Job Results:
Ligand
Structure
Job ID
baacf95062c374f8ce5148662fe61a2e
Job name
NA
Time
2026-02-27 15:12:55
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Chromodomain-helicase-DNA-binding protein 1 | 4O42 | 4.28 | |
Target general information Gen name CHD1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family SNF2/RAD54 helicase family Biochemical class Dna binding protein / viral protein Function ATP binding.ATP-dependent DNA helicase activity.DNA binding.Methylated histone binding. Related diseases Pilarowski-Bjornsson syndrome (PILBOS) [MIM:617682]: An autosomal dominant disorder characterized by developmental delay, speech apraxia, intellectual disability, autism, and facial dysmorphic features. Some patients may have seizures. {ECO:0000269|PubMed:28866611}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O60341-1; B2BUF1; P28799; O76024 EC number 3.6.4.12 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Chromatin regulator; Cytoplasm; Disease variant; DNA-binding; Helicase; Hydrolase; Intellectual disability; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 20969.1 Length 180 Aromaticity 0.12 Instability index 46.35 Isoelectric point 5.88 Charge (pH=7) -2.83 2D Binding mode Binding energy (Kcal/mol) -5.83  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence EFETIERFMDCRIGRKGATGATTTIYAVEADGDPNAGFEKNKEPGEIQYLIKWKGWSHIHNTWETEETLKQQNVRGMKKLDNYKKKDQETKRWLKNASPEDVEYYNCQQELTDDLHKQYQIVERIIAHSNQKSAAGYPDYYCKWQGLPYSECSWEDGALISKKFQACIDEYFSRTARSXV Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Argininosuccinate synthase | 2NZ2 | 4.28 | |
Target general information Gen name ASS1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ASS Protein family Argininosuccinate synthase family, Type 1 subfamily Biochemical class Ligase Function Amino acid binding.Argininosuccinate synthase activity.ATP binding.Identical protein binding.RNA binding. Related diseases Citrullinemia 1 (CTLN1) [MIM:215700]: The classic form of citrullinemia, an autosomal recessive disease characterized primarily by elevated serum and urine citrulline levels. Ammonia intoxication is another manifestation. It is a disorder of the urea cycle, usually manifesting in the first few days of life. Affected infants appear normal at birth, but as ammonia builds up in the body they present symptoms such as lethargy, poor feeding, vomiting, seizures and loss of consciousness. Less commonly, a milder form can develop later in childhood or adulthood. {ECO:0000269|PubMed:11708871, ECO:0000269|PubMed:11941481, ECO:0000269|PubMed:12815590, ECO:0000269|PubMed:14680976, ECO:0000269|PubMed:15863597, ECO:0000269|PubMed:16475226, ECO:0000269|PubMed:18473344, ECO:0000269|PubMed:19006241, ECO:0000269|PubMed:1943692, ECO:0000269|PubMed:2358466, ECO:0000269|PubMed:23611581, ECO:0000269|PubMed:24889030, ECO:0000269|PubMed:25179242, ECO:0000269|PubMed:27287393, ECO:0000269|PubMed:28111830, ECO:0000269|PubMed:7977368}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00125; DB00128; DB00171; DB00155 Interacts with P10398; P00966; Q9HBL8; Q9NVM4 EC number 6.3.4.5 Uniprot keywords 3D-structure; Acetylation; Amino-acid biosynthesis; Arginine biosynthesis; ATP-binding; Cytoplasm; Direct protein sequencing; Disease variant; Ligase; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Urea cycle Protein physicochemical properties Chain ID A Molecular weight (Da) 41098.8 Length 364 Aromaticity 0.1 Instability index 36.56 Isoelectric point 8.4 Charge (pH=7) 3.55 2D Binding mode Binding energy (Kcal/mol) -5.84  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence KGSVVLAYSGGLDTSCILVWLKEQGYDVIAYLANIGQKEDFEEARKKALKLGAKKVFIEDVSREFVEEFIWPAIQSSALYEDRYLLGTSLARPCIARKQVEIAQREGAKYVSHGATGKGNDQVRFELSCYSLAPQIKVIAPWRMPEFYNRFKRNDLMEYAKQHGIPIPVTPKNPWSMDENLMHISYEAGILENPKNQAPPGLYTKTQDPAKAPNTPDILEIEFKKGVPVKVTNVKDGTTHQTSLELFMYLNEVAGKHGVGRIDIVENRFIGMKSRGIYETPAGTILYHAHLDIEAFTMDREVRKIKQGLGLKFAELVYTGFWHSPECEFVRHCIAKSQERVEGKVQVSVLKGQVYILGRESPLS Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | mRNA-capping enzyme | 2C46 | 4.28 | |
Target general information Gen name RNGTT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CAP1A Protein family Non-receptor class of the protein-tyrosine phosphatase family; Eukaryotic GTase family Biochemical class Transferase Function GTP binding.MRNA guanylyltransferase activity.Polynucleotide 5'-phosphatase activity.Protein tyrosine/serine/threonine phosphatase activity.Protein tyrosine phosphatase activity.RNA guanylyltransferase activity.Triphosphatase activity. Related diseases Atrial fibrillation, familial, 14 (ATFB14) [MIM:615378]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:19808477}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Genetic variations in SCN2B may be involved in Brugada syndrome (PubMed:23559163). This tachyarrhythmia is characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:23559163}. Drugs (DrugBank ID) NA Interacts with Q92624; P16333-1 EC number 2.7.7.50; 3.6.1.74 Uniprot keywords 3D-structure; Alternative splicing; GTP-binding; Host-virus interaction; Hydrolase; mRNA capping; mRNA processing; Multifunctional enzyme; Nucleotide-binding; Nucleotidyltransferase; Nucleus; Protein phosphatase; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 21849.8 Length 189 Aromaticity 0.11 Instability index 53.71 Isoelectric point 5.89 Charge (pH=7) -2.91 2D Binding mode Binding energy (Kcal/mol) -5.83  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence NKIPPRWLNCPRRGQPVAGRFLPLKTMLGPRYDSQVAEENRFHPSMLSNYLKSVKMGLLVDLTNTSRFYDRNDIEKEGIKYIKLQCKGHGECPTTENTETFIRLCERFELIGVHCTHGFNRTGFLICAFLVEKMDWSIEAAVATFAQARPPGIYKGDYLKELFRRYGDIEEAPPPPLLPDWCFEDDEDE Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Natriuretic peptides B | 1YK1 | 4.28 | |
