Job Results:
Ligand
Structure
Job ID
22a4f6254c6ea1b7fba986d94e380114
Job name
NA
Time
2026-02-27 16:46:57
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Fatty acid synthase (FASN) | 3TJM | 6.11 | |
Target general information Gen name FASN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Yeast fatty acid synthase; Fatty-acyl-CoA synthase; Fatty acyl-CoA synthetase enzyme; FAS Protein family NA Biochemical class Acyltransferase Function Fatty acid synthetase catalyzes the formation of long-chain fatty acids from acetyl-CoA, malonyl-CoA and NADPH. This multifunctional protein has 7 catalytic activities as an acyl carrier protein. Related diseases Glycine encephalopathy 2 (GCE2) [MIM:620398]: A form of glycine encephalopathy, a metabolic disorder characterized by a high concentration of glycine in the body fluids. Affected individuals typically have severe neurological symptoms, including seizure, lethargy, and muscular hypotonia soon after birth. Most of them die within the neonatal period. Atypical cases have later disease onset and less severely affected psychomotor development. {ECO:0000269|PubMed:10873393, ECO:0000269|PubMed:11286506, ECO:0000269|PubMed:16051266, ECO:0000269|PubMed:26371980, ECO:0000269|PubMed:28244183, ECO:0000269|PubMed:8005589, ECO:0000269|PubMed:9600239, ECO:0000269|PubMed:9621520}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01034; DB01083 Interacts with Q15848; Q16665; P42858; Q8IV20; Q8TBB1; PRO_0000045603 [Q99IB8] EC number EC 2.3.1.85 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Isopeptide bond; Lipid biosynthesis; Lipid metabolism; Lyase; Multifunctional enzyme; NAD; NADP; Oxidoreductase; Phosphopantetheine; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; S-nitrosylation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30174.9 Length 275 Aromaticity 0.09 Instability index 43.28 Isoelectric point 5.92 Charge (pH=7) -5.4 2D Binding mode Binding energy (Kcal/mol) -8.33 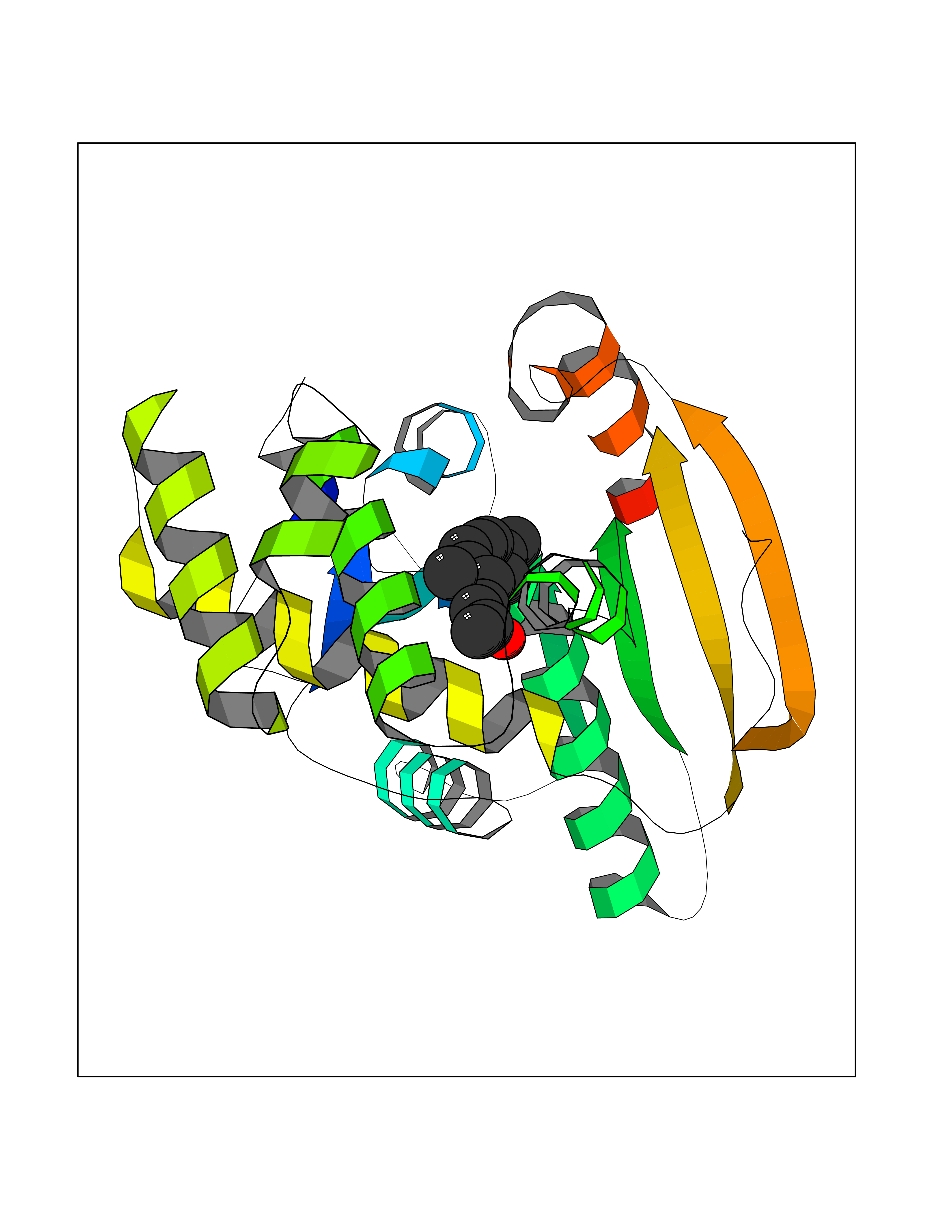 Molscript Map 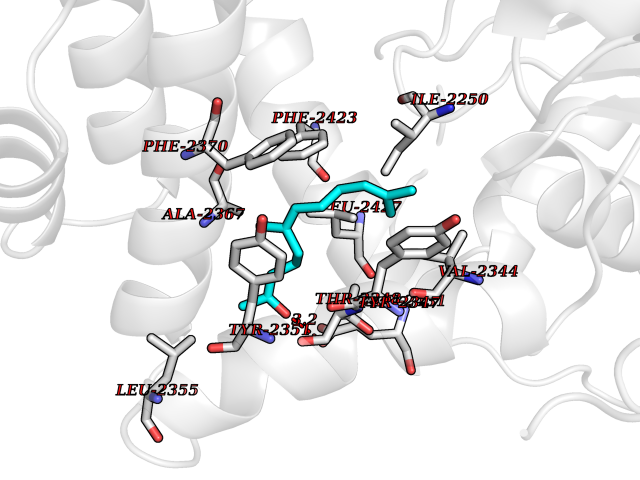 Pymol Map 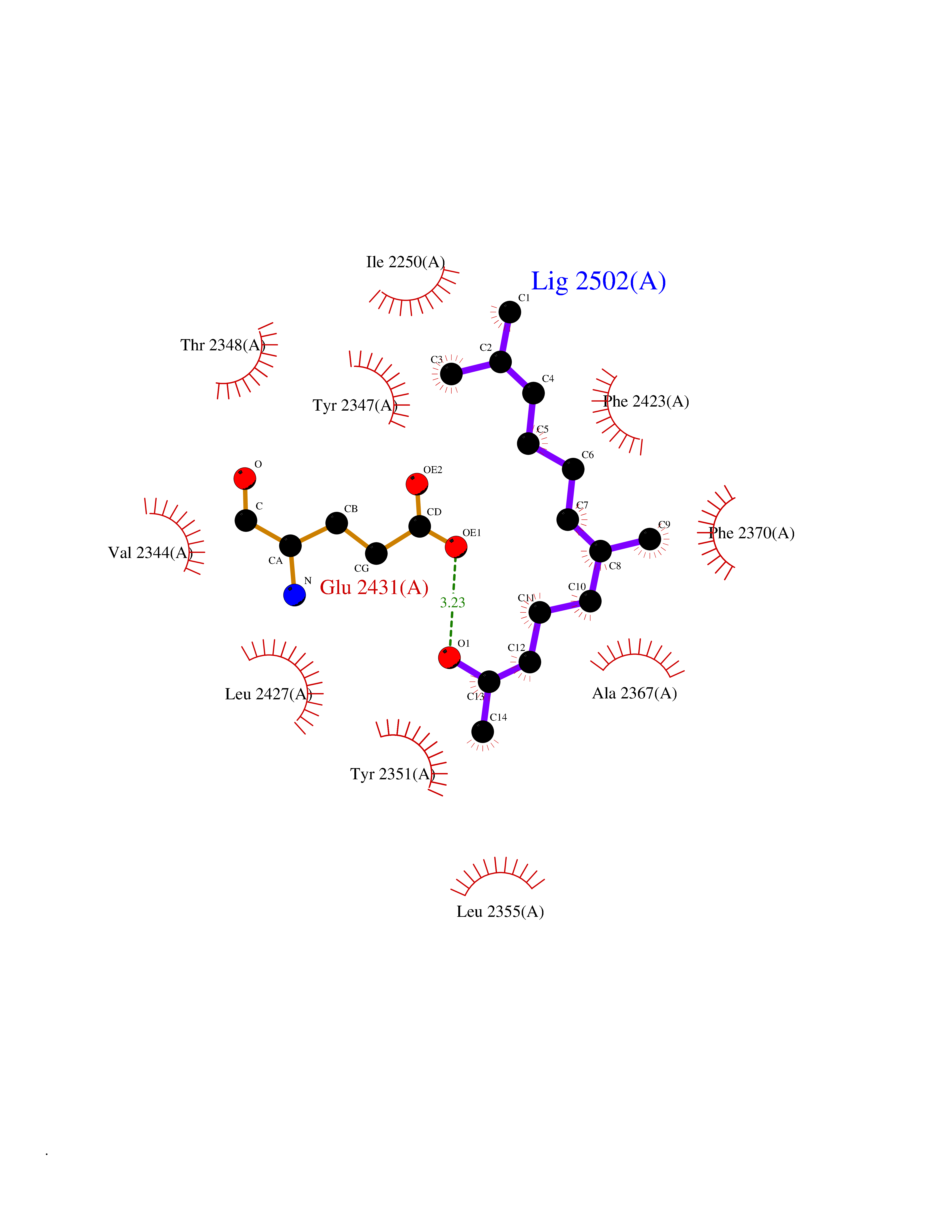 Ligplot Map 3D Binding mode Sequence NLRSLLVNPEGPTLMRLNSVQSSERPLFLVHPIEGSTTVFHSLASRLSIPTYGLQCTRAAPLDSIHSLAAYYIDCIRQVQPEGPYRVAGYSYGACVAFEMCSQLQAQQSPAPTHNSLFLFDGSPTYVLAYTGSYRAKLTPGCEAEAETEAICFFVQQFTDMEHNRVLEALLPLKGLEERVAAAVDLIIKSHQGLDRQELSFAARSFYYKLRAAEQYTPKAKYHGNVMLLRAAAGADYNLSQVCDGKVSVHVIEGDHATLLEGSGLESIISIIHSS Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Plasmodium Dihydroorotate dehydrogenase (Malaria DHOdehase) | 1TV5 | 6.11 | |
Target general information Gen name Malaria DHOdehase Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID Synonyms PFF0160c; Mitochondrially bound dihydroorotate-ubiqui oxidoreductase; Dihydroorotate oxidase of Plasmodium falciparum; Dihydroorotate dehydrogenase of Plasmodium falciparum; DHOdehase of Plasmodium fa Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 41846.8 Length 371 Aromaticity 0.1 Instability index 37.25 Isoelectric point 8.21 Charge (pH=7) 3.13 2D Binding mode Binding energy (Kcal/mol) -8.33 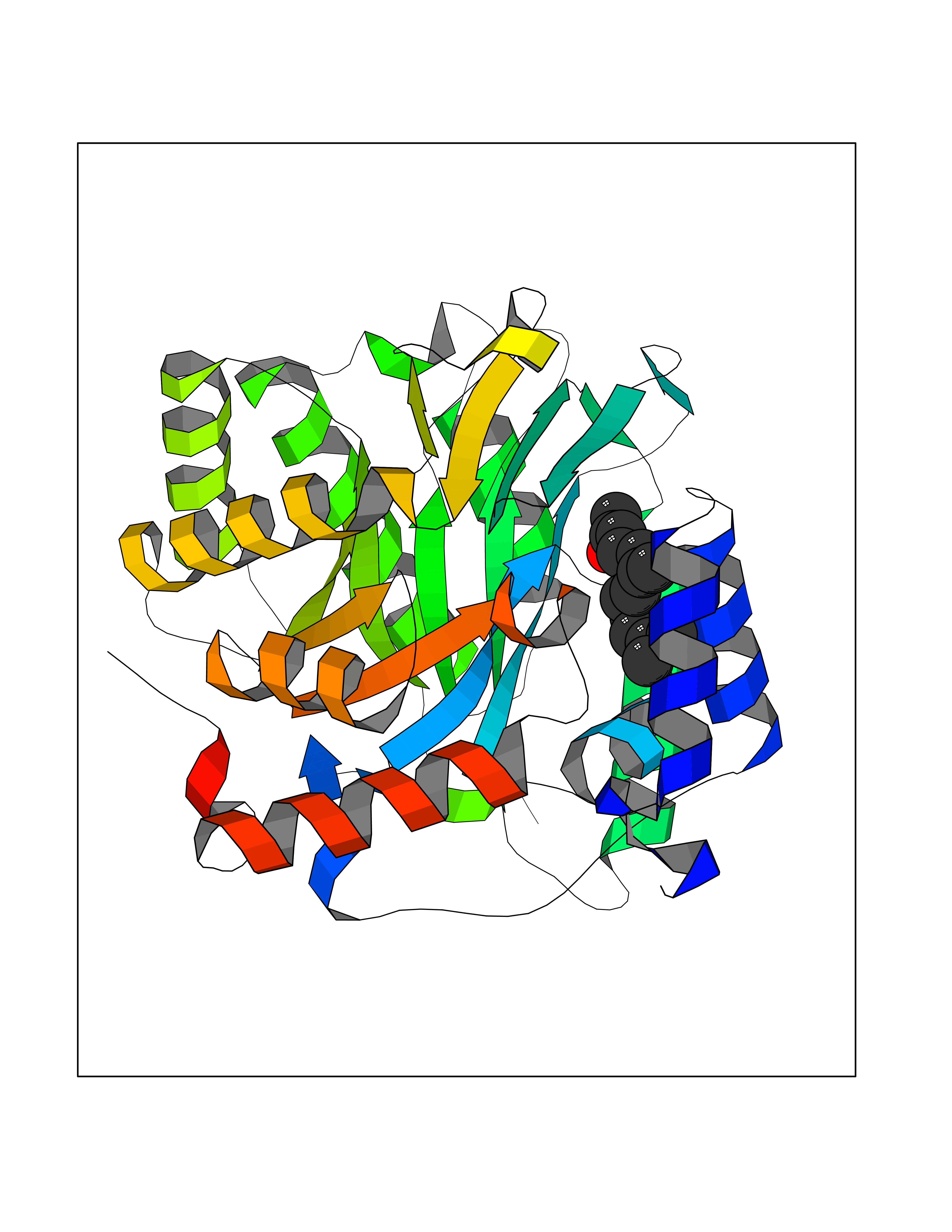 Molscript Map 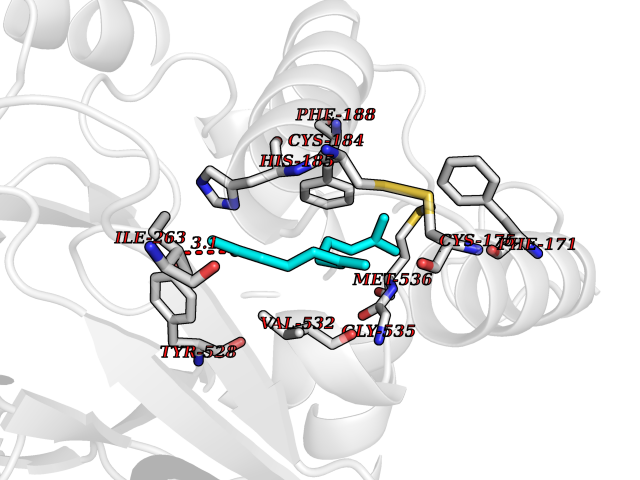 Pymol Map 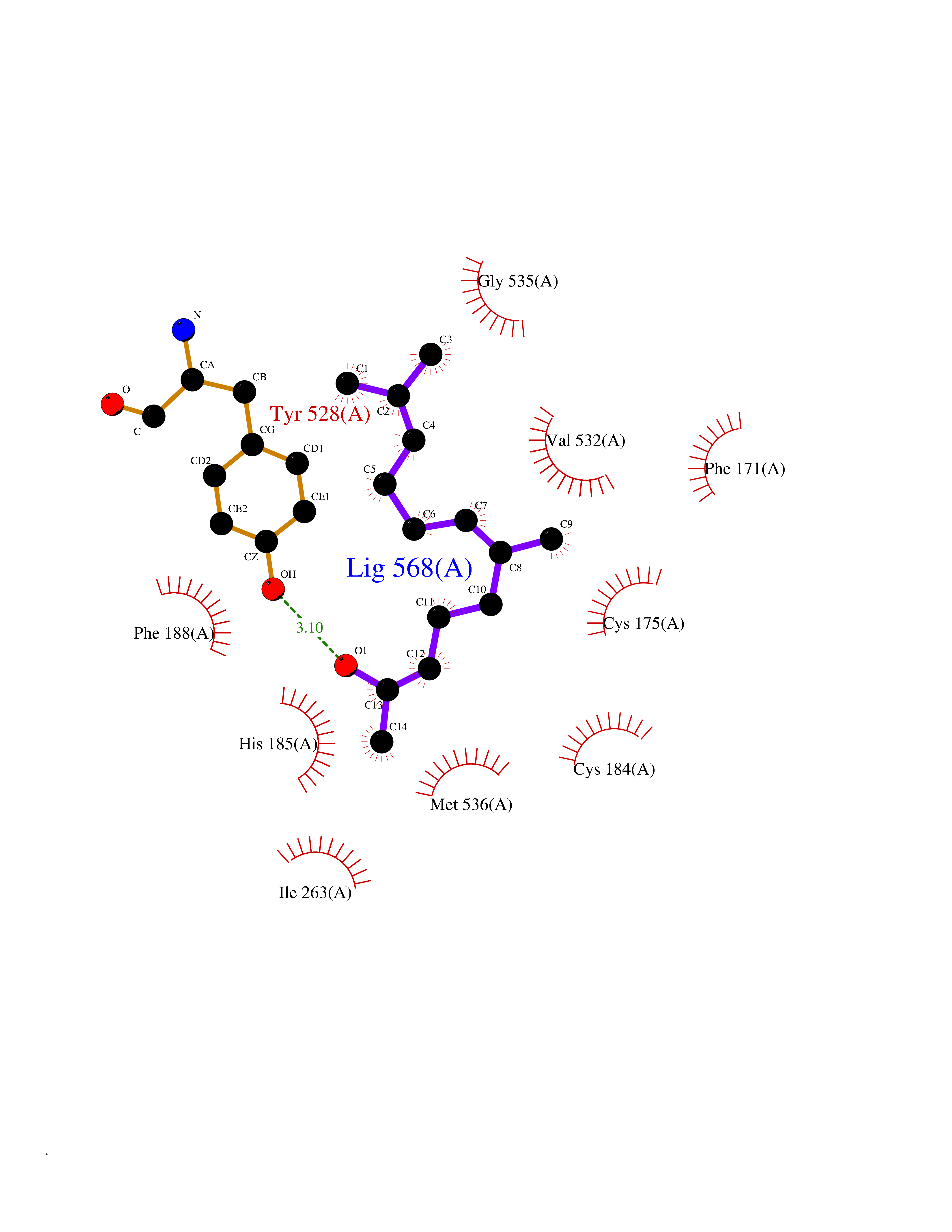 Ligplot Map 3D Binding mode Sequence FESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKH Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Dipeptidyl peptidase 8 (DPP-8) | 6EOP | 6.11 | |
Target general information Gen name DPP8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Prolyl dipeptidase DPP8; MSTP141; MSTP135; MSTP097; Dipeptidyl peptidase VIII; Dipeptidyl peptidase IV-related protein 1; DPRP1; DPRP-1; DPP VIII; DP8 Protein family Peptidase S9B family, DPPIV subfamily Biochemical class Peptidase Function Dipeptidyl peptidase that cleaves off N-terminal dipeptides from proteins having a Pro or Ala residue at position 2. Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.14.5 Uniprot keywords 3D-structure; Alternative splicing; Aminopeptidase; Apoptosis; Cytoplasm; Hydrolase; Protease; Proteomics identification; Reference proteome; Serine protease Protein physicochemical properties Chain ID A,D Molecular weight (Da) 97764.9 Length 849 Aromaticity 0.12 Instability index 47.71 Isoelectric point 5.69 Charge (pH=7) -21.66 2D Binding mode Binding energy (Kcal/mol) -8.33 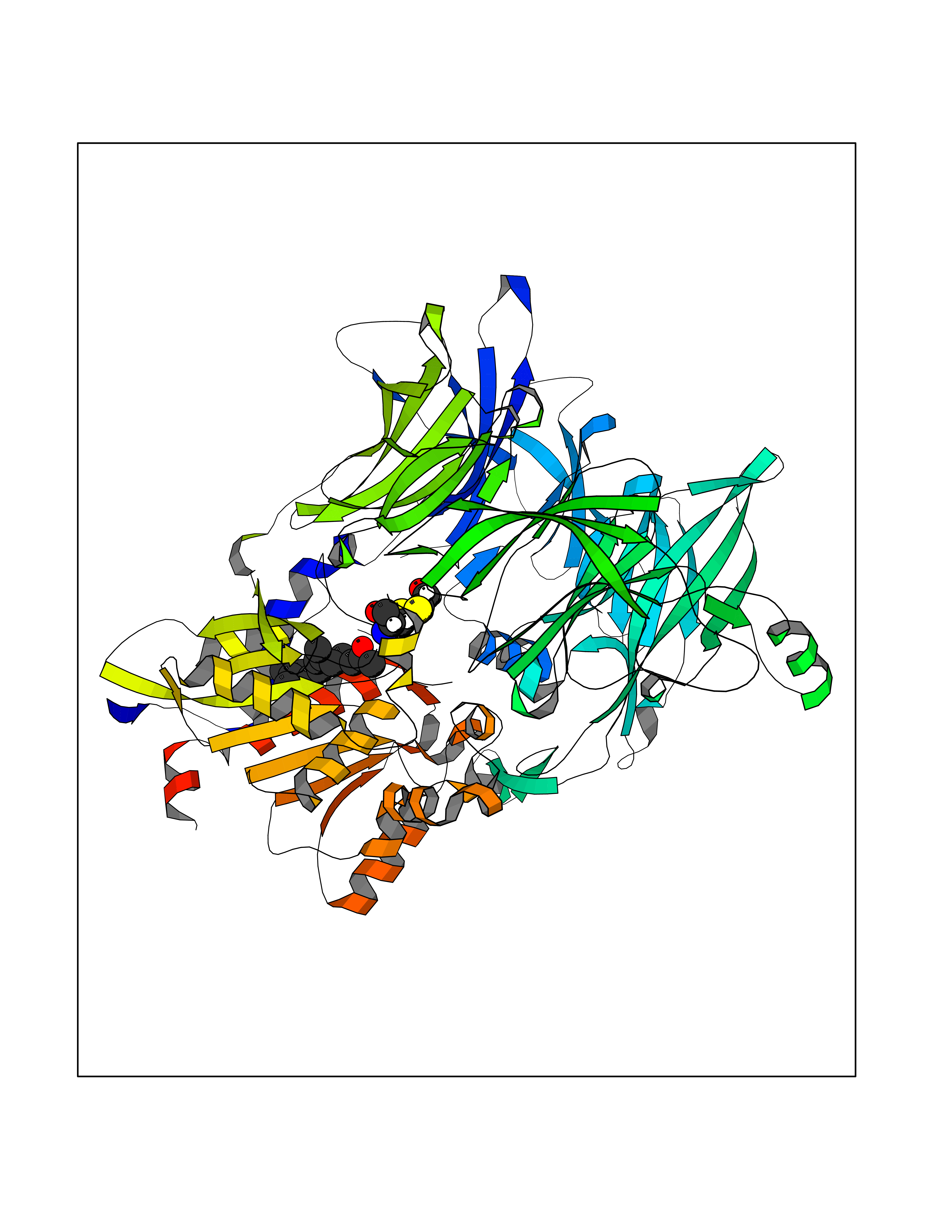 Molscript Map 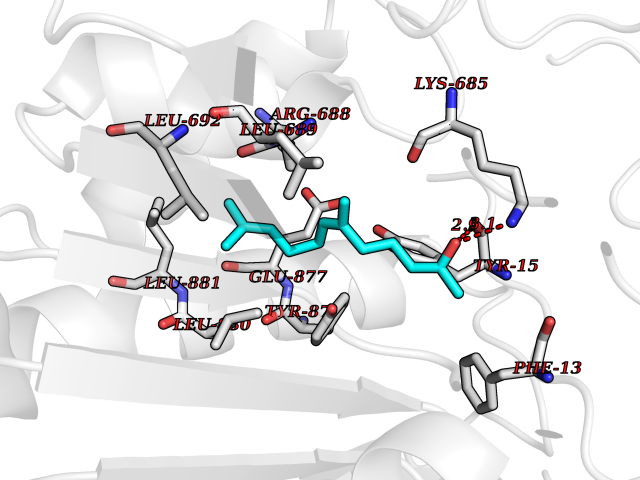 Pymol Map 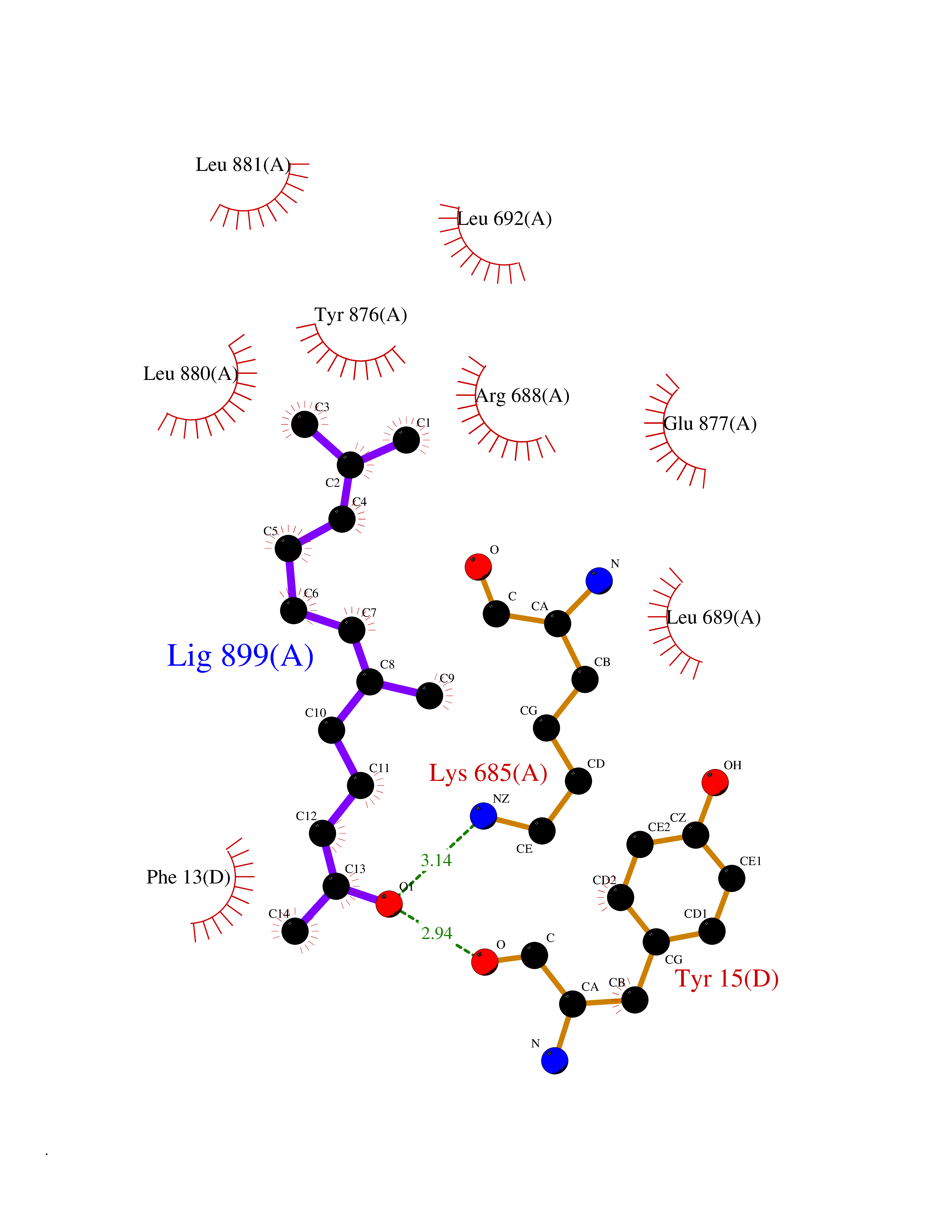 Ligplot Map 3D Binding mode Sequence LEPFYVERYSWSQLKKLLADTRKYHGYMMAKAPHDFMFVKRNDPDGPHSDRIYYLAMSNRENTLFYSEIPKTINRAAVLMLSWKPLLDLFQYSREEELLRERKRIGTVGIASYDYHQGSGTFLFQAGSGIYHVKDGGPQGFTQQPLRPNLVETSCPNIRMDPKLCPADPDWIAFIHSNDIWISNIVTREERRLTYVHNELANMEEDARSAGVATFVLQEEFDRYSGYWWCPKAETTPSGGKILRILYEENDESEVEIIHVTSPMLETRRADSFRYPKTGTANPKVTFKMSEIMIDAEGRIIDVIDKELIQPFEILFEGVEYIARAGWTPEGKYAWSILLDRSQTRLQIVLISPELFIPVEDDVMERQRLIESVPDSVTPLIIYEETTDIWINIHDIFHVFPQSHEEEIEFIFASECKTGFRHLYKITSILKESKYKRSSGGLPAPSDFKCPIKEEIAITSGEWEVLGRHGSNIQVDEVRRLVYFEGTKDSPLEHHLYVVSYVNPGEVTRLTDRGYSHSCCISQHCDFFISKYSNQKNPHCVSLYKLSSPEDDPTCKTKEFWATILDSAGPLPDYTPPEIFSFESTTGFTLYGMLYKPHDLQPGKKYPTVLFIYGGPQVQLVNNRFKGVKYFRLNTLASLGYVVVVIDNRGSXHRGLKFEGAFKYKMGQIEIDDQVEGLQYLASRYDFIDLDRVGIHGWSYGGYLSLMALMQRSDIFRVAIAGAPVTLWIFYDTGYTERYMGHPDQNEQGYYLGSVAMQAEKFPSEPNRLLLLHGFLDENVHFAHTSILLSFLVRAGKPYDLQIYPQERHSIRVPESGEHYELHLLHYLQENLGSRIAALKVSLRFLYEG Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Cholesterol 24-hydroxylase (CYP46A1) | 3MDR | 6.11 | |
Target general information Gen name CYP46A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cytochrome P450 46A1; CYP46; Cholesterol 24S-hydroxylase; Cholesterol 24-monooxygenase; CH24H Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function Primarily catalyzes the hydroxylation (with S stereochemistry) at C-24 of cholesterol side chain, triggering cholesterol diffusion out of neurons and its further degradation. By promoting constant cholesterol elimination in neurons, may activate the mevalonate pathway and coordinate the synthesis of new cholesterol and nonsterol isoprenoids involved in synaptic activity and learning. Further hydroxylates cholesterol derivatives and hormone steroids on both the ring and side chain of these molecules, converting them into active oxysterols involved in lipid signaling and biosynthesis. Acts as an epoxidase converting cholesta-5,24-dien-3beta-ol/desmosterol into (24S),25-epoxycholesterol, an abundant lipid ligand of nuclear NR1H2 and NR1H3 receptors shown to promote neurogenesis in developing brain. May also catalyze the oxidative metabolism of xenobiotics, such as clotrimazole. P450 monooxygenase that plays a major role in cholesterol homeostasis in the brain. Related diseases Spinocerebellar ataxia, autosomal recessive, with axonal neuropathy 1 (SCAN1) [MIM:607250]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCAN1 is an autosomal recessive cerebellar ataxia (ARCA) associated with peripheral axonal motor and sensory neuropathy, distal muscular atrophy, pes cavus and steppage gait as seen in Charcot-Marie-Tooth neuropathy. All affected individuals have normal