Job Results:
Ligand
Structure
Job ID
6365174e5cc200e6a930804dda23a0ba
Job name
NA
Time
2026-02-27 16:33:59
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Zinc finger protein Helios (IKZF2) | 7LPS | 4.39 | |
Target general information Gen name IKZF2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ikaros family zinc finger protein 2 Protein family Ikaros C2H2-type zinc-finger protein family Biochemical class NA Function Associates with Ikaros at centromeric heterochromatin. Related diseases Developmental and epileptic encephalopathy 25, with amelogenesis imperfecta (DEE25) [MIM:615905]: An autosomal recessive disease characterized by subclinical seizures appearing in the first days of life, evolving to severe epileptic disease. Affected individuals have profound or severe delayed development with lack of speech, and most patients do not acquire the ability to sit. Additional variable features include axial hypotonia, peripheral hypertonia, and abnormal involuntary movements such as dystonia and choreoathetosis. Dental abnormalities, including delayed eruption, hypodontia, tooth hypoplasia, yellow discoloration, thin enamel, and enamel chipping are observed in most patients. {ECO:0000269|PubMed:24995870, ECO:0000269|PubMed:26384929, ECO:0000269|PubMed:30054523}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P29972; P56545; P56545-3; Q17RB8; P09022; Q8N8B7-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID B,C Molecular weight (Da) 47006.6 Length 410 Aromaticity 0.09 Instability index 44.28 Isoelectric point 7.23 Charge (pH=7) 0.69 2D Binding mode Binding energy (Kcal/mol) -5.98 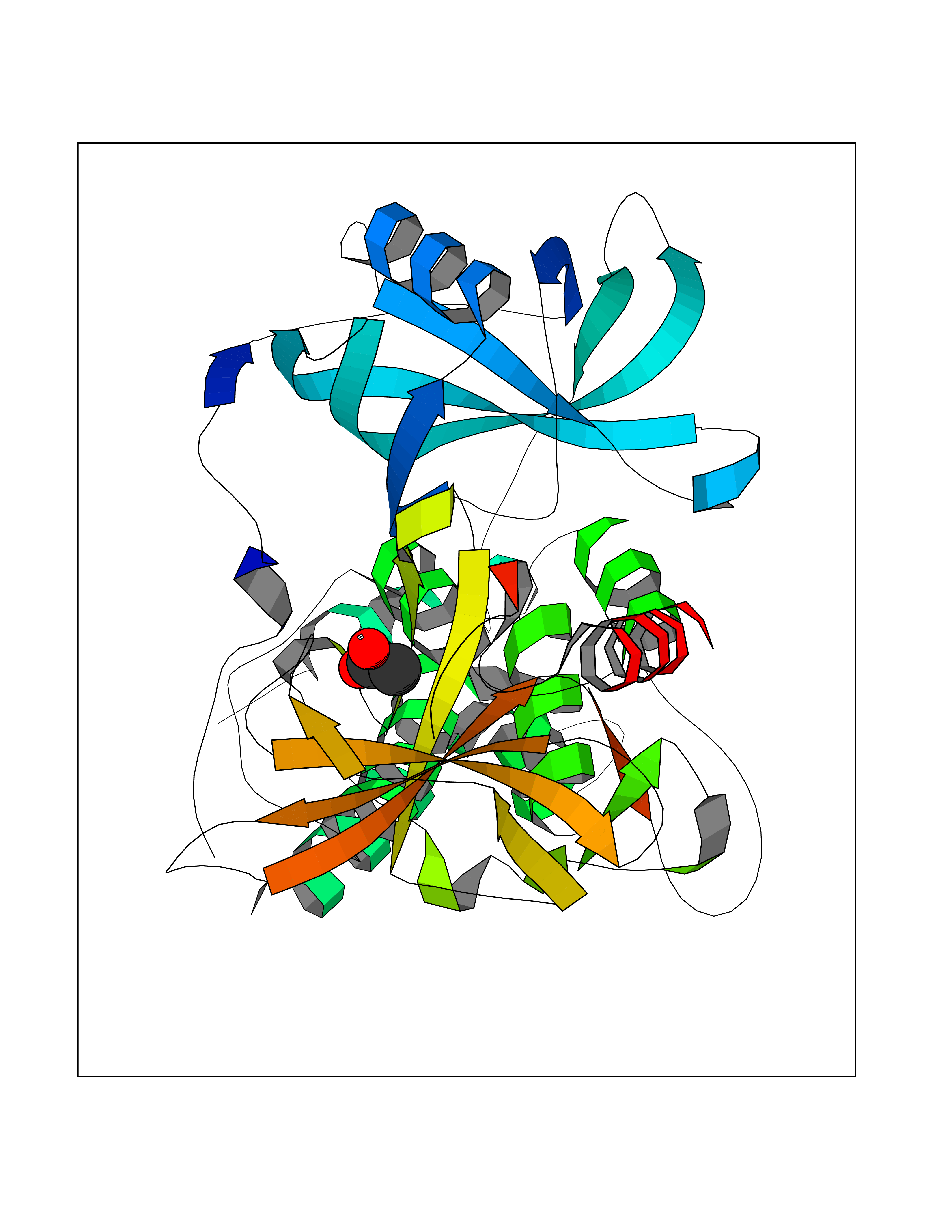 Molscript Map 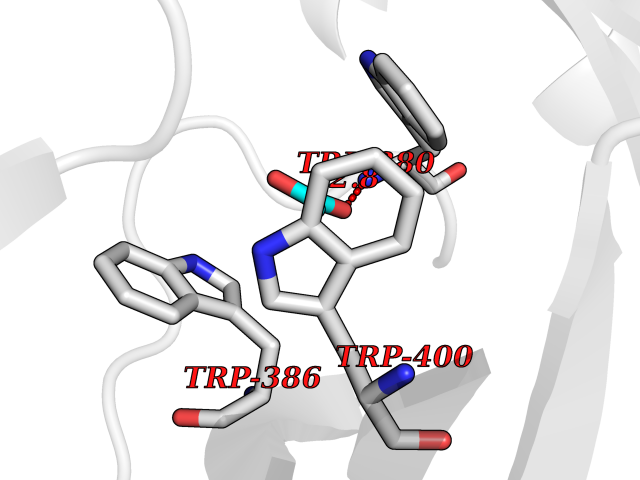 Pymol Map 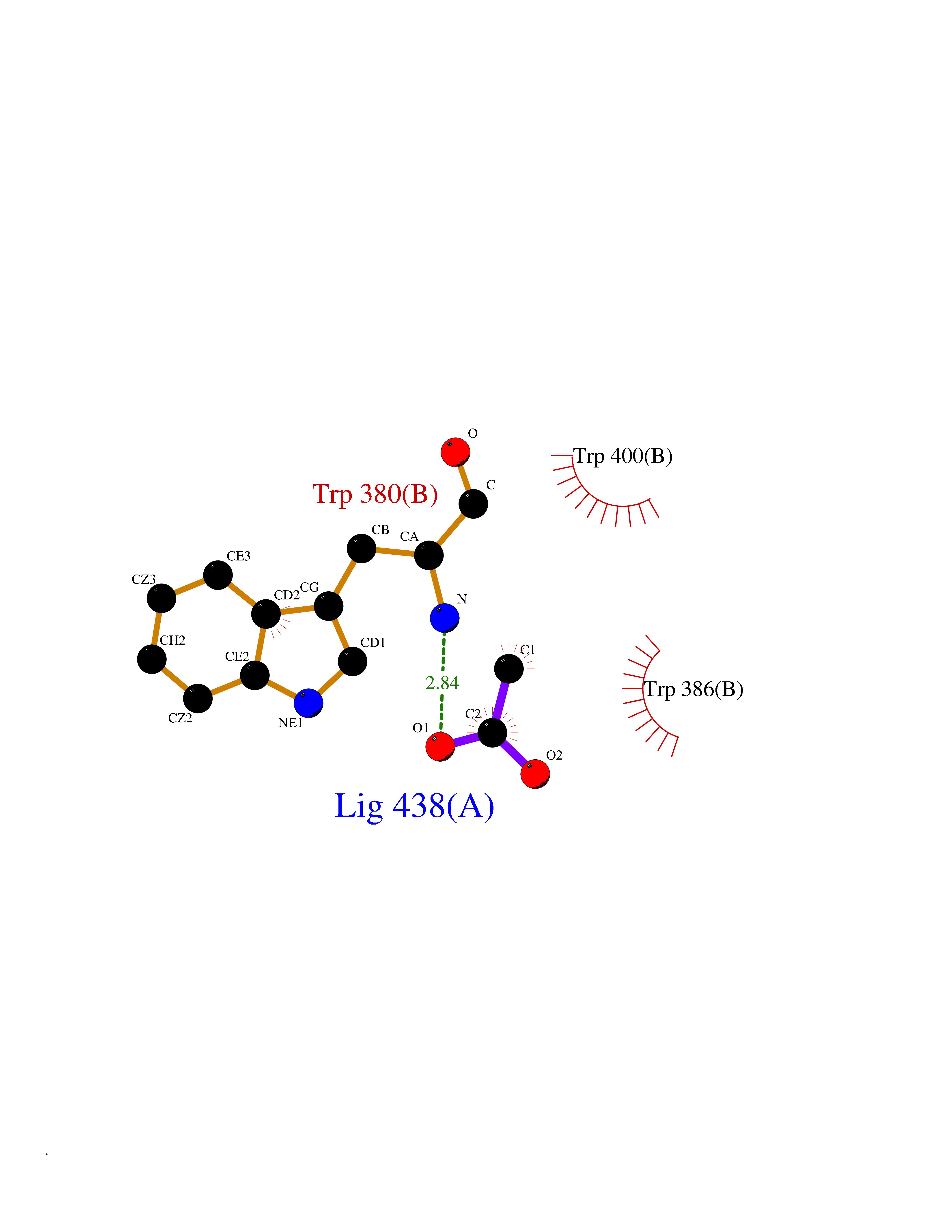 Ligplot Map 3D Binding mode Sequence INFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEQDFGIEIVKVKAIGRQRFKVLELRTQSDGIQQAKVQILPECVLPSTMSAVQLESLNKCQIFPCSYKWWQKYQKRKFHCANLTSWPRWLYSLYDAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPDGERPFHCNQCGASFTQKGNLLRHIKLHSG Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Glucose-dependent insulinotropic receptor (GPR119) | 7XZ6 | 4.39 | |
Target general information Gen name GPR119 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GPR119; G-protein coupled receptor 119 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for the endogenous fatty-acid ethanolamide oleoylethanolamide (OEA) and lysophosphatidylcholine (LPC). Functions as a glucose-dependent insulinotropic receptor. The activity of this receptor is mediated by G proteins which activate adenylate cyclase. Seems to act through a G(s) mediated pathway. Related diseases Developmental and epileptic encephalopathy 24 (DEE24) [MIM:615871]: A disease characterized by early-onset seizures, intellectual disability of varying degrees, and behavioral disturbances or autistic features in most individuals. {ECO:0000269|PubMed:24747641, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Generalized epilepsy with febrile seizures plus 10 (GEFSP10) [MIM:618482]: An autosomal dominant neurologic disorder with incomplete penetrance, characterized by variable types of seizures including absence, tonic-clonic, febrile, focal, and eyelid myoclonia. Some patients have normal neurologic development. Others have mild-to-moderate intellectual disability or autism spectrum disorder. {ECO:0000269|PubMed:29936235, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05166 Interacts with Q12797-6 EC number NA Uniprot keywords 3D-structure; Cell membrane; G-protein coupled receptor; Lipid-binding; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32134.1 Length 292 Aromaticity 0.12 Instability index 34.96 Isoelectric point 9.12 Charge (pH=7) 8.03 2D Binding mode Binding energy (Kcal/mol) -5.99 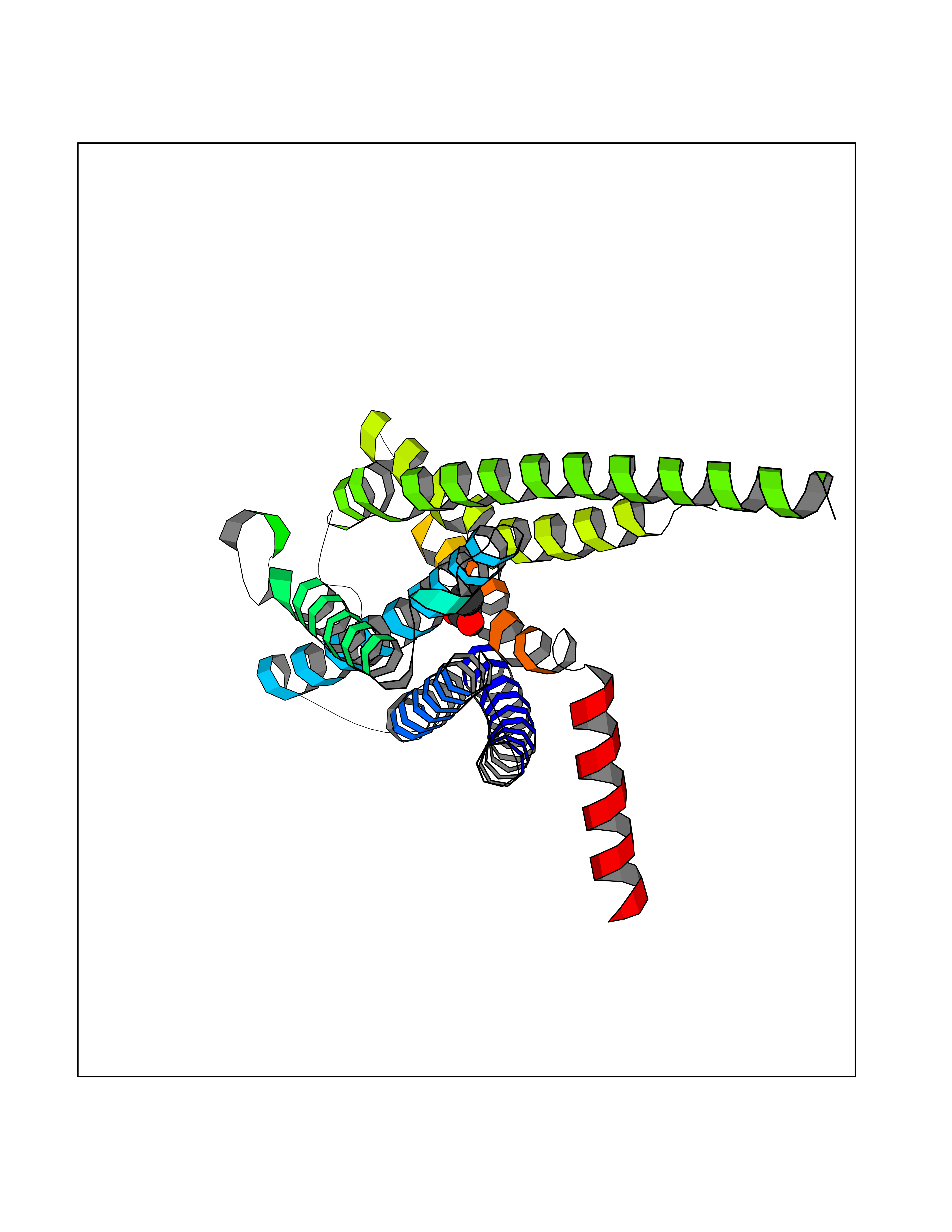 Molscript Map 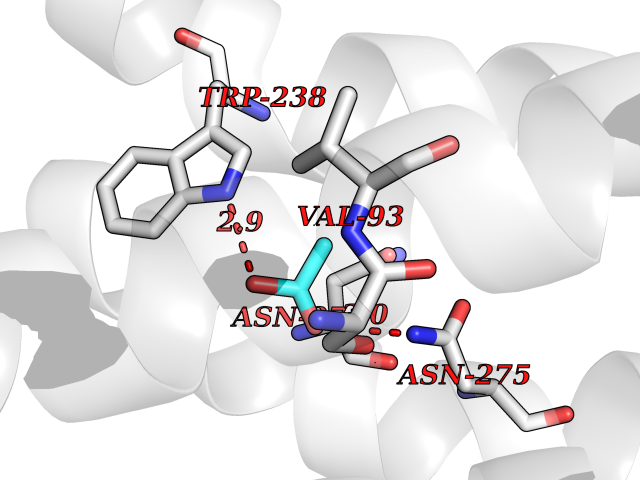 Pymol Map 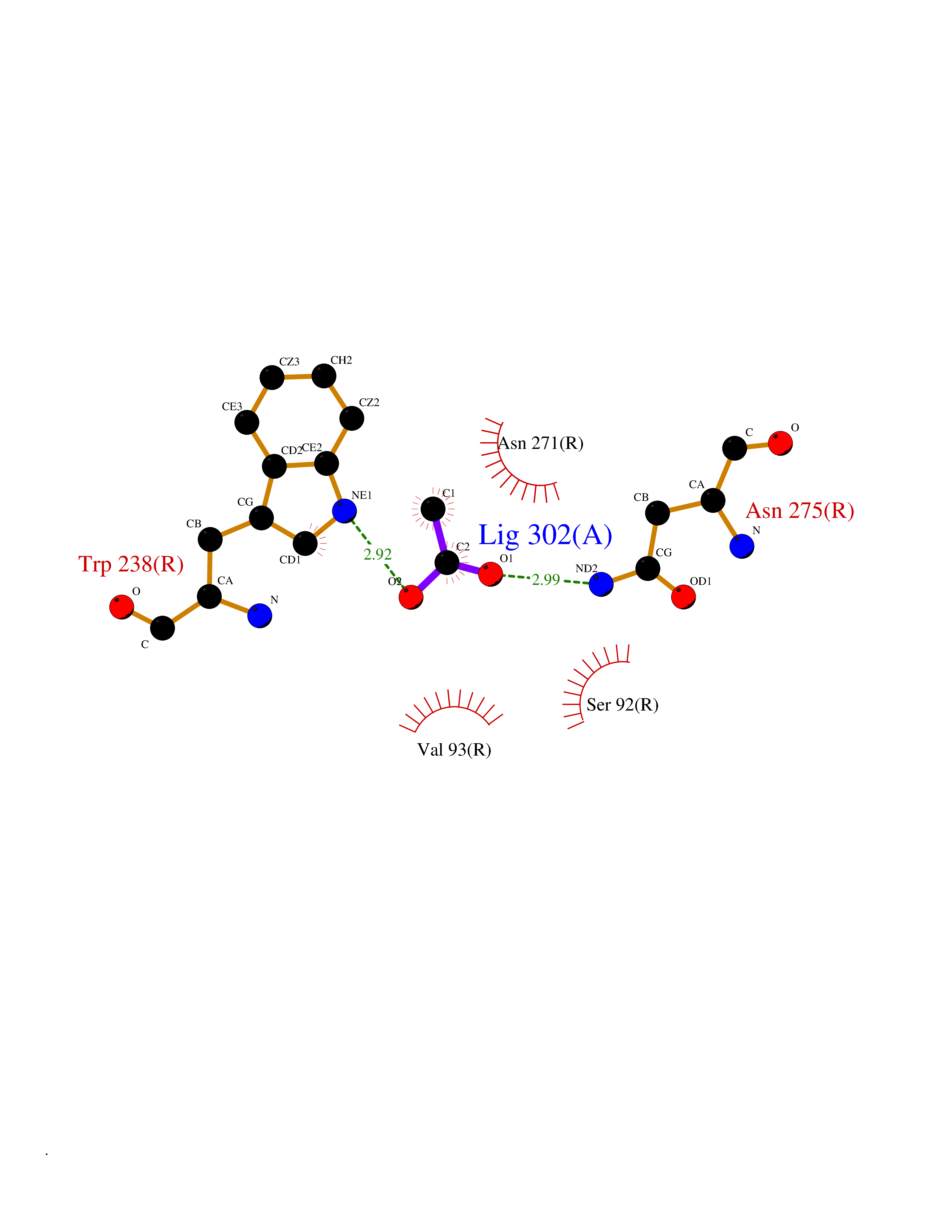 Ligplot Map 3D Binding mode Sequence MESSFSFGVILAVLASLIIATNTLVAVAVLLLIHKNDGVSLCFTLNLAVADTLIGVAISGLLTDQLSSPSRPTQKTLCSLRMAFVTSSAAASVLTVMLITFDRYLAIKQPFRYLKIMSGFVAGACIAGLWLVSYLIGFLPLGIPMFQQTAYKGQCSFFAVFHPHFVLTLSCVGFFPAMLLFVFFYCDMLKIASMHSQQIRKMEHAGAMAGSDFKALRTVSVLIGSFALSWTPFLITGIVQVACQECHLYLVLERYLWLLGVGNSLLNPLIYAYWQKEVRLQLYHMALGVKKV Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Cardiac phospholamban (PLN) | 7E0Z | 4.39 | |
Target general information Gen name PLN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Phospholamban; PLN; PLB Protein family Phospholamban family Biochemical class NA Function Reversibly inhibits the activity of ATP2A2 in cardiac sarcoplasmic reticulum by decreasing the apparent affinity of the ATPase for Ca(2+). Modulates the contractility of the heart muscle in response to physiological stimuli via its effects on ATP2A2. Modulatescalcium re-uptake during muscle relaxation and plays an important role in calcium homeostasis in the heart muscle. The degree of ATP2A2 inhibition depends on the oligomeric state of PLN. ATP2A2 inhibition is alleviated by PLN phosphorylation. Related diseases Cardiomyopathy, dilated, 1P (CMD1P) [MIM:609909]: A disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death. {ECO:0000269|PubMed:12610310, ECO:0000269|PubMed:16432188, ECO:0000269|PubMed:22137083, ECO:0000269|PubMed:22427649, ECO:0000269|PubMed:22707725}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Cardiomyopathy, familial hypertrophic, 18 (CMH18) [MIM:613874]: A hereditary heart disorder characterized by ventricular hypertrophy, which is usually asymmetric and often involves the interventricular septum. The symptoms include dyspnea, syncope, collapse, palpitations, and chest pain. They can be readily provoked by exercise. The disorder has inter- and intrafamilial variability ranging from