Job Results:
Ligand
Structure
Job ID
634b273143b25dfaacd81a63c2250095
Job name
NA
Time
2026-02-27 17:35:01
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Cyclin-dependent kinase 4 | 2W96 | 5.35 | |
Target general information Gen name CDK4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, CDC2/CDKX subfamily Biochemical class Cell cycle Function ATP binding.Cyclin binding.Cyclin-dependent protein serine/threonine kinase activity.Cyclin-dependent protein serine/threonine kinase regulator activity.Protein complex binding. Related diseases Melanoma, cutaneous malignant 3 (CMM3) [MIM:609048]: A malignant neoplasm of melanocytes, arising de novo or from a pre-existing benign nevus, which occurs most often in the skin but may also involve other sites. {ECO:0000269|PubMed:7652577, ECO:0000269|PubMed:8528263, ECO:0000269|PubMed:9311594, ECO:0000269|PubMed:9425228}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12001; DB03496; DB12010; DB09073; DB02733; DB11730; DB15442 Interacts with Q9UH17; P24385; P30279; P30281; Q16543; P50613; P38936; P46527; P49918; P42771; P42772; P42773; P55273; Q9UJC3; P08238; Q9UKT9; Q0VD86; P01106; Q9ULD0; P28749; Q08999; P09936; Q8N720 EC number 2.7.11.22 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cell cycle; Cell division; Cytoplasm; Disease variant; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID B Molecular weight (Da) 30138.4 Length 267 Aromaticity 0.09 Instability index 36.2 Isoelectric point 5.78 Charge (pH=7) -5.83 2D Binding mode Binding energy (Kcal/mol) -7.29 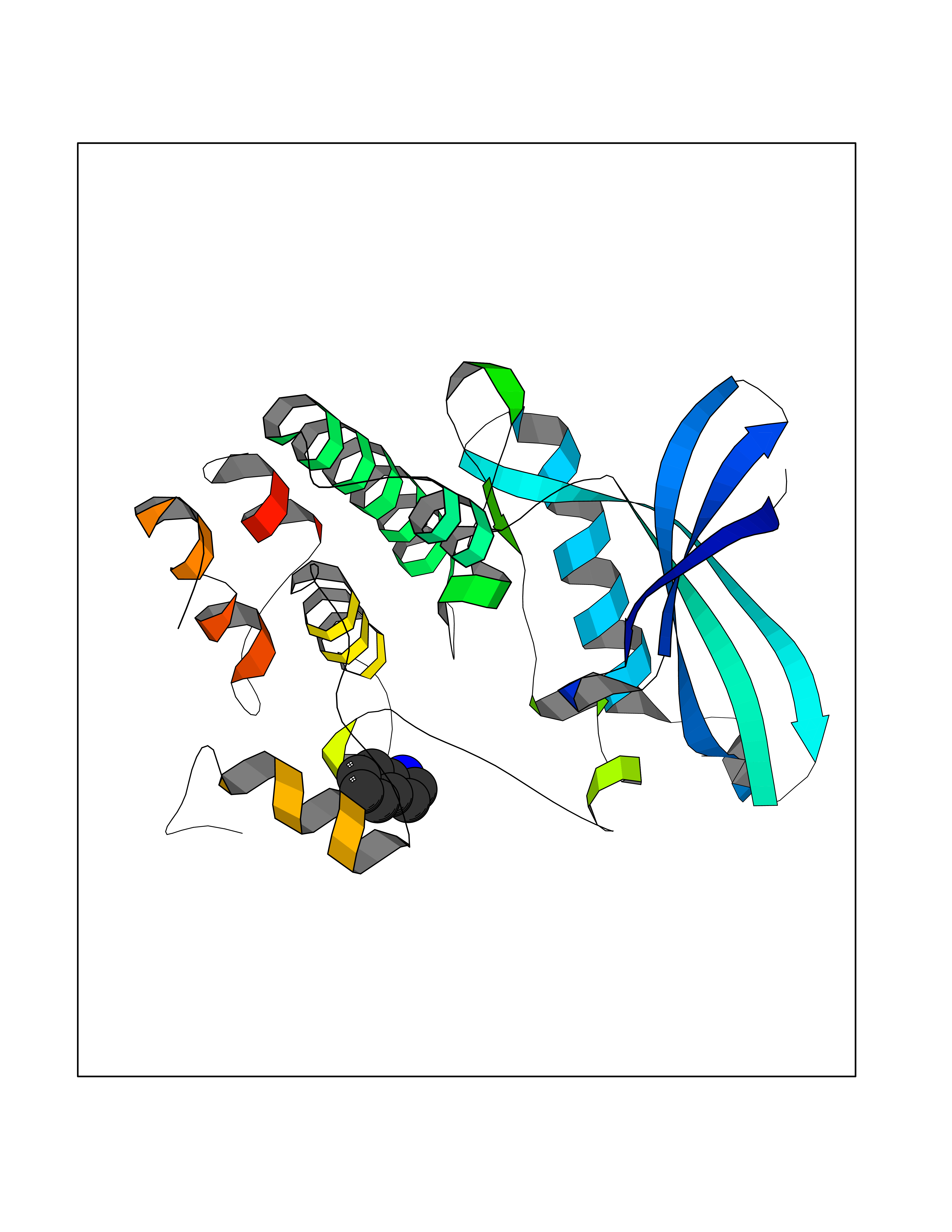 Molscript Map 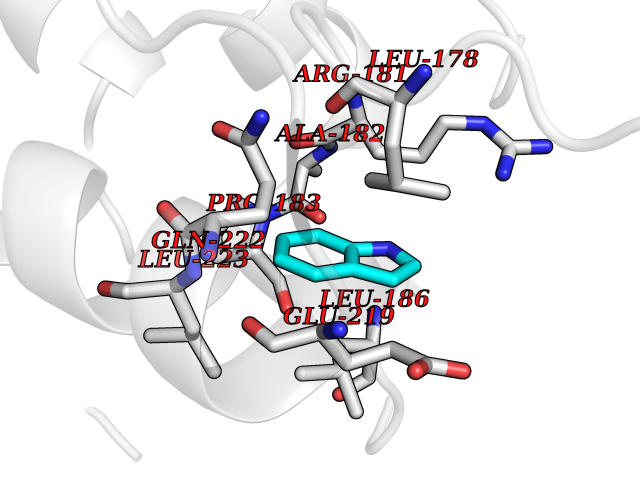 Pymol Map 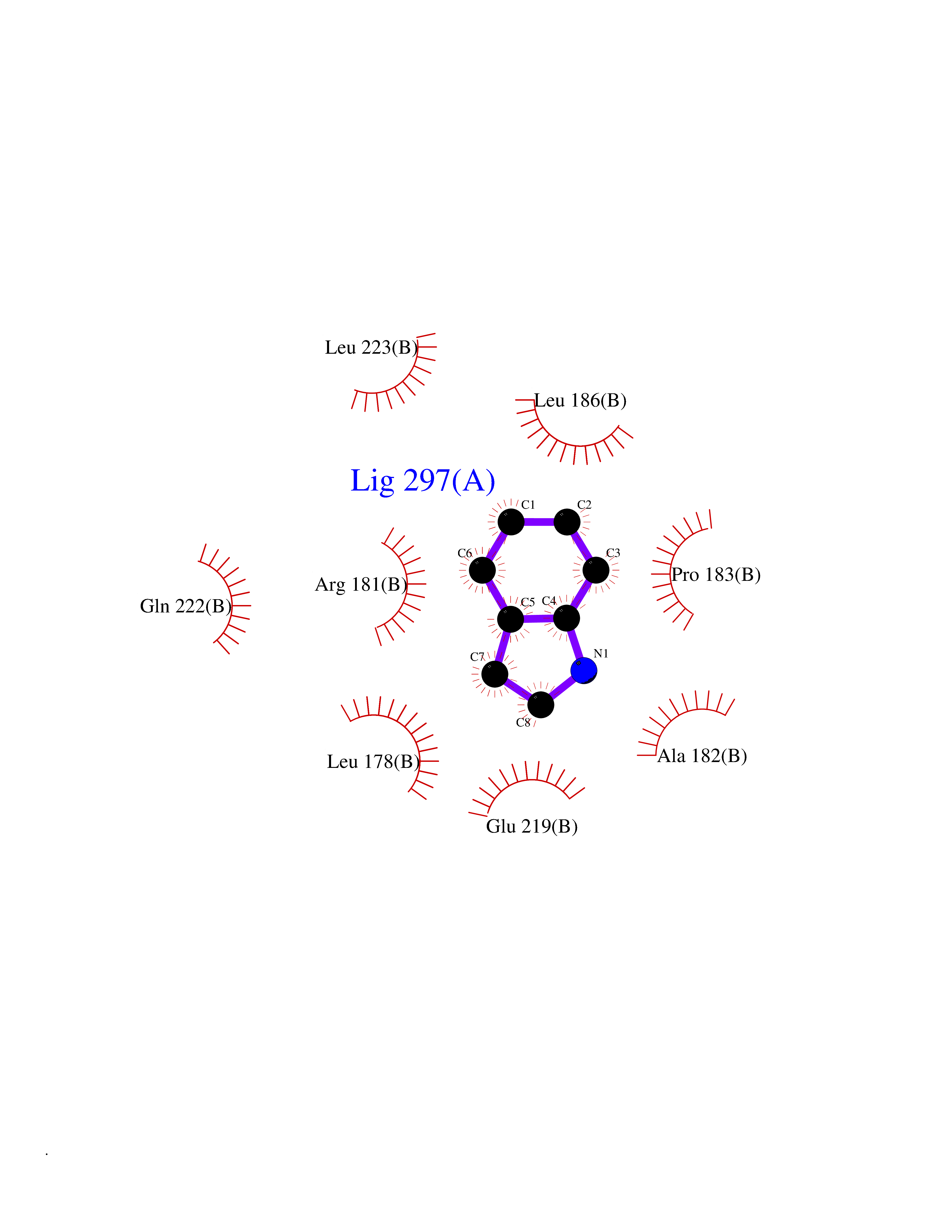 Ligplot Map 3D Binding mode Sequence SRYEPVAEIGVGAYGTVYKARDPHSGHFVALKSVRVPNGEEGLPISTVREVALLRRLEAFEHPNVVRLMDVCATSRTDREIKVTLVFEHVDQDLRTYLDKAPPPGLPAETIKDLMRQFLRGLDFLHANCIVHRDLKPENILVTSGGTVKLADFGLARIYSYQMALDPVVVTLWYRAPEVLLQSTYATPVDMWSVGCIFAEMFRRKPLFCGNSEADQLGKIFDLIGLPPEDDWVPEMEESGAQLLLEMLTFNPHKRISAFRALQHSYL Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Diamine oxidase (AOC1) | 3HIG | 5.35 | |
Target general information Gen name AOC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Kidney amine oxidase; KAO; Histaminase; Amiloride-binding protein; AOC1; ABP Protein family Copper/topaquinone oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the degradation of compounds such as putrescine, histamine, spermine, and spermidine, substances involved in allergic and immune responses, cell proliferation, tissue differentiation, tumor formation, and possibly apoptosis. Placental DAO is thought to play a role in the regulation of the female reproductive function. Related diseases Lichtenstein-Knorr syndrome (LIKNS) [MIM:616291]: An autosomal recessive neurologic disorder characterized by progressive cerebellar ataxia and severe progressive sensorineural hearing loss. {ECO:0000269|PubMed:25205112, ECO:0000269|PubMed:30237576}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00594; DB01373; DB09130; DB03608; DB05383 Interacts with Q15038; O75593; Q8IUC2; Q96HA8; Q7Z3K3; Q6ZRY4; Q01085-2; O43711; Q96K80 EC number EC 1.4.3.22 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Copper; Direct protein sequencing; Disulfide bond; Glycoprotein; Heparin-binding; Membrane; Metal-binding; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal; TPQ Protein physicochemical properties Chain ID A,B Molecular weight (Da) 69037.3 Length 607 Aromaticity 0.13 Instability index 43.33 Isoelectric point 6.52 Charge (pH=7) -4.61 2D Binding mode Binding energy (Kcal/mol) -7.29  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence RKAGVFSDLSNQELKAVHSFLWSKKELRLQPSSTTTMAKNTVFLIEMLLPKKYHVLRFLDKGERHPVREARAVIFFGDQEHPNVTEFAVGPLPGPCYMRALSPRPGYQSSWASRPISTAEYALLYHTLQEATKPLHQFFLNTTGFSFQDCHDRCLAFTDVAPRGVASGQRRSWLIIQRYVEGYFLHPTGLELLVDHGSTDAGHWAVEQVWYNGKFYGSPEELARKYADGEVDVVVLEPPLFSSLVQPHGPRFRLEGNAVLYGGWSFAFRLRSSSGLQVLNVHFGGERIAYEVSVQEAVALYGGHTPAGMQTKYLDVGWGLGSVTHELAPGIDCPETATFLDTFHYYDADDPVHYPRALCLFEMPTGVPLKGQVLVLRTTSTVYNXDYIWDFIFYPNGVMEAKMHATGYVHATFYTPEGLRHGTRLHTHLIGNIHTHLVHYRVDLDVAGTKNSFQTLQYSWERQAAFRFKRKLPKYLLFTSPQENPWGHKRSYRLQIHSMADQVLPPGWQEEQAITWARYPLAVTKYRESELCSSSIYHQNDPWDPPVVFEQFLHNNENIENEDLVAWVTVGFLHIPHSEDIPNTATPGNSVGFLLRPFNFNGTYRPV Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | S-adenosylmethionine decarboxylase proenzyme (AMD1) | 1JL0 | 5.35 | |
Target general information Gen name AMD1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SamDC; S-adenosylmethioninedecarboxylase; AdoMetDC; AMD Protein family Eukaryotic AdoMetDC family Biochemical class Carbon-carbon lyase Function Promotes maintenance and self-renewal of embryonic stem cells, by maintaining spermine levels. Essential for biosynthesis of the polyamines spermidine and spermine. Related diseases Niemann-Pick disease A (NPDA) [MIM:257200]: An early-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Niemann-Pick disease type A is a primarily neurodegenerative disorder characterized by onset within the first year of life, intellectual disability, digestive disorders, failure to thrive, major hepatosplenomegaly, and severe neurologic symptoms. The severe neurological disorders and pulmonary infections lead to an early death, often around the age of four. Clinical features are variable. A phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. {ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1391960, ECO:0000269|PubMed:15221801, ECO:0000269|PubMed:15877209, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:1718266, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:2023926, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430884, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:8680412, ECO:0000269|PubMed:8693491, ECO:0000269|PubMed:9266408, ECO:0000269|PubMed:9660788}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Niemann-Pick disease B (NPDB) [MIM:607616]: A late-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Clinical signs involve only visceral organs. The most constant sign is hepatosplenomegaly which can be associated with pulmonary symptoms. Patients remain free of neurologic manifestations. However, a phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. In Niemann-Pick disease type B, onset of the first symptoms occurs in early childhood and patients can survive into adulthood. {ECO:0000269|PubMed:12369017, ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1301192, ECO:0000269|PubMed:15241805, ECO:0000269|PubMed:16010684, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:16472269, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:1885770, ECO:0000269|PubMed:19050888, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:21098024, ECO:0000269|PubMed:21621718, ECO:0000269|PubMed:22613662, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430512, ECO:0000269|PubMed:25920558, ECO:0000269|PubMed:26084044, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:27659707, ECO:0000269|PubMed:8051942, ECO:0000269|PubMed:8664904}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08163; DB00118; DB01917 Interacts with P17707; Q96A98; Q8WY91 EC number EC 4.1.1.50 Uniprot keywords 3D-structure; Alternative splicing; Autocatalytic cleavage; Decarboxylase; Direct protein sequencing; Lyase; Phosphoprotein; Polyamine biosynthesis; Proteomics identification; Pyruvate; Reference proteome; S-adenosyl-L-methionine; Schiff base; Spermidine biosynthesis; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 35790.5 Length 311 Aromaticity 0.14 Instability index 39.47 Isoelectric point 6.03 Charge (pH=7) -2.01 2D Binding mode Binding energy (Kcal/mol) -7.29  Molscript Map 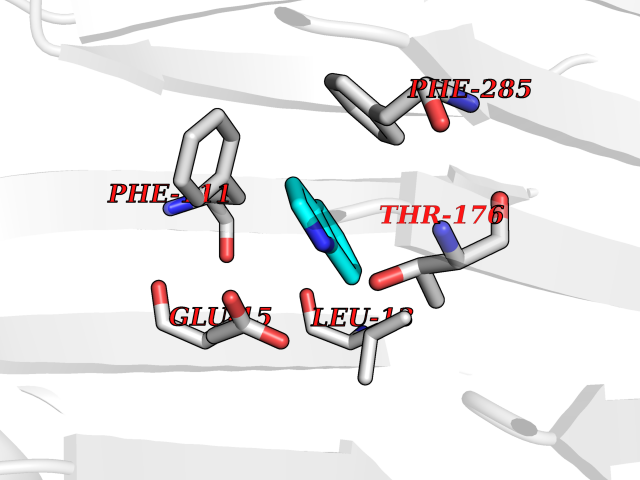 Pymol Map 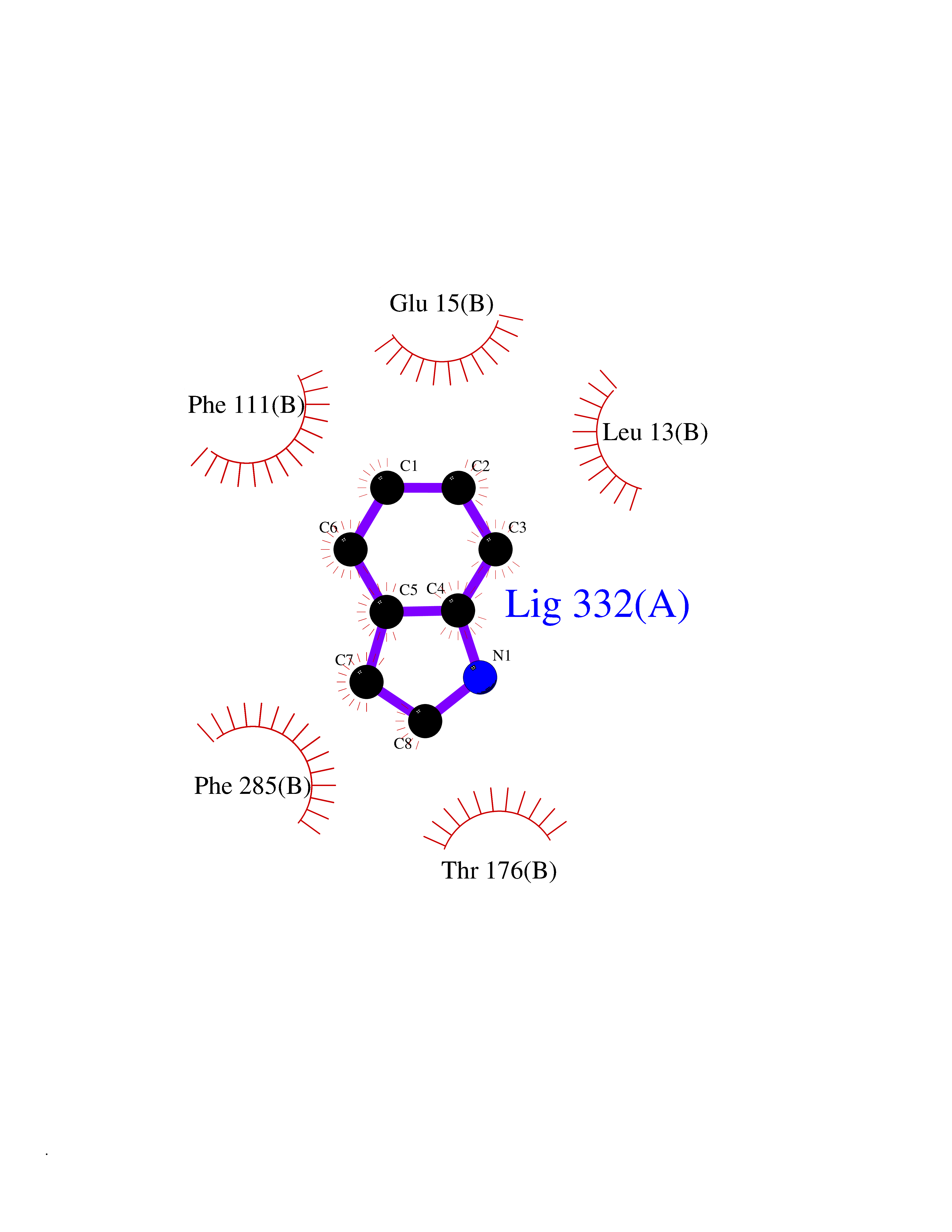 Ligplot Map 3D Binding mode Sequence HFFEGTEKLLEVWFSRQGSGDLRTIPRSEWDILLKDVQCSIISVTKTDKQEAYVLSESSMFVSKRRFILKTCGTTLLLKALVPLLKLARDYSGFDSIQSFFYSRKNFMKPSHQGYPHRNFQEEIEFLNAIFPNGAGYCMGRMNSDCWYLYTLDFRVISQPDQTLEILMSELDPAVMDQFYMKDGVTAKDVTRESGIRDLIPGSVIDATMFNPCGYSMNGMKSDGTYWTIAITPEPEFSYVSFETNLSQTSYDDLIRKVVEVFKPGKFVTTLFVNQSSKCPQKIEGFKRLDCQSAMFNDYNFVFTSFAKKQQ Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Monoamine oxidase type B (MAO-B) | 2V5Z | 5.35 | |