intelligence. {ECO:0000269|PubMed:12244316, ECO:0000269|PubMed:15647511, ECO:0000269|PubMed:15920477, ECO:0000269|PubMed:16141202, ECO:0000269|PubMed:17948061}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.14.14.25 Uniprot keywords 3D-structure; Alternative splicing; Cell projection; Cholesterol metabolism; Endoplasmic reticulum; Heme; Iron; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism; Synapse; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 48977 Length 427 Aromaticity 0.1 Instability index 49.59 Isoelectric point 9.04 Charge (pH=7) 6.25 2D Binding mode Binding energy (Kcal/mol) -8.33 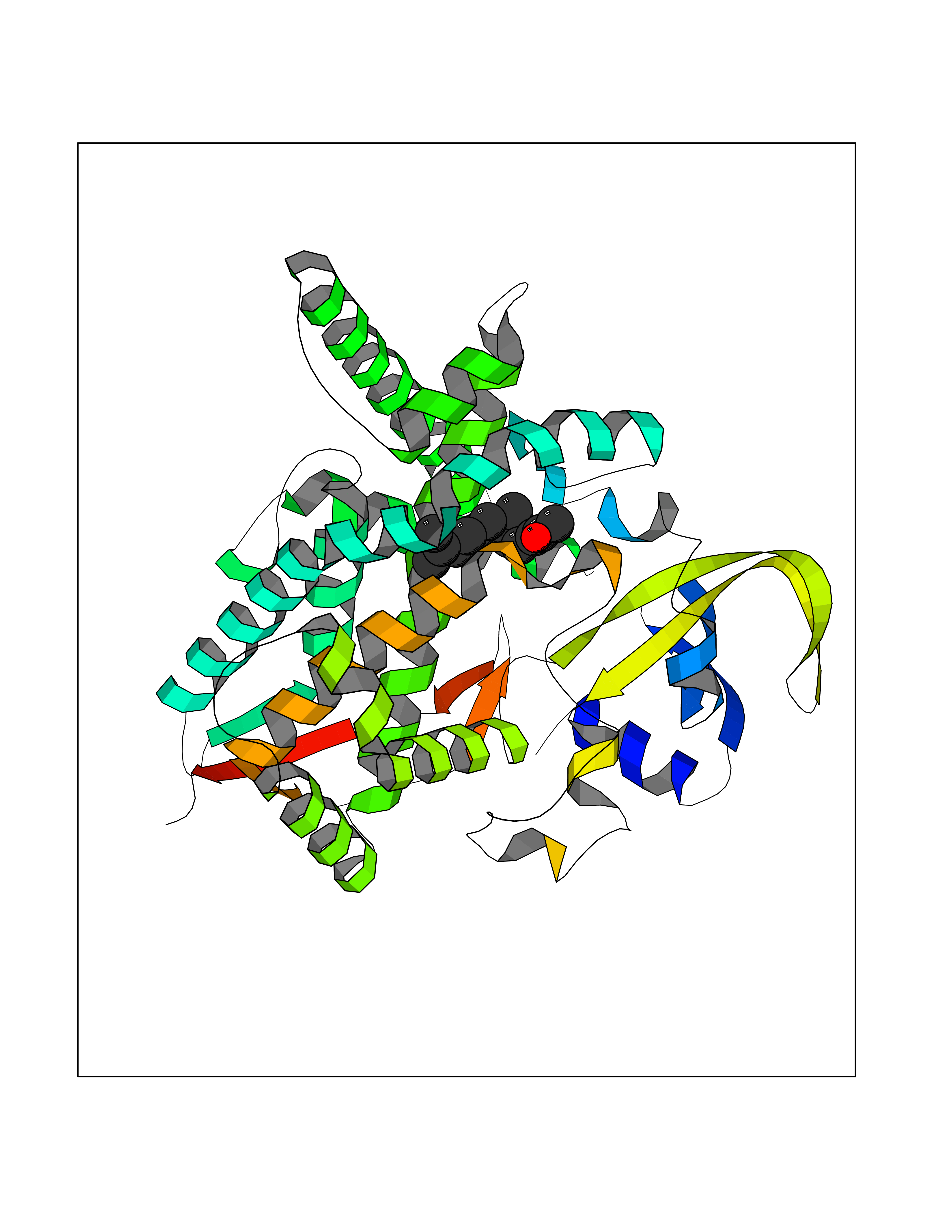 Molscript Map 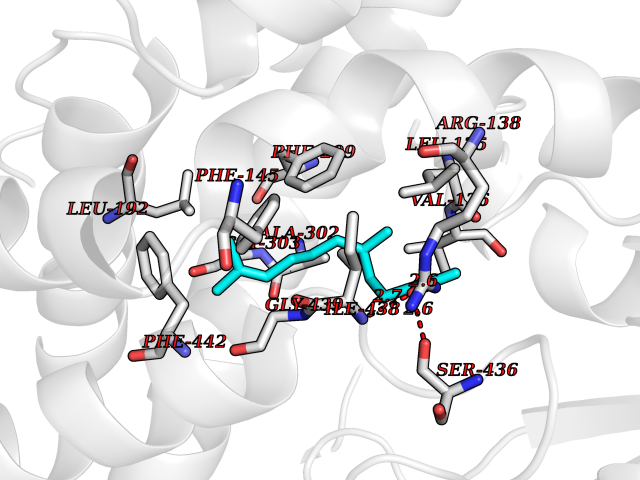 Pymol Map 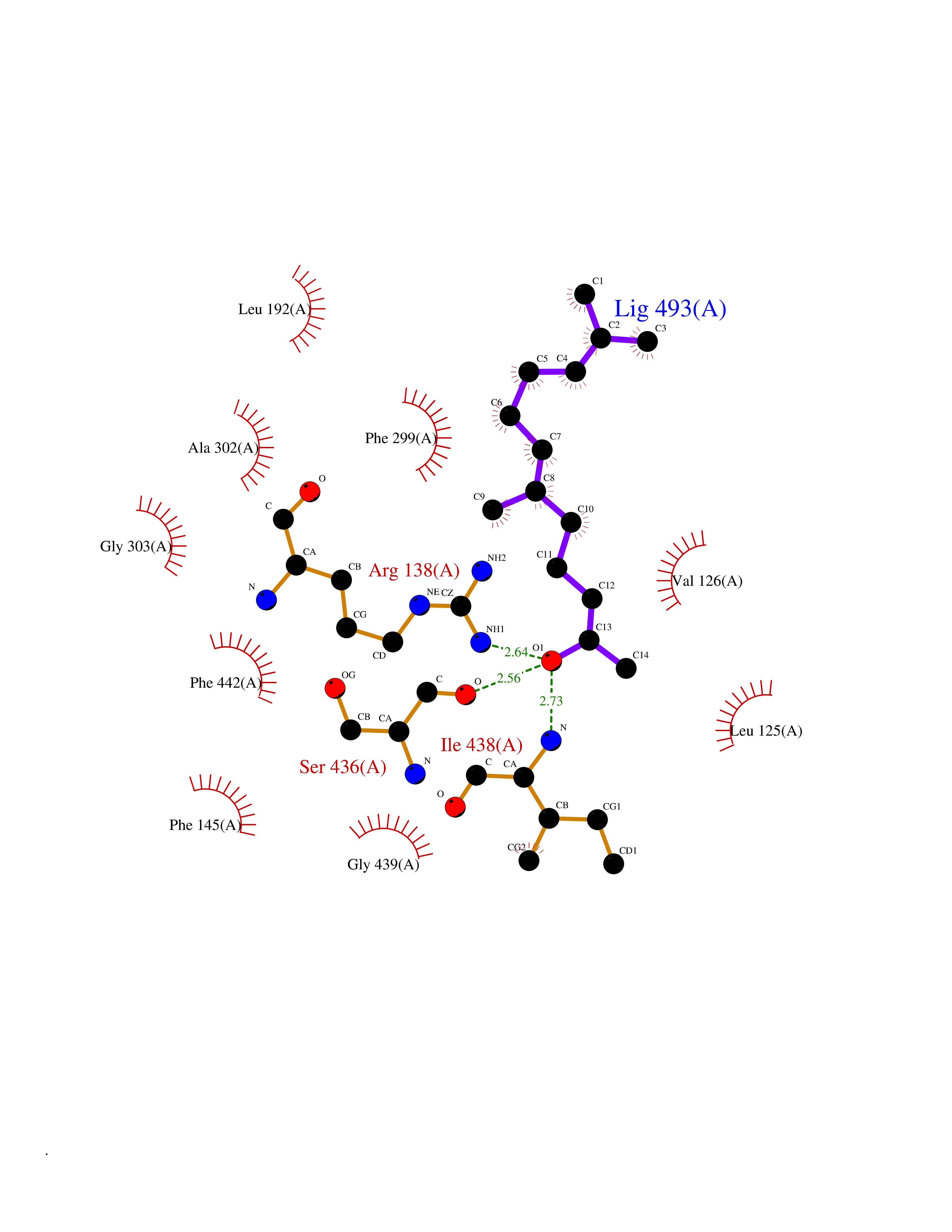 Ligplot Map 3D Binding mode Sequence RVLQDVFLDWAKKYGPVVRVNVFHKTSVIVTSPESVKKFLMSTKYNKDSKMYRALQTVFGERLFGQGLVSECNYERWHKQRRVIDLAFSRSSLVSLMETFNEKAEQLVEILEAKADGQTPVSMQDMLTYTAMDILAKAAFGMETSMLLGAQKPLSQAVKLMLEGITASRNTKRKQLREVRESIRFLRQVGRDWVQRRREALKRGEEVPADILTQILKAEEGAQDDEGLLDNFVTFFIAGHETSANHLAFTVMELSRQPEIVARLQAEVDEVIGSKRYLDFEDLGRLQYLSQVLKESLRLYPPAWGTFRLLEEETLIDGVRVPGNTPLLFSTYVMGRMDTYFEDPLTFNPDRFGPGAPKPRFTYFPFSLGHRSCIGQQFAQMEVKVVMAKLLQRLEFRLVPGQRFGLQEQATLKPLDPVLCTLRPRGW Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Dual-specificity tyrosine-phosphorylation regulated kinase 2 (DYRK2) | 6HDR | 6.11 | |
Target general information Gen name DYRK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dual specificity tyrosine-phosphorylation-regulated kinase 2 Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, MNB/DYRK subfamily Biochemical class Kinase Function Functions in part via its role in ubiquitin-dependent proteasomal protein degradation. Functions downstream of ATM and phosphorylates p53/TP53 at 'Ser-46', and thereby contributes to the induction of apoptosis in response to DNA damage. Phosphorylates NFATC1, and thereby inhibits its accumulation in the nucleus and its transcription factor activity. Phosphorylates EIF2B5 at 'Ser-544', enabling its subsequent phosphorylation and inhibition by GSK3B. Likewise, phosphorylation of NFATC1, CRMP2/DPYSL2 and CRMP4/DPYSL3 promotes their subsequent phosphorylation by GSK3B. May play a general role in the priming of GSK3 substrates. Inactivates GYS1 by phosphorylation at 'Ser-641', and potentially also a second phosphorylation site, thus regulating glycogen synthesis. Mediates EDVP E3 ligase complex formation and is required for the phosphorylation and subsequent degradation of KATNA1. Phosphorylates TERT at 'Ser-457', promoting TERT ubiquitination by the EDVP complex. Phosphorylates SIAH2, and thereby increases its ubiquitin ligase activity. Promotes the proteasomal degradation of MYC and JUN, and thereby regulates progress through the mitotic cell cycle and cell proliferation. Promotes proteasomal degradation of GLI2 and GLI3, and thereby plays a role in smoothened and sonic hedgehog signaling. Plays a role in cytoskeleton organization and neurite outgrowth via its phosphorylation of DCX and DPYSL2. Phosphorylates CRMP2/DPYSL2, CRMP4/DPYSL3, DCX, EIF2B5, EIF4EBP1, GLI2, GLI3, GYS1, JUN, MDM2, MYC, NFATC1, p53/TP53, TAU/MAPT and KATNA1. Can phosphorylate histone H1, histone H3 and histone H2B (in vitro). Can phosphorylate CARHSP1 (in vitro). Serine/threonine-protein kinase involved in the regulation of the mitotic cell cycle, cell proliferation, apoptosis, organization of the cytoskeleton and neurite outgrowth. Related diseases Bone marrow failure and diabetes mellitus syndrome (BMFDMS) [MIM:620044]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFDMS is an autosomal recessive form characterized by various degrees of bone marrow failure, ranging from dyserythropoiesis to bone marrow aplasia, with onset in infancy or early childhood, and non-autoimmune insulin-dependent diabetes mellitus appearing in the first or second decades. Many patients show pigmentary skin abnormalities and short stature. {ECO:0000269|PubMed:28073829, ECO:0000269|PubMed:35611808, ECO:0000269|PubMed:35931051}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NR20; Q13422; Q9BQD3; Q9BRK4; P23497; O43379; P62258; Q96C00 EC number EC 2.7.12.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cytoplasm; Kinase; Magnesium; Manganese; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID A Molecular weight (Da) 46422.1 Length 407 Aromaticity 0.09 Instability index 44.91 Isoelectric point 9.09 Charge (pH=7) 12.37 2D Binding mode Binding energy (Kcal/mol) -8.34  Molscript Map 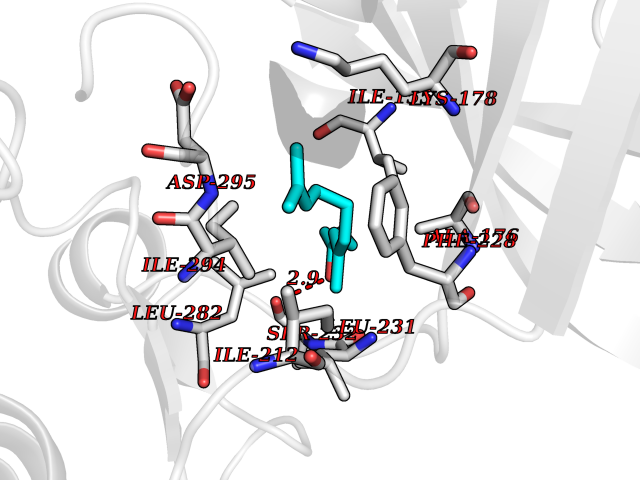 Pymol Map 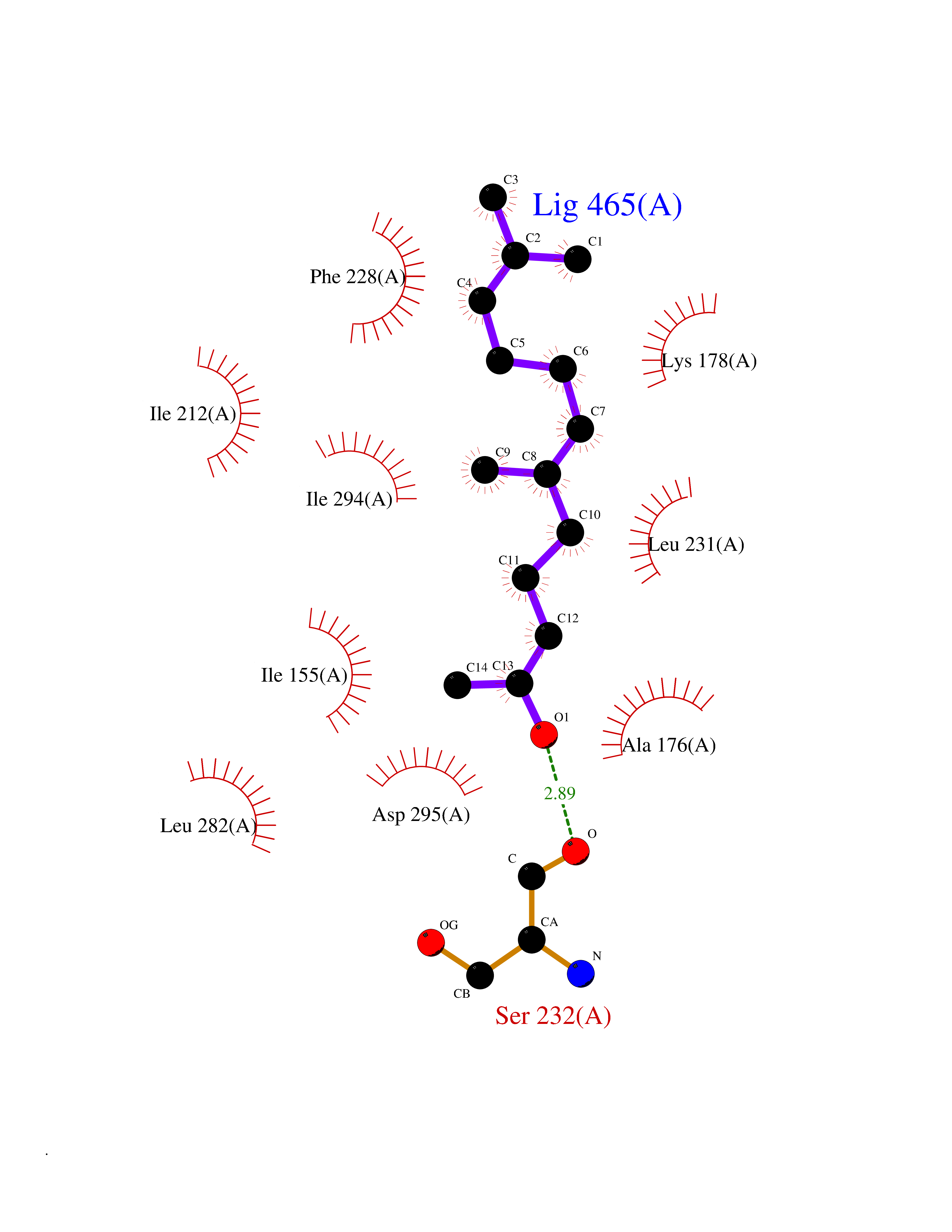 Ligplot Map 3D Binding mode Sequence HHHSXGVDLGTENLYFQSMGKVKATPMTPEQAMKQYMQKLTAFEHHEIFSYPEIYFLGLNAKKRQGMTGGPNNGGYDDDQGSYVQVPHDHVAYRYEVLKVIGKGSFGQVVKAYDHKVHQHVALKMVRNEKRFHRQAAEEIRILEHLRKQDKDNTMNVIHMLENFTFRNHICMTFELLSMNLYELIKKNKFQGFSLPLVRKFAHSILQCLDALHKNRIIHCDLKPENILLKQQGRSGIKVIDFGSSCYEHQRVYTXIQSRFYRAPEVILGARYGMPIDMWSLGCILAELLTGYPLLPGEDEGDQLACMIELLGMPSQKLLDASKRAKNFVSXKGYPRYCTVTTLSDVVLNGGRSRRGKLRGPPESREWGNALKGCDDPLFLDFLKQCLEWDPAVRMTPGQALRHPWLR Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Peptostreptococcal albumin-binding protein | 2VDB | 6.10 | |
Target general information Gen name pab Organism Finegoldia magna (Peptostreptococcus magnus) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Protein binding Function Binds serum albumin. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB03600; DB00788 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell wall; Peptidoglycan-anchor; Secreted; Signal Protein physicochemical properties Chain ID B Molecular weight (Da) 21751.6 Length 189 Aromaticity 0.1 Instability index 49.17 Isoelectric point 5.44 Charge (pH=7) -6.7 2D Binding mode Binding energy (Kcal/mol) -8.31  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence EVAHRFKDLGEENFKALVLIAFAQYLQQCPFEDHVKLVNEVTEFAKTCVADESAENCDKSLHTLFGDKLCTVATLEMADCCAKQEPERNECFLQHKDDNPNLPRLVRPEVDVMCTAFHDNEETFLKKYLYEIARRHPYFYAPELLFFAKRYKAAFTECCQAADKAACLLPKLDELRDEGKASSAKQRLK Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Neutrophil gelatinase-associated lipocalin (LCN2) | 5NKN | 6.10 | |
Target general information Gen name LCN2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p25; Siderocalin LCN2; Oncogene 24p3; NGAL; Lipocalin-2; LCN2; 25 kDa alpha-2-microglobulin-related subunit of MMP-9 Protein family Calycin superfamily, Lipocalin family Biochemical class Calycin family Function Iron-trafficking protein involved in multiple processes such as apoptosis, innate immunity and renal development. Binds iron through association with 2,5-dihydroxybenzoic acid (2,5- DHBA), a siderophore that shares structural similarities withbacterial enterobactin, and delivers or removes iron from the cell, depending on the context. Iron-bound form (holo-24p3) is internalized following binding to the SLC22A17 (24p3R) receptor, leading to release of iron and subsequent increase of intracellular iron concentration. In contrast, association of the iron-free form (apo-24p3) with the SLC22A17 (24p3R) receptor is followed by association with an intracellular siderophore, iron chelation and iron transfer to the extracellular medium, thereby reducing intracellular iron concentration. Involved in apoptosis due to interleukin-3 (IL3) deprivation: iron-loaded