benign to malignant forms with high risk of cardiac failure and sudden cardiac death. {ECO:0000269|PubMed:12705874}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q3SXY8; P07307-3; O15342; Q9BXK5; Q13323; P19397; O95471; Q9UHP7-3; Q7Z7G2; O43889-2; Q96BA8; Q09013; Q92838; Q9GZR5; Q5JX71; Q14318; Q8TBE3; P48165; Q8TDT2; O60883; Q8TED1; P31937; Q7Z5P4; P43628; Q5T700; Q8N112; Q9GZY8-5; Q6N075; Q99735; O14880; Q9GZW8; Q9H2K0; P15941-11; Q8TBJ4; O95197-3; Q9NR31; A0A0S2Z4U3; Q9Y3P8; Q15849; Q8IWU4; O95436-2; Q9NQQ7-3; Q9NP94; Q9HBV2; Q9NPE6; Q16623; P32856-2; Q9BVX2; Q7Z7N9; Q6UW68; Q9NWC5; Q96B21; Q4KMG9; Q8N661; O15393-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Cardiomyopathy; Disease variant; Endoplasmic reticulum; Lipoprotein; Membrane; Mitochondrion; Palmitate; Phosphoprotein; Proteomics identification; Reference proteome; Sarcoplasmic reticulum; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 40393 Length 349 Aromaticity 0.13 Instability index 35.1 Isoelectric point 8.98 Charge (pH=7) 6.56 2D Binding mode Binding energy (Kcal/mol) -5.99 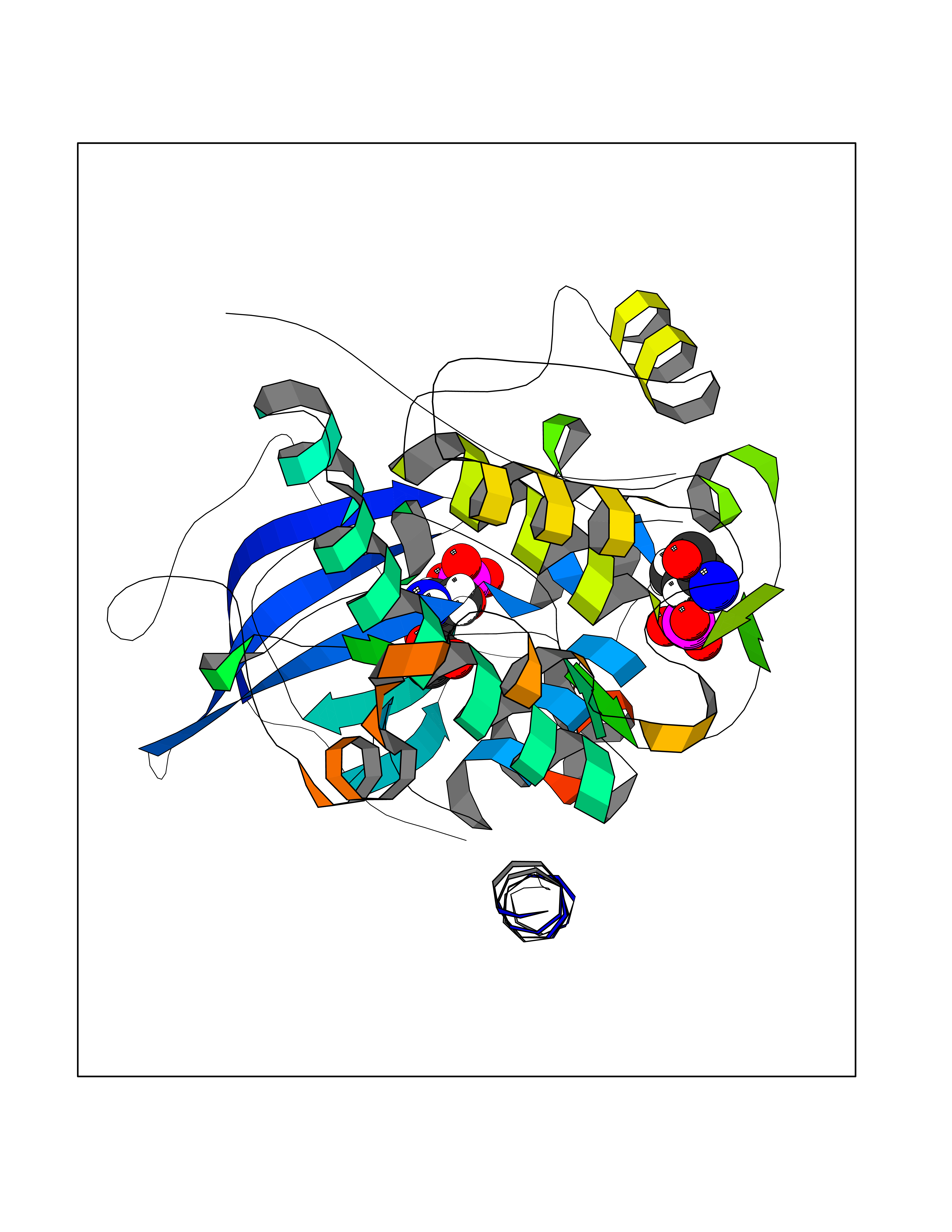 Molscript Map 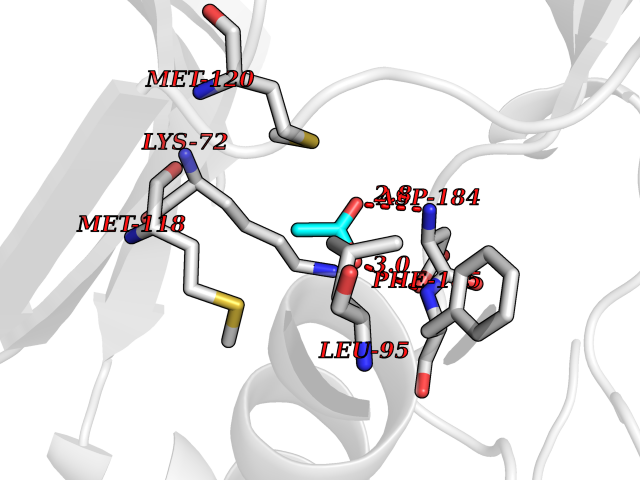 Pymol Map 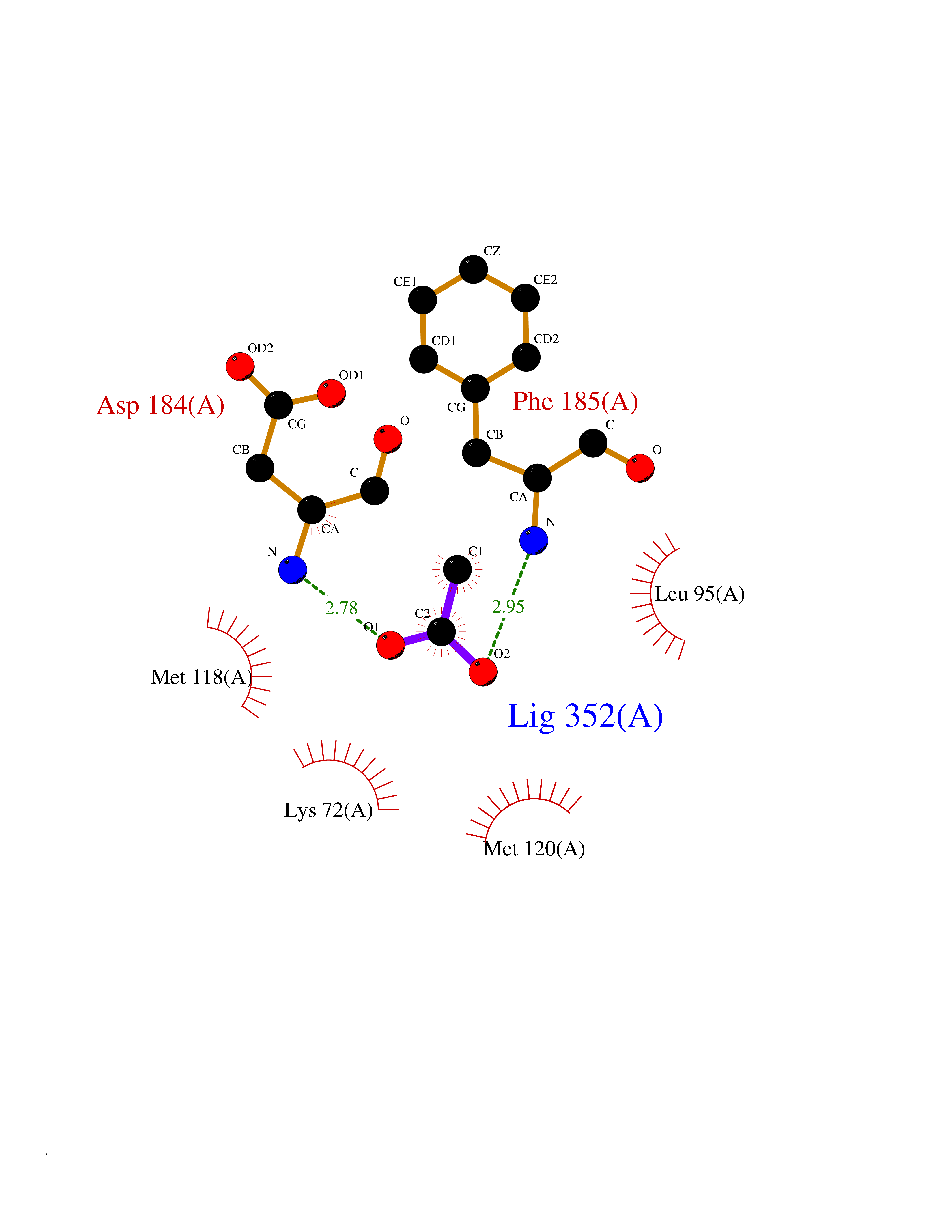 Ligplot Map 3D Binding mode Sequence ESVKEFLAKAKEDFLKKWETPQNTAQLDQFDRIKTLGTGSFGRVMLVKHKESGNHYAMKILDKQKVVKLKQIEHTLNEKRILQAVNFPFLVKLEFSFKDNSNLYMVMEYVAGGEMFSHLRRIGRFSEPHARFYAAQIVLTFEYLHSLDLIYRDLKPENLLIDQQGYIQVTDFGFAKRVKGRTWXLCGTPEYLAPEIILSKGYNKAVDWWALGVLIYEMAAGYPPFFADQPIQIYEKIVSGKVRFPSHFSSDLKDLLRNLLQVDLTKRFGNLKNGVNDIKNHKWFATTDWIAIYQRKVEAPFIPKFKGPGDTSNFDDYEEEEIRVXINEKCGKEFTEFTRSAIRRASTIE Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Chromodomain-helicase-DNA-binding protein 1 | 4O42 | 4.38 | |
Target general information Gen name CHD1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family SNF2/RAD54 helicase family Biochemical class Dna binding protein / viral protein Function ATP binding.ATP-dependent DNA helicase activity.DNA binding.Methylated histone binding. Related diseases Pilarowski-Bjornsson syndrome (PILBOS) [MIM:617682]: An autosomal dominant disorder characterized by developmental delay, speech apraxia, intellectual disability, autism, and facial dysmorphic features. Some patients may have seizures. {ECO:0000269|PubMed:28866611}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O60341-1; B2BUF1; P28799; O76024 EC number 3.6.4.12 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Chromatin regulator; Cytoplasm; Disease variant; DNA-binding; Helicase; Hydrolase; Intellectual disability; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 20969.1 Length 180 Aromaticity 0.12 Instability index 46.35 Isoelectric point 5.88 Charge (pH=7) -2.83 2D Binding mode Binding energy (Kcal/mol) -5.97  Molscript Map 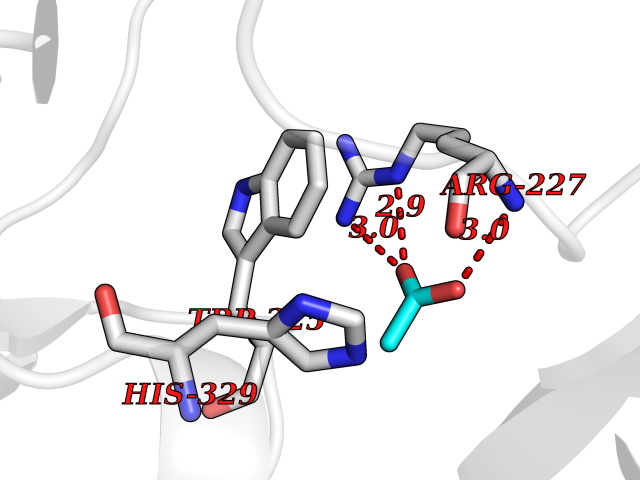 Pymol Map 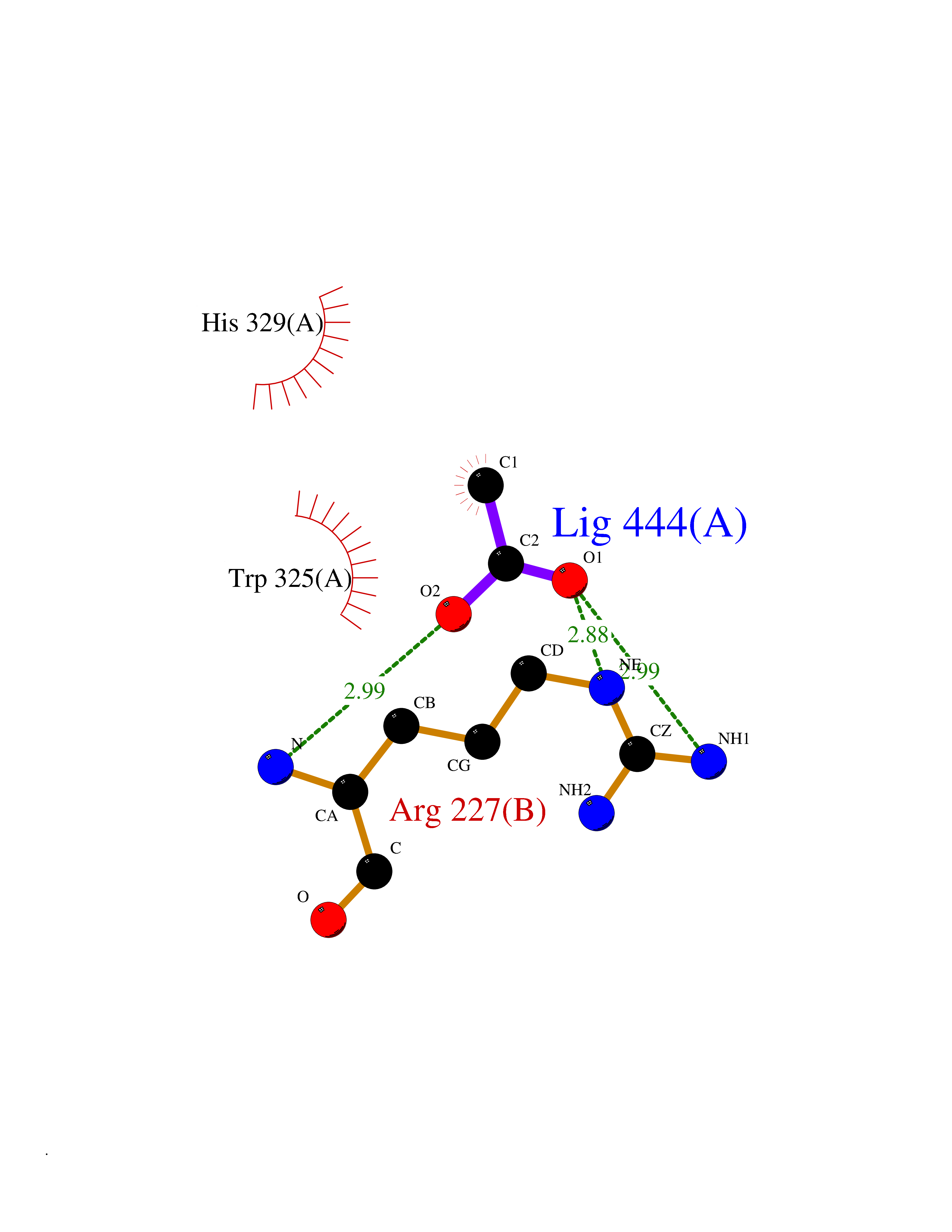 Ligplot Map 3D Binding mode Sequence EFETIERFMDCRIGRKGATGATTTIYAVEADGDPNAGFEKNKEPGEIQYLIKWKGWSHIHNTWETEETLKQQNVRGMKKLDNYKKKDQETKRWLKNASPEDVEYYNCQQELTDDLHKQYQIVERIIAHSNQKSAAGYPDYYCKWQGLPYSECSWEDGALISKKFQACIDEYFSRTARSXV Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Histone deacetylase 3 (HDAC3) | 4A69 | 4.38 | |
Target general information Gen name HDAC3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SMAP45; RPD32; RPD3-2; HD3 Protein family Histone deacetylase family, HD type 1 subfamily Biochemical class Carbon-nitrogen hydrolase Function Gives a tag for epigenetic repression and plays an important role in transcriptional regulation, cell cycle progression and developmental events. Histone deacetylases act via the formation of large multiprotein complexes. Participates in the BCL6 transcriptional repressor activity by deacetylating the H3 'Lys-27' (H3K27) on enhancer elements, antagonizing EP300 acetyltransferase activity and repressing proximal gene expression. Probably participates in the regulation of transcription through its binding to the zinc-finger transcription factor YY1; increases YY1 repression activity. Required to repress transcription of the POU1F1 transcription factor. Acts as a molecular chaperone for shuttling phosphorylated NR2C1 to PML bodies for sumoylation. Contributes, together with XBP1 isoform 1, to the activation of NFE2L2-mediated HMOX1 transcription factor gene expression in a PI(3)K/mTORC2/Akt-dependent signaling pathway leading to endothelial cell (EC) survival under disturbed flow/oxidative stress. Regulates both the transcriptional activation and repression phases of the circadian clock in a deacetylase activity-independent manner. During the activation phase, promotes the accumulation of ubiquitinated ARNTL/BMAL1 at the E-boxes and during the repression phase, blocks FBXL3-mediated CRY1/2 ubiquitination and promotes the interaction of CRY1 and ARNTL/BMAL1. The NCOR1-HDAC3 complex regulates the circadian expression of the core clock gene ARTNL/BMAL1 and the genes involved in lipid metabolism in the liver. Responsible for the deacetylation of lysine residues on the N-terminal part of the core histones (H2A, H2B, H3 and H4), and some other non-histone substrates. Related diseases Cocoon syndrome (COCOS) [MIM:613630]: A lethal syndrome characterized by multiple fetal malformations including defective face and seemingly absent limbs, which are bound to the trunk and encased under the skin. {ECO:0000269|PubMed:20961246}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Bartsocas-Papas syndrome 2 (BPS2) [MIM:619339]: An autosomal recessive, severe form of popliteal pterygium syndrome. Popliteal pterygia syndromes have considerable variability in severity and in the associated phenotypic features but they are all characterized by cutaneous webbing across one or more major joints, cleft lip and/or palate, syndactyly, and genital