Job Results:
Ligand
Structure
Job ID
6a37619723fa756e073677fca12f4afa
Job name
NA
Time
2024-07-14 16:31:23
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Carbonic anhydrase VII (CA-VII) | 3ML5 | 7.19 | |
Target general information Gen name CA7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Carbonic anhydrase 7; Carbonate dehydratase VII Protein family Alpha-carbonic anhydrase family Biochemical class Alpha-carbonic anhydrase Function Reversible hydration of carbon dioxide. Related diseases Neurodegeneration with brain iron accumulation 8 (NBIA8) [MIM:617917]: A neurodegenerative disorder associated with iron accumulation, primarily in the basal ganglia. Disease onset is in early childhood. Clinical features include speech delay, progressive cerebellar ataxia, unbalanced gait, and loss of ambulation. NBIA8 transmission pattern is consistent with autosomal recessive inheritance. {ECO:0000269|PubMed:29395073}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00819; DB00562; DB00606; DB01144; DB08846; DB00311; DB00774; DB00703; DB00909 Interacts with NA EC number EC 4.2.1.1 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Lyase; Metal-binding; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 29393.6 Length 262 Aromaticity 0.11 Instability index 40.14 Isoelectric point 7 Charge (pH=7) -0.01 2D Binding mode Binding energy (Kcal/mol) -8.05  Molscript Map 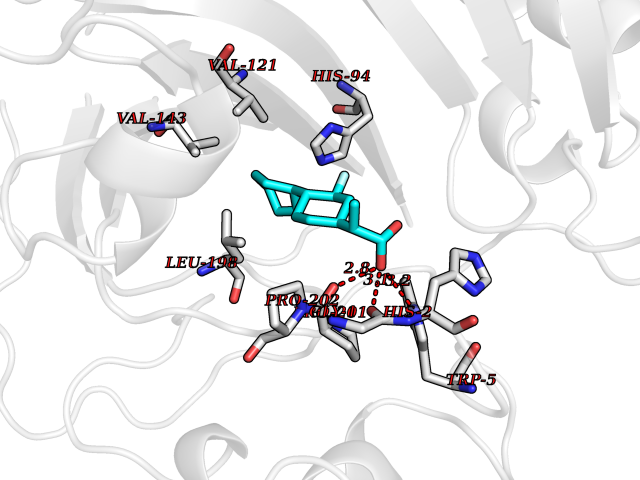 Pymol Map 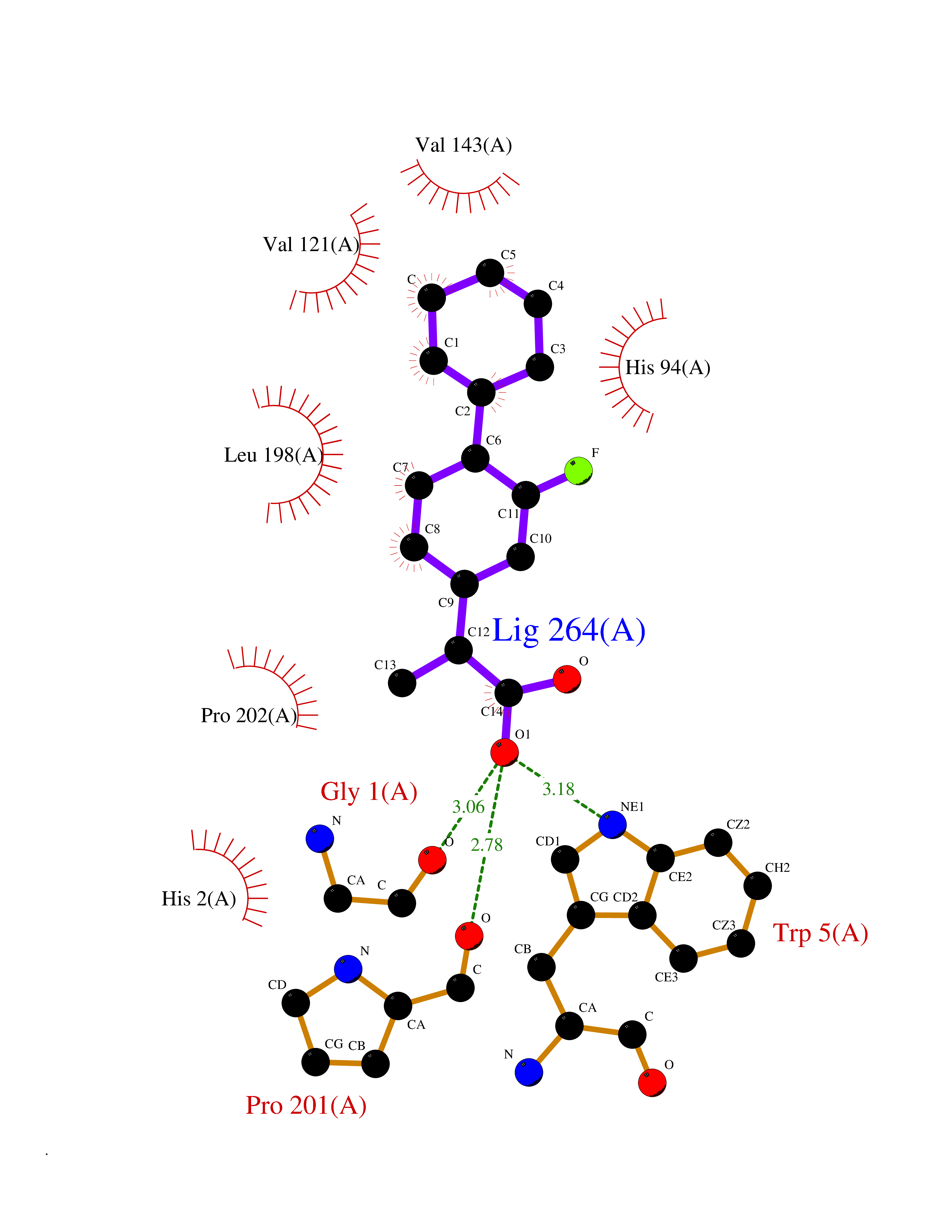 Ligplot Map 3D Binding mode Sequence GHHGWGYGQDDGPSHWHKLYPIAQGDRQSPINIISSQAVYSPSLQPLELSYEACMSLSITNNGHSVQVDFNDSDDRTVVTGGPLEGPYRLKQFHFHWGKKHDVGSEHTVDGKSFPSELHLVHWNAKKYSTFGEAASAPDGLAVVGVFLETGDEHPSMNRLTDALYMVRFKGTKAQFSCFNPKSLLPASRHYWTYPGSLTTPPLSESVTWIVLREPISISERQMGKFRSLLFTSEDDERIHMVNNFRPPQPLKGRVVKASFRA Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Myeloperoxidase (MPO) | 4DL1 | 7.19 | |
Target general information Gen name MPO Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MPO Protein family Peroxidase family, XPO subfamily Biochemical class Peroxidases Function Part of the host defense system of polymorphonuclear leukocytes. It is responsible for microbicidal activity against a wide range of organisms. In the stimulated PMN, MPO catalyzes the production of hypohalous acids, primarily hypochlorous acidin physiologic situations, and other toxic intermediates that greatly enhance PMN microbicidal activity. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06111; DB00233; DB00006; DB02300; DB06774; DB00958; DB06468; DB00833; DB00535; DB00515; DB00847; DB00250; DB05161; DB01225; DB00583; DB01065; DB00461; DB04821; DB00104; DB00526; DB00550; DB00208; DB06823; DB00500; DB04827 Interacts with P27918; Q9UNE7 EC number EC 1.11.2.2 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Heme; Hydrogen peroxide; Iron; Lysosome; Metal-binding; Oxidation; Oxidoreductase; Peroxidase; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID A,B,E,F,I,J,M,N Molecular weight (Da) 53052.6 Length 466 Aromaticity 0.08 Instability index 40.64 Isoelectric point 9.48 Charge (pH=7) 15.12 2D Binding mode Binding energy (Kcal/mol) -7.89 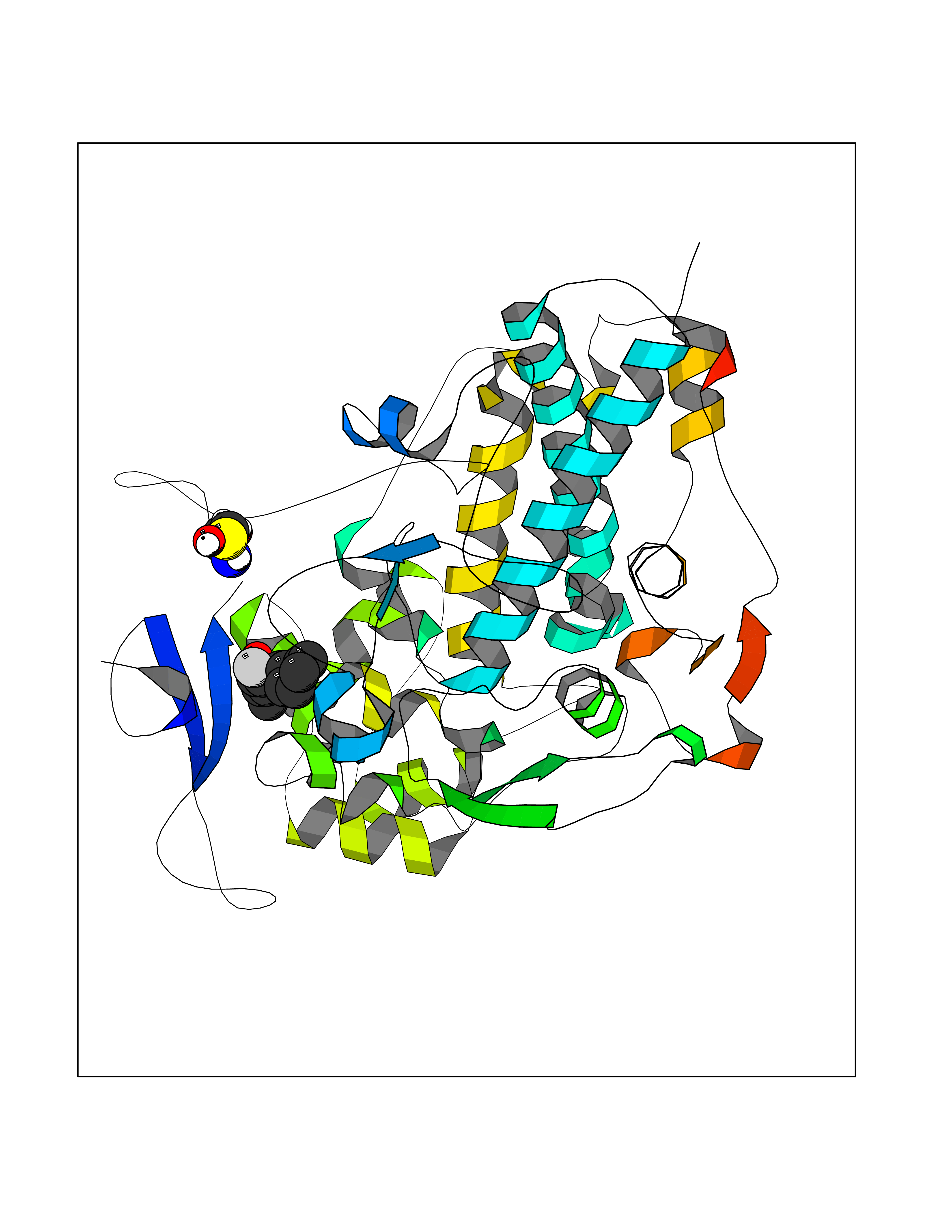 Molscript Map 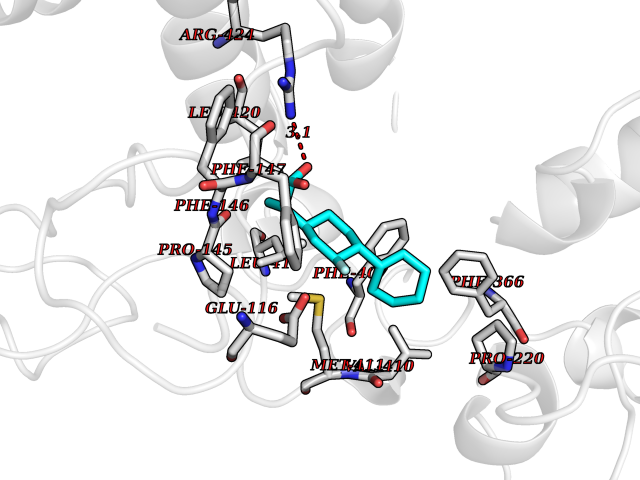 Pymol Map 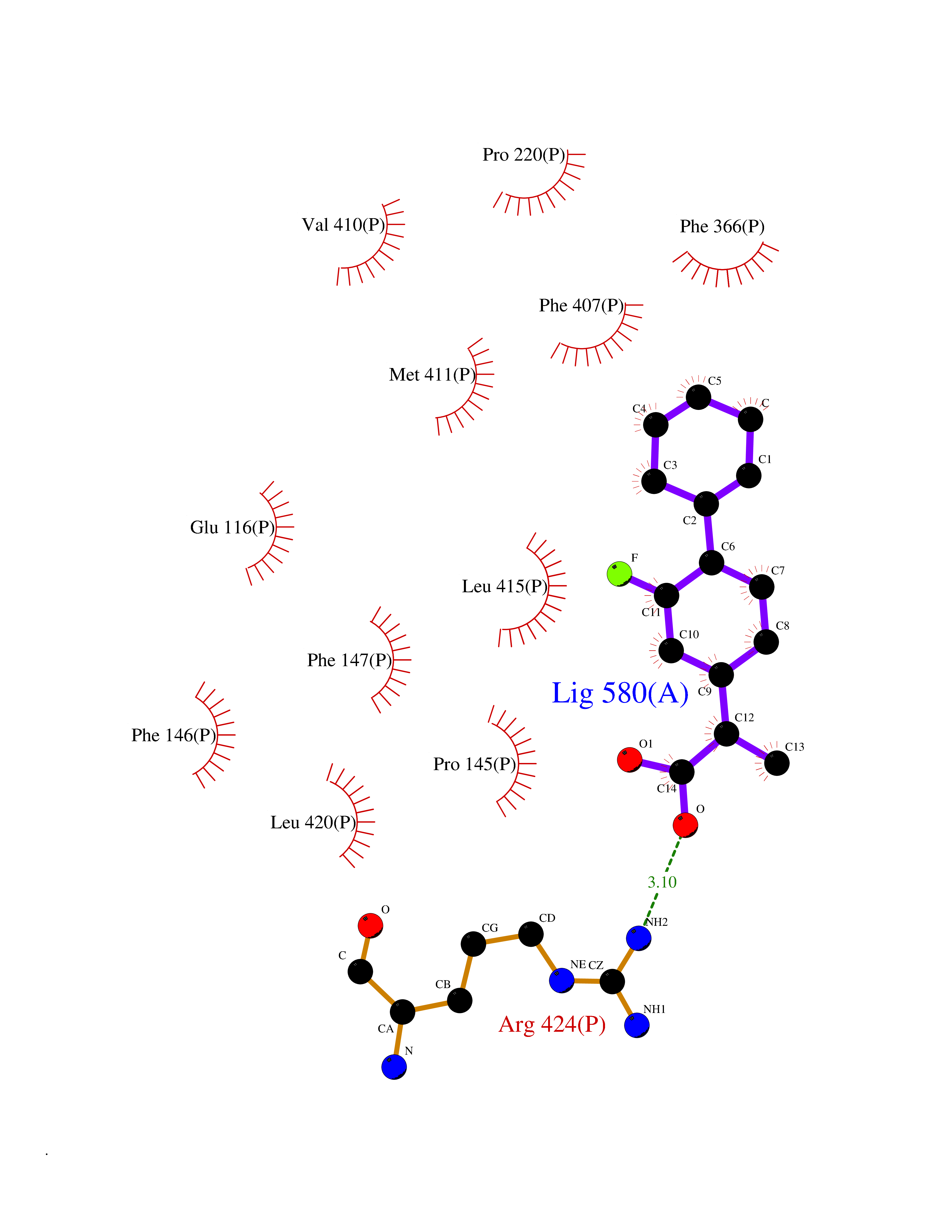 Ligplot Map 3D Binding mode Sequence VNCETSCVQQPPCFPLKIPPNDPRIKNQADCIPFFRSXPACPGSNITIRNQINALTSFVDASMVYGSEEPLARNLRNMSNQLGLLAVNQRFQDNGRALLPFDNLHDDPCLLTNRSARIPCFLAGDTRSSEMPELTSMHTLLLREHNRLATELKSLNPRWDGERLYQEARKIVGAMVQIITYRDYLPLVLGPTAMRKYLPTYRSYNDSVDPRIANVFTNAFRYGHTLIQPFMFRLDNRYQPMEPNPRVPLSRVFFASWRVVLEGGIDPILRGLMATPAKLNRQNQIAVDEIRERLFEQVMRIGLDLPALNMQRSRDHGLPGYNAWRRFCGLPQPETVGQLGTVLRNLKLARKLMEQYGTPNNIDIWMGGVSEPLKRKGRVGPLLACIIGTQFRKLRDGDRFWWENEGVFSMQQRQALAQISLPRIICDNTGITTVSKNNIFMSNSYPRDFVNCSTLPALNLASWREA Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Serine/threonine-protein kinase cot (COT) | 4Y85 | 7.19 | |
Target general information Gen name MAP3K8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tumor progression locus 2; TPL-2; Proto-oncogene c-Cot; Mitogen-activated protein kinase kinase kinase 8; ESTF; Cancer Osaka thyroid oncogene; COT Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, MAP kinase kinase kinase subfamily Biochemical class NA Function Required for lipopolysaccharide (LPS)-induced, TLR4-mediated activation of the MAPK/ERK pathway in macrophages, thus being critical for production of the proinflammatory cytokine TNF-alpha (TNF) during immune responses. Involved in the regulation of T-helper cell differentiation and IFNG expression in T-cells. Involved in mediating host resistance to bacterial infection through negative regulation of type I interferon (IFN) production. In vitro, activates MAPK/ERK pathway in response to IL1 in an IRAK1-independent manner, leading to up-regulation of IL8 and CCL4. Transduces CD40 and TNFRSF1A signals that activate ERK in B-cells and macrophages, and thus may play a role in the regulation of immunoglobulin production. May also play a role in the transduction of TNF signals that activate JNK and NF-kappa-B in some cell types. In adipocytes, activates MAPK/ERK pathway in an IKBKB-dependent manner in response to IL1B and TNF, but not insulin, leading to induction of lipolysis. Plays a role in the cell cycle. Isoform 1 shows some transforming activity, although it is much weaker than that of the activated oncogenic variant. Related diseases Hyperinsulinemic hypoglycemia, familial, 2 (HHF2) [MIM:601820]: A form of hyperinsulinemic hypoglycemia, a clinically and genetically heterogeneous disorder characterized by inappropriate insulin secretion from the pancreatic beta-cells in the presence of low blood glucose levels. HHF2 is a common cause of persistent hypoglycemia in infancy. Unless early and aggressive intervention is undertaken, brain damage from recurrent episodes of hypoglycemia may occur. HHF2 inheritance can be autosomal dominant or autosomal recessive. {ECO:0000269|PubMed:10204114, ECO:0000269|PubMed:12364426, ECO:0000269|PubMed:15562009, ECO:0000269|PubMed:15579781, ECO:0000269|PubMed:15807877, ECO:0000269|PubMed:15998776, ECO:0000269|PubMed:16332676, ECO:0000269|PubMed:16357843, ECO:0000269|PubMed:18596924, ECO:0000269|PubMed:19357197, ECO:0000269|PubMed:7847376, ECO:0000269|PubMed:8923010}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Diabetes mellitus, permanent neonatal, 2 (PNDM2) [MIM:618856]: A form of permanent neonatal