Target general information Gen name MAOB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MAO-B; Amine oxidase [flavin-containing] B Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. MAOB preferentially degrades benzylamine and phenylethylamine. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB08176; DB02211; DB08516; DB08480; DB01472; DB04307; DB07512; DB07513; DB00915; DB00182; DB06698; DB04889; DB00215; DB09130; DB04147; DB00988; DB01363; DB00668; DB01175; DB02509; DB03147; DB14914; DB00614; DB04818; DB02095; DB01247; DB00601; DB01577; DB01442; DB01171; DB08082; DB02643; DB04677; DB03894; DB08804; DB04820; DB00184; DB04821; DB12612; DB01626; DB00780; DB00191; DB00388; DB01132; DB00721; DB01168; DB01367; DB09363; DB06654; DB01037; DB01104; DB14569; DB09042; DB00752; DB16446; DB09185; DB04832; DB00909 Interacts with P55212; P28329-3; Q8NI60; Q5RI15; Q92915-2; P22607; Q53GS7; P06396; P01112; O14901; P13473-2; P21397; Q9BVL2; O75400-2; P62826; Q6NTF9-3; Q9Y371; Q7Z699; Q9UMX0; Q9Y649 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; FAD; Flavoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Oxidoreductase; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56019.9 Length 494 Aromaticity 0.09 Instability index 34.81 Isoelectric point 6.51 Charge (pH=7) -2.2 2D Binding mode Binding energy (Kcal/mol) -7.3 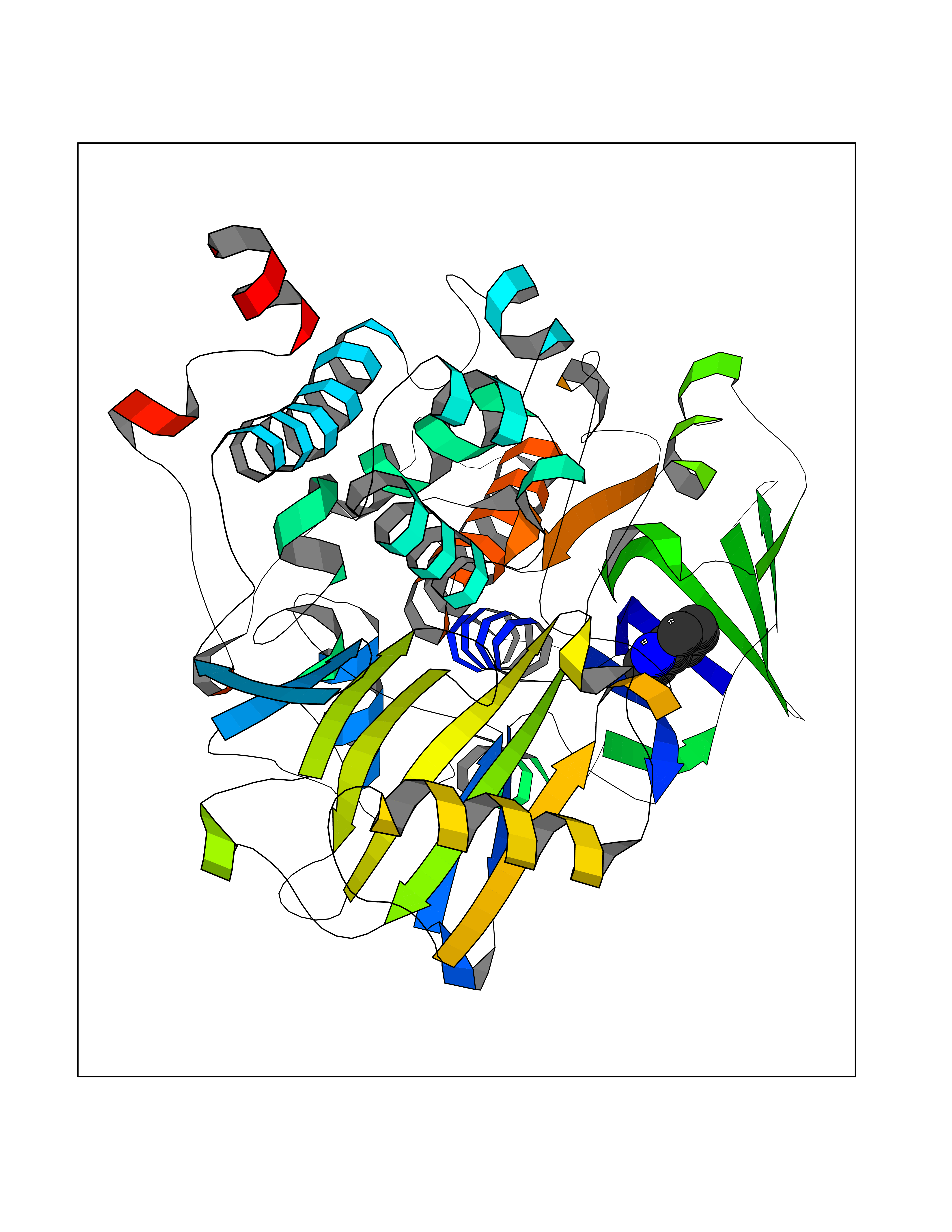 Molscript Map 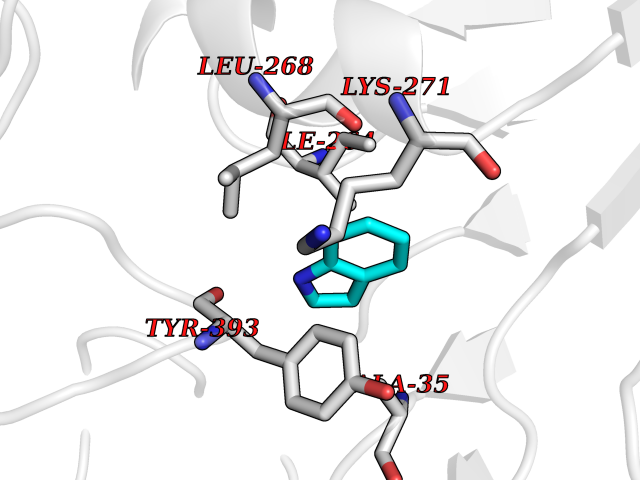 Pymol Map 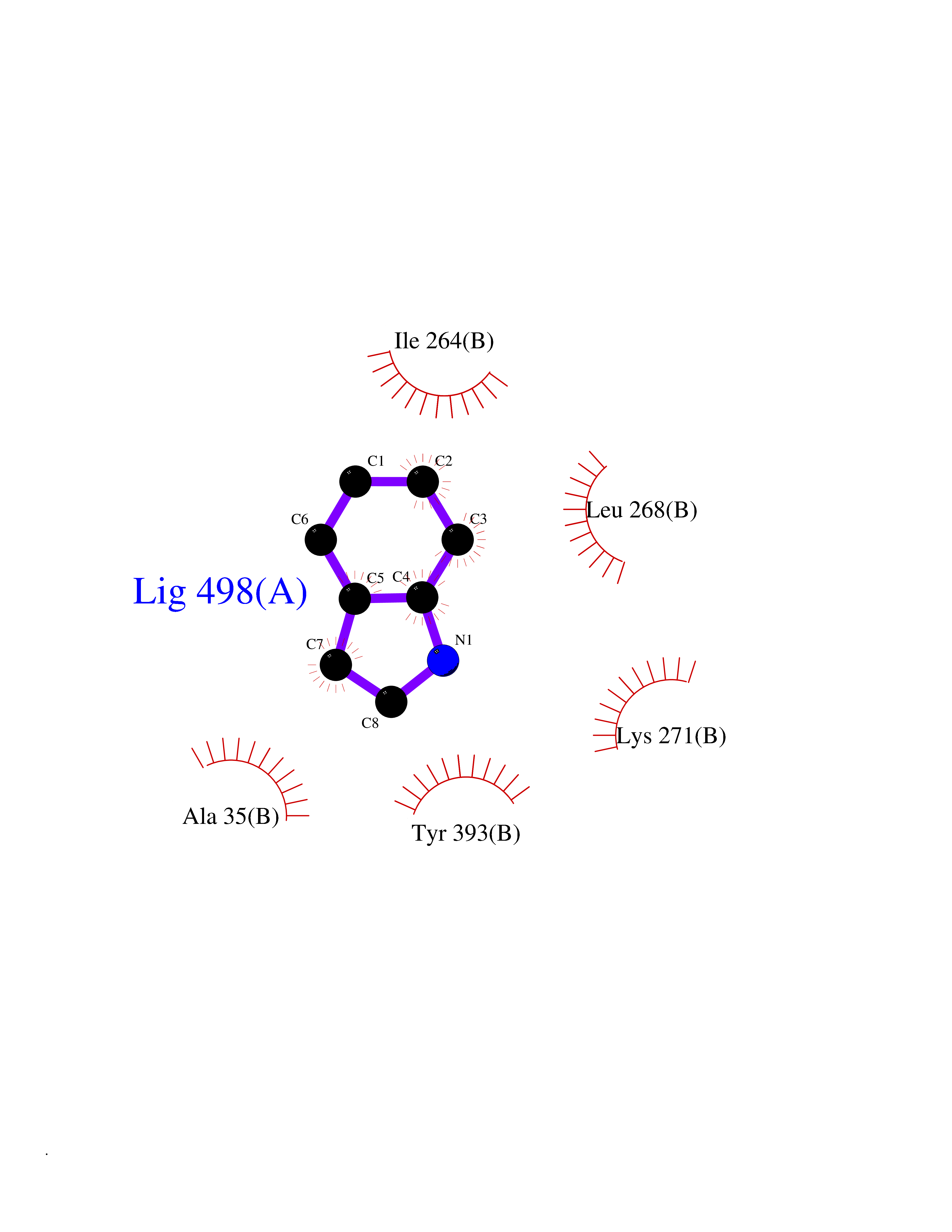 Ligplot Map 3D Binding mode Sequence NKCDVVVVGGGISGMAAAKLLHDSGLNVVVLEARDRVGGRTYTLRNQKVKYVDLGGSYVGPTQNRILRLAKELGLETYKVNEVERLIHHVKGKSYPFRGPFPPVWNPITYLDHNNFWRTMDDMGREIPSDAPWKAPLAEEWDNMTMKELLDKLCWTESAKQLATLFVNLCVTAETHEVSALWFLWYVKQCGGTTRIISTTNGGQERKFVGGSGQVSERIMDLLGDRVKLERPVIYIDQTRENVLVETLNHEMYEAKYVISAIPPTLGMKIHFNPPLPMMRNQMITRVPLGSVIKCIVYYKEPFWRKKDYCGTMIIDGEEAPVAYTLDDTKPEGNYAAIMGFILAHKARKLARLTKEERLKKLCELYAKVLGSLEALEPVHYEEKNWCEEQYSGGCYTTYFPPGILTQYGRVLRQPVDRIYFAGTETATHWSGYMEGAVEAGERAAREILHAMGKIPEDEIWQSEPESVDVPAQPITTTFLERHLPSVPGLLRLI Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | 2-hydroxy-6-oxo-7-methylocta-2,4-dienoate hydrolase | 1UK8 | 5.35 | |
Target general information Gen name cumD Organism Pseudomonas fluorescens Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function Hydrolase activity. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03741; DB03793; DB03568; DB02531; DB03750; DB02406; DB03766 Interacts with NA EC number NA Uniprot keywords 3D-structure; Hydrolase Protein physicochemical properties Chain ID A Molecular weight (Da) 30307.9 Length 271 Aromaticity 0.1 Instability index 37.49 Isoelectric point 5.02 Charge (pH=7) -11.58 2D Binding mode Binding energy (Kcal/mol) -7.3  Molscript Map 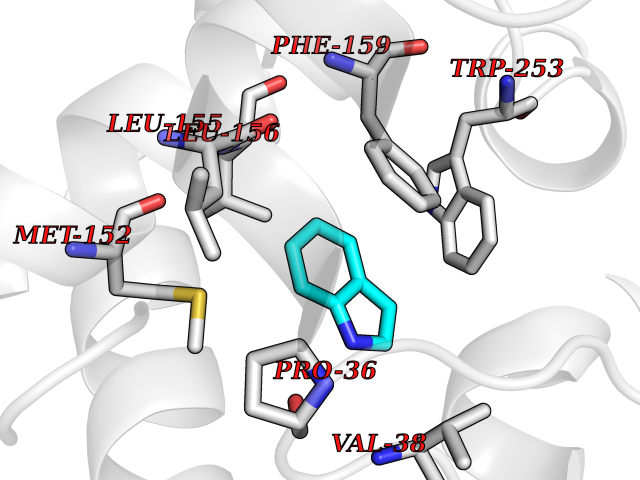 Pymol Map 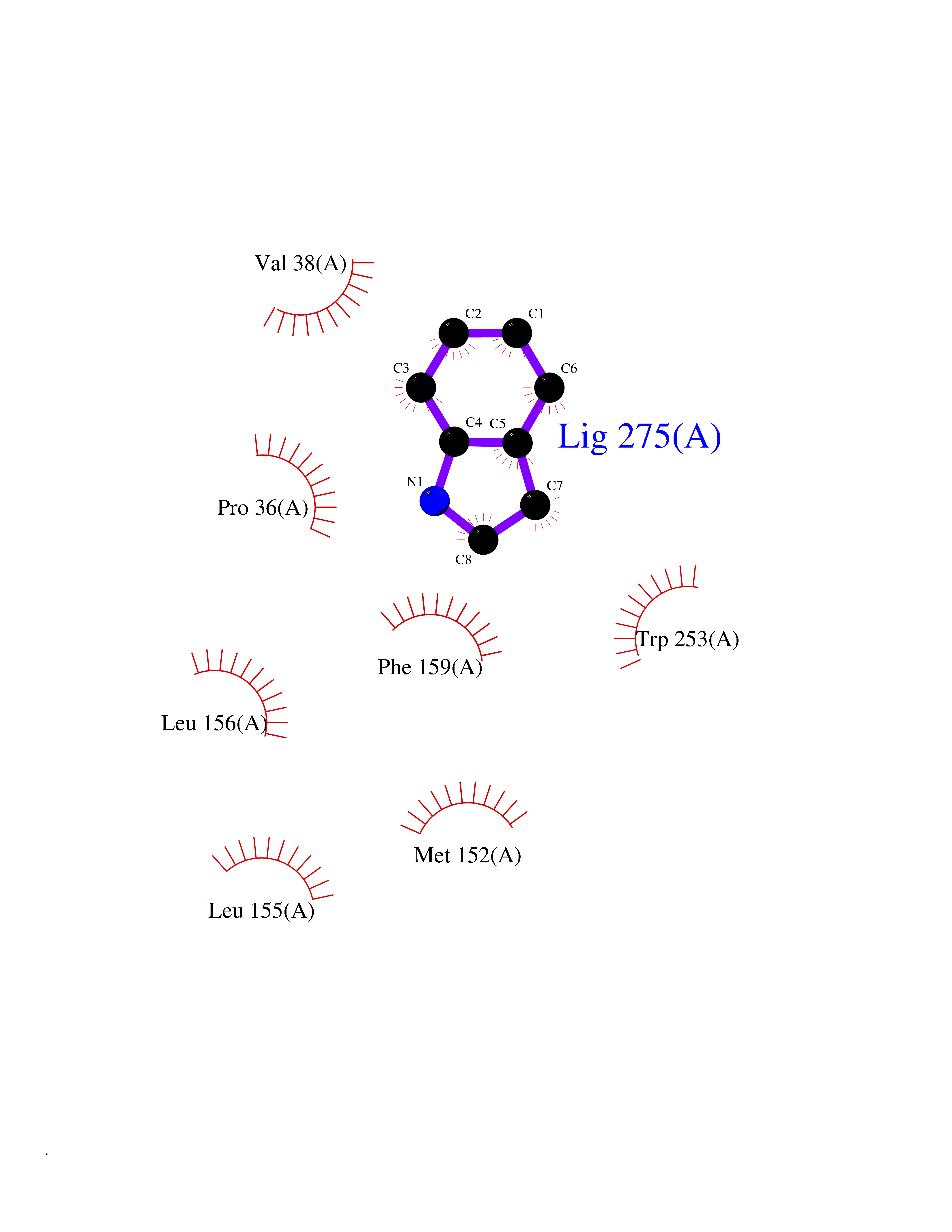 Ligplot Map 3D Binding mode Sequence NLEIGKSILAAGVLTNYHDVGEGQPVILIHGSGPGVSAYANWRLTIPALSKFYRVIAPDMVGFGFTDRPENYNYSKDSWVDHIIGIMDALEIEKAHIVGNAFGGGLAIATALRYSERVDRMVLMGAAGTRFDVTEGLNAVWGYTPSIENMRNLLDIFAYDRSLVTDELARLRYEASIQPGFQESFSSMFPEPRQRWIDALASSDEDIKTLPNETLIIHGREDQVVPLSSSLRLGELIDRAQLHVFGRCGHWTQIEQTDRFNRLVVEFFNEA Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Peroxisome proliferator-activated receptor alpha (PPARA) | 3VI8 | 5.35 | |
Target general information Gen name PPARA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Peroxisome proliferater-activated receptor alpha; PPARalpha; PPAR-alpha; PPAR; Nuclear receptor subfamily 1 group C member 1; NR1C1 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Key regulator of lipid metabolism. Activated by the endogenous ligand 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine (16:0/18:1-GPC). Activated by oleylethanolamide, a naturally occurring lipid that regulates satiety. Receptor for peroxisome proliferators such as hypolipidemic drugs and fatty acids. Regulates the peroxisomal beta-oxidation pathway of fatty acids. Functions as transcription activator for the ACOX1 and P450 genes. Transactivation activity requires heterodimerization with RXRA and is antagonized by NR2C2. May be required for the propagation of clock information to metabolic pathways regulated by PER2. Ligand-activated transcription factor. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08915; DB00132; DB01118; DB04557; DB01393; DB04519; DB05416; DB09064; DB09006; DB00636; DB09213; DB03756; DB05187; DB06521; DB01039; DB13873; DB00573; DB13961; DB02266; DB01241; DB07215; DB01050; DB00159; DB07724; DB00328; DB12007; DB03017; DB12961; DB06510; DB08231; DB11605; DB01890; DB04224; DB11133; DB03796; DB02746; DB01708; DB06533; DB04971; DB02709; DB00412; DB09422; DB03193; DB06536; DB00197; DB00313 Interacts with P02768-3; P55212; P45973; P06307; Q3L8U1-3; G5E9A7; P22607; P62993; Q14957; P06396; P42858; Q8WXH2; P13473-2; O75376; Q13133; A0A6Q8PF08; P54725; P62826; Q7Z699; P37173; P55072; P55055-1; Q13133 EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Biological rhythms; DNA-binding; Lipid-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 29322.1 Length 258 Aromaticity 0.09 Instability index 35.53 Isoelectric point 6.09 Charge (pH=7) -3.57 2D Binding mode Binding energy (Kcal/mol) -7.29 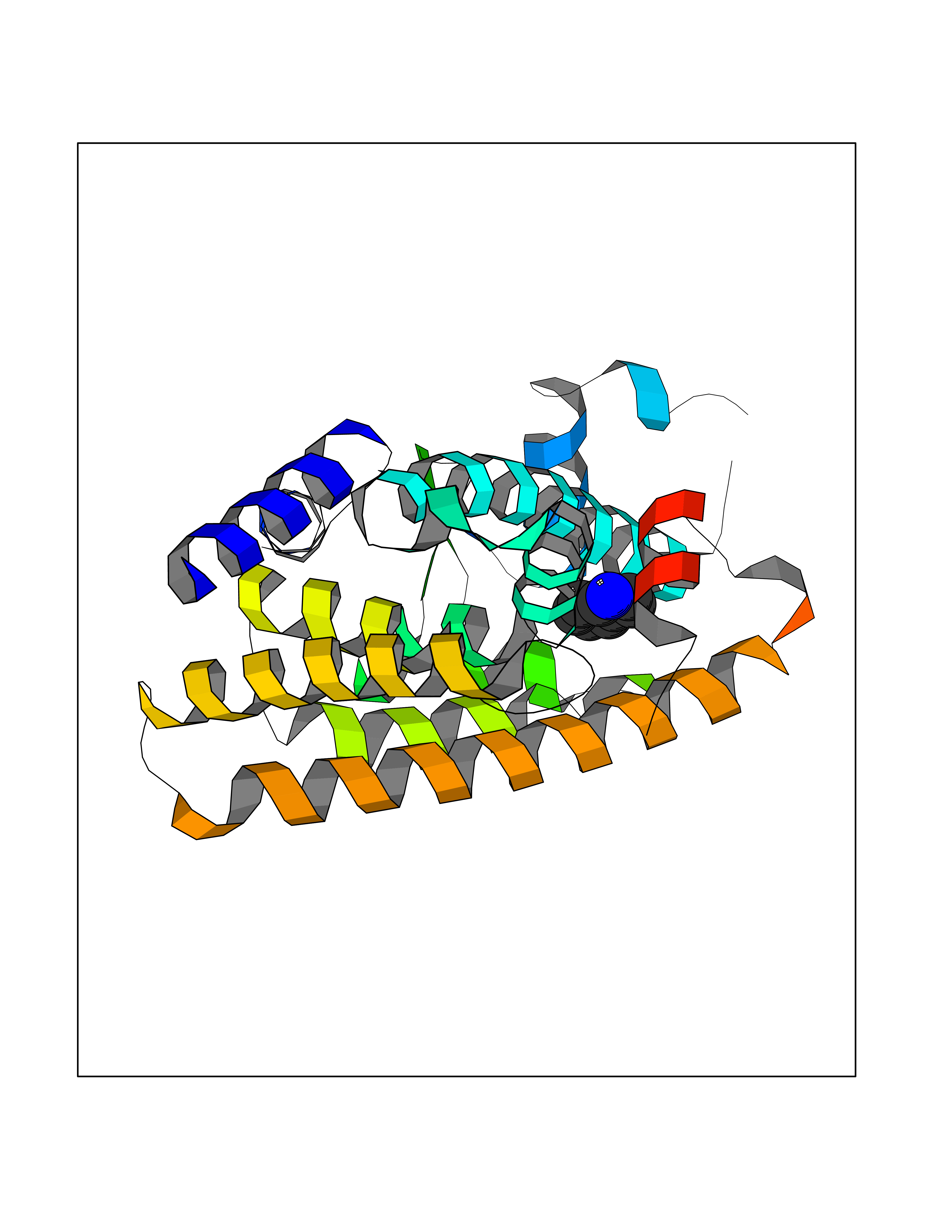 Molscript Map 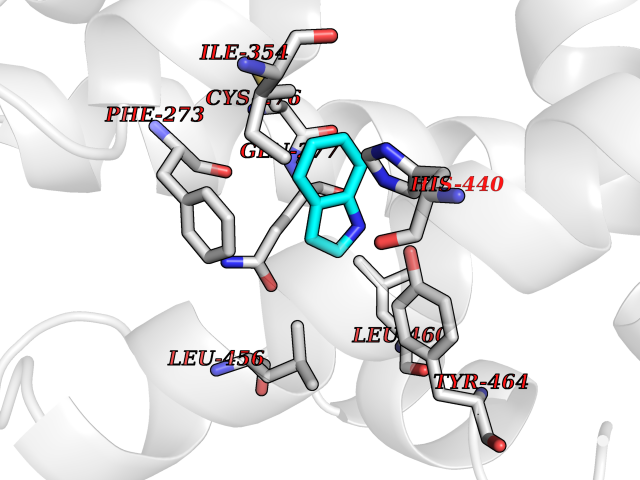 Pymol Map 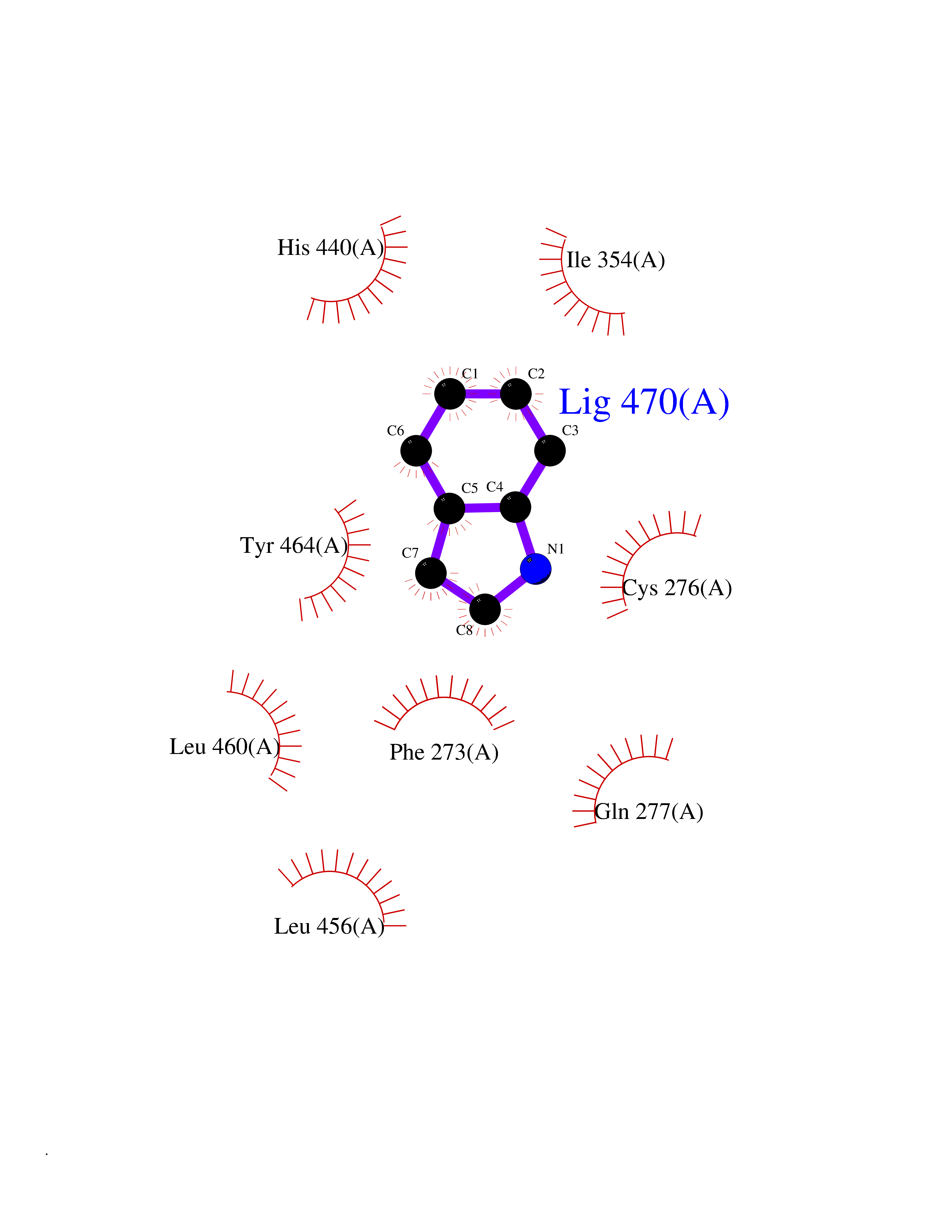 Ligplot Map 3D Binding mode Sequence DLKSLAKRIYEAYLKNFNMNKVKARVILSPFVIHDMETLCMAEKTLVAKLVANGNKEAEVRIFHCCQCTSVETVTELTEFAKAIPGFANLDLNDQVTLLKYGVYEAIFAMLSSVMNKDGMLVAYGNGFITREFLKSLRKPFCDIMEPKFDFAMKFNALELDDSDISLFVAAIICCGDRPGLLNVGHIEKMQEGIVHVLRLHLQSNHPDDIFLFPKLLQKMADLRQLVTEHAQLVQIIKKTESDAALHPLLQEIYRDMY Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Cyclopropane mycolic acid synthase MmaA2 | 1TPY | 5.35 | |
Target general information Gen name mmaA2 Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID NA Synonyms Rv0644c;mma2 Protein family CFA/CMAS family Biochemical class Transferase Function Cyclopropane-fatty-acyl-phospholipid synthase activity.Methyltransferase activity. Related diseases Oocyte/zygote/embryo maturation arrest 16 (OZEMA16) [MIM:617234]: A rare cause of female primary infertility. In affected women, ovulation and fertilization proceed normally but embryos are arrested at early stages of development. Inheritance is autosomal recessive. {ECO:0000269|PubMed:27545678}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01718; DB01752 Interacts with NA EC number 2.1.1.79 Uniprot keywords 3D-structure; Acetylation; Lipid biosynthesis; Lipid metabolism; Methyltransferase; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 32493.6 Length 285 Aromaticity 0.1 Instability index 43.61 Isoelectric point 5.53 Charge (pH=7) -10.17 2D Binding mode Binding energy (Kcal/mol) -7.3 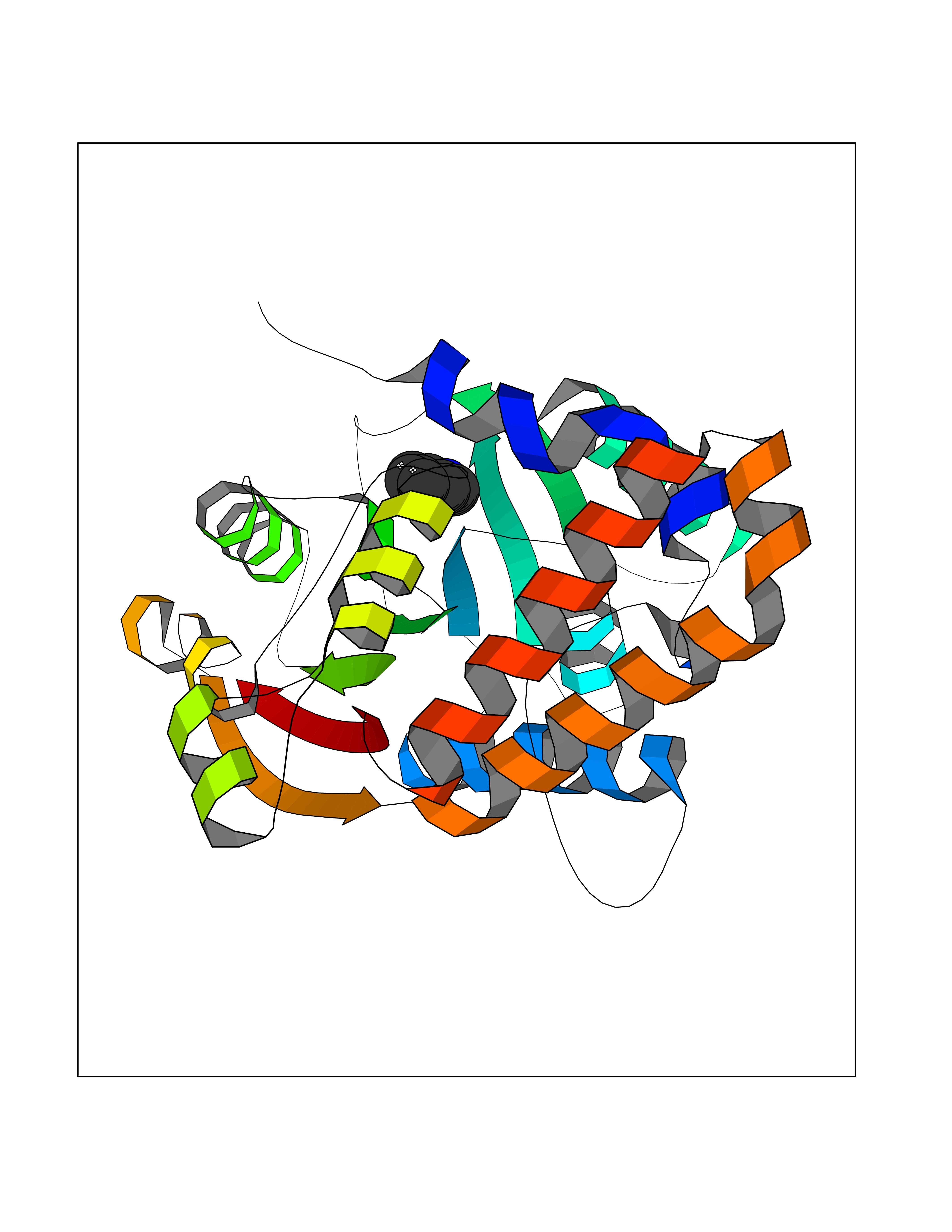 Molscript Map 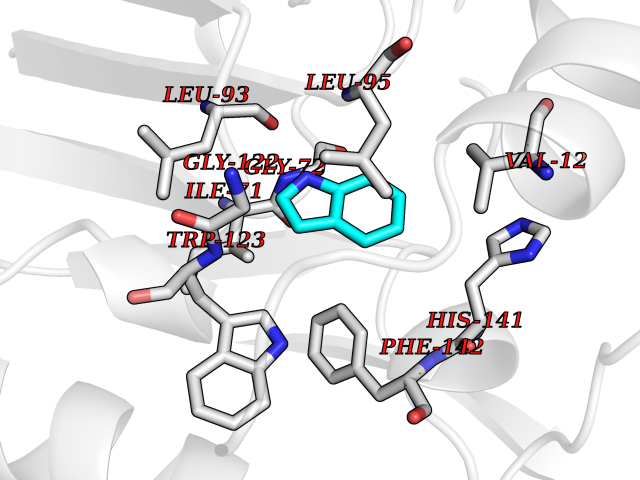 Pymol Map 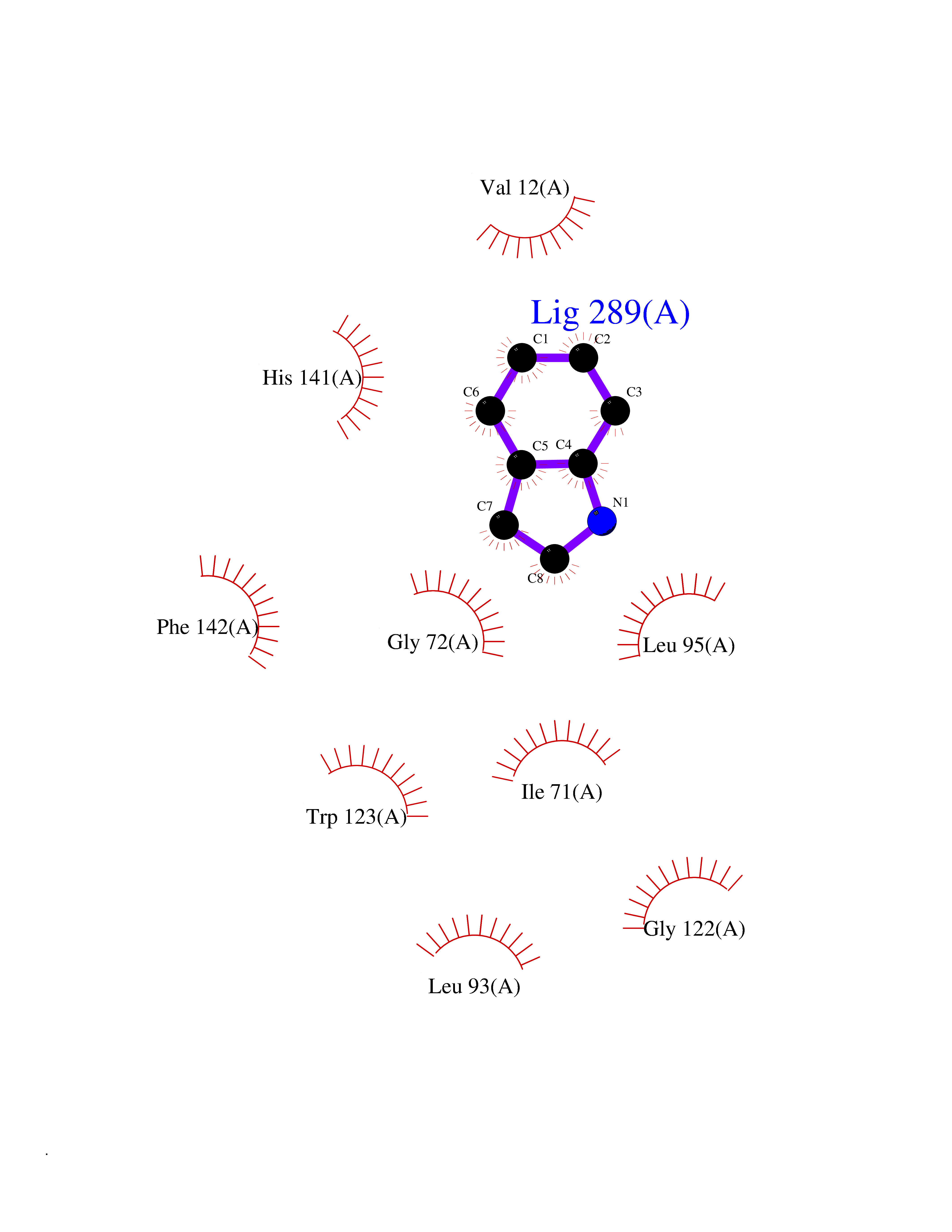 Ligplot Map 3D Binding mode Sequence NDLTPHFEDVQAHYDLSDDFFRLFLDPTQTYSCAHFEREDMTLEEAQIAKIDLALGKLGLQPGMTLLDIGCGWGATMRRAIAQYDVNVVGLTLSKNQAAHVQKSFDEMDTPRDRRVLLAGWEQFNEPVDRIVSIGAFEHFGHDRHADFFARAHKILPPDGVLLLHTITGLTRQQMVDHGLPLTLWLARFLKFIATEIFPGGQPPTIEMVEEQSAKTGFTLTRRQSLQPHYARTLDLWAEALQEHKSEAIAIQSEEVYERYMKYLTGCAKLFRVGYIDVNQFTLAK Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Scavenger decapping enzyme DcpS (DCPS) | 1ST4 | 5.35 | |