malformations. {ECO:0000269|PubMed:25691407}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12565; DB05015; DB01262; DB11841; DB12645; DB11830; DB06603; DB06819; DB05223; DB03766; DB12847; DB06176; DB00313; DB02546 Interacts with O43823; Q9ULX6; Q9UKG1; P24385; P23771; Q13227; Q13547; O60341; Q969R5; P43356; Q9UIS9; P01106; O75376; Q9Y618; Q15466; P48552; P00558; P00558-1; P60510; Q15022; O09106; P08393; Q60974; Q8CBD1; P12504 EC number EC 3.5.1.98 Uniprot keywords 3D-structure; Alternative splicing; Biological rhythms; Chromatin regulator; Chromosome; Cytoplasm; Host-virus interaction; Hydrolase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 50376.9 Length 437 Aromaticity 0.14 Instability index 29.95 Isoelectric point 6.1 Charge (pH=7) -6.84 2D Binding mode Binding energy (Kcal/mol) -5.97  Molscript Map  Pymol Map 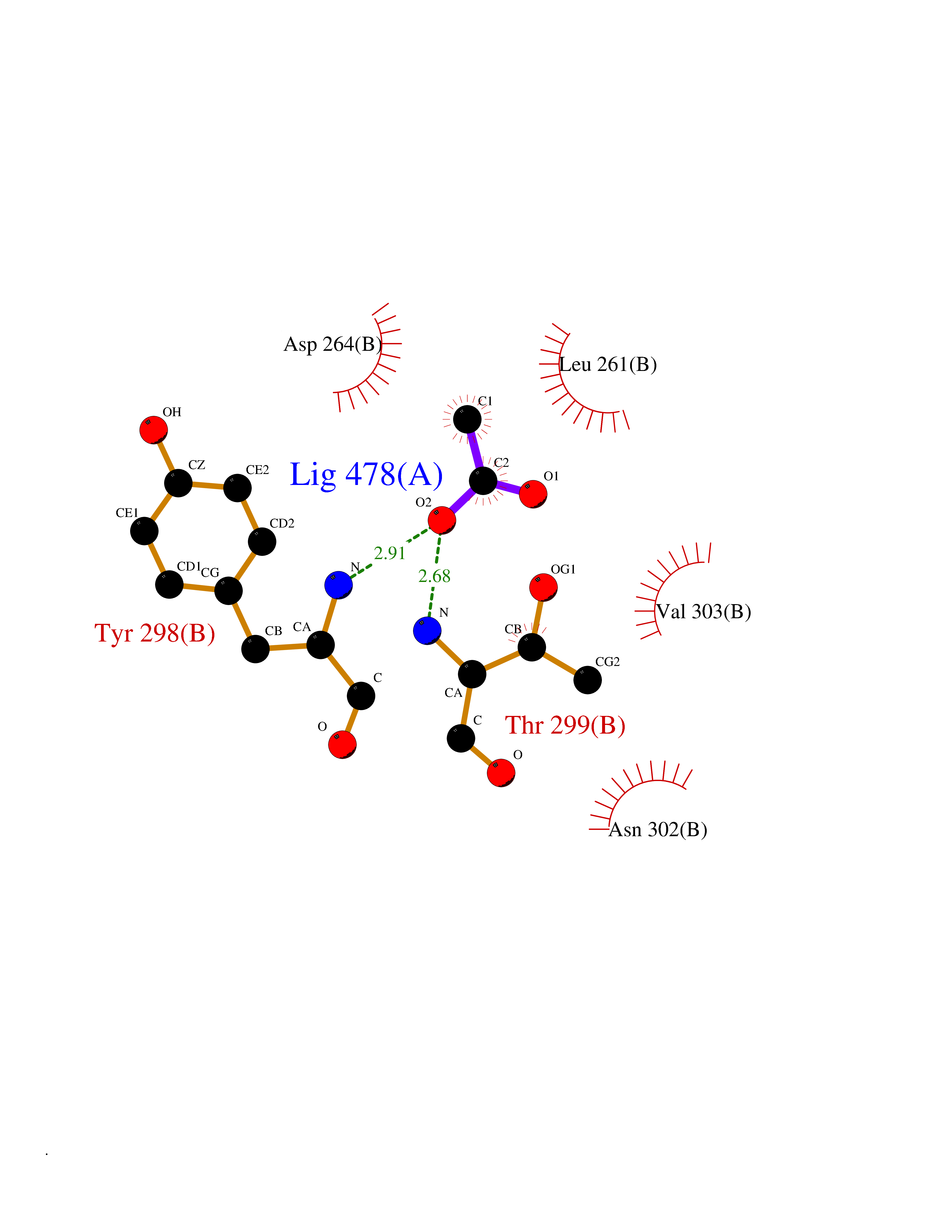 Ligplot Map 3D Binding mode Sequence KFINMNGLMADPMKVYKDRQVMNMWSEQEKETFREKFMQHPKNFGLIASFLERKTVAECVLYYYLTKKAKTVAYFYDPDVGNFHYGAGHPMKPHRLALTHSLVLHYGLYKKMIVFKPYQASQHDMCRFHSEDYIDFLQRVSPTNMQGFTKSLNAFNVGDDCPVFPGLFEFCSRYTGASLQGATQLNNKICDIAINWAGGLHHAKKFEASGFCYVNDIVIGILELLKYHPRVLYIDIDIHHGDGVQEAFYLTDRVMTVSFHKYGNYFFPGTGDMYEVGAESGRYYCLNVPLRDGIDDQSYKHLFQPVINQVVDFYQPTCIVLQCGADSLGCDRLGCFNLSIRGHGECVEYVKSFNIPLLVLGGGGYTVRNVARCWTYETSLLVEEAISEELPYSEYFEYFAPDFTLHPDVSTRIENQNSRQYLDQIRQTIFENLKMLN Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Dopamine beta-hydroxylase | 4ZEL | 4.38 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -5.98 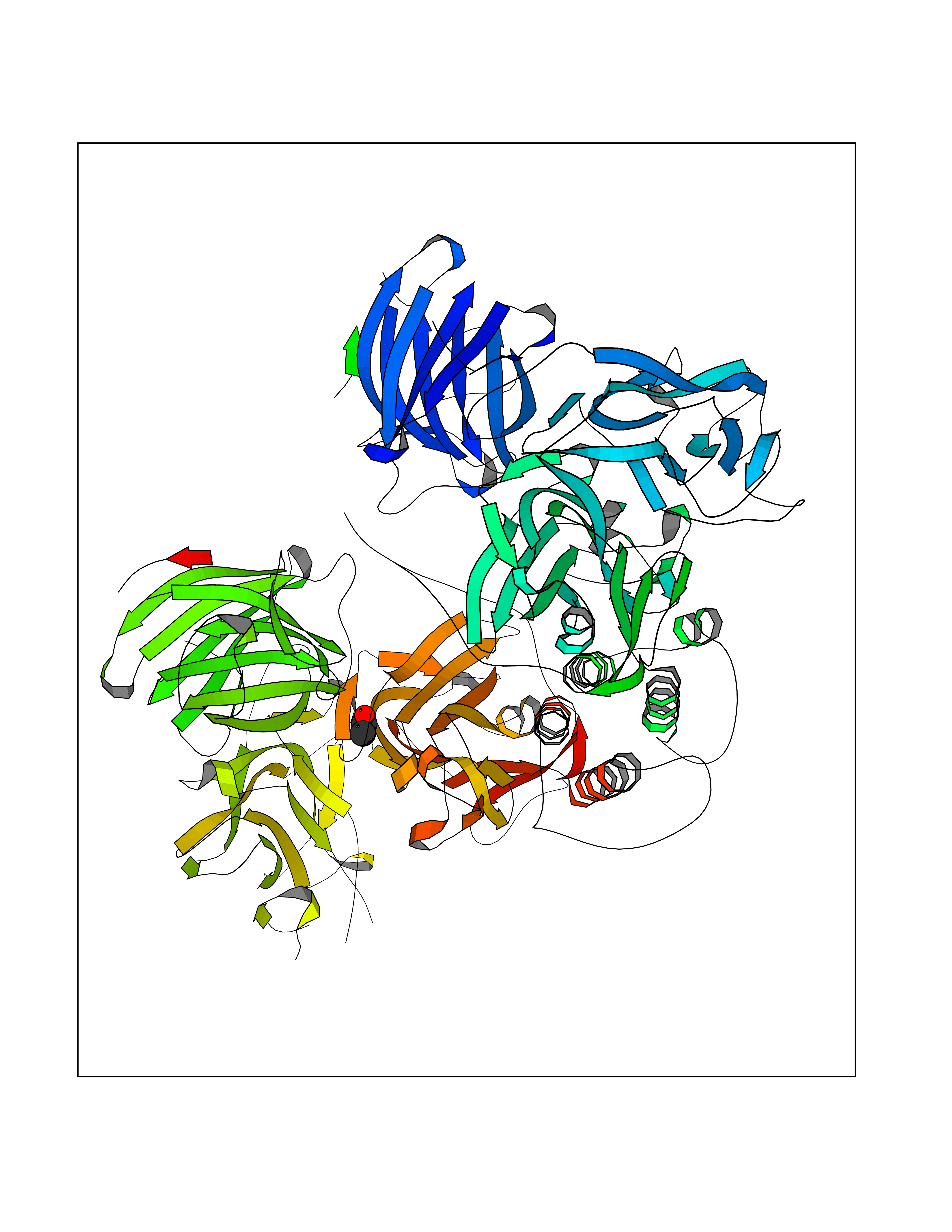 Molscript Map 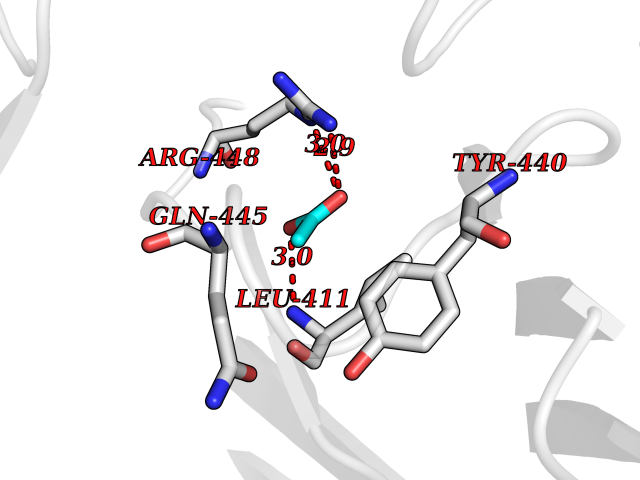 Pymol Map 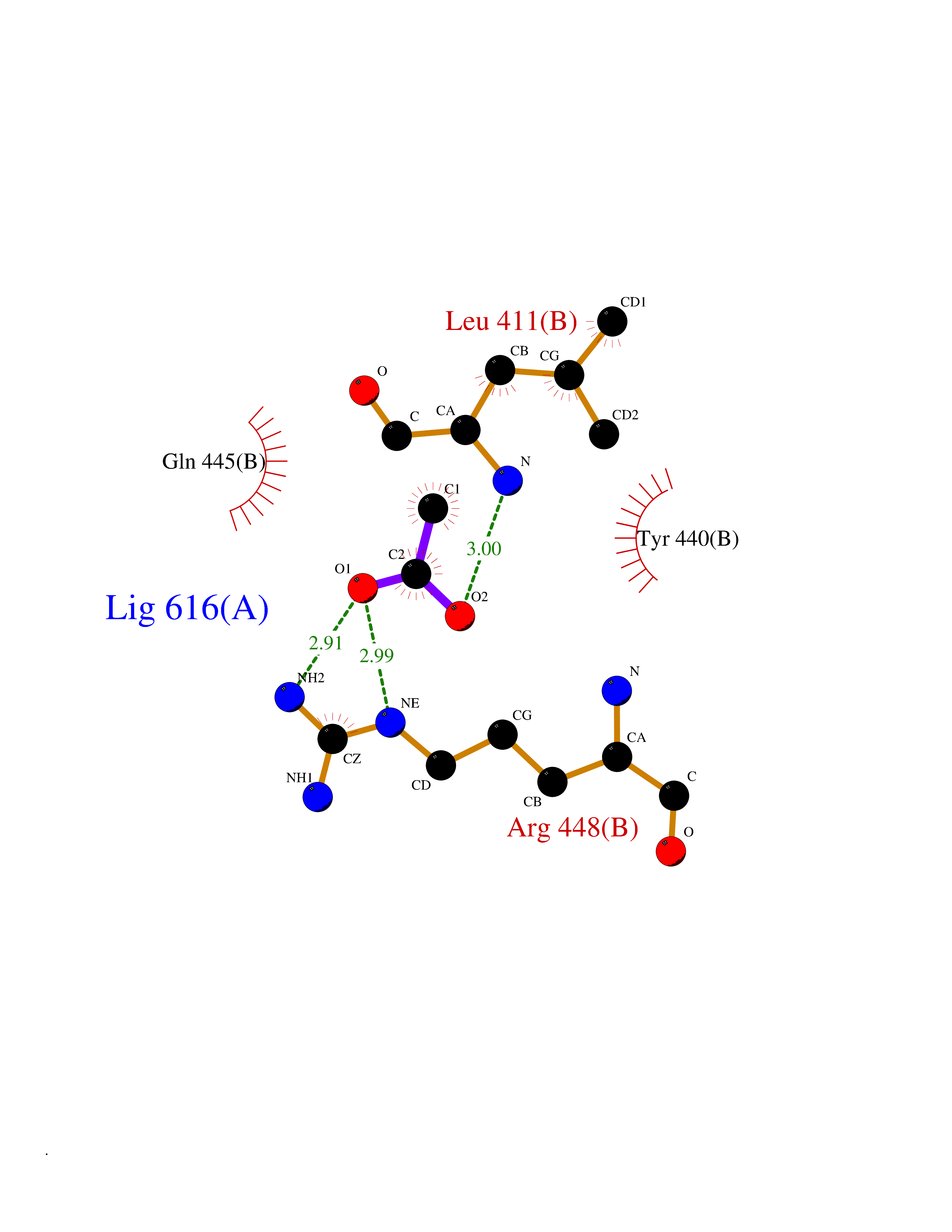 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | p-hydroxybenzoate hydroxylase | 1K0I | 4.38 | |
Target general information Gen name pobA Organism Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Uniprot ID TTD ID NA Synonyms PA0247 Protein family Aromatic-ring hydroxylase family Biochemical class Hydrolase Function 4-hydroxybenzoate 3-monooxygenase activity.FAD binding.Flavin adenine dinucleotide binding. Related diseases Immunodeficiency, common variable, 12, with autoimmunity (CVID12) [MIM:616576]: A primary immunodeficiency characterized by hypogammaglobulinemia and recurrent bacterial infections. About half of patients develop autoimmune features, including cytopenia, as well as generalized inflammation and lymphoproliferation manifest as lymphadenopathy or hepatosplenomegaly. {ECO:0000269|PubMed:26279205}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02839; DB04242; DB03482; DB02362; DB03147 Interacts with NA EC number 1.14.13.2 Uniprot keywords 3D-structure; Aromatic hydrocarbons catabolism; FAD; Flavoprotein; Monooxygenase; NADP; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 44295 Length 394 Aromaticity 0.09 Instability index 37.66 Isoelectric point 6.2 Charge (pH=7) -3.67 2D Binding mode Binding energy (Kcal/mol) -5.98  Molscript Map 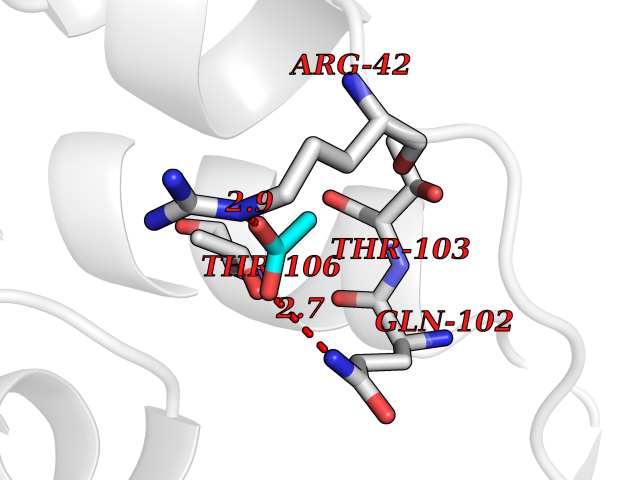 Pymol Map 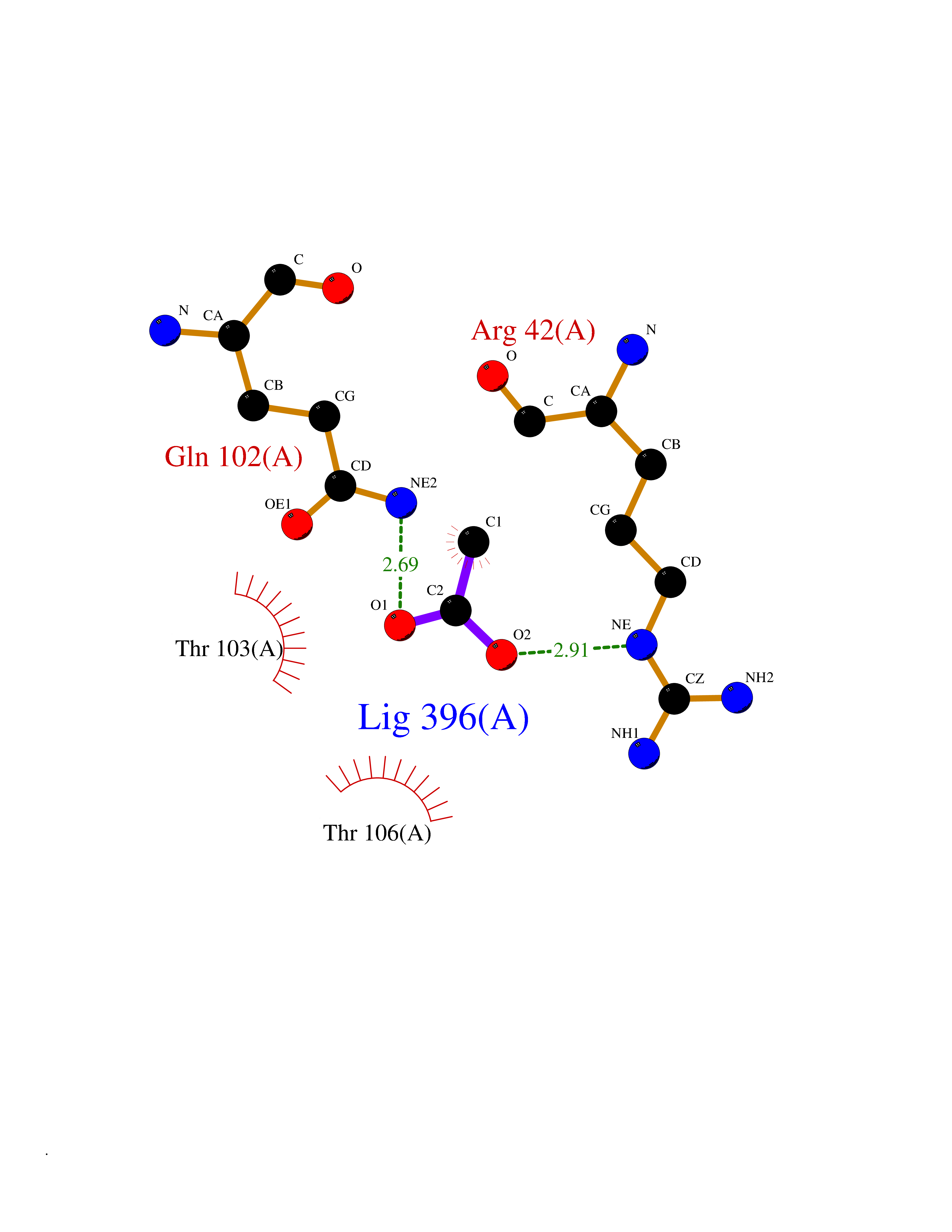 Ligplot Map 3D Binding mode Sequence MKTQVAIIGAGPSGLLLGQLLHKAGIDNVILERQTPDYVLGRIRAGVLEQGMVDLLREAGVDRRMARDGLVHEGVEIAFAGQRRRIDLKRLSGGKTVTVYGQTEVTRDLMEAREACGATTVYQAAEVRLHDLQGERPYVTFERDGERLRLDCDYIAGCDGFHGISRQSIPAERLKVFERVYPFGWLGLLADTPPVSHELIYANHPRGFALCSQRSATRSQYYVQVPLSEKVEDWSDERFWTELKARLPSEVAEKLVTGPSLEKSIAPLRSFVVEPMQHGRLFLAGDAAHIVPPTGAKGLNLAASDVSTLYRLLLKAYREGRGELLERYSAICLRRIWKAERFSWWMTSVLHRFPDTDAFSQRIQQTELEYYLGSEAGLATIAENYVGLPYEEIE Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Nitric-oxide synthase brain (NOS1) | 5ADF | 4.38 | |
Target general information Gen name NOS1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Peptidyl-cysteine S-nitrosylase NOS1; Nitric oxide synthase, brain; Neuronal NOS; NOS, type I; NOS type I; NNOS; NC-NOS; N-NOS; BNOS Protein family NOS family Biochemical class Paired donor oxygen oxidoreductase Function In the brain and peripheral nervous system, NO displays many properties of a neurotransmitter. Probably has nitrosylase activity and mediates cysteine S-nitrosylation of cytoplasmic target proteins such SRR. Produces nitric oxide (NO) which is a messenger molecule with diverse functions throughout the body. Related diseases Variation Asp-298 in NOS3 may be associated with susceptibility to coronary spasm. {ECO:0000269|PubMed:11740345, ECO:0000269|PubMed:9737779}. Drugs (DrugBank ID) DB02143; DB02727; DB01997; DB03892; DB02207; DB03710; DB00155; DB00843; DB00997; DB03147; DB03247; DB01942; DB01221; DB02077; DB01821; DB09241; DB03144; DB03449; DB02044; DB02644; DB08019; DB08018; DB02027; DB03461; DB04223; DB06096; DB02991; DB03707 Interacts with Q08AM6 EC number EC 1.14.13.39 Uniprot keywords 3D-structure; Alternative splicing; Calmodulin-binding; Cell membrane; Cell projection; FAD; Flavoprotein; FMN; Heme; Iron; Membrane; Metal-binding; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Synapse; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34875.7 Length 299 Aromaticity 0.1 Instability index 42.94 Isoelectric point 5.96 Charge (pH=7) -6.25 2D Binding mode Binding energy (Kcal/mol) -5.98 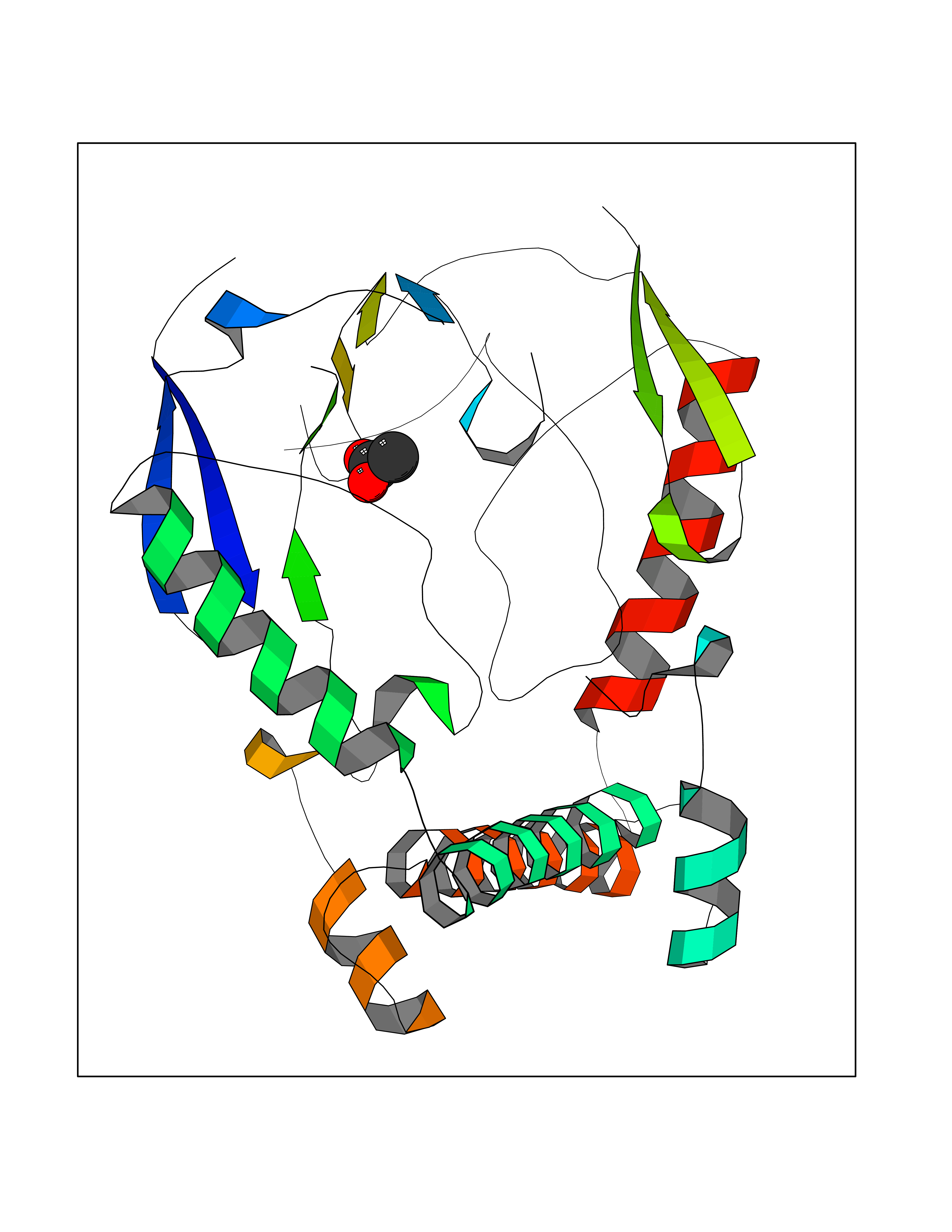 Molscript Map  Pymol Map 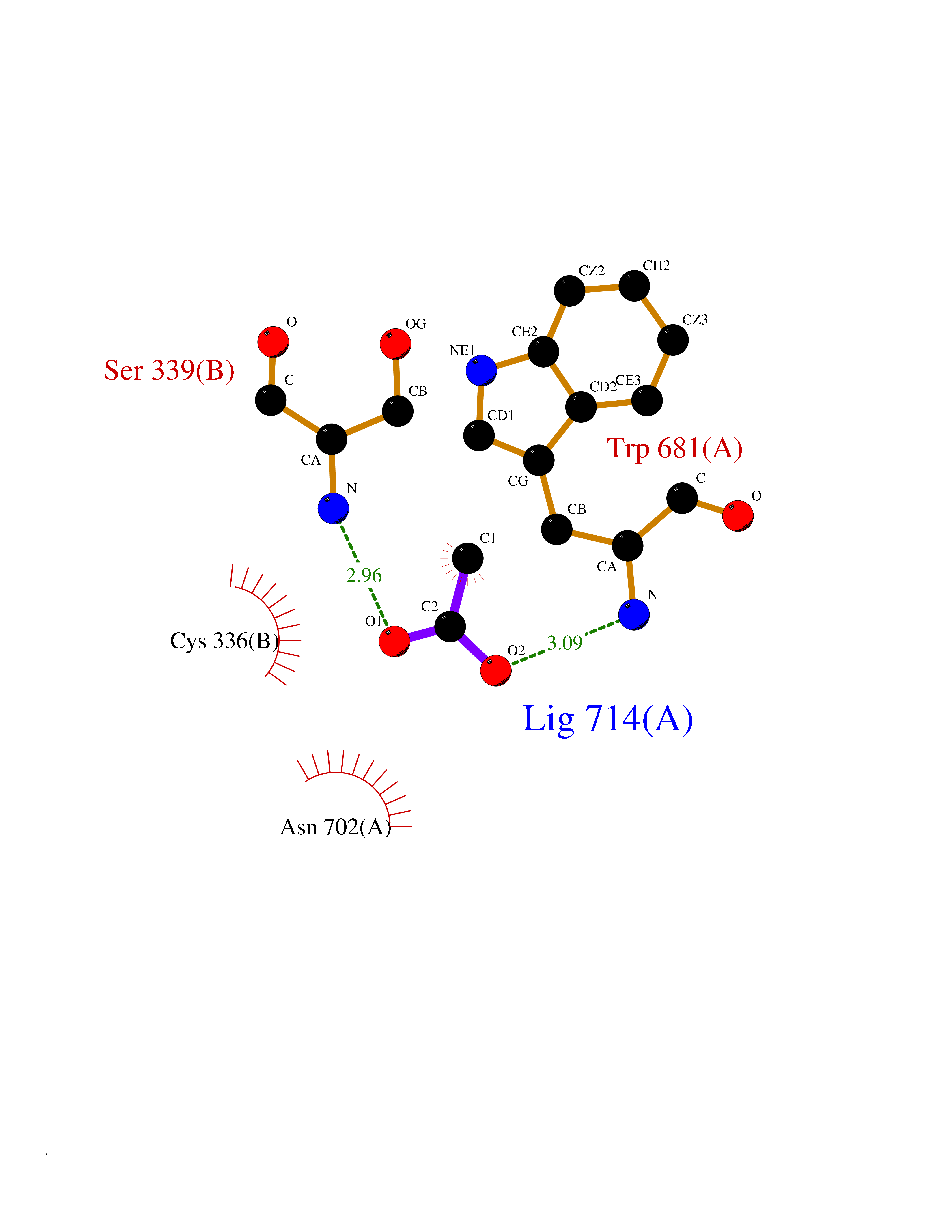 Ligplot Map 3D Binding mode Sequence CPRFLKVKNWETEVVLTDTLHLKSTLETGCTEYICMGSIMHPRDYCDNSRYNILEEVAKKMNLDMRKTSSLWKDQALVEINIAVLYSFQSDKVTIVDHHSATESFIKHMENEYRCRGGCPADWVWIVPPMSGSITPVFHQEMLNYRLRFLKVKNWETEVVLTDTLHLKSTLETGCTEYICMGSIMHPRDYCDNSRYNILEEVAKKMNLDMRKTSSLWKDQALVEINIAVLYSFQSDKVTIVDHHSATESFIKHMENEYRCRGGCPADWVWIVPPMSGSITPVFHQEMLNYRLTPSFEYQ Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Pyruvate kinase PKLR | 4IP7 | 4.38 | |
