Job Results:
Ligand
Structure
Job ID
9d23599582ab75b793872ebb630d3299
Job name
NA
Time
2026-02-27 13:43:17
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Nischarin | 3P0C | 5.20 | |
Target general information Gen name NISCH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms KIAA0975;IRAS Protein family NA Biochemical class Signaling protein Function Identical protein binding.Integrin binding.Phosphatidylinositol binding. Related diseases Dyskinesia, limb and orofacial, infantile-onset (IOLOD) [MIM:616921]: An autosomal recessive, early-onset hyperkinetic movement disorder characterized by axial hypotonia, dyskinesia of the limbs and trunk, orofacial dyskinesia, drooling, and dysarthria. The severity of the hyperkinesis is variable. {ECO:0000269|PubMed:27058446}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Striatal degeneration, autosomal dominant 2 (ADSD2) [MIM:616922]: An autosomal dominant disorder characterized by striatal degeneration and dysfunction of basal ganglia, resulting in hyperkinesis. {ECO:0000269|PubMed:27058447}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08838; DB09242; DB15133; DB00697 Interacts with Q9Y2I1; P51151; P61107; P05714 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Apoptosis; Cell membrane; Coiled coil; Cytoplasm; Endosome; Leucine-rich repeat; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat Protein physicochemical properties Chain ID A,B Molecular weight (Da) 25709.3 Length 219 Aromaticity 0.11 Instability index 13.52 Isoelectric point 8.57 Charge (pH=7) 3.01 2D Binding mode Binding energy (Kcal/mol) -7.09 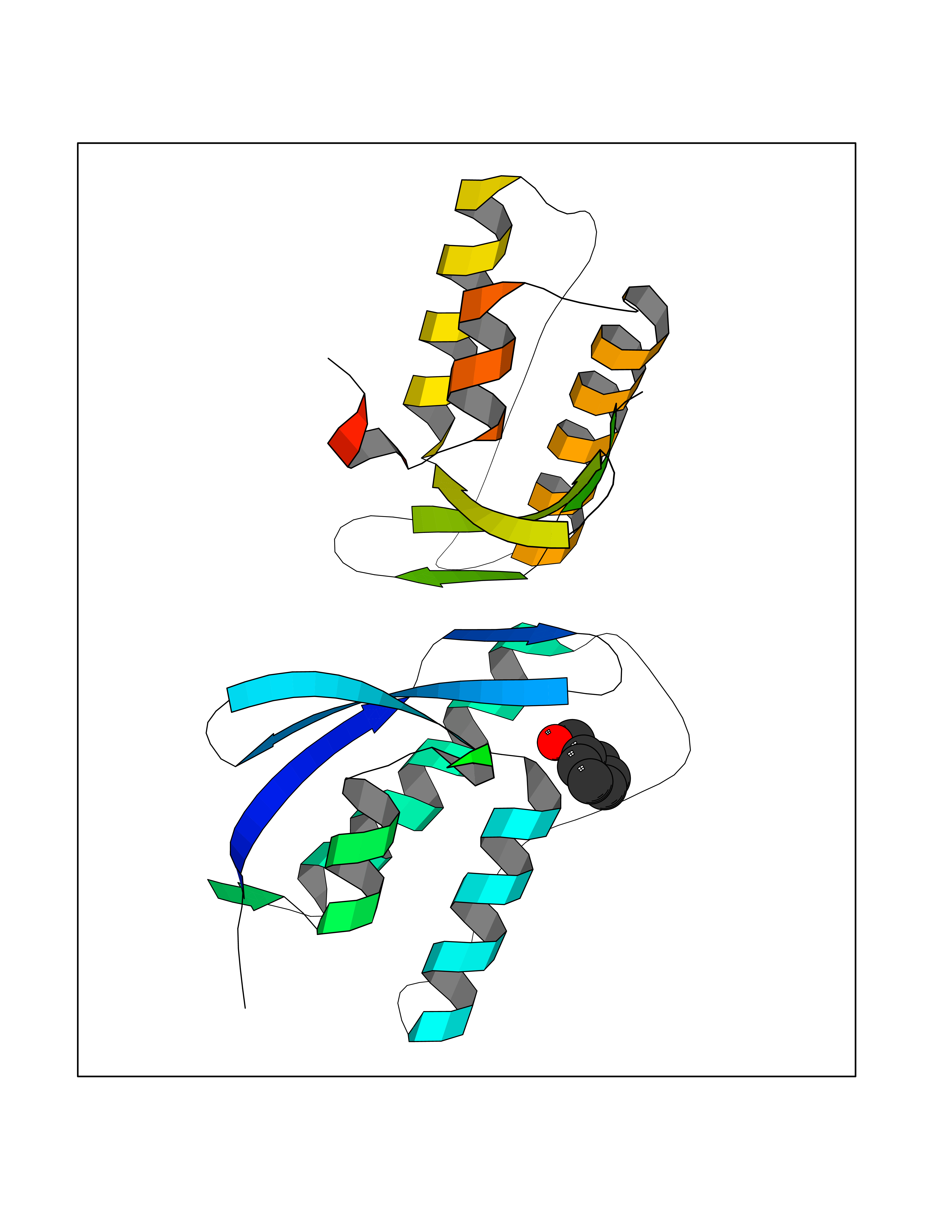 Molscript Map 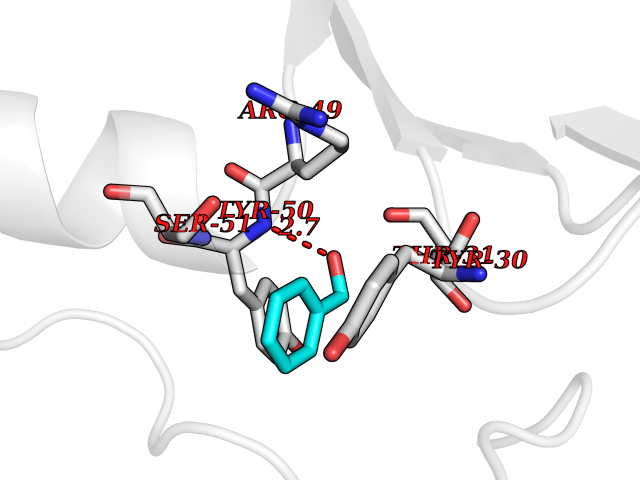 Pymol Map 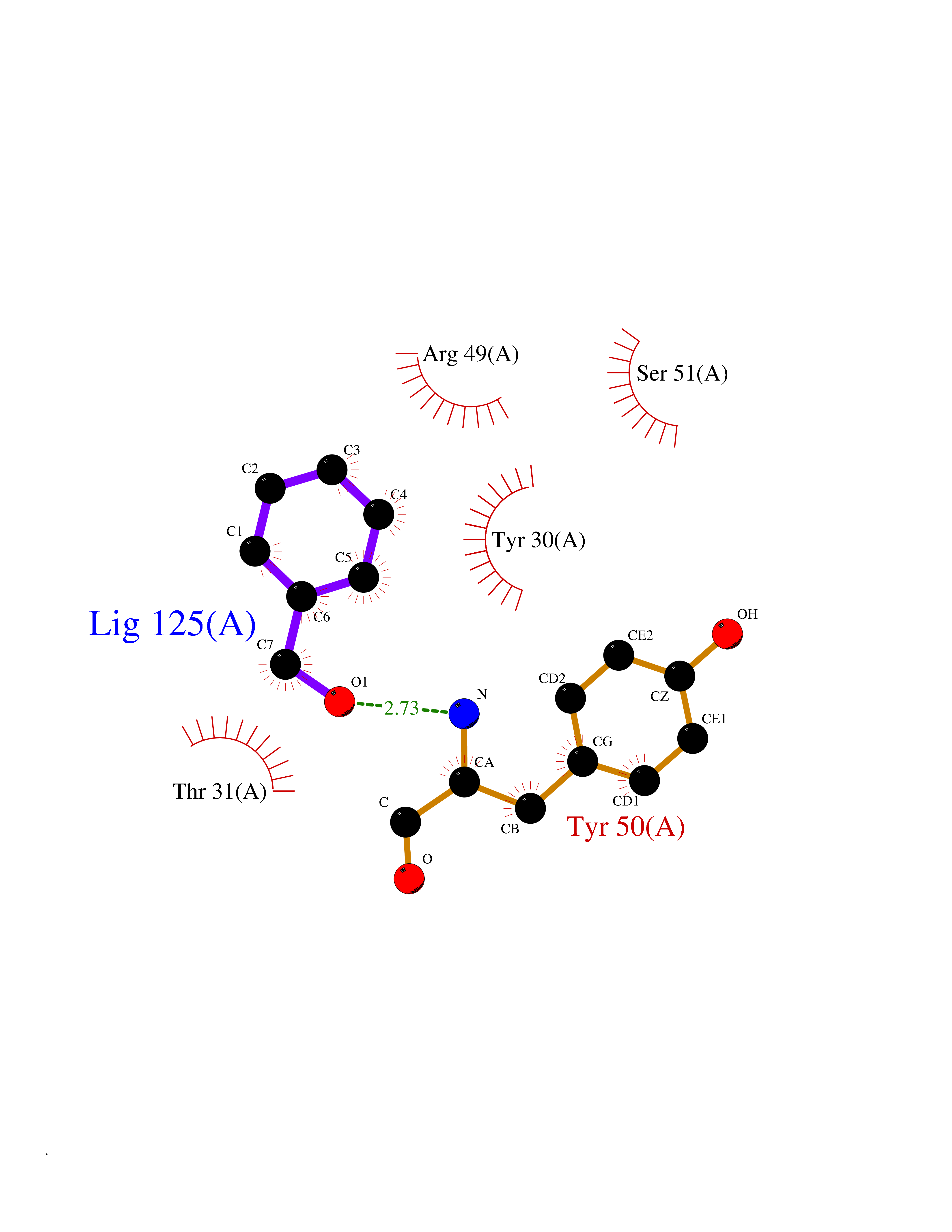 Ligplot Map 3D Binding mode Sequence NLYFQSMEARVVGSELVDTYTVYIIQVTDGSHEWTVKHRYSDFHDLHEKLVAERKIDKNLLPPKKIIGKNSRSLVEKREKDLEVYLQKLLAAFPGVTPRVLAHFLHFHFYESMEARVVGSELVDTYTVYIIQVTDGSHEWTVKHRYSDFHDLHEKLVAERKIDKNLLPPKKIIGKNSRSLVEKREKDLEVYLQKLLAAFPGVTPRVLAHFLHFHFYEIN Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Pseudomonas Transcriptional activator protein LasR (Pseudo LasR) | 3IX3 | 5.20 | |
Target general information Gen name Pseudo LasR Organism Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Uniprot ID TTD ID Synonyms NA Protein family Autoinducer-regulated transcriptional regulatory protein family Biochemical class NA Function Transcriptional activator of elastase structural gene (LasB). Binds to the PAI autoinducer. Related diseases Growth hormone deficiency, isolated, 1A (IGHD1A) [MIM:262400]: An autosomal recessive, severe deficiency of growth hormone leading to dwarfism. Patients often develop antibodies to administered growth hormone. {ECO:0000269|PubMed:8364549}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 1B (IGHD1B) [MIM:612781]: An autosomal recessive deficiency of growth hormone leading to short stature. Patients have low but detectable levels of growth hormone, significantly retarded bone age, and a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:12655557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Kowarski syndrome (KWKS) [MIM:262650]: A syndrome clinically characterized by short stature associated with bioinactive growth hormone, normal or slightly increased growth hormone secretion, pathologically low insulin-like growth factor 1 levels, and normal catch-up growth on growth hormone replacement therapy. {ECO:0000269|PubMed:17519310, ECO:0000269|PubMed:8552145, ECO:0000269|PubMed:9276733}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 2 (IGHD2) [MIM:173100]: An autosomal dominant deficiency of growth hormone leading to short stature. Clinical severity is variable. Patients have a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:11502836, ECO:0000269|PubMed:9152628}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08324 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; DNA-binding; Quorum sensing; Reference proteome; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 18305.5 Length 163 Aromaticity 0.12 Instability index 46.52 Isoelectric point 5.19 Charge (pH=7) -6.78 2D Binding mode Binding energy (Kcal/mol) -7.1 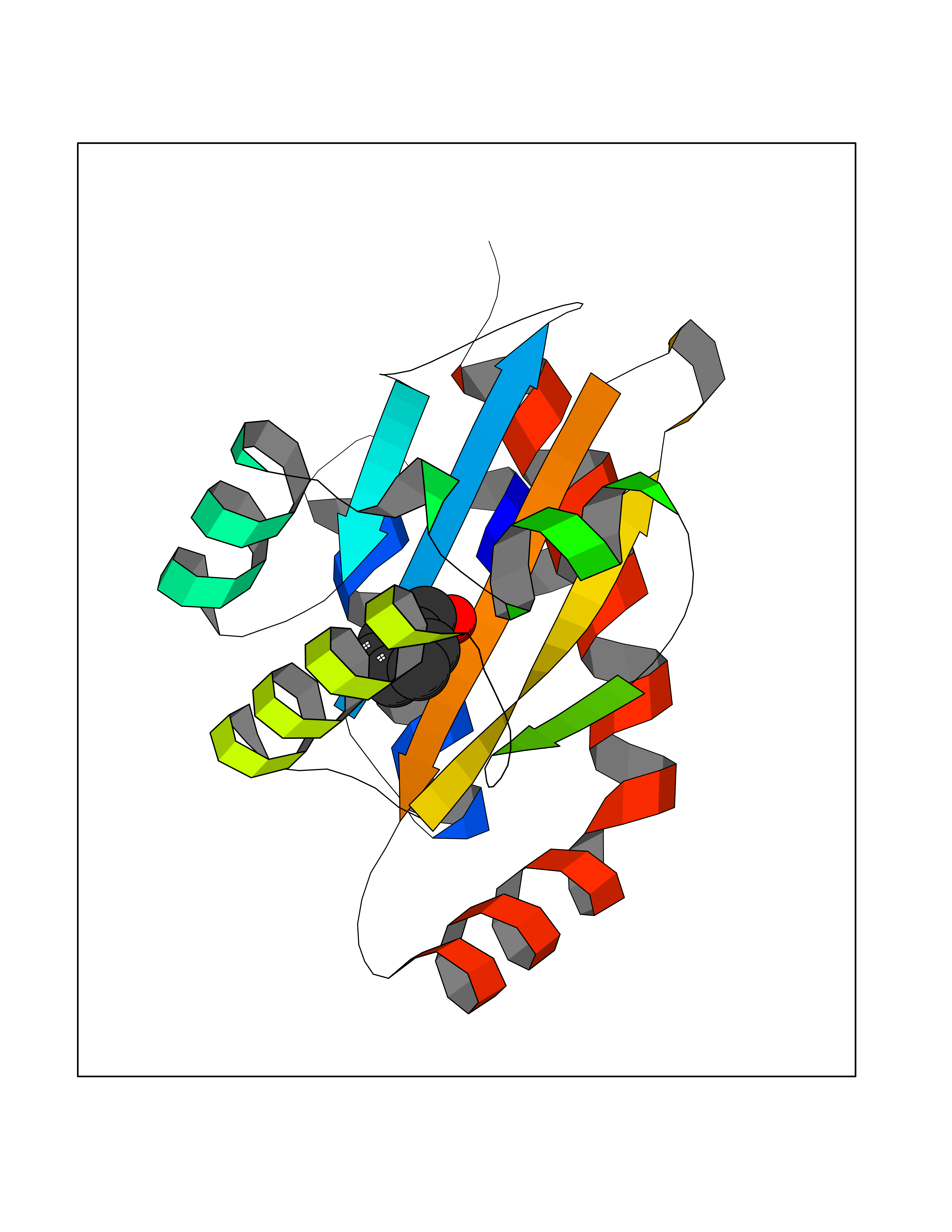 Molscript Map 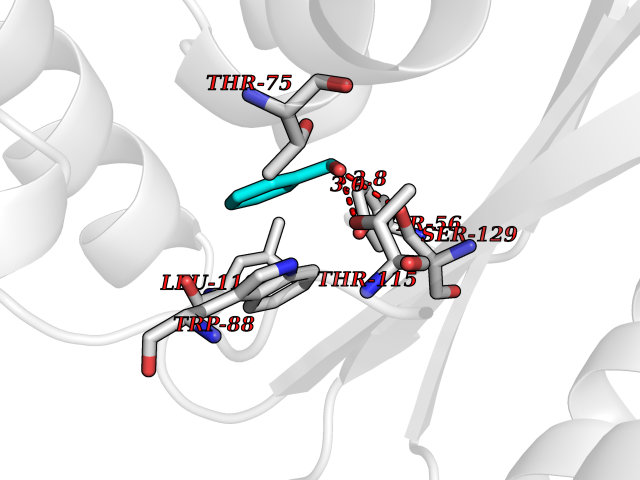 Pymol Map 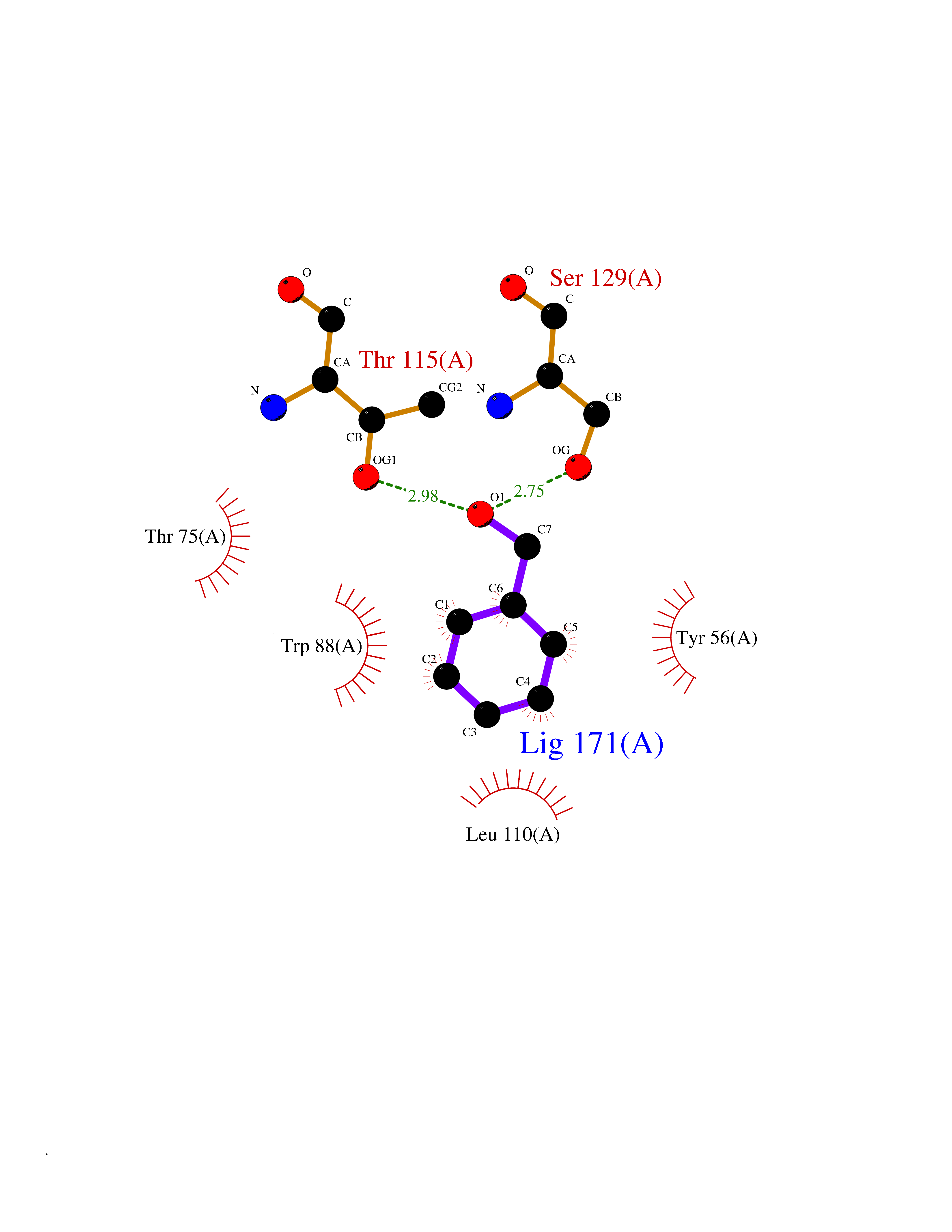 Ligplot Map 3D Binding mode Sequence FLELERSSGKLEWSAILQKMASDLGFSKILFGLLPKDSQDYENAFIVGNYPAAWREHYDRAGYARVDPTVSHCTQSVLPIFWEPSIYQTRKQHEFFEEASAAGLVYGLTMPLHGARGELGALSLSVEAENRAEANRFMESVLPTLWMLKDYALQSGAGLAFEH Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Haemophilus influenzae NadR protein (Hae-influ nadR) | 1LW7 | 5.20 | |
