Job Results:
Ligand
Structure
Job ID
60cfc8edcb03d90540341cd2f0373921
Job name
NA
Time
2026-03-02 09:52:38
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | DNA-(apurinic or apyrimidinic site) lyase | 4QHE | 4.38 | |
Target general information Gen name APEX1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms APE;APX;APE1;APEX;HAP1;REF1 Protein family DNA repair enzymes AP/ExoA family Biochemical class Lyase Function 3'-5' exonuclease activity.Chromatin DNA binding.Class I DNA-(apurinic or apyrimidinic site) lyase activity.Class III/IV DNA-(apurinic or apyrimidinic site) lyase activity.Damaged DNA binding.DNA-(apurinic or apyrimidinic site) lyase activity.DNA binding.Double-stranded DNA 3'-5' exodeoxyribonuclease activity.Double-stranded DNA exodeoxyribonuclease activity.Double-stranded telomeric DNA binding.Endodeoxyribonuclease activity.Endonuclease activity.Metal ion binding.NF-kappaB binding.Oxidoreductase activity.Phosphodiesterase I activity.Phosphoric diester hydrolase activity.Protein complex binding.RNA binding.RNA-DNA hybrid ribonuclease activity.Site-specific endodeoxyribonuclease activity, specific for altered base.Transcription coactivator activity.Transcription corepressor activity.Uracil DNA N-glycosylase activity. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB04967 Interacts with Q09472; Q8N4N3; Q16236; Q96EB6; O88846 EC number 3.1.11.2; 3.1.21.- Uniprot keywords 3D-structure; Acetylation; Activator; Cleavage on pair of basic residues; Cytoplasm; Direct protein sequencing; Disulfide bond; DNA damage; DNA recombination; DNA repair; DNA-binding; Endonuclease; Endoplasmic reticulum; Exonuclease; Hydrolase; Magnesium; Metal-binding; Mitochondrion; Nuclease; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; RNA-binding; S-nitrosylation; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 31556.6 Length 281 Aromaticity 0.1 Instability index 44.46 Isoelectric point 7.17 Charge (pH=7) 0.3 2D Binding mode Binding energy (Kcal/mol) -5.97  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ASEGPALYEDPPDQKTSPSGKPATLKICSWNVDGLRAWIKKKGLDWVKEEAPDILCLQETKCSENKLPAELQELPGLSHQYWSAPSDKEGYSGVGLLSRQAPLKVSYGIGDEEHDQEGRVIVAEFDSFVLVTAYVPNAGRGLVRLEYRQRWDEAFRKFLKGLASRKPLVLCGDLNVAHEEIDLRNPKGNKKNAGFTPQERQGFGELLQAVPLADSFRHLYPNTPYAYTFWTYMMNARSKNVGWRLDYFLLSHSLLPALCDSKIRSKALGSDHCPITLYLAL Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Neuronal acetylcholine receptor subunit alpha-3 | 4ZK4 | 4.38 | |
Target general information Gen name CHRNA3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NACHRA3 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Acetylcholine binding protein Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ligand-gated ion channel activity.Serotonin-gated cation-selective channel activity. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46391.5 Length 408 Aromaticity 0.12 Instability index 30.23 Isoelectric point 4.6 Charge (pH=7) -22.73 2D Binding mode Binding energy (Kcal/mol) -5.98 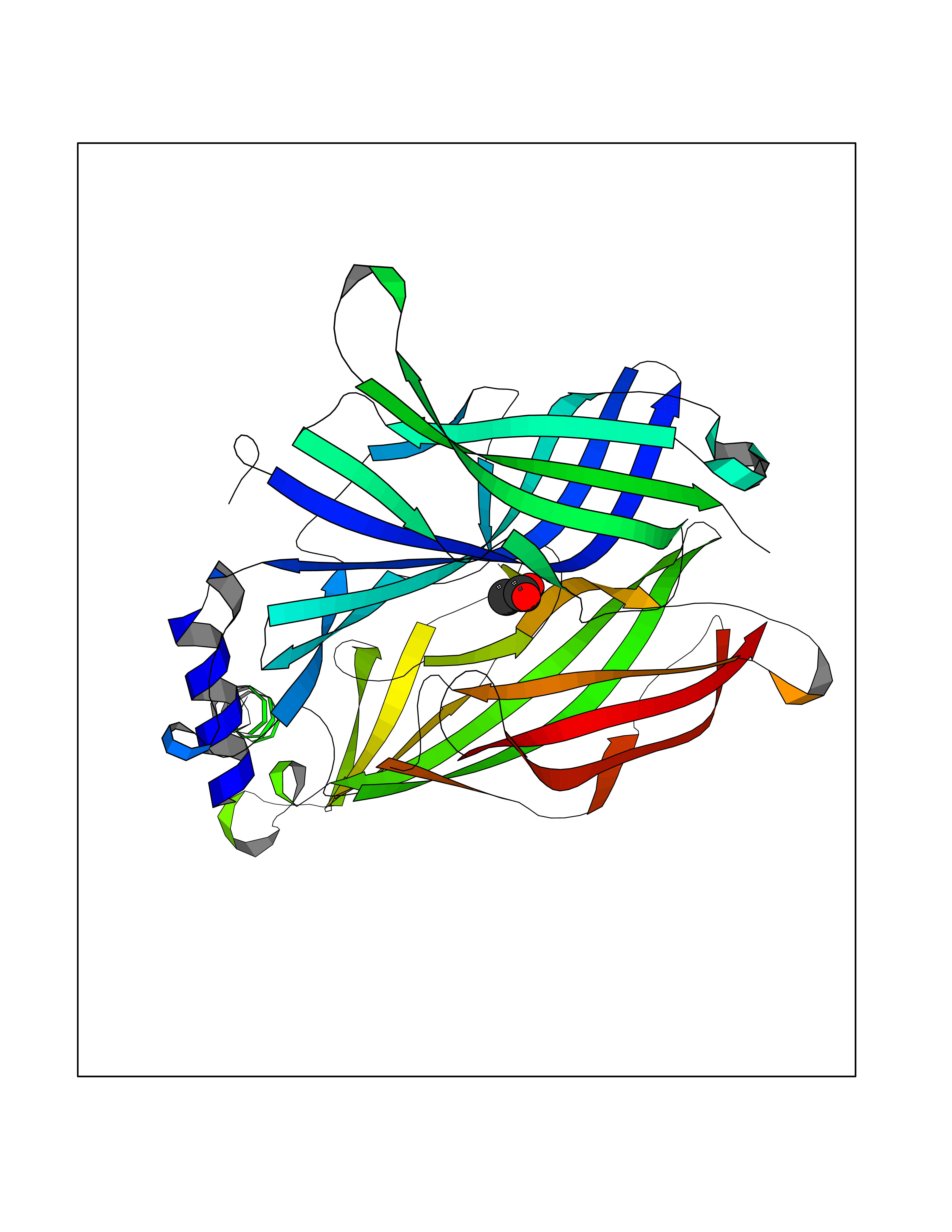 Molscript Map 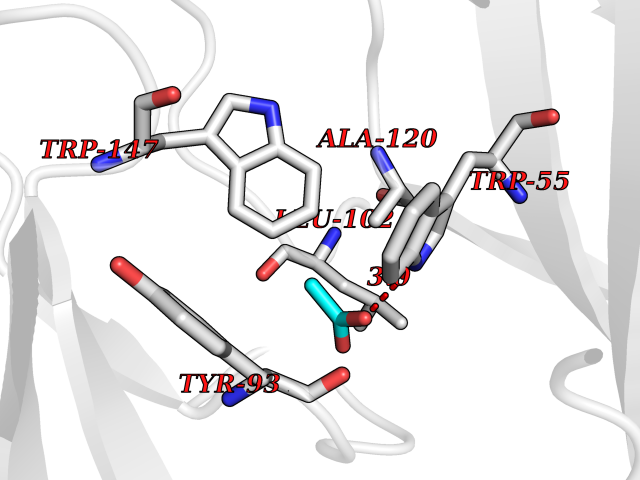 Pymol Map 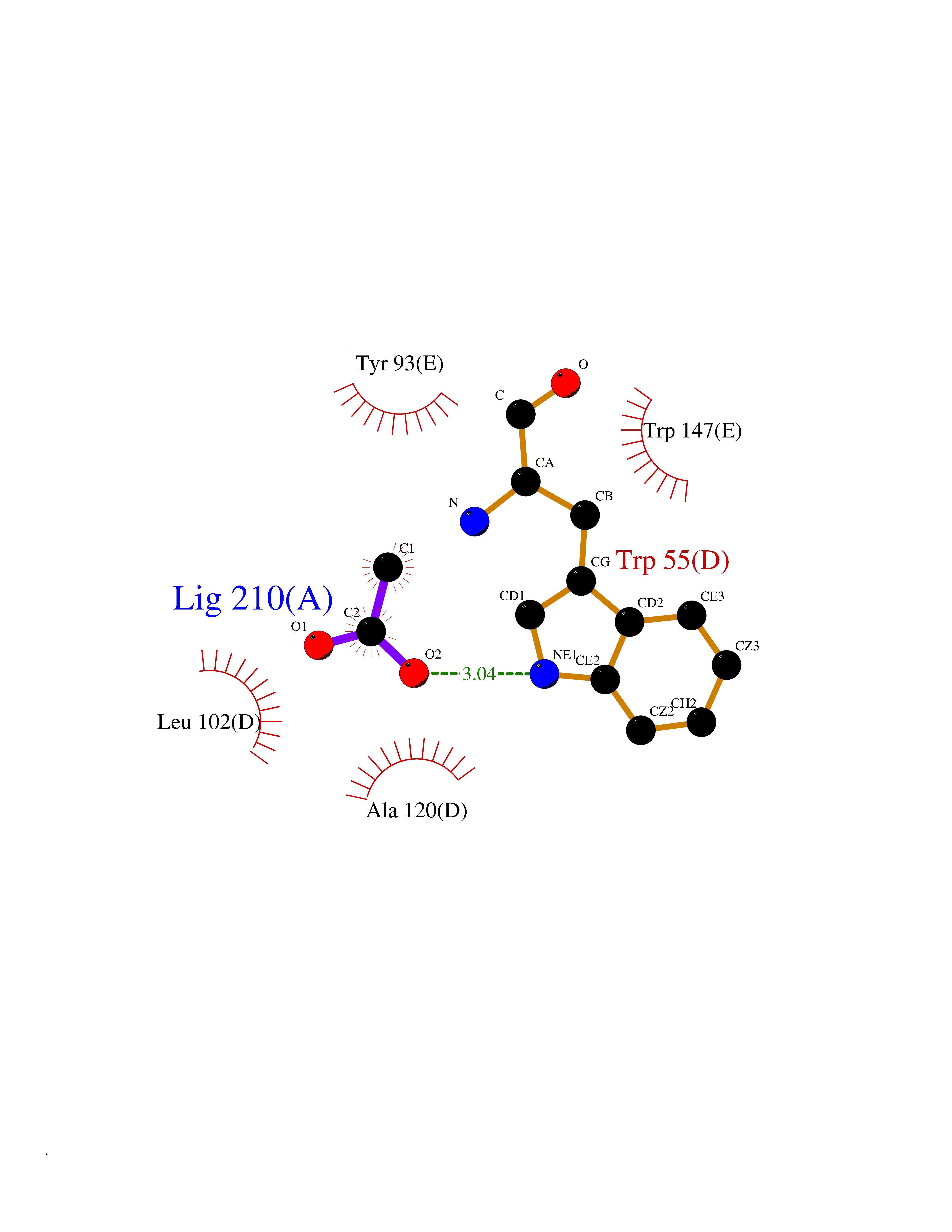 Ligplot Map 3D Binding mode Sequence LHSQANLMRLKSDLFYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRERRLHSQANLMRLKSDLFNRYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRE Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Xanthine dehydrogenase/oxidase (XDH) | 2E1Q | 4.38 | |
Target general information Gen name XDH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Xanthine oxidase; Xanthine dehydrogenase; XDHA Protein family Xanthine dehydrogenase family Biochemical class CH/CH(2) oxidoreductase Function Catalyzes the oxidation of hypoxanthine to xanthine. Catalyzes the oxidation of xanthine to uric acid. Contributes to the generation of reactive oxygen species. Has also low oxidase activity towards aldehydes (in vitro). Key enzyme in purine degradation. Related diseases Xanthinuria 1 (XAN1) [MIM:278300]: A disorder characterized by excretion of very large amounts of xanthine in the urine and a tendency to form xanthine stones. Uric acid is strikingly diminished in serum and urine. XAN1 is due to isolated xanthine dehydrogenase deficiency. Patients can metabolize allopurinol. {ECO:0000269|PubMed:10844591, ECO:0000269|PubMed:11379872, ECO:0000269|PubMed:14551354, ECO:0000269|PubMed:9153281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00640; DB00041; DB00437; DB00993; DB00958; DB01136; DB00856; DB00515; DB00746; DB03328; DB00997; DB03516; DB12466; DB04854; DB03147; DB04335; DB01020; DB00583; DB00170; DB01033; DB00157; DB03841; DB00336; DB01250; DB05262; DB06478; DB01168; DB00339; DB00127; DB01685; DB00831 Interacts with Q9Y3R0-3 EC number NA Uniprot keywords 2Fe-2S; 3D-structure; Cytoplasm; Disease variant; Disulfide bond; FAD; Flavoprotein; Iron; Iron-sulfur; Metal-binding; Molybdenum; NAD; Oxidoreductase; Peroxisome; Proteomics identification; Reference proteome; Secreted Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 143697 Length 1307 Aromaticity 0.08 Instability index 37.9 Isoelectric point 8.01 Charge (pH=7) 7.07 2D Binding mode Binding energy (Kcal/mol) -5.98 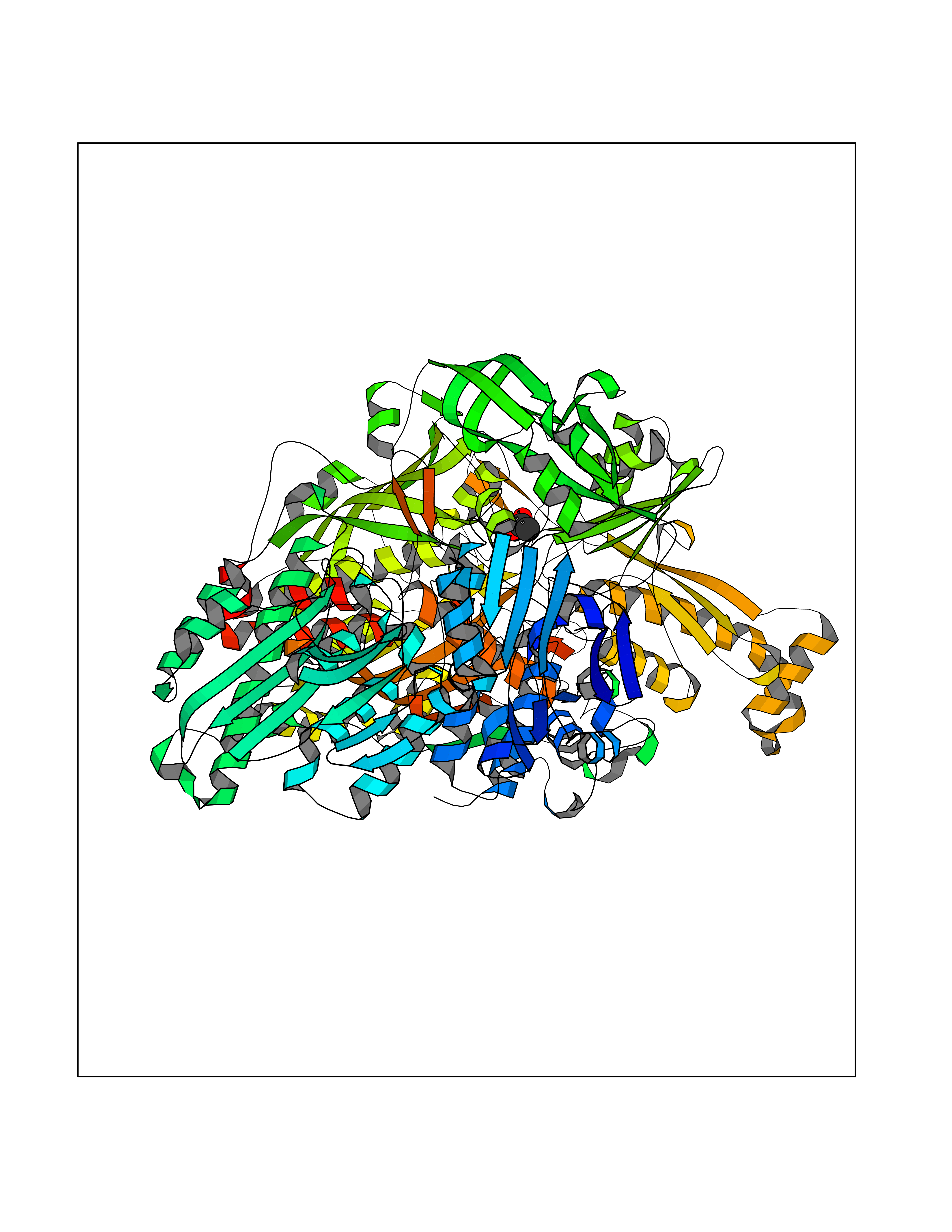 Molscript Map 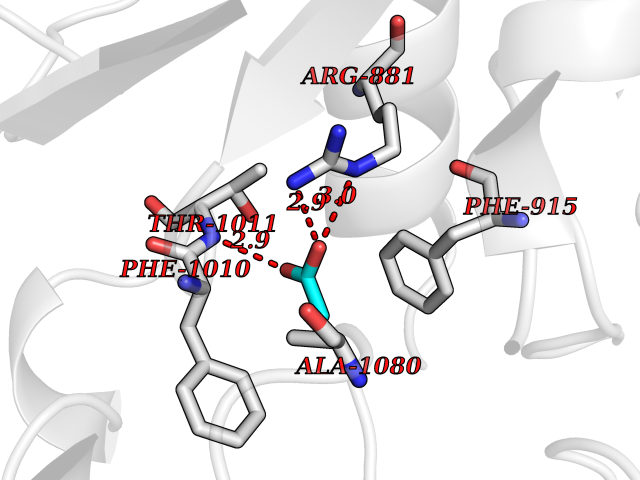 Pymol Map 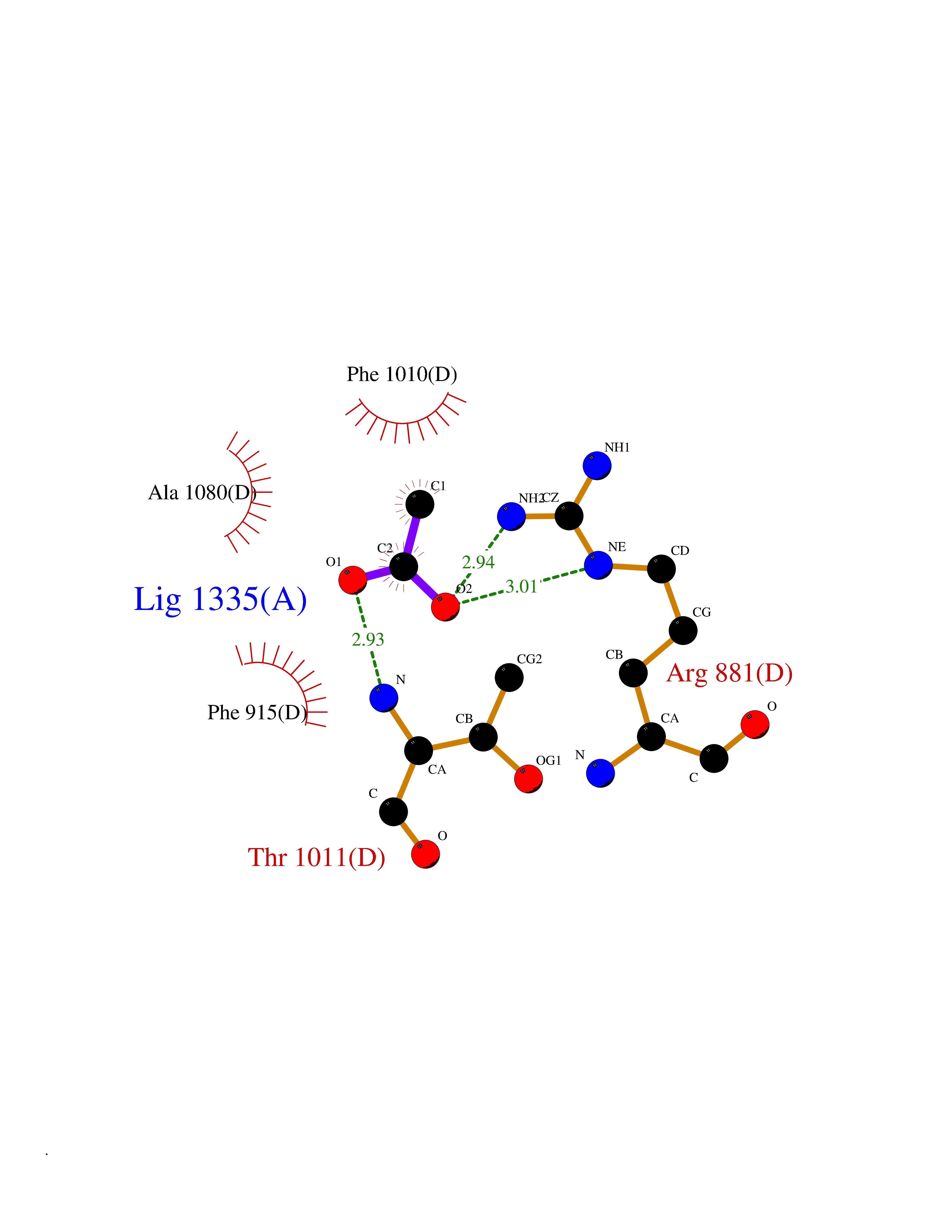 Ligplot Map 3D Binding mode Sequence ADKLVFFVNGRKVVEKNADPETTLLAYLRRKLGLSGTKLGCGEGGCGACTVMLSKYDRLQNKIVHFSANACLAPICSLHHVAVTTVEGIGSTKTRLHPVQERIAKSHGSQCGFCTPGIVMSMYTLLRNQPEPTMEEIENAFQGNLCRCTGYRPILQGFRTFARDGSPSLFKPEEFTPLDPTQEPIFPPELLRLKDTPRKQLRFEGERVTWIQASTLKELLDLKAQHPDAKLVVGNTEIGIEMKFKNMLFPMIVCPAWIPELNSVEHGPDGISFGAACPLSIVEKTLVDAVAKLPAQKTEVFRGVLEQLRWFAGKQVKSVASVGGNIITASPISDLNPVFMASGAKLTLVSRGTRRTVQMDHTFFPGYRKTLLSPEEILLSIEIPYSREGEYFSAFKQASRREDDIAKVTSGMRVLFKPGTTEVQELALCYGGMANRTISALKTTQRQLSKLWKEELLQDVCAGLAEELHLPPDAPGGMVDFRCTLTLSFFFKFYLTVLQKLGQENLEDKCGKLDPTFASATLLFQKDPPADVQLFQEVPKGQSEEDMVGRPLPHLAADMQASGEAVYCDDIPRYENELSLRLVTSTRAHAKIKSIDTSEAKKVPGFVCFISADDVPGSNITGICNDETVFAKDKVTCVGHIIGAVVADTPEHTQRAAQGVKITYEELPAIITIEDAIKNNSFYGPELKIEKGDLKKGFSEADNVVSGEIYIGGQEHFYLETHCTIAVPKGEAGEMELFVSTQNTMKTQSFVAKMLGVPANRIVVRVKRMGGGFGGKVTRSTVVSTAVALAAYKTGRPVRCMLDRDEDMLITGGRHPFLARYKVGFMKTGTVVALEVDHFSNVGNTQDLSQSIMERALFHMDNCYKIPNIRGTGRLCKTNLPSNTAFRGFGGPQGMLIAECWMSEVAVTCGMPAEEVRRKNLYKEGDLTHFNQKLEGFTLPRCWEECLASSQYHARKSEVDKFNKENCWKKRGLCIIPTKFGISFTVPFLNQAGALLHVYTDGSVLLTHGGTEMGQGLHTKMVQVASRALKIPTSKIYISETSTNTVPNTSPTAASVSADLNGQAVYAACQTILKRLEPYKKKNPSGSWEDWVTAAYMDTVSLSATGFYRTPNLGYSFETNSGNPFHYFSYGVACSEVEIDCLTGDHKNLRTDIVMDVGSSLNPAIDIGQVEGAFVQGLGLFTLEELHYSPEGSLHTRGPSTYKIPAFGSIPIEFRVSLLRDCPNKKAIYASKAVGEPPLFLAASIFFAIKDAIRAARAQHTGNNVKELFRLDSPATPEKIRNACVDKFTTLCVTGVPENCKPWSVRV Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | 2-hydroxy-6-oxo-7-methylocta-2,4-dienoate hydrolase | 1UK8 | 4.38 | |
