Job Results:
Ligand
Structure
Job ID
0006599cba9dbe83f9144f4d36fa70dc
Job name
NA
Time
2024-05-29 06:52:44
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 81 | Gamma-aminobutyric acid type B receptor subunit 1 | 4MS4 | 7.90 | |
Target general information Gen name GABBR1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms GPRC3B;GPR51 Protein family G-protein coupled receptor 3 family, GABA-B receptor subfamily Biochemical class Signaling protein / antagonist Function G-protein coupled GABA receptor activity. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08891; DB08892; DB00181; DB00363; DB02530; DB05010; DB09072 Interacts with Q9UBS5; Q9UBS5-2; P46459; Q86UR5 EC number NA Uniprot keywords 3D-structure; Cell membrane; Coiled coil; Direct protein sequencing; Disease variant; Disulfide bond; Epilepsy; G-protein coupled receptor; Glycoprotein; Intellectual disability; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 46502.1 Length 408 Aromaticity 0.12 Instability index 50.05 Isoelectric point 5.78 Charge (pH=7) -5.62 2D Binding mode Binding energy (Kcal/mol) -9.51 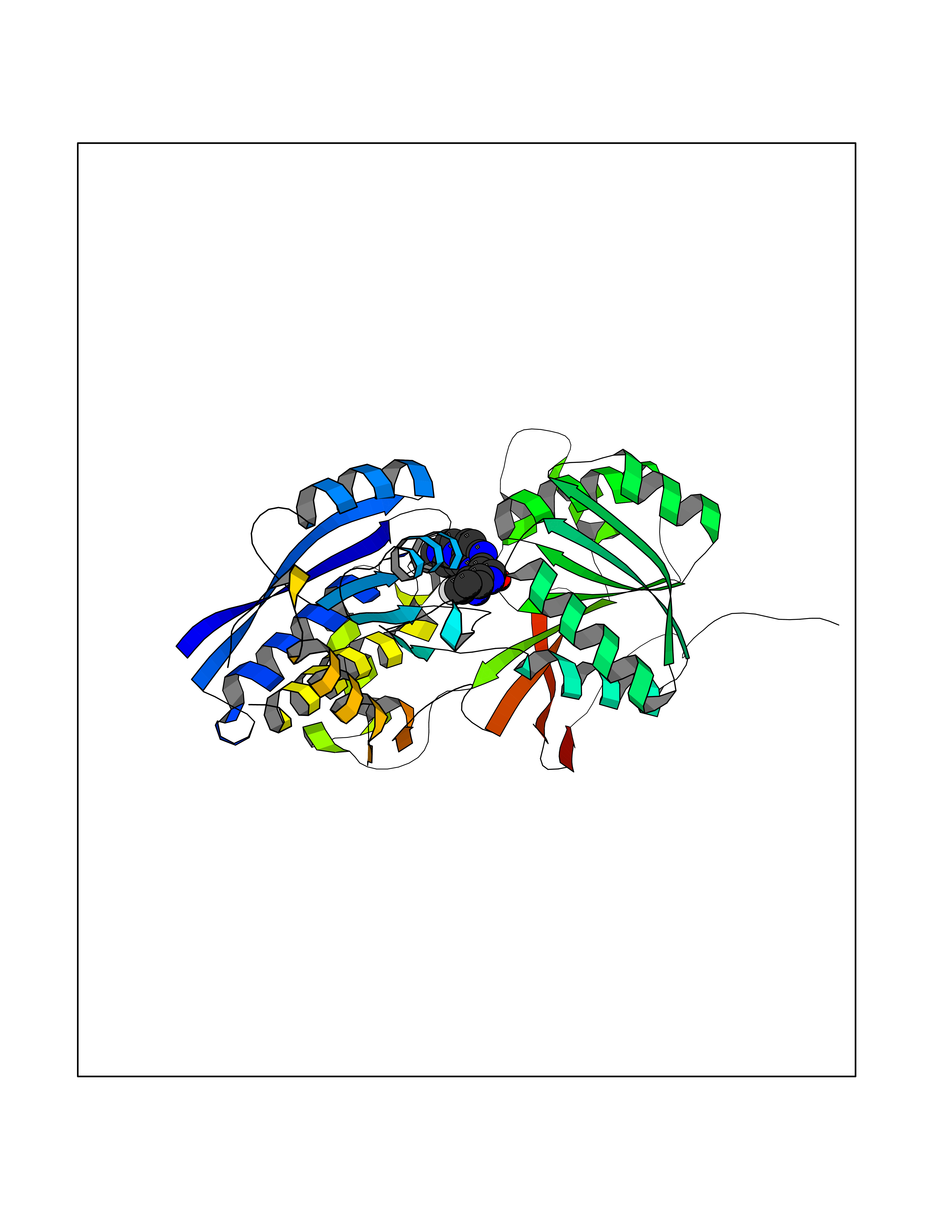 Molscript Map 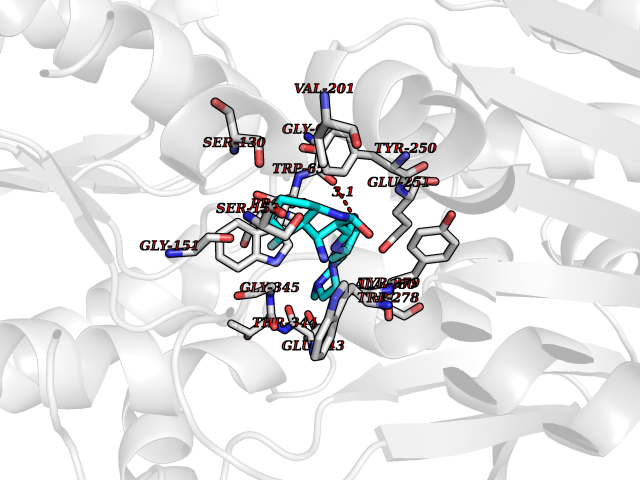 Pymol Map 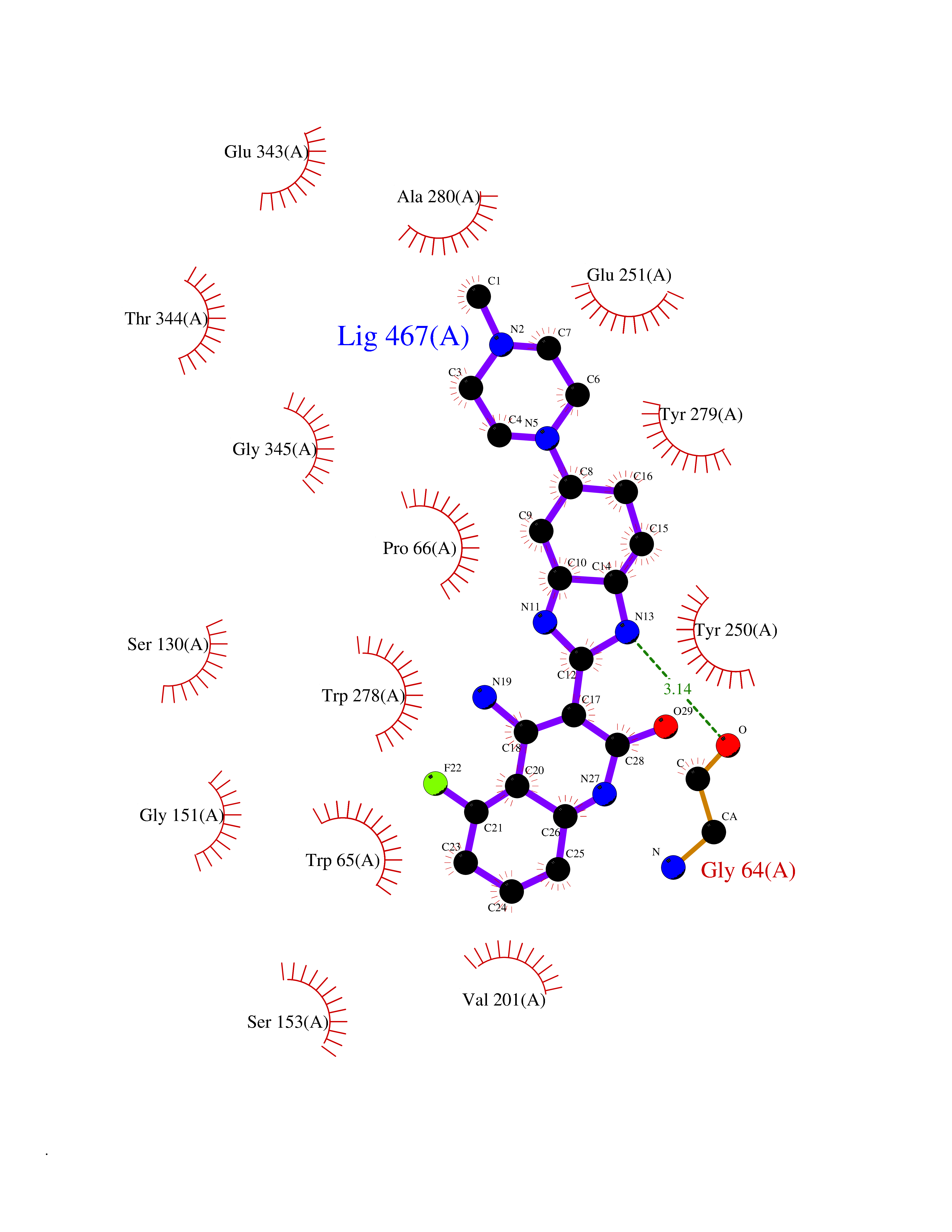 Ligplot Map 3D Binding mode Sequence RRAVYIGALFPMSGGWPGGQACQPAVEMALEDVNSRRDILPDYELKLIHHDSKCDPGQATKYLYELLYNDPIKIILMPGCSSVSTLVAEAARMWNLIVLSYGSSSPALSNRQRFPTFFRTHPSATLHNPTRVKLFEKWGWKKIATIQQTTEVFTSTLDDLEERVKEAGIEITFRQSFFSDPAVPVKNLKRQDARIIVGLFYETEARKVFCEVYKERLFGKKYVWFLIGWYADNWFKIYDPSINCTVDEMTEAVEGHITTEIVMLNPANTRSISNMTSQEFVEKLTKRLKRHPEETGGFQEAPLAYDAIWALALALNKTSRLEDFNYNNQTITDQIYRAMNSSSFEGVSGHVVFDASGSRMAWTLIEQLQGGSYKKIGYYDSTKDDLSWSKTDKWIGGSPPADDYKDDD Hydrogen bonds contact Hydrophobic contact | ||||
| 82 | Corynebacterium Pup-protein ligase (Cory pafA) | 4BJR | 7.90 | |
Target general information Gen name Cory pafA Organism Corynebacterium glutamicum (strain ATCC 13032 / DSM 20300 / JCM 1318 / BCRC 11384 / CCUG 27702 / LMG 3730 / NBRC 12168 / NCIMB 10025 / NRRL B-2784 / 534) Uniprot ID TTD ID Synonyms Pup-conjugating enzyme; Pup--protein ligase; Proteasome accessory factor A Protein family Pup ligase/Pup deamidase family, Pup-conjugating enzyme subfamily Biochemical class NA Function Catalyzes the covalent attachment of the prokaryotic ubiquitin-like protein modifier Pup to the proteasomal substrate proteins, thereby targeting them for proteasomal degradation. This tagging system is termed pupylation. The ligation reaction involves the side-chain carboxylate of the C-terminal glutamate of Pup and the side-chain amino group of a substrate lysine. Related diseases Anemia, non-spherocytic hemolytic, due to G6PD deficiency (NSHA) [MIM:300908]: A disease characterized by G6PD deficiency, acute hemolytic anemia, fatigue, back pain, and jaundice. In most patients, the disease is triggered by an exogenous agent, such as some drugs, food, or infection. Increased unconjugated bilirubin, lactate dehydrogenase, and reticulocytosis are markers of the disorder. Although G6PD deficiency can be life-threatening, most patients are asymptomatic throughout their life. {ECO:0000269|PubMed:12524354, ECO:0000269|PubMed:1303180, ECO:0000269|PubMed:1303182, ECO:0000269|PubMed:1536798, ECO:0000269|PubMed:1611091, ECO:0000269|PubMed:1889820, ECO:0000269|PubMed:1945893, ECO:0000269|PubMed:20007901, ECO:0000269|PubMed:26479991, ECO:0000269|PubMed:2836867, ECO:0000269|PubMed:2912069, ECO:0000269|PubMed:30988594, ECO:0000269|PubMed:38066190, ECO:0000269|PubMed:7858267, ECO:0000269|PubMed:7959695, ECO:0000269|PubMed:8193373, ECO:0000269|PubMed:8490627, ECO:0000269|PubMed:8533762, ECO:0000269|PubMed:8733135, ECO:0000269|PubMed:9452072}. The disease is caused by variants affecting the gene represented in this entry. Deficiency of G6PD is associated with hemolytic anemia in two different situations. First, in areas in which malaria has been endemic, G6PD-deficiency alleles have reached high frequencies (1% to 50%) and deficient individuals, though essentially asymptomatic in the steady state, have a high risk of acute hemolytic attacks. Secondly, sporadic cases of G6PD deficiency occur at a very low frequencies, and they usually present a more severe phenotype. Several types of NSHA are recognized. Class-I variants are associated with severe NSHA; class-II have an activity <10% of normal; class-III have an activity of 10% to 60% of normal; class-IV have near normal activity. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; ATP-binding; Ligase; Magnesium; Metal-binding; Nucleotide-binding; Reference proteome; Ubl conjugation pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 110572 Length 994 Aromaticity 0.07 Instability index 42.33 Isoelectric point 5.26 Charge (pH=7) -34.19 2D Binding mode Binding energy (Kcal/mol) -8.59 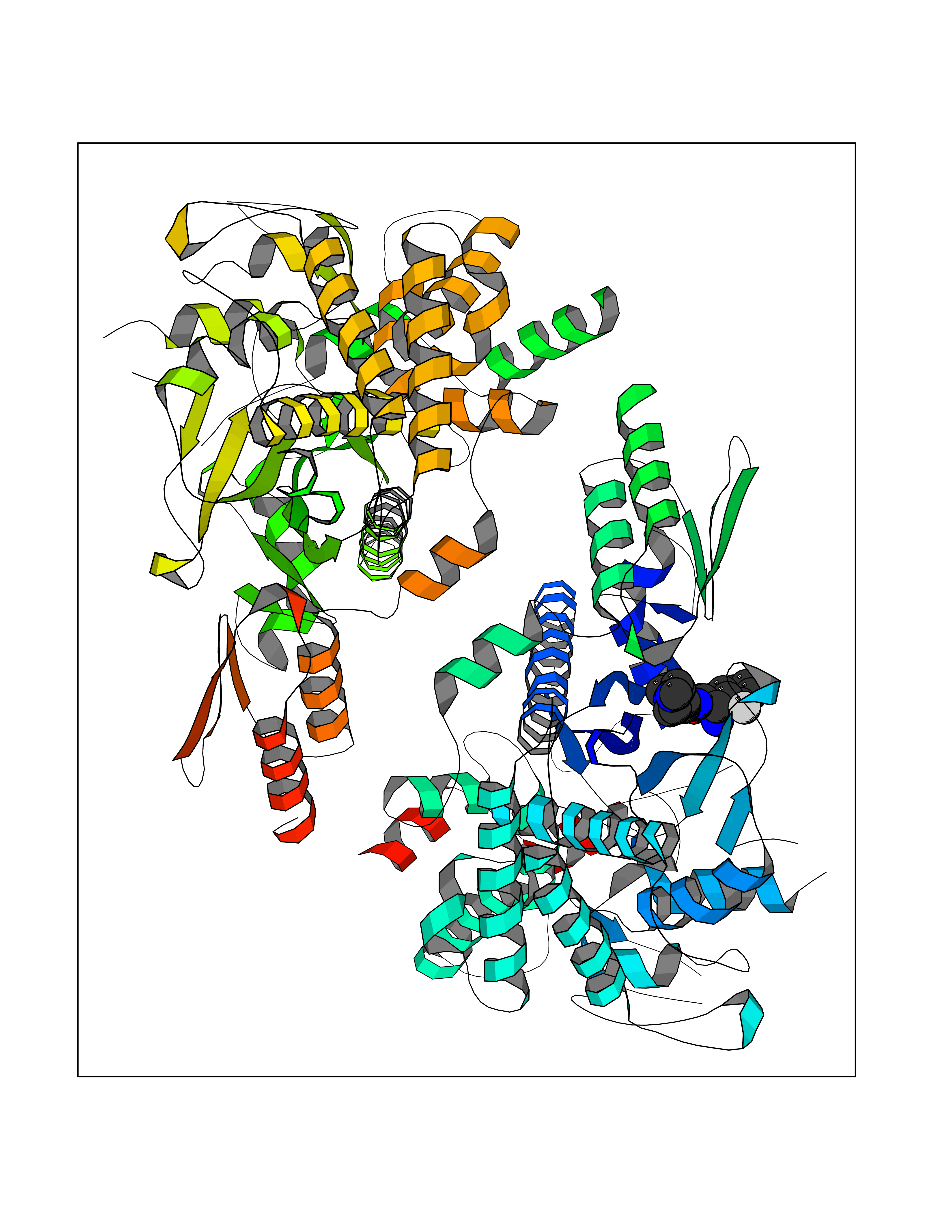 Molscript Map 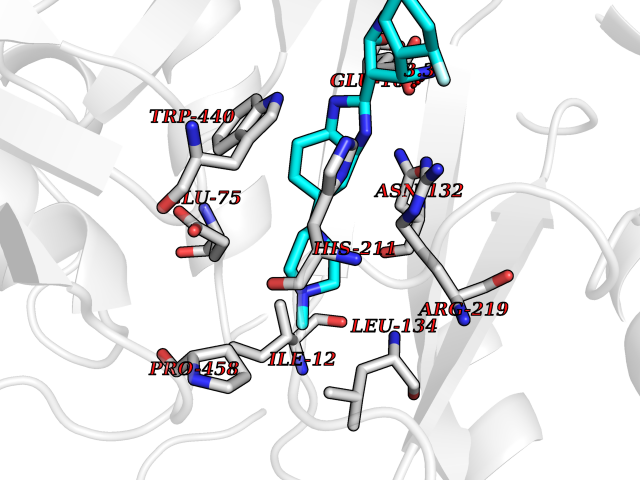 Pymol Map 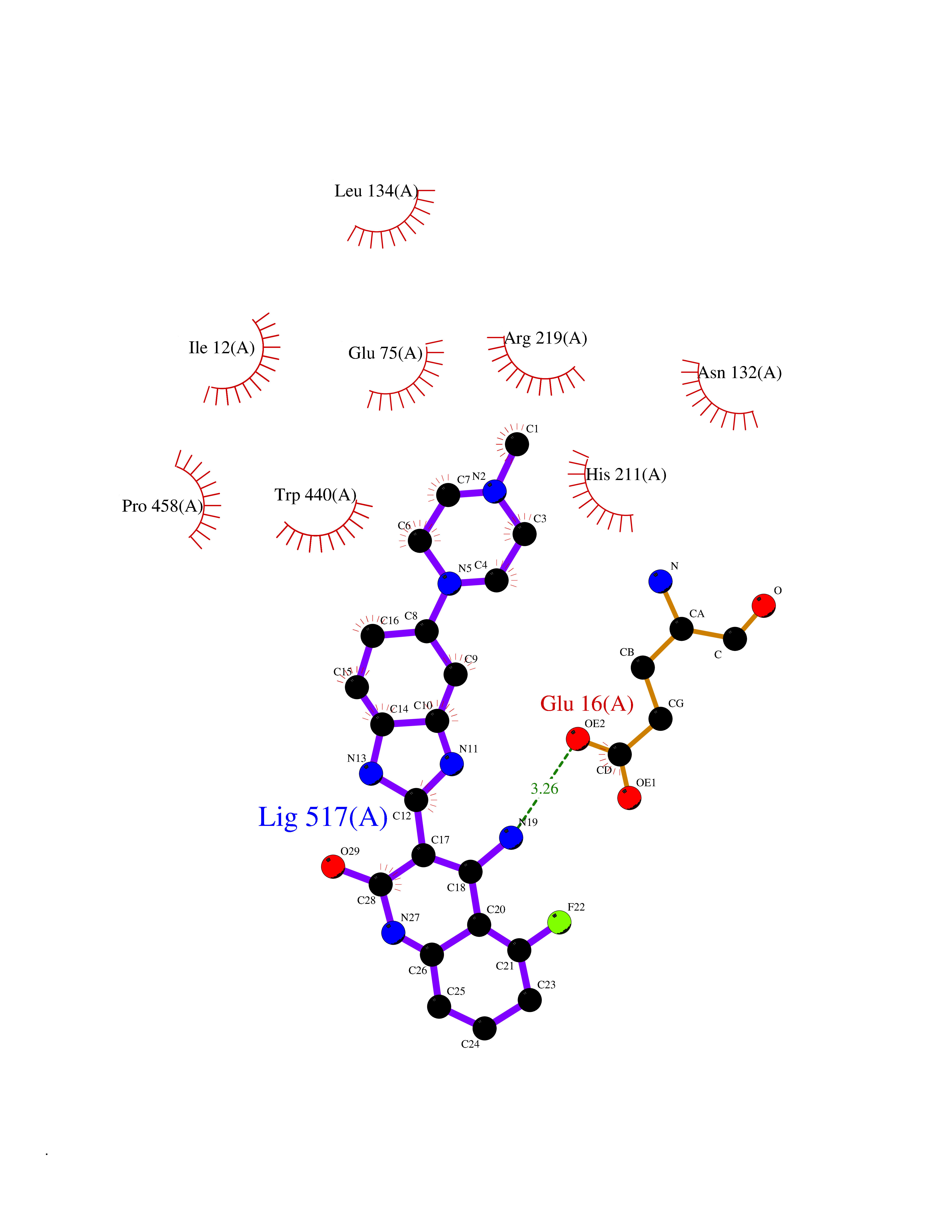 Ligplot Map 3D Binding mode Sequence TVESALTRRIMGIETEYGLTFVDGDSKKLRPDEIARRMFRPIVEKYSSSNIFIPNGSRLYLNVGSHPEYATAECDNLTQLINFEKAGDVIADRMAVDAEESLAKEDIAGQVYLFKNNVDSVGNSYGCHENYLVGRSMPLKALGKRLMPFLITRQLICGAGRIHHPNPSFPLGYCISQRSDHVWEGVSSASRPIINTRDEPHADSHSYRRLHVIVGDANMAEPSIALKVGSTLLVLEMIEADFGLPSLELANDIASIREISRDATGSTLLSLKDGTTMTALQIQQVVFEHASKWLEQRPEPEFSGTSNTEMARVLDLWGRMLKAIESGDFSEVDTEIDWVIKKKLIDRFIQRGNLGLDDPKLAQVDLTYHDIRPGRGLFSVLQSRGMIKRWTTDEAILAAVDTAPDTTRAHLRGRILKAADTLGVPVTVDWMRHKVNRPEPQSVELGDPFSAVNSEVDQLIEYMTVHAGSASGTSLLDEIDGLLENNAEEFVRSYVQKGGETVESALTRRIMGIETEYGLTFVDGDSKKLRPDEIARRMFRPIVEKYSSSNIFIPNGSRLYLNVGSHPEYATAECDNLTQLINFEKAGDVIADRMAVDAEESLAKEDIAGQVYLFKNNVDSVGNSYGCHENYLVGRSMPLKALGKRLMPFLITRQLICGAGRIHHPNPSFPLGYCISQRSDHVWEGVSSASRPIINTRDEPHADSHSYRRLHVIVGDANMAEPSIALKVGSTLLVLEMIEADFGLPSLELANDIASIREISRDATGSTLLSLKDGTTMTALQIQQVVFEHASKWLEQRPEPEFSGTSNTEMARVLDLWGRMLKAIESGDFSEVDTEIDWVIKKKLIDRFIQRGNLGLDDPKLAQVDLTYHDIRPGRGLFSVLQSRGMIKRWTTDEAILAAVDTAPDTTRAHLRGRILKAADTLGVPVTVDWMRHKVNRPEPQSVELGDPFSAVNSEVDQLIEYMTVHASLLDEIDGLLENNAEEFVRSYVQKGGE Hydrogen bonds contact Hydrophobic contact | ||||
| 83 | HMG-CoA reductase (HMGCR) | 2R4F | 7.89 | |
