSTHTMAPWVKRCFLHKLPTFLFMKRRQDVQEALEGVSFIAQHMKNDDEDQSVVEDWKYVAMVVDRLFLWVFMFVCVLGTVGLFLP Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Neuronal acetylcholine receptor alpha-3/beta-4 (CHRNA3/B4) | 6PV7 | 4.73 | |
Target general information Gen name CHRNA3-CHRNB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Neuronal acetylcholine receptor Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Neurotransmitter receptor Function A type of nicotinic acetylcholine receptor, consisting of 3 and 4 subunits. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89661.2 Length 775 Aromaticity 0.12 Instability index 35.11 Isoelectric point 6.07 Charge (pH=7) -5.67 2D Binding mode Binding energy (Kcal/mol) -6.45 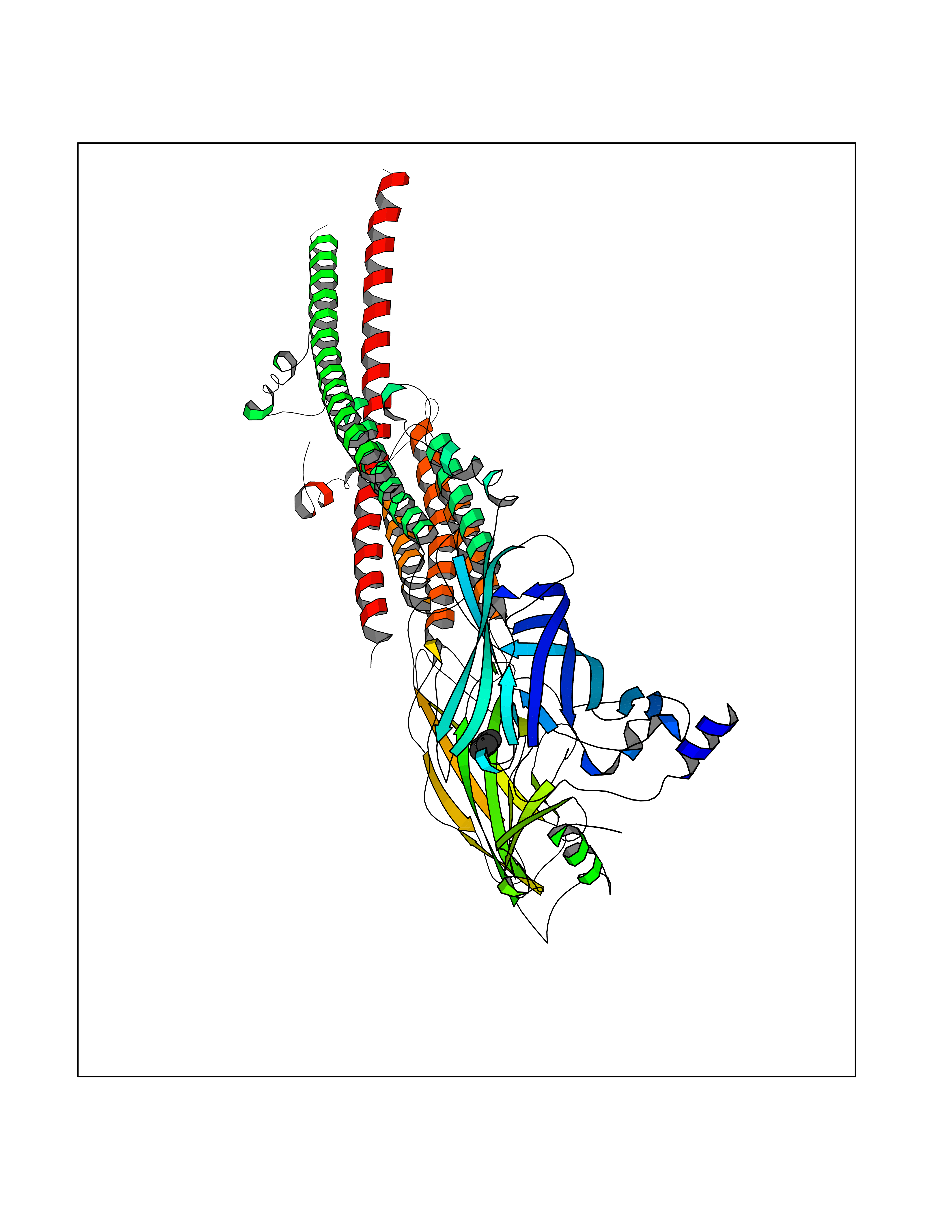 Molscript Map 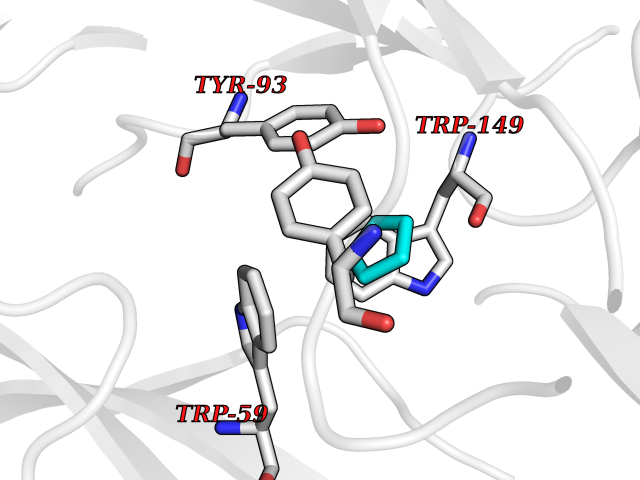 Pymol Map 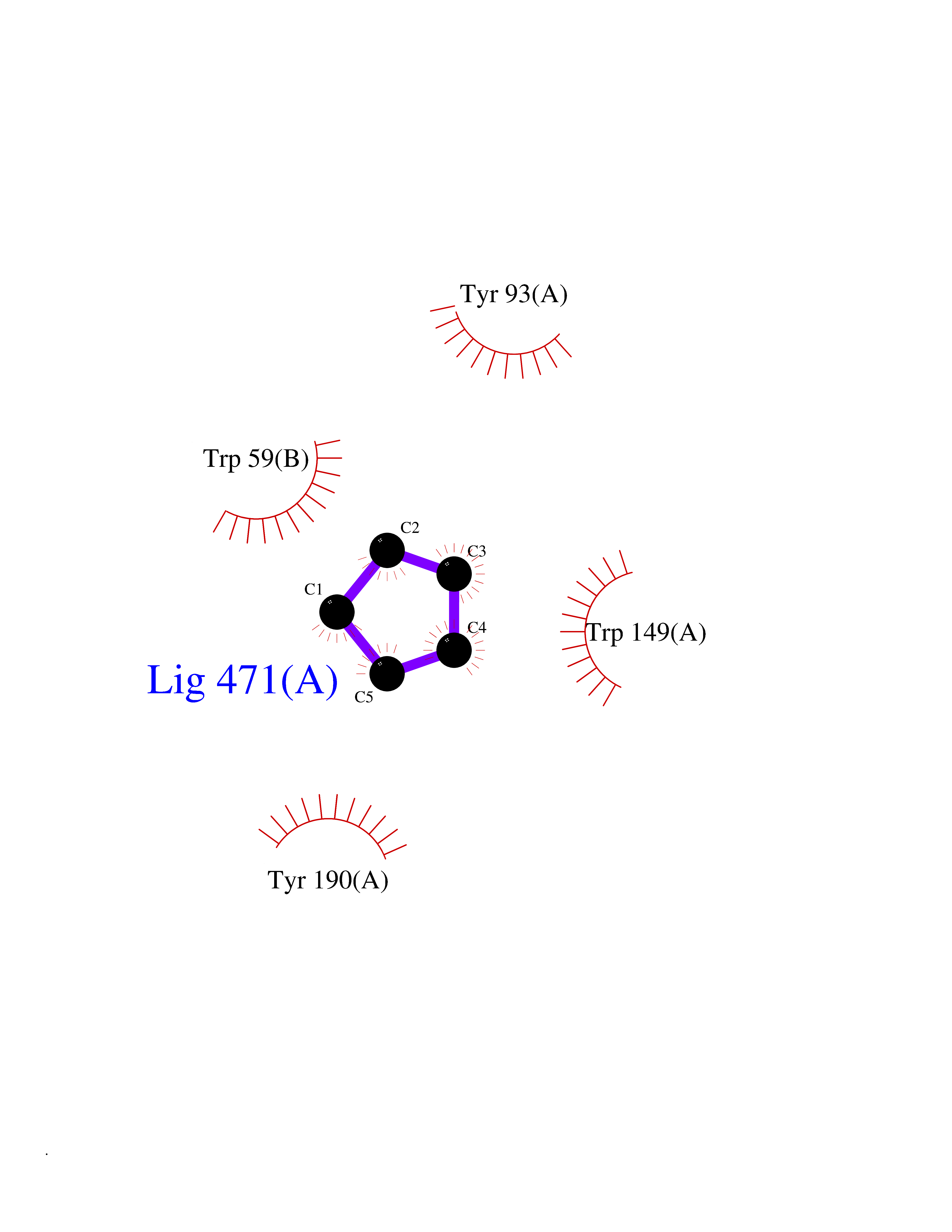 Ligplot Map 3D Binding mode Sequence SEAEHRLFERLFEDYNEIIRPVANVSDPVIIHFEVSMSQLVKVDEVNQIMETNLWLKQIWNDYKLKWNPSDYGGAEFMRVPAQKIWKPDIVLYNNAVGDFQVDDKTKALLKYTGEVTWIPPAIFKSSCKIDVTYFPFDYQNCTMKFGSWSYDKAKIDLVLIGSSMNLKDYWESGEWAIIKAPGYKHDIKYNCCEEIYPDITYSLYIRRLPLFYTINLIIPCLLISFLTVLVFYLPSDCGEKVTLCISVLLSLTVFLLVITETIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHYRTPTTHTMPSWVKTVFLNLLPRVMFMTRIKEAIQSVKYIAENMKAQNEAKEIQDDWKYVAMVIDRIFLWVFTLVCILGTAGLFLQPLMRVANAEEKLMDDLLNKTRYNNLIRPATSSSQLISIKLQLSLAQLISVNEREQIMTTNVWLKQEWTDYRLTWNSSRYEGVNILRIPAKRIWLPDIVLYNNADGTYEVSVYTNLIVRSNGSVLWLPPAIYKSACKIEVKYFPFDQQNCTLKFRSWTYDHTEIDMVLMTPTASMDDFTPSGEWDIVALPGRRTVNPQDPSYVDVTYDFIIKRKPLFYTINLIIPCVLTTLLAILVFYLPSDCGEKMTLCISVLLALTFFLLLISKIVPPTSLDVPLIGKYLMFTMVLVTFSIVTSVCVLNVHHRSPSTHTMAPWVKRCFLHKLPTFLFMKRRQDVQEALEGVSFIAQHMKNDDEDQSVVEDWKYVAMVVDRLFLWVFMFVCVLGTVGLFLP Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Casein kinase II alpha prime (CSNK2A2) | 5YF9 | 4.73 | |
Target general information Gen name CSNK2A2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Casein kinase II subunit alpha'; CK2A2; CK II alpha' Protein family Protein kinase superfamily, Ser/Thr protein kinase family, CK2 subfamily Biochemical class Kinase Function Regulates numerous cellular processes, such as cell cycle progression, apoptosis and transcription, as well as viral infection. May act as a regulatory node which integrates and coordinates numerous signals leading to an appropriate cellular response. During mitosis, functions as a component of the p53/TP53-dependent spindle assembly checkpoint (SAC) that maintains cyclin-B-CDK1 activity and G2 arrest in response to spindle damage. Also required for p53/TP53-mediated apoptosis, phosphorylating 'Ser-392' of p53/TP53 following UV irradiation. Can also negatively regulate apoptosis. Phosphorylates the caspases CASP9 and CASP2 and the apoptotic regulator NOL3. Phosphorylation protects CASP9 from cleavage and activation by CASP8, and inhibits the dimerization of CASP2 and activation of CASP8. Regulates transcription by direct phosphorylation of RNA polymerases I, II, III and IV. Also phosphorylates and regulates numerous transcription factors including NF-kappa-B, STAT1, CREB1, IRF1, IRF2, ATF1, SRF, MAX, JUN, FOS, MYC and MYB. Phosphorylates Hsp90 and its co-chaperones FKBP4 and CDC37, which is essential for chaperone function. Regulates Wnt signaling by phosphorylating CTNNB1 and the transcription factor LEF1. Acts as an ectokinase that phosphorylates several extracellular proteins. During viral infection, phosphorylates various proteins involved in the viral life cycles of EBV, HSV, HBV, HCV, HIV, CMV and HPV. Catalytic subunit of a constitutively active serine/threonine-protein kinase complex that phosphorylates a large number of substrates containing acidic residues C-terminal to the phosphorylated serine or threonine. Related diseases Neurodevelopmental disorder with language delay and seizures (NEDLDS) [MIM:619908]: An autosomal recessive disorder characterized by global developmental delay, intellectual disability, speech delay, and seizures. Additional features may include axial hypotonia, peripheral hypertonia, hypothyroidism, and non-specific dysmorphic features or brain imaging abnormalities. {ECO:0000269|PubMed:35240055}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07546; DB12010 Interacts with Q8NDY6; P68400; P67870; O60282; Q9NRD5; Q8WV44; Q9BS34 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; Apoptosis; ATP-binding; Cell cycle; Cytoplasm; Kinase; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transcription; Transcription regulation; Transferase; Wnt signaling pathway Protein physicochemical properties Chain ID X Molecular weight (Da) 38039.2 Length 319 Aromaticity 0.13 Instability index 43.61 Isoelectric point 8.29 Charge (pH=7) 3.03 2D Binding mode Binding energy (Kcal/mol) -6.45 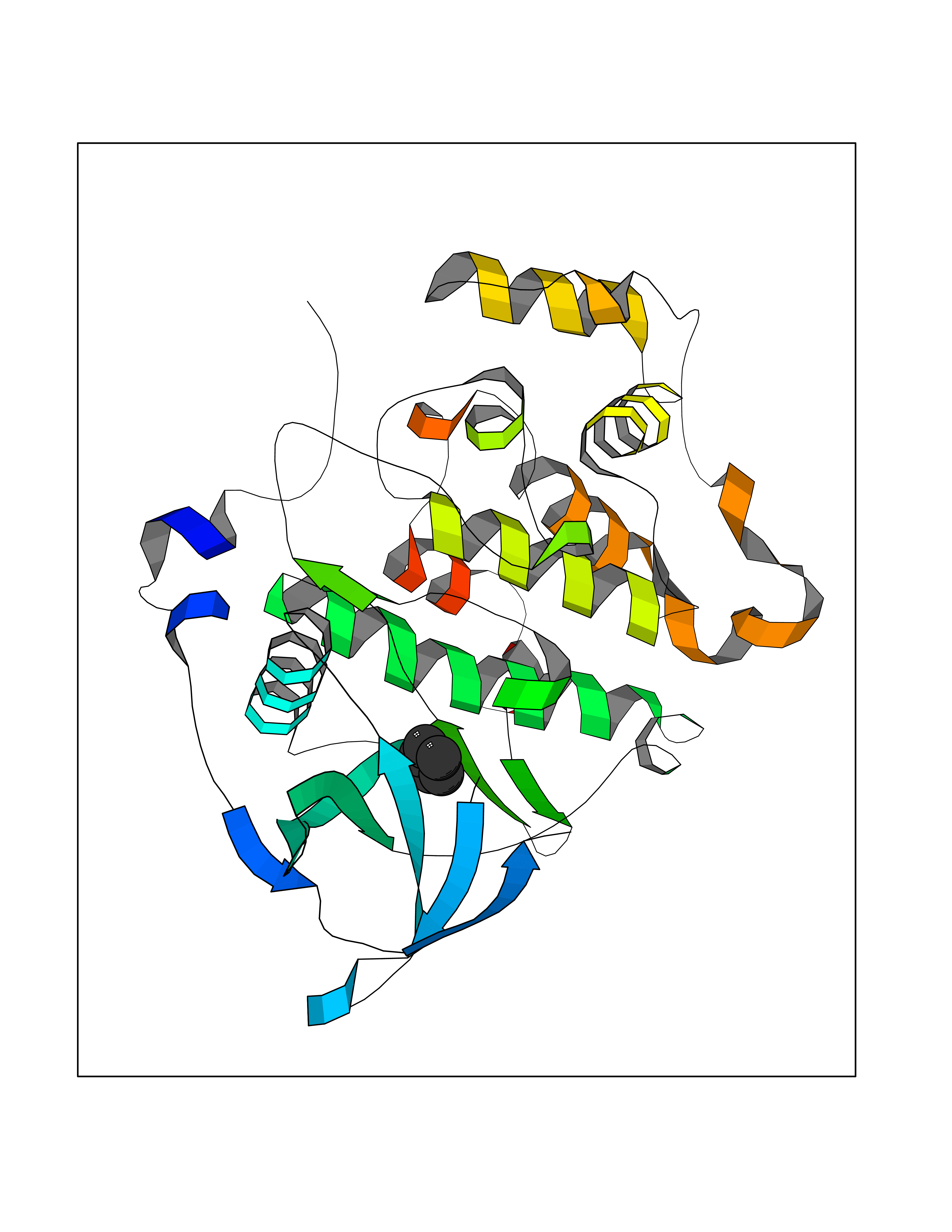 Molscript Map 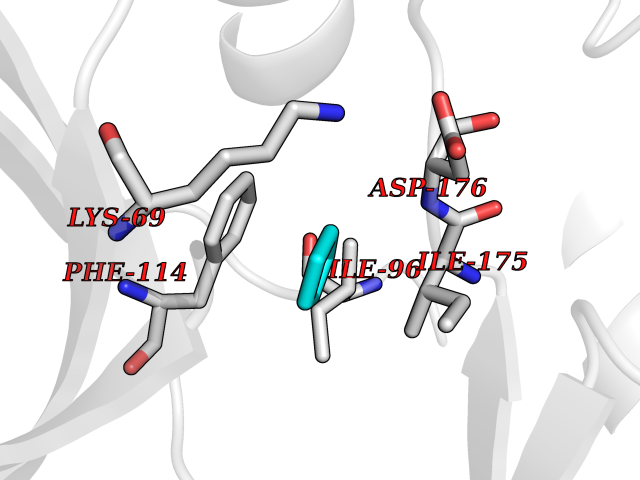 Pymol Map 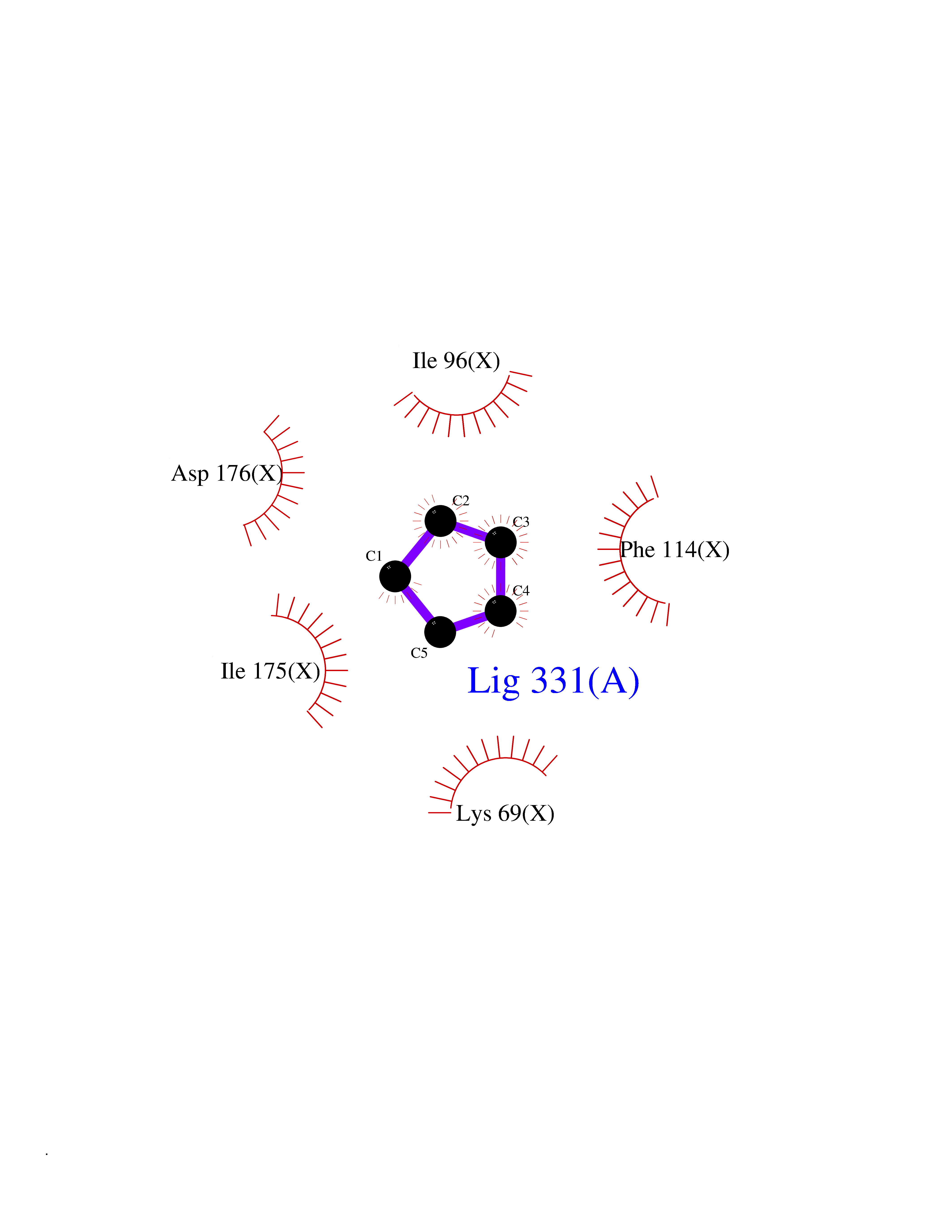 Ligplot Map 3D Binding mode Sequence AGSRARVYAEVNSLRSREYWDYEAHVPSWGNQDDYQLVRKLGEVFEAINITNNERVVVKILKPVKKKKIKREVKILENLRGGTNIIKLIDTVKDPVSKTPALVFEYINNTDFKQLYQILTDFDIRFYMYELLKALDYCHSKGIMHRDVKPHNVMIDHQQKKLRLIDWGLAEFYHPAQEYNVRVASRYFKGPELLVDYQMYDYSLDMWSLGCMLASMIFRREPFFHGQDNYDQLVRIAKVLGTEELYGYLKKYHIDLDPHFNDILGQHSRKRWENFIHSENRHLVSPEALDLLDKLLRYDHQQRLTAKEAMEHPYFYPVV Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Squalene synthetase (FDFT1) | 3WCM | 4.73 | |