Target general information Gen name DCPS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Scavenger mRNA-decapping enzyme DcpS; Histidine triad protein member5; Hint-related 7meGMP-directed hydrolase; HINT-5; DCS-1; DCPS Protein family HIT family Biochemical class Acid anhydrides hydrolase Function Decapping scavenger enzyme that catalyzes the cleavage of a residual cap structure following the degradation of mRNAs by the 3'->5' exosome-mediated mRNA decay pathway. Hydrolyzes cap analog structures like 7-methylguanosine nucleoside triphosphate (m7GpppG) with up to 10 nucleotide substrates (small capped oligoribonucleotides) and specifically releases 5'-phosphorylated RNA fragments and 7-methylguanosine monophosphate (m7GMP). Cleaves cap analog structures like tri-methyl guanosine nucleoside triphosphate (m3(2,2,7)GpppG) with very poor efficiency. Does not hydrolyze unmethylated cap analog (GpppG) and shows no decapping activity on intact m7GpppG-capped mRNA molecules longer than 25 nucleotides. Does not hydrolyze 7-methylguanosine diphosphate (m7GDP) to m7GMP (PubMed:22985415). May also play a role in the 5'->3 mRNA decay pathway; m7GDP, the downstream product released by the 5'->3' mRNA mediated decapping activity, may be also converted by DCPS to m7GMP (PubMed:14523240). Binds to m7GpppG and strongly to m7GDP. Plays a role in first intron splicing of pre- mRNAs. Inhibits activation-induced cell death. Related diseases Al-Raqad syndrome (ARS) [MIM:616459]: A syndrome characterized by delayed psychomotor development, moderate to severe intellectual disability, poor or absent speech, microcephaly, congenital hypotonia, and severe growth delay. {ECO:0000269|PubMed:25701870, ECO:0000269|PubMed:25712129}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07644; DB07643; DB07642; DB03593; DB01960; DB01649; DB03958 Interacts with Q96C86; P52292; O15131; O60684 EC number EC 3.6.1.59 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Disease variant; Hydrolase; Intellectual disability; mRNA processing; mRNA splicing; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID B,A Molecular weight (Da) 69192.9 Length 597 Aromaticity 0.09 Instability index 54.62 Isoelectric point 6.12 Charge (pH=7) -9.94 2D Binding mode Binding energy (Kcal/mol) -7.3  Molscript Map 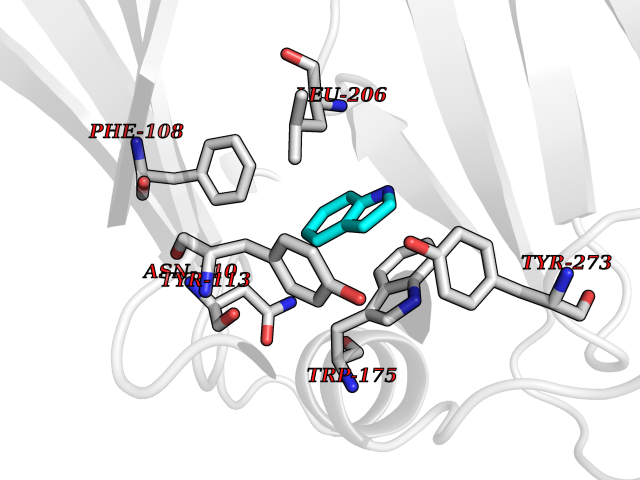 Pymol Map 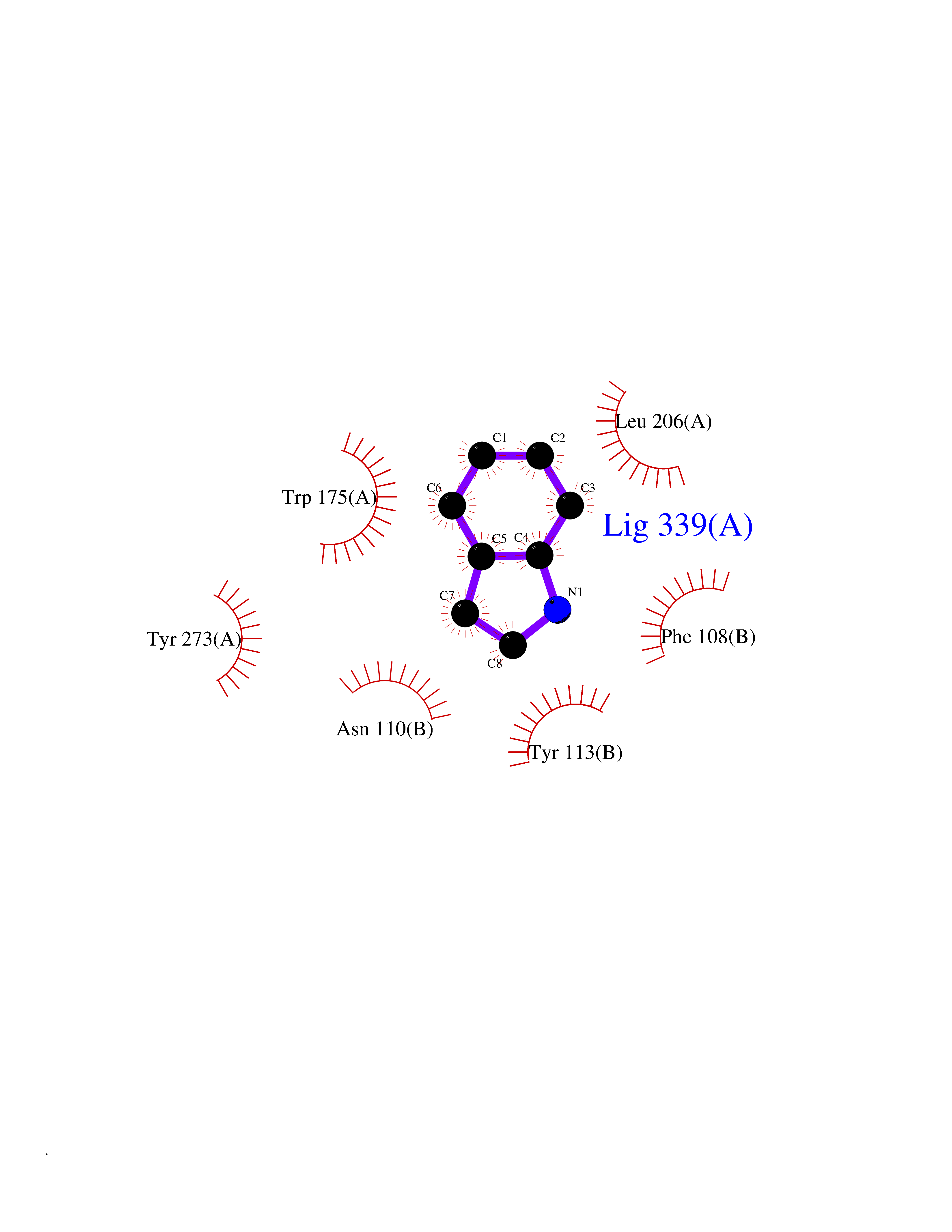 Ligplot Map 3D Binding mode Sequence VRLPFSGFRLQKVLRESARDKIIFLHGKVNEASGDGDGEDAVVILEKTPFQVEQVAQLLTGSPELQLQFSNDIYSTYHLFPPRQLNDVKTTVVYPATEKHLQKYLRQDLRLIRETGDDYRNITLPHLESQSLSIQWVYNILDKKAEADRIVFENPDPSDGFVLIPDLKWNQQQLDDLYLIAICHRRGIRSLRDLTPEHLPLLRNILHQGQEAILQRYRMKGDHLRVYLHYLPSYYHLNVHFTALGFEAPGSGVERAHLLAEVIENLECDPRHYQQRTLTFALRADDPLLKLLQEAQQAPVRLPFSGFRLQKVLRESARDKIIFLHGKVNEASGDGDGEDAVVILEKTPFQVEQVAQLLTGSPELQLQFSNDIYSTYHLFPPRQLNDVKTTVVYPATEKHLQKYLRQDLRLIRETGDDYRNITLPHLESQSLSIQWVYNILDKKAEADRIVFENPDPSDGFVLIPDLKWNQQQLDDLYLIAICHRRGIRSLRDLTPEHLPLLRNILHQGQEAILQRYRMKGDHLRVYLHYLPSYYHLNVHFTALGFEAPGSGVERAHLLAEVIENLECDPRHYQQRTLTFALRADDPLLKLLQEAQQS Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Caspase-7 (CASP7) | 1SHJ | 5.35 | |
Target general information Gen name CASP7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MCH3; ICE-like apoptotic protease 3; ICE-LAP3; CMH-1; CASP-7; Apoptotic protease Mch-3 Protein family Peptidase C14A family Biochemical class Peptidase Function Cleaves and activates sterol regulatory element binding proteins (SREBPs). Proteolytically cleaves poly(ADP-ribose) polymerase (PARP) at a '216-Asp-|-Gly-217' bond. Overexpression promotes programmed cell death. Involved in the activation cascade of caspases responsible for apoptosis execution. Related diseases Pregnancy loss, recurrent, 3 (RPRGL3) [MIM:614391]: A common complication of pregnancy, resulting in spontaneous abortion before the fetus has reached viability. The term includes all miscarriages from the time of conception until 24 weeks of gestation. Recurrent pregnancy loss is defined as 3 or more consecutive spontaneous abortions. {ECO:0000269|PubMed:17339269}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05408; DB03384; DB06255 Interacts with Q13490; P83105; P42858; Q8N4N3-2; P43364; Q16236; Q9GZT8; Q13177; P27986-2; P21673; Q86WV1-2; P17405; P98170 EC number EC 3.4.22.60 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; Apoptosis; Cytoplasm; Hydrolase; Nucleus; Phosphoprotein; Protease; Proteomics identification; Reference proteome; RNA-binding; Secreted; Thiol protease; Ubl conjugation; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 47441.5 Length 417 Aromaticity 0.11 Instability index 20.98 Isoelectric point 8.38 Charge (pH=7) 6.12 2D Binding mode Binding energy (Kcal/mol) -7.3 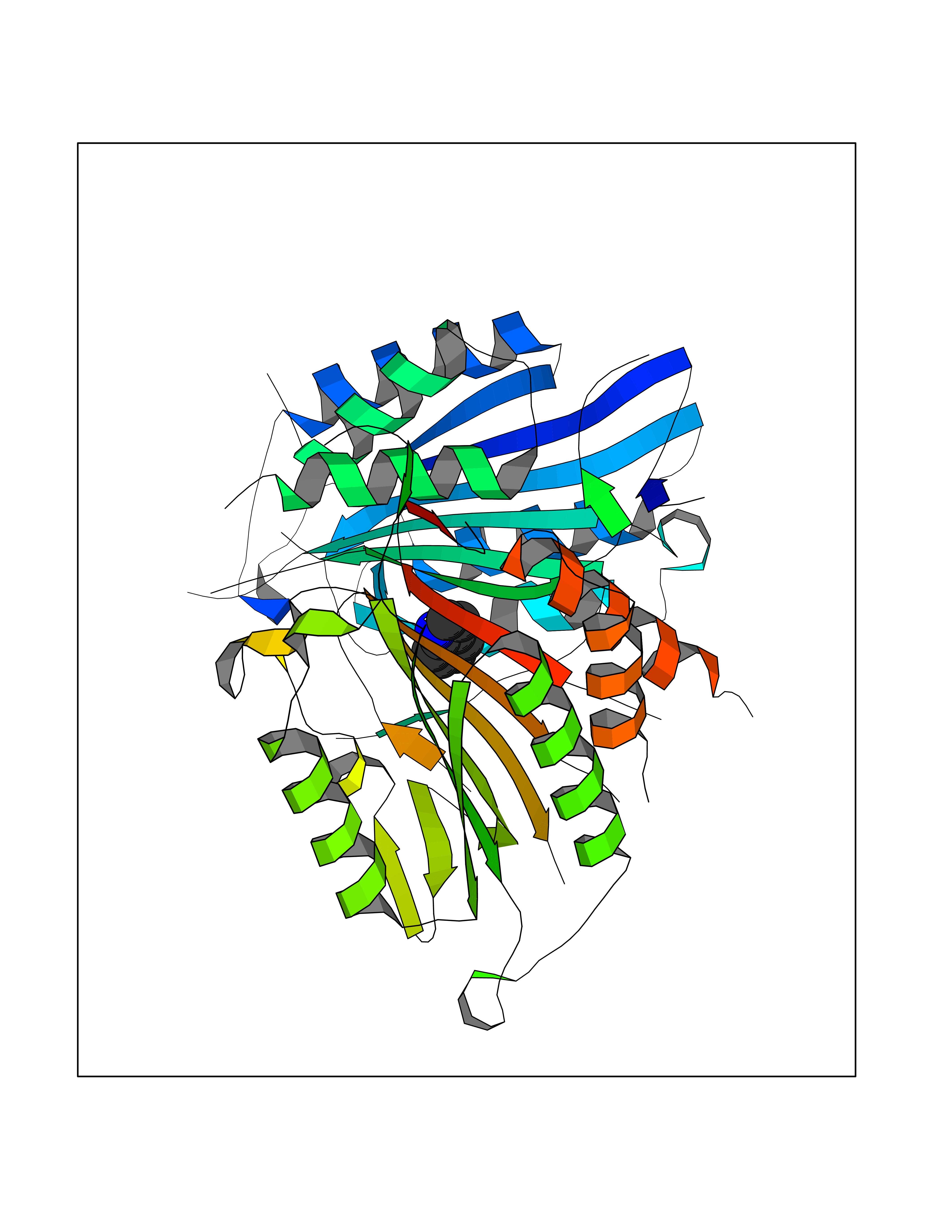 Molscript Map 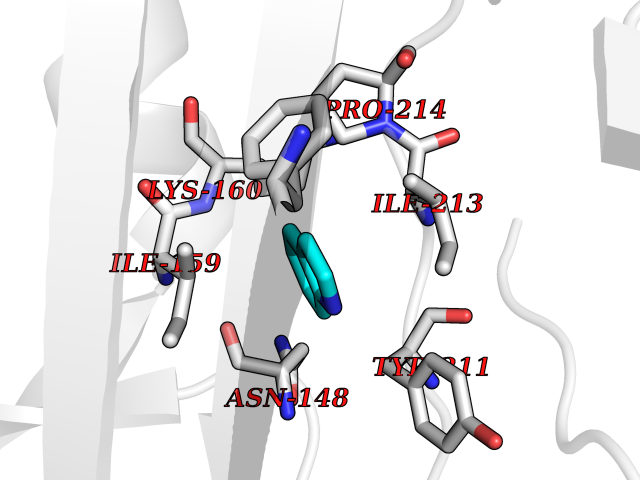 Pymol Map 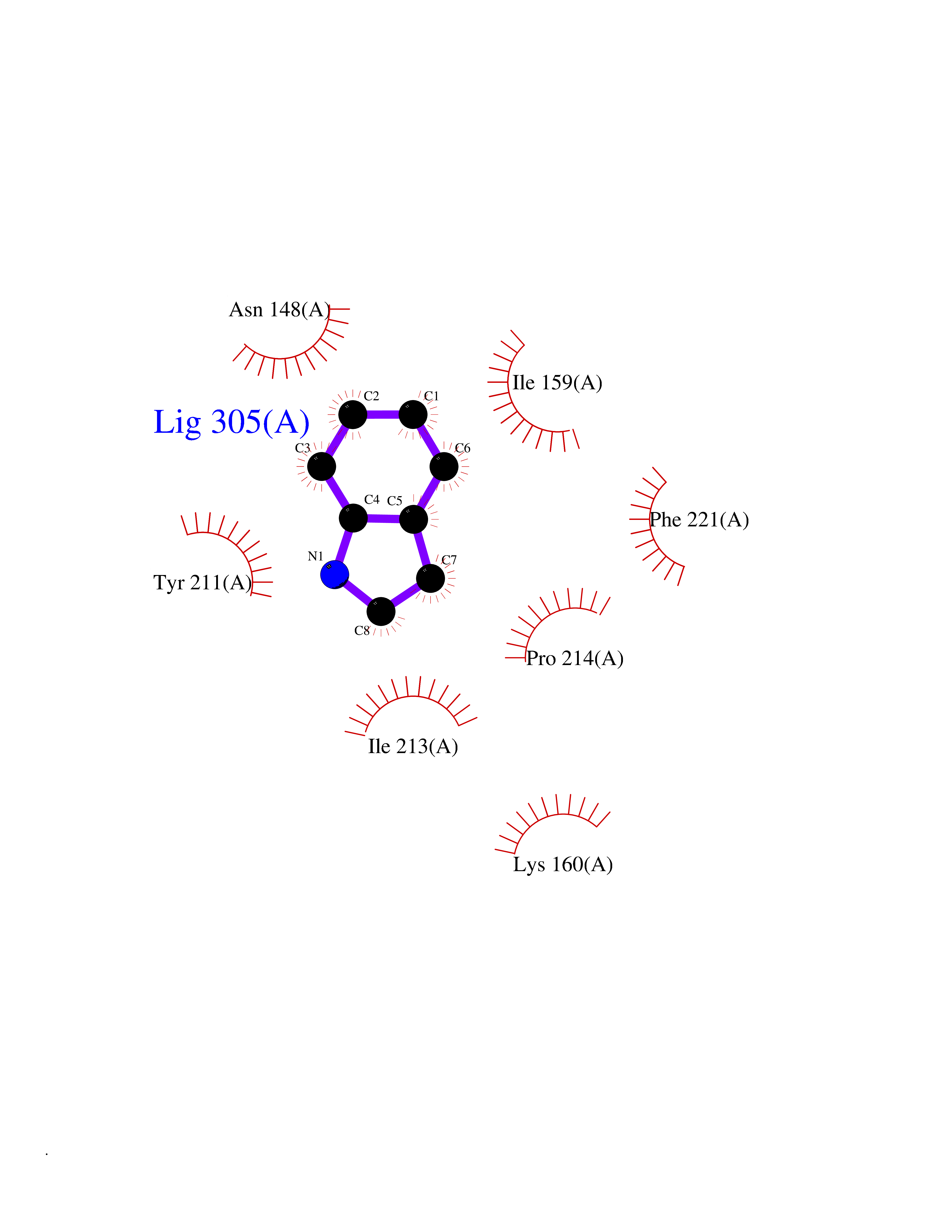 Ligplot Map 3D Binding mode Sequence TYQYNMNFEKLGKCIIINNKNFDKVTGMGVRNGTDKDAEALFKCFRSLGFDVIVYNDCSCAKMQDLLKKASEEDHTNAACFACILLSHGEENVIYGKDGVTPIKDLTAHFRGARCKTLLEKPKLFFIQACRGTEPRYKIPVEADFLFAYSTVRGSWFVQALCSILEEHGKDLEIMQILTRVNDRVARHFKKQIPCVVSMLTKELYFSQVPTYQYNMNFEKLGKCIIINNKNFDKVTGMGVRNGTDKDAEALFKCFRSLGFDVIVYNDCSCAKMQDLLKKASEEDHTNAACFACILLSHGEENVIYGKDGVTPIKDLTAHFRGARCKTLLEKPKLFFIQACRGPRYKIPVEADFLFAYSTVPGSWFVQALCSILEEHGKDLEIMQILTRVNDRVARHFESKQIPCVVSMLTKELYFSQ Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Protein arginine methyltransferase 3 (PRMT3) | 3SMQ | 5.35 | |
Target general information Gen name PRMT3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein arginine N-methyltransferase 3; Heterogeneous nuclear ribonucleoprotein methyltransferase-like protein 3; HRMT1L3 Protein family Class I-like SAM-binding methyltransferase superfamily, Protein arginine N-methyltransferase family Biochemical class NA Function Methylates (mono and asymmetric dimethylation) the guanidino nitrogens of arginyl residues in some proteins. Related diseases May be involved in T-cell exhaustion associated with chronic viral infections such as with human immunodeficiency virus (HIV) and hepatitic C virus (HCV). {ECO:0000269|PubMed:19001139, ECO:0000269|PubMed:19587053}.; DISEASE: T-cell lymphoma, subcutaneous panniculitis-like (SPTCL) [MIM:618398]: An uncommon form of T-cell non-Hodgkin lymphoma, in which cytotoxic CD8+ T-cells infiltrate subcutaneous adipose tissue, and rimming adipocytes in a lace-like pattern. Affected individuals typically present with multiple subcutaneous nodules, systemic B-cell symptoms, and, in a subset of cases, autoimmune disorders, most commonly systemic lupus erythematosus. A subset of patients develop hemophagocytic lymphohistiocytosis. SPTCL transmission pattern is consistent with autosomal recessive inheritance with incomplete penetrance. {ECO:0000269|PubMed:30374066, ECO:0000269|PubMed:30792187, ECO:0000269|Ref.2}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01752 Interacts with Q8N7W2-2; Q6FHY5; Q8WWB5; P15880 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Metal-binding; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 33627.7 Length 299 Aromaticity 0.1 Instability index 32.52 Isoelectric point 6.86 Charge (pH=7) -0.41 2D Binding mode Binding energy (Kcal/mol) -7.29 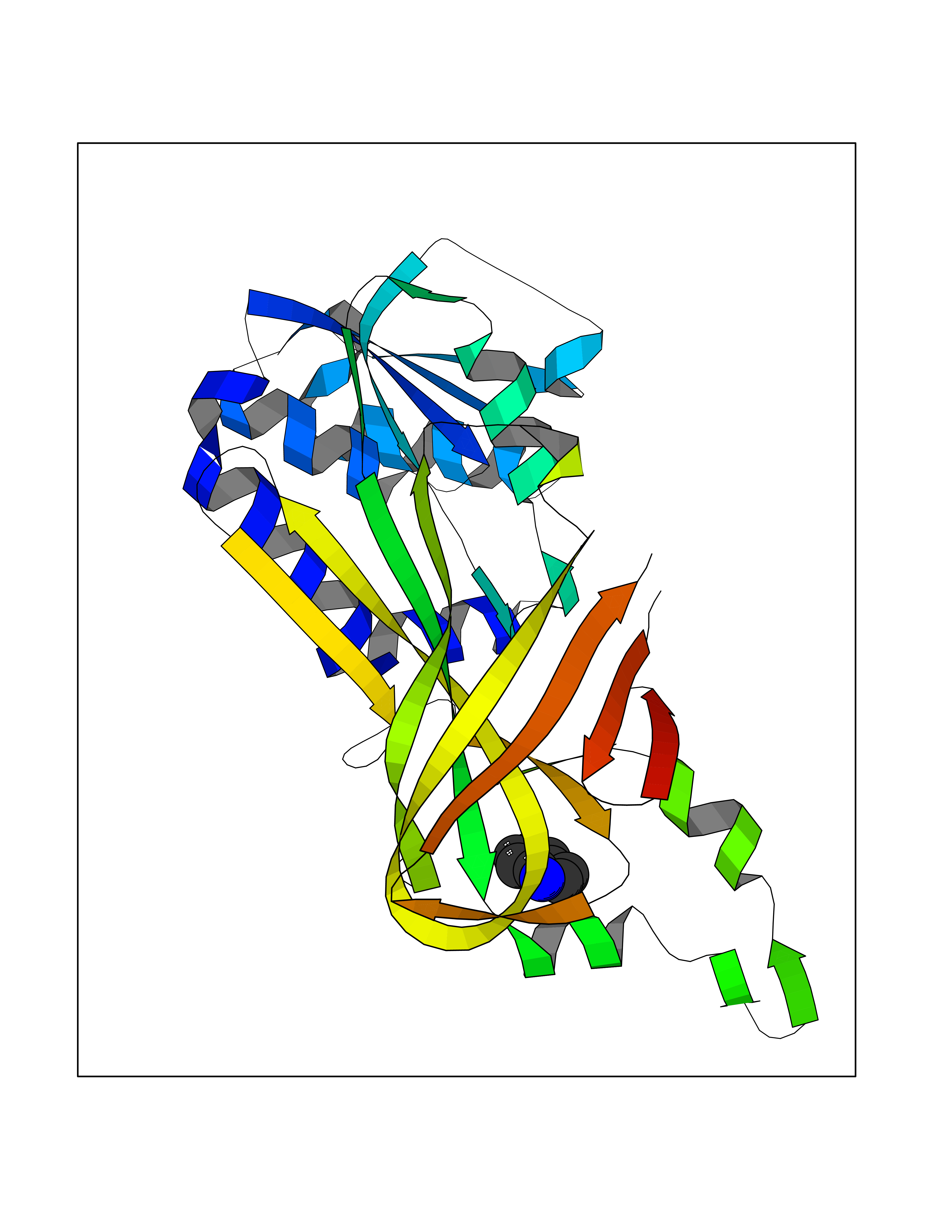 Molscript Map 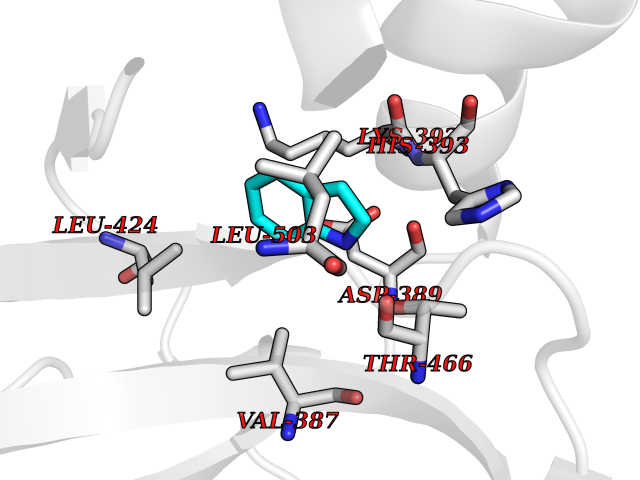 Pymol Map 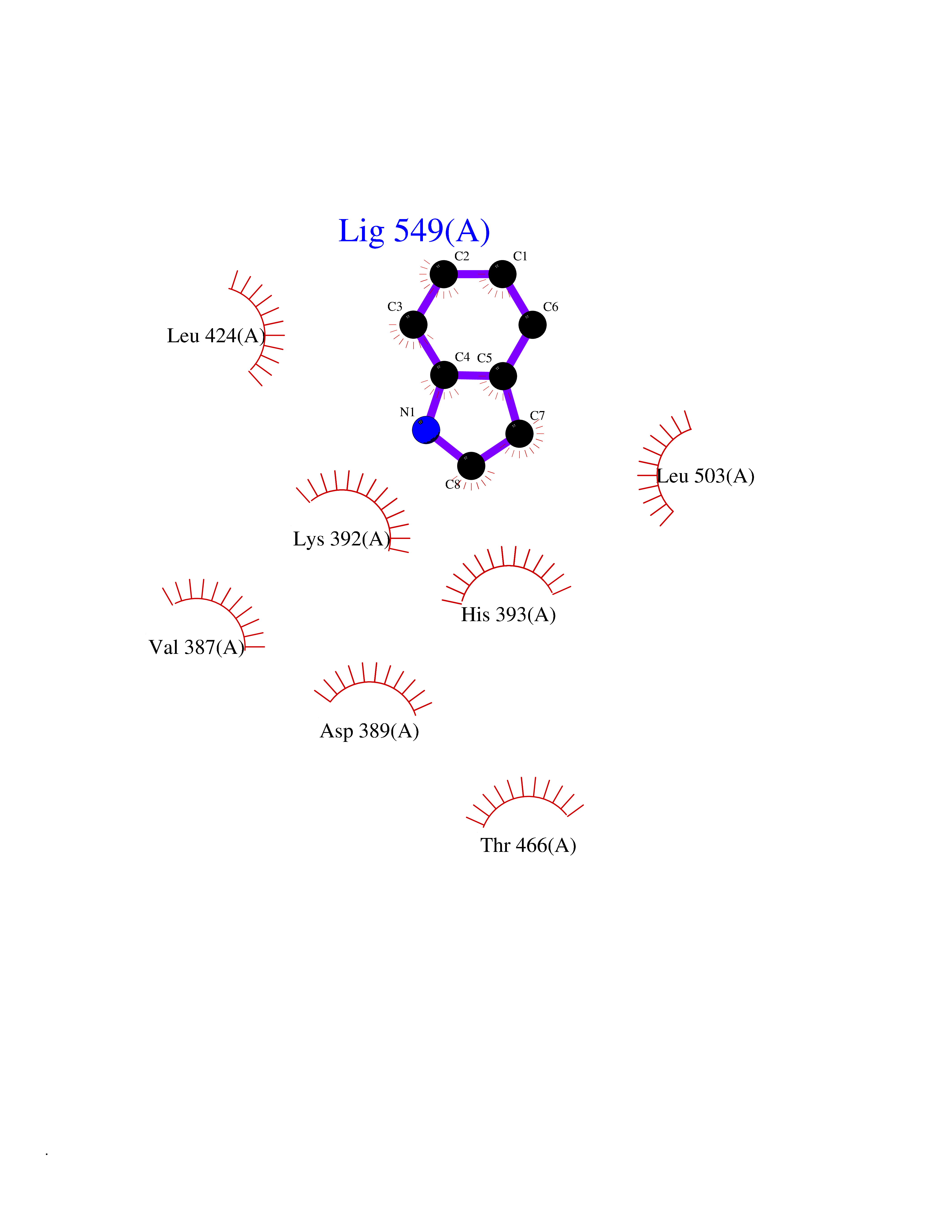 Ligplot Map 3D Binding mode Sequence HYGIHEEMLKDKIRTESYRDFIYQNPHIFKDKVVLDVGCGTGILSMFAAKAGAKKVLGVDQSEILYQAMDIIRLNKLEDTITLIKGKIEEVHLPVEKVDVIISEWMGYFLLFESMLDSVLYAKNKYLAKGGSVYPDICTISLVAVSDVNKHADRIAFWDDVYGFKMSCMKKAVIPEAVVEVLDPKTLISEPCGIKHIDCHTTSISDLEFSSDFTLKITRTSMCTAIAGYFDIYFEKNCHNRVVFSTGPQSTKTHWKQTVFLLEKPFSVKAGEALKGKVTVHKSLTVTLTLNNSTQTYGL Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Dihydrofolate synthase/folylpolyglutamate synthase | 1W78 | 5.34 | |
Target general information Gen name folC Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms dedC;JW2312;b2315 Protein family Folylpolyglutamate synthase family Biochemical class Synthase Function ATP binding.Dihydrofolate synthase activity.Metal ion binding.Tetrahydrofolylpolyglutamate synthase activity. Related diseases Complement hyperactivation, angiopathic thrombosis, and protein-losing enteropathy (CHAPLE) [MIM:226300]: An autosomal recessive disease characterized by abdominal pain and diarrhea, primary intestinal lymphangiectasia, edema due to hypoproteinemia, malabsorption, and less frequently, bowel inflammation, recurrent infections, and angiopathic thromboembolic disease. Patients' T lymphocytes show increased complement activation causing surface deposition of complement and the generation of soluble C5a. {ECO:0000269|PubMed:28657829, ECO:0000269|PubMed:28657861}. The disease is caused by variants affecting the gene represented in this entry. CHAPLE is caused by biallelic mutations in the CD55 gene. Drugs (DrugBank ID) DB02437; DB03830 Interacts with NA EC number 6.3.2.12; 6.3.2.17 Uniprot keywords 3D-structure; ATP-binding; Folate biosynthesis; Ligase; Magnesium; Metal-binding; Nucleotide-binding; One-carbon metabolism; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 44392.8 Length 414 Aromaticity 0.07 Instability index 29.47 Isoelectric point 5.2 Charge (pH=7) -15.5 2D Binding mode Binding energy (Kcal/mol) -7.28 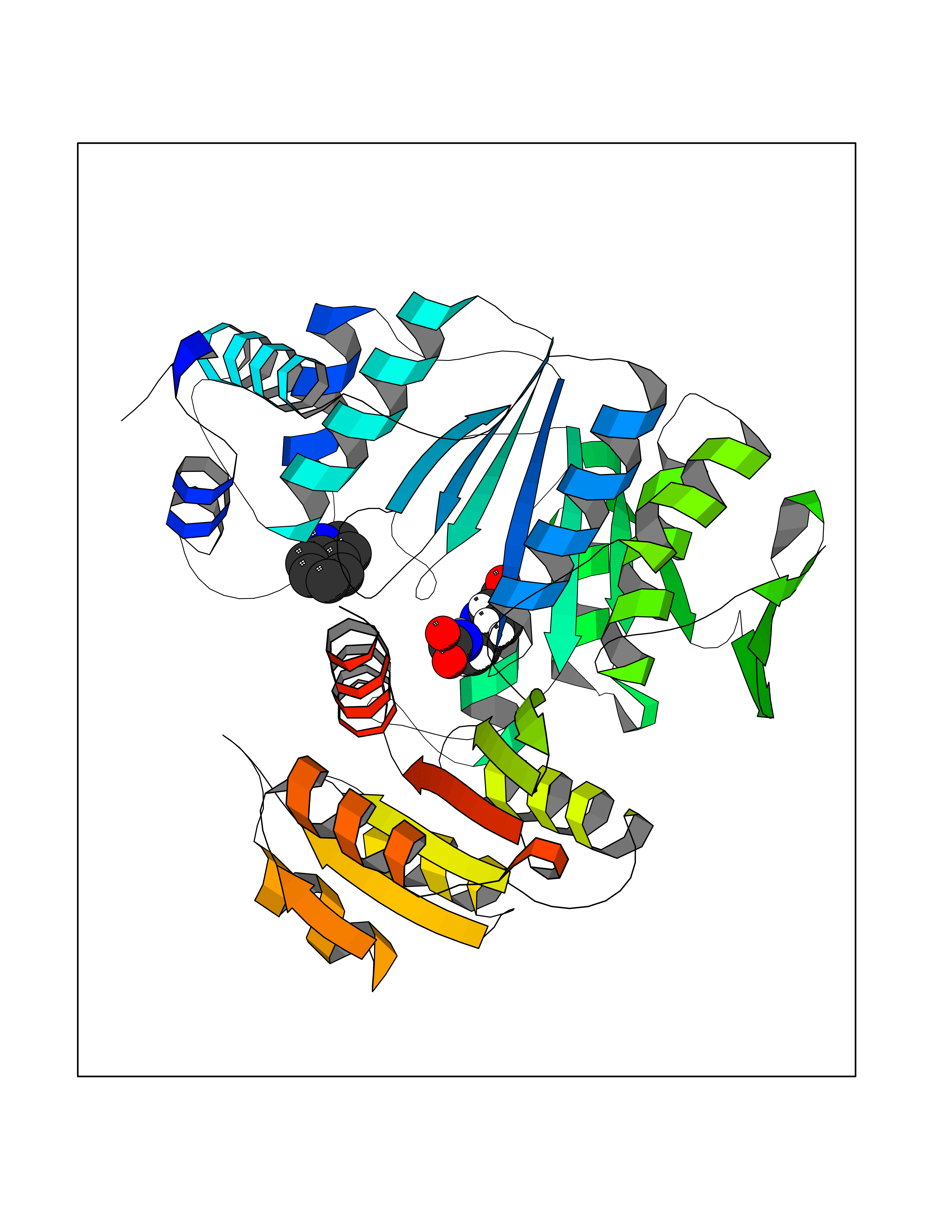 Molscript Map 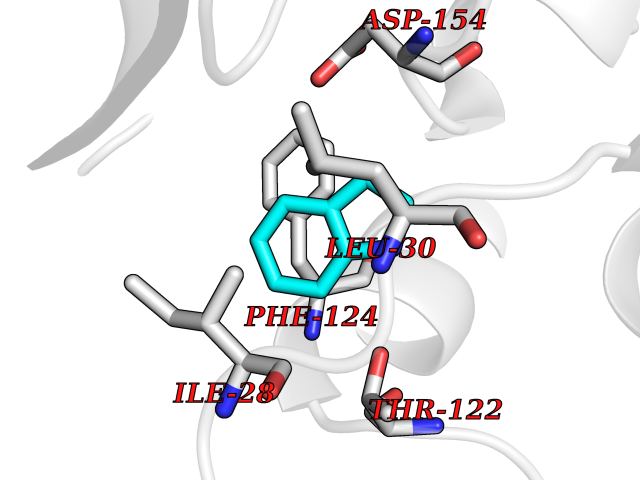 Pymol Map 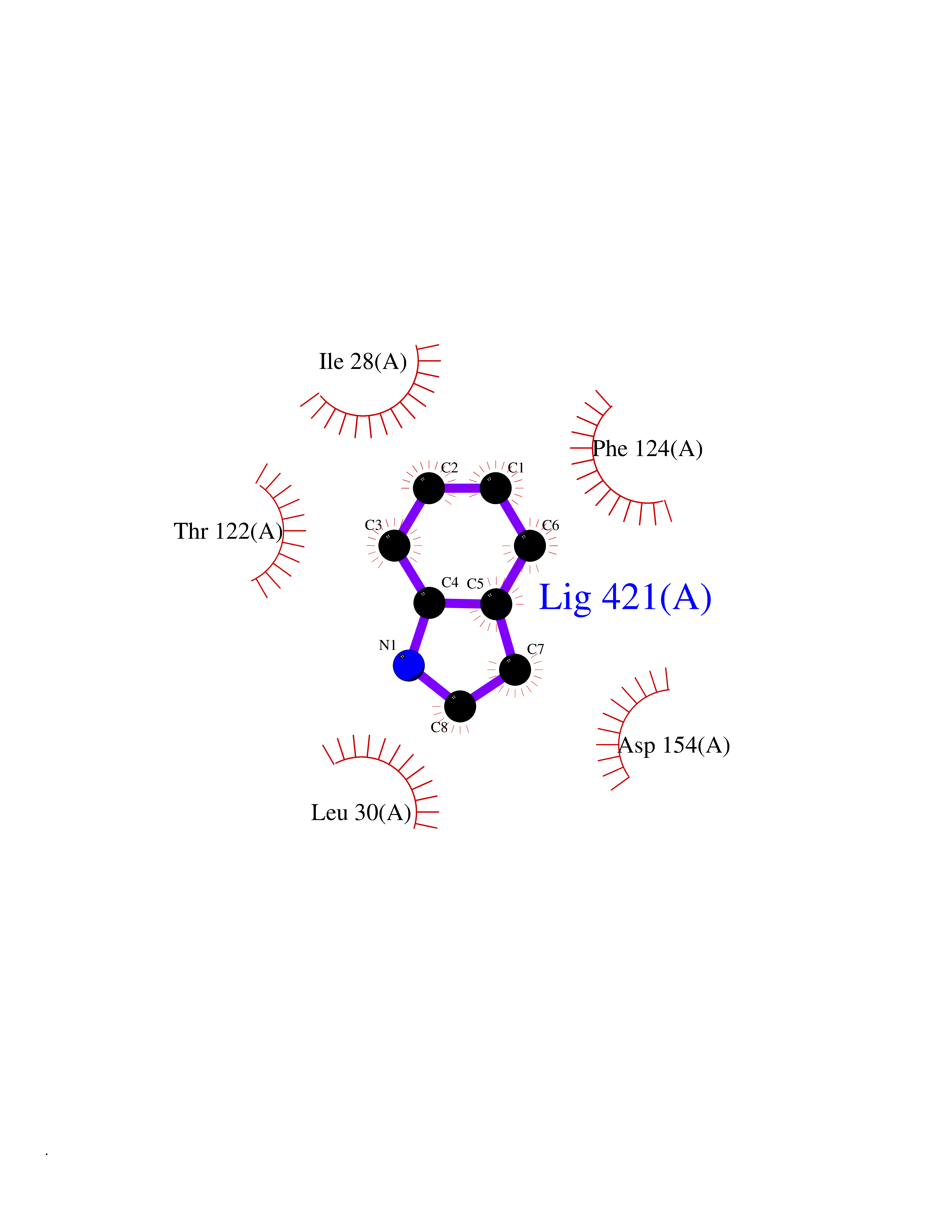 Ligplot Map 3D Binding mode Sequence TPQAASPLASWLSYLENLHSKTIDLGLERVSLVAARLGVLKPAPFVFTVAGTNGKGTTCRTLESILMAAGYKVGVYSSPHLVRYTERVRVQGQELPESAHTASFAEIESARGDISLTYFEYGTLSALWLFKQAQLDVVILEVGLGGRLDATNIVDADVAVVTSIALDHTDWLGPDRESIGREXAGIFRSEKPAIVGEPEMPSTIADVAQEKGALLQRRGVEWNYSVTDHDWAFSDAHGTLENLPLPLVPQPNAATALAALRASGLEVSENAIRDGIASAILPGRFQIVSESPRVIFDVAHNPHAAEYLTGRMKALPKNGRVLAVIGMLHDKDIAGTLAWLKSVVDDWYCAPLEGPRGATAEQLLEHLGNGKSFDSVAQAWDAAMADAKAEDTVLVCGSFHTVAHVMEVIDARRS Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | PI3-kinase alpha (PIK3CA) | 4L23 | 5.34 | |