Target general information Gen name NPPB Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Natriuretic peptide family Biochemical class Hormone / growth factor receptor Function Diuretic hormone activity.Hormone activity.Peptide hormone receptor binding.Receptor binding. Related diseases Multiple fibroadenomas of the breast (MFAB) [MIM:615554]: A benign breast disease marked by lobuloalveolar growth with abnormally high proliferation of the epithelium, and characterized by the presence of more than 3 fibroadenomas in one breast. Fibroadenomas are adenomas containing fibrous tissue. {ECO:0000269|PubMed:18779591}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperprolactinemia (HPRL) [MIM:615555]: A disorder characterized by increased levels of prolactin in the blood not associated with gestation or the puerperium. HPRL may result in infertility, hypogonadism, and galactorrhea. {ECO:0000269|PubMed:24195502}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01136; DB06412 Interacts with A8MQ03; P57678; Q6A162; P60411; Q7Z3S9; P25788; Q9UJW9 EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Disulfide bond; Glycoprotein; Hormone; Pharmaceutical; Proteoglycan; Proteomics identification; Reference proteome; Secreted; Signal; Vasoactive; Vasodilator Protein physicochemical properties Chain ID E Molecular weight (Da) 46353.1 Length 415 Aromaticity 0.1 Instability index 37.91 Isoelectric point 5.51 Charge (pH=7) -12.09 2D Binding mode Binding energy (Kcal/mol) -5.84  Molscript Map  Pymol Map 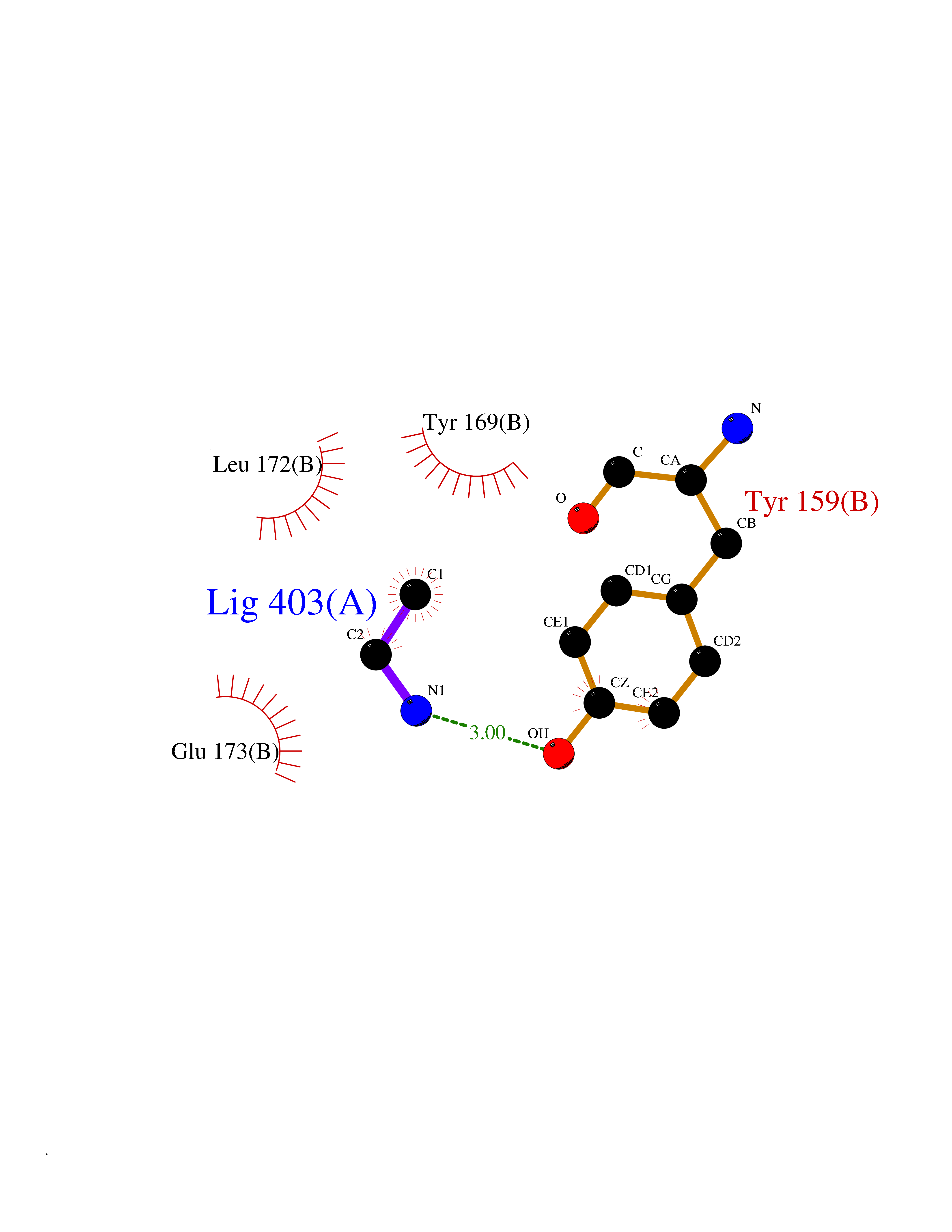 Ligplot Map 3D Binding mode Sequence GCFGRKMDRISSSSGLGCKVLALPPQKIEVLVLLPQDDSYLFSLTRVRPAIEYALRSVEGLLPPGTRFQVAYEDSDCGNRALFSLVDRVAAARGAKPDLILGPVCEYAAAPVARLASHWDLPMLSAGALAAGFQHKDSEYSHLTRVAPAYAKMGEMMLALFRHHHWSRAALVYSDDKLERNCYFTLEGVHEVFQEEGLHTSIYSFDETKDLDLEDIVRNIQASERVVIMCASSDTIRSIMLVAHRHGMTSGDYAFFNIELFNSSSYGDGSWKRGDKHDFEAKQAYSSLQTVTLLRTVKPEFEKFSMEVKSSVEKQGLNMEDYVNMFVEGFHDAILLYVLALHEVLRAGYSKKDGGKIIQQTWNRTFEGIAGQVSIDANGDRYGDFSVIAMTDVEAGTQEVIGDYFGKEGRFEMRP Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | UDP-galactopyranose mutase | 1I8T | 4.28 | |
Target general information Gen name glf Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b2036;JW2021;yefE Protein family UDP-galactopyranose/dTDP-fucopyranose mutase family Biochemical class Isomerase Function Flavin adenine dinucleotide binding.UDP-galactopyranose mutase activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB03147 Interacts with P11868 EC number 5.4.99.9 Uniprot keywords 3D-structure; Direct protein sequencing; FAD; Flavoprotein; Isomerase; Lipopolysaccharide biosynthesis; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42965.3 Length 367 Aromaticity 0.14 Instability index 32.48 Isoelectric point 6.62 Charge (pH=7) -1.52 2D Binding mode Binding energy (Kcal/mol) -5.84  Molscript Map 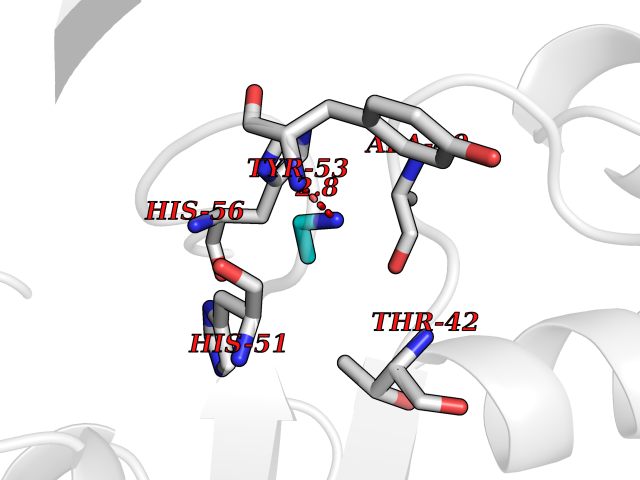 Pymol Map 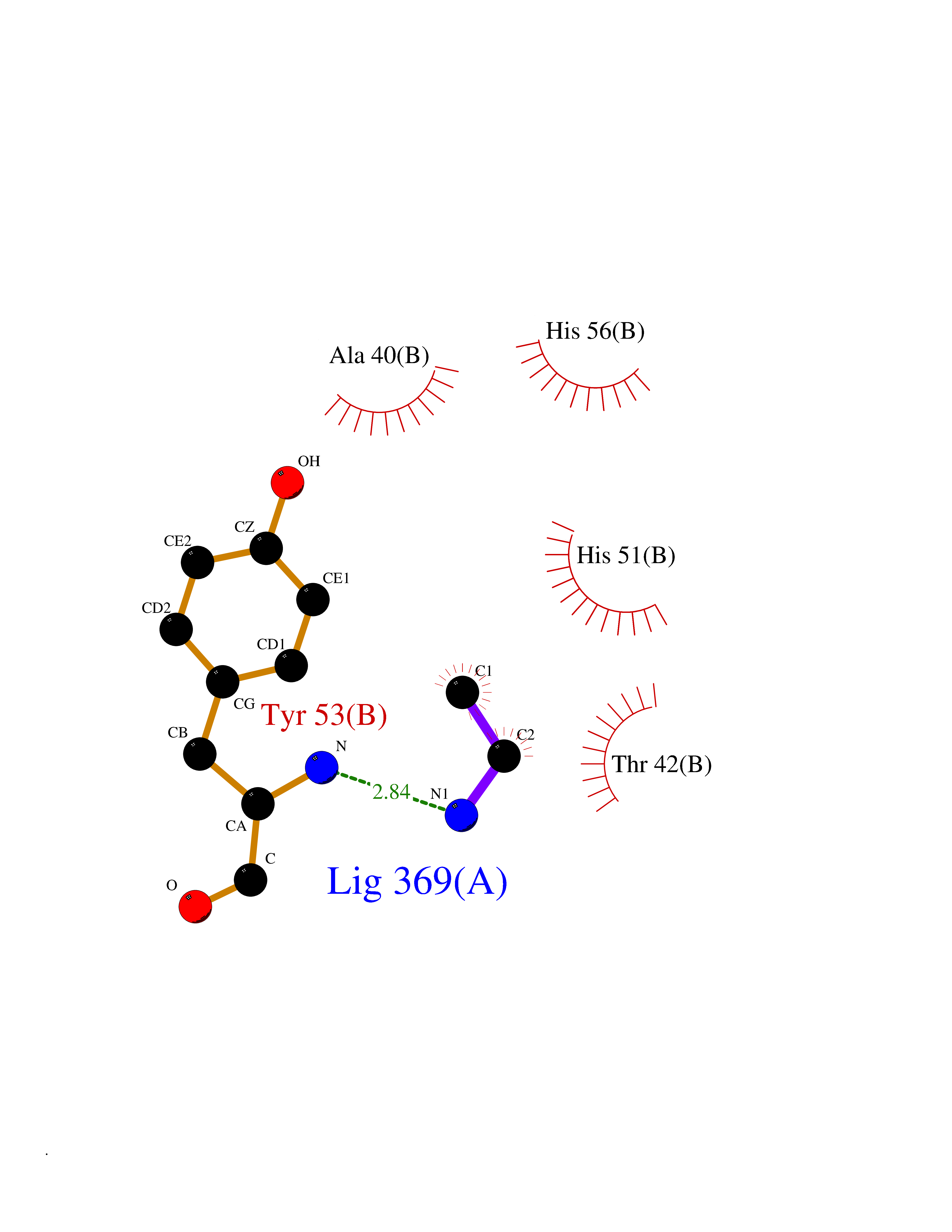 Ligplot Map 3D Binding mode Sequence MYDYIIVGSGLFGAVCANELKKLNKKVLVIEKRNHIGGNAYTEDCEGIQIHKYGAHIFHTNDKYIWDYVNDLVEFNRFTNSPLAIYKDKLFNLPFNMNTFHQMWGVKDPQEAQNIINAQKKKYGDKVPENLEEQAISLVGEDLYQALIKGYTEKQWGRSAKELPAFIIKRIPVRFTFDNNYFSDRYQGIPVGGYTKLIEKMLEGVDVKLGIDFLKDKDSLASKAHRIIYTGPIDQYFDYRFGALEYRSLKFETERHEFPNFQGNAVINFTDANVPYTRIIEHKHFDYVETKHTVVTKEYPLEWKVGDEPYYPVNDNKNMELFKKYRELASREDKVIFGGRLAEYKYYDMHQVISAALYQVKNIMSTD Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Lysine--tRNA ligase | 4YCU | 4.28 | |
Target general information Gen name KARS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms KARS;KIAA0070 Protein family Class-II aminoacyl-tRNA synthetase family Biochemical class ligase / ligase inhibitor Function Amino acid binding.ATP adenylyltransferase activity.ATP binding.Identical protein binding.Lysine-tRNA ligase activity.Protein homodimerization activity.TRNA binding. Related diseases Charcot-Marie-Tooth disease, recessive intermediate B (CMTRIB) [MIM:613641]: A form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Recessive intermediate forms of Charcot-Marie-Tooth disease are characterized by clinical and pathologic features intermediate between demyelinating and axonal peripheral neuropathies, and motor median nerve conduction velocities ranging from 25 to 45 m/sec. {ECO:0000269|PubMed:20920668}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, autosomal recessive, 89 (DFNB89) [MIM:613916]: A form of non-syndromic deafness characterized by bilateral, prelingual, moderate to severe hearing loss affecting all frequencies. {ECO:0000269|PubMed:23768514}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, congenital, and adult-onset progressive leukoencephalopathy (DEAPLE) [MIM:619196]: An autosomal recessive, complex neurodegenerative disorder characterized by congenital sensorineural deafness, and progressive motor and cognitive decline apparent in young adulthood. Brain imaging shows diffuse white matter abnormalities affecting various brain regions, consistent with a progressive leukoencephalopathy. More variable additional features may include visual impairment and axonal peripheral neuropathy. Premature death may occurr in some patients. {ECO:0000269|PubMed:28887846, ECO:0000269|PubMed:30737337, ECO:0000269|PubMed:31116475}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Leukoencephalopathy, progressive, infantile-onset, with or without deafness (LEPID) [MIM:619147]: An autosomal recessive, complex neurodegenerative disorder apparent from infancy. LEPID is characterized by early-onset progressive leukoencephalopathy with brainstem and spinal cord calcifications, sensorineural deafness in most patients, global developmental delay with cognitive impairment and poor or absent speech, developmental regression, and neurologic deterioration. Additional more variable features may include poor overall growth with microcephaly, seizures, visual loss, microcytic anemia, and hepatic enlargement or abnormal liver enzymes. Premature death is common. {ECO:0000269|PubMed:25330800, ECO:0000269|PubMed:29615062, ECO:0000269|PubMed:30252186, ECO:0000269|PubMed:30715177}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00123 Interacts with Q13155; P07814; Q15046; P08865; P00441; Q13155 EC number 2.7.7.-; 6.1.1.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminoacyl-tRNA synthetase; ATP-binding; Cell membrane; Charcot-Marie-Tooth disease; Cytoplasm; Deafness; Direct protein sequencing; Disease variant; Host-virus interaction; Intellectual disability; Ligase; Membrane; Mitochondrion; Neurodegeneration; Neuropathy; Non-syndromic deafness; Nucleotide-binding; Nucleus; Phosphoprotein; Protein biosynthesis; Proteomics identification; Reference proteome; Secreted; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 37110.4 Length 323 Aromaticity 0.1 Instability index 50.17 Isoelectric point 4.92 Charge (pH=7) -17.53 2D Binding mode Binding energy (Kcal/mol) -5.84  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence IIRSKIITYIRSFLDELGFLEIETPMMNIIPGGAVAKPFITYHNELDMNLYMRIAPELYHKMLVVGGIDRVYEIGRQFRNEGIDLTHNPEFTTCEFYMAYADYHDLMEITEKMVSGMVKHITGSYKVTYHPDGPEGQAYDVDFTPPFRRINMVEELEKALGMKLPETNLFETEETRKILDDICVAKAVECPPPRTTARLLDKLVGEFLEVTCINPTFICDHPQIMSPLAKWHRSKEGLTERFELFVMKKEICNAYTELNDPMRQRQLFEEQAKAKAAGDDEAMFIDENFCTALEYGLPPTAGWGMGIDRVAMFLTDSNNIKEV Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Trypanosoma Cruzipain (Trypano CYSP) | 1EWM | 4.28 | |