Target general information Gen name Hae-influ nadR Organism Haemophilus influenzae (strain ATCC 51907 / DSM 11121 / KW20 / Rd) Uniprot ID TTD ID Synonyms nadR; Transcriptional regulator nadR Protein family Bacterial NMN adenylyltransferase family; Bacterial RNK family Biochemical class Nicotinamide ribonucleoside uptake permease Function This enzyme has twoactivities: nicotinamide mononucleotide (NMN) adenylyltransferase and ribosylnicotinamide (RN) kinase. The RN kinase activity catalyzes the phosphorylation of RN to form nicotinamide ribonucleotide. The NMN adenylyltransferase activity catalyzes the transfer of the AMP moiety of ATP to nicotinamide ribonucleotide to form NAD(+). Related diseases Involved in the epigenetic regulation of ESR1 expression in breast cancer in a TFAP2C, IFI16 and HDAC4/5/6-dependent manner. {ECO:0000269|PubMed:24413532}. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative initiation; ATP-binding; Cell membrane; Cytoplasm; Kinase; Membrane; Multifunctional enzyme; NAD; Nucleotide-binding; Pyridine nucleotide biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 39581.5 Length 344 Aromaticity 0.14 Instability index 41.39 Isoelectric point 6.94 Charge (pH=7) -0.18 2D Binding mode Binding energy (Kcal/mol) -7.09  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence EKKVGVIFGKFYPVHTGHINXIYEAFSKVDELHVIVCSDTVRDLKLFYDSKXKRXPTVQDRLRWXQQIFKYQKNQIFIHHLVEDGIPSYPNGWQSWSEAVKTLFHEKHFEPSIVFSSEPQDKAPYEKYLGLEVSLVDPDRTFFNVSATKIRTTPFQYWKFIPKEARPFFAKTVAILGGESSGKSVLVNKLAAVFNTTSAWEYGREFVFEKLGGDEQAMQYSDYPQXALGHQRYIDYAVRHSHKIAFIDTDFITTQAFCIQYEGKAHPFLDSXIKEYPFDVTILLKNNTEQKQRQQFQQLLKKLLDKYKVPYIEIESPSYLDRYNQVKAVIEKVLNEEEISELQN Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Proto-oncogene c-Met (MET) | 3DKC | 5.20 | |
Target general information Gen name MET Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine-protein kinase Met; Scatter factor receptor; SF receptor; Met proto-oncogene tyrosine kinase; Hepatocyte growth factor receptor; HGF/SF receptor; HGF-SF receptor; HGF receptor; C-met; C-Met r Protein family Protein kinase superfamily, Tyr protein kinase family Biochemical class Kinase Function Regulates many physiological processes including proliferation, scattering, morphogenesis and survival. Ligand binding at the cell surface induces autophosphorylation of MET on its intracellular domain that provides docking sites for downstream signaling molecules. Following activation by ligand, interacts with the PI3-kinase subunit PIK3R1, PLCG1, SRC, GRB2, STAT3 or the adapter GAB1. Recruitment of these downstream effectors by MET leads to the activation of several signaling cascades including the RAS-ERK, PI3 kinase-AKT, or PLCgamma-PKC. The RAS-ERK activation is associated with the morphogenetic effects while PI3K/AKT coordinates prosurvival effects. During embryonic development, MET signaling plays a role in gastrulation, development and migration of muscles and neuronal precursors, angiogenesis and kidney formation. In adults, participates in wound healing as well as organ regeneration and tissue remodeling. Promotes also differentiation and proliferation of hematopoietic cells. May regulate cortical bone osteogenesis. Receptor tyrosine kinase that transduces signals from the extracellular matrix into the cytoplasm by binding to hepatocyte growth factor/HGF ligand. Related diseases Activation of MET after rearrangement with the TPR gene produces an oncogenic protein.; DISEASE: Defects in MET may be associated with gastric cancer.; DISEASE: Hepatocellular carcinoma (HCC) [MIM:114550]: A primary malignant neoplasm of epithelial liver cells. The major risk factors for HCC are chronic hepatitis B virus (HBV) infection, chronic hepatitis C virus (HCV) infection, prolonged dietary aflatoxin exposure, alcoholic cirrhosis, and cirrhosis due to other causes. {ECO:0000269|PubMed:9927037}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Renal cell carcinoma papillary (RCCP) [MIM:605074]: A subtype of renal cell carcinoma tending to show a tubulo-papillary architecture formed by numerous, irregular, finger-like projections of connective tissue. Renal cell carcinoma is a heterogeneous group of sporadic or hereditary carcinoma derived from cells of the proximal renal tubular epithelium. {ECO:0000269|PubMed:10327054, ECO:0000269|PubMed:10417759, ECO:0000269|PubMed:10433944, ECO:0000269|PubMed:9140397, ECO:0000269|PubMed:9563489}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: A common allele in the promoter region of the MET shows genetic association with susceptibility to autism in some families. Functional assays indicate a decrease in MET promoter activity and altered binding of specific transcription factor complexes.; DISEASE: MET activating mutations may be involved in the development of a highly malignant, metastatic syndrome known as cancer of unknown primary origin (CUP) or primary occult malignancy. Systemic neoplastic spread is generally a late event in cancer progression. However, in some instances, distant dissemination arises at a very early stage, so that metastases reach clinical relevance before primary lesions. Sometimes, the primary lesions cannot be identified in spite of the progresses in the diagnosis of malignancies.; DISEASE: Deafness, autosomal recessive, 97 (DFNB97) [MIM:616705]: A form of non-syndromic sensorineural hearing loss with prelingual onset. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. {ECO:0000269|PubMed:25941349}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Osteofibrous dysplasia (OSFD) [MIM:607278]: A congenital disorder of osteogenesis characterized by non-neoplastic, radiolucent lesions that affect the cortical bone immediately under the periosteum. It usually manifests as a painless swelling or anterior bowing of the long bones, most commonly the tibia and fibula. {ECO:0000269|PubMed:26637977}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Disease-associated variants identified in 4 families cause the deletion of exon 14. This results in the exclusion of an ubiquitination target site within the cytoplasmic domain, hence in protein stabilization. The persistent presence of MET at the cell surface in conditions of ligand-dependent activation retards osteoblastic differentiation. {ECO:0000269|PubMed:26637977}.; DISEASE: Arthrogryposis, distal, 11 (DA11) [MIM:620019]: A form of distal arthrogryposis, a disease characterized by congenital joint contractures that mainly involve two or more distal parts of the limbs, in the absence of a primary neurological or muscle disease. DA11 is an autosomal dominant form characterized mainly by camptodactyly. Other features include absent flexion creases and limited forearm supination. {ECO:0000269|PubMed:30777867}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06896; DB08791; DB06997; DB07969; DB08079; DB16695; DB12742; DB12267; DB08875; DB11791; DB08865; DB12010; DB02152; DB07369; DB06995; DB06314; DB01268; DB15133; DB12200; DB11800 Interacts with P22681; Q96EY1; Q96EY1-2; P00533; P09769; P14210; P14210-6; O15357; P35968; P06239; P07948; P08581; P41218; P15941; P16333; O43639; Q16288; P27986; O00459; Q92569; P19174; O43157; O15031; Q9ULL4; Q8TCU6; P18031; Q06124; P23467; Q12913; Q16827; P20936; Q9UQQ2; O60880; O14796; Q9NP31; Q8N5H7; Q15464; P29353; P98077; Q6S5L8; Q96IW2; Q9H6Q3; O75159; O14544; P12931; Q9ULZ2; P43405; P42680; Q9HBL0; Q63HR2; Q68CZ2; Q9UKW4; P07947; P43403; Q08048; P0DQD2; P35918; Q00944 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Chromosomal rearrangement; Deafness; Disease variant; Disulfide bond; Glycoprotein; Kinase; Membrane; Non-syndromic deafness; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Secreted; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 35229.5 Length 312 Aromaticity 0.1 Instability index 37.98 Isoelectric point 7.79 Charge (pH=7) 1.93 2D Binding mode Binding energy (Kcal/mol) -7.1  Molscript Map  Pymol Map 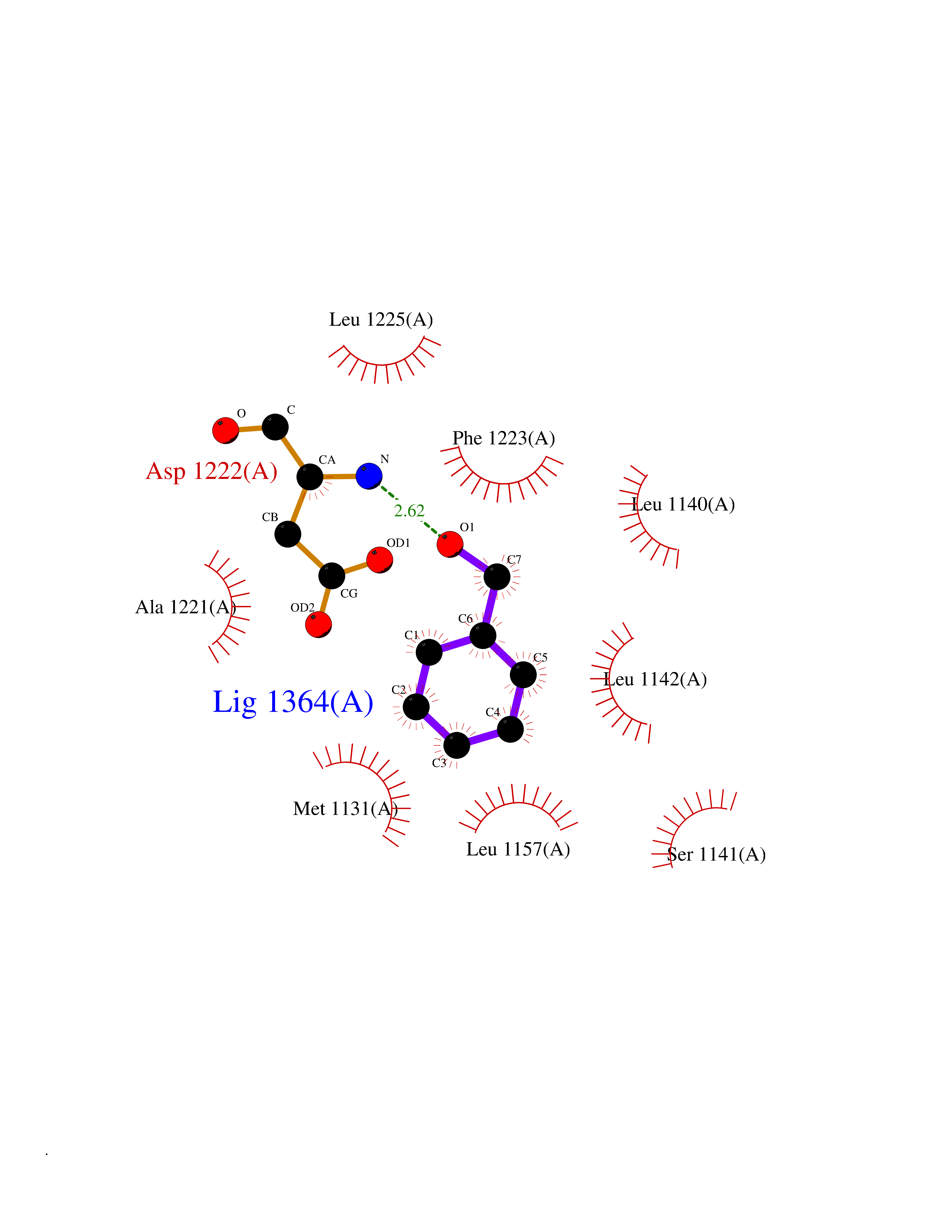 Ligplot Map 3D Binding mode Sequence VHIDLSALNPELVQAVQHVVIGPSSLIVHFNEVIGRGHFGCVYHGTLLDNDGKKIHCAVKSLNRITDIGEVSQFLTEGIIMKDFSHPNVLSLLGICLRSEGSPLVVLPYMKHGDLRNFIRNETHNPTVKDLIGFGLQVAKGMKFLASKKFVHRDLAARNCMLDEKFTVKVADFGLARDMYDKEFDSVHNKTGAKLPVKWMALESLQTQKFTTKSDVWSFGVLLWELMTRGAPPYPDVNTFDITVYLLQGRRLLQPEYCPDPLYEVMLKCWHPKAEMRPSFSELVSRISAIFSTFIGEHYVHVNATYVNVKEG Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Toll-like receptor 1 (TLR1) | 2Z7X | 5.20 | |
Target general information Gen name TLR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Toll/interleukin-1 receptor-like protein; TIL; KIAA0012; CD281 Protein family Toll-like receptor family Biochemical class Toll-like receptor Function Specifically recognizes diacylated and triacylated lipopeptides. Cooperates with TLR2 to mediate the innate immune response to bacterial lipoproteins or lipopeptides. Forms the activation cluster TLR2:TLR1:CD14 in response to triacylated lipopeptides, this cluster triggers signaling from the cell surface and subsequently is targeted to the Golgi in a lipid-raft dependent pathway. Acts via MYD88 and TRAF6, leading to NF-kappa-B activation, cytokine secretion and the inflammatory response. Participates in the innate immune response to microbial agents. Related diseases Hao-Fountain syndrome (HAFOUS) [MIM:616863]: An autosomal dominant neurodevelopmental disorder characterized by global