Job Results:
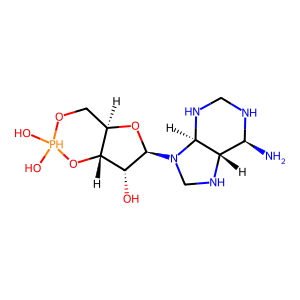
Ligand
Structure
Job ID
e848ab91d8ae28235d6e66c6c8c71011
Job name
NA
Time
2024-09-30 12:19:50
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Ubiquitin carboxyl-terminal hydrolase 30 (USP30) | 5OHK | 6.44 | |
Target general information Gen name USP30 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Deubiquitinating enzyme 30; Ubiquitin thioesterase 30; Ubiquitin-specific-processing protease 30; Ub-specific protease 30 Protein family Peptidase C19 family Biochemical class Peptidase Function Deubiquitinating enzyme tethered to the mitochondrial outer membrane that acts as a key inhibitor of mitophagy by counteracting the action of parkin (PRKN): hydrolyzes ubiquitin attached by parkin on target proteins, such as RHOT1/MIRO1 and TOMM20, thereby blocking parkin's ability to drive mitophagy. Preferentially cleaves 'Lys-6'- and 'Lys-11'-linked polyubiquitin chains, 2 types of linkage that participate in mitophagic signaling. Does not cleave efficiently polyubiquitin phosphorylated at 'Ser-65'. Acts as negative regulator of mitochondrial fusion by mediating deubiquitination of MFN1 and MFN2 (By similarity). Related diseases Pseudohypoaldosteronism 2C (PHA2C) [MIM:614492]: An autosomal dominant disorder characterized by severe hypertension, hyperkalemia, hyperchloremia, mild hyperchloremic metabolic acidosis in some cases, and correction of physiologic abnormalities by thiazide diuretics. {ECO:0000269|PubMed:11498583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuropathy, hereditary sensory and autonomic, 2A (HSAN2A) [MIM:201300]: A form of hereditary sensory and autonomic neuropathy, a genetically and clinically heterogeneous group of disorders characterized by degeneration of dorsal root and autonomic ganglion cells, and by sensory and/or autonomic abnormalities. HSAN2A is an autosomal recessive disorder characterized by impairment of pain, temperature and touch sensation, onset of symptoms in infancy or early childhood, occurrence of distal extremity pathologies (paronychia, whitlows, ulcers, and Charcot joints), frequent amputations, sensory loss that affects all modalities of sensation (lower and upper limbs and perhaps the trunk as well), absence or diminution of tendon reflexes (usually in all limbs), minimal autonomic dysfunction, absence of sensory nerve action potentials, and virtual absence of myelinated fibers with decreased numbers of unmyelinated fibers in sural nerves. {ECO:0000269|PubMed:15060842, ECO:0000269|PubMed:15911806, ECO:0000269|PubMed:18521183}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P54253; Q6NTF9-3 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Hydrolase; Isopeptide bond; Membrane; Mitochondrion; Mitochondrion outer membrane; Protease; Proteomics identification; Reference proteome; Thiol protease; Transmembrane; Transmembrane helix; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 43456.9 Length 379 Aromaticity 0.09 Instability index 50.2 Isoelectric point 6.43 Charge (pH=7) -4.37 2D Binding mode Binding energy (Kcal/mol) -6.53 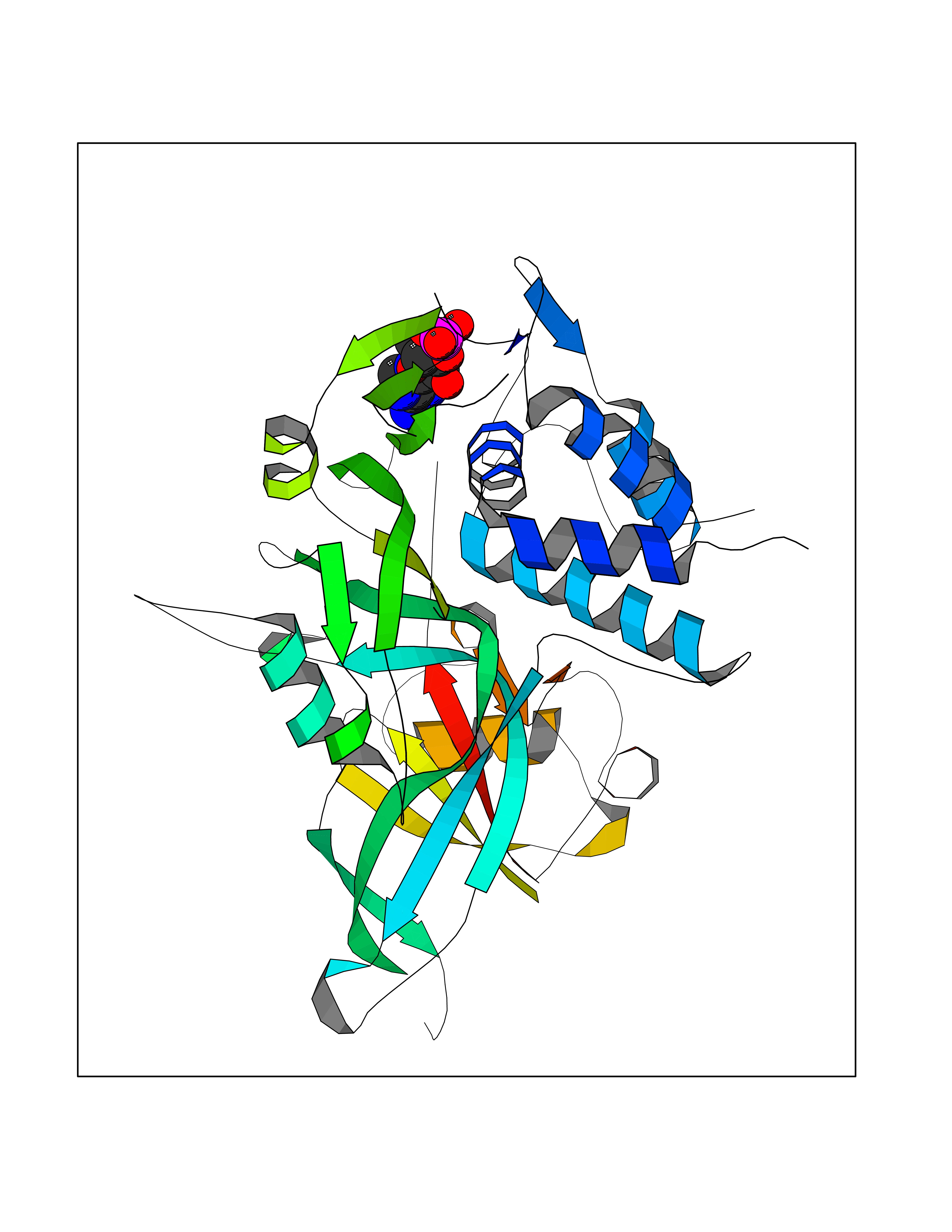 Molscript Map 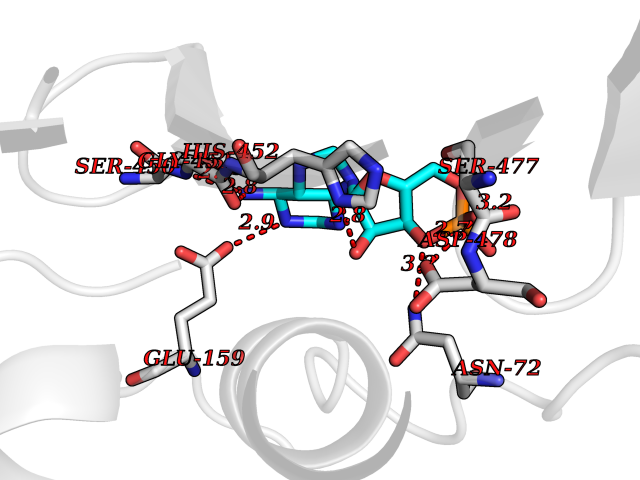 Pymol Map 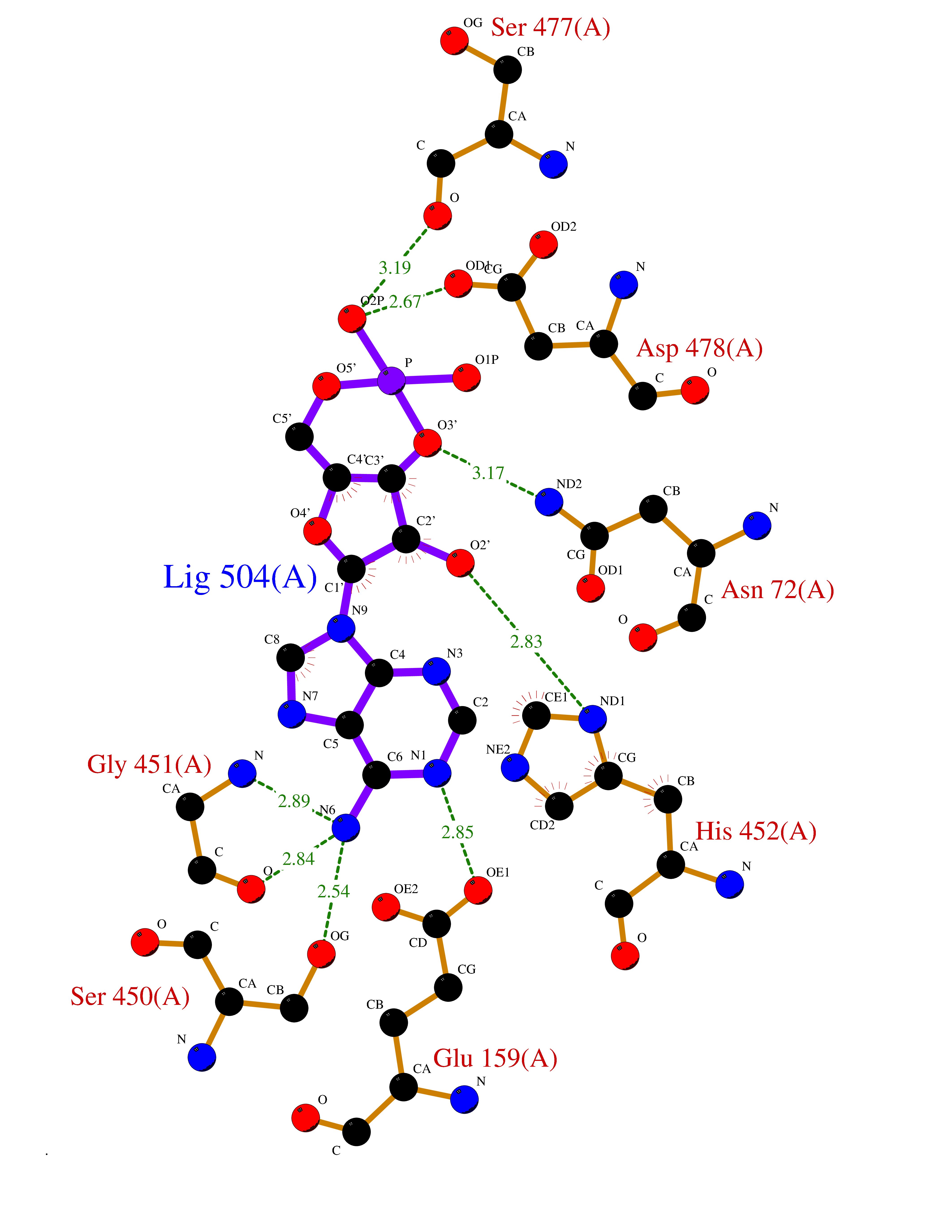 Ligplot Map 3D Binding mode Sequence KGLVPGLVNLGNTCFMNSLLQGLSACPAFIRWLEEFTSQYSRQYLSLTLLHLLKALSCQEVTDDEVLDASCLLDVLRMYRWQISSFEEQDAHELFHVITSSLEDERDGSGSHWKSQHPFHGRLTSNMVCKHCEHQSPVRFDTFDSLSLSIPAATWGHPLTLDHCLHHFISSESVRDVVCDNCTKRTTFVKQLKLGKLPQCLCIHLQRLSWSSHGTPLKRHEHVQFNEDLSMDEYKYHSNASTYLFRLMAVVVHHGDMHSGHFVTYRRSPPSSNQWLWVSDDTVRKASLQEVLSSSAYLLFYERVMQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRG Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Urokinase plasminogen activator surface receptor (PLAUR) | 2FD6 | 6.43 | |
Target general information Gen name PLAUR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Urokinase-type plasminogen activator receptor; Urokinase plasminogen activator receptor; Monocyte activation antigenMo3; CD87 antigen; CD87 Protein family NA Biochemical class NA Function Plays a role in localizing and promoting plasmin formation. Mediates the proteolysis-independent signal transduction activation effects of U-PA. It is subject to negative-feedback regulation by U-PA which cleaves it into an inactive form. Acts as a receptor for urokinase plasminogen activator. Related diseases Limb pelvis hypoplasia aplasia syndrome (LPHAS) [MIM:276820]: A syndrome of severe deficiency of the extremities due to hypo- or aplasia of one or more long bones of one or more limbs. Pelvic manifestations include hip dislocation, hypoplastic iliac bone and aplastic pubic bones. Thoracic deformity, unusual facies and genitourinary anomalies can be present. {ECO:0000269|PubMed:16826533, ECO:0000269|PubMed:17431918, ECO:0000269|PubMed:20949531, ECO:0000269|PubMed:21271649, ECO:0000269|PubMed:27638328}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fuhrmann syndrome (FUHRS) [MIM:228930]: Distinct limb-malformation disorder characterized also by various degrees of limb aplasia/hypoplasia and joint dysplasia. {ECO:0000269|PubMed:16826533}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06245; DB00031; DB00013; DB05476 Interacts with P03950; P00749; P54252; P06396; A0A6Q8PF08; Q7Z699 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell junction; Cell membrane; Cell projection; Direct protein sequencing; Disulfide bond; Glycoprotein; GPI-anchor; Lipoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Repeat; Secreted; Signal Protein physicochemical properties Chain ID H Molecular weight (Da) 22907.4 Length 212 Aromaticity 0.12 Instability index 38.8 Isoelectric point 7.82 Charge (pH=7) 1.16 2D Binding mode Binding energy (Kcal/mol) -6.5 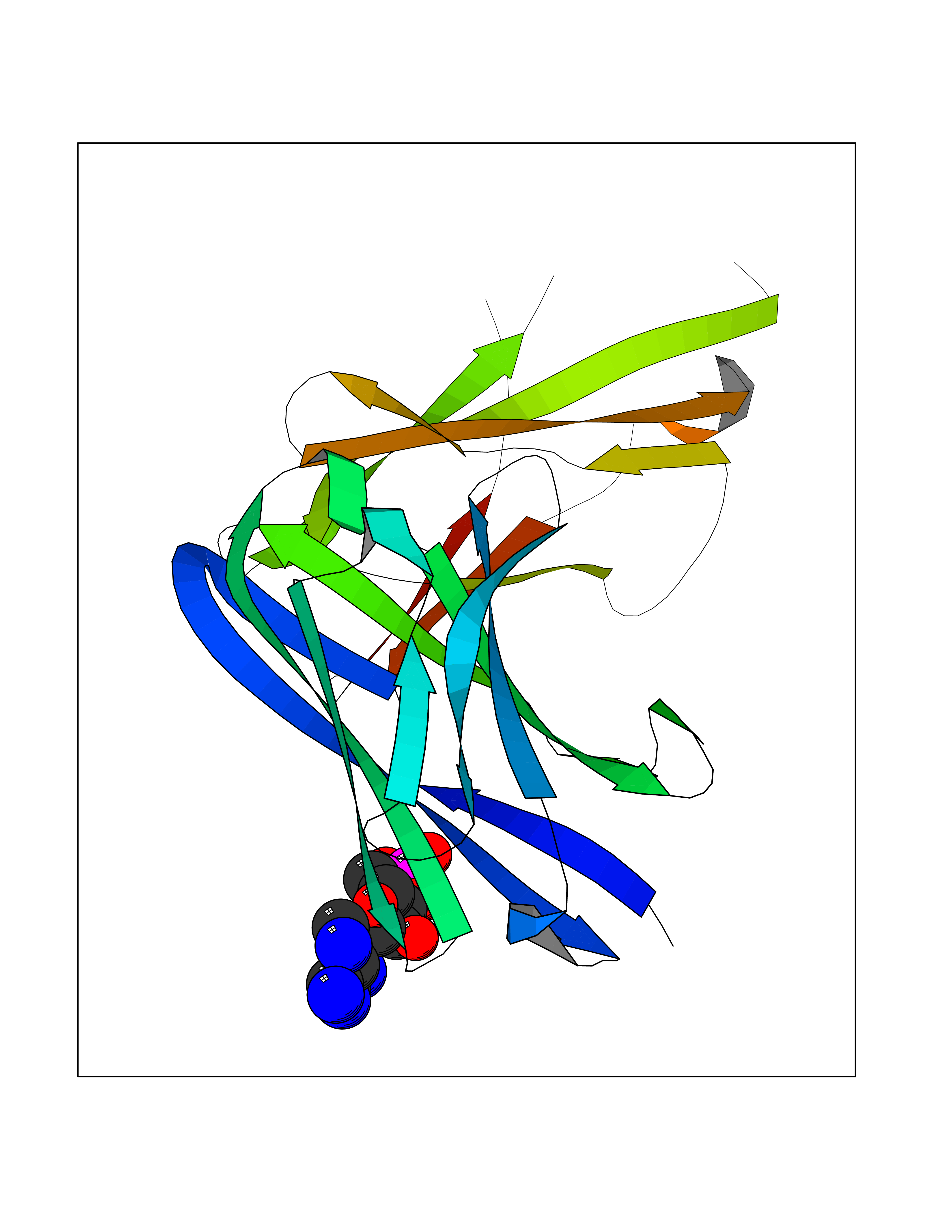 Molscript Map 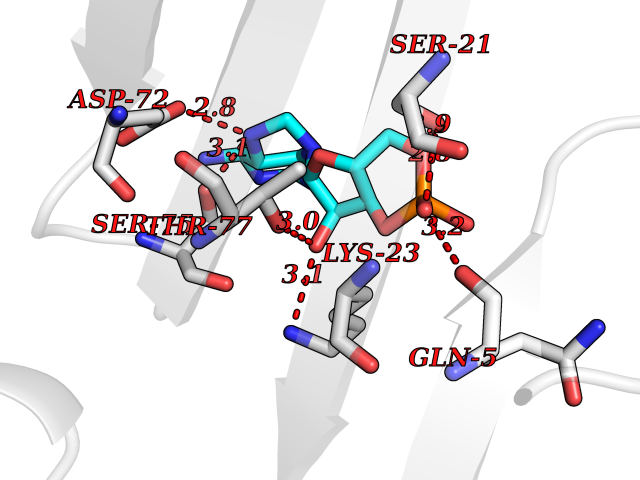 Pymol Map 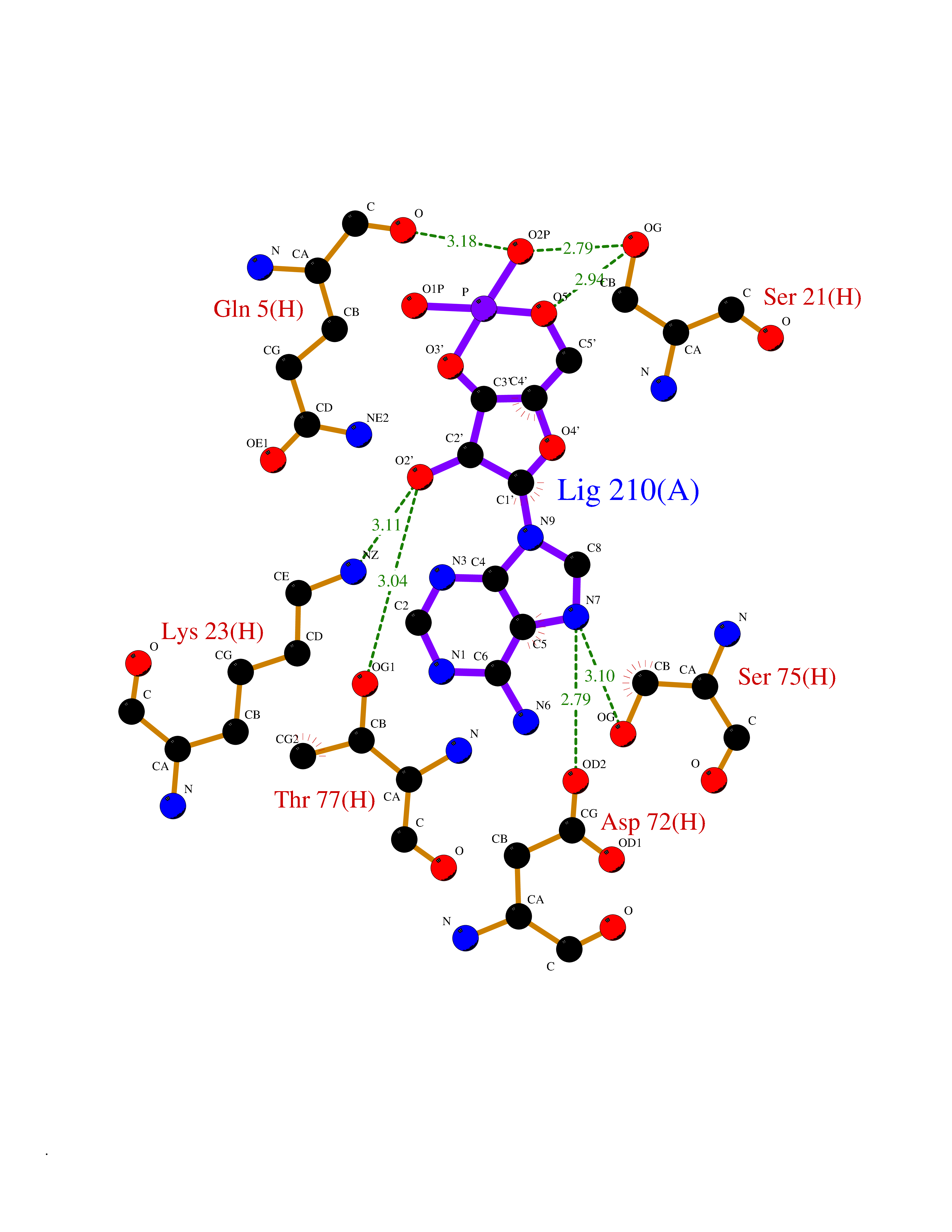 Ligplot Map 3D Binding mode Sequence GVKLQQSGPEVVKPGASVKISCKASGYSFTNFYIHWVKQRPGQGLEWIGWIFHGSDNTEYNEKFKDKATLTADTSSSTAYMQLSSLTSEDSAVYFCARWGPHWYFDVWGQGTTVTVSSAKTTPPSVYPLAPNSMVTLGCLVKGYFPEPVTVTWNSGSLSSGVHTFPAVLQSDLYTLSSSVTVPSSTWPSETVTCNVAHPASSTKVDKKIAAA Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Platelet glycoprotein VI (GP6) | 5OU7 | 6.43 | |