Target general information Gen name cumD Organism Pseudomonas fluorescens Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function Hydrolase activity. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03741; DB03793; DB03568; DB02531; DB03750; DB02406; DB03766 Interacts with NA EC number NA Uniprot keywords 3D-structure; Hydrolase Protein physicochemical properties Chain ID A Molecular weight (Da) 30307.9 Length 271 Aromaticity 0.1 Instability index 37.49 Isoelectric point 5.02 Charge (pH=7) -11.58 2D Binding mode Binding energy (Kcal/mol) -5.97  Molscript Map 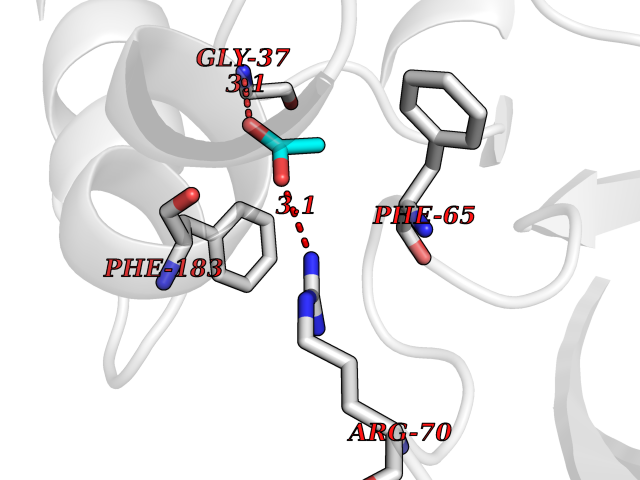 Pymol Map  Ligplot Map 3D Binding mode Sequence NLEIGKSILAAGVLTNYHDVGEGQPVILIHGSGPGVSAYANWRLTIPALSKFYRVIAPDMVGFGFTDRPENYNYSKDSWVDHIIGIMDALEIEKAHIVGNAFGGGLAIATALRYSERVDRMVLMGAAGTRFDVTEGLNAVWGYTPSIENMRNLLDIFAYDRSLVTDELARLRYEASIQPGFQESFSSMFPEPRQRWIDALASSDEDIKTLPNETLIIHGREDQVVPLSSSLRLGELIDRAQLHVFGRCGHWTQIEQTDRFNRLVVEFFNEA Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Metabotropic glutamate receptor 3 (mGluR3) | 4XAR | 4.38 | |
Target general information Gen name GRM3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms mGLUR3; Group III metabotropic glutamate receptor; GPRC1C Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling inhibits adenylate cyclase activity. G-protein coupled receptor for glutamate. Related diseases Paramyotonia congenita (PMC) [MIM:168300]: An autosomal dominant channelopathy characterized by myotonia, increased by exposure to cold, intermittent flaccid paresis, not necessarily dependent on cold or myotonia, lability of serum potassium, non-progressive nature and lack of atrophy or hypertrophy of muscles. In some patients, myotonia is not increased by cold exposure (paramyotonia without cold paralysis). Patients may have a combination phenotype of PMC and HYPP. {ECO:0000269|PubMed:10369308, ECO:0000269|PubMed:10727489, ECO:0000269|PubMed:1310898, ECO:0000269|PubMed:1316765, ECO:0000269|PubMed:1338909, ECO:0000269|PubMed:15318338, ECO:0000269|PubMed:15790667, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:18166706, ECO:0000269|PubMed:18690054, ECO:0000269|PubMed:19077043, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:8242056, ECO:0000269|PubMed:8308722, ECO:0000269|PubMed:8388676, ECO:0000269|PubMed:8580427}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hypokalemic 2 (HOKPP2) [MIM:613345]: An autosomal dominant disorder manifested by episodic flaccid generalized muscle weakness associated with falls of serum potassium levels. {ECO:0000269|PubMed:10599760, ECO:0000269|PubMed:10851391, ECO:0000269|PubMed:10944223, ECO:0000269|PubMed:11558801, ECO:0000269|PubMed:11591859, ECO:0000269|PubMed:16890191, ECO:0000269|PubMed:17898326, ECO:0000269|PubMed:18162704, ECO:0000269|PubMed:19118277, ECO:0000269|PubMed:20522878, ECO:0000269|PubMed:21043388, ECO:0000269|PubMed:24549961}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hyperkalemic (HYPP) [MIM:170500]: An autosomal dominant channelopathy characterized by episodic flaccid generalized muscle weakness associated with high levels of serum potassium. Concurrence of myotonia is found in HYPP patients. {ECO:0000269|PubMed:1659668, ECO:0000269|PubMed:1659948, ECO:0000269|PubMed:20076800}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis normokalemic (NKPP) [MIM:170500]: A disorder closely related to hyperkalemic periodic paralysis, but marked by a lack of alterations in potassium levels during attacks of muscle weakness. {ECO:0000269|PubMed:15596759, ECO:0000269|PubMed:18046642, ECO:0000269|PubMed:20522878}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myotonia SCN4A-related (MYOSCN4A) [MIM:608390]: A phenotypically highly variable myotonia aggravated by potassium loading, and sometimes by cold. Myotonia is characterized by sustained muscle tensing that prevents muscles from relaxing normally. It causes muscle stiffness that can interfere with movement. In some people the stiffness is very mild, while in other cases it may be severe enough to interfere with walking, running, and other activities of daily life. Myotonia SCN4A-related includes myotonia permanens and myotonia fluctuans. In myotonia permanens, the myotonia is generalized and there is a hypertrophy of the muscle, particularly in the neck and the shoulder. Attacks of severe muscle stiffness of the thoracic muscles may be life threatening due to impaired ventilation. In myotonia fluctuans, the muscle stiffness may fluctuate from day to day, provoked by exercise. {ECO:0000269|PubMed:10218481, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:16832098, ECO:0000269|PubMed:17212350, ECO:0000269|PubMed:17998485, ECO:0000269|PubMed:18203179, ECO:0000269|PubMed:18337100, ECO:0000269|PubMed:19015483, ECO:0000269|PubMed:19347921, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:27653901, ECO:0000269|PubMed:8058156, ECO:0000269|PubMed:9392583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 16 (CMS16) [MIM:614198]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS16 is characterized by fatigable generalized weakness and recurrent attacks of respiratory and bulbar paralysis since birth. The fatigable weakness involves lid-elevator, external ocular, facial, limb and truncal muscles and an decremental response of the compound muscle action potential on repetitive stimulation. {ECO:0000269|PubMed:12766226, ECO:0000269|PubMed:25707578, ECO:0000269|PubMed:26659129}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22A, classic (CMYO22A) [MIM:620351]: A form of congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22A is an autosomal recessive form characterized by fetal hypokinesia, polyhydramnios, and severe neonatal hypotonia associated with respiratory insufficiency. Affected individuals who survive the neonatal period have delayed motor development, difficulty walking, proximal muscle weakness of the upper and lower limbs, facial and neck muscle weakness, easy fatigability, and mild limb contractures or foot deformities. {ECO:0000269|PubMed:26700687, ECO:0000269|PubMed:28262468, ECO:0000269|PubMed:36090556}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22B, severe fetal (CMYO22B) [MIM:620369]: A severe congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22B is an autosomal recessive form characterized by onset in utero. Affected individuals