developmental delay, varying degrees of intellectual disability, autism spectrum disorder, poor or absent speech, and mild facial dysmorphism. Most patients develop seizures. Additional variable features include hypotonia, hypogonadism in males, and ocular anomalies. {ECO:0000269|PubMed:26365382, ECO:0000269|PubMed:30679821}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q15399; Q9BXR5; O60603; Q9Y2C9 EC number NA Uniprot keywords 3D-structure; Cell membrane; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Glycoprotein; Golgi apparatus; Immunity; Inflammatory response; Innate immunity; Leucine-rich repeat; Membrane; NAD; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,C Molecular weight (Da) 62767.4 Length 555 Aromaticity 0.07 Instability index 38.79 Isoelectric point 6.85 Charge (pH=7) -0.55 2D Binding mode Binding energy (Kcal/mol) -7.1 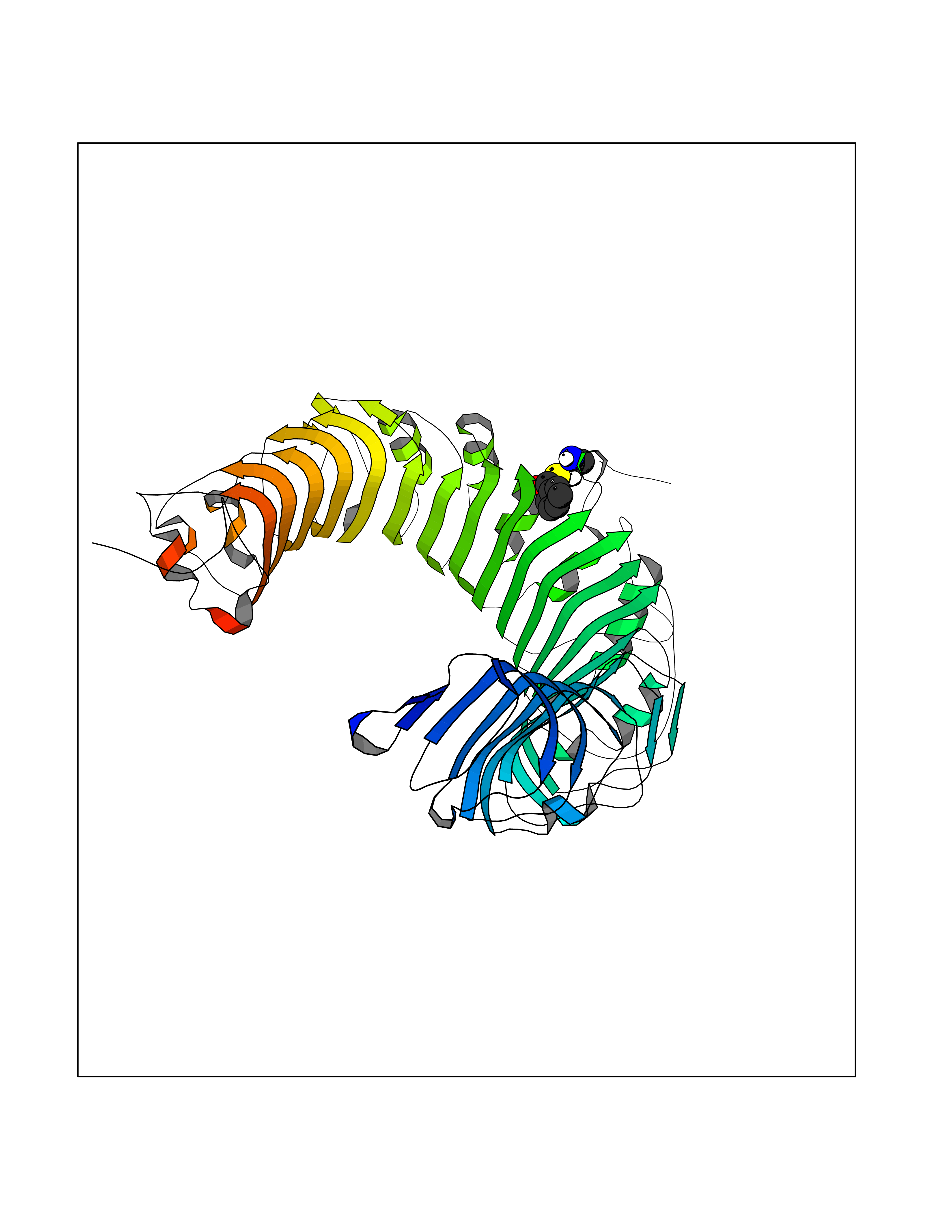 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SLSCDRNGICKGSSGSLNSIPSGLTEAVKSLDLSNNRITYISNSDLQRCVNLQALVLTSNGINTIEEDSFSSLGSLEHLDLSYNYLSNLSSSWFKPLSSLTFLNLLGNPYKTLGETSLFSHLTKLQILRVGNMDTFTKIQRKDFAGLTFLEELEIDASDLQSYEPKSLKSIQNVSHLILHMKQHILLLEIFVDVTSSVECLELRDTDLDTFHFSELSTGETNSLIKKFTFRNVKITDESLFQVMKLLNQISGLLELEFDDCTLNGVGNFRASDNDRVIDPGKVETLTIRRLHIPRFYLFYDLSTLYSLTERVKRITVENSKVFLVPCLLSQHLKSLEYLDLSENLMVEEYLKNSACEDAWPSLQTLILRQNHLASLEKTGETLLTLKNLTNIDISKNSFHSMPETCQWPEKMKYLNLSSTRIHSVTGCIPKTLEILDVSNNNLNLFSLNLPQLKELYISRNKLMTLPDASLLPMLLVLKISRNQLKSVPDGIFDRLTSLQKIWLHTNPWDCSCPRIDYLSRWLNKNSQKEQGSAKCSGSGKPVRSIICPXSKKKK Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Dipeptidyl peptidase 9 (DPP-9) | 6EOR | 5.20 | |
Target general information Gen name DPP9 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dipeptidyl peptidase-like protein 9; Dipeptidyl peptidase IX; Dipeptidyl peptidase IV-related protein 2; DPRP2; DPRP-2; DPP IX; DPLP9; DP9 Protein family Peptidase S9B family, DPPIV subfamily Biochemical class Peptidase Function Dipeptidyl peptidase that cleaves off N-terminal dipeptides from proteins having a Pro or Ala residue at position 2. Related diseases Hatipoglu immunodeficiency syndrome (HATIS) [MIM:620331]: An autosomal recessive immunologic disorder manifesting in infancy or early childhood, and characterized by failure to thrive, short stature, skin pigmentation abnormalities, pancytopenia, and susceptibility to recurrent infections. {ECO:0000269|PubMed:36112693}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NXR5; Q86TI2; Q6NUP5; P46379-2; Q8WUW1; Q96A83-2; O75190-2; O14645; Q01658; P29692-2; Q06787-7; Q9Y5Q9; O14901; Q9BVL2; Q96CV9; Q06830; P14678-2; P49458; Q11203; Q13148; P14927 EC number EC 3.4.14.5 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminopeptidase; Cytoplasm; Disease variant; Hydrolase; Nucleus; Protease; Proteomics identification; Reference proteome; Serine protease Protein physicochemical properties Chain ID A Molecular weight (Da) 92797.4 Length 808 Aromaticity 0.12 Instability index 37.45 Isoelectric point 6.34 Charge (pH=7) -8.98 2D Binding mode Binding energy (Kcal/mol) -7.09 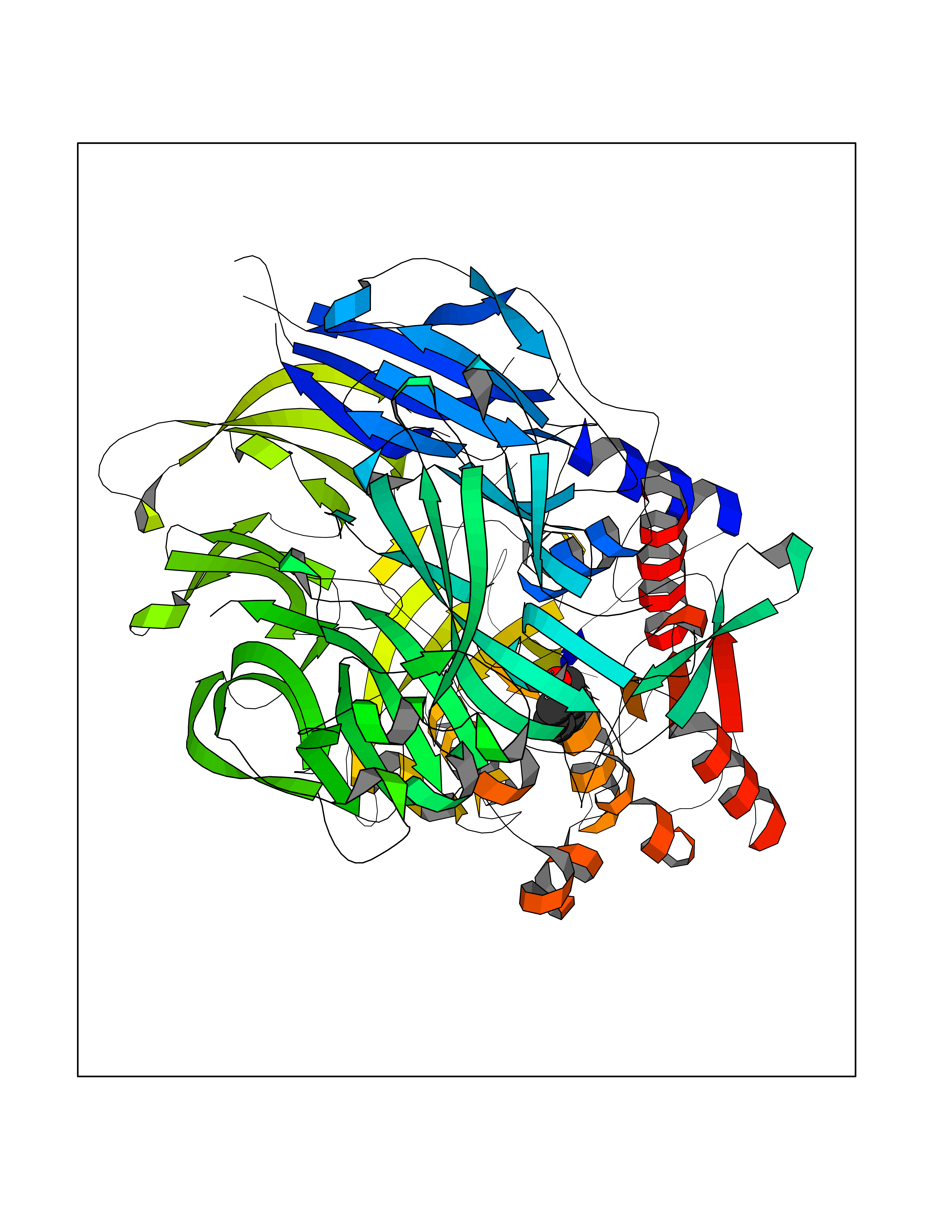 Molscript Map 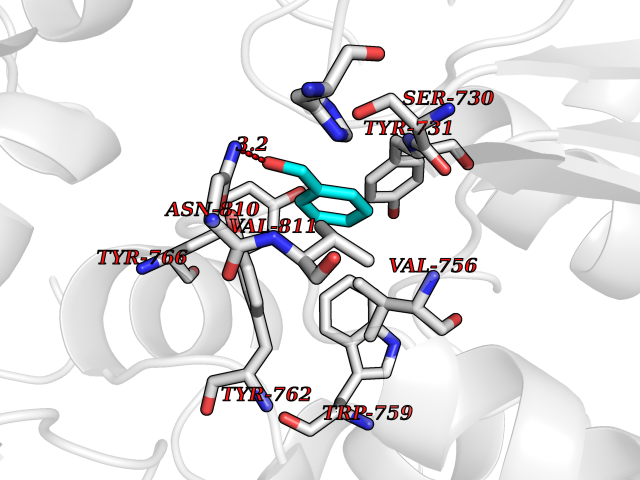 Pymol Map 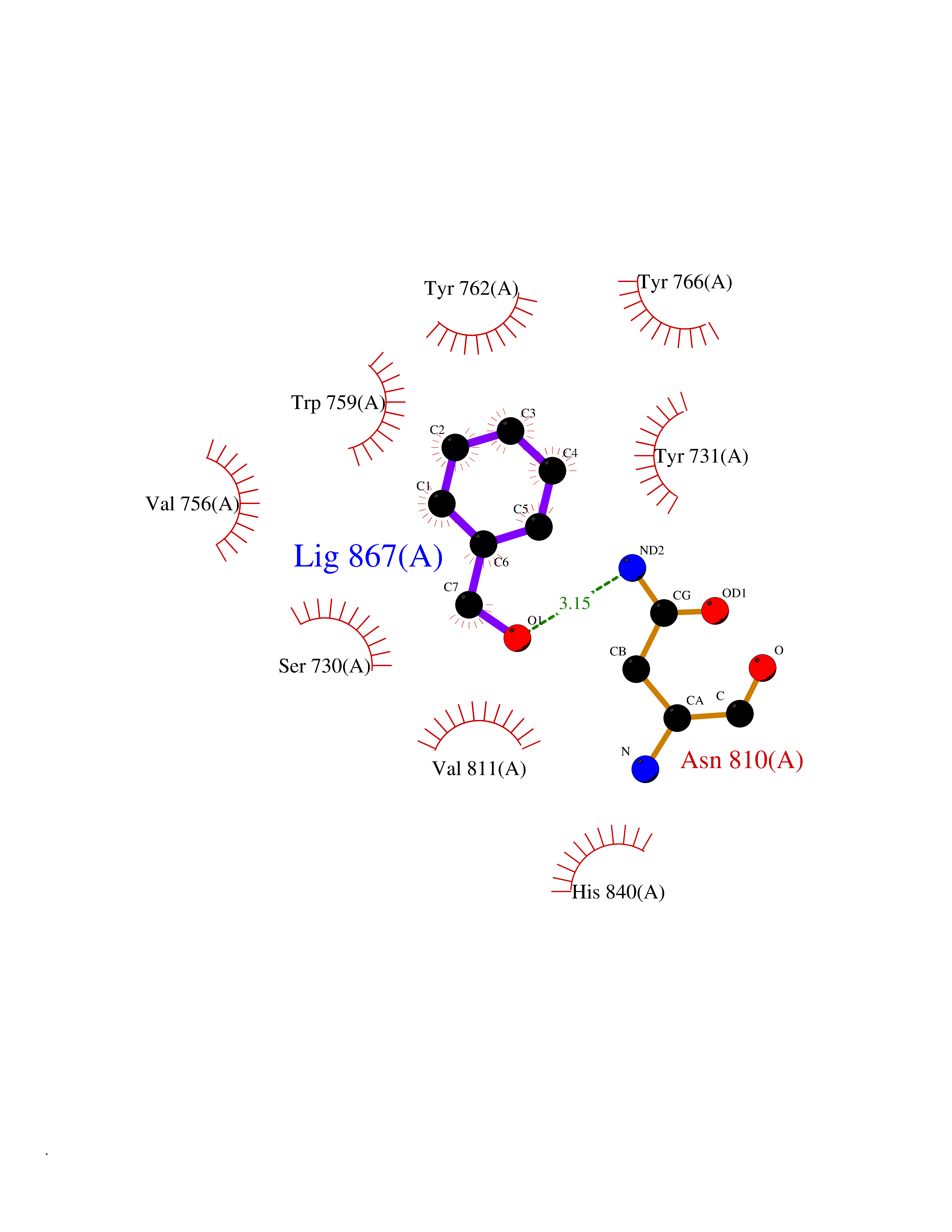 Ligplot Map 3D Binding mode Sequence AARFQVQKHSWDGLRSIIHGSRKAPHDFQFVQKSGPHSHRLYYLGMPYRENSLLYSEIPKLLLSWKQMLDHFQATPHHGVYSREEELLRERKRLGVFGITSYDFHSESGLFLFQASNSLFHCRDGGKNGFMVSPMKPLEIKTQCSGPRMDPKICPADPAFFSFINNSDLWVANIETGEERRLTFCHQNVLDDPKSAGVATFVIQEEFDRFTGYWWCPTASWEGLKTLRILYEEVDESEVEVIHVPSPALEERKTDSYRYPRTGSKNPKIALKLAEFQTDSQGKIVSTQEKELVQPFSSLFPKVEYIARAGWTRDGKYAWAMFLDRPQQWLQLVLLPPALFIPSTENEEQRLASARAVPRNVQPYVVYEEVTNVWINVHDIFYPFPQLCFLRANECKTGFCHLYKVTAVLKSQGYDWSEPFSPGEDEFKCPIKEEIALTSGEWEVLARHGSKIWVNEETKLVYFQGTKDTPLEHHLYVVSYEAAGEIVRLTTPGFSHSCSMSQNFDMFVSHYSSVSTPPCVHVYKLSGPDDDPLHKQPRFWASMMEADYVPPEIFHFHTRSDVRLYGMIYKPHALQPGKKHPTVLFVYGGPQVQLVNNSFKGIKYLRLNTLASLGYAVVVIDGRGSCQRGLRFEGALKNQMGQVEIEDQVEGLQFVAEKYGFIDLSRVAIHGWSYGGFLSLMGLIHKPQVFKVAIAGAPVTVWMAYDTGYTERYMDVPENNQHGYEAGSVALHVEKLPNEPNRLLILHGFLDENVHFFHTNFLVSQLIRAGKPYQLQIYPNERHSIRCPESGEHYEVTLLHFLQEYLHH Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | RAC-beta serine/threonine-protein kinase (AKT2) | 3D0E | 5.20 | |