Target general information Gen name PKLR Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PKL;PK1 Protein family Pyruvate kinase family Biochemical class Transferase Function ATP binding.Kinase activity.Magnesium ion binding.Potassium ion binding.Pyruvate kinase activity. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02726; DB00787; DB04551; DB16236; DB00119 Interacts with Q9UBL6-2 EC number 2.7.1.40 Uniprot keywords 3D-structure; Allosteric enzyme; Alternative splicing; ATP-binding; Disease variant; Glycolysis; Hereditary hemolytic anemia; Kinase; Magnesium; Manganese; Metal-binding; Nucleotide-binding; Phosphoprotein; Potassium; Proteomics identification; Pyruvate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 45695.1 Length 421 Aromaticity 0.06 Instability index 34.44 Isoelectric point 6.88 Charge (pH=7) -0.35 2D Binding mode Binding energy (Kcal/mol) -5.97  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GTAFFQQQQLPAAMADTFLEHLCLLDIDSEPVAARSTSIIATIGPASRSVERLKEMIKAGMNIARLNFSHGSHEYHAESIANVREAVESFSPLSYRPVAIALDTKGPEIGLSEQDVRDLRFGVEHGVDIVFASFVRKASDVAAVRAALGPEGHGIKIISKIENHEGVKRFDEILEVSDGIMVARGDLGIEIPAEKVFLAQKMMIGRCNLAGKPVVCATQMLESMITKPRPTRAETSDVANAVLDGADCIMLSGETAKGNFPVEAVKMQHAIAREAEAAVYHRQLFEELRRAAPLSRDPTEVTAIGAVEAAFKCCAAAIIVLTTTGRSAQLLSRYRPRAAVIAVTRSAQAARQVHLCRGVFPLLYREPPEAIWADDVDRRVQFGIESGKLRGFLRVGDLVIVVTGWRPGSGYTNIMRVLSIS Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Low molecular weight phosphotyrosine protein phosphatase | 5KQL | 4.38 | |
Target general information Gen name ACP1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Low molecular weight phosphotyrosine protein phosphatase family Biochemical class hydrolase / hydrolase inhibitor Function Acid phosphatase activity.Non-membrane spanning protein tyrosine phosphatase activity. Related diseases Waardenburg syndrome 4A (WS4A) [MIM:277580]: A disorder characterized by the association of Waardenburg features (depigmentation and deafness) with the absence of enteric ganglia in the distal part of the intestine (Hirschsprung disease). {ECO:0000269|PubMed:12189494, ECO:0000269|PubMed:8634719}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hirschsprung disease 2 (HSCR2) [MIM:600155]: A disorder of neural crest development characterized by absence of enteric ganglia along a variable length of the intestine. It is the most common cause of congenital intestinal obstruction. Early symptoms range from complete acute neonatal obstruction, characterized by vomiting, abdominal distention and failure to pass stool, to chronic constipation in the older child. {ECO:0000269|PubMed:11471546, ECO:0000269|PubMed:28236341, ECO:0000269|PubMed:8001158, ECO:0000269|PubMed:8630503, ECO:0000269|PubMed:8852660}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: ABCD syndrome (ABCDS) [MIM:600501]: An autosomal recessive syndrome characterized by albinism, black lock at temporal occipital region, bilateral deafness, aganglionosis of the large intestine and total absence of neurocytes and nerve fibers in the small intestine. {ECO:0000269|PubMed:11891690}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Heterozygous mutations in EDNRB may be responsible for Waardenburg syndrome 2, an autosomal dominant disorder characterized by sensorineural deafness and pigmentary disturbances. {ECO:0000269|PubMed:28236341}. Drugs (DrugBank ID) DB04214; DB00173 Interacts with Q96CV9 EC number 3.1.3.2; 3.1.3.48 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Hydrolase; Phosphoprotein; Protein phosphatase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 17582.8 Length 154 Aromaticity 0.09 Instability index 50.52 Isoelectric point 7 Charge (pH=7) -0 2D Binding mode Binding energy (Kcal/mol) -5.98  Molscript Map 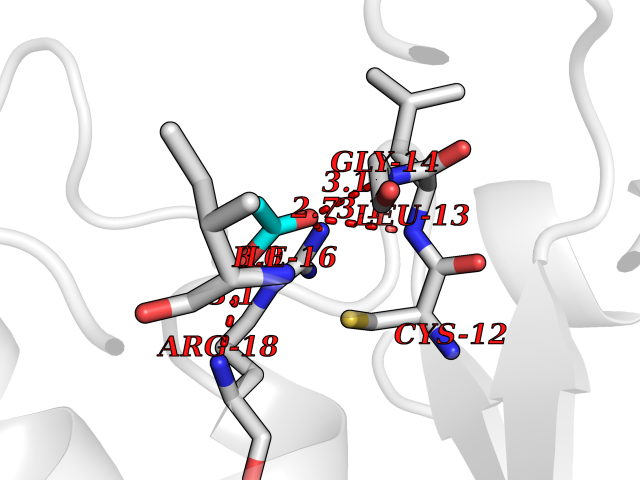 Pymol Map 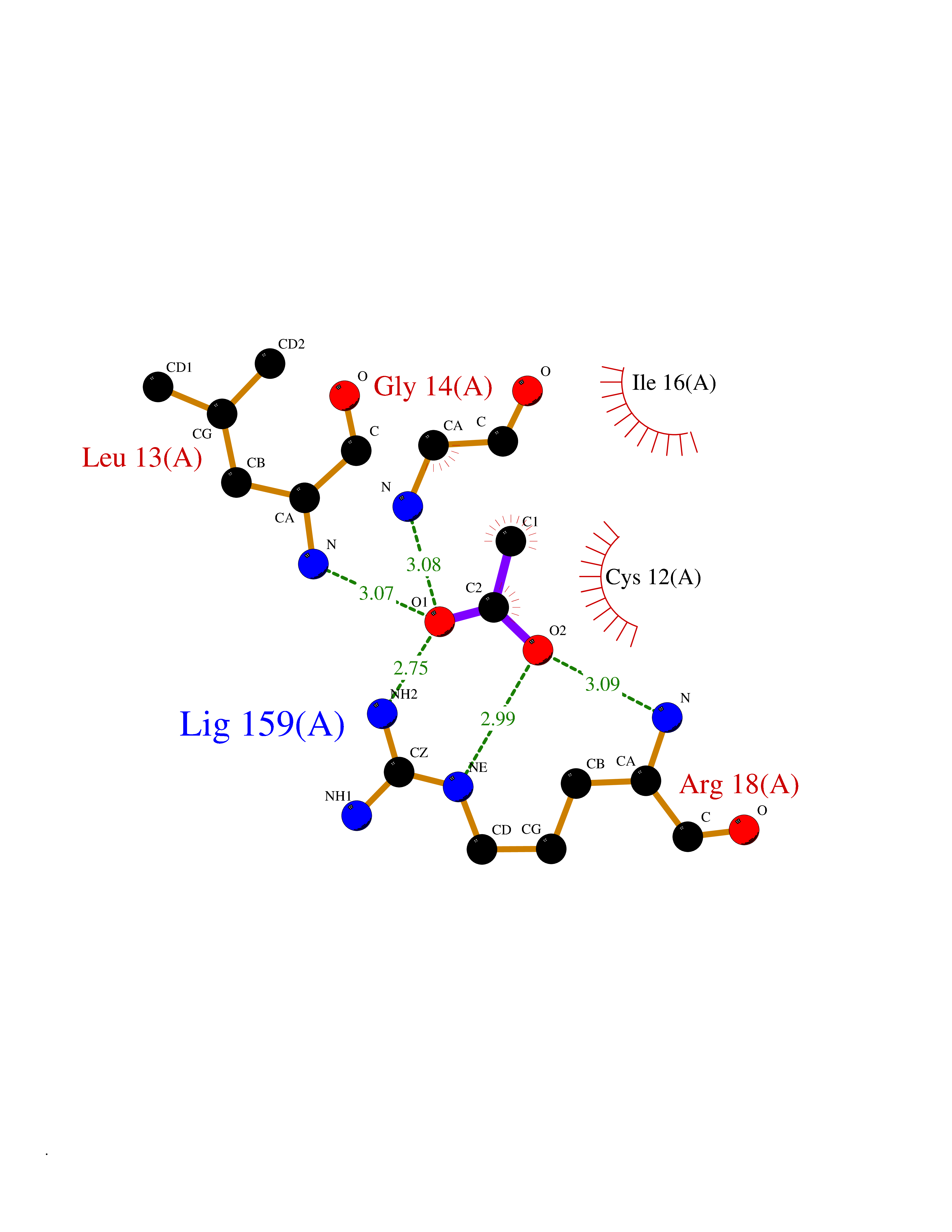 Ligplot Map 3D Binding mode Sequence ATKSVLFVCLGNICRSPIAEAVFRKLVTDQNISENWRVDSAATSGYEIGNPPDYRGQSCMKRHGIPMSHVARQITKEDFATFDYILCMDESNLRDLNRKSNQVKTCKAKIELLGSYDPQKQLIIEDPYYGNDSDFETVYQQCVRCCRAFLEKAH Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Cystathionine gamma-lyase (CTH) | 3COG | 4.38 | |
Target general information Gen name CTH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Gamma-cystathionase; Cysteine-protein sulfhydrase Protein family Trans-sulfuration enzymes family Biochemical class NA Function Catalyzes the last step in the trans-sulfuration pathway from methionine to cysteine. Has broad substrate specificity. Converts cystathionine to cysteine, ammonia and 2-oxobutanoate. Converts two cysteine molecules to lanthionine and hydrogen sulfide. Can also accept homocysteine as substrate. Specificity depends on the levels of the endogenous substrates. Generates the endogenous signaling molecule hydrogen sulfide (H2S), and so contributes to the regulation of blood pressure. Acts as a cysteine-protein sulfhydrase by mediating sulfhydration of target proteins: sulfhydration consists of converting -SH groups into -SSH on specific cysteine residues of target proteins such as GAPDH, PTPN1 and NF-kappa-B subunit RELA, thereby regulating their function. Related diseases Cystathioninuria (CSTNU) [MIM:219500]: Autosomal recessive phenotype characterized by abnormal accumulation of plasma cystathionine, leading to increased urinary excretion. {ECO:0000269|PubMed:12574942, ECO:0000269|PubMed:18476726}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02328; DB03928; DB00151; DB04217; DB00114 Interacts with P32929; Q96NT3; Q96NT3-2; Q96HA8; Q6P9E2 EC number EC 4.4.1.1 Uniprot keywords 3D-structure; Alternative splicing; Amino-acid biosynthesis; Calmodulin-binding; Cysteine biosynthesis; Cytoplasm; Disease variant; Lipid metabolism; Lyase; Proteomics identification; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 86026 Length 782 Aromaticity 0.08 Instability index 32.4 Isoelectric point 6.27 Charge (pH=7) -9.46 2D Binding mode Binding energy (Kcal/mol) -5.98  