Target general information Gen name GP6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Glycoprotein 6; GPVI Protein family NA Biochemical class NA Function Collagen receptor involved in collagen-induced platelet adhesion and activation. Plays a key role in platelet procoagulant activity and subsequent thrombin and fibrin formation. This procoagulant function may contribute to arterial and venous thrombus formation. The signaling pathway involves the FcR gamma-chain, the Src kinases (likely FYN or LYN) and SYK, the adapter protein LAT and leads to the activation of PLCG2. Related diseases Bleeding disorder, platelet-type, 11 (BDPLT11) [MIM:614201]: A mild to moderate bleeding disorder caused by defective platelet activation and aggregation in response to collagen. {ECO:0000269|PubMed:19549989, ECO:0000269|PubMed:19552682}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P06241; P07948; P06241; P07948 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Blood coagulation; Cell membrane; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hemostasis; Immunoglobulin domain; Membrane; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 19027.4 Length 173 Aromaticity 0.11 Instability index 37.14 Isoelectric point 8.68 Charge (pH=7) 2.52 2D Binding mode Binding energy (Kcal/mol) -6.82 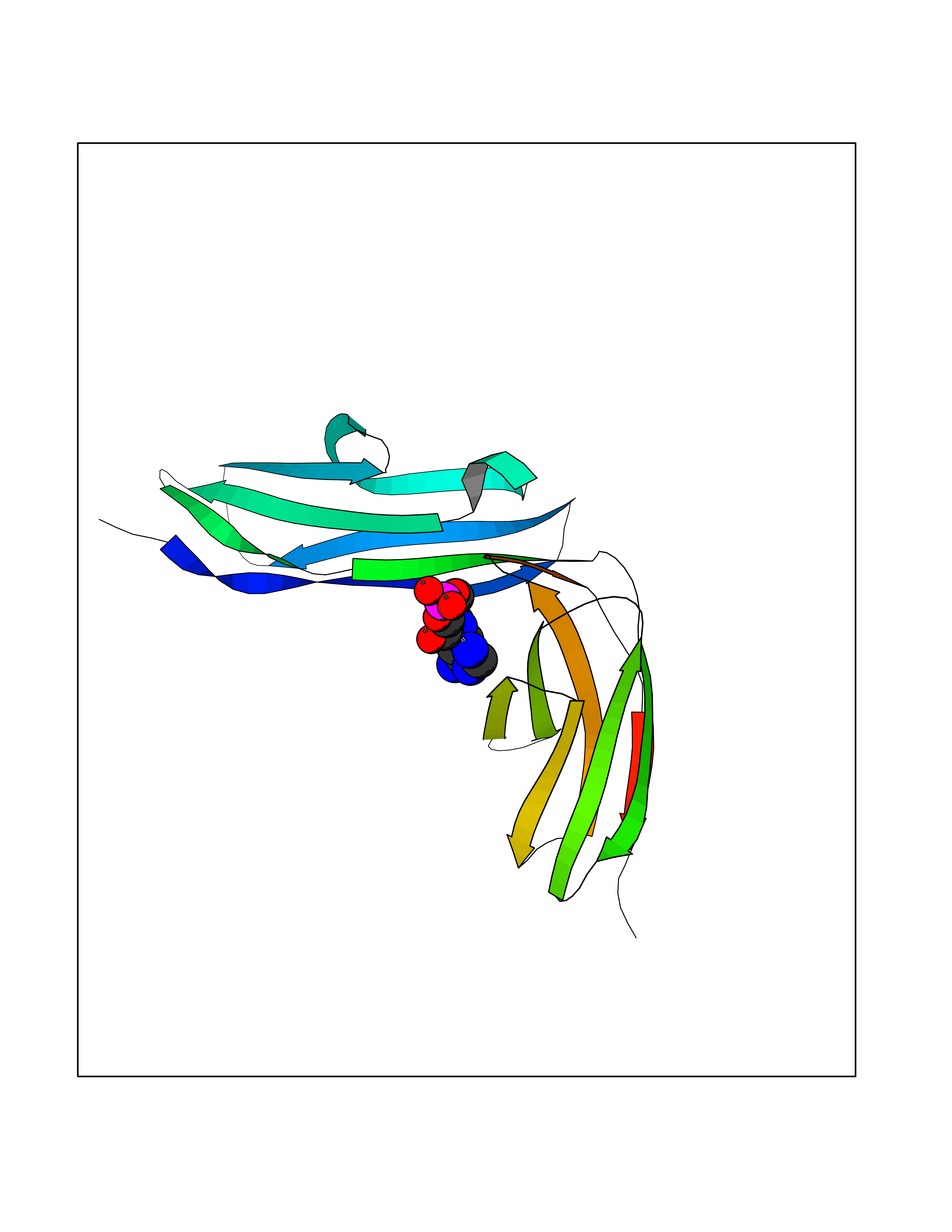 Molscript Map 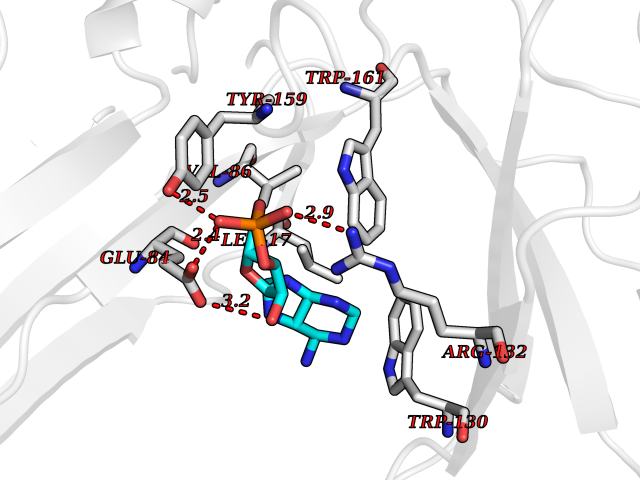 Pymol Map 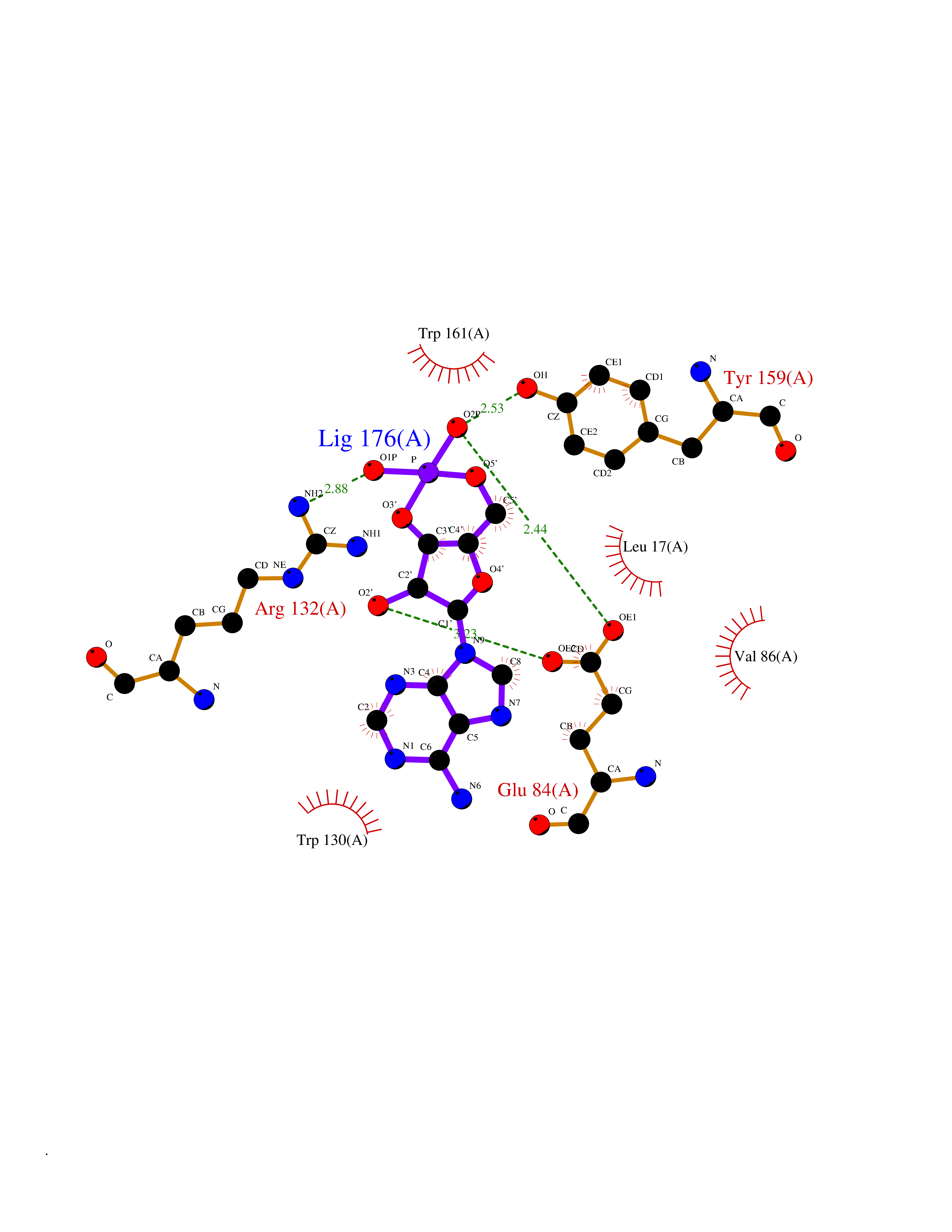 Ligplot Map 3D Binding mode Sequence SGPLPKPSLQALPSSLVPLEKPVTLRCQGPPGVDLYRLEKLSSSRYQDQAVLFIPAMKRSLAGRYRCSYQNGSLWSLPSDQLELVATGVFAKPSLSAQPGSGGDVTLQCQTRYGFDQFALYKEGDPERWYRASFPIITVTAAHSGTYRCYSFSSRDPYLWSAPSDPLELVVTG Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Fatty acid-binding protein 5 (FABP5) | 5UR9 | 6.43 | |
Target general information Gen name FABP5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Psoriasis-associated fatty acid-binding protein homolog; PA-FABP; Fatty acid-binding protein, epidermal; Fatty Acid BindingProtein mal1; Epidermal-type fatty acid-binding protein; E-FABP Protein family Calycin superfamily, Fatty-acid binding protein (FABP) family Biochemical class Fatty acid binding protein Function Intracellular carrier for long-chain fatty acids and related active lipids, such as the endocannabinoid, that regulates the metabolism and actions of the ligands they bind. In addition to the cytosolic transport, selectively delivers specific fatty acids from the cytosol to the nucleus, wherein they activate nuclear receptors. Delivers retinoic acid to the nuclear receptor peroxisome proliferator-activated receptor delta; which promotes proliferation and survival. May also serve as a synaptic carrier of endocannabinoid at central synapses and thus controls retrograde endocannabinoid signaling. Modulates inflammation by regulating PTGES induction via NF-kappa-B activation, and prostaglandin E2 (PGE2) biosynthesis during inflammation. May be involved in keratinocyte differentiation. Related diseases Erythrocytosis, familial, 4 (ECYT4) [MIM:611783]: An autosomal dominant disorder characterized by elevated serum hemoglobin and hematocrit, and normal platelet and leukocyte counts. {ECO:0000269|PubMed:18184961, ECO:0000269|PubMed:18378852, ECO:0000269|PubMed:19208626, ECO:0000269|PubMed:22367913}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03796 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Disulfide bond; Lipid transport; Lipid-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Secreted; Synapse; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 15033.1 Length 134 Aromaticity 0.06 Instability index 28.26 Isoelectric point 6.83 Charge (pH=7) -0.14 2D Binding mode Binding energy (Kcal/mol) -7.43  Molscript Map 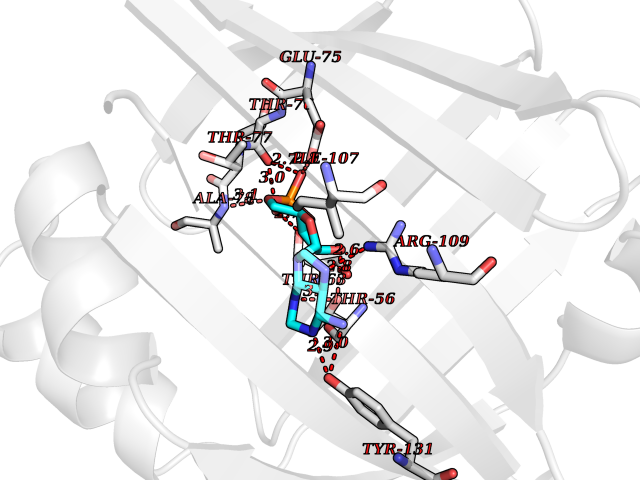 Pymol Map 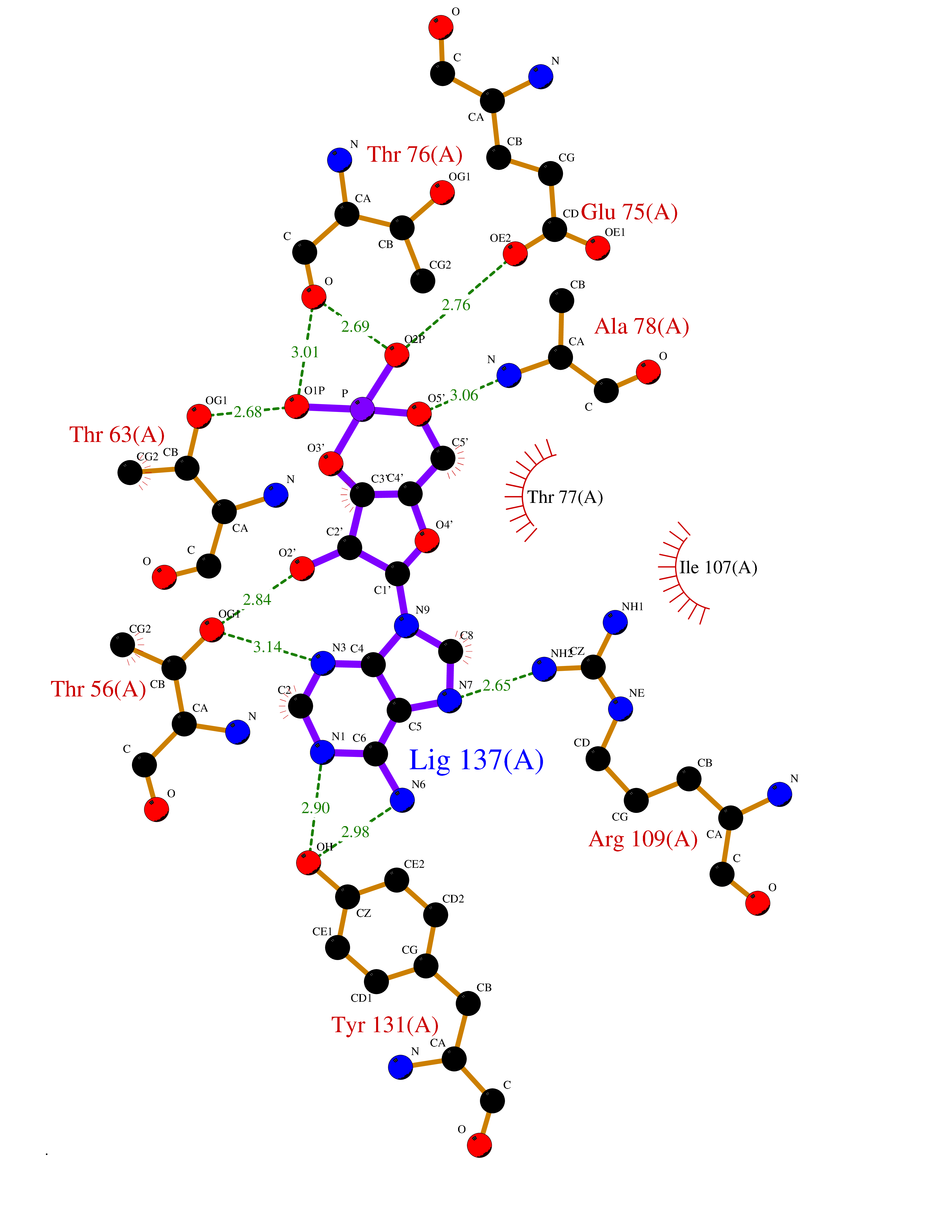 Ligplot Map 3D Binding mode Sequence ATVQQLEGRWRLVDSKGFDEYMKELGVGIALRKMGAMAKPDCIITCDGKNLTIKTESTLKTTQFSCTLGEKFEETTADGRKTQTVCNFTDGALVQHQEWDGKESTITRKLKDGKLVVECVMNNVTCTRIYEKVE Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | ATP-binding cassette transporter G1 (ABCG1) | 7FDV | 6.42 | |
Target general information Gen name ABCG1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms White protein homolog; WHT1; ATP-binding cassette transporter 8; ATP-binding cassette sub-family G member 1; ABC8 Protein family ABC transporter superfamily, ABCG family, Eye pigment precursor importer (TC 3.A.1.204) subfamily Biochemical class ABC transporter Function Catalyzes the efflux of phospholipids such as sphingomyelin, cholesterol and its oxygenated derivatives like 7beta-hydroxycholesterol and this transport is coupled to hydrlysis of ATP. The lipid efflux is ALB-dependent. Is an active component of the macrophage lipid export complex. Could also be involved in intracellular lipid transport processes. The role in cellular lipid homeostasis may not be limited to macrophages. Prevents cell death by transporting cytotoxic 7beta-hydroxycholesterol. Related diseases Faundes-Banka syndrome (FABAS) [MIM:619376]: An autosomal dominant disorder characterized by variable combinations of developmental delay, microcephaly, micrognathia and dysmorphic features. {ECO:0000269|PubMed:33547280}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00171; DB00163 Interacts with Q9H172; Q13520; P13473-2; O75400-2; Q9Y371 EC number NA Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Endoplasmic reticulum; Golgi apparatus; Lipid transport; Lipoprotein; Membrane; Nucleotide-binding; Palmitate; Proteomics identification; Reference proteome; Translocase; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 59054.9 Length 529 Aromaticity 0.12 Instability index 32.7 Isoelectric point 8.45 Charge (pH=7) 5.45 2D Binding mode Binding energy (Kcal/mol) -6.57  Molscript Map 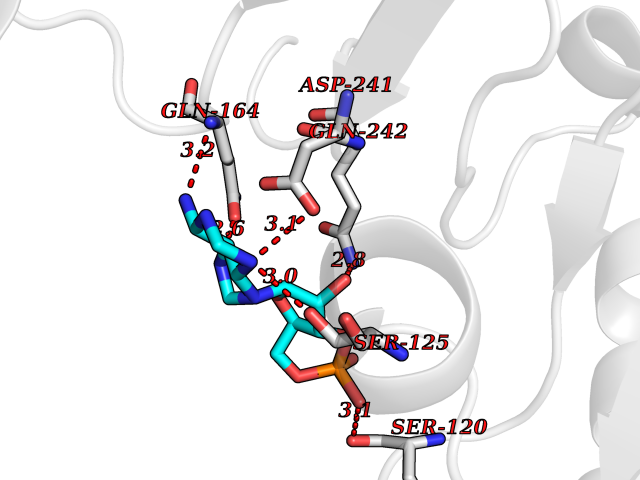 Pymol Map 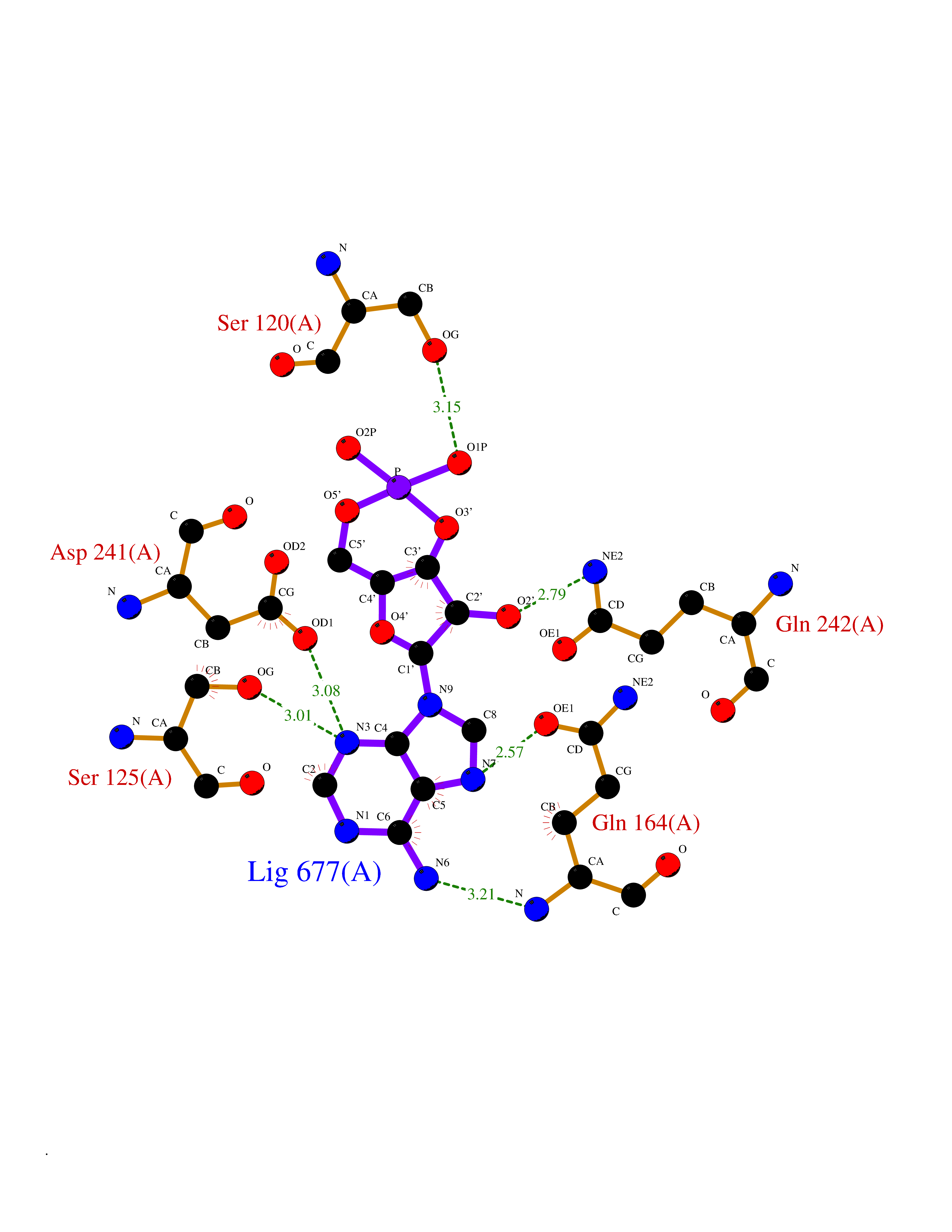 Ligplot Map 3D Binding mode Sequence RAAVNIEFRDLSYSVPGYKTLLKGISGKFNSGELVAIMGPSGAGKSTLMNILAGYRETGMKGAVLINGLPRDLRCFRKVSCYIMQDDMLLPHLTVQEAMMVSAHLKLQEKDEGRREMVKEILTALGLLSCANTRTGSLSGGQRKRLAIALELVNNPPVMFFDQPTSGLDSASCFQVVSLMKGLAQGGRSIICTIHQPSAKLFELFDQLYVLSQGQCVYRGKVCNLVPYLRDLGLNCPTYHNPADFVMEVASGEYCLTQFCILFKRTFLSIMRDSVLTHLRITSHIGIGLLIGLLYLGIGNEAKKVLSNSGFLFFSMLFLMFAALMPTVLTFPLEMGVFLREHLNYWYSLKAYYLAKTMADVPFQIMFPVAYCSIVYWMTSQPSDAVRFVLFAALGTMTSLVAQSLGLLIGAASTSLQVATFVGPVTAIPVLLFSGFFVSFDTIPTYLQWMSYISYVRYGFEGVILSIYGLDREDLHCDIDETCHFQKSEAILRELDVENAKLYLDFIVLGIFFISLRLIAYFVLRYKIR Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | CAAX farnesyltransferase beta (FNTB) | 1SA4 | 6.42 | |