form increases intracellular iron concentration without promoting apoptosis, while iron-free form decreases intracellular iron levels, inducing expression of the proapoptotic protein BCL2L11/BIM, resulting in apoptosis. Involved in innate immunity, possibly by sequestrating iron, leading to limit bacterial growth. . Related diseases Pseudovaginal perineoscrotal hypospadias (PPSH) [MIM:264600]: A form of male pseudohermaphroditism in which 46,XY males show ambiguous genitalia at birth, including perineal hypospadias and a blind perineal pouch, and develop masculinization at puberty. The name of the disorder stems from the finding of a blind-ending perineal opening resembling a vagina and a severely hypospadiac penis with the urethra opening onto the perineum. {ECO:0000269|PubMed:10718838, ECO:0000269|PubMed:10898110, ECO:0000269|PubMed:10999800, ECO:0000269|PubMed:12843198, ECO:0000269|PubMed:15064320, ECO:0000269|PubMed:1522235, ECO:0000269|PubMed:15528927, ECO:0000269|PubMed:15770495, ECO:0000269|PubMed:16098368, ECO:0000269|PubMed:16181229, ECO:0000269|PubMed:7554313, ECO:0000269|PubMed:8626825, ECO:0000269|PubMed:8768837, ECO:0000269|PubMed:9208814, ECO:0000269|PubMed:9745434, ECO:0000269|PubMed:9843052}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02710; DB01672; DB01926; DB04043; DB01631; DB04476 Interacts with P49419-2; Q9NXW9; Q8WXI3; Q12797-6; Q9BXY8; Q96LC9; P49069; P24863; Q9UKJ5; Q9H1P6; Q9H6B4; O14595; Q08426; Q6NZ36-4; B3EWG5; Q7Z4H3; Q6ISS4; Q5TA76; P80188; Q9UIQ6-2; Q9Y6Y9; Q96JG8; Q8IXL7-2; Q969H8; Q969S2; Q17RF5; P07237; P13667; Q96FA3; Q9NRD5; Q13526; Q9UGP5-2; Q12837; P54646; Q86Y79; O60895; Q9BWG6; P60059; O43765; Q96EQ0; Q8IYX1; Q9UL33-2; P20396; O43715; Q13049; Q99816; Q5W5X9-3; Q99757; P57075-2; Q969M7; Q9UMX0; Q9UHD9; P15692-12; Q14119; Q9Y6T4; Q9H0D6; O96006; A0A1U9X8X8 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Glycoprotein; Immunity; Innate immunity; Ion transport; Iron; Iron transport; Proteomics identification; Pyrrolidone carboxylic acid; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 19748.4 Length 172 Aromaticity 0.13 Instability index 30.73 Isoelectric point 7.71 Charge (pH=7) 0.72 2D Binding mode Binding energy (Kcal/mol) -8.32  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SDLIPAPPLSKVPLQQNFQDNQFHGKWYVVGVAGNGFLREDKDPIKMAATIYELKEDKSYNVTFQKFPMKKCQYMTDTLVPGSQPGEFTLGNIKSEPGYTSWLVRVVSTNYNQHAMVFFKAVQQNREDFFITLYGRTKELTSELKENFIRFSKSLGLPENHIVFPVPIDQCI Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | "Periplasmic trehalase (EC 3.2.1.28) (Alpha,alpha-trehalase) (Alpha,alpha-trehalose glucohydrolase) (Tre37A)" | 2JG0 | 6.09 | |
Target general information Gen name treA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1186;osmA;b1197 Protein family Glycosyl hydrolase 37 family Biochemical class NA Function Provides the cells with the ability to utilize trehalose at high osmolarity by splitting it into glucose molecules that can subsequently be taken up by the phosphotransferase-mediated uptake system. Related diseases SRC kinase activity has been shown to be increased in several tumor tissues and tumor cell lines such as colon carcinoma cells. {ECO:0000269|PubMed:2498394, ECO:0000269|PubMed:3093483}.; DISEASE: Thrombocytopenia 6 (THC6) [MIM:616937]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC6 is an autosomal dominant form. Affected individuals may also have bone abnormalities and an increased risk for myelofibrosis. {ECO:0000269|PubMed:26936507}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.2.1.28 Uniprot keywords 3D-structure; Direct protein sequencing; Glycosidase; Hydrolase; Periplasm; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 57508.9 Length 507 Aromaticity 0.11 Instability index 48.32 Isoelectric point 5.48 Charge (pH=7) -10.13 2D Binding mode Binding energy (Kcal/mol) -8.3  Molscript Map  Pymol Map 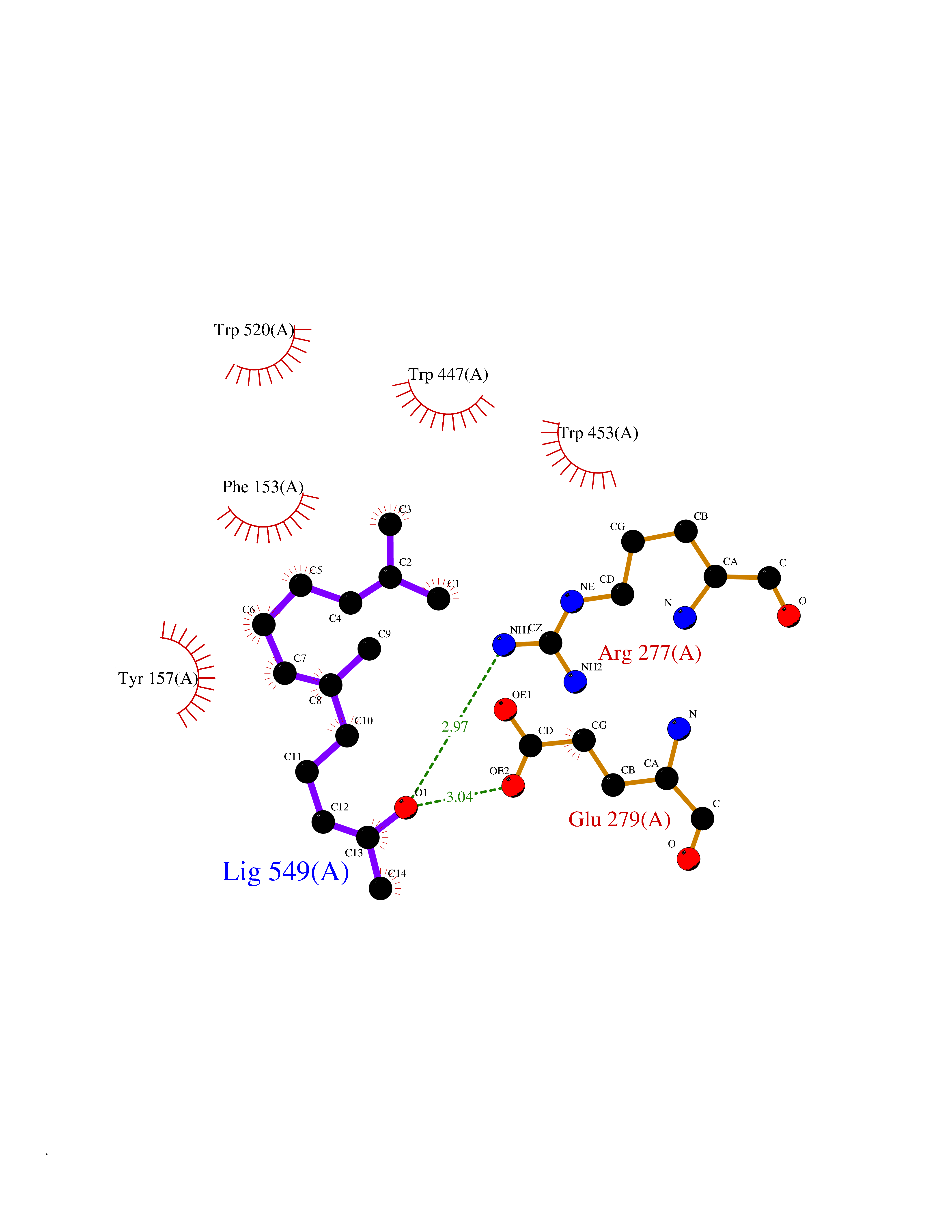 Ligplot Map 3D Binding mode Sequence PQPPDILLGPLFNDVQNAKLFPDQKTFADAVPNSDPLMILADYRMQQNQSGFDLRHFVNVNFTLPKYVPPEGQSLREHIDGLWPVLTRSTENTEKWDSLLPLPEPYVVPGGRFREVYYWDSYFTMLGLAESGHWDKVADMVANFAHEIDTYGHIPNGNRSYYLSRSQPPFFALMVELLAQHEGDAALKQYLPQMQKEYAYWMDGVENLQAGQQEKRVVKLQDGTLLNRYWDDRDTPRPESWVEDIATAKSNPNRPATEIYRDLRSAAASGWDFSSRWMDNPQQLNTLRTTSIVPVDLNSLMFKMEKILARASKAAGDNAMANQYETLANARQKGIEKYLWNDQQGWYADYDLKSHKVRNQLTAAALFPLYVNAAAKDRANKMATATKTHLLQPGGLNTTSVKSGQQWDAPNGWAPLQWVATEGLQNYGQKEVAMDISWHFLTNVQHTYDREKKLVEKYDVSTTGTGGGGGEYPLQDGFGWTNGVTLKMLDLICPKEQPCDNVPATRP Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Glutamate receptor ionotropic NMDA 2A (NMDAR2A) | 5KCJ | 6.09 | |
Target general information Gen name GRIN2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NR2A; NMDA receptor NR2A; N-methyl D-aspartate receptor subtype 2A; HNR2A; Glutamate receptor ionotropic, NMDA 2A; Glutamate [NMDA] receptor subunit epsilon-1; GluN2A Protein family Glutamate-gated ion channel (TC 1.A.10.1) family, NR1/GRIN1 subfamily Biochemical class Glutamate-gated ion channel Function Channel activation requires binding of the neurotransmitter glutamate to the epsilon subunit, glycine binding to the zeta subunit, plus membrane depolarization to eliminate channel inhibition by Mg(2+). Sensitivity to glutamate and channel kinetics depend on the subunit composition; channels containing GRIN1 and GRIN2A have higher sensitivity to glutamate and faster kinetics than channels formed by GRIN1 and GRIN2B. Contributes to the slow phase of excitatory postsynaptic current, long-term synaptic potentiation, and learning. Component of NMDA receptor complexes that function as heterotetrameric, ligand-gated ion channels with high calcium permeability and voltage-dependent sensitivity to magnesium. Related diseases Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal dominant (NDHMSD) [MIM:614254]: An autosomal dominant neurodevelopmental disorder characterized by severe intellectual disability and developmental delay, absent speech, muscular hypotonia, dyskinesia, and hyperkinetic movements. Cortical blindness, cerebral atrophy, and seizures are present in some patients. {ECO:0000269|PubMed:21376300, ECO:0000269|PubMed:25167861, ECO:0000269|PubMed:25864721, ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28095420, ECO:0000269|PubMed:28228639, ECO:0000269|PubMed:28389307, ECO:0000269|PubMed:38538865}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal recessive (NDHMSR) [MIM:617820]: An autosomal recessive neurodevelopmental disorder characterized by severe intellectual disability and psychomotor developmental delay, involuntary and stereotypic movements, spasticity, and inability to walk without support. Intractable seizures manifest in some patients. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28051072}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 101 (DEE101) [MIM:619814]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE101 is an autosomal recessive, severe form characterized by onset of seizures in early infancy. Death in infancy may occur. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:34611970}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01931; DB00659; DB06151; DB08838; DB01238; DB00289; DB05824; DB04620; DB03929; DB00647; DB00843; DB00228; DB11823; DB13146; DB06741; DB00142; DB00874; DB08954; DB06738; DB09409; DB09481; DB01043; DB00454; DB00333; DB04896; DB01173; DB00312; DB01174; DB01708; DB00418; DB00193 Interacts with P05067; P35637; Q12879-1; Q13224; Q62936 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Intellectual disability; Ion channel; Ion transport; Ligand-gated ion channel; Magnesium; Membrane; Metal-binding; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Zinc Protein physicochemical properties Chain ID B Molecular weight (Da) 53395.6 Length 469 Aromaticity 0.11 Instability index 29.84 Isoelectric point 8.72 Charge (pH=7) 5.65 2D Binding mode Binding energy (Kcal/mol) -8.3 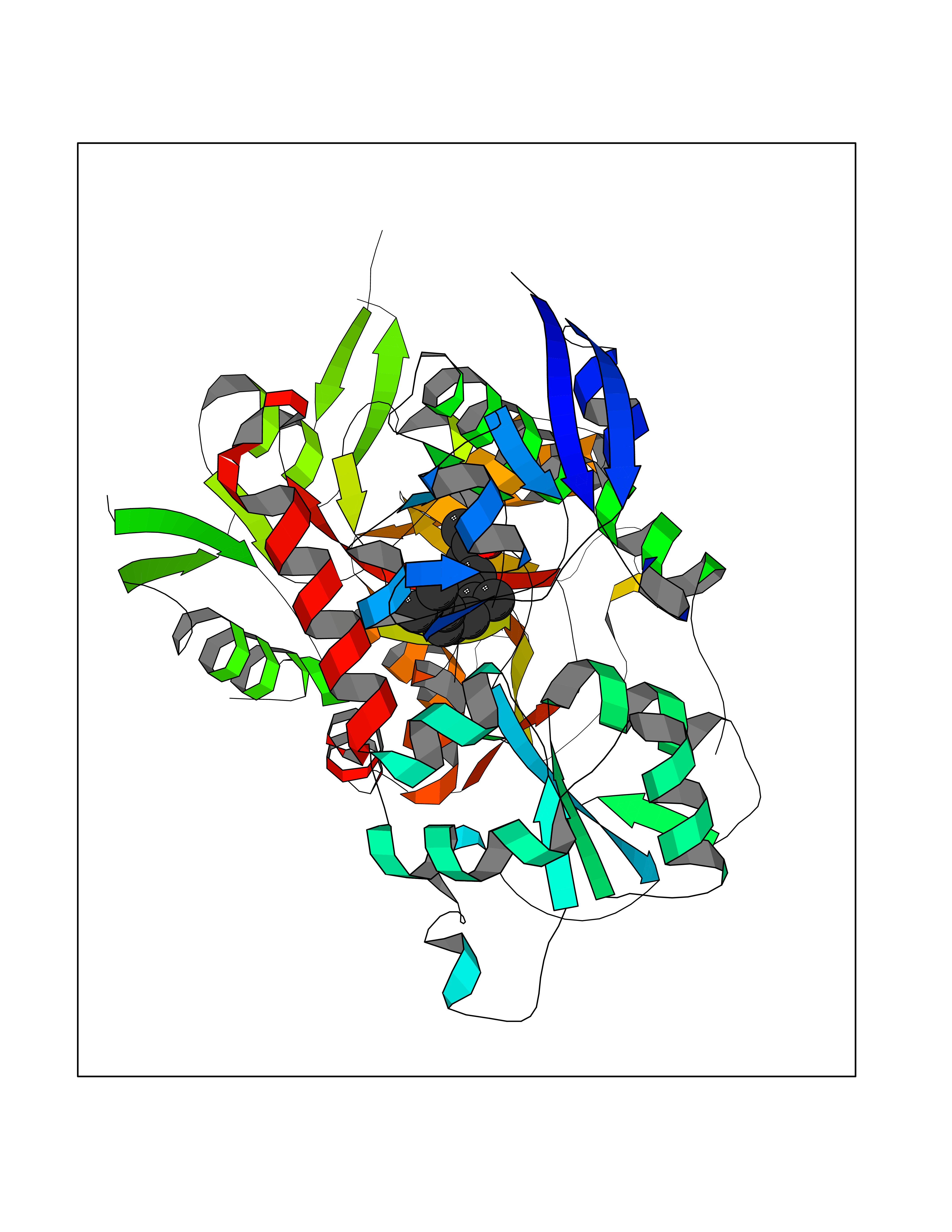 Molscript Map 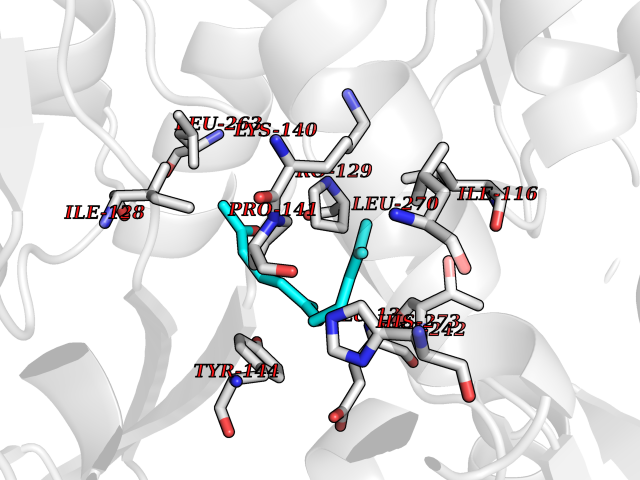 Pymol Map 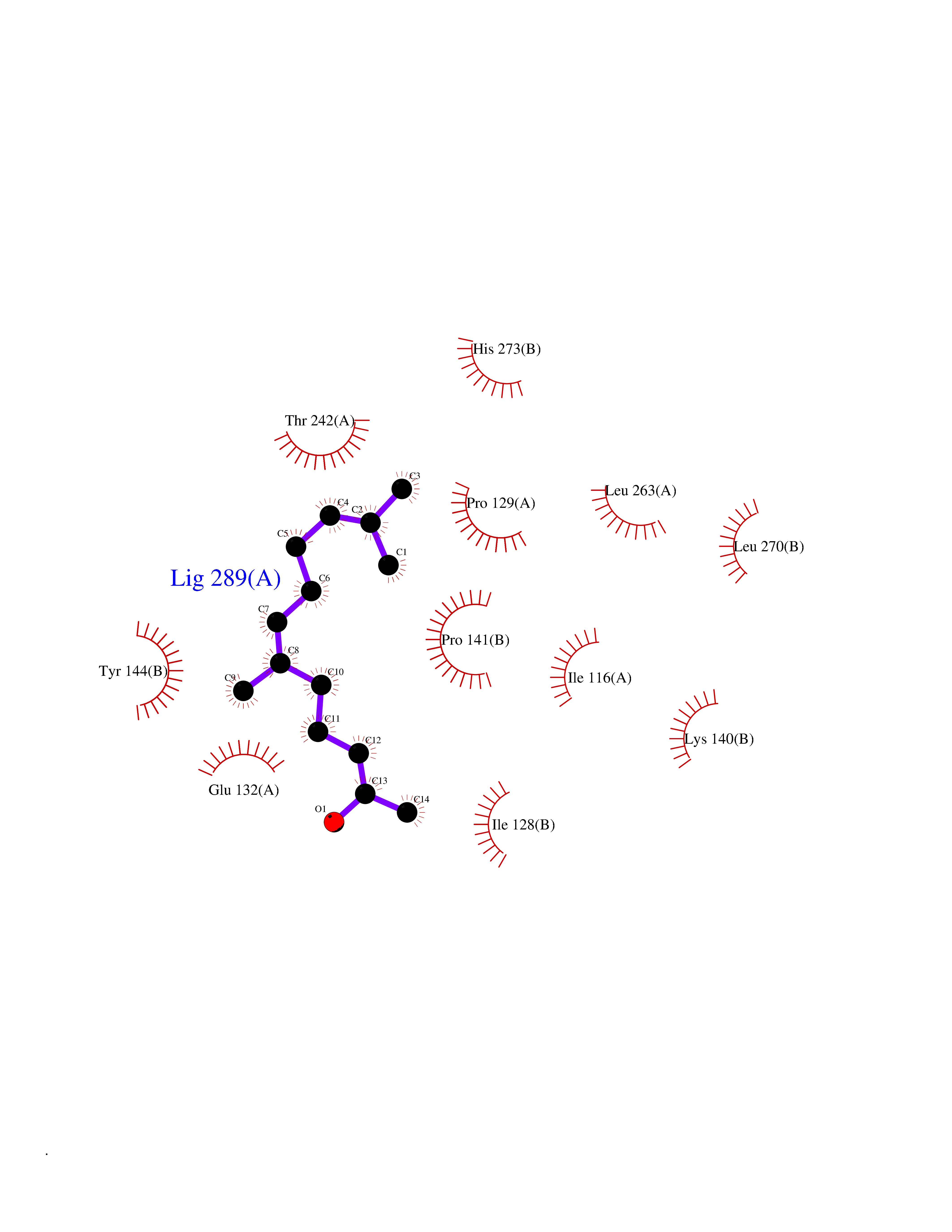 Ligplot Map 3D Binding mode Sequence DNHLSIVTLEEAPFVILKKLSRTVKFTYDLYLVTNGKHGKKVNNVWNGMIGEVVYQRAVMAVGSLTINEERSEVVDFSVPFVETGISVMVSRGTQVTGLSDKKFQRPHDYSPPFRFGTVPNGSTERNIRNNYPYMHQYMTKFNQKGVEDALVSLKTGKLDAFIYDAAVLNYKAGRDEGCKLVTIGSGYIFATTGYGIALQKGSPWKRQIDLALLQFVGDGEMEELETLWLTGICTRLKIVTIHQEPFVYYGFCIDLLIKLARTMNFTYEVHLVADGKFGTQERVNKKEWNGMMGELLSGQADMIVAPLTINNERAQYIEFSKPFKYQGLTILVKKGTRITGINDPRLRNPSDKFIYATVKQSSVDIYFRRQVELSTMYRHMEKHNYESAAEAIQAVRDNKLHAFIWDSAVLEFEASQKCDLVTTGELFFRSGFGIGMRKDSPWKQNVSLSILKSHENGFMEDLDKTWVR Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Cannabinoid receptor 2 (CB2) | 6PT0 | 6.09 | |
Target general information Gen name CNR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hCB2; Cannabinoid CB2 receptor; CX5; CB2B; CB2A; CB-2 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function May function in inflammatory response, nociceptive transmission and bone homeostasis. Heterotrimeric G protein-coupled receptor for endocannabinoid 2-arachidonoylglycerol mediating inhibition of adenylate cyclase. Related diseases Factor V deficiency (FA5D) [MIM:227400]: A blood coagulation disorder leading to a hemorrhagic diathesis known as parahemophilia. {ECO:0000269|PubMed:10942390, ECO:0000269|PubMed:12393490}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thrombophilia due to activated protein C resistance (THPH2) [MIM:188055]: A hemostatic disorder due to defective degradation of factor V by activated protein C. It is characterized by a poor anticoagulant response to activated protein C resulting in tendency to thrombosis. {ECO:0000269|PubMed:10391209, ECO:0000269|PubMed:10942390, ECO:0000269|PubMed:11435304, ECO:0000269|PubMed:11858490, ECO:0000269|PubMed:14617013, ECO:0000269|PubMed:14695241, ECO:0000269|PubMed:16710414, ECO:0000269|PubMed:8164741, ECO:0000269|PubMed:9454742}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Budd-Chiari syndrome (BDCHS) [MIM:600880]: A syndrome caused by obstruction of hepatic venous outflow involving either the hepatic veins or the terminal segment of the inferior vena cava. Obstructions are generally caused by thrombosis and lead to hepatic congestion and ischemic necrosis. Clinical manifestations observed in the majority of patients include hepatomegaly, right upper quadrant pain and abdominal ascites. Budd-Chiari syndrome is associated with a combination of disease states including primary myeloproliferative syndromes and thrombophilia due to factor V Leiden, protein C deficiency and antithrombin III deficiency. Budd-Chiari syndrome is a rare but typical complication in patients with polycythemia vera. {ECO:0000269|PubMed:9245936}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Ischemic stroke (ISCHSTR) [MIM:601367]: A stroke is an acute neurologic event leading to death of neural tissue of the brain and resulting in loss of motor, sensory and/or cognitive function. Ischemic strokes, resulting from vascular occlusion, is considered to be a highly complex disease consisting of a group of heterogeneous disorders with multiple genetic and environmental risk factors. {ECO:0000269|PubMed:15534175}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Pregnancy loss, recurrent, 1 (RPRGL1) [MIM:614389]: A common complication of pregnancy, resulting in spontaneous abortion before the fetus has reached viability. The term includes all miscarriages from the time of conception until 24 weeks of gestation. Recurrent pregnancy loss is defined as 3 or more consecutive spontaneous abortions. {ECO:0000269|PubMed:11018168}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09061; DB00470; DB06202; DB14009; DB00486; DB14011; DB02955; DB16321; DB11755 Interacts with Q9UKJ8; Q15848; Q9NRZ5; P13236; P21964; Q14802-3; Q8N387; Q8IXM6; I3L0A0; Q96AA3; Q9Y6D0; Q6ICL7; Q9NP94; Q13501; Q96HH6; Q969S6; Q9NWH2; Q9H2L4; Q8N2M4; Q6ZT21; Q5TGU0; Q9Y548; Q9BSR8; Q96EC8 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cell projection; G-protein coupled receptor; Glycoprotein; Inflammatory response; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32999.2 Length 298 Aromaticity 0.11 Instability index 30.98 Isoelectric point 9.49 Charge (pH=7) 14.35 2D Binding mode Binding energy (Kcal/mol) -8.31  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MKDYMILSGPQKTAVAVLCTLLGLLSALENVAVLYLILSSHQLRRKPSYLFIGSLAGADFLASVVFACSFVNFHVFHGVDSKAVFLLKIGSVTMTFTASVGSLLLTAIDRYLCLRYPPSYKALLTRGRALVTLGIMWVLSALVSYLPLMGWTCCPRPCSELFPLIPNDYLLSWLLFIAFLFSGIIYTYGHVLWKAHQHVASLSGHQDRQVPGMARMRLDVRLAKTLGLVLAVLLICWFPVLALMAHSLATTLSDQVKKAFAFCSMLCLINSMVNPVIYALRSGEIRSSAHHCLAHWKK Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Neuronal acetylcholine receptor alpha-2 (CHRNA2) | 5FJV | 6.09 | |
Target general information Gen name CHRNA2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CHRNA2 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-2/CHRNA2 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane. Related diseases Epilepsy, nocturnal frontal lobe, 4 (ENFL4) [MIM:610353]: An autosomal dominant focal epilepsy characterized by nocturnal seizures associated with fear sensation, tongue movements, and nocturnal wandering, closely resembling nightmares and sleep walking. {ECO:0000269|PubMed:16826524}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Seizures, benign familial infantile, 6 (BFIS6) [MIM:610353]: A form of benign familial infantile epilepsy, a neurologic disorder characterized by afebrile seizures occurring in clusters during the first year of life, without neurologic sequelae. BFIS6 inheritance is autosomal dominant. {ECO:0000269|PubMed:25847220}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00732; DB00237; DB00411; DB00565; DB01245; DB00514; DB01135; DB07720; DB00898; DB00472; DB00483; DB08960; DB00657; DB01336; DB00416; DB01226; DB00184; DB01337; DB01338; DB00721; DB00728; DB05740; DB00202; DB01199; DB01339 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 48323.4 Length 413 Aromaticity 0.15 Instability index 32 Isoelectric point 5.69 Charge (pH=7) -6.58 2D Binding mode Binding energy (Kcal/mol) -8.31  Molscript Map 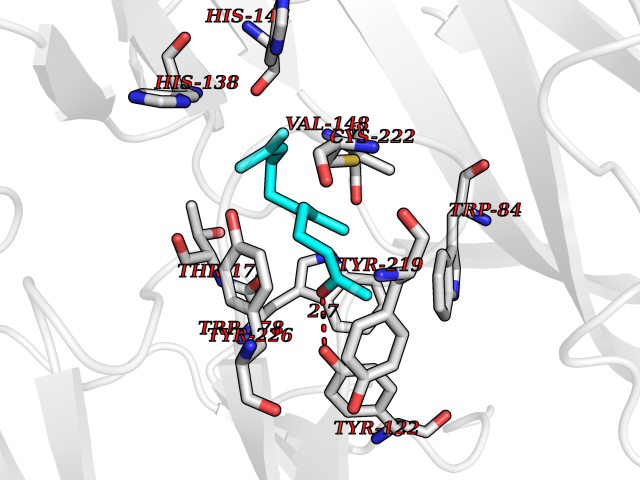 Pymol Map 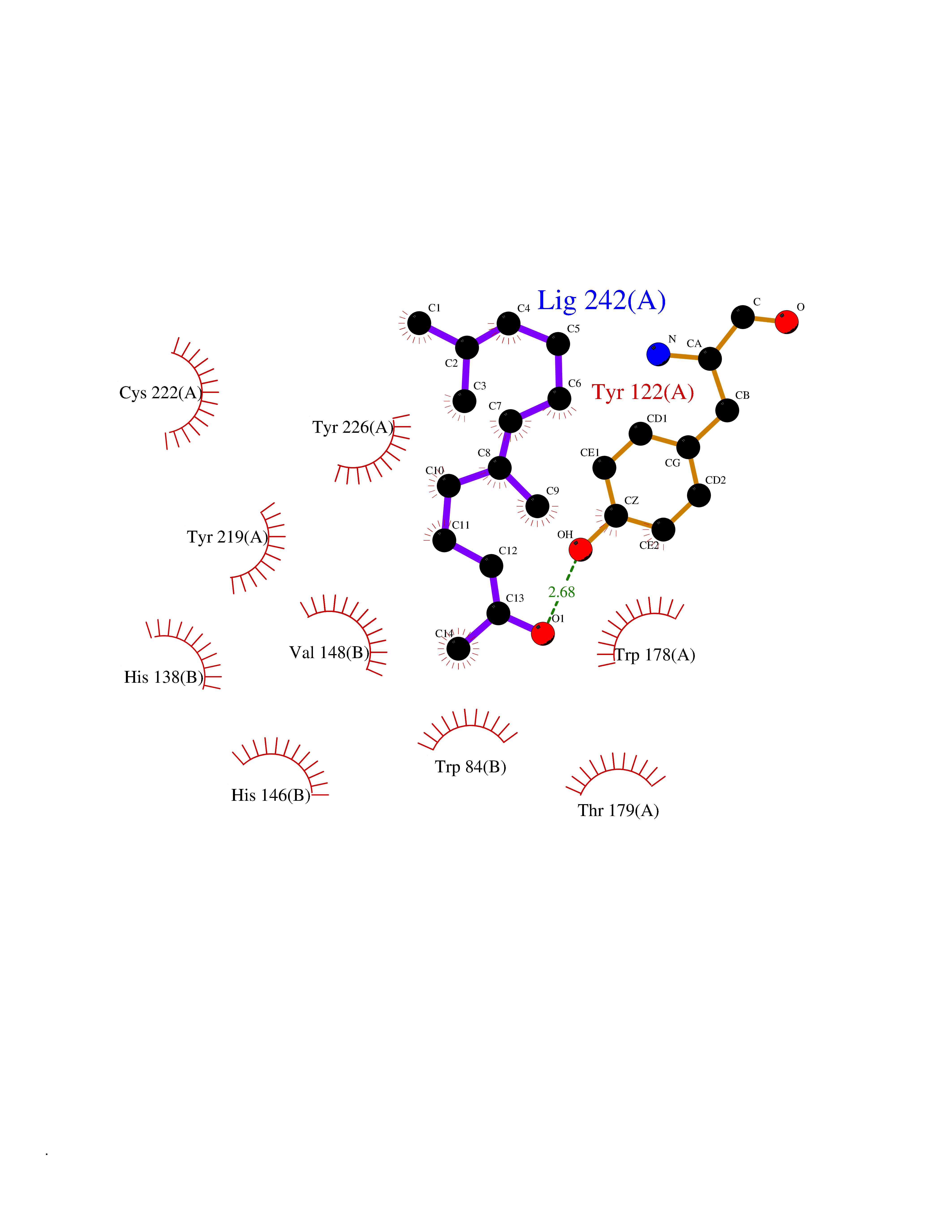 Ligplot Map 3D Binding mode Sequence DRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLP Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Hyperpolarization cyclic nucleotide-gated channel 2 (HCN2) | 3U10 | 6.09 | |