diabetes mellitus, a type of diabetes characterized by onset of persistent hyperglycemia within the first six months of life. Initial clinical manifestations include intrauterine growth retardation, hyperglycemia, glycosuria, osmotic polyuria, severe dehydration, and failure to thrive. Some PNDM2 patients may also have developmental delay, muscle weakness, epilepsy and dysmorphic features. PNDM2 transmission pattern is consistent with autosomal dominant inheritance. {ECO:0000269|PubMed:15115830, ECO:0000269|PubMed:15292329, ECO:0000269|PubMed:15448106, ECO:0000269|PubMed:15448107, ECO:0000269|PubMed:15580558, ECO:0000269|PubMed:15583126, ECO:0000269|PubMed:16609879, ECO:0000269|PubMed:16731833, ECO:0000269|PubMed:17213273, ECO:0000269|PubMed:17652641, ECO:0000269|PubMed:17855752, ECO:0000269|PubMed:20022885, ECO:0000269|PubMed:28842488}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Transient neonatal diabetes mellitus 3 (TNDM3) [MIM:610582]: Neonatal diabetes mellitus, defined as insulin-requiring hyperglycemia within the first month of life, is a rare entity. In about half of the neonates, diabetes is transient and resolves at a median age of 3 months, whereas the rest have a permanent form of diabetes. In a significant number of patients with transient neonatal diabetes mellitus, diabetes type 2 appears later in life. The onset and severity of TNDM3 is variable with childhood-onset diabetes, gestational diabetes or adult-onset diabetes described. {ECO:0000269|PubMed:15718250, ECO:0000269|PubMed:15784703}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in KCNJ11 may contribute to non-insulin-dependent diabetes mellitus (NIDDM), also known as diabetes mellitus type 2.; DISEASE: Maturity-onset diabetes of the young 13 (MODY13) [MIM:616329]: A form of diabetes that is characterized by an autosomal dominant mode of inheritance, onset in childhood or early adulthood (usually before 25 years of age), a primary defect in insulin secretion and frequent insulin-independence at the beginning of the disease. {ECO:0000269|PubMed:22701567}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P08238; P19838; Q00653; Q13526; Q8NFZ5 EC number EC 2.7.11.25 Uniprot keywords 3D-structure; Alternative initiation; ATP-binding; Cell cycle; Cytoplasm; Immunity; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 34773.8 Length 307 Aromaticity 0.09 Instability index 39.24 Isoelectric point 6.68 Charge (pH=7) -1.2 2D Binding mode Binding energy (Kcal/mol) -8.69 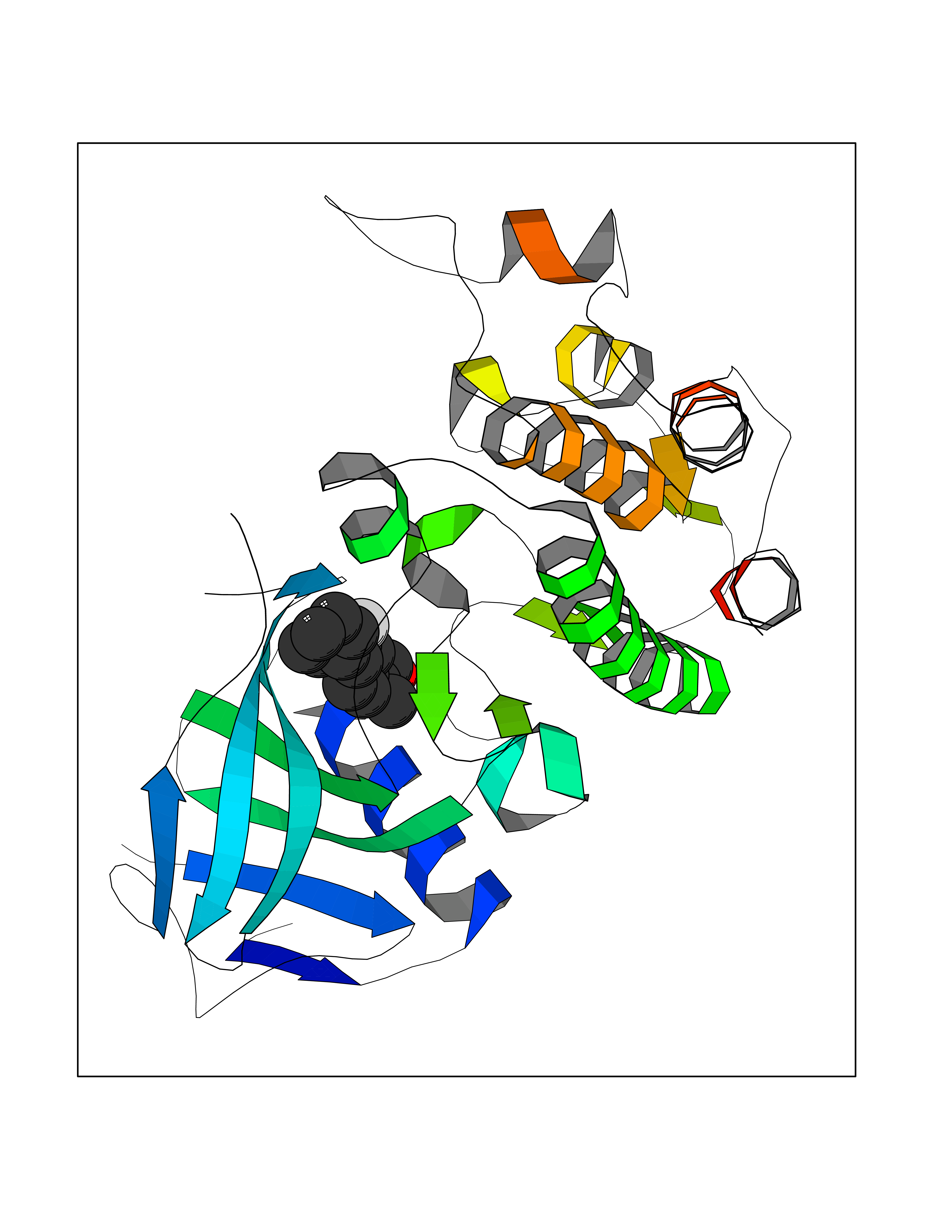 Molscript Map 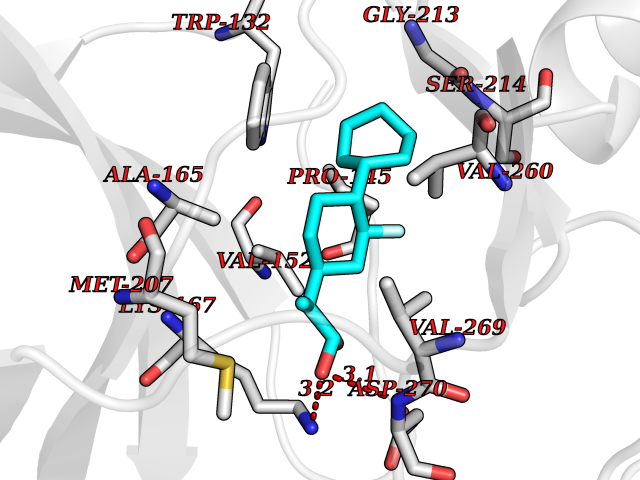 Pymol Map 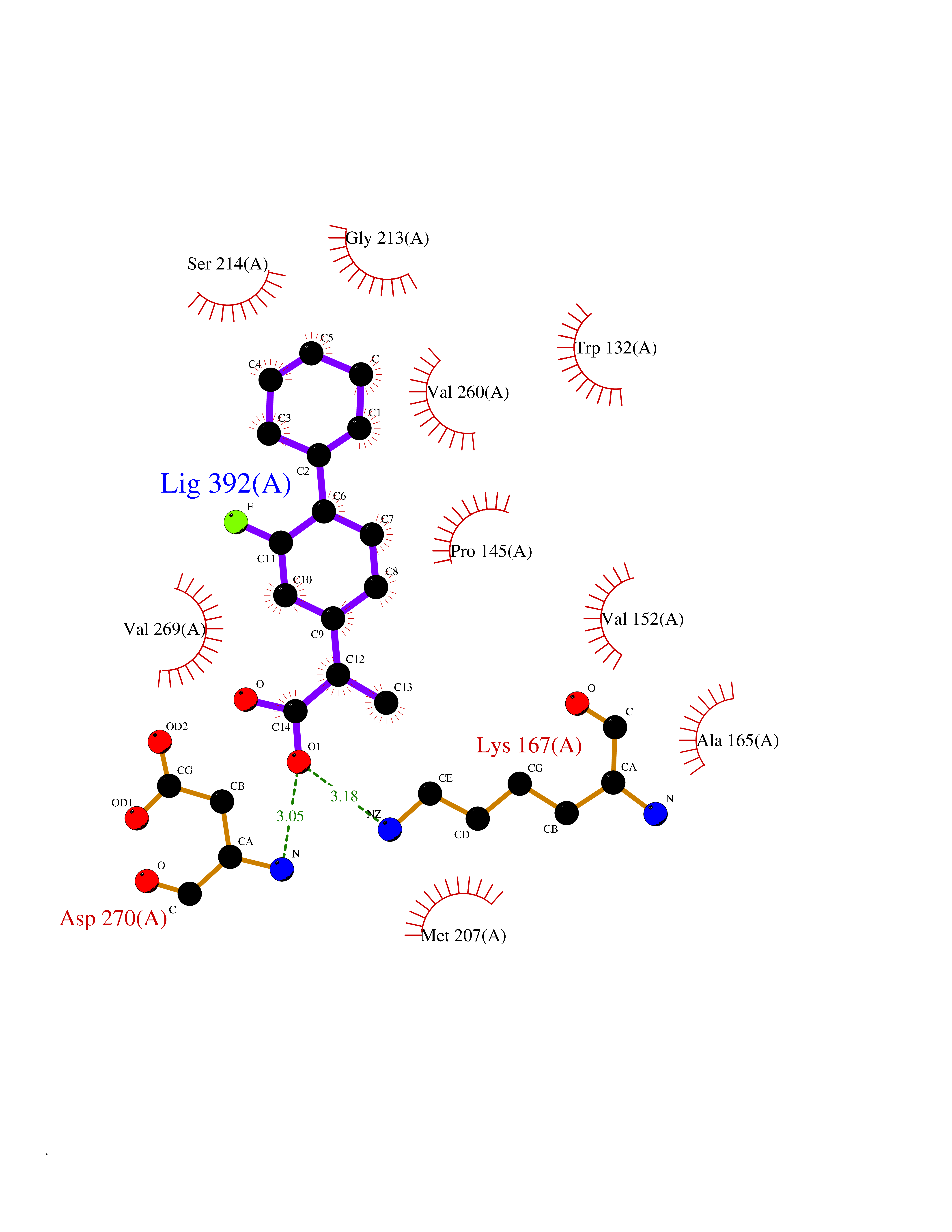 Ligplot Map 3D Binding mode Sequence LSSVRYGTVEDLLAFANHISNTPQESGILLNMVITPQNGRYQIDSDVLLIPWKLTYRNIFIPRGAFGKVYLAQDIKTKKRMACKLIPVDQFKPSDVEIQACFRHENIAELYGAVLWGETVHLFMEAGEGGSVLEKLESCGPMREFEIIWVTKHVLKGLDFLHSKKVIHHDIKPSNIVFMSTKAVLVDFGLSVQMTEDVYFPKDLRGTEIYMSPEVILCRGHSTKADIYSLGATLIHMQTGTPPWVKRYPRSAYPSYLYIIHKQAPPLEDIADDCSPGMRELIEASLERNPNHRPRAADLLKHEALNP Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Cytidine deaminase (CDA) | 1MQ0 | 7.19 | |
Target general information Gen name CDA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cytidine aminohydrolase; CDA Protein family Cytidine and deoxycytidylate deaminase family Biochemical class Carbon-nitrogen hydrolase Function This enzyme scavenges exogenous and endogenous cytidine and 2'-deoxycytidine for UMP synthesis. Related diseases Immunodeficiency 35 (IMD35) [MIM:611521]: A primary immunodeficiency characterized by recurrent skin abscesses, pneumonia, and highly elevated serum IgE. {ECO:0000269|PubMed:17088085}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03185; DB00928; DB01101; DB15694; DB00987; DB01262; DB00441 Interacts with Q96EY9; Q96B67; P32320; Q96D03; Q9BVV2; O15353; Q9H8Y8; Q0VD86; Q3LI64; Q8IUC2; Q8TBB1; Q96HA8; Q96CS7; P25786; P43115-12; O00560; O43167 EC number EC 3.5.4.5 Uniprot keywords 3D-structure; Hydrolase; Metal-binding; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 28728.6 Length 260 Aromaticity 0.1 Instability index 48.2 Isoelectric point 4.75 Charge (pH=7) -10.11 2D Binding mode Binding energy (Kcal/mol) -8.38 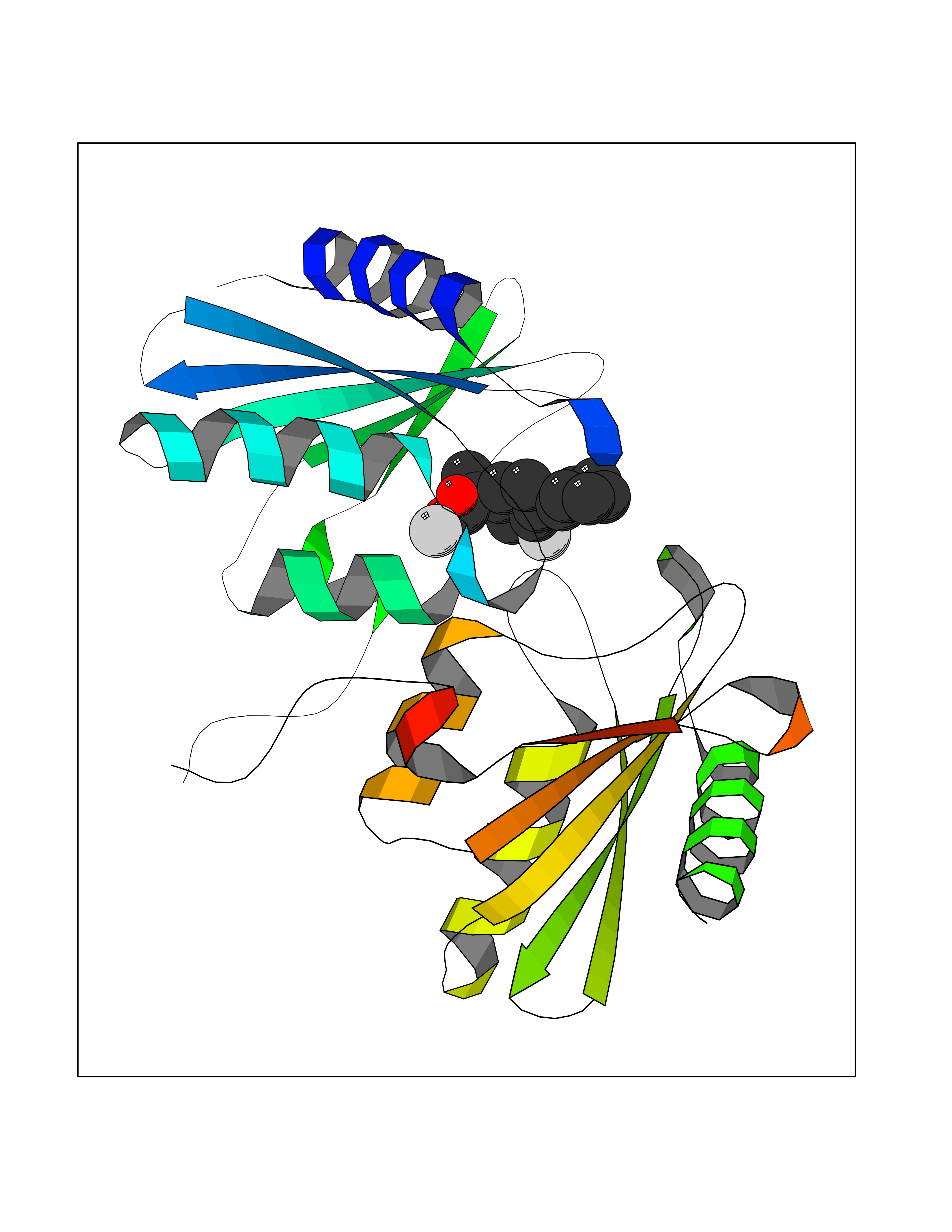 Molscript Map 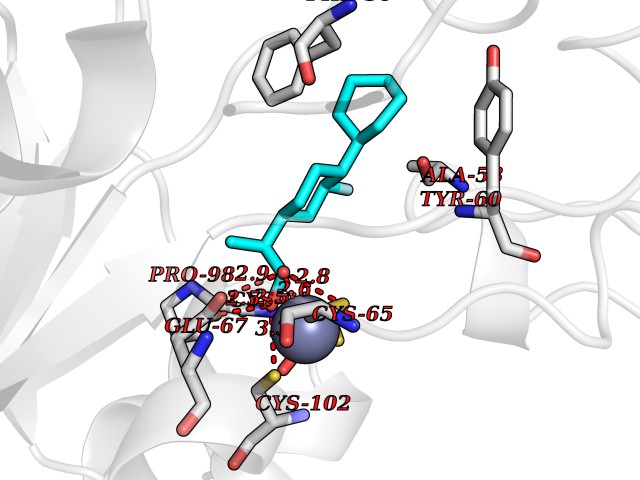 Pymol Map 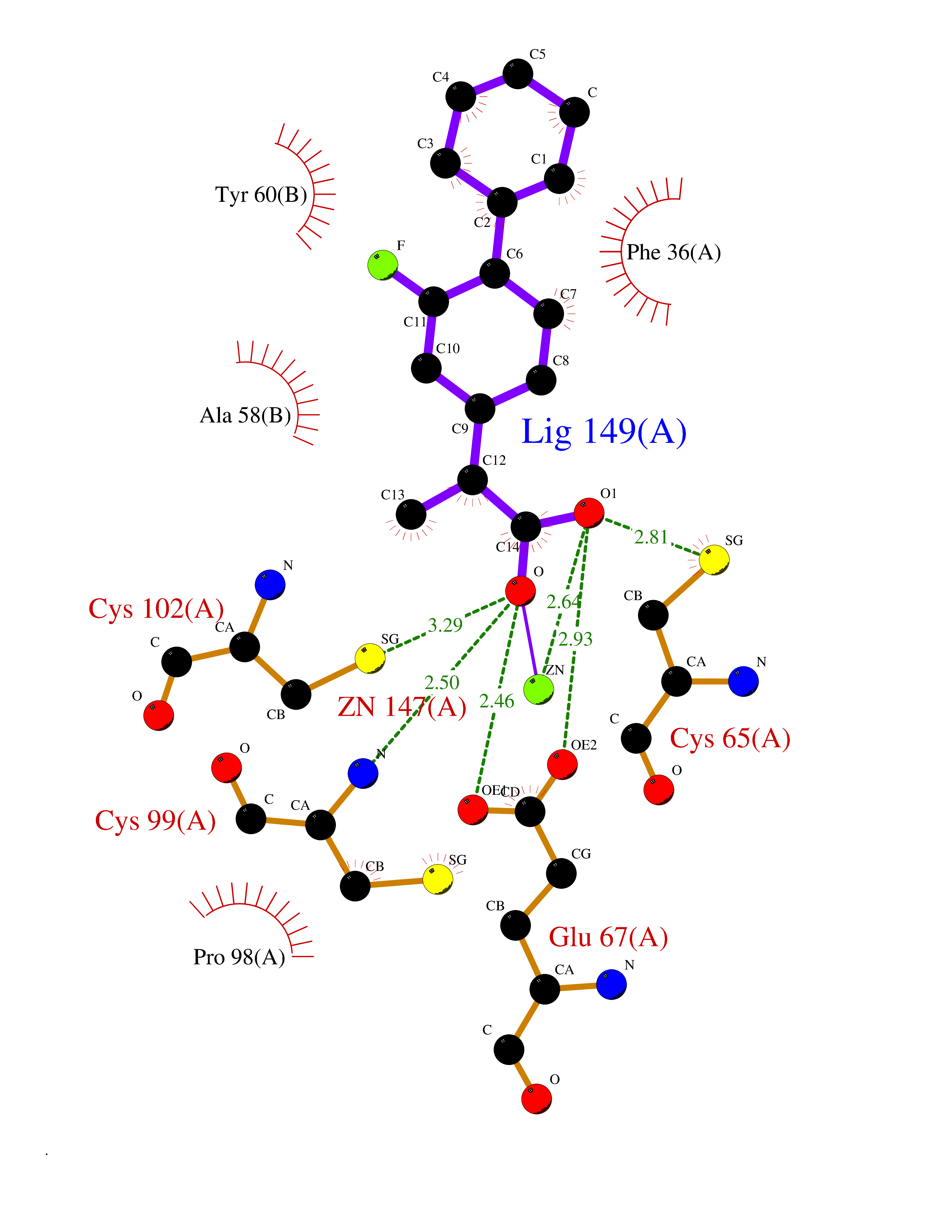 Ligplot Map 3D Binding mode Sequence ECVQQLLVCSQEAKQSAYCPYSHFPVGAALLTQEGRIFKGCNIENACYPLGICAERTAIQKAVSEGYKDFRAIAIASDMQDDFISPCGACRQVMREFGTNWPVYMTKPDGTYIVMTVQELLPSSFGPEDLECVQQLLVCSQEAKQSAYCPYSHFPVGAALLTQEGRIFKGCNIENACYPLGICAERTAIQKAVSEGYKDFRAIAIASDMQDDFISPCGACRQVMREFGTNWPVYMTKPDGTYIVMTVQELLPSSFGPEDL Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Ephrin type-A receptor 7 (EPHA7) | 3DKO | 7.18 | |
Target general information Gen name EPHA7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hEK11; EPH-like kinase 11; EPH homology kinase 3; EK11; EHK3; EHK-3 Protein family Protein kinase superfamily, Tyr protein kinase family, Ephrin receptor subfamily Biochemical class Kinase Function The signaling pathway downstream of the receptor is referred to as forward signaling while the signaling pathway downstream of the ephrin ligand is referred to as reverse signaling. Among GPI-anchored ephrin-A ligands, EFNA5 is a cognate/functional ligand for EPHA7 and their interaction regulates brain development modulating cell-cell adhesion and repulsion. Has a repellent activity on axons and is for instance involved in the guidance of corticothalamic axons and in the proper topographic mapping of retinal axons to the colliculus. May also regulate brain development through a caspase(CASP3)-dependent proapoptotic activity. Forward signaling may result in activation of components of the ERK signaling pathway including MAP2K1, MAP2K2, MAPK1 AND MAPK3 which are phosphorylated upon activation of EPHA7. Receptor tyrosine kinase which binds promiscuously GPI-anchored ephrin-A family ligands residing on adjacent cells, leading to contact-dependent bidirectional signaling into neighboring cells. Related diseases WHIM syndrome 2 (WHIMS2) [MIM:619407]: An autosomal recessive form of WHIM syndrome, a primary immunodeficiency disorder characterized by warts, hypogammaglobulinemia, infections, and myelokathexis. Myelokathexis is a unique form of non-cyclic severe congenital neutropenia caused by accumulation of mature and degenerating neutrophils in the bone marrow. Monocytopenia and lymphopenia, especially