Target general information Gen name Trypano CYSP Organism Trypanosoma cruzi Uniprot ID TTD ID Synonyms Cruzaine; Major cysteine proteinase Protein family Peptidase C1 family Biochemical class Peptidase Function The cysteine protease may play an important role in the development and differentiation of the parasites at several stages of their life cycle. Related diseases Sick sinus syndrome 2 (SSS2) [MIM:163800]: The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS2 onset is in utero or at birth. {ECO:0000269|PubMed:15123648, ECO:0000269|PubMed:16407510, ECO:0000269|PubMed:20662977, ECO:0000269|PubMed:23103389}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 8 (BRGDA8) [MIM:613123]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:19165230}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Epilepsy, idiopathic generalized 18 (EIG18) [MIM:619521]: An autosomal dominant form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. EIG18 is characterized by onset of myoclonic seizures in infancy. Although the seizures remit, some patients may have later speech or cognitive impairment. {ECO:0000269|PubMed:30127718}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02200; DB02051; DB01871; DB01810; DB02128; DB03536; DB04427; DB03691; DB04502; DB03573 Interacts with NA EC number EC 3.4.22.51 Uniprot keywords 3D-structure; Autocatalytic cleavage; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Protease; Signal; Thiol protease; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 22703 Length 215 Aromaticity 0.09 Instability index 28.98 Isoelectric point 4.37 Charge (pH=7) -13.9 2D Binding mode Binding energy (Kcal/mol) -5.84  Molscript Map 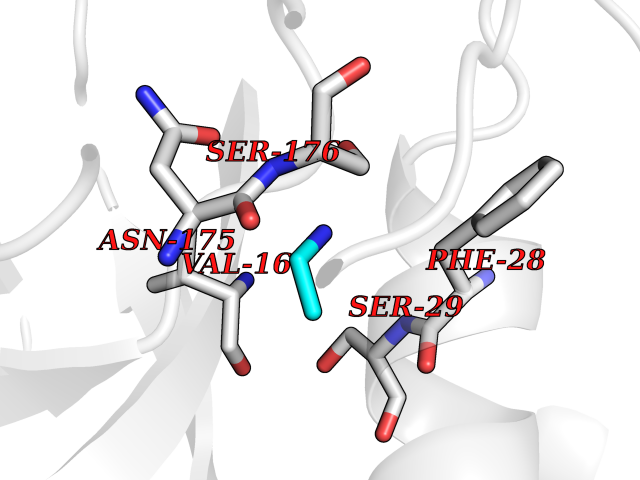 Pymol Map 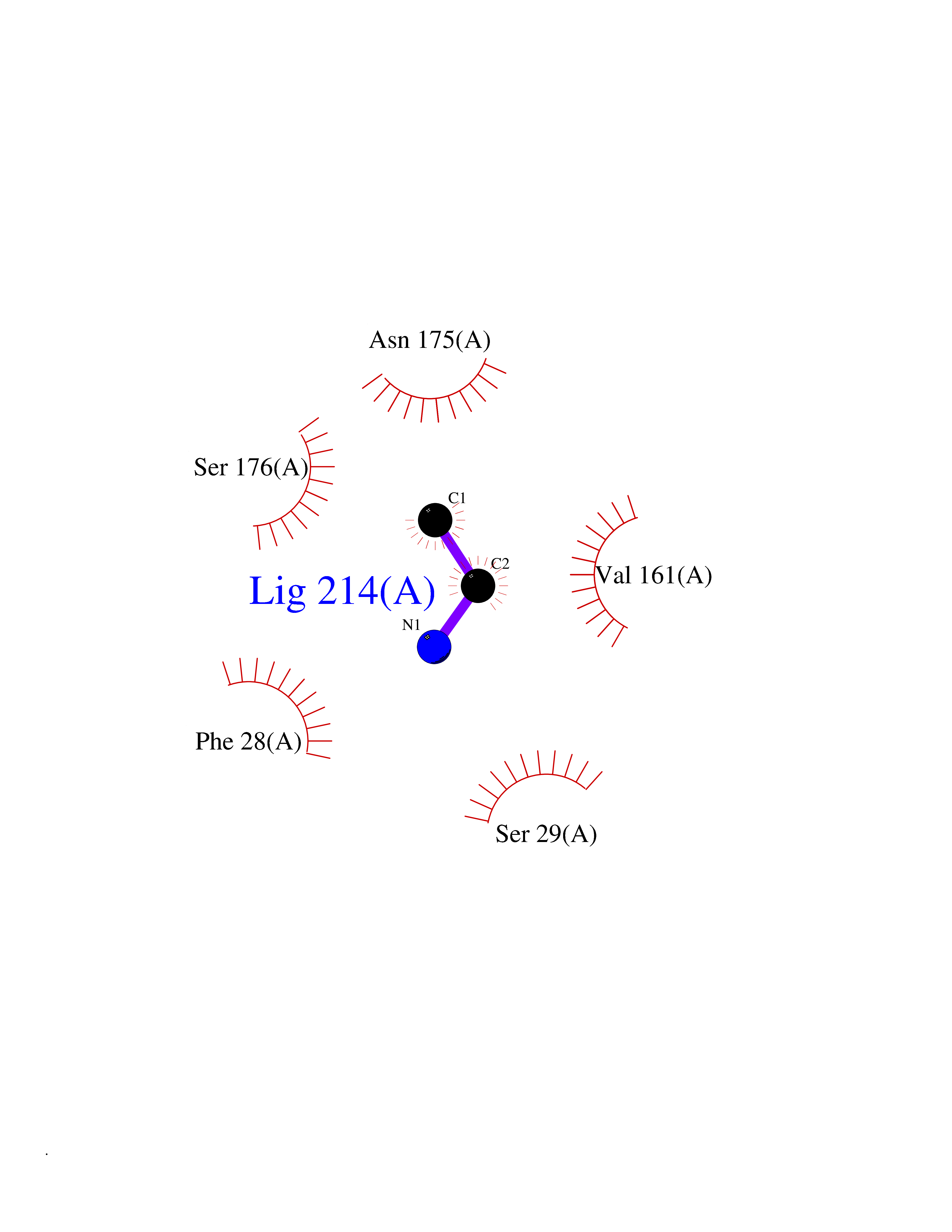 Ligplot Map 3D Binding mode Sequence APAAVDWRARGAVTAVKDQGQCGSCWAFSAIGNVECQWFLAGHPLTNLSEQMLVSCDKTDSGCSGGLMNNAFEWIVQENNGAVYTEDSYPYASGEGISPPCTTSGHTVGATITGHVELPQDEAQIAAWLAVNGPVAVAVDASSWMTYTGGVMTSCVSEQLDHGVLLVGYNDSAAVPYWIIKNSWTTQWGEEGYIRIAKGSNQCLVKEEASSAVVG Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | N-acetylmannosamine kinase (GNE) | 4ZHT | 4.28 | |
Target general information Gen name GNE Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UDPGlcNAc2epimerase/ManAc kinase; GNE; Bifunctional UDPNacetylglucosamine 2epimerase/Nacetylmannosamine kinase Protein family UDP-N-acetylglucosamine 2-epimerase family; ROK (NagC/XylR) family Biochemical class Kinase Function Regulates and initiates biosynthesis of N- acetylneuraminic acid (NeuAc), a precursor of sialic acids. Plays an essential role in early development. Required for normal sialylation in hematopoietic cells. Sialylation is implicated in cell adhesion, signal transduction, tumorigenicity and metastatic behavior of malignant cells. {ECO:0000250, ECO:0000269|PubMed:10334995}. Related diseases Sialuria (SIALURIA) [MIM:269921]: In sialuria, free sialic acid accumulates in the cytoplasm and gram quantities of neuraminic acid are secreted in the urine. The metabolic defect involves lack of feedback inhibition of UDP-GlcNAc 2-epimerase by CMP-Neu5Ac, resulting in constitutive overproduction of free Neu5Ac. Clinical features include variable degrees of developmental delay, coarse facial features and hepatomegaly. Sialuria inheritance is autosomal dominant. {ECO:0000269|PubMed:10330343, ECO:0000269|PubMed:10356312, ECO:0000269|PubMed:11326336, ECO:0000269|PubMed:2808337}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Nonaka myopathy (NM) [MIM:605820]: An autosomal recessive myopathy characterized by early adult onset and progressive distal muscle weakness that preferentially affects the anterior tibial muscles, usually sparing the quadriceps femoris. Some individuals may have involvement of the upper limbs or proximal muscles. Muscle biopsy reveals presence of rimmed vacuoles. {ECO:0000269|PubMed:11528398, ECO:0000269|PubMed:11916006, ECO:0000269|PubMed:12177386, ECO:0000269|PubMed:12325084, ECO:0000269|PubMed:12409274, ECO:0000269|PubMed:12473753, ECO:0000269|PubMed:12473769, ECO:0000269|PubMed:12473780, ECO:0000269|PubMed:12497639, ECO:0000269|PubMed:12811782, ECO:0000269|PubMed:12913203, ECO:0000269|PubMed:14707127, ECO:0000269|PubMed:15146476, ECO:0000269|PubMed:16503651}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thrombocytopenia 12 with or without myopathy (THC12) [MIM:620757]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC12 is an autosomal recessive form manifesting from infancy or early childhood with bleeding episodes. Clinical features include petechiae, easy bruising, epistaxis, hematomas, menorrhagia, and increased bleeding after trauma or surgery. Rare patients may have thrombocytopenia without bleeding. Some affected individuals have myopathic features, usually apparent in the second or third decades of life. {ECO:0000269|PubMed:25257349, ECO:0000269|PubMed:30171045, ECO:0000269|PubMed:33198675, ECO:0000269|PubMed:34788986, ECO:0000269|PubMed:34858435, ECO:0000269|PubMed:35052006, ECO:0000269|PubMed:38237079}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P12814; Q6UY14-3; Q969Y2; Q15323; P60370; P60409; P60410; P60411; Q9BQ66; P26371; Q9BYQ4; Q7Z3S9; O43597; Q6UY14-3; Q6P5X5; P27918; A8MQ03; Q16610; Q9UHF1; P28799; P49639; Q5T749; Q15323; O76011; Q6A162; P78385; P78386; O43790; Q07627; Q8IUG1; P60409; P60410; Q8IUC1; P60328; Q52LG2; Q3SY46; Q9BYP8; Q3LHN2; Q3SYF9; Q9BYR8; Q9BYR6; Q9BYQ7; Q9BYQ6; Q9BYR3; P26371; Q3LI64; Q3LI66; Q3LI67; Q9BYQ4; Q9BYQ3; Q9BYQ0; Q99750; Q8IV28; P0DPK4; O15496; O43609; O43610; P14373; Q8IWZ5; Q15654; O14817; Q2TAL6; Q9BRX9; O76024; Q9NZC7-5 EC number NA Uniprot keywords 3D-structure; Allosteric enzyme; Alternative splicing; ATP-binding; Cytoplasm; Disease variant; Hydrolase; Kinase; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 41589.2 Length 384 Aromaticity 0.07 Instability index 35.07 Isoelectric point 7.05 Charge (pH=7) 0.19 2D Binding mode Binding energy (Kcal/mol) -5.83 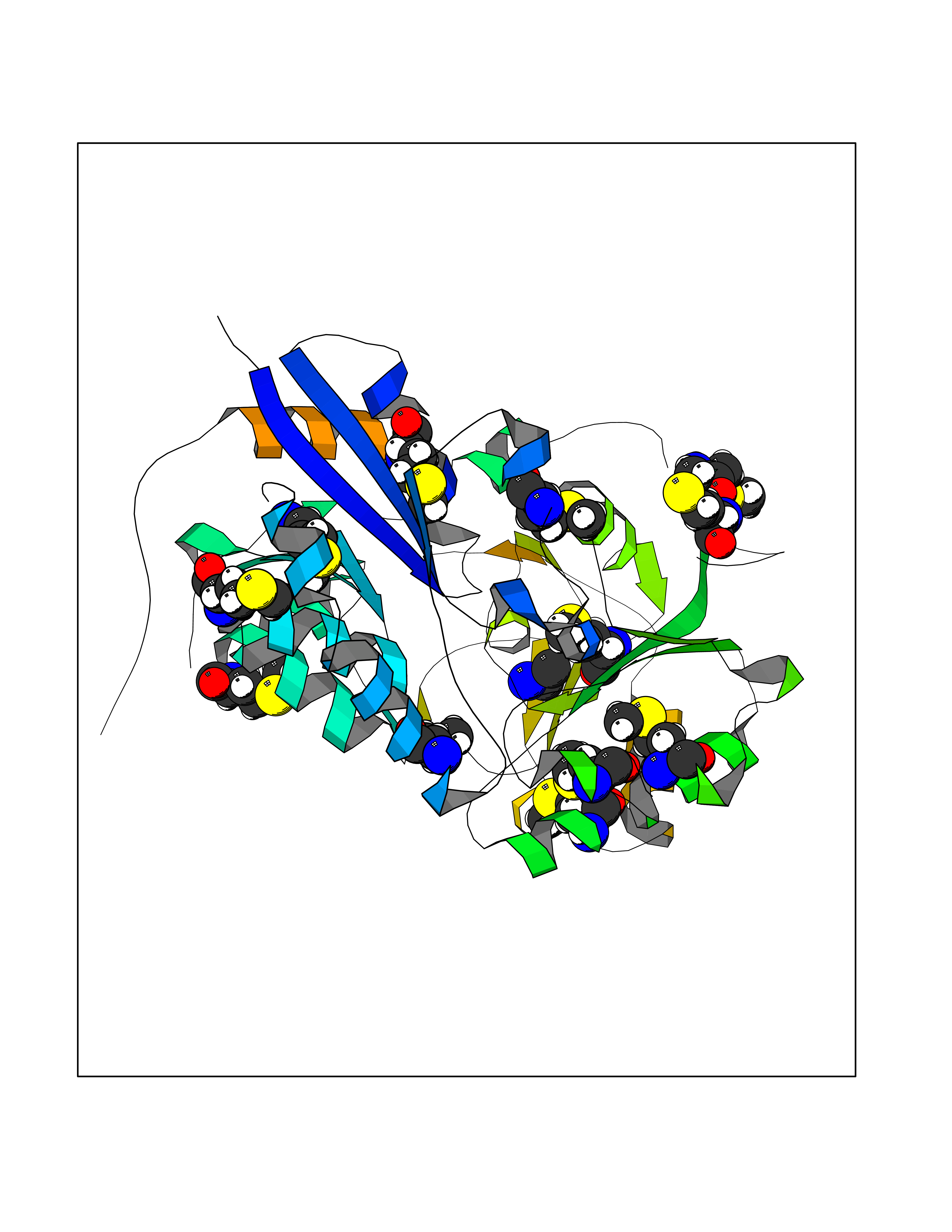 Molscript Map 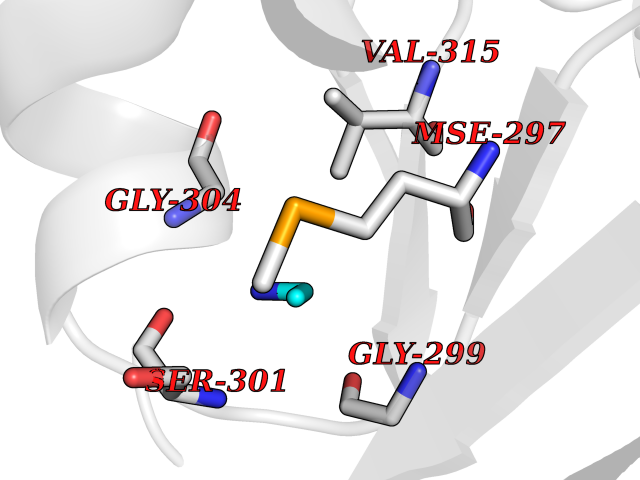 Pymol Map  Ligplot Map 3D Binding mode Sequence NRKLRVCVATCNRADYSKLAPIXFGIKTEPEFFELDVVVLGSHLIDDYGNTYRXIEQDDFDINTRLHTIVRGEDEAAXVESVGLALVKLPDVLNRLKPDIXIVHGDRFDALALATSAALXNIRILHIEGGEVSGTIDDSIRHAITKLAHYHVCCTRSAEQHLISXCEDHDRILLAGCPSYDKLLSAKNKDYXSIIRXWLGDDVKSKDYIVALQHPVTTDIKHSIKXFELTLDALISFNKRTLVLFPNIDAGSKEXVRVXRKKGIEHHPNFRAVKHVPFDQFIQLVAHAGCXIGNSSCGVREVGAFGTPVINLGTRQIGRETGENVLHVRDADTQDKILQALHLQFGKQYPCSKIYGDGNAVPRILKFLKSIDLQEPLQKKFCFP Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | ADP-ribosylation factor 1 (ARF1) | 1HUR | 4.28 | |
Target general information Gen name ARF1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PVNH8 Protein family Small GTPase superfamily, Arf family Biochemical class Small GTPase Function Modulates vesicle budding and uncoating within the Golgi complex. Deactivation induces the redistribution of the entire Golgi complex to the endoplasmic reticulum, suggesting a crucial role in protein trafficking. In its GTP-bound form, its triggers the association with coat proteins with the Golgi membrane. The hydrolysis of ARF1-bound GTP, which is mediated by ARFGAPs proteins, is required for dissociation of coat proteins from Golgi membranes and vesicles. The GTP-bound form interacts with PICK1 to limit PICK1-mediated inhibition of Arp2/3 complex activity; the function is linked to AMPA receptor (AMPAR) trafficking, regulation of synaptic plasicity of excitatory synapses and spine shrinkage during long-term depression (LTD). GTP-binding protein involved in protein trafficking among different compartments. Related diseases Periventricular nodular heterotopia 8 (PVNH8) [MIM:618185]: A form of periventricular nodular heterotopia, a disorder resulting from a defect in the pattern of neuronal migration in which ectopic collections of neurons lie along the lateral ventricles of the brain or just beneath, contiguously or in isolated patches. PVNH8 is an autosomal dominant disease characterized by developmental disabilities, speech delay, seizures and attention deficit-hyperactivity disorder. {ECO:0000269|PubMed:28868155}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02774; DB07348; DB09093; DB09462; DB04121; DB04315; DB04137; DB08231 Interacts with P53367; P53365; Q99418; P46940; O60271 EC number NA Uniprot keywords 3D-structure; Acetylation; Direct protein sequencing; Disease variant; ER-Golgi transport; Golgi apparatus; GTP-binding; Hydrolase; Lipoprotein; Membrane; Myristate; Nucleotide-binding; Protein transport; Proteomics identification; Reference proteome; Synapse; Synaptosome; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 41112.6 Length 360 Aromaticity 0.09 Instability index 28.3 Isoelectric point 6.38 Charge (pH=7) -1.68 2D Binding mode Binding energy (Kcal/mol) -5.83  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GNIFANLFKGLFGKKEMRILMVGLDAAGKTTILYKLKLGEIVTTIPTIGFNVETVEYKNISFTVWDVGGQDKIRPLWRHYFQNTQGLIFVVDSNDRERVNEAREELMRMLAEDELRDAVLLVFANKQDLPNAMNAAEITDKLGLHSLRHRNWYIQATCATSGDGLYEGLDWLSNQLRNQKGNIFANLFKGLFGKKEMRILMVGLDAAGKTTILYKLKLGEIVTTIPTIGFNVETVEYKNISFTVWDVGGQDKIRPLWRHYFQNTQGLIFVVDSNDRERVNEAREELMRMLAEDELRDAVLLVFANKQDLPNAMNAAEITDKLGLHSLRHRNWYIQATCATSGDGLYEGLDWLSNQLRNQK Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Lysine-specific demethylase 6B (KDM6B) | 6F6D | 4.28 | |
Target general information Gen name KDM6B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine demethylase 6B; KIAA0346; Jumonji domain-containing protein 3; JmjC domain-containing protein 3; JMJD3 Protein family UTX family Biochemical class NA Function Histone demethylase that specifically demethylates 'Lys-27' of histone H3, thereby playing a central role in histone code. Demethylates trimethylated and dimethylated H3 'Lys-27'. Plays a central role in regulation of posterior development, by regulating HOX gene expression. Involved in inflammatory response by participating in macrophage differentiation in case of inflammation by regulating gene expression and macrophage differentiation. Plays a demethylase-independent role in chromatin remodeling to regulate T-box family member-dependent gene expression by acting as a link between T-box factors and the SMARCA4-containing SWI/SNF remodeling complex (By similarity). Related diseases Stolerman neurodevelopmental syndrome (NEDSST) [MIM:618505]: An autosomal dominant disorder characterized by global developmental delay, variable intellectual disability, poor language acquisition, and dysmorphic facial features including a prominent nasal bridge and coarse features. Some patients manifest autism spectrum disorder. Musculoskeletal features may be present and include widened and thickened hands and fingers, joint hypermobility, clinodactyly of the fifth fingers, and toe syndactyly. {ECO:0000269|PubMed:31124279}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P03372 EC number EC 1.14.11.- Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Dioxygenase; Disease variant; Inflammatory response; Intellectual disability; Iron; Isopeptide bond; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 53008.8 Length 467 Aromaticity 0.1 Instability index 44.23 Isoelectric point 8.37 Charge (pH=7) 4.9 2D Binding mode Binding energy (Kcal/mol) -5.84  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ESYLSPAQSVKPKINTEEKLPREKLNPPTPSIYLESKRDAFSPVLLQFCTDPRNPITVIRGLAGSLRLNLGLFSTKTLVEASGEHTVEVRTQVQQPSDENWDLTGTRQIWPCESSRSHTTIAKYAQYQASSFQESHIIKFGTNIDLSDAKRWKPQLQELLKLPAFMRVTSTGNMLSHVGHTILGMNTVQLYMKVPGSRTPGHQENNNFCSVNINIGPGDCEWFAVHEHYWETISAFCDRHGVDYLTGSWWPILDDLYASNIPVYRFVQRPGDLVWINAGTVHWVQATGWCNNIAWNVGPLTAYQYQLALERYEWNEVKNVKSIVPMIHVSWNVARTVKISDPDLFKMIKFCLLQSMKHCQVQRESLVRAGKKIAYQGRVKDEPAYYCNECDVEVFNILFVTSENGSRNTYLVHCEGCARRRSAGLQGVVVLEQYRTEELAQAYDAFTLAPRIQLMTKAARKSAPATG Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Mycobacterium Decaprenylphosphoryl-beta-D-ribose oxidase (McyB dprE1) | 4P8N | 4.28 | |
Target general information Gen name McyB dprE1 Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID Synonyms FAD-dependent decaprenylphosphoryl-beta-D-ribofuranose 2-oxidase; Decaprenylphosphoryl-beta-D-ribose oxidase; Decaprenylphosphoryl-beta-D-ribose 2-epimerase flavoprotein subunit; Decaprenylphosphoryl- Protein family DprE1 family Biochemical class NA Function Component of the DprE1-DprE2 complex that catalyzes the 2-step epimerization of decaprenyl-phospho-ribose (DPR) to decaprenyl-phospho-arabinose (DPA), a key precursor that serves as the arabinose donor required for the synthesis of cell-wall arabinans. DprE1 catalyzes the first step of epimerization, namely FAD-dependent oxidation of the C2' hydroxyl of DPR to yield the keto intermediate decaprenyl-phospho-2'-keto-D-arabinose (DPX). The intermediate DPX is then transferred to DprE2 subunit of the epimerase complex, most probably through a 'substrate channel' at the interface of DprE1-DprE2 complex. Can also use farnesyl-phosphoryl-beta-D-ribofuranose (FPR) as substrate in vitro. Appears to be essential for the growth and survival of M.tuberculosis. Related diseases Neuronopathy, distal hereditary motor, autosomal recessive 8 (HMNR8) [MIM:618912]: An autosomal recessive disorder characterized by motor axonal neuropathy, slowly progressive distal muscle weakness mainly affecting the lower limbs, difficulty walking, and increased serum sorbitol. Additional variable features are distal sensory impairment, upper limb tremor, scoliosis, and mild hearing loss. {ECO:0000269|PubMed:32367058, ECO:0000269|PubMed:33314640, ECO:0000269|PubMed:33397963}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.1.98.3 Uniprot keywords 3D-structure; Antibiotic resistance; Cell wall biogenesis/degradation; FAD; Flavoprotein; Oxidoreductase; Periplasm; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 49442.7 Length 454 Aromaticity 0.09 Instability index 28.24 Isoelectric point 7.29 Charge (pH=7) 0.64 2D Binding mode Binding energy (Kcal/mol) -5.84  Molscript Map 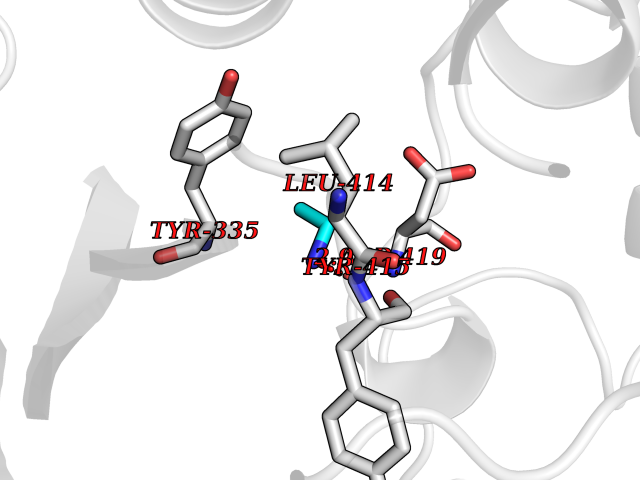 Pymol Map 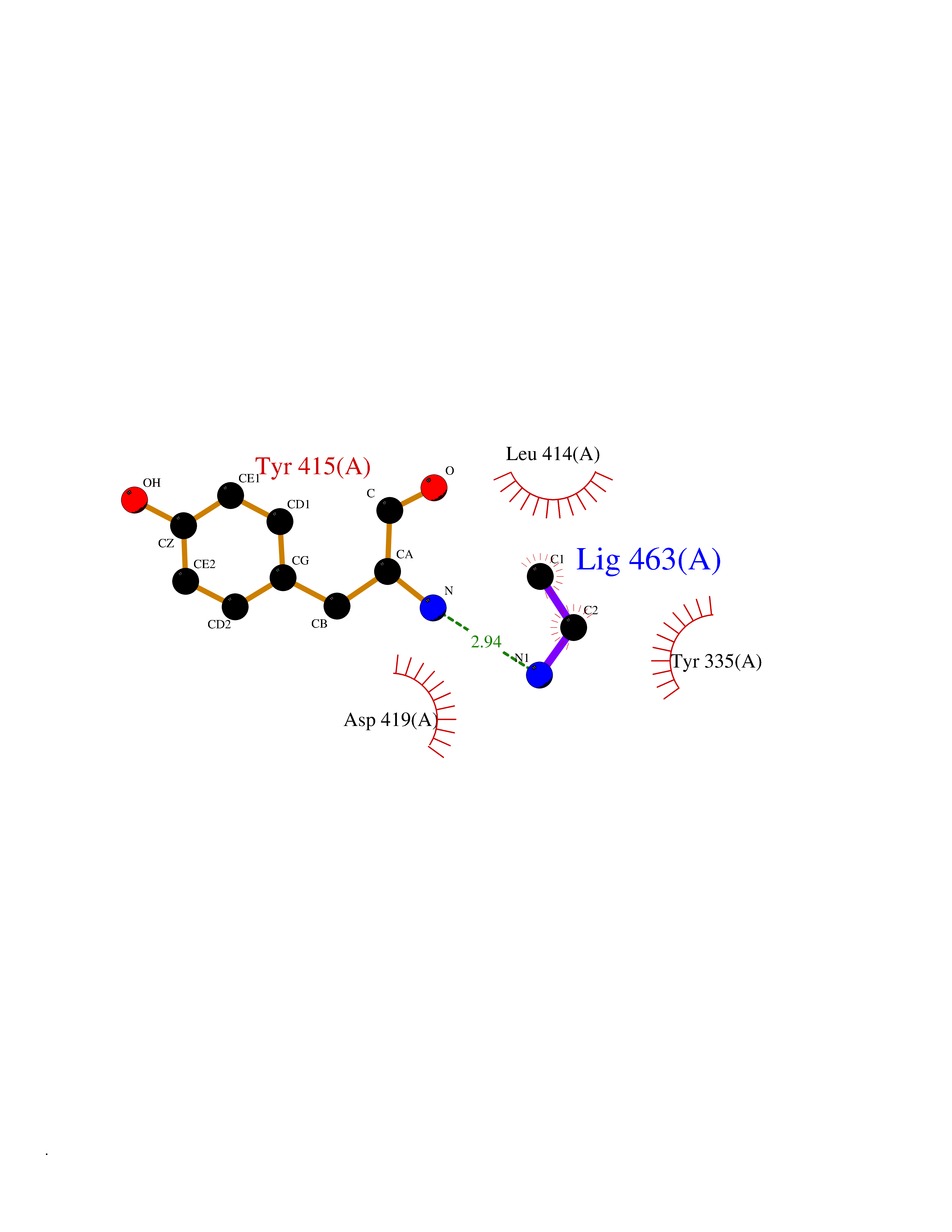 Ligplot Map 3D Binding mode Sequence VGATTTATRLTGWGRTAPSVANVLRTPDAEMIVKAVARVAESGGGRGAIARGLGRSYGDNAQNGGGLVIDMTPLNTIHSIDADTKLVDIDAGVNLDQLMKAALPFGLWVPVLPGTRQVTVGGAIACDIHGKNHHSAGSFGNHVRSMDLLTADGEIRHLTPTGEDAELFWATVGGNGLTGIIMRATIEMTPTSTAYFIADGDVTASLDETIALHSDGSEARYTYSSAWFDAISAPPKLGRAAVSRGRLATVEQLPAKLRSEPLKFDAPQLLTLPDVFPNGLANKYTFGPIGELWYRKSGTYRGKVQNLTQFYHPLDMFGEWNRAGFLQYQFVIPTEAVDEFKKIIGVIQASGHYSFLNVFKLFGPRNQAPLSFPIPGWNICVDFPIKDGLGKFVSELDRRVLEFGGRLYTAKDSRTTAETFHAMYPRVDEWISVRRKVDPLRVFASDMARRLELL Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Dihydrodiol dehydrogenase type I (AKR1C3) | 2F38 | 4.28 | |
Target general information Gen name AKR1C3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Trans-1,2-dihydrobenzene-1,2-diol dehydrogenase; Testosterone 17-beta-dehydrogenase 5; Prostaglandin F synthase; PGFS; KIAA0119; Indanol dehydrogenase; HSD17B5; HA1753; Dihydrodiol dehydrogenase 3; DD Protein family Aldo/keto reductase family Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the conversion of aldehydes and ketones to alcohols. Catalyzes the reduction of prostaglandin (PG) D2, PGH2 and phenanthrenequinone (PQ) and the oxidation of 9-alpha,11-beta-PGF2 to PGD2. Functions as a bi-directional 3-alpha-, 17-beta- and 20-alpha HSD. Can interconvert active androgens, estrogens and progestins with their cognate inactive metabolites. Preferentially transforms androstenedione (4-dione) to testosterone. Related diseases Neurodevelopmental disorder with language impairment and behavioral abnormalities (NEDLIB) [MIM:618917]: A neurodevelopmental disorder characterized by global developmental delay, impaired intellectual development, poor or absent speech, and behavioral abnormalities, such as autism spectrum disorder, repetitive behaviors, and hyperactivity. Some patients develop seizures and manifest developmental regression. {ECO:0000269|PubMed:31300657}. The disease is caused by variants affecting the gene represented in this entry. The genetic variation producing the missense variant p.Q607E, associated with NEDLIB, is predicted to deeply affect RNA editing. In a physiological context, the adenosine (A) residue of the original glutamine (Q) codon CAG is post-transcriptionaly edited to inosine (I) by ADAR2, leading to a codon recognized by the ribosome as arginine (R). The glutamate (E) codon GAG, resulting from the genetic variation, is predicted to be edited 90% less than the normal CAG codon. If edited, the codon GIG would be translated as p.Q607G. {ECO:0000305|PubMed:31300657}. Drugs (DrugBank ID) DB07700; DB01561; DB01536; DB00997; DB01039; DB02266; DB13751; DB00328; DB06077; DB00959; DB00157; DB03461; DB09074; DB00776; DB02056; DB01698; DB02901 Interacts with P17516 EC number EC 1.-.-.- Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Lipid metabolism; NAD; NADP; Oxidoreductase; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 36382.2 Length 319 Aromaticity 0.09 Instability index 47.73 Isoelectric point 8.1 Charge (pH=7) 2.51 2D Binding mode Binding energy (Kcal/mol) -5.83  Molscript Map 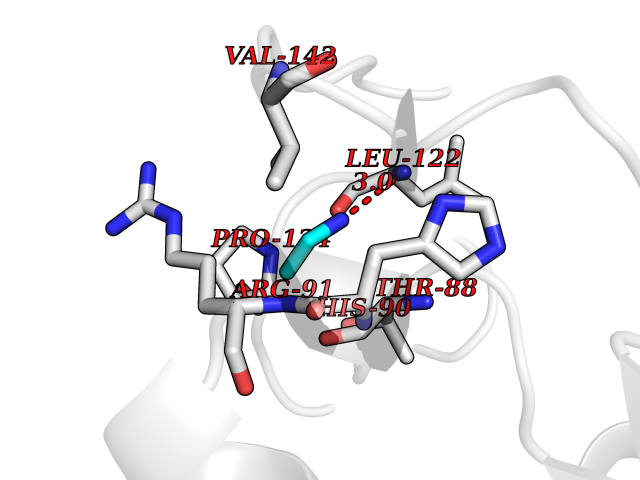 Pymol Map 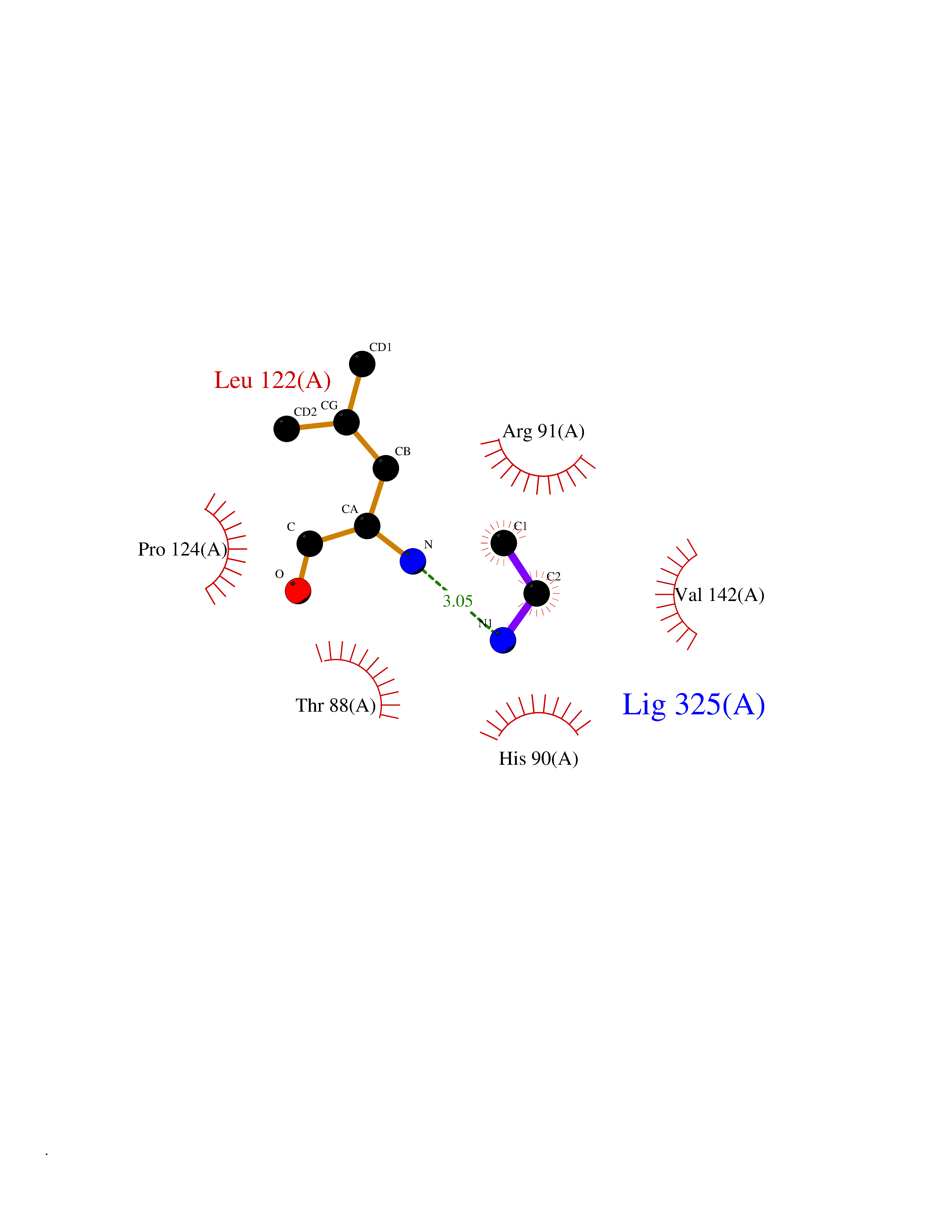 Ligplot Map 3D Binding mode Sequence QQCVKLNDGHFMPVLGFGTYAPPEVPRSKALEVTKLAIEAGFRHIDSAHLYNNEEQVGLAIRSKIADGSVKREDIFYTSKLWSTFHRPELVRPALENSLKKAQLDYVDLYLIHSPMSLKPGEELSPTDENGKVIFDIVDLCTTWEAMEKCKDAGLAKSIGVSNFNRRQLEMILNKPGLKYKPVCNQVECHPYFNRSKLLDFCKSKDIVLVAYSALGSQRDKRWVDPNSPVLLEDPVLCALAKKHKRTPALIALRYQLQRGVVVLAKSYNEQRIRQNVQVFEFQLTAEDMKAIDGLDRNLHYFNSDSFASHPNYPYSDEY Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Ubiquitin thioesterase L3 (UCHL3) | 1XD3 | 4.28 | |
Target general information Gen name UCHL3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ubiquitin carboxyl-terminal hydrolase isozyme L3; UCH-L3 Protein family Peptidase C12 family Biochemical class Peptidase Function Thiol protease that recognizes and hydrolyzes a peptide bond at the C-terminal glycine of either ubiquitin or NEDD8. Has a 10-fold preference for Arg and Lys at position P3", and exhibits a preference towards 'Lys-48'-linked ubiquitin chains. Deubiquitinates ENAC in apical compartments, thereby regulating apical membrane recycling. Indirectly increases the phosphorylation of IGFIR, AKT and FOXO1 and promotes insulin-signaling and insulin-induced adipogenesis. Required for stress-response retinal, skeletal muscle and germ cell maintenance. May be involved in working memory. Can hydrolyze UBB(+1), a mutated form of ubiquitin which is not effectively degraded by the proteasome and is associated with neurogenerative disorders. Deubiquitinating enzyme (DUB) that controls levels of cellular ubiquitin through processing of ubiquitin precursors and ubiquitinated proteins. Related diseases Epilepsy, familial focal, with variable foci 4 (FFEVF4) [MIM:617935]: An autosomal dominant form of epilepsy characterized by focal seizures arising from different cortical regions, including the temporal, frontal, parietal, and occipital lobes. Seizure types commonly include temporal lobe epilepsy, frontal lobe epilepsy, and nocturnal frontal lobe epilepsy. Some patients may have intellectual disability or autism spectrum disorders. Seizure onset usually occurs in the first or second decades, although later onset has been reported, and there is phenotypic variability within families. A subset of patients have structural brain abnormalities. Penetrance of the disorder is incomplete. FFEVF4 is characterized by onset of focal seizures in the first years of life. {ECO:0000269|PubMed:24157691, ECO:0000269|PubMed:28235671}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 62 (DEE62) [MIM:617938]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE62 is characterized by onset of seizures in the first year of life. {ECO:0000269|PubMed:29466837}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9H078; G5E9A7; Q15797; Q7Z699 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Cytoplasm; Hydrolase; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Thiol protease; Ubl conjugation pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34540.8 Length 304 Aromaticity 0.07 Instability index 39.91 Isoelectric point 5.01 Charge (pH=7) -17.24 2D Binding mode Binding energy (Kcal/mol) -5.84  Molscript Map 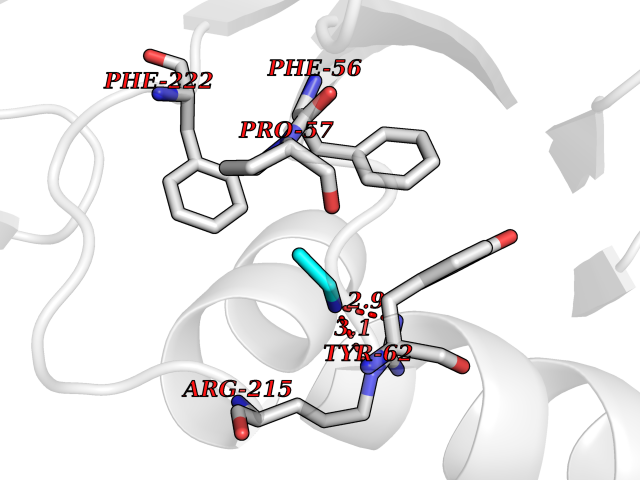 Pymol Map 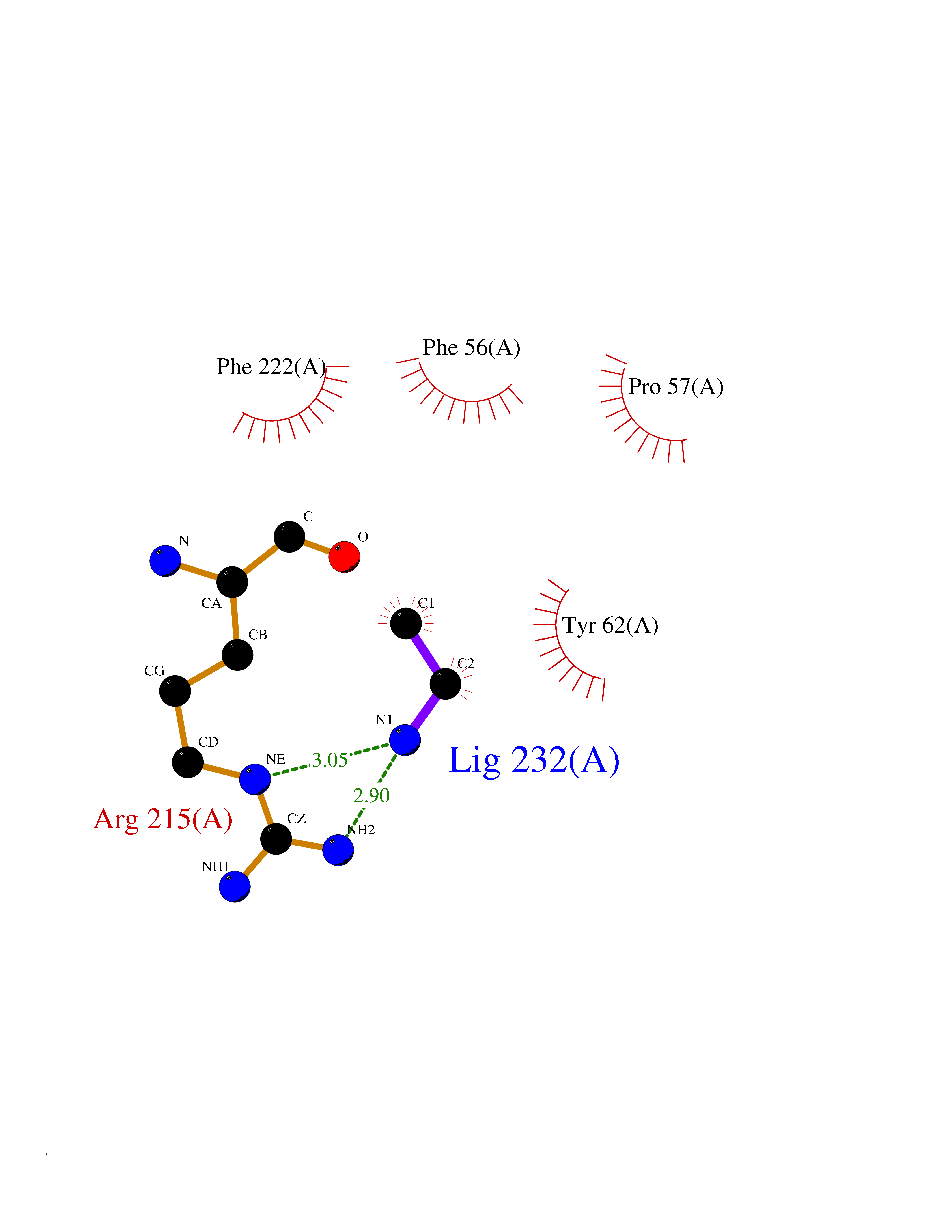 Ligplot Map 3D Binding mode Sequence EGQRWLPLEANPEVTNQFLKQLGLHPNWQFVDVYGMDPELLSMVPRPVCAVLLLFPITEKYEVFRTEEEEKIKSQGQDVTSSVYFMKQTISNACGTIGLIHAIANNKDKMHFESGSTLKKFLEESVSMSPEERARYLENYDAIRVTHETSAHEGQTEAPSIDEKVDLHFIALVHVDGHLYELDGRKPFPINHGETSDETLLEDAIEVCKKFMERDPDELRFNAIALSAAMQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRG Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Hepatitis A virus cellular receptor 2 (TIM3) | 7M3Z | 4.28 | |
Target general information Gen name Hepatitis A virus HAVCR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms TIMD3; TIMD-3; TIM3; TIM-3; T-cell membrane protein 3; T-cell immunoglobulin mucin receptor 3; T-cell immunoglobulin and mucin domain-containing protein 3; HAVcr-2; HAVCR2; CD366 Protein family Immunoglobulin superfamily, TIM family Biochemical class Immunoglobulin Function Generally accepted to have an inhibiting function. Reports on stimulating functions suggest that the activity may be influenced by the cellular context and/or the respective ligand. Regulates macrophage activation. Inhibits T-helper type 1 lymphocyte (Th1)-mediated auto- and alloimmune responses and promotes immunological tolerance. In CD8+ cells attenuates TCR-induced signaling, specifically by blocking NF-kappaB and NFAT promoter activities resulting in the loss of IL-2 secretion. The function may implicate its association with LCK proposed to impair phosphorylation of TCR subunits, and/or LGALS9-dependent recruitment of PTPRC to the immunological synapse. In contrast, shown to activate TCR-induced signaling in T-cells probably implicating ZAP70, LCP2, LCK and FYN. Expressed on Treg cells can inhibit Th17 cell responses. Receptor for LGALS9. Binding to LGALS9 is believed to result in suppression of T-cell responses; the resulting apoptosis of antigen-specific cells may implicate HAVCR2 phosphorylation and disruption of its association with BAG6. Binding to LGALS9 is proposed to be involved in innate immune response to intracellular pathogens. Expressed on Th1 cells interacts with LGALS9 expressed on Mycobacterium tuberculosis-infected macrophages to stimulate antibactericidal activity including IL-1 beta secretion and to restrict intracellular bacterial growth. However, the function as receptor for LGALS9 has been challenged. Also reported to enhance CD8+ T-cell responses to an acute infection such as by Listeria monocytogenes. Receptor for phosphatidylserine (PtSer); PtSer-binding is calcium-dependent. May recognize PtSer on apoptotic cells leading to their phagocytosis. Mediates the engulfment of apoptotic cells by dendritic cells. Expressed on T-cells, promotes conjugation but not engulfment of apoptotic cells. Expressed on dendritic cells (DCs) positively regulates innate immune response and in synergy with Toll-like receptors promotes secretion of TNF-alpha. In tumor-imfiltrating DCs suppresses nucleic acid-mediated innate immune repsonse by interaction with HMGB1 and interfering with nucleic acid-sensing and trafficking of nucleid acids to endosomes. Expressed on natural killer (NK) cells acts as a coreceptor to enhance IFN-gamma production in response to LGALS9. In contrast, shown to suppress NK cell-mediated cytotoxicity. Negatively regulates NK cell function in LPS-induced endotoxic shock. Cell surface receptor implicated in modulating innate and adaptive immune responses. Related diseases May be involved in T-cell exhaustion associated with chronic viral infections such as with human immunodeficiency virus (HIV) and hepatitic C virus (HCV). {ECO:0000269|PubMed:19001139, ECO:0000269|PubMed:19587053}.; DISEASE: T-cell lymphoma, subcutaneous panniculitis-like (SPTCL) [MIM:618398]: An uncommon form of T-cell non-Hodgkin lymphoma, in which cytotoxic CD8+ T-cells infiltrate subcutaneous adipose tissue, and rimming adipocytes in a lace-like pattern. Affected individuals typically present with multiple subcutaneous nodules, systemic B-cell symptoms, and, in a subset of cases, autoimmune disorders, most commonly systemic lupus erythematosus. A subset of patients develop hemophagocytic lymphohistiocytosis. SPTCL transmission pattern is consistent with autosomal recessive inheritance with incomplete penetrance. {ECO:0000269|PubMed:30374066, ECO:0000269|PubMed:30792187, ECO:0000269|Ref.2}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P13688; Q96IW7; Q8N2M4 EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Alternative splicing; Cell junction; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Immunity; Immunoglobulin domain; Inflammatory response; Innate immunity; Membrane; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 12286.9 Length 109 Aromaticity 0.12 Instability index 26.55 Isoelectric point 5.04 Charge (pH=7) -2.58 2D Binding mode Binding energy (Kcal/mol) -5.83  Molscript Map 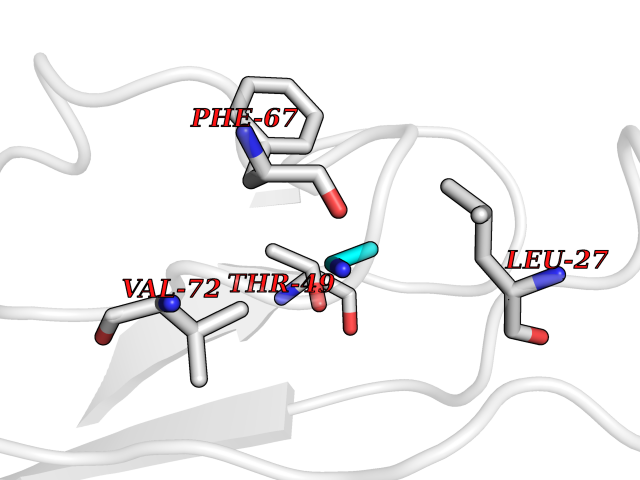 Pymol Map 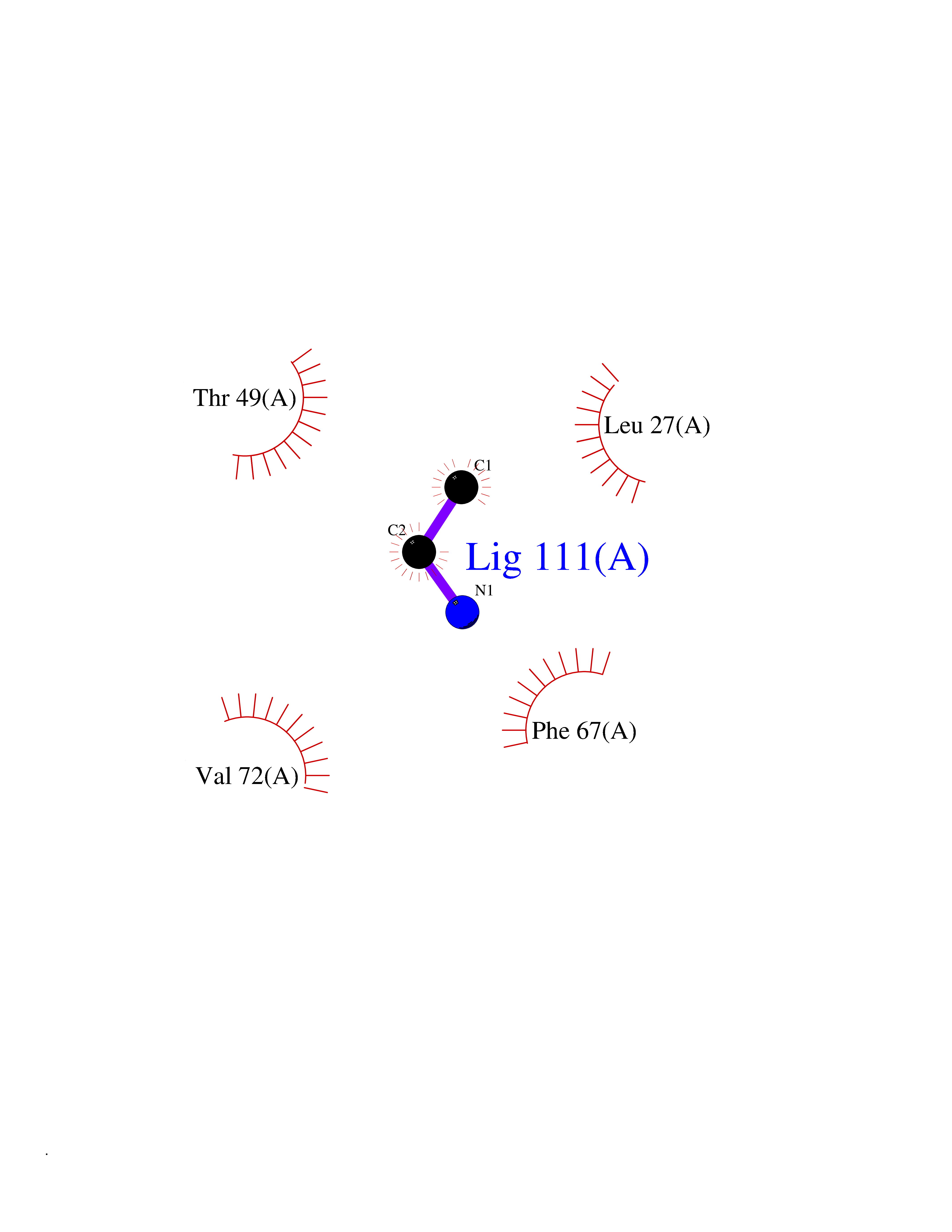 Ligplot Map 3D Binding mode Sequence SEVEYRAEVGQNAYLPCFYTPAAPGNLVPVCWGKGACPVFECGNVVLRTDERDVNYWTSRYWLNGDFRKGDVSLTIENVTLADSGIYCCRIQIPGIMNDEKFNLKLVIK Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Complement C1s component (C1S) | 1ELV | 4.28 | |
Target general information Gen name C1S Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Complement component 1 subcomponent s; Complement C1s subcomponent; C1-esterase; C1 esterase Protein family Peptidase S1 family Biochemical class Peptidase Function C1r activates C1s so that it can, in turn, activate C2 and C4. C1s B chain is a serine protease that combines with C1q and C1r to form C1, the first component of the classical pathway of the complement system. Related diseases Complement component C1s deficiency (C1SD) [MIM:613783]: A rare defect resulting in C1 deficiency and impaired activation of the complement classical pathway. C1 deficiency generally leads to severe immune complex disease with features of systemic lupus erythematosus and glomerulonephritis. {ECO:0000269|PubMed:11390518}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ehlers-Danlos syndrome, periodontal type, 2 (EDSPD2) [MIM:617174]: A form of Ehlers-Danlos syndrome, a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDSPD2 is characterized by the association of typical features of Ehlers-Danlos syndrome with gingival recession and severe early-onset periodontal disease, leading to premature loss of permanent teeth. EDSPD2 transmission pattern is consistent with autosomal dominant inheritance. {ECO:0000269|PubMed:27745832}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02371; DB09228; DB09130; DB12831; DB06404; DB14996; DB01593; DB14487; DB14533; DB14548 Interacts with P00736; P09871; P06681; O43889-2; Q9H6H4; P05155 EC number EC 3.4.21.42 Uniprot keywords 3D-structure; Calcium; Complement pathway; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Ehlers-Danlos syndrome; Glycoprotein; Hydrolase; Hydroxylation; Immunity; Innate immunity; Metal-binding; Protease; Proteomics identification; Reference proteome; Repeat; Serine protease; Signal; Sushi Protein physicochemical properties Chain ID A Molecular weight (Da) 33278.6 Length 303 Aromaticity 0.1 Instability index 33.69 Isoelectric point 5.16 Charge (pH=7) -7.95 2D Binding mode Binding energy (Kcal/mol) -5.84 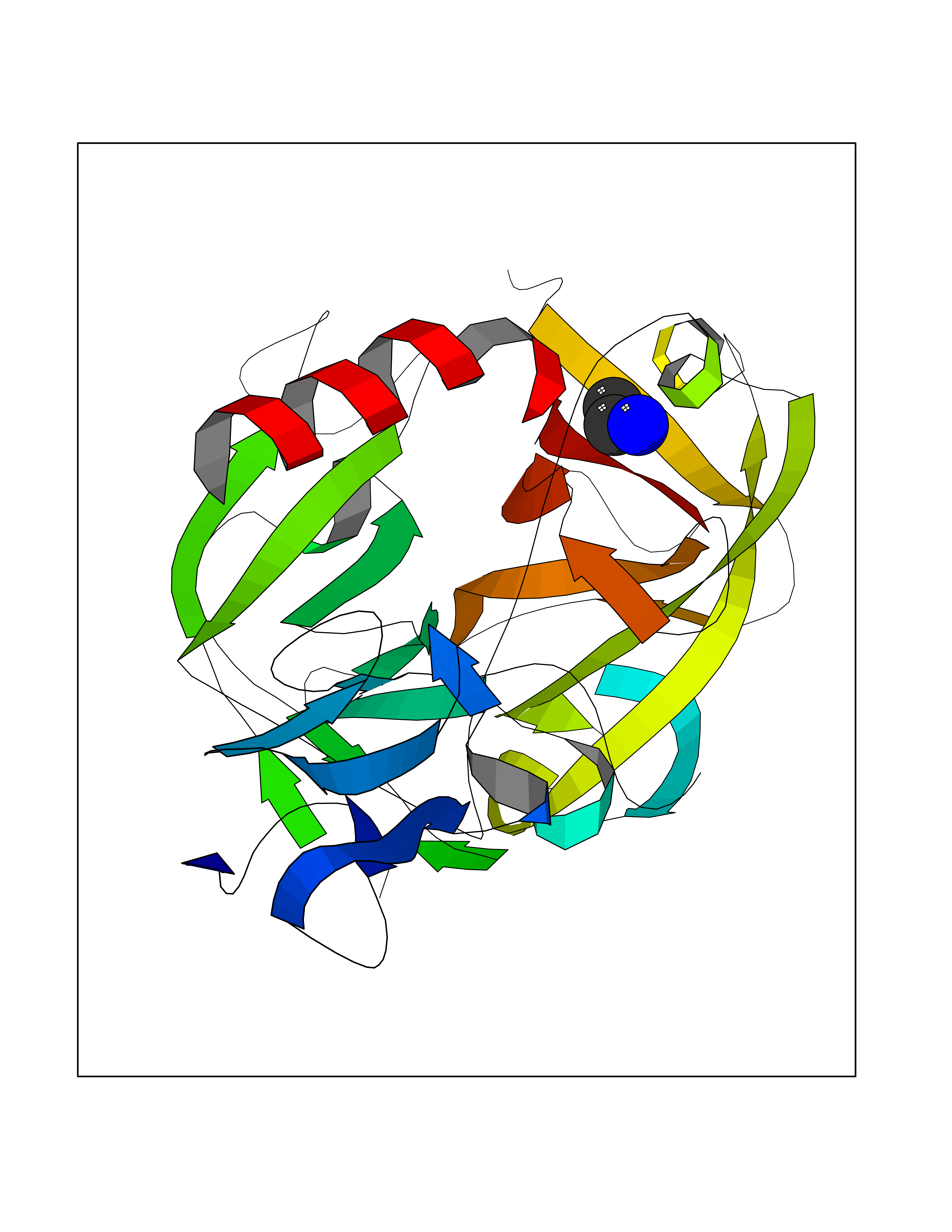 Molscript Map  Pymol Map 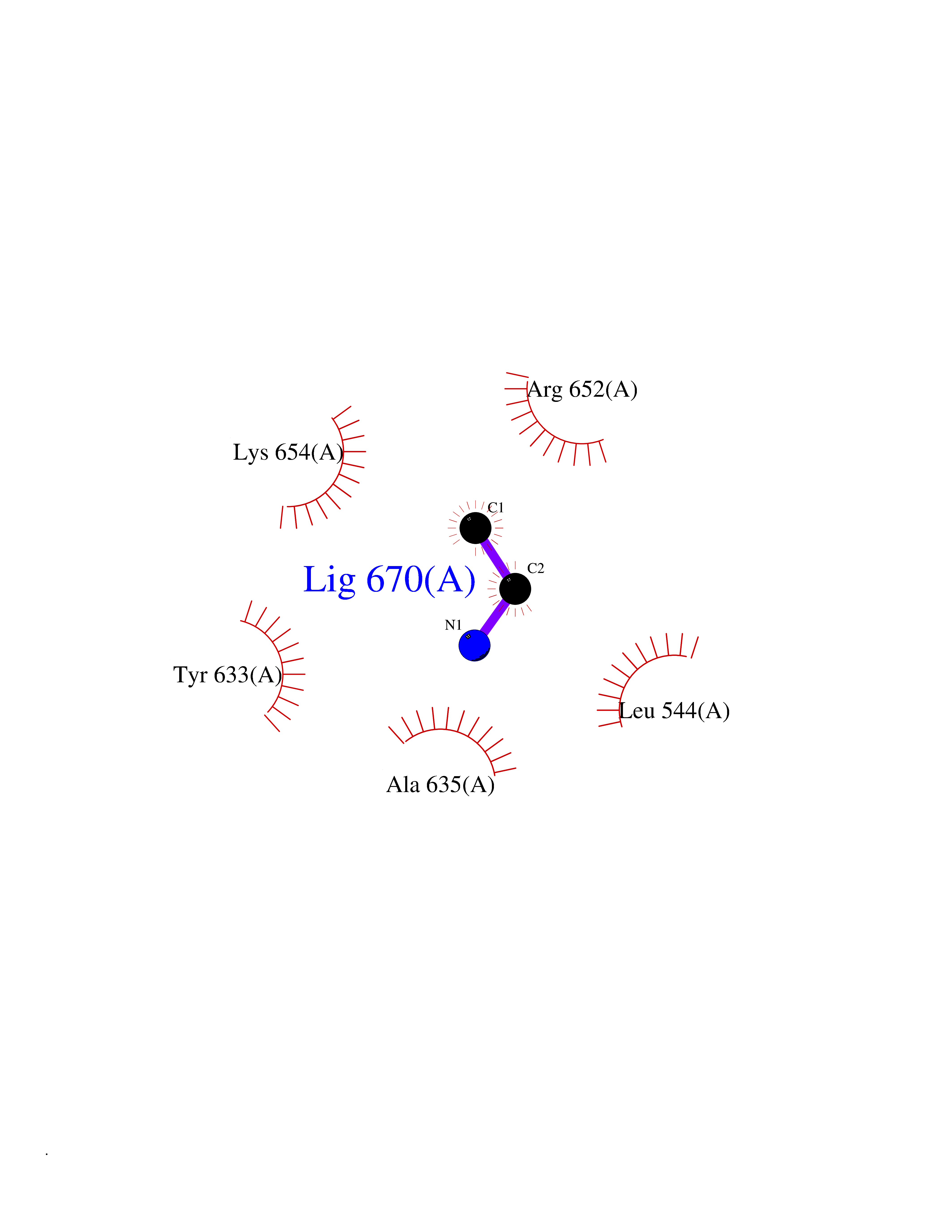 Ligplot Map 3D Binding mode Sequence LDCGIPESIENGKVEDPESTLFGSVIRYTCEEPYYYMEGGGEYHCAGNGSWVNEVLGPELPKCVPVCGVPREPFIIGGSDADIKNFPWQVFFDNPWAGGALINEYWVLTAAHVVEGNREPTMYVGSTSVQKMLTPEHVFIHPGWKLLAVPEGRTNFDNDIALVRLKDPVKMGPTVSPICLPGTSSDYNLMDGDLGLISGWGRTEKRDRAVRLKAARLPVAPLRKCKEVAYVFTPNMICAGGEKGMDSCKGDSGGAFAVQDPNDKTKFYAAGLVSWGPQCGTYGLYTRVKNYVDWIMKTMQENS Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | CDC7-related kinase (CDC7) | 6YA6 | 4.28 | |
Target general information Gen name CDC7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms huCdc7; HsCdc7; Cell division cycle 7related protein kinase; Cell division cycle 7-related protein kinase; CDC7related kinase; CDC7L1 Protein family Protein kinase superfamily, Ser/Thr protein kinase family, CDC7 subfamily Biochemical class Kinase Function Can phosphorylates MCM2 and MCM3. Seems to phosphorylate critical substrates that regulate the G1/S phase transition and/or DNA replication. Related diseases Blistering, acantholytic, of oral and laryngeal mucosa (ABOLM) [MIM:619226]: An autosomal recessive disorder characterized by recurrent, suprabasal acantholytic blisters in the oral and laryngeal mucosa. Skin, conjunctival and genital mucosa, nail folds, and nails are unaffected. Normal structure is observed in the scalp epidermis and hair follicle. {ECO:0000269|PubMed:30528827}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q8N9N5-2; Q13137; Q9UBU7; Q9UBU7-1; P51114-2; Q9UKD1; Q08379; Q9NYA3; Q6NT76; P42858; Q13422-7; Q9UKT9; Q9H2S9; Q9Y250; Q9BTE3; Q6FHY5; Q5JR59-3; Q14140; O75886; P55061; P14373; O94972; Q2TAA8; P0CW01; Q9Y6T4; Q96DT7-3; Q9HCK0; Q8NAP8 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell cycle; Cell division; Isopeptide bond; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 38430.1 Length 344 Aromaticity 0.08 Instability index 37.74 Isoelectric point 8.48 Charge (pH=7) 4.45 2D Binding mode Binding energy (Kcal/mol) -5.84  Molscript Map 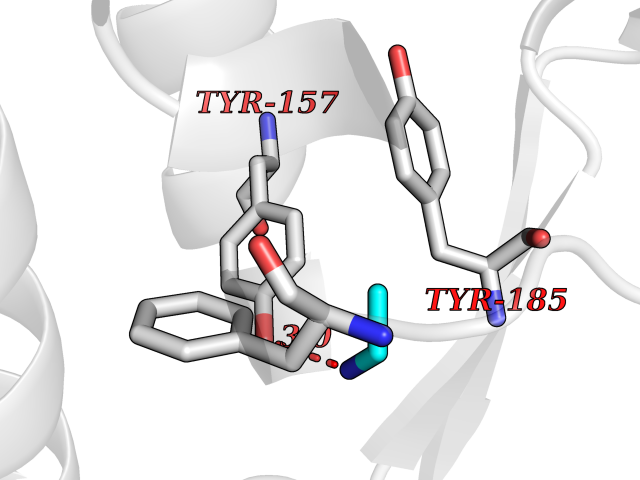 Pymol Map 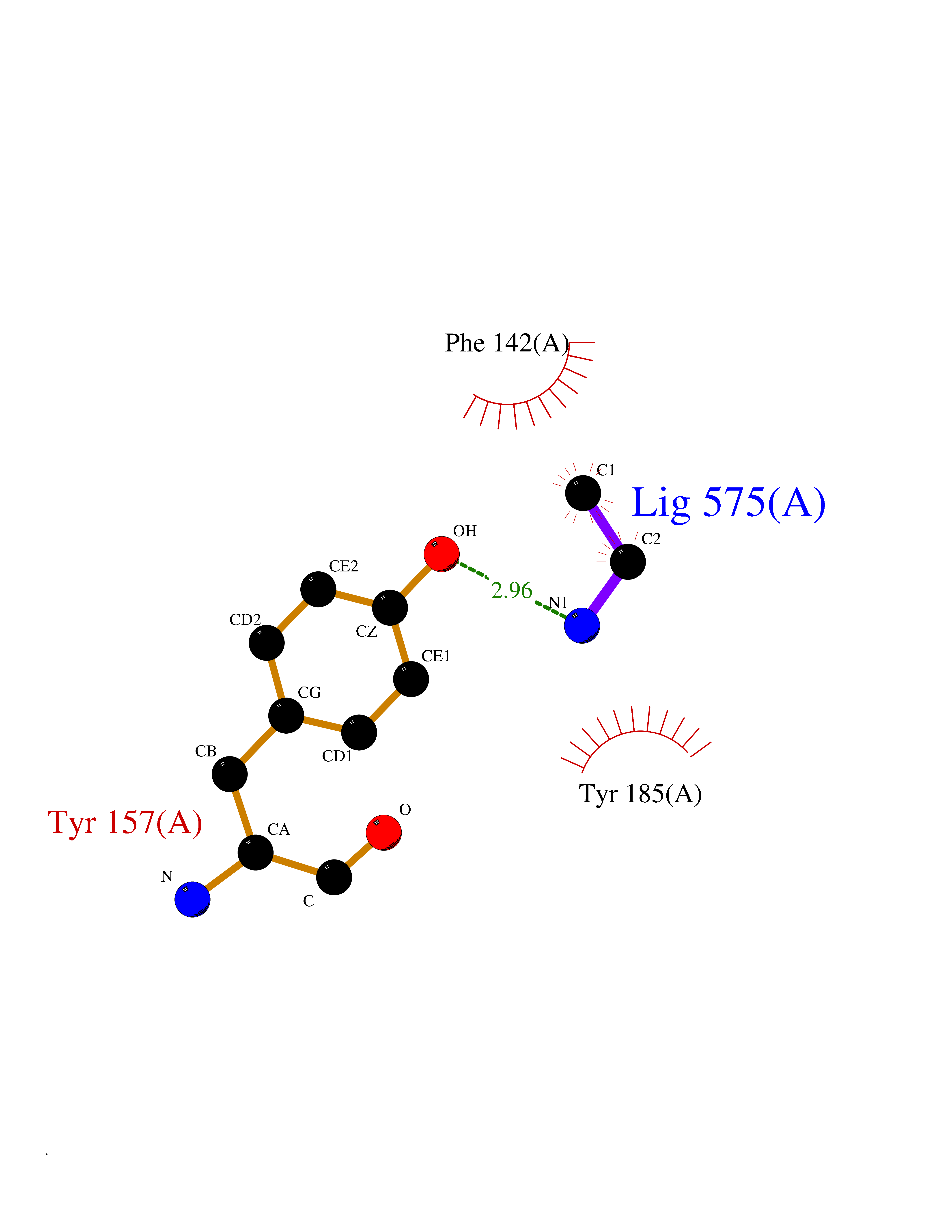 Ligplot Map 3D Binding mode Sequence AGVKKDIEKLYEAVPQLSNVFKIEDKIGEGTFSSVYLATAQLQVGPEEKIALKHLIPTSHPIRIAAELQCLTVAGGQDNVMGVKYCFRKNDHVVIAMPYLEHESFLDILNSLSFQEVREYMLNLFKALKRIHQFGIVHRDVKPSNFLYNRRLKKYALVDFGLAQGTHDTKIELLKFVQPASLTCDCYATDKVCSICLSRRQQVAPRAGTPGFRAPEVLTKCPNQTTAIDMWSAGVIFLSLLSGRYPFYKASDDLTALAQIMTIRGSRETIQAAKTFGKSILCSKEVPAQDLRKLCERLRGAGAGGWNEVPDEAYDLLDKLLDLNPASRITAEEALLHPFFKDMS Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | "15-cis-phytoene desaturase, chloroplastic/chromoplastic (EC 1.3.5.5) (Phytoene dehydrogenase) (Phytoene desaturase)" | 5MOG | 4.27 | |
Target general information Gen name PDS1 Organism Oryza sativa subsp. indica (Rice) Uniprot ID TTD ID NA Synonyms PDS;OsI_010044 Protein family Carotenoid/retinoid oxidoreductase family Biochemical class NA Function Converts phytoene into zeta-carotene via the intermediary of phytofluene by the symmetrical introduction of two double bonds at the C-11 and C-11' positions of phytoene with a concomitant isomerization of two neighboring double bonds at the C9 and C9' positions from trans to cis. Active with decylplastoquinone (DPQ) as substrate (PubMed:26147209, PubMed:29176862). Also active with other benzoquinones, which are strongly preferred over naphthoquinones as substrates (PubMed:26147209). {ECO:0000269|PubMed:26147209, ECO:0000269|PubMed:29176862}." Related diseases NA Drugs (DrugBank ID) NA Interacts with NA EC number 1.3.5.5 Uniprot keywords 3D-structure; Carotenoid biosynthesis; Chloroplast; Chromoplast; Direct protein sequencing; FAD; Flavoprotein; Membrane; Oxidoreductase; Plastid; Reference proteome; Transit peptide Protein physicochemical properties Chain ID E Molecular weight (Da) 52485.1 Length 466 Aromaticity 0.1 Instability index 45.53 Isoelectric point 5.93 Charge (pH=7) -5.81 2D Binding mode Binding energy (Kcal/mol) -5.82 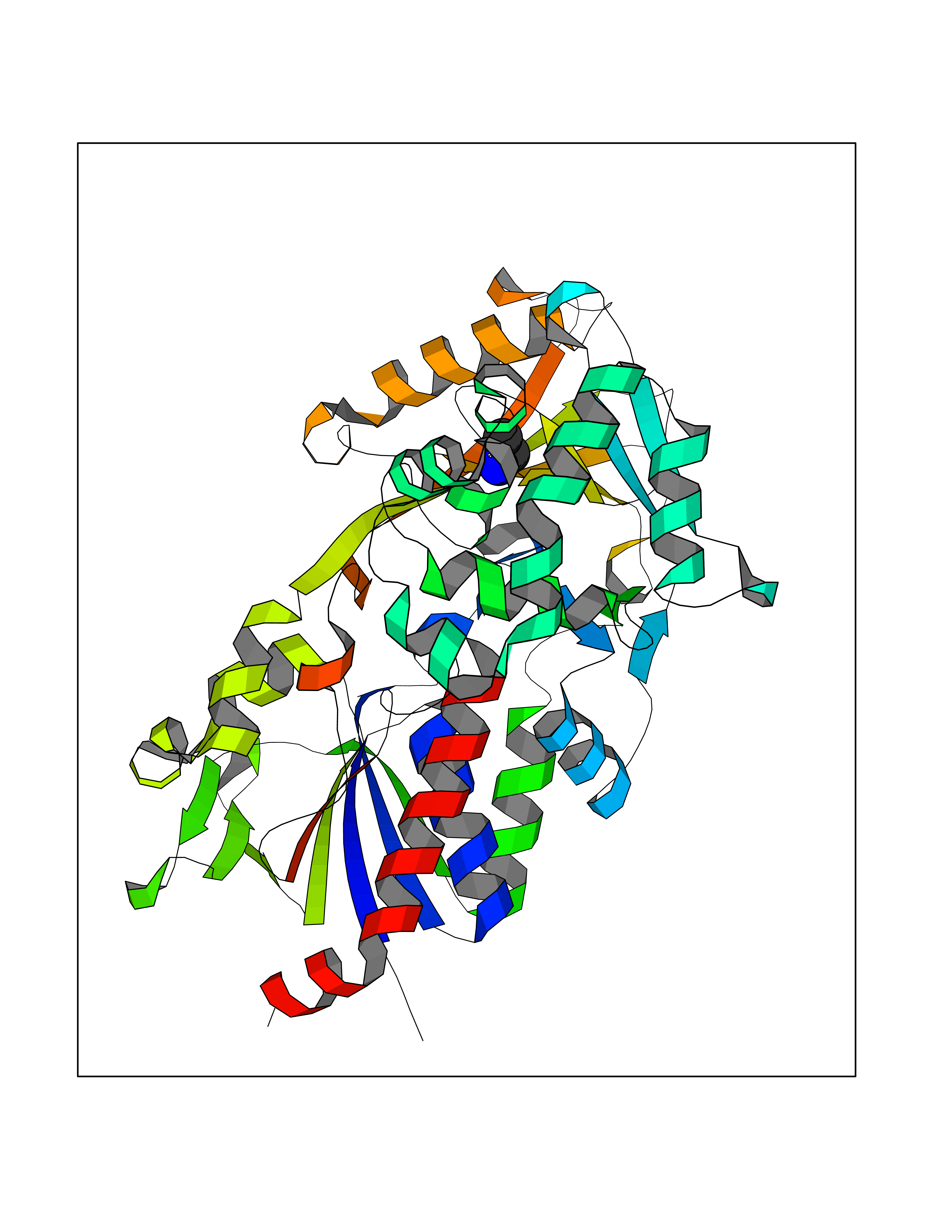 Molscript Map 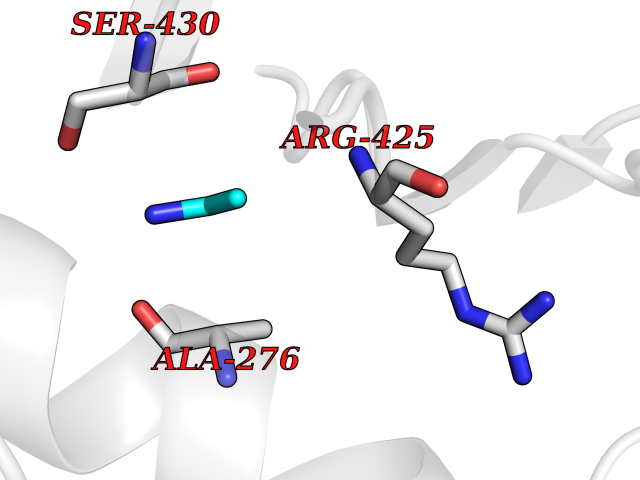 Pymol Map 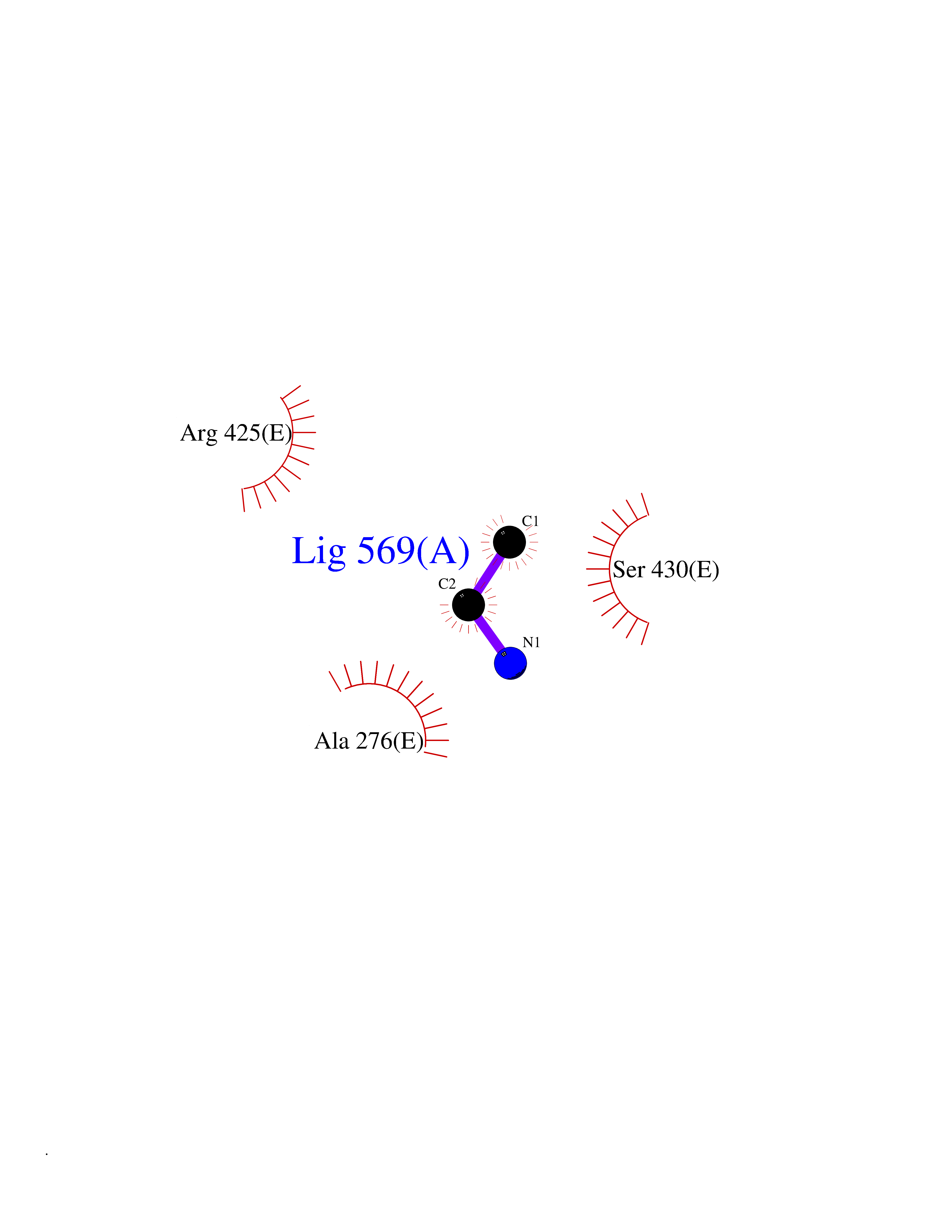 Ligplot Map 3D Binding mode Sequence TKPLQVVIAGAGLAGLSTAKYLADAGHKPILLEARDVLGGKIAAWKDEDGDWYETGLHIFFGAYPNIQNLFGELGINDRLQWKEHSMIFAMPNKPGEFSRFDFPETLPAPLNGIWAILRNNEMLTWPEKVKFALGLLPAMVGGQAYVEAQDGFTVSEWMKKQGVPDRVNDEVFIAMSKALNFINPDELSMQCILIALNRFLQEKHGSKMAFLDGNPPERLCMPIVDHVRSLGGEVRLNSRIQKIELNPDGTVKHFALTDGTQITGDAYVFATPVDILKLLVPQEWKEISYFKKLEKLVGVPVINVHIWFDRKLKNTYDHLLFSRSSLLSVYADMSVTCKEYYDPNRSMLELVFAPAEEWVGRSDTEIIEATMQELAKLFPDEIAADQSKAKILKYHVVKTPRSVYKTIPDCEPCRPLQRSPIEGFYLAGDYTKQKYLASMEGAVLSGKLCAQSVVEDYKMLSRRSL Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Folate receptor beta (FOLR2) | 4KN0 | 4.27 | |
Target general information Gen name FOLR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Placental folate-binding protein; Folate receptor, fetal/placental; Folate receptor type-beta; Folate receptor 2; FR-beta; FOLR2 Protein family Folate receptor family Biochemical class Folate receptor Function Binds to folate and reduced folic acid derivatives and mediates delivery of 5-methyltetrahydrofolate and folate analogs into the interior of cells. Has high affinity for folate and folic acid analogs at neutral pH. Exposure to slightly acidic pH after receptor endocytosis triggers a conformation change that strongly reduces its affinity for folates and mediates their release. Related diseases Acute hepatic porphyria (AHEPP) [MIM:612740]: A form of porphyria. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. AHP is characterized by attacks of gastrointestinal disturbances, abdominal colic, paralyses and peripheral neuropathy. Most attacks are precipitated by drugs, alcohol, caloric deprivation, infections, or endocrine factors. {ECO:0000269|PubMed:10706561, ECO:0000269|PubMed:1309003, ECO:0000269|PubMed:1569184, ECO:0000269|PubMed:17236137, ECO:0000269|PubMed:2063868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00158; DB00563; DB05168 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Direct protein sequencing; Disulfide bond; Folate-binding; Glycoprotein; GPI-anchor; Lipoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23841.6 Length 205 Aromaticity 0.12 Instability index 56.78 Isoelectric point 7.92 Charge (pH=7) 2.58 2D Binding mode Binding energy (Kcal/mol) -5.83 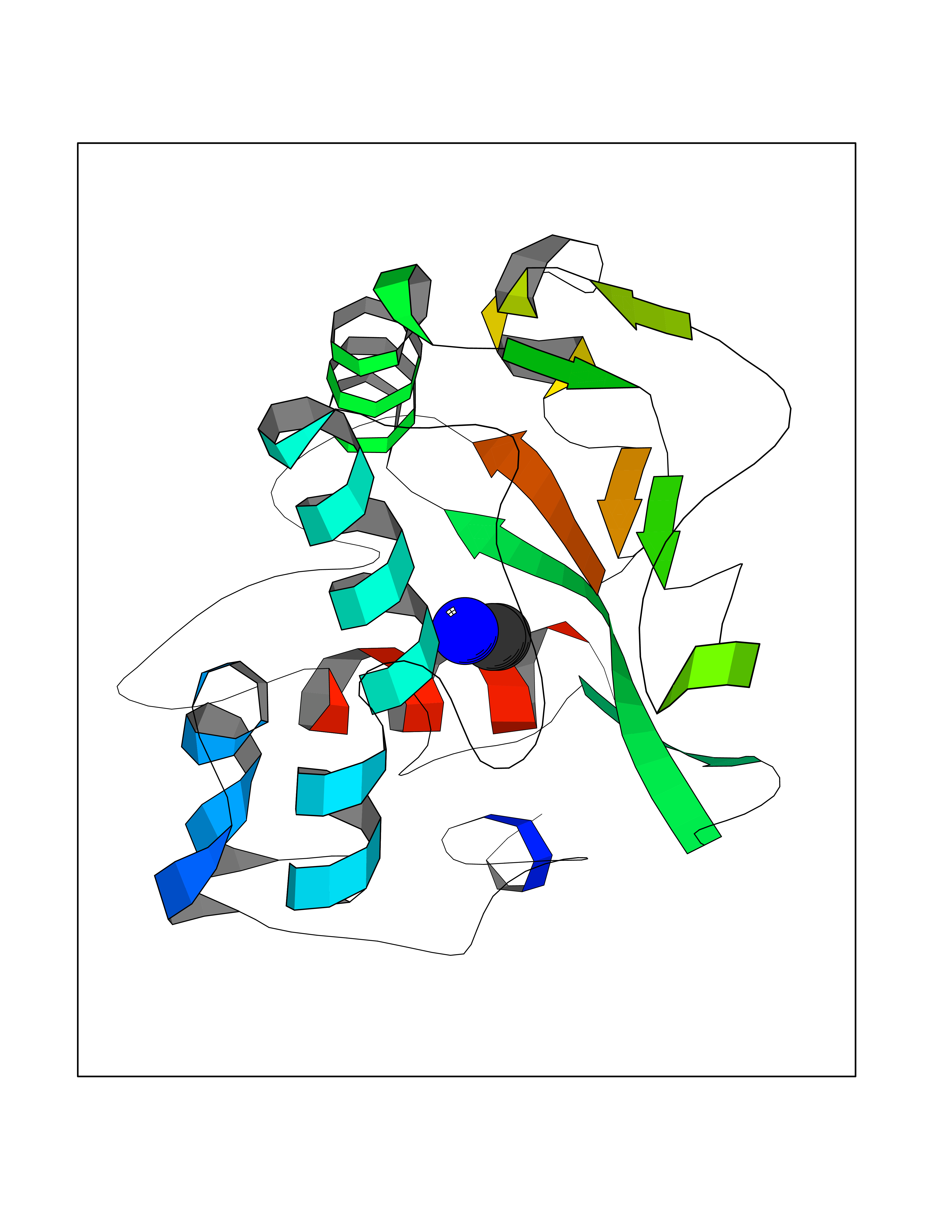 Molscript Map 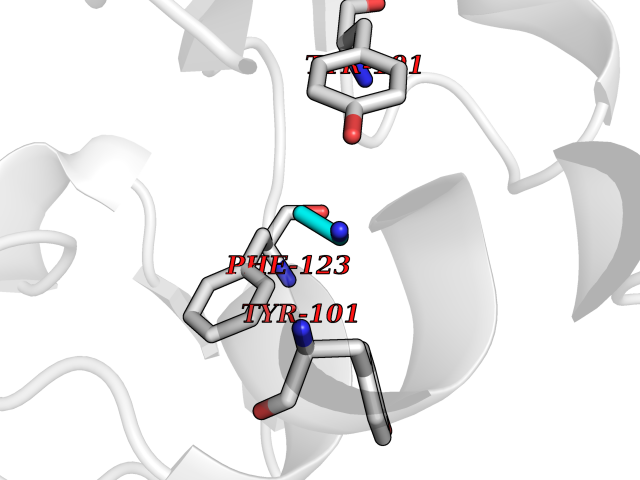 Pymol Map 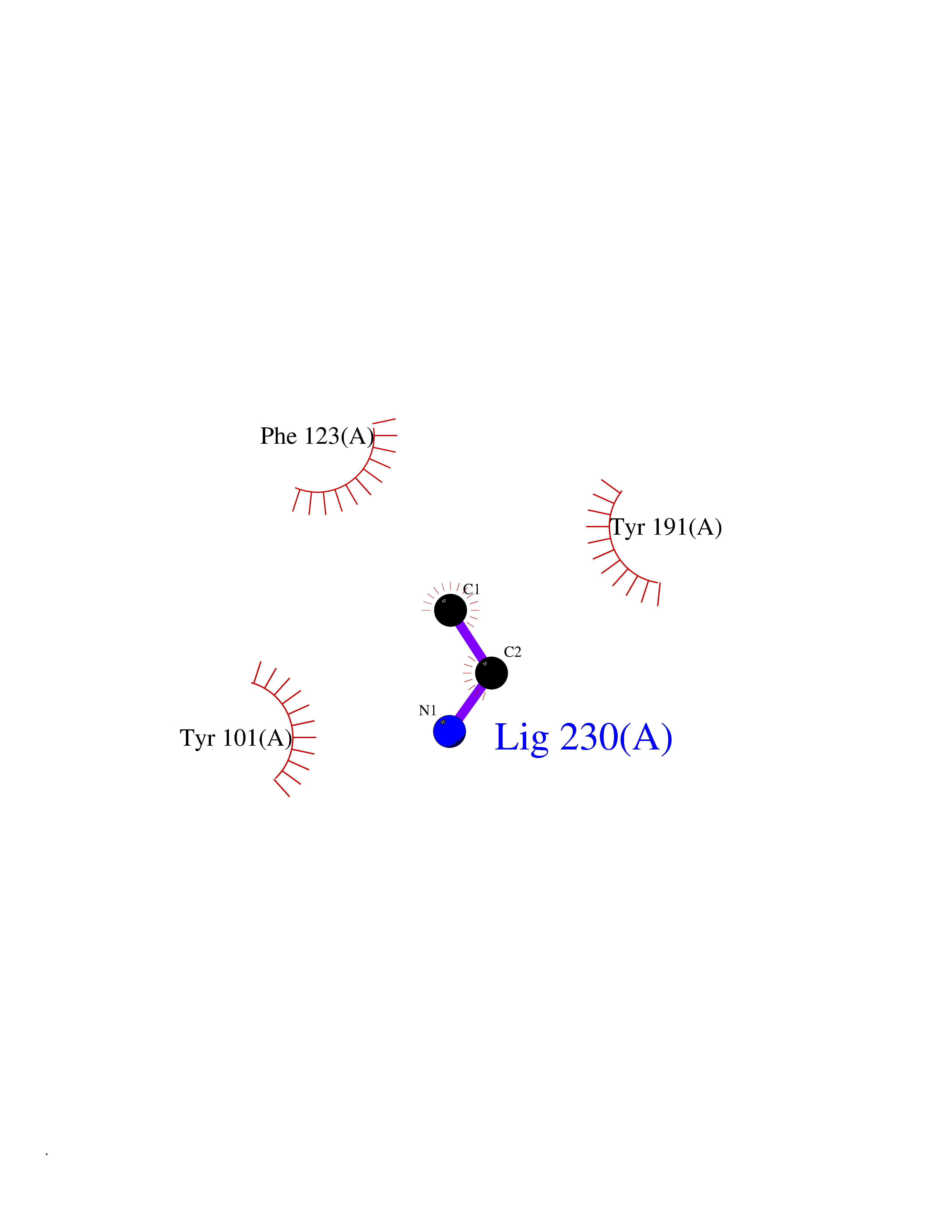 Ligplot Map 3D Binding mode Sequence RTDLLNVCMDAKHHKTKPGPEDKLHDQCSPWKKNACCTASTSQELHKDTSRLYNFNWDHCGKMEPACKRHFIQDTCLYECSPNLGPWIQQVNQSWRKERFLDVPLCKEDCQRWWEDCHTSHTCKSNWHRGWDWTSGVNKCPAGALCRTFESYFPTPAALCEGLWSHSYKVSNYSRGSGRCIQMWFDSAQGNPNEEVARFYAAAMH Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | 5'-methylthioadenosine/S-adenosylhomocysteine nucleosidase | 4WKC | 4.27 | |
Target general information Gen name mtnN Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms pfs;b0159;yadA;JW0155;mtn Protein family PNP/UDP phosphorylase family, MtnN subfamily Biochemical class hydrolase / hydrolase inhibitor Function Adenosylhomocysteine nucleosidase activity.Identical protein binding.Methylthioadenosine nucleosidase activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02158; DB08606; DB02933; DB00173; DB02281 Interacts with P0AF12 EC number 3.2.2.9 Uniprot keywords 3D-structure; Amino-acid biosynthesis; Hydrolase; Methionine biosynthesis; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 24353.7 Length 232 Aromaticity 0.05 Instability index 22.1 Isoelectric point 5.09 Charge (pH=7) -9.9 2D Binding mode Binding energy (Kcal/mol) -5.82  Molscript Map 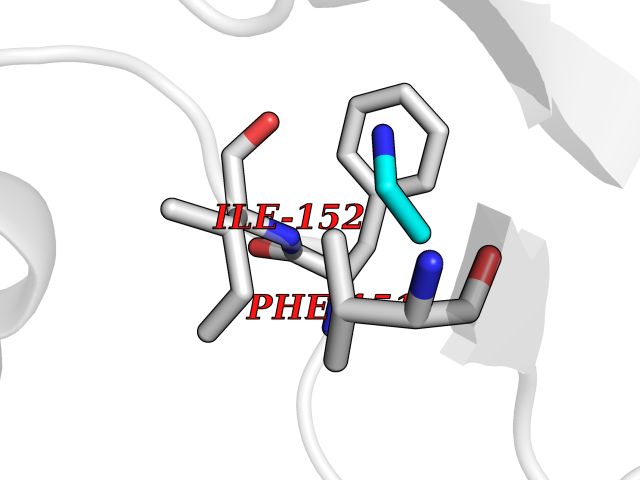 Pymol Map  Ligplot Map 3D Binding mode Sequence MKIGIIGAMEEEVTLLRDKIENRQTISLGGCEIYTGQLNGTEVALLKSGIGKVAAALGATLLLEHCKPDVIINTGSAGGLAPTLKVGDIVVSDEARYHDADVTAFGYEYGQLPGCPAGFKADDKLIAAAEACIAELNLNAVRGLIVSGDAFINGSVGLAKIRHNFPQAIAVEMEATAIAHVCHNFNVPFVVVRAISDVADQQSHLSFDEFLAVAAKQSSLMVESLVQKLAHG Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Ornithine decarboxylase (ODC1) | 2OO0 | 4.27 | |
Target general information Gen name ODC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ODC Protein family Orn/Lys/Arg decarboxylase class-II family Biochemical class Carbon-carbon lyase Function Polyamines are essential for cell proliferation and are implicated in cellular processes, ranging from DNA replication to apoptosis. Catalyzes the first and rate-limiting step of polyamine biosynthesis that converts ornithine into putrescine, which is the precursor for the polyamines, spermidine and spermine. Related diseases Bachmann-Bupp syndrome (BABS) [MIM:619075]: An autosomal dominant disorder characterized by global developmental delay, alopecia, absolute or relative macrocephaly, and facial dysmorphism. Neuroimaging shows white matter abnormalities, prominent Virchow-Robin spaces, periventricular cysts, and abnormalities of the corpus callosum. {ECO:0000269|PubMed:30239107, ECO:0000269|PubMed:30475435}. The disease is caused by variants affecting the gene represented in this entry. BABS is due to truncating variants that lead to a gain of function. This phenomenon apparently results from truncation proximal to or involving the C-terminal region of ODC1 protein, distal enough to allow escape from nonsense-mediated decay. A gain of function is corroborated by elevated plasma levels of N-acetylputrescine, with otherwise normal polyamine levels, in affected individuals. {ECO:0000269|PubMed:30475435}. Drugs (DrugBank ID) DB06243; DB04263; DB03856; DB04083; DB02824; DB01917; DB00114; DB02209; DB00203; DB00127; DB00313 Interacts with Q9H8Y8; Q92993; Q9UMX2; Q9UMX2-2 EC number EC 4.1.1.17 Uniprot keywords 3D-structure; Decarboxylase; Disease variant; Hypotrichosis; Lyase; Phosphoprotein; Polyamine biosynthesis; Proteomics identification; Pyridoxal phosphate; Reference proteome; S-nitrosylation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 45682.9 Length 410 Aromaticity 0.11 Instability index 40.93 Isoelectric point 5.61 Charge (pH=7) -6.68 2D Binding mode Binding energy (Kcal/mol) -5.82  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LMNNFGNEEFDCHFLDEGFTAKDILDQKINEVSSSDDKDAFYVADLGDILKKHLRWLKALPRVTPFYAVKCNDSKAIVKTLAATGTGFDCASKTEIQLVQSLGVPPERIIYANPCKQVSQIKYAANNGVQMMTFDSEVELMKVARAHPKAKLVLRIATDDSKAVCRLSVKFGATLRTSRLLLERAKELNIDVVGVSFHVGSGCTDPETFVQAISDARCVFDMGAEVGFSMYLLDIGGGFPGSEDVKLKFEEITGVINPALDKYFPSDSGVRIIAEPGRYYVASAFTLAVNIIAKKIVLEQTFMYYVNDGVYGSFNCILYDHAHVKPLLQKRPKPDEKYYSSSIWGPTCDGLDRIVERCDLPEMHVGDWMLFENMGAYTVAAASTFNGFQRPTIYYVMSGPAWQLMQQFQN Hydrogen bonds contact Hydrophobic contact | ||||