show fetal akinesia, and develop fetal hydrops with pulmonary hypoplasia, severe joint contractures, and generalized muscle hypoplasia. Death occurs in utero or soon after birth. {ECO:0000269|PubMed:26700687}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05096 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50355.5 Length 445 Aromaticity 0.11 Instability index 38.26 Isoelectric point 6.52 Charge (pH=7) -1.53 2D Binding mode Binding energy (Kcal/mol) -5.97  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence RREIKIEGDLVLGGLFPINEKGTGTEECGRINEDRGIQRLEAMLFAIDEINKDDYLLPGVKLGVHILDTCSRDTYALEQSLEFVRASLLLIAGVIGGSYSSVSIQVANLLRLFQIPQISYASTSAKLSDKSRYDYFARTVPPDFYQAKAMAEILRFFNWTYVSTVASEGDYGETGIEAFEQEARLRNISIATAEKVGRSNIRKSYDSVIRELLQKPNARVVVLFMRSDDSRELIAAASRANASFTWVASDGWGAQESIIKGSEHVAYGAITLELASQPVRQFDRYFQSLNPYNNHRNPWFRDFWEQKFQCSLRVCDKHLAIDSSNYEQESKIMFVVNAVYAMAHALHKMQRTLCPNTTKLCDAMKILDGKKLYKDYLLKINFTAPDADSIVKFDTFGDGMGRYNVFNFQNVGGKYSYLKVGHWAETLSLDVNSIHWSRNSVPTSE Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Protoporphyrinogen oxidase (PPOX) | 3NKS | 4.38 | |
Target general information Gen name PPOX Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PPO Protein family Protoporphyrinogen/coproporphyrinogen oxidase family, Protoporphyrinogen oxidase subfamily Biochemical class NA Function Catalyzes the 6-electron oxidation of protoporphyrinogen-IX to form protoporphyrin-IX. Related diseases Variegate porphyria (VP) [MIM:176200]: A form of porphyria. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. Variegate porphyria is an acute hepatic form characterized by partial reduction of protoporphyrinogen oxidase activity, increased photosensitivity, skin blistering and scarring of sun-exposed areas, skin hyperpigmentation, abdominal pain, and neuropsychiatric symptoms. High fecal levels of protoporphyrin and coproporphyrin, increased urine uroporphyrins and iron overload are typical markers of the disease. Inheritance is autosomal dominant with incomplete penetrance. {ECO:0000269|PubMed:10486317, ECO:0000269|PubMed:11074242, ECO:0000269|PubMed:11102990, ECO:0000269|PubMed:11348478, ECO:0000269|PubMed:11350188, ECO:0000269|PubMed:11474578, ECO:0000269|PubMed:12380696, ECO:0000269|PubMed:12655566, ECO:0000269|PubMed:12859407, ECO:0000269|PubMed:12922165, ECO:0000269|PubMed:14669009, ECO:0000269|PubMed:16433813, ECO:0000269|PubMed:16621625, ECO:0000269|PubMed:16922948, ECO:0000269|PubMed:16947091, ECO:0000269|PubMed:18350656, ECO:0000269|PubMed:18570668, ECO:0000269|PubMed:19320019, ECO:0000269|PubMed:21048046, ECO:0000269|PubMed:23430901, ECO:0000269|PubMed:23467411, ECO:0000269|PubMed:24073655, ECO:0000269|PubMed:8817334, ECO:0000269|PubMed:8852667, ECO:0000269|PubMed:9763307}. The disease is caused by variants affecting the gene represented in this entry. Mutations leading to severe PPOX deficiency cause the rare homozygous variant form of VP. Missense mutations that preserve 10%-25% of wild-type activity may not cause clinically overt VP in heterozygotes (PubMed:9811936). Mutations with intermediate effect on catalytic activity may cause VP, but with a low clinical penetrance (PubMed:10486317). {ECO:0000269|PubMed:10486317, ECO:0000269|PubMed:9811936}.; DISEASE: Variegate porphyria, childhood-onset (VPCO) [MIM:620483]: An autosomal recessive form of variegate porphyria, a disorder of heme biosynthesis that results from diminished activity of protoporphyrinogen oxidase. VPCO is characterized by severe protoporphyrinogen oxidase deficiency, onset of photosensitization by porphyrins in early childhood, skin scarring and hyperpigmentation, and skeletal abnormalities of the hand. Additional variable features are short stature, impaired intellectual development, and seizures. VPCO patients rarely experience acute neuropsychiatric or abdominal attacks. {ECO:0000269|PubMed:10870850, ECO:0000269|PubMed:11286631, ECO:0000269|PubMed:33159949, ECO:0000269|PubMed:8673113, ECO:0000269|PubMed:9541112, ECO:0000269|PubMed:9811936}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.3.3.4 Uniprot keywords 3D-structure; Disease variant; FAD; Flavoprotein; Heme biosynthesis; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Porphyrin biosynthesis; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 49470.2 Length 465 Aromaticity 0.05 Instability index 48.23 Isoelectric point 7.8 Charge (pH=7) 1.98 2D Binding mode Binding energy (Kcal/mol) -5.98 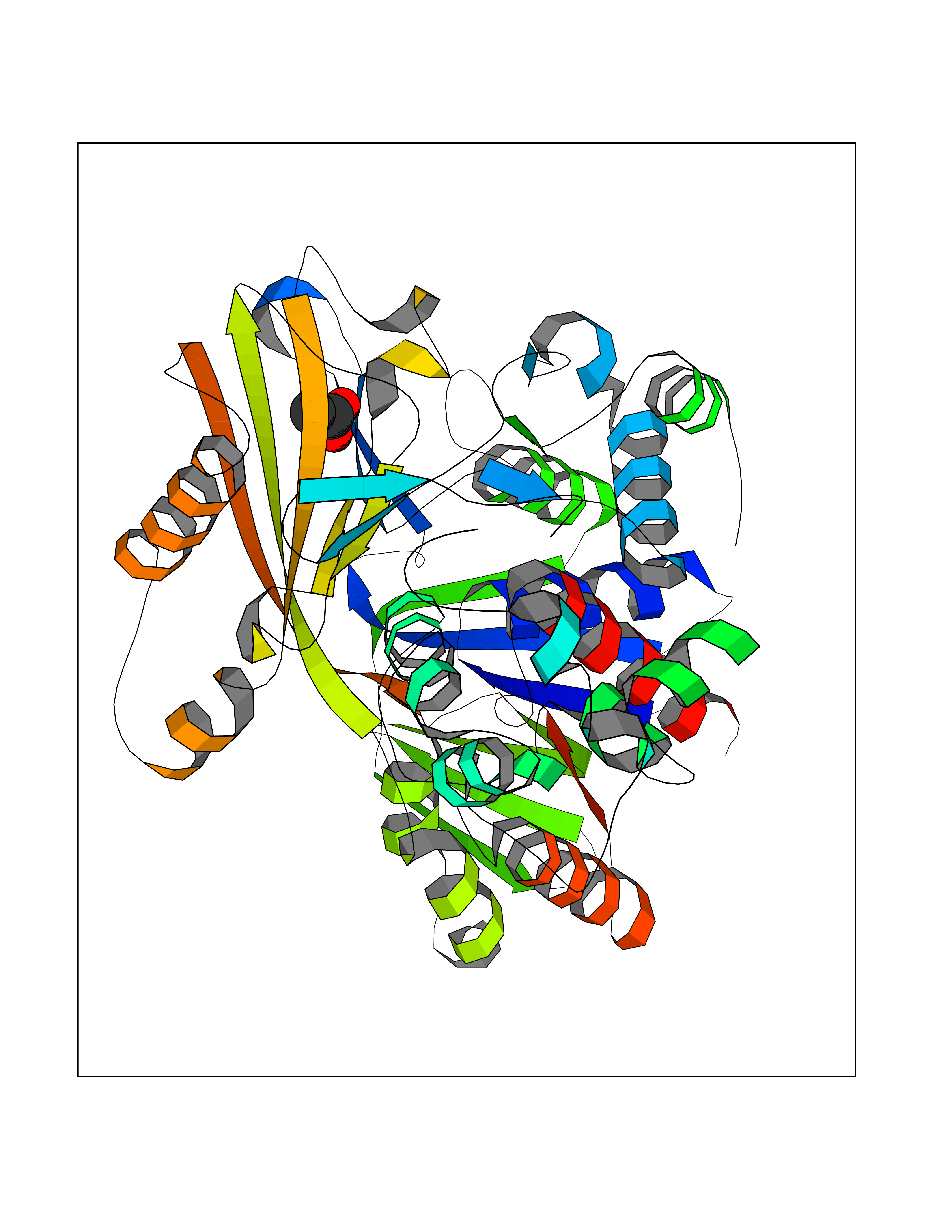 Molscript Map 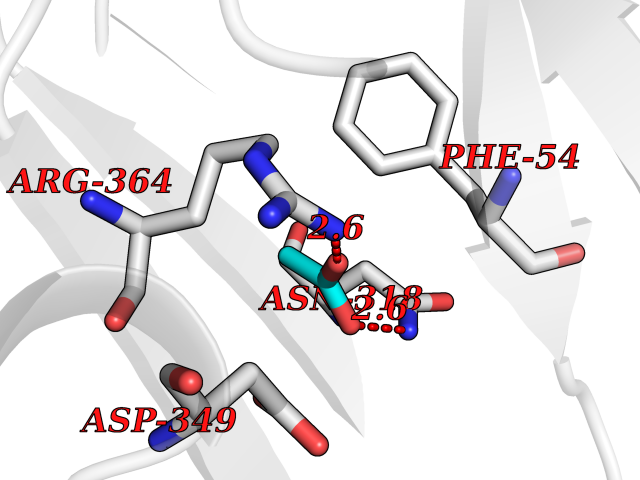 Pymol Map 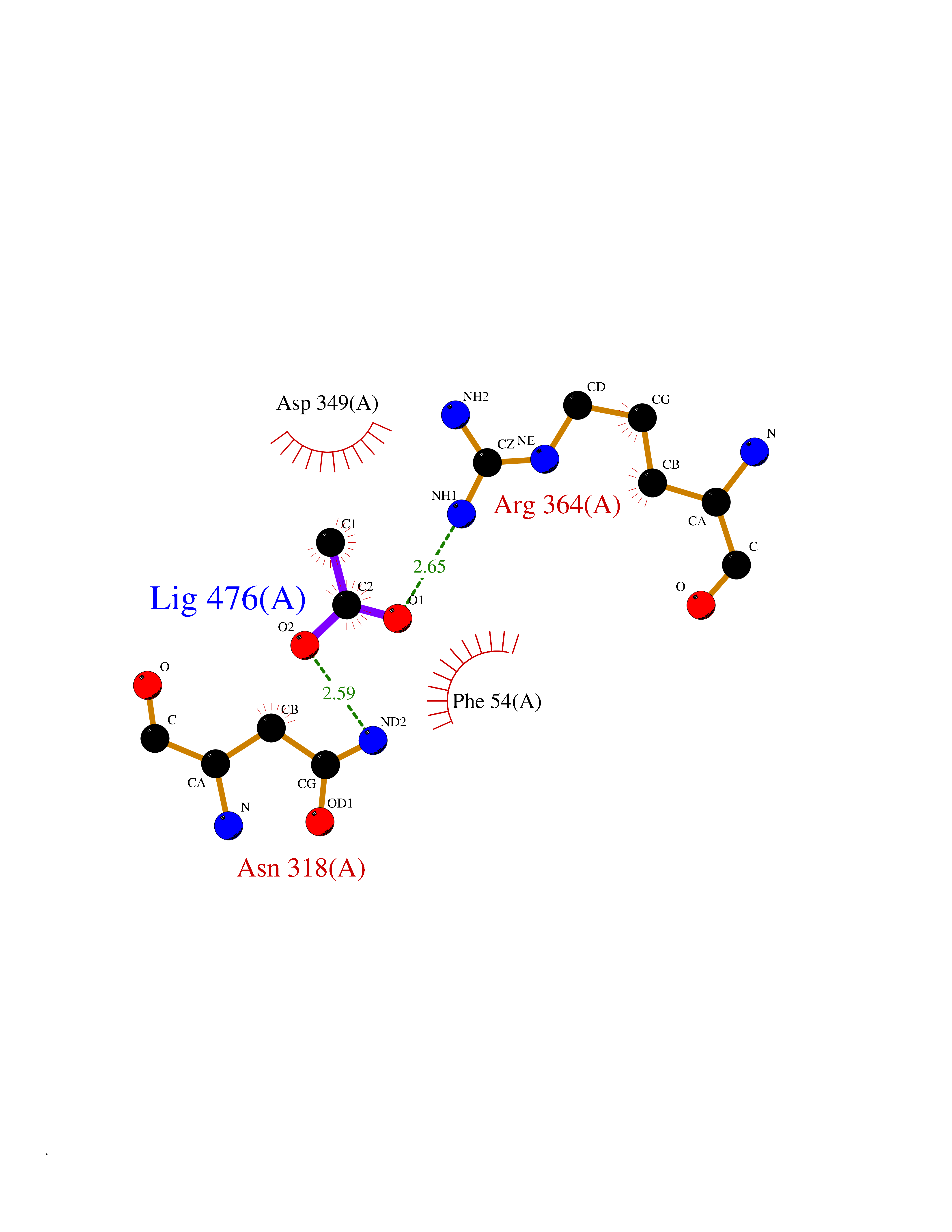 Ligplot Map 3D Binding mode Sequence GRTVVVLGGGISGLAASYHLSRAPCPPKVVLVESSERLGGWIRSVRGPNGAIFELGPRGIRPAGALGARTLLLVSELGLDSEVLPVRGDHPAAQNRFLYVGGALHALPTGLRGPSPPFSKPLFWAGLRELTKPRGKEPDETVHSFAQRRLGPEVASLAMDSLCRGVFAGNSRELSIRSCFPSLFQAEQTHRSILLGLLLGQPDSALIRQALAERWSQWSLRGGLEMLPQALETHLTSRGVSVLRGQPVCGLSLQAEGRWKVSLRDSSLEADHVISAIPASVLSELLPAEAAPLARALSAITAVSVAVVNLQYQGAHLPVQGFGHLVPSSEDPGVLGIVYDSVAFPEQDGSPPGLRVTVMLGGSWLQTLEASGCVLSQELFQQRAQEAAATQLGLKEMPSHCLVHLHKNCIPQYTLGHWQKLESARQFLTAHRLPLTLAGASYEGVAVNDCIESGRQAAVSVLGTE Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | SF3b complex (SF3b) | 3LQV | 4.38 | |
Target general information Gen name SF3B1-SF3B2-SF3B3-SF3B4-SF3B5-SF3B6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NA Protein family SF3B1 family Biochemical class NA Function NA Related diseases Developmental and epileptic encephalopathy 24 (DEE24) [MIM:615871]: A disease characterized by early-onset seizures, intellectual disability of varying degrees, and behavioral disturbances or autistic features in most individuals. {ECO:0000269|PubMed:24747641, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Generalized epilepsy with febrile seizures plus 10 (GEFSP10) [MIM:618482]: An autosomal dominant neurologic disorder with incomplete penetrance, characterized by variable types of seizures including absence, tonic-clonic, febrile, focal, and eyelid myoclonia. Some patients have normal neurologic development. Others have mild-to-moderate intellectual disability or autism spectrum disorder. {ECO:0000269|PubMed:29936235, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14017 Interacts with Q13936; Q96I25; Q15428; Q9Y3B4; O43719; Q96I25; Q9Y3B4 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Citrullination; Isopeptide bond; mRNA processing; mRNA splicing; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; RNA-binding; Spliceosome; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 13222 Length 113 Aromaticity 0.12 Instability index 27.56 Isoelectric point 9.22 Charge (pH=7) 3.86 2D Binding mode Binding energy (Kcal/mol) -5.98 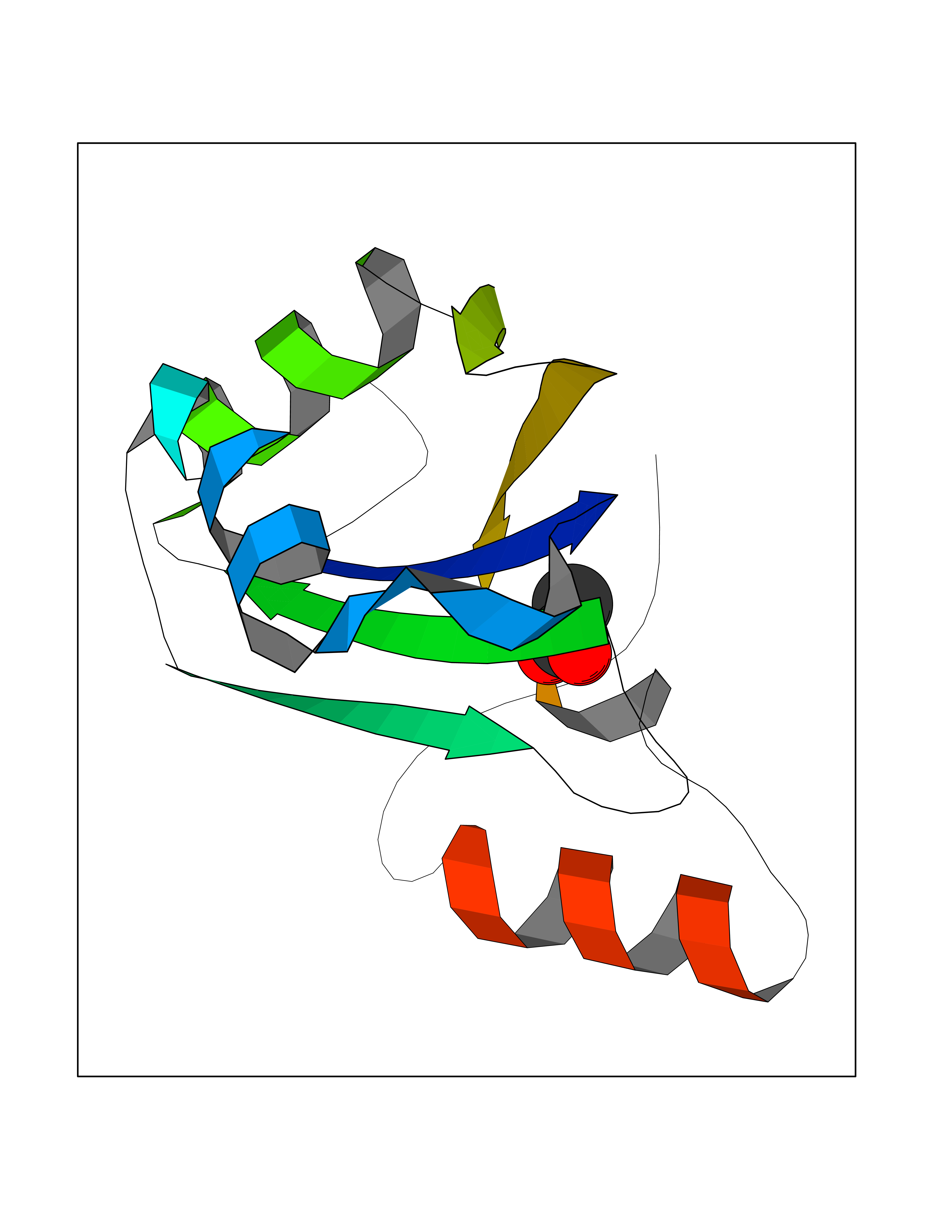 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LPPEVNRILYIRNLPYKITAEEMYDIFGKYGPIRQIRVGNTPETRGTAYVVYEDIFDAKNAVDHLSGFNVSNRYLVVLYYNANRAFQKMDTKKKEEQLKLLKEKYGINTDPPK Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Aggrecanase (ADAMTS5) | 3HY7 | 4.38 | |
Target general information Gen name ADAMTS5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aggrecanase-2; ADMP-2; ADAMTS5; ADAMTS-5; ADAM-TS5; ADAM-TS 5; ADAM-TS 11; A disintegrin and metalloproteinase with thrombospondinmotifs 5 Protein family NA Biochemical class Peptidase Function Cleaves aggrecan, a cartilage proteoglycan, and may be involved in its turnover. May play an important role in the destruction of aggrecan in arthritic diseases. May play a role in proteolytic processing mostly during the peri-implantation period. Related diseases Mucopolysaccharidosis 1H (MPS1H) [MIM:607014]: A severe form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1H usually present, within the first year of life, a combination of hepatosplenomegaly, skeletal deformities, corneal clouding and severe intellectual disability. Obstructive airways disease, respiratory infection and cardiac complications usually result in death before 10 years of age. {ECO:0000269|PubMed:10466419, ECO:0000269|PubMed:10735634, ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:1301941, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:19396826, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:24036510, ECO:0000269|PubMed:31194252, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:7951228, ECO:0000269|PubMed:8019563, ECO:0000269|PubMed:8328452, ECO:0000269|PubMed:8401515, ECO:0000269|Ref.20}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 1H/S (MPS1H/S) [MIM:607015]: A form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. MPS1H/S represents an intermediate phenotype of the MPS1 clinical spectrum. It is characterized by relatively little neurological involvement, but most of the somatic symptoms described for severe MPS1 develop in the early to mid-teens, causing considerable loss of mobility. {ECO:0000269|PubMed:10466419, ECO:0000269|PubMed:10735634, ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:8401515}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 1S (MPS1S) [MIM:607016]: A mild form of mucopolysaccharidosis type 1, a rare lysosomal storage disease characterized by progressive physical deterioration with urinary excretion of dermatan sulfate and heparan sulfate. Patients with MPS1S may have little or no neurological involvement, normal stature and life span, but present development of joints stiffness, mild hepatosplenomegaly, aortic valve disease and corneal clouding. {ECO:0000269|PubMed:12559846, ECO:0000269|PubMed:15300847, ECO:0000269|PubMed:19396826, ECO:0000269|PubMed:21394825, ECO:0000269|PubMed:25256405, ECO:0000269|PubMed:7550232, ECO:0000269|PubMed:7550242, ECO:0000269|PubMed:8213840}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06837; DB03880; DB06945 Interacts with P13608 EC number EC 3.4.24.- Uniprot keywords 3D-structure; Cleavage on pair of basic residues; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 23997.8 Length 217 Aromaticity 0.06 Instability index 46.68 Isoelectric point 5.82 Charge (pH=7) -8.36 2D Binding mode Binding energy (Kcal/mol) -5.97  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SRARQVELLLVADASMARKYGRGLQHYLLTLASIANRLYSHASIENHIRLAVVKVVVLGDKDKSLEVSKNAATTLKNFCKWQHQHNQLGDDHEEHYDAAILFTREDLCGHHSCDTLGMADVGTICSPERSCAVIEDDGLHAAFTVAHEIGHLLGLSHDDSKFCEETFGSTEDKRLMSSILTSIDASKPWSKCTSATITEFLDDGHGNCLLDLPRKQI Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Ornithine delta-aminotransferase (OAT) | 2OAT | 4.37 | |
Target general information Gen name OAT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ornithine--oxo-acid aminotransferase; Ornithine aminotransferase, mitochondrial Protein family Class-III pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transaminase Function Catalyzes the transfer of the delta-amino group from L-ornithine. Related diseases Hyperornithinemia with gyrate atrophy of choroid and retina (HOGA) [MIM:258870]: A disorder clinically characterized by a triad of progressive chorioretinal degeneration, early cataract formation, and type II muscle fiber atrophy. Characteristic chorioretinal atrophy with progressive constriction of the visual fields leads to blindness at the latest during the sixth decade of life. Patients generally have normal intelligence. {ECO:0000269|PubMed:1612597, ECO:0000269|PubMed:1737786, ECO:0000269|PubMed:23076989, ECO:0000269|PubMed:2793865, ECO:0000269|PubMed:3375240, ECO:0000269|PubMed:7668253, ECO:0000269|PubMed:7887415}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02821; DB02054; DB00129; DB00114 Interacts with P05067 EC number EC 2.6.1.13 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminotransferase; Direct protein sequencing; Disease variant; Mitochondrion; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase; Transit peptide Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 44807.9 Length 404 Aromaticity 0.09 Instability index 26.67 Isoelectric point 5.72 Charge (pH=7) -6.54 2D Binding mode Binding energy (Kcal/mol) -5.96  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GPPTSDDIFEREYKYGAHNYHPLPVALERGKGIYLWDVEGRKYFDFLSSYSAVNQGHCHPKIVNALKSQVDKLTLTSRAFYNNVLGEYEEYITKLFNYHKVLPMNTGVEAGETACKLARKWGYTVKGIQKYKAKIVFAAGNFWGRTLSAISSSTDPTSYDGFGPFMPGFDIIPYNDLPALERALQDPNVAAFMVEPIQGEAGVVVPDPGYLMGVRELCTRHQVLFIADEIQTGLARTGRWLAVDYENVRPDIVLLGKALSGGLYPVSAVLCDDDIMLTIKPGEHGSTYGGNPLGCRVAIAALEVLEEENLAENADKLGIILRNELMKLPSDVVTAVRGKGLLNAIVIKETKDWDAWKVCLRLRDNGLLAKPTHGDIIRFAPPLVIKEDELRESIEIINKTILSF Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Tissue-type plasminogen activator (PLAT) | 1RTF | 4.37 | |
Target general information Gen name PLAT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms TPA; T-plasminogen activator; T-PA; Reteplase; Alteplase Protein family Peptidase S1 family Biochemical class Peptidase Function By controlling plasmin-mediated proteolysis, it plays an important role in tissue remodeling and degradation, in cell migration and many other physiopathological events. Plays a direct role in facilitating neuronal migration. Converts the abundant, but inactive, zymogen plasminogen to plasmin by hydrolyzing a single Arg-Val bond in plasminogen. Related diseases Increased activity of TPA results in increased fibrinolysis of fibrin blood clots that is associated with excessive bleeding. Defective release of TPA results in hypofibrinolysis that can lead to thrombosis or embolism. {ECO:0000269|PubMed:1762144}. Drugs (DrugBank ID) DB07684; DB00513; DB09228; DB09213; DB06404; DB01088; DB16701 Interacts with P05155 EC number EC 3.4.21.68 Uniprot keywords 3D-structure; Alternative splicing; Cleavage on pair of basic residues; Direct protein sequencing; Disulfide bond; EGF-like domain; Glycoprotein; Hydrolase; Kringle; Pharmaceutical; Plasminogen activation; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID B Molecular weight (Da) 26271.7 Length 234 Aromaticity 0.09 Instability index 40.44 Isoelectric point 5.83 Charge (pH=7) -5.54 2D Binding mode Binding energy (Kcal/mol) -5.96 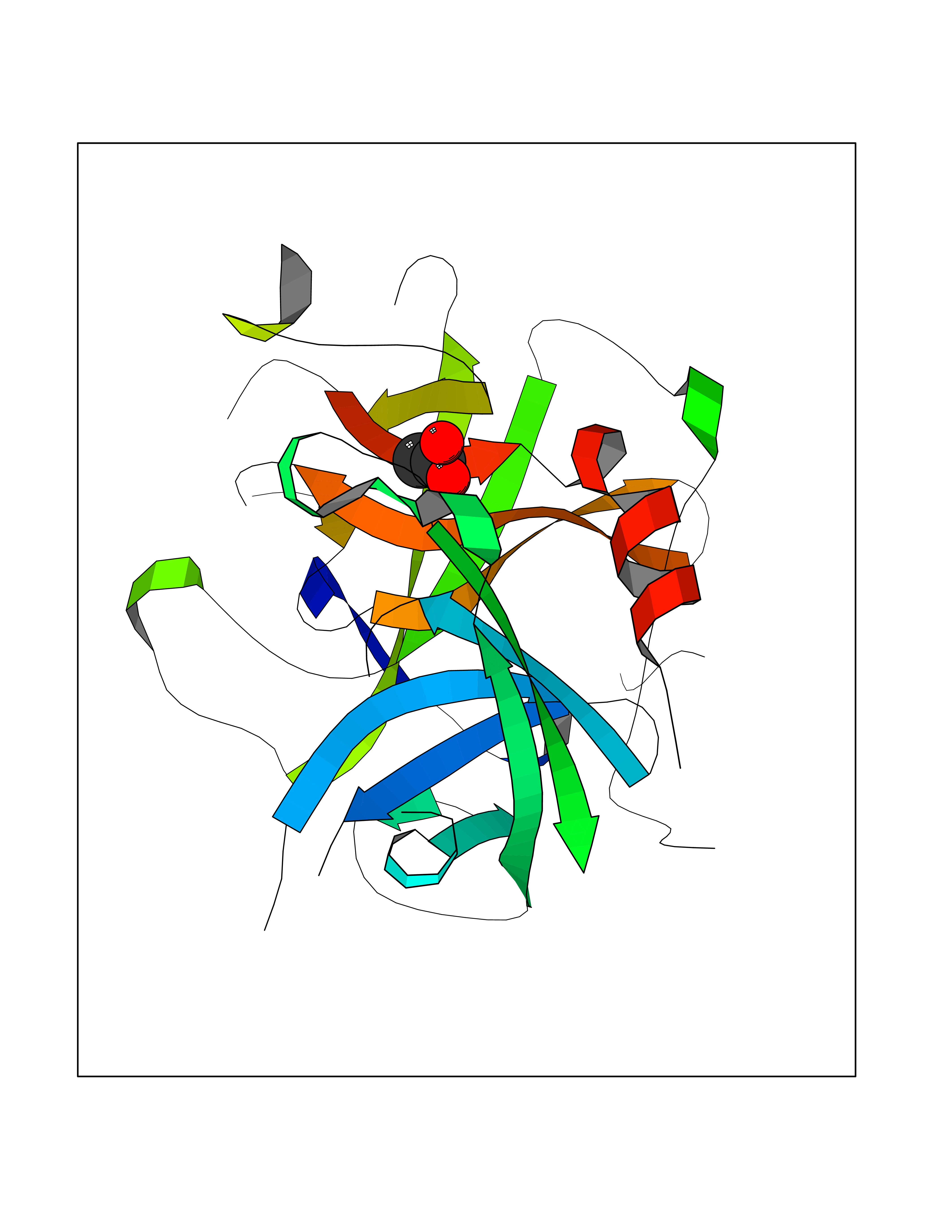 Molscript Map 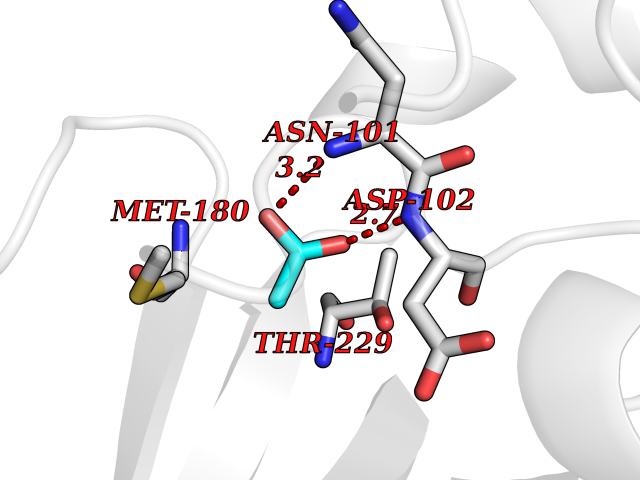 Pymol Map 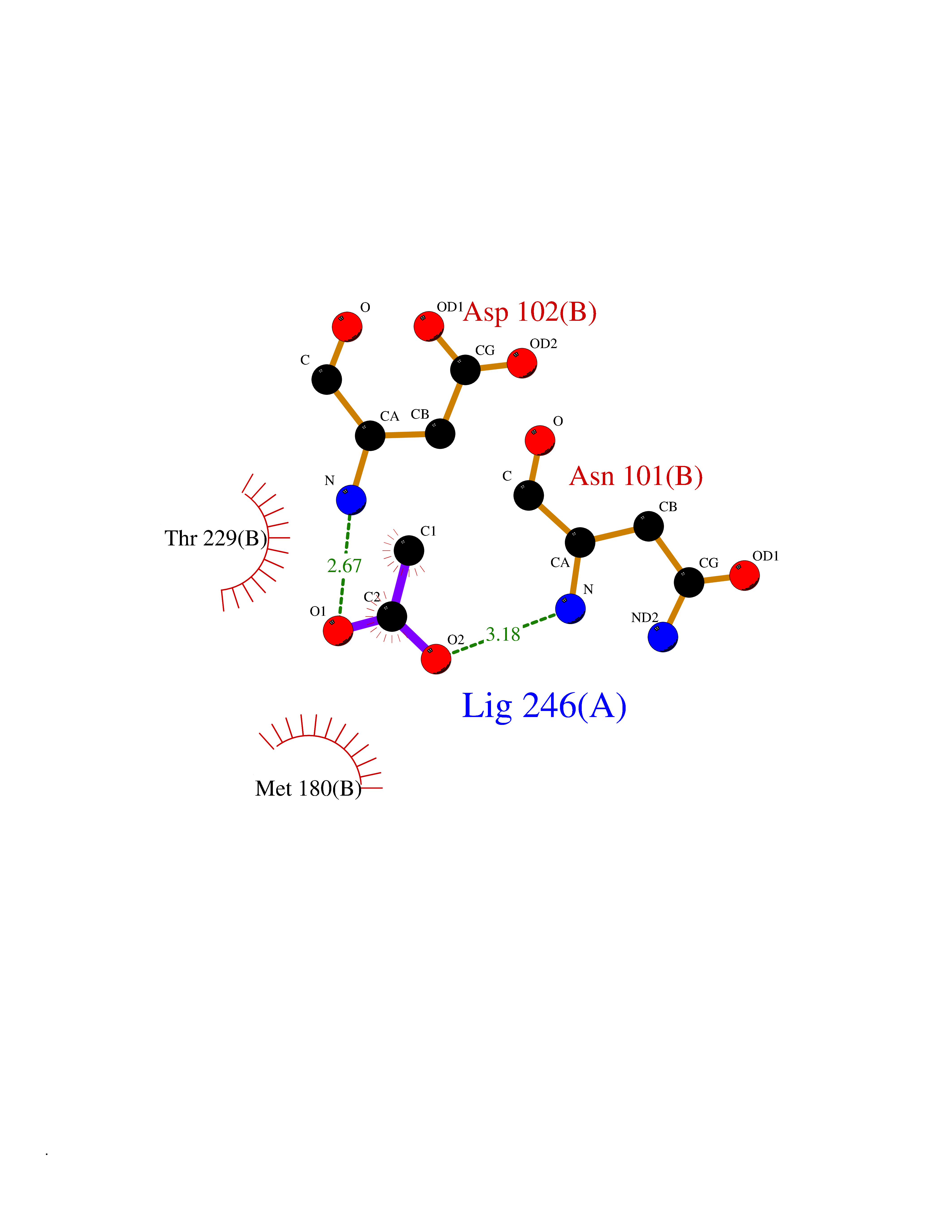 Ligplot Map 3D Binding mode Sequence CGLRQYIKGGLFADIASHPWQAAIFAKGERFLCGGILISSCWILSAAHCFPPHHLTVILGRTYRVVPGEEEQKFEVEKYIVHKEFDDDTYDNDIALLQLKRCAQESSVVRTVCLPPADLQLPDWTECELSGYGKHEALSPFYSERLKEAHVRLYPSSRCQHLLNRTVTDNMLCAGDNLHDACQGDSGGPLVCLNDGRMTLVGIISWGLGCQKDVPGVYTKVTNYLDWIRDNMRP Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Ornithine transcarbamylase (OTC) | 1OTH | 4.37 | |
Target general information Gen name OTC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms OTCase; Ornithine carbamoyltransferase, mitochondrial Protein family Aspartate/ornithine carbamoyltransferase superfamily, OTCase family Biochemical class NA Function Catalyzes the second step of the urea cycle, the condensation of carbamoyl phosphate with L-ornithine to form L-citrulline. The urea cycle ensures the detoxification of ammonia by converting it to urea for excretion. Related diseases Ornithine carbamoyltransferase deficiency (OTCD) [MIM:311250]: An X-linked disorder of the urea cycle which causes a form of hyperammonemia. Mutations with no residual enzyme activity are always expressed in hemizygote males by a very severe neonatal hyperammonemic coma that generally proves to be fatal. Heterozygous females are either asymptomatic or express orotic aciduria spontaneously or after protein intake. The disorder is treatable with supplemental dietary arginine and low protein diet. The arbitrary classification of patients into the 'neonatal' group (clinical hyperammonemia in the first few days of life) and 'late' onset (clinical presentation after the neonatal period) has been used to differentiate severe from mild forms. {ECO:0000269|PubMed:10070627, ECO:0000269|PubMed:10502831, ECO:0000269|PubMed:10737985, ECO:0000269|PubMed:11793483, ECO:0000269|PubMed:1480464, ECO:0000269|PubMed:1671317, ECO:0000269|PubMed:1721894, ECO:0000269|PubMed:2347583, ECO:0000269|PubMed:2474822, ECO:0000269|PubMed:2556444, ECO:0000269|PubMed:3170748, ECO:0000269|PubMed:7474905, ECO:0000269|PubMed:7951259, ECO:0000269|PubMed:8019569, ECO:0000269|PubMed:8081373, ECO:0000269|PubMed:8081398, ECO:0000269|PubMed:8099056, ECO:0000269|PubMed:8112735, ECO:0000269|PubMed:8530002, ECO:0000269|PubMed:8807340, ECO:0000269|PubMed:8830175, ECO:0000269|PubMed:8956038, ECO:0000269|PubMed:8956045, ECO:0000269|PubMed:9065786, ECO:0000269|PubMed:9143919, ECO:0000269|PubMed:9266388, ECO:0000269|PubMed:9286441, ECO:0000269|PubMed:9452024, ECO:0000269|PubMed:9452049, ECO:0000269|PubMed:9452065, ECO:0000269|Ref.32, ECO:0000269|Ref.43}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00155; DB02011; DB04185; DB00129 Interacts with NA EC number EC 2.1.3.3 Uniprot keywords 3D-structure; Acetylation; Amino-acid biosynthesis; Arginine biosynthesis; Disease variant; Mitochondrion; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Transit peptide; Urea cycle Protein physicochemical properties Chain ID A Molecular weight (Da) 36060.2 Length 321 Aromaticity 0.08 Instability index 36.44 Isoelectric point 7.87 Charge (pH=7) 1.48 2D Binding mode Binding energy (Kcal/mol) -5.96 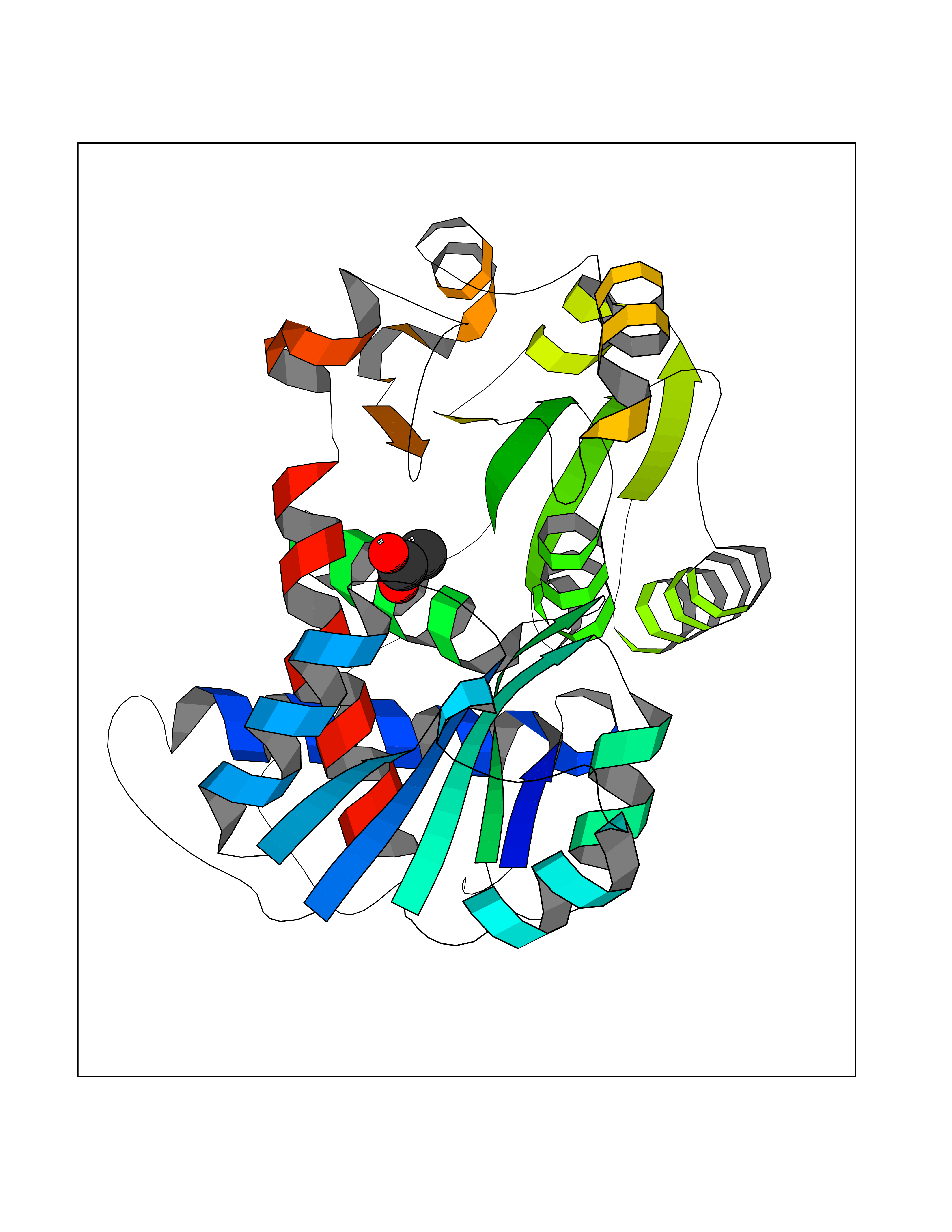 Molscript Map 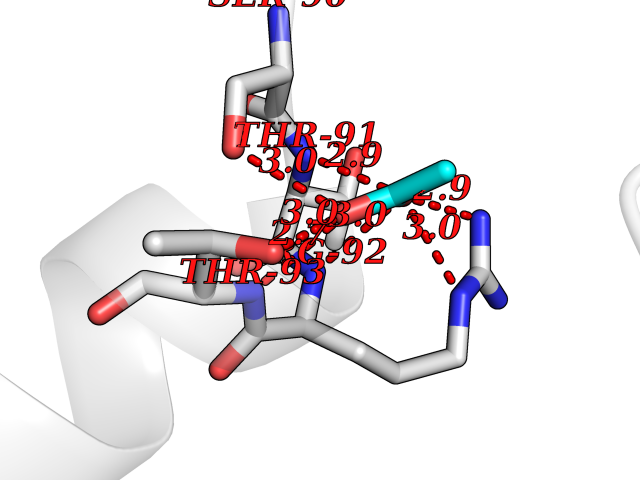 Pymol Map 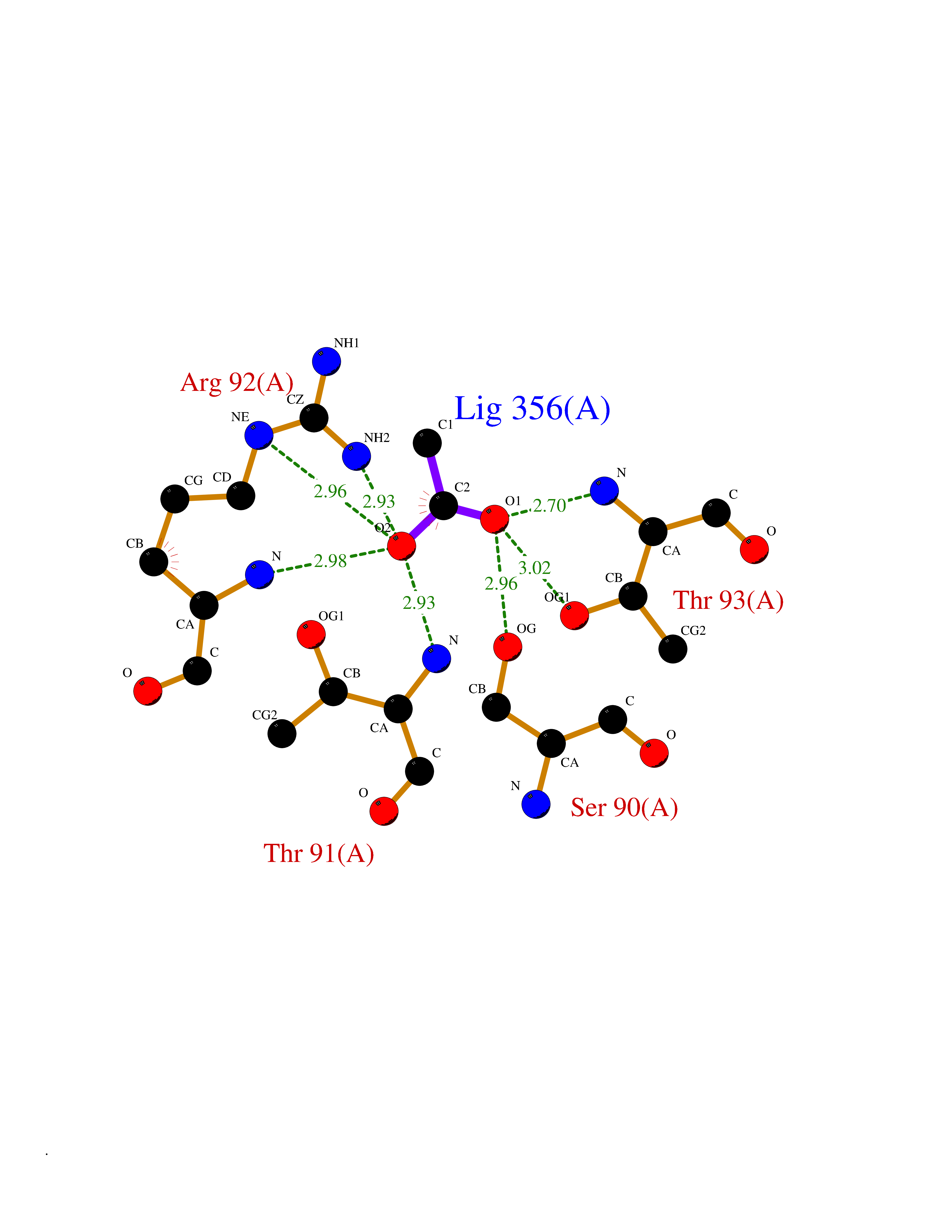 Ligplot Map 3D Binding mode Sequence KVQLKGRDLLTLKNFTGEEIKYMLWLSADLKFRIKQKGEYLPLLQGKSLGMIFEKRSTRTRLSTETGFALLGGHPCFLTTQDIHLGVNESLTDTARVLSSMADAVLARVYKQSDLDTLAKEASIPIINGLSDLYHPIQILADYLTLQEHYSSLKGLTLSWIGDGNNILHSIMMSAAKFGMHLQAATPKGYEPDASVTKLAEQYAKENGTKLLLTNDPLEAAHGGNVLITDTWISMGREEEKKKRLQAFQGYQVTMKTAKVAASDWTFLHCLPRKPEEVDDEVFYSPRSLVFPEAENRKWTIMAVMVSLLTDYSPQLQKPKF Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Exotoxin A | 1XK9 | 4.37 | |
Target general information Gen name eta Organism Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Uniprot ID TTD ID NA Synonyms PA1148 Protein family NA Biochemical class Transferase Function NAD+-diphthamide ADP-ribosyltransferase activity. Related diseases A chromosomal aberration involving TOP1 is found in a form of therapy-related myelodysplastic syndrome. Translocation t(11;20)(p15;q11) with NUP98. {ECO:0000269|PubMed:10556215}. Drugs (DrugBank ID) DB02701; DB08348 Interacts with NA EC number 2.4.2.36 Uniprot keywords 3D-structure; Direct protein sequencing; Disulfide bond; Glycosyltransferase; NAD; Nucleotidyltransferase; Reference proteome; Signal; Toxin; Transferase; Virulence Protein physicochemical properties Chain ID A,B Molecular weight (Da) 43735.4 Length 402 Aromaticity 0.09 Instability index 39.34 Isoelectric point 4.92 Charge (pH=7) -15.55 2D Binding mode Binding energy (Kcal/mol) -5.96  Molscript Map 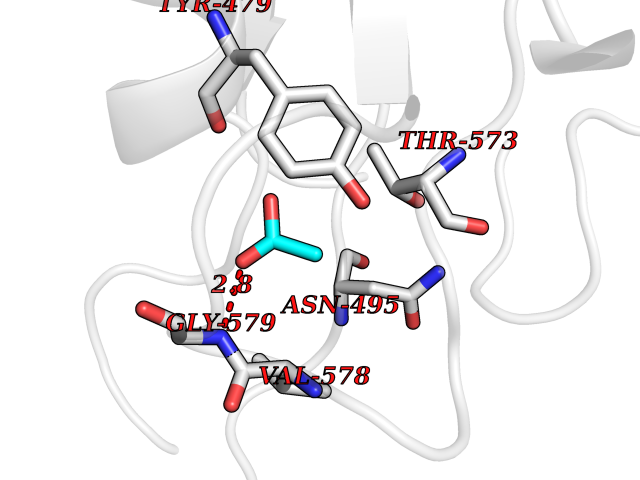 Pymol Map  Ligplot Map 3D Binding mode Sequence EFLGDGGDVSFSTRGTQNWTVERLLQAHRQLEERGYVFVGYHGTFLEAAQSIVFGGVRARSQDLDAIWRGFYIAGDPALAYGYAQDQEPDARGRIRNGALLRVYVPRSSLPGFYRTSLTLAAPEAAGEVERLIGHPLPLRLDAITGPEEEGGRLETILGWPLAERTVVIPSAIPTDPRNVGGDLDPSSIPDKEQAISALPDYASEFLGDGGDVSFSTRGTQNWTVERLLQAHRQLEERGYVFVGYHGTFLEAAQSIVFGGVRARIWRGFYIAGDPALAYGYAQDQEPDARGRIRNGALLRVYVPRSSLPGFYRTSLTLAAPEAAGEVERLIGHPLPLRLDAITGPEEEGGRLETILGWPLAERTVVIPSAIPTDPRNVGGDLDPSSIPDKEQAISALPDYAS Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Aldosterone synthase (CYP11B2) | 4DVQ | 4.37 | |
Target general information Gen name CYP11B2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Steroid 18hydroxylase; Cytochrome P450C18; Cytochrome P450Aldo; Cytochrome P450 11B2, mitochondrial; CYPXIB2; CYP11B2; Aldosteronesynthesizing enzyme; ALDOS Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function Preferentially catalyzes the conversion of 11- deoxycorticosterone to aldosterone via corticosterone and 18- hydroxycorticosterone. Related diseases Corticosterone methyloxidase 1 deficiency (CMO-1 deficiency) [MIM:203400]: Autosomal recessive disorder of aldosterone biosynthesis. There are two biochemically different forms of selective aldosterone deficiency be termed corticosterone methyloxidase (CMO) deficiency type 1 and type 2. In CMO-1 deficiency, aldosterone is undetectable in plasma, while its immediate precursor, 18-hydroxycorticosterone, is low or normal. {ECO:0000269|PubMed:11238478, ECO:0000269|PubMed:8439335, ECO:0000269|PubMed:9177280}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Corticosterone methyloxidase 2 deficiency (CMO-2 deficiency) [MIM:610600]: Autosomal recessive disorder of aldosterone biosynthesis. In CMO-2 deficiency, aldosterone can be low or normal, but at the expense of increased secretion of 18-hydroxycorticosterone. Consequently, patients have a greatly increased ratio of 18-hydroxycorticosterone to aldosterone and a low ratio of corticosterone to 18-hydroxycorticosterone in serum. {ECO:0000269|PubMed:12788848, ECO:0000269|PubMed:1346492, ECO:0000269|PubMed:1594605, ECO:0000269|PubMed:9625333, ECO:0000269|PubMed:9814506}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04630; DB00700; DB00292; DB00741; DB14539; DB14540; DB14543; DB14545; DB14544; DB05667; DB01011; DB01388; DB11837; DB00421; DB06281 Interacts with NA EC number EC 1.14.15.4 Uniprot keywords 3D-structure; Disease variant; Heme; Iron; Lipid metabolism; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Steroidogenesis; Transit peptide Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H,I,J,K,L Molecular weight (Da) 52334.4 Length 454 Aromaticity 0.1 Instability index 44.09 Isoelectric point 8.81 Charge (pH=7) 6.34 2D Binding mode Binding energy (Kcal/mol) -5.96  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LPFEAMPQHPGNRWLRLLQIWREQGYEHLHLEMHQTFQELGPIFRYPRMVCVMLPEDVEKLQQVDSLHPCRMILEPWVAYRQHRGHKCGVFLLNGPEWRFNRLRLNPDVLSPKAVQRFLPMVDAVARDFSQALKKKVLQNARGSLTLDVQPSIFHYTIEASNLALFGERLGLVGHSPSSASLNFLHALEVMFKSTVQLMFMPRSLSRWISPKVWKEHFEAWDCIFQYGDNCIQKIYQELAFNRPQHYTGIVAELLLKAELSLEAIKANSMELTAGSVDTTAFPLLMTLFELARNPDVQQILRQESLAAAASISEHPQKATTELPLLRAALKETLRLYPVGLFLERVVSSDLVLQNYHIPAGTLVQVFLYSLGRNAALFPRPERYNPQRWLDHVPFGFGMRQCLGRRLAEAEMLLLLHHVLKHFLVETLTQEDIKMVYSFILRPGTSPLLTFRAI Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Liver carboxylesterase (CES1) | 2H7C | 4.37 | |
Target general information Gen name CES1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine esterase 1; Monocyte/macrophage serine esterase; Human carboxylesterase 1; HMSE; HCE1; CES1; Brain carboxylesterase hBr1; Acyl coenzyme A:cholesterol acyltransferase Protein family Type-B carboxylesterase/lipase family Biochemical class Carboxylic ester hydrolase Function Involved in the detoxification of xenobiotics and in the activation of ester and amide prodrugs. Hydrolyzes aromatic and aliphatic esters, but has no catalytic activity toward amides or a fatty acyl coa ester. Related diseases Protoporphyria, erythropoietic, 1 (EPP1) [MIM:177000]: An autosomal recessive form of porphyria with onset usually before age 10 years. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. Erythropoietic protoporphyria is marked by excessive protoporphyrin in erythrocytes, plasma, liver and feces, and by widely varying photosensitive skin changes ranging from a burning or pruritic sensation to erythema, edema and wheals. {ECO:0000269|PubMed:10942404, ECO:0000269|PubMed:11375302, ECO:0000269|PubMed:12063482, ECO:0000269|PubMed:12601550, ECO:0000269|PubMed:1376018, ECO:0000269|PubMed:15286165, ECO:0000269|PubMed:17196862, ECO:0000269|PubMed:1755842, ECO:0000269|PubMed:7910885, ECO:0000269|PubMed:8757534, ECO:0000269|PubMed:9211198, ECO:0000269|PubMed:9585598, ECO:0000269|PubMed:9740232}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07821; DB08224; DB03056; DB06442; DB01086; DB09061; DB14737; DB01101; DB02659; DB01410; DB00758; DB00907; DB04838; DB06695; DB00647; DB01452; DB00470; DB01039; DB14845; DB00875; DB11181; DB02161; DB00328; DB00762; DB00583; DB14009; DB00454; DB06693; DB00688; DB03721; DB01183; DB00198; DB09269; DB01599; DB09342; DB00881; DB14761; DB12404; DB06201; DB11362; DB00641; DB00382; DB00675; DB12095; DB09299; DB04795; DB00519; DB16349 Interacts with Q8IVF2-3; A8MQ03; Q15125; Q5T7V8; Q5T749; Q13113; Q9NRQ2; Q9NR31; O95231 EC number EC 3.1.1.1 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Direct protein sequencing; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Hydrolase; Lipid droplet; Lipid metabolism; Phosphoprotein; Proteomics identification; Reference proteome; Serine esterase; Signal Protein physicochemical properties Chain ID A,B,C,D,E,F Molecular weight (Da) 58522.7 Length 531 Aromaticity 0.09 Instability index 35.86 Isoelectric point 6.05 Charge (pH=7) -5.56 2D Binding mode Binding energy (Kcal/mol) -5.96 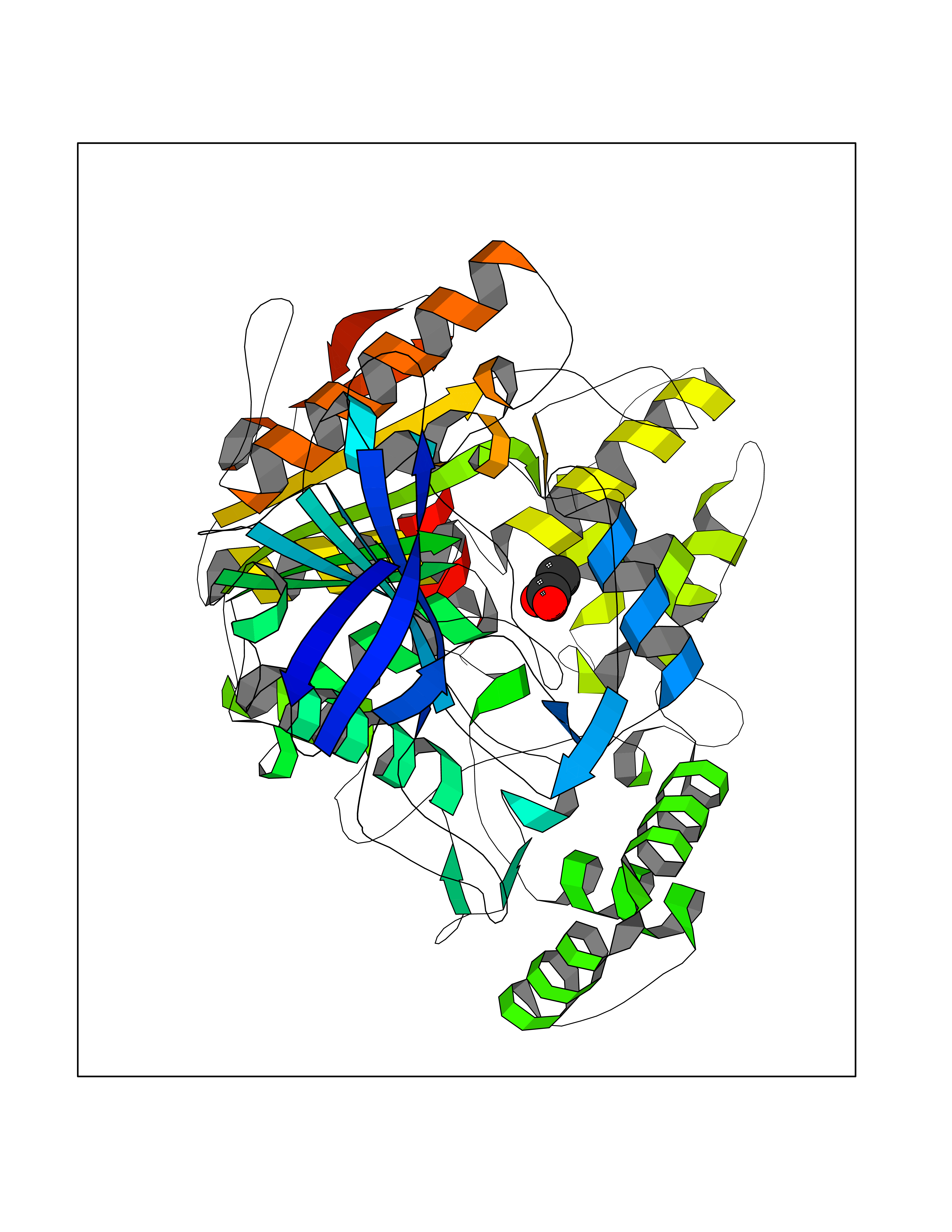 Molscript Map 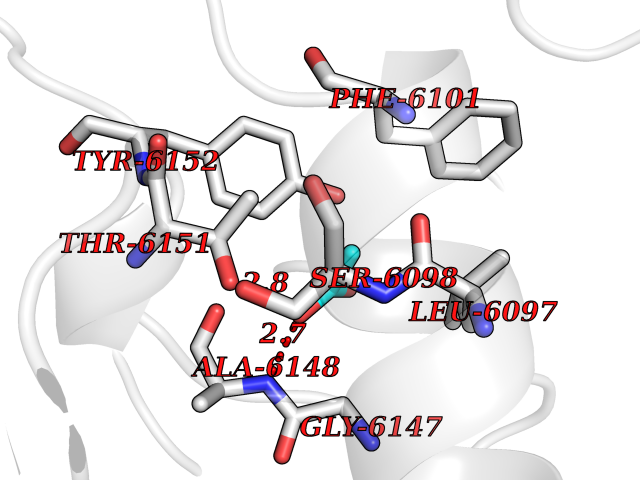 Pymol Map 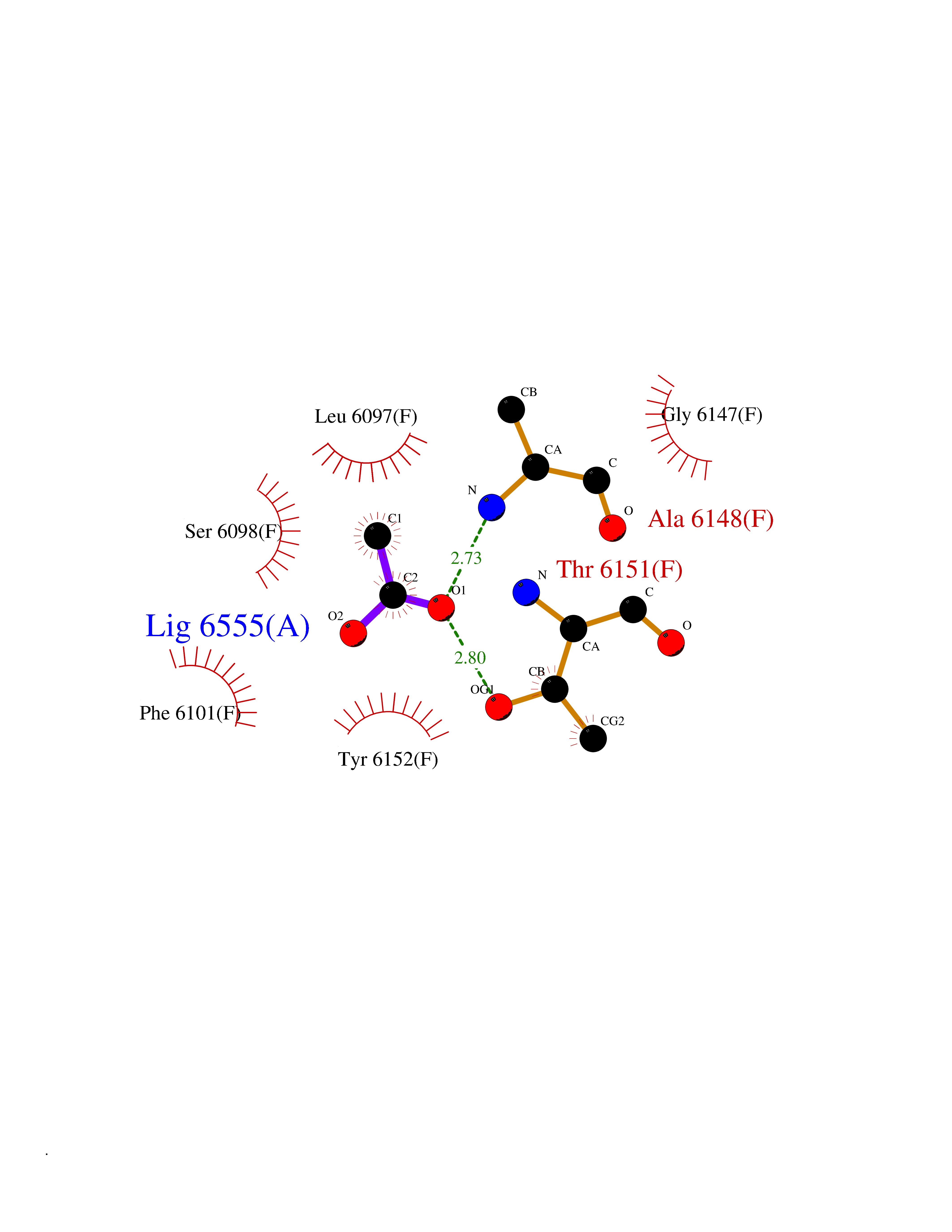 Ligplot Map 3D Binding mode Sequence SPPVVDTVHGKVLGKFVSLEGFAQPVAIFLGIPFAKPPLGPLRFTPPQPAEPWSFVKNATSYPPMCTQDPKAGQLLSELFTNRKENIPLKLSEDCLYLNIYTPADLTKKNRLPVMVWIHGGGLMVGAASTYDGLALAAHENVVVVTIQYRLGIWGFFSTGDEHSRGNWGHLDQVAALRWVQDNIASFGGNPGSVTIFGESAGGESVSVLVLSPLAKNLFHRAISESGVALTSVLVKKGDVKPLAEQIAITAGCKTTTSAVMVHCLRQKTEEELLETTLKMKFLSLDLQGDPRESQPLLGTVIDGMLLLKTPEELQAERNFHTVPYMVGINKQEFGWLIPMLMSYPLSEGQLDQKTAMSLLWKSYPLVCIAKELIPEATEKYLGGTDDTVKKKDLFLDLIADVMFGVPSVIVARNHRDAGAPTYMYEFQYRPSFSSDMKPKTVIGDHGDELFSVFGAPFLKEGASEEEIRLSKMVMKFWANFARNGNPNGEGLPHWPEYNQKEGYLQIGANTQAAQKLKDKEVAFWTNLFAK Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Multifunctional protein ADE2 | 2H31 | 4.37 | |
Target general information Gen name PAICS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ADE2;PAIS;AIRC Protein family SAICAR synthetase family; AIR carboxylase family, Class II subfamily Biochemical class Ligase Function ATP binding.Cadherin binding.Identical protein binding.Phosphoribosylaminoimidazole carboxylase activity.Phosphoribosylaminoimidazolesuccinocarboxamide synthase activity. Related diseases Phosphoribosylaminoimidazole carboxylase deficiency (PAICSD) [MIM:619859]: An autosomal recessive inborn error of purine metabolism, clinically characterized by multiple congenital anomalies and early neonatal death. {ECO:0000269|PubMed:31600779}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00128 Interacts with Q16543; P51116; Q969R5; Q8TBB1; Q6ZVK8; P22234; O75928-2; C9JJ79; Q9Y4B4; P78317; Q7KZS0; Q9UBW7 EC number 4.1.1.21; 6.3.2.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Decarboxylase; Direct protein sequencing; Disease variant; Ligase; Lyase; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Proteomics identification; Purine biosynthesis; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 41862.7 Length 386 Aromaticity 0.08 Instability index 34.99 Isoelectric point 7.02 Charge (pH=7) 0.04 2D Binding mode Binding energy (Kcal/mol) -5.96 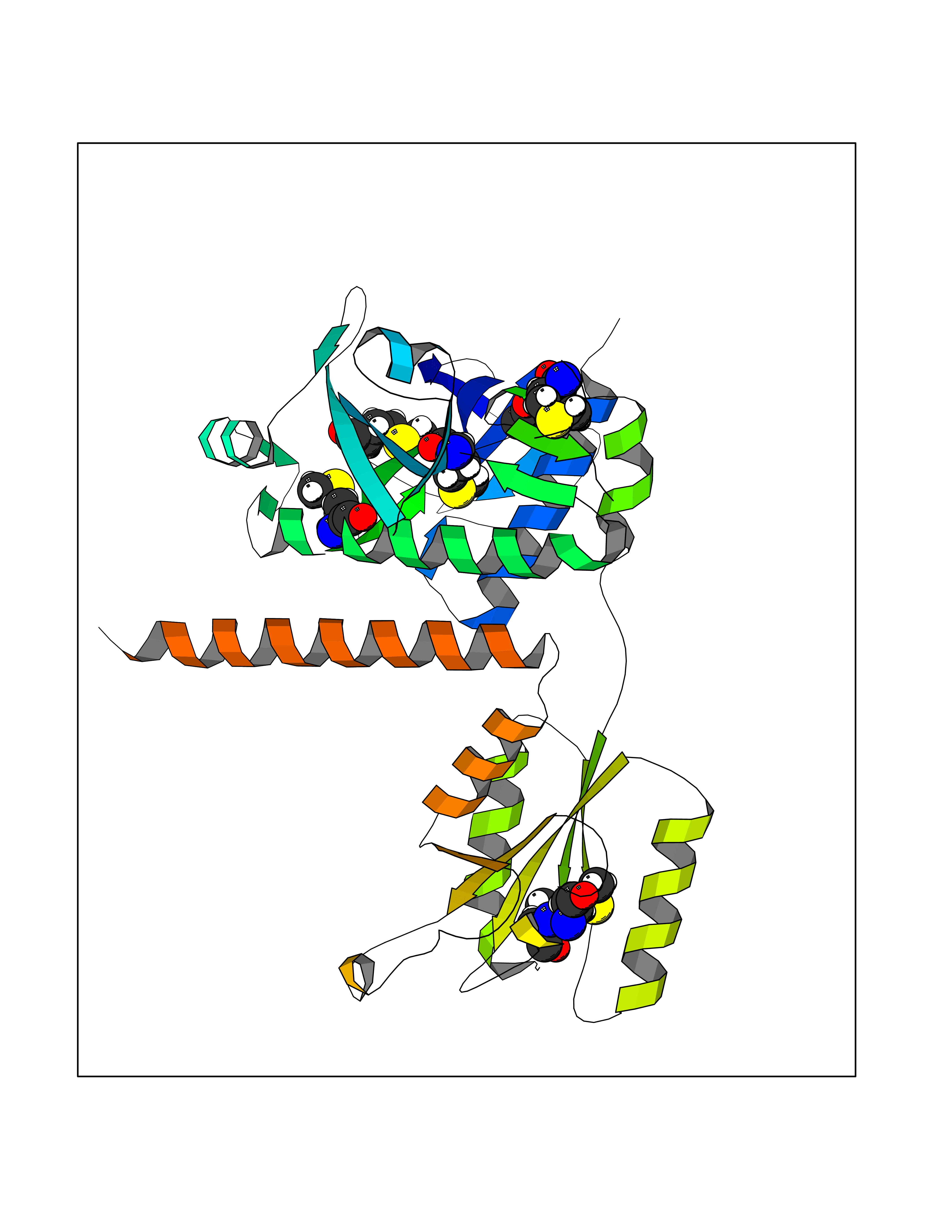 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LNIGKKLYEGKTKEVYELLDSPGKVLLQSKGKAAISNKITSCIFQLLQEAGIKTAFTRKCGETAFIAPQCEXIPIEWVCRRIATGSFLKRNPGVKEGYKFYPPKVELFFKDDANNDPQWSEEQLIAAKFCFAGLLIGQTEVDIXSHATQAIFEILEKSWLPQNCTLVDXKIEFGVDVTTKEIVLADVIDNDSWRLWPSGPEGLQXVKKNFEWVAERVELLLKSESQCRVVVLXGSTSDLGHCEKIKKACGNFGIPCELRVTSAHKGPDETLRIKAEYEGDGIPTVFVAVAGRSNGLGPVXSGNTAYPVISCPPLTPDWGVQDVWSSLRLPSGLGCSTVLSPEGSAQFAAQIFGLSNHLVWSKLRASILNTWISLKQADKKIRECNL Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | 4-hydroxyphenylpyruvate dioxygenase | 3ISQ | 4.37 | |
Target general information Gen name HPD Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PPD Protein family 4HPPD family Biochemical class Oxidoreductase Function 4-hydroxyphenylpyruvate dioxygenase activity.Metal ion binding. Related diseases Tyrosinemia 3 (TYRSN3) [MIM:276710]: An inborn error of metabolism characterized by elevations of tyrosine in the blood and urine, seizures and mild intellectual disability. {ECO:0000269|PubMed:10942115, ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hawkinsinuria (HWKS) [MIM:140350]: An inborn error of tyrosine metabolism characterized by failure to thrive, persistent metabolic acidosis, fine and sparse hair, and excretion of the unusual cyclic amino acid metabolite, hawkinsin, in the urine. {ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02850; DB00348 Interacts with NA EC number 1.13.11.27 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Dioxygenase; Disease variant; Endoplasmic reticulum; Golgi apparatus; Intellectual disability; Iron; Membrane; Metal-binding; Oxidoreductase; Phenylalanine catabolism; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Tyrosine catabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 43164.8 Length 376 Aromaticity 0.11 Instability index 32.38 Isoelectric point 6.73 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -5.96  Molscript Map 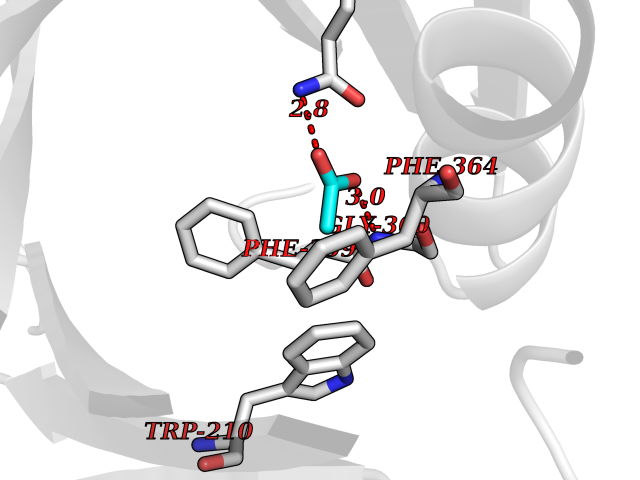 Pymol Map 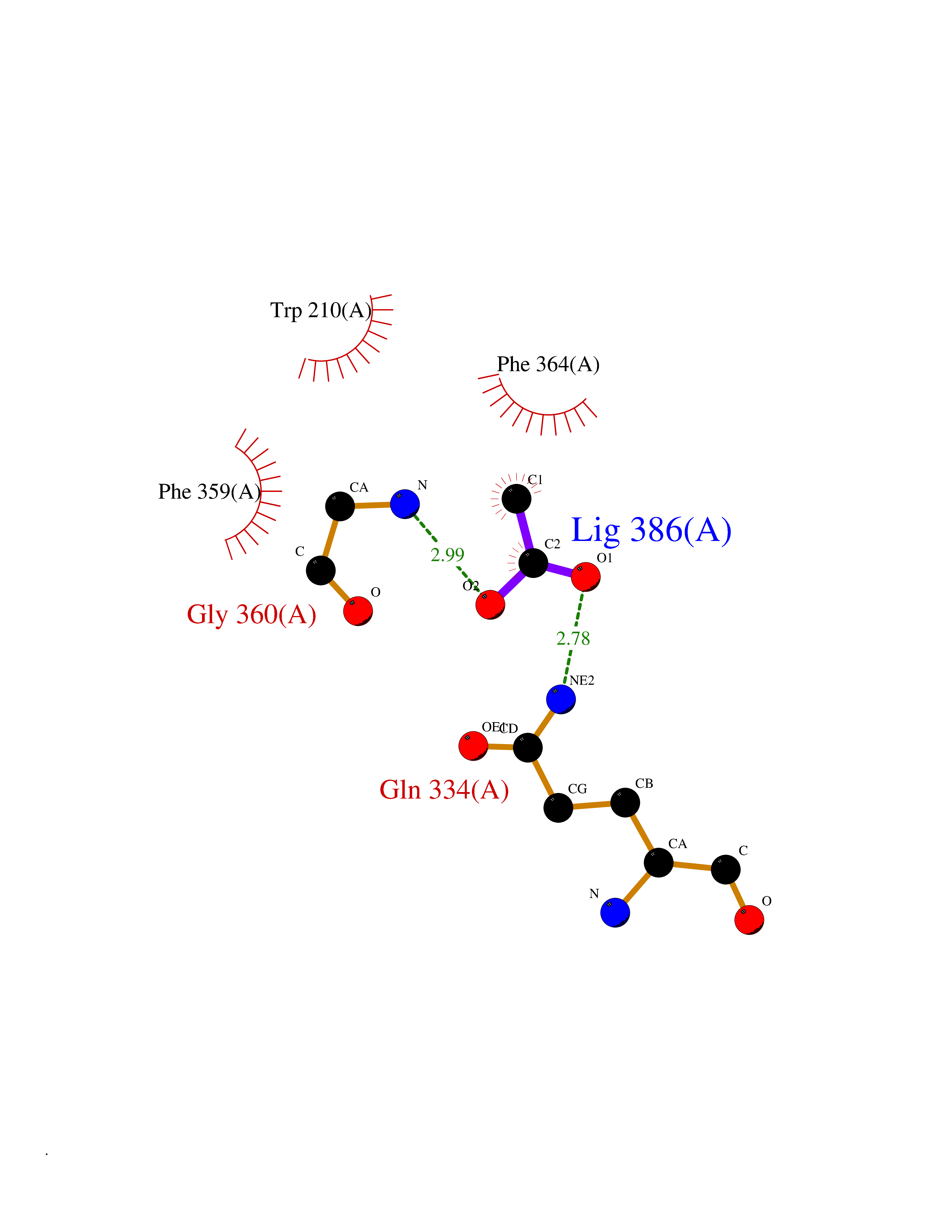 Ligplot Map 3D Binding mode Sequence AKPERGRFLHFHSVTFWVGNAKQAASFYCSKMGFEPLAYRGLETGSREVVSHVIKQGKIVFVLSSALNPWNKEMGDHLVKHGDGVKDIAFEVEDCDYIVQKARERGAKIMREPWVEQDKFGKVKFAVLQTYGDTTHTLVEKMNYIGQFLPGYEAPAFMDPLLPKLPKCSLEMIDHIVGNQPDQEMVSASEWYLKNLQFHRFWSVDDTQVHTEYSSLRSIVVANYEESIKMPINEPAPGKKKSQIQEYVDYNGGAGVQHIALKTEDIITAIRHLRERGLEFLSVPSTYYKQLREKLKTAKIKVKENIDALEELKILVDYDEKGYLLQIFTKPVQDRPTLFLEVIQRHNHQGFGAGNFNSLFKAFEEEQNLRGNLTNM Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Caspase-3 (CASP3) | 2XYG | 4.37 | |
Target general information Gen name CASP3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Yama protein; SREBP cleavage activity 1; SCA-1; Protein Yama; Cysteine protease CPP32; Caspase 3; CPP32; CPP-32; CASP-3; Apopain Protein family Peptidase C14A family Biochemical class Peptidase Function At the onset of apoptosis it proteolytically cleaves poly(ADP-ribose) polymerase (PARP) at a '216-Asp-|-Gly-217' bond. Cleaves and activates sterol regulatory element binding proteins (SREBPs) between the basic helix-loop-helix leucine zipper domain and the membrane attachment domain. Cleaves and activates caspase-6, -7 and -9. Involved in the cleavage of huntingtin. Triggers cell adhesion in sympathetic neurons through RET cleavage. Involved in the activation cascade of caspases responsible for apoptosis execution. Related diseases Smith-Kingsmore syndrome (SKS) [MIM:616638]: An autosomal dominant syndrome characterized by intellectual disability, macrocephaly, seizures, umbilical hernia, and facial dysmorphic features. {ECO:0000269|PubMed:25851998, ECO:0000269|PubMed:26542245, ECO:0000269|PubMed:27830187}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Focal cortical dysplasia 2 (FCORD2) [MIM:607341]: A form of focal cortical dysplasia, a malformation of cortical development that results in medically refractory epilepsy in the pediatric population and in adults. FCORD2 is a severe form, with onset usually in childhood, characterized by disrupted cortical lamination and specific cytological abnormalities. It is classified in 2 subtypes: type IIA characterized by dysmorphic neurons and lack of balloon cells; type IIB with dysmorphic neurons and balloon cells. {ECO:0000269|PubMed:25799227, ECO:0000269|PubMed:25878179, ECO:0000269|PubMed:26018084, ECO:0000269|PubMed:27830187}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08498; DB08497; DB08213; DB06862; DB08251; DB03124; DB08229; DB00945; DB05408; DB13751; DB06255; DB07696; DB01017; DB08499; DB12843; DB13048; DB00282; DB12709 Interacts with O43823; Q9Y243; P05067; P54252; P55212; P55211; Q14203-5; P42858; Q00987; O60551; P09874; Q5JUK2; P10599; Q9BYP7; P98170 EC number EC 3.4.22.56 Uniprot keywords 3D-structure; Acetylation; Apoptosis; Cytoplasm; Direct protein sequencing; Hydrolase; Phosphoprotein; Protease; Proteomics identification; Reference proteome; S-nitrosylation; Thiol protease; Ubl conjugation; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 27483.1 Length 239 Aromaticity 0.11 Instability index 38.09 Isoelectric point 8.39 Charge (pH=7) 3.03 2D Binding mode Binding energy (Kcal/mol) -5.95  Molscript Map 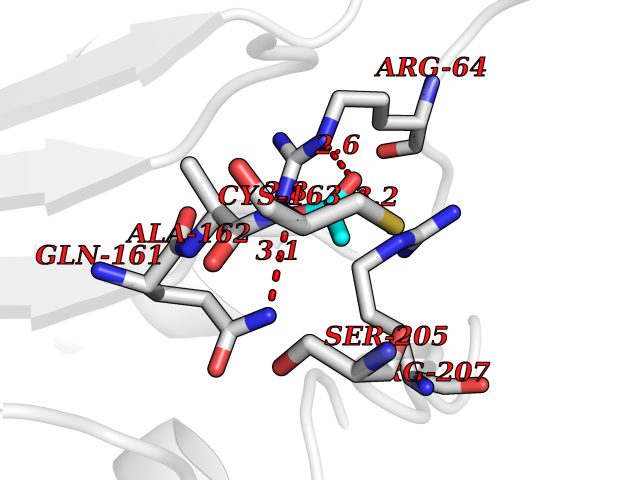 Pymol Map 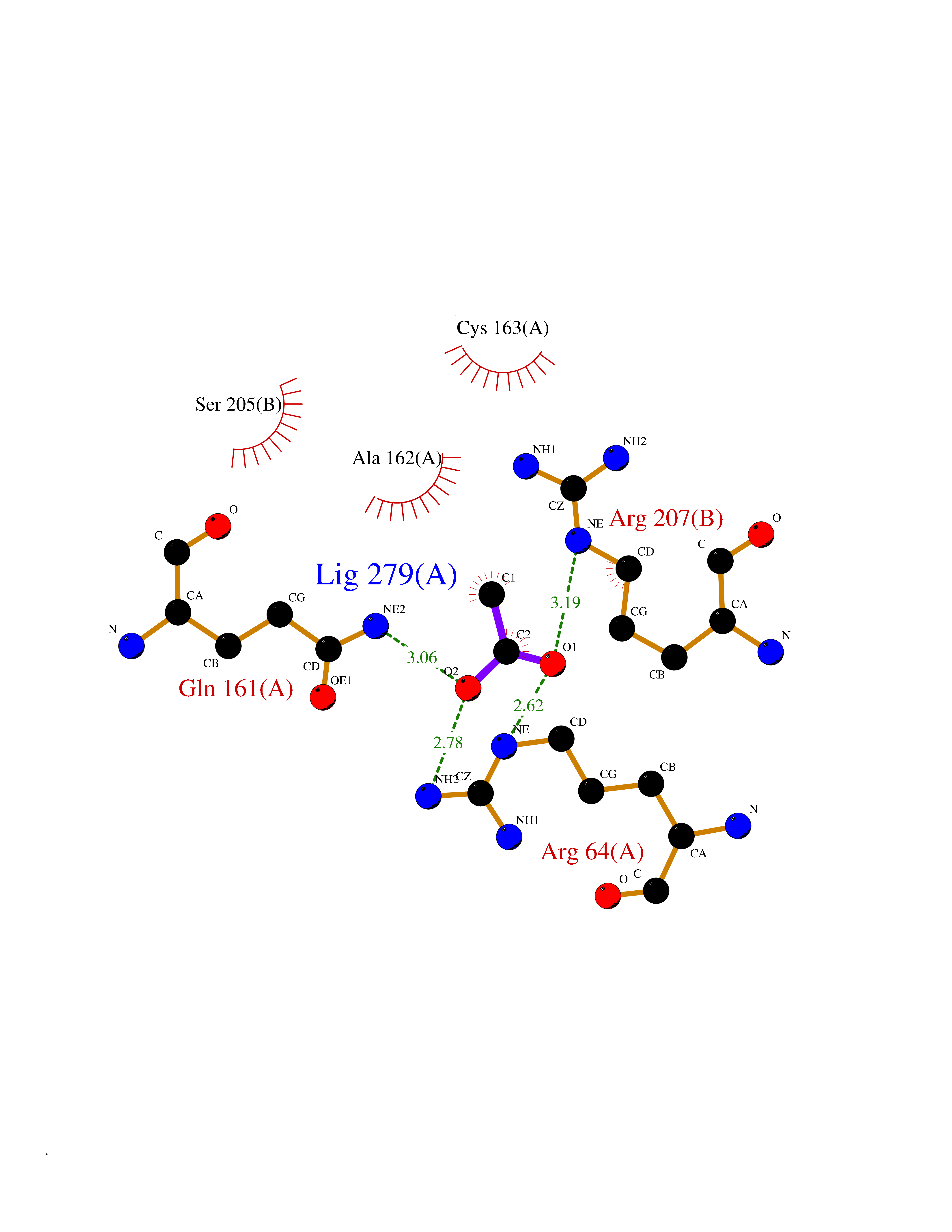 Ligplot Map 3D Binding mode Sequence SGISLDNSYKMDYPEMGLCIIINNKNFHKSTGMTSRSGTDVDAANLRETFRNLKYEVRNKNDLTREEIVELMRDVSKEDHSKRSSFVCVLLSHGEEGIIFGTNGPVDLKKITNFFRGDRCRSLTGKPKLFIIQACRGTELDCGIETHKIPVEADFLYAYSTAPGYYSWRNSKDGSWFIQSLCAMLKQYADKLEFMHILTRVNRKVATEFESFSFDATFHAKKQIPCIVSMLTKELYFYH Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Folic acid synthesis protein FOL1 | 2BMB | 4.37 | |
Target general information Gen name FOL1 Organism Saccharomyces cerevisiae (strain ATCC 204508 / S288c) (Baker's yeast) Uniprot ID TTD ID NA Synonyms N0848;YNL256W Protein family DHNA family; HPPK family; DHPS family Biochemical class Transferase Function 2-amino-4-hydroxy-6-hydroxymethyldihydropteridine diphosphokinase activity.7,8-dihydromonapterin aldolase activity.ATP binding.Dihydroneopterin aldolase activity.Dihydropteroate synthase activity.Kinase activity.Metal ion binding. Related diseases LIMK1 is located in the Williams-Beuren syndrome (WBS) critical region. WBS results from a hemizygous deletion of several genes on chromosome 7q11.23, thought to arise as a consequence of unequal crossing over between highly homologous low-copy repeat sequences flanking the deleted region. Drugs (DrugBank ID) DB00634 Interacts with NA EC number 2.5.1.15; 2.7.6.3; 4.1.2.25 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Folate biosynthesis; Kinase; Lyase; Magnesium; Membrane; Metal-binding; Mitochondrion; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 58205.2 Length 513 Aromaticity 0.08 Instability index 42.41 Isoelectric point 5.92 Charge (pH=7) -8.31 2D Binding mode Binding energy (Kcal/mol) -5.96 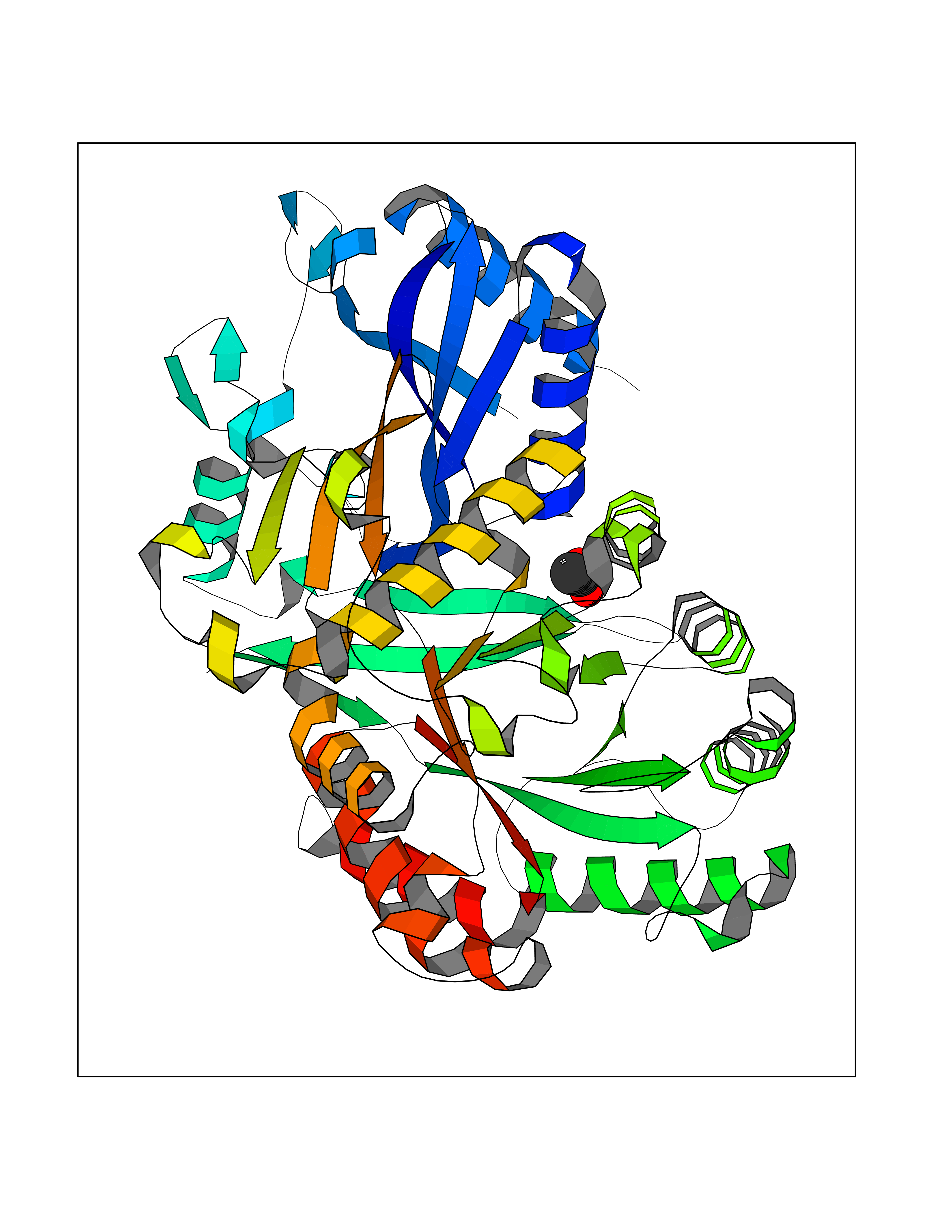 Molscript Map 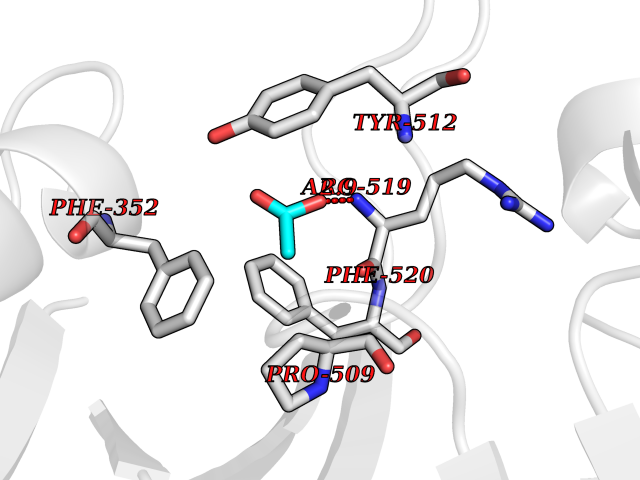 Pymol Map 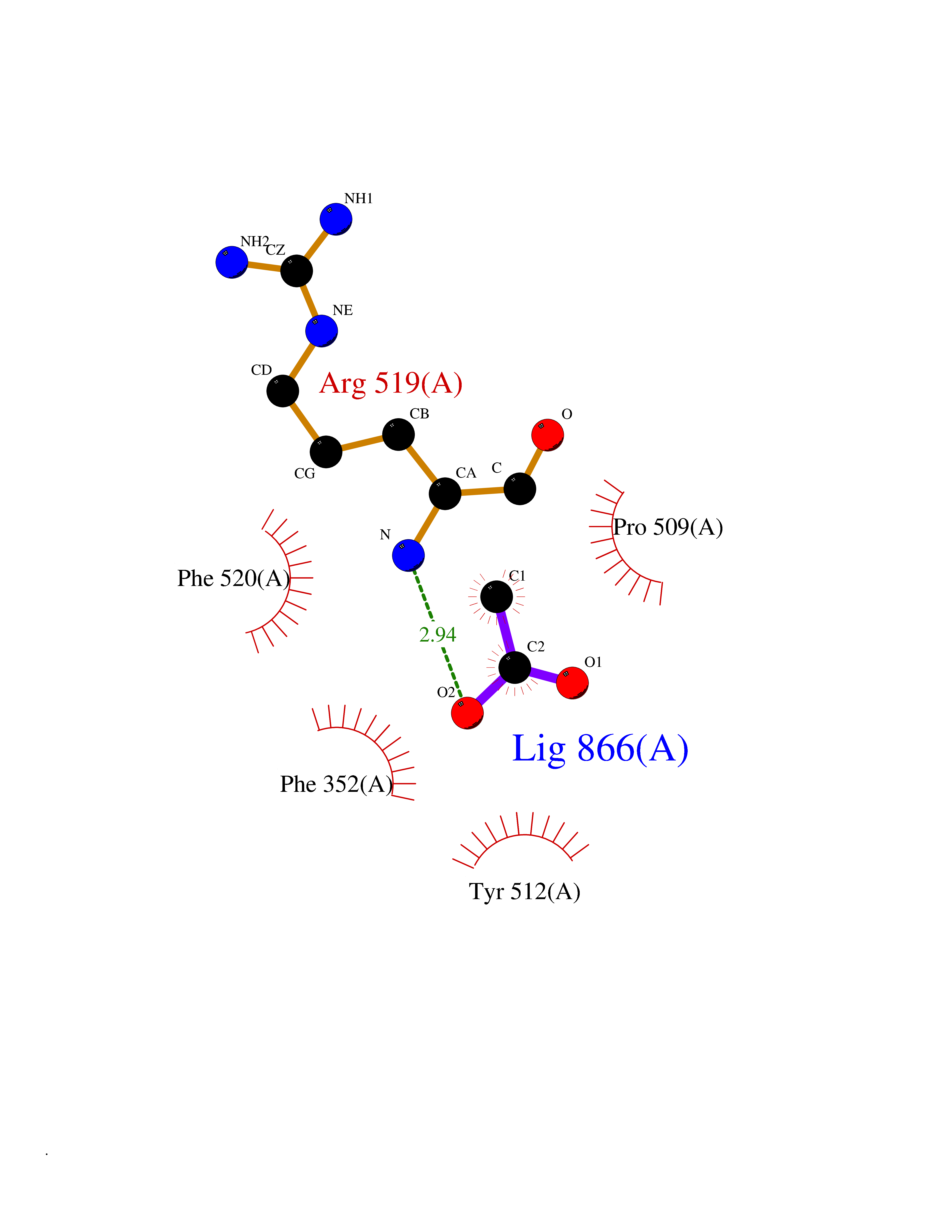 Ligplot Map 3D Binding mode Sequence SWKRAFLAFGSNIGDRFKHIQMALQLLSREKTVKLRNISSIFESEPMYFKDQTPFMNGCVEVETLLTPSELLKLCKKIEYEELQRTIDLDIVMFLNSAGEDIIVNEPDLNIPHPRMLERTFVLEPLCELISPVHLHPVTAEPIVDHLKQLYDKQHDEDTLWKLVPLPYRSGVEPRFLKFKTATKTNRITVSPTYIMAIFNATPDSFSDGGEHFADIESQLNDIIKLCKDALYLHESVIIDVGGCSTRPNSIQASEEEEIRRSIPLIKAIRESTELPQDKVILSIDTYRSNVAKEAIKVGVDIINDISGGLFDSNMFAVIAENPEICYILSHTRGDISTMNRLAHYENFALGDSIQQEFVHNTDIQQLDDLKDKTVLIRNVGQEIGERYIKAIDNGVKRWQILIDPGLGFAKTWKQNLQIIRHIPILKNYSFTMNSNNSQVYVNLRNMPVLLGPSRKKFIGHITKDVDAKQRDFATGAVVASCIGFGSDMVRVHDVKNCSKSIKLADAIYKGLE Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Dihydroorotate dehydrogenase (quinone), mitochondrial | 4CQ8 | 4.37 | |
Target general information Gen name PFF0160c Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID NA Synonyms NA Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class Oxidoreductase Function Dihydroorotate dehydrogenase activity. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42573.5 Length 378 Aromaticity 0.1 Instability index 36.63 Isoelectric point 8.21 Charge (pH=7) 3.17 2D Binding mode Binding energy (Kcal/mol) -5.97 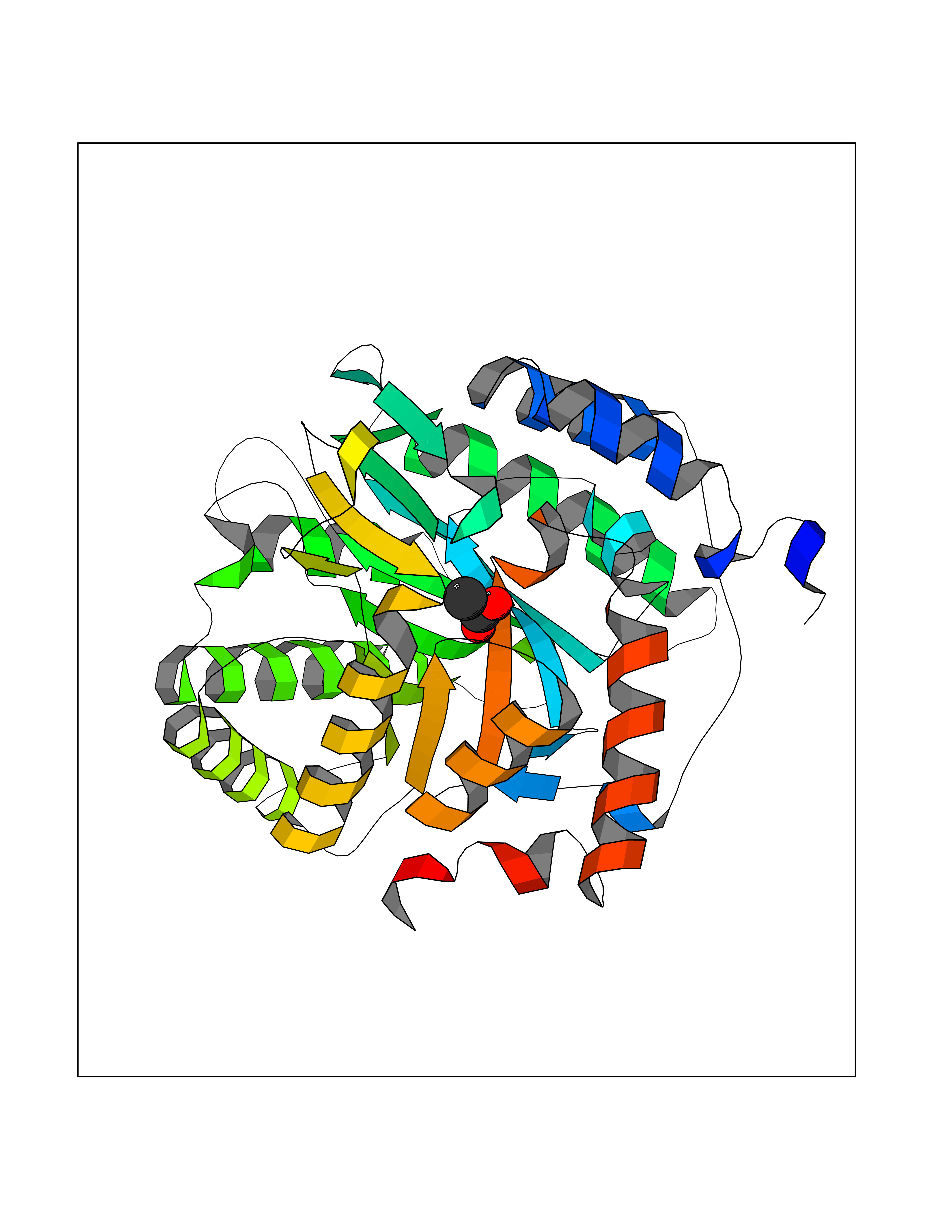 Molscript Map 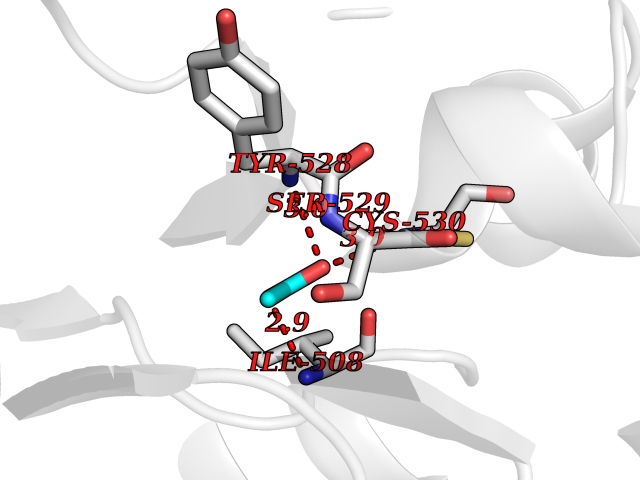 Pymol Map 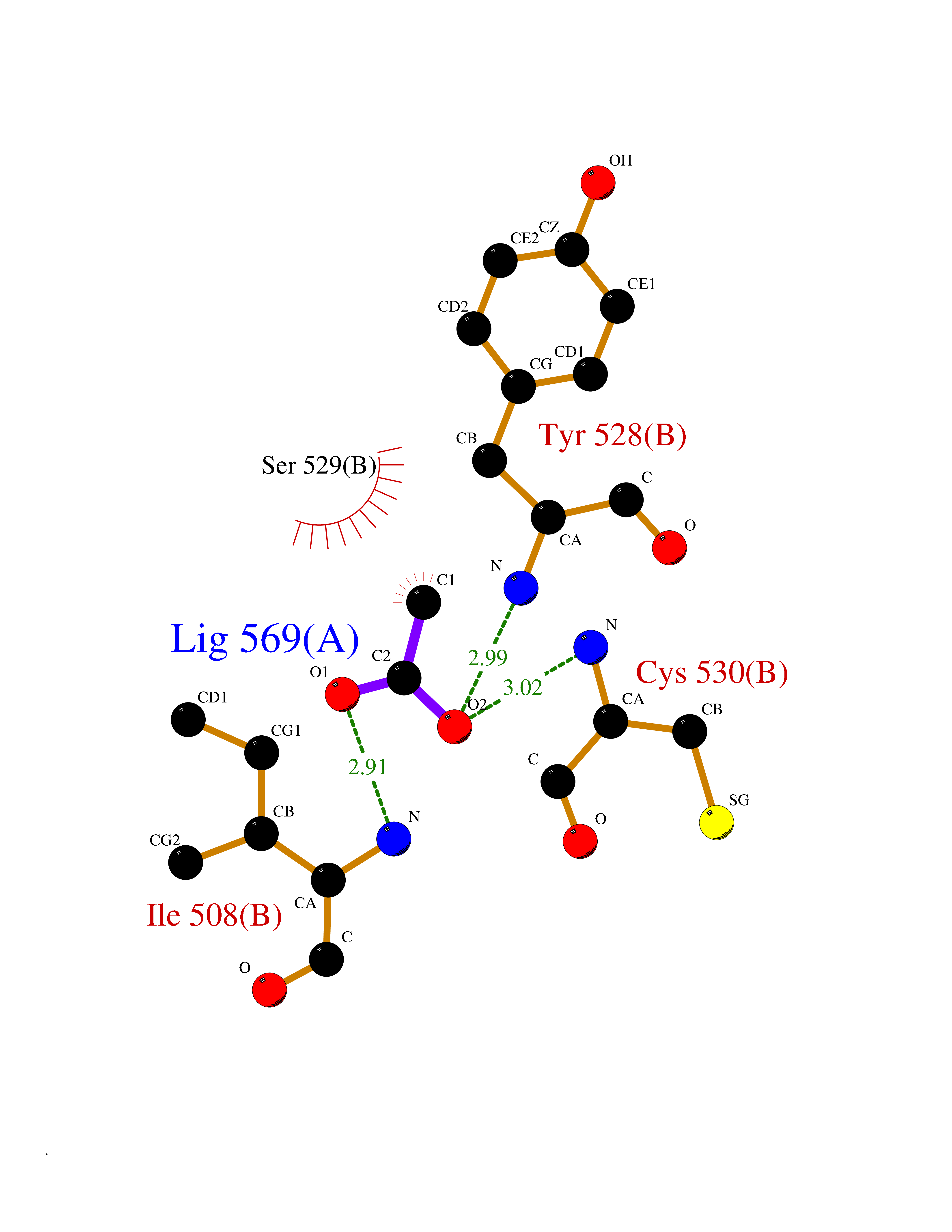 Ligplot Map 3D Binding mode Sequence ADPFESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEKNNFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKHS Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Histone deacetylase 2 (HDAC2) | 4LY1 | 4.37 | |
Target general information Gen name HDAC2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HD2 Protein family Histone deacetylase family, HD type 1 subfamily Biochemical class Carbon-nitrogen hydrolase Function Gives a tag for epigenetic repression and plays an important role in transcriptional regulation, cell cycle progression and developmental events. Histone deacetylases act via the formation of large multiprotein complexes. Forms transcriptional repressor complexes by associating with MAD, SIN3, YY1 and N-COR. Interacts in the late S-phase of DNA-replication with DNMT1 in the other transcriptional repressor complex composed of DNMT1, DMAP1, PCNA, CAF1. Deacetylates TSHZ3 and regulates its transcriptional repressor activity. Component of a RCOR/GFI/KDM1A/HDAC complex that suppresses, via histone deacetylase (HDAC) recruitment, a number of genes implicated in multilineage blood cell development. May be involved in the transcriptional repression of circadian target genes, such as PER1, mediated by CRY1 through histone deacetylation. Involved in MTA1-mediated transcriptional corepression of TFF1 and CDKN1A. Responsible for the deacetylation of lysine residues on the N-terminal part of the core histones (H2A, H2B, H3 and H4). Related diseases Ventricular tachycardia, catecholaminergic polymorphic, 1, with or without atrial dysfunction and/or dilated cardiomyopathy (CPVT1) [MIM:604772]: An arrhythmogenic disorder characterized by stress-induced, bidirectional ventricular tachycardia that may degenerate into cardiac arrest and cause sudden death. Patients present with recurrent syncope, seizures, or sudden death after physical activity or emotional stress. CPVT1 inheritance is autosomal dominant. {ECO:0000269|PubMed:11157710, ECO:0000269|PubMed:11159936, ECO:0000269|PubMed:11208676, ECO:0000269|PubMed:12093772, ECO:0000269|PubMed:12106942, ECO:0000269|PubMed:14571276, ECO:0000269|PubMed:15046072, ECO:0000269|PubMed:15046073, ECO:0000269|PubMed:15466642, ECO:0000269|PubMed:15544015, ECO:0000269|PubMed:16188589, ECO:0000269|PubMed:24793461, ECO:0000269|PubMed:25372681, ECO:0000269|PubMed:27733687}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ventricular arrhythmias due to cardiac ryanodine receptor calcium release deficiency syndrome (VACRDS) [MIM:115000]: An autosomal dominant arrhythmogenic disorder characterized by syncope, cardiac arrest and/or sudden unexpected death, often in association with physical exertion or acute emotional stress. Patients who survive manifest polymorphic ventricular tachycardia and ventricular fibrillation. Unlike typical catecholaminergic ventricular tachycardia, arrhythmias are not reproducible on exercise stress testing or adrenaline challenge. {ECO:0000269|PubMed:12093772, ECO:0000269|PubMed:17984046, ECO:0000269|PubMed:33536282}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12565; DB01223; DB01076; DB05015; DB01262; DB11841; DB01095; DB12645; DB00227; DB11830; DB01303; DB06603; DB06819; DB05223; DB00175; DB03766; DB12847; DB06176; DB00641; DB00277; DB09091; DB00313; DB02546 Interacts with Q9C0K0; Q9HCU9; P68400; Q9UER7; P51610; Q13547; Q9UIS9; Q13330; P01106; P06748; P48382; Q96ST3; O95863; Q9HD15; O43463; Q9H3M7; Q92618; Q17R98; Q2HR82; PRO_0000449623 [P0DTD1] EC number EC 3.5.1.98 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Biological rhythms; Chromatin regulator; Cytoplasm; Hydrolase; Isopeptide bond; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; S-nitrosylation; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 42020.5 Length 366 Aromaticity 0.13 Instability index 29.52 Isoelectric point 6.52 Charge (pH=7) -2.16 2D Binding mode Binding energy (Kcal/mol) -5.96  Molscript Map 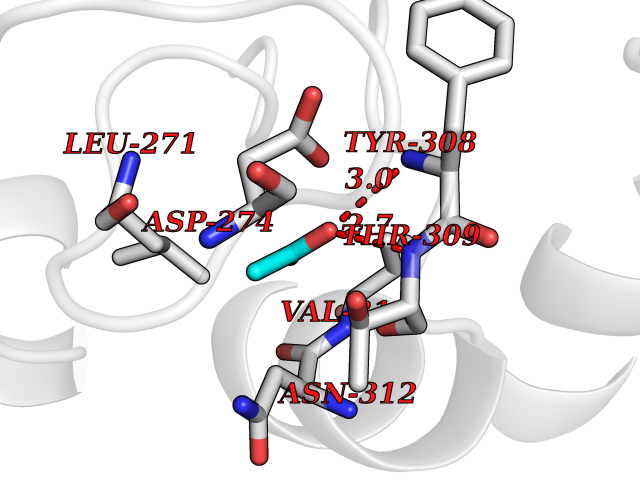 Pymol Map 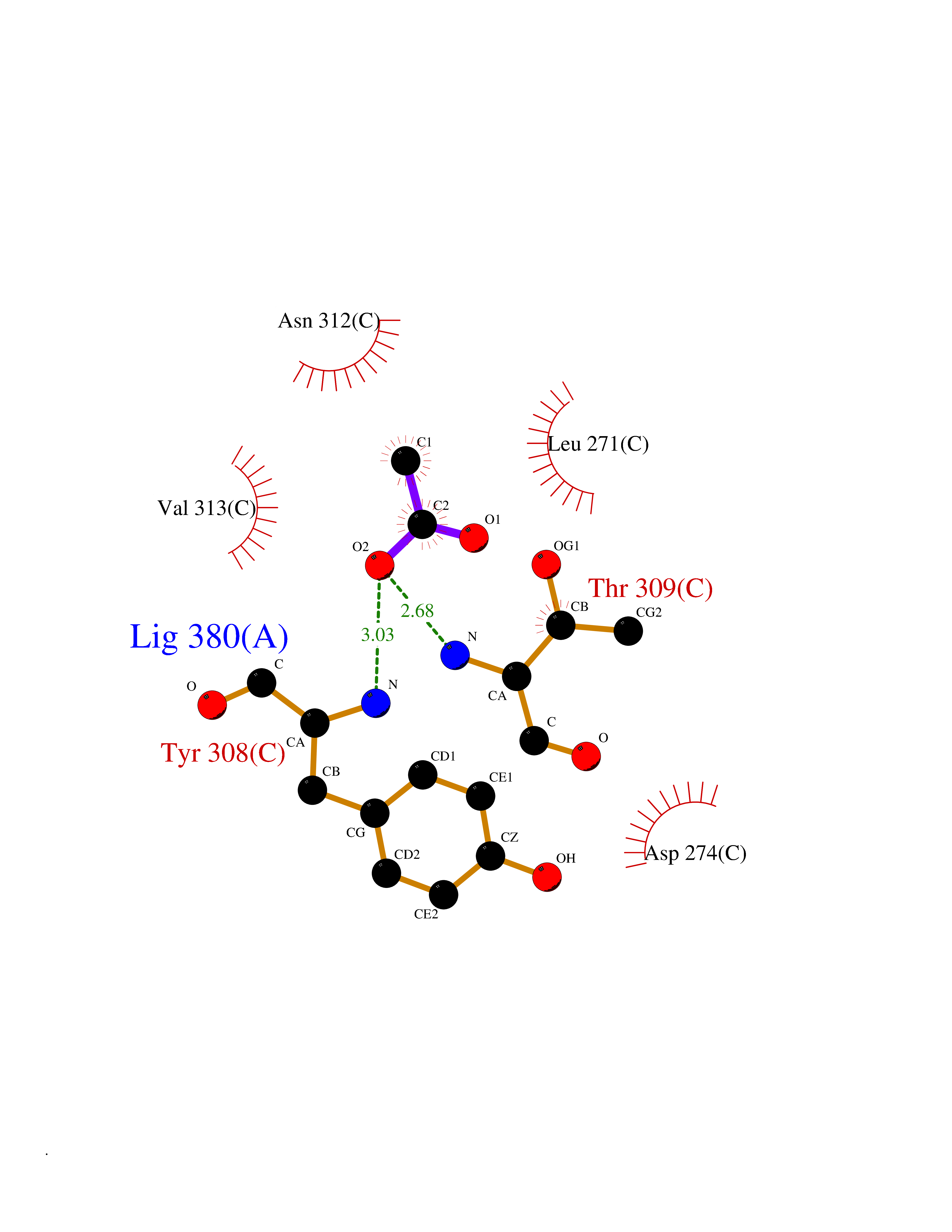 Ligplot Map 3D Binding mode Sequence KKKVCYYYDGDIGNYYYGQGHPMKPHRIRMTHNLLLNYGLYRKMEIYRPHKATAEEMTKYHSDEYIKFLRSIRPDNMSEYSKQMQRFNVGEDCPVFDGLFEFCQLSTGGSVAGAVKLNRQQTDMAVNWAGGLHHAKKSEASGFCYVNDIVLAILELLKYHQRVLYIDIDIHHGDGVEEAFYTTDRVMTVSFHKYGEYFPGTGDLRDIGAGKGKYYAVNFPMRDGIDDESYGQIFKPIISKVMEMYQPSAVVLQCGADSLSGDRLGCFNLTVKGHAKCVEVVKTFNLPLLMLGGGGYTIRNVARCWTYETAVALDCEIPNELPYNDYFEYFGPDFKLHISPSNMTNQNTPEYMEKIKQRLFENLRML Hydrogen bonds contact Hydrophobic contact | ||||