Target general information Gen name FNTB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAS proteins prenyltransferasebeta; FTase-beta; FNTB; CAAX farnesyltransferase beta subunit Protein family Protein prenyltransferase subunit beta family Biochemical class Alkyl aryl transferase Function Essential subunit of the farnesyltransferase complex. Catalyzes the transfer of a farnesyl moiety from farnesyl diphosphate to a cysteine at the fourth position from the C- terminus of several proteins having the C-terminal sequence Cys- aliphatic-aliphatic-X. Related diseases Lethal congenital contracture syndrome 2 (LCCS2) [MIM:607598]: A form of lethal congenital contracture syndrome, an autosomal recessive disorder characterized by degeneration of anterior horn neurons, extreme skeletal muscle atrophy, and congenital non-progressive joint contractures (arthrogryposis). The contractures can involve the upper or lower limbs and/or the vertebral column, leading to various degrees of flexion or extension limitations evident at birth. LCCS2 patients manifest craniofacial/ocular findings, lack of hydrops, multiple pterygia, and fractures, as well as a normal duration of pregnancy and a unique feature of a markedly distended urinary bladder (neurogenic bladder defect). The phenotype suggests a spinal cord neuropathic etiology. {ECO:0000269|PubMed:17701904}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythroleukemia, familial (FERLK) [MIM:133180]: An autosomal dominant myeloproliferative disorder characterized by neoplastic proliferation of erythroblastic and myeloblastic elements with atypical erythroblasts and myeloblasts in the peripheral blood. Disease penetrance is incomplete. {ECO:0000269|PubMed:27416908}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Visceral neuropathy, familial, 1, autosomal recessive (VSCN1) [MIM:243180]: An autosomal recessive disorder characterized by intestinal dysmotility due to aganglionosis (Hirschsprung disease), hypoganglionosis, and/or chronic intestinal pseudoobstruction. Additional variable features are progressive peripheral neuropathy, arthrogryposis, hypoplasia or aplasia of the olfactory bulb and of the external auditory canals, microtia or anotia, and facial dysmorphism. Some patients present structural cardiac anomalies and arthrogryposis with multiple pterygia. {ECO:0000269|PubMed:33497358}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07216; DB08674; DB08676; DB06953; DB07771; DB07895; DB04893; DB07780; DB07841; DB07227; DB06448; DB04960 Interacts with P21549; Q9BWW8; Q8N5M1; Q8N4L8; G5E9A7; P49354; P31273; A5PL33-2; Q9H0W8; Q7Z699; O43711 EC number EC 2.5.1.58 Uniprot keywords 3D-structure; Alternative splicing; Lipid metabolism; Metal-binding; Phosphoprotein; Prenyltransferase; Proteomics identification; Reference proteome; Repeat; Transferase; Zinc Protein physicochemical properties Chain ID B,A Molecular weight (Da) 83751.2 Length 725 Aromaticity 0.11 Instability index 46.44 Isoelectric point 5.61 Charge (pH=7) -20.1 2D Binding mode Binding energy (Kcal/mol) -6.83  Molscript Map 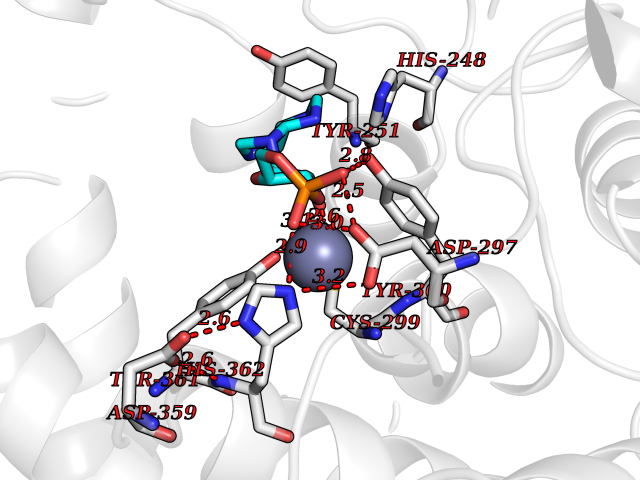 Pymol Map 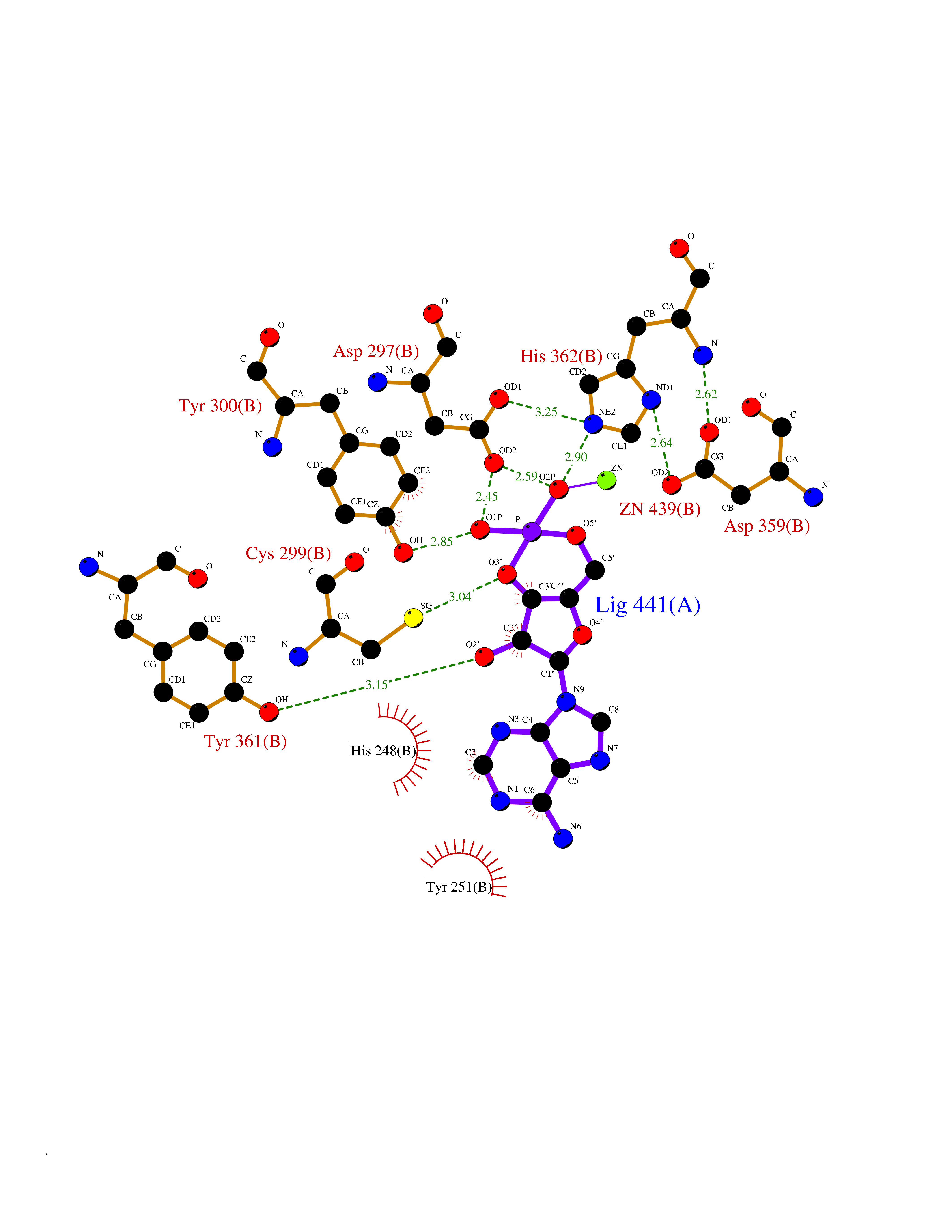 Ligplot Map 3D Binding mode Sequence FVSLDSPSYVLYRDRAEWADIDPVPQNDGPNPVVQIIYSDKFRDVYDYFRAVLQRDERSERAFKLTRDAIELNAANYTVWHFRRVLLKSLQKDLHEEMNYITAIIEEQPKNYQVWHHRRVLVEWLRDPSQELEFIADILNQDAKNYHAWQHRQWVIQEFKLWDNELQYVDQLLKEDVRNNSVWNQRYFVISNTTGYNDRAVLEREVQYTLEMIKLVPHNESAWNYLKGILQDRGLSKYPNLLNQLLDLQPSHSSPYLIAFLVDIYEDMLENQCDNKEDILNKALELCEILAKEKDTIRKEYWRYIGRSLQSKHSTSSPVWSEPLYSLRPEHARERLQDDSVETVTSIEQAKVEEKIQEVFSSYKFNHLVPRLVLQREKHFHYLKRGLRQLTDAYECLDASRPWLCYWILHSLELLDEPIPQIVATDVCQFLELCQSPEGGFGGGPGQYPHLAPTYAAVNALCIIGTEEAYDIINREKLLQYLYSLKQPDGSFLMHVGGEVDVRSAYCAASVASLTNIITPDLFEGTAEWIARCQNWEGGIGGVPGMEAHGGYTFCGLAALVILKRERSLNLKSLLQWVTSRQMRFEGGFQGRCNKLVDGCYSFWQAGLLPLLHRALHAQGDPALSMSHWMFHQQALQEYILMCCQCPAGGLLDKPGKSRDFYHTCYCLSGLSIAQHFGSGAMLHDVVLGVPENALQPTHPVYNIGPDKVIQATTYFLQKPVPGFE Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Gamma-aminobutyric acid type B receptor subunit 1 | 4MS4 | 6.41 | |
Target general information Gen name GABBR1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms GPRC3B;GPR51 Protein family G-protein coupled receptor 3 family, GABA-B receptor subfamily Biochemical class Signaling protein / antagonist Function G-protein coupled GABA receptor activity. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08891; DB08892; DB00181; DB00363; DB02530; DB05010; DB09072 Interacts with Q9UBS5; Q9UBS5-2; P46459; Q86UR5 EC number NA Uniprot keywords 3D-structure; Cell membrane; Coiled coil; Direct protein sequencing; Disease variant; Disulfide bond; Epilepsy; G-protein coupled receptor; Glycoprotein; Intellectual disability; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 46502.1 Length 408 Aromaticity 0.12 Instability index 50.05 Isoelectric point 5.78 Charge (pH=7) -5.62 2D Binding mode Binding energy (Kcal/mol) -8.18 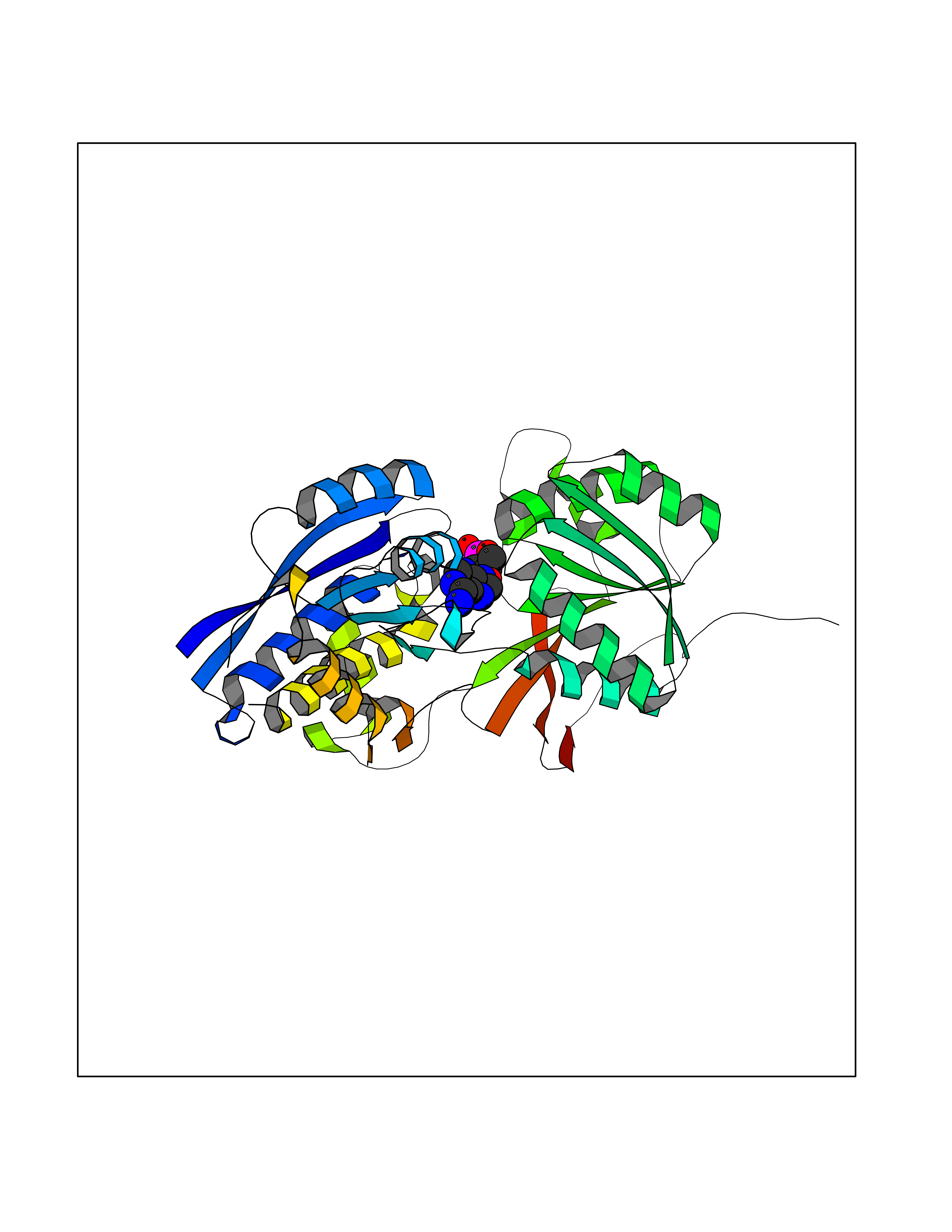 Molscript Map 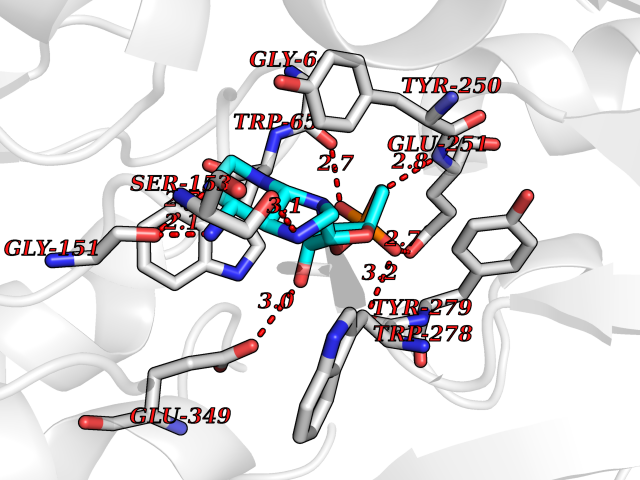 Pymol Map 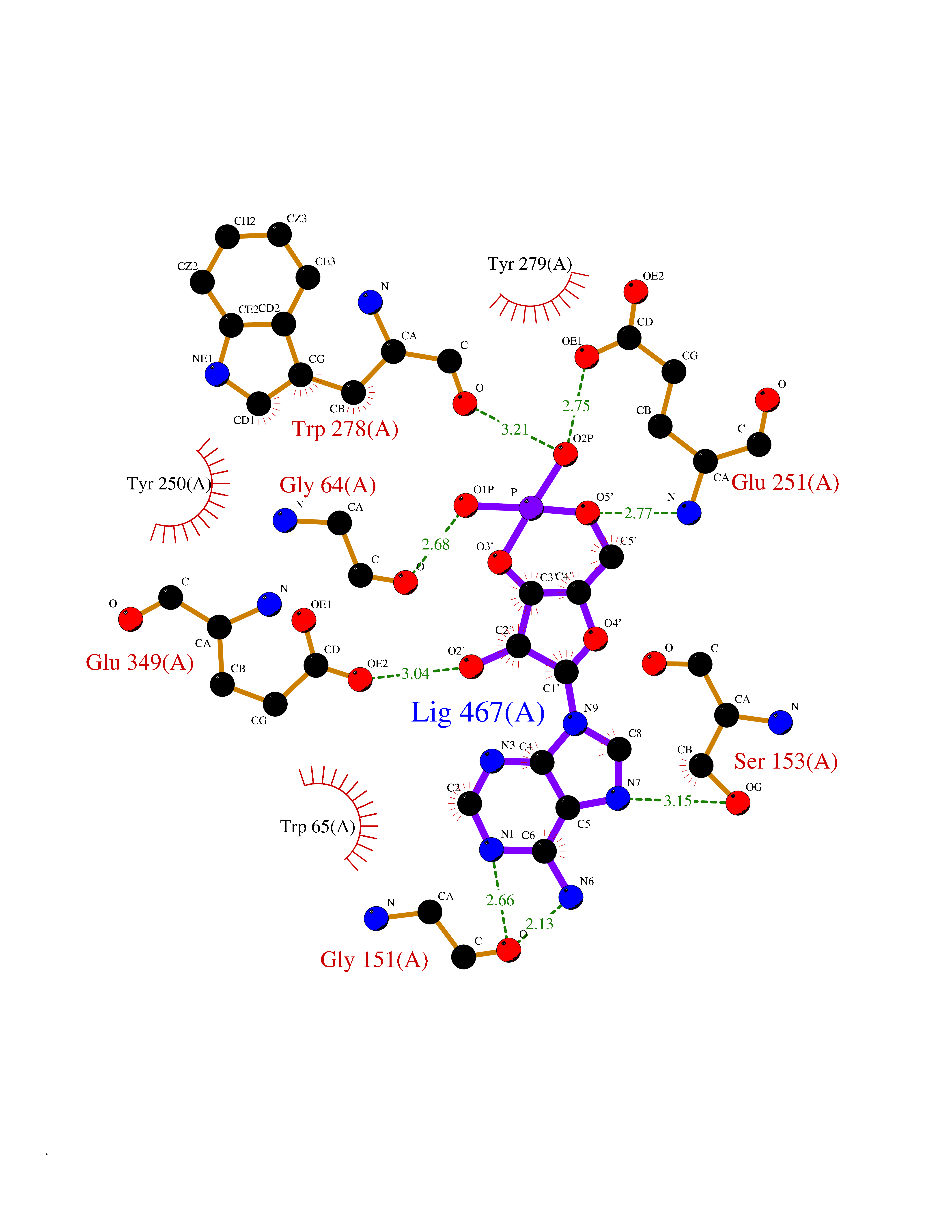 Ligplot Map 3D Binding mode Sequence RRAVYIGALFPMSGGWPGGQACQPAVEMALEDVNSRRDILPDYELKLIHHDSKCDPGQATKYLYELLYNDPIKIILMPGCSSVSTLVAEAARMWNLIVLSYGSSSPALSNRQRFPTFFRTHPSATLHNPTRVKLFEKWGWKKIATIQQTTEVFTSTLDDLEERVKEAGIEITFRQSFFSDPAVPVKNLKRQDARIIVGLFYETEARKVFCEVYKERLFGKKYVWFLIGWYADNWFKIYDPSINCTVDEMTEAVEGHITTEIVMLNPANTRSISNMTSQEFVEKLTKRLKRHPEETGGFQEAPLAYDAIWALALALNKTSRLEDFNYNNQTITDQIYRAMNSSSFEGVSGHVVFDASGSRMAWTLIEQLQGGSYKKIGYYDSTKDDLSWSKTDKWIGGSPPADDYKDDD Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Endocytic receptor Endo180 (MRC2) | 5E4K | 6.40 | |