Target general information Gen name PIK3CA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p110alpha; Serine/threonine protein kinase PIK3CA; PtdIns3kinase subunit p110alpha; PtdIns3kinase subunit alpha; PtdIns-3-kinase subunit p110-alpha; PtdIns-3-kinase subunit alpha; Phosphoinositide3kin Protein family PI3K p85 subunit family Biochemical class Kinase Function Phosphoinositide-3-kinase (PI3K) that phosphorylates PtdIns (Phosphatidylinositol), PtdIns4P (Phosphatidylinositol 4-phosphate) and PtdIns(4,5)P2 (Phosphatidylinositol 4,5-bisphosphate) to generate phosphatidylinositol 3,4,5-trisphosphate (PIP3). PIP3 plays a key role by recruiting PH domain-containing proteins to the membrane, including AKT1 and PDPK1, activating signaling cascades involved in cell growth, survival, proliferation, motility and morphology. Participates in cellular signaling in response to various growth factors. Involved in the activation of AKT1 upon stimulation by receptor tyrosine kinases ligands such as EGF, insulin, IGF1, VEGFA and PDGF. Involved in signaling via insulin-receptor substrate (IRS) proteins. Essential in endothelial cell migration during vascular development through VEGFA signaling, possibly by regulating RhoA activity. Required for lymphatic vasculature development, possibly by binding to RAS and by activation by EGF and FGF2, but not by PDGF. Regulates invadopodia formation through the PDPK1-AKT1 pathway. Participates in cardiomyogenesis in embryonic stem cells through a AKT1 pathway. Participates in vasculogenesis in embryonic stem cells through PDK1 and protein kinase C pathway. Also has serine-protein kinase activity: phosphorylates PIK3R1 (p85alpha regulatory subunit), EIF4EBP1 and HRAS. Plays a role in the positive regulation of phagocytosis and pinocytosis. Related diseases Agammaglobulinemia 7, autosomal recessive (AGM7) [MIM:615214]: A primary immunodeficiency characterized by profoundly low or absent serum antibodies and low or absent circulating B-cells due to an early block of B-cell development. Affected individuals develop severe infections in the first years of life. {ECO:0000269|PubMed:22351933}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: SHORT syndrome (SHORTS) [MIM:269880]: A rare, multisystem disease characterized by short stature, anomalies of the anterior chamber of the eye, characteristic facial features such as triangular facies, lack of facial fat, and hypoplastic nasal alae with overhanging columella, partial lipodystrophy, hernias, hyperextensibility, and delayed dentition. The clinical phenotype can include insulin resistance, nephrocalcinosis, and hearing deficits. Developmental milestones and cognition are normal. {ECO:0000269|PubMed:23810378, ECO:0000269|PubMed:23810379, ECO:0000269|PubMed:23810382}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Immunodeficiency 36 with lymphoproliferation (IMD36) [MIM:616005]: A primary immunodeficiency characterized by impaired B-cell function, hypogammaglobulinemia and recurrent infections. {ECO:0000269|PubMed:25133428}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06486; DB05210; DB08059 Interacts with Q8IZP0; P42684; P10275; P30530; P22681; P10747; Q13111; Q8IY22; P16410; Q9Y2H0; P00533; P41970; P04626; P21860; P48023; P11362; P17948; P36888; Q13480; Q13322; P62993; P42858; Q9Y6W8; P08069; P06213; P35568; Q9Y4H2; P10721; Q86VI4-3; O43561; P29376; Q92918; P45983; P08581; Q8WX92; P04629; P09619; P42336; P42338; O00329; O00459; Q13905; P26373; P19793; Q9UPX8; Q9H0K1; Q96EB6; Q07889; P12931; P30874; P58753; O15455; Q15661; Q9ULW0; Q9UKW4; P35570; P03496; Q6PFX7; Q8BM65-4; Q99152; P0DOJ9; P01023; Q9NY61; O95704; P05067; P51451; Q9UQM7; P29466-3; P55210; P06493; P20674; P09172; P50570-2; P01100; Q8TB36; P14136; P62993; P42858; P35568; P05556; P05412; P07948; Q13387-4; P41227; P01111; Q9Y6R0; P42336; P42338; O00329; P17612; P49810; P63000; P51812; P23443-4; P34741; Q9NP31; P84022; P12931; Q9UNE7; Q15583-2; O60784-2; O14656-2; P02766; P42681; P60604; Q9UBQ0-2; O76024; Q8IUH5 EC number EC 2.7.1.153 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Disease variant; Dwarfism; Host-virus interaction; Phosphoprotein; Protein transport; Proteomics identification; Reference proteome; Repeat; SH2 domain; SH3 domain; Stress response; Transport; Ubl conjugation Protein physicochemical properties Chain ID B Molecular weight (Da) 73365.8 Length 625 Aromaticity 0.1 Instability index 48.48 Isoelectric point 7.81 Charge (pH=7) 3.22 2D Binding mode Binding energy (Kcal/mol) -7.28 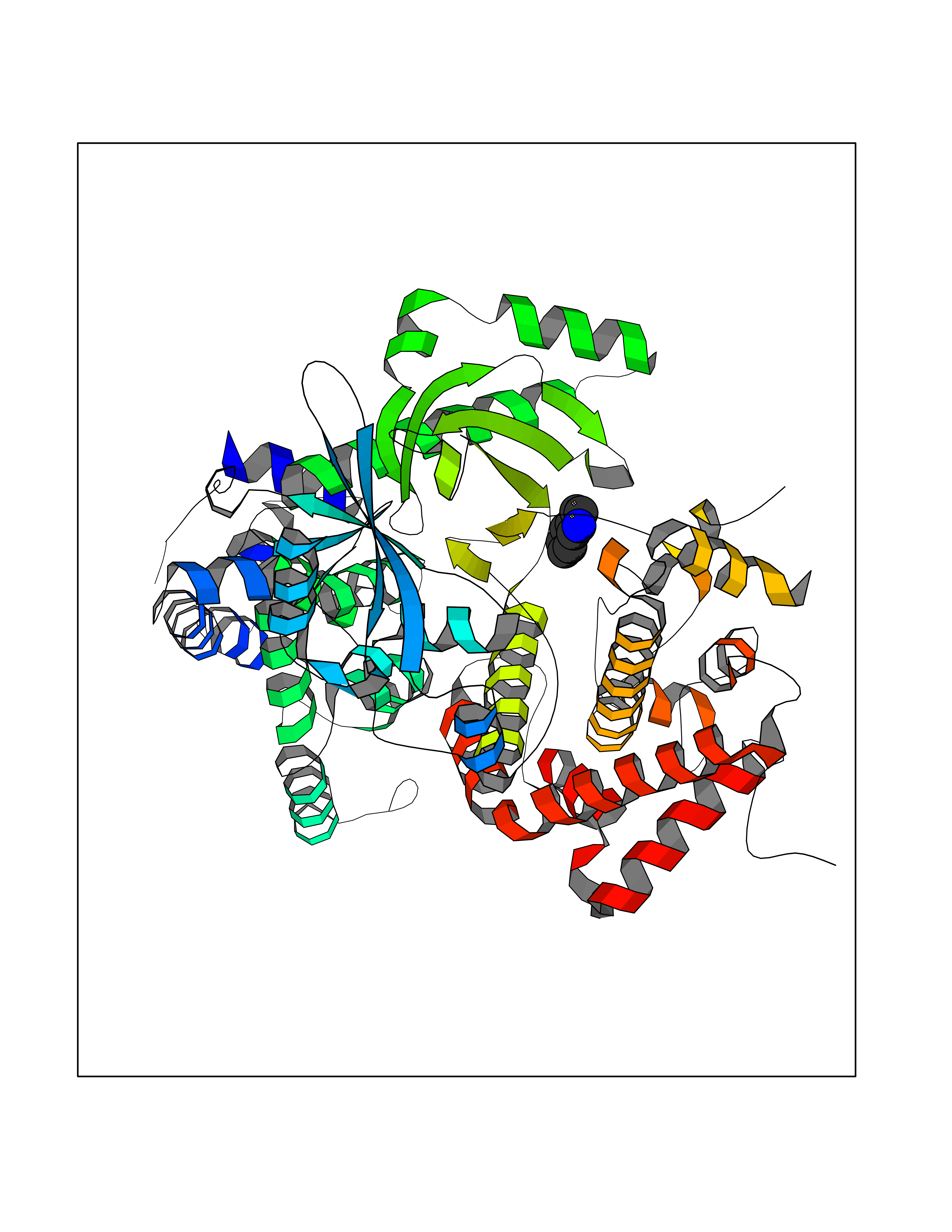 Molscript Map 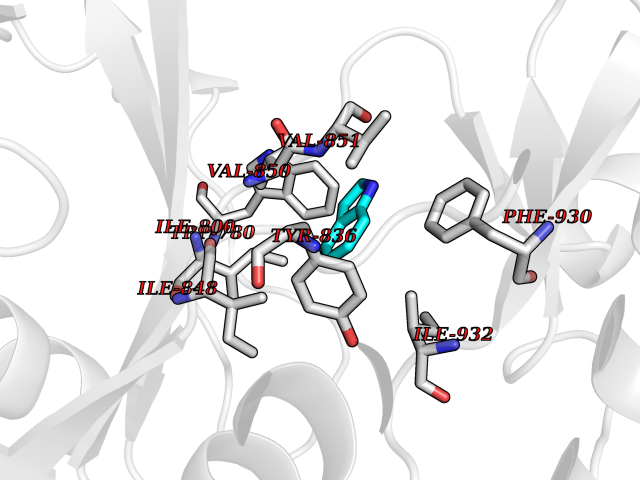 Pymol Map 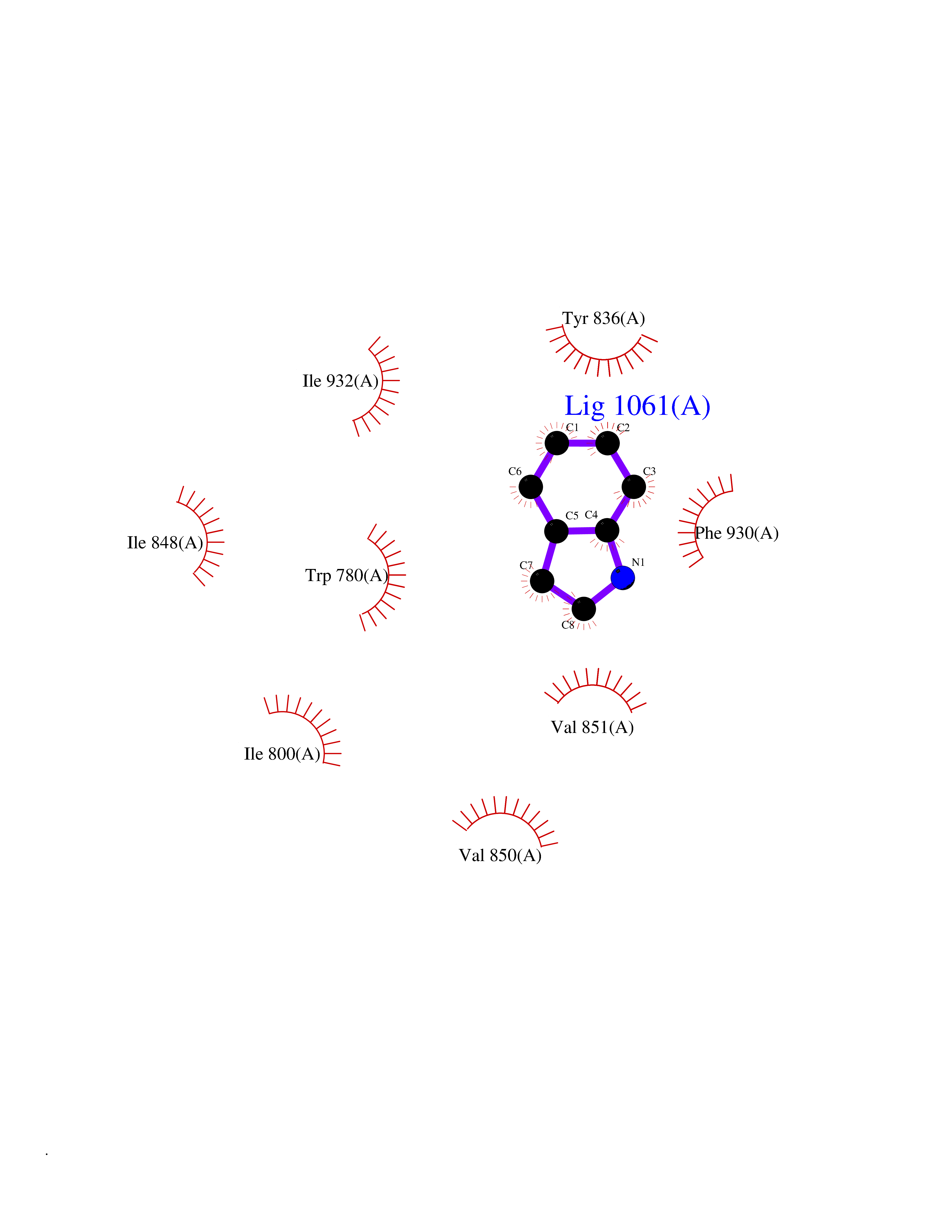 Ligplot Map 3D Binding mode Sequence ILNREIGFAIGMPVCEFDMVKDPEVQDFRRNILNVCKEAVDLRDLNSPHSRAMYVYPPNVESSPELPKHIYNKLDKGQIIVVIWVIVSPNNDKQKYTLKINHDCVPEQVIAEAIRKYILKVCGCDEYFLEKYPLSQYKYIRSCIMLGRMPNLMLMAKESLYSQLPMDCFTMPSYSLLDCNYPDPMVRGFAVRCLEKYLTDDKLSQYLIQLVQVLKYEQYLDNLLVRFLLKKALTNQRIGHFFFWHLKSEMHNKTVSQRFGLLLESYCRACGMYLKHLNRQVEAMEKLINLTDILKQEKKDETQKVQMKFLVEQMRRPDFMDALQGFLSPLNPAHQLGNLRLEECRIMSSAKRPLWLNWENPDIMSELLFQNNEIIFKNGDDLRQDMLTLQIIRIMENIWQNQGLDLRMLPYGCLSIGDCVGLIEVVRNSHTIMQIQCKNSHTLHQWLKDKNKGEIYDAAIDLFTRSCAGYCVATFILGIGDRHNSNIMVKDDGQLFHIDFGHFLDHKKKKFGYKRERVPFVLTQDFLIVISKGAQECTKTREFERFQEMCYKAYLAIRQHANLFINLFSMMLGSGMPELQSFDDIAYIRKTLALDKTEQEALEYFMKQMNDAHHGGWTTKMDWIF Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Mycobacterium Membrane protein mmpL3 (MycB mmpL3) | 7NVH | 5.34 | |
Target general information Gen name MycB mmpL3 Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID Synonyms Trehalose monomycolate exporter MmpL3; TMM exporter MmpL3 Protein family Resistance-nodulation-cell division (RND) (TC 2.A.6) family, MmpL subfamily Biochemical class NA Function Transports trehalose monomycolate (TMM) across the inner membrane. Could also be part of a heme-iron acquisition system. Related diseases Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:8955068}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:17332249}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 3 (NS3) [MIM:609942]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:16474405, ECO:0000269|PubMed:16773572, ECO:0000269|PubMed:17056636, ECO:0000269|PubMed:17468812, ECO:0000269|PubMed:19396835, ECO:0000269|PubMed:20949621}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Gastric cancer (GASC) [MIM:613659]: A malignant disease which starts in the stomach, can spread to the esophagus or the small intestine, and can extend through the stomach wall to nearby lymph nodes and organs. It also can metastasize to other parts of the body. The term gastric cancer or gastric carcinoma refers to adenocarcinoma of the stomach that accounts for most of all gastric malignant tumors. Two main histologic types are recognized, diffuse type and intestinal type carcinomas. Diffuse tumors are poorly differentiated infiltrating lesions, resulting in thickening of the stomach. In contrast, intestinal tumors are usually exophytic, often ulcerating, and associated with intestinal metaplasia of the stomach, most often observed in sporadic disease. {ECO:0000269|PubMed:14534542, ECO:0000269|PubMed:3034404, ECO:0000269|PubMed:7773929}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in KRAS are a cause of pylocytic astrocytoma (PA). Pylocytic astrocytomas are neoplasms of the brain and spinal cord derived from glial cells which vary from histologically benign forms to highly anaplastic and malignant tumors. {ECO:0000269|PubMed:16247081}.; DISEASE: Cardiofaciocutaneous syndrome 2 (CFC2) [MIM:615278]: A form of cardiofaciocutaneous syndrome, a multiple congenital anomaly disorder characterized by a distinctive facial appearance, heart defects and intellectual disability. Heart defects include pulmonic stenosis, atrial septal defects and hypertrophic cardiomyopathy. Some affected individuals present with ectodermal abnormalities such as sparse, friable hair, hyperkeratotic skin lesions and a generalized ichthyosis-like condition. Typical