Target general information Gen name FDFT1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Squalene synthase; SS; SQS; Farnesyl-diphosphate farnesyltransferase; FPP:FPP farnesyltransferase Protein family Phytoene/squalene synthase family Biochemical class Alkyl aryl transferase Function Participates in the isoprenoid biosynthetic pathway, catalyzing a two-step reaction in which two identical molecules of farnesyl pyrophosphate (FPP) are converted into squalene, with the consumption of NADPH. Related diseases Squalene synthase deficiency (SQSD) [MIM:618156]: An autosomal recessive disorder characterized by profound developmental delay, brain abnormalities, 2/3 syndactyly of the toes, facial dysmorphisms, low total and LDL-cholesterol, and abnormal urine organic acids. {ECO:0000269|PubMed:29909962}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05317 Interacts with Q13520; Q3SXY8; P04233-2; P11912; O75503; O43889-2; Q9GZR5; Q5JX71; P48165; Q8TDT2; Q8N5M9; Q6IBW4-4; Q96RD7; Q14973; Q9NQQ7-3; Q96MV1; Q9Y320 EC number EC 2.5.1.21 Uniprot keywords 3D-structure; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Endoplasmic reticulum; Lipid biosynthesis; Lipid metabolism; Magnesium; Membrane; Metal-binding; Multifunctional enzyme; NAD; NADP; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transferase; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 37860 Length 329 Aromaticity 0.1 Instability index 40.19 Isoelectric point 5.47 Charge (pH=7) -7.65 2D Binding mode Binding energy (Kcal/mol) -6.45 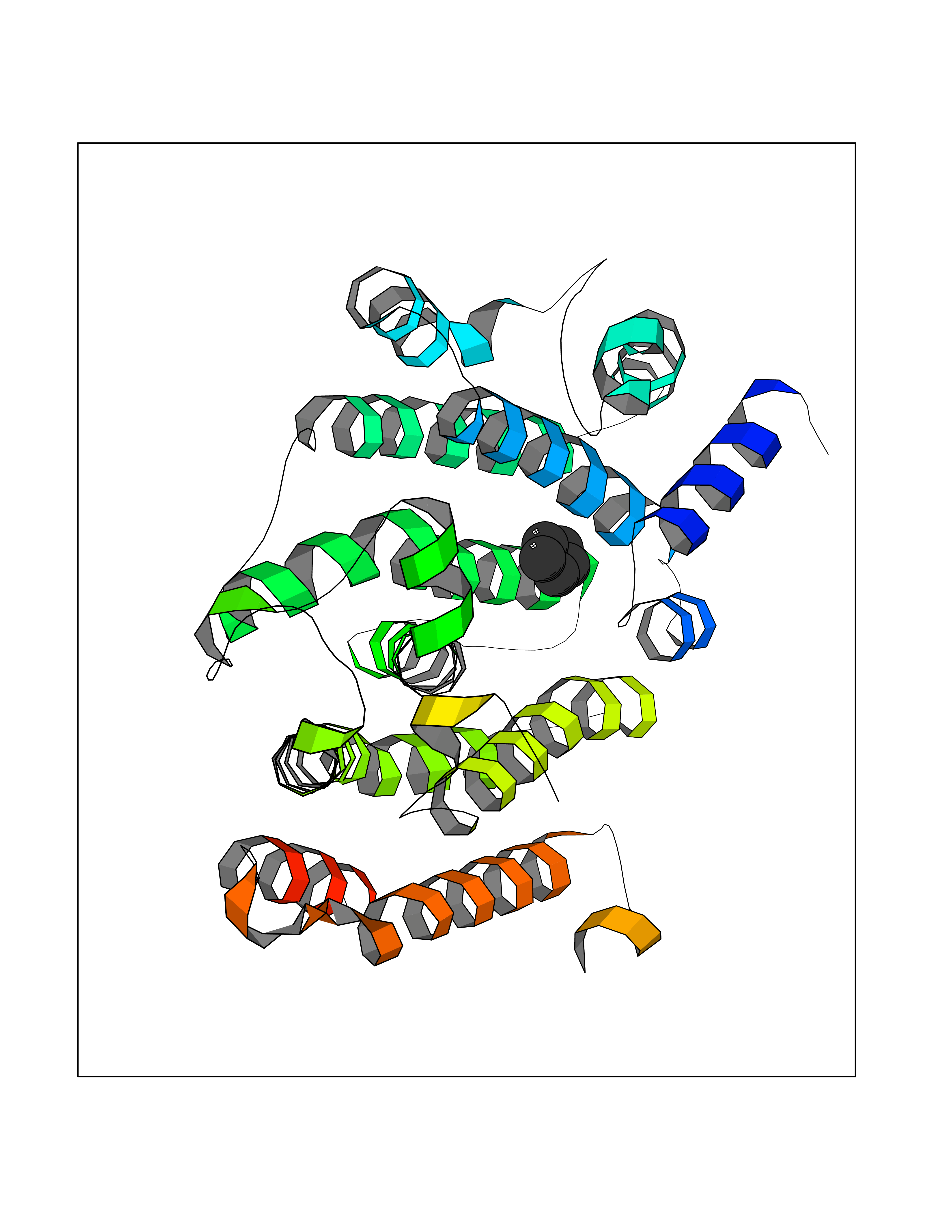 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LSSSLKTCYKYLNQTSRSFAAVIQALDGEMRNAVCIFYLVLRALDTLEDDMTISVEKKVPLLHNFHSFLYQPDWRFMESKEKDRQVLEDFPTISLEFRNLAEKYQTVIADICRRMGIGMAEFLDKHVTSEQEWDKYCHYVAGLVGIGLSRLFSASEFEDPLVGEDTERANSMGLFLQKTNIIRDYLEDQQGGREFWPQEVWSRYVKKLGDFALPENIDLAVQCLNELITNALHHIPDVITYLSRLRNQSVFNFCAIPQVMAIATLAACYNNQQVFKGAVLIVTLMMDATNMPAVKAIIYQYMEEIYHRIPDSNPSSSKTRQIISTIRTQ Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Histone-lysine N-methyltransferase (HLNM) | 3QOW | 4.73 | |
Target general information Gen name DOT1L Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine N-methyltransferase 4; KMT4; KIAA1814; Histone-lysine N-methyltransferase, H3 lysine-79 specific; Histone H3-K79 methyltransferase; H3-K79-HMTase; DOT1-like protein Protein family Class I-like SAM-binding methyltransferase superfamily, DOT1 family Biochemical class Methyltransferase Function Histone methyltransferase. Methylates 'Lys-79' of histone H3. Nucleosomes are preferred as substrate compared to free histones. Binds to DNA. Related diseases Defects in DOTL1 are associated with an autosomal dominant form of global developmental delay and intellectual disability, with or without one or more major congenital anomalies (PubMed:37827158). The patient phenotypes are characterized by central nervous system (CNS) dysfunction, such as mild motor delay and significant speech and language delay, and a range of congenital anomalies, including brain structural anomalies, cardiac defects, varied urogenital features and growth restriction (PubMed:37827158). Variants may cause a gain-of-function effect leading to an increase in cellular H3K79 methylation levels (PubMed:37827158). {ECO:0000269|PubMed:37827158}. Drugs (DrugBank ID) NA Interacts with Q03111; P42568 EC number EC 2.1.1.43 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Disease variant; DNA-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 37367.1 Length 322 Aromaticity 0.11 Instability index 32.71 Isoelectric point 6.03 Charge (pH=7) -5.25 2D Binding mode Binding energy (Kcal/mol) -6.45  Molscript Map  Pymol Map 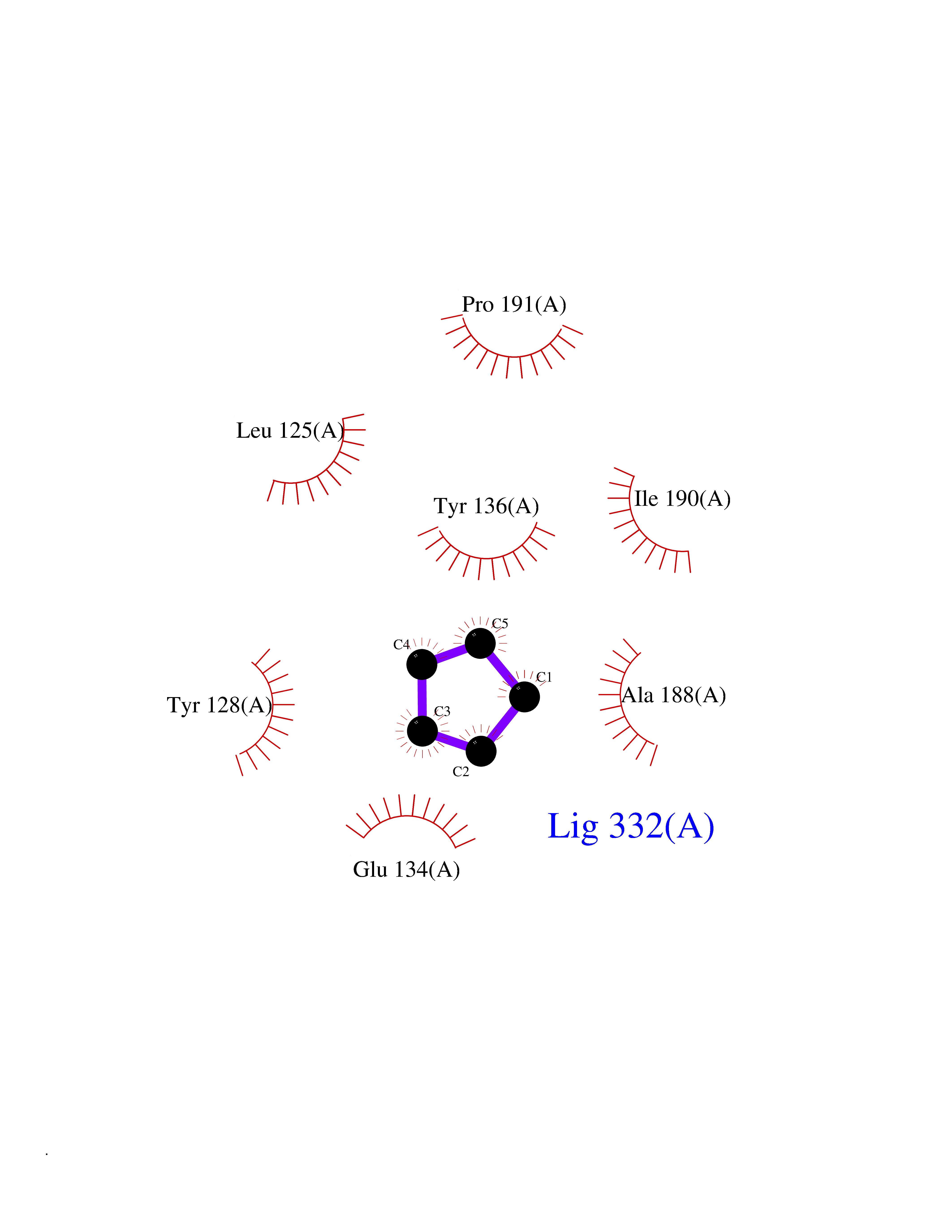 Ligplot Map 3D Binding mode Sequence KLELRLKSPVGAEPAVYPWPLPVYDKHHDAAHEIIETIRWVCEEIPDLKLAMENYLIDYDTKSFESMQRLCDKYNRAIDSIHQLWKGTTQPMKLNTRPSTGLLRHILQQVYNHSVTDPEKLNNYEPFSPEVYGETSFDLVAQMIDEIKMTDDDLFVDLGSGVGQVVLQVAAATNCKHHYGVEKADIPAKYAETMDREFRKWMKWYGKKHAEYTLERGDFLSEEWRERIANTSVIFVNNFAFGPEVDHQLKERFANMKEGGRIVSSKPFAPLNFRINSRNLSDIGTIMRVVELSPLKWTGKPVSYYLHTIDRTILENYFSSLK Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | ATP-binding cassette transporter C8 (ABCC8) | 6C3O | 4.73 | |
Target general information Gen name ABCC8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Sulfonylurea receptor 1; SUR1-type K(ATP) channel; SUR1; Kir6.2/SUR1; ATPbinding cassette subfamily C member 8; ABCC8 Protein family ABC transporter superfamily, ABCC family, Conjugate transporter (TC 3.A.1.208) subfamily Biochemical class ABC transporter Function Putative subunit of the beta-cell ATP-sensitive potassium channel (KATP). Regulator of ATP-sensitive K(+) channels and insulin release. Related diseases Leucine-induced hypoglycemia (LIH) [MIM:240800]: Rare cause of hypoglycemia and is described as a condition in which symptomatic hypoglycemia is provoked by high protein feedings. Hypoglycemia is also elicited by administration of oral or intravenous infusions of a single amino acid, leucine. {ECO:0000269|PubMed:15356046}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperinsulinemic hypoglycemia, familial, 1 (HHF1) [MIM:256450]: A form of hyperinsulinemic hypoglycemia, a clinically and genetically heterogeneous disorder characterized by inappropriate insulin secretion from the pancreatic beta-cells in the presence of low blood glucose levels. HHF1 is the most common cause of persistent hypoglycemia in infancy. Unless early and aggressive intervention is undertaken, brain damage from recurrent episodes of hypoglycemia may occur. HHF1 inheritance can be autosomal dominant or autosomal recessive. {ECO:0000269|PubMed:10202168, ECO:0000269|PubMed:10204114, ECO:0000269|PubMed:10334322, ECO:0000269|PubMed:10615958, ECO:0000269|PubMed:11018078, ECO:0000269|PubMed:11226335, ECO:0000269|PubMed:11867634, ECO:0000269|PubMed:12364426, ECO:0000269|PubMed:12941782, ECO:0000269|PubMed:15562009, ECO:0000269|PubMed:15579781, ECO:0000269|PubMed:15807877, ECO:0000269|PubMed:16357843, ECO:0000269|PubMed:16429405, ECO:0000269|PubMed:24814349, ECO:0000269|PubMed:25720052, ECO:0000269|PubMed:8650576, ECO:0000269|PubMed:8751851, ECO:0000269|PubMed:8923011, ECO:0000269|PubMed:9618169, ECO:0000269|PubMed:9648840, ECO:0000269|PubMed:9769320}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Diabetes mellitus, permanent neonatal, 3 (PNDM3) [MIM:618857]: A form of permanent neonatal diabetes mellitus, a type of diabetes characterized by onset of persistent hyperglycemia within the first six months of life. Initial clinical manifestations include intrauterine growth retardation, hyperglycemia, glycosuria, osmotic polyuria, severe dehydration, and failure to thrive. Some PNDM3 patients may also have developmental delay, muscle weakness, and epilepsy. PNDM3 transmission pattern is consistent with autosomal dominant or autosomal recessive inheritance. {ECO:0000269|PubMed:16613899, ECO:0000269|PubMed:16885549, ECO:0000269|PubMed:17213273, ECO:0000269|PubMed:17668386}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Transient neonatal diabetes mellitus 2 (TNDM2) [MIM:610374]: Neonatal diabetes is a form of diabetes mellitus defined by the onset of mild-to-severe hyperglycemia within the first months of life. Transient neonatal diabetes remits early, with a possible relapse during adolescence. {ECO:0000269|PubMed:16885549}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00171; DB00672; DB01120; DB00222; DB01067; DB01251; DB01016; DB01382; DB01252; DB00731; DB00912; DB00839; DB01124 Interacts with Q14654 EC number NA Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Diabetes mellitus; Disease variant; Glycoprotein; Membrane; Nucleotide-binding; Proteomics identification; Receptor; Reference proteome; Repeat; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID E Molecular weight (Da) 144112 Length 1290 Aromaticity 0.1 Instability index 32.98 Isoelectric point 8.87 Charge (pH=7) 19.54 2D Binding mode Binding energy (Kcal/mol) -6.45  Molscript Map 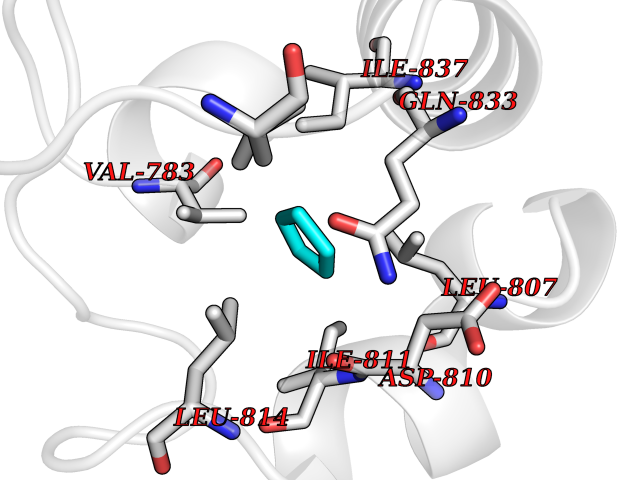 Pymol Map 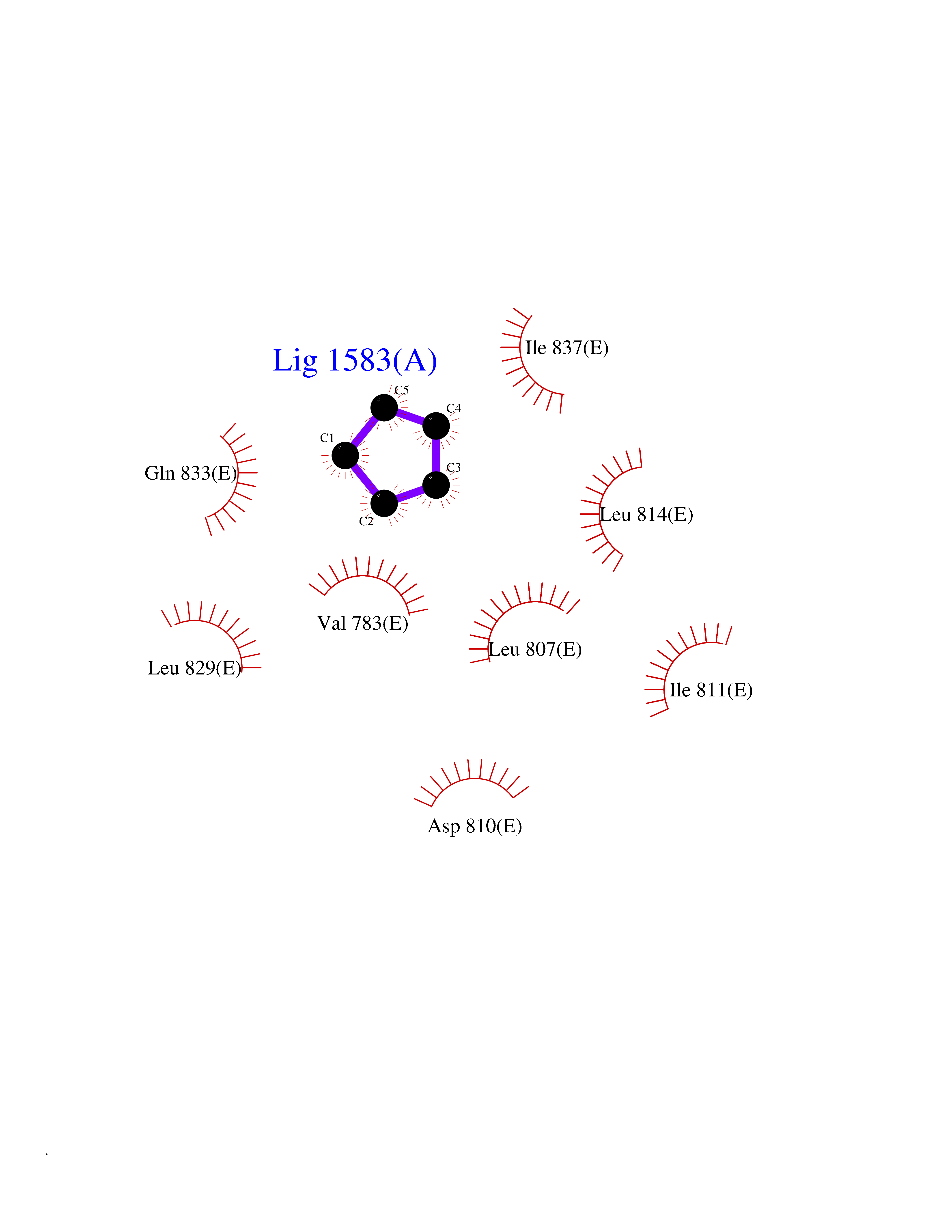 Ligplot Map 3D Binding mode Sequence PLAFCGSENHSAAYRVDQGVLNNGCFVDALNVVPHVFLLFITFPILFIGWGFPGHNLRWILTFMLLFVLVCEIAEGILSDGVTESHHLHLYMPAGMAFMAAVTSVVYYHNIETSNFPKLLIALLVYWTLAFITKTIKFVKFLDHAIGFSQLRFCLTGLLVILYGMLLLVEVNVIRVNYQRLCEAFDAQVRKDAIWQALSHAFGRRLVLSSTFRILADLLGFAGPLCIFGIVDHLGKANAYVLAVLLFLALLLQRTFLQASYYVAIETGINLRGAIQTKIYNKIMHLSTSNLSMGEMTAGQICNLVAIDTNQLMWFFFLCPNLWAMPVQIIVGVILLYYILGVSALIGAAVIILLAPVQYFVATKLSQAQRSTLEYSNERLKQTNEMLRGIKLLKLYAWENIFRTRVETTRRKEMTSLRAFAIYTSISIFMNTAIPIAAVLITFVGHVPSVAFASLSLFHILVTPLFLLSSVVRSTVKALVSVQKLSEFLSSAEIRCCVQIMGGYFTWTPDGIPTLSNITIRIPRGQLTMIVGQVGCGKSSLLLAALGEMQKVSGAVFWSRGPVAYASQKPWLLNATVEENIIFESPFNKQRYKMVIEACSLQPDIDILPHGDQTQIGERGINLSGGQRQRISVARALYQHANVVFLDDPFSALDIHLSDHLMQAGILELLRDDKRTVVLVTHKLQYLPHADWIIAMKDGTIQREGTLKDFQRSECQLFEHWKTLEIPWRACAKYLSSAGILLLSLLVFSQLLKHMVLVAIDYWLAKWTDTVYAMVFTVLCSLGIVLCLVTSVTVEWTGLKVAKRLHRSLLNRIILAPMRFFETTPLGSILNRFSSDCNTIDQHIPSTLECLSRSTLLCVSALAVISYVTPVFLVALLPLAIVCYFIQKYFRVASRDLQQLDDTTQLPLLSHFAETVEGLTTIRAFRYEARFQQKLLEYTDSNNIASLFLTAANRWLEVRMEYIGACVVLIAAVTSISNSLHRELSAGLVGLGLTYALMVSNYLNWMVRNLADMELQLGAVKRIHGLLKTEAESYEGLLAPSLIPKNWPDQGKIQIQNLSVRYDSSLKPVLKHVNALIAPGQKIGICGRTGSGKSSFSLAFFRMVDTFEGHIIIDGIDIAKLPLHTLRSRLSIILQDPVLFSGTIRFNLDPERKCSDSTLWEALEIAQLKLVVKALPGGLDAIITEGGENFSQGQRQLFCLARAFVRKTSIFIMDEATASIDMATENILQKVVMTAFADRTVVTIAHRVHTILSADLVIVLKRGAILEFDKPEKLLSRKDSVFASFVRADK Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Retinaldehyde-binding protein 1 | 3HX3 | 4.72 | |
Target general information Gen name RLBP1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CRALBP Protein family NA Biochemical class Transport protein Function 11-cis retinal binding.Retinol binding.Transporter activity. Related diseases Bothnia retinal dystrophy (BRD) [MIM:607475]: A type of retinitis punctata albescens. Affected individuals show night blindness from early childhood with features consistent with retinitis punctata albescens and macular degeneration. {ECO:0000269|PubMed:10102298}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rod-cone dystrophy Newfoundland (NFRCD) [MIM:607476]: A rod-cone dystrophy reminiscent of retinitis punctata albescens but with a substantially lower age at onset and more-rapid and distinctive progression. Rod-cone dystrophies results from initial loss of rod photoreceptors, later followed by cone photoreceptors loss. {ECO:0000269|PubMed:11868161}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Retinitis punctata albescens (RPA) [MIM:136880]: A form of fleck retina disease characterized by aggregation of white flecks posteriorly in the retina, causing night blindness and delayed dark adaptation. It differs from fundus albipunctatus in being progressive and evolving to generalized atrophy of the retina. {ECO:0000269|PubMed:10102299, ECO:0000269|PubMed:11453974, ECO:0000269|PubMed:9326942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00162 Interacts with Q9P2G9-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Disease variant; Proteomics identification; Reference proteome; Retinol-binding; Sensory transduction; Transport; Vision Protein physicochemical properties Chain ID A Molecular weight (Da) 28328.6 Length 250 Aromaticity 0.14 Instability index 52.64 Isoelectric point 4.96 Charge (pH=7) -9.87 2D Binding mode Binding energy (Kcal/mol) -6.43 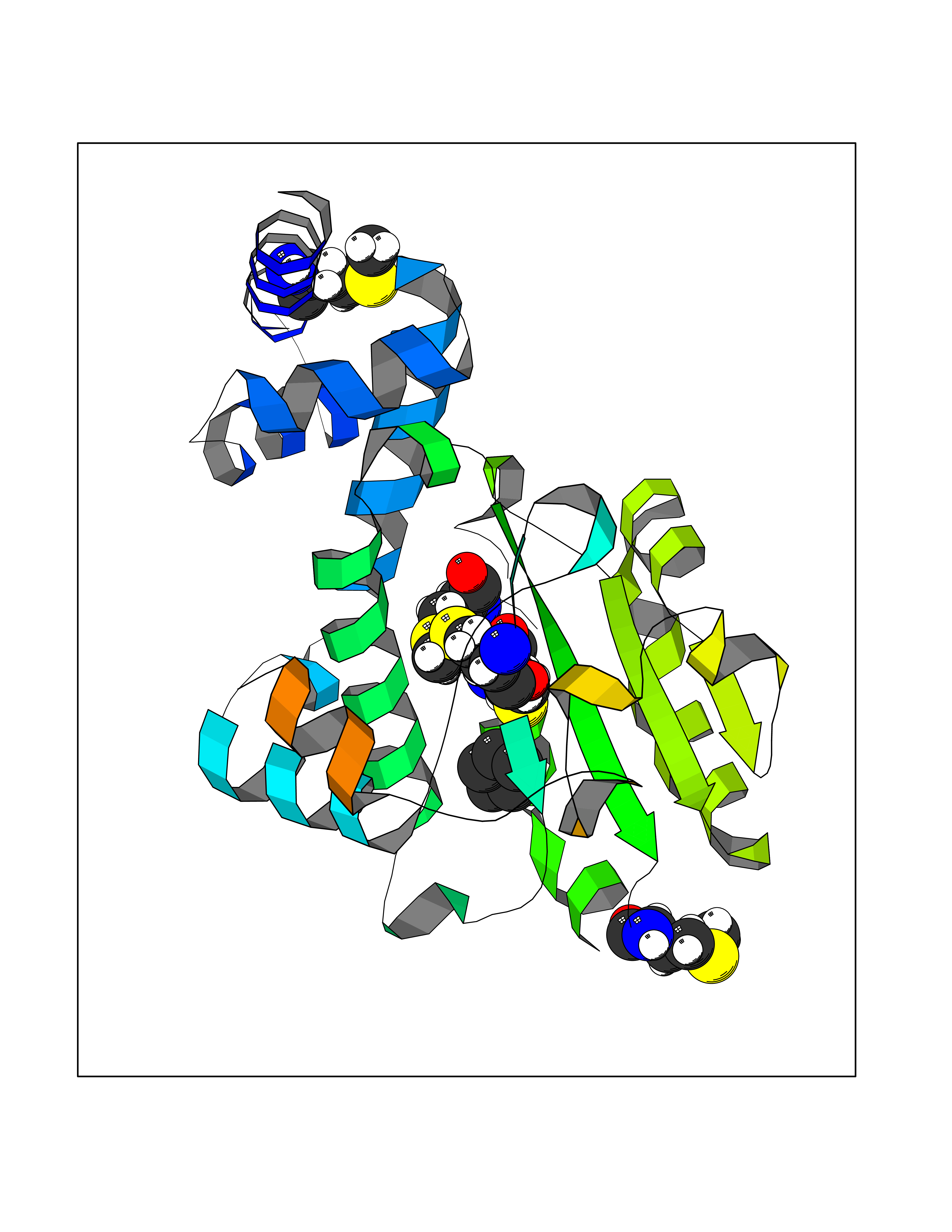 Molscript Map 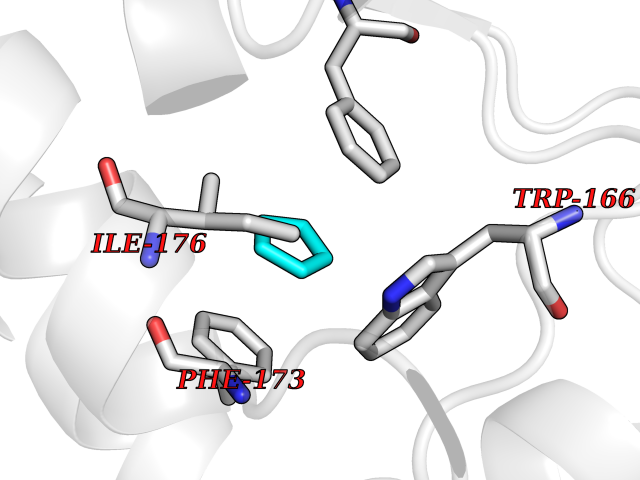 Pymol Map 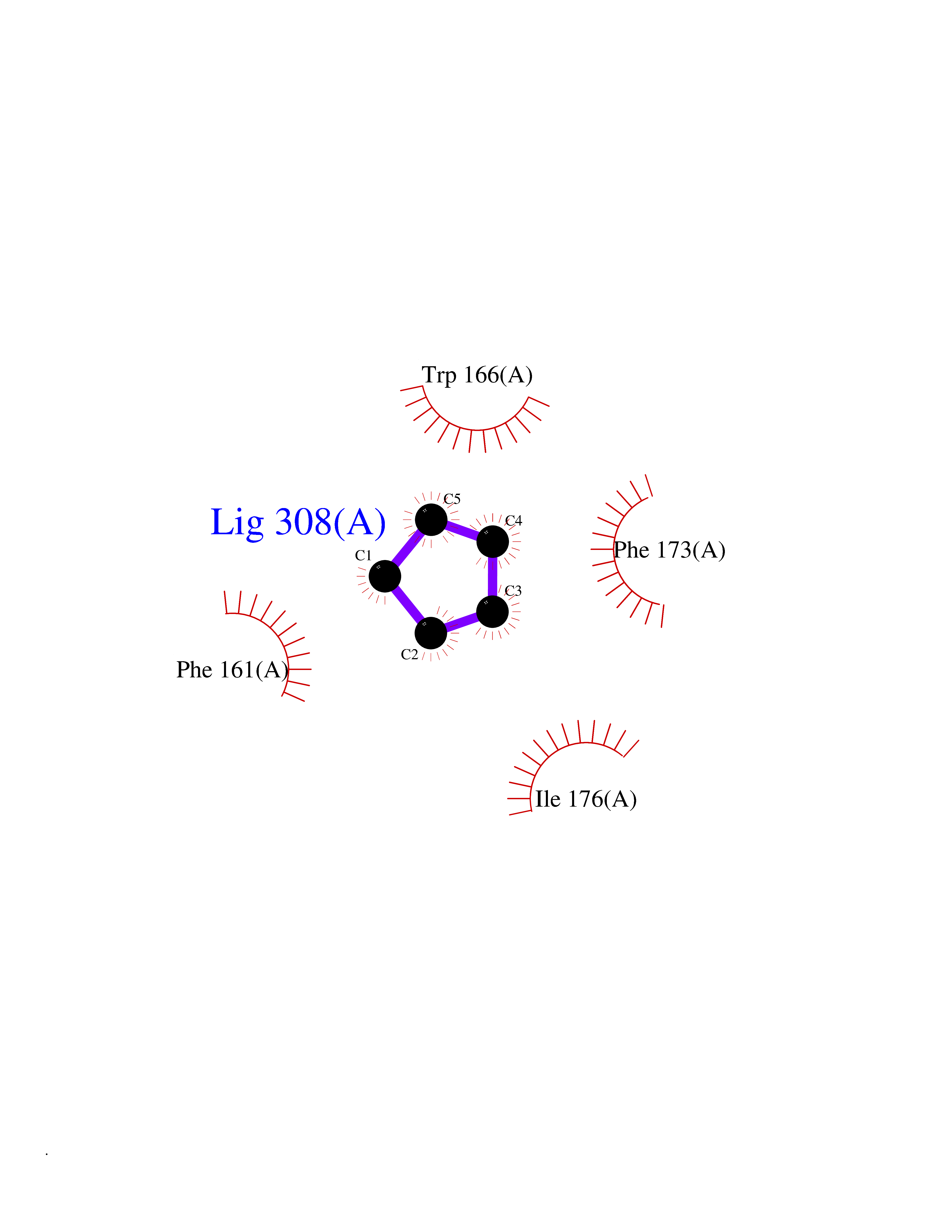 Ligplot Map 3D Binding mode Sequence ETREEAVRELQEXVQAQAASGEELAVAVAERVQEKDSGFFLRFIRARKFNVGRAYELLRGYVNFRLQYPELFDSLSPEAVRCTIEAGYPGVLSSRDKYGRVVXLFNIENWQSQEITFDEILQAYCFILEKLLENEETQINGFCIIENFKGFTXQQAASLRTSDLRKXVDXLQDSFPAWFKAIHFIHQPWYFTTTYNVVKPFLKSKLLERVFVHGDDLSGFYQEIDENILPSDFGGTLPKYDGKAVAEQLF Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Leukotriene A-4 hydrolase (LTA4H) | 3U9W | 4.72 | |
Target general information Gen name LTA4H Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Leukotriene A4 hydrolase; Leukotriene A(4)Leukotriene A-4 hydrolase hydrolase; Leukotriene A(4) hydrolase; LTA4; LTA-H; LTA-4hydrolase; LTA-4 hydrolase Protein family Peptidase M1 family Biochemical class Ether bond hydrolase Function Has also aminopeptidase activity. Epoxide hydrolase that catalyzes the final step in the biosynthesis of the proinflammatory mediator leukotriene B4. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07102; DB06917; DB07258; DB07094; DB07259; DB02352; DB07292; DB07104; DB06828; DB08466; DB01197; DB05177; DB03366; DB08040; DB06851; DB02062; DB07099; DB07260; DB07196; DB11781; DB03424; DB07237 Interacts with Q9BSI4 EC number EC 3.3.2.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Hydrolase; Leukotriene biosynthesis; Lipid metabolism; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 68927 Length 608 Aromaticity 0.1 Instability index 38.84 Isoelectric point 5.87 Charge (pH=7) -9.86 2D Binding mode Binding energy (Kcal/mol) -6.44 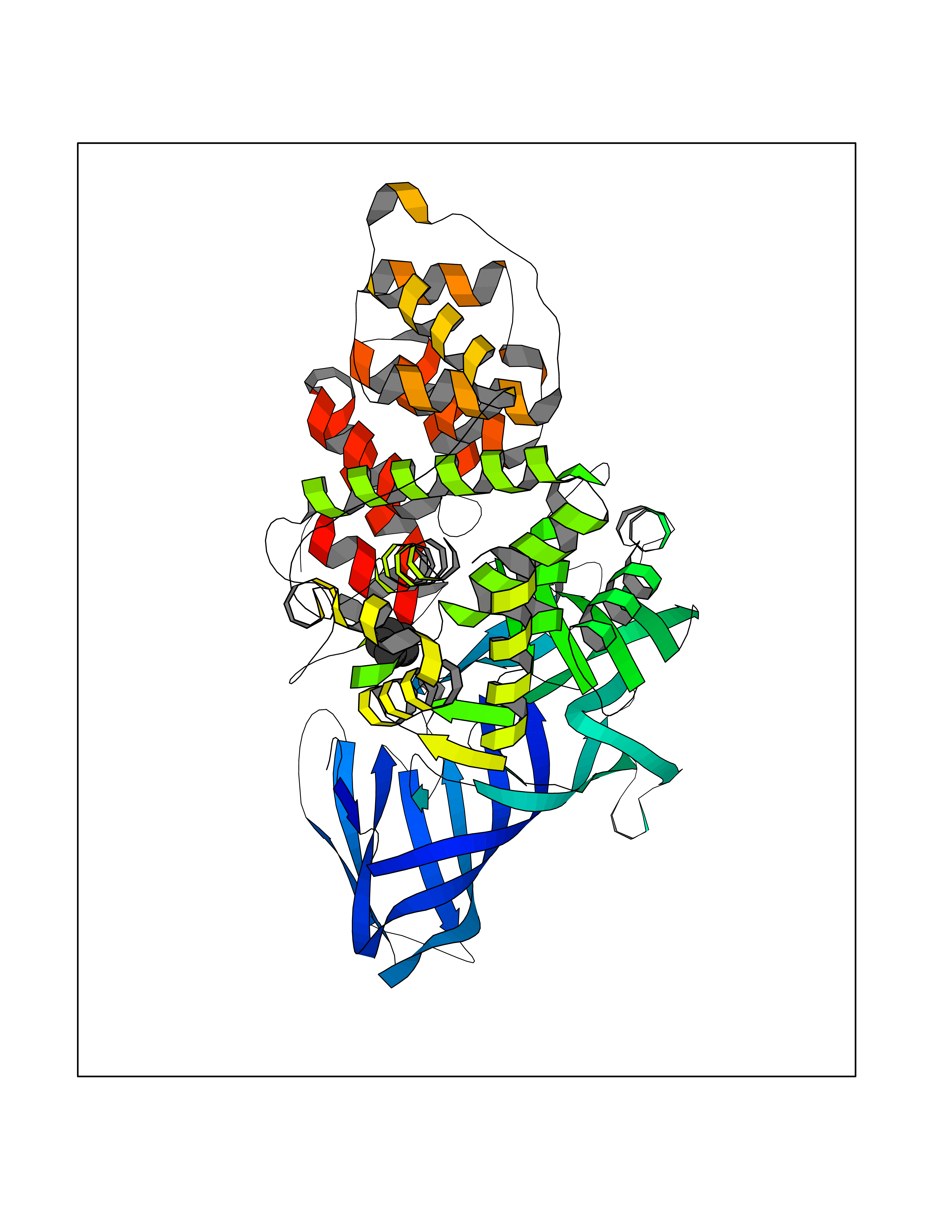 Molscript Map 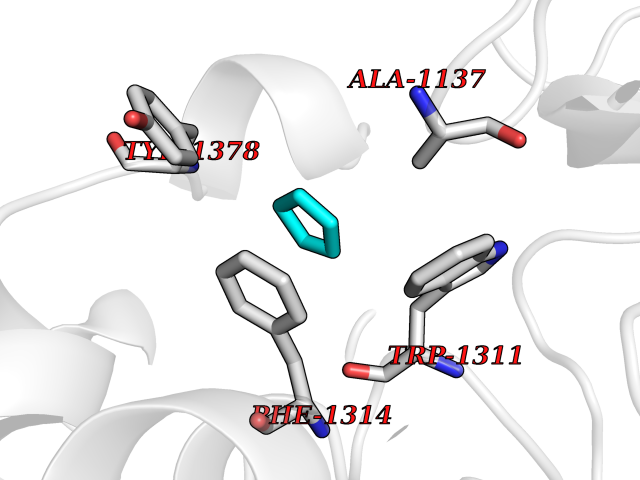 Pymol Map 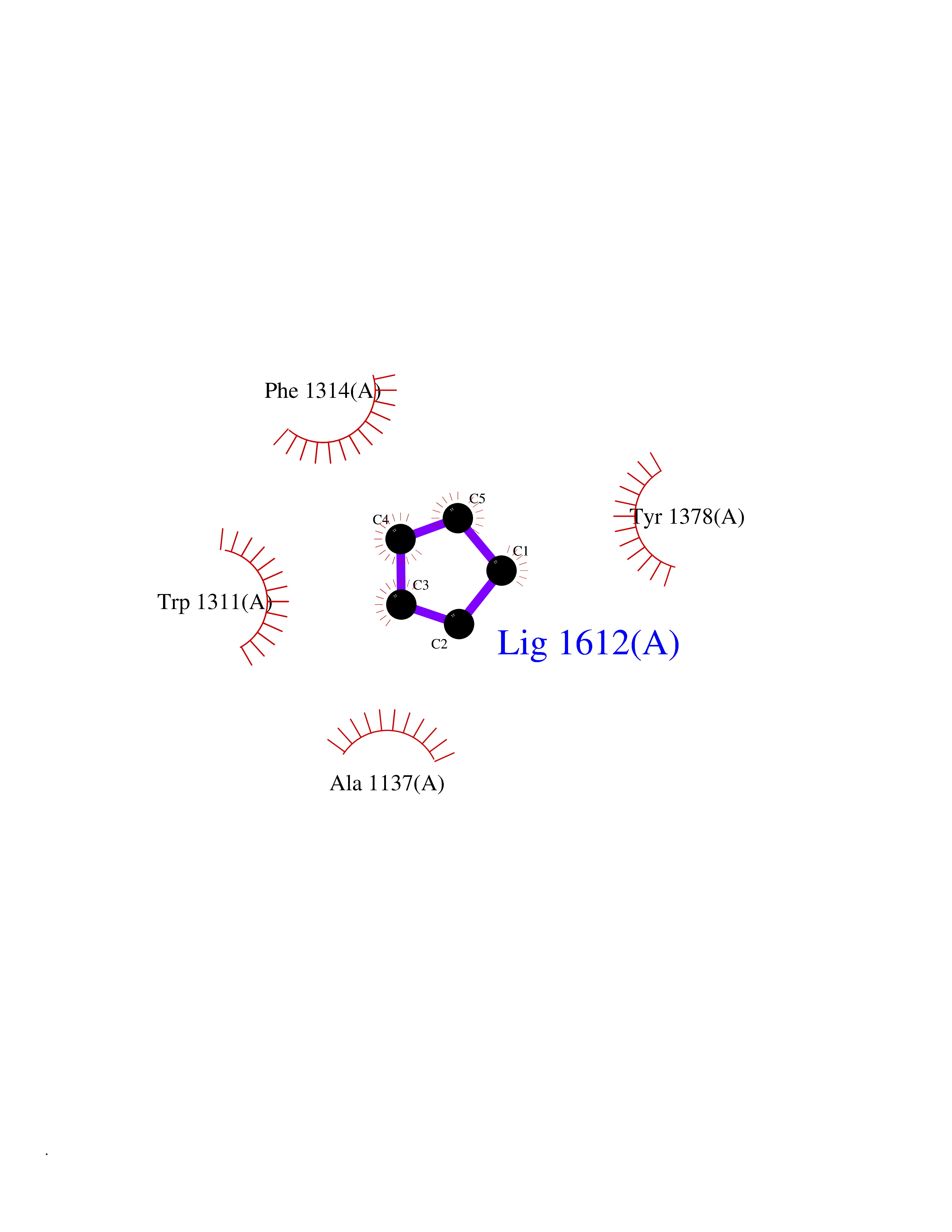 Ligplot Map 3D Binding mode Sequence IVDTCSLASPASVCRTKHLHLRCSVDFTRRTLTGTAALTVQSQEDNLRSLVLDTKDLTIEKVVINGQEVKYALGERQSYKGSPMEISLPIALSKNQEIVIEISFETSPKSSALQWLTPEQTSGKEHPYLFSQCQAIHCRAILPCQDTPSVKLTYTAEVSVPKELVALMSAIRDGETPDPEDPSRKIYKFIQKVPIPCYLIALVVGALESRQIGPRTLVWSEKEQVEKSAYEFSETESMLKIAEDLGGPYVWGQYDLLVLPPSFPYGGMENPCLTFVTPTLLAGDKSLSNVIAHEISHSWTGNLVTNKTWDHFWLNEGHTVYLERHICGRLFGEKFRHFNALGGWGELQNSVKTFGETHPFTKLVVDLTDIDPDVAYSSVPYEKGFALLFYLEQLLGGPEIFLGFLKAYVEKFSYKSITTDDWKDFLYSYFKDKVDVLNQVDWNAWLYSPGLPPIKPNYDMTLTNACIALSQRWITAKEDDLNSFNATDLKDLSSHQLNEFLAQTLQRAPLPLGHIKRMQEVYNFNAINNSEIRFRWLRLCIQSKWEDAIPLALKMATEQGRMKFTRPLFKDLAAFDKSHDQAVRTYQEHKASMHPVTAMLVGKDLKVD Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Interferon gamma | 1FYH | 4.72 | |
Target general information Gen name IFNG Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Type II (or gamma) interferon family Biochemical class Immune system Function Cytokine activity.Interferon-gamma receptor binding. Related diseases Aplastic anemia (AA) [MIM:609135]: A form of anemia in which the bone marrow fails to produce adequate numbers of peripheral blood elements. It is characterized by peripheral pancytopenia and marrow hypoplasia. {ECO:0000269|PubMed:15327519}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Immunodeficiency 69 (IMD69) [MIM:618963]: A form of Mendelian susceptibility to mycobacterial disease, a rare condition caused by impairment of interferon-gamma mediated immunity. It is characterized by predisposition to illness caused by moderately virulent mycobacterial species, such as Bacillus Calmette-Guerin (BCG) vaccine, environmental non-tuberculous mycobacteria, and by the more virulent Mycobacterium tuberculosis. Other microorganisms rarely cause severe clinical disease in individuals with susceptibility to mycobacterial infections. Clinical outcome severity depends on the degree of impairment of interferon-gamma mediated immunity. IMD69 is an autosomal recessive disorder manifesting with fever, hepatosplenomegaly, leukocytosis, and thrombocytosis during the acute infection. {ECO:0000269|PubMed:32163377}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14724; DB05111; DB10770; DB10772; DB01296; DB01250; DB05110 Interacts with P15260; Q66793 EC number NA Uniprot keywords 3D-structure; Antiviral defense; Cleavage on pair of basic residues; Cytokine; Direct protein sequencing; Glycoprotein; Growth regulation; Pharmaceutical; Proteomics identification; Pyrrolidone carboxylic acid; Reference proteome; Secreted; Signal Protein physicochemical properties Chain ID A,D Molecular weight (Da) 28390 Length 242 Aromaticity 0.12 Instability index 18.95 Isoelectric point 8.42 Charge (pH=7) 1.71 2D Binding mode Binding energy (Kcal/mol) -6.44  Molscript Map 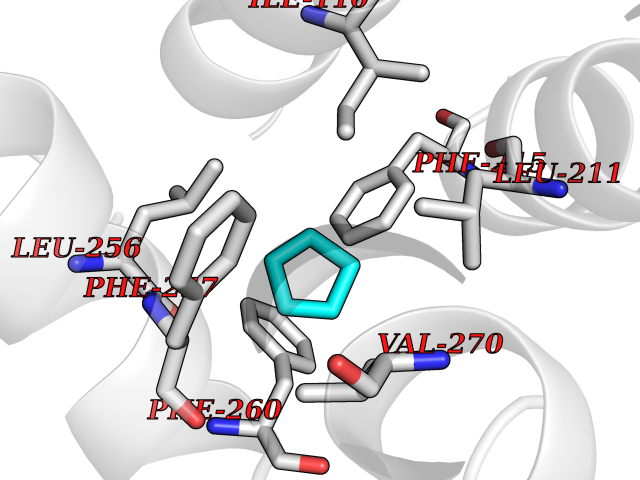 Pymol Map  Ligplot Map 3D Binding mode Sequence MQDPYVKEAENLKKYFNAGHSDVADNGTLFLGILKNWKEESDRKIMQSQIVSFYFKLFKNFKDDQSIQKSVETIKEDMNVKFFNSNKKKRDDFEKLTNYSVTDLNVQRKAIDELIQVMAELGANVSGEFVKEAENLKKYFNDNGTLFLGILKNWKEESDRKIMQSQIVSFYFKLFKNFKDDQSIQKSVETIKEDMNVKFFNSNKKKRDDFEKLTNYSVTDLNVQRKAIHELIQVMAELSPAA Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | 4-cresol dehydrogenase [hydroxylating] flavoprotein subunit | 1WVF | 4.72 | |
Target general information Gen name pchF Organism Pseudomonas putida (Arthrobacter siderocapsulatus) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Oxidoreductase Function 4-cresol dehydrogenase (hydroxylating) activity.Flavin adenine dinucleotide binding.Oxidoreductase activity, acting on CH-OH group of donors. Related diseases Dihydrolipoamide dehydrogenase deficiency (DLDD) [MIM:246900]: An autosomal recessive metabolic disorder characterized biochemically by a combined deficiency of the branched-chain alpha-keto acid dehydrogenase complex (BCKDC), pyruvate dehydrogenase complex (PDC), and alpha-ketoglutarate dehydrogenase complex (KGDC). Clinically, affected individuals have lactic acidosis and neurologic deterioration due to sensitivity of the central nervous system to defects in oxidative metabolism. {ECO:0000269|PubMed:10448086, ECO:0000269|PubMed:11687750, ECO:0000269|PubMed:12925875, ECO:0000269|PubMed:15712224, ECO:0000269|PubMed:16442803, ECO:0000269|PubMed:16770810, ECO:0000269|PubMed:17404228, ECO:0000269|PubMed:20160912, ECO:0000269|PubMed:8506365, ECO:0000269|PubMed:8968745, ECO:0000269|PubMed:9540846, ECO:0000269|PubMed:9934985}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.17.9.1 Uniprot keywords 3D-structure; Direct protein sequencing; FAD; Flavoprotein; Oxidoreductase; Plasmid Protein physicochemical properties Chain ID A Molecular weight (Da) 57240.8 Length 515 Aromaticity 0.1 Instability index 30.94 Isoelectric point 6.06 Charge (pH=7) -4.42 2D Binding mode Binding energy (Kcal/mol) -6.44  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence AVLPKGVTQGEFNKAVQKFRALLGDDNVLVESDQLVPYNKIMMPVENAAHAPSAAVTATTVEQVQGVVKICNEHKIPIWTISTGRNFGYGSAAPVQRGQVILDLKKMNKIIKIDPEMCYALVEPGVTFGQMYDYIQENNLPVMLSFSAPSAIAGPVGNTMDRGVGYTPYGEHFMMQCGMEVVLANGDVYRTGMGGVPGSNTWQIFKWGYGPTLDGMFTQANYGICTKMGFWLMPKPPVFKPFEVIFEDEADIVEIVDALRPLRMSNTIPNSVVIASTLWEAGSAHLTRAQYTTEPGHTPDSVIKQMQKDTGMGAWNLYAALYGTQEQVDVNWKIVTDVFKKLGKGRIVTQEEAGDTQPFKYRAQLMSGVPNLQEFGLYNWRGGGGSMWFAPVSEARGSECKKQAAMAKRVLHKYGLDYVAEFIVAPRDMHHVIDVLYDRTNPEETKRADACFNELLDEFEKEGYAVYRVNTRFQDRVAQSYGPVKRKLEHAIKRAVDPNNILAPGRSGIDLNNDF Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Retinoic acid receptor beta (RARB) | 4DM6 | 4.72 | |
Target general information Gen name RARB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-epsilon; RAR-beta; Nuclear receptor subfamily 1 group B member 2; NR1B2; HBV-activated protein; HAP Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RXR/RAR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence or presence of hormone ligand, acts mainly as an activator of gene expression due to weak binding to corepressors. In concert with RARG, required for skeletal growth, matrix homeostasis and growth plate function. Receptor for retinoic acid. Related diseases Microphthalmia, syndromic, 12 (MCOPS12) [MIM:615524]: A form of microphthalmia, a disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues (anophthalmia). In many cases, microphthalmia/anophthalmia occurs in association with syndromes that include non-ocular abnormalities. MCOPS12 patients manifest variable features, including diaphragmatic hernia, pulmonary hypoplasia, and cardiac abnormalities. {ECO:0000269|PubMed:24075189, ECO:0000269|PubMed:27120018}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00459; DB00210; DB00523; DB02877; DB00926; DB05785; DB04942; DB00799; DB00755; DB12808 Interacts with O95273; P50222; Q9UBK2; P62195; P28702; P28702-3; P48443; P03255 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Microphthalmia; Nucleus; Phosphoprotein; Proto-oncogene; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 25904.1 Length 229 Aromaticity 0.06 Instability index 44.34 Isoelectric point 7.55 Charge (pH=7) 0.73 2D Binding mode Binding energy (Kcal/mol) -6.44  Molscript Map 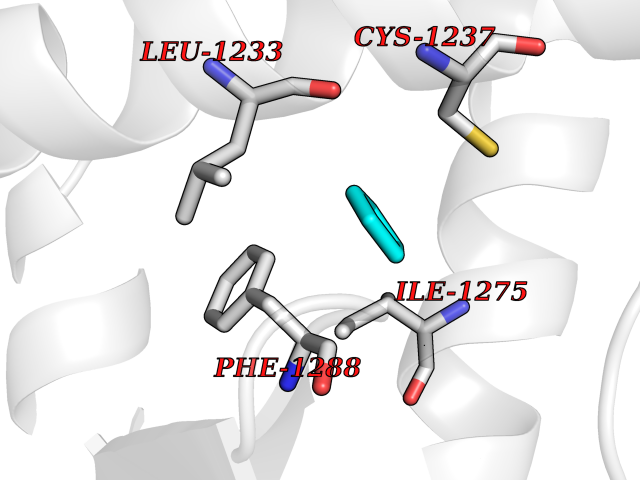 Pymol Map 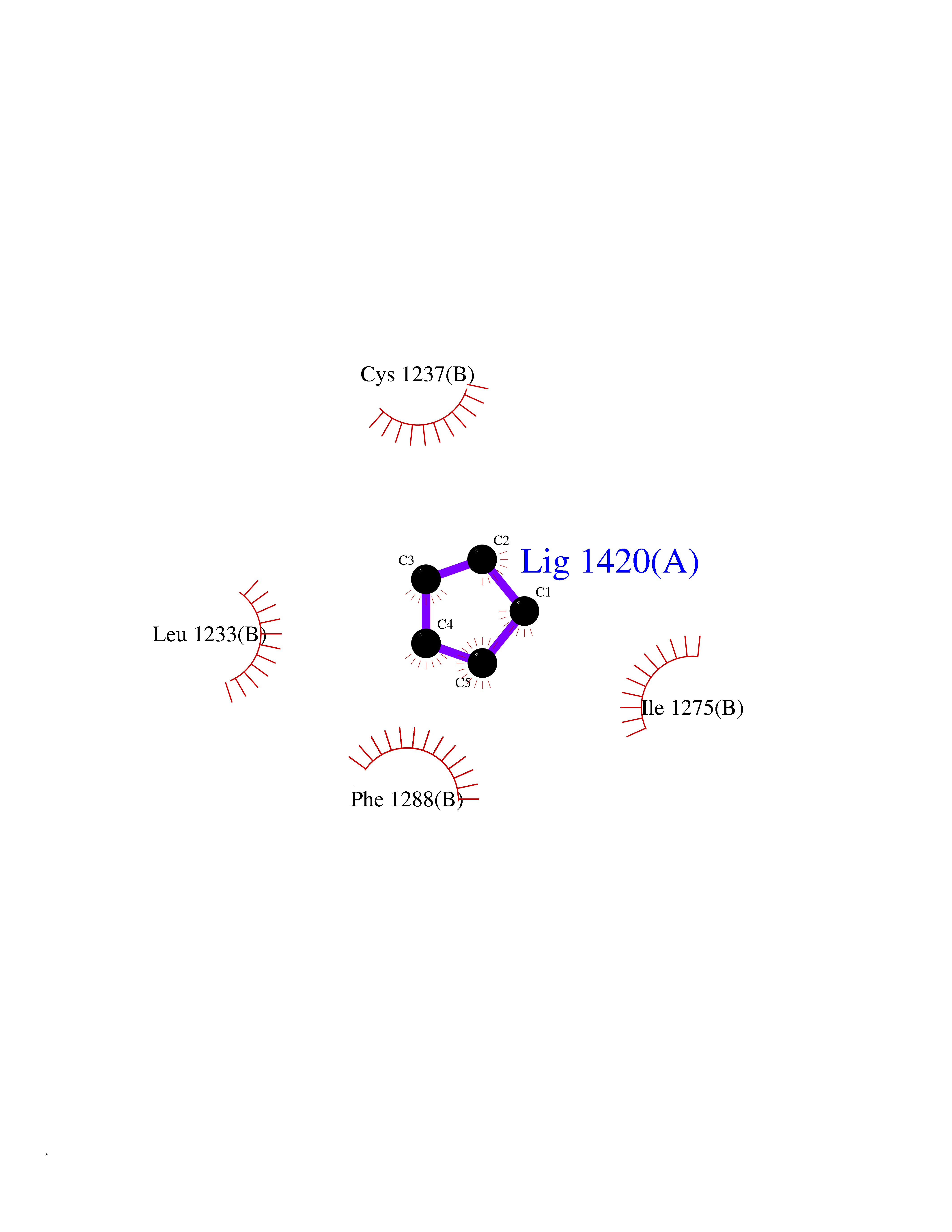 Ligplot Map 3D Binding mode Sequence TEKIRKAHQETFPSLCQLGKYTTNSSADHRVRLDLGLWDKFSELATKCIIKIVEFAKRLPGFTGLTIADQITLLKAACLDILILRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFTFANQLLPLEMDDTETGLLSAICLICGDRQDLEEPTKVDKLQEPLLEALKIYIRKRRPSKPHMFPKILMKITDLRSISAKGAERVITLKMEIPGSMPPLIQEMLEN Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | 4-hydroxyphenylpyruvate dioxygenase | 3ISQ | 4.72 | |
Target general information Gen name HPD Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PPD Protein family 4HPPD family Biochemical class Oxidoreductase Function 4-hydroxyphenylpyruvate dioxygenase activity.Metal ion binding. Related diseases Tyrosinemia 3 (TYRSN3) [MIM:276710]: An inborn error of metabolism characterized by elevations of tyrosine in the blood and urine, seizures and mild intellectual disability. {ECO:0000269|PubMed:10942115, ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hawkinsinuria (HWKS) [MIM:140350]: An inborn error of tyrosine metabolism characterized by failure to thrive, persistent metabolic acidosis, fine and sparse hair, and excretion of the unusual cyclic amino acid metabolite, hawkinsin, in the urine. {ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02850; DB00348 Interacts with NA EC number 1.13.11.27 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Dioxygenase; Disease variant; Endoplasmic reticulum; Golgi apparatus; Intellectual disability; Iron; Membrane; Metal-binding; Oxidoreductase; Phenylalanine catabolism; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Tyrosine catabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 43164.8 Length 376 Aromaticity 0.11 Instability index 32.38 Isoelectric point 6.73 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -6.43 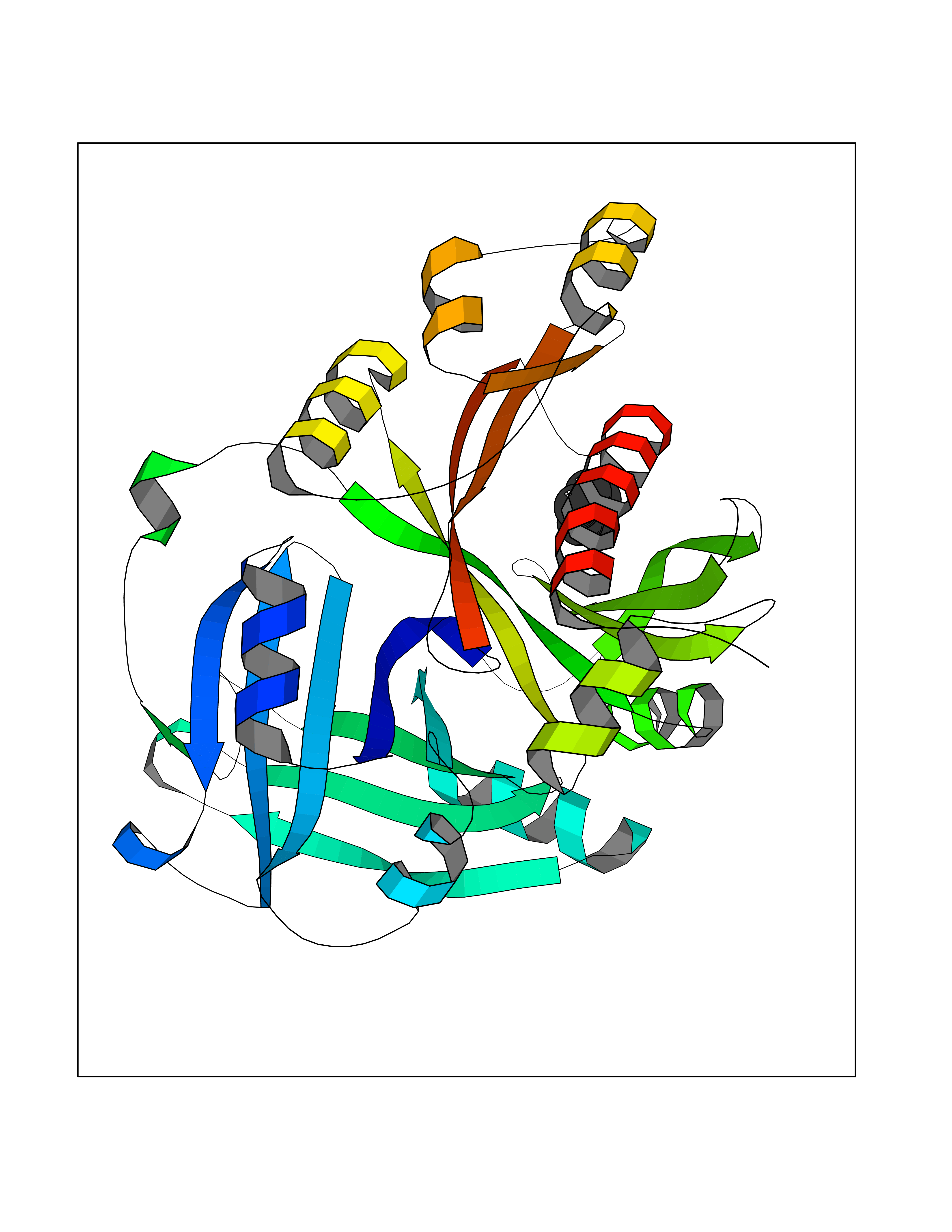 Molscript Map 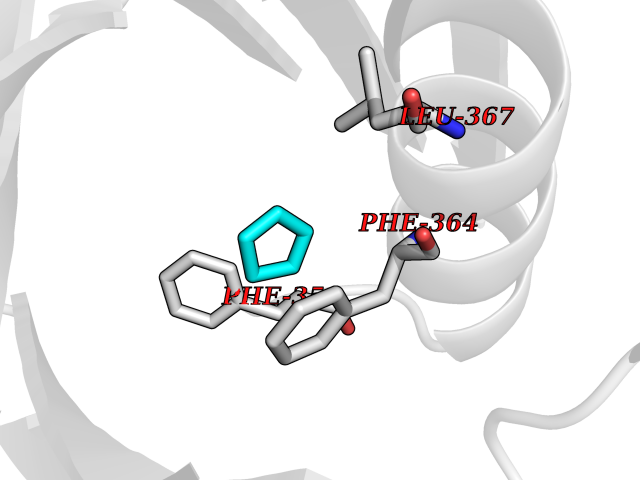 Pymol Map 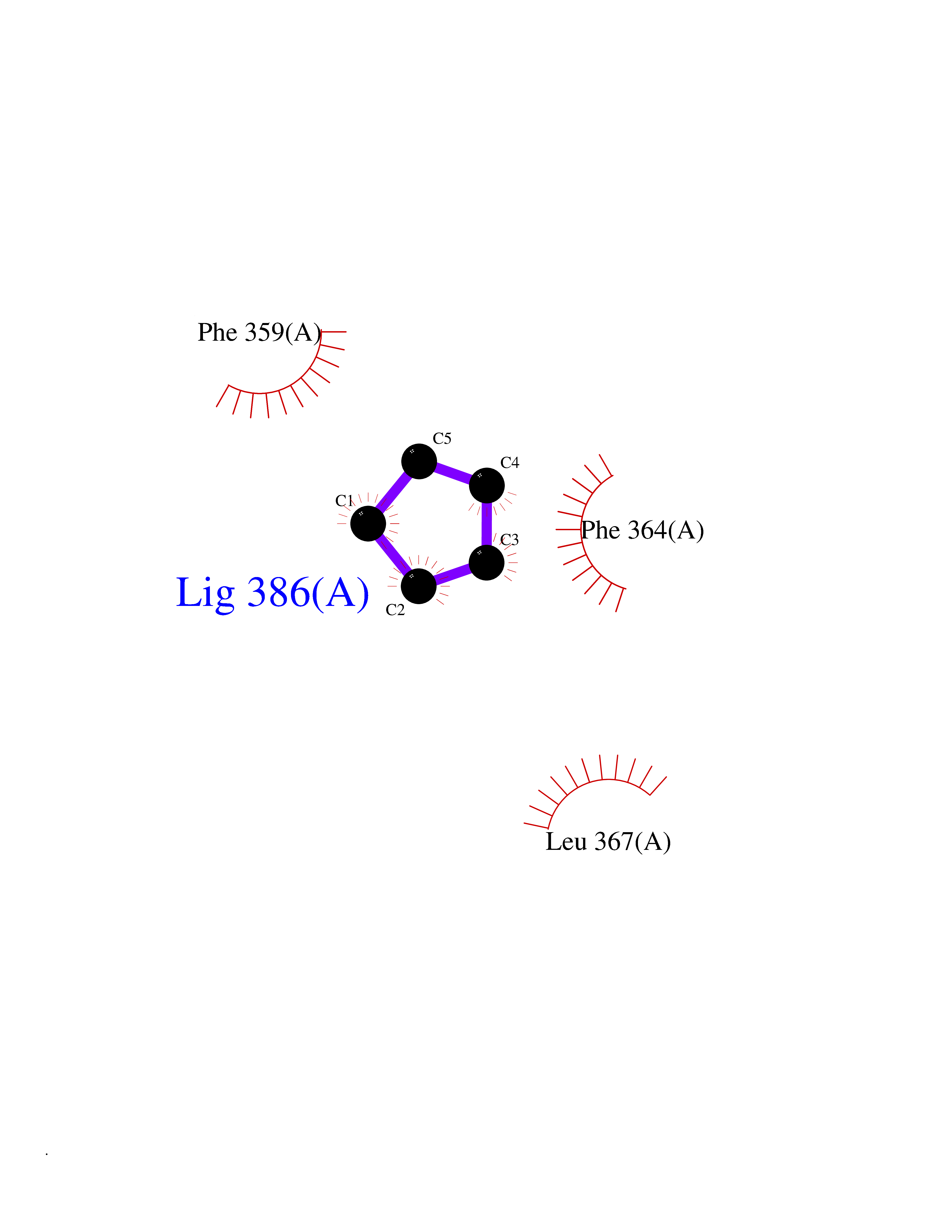 Ligplot Map 3D Binding mode Sequence AKPERGRFLHFHSVTFWVGNAKQAASFYCSKMGFEPLAYRGLETGSREVVSHVIKQGKIVFVLSSALNPWNKEMGDHLVKHGDGVKDIAFEVEDCDYIVQKARERGAKIMREPWVEQDKFGKVKFAVLQTYGDTTHTLVEKMNYIGQFLPGYEAPAFMDPLLPKLPKCSLEMIDHIVGNQPDQEMVSASEWYLKNLQFHRFWSVDDTQVHTEYSSLRSIVVANYEESIKMPINEPAPGKKKSQIQEYVDYNGGAGVQHIALKTEDIITAIRHLRERGLEFLSVPSTYYKQLREKLKTAKIKVKENIDALEELKILVDYDEKGYLLQIFTKPVQDRPTLFLEVIQRHNHQGFGAGNFNSLFKAFEEEQNLRGNLTNM Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Dual specificity mitogen-activated protein kinase kinase 1 | 3EQC | 4.72 | |
Target general information Gen name MAP2K1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PRKMK1;MEK1 Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, MAP kinase kinase subfamily Biochemical class Transferase Function ATP binding.MAP kinase kinase activity.Protein C-terminus binding.Protein kinase activity.Protein N-terminus binding.Protein serine/threonine/tyrosine kinase activity.Protein serine/threonine kinase activator activity.Protein serine/threonine kinase activity.Protein tyrosine kinase activity.Signal transducer, downstream of receptor, with protein tyrosine phosphatase activity. Related diseases Cardiofaciocutaneous syndrome 3 (CFC3) [MIM:615279]: A form of cardiofaciocutaneous syndrome, a multiple congenital anomaly disorder characterized by a distinctive facial appearance, heart defects and intellectual disability. Heart defects include pulmonic stenosis, atrial septal defects and hypertrophic cardiomyopathy. Some affected individuals present with ectodermal abnormalities such as sparse, friable hair, hyperkeratotic skin lesions and a generalized ichthyosis-like condition. Typical facial features are similar to Noonan syndrome. They include high forehead with bitemporal constriction, hypoplastic supraorbital ridges, downslanting palpebral fissures, a depressed nasal bridge, and posteriorly angulated ears with prominent helices. Distinctive features of CFC3 include macrostomia and horizontal shape of palpebral fissures. {ECO:0000269|PubMed:16439621, ECO:0000269|PubMed:18042262}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melorheostosis, isolated (MEL) [MIM:155950]: A sclerosing bone disorder characterized by hyperostosis of the cortex of tubular bones, frequently involving one limb. The lesions may be accompanied by abnormalities of adjacent soft tissue, joint contractures, sclerodermatous skin lesions, muscle atrophy, or hemangioma. {ECO:0000269|PubMed:29643386}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06892; DB07046; DB08208; DB03115; DB11967; DB06616; DB05239; DB02152; DB07101; DB08130; DB14904; DB11689; DB08911 Interacts with Q8N9N5; Q8N9N5-2; Q9NR09; P15056; Q9Y297; O15519-1; P28482; P27361; Q13526; Q9H8W4; P04049; Q8WWU5-7; Q86Y07; Q86Y07-1; P46937 EC number 2.7.12.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cardiomyopathy; Cytoplasm; Cytoskeleton; Direct protein sequencing; Disease variant; Ectodermal dysplasia; Intellectual disability; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 34949.2 Length 312 Aromaticity 0.07 Instability index 45.3 Isoelectric point 5.96 Charge (pH=7) -4.47 2D Binding mode Binding energy (Kcal/mol) -6.44 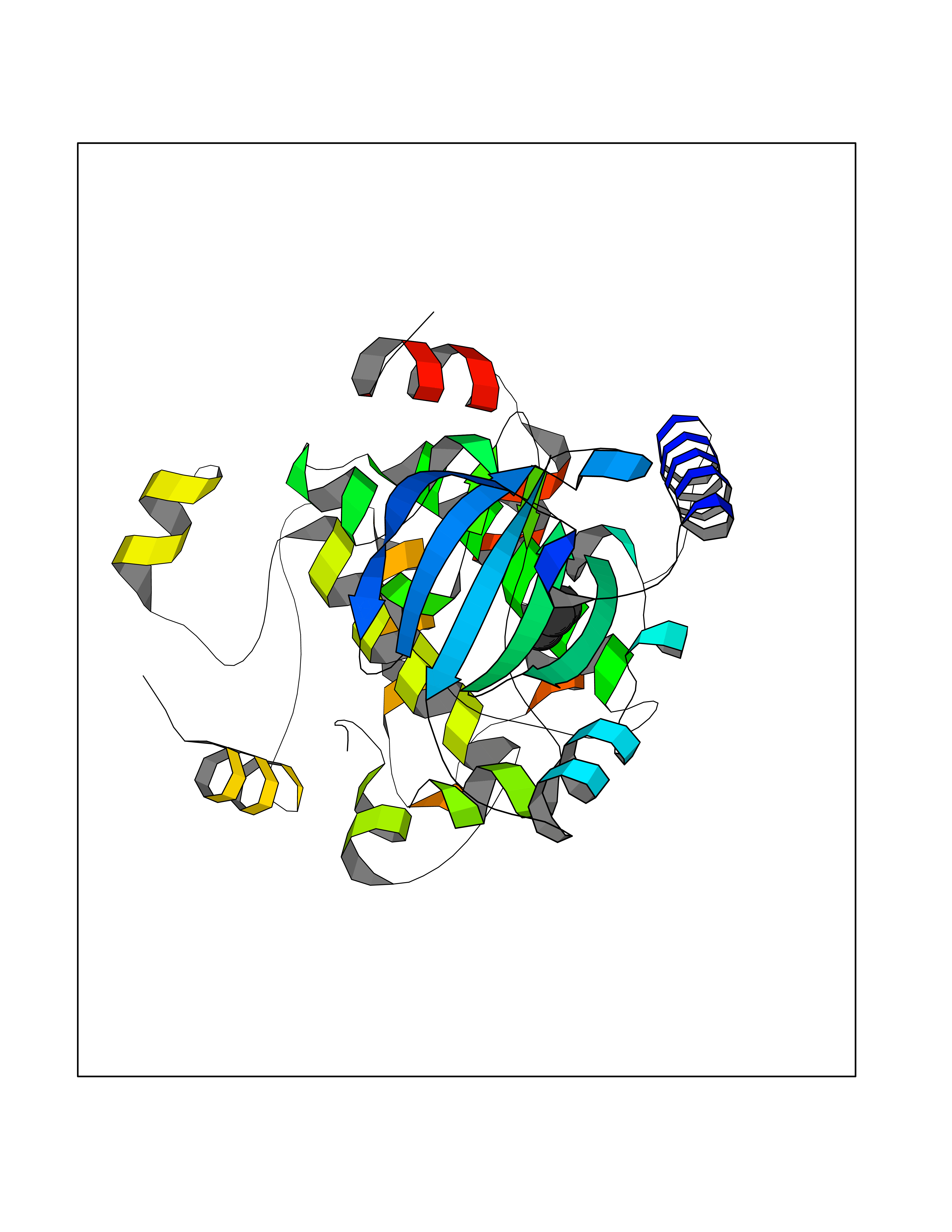 Molscript Map 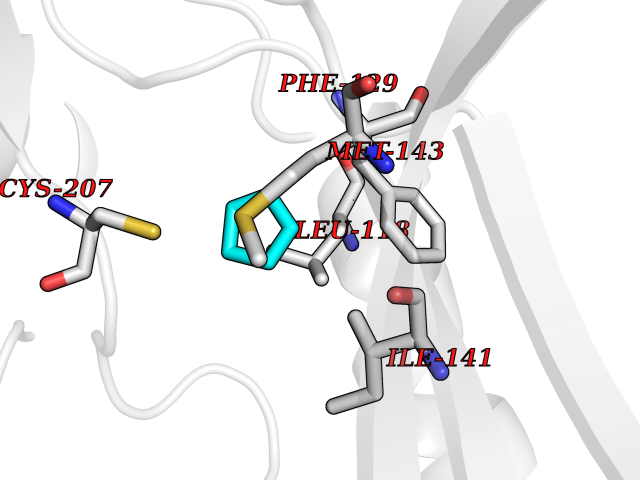 Pymol Map 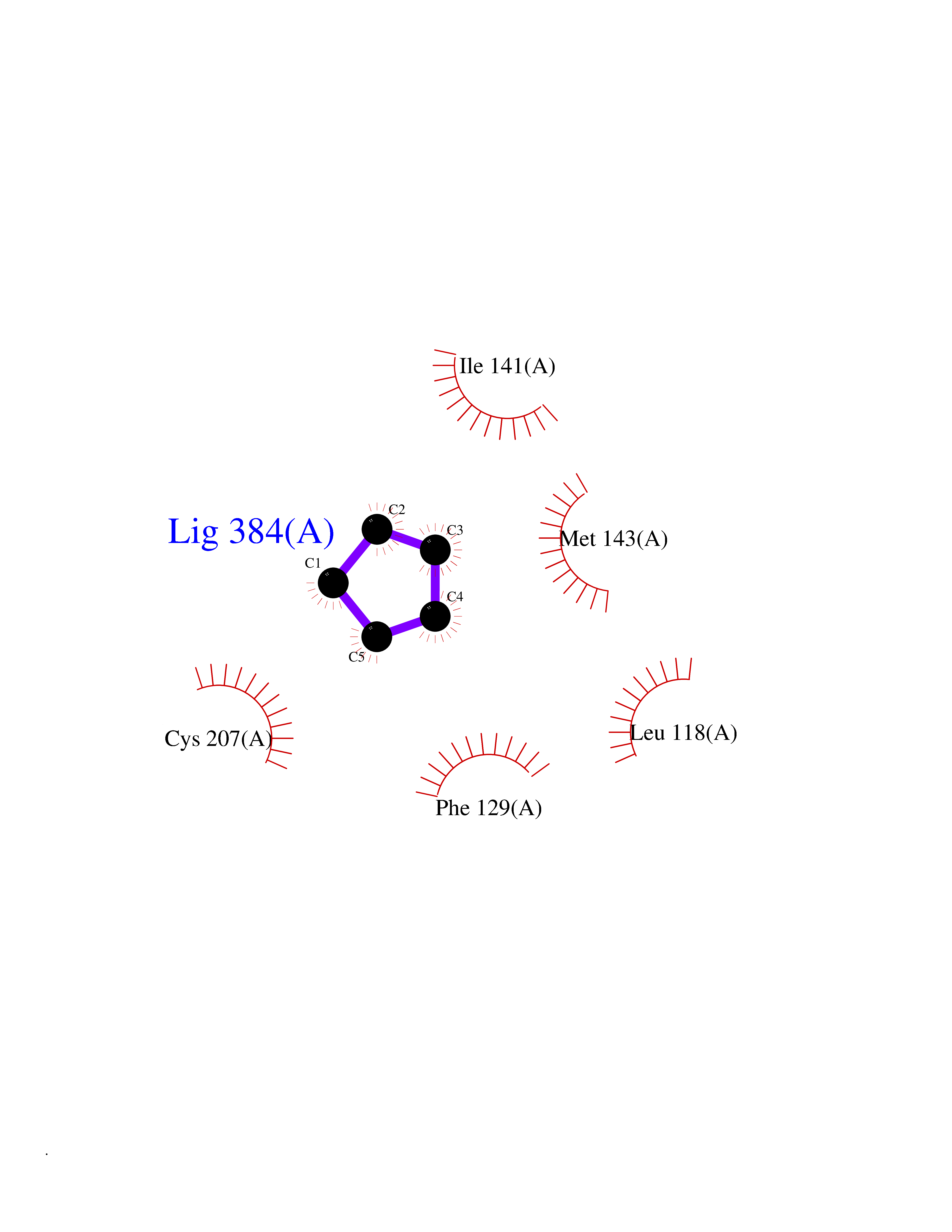 Ligplot Map 3D Binding mode Sequence ELELDEQQRKRLEAFLTQKQKVGELKDDDFEKISELGAGNGGVVFKVSHKPSGLVMARKLIHLEIKPAIRNQIIRELQVLHECNSPYIVGFYGAFYSDGEISICMEHMDGGSLDQVLKKAGRIPEQILGKVSIAVIKGLTYLREKHKIMHRDVKPSNILVNSRGEIKLCDFGVSGQLIDSMAVGTRSYMSPERLQGTHYSVQSDIWSMGLSLVEMAVGRYPIPPPDAKELELMFGCPMAIFELLDYIVNEPPPKLPSGVFSLEFQDFVNKCLIKNPAERADLKQLMVHAFIKRSDAEEVDFAGWLCSTIGLN Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | 2-oxopropyl-CoM reductase, carboxylating | 1MO9 | 4.72 | |
Target general information Gen name xecC Organism Xanthobacter autotrophicus (strain ATCC BAA-1158 / Py2) Uniprot ID TTD ID NA Synonyms Xaut_4867 Protein family Class-I pyridine nucleotide-disulfide oxidoreductase family Biochemical class Oxidoreductase Function 2-oxopropyl-CoM reductase (carboxylating) activity.Flavin adenine dinucleotide binding. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB03163; DB03147 Interacts with NA EC number 1.8.1.5 Uniprot keywords 3D-structure; Disulfide bond; FAD; Flavoprotein; NADP; Oxidoreductase; Plasmid; Redox-active center; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 114413 Length 1044 Aromaticity 0.08 Instability index 25.66 Isoelectric point 5.68 Charge (pH=7) -21.74 2D Binding mode Binding energy (Kcal/mol) -6.44 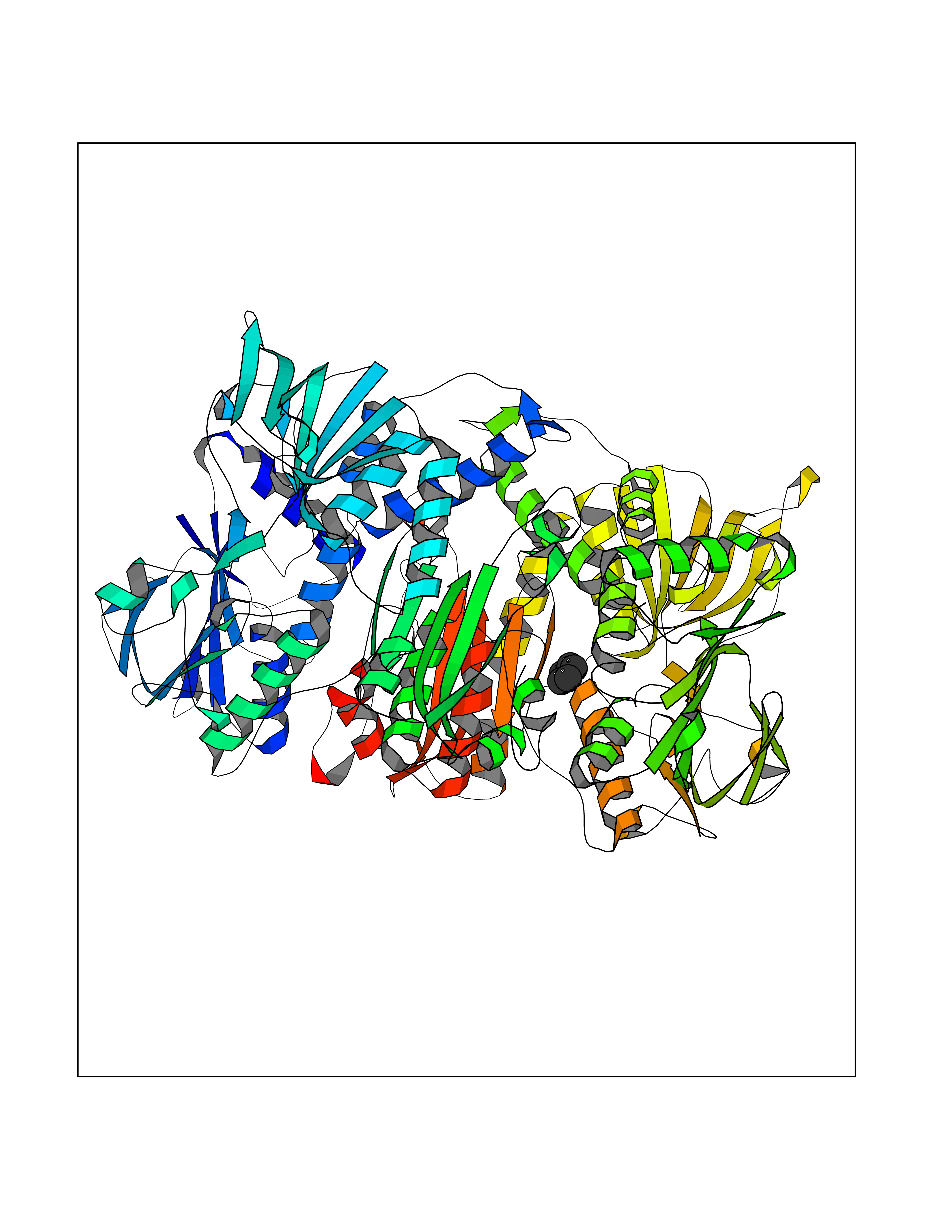 Molscript Map 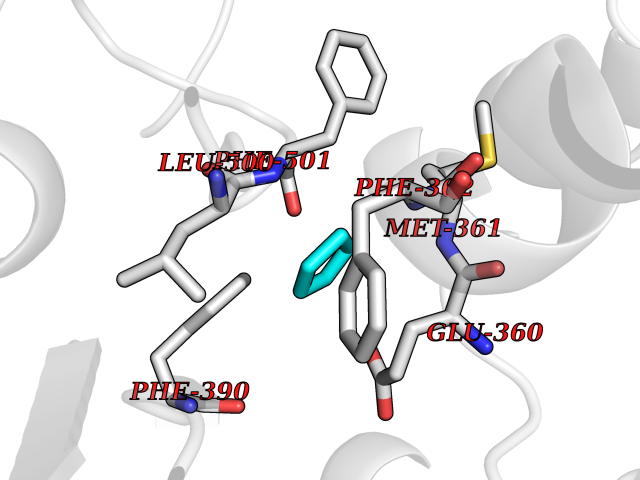 Pymol Map 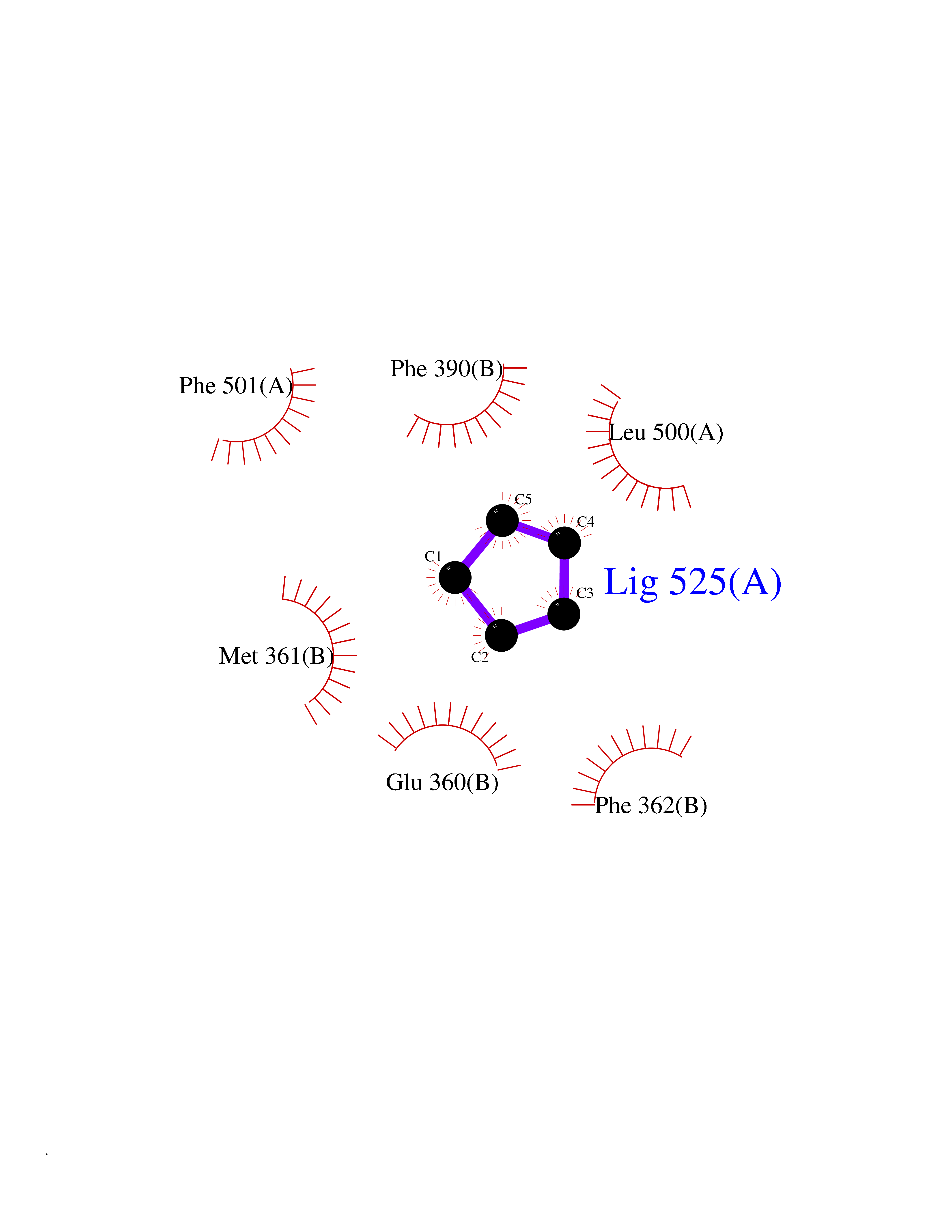 Ligplot Map 3D Binding mode Sequence KVWNARNDHLTINQWATRIDEILEAPDGGEVIYNVDENDPREYDAIFIGGGAAGRFGSAYLRAMGGRQLIVDRWPFLGGSCPHNACVPHHLFSDCAAELMLARTFSGQYWFPDMTEKVVGIKEVVDLFRAGRNGPHGIMNFQSKEQLNLEYILNCPAKVIDNHTVEAAGKVFKAKNLILAVGAGPGTLDVPGVNAKGVFDHATLVEELDYEPGSTVVVVGGSKTAVEYGCFFNATGRRTVMLVRTEPLKLIKDNETRAYVLDRMKEQGMEIISGSNVTRIEEDANGRVQAVVAMTPNGEMRIETDFVFLGLGEQPRSAELAKILGLDLGPKGEVLVNEYLQTSVPNVYAVGDLIGGPMEMFKARKSGCYAARNVMGEKISYTPKNYPDFLHTHYEVSFLGMGEEEARAAGHEIVTIKMPPDTENGLNVALPASDRTMLYAFGKGTAHMSGFQKIVIDAKTRKVLGAHHVGYGAKDAFQYLNVLIKQGLTVDELGDMDELFLNPTHFIQLSRLRAGSKNLVSLKVWNARNDHLTINQWATRIDEILEAPDGGEVIYNVDENDPREYDAIFIGGGAAGRFGSAYLRAMGGRQLIVDRWPFLGGSCPHNACVPHHLFSDCAAELMLARTFSGQYWFPDMTEKVVGIKEVVDLFRAGRNGPHGIMNFQSKEQLNLEYILNCPAKVIDNHTVEAAGKVFKAKNLILAVGAGPGTLDVPGVNAKGVFDHATLVEELDYEPGSTVVVVGGSKTAVEYGCFFNATGRRTVMLVRTEPLKLIKDNETRAYVLDRMKEQGMEIISGSNVTRIEEDANGRVQAVVAMTPNGEMRIETDFVFLGLGEQPRSAELAKILGLDLGPKGEVLVNEYLQTSVPNVYAVGDLIGGPMEMFKARKSGCYAARNVMGEKISYTPKNYPDFLHTHYEVSFLGMGEEEARAAGHEIVTIKMPPDTENGLNVALPASDRTMLYAFGKGTAHMSGFQKIVIDAKTRKVLGAHHVGYGAKDAFQYLNVLIKQGLTVDELGDMDELFLNPTHFIQLSRLRAGSKNLVSL Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Macrophage-stimulating protein receptor (RON) | 3PLS | 4.72 | |
Target general information Gen name MST1R Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p185Ron; Proteintyrosine kinase 8; Macrophagestimulating protein receptor beta chain; Macrophagestimulating protein receptor; MST1R; MSP receptor; CDw136; CD136 Protein family Protein kinase superfamily, Tyr protein kinase family Biochemical class Kinase Function Receptor tyrosine kinase that transduces signals from the extracellular matrix into the cytoplasm by binding to MST1 ligand. Regulates many physiological processes including cell survival, migration and differentiation. Ligand binding at the cell surface induces autophosphorylation of RON on its intracellular domain that provides docking sites for downstream signaling molecules. Following activation by ligand, interacts with the PI3-kinase subunit PIK3R1, PLCG1 or the adapter GAB1. Recruitment of these downstream effectors by RON leads to the activation of several signaling cascades including the RAS-ERK, PI3 kinase-AKT, or PLCgamma-PKC. RON signaling activates the wound healing response by promoting epithelial cell migration, proliferation as well as survival at the wound site. Plays also a role in the innate immune response by regulating the migration and phagocytic activity of macrophages. Alternatively, RON can also promote signals such as cell migration and proliferation in response to growth factors other than MST1 ligand. Related diseases Nasopharyngeal carcinoma, 3 (NPCA3) [MIM:617075]: A form of nasopharyngeal carcinoma, a malignant neoplasm that originates in the nasopharyngeal epithelium and includes 4 subtypes: keratinizing squamous cell, non-keratinizing, basaloid squamous cell, and papillary adenocarcinoma. {ECO:0000269|PubMed:26951679}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08865; DB12010 Interacts with P26927; O43157; O15031; Q9ULL4 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cleavage on pair of basic residues; Disease variant; Disulfide bond; Glycoprotein; Immunity; Innate immunity; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 34204.1 Length 298 Aromaticity 0.09 Instability index 38.81 Isoelectric point 6.37 Charge (pH=7) -3.11 2D Binding mode Binding energy (Kcal/mol) -6.44 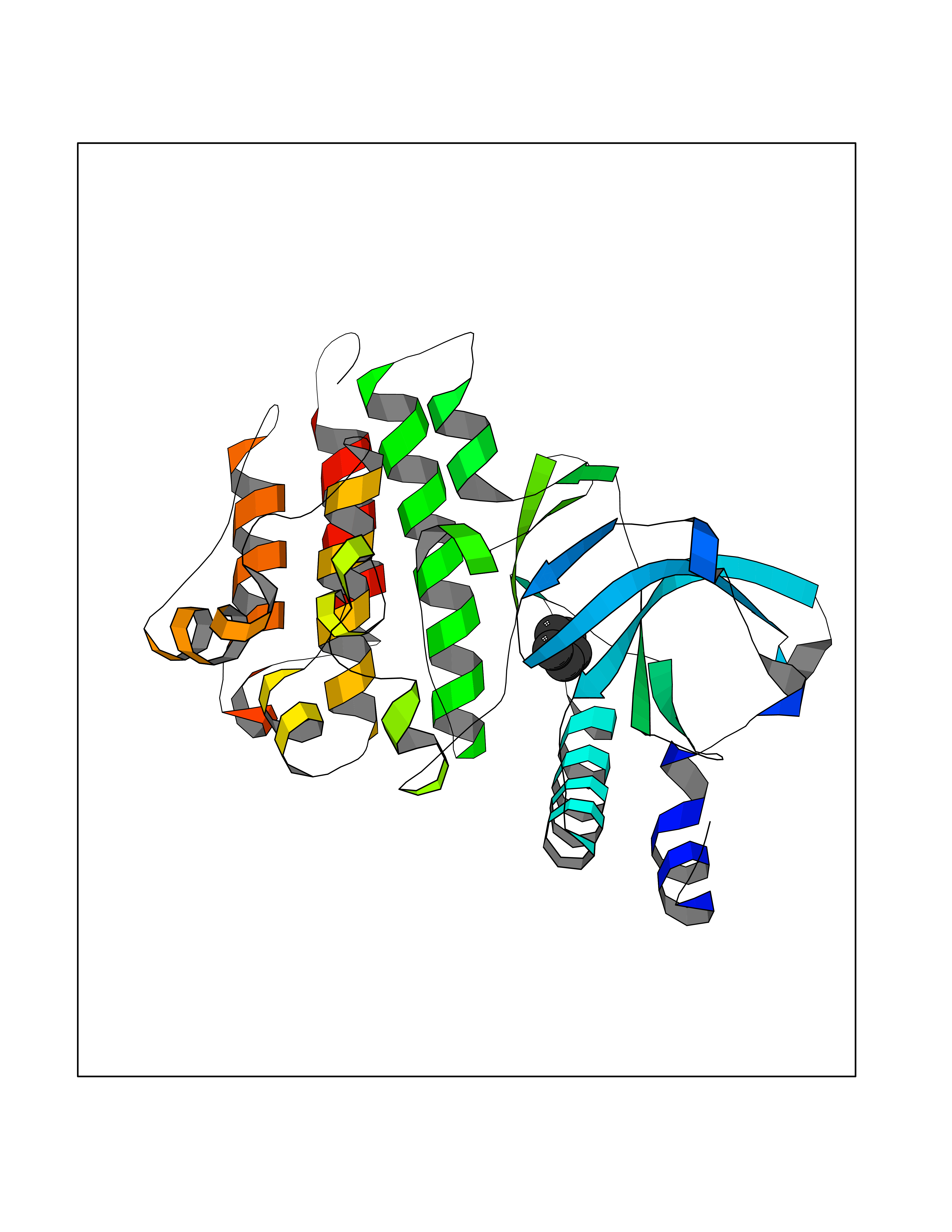 Molscript Map 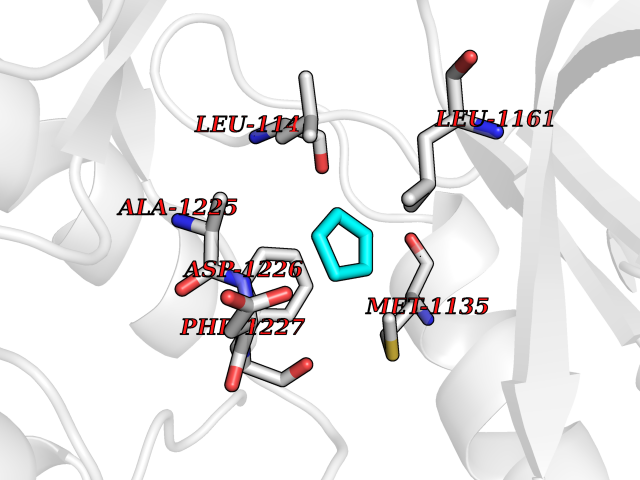 Pymol Map 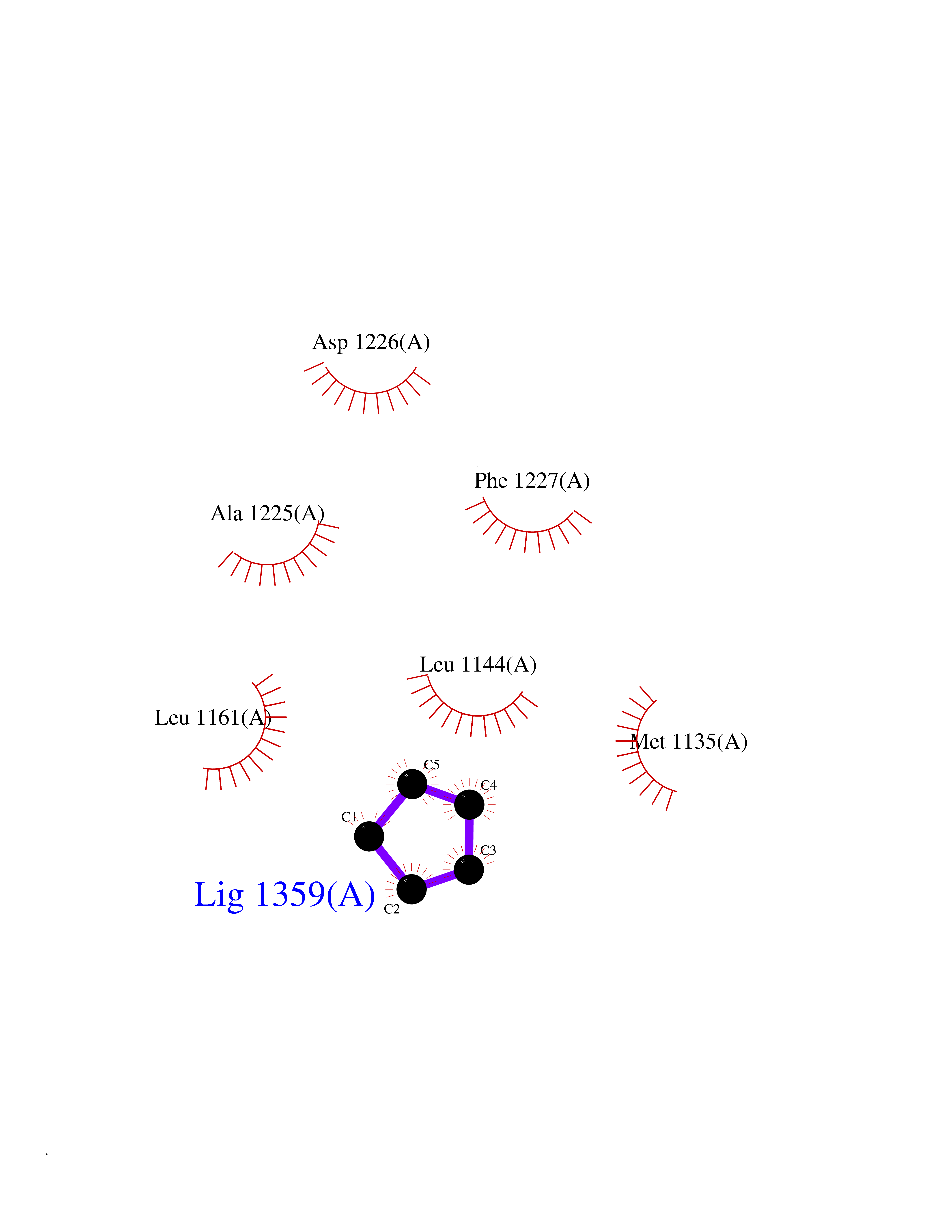 Ligplot Map 3D Binding mode Sequence RDLDSALLAEVKDVLIPHERVVTHSDRVIGKGHFGVVYHGEYIDQAQNRIQCAIKSLSRITEMQQVEAFLREGLLMRGLNHPNVLALIGIMLPPEGLPHVLLPYMCHGDLLQFIRSPQRNPTVKDLISFGLQVARGMEYLAEQKFVHRDLAARNCMLDESFTVKVADFGLARDILDREYYSVQQHRHARLPVKWTALESLQTYRFTTKSDVWSFGVLLWELLTRGAPPYRHIDPFDLTHFLAQGRRLPQPEYCPDSLYQVMQQCWEADPAVRPTFRVLVGEVEQIVSALLGDHYVQLP Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Pseudomonas Transcriptional activator protein LasR (Pseudo LasR) | 3IX3 | 4.72 | |
Target general information Gen name Pseudo LasR Organism Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Uniprot ID TTD ID Synonyms NA Protein family Autoinducer-regulated transcriptional regulatory protein family Biochemical class NA Function Transcriptional activator of elastase structural gene (LasB). Binds to the PAI autoinducer. Related diseases Growth hormone deficiency, isolated, 1A (IGHD1A) [MIM:262400]: An autosomal recessive, severe deficiency of growth hormone leading to dwarfism. Patients often develop antibodies to administered growth hormone. {ECO:0000269|PubMed:8364549}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 1B (IGHD1B) [MIM:612781]: An autosomal recessive deficiency of growth hormone leading to short stature. Patients have low but detectable levels of growth hormone, significantly retarded bone age, and a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:12655557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Kowarski syndrome (KWKS) [MIM:262650]: A syndrome clinically characterized by short stature associated with bioinactive growth hormone, normal or slightly increased growth hormone secretion, pathologically low insulin-like growth factor 1 levels, and normal catch-up growth on growth hormone replacement therapy. {ECO:0000269|PubMed:17519310, ECO:0000269|PubMed:8552145, ECO:0000269|PubMed:9276733}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 2 (IGHD2) [MIM:173100]: An autosomal dominant deficiency of growth hormone leading to short stature. Clinical severity is variable. Patients have a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:11502836, ECO:0000269|PubMed:9152628}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08324 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; DNA-binding; Quorum sensing; Reference proteome; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 18305.5 Length 163 Aromaticity 0.12 Instability index 46.52 Isoelectric point 5.19 Charge (pH=7) -6.78 2D Binding mode Binding energy (Kcal/mol) -6.44 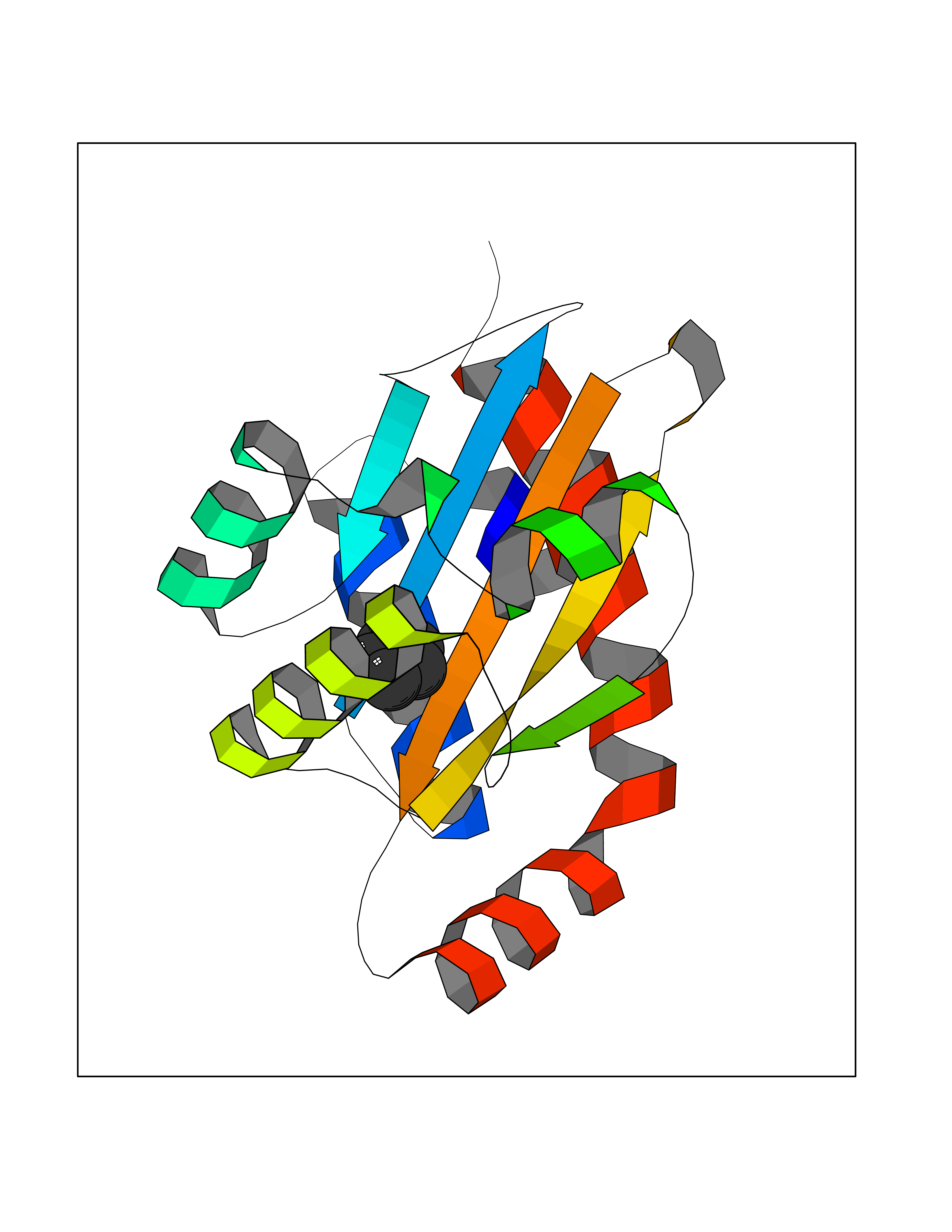 Molscript Map 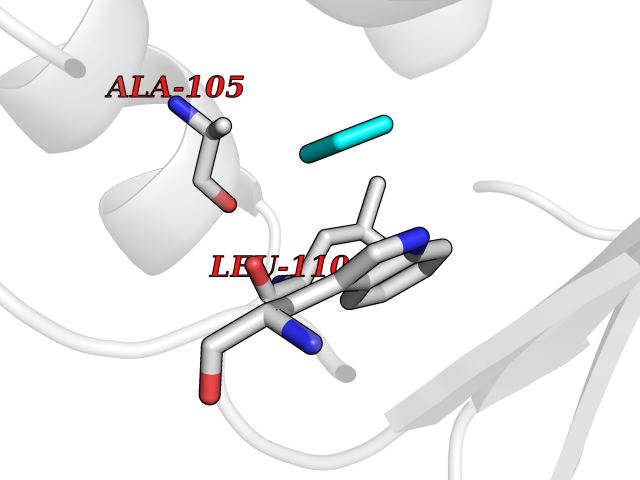 Pymol Map 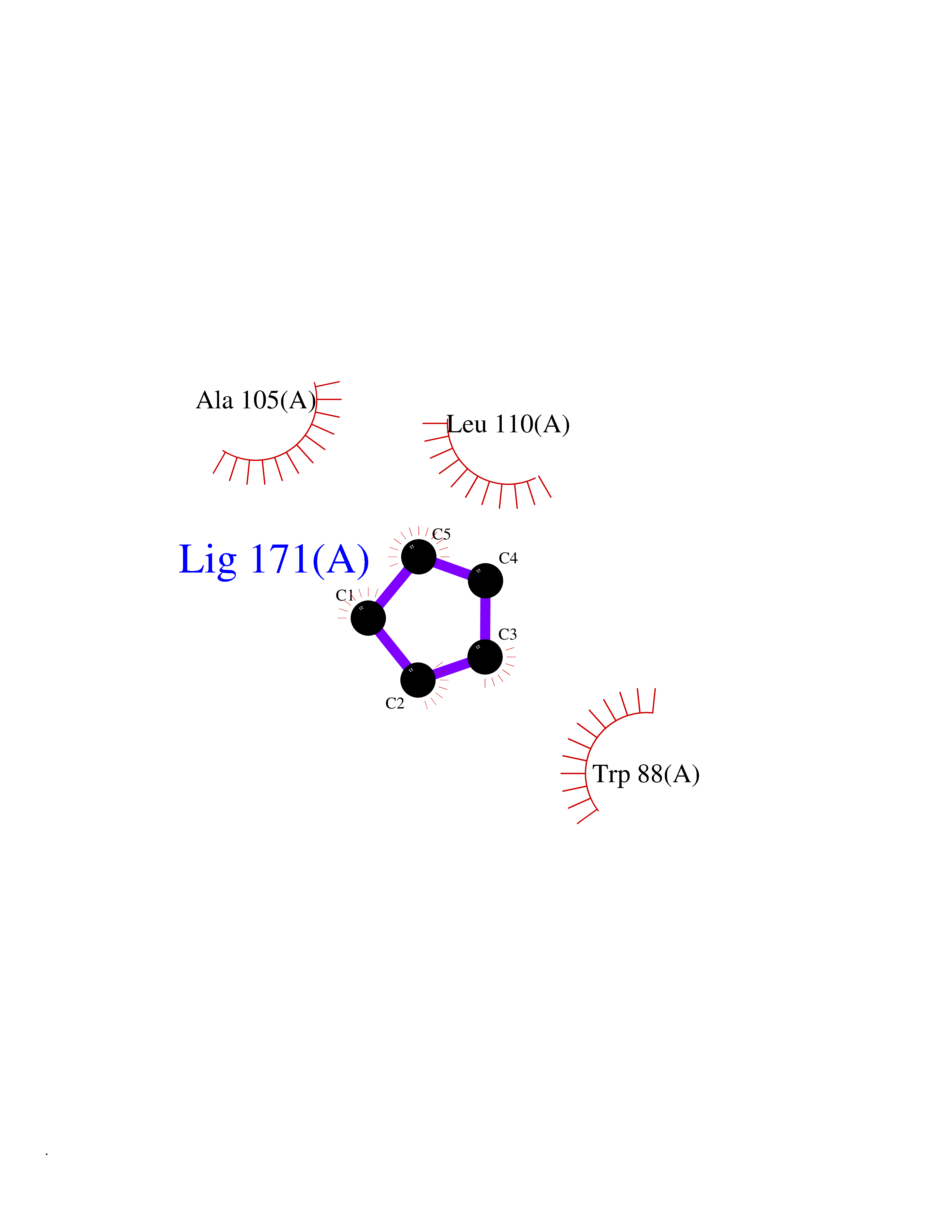 Ligplot Map 3D Binding mode Sequence FLELERSSGKLEWSAILQKMASDLGFSKILFGLLPKDSQDYENAFIVGNYPAAWREHYDRAGYARVDPTVSHCTQSVLPIFWEPSIYQTRKQHEFFEEASAAGLVYGLTMPLHGARGELGALSLSVEAENRAEANRFMESVLPTLWMLKDYALQSGAGLAFEH Hydrogen bonds contact Hydrophobic contact | ||||