Target general information Gen name AKT2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAC-PK-beta; Protein kinase B beta; Protein kinase Akt-2; PKB beta Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family, RAC subfamily Biochemical class Kinase Function AKT2 is one of 3 closely related serine/threonine-protein kinases (AKT1, AKT2 and AKT3) called the AKT kinase, and which regulate many processes including metabolism, proliferation, cell survival, growth and angiogenesis. This is mediated through serine and/or threonine phosphorylation of a range of downstream substrates. Over 100 substrate candidates have been reported so far, but for most of them, no isoform specificity has been reported. AKT is responsible of the regulation of glucose uptake by mediating insulin-induced translocation of the SLC2A4/GLUT4 glucose transporter to the cell surface. Phosphorylation of PTPN1 at 'Ser-50' negatively modulates its phosphatase activity preventing dephosphorylation of the insulin receptor and the attenuation of insulin signaling. Phosphorylation of TBC1D4 triggers the binding of this effector to inhibitory 14-3-3 proteins, which is required for insulin-stimulated glucose transport. AKT regulates also the storage of glucose in the form of glycogen by phosphorylating GSK3A at 'Ser-21' and GSK3B at 'Ser-9', resulting in inhibition of its kinase activity. Phosphorylation of GSK3 isoforms by AKT is also thought to be one mechanism by which cell proliferation is driven. AKT regulates also cell survival via the phosphorylation of MAP3K5 (apoptosis signal-related kinase). Phosphorylation of 'Ser-83' decreases MAP3K5 kinase activity stimulated by oxidative stress and thereby prevents apoptosis. AKT mediates insulin-stimulated protein synthesis by phosphorylating TSC2 at 'Ser-939' and 'Thr-1462', thereby activating mTORC1 signaling and leading to both phosphorylation of 4E-BP1 and in activation of RPS6KB1. AKT is involved in the phosphorylation of members of the FOXO factors (Forkhead family of transcription factors), leading to binding of 14-3-3 proteins and cytoplasmic localization. In particular, FOXO1 is phosphorylated at 'Thr-24', 'Ser-256' and 'Ser-319'. FOXO3 and FOXO4 are phosphorylated on equivalent sites. AKT has an important role in the regulation of NF-kappa-B-dependent gene transcription and positively regulates the activity of CREB1 (cyclic AMP (cAMP)-response element binding protein). The phosphorylation of CREB1 induces the binding of accessory proteins that are necessary for the transcription of pro-survival genes such as BCL2 and MCL1. AKT phosphorylates 'Ser-454' on ATP citrate lyase (ACLY), thereby potentially regulating ACLY activity and fatty acid synthesis. Activates the 3B isoform of cyclic nucleotide phosphodiesterase (PDE3B) via phosphorylation of 'Ser-273', resulting in reduced cyclic AMP levels and inhibition of lipolysis. Phosphorylates PIKFYVE on 'Ser-318', which results in increased PI(3)P-5 activity. The Rho GTPase-activating protein DLC1 is another substrate and its phosphorylation is implicated in the regulation cell proliferation and cell growth. AKT plays a role as key modulator of the AKT-mTOR signaling pathway controlling the tempo of the process of newborn neurons integration during adult neurogenesis, including correct neuron positioning, dendritic development and synapse formation. Signals downstream of phosphatidylinositol 3-kinase (PI(3)K) to mediate the effects of various growth factors such as platelet-derived growth factor (PDGF), epidermal growth factor (EGF), insulin and insulin-like growth factor I (IGF-I). AKT mediates the antiapoptotic effects of IGF-I. Essential for the SPATA13-mediated regulation of cell migration and adhesion assembly and disassembly. May be involved in the regulation of the placental development. Related diseases Defects in AKT2 are a cause of susceptibility to breast cancer (BC). AKT2 promotes metastasis of tumor cells without affecting the latency of tumor development. May play a role in glioblastoma cell survival (PubMed:20167810). {ECO:0000269|PubMed:20167810}.; DISEASE: Type 2 diabetes mellitus (T2D) [MIM:125853]: A multifactorial disorder of glucose homeostasis caused by a lack of sensitivity to insulin. Affected individuals usually have an obese body habitus and manifestations of a metabolic syndrome characterized by diabetes, insulin resistance, hypertension and hypertriglyceridemia. The disease results in long-term complications that affect the eyes, kidneys, nerves, and blood vessels. {ECO:0000269|PubMed:15166380, ECO:0000269|PubMed:19164855}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Hypoinsulinemic hypoglycemia with hemihypertrophy (HIHGHH) [MIM:240900]: A disorder characterized by hypoglycemia, low insulin levels, low serum levels of ketone bodies and branched-chain amino acids, left-sided hemihypertrophy, neonatal macrosomia, reduced consciousness and hypoglycemic seizures. {ECO:0000269|PubMed:21979934}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08073; DB07859; DB12218; DB07947; DB07812 Interacts with P31749; P49841; P08238; Q6FHY5; Q9NRD5; Q04864-2; O60504; P53804; Q9C0C9; P08670; Q15118-1 