facial features are similar to Noonan syndrome. They include high forehead with bitemporal constriction, hypoplastic supraorbital ridges, downslanting palpebral fissures, a depressed nasal bridge, and posteriorly angulated ears with prominent helices. CFC2 patients often do not have the skin abnormalities, such as ichthyosis, hyperkeratosis, and hemangioma observed in CFC1. {ECO:0000269|PubMed:16474404, ECO:0000269|PubMed:16474405, ECO:0000269|PubMed:17056636, ECO:0000269|PubMed:20949621, ECO:0000269|PubMed:21797849}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: KRAS mutations are involved in cancer development. {ECO:0000269|PubMed:14534542, ECO:0000269|PubMed:1553789, ECO:0000269|PubMed:16533793, ECO:0000269|PubMed:24623306, ECO:0000269|PubMed:3034404, ECO:0000269|PubMed:3627975, ECO:0000269|PubMed:6092920, ECO:0000269|PubMed:6695174, ECO:0000269|PubMed:7773929}.; DISEASE: Oculoectodermal syndrome (OES) [MIM:600268]: A syndrome characterized by the association of epibulbar dermoids and aplasia cutis congenita. Affected individuals show multiple, asymmetric, atrophic, non-scarring and hairless regions that may be associated with hamartomas. Ectodermal changes include linear hyperpigmentation that may follow the lines of Blaschko and rarely epidermal nevus-like lesions. Epibulbar dermoids may be uni-or bilateral. Additional ocular anomalies such as skin tags of the upper eyelid, rarely optic nerve or retinal changes, and microphthalmia can be present. The phenotypic expression is highly variable, and various other abnormalities have occasionally been reported including growth failure, lymphedema, cardiovascular defects, as well as neurodevelopmental symptoms like developmental delay, epilepsy, learning difficulties, and behavioral abnormalities. Benign tumor-like lesions such as nonossifying fibromas of the long bones and giant cell granulomas of the jaws have repeatedly been observed and appear to be age-dependent, becoming a common manifestation in individuals aged 5 years or older. {ECO:0000269|PubMed:25808193, ECO:0000269|PubMed:26970110, ECO:0000269|PubMed:30891959}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Schimmelpenning-Feuerstein-Mims syndrome (SFM) [MIM:163200]: A disease characterized by sebaceous nevi, often on the face, associated with variable ipsilateral abnormalities of the central nervous system, ocular anomalies, and skeletal defects. Many oral manifestations have been reported, not only including hypoplastic and malformed teeth, and mucosal papillomatosis, but also ankyloglossia, hemihyperplastic tongue, intraoral nevus, giant cell granuloma, ameloblastoma, bone cysts, follicular cysts, oligodontia, and odontodysplasia. Sebaceous nevi follow the lines of Blaschko and these can continue as linear intraoral lesions, as in mucosal papillomatosis. {ECO:0000269|PubMed:30891959}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Cell wall biogenesis/degradation; Lipid transport; Membrane; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 77237 Length 717 Aromaticity 0.09 Instability index 33.34 Isoelectric point 8.65 Charge (pH=7) 4.3 2D Binding mode Binding energy (Kcal/mol) -7.29 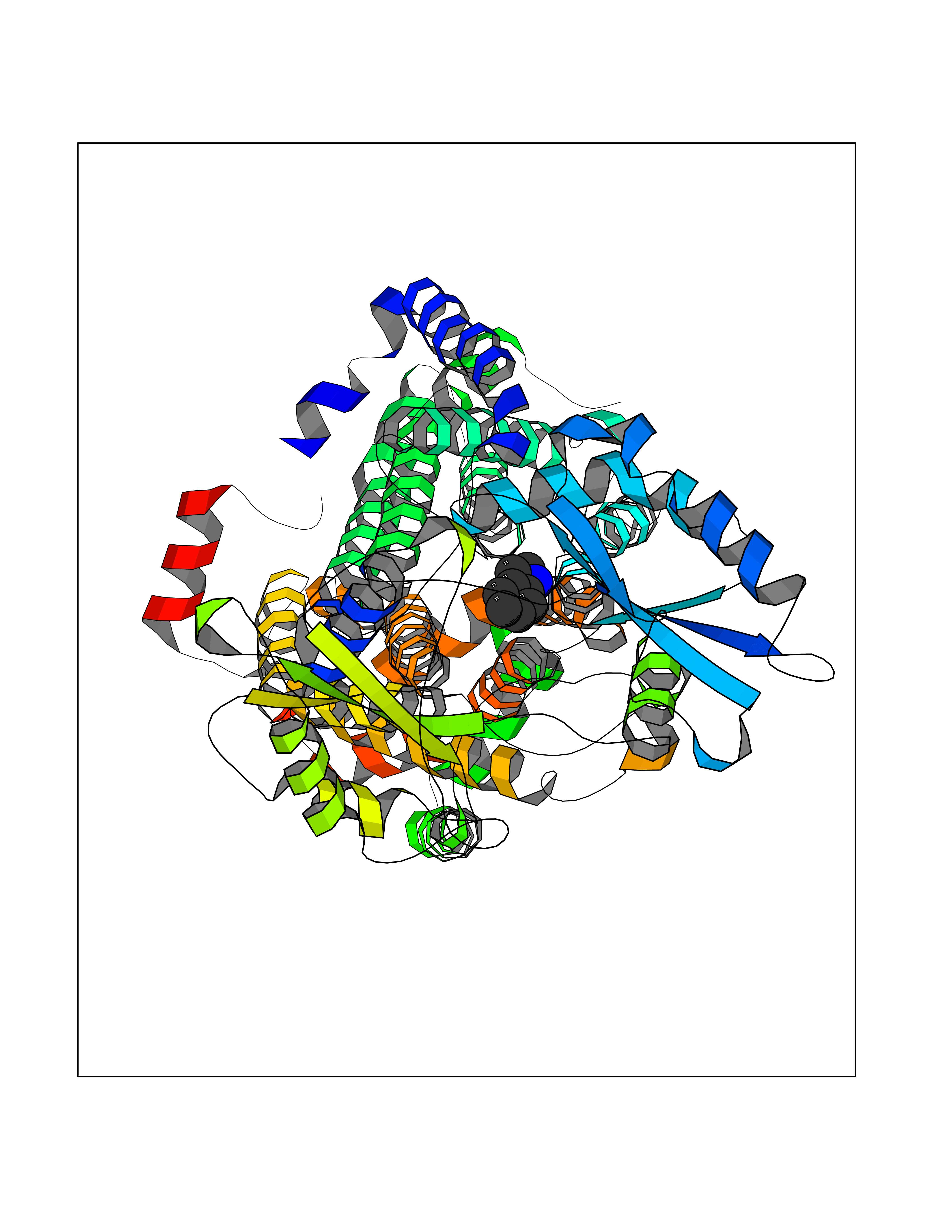 Molscript Map 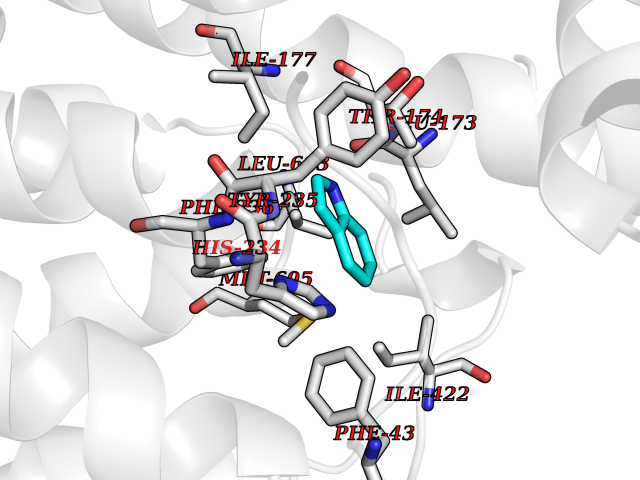 Pymol Map 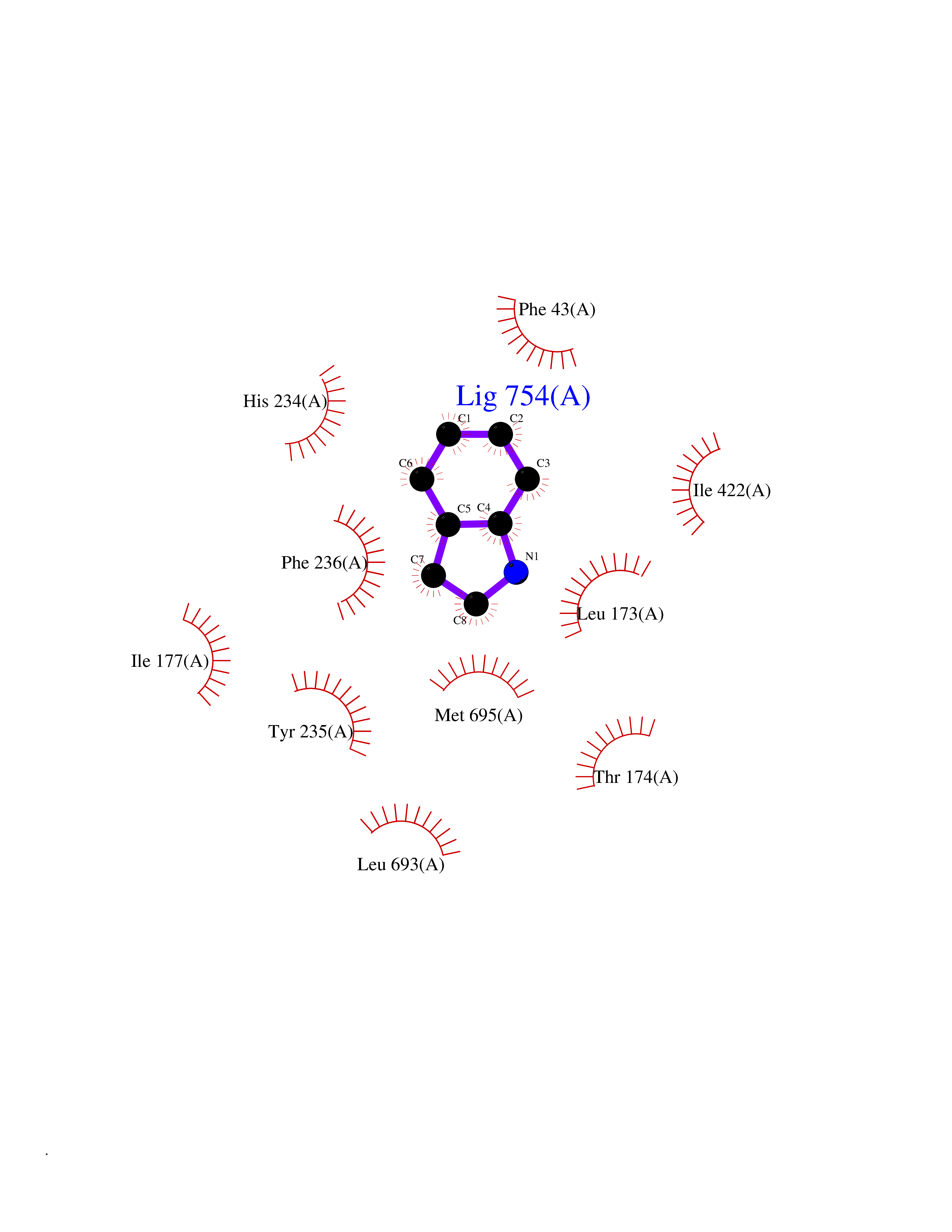 Ligplot Map 3D Binding mode Sequence MFAWWGRTVYRYRFIVIGVMVALCLGGGVFGLSLGKHVTQSGFYDDGSQSVQASVLGDQVYGRDRSGHIVAIFQAPAGKTVDDPAWSKKVVDELNRFQQDHPDQVLGWAGYLRASQATGMATADKKYTFVSIPLKGDDDDTILNNYKAIAPDLQRLDGGTVKLAGLQPVAEALTGTIATDQRRMEVLALPLVAVVLFFVFGGVIAAGLPVMVGGLCIAGALGIMRFLAIFGPVHYFAQPVVSLIGLGIAIDYGLFIVSRFREEIAEGYDTETAVRRTVITAGRTVTFSAVLIVASAIGLLLFPQGFLKSLTYATIASVMLSAILSITVLPACLGILGKHVDAEEVEAGFWGKLVNRVMKRPVLFAAPIVIIMILLIIPVGKLSLGGISEKYLPPTNSVRQAQEEFDKLFPGYRTNPLTLVIQTSNHQPVTDAQIADIRSKAMAIGGFIEPDNDPANMWQERAYAVGASKDPSVRVLQNGLINPADASKKLTELRAITPPKGITVLVGGTPALELDSIHGLFAKMPLMVVILLTTTIVLMFLAFGSVVLPIKATLMSALTLGSTMGILTWIFVDGHFSKWLNFTPTPLTAPVIGLIIALVFGLSTDYEVFLVSRMVEARERGMSTQEAIRIGTAATGRIITAAALIVAVVAGAFVFSDLVMMKYLAFGLMAALLLDATVVRMFLVPSVMKLLGDDCWWAPRWARRLQTRIGLGEIHLP Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Caspase-6 (CASP6) | 4NBL | 5.34 | |
Target general information Gen name CASP6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MCH2; Caspase-6 subunit p18; Caspase-6 subunit p11; CASP-6; Apoptotic protease Mch-2 Protein family Peptidase C14A family Biochemical class Peptidase Function Cleaves poly(ADP-ribose) polymerase in vitro, as well as lamins. Overexpression promotes programmed cell death. Involved in the activation cascade of caspases responsible for apoptosis execution. Related diseases Growth hormone deficiency, isolated, 1A (IGHD1A) [MIM:262400]: An autosomal recessive, severe deficiency of growth hormone leading to dwarfism. Patients often develop antibodies to administered growth hormone. {ECO:0000269|PubMed:8364549}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 1B (IGHD1B) [MIM:612781]: An autosomal recessive deficiency of growth hormone leading to short stature. Patients have low but detectable levels of growth hormone, significantly retarded bone age, and a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:12655557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Kowarski syndrome (KWKS) [MIM:262650]: A syndrome clinically characterized by short stature associated with bioinactive growth hormone, normal or slightly increased growth hormone secretion, pathologically low insulin-like growth factor 1 levels, and normal catch-up growth on growth hormone replacement therapy. {ECO:0000269|PubMed:17519310, ECO:0000269|PubMed:8552145, ECO:0000269|PubMed:9276733}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 2 (IGHD2) [MIM:173100]: An autosomal dominant deficiency of growth hormone leading to short stature. Clinical severity is variable. Patients have a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:11502836, ECO:0000269|PubMed:9152628}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9Y614; Q6DHV7-2; Q6UY14-3; Q96MA6; Q5T2L2; Q96Q83-2; Q9Y303-2; Q9NU02; P09525; P06727; Q8WW27; Q66PJ3-4; Q6XD76; P18848; Q9H0Y0; Q14032; P54687-4; P06276; Q9NSI6-4; Q96Q07-2; Q9H0W9-3; Q9NQ89; Q13901; Q3SXR2; Q8N1A6; P17655; P20807-4; P42574; P55212; O00257-3; P24863; Q9NNX6-10; Q9UJX2; P42773; O95674; Q494V2-2; Q8WUX9; Q9Y3D0; Q8N365; Q3SX64; Q99966; P09496-2; Q6PJW8-3; Q96BR5; P02458-1; Q9UGL9; Q9UKG9-2; P26998; P35222; Q53TN4; P61962; O60479; Q96EY1-3; Q92782-2; Q9BPU6; A0AVK6; Q658K8; O00303; Q13347; O00472; O00423; Q6NXG1-3; Q49AJ0-4; Q8N128-2; Q8IZU1; Q6ZNL6; Q9NSA1; Q06547-3; Q49A26-4; Q9HAV0; Q6NXT2; Q9BT25; Q9NRZ9-6; Q96EW2-2; P42858; Q8N6M8-2; Q92613; P0C870; Q9UK76; Q8N5Z5; Q8TBB5-2; Q9UH77; Q8N4N3-2; Q5JUW0-3; Q8N1A0; P13473-2; Q6DKI2; Q9H2C1; Q8N0U6; Q9Y234; Q8TBB1; Q1L5Z9; Q96JB6; Q16609; Q8IYG6; P0DP58-2; Q969L2; P27338; A6NJ78-4; Q96C03-3; Q8N5J2-3; A0A0A0MR05; P34949-2; Q9BV20; Q6IN84-2; A2RUH7; P01106; Q9H7X0; Q15742-2; Q9UJ70-2; Q8NDH3-5; Q96HA8; P36639-4; Q8NFH4; Q8NFH3; Q7Z3B4; Q6N063-2; Q6GQQ9-2; Q9H8K7; Q99447; P27815-4; O15534; Q9BUL5; Q00169; P48739; P61925; Q58EX7-2; O60664; Q14181; P0DPB6; P36954; Q07869; O60927; Q6ZMI0-5; P54619; Q8NCQ7-2; P41222; P29074; Q8WUD1-2; Q5R372-9; Q9HD47-3; Q09028; Q04206; P47804-3; Q15382; Q06587; Q8N5U6; P62701; Q66K80; Q01826; O15126; P22307-3; Q9BRK5; Q9NTN9-3; P01011; Q15393; Q9NR46; Q9BZQ2; O60902-3; Q86US8; P37840; Q96H20; Q13573; Q7Z6I5; Q496A3; Q9C004; Q5W111-2; Q96BD6; Q92797-2; O60506-4; O15273; Q86WV5; Q96A09; P54274-2; P22735; O43548; Q9NQ88; Q9UIK5-2; Q53NU3; P04637; Q12888; P36406; Q86WT6-2; Q13885; P49459; Q9P1Q0-4; Q9NX94; Q8NA23-2; Q9BQA1; O00755; O95070; O43829; Q8IWT0-2; Q53FD0-2; Q05CR2; Q96JL9-2; Q96LX8; Q3KNS6-3; Q5JTY5; A0A384MDV8; B7Z3E8; Q86V28 EC number EC 3.4.22.59 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; Autocatalytic cleavage; Cytoplasm; Hydrolase; Lipoprotein; Nucleus; Palmitate; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Thiol protease; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 57170 Length 500 Aromaticity 0.12 Instability index 31.33 Isoelectric point 8.05 Charge (pH=7) 4.52 2D Binding mode Binding energy (Kcal/mol) -7.29 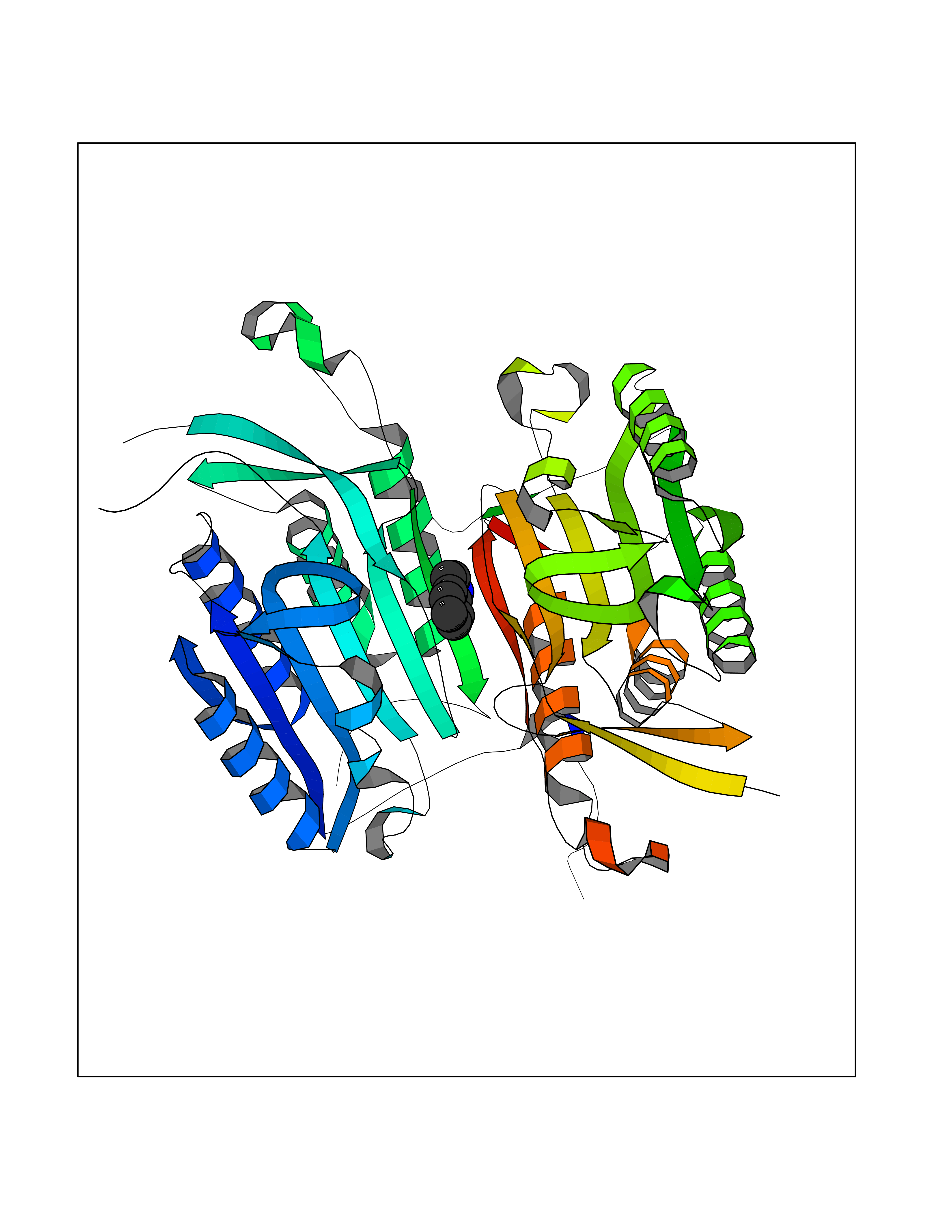 Molscript Map 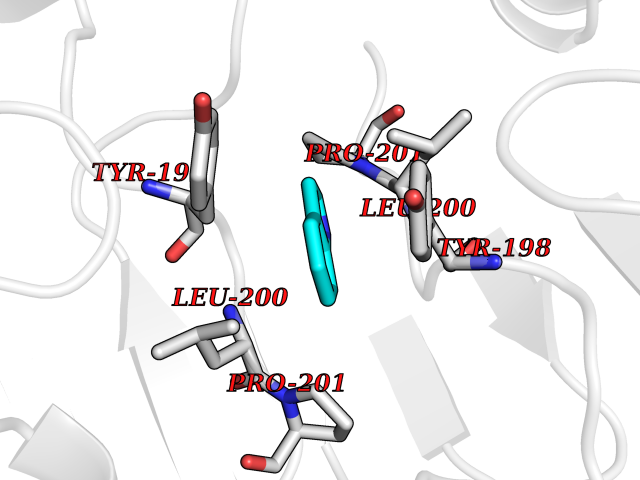 Pymol Map 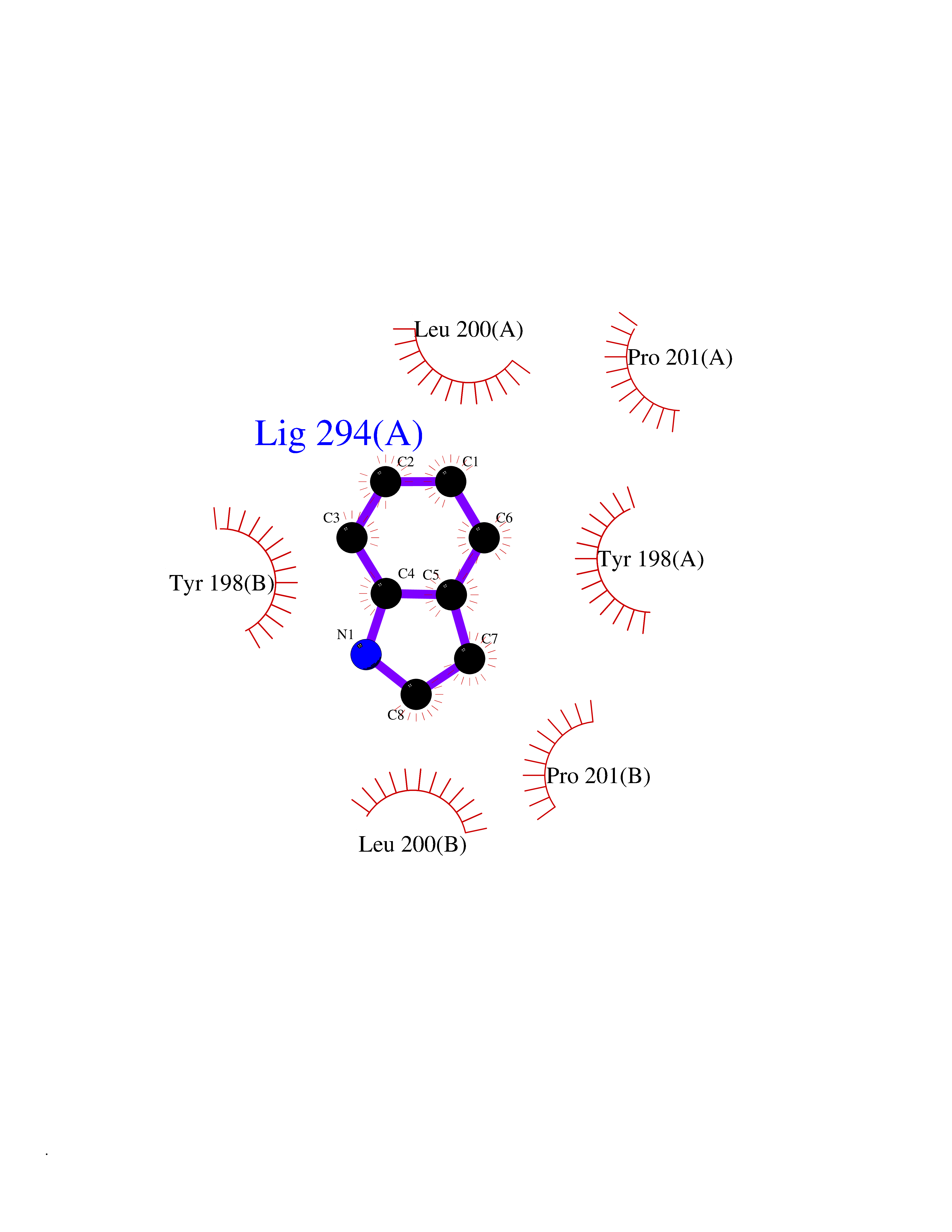 Ligplot Map 3D Binding mode Sequence AFYKREMFDPAEKYKMDHRRRGIALIFNHERFFWHLTLPERRGTCADRDNLTRRFSDLGFEVKCFNDLKAEELLLKIHEVSTVSHADADCFVCVFLSHGEGNHIYAYDAKIEIQTLTGLFKGDKCHSLVGKPKIFIIQAARGNQHDVPVIPDTNITEVDAASVYTLPAGADFLMCYSVAEGYYSHRETVNGSWYIQDLCEMLGKYGSSLEFTELLTLVNRKVSQRRVDFCKDPSAIGKKQVPCFASMLTKKLHFFPKSMFDPAEKYKMDHRRRGIALIFNHERFFWHLTLPERRGTCADRDNLTRRFSDLGFEVKCFNDLKAEELLLKIHEVSTVSHADADCFVCVFLSHGEGNHIYAYDAKIEIQTLTGLFKGDKCHSLVGKPKIFIIQAARGNTNITEVDAASVYTLPAGADFLMCYSVAEGYYSHRETVNGSWYIQDLCEMLGKYGSSLEFTELLTLVNRKVSQRRVDFCKDPSAIGKKQVPCFASMLTKKLHFFPK Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | DNA mismatch repair protein MSH2 (MSH2) | 3THX | 5.34 | |