Target general information Gen name HCN2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channel 2; Brain cyclic nucleotide-gated channel 2; BCNG2; BCNG-2 Protein family Potassium channel HCN family Biochemical class Voltage-gated ion channel Function Contributes to the native pacemaker currents in heart (If) and in neurons (Ih). Can also transport ammonium in the distal nephron. Produces a large instantaneous current. Modulated by intracellular chloride ions and pH; acidic pH shifts the activation to more negative voltages. Hyperpolarization-activated ion channel exhibiting weak selectivity for potassium over sodium ions. Related diseases Epilepsy, idiopathic generalized 17 (EIG17) [MIM:602477]: A form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. Both autosomal dominant and autosomal recessive EIG17 inheritance have been reported. {ECO:0000269|PubMed:22131395, ECO:0000269|PubMed:29064616}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Febrile seizures, familial, 2 (FEB2) [MIM:602477]: Seizures associated with febrile episodes in childhood without any evidence of intracranial infection or defined pathologic or traumatic cause. It is a common condition, affecting 2-5% of children aged 3 months to 5 years. The majority are simple febrile seizures (generally defined as generalized onset, single seizures with a duration of less than 30 minutes). Complex febrile seizures are characterized by focal onset, duration greater than 30 minutes, and/or more than one seizure in a 24 hour period. The likelihood of developing epilepsy following simple febrile seizures is low. Complex febrile seizures are associated with a moderately increased incidence of epilepsy. FEB2 transmission pattern is consistent with autosomal dominant inheritance. {ECO:0000269|PubMed:24324597}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02527; DB02315; DB09083 Interacts with Q9UL51; Q4ACU6-1 EC number NA Uniprot keywords 3D-structure; Ammonia transport; cAMP; cAMP-binding; Cell membrane; Disease variant; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Lipoprotein; Membrane; Methylation; Nucleotide-binding; Palmitate; Phosphoprotein; Potassium; Potassium channel; Potassium transport; Proteomics identification; Reference proteome; Sodium; Sodium channel; Sodium transport; Transmembrane; Transmembrane helix; Transport; Voltage-gated channel Protein physicochemical properties Chain ID A Molecular weight (Da) 23672.9 Length 202 Aromaticity 0.12 Instability index 38.05 Isoelectric point 8.85 Charge (pH=7) 4.11 2D Binding mode Binding energy (Kcal/mol) -8.3 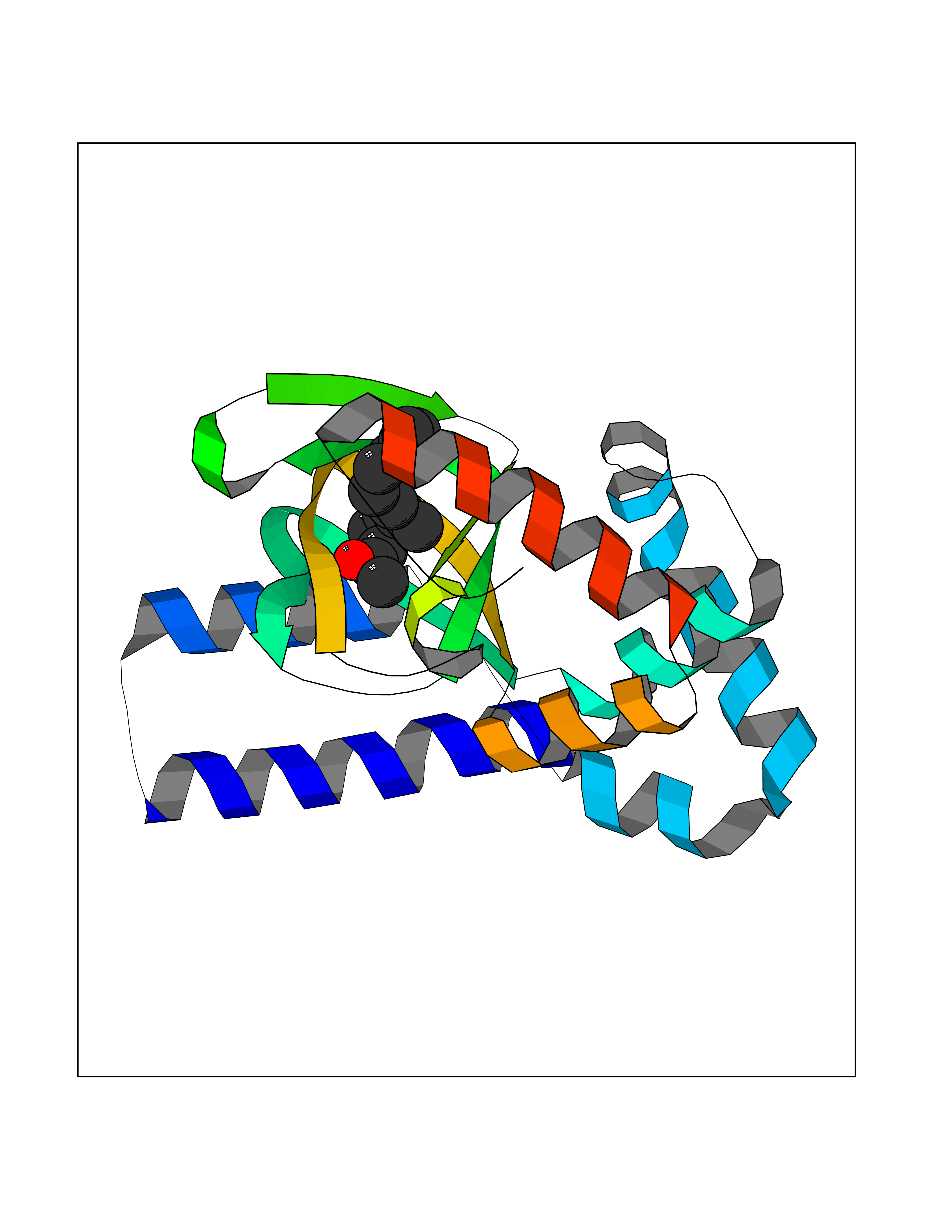 Molscript Map  Pymol Map 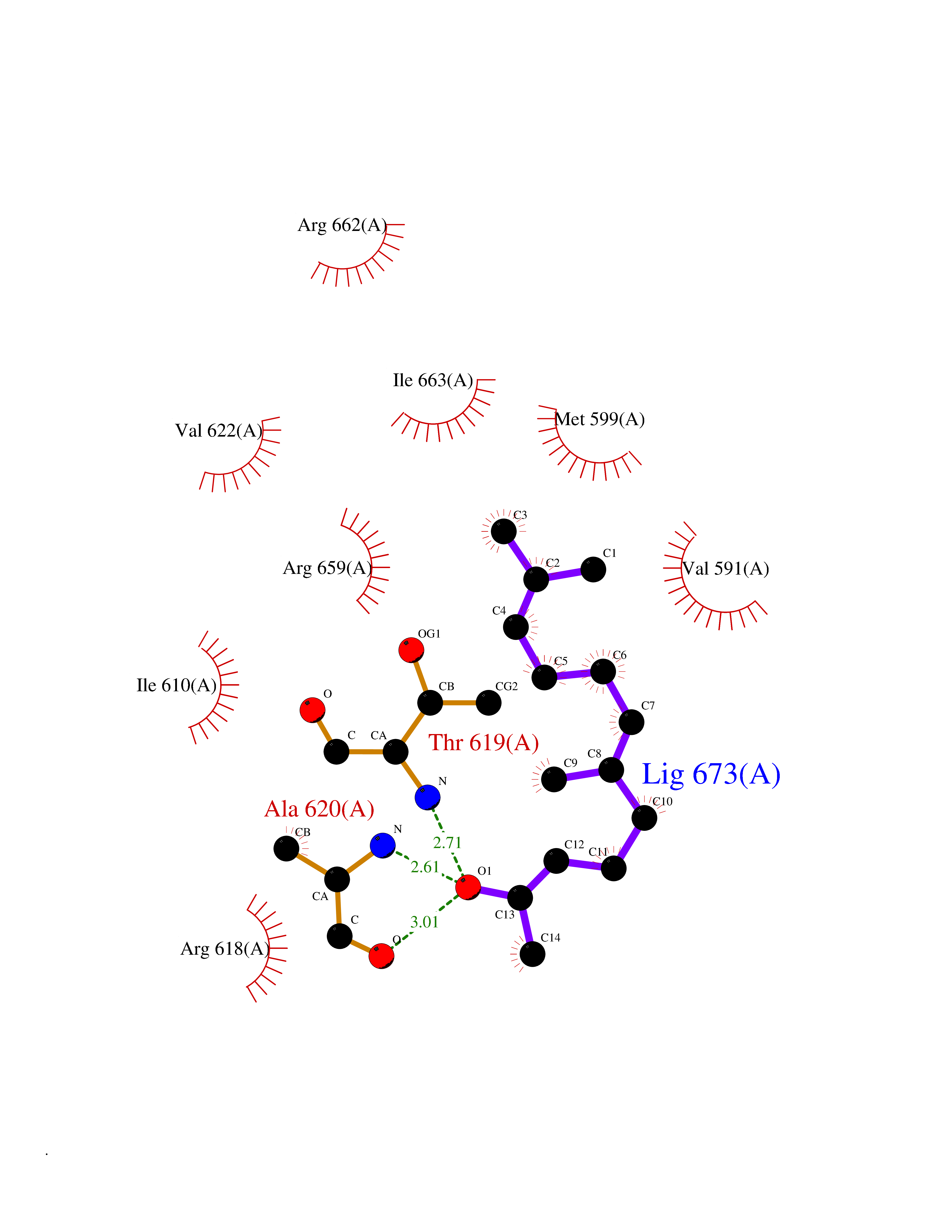 Ligplot Map 3D Binding mode Sequence DSSRRQYQEKYKQVEQYMSFHKLPADFRQKIHDYYEHRYQGKMFDEDSILGELNGPLREEIVNFNCRKLVASMPLFANADPNFVTAMLTKLKFEVFQPGDYIIREGTIGKKMYFIQHGVVSVLTKGNKEMKLSDGSYFGEICLLTRGRRTASVRADTYCRLYSLSVDNFNEVLEEYPMMRRAFETVAIDRLDRIGKKNSILL Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Acetyl-CoA carboxylase 2 (ACACB) | 3GID | 6.08 | |
Target general information Gen name ACACB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Acetyl-Coenzyme A carboxylase beta; Acetyl CoA carboxylase beta; ACCbeta; ACC2; ACC-beta; ACACB Protein family NA Biochemical class Carbon-carbon ligase Function Catalyzes the ATP-dependent carboxylation of acetyl-CoA to malonyl-CoA. Carries out three functions: biotin carboxyl carrier protein, biotin carboxylase and carboxyltransferase. Involved in inhibition of fatty acid and glucose oxidation and enhancement of fat storage. May play a role in regulation of mitochondrial fatty acid oxidation through malonyl- CoA-dependent inhibition of carnitine palmitoyltransferase 1. Related diseases Spinocerebellar ataxia 6 (SCA6) [MIM:183086]: Spinocerebellar ataxia is a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA6 is an autosomal dominant cerebellar ataxia (ADCA), mainly caused by expansion of a CAG repeat in the coding region of CACNA1A. There seems to be a correlation between the repeat number and earlier onset of the disorder. {ECO:0000269|PubMed:16325861, ECO:0000269|PubMed:20682717, ECO:0000269|PubMed:29053796, ECO:0000269|PubMed:8988170, ECO:0000269|PubMed:9302278, ECO:0000269|PubMed:9345107}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Migraine, familial hemiplegic, 1 (FHM1) [MIM:141500]: A subtype of migraine with aura associated with ictal hemiparesis and, in some families, cerebellar ataxia and atrophy. Migraine is a disabling symptom complex of periodic headaches, usually temporal and unilateral. Headaches are often accompanied by irritability, nausea, vomiting and photophobia, preceded by constriction of the cranial arteries. Migraine with aura is characterized by recurrent attacks of reversible neurological symptoms (aura) that precede or accompany the headache. Aura may include a combination of sensory disturbances, such as blurred vision, hallucinations, vertigo, numbness and difficulty in concentrating and speaking. {ECO:0000269|PubMed:10408532, ECO:0000269|PubMed:11409427, ECO:0000269|PubMed:11439943, ECO:0000269|PubMed:15032980, ECO:0000269|PubMed:18400034, ECO:0000269|PubMed:24836863, ECO:0000269|PubMed:26716990, ECO:0000269|PubMed:28900389, ECO:0000269|PubMed:8898206}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Episodic ataxia 2 (EA2) [MIM:108500]: An autosomal dominant disorder characterized by acetozolamide-responsive attacks of ataxia, migraine-like symptoms, interictal nystagmus, and cerebellar atrophy. {ECO:0000269|PubMed:10408533, ECO:0000269|PubMed:10987655, ECO:0000269|PubMed:11176968, ECO:0000269|PubMed:11723274, ECO:0000269|PubMed:12420090, ECO:0000269|PubMed:14718690, ECO:0000269|PubMed:15173248, ECO:0000269|PubMed:15293273, ECO:0000269|PubMed:18602318, ECO:0000269|PubMed:19232643, ECO:0000269|PubMed:20129625, ECO:0000269|PubMed:21696515, ECO:0000269|PubMed:38221525, ECO:0000269|PubMed:8898206, ECO:0000269|PubMed:9302278}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 42 (DEE42) [MIM:617106]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE42 inheritance is autosomal dominant. {ECO:0000269|PubMed:27250579, ECO:0000269|PubMed:27476654}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03781; DB00173; DB00121; DB07870; DB02859 Interacts with O00763; P50747; Q9CQ20 EC number EC 6.4.1.2 Uniprot keywords 3D-structure; Allosteric enzyme; Alternative splicing; ATP-binding; Biotin; Fatty acid biosynthesis; Fatty acid metabolism; Ligase; Lipid biosynthesis; Lipid metabolism; Magnesium; Manganese; Metal-binding; Mitochondrion; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transit peptide Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42825.6 Length 385 Aromaticity 0.09 Instability index 35.24 Isoelectric point 5.9 Charge (pH=7) -5.45 2D Binding mode Binding energy (Kcal/mol) -8.3 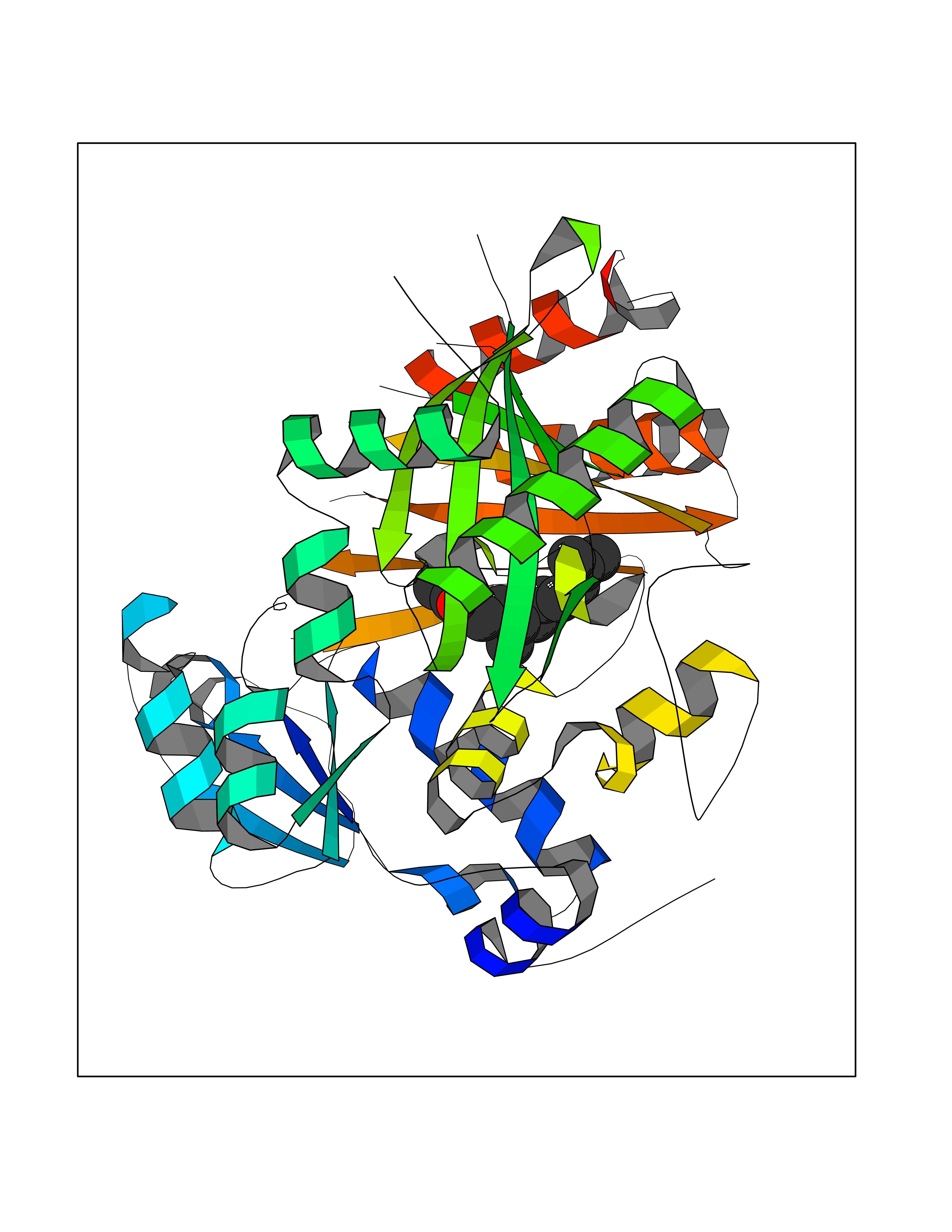 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence DFTVASPAEFVTRFGGDRVIEKVLIANNGIAAVKCMRSIRRWAYEMFRNERAIRFVVMVTPEDLKANAEYIKMADHYVPVPGGPNNNNYANVELIVDIAKRIPVQAVWAGWGHASENPKLPELLCKNGVAFLGPPSEAMWALGDKIASTVVAQTLQVPTLPHARHLEVQILADQYGNAVSLFGRDCSIQQKIVEEAPATIAPLAIFEFMEQCAIRLAKTVGYVSAGTVEYLYSQDGSFHFLELNPRLQVEHPCTEMIADVNLPAAQLQIAMGVPLHRLKDIRLLYGESPWGVTPISFETPSNPPLARGHVIAARITSGTVQELNFRSSKNVWGYFSVQFGHCFSWGENREEAISNMVVALKELSIRRTTVEYLINLLETESFQNN Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Dihydroorotate dehydrogenase (quinone), mitochondrial | 4CQ8 | 6.08 | |