Molscript Map 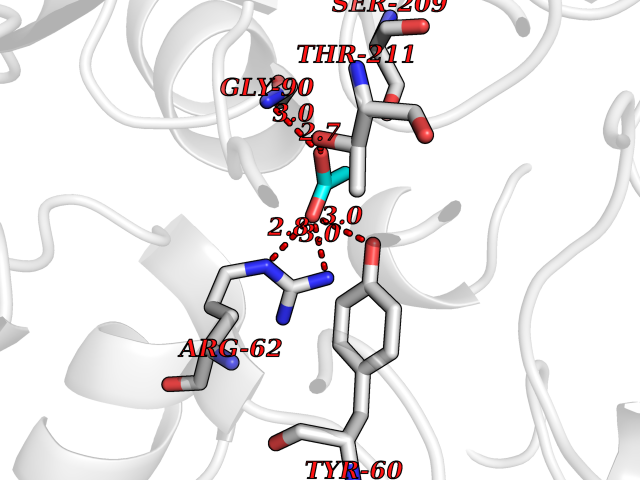 Pymol Map 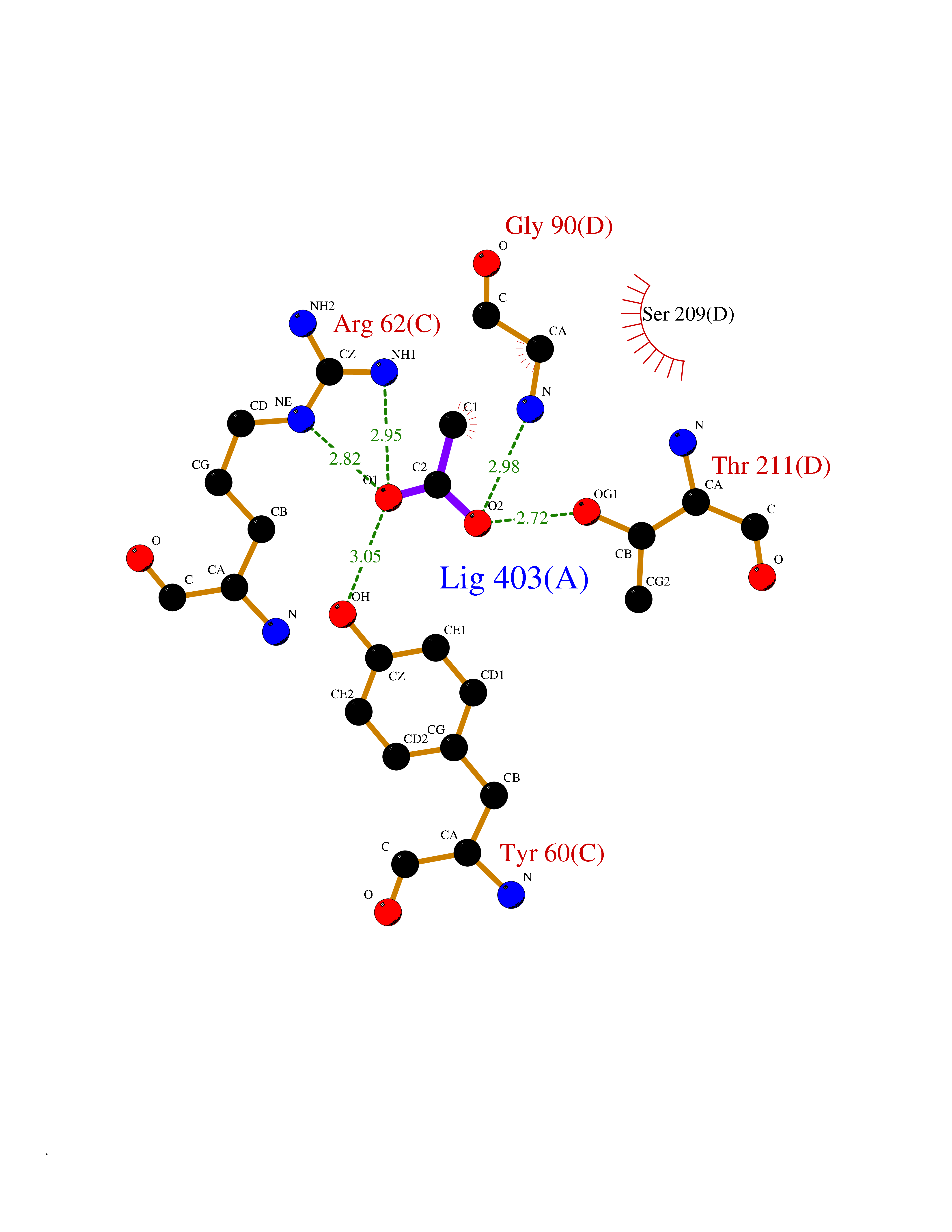 Ligplot Map 3D Binding mode Sequence GFLPHFQHFATQAIHVGQDPEQWTSRAVVPPISLSTTFKQGAPGQHSGFEYSRSGNPTRNCLEKAVAALDGAKYCLAFASGLAATVTITHLLKAGDQIICMDDVYGGTNRYFRQVASEFGLKISFVDCSKIKLLEAAITPETKLVWIETPTNPTQKVIDIEGCAHIVHKHGDIILVVDNTFMSPYFQRPLALGADISMYSATKYMNGHSDVVMGLVSVNCESLHNRLRFLQNSLGAVPSPIDCYLCNRGLKTLHVRMEKHFKNGMAVAQFLESNPWVEKVIYPGLPSHPQHELVKRQCTGCTGMVTFYIKGTLQHAEIFLKNLKLFTLAESLGGFESLAELPAIMTHASVLKNDRDVLGISDTLIRLSVGLEDEEDLLEDLDQALKAAHPPSGFLPHFQHFATQAIHVGQDPEQWTSRAVVPPISLSTTFKQGAPGQGFEYSRSGNPTRNCLEKAVAALDGAKYCLAFASGLAATVTITHLLKAGDQIICMDDVYGGTNRYFRQVASEFGLKISFVDCSKIKLLEAAITPETKLVWIETPTNPTQKVIDIEGCAHIVHKHGDIILVVDNTFMSPYFQRPLALGADISMYSATKYMNGHSDVVMGLVSVNCESLHNRLRFLQNSLGAVPSPIDCYLCNRGLKTLHVRMEKHFKNGMAVAQFLESNPWVEKVIYPGLPSHPQHELVKRQCTGCTGMVTFYIKGTLQHAEIFLKNLKLFTLAESLGGFESLAELPAIMTHASVLKNDRDVLGISDTLIRLSVGLEDEEDLLEDLDQALKAAHPPS Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | 2-hydroxy-6-oxo-7-methylocta-2,4-dienoate hydrolase | 1UK8 | 4.38 | |
Target general information Gen name cumD Organism Pseudomonas fluorescens Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function Hydrolase activity. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03741; DB03793; DB03568; DB02531; DB03750; DB02406; DB03766 Interacts with NA EC number NA Uniprot keywords 3D-structure; Hydrolase Protein physicochemical properties Chain ID A Molecular weight (Da) 30307.9 Length 271 Aromaticity 0.1 Instability index 37.49 Isoelectric point 5.02 Charge (pH=7) -11.58 2D Binding mode Binding energy (Kcal/mol) -5.97  Molscript Map 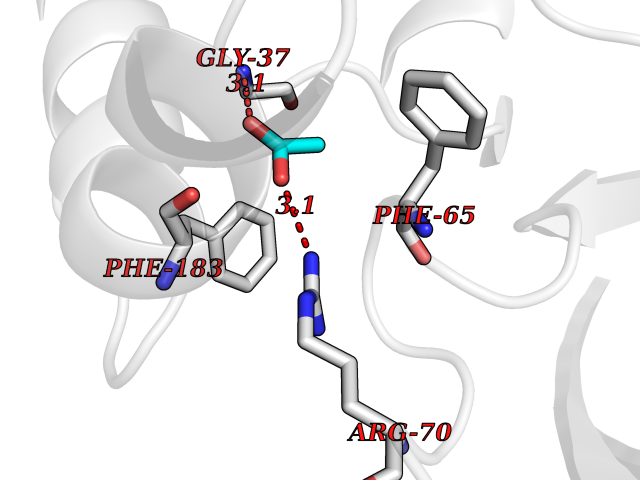 Pymol Map 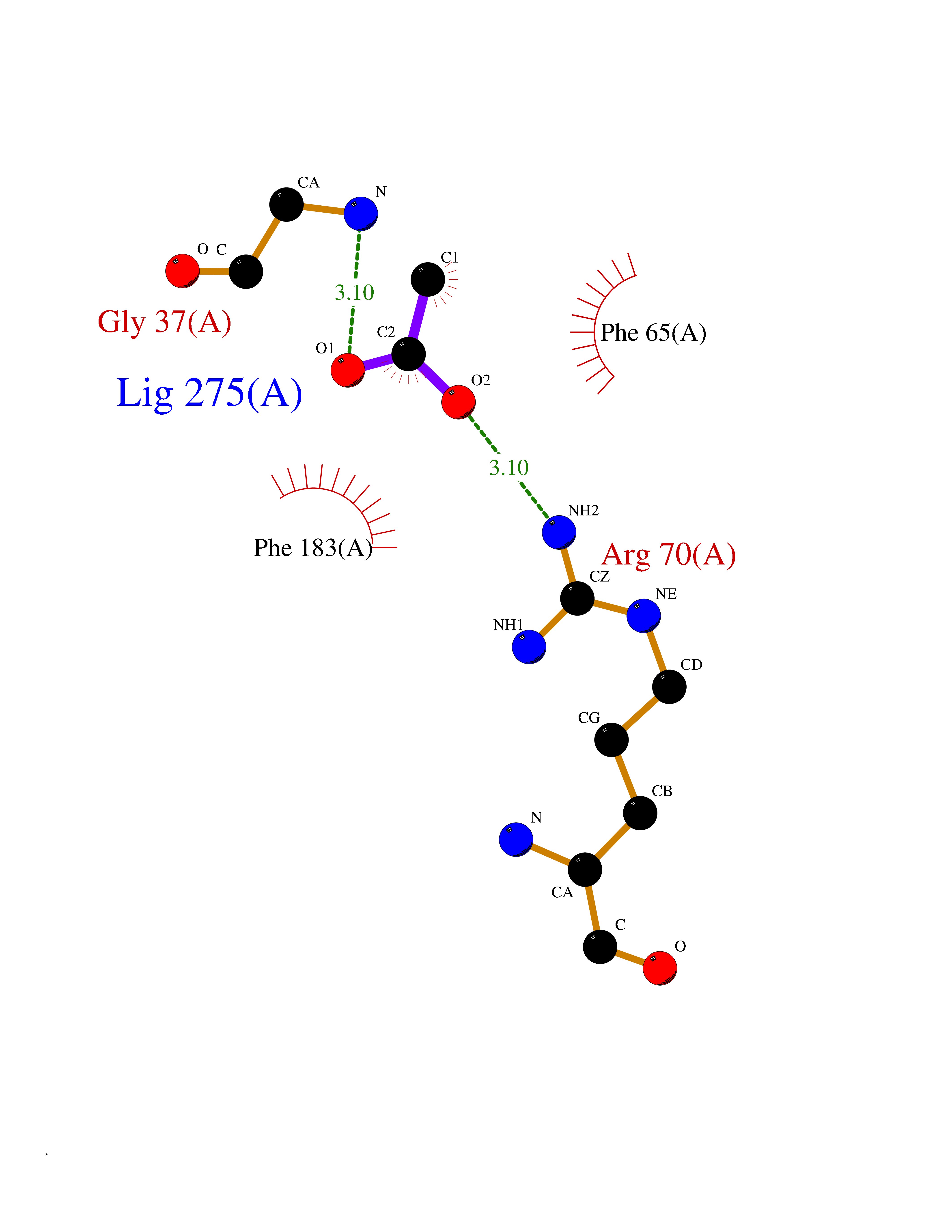 Ligplot Map 3D Binding mode Sequence NLEIGKSILAAGVLTNYHDVGEGQPVILIHGSGPGVSAYANWRLTIPALSKFYRVIAPDMVGFGFTDRPENYNYSKDSWVDHIIGIMDALEIEKAHIVGNAFGGGLAIATALRYSERVDRMVLMGAAGTRFDVTEGLNAVWGYTPSIENMRNLLDIFAYDRSLVTDELARLRYEASIQPGFQESFSSMFPEPRQRWIDALASSDEDIKTLPNETLIIHGREDQVVPLSSSLRLGELIDRAQLHVFGRCGHWTQIEQTDRFNRLVVEFFNEA Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | 2-hydroxy-6-oxo-7-methylocta-2,4-dienoate hydrolase | 1UK8 | 4.38 | |
Target general information Gen name cumD Organism Pseudomonas fluorescens Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function Hydrolase activity. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03741; DB03793; DB03568; DB02531; DB03750; DB02406; DB03766 Interacts with NA EC number NA Uniprot keywords 3D-structure; Hydrolase Protein physicochemical properties Chain ID A Molecular weight (Da) 30307.9 Length 271 Aromaticity 0.1 Instability index 37.49 Isoelectric point 5.02 Charge (pH=7) -11.58 2D Binding mode Binding energy (Kcal/mol) -5.97  Molscript Map 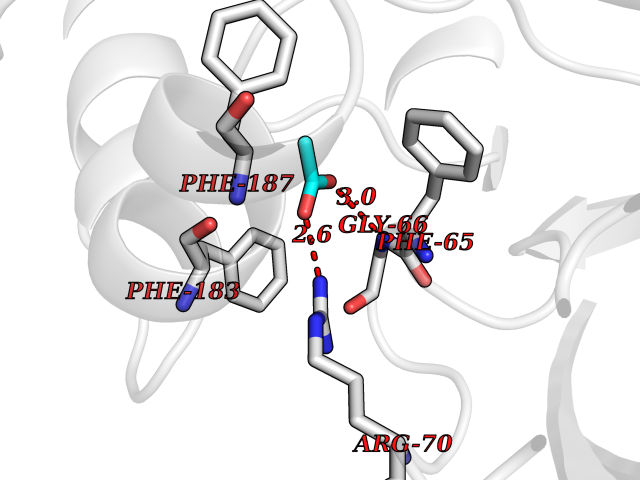 Pymol Map 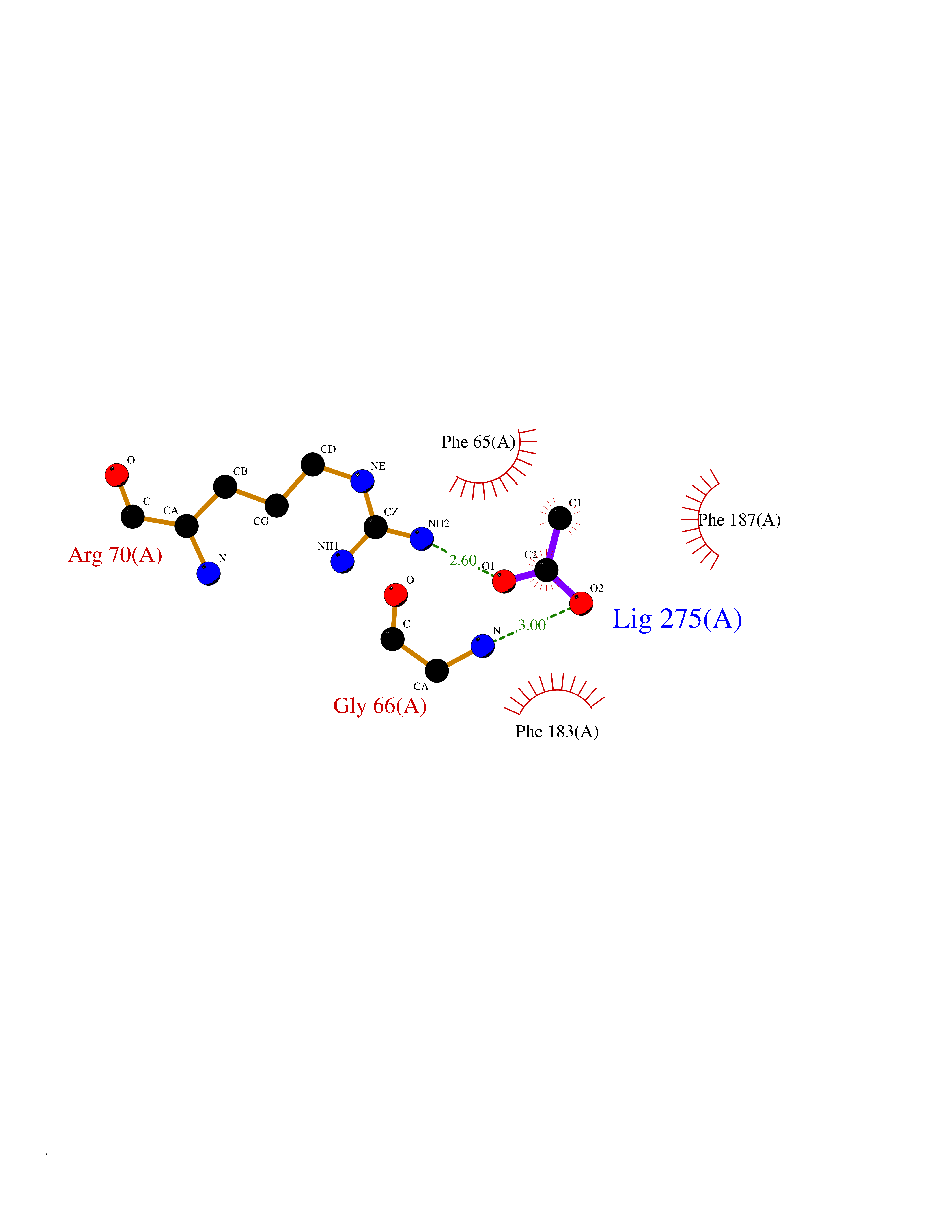 Ligplot Map 3D Binding mode Sequence NLEIGKSILAAGVLTNYHDVGEGQPVILIHGSGPGVSAYANWRLTIPALSKFYRVIAPDMVGFGFTDRPENYNYSKDSWVDHIIGIMDALEIEKAHIVGNAFGGGLAIATALRYSERVDRMVLMGAAGTRFDVTEGLNAVWGYTPSIENMRNLLDIFAYDRSLVTDELARLRYEASIQPGFQESFSSMFPEPRQRWIDALASSDEDIKTLPNETLIIHGREDQVVPLSSSLRLGELIDRAQLHVFGRCGHWTQIEQTDRFNRLVVEFFNEA Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Mycobacterium Serine/threonine-protein kinase PknB (MycB pknB) | 2FUM | 4.38 | |
Target general information Gen name MycB pknB Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID Synonyms pknB; Serine/threonineprotein kinase PknB Protein family Protein kinase superfamily, Ser/Thr protein kinase family Biochemical class NA Function Key component of a signal transduction pathway that regulates cell growth and cell division via phosphorylation of target proteins such as GarA, GlmU, PapA5, PbpA, FhaB (Rv0019c), FhaA(Rv0020c), MviN, PstP, EmbR, Rv1422, Rv1747 and RseA. Shows a strong preference for Thr versus Ser as the phosphoacceptor. Related diseases Immunodeficiency 67 (IMD67) [MIM:607676]: An autosomal recessive primary immunodeficiency characterized by recurrent, life-threatening systemic and invasive bacterial infections beginning in infancy or early childhood. {ECO:0000269|PubMed:12637671, ECO:0000269|PubMed:12925671, ECO:0000269|PubMed:16950813, ECO:0000269|PubMed:17878374, ECO:0000269|PubMed:19663824, ECO:0000269|PubMed:21057262, ECO:0000269|PubMed:24316379}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02930; DB03909; DB12010 Interacts with P71590; P9WJA9; P9WI81 EC number NA Uniprot keywords 3D-structure; Acetylation; ATP-binding; Cell membrane; Direct protein sequencing; Kinase; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Phosphoprotein; Reference proteome; Repeat; Serine/threonine-protein kinase; Transferase; Transmembrane; Transmembrane helix; Virulence Protein physicochemical properties Chain ID A Molecular weight (Da) 28718.1 Length 263 Aromaticity 0.06 Instability index 32.29 Isoelectric point 5.44 Charge (pH=7) -9.7 2D Binding mode Binding energy (Kcal/mol) -5.98  Molscript Map  Pymol Map 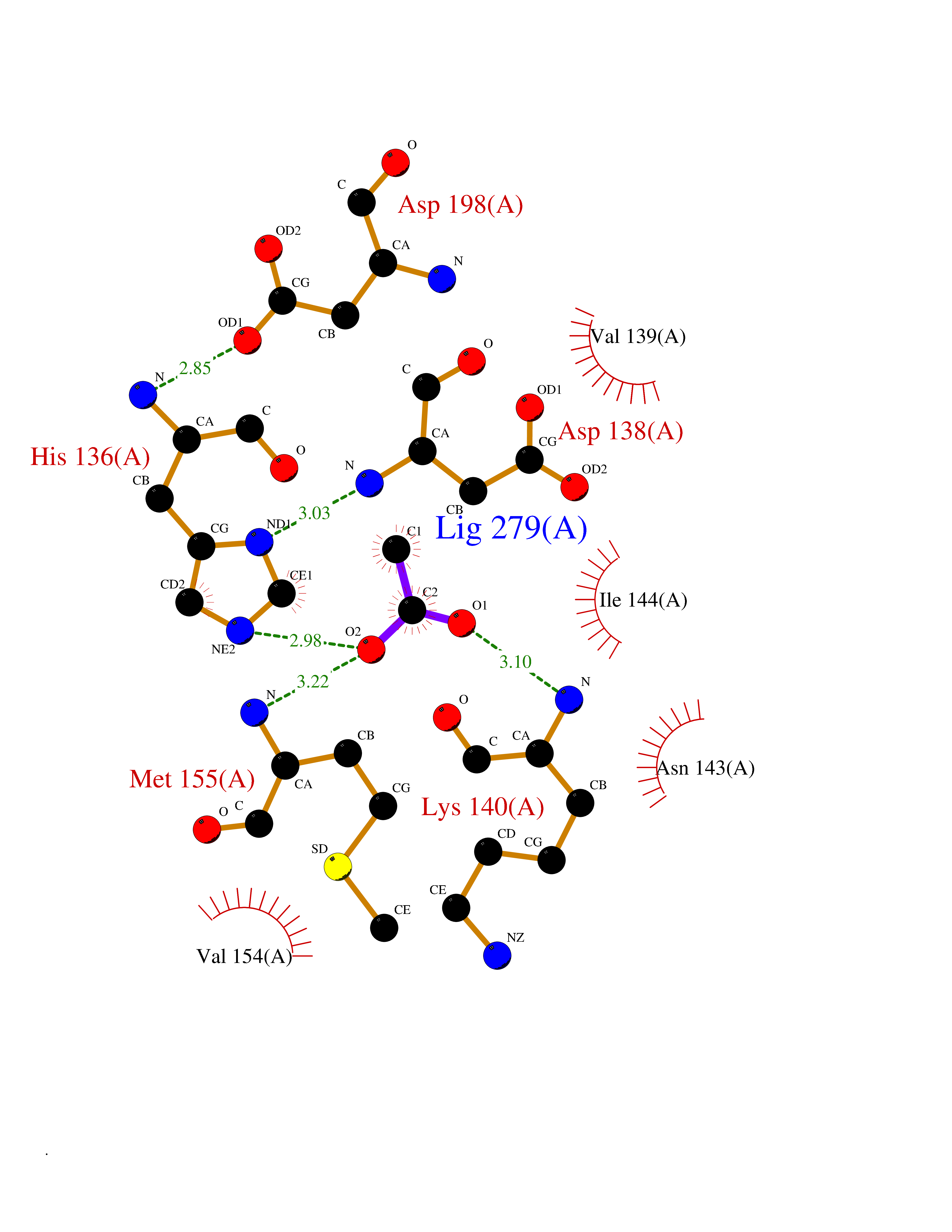 Ligplot Map 3D Binding mode Sequence TPSHLSDRYELGEILGFGGMSEVHLARDLRLHRDVAVKVLRADLARDPSFYLRFRREAQNAAALNHPAIVAVYDTGEAETPAGPLPYIVMEYVDGVTLRDIVHTEGPMTPKRAIEVIADACQALNFSHQNGIIHRDVKPANIMISATNAVKVMDFGIARAIADGTAQYLSPEQARGDSVDARSDVYSLGCVLYEVLTGEPPFTGDSPVSVAYQHVREDPIPPSARHEGLSADLDAVVLKALAKNPENRYQTAAEMRADLVRVH Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Neuronal acetylcholine receptor alpha-4 (CHRNA4) | 6CNJ | 4.38 | |