Target general information Gen name HMGCR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms 3-hydroxy-3-methylglutaryl-coenzyme A reductase Protein family HMG-CoA reductase family Biochemical class CH-OH donor oxidoreductase Function Transmembrane glycoprotein that is the rate-limiting enzyme in cholesterol biosynthesis as well as in the biosynthesis of nonsterol isoprenoids that are essential for normal cell function including ubiquinone and geranylgeranyl proteins. Related diseases Muscular dystrophy, limb-girdle, autosomal recessive 28 (LGMDR28) [MIM:620375]: An autosomal recessive form of limb girdle muscular dystrophy, a group of genetically heterogeneous muscular disorders that share proximal muscle weakness as the major attribute. Most limb girdle muscular dystrophies present with elevated creatinine kinase and myopathic electromyographic features. Disease is usually progressive to a variable degree, ranging from minor disability to complete inability to ambulate, and can involve the large proximal muscles, as well as axial and facial muscles. Different disease forms may exhibit skeletal muscle hypertrophy, kyphoscoliosis, and contractures or involve other muscle groups and manifest with distal weakness, cardiomyopathy, dysphagia, and respiratory difficulties. LGMDR28 is characterized by progressive muscle weakness affecting the proximal and axial muscles of the upper and lower limbs, and highly variable age at onset. Most patients have limited ambulation or become wheelchair-bound within a few decades, and respiratory insufficiency commonly occurs. {ECO:0000269|PubMed:36745799, ECO:0000269|PubMed:37167966}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03169; DB04447; DB01076; DB09061; DB00439; DB01992; DB01095; DB00227; DB14009; DB04377; DB06693; DB14011; DB00157; DB03461; DB08860; DB00175; DB01098; DB00641; DB05317; DB09270 Interacts with Q9Y5Z9; Q9Y5Z9-1 EC number EC 1.1.1.34 Uniprot keywords 3D-structure; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Disease variant; Endoplasmic reticulum; Glycoprotein; Isopeptide bond; Limb-girdle muscular dystrophy; Lipid biosynthesis; Lipid metabolism; Membrane; NADP; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 84796.9 Length 798 Aromaticity 0.05 Instability index 47.61 Isoelectric point 6.2 Charge (pH=7) -3.71 2D Binding mode Binding energy (Kcal/mol) -7.69 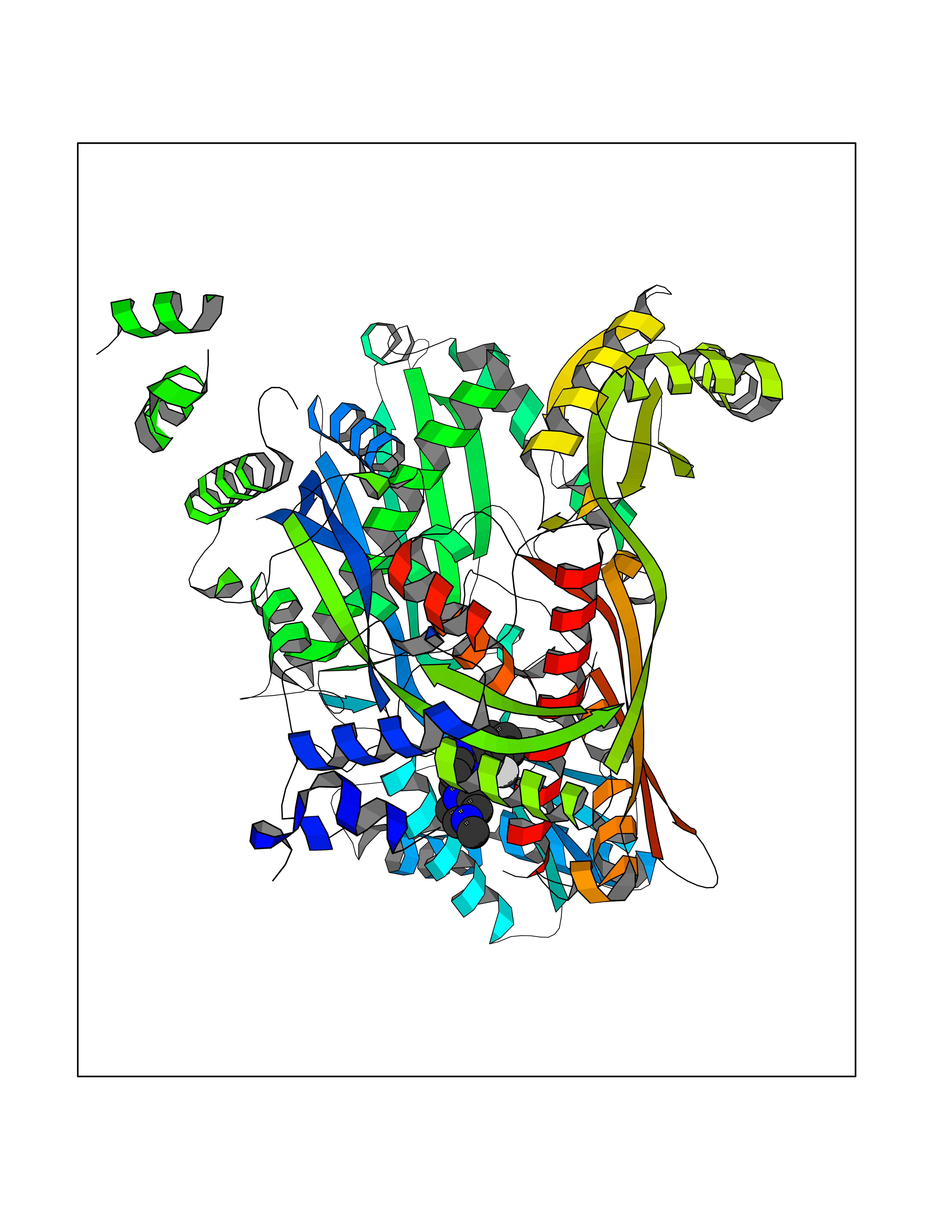 Molscript Map 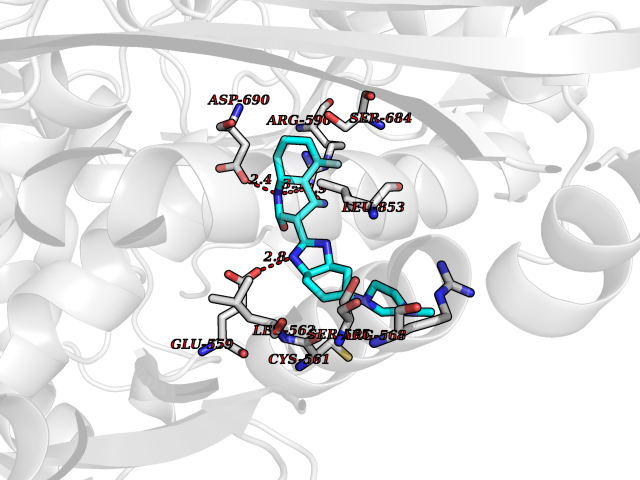 Pymol Map 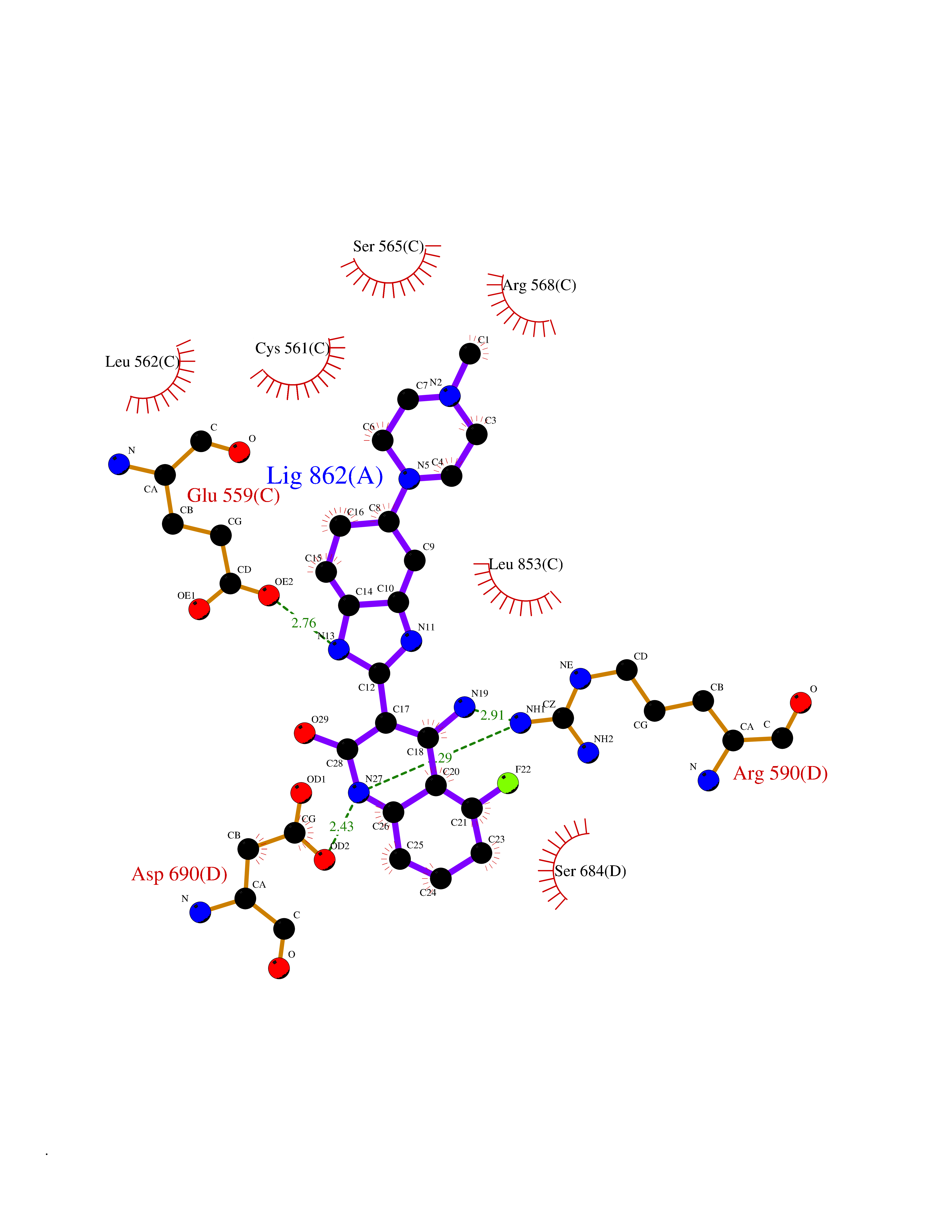 Ligplot Map 3D Binding mode Sequence GAKFLSDAEIIQLVNETLIETHERGVSIRRQLLSKKLSEPSSLQYLPYRDYNYSLVMGACCENVIGYMPIPVGVAGPLCLDEKEFQVPMATTEGCLVASTNRGCRAIGLGGGASSRVLADGMTRGPVVRLPRACDSAEVKAWLETSEGFAVIKEAFDSTSRFARLQKLHTSIAGRNLYIRFQSRSGDAMGMNMISKGTEKALSKLHEYFPEMQILAVSGNYCTDKKPAAINWIEGRGKSVVCEAVIPAKVVREVLKTTTEAMIEVNINKNLVGSAMAGSIGGYNAHAANIVTAIYIACGQDAAQNVGSSNCITLMEASGPTNEDLYISCTMPSIEIGTVGGGTNLLPQQACLQMLGVQGACKDNPGENARQLARIVCGTVMAGELSLMAALAAGPNEECLQILGNGAKFLSDAEIIQLVETLIETHERGVSIRRQLLSKKLSEPSSLQYLPYRDYNYSLVMGACCENVIGYMPIPVGVAGPLCLDEKEFQVPMATTEGCLVASTNRGCRAIGLGGGASSRVLADGMTRGPVVRLPRACDSAEVKAWLETSEGFAVIKEAFDSTSRFARLQKLHTSIAGRNLYIRFQSRSGDAMGMNMISKGTEKALSKLHEYFPEMQILAVSGNYCTDKKPAAINWIEGRGKSVVCEAVIPAKVVREVLKTTTEAMIEVNINKNLVGSAMAGSIGGYNAHAANIVTAIYIACGQDAAQNVGSSNCITLMEASGPTNEDLYISCTMPSIEIGTVGGGTNLLPQQACLQMLGVQGACKDNPGENARQLARIVCGTVMAGELSLMAALAAG Hydrogen bonds contact Hydrophobic contact | ||||
| 84 | NEDD8-activating enzyme E1C (UBA3) | 3GZN | 7.89 | |
Target general information Gen name UBA3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ubiquitin-like modifier-activating enzyme 3; Ubiquitin-activating enzyme E1C; Ubiquitin-activating enzyme 3; UBE1C; NEDD8-activating enzyme E1 catalytic subunit Protein family Ubiquitin-activating E1 family, UBA3 subfamily Biochemical class Carbon-sulfur ligase Function E1 activates NEDD8 by first adenylating its C-terminal glycine residue with ATP, thereafter linking this residue to the side chain of the catalytic cysteine, yielding a NEDD8-UBA3 thioester and free AMP. E1 finally transfers NEDD8 to the catalytic cysteine of UBE2M. Down-regulates steroid receptor activity. Necessary for cell cycle progression. Catalytic subunit of the dimeric UBA3-NAE1 E1 enzyme. Related diseases Galactosemia 3 (GALAC3) [MIM:230350]: A form of galactosemia, an inborn error of galactose metabolism typically manifesting in the neonatal period, after ingestion of galactose, with jaundice, hepatosplenomegaly, hepatocellular insufficiency, food intolerance, hypoglycemia, renal tubular dysfunction, muscle hypotonia, sepsis and cataract. GALAC3 is an autosomal recessive form caused by galactose epimerase deficiency. It can manifest as benign, peripheral form with mild symptoms and enzymatic deficiency in circulating blood cells only. A second form, known as generalized epimerase deficiency, is characterized by undetectable levels of enzyme activity in all tissues and severe clinical features, including restricted growth and intellectual disability. {ECO:0000269|PubMed:11279193, ECO:0000269|PubMed:11903335, ECO:0000269|PubMed:15639193, ECO:0000269|PubMed:16301867, ECO:0000269|PubMed:16302980, ECO:0000269|PubMed:9326324, ECO:0000269|PubMed:9538513, ECO:0000269|PubMed:9973283}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thrombocytopenia 13, syndromic (THC13) [MIM:620776]: An autosomal recessive form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC13 patients have enlarged, gray platelets with defective function. Some affected individuals have leukopenia or anemia and pancytopenia. Additional variable features include mitral valve malformations, pyloric stenosis, and impaired intellectual development. {ECO:0000269|PubMed:30247636, ECO:0000269|PubMed:33510604, ECO:0000269|PubMed:34159722, ECO:0000269|PubMed:36395340}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P02649; P05067; Q96FN4; Q9BVJ7; O14732; Q13564; P54727; Q8TBC4; P61081 EC number EC 6.2.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cell cycle; Ligase; Nucleotide-binding; Proteomics identification; Reference proteome; Ubl conjugation pathway Protein physicochemical properties Chain ID B,I Molecular weight (Da) 57143 Length 508 Aromaticity 0.08 Instability index 45.25 Isoelectric point 5.82 Charge (pH=7) -8.21 2D Binding mode Binding energy (Kcal/mol) -9.09 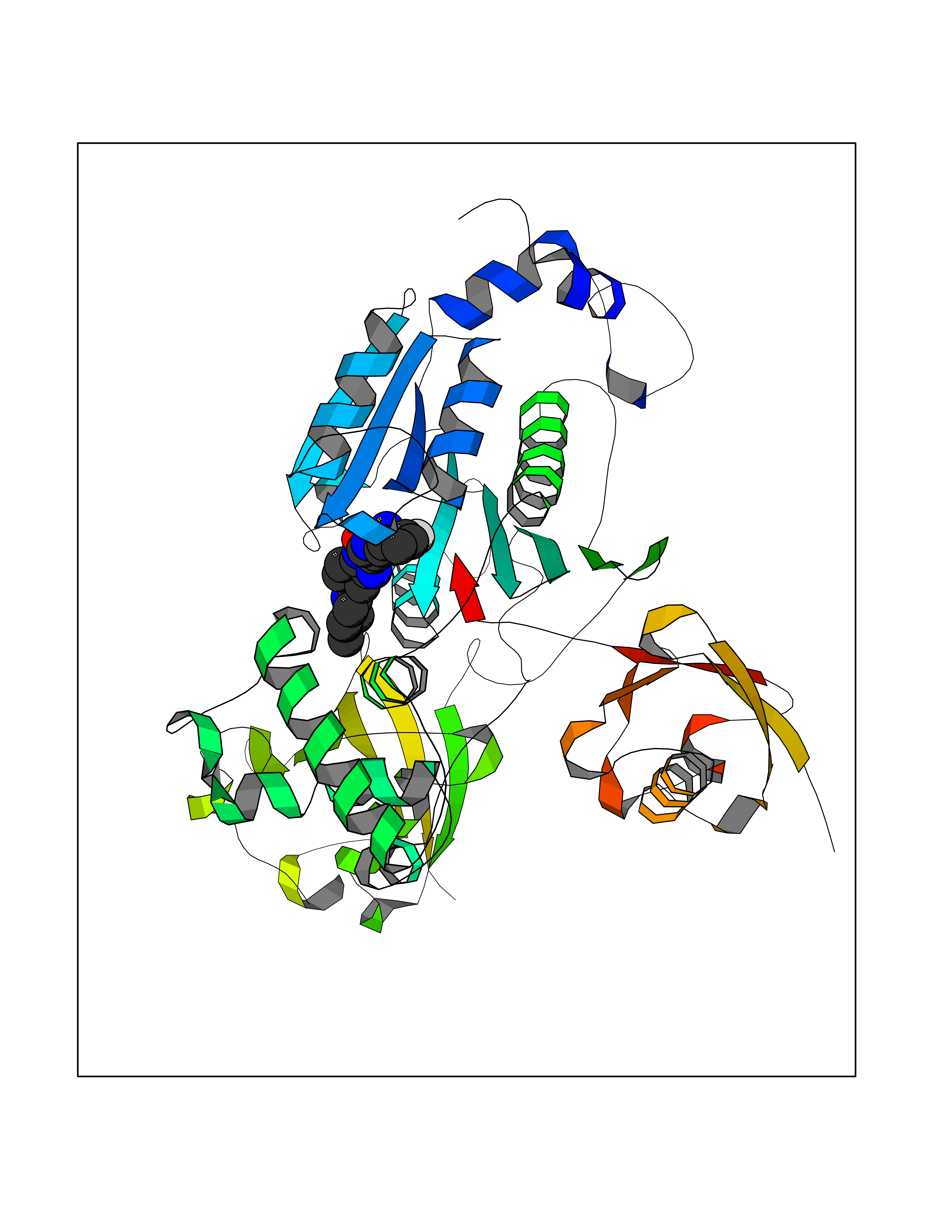 Molscript Map 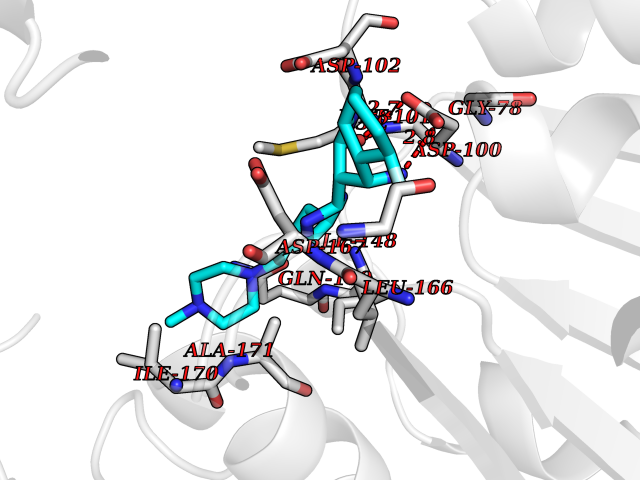 Pymol Map 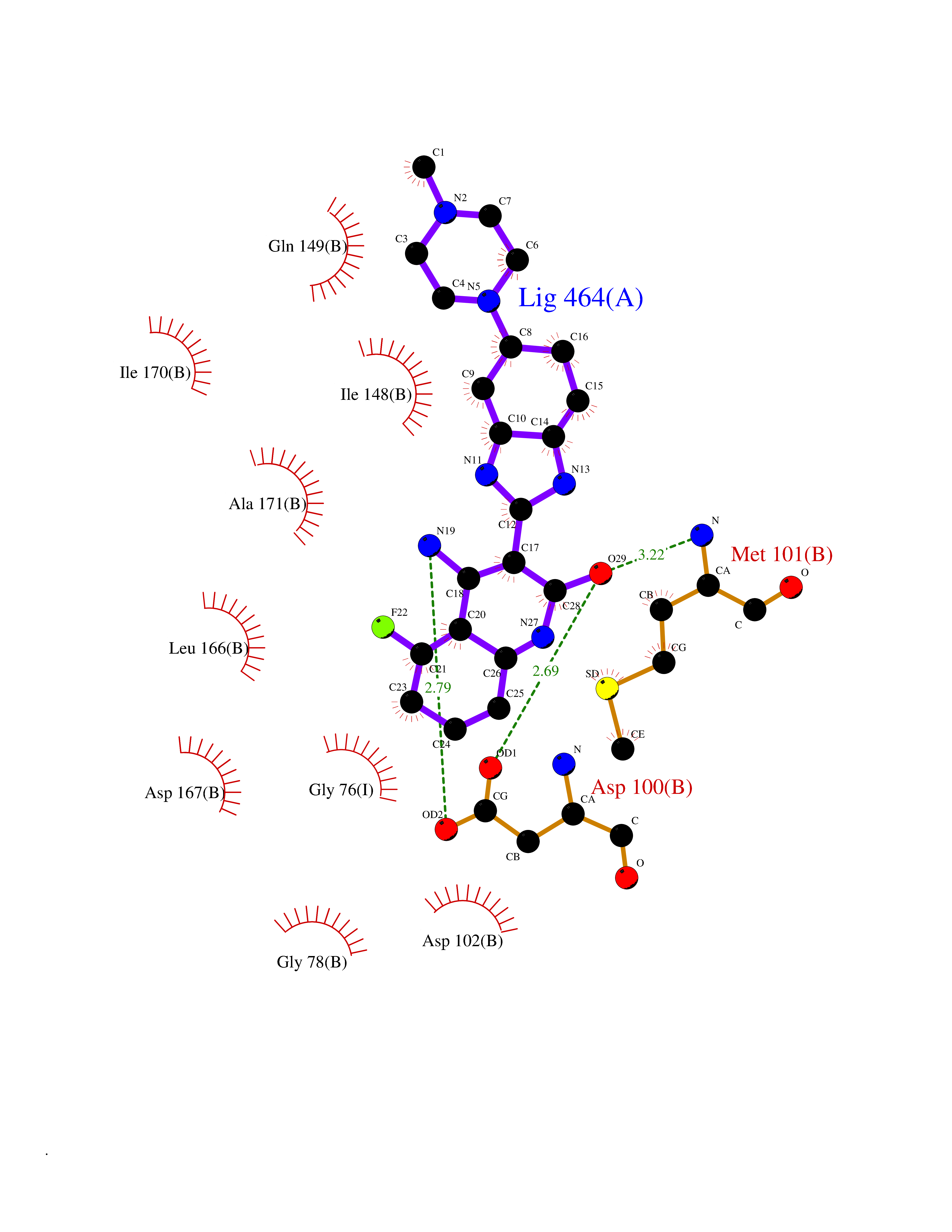 Ligplot Map 3D Binding mode Sequence DWEGRWNHVKKFLERSGPFTHPDFEPSTESLQFLLDTCKVLVIGAGGLGCELLKNLALSGFRQIHVIDMDTIDVSNLNRQFLFRPKDIGRPKAEVAAEFLNDRVPNCNVVPHFNKIQDFNDTFYRQFHIIVCGLDSIIARRWINGMLISLLNYEDGVLDPSSIVPLIDGGTEGFKGNARVILPGMTACIECTLELYPPQVNFPMCTIASMPRLPEHCIEYVRMLQWPKEQPFGEGVPLDGDDPEHIQWIFQKSLERASQYNIRGVTYRLTQGVVKRIIPAVASTNAVIAAVCATEVFKIATSAYIPLNNYLVFNDVDGLYTYTFEAERKENCPACSQLPQNIQFSPSAKLQEVLDYLTNSASLQMKSPAITATLEGKNRTLYLQSVTSIEERTRPNLSKLKELGLVDGQELAVADVTTPQTVLFKLHFTHHHMLIKVKTLTGKEIEIDIEPTDKVERIKERVEEKEGIPPQQQRLIYSGKQMNDEKTAADYKILGGSVLHLVLALRGG Hydrogen bonds contact Hydrophobic contact | ||||
| 85 | Caterpiller protein 1.1 (NLRP3) | 7PZC | 7.89 | |