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Apoptosis; ATP-binding; Carbohydrate metabolism; Cell membrane; Cytoplasm; Developmental protein; Diabetes mellitus; Disease variant; Disulfide bond; Endosome; Glucose metabolism; Glycogen biosynthesis; Glycogen metabolism; Glycoprotein; Kinase; Manganese; Membrane; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Reference proteome; Serine/threonine-protein kinase; Sugar transport; Transferase; Translation regulation; Transport; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 37380.5 Length 324 Aromaticity 0.12 Instability index 29.68 Isoelectric point 6.19 Charge (pH=7) -3.43 2D Binding mode Binding energy (Kcal/mol) -7.1 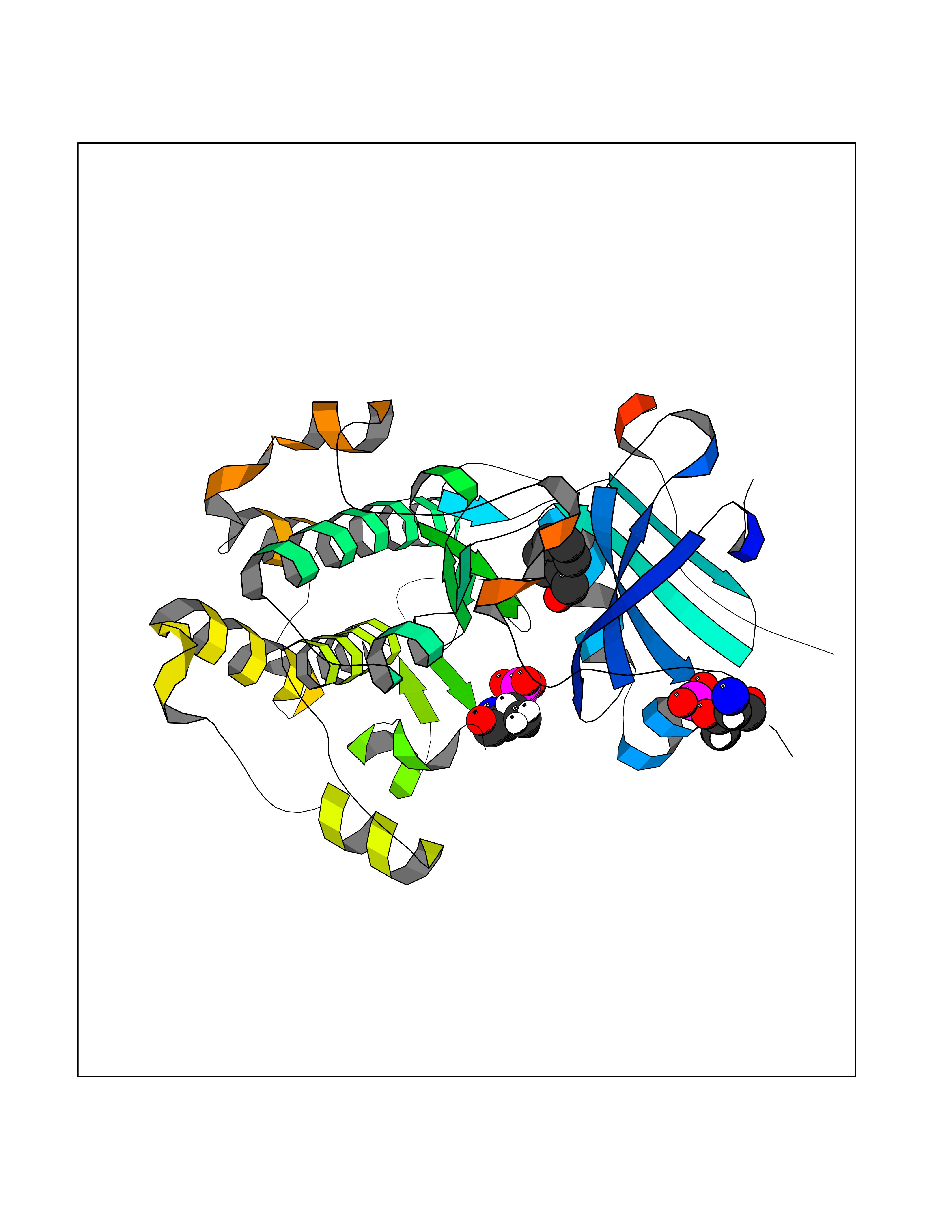 Molscript Map 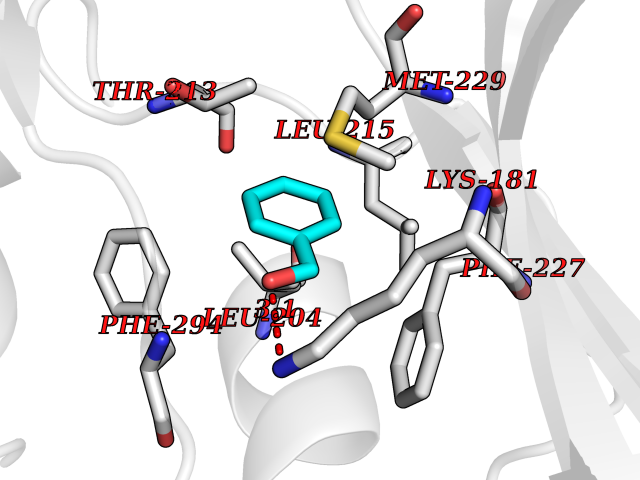 Pymol Map 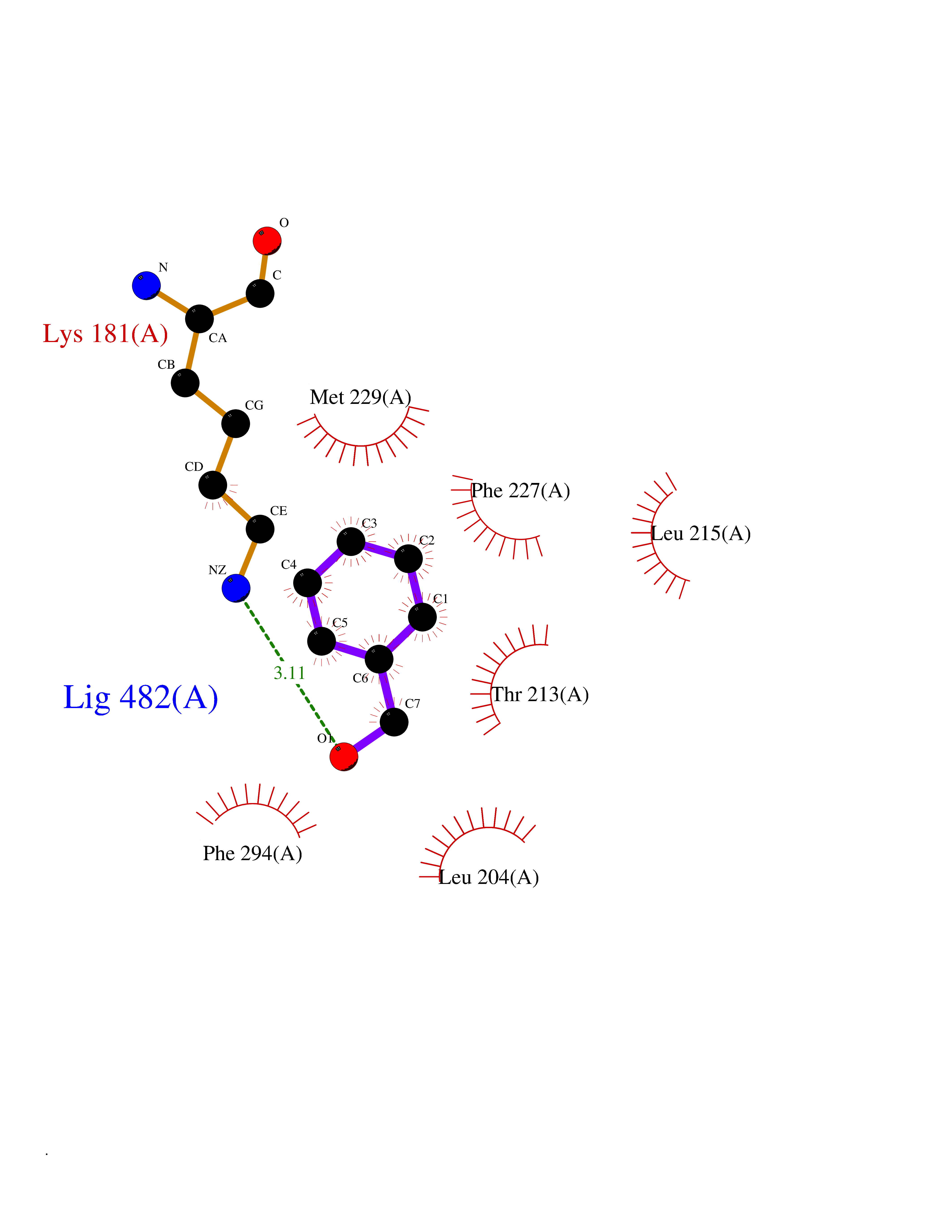 Ligplot Map 3D Binding mode Sequence KVTMNDFDYLKLLGKGTFGKVILVREKATGRYYAMKILRKEVIIAKDEVAHTVTESRVLQNTRHPFLTALKYAFQTHDRLCFVMEYANGGELFFHLSRERVFTEERARFYGAEIVSALEYLHSRDVVYRDIKLENLMLDKDGHIKITDFGLCKEGISDGATMKXFCGTPEYLAPEVLEDNDYGRAVDWWGLGVVMYEMMCGRLPFYNQDHERLFELILMEEIRFPRTLSPEAKSLLAGLLKKDPKQRLGGGPSDAKEVMEHRFFLSINWQDVVQKKLLPPFKPQVTSEVDTRYFDDEFTAQSITIXPPDQRTHFPQFDYSASIR Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Tyrosine-protein kinase BTK (ATK) | 4RFZ | 5.19 | |
Target general information Gen name BTK Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Bruton's tyrosine kinase; Bruton tyrosine kinase; BPK; B-cell progenitor kinase; B cell progenitor kinase; Agammaglobulinemia tyrosine kinase; Agammaglobulinaemia tyrosine kinase; AGMX1 Protein family Protein kinase superfamily, Tyr protein kinase family, TEC subfamily Biochemical class Kinase Function Binding of antigen to the B-cell antigen receptor (BCR) triggers signaling that ultimately leads to B-cell activation. After BCR engagement and activation at the plasma membrane, phosphorylates PLCG2 at several sites, igniting the downstream signaling pathway through calcium mobilization, followed by activation of the protein kinase C (PKC) family members. PLCG2 phosphorylation is performed in close cooperation with the adapter protein B-cell linker protein BLNK. BTK acts as a platform to bring together a diverse array of signaling proteins and is implicated in cytokine receptor signaling pathways. Plays an important role in the function of immune cells of innate as well as adaptive immunity, as a component of the Toll-like receptors (TLR) pathway. The TLR pathway acts as a primary surveillance system for the detection of pathogens and are crucial to the activation of host defense. Especially, is a critical molecule in regulating TLR9 activation in splenic B-cells. Within the TLR pathway, induces tyrosine phosphorylation of TIRAP which leads to TIRAP degradation. BTK plays also a critical role in transcription regulation. Induces the activity of NF-kappa-B, which is involved in regulating the expression of hundreds of genes. BTK is involved on the signaling pathway linking TLR8 and TLR9 to NF-kappa-B. Transiently phosphorylates transcription factor GTF2I on tyrosine residues in response to BCR. GTF2I then translocates to the nucleus to bind regulatory enhancer elements to modulate gene expression. ARID3A and NFAT are other transcriptional target of BTK. BTK is required for the formation of functional ARID3A DNA-binding complexes. There is however no evidence that BTK itself binds directly to DNA. BTK has a dual role in the regulation of apoptosis. Non-receptor tyrosine kinase indispensable for B lymphocyte development, differentiation and signaling. Related diseases X-linked agammaglobulinemia (XLA) [MIM:300755]: Humoral immunodeficiency disease which results in developmental defects in the maturation pathway of B-cells. Affected boys have normal levels of pre-B-cells in their bone marrow but virtually no circulating mature B-lymphocytes. This results in a lack of immunoglobulins of all classes and leads to recurrent bacterial infections like otitis, conjunctivitis, dermatitis, sinusitis in the first few years of life, or even some patients present overwhelming sepsis or meningitis, resulting in death in a few hours. Treatment in most cases is by infusion of intravenous immunoglobulin. {ECO:0000269|PubMed:10220140, ECO:0000269|PubMed:10612838, ECO:0000269|PubMed:10678660, ECO:0000269|PubMed:7627183, ECO:0000269|PubMed:7633420, ECO:0000269|PubMed:7633429, ECO:0000269|PubMed:7711734, ECO:0000269|PubMed:7809124, ECO:0000269|PubMed:7849006, ECO:0000269|PubMed:7849697, ECO:0000269|PubMed:7849721, ECO:0000269|PubMed:7880320, ECO:0000269|PubMed:7897635, ECO:0000269|PubMed:8013627, ECO:0000269|PubMed:8162018, ECO:0000269|PubMed:8162056, ECO:0000269|PubMed:8594569, ECO:0000269|PubMed:8634718, ECO:0000269|PubMed:8695804, ECO:0000269|PubMed:8723128, ECO:0000269|PubMed:8834236, ECO:0000269|PubMed:9016530, ECO:0000269|PubMed:9260159, ECO:0000269|PubMed:9280283, ECO:0000269|PubMed:9445504, ECO:0000269|PubMed:9545398}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 