Target general information Gen name PFF0160c Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID NA Synonyms NA Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class Oxidoreductase Function Dihydroorotate dehydrogenase activity. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42573.5 Length 378 Aromaticity 0.1 Instability index 36.63 Isoelectric point 8.21 Charge (pH=7) 3.17 2D Binding mode Binding energy (Kcal/mol) -8.3 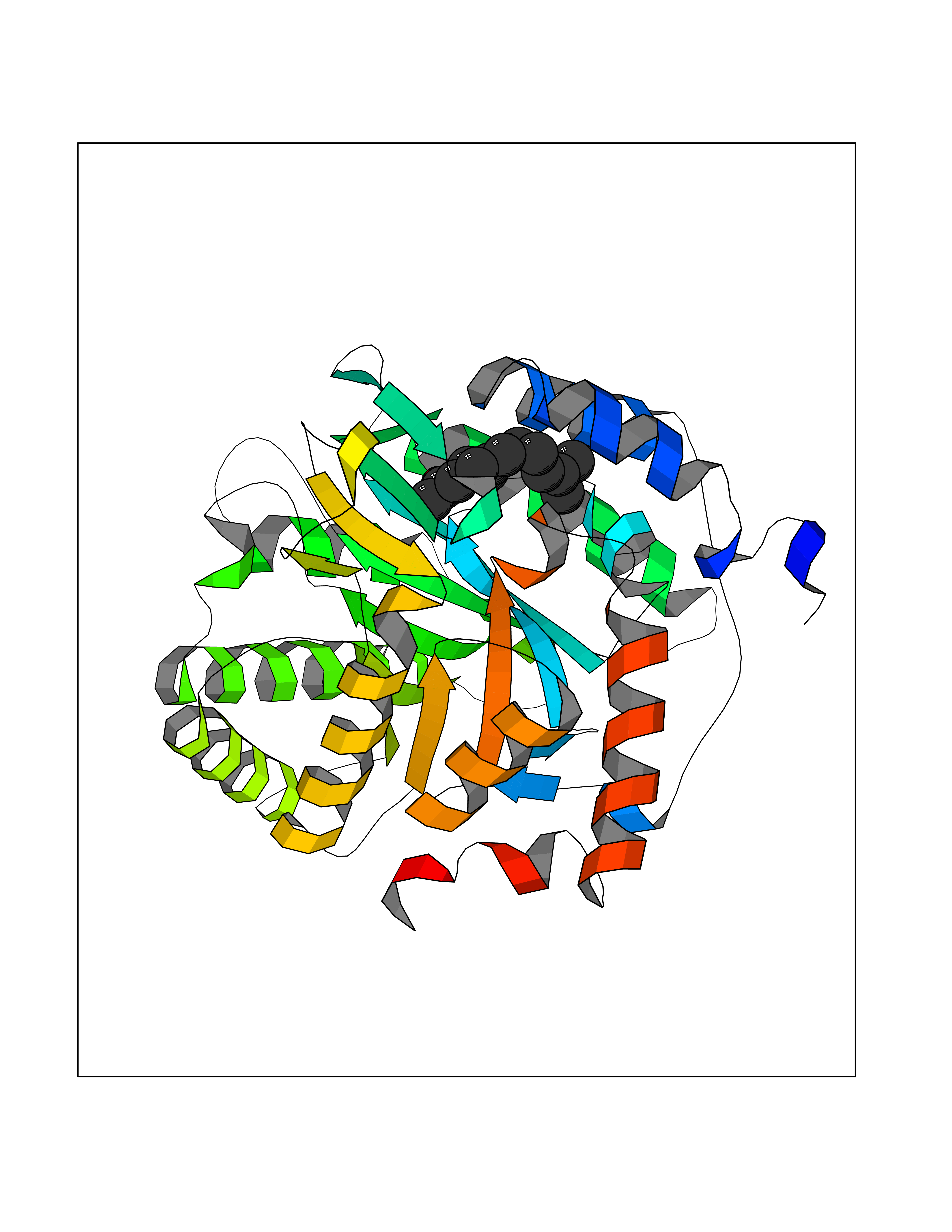 Molscript Map 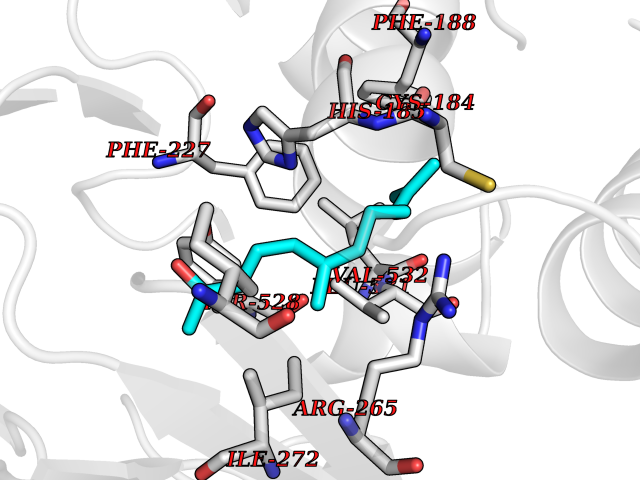 Pymol Map 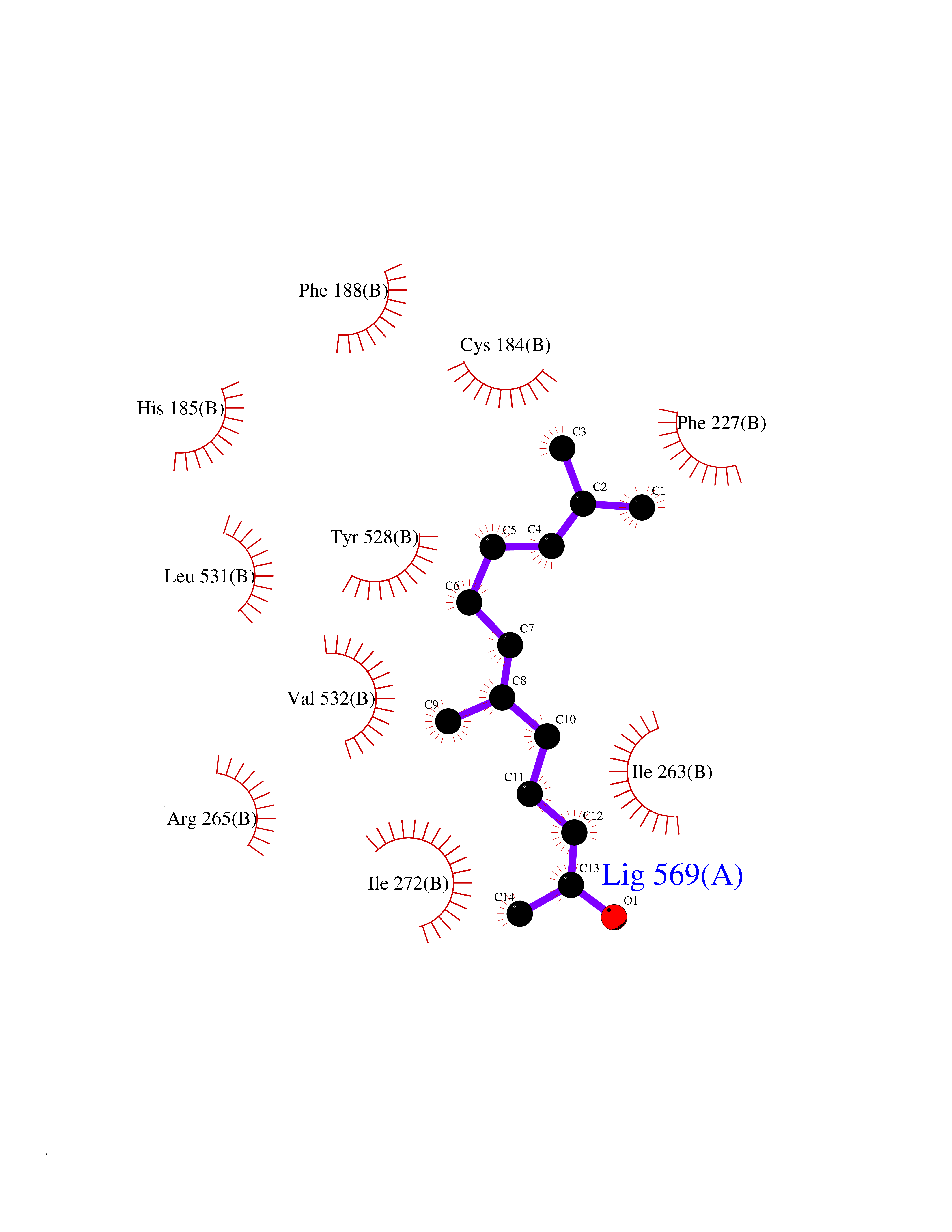 Ligplot Map 3D Binding mode Sequence ADPFESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEKNNFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKHS Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Opioid receptor sigma 1 (OPRS1) | 5HK1 | 6.08 | |
Target general information Gen name SIGMAR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hSigmaR1; Sigma1R; Sigma1-receptor; Sigma non-opioid intracellular receptor 1; Sigma 1-type opioid receptor; SRBP; SR31747-binding protein; SR31747 binding protein 1; SR-BP; SIG-1R; Opioid receptor, s Protein family ERG2 family Biochemical class GPCR rhodopsin Function Involved in the regulation of different receptors it plays a role in BDNF signaling and EGF signaling. Also regulates ion channels like the potassium channel and could modulate neurotransmitter release. Plays a role in calcium signaling through modulation together with ANK2 of the ITP3R-dependent calcium efflux at the endoplasmic reticulum. Plays a role in several other cell functions including proliferation, survival and death. Originally identified for its ability to bind various psychoactive drugs it is involved in learning processes, memory and mood alteration. Necessary for proper mitochondrial axonal transport in motor neurons, in particular the retrograde movement of mitochondria. Plays a role in protecting cells against oxidative stress-induced cell death via its interaction with RNF112. Functions in lipid transport from the endoplasmic reticulum and is involved in a wide array of cellular functions probably through regulation of the biogenesis of lipid microdomains at the plasma membrane. Related diseases Amyotrophic lateral sclerosis 16, juvenile (ALS16) [MIM:614373]: A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. {ECO:0000269|PubMed:21842496}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal recessive 2 (HMNR2) [MIM:605726]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. HMNR2 is characterized by onset of distal muscle weakness and wasting affecting the lower and upper limbs in the first decade. {ECO:0000269|PubMed:26078401, ECO:0000269|PubMed:27629094}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00321; DB09014; DB00907; DB00514; DB01488; DB00574; DB00502; DB00956; DB00704; DB00540; DB06174; DB00652; DB11186; DB03575; DB05316; DB01708; DB00409; DB01104 Interacts with Q92847-1; Q99720-1; O00213-2; P17612; P50454; P37173 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Amyotrophic lateral sclerosis; Cell junction; Cell membrane; Cell projection; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Lipid droplet; Lipid transport; Membrane; Neurodegeneration; Neuropathy; Nucleus; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 20805.3 Length 185 Aromaticity 0.14 Instability index 31.72 Isoelectric point 5.44 Charge (pH=7) -6.63 2D Binding mode Binding energy (Kcal/mol) -8.3 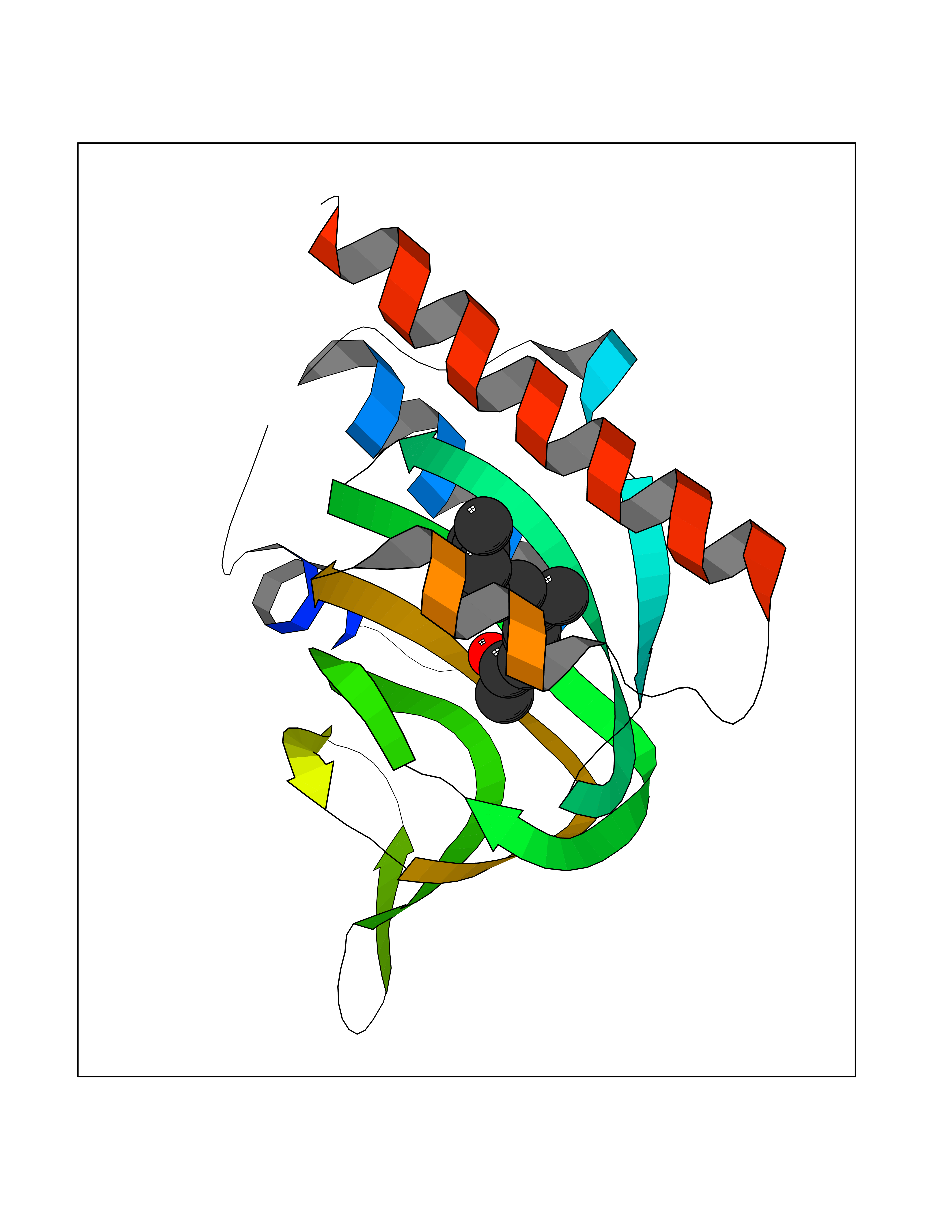 Molscript Map 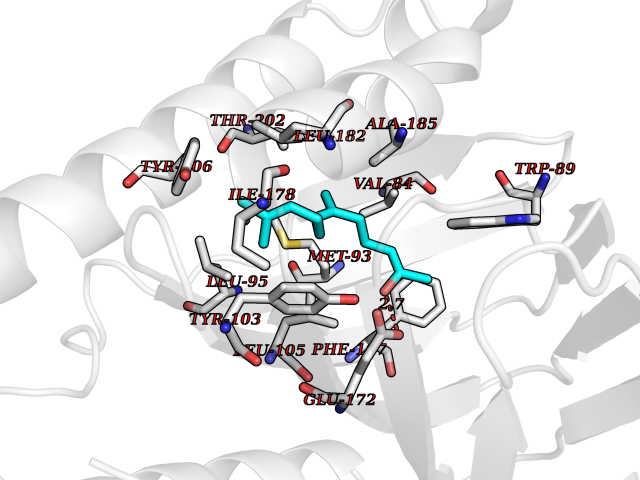 Pymol Map 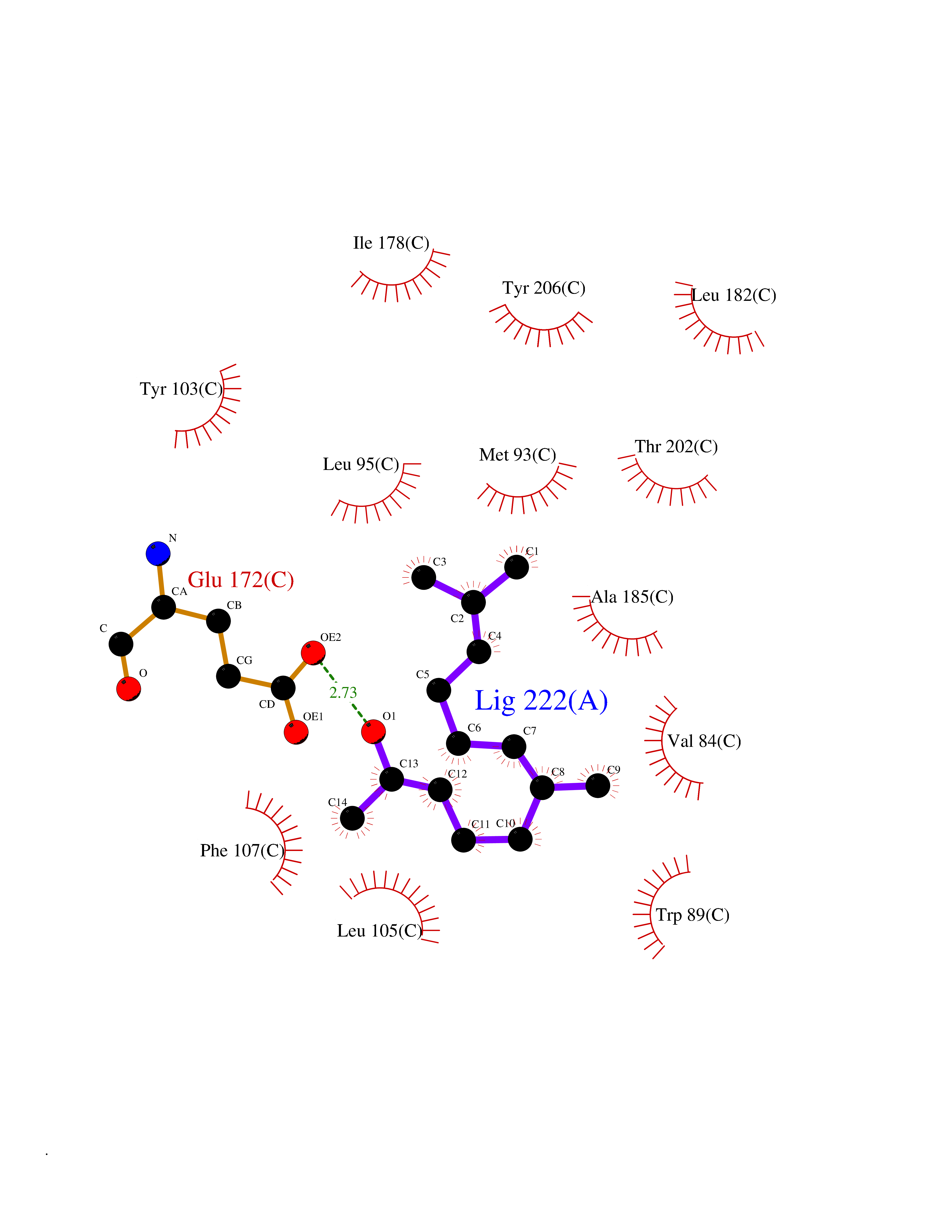 Ligplot Map 3D Binding mode Sequence VFQREEIAQLARQYAGLDHELAFSRLIVELRRLHPGHVLPDEELQWVFVNAGGWMGAMCLLHASLSEYVLLFGTALGSRGHSGRYWAEISDTIISGTFHQWREGTTKSEVFYPGETVVHGPGEATAVEWGPNTWMVEYGRGVIPSTLAFALADTVFSTQDFLTLFYTLRSYARGLRLELTTYLFG Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | Cholestenol delta-isomerase (EBP) | 6OHU | 6.08 | |
Target general information Gen name EBP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Emopamilbinding protein; EBP; Delta(8)Delta(7) sterol isomerase; D8D7 sterol isomerase; 3betahydroxysteroidDelta(8),Delta(7)isomerase Protein family EBP family Biochemical class Intramolecular oxidoreductases Function Catalyzes the conversion of Delta(8)-sterols to their corresponding Delta(7)-isomers. Related diseases Chondrodysplasia punctata 2, X-linked dominant (CDPX2) [MIM:302960]: A clinically and genetically heterogeneous disorder characterized by punctiform calcification of the bones. The key clinical features of CDPX2 are chondrodysplasia punctata, linear ichthyosis, cataracts and short stature. CDPX2 is a rare disorder of defective cholesterol biosynthesis, biochemically characterized by an increased amount of 8-dehydrocholesterol and cholest-8(9)-en-3-beta-ol in the plasma and tissues. {ECO:0000269|PubMed:10391218, ECO:0000269|PubMed:10391219, ECO:0000269|PubMed:10942423, ECO:0000269|PubMed:11493318, ECO:0000269|PubMed:18176751, ECO:0000269|PubMed:25814754}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: MEND syndrome (MEND) [MIM:300960]: An X-linked recessive disorder associated with a defect in sterol biosynthesis. Disease manifestations and severity are highly variable. Clinical features include intellectual disability, short stature, scoliosis, digital abnormalities, cataracts, and dermatologic abnormalities. {ECO:0000269|PubMed:12503101, ECO:0000269|PubMed:20949533, ECO:0000269|PubMed:24459067, ECO:0000269|PubMed:24700572}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00675 Interacts with O95870; Q86W74-2; Q13520; Q3SXY8; Q8N6S5; Q9H2C2; Q9HD20-3; O95393; Q12983; Q8WVV5; P06681; O14523; Q8WVX3-2; P01031; Q6UWT4; Q9P0B6; Q8NHW4; P25942; Q07108; P60033; O14735; P23141-3; Q9H9P2; Q8NHS1; O43889-2; Q96BA8; P49447; O43169; P78329; P81534; Q9H1M4; Q96LL9; Q15125; Q08426; Q9BV81; P54849; Q9UKR5; Q9Y282; Q7L5A8; Q5JX71; Q96IV6; Q9UGM5; Q9Y3D6; Q14318; Q9BWH2; Q14802-3; Q9H0Q3; Q8WWP7; Q96F15; P29033; O95452; O14653; Q8TDT2; P02724; P30519; Q7Z5P4; Q9Y5U9; Q9Y5U4; Q8N5M9; Q5T700; Q68G75; Q7L5N7; Q7Z4F1; Q96AG4; Q16873; Q6ZSS7; P50281; Q5J8X5; Q9UHE5; O95167; Q9NX14; Q99519; Q92982; Q9NZG7; Q16617; Q8IXM6; Q2M2E3; Q9P0S3; Q53FV1; Q8N138; Q7RTS5; Q9Y342; Q04941; Q8IY26; Q01453; P54315; P43378; P15151; Q8N8N0; Q5QGT7; Q96GQ5; Q9NTJ5; Q969E2; O75396; Q9Y6X1; Q8N6R1; Q9BWM7; Q8TD22; Q8IWU4; Q96G79; Q2M3R5; Q9NVC3; P08195-4; Q96JW4; Q6P1K1; Q0VAQ4; Q9NRQ5; B2RUZ4; Q9BZL3; Q6UX34; Q86Y82; P61266; Q13190; O43752; O15400; Q9UNK0; O43759-2; P57105; Q8N2H4; Q96BZ9; P07204; O14925; Q96CP7; Q96MV1; P55061; Q9NV29; P17152; Q9BXJ8; A0PK00; Q9NV12; Q9BVK8; Q9NUH8; Q9P0S9; Q9NRX6; Q8N511; Q969S6; Q9BTX3; A2RU14; Q9H0R3; Q8NBD8; Q8WW34-2; Q9NWH2; Q9BU79; Q8TBM7; Q69YG0; Q9NW97; Q9H2L4; Q6PI78; Q8N2M4; Q8N661; Q5BJF2; Q9NSU2-1; A0AVG3; Q5TGU0; A5PKU2; Q9Y385; Q9Y5Z9; Q53HI1; Q9H1C4; Q9NZ43; P23763-3; P63027; Q15836; O75379; Q9P0L0; O95292; O95070; Q9Y548; Q9BSR8; Q96EC8; Q6UX98; O95159 EC number EC 5.3.3.5 Uniprot keywords 3D-structure; Acetylation; Cataract; Cholesterol biosynthesis; Cholesterol metabolism; Cytoplasmic vesicle; Disease variant; Dwarfism; Endoplasmic reticulum; Ichthyosis; Isomerase; Lipid biosynthesis; Lipid metabolism; Membrane; Nucleus; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 24012.7 Length 206 Aromaticity 0.17 Instability index 42.52 Isoelectric point 6.19 Charge (pH=7) -3.19 2D Binding mode Binding energy (Kcal/mol) -8.3 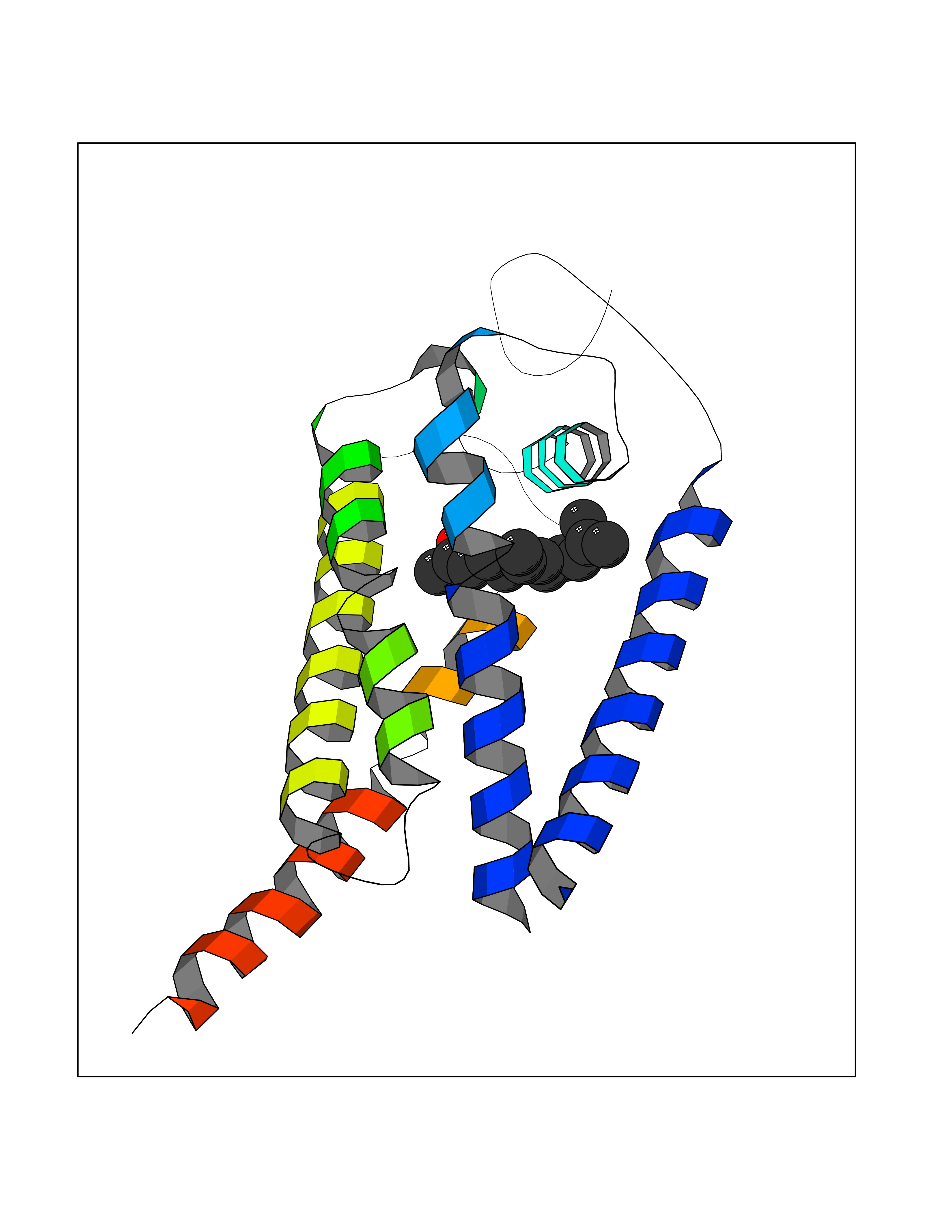 Molscript Map 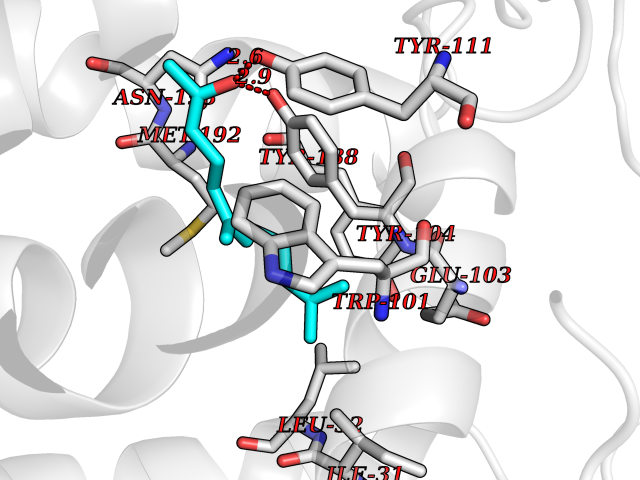 Pymol Map 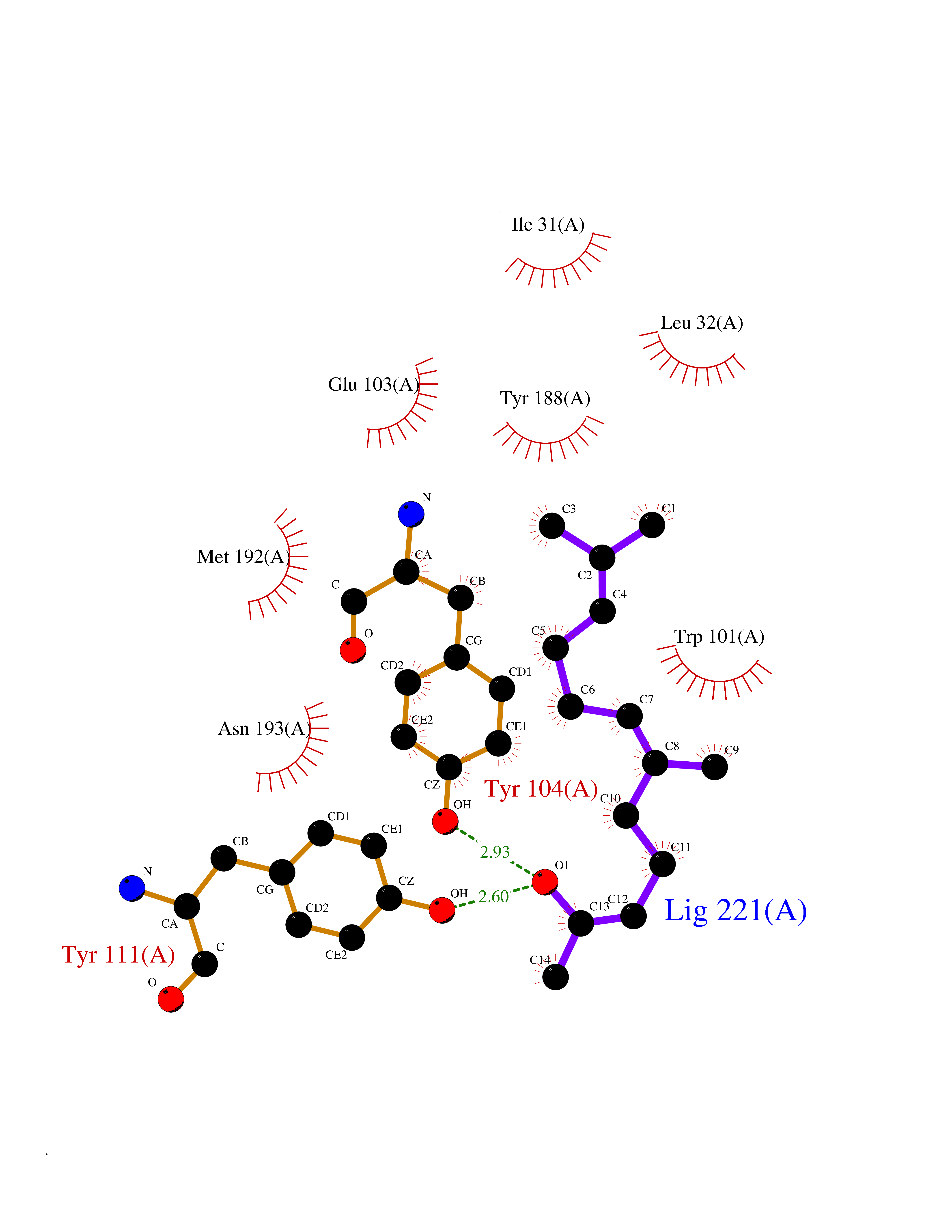 Ligplot Map 3D Binding mode Sequence PLHPYWPQHLRLDNFVPNDRPTWHILAGLFSVTGVLVVTTWLLSGRTWRRLSLCWFAVCGFIHLVIEGWFVLYYEDLLGDQAFLSQLWKEYAKGDSRYILGDNFTVCMETITACLWGPLSLWVVIAFLRQHPLRFILQLVVSVGQIYGDVLYFLTEHRDGFQHGELGHPLYFWFYFVFMNALWLVLPGVLVLDAVKHLTHAQSTLD Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Lysophosphatidic acid receptor 1 (LPAR1) | 7TD0 | 6.08 | |