Target general information Gen name MSH2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hMSH2; MutS protein homolog 2; Mismatch repair gene Msh2 Protein family DNA mismatch repair MutS family Biochemical class NA Function Forms two different heterodimers: MutS alpha (MSH2-MSH6 heterodimer) and MutS beta (MSH2-MSH3 heterodimer) which binds to DNA mismatches thereby initiating DNA repair. When bound, heterodimers bend the DNA helix and shields approximately 20 base pairs. MutS alpha recognizes single base mismatches and dinucleotide insertion-deletion loops (IDL) in the DNA. MutS beta recognizes larger insertion-deletion loops up to 13 nucleotides long. After mismatch binding, MutS alpha or beta forms a ternary complex with the MutL alpha heterodimer, which is thought to be responsible for directing the downstream MMR events, including strand discrimination, excision, and resynthesis. Recruits DNA helicase MCM9 to chromatin which unwinds the mismatch containg DNA strand. ATP binding and hydrolysis play a pivotal role in mismatch repair functions. The ATPase activity associated with MutS alpha regulates binding similar to a molecular switch: mismatched DNA provokes ADP-->ATP exchange, resulting in a discernible conformational transition that converts MutS alpha into a sliding clamp capable of hydrolysis-independent diffusion along the DNA backbone. This transition is crucial for mismatch repair. MutS alpha may also play a role in DNA homologous recombination repair. In melanocytes may modulate both UV-B-induced cell cycle regulation and apoptosis. Component of the post-replicative DNA mismatch repair system (MMR). Related diseases Lynch syndrome 1 (LYNCH1) [MIM:120435]: A form of Lynch syndrome, an autosomal dominant disease associated with marked increase in cancer susceptibility. It is characterized by a familial predisposition to early-onset colorectal carcinoma (CRC) and extra-colonic tumors of the gastrointestinal, urological and female reproductive tracts. Lynch syndrome is reported to be the most common form of inherited colorectal cancer in the Western world. Clinically, it is often divided into two subgroups. Type I is characterized by hereditary predisposition to colorectal cancer, a young age of onset, and carcinoma observed in the proximal colon. Type II is characterized by increased risk for cancers in certain tissues such as the uterus, ovary, breast, stomach, small intestine, skin, and larynx in addition to the colon. Diagnosis of classical Lynch syndrome is based on the Amsterdam criteria: 3 or more relatives affected by colorectal cancer, one a first degree relative of the other two; 2 or more generation affected; 1 or more colorectal cancers presenting before 50 years of age; exclusion of hereditary polyposis syndromes. The term 'suspected Lynch syndrome' or 'incomplete Lynch syndrome' can be used to describe families who do not or only partially fulfill the Amsterdam criteria, but in whom a genetic basis for colon cancer is strongly suspected. {ECO:0000269|PubMed:10375096, ECO:0000269|PubMed:10386556, ECO:0000269|PubMed:10528862, ECO:0000269|PubMed:10573010, ECO:0000269|PubMed:10612836, ECO:0000269|PubMed:10777691, ECO:0000269|PubMed:10829038, ECO:0000269|PubMed:11726306, ECO:0000269|PubMed:11870161, ECO:0000269|PubMed:11920458, ECO:0000269|PubMed:12112654, ECO:0000269|PubMed:12124176, ECO:0000269|PubMed:12132870, ECO:0000269|PubMed:12200596, ECO:0000269|PubMed:12362047, ECO:0000269|PubMed:12373605, ECO:0000269|PubMed:12655564, ECO:0000269|PubMed:12655568, ECO:0000269|PubMed:12658575, ECO:0000269|PubMed:14635101, ECO:0000269|PubMed:15046096, ECO:0000269|PubMed:15300854, ECO:0000269|PubMed:15342696, ECO:0000269|PubMed:15365995, ECO:0000269|PubMed:15613555, ECO:0000269|PubMed:15870828, ECO:0000269|PubMed:15896463, ECO:0000269|PubMed:15991316, ECO:0000269|PubMed:15996210, ECO:0000269|PubMed:16451135, ECO:0000269|PubMed:17101317, ECO:0000269|PubMed:17128465, ECO:0000269|PubMed:18561205, ECO:0000269|PubMed:18625694, ECO:0000269|PubMed:18781619, ECO:0000269|PubMed:18822302, ECO:0000269|PubMed:18951462, ECO:0000269|PubMed:21120944, ECO:0000269|PubMed:22102614, ECO:0000269|PubMed:22371642, ECO:0000269|PubMed:7874129, ECO:0000269|PubMed:8261515, ECO:0000269|PubMed:8700523, ECO:0000269|PubMed:8797773, ECO:0000269|PubMed:8872463, ECO:0000269|PubMed:9048925, ECO:0000269|PubMed:9240418, ECO:0000269|PubMed:9298827, ECO:0000269|PubMed:9311737, ECO:0000269|PubMed:9419403, ECO:0000269|PubMed:9559627, ECO:0000269|PubMed:9621522, ECO:0000269|PubMed:9718327, ECO:0000269|PubMed:9889267}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Muir-Torre syndrome (MRTES) [MIM:158320]: Rare autosomal dominant disorder characterized by sebaceous neoplasms and visceral malignancy. {ECO:0000269|PubMed:7713503}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Endometrial cancer (ENDMC) [MIM:608089]: A malignancy of endometrium, the mucous lining of the uterus. Most endometrial cancers are adenocarcinomas, cancers that begin in cells that make and release mucus and other fluids. {ECO:0000305|PubMed:11306449, ECO:0000305|PubMed:21642682}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Mismatch repair cancer syndrome 2 (MMRCS2) [MIM:619096]: An autosomal recessive form of mismatch repair cancer syndrome, a childhood cancer predisposition syndrome encompassing a broad tumor spectrum. This includes hematological malignancies, central nervous system tumors, Lynch syndrome-associated malignancies such as colorectal tumors as well as multiple intestinal polyps, embryonic tumors and rhabdomyosarcoma. Multiple cafe-au-lait macules, a feature reminiscent of neurofibromatosis type 1, are often found as first manifestation of the underlying cancer. {ECO:0000269|PubMed:12549480, ECO:0000269|PubMed:16372347}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Colorectal cancer (CRC) [MIM:114500]: A complex disease characterized by malignant lesions arising from the inner wall of the large intestine (the colon) and the rectum. Genetic alterations are often associated with progression from premalignant lesion (adenoma) to invasive adenocarcinoma. Risk factors for cancer of the colon and rectum include colon polyps, long-standing ulcerative colitis, and genetic family history. {ECO:0000269|PubMed:12792735, ECO:0000269|PubMed:14504054, ECO:0000269|PubMed:15996210, ECO:0000269|PubMed:9559627}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q92624; Q9UQ84-1; P09429; P20585; P52701; Q8IY92; P39875 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Chromosome; Disease variant; DNA damage; DNA repair; DNA-binding; Hereditary nonpolyposis colorectal cancer; Isopeptide bond; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Tumor suppressor; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 97883.4 Length 868 Aromaticity 0.08 Instability index 36.78 Isoelectric point 5.79 Charge (pH=7) -10.09 2D Binding mode Binding energy (Kcal/mol) -7.28 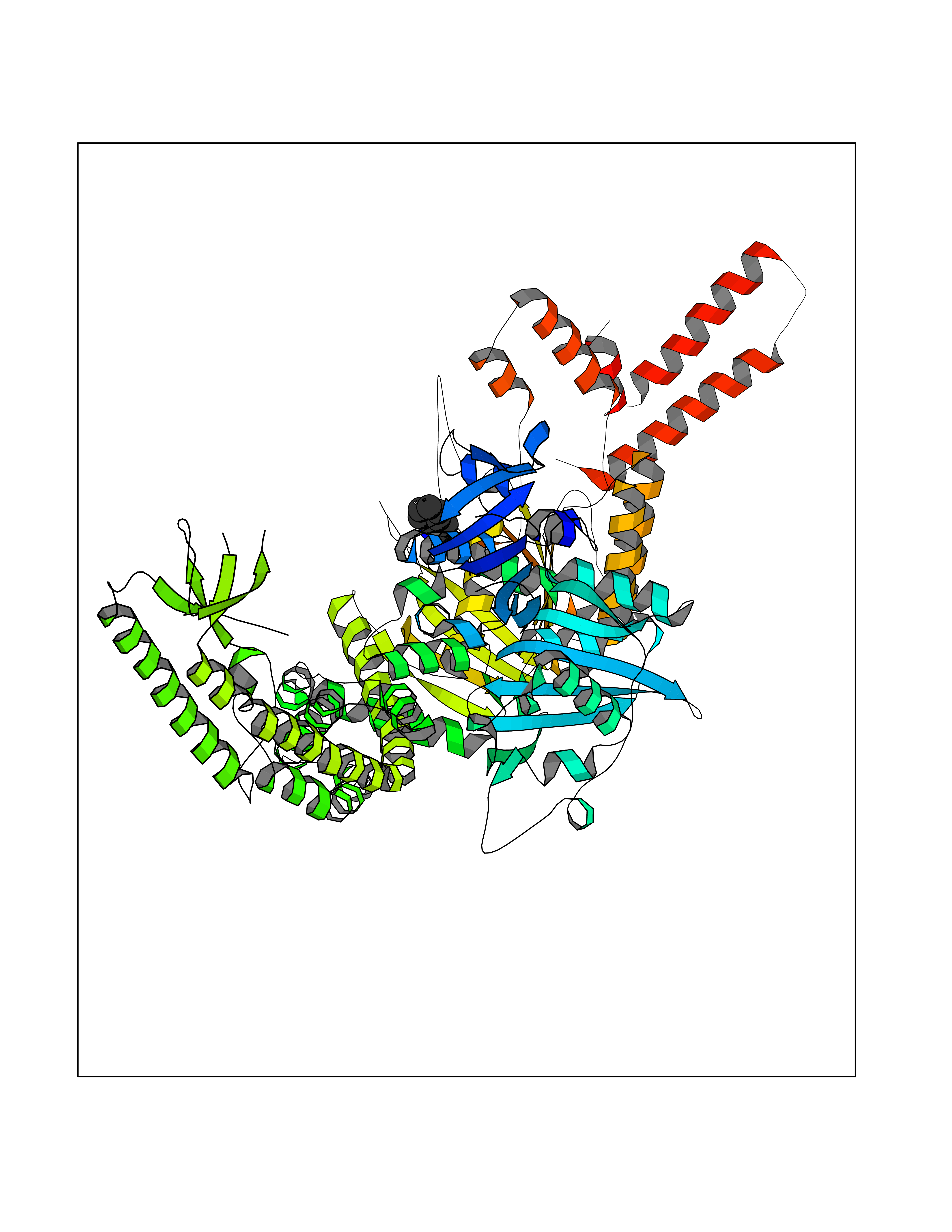 Molscript Map 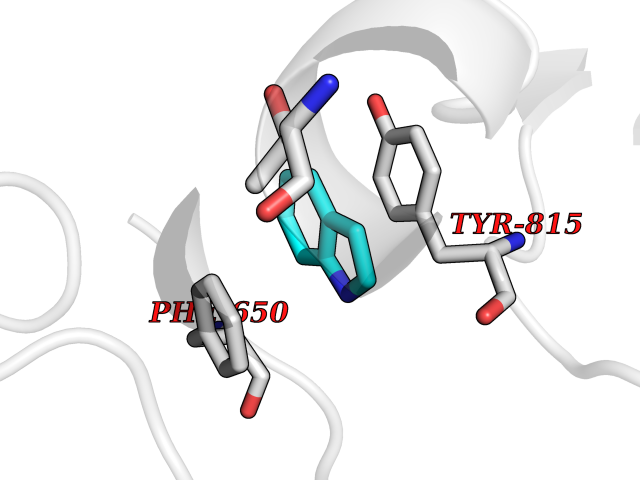 Pymol Map 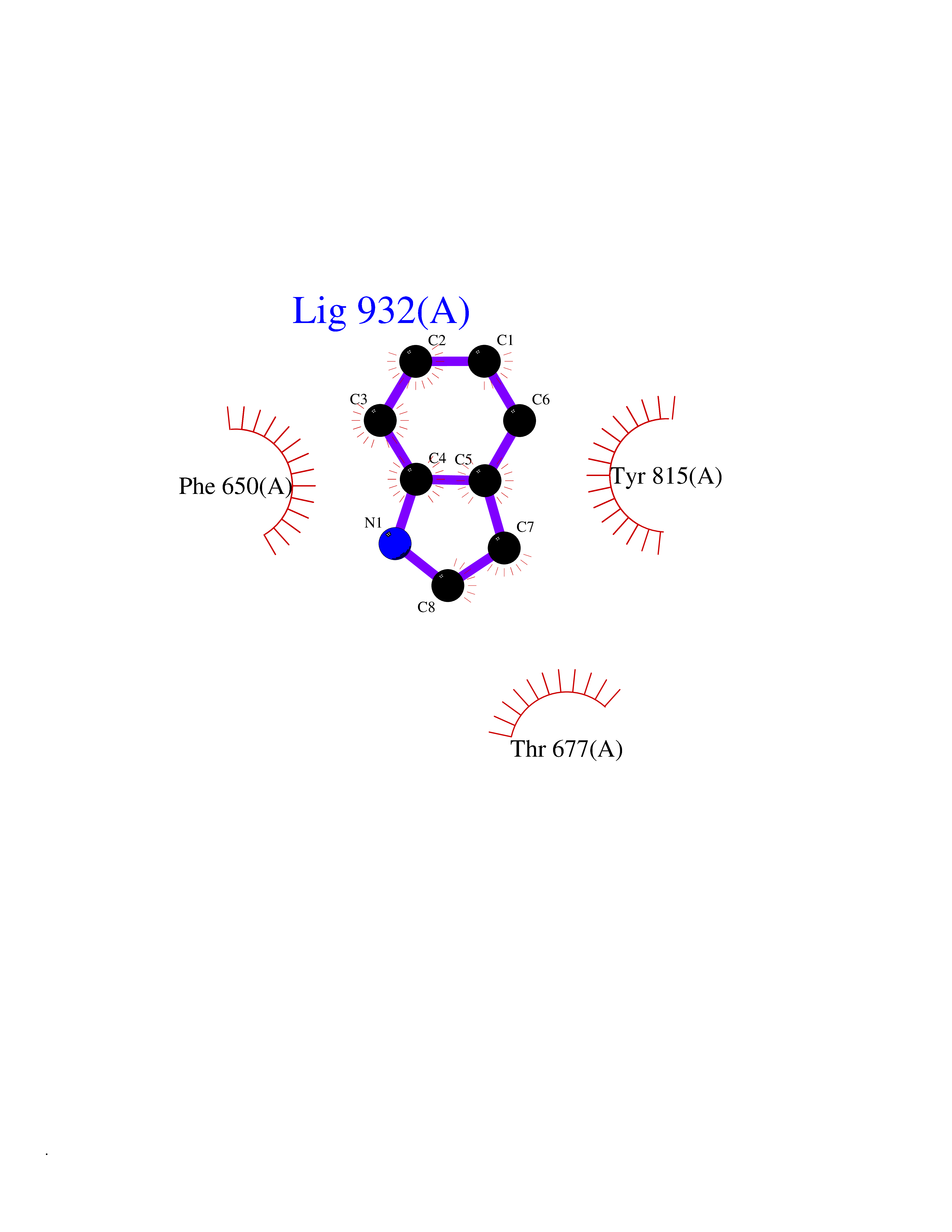 Ligplot Map 3D Binding mode Sequence ESAAEVGFVRFFQGMPEKPTTTVRLFDRGDFYTAHGEDALLAAREVFKTQGVIKYMGPAGAKNLQSVVLSKMNFESFVKDLLLVRQYRVEVYKNRASKENDWYLAYKASPGNLSQFEDILFIGVVGVKMSAVDGQRQVGVGYVDSIQRKLGLCEFPDNDQFSNLEALLIQIGPKECVLPGGETAGDMGKLRQIIQRGGILITERKKADFSTKDIYQDLNRLLKGKKGEQMNSAVLPEMENQVAVSSLSAVIKFLELLSDDSNFGQFELTTFDFSQYMKLDIAAVRALNLFQQSLAALLNKCKTPQGQRLVNQWIKQPLMDKNRIEERLNLVEAFVEDAELRQTLQEDLLRRFPDLNRLAKKFQRQAANLQDCYRLYQGINQLPNVIQALEKHEGKHQKLLLAVFVTPLTDLRSDFSKFQEMIETTLDMDQVENHEFLVKPSFDPNLSELREIMNDLEKKMQSTLISAARDLGLDPGKQIKLDSSAGYYFRVTCKEEKVLRNNKNFSTVDIQGVKFTNSKLTSLNEEYTKNKTEYEEAQDAIVKEIVNISSGYVEPMQTLNDVLAQLDAVVSFAHVSNGAPVPYVRPAILEKGQGRIILKASRHACVEVQIAFIPNDVYFEKDKQMFHIITGPNMGGKSTYIRQTGVIVLMAQIGCFVPCESAEVSIVDCILARVGSTFMAEMLETASILRSATKDSLIIIDELGRGTSTYDGFGLAWAISEYIATKIGAFCMFATHFHELTALANQIPTVNNLHVTALTTEETLTMLYQVKKGVCDQSFGIHVAELANFPKHVIECAKQKALELEEFQYKCYLEREQGEKIIQEFLSKVKQMPFTEMSEENITIKLKQLKAEVIAKNNSFVNEIISRI Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | Hepatitis B virus Capsid protein (HBV C) | 7PZL | 5.34 | |