Target general information Gen name MRC2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Urokinase-type plasminogen activator receptor-associated protein; Urokinase receptor-associated protein; UPARAP; UPAR-associated protein; Macrophage mannose receptor 2; KIAA0709; Endocytic receptor 18 Protein family NA Biochemical class NA Function May play a role as endocytotic lectin receptor displaying calcium-dependent lectin activity. Internalizes glycosylated ligands from the extracellular space for release in an endosomal compartment via clathrin-mediated endocytosis. May be involved in plasminogen activation system controlling the extracellular level of PLAUR/PLAU, and thus may regulate protease activity at the cell surface. May contribute to cellular uptake, remodeling and degradation of extracellular collagen matrices. May play a role during cancer progression as well as in other chronic tissue destructive diseases acting on collagen turnover. May participate in remodeling of extracellular matrix cooperating with the matrix metalloproteinases (MMPs). Related diseases Lymphatic malformation 8 (LMPHM8) [MIM:618773]: A form of primary lymphedema, a disease characterized by swelling of body parts due to developmental anomalies and functional defects of the lymphatic system. Adult patients with lymphedema may suffer from recurrent local infections. Impaired lymphatic drainage in the fetus can develop into hydrops fetalis, a severe condition characterized by excessive fluid accumulation in more than two fetal extra-vascular compartments and body cavities, placental enlargement and edema, pericardial or pleural effusion, or ascites. LMPHM8 is an autosomal recessive form characterized by onset in utero and fetal death due to non-immune hydrops fetalis. {ECO:0000269|PubMed:30115739}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P02454 EC number NA Uniprot keywords 3D-structure; Calcium; Direct protein sequencing; Disulfide bond; Endocytosis; Glycoprotein; Isopeptide bond; Lectin; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 48666.8 Length 424 Aromaticity 0.12 Instability index 54.45 Isoelectric point 5.01 Charge (pH=7) -18.38 2D Binding mode Binding energy (Kcal/mol) -6.78 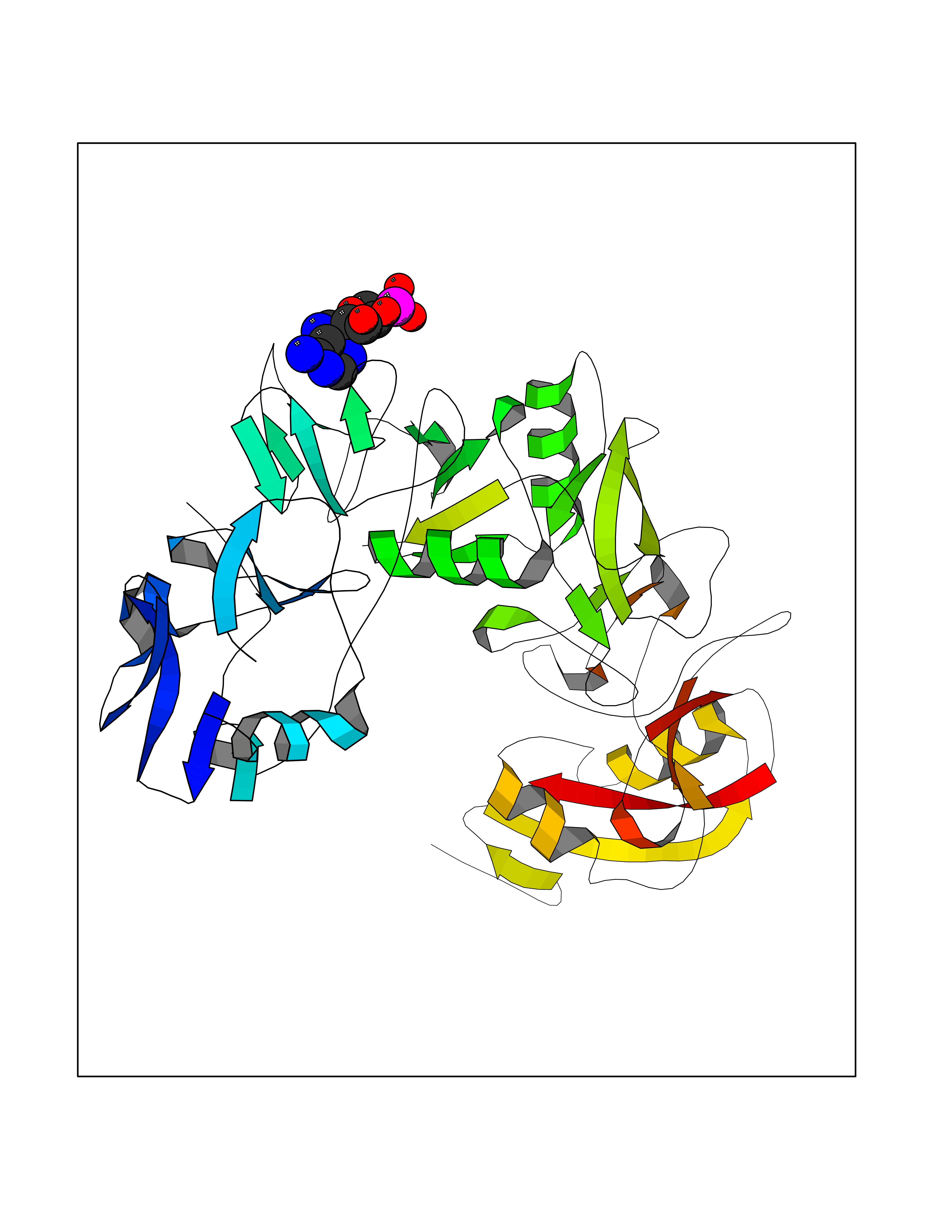 Molscript Map 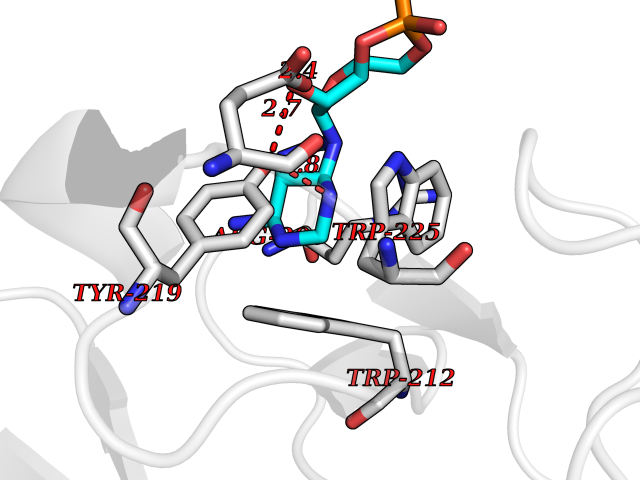 Pymol Map 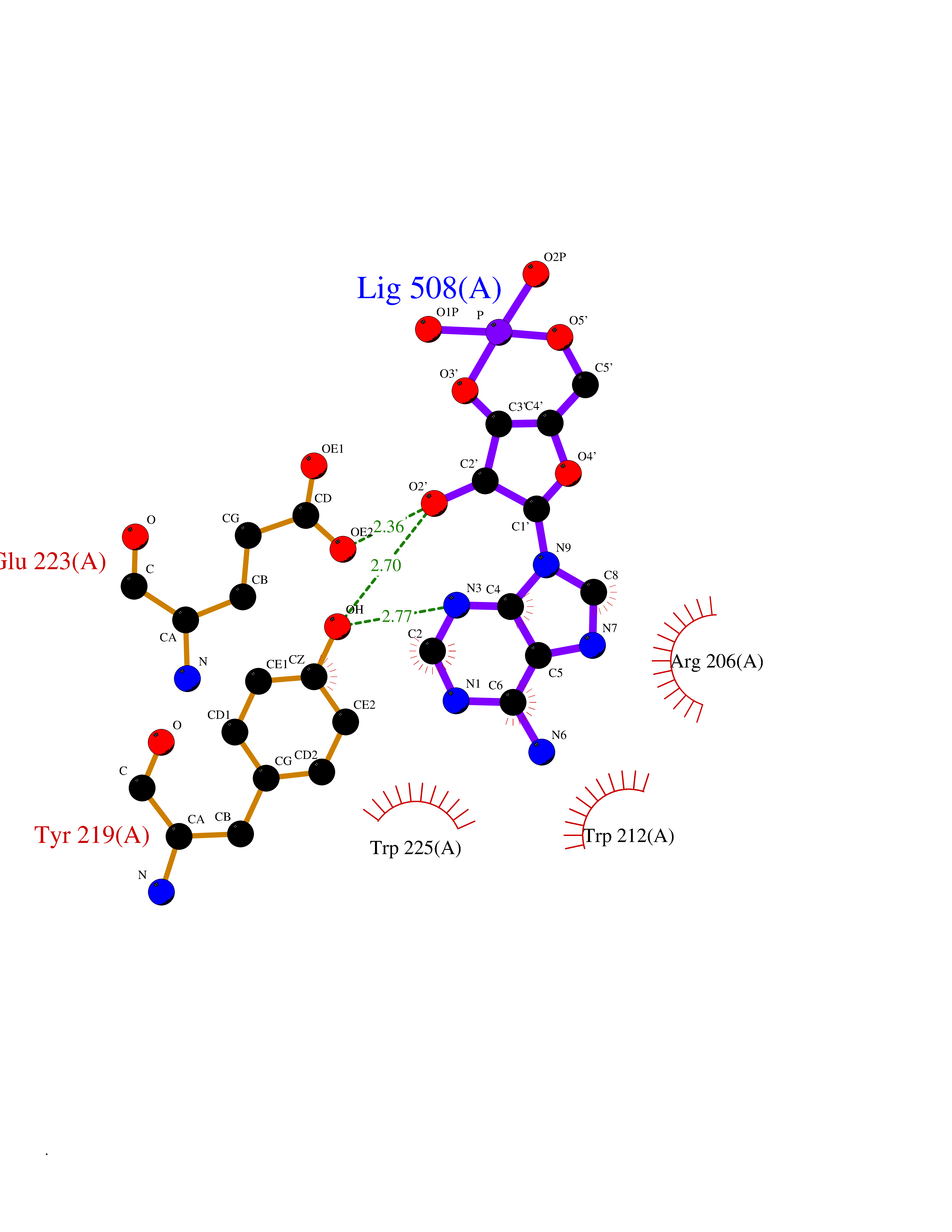 Ligplot Map 3D Binding mode Sequence ALPEPNIFLIFSHGLQGCLEAQGGQVRVTPACNTSLPAQRWKWVSRNRLFNLGTMQCLGTASLGMYECDREALNLRWHCRTLGDQLSLLLQWRIYGSEEDLCALPYHEVYTIQGNSHGKPCTIPFKYDNQWFHGCTSTGREDGHLWCATTQDYGKDERWGFCPIKSNDCETFWDKDQLTDSCYQFNFQSTLSWREAWASCEQQGADLLSITEIHEQTYINGLLTGYSSTLWIGLNDLDTSGGWQWSDNSPLKYLNWESDQPDNPSEENCGVIRTESSGGWQNRDCSIALPYVCKKKPVECEPSWQPFQGHCYRLQAEKRSWQESKKACLRGGGDLVSIHSMAELEFITKQIKQEVEELWIGLNDLKLQMNFEWSDGSLVSFTHWHPFEPNNFRDSLEDCVTIWGPEGRWNDSPCNQSLPSICKK Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Phosphodiesterase 3A (PDE3A) | 7EG0 | 6.39 | |
Target general information Gen name PDE3A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cGMP-inhibited 3',5'-cyclic phosphodiesterase A; Phosphodiesterase 3A; Cyclic GMP-inhibited phosphodiesterase A; Cyclic GMP inhibited phosphodiesterase A; CGI-PDE A Protein family Cyclic nucleotide phosphodiesterase family, PDE3 subfamily Biochemical class Phosphoric diester hydrolase Function Cyclic nucleotide phosphodiesterase with a dual-specificity for the second messengers cAMP and cGMP, which are key regulators of many important physiological processes. Related diseases Hypertension and brachydactyly syndrome (HTNB) [MIM:112410]: A syndrome characterized by brachydactyly type E, severe salt-independent but age-dependent hypertension, an increased fibroblast growth rate, neurovascular contact at the rostral-ventrolateral medulla, and altered baroreflex blood pressure regulation. It results in death from stroke before age 50 years when untreated. Brachydactyly type E is characterized by shortening of the fingers mainly in the metacarpals and metatarsals. {ECO:0000269|PubMed:25961942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01223; DB01427; DB00261; DB00201; DB01166; DB04880; DB05266; DB00922; DB00235; DB01303; DB00277; DB08811; DB09283 Interacts with Q9Y6D6; Q9Y6D5 EC number EC 3.1.4.17 Uniprot keywords 3D-structure; cAMP; cGMP; Cytoplasm; Direct protein sequencing; Disease variant; Hydrolase; Isopeptide bond; Manganese; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 108171 Length 939 Aromaticity 0.11 Instability index 39.75 Isoelectric point 6.53 Charge (pH=7) -4.15 2D Binding mode Binding energy (Kcal/mol) -7.06 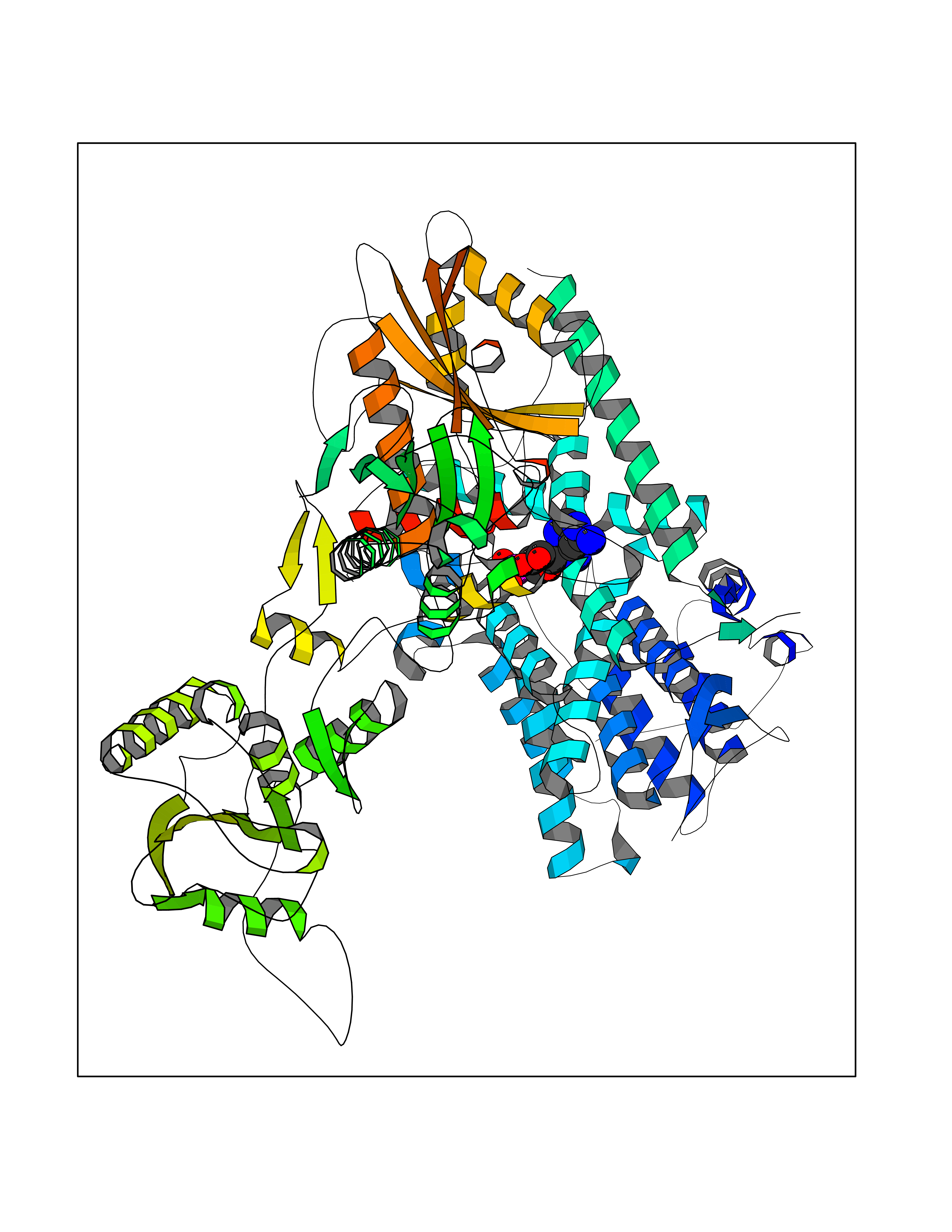 Molscript Map 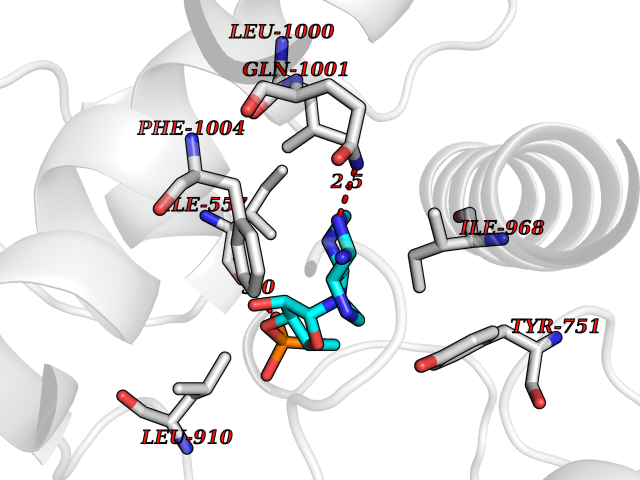 Pymol Map 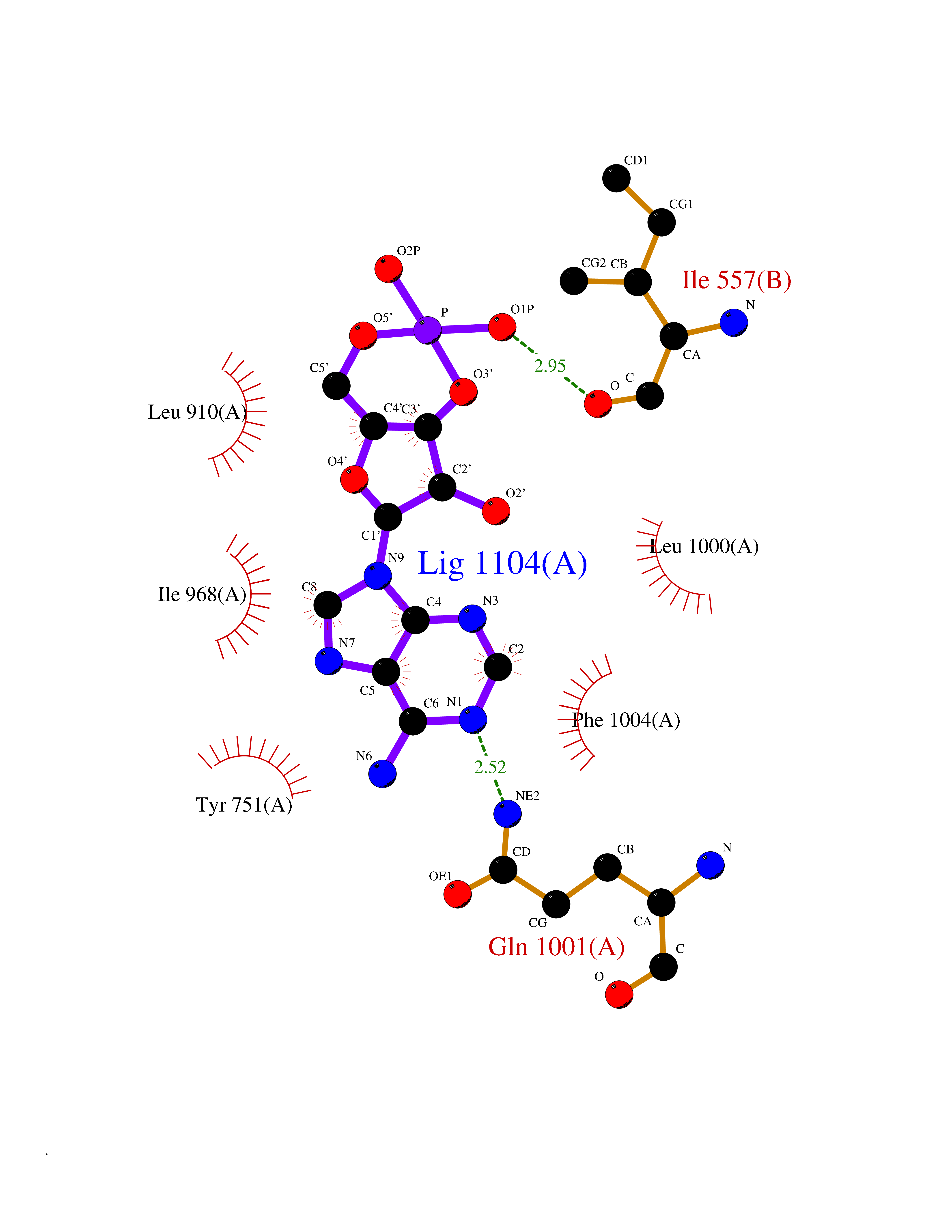 Ligplot Map 3D Binding mode Sequence KPILAPEPLVMDNLDSIMEQLNTWNFPIFDLVENIGRKCGRILSQVSYRLFEDMGLFEAFKIPIREFMNYFHALEIGYRDIPYHNRIHATDVLHAVWYLTTQPIPGLSTVGYVFSKTYNVTDDKYGCLSGNIPALELMALYVAAAMHDYDHPGRTNAFLVATSAPQAVLYNDRSVLENHHAAAAWNLFMSRPEYNFLINLDHVEFKHFRFLVIEAILATDLKKHFDFVAKFNGKVNDDVGIDWTNENDRLLVCQMCIKLADINGPAKCKELHLQWTDGIVNEFYEQGDEEASLGLPISPFMDRSAPQLANLQESFISHIVGPLCNSYDSAGLMPGKWVEKIYCQITQHLLQNHKMWKKVIEEEQRLAGIENQNISVDLETNYAELVLDVGRVTLGENSRKKMKDCKLRKKQNESVSRAMCALLNSGGGVIKAEIENEDYSYTKDGIGLDLENSFSNILLFVPEYLDFMQNGNYFLIFVKSWSLNTSGLRITTLSSNLYKRDITSAKVMNATAALEFLKDMKKTRGRLYLRPELLAKRPCVDIQEENNMKALAGVFFDRTELDRKEKLTFTESTHVEIKNFSTERLLQRIKEILPQYVSAFANTDGGYLFIGLNEDKEIIGFKAEMSDLDDLEREIEKSIRKMPVHHFCMEKKKINYSCKFLGVYDKGSLCGYVCALRVERFCCAVFAKEPDSWHVKDNRVMQLTRKEWIQFMVEAEPKFSSAYEEVISQINTSLPAPHSWPLLEWQRQRHHCPGLSGRITYTPENLCRKLFLQHEGLKQLICEEMSSVRKGSLIFSRSWSVDLGLQENHKVLCDALLISQDSPPVLYTFHMVQDEEFKGYSTQTALTLKQKLAKIGGYTKKVCVMTKIFYLSPEGMTSCQYDLRSQVIYPESYYFTRRKYLLKALFKALKRLKSLRDQFSFAENLYQIIGIDCFQKNDK Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Voltage-gated calcium channel alpha Cav3.3 (CACNA1I) | 7WLL | 6.39 | |
Target general information Gen name CACNA1I Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Voltage-gated calcium channel subunit alpha Cav3.3; Voltage-dependent T-type calcium channel subunit alpha-1I; KIAA1120; Ca(v)3.3 Protein family Calcium channel alpha-1 subunit (TC 1.A.1.11) family, CACNA1I subfamily Biochemical class Voltage-gated ion channel Function Voltage-sensitive calcium channels (VSCC) mediate the entry of calcium ions into excitable cells and are also involved in a variety of calcium-dependent processes, including muscle contraction, hormone or neurotransmitter release, gene expression, cell motility, cell division and cell death. This channel gives rise to T-type calcium currents. T-type calcium channels belong to the "low-voltage activated (LVA)" group and are strongly blocked by nickel and mibefradil. A particularity of this type of channels is an opening at quite negative potentials, and a voltage-dependent inactivation. T-type channels serve pacemaking functions in both central neurons and cardiac nodal cells and support calcium signaling in secretory cells and vascular smooth muscle. They may also be involved in the modulation of firing patterns of neurons which is important for information processing as well as in cell growth processes. Gates in voltage ranges similar to, but higher than alpha 1G or alpha 1H (By similarity). Related diseases Neurodevelopmental disorder with speech impairment and with or without