Target general information Gen name LPAR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysophosphatidic acid receptor Edg-2; LPA1; LPA-1; LPA receptor 1; EDG2; EDG 2 receptor Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Plays a role in the reorganization of the actin cytoskeleton, cell migration, differentiation and proliferation, and thereby contributes to the responses to tissue damage and infectious agents. Activates downstream signaling cascades via the G(i)/G(o), G(12)/G(13), and G(q) families of heteromeric G proteins. Signaling inhibits adenylyl cyclase activity and decreases cellular cAMP levels. Signaling triggers an increase of cytoplasmic Ca(2+) levels. Activates RALA; this leads to the activation of phospholipase C (PLC) and the formation of inositol 1,4,5-trisphosphate. Signaling mediates activation of down-stream MAP kinases. Contributes to the regulation of cell shape. Promotes Rho-dependent reorganization of the actin cytoskeleton in neuronal cells and neurite retraction. Promotes the activation of Rho and the formation of actin stress fibers. Promotes formation of lamellipodia at the leading edge of migrating cells via activation of RAC1. Through its function as lysophosphatidic acid receptor, plays a role in chemotaxis and cell migration, including responses to injury and wounding. Plays a role in triggering inflammation in response to bacterial lipopolysaccharide (LPS) via its interaction with CD14. Promotes cell proliferation in response to lysophosphatidic acid. Required for normal skeleton development. May play a role in osteoblast differentiation. Required for normal brain development. Required for normal proliferation, survival and maturation of newly formed neurons in the adult dentate gyrus. Plays a role in pain perception and in the initiation of neuropathic pain. Receptor for lysophosphatidic acid (LPA). Related diseases Anterior segment dysgenesis 6 (ASGD6) [MIM:617315]: A form of anterior segment dysgenesis, a group of defects affecting anterior structures of the eye including cornea, iris, lens, trabecular meshwork, and Schlemm canal. Anterior segment dysgeneses result from abnormal migration or differentiation of the neural crest derived mesenchymal cells that give rise to components of the anterior chamber during eye development. Different anterior segment anomalies may exist alone or in combination, including iris hypoplasia, enlarged or reduced corneal diameter, corneal vascularization and opacity, posterior embryotoxon, corectopia, polycoria, abnormal iridocorneal angle, ectopia lentis, and anterior synechiae between the iris and posterior corneal surface. Clinical conditions falling within the phenotypic spectrum of anterior segment dysgeneses include aniridia, Axenfeld anomaly, Reiger anomaly/syndrome, Peters anomaly, and iridogoniodysgenesis. ASGD6 patients predominantly manifest Peters anomaly. Peters anomaly consists of corneal leukoma, defects in the posterior structures of the cornea such as absence of the posterior corneal stroma and Descemet membrane, and a variable degree of iridocorneal and/or keratolenticular adhesions. Over 50% of patients develop glaucoma in childhood. {ECO:0000269|PubMed:11403040}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glaucoma 3, primary congenital, A (GLC3A) [MIM:231300]: An autosomal recessive form of primary congenital glaucoma (PCG). PCG is characterized by marked increase of intraocular pressure at birth or early childhood, large ocular globes (buphthalmos) and corneal edema. It results from developmental defects of the trabecular meshwork and anterior chamber angle of the eye that prevent adequate drainage of aqueous humor. {ECO:0000269|PubMed:10227395, ECO:0000269|PubMed:10655546, ECO:0000269|PubMed:11184479, ECO:0000269|PubMed:11527932, ECO:0000269|PubMed:11774072, ECO:0000269|PubMed:11980847, ECO:0000269|PubMed:12036985, ECO:0000269|PubMed:12525557, ECO:0000269|PubMed:14635112, ECO:0000269|PubMed:14640114, ECO:0000269|PubMed:15255109, ECO:0000269|PubMed:15342693, ECO:0000269|PubMed:15475877, ECO:0000269|PubMed:16490498, ECO:0000269|PubMed:16688110, ECO:0000269|PubMed:16735994, ECO:0000269|PubMed:16862072, ECO:0000269|PubMed:18470941, ECO:0000269|PubMed:9463332, ECO:0000269|PubMed:9497261}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Glaucoma 1, open angle, A (GLC1A) [MIM:137750]: A form of primary open angle glaucoma (POAG). POAG is characterized by a specific pattern of optic nerve and visual field defects. The angle of the anterior chamber of the eye is open, and usually the intraocular pressure is increased. However, glaucoma can occur at any intraocular pressure. The disease is generally asymptomatic until the late stages, by which time significant and irreversible optic nerve damage has already taken place. {ECO:0000269|PubMed:11774072}. The gene represented in this entry acts as a disease modifier. Digenic mutations in CYP1B1 and MYOC have been found in a family segregating both primary adult-onset and juvenile forms of open angle glaucoma (PubMed:11774072). All affected family members with mutations in both MYOC and CYP1B1 had juvenile glaucoma, whereas those with only the MYOC mutation had the adult-onset form (PubMed:11774072). {ECO:0000269|PubMed:11774072}. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; Endosome; G-protein coupled receptor; Glycoprotein; Lipid-binding; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 33201.3 Length 290 Aromaticity 0.14 Instability index 34.12 Isoelectric point 8.86 Charge (pH=7) 6.12 2D Binding mode Binding energy (Kcal/mol) -8.29 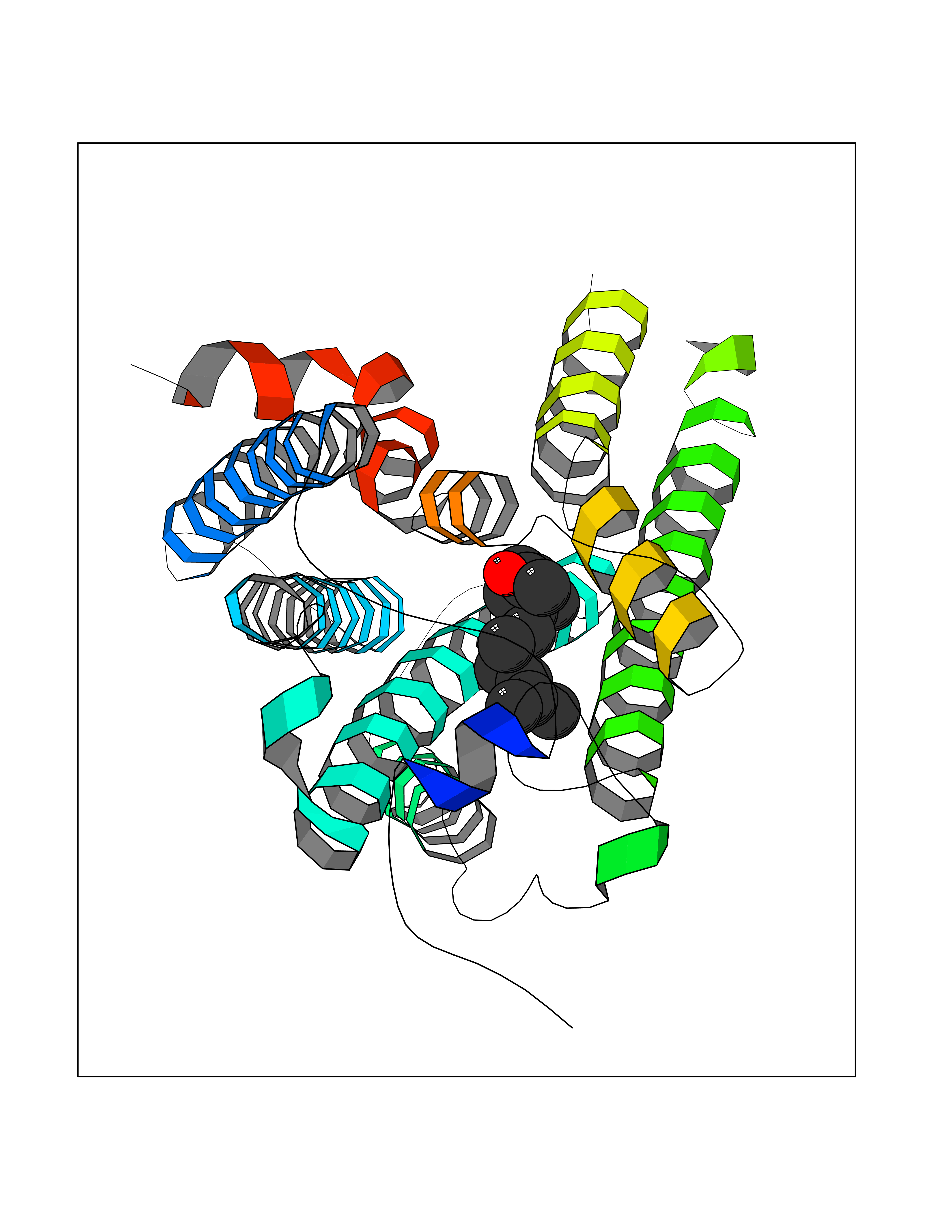 Molscript Map  Pymol Map 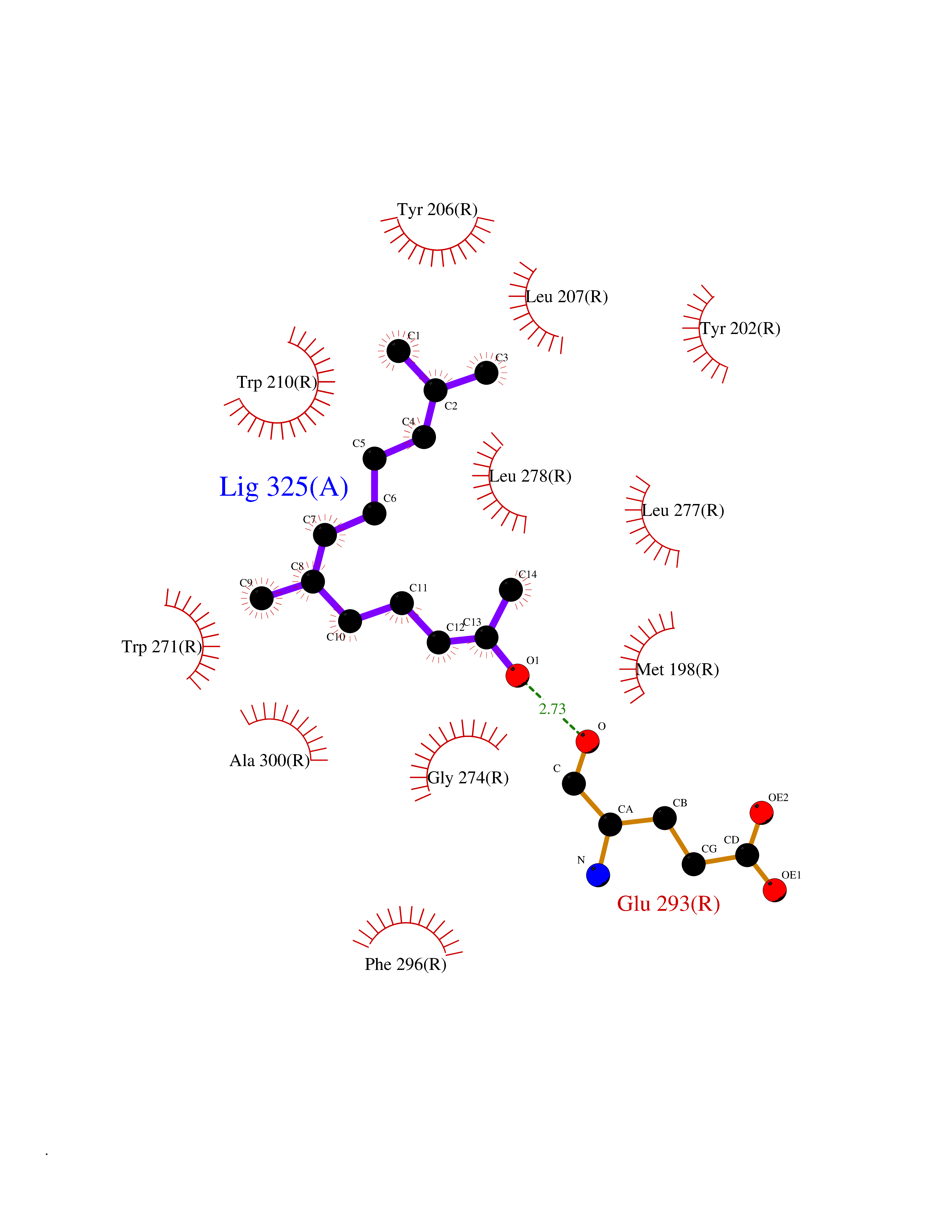 Ligplot Map 3D Binding mode Sequence QCFYNESIAFFYNRSGKHLATEWNTVSKLVMGLGITVCIFIMLANLLVMVAIYVNRRFHFPIYYLMANLAAADFFAGLAYFYLMFNTGPNTRRLTVSTWLLRQGLIDTSLTASVANLLAIAIERHITVFRMQLHTRMSNRRVVVVIVVIWTMAIVMGAIPSVGWNCICDIENCSNMAPLYSDSYLVFWAIFNLVTFVVMVVLYAHIFGYVRQRTMRMDTMMSLLKTVVIVLGAFIICWTPGLVLLLLDVCCPQCDVLAYEKFFLLLAEFNSAMNPIIYSYRDKEMSATFR Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Cystathionine gamma-lyase (CTH) | 3COG | 6.07 | |