3, with agammaglobulinemia (IGHD3) [MIM:307200]: An X-linked recessive disorder characterized by growth hormone deficiency, short stature, delayed bone age, agammaglobulinemia with markedly reduced numbers of B cells, and good response to treatment with growth hormone. {ECO:0000269|PubMed:8013627}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15327; DB11703; DB15347; DB01254; DB15170; DB14785; DB12010; DB09053; DB01863; DB17472; DB14924; DB11764; DB15227; DB16657; DB05204; DB15035 Interacts with Q13444; Q99856; Q8WV28; Q06187; P78347; P08238; Q9BVA0; P21145; P50222; Q04759; O60239; P42768 EC number EC 2.7.10.2 Uniprot keywords 3D-structure; Acetylation; Adaptive immunity; Alternative promoter usage; Apoptosis; ATP-binding; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Dwarfism; Immunity; Innate immunity; Kinase; Lipid-binding; Membrane; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; SH2 domain; SH3 domain; Transcription; Transcription regulation; Transferase; Tyrosine-protein kinase; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 30491.7 Length 263 Aromaticity 0.12 Instability index 45.93 Isoelectric point 5.39 Charge (pH=7) -7.65 2D Binding mode Binding energy (Kcal/mol) -7.08 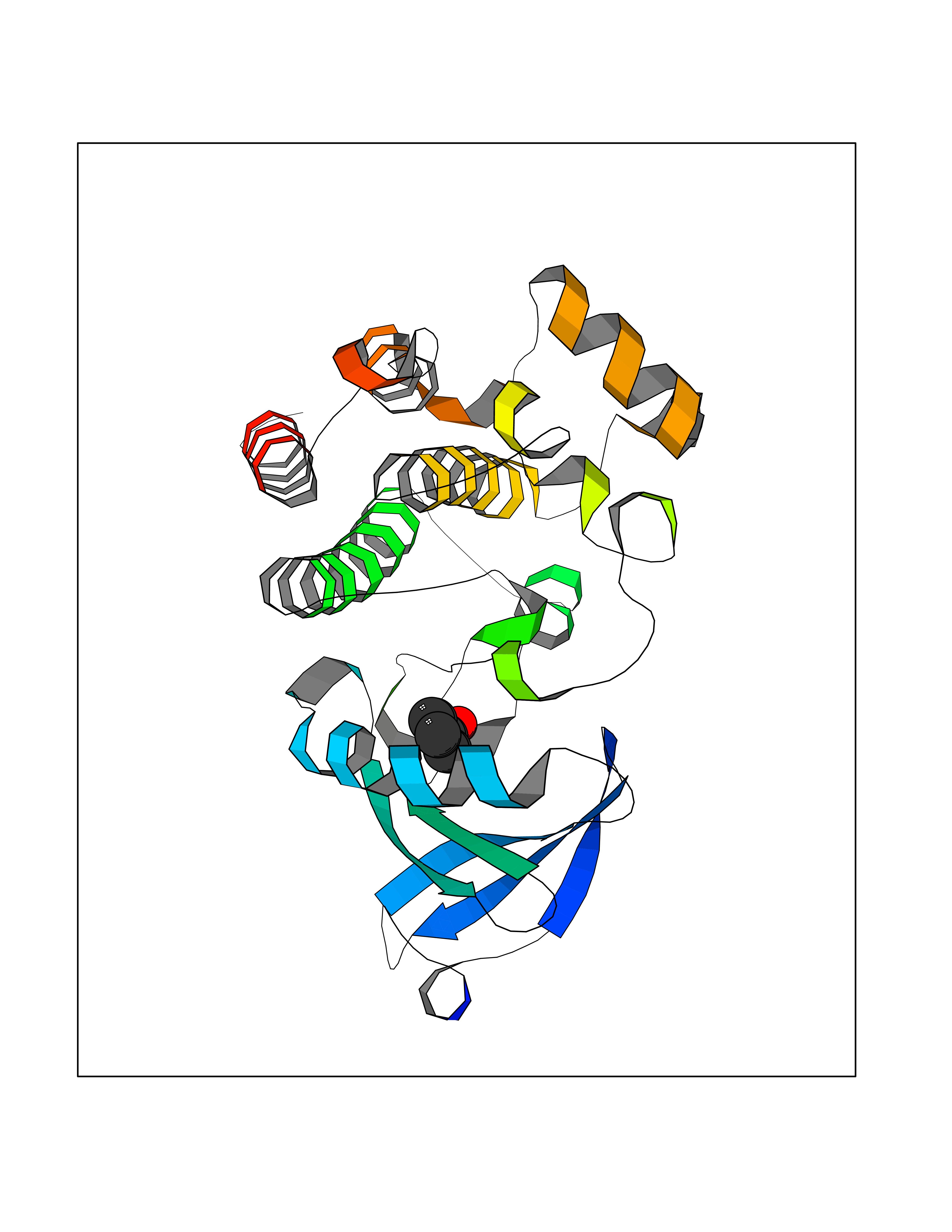 Molscript Map 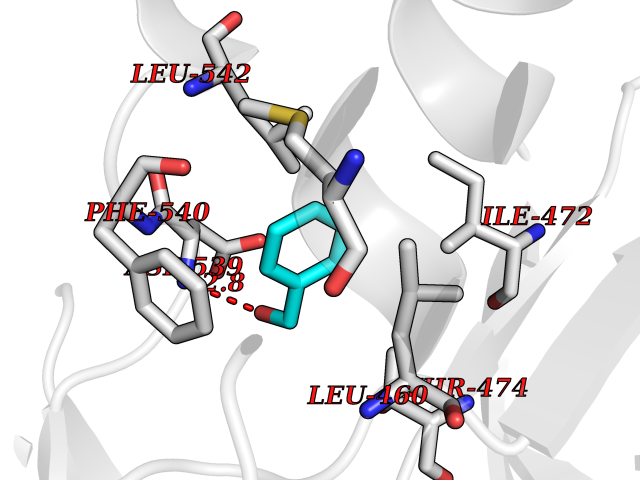 Pymol Map 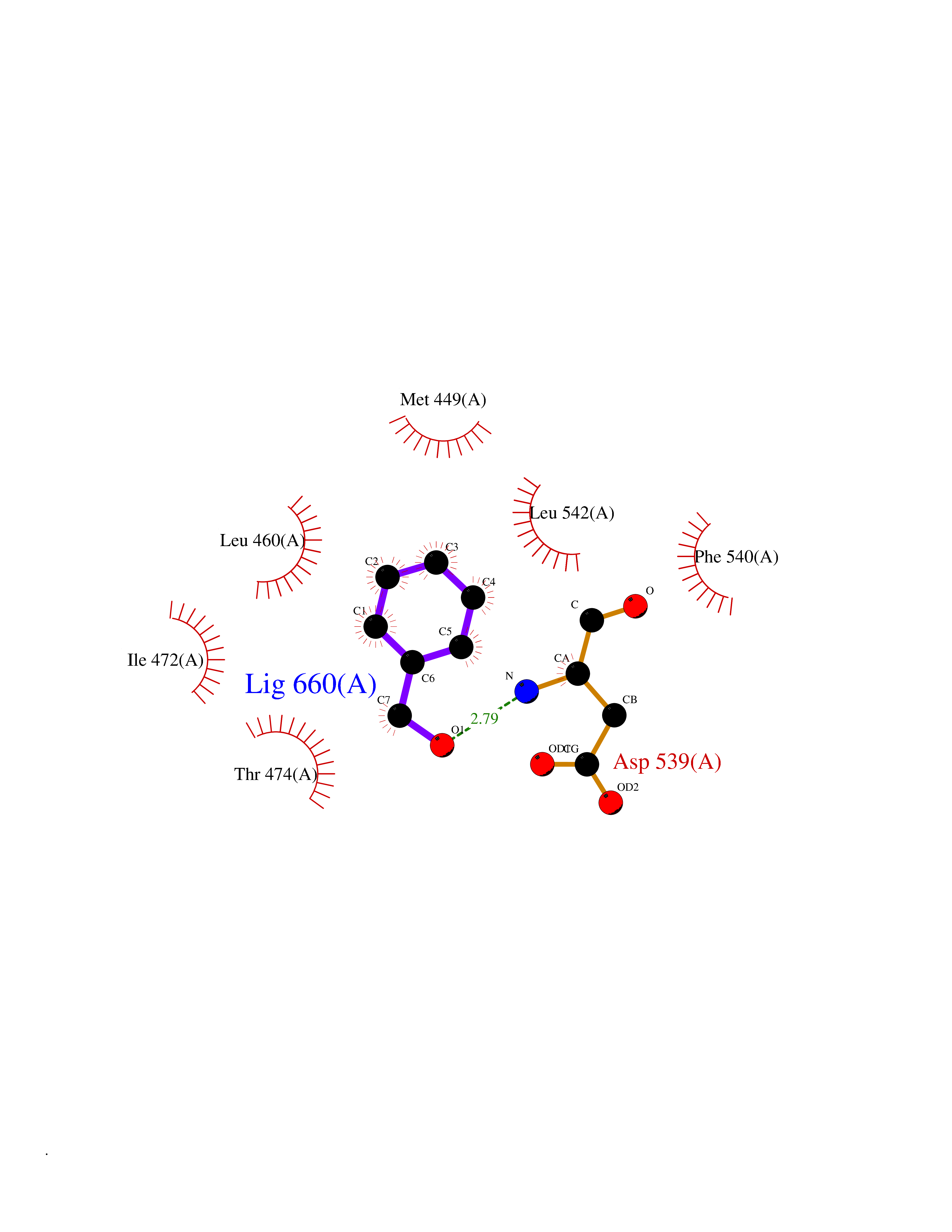 Ligplot Map 3D Binding mode Sequence EIDPKDLTFLKELGTGQFGVVKYGKWRGQYDVAIKMIKEGSMSEDEFIEEAKVMMNLSHEKLVQLYGVCTKQRPIFIITEYMANGCLLNYLREARHAFQTQQLLEMCKDVCEAMEYLESKQFLHRDLAARNCLVNDQGVVKVSDFGLSRYVLDDEYTSSVGSKFPVRWSPPEVLMYSKFSSKSDIWAFGVLMWEIYSLGKMPYERFTNSETAEHIAQGLRLYRPHLASAAVYTIMYSCWHEKADERPTFKILLSNILDVMDEE Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Lecithin-cholesterol acyltransferase (LCAT) | 6MVD | 5.19 | |
Target general information Gen name LCAT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Phospholipidcholesterolacyltransferase; Phospholipid-cholesterol acyltransferase; Phosphatidylcholinesterol acyltransferase; Phosphatidylcholine-sterol acyltransferase Protein family AB hydrolase superfamily, Lipase family Biochemical class Acyltransferase Function Synthesized mainly in the liver and secreted into plasma where it converts cholesterol and phosphatidylcholines (lecithins) to cholesteryl esters and lysophosphatidylcholines on the surface of high and low density lipoproteins (HDLs and LDLs). The cholesterol ester is then transported back to the liver. Has a preference for plasma 16:0-18:2 or 18:O-18:2 phosphatidylcholines. Also produced in the brain by primary astrocytes, and esterifies free cholesterol on nascent APOE-containing lipoproteins secreted from glia and influences cerebral spinal fluid (CSF) APOE- and APOA1 levels. Together with APOE and the cholesterol transporter ABCA1, plays a key role in the maturation of glial-derived, nascent lipoproteins. Required for remodeling high-density lipoprotein particles into their spherical forms. Central enzyme in the extracellular metabolism of plasma lipoproteins. Related diseases Lecithin-cholesterol acyltransferase deficiency (LCATD) [MIM:245900]: A disorder of lipoprotein metabolism characterized by inadequate esterification of plasmatic cholesterol. Two clinical forms are recognized: complete LCAT deficiency and fish-eye disease. LCATD is generally referred to the complete form which is associated with absence of both alpha and beta LCAT activities resulting in esterification anomalies involving both HDL (alpha-LCAT activity) and LDL (beta-LCAT activity). It causes a typical triad of diffuse corneal opacities, target cell hemolytic anemia, and proteinuria with renal failure. {ECO:0000269|PubMed:11423760, ECO:0000269|PubMed:12957688, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:16051254, ECO:0000269|PubMed:16216249, ECO:0000269|PubMed:1681161, ECO:0000269|PubMed:1859405, ECO:0000269|PubMed:2370048, ECO:0000269|PubMed:7607641, ECO:0000269|PubMed:7711728, ECO:0000269|PubMed:8318557, ECO:0000269|PubMed:8432868, ECO:0000269|PubMed:8807342, ECO:0000269|PubMed:9007616, ECO:0000269|PubMed:9741700}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fish-eye disease (FED) [MIM:136120]: A disorder of lipoprotein metabolism due to partial lecithin-cholesterol acyltransferase deficiency that affects only alpha-LCAT