seizures (NEDSIS) [MIM:620114]: An autosomal dominant disorder with variable manifestations. Severely affected individuals have profound global developmental delay, hypotonia, delayed or absent walking, absent speech, feeding difficulties, cortical visual impairment, and onset of hyperexcitability and seizures in the first months or years of life. Some patients manifest a milder phenotype characterized by mild to moderate cognitive impairment and mild speech delay, usually without seizures. {ECO:0000269|PubMed:33704440}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01118; DB00381; DB09231; DB13746; DB11148; DB11093; DB11348; DB14481; DB09061; DB00568; DB09235; DB00228; DB00153; DB04841; DB09238; DB14009; DB01388; DB14011; DB00622; DB01115; DB06712; DB06152; DB00617; DB09498; DB09089; DB00661; DB00909 Interacts with Q8NEC5; Q96P56 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Calcium channel; Calcium transport; Disease variant; Glycoprotein; Intellectual disability; Ion channel; Ion transport; Membrane; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transmembrane; Transmembrane helix; Transport; Voltage-gated channel Protein physicochemical properties Chain ID A Molecular weight (Da) 130104 Length 1135 Aromaticity 0.12 Instability index 41.15 Isoelectric point 8.25 Charge (pH=7) 8.02 2D Binding mode Binding energy (Kcal/mol) -6.87 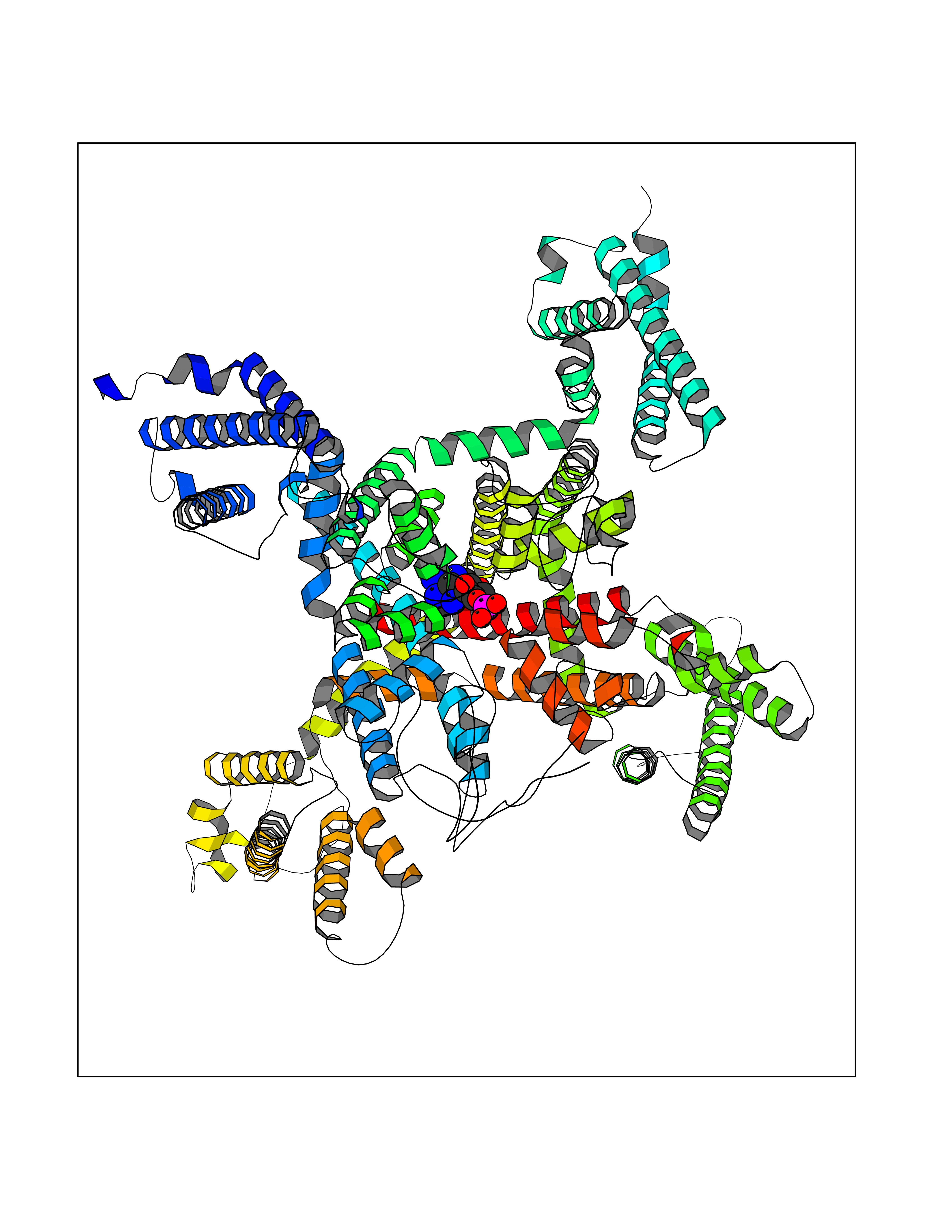 Molscript Map 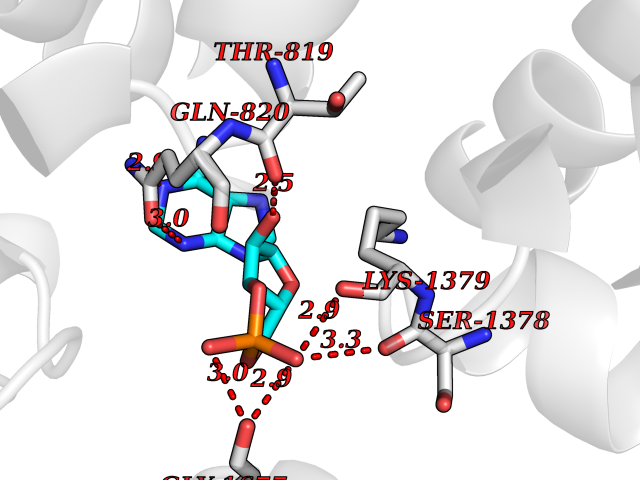 Pymol Map 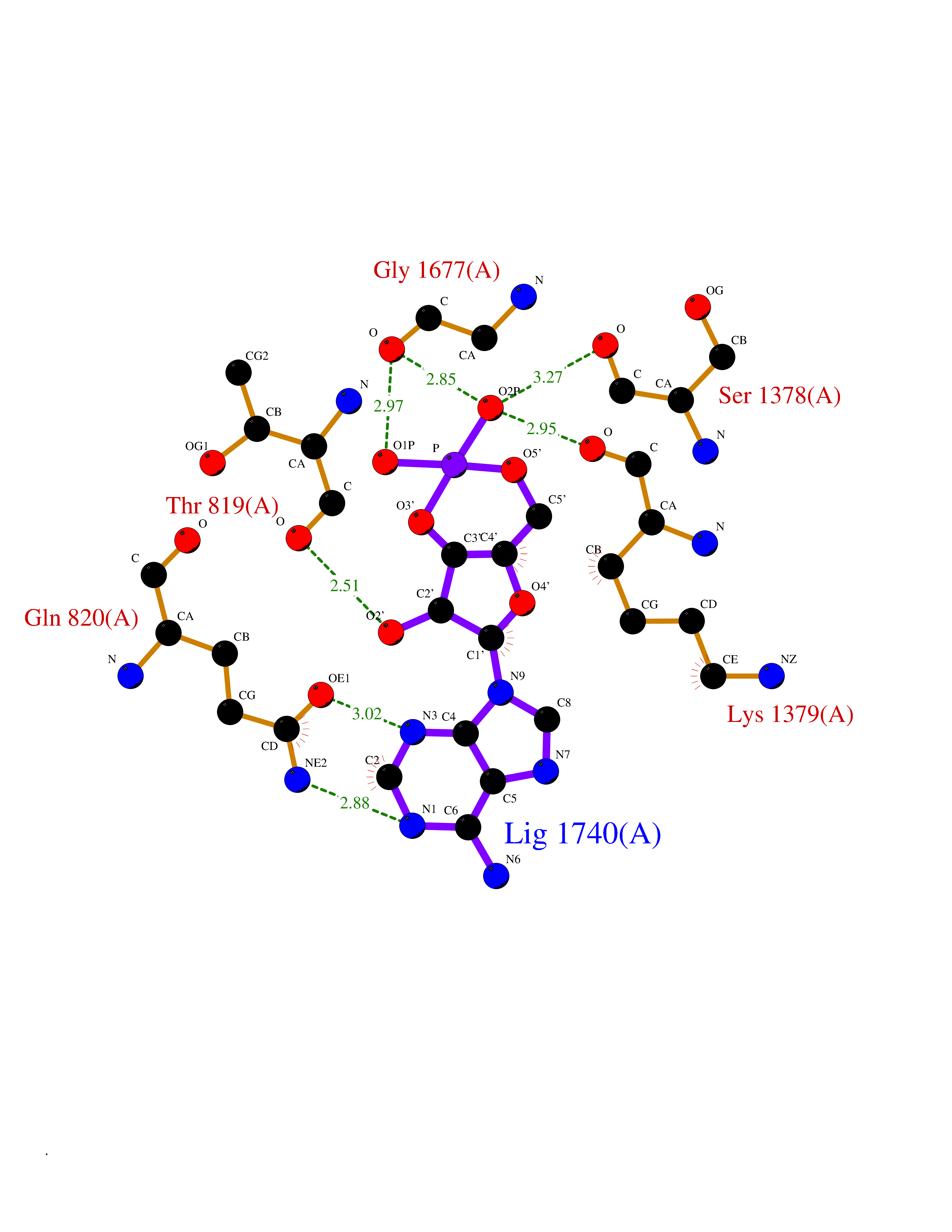 Ligplot Map 3D Binding mode Sequence TSPRNWCIKMVCNPWFECVSMLVILLNCVTLGMYQPCDDMDCLSDRCKILQVFDDFIFIFFAMEMVLKMVALGIFGKKCYLGDTWNRLDFFIVMAGMVEYSLDLQNINLSAIRTVRVLRPLKAINRVPSMRILVNLLLDTLPMLGNVLLLCFFVFFIFGIIGVQLWAGLLRNRCFLEENFTIQGDVALPPYYQPEEDDEMPFICSLSGDNGIMGCHEIPPLKCVNWNRYYNVCRTGSANPHKGAINFDNIGYAWIVIFQVITLEGWVEIMYYVMDAHSFYNFIYFILLIIVGSFFMINLCLVVIATQFSETKQREHRLMLRETRAKLRGIVDSKYFNRGIMMAILVNTVSMGIEHHEQPEELTNILEICNVVFTSMFALEMILKLAAFGLFDYLRNPYNIFDSIIVIISIWEIVGQADGGLSVLRTFRLLRVLKLVRFMPALRRQLVVLMKTMDNVATFCMLLMLFIFIFSILGMHIFGCKFSLRTDTGDTVPDRKNFDSLLWAIVTVFQILTQEDWNVVLYNGMASTSPWASLYFVALMTFGNYVLFNLLVAILVEGFQQTIIAHKLFDYVVLAFIFLNCITIALERPQIEAGSTERIFLTVSNYIFTAIFVGEMTLKVVSLGLYFGEQAYLRSSWNVLDGFLVFVSIIDIVVSLASAGGAKILGVLRVLRLLRTLRPLRVISRAPGLKLVVETLISSLKPIGNIVLICCAFFIIFGILGVQLFKGKFYHCLGVDTRNITNRSDCMAANYRWVHHKYNFDNLGQALMSLFVLASKDGWVNIMYNGLDAVAVDQQPVTNHNPWMLLYFISFLLIVSFFVLNMFVGVVVENFHKCRQHQEAEEARRREEKRLRRLEKKRRKAQRLPYYATYCHTRLLIHSMCTSHYLDIFITFIICLNVVTMSLEHYNQPTSLETALKYCNYMFTTVFVLEAVLKLVAFGLRRFFKDRWNQLDLAIVLLSVMGITLEEIEINAALPINPTIIRIMRVLRIARVLKLLKMATGMRALLDTVVQALPQVGNLGLLFMLLFFIYAALGVELFGKLVCNDENPCEGMSRHATFENFGMAFLTLFQVSTGDNWNGIMKDTLRDCTHDERSCLSSLQFVSPLYFVSFVLTAQFVLINVVVAVLMKHLDDSNK Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Adrenergic receptor beta-2 (ADRB2) | 2RH1 | 6.38 | |
Target general information Gen name ADRB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Beta-2 adrenoreceptor; Beta-2 adrenoceptor; Beta-2 adrenergic receptor; B2AR; ADRB2R Protein family G-protein coupled receptor 1 family, Adrenergic receptor subfamily, ADRB2 sub-subfamily Biochemical class GPCR rhodopsin Function The beta-2-adrenergic receptor binds epinephrine with an approximately 30-fold greater affinity than it does norepinephrine. Beta-adrenergic receptors mediate the catecholamine-induced activation of adenylate cyclase through the action of G proteins. Related diseases Cortical dysplasia, complex, with other brain malformations 6 (CDCBM6) [MIM:615771]: A disorder of aberrant neuronal migration and disturbed axonal guidance. Affected individuals have microcephaly, ataxia, and severe delayed psychomotor development. Brain imaging shows variable malformations of cortical development, including white matter streaks, dysmorphic basal ganglia, corpus callosum abnormalities, brainstem and cerebellar hypoplasia, cortical dysplasia, polymicrogyria. {ECO:0000269|PubMed:23246003}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Skin creases, congenital symmetric circumferential, 1 (CSCSC1) [MIM:156610]: An autosomal dominant disease characterized by multiple, symmetric, circumferential rings of folded skin, affecting primarily the limbs. Affected individuals also exhibit intellectual disability, cleft palate, and dysmorphic features. {ECO:0000269|PubMed:26637975}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07543; DB01193; DB00866; DB01118; DB00182; DB01102; DB01274; DB01238; DB09204; DB06216; DB00335; DB01408; DB05590; DB09013; DB00195; DB00217; DB01295; DB00612; DB00901; DB08807; DB06726; DB08808; DB00248; DB00521; DB01136; DB04846; DB01407; DB00785; DB01151; DB11273; DB13345; DB00449; DB11278; DB00841; DB09273; DB06262; DB01363; DB01364; DB00668; DB01049; DB11587; DB01288; DB00983; DB05039; DB00221; DB01064; DB00598; DB01210; DB13139; DB01365; DB13624; DB01214; DB00264; DB01203; DB05849; DB04861; DB00368; DB00540; DB00334; DB09080; DB00816; DB01580; DB00715; DB01359; DB00925; DB00397; DB00960; DB01291; DB01366; DB01182; DB00571; DB06814; DB00852; DB01917; DB11124; DB00867; DB01001; DB00938; DB00489; DB03566; DB00127; DB00871; DB00373; DB00726; DB12248; DB09082; DB09185 Interacts with P30542; P07550; P32121; Q96B67; Q9UII2; Q9ULD4-2; Q9NSI6-4; Q5M9N0-2; A0AVK6; Q658K8; O00472; Q15910-2; Q15486; P61978; Q5TCQ9; Q99685; O14745; Q9NR21-5; Q8WVD3; Q9H0X6; Q13573; P12931; Q5T0J7-2; Q8N0U2 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; Endosome; G-protein coupled receptor; Glycoprotein; Golgi apparatus; Hydroxylation; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32266.1 Length 282 Aromaticity 0.15 Instability index 36.1 Isoelectric point 8.02 Charge (pH=7) 2.1 2D Binding mode Binding energy (Kcal/mol) -6.5 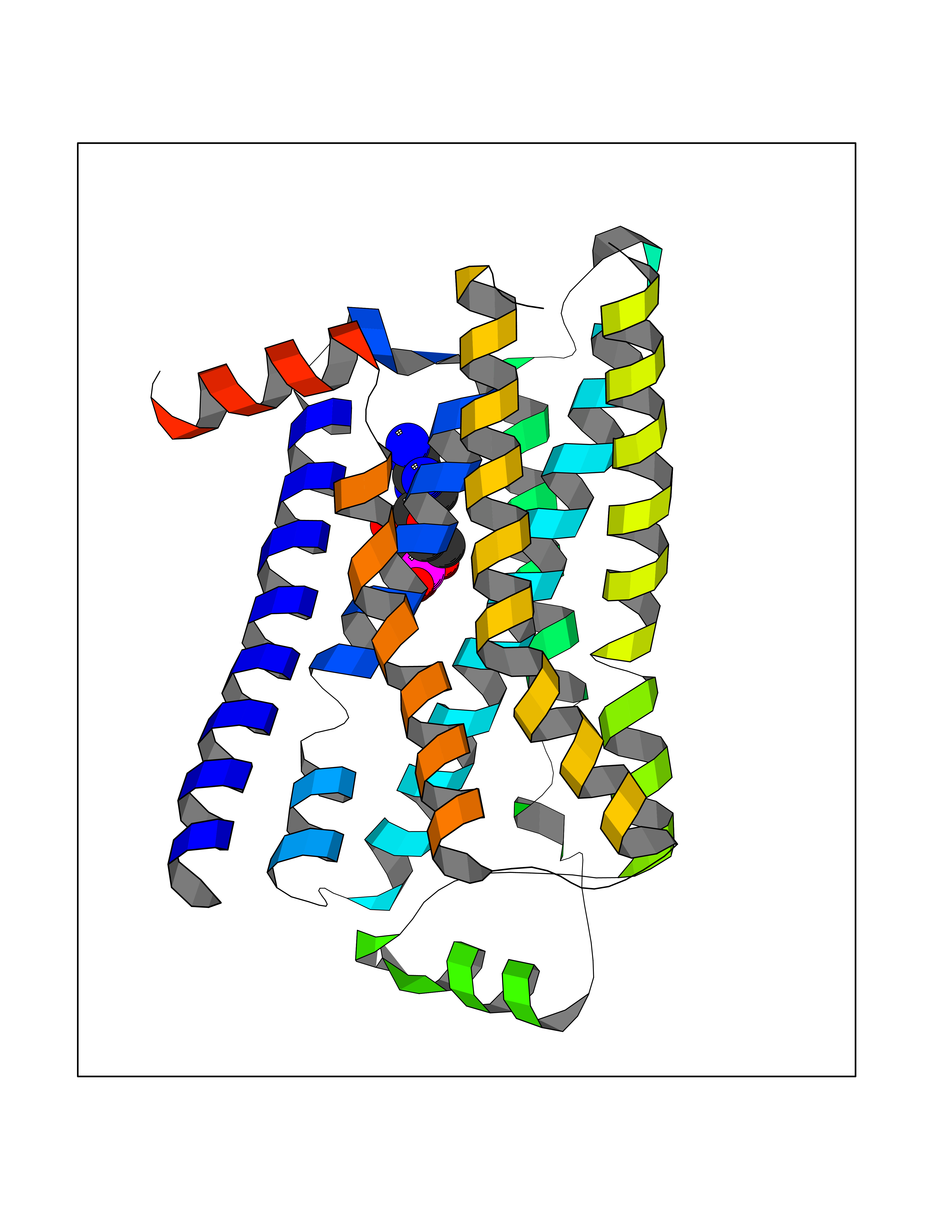 Molscript Map 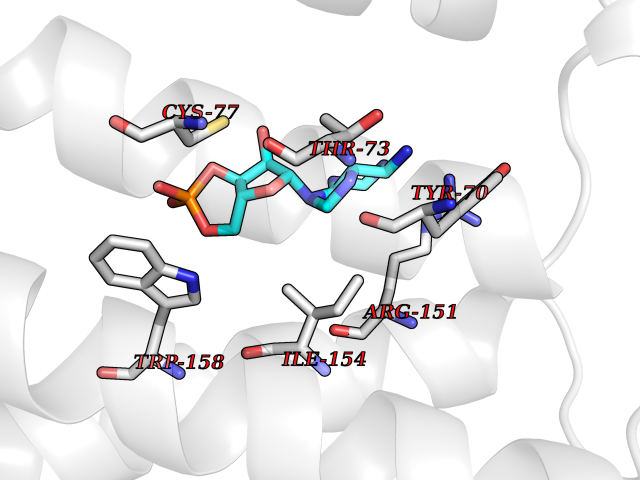 Pymol Map 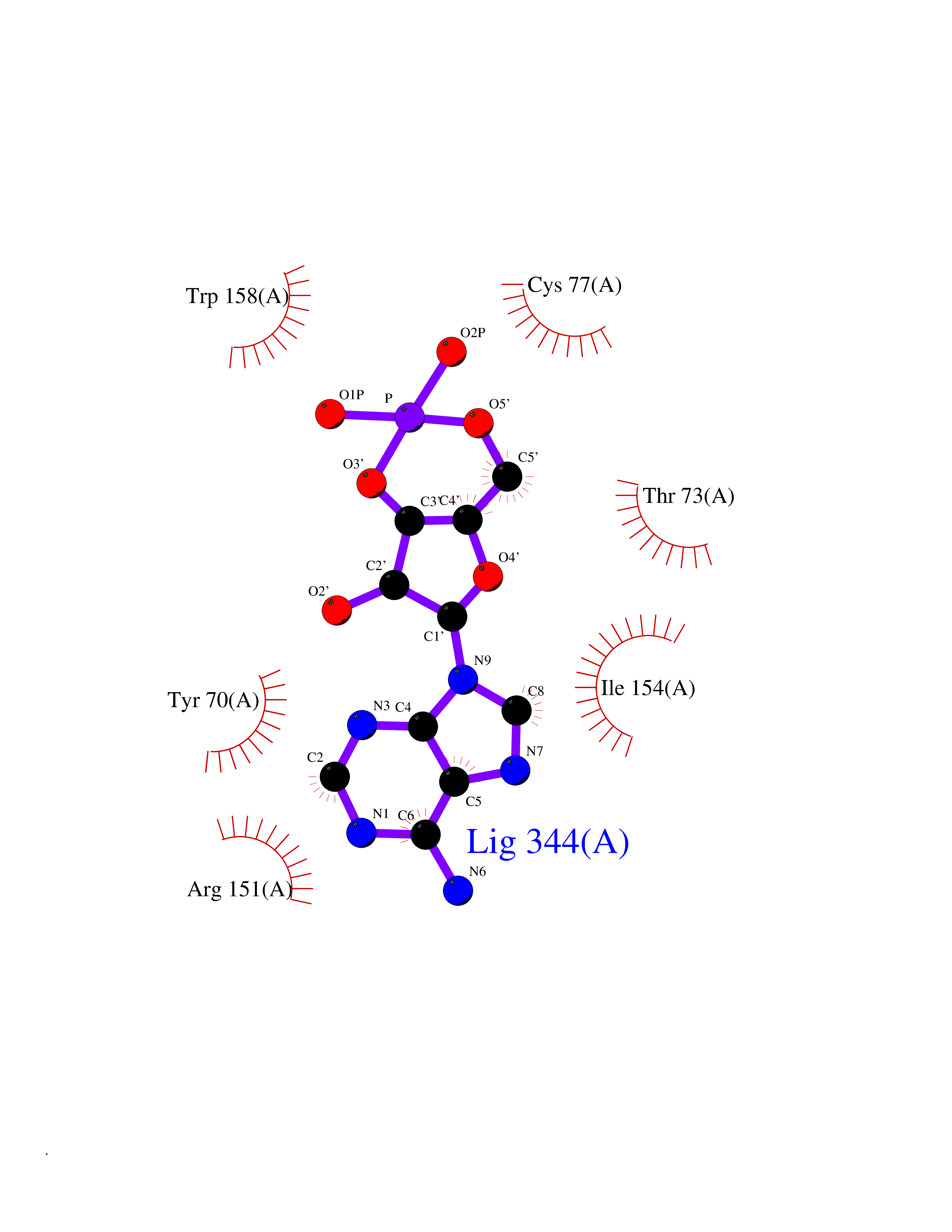 Ligplot Map 3D Binding mode Sequence DEVWVVGMGIVMSLIVLAIVFGNVLVITAIAKFERLQTVTNYFITSLACADLVMGLAVVPFGAAHILMKMWTFGNFWCEFWTSIDVLCVTASIETLCVIAVDRYFAITSPFKYQSLLTKNKARVIILMVWIVSGLTSFLPIQMHWYRATHQEAINCYAEETCCDFFTNQAYAIASSIVSFYVPLVIMVFVYSRVFQEAKRQLKFCLKEHKALKTLGIIMGTFTLCWLPFFIVNIVHVIQDNLIRKEVYILLNWIGYVNSGFNPLIYCRSPDFRIAFQELLCL Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Oxygen-insensitive NADPH nitroreductase | 3QDL | 6.37 | |
Target general information Gen name rdxA Organism Helicobacter pylori (strain ATCC 700392 / 26695) (Campylobacter pylori) Uniprot ID TTD ID NA Synonyms HP_0954 Protein family Nitroreductase family Biochemical class Oxidoreductase Function Oxidoreductase activity. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB00916 Interacts with NA EC number 1.-.-.- Uniprot keywords 3D-structure; Antibiotic resistance; NADP; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 40094.3 Length 352 Aromaticity 0.08 Instability index 55.15 Isoelectric point 6.72 Charge (pH=7) -0.86 2D Binding mode Binding energy (Kcal/mol) -7.46 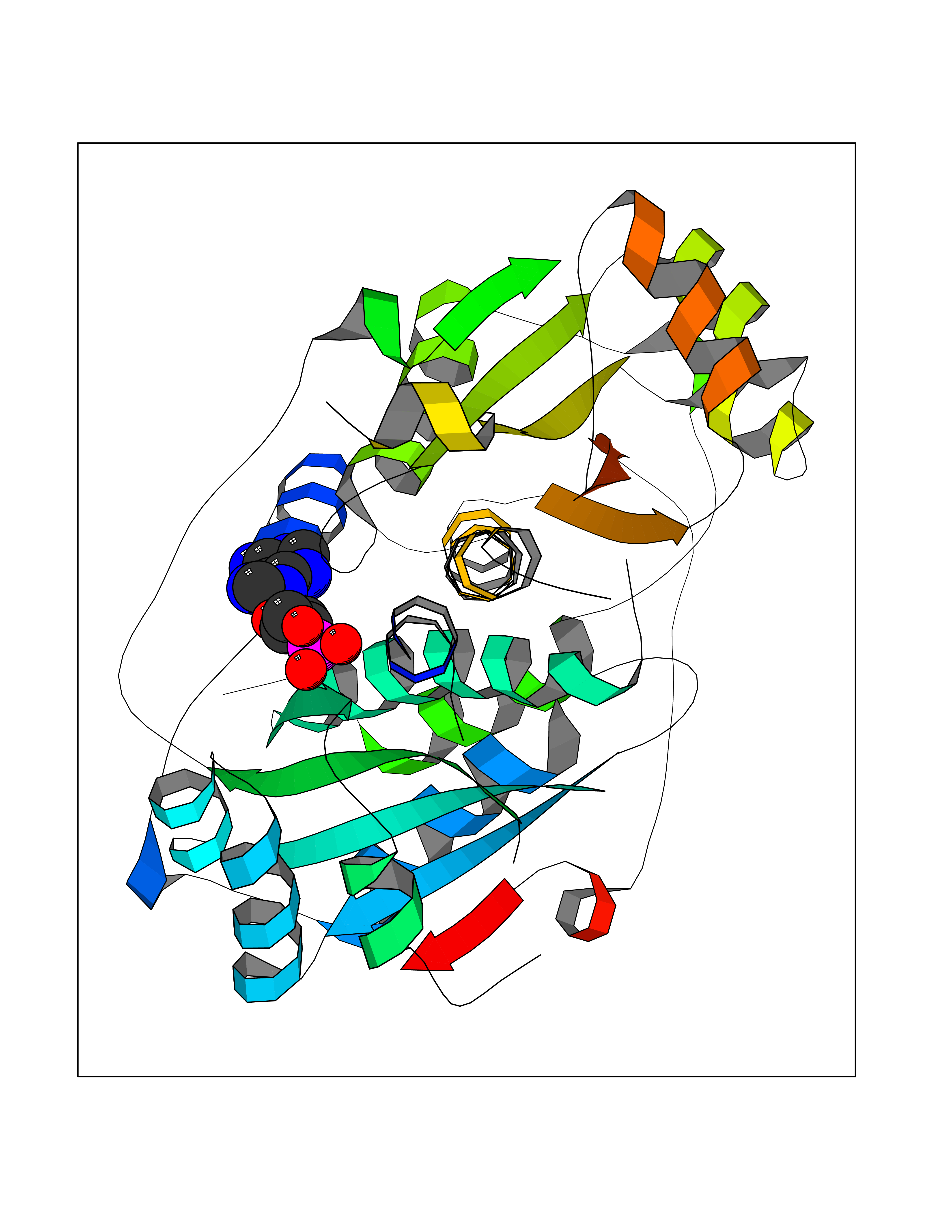 Molscript Map 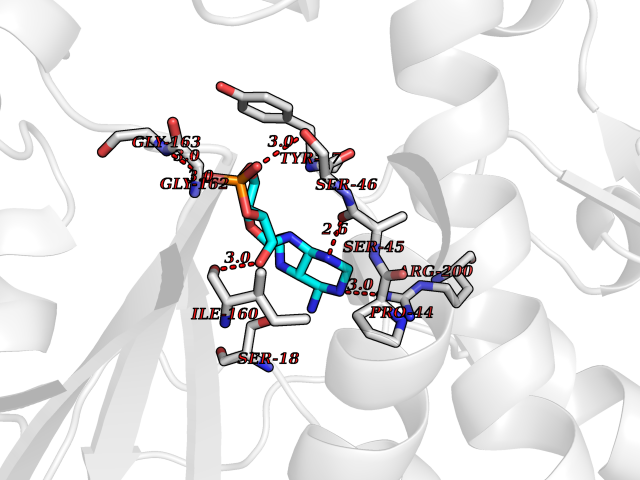 Pymol Map 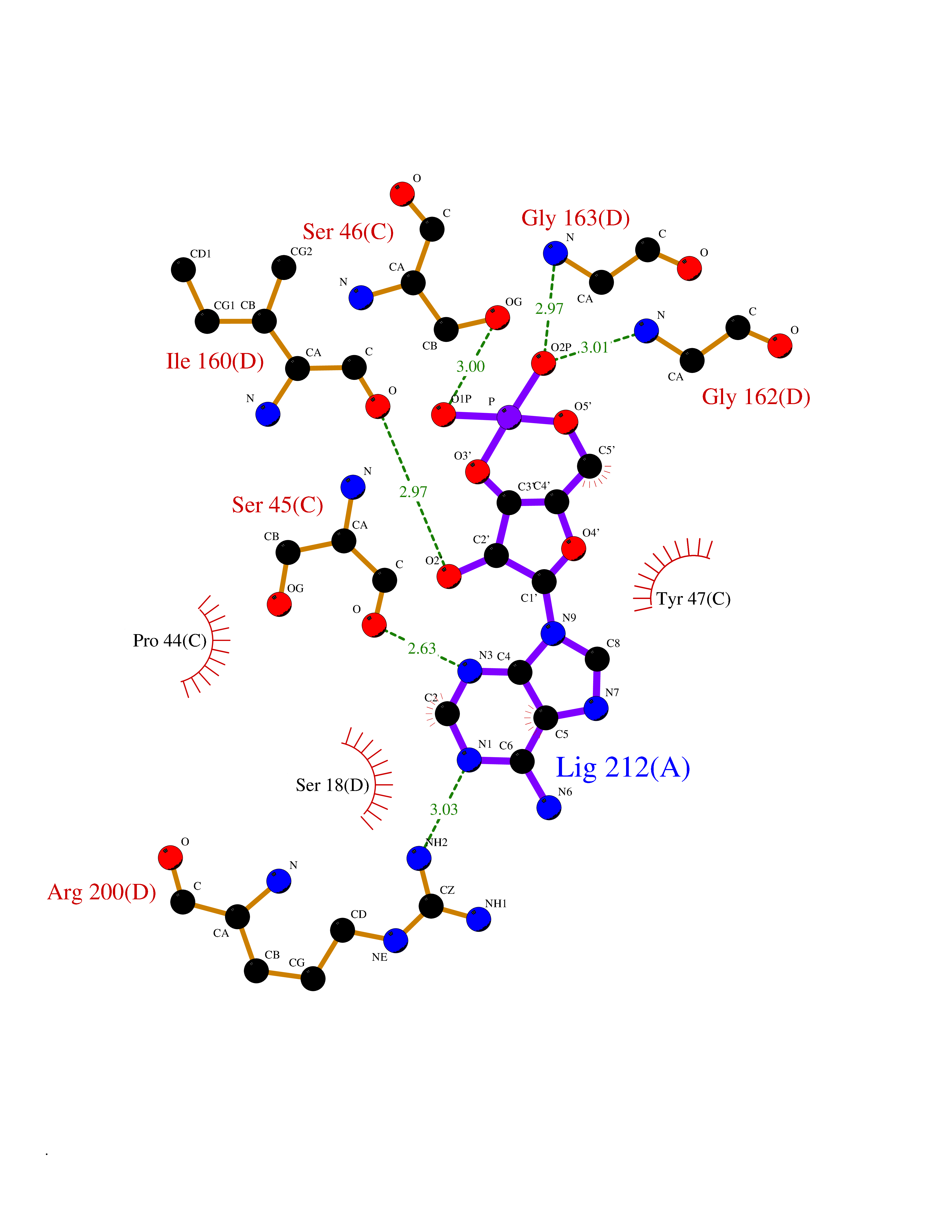 Ligplot Map 3D Binding mode Sequence MQRLESYILMKFLDQEKRRQLLNERHSCKMFDSHYEFSSTELEEIAEIARLSPSSYNTQPWHFVMVTDKDLKKQIAAHSYFNEEMIKSASALMVVCSLSYILEQCYIAVGQICMGVSLMGLDSCIIGGFDPLKVGEVLEERINPKIACLIALGKRVAEASQKSRKSKVDAITWLMKFLDQEKRRQLLNERHSCKMFDSHYEFSSTELEEIAEIARLSPSSYNTQPWHFVMVTDKDLKKQIAAHSYFNEEMIKSASALMVVCSLRPSELLPMQRLESYILEQCYIAVGQICMGVSLMGLDSCIIGGFDPLKVGEVLEERINKPKIACLIALGKRVAEASQKSRKSKVDAITWL Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Carbonic anhydrase IV (CA-IV) | 3FW3 | 6.37 | |
Target general information Gen name CA4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Carbonic anhydrase 4; Carbonate dehydratase IV; CAIV Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function May stimulate the sodium/bicarbonate transporter activity of SLC4A4 that acts in pH homeostasis. It is essential for acid overload removal from the retina and retina epithelium, and acid release in the choriocapillaris in the choroid. Reversible hydration of carbon dioxide. Related diseases Retinitis pigmentosa 17 (RP17) [MIM:600852]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:15563508, ECO:0000269|PubMed:17652713, ECO:0000269|PubMed:20450258}. The disease is caused by variants affecting the gene represented in this entry. Defective acid overload removal from retina and retinal epithelium, due to mutant CA4, is responsible for photoreceptor degeneration, indicating that impaired pH homeostasis is the most likely cause underlying the RP17 phenotype. Drugs (DrugBank ID) DB00819; DB00436; DB00562; DB01194; DB00606; DB01144; DB00869; DB08846; DB00311; DB00774; DB00703; DB00232; DB09460; DB00273; DB01021; DB00909 Interacts with NA EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; GPI-anchor; Lipoprotein; Lyase; Membrane; Metal-binding; Proteomics identification; Reference proteome; Retinitis pigmentosa; Signal; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 27055.7 Length 235 Aromaticity 0.09 Instability index 44.3 Isoelectric point 6.87 Charge (pH=7) -0.36 2D Binding mode Binding energy (Kcal/mol) -6.7 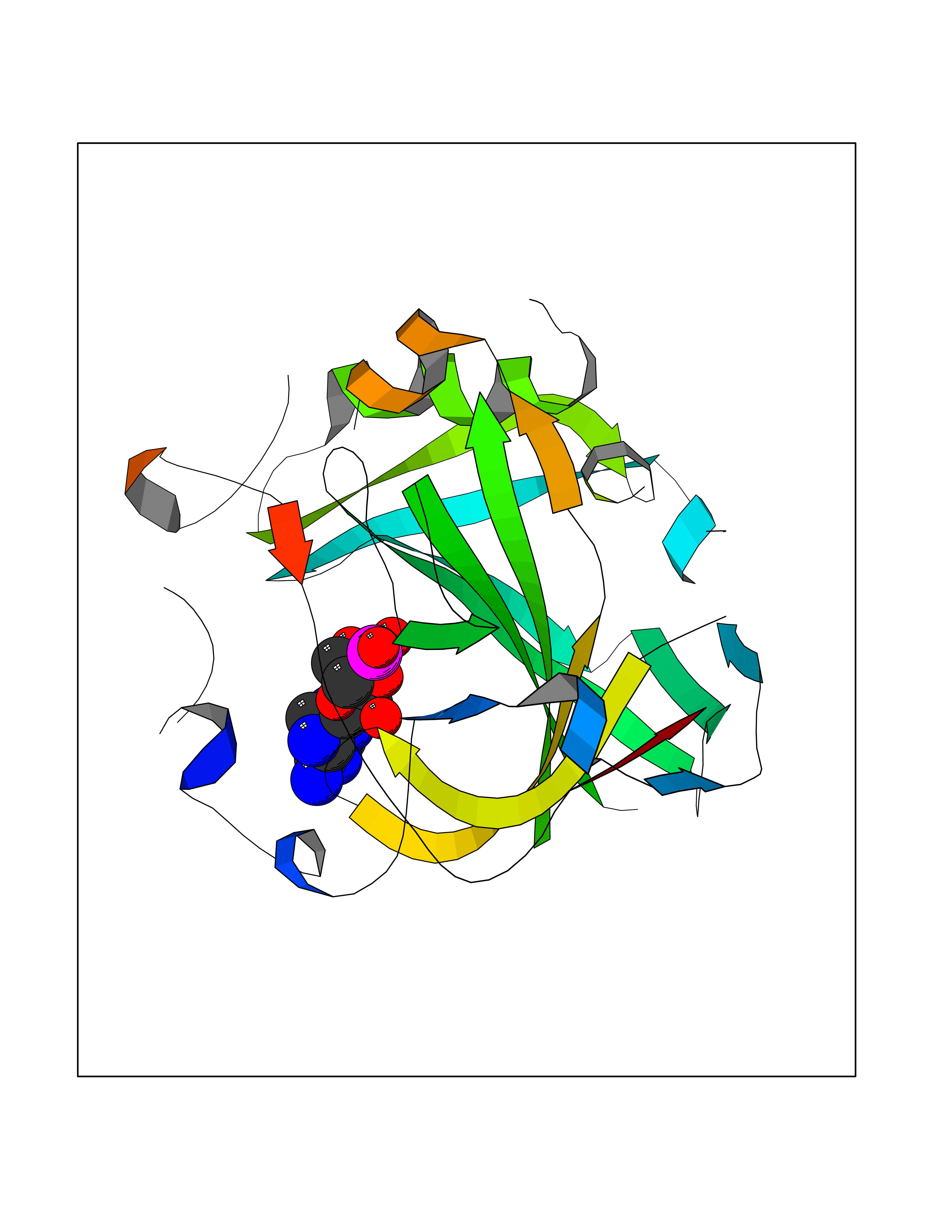 Molscript Map 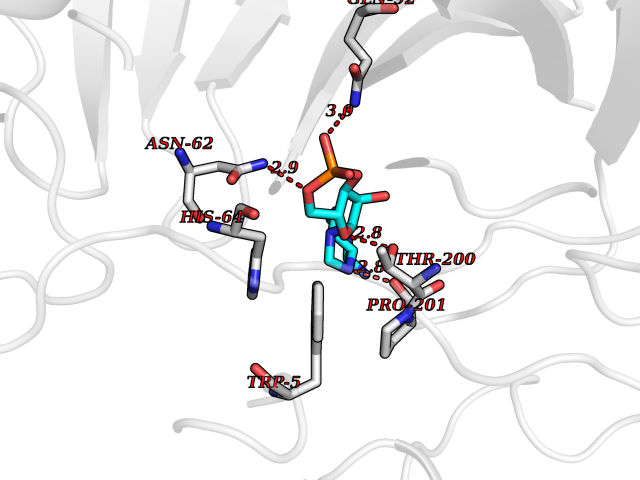 Pymol Map 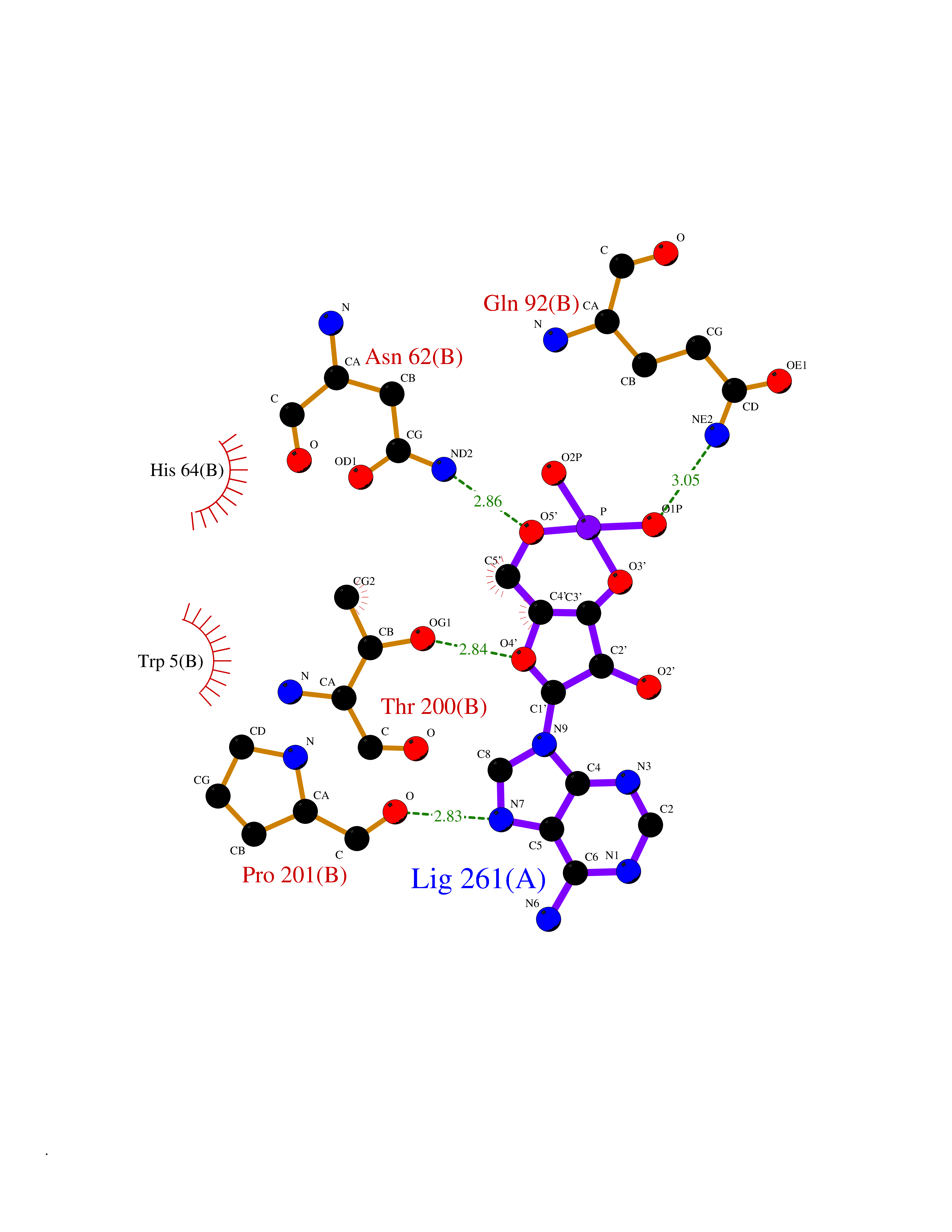 Ligplot Map 3D Binding mode Sequence HWCYEVQLVPVKWGGNCQKDRQSPINIVTTKAKVDKKLGRFFFGYDKKQTWTVQNNGHSVMMLLENKASISGGGLPAPYQAKQLHLHWSDLPYKGSEHSLDGEHFAMEMHIVHEKEEIAVLAFLVEATQVNEGFQPLVEALSNIPKPEMSTTMAESSLLDLLPEEKRHYFRYLGSLTTPTCDEKVVWTVFREPIQLHREQILAFQKLYYDKEQTVSMKDNVRPLQQLGQRTVIKS Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Solute carrier family 12 member 4 (SLC12A4) | 7AIP | 6.37 | |
Target general information Gen name SLC12A4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hKCC1; KCC1; Erythroid K-Cl cotransporter 1; Electroneutral potassium-chloride cotransporter 1 Protein family SLC12A transporter family, K/Cl co-transporter subfamily Biochemical class NA Function Mediates electroneutral potassium-chloride cotransport when activated by cell swelling. May contribute to cell volume homeostasis in single cells. May be involved in the regulation of basolateral Cl(-) exit in NaCl absorbing epithelia (By similarity). Isoform 4 has no transport activity. Related diseases Olmsted syndrome 1 (OLMS1) [MIM:614594]: An autosomal dominant, rare congenital disorder characterized by bilateral mutilating palmoplantar keratoderma and periorificial keratotic plaques with severe itching at all lesions. Diffuse alopecia, constriction of digits, and onychodystrophy have also been reported. Infections and squamous cell carcinomas can arise on the keratotic areas. The digital constriction may progress to autoamputation of fingers and toes. {ECO:0000269|PubMed:22405088, ECO:0000269|PubMed:22835024}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Palmoplantar keratoderma, non-epidermolytic, focal 2 (FNEPPK2) [MIM:616400]: A dermatological disorder characterized by non-epidermolytic, abnormal thickening of the skin on the palms and soles. Focal palmoplantar keratoderma consists of localized areas of hyperkeratosis located mainly on pressure points and sites of recurrent friction. {ECO:0000269|PubMed:25285920}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00887; DB00761 Interacts with Q9Y2T2; Q9BYD5; A8MQ03; P59991; Q3LHN2; Q8IYB1; Q8N9M5; Q8N720 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Chloride; Disulfide bond; Glycoprotein; Ion transport; Membrane; Phosphoprotein; Potassium; Potassium transport; Proteomics identification; Reference proteome; Symport; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 95490.2 Length 867 Aromaticity 0.11 Instability index 40.85 Isoelectric point 8.2 Charge (pH=7) 5.32 2D Binding mode Binding energy (Kcal/mol) -7.26 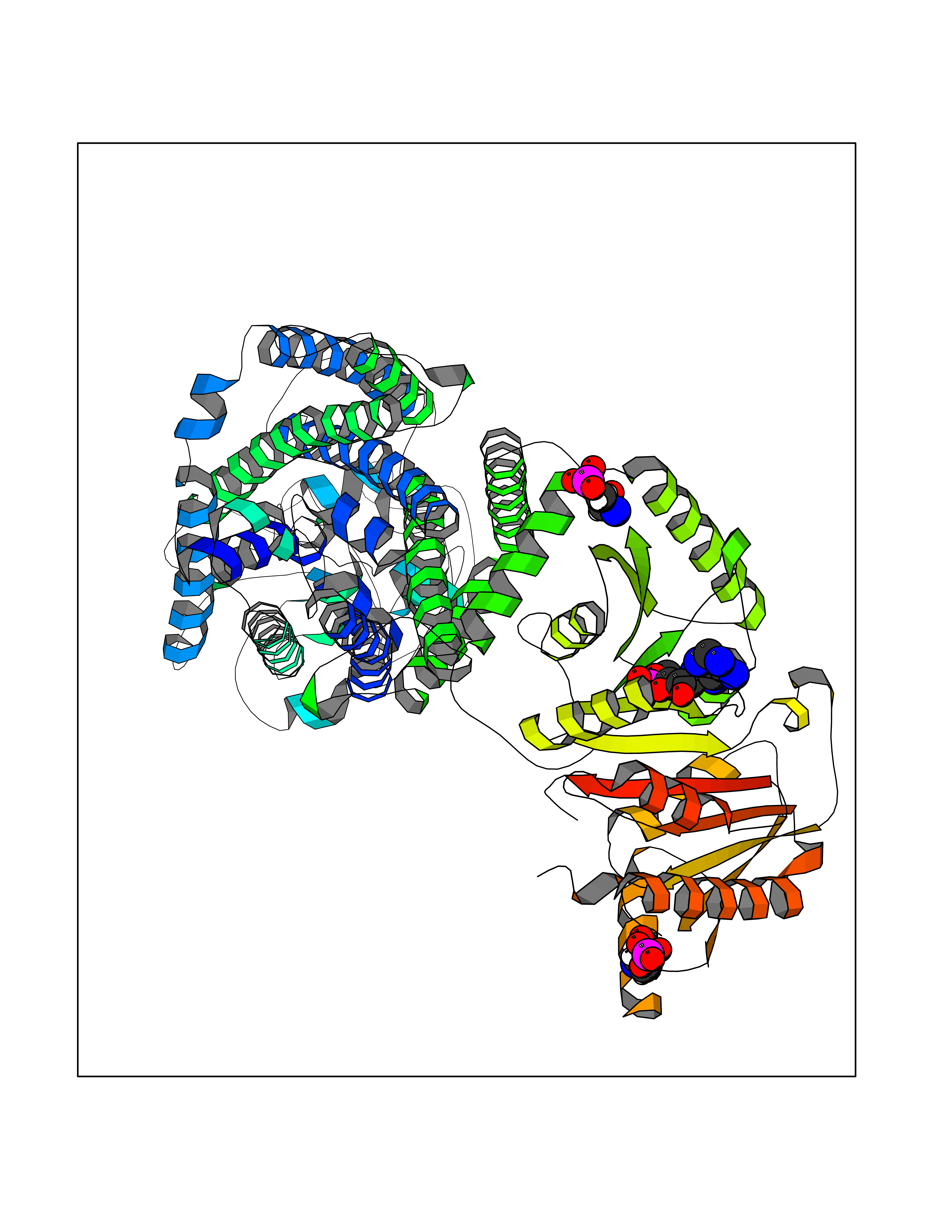 Molscript Map 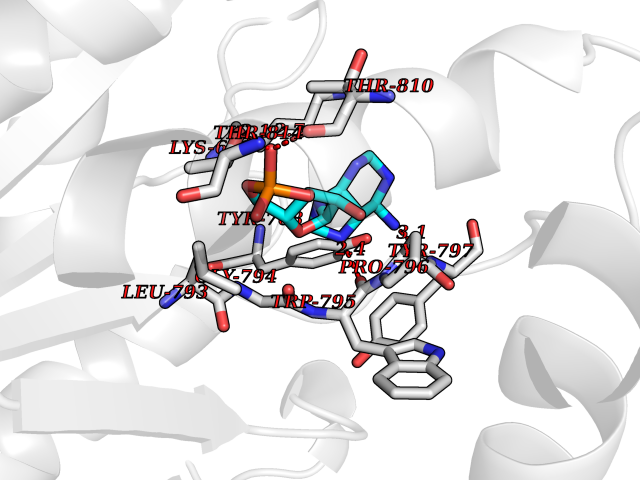 Pymol Map 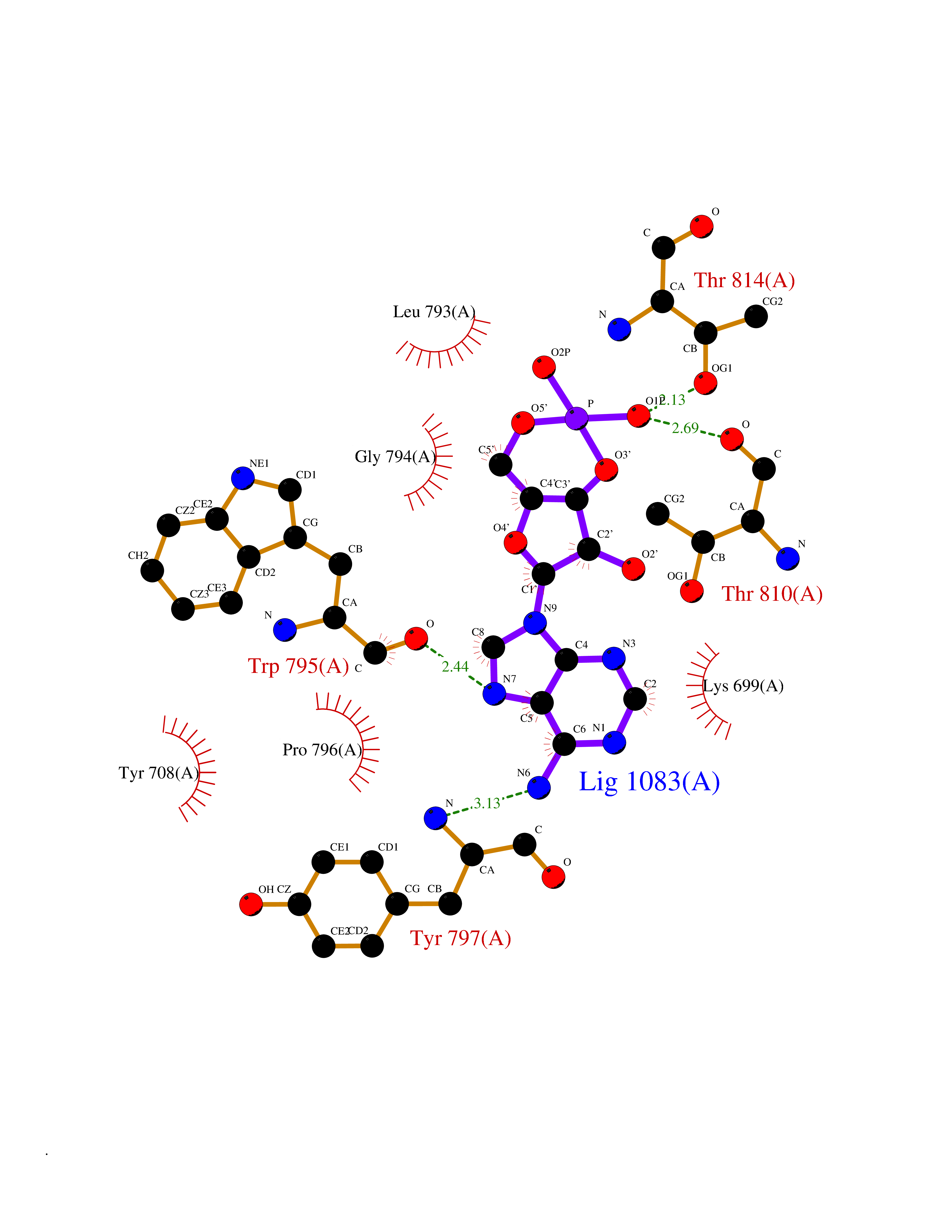 Ligplot Map 3D Binding mode Sequence PSMGTLMGVYLPCLQNIFGVILFLRLTWMVGTAGVLQALLIVLICCCCTLLTAISMSAIATNGVVPAGGSYFMISRSLGPEFGGAVGLCFYLGTTFAAAMYILGAIEILLTYIAPPAAIFYSNATLNNMRVYGTIFLTFMTLVVFVGVKYVNKFASLFLACVIISILSIYAGGIKSIFDPPVFPVCMLGNRTLSRDQFDICAKTAVVDNETVATQLWSFFCHSPNLTTDSCDPYFMLNNVTEIPGIPGAAAGVLQENLWSAYLEKGDIVEKHGLPSADAPSLKESLPLYVVADIATSFTVLVGIFFPSVTGIMAGSNRSGDLRDAQKSIPVGTILAIITTSLVYFSSVVLFGACIEGVVLRDKYGDGVSRNLVVGTLAWPSPWVIVIGSFFSTCGAGLQSLTGAPRLLQAIAKDNIIPFLRVFGHGKVNGEPTWALLLTALIAELGILIASLDMVAPILSMFFLMCYLFVNLACAVQTLLRTPNWRPRFKYYHWALSFLGMSLCLALMFVSSWYYALVAMLIAGMIYKYIEYQGAEKEWGDGIRGLSLSAARYALLRLEEGPPHTKNWRPQLLVLLKLDEDLHVKYPRLLTFASQLKAGKGLTIVGSVIQGXFLESYGEAQAAEQTIKNMMEIEKVKGFCQVVVASKVREGLAHLIQSCGLGGMRHNSVVLGWPYGWRQSEDPRAWKTFIDTVRCTTAAHLALLVPKNIAFYPSNHERYLEGHIDVWWIVHDGGMLMLLPFLLRQHKVWRKCRMRIFTVNSIQMKKDLAVFLYHLRLEAEVEVVEMHNXDISAYTYERTSNVRRMHTAVKLNEVIVTRSHDARLVLLNMPGPPRNSEGDENYMEFLEVLTEGLERVLLVRGGGREVI Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Bifunctional dihydrofolate reductase-thymidylate synthase | 1J3K | 6.36 | |
