Job Results:
Ligand
Structure
Job ID
ea4aeeea43c0082ff76353d76181fa87
Job name
NA
Time
2024-06-15 15:52:32
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Thyroid hormone receptor beta (THRB) | 1N46 | 6.06 | |
Target general information Gen name THRB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms c-erbA-beta; c-erbA-2; THR1; Nuclear receptor subfamily 1 group A member 2; NR1A2; ERBA2 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function High affinity receptor for thyroid hormones, including triiodothyronine and thyroxine. Nuclear hormone receptor that can act as a repressor or activator of transcription. Related diseases Thyroid hormone resistance, generalized, autosomal dominant (GRTHD) [MIM:188570]: An autosomal dominant disease characterized by high levels of circulating thyroid hormones (T3-T4), goiter, abnormal mental functions, increased susceptibility to infections, abnormal growth and bone maturation, tachycardia and deafness. Affected individuals may also have attention deficit-hyperactivity disorders (ADHD) and language difficulties. Patients have normal or slightly elevated thyroid stimulating hormone (TSH). {ECO:0000269|PubMed:10660344, ECO:0000269|PubMed:12511610, ECO:0000269|PubMed:12554782, ECO:0000269|PubMed:1314846, ECO:0000269|PubMed:1324420, ECO:0000269|PubMed:1563081, ECO:0000269|PubMed:1587388, ECO:0000269|PubMed:1619012, ECO:0000269|PubMed:1661299, ECO:0000269|PubMed:16804041, ECO:0000269|PubMed:1846005, ECO:0000269|PubMed:19268523, ECO:0000269|PubMed:2153155, ECO:0000269|PubMed:2510172, ECO:0000269|PubMed:7833659, ECO:0000269|PubMed:8175986, ECO:0000269|PubMed:8514853, ECO:0000269|PubMed:8664910, ECO:0000269|PubMed:8889584}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid hormone resistance, generalized, autosomal recessive (GRTHR) [MIM:274300]: An autosomal recessive disorder characterized by goiter, clinical euthyroidism, end-organ unresponsiveness to thyroid hormone, abnormal growth and bone maturation, and deafness. Patients also have high levels of circulating thyroid hormones, with elevated thyroid stimulating hormone. {ECO:0000269|PubMed:1653889}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Selective pituitary thyroid hormone resistance (PRTH) [MIM:145650]: Variant form of thyroid hormone resistance and is characterized by clinical hyperthyroidism, with elevated free thyroid hormones, but inappropriately normal serum TSH. Unlike GRTH, where the syndrome usually segregates with a dominant allele, the mode of inheritance in PRTH has not been established. {ECO:0000269|PubMed:7528740, ECO:0000269|PubMed:8381821}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08085; DB03181; DB02106; DB01118; DB00509; DB05035; DB03788; DB03176; DB00451; DB00279; DB01583; DB05192; DB07425; DB09100; DB03604 Interacts with Q60974; Q9Y618 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Deafness; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 27235.4 Length 239 Aromaticity 0.09 Instability index 43.29 Isoelectric point 5.42 Charge (pH=7) -8.55 2D Binding mode Binding energy (Kcal/mol) -6.95  Molscript Map 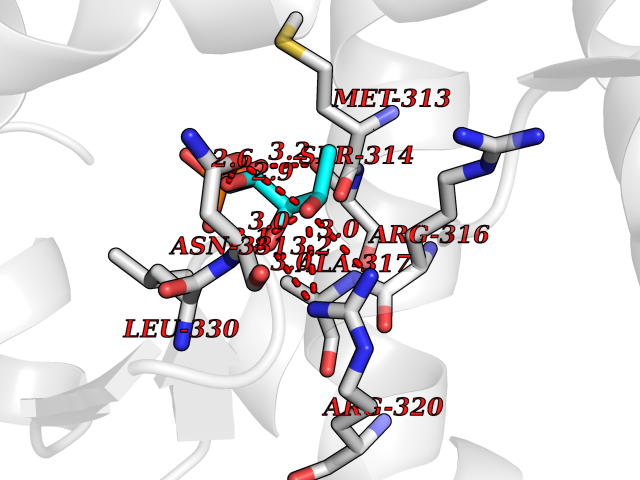 Pymol Map 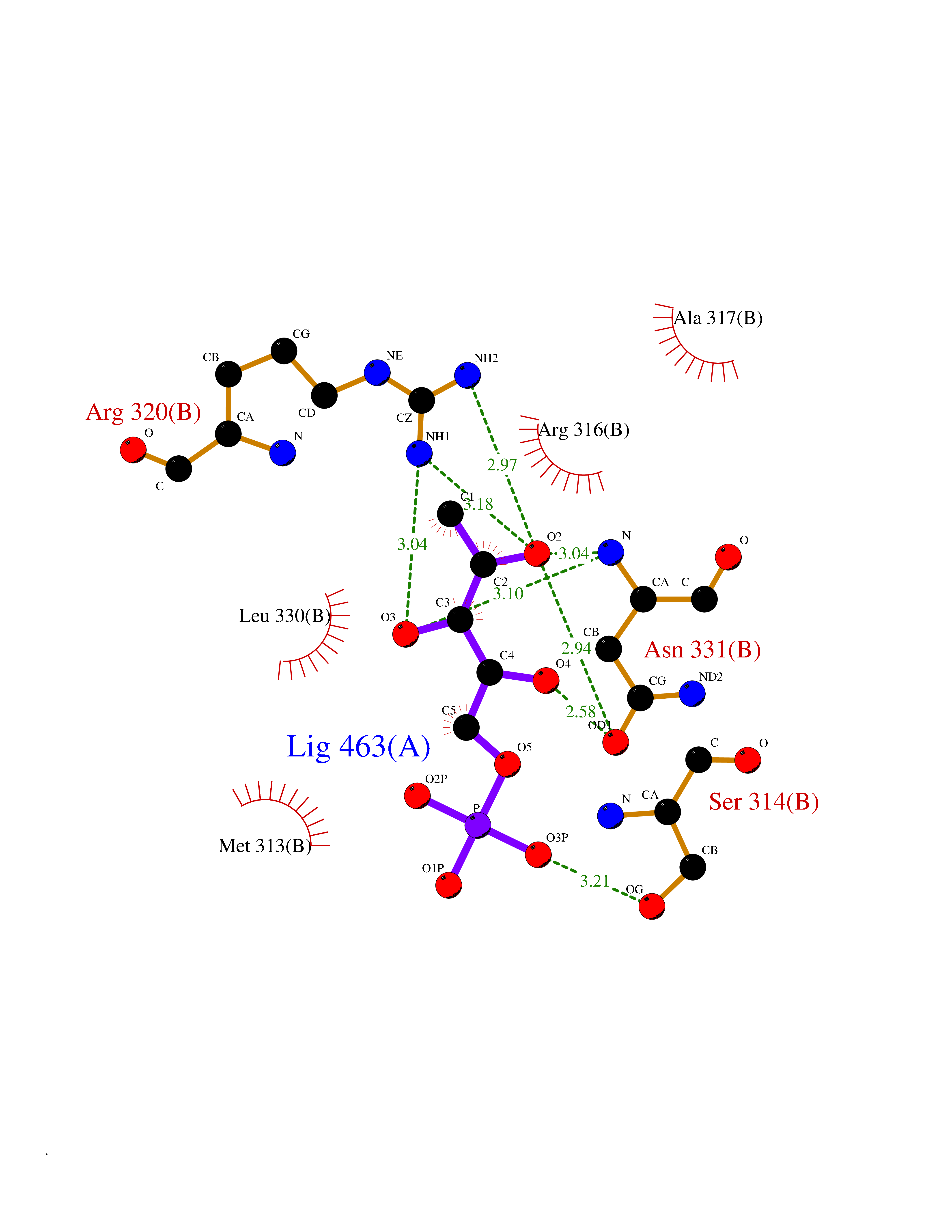 Ligplot Map 3D Binding mode Sequence KPEPTDEEWELIKTVTEAHVATNAQWKQKRKFLPEDIGQAKVDLEAFSHFTKIITPAITRVVDFAKKLPMFCELPCEDQIILLKGCCMEIMSLRAAVRYDPESETLTLNGEMAVTRGQLKNGGLGVVSDAIFDLGMSLSSFNLDDTEVALLQAVLLMSSDRPGLACVERIEKYQDSFLLAFEHYINYRKHHVTHFWPKLLMKVTDLRMIGACHASRFLHMKVECPTELFPPLFLEVFED Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | 4-hydroxyphenylpyruvate dioxygenase (EC 1.13.11.27) (4-hydroxyphenylpyruvic acid oxidase) (4HPPD) (HPD) (HPPDase) | 5DHW | 6.05 | |
Target general information Gen name HPD Organism Arabidopsis thaliana (Mouse-ear cress) Uniprot ID TTD ID NA Synonyms At1g06570;PDS1;F12K11.9 Protein family 4HPPD family Biochemical class NA Function Catalyzes the conversion of 4-hydroxyphenylpyruvic acid to homogentisic acid, one of the steps in tyrosine catabolism. {ECO:0000269|PubMed:15301540}." Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P93836 EC number 1.13.11.27 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Dioxygenase; Iron; Metal-binding; Oxidoreductase; Phenylalanine catabolism; Reference proteome; Repeat; Tyrosine catabolism Protein physicochemical properties Chain ID B Molecular weight (Da) 41240.4 Length 371 Aromaticity 0.11 Instability index 47.03 Isoelectric point 6.09 Charge (pH=7) -4.66 2D Binding mode Binding energy (Kcal/mol) -6.81 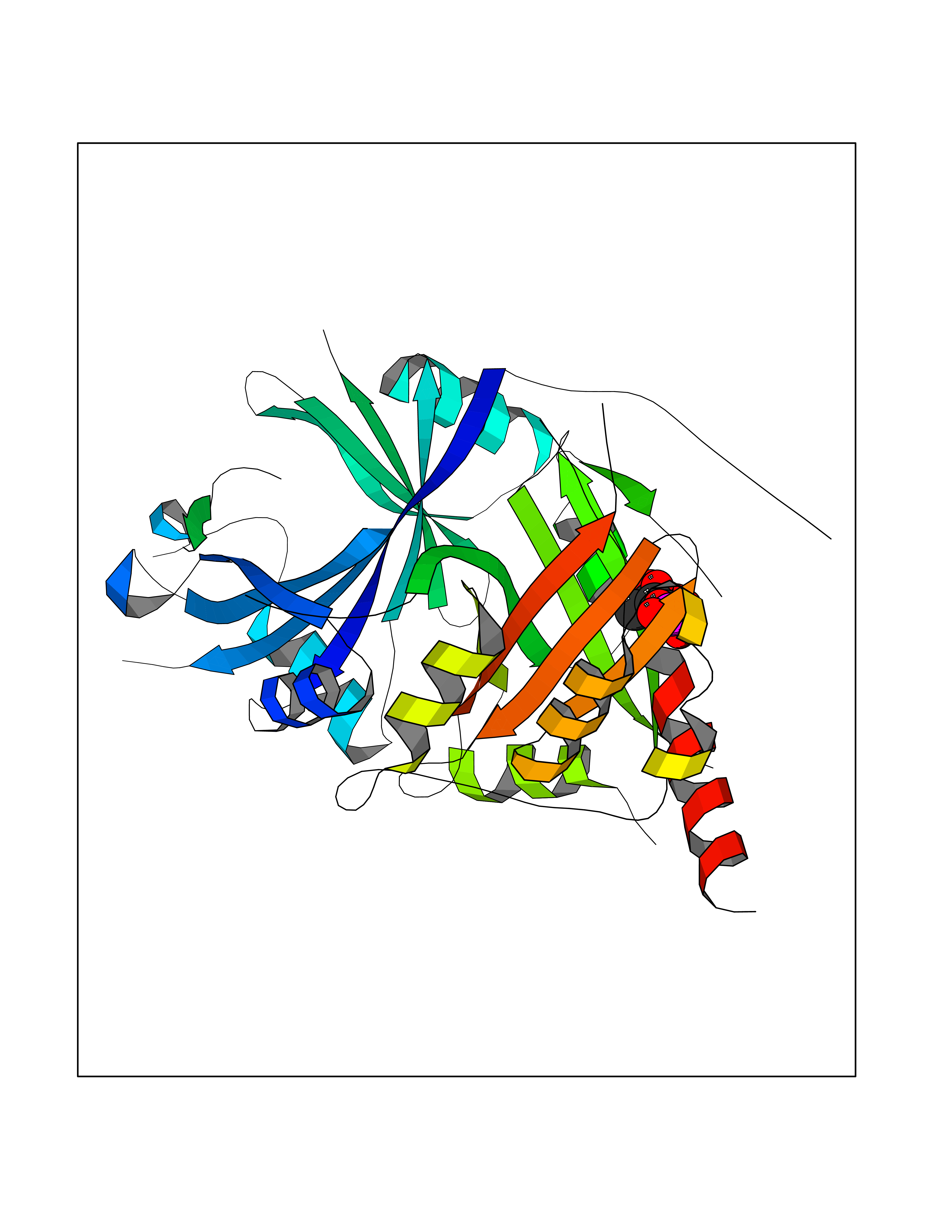 Molscript Map 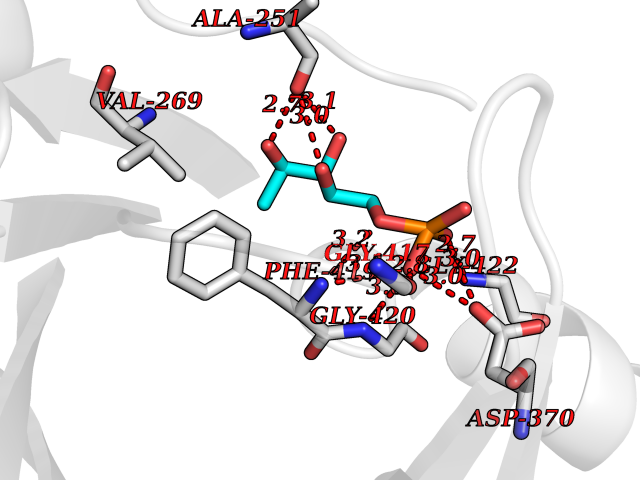 Pymol Map 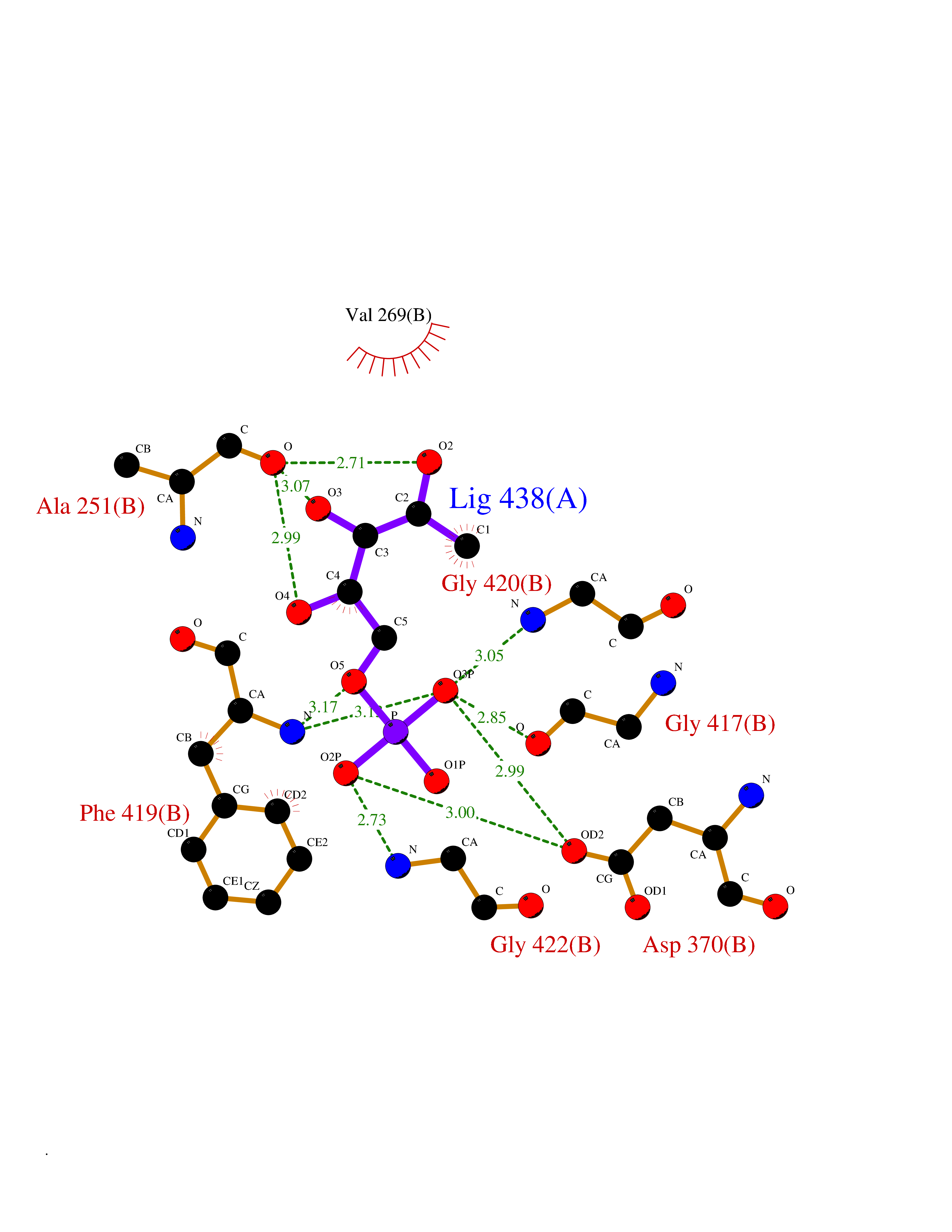 Ligplot Map 3D Binding mode Sequence SKFVRKNPKSDKFKVKRFHHIEFWCGDATNVARRFSWGLGMRFSAKSDLSTGNMVHASYLLTSGDLRFLFTAPYSPSPTTTASIPSFDHGSCRSFFSSHGLGVRAVAIEVEDAESAFSISVANGAIPSSPPIVLNEAVTIAEVKLYGDVVLRYVSYKFLPGFERVEFPLDYGIRRLDHAVGNVPELGPALTYVAGFTGFHQFAEFESGLNSAVLASNDEMVLLPINEPVHGRKSQIQTYLEHNEGAGLQHLALMSEDIFRTLREMRKRSSIGGFDFMPSPPPTYYQNLKKRVGDVLSDDQIKECEELGILVDRDDQGTLLQIFTKPLGDRPTIFIEIIQRVGCMMYQSGGCGGFGKGNFSELFKSIEEYEK Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | Streptogramin A acetyltransferase | 3DHO | 6.05 | |
Target general information Gen name vatD Organism Enterococcus faecium (Streptococcus faecium) Uniprot ID TTD ID NA Synonyms satA Protein family Transferase hexapeptide repeat family Biochemical class Transferase Function Transferase activity, transferring acyl groups. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01992; DB01764; DB01669 Interacts with NA EC number 2.3.1.- Uniprot keywords 3D-structure; Acyltransferase; Antibiotic resistance; Repeat; Transferase Protein physicochemical properties Chain ID A,B,C,D,E,F Molecular weight (Da) 68559.3 Length 609 Aromaticity 0.09 Instability index 29.65 Isoelectric point 5.39 Charge (pH=7) -11.69 2D Binding mode Binding energy (Kcal/mol) -6.78 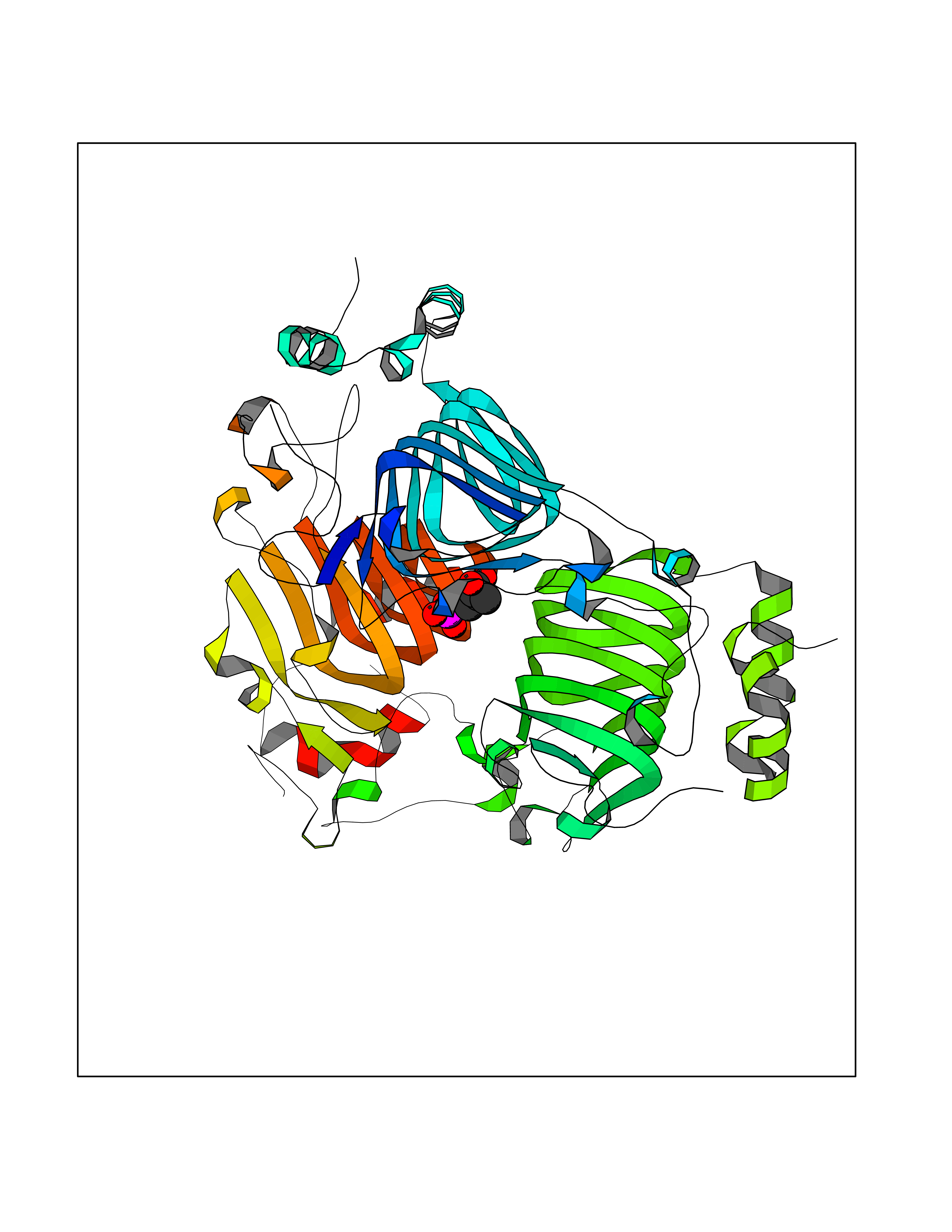 Molscript Map 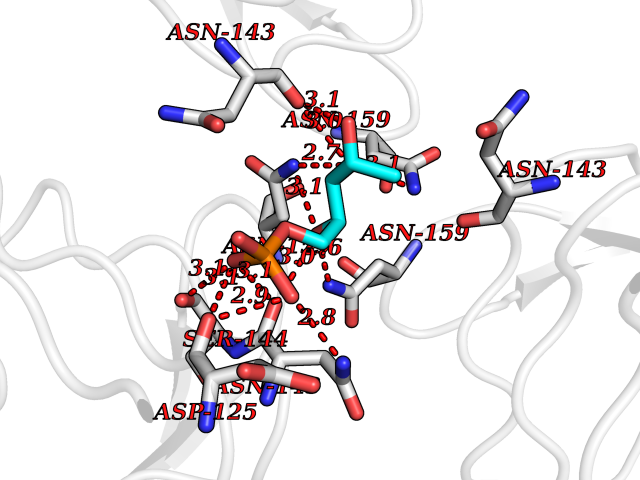 Pymol Map 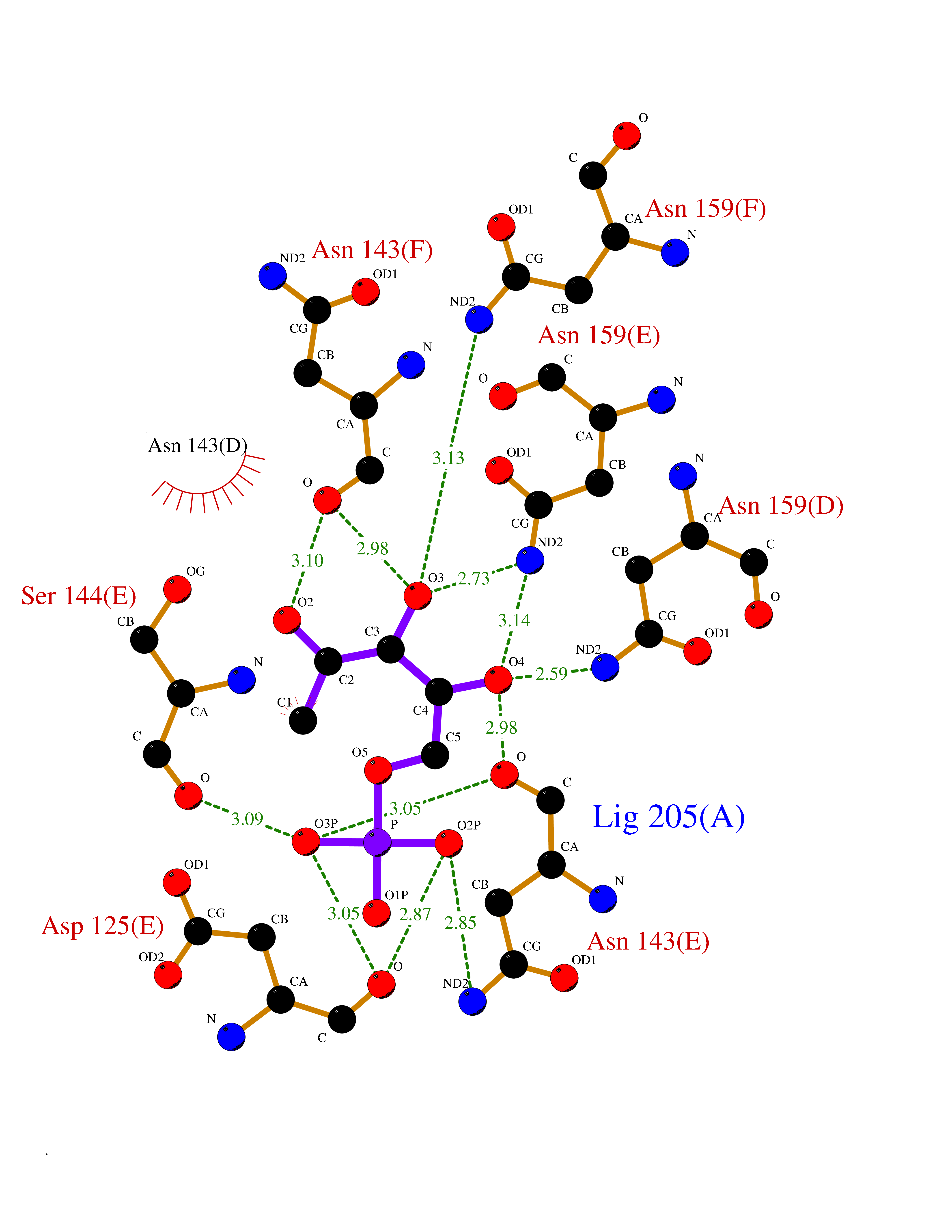 Ligplot Map 3D Binding mode Sequence MGPNPMKMYPIEGNKSVQFIKPILEKLENVEVGEYSYYDSKNGETFDKQILYHYPILNDKLKIGKFCSIGPGVTIIMNGANHRMDGSTYPFNLFGNGWEKHMPKLDQLPIKGDTIIGNDVWIGKDVVIMPGVKIGDGAIVAANSVVVKDIAPYMLAGGNPANEIKQRFDQDTINQLLDIKWWNWPIDIINENIDKILDNSIIRMGPNPMKMYPIEGNKSVQFIKPILEKLENVEVGEYSYYDSKNGETFDKQILYHYPILNDKLKIGKFCSIGPGVTIIMNGANHRMDGSTYPFNLFGNGWEKHMPKLDQLPIKGDTIIGNDVWIGKDVVIMPGVKIGDGAIVAANSVVVKDIAPYMLAGGNPANEIKQRFDQDTINQLLDIKWWNWPIDIINENIDKILDNSIIRMGPNPMKMYPIEGNKSVQFIKPILEKLENVEVGEYSYYDSKNGETFDKQILYHYPILNDKLKIGKFCSIGPGVTIIMNGANHRMDGSTYPFNLFGNGWEKHMPKLDQLPIKGDTIIGNDVWIGKDVVIMPGVKIGDGAIVAANSVVVKDIAPYMLAGGNPANEIKQRFDQDTINQLLDIKWWNWPIDIINENIDKILDNSIIR Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | Interleukin 3 receptor alpha (IL3RA) | 5UWC | 6.04 | |