B lymphopenia, also commonly occur. There is significant phenotypic variation among patients, such that some individuals may have an incomplete form of the disorder in which one or more of the classic tetrad features are not present. {ECO:0000269|PubMed:24777453}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07970; DB12010 Interacts with P07333; P52803; P29317; P29320; P29323; P54760; Q16288; P52793 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cell membrane; Developmental protein; Glycoprotein; Kinase; Membrane; Neurogenesis; Nucleotide-binding; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 31250.7 Length 275 Aromaticity 0.09 Instability index 28.85 Isoelectric point 6.44 Charge (pH=7) -1.92 2D Binding mode Binding energy (Kcal/mol) -8.37 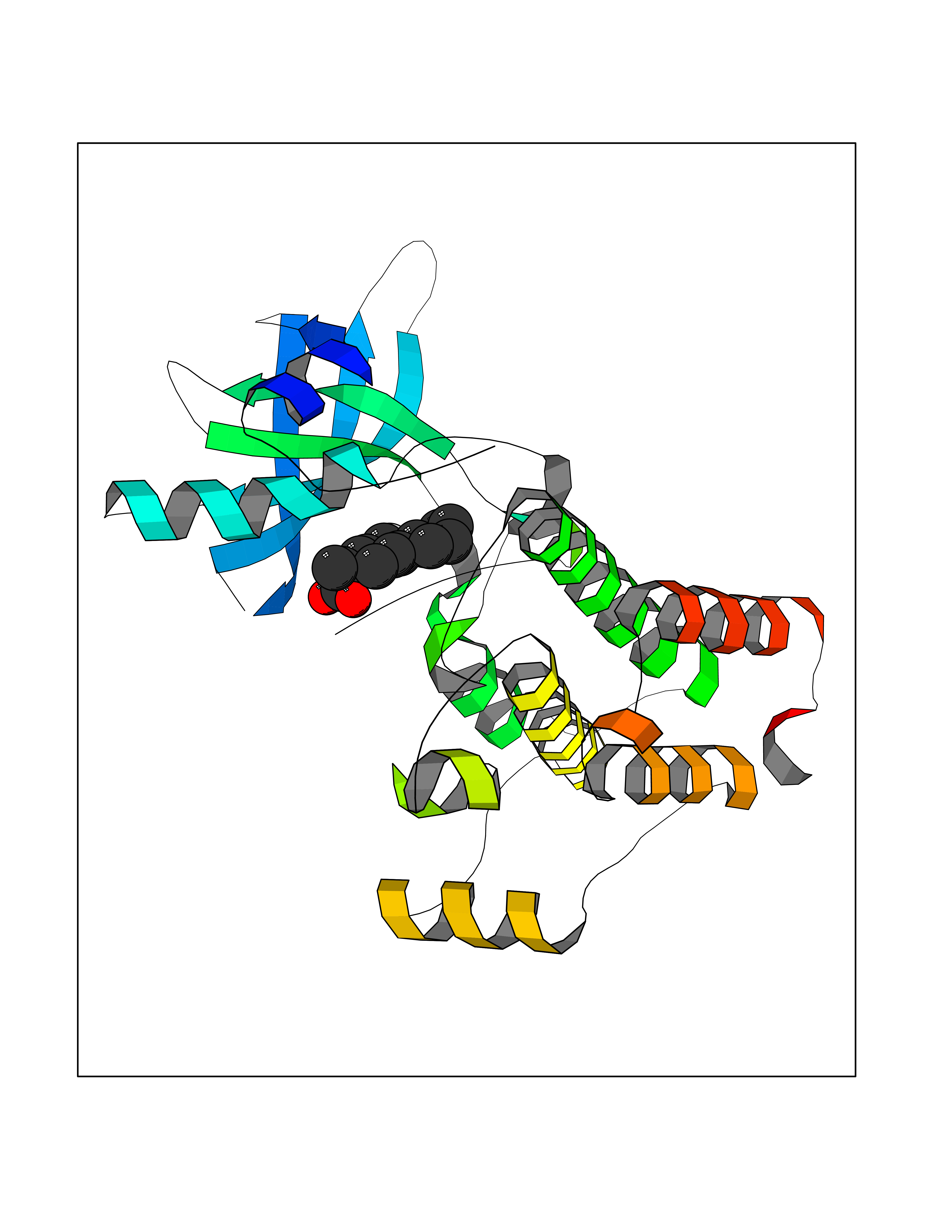 Molscript Map 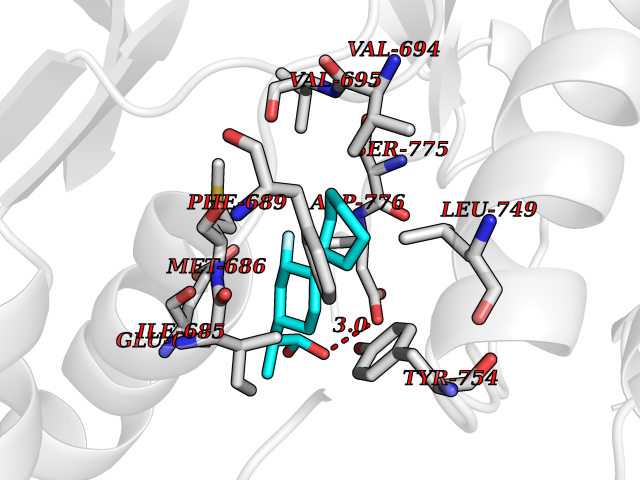 Pymol Map 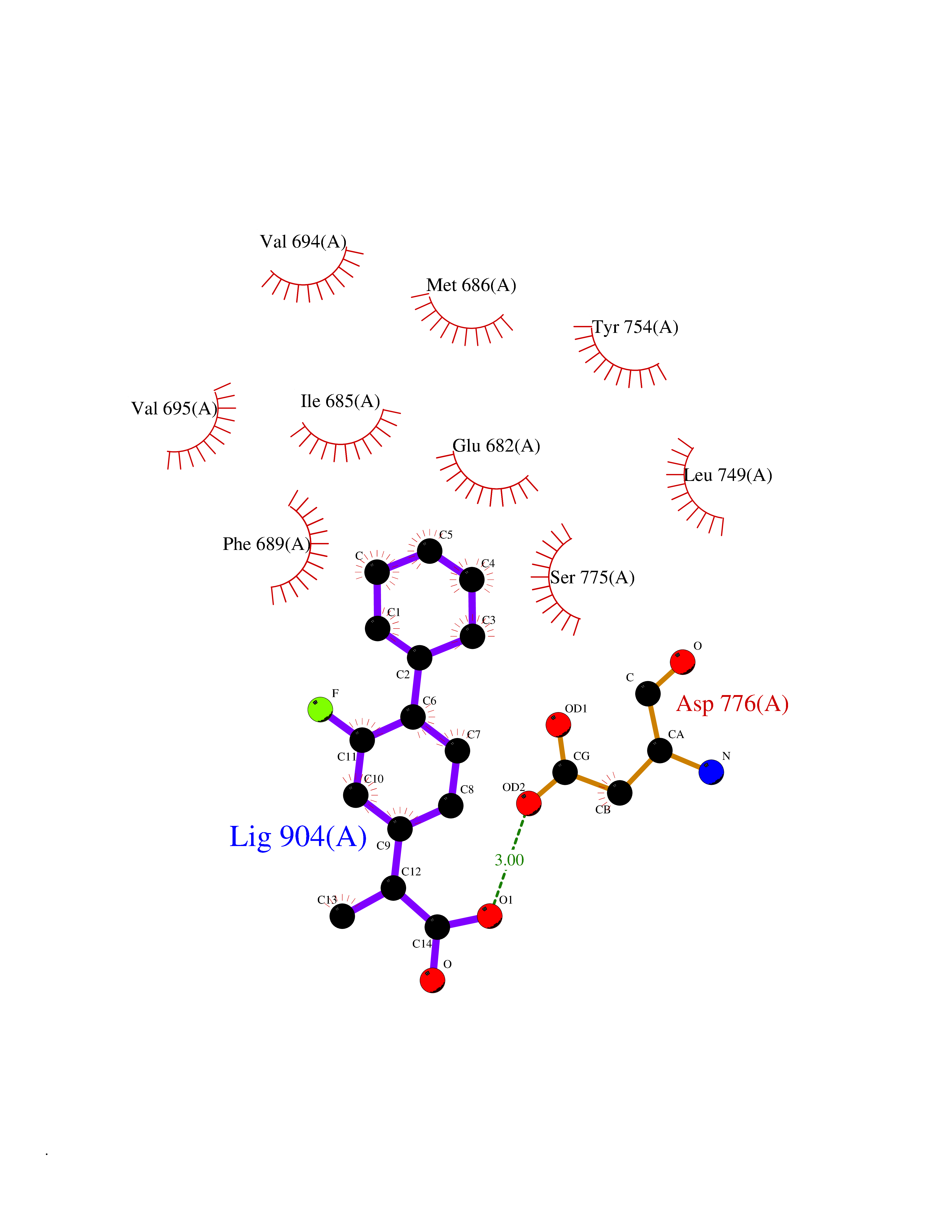 Ligplot Map 3D Binding mode Sequence TYIDPETYEDPNRAVHQFAKELDASCIKIERVIGAEFGEVCSGRLKLPGKRDVAVAIKTLKVGYTEKQRRDFLCEASIMGQFDHPNVVHLEGVVTRGKPVMIVIEFMENGALDAFLRKHDGQFTVIQLVGMLRGIAAGMRYLADMGYVHRDLAARNILVNSNLVCKVSDFGPVRWTAPEAIQYRKFTSASDVWSYGIVMWEVMSYGERPYWDMSNQDVIKAIEEGYRLPAPMDCPAGLHQLMLDCWQKERAERPKFEQIVGILDKMIRNPNSAHH Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Bone morphogenetic protein 1 (BMP1) | 6BTQ | 7.18 | |
Target general information Gen name BMP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Procollagen C-proteinase; PCP protein; PCOLC; Mammalian tolloid protein; MTld; BMP-1 Protein family NA Biochemical class Peptidase Function Induces cartilage and bone formation. May participate in dorsoventral patterning during early development by cleaving chordin (CHRD). Responsible for the proteolytic activation of lysyl oxidase LOX. Cleaves the C-terminal propeptides of procollagen I, II and III. Related diseases Osteogenesis imperfecta 13 (OI13) [MIM:614856]: An autosomal recessive form of osteogenesis imperfecta, a disorder of bone formation characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. OI13 is characterized by normal teeth, faint blue sclerae, severe growth deficiency, severe bone deformity, and recurrent fractures affecting both upper and lower limbs. {ECO:0000269|PubMed:22052668, ECO:0000269|PubMed:22482805, ECO:0000269|PubMed:25402547}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P13497; Q9H2X0; P20908; O14793; Q15113; P07585; P97299 EC number EC 3.4.24.19 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Chondrogenesis; Cleavage on pair of basic residues; Cytokine; Developmental protein; Differentiation; Disease variant; Disulfide bond; EGF-like domain; Extracellular matrix; Glycoprotein; Golgi apparatus; Growth factor; Hydrolase; Metal-binding; Metalloprotease; Methylation; Osteogenesis; Osteogenesis imperfecta; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 22934.8 Length 202 Aromaticity 0.11 Instability index 36.61 Isoelectric point 8.75 Charge (pH=7) 4.46 2D Binding mode Binding energy (Kcal/mol) -7.95 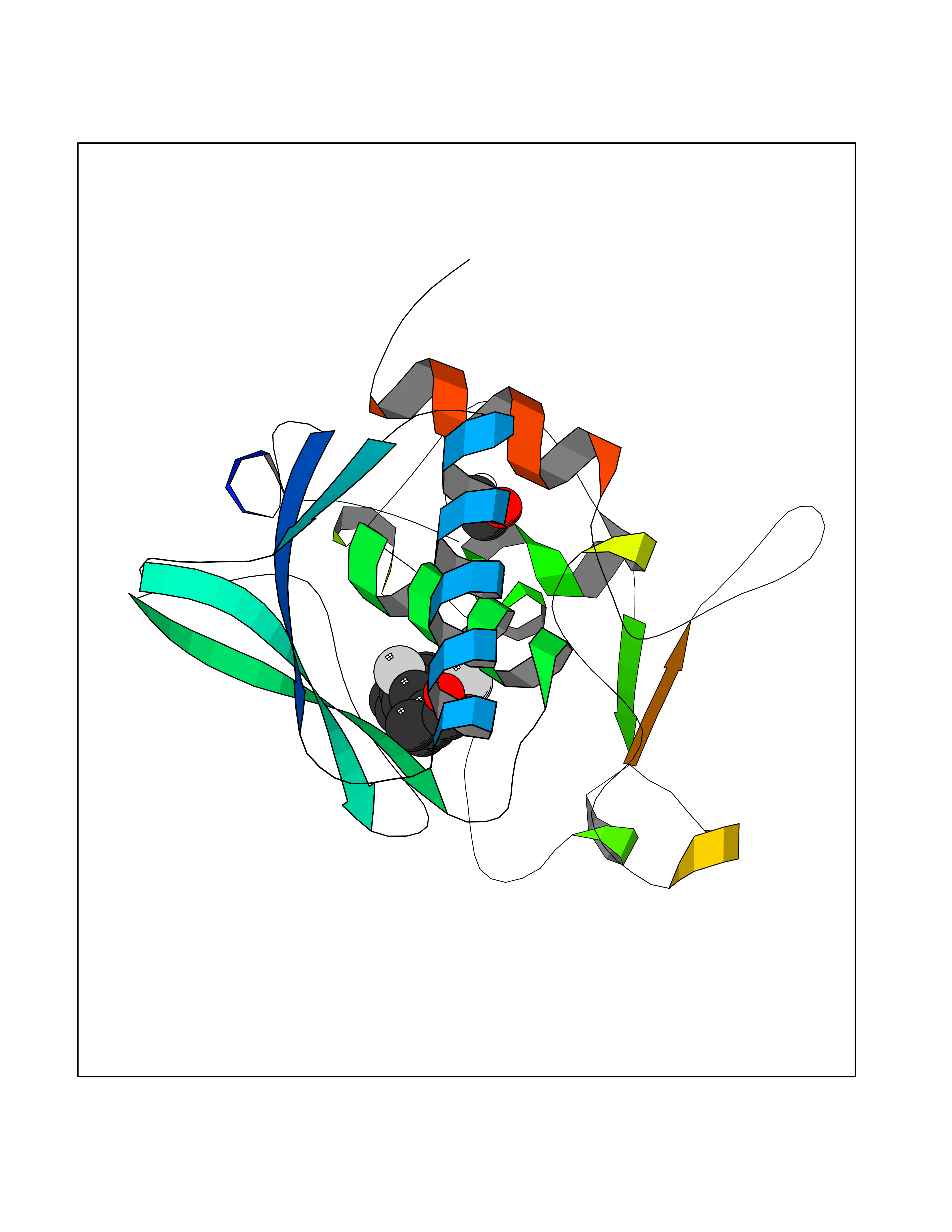 Molscript Map 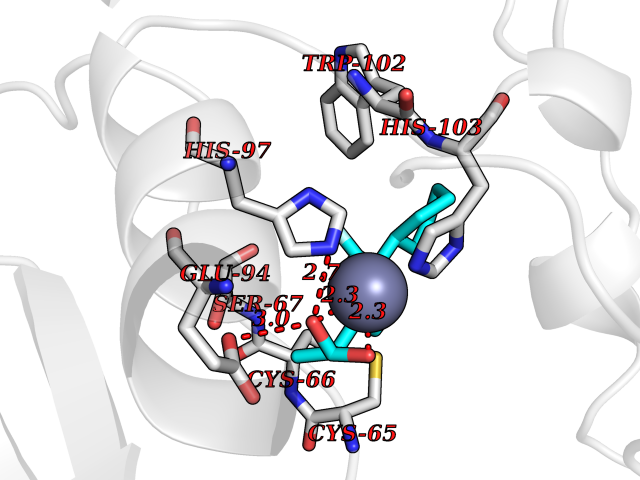 Pymol Map 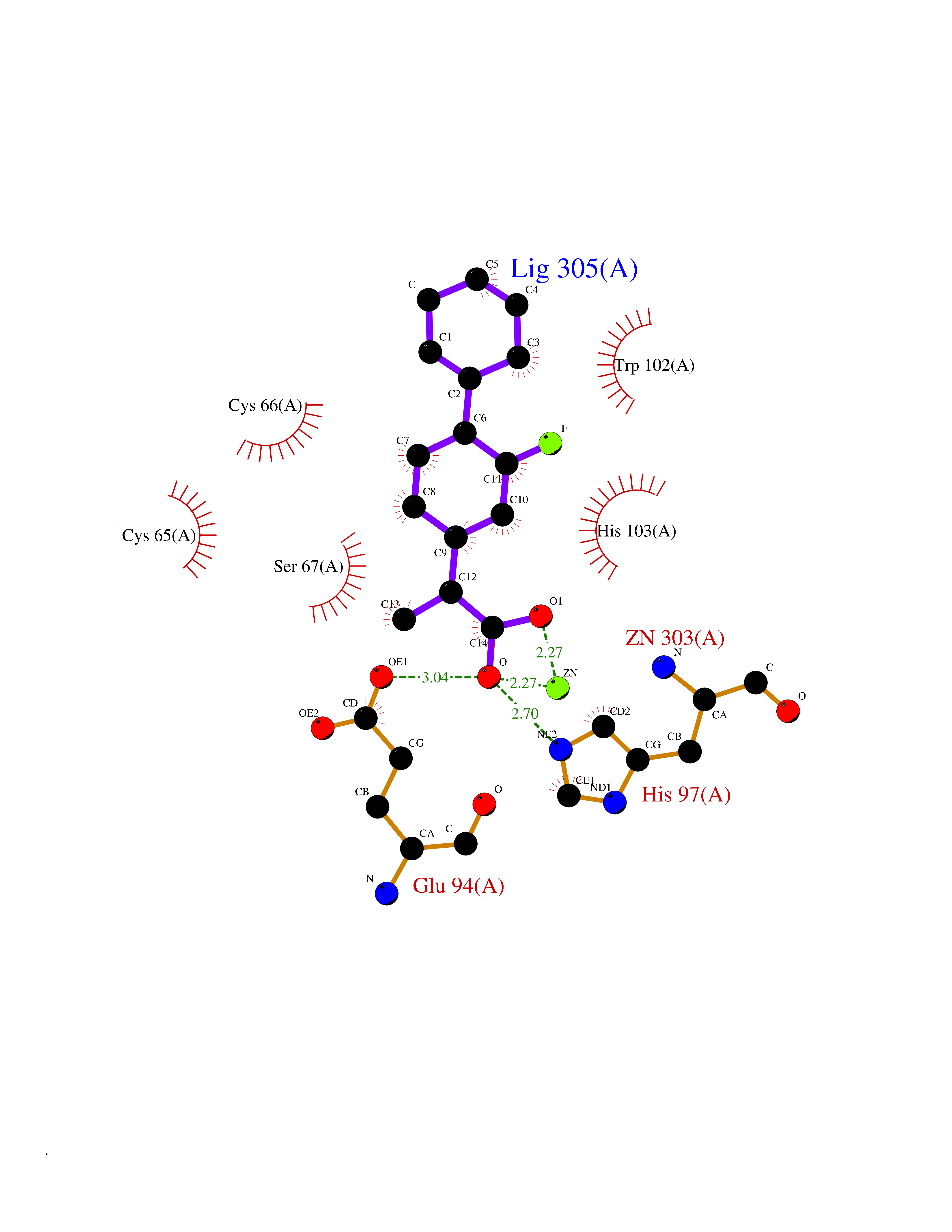 Ligplot Map 3D Binding mode Sequence XAATSRPERVWPDGVIPFVIGGNFTGSQRAVFRQAMRHWEKHTCVTFLERTDEDSYIVFTYRPCGCCSYVGRRGGGPQAISIGKNCDKFGIVVHELGHVVGFWHEHTRPDRDRHVSIVRENIQPGQEYNFLKMEPQEVESLGETYDFDSIMHYARNTFSRGIFLDTIVPKYEVNGVKPPIGQRTRLSKGDIAQARKLYKCPA Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | T-cell surface glycoprotein CD1a (CD1A) | 6NUX | 7.18 | |
Target general information Gen name CD1A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hTa1 thymocyteantigen; hTa1 thymocyte antigen; T-cell surfaceantigen T6/Leu-6; T-cell surface antigen T6/Leu-6; CD1a Protein family NA Biochemical class Immunoglobulin Function Antigen-presenting protein that binds self and non-self lipid and glycolipid antigens and presents them to T-cell receptors on natural killer T-cells. Related diseases Pulmonary hypertension, primary, 1 (PPH1) [MIM:178600]: A rare disorder characterized by plexiform lesions of proliferating endothelial cells in pulmonary arterioles. The lesions lead to elevated pulmonary arterial pression, right ventricular failure, and death. The disease can occur from infancy throughout life and it has a mean age at onset of 36 years. Penetrance is reduced. Although familial pulmonary hypertension is rare, cases secondary to known etiologies are more common and include those associated with the appetite-suppressant drugs. {ECO:0000269|PubMed:10903931, ECO:0000269|PubMed:10973254, ECO:0000269|PubMed:11015450, ECO:0000269|PubMed:11115378, ECO:0000269|PubMed:12045205, ECO:0000269|PubMed:12358323, ECO:0000269|PubMed:15965979, ECO:0000269|PubMed:24936649, ECO:0000269|PubMed:25187962, ECO:0000269|PubMed:28507310}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pulmonary venoocclusive disease 1, autosomal dominant (PVOD1) [MIM:265450]: A disease characterized by widespread fibrous obstruction and intimal thickening of septal veins and preseptal venules, a low diffusing capacity for carbon monoxide, occult alveolar hemorrhage, and nodular ground-glass opacities, septal lines and lymph node enlargement showed by high-resolution computed tomography of the chest. It is frequently associated with pulmonary capillary dilatation and proliferation, and is a rare and devastating cause of pulmonary hypertension. {ECO:0000269|PubMed:12446270, ECO:0000269|PubMed:16429395}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00098 Interacts with P61769 EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Cell membrane; Disulfide bond; Endosome; Glycoprotein; Immunity; Immunoglobulin domain; Membrane; Proteomics identification; Reference proteome; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 30867.3 Length 270 Aromaticity 0.13 Instability index 37.45 Isoelectric point 6.16 Charge (pH=7) -4.5 2D Binding mode Binding energy (Kcal/mol) -8.69 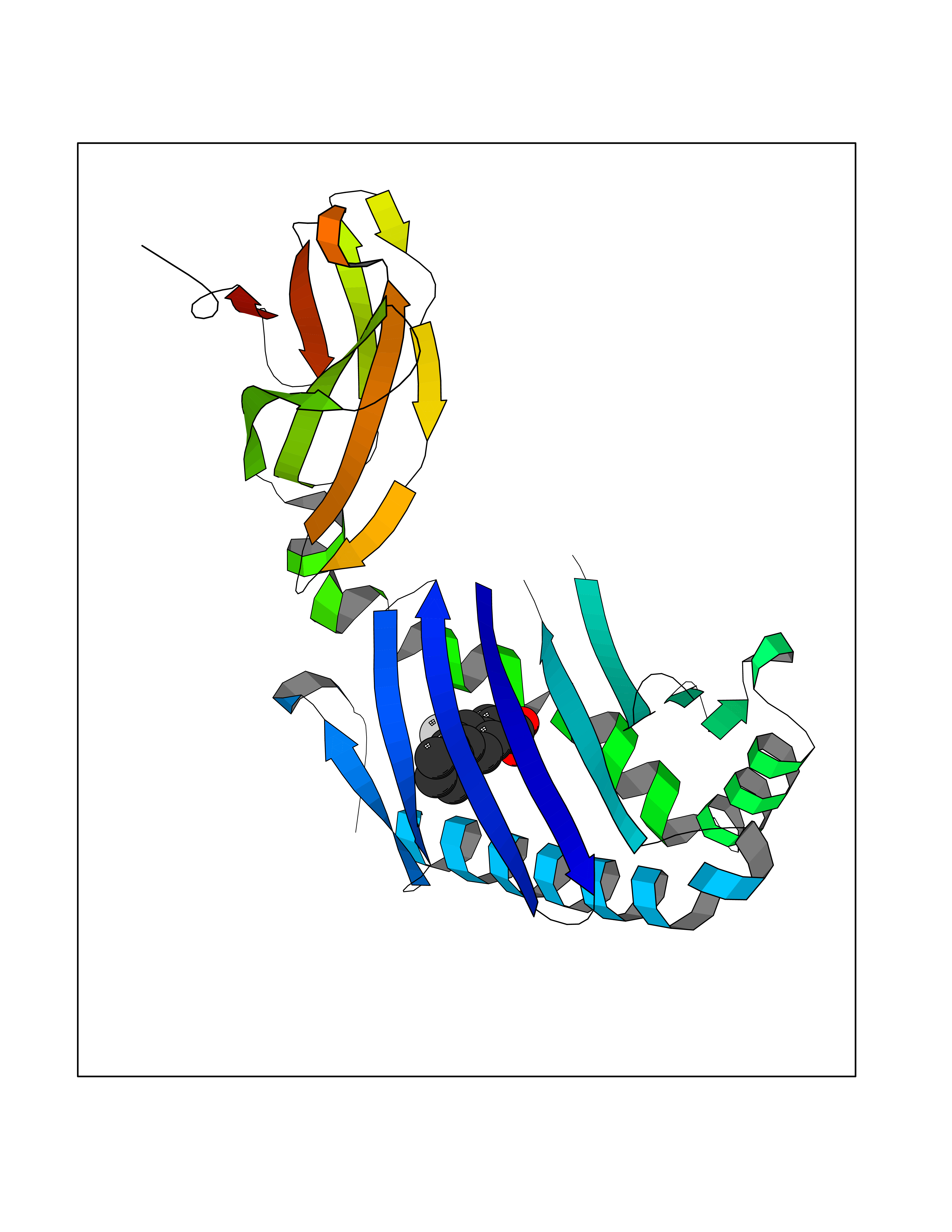 Molscript Map 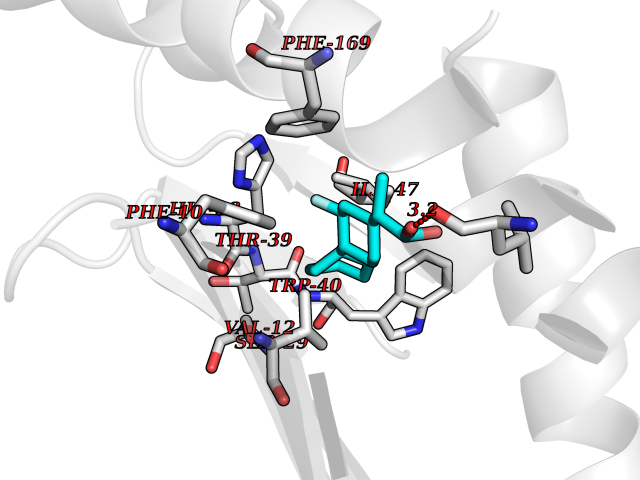 Pymol Map 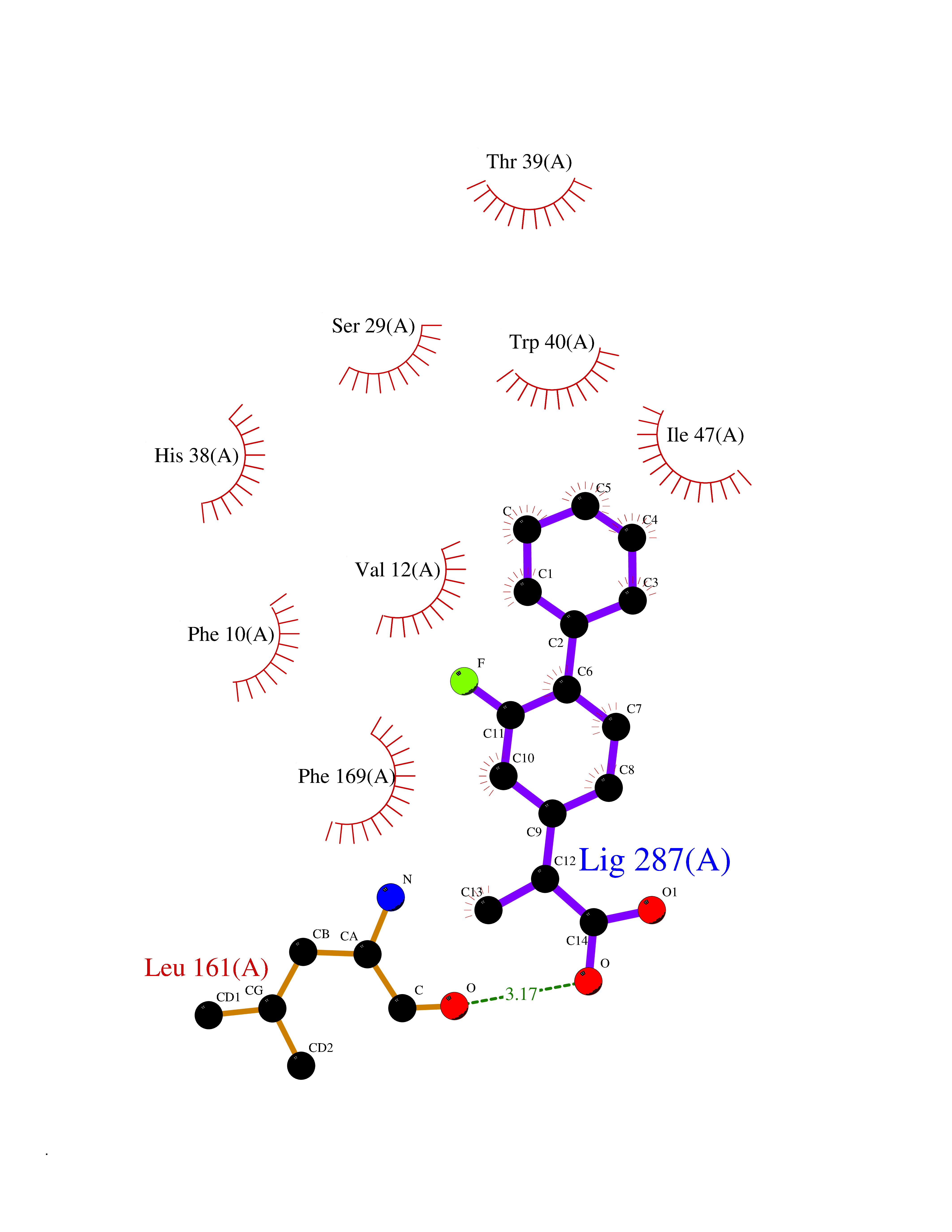 Ligplot Map 3D Binding mode Sequence SFHVIWIASFYNHSWKQNLVSGWLSDLQTHTWDSNSSTIVFLWPWSRGNFSNEWKELETLFRIRTIRSFEGIRRYAHELQFEYPFEIQVTGGCESGSFLQLAYQGSDFVSFQNNSWLPYPVAGNMAKHFCKVLNQNQHENDITHNLLSDTCPRFILGLLDAGKAHLQRQVKPEAWLSHGPSPGPGHLQLVCHVSGFYPKPVWVMWMRGEQEQQGTQRGDILPSADGTWYLRATLEVAAGEAADLSCRVKHSSLEGQDIVLYWEGSLVPRG Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Histidine decarboxylase (HDC) | 4E1O | 7.18 | |
Target general information Gen name HDC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Human histidine decarboxylase Protein family Group II decarboxylase family Biochemical class Carbon-carbon lyase Function Catalyzes the biosynthesis of histamine from histidine. Related diseases Corticosterone methyloxidase 1 deficiency (CMO-1 deficiency) [MIM:203400]: Autosomal recessive disorder of aldosterone biosynthesis. There are two biochemically different forms of selective aldosterone deficiency be termed corticosterone methyloxidase (CMO) deficiency type 1 and type 2. In CMO-1 deficiency, aldosterone is undetectable in plasma, while its immediate precursor, 18-hydroxycorticosterone, is low or normal. {ECO:0000269|PubMed:11238478, ECO:0000269|PubMed:8439335, ECO:0000269|PubMed:9177280}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Corticosterone methyloxidase 2 deficiency (CMO-2 deficiency) [MIM:610600]: Autosomal recessive disorder of aldosterone biosynthesis. In CMO-2 deficiency, aldosterone can be low or normal, but at the expense of increased secretion of 18-hydroxycorticosterone. Consequently, patients have a greatly increased ratio of 18-hydroxycorticosterone to aldosterone and a low ratio of corticosterone to 18-hydroxycorticosterone in serum. {ECO:0000269|PubMed:12788848, ECO:0000269|PubMed:1346492, ECO:0000269|PubMed:1594605, ECO:0000269|PubMed:9625333, ECO:0000269|PubMed:9814506}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00117; DB00114 Interacts with Q86UW9 EC number EC 4.1.1.22 Uniprot keywords 3D-structure; Alternative splicing; Catecholamine biosynthesis; Decarboxylase; Lyase; Proteomics identification; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A,B,C,D,E,F Molecular weight (Da) 107706 Length 956 Aromaticity 0.1 Instability index 55.17 Isoelectric point 6.23 Charge (pH=7) -9.63 2D Binding mode Binding energy (Kcal/mol) -8.8  Molscript Map 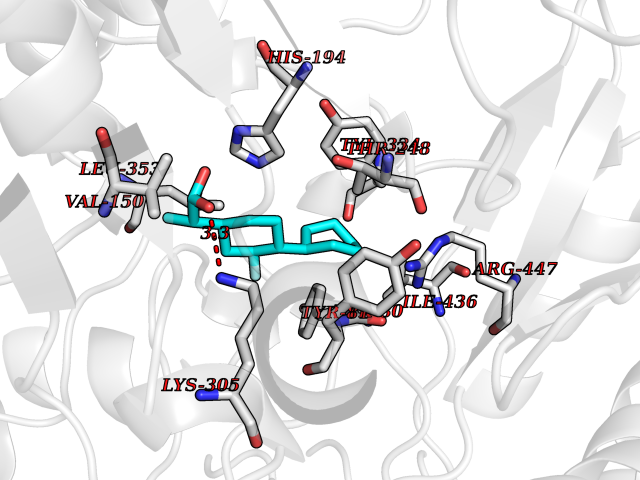 Pymol Map 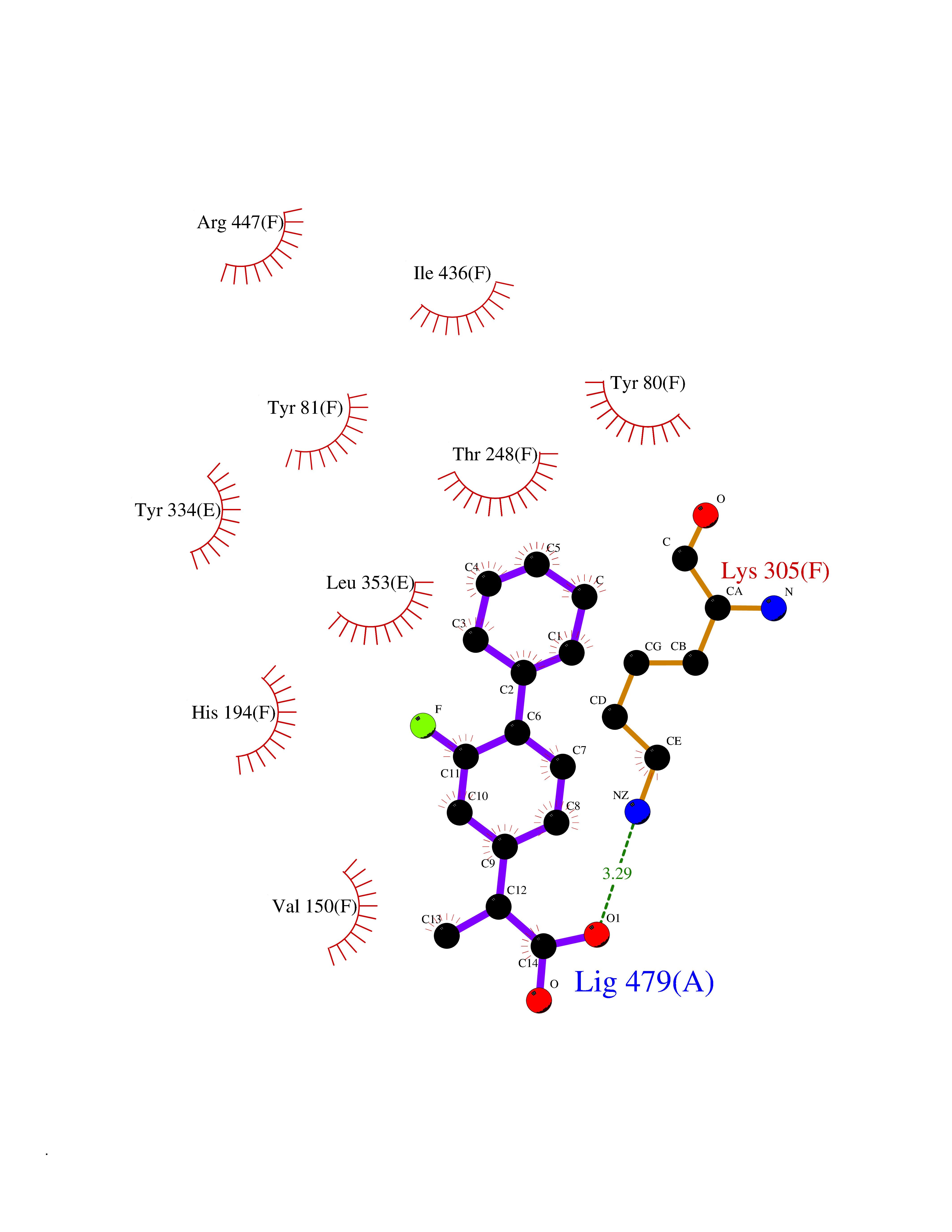 Ligplot Map 3D Binding mode Sequence GSMEPEEYRERGREMVDYICQYLSTVRERRVTPDVQPGYLRAQLPESAPEDPDSWDSIFGDIERIIMPGVVHWQSPHMHAYYPALTSWPSLLGDMLADAINCLGFTWASSPACTELEMNVMDWLAKMLGLPEHFLHHHPSSQGGGVLQSTVSESTLIALLAARKNKILEMKTSEPDADESSLNARLVAYASDQAHSSVEKAGLISLVKMKFLPVDDNFSLRGEALQKAIEEDKQRGLVPVFVCATLGTTGVCAFDXLSELGPICAREGLWLHIDAAYAGTAFLCPEFRGFLKGIEYADSFTFNPSKWMMVHFDCTGFWVKDKYKLQQTFSVNPIYLRHANSGVATDFMHWQIPLSRRFRSVKLWFVIRSFGVKNLQAHVRHGTEMAKYFESLVRNDPSFEIPAKRHLGLVVFRLKGPNSLTENVLKEIAKAGRLFLIPATIQDKLIIRFTVTSQFTTRDDILRDWNLIRDAATLILSQGSMEPEEYRERGREMVDYICQYLSTVRERRVTPDVQPGYLRAQLPESAPEDPDSWDSIFGDIERIIMPGVVHWQSPHMHAYYPALTSWPSLLGDMLADAINCLGFTWASSPACTELEMNVMDWLAKMLGLPEHFLHHHPSSQGGGVLQSTVSESTLIALLAARKNKILEMKTSEPDADESSLNARLVAYASDQAHSSVEKAGLISLVKMKFLPVDDNFSLRGEALQKAIEEDKQRGLVPVFVCATLGTTGVCAFDXLSELGPICAREGLWLHIDAAYAGTAFLCPEFRGFLKGIEYADSFTFNPSKWMMVHFDCTGFWVKDKYKLQQTFSVNPIYLRHANSGVATDFMHWQIPLSRRFRSVKLWFVIRSFGVKNLQAHVRHGTEMAKYFESLVRNDPSFEIPAKRHLGLVVFRLKGPNSLTENVLKEIAKAGRLFLIPATIQDKLIIRFTVTSQFTTRDDILRDWNLIRDAATLILSQ Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Janus kinase 1 (JAK-1) | 4E5W | 7.17 | |
Target general information Gen name JAK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine-protein kinase JAK1; JAK1B; JAK1A Protein family Protein kinase superfamily, Tyr protein kinase family, JAK subfamily Biochemical class Kinase Function Kinase partner for the interleukin (IL)-2 receptor as well as interleukin (IL)-10 receptor. Tyrosine kinase of the non-receptor type, involved in the IFN-alpha/beta/gamma signal pathway. Related diseases Autoinflammation, immune dysregulation, and eosinophilia (AIIDE) [MIM:618999]: An autosomal dominant disorder characterized by immune dysregulation, severe atopic dermatitis, and chronic gastrointestinal inflammation. Additional features include asthma, food or environmental allergies, as well as poor overall growth with short stature. {ECO:0000269|PubMed:28111307, ECO:0000269|PubMed:32750333}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04716; DB14973; DB11817; DB12500; DB14845; DB12010; DB16756; DB11763; DB02375; DB15822; DB08877; DB08895; DB15091 Interacts with P04626; P48551; P40189-1; O60674; Q99650; P42224; P40763 EC number EC 2.7.10.2 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Disease variant; Kinase; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; SH2 domain; Transferase; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 32050.7 Length 280 Aromaticity 0.09 Instability index 44.51 Isoelectric point 8.07 Charge (pH=7) 2.39 2D Binding mode Binding energy (Kcal/mol) -8.42 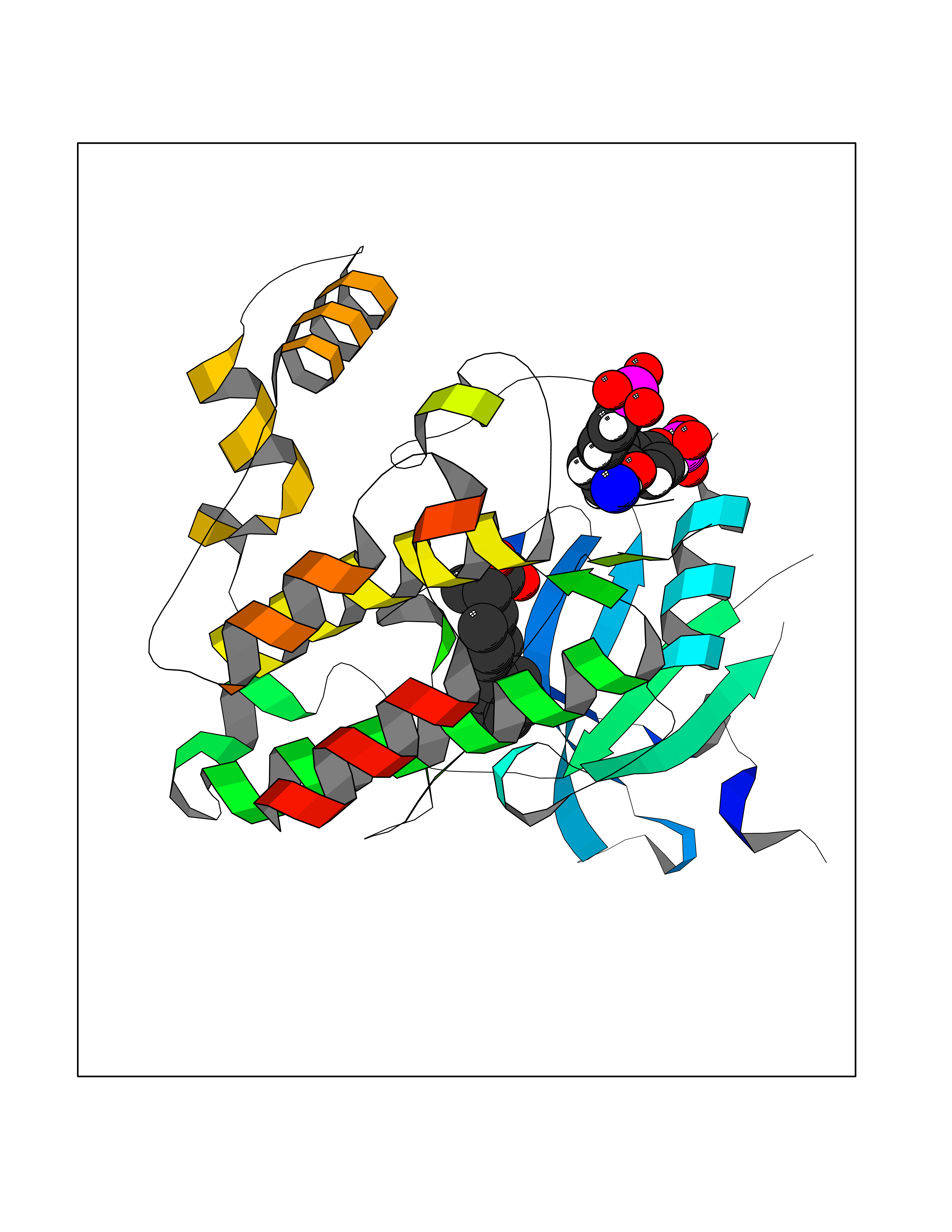 Molscript Map 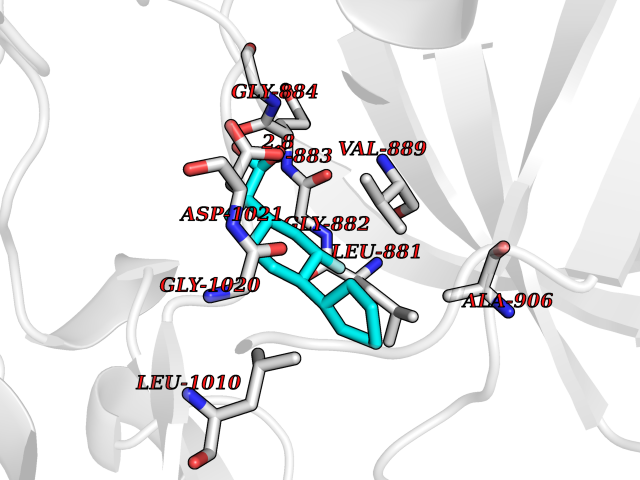 Pymol Map 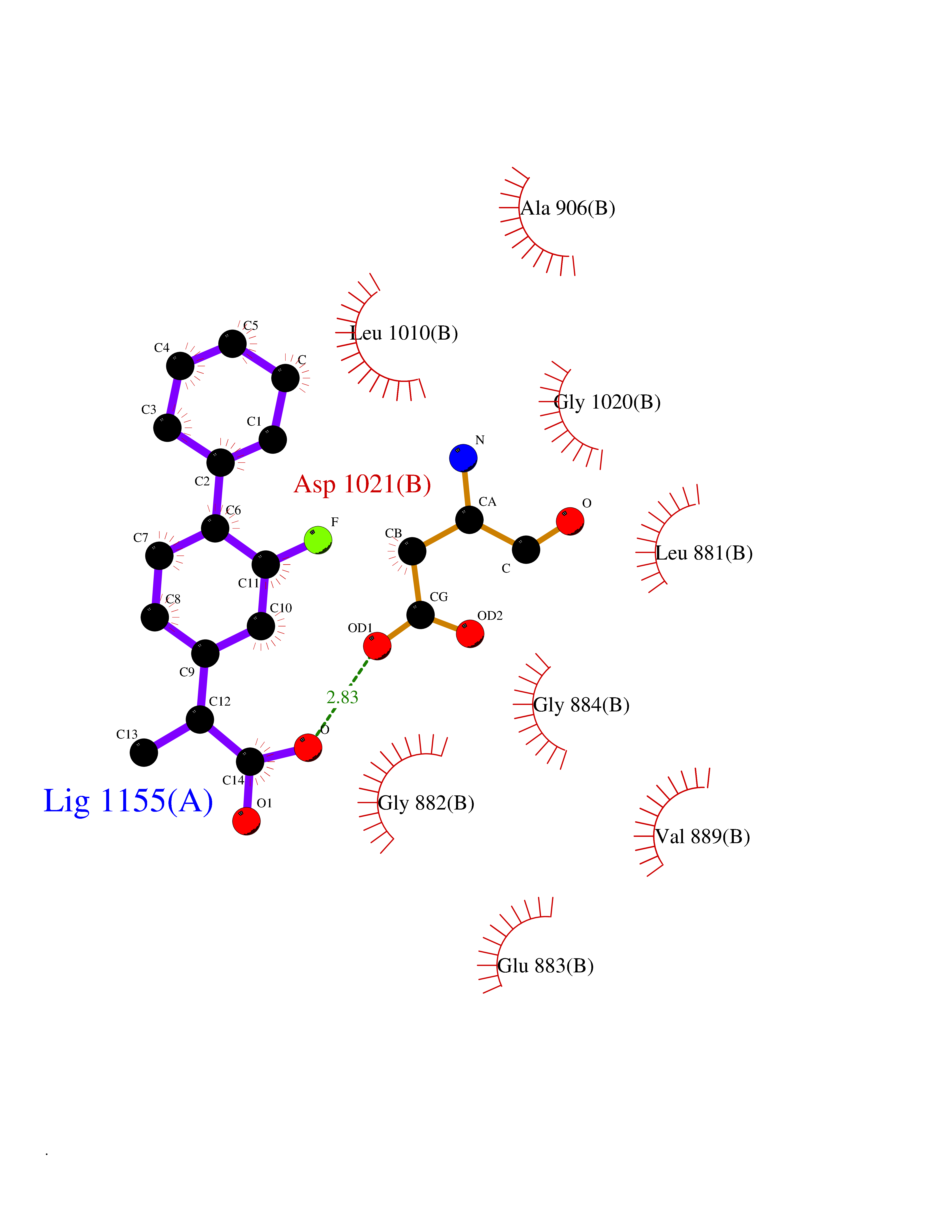 Ligplot Map 3D Binding mode Sequence VDPTHFEKRFLKRIRDLGEGHFGKVELCRYDPEGDNTGEQVAVKSLKPNHIADLKKEIEILRNLYHENIVKYKGICTENGIKLIMEFLPSGSLKEYLPKNKNKINLKQQLKYAVQICKGMDYLGSRQYVHRDLAARNVLVESEHQVKIGDFGLTKAIEKEXXTVKDDRDSPVFWYAPECLMQSKFYIASDVWSFGVTLHELLTYCDSDSSPMALFLKMIGPTHGQMTVTRLVNTLKEGKRLPCPPNCPDEVYQLMRKCWEFQPSNRTSFQNLIEGFEALL Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | MEK kinase kinase 3 (MAP4K3) | 5J5T | 7.17 | |
Target general information Gen name MAP4K3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAB8IPL1; Mitogen-activated protein kinase kinase kinase kinase 3; MEKKK 3; MAPK/ERK kinase kinase kinase 3; Germinal center kinase-related protein kinase; GLK Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, STE20 subfamily Biochemical class Kinase Function Appears to act upstream of the JUN N-terminal pathway. May play a role in the response to environmental stress. Related diseases A chromosomal aberration involving NPM1 is found in a form of non-Hodgkin lymphoma. Translocation t(2;5)(p23;q35) with ALK. The resulting chimeric NPM1-ALK protein homodimerize and the kinase becomes constitutively activated. {ECO:0000269|PubMed:8122112, ECO:0000269|PubMed:8633037}.; DISEASE: A chromosomal aberration involving NPM1 is found in a form of acute promyelocytic leukemia. Translocation t(5;17)(q32;q11) with RARA. {ECO:0000269|PubMed:8562957}.; DISEASE: A chromosomal aberration involving NPM1 is a cause of myelodysplastic syndrome (MDS). Translocation t(3;5)(q25.1;q34) with MLF1. {ECO:0000269|PubMed:8570204}.; DISEASE: Defects in NPM1 are associated with acute myelogenous leukemia (AML). Mutations in exon 12 affecting the C-terminus of the protein are associated with an aberrant cytoplasmic location. {ECO:0000269|PubMed:15659725}. Drugs (DrugBank ID) DB12010 Interacts with P62993; Q13094; Q9Y4K4; Q04759; Q02111 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Kinase; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 33570.4 Length 298 Aromaticity 0.09 Instability index 40.61 Isoelectric point 6.85 Charge (pH=7) -0.5 2D Binding mode Binding energy (Kcal/mol) -8.67  Molscript Map 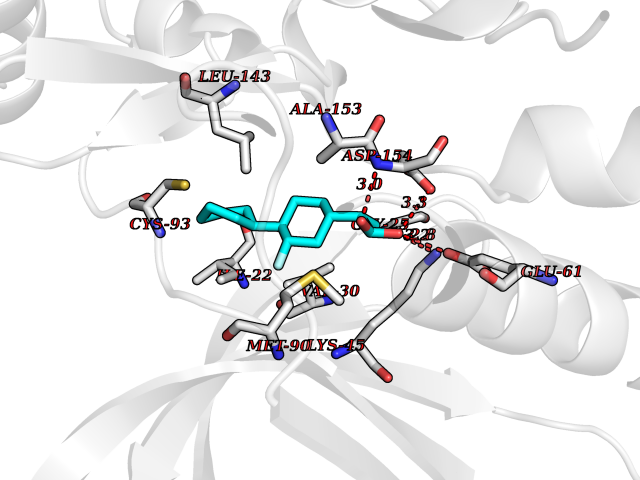 Pymol Map 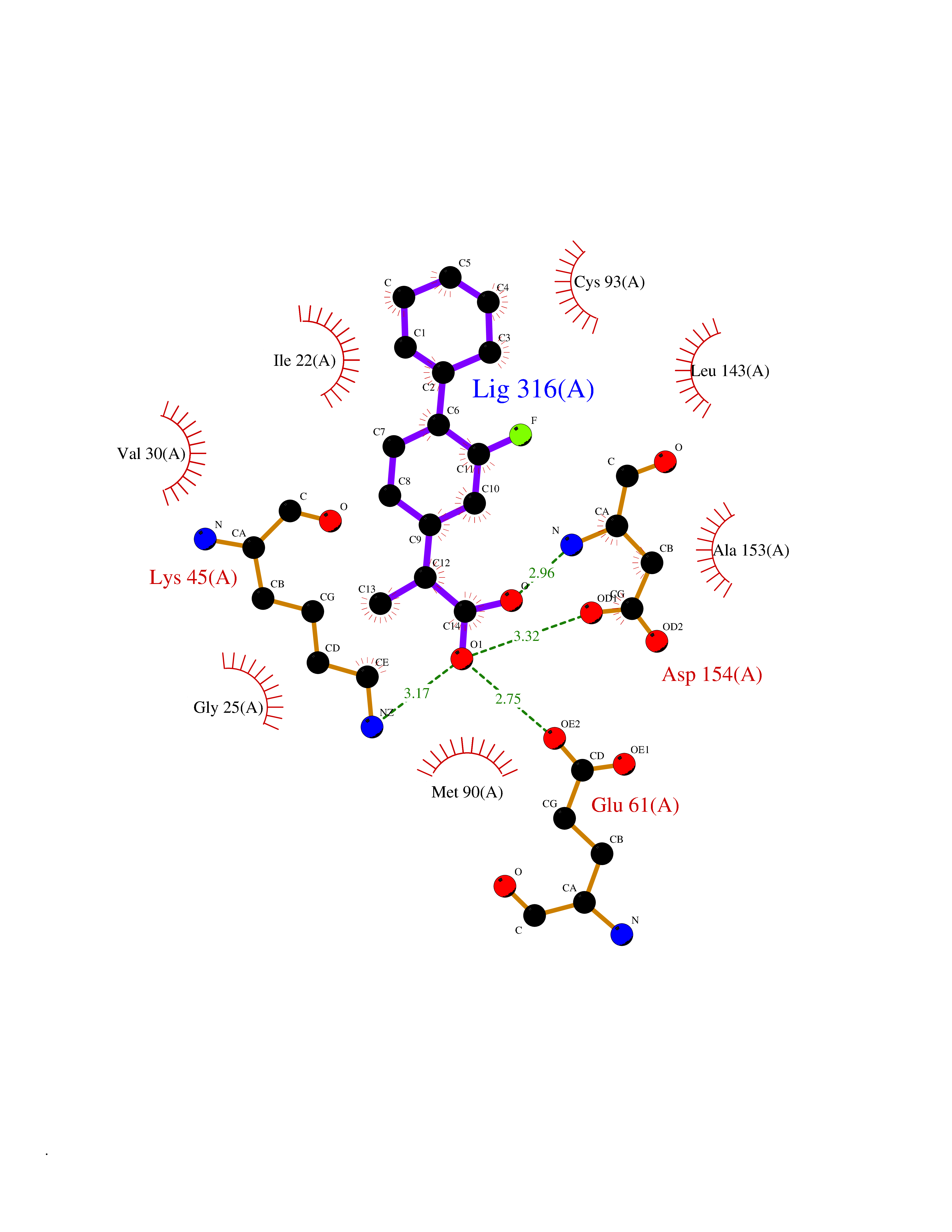 Ligplot Map 3D Binding mode Sequence SQEDFELIQRIGSGTYGDVYKARNVNTGELAAIKVIKLEPGEDFAVVQQEIIMMKDCKHPNIVAYFGSYLRRDKLWICMEFCGGGSLQDIYHVTGPLSELQIAYVSRETLQGLYYLHSKGKMHRDIKGANILLXDNGHVKLADFGVSAQITATIAAFIGTPYWMAPEVAAVERKGGYNQLCDLWAVGITAIELAELQPPMFDLHPMRALFLMTKSNFQPPKLKDKMKWSNSFHHFVKMALTKNPKKRPTAEKLLQHPFVTQHLTRSLAIELLDKVNNPSTYHDFDDDDPEPLVAVPHR Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Oxalosuccinate decarboxylase (IDH1) | 6ADG | 7.16 | |
Target general information Gen name IDH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PICD; NADP(+)-specific ICDH; Isocitrate dehydrogenase [NADP] cytoplasmic; IDP; IDH; Cytosolic NADP-isocitrate dehydrogenase Protein family Isocitrate and isopropylmalate dehydrogenases family Biochemical class Short-chain dehydrogenases reductase Function Catalyses the NADPH-dependent reduction of alpha-ketoglutarate to R(-)-2-hydroxyglutarate (2HG). Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:19117336, ECO:0000269|PubMed:19935646}. The gene represented in this entry is involved in disease pathogenesis. Mutations affecting Arg-132 are tissue-specific, and suggest that this residue plays a unique role in the development of high-grade gliomas. Mutations of Arg-132 to Cys, His, Leu or Ser abolish magnesium binding and abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. Elevated levels of R(-)-2-hydroxyglutarate are correlated with an elevated risk of malignant brain tumors. {ECO:0000269|PubMed:19935646}.; DISEASE: Genetic variations are associated with cartilaginous tumors such as enchondroma or chondrosarcoma. Mutations of Arg-132 to Cys, Gly or His abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. {ECO:0000269|PubMed:26161668}. Drugs (DrugBank ID) DB09374; DB01727; DB14568; DB03461; DB16267 Interacts with P0DP23; P27797; P36957; O75874; Q8TDX7; P16284; P17612; P50454; P37173; Q05086-3 EC number EC 1.1.1.42 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Glyoxylate bypass; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; Tricarboxylic acid cycle Protein physicochemical properties Chain ID A,B Molecular weight (Da) 92711.7 Length 823 Aromaticity 0.1 Instability index 26.74 Isoelectric point 6.42 Charge (pH=7) -4.48 2D Binding mode Binding energy (Kcal/mol) -8.86  Molscript Map 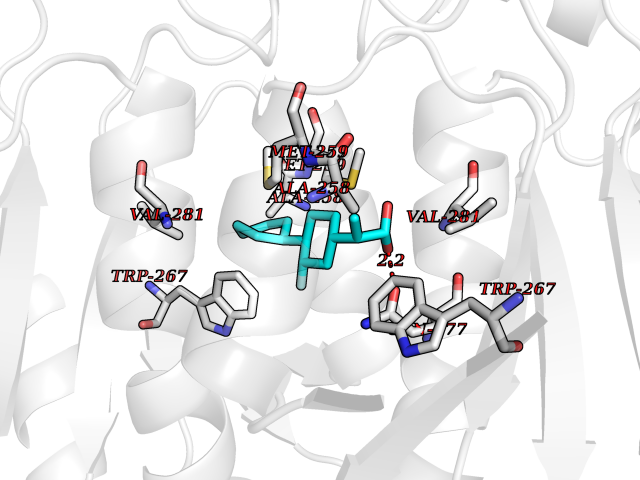 Pymol Map 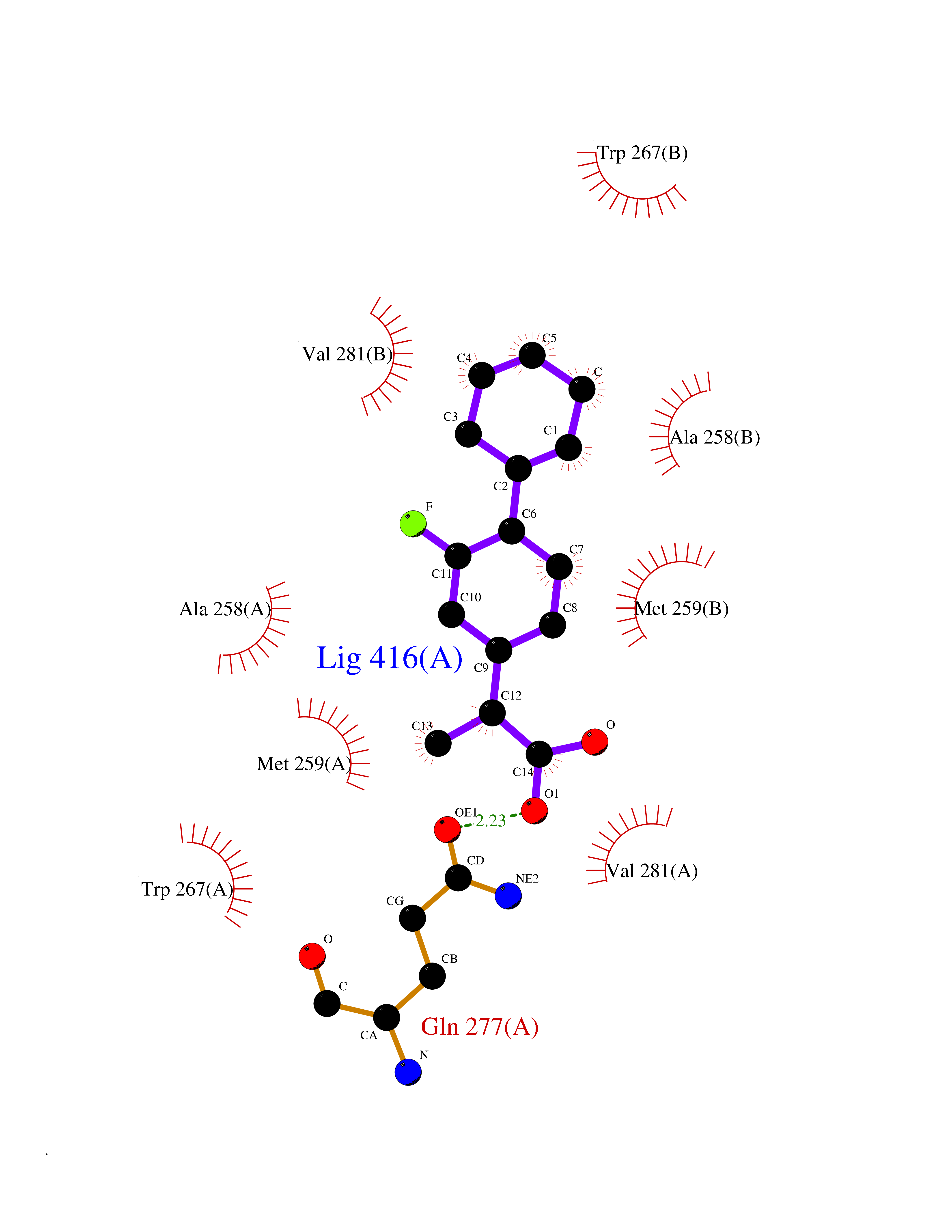 Ligplot Map 3D Binding mode Sequence KKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAKLKKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAK Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Mutated oxalosuccinate decarboxylase (mIDH1) | 6ADG | 7.16 | |
Target general information Gen name IDH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PICD (mutated); Oxalosuccinate decarboxylase (mutated); NADP(+)-specific ICDH (mutated); Isocitrate dehydrogenase [NADP] cytoplasmic (mutated); IDP (mutated); IDH (mutated); Cytosolic NADP-isocitrate Protein family Isocitrate and isopropylmalate dehydrogenases family Biochemical class Short-chain dehydrogenases reductase Function Catalyses the NADPH-dependent reduction of alpha-ketoglutarate to R(-)-2-hydroxyglutarate (2HG). Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:19117336, ECO:0000269|PubMed:19935646}. The gene represented in this entry is involved in disease pathogenesis. Mutations affecting Arg-132 are tissue-specific, and suggest that this residue plays a unique role in the development of high-grade gliomas. Mutations of Arg-132 to Cys, His, Leu or Ser abolish magnesium binding and abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. Elevated levels of R(-)-2-hydroxyglutarate are correlated with an elevated risk of malignant brain tumors. {ECO:0000269|PubMed:19935646}.; DISEASE: Genetic variations are associated with cartilaginous tumors such as enchondroma or chondrosarcoma. Mutations of Arg-132 to Cys, Gly or His abolish the conversion of isocitrate to alpha-ketoglutarate. Instead, alpha-ketoglutarate is converted to R(-)-2-hydroxyglutarate. {ECO:0000269|PubMed:26161668}. Drugs (DrugBank ID) DB09374; DB01727; DB14568; DB03461; DB16267 Interacts with P0DP23; P27797; P36957; O75874; Q8TDX7; P16284; P17612; P50454; P37173; Q05086-3 EC number EC 1.1.1.42 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Glyoxylate bypass; Magnesium; Manganese; Metal-binding; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; Tricarboxylic acid cycle Protein physicochemical properties Chain ID A,B Molecular weight (Da) 92711.7 Length 823 Aromaticity 0.1 Instability index 26.74 Isoelectric point 6.42 Charge (pH=7) -4.48 2D Binding mode Binding energy (Kcal/mol) -8.89 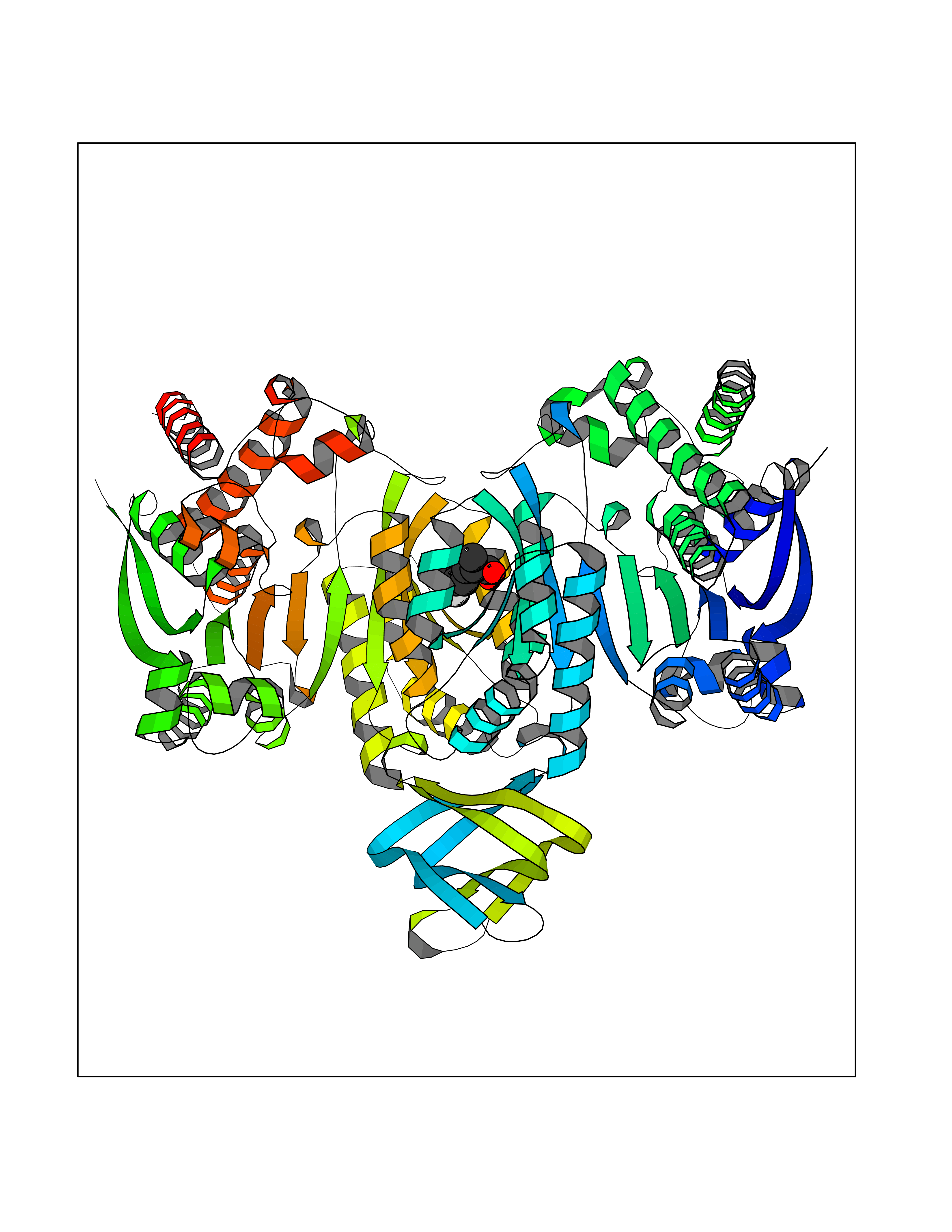 Molscript Map 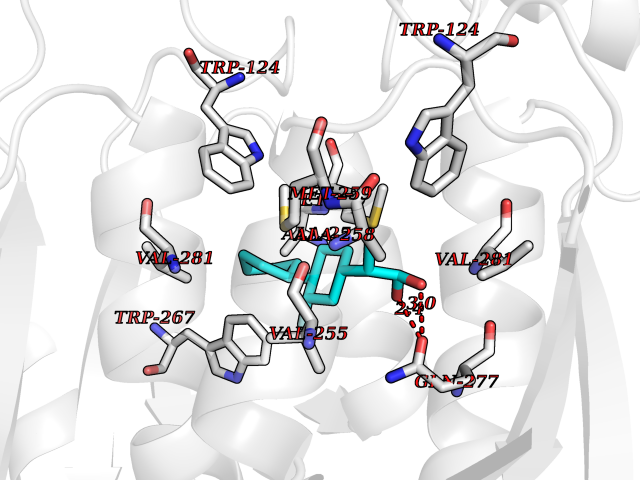 Pymol Map 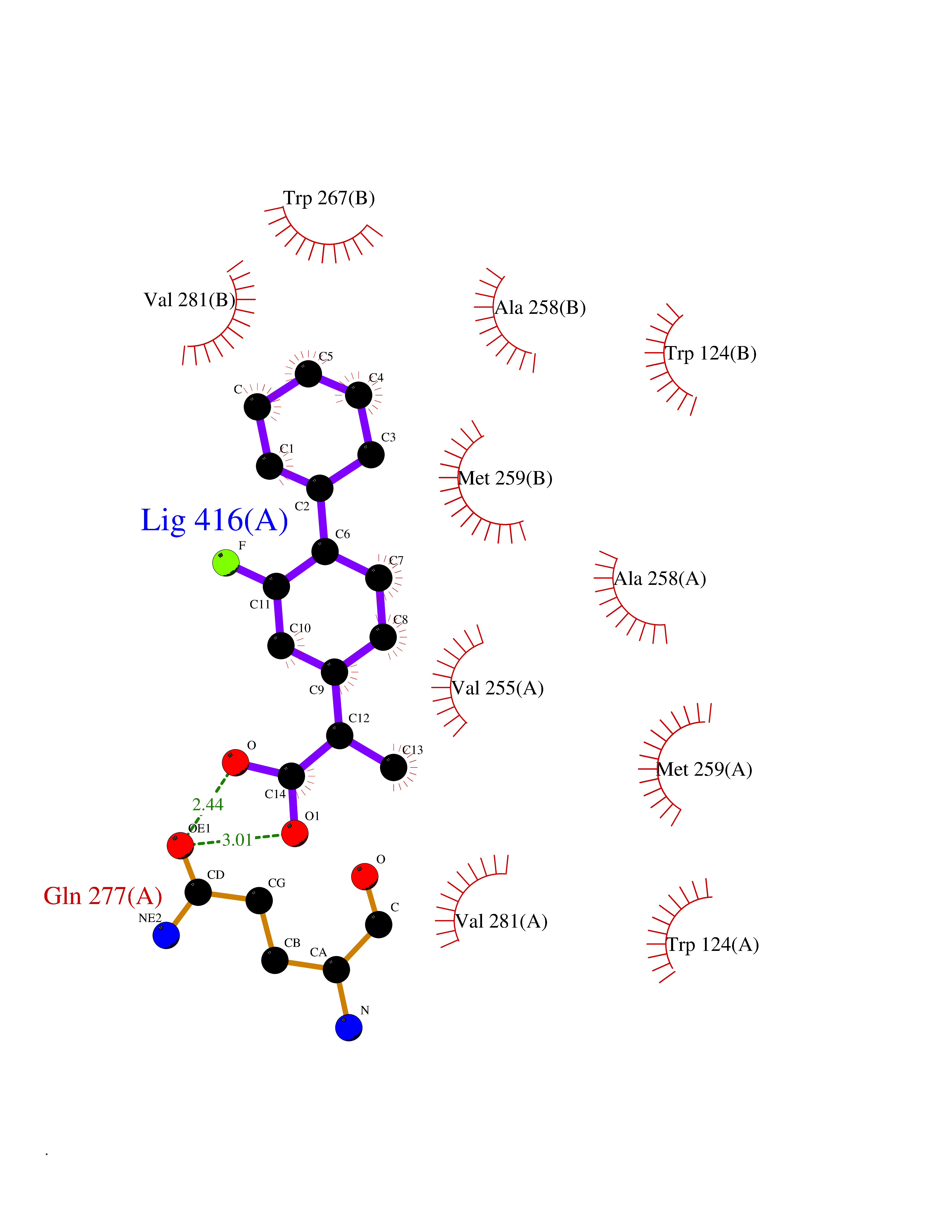 Ligplot Map 3D Binding mode Sequence KKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAKLKKISGGSVVEMQGDEMTRIIWELIKEKLIFPYVELDLHSYDLGIENRDATNDQVTKDAAEAIKKHNVGVKCATITPDEKRVEEFKLKQMWKSPNGTIRNILGGTVFREAIICKNIPRLVSGWVKPIIIGHHAYGDQYRATDFVVPGPGKVEITYTPSDGTQKVTYLVHNFEEGGGVAMGMYNQDKSIEDFAHSSFQMALSKGWPLYLSTKNTILKKYDGRFKDIFQEIYDKQYKSQFEAQKIWYEHRLIDDMVAQAMKSEGGFIWACKNYDGDVQSDSVAQGYGSLGMMTSVLVCPDGKTVEAEAAHGTVTRHYRMYQKGQETSTNPIASIFAWTRGLAHRAKLDNNKELAFFANALEEVSIETIEAGFMTKDLAACIKGLPNVQRSDYLNTFEFMDKLGENLKIKLAQAK Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | HMG-CoA reductase (HMGCR) | 2R4F | 7.16 | |
Target general information Gen name HMGCR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms 3-hydroxy-3-methylglutaryl-coenzyme A reductase Protein family HMG-CoA reductase family Biochemical class CH-OH donor oxidoreductase Function Transmembrane glycoprotein that is the rate-limiting enzyme in cholesterol biosynthesis as well as in the biosynthesis of nonsterol isoprenoids that are essential for normal cell function including ubiquinone and geranylgeranyl proteins. Related diseases Muscular dystrophy, limb-girdle, autosomal recessive 28 (LGMDR28) [MIM:620375]: An autosomal recessive form of limb girdle muscular dystrophy, a group of genetically heterogeneous muscular disorders that share proximal muscle weakness as the major attribute. Most limb girdle muscular dystrophies present with elevated creatinine kinase and myopathic electromyographic features. Disease is usually progressive to a variable degree, ranging from minor disability to complete inability to ambulate, and can involve the large proximal muscles, as well as axial and facial muscles. Different disease forms may exhibit skeletal muscle hypertrophy, kyphoscoliosis, and contractures or involve other muscle groups and manifest with distal weakness, cardiomyopathy, dysphagia, and respiratory difficulties. LGMDR28 is characterized by progressive muscle weakness affecting the proximal and axial muscles of the upper and lower limbs, and highly variable age at onset. Most patients have limited ambulation or become wheelchair-bound within a few decades, and respiratory insufficiency commonly occurs. {ECO:0000269|PubMed:36745799, ECO:0000269|PubMed:37167966}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03169; DB04447; DB01076; DB09061; DB00439; DB01992; DB01095; DB00227; DB14009; DB04377; DB06693; DB14011; DB00157; DB03461; DB08860; DB00175; DB01098; DB00641; DB05317; DB09270 Interacts with Q9Y5Z9; Q9Y5Z9-1 EC number EC 1.1.1.34 Uniprot keywords 3D-structure; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Disease variant; Endoplasmic reticulum; Glycoprotein; Isopeptide bond; Limb-girdle muscular dystrophy; Lipid biosynthesis; Lipid metabolism; Membrane; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 84796.9 Length 798 Aromaticity 0.05 Instability index 47.61 Isoelectric point 6.2 Charge (pH=7) -3.71 2D Binding mode Binding energy (Kcal/mol) -7.33 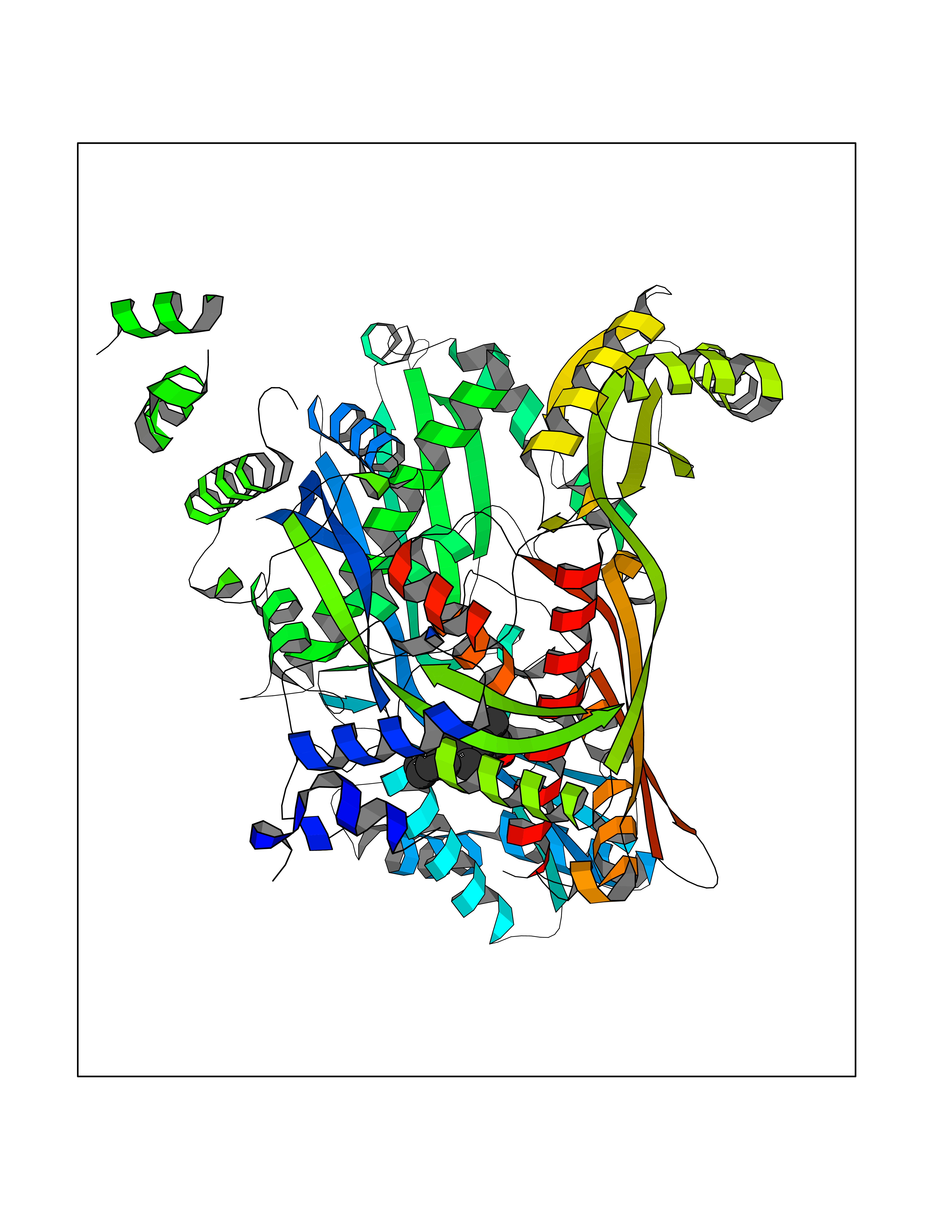 Molscript Map 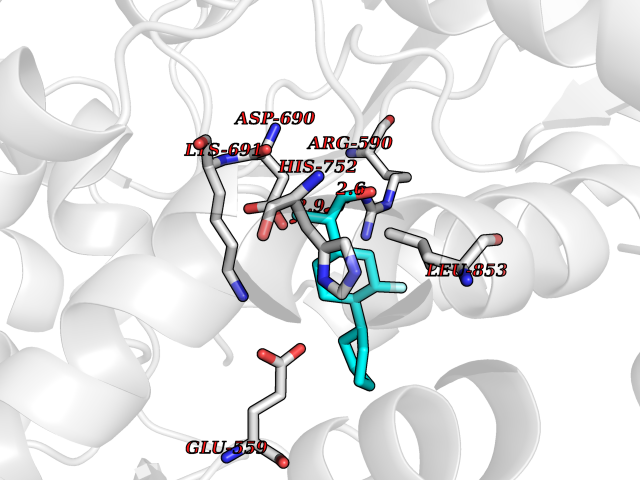 Pymol Map 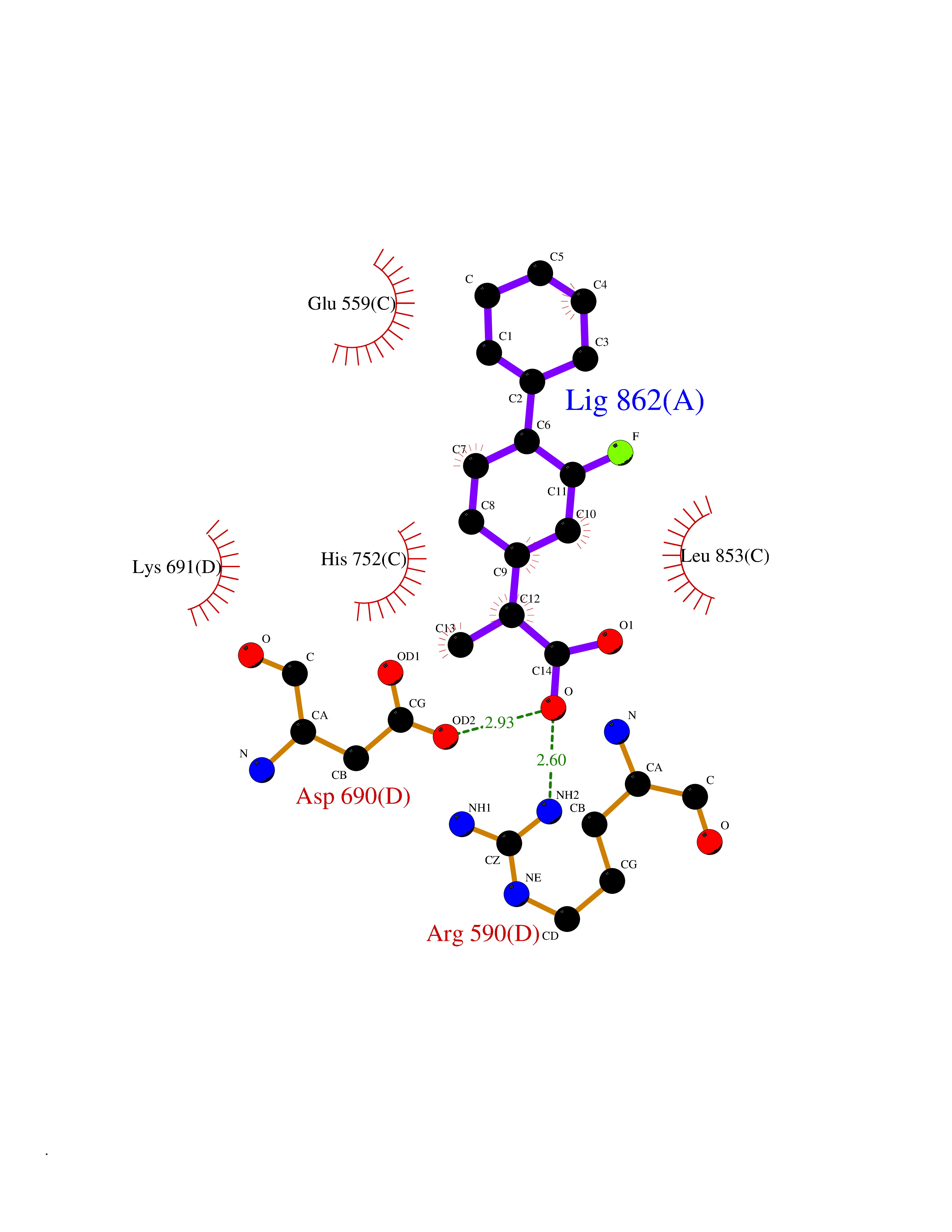 Ligplot Map 3D Binding mode Sequence GAKFLSDAEIIQLVNETLIETHERGVSIRRQLLSKKLSEPSSLQYLPYRDYNYSLVMGACCENVIGYMPIPVGVAGPLCLDEKEFQVPMATTEGCLVASTNRGCRAIGLGGGASSRVLADGMTRGPVVRLPRACDSAEVKAWLETSEGFAVIKEAFDSTSRFARLQKLHTSIAGRNLYIRFQSRSGDAMGMNMISKGTEKALSKLHEYFPEMQILAVSGNYCTDKKPAAINWIEGRGKSVVCEAVIPAKVVREVLKTTTEAMIEVNINKNLVGSAMAGSIGGYNAHAANIVTAIYIACGQDAAQNVGSSNCITLMEASGPTNEDLYISCTMPSIEIGTVGGGTNLLPQQACLQMLGVQGACKDNPGENARQLARIVCGTVMAGELSLMAALAAGPNEECLQILGNGAKFLSDAEIIQLVETLIETHERGVSIRRQLLSKKLSEPSSLQYLPYRDYNYSLVMGACCENVIGYMPIPVGVAGPLCLDEKEFQVPMATTEGCLVASTNRGCRAIGLGGGASSRVLADGMTRGPVVRLPRACDSAEVKAWLETSEGFAVIKEAFDSTSRFARLQKLHTSIAGRNLYIRFQSRSGDAMGMNMISKGTEKALSKLHEYFPEMQILAVSGNYCTDKKPAAINWIEGRGKSVVCEAVIPAKVVREVLKTTTEAMIEVNINKNLVGSAMAGSIGGYNAHAANIVTAIYIACGQDAAQNVGSSNCITLMEASGPTNEDLYISCTMPSIEIGTVGGGTNLLPQQACLQMLGVQGACKDNPGENARQLARIVCGTVMAGELSLMAALAAG Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Pol polyprotein | 5KAO | 7.16 | |
Target general information Gen name pol Organism Human immunodeficiency virus type 1 (HIV-1) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase / hydrolase inhibitor Function Aspartic-type endopeptidase activity. Related diseases Oocyte/zygote/embryo maturation arrest 16 (OZEMA16) [MIM:617234]: A rare cause of female primary infertility. In affected women, ovulation and fertilization proceed normally but embryos are arrested at early stages of development. Inheritance is autosomal recessive. {ECO:0000269|PubMed:27545678}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00701; DB01072; DB04887; DB01264; DB01319; DB00224; DB01601; DB00503; DB01232; DB00932 Interacts with NA EC number NA Uniprot keywords 3D-structure; Aspartyl protease; Hydrolase; Protease Protein physicochemical properties Chain ID A,B Molecular weight (Da) 21411 Length 198 Aromaticity 0.05 Instability index 42.78 Isoelectric point 9.45 Charge (pH=7) 4.15 2D Binding mode Binding energy (Kcal/mol) -7.7  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence PQVTLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYDQILIEIAGHKAIGTVLVGPTPVNIIGRNLLTQIGATLNFPQVTLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYDQILIEIAGHKAIGTVLVGPTPVNIIGRNLLTQIGATLNF Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Bacterial DD-carboxypeptidase (Bact vanYB) | 5ZHW | 7.15 | |
Target general information Gen name Bact vanYB Organism Enterococcus faecalis (strain ATCC 700802 / V583) Uniprot ID TTD ID Synonyms vanYB; DD-peptidase; DD-carboxypeptidase; D-alanyl-D-alanine carboxypeptidase-transpeptidase Protein family Peptidase M15B family Biochemical class Peptidase Function Vancomycin-inducible, penicillin-resistant, DD- carboxypeptidase that hydrolyzes depsipeptide- and D-alanyl-D- alanine-containing peptidoglycan precursors. Insensitive to beta- lactams. Related diseases Brachyolmia 3 (BCYM3) [MIM:113500]: A form of brachyolmia, a clinically and genetically heterogeneous skeletal dysplasia primarily affecting the spine and characterized by a short trunk, short stature, and platyspondyly. BCYM3 is an autosomal dominant form with severe scoliosis with or without kyphosis, and flattened irregular cervical vertebrae. {ECO:0000269|PubMed:18587396}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondylometaphyseal dysplasia Kozlowski type (SMDK) [MIM:184252]: A form of spondylometaphyseal dysplasia, a group of short stature disorders distinguished by abnormalities in the vertebrae and the metaphyses of the tubular bones. It is characterized by postnatal dwarfism, significant scoliosis and mild metaphyseal abnormalities in the pelvis. The vertebrae exhibit platyspondyly and overfaced pedicles. {ECO:0000269|PubMed:19232556, ECO:0000269|PubMed:20577006, ECO:0000269|PubMed:22702953}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Metatropic dysplasia (MTD) [MIM:156530]: A severe spondyloepimetaphyseal dysplasia characterized by short limbs with limitation and enlargement of joints and usually severe kyphoscoliosis. Radiologic features include severe platyspondyly, severe metaphyseal enlargement and shortening of long bones. {ECO:0000269|PubMed:19232556, ECO:0000269|PubMed:20425821, ECO:0000269|PubMed:20577006, ECO:0000269|PubMed:22702953, ECO:0000269|PubMed:26249260, ECO:0000269|Ref.6}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal dominant 8 (HMND8) [MIM:600175]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. {ECO:0000269|PubMed:20037588, ECO:0000269|PubMed:22526352, ECO:0000269|PubMed:22702953}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, axonal, 2C (CMT2C) [MIM:606071]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:20037586, ECO:0000269|PubMed:20037587, ECO:0000269|PubMed:20037588, ECO:0000269|PubMed:21115951, ECO:0000269|PubMed:21288981, ECO:0000269|PubMed:22702953, ECO:0000269|PubMed:25256292}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Scapuloperoneal spinal muscular atrophy (SPSMA) [MIM:181405]: A clinically variable neuromuscular disorder characterized by neurogenic scapuloperoneal amyotrophy, laryngeal palsy, congenital absence of muscles, progressive scapuloperoneal atrophy and progressive distal weakness and amyotrophy. {ECO:0000269|PubMed:20037587, ECO:0000269|PubMed:22702953}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepiphyseal dysplasia Maroteaux type (SEDM) [MIM:184095]: A clinically variable spondyloepiphyseal dysplasia with manifestations limited to the musculoskeletal system. Clinical features include short stature, brachydactyly, platyspondyly, short and stubby hands and feet, epiphyseal hypoplasia of the large joints, and iliac hypoplasia. Intelligence is normal. {ECO:0000269|PubMed:20503319, ECO:0000269|PubMed:22702953}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Parastremmatic dwarfism (PSTD) [MIM:168400]: A bone dysplasia characterized by severe dwarfism, kyphoscoliosis, distortion and bowing of the extremities, and contractures of the large joints. Radiographically, the disease is characterized by a combination of decreased bone density, bowing of the long bones, platyspondyly and striking irregularities of endochondral ossification with areas of calcific stippling and streaking in radiolucent epiphyses, metaphyses and apophyses. {ECO:0000269|PubMed:20503319}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Digital arthropathy-brachydactyly, familial (FDAB) [MIM:606835]: A disorder characterized by irregularities in the proximal articular surfaces of the distal interphalangeal joints of the hand. Individuals appear normal at birth, with no clinical or radiographic evidence of a developmental skeletal dysplasia. The earliest changes appear during the first decade of life. By adulthood, all interphalangeal, metacarpophalangeal, and metatarsophalangeal joints are affected by a deforming, painful osteoarthritis. The remainder of the skeleton is clinically and radiographically unaffected. {ECO:0000269|PubMed:21964574}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Avascular necrosis of the femoral head, primary 2 (ANFH2) [MIM:617383]: A disease characterized by mechanical failure of the subchondral bone, and degeneration of the hip joint. It usually leads to destruction of the hip joint in the third to fifth decade of life. The clinical manifestations, such as pain on exertion, a limping gait, and a discrepancy in leg length, cause considerable disability. {ECO:0000269|PubMed:27330106}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.16.4 Uniprot keywords 3D-structure; Antibiotic resistance; Carboxypeptidase; Cell membrane; Cell shape; Cell wall biogenesis/degradation; Hydrolase; Membrane; Metal-binding; Peptidoglycan synthesis; Protease; Reference proteome; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 20547.5 Length 181 Aromaticity 0.11 Instability index 33.61 Isoelectric point 4.81 Charge (pH=7) -10.78 2D Binding mode Binding energy (Kcal/mol) -8.3  Molscript Map 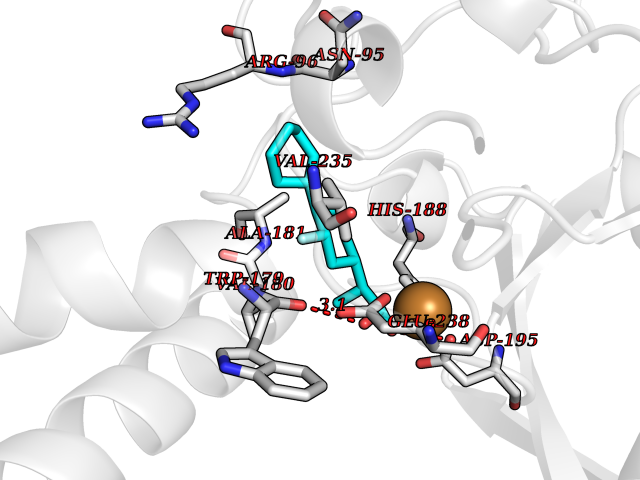 Pymol Map 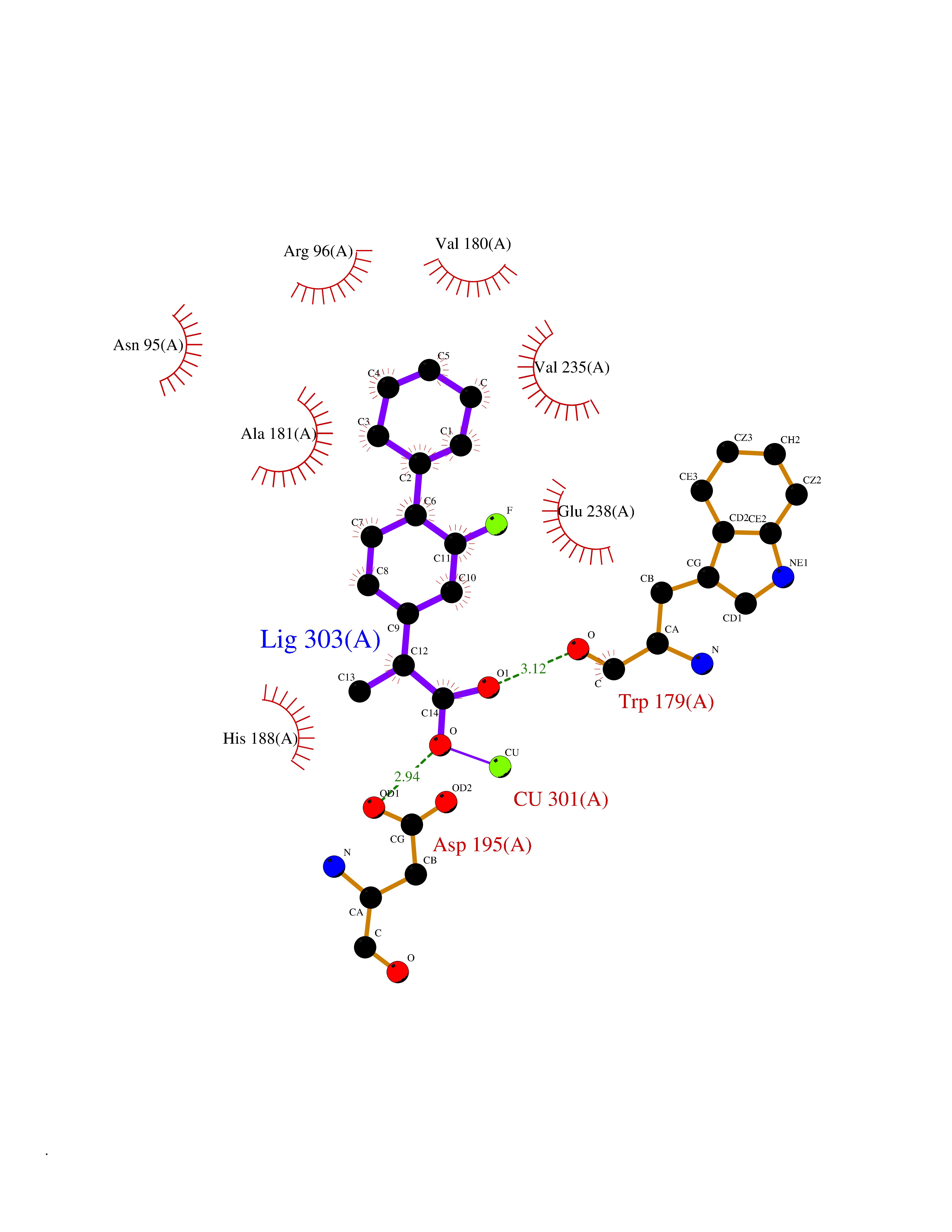 Ligplot Map 3D Binding mode Sequence EWSLILVNRQNPIPAQYDVELEQLSNGERIDIRISPYLQDLFDAARADGVYPIVASGYRTTEKQQEIXDEKVAEYKAKGYTSAQAKAEAETWVAVPGTSEHQLGLAVDINADGIHSTGNEVYRWLDENSYRFGFIRRYPPDKTEITGVSNEPWHYRYVGIEAATKIYHQGLCLEEYLNTEK Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Zinc finger protein Helios (IKZF2) | 7LPS | 7.14 | |
Target general information Gen name IKZF2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ikaros family zinc finger protein 2 Protein family Ikaros C2H2-type zinc-finger protein family Biochemical class NA Function Associates with Ikaros at centromeric heterochromatin. Related diseases Developmental and epileptic encephalopathy 25, with amelogenesis imperfecta (DEE25) [MIM:615905]: An autosomal recessive disease characterized by subclinical seizures appearing in the first days of life, evolving to severe epileptic disease. Affected individuals have profound or severe delayed development with lack of speech, and most patients do not acquire the ability to sit. Additional variable features include axial hypotonia, peripheral hypertonia, and abnormal involuntary movements such as dystonia and choreoathetosis. Dental abnormalities, including delayed eruption, hypodontia, tooth hypoplasia, yellow discoloration, thin enamel, and enamel chipping are observed in most patients. {ECO:0000269|PubMed:24995870, ECO:0000269|PubMed:26384929, ECO:0000269|PubMed:30054523}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P29972; P56545; P56545-3; Q17RB8; P09022; Q8N8B7-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID B,C Molecular weight (Da) 47006.6 Length 410 Aromaticity 0.09 Instability index 44.28 Isoelectric point 7.23 Charge (pH=7) 0.69 2D Binding mode Binding energy (Kcal/mol) -9.07 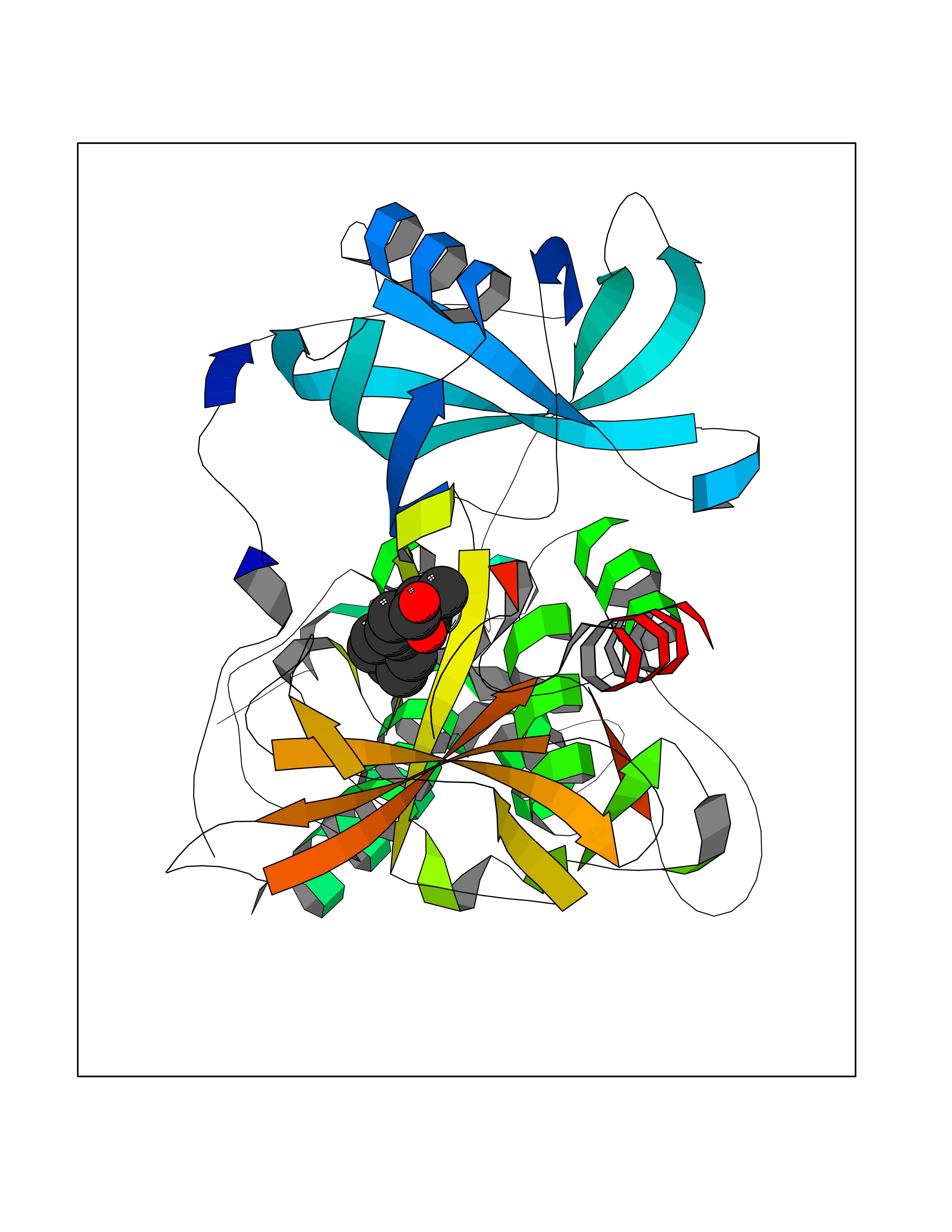 Molscript Map 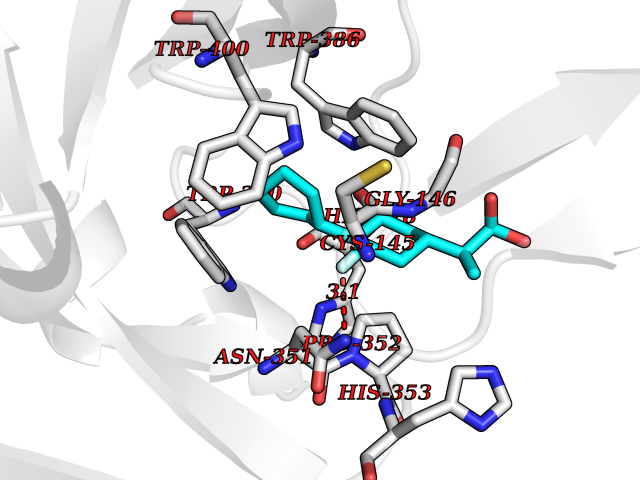 Pymol Map  Ligplot Map 3D Binding mode Sequence INFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEQDFGIEIVKVKAIGRQRFKVLELRTQSDGIQQAKVQILPECVLPSTMSAVQLESLNKCQIFPCSYKWWQKYQKRKFHCANLTSWPRWLYSLYDAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPDGERPFHCNQCGASFTQKGNLLRHIKLHSG Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Isoleucine--tRNA ligase | 1FFY | 7.14 | |
Target general information Gen name ileS Organism Staphylococcus aureus Uniprot ID TTD ID NA Synonyms NA Protein family Class-I aminoacyl-tRNA synthetase family, IleS type 1 subfamily Biochemical class Ligase / rna Function Aminoacyl-tRNA editing activity.ATP binding.Isoleucine-tRNA ligase activity.Metal ion binding.TRNA binding. Related diseases Charcot-Marie-Tooth disease, axonal, 2D (CMT2D) [MIM:601472]: A dominant axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:12690580, ECO:0000269|PubMed:17035524, ECO:0000269|PubMed:17101916, ECO:0000269|PubMed:17663003, ECO:0000269|PubMed:20169446, ECO:0000269|PubMed:24604904, ECO:0000269|PubMed:25168514, ECO:0000269|PubMed:26244500, ECO:0000269|PubMed:26503042, ECO:0000269|PubMed:31173493}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal dominant 5 (HMND5) [MIM:600794]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. {ECO:0000269|PubMed:12690580, ECO:0000269|PubMed:17035524, ECO:0000269|PubMed:23279345, ECO:0000269|PubMed:24627108, ECO:0000269|PubMed:26503042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinal muscular atrophy, infantile, James type (SMAJI) [MIM:619042]: An autosomal dominant form of spinal muscular atrophy, a group of neuromuscular disorders characterized by degeneration of the anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. SMAJI is a severe disease characterized by hypotonia manifesting in the first weeks or months of life, delayed motor development, motor regression, and muscle weakness and atrophy primarily affecting distal muscles. Additional variable features include feeding difficulties, poor overall growth, foot deformities, kyphosis, hyperlordosis, scoliosis, vocal cord dysfunction, and respiratory insufficiency. {ECO:0000269|PubMed:32181591}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00410 Interacts with NA EC number 6.1.1.5 Uniprot keywords 3D-structure; Aminoacyl-tRNA synthetase; ATP-binding; Cytoplasm; Direct protein sequencing; Ligase; Metal-binding; Nucleotide-binding; Protein biosynthesis; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 79061.6 Length 685 Aromaticity 0.1 Instability index 32.06 Isoelectric point 5.43 Charge (pH=7) -20.76 2D Binding mode Binding energy (Kcal/mol) -7.91  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MDYEKTLLMPKTDFPMRGGLPNKEPQIQEKWDAEDQYHKALEKNKGNETFILHDGPPYANGNLHMGHALNKILKDFIVRYKTMQGFYAPYVPGWDTHGLPIEQALTKKGVDRKKMSTAEFREKCKEFALEQIELQKKDFRRLGVRGDFNDPYITLKPEYEAAQIRIFGEMADKGLIYKQWFASISKVRQDILDAIENTNFKVNWGKTRIYNMVRDRGEWVISRQRVWGVPLPVFYAENGEIIMTKETVNHVADLFAEHGSNIWFEREAKDLLPEGFTHPGSPNGTFTKETDIMDVWFDSGSSHRGVLETRPELSFPADMYLEGSDQYRGWFNSSITTSVATRGVSPYKFLLSHGFVMDGEGKKMSKSLGNVIVPDQVVKQKGADIARLWVSSTDYLADVRISDEILKQTSDDYRKIRNTLRFMLGNINDFNPDTDSIPESELLEVDRYLLNRLREFTASTINNYENFDYLNIYQEVQNFINVELSNFYLDYGKDILYIEQRDSHIRRSMQTVLYQILVDMTKLLAPILVHTAEEVWSHTPHVKEESVHLADMPKVVEVDQALLDKWRTFMNLRDDVNRALETARNEKVIGKSLEAKVTIASNDKFNASEFLTSFDALHQLFIVSQVKVVDKLDDQATAYEHGDIVIEHADGEKCERCWNYSEDLGAVDELTHLCPRCQQVVKSLV Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Lysine--tRNA ligase | 4YCU | 7.13 | |
Target general information Gen name KARS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms KARS;KIAA0070 Protein family Class-II aminoacyl-tRNA synthetase family Biochemical class ligase / ligase inhibitor Function Amino acid binding.ATP adenylyltransferase activity.ATP binding.Identical protein binding.Lysine-tRNA ligase activity.Protein homodimerization activity.TRNA binding. Related diseases Charcot-Marie-Tooth disease, recessive intermediate B (CMTRIB) [MIM:613641]: A form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Recessive intermediate forms of Charcot-Marie-Tooth disease are characterized by clinical and pathologic features intermediate between demyelinating and axonal peripheral neuropathies, and motor median nerve conduction velocities ranging from 25 to 45 m/sec. {ECO:0000269|PubMed:20920668}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, autosomal recessive, 89 (DFNB89) [MIM:613916]: A form of non-syndromic deafness characterized by bilateral, prelingual, moderate to severe hearing loss affecting all frequencies. {ECO:0000269|PubMed:23768514}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, congenital, and adult-onset progressive leukoencephalopathy (DEAPLE) [MIM:619196]: An autosomal recessive, complex neurodegenerative disorder characterized by congenital sensorineural deafness, and progressive motor and cognitive decline apparent in young adulthood. Brain imaging shows diffuse white matter abnormalities affecting various brain regions, consistent with a progressive leukoencephalopathy. More variable additional features may include visual impairment and axonal peripheral neuropathy. Premature death may occurr in some patients. {ECO:0000269|PubMed:28887846, ECO:0000269|PubMed:30737337, ECO:0000269|PubMed:31116475}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Leukoencephalopathy, progressive, infantile-onset, with or without deafness (LEPID) [MIM:619147]: An autosomal recessive, complex neurodegenerative disorder apparent from infancy. LEPID is characterized by early-onset progressive leukoencephalopathy with brainstem and spinal cord calcifications, sensorineural deafness in most patients, global developmental delay with cognitive impairment and poor or absent speech, developmental regression, and neurologic deterioration. Additional more variable features may include poor overall growth with microcephaly, seizures, visual loss, microcytic anemia, and hepatic enlargement or abnormal liver enzymes. Premature death is common. {ECO:0000269|PubMed:25330800, ECO:0000269|PubMed:29615062, ECO:0000269|PubMed:30252186, ECO:0000269|PubMed:30715177}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00123 Interacts with Q13155; P07814; Q15046; P08865; P00441; Q13155 EC number 2.7.7.-; 6.1.1.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminoacyl-tRNA synthetase; ATP-binding; Cell membrane; Charcot-Marie-Tooth disease; Cytoplasm; Deafness; Direct protein sequencing; Disease variant; Host-virus interaction; Intellectual disability; Ligase; Membrane; Mitochondrion; Neurodegeneration; Neuropathy; Non-syndromic deafness; Nucleotide-binding; Nucleus; Phosphoprotein; Protein biosynthesis; Proteomics identification; Reference proteome; Secreted; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 37110.4 Length 323 Aromaticity 0.1 Instability index 50.17 Isoelectric point 4.92 Charge (pH=7) -17.53 2D Binding mode Binding energy (Kcal/mol) -8.41 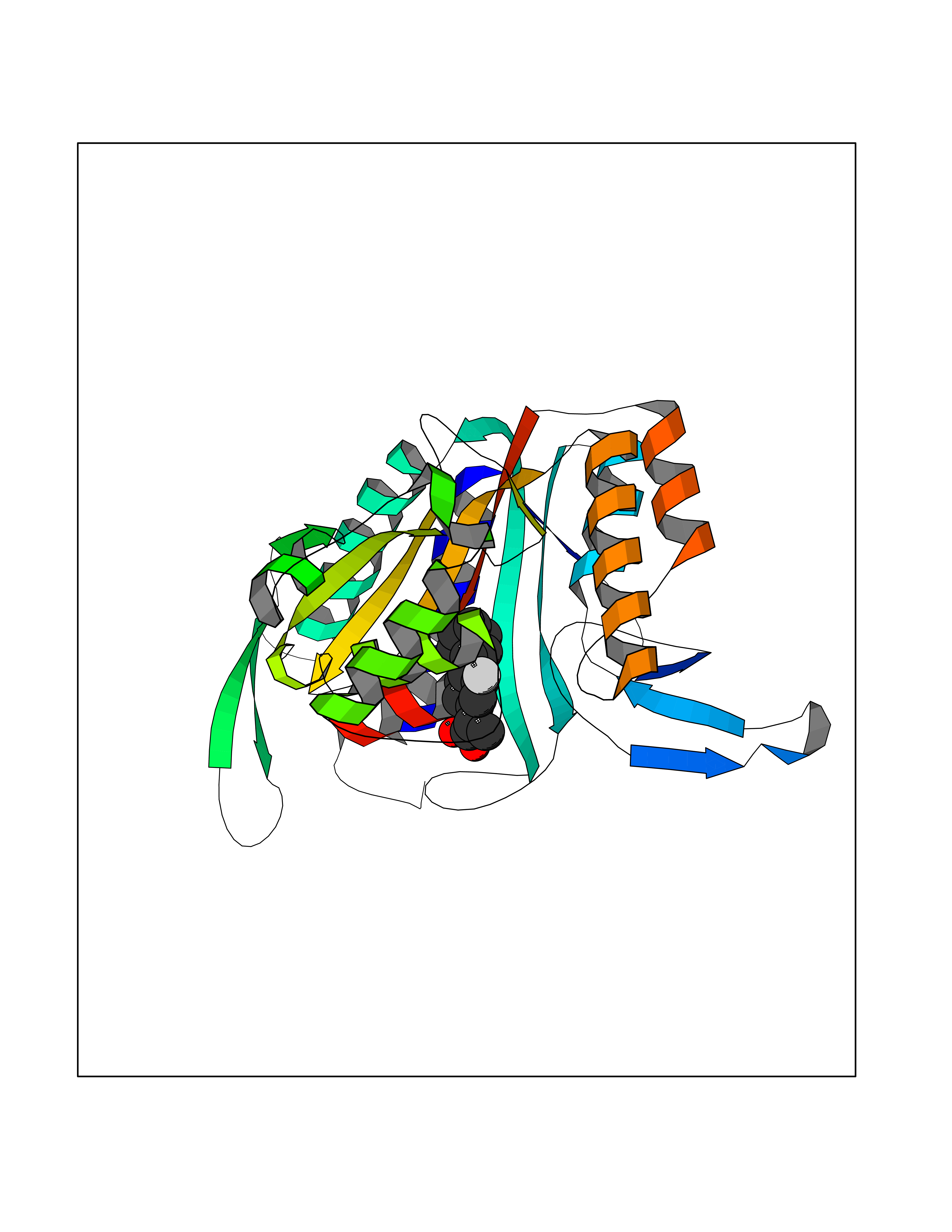 Molscript Map 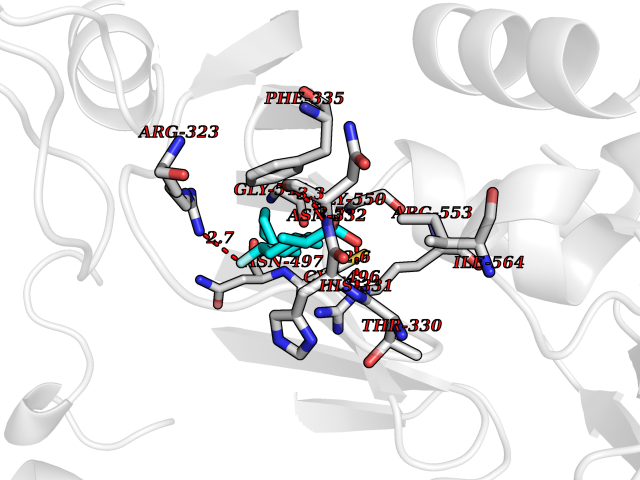 Pymol Map 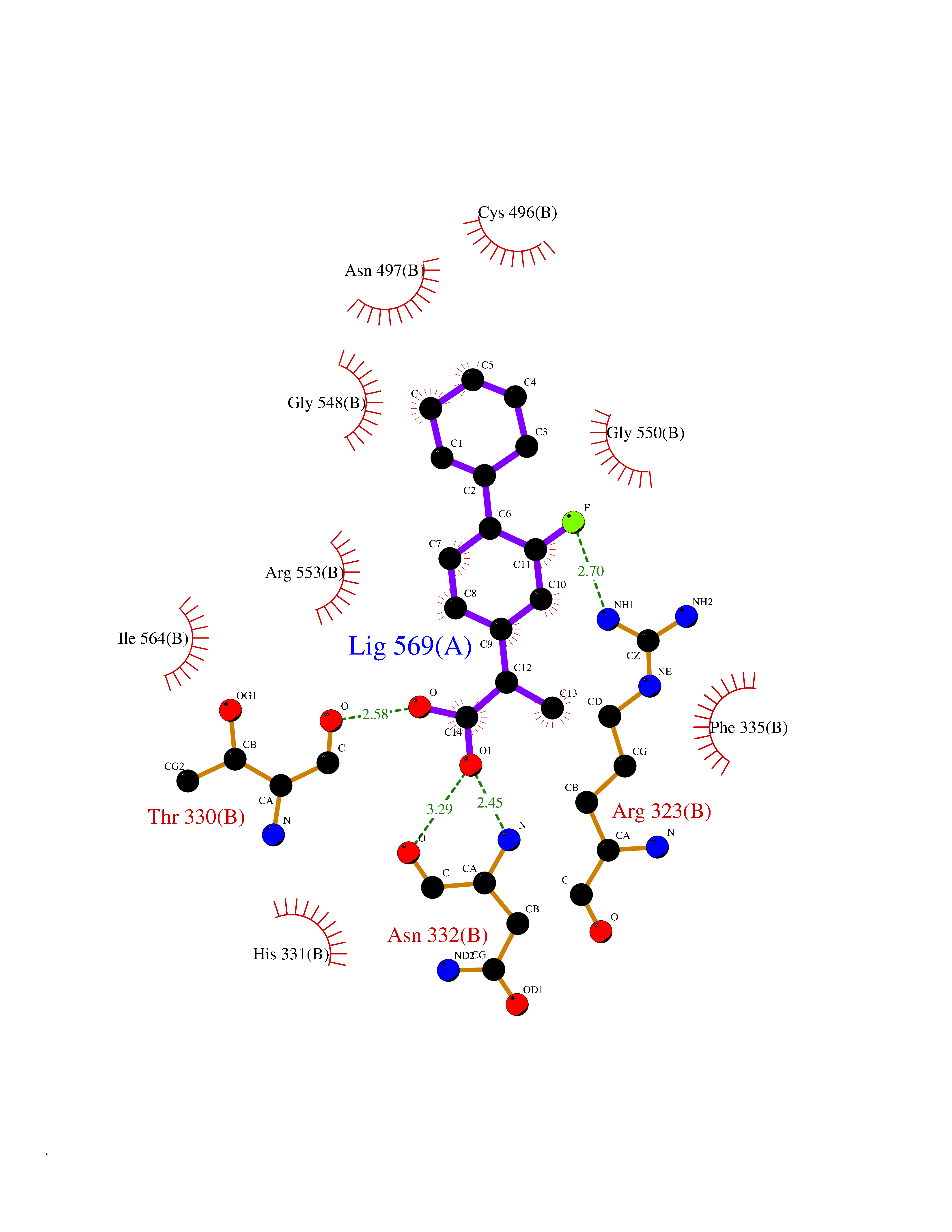 Ligplot Map 3D Binding mode Sequence IIRSKIITYIRSFLDELGFLEIETPMMNIIPGGAVAKPFITYHNELDMNLYMRIAPELYHKMLVVGGIDRVYEIGRQFRNEGIDLTHNPEFTTCEFYMAYADYHDLMEITEKMVSGMVKHITGSYKVTYHPDGPEGQAYDVDFTPPFRRINMVEELEKALGMKLPETNLFETEETRKILDDICVAKAVECPPPRTTARLLDKLVGEFLEVTCINPTFICDHPQIMSPLAKWHRSKEGLTERFELFVMKKEICNAYTELNDPMRQRQLFEEQAKAKAAGDDEAMFIDENFCTALEYGLPPTAGWGMGIDRVAMFLTDSNNIKEV Hydrogen bonds contact Hydrophobic contact | ||||