Job Results:
Ligand
Structure
Job ID
b5aa43c6924a21fc75a6162a600078fc
Job name
NA
Time
2026-03-04 17:25:26
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Pseudomonas Transcriptional activator protein LasR (Pseudo LasR) | 3IX3 | 8.30 | |
Target general information Gen name Pseudo LasR Organism Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Uniprot ID TTD ID Synonyms NA Protein family Autoinducer-regulated transcriptional regulatory protein family Biochemical class NA Function Transcriptional activator of elastase structural gene (LasB). Binds to the PAI autoinducer. Related diseases Growth hormone deficiency, isolated, 1A (IGHD1A) [MIM:262400]: An autosomal recessive, severe deficiency of growth hormone leading to dwarfism. Patients often develop antibodies to administered growth hormone. {ECO:0000269|PubMed:8364549}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 1B (IGHD1B) [MIM:612781]: An autosomal recessive deficiency of growth hormone leading to short stature. Patients have low but detectable levels of growth hormone, significantly retarded bone age, and a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:12655557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Kowarski syndrome (KWKS) [MIM:262650]: A syndrome clinically characterized by short stature associated with bioinactive growth hormone, normal or slightly increased growth hormone secretion, pathologically low insulin-like growth factor 1 levels, and normal catch-up growth on growth hormone replacement therapy. {ECO:0000269|PubMed:17519310, ECO:0000269|PubMed:8552145, ECO:0000269|PubMed:9276733}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 2 (IGHD2) [MIM:173100]: An autosomal dominant deficiency of growth hormone leading to short stature. Clinical severity is variable. Patients have a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:11502836, ECO:0000269|PubMed:9152628}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08324 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; DNA-binding; Quorum sensing; Reference proteome; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 18305.5 Length 163 Aromaticity 0.12 Instability index 46.52 Isoelectric point 5.19 Charge (pH=7) -6.78 2D Binding mode Binding energy (Kcal/mol) -11.32  Molscript Map 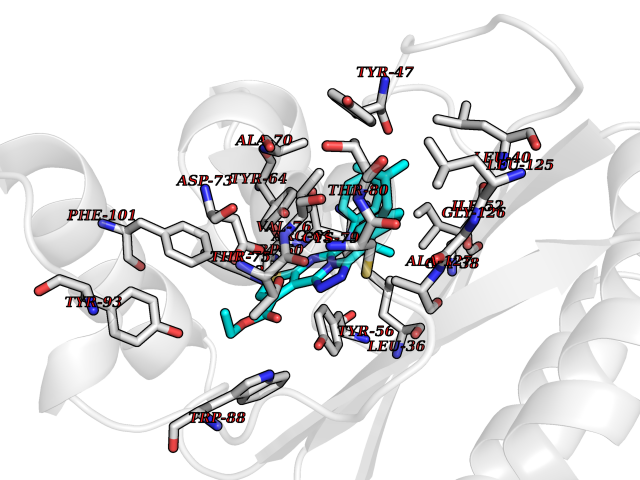 Pymol Map 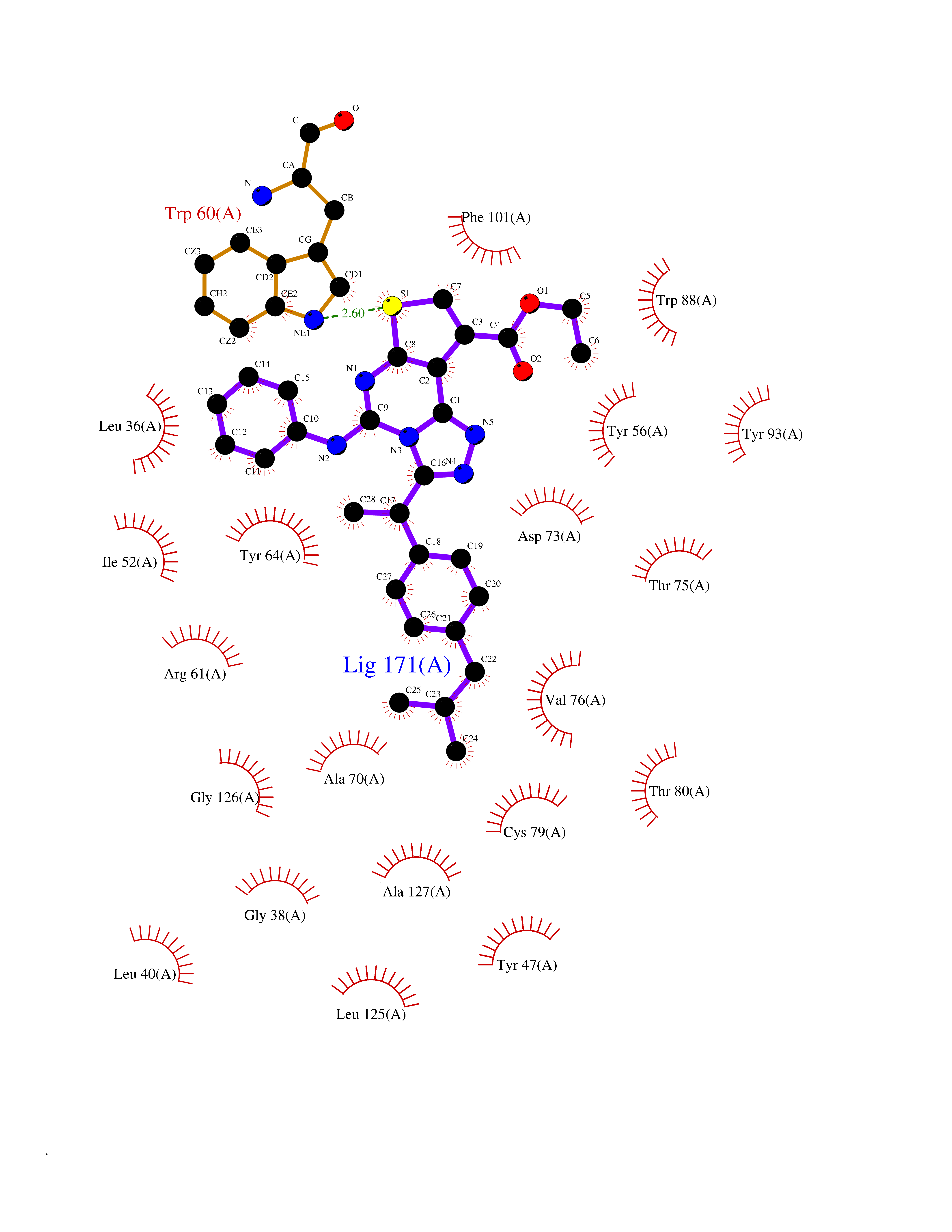 Ligplot Map 3D Binding mode Sequence FLELERSSGKLEWSAILQKMASDLGFSKILFGLLPKDSQDYENAFIVGNYPAAWREHYDRAGYARVDPTVSHCTQSVLPIFWEPSIYQTRKQHEFFEEASAAGLVYGLTMPLHGARGELGALSLSVEAENRAEANRFMESVLPTLWMLKDYALQSGAGLAFEH Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Staphylococcus Enoyl ACP reductase (Stap-coc fabI) | 6YUR | 8.30 | |
Target general information Gen name Stap-coc fabI Organism Staphylococcus aureus (strain NCTC 8325 / PS 47) Uniprot ID TTD ID Synonyms NADPHdependent enoylACP reductase; Enoyl[acylcarrierprotein] reductase [NADPH] FabI Protein family Short-chain dehydrogenases/reductases (SDR) family, FabI subfamily Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the reduction of a carbon-carbon double bond in an enoyl moiety that is covalently linked to an acyl carrier protein (ACP). It has a preference for a long chain (C12) substrate compared to the shorter (C4) acyl group. Involved in the elongation cycle of fatty acid which are used in the lipid metabolism. Related diseases Amyloidosis, hereditary systemic 4, Finnish type (AMYLD4) [MIM:105120]: A form of hereditary systemic amyloidosis, a disorder characterized by amyloid deposition in multiple tissues resulting in a wide clinical spectrum. AMYLD4 is due to gelsolin amyloid deposition and is typically characterized by cranial neuropathy and lattice corneal dystrophy. Most patients have modest involvement of internal organs, but severe systemic disease can develop in some individuals causing peripheral polyneuropathy, amyloid cardiomyopathy, and nephrotic syndrome leading to renal failure. AMYLD4 is usually inherited in an autosomal dominant pattern. However, homozygotes with a more severe phenotype have also been reported. {ECO:0000269|PubMed:1338910, ECO:0000269|PubMed:19666512, ECO:0000269|PubMed:2176481}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; NAD; NADP; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A,C Molecular weight (Da) 55674.6 Length 510 Aromaticity 0.07 Instability index 33.86 Isoelectric point 5.64 Charge (pH=7) -9.3 2D Binding mode Binding energy (Kcal/mol) -11.33  Molscript Map 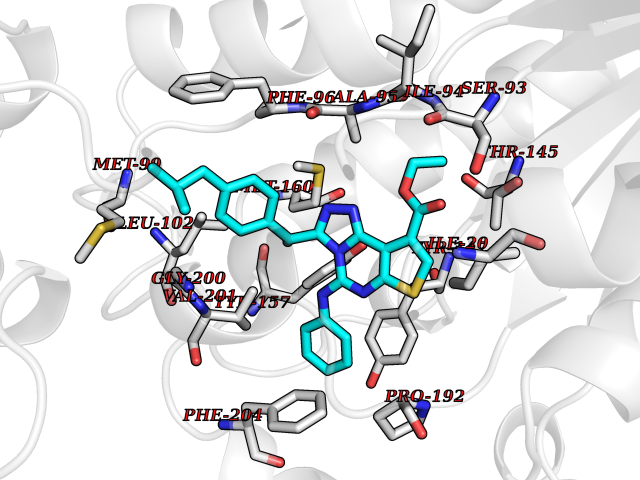 Pymol Map 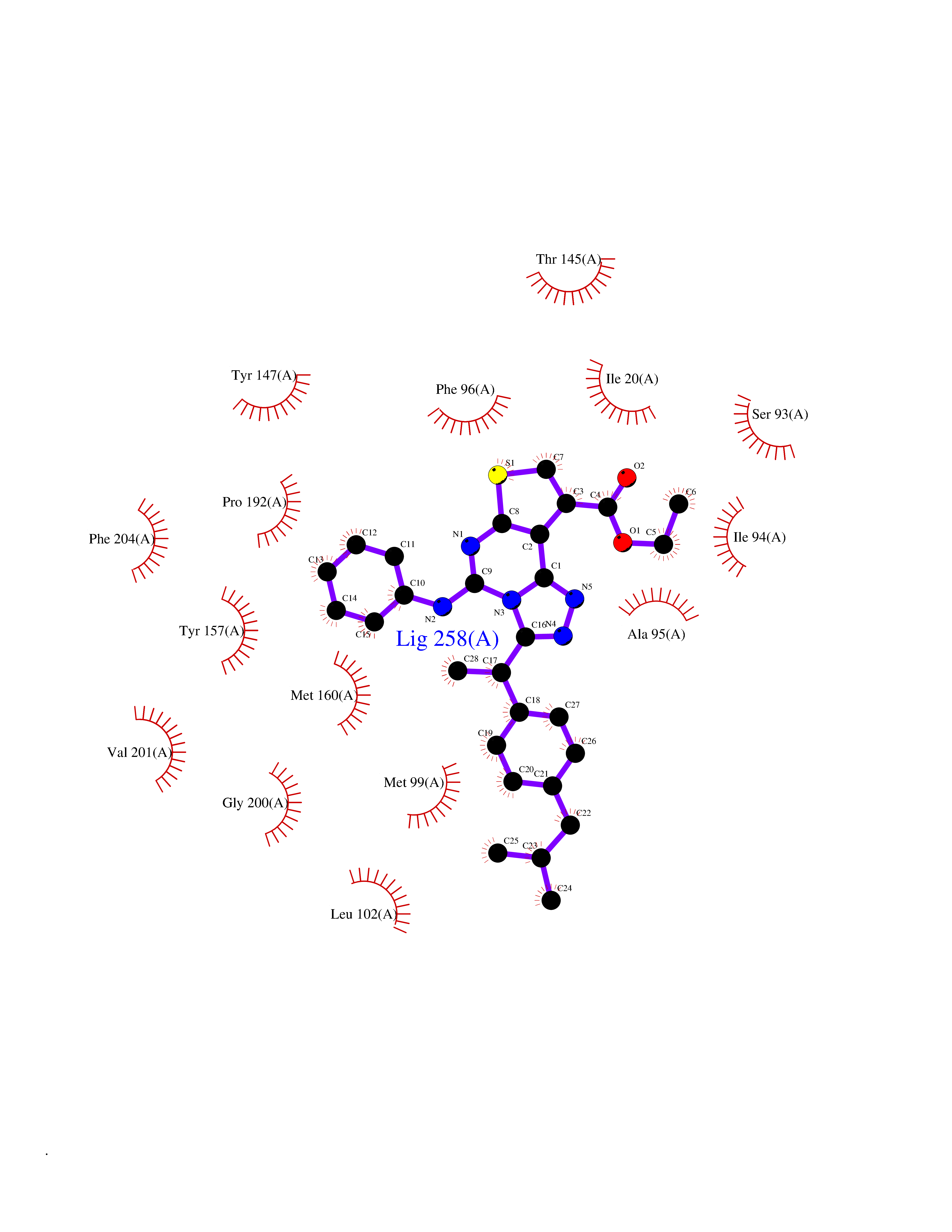 Ligplot Map 3D Binding mode Sequence VNLENKTYVIMGIANKRSIAFGVAKVLDQLGAKLVFTYRKERSRKELEKLLEQLNQPEAHLYQIDVQSDEEVINGFEQIGKDVGNIDGVYHSIAFANMEDLRGRFSETSREGFLLAQDISSYSLTIVAHEAKKLMPEGGSIVATTYLGGEFAVQNYNVMGVAKASLEANVKYLALDLGPDNIRVNAISAGPIRTLSAKGVGGFNTILKEIEERAPLKRNVDQVEVGKTAAYLLSDLSSGVTGENIHVDSGFHAIKVNLENKTYVIMGIANKRSIAFGVAKVLDQLGAKLVFTYRKERSRKELEKLLEQLNQPEAHLYQIDVQSDEEVINGFEQIGKDVGNIDGVYHSIAFANMEDLRGRFSETSREGFLLAQDISSYSLTIVAHEAKKLMPEGGSIVATTYLGGEFAVQNYNVMGVAKASLEANVKYLALDLGPDNIRVNAISAGPIRTLSAKGVGGFNTILKEIEERAPLKRNVDQVEVGKTAAYLLSDLSSGVTGENIHVDSGFHAIK Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Ecdysone receptor (20-hydroxy-ecdysone receptor) (EcRH) (Ecdysteroid receptor) (Nuclear receptor subfamily 1 group H member 1) | 3IXP | 8.29 | |
Target general information Gen name NA Organism Helicoverpa armigera (Cotton bollworm) (Heliothis armigera) Uniprot ID TTD ID NA Synonyms NA Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class NA Function NA Related diseases NA Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; DNA-binding; Metal-binding; Nucleus; Receptor; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,D Molecular weight (Da) 52799.1 Length 462 Aromaticity 0.09 Instability index 51.97 Isoelectric point 8.09 Charge (pH=7) 2.37 2D Binding mode Binding energy (Kcal/mol) -11.3  Molscript Map 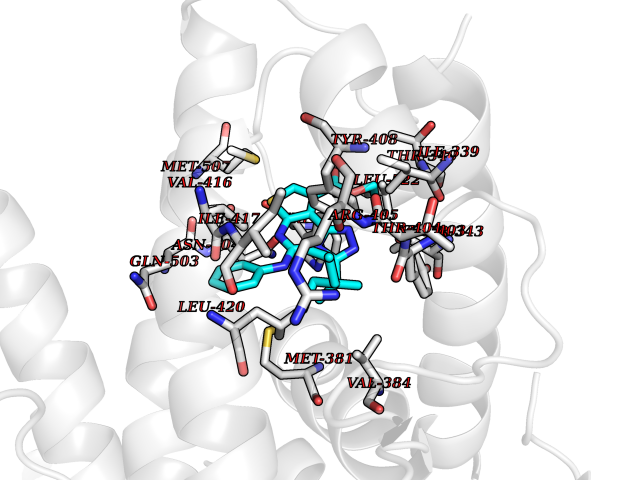 Pymol Map 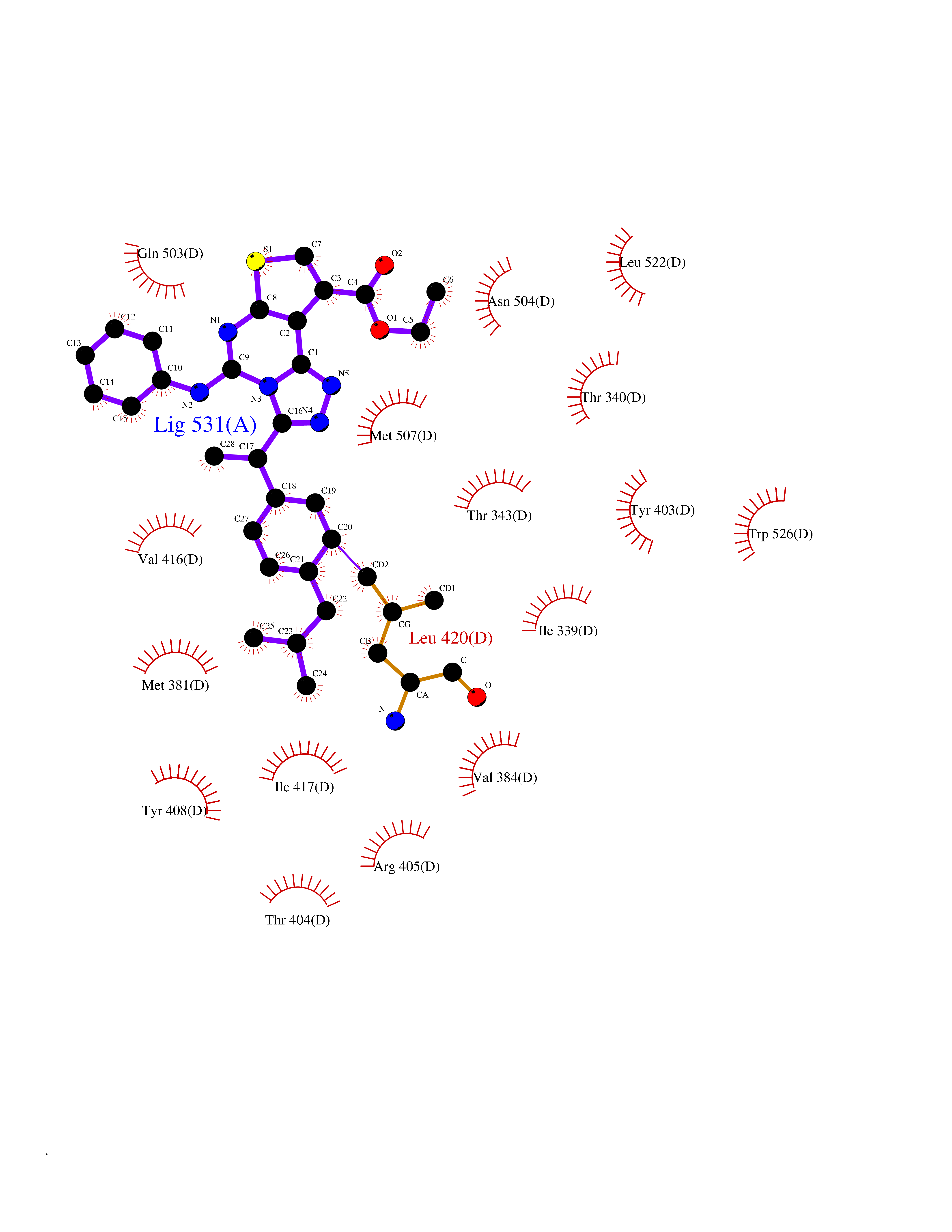 Ligplot Map 3D Binding mode Sequence QELSIERLLEMESLVADPSEEFQFLRVGPDSNVPPKFRAPVSSLCQIGNKQIAALVVWARDIPHFSQLEMEDQILLIKGSWNELLLFAIAWRSMEFLTSPPQLMCLMPGMTLHRNSALQAGVGQIFDRVLSELSLKMRTLRVDQAEYVALKAIILLNPDVKGLKNRQEVEVLREKMFLCLDEYCRRSRSSEEGRFAALLLRLPALRSISLKSFEHLFFFHLVADTSIAGYIRDALRNHAPPIVPPLTANQKSLIARLVYYQEGYEQMPFRQITEMTILTVQLIVEFAKGLPGFSKISQSDQITLLKACSSEVMMLRVARRYDAATDSVLFANNQAYTRDNYRKAGMAYVIEDLLHFCRCMYSMMMDNVHYALLTAIVIFSDRPGLEQPSLVEEIQRYYLNTLRVYILNQNSASPRSAVIFGKILGILTEIRTLGMQNSNMCISLKLKNRKLPPFLEEIWDVA Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Acetylcholinesterase (AChE) (EC 3.1.1.7) | 1GPK | 8.29 | |
Target general information Gen name ache Organism Tetronarce californica (Pacific electric ray) (Torpedo californica) Uniprot ID TTD ID NA Synonyms NA Protein family Type-B carboxylesterase/lipase family Biochemical class NA Function Terminates signal transduction at the neuromuscular junction by rapid hydrolysis of the acetylcholine released into the synaptic cleft. May be involved in cell-cell interactions. Related diseases Noonan syndrome 5 (NS5) [MIM:611553]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:17603482, ECO:0000269|PubMed:17603483, ECO:0000269|PubMed:20683980}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: LEOPARD syndrome 2 (LPRD2) [MIM:611554]: A disorder characterized by lentigines, electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonic stenosis, abnormalities of genitalia, retardation of growth, and sensorineural deafness. {ECO:0000269|PubMed:17603483}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Cardiomyopathy, dilated, 1NN (CMD1NN) [MIM:615916]: A disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death. {ECO:0000269|PubMed:24777450}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.1.1.7 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Direct protein sequencing; Disulfide bond; Glycoprotein; GPI-anchor; Hydrolase; Lipoprotein; Membrane; Neurotransmitter degradation; Serine esterase; Signal; Synapse Protein physicochemical properties Chain ID A Molecular weight (Da) 59779.2 Length 529 Aromaticity 0.12 Instability index 48.49 Isoelectric point 5.8 Charge (pH=7) -8.48 2D Binding mode Binding energy (Kcal/mol) -11.31 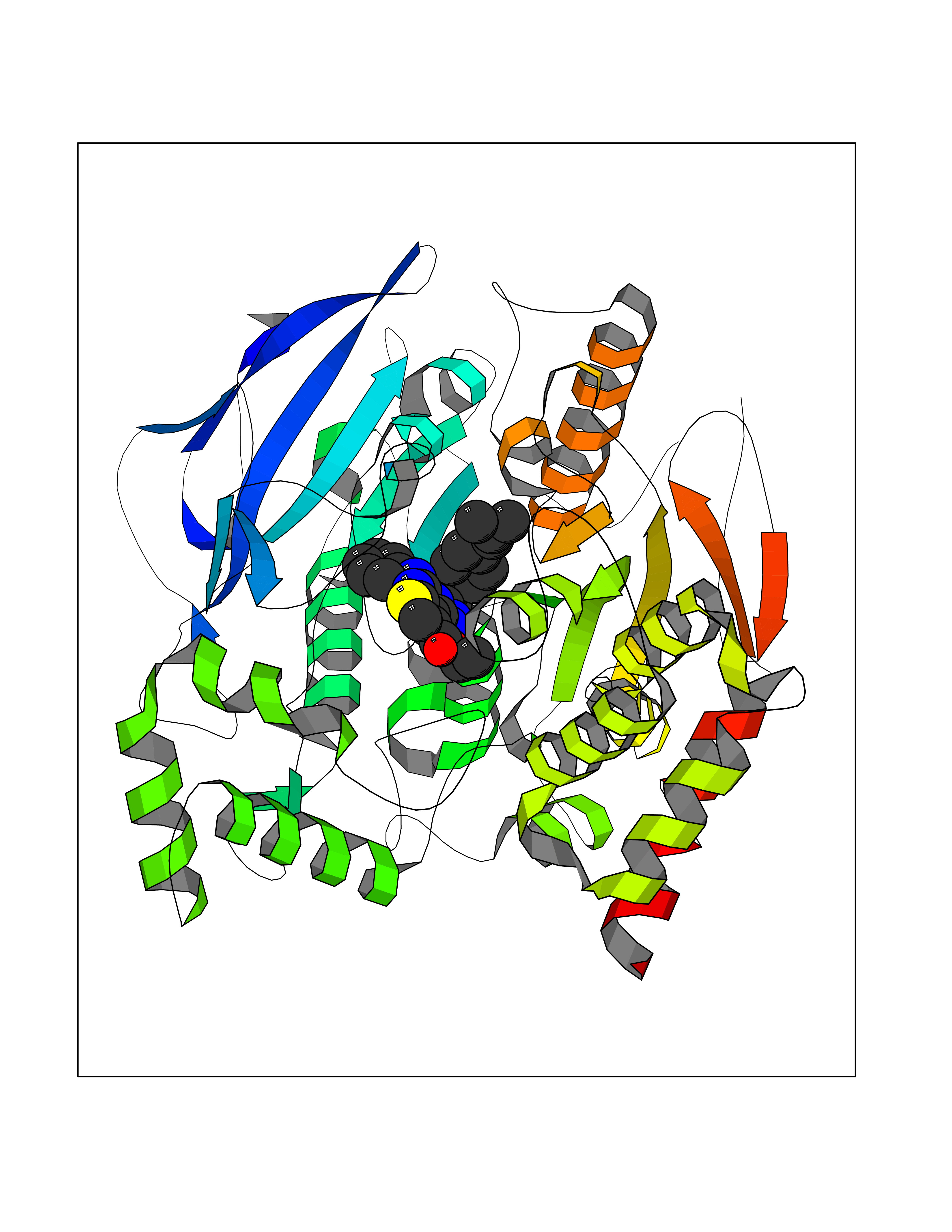 Molscript Map 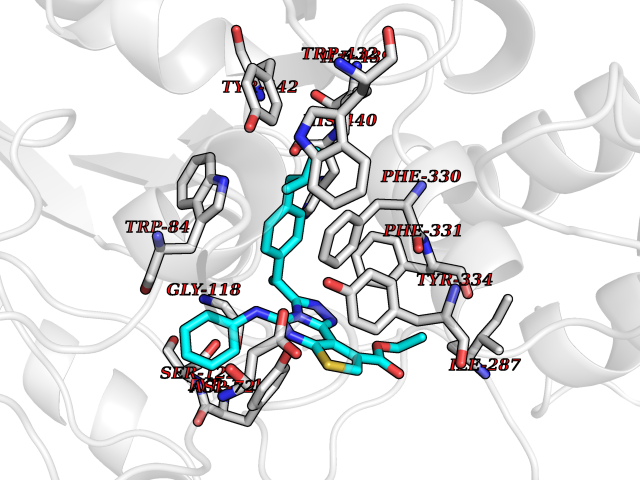 Pymol Map 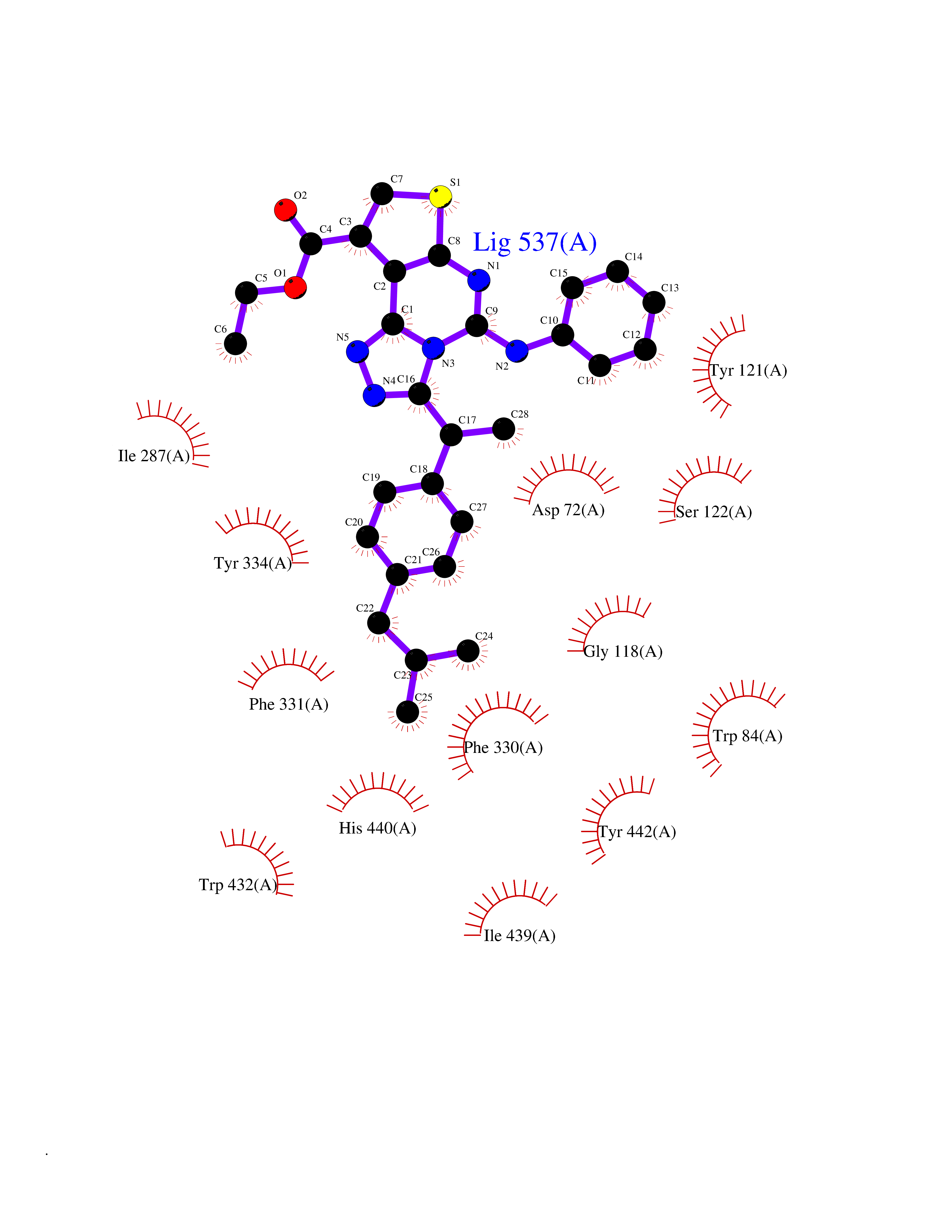 Ligplot Map 3D Binding mode Sequence SELLVNTKSGKVMGTRVPVLSSHISAFLGIPFAEPPVGNMRFRRPEPKKPWSGVWNASTYPNNCQQYVDEQFPGFSGSEMWNPNREMSEDCLYLNIWVPSPRPKSTTVMVWIYGGGFYSGSSTLDVYNGKYLAYTEEVVLVSLSYRVGAFGFLALHGSQEAPGNVGLLDQRMALQWVHDNIQFFGGDPKTVTIFGESAGGASVGMHILSPGSRDLFRRAILQSGSPNCPWASVSVAEGRRRAVELGRNLNCNLNSDEELIHCLREKKPQELIDVEWNVLPFDSIFRFSFVPVIDGEFFPTSLESMLNSGNFKKTQILLGVNKDEGSFFLLYGAPGFSKDSESKISREDFMSGVKLSVPHANDLGLDAVTLQYTDWMDDNNGIKNRDGLDDIVGDHNVICPLMHFVNKYTKFGNGTYLYFFNHRASNLVWPEWMGVIHGYEIEFVFGLPLVKELNYTAEEEALSRRIMHYWATFAKTGNPNEPESKWPLFTTKEQKFIDLNTEPMKVHQRLRVQMCVFWNQFLPKLLNAT Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Thiopurine S-methyltransferase | 2BZG | 8.28 | |
Target general information Gen name TPMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, TPMT family Biochemical class Transferase Function Thiopurine S-methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00993; DB00436; DB01327; DB01033; DB01250; DB01021 Interacts with Q8TAP4-4; Q15047-2; P61981 EC number 2.1.1.67 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 25971.5 Length 229 Aromaticity 0.12 Instability index 32.58 Isoelectric point 6.74 Charge (pH=7) -0.6 2D Binding mode Binding energy (Kcal/mol) -11.29 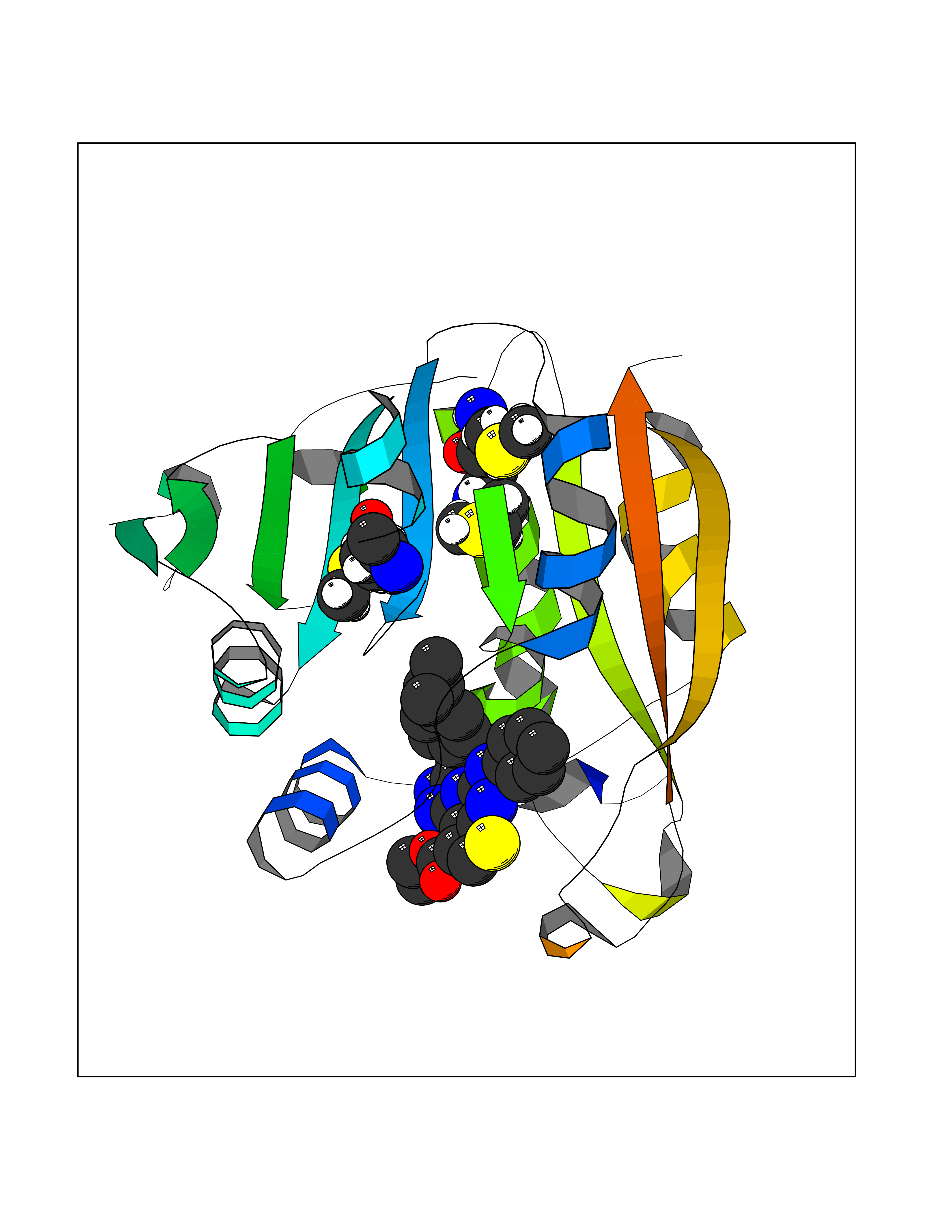 Molscript Map 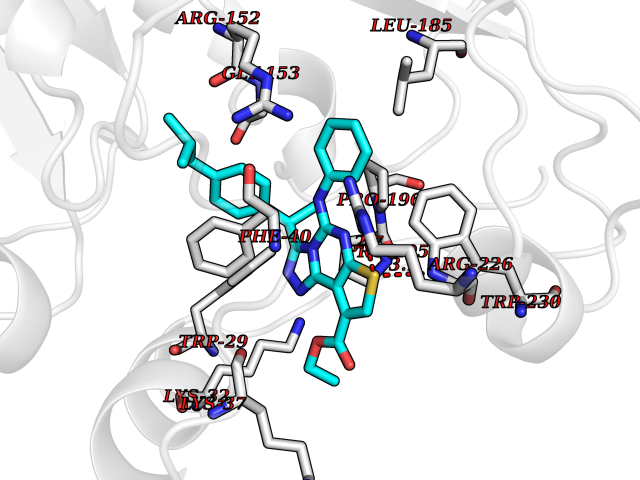 Pymol Map 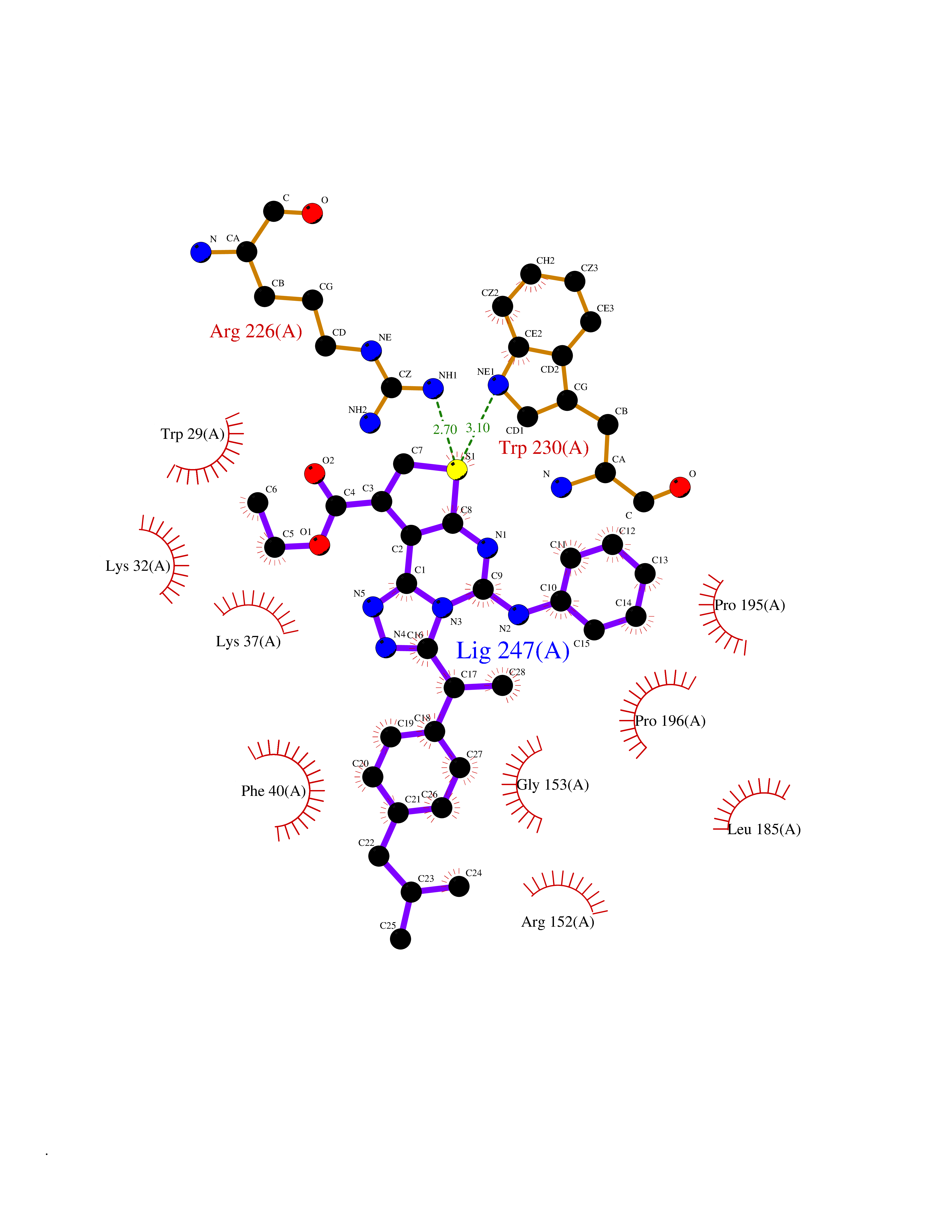 Ligplot Map 3D Binding mode Sequence EVQKNQVLTLEEWQDKWVNGKTAFHQEQGHQLLKKHLDTFLKGKSGLRVFFPLCGKAVEXKWFADRGHSVVGVEISELGIQEFFTEQNLSYSEEPITEIPGTKVFKSSSGNISLYCCSIFDLPRTNIGKFDXIWDRGALVAINPGDRKCYADTXFSLLGKKFQYLLCVLSYDPTKHPGPPFYVPHAEIERLFGKICNIRCLEKVDAFEERHKSWGIDCLFEKLYLLTEK Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Dihydroorotate dehydrogenase (DHODH) | 4OQV | 8.28 | |
Target general information Gen name DHODH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dihydroorotate oxidase; Dihydroorotate dehydrogenase (quinone), mitochondrial; DHOdehase; DHODH Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Postaxial acrofacial dysostosis (POADS) [MIM:263750]: POADS is characterized by severe micrognathia, cleft lip and/or palate, hypoplasia or aplasia of the posterior elements of the limbs, coloboma of the eyelids and supernumerary nipples. POADS is a very rare disorder: only 2 multiplex families, each consisting of 2 affected siblings born to unaffected, nonconsanguineous parents, have been described among a total of around 30 reported cases. {ECO:0000269|PubMed:19915526}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07559; DB07561; DB08172; DB08169; DB07443; DB07978; DB07975; DB04281; DB08249; DB07977; DB07976; DB04583; DB08008; DB01117; DB03523; DB03480; DB02613; DB04147; DB03247; DB01097; DB06481; DB08006; DB02262; DB05125; DB08880; DB07646 Interacts with Q6ZMZ0; P49638 EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Disease variant; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Proteomics identification; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 38341.4 Length 353 Aromaticity 0.05 Instability index 39.27 Isoelectric point 9.28 Charge (pH=7) 5.52 2D Binding mode Binding energy (Kcal/mol) -11.29  Molscript Map 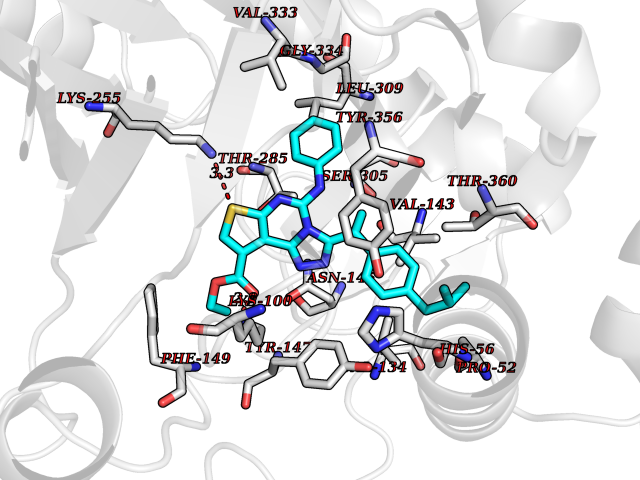 Pymol Map 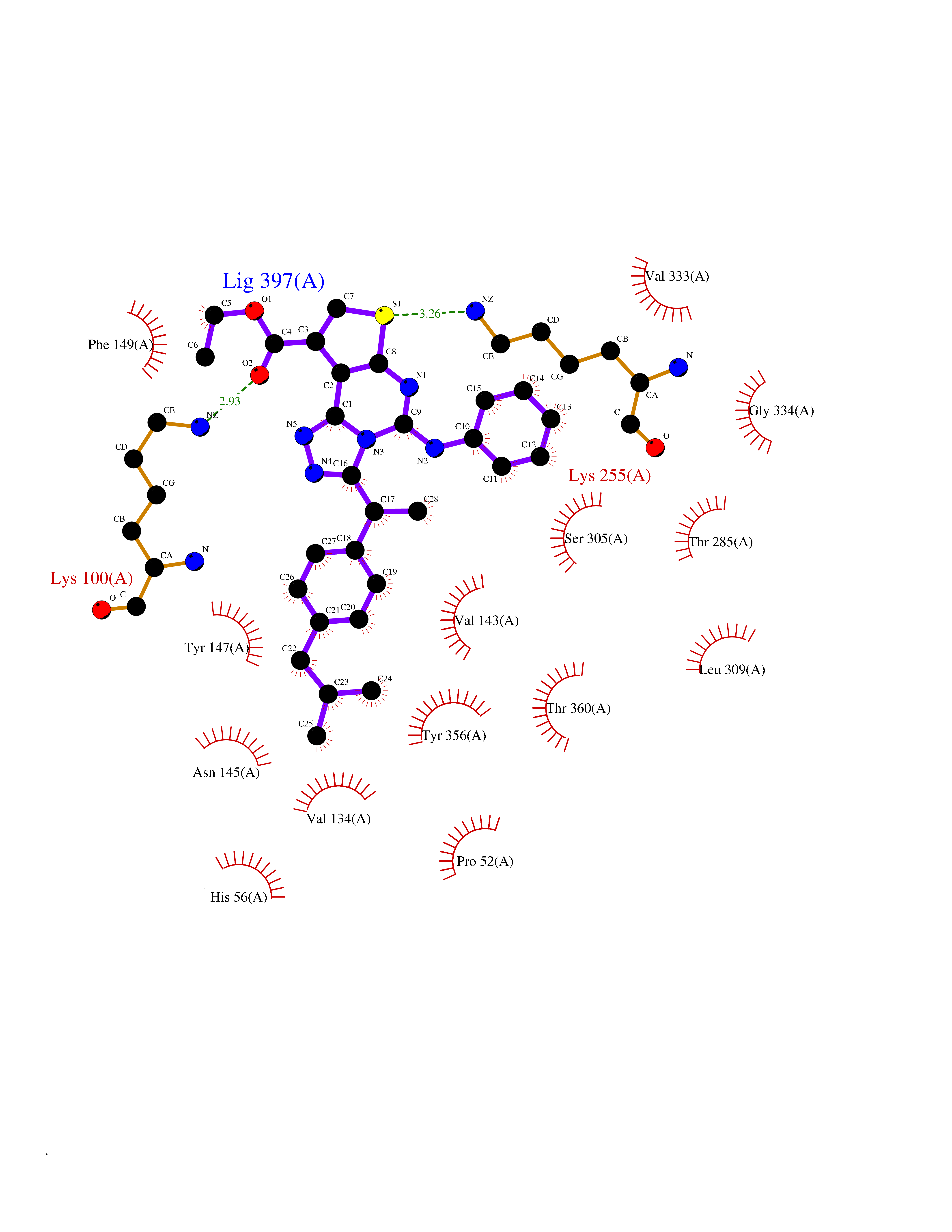 Ligplot Map 3D Binding mode Sequence DERFYAEHLMPTLQGLLDPESAHRLAVRFTSLGLLPRARFQDSDMLEVRVLGHKFRNPVGIAAGFDKHGEAVDGLYKMGFGFVEIGSVTPKPQEGNPRPRVFRLPEDQAVINRYGFNSHGLSVVEHRLRARQQKQAKLTEDGLPLGVNLGKNKTSVDAAEDYAEGVRVLGPLADYLVVNVSSPGKAELRRLLTKVLQERDGLRRVHRPAVLVKIAPDLTSQDKEDIASVVKELGIDGLIVTNTTVSRPAGLQGALRSETGGLSGKPLRDLSTQTIREMYALTQGRVPIIGVGGVSSGQDALEKIRAGASLVQLYTALTFWGPPVVGKVKRELEALLKEQGFGGVTDAIGADHR Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Bile acid receptor | 4QE6 | 8.28 | |
Target general information Gen name NR1H4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms HRR1;BAR;RIP14;FXR Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Transcription Function Bile acid binding.Bile acid receptor activity.Chenodeoxycholic acid binding.Ligand-dependent nuclear receptor binding.Retinoid X receptor binding.RNA polymerase II distal enhancer sequence-specific DNA binding.RNA polymerase II regulatory region sequence-specific DNA binding.RNA polymerase II transcription factor activity, ligand-activated sequence-specific DNA binding.Sequence-specific DNA binding.Steroid hormone receptor activity.Thyroid hormone receptor activity.Transcriptional activator activity, RNA polymerase II core promoter proximal region sequence-specific binding.Transcriptional activator activity, RNA polymerase II transcription factor binding.Transcription coactivator activity.Transcription corepressor activity.Transcription factor activity, RNA polymerase II distal enhancer sequence-specific binding.Transcription factor activity, sequence-specific DNA binding.Transcription regulatory region sequence-specific DNA binding.Zinc ion binding. Related diseases May be involved in intrahepatic cholestasis of pregnancy. {ECO:0000305|PubMed:17681172}.; DISEASE: May be involved in cholesterol cholelithiasis. {ECO:0000305|PubMed:17931734}.; DISEASE: Cholestasis, progressive familial intrahepatic, 5 (PFIC5) [MIM:617049]: A disorder characterized by early onset of cholestasis that progresses to hepatic fibrosis, cirrhosis, and end-stage liver disease before adulthood. PFIC5 is an autosomal recessive, severe form characterized by onset of intralobular cholestasis in the neonatal period. {ECO:0000269|PubMed:26888176}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08220; DB00132; DB04557; DB06777; DB02659; DB03619; DB02509; DB02545; DB11605; DB16255; DB05990; DB04348; DB16343; DB01586 Interacts with Q15788; O75151; Q8WTS6; P28702; P28702-3; P48443; Q15788; P78527; P03372 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative promoter usage; Alternative splicing; Disease variant; DNA-binding; Immunity; Inflammatory response; Innate immunity; Intrahepatic cholestasis; Isopeptide bond; Metal-binding; Methylation; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repressor; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 28271.2 Length 241 Aromaticity 0.08 Instability index 47.06 Isoelectric point 5.39 Charge (pH=7) -11.24 2D Binding mode Binding energy (Kcal/mol) -11.29  Molscript Map 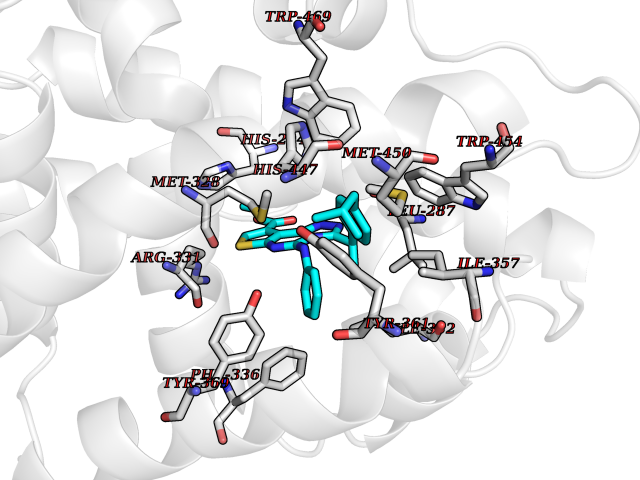 Pymol Map 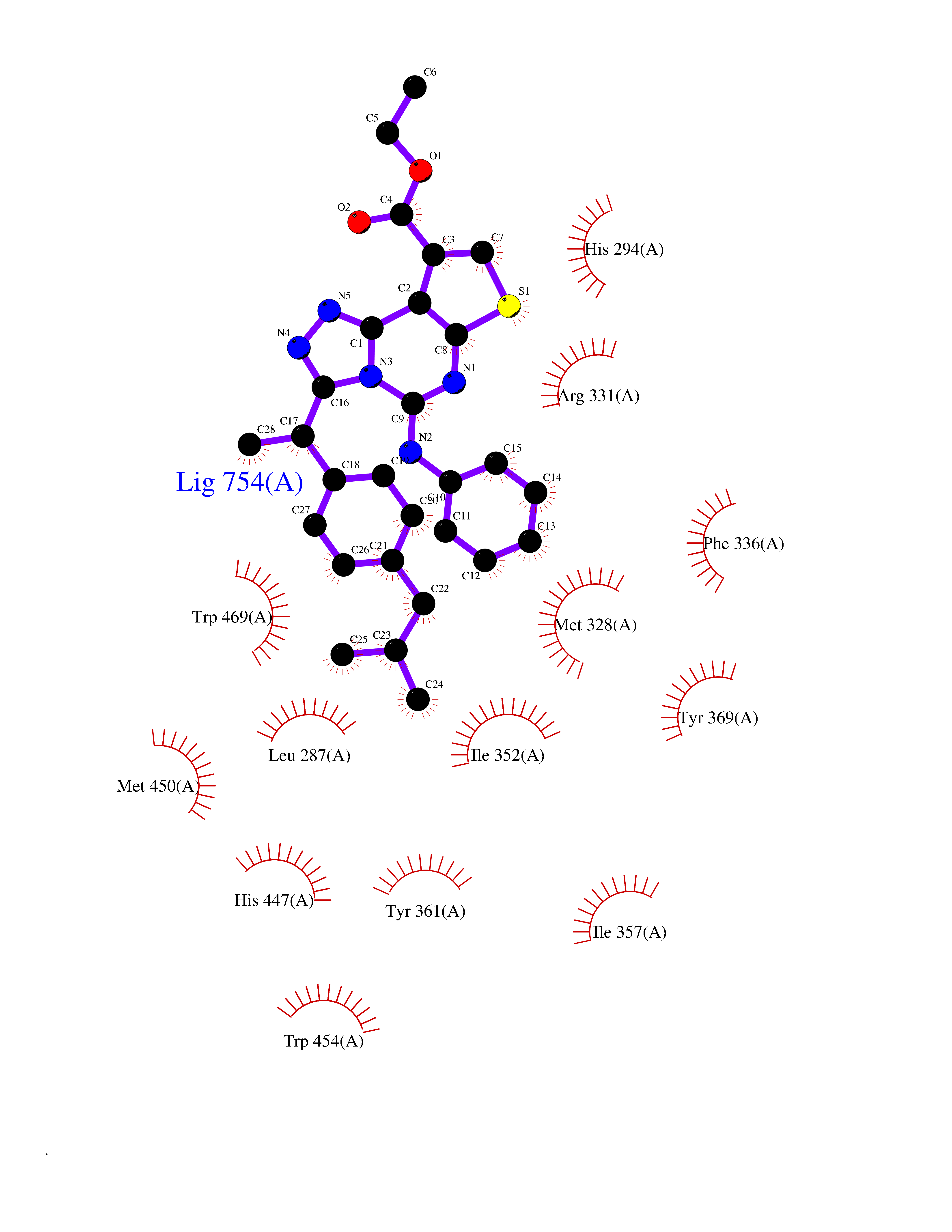 Ligplot Map 3D Binding mode Sequence HMELTPDQQTLLHFIMDSYNKQRMPQEITNKILKEEFSAEENFLILTEMATNHVQVLVEFTKKLPGFQTLDHEDQIALLKGSAVEAMFLRSAEIFNKSGHSDLLEERIRNSGISDEYITPMFSFYKSIGELKMTQEEYALLTAIVILSPDRQYIKDREAVEKLQEPLLDVLQKLCKIHQPENPQHFACLLGRLTELRTFNHHHAEMLMSWRVNDHKFTPLLCEIWDVQKENALLRYLLDKD Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Cytochrome P450 1A2 | 2HI4 | 8.27 | |
Target general information Gen name CYP1A2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Aromatase activity.Caffeine oxidase activity.Demethylase activity.Electron carrier activity.Enzyme binding.Heme binding.Iron ion binding.Monooxygenase activity.Oxidoreductase activity.Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, reduced flavin or flavoprotein as one donor, and incorporation of one atom of oxygen.Oxygen binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08496; DB01667; DB14132; DB04356; DB02489; DB11932; DB12001; DB05812; DB13573; DB01418; DB00316; DB15568; DB06594; DB00518; DB05396; DB00969; DB07453; DB01424; DB01223; DB01118; DB00321; DB00261; DB01217; DB01435; DB06605; DB05676; DB06413; DB06216; DB01072; DB15011; DB06442; DB06626; DB00993; DB00972; DB13203; DB05015; DB16703; DB06769; DB01086; DB06770; DB06771; DB06732; DB00195; DB04889; DB11967; DB13975; DB00188; DB12151; DB01558; DB14018; DB13812; DB00201; DB09061; DB14737; DB11791; DB06774; DB00564; DB06016; DB01136; DB12814; DB00477; DB00356; DB01166; DB00501; DB01012; DB00568; DB00827; DB00537; DB00215; DB12499; DB14025; DB00349; DB01242; DB00575; DB00758; DB00363; DB00286; DB11672; DB14635; DB00924; DB08912; DB00851; DB06292; DB01254; DB01609; DB01151; DB16650; DB12161; DB01191; DB00633; DB11994; DB00586; DB11511; DB12945; DB00280; DB01184; DB09167; DB05928; DB01142; DB09273; DB00470; DB00476; DB00625; DB15444; DB06210; DB13874; DB11718; DB00467; DB11404; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB04574; DB13592; DB00330; DB00898; DB00977; DB00773; DB01628; DB00927; DB04854; DB01482; DB00574; DB12265; DB15669; DB01195; DB08972; DB04841; DB00544; DB00472; DB00499; DB00176; DB01320; DB00998; DB14029; DB06160; DB01044; DB01241; DB01155; DB01645; DB01381; DB00986; DB00365; DB00400; DB05708; DB00629; DB00502; DB01094; DB14999; DB04076; DB11737; DB00619; DB00458; DB11564; DB01306; DB09456; DB09564; DB01307; DB00047; DB01309; DB00030; DB00046; DB11567; DB00071; DB11568; DB05258; DB00034; DB00105; DB15131; DB00011; DB00018; DB00069; DB00060; DB00068; DB00033; DB00951; DB11757; DB09570; DB01026; DB01097; DB16217; DB09078; DB01002; DB05667; DB00281; DB12406; DB09198; DB04948; DB00978; DB06448; DB16220; DB01601; DB00455; DB04871; DB06077; DB01283; DB00772; DB00934; DB06234; DB14009; DB00784; DB01065; DB00170; DB00454; DB00532; DB00333; DB00763; DB00553; DB01028; DB09241; DB01233; DB00379; DB06148; DB01388; DB06595; DB00370; DB16236; DB00745; DB11763; DB00218; DB06510; DB14011; DB00461; DB00607; DB00779; DB00788; DB06600; DB00238; DB06803; DB00184; DB01115; DB11793; DB00435; DB05115; DB00717; DB01059; DB00540; DB05990; DB01165; DB00334; DB16267; DB00338; DB00904; DB11632; DB11443; DB01173; DB11837; DB09330; DB01303; DB11697; DB00377; DB00715; DB06589; DB11774; DB00487; DB00008; DB00022; DB09122; DB13634; DB00806; DB11198; DB08883; DB00850; DB03783; DB01174; DB00388; DB00252; DB11450; DB01100; DB13823; DB04951; DB17472; DB11642; DB08910; DB15822; DB01058; DB01087; DB00794; DB00420; DB09288; DB01182; DB06479; DB00818; DB00571; DB13449; DB11892; DB04216; DB00908; DB00468; DB01129; DB00980; DB09290; DB00863; DB01367; DB00409; DB02709; DB13174; DB01045; DB11753; DB00740; DB14924; DB00503; DB00533; DB01656; DB15119; DB00268; DB00296; DB00412; DB00817; DB12332; DB13772; DB06654; DB11491; DB00418; DB01037; DB11689; DB06290; DB13261; DB15093; DB00052; DB00398; DB01208; DB09118; DB00428; DB06820; DB00382; DB00675; DB06083; DB09071; DB05488; DB09256; DB01079; DB01405; DB00857; DB08880; DB11712; DB01412; DB00277; DB00730; DB01623; DB00208; DB06137; DB00697; DB01056; DB06264; DB00752; DB00384; DB12245; DB00831; DB15442; DB00440; DB00685; DB08867; DB14989; DB13609; DB06235; DB00313; DB08881; DB00661; DB09185; DB12026; DB00682; DB02134; DB00549; DB00744; DB00315; DB00425; DB09225; DB09120 Interacts with O95870 EC number 1.14.14.1; 4.2.1.152 Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Fatty acid metabolism; Glycoprotein; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54475 Length 480 Aromaticity 0.1 Instability index 40.43 Isoelectric point 9.16 Charge (pH=7) 9.89 2D Binding mode Binding energy (Kcal/mol) -11.28 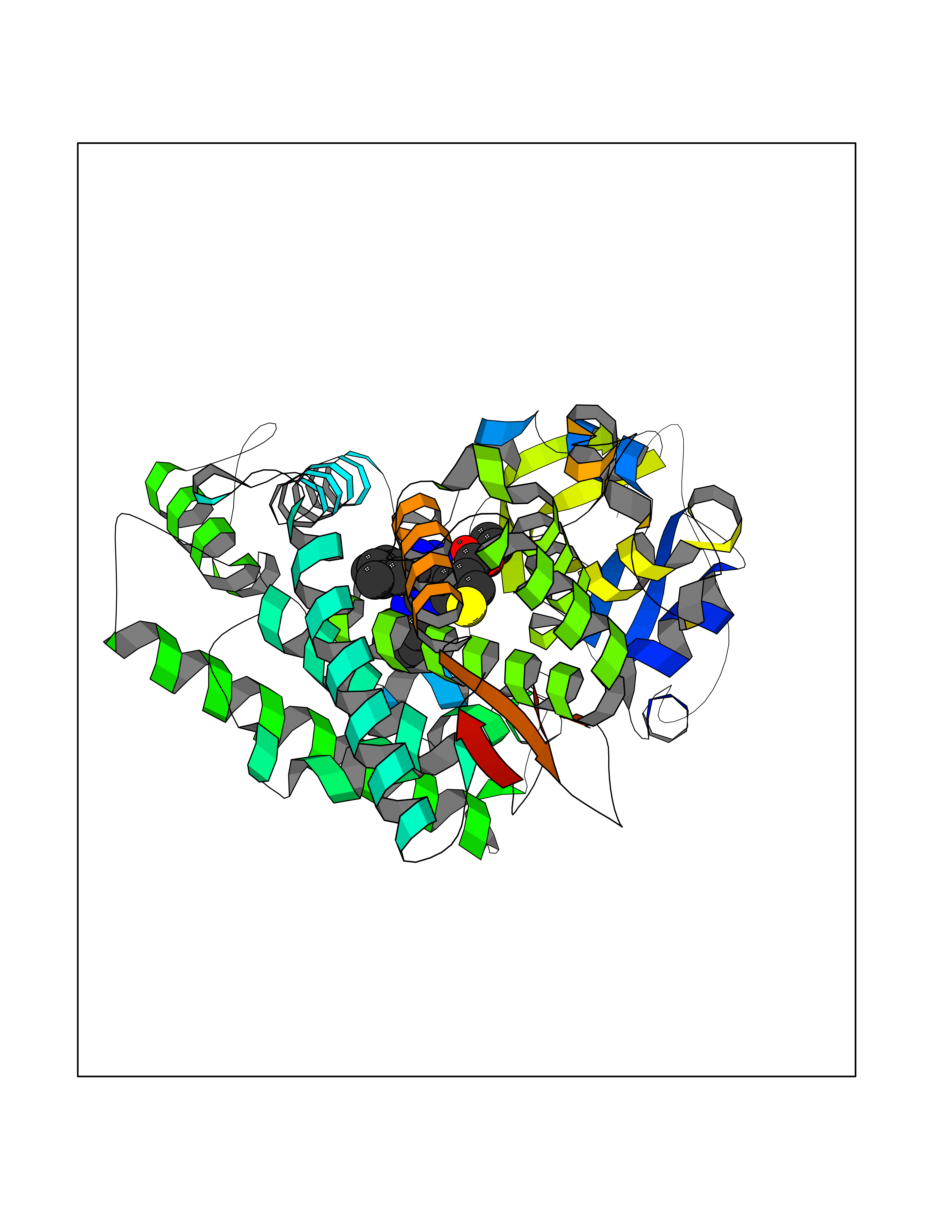 Molscript Map 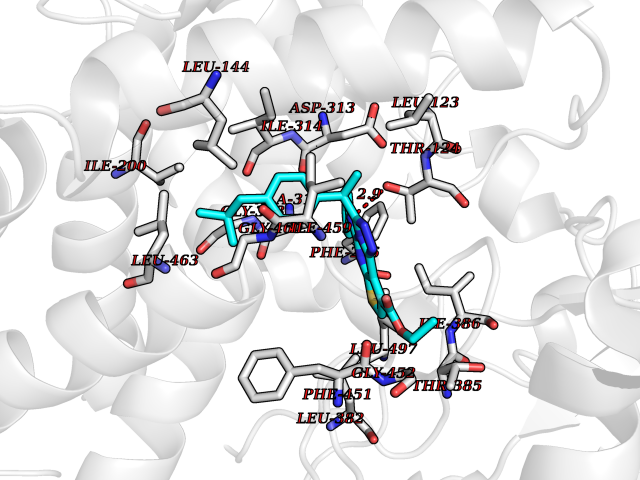 Pymol Map 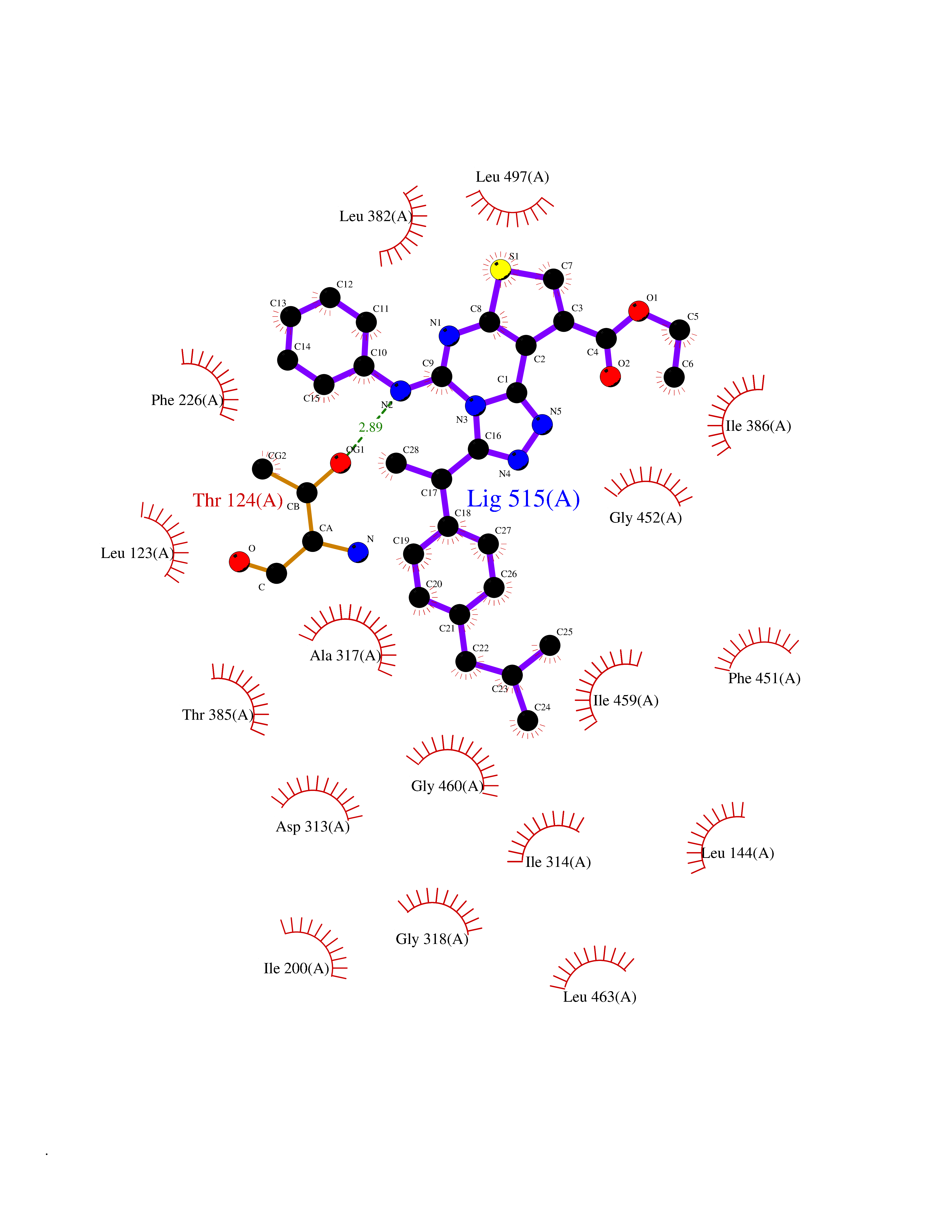 Ligplot Map 3D Binding mode Sequence RVPKGLKSPPEPWGWPLLGHVLTLGKNPHLALSRMSQRYGDVLQIRIGSTPVLVLSRLDTIRQALVRQGDDFKGRPDLYTSTLITDGQSLTFSTDSGPVWAARRRLAQNALNTFSIASDPASSSSCYLEEHVSKEAKALISRLQELMAGPGHFDPYNQVVVSVANVIGAMCFGQHFPESSDEMLSLVKNTHEFVETASSGNPLDFFPILRYLPNPALQRFKAFNQRFLWFLQKTVQEHYQDFDKNSVRDITGALFKHSKKGPRASGNLIPQEKIVNLVNDIFGAGFDTVTTAISWSLMYLVTKPEIQRKIQKELDTVIGRERRPRLSDRPQLPYLEAFILETFRHSSFLPFTIPHSTTRDTTLNGFYIPKKCCVFVNQWQVNHDPELWEDPSEFRPERFLTADGTAINKPLSEKMMLFGMGKRRCIGEVLAKWEIFLFLAILLQQLEFSVPPGVKVDLTPIYGLTMKHARCEHVQARRFS Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Soluble epoxide hydrolase (EPHX2) | 1ZD3 | 8.27 | |
Target general information Gen name EPHX2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Bifunctional epoxide hydrolase 2 Protein family AB hydrolase superfamily, Epoxide hydrolase family Biochemical class Ether bond hydrolase Function Bifunctional enzyme. The C-terminal domain has epoxide hydrolase activity and acts on epoxides (alkene oxides, oxiranes) and arene oxides. Plays a role in xenobiotic metabolism by degrading potentially toxic epoxides (By similarity). Also determines steady-state levels of physiological mediators. The N-terminal domain has lipid phosphatase activity, with the highest activity towards threo-9,10-phosphonooxy-hydroxy-octadecanoic acid, followed by erythro-9,10-phosphonooxy-hydroxy-octadecanoic acid, 12-phosphonooxy-octadec-9Z-enoic acid and 12-phosphonooxy-octadec-9E-enoic acid. Related diseases Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:17332249}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 6 (NS6) [MIM:613224]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:19966803}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: RAS-associated autoimmune leukoproliferative disorder (RALD) [MIM:614470]: A disorder of apoptosis, characterized by chronic accumulation of non-malignant lymphocytes, defective lymphocyte apoptosis, and an increased risk for the development of hematologic malignancies. {ECO:0000269|PubMed:17517660}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanocytic nevus syndrome, congenital (CMNS) [MIM:137550]: A syndrome characterized by congenital pigmentary skin lesions which can occur at any site and can cover most of the body surface. These lesions may or may not be hairy. Congenital melanocytic nevi are associated with neuromelanosis (the presence of melanin-producing cells within the brain parenchyma or leptomeninges). Less commonly they are associated with malignant melanoma in childhood, both in the skin and the central nervous system. CMNS patients also tend to have a characteristic facial appearance, including wide or prominent forehead, periorbital fullness, small short nose with narrow nasal bridge, round face, full cheeks, prominent premaxilla, and everted lower lip. {ECO:0000269|PubMed:18633438, ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanosis, neurocutaneous (NCMS) [MIM:249400]: A rare congenital disease characterized by the presence of giant or multiple melanocytic nevi on the skin, foci of melanin-producing cells within the brain parenchyma, and infiltration of leptomeninges by abnormal melanin deposits. Neurologic abnormalities include seizures, hydrocephalus, arachnoid cysts, tumors, and syringomyelia. Some patients may develop malignant melanoma. {ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Keratinocytic non-epidermolytic nevus (KNEN) [MIM:162900]: Epidermal nevi of the common, non-organoid and non-epidermolytic type are benign skin lesions and may vary in their extent from a single (usually linear) lesion to widespread and systematized involvement. They may be present at birth or develop early during childhood. {ECO:0000269|PubMed:22499344}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid cancer, non-medullary, 2 (NMTC2) [MIM:188470]: A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms. {ECO:0000269|PubMed:12727991}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08257; DB08258; DB08259; DB06345; DB12610; DB08256; DB02029; DB04213; DB03677 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aromatic hydrocarbons catabolism; Cytoplasm; Detoxification; Direct protein sequencing; Hydrolase; Lipid metabolism; Lipoprotein; Magnesium; Metal-binding; Multifunctional enzyme; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 61744.9 Length 547 Aromaticity 0.09 Instability index 43.97 Isoelectric point 5.81 Charge (pH=7) -7.76 2D Binding mode Binding energy (Kcal/mol) -11.28 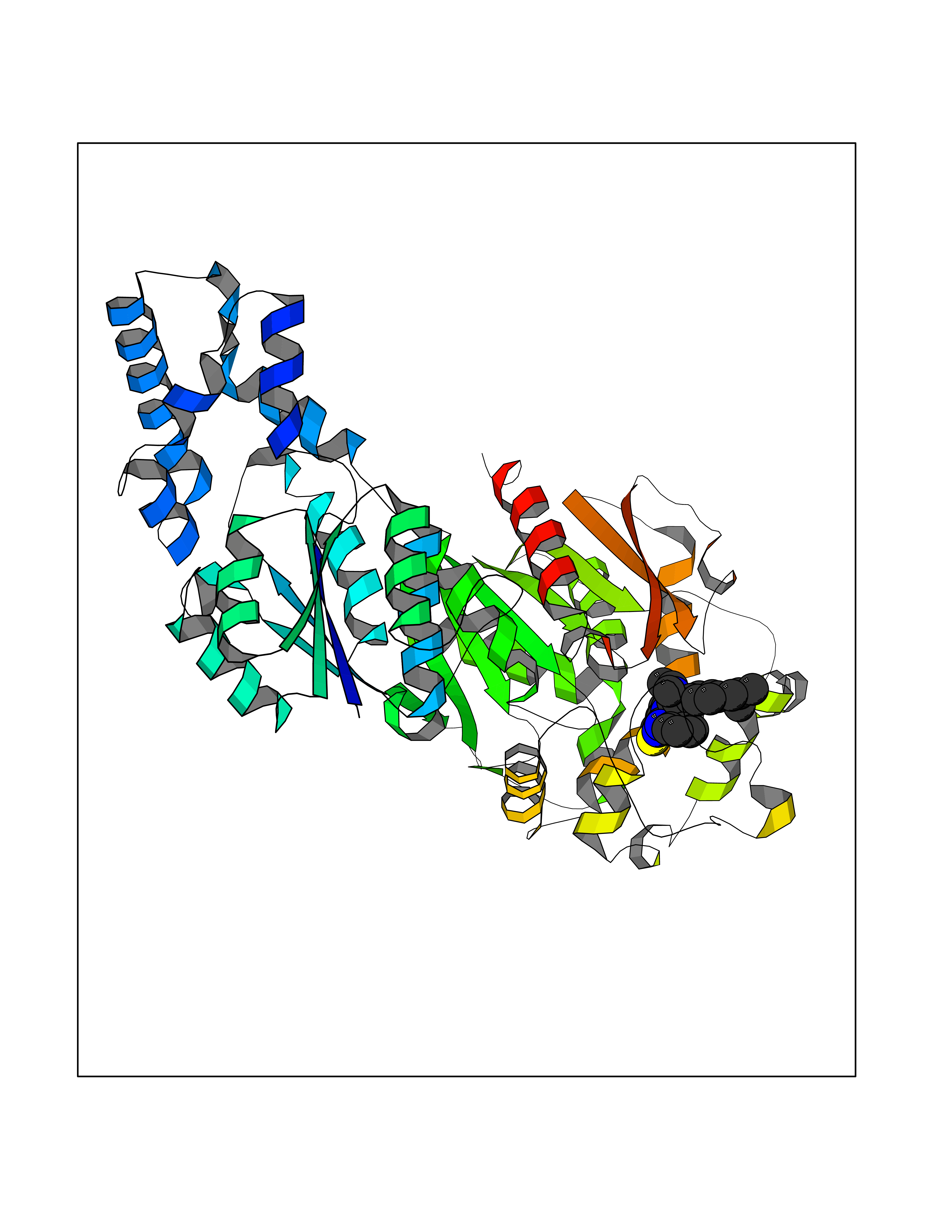 Molscript Map 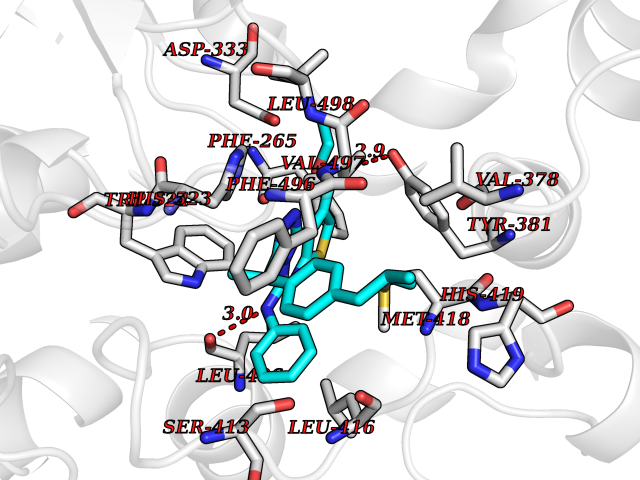 Pymol Map 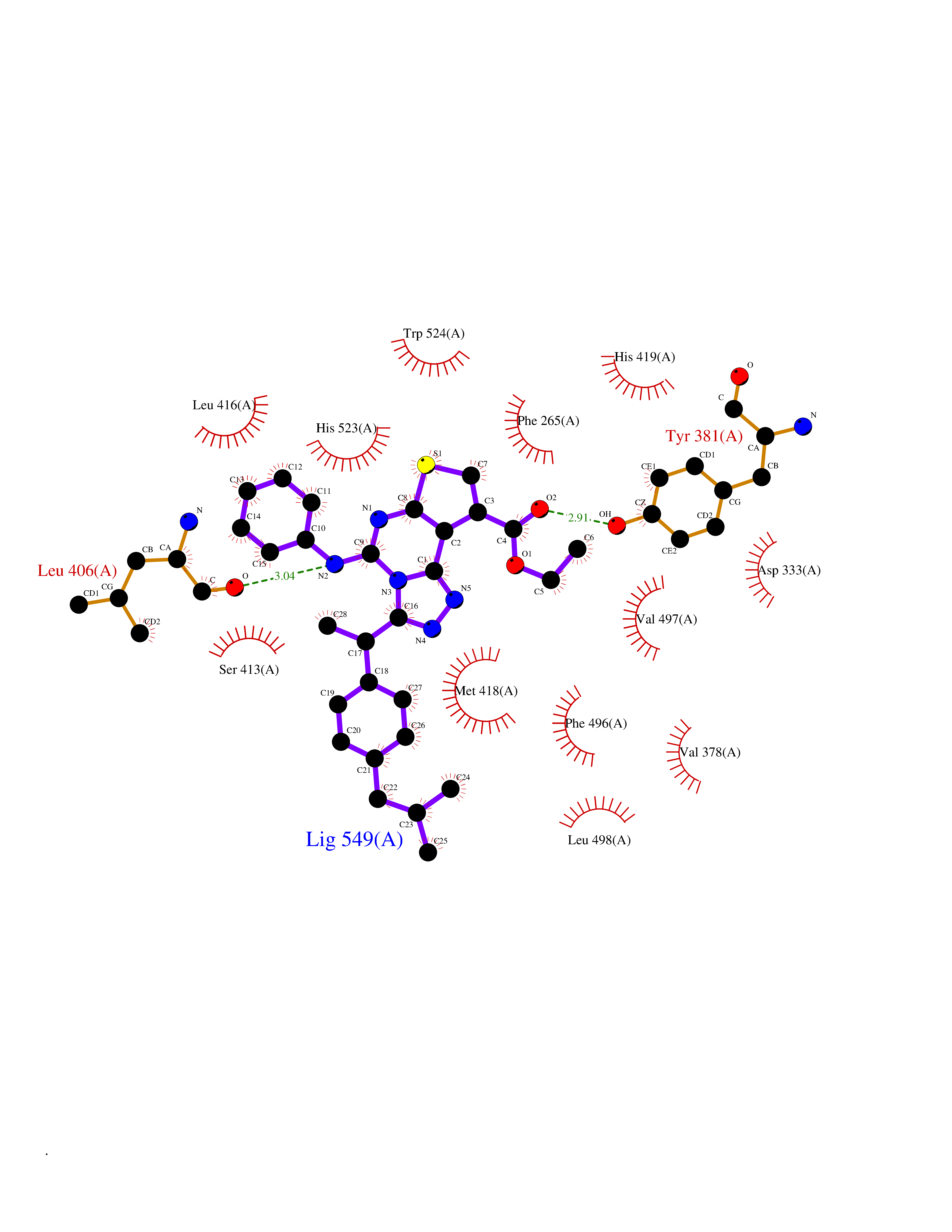 Ligplot Map 3D Binding mode Sequence TLRAAVFDLDGVLALPAVFGVLGRTEEALALPRGLLNDAFQKGGPEGATTRLMKGEITLSQWIPLMEENCRKCSETAKVCLPKNFSIKEIFDKAISARKINRPMLQAALMLRKKGFTTAILTNTWLDDRAERDGLAQLMCELKMHFDFLIESCQVGMVKPEPQIYKFLLDTLKASPSEVVFLDDIGANLKPARDLGMVTILVQDTDTALKELEKVTGIQLLNTPAPLPTSCNPSDMSHGYVTVKPRVRLHFVELGSGPAVCLCHGFPESWYSWRYQIPALAQAGYRVLAMDMKGYGESSAPPEIEEYCMEVLCKEMVTFLDKLGLSQAVFIGHDWGGMLVWYMALFYPERVRAVASLNTPFIPANPNMSPLESIKANPVFDYQLYFQEPGVAEAELEQNLSRTFKSLFRASDESVLSMHKVCEAGGLFVNSPEEPSLSRMVTEEEIQFYVQQFKKSGFRGPLNWYRNMERNWKWACKSLGRKILIPALMVTAEKDFVLVPQMSQHMEDWIPHLKRGHIEDCGHWTQMDKPTEVNQILIKWLDSDARN Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Metabotropic glutamate receptor 2 (mGluR2) | 7E9G | 8.27 | |
Target general information Gen name GRM2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms mGLUR2; Group II metabotropic glutamate receptor; Glutamate receptor mGLU2; GPRC1B Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors, such as adenylate cyclase. Signaling inhibits adenylate cyclase activity. May mediate suppression of neurotransmission or may be involved in synaptogenesis or synaptic stabilization. G-protein coupled receptor for glutamate. Related diseases Oocyte/zygote/embryo maturation arrest 21 (OZEMA21) [MIM:620610]: An autosomal dominant, female infertility disorder characterized by zygote development arrest due to failure of pronuclei fusion. {ECO:0000269|PubMed:33948904, ECO:0000269|PubMed:33953335}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05096 Interacts with Q5T8D3-2; Q9BYF1; Q13520; Q13323; Q8WV48; P57739; O95484; Q7Z7G2; P00387; P27487; P28223-1; Q5SR56; O14880; Q8N4V1; Q58DX5; Q13113; Q9NR31; Q8IWU4; Q9H2H9; P27105; Q8N3G9; Q96Q45-2; Q9NWD8; Q8WUV1; Q9UMX0-2; P0DTC2 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cell projection; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 85146.2 Length 769 Aromaticity 0.11 Instability index 36.84 Isoelectric point 8.48 Charge (pH=7) 9.7 2D Binding mode Binding energy (Kcal/mol) -11.28 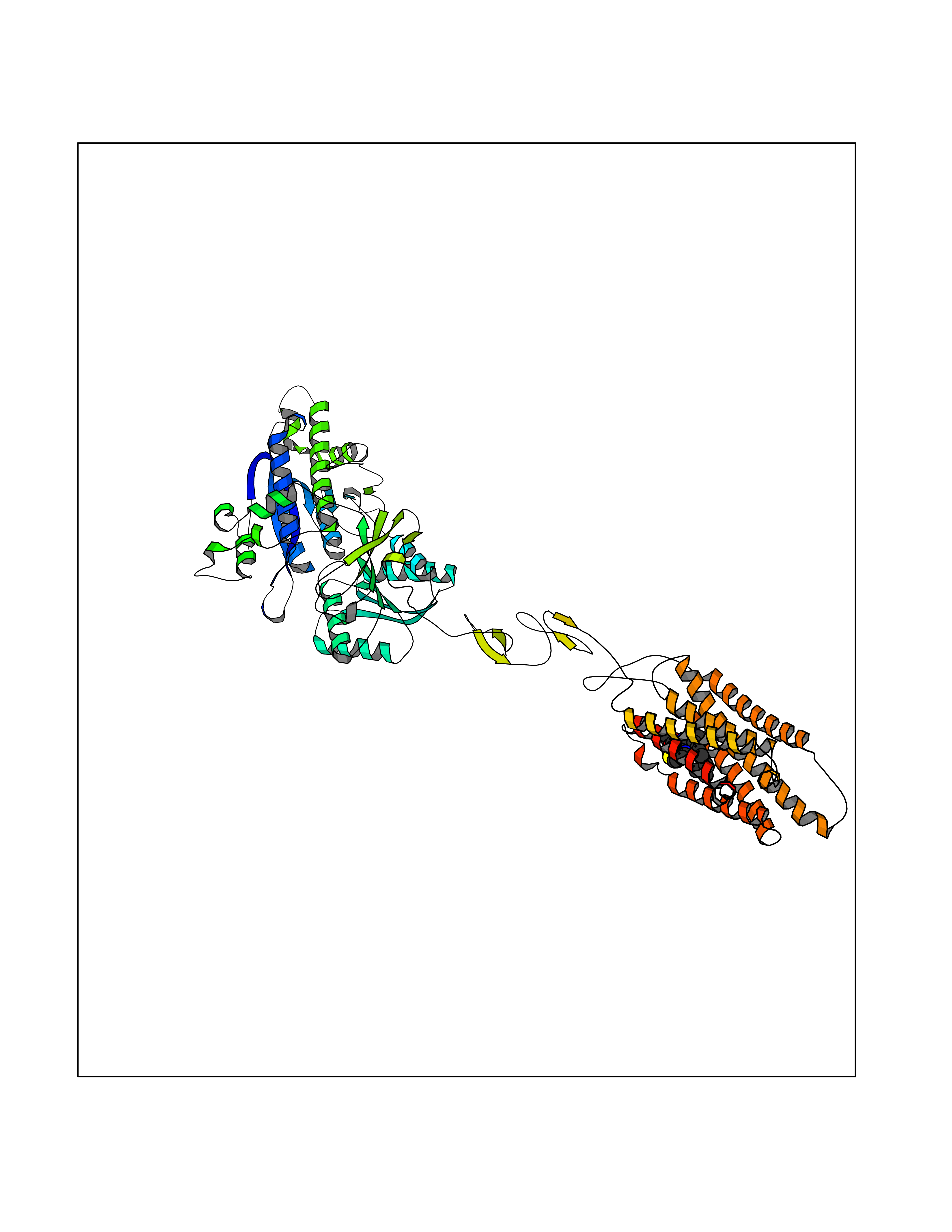 Molscript Map 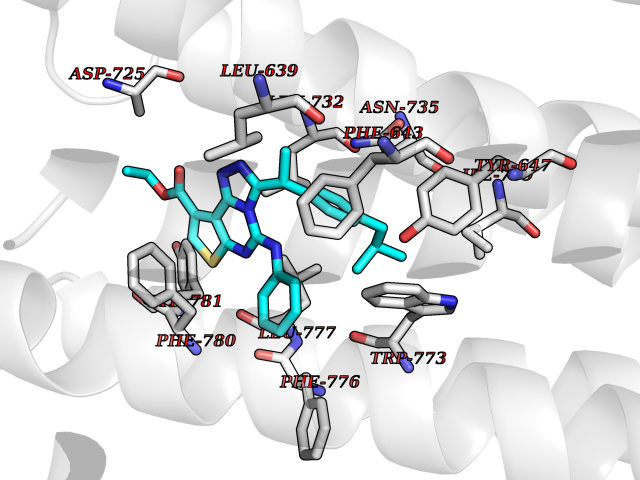 Pymol Map 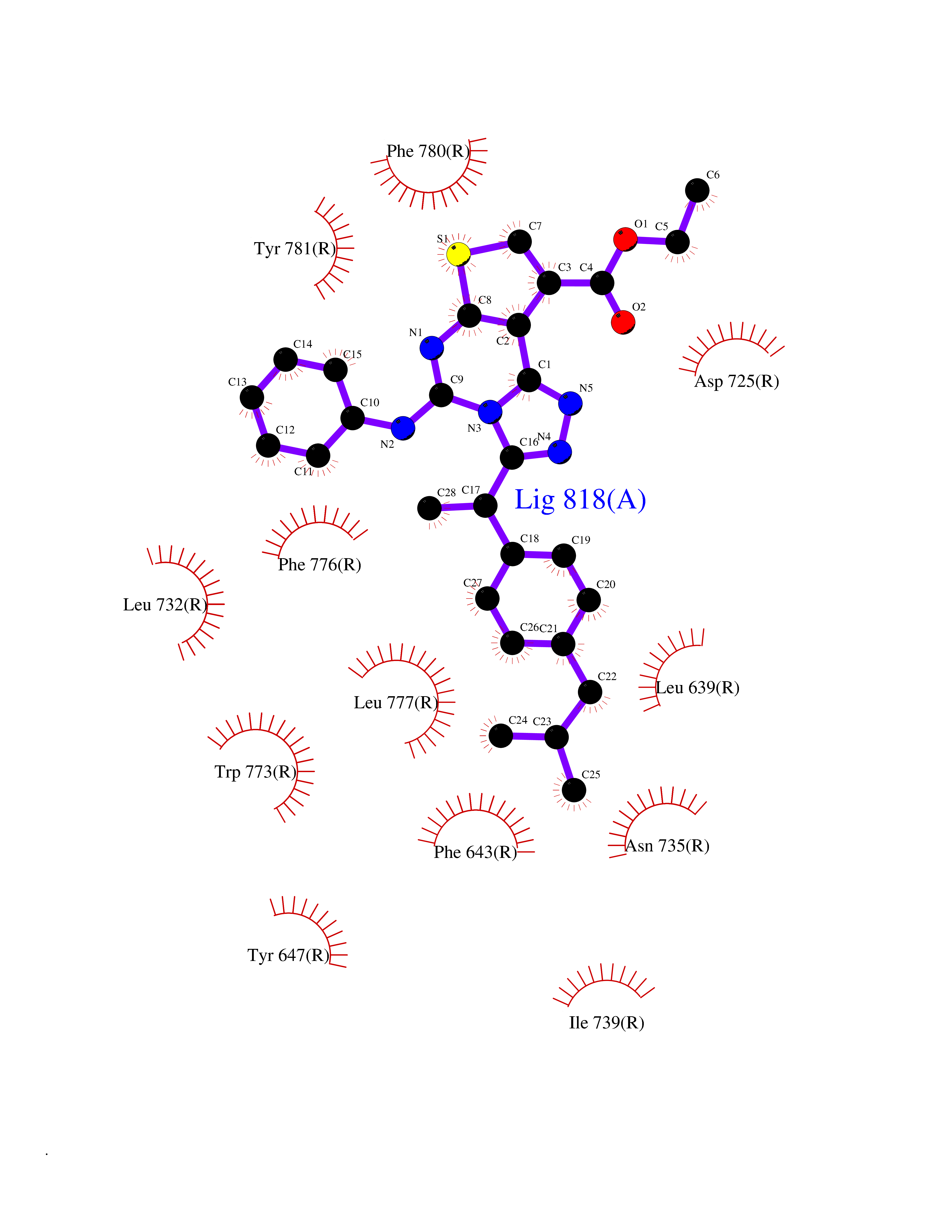 Ligplot Map 3D Binding mode Sequence KKVLTLEGDLVLGGLFPVHQKGGPAEDCGPVNEHRGIQRLEAMLFALDRINRDPHLLPGVRLGAHILDSCSKDTHALEQALDFVRASLITGVIGGSYSDVSIQVANLLRLFQIPQISYASTSAKLSDKSRYDYFARTVPPDFFQAKAMAEILRFFNWTYVSTVASEGDYGETGIEAFELEARARNICVATSEKVGRAMSRAAFEGVVRALLQKPSARVAVLFTRSEDARELLAASQRLNASFTWVASDGWGALESVVAGSEGAAEGAITIELASYPISDFASYFQSLDPWNNSRNPWFREFWEQRFRCSFRQRDCAAHSLRAVPFEQESKIMFVVNAVYAMAHALHNMHRALCPNTTRLCDAMRPVNGRRLYKDFVLNVKFDAPFRPADTHNEVRFDRFGDGIGRYNIFTYLRAGSGRYRYQKVGYWAEGLTLDTSLIPWASPSAGPLPASRCSEPCLQNEVKSVQPGEVCCWLCIPCQPYEYRLDEFTCADCGLGYWPNASLTGCFELPQEYIRWGDAWAVGPVTIACLGALATLFVLGVFVRHNATPVVKAAGRELCYILLGGVFLCYCMTFIFIAKPSTAVCTLRRLGLGTAFSVCYSALLTKTNRIARIFGGAREGAQRPRFISPASQVAICLALISGQLLIVVAWLVVEAPGTGKETAPERREVVTLRCNHRDASMLGSLAYNVLLIALCTLYAFKTRKCPENFNEAKFIGFTMYTTCIIWLAFLPIFYVTSSDYRVQTTTMCVSVSLSGSVVLGCLFAPKLHI Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Receptor-type protein-tyrosine phosphatase zeta (PTPRZ1) | 5H08 | 8.27 | |
Target general information Gen name PTPRZ1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Receptor protein tyrosine phosphatase zeta; R-PTP-zeta; PTPRZ1 Protein family Protein-tyrosine phosphatase family, Receptor class 5 subfamily Biochemical class Phosphoric monoester hydrolase Function Protein tyrosine phosphatase that negatively regulates oligodendrocyte precursor proliferation in the embryonic spinal cord. Required for normal differentiation of the precursor cells into mature, fully myelinating oligodendrocytes. May play a role in protecting oligondendrocytes against apoptosis. May play a role in the establishment of contextual memory, probably via the dephosphorylation of proteins that are part of important signaling cascades. Related diseases Optic atrophy 1 (OPA1) [MIM:165500]: A condition that features progressive visual loss in association with optic atrophy. Atrophy of the optic disk indicates a deficiency in the number of nerve fibers which arise in the retina and converge to form the optic disk, optic nerve, optic chiasm and optic tracts. OPA1 is characterized by an insidious onset of visual impairment in early childhood with moderate to severe loss of visual acuity, temporal optic disk pallor, color vision deficits, and centrocecal scotoma of variable density. {ECO:0000269|PubMed:11017079, ECO:0000269|PubMed:11017080, ECO:0000269|PubMed:11440988, ECO:0000269|PubMed:11440989, ECO:0000269|PubMed:11810270, ECO:0000269|PubMed:12036970, ECO:0000269|PubMed:12566046, ECO:0000269|PubMed:14961560, ECO:0000269|PubMed:15948788, ECO:0000269|PubMed:16513463, ECO:0000269|PubMed:16617242, ECO:0000269|PubMed:18204809, ECO:0000269|PubMed:18360822, ECO:0000269|PubMed:19319978, ECO:0000269|PubMed:19325939, ECO:0000269|PubMed:19969356, ECO:0000269|PubMed:20185555, ECO:0000269|PubMed:22382025, ECO:0000269|PubMed:22857269, ECO:0000269|PubMed:23401657}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Dominant optic atrophy plus syndrome (DOA+) [MIM:125250]: A neurologic disorder characterized most commonly by an insidious onset of visual loss and sensorineural hearing loss in childhood with variable presentation of other clinical manifestations including progressive external ophthalmoplegia, muscle cramps, hyperreflexia, and ataxia. There appears to be a wide range of intermediate phenotypes. {ECO:0000269|PubMed:15531309, ECO:0000269|PubMed:16240368, ECO:0000269|PubMed:18065439, ECO:0000269|PubMed:18158317, ECO:0000269|PubMed:18195150, ECO:0000269|PubMed:20185555, ECO:0000269|PubMed:21112924, ECO:0000269|PubMed:23387428}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Behr syndrome (BEHRS) [MIM:210000]: An autosomal recessive syndrome characterized by optic atrophy beginning in early childhood associated with ataxia, pyramidal signs, spasticity, intellectual disability, and posterior column sensory loss. The ataxia, spasticity, and muscle contractures, mainly of the hip adductors, hamstrings, and soleus, are progressive and become more prominent in the second decade. {ECO:0000269|PubMed:21636302, ECO:0000269|PubMed:25012220, ECO:0000269|PubMed:25146916}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mitochondrial DNA depletion syndrome 14, cardioencephalomyopathic type (MTDPS14) [MIM:616896]: An autosomal recessive mitochondrial disorder characterized by lethal infantile encephalopathy, hypertrophic cardiomyopathy and optic atrophy. Skeletal muscle biopsies show significant mtDNA depletion and abnormal mitochondria. {ECO:0000269|PubMed:26561570}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9UM73; Q12860 EC number EC 3.1.3.48 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Hydrolase; Membrane; Phosphoprotein; Protein phosphatase; Proteoglycan; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 32208.1 Length 282 Aromaticity 0.11 Instability index 38.98 Isoelectric point 7.35 Charge (pH=7) 0.89 2D Binding mode Binding energy (Kcal/mol) -11.28 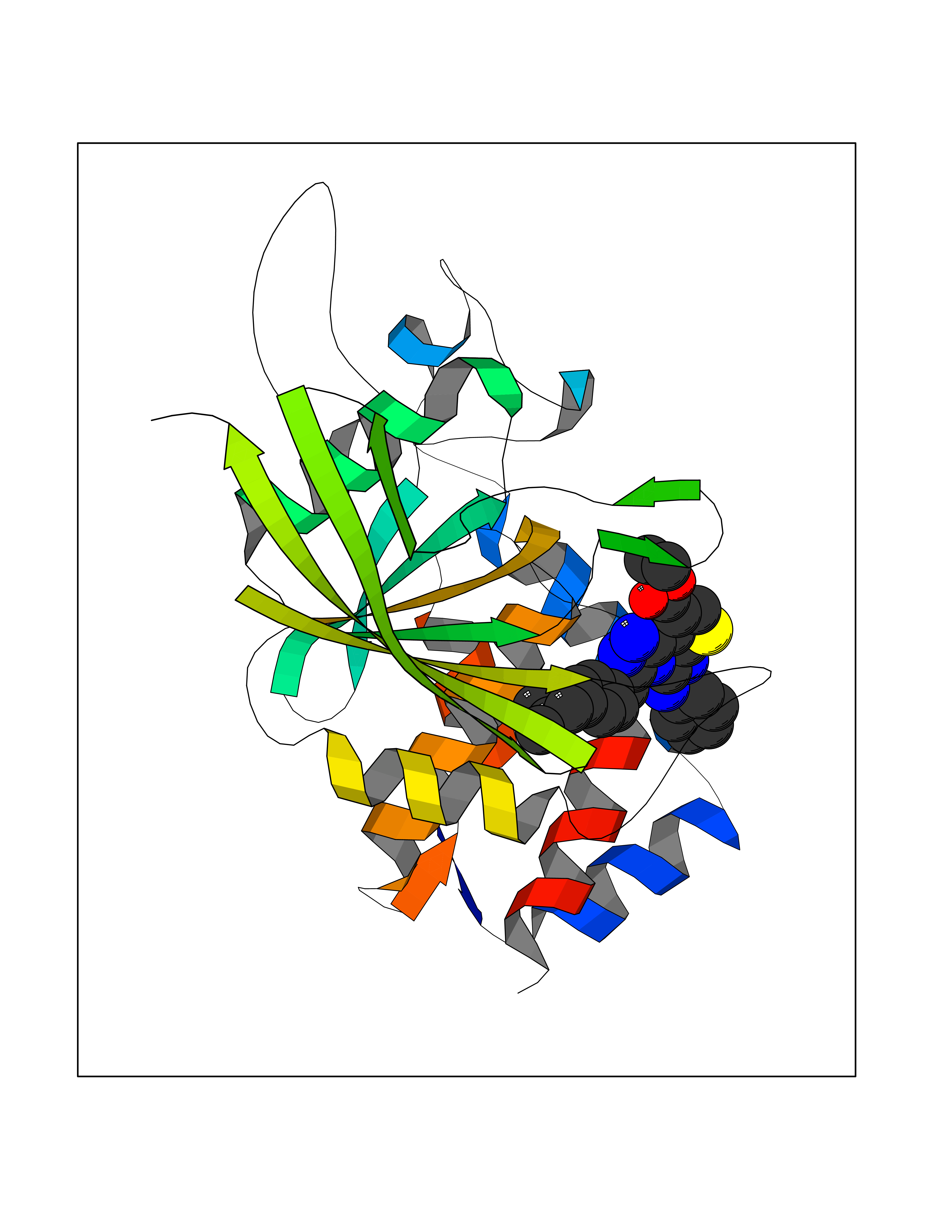 Molscript Map 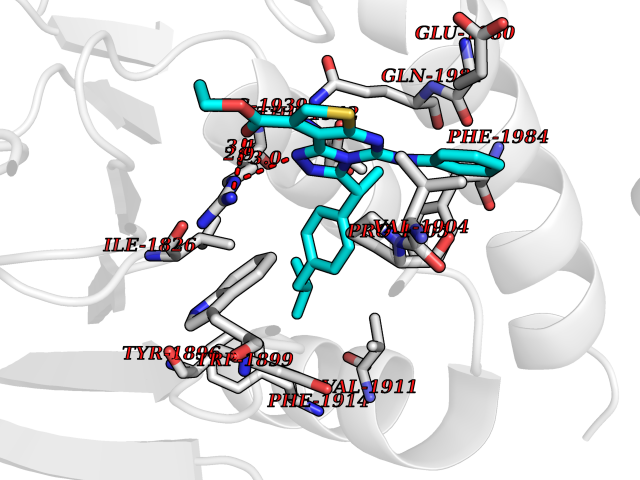 Pymol Map 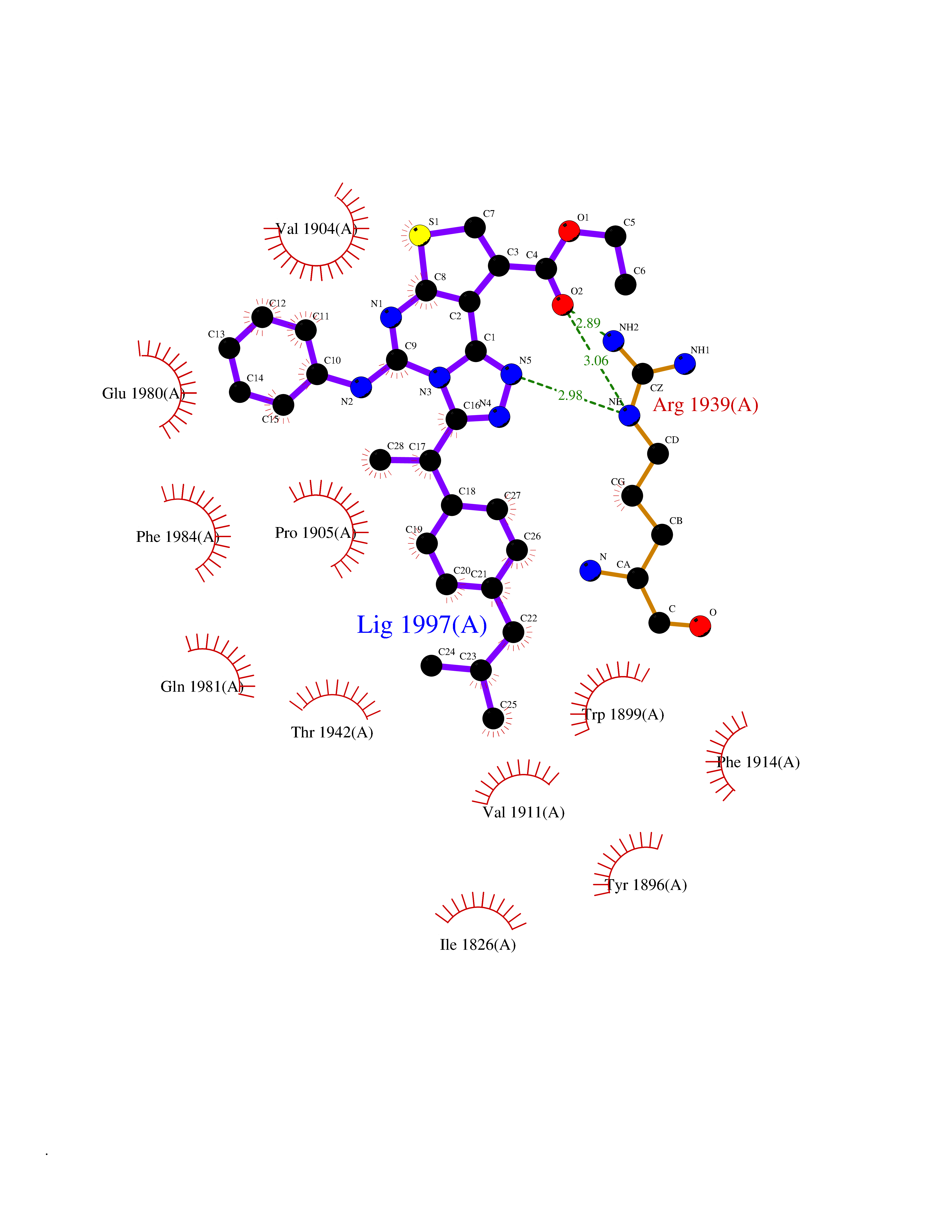 Ligplot Map 3D Binding mode Sequence GPAIPIKHFPKHVADLHASSGFTEEFEEVQSCTVDLGITADSSNHPDNKHKNRYINIVAYDHSRVKLAQLAEKDGKLTDYINANYVDGYNRPKAYIAAQGPLKSTAEDFWRMIWEHNVEVIVMITNLVEKGRRKCDQYWPADGSEEYGNFLVTQKSVQVLAYYTVRNFTLRNTKIRVVTQYHYTQWPDMGVPEYSLPVLTFVRKAAYAKRHAVGPVVVHCSAGVGRTGTYIVLDSMLQQIQHEGTVNIFGFLKHIRSQRNYLVQTEEQYVFIHDTLVEAILS Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Monoacylglycerol lipase ABHD6 (ABHD6) | 7OTS | 8.26 | |
Target general information Gen name ABHD6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Abhydrolase domain-containing protein 6; 2-arachidonoylglycerol hydrolase Protein family AB hydrolase superfamily Biochemical class Carboxylic ester hydrolase Function Lipase that preferentially hydrolysis medium-chain saturated monoacylglycerols including 2-arachidonoylglycerol. Through 2-arachidonoylglycerol degradation may regulate endocannabinoid signaling pathways. Also has a lysophosphatidyl lipase activity with a preference for lysophosphatidylglycerol among other lysophospholipids. Also able to degrade bis(monoacylglycero)phosphate (BMP) and constitutes the major enzyme for BMP catabolism. BMP, also known as lysobisphosphatidic acid, is enriched in late endosomes and lysosomes and plays a key role in the formation of intraluminal vesicles and in lipid sorting. Related diseases Blistering, acantholytic, of oral and laryngeal mucosa (ABOLM) [MIM:619226]: An autosomal recessive disorder characterized by recurrent, suprabasal acantholytic blisters in the oral and laryngeal mucosa. Skin, conjunctival and genital mucosa, nail folds, and nails are unaffected. Normal structure is observed in the scalp epidermis and hair follicle. {ECO:0000269|PubMed:30528827}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9Y3D6; Q5QGT7; Q969E2; Q9NYZ1 EC number EC 3.1.1.23 Uniprot keywords 3D-structure; Endosome; Hydrolase; Lipid metabolism; Lysosome; Membrane; Mitochondrion; Proteomics identification; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 33054 Length 292 Aromaticity 0.08 Instability index 33.63 Isoelectric point 8.68 Charge (pH=7) 5.47 2D Binding mode Binding energy (Kcal/mol) -11.26  Molscript Map 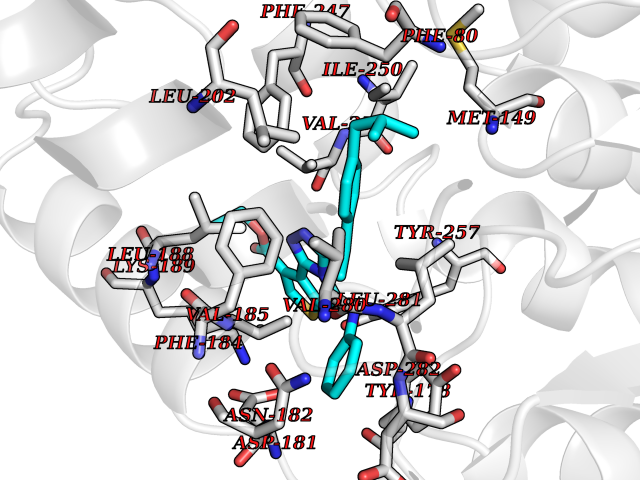 Pymol Map 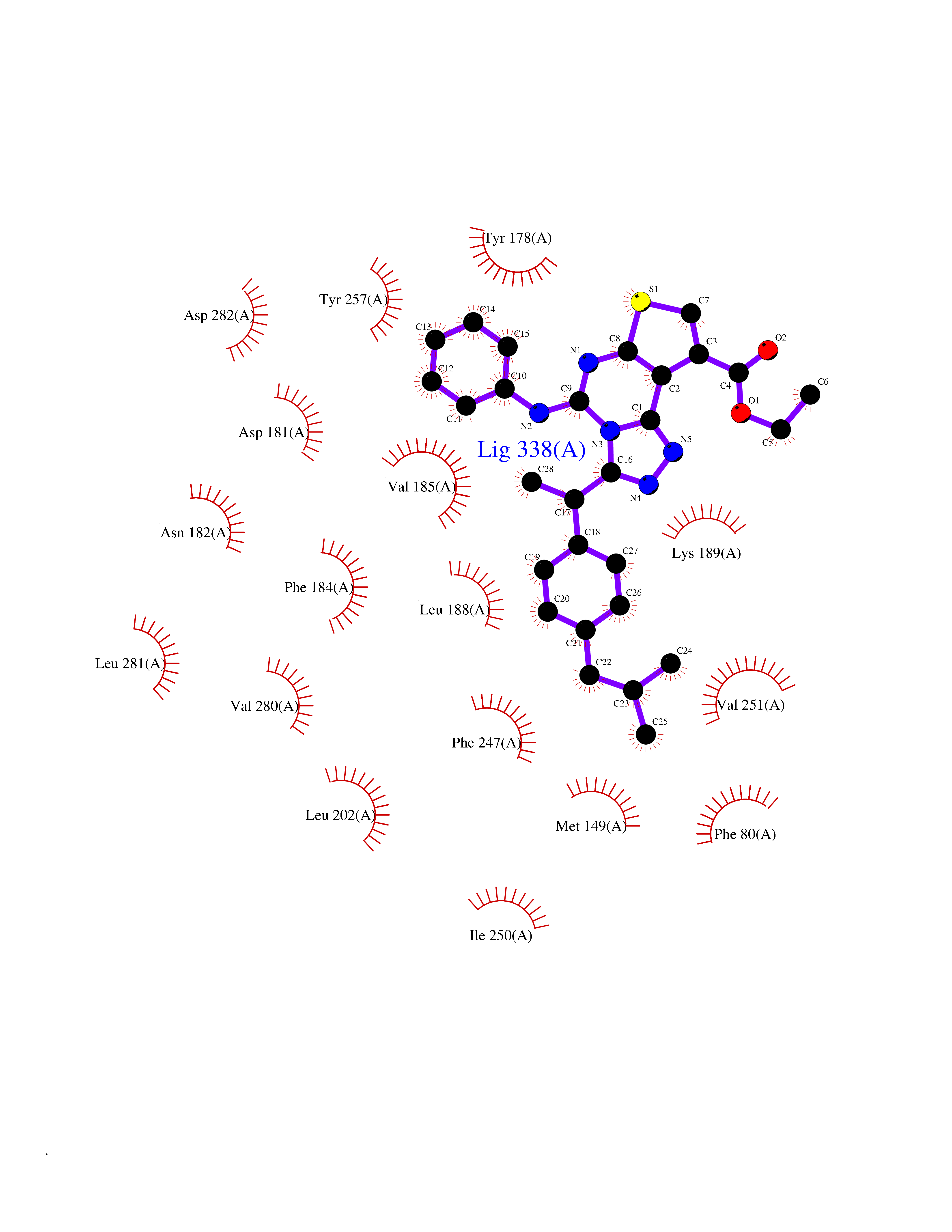 Ligplot Map 3D Binding mode Sequence TLGMQVRYVHHEDYQFCYSFRGRPGHKPSILMLHGFSAHKDMWLSVVKFLPKNLHLVCVDMPGHEGTTRSSLDDLSIDGQVKRIHQFVECLKLNKKPFHLVGTAMGGQVAGVYAAYYPSDVSSLCLVCPAGLQYSTDNQFVQRLKELQGSAAVEKIPLIPSTPEEMSEMLQLCSYVRFKVPQQILQGLVDVRIPHNNFYRKLFLEIVSEKSRYSLHQNMDKIKVPTQIIWGKQDQVLDVSGADMLAKSIANCQVELLENCGHSVVMERPRKTAKLIIDFLASVHNTDNNKKL Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | "15-cis-phytoene desaturase, chloroplastic/chromoplastic (EC 1.3.5.5) (Phytoene dehydrogenase) (Phytoene desaturase)" | 5MOG | 8.25 | |
Target general information Gen name PDS1 Organism Oryza sativa subsp. indica (Rice) Uniprot ID TTD ID NA Synonyms PDS;OsI_010044 Protein family Carotenoid/retinoid oxidoreductase family Biochemical class NA Function Converts phytoene into zeta-carotene via the intermediary of phytofluene by the symmetrical introduction of two double bonds at the C-11 and C-11' positions of phytoene with a concomitant isomerization of two neighboring double bonds at the C9 and C9' positions from trans to cis. Active with decylplastoquinone (DPQ) as substrate (PubMed:26147209, PubMed:29176862). Also active with other benzoquinones, which are strongly preferred over naphthoquinones as substrates (PubMed:26147209). {ECO:0000269|PubMed:26147209, ECO:0000269|PubMed:29176862}." Related diseases NA Drugs (DrugBank ID) NA Interacts with NA EC number 1.3.5.5 Uniprot keywords 3D-structure; Carotenoid biosynthesis; Chloroplast; Chromoplast; Direct protein sequencing; FAD; Flavoprotein; Membrane; Oxidoreductase; Plastid; Reference proteome; Transit peptide Protein physicochemical properties Chain ID E Molecular weight (Da) 52485.1 Length 466 Aromaticity 0.1 Instability index 45.53 Isoelectric point 5.93 Charge (pH=7) -5.81 2D Binding mode Binding energy (Kcal/mol) -11.26  Molscript Map 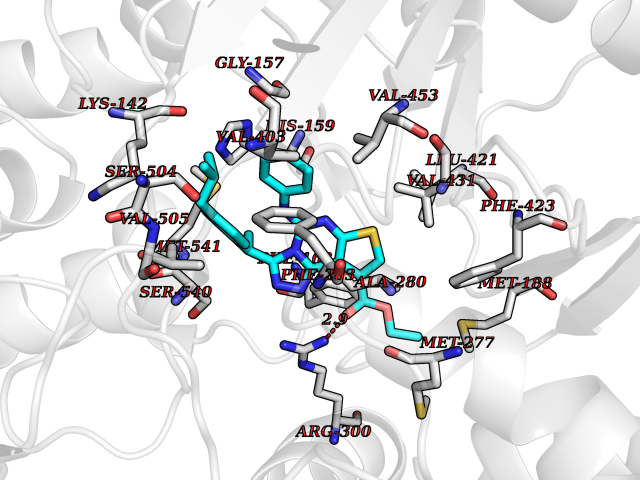 Pymol Map 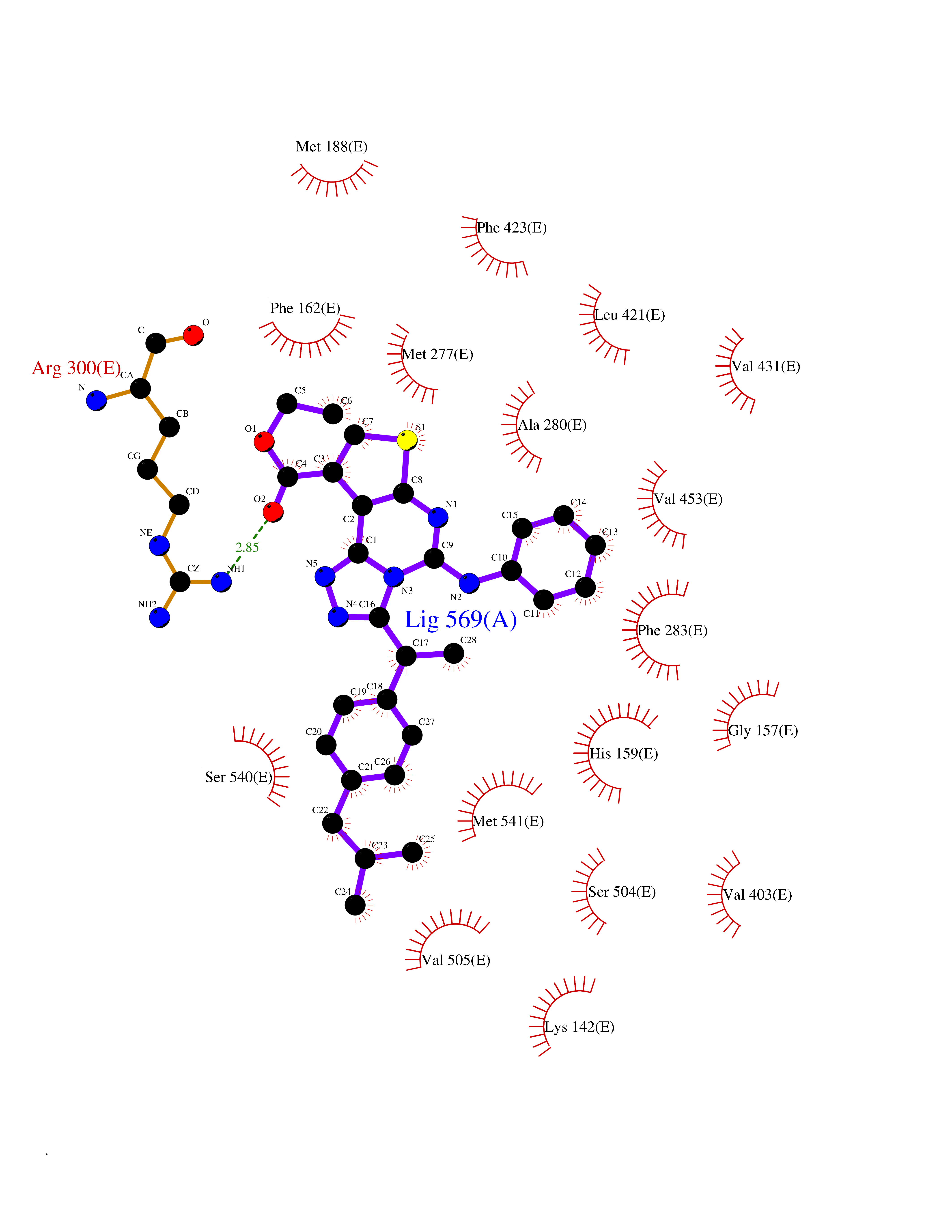 Ligplot Map 3D Binding mode Sequence TKPLQVVIAGAGLAGLSTAKYLADAGHKPILLEARDVLGGKIAAWKDEDGDWYETGLHIFFGAYPNIQNLFGELGINDRLQWKEHSMIFAMPNKPGEFSRFDFPETLPAPLNGIWAILRNNEMLTWPEKVKFALGLLPAMVGGQAYVEAQDGFTVSEWMKKQGVPDRVNDEVFIAMSKALNFINPDELSMQCILIALNRFLQEKHGSKMAFLDGNPPERLCMPIVDHVRSLGGEVRLNSRIQKIELNPDGTVKHFALTDGTQITGDAYVFATPVDILKLLVPQEWKEISYFKKLEKLVGVPVINVHIWFDRKLKNTYDHLLFSRSSLLSVYADMSVTCKEYYDPNRSMLELVFAPAEEWVGRSDTEIIEATMQELAKLFPDEIAADQSKAKILKYHVVKTPRSVYKTIPDCEPCRPLQRSPIEGFYLAGDYTKQKYLASMEGAVLSGKLCAQSVVEDYKMLSRRSL Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Pregnane X receptor (NR1I2) | 1NRL | 8.25 | |
Target general information Gen name NR1I2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Steroid and xenobiotic receptor; SXR; PXR; Orphan nuclear receptor PXR; Orphan nuclear receptor PAR1; Nuclear receptor subfamily 1 group I member 2 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Transcription factor that activates the transcription of multiple genes involved in the metabolism and secretion of potentially harmful xenobiotics, drugs and endogenous compounds. Activated by the antibiotic rifampicin and various plant metabolites, such as hyperforin, guggulipid, colupulone, and isoflavones. Response to specific ligands is species-specific. Activated by naturally occurring steroids, such as pregnenolone and progesterone. Binds to a response element in the promoters of the CYP3A4 and ABCB1/MDR1 genes. Nuclear receptor that binds and is activated by variety of endogenous and xenobiotic compounds. Related diseases Atrial fibrillation, familial, 14 (ATFB14) [MIM:615378]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:19808477}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Genetic variations in SCN2B may be involved in Brugada syndrome (PubMed:23559163). This tachyarrhythmia is characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:23559163}. Drugs (DrugBank ID) DB04468; DB14001; DB01393; DB00564; DB06777; DB01068; DB00257; DB00531; DB14002; DB01234; DB14649; DB00255; DB01248; DB05928; DB01127; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00977; DB00754; DB01039; DB13873; DB00499; DB01645; DB07931; DB01892; DB01181; DB01026; DB00431; DB00532; DB00849; DB01110; DB00834; DB11605; DB01115; DB00239; DB01229; DB00312; DB04930; DB01174; DB04824; DB00252; DB12582; DB01708; DB02789; DB11087; DB04216; DB02709; DB01045; DB01220; DB08864; DB00503; DB00421; DB04466; DB01138; DB00675; DB07080; DB08604; DB13179; DB00163; DB00682 Interacts with P08238; Q15788 EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 31908.8 Length 275 Aromaticity 0.11 Instability index 46.37 Isoelectric point 6.78 Charge (pH=7) -0.54 2D Binding mode Binding energy (Kcal/mol) -11.25  Molscript Map 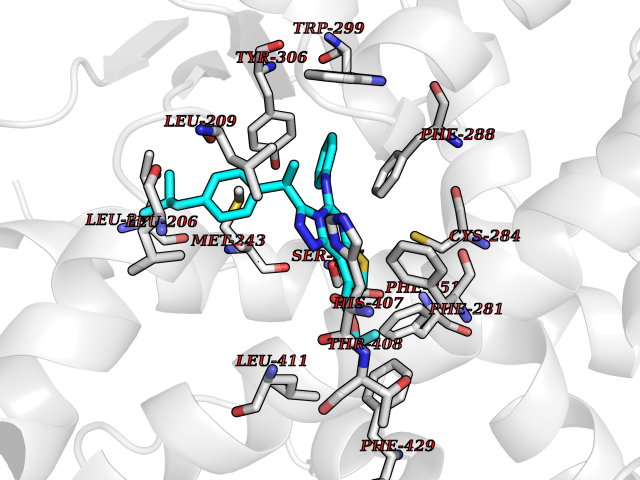 Pymol Map 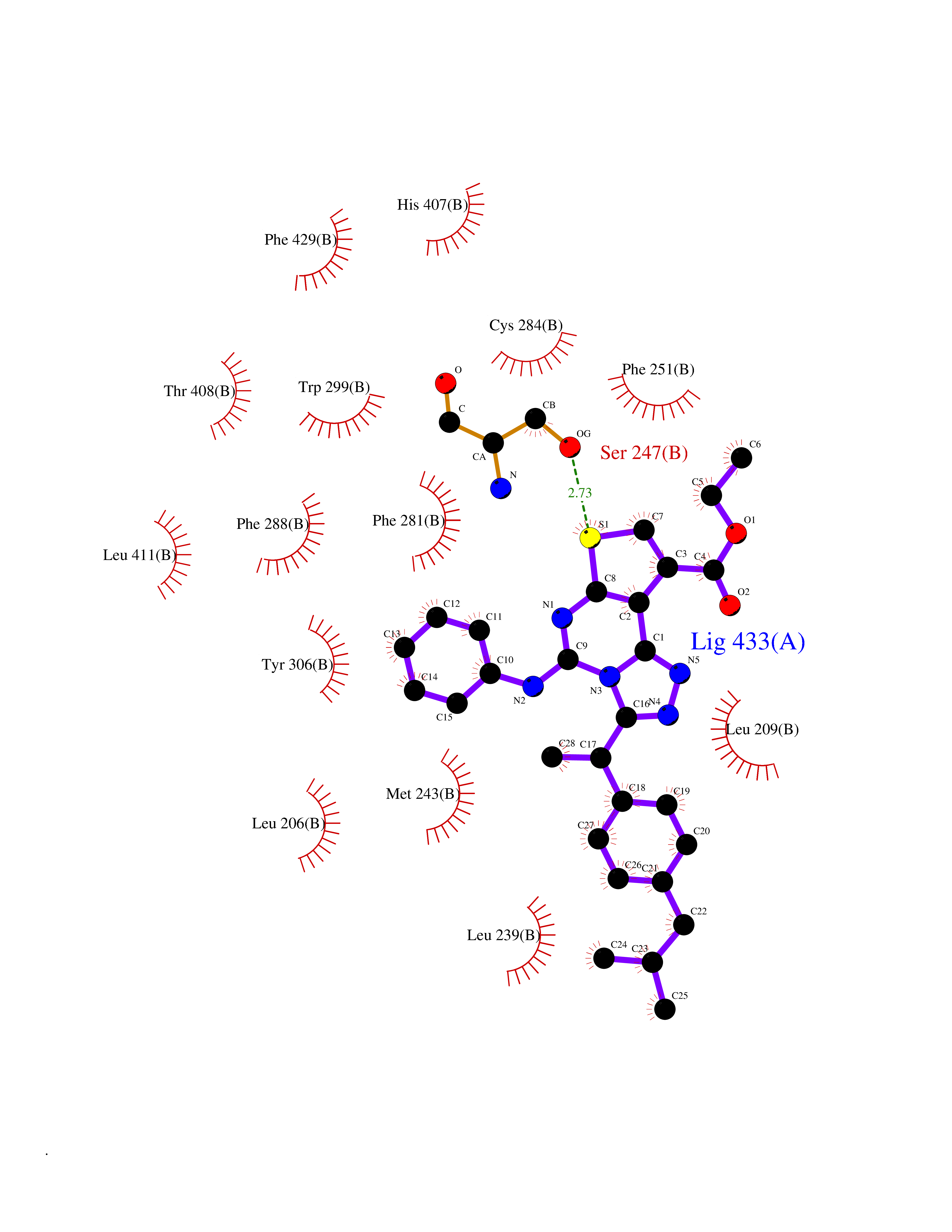 Ligplot Map 3D Binding mode Sequence GLTEEQRMMIRELMDAQMKTFDTTFSHFKNFRLPGVREEAAKWSQVRKDLCSLKVSLQLRGEDGSVWNYKPPADSGGKEIFSLLPHMADMSTYMFKGIISFAKVISYFRDLPIEDQISLLKGAAFELCQLRFNTVFNAETGTWECGRLSYCLEDTAGGFQQLLLEPMLKFHYMLKKLQLHEEEYVLMQAISLFSPDRPGVLQHRVVDQLQEQFAITLKSYIECNRPQPAHRFLFLKIMAMLTELRSINAQHTQRLLRIQDIHPFATPLMQELFGI Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Vitamin K-dependent protein C (PROC) | 1LQV | 8.24 | |
Target general information Gen name PROC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin K-dependent protein C light chain; Vitamin K-dependent protein C heavy chain; PROC; Blood coagulation factor XIV; Autoprothrombin IIA; Anticoagulant protein C; Activation peptide Protein family Peptidase S1 family Biochemical class Peptidase Function Protein C is avitamin K-dependent serine protease that regulates blood coagulation by inactivating factors Va and VIIIa in the presence of calcium ions and phospholipids. Related diseases Thrombophilia due to protein C deficiency, autosomal dominant (THPH3) [MIM:176860]: A hemostatic disorder characterized by impaired regulation of blood coagulation and a tendency to recurrent venous thrombosis. Individuals with decreased amounts of protein C are classically referred to as having type I protein C deficiency and those with normal amounts of a functionally defective protein as having type II deficiency. {ECO:0000269|PubMed:1301959, ECO:0000269|PubMed:1347706, ECO:0000269|PubMed:1511989, ECO:0000269|PubMed:1868249, ECO:0000269|PubMed:2437584, ECO:0000269|PubMed:25618265, ECO:0000269|PubMed:25748729, ECO:0000269|PubMed:2602169, ECO:0000269|PubMed:7792728, ECO:0000269|PubMed:7865674, ECO:0000269|PubMed:8292730, ECO:0000269|PubMed:8398832, ECO:0000269|PubMed:8499568, ECO:0000269|PubMed:8560401, ECO:0000269|PubMed:8829639, ECO:0000269|PubMed:9798967}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thrombophilia due to protein C deficiency, autosomal recessive (THPH4) [MIM:612304]: A hemostatic disorder characterized by impaired regulation of blood coagulation and a tendency to recurrent venous thrombosis. It results in a thrombotic condition that can manifest as a severe neonatal disorder or as a milder disorder with late-onset thrombophilia. The severe form leads to neonatal death through massive neonatal venous thrombosis. Often associated with ecchymotic skin lesions which can turn necrotic called purpura fulminans, this disorder is very rare. {ECO:0000269|PubMed:1511988, ECO:0000269|PubMed:1593215, ECO:0000269|PubMed:1611081, ECO:0000269|PubMed:25618265, ECO:0000269|PubMed:7841323, ECO:0000269|PubMed:7841324, ECO:0000269|PubMed:7878626}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB13192; DB00025; DB09131; DB09332; DB13998; DB00170; DB13999; DB13149; DB00464; DB14738 Interacts with A8MQ03; P51511 EC number EC 3.4.21.69 Uniprot keywords 3D-structure; Alternative splicing; Blood coagulation; Calcium; Cleavage on pair of basic residues; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Endoplasmic reticulum; Gamma-carboxyglutamic acid; Glycoprotein; Golgi apparatus; Hemostasis; Hydrolase; Hydroxylation; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Thrombophilia; Zymogen Protein physicochemical properties Chain ID C,D Molecular weight (Da) 45326 Length 411 Aromaticity 0.12 Instability index 40.68 Isoelectric point 7.07 Charge (pH=7) 0.29 2D Binding mode Binding energy (Kcal/mol) -11.24  Molscript Map 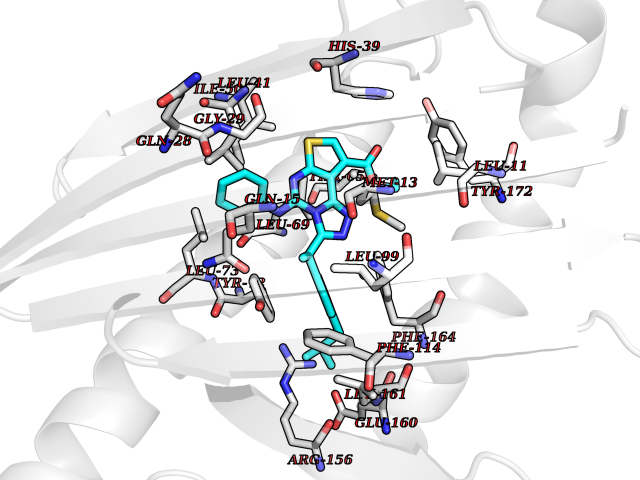 Pymol Map 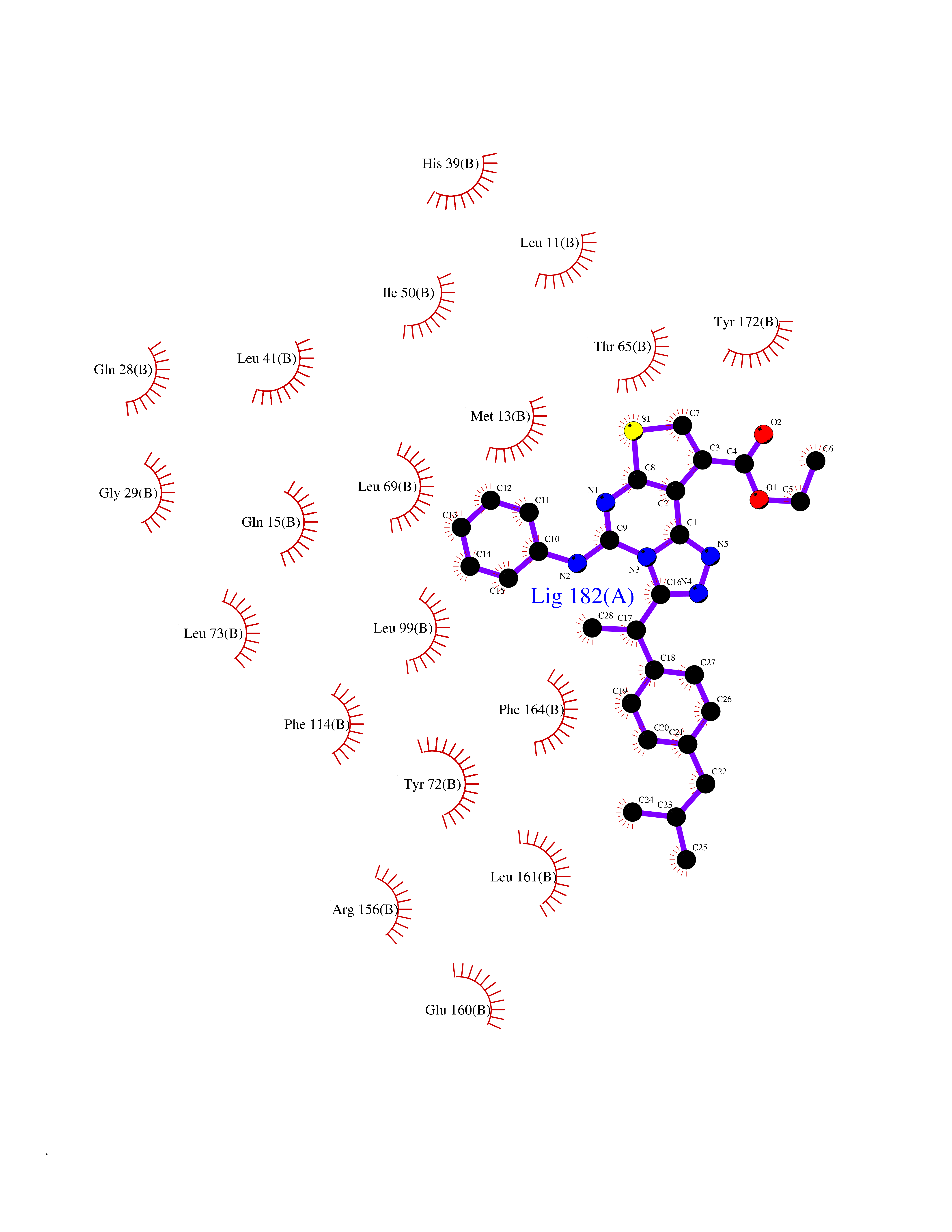 Ligplot Map 3D Binding mode Sequence GLQRLHMLQISYFRDPYHVWYQGNASLGGHLTHVLEGPDTNTTIIQLQPLQEPESWARTQSGLQSYLLQFHGLVRLVHQERTLAFPLTIRCFLGCELPPEGSRAHVFFEVAVNGSSFVSFRPERALWQADTQVTSGVVTFTLQQLNAYNRTRYELREFLEDTCVQYVQKHISANSFLXXLRHSSLXRXCIXXICDFXXAKXIFQNANSFLXXLRHSSLXRXCIXXICDFXXAKXIFQNLQRLHMLQISYFRDPYHVWYQGNASLGGHLTHVLEGPDTNTTIIQLQPLQEPESWARTQSGLQSYLLQFHGLVRLVHQERTLAFPLTIRCFLGCELPPEGSRAHVFFEVAVNGSSFVSFRPERALWQADTQVTSGVVTFTLQQLNAYNRTRYELREFLEDTCVQYVQKHISAE Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | Apoptotic protease-activating factor 1 | 1Z6T | 8.24 | |
Target general information Gen name APAF1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms KIAA0413 Protein family NA Biochemical class Apoptosis Function ADP binding.ATP binding.Cysteine-type endopeptidase activator activity involved in apoptotic process.Heat shock protein binding.Identical protein binding.Nucleotide binding. Related diseases Pilarowski-Bjornsson syndrome (PILBOS) [MIM:617682]: An autosomal dominant disorder characterized by developmental delay, speech apraxia, intellectual disability, autism, and facial dysmorphic features. Some patients may have seizures. {ECO:0000269|PubMed:28866611}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00171 Interacts with P55211; P99999; P62136; P62258 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Calcium; Cytoplasm; Direct protein sequencing; Nucleotide-binding; Proteomics identification; Reference proteome; Repeat; WD repeat Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 55520.6 Length 482 Aromaticity 0.09 Instability index 51.43 Isoelectric point 6.61 Charge (pH=7) -1.99 2D Binding mode Binding energy (Kcal/mol) -11.24 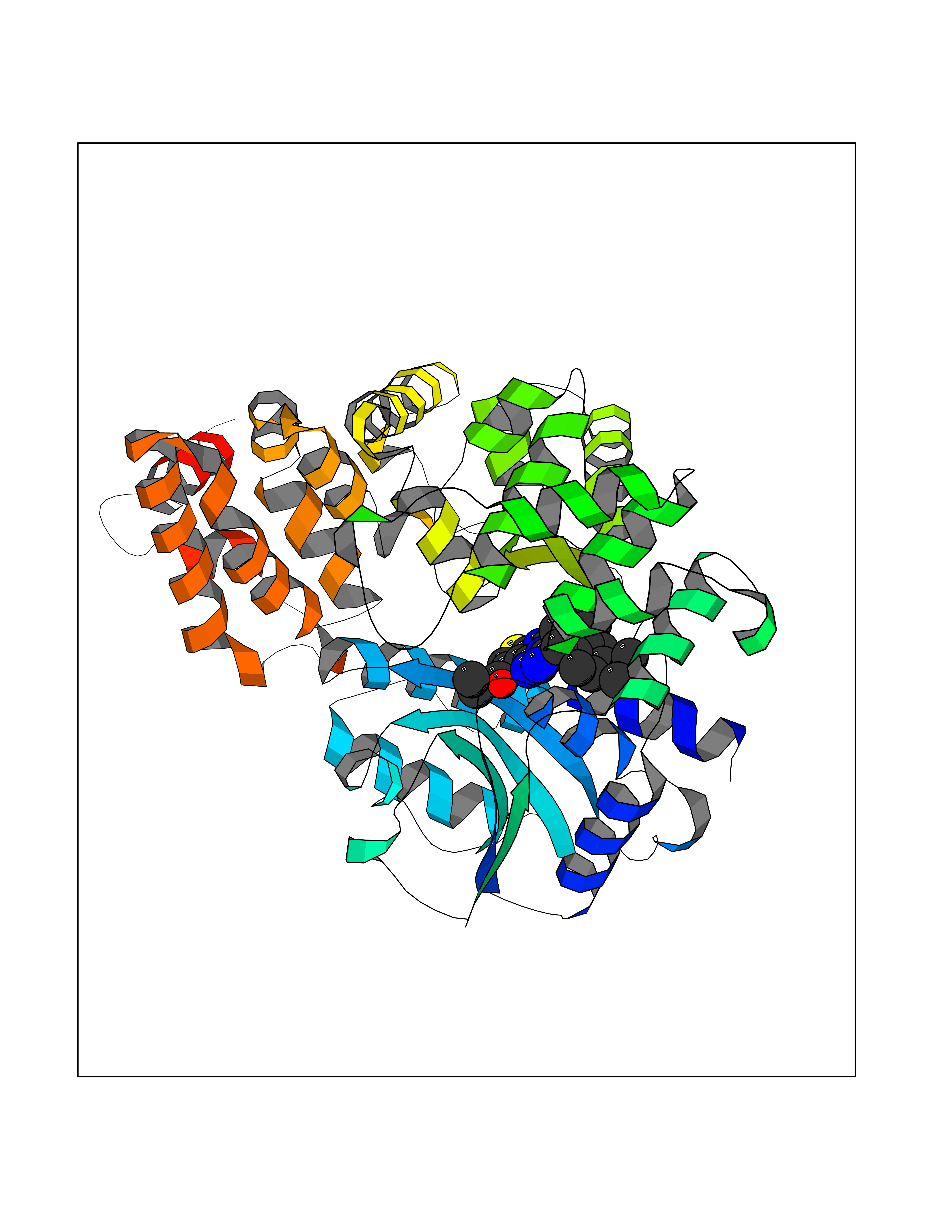 Molscript Map 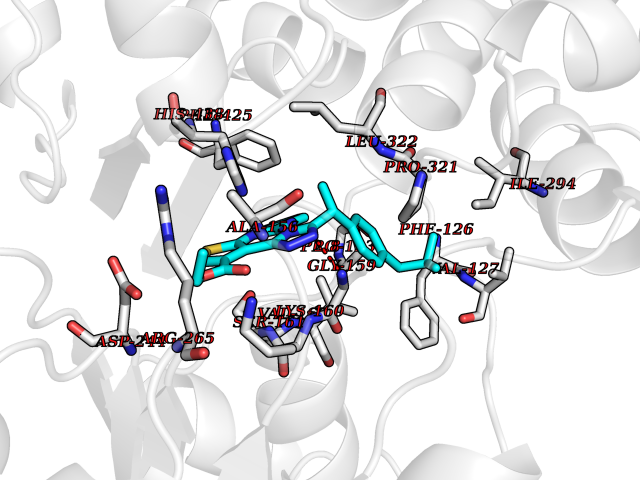 Pymol Map 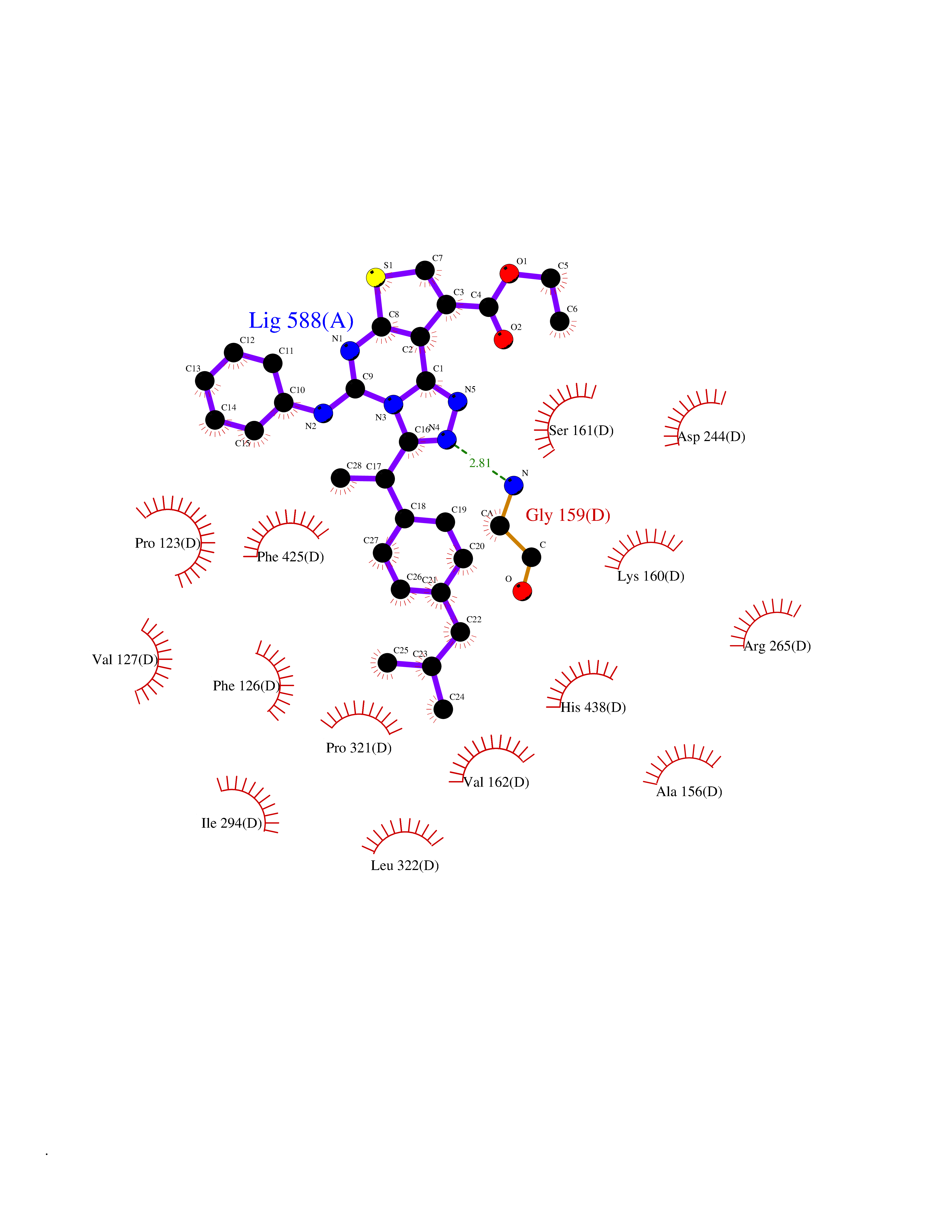 Ligplot Map 3D Binding mode Sequence GITSYVRTVLCEGGVPQRPVVFVTRKKLVNAIQQKLSKLKGEPGWVTIHGMAGCGKSVLAAEAVRDHSLLEGCFPGGVHWVSVGKQDKSGLLMKLQNLCTRLDQDESFSQRLPLNIEEAKDRLRILMLRKHPRSLLILDDVWDSWVLKAFDSQCQILLTTRDKSVTDSVMGPKYVVPVESSLGKEKGLEILSLFVNMKKADLPEQAHSIIKECKGSPLVVSLIGALLRDFPNRWEYYLKQLQNKQFKRIRKSSSYDYEALDEAMSISVEMLREDIKDYYTDLSILQKDVKVPTKVLCILWDMETEEVEDILQEFVNKSLLFCDRNGKSFRYYLHDLQVDFLTEKNCSQLQDLHKKIITQFQRYHQPHTLSPDQEDCMYWYNFLAYHMASAKMHKELCALMFSLDWIKAKTELVGPAHLIHEFVEYRHILDEKDCAVSENFQEFLSLNGHLLGRQPFPNIVQLGLCEPETSEVYQQAKLQAKQ Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | 3-oxoacyl-[acyl-carrier-protein] synthase 2 | 3I8P | 8.24 | |
Target general information Gen name fabF Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1081;b1095;fabJ Protein family Thiolase-like superfamily, Beta-ketoacyl-ACP synthases family Biochemical class Transferase Function 3-oxoacyl-[acyl-carrier-protein] synthase activity.Beta-ketoacyl-acyl-carrier-protein synthase II activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08366; DB01034; DB03017; DB08407 Interacts with P0A6Y8 EC number 2.3.1.179 Uniprot keywords 3D-structure; Acyltransferase; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 42794.9 Length 411 Aromaticity 0.06 Instability index 31.41 Isoelectric point 5.72 Charge (pH=7) -7.34 2D Binding mode Binding energy (Kcal/mol) -11.25 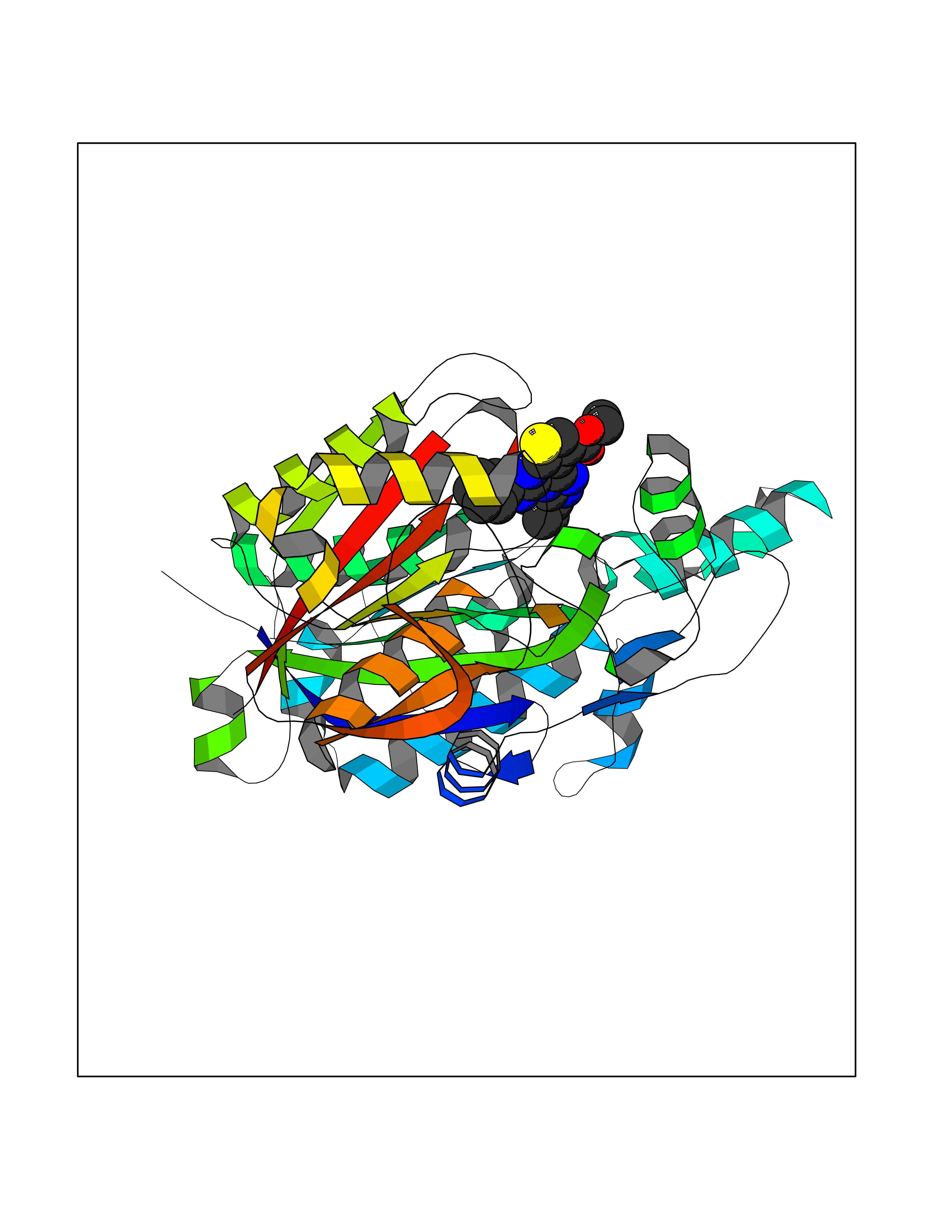 Molscript Map 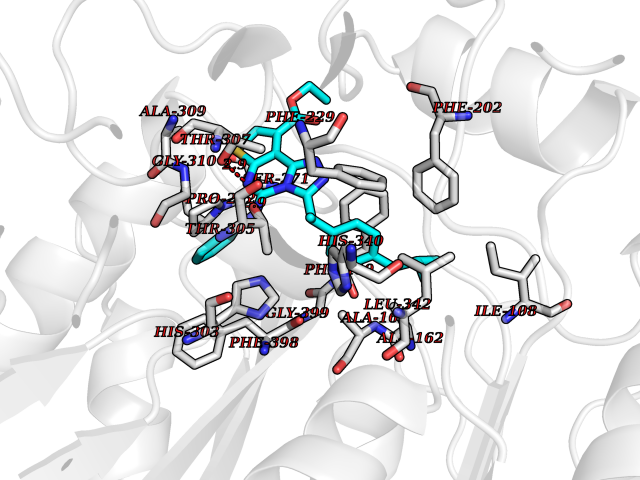 Pymol Map 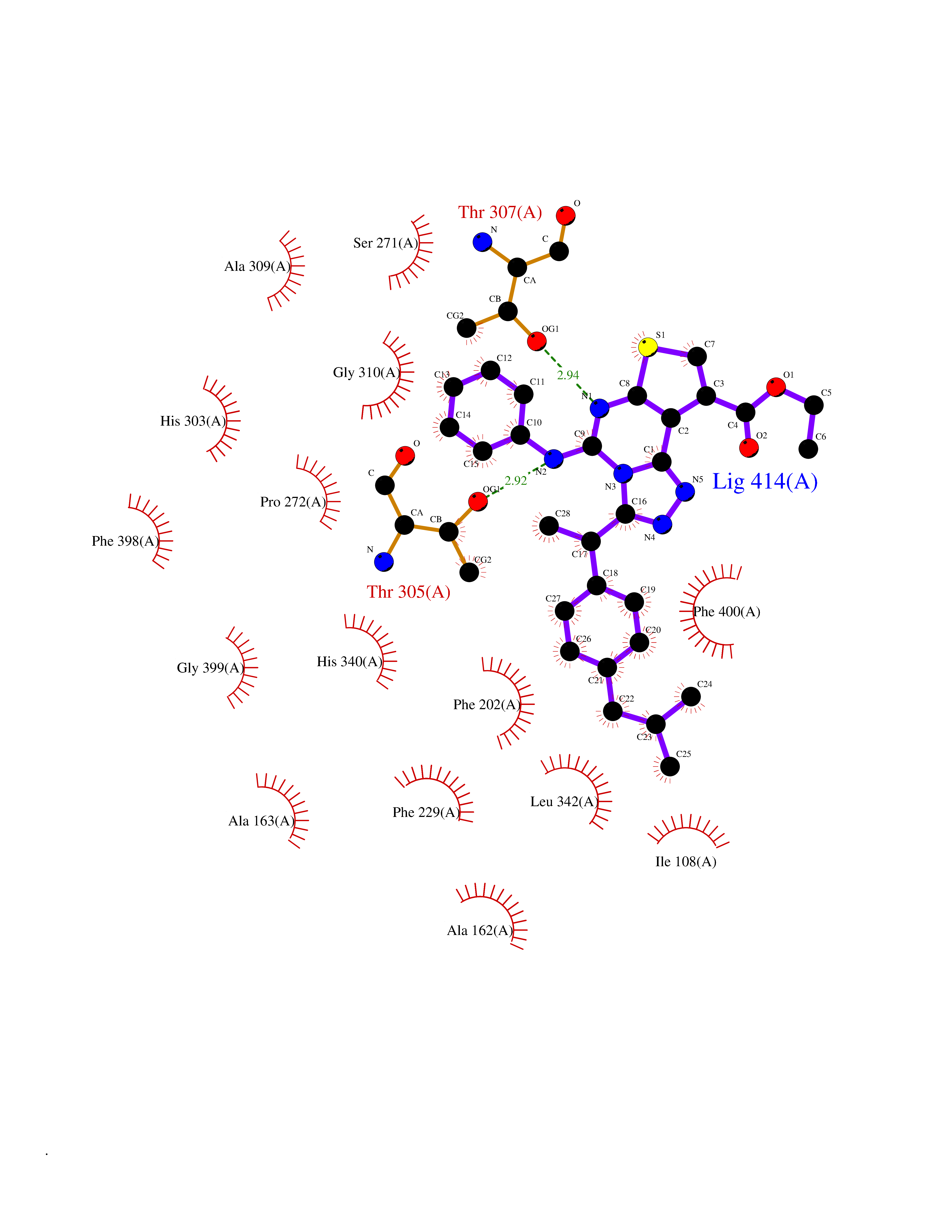 Ligplot Map 3D Binding mode Sequence KRRVVVTGLGMLSPVGNTVESTWKALLAGQSGISLIDHFDTSAYATKFAGLVKDFNCEDIISRKEQRKMDAFIQYGIVAGVQAMQDSGLEITEENATRIGAAIGSGIGGLGLIEENHTSLMNGGPRKISPFFVPSTIVNMVAGHLTIMYGLRGPSISIATAATSGVHNIGHAARIIAYGDADVMVAGGAEKASTPLGVGGFGAARALSTRNDNPQAASRPWDKERDGFVLGDGAGMLVLEEYEHAKKRGAKIYAELVGFGMSSDAYHMTSPPENGAGAALAMANALRDAGIEASQIGYVNAHGTSTPAGDKAEAQAVKTIFGEAASRVLVSSTKSMTGHLLGAAGAVESIYSILALRDQAVPPTINLDNPDEGCDLDFVPHEARQVSGMEYTLCNSFGFGGTNGSLIFKKI Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Cyclopropane mycolic acid synthase MmaA2 | 1TPY | 8.24 | |
Target general information Gen name mmaA2 Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID NA Synonyms Rv0644c;mma2 Protein family CFA/CMAS family Biochemical class Transferase Function Cyclopropane-fatty-acyl-phospholipid synthase activity.Methyltransferase activity. Related diseases Oocyte/zygote/embryo maturation arrest 16 (OZEMA16) [MIM:617234]: A rare cause of female primary infertility. In affected women, ovulation and fertilization proceed normally but embryos are arrested at early stages of development. Inheritance is autosomal recessive. {ECO:0000269|PubMed:27545678}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01718; DB01752 Interacts with NA EC number 2.1.1.79 Uniprot keywords 3D-structure; Acetylation; Lipid biosynthesis; Lipid metabolism; Methyltransferase; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 32493.6 Length 285 Aromaticity 0.1 Instability index 43.61 Isoelectric point 5.53 Charge (pH=7) -10.17 2D Binding mode Binding energy (Kcal/mol) -11.24 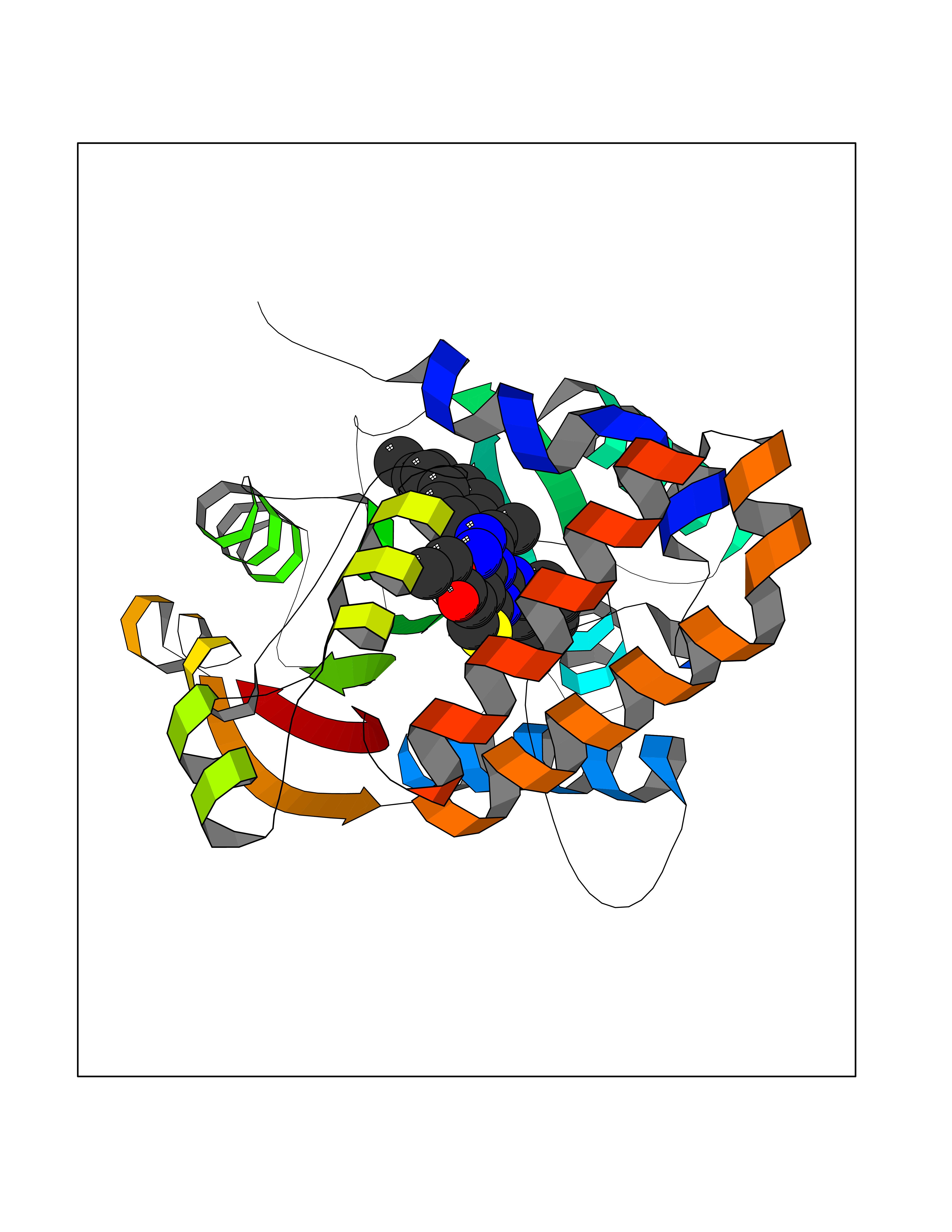 Molscript Map 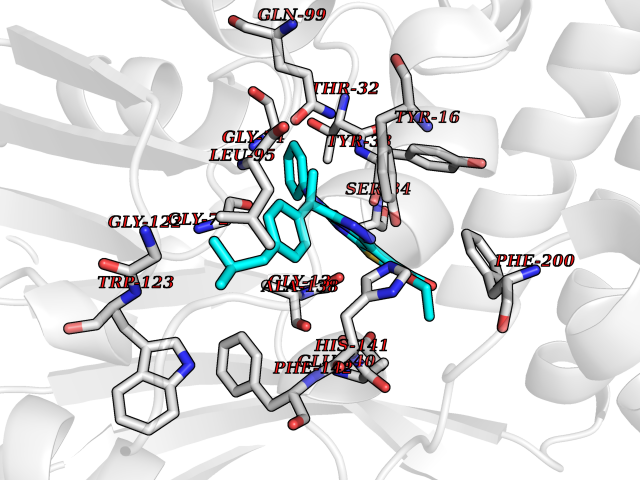 Pymol Map 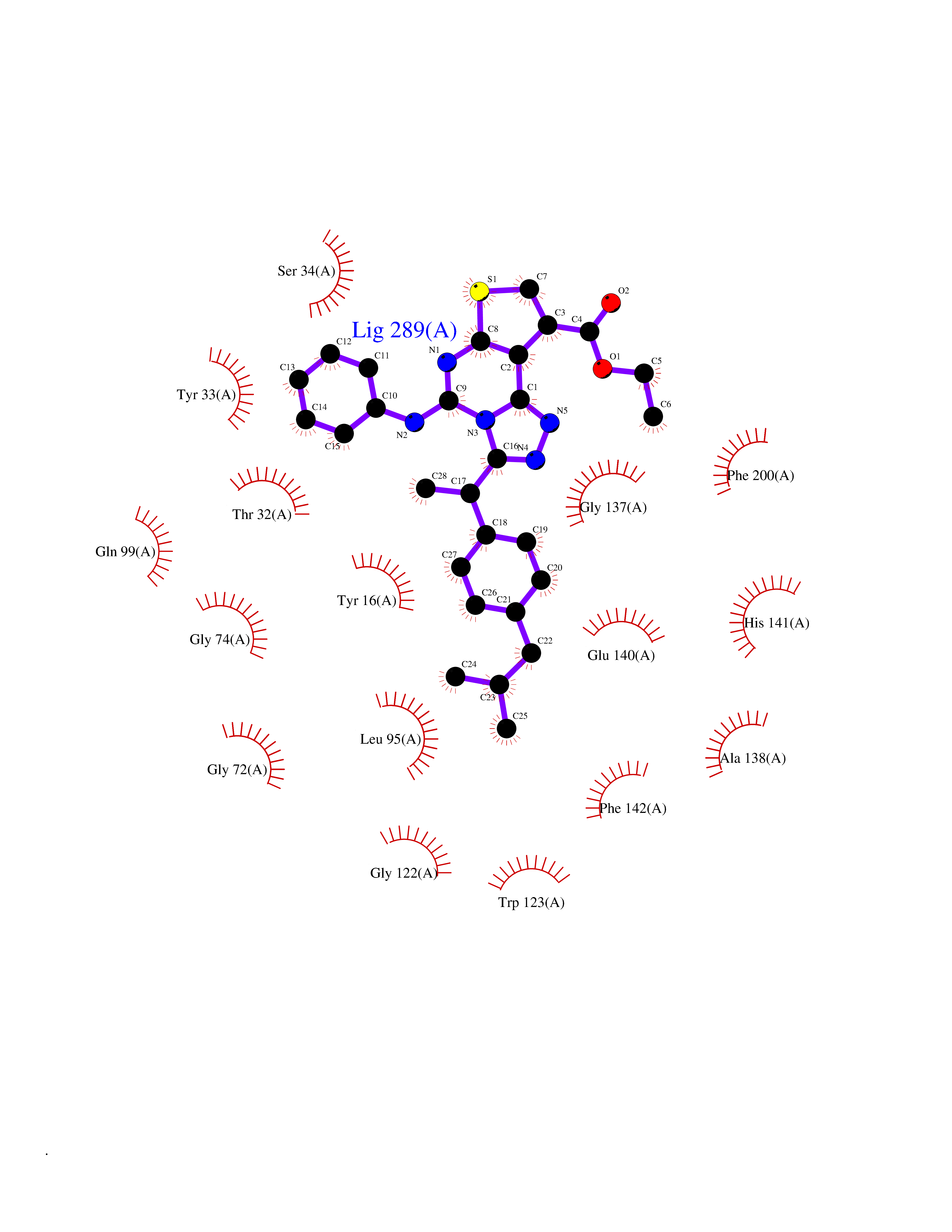 Ligplot Map 3D Binding mode Sequence NDLTPHFEDVQAHYDLSDDFFRLFLDPTQTYSCAHFEREDMTLEEAQIAKIDLALGKLGLQPGMTLLDIGCGWGATMRRAIAQYDVNVVGLTLSKNQAAHVQKSFDEMDTPRDRRVLLAGWEQFNEPVDRIVSIGAFEHFGHDRHADFFARAHKILPPDGVLLLHTITGLTRQQMVDHGLPLTLWLARFLKFIATEIFPGGQPPTIEMVEEQSAKTGFTLTRRQSLQPHYARTLDLWAEALQEHKSEAIAIQSEEVYERYMKYLTGCAKLFRVGYIDVNQFTLAK Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | 5-HT 2A receptor (HTR2A) | 7WC7 | 8.24 | |
Target general information Gen name HTR2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serotonin receptor 2A; HTR2; 5-hydroxytryptamine receptor 2A; 5-HT-2A; 5-HT-2 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function G-protein coupled receptor for 5-hydroxytryptamine (serotonin). Also functions as a receptor for various drugs and psychoactive substances, including mescaline, psilocybin, 1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane (DOI) and lysergic acid diethylamide (LSD). Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Beta-arrestin family members inhibit signaling via G proteins and mediate activation of alternative signaling pathways. Signaling activates phospholipase C and a phosphatidylinositol-calcium second messenger system that modulates the activity of phosphatidylinositol 3-kinase and promotes the release of Ca(2+) ions from intracellular stores. Affects neural activity, perception, cognition and mood. Plays a role in the regulation of behavior, including responses to anxiogenic situations and psychoactive substances. Plays a role in intestinal smooth muscle contraction, and may play a role in arterial vasoconstriction. Related diseases Heinz body anemias (HEIBAN) [MIM:140700]: Form of non-spherocytic hemolytic anemia of Dacie type 1. After splenectomy, which has little benefit, basophilic inclusions called Heinz bodies are demonstrable in the erythrocytes. Before splenectomy, diffuse or punctate basophilia may be evident. Most of these cases are probably instances of hemoglobinopathy. The hemoglobin demonstrates heat lability. Heinz bodies are observed also with the Ivemark syndrome (asplenia with cardiovascular anomalies) and with glutathione peroxidase deficiency. {ECO:0000269|PubMed:186485, ECO:0000269|PubMed:2599881, ECO:0000269|PubMed:6259091, ECO:0000269|PubMed:8704193}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Beta-thalassemia (B-THAL) [MIM:613985]: A form of thalassemia. Thalassemias are common monogenic diseases occurring mostly in Mediterranean and Southeast Asian populations. The hallmark of beta-thalassemia is an imbalance in globin-chain production in the adult HbA molecule. Absence of beta chain causes beta(0)-thalassemia, while reduced amounts of detectable beta globin causes beta(+)-thalassemia. In the severe forms of beta-thalassemia, the excess alpha globin chains accumulate in the developing erythroid precursors in the marrow. Their deposition leads to a vast increase in erythroid apoptosis that in turn causes ineffective erythropoiesis and severe microcytic hypochromic anemia. Clinically, beta-thalassemia is divided into thalassemia major which is transfusion dependent, thalassemia intermedia (of intermediate severity), and thalassemia minor that is asymptomatic. {ECO:0000269|PubMed:12144064, ECO:0000269|PubMed:12149194, ECO:0000269|PubMed:15481886, ECO:0000269|PubMed:2399911, ECO:0000269|PubMed:6166632, ECO:0000269|PubMed:7693620}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Sickle cell disease (SKCA) [MIM:603903]: Characterized by abnormally shaped red cells resulting in chronic anemia and periodic episodes of pain, serious infections and damage to vital organs. Normal red blood cells are round and flexible and flow easily through blood vessels, but in sickle cell anemia, the abnormal hemoglobin (called Hb S) causes red blood cells to become stiff. They are C-shaped and resembles a sickle. These stiffer red blood cells can led to microvascular occlusion thus cutting off the blood supply to nearby tissues. {ECO:0000269|PubMed:1195378, ECO:0000269|PubMed:13464827, ECO:0000269|PubMed:16001361, ECO:0000269|PubMed:24100324, ECO:0000269|Ref.10}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Beta-thalassemia, dominant, inclusion body type (B-THALIB) [MIM:603902]: An autosomal dominant form of beta thalassemia characterized by moderate anemia, lifelong jaundice, cholelithiasis and splenomegaly, marked morphologic changes in the red cells, erythroid hyperplasia of the bone marrow with increased numbers of multinucleate red cell precursors, and the presence of large inclusion bodies in the normoblasts, both in the marrow and in the peripheral blood after splenectomy. {ECO:0000269|PubMed:1971109}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB13940; DB01537; DB14010; DB01614; DB06288; DB00321; DB00543; DB08927; DB04599; DB05227; DB00714; DB01238; DB14185; DB06216; DB05687; DB09223; DB09128; DB01200; DB09016; DB00248; DB09061; DB06016; DB00477; DB01239; DB08810; DB00604; DB01242; DB00363; DB00924; DB00434; DB06512; DB01151; DB11273; DB13345; DB00320; DB01488; DB00843; DB09167; DB06446; DB01142; DB05492; DB00751; DB12177; DB01049; DB00696; DB01175; DB06678; DB09194; DB00574; DB04908; DB00875; DB00623; DB04842; DB12141; DB00502; DB05079; DB04946; DB00458; DB01221; DB12465; DB00555; DB00589; DB09195; DB00408; DB06077; DB08815; DB00934; DB14009; DB00933; DB01403; DB00247; DB06148; DB01454; DB00805; DB00370; DB01442; DB01618; DB13948; DB14011; DB08804; DB01149; DB12555; DB00540; DB06229; DB00334; DB01267; DB00715; DB01186; DB08922; DB05316; DB09286; DB01621; DB06153; DB00420; DB00777; DB01224; DB00409; DB00734; DB12693; DB12163; DB08839; DB06144; DB09304; DB01079; DB00679; DB01623; DB13025; DB00656; DB00726; DB16351; DB06109; DB01392; DB00246; DB00315; DB09225; DB01624 Interacts with P28223; P41595; P28335; Q14416; P28335-1; P18654 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Behavior; Cell membrane; Cell projection; Cytoplasmic vesicle; Disulfide bond; G-protein coupled receptor; Glycoprotein; Host cell receptor for virus entry; Host-virus interaction; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 40088.9 Length 358 Aromaticity 0.11 Instability index 29.25 Isoelectric point 6.79 Charge (pH=7) -0.21 2D Binding mode Binding energy (Kcal/mol) -11.24 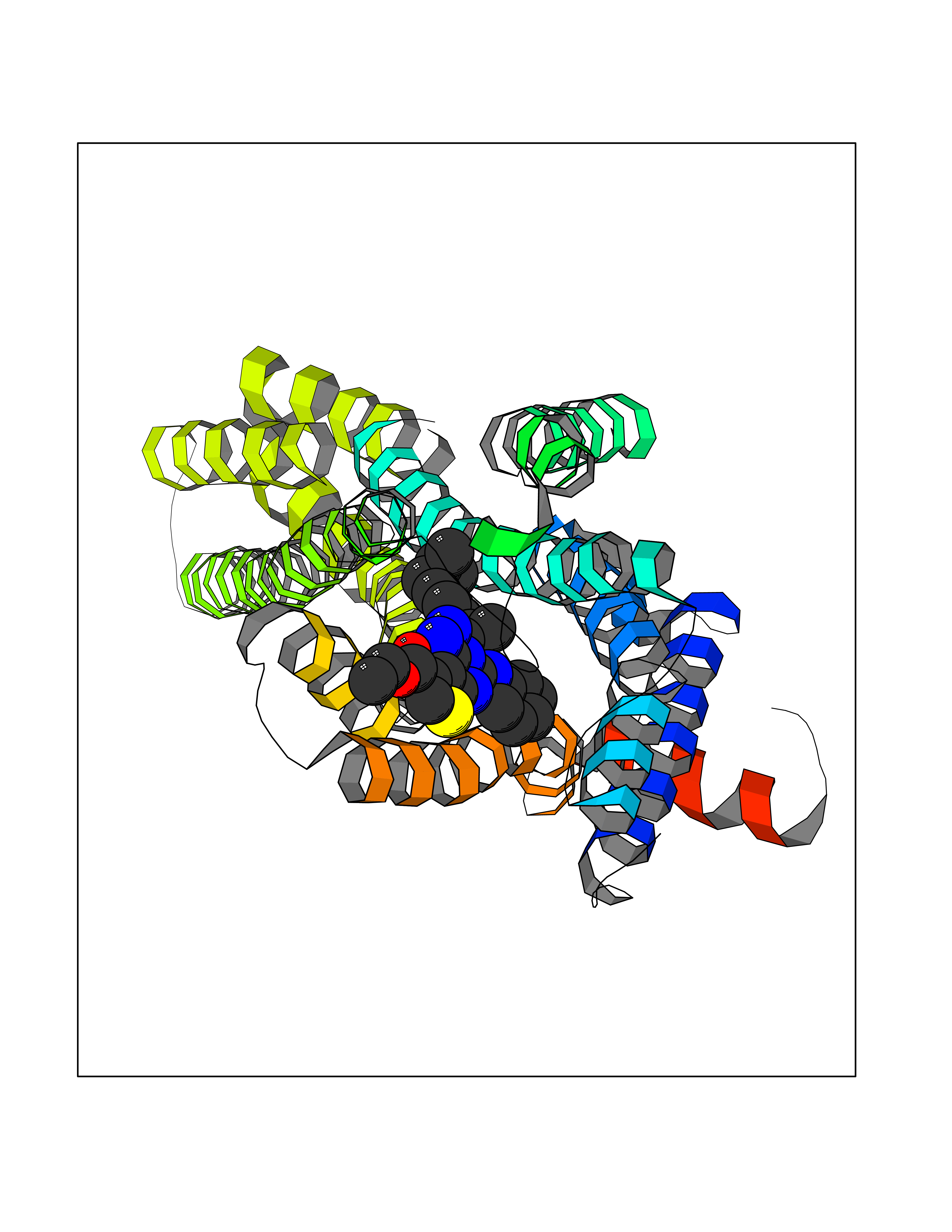 Molscript Map 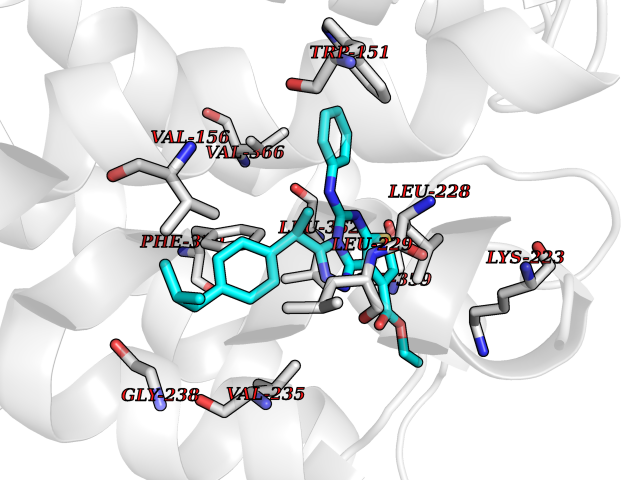 Pymol Map 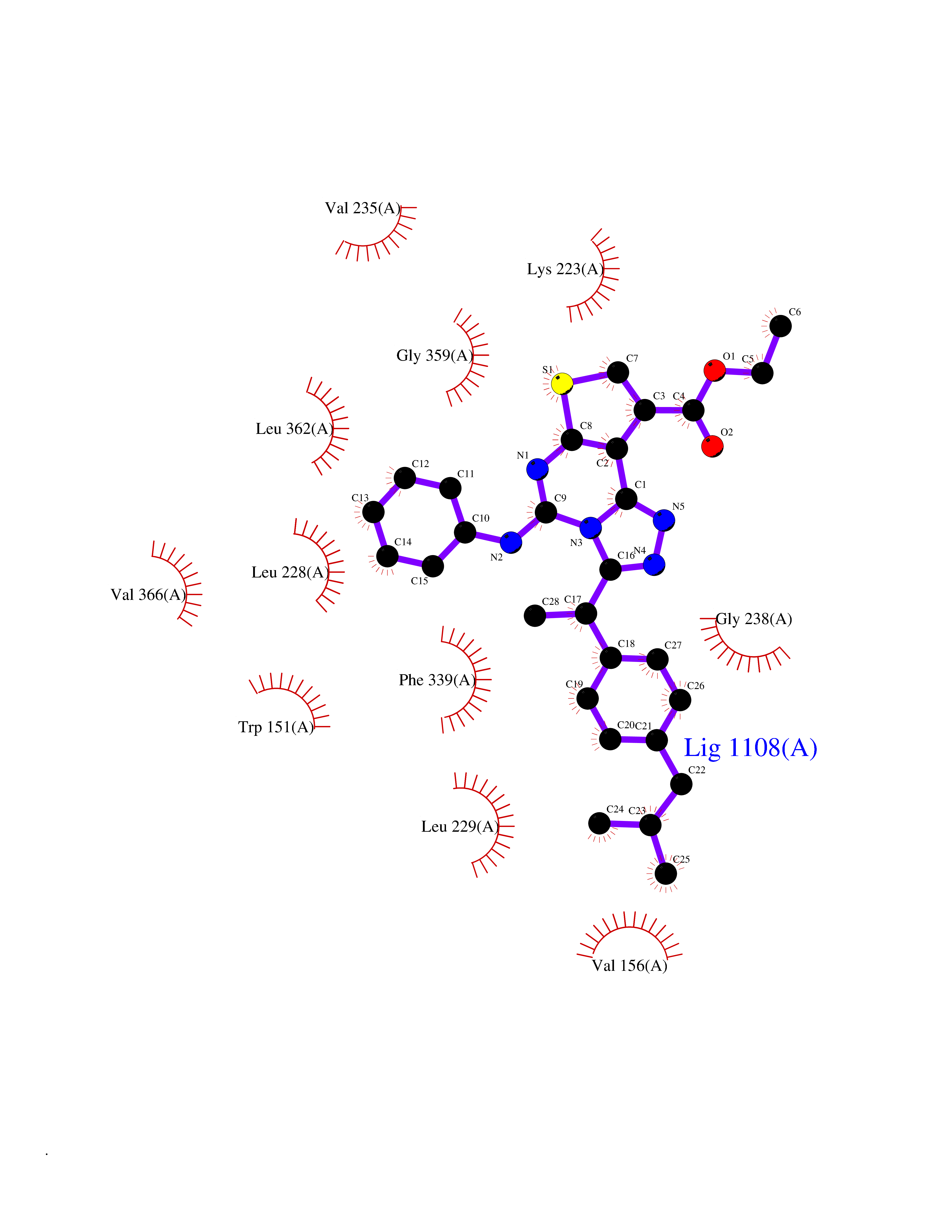 Ligplot Map 3D Binding mode Sequence LQEKNWSALLTAVVIILTIAGNILVIMAVSLEKKLQNATNYFLMSLAIADMLLGFLVMPVSMLTILYGYRWPLPSKLCAVWIYLDVLFSTAKIWHLCAISLDRYVAIQNPSRTKAFLKIIAVWTISVGISMPIPVFGLQDDSKVFKEGSCLLADDNFVLIGSFVSFFIPLTIMVITYFLTIKSLQKEAADLEDNWETLNDNLKVIEKADNAAQVKDALTKMRAAALDADILVGQIDDALKLANEGKVKEAQAAAEQLKTTINAYIQKYGQSISNEQKACKVLGIVFFLFVVMWCPFFITNIMAVICKESCNEDVIGALLNVFVWIGYLNSAVNPLVYTLFNKTYRSAFSRYIQCQYKE Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Serine--pyruvate aminotransferase | 5HHY | 8.23 | |
Target general information Gen name AGXT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms AGT1;SPAT Protein family Class-V pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transferase Function Alanine-glyoxylate transaminase activity.Amino acid binding.Identical protein binding.Protein homodimerization activity.Protein self-association.Pyridoxal phosphate binding.Receptor binding.Serine-pyruvate transaminase activity.Transaminase activity. Related diseases Hyperoxaluria primary 1 (HP1) [MIM:259900]: An inborn error of glyoxylate metabolism characterized by increased excretion of oxalate and glycolate, and progressive tissue accumulation of insoluble calcium oxalate. Affected individuals are at risk for nephrolithiasis, nephrocalcinosis and early onset end-stage renal disease. {ECO:0000269|PubMed:10394939, ECO:0000269|PubMed:10453743, ECO:0000269|PubMed:10541294, ECO:0000269|PubMed:10862087, ECO:0000269|PubMed:10960483, ECO:0000269|PubMed:12559847, ECO:0000269|PubMed:12777626, ECO:0000269|PubMed:1301173, ECO:0000269|PubMed:1349575, ECO:0000269|PubMed:15253729, ECO:0000269|PubMed:15849466, ECO:0000269|PubMed:15961946, ECO:0000269|PubMed:15963748, ECO:0000269|PubMed:16971151, ECO:0000269|PubMed:1703535, ECO:0000269|PubMed:17495019, ECO:0000269|PubMed:2039493, ECO:0000269|PubMed:23229545, ECO:0000269|PubMed:24055001, ECO:0000269|PubMed:24934730, ECO:0000269|PubMed:26149463, ECO:0000269|PubMed:8101040, ECO:0000269|PubMed:9192270, ECO:0000269|PubMed:9604803}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08060; DB00160; DB02079; DB00145; DB04083; DB00114; DB00133 Interacts with Q5BKX5-3; P21549; P0C7T5; Q9NR55; Q9H5F2; Q9UKJ5; A8MQ03; O43281-2; A1KXE4-2; Q5TD97; P49356; P53539; P49639; Q15323; O76011; O76014; Q6A162; Q07627; P60328; Q9BYR8; Q3LI67; Q8IUC2; Q9BYQ4; O60711; Q99750; Q5VZ52; P0DPK4; O43482; P50542-1; O15496; Q6ZR37; Q9NQX0; Q8HWS3; Q5W111-2; Q8WVR3; Q6DKK2 EC number 2.6.1.44; 2.6.1.51 Uniprot keywords 3D-structure; Acetylation; Aminotransferase; Disease variant; Peroxisome; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84537 Length 771 Aromaticity 0.07 Instability index 36.81 Isoelectric point 8.42 Charge (pH=7) 6.81 2D Binding mode Binding energy (Kcal/mol) -11.22  Molscript Map  Pymol Map 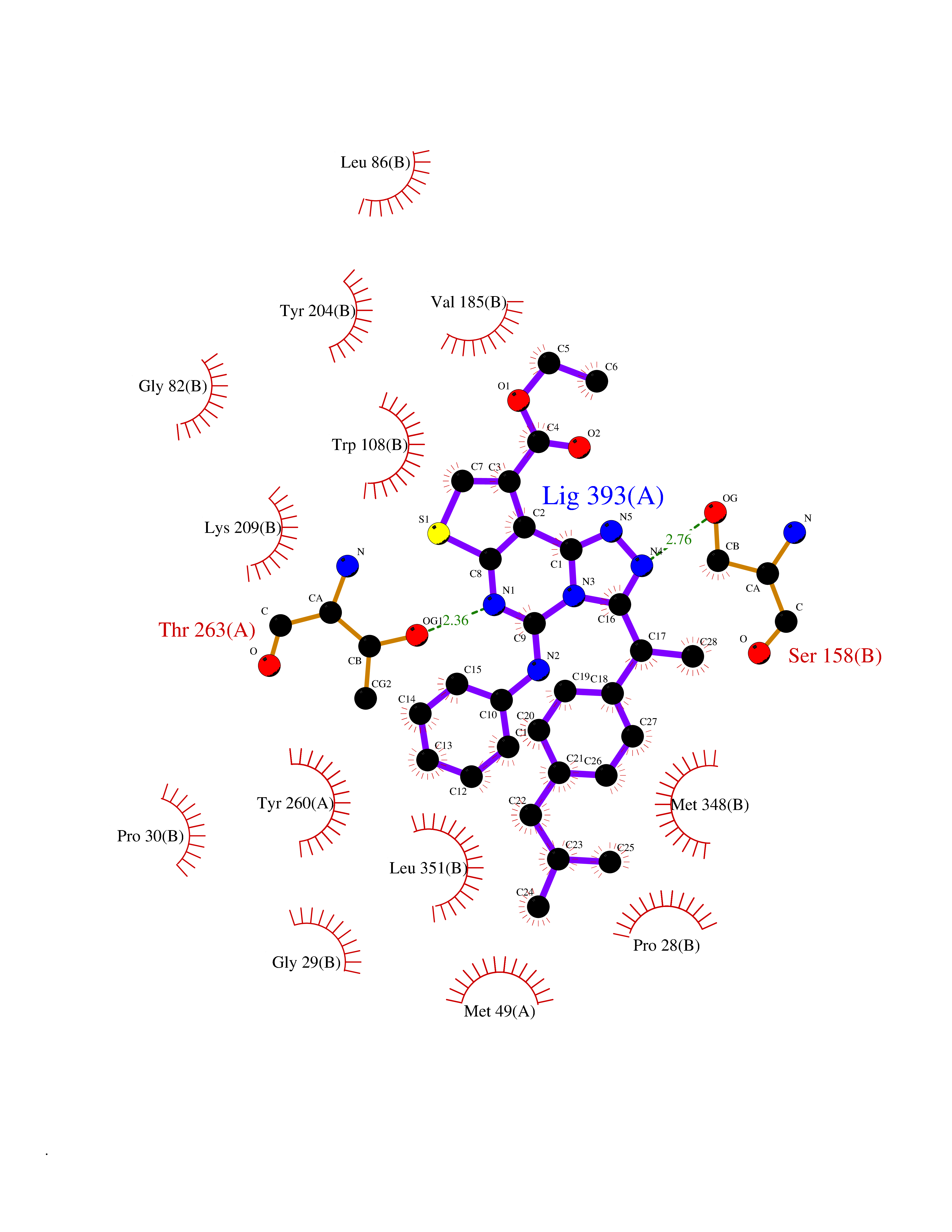 Ligplot Map 3D Binding mode Sequence LLVTPPKALLKPLSIPNQLLLGPGPSNLPPRIMAAGGLQMIGSMSKDMYQIMDEIKEGIQYVFQTRNPLTLVISGSGHCALEAALVNVLEPGDSFLVGANGIWGQRAVDIGERIGARVHPMTKDPGGHYTLQEVEEGLAQHKPVLLFLTHGESSTGVLQPLDGFGELCHRYKCLLLVDSVASLGGTPLYMDRQGIDILYSGSQKALNAPPGTSLISFSDKAKKKMYSRKTKPFSFYLDIKWLANFWGCDDQPRMYHHTIPVISLYSLRESLALIAEQGLENSWRQHREAAAYLHGRLQALGLQLFVKDPALRLPTVTTVAVPAGYDWRDIVSYVIDHFDIEIMGGLGPSTGKVLRIGLLGCNATRENVDRVTEALRAALQHCPKKLLVTPPKALLKPLSIPNQLLLGPGPSNLPPRIMAAGGLQMIGSMSKDMYQIMDEIKEGIQYVFQTRNPLTLVISGSGHCALEAALVNVLEPGDSFLVGANGIWGQRAVDIGERIGARVHPMTKDPGGHYTLQEVEEGLAQHKPVLLFLTHGESSTGVLQPLDGFGELCHRYKCLLLVDSVASLGGTPLYMDRQGIDILYSGSQKALNAPPGTSLISFSDKAKKKMYSRKTKPFSFYLDIKWLANFWGCDDQPRMYHHTIPVISLYSLRESLALIAEQGLENSWRQHREAAAYLHGRLQALGLQLFVKDPALRLPTVTTVAVPAGYDWRDIVSYVIDHFDIEIMGGLGPSTGKVLRIGLLGCNATRENVDRVTEALRAALQHCPKKK Hydrogen bonds contact Hydrophobic contact | ||||