Target general information Gen name CHRNA4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nicotinic acetylcholine receptor alpha4; CHRNA4; Alpha-4 nAChR Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-4/CHRNA4 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasmamembrane permeable to sodium ions. Related diseases Epilepsy, nocturnal frontal lobe, 1 (ENFL1) [MIM:600513]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:10563623, ECO:0000269|PubMed:14623738, ECO:0000269|PubMed:7550350}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01351; DB01352; DB00572; DB01483; DB00237; DB00241; DB01353; DB00564; DB00565; DB09028; DB01245; DB00514; DB01496; DB07720; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00898; DB01354; DB01355; DB00753; DB00657; DB00333; DB00463; DB00849; DB00184; DB00312; DB01174; DB00981; DB05458; DB00794; DB05740; DB00747; DB00418; DB00202; DB00306; DB00599; DB01273 Interacts with Q6UY14-3; P05067; P83916; Q6UXH1-1; Q6UXH1-3; P20042; Q9NZR2; Q92673; P17787 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84601.2 Length 728 Aromaticity 0.13 Instability index 39.72 Isoelectric point 5.86 Charge (pH=7) -9.84 2D Binding mode Binding energy (Kcal/mol) -5.97  Molscript Map 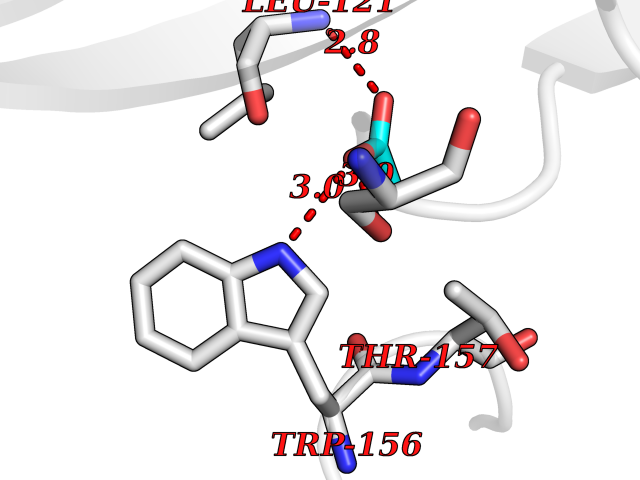 Pymol Map 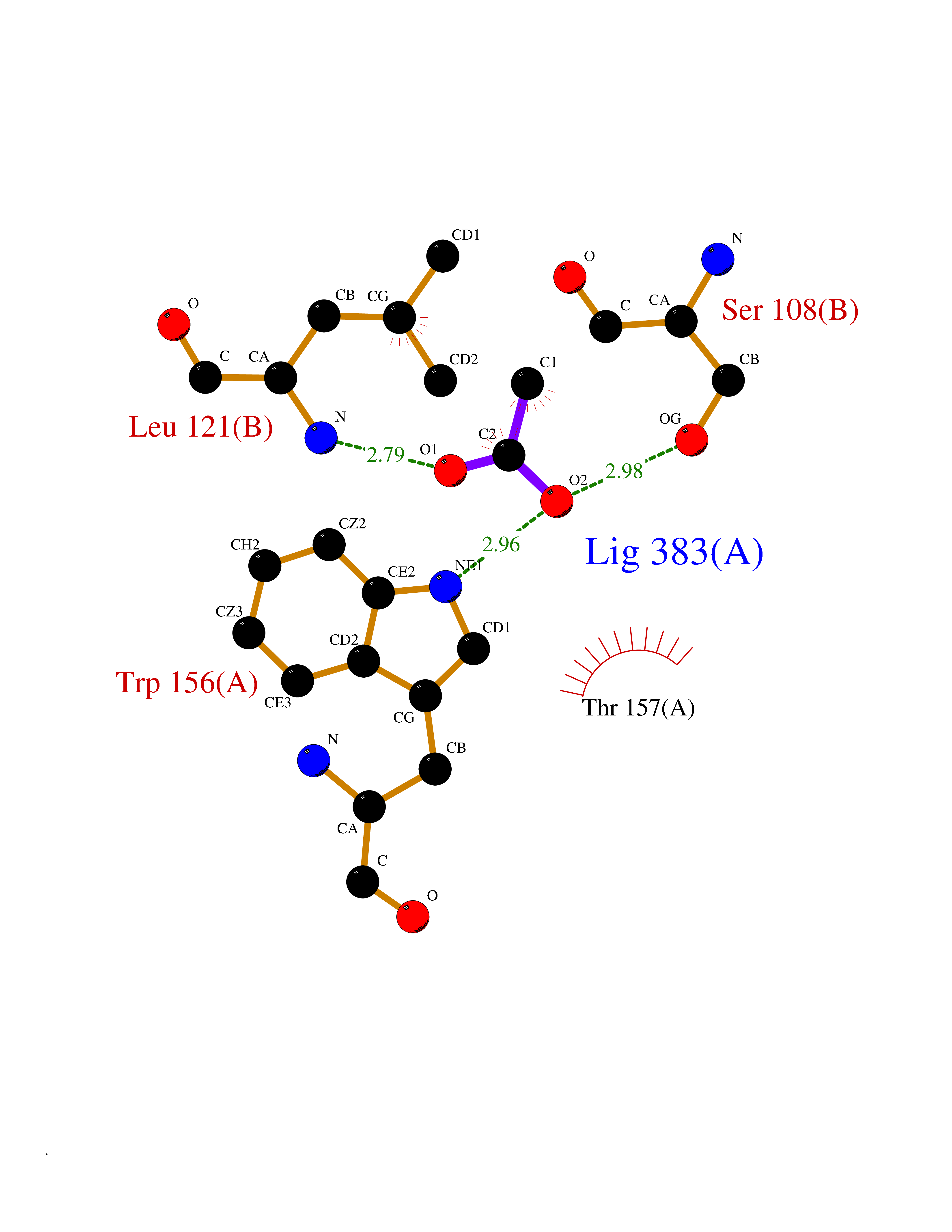 Ligplot Map 3D Binding mode Sequence ETRAHAEERLLKKLFSGYNKWSRPVANISDVVLVRFGLSIAQLIDVDEKNQMMTTNVWVKQEWHDYKLRWDPADYENVTSIRIPSELIWRPDIVLYNNADGDFAVTHLTKAHLFHDGRVQWTPPAIYKSSCSIDVTFFPFDQQNCTMKFGSWTYDKAKIDLVNMHSRVDQLDFWESGEWVIVDAVGTYNTRKYECCAEIYPDITYAFVIRRLPLFYTINLIIPCLLISCLTVLVFYLPSECGEKITLCISVLLSLTVFLLLITEIIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHHRSPRTHTMPTWVRRVFLDIVPRLLLMKRFERSVKEDWKYVAMVIDRIFLWMFIIVCLLGTVGLFLPPWDTEERLVEHLLDPSRYNKLIRPATNGSELVTVQLMVSLAQLISVHEREQIMTTNVWLTQEWEDYRLTWKPEEFDNMKKVRLPSKHIWLPDVVLYNNADGMYEVSFYSNAVVSYDGSIFWLPPAIYKSACKIEVKHFPFDQQNCTMKFRSWTYDRTEIDLVLKSEVASLDDFTPSGEWDIVALPGRRNENPDDSTYVDITYDFIIRRKPLFYTINLIIPCVLITSLAILVFYLPSDCGEKMTLCISVLLALTVFLLLISKIVPPTSLDVPLVGKYLMFTMVLVTFSIVTSVCVLNVHHRSPTTHTMAPWVKVVFLEKLPALLFMQQSVSEDWKYVAMVIDRLFLWIFVFVCVFGTIGMF Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | RAC-beta serine/threonine-protein kinase (AKT2) | 3D0E | 4.38 | |
Target general information Gen name AKT2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAC-PK-beta; Protein kinase B beta; Protein kinase Akt-2; PKB beta Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family, RAC subfamily Biochemical class Kinase Function AKT2 is one of 3 closely related serine/threonine-protein kinases (AKT1, AKT2 and AKT3) called the AKT kinase, and which regulate many processes including metabolism, proliferation, cell survival, growth and angiogenesis. This is mediated through serine and/or threonine phosphorylation of a range of downstream substrates. Over 100 substrate candidates have been reported so far, but for most of them, no isoform specificity has been reported. AKT is responsible of the regulation of glucose uptake by mediating insulin-induced translocation of the SLC2A4/GLUT4 glucose transporter to the cell surface. Phosphorylation of PTPN1 at 'Ser-50' negatively modulates its phosphatase activity preventing dephosphorylation of the insulin receptor and the attenuation of insulin signaling. Phosphorylation of TBC1D4 triggers the binding of this effector to inhibitory 14-3-3 proteins, which is required for insulin-stimulated glucose transport. AKT regulates also the storage of glucose in the form of glycogen by phosphorylating GSK3A at 'Ser-21' and GSK3B at 'Ser-9', resulting in inhibition of its kinase activity. Phosphorylation of GSK3 isoforms by AKT is also thought to be one mechanism by which cell proliferation is driven. AKT regulates also cell survival via the phosphorylation of MAP3K5 (apoptosis signal-related kinase). Phosphorylation of 'Ser-83' decreases MAP3K5 kinase activity stimulated by oxidative stress and thereby prevents apoptosis. AKT mediates insulin-stimulated protein synthesis by phosphorylating TSC2 at 'Ser-939' and 'Thr-1462', thereby activating mTORC1 signaling and leading to both phosphorylation of 4E-BP1 and in activation of RPS6KB1. AKT is involved in the phosphorylation of members of the FOXO factors (Forkhead family of transcription factors), leading to binding of 14-3-3 proteins and cytoplasmic localization. In particular, FOXO1 is phosphorylated at 'Thr-24', 'Ser-256' and 'Ser-319'. FOXO3 and FOXO4 are phosphorylated on equivalent sites. AKT has an important role in the regulation of NF-kappa-B-dependent gene transcription and positively regulates the activity of CREB1 (cyclic AMP (cAMP)-response element binding protein). The phosphorylation of CREB1 induces the binding of accessory proteins that are necessary for the transcription of pro-survival genes such as BCL2 and MCL1. AKT phosphorylates 'Ser-454' on ATP citrate lyase (ACLY), thereby potentially regulating ACLY activity and fatty acid synthesis. Activates the 3B isoform of cyclic nucleotide phosphodiesterase (PDE3B) via phosphorylation of 'Ser-273', resulting in reduced cyclic AMP levels and inhibition of lipolysis. Phosphorylates PIKFYVE on 'Ser-318', which results in increased PI(3)P-5 activity. The Rho GTPase-activating protein DLC1 is another substrate and its phosphorylation is implicated in the regulation cell proliferation and cell growth. AKT plays a role as key modulator of the AKT-mTOR signaling pathway controlling the tempo of the process of newborn neurons integration during adult neurogenesis, including correct neuron positioning, dendritic development and synapse formation. Signals downstream of phosphatidylinositol 3-kinase (PI(3)K) to mediate the effects of various growth factors such as platelet-derived growth factor (PDGF), epidermal growth factor (EGF), insulin and insulin-like growth factor I (IGF-I). AKT mediates the antiapoptotic effects of IGF-I. Essential for the SPATA13-mediated regulation of cell migration and adhesion assembly and disassembly. May be involved in the regulation of the placental development. Related diseases Defects in AKT2 are a cause of susceptibility to breast cancer (BC). AKT2 promotes