Target general information Gen name N/A Organism Plasmodium falciparum (isolate K1 / Thailand) Uniprot ID TTD ID NA Synonyms NA Protein family Dihydrofolate reductase family; Thymidylate synthase family Biochemical class Oxidoreductase Function Dihydrofolate reductase activity.Thymidylate synthase activity. Related diseases Acute hepatic porphyria (AHEPP) [MIM:612740]: A form of porphyria. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. AHP is characterized by attacks of gastrointestinal disturbances, abdominal colic, paralyses and peripheral neuropathy. Most attacks are precipitated by drugs, alcohol, caloric deprivation, infections, or endocrine factors. {ECO:0000269|PubMed:10706561, ECO:0000269|PubMed:1309003, ECO:0000269|PubMed:1569184, ECO:0000269|PubMed:17236137, ECO:0000269|PubMed:2063868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01131; DB00205; DB01299 Interacts with NA EC number 1.5.1.3; 2.1.1.45 Uniprot keywords 3D-structure; Methyltransferase; Multifunctional enzyme; NADP; Nucleotide biosynthesis; One-carbon metabolism; Oxidoreductase; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 61720.6 Length 525 Aromaticity 0.13 Instability index 33.9 Isoelectric point 8.79 Charge (pH=7) 11.61 2D Binding mode Binding energy (Kcal/mol) -7.27 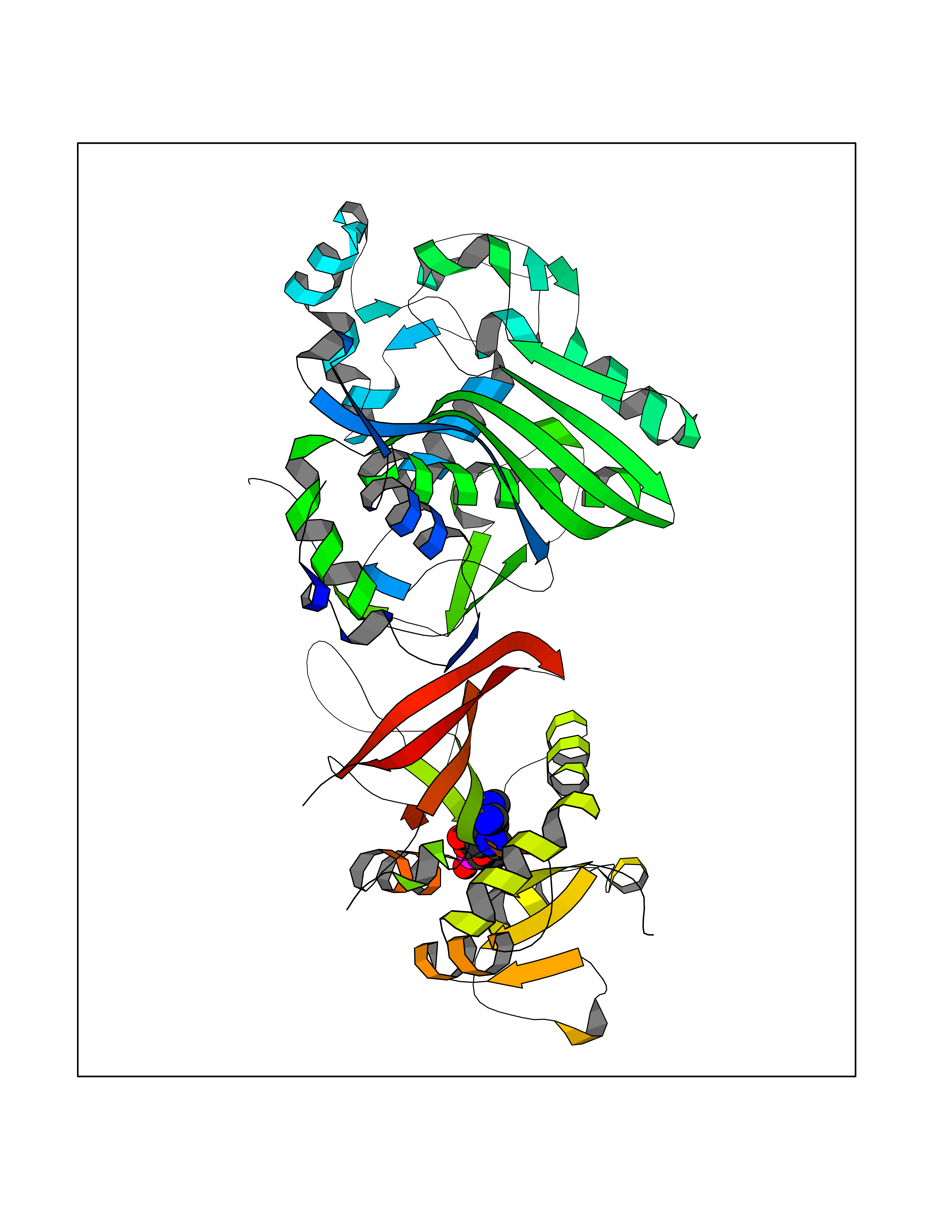 Molscript Map 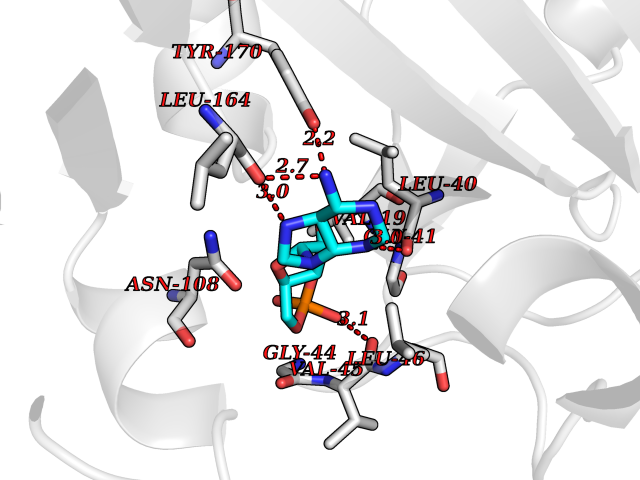 Pymol Map 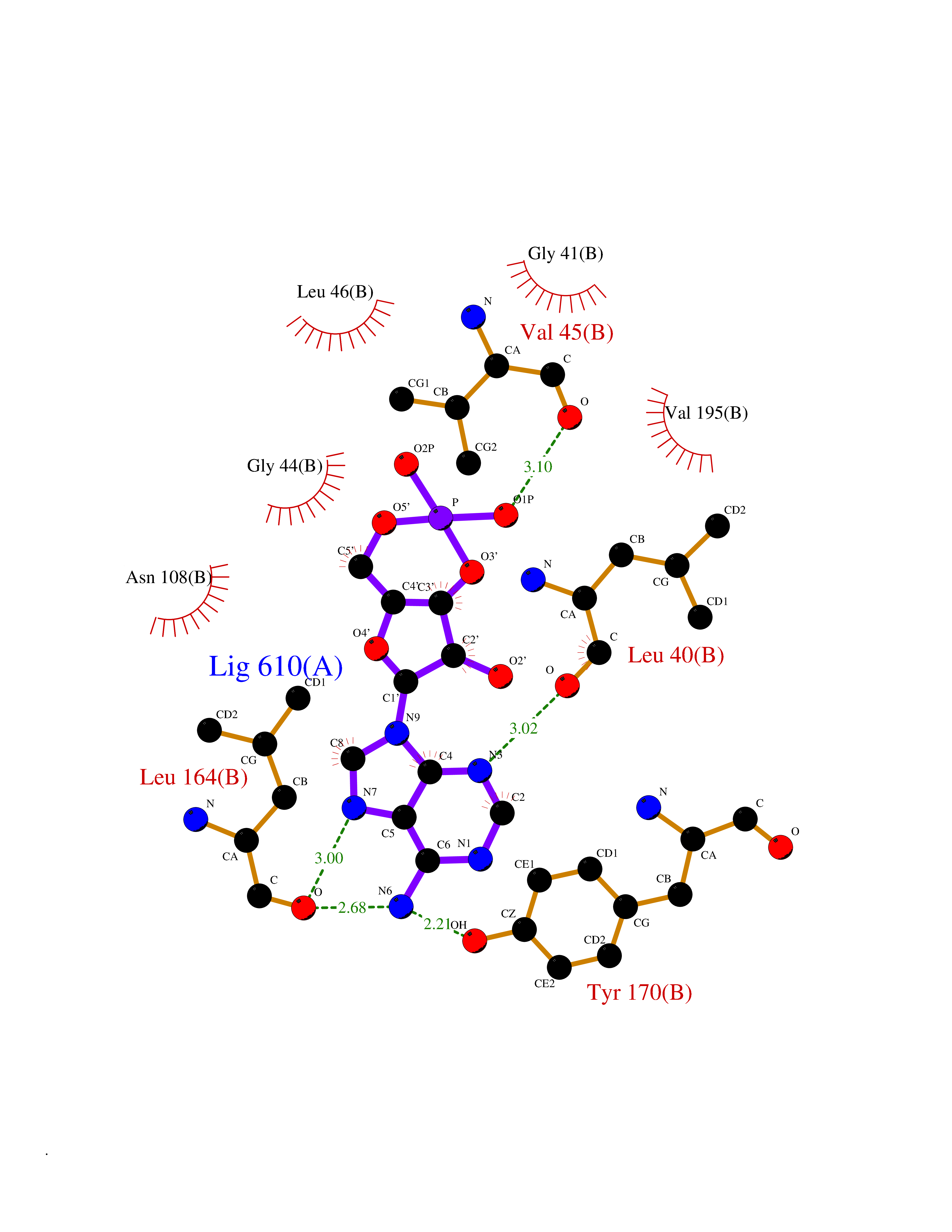 Ligplot Map 3D Binding mode Sequence NSIHPNDFQIYNSLKYKYHPEYQYLNIIYDIMMNGNKQSDRTGVGVLSKFGYIMKFDLSQYFPLLTTKKLFLRGIIEELLWFIRGETNGNTLLNKNVRIWEANGTREFLDNRKLFHREVNDLGPIYGFQWRHFGAEYTNMYDNYENKGVDQLKNIINLIKNDPTSRRILLCAWNVKDLDQMALPPCHILCQFYVFDGKLSCIMYQRSCDLGLGVPFNIASYSIFTHMIAQVCNLQPAQFIHVLGNAHVYNNHIDSLKIQLNRIPYPFPTLKLNPDIKNIEDFTISDFTIQNYVHHEKISMDMAAMMEQVCDVFDIYAICACCKVESKNEGKKNEVFNNYTFRGLGNKGVLPWKCISLDMKYFRAVTTYVNESKYEKLKYKRCKYLPNSKKLQNVVVMGRTNWESIPKKFKPLSNRINVILSRTLKKEDFDEDVYIINKVEDLIVLLGKLNYYKCFILGGSVVYQEFLEKKLIKKIYFTRINSTYECDVFFPEINENEYQIISVSDVYTSNNTTLDFIIYKKTNNK Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Activin receptor type IIA (ACVR2A) | 3Q4T | 6.36 | |
Target general information Gen name ACVR2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Activin receptor type2A; Activin receptor type-2A; ACVR2; ACTRIIA; ACTR-IIA Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family, TGFB receptor subfamily Biochemical class Kinase Function Type II receptors phosphorylate and activate type I receptors which autophosphorylate, then bind and activate SMAD transcriptional regulators. Receptor for activin A, activin B and inhibin A. Mediates induction of adipogenesis by GDF6. On ligand binding, forms a receptor complex consisting of two type II and two type I transmembrane serine/threonine kinases. Related diseases Periventricular nodular heterotopia 8 (PVNH8) [MIM:618185]: A form of periventricular nodular heterotopia, a disorder resulting from a defect in the pattern of neuronal migration in which ectopic collections of neurons lie along the lateral ventricles of the brain or just beneath, contiguously or in isolated patches. PVNH8 is an autosomal dominant disease characterized by developmental disabilities, speech delay, seizures and attention deficit-hyperactivity disorder. {ECO:0000269|PubMed:28868155}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12118 Interacts with NA EC number EC 2.7.11.30 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Disulfide bond; Glycoprotein; Kinase; Magnesium; Manganese; Membrane; Metal-binding; Nucleotide-binding; Proteomics identification; Receptor; Reference proteome; Serine/threonine-protein kinase; Signal; Transferase; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 34599.3 Length 305 Aromaticity 0.09 Instability index 44.48 Isoelectric point 5.7 Charge (pH=7) -8.29 2D Binding mode Binding energy (Kcal/mol) -7.52 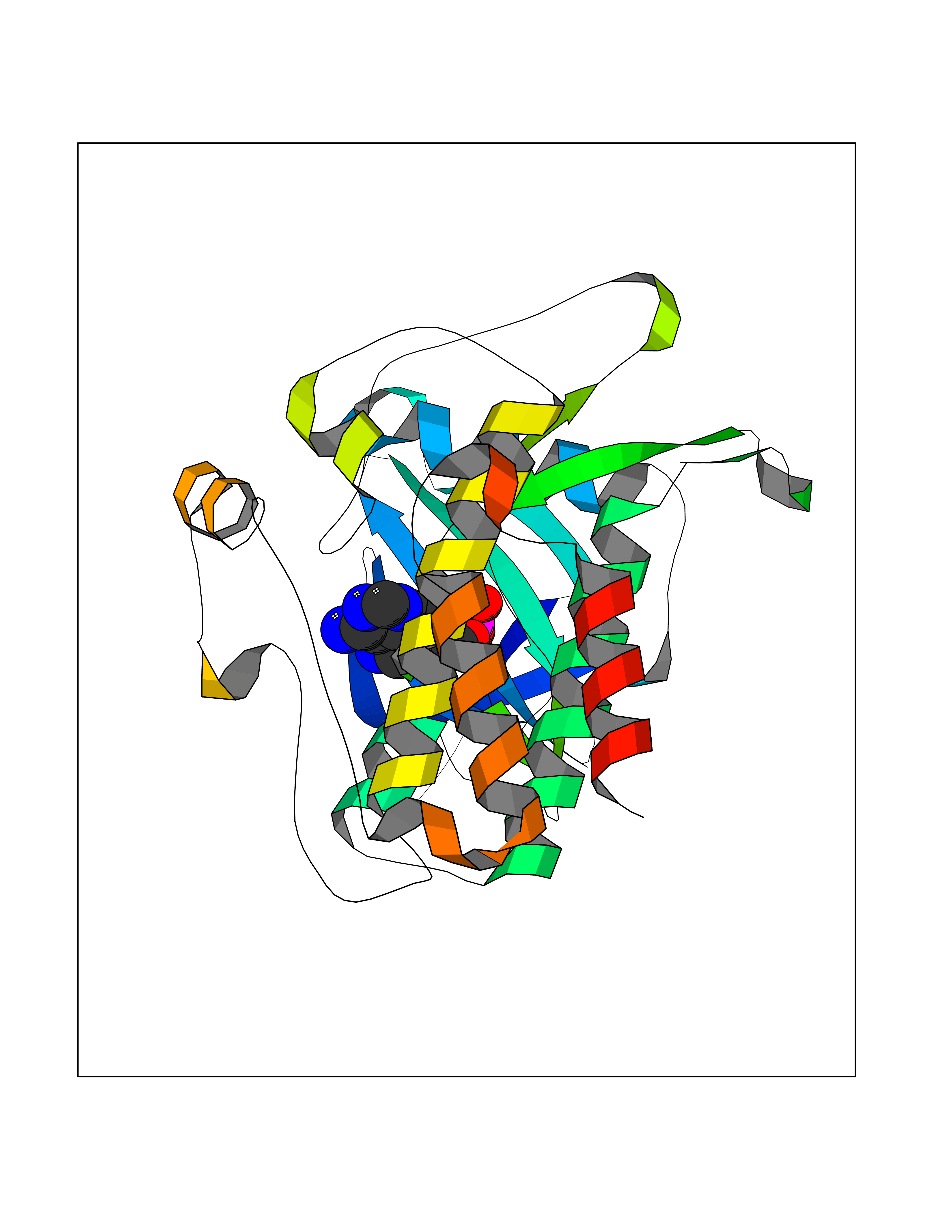 Molscript Map 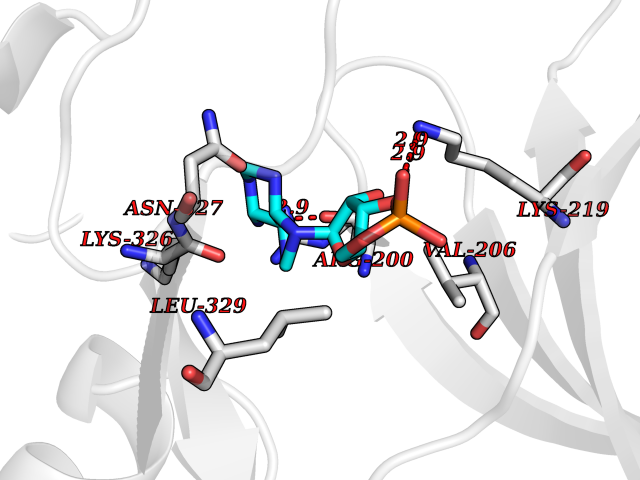 Pymol Map 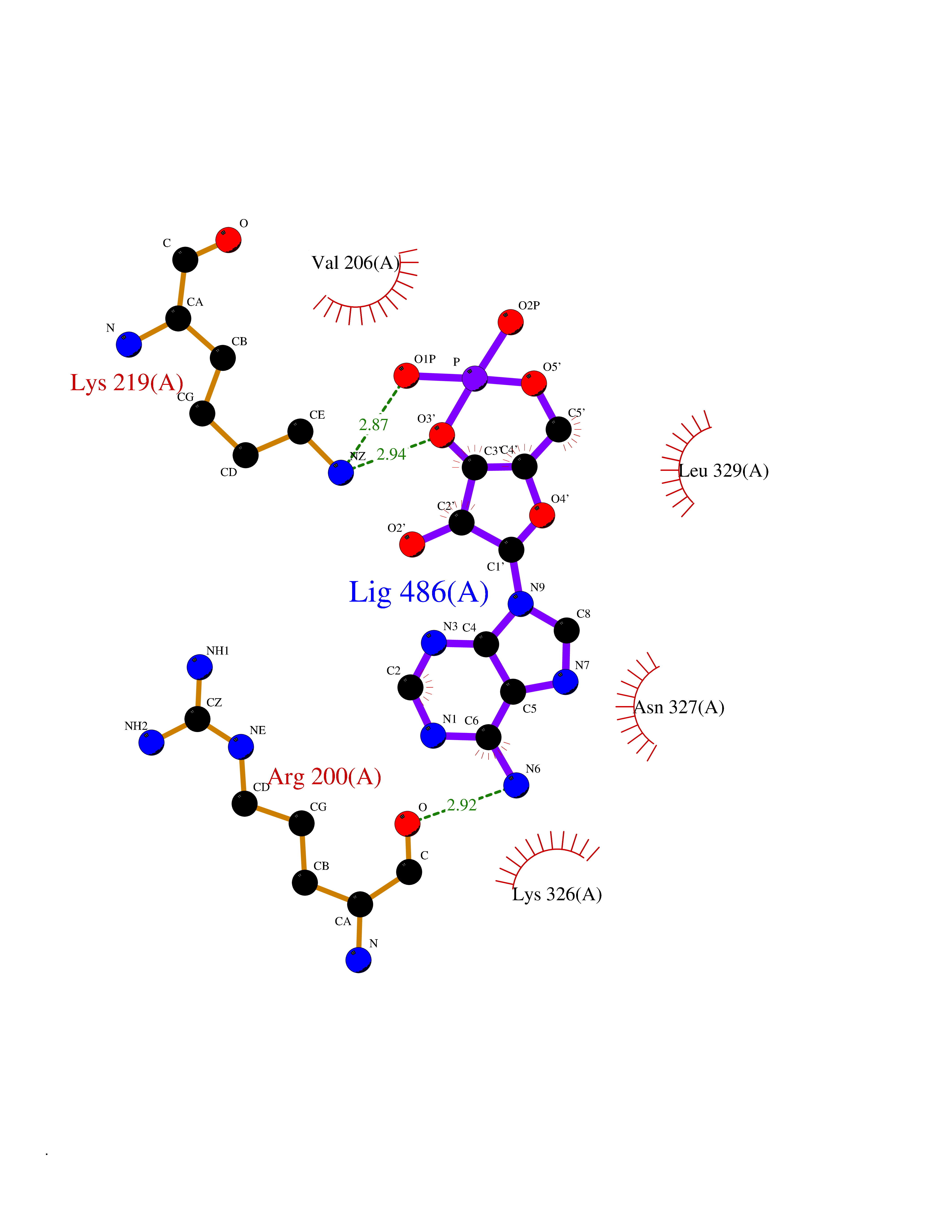 Ligplot Map 3D Binding mode Sequence LGTENLYFQSMPLQLLEVKARGRFGCVWKAQLLNEYVAVKIFPIQDKQSWQNEYEVYSLPGMKHENILQFIGAEKRGTSVDVDLWLITAFHEKGSLSDFLKANVVSWNELCHIAETMARGLAYLHEDIPGLKDGHKPAISHRDIKSKNVLLKNNLTACIADFGLALKFEAGKSAGDTHGQVGTRRYMAPEVLEGAINFQRDAFLRIDMYAMGLVLWELASRCTAADGPVDEYMLPFEEEIGQHPSLEDMQEVVVHKKKRPVLRDYWQKHAGMAMLCETIEECWDHDAEARLSAGCVGERITQMQR Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | MEK kinase kinase 4 (MAP4K4) | 4U40 | 6.35 | |
Target general information Gen name MAP4K4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nckinteracting kinase; Nck-interacting kinase; Mitogenactivated protein kinase kinase kinase kinase 4; Mitogen-activated protein kinase kinase kinase kinase 4; MEKKK 4; MAPK/ERK kinase kinase kinase 4 Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, STE20 subfamily Biochemical class Kinase Function Appears to act upstream of the JUN N-terminal pathway. Phosphorylates SMAD1 on Thr-322. Serine/threonine kinase that may play a role in the response to environmental stress and cytokines such as TNF-alpha. Related diseases Costello syndrome (CSTLO) [MIM:218040]: A rare condition characterized by prenatally increased growth, postnatal growth deficiency, intellectual disability, distinctive facial appearance, cardiovascular abnormalities (typically pulmonic stenosis, hypertrophic cardiomyopathy and/or atrial tachycardia), tumor predisposition, skin and musculoskeletal abnormalities. {ECO:0000269|PubMed:16170316, ECO:0000269|PubMed:16329078, ECO:0000269|PubMed:16443854, ECO:0000269|PubMed:17054105, ECO:0000269|PubMed:18039947, ECO:0000269|PubMed:18247425, ECO:0000269|PubMed:19995790}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy with excess of muscle spindles (CMEMS) [MIM:218040]: Variant of Costello syndrome. {ECO:0000269|PubMed:17412879}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid cancer, non-medullary, 2 (NMTC2) [MIM:188470]: A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms. {ECO:0000269|PubMed:12727991}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Mutations which change positions 12, 13 or 61 activate the potential of HRAS to transform cultured cells and are implicated in a variety of human tumors. {ECO:0000269|PubMed:3670300}.; DISEASE: Bladder cancer (BLC) [MIM:109800]: A malignancy originating in tissues of the urinary bladder. It often presents with multiple tumors appearing at different