activity. FED is characterized by low plasma HDL and corneal opacities due to accumulation of cholesterol deposits in the cornea ('fish-eye'). {ECO:0000269|PubMed:1516702, ECO:0000269|PubMed:1571050, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:1737840, ECO:0000269|PubMed:21901787, ECO:0000269|PubMed:8620346, ECO:0000269|PubMed:9261271}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P02647; O76024 EC number EC 2.3.1.43 Uniprot keywords 3D-structure; Acyltransferase; Cholesterol metabolism; Corneal dystrophy; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lipid metabolism; Proteomics identification; Reference proteome; Secreted; Signal; Steroid metabolism; Sterol metabolism; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 42715.4 Length 376 Aromaticity 0.12 Instability index 42.05 Isoelectric point 5.69 Charge (pH=7) -9.12 2D Binding mode Binding energy (Kcal/mol) -7.08  Molscript Map 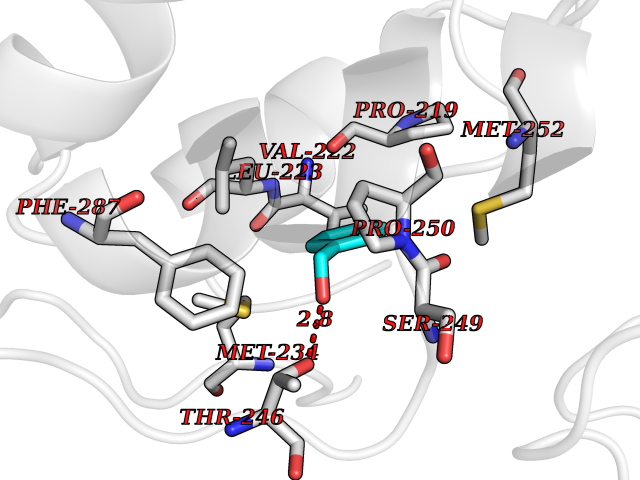 Pymol Map 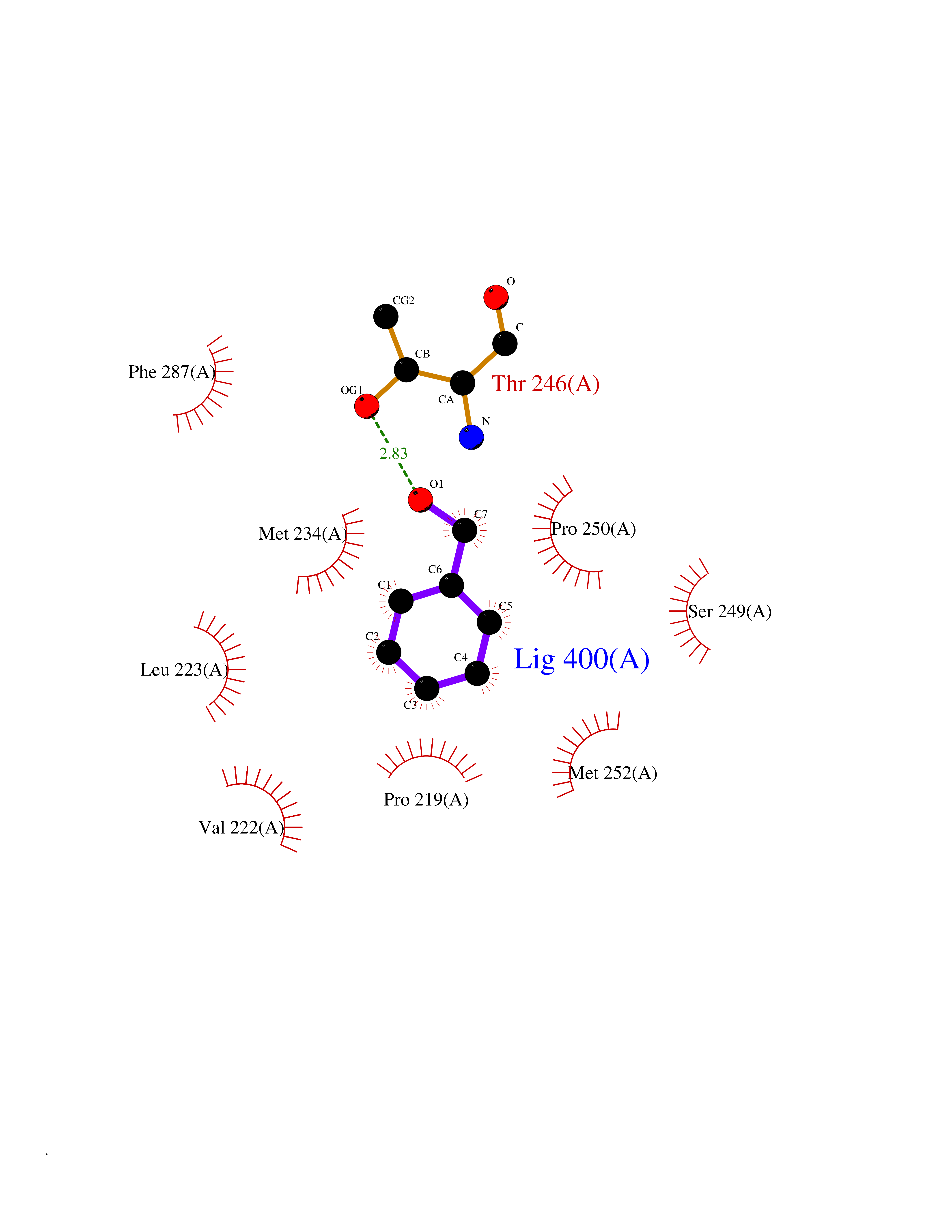 Ligplot Map 3D Binding mode Sequence HTRPVILVPGCLGNQLEAKLDKPDVVNWMCYRKTEDFFTIWLDLNMFLPLGVDCWIDNTRVVYNRSSGLVSNAPGVQIRVPGFGKTYSVEYLDSSKLAGYLHTLVQNLVNNGYVRDETVRAAPYDWRLEPGQQEEYYRKLAGLVEEMHAAYGKPVFLIGHSLGCLHLLYFLLRQPQAWKDRFIDGFISLGAPWGGSIKPMLVLASGDNQGIPIMSSIKEEQRITTTSPWMFPSRMAWPEDHVFISTPSFNYTGRDFQRFFADLHFEEGWYMWLQSRDLLAGLPAPGVEVYCLYGVGLPTPRTYIYDHGFPYTDPVGVLYEDGDDTVATRSTELCGLWQGRQPQPVHLLPLHGIQHLNMVFSNLTLEHINAILLGAH Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Ubiquitin carboxyl-terminal hydrolase 14 (USP14) | 6IIK | 5.19 | |
Target general information Gen name USP14 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ubiquitin-specific-processing protease 14; Ubiquitin thioesterase 14; TGT; Deubiquitinating enzyme 14 Protein family Peptidase C19 family, USP14/UBP6 subfamily Biochemical class Peptidase Function Ensures the regeneration of ubiquitin at the proteasome. Is a reversibly associated subunit of the proteasome and a large fraction of proteasome-free protein exists within the cell. Required for the degradation of the chemokine receptor CXCR4 which is critical for CXCL12-induced cell chemotaxis. Serves also as a physiological inhibitor of endoplasmic reticulum-associated degradation (ERAD) under the non-stressed condition by inhibiting the degradation of unfolded endoplasmic reticulum proteins via interaction with ERN1. Indispensable for synaptic development and function at neuromuscular junctions (NMJs). Plays a role in the innate immune defense against viruses by stabilizing the viral DNA sensor CGAS and thus inhibiting its autophagic degradation. Proteasome-associated deubiquitinase which releases ubiquitin from the proteasome targeted ubiquitinated proteins. Related diseases Hypophosphatemic rickets, autosomal dominant (ADHR) [MIM:193100]: A disease characterized by isolated renal phosphate wasting, hypophosphatemia, and inappropriately normal 1,25-dihydroxyvitamin D3 (calcitriol) levels. Patients frequently present with bone pain, rickets, and tooth abscesses. {ECO:0000269|PubMed:11062477, ECO:0000269|PubMed:11409890, ECO:0000269|PubMed:16638743}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Tumoral calcinosis, hyperphosphatemic, familial, 2 (HFTC2) [MIM:617993]: A form of hyperphosphatemic tumoral calcinosis, a rare autosomal recessive metabolic disorder that manifests with hyperphosphatemia and massive calcium deposits in the skin and subcutaneous tissues. Some patients have recurrent, transient, painful swellings of the long bones associated with the radiographic findings of periosteal reaction and cortical hyperostosis and absence of skin involvement. {ECO:0000269|PubMed:15590700, ECO:0000269|PubMed:16030159, ECO:0000269|PubMed:16151858, ECO:0000269|PubMed:24680727}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12695 Interacts with Q08209 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell membrane; Cytoplasm; Hydrolase; Immunity; Innate immunity; Membrane; Phosphoprotein; Protease; Proteasome; Proteomics identification; Reference proteome; Thiol protease; Ubl conjugation pathway Protein physicochemical properties Chain ID A Molecular weight (Da) 38476.7 Length 335 Aromaticity 0.1 Instability index 61.05 Isoelectric point 5.6 Charge (pH=7) -4.84 2D Binding mode Binding energy (Kcal/mol) -7.08  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ELPCGLTNLGNTCYMNATVQCIRSVPELKDALKRYAGALRASGEMASAQYITAALRDLFDSMDKTSSSIPPIILLQFLHMAFPQFAEKGEQGQYLQQDANECWIQMMRVLQQKLEAIEDKSLIDQFFGVEFETTMKCTESEEEEVTKGKENQLQLSCFINQEVKYLFTGLKLRLQEEITKQSPTLQRNALYIKSSKISRLPAYLTIQMVRFFNAKVLKDVKFPLMLDMYELCTPELQEKMVSFRSKFKDLYEPFSFADDIGSNNCGYYDLQAVLTHQGRSSSSGHYVSWVKRKQDEWIKFDDDKVSIVTPEDