Job Results:
Ligand
Structure
Job ID
b66da62373d29d285a275497218b65f3
Job name
NA
Time
2024-11-27 22:32:54
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Macrophage colony-stimulating factor 1 receptor (CSF1R) | 2I1M | 7.97 | |
Target general information Gen name CSF1R Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Proto-oncogene c-Fms; M-CSF-R; FMS; CSF-1R; CSF-1-R; CSF-1 receptor; CD115 Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Promotes the release of proinflammatory chemokines in response to IL34 and CSF1, and thereby plays an important role in innate immunity and in inflammatory processes. Plays an important role in the regulation of osteoclast proliferation and differentiation, the regulation of bone resorption, and is required for normal bone and tooth development. Required for normal male and female fertility, and for normal development of milk ducts and acinar structures in the mammary gland during pregnancy. Promotes reorganization of the actin cytoskeleton, regulates formation of membrane ruffles, cell adhesion and cell migration, and promotes cancer cell invasion. Activates several signaling pathways in response to ligand binding. Phosphorylates PIK3R1, PLCG2, GRB2, SLA2 and CBL. Activation of PLCG2 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate, that then lead to the activation of protein kinase C family members, especially PRKCD. Phosphorylation of PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, leads to activation of the AKT1 signaling pathway. Activated CSF1R also mediates activation of the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1, and of the SRC family kinases SRC, FYN and YES1. Activated CSF1R transmits signals both via proteins that directly interact with phosphorylated tyrosine residues in its intracellular domain, or via adapter proteins, such as GRB2. Promotes activation of STAT family members STAT3, STAT5A and/or STAT5B. Promotes tyrosine phosphorylation of SHC1 and INPP5D/SHIP-1. Receptor signaling is down-regulated by protein phosphatases, such as INPP5D/SHIP-1, that dephosphorylate the receptor and its downstream effectors, and by rapid internalization of the activated receptor. Tyrosine-protein kinase that acts as cell-surface receptor for CSF1 and IL34 and plays an essential role in the regulation of survival, proliferation and differentiation of hematopoietic precursor cells, especially mononuclear phagocytes, such as macrophages and monocytes. Related diseases Aberrant expression of CSF1 or CSF1R can promote cancer cell proliferation, invasion and formation of metastases. Overexpression of CSF1 or CSF1R is observed in a significant percentage of breast, ovarian, prostate, and endometrial cancers.; DISEASE: Aberrant expression of CSF1 or CSF1R may play a role in inflammatory diseases, such as rheumatoid arthritis, glomerulonephritis, atherosclerosis, and allograft rejection.; DISEASE: Leukoencephalopathy, hereditary diffuse, with spheroids 1 (HDLS1) [MIM:221820]: An autosomal dominant adult-onset rapidly progressive neurodegenerative disorder characterized by variable behavioral, cognitive, and motor changes. Patients often die of dementia within 6 years of onset. Brain imaging shows patchy abnormalities in the cerebral white matter, predominantly affecting the frontal and parietal lobes. {ECO:0000269|PubMed:22197934, ECO:0000269|PubMed:23408870, ECO:0000269|PubMed:24336230, ECO:0000269|PubMed:24532199}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brain abnormalities, neurodegeneration, and dysosteosclerosis (BANDDOS) [MIM:618476]: An autosomal recessive disease with variable manifestations. Main features are brain malformations with calcifying leukoencephalopathy, progressive neurodegeneration, and bone sclerotic features. The age at onset ranges from infancy to early adulthood. Neurologic features include loss of previous motor and language skills, cognitive impairment, spasticity, and focal seizures. Brain imaging shows periventricular white matter abnormalities and calcifications, large cisterna magna or Dandy-Walker malformation, and sometimes agenesis of the corpus callosum. {ECO:0000269|PubMed:30982608, ECO:0000269|PubMed:30982609}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07167; DB07202; DB12147; DB12010; DB00619; DB06080; DB12978; DB01268 Interacts with P09603; Q15375; P29323; Q6ZMJ4-1 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Immunity; Immunoglobulin domain; Inflammatory response; Innate immunity; Kinase; Membrane; Neurodegeneration; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 35082.9 Length 311 Aromaticity 0.11 Instability index 44.6 Isoelectric point 8.13 Charge (pH=7) 2.42 2D Binding mode Binding energy (Kcal/mol) -8.74 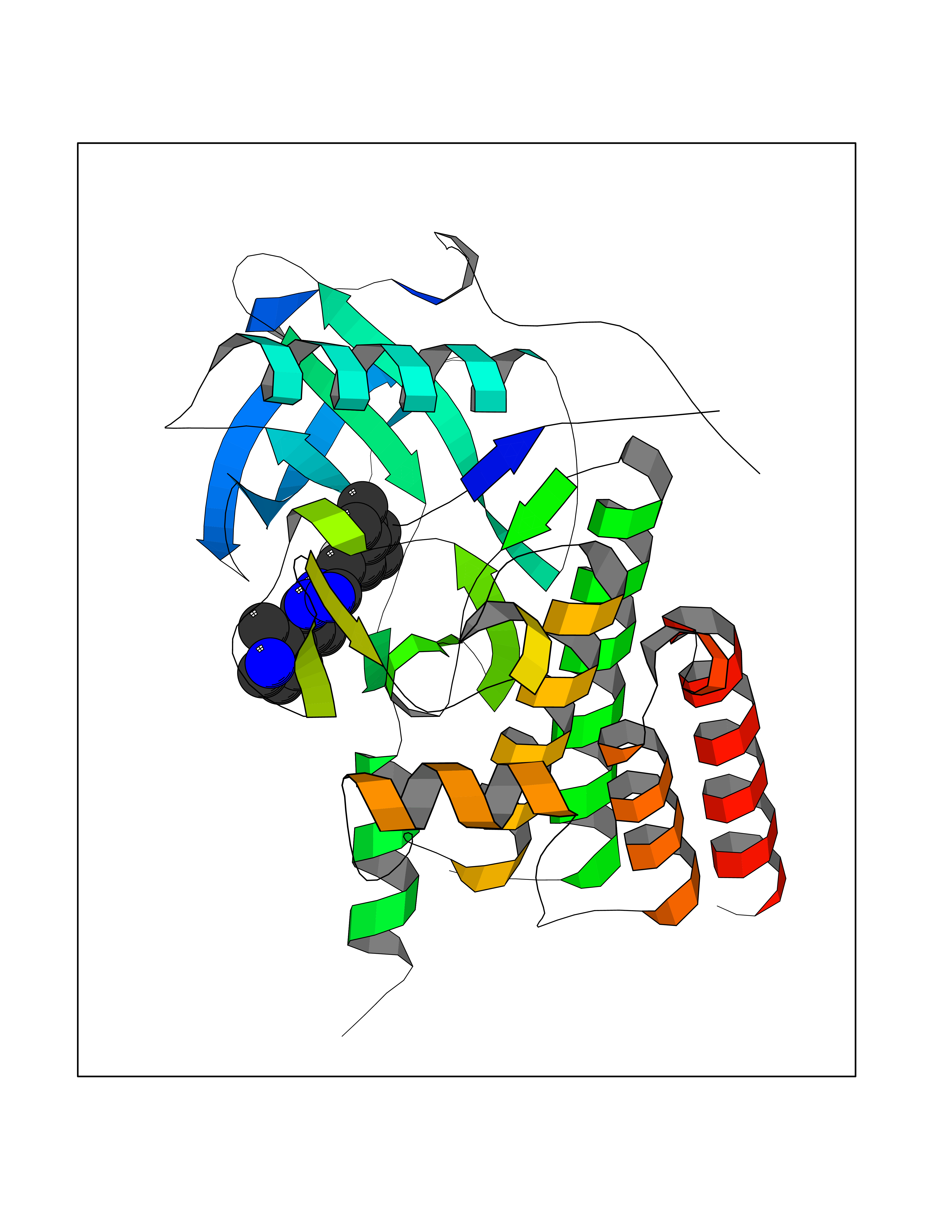 Molscript Map 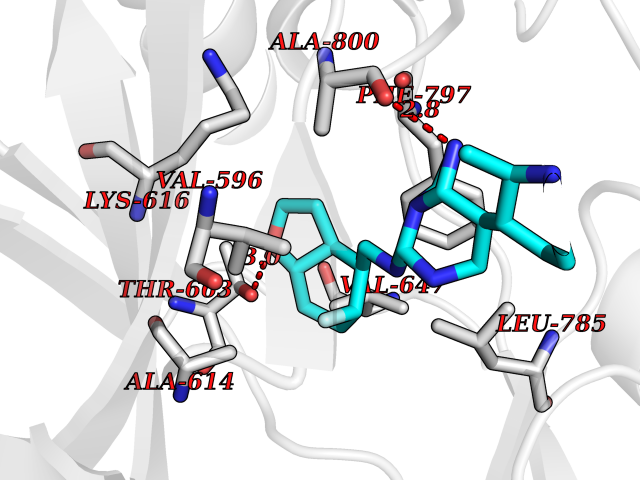 Pymol Map 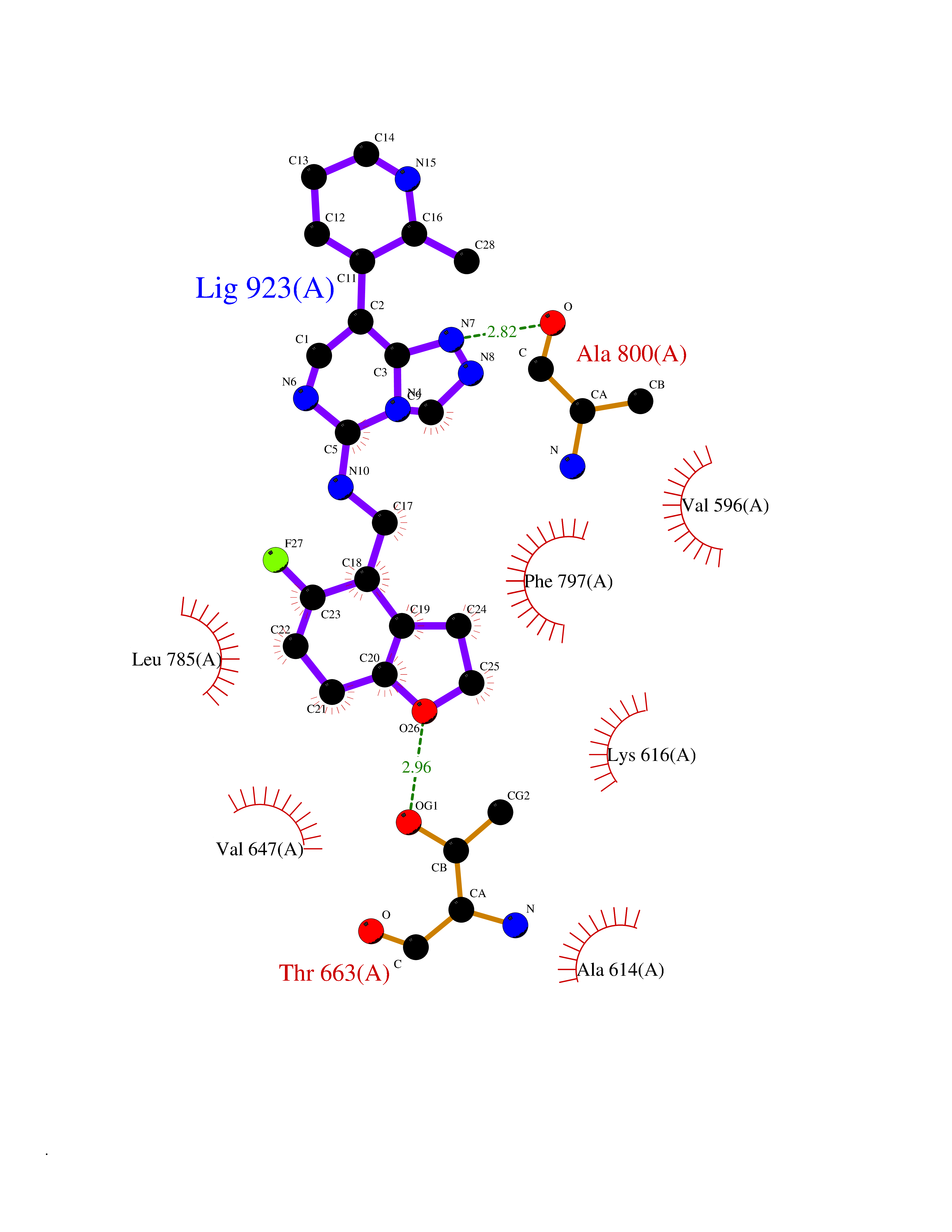 Ligplot Map 3D Binding mode Sequence QVRWKIIESYNSYTFIDPTQLPYNEKWEFPRNNLQFGKTLGAGAFGKVVEATAFGLGKEDAVLKVAVKMLKSTAHADEKEALMSELKIMSHLGQHENIVNLLGACTHGGPVLVITEYCCYGDLLNFLRRKSRVLETDSTASTRDLLHFSSQVAQGMAFLASKNCIHRDVAARNVLLTNGHVAKIGDFGLARDIMNDSNYIVKGNARLPVKWMAPESIFDCVYTVQSDVWSYGILLWEIFSLGLNPYPGILVNSKFYKLVKDGYQMAQPAFAPKNIYSIMQACWALEPTHRPTFQQICSFLQEQAQEDRRER Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Voltage-gated sodium channel alpha Nav1.7 (SCN9A) | 7W9M | 7.97 | |
Target general information Gen name SCN9A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hNE-Na; Voltage-gated sodium channel subunit alpha Nav1.7; Sodium channel proteintype IX subunit alpha; Sodium channel proteintype 9 subunit alpha; Sodium channel protein type IX subunit alpha; Sodium Protein family Sodium channel (TC 1.A.1.10) family, Nav1.7/SCN9A subfamily Biochemical class Voltage-gated ion channel Function Assuming opened or closed conformations in response to the voltage difference across the membrane, the protein forms a sodium-selective channel through which Na(+) ions may pass in accordance with their electrochemical gradient. It is a tetrodotoxin-sensitive Na(+) channel isoform. Plays a role in pain mechanisms, especially in the development of inflammatory pain. Mediates the voltage-dependent sodium ion permeability of excitable membranes. Related diseases Primary erythermalgia (PERYTHM) [MIM:133020]: Autosomal dominant disease characterized by recurrent episodes of severe pain associated with redness and warmth in the feet or hands. {ECO:0000269|PubMed:14985375, ECO:0000269|PubMed:15385606, ECO:0000269|PubMed:15955112, ECO:0000269|PubMed:15958509, ECO:0000269|PubMed:16216943, ECO:0000269|PubMed:16392115, ECO:0000269|PubMed:16702558, ECO:0000269|PubMed:16988069, ECO:0000269|PubMed:18945915, ECO:0000269|PubMed:19369487, ECO:0000269|PubMed:24311784}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Indifference to pain, congenital, autosomal recessive (CIP) [MIM:243000]: A disorder characterized by congenital inability to perceive any form of pain, in any part of the body. All other sensory modalities are preserved and the peripheral and central nervous systems are apparently intact. Patients perceive the sensations of touch, warm and cold temperature, proprioception, tickle and pressure, but not painful stimuli. There is no evidence of a motor or sensory neuropathy, either axonal or demyelinating. {ECO:0000269|PubMed:20635406}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Paroxysmal extreme pain disorder (PEXPD) [MIM:167400]: An autosomal dominant paroxysmal disorder of pain and autonomic dysfunction. The distinctive features are paroxysmal episodes of burning pain in the rectal, ocular, and mandibular areas accompanied by autonomic manifestations such as skin flushing. {ECO:0000269|PubMed:17145499, ECO:0000269|PubMed:18945915, ECO:0000269|PubMed:25285947}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09088; DB13746; DB05541; DB00564; DB01161; DB00907; DB13269; DB13961; DB06218; DB00555; DB00281; DB00776; DB11186; DB09345; DB01069; DB09342; DB00243; DB06201; DB09085; DB00273; DB00313; DB00909 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Cell projection; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Membrane; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Sodium; Sodium channel; Sodium transport; Transmembrane; Transmembrane helix; Transport; Ubl conjugation; Voltage-gated channel Protein physicochemical properties Chain ID A Molecular weight (Da) 162402 Length 1418 Aromaticity 0.14 Instability index 35.66 Isoelectric point 6.72 Charge (pH=7) -1.33 2D Binding mode Binding energy (Kcal/mol) -7.88  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GPQSFVHFTKQSLALIEQRIAERKSKEPKPSSDLEAGKQLPFIYGDIPPGMVSEPLEDLDPYYADKKTFIVLNKGKTIFRFNATPALYMLSPFSPLRRISIKILVHSLFSMLIMCTILTNCIFMTMNNPPDWTKNVEYTFTGIYTFESLVKILARGFCVGEFTFLRDPWNWLDFVVIVFAYLTEFVNNVSALRTFRVLRALKTISVIPGLKTIVGALIQSVKKLSDVMILTVFCLSVFALIGLQLFMGNLKHKCFRNSLENNETLESIMNTLESEEDFRKYFYYLEGSKDALLCGFSTDSGQCPEGYTCVKIGRNPDYGYTSFDTFSWAFLALFRLMTQDYWENLYQQTLRAAGKTYMIFFVVVIFLGSFYLINLILAVVAMAYKEQNQANIEEAKQKELEFQQMLDRLKKEQEPYWIKFKKCIYFIVMDPFVDLAITICIVLNTLFMAMEHHPMTEEFKNVLAIGNLVFTGIFAAEMVLKLIAMDPYEYFQVGWNIFDSLIVTLSLVELFLADVEGLSVLRSFRLLRVFKLAKSWPTLNMLIKIIGNSVGALGNLTLVLAIIVFIFAVVGMQLFGKSYKECVCKINDDCTLPRWHMNDFFHSFLIVFRVLCGEWIETMWDCMEVAGQAMCLIVYMMVMVIGNLVVLNLFLALLLSSFSSDNLTAIEEDPDANNLQIAVTRIKKGINYVKQTLREFILKAFGKIWWNIRKTCYKIVEHSWFESFIVLMILLSSGALAFEDIYIERKKTIKIILEYADKIFTYIFILEMLLKWIAYGYKTYFTNAWCWLDFLIVDVSLVTLVANTLGYSDLGPIKSLRTLRALRPLRALSRFEGMRVVVNALIGAIPSIMNVLLVCLIFWLIFSIMGVNLFAGKFYECINTTDGSRFPASQVPNRSECFALMNVSQNVRWKNLKVNFDNVGLGYLSLLQVATFKGWTIIMYAAVDSVNVDKQPKYEYSLYMYIYFVVFIIFGSFFTLNLFIGVIIDNFNQQKKKLGGQDIFMTEEQKKYYNAMKKLGSKKPQKPIPRPGNKIQGCIFDLVTNQAFDISIMVLICLNMVTMMVEKEGQSQHMTEVLYWINVVFIILFTGECVLKLISLRHYYFTVGWNIFDFVVVIISIVGMFLADLIETYFVSPTLFRVIRLARIGRILRLVKGAKGIRTLLFALMMSLPALFNIGLLLFLVMFIYAIFGMSNFAYVKKEDGINDMFNFETFGNSMICLFQITTSAGWDGLLAPILNSKPPDCDPKKVHPGSSVEGDCGNPSVGIFYFVSYIIISFLVVVNMYIAVILENFSVATEESTEPLSEDDFEMFYEVWEKFDPDATQFIEFSKLSDFAAALDPPLLIAKPNKVQLIAMDLPMVSGDRIHCLDILFAFTKRVLGESGEMDSLRSQMEERFMSANPSKVSYEPITTTLKRKQEDV Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Platelet-derived growth factor receptor alpha (PDGFRA) | 5K5X | 7.97 | |
Target general information Gen name PDGFRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RHEPDGFRA; Platelet-derived growth factor receptor 2; Platelet-derived growth factor alpha receptor; PDGFR2; PDGFR-alpha; PDGFR-2; PDGF-R-alpha; CD140a antigen; CD140a; CD140 antigen-like family membe Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Depending on the context, promotes or inhibits cell proliferation and cell migration. Plays an important role in the differentiation of bone marrow-derived mesenchymal stem cells. Required for normal skeleton development and cephalic closure during embryonic development. Required for normal development of the mucosa lining the gastrointestinal tract, and for recruitment of mesenchymal cells and normal development of intestinal villi. Plays a role in cell migration and chemotaxis in wound healing. Plays a role in platelet activation, secretion of agonists from platelet granules, and in thrombin-induced platelet aggregation. Binding of its cognate ligands - homodimeric PDGFA, homodimeric PDGFB, heterodimers formed by PDGFA and PDGFB or homodimeric PDGFC -leads to the activation of several signaling cascades; the response depends on the nature of the bound ligand and is modulated by the formation of heterodimers between PDGFRA and PDGFRB. Phosphorylates PIK3R1, PLCG1, and PTPN11. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate, mobilization of cytosolic Ca(2+) and the activation of protein kinase C. Phosphorylates PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, and thereby mediates activation of the AKT1 signaling pathway. Mediates activation of HRAS and of the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1. Promotes activation of STAT family members STAT1, STAT3 and STAT5A and/or STAT5B. Receptor signaling is down-regulated by protein phosphatases that dephosphorylate the receptor and its down-stream effectors, and by rapid internalization of the activated receptor. Tyrosine-protein kinase that acts as a cell-surface receptor for PDGFA, PDGFB and PDGFC and plays an essential role in the regulation of embryonic development, cell proliferation, survival and chemotaxis. Related diseases A chromosomal aberration involving PDGFRA is found in some cases of hypereosinophilic syndrome. Interstitial chromosomal deletion del(4)(q12q12) causes the fusion of FIP1L1 and PDGFRA (FIP1L1-PDGFRA). Mutations that cause overexpression and/or constitutive activation of PDGFRA may be a cause of hypereosinophilic syndrome. {ECO:0000269|PubMed:12808148}.; DISEASE: Gastrointestinal stromal tumor (GIST) [MIM:606764]: Common mesenchymal neoplasms arising in the gastrointestinal tract, most often in the stomach. They are histologically, immunohistochemically, and genetically different from typical leiomyomas, leiomyosarcomas, and schwannomas. Most GISTs are composed of a fairly uniform population of spindle-shaped cells. Some tumors are dominated by epithelioid cells or contain a mixture of spindle and epithelioid morphologies. Primary GISTs in the gastrointestinal tract commonly metastasize in the omentum and mesenteries, often as multiple nodules. However, primary tumors may also occur outside of the gastrointestinal tract, in other intra-abdominal locations, especially in the omentum and mesentery. {ECO:0000269|PubMed:12522257, ECO:0000269|PubMed:15928335}. The gene represented in this entry may be involved in disease pathogenesis. Mutations causing PDGFRA constitutive activation have been found in gastrointestinal stromal tumors lacking KIT mutations (PubMed:12522257). {ECO:0000269|PubMed:12522257}.; DISEASE: GIST-plus syndrome (GISTPS) [MIM:175510]: A disorder characterized by multiple mesenchymal tumors of the gastrointestinal tract, including gastrointestinal stromal tumor, inflammatory fibroid polyps, and fibroid tumors. Additional features are coarse facies and skin, broad hands and feet, and premature tooth loss. GISTPS is an autosomal dominant disease with incomplete penetrance. Gastrointestinal stromal tumor and inflammatory fibroid polyps may also occur in isolation. {ECO:0000269|PubMed:14699510, ECO:0000269|PubMed:17087943, ECO:0000269|PubMed:25975287}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12742; DB00102; DB12147; DB10772; DB12010; DB00619; DB09078; DB06595; DB09079; DB06043; DB06589; DB08901; DB08896; DB14840; DB01268; DB11800; DB05146 Interacts with P46108; P46109; P00533; Q8N6L0; P04085; P01127; Q9NRA1; P31947; P62258; Q9NRA1-1; A8T7D5; P05067; Q8IY26 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cell projection; Chemotaxis; Developmental protein; Disease variant; Disulfide bond; Glycoprotein; Golgi apparatus; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 39294.8 Length 345 Aromaticity 0.11 Instability index 46.74 Isoelectric point 6.6 Charge (pH=7) -1.38 2D Binding mode Binding energy (Kcal/mol) -7.21  Molscript Map 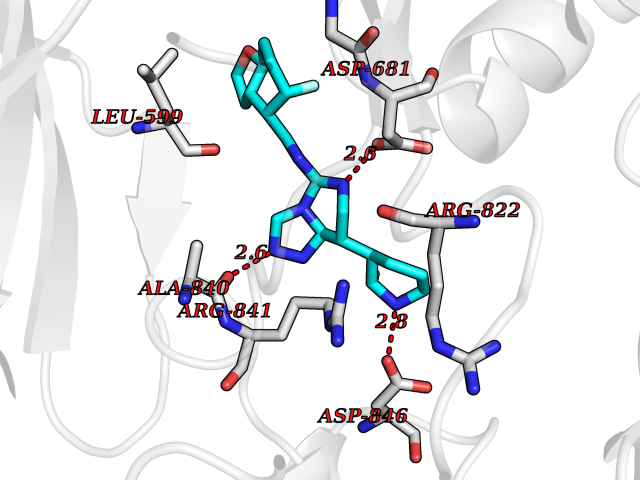 Pymol Map 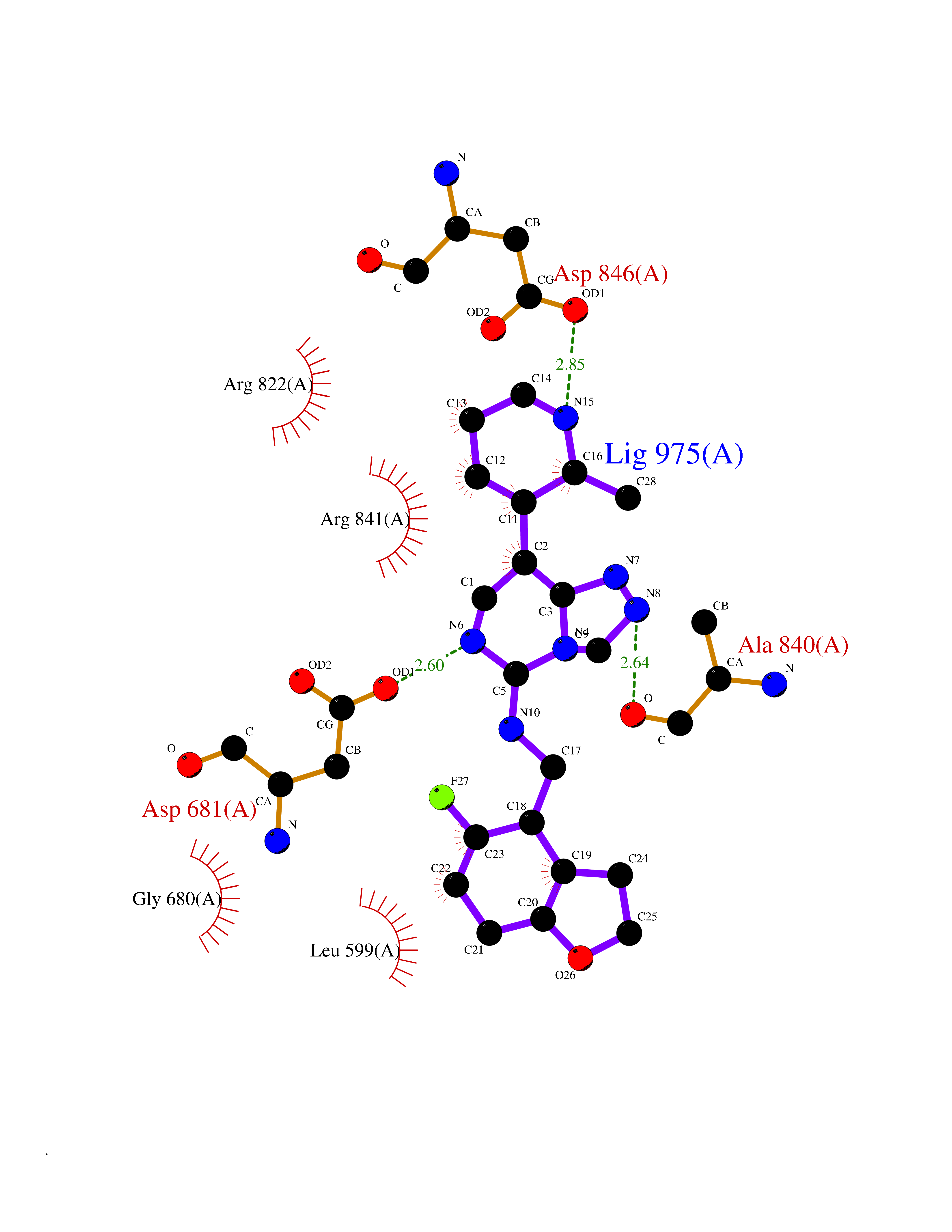 Ligplot Map 3D Binding mode Sequence RYEIRWRVIESISPDGHEYIYVDPMQLPYDSRWEFPRDGLVLGRVLGSGAFGKVVEGTAYGLSRSQPVMKVAVKMLKPTARSSEKQALMSELKIMTHLGPHLNIVNLLGACTKSGPIYIITEYCFYGDLVNYLHKNRDSFLSHSMLDSEVKNLLSDDNSEGLTLLDLLSFTYQVARGMEFLASKNCVHRDLAARNVLLAQGKIVKICDFGLARDIMHDSNYVSKGSTFLPVKWMAPESIFDNLYTTLSDVWSYGILLWEIFSLGGTPYPGMMVDSTFYNKIKSGYRMAKPDHATSEVYEIMVKCWNSEPEKRPSFYHLSEIVENLLPGQYKKSYEKIHLDFLKSD Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Eukaryotic translation initiation factor 2-alpha kinase 4 (EIF2AK4) | 7QQ6 | 7.96 | |
Target general information Gen name EIF2AK4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms eIF-2-alpha kinase GCN2; KIAA1338; GCN2-like protein; GCN2 Protein family Protein kinase superfamily, Ser/Thr protein kinase family, GCN2 subfamily Biochemical class NA Function Metabolic-stress sensing protein kinase that phosphorylates the alpha subunit of eukaryotic translation initiation factor 2 (eIF-2-alpha/EIF2S1) on 'Ser-52' in response to low amino acid availability. Plays a role as an activator of the integrated stress response (ISR) required for adapatation to amino acid starvation. Converts phosphorylated eIF-2-alpha/EIF2S1 either to a competitive inhibitor of the translation initiation factor eIF-2B, leading to a global protein synthesis repression, and thus to a reduced overall utilization of amino acids, or to a translational initiation activation of specific mRNAs, such as the transcriptional activator ATF4, and hence allowing ATF4-mediated reprogramming of amino acid biosynthetic gene expression to alleviate nutrient depletion. Binds uncharged tRNAs (By similarity). Involved in cell cycle arrest by promoting cyclin D1 mRNA translation repression after the unfolded protein response pathway (UPR) activation or cell cycle inhibitor CDKN1A/p21 mRNA translation activation in response to amino acid deprivation. Plays a role in the consolidation of synaptic plasticity, learning as well as formation of long-term memory. Plays a role in neurite outgrowth inhibition. Plays a proapoptotic role in response to glucose deprivation. Promotes global cellular protein synthesis repression in response to UV irradiation independently of the stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) and p38 MAPK signaling pathways (By similarity). Plays a role in the antiviral response against alphavirus infection; impairs early viral mRNA translation of the incoming genomic virus RNA, thus preventing alphavirus replication (By similarity). Related diseases Pulmonary venoocclusive disease 2, autosomal recessive (PVOD2) [MIM:234810]: A disease characterized by widespread fibrous obstruction and intimal thickening of septal veins and preseptal venules, a low diffusing capacity for carbon monoxide, occult alveolar hemorrhage, and nodular ground-glass opacities, septal lines and lymph node enlargement showed by high-resolution computed tomography of the chest. It is frequently associated with pulmonary capillary dilatation and proliferation, and is a rare and devastating cause of pulmonary hypertension. {ECO:0000269|PubMed:24135949, ECO:0000269|PubMed:24292273}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with NA EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; Activation of host autophagy by virus; Activator; Adaptive immunity; Alternative splicing; Antiviral defense; ATP-binding; Cell cycle; Coiled coil; Cytoplasm; Disease variant; Growth arrest; Host-virus interaction; Immunity; Kinase; Neurogenesis; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; RNA-binding; Serine/threonine-protein kinase; Stress response; Transferase; Translation regulation; tRNA-binding Protein physicochemical properties Chain ID A Molecular weight (Da) 31090.3 Length 269 Aromaticity 0.1 Instability index 42.78 Isoelectric point 7.75 Charge (pH=7) 1.43 2D Binding mode Binding energy (Kcal/mol) -8.9 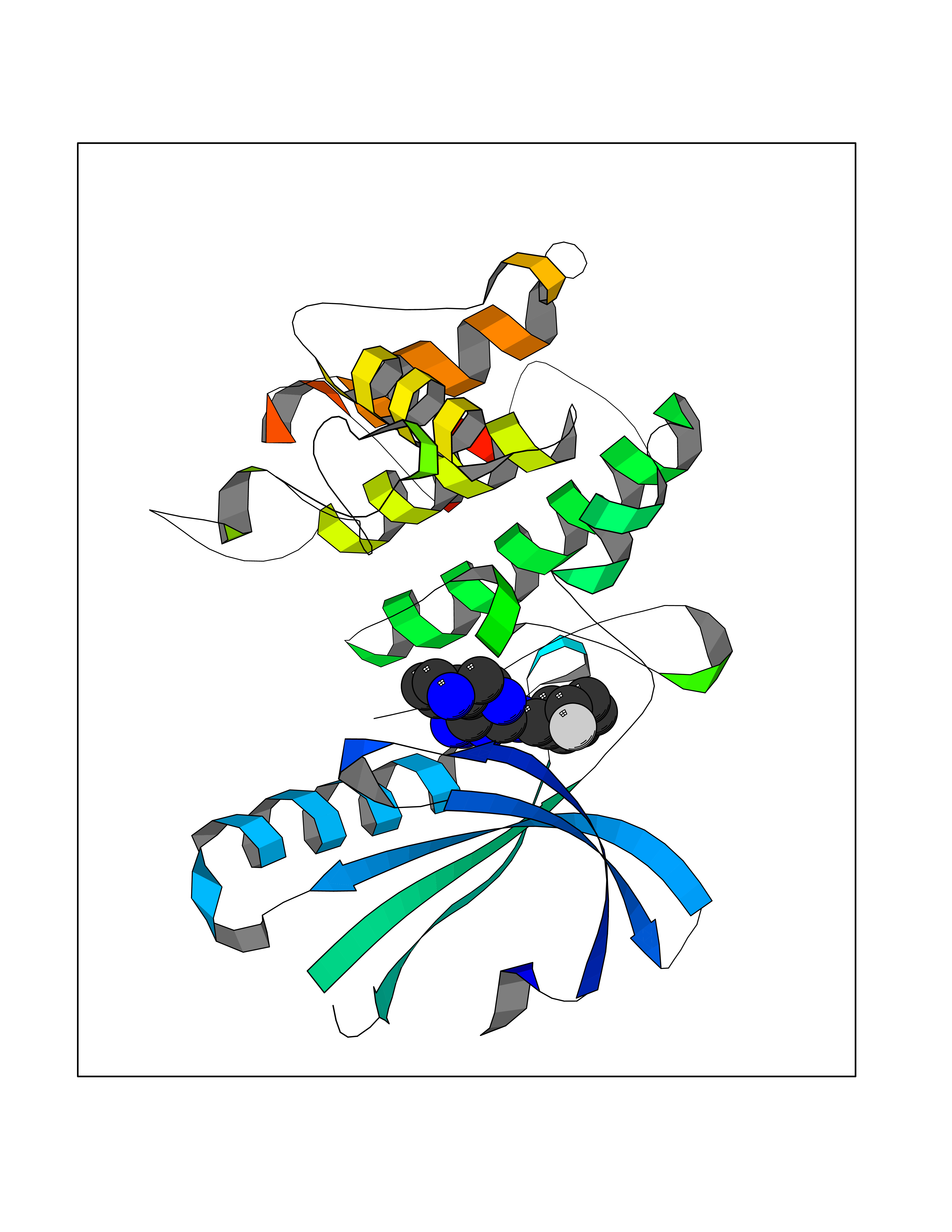 Molscript Map 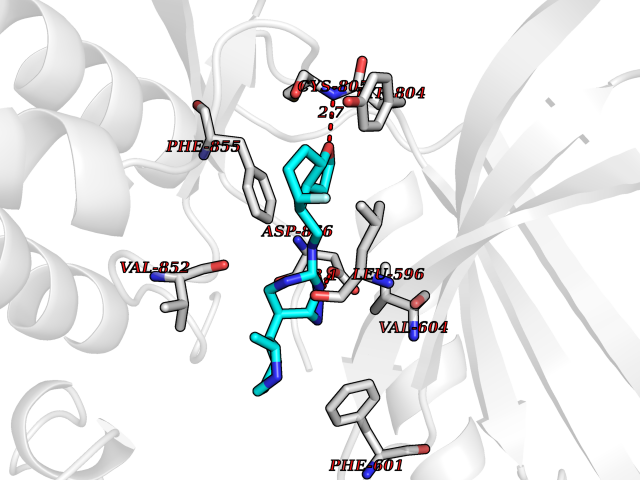 Pymol Map  Ligplot Map 3D Binding mode Sequence SRYFIEFEELQLLGKGAFGAVIKVQNKLDGCCYAVKRIPINPASRQFRRIKGEVTLLSRLHHENIVRYYNAWIERHERPVHYLYIQMEYCEKSTLRDTIDQGLYRDTVRLWRLFREILDGLAYIHEKGMIHRNLKPVNIFLDSDDHVKIGDFGLIKSDPSGHLTGMVGTALYVSPEVQGSTKSAYNQKVDLFSLGIIFFEMSYHPMVTASERIFVLNQLRDPTSPKFPEDFDDGEHAKQKSVISWLLNHDPAKRPTATELLKSELLPPP Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Mycobacterium Isocitrate lyase (MycB icl) | 1F8M | 7.95 | |
Target general information Gen name MycB icl Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID Synonyms Isocitratase; Isocitrase; ICL Protein family Isocitrate lyase/PEP mutase superfamily, Isocitrate lyase family Biochemical class Carbon-carbon lyase Function Catalyzes the formation of succinate and glyoxylate from isocitrate, a key step of the glyoxylate cycle. May be involved in the assimilation of one-carbon compounds via the isocitrate lyase- positive serine pathway. Related diseases Intellectual developmental disorder, autosomal recessive 80, with variant lissencephaly (MRT80) [MIM:620653]: An autosomal recessive disorder characterized by global developmental delay, mildly to moderately impaired intellectual development, attention deficit-hyperactivity disorder, hypotonia, seizure, poor social skills, and autistic traits. Brain imaging shows fronto-temporal lissencephaly and pachygyria. {ECO:0000269|PubMed:37880421}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04343 Interacts with NA EC number EC 4.1.3.1 Uniprot keywords 3D-structure; Glyoxylate bypass; Isopeptide bond; Lyase; Magnesium; Manganese; Metal-binding; Reference proteome; Tricarboxylic acid cycle; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 93759.5 Length 854 Aromaticity 0.09 Instability index 28.02 Isoelectric point 4.98 Charge (pH=7) -34.53 2D Binding mode Binding energy (Kcal/mol) -7.51  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ASVVGTPKSAEQIQQEWDTNPRWKDVTRTYSAEDVVALQGSVVEEHTLARRGAEVLWEQLHDLEWVNALGALTGNMAVQQVRAGLKAIYLSGWQVAGDANLSGHTYPDQSLYPANSVPQVVRRINNALQRADQIAKIEGDTSVENWLAPIVADGEAGFGGALNVYELQKALIAAGVAGSHWEDQLASEKKCGHLGGKVLIPTQQHIRTLTSARLAADVADVPTVVIARTDAEAATLITSDVDERDQPFITGERTREGFYRTKNGIEPCIARAKAYAPFADLIWMETGTPDLEAARQFSEAVKAEYPDQMLAYNCSPSFNWKKHLDDATIAKFQKELAAMGFKFQFITLAGFHALNYSMFDLAYGYAQNQMSAYVELQEREFAAEERGYTATKHQREVGAGYFDRIATTVDPNSSTTALTGSTEEGQFASVVGTPKSAEQIQQEWDTNPRWKDVTRTYSAEDVVALQGSVVEEHTLARRGAEVLWEQLHDLEWVNALGALTGNMAVQQVRAGLKAIYLSGWQVAGDANLSGHTYPDQSLYPANSVPQVVRRINNALQRADQIAKIEGDTSVENWLAPIVADGEAGFGGALNVYELQKALIAAGVAGSHWEDQLASEKKCGHLGGKVLIPTQQHIRTLTSARLAADVADVPTVVIARTDAEAATLITSDVDERDQPFITGERTREGFYRTKNGIEPCIARAKAYAPFADLIWMETGTPDLEAARQFSEAVKAEYPDQMLAYNCSPSFNWKKHLDDATIAKFQKELAAMGFKFQFITLAGFHALNYSMFDLAYGYAQNQMSAYVELQEREFAAEERGYTATKHQREVGAGYFDRIATTVDPNSSTTALTGSTEEGQF Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Corynebacterium Pup-protein ligase (Cory pafA) | 4BJR | 7.95 | |
Target general information Gen name Cory pafA Organism Corynebacterium glutamicum (strain ATCC 13032 / DSM 20300 / JCM 1318 / BCRC 11384 / CCUG 27702 / LMG 3730 / NBRC 12168 / NCIMB 10025 / NRRL B-2784 / 534) Uniprot ID TTD ID Synonyms Pup-conjugating enzyme; Pup--protein ligase; Proteasome accessory factor A Protein family Pup ligase/Pup deamidase family, Pup-conjugating enzyme subfamily Biochemical class NA Function Catalyzes the covalent attachment of the prokaryotic ubiquitin-like protein modifier Pup to the proteasomal substrate proteins, thereby targeting them for proteasomal degradation. This tagging system is termed pupylation. The ligation reaction involves the side-chain carboxylate of the C-terminal glutamate of Pup and the side-chain amino group of a substrate lysine. Related diseases Anemia, non-spherocytic hemolytic, due to G6PD deficiency (NSHA) [MIM:300908]: A disease characterized by G6PD deficiency, acute hemolytic anemia, fatigue, back pain, and jaundice. In most patients, the disease is triggered by an exogenous agent, such as some drugs, food, or infection. Increased unconjugated bilirubin, lactate dehydrogenase, and reticulocytosis are markers of the disorder. Although G6PD deficiency can be life-threatening, most patients are asymptomatic throughout their life. {ECO:0000269|PubMed:12524354, ECO:0000269|PubMed:1303180, ECO:0000269|PubMed:1303182, ECO:0000269|PubMed:1536798, ECO:0000269|PubMed:1611091, ECO:0000269|PubMed:1889820, ECO:0000269|PubMed:1945893, ECO:0000269|PubMed:20007901, ECO:0000269|PubMed:26479991, ECO:0000269|PubMed:2836867, ECO:0000269|PubMed:2912069, ECO:0000269|PubMed:30988594, ECO:0000269|PubMed:38066190, ECO:0000269|PubMed:7858267, ECO:0000269|PubMed:7959695, ECO:0000269|PubMed:8193373, ECO:0000269|PubMed:8490627, ECO:0000269|PubMed:8533762, ECO:0000269|PubMed:8733135, ECO:0000269|PubMed:9452072}. The disease is caused by variants affecting the gene represented in this entry. Deficiency of G6PD is associated with hemolytic anemia in two different situations. First, in areas in which malaria has been endemic, G6PD-deficiency alleles have reached high frequencies (1% to 50%) and deficient individuals, though essentially asymptomatic in the steady state, have a high risk of acute hemolytic attacks. Secondly, sporadic cases of G6PD deficiency occur at a very low frequencies, and they usually present a more severe phenotype. Several types of NSHA are recognized. Class-I variants are associated with severe NSHA; class-II have an activity <10% of normal; class-III have an activity of 10% to 60% of normal; class-IV have near normal activity. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; ATP-binding; Ligase; Magnesium; Metal-binding; Nucleotide-binding; Reference proteome; Ubl conjugation pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 110572 Length 994 Aromaticity 0.07 Instability index 42.33 Isoelectric point 5.26 Charge (pH=7) -34.19 2D Binding mode Binding energy (Kcal/mol) -8.23 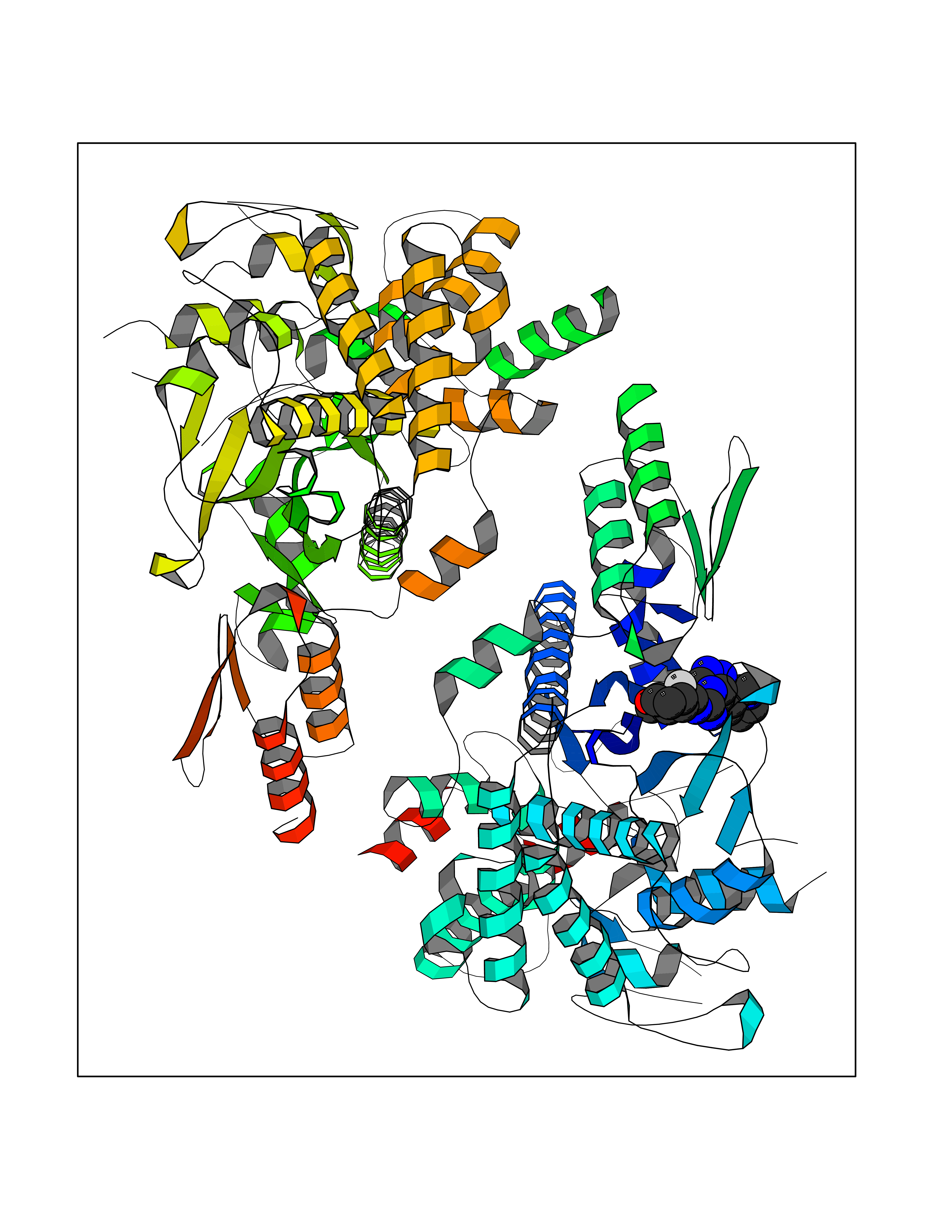 Molscript Map 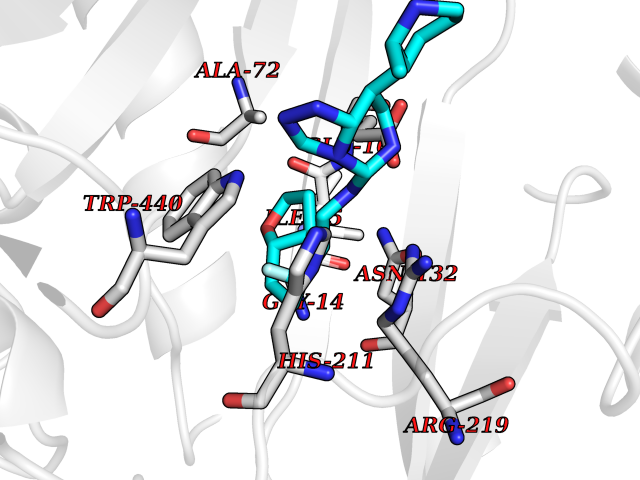 Pymol Map 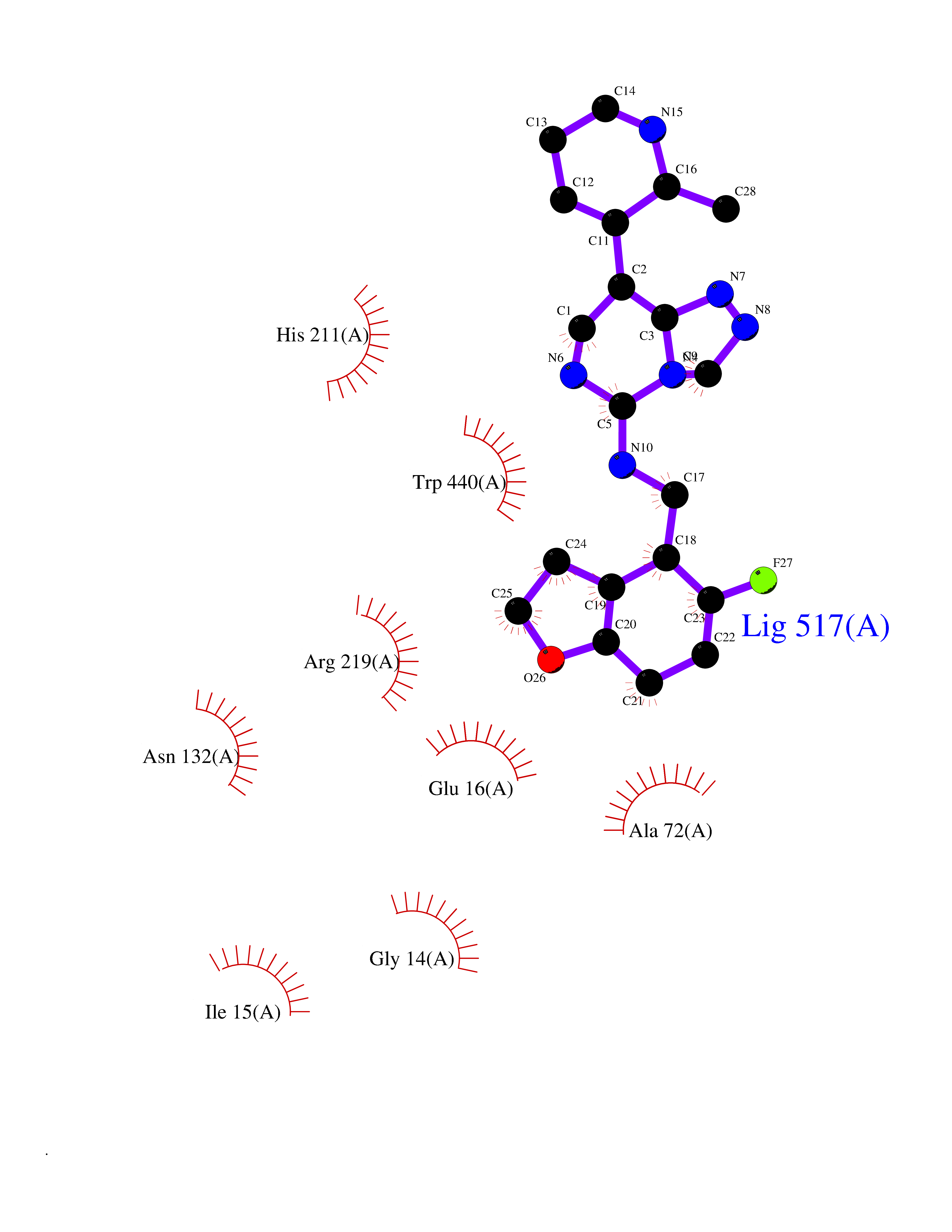 Ligplot Map 3D Binding mode Sequence TVESALTRRIMGIETEYGLTFVDGDSKKLRPDEIARRMFRPIVEKYSSSNIFIPNGSRLYLNVGSHPEYATAECDNLTQLINFEKAGDVIADRMAVDAEESLAKEDIAGQVYLFKNNVDSVGNSYGCHENYLVGRSMPLKALGKRLMPFLITRQLICGAGRIHHPNPSFPLGYCISQRSDHVWEGVSSASRPIINTRDEPHADSHSYRRLHVIVGDANMAEPSIALKVGSTLLVLEMIEADFGLPSLELANDIASIREISRDATGSTLLSLKDGTTMTALQIQQVVFEHASKWLEQRPEPEFSGTSNTEMARVLDLWGRMLKAIESGDFSEVDTEIDWVIKKKLIDRFIQRGNLGLDDPKLAQVDLTYHDIRPGRGLFSVLQSRGMIKRWTTDEAILAAVDTAPDTTRAHLRGRILKAADTLGVPVTVDWMRHKVNRPEPQSVELGDPFSAVNSEVDQLIEYMTVHAGSASGTSLLDEIDGLLENNAEEFVRSYVQKGGETVESALTRRIMGIETEYGLTFVDGDSKKLRPDEIARRMFRPIVEKYSSSNIFIPNGSRLYLNVGSHPEYATAECDNLTQLINFEKAGDVIADRMAVDAEESLAKEDIAGQVYLFKNNVDSVGNSYGCHENYLVGRSMPLKALGKRLMPFLITRQLICGAGRIHHPNPSFPLGYCISQRSDHVWEGVSSASRPIINTRDEPHADSHSYRRLHVIVGDANMAEPSIALKVGSTLLVLEMIEADFGLPSLELANDIASIREISRDATGSTLLSLKDGTTMTALQIQQVVFEHASKWLEQRPEPEFSGTSNTEMARVLDLWGRMLKAIESGDFSEVDTEIDWVIKKKLIDRFIQRGNLGLDDPKLAQVDLTYHDIRPGRGLFSVLQSRGMIKRWTTDEAILAAVDTAPDTTRAHLRGRILKAADTLGVPVTVDWMRHKVNRPEPQSVELGDPFSAVNSEVDQLIEYMTVHASLLDEIDGLLENNAEEFVRSYVQKGGE Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Pseudomonas Phosphomannomutase/phosphoglucomutase (Pseudo algC) | 1P5D | 7.95 | |
Target general information Gen name Pseudo algC Organism Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Uniprot ID TTD ID Synonyms algC; PMM/PGM; PMM / PGM Protein family Phosphohexose mutase family Biochemical class Intramolecular transferases Function Highly reversible phosphoryltransferase. The phosphomannomutase activity produces a precursor for alginate polymerization, the alginate layer causes a mucoid phenotype and provides a protective barrier against host immune defenses and antibiotics. Also involved in core lipopolysaccaride (LPS) biosynthesis due to its phosphoglucomutase activity. Essential for rhamnolipid production, an exoproduct correlated with pathogenicity (PubMed:10481091). Required for biofilm production. The reaction proceeds via 2 processive phosphoryl transferase reactions; first from enzyme-phospho-Ser-108 to the substrate (generatinga bisphosphorylated substrate intermediate and a dephosphorylated enzyme), a 180 degree rotation of the intermediate (probably aided by movement of domain 4), and subsequent transfer of phosphate back to the enzyme (PubMed:11716469, PubMed:16880541, PubMed:16595672, PubMed:22242625). Related diseases Intellectual developmental disorder, X-linked, syndromic, Claes-Jensen type (MRXSCJ) [MIM:300534]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRXSCJ patients manifest intellectual disability associated with variable features such as slowly progressive spastic paraplegia, seizures, facial dysmorphism. {ECO:0000269|PubMed:15586325, ECO:0000269|PubMed:16538222, ECO:0000269|PubMed:16541399, ECO:0000269|PubMed:17320160, ECO:0000269|PubMed:17468742, ECO:0000269|PubMed:23356856, ECO:0000269|PubMed:25666439}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02007; DB02843; DB02900; DB02867; DB04522 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alginate biosynthesis; Direct protein sequencing; Isomerase; Lipopolysaccharide biosynthesis; Magnesium; Metal-binding; Multifunctional enzyme; Phosphoprotein; Reference proteome; Virulence Protein physicochemical properties Chain ID X Molecular weight (Da) 49419.9 Length 455 Aromaticity 0.07 Instability index 28.92 Isoelectric point 5.11 Charge (pH=7) -12.7 2D Binding mode Binding energy (Kcal/mol) -7.88 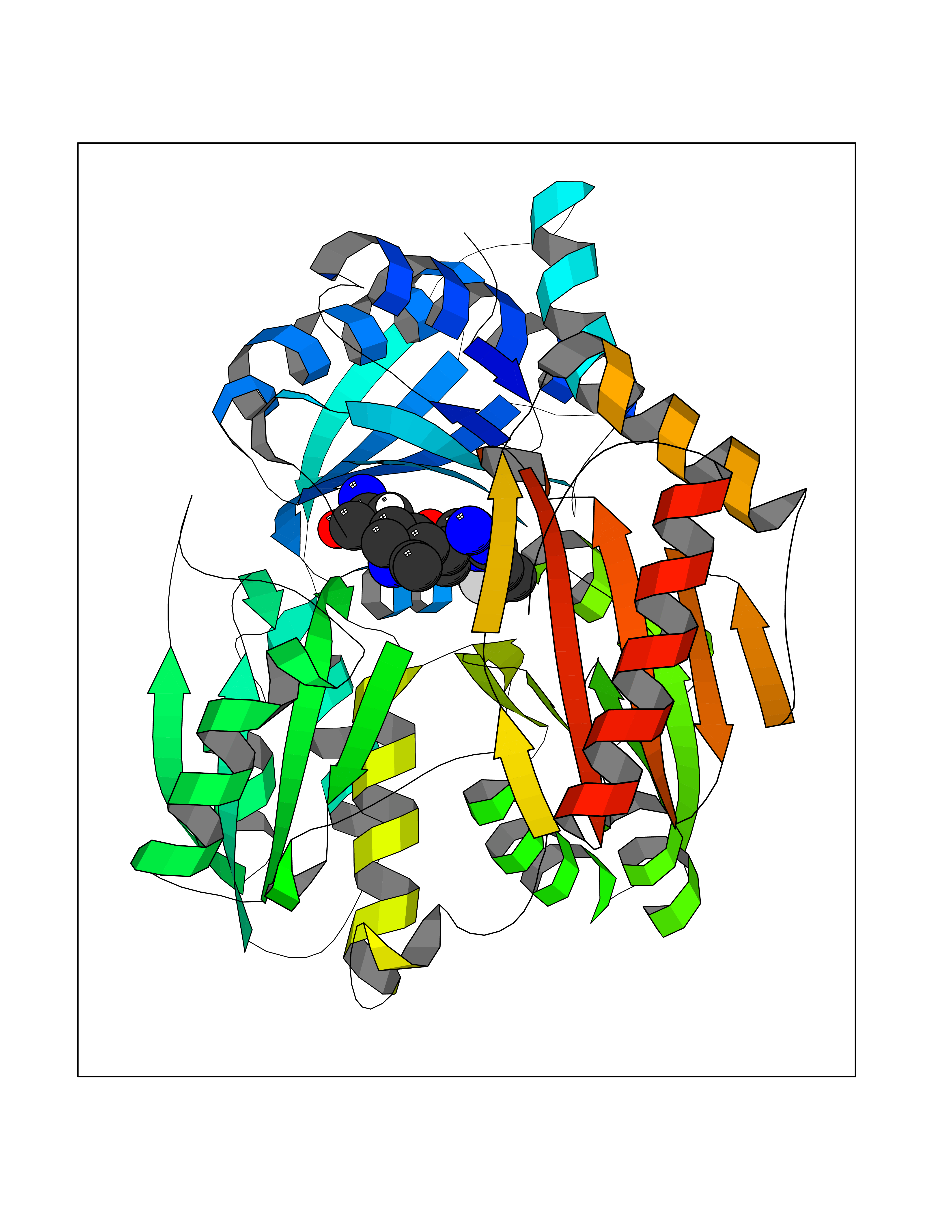 Molscript Map 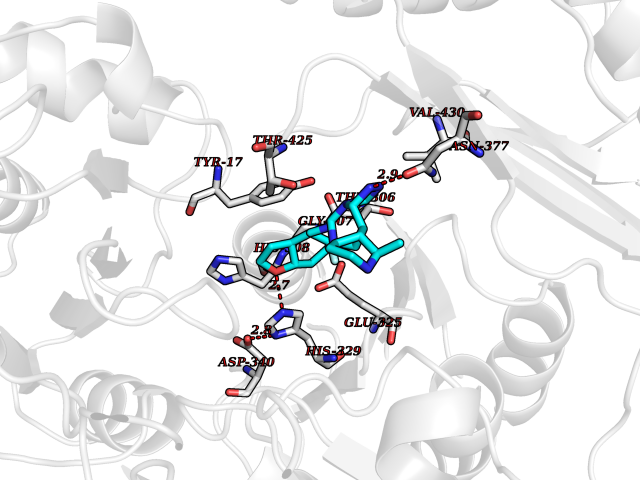 Pymol Map 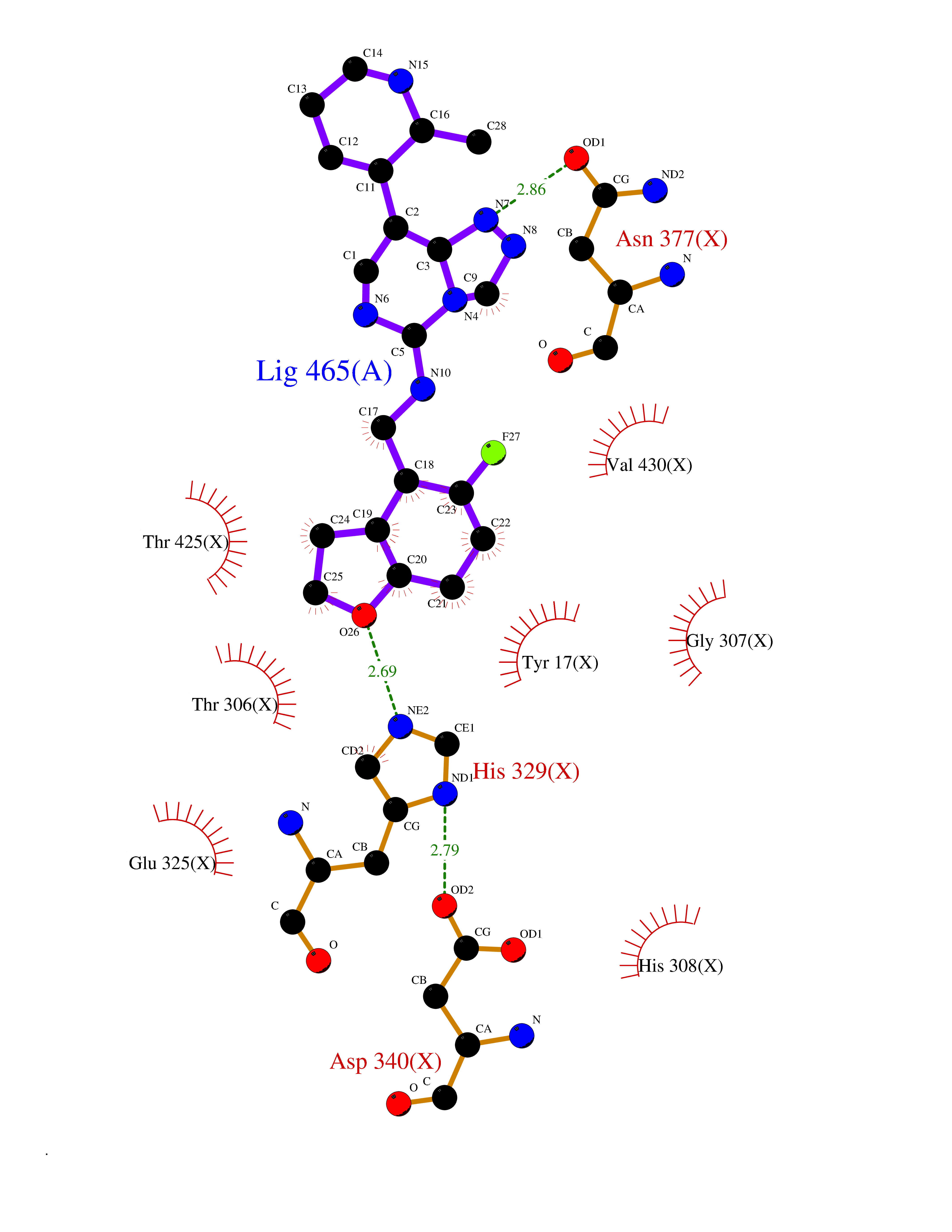 Ligplot Map 3D Binding mode Sequence LPASIFRAYDIRGVVGDTLTAETAYWIGRAIGSESLARGEPCVAVGRDGRLSGPELVKQLIQGLVDCGCQVSDVGMVPTPVLYYAANVLEGKSGVMLTGXHNPPDYNGFKIVVAGETLANEQIQALRERIEKNDLASGVGSVEQVDILPRYFKQIRDDIAMAKPMKVVVDCGNGVAGVIAPQLIEALGCSVIPLYCEVDGNFPNHHPDPGKPENLKDLIAKVKAENADLGLAFDGDGDRVGVVTNTGTIIYPDRLLMLFAKDVVSRNPGADIIFDVKCTRRLIALISGYGGRPVMWKTGHSLIKKKMKETGALLAGEMSGHVFFKERWFGFDDGIYSAARLLEILSQDQRDSEHVFSAFPSDISTPEINITVTEDSKFAIIEALQRDAQWGEGNITTLDGVRVDYPKGWGLVRASNTTPVLVLRFEADTEEELERIKTVFRNQLKAVDSSLPVPF Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Glutamate carboxypeptidase III (NAALAD2) | 3FED | 7.95 | |
Target general information Gen name NAALAD2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NAALADase II; NAALAD2 Protein family Peptidase M28 family, M28B subfamily Biochemical class Peptidase Function Has N-acetylated-alpha-linked-acidic dipeptidase (NAALADase) activity. Also exhibits a dipeptidyl-peptidase IV type activity. Inactivate the peptide neurotransmitter N- acetylaspartylglutamate. Related diseases Dystonia 1, torsion, autosomal dominant (DYT1) [MIM:128100]: A primary torsion dystonia, and the most common and severe form. Dystonia is defined by the presence of sustained involuntary muscle contractions, often leading to abnormal postures. Dystonia type 1 is characterized by involuntary, repetitive, sustained muscle contractions or postures involving one or more sites of the body, in the absence of other neurological symptoms. Typically, symptoms develop first in an arm or leg in middle to late childhood and progress in approximately 30% of patients to other body regions (generalized dystonia) within about five years. 'Torsion' refers to the twisting nature of body movements observed in DYT1, often affecting the trunk. Distribution and severity of symptoms vary widely between affected individuals, ranging from mild focal dystonia to severe generalized dystonia, even within families. {ECO:0000269|PubMed:14970196, ECO:0000269|PubMed:15505207, ECO:0000269|PubMed:16361107, ECO:0000269|PubMed:17428918, ECO:0000269|PubMed:18167355, ECO:0000269|PubMed:18477710, ECO:0000269|PubMed:18827015, ECO:0000269|PubMed:19955557, ECO:0000269|PubMed:20169475, ECO:0000269|PubMed:21102408, ECO:0000269|PubMed:24930953, ECO:0000269|PubMed:27490483, ECO:0000269|PubMed:9288096}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Arthrogryposis multiplex congenita 5 (AMC5) [MIM:618947]: A form of arthrogryposis multiplex congenita, a developmental condition characterized by multiple joint contractures resulting from reduced or absent fetal movements. AMC5 is an autosomal recessive form characterized by severe congenital contractures, developmental delay, strabismus and tremor. {ECO:0000269|PubMed:28516161, ECO:0000269|PubMed:29053766, ECO:0000269|PubMed:30244176}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9UHD4; Q6NTF9-3; B2RUZ4; O76024 EC number EC 3.4.17.21 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Carboxypeptidase; Cell membrane; Dipeptidase; Glycoprotein; Hydrolase; Membrane; Metal-binding; Metalloprotease; Multifunctional enzyme; Protease; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 77761.6 Length 690 Aromaticity 0.12 Instability index 39.42 Isoelectric point 8.48 Charge (pH=7) 4.65 2D Binding mode Binding energy (Kcal/mol) -8.56 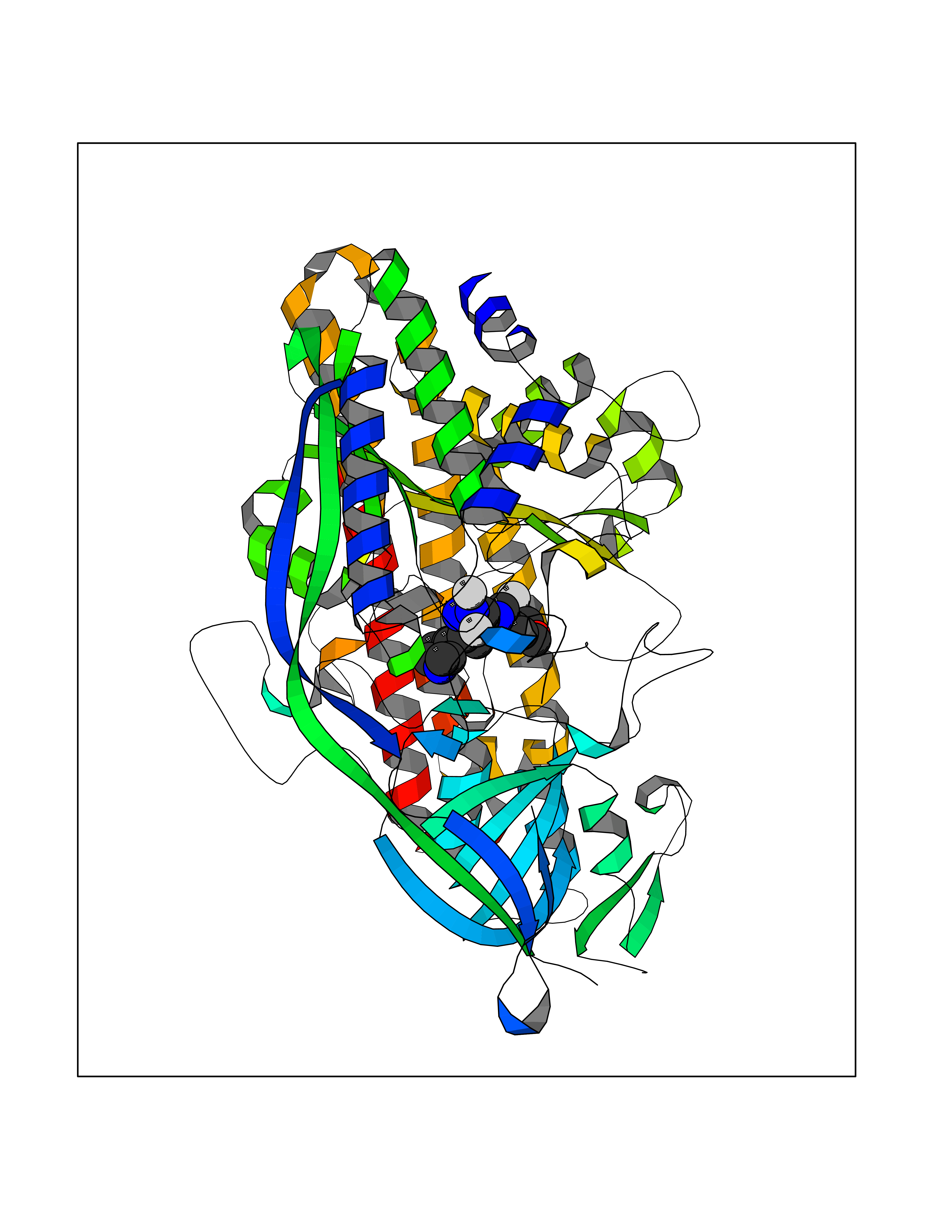 Molscript Map 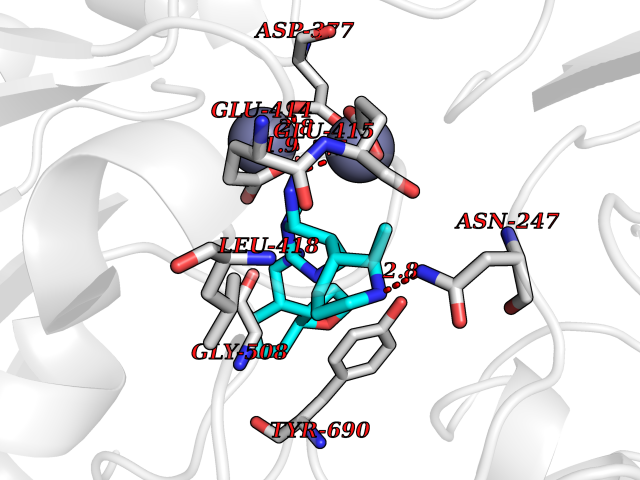 Pymol Map 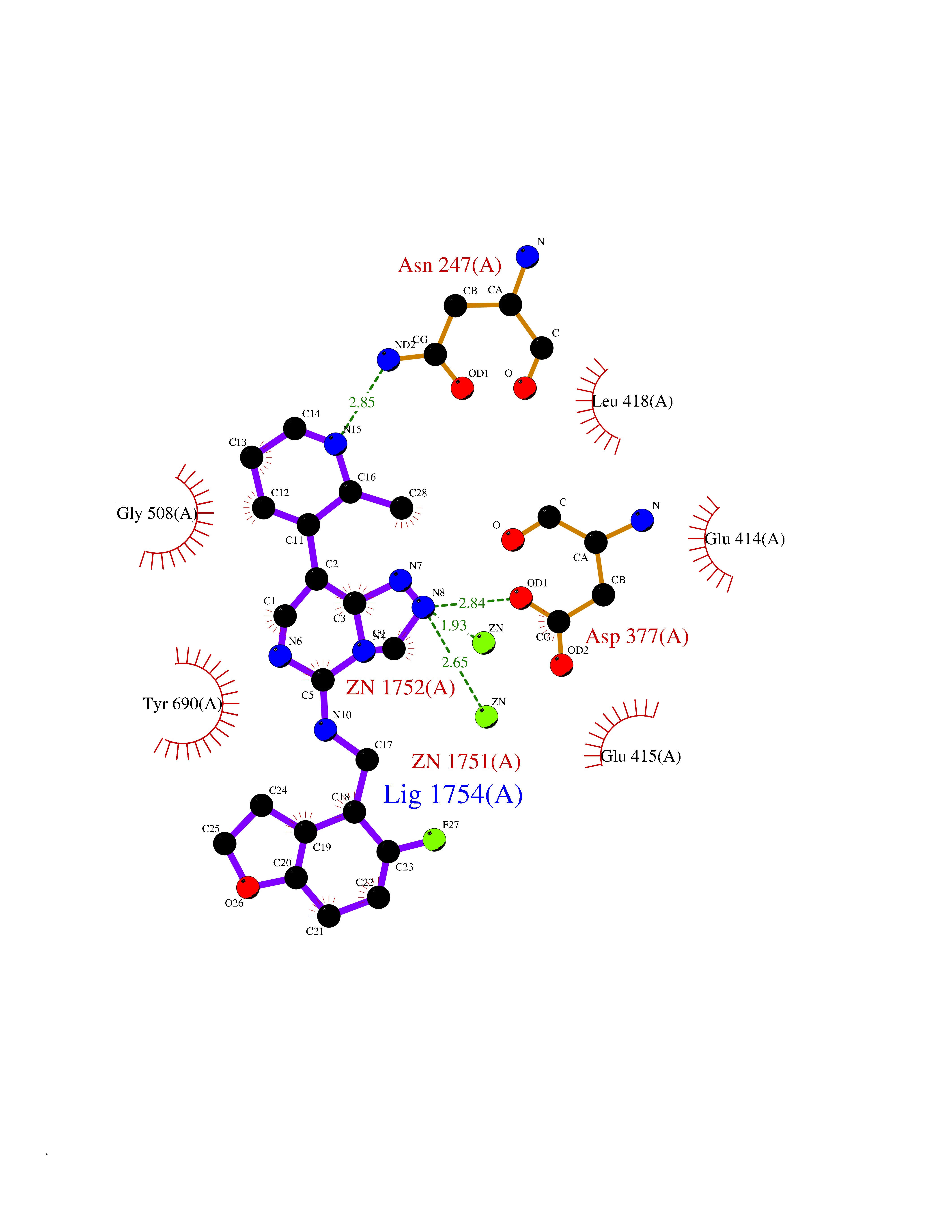 Ligplot Map 3D Binding mode Sequence SIRWKLVSEMKAENIKSFLRSFTKLPHLAGTEQNFLLAKKIQTQWKKFGLDSAKLVHYDVLLSYPNETNANYISIVDEHETEIFKTSPPPDGYENVTNIVPPYNAFSAQGMPEGDLVYVNYARTEDFFKLEREMGINCTGKIVIARYGKIFRGNKVKNAMLAGAIGIILYSDPADYFAPEVQPYPKGWNLPGTAAQRGNVLNLNGAGDPLTPGYPAKEYTFRLDVEEGVGIPRIPVHPIGYNDAEILLRYLGGIAPPDKSWKGALNVSYSIGPGFTGSSFRKVRMHVYNINKITRIYNVVGTIRGSVEPDRYVILGGHRDSWVFGAIDPTSGVAVLQEIARSFGKLMSKGWRPRRTIIFASWDAEEFGLLGSTEWAEENVKILQERSIAYINSDSSIEGNYTLRVDCTPLLYQLVYKLTKEIPSPDDGFESKSLYESWLEKDPSPENKNLPRINKLGSGSDFEAYFQRLGIASGRARYTKNKKTDKYSSYPVYHTIYETFELVEKFYDPTFKKQLSVAQLRGALVYELVDSKIIPFNIQDYAEALKNYAASIYNLSKKHDQQLTDHGVSFDSLFSAVKNFSEAASDFHKRLIQVDLNNPIAVRMMNDQLMLLERAFIDPLGLPGKLFYRHIIFAPSSHNKYAGESFPGIYDAIFDIENKANSRLAWKEVKKHISIAAFTIQAAAGTLKEV Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Fungal Isoleucyl t-RNA synthetase (Fung ILS1) | 7D5C | 7.94 | |
Target general information Gen name Fung ILS1 Organism Saccharomyces cerevisiae (strain ATCC 204508 / S288c) (Baker's yeast) Uniprot ID TTD ID Synonyms Fung Isoleucyl-tRNA synthetase; Fung Isoleucine--tRNA ligase, cytoplasmic; Fung IleRS Protein family Class-I aminoacyl-tRNA synthetase family Biochemical class Carbon-oxygen ligase Function Has aminoacyl-tRNA editing and isoleucine-tRNA ligase activity. Related diseases Intellectual developmental disorder with dysmorphic facies and ptosis (IDDDFP) [MIM:617333]: An autosomal dominant neurodevelopmental disorder characterized by delayed psychomotor development, intellectual disability, delayed language, and facial dysmorphisms, most notably ptosis. Additional features may include poor growth, hypotonia, and seizures. {ECO:0000269|PubMed:27939639, ECO:0000269|PubMed:27939640}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 6.1.1.5 Uniprot keywords 3D-structure; Aminoacyl-tRNA synthetase; ATP-binding; Cytoplasm; Direct protein sequencing; Isopeptide bond; Ligase; Nucleotide-binding; Phosphoprotein; Protein biosynthesis; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 105860 Length 919 Aromaticity 0.12 Instability index 37.01 Isoelectric point 5.73 Charge (pH=7) -14.6 2D Binding mode Binding energy (Kcal/mol) -9.33 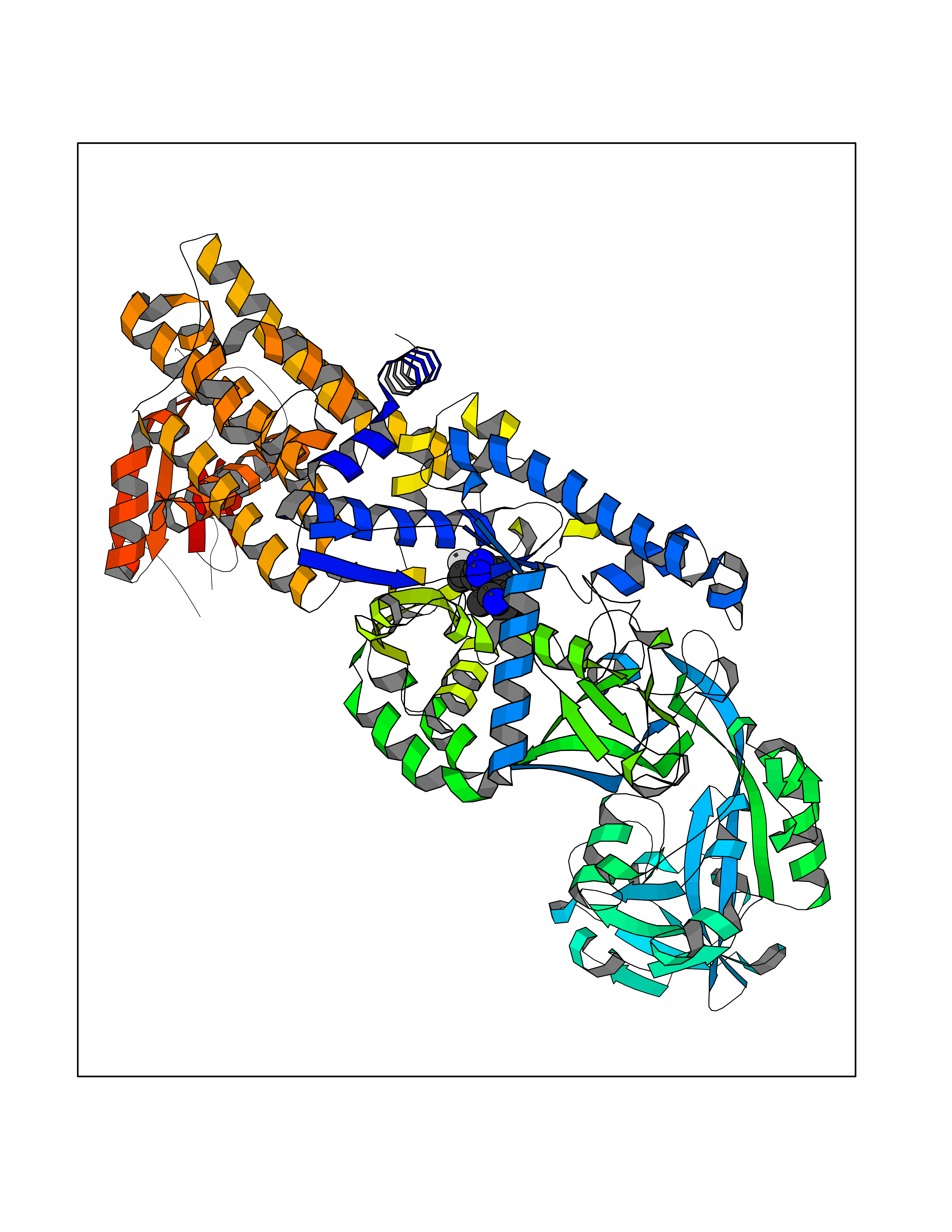 Molscript Map 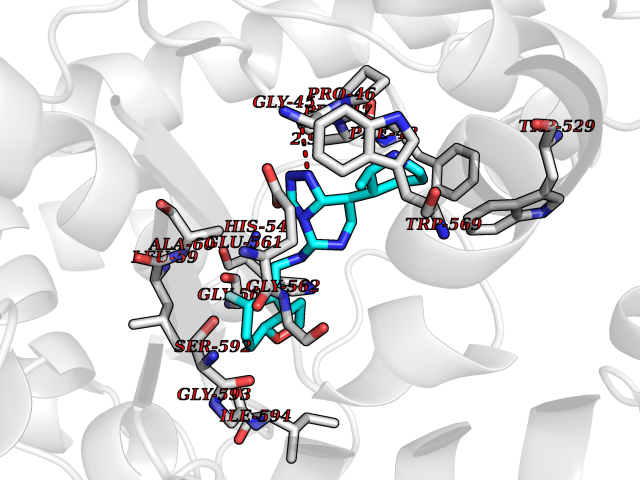 Pymol Map 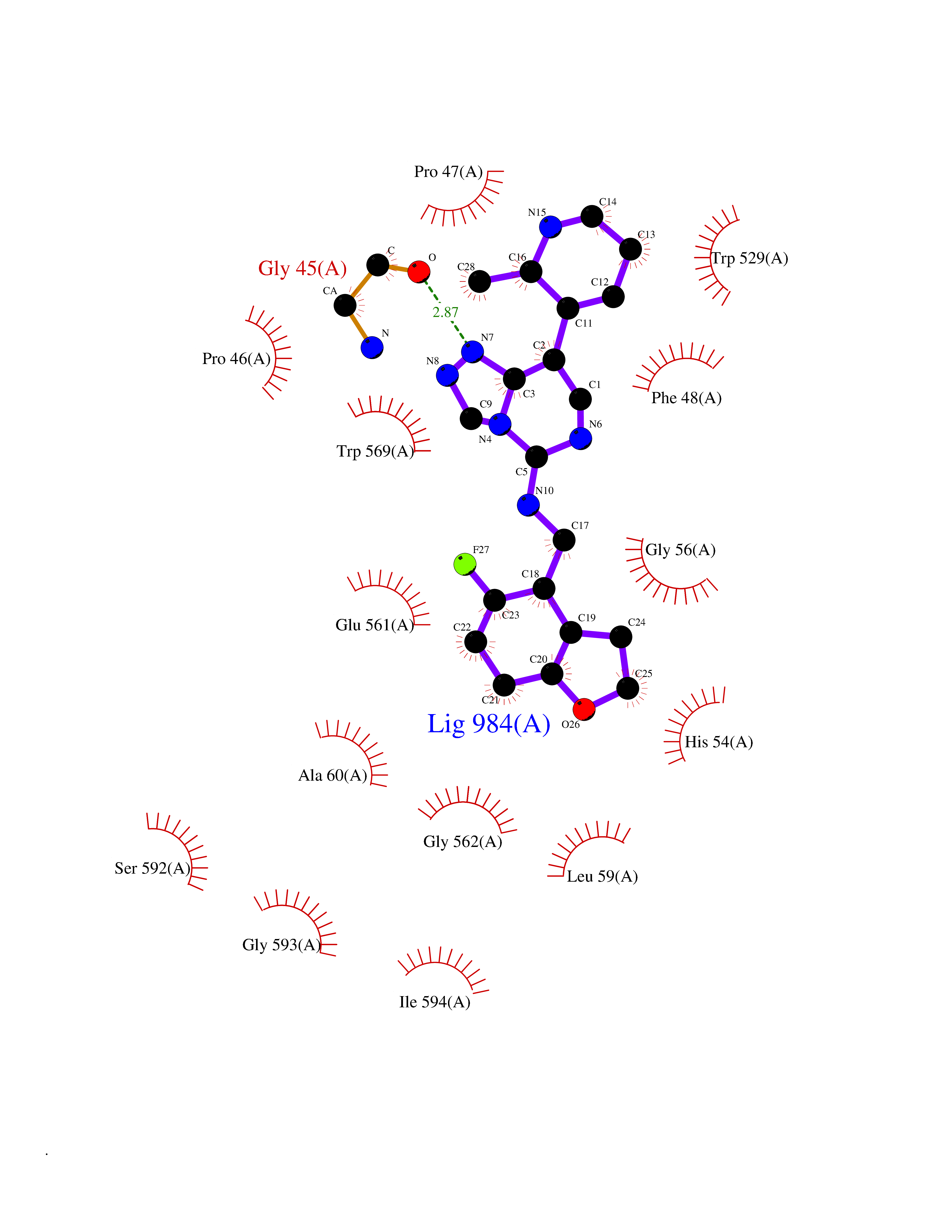 Ligplot Map 3D Binding mode Sequence FSFPKEEEKVLSLWDEIDAFHTSLELTKDKPEFSFFDGPPFATGTPHYGHILASTIKDIVPRYATMTGHHVERRFGWDTHGVPIEHIIDKKLGITGKDDVFKYGLENYNNECRSIVMTYASDWRKTIGRLGRWIDFDNDYKTMYPSFMESTWWAFKQLHEKGQVYRGFKVMPYSTGLTTPLSNFEAQQNYKDVNDPAVTIGFNVIGQEKTQLVAWTTTPWTLPSNLSLCVNADFEYVKIYDETRDRYFILLESLIKTLYKKPKNEKYKIVEKIKGSDLVGLKYEPLFPYFAEQFHETAFRVISDDYVTSDSGTGIVHNAPAFGEEDNAACLKNGVISEDSVLPNAIDDLGRFTKDVPDFEGVYVKDADKLIIKYLTNTGNLLLASQIRHSYPFCWRSDTPLLYRSVPAWFVRVKNIVPQMLDSVMKSHWVPNTIKEKRFANWIANARDWNVSRNRYWGTPIPLWVSDDFEEVVCVGSIKELEELTGVRNITDLHRDVIDKLTIPSKQGKGDLKRIEEVFDCWFESGSMPYASQHYPFENTEKFDERVPANFISEGLDQTRGWFYTLAVLGTHLFGSVPYKNVIVSGIVLAADGRKMSKSLKNYPDPSIVLNKYGADALRLYLINSPVLKAESLKFKEEGVKEVVSKVLLPWWNSFKFLDGQIALLKKMSNIDFQYDDSVKSDNVMDRWILASMQSLVQFIHEEMGQYKLYTVVPKLLNFIDELTNWYIRFNRRRLKGENGVEDCLKALNSLFDALFTFVRAMAPFTPFLSESIYLRLKEYIPEAVLAKYGKDGRSVHFLSYPVVKKEYFDEAIETAVSRMQSVIDLGRNIREKKTISLKTPLKTLVILHSDESYLKDVEALKNYIIEELNVRDVVITSDEAKYGVEYRGLPESAVQAGQETRTDQDVLIIMDTNIYSEL Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Endothelin receptor type B | 5GLI | 7.94 | |
Target general information Gen name EDNRB Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ETRB Protein family G-protein coupled receptor 1 family, Endothelin receptor subfamily, EDNRB sub-subfamily Biochemical class Signaling protein Function Endothelin receptor activity.Peptide hormone binding.Type 1 angiotensin receptor binding. Related diseases Waardenburg syndrome 4A (WS4A) [MIM:277580]: A disorder characterized by the association of Waardenburg features (depigmentation and deafness) with the absence of enteric ganglia in the distal part of the intestine (Hirschsprung disease). {ECO:0000269|PubMed:12189494, ECO:0000269|PubMed:8634719}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hirschsprung disease 2 (HSCR2) [MIM:600155]: A disorder of neural crest development characterized by absence of enteric ganglia along a variable length of the intestine. It is the most common cause of congenital intestinal obstruction. Early symptoms range from complete acute neonatal obstruction, characterized by vomiting, abdominal distention and failure to pass stool, to chronic constipation in the older child. {ECO:0000269|PubMed:11471546, ECO:0000269|PubMed:28236341, ECO:0000269|PubMed:8001158, ECO:0000269|PubMed:8630503, ECO:0000269|PubMed:8852660}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: ABCD syndrome (ABCDS) [MIM:600501]: An autosomal recessive syndrome characterized by albinism, black lock at temporal occipital region, bilateral deafness, aganglionosis of the large intestine and total absence of neurocytes and nerve fibers in the small intestine. {ECO:0000269|PubMed:11891690}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Heterozygous mutations in EDNRB may be responsible for Waardenburg syndrome 2, an autosomal dominant disorder characterized by sensorineural deafness and pigmentary disturbances. {ECO:0000269|PubMed:28236341}. Drugs (DrugBank ID) DB06403; DB00559; DB06460; DB06138; DB08932; DB06268; DB06558 Interacts with P05305 EC number NA Uniprot keywords 3D-structure; Albinism; Alternative splicing; Cell membrane; Deafness; Disease variant; Disulfide bond; G-protein coupled receptor; Glycoprotein; Hirschsprung disease; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix; Waardenburg syndrome Protein physicochemical properties Chain ID A Molecular weight (Da) 35251 Length 312 Aromaticity 0.12 Instability index 32.82 Isoelectric point 9.08 Charge (pH=7) 11.97 2D Binding mode Binding energy (Kcal/mol) -7.79 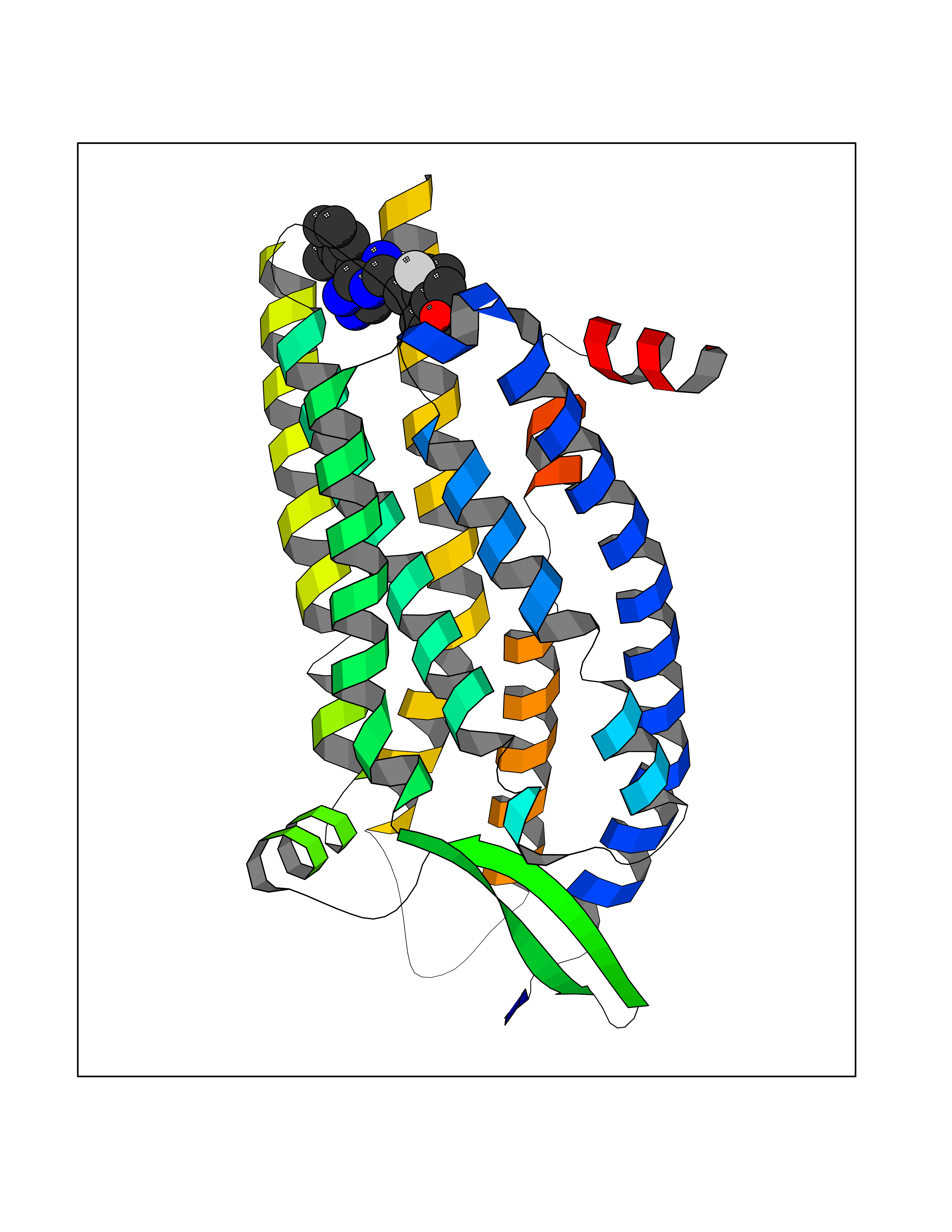 Molscript Map 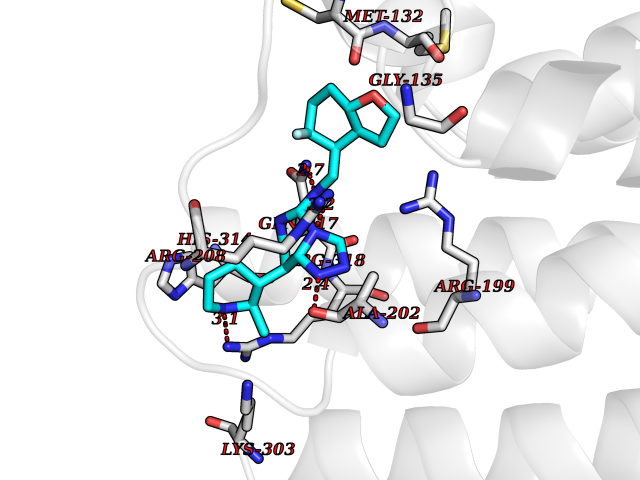 Pymol Map 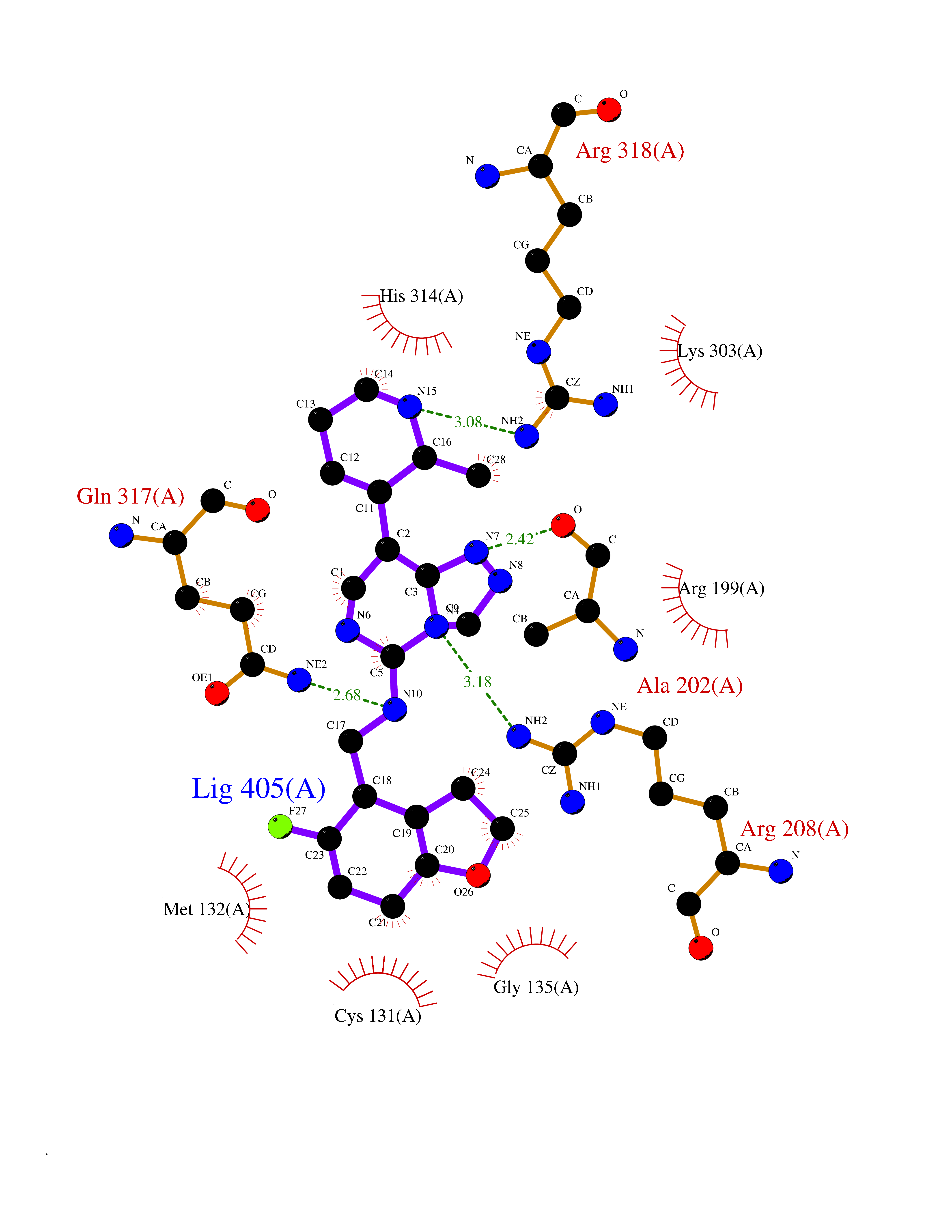 Ligplot Map 3D Binding mode Sequence ISPPPCQGPIEIKETFKYINTVVSCLVFVLGIIGNSTLLYIIYKNKCMRNGPNILIASLALGDLLHIVIAIPINVYKLLAEDWPFGAEMCKLVPFIQKASVGITVLSLCALSIDRYRAVASWSRIKGIGVPKWTAVEIVLIWVVSVVLAVPEAIGFDIITMDYKGSYLRICLLHPVQKTAFMQFYATAKDWWLFSFYFCLPLAITAFFYTLMTCEMLRKLNDHLKQRREVAKTVFCLVLVFALCWLPLHLARILKLTLYNQNDPNRCELLSFLLVLDYIGINMASLNSCANPIALYLVSKRFKNAFKSALCC Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Threonine--tRNA ligase, cytoplasmic | 4HWT | 7.93 | |
Target general information Gen name TARS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms TARS Protein family Class-II aminoacyl-tRNA synthetase family Biochemical class Ligase / ligase inhibitor Function ATP binding.Protein homodimerization activity.Threonine-tRNA ligase activity.TRNA binding. Related diseases Trichothiodystrophy 7, non-photosensitive (TTD7) [MIM:618546]: A form of trichothiodystrophy, a disease characterized by sulfur-deficient brittle hair and multisystem variable abnormalities. The spectrum of clinical features varies from mild disease with only hair involvement to severe disease with cutaneous, neurologic and profound developmental defects. Ichthyosis, intellectual and developmental disabilities, decreased fertility, abnormal characteristics at birth, ocular abnormalities, short stature, and infections are common manifestations. There are both photosensitive and non-photosensitive forms of the disorder. TTD7 patients do not manifest cutaneous photosensitivity. They have cysteine- and threonine-deficient hair with alternating light and dark 'tiger-tail' banding pattern observed under polarization microscopy. Inheritance pattern is autosomal recessive. {ECO:0000269|PubMed:31374204}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00156 Interacts with Q9BPX7; Q96CV9; O43704; A2RTX5 EC number 6.1.1.3 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminoacyl-tRNA synthetase; ATP-binding; Cytoplasm; Disease variant; Ligase; Nucleotide-binding; Phosphoprotein; Protein biosynthesis; Proteomics identification; Reference proteome; RNA-binding; tRNA-binding; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 33945.3 Length 290 Aromaticity 0.13 Instability index 41.57 Isoelectric point 6.29 Charge (pH=7) -3.28 2D Binding mode Binding energy (Kcal/mol) -8.15 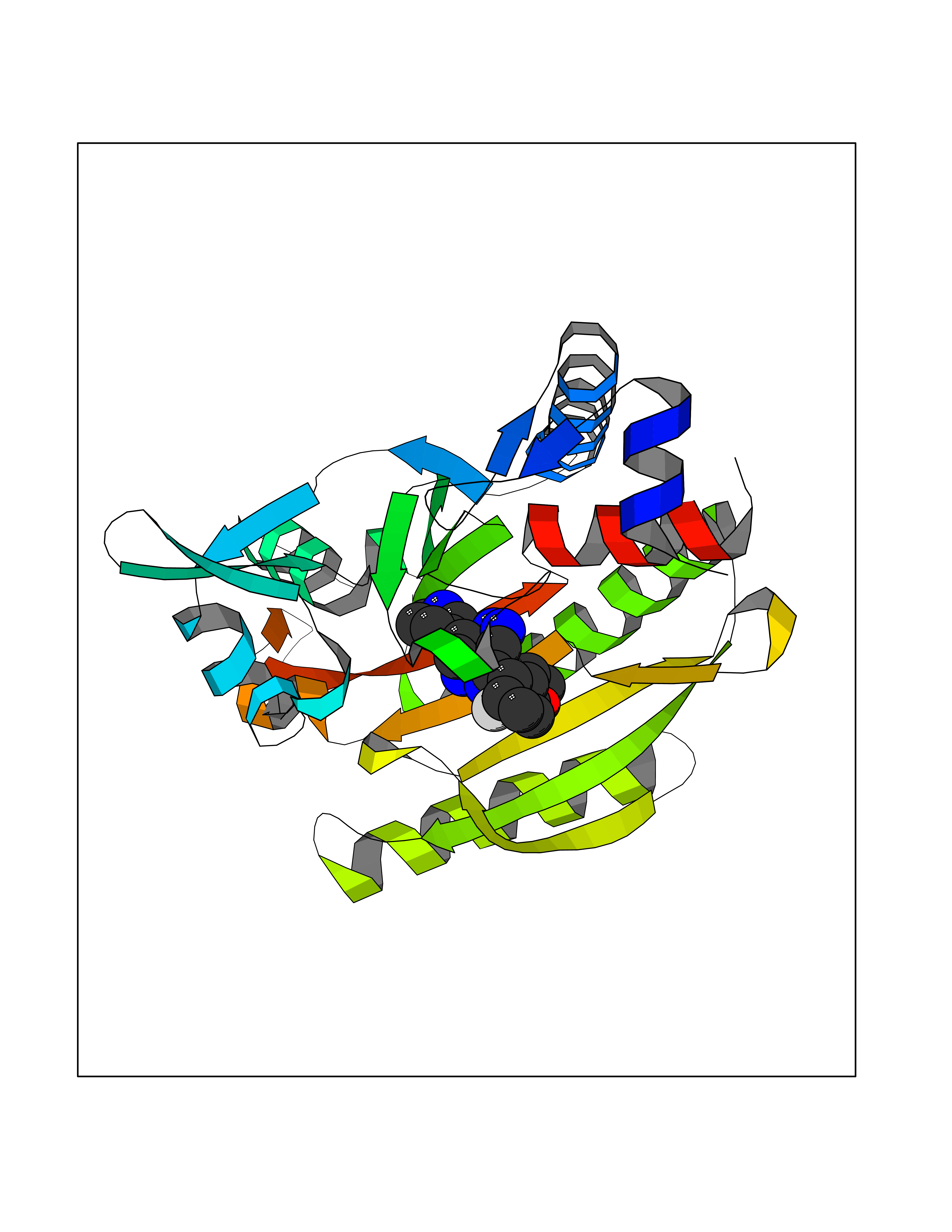 Molscript Map 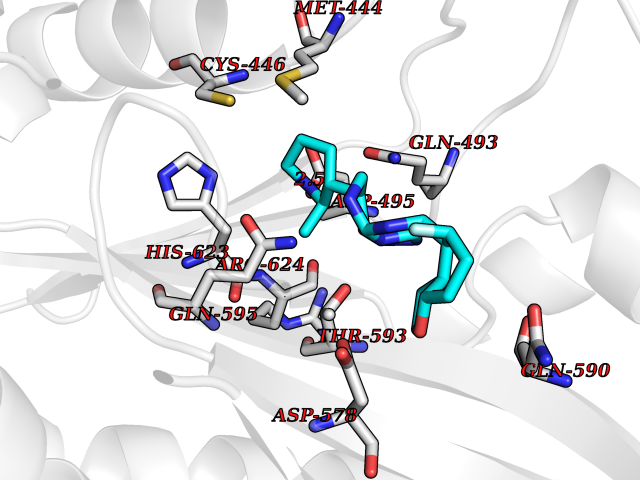 Pymol Map 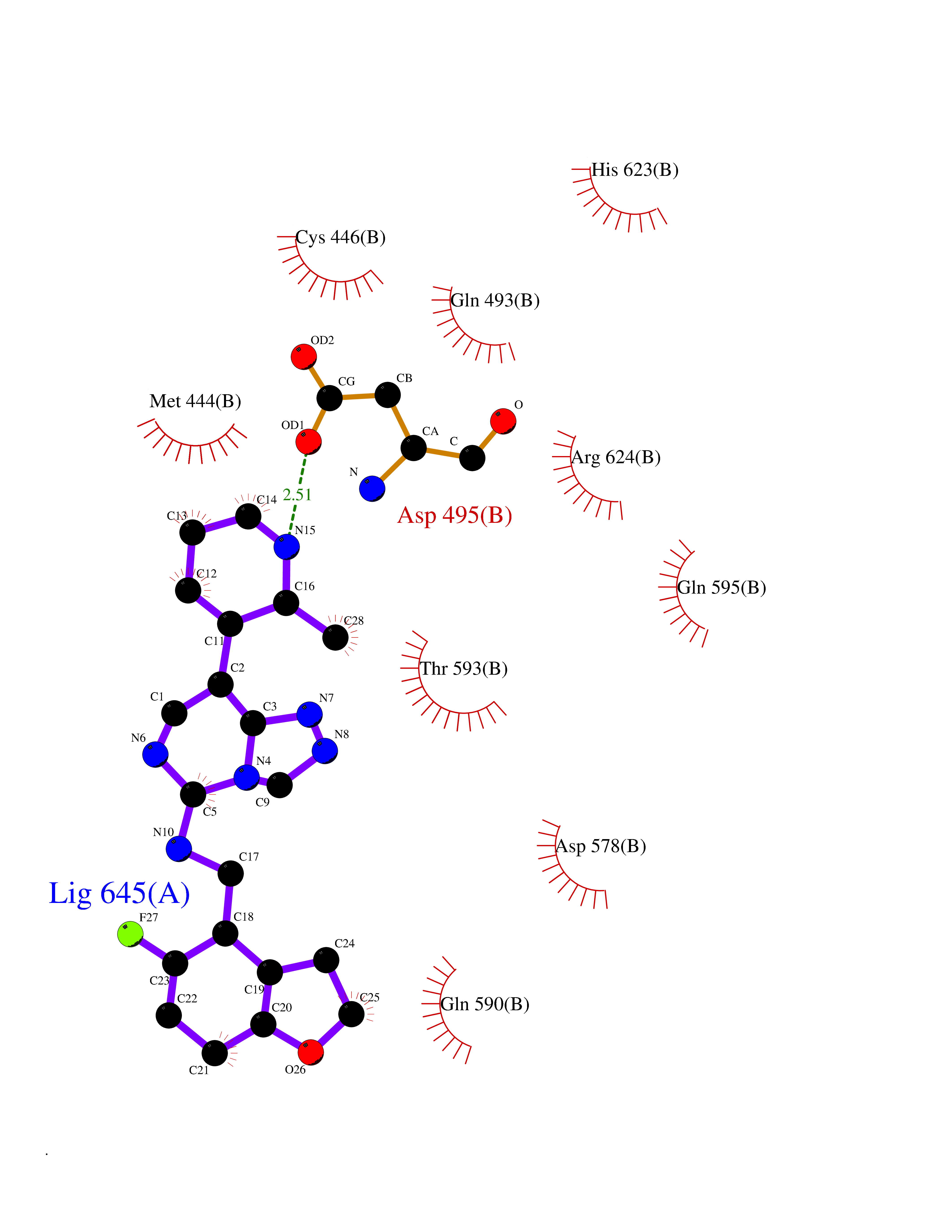 Ligplot Map 3D Binding mode Sequence RDHRKIGRDQELYFFHELSPGSCFFLPKGAYIYNALIEFIRSEYRKRGFQEVVTPNIFNSRLWMTSGHWQHYSENMFSFEVEKELFALKPMNCPGHCLMFDHRPRSWRELPLRLADFGVLHRNELSGALTGLTRVRRFQQDDAHIFCAMEQIEDEIKGCLDFLRTVYSVFGFSFKLNLSTRPEKFLGDIEVWDQAEKQLENSLNEFGEKWELNSGDGAFYGPKIDIQIKDAIGRYHQCATIQLDFQLPIRFNLTYVSHDGDDKKRPVIVHRAILGSVERMIAILTENYGG Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Thymidylate synthase (TYMS) | 1HVY | 7.93 | |
Target general information Gen name TYMS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms TSase; TS Protein family Thymidylate synthase family Biochemical class Methyltransferase Function Contributes to the de novo mitochondrial thymidylate biosynthesis pathway. Related diseases Dyskeratosis congenita, digenic (DKCD) [MIM:620040]: A form of dyskeratosis congenita, a rare multisystem disorder caused by defective telomere maintenance. It is characterized by progressive bone marrow failure, and the clinical triad of reticulated skin hyperpigmentation, nail dystrophy, and mucosal leukoplakia. Common but variable features include premature graying, aplastic anemia, low platelets, osteoporosis, pulmonary fibrosis, and liver fibrosis among others. Early mortality is often associated with bone marrow failure, infections, fatal pulmonary complications, or malignancy. DKCD transmission pattern is consistent with digenic inheritance. {ECO:0000269|PubMed:35931051}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry. TYMS germline variants in the presence of a common ENOSF1 haplotype (defined by rs699517, rs2790 and rs1512643) result in severe thymidylate synthase deficiency and disease. The pathogenic mechanism involves increased expression of ENOSF1 relative to TYMS, and post-transcriptional inhibition of TYMS translation through ENOSF1-TYMS RNA-RNA interactions. {ECO:0000269|PubMed:35931051}. Drugs (DrugBank ID) DB03541; DB07577; DB08734; DB05308; DB01101; DB03800; DB00322; DB00544; DB00441; DB00563; DB08479; DB08478; DB05457; DB00642; DB06813; DB00293; DB04530; DB09256; DB09327; DB05116; DB01643; DB00432 Interacts with NA EC number EC 2.1.1.45 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Direct protein sequencing; Disease variant; Dyskeratosis congenita; Isopeptide bond; Membrane; Methyltransferase; Mitochondrion; Mitochondrion inner membrane; Nucleotide biosynthesis; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 32889.3 Length 287 Aromaticity 0.1 Instability index 40.61 Isoelectric point 6.57 Charge (pH=7) -1.26 2D Binding mode Binding energy (Kcal/mol) -8.29  Molscript Map 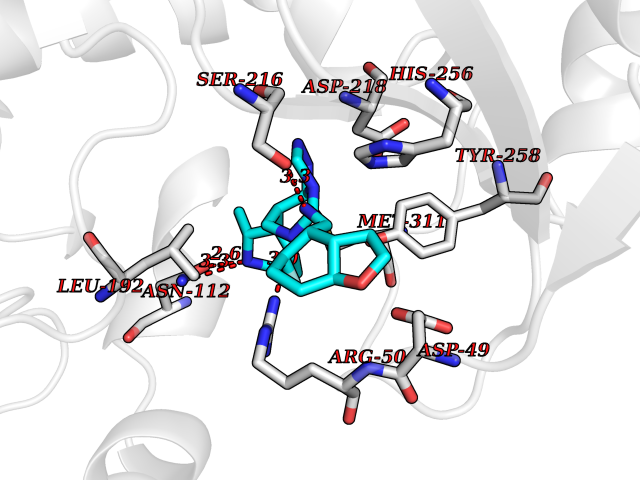 Pymol Map 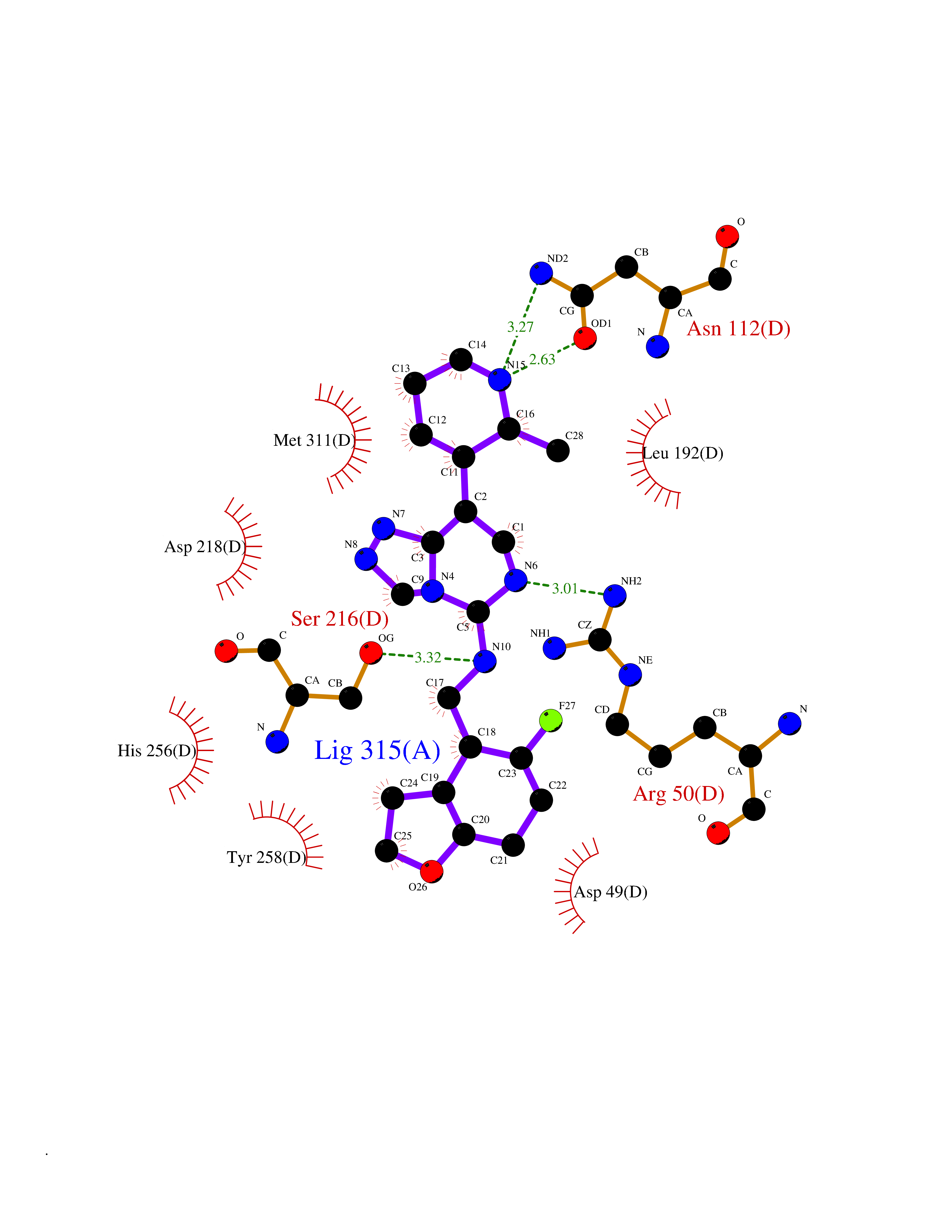 Ligplot Map 3D Binding mode Sequence PPHGELQYLGQIQHILRGVRKDDRTGTGTLSVFGMQARYSLRDEFPLLTTKRVFWKGVLEELLWFIKGSTNAKELSSKGVKIWDANGSRDFLDSLGFSTREEGDLGPVYGFQWRHFGAEYRDMESDYSGQGVDQLQRVIDTIKTNPDDRRIIMCAWNPRDLPLMALPPCHALCQFYVVNSELSCQLYQRSGDMGLGVPFNIASYALLTYMIAHITGLKPGDFIHTLGDAHIYLNHIEPLKIQLQREPRPFPKLRILRKVEKIDDFKAEDFQIEGYNPHPTIKMEMAV Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Monoamine oxidase type A (MAO-A) | 2Z5Y | 7.92 | |
Target general information Gen name MAOA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Monoamine oxidase A; Amine oxidase [flavin-containing] A Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function MAOA preferentially oxidizes biogenic amines such as 5-hydroxytryptamine (5-HT), norepinephrine and epinephrine. Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. Related diseases Brunner syndrome (BRNRS) [MIM:300615]: A form of X-linked non-dysmorphic mild intellectual disability. Male patients are affected by borderline intellectual deficit and exhibit abnormal behavior, including disturbed regulation of impulsive aggression. Obligate female carriers have normal intelligence and behavior. {ECO:0000269|PubMed:8211186}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01472; DB00918; DB00182; DB06698; DB04889; DB13876; DB01445; DB06774; DB00215; DB04017; DB09130; DB05205; DB07641; DB00988; DB01363; DB00668; DB12329; DB01175; DB03147; DB14914; DB00614; DB01381; DB07919; DB04818; DB01247; DB00601; DB01577; DB00805; DB01442; DB01171; DB08804; DB00952; DB04820; DB00184; DB04821; DB06412; DB01626; DB00780; DB00191; DB00388; DB00397; DB09244; DB04850; DB00721; DB01168; DB00571; DB00852; DB09363; DB00140; DB00953; DB06654; DB01037; DB01104; DB00669; DB14569; DB09042; DB00624; DB13943; DB13944; DB13946; DB09245; DB00752; DB15328; DB09185; DB04832; DB00315; DB00909 Interacts with P27338 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Catecholamine metabolism; Direct protein sequencing; Disease variant; FAD; Flavoprotein; Intellectual disability; Membrane; Mitochondrion; Mitochondrion outer membrane; Neurotransmitter degradation; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 58195.3 Length 513 Aromaticity 0.11 Instability index 34.97 Isoelectric point 7.98 Charge (pH=7) 2.87 2D Binding mode Binding energy (Kcal/mol) -10.03  Molscript Map 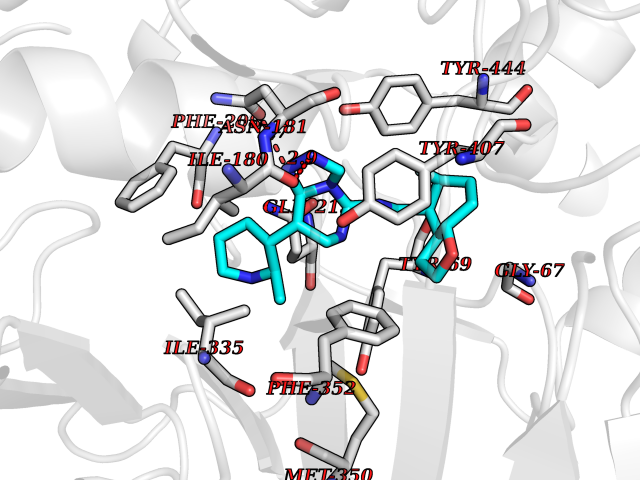 Pymol Map 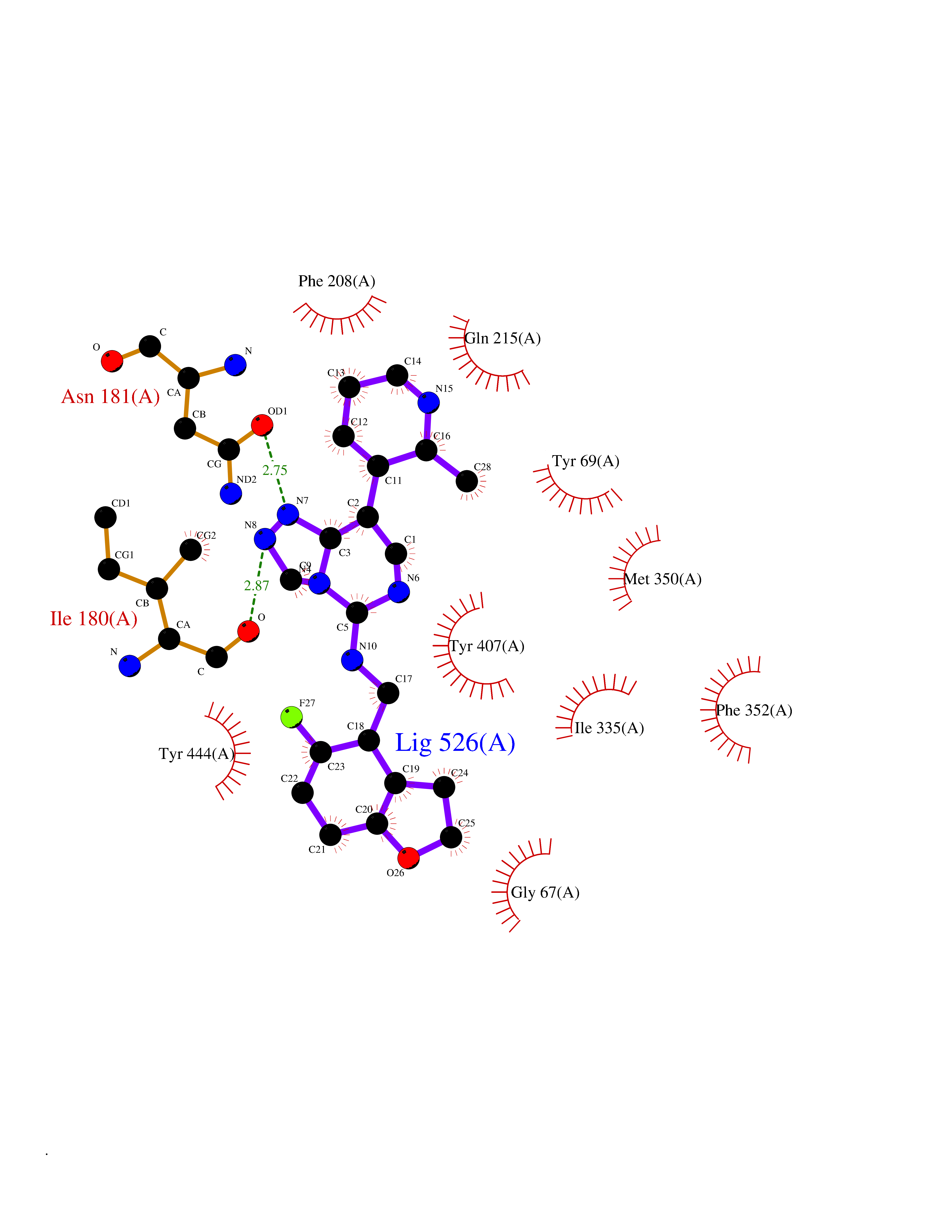 Ligplot Map 3D Binding mode Sequence HMFDVVVIGGGISGLSAAKLLTEYGVSVLVLEARDRVGGRTYTIRNEHVDYVDVGGAYVGPTQNRILRLSKELGIETYKVNVSERLVQYVKGKTYPFRAAFPPVWNPIAYLDYNNLWRTIDNMGKEIPTDAPWEAQHADKWDKMTMKELIDKICWTKTARRFAYLFVNINVTSEPHEVSALWFLWYVKQCGGTTRIFSVTNGGQERKFVGGSGQVSERIMDLLGDQVKLNHPVTHVDQSSDNIIIETLNHEHYECKYVINAIPPTLTAKIHFRPELPAERNQLIQRLPMGAVIKCMMYYKEAFWKKKDYCGCMIIEDEDAPISITLDDTKPDGSLPAIMGFILARKADRLAKLHKEIRKKKICELYAKVLGSQEALHPVHYEEKNWCEEQYSGGCYTAYFPPGIMTQYGRVIRQPVGRIFFAGTETATKWSGYMEGAVEAGERAAREVLNGLGKVTEKDIWVQEPESKDVPAVEITHTFWERNLPSVSGLLKIIGFSTSVTALGFVLYKYKLL Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Aspartyl aminopeptidase (DNPEP) | 4DYO | 7.92 | |
Target general information Gen name DNPEP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms DAP; ASPEP Protein family Peptidase M18 family Biochemical class Peptidase Function Likely to play an important role in intracellular protein and peptide metabolism. Aminopeptidase with specificity towards an acidic amino acid at the N-terminus. Related diseases Cohen-Gibson syndrome (COGIS) [MIM:617561]: An autosomal dominant overgrowth disorder characterized by accelerated osseous maturation, advanced bone age, skeletal abnormalities including flaring of the metaphyses of the long bones, large hands with long fingers and camptodactyly, scoliosis, cervical spine anomalies, dysmorphic facial features, and variable intellectual disability. {ECO:0000269|PubMed:25787343, ECO:0000269|PubMed:27193220, ECO:0000269|PubMed:27868325, ECO:0000269|PubMed:28229514, ECO:0000269|PubMed:28475857}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00142 Interacts with Q9ULA0; Q8TBB1; Q00013; Q9UPN6 EC number EC 3.4.11.21 Uniprot keywords 3D-structure; Acetylation; Alternative initiation; Aminopeptidase; Cytoplasm; Hydrolase; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 50574.6 Length 457 Aromaticity 0.07 Instability index 55.73 Isoelectric point 7.73 Charge (pH=7) 2.3 2D Binding mode Binding energy (Kcal/mol) -7.64 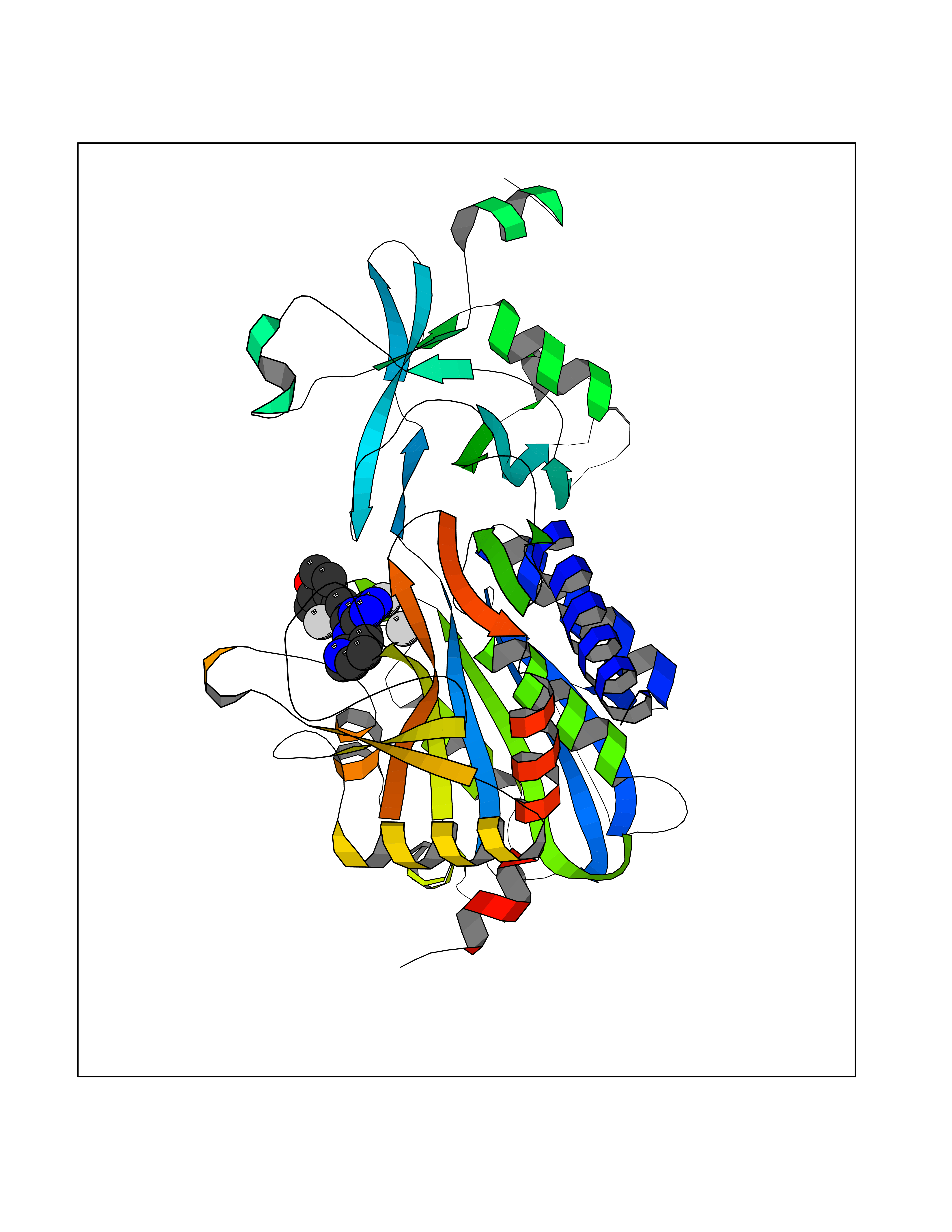 Molscript Map 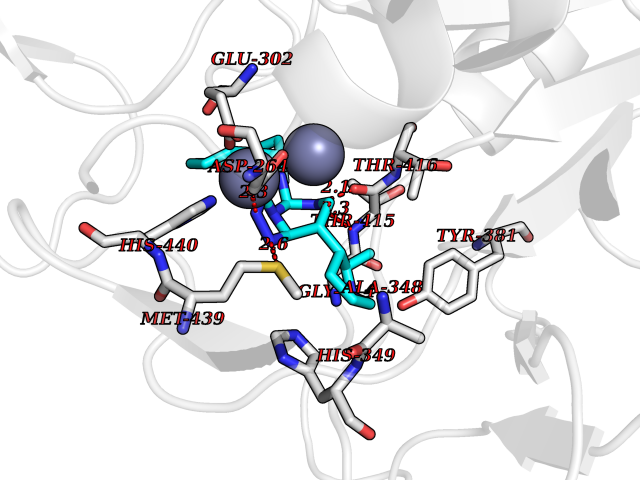 Pymol Map 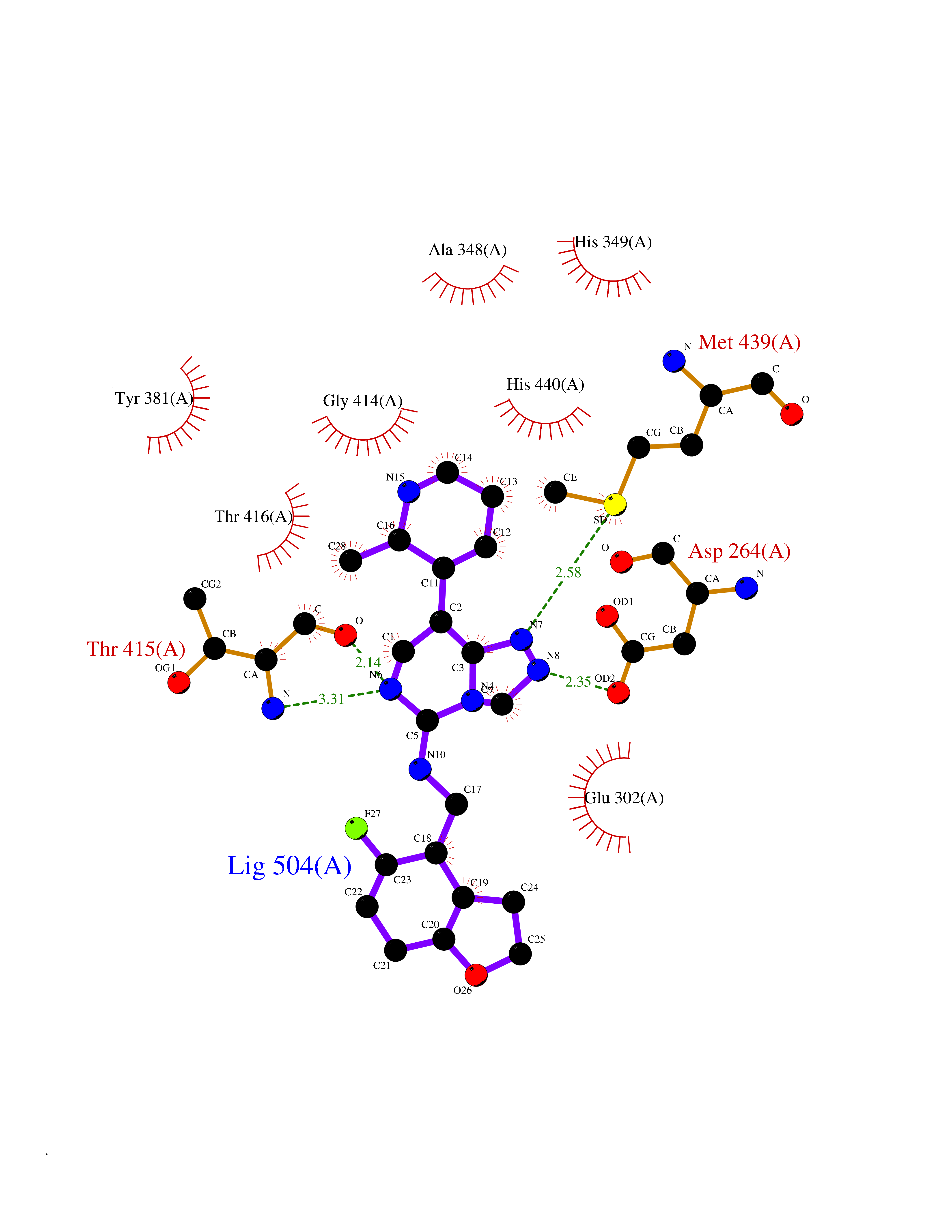 Ligplot Map 3D Binding mode Sequence GKARKEAVQTAAKELLKFVNRSPSPFHAVAECRNRLLQAGFSELKETEKWNIKPESKYFMTRNSSTIIAFAVGGQYVPGNGFSLIGAHTDSPCLRVKRRSRRSQVGFQQVGVETYGGGIWSTWFDRDLTLAGRVIVKCPTSGRLEQQLVHVERPILRIPHLAIHLQRNINENFGPNTEMHLVPILATAIQEELEKGTERHHSVLMSLLCAHLGLSPKDIVEMELCLADTQPAVLGGAYDEFIFAPRLDNLHSCFCALQALIDSCAGPGSLATEPHVRMVTLYDNEEVGSESAQGAQSLLTELVLRRISASCQHPTAFEEAIPKSFMISADMAHAVHPNYLDKHEENHRPLFHKGPVIKVNSKQRYASNAVSEALIREVANKVKVPLQDLMVRNDTPCGTTIGPILASRLGLRVLDLGSPQLAMHSIREMACTTGVLQTLTLFKGFFELFPSLAENLY Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | ERK activator kinase 2 (MEK2) | 1S9I | 7.92 | |
Target general information Gen name MAP2K2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PRKMK2; MKK2; MEK 2; MAPKK 2; MAPK/ERK kinase 2; MAP kinase kinase 2; Dual specificity mitogenactivated protein kinase kinase 2; Dual specificity mitogen-activated protein kinase kinase 2 Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, MAP kinase kinase subfamily Biochemical class Kinase Function Activates the ERK1 and ERK2 MAP kinases. Catalyzes the concomitant phosphorylation of a threonine and a tyrosine residue in a Thr-Glu-Tyr sequence located in MAP kinases. Related diseases Cardiofaciocutaneous syndrome 4 (CFC4) [MIM:615280]: A form of cardiofaciocutaneous syndrome, a multiple congenital anomaly disorder characterized by a distinctive facial appearance, heart defects and intellectual disability. Heart defects include pulmonic stenosis, atrial septal defects and hypertrophic cardiomyopathy. Some affected individuals present with ectodermal abnormalities such as sparse, friable hair, hyperkeratotic skin lesions and a generalized ichthyosis-like condition. Typical facial features are similar to Noonan syndrome. They include high forehead with bitemporal constriction, hypoplastic supraorbital ridges, downslanting palpebral fissures, a depressed nasal bridge, and posteriorly angulated ears with prominent helices. {ECO:0000269|PubMed:16439621, ECO:0000269|PubMed:18042262, ECO:0000269|PubMed:20358587}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11967; DB06616; DB12010; DB14904; DB11689; DB08911 Interacts with P05067; P10398; Q96II5; P15056; O95273; Q12959; P61978-2; Q8IVT5; P00540; P04049 EC number EC 2.7.12.2 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Cardiomyopathy; Cytoplasm; Direct protein sequencing; Disease variant; Ectodermal dysplasia; Intellectual disability; Kinase; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 33960.9 Length 303 Aromaticity 0.07 Instability index 45.61 Isoelectric point 6.29 Charge (pH=7) -2.53 2D Binding mode Binding energy (Kcal/mol) -8.98 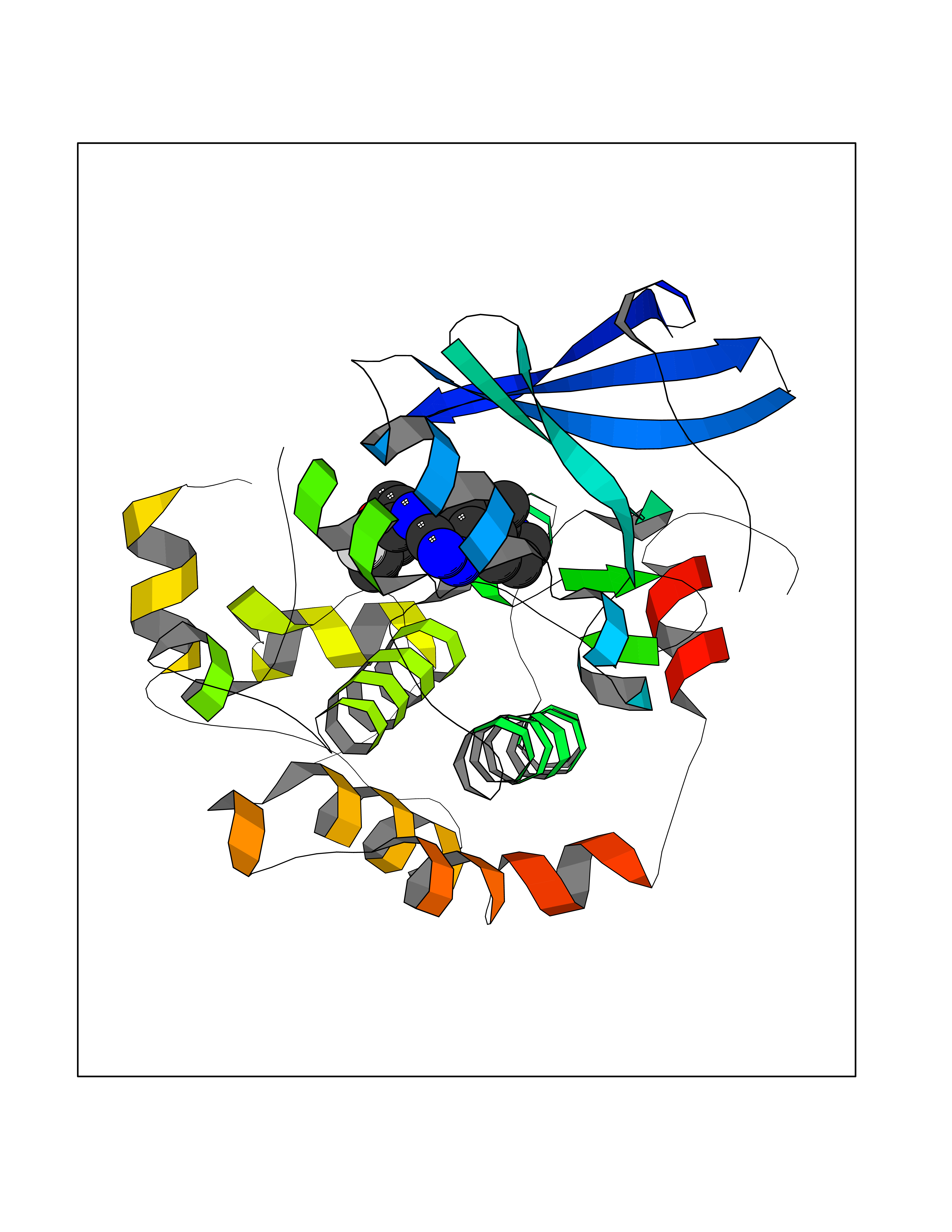 Molscript Map 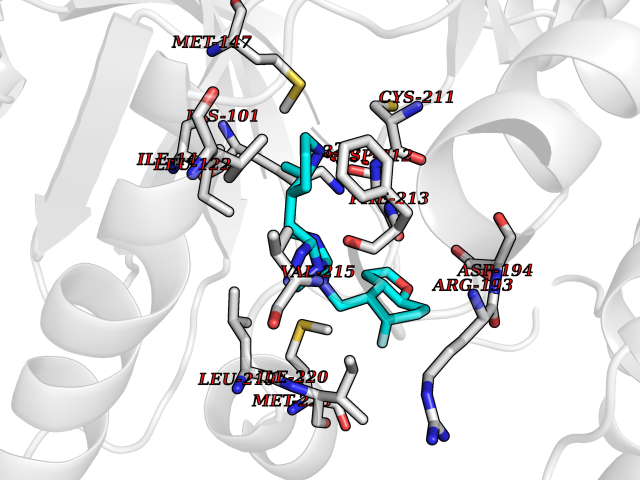 Pymol Map 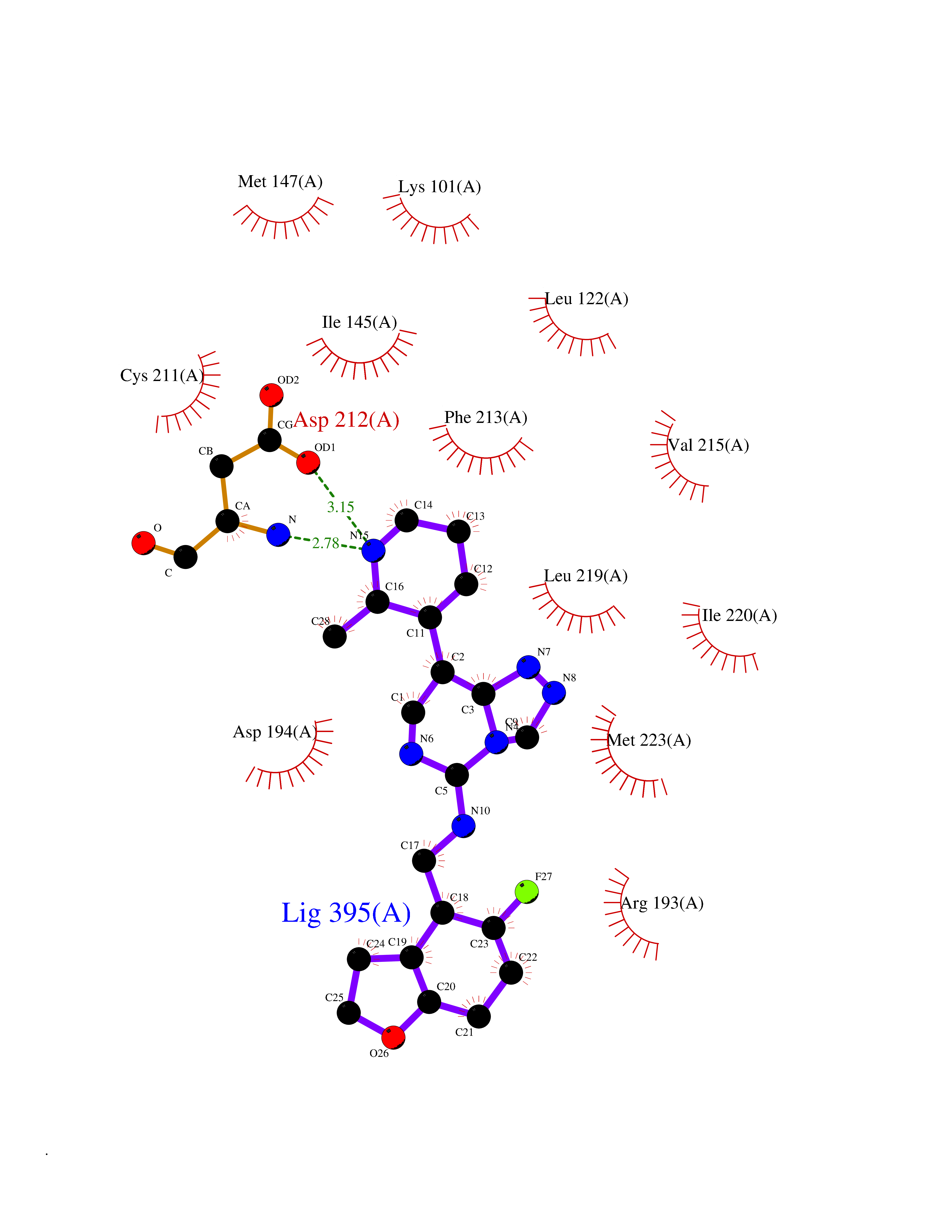 Ligplot Map 3D Binding mode Sequence QKAKVGELKDDDFERISELGAGNGGVVTKVQHRPSGLIMARKLIHLEIKPAIRNQIIRELQVLHECNSPYIVGFYGAFYSDGEISICMEHMDGGSLDQVLKEAKRIPEEILGKVSIAVLRGLAYLREKHQIMHRDVKPSNILVNSRGEIKLCDFGVSGQLIDSMVGTRSYMAPERLQGTHYSVQSDIWSMGLSLVELAVGRYPIPPPDAKELEAIFGRPVVDRPAMAIFELLDYIVNEPPPKLPNGVFTPDFQEFVNKCLIKNPAERADLKMLTNHTFIKRSEVEEVDFAGWLCKTLRLNQPG Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Monocarboxylate transporter 1 (SLC16A1) | 6LZ0 | 7.92 | |
Target general information Gen name SLC16A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Solute carrier family 16 member 1; MCT1; MCT 1 Protein family Major facilitator superfamily, Monocarboxylate porter (TC 2.A.1.13) family Biochemical class Major facilitator Function Catalyzes the rapid transport across the plasma membrane of many monocarboxylates such as lactate, pyruvate, branched-chain oxo acids derived from leucine, valine and isoleucine, and the ketone bodies acetoacetate, beta-hydroxybutyrate and acetate. Depending on the tissue and on cicumstances, mediates the import or export of lactic acid and ketone bodies. Required for normal nutrient assimilation, increase of white adipose tissue and body weight gain when on a high-fat diet. Plays a role in cellular responses to a high-fat diet by modulating the cellular levels of lactate and pyruvate, small molecules that contribute to the regulation of central metabolic pathways and insulin secretion, with concomitant effects on plasma insulin levels and blood glucose homeostasis. Proton-coupled monocarboxylate transporter. Related diseases Symptomatic deficiency in lactate transport (SDLT) [MIM:245340]: Deficiency of lactate transporter may result in an acidic intracellular environment created by muscle activity with consequent degeneration of muscle and release of myoglobin and creatine kinase. This defect might compromise extreme performance in otherwise healthy individuals. {ECO:0000269|PubMed:10590411}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperinsulinemic hypoglycemia, familial, 7 (HHF7) [MIM:610021]: A form of hyperinsulinemic hypoglycemia, a clinically and genetically heterogeneous disorder characterized by inappropriate insulin secretion from the pancreatic beta-cells in the presence of low blood glucose levels. HHF7 features include exercise-induced hyperinsulinism, loss of consciousness due to hypoglycemia, and hypoglycemic seizures. HHF7 inheritance is autosomal dominant. {ECO:0000269|PubMed:17701893}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Monocarboxylate transporter 1 deficiency (MCT1D) [MIM:616095]: A metabolic disorder characterized by recurrent ketoacidosis, a pathologic state due to ketone formation exceeding ketone utilization. The clinical consequences of ketoacidosis are vomiting, osmotic diuresis, dehydration, and Kussmaul breathing. The condition may progress to decreased consciousness and, ultimately, death. {ECO:0000269|PubMed:25390740}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03166; DB01762; DB03773; DB04074; DB00345; DB00415; DB08892; DB03793; DB03066; DB07767; DB00529; DB01440; DB00142; DB04398; DB09338; DB00563; DB00731; DB00627; DB04552; DB00175; DB01032; DB00119; DB04216; DB00936; DB04348; DB00313 Interacts with Q66PJ3-4; Q92782-2; Q9UH65 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Membrane; Phosphoprotein; Proteomics identification; Reference proteome; Symport; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 40405.6 Length 375 Aromaticity 0.15 Instability index 32.61 Isoelectric point 9.12 Charge (pH=7) 10.98 2D Binding mode Binding energy (Kcal/mol) -8.8 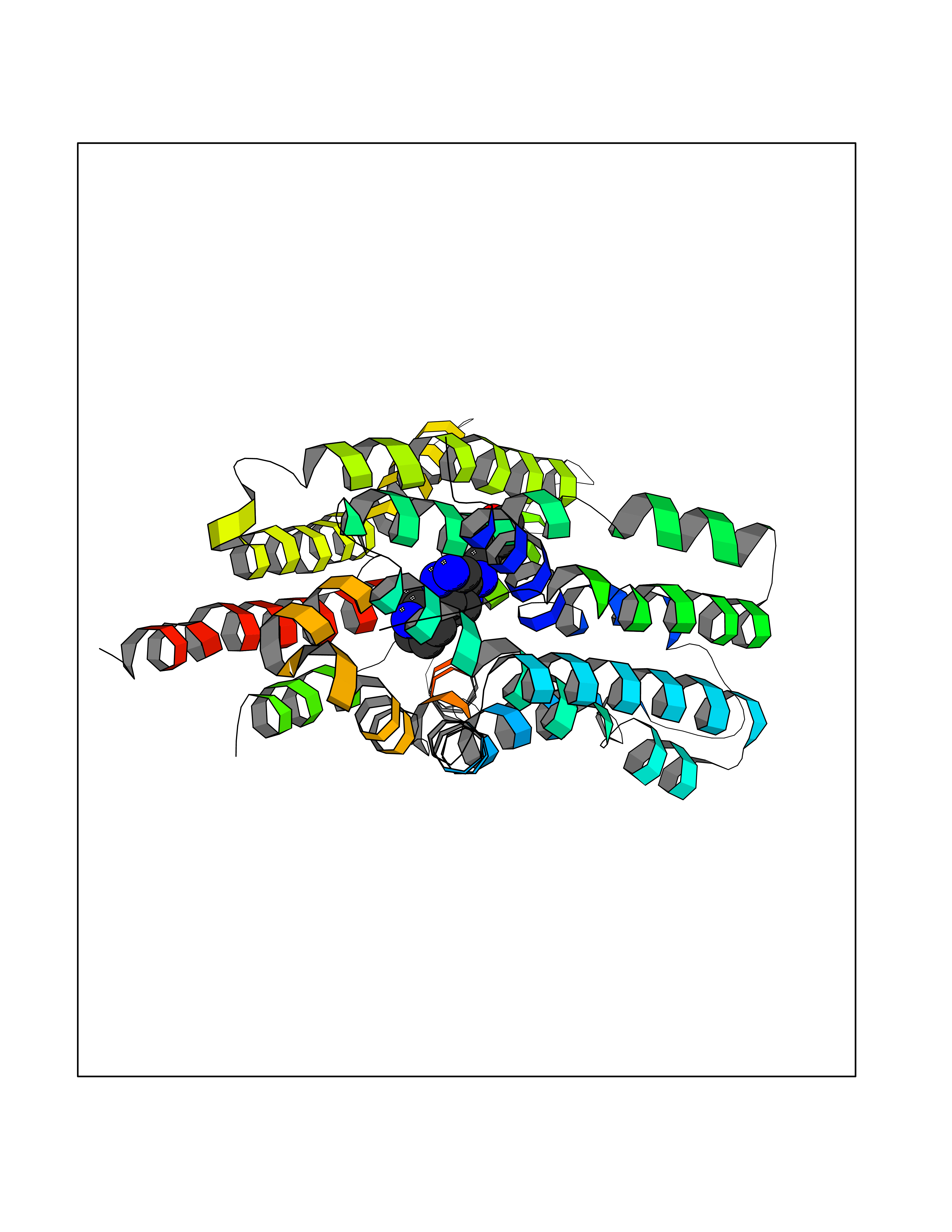 Molscript Map 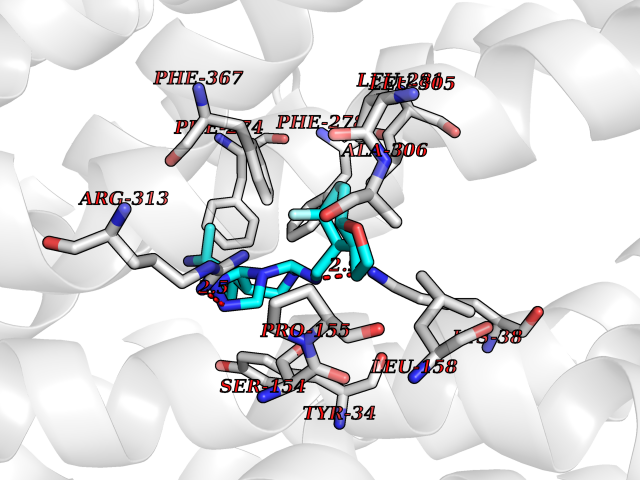 Pymol Map 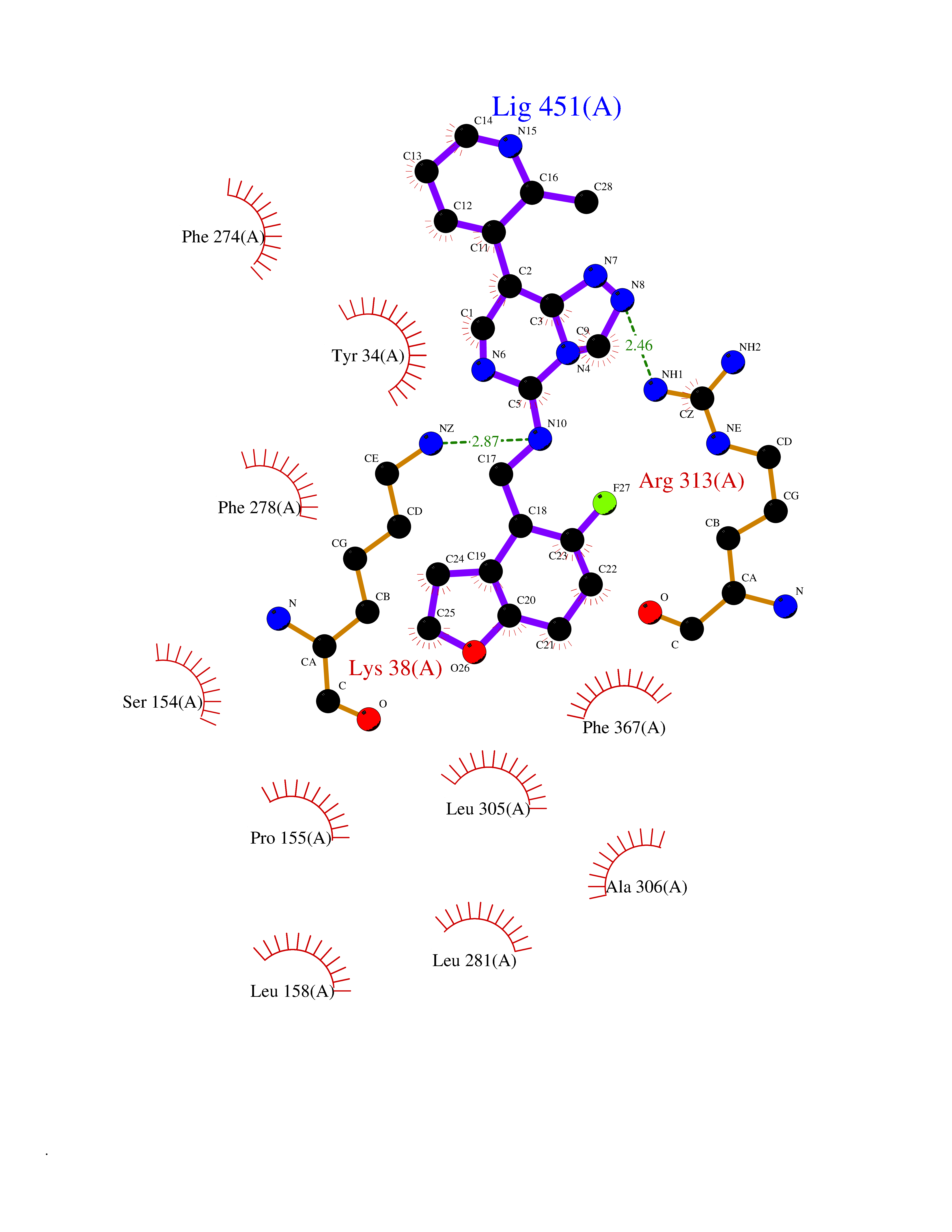 Ligplot Map 3D Binding mode Sequence GGWGWAVVIGAFISIGFSYAFPKSITVFFKEIEGIFHATTSEVSWISSIMLAVMYGGGPISSILVNKYGSRIVMIVGGCLSGCGLIAASFCNTVQQLYVCIGVIGGLGLAFNLNPALTMIGKYFYKRRPLANGLAMAGSPVFLCTLAPLNQVFFGIFGWRGSFLILGGLLLNCCVAGALMRPIGPHRGFLLYLSGNVIMFFGLFAPLVFLSSYGKSQHYSSEKSAFLLSILAFVDMVARPSMGLVANTKPIRPRIQYFFAASVVANGVCHMLAPLSTTYVGFCVYAGFFGFAFGWLSSVLFETLMDLVGPQRFSSAVGLVTIVECCPVLLGPPLLGRLNDMYGDYKYTYWACGVVLIISGIYLFIGMGINYRLLA Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Phosphodiesterase 3B (PDE3B) | 1SOJ | 7.92 | |
Target general information Gen name PDE3B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cGMPinhibited 3',5'cyclic phosphodiesterase B; cGMP-inhibited 3',5'-cyclic phosphodiesterase B; Cyclic GMPinhibited phosphodiesterase B; Cyclic GMP-inhibited phosphodiesterase B; CGIPDE1; CGIPDE B; CG Protein family Cyclic nucleotide phosphodiesterase family, PDE3 subfamily Biochemical class Phosphoric diester hydrolase Function May play a role in fat metabolism. Regulates cAMP binding of RAPGEF3. Through simultaneous binding to RAPGEF3 and PIK3R6 assembles a signaling complex in which the PI3K gamma complex is activated by RAPGEF3 and which is involved in angiogenesis. Cyclic nucleotide phosphodiesterase with a dual-specificity for the second messengers cAMP and cGMP, which are key regulators of many important physiological processes. Related diseases Erythrocytosis, familial, 4 (ECYT4) [MIM:611783]: An autosomal dominant disorder characterized by elevated serum hemoglobin and hematocrit, and normal platelet and leukocyte counts. {ECO:0000269|PubMed:18184961, ECO:0000269|PubMed:18378852, ECO:0000269|PubMed:19208626, ECO:0000269|PubMed:22367913}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07954; DB01640; DB01427; DB00201; DB01970; DB01113; DB09283 Interacts with O60760; Q8TBB1; P48736; Q5UE93; O95398 EC number EC 3.1.4.17 Uniprot keywords 3D-structure; Alternative splicing; Angiogenesis; cAMP; cGMP; Hydrolase; Magnesium; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 41872.2 Length 364 Aromaticity 0.11 Instability index 48.97 Isoelectric point 5.64 Charge (pH=7) -11.84 2D Binding mode Binding energy (Kcal/mol) -9 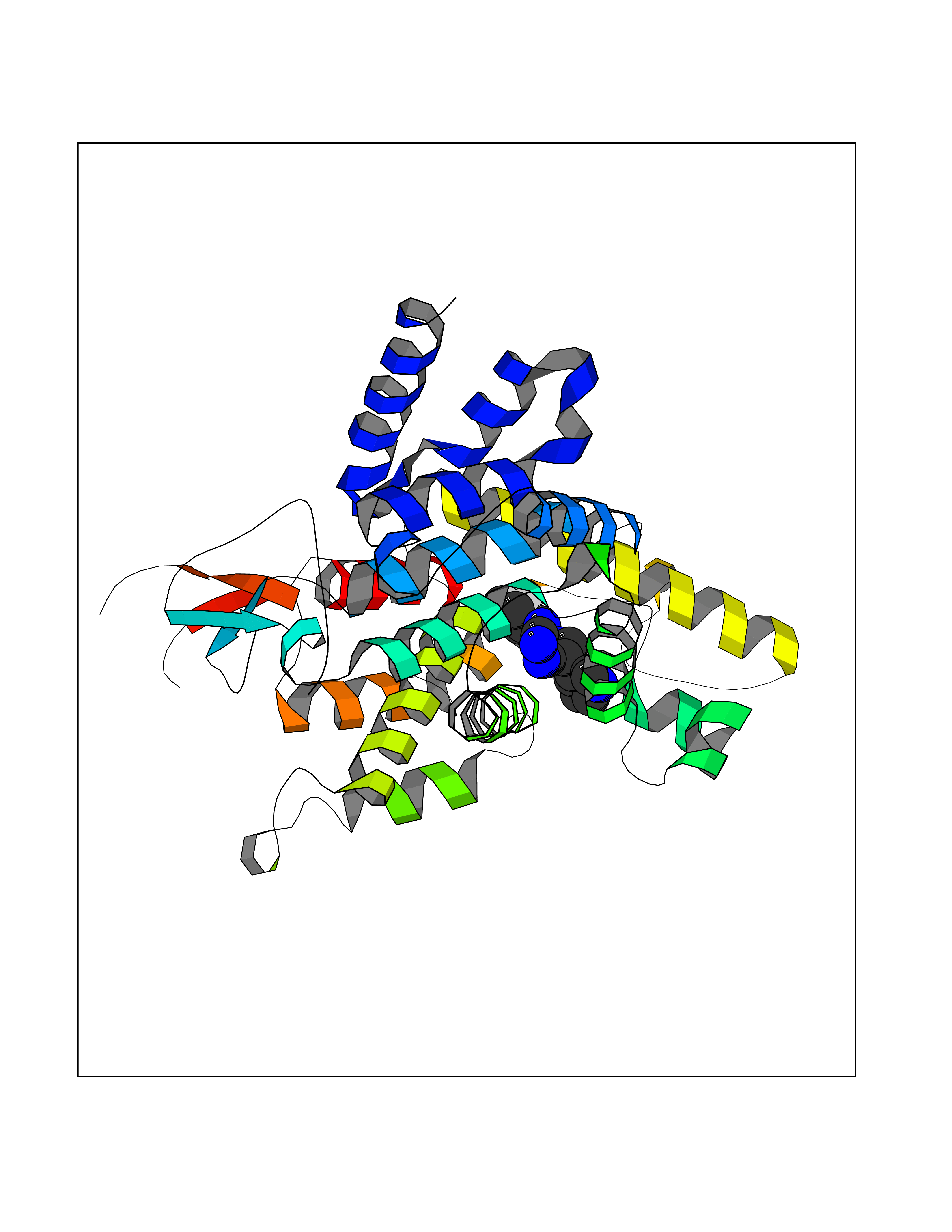 Molscript Map 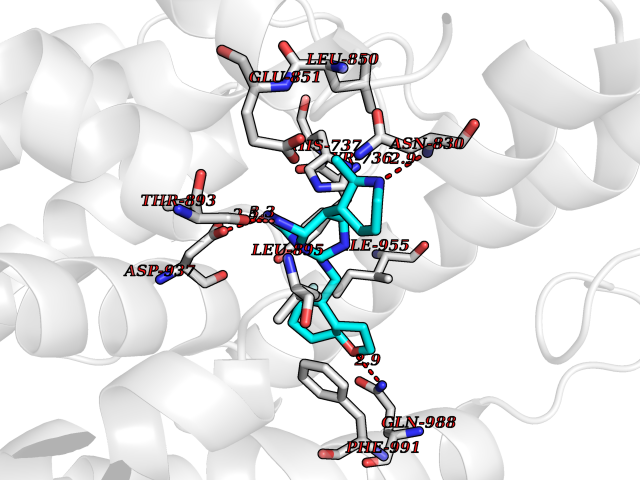 Pymol Map 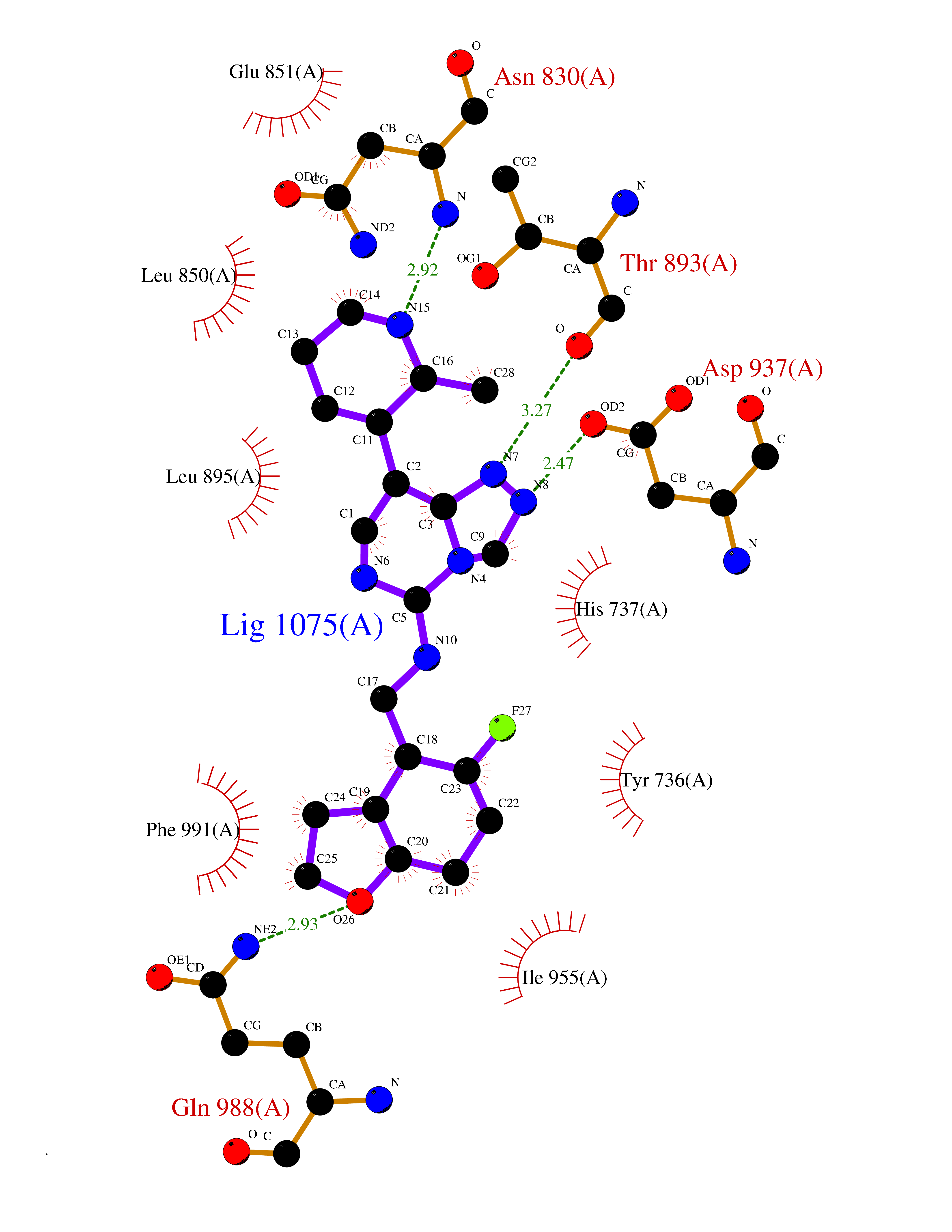 Ligplot Map 3D Binding mode Sequence LDLILVEEYDSLIEKMSNWNFPIFELVEKMGEKSGRILSQVMYTLFQDTGLLEIFKIPTQQFMNYFRALENGYRDIPYHNRIHATDVLHAVWYLTTRPVPGLQQIHNGRIAYISSKSCSNPDESYGCLSSNIPALELMALYVAAAMHDYDHPGRTNAFLVATNAPQAVLYNDRSVLENHHAASAWNLYLSRPEYNFLLHLDHVEFKRFRFLVIEAILATDLKKHFDFLAEFNAKANDVNSNGIEWSNENDRLLVCQVCIKLADINGPAKVRDLHLKWTEGIVNEFYEQGDEEANLGLPISPFMDRSSPQLAKLQESFITHIVGPLCNSYDAAGLLPGQWLEAESRRRIFCQLMHHLTENHKIWK Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Notch-1 receptor (NOTCH1) | 5L0R | 7.91 | |
Target general information Gen name NOTCH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hN1; Translocationassociated notch protein TAN1; Translocation-associated notch protein TAN-1; TAN1; Notch 1 intracellular domain; Notch 1; Neurogenic locus notch homolog protein 1; NICD Protein family NOTCH family Biochemical class Notch protein Function Upon ligand activation through the released notch intracellular domain (NICD) it forms a transcriptional activator complex with RBPJ/RBPSUH and activates genes of the enhancer of split locus. Affects the implementation of differentiation, proliferation and apoptotic programs. Involved in angiogenesis; negatively regulates endothelial cell proliferation and migration and angiogenic sprouting. Involved in the maturation of both CD4(+) and CD8(+) cells in the thymus. Important for follicular differentiation and possibly cell fate selection within the follicle. During cerebellar development, functions as a receptor for neuronal DNER and is involved in the differentiation of Bergmann glia. Represses neuronal and myogenic differentiation. May play an essential role in postimplantation development, probably in some aspect of cell specification and/or differentiation. May be involved in mesoderm development, somite formation and neurogenesis. May enhance HIF1A function by sequestering HIF1AN away from HIF1A. Required for the THBS4 function in regulating protective astrogenesis from the subventricular zone (SVZ) niche after injury. Involved in determination of left/right symmetry by modulating the balance between motile and immotile (sensory) cilia at the left-right organiser (LRO). Functions as a receptor for membrane-bound ligands Jagged-1 (JAG1), Jagged-2 (JAG2) and Delta-1 (DLL1) to regulate cell-fate determination. Related diseases Aortic valve disease 1 (AOVD1) [MIM:109730]: A common defect in the aortic valve in which two rather than three leaflets are present. It is often associated with aortic valve calcification, stenosis and insufficiency. In extreme cases, the blood flow may be so restricted that the left ventricle fails to grow, resulting in hypoplastic left heart syndrome. {ECO:0000269|PubMed:16025100}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Adams-Oliver syndrome 5 (AOS5) [MIM:616028]: A form of Adams-Oliver syndrome, a disorder characterized by the congenital absence of skin (aplasia cutis congenita) in combination with transverse limb defects. Aplasia cutis congenita can be located anywhere on the body, but in the vast majority of the cases, it is present on the posterior parietal region where it is often associated with an underlying defect of the parietal bones. Limb abnormalities are typically limb truncation defects affecting the distal phalanges or entire digits (true ectrodactyly). Only rarely, metatarsals/metacarpals or more proximal limb structures are also affected. Apart from transverse limb defects, syndactyly, most commonly of second and third toes, can also be observed. The clinical features are highly variable and can also include cardiovascular malformations, brain abnormalities and vascular defects such as cutis marmorata and dilated scalp veins. {ECO:0000269|PubMed:25132448}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q13315; Q969H0; Q16665; P78504; O60341; Q8N423; Q92585; P19838; P46531; Q13153; Q13526; Q06330; Q06330-6; Q13573; P98170; Q8IZL2; Q96JK9 EC number NA Uniprot keywords 3D-structure; Activator; Angiogenesis; ANK repeat; Calcium; Cell membrane; Developmental protein; Differentiation; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Endosome; Glycoprotein; Hydroxylation; Isopeptide bond; Membrane; Metal-binding; Notch signaling pathway; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transcription; Transcription regulation; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 46121.2 Length 394 Aromaticity 0.13 Instability index 39.95 Isoelectric point 7.22 Charge (pH=7) 0.63 2D Binding mode Binding energy (Kcal/mol) -9.53  Molscript Map 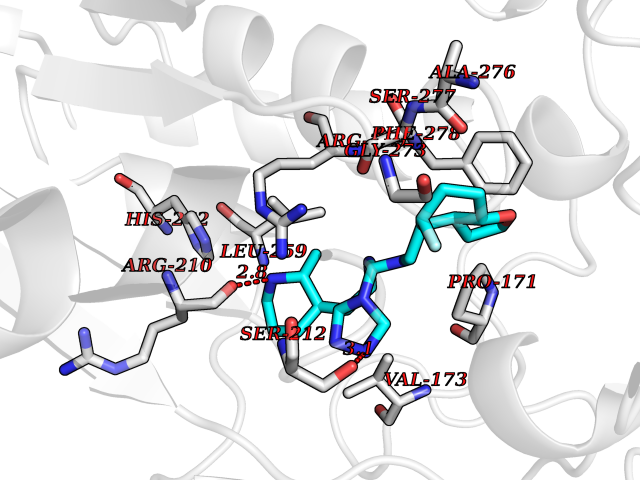 Pymol Map 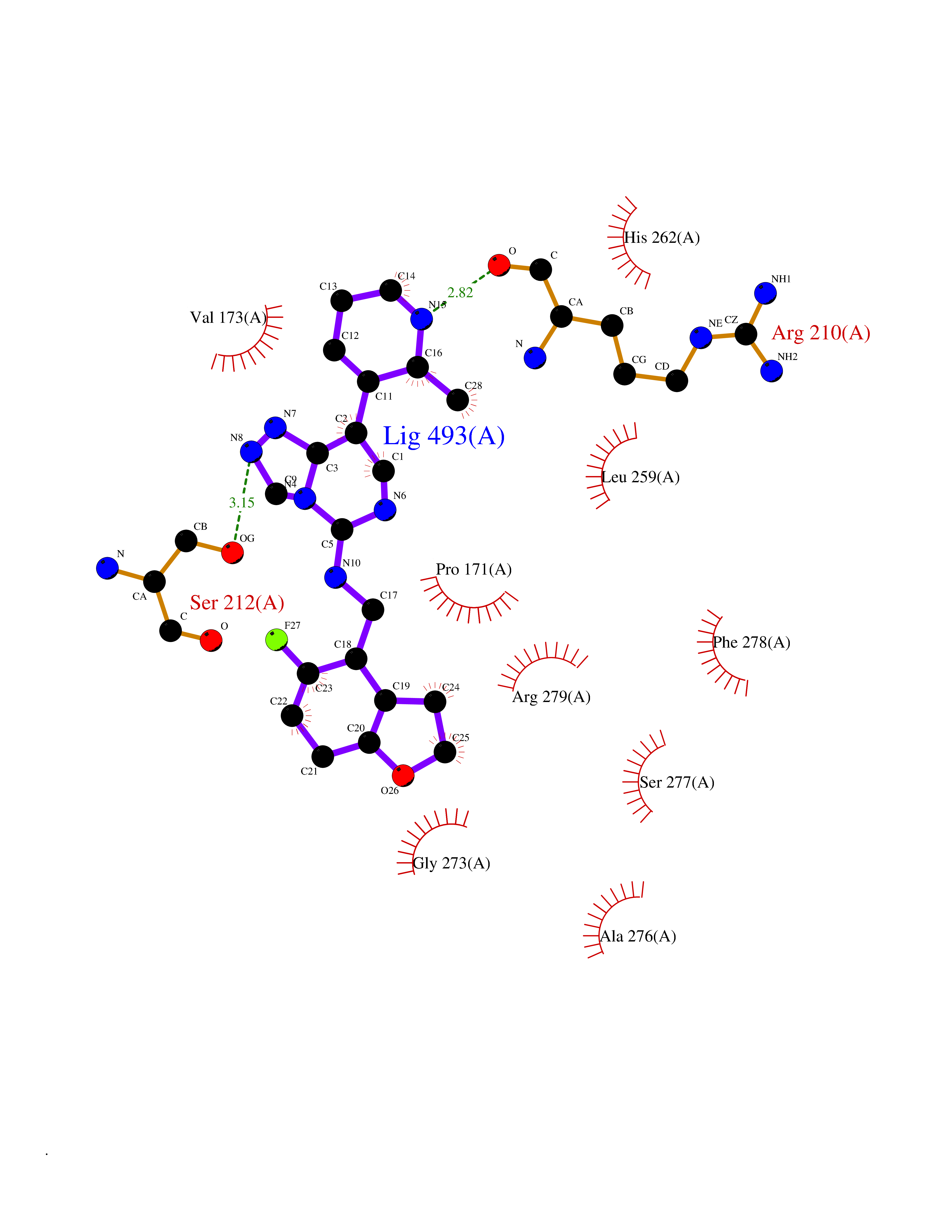 Ligplot Map 3D Binding mode Sequence KWKVFIDQINRSLENYEPCSSQNCSCYHGVIEEDLTPFRGGISRKMMAEVVRRKLGTHYQITKNRLYRENDCMFPSRCSGVEHFILEVIGRLPDMEMVINVRDYPQVPKWMEPAIPVFSFSKTSEYHDIMYPAWTFWEGGPAVWPIYPTGLGRWDLFREDLVRSAAQWPWKKKNSTAYFRGSRTSPERDPLILLSRKNPKLVDAEYTKNQAWKSMKDTLGKPAAKDVHLVDHCKYKYLFNFRGVAASFRFKHLFLCGSLVFHVGDEWLEFFYPQLKPWVHYIPVKTDLSNVQELLQFVKANDDVAQEIAERGSQFIRNHLQMDDITCYWENLLSEYSKFLSYNVTRRKGYDQIIPVNECVSNPCQNDATCLDQIGEFQCICMPGYEGVHCEVNT Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Proto-oncogene c-Met (MET) | 3DKC | 7.91 | |
Target general information Gen name MET Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine-protein kinase Met; Scatter factor receptor; SF receptor; Met proto-oncogene tyrosine kinase; Hepatocyte growth factor receptor; HGF/SF receptor; HGF-SF receptor; HGF receptor; C-met; C-Met r Protein family Protein kinase superfamily, Tyr protein kinase family Biochemical class Kinase Function Regulates many physiological processes including proliferation, scattering, morphogenesis and survival. Ligand binding at the cell surface induces autophosphorylation of MET on its intracellular domain that provides docking sites for downstream signaling molecules. Following activation by ligand, interacts with the PI3-kinase subunit PIK3R1, PLCG1, SRC, GRB2, STAT3 or the adapter GAB1. Recruitment of these downstream effectors by MET leads to the activation of several signaling cascades including the RAS-ERK, PI3 kinase-AKT, or PLCgamma-PKC. The RAS-ERK activation is associated with the morphogenetic effects while PI3K/AKT coordinates prosurvival effects. During embryonic development, MET signaling plays a role in gastrulation, development and migration of muscles and neuronal precursors, angiogenesis and kidney formation. In adults, participates in wound healing as well as organ regeneration and tissue remodeling. Promotes also differentiation and proliferation of hematopoietic cells. May regulate cortical bone osteogenesis. Receptor tyrosine kinase that transduces signals from the extracellular matrix into the cytoplasm by binding to hepatocyte growth factor/HGF ligand. Related diseases Activation of MET after rearrangement with the TPR gene produces an oncogenic protein.; DISEASE: Defects in MET may be associated with gastric cancer.; DISEASE: Hepatocellular carcinoma (HCC) [MIM:114550]: A primary malignant neoplasm of epithelial liver cells. The major risk factors for HCC are chronic hepatitis B virus (HBV) infection, chronic hepatitis C virus (HCV) infection, prolonged dietary aflatoxin exposure, alcoholic cirrhosis, and cirrhosis due to other causes. {ECO:0000269|PubMed:9927037}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Renal cell carcinoma papillary (RCCP) [MIM:605074]: A subtype of renal cell carcinoma tending to show a tubulo-papillary architecture formed by numerous, irregular, finger-like projections of connective tissue. Renal cell carcinoma is a heterogeneous group of sporadic or hereditary carcinoma derived from cells of the proximal renal tubular epithelium. {ECO:0000269|PubMed:10327054, ECO:0000269|PubMed:10417759, ECO:0000269|PubMed:10433944, ECO:0000269|PubMed:9140397, ECO:0000269|PubMed:9563489}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: A common allele in the promoter region of the MET shows genetic association with susceptibility to autism in some families. Functional assays indicate a decrease in MET promoter activity and altered binding of specific transcription factor complexes.; DISEASE: MET activating mutations may be involved in the development of a highly malignant, metastatic syndrome known as cancer of unknown primary origin (CUP) or primary occult malignancy. Systemic neoplastic spread is generally a late event in cancer progression. However, in some instances, distant dissemination arises at a very early stage, so that metastases reach clinical relevance before primary lesions. Sometimes, the primary lesions cannot be identified in spite of the progresses in the diagnosis of malignancies.; DISEASE: Deafness, autosomal recessive, 97 (DFNB97) [MIM:616705]: A form of non-syndromic sensorineural hearing loss with prelingual onset. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. {ECO:0000269|PubMed:25941349}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Osteofibrous dysplasia (OSFD) [MIM:607278]: A congenital disorder of osteogenesis characterized by non-neoplastic, radiolucent lesions that affect the cortical bone immediately under the periosteum. It usually manifests as a painless swelling or anterior bowing of the long bones, most commonly the tibia and fibula. {ECO:0000269|PubMed:26637977}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Disease-associated variants identified in 4 families cause the deletion of exon 14. This results in the exclusion of an ubiquitination target site within the cytoplasmic domain, hence in protein stabilization. The persistent presence of MET at the cell surface in conditions of ligand-dependent activation retards osteoblastic differentiation. {ECO:0000269|PubMed:26637977}.; DISEASE: Arthrogryposis, distal, 11 (DA11) [MIM:620019]: A form of distal arthrogryposis, a disease characterized by congenital joint contractures that mainly involve two or more distal parts of the limbs, in the absence of a primary neurological or muscle disease. DA11 is an autosomal dominant form characterized mainly by camptodactyly. Other features include absent flexion creases and limited forearm supination. {ECO:0000269|PubMed:30777867}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06896; DB08791; DB06997; DB07969; DB08079; DB16695; DB12742; DB12267; DB08875; DB11791; DB08865; DB12010; DB02152; DB07369; DB06995; DB06314; DB01268; DB15133; DB12200; DB11800 Interacts with P22681; Q96EY1; Q96EY1-2; P00533; P09769; P14210; P14210-6; O15357; P35968; P06239; P07948; P08581; P41218; P15941; P16333; O43639; Q16288; P27986; O00459; Q92569; P19174; O43157; O15031; Q9ULL4; Q8TCU6; P18031; Q06124; P23467; Q12913; Q16827; P20936; Q9UQQ2; O60880; O14796; Q9NP31; Q8N5H7; Q15464; P29353; P98077; Q6S5L8; Q96IW2; Q9H6Q3; O75159; O14544; P12931; Q9ULZ2; P43405; P42680; Q9HBL0; Q63HR2; Q68CZ2; Q9UKW4; P07947; P43403; Q08048; P0DQD2; P35918; Q00944 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Chromosomal rearrangement; Deafness; Disease variant; Disulfide bond; Glycoprotein; Kinase; Membrane; Non-syndromic deafness; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Secreted; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 35229.5 Length 312 Aromaticity 0.1 Instability index 37.98 Isoelectric point 7.79 Charge (pH=7) 1.93 2D Binding mode Binding energy (Kcal/mol) -9.1  Molscript Map 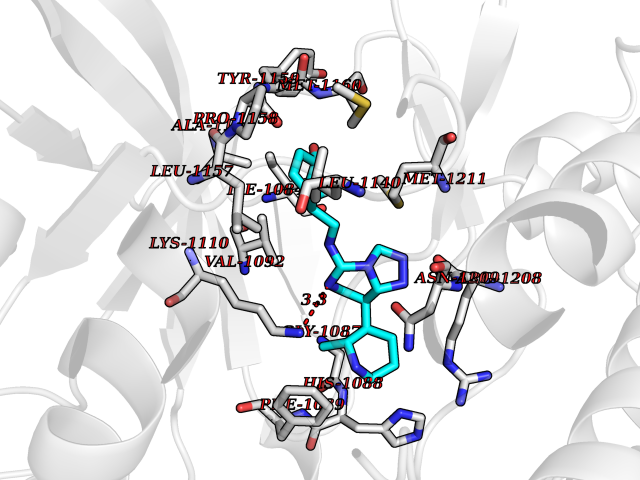 Pymol Map 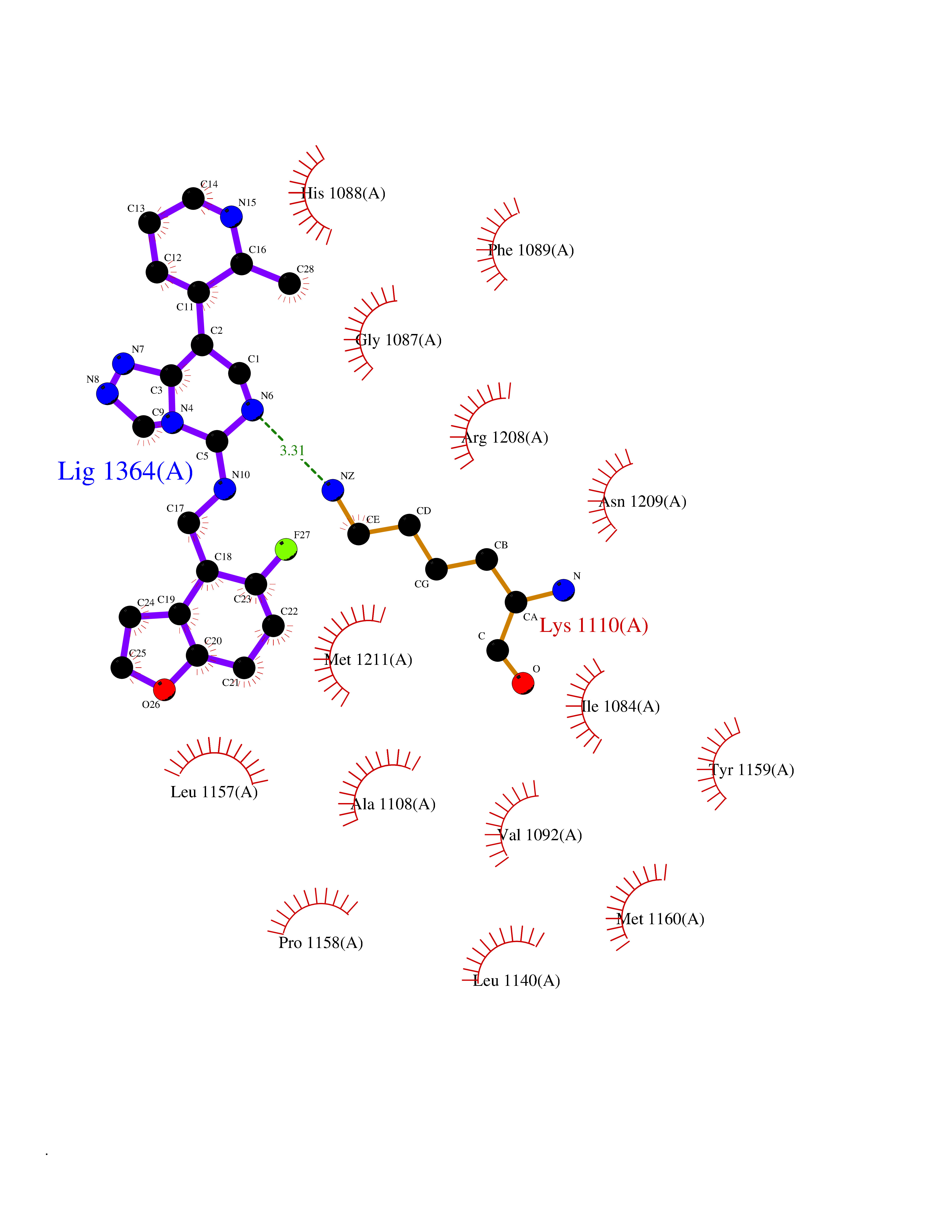 Ligplot Map 3D Binding mode Sequence VHIDLSALNPELVQAVQHVVIGPSSLIVHFNEVIGRGHFGCVYHGTLLDNDGKKIHCAVKSLNRITDIGEVSQFLTEGIIMKDFSHPNVLSLLGICLRSEGSPLVVLPYMKHGDLRNFIRNETHNPTVKDLIGFGLQVAKGMKFLASKKFVHRDLAARNCMLDEKFTVKVADFGLARDMYDKEFDSVHNKTGAKLPVKWMALESLQTQKFTTKSDVWSFGVLLWELMTRGAPPYPDVNTFDITVYLLQGRRLLQPEYCPDPLYEVMLKCWHPKAEMRPSFSELVSRISAIFSTFIGEHYVHVNATYVNVKEG Hydrogen bonds contact Hydrophobic contact | ||||