Target general information Gen name CTH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Gamma-cystathionase; Cysteine-protein sulfhydrase Protein family Trans-sulfuration enzymes family Biochemical class NA Function Catalyzes the last step in the trans-sulfuration pathway from methionine to cysteine. Has broad substrate specificity. Converts cystathionine to cysteine, ammonia and 2-oxobutanoate. Converts two cysteine molecules to lanthionine and hydrogen sulfide. Can also accept homocysteine as substrate. Specificity depends on the levels of the endogenous substrates. Generates the endogenous signaling molecule hydrogen sulfide (H2S), and so contributes to the regulation of blood pressure. Acts as a cysteine-protein sulfhydrase by mediating sulfhydration of target proteins: sulfhydration consists of converting -SH groups into -SSH on specific cysteine residues of target proteins such as GAPDH, PTPN1 and NF-kappa-B subunit RELA, thereby regulating their function. Related diseases Cystathioninuria (CSTNU) [MIM:219500]: Autosomal recessive phenotype characterized by abnormal accumulation of plasma cystathionine, leading to increased urinary excretion. {ECO:0000269|PubMed:12574942, ECO:0000269|PubMed:18476726}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02328; DB03928; DB00151; DB04217; DB00114 Interacts with P32929; Q96NT3; Q96NT3-2; Q96HA8; Q6P9E2 EC number EC 4.4.1.1 Uniprot keywords 3D-structure; Alternative splicing; Amino-acid biosynthesis; Calmodulin-binding; Cysteine biosynthesis; Cytoplasm; Disease variant; Lipid metabolism; Lyase; Proteomics identification; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 86026 Length 782 Aromaticity 0.08 Instability index 32.4 Isoelectric point 6.27 Charge (pH=7) -9.46 2D Binding mode Binding energy (Kcal/mol) -8.28 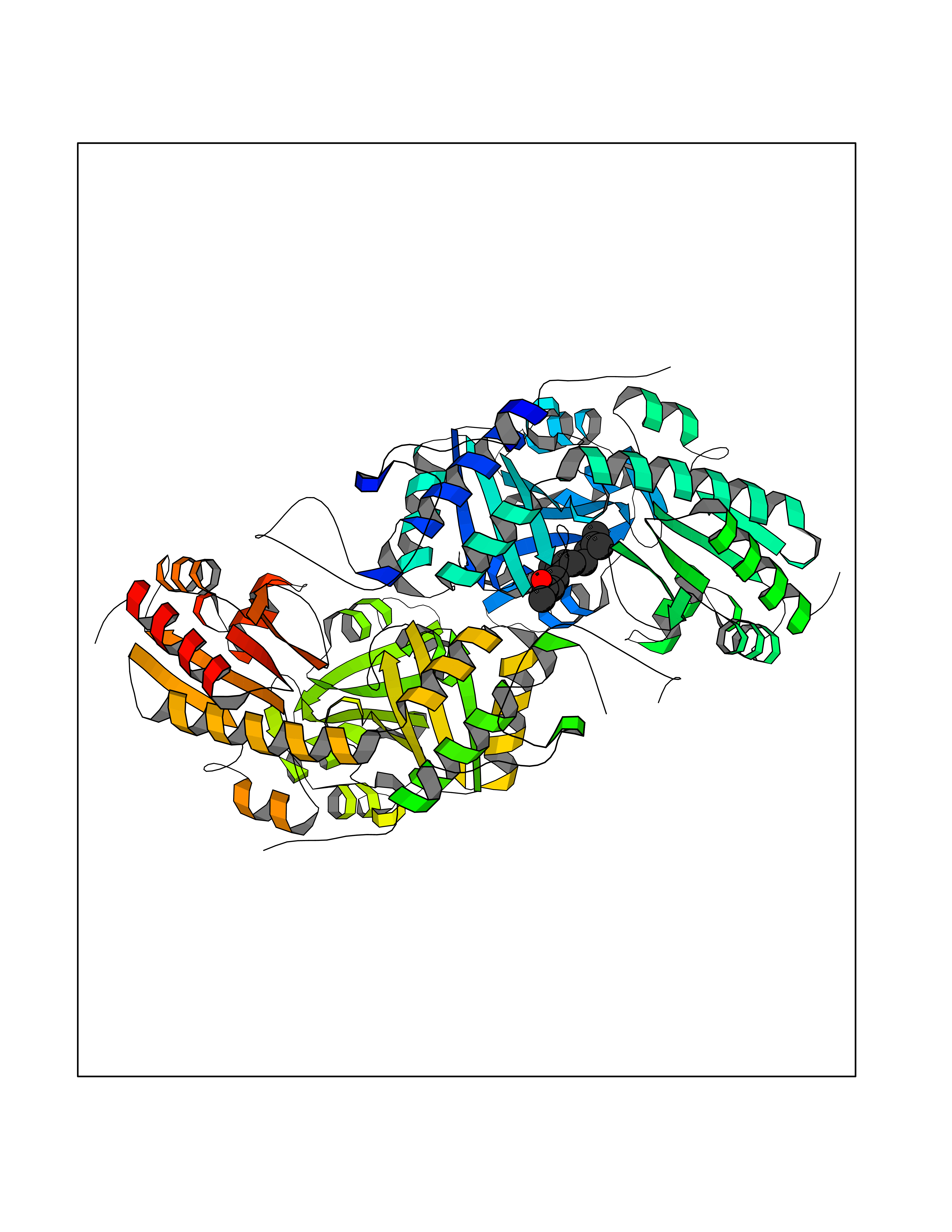 Molscript Map 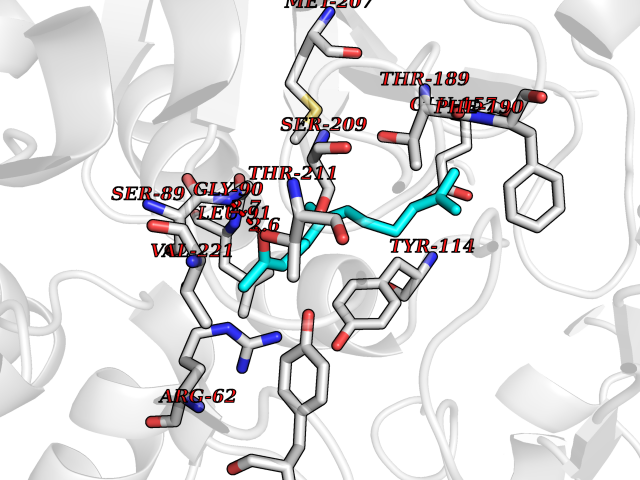 Pymol Map 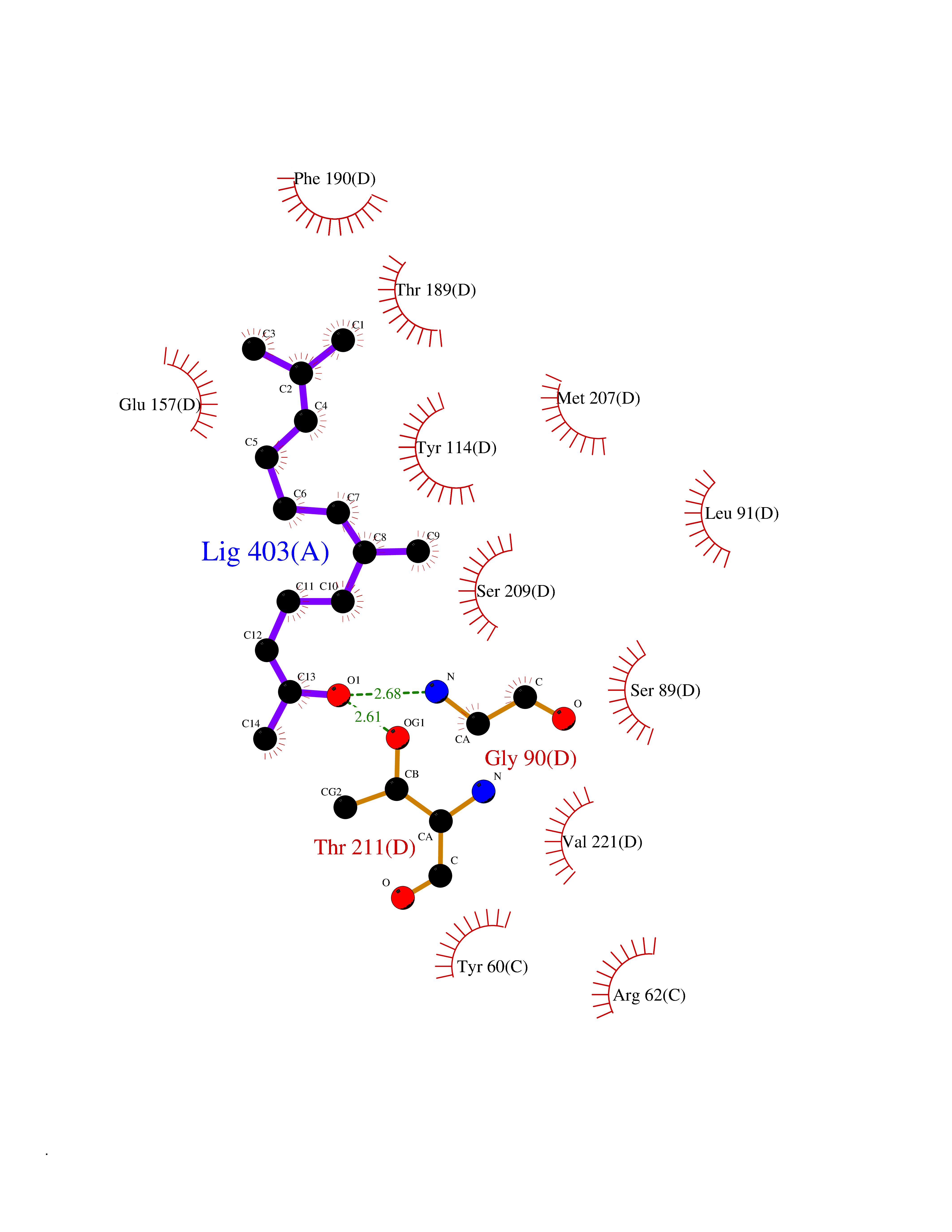 Ligplot Map 3D Binding mode Sequence GFLPHFQHFATQAIHVGQDPEQWTSRAVVPPISLSTTFKQGAPGQHSGFEYSRSGNPTRNCLEKAVAALDGAKYCLAFASGLAATVTITHLLKAGDQIICMDDVYGGTNRYFRQVASEFGLKISFVDCSKIKLLEAAITPETKLVWIETPTNPTQKVIDIEGCAHIVHKHGDIILVVDNTFMSPYFQRPLALGADISMYSATKYMNGHSDVVMGLVSVNCESLHNRLRFLQNSLGAVPSPIDCYLCNRGLKTLHVRMEKHFKNGMAVAQFLESNPWVEKVIYPGLPSHPQHELVKRQCTGCTGMVTFYIKGTLQHAEIFLKNLKLFTLAESLGGFESLAELPAIMTHASVLKNDRDVLGISDTLIRLSVGLEDEEDLLEDLDQALKAAHPPSGFLPHFQHFATQAIHVGQDPEQWTSRAVVPPISLSTTFKQGAPGQGFEYSRSGNPTRNCLEKAVAALDGAKYCLAFASGLAATVTITHLLKAGDQIICMDDVYGGTNRYFRQVASEFGLKISFVDCSKIKLLEAAITPETKLVWIETPTNPTQKVIDIEGCAHIVHKHGDIILVVDNTFMSPYFQRPLALGADISMYSATKYMNGHSDVVMGLVSVNCESLHNRLRFLQNSLGAVPSPIDCYLCNRGLKTLHVRMEKHFKNGMAVAQFLESNPWVEKVIYPGLPSHPQHELVKRQCTGCTGMVTFYIKGTLQHAEIFLKNLKLFTLAESLGGFESLAELPAIMTHASVLKNDRDVLGISDTLIRLSVGLEDEEDLLEDLDQALKAAHPPS Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Nitric-oxide synthase inducible (NOS2) | 3E7G | 6.07 | |
Target general information Gen name NOS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms iNOS; Peptidyl-cysteine S-nitrosylase NOS2; Nitric oxide synthase, inducible; NOS2A; NOS type II; Inducible NOS; Inducible NO synthase; Hepatocyte NOS; HEP-NOS Protein family NOS family Biochemical class Paired donor oxygen oxidoreductase Function Produces nitric oxide (NO) which is a messenger molecule with diverse functions throughout the body. In macrophages, NO mediates tumoricidal and bactericidal actions. Also has nitrosylase activity and mediates cysteine S-nitrosylation of cytoplasmic target proteins such PTGS2/COX2 (By similarity). As component of the iNOS-S100A8/9 transnitrosylase complex involved in the selective inflammatory stimulus-dependent S-nitrosylation of GAPDH on 'Cys-247' implicated in regulation of the GAIT complex activity and probably multiple targets including ANXA5, EZR, MSN and VIM. Involved in inflammation, enhances the synthesis of proinflammatory mediators such as IL6 and IL8. Related diseases Cerebellar ataxia, impaired intellectual development, and dysequilibrium syndrome 3 (CAMRQ3) [MIM:613227]: An autosomal recessive, congenital cerebellar ataxia associated with dysarthia, quadrupedal gait and intellectual disability. {ECO:0000269|PubMed:19461874}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07003; DB07007; DB07011; DB07405; DB08750; DB01997; DB07029; DB07008; DB08214; DB07002; DB01835; DB06879; DB04534; DB03100; DB02207; DB00125; DB00155; DB01234; DB14649; DB11327; DB00997; DB07306; DB07388; DB05252; DB01381; DB03366; DB05214; DB04400; DB09237; DB00244; DB01110; DB01017; DB03144; DB01686; DB03449; DB06916; DB07318; DB07389; DB02044; DB02644; DB05383; DB02234; DB03953; DB02462; DB08814 Interacts with P04406 EC number EC 1.14.13.39 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Calmodulin-binding; Cytoplasm; FAD; Flavoprotein; FMN; Heme; Iron; Metal-binding; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 48633 Length 421 Aromaticity 0.12 Instability index 46.5 Isoelectric point 6.75 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -8.28 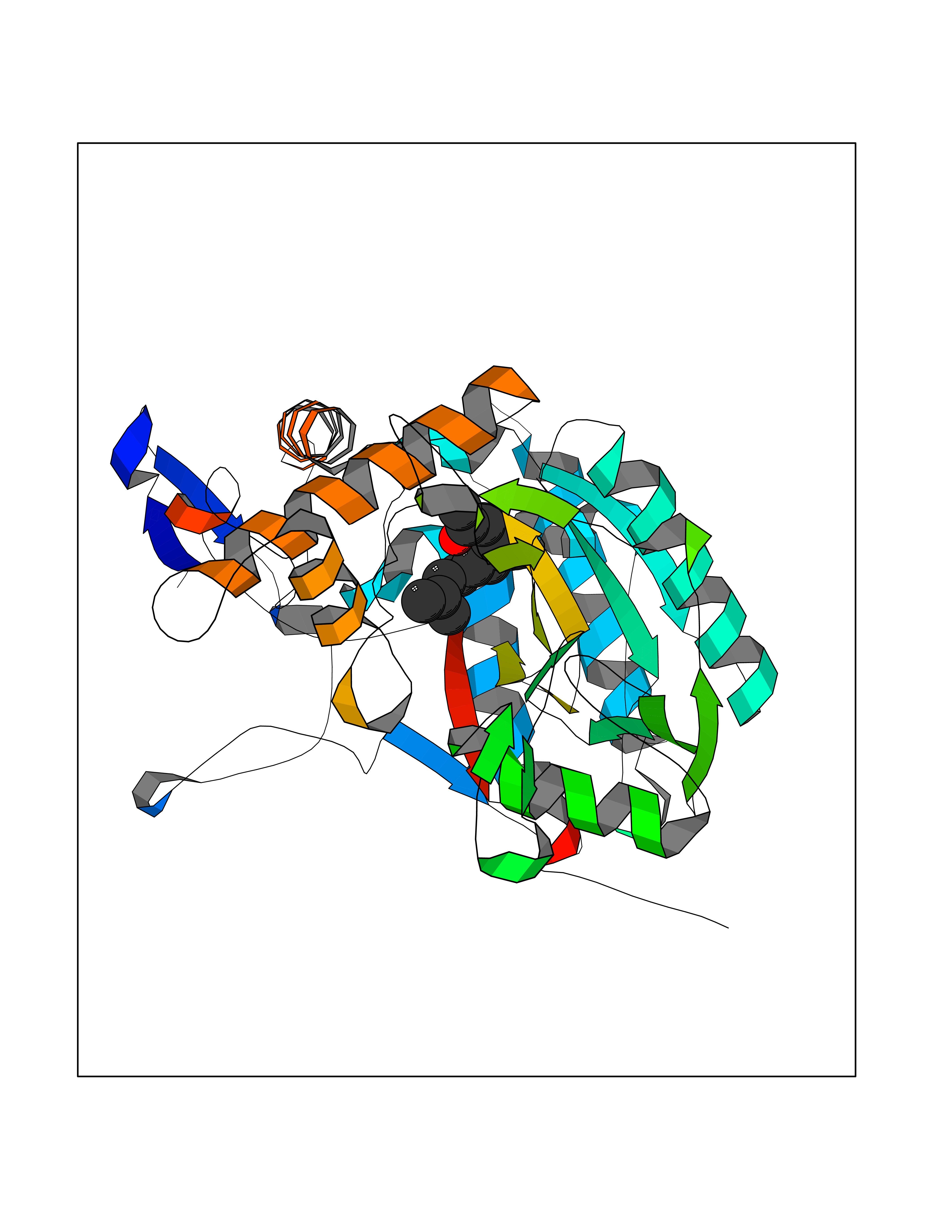 Molscript Map 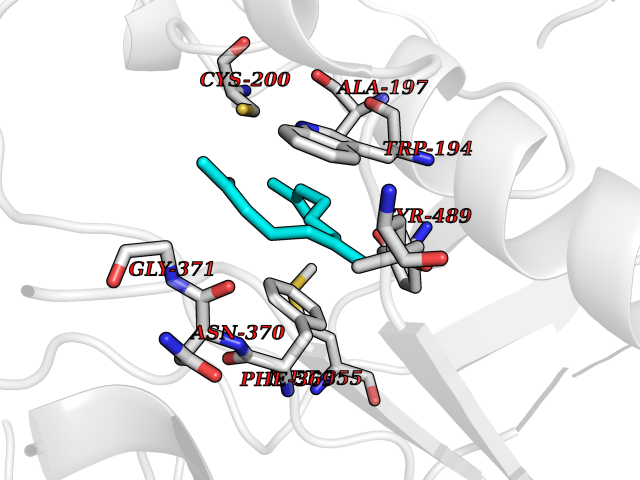 Pymol Map 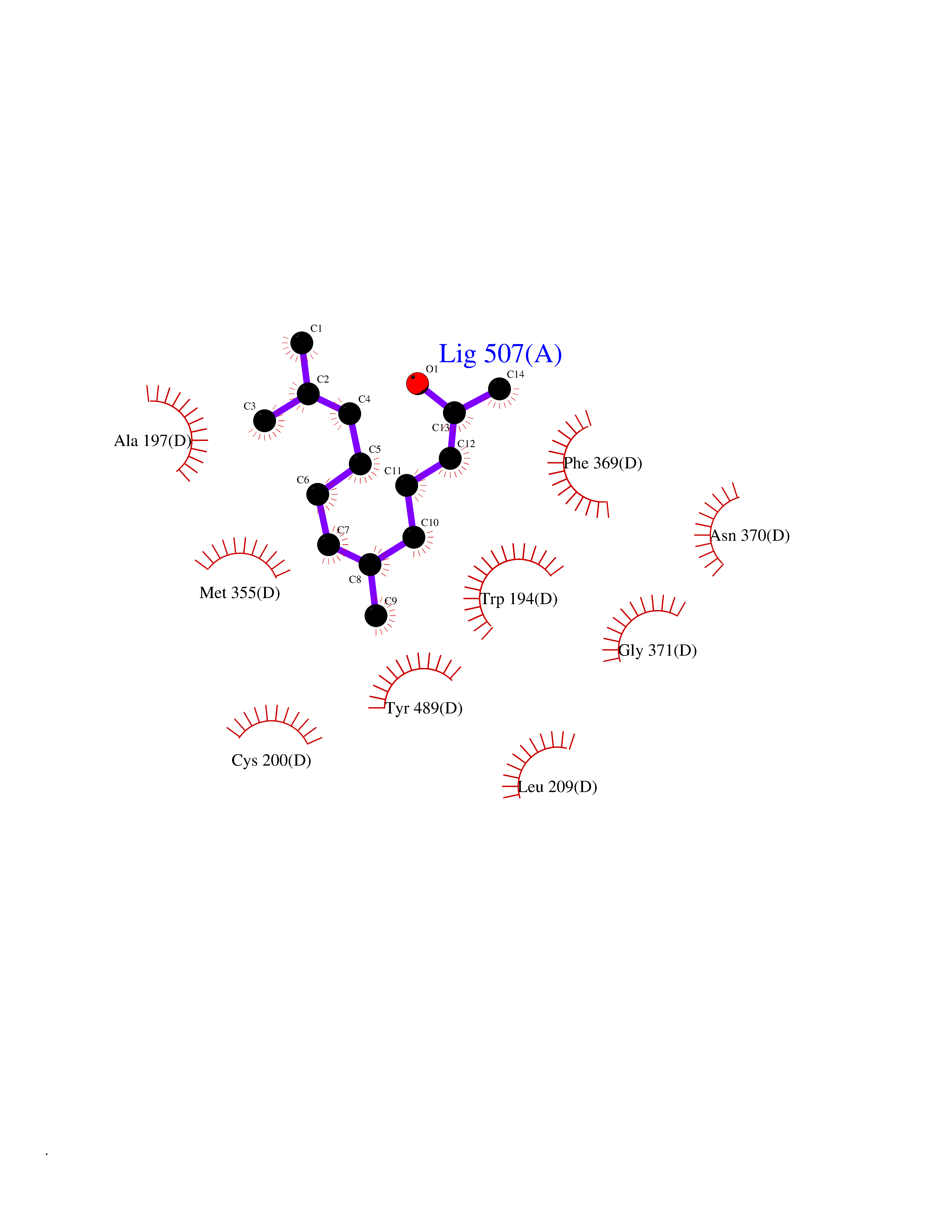 Ligplot Map 3D Binding mode Sequence RHVRIKNWGSGMTFQDTLHHKAKGILTCRSKSCLGSIMTPKSLTRGPRDKPTPPDELLPQAIEFVNQYYGSFKEAKIEEHLARVEAVTKEIETTGTYQLTGDELIFATKQAWRNAPRCIGRIQWSNLQVFDARSCSTAREMFEHICRHVRYSTNNGNIRSAITVFPQRSDGKHDFRVWNAQLIRYAGYQMPDGSIRGDPANVEFTQLCIDLGWKPKYGRFDVVPLVLQANGRDPELFEIPPDLVLEVAMEHPKYEWFRELELKWYALPAVANMLLEVGGLEFPGCPFNGWYMGTEIGVRDFCDVQRYNILEEVGRRMGLETHKLASLWKDQAVVEINIAVLHSFQKQNVTIMDHHSAAESFMKYMQNEYRSRGGCPADWIWLVPPMSGSITPVFHQEMLNYVLSPFYYYQVEAWKTHVWQD Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Cyclopropane mycolic acid synthase MmaA2 | 1TPY | 6.07 | |
Target general information Gen name mmaA2 Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID NA Synonyms Rv0644c;mma2 Protein family CFA/CMAS family Biochemical class Transferase Function Cyclopropane-fatty-acyl-phospholipid synthase activity.Methyltransferase activity. Related diseases Oocyte/zygote/embryo maturation arrest 16 (OZEMA16) [MIM:617234]: A rare cause of female primary infertility. In affected women, ovulation and fertilization proceed normally but embryos are arrested at early stages of development. Inheritance is autosomal recessive. {ECO:0000269|PubMed:27545678}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01718; DB01752 Interacts with NA EC number 2.1.1.79 Uniprot keywords 3D-structure; Acetylation; Lipid biosynthesis; Lipid metabolism; Methyltransferase; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 32493.6 Length 285 Aromaticity 0.1 Instability index 43.61 Isoelectric point 5.53 Charge (pH=7) -10.17 2D Binding mode Binding energy (Kcal/mol) -8.28 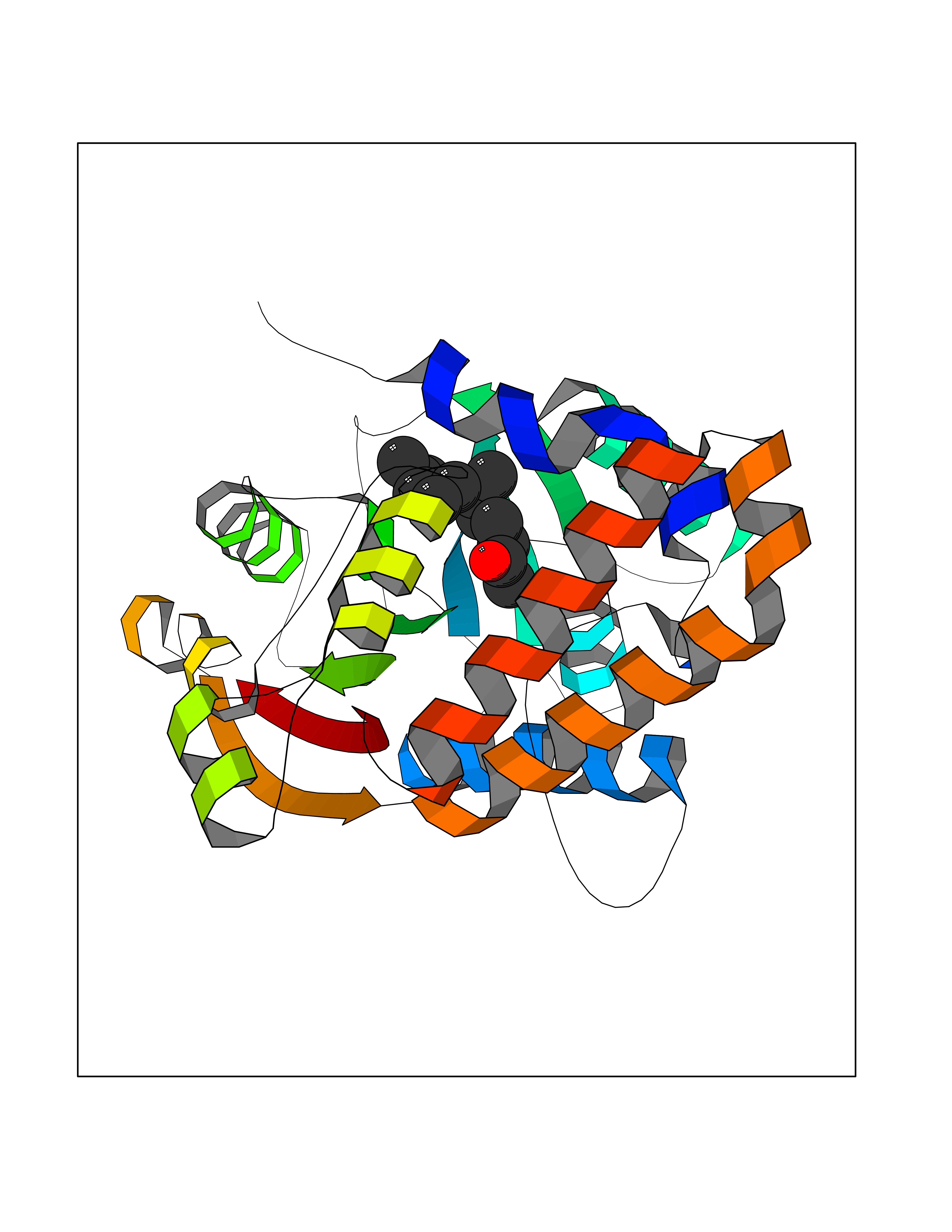 Molscript Map 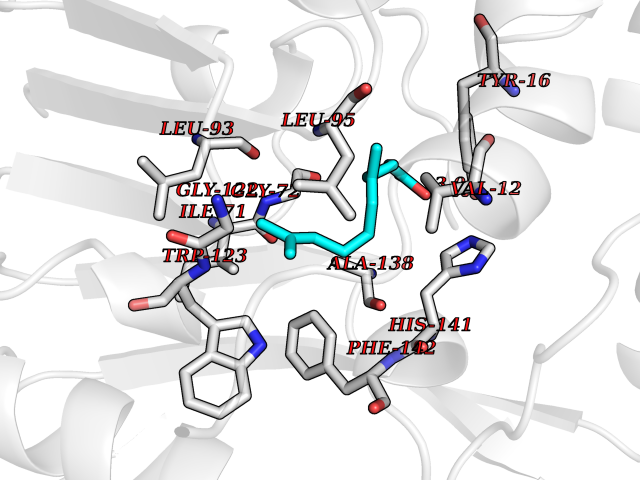 Pymol Map 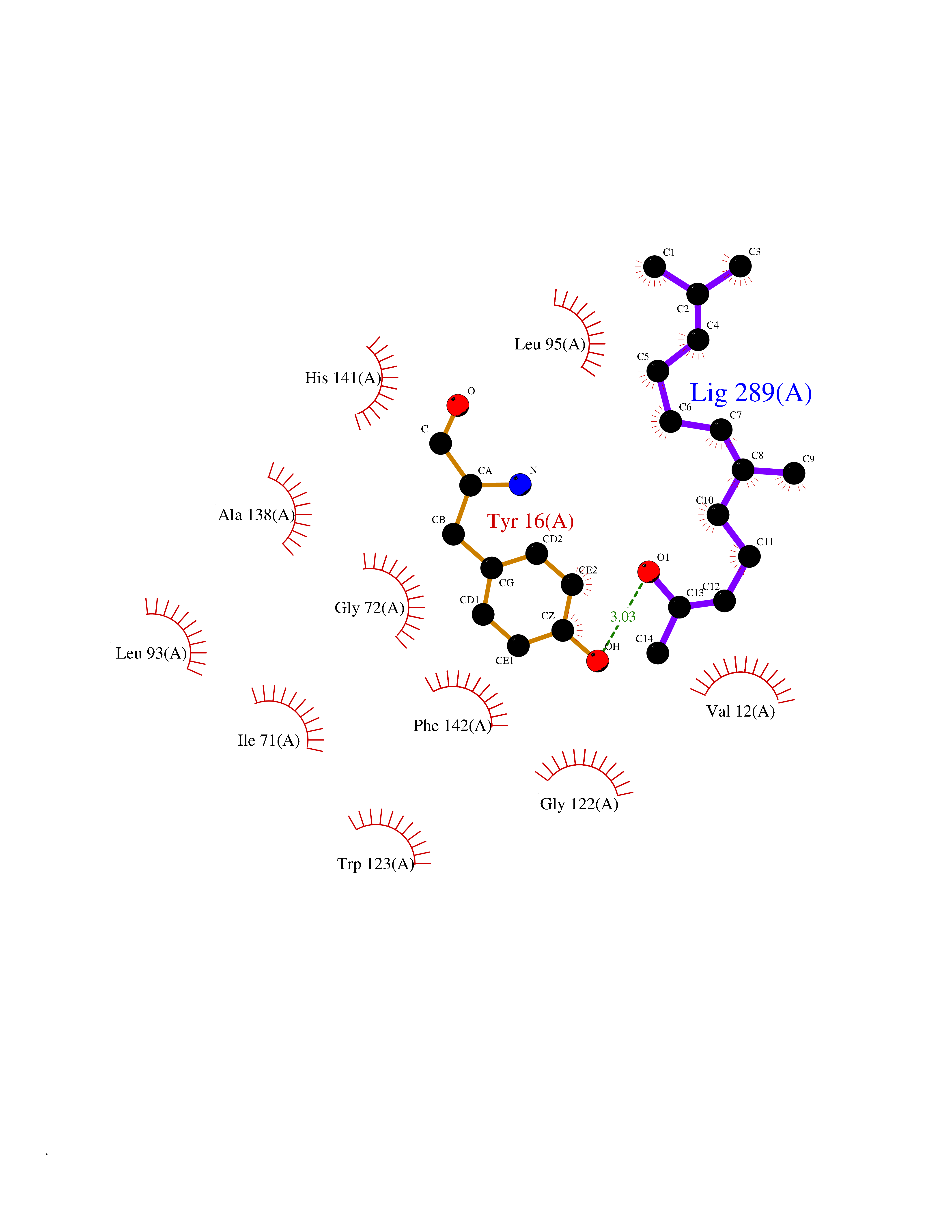 Ligplot Map 3D Binding mode Sequence NDLTPHFEDVQAHYDLSDDFFRLFLDPTQTYSCAHFEREDMTLEEAQIAKIDLALGKLGLQPGMTLLDIGCGWGATMRRAIAQYDVNVVGLTLSKNQAAHVQKSFDEMDTPRDRRVLLAGWEQFNEPVDRIVSIGAFEHFGHDRHADFFARAHKILPPDGVLLLHTITGLTRQQMVDHGLPLTLWLARFLKFIATEIFPGGQPPTIEMVEEQSAKTGFTLTRRQSLQPHYARTLDLWAEALQEHKSEAIAIQSEEVYERYMKYLTGCAKLFRVGYIDVNQFTLAK Hydrogen bonds contact Hydrophobic contact | ||||