Target general information Gen name IL3RA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Interleukin-3 receptor subunit alpha; IL3R; IL-3RA; IL-3R-alpha; IL-3R subunit alpha; IL-3 receptor subunit alpha; CD123 antigen; CD123 Protein family Type I cytokine receptor family, Type 5 subfamily Biochemical class Cytokine receptor Function This is a receptor for interleukin-3. Related diseases Microcephaly and chorioretinopathy, autosomal recessive, 2 (MCCRP2) [MIM:616171]: A severe disorder characterized by microcephaly, delayed psychomotor development, growth retardation with dwarfism, and ocular abnormalities. {ECO:0000269|PubMed:25344692}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00020; DB14731 Interacts with O95393; O14523; Q8N6F1-2; P54852; Q8WWP7; Q8N5M9; Q92993; Q8TAP4-4; Q9NZG7; Q9NXK6; P17252; Q9Y6X1; Q15047-2; Q0VAQ4; Q8N2H4; Q5BJH2-2; Q9BVK8; Q8TBM7; Q6PI78; A5PKU2; O95183; P61981; P08700 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Disulfide bond; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID G Molecular weight (Da) 29237 Length 253 Aromaticity 0.12 Instability index 46.45 Isoelectric point 9.01 Charge (pH=7) 8.21 2D Binding mode Binding energy (Kcal/mol) -5.99 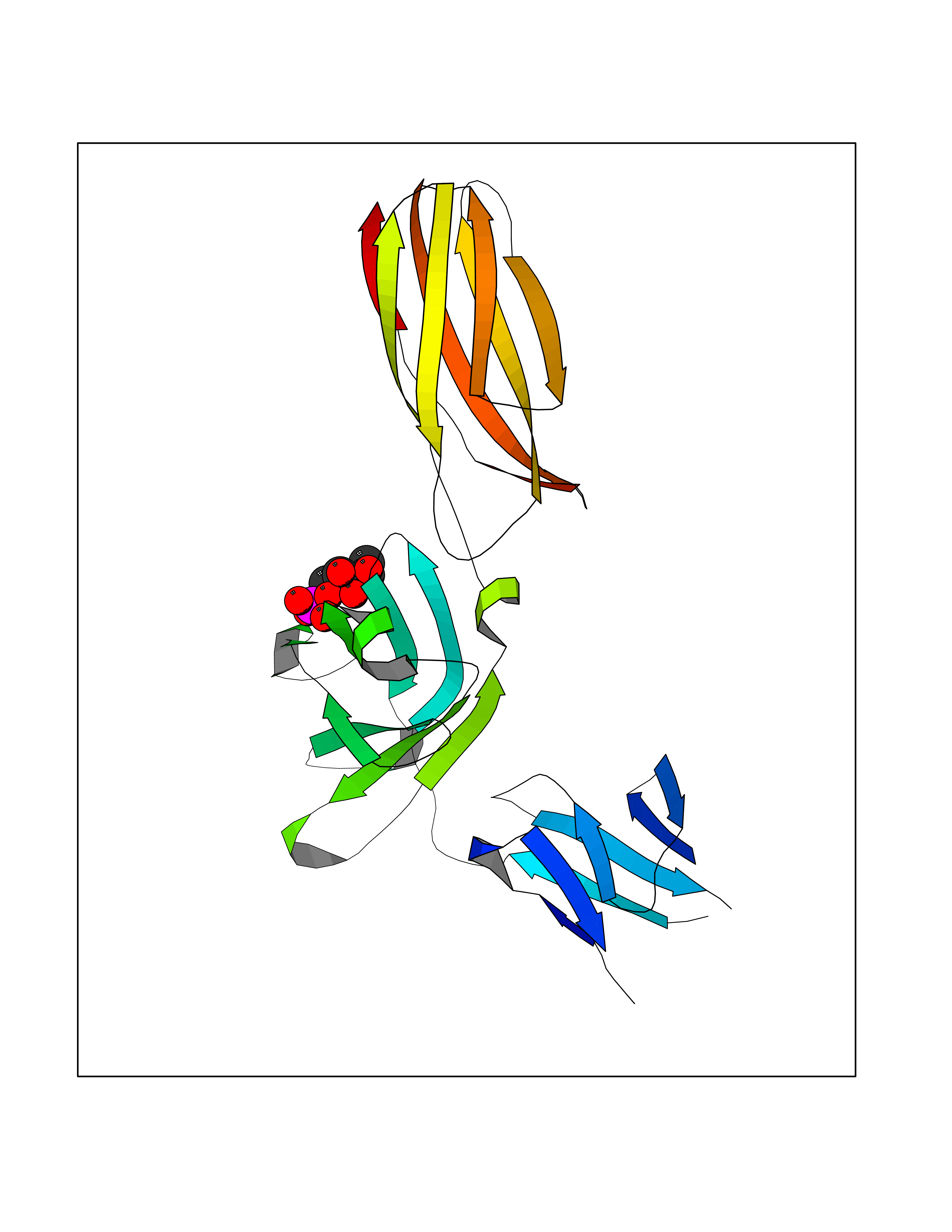 Molscript Map 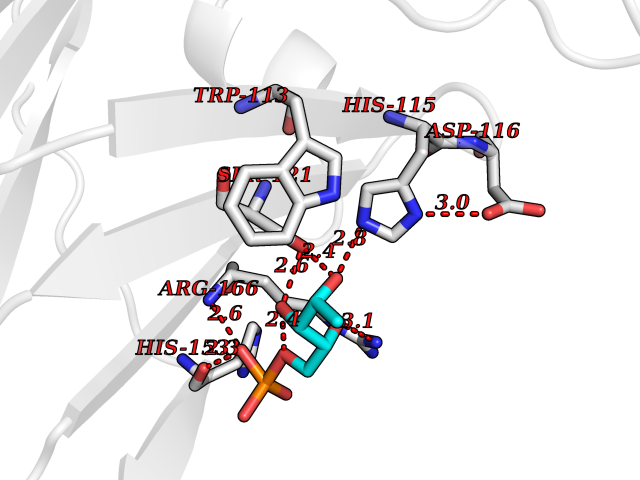 Pymol Map 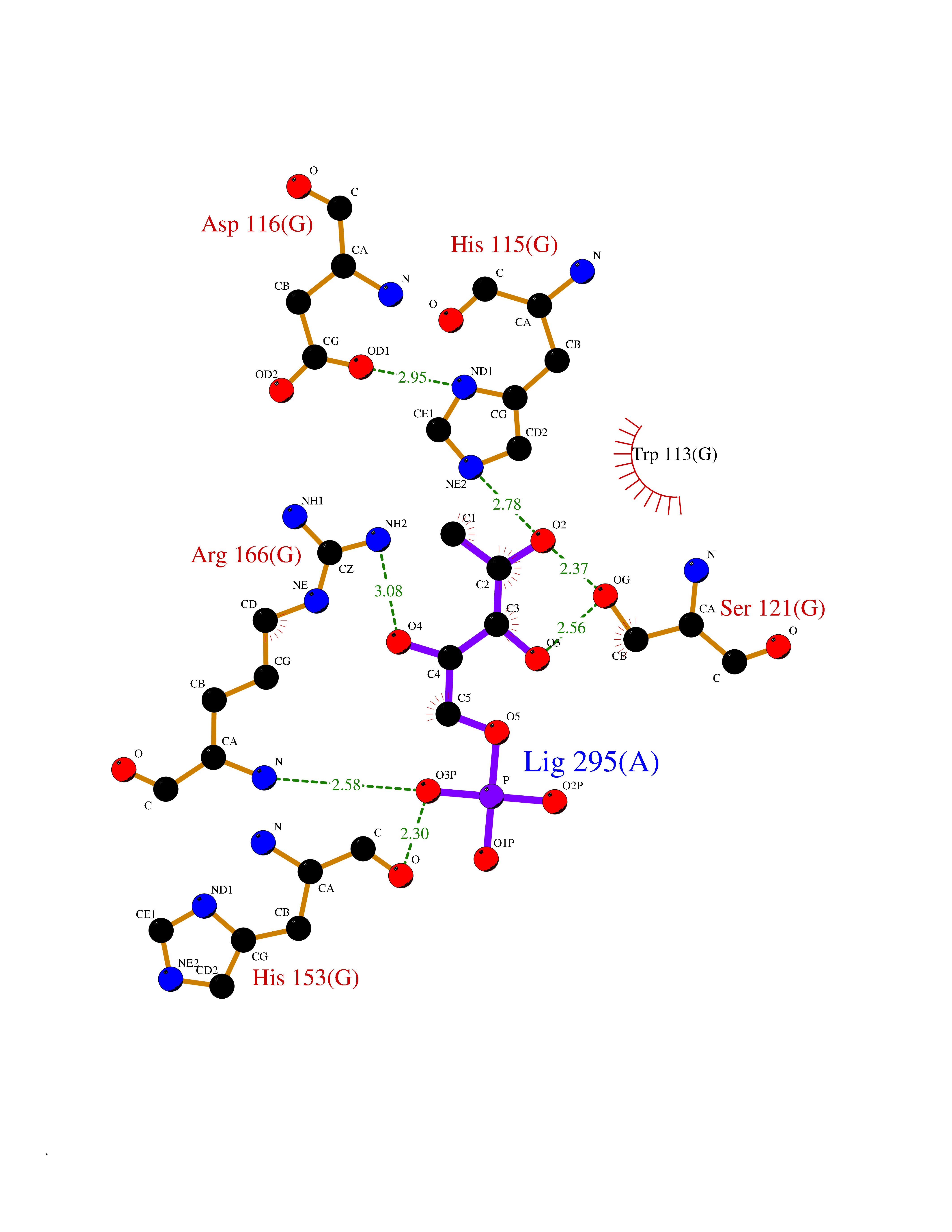 Ligplot Map 3D Binding mode Sequence ITNLRMKAKAQQLTWECVKDADYSMPAVNNSYCQFGAISLCEVTNYTVRVSTWILFPENSGKPWAGAENLTCWIHDVDFLSCSWAVGPGAPADVQYDLYLNVANRRQQYECLHYKTDAQGTRIGCRFDDISRLSSGSQSSHILVRGRSAAFGIPCTDKFVVFSQIEILTPPQMTAKCNKTHSFMHWKMRSHFNRKFRYELQIQKRMQPVITEQVRDRTSFQLLNPGTYTVQIRARERVYEFLSAWSTPQRFEC Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | D-alanyl-D-alanine carboxypeptidase DacC | 3ITA | 6.04 | |
Target general information Gen name dacC Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b0839;JW0823 Protein family Peptidase S11 family Biochemical class Hydrolase Function Carboxypeptidase activity.Endopeptidase activity.Penicillin binding.Serine-type D-Ala-D-Ala carboxypeptidase activity. Related diseases Charcot-Marie-Tooth disease, axonal, 2T (CMT2T) [MIM:617017]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:26991897, ECO:0000269|PubMed:27588448}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinocerebellar ataxia 43 (SCA43) [MIM:617018]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA43 is a slowly progressive, autosomal dominant form. {ECO:0000269|PubMed:27583304}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01602; DB00578; DB09319; DB00671; DB00274; DB01329; DB01331; DB00430; DB01332; DB09050; DB01000; DB00303 Interacts with NA EC number 3.4.16.4 Uniprot keywords 3D-structure; Carboxypeptidase; Cell inner membrane; Cell membrane; Cell shape; Cell wall biogenesis/degradation; Direct protein sequencing; Hydrolase; Membrane; Peptidoglycan synthesis; Protease; Reference proteome; Signal Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 22829.7 Length 214 Aromaticity 0.07 Instability index 21.24 Isoelectric point 6.36 Charge (pH=7) -0.93 2D Binding mode Binding energy (Kcal/mol) -6.39  Molscript Map 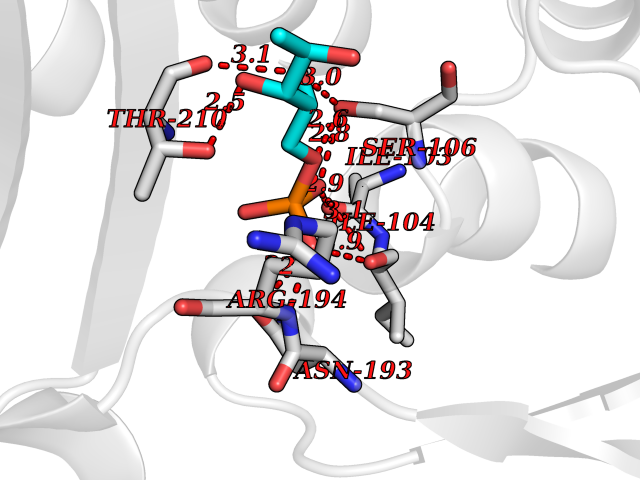 Pymol Map 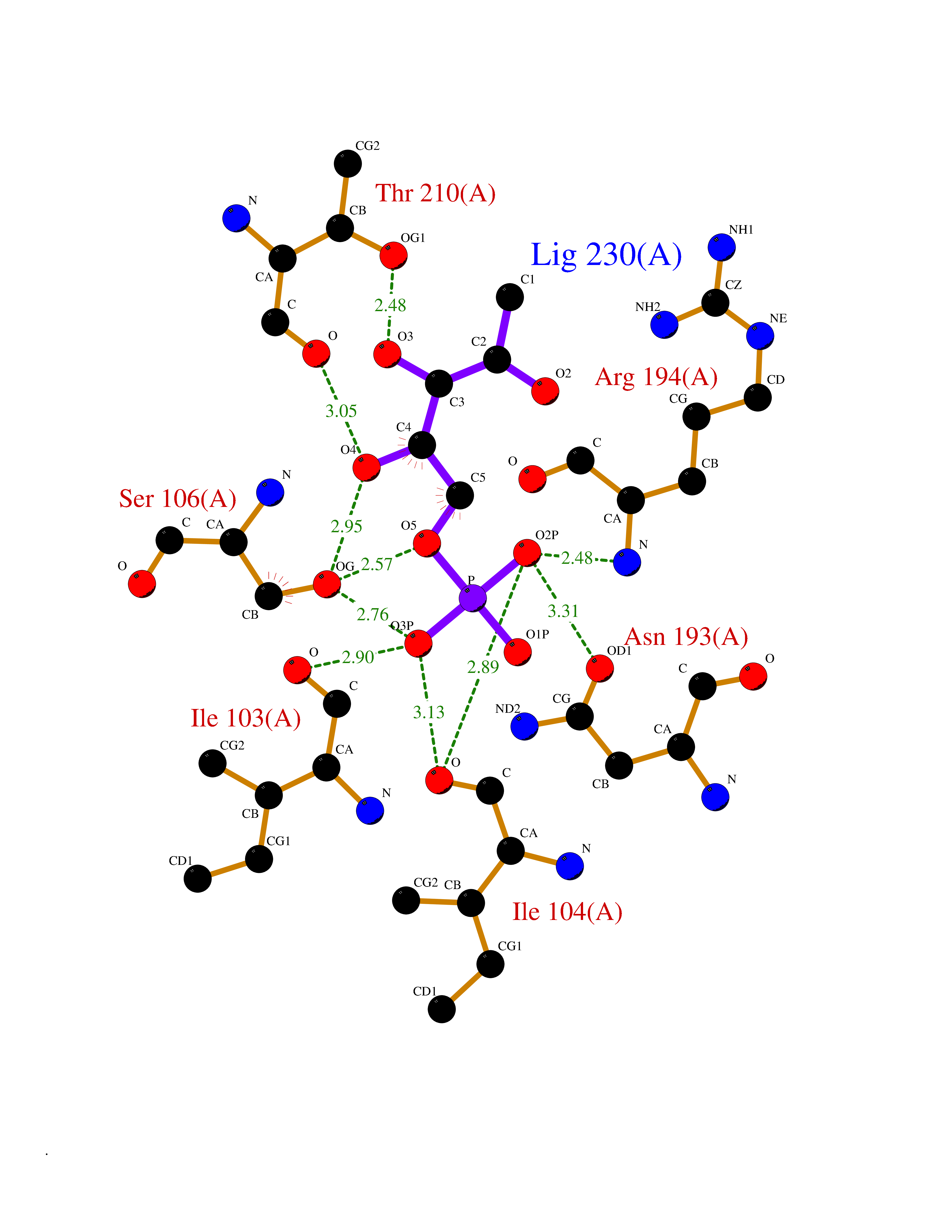 Ligplot Map 3D Binding mode Sequence AWILMDYASGKVLAEGNADEKLDPASLTKIMTSYVVGQALKADKIKLTDMVTVGKDAWATGNPALRGSSVMFLKPGDQVSVADLNKGVIIQSGNDACIALADYVAGSQESFIGLMNGYAKKLGLTNTTFQTVHGLDAPGQFSTARDMALLGKALIHDVPEEYAIHKEKEFTFNKIRQPNRNRLLWSSNLNVDGMKTGTTAGAGYNLVASATQGD Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Hyperpolarization cyclic nucleotide-gated channel 4 (HCN4) | 3OTF | 6.04 | |
Target general information Gen name HCN4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channel 4 Protein family Potassium channel HCN family Biochemical class Voltage-gated ion channel Function Contributes to the native pacemaker currents in heart (If) that regulate the rhythm of heart beat. May contribute to the native pacemaker currents in neurons (Ih). May mediate responses to sour stimuli. Hyperpolarization-activated ion channel with very slow activation and inactivation exhibiting weak selectivity for potassium over sodium ions. Related diseases Sick sinus syndrome 2 (SSS2) [MIM:163800]: The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS2 onset is in utero or at birth. {ECO:0000269|PubMed:15123648, ECO:0000269|PubMed:16407510, ECO:0000269|PubMed:20662977, ECO:0000269|PubMed:23103389}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 8 (BRGDA8) [MIM:613123]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:19165230}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Epilepsy, idiopathic generalized 18 (EIG18) [MIM:619521]: An autosomal dominant form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. EIG18 is characterized by onset of myoclonic seizures in infancy. Although the seizures remit, some patients may have later speech or cognitive impairment. {ECO:0000269|PubMed:30127718}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O60741; Q9Y3Q4 EC number NA Uniprot keywords 3D-structure; Brugada syndrome; cAMP; cAMP-binding; Cell membrane; Disease variant; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Nucleotide-binding; Phosphoprotein; Potassium; Potassium channel; Potassium transport; Proteomics identification; Reference proteome; Sodium; Sodium channel; Sodium transport; Transmembrane; Transmembrane helix; Transport; Voltage-gated channel Protein physicochemical properties Chain ID A Molecular weight (Da) 23211.2 Length 197 Aromaticity 0.12 Instability index 42.69 Isoelectric point 8.67 Charge (pH=7) 3.11 2D Binding mode Binding energy (Kcal/mol) -6.9 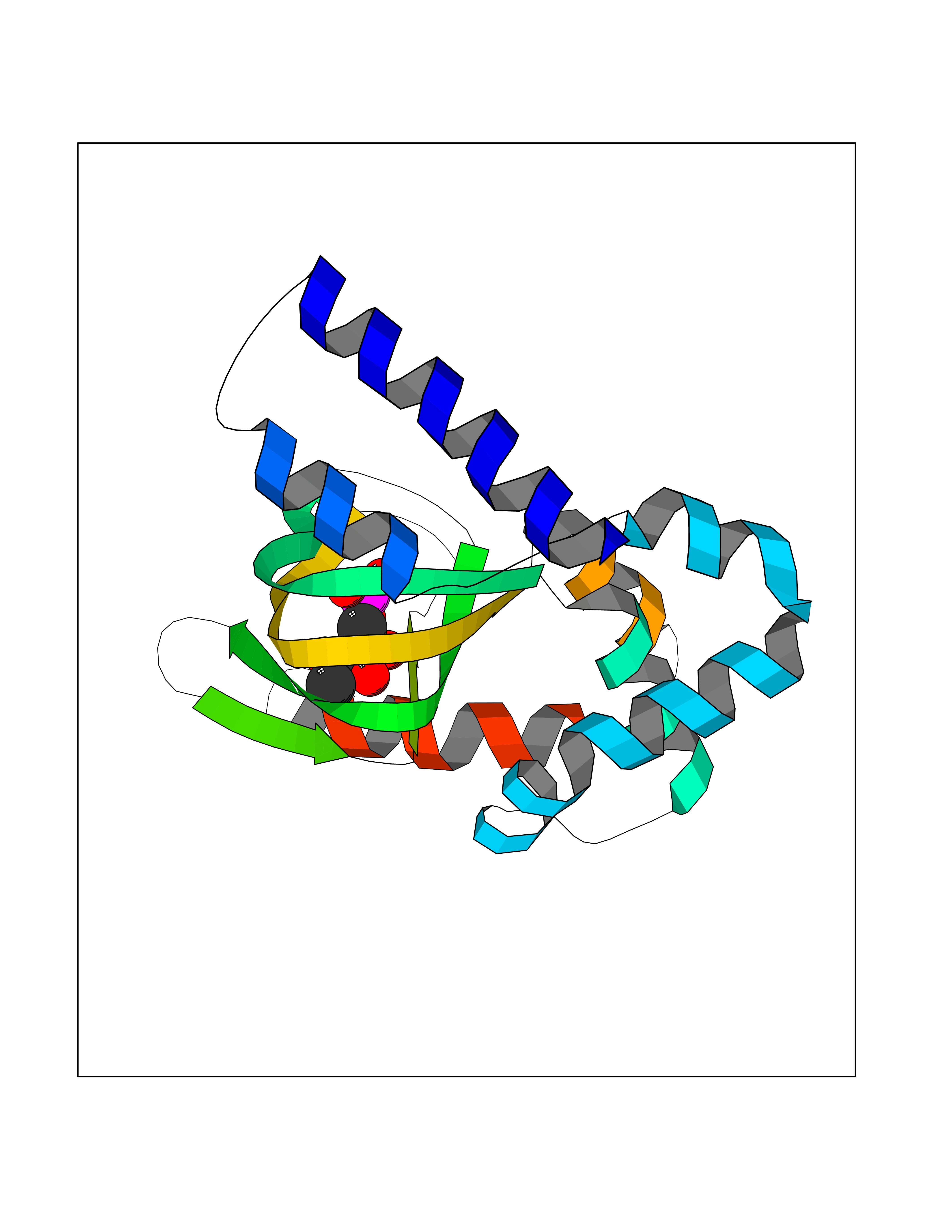 Molscript Map 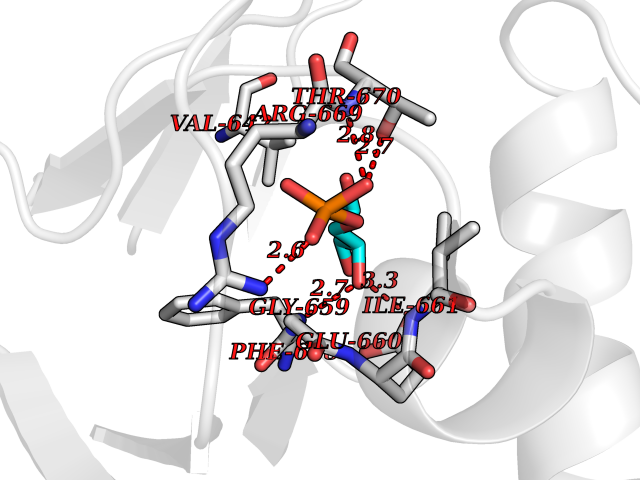 Pymol Map 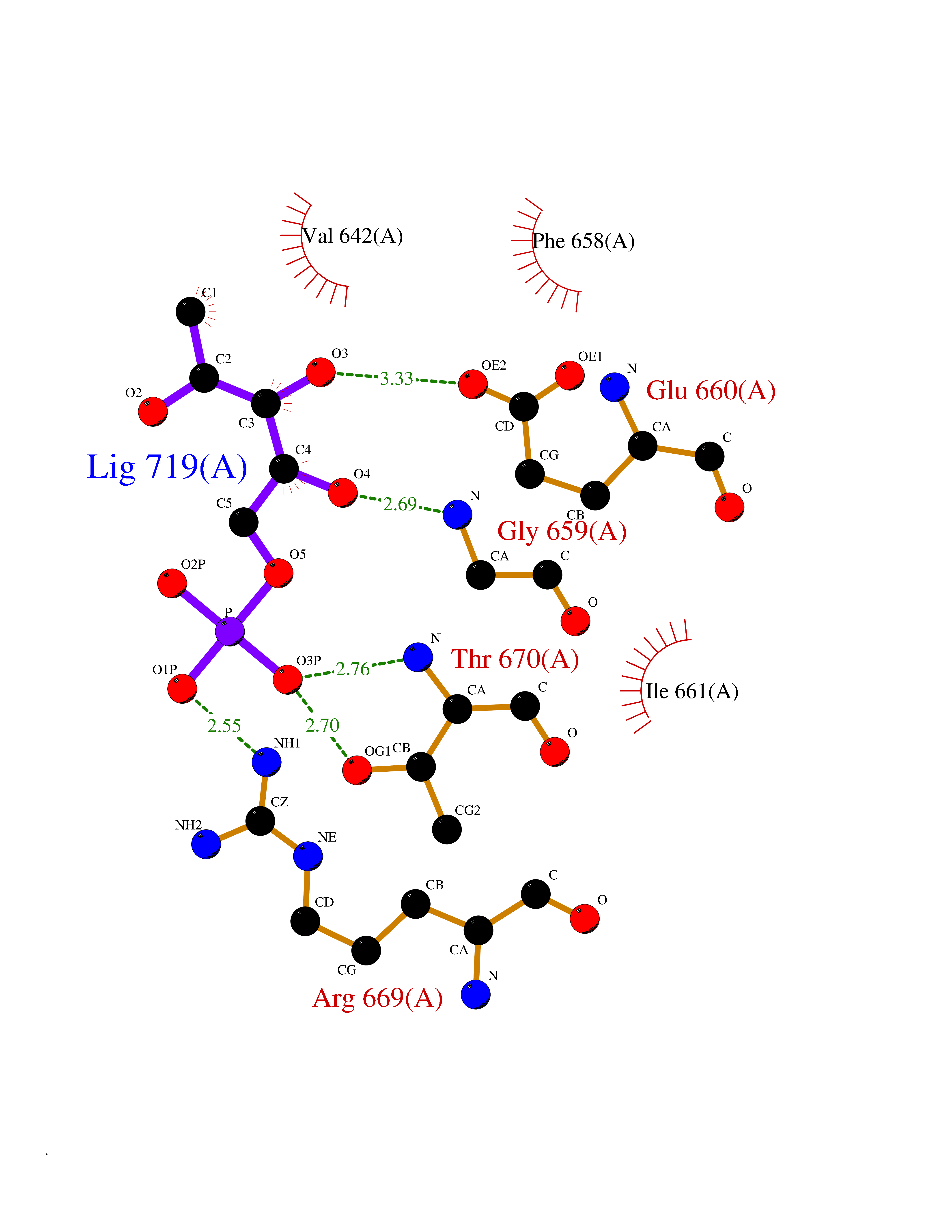 Ligplot Map 3D Binding mode Sequence DSSRRQYQEKYKQVEQYMSFHKLPPDTRQRIHDYYEHRYQGKMFDEESILGELSEPLREEIINFNCRKLVASMPLFANADPNFVTSMLTKLRFEVFQPGDYIIREGTIGKKMYFIQHGVVSVLTKGNKETKLADGSYFGEICLLTRGRRTASVRADTYCRLYSLSVDNFNEVLEEYPMMRRAFETVALDRLDRIGKK Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Inosine-5'-monophosphate dehydrogenase 1 (IMPDH1) | 1JCN | 6.03 | |