times and at different sites in the bladder. Most bladder cancers are transitional cell carcinomas that begin in cells that normally make up the inner lining of the bladder. Other types of bladder cancer include squamous cell carcinoma (cancer that begins in thin, flat cells) and adenocarcinoma (cancer that begins in cells that make and release mucus and other fluids). Bladder cancer is a complex disorder with both genetic and environmental influences. {ECO:0000269|PubMed:6298635, ECO:0000269|PubMed:6844927}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Schimmelpenning-Feuerstein-Mims syndrome (SFM) [MIM:163200]: A disease characterized by sebaceous nevi, often on the face, associated with variable ipsilateral abnormalities of the central nervous system, ocular anomalies, and skeletal defects. Many oral manifestations have been reported, not only including hypoplastic and malformed teeth, and mucosal papillomatosis, but also ankyloglossia, hemihyperplastic tongue, intraoral nevus, giant cell granuloma, ameloblastoma, bone cysts, follicular cysts, oligodontia, and odontodysplasia. Sebaceous nevi follow the lines of Blaschko and these can continue as linear intraoral lesions, as in mucosal papillomatosis. {ECO:0000269|PubMed:22683711}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with Q9NRI5; Q9H0R5; Q8N4C8 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cytoplasm; Kinase; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 33865.6 Length 295 Aromaticity 0.08 Instability index 42.63 Isoelectric point 8.47 Charge (pH=7) 3.5 2D Binding mode Binding energy (Kcal/mol) -7.35 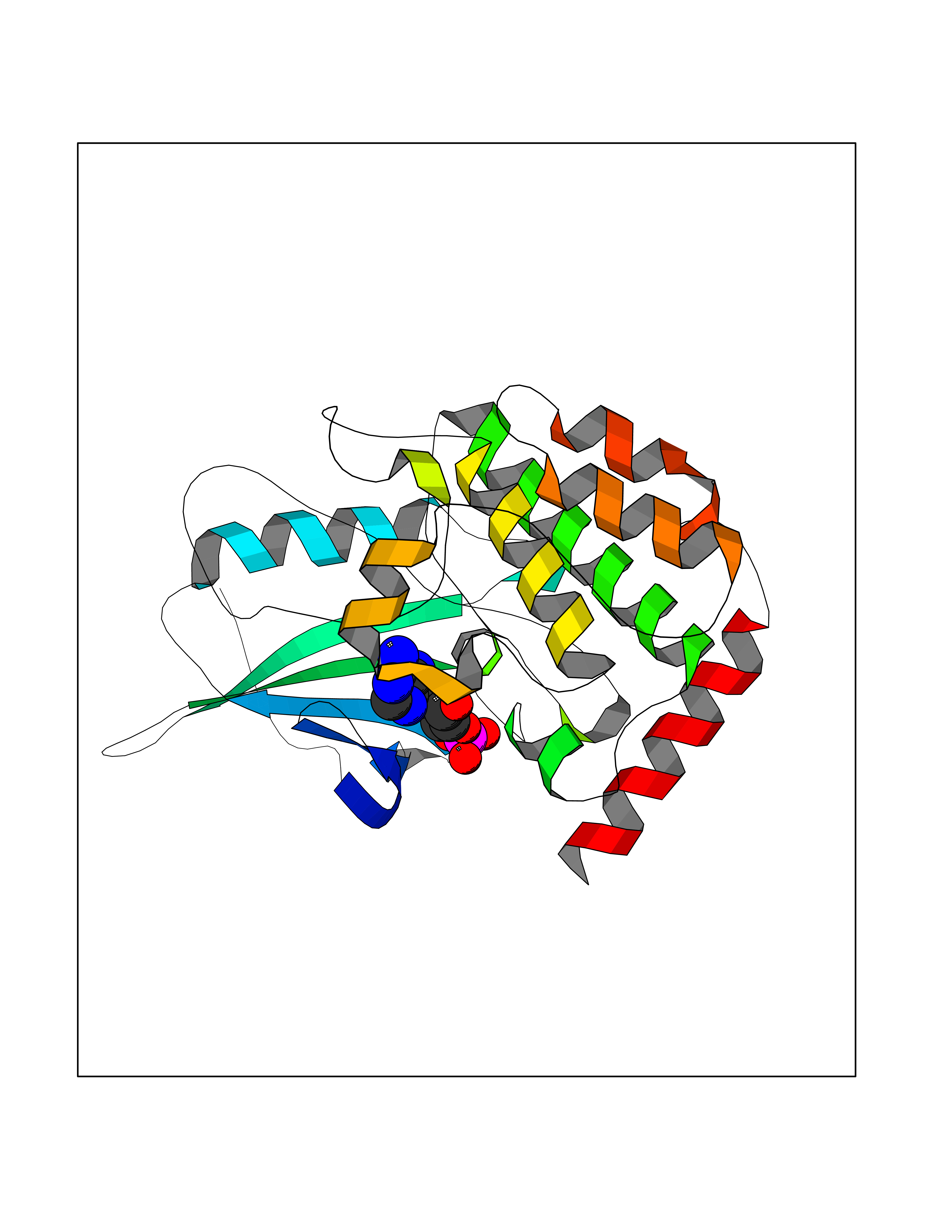 Molscript Map 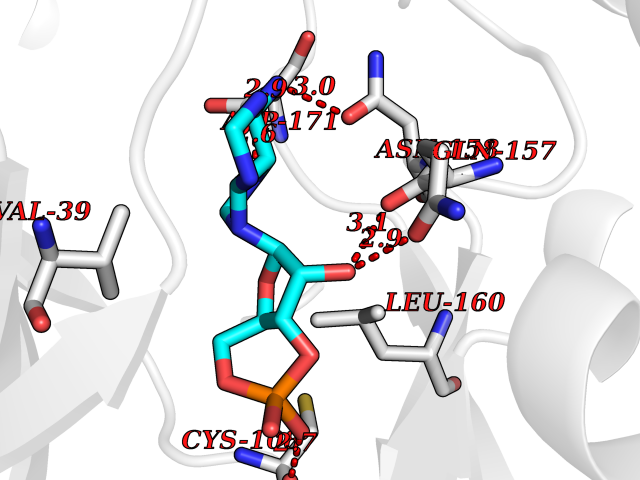 Pymol Map 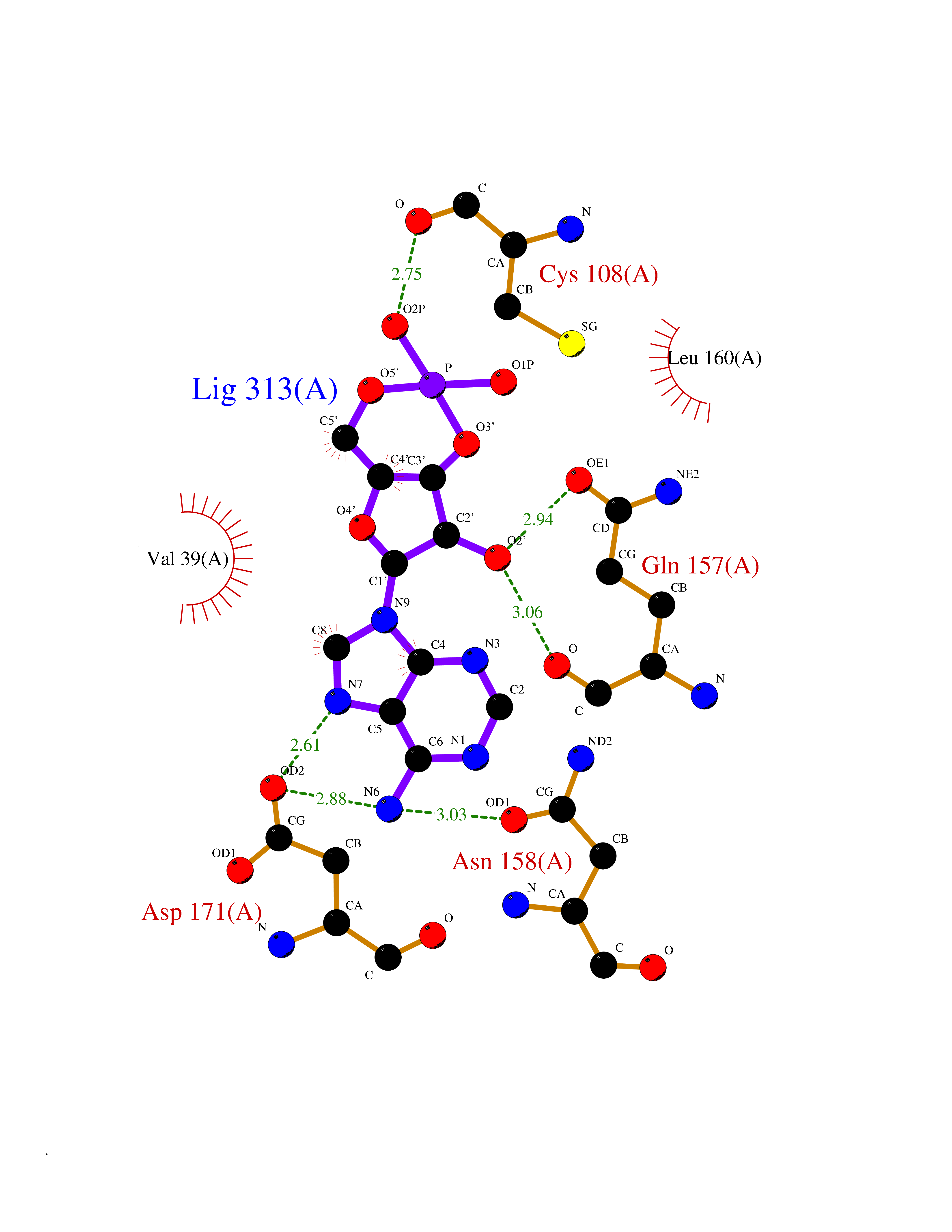 Ligplot Map 3D Binding mode Sequence SLRDPAGIFELVEVVGNGTYGQVYKGRHVKTGQLAAIKVMDVTEDEEEEIKLEINMLKKYSHHRNIATYYGAFIKKSPPGHDDQLWLVMEFCGAGSITDLVKNTKGNTLKEDWIAYISREILRGLAHLHIHHVIHRDIKGQNVLLTENAEVKLVDFGVSAQLDRTVGRRNTFIGTPYWMAPEVIACDENPDATYDYRSDLWSCGITAIEMAEGAPPLCDMHPMRALFLIPRNPPPRLKSKKWSKKFFSFIEGCLVKNYMQRPSTEQLLKHPFIRDQPNERQVRIQLKDHIDRTRK Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Corynebacterium Pup-protein ligase (Cory pafA) | 4BJR | 6.35 | |
Target general information Gen name Cory pafA Organism Corynebacterium glutamicum (strain ATCC 13032 / DSM 20300 / JCM 1318 / BCRC 11384 / CCUG 27702 / LMG 3730 / NBRC 12168 / NCIMB 10025 / NRRL B-2784 / 534) Uniprot ID TTD ID Synonyms Pup-conjugating enzyme; Pup--protein ligase; Proteasome accessory factor A Protein family Pup ligase/Pup deamidase family, Pup-conjugating enzyme subfamily Biochemical class NA Function Catalyzes the covalent attachment of the prokaryotic ubiquitin-like protein modifier Pup to the proteasomal substrate proteins, thereby targeting them for proteasomal degradation. This tagging system is termed pupylation. The ligation reaction involves the side-chain carboxylate of the C-terminal glutamate of Pup and the side-chain amino group of a substrate lysine. Related diseases Anemia, non-spherocytic hemolytic, due to G6PD deficiency (NSHA) [MIM:300908]: A disease characterized by G6PD deficiency, acute hemolytic anemia, fatigue, back pain, and jaundice. In most patients, the disease is triggered by an exogenous agent, such as some drugs, food, or infection. Increased unconjugated bilirubin, lactate dehydrogenase, and reticulocytosis are markers of the disorder. Although G6PD deficiency can be life-threatening, most patients are asymptomatic throughout their life. {ECO:0000269|PubMed:12524354, ECO:0000269|PubMed:1303180, ECO:0000269|PubMed:1303182, ECO:0000269|PubMed:1536798, ECO:0000269|PubMed:1611091, ECO:0000269|PubMed:1889820, ECO:0000269|PubMed:1945893, ECO:0000269|PubMed:20007901, ECO:0000269|PubMed:26479991, ECO:0000269|PubMed:2836867, ECO:0000269|PubMed:2912069, ECO:0000269|PubMed:30988594, ECO:0000269|PubMed:38066190, ECO:0000269|PubMed:7858267, ECO:0000269|PubMed:7959695, ECO:0000269|PubMed:8193373, ECO:0000269|PubMed:8490627, ECO:0000269|PubMed:8533762, ECO:0000269|PubMed:8733135, ECO:0000269|PubMed:9452072}. The disease is caused by variants affecting the gene represented in this entry. Deficiency of G6PD is associated with hemolytic anemia in two different situations. First, in areas in which malaria has been endemic, G6PD-deficiency alleles have reached high frequencies (1% to 50%) and deficient individuals, though essentially asymptomatic in the steady state, have a high risk of acute hemolytic attacks. Secondly, sporadic cases of G6PD deficiency occur at a very low frequencies, and they usually present a more severe phenotype. Several types of NSHA are recognized. Class-I variants are associated with severe NSHA; class-II have an activity <10% of normal; class-III have an activity of 10% to 60% of normal; class-IV have near normal activity. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; ATP-binding; Ligase; Magnesium; Metal-binding; Nucleotide-binding; Reference proteome; Ubl conjugation pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 110572 Length 994 Aromaticity 0.07 Instability index 42.33 Isoelectric point 5.26 Charge (pH=7) -34.19 2D Binding mode Binding energy (Kcal/mol) -7.43 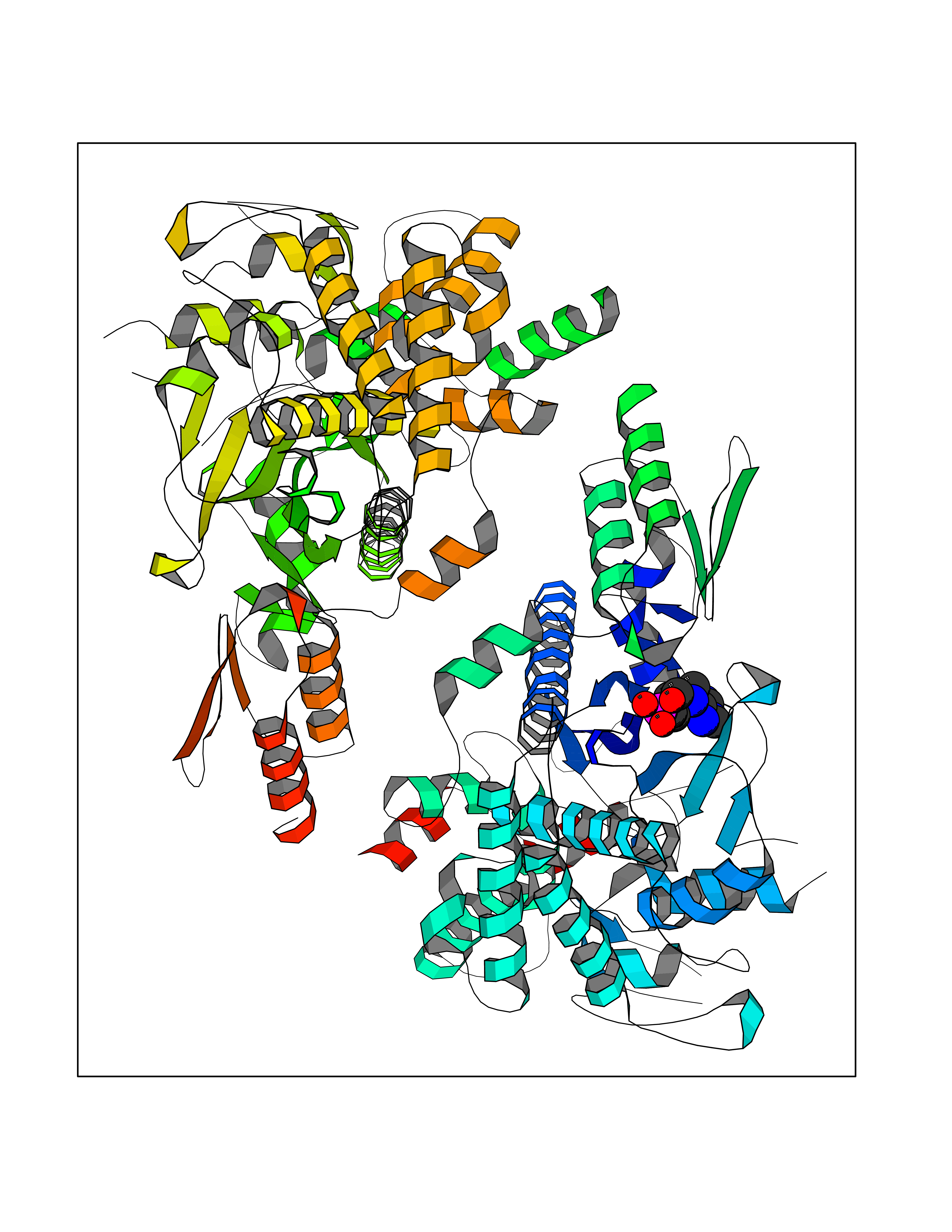 Molscript Map 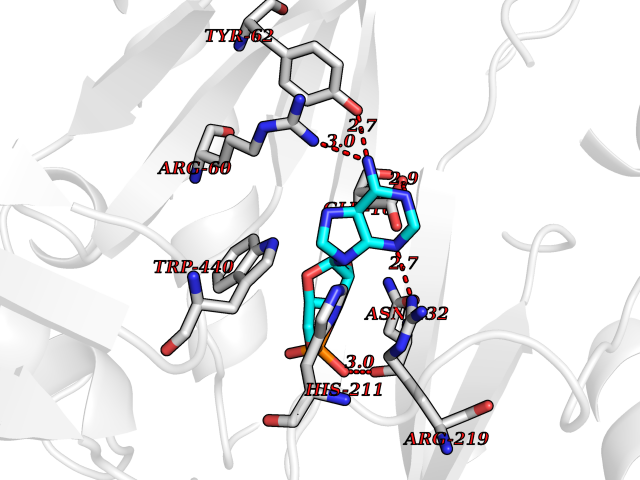 Pymol Map 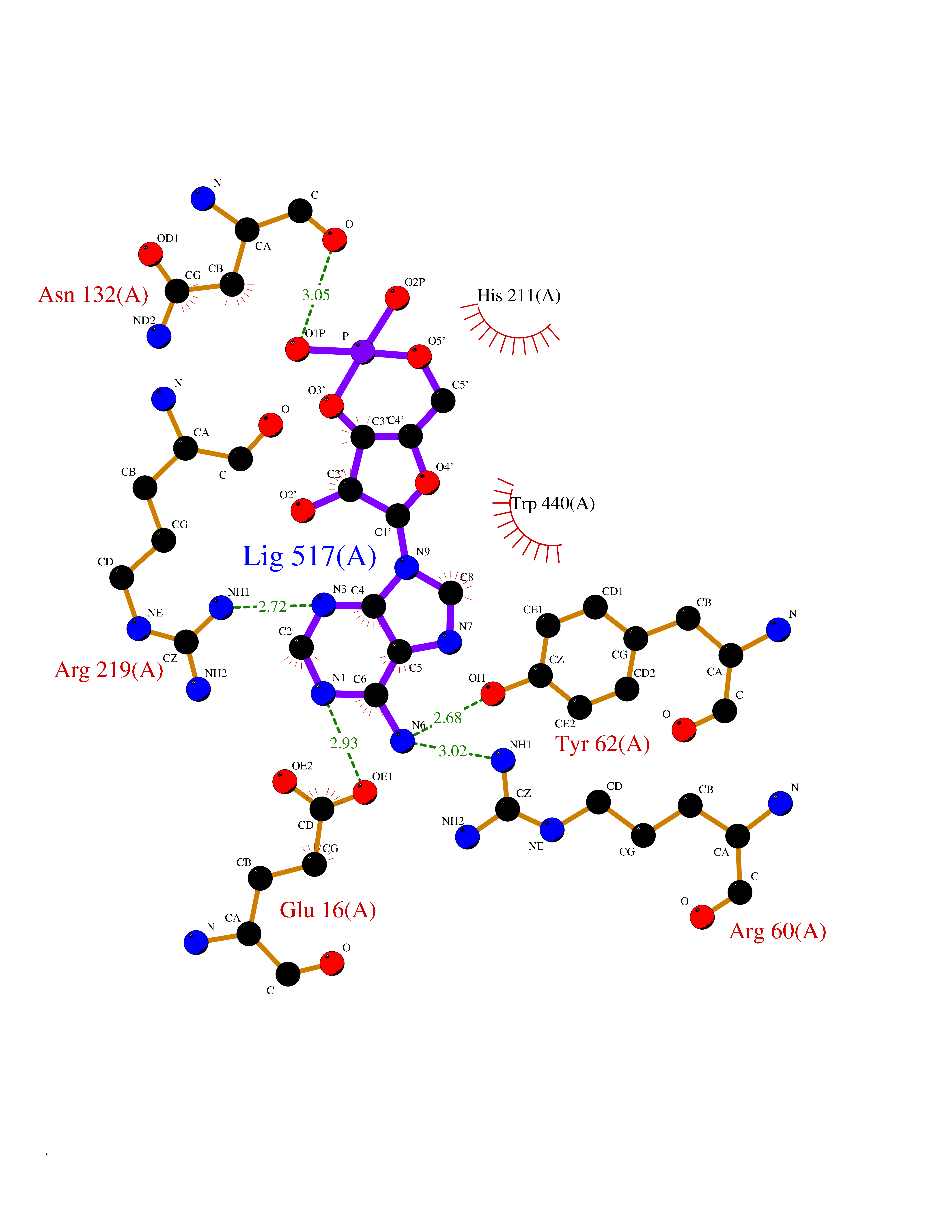 Ligplot Map 3D Binding mode Sequence TVESALTRRIMGIETEYGLTFVDGDSKKLRPDEIARRMFRPIVEKYSSSNIFIPNGSRLYLNVGSHPEYATAECDNLTQLINFEKAGDVIADRMAVDAEESLAKEDIAGQVYLFKNNVDSVGNSYGCHENYLVGRSMPLKALGKRLMPFLITRQLICGAGRIHHPNPSFPLGYCISQRSDHVWEGVSSASRPIINTRDEPHADSHSYRRLHVIVGDANMAEPSIALKVGSTLLVLEMIEADFGLPSLELANDIASIREISRDATGSTLLSLKDGTTMTALQIQQVVFEHASKWLEQRPEPEFSGTSNTEMARVLDLWGRMLKAIESGDFSEVDTEIDWVIKKKLIDRFIQRGNLGLDDPKLAQVDLTYHDIRPGRGLFSVLQSRGMIKRWTTDEAILAAVDTAPDTTRAHLRGRILKAADTLGVPVTVDWMRHKVNRPEPQSVELGDPFSAVNSEVDQLIEYMTVHAGSASGTSLLDEIDGLLENNAEEFVRSYVQKGGETVESALTRRIMGIETEYGLTFVDGDSKKLRPDEIARRMFRPIVEKYSSSNIFIPNGSRLYLNVGSHPEYATAECDNLTQLINFEKAGDVIADRMAVDAEESLAKEDIAGQVYLFKNNVDSVGNSYGCHENYLVGRSMPLKALGKRLMPFLITRQLICGAGRIHHPNPSFPLGYCISQRSDHVWEGVSSASRPIINTRDEPHADSHSYRRLHVIVGDANMAEPSIALKVGSTLLVLEMIEADFGLPSLELANDIASIREISRDATGSTLLSLKDGTTMTALQIQQVVFEHASKWLEQRPEPEFSGTSNTEMARVLDLWGRMLKAIESGDFSEVDTEIDWVIKKKLIDRFIQRGNLGLDDPKLAQVDLTYHDIRPGRGLFSVLQSRGMIKRWTTDEAILAAVDTAPDTTRAHLRGRILKAADTLGVPVTVDWMRHKVNRPEPQSVELGDPFSAVNSEVDQLIEYMTVHASLLDEIDGLLENNAEEFVRSYVQKGGE Hydrogen bonds contact Hydrophobic contact | ||||