Target general information Gen name HBV C Organism Hepatitis B virus genotype D subtype ayw (isolate France/Tiollais/1979) (HBV-D) Uniprot ID TTD ID Synonyms Core antigen; Core protein; HBcAg; p21.5 Protein family Orthohepadnavirus core antigen family Biochemical class NA Function Self assembles to form an icosahedral capsid. Most capsid appear to be large particles with an icosahedral symmetry of T=4 and consist of 240 copies of capsid protein, though a fraction forms smaller T=3 particles consisting of 180 capsid proteins. Entering capsid are transported along microtubules to the nucleus. Phosphorylation of the capsid is thought to induce exposure of nuclear localization signal in the C-terminal portion of the capsid protein that allows binding to the nuclear pore complex via the importin (karyopherin-) alpha and beta. Capsids are imported in intact form through the nuclear pore into the nuclear basket, where it probably binds NUP153. Only capsids that contain the mature viral genome can release the viral DNA and capsid protein into the nucleoplasm. Immature capsids get stucked in the basket. Capsids encapsulate the pre-genomic RNA and the P protein. Pre-genomic RNA is reverse transcribed into DNA while the capsid is still in the cytoplasm. The capsid can then either be directed to the nucleus, providing more genome for transcription, or bud through the endoplasmic reticulum to provide new virions. Related diseases Oocyte/zygote/embryo maturation arrest 14 (OZEMA14) [MIM:620276]: An autosomal recessive female infertility disorder characterized by oocyte maturation arrest, fertilization failure, and/or early embryonic arrest. {ECO:0000269|PubMed:32666501, ECO:0000269|PubMed:33683667, ECO:0000269|PubMed:33898437, ECO:0000269|PubMed:34218387}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative initiation; Capsid protein; Cytoplasmic inwards viral transport; DNA-binding; Host cytoplasm; Host-virus interaction; Microtubular inwards viral transport; Phosphoprotein; Reference proteome; Repeat; RNA-binding; T=4 icosahedral capsid protein; Viral penetration into host nucleus; Virion; Virus entry into host cell Protein physicochemical properties Chain ID A,B Molecular weight (Da) 32388.9 Length 286 Aromaticity 0.12 Instability index 42.05 Isoelectric point 5.06 Charge (pH=7) -11.81 2D Binding mode Binding energy (Kcal/mol) -7.29 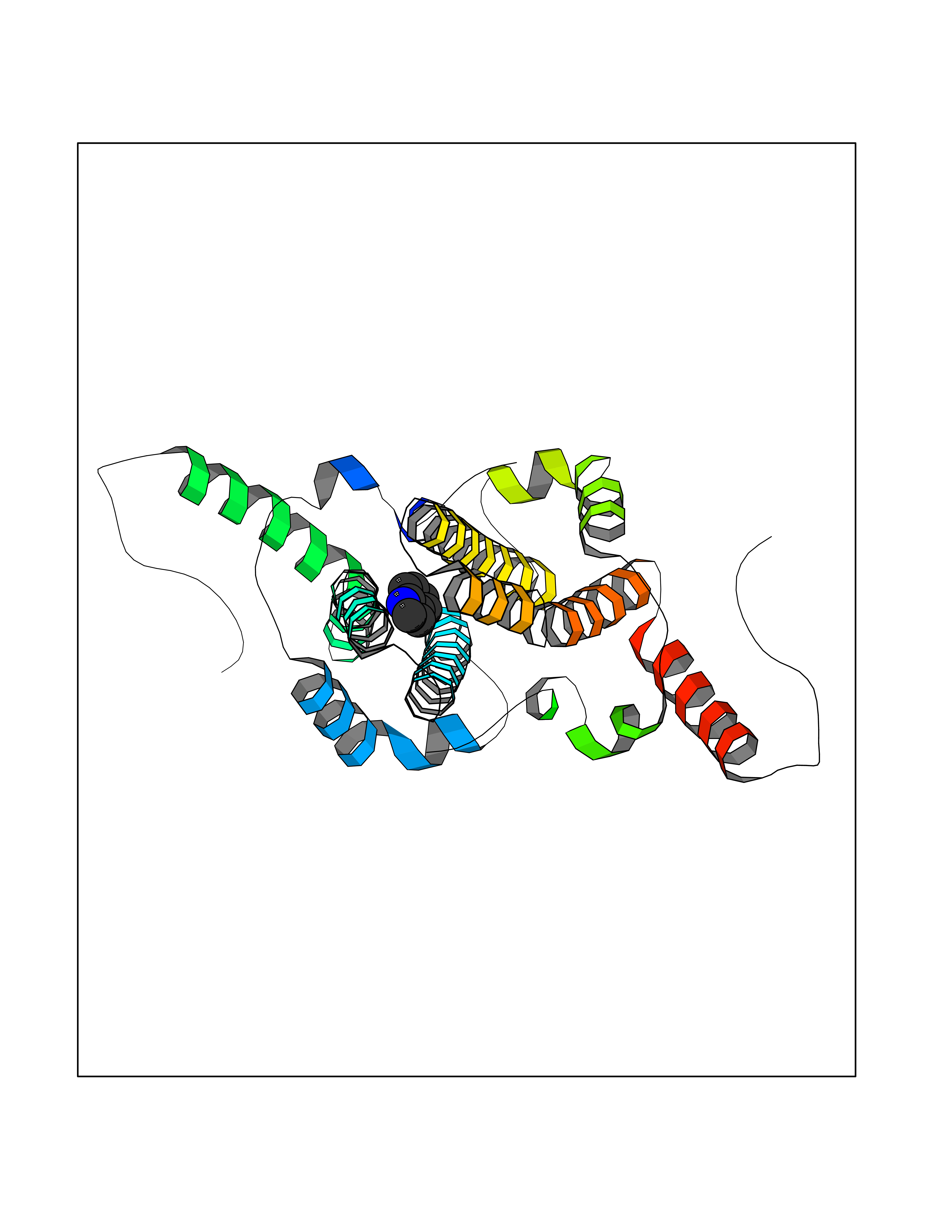 Molscript Map 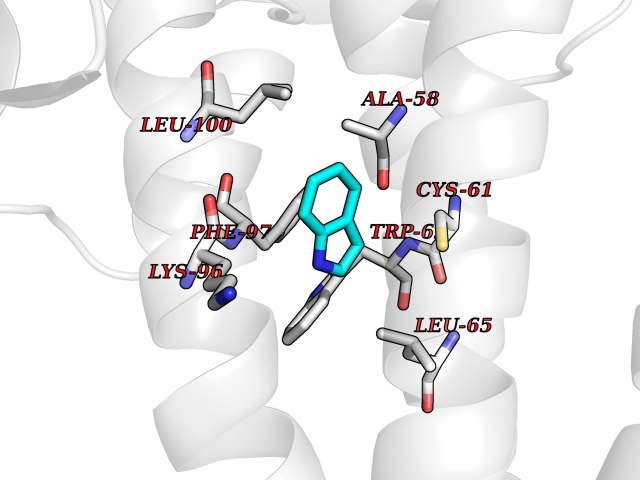 Pymol Map 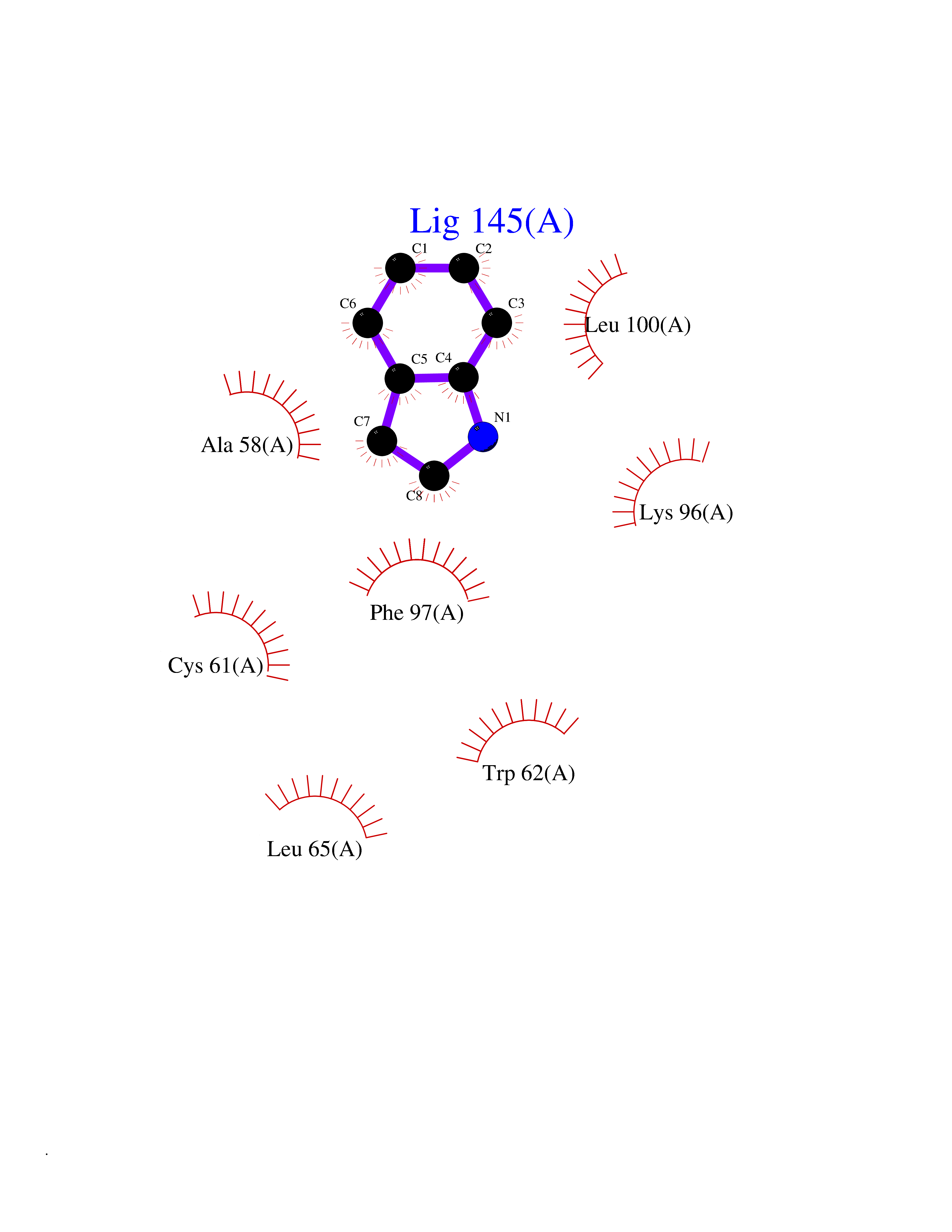 Ligplot Map 3D Binding mode Sequence MDIDPYKEFGATVELLSFLPSDFFPSVRDLLDTASALYREALESPEHCSPHHTALRQAIVCWGELMTLATWVGVNLEDPASRDLVVSYVNTNMGLKFRQLLWFHISCLTFGRETVIEYLVSFGVWIRTPPAYRPPNAPILSTLMDIDPYKEFGATVELLSFLPSDFFPSVRDLLDTASALYREALESPEHCSPHHTALRQAIVCWGELMTLATWVGVNLEDPASRDLVVSYVNTNMGLKFRQLLWFHISCLTFGRETVIEYLVSFGVWIRTPPAYRPPNAPILSTL Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Calcium-activated potassium channel KCa2.2 (KCNN2) | 5V02 | 5.34 | |
Target general information Gen name KCNN2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Small conductance calcium-activated potassium channel protein 2; SKCa2; SKCa 2; SK2; KCa2.2 Protein family Potassium channel KCNN family, KCa2.2/KCNN2 subfamily Biochemical class Voltage-gated ion channel Function Forms a voltage-independent potassium channel activated by intracellular calcium. Activation is followed by membrane hyperpolarization. Thought to regulate neuronal excitability by contributing to the slow component of synaptic afterhyperpolarization. The channel is blocked by apamin. Related diseases Dystonia 34, myoclonic (DYT34) [MIM:619724]: A form of dystonia, a disorder defined by the presence of sustained involuntary muscle contraction, often leading to abnormal postures. DYT34 is an autosomal dominant form characterized by childhood-onset dystonia predominantly affecting hands and neck, with a fast tremor with superimposed myoclonus and, in some individuals, subtle cerebellar signs. {ECO:0000269|PubMed:32212350}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Neurodevelopmental disorder with or without variable movement or behavioral abnormalities (NEDMAB) [MIM:619725]: An autosomal dominant disorder characterized by motor and language developmental delay, intellectual disability often associated with early-onset movement disorders comprising cerebellar ataxia and/or extrapyramidal symptoms. Other variable features include autism spectrum disorder or autistic features and epilepsy. {ECO:0000269|PubMed:33242881}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02587; DB01110; DB01054; DB00721; DB16733; DB00867; DB09089 Interacts with P35609 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calmodulin-binding; Cytoplasm; Disease variant; Dystonia; Intellectual disability; Ion channel; Ion transport; Membrane; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID B,R Molecular weight (Da) 26727.8 Length 233 Aromaticity 0.06 Instability index 24.91 Isoelectric point 5.01 Charge (pH=7) -11.73 2D Binding mode Binding energy (Kcal/mol) -7.29 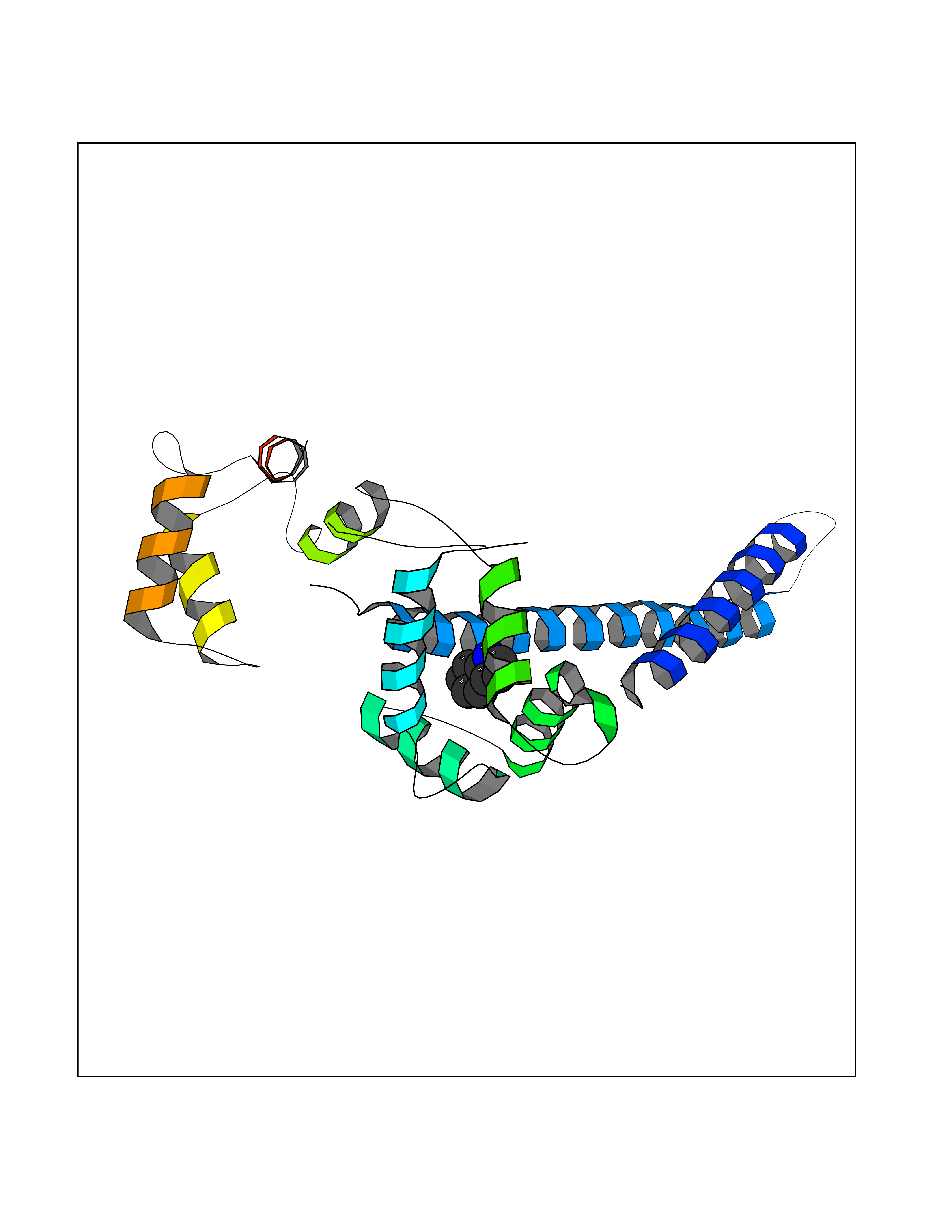 Molscript Map 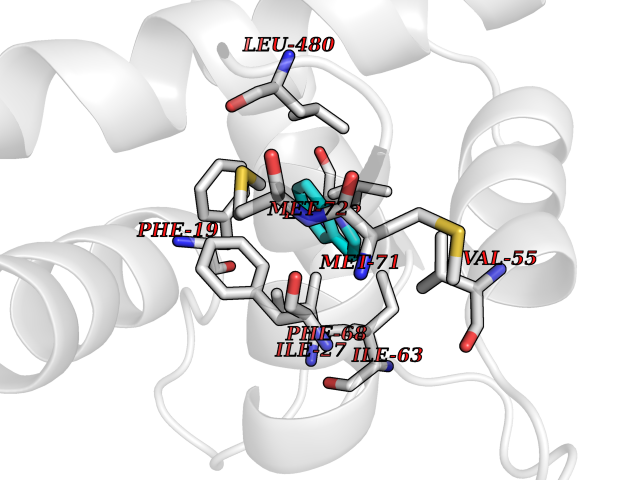 Pymol Map 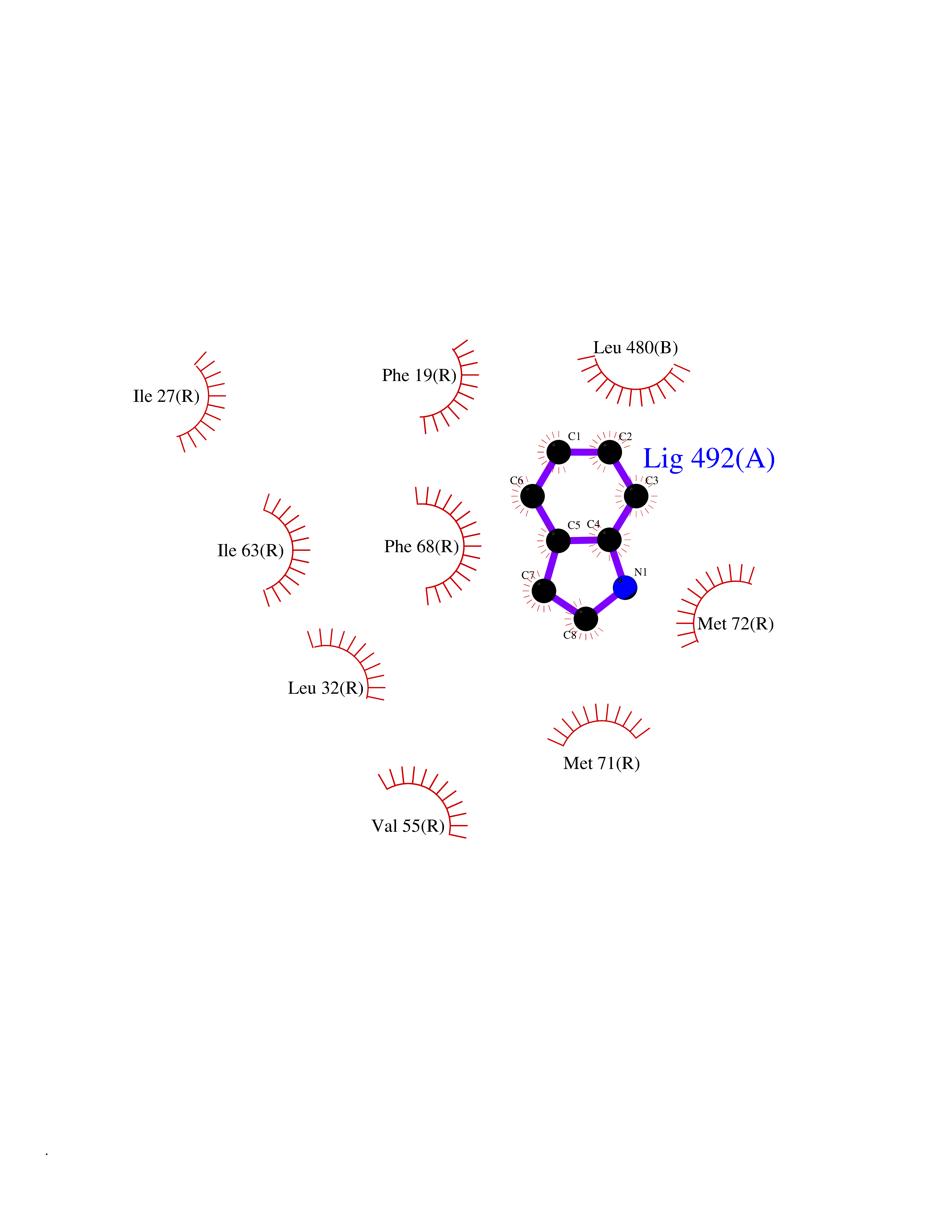 Ligplot Map 3D Binding mode Sequence GRKLELTKADTQLTKRVKNAAANVLRETWLIYKNTKLVKKIDHAKVRKHQRKFLQAIHQLRSVKMEQRKLNDQANTLVDLAKTQLEHDQLTEEQIAEFKEAFSLFDKDGDGTITTKELGTVMRSLGQNPTEAELQDMINEVDADGNGTIDFPEFLTMMARKMKDTDSEEEIREAFRVFDKDGNGYISAAELRHVMTNLGEKLTDEEVDEMIREADIDGDGQVNYEEFVQMMTA Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Cannabinoid receptor 1 (CB1) | 5U09 | 5.33 | |
Target general information Gen name CNR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cannabinoid CB1 receptor; CNR; CB-R; CANN6 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Mediates many cannabinoid-induced effects, acting, among others, on food intake, memory loss, gastrointestinal motility, catalepsy, ambulatory activity, anxiety, chronic pain. Signaling typically involves reduction in cyclic AMP. In the hypothalamus, may have a dual effect on mitochondrial respiration depending upon the agonist dose and possibly upon the cell type. Increases respiration at low doses, while decreases respiration at high doses. At high doses, CNR1 signal transduction involves G-protein alpha-i protein activation and subsequent inhibition of mitochondrial soluble adenylate cyclase, decrease in cyclic AMP concentration, inhibition of protein kinase A (PKA)-dependent phosphorylation of specific subunits of the mitochondrial electron transport system, including NDUFS2. In the hypothalamus, inhibits leptin-induced reactive oxygen species (ROS) formation and mediates cannabinoid-induced increase in SREBF1 and FASN gene expression. In response to cannabinoids, drives the release of orexigenic beta-endorphin, but not that of melanocyte-stimulating hormone alpha/alpha-MSH, from hypothalamic POMC neurons, hence promoting food intake. In the hippocampus, regulates cellular respiration and energy production in response to cannabinoids. Involved in cannabinoid-dependent depolarization-induced suppression of inhibition (DSI), a process in which depolarization of CA1 postsynaptic pyramidal neurons mobilizes eCBs, which retrogradely activate presynaptic CB1 receptors, transiently decreasing GABAergic inhibitory neurotransmission. Also reduces excitatory synaptic transmission. In superior cervical ganglions and cerebral vascular smooth muscle cells, inhibits voltage-gated Ca(2+) channels in a constitutive, as well as agonist-dependent manner. In cerebral vascular smooth muscle cells, cannabinoid-induced inhibition of voltage-gated Ca(2+) channels leads to vasodilation and decreased vascular tone. Induces leptin production in adipocytes and reduces LRP2-mediated leptin clearance in the kidney, hence participating in hyperleptinemia. In adipose tissue, CNR1 signaling leads to increased expression of SREBF1, ACACA and FASN genes. In the liver, activation by endocannabinoids leads to increased de novo lipogenesis and reduced