Target general information Gen name IMPDH1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Superoxide-inducible protein 12; SOI12; Probable inosine-5'-monophosphate dehydrogenase IMD1; NAD-dependent inosine monophosphate dehydrogenase; Inosine dehydrogenase; IMPDH-I; IMPDH 1; IMPDH; IMPD1; Protein family IMPDH/GMPR family Biochemical class CH-OH donor oxidoreductase Function Could also have a single-stranded nucleic acid-binding activity and could play a role in RNA and/or DNA metabolism. It may also have a role in the development of malignancy and the growth progression of some tumors. Catalyzes the conversion of inosine 5'-phosphate (IMP) to xanthosine 5'-phosphate (XMP), the first committed and rate-limiting step in the de novo synthesis of guanine nucleotides, and therefore plays an important role in the regulation of cell growth. Related diseases Retinitis pigmentosa 10 (RP10) [MIM:180105]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:11875049, ECO:0000269|PubMed:11875050, ECO:0000269|PubMed:16384941}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Leber congenital amaurosis 11 (LCA11) [MIM:613837]: A severe dystrophy of the retina, typically becoming evident in the first years of life. Visual function is usually poor and often accompanied by nystagmus, sluggish or near-absent pupillary responses, photophobia, high hyperopia and keratoconus. {ECO:0000269|PubMed:16384941}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03948; DB00993; DB01033; DB00688; DB01024; DB00157; DB00811; DB06408; DB06103 Interacts with Q96D03; P20839-3; P12268; O75928-2; Q9Y4B4; P78317; Q7KZS0 EC number EC 1.1.1.205 Uniprot keywords 3D-structure; Alternative splicing; CBS domain; Cytoplasm; Direct protein sequencing; Disease variant; DNA-binding; GMP biosynthesis; Leber congenital amaurosis; Metal-binding; Methylation; NAD; Nucleus; Oxidoreductase; Phosphoprotein; Potassium; Proteomics identification; Purine biosynthesis; Reference proteome; Repeat; Retinitis pigmentosa; RNA-binding Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42447.5 Length 395 Aromaticity 0.06 Instability index 33.17 Isoelectric point 5.61 Charge (pH=7) -6.1 2D Binding mode Binding energy (Kcal/mol) -6.76  Molscript Map 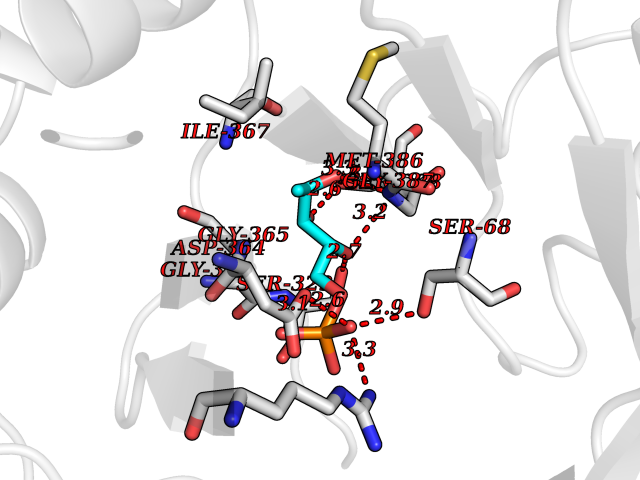 Pymol Map 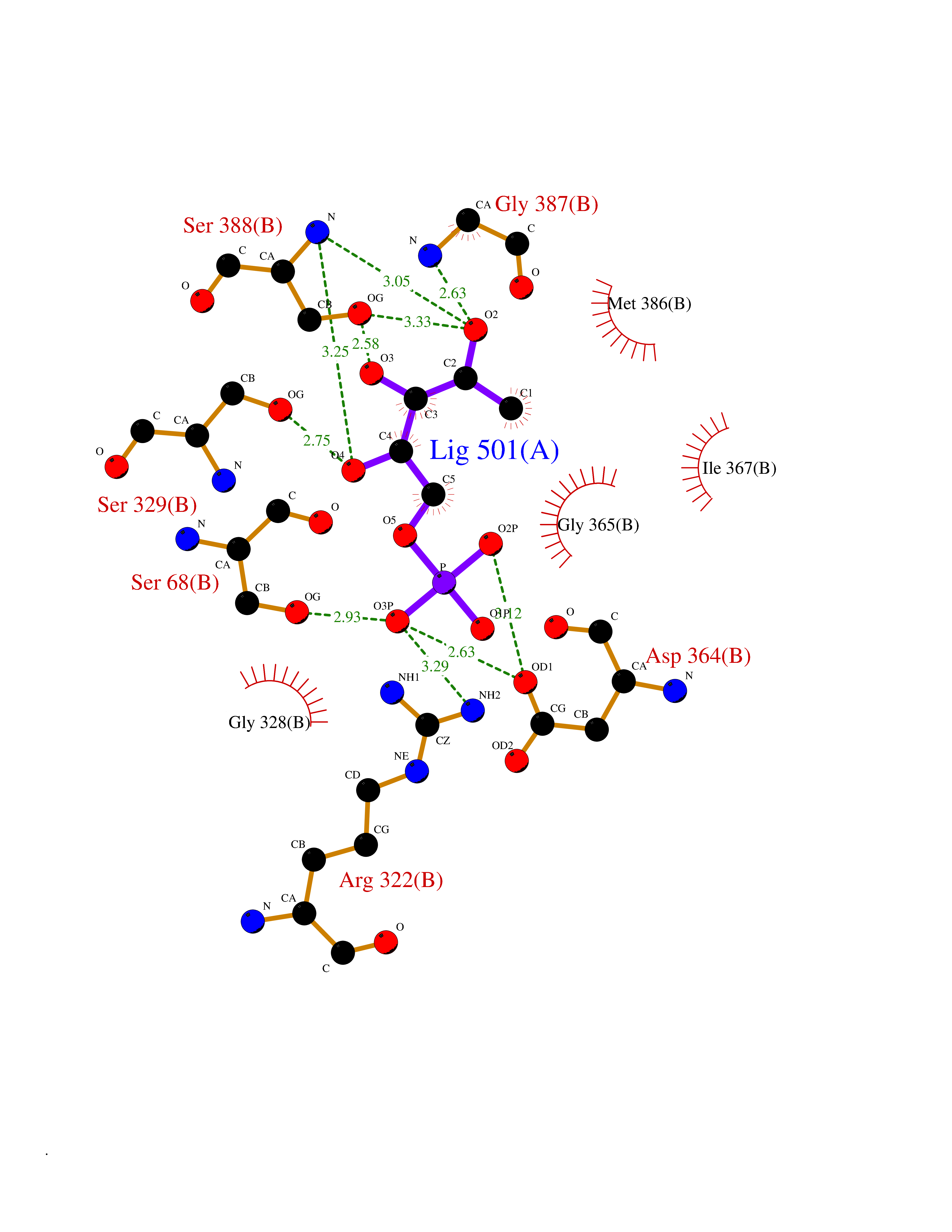 Ligplot Map 3D Binding mode Sequence TGYVPEDGLTAQQLFASADDLTYNDFLILPGFIDFIADEVDLTSALTRKITLKTPLISSPMDTVTEADMAIAMALMGGIGFIHHNCTPEFQANEVRKVKNFEQGFITDPVVLSPGIPITEVGIVTSRDIDPRIELVVAPAGVTLKEANEILQRSKKGKLPIVNDCDELVRTDLKKNRDYPLASKDSQKQLLCGAAVGTREDDKYRLDLLTQAGVDVIVLDSSQGNSVYQIAMVHYIKQKYPHLQVIGGNVVTAAQAKNLIDAGVDGLRVGMGCGSICITQEVMACGRPQGTAVYKVAEYARRFGVPIIADGGIQTVGHVVKALALGASTVMMGSLLAATTEAPGEKGSIQKFVPYLIAGIQHGCQDIGARSLSVLRSMMYSGELKFEKRTMSAQI Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | SPRY domain-containing SOCS box protein 2 (SSB-2) (Gene-rich cluster protein C9) | 3EMW | 6.02 | |
Target general information Gen name SPSB2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms GRCC9;SSB2 Protein family SPSB family Biochemical class NA Function Substrate recognition component of a SCF-like ECS (Elongin BC-CUL2/5-SOCS-box protein) E3 ubiquitin-protein ligase complex which mediates the ubiquitination and subsequent proteasomal degradation of target proteins (PubMed:15601820, PubMed:21199876). Negatively regulates nitric oxide (NO) production and limits cellular toxicity in activated macrophages by mediating the ubiquitination and proteasomal degradation of NOS2 (PubMed:21199876). Acts as a bridge which links NOS2 with the ECS E3 ubiquitin ligase complex components ELOC and CUL5 (PubMed:21199876). {ECO:0000269|PubMed:15601820, ECO:0000269|PubMed:21199876}." Related diseases Intellectual developmental disorder, autosomal recessive 2 (MRT2) [MIM:607417]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT2 patients display mild intellectual disability with a standard IQ ranged from 50 to 70. IQ scores are lower in males than females. Developmental milestones are mildly delayed. There are no dysmorphic or autistic features. {ECO:0000269|PubMed:15557513, ECO:0000269|PubMed:28143899}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with G5E9A7; Q15369; Q53EP0-3; O60333-2; Q16656-4; Q96IZ0; P16284; Q92569; O60260-5; Q99873; Q6P9E2; Q9Y3C5; Q96GM5; P61086; P08670; P09052 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Host-virus interaction; Proteomics identification; Reference proteome; Ubl conjugation pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 22703.1 Length 207 Aromaticity 0.09 Instability index 50.84 Isoelectric point 5.98 Charge (pH=7) -2.79 2D Binding mode Binding energy (Kcal/mol) -6.44  Molscript Map 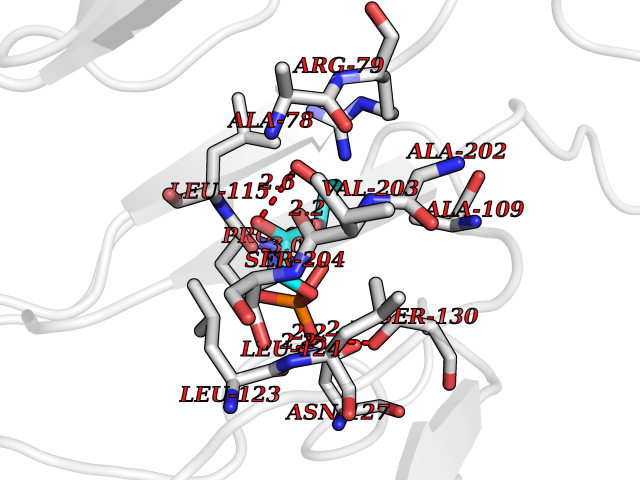 Pymol Map 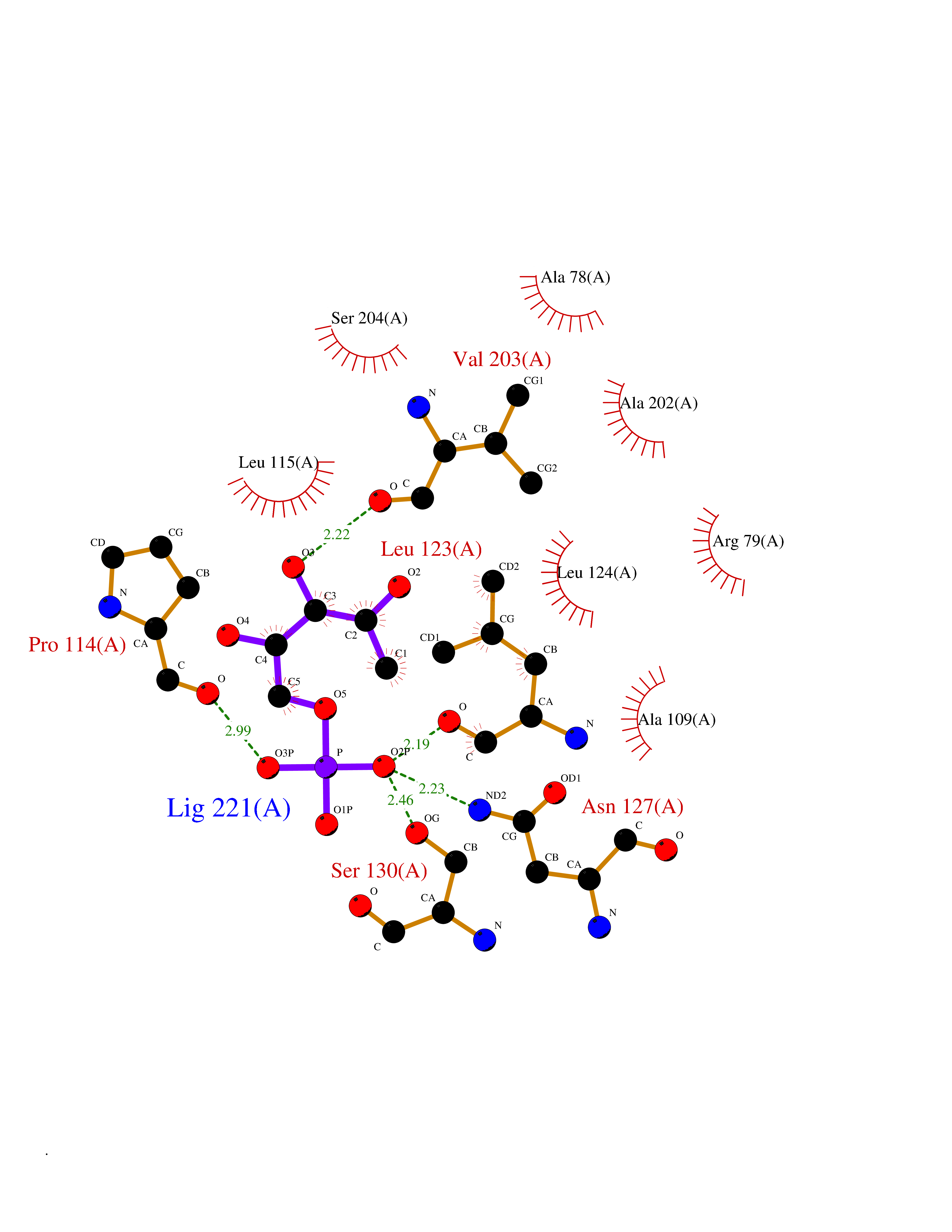 Ligplot Map 3D Binding mode Sequence LYFQSMPEGLEELLSAPPPDLGAQRRHGWNPKDCSENIEVKEGGLYFERRPVAQSTDGARGKRGYSRGLHAWEISWPLEQRGTHAVVGVATALAPLQTDHYAALLGSNSESWGWDIGRGKLYHQSKGPGAPQYPAGTQGEQLEVPERLLVVLDMEEGTLGYAIGGTYLGPAFRGLKGRTLYPAVSAVWGQCQVRIRYLGEDINNNNN Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | "Periplasmic trehalase (EC 3.2.1.28) (Alpha,alpha-trehalase) (Alpha,alpha-trehalose glucohydrolase) (Tre37A)" | 2JG0 | 6.02 | |