Target general information Gen name NLRP3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PYRIN-containing APAF1-like protein 1; PYPAF1; NALP3; NACHT, LRR and PYD domains-containing protein 3; Cryopyrin; Cold-induced autoinflammatory syndrome 1 protein; CLR1.1; CIAS1; C1orf7; Angiotensin/v Protein family NLRP family Biochemical class NA Function In response to pathogens and other damage-associated signals, initiates the formation of the inflammasome polymeric complex, made of NLRP3, PYCARD and CASP1 (and possibly CASP4 and CASP5). Recruitment of proCASP1 to the inflammasome promotes its activation and CASP1-catalyzed IL1B and IL18 maturation and secretion in the extracellular milieu. Activation of NLRP3 inflammasome is also required for HMGB1 secretion. The active cytokines and HMGB1 stimulate inflammatory responses. Inflammasomes can also induce pyroptosis, an inflammatory form of programmed cell death. Under resting conditions, NLRP3 is autoinhibited. NLRP3 activation stimuli include extracellular ATP, reactive oxygen species, K(+) efflux, crystals of monosodium urate or cholesterol, amyloid-beta fibers, environmental or industrial particles and nanoparticles, cytosolic dsRNA, etc. However, it is unclear what constitutes the direct NLRP3 activator. Activation in presence of cytosolic dsRNA is mediated by DHX33. Independently of inflammasome activation, regulates the differentiation of T helper 2 (Th2) cells and has a role in Th2 cell-dependent asthma and tumor growth. During Th2 differentiation, required for optimal IRF4 binding to IL4 promoter and for IRF4-dependent IL4 transcription. Binds to the consensus DNA sequence 5'-GRRGGNRGAG-3'. May also participate in the transcription of IL5, IL13, GATA3, CCR3, CCR4 and MAF. As the sensor component of the NLRP3 inflammasome, plays a crucial role in innate immunity and inflammation. Related diseases Familial cold autoinflammatory syndrome 1 (FCAS1) [MIM:120100]: A rare autosomal dominant systemic inflammatory disease characterized by recurrent episodes of maculopapular rash associated with arthralgias, myalgias, fever and chills, swelling of the extremities, and conjunctivitis after generalized exposure to cold. Rarely, some patients may also develop late-onset renal amyloidosis. {ECO:0000269|PubMed:11687797, ECO:0000269|PubMed:11992256, ECO:0000269|PubMed:12355493, ECO:0000269|PubMed:12522564, ECO:0000269|PubMed:15593220, ECO:0000269|PubMed:17284928, ECO:0000269|PubMed:24952504}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Muckle-Wells syndrome (MWS) [MIM:191900]: A hereditary periodic fever syndrome characterized by fever, chronic recurrent urticaria, arthralgias, progressive sensorineural deafness, and reactive renal amyloidosis. The disease may be severe if generalized reactive amyloidosis occurs. {ECO:0000269|PubMed:11687797, ECO:0000269|PubMed:11992256, ECO:0000269|PubMed:12355493, ECO:0000269|PubMed:15593220, ECO:0000269|PubMed:24952504}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Chronic infantile neurologic cutaneous and articular syndrome (CINCA) [MIM:607115]: Rare congenital inflammatory disorder characterized by a triad of neonatal onset of cutaneous symptoms, chronic meningitis, and joint manifestations with recurrent fever and inflammation. {ECO:0000269|PubMed:12032915, ECO:0000269|PubMed:12483741, ECO:0000269|PubMed:14630794, ECO:0000269|PubMed:15231984, ECO:0000269|PubMed:15334500, ECO:0000269|PubMed:15593220, ECO:0000269|PubMed:24952504, ECO:0000269|PubMed:31086329}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Keratoendothelitis fugax hereditaria (KEFH) [MIM:148200]: An autosomal dominant corneal disease that periodically, and fleetingly, affects the corneal endothelium, stroma, and vision, eventually leading to central corneal stromal opacities in some patients. The disease is characterized by unilateral attacks of ocular pain, pericorneal injection, and photophobia. The acute symptoms vanish in 1-2 days but vision remains blurry for several weeks. The attacks start at the age of 3-12 years and can affect either eye. They generally decrease in frequency and get milder with age. {ECO:0000269|PubMed:29366613, ECO:0000269|PubMed:35559676}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, autosomal dominant, 34, with or without inflammation (DFNA34) [MIM:617772]: A form of sensorineural hearing loss. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. DFNA34 is a postlingual, slowly progressive form with variable severity and variable additional features. Some DFNA34 patients have autoinflammatory manifestations. {ECO:0000269|PubMed:28847925}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P27797; P36957; P19525; Q7Z434; Q8TDX7; Q96P20; Q9ULZ3; Q80H93; P0DTC9; Q9ES74; Q8TDX7-1 EC number NA Uniprot keywords 3D-structure; Activator; ADP-ribosylation; Alternative splicing; Amyloidosis; ATP-binding; Cytoplasm; Cytoskeleton; Deafness; Disease variant; Disulfide bond; Endoplasmic reticulum; Golgi apparatus; Hydrolase; Immunity; Inflammasome; Inflammatory response; Innate immunity; Isopeptide bond; Leucine-rich repeat; Lipoprotein; Membrane; Mitochondrion; Non-syndromic deafness; Nucleotide-binding; Nucleus; Palmitate; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Secreted; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 115171 Length 1010 Aromaticity 0.08 Instability index 45.92 Isoelectric point 6.17 Charge (pH=7) -11.22 2D Binding mode Binding energy (Kcal/mol) -9.21  Molscript Map 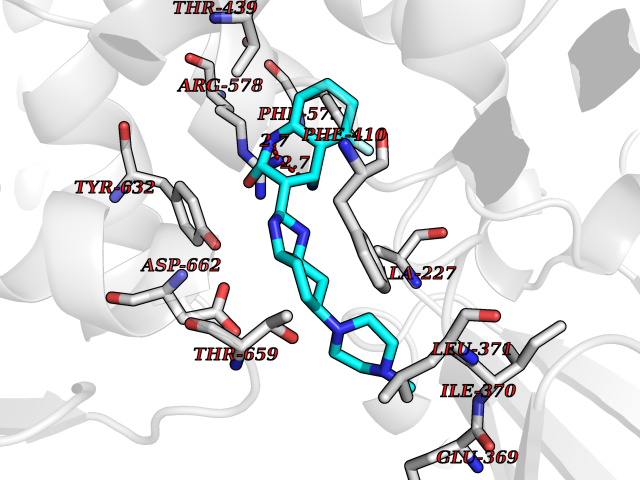 Pymol Map 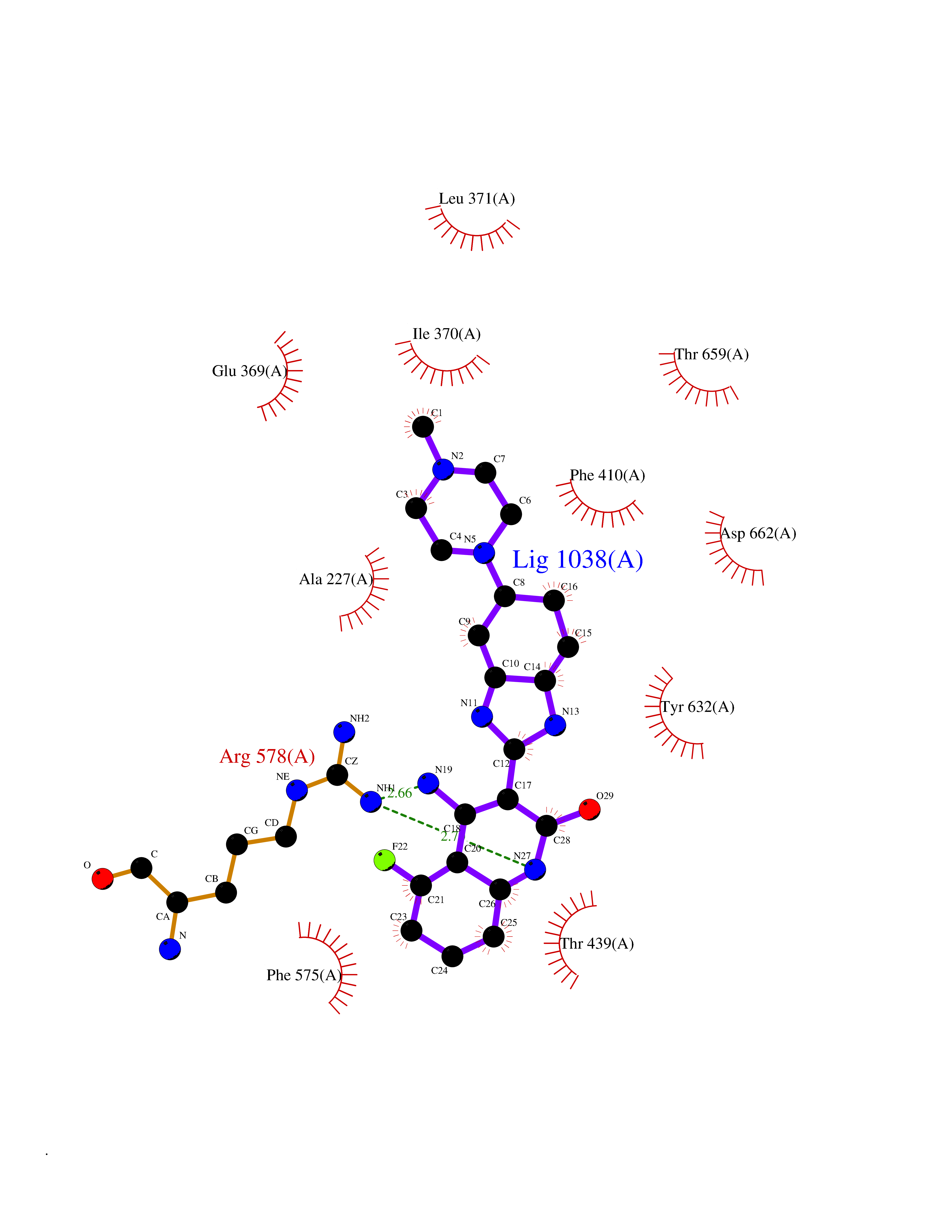 Ligplot Map 3D Binding mode Sequence MASTRCKLARYLEDLEDVDLKKFKMHLEDYPPQKGCIPLPRGQTEKADHVDLATLMIDFNGEEKAWAMAVWIFAAINRRDLYEKAKRDEPKWGSDNARVSNPTVICQEDSIEEEWMGLLEYLSRISICKMKKDYRKKYRKYVRSRFQCIEESVSLNKRYTRLRLIKEHRSQSPVSPIKMELLFDPDDEHSEPVHTVVFQGAAGIGKTILARKMMLDWASGTLYQDRFDYLFYIHCREVSLVTQRSLGDLIMSCCPDPNPPIHKIVRKPSRILFLMDGFDELQGAFDEHIGPLCTDWQKAERGDILLSSLIRKKLLPEASLLITTRPVALEKLQHLLDHPRHVEILGFSEAKRKEYFFKYFSDEAQARAAFSLIQENEVLFTMCFIPLVCWIVCTGLKQQMESGKSLAQTSKTTTAVYVFFLSSLLQPRGGSQEHGLCAHLWGLCSLAADGIWNQKILFEESDLRNHGLQKADVSAFLRMNLFQKEVDCEKFYSFIHMTFQEFFAAMYYLLEEEKEGRTNVPGSRLKLPSRDVTVLLENYGKFEKGYLIFVVRFLFGLVNQERTSYLEKKLSCKISQQIRLELLKWIEVKAKAKKLQIQPSQLELFYCLYEMQEEDFVQRAMDYFPKIEINLSTRMDHMVSSFCIENCHRVESLSLGFLHNMPKEEEEEEKEGRHLDMVQCVLPSSSHAACSHGLVNSHLTSSFCRGLFSVLSTSQSLTELDLSDNSLGDPGMRVLCETLQHPGCNIRRLWLGRCGLSHECCFDISLVLSSNQKLVELDLSDNALGDFGIRLLCVGLKHLLCNLKKLWLVSCCLTSACCQDLASVLSTSHSLTRLYVGENALGDSGVAILCEKAKNPQCNLQKLGLVNSGLTSVCCSALSSVLSTNQNLTHLYLRGNTLGDKGIKLLCEGLLHPDCKLQVLELDNCNLTSHCCWDLSTLLTSSQSLRKLSLGNNDLGDLGVMMFCEVLKQQSCLLQNLGLSEMYFNYETKSALETLQEEKPELTVVFEPSW Hydrogen bonds contact Hydrophobic contact | ||||
| 86 | Histone acetyltransferase KAT2B (KAT2B) | 1CM0 | 7.88 | |
Target general information Gen name KAT2B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Spermidine acetyltransferase KAT2B; PCAF; P300/CBP-associated factor; P/CAF; Lysine acetyltransferase 2B; Histone acetyltransferase PCAF; Histone acetylase PCAF Protein family Acetyltransferase family, GCN5 subfamily Biochemical class Acyltransferase Function Has significant histone acetyltransferase activity with core histones (H3 and H4), and also with nucleosome core particles. Also acetylates non-histone proteins, such as ACLY, PLK4 and TBX5. Inhibits cell-cycle progression and counteracts the mitogenic activity of the adenoviral oncoprotein E1A. Acts as a circadian transcriptional coactivator which enhances the activity of the circadian transcriptional activators: NPAS2-ARNTL/BMAL1 and CLOCK-ARNTL/BMAL1 heterodimers. Involved in heart and limb development by mediating acetylation of TBX5, acetylation regulating nucleocytoplasmic shuttling of TBX5. Acts as a negative regulator of centrosome amplification by mediating acetylation of PLK4. Also acetylates spermidine. Functions as a histone acetyltransferase (HAT) to promote transcriptional activation. Related diseases Defects in KAT2B has been found in a patient with isolated coloboma, a defect of the eye characterized by the absence of ocular structures due to abnormal morphogenesis of the optic cup and stalk, and the fusion of the fetal fissure (optic fissure). Isolated colobomas may be associated with an abnormally small eye (microphthalmia) or small cornea. {ECO:0000269|PubMed:28493397}. Drugs (DrugBank ID) DB08186; DB01992; DB08291 Interacts with O60566; Q92793; Q96KQ7; Q09472; Q16665; Q9Y5W3; Q96EB6; Q8IXJ6; Q7Z699; Q16594; Q15672; P22415; P28033; P03129; P02299; P84040; P0DTC9; P59595; O88898-2; P03255; P03255-2 EC number EC 2.3.1.48 Uniprot keywords 3D-structure; Activator; Acyltransferase; Biological rhythms; Bromodomain; Cell cycle; Cytoplasm; Cytoskeleton; Disease variant; Host-virus interaction; Nucleus; Proteomics identification; Reference proteome; Transcription; Transcription regulation; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 18743.8 Length 162 Aromaticity 0.12 Instability index 25.09 Isoelectric point 9.44 Charge (pH=7) 9.34 2D Binding mode Binding energy (Kcal/mol) -8.58  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence KVIEFHVVGNSLNQKPNKKILMWLVGLQNVFSHQLPRMPKEYITRLVFDPKHKTLALIKDGRVIGGICFRMFPSQGFTEIVFCAVTSNEQVKGYGTHLMNHLKEYHIKHDILNFLTYADEYAIGYFKKQGFSKEIKIPKTKYVGYIKDYEGATLMGCELNPR Hydrogen bonds contact Hydrophobic contact | ||||
| 87 | Wnt-7a protein (WNT7A) | 4UZQ | 7.88 | |
Target general information Gen name WNT7A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein Wnt-7a Protein family Wnt family Biochemical class NA Function Plays an important role in embryonic development, including dorsal versus ventral patterning during limb development, skeleton development and urogenital tract development. Required for central nervous system (CNS) angiogenesis and blood-brain barrier regulation. Required for normal, sexually dimorphic development of the Mullerian ducts, and for normal fertility in both sexes. Required for normal neural stem cell proliferation in the hippocampus dentate gyrus. Required for normal progress through the cell cycle in neural progenitor cells, for self-renewal of neural stem cells, and for normal neuronal differentiation and maturation. Promotes formation of synapses via its interaction with FZD5. Ligand for members of the frizzled family of seven transmembrane receptors that functions in the canonical Wnt/beta-catenin signaling pathway. Related diseases Limb pelvis hypoplasia aplasia syndrome (LPHAS) [MIM:276820]: A syndrome of severe deficiency of the extremities due to hypo- or aplasia of one or more long bones of one or more limbs. Pelvic manifestations include hip dislocation, hypoplastic iliac bone and aplastic pubic bones. Thoracic deformity, unusual facies and genitourinary anomalies can be present. {ECO:0000269|PubMed:16826533, ECO:0000269|PubMed:17431918, ECO:0000269|PubMed:20949531, ECO:0000269|PubMed:21271649, ECO:0000269|PubMed:27638328}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fuhrmann syndrome (FUHRS) [MIM:228930]: Distinct limb-malformation disorder characterized also by various degrees of limb aplasia/hypoplasia and joint dysplasia. {ECO:0000269|PubMed:16826533}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P55212; P22607; P06396; P13473-2; Q9UMX0; Q9Y5W5; Q5T9L3; Q9Z0J1 EC number NA Uniprot keywords 3D-structure; Developmental protein; Disease variant; Disulfide bond; Extracellular matrix; Glycoprotein; Lipoprotein; Proteomics identification; Reference proteome; Secreted; Signal; Wnt signaling pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 40475.5 Length 356 Aromaticity 0.1 Instability index 50.49 Isoelectric point 7.67 Charge (pH=7) 1.62 2D Binding mode Binding energy (Kcal/mol) -8.42  Molscript Map  Pymol Map 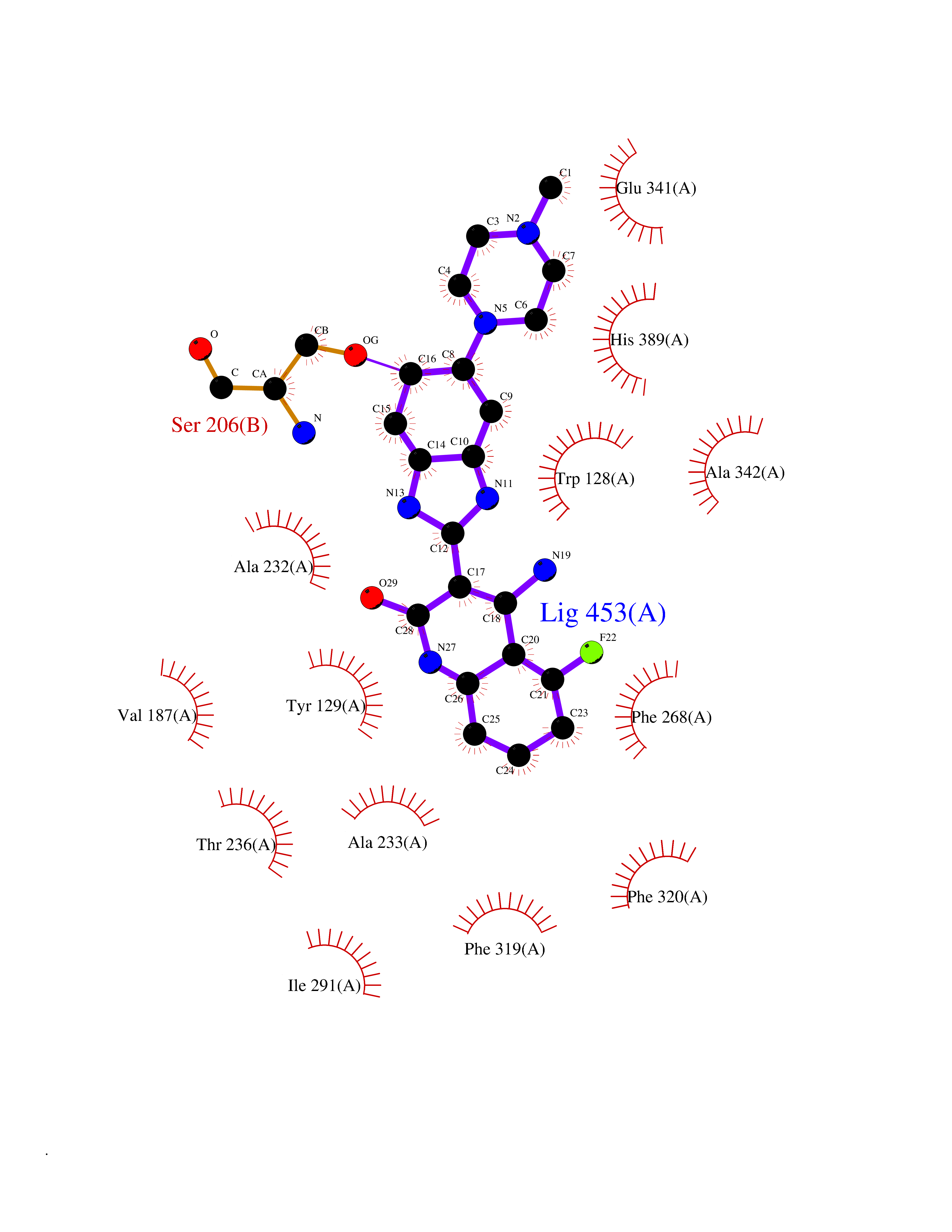 Ligplot Map 3D Binding mode Sequence EDLRLHLLLNTSVTCNDGSPAGYYLKESRGSRRWLLFLEGGWYCFNRENCDSRYDTMRRLMSSRDWPRTRTGTGILSSQPEENPYWWNANMVFIPYCSSDVWSGASSKSEKNEYAFMGALIIQEVVRELLGRGLSGAKVLLLAGSAAGGTGVLLNVDRVAEQLEKLGYPAIQVRGLADSGWFLDNKQYRHTDCVDTITCAPTEAIRRGIRYWNGVVPERCRRQFQEGEEWNCFFGYKVYPTLRSPVFVVQWLFDEAQLTVDNVHLTGQPVQEGLRLYIQNLGRELRHTLKDVPASFAPACLSHEIIIRSHWTDVQVKGTSLPRALHCWDRSLHKGCPVHLVDSCPWPHCNPSCPTS Hydrogen bonds contact Hydrophobic contact | ||||
| 88 | Pol polyprotein | 5KAO | 7.87 | |
Target general information Gen name pol Organism Human immunodeficiency virus type 1 (HIV-1) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase / hydrolase inhibitor Function Aspartic-type endopeptidase activity. Related diseases Oocyte/zygote/embryo maturation arrest 16 (OZEMA16) [MIM:617234]: A rare cause of female primary infertility. In affected women, ovulation and fertilization proceed normally but embryos are arrested at early stages of development. Inheritance is autosomal recessive. {ECO:0000269|PubMed:27545678}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00701; DB01072; DB04887; DB01264; DB01319; DB00224; DB01601; DB00503; DB01232; DB00932 Interacts with NA EC number NA Uniprot keywords 3D-structure; Aspartyl protease; Hydrolase; Protease Protein physicochemical properties Chain ID A,B Molecular weight (Da) 21411 Length 198 Aromaticity 0.05 Instability index 42.78 Isoelectric point 9.45 Charge (pH=7) 4.15 2D Binding mode Binding energy (Kcal/mol) -8.6  Molscript Map 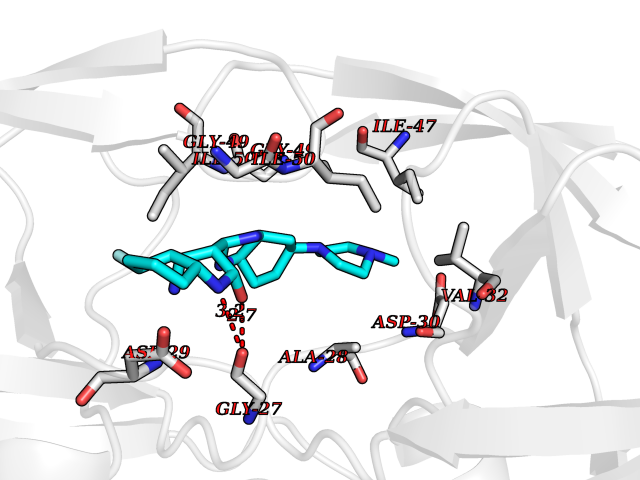 Pymol Map 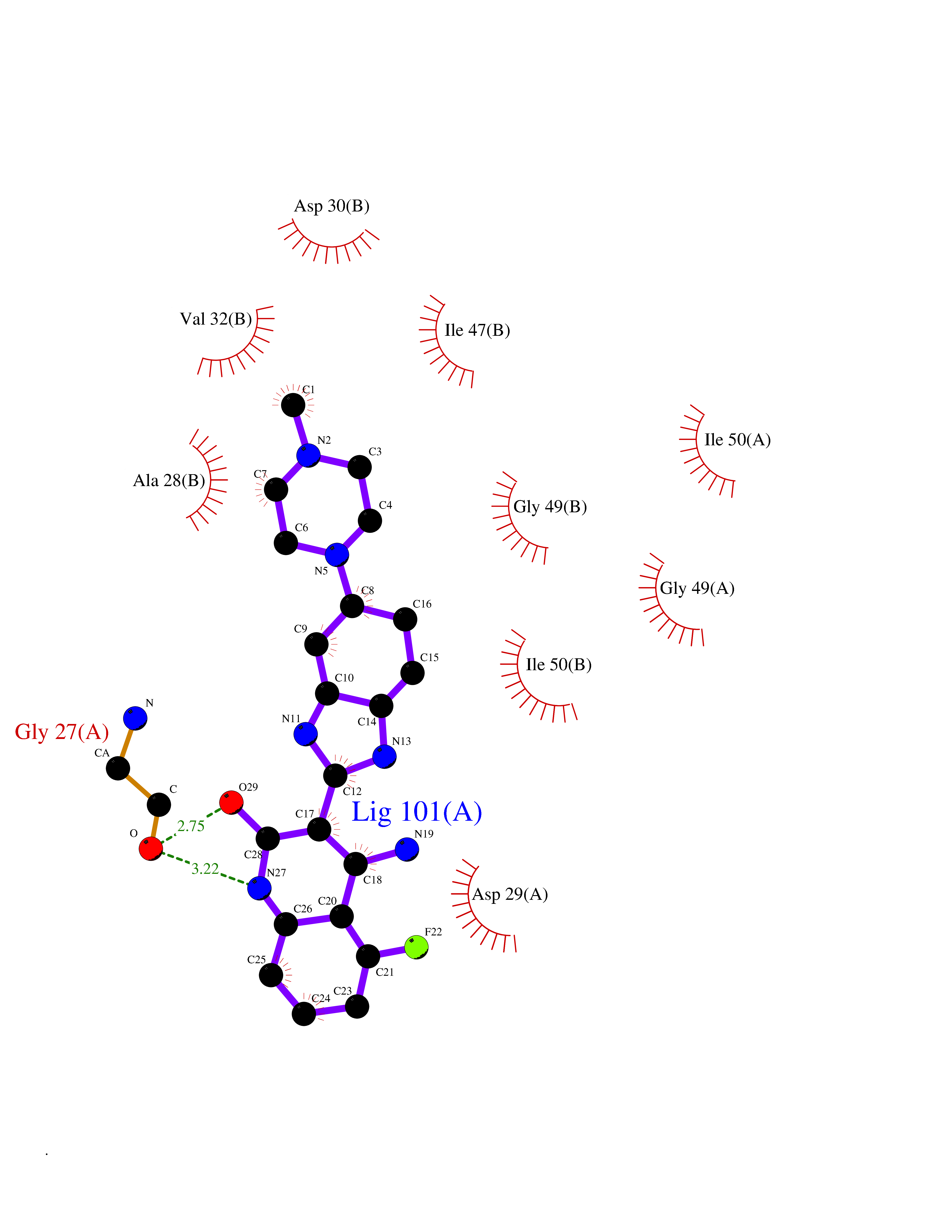 Ligplot Map 3D Binding mode Sequence PQVTLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYDQILIEIAGHKAIGTVLVGPTPVNIIGRNLLTQIGATLNFPQVTLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYDQILIEIAGHKAIGTVLVGPTPVNIIGRNLLTQIGATLNF Hydrogen bonds contact Hydrophobic contact | ||||
| 89 | Monoamine oxidase type A (MAO-A) | 2Z5Y | 7.87 | |
Target general information Gen name MAOA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Monoamine oxidase A; Amine oxidase [flavin-containing] A Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function MAOA preferentially oxidizes biogenic amines such as 5-hydroxytryptamine (5-HT), norepinephrine and epinephrine. Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. Related diseases Brunner syndrome (BRNRS) [MIM:300615]: A form of X-linked non-dysmorphic mild intellectual disability. Male patients are affected by borderline intellectual deficit and exhibit abnormal behavior, including disturbed regulation of impulsive aggression. Obligate female carriers have normal intelligence and behavior. {ECO:0000269|PubMed:8211186}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01472; DB00918; DB00182; DB06698; DB04889; DB13876; DB01445; DB06774; DB00215; DB04017; DB09130; DB05205; DB07641; DB00988; DB01363; DB00668; DB12329; DB01175; DB03147; DB14914; DB00614; DB01381; DB07919; DB04818; DB01247; DB00601; DB01577; DB00805; DB01442; DB01171; DB08804; DB00952; DB04820; DB00184; DB04821; DB06412; DB01626; DB00780; DB00191; DB00388; DB00397; DB09244; DB04850; DB00721; DB01168; DB00571; DB00852; DB09363; DB00140; DB00953; DB06654; DB01037; DB01104; DB00669; DB14569; DB09042; DB00624; DB13943; DB13944; DB13946; DB09245; DB00752; DB15328; DB09185; DB04832; DB00315; DB00909 Interacts with P27338 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Catecholamine metabolism; Direct protein sequencing; Disease variant; FAD; Flavoprotein; Intellectual disability; Membrane; Mitochondrion; Mitochondrion outer membrane; Neurotransmitter degradation; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 58195.3 Length 513 Aromaticity 0.11 Instability index 34.97 Isoelectric point 7.98 Charge (pH=7) 2.87 2D Binding mode Binding energy (Kcal/mol) -9.77 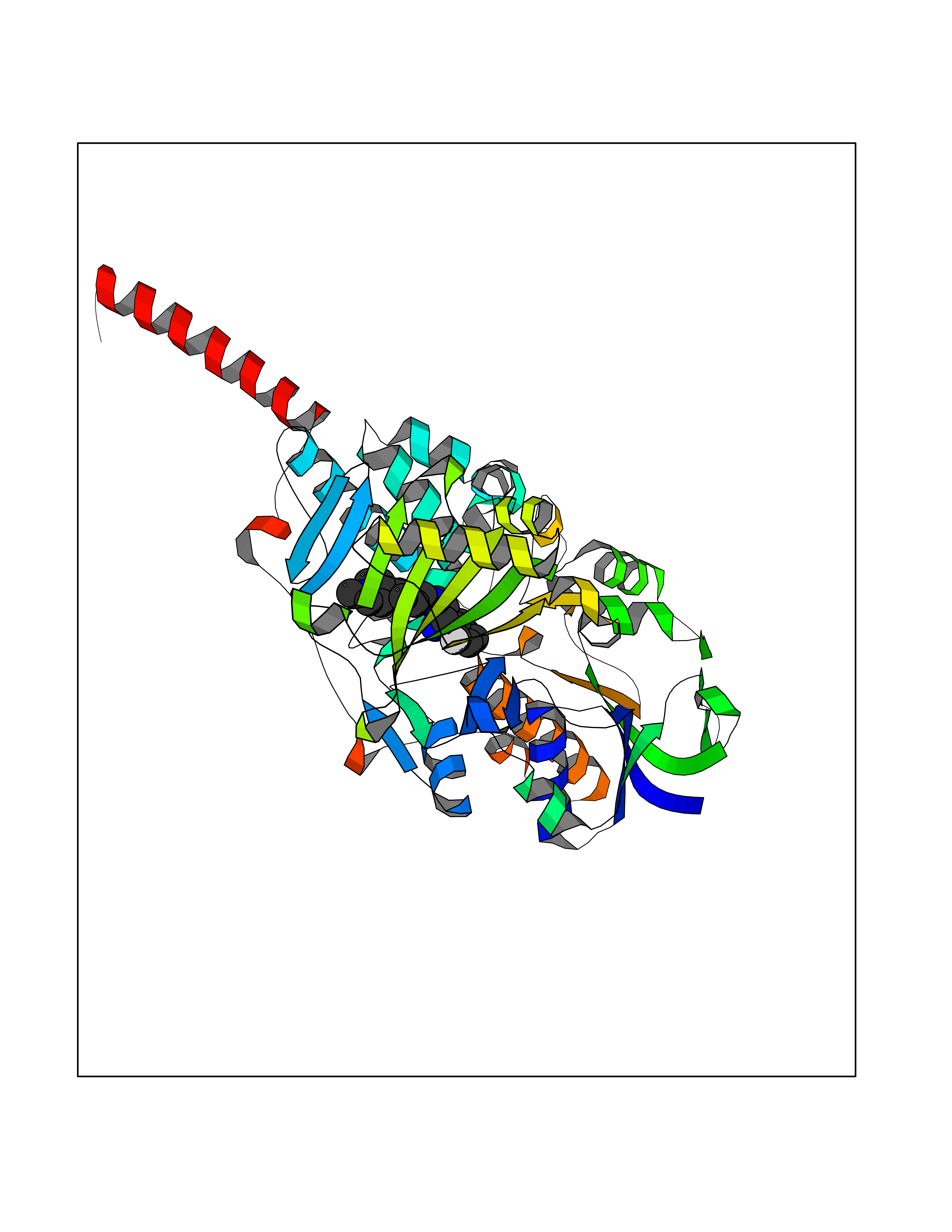 Molscript Map 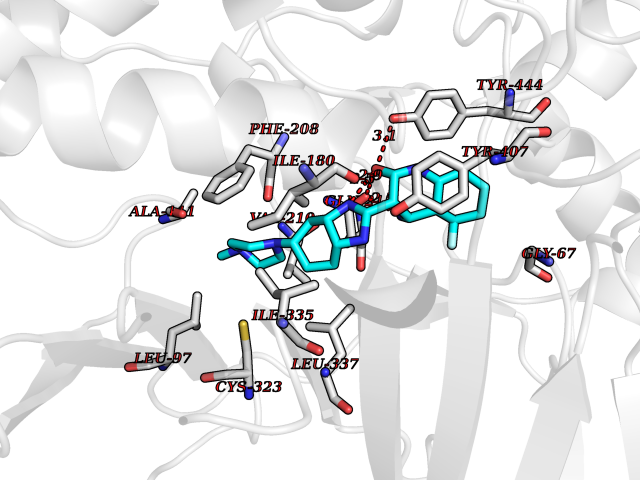 Pymol Map 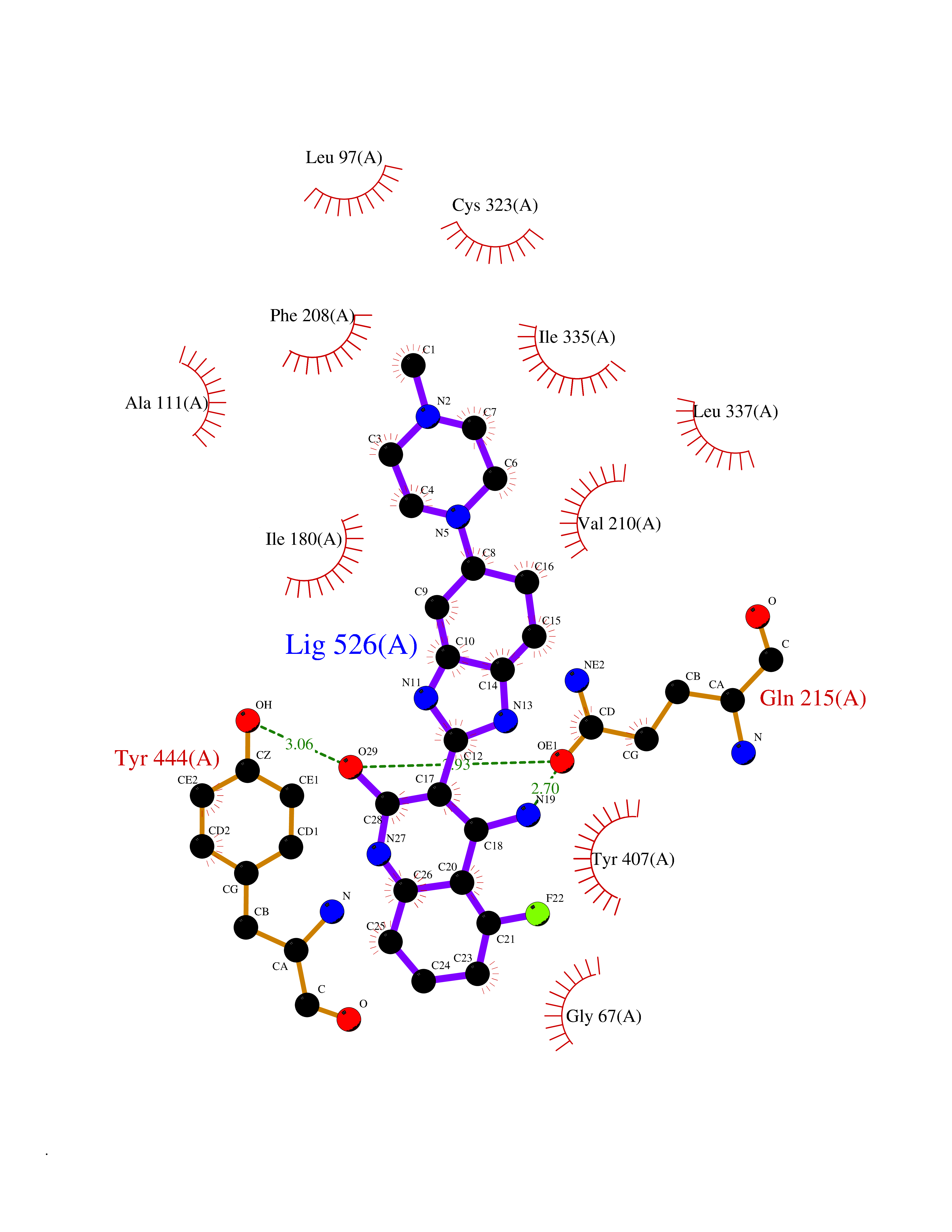 Ligplot Map 3D Binding mode Sequence HMFDVVVIGGGISGLSAAKLLTEYGVSVLVLEARDRVGGRTYTIRNEHVDYVDVGGAYVGPTQNRILRLSKELGIETYKVNVSERLVQYVKGKTYPFRAAFPPVWNPIAYLDYNNLWRTIDNMGKEIPTDAPWEAQHADKWDKMTMKELIDKICWTKTARRFAYLFVNINVTSEPHEVSALWFLWYVKQCGGTTRIFSVTNGGQERKFVGGSGQVSERIMDLLGDQVKLNHPVTHVDQSSDNIIIETLNHEHYECKYVINAIPPTLTAKIHFRPELPAERNQLIQRLPMGAVIKCMMYYKEAFWKKKDYCGCMIIEDEDAPISITLDDTKPDGSLPAIMGFILARKADRLAKLHKEIRKKKICELYAKVLGSQEALHPVHYEEKNWCEEQYSGGCYTAYFPPGIMTQYGRVIRQPVGRIFFAGTETATKWSGYMEGAVEAGERAAREVLNGLGKVTEKDIWVQEPESKDVPAVEITHTFWERNLPSVSGLLKIIGFSTSVTALGFVLYKYKLL Hydrogen bonds contact Hydrophobic contact | ||||
| 90 | Phosphodiesterase 3A (PDE3A) | 7EG0 | 7.86 | |
Target general information Gen name PDE3A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cGMP-inhibited 3',5'-cyclic phosphodiesterase A; Phosphodiesterase 3A; Cyclic GMP-inhibited phosphodiesterase A; Cyclic GMP inhibited phosphodiesterase A; CGI-PDE A Protein family Cyclic nucleotide phosphodiesterase family, PDE3 subfamily Biochemical class Phosphoric diester hydrolase Function Cyclic nucleotide phosphodiesterase with a dual-specificity for the second messengers cAMP and cGMP, which are key regulators of many important physiological processes. Related diseases Hypertension and brachydactyly syndrome (HTNB) [MIM:112410]: A syndrome characterized by brachydactyly type E, severe salt-independent but age-dependent hypertension, an increased fibroblast growth rate, neurovascular contact at the rostral-ventrolateral medulla, and altered baroreflex blood pressure regulation. It results in death from stroke before age 50 years when untreated. Brachydactyly type E is characterized by shortening of the fingers mainly in the metacarpals and metatarsals. {ECO:0000269|PubMed:25961942}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01223; DB01427; DB00261; DB00201; DB01166; DB04880; DB05266; DB00922; DB00235; DB01303; DB00277; DB08811; DB09283 Interacts with Q9Y6D6; Q9Y6D5 EC number EC 3.1.4.17 Uniprot keywords 3D-structure; cAMP; cGMP; Cytoplasm; Direct protein sequencing; Disease variant; Hydrolase; Isopeptide bond; Manganese; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 108171 Length 939 Aromaticity 0.11 Instability index 39.75 Isoelectric point 6.53 Charge (pH=7) -4.15 2D Binding mode Binding energy (Kcal/mol) -9.25 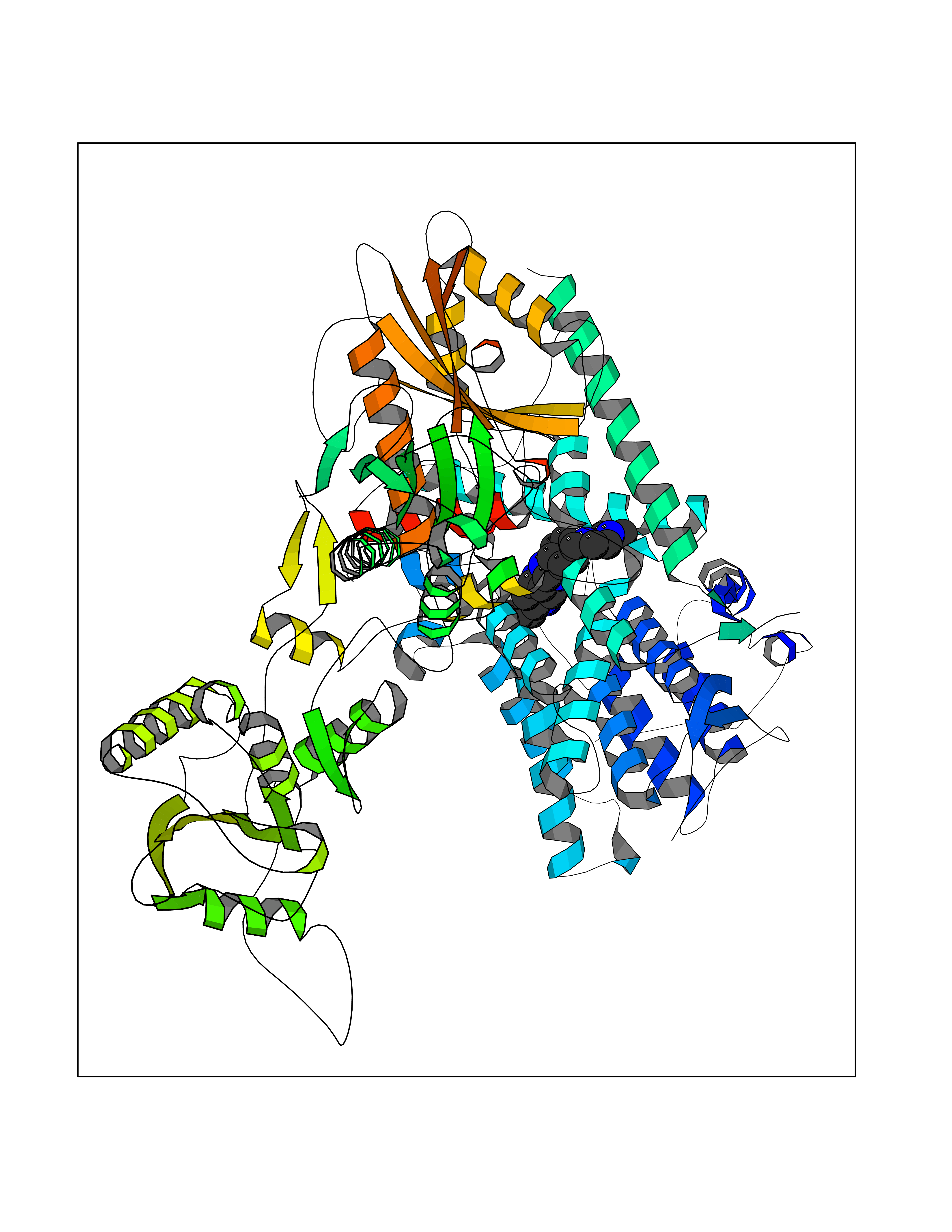 Molscript Map 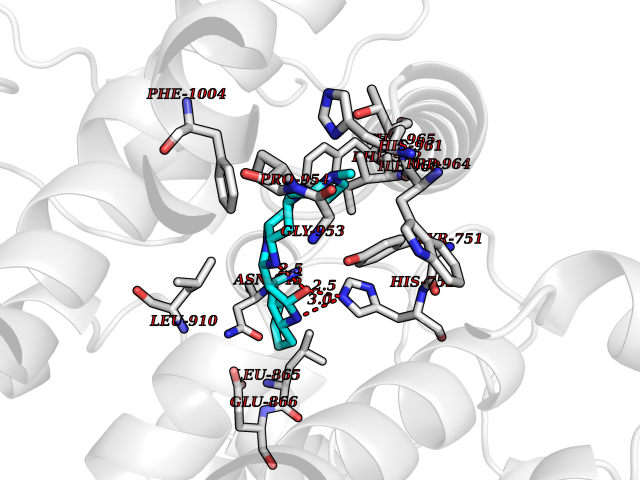 Pymol Map 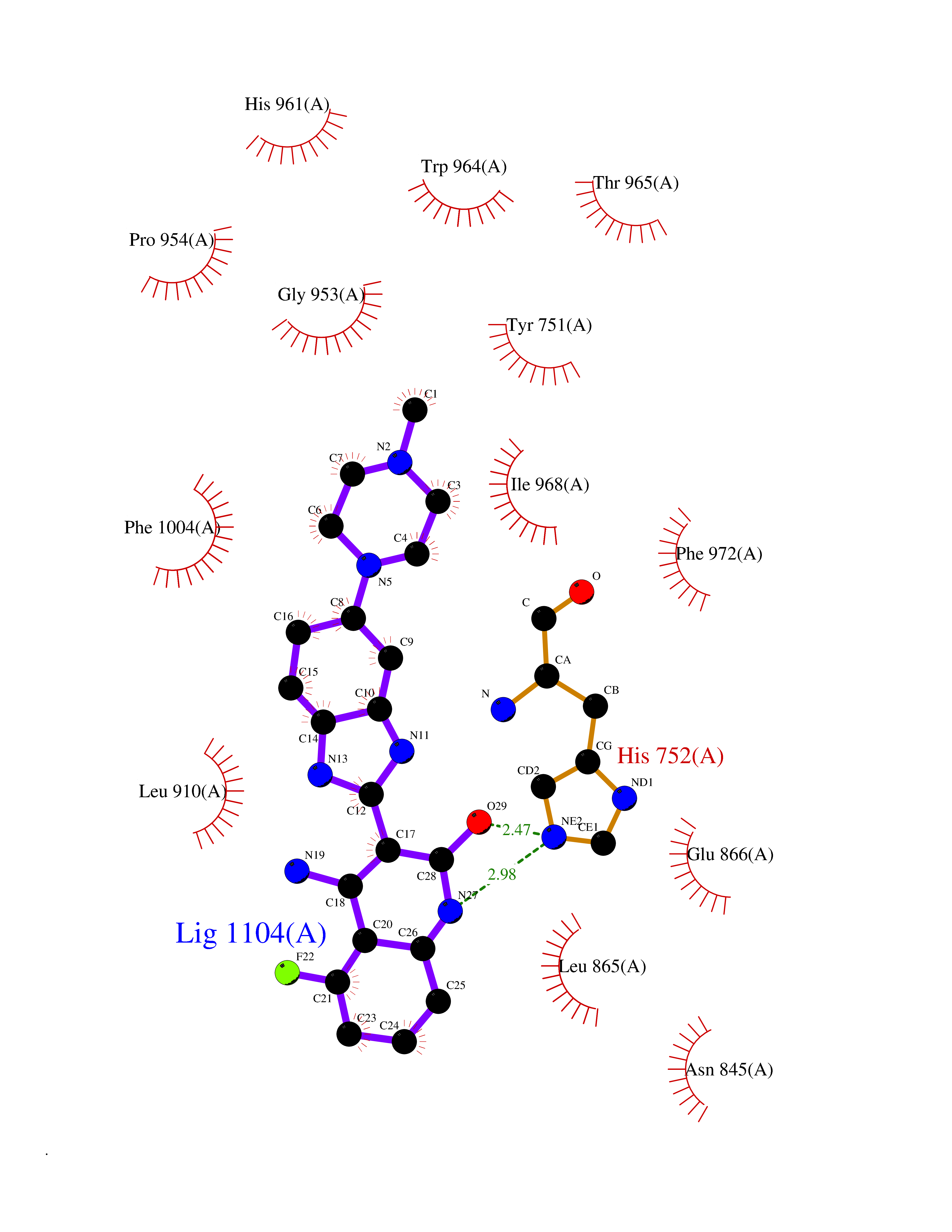 Ligplot Map 3D Binding mode Sequence KPILAPEPLVMDNLDSIMEQLNTWNFPIFDLVENIGRKCGRILSQVSYRLFEDMGLFEAFKIPIREFMNYFHALEIGYRDIPYHNRIHATDVLHAVWYLTTQPIPGLSTVGYVFSKTYNVTDDKYGCLSGNIPALELMALYVAAAMHDYDHPGRTNAFLVATSAPQAVLYNDRSVLENHHAAAAWNLFMSRPEYNFLINLDHVEFKHFRFLVIEAILATDLKKHFDFVAKFNGKVNDDVGIDWTNENDRLLVCQMCIKLADINGPAKCKELHLQWTDGIVNEFYEQGDEEASLGLPISPFMDRSAPQLANLQESFISHIVGPLCNSYDSAGLMPGKWVEKIYCQITQHLLQNHKMWKKVIEEEQRLAGIENQNISVDLETNYAELVLDVGRVTLGENSRKKMKDCKLRKKQNESVSRAMCALLNSGGGVIKAEIENEDYSYTKDGIGLDLENSFSNILLFVPEYLDFMQNGNYFLIFVKSWSLNTSGLRITTLSSNLYKRDITSAKVMNATAALEFLKDMKKTRGRLYLRPELLAKRPCVDIQEENNMKALAGVFFDRTELDRKEKLTFTESTHVEIKNFSTERLLQRIKEILPQYVSAFANTDGGYLFIGLNEDKEIIGFKAEMSDLDDLEREIEKSIRKMPVHHFCMEKKKINYSCKFLGVYDKGSLCGYVCALRVERFCCAVFAKEPDSWHVKDNRVMQLTRKEWIQFMVEAEPKFSSAYEEVISQINTSLPAPHSWPLLEWQRQRHHCPGLSGRITYTPENLCRKLFLQHEGLKQLICEEMSSVRKGSLIFSRSWSVDLGLQENHKVLCDALLISQDSPPVLYTFHMVQDEEFKGYSTQTALTLKQKLAKIGGYTKKVCVMTKIFYLSPEGMTSCQYDLRSQVIYPESYYFTRRKYLLKALFKALKRLKSLRDQFSFAENLYQIIGIDCFQKNDK Hydrogen bonds contact Hydrophobic contact | ||||
| 91 | Phenylethanolamine N-methyltransferase (PNMT) | 2G72 | 7.86 | |
Target general information Gen name PNMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNMTase; PENT; Noradrenaline N-methyltransferase Protein family Class I-like SAM-binding methyltransferase superfamily, NNMT/PNMT/TEMT family Biochemical class NA Function Converts noradrenaline to adrenaline. Related diseases A chromosomal aberration involving TRIM24/TIF1 is found in papillary thyroid carcinomas (PTCs). Translocation t(7;10)(q32;q11) with RET. The translocation generates the TRIM24/RET (PTC6) oncogene. {ECO:0000269|PubMed:10439047}. Drugs (DrugBank ID) DB08129; DB08128; DB07739; DB07798; DB07747; DB03468; DB08550; DB03824; DB04273; DB07906; DB07597; DB09571; DB00968; DB08631; DB01752; DB08654 Interacts with Q9P2G9-2; Q8TBB1 EC number EC 2.1.1.28 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 29198.9 Length 264 Aromaticity 0.09 Instability index 54.33 Isoelectric point 5.91 Charge (pH=7) -3.69 2D Binding mode Binding energy (Kcal/mol) -10  Molscript Map 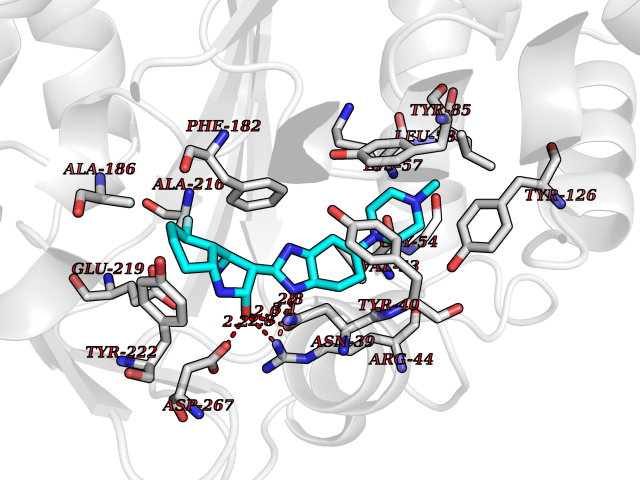 Pymol Map 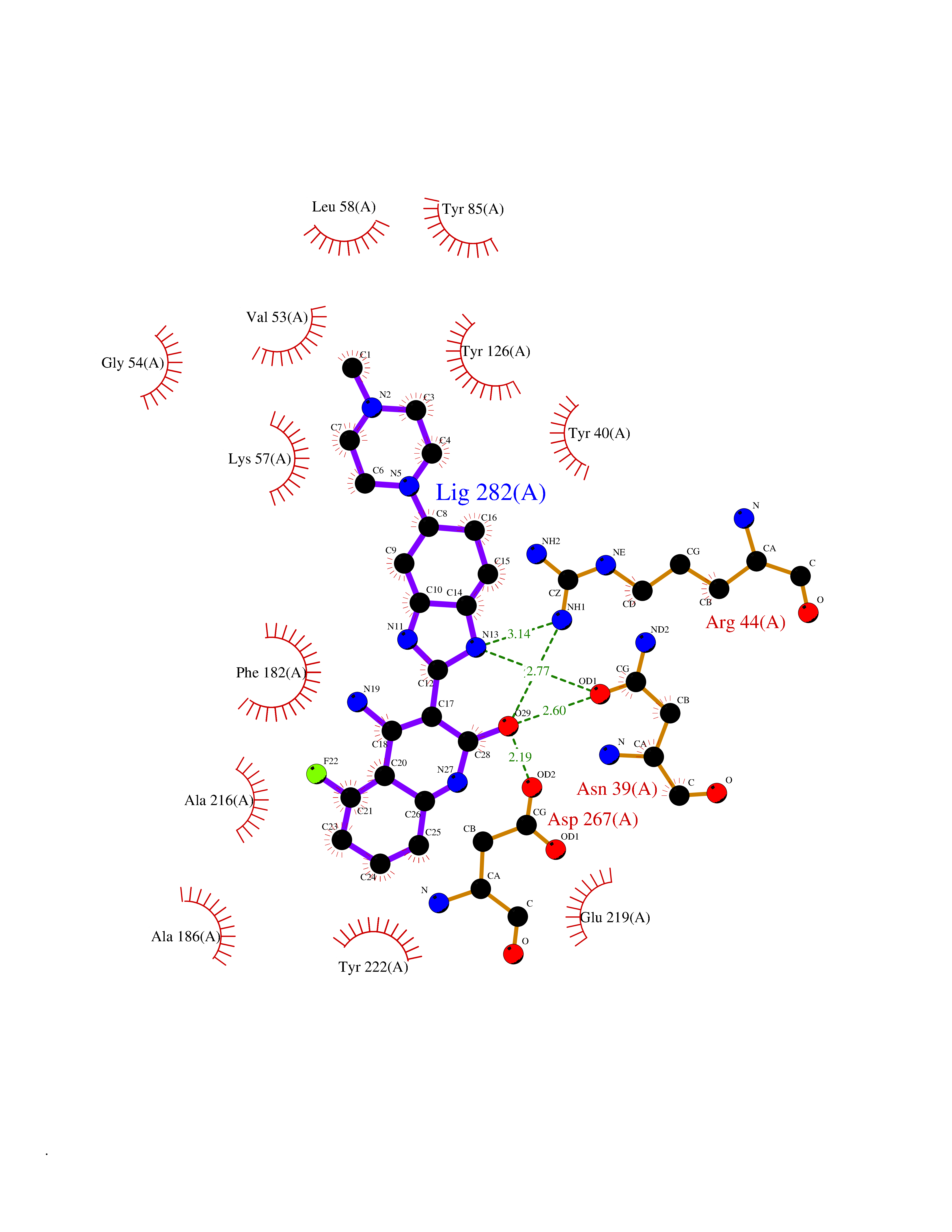 Ligplot Map 3D Binding mode Sequence APGQAAVASAYQRFEPRAYLRNNYAPPRGDLCNPNGVGPWKLRCLAQTFATGEVSGRTLIDIGSGPTVYQLLSACSHFEDITMTDFLEVNRQELGRWLQEEPGAFNWSMYSQHACLIEGKGECWQDKERQLRARVKRVLPIDVHQPQPLGAGSPAPLPADALVSAFCLEAVSPDLASFQRALDHITTLLRPGGHLLLIGALEESWYLAGEARLTVVPVSEEEVREALVRSGYKVRDLRTYIMPAHLQTGVDDVKGVFFAWAQKV Hydrogen bonds contact Hydrophobic contact | ||||
| 92 | Guanine nucleotide-binding protein G(I)/G(S)/G(O) subunit gamma-2 | 5HE0 | 7.86 | |
Target general information Gen name GNG2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family G protein gamma family Biochemical class Transferase / transferase inhibitor Function G-protein beta-subunit binding.Signal transducer activity. Related diseases HDAC4 point mutations and chromosomal microdeletions encompassing this gene have been found in patients with brachydactyly and intellectual disability syndrome (PubMed:20691407, PubMed:23188045, PubMed:24715439). However, HDAC4 haploinsufficiency is not fully penetrant and multiple genes may contribute to manifestation of the full phenotypic spectrum (PubMed:23188045, PubMed:24715439). {ECO:0000269|PubMed:20691407, ECO:0000269|PubMed:23188045, ECO:0000269|PubMed:24715439}.; DISEASE: Neurodevelopmental disorder with central hypotonia and dysmorphic facies (NEDCHF) [MIM:619797]: An autosomal dominant disease characterized by global developmental delay, impaired intellectual development, seizures, distinctive facial features, scoliosis, delayed closure of the anterior fontanel, and non-specific brain abnormalities. {ECO:0000269|PubMed:33537682}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01159 Interacts with P62873 EC number NA Uniprot keywords 3D-structure; Acetylation; Cell membrane; Host-virus interaction; Lipoprotein; Membrane; Methylation; Prenylation; Proteomics identification; Reference proteome; Transducer Protein physicochemical properties Chain ID G Molecular weight (Da) 37822.3 Length 331 Aromaticity 0.09 Instability index 52.96 Isoelectric point 7.77 Charge (pH=7) 2.02 2D Binding mode Binding energy (Kcal/mol) -8.49 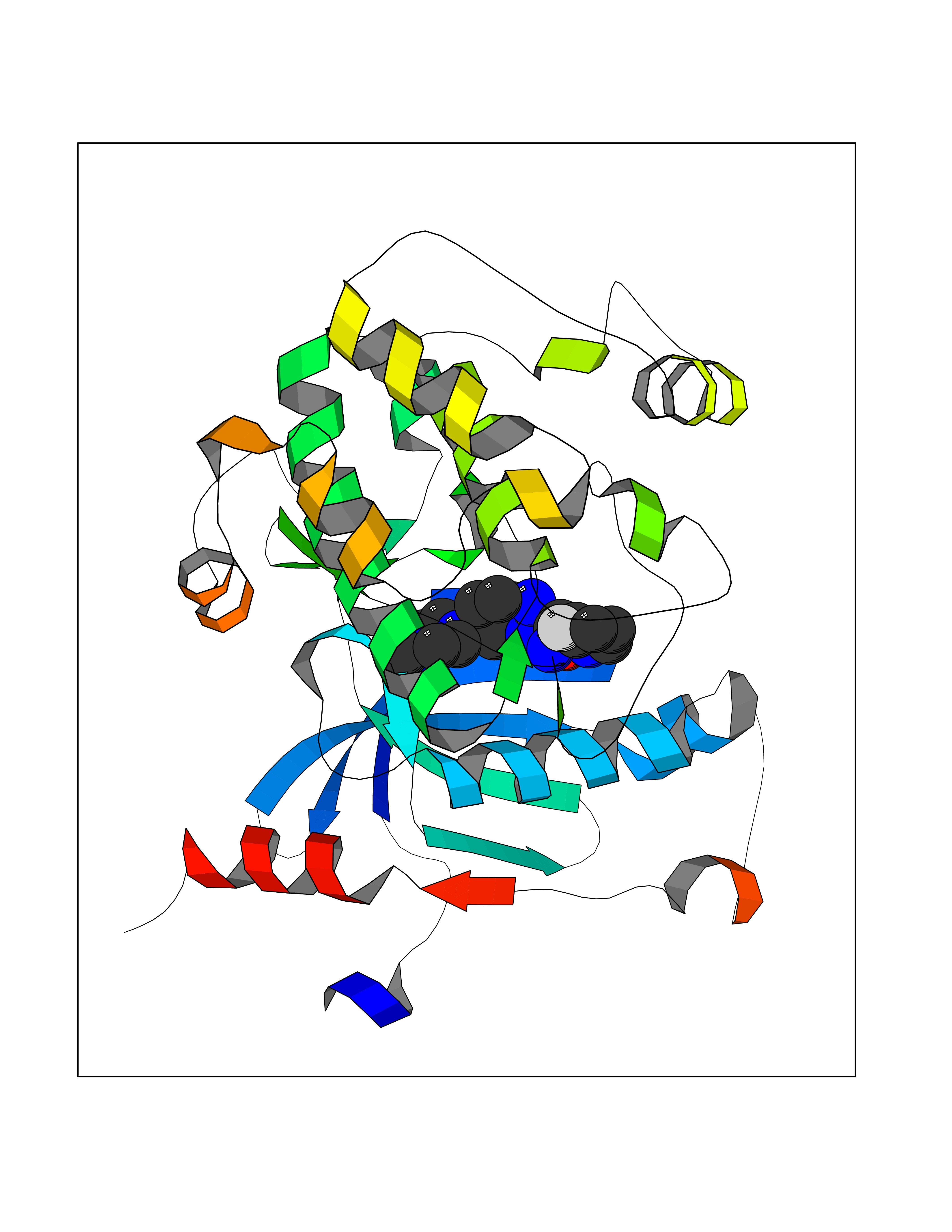 Molscript Map 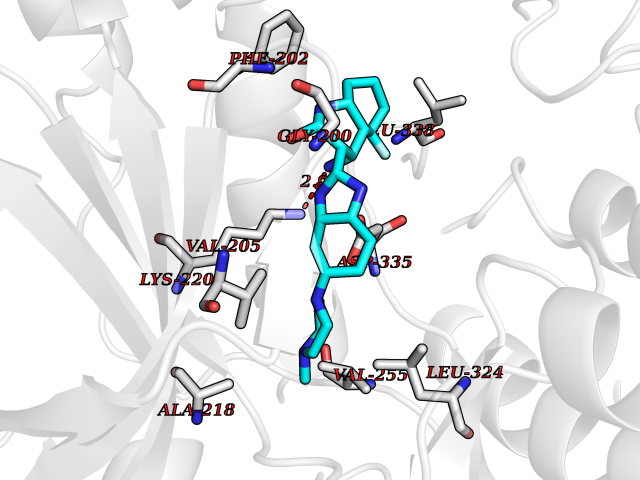 Pymol Map 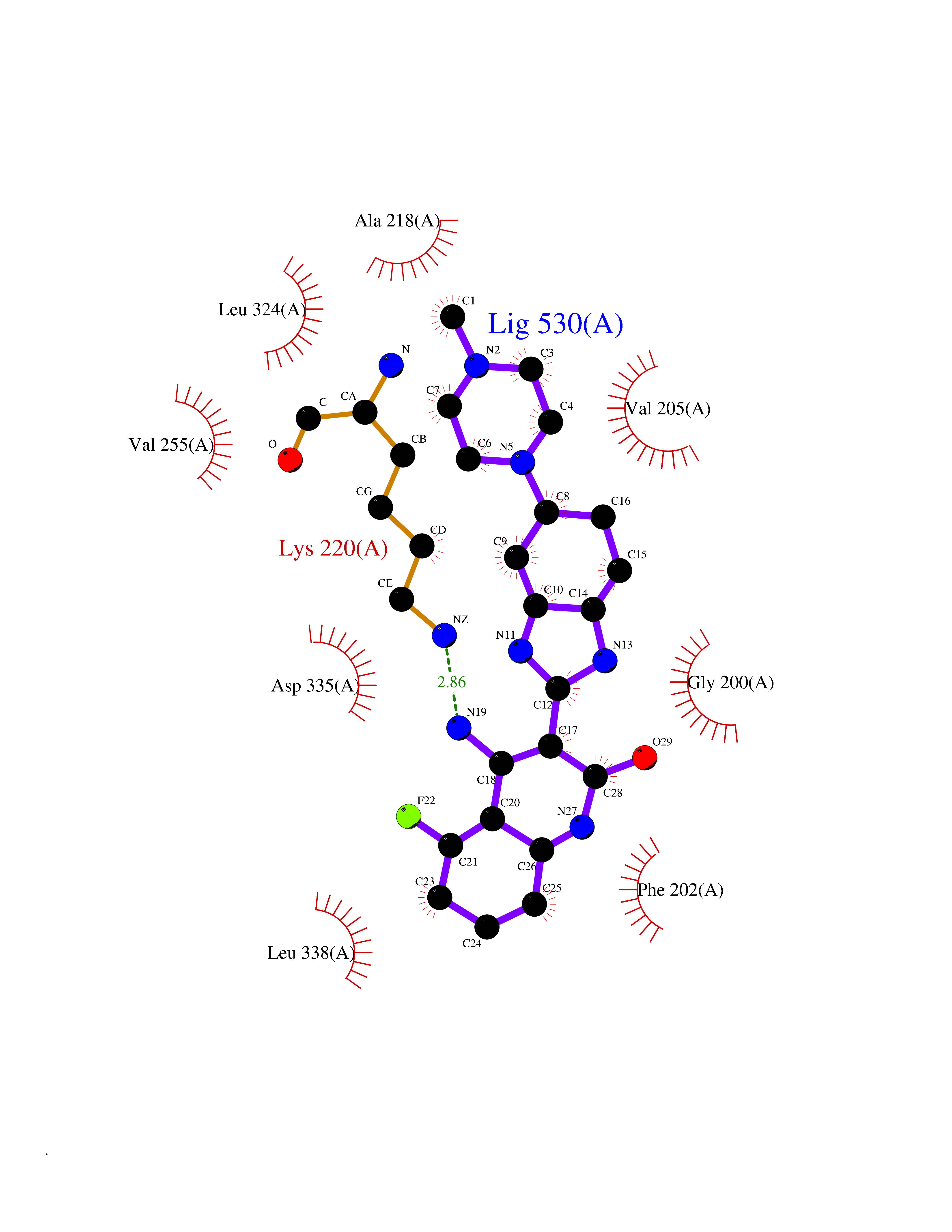 Ligplot Map 3D Binding mode Sequence VELNIHLTMNDFSVHRIIGRGGFGEVYGCRKADTGKMYAMKCLDKKRIKMKQGETLALNERIMLSLVSTGDCPFIVCMSYAFHTPDKLSFILDLMNGGDLHYHLSQHGVFSEADMRFYAAEIILGLEHMHNRFVVYRDLKPANILLDEHGHVRISDLGLACDFSKKKPHASVGTHGYMAPEVLQKGVAYDSSADWFSLGCMLFKLLRGHSPFRQHKTKDKHEIDRMTLTMAVELPDSFSPELRSLLEGLLQRDVNRRLGCLGRGAQEVKESPFFRSLDWQMVFLQKYPPPLIPPRGKGIKLLDSDQELYRNFPLTISERWQQEVAETVFDT Hydrogen bonds contact Hydrophobic contact | ||||
| 93 | Transferrin (TF) | 1RYO | 7.85 | |
Target general information Gen name TF Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Siderophilin; Serotransferrin; PRO1400; Beta-1 metal-binding globulin Protein family Transferrin family Biochemical class Transferrin Function It is responsible for the transport of iron from sites of absorption and heme degradation to those of storage and utilization. Serum transferrin may also have a further role in stimulating cell proliferation. Transferrins are iron binding transport proteins which can bind two Fe(3+) ions in association with the binding of an anion, usually bicarbonate. Related diseases Atransferrinemia (ATRAF) [MIM:209300]: A rare autosomal recessive disorder characterized by abnormal synthesis of transferrin leading to iron overload and microcytic hypochromic anemia. {ECO:0000269|PubMed:11110675, ECO:0000269|PubMed:15466165}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01370; DB14517; DB14518; DB01294; DB14526; DB14527; DB11136; DB14528; DB14529; DB14530; DB00515; DB09130; DB11397; DB13949; DB14490; DB14491; DB14488; DB14501; DB14489; DB13257; DB06215; DB06784; DB05260; DB01592; DB00893; DB00677; DB06757; DB11182; DB14520; DB01593; DB14487; DB14533; DB14548 Interacts with O43315; O00501; Q7Z7G2; Q9GZR5; Q9Y282; Q96KR6; P01350; P08034; Q8NBJ4; O15529; Q8TED1; Q7Z5P4; A8MZ59; O15173; Q96TC7; Q3KNW5; Q9BXS9-3; Q99523; O43278-2; Q8N9I0; P02786; Q4KMG9; Q9K0U9; Q09057; Q9K0V0; P02786 EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Ion transport; Iron; Iron transport; Metal-binding; Methylation; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 35854.5 Length 324 Aromaticity 0.1 Instability index 34.17 Isoelectric point 7.58 Charge (pH=7) 1.42 2D Binding mode Binding energy (Kcal/mol) -9.14 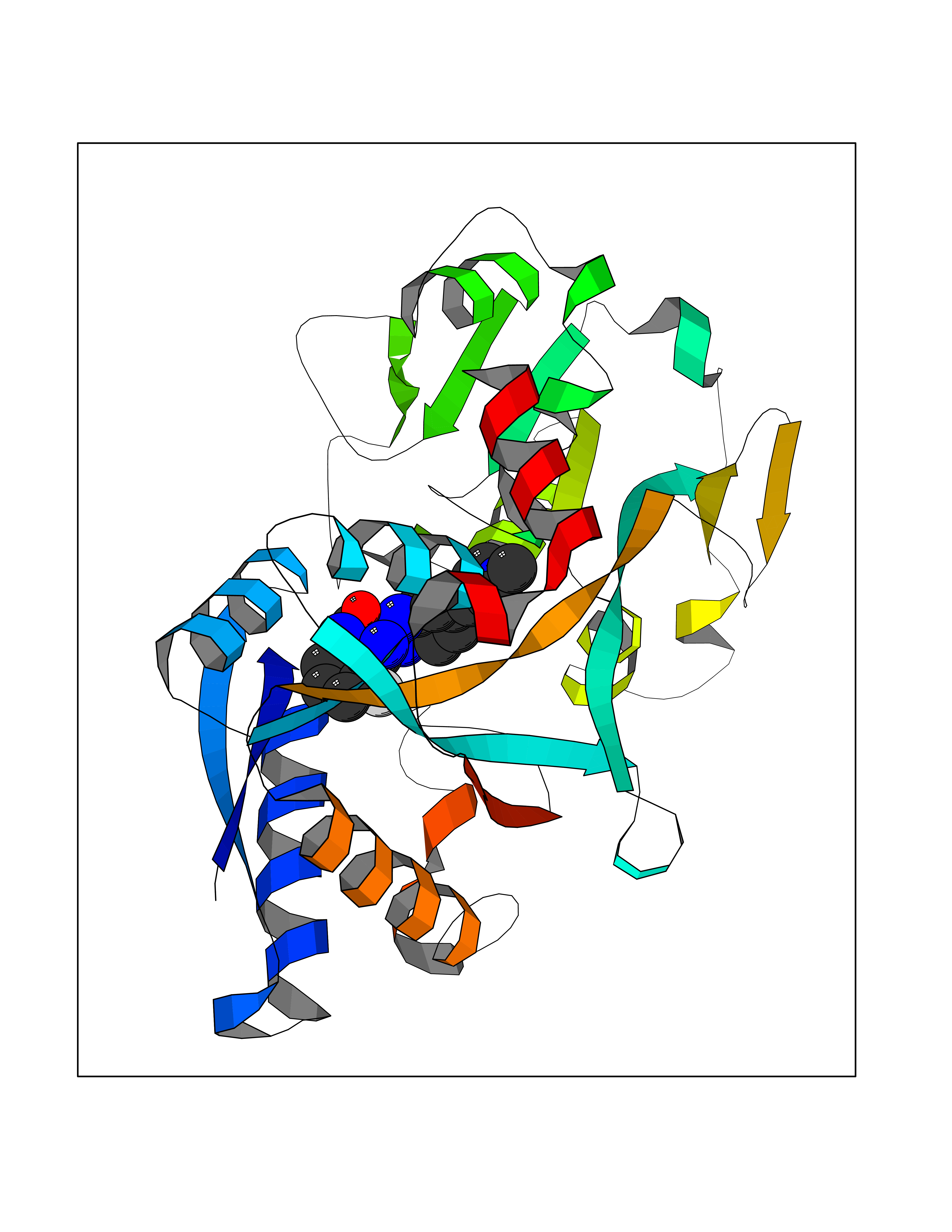 Molscript Map 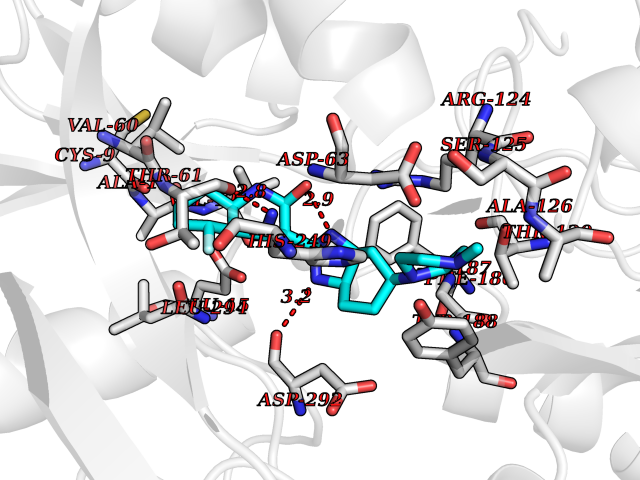 Pymol Map 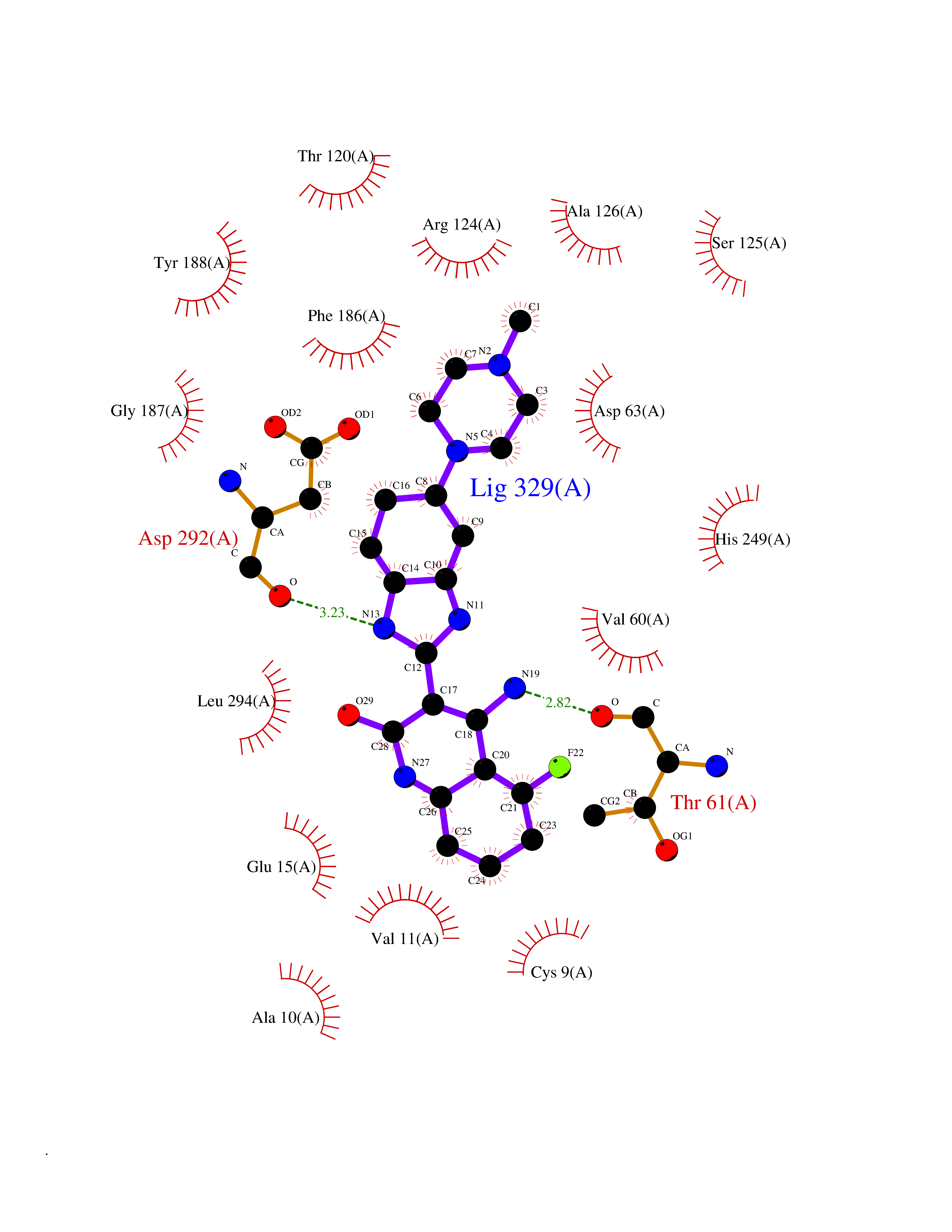 Ligplot Map 3D Binding mode Sequence KTVRWCAVSEHEATKCQSFRDHMKSVIPSDGPSVACVKKASYLDCIRAIAANEADAVTLDAGLVYDAYLAPNNLKPVVAEFYGSKEDPQTFYYAVAVVKKDSGFQMNQLRGKKSCHTGLGRSAGWNIPIGLLYCDLPEPRKPLEKAVANFFSGSCAPCADGTDFPQLCQLCPGCGCSTLNQYFGYSGAFKCLKDGAGDVAFVKHSTIFENLANKADRDQYELLCLDNTRKPVDEYKDCHLAQVPSHTVVARSMGGKEDLIWELLNQAQEHFGKDKSKEFQLFSSPHGKDLLFKDSAHGFLKVPPRMDAKMYLGYEYVTAIRNLR Hydrogen bonds contact Hydrophobic contact | ||||
| 94 | Sphingosine-1-phosphate receptor 2 (S1PR2) | 7T6B | 7.85 | |
Target general information Gen name S1PR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Sphingosine 1-phosphate receptor Edg-5; S1PR2; S1P2; S1P receptor Edg-5; S1P receptor 2; Endothelial differentiation G-protein coupled receptor 5 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for the lysosphingolipid sphingosine 1- phosphate (S1P). S1P is a bioactive lysophospholipid that elicits diverse physiological effect on most types of cells and tissues. When expressed in rat HTC4 hepatoma cells, is capable of mediating S1P-induced cell proliferation and suppression of apoptosis. Related diseases Deafness, autosomal recessive, 68 (DFNB68) [MIM:610419]: A form of non-syndromic sensorineural hearing loss. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. {ECO:0000269|PubMed:26805784}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P16144; Q9JK11-1 EC number NA Uniprot keywords 3D-structure; Cell membrane; Deafness; Disease variant; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Non-syndromic deafness; Palmitate; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 28917.3 Length 264 Aromaticity 0.11 Instability index 38.95 Isoelectric point 9.11 Charge (pH=7) 9.27 2D Binding mode Binding energy (Kcal/mol) -8.8  Molscript Map 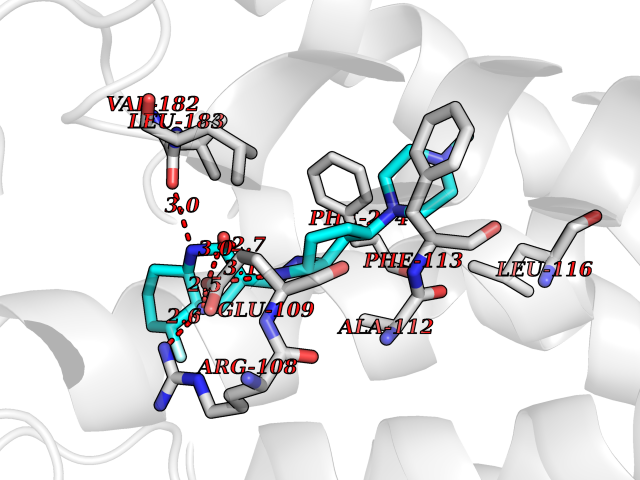 Pymol Map 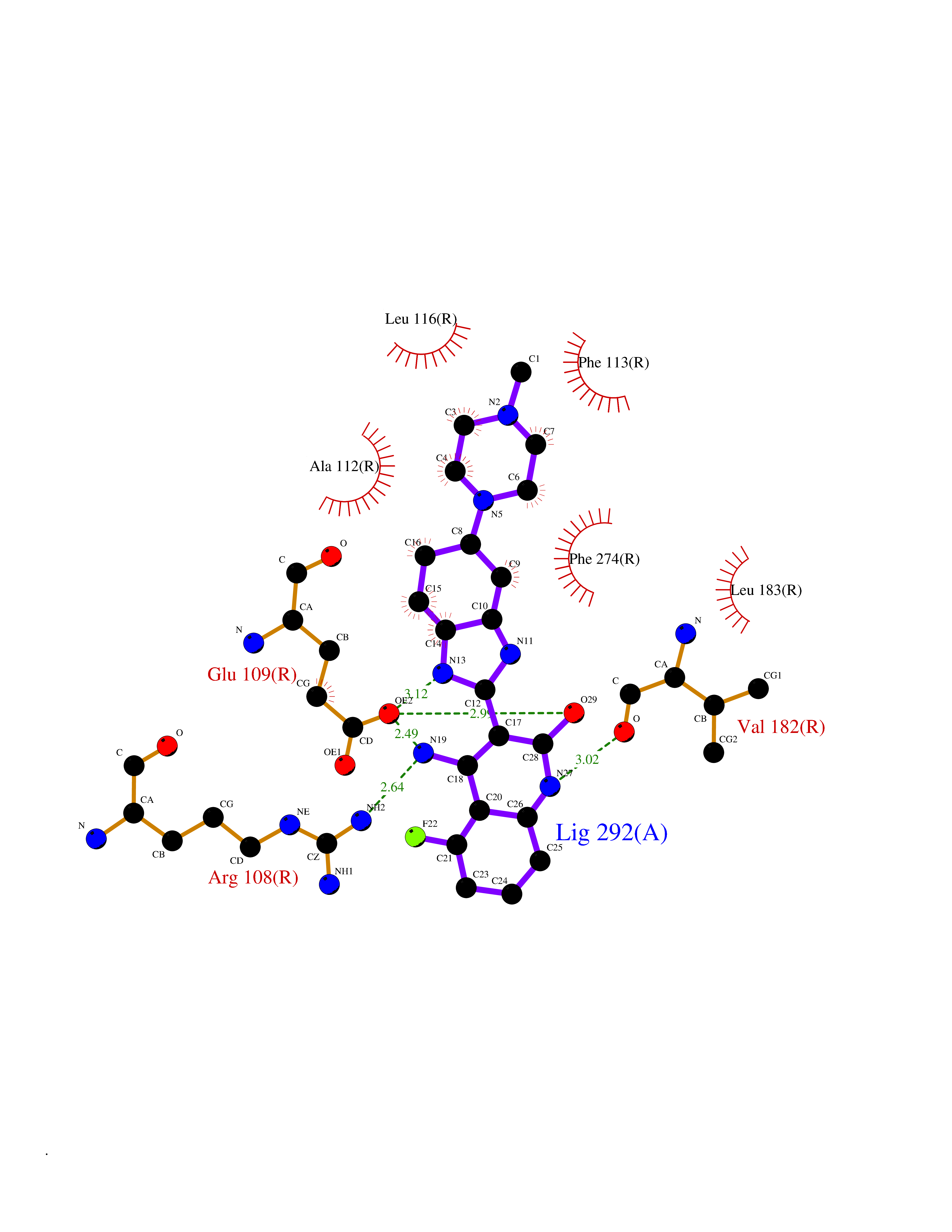 Ligplot Map 3D Binding mode Sequence NKVQEHYNYTKTSRQVASAFIVILCCAIVVENLLVLIAVARNSKFHSAMYLFLGNLAASDLLAGVAFVANTLLSGSVTLRLTPVQWFAREGSAFITLSASVFSLLAIAIERHVAIAKVKLYGSDKSCRMLLLIGASWLISLVLGGLPILGWNCLGHLEACSTVLPLYAKHYVLCVVTIFSIILLAIVALYVRIYCVVRSSQTLALLKTVTIVLGVFIVCWLPAFSILLLDYACPVHSCPILYKAHYFFAVSTLNSLLNPVIYTW Hydrogen bonds contact Hydrophobic contact | ||||
| 95 | Platelet-derived growth factor receptor alpha (PDGFRA) | 5K5X | 7.84 | |
Target general information Gen name PDGFRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RHEPDGFRA; Platelet-derived growth factor receptor 2; Platelet-derived growth factor alpha receptor; PDGFR2; PDGFR-alpha; PDGFR-2; PDGF-R-alpha; CD140a antigen; CD140a; CD140 antigen-like family membe Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Depending on the context, promotes or inhibits cell proliferation and cell migration. Plays an important role in the differentiation of bone marrow-derived mesenchymal stem cells. Required for normal skeleton development and cephalic closure during embryonic development. Required for normal development of the mucosa lining the gastrointestinal tract, and for recruitment of mesenchymal cells and normal development of intestinal villi. Plays a role in cell migration and chemotaxis in wound healing. Plays a role in platelet activation, secretion of agonists from platelet granules, and in thrombin-induced platelet aggregation. Binding of its cognate ligands - homodimeric PDGFA, homodimeric PDGFB, heterodimers formed by PDGFA and PDGFB or homodimeric PDGFC -leads to the activation of several signaling cascades; the response depends on the nature of the bound ligand and is modulated by the formation of heterodimers between PDGFRA and PDGFRB. Phosphorylates PIK3R1, PLCG1, and PTPN11. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate, mobilization of cytosolic Ca(2+) and the activation of protein kinase C. Phosphorylates PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, and thereby mediates activation of the AKT1 signaling pathway. Mediates activation of HRAS and of the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1. Promotes activation of STAT family members STAT1, STAT3 and STAT5A and/or STAT5B. Receptor signaling is down-regulated by protein phosphatases that dephosphorylate the receptor and its down-stream effectors, and by rapid internalization of the activated receptor. Tyrosine-protein kinase that acts as a cell-surface receptor for PDGFA, PDGFB and PDGFC and plays an essential role in the regulation of embryonic development, cell proliferation, survival and chemotaxis. Related diseases A chromosomal aberration involving PDGFRA is found in some cases of hypereosinophilic syndrome. Interstitial chromosomal deletion del(4)(q12q12) causes the fusion of FIP1L1 and PDGFRA (FIP1L1-PDGFRA). Mutations that cause overexpression and/or constitutive activation of PDGFRA may be a cause of hypereosinophilic syndrome. {ECO:0000269|PubMed:12808148}.; DISEASE: Gastrointestinal stromal tumor (GIST) [MIM:606764]: Common mesenchymal neoplasms arising in the gastrointestinal tract, most often in the stomach. They are histologically, immunohistochemically, and genetically different from typical leiomyomas, leiomyosarcomas, and schwannomas. Most GISTs are composed of a fairly uniform population of spindle-shaped cells. Some tumors are dominated by epithelioid cells or contain a mixture of spindle and epithelioid morphologies. Primary GISTs in the gastrointestinal tract commonly metastasize in the omentum and mesenteries, often as multiple nodules. However, primary tumors may also occur outside of the gastrointestinal tract, in other intra-abdominal locations, especially in the omentum and mesentery. {ECO:0000269|PubMed:12522257, ECO:0000269|PubMed:15928335}. The gene represented in this entry may be involved in disease pathogenesis. Mutations causing PDGFRA constitutive activation have been found in gastrointestinal stromal tumors lacking KIT mutations (PubMed:12522257). {ECO:0000269|PubMed:12522257}.; DISEASE: GIST-plus syndrome (GISTPS) [MIM:175510]: A disorder characterized by multiple mesenchymal tumors of the gastrointestinal tract, including gastrointestinal stromal tumor, inflammatory fibroid polyps, and fibroid tumors. Additional features are coarse facies and skin, broad hands and feet, and premature tooth loss. GISTPS is an autosomal dominant disease with incomplete penetrance. Gastrointestinal stromal tumor and inflammatory fibroid polyps may also occur in isolation. {ECO:0000269|PubMed:14699510, ECO:0000269|PubMed:17087943, ECO:0000269|PubMed:25975287}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12742; DB00102; DB12147; DB10772; DB12010; DB00619; DB09078; DB06595; DB09079; DB06043; DB06589; DB08901; DB08896; DB14840; DB01268; DB11800; DB05146 Interacts with P46108; P46109; P00533; Q8N6L0; P04085; P01127; Q9NRA1; P31947; P62258; Q9NRA1-1; A8T7D5; P05067; Q8IY26 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cell projection; Chemotaxis; Developmental protein; Disease variant; Disulfide bond; Glycoprotein; Golgi apparatus; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 39294.8 Length 345 Aromaticity 0.11 Instability index 46.74 Isoelectric point 6.6 Charge (pH=7) -1.38 2D Binding mode Binding energy (Kcal/mol) -8.42 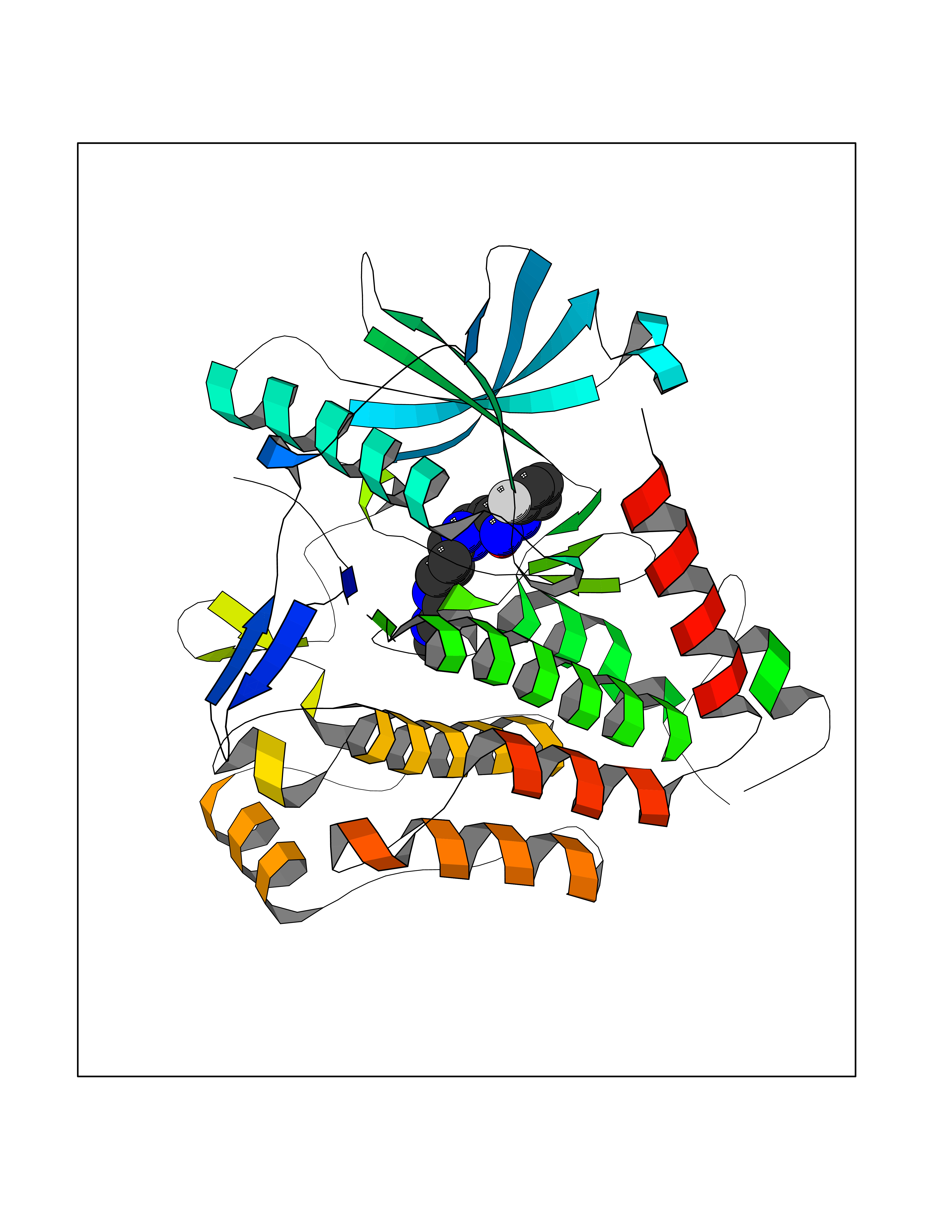 Molscript Map 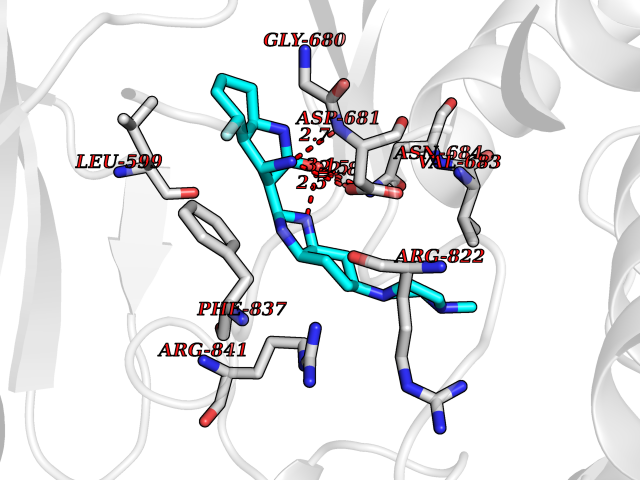 Pymol Map 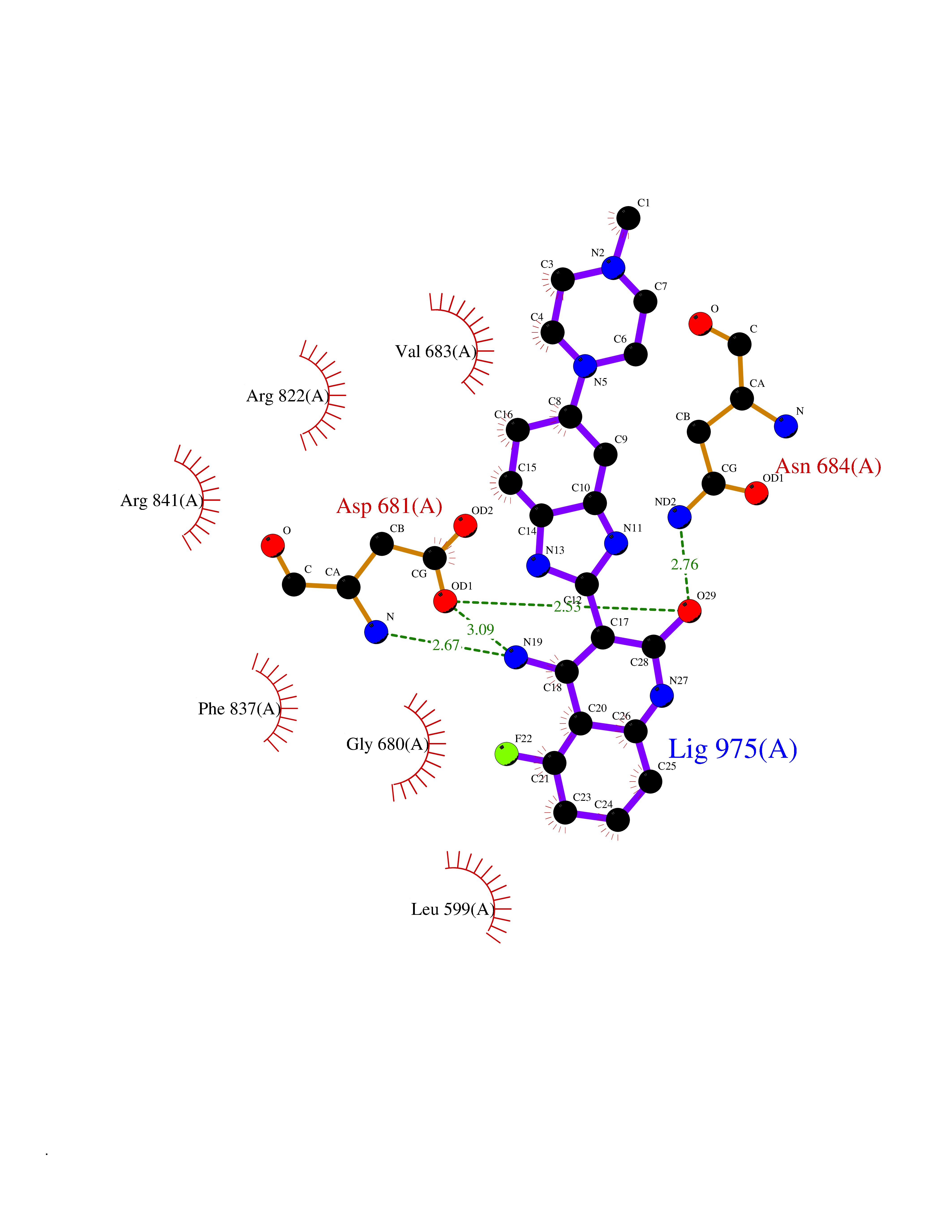 Ligplot Map 3D Binding mode Sequence RYEIRWRVIESISPDGHEYIYVDPMQLPYDSRWEFPRDGLVLGRVLGSGAFGKVVEGTAYGLSRSQPVMKVAVKMLKPTARSSEKQALMSELKIMTHLGPHLNIVNLLGACTKSGPIYIITEYCFYGDLVNYLHKNRDSFLSHSMLDSEVKNLLSDDNSEGLTLLDLLSFTYQVARGMEFLASKNCVHRDLAARNVLLAQGKIVKICDFGLARDIMHDSNYVSKGSTFLPVKWMAPESIFDNLYTTLSDVWSYGILLWEIFSLGGTPYPGMMVDSTFYNKIKSGYRMAKPDHATSEVYEIMVKCWNSEPEKRPSFYHLSEIVENLLPGQYKKSYEKIHLDFLKSD Hydrogen bonds contact Hydrophobic contact | ||||
| 96 | Threonine--tRNA ligase, cytoplasmic | 4HWT | 7.84 | |
Target general information Gen name TARS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms TARS Protein family Class-II aminoacyl-tRNA synthetase family Biochemical class Ligase / ligase inhibitor Function ATP binding.Protein homodimerization activity.Threonine-tRNA ligase activity.TRNA binding. Related diseases Trichothiodystrophy 7, non-photosensitive (TTD7) [MIM:618546]: A form of trichothiodystrophy, a disease characterized by sulfur-deficient brittle hair and multisystem variable abnormalities. The spectrum of clinical features varies from mild disease with only hair involvement to severe disease with cutaneous, neurologic and profound developmental defects. Ichthyosis, intellectual and developmental disabilities, decreased fertility, abnormal characteristics at birth, ocular abnormalities, short stature, and infections are common manifestations. There are both photosensitive and non-photosensitive forms of the disorder. TTD7 patients do not manifest cutaneous photosensitivity. They have cysteine- and threonine-deficient hair with alternating light and dark 'tiger-tail' banding pattern observed under polarization microscopy. Inheritance pattern is autosomal recessive. {ECO:0000269|PubMed:31374204}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00156 Interacts with Q9BPX7; Q96CV9; O43704; A2RTX5 EC number 6.1.1.3 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminoacyl-tRNA synthetase; ATP-binding; Cytoplasm; Disease variant; Ligase; Nucleotide-binding; Phosphoprotein; Protein biosynthesis; Proteomics identification; Reference proteome; RNA-binding; tRNA-binding; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 33945.3 Length 290 Aromaticity 0.13 Instability index 41.57 Isoelectric point 6.29 Charge (pH=7) -3.28 2D Binding mode Binding energy (Kcal/mol) -8.45 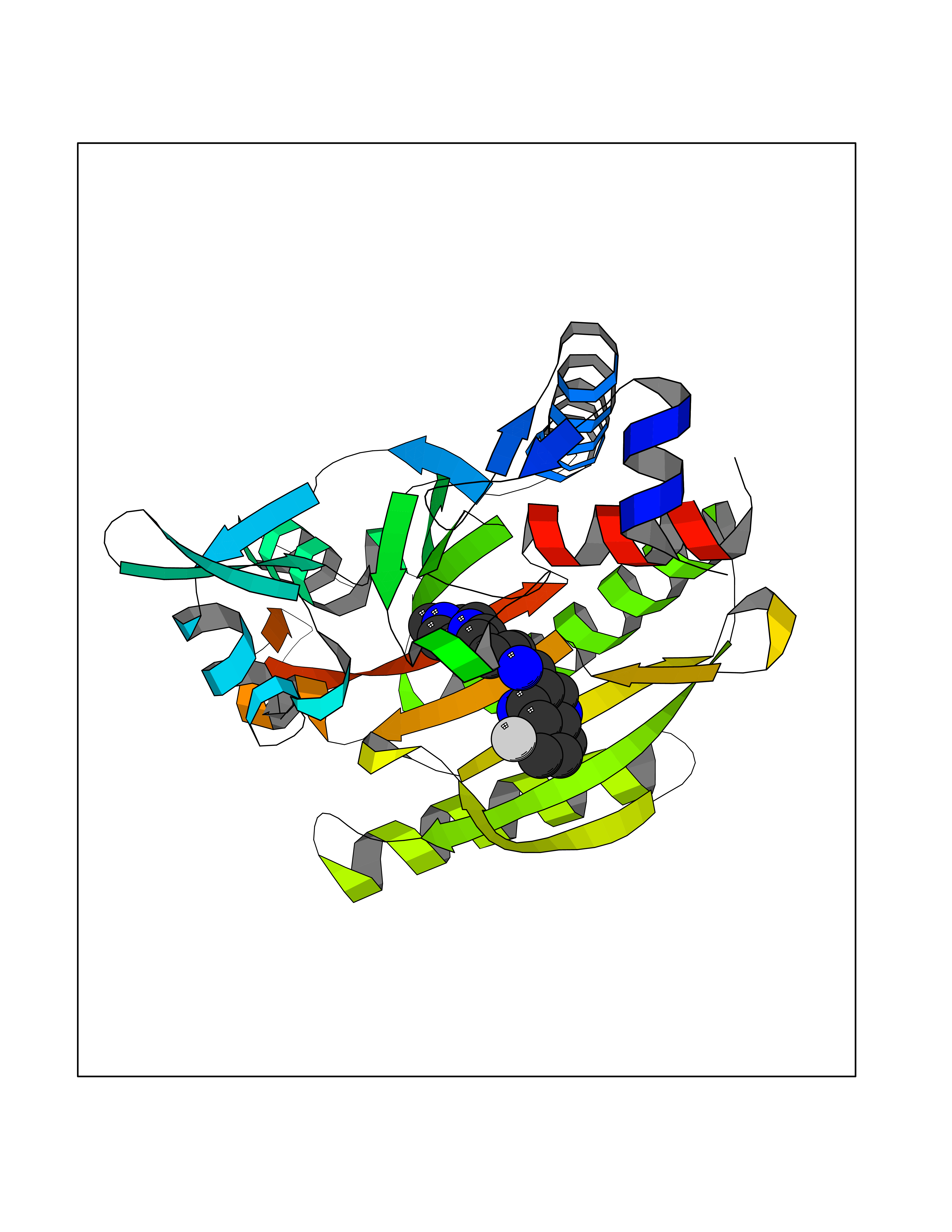 Molscript Map 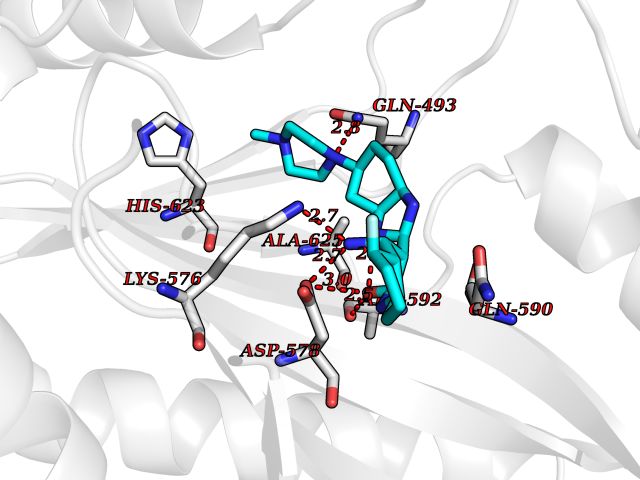 Pymol Map 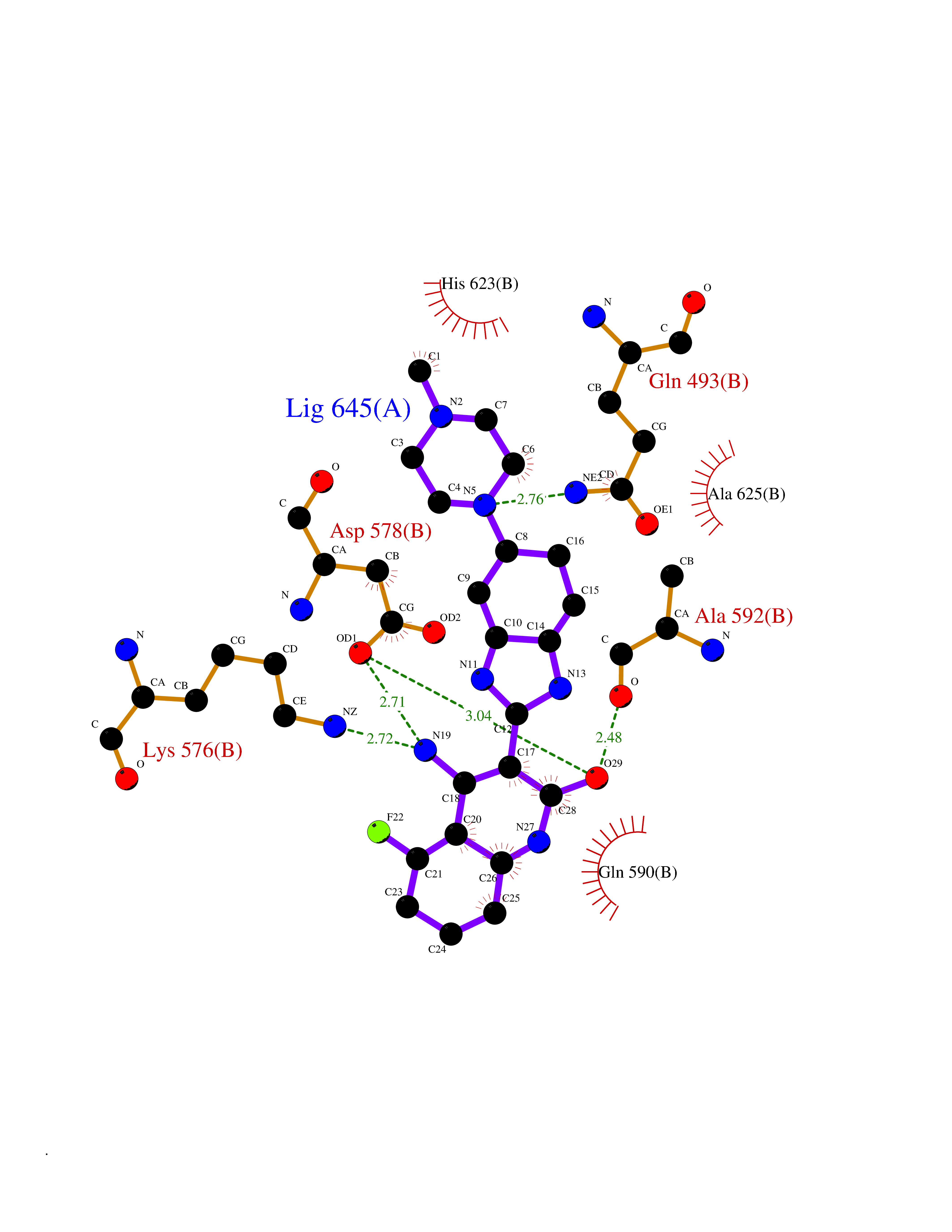 Ligplot Map 3D Binding mode Sequence RDHRKIGRDQELYFFHELSPGSCFFLPKGAYIYNALIEFIRSEYRKRGFQEVVTPNIFNSRLWMTSGHWQHYSENMFSFEVEKELFALKPMNCPGHCLMFDHRPRSWRELPLRLADFGVLHRNELSGALTGLTRVRRFQQDDAHIFCAMEQIEDEIKGCLDFLRTVYSVFGFSFKLNLSTRPEKFLGDIEVWDQAEKQLENSLNEFGEKWELNSGDGAFYGPKIDIQIKDAIGRYHQCATIQLDFQLPIRFNLTYVSHDGDDKKRPVIVHRAILGSVERMIAILTENYGG Hydrogen bonds contact Hydrophobic contact | ||||
| 97 | Ubiquitin-activating enzyme E1 (UBAE1) | 6DC6 | 7.84 | |
Target general information Gen name UBA1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ubiquitin-like modifier-activating enzyme 1; UBE1; Protein A1S9; A1S9T Protein family Ubiquitin-activating E1 family Biochemical class NA Function Catalyzes the first step in ubiquitin conjugation to mark cellular proteins for degradation through the ubiquitin-proteasome system. Activates ubiquitin by first adenylating its C-terminal glycine residue with ATP, and thereafter linking this residue to the side chain of a cysteine residue in E1, yielding a ubiquitin-E1 thioester and free AMP. Essential for the formation of radiation-induced foci, timely DNA repair and for response to replication stress. Promotes the recruitment of TP53BP1 and BRCA1 at DNA damage sites. Related diseases Spinal muscular atrophy X-linked 2 (SMAX2) [MIM:301830]: A lethal infantile form of spinal muscular atrophy, a neuromuscular disorder characterized by degeneration of the anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. Clinical features include hypotonia, areflexia, and multiple congenital contractures. {ECO:0000269|PubMed:18179898, ECO:0000269|PubMed:23518311}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: VEXAS syndrome (VEXAS) [MIM:301054]: A sporadic, often fatal, treatment-refractory inflammatory syndrome that develops in late adulthood. Clinical features include fevers, cytopenias, characteristic vacuoles in myeloid and erythroid precursor cells, dysplastic bone marrow, neutrophilic cutaneous and pulmonary inflammation, chondritis, and vasculitis. The disease affects only males and is associated with de novo somatic mutations. {ECO:0000269|PubMed:33108101}. The disease is caused by variants affecting the gene represented in this entry. Somatic variants affecting the initiator methionine of isoform 2 are recurrently found in VEXAS patients. These variants cause loss of isoform 2 and production of a shorter isoform with strongly reduced enzymatic activity from a downstream methionine (Met-67). {ECO:0000269|PubMed:33108101}. Drugs (DrugBank ID) DB04119; DB04216 Interacts with Q9H2C0; P42858; Q96FW1; Q9BUZ4; P63279; O00308; Q76353 EC number EC 6.2.1.45 Uniprot keywords 3D-structure; Acetylation; Alternative initiation; ATP-binding; Cytoplasm; Direct protein sequencing; Disease variant; Ligase; Mitochondrion; Neurodegeneration; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 119604 Length 1068 Aromaticity 0.09 Instability index 36.15 Isoelectric point 5.4 Charge (pH=7) -26.22 2D Binding mode Binding energy (Kcal/mol) -8.36  Molscript Map 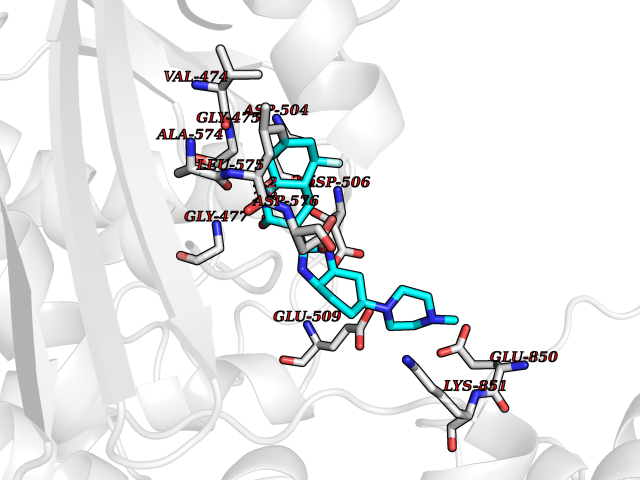 Pymol Map 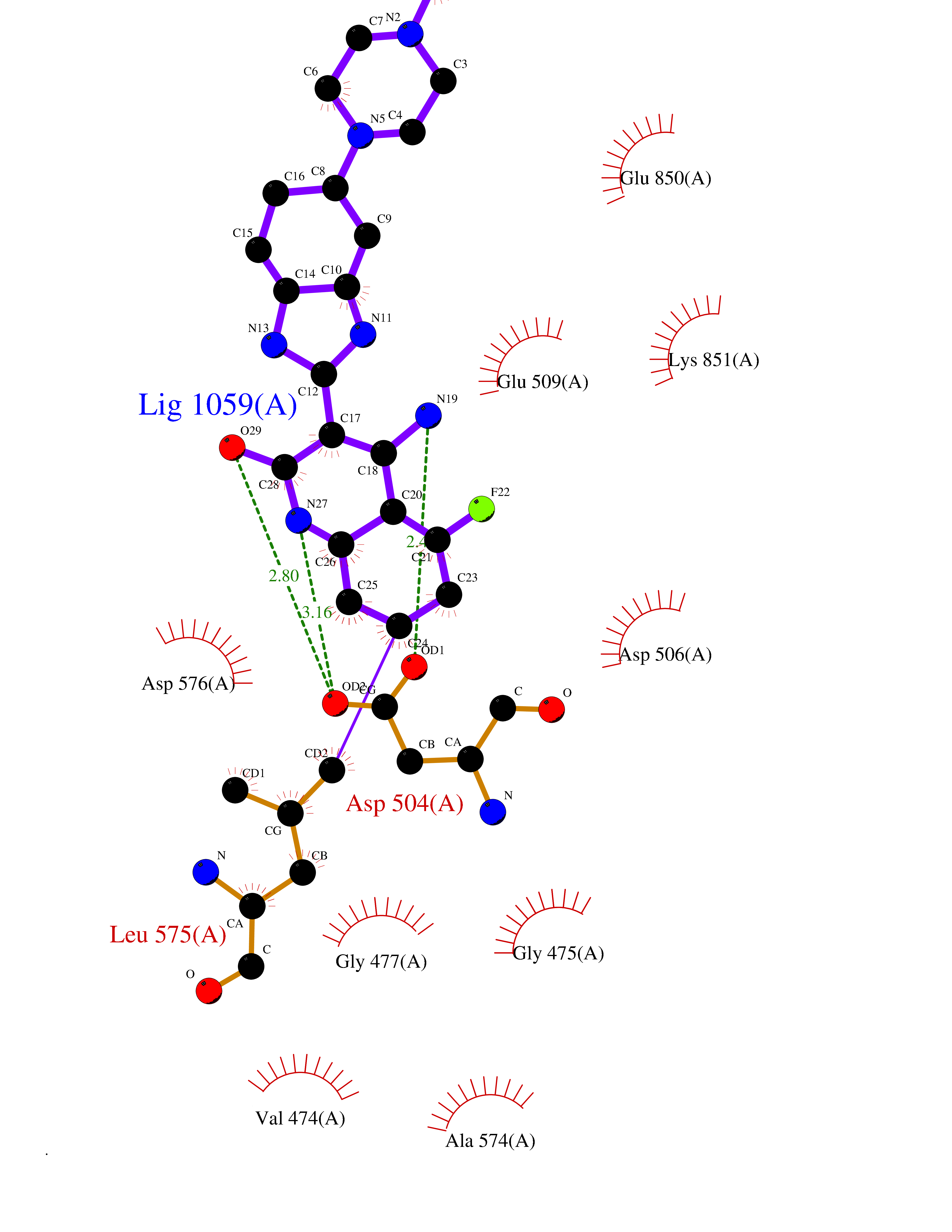 Ligplot Map 3D Binding mode Sequence DIDEGLYSRQLYVLGHEAMKRLQTSSVLVSGLRGLGVEIAKNIILGGVKAVTLHDQGTAQWADLSSQFYLREEDIGKNRAEVSQPRLAELNSYVPVTAYTGPLVEDFLSGFQVVVLTNTPLEDQLRVGEFCHNRGIKLVVADTRGLFGQLFCDFGEEMILTDSNGEQPLSAMVSMVTKDNPGVVTCLDEARHGFESGDFVSFSEVQGMVELNGNQPMEIKVLGPYTFSICDTSNFSDYIRGGIVSQVKVPKKISFKSLVASLAEPDFVVTDFAKFSRPAQLHIGFQALHQFCAQHGRPPRPRNEEDAAELVALAQAVNARALPAVQQNNLDEDLIRKLAYVAAGDLAPINAFIGGLAAQEVMKACSGKFMPIMQWLYFDALECLPEDKEVLTEDKCLQRQNRYDGQVAVFGSDLQEKLGKQKYFLVGAGAIGCELLKNFAMIGLGCGEGGEIIVTDMDTIEKSNLNRQFLFRPWDVTKLKSDTAAAAVRQMNPHIRVTSHQNRVGPDTERIYDDDFFQNLDGVANALDNVDARMYMDRRCVYYRKPLLESGTLGTKGNVQVVIPFLTESYSSSQDPPEKSIPIATLKNFPNAIEHTLQWARDEFEGLFKQPAENVNQYLTDPKFVERTLRLAGTQPLEVLEAVQRSLVLQRPQTWADCVTWACHHWHTQYSNNIRQLLHNFPPDQLTSSGAPFWSGPKRCPHPLTFDVNNPLHLDYVMAAANLFAQTYGLTGSQDRAAVATFLQSVQVPEFTPKSVDDSRLEELKATLPSPDKLPGFKMYPIDFEKDDDSNFHMDFIVAASNLRAENYDIPSADRHKSKLIAGKIIPAIATTTAAVVGLVCLELYKVVQGHRQLDSYKNGFLNLALPFFGFSEPLAAPRHQYYNQEWTLWDRFEVQGLQPNGEEMTLKQFLDYFKTEHKLEITMLSQGVSMLYSFFMPAAKLKERLDQPMTEIVSRVSKRKLGRHVRALVLELCCNDESGEDVEVPYVRYTIMQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRGG Hydrogen bonds contact Hydrophobic contact | ||||
| 98 | Adenosine A2a receptor (ADORA2A) | 5IU4 | 7.84 | |
Target general information Gen name ADORA2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Adenosine receptor A2a; ADORA2; A2a Adenosine receptor; A(2A) adenosine receptor Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function The activity of this receptor is mediated by G proteins which activate adenylyl cyclase. Receptor for adenosine. Related diseases Intellectual developmental disorder, autosomal recessive 59 (MRT59) [MIM:617323]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. {ECO:0000269|PubMed:26416544}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08770; DB14132; DB00640; DB05009; DB05191; DB04853; DB00201; DB04932; DB09273; DB00651; DB00824; DB11757; DB17080; DB00555; DB00358; DB00683; DB01303; DB00806; DB06213; DB01412; DB00277 Interacts with P30542; P29274; P29275; O15155; P21554; Q99418; O15354; Q7Z6G3; O43759-2; Q13107; Q5T9L3-1; P31424-1 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32419.4 Length 296 Aromaticity 0.12 Instability index 39.04 Isoelectric point 8.77 Charge (pH=7) 7.19 2D Binding mode Binding energy (Kcal/mol) -8.72 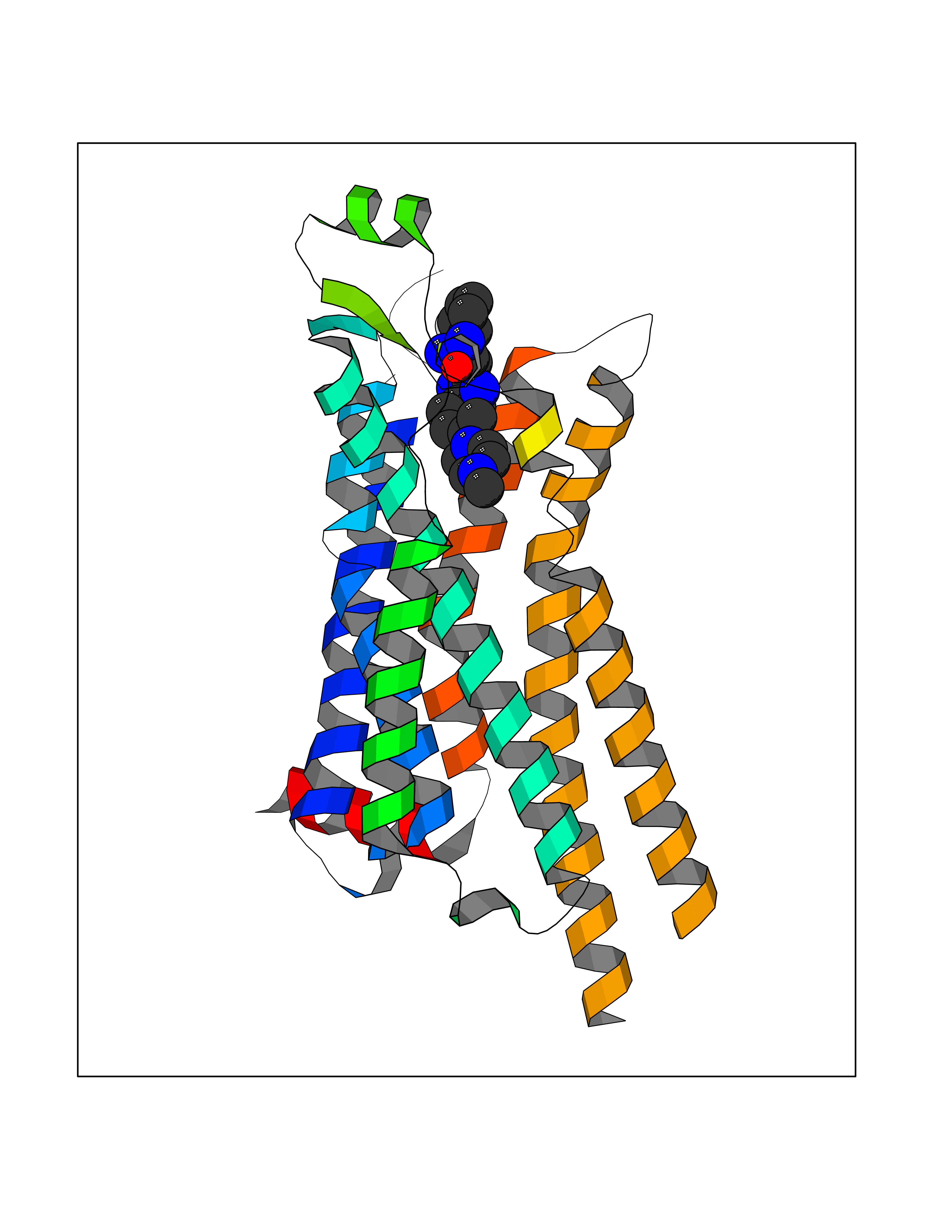 Molscript Map 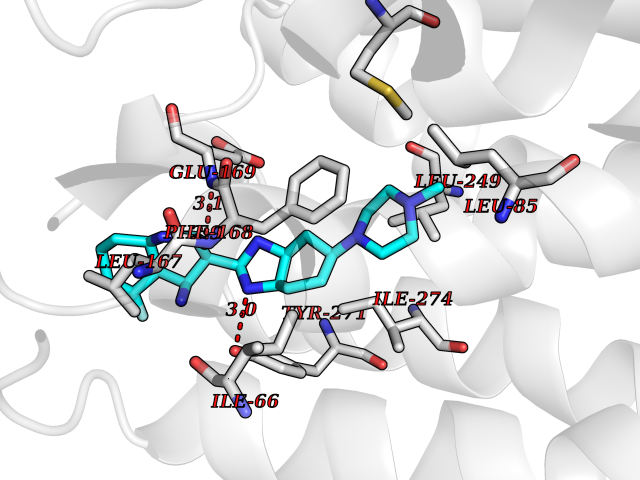 Pymol Map 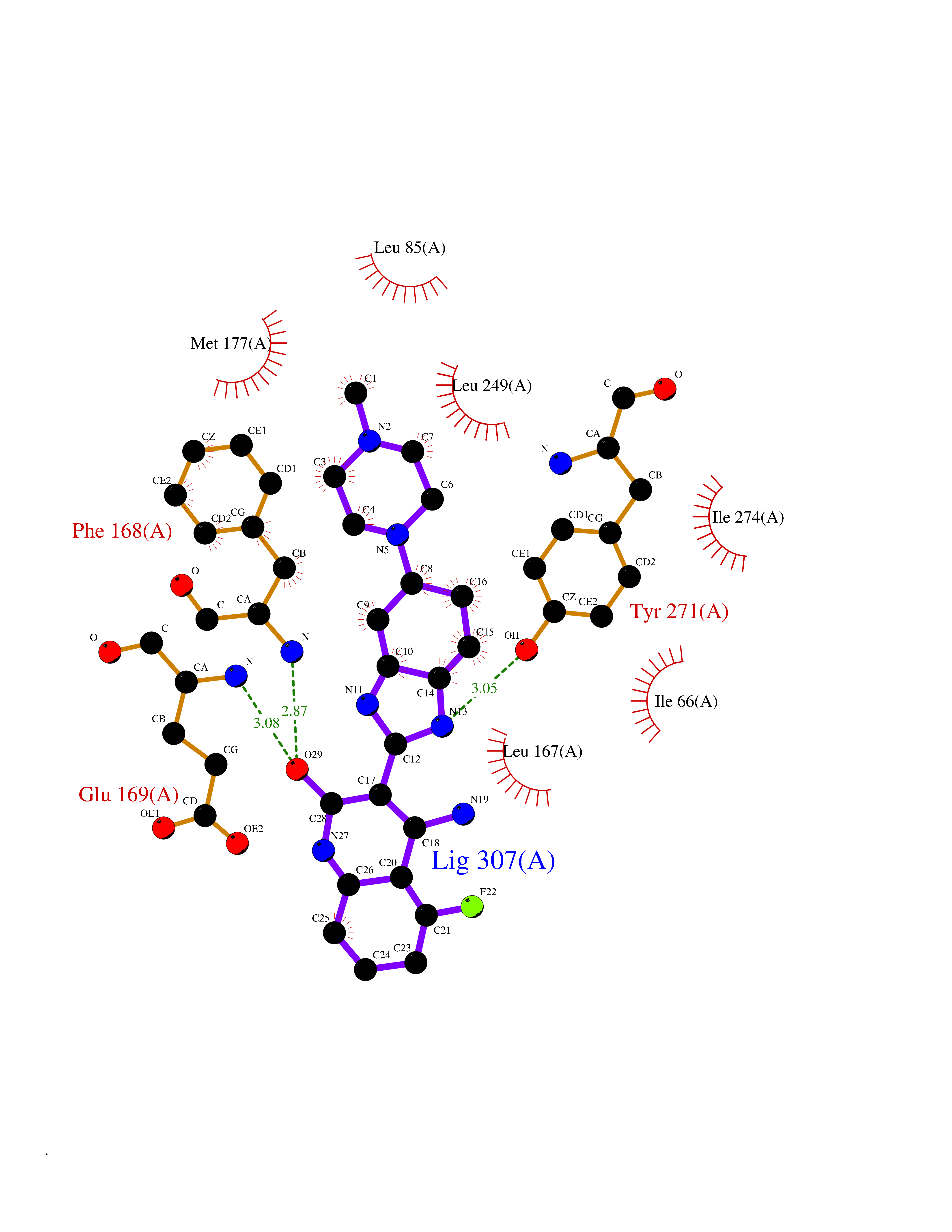 Ligplot Map 3D Binding mode Sequence APPIMGSSVYITVELAIAVLAILGNVLVCWAVWLNSNLQNVTNYFVVSLAAADILVGVLAIPFAITISTGFCAACHGCLFIACFVLVLAQSSIFSLLAIAIDRYIAIAIPLRYNGLVTGTRAAGIIAICWVLSFAIGLTPMLGWNNCGQPKEGKAHSQGCGEGQVACLFEDVVPMNYMVYFNFFACVLVPLLLMLGVYLRIFAAARRQLERARSTLQKEVHAAKSAAIIAGLFALCWLPLHIINCFTFFCPDCSHAPLWLMYLAIVLAHTNSVVNPFIYAYRIREFRQTFRKIIRS Hydrogen bonds contact Hydrophobic contact | ||||
| 99 | Prostaglandin D2 receptor 2 (PTGDR2) | 6D26 | 7.84 | |
Target general information Gen name PTGDR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PTGDR2; Chemoattractant receptor-homologous molecule expressed on Th2 cells; CD294 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for prostaglandin D2 (PGD2). Coupled to the G(i)-protein. Receptor activation may result in pertussis toxin- sensitive decreases in cAMP levels and Ca(2+) mobilization. PI3K signaling is also implicated in mediating PTGDR2 effects. PGD2 induced receptor internalization. CRTH2 internalization can be regulated by diverse kinases such as, PKC, PKA, ADRBK1/GRK2, GPRK5/GRK5 and GRK6. Receptoractivation is responsible, at least in part, in immune regulation and allergic/inflammation responses. Related diseases Neurodevelopmental disorder with seizures, hypotonia, and brain imaging abnormalities (NEDSHBA) [MIM:618922]: An autosomal recessive neurodevelopmental disorder characterized by global developmental delay, hypotonia, severe to profound intellectual disability, early-onset epilepsy, and microcephaly. Neuroimaging shows cerebral atrophy, thin corpus callosum and hypomyelination in a majority of cases. Death in childhood may occur. {ECO:0000269|PubMed:27435318, ECO:0000269|PubMed:28097321, ECO:0000269|PubMed:32286009, ECO:0000269|PubMed:33476302, ECO:0000269|PubMed:33500274}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00770; DB12789; DB00917; DB01088; DB00328; DB02056; DB13036; DB00605; DB04828 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 49740.6 Length 447 Aromaticity 0.1 Instability index 37.89 Isoelectric point 10.13 Charge (pH=7) 21.88 2D Binding mode Binding energy (Kcal/mol) -9.07  Molscript Map 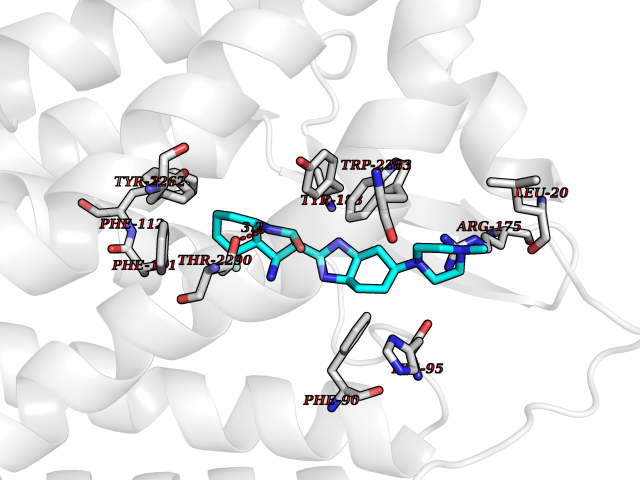 Pymol Map 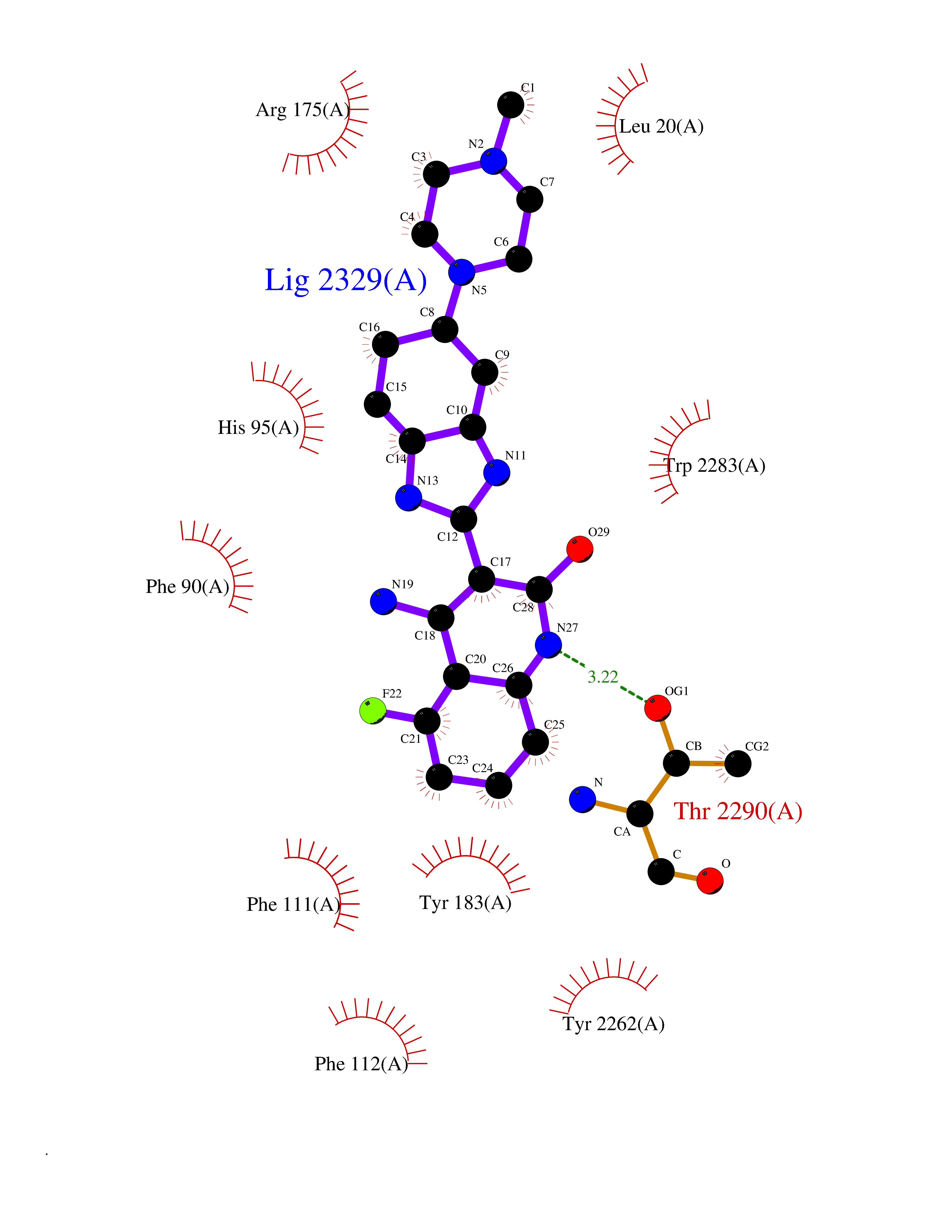 Ligplot Map 3D Binding mode Sequence ATLKPLCPILEQMSRLQSHSATSIRYIDHAAVLLHGLASLLGLVENGVILFVVGCRMRQTVVTTWVLHLALSDLLASASLPFFTYFLAVGHSWELGTTFCKLHSSIFFLNMFASGFLLSAISLDRCLQVVRPVWAQNHRTVAAAHKVCLVLWALAVLNTVPYFVFRDTISRLDGRIMCYYNVLLLNPGPDRDATCNSRQAALAVSKFLLAFLVPLAIIASSHAAVSLRLQHRADLGLQHRNIFEMLRIDEGGGSGGDEAEKLFNQDVDAAVRGILRNAKLKPVYDSLDAVRRAALINMVFQMGETGVAGFTNSLRMLQQKRWDEAAVNLAKSRWYNQTPNRAKRVITTFRTGTWDAYRRRPGRFVRLVAAVVAAFALCWGPYHVFSLLEARAHANPGLRPLVWRGLPFVTSLAFFNSVANPVLYVLTXPDMLRKLRRSLRTVLESVL Hydrogen bonds contact Hydrophobic contact | ||||