fatty acid catabolism, associated with increased expression of SREBF1/SREBP-1, GCK, ACACA, ACACB and FASN genes. May also affect de novo cholesterol synthesis and HDL-cholesteryl ether uptake. Peripherally modulates energy metabolism. In high carbohydrate diet-induced obesity, may decrease the expression of mitochondrial dihydrolipoyl dehydrogenase/DLD in striated muscles, as well as that of selected glucose/ pyruvate metabolic enzymes, hence affecting energy expenditure through mitochondrial metabolism. In response to cannabinoid anandamide, elicits a proinflammatory response in macrophages, which involves NLRP3 inflammasome activation and IL1B and IL18 secretion. In macrophages infiltrating pancreatic islets, this process may participate in the progression of type-2 diabetes and associated loss of pancreatic beta-cells. G-protein coupled receptor for endogenous cannabinoids (eCBs), including N-arachidonoylethanolamide (also called anandamide or AEA) and 2-arachidonoylglycerol (2-AG), as well as phytocannabinoids, such as delta(9)-tetrahydrocannabinol (THC). Related diseases Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:18177726}. The protein represented in this entry may be involved in disease pathogenesis. May contribute to the development of diet-induced obesity and several obesity-associated features, such as dyslipidemia and liver steatosis, regulating peripheral lipogenesis, energy expenditure and feeding behavior. CNR1 inverse agonists have been shown to reduce body weight and improve metabolic abnormalities in obese subjects, although adverse neuropsychiatric effects, including anxiety, irritability, and depressed mood, halted their therapeutic development (PubMed:18177726). In obese mice, peripherally restricted CNR1 inverse agonists have been shown to normalize metabolic abnormalities, including insulin resistance and fatty liver, and to reverse leptin resistance. {ECO:0000269|PubMed:18177726}.; DISEASE: Dysfunction of the endogenous cannabinoid system including CNR1 has been implicated in the pathogenesis of a number of central nervous system disorders, including Huntington disease, Parkinson disease, and Alzheimer disease (PubMed:32549916). In post-mortem brains from Huntington disease patients, a progressive CNR1 loss has been observed in the caudate nucleus, putamen, and substantia nigra pars reticulata, and altered expression and abnormal endocannabinoid levels precede motor symptoms in a disease mouse model (PubMed:10828533, PubMed:19524019, PubMed:8255419). In Parkinson disease, low CNR1 expression in mid-superior frontal gyrus and mid-cingulate cortex has been associated with poor mind, poor executive functioning and poor episode memory, while patients with more severe visuospatial dysfunction showed decreased receptor availability in the precuneus, mid-cingulate, supplementary motor cortex, inferior orbitofrontal gyrus and thalamus (PubMed:31342135). In an animal model for Alzheimer disease, CNR1 heterozygous deletion has been associated with decreased levels of postsynaptic density protein 95 (DLG4/PSD95) and accelerated memory impairment, suggesting synaptic dysfunction and a crucial role for CNR1 in the progression of disease symptoms (PubMed:10828533, PubMed:19524019, PubMed:30096288, PubMed:31342135, PubMed:8255419). {ECO:0000269|PubMed:10828533, ECO:0000269|PubMed:19524019, ECO:0000269|PubMed:30096288, ECO:0000269|PubMed:31342135, ECO:0000269|PubMed:32549916, ECO:0000269|PubMed:8255419}. Drugs (DrugBank ID) DB05750; DB09061; DB00470; DB14009; DB00486; DB14011; DB11745; DB09288; DB02955; DB06155; DB05077; DB11755; DB05201 Interacts with P29274; P21554 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Cell projection; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Neurodegeneration; Obesity; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 32070.3 Length 282 Aromaticity 0.13 Instability index 40.15 Isoelectric point 9.16 Charge (pH=7) 9.36 2D Binding mode Binding energy (Kcal/mol) -7.27 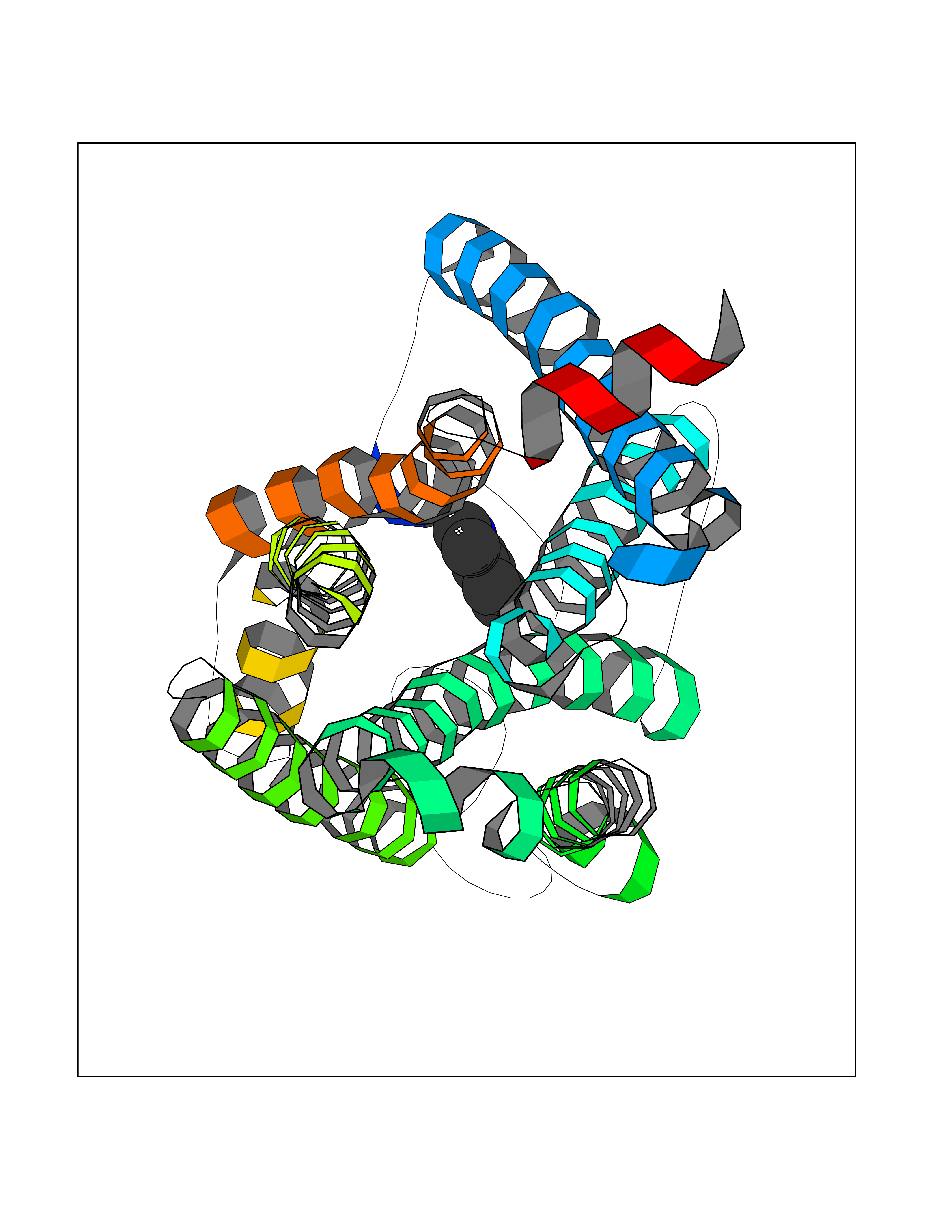 Molscript Map 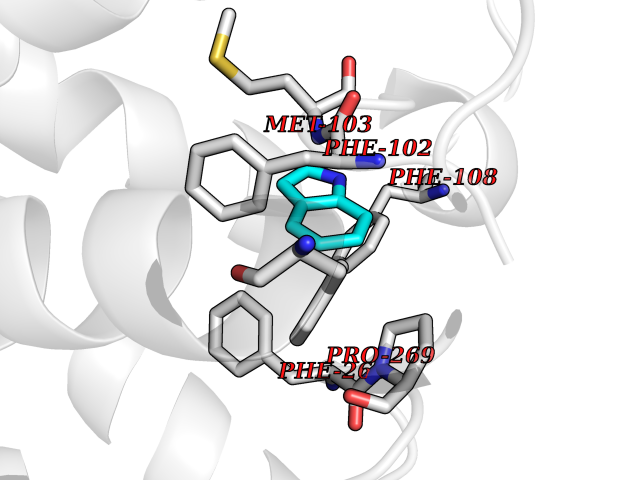 Pymol Map 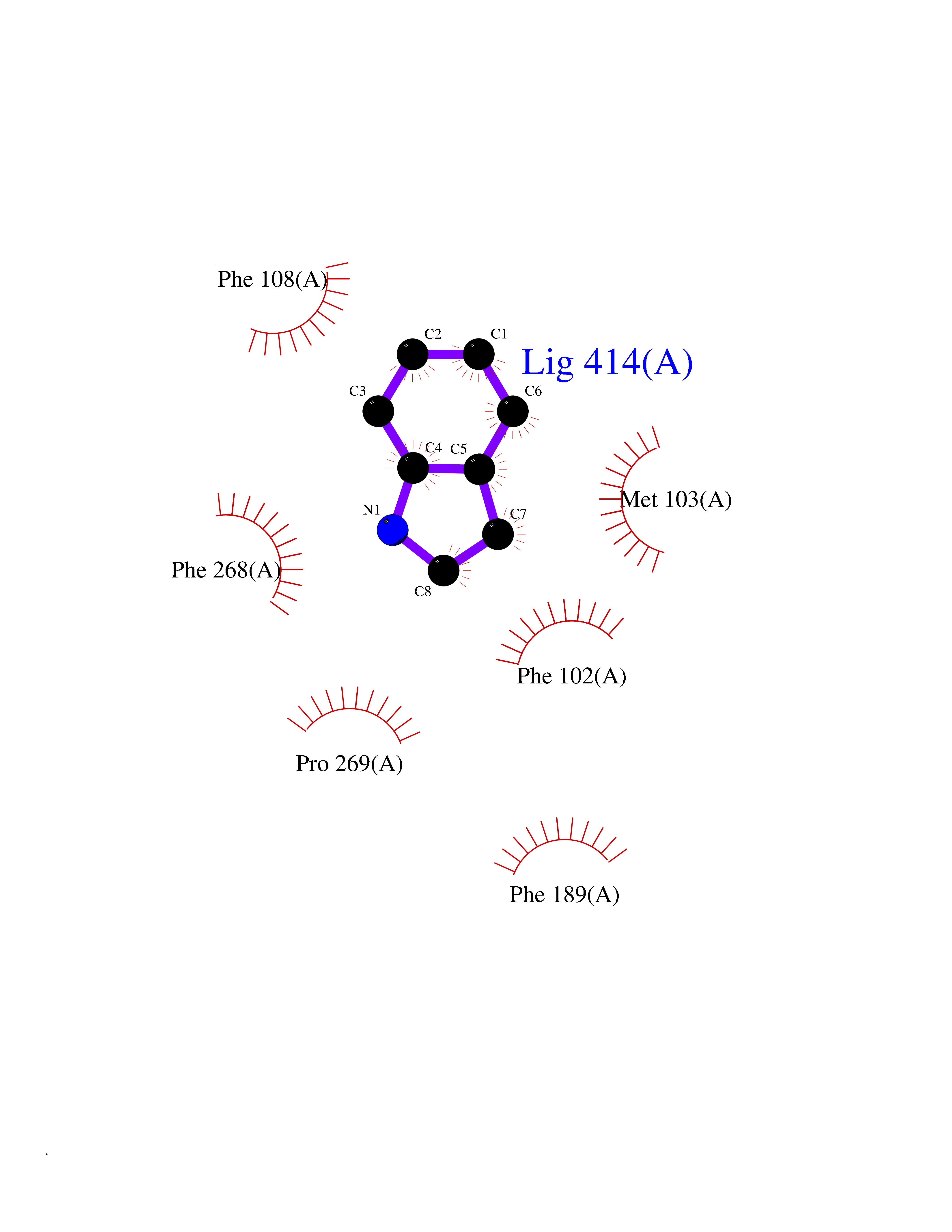 Ligplot Map 3D Binding mode Sequence ENFMDIECFMVLNPSQQLAIAVLSLTLGTFTVLENLLVLCVILHSRSLRCRPSYHFIGSLAVADLLGSVIFVYSFIDFHVFHRKDSRNVFLFKLGGVTASFTASVGSLFLAAIDRYISIHRPLAYKRIVTRPKAVVAFCLMWTIAIVIAVLPLLGWNCEKLQSVCSDIFPHIDETYLMFWIGVTSVLLLFIVYAYMYILWKADQARMDIRLAKTLVLILVVLIICWGPLLAIMVYDVFGKMNKLIKTVFAFCSMLCLLNSTVNPIIYALRSKDLRHAFRSMF Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Neutrophil collagenase | 4QKZ | 5.33 | |
Target general information Gen name MMP8 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CLG1 Protein family Peptidase M10A family Biochemical class Hydrolase / hydrolase inhibitor Function Metalloendopeptidase activity.Serine-type endopeptidase activity.Zinc ion binding. Related diseases Protoporphyria, erythropoietic, 1 (EPP1) [MIM:177000]: An autosomal recessive form of porphyria with onset usually before age 10 years. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. Erythropoietic protoporphyria is marked by excessive protoporphyrin in erythrocytes, plasma, liver and feces, and by widely varying photosensitive skin changes ranging from a burning or pruritic sensation to erythema, edema and wheals. {ECO:0000269|PubMed:10942404, ECO:0000269|PubMed:11375302, ECO:0000269|PubMed:12063482, ECO:0000269|PubMed:12601550, ECO:0000269|PubMed:1376018, ECO:0000269|PubMed:15286165, ECO:0000269|PubMed:17196862, ECO:0000269|PubMed:1755842, ECO:0000269|PubMed:7910885, ECO:0000269|PubMed:8757534, ECO:0000269|PubMed:9211198, ECO:0000269|PubMed:9585598, ECO:0000269|PubMed:9740232}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07772; DB07713; DB07397; DB02326; DB03207; DB02953; DB08476; DB07900; DB03622; DB03880; DB08028; DB03636; DB00786; DB08403; DB06971 Interacts with NA EC number 3.4.24.34 Uniprot keywords 3D-structure; Calcium; Collagen degradation; Direct protein sequencing; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 18096.6 Length 163 Aromaticity 0.12 Instability index 27.13 Isoelectric point 4.64 Charge (pH=7) -11.95 2D Binding mode Binding energy (Kcal/mol) -7.27 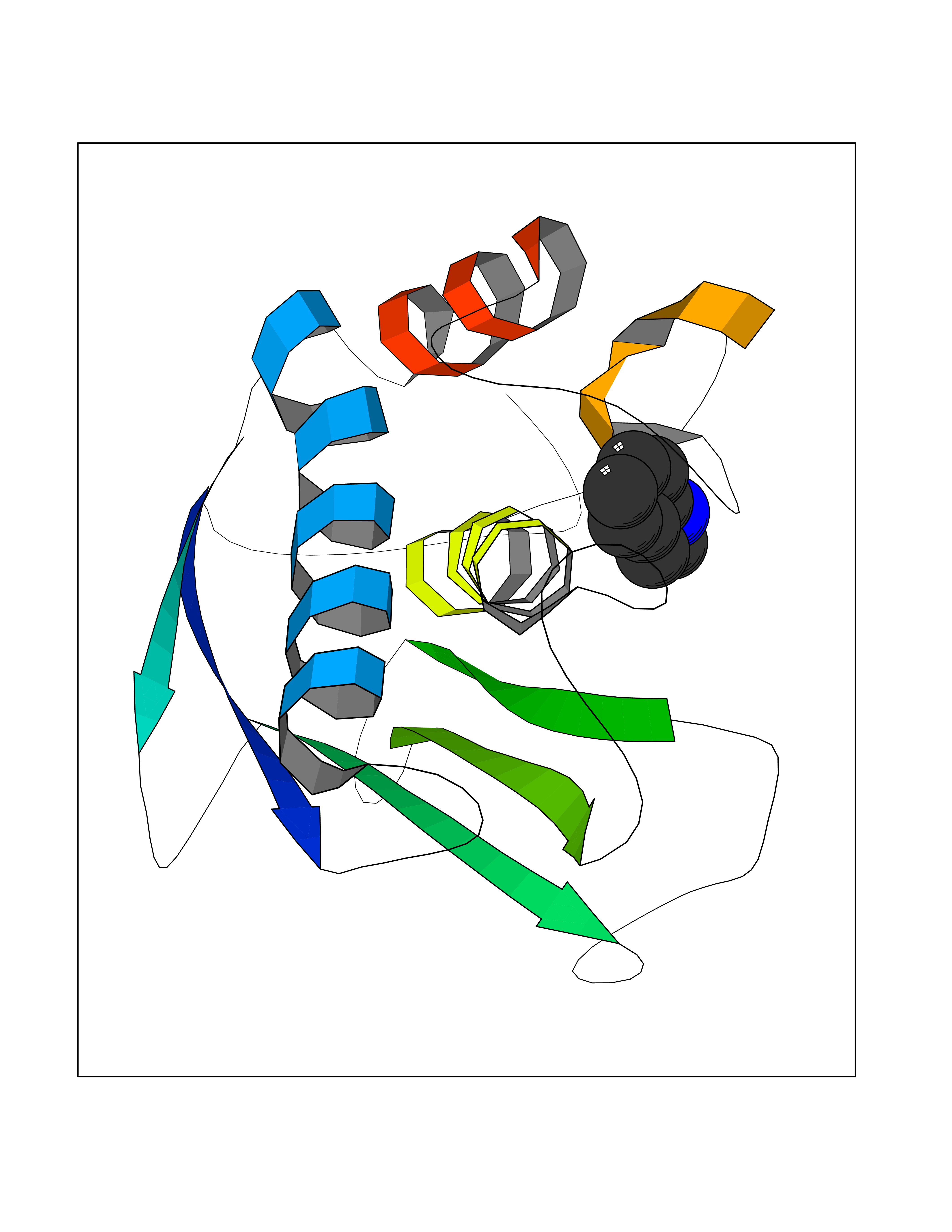 Molscript Map 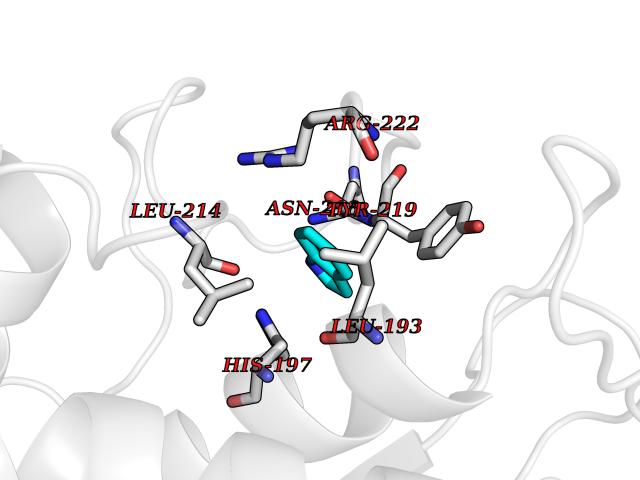 Pymol Map 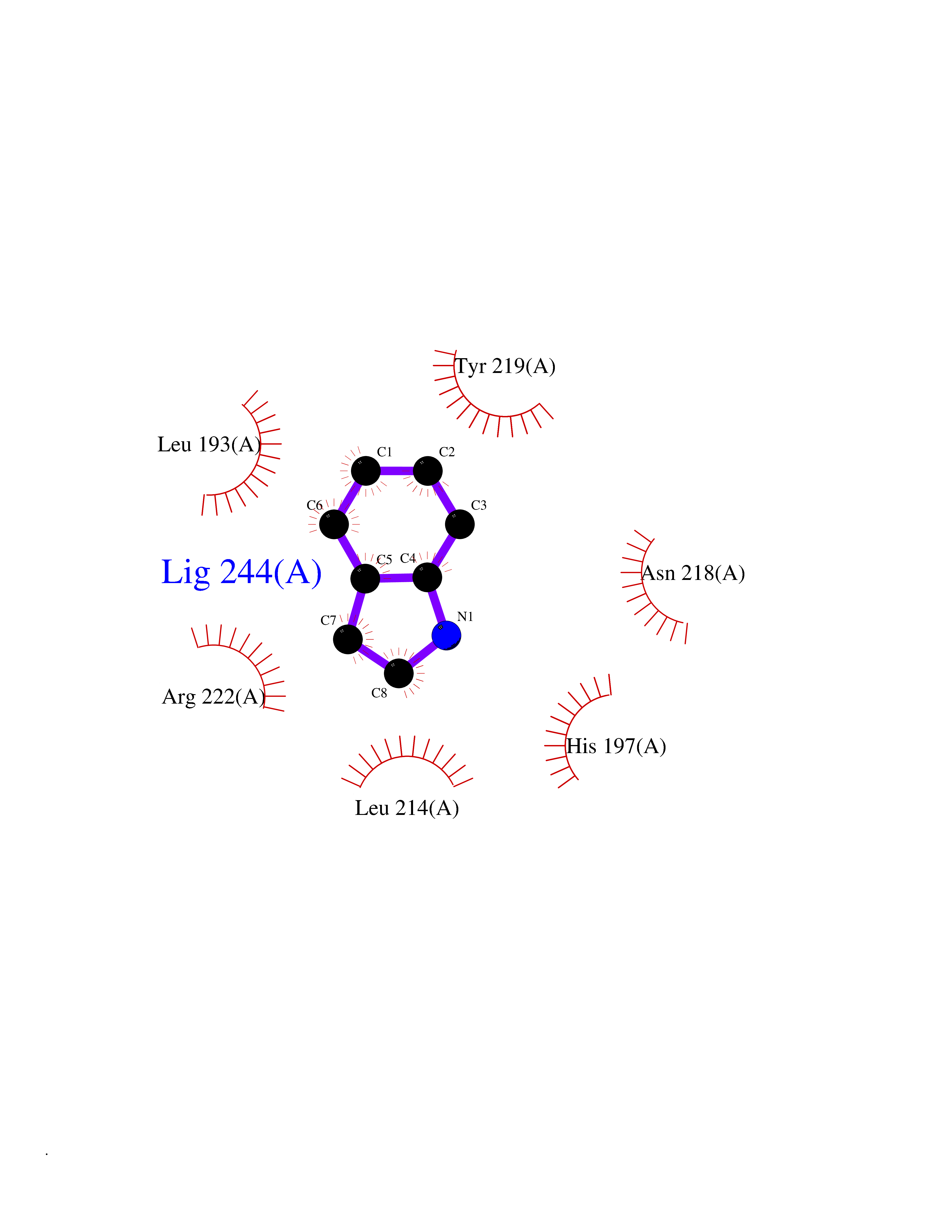 Ligplot Map 3D Binding mode Sequence MLTPGNPKWERTNLTYRIRNYTPQLSEAEVERAIKDAFELWSVASPLIFTRISQGEADINIAFYQRDHGDNSPFDGPNGILAHAFQPGQGIGGDAHFDAEETWTNTSANYNLFLVAAHEFGHSLGLAHSSDPGALMYPNYAFRETSNYSLPQDDIDGIQAIYG Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | SEC14-like protein 3 | 4UYB | 5.33 | |
Target general information Gen name SEC14L3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms TAP2 Protein family NA Biochemical class Transport protein Function Lipid binding.Transporter activity. Related diseases Chondrodysplasia with platyspondyly, distinctive brachydactyly, hydrocephaly, and microphthalmia (CDP-PBHM) [MIM:300863]: A disease characterized by chondrodysplasia, severe platyspondyly, hydrocephaly, and facial features with microphthalmia. Bone abnormalities include a distinctive metaphyseal cupping of the metacarpals, metatarsals, and phalanges. Affected females show a milder phenotype with small stature, sometimes associated with body asymmetry and mild intellectual disability. {ECO:0000269|PubMed:20181727}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB14001; DB14002; DB11635; DB11251; DB00163 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Lipid-binding; Proteomics identification; Reference proteome; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 46148.7 Length 401 Aromaticity 0.1 Instability index 45.19 Isoelectric point 5.79 Charge (pH=7) -5.94 2D Binding mode Binding energy (Kcal/mol) -7.27  Molscript Map 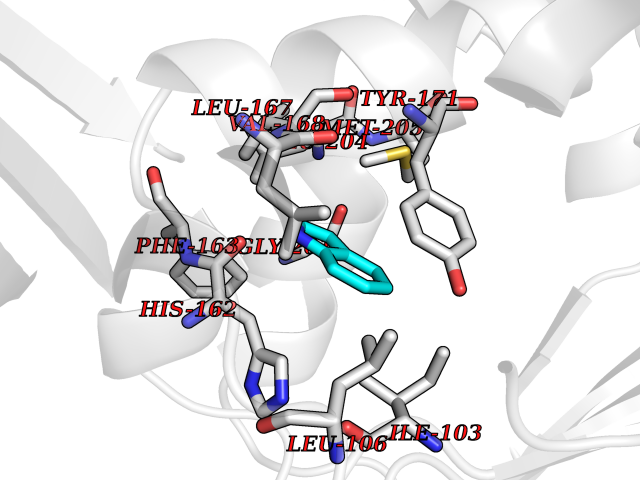 Pymol Map 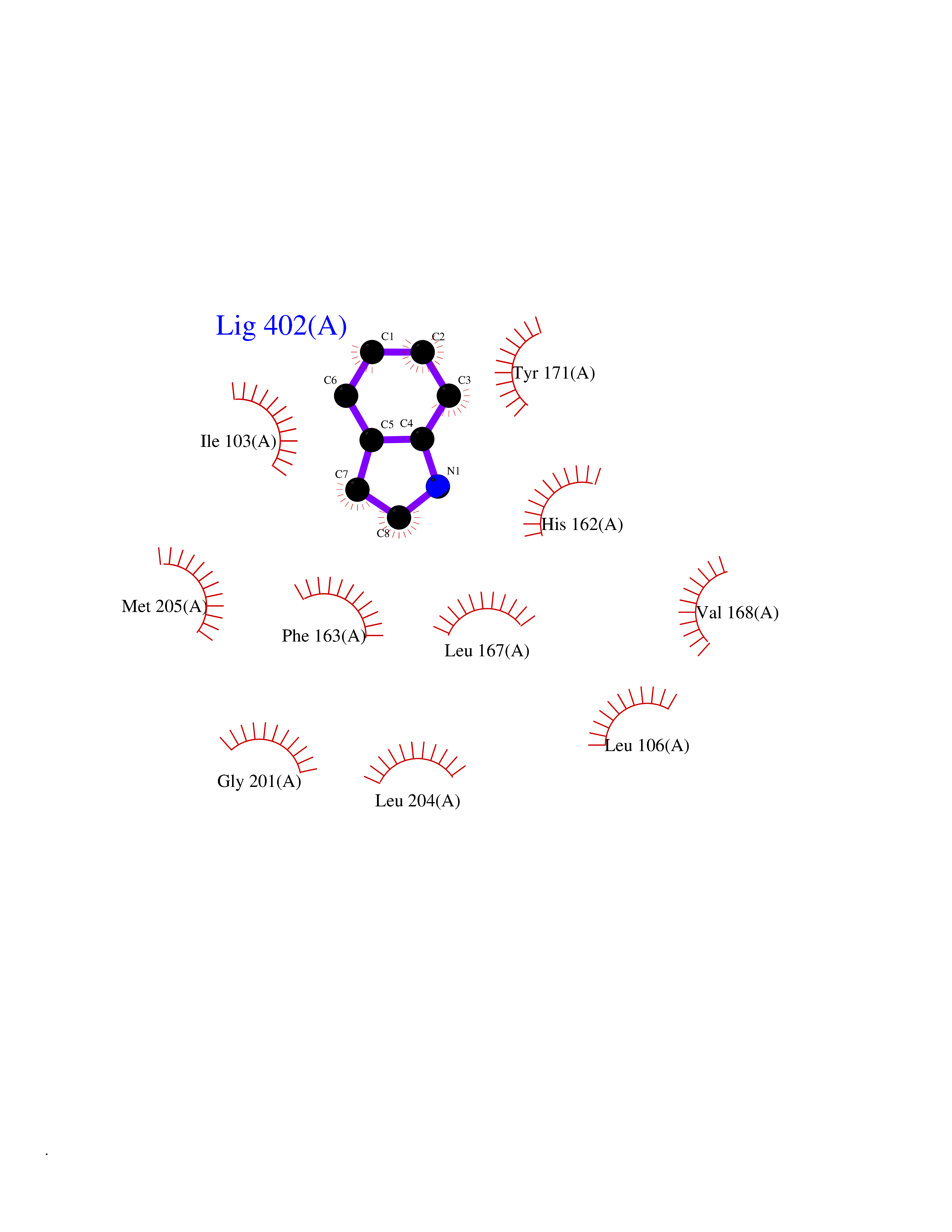 Ligplot Map 3D Binding mode Sequence SMSGRVGDLSPKQAETLAKFRENVQDVLPALPNPDDYFLLRWLRARNFDLQKSEALLRKYMEFRKTMDIDHILDWQPPEVIQKYMPGGLCGYDRDGCPVWYDIIGPLDPKGLLFSVTKQDLLKTKMRDCERILHECDLQTERLGKKIETIVMIFDCEGLGLKHFWKPLVEVYQEFFGLLEENYPETLKFMLIVKATKLFPVGYNLMKPFLSEDTRRKIIVLGNNWKEGLLKLISPEELPAQFGGTLTDPDGNPKCLTKINYGGEIPKSMYVRDQVKTQYEHSVQINRGSSHQVEYEILFPGCVLRWQFSSDGADIGFGVFLKTKMGERQRAGEMTEVLPSQRYNAHMVPEDGNLTCSEAGVYVLRFDNTYSFVHAKKVSFTVEVLLPDEGMQKYDKELTPV Hydrogen bonds contact Hydrophobic contact | ||||