metastasis of tumor cells without affecting the latency of tumor development. May play a role in glioblastoma cell survival (PubMed:20167810). {ECO:0000269|PubMed:20167810}.; DISEASE: Type 2 diabetes mellitus (T2D) [MIM:125853]: A multifactorial disorder of glucose homeostasis caused by a lack of sensitivity to insulin. Affected individuals usually have an obese body habitus and manifestations of a metabolic syndrome characterized by diabetes, insulin resistance, hypertension and hypertriglyceridemia. The disease results in long-term complications that affect the eyes, kidneys, nerves, and blood vessels. {ECO:0000269|PubMed:15166380, ECO:0000269|PubMed:19164855}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Hypoinsulinemic hypoglycemia with hemihypertrophy (HIHGHH) [MIM:240900]: A disorder characterized by hypoglycemia, low insulin levels, low serum levels of ketone bodies and branched-chain amino acids, left-sided hemihypertrophy, neonatal macrosomia, reduced consciousness and hypoglycemic seizures. {ECO:0000269|PubMed:21979934}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08073; DB07859; DB12218; DB07947; DB07812 Interacts with P31749; P49841; P08238; Q6FHY5; Q9NRD5; Q04864-2; O60504; P53804; Q9C0C9; P08670; Q15118-1 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Apoptosis; ATP-binding; Carbohydrate metabolism; Cell membrane; Cytoplasm; Developmental protein; Diabetes mellitus; Disease variant; Disulfide bond; Endosome; Glucose metabolism; Glycogen biosynthesis; Glycogen metabolism; Glycoprotein; Kinase; Manganese; Membrane; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Reference proteome; Serine/threonine-protein kinase; Sugar transport; Transferase; Translation regulation; Transport; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 37380.5 Length 324 Aromaticity 0.12 Instability index 29.68 Isoelectric point 6.19 Charge (pH=7) -3.43 2D Binding mode Binding energy (Kcal/mol) -5.97 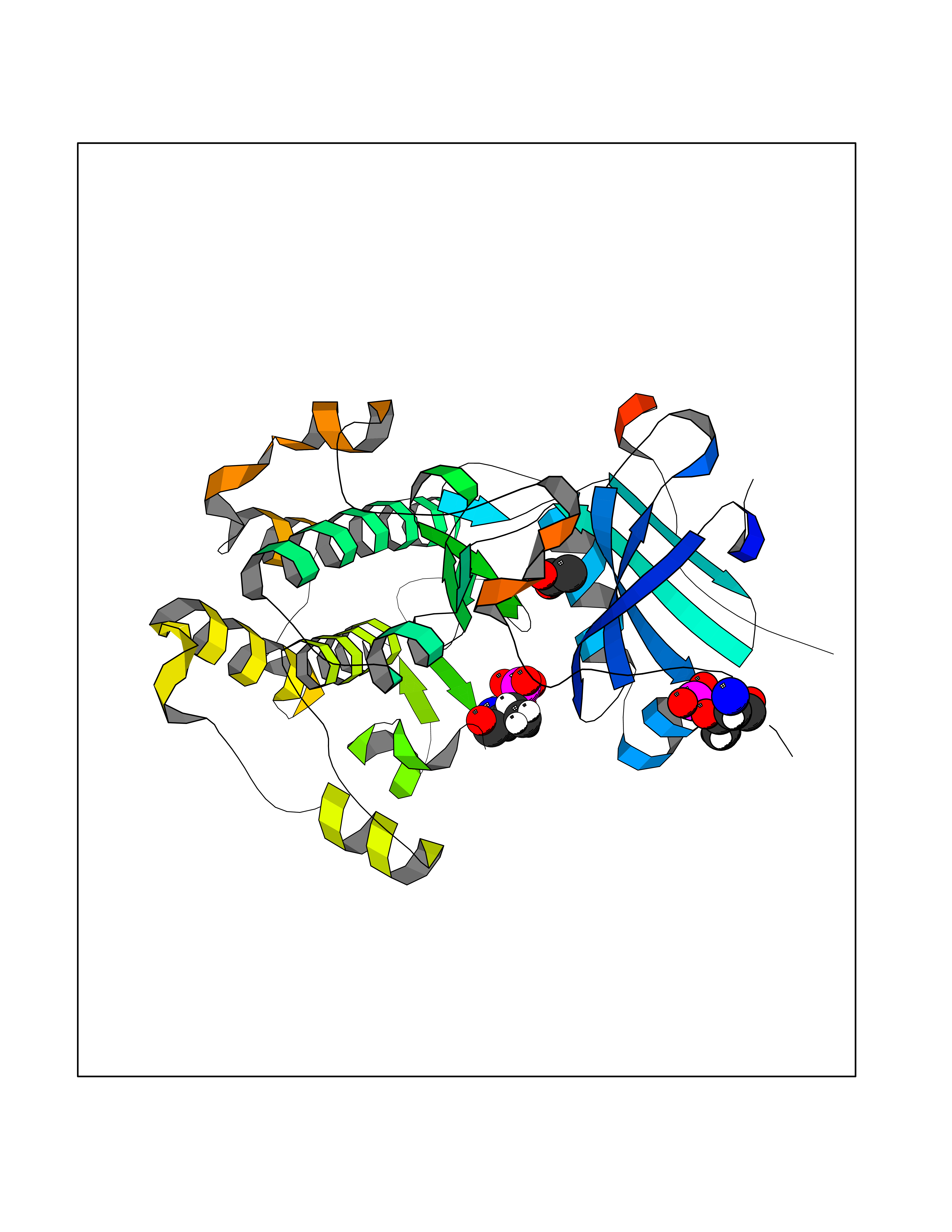 Molscript Map 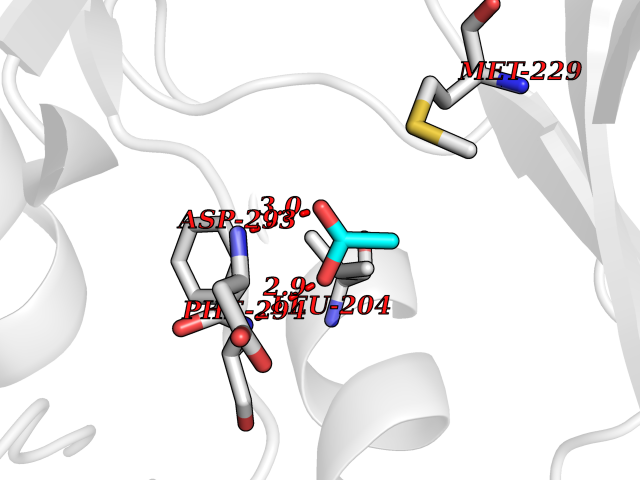 Pymol Map 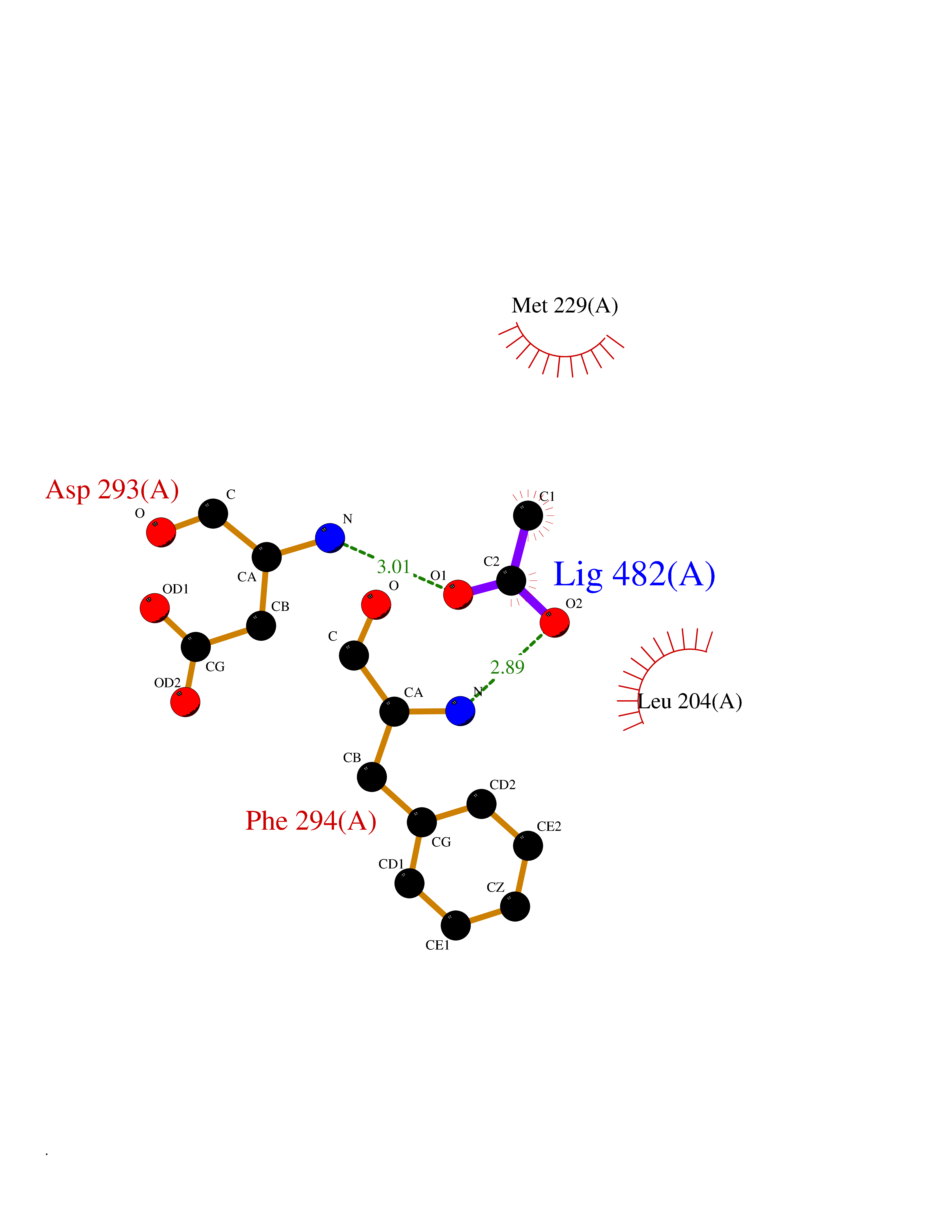 Ligplot Map 3D Binding mode Sequence KVTMNDFDYLKLLGKGTFGKVILVREKATGRYYAMKILRKEVIIAKDEVAHTVTESRVLQNTRHPFLTALKYAFQTHDRLCFVMEYANGGELFFHLSRERVFTEERARFYGAEIVSALEYLHSRDVVYRDIKLENLMLDKDGHIKITDFGLCKEGISDGATMKXFCGTPEYLAPEVLEDNDYGRAVDWWGLGVVMYEMMCGRLPFYNQDHERLFELILMEEIRFPRTLSPEAKSLLAGLLKKDPKQRLGGGPSDAKEVMEHRFFLSINWQDVVQKKLLPPFKPQVTSEVDTRYFDDEFTAQSITIXPPDQRTHFPQFDYSASIR Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | SF3b complex (SF3b) | 3LQV | 4.38 | |
Target general information Gen name SF3B1-SF3B2-SF3B3-SF3B4-SF3B5-SF3B6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NA Protein family SF3B1 family Biochemical class NA Function NA Related diseases Developmental and epileptic encephalopathy 24 (DEE24) [MIM:615871]: A disease characterized by early-onset seizures, intellectual disability of varying degrees, and behavioral disturbances or autistic features in most individuals. {ECO:0000269|PubMed:24747641, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Generalized epilepsy with febrile seizures plus 10 (GEFSP10) [MIM:618482]: An autosomal dominant neurologic disorder with incomplete penetrance, characterized by variable types of seizures including absence, tonic-clonic, febrile, focal, and eyelid myoclonia. Some patients have normal neurologic development. Others have mild-to-moderate intellectual disability or autism spectrum disorder. {ECO:0000269|PubMed:29936235, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14017 Interacts with Q13936; Q96I25; Q15428; Q9Y3B4; O43719; Q96I25; Q9Y3B4 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Citrullination; Isopeptide bond; mRNA processing; mRNA splicing; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; RNA-binding; Spliceosome; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 13222 Length 113 Aromaticity 0.12 Instability index 27.56 Isoelectric point 9.22 Charge (pH=7) 3.86 2D Binding mode Binding energy (Kcal/mol) -5.97 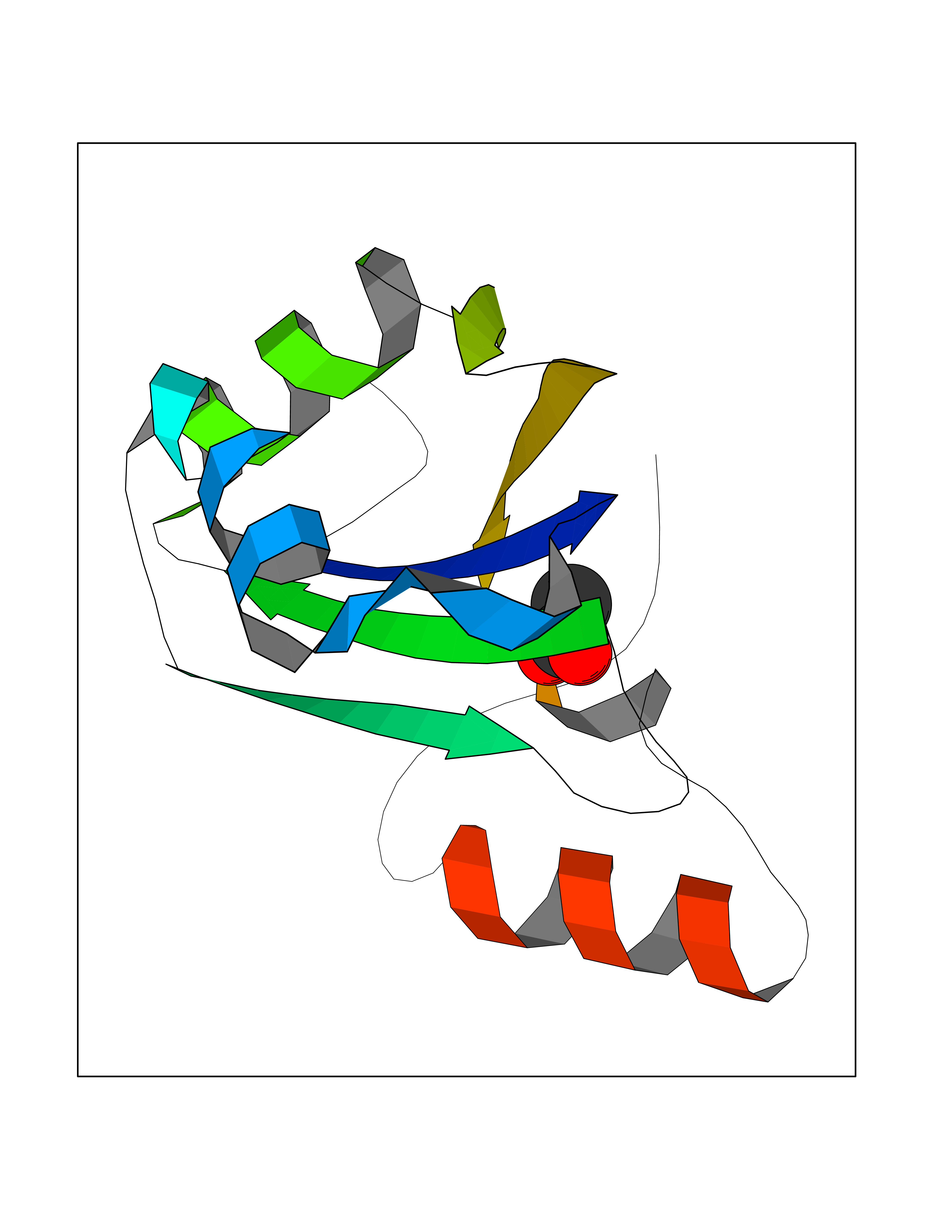 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LPPEVNRILYIRNLPYKITAEEMYDIFGKYGPIRQIRVGNTPETRGTAYVVYEDIFDAKNAVDHLSGFNVSNRYLVVLYYNANRAFQKMDTKKKEEQLKLLKEKYGINTDPPK Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Casein kinase I epsilon (CSNK1E) | 4HNI | 4.38 | |
Target general information Gen name CSNK1E Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Casein kinase I isoform epsilon; CKIe; CKI-epsilon Protein family Protein kinase superfamily, CK1 Ser/Thr protein kinase family, Casein kinase I subfamily Biochemical class Kinase Function Can phosphorylate a large number of proteins. Participates in Wnt signaling. Phosphorylates DVL1 and DVL2. Central component of the circadian clock. In balance with PP1, determines the circadian period length, through the regulation of the speed and rhythmicity of PER1 and PER2 phosphorylation. Controls PER1 and PER2 nuclear transport and degradation. Inhibits cytokine-induced granuloytic differentiation. Casein kinases are operationally defined by their preferential utilization of acidic proteins such as caseins as substrates. Related diseases Truncation of the 3'-untranslated (3'-UTR) region of CD274 transcripts leads to elevated expression of CD274 in multiple cancers including T-cell leukemia, diffuse large B-cell lymphoma and stomach adenocarcinoma (PubMed:27281199). Disruption of 3'-UTR region is caused by structural variants that stabilize CD274 transcripts, leading to overexpression (PubMed:27281199). Increased expression in tumors promotes immune evasion and tumor cell growth by allowing malignant cells to escape destruction by the immune system (PubMed:27281199). {ECO:0000269|PubMed:27281199}. Drugs (DrugBank ID) DB06195; DB14989 Interacts