Target general information Gen name treA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1186;osmA;b1197 Protein family Glycosyl hydrolase 37 family Biochemical class NA Function Provides the cells with the ability to utilize trehalose at high osmolarity by splitting it into glucose molecules that can subsequently be taken up by the phosphotransferase-mediated uptake system. Related diseases SRC kinase activity has been shown to be increased in several tumor tissues and tumor cell lines such as colon carcinoma cells. {ECO:0000269|PubMed:2498394, ECO:0000269|PubMed:3093483}.; DISEASE: Thrombocytopenia 6 (THC6) [MIM:616937]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC6 is an autosomal dominant form. Affected individuals may also have bone abnormalities and an increased risk for myelofibrosis. {ECO:0000269|PubMed:26936507}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.2.1.28 Uniprot keywords 3D-structure; Direct protein sequencing; Glycosidase; Hydrolase; Periplasm; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 57508.9 Length 507 Aromaticity 0.11 Instability index 48.32 Isoelectric point 5.48 Charge (pH=7) -10.13 2D Binding mode Binding energy (Kcal/mol) -6.86  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence PQPPDILLGPLFNDVQNAKLFPDQKTFADAVPNSDPLMILADYRMQQNQSGFDLRHFVNVNFTLPKYVPPEGQSLREHIDGLWPVLTRSTENTEKWDSLLPLPEPYVVPGGRFREVYYWDSYFTMLGLAESGHWDKVADMVANFAHEIDTYGHIPNGNRSYYLSRSQPPFFALMVELLAQHEGDAALKQYLPQMQKEYAYWMDGVENLQAGQQEKRVVKLQDGTLLNRYWDDRDTPRPESWVEDIATAKSNPNRPATEIYRDLRSAAASGWDFSSRWMDNPQQLNTLRTTSIVPVDLNSLMFKMEKILARASKAAGDNAMANQYETLANARQKGIEKYLWNDQQGWYADYDLKSHKVRNQLTAAALFPLYVNAAAKDRANKMATATKTHLLQPGGLNTTSVKSGQQWDAPNGWAPLQWVATEGLQNYGQKEVAMDISWHFLTNVQHTYDREKKLVEKYDVSTTGTGGGGGEYPLQDGFGWTNGVTLKMLDLICPKEQPCDNVPATRP Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Phosphodiesterase 10A (PDE10) | 2OUR | 6.02 | |
Target general information Gen name PDE10A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cAMP and cAMPinhibited cGMP 3',5'cyclic phosphodiesterase 10A; cAMP and cAMP-inhibited cGMP 3',5'-cyclic phosphodiesterase 10A Protein family Cyclic nucleotide phosphodiesterase family Biochemical class Phosphoric diester hydrolase Function Can hydrolyze both cAMP and cGMP, but has higher affinity for cAMP and is more efficient with cAMP as substrate. May play a critical role in regulating cAMP and cGMP levels in the striatum, a region of the brain that contributes to the control of movement and cognition. Plays a role in signal transduction by regulating the intracellular concentration of cyclic nucleotides. Related diseases Dyskinesia, limb and orofacial, infantile-onset (IOLOD) [MIM:616921]: An autosomal recessive, early-onset hyperkinetic movement disorder characterized by axial hypotonia, dyskinesia of the limbs and trunk, orofacial dyskinesia, drooling, and dysarthria. The severity of the hyperkinesis is variable. {ECO:0000269|PubMed:27058446}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Striatal degeneration, autosomal dominant 2 (ADSD2) [MIM:616922]: An autosomal dominant disorder characterized by striatal degeneration and dysfunction of basal ganglia, resulting in hyperkinesis. {ECO:0000269|PubMed:27058447}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08384; DB08386; DB08383; DB08389; DB00201; DB00975; DB08387; DB01113; DB08391; DB08811; DB09283; DB08814 Interacts with NA EC number EC 3.1.4.17 Uniprot keywords 3D-structure; Allosteric enzyme; Alternative initiation; Alternative splicing; cAMP; cAMP-binding; cGMP; cGMP-binding; Cytoplasm; Disease variant; Hydrolase; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat Protein physicochemical properties Chain ID A,B Molecular weight (Da) 37469.9 Length 324 Aromaticity 0.1 Instability index 48.52 Isoelectric point 6.28 Charge (pH=7) -4.08 2D Binding mode Binding energy (Kcal/mol) -6.67  Molscript Map  Pymol Map 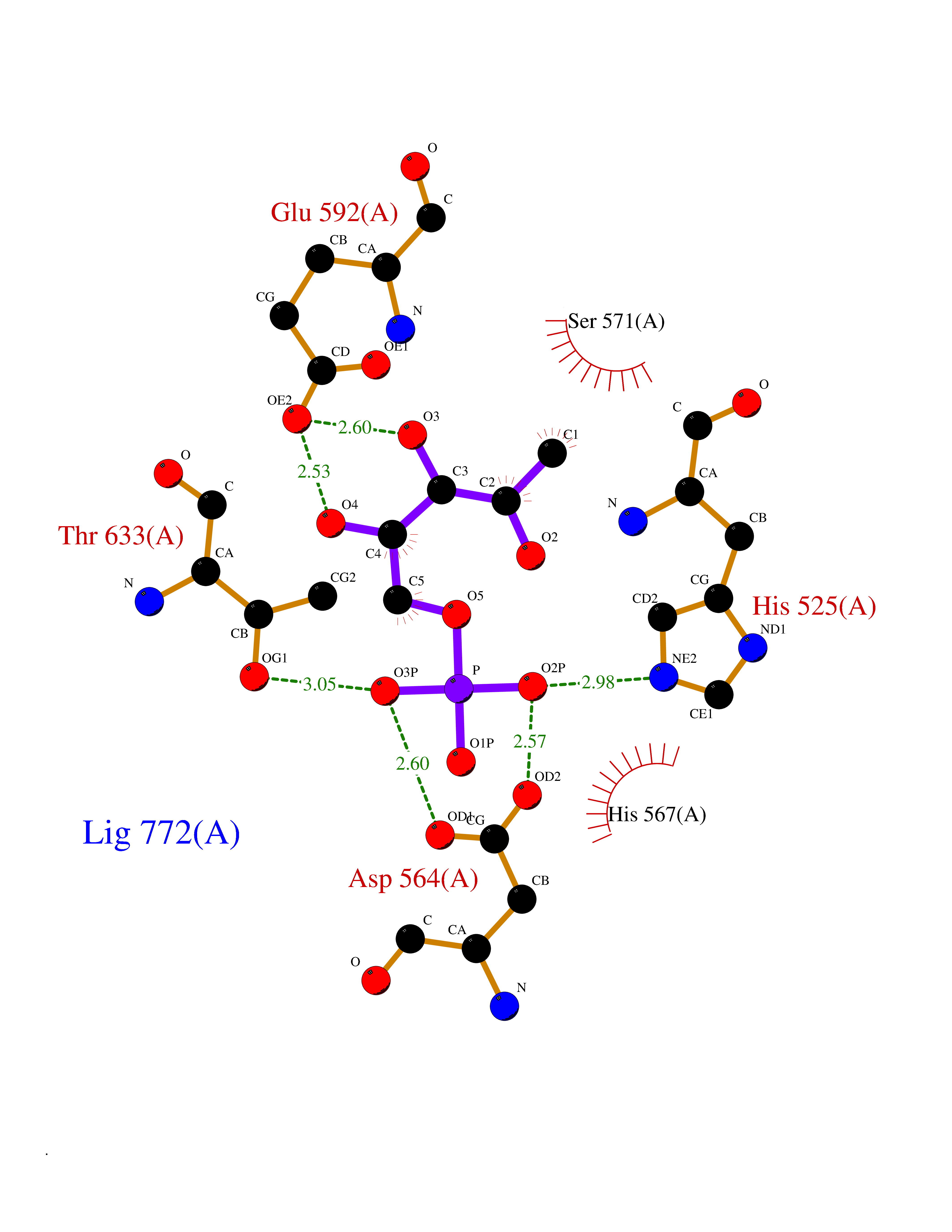 Ligplot Map 3D Binding mode Sequence HMSICTSEEWQGLMQFTLPVRLCKEIELFHFDIGPFENMWPGIFVYMVHRSCGTSCFELEKLCRFIMSVKKNYRRVPYHNWKHAVTVAHCMYAILQNNHTLFTDLERKGLLIACLCHDLDHRGFSNSYLQKFDHPLAALYSTSTMEQHHFSQTVSILQLEGHNIFSTLSSSEYEQVLEIIRKAIIATDLALYFGNRKQLEEMYQTGSLNLNNQSHRDRVIGLMMTACALCSVTKLWPVTKLTANDIYAEFWAEGDEMKKLGIQPIPMMDRDKKDEVPQGQLGFYNAVAIPCYTTLTQILPPTEPLLKACRDNLSQWEKVIRGEE Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Aspartyl aminopeptidase (DNPEP) | 4DYO | 6.02 | |
Target general information Gen name DNPEP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms DAP; ASPEP Protein family Peptidase M18 family Biochemical class Peptidase Function Likely to play an important role in intracellular protein and peptide metabolism. Aminopeptidase with specificity towards an acidic amino acid at the N-terminus. Related diseases Cohen-Gibson syndrome (COGIS) [MIM:617561]: An autosomal dominant overgrowth disorder characterized by accelerated osseous maturation, advanced bone age, skeletal abnormalities including flaring of the metaphyses of the long bones, large hands with long fingers and camptodactyly, scoliosis, cervical spine anomalies, dysmorphic facial features, and variable intellectual disability. {ECO:0000269|PubMed:25787343, ECO:0000269|PubMed:27193220, ECO:0000269|PubMed:27868325, ECO:0000269|PubMed:28229514, ECO:0000269|PubMed:28475857}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00142 Interacts with Q9ULA0; Q8TBB1; Q00013; Q9UPN6 EC number EC 3.4.11.21 Uniprot keywords 3D-structure; Acetylation; Alternative initiation; Aminopeptidase; Cytoplasm; Hydrolase; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 50574.6 Length 457 Aromaticity 0.07 Instability index 55.73 Isoelectric point 7.73 Charge (pH=7) 2.3 2D Binding mode Binding energy (Kcal/mol) -6.88 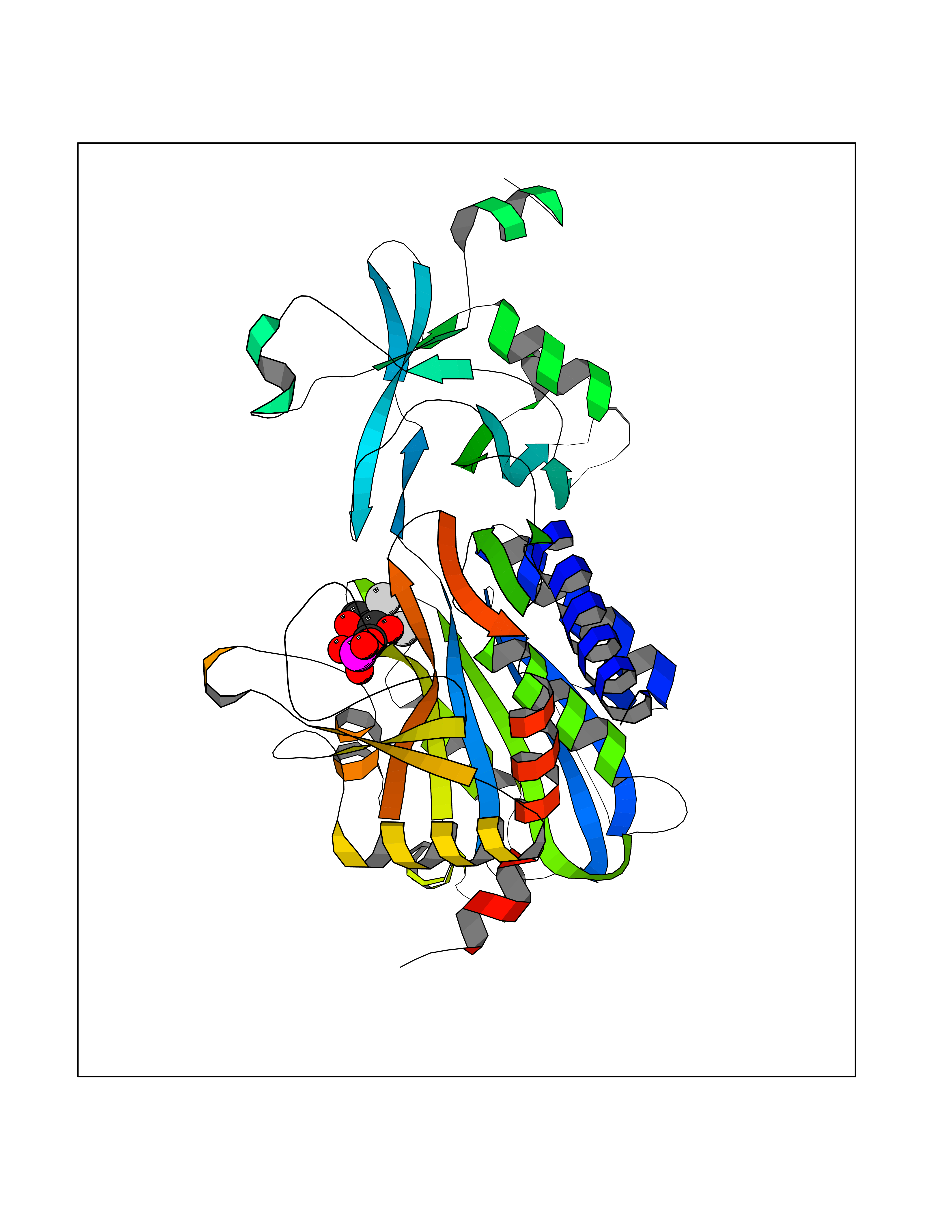 Molscript Map  Pymol Map 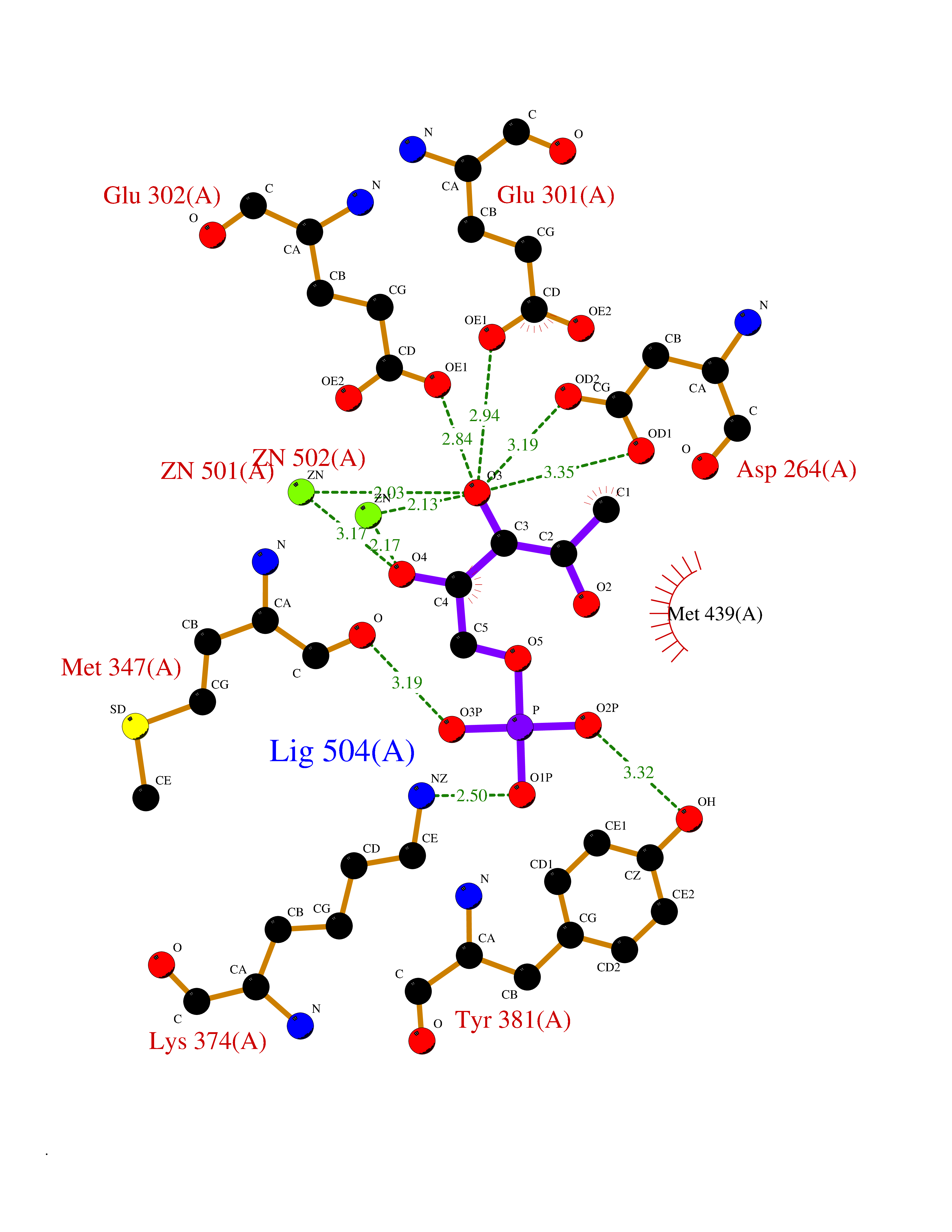 Ligplot Map 3D Binding mode Sequence GKARKEAVQTAAKELLKFVNRSPSPFHAVAECRNRLLQAGFSELKETEKWNIKPESKYFMTRNSSTIIAFAVGGQYVPGNGFSLIGAHTDSPCLRVKRRSRRSQVGFQQVGVETYGGGIWSTWFDRDLTLAGRVIVKCPTSGRLEQQLVHVERPILRIPHLAIHLQRNINENFGPNTEMHLVPILATAIQEELEKGTERHHSVLMSLLCAHLGLSPKDIVEMELCLADTQPAVLGGAYDEFIFAPRLDNLHSCFCALQALIDSCAGPGSLATEPHVRMVTLYDNEEVGSESAQGAQSLLTELVLRRISASCQHPTAFEEAIPKSFMISADMAHAVHPNYLDKHEENHRPLFHKGPVIKVNSKQRYASNAVSEALIREVANKVKVPLQDLMVRNDTPCGTTIGPILASRLGLRVLDLGSPQLAMHSIREMACTTGVLQTLTLFKGFFELFPSLAENLY Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | S-nitrosoglutathione reductase (CBR1) | 3BHJ | 6.01 | |
Target general information Gen name CBR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ProstaglandinE(2) 9reductase; Prostaglandin 9ketoreductase; NADPHdependent carbonyl reductase 1; Carbonyl reductase [NADPH] 1; CBR1; 15hydroxyprostaglandin dehydrogenase [NADP(+)] Protein family Short-chain dehydrogenases/reductases (SDR) family Biochemical class Short-chain dehydrogenases reductase Function NADPH-dependent reductase with broad substratespecificity. Catalyzes the reduction of a wide variety of carbonyl compounds including quinones, prostaglandins, menadione, plus various xenobiotics. Catalyzes the reduction of the antitumor anthracyclines doxorubicin and daunorubicin to the cardiotoxic compounds doxorubicinol and daunorubicinol. Can convert prostaglandin E2 to prostaglandin F2-alpha. Can bind glutathione, which explains its higher affinity for glutathione-conjugated substrates. Catalyzes the reduction of S-nitrosoglutathione. Related diseases Growth hormone deficiency, isolated, 4 (IGHD4) [MIM:618157]: An autosomal recessive deficiency of growth hormone leading to early and severe growth failure and short stature. Patients have low but detectable levels of growth hormone, significantly retarded bone age, and a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:10084571, ECO:0000269|PubMed:11232012, ECO:0000269|PubMed:12534354, ECO:0000269|PubMed:8528260, ECO:0000269|PubMed:9467553}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03556; DB04463; DB00414; DB06263; DB11672; DB14635; DB00694; DB12161; DB00997; DB01039; DB00502; DB03394; DB09212; DB01046; DB00776; DB04216; DB02709; DB01698; DB05197; DB04844 Interacts with O75828 EC number EC 1.1.1.184 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Lipid metabolism; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 30156.3 Length 275 Aromaticity 0.06 Instability index 43.19 Isoelectric point 8.55 Charge (pH=7) 2.98 2D Binding mode Binding energy (Kcal/mol) -6.54 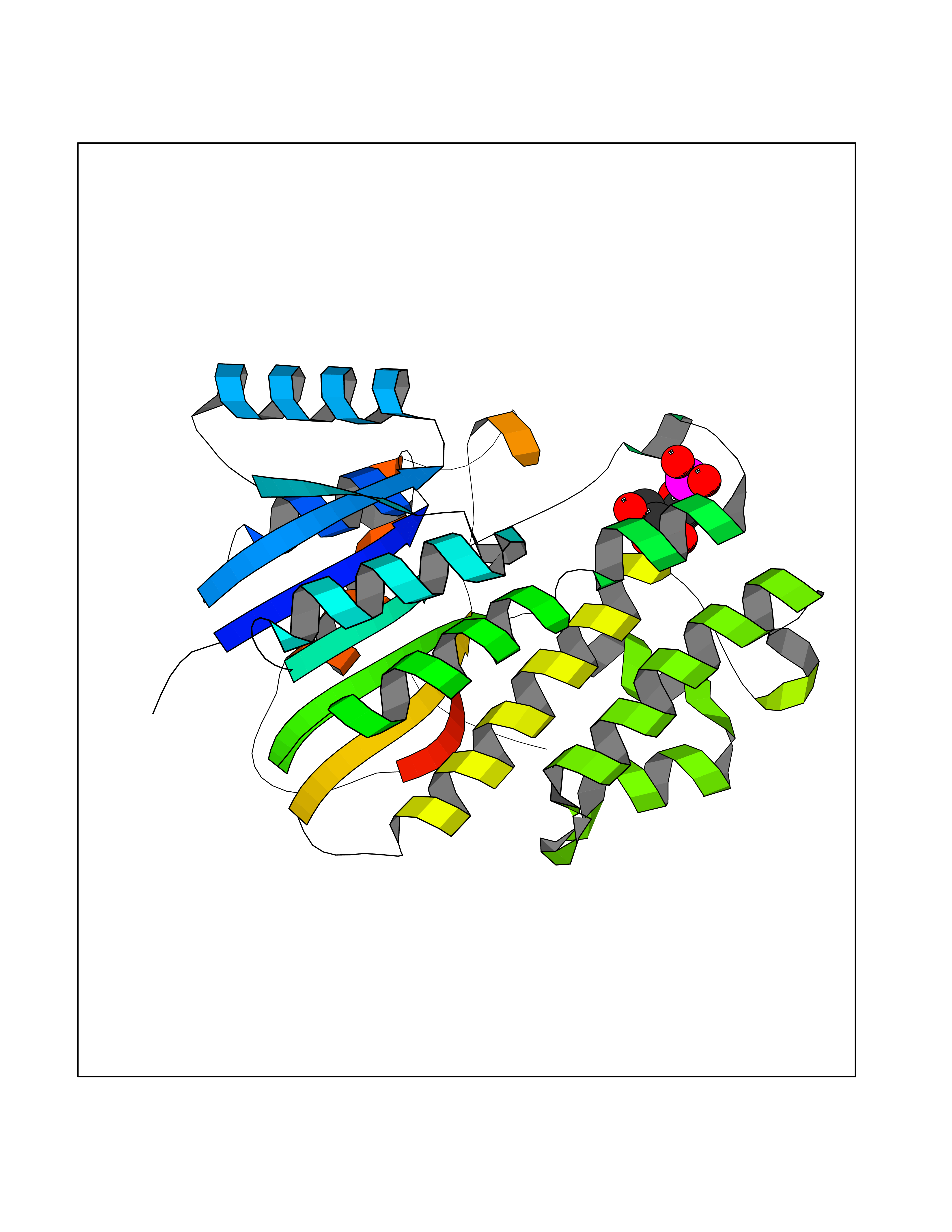 Molscript Map 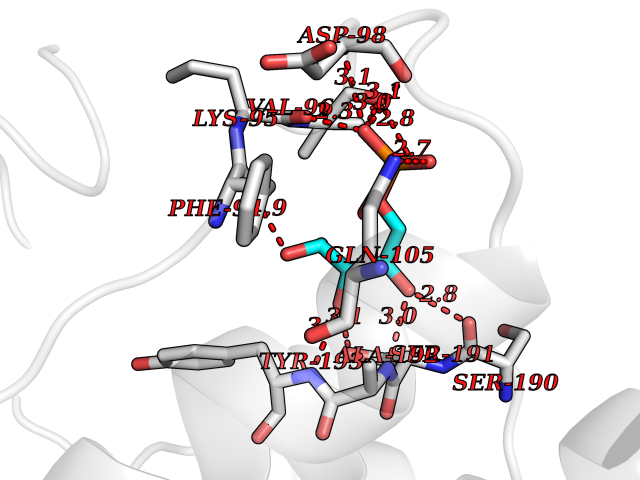 Pymol Map 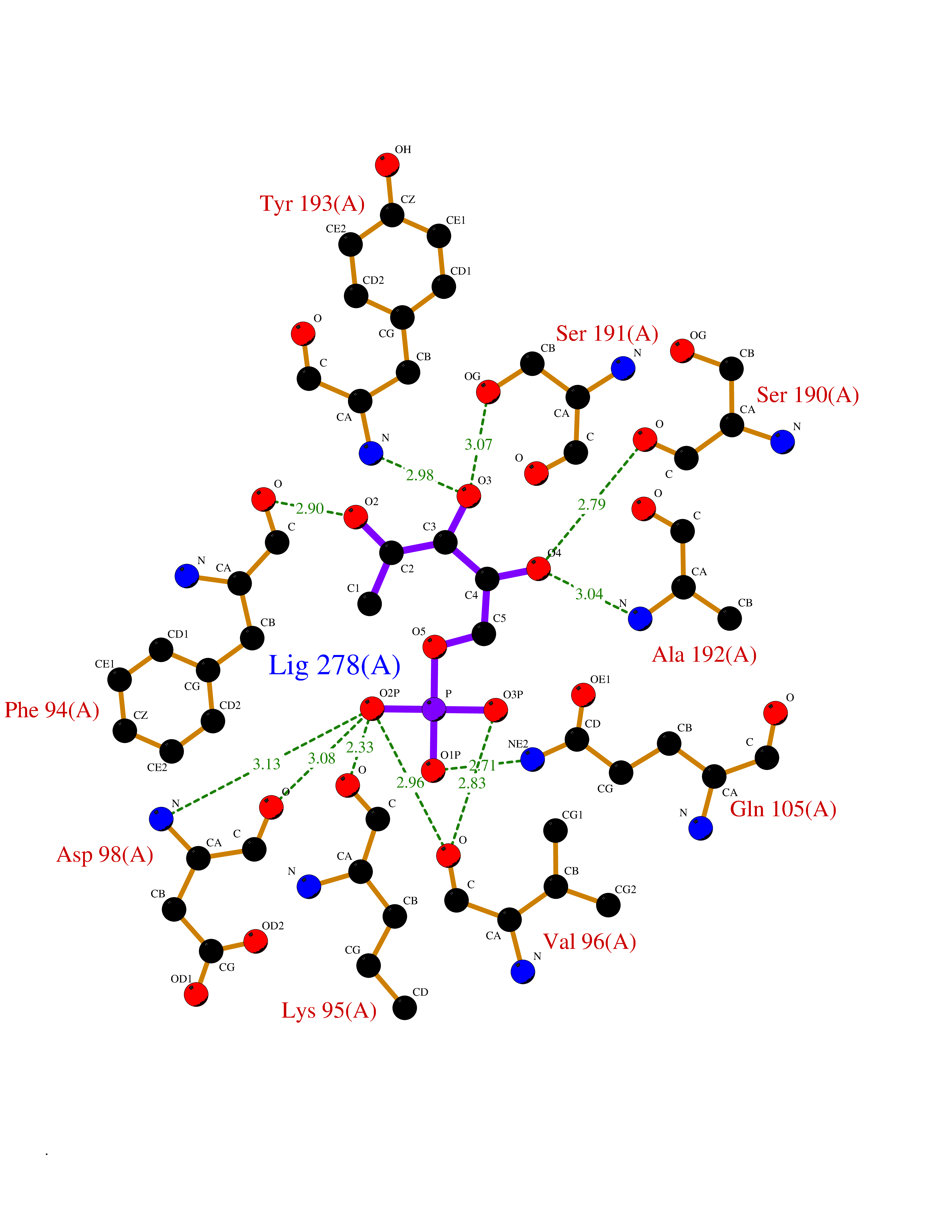 Ligplot Map 3D Binding mode Sequence SGIHVALVTGGNKGIGLAIVRDLCRLFSGDVVLTARDVTRGQAAVQQLQAEGLSPRFHQLDIDDLQSIRALRDFLRKEYGGLDVLVNNAGIAFKVADPTPFHIQAEVTMKTNFFGTRDVCTELLPLIKPQGRVVNVSSIMSVRALKSCSPELQQKFRSETITEEELVGLMNKFVEDTKKGVHQKEGWPSSAYGVTKIGVTVLSRIHARKLSEQRKGDKILLNACCPGWVRTDMAGPKATKSPEEGAETPVYLALLPPDAEGPHGQFVSEKRVEQW Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Pyruvate dehydrogenase E1 component subunit beta, mitochondrial | 2OZL | 6.01 | |
Target general information Gen name PDHB Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PHE1B Protein family NA Biochemical class Oxidoreductase Function Pyruvate dehydrogenase (acetyl-transferring) activity.Pyruvate dehydrogenase activity. Related diseases Pyruvate dehydrogenase E1-beta deficiency (PDHBD) [MIM:614111]: An enzymatic defect causing primary lactic acidosis in children. It is associated with a broad clinical spectrum ranging from fatal lactic acidosis in the newborn to chronic neurologic dysfunction with structural abnormalities in the central nervous system without systemic acidosis. {ECO:0000269|PubMed:15138885}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00157; DB00119 Interacts with P10515; P08559; P08559-1 EC number 1.2.4.1 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Carbohydrate metabolism; Direct protein sequencing; Disease variant; Glucose metabolism; Metal-binding; Mitochondrion; Oxidoreductase; Phosphoprotein; Potassium; Proteomics identification; Pyruvate; Reference proteome; Thiamine pyrophosphate; Transit peptide; Tricarboxylic acid cycle Protein physicochemical properties Chain ID B,D Molecular weight (Da) 36554.4 Length 329 Aromaticity 0.09 Instability index 29.78 Isoelectric point 6.66 Charge (pH=7) -0.99 2D Binding mode Binding energy (Kcal/mol) -7.12 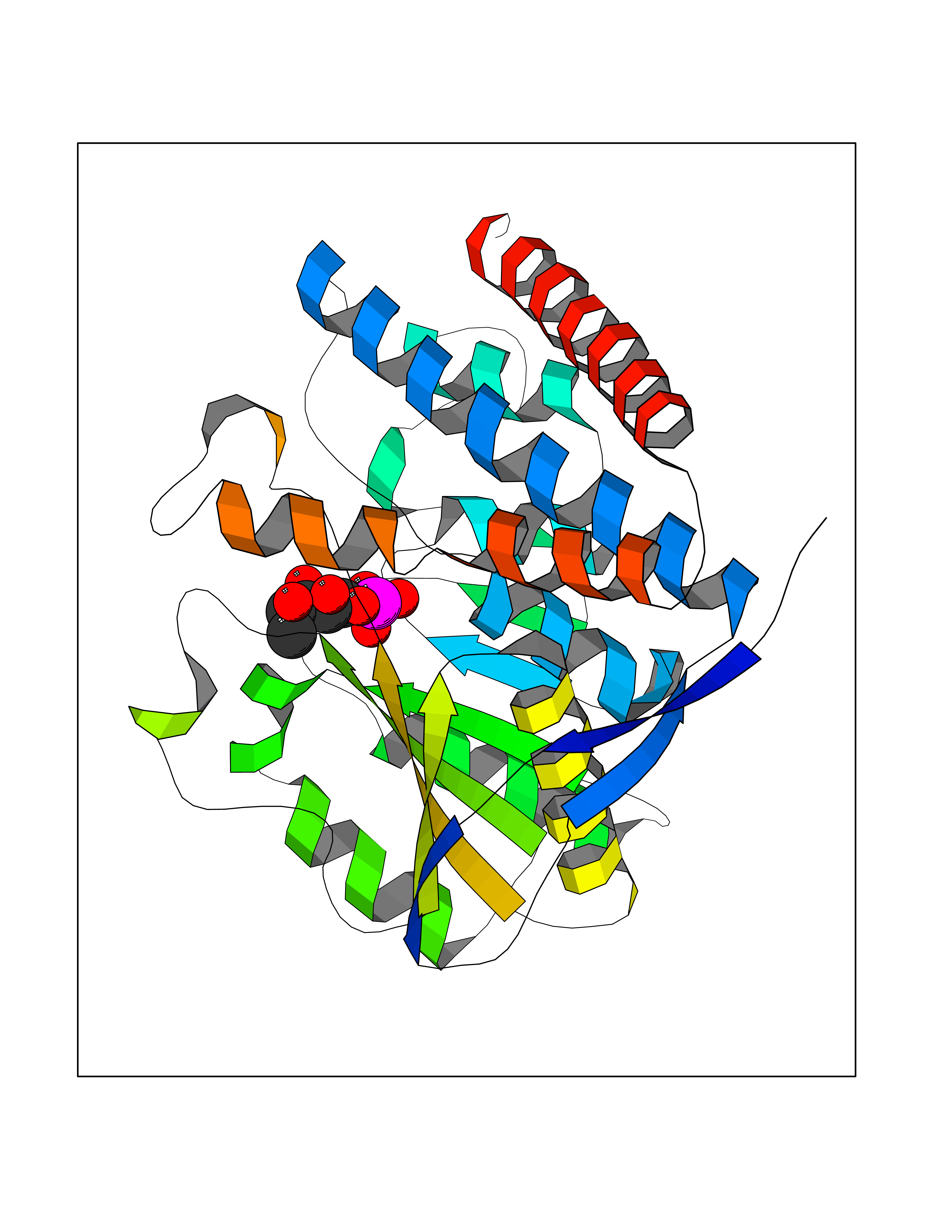 Molscript Map 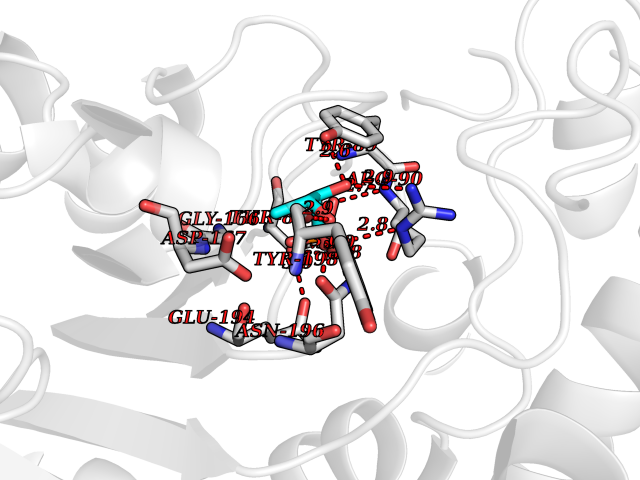 Pymol Map 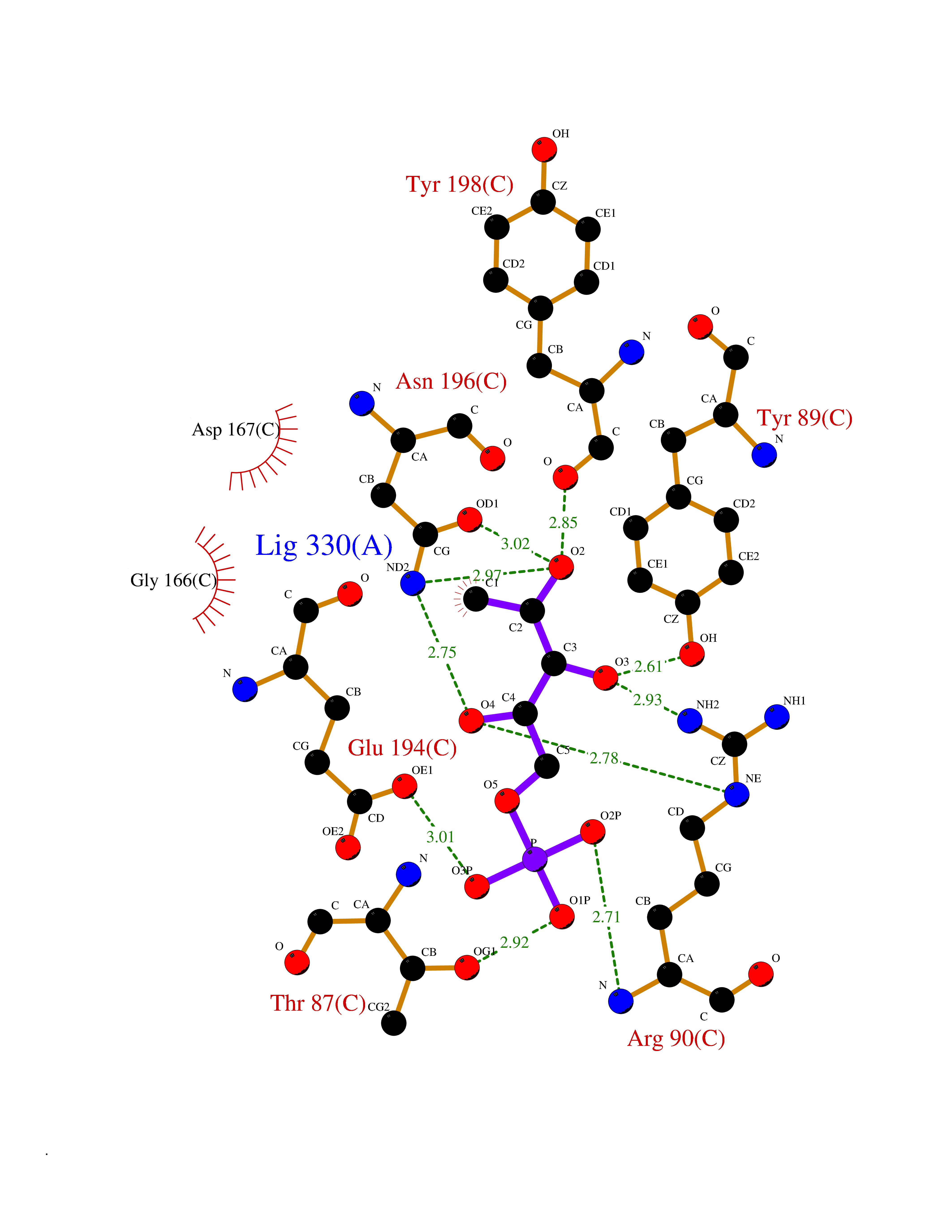 Ligplot Map 3D Binding mode Sequence SFANDATFEIKKCDLHRLEEGPPVTTVLTREDGLKYYRMMQTVRRMELKADQLYKQKIIRGFCHLCDGQEACCVGLEAGINPTDHLITAYRAHGFTFTRGLSVREILAELTGRKGGCAKGKGGSMHMYAKNFYGGNGIVGAQVPLGAGIALACKYNGKDEVCLTLYGDGAANQGQIFEAYNMAALWKLPCIFICENNRYGMGTSVERAAASTDYYKRGDFIPGLRVDGMDILCVREATRFAAAYCRSGKGPILMELQTYRYHGHEMSDPGVSYRTREEIQEVRSKSDPIMLLKDRMVNSNLASVEELKEIDVEVRKEIEDAAQFATADP Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Fatty acid synthase (FASN) | 3TJM | 6.01 | |
Target general information Gen name FASN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Yeast fatty acid synthase; Fatty-acyl-CoA synthase; Fatty acyl-CoA synthetase enzyme; FAS Protein family NA Biochemical class Acyltransferase Function Fatty acid synthetase catalyzes the formation of long-chain fatty acids from acetyl-CoA, malonyl-CoA and NADPH. This multifunctional protein has 7 catalytic activities as an acyl carrier protein. Related diseases Glycine encephalopathy 2 (GCE2) [MIM:620398]: A form of glycine encephalopathy, a metabolic disorder characterized by a high concentration of glycine in the body fluids. Affected individuals typically have severe neurological symptoms, including seizure, lethargy, and muscular hypotonia soon after birth. Most of them die within the neonatal period. Atypical cases have later disease onset and less severely affected psychomotor development. {ECO:0000269|PubMed:10873393, ECO:0000269|PubMed:11286506, ECO:0000269|PubMed:16051266, ECO:0000269|PubMed:26371980, ECO:0000269|PubMed:28244183, ECO:0000269|PubMed:8005589, ECO:0000269|PubMed:9600239, ECO:0000269|PubMed:9621520}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01034; DB01083 Interacts with Q15848; Q16665; P42858; Q8IV20; Q8TBB1; PRO_0000045603 [Q99IB8] EC number EC 2.3.1.85 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Isopeptide bond; Lipid biosynthesis; Lipid metabolism; Lyase; Multifunctional enzyme; NAD; NADP; Oxidoreductase; Phosphopantetheine; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; S-nitrosylation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30174.9 Length 275 Aromaticity 0.09 Instability index 43.28 Isoelectric point 5.92 Charge (pH=7) -5.4 2D Binding mode Binding energy (Kcal/mol) -6.61 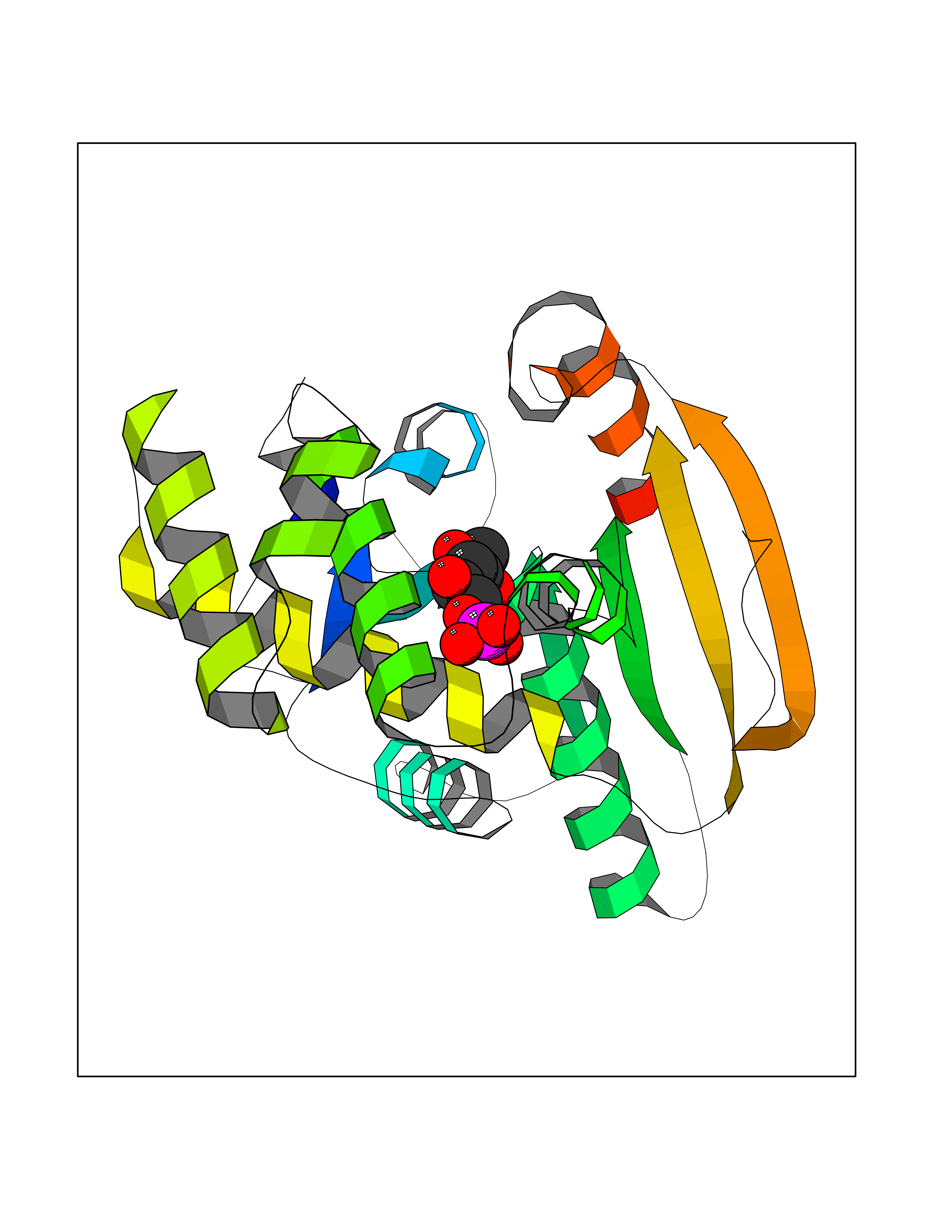 Molscript Map 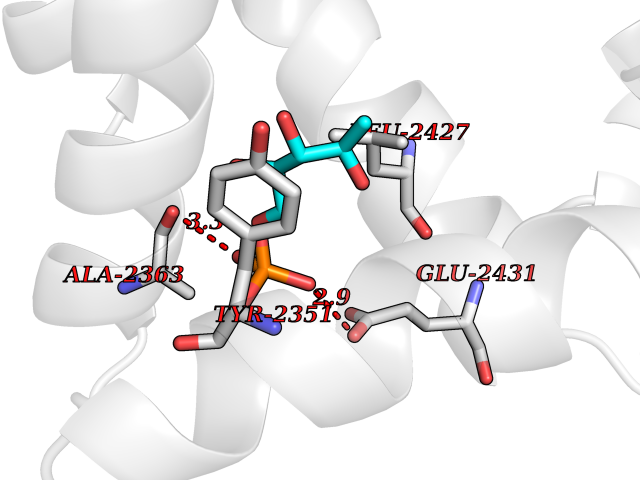 Pymol Map 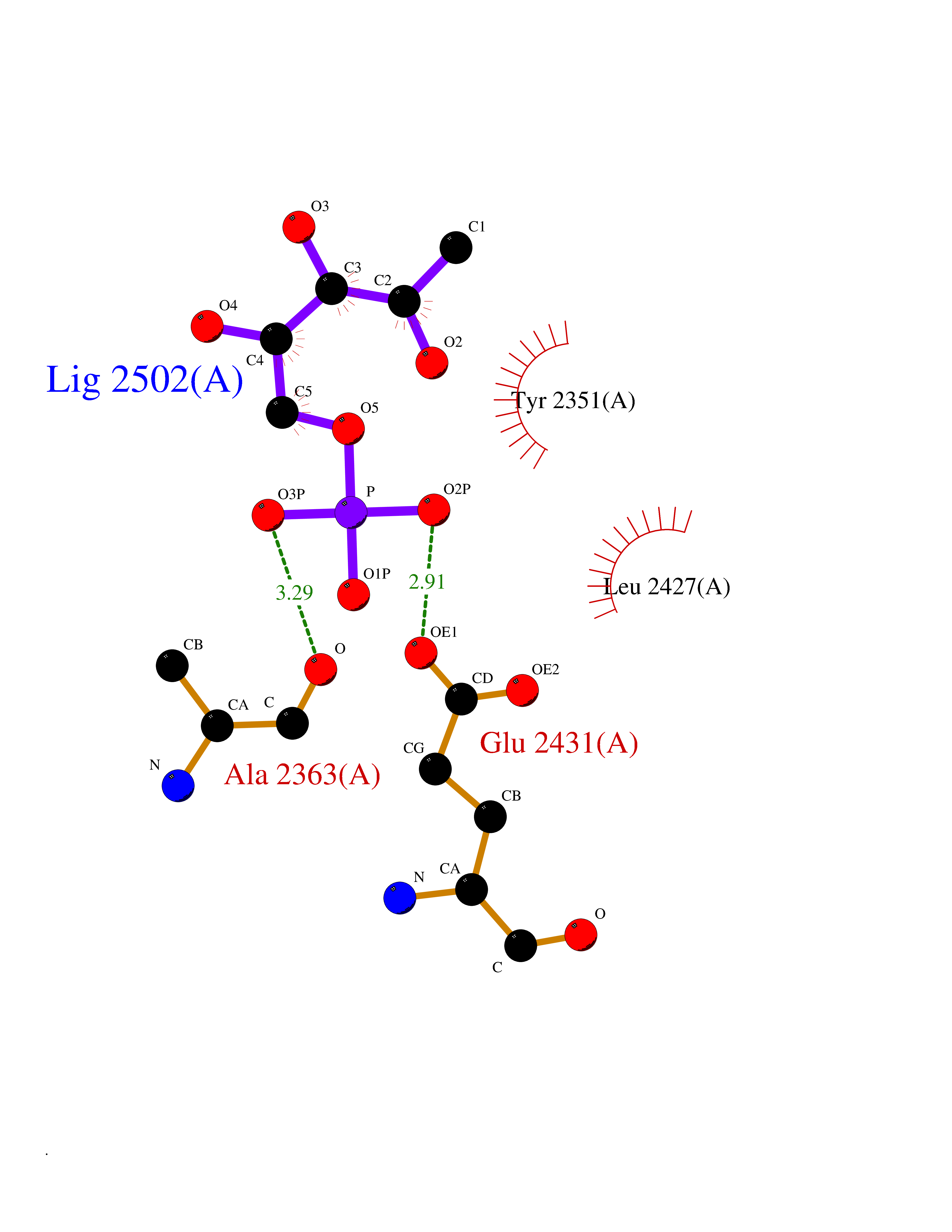 Ligplot Map 3D Binding mode Sequence NLRSLLVNPEGPTLMRLNSVQSSERPLFLVHPIEGSTTVFHSLASRLSIPTYGLQCTRAAPLDSIHSLAAYYIDCIRQVQPEGPYRVAGYSYGACVAFEMCSQLQAQQSPAPTHNSLFLFDGSPTYVLAYTGSYRAKLTPGCEAEAETEAICFFVQQFTDMEHNRVLEALLPLKGLEERVAAAVDLIIKSHQGLDRQELSFAARSFYYKLRAAEQYTPKAKYHGNVMLLRAAAGADYNLSQVCDGKVSVHVIEGDHATLLEGSGLESIISIIHSS Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Cholesterol oxidase | 4REK | 6.01 | |
Target general information Gen name choA Organism Streptomyces sp. (strain SA-COO) Uniprot ID TTD ID NA Synonyms NA Protein family GMC oxidoreductase family Biochemical class Oxidoreductase Function Cholesterol oxidase activity.Flavin adenine dinucleotide binding.Steroid delta-isomerase activity. Related diseases Bothnia retinal dystrophy (BRD) [MIM:607475]: A type of retinitis punctata albescens. Affected individuals show night blindness from early childhood with features consistent with retinitis punctata albescens and macular degeneration. {ECO:0000269|PubMed:10102298}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rod-cone dystrophy Newfoundland (NFRCD) [MIM:607476]: A rod-cone dystrophy reminiscent of retinitis punctata albescens but with a substantially lower age at onset and more-rapid and distinctive progression. Rod-cone dystrophies results from initial loss of rod photoreceptors, later followed by cone photoreceptors loss. {ECO:0000269|PubMed:11868161}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Retinitis punctata albescens (RPA) [MIM:136880]: A form of fleck retina disease characterized by aggregation of white flecks posteriorly in the retina, causing night blindness and delayed dark adaptation. It differs from fundus albipunctatus in being progressive and evolving to generalized atrophy of the retina. {ECO:0000269|PubMed:10102299, ECO:0000269|PubMed:11453974, ECO:0000269|PubMed:9326942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147; DB02332 Interacts with NA EC number 1.1.3.6; 5.3.3.1 Uniprot keywords 3D-structure; Cholesterol metabolism; Direct protein sequencing; FAD; Flavoprotein; Isomerase; Lipid metabolism; Oxidoreductase; Secreted; Signal; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54367.8 Length 498 Aromaticity 0.1 Instability index 30.62 Isoelectric point 6.69 Charge (pH=7) -0.71 2D Binding mode Binding energy (Kcal/mol) -6.7  Molscript Map 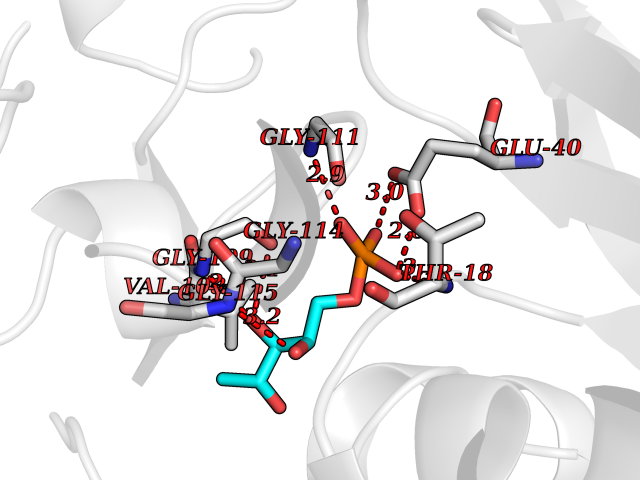 Pymol Map 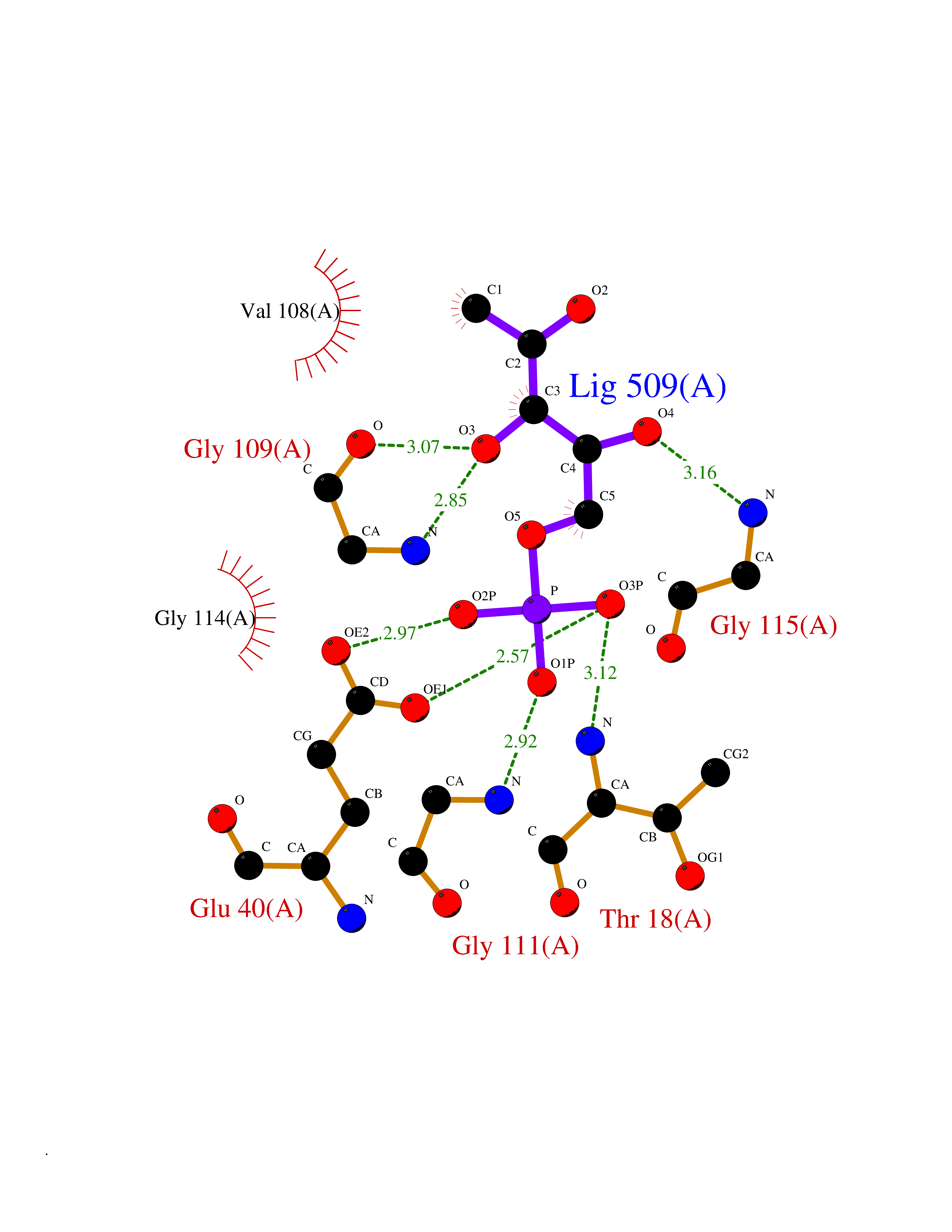 Ligplot Map 3D Binding mode Sequence GYVPAVVIGTGYGAAVSALRLGEAGVQTLMLEMGQLWNQPGPDGNIFCGMLNPDKRSSWFKNRTEAPLGSFLWLDVVNRNIDPYAGVLDRVNYDQMSVYVGRGVGGGSLVNGGMAVEPKRSYFEEILPRVDSSEMYDRYFPRANSMLRVNHIDTKWFEDTEWYKFARVSREQAGKAGLGTVFVPNVYDFGYMQREAAGEVPKSALATEVIYGNNHGKQSLDKTYLAAALGTGKVTIQTLHQVKTIRQTKDGGYALTVEQKDTDGKLLATKEISCRYLFLGAGSLGSTELLVRARDTGTLPNLNSEVGAGWGPNGNIMTARANHMWNPTGAHQSSIPALGIDAWDNSDSSVFAEIAPMPAGLETWVSLYLAITKNPQRGTFVYDAATDRAKLNWTRDQNAPAVNAAKALFDRINKANGTIYRYDLFGTQLKAFADDFCYHPLGGCVLGKATDDYGRVAGYKNLYVTDGSLIPGSVGVNPFVTITALAERNVERIIKQDV Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Ribonucleoside-diphosphate reductase subunit M2 B | 4DJN | 6.00 | |
Target general information Gen name RRM2B Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms P53R2 Protein family Ribonucleoside diphosphate reductase small chain family Biochemical class Oxidoreductase Function Metal ion binding.Ribonucleoside-diphosphate reductase activity, thioredoxin disulfide as acceptor. Related diseases Mitochondrial DNA depletion syndrome 8A (MTDPS8A) [MIM:612075]: A disorder due to mitochondrial dysfunction characterized by various combinations of neonatal hypotonia, neurological deterioration, respiratory distress, lactic acidosis, and renal tubulopathy. {ECO:0000269|PubMed:17486094, ECO:0000269|PubMed:18504129}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mitochondrial DNA depletion syndrome 8B (MTDPS8B) [MIM:612075]: A disease due to mitochondrial dysfunction and characterized by ophthalmoplegia, ptosis, gastrointestinal dysmotility, cachexia, peripheral neuropathy. {ECO:0000269|PubMed:19667227}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Progressive external ophthalmoplegia with mitochondrial DNA deletions, autosomal dominant, 5 (PEOA5) [MIM:613077]: A disorder characterized by progressive weakness of ocular muscles and levator muscle of the upper eyelid. In a minority of cases, it is associated with skeletal myopathy, which predominantly involves axial or proximal muscles and which causes abnormal fatigability and even permanent muscle weakness. Ragged-red fibers and atrophy are found on muscle biopsy. A large proportion of chronic ophthalmoplegias are associated with other symptoms, leading to a multisystemic pattern of this disease. Additional symptoms are variable, and may include cataracts, hearing loss, sensory axonal neuropathy, ataxia, depression, hypogonadism, and parkinsonism. {ECO:0000269|PubMed:19664747}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Rod-cone dystrophy, sensorineural deafness, and Fanconi-type renal dysfunction (RCDFRD) [MIM:268315]: An autosomal recessive disease characterized by visual impairment due to rod-cone dystrophy, sensorineural hearing loss, and Fanconi-type renal dysfunction resulting in rickets-like skeletal changes. Death may occur in childhood or young adulthood due to renal failure. Disease onset is before age 5 years. {ECO:0000269|PubMed:32827185}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00242 Interacts with Q13315; Q00987; O43929; Q9H4P4; Q7LG56; Q00987 EC number 1.17.4.1 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Deafness; Deoxyribonucleotide synthesis; Disease variant; DNA damage; DNA repair; Iron; Metal-binding; Neuropathy; Nucleus; Oxidoreductase; Primary mitochondrial disease; Progressive external ophthalmoplegia; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 32464.8 Length 287 Aromaticity 0.15 Instability index 44.18 Isoelectric point 5.48 Charge (pH=7) -5.86 2D Binding mode Binding energy (Kcal/mol) -6.17 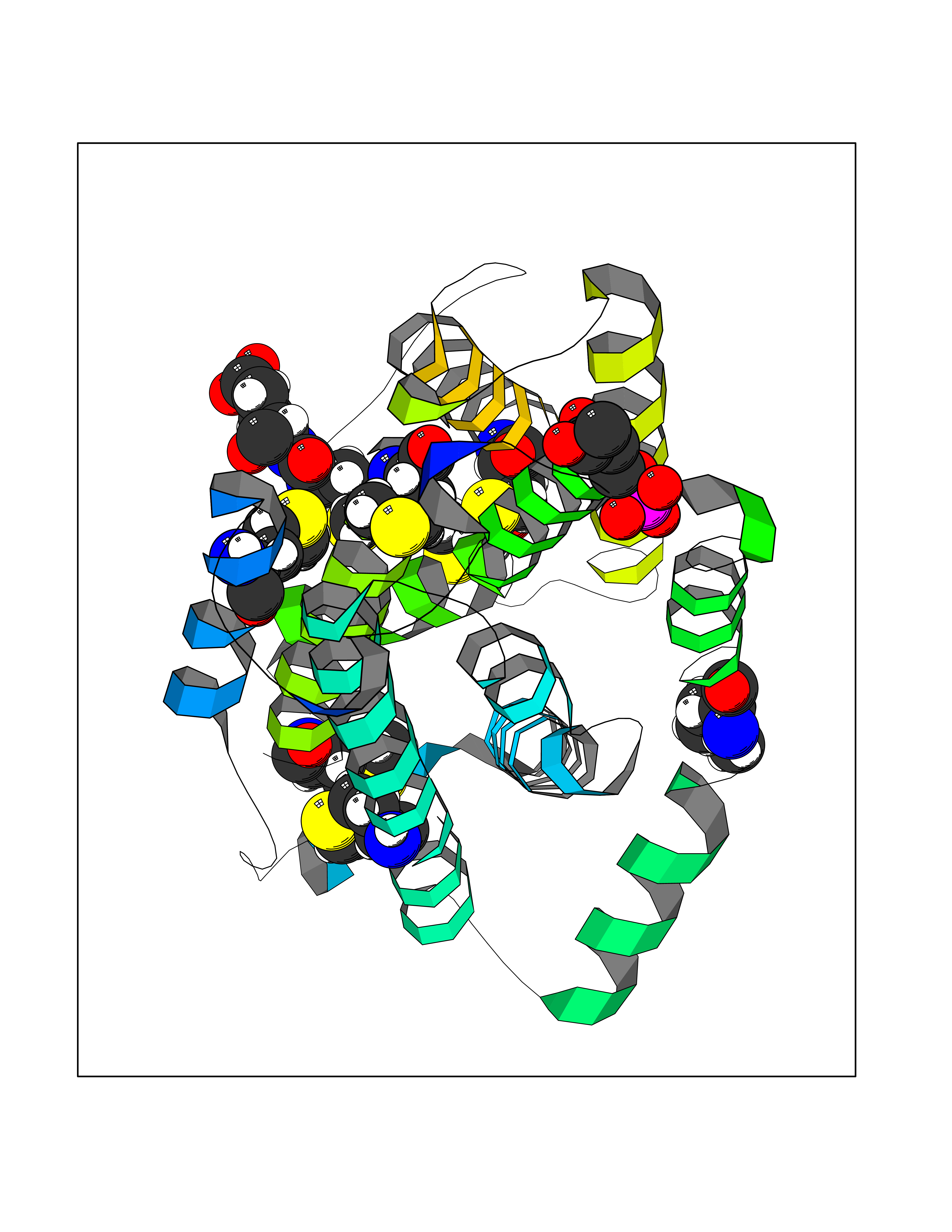 Molscript Map 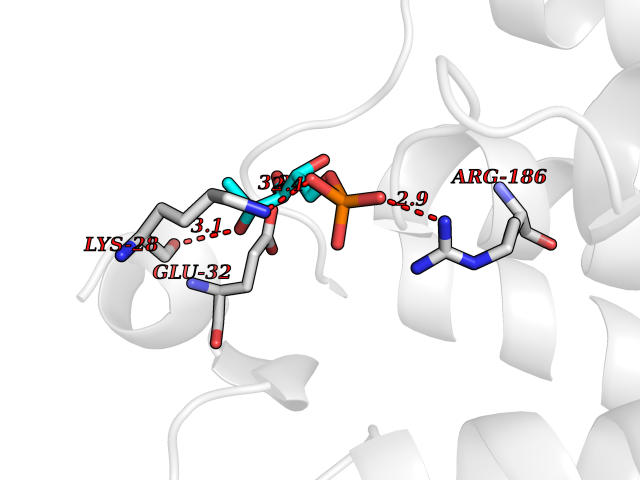 Pymol Map 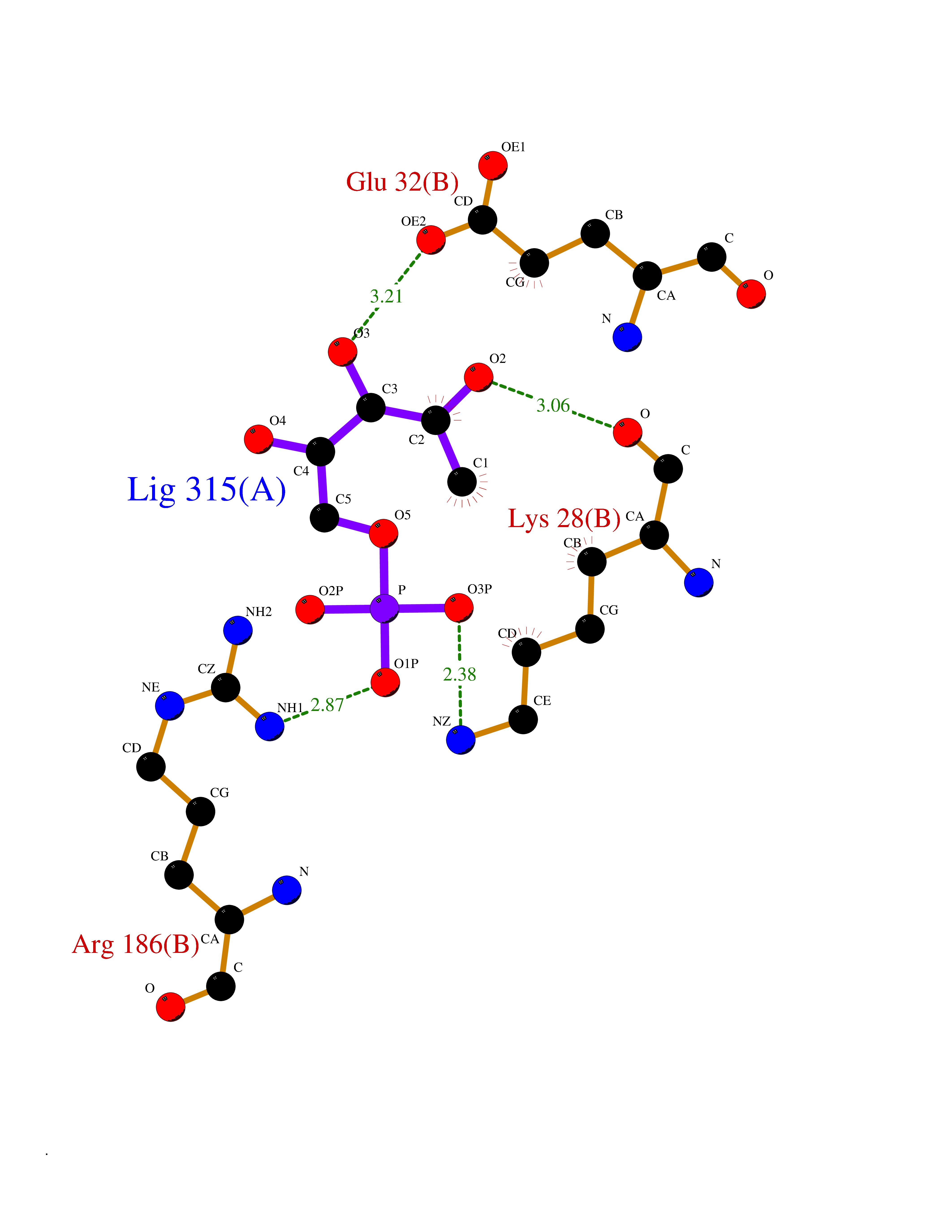 Ligplot Map 3D Binding mode Sequence IKSNEEPLLRKSSRRFVIFPIQYPDIWKXYKQAQASFWTAEEVDLSKDLPHWNKLKADEKYFISHILAFFAASDGIVNENLVERFSQEVQVPEARCFYGFQILIENVHSEXYSLLIDTYIRDPKKREFLFNAIETXPYVKKKADWALRWIADRKSTFGERVVAFAAVEGVFFSGSFAAIFWLKKRGLXPGLTFSNELISRDEGLHCDFACLXFQYLVNKPSEERVREIIVDAVKIEQEFLTEALPVGLIGXNCILXKQYIEFVADRLLVELGFSKVFQAENPFDFXE Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Myosin-7 (MYH7) | 4DB1 | 6.00 | |
Target general information Gen name MYH7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Myosin heavy chain, cardiac muscle beta isoform; Myosin heavy chain slow isoform; Myosin heavy chain 7; MyHCslow; MyHCbeta; MYH7 Protein family TRAFAC class myosin-kinesin ATPase superfamily, Myosin family Biochemical class TRAFAC class myosin-kinesin ATPase Function Muscle contraction. Related diseases Cardiomyopathy, familial hypertrophic, 1 (CMH1) [MIM:192600]: A hereditary heart disorder characterized by ventricular hypertrophy, which is usually asymmetric and often involves the interventricular septum. The symptoms include dyspnea, syncope, collapse, palpitations, and chest pain. They can be readily provoked by exercise. The disorder has inter- and intrafamilial variability ranging from benign to malignant forms with high risk of cardiac failure and sudden cardiac death. {ECO:0000269|PubMed:10065021, ECO:0000269|PubMed:10329202, ECO:0000269|PubMed:10521296, ECO:0000269|PubMed:10563488, ECO:0000269|PubMed:10679957, ECO:0000269|PubMed:10862102, ECO:0000269|PubMed:11113006, ECO:0000269|PubMed:11133230, ECO:0000269|PubMed:11214007, ECO:0000269|PubMed:11424919, ECO:0000269|PubMed:11733062, ECO:0000269|PubMed:11861413, ECO:0000269|PubMed:11968089, ECO:0000269|PubMed:12081993, ECO:0000269|PubMed:12566107, ECO:0000269|PubMed:12590187, ECO:0000269|PubMed:12707239, ECO:0000269|PubMed:12818575, ECO:0000269|PubMed:12820698, ECO:0000269|PubMed:12951062, ECO:0000269|PubMed:12974739, ECO:0000269|PubMed:12975413, ECO:0000269|PubMed:1417858, ECO:0000269|PubMed:15358028, ECO:0000269|PubMed:15483641, ECO:0000269|PubMed:1552912, ECO:0000269|PubMed:15563892, ECO:0000269|PubMed:15856146, ECO:0000269|PubMed:15858117, ECO:0000269|PubMed:16199542, ECO:0000269|PubMed:16267253, ECO:0000269|PubMed:1638703, ECO:0000269|PubMed:16650083, ECO:0000269|PubMed:16938236, ECO:0000269|PubMed:17095604, ECO:0000269|PubMed:17372140, ECO:0000269|PubMed:18175163, ECO:0000269|PubMed:18403758, ECO:0000269|PubMed:1975517, ECO:0000269|PubMed:25182012, ECO:0000269|PubMed:7581410, ECO:0000269|PubMed:7731997, ECO:0000269|PubMed:7848441, ECO:0000269|PubMed:7874131, ECO:0000269|PubMed:7909436, ECO:0000269|PubMed:8250038, ECO:0000269|PubMed:8254035, ECO:0000269|PubMed:8268932, ECO:0000269|PubMed:8282798, ECO:0000269|PubMed:8343162, ECO:0000269|PubMed:8435239, ECO:0000269|PubMed:8483915, ECO:0000269|PubMed:8533830, ECO:0000269|PubMed:8655135, ECO:0000269|PubMed:8899546, ECO:0000269|PubMed:9544842, ECO:0000269|PubMed:9822100, ECO:0000269|PubMed:9829907}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 7A, myosin storage, autosomal dominant (CMYO7A) [MIM:608358]: A skeletal muscle disorder characterized by prominent axial and proximal weakening, spinal stiffness, severe scoliosis, with or without respiratory and cardiac involvement. The age at symptom onset can range from early childhood to late adulthood, and disease severity ranges from asymptomatic to severe muscular weakness and respiratory insufficiency. Histopathological examination shows variable findings including subsarcolemmal hyaline bodies in type 1 fibers. {ECO:0000269|PubMed:14520662, ECO:0000269|PubMed:15136674, ECO:0000269|PubMed:16684601, ECO:0000269|PubMed:17336526}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Cardiomyopathy, dilated, 1S (CMD1S) [MIM:613426]: A disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death. {ECO:0000269|PubMed:11106718, ECO:0000269|PubMed:12379228, ECO:0000269|PubMed:15769782, ECO:0000269|PubMed:18506004, ECO:0000269|PubMed:21127202, ECO:0000269|PubMed:21846512}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myopathy, distal, 1 (MPD1) [MIM:160500]: A muscular disorder characterized by early-onset selective weakness of the great toe and ankle dorsiflexors, followed by weakness of the finger extensors. Mild proximal weakness occasionally develops years later after the onset of the disease. {ECO:0000269|PubMed:15322983, ECO:0000269|PubMed:17548557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 7B, myosin storage, autosomal recessive (CMYO7B) [MIM:255160]: A skeletal muscle disorder characterized by the onset of scapuloperoneal muscle weakness in early childhood or young adulthood. Affected individuals have difficulty walking, steppage gait, and scapular winging due to shoulder girdle involvement. The severity and progression of the disorder is highly variable. Most patients develop respiratory insufficiency and restrictive lung disease. Some develop hypertrophic cardiomyopathy. Histopathological examination shows variable findings including subsarcolemmal hyaline bodies in type 1 fibers. {ECO:0000269|PubMed:25666907}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Left ventricular non-compaction 5 (LVNC5) [MIM:613426]: A form of left ventricular non-compaction, a cardiomyopathy due to myocardial morphogenesis arrest and characterized by a hypertrophic left ventricle, a severely thickened 2-layered myocardium, numerous prominent trabeculations, deep intertrabecular recesses, and poor systolic function. Clinical manifestations are variable. Some affected individuals experience no symptoms at all, others develop heart failure. In some cases, left ventricular non-compaction is associated with other congenital heart anomalies. LVNC5 is an autosomal dominant condition. {ECO:0000269|PubMed:18506004}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry. Drugs (DrugBank ID) DB08378; DB14921 Interacts with Q5SYC1; Q9BQD3; Q96DD0 EC number NA Uniprot keywords 3D-structure; Actin-binding; ATP-binding; Calmodulin-binding; Cardiomyopathy; Coiled coil; Cytoplasm; Disease variant; Methylation; Motor protein; Muscle protein; Myosin; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Thick filament Protein physicochemical properties Chain ID A Molecular weight (Da) 81702.7 Length 716 Aromaticity 0.11 Instability index 36.28 Isoelectric point 6.43 Charge (pH=7) -3.73 2D Binding mode Binding energy (Kcal/mol) -6.95 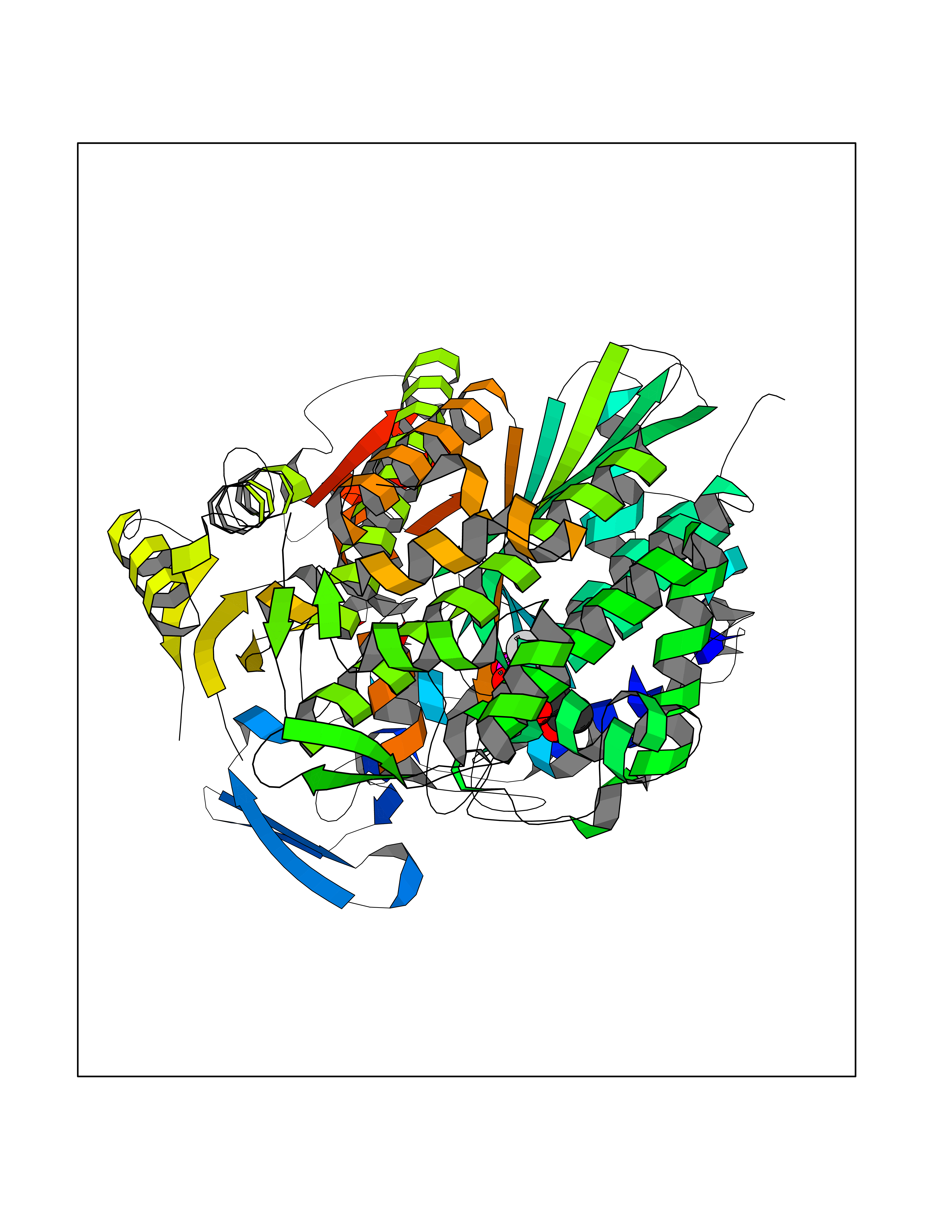 Molscript Map 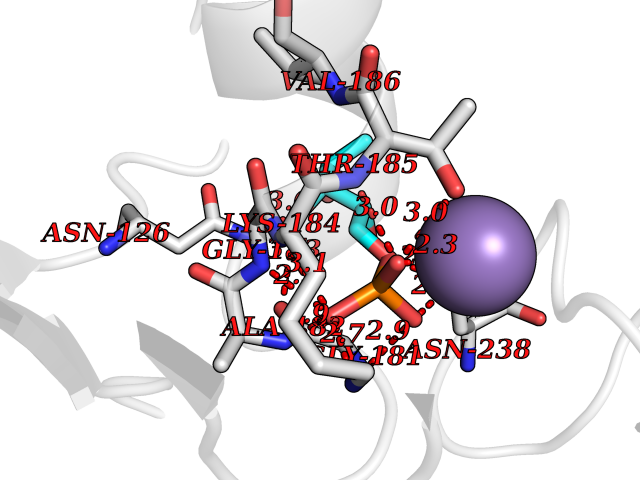 Pymol Map 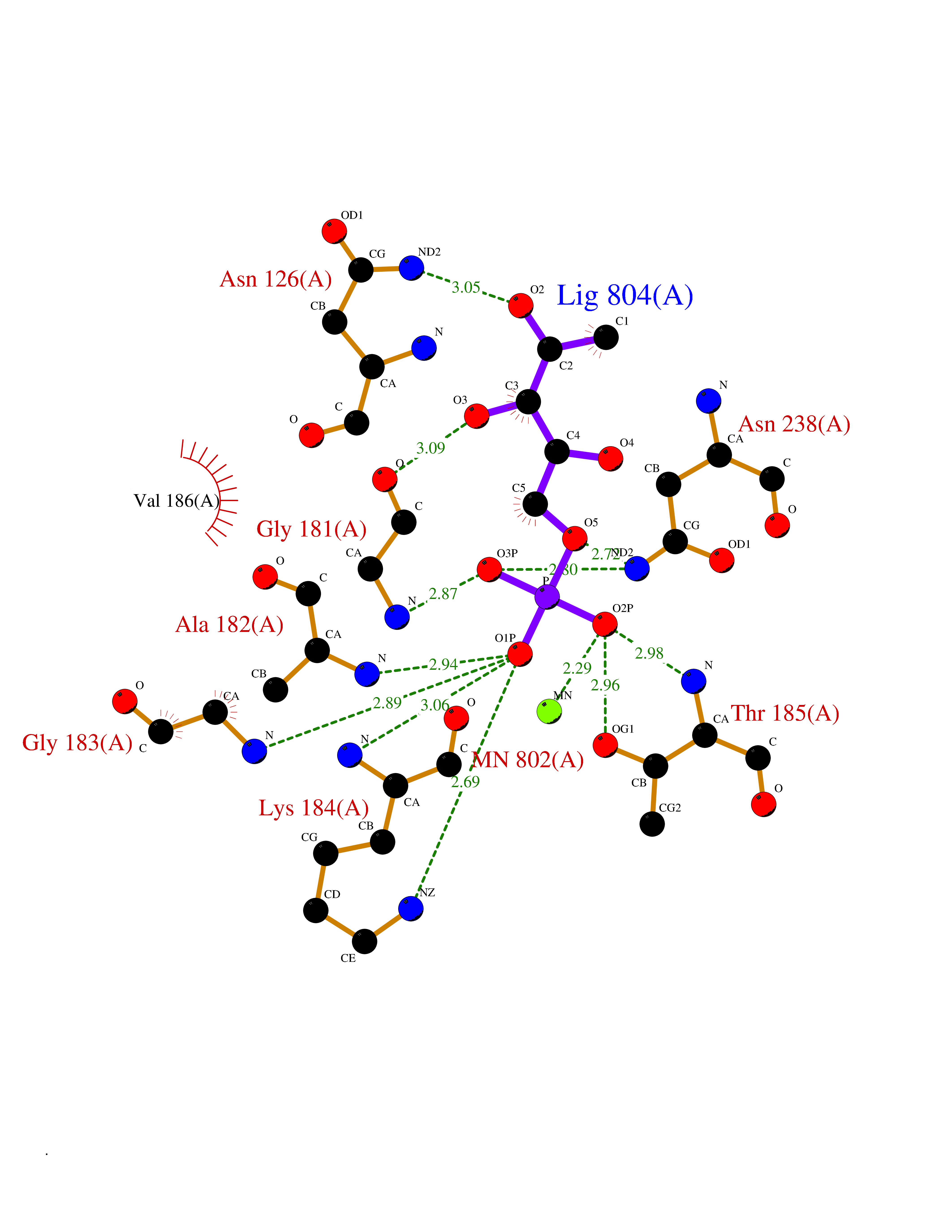 Ligplot Map 3D Binding mode Sequence GGDSEMAVFGAAAPYLRKSEKERLEAQTRPFDLKKDVFVPDDKQEFVKAKIVSREGGKVTAETEYGKTVTVKEDQVMQQNPPKFDKIEDMAMLTFLHEPAVLYNLKDRYGSWMIYTYSGLFCVTVNPYKWLPVYTPEVVAAYRGKKRSEAPPHIFSISDNAYQYMLTDRENQSILITGESGAGKTVNTKRVIQYFAVIAAIGDRGKGTLEDQIIQANPALEAFGNAKTVRNDNSSRFGKFIRIHFGATGKLASADIETYLLEKSRVIFQLKAERDYHIFYQILSNKKPELLDMLLITNNPYDYAFISQGETTVASIDDAEELMATDNAFDVLGFTSEEKNSMYKLTGAIMHFGNMKFKLKQREEQAEPDGTEEADKSAYLMGLNSADLLKGLCHPRYVTKGQNVQQVIYATGALAKAVYERMFNWMVTRINATLETKQPRQYFIGVLDIAGFEIFDFNSFEQLCINFTNEKLQQFFNHHMFVLEQEEYKKEGIEWTFIDFGMDLQACIDLIEKPMGIMSILEEECMFPKATDMTFKAKLFDNHLGKSANFQKPRNPEAHFSLIHYAGIVDYNIIGWLQKNKDPLNETVVGLYQKSSLKLLSTLFANYAFQTVSALHRENLNKLMTNLRSTHPHFVRCIIPNETKSPGVMDNPLVMHQLRCNGVLEGIRICRKGFPNRILYAEKLLSSLDIDHNQYKFGHTKVFFKAGLLGLLEEMR Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Serine hydroxymethyltransferase, cytosolic | 1BJ4 | 6.00 | |
Target general information Gen name SHMT1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family SHMT family Biochemical class Transferase Function Glycine hydroxymethyltransferase activity.Identical protein binding.L-allo-threonine aldolase activity.MRNA 5'-UTR binding.Protein homodimerization activity.Pyridoxal phosphate binding.Serine binding.Translation repressor activity, nucleic acid binding. Related diseases Deafness, autosomal dominant, 77 (DFNA77) [MIM:618915]: A form of non-syndromic deafness characterized by adult onset of bilateral, postlingual, mild-to-severe sensorineural hearing loss. Sensorineural hearing loss results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. {ECO:0000269|PubMed:31273342}. The gene represented in this entry is involved in disease pathogenesis. Drugs (DrugBank ID) DB02800; DB00145; DB01055; DB02824; DB00114; DB00116; DB02067 Interacts with P26196; Q9H8Y8; P50213; P45984; Q99750; P34896; P0DMM9 EC number 2.1.2.1 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; One-carbon metabolism; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 51456.1 Length 470 Aromaticity 0.08 Instability index 37.11 Isoelectric point 6.78 Charge (pH=7) -0.97 2D Binding mode Binding energy (Kcal/mol) -6.46  Molscript Map 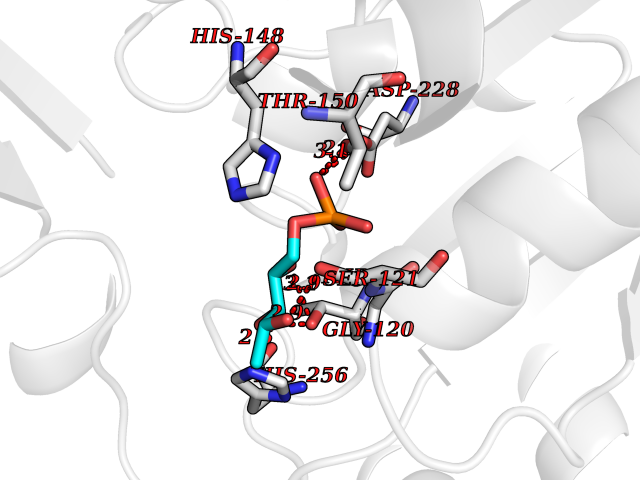 Pymol Map 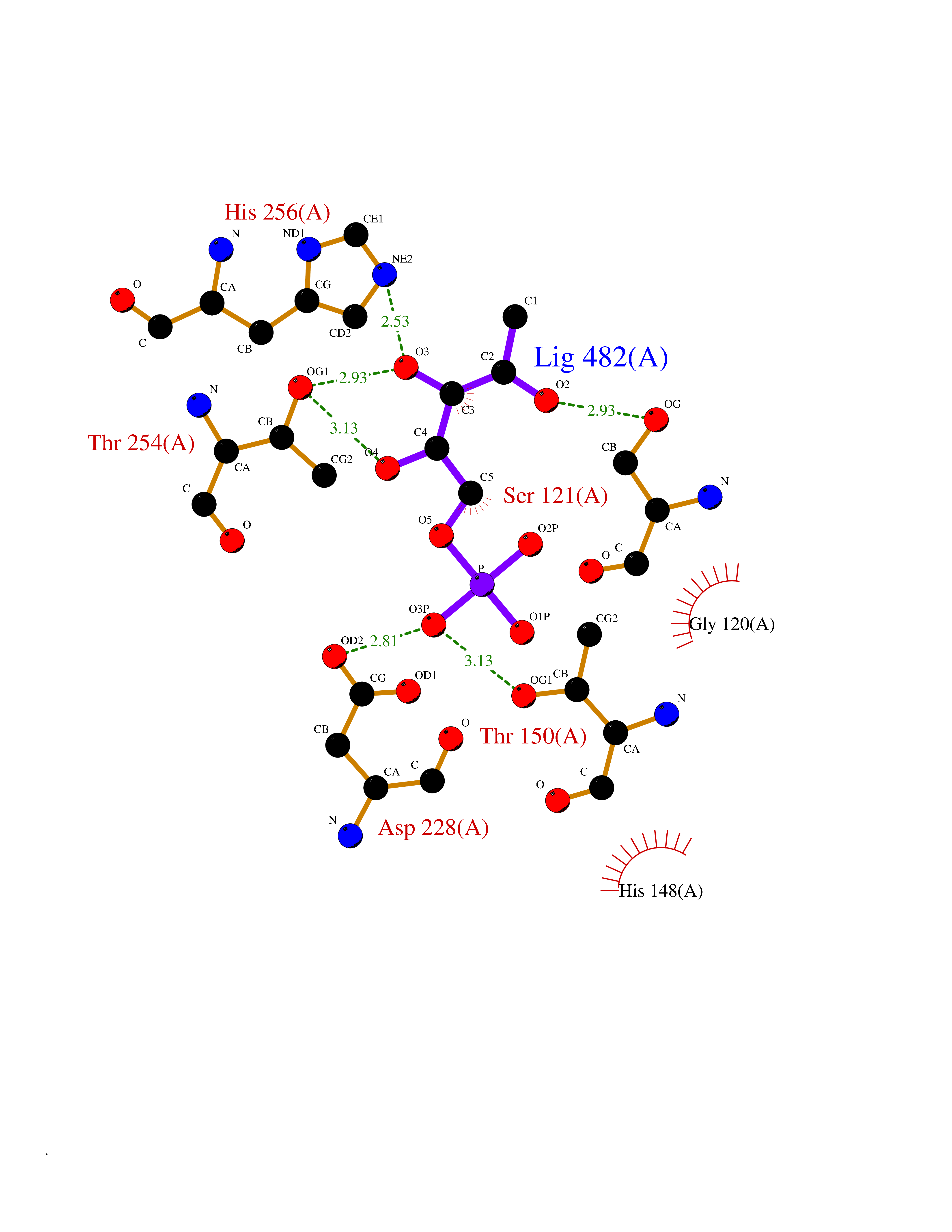 Ligplot Map 3D Binding mode Sequence DADLWSSHDAMLAQPLKDSDVEVYNIIKKESNRQRVGLELIASENFASRAVLEALGSCLNNKYSEGYPGQRYYGGTEFIDELETLCQKRALQAYKLDPQCWGVNVQPYSGSPANFAVYTALVEPHGRIMGLDLPDGGHLTHGFMTDKKKISATSIFFESMPYKVNPDTGYINYDQLEENARLFHPKLIIAGTSCYSRNLEYARLRKIADENGAYLMADMAHISGLVAAGVVPSPFEHCHVVTTTTHKTLRGCRAGMIFYRKGVKSVDPATGKEILYNLESLINSAVFPGLQGGPHNHAIAGVAVALKQAMTLEFKVYQHQVVANCRALSEALTELGYKIVTGGSDNHLILVDLRSKGTDGGRAEKVLEACSIACNKNTCPGDRSALRPSGLRLGTPALTSRGLLEKDFQKVAHFIHRGIELTLQIQSDTGVAATLKEFKERLAGDKYQAAVQALREEVESFASLFPLPGL Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Voltage-gated calcium channel alpha Cav3.1 (CACNA1G) | 6KZP | 6.00 | |
Target general information Gen name CACNA1G Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Voltage-gated calcium channel alpha subunit Cav3.1; Voltage-dependent T-type calcium channel; NBR13; Cav3.1c; CACNA1G Protein family Calcium channel alpha-1 subunit (TC 1.A.1.11) family, CACNA1G subfamily Biochemical class Voltage-gated ion channel Function Voltage-sensitive calcium channels (vscc) mediate the entry of calcium ions into excitable cells and are also involved in a variety of calcium-dependent processes, including muscle contraction, hormone or neurotransmitter release and gene expression. Related diseases Spinocerebellar ataxia 42 (SCA42) [MIM:616795]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA42 is a slowly progressive, autosomal dominant form with variable severity. {ECO:0000269|PubMed:26456284, ECO:0000269|PubMed:26715324}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinocerebellar ataxia 42, early-onset, severe, with neurodevelopmental deficits (SCA42ND) [MIM:618087]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA42ND is an early-onset, severe form associated with motor and cognitive impairment, cerebellar atrophy as well as variable features such as facial dysmorphisms, digital anomalies, microcephaly and epilepsy. SCA42ND inheritance is autosomal dominant. {ECO:0000269|PubMed:29878067}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09227; DB09231; DB13746; DB11148; DB11093; DB11348; DB14481; DB09061; DB00568; DB09235; DB00228; DB00153; DB00593; DB04841; DB09238; DB14009; DB05246; DB01388; DB14011; DB00622; DB01115; DB06712; DB09089; DB00347; DB00661; DB00909 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Calcium channel; Calcium transport; Cell membrane; Cytoplasm; Disease variant; Glycoprotein; Ion channel; Ion transport; Membrane; Neurodegeneration; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Spinocerebellar ataxia; Transmembrane; Transmembrane helix; Transport; Voltage-gated channel Protein physicochemical properties Chain ID A Molecular weight (Da) 116116 Length 1014 Aromaticity 0.13 Instability index 33.06 Isoelectric point 6.61 Charge (pH=7) -2.04 2D Binding mode Binding energy (Kcal/mol) -6.51 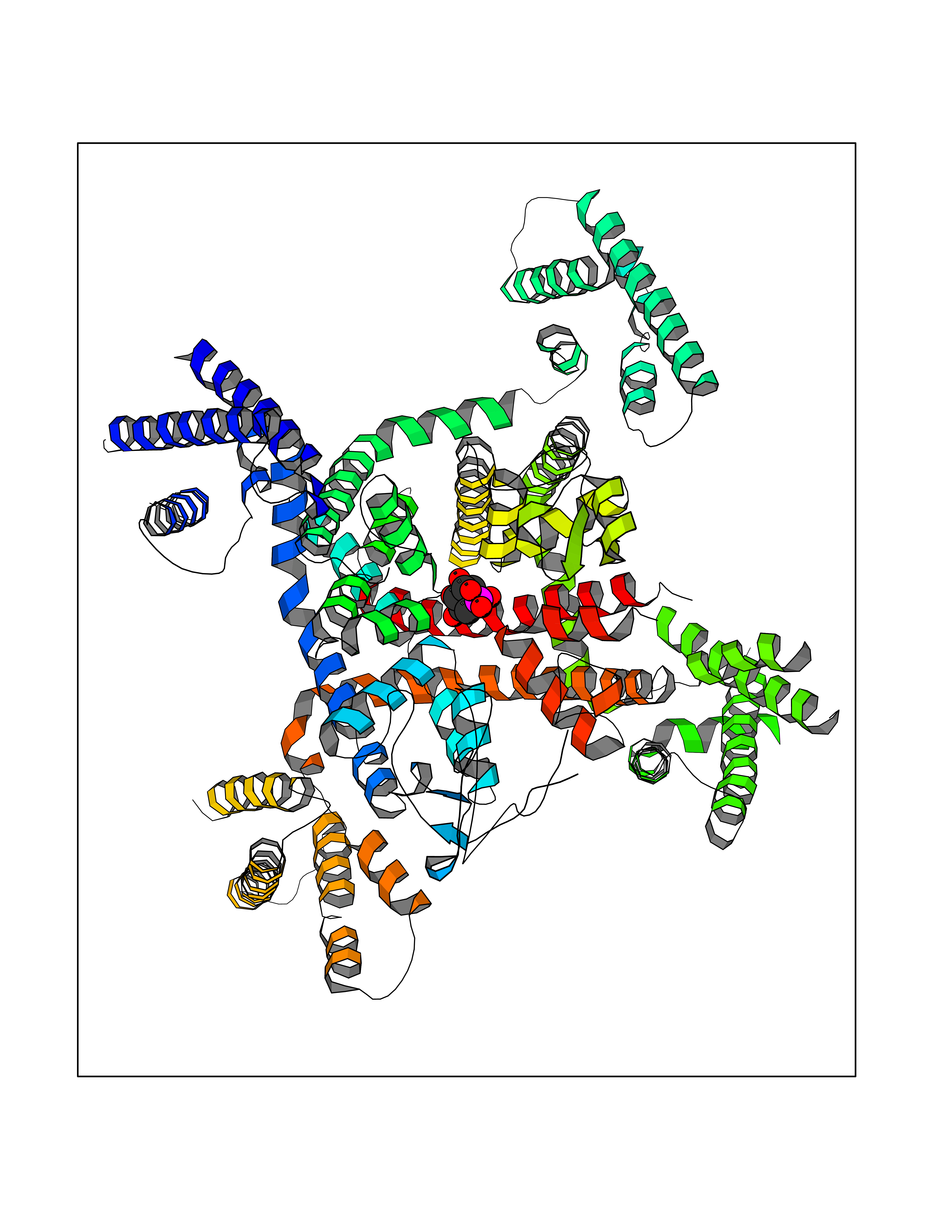 Molscript Map 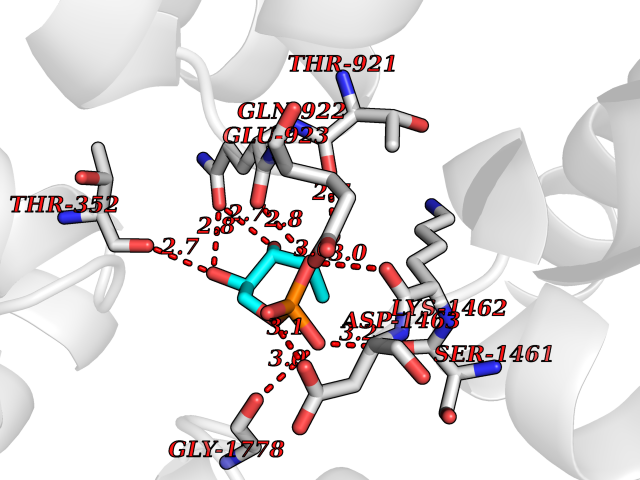 Pymol Map  Ligplot Map 3D Binding mode Sequence NPWFERISMLVILLNCVTLGMFRPCEDIACDSQRCRILQAFDDFIFAFFAVEMVVKMVAGDTWNRLDFFIVIAGMLEYSLDLQNVSFSAVRTVRVLRPLRAINRVPSMRILVTLLLDTLPMLGNVLLLCFFVFFIFGIVGVQLWAGLLRNRCFLPENFSLPLSVDLERYYQTENEDESPFICSQPRENGMRSCRSVPTLRCVNWNQYYTNCSAGEHNPFKGAINFDNIGYAWIAIFQVITLEGWVDIMYFVMDAHSFYNFIYFILLIIVGSFFMINLCLVVIATQFSETKQREIVDSKYFGRGIMIAILVNTLSMGIEYHEQPEELTNALEISNIVFTSLFALEMLLKLLVYGPFGYIKNPYNIFDGVIVVISVWEIVSVLRTFRLMRVLKLVRFLPALQRQLVVLMKTMDNVATFCMLLMLFIFIFSILGMHLFGCKFASLPDRKNFDSLLWAIVTVFQILTQEDWNKVLYNGMASTSSWAALYFIALMTFGNYVLFNLLVAILVEGFQFRLLCHRIITHKMFDHVVLVIIFLNCITIAMERPKIDPHSAERIFLTLSNYIFTAVFLAEMTVKVVALGSSWNVLDGLLVLISVIDILVSMVSKILGMLRVLRLLRTLRPLRVISRAQGLKLVVETLMSSLKPIGNIVVICCAFFIIFGILGVQLFKGKFFVCQGEDTRNITNKSDCAEASYRWVRHKYNFDNLGQALMSLFVLASKDGWVDIMYDGLDAVGVDQQPIMNHNPWMLLYFISFLLIVAFFVLNMFVGVVVENFHYLDLFITGVIGLNVVTMAMEHYQQPQILDEALKICNYIFTVIFVLESVFKLVAFGFRRFFQDRWNQLDLAIVLLSIMGITLEEIEVNASLPINPTIIRIMRVLRIARVLKLLKMAVGMRALLDTVMQALPQVGNLGLLFMLLFFIFAALGVELFGDLECDETHPCEGLGRHATFRNFGMAFLTLFRVSTGDNWNGIMKDTLRDYNTVISPIYFVSFVLTAQFVLVNVVIAVLMKHLEESNK Hydrogen bonds contact Hydrophobic contact | ||||