with P25054; O15169; O14640; O14641; Q92997; Q9BQ89; Q1W6H9; Q86UY5; P08238; P23508; Q00987; Q16625; O15055; O75382; P62258; Q04917; Q5T7W0; O70239; Q60838 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; ATP-binding; Biological rhythms; Cytoplasm; Kinase; Methylation; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 32897.6 Length 284 Aromaticity 0.13 Instability index 40.77 Isoelectric point 9.3 Charge (pH=7) 11.34 2D Binding mode Binding energy (Kcal/mol) -5.97  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LRVGNKYRLGRKIGSGSFGDIYLGANIASGEEVAIKLECVHIESKFYKMMQGGVGIPSIKWCGAEGDYNVMVMELLGPSLEDLFNFCSRKFSLKTVLLLADQMISRIEYIHSKNFIHRDVKPDNFLMGLGKKGNLVYIIDFGLAKKYRDARTHQHIPYRENKNLTGTARYASINTHLGIEQSRRDDLESLGYVLMYFNLGSLPWQGLKAATKRQKYERISEKKMSTPIEVLCKGYPSEFSTYLNFCRSLRFDDKPDYSYLRQLFRNLFHRQGFSYDYVFDWNML Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Glutamate receptor ionotropic kainate 1 (GRIK1) | 3FV2 | 4.37 | |
Target general information Gen name GRIK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Glutamate receptor 5; GluR5 kainate receptor; GluR5; GluR-5; GRIK1; Excitatory amino acid receptor 3; EAA3 Protein family Glutamate-gated ion channel (TC 1.A.10.1) family, GRIK1 subfamily Biochemical class Glutamate-gated ion channel Function Ionotropic glutamate receptor. L-glutamate acts as an excitatory neurotransmitter at many synapses in the central nervous system. Binding of the excitatory neurotransmitter L- glutamate induces a conformation change, leading tothe opening of the cation channel, and thereby converts the chemical signal to an electrical impulse. The receptor then desensitizes rapidly and enters a transient inactive state, characterized by the presence of bound agonist. May be involved in the transmission of light information from the retina to the hypothalamus. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB00237; DB00142; DB06354; DB00273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; RNA editing; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 29057.1 Length 256 Aromaticity 0.1 Instability index 24.38 Isoelectric point 8.3 Charge (pH=7) 1.97 2D Binding mode Binding energy (Kcal/mol) -5.95 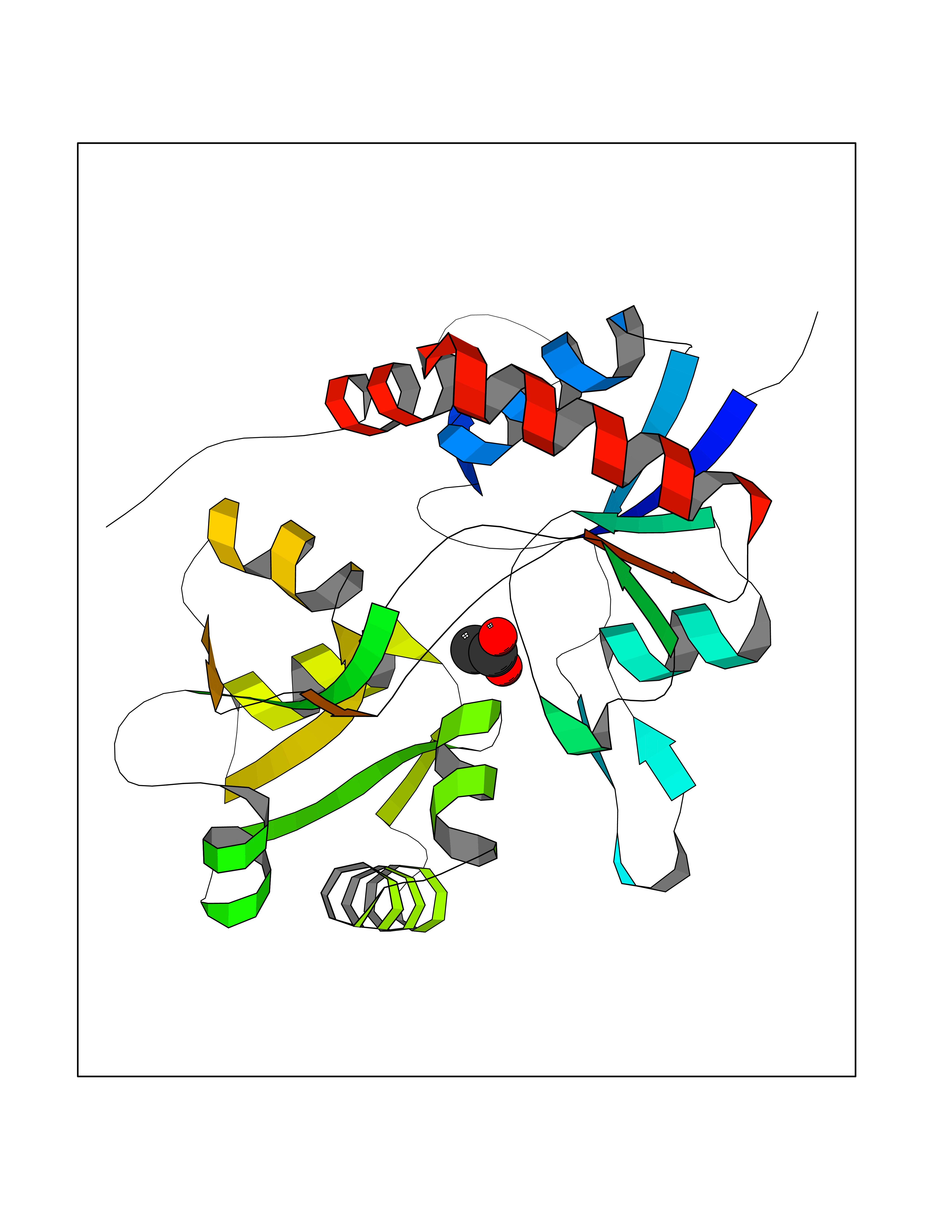 Molscript Map 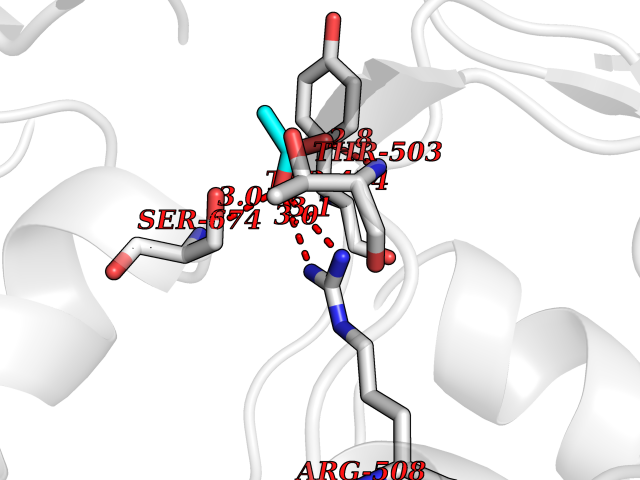 Pymol Map 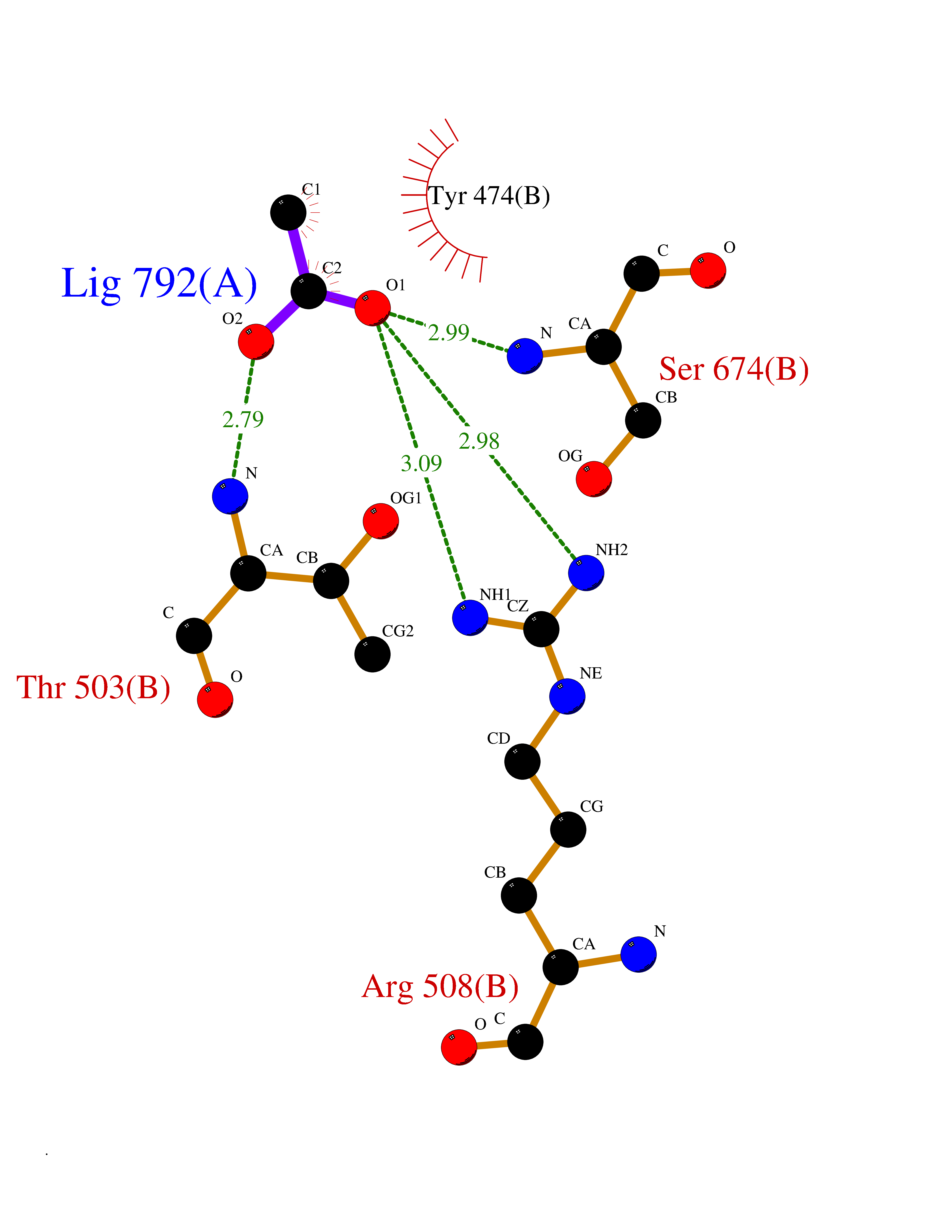 Ligplot Map 3D Binding mode Sequence ANRTLIVTTILEEPYVMYRKSDKPLYGNDRFEGYCLDLLKELSNILGFIYDVKLVPDGKYGAQNDKGEWNGMVKELIDHRADLAVAPLTITYVREKVIDFSKPFMTLGISILYRKGTPIDSADDLAKQTKIEYGAVRDGSTMTFFKKSKISTYEKMWAFMSSRQQTALVRNSDEGIQRVLTTDYALLMESTSIEYVTQRNCNLTQIGGLIDSKGYGVGTPIGSPYRDKITIAILQLQEEGKLHMMKEKWWRGNGCP Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Caspase-3 (CASP3) | 2XYG | 4.37 | |
Target general information Gen name CASP3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Yama protein; SREBP cleavage activity 1; SCA-1; Protein Yama; Cysteine protease CPP32; Caspase 3; CPP32; CPP-32; CASP-3; Apopain Protein family Peptidase C14A family Biochemical class Peptidase Function At the onset of apoptosis it proteolytically cleaves poly(ADP-ribose) polymerase (PARP) at a '216-Asp-|-Gly-217' bond. Cleaves and activates sterol regulatory element binding proteins (SREBPs) between the basic helix-loop-helix leucine zipper domain and the membrane attachment domain. Cleaves and activates caspase-6, -7 and -9. Involved in the cleavage of huntingtin. Triggers cell adhesion in sympathetic neurons through RET cleavage. Involved in the activation cascade of caspases responsible for apoptosis execution. Related diseases Smith-Kingsmore syndrome (SKS) [MIM:616638]: An autosomal dominant syndrome characterized by intellectual disability, macrocephaly, seizures, umbilical hernia, and facial dysmorphic features. {ECO:0000269|PubMed:25851998, ECO:0000269|PubMed:26542245, ECO:0000269|PubMed:27830187}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Focal cortical dysplasia 2 (FCORD2) [MIM:607341]: A form of focal cortical dysplasia, a malformation of cortical development that results in medically refractory epilepsy in the pediatric population and in adults. FCORD2 is a severe form, with onset usually in childhood, characterized by disrupted cortical lamination and specific cytological abnormalities. It is classified in 2 subtypes: type IIA characterized by dysmorphic neurons and lack of balloon cells; type IIB with dysmorphic neurons and balloon cells. {ECO:0000269|PubMed:25799227, ECO:0000269|PubMed:25878179, ECO:0000269|PubMed:26018084, ECO:0000269|PubMed:27830187}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08498; DB08497; DB08213; DB06862; DB08251; DB03124; DB08229; DB00945; DB05408; DB13751; DB06255; DB07696; DB01017; DB08499; DB12843; DB13048; DB00282; DB12709 Interacts with O43823; Q9Y243; P05067; P54252; P55212; P55211; Q14203-5; P42858; Q00987; O60551; P09874; Q5JUK2; P10599; Q9BYP7; P98170 EC number EC 3.4.22.56 Uniprot keywords 3D-structure; Acetylation; Apoptosis; Cytoplasm; Direct protein sequencing; Hydrolase; Phosphoprotein; Protease; Proteomics identification; Reference proteome; S-nitrosylation; Thiol protease; Ubl conjugation; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 27483.1 Length 239 Aromaticity 0.11 Instability index 38.09 Isoelectric point 8.39 Charge (pH=7) 3.03 2D Binding mode Binding energy (Kcal/mol) -5.96 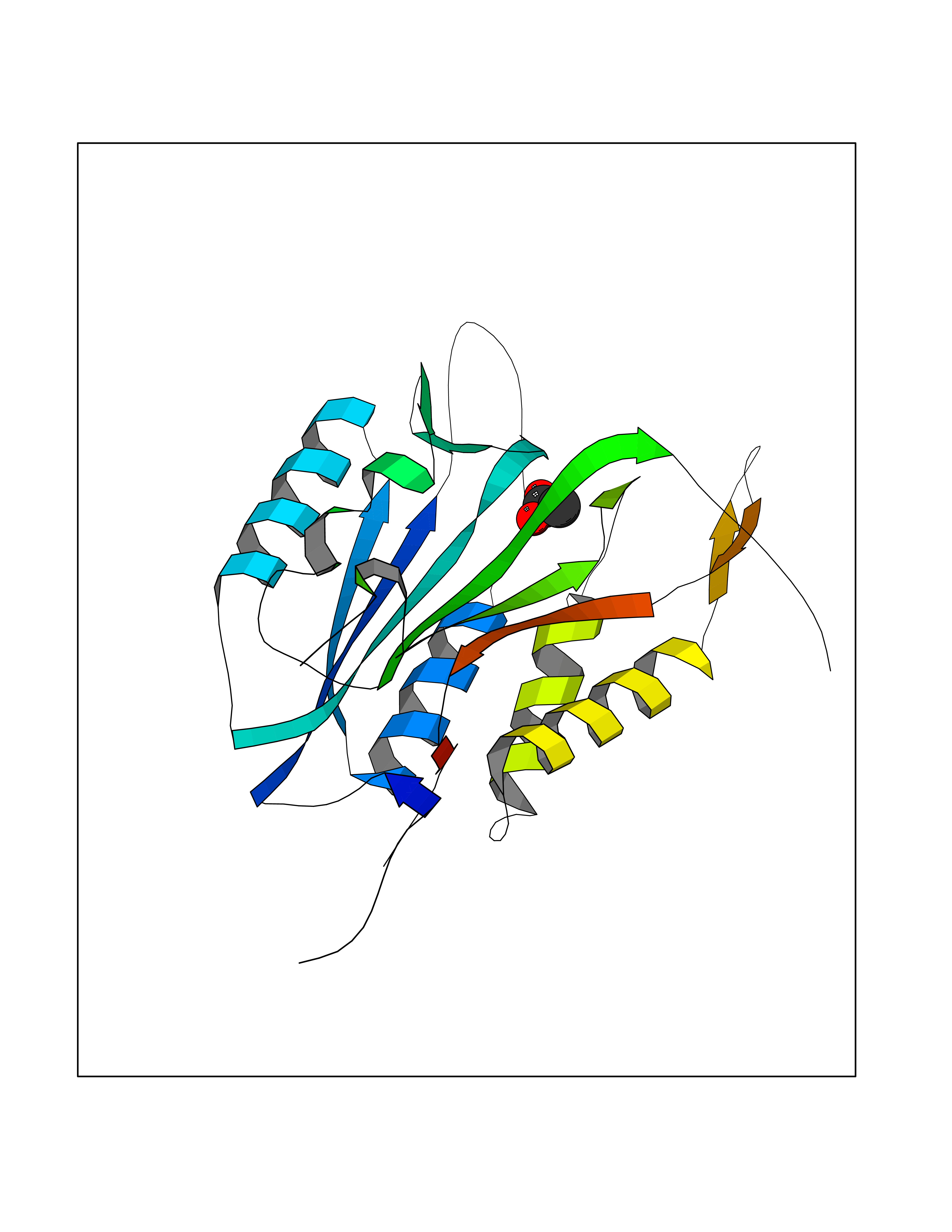 Molscript Map 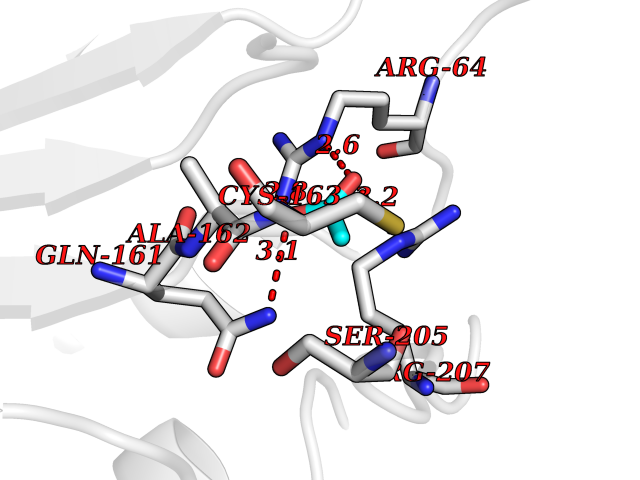 Pymol Map 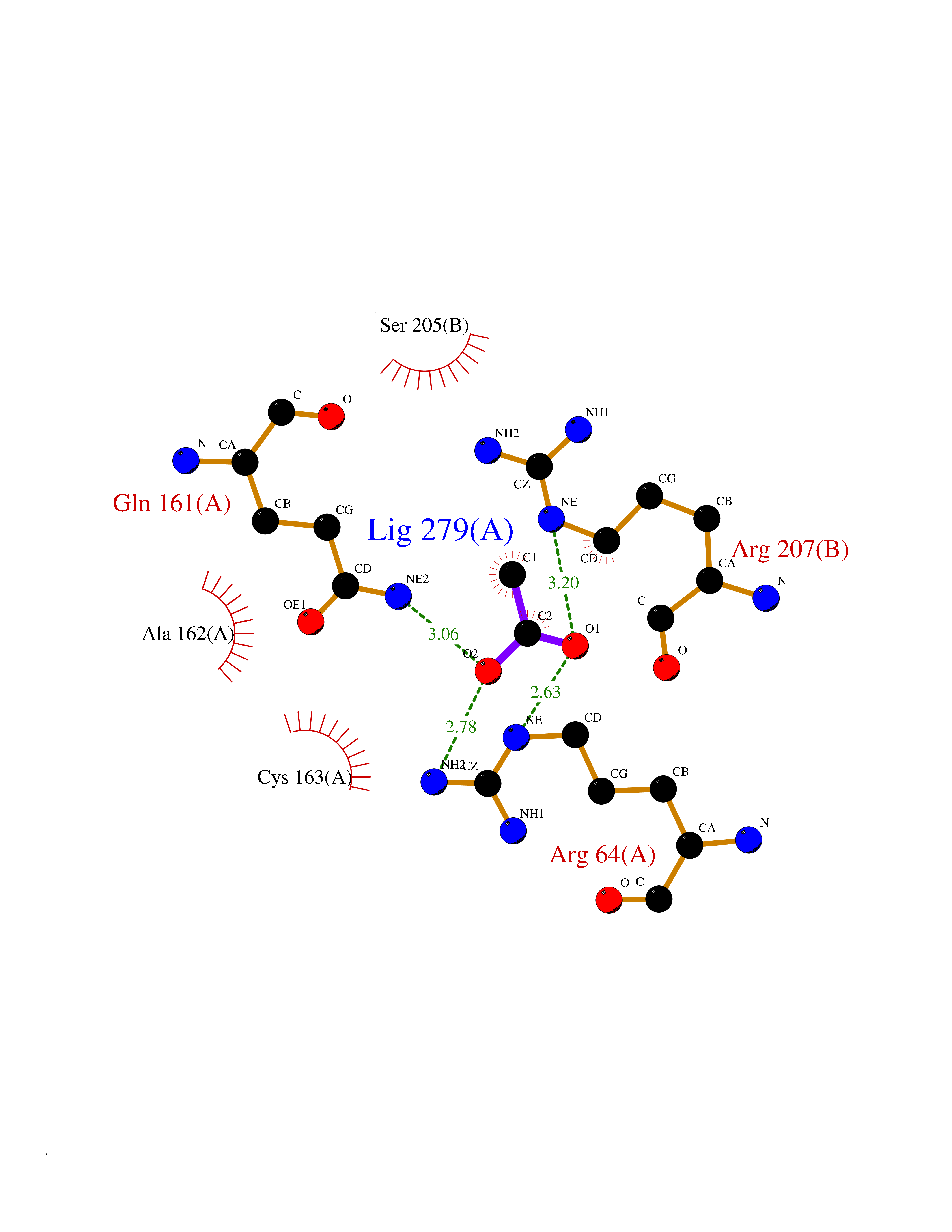 Ligplot Map 3D Binding mode Sequence SGISLDNSYKMDYPEMGLCIIINNKNFHKSTGMTSRSGTDVDAANLRETFRNLKYEVRNKNDLTREEIVELMRDVSKEDHSKRSSFVCVLLSHGEEGIIFGTNGPVDLKKITNFFRGDRCRSLTGKPKLFIIQACRGTELDCGIETHKIPVEADFLYAYSTAPGYYSWRNSKDGSWFIQSLCAMLKQYADKLEFMHILTRVNRKVATEFESFSFDATFHAKKQIPCIVSMLTKELYFYH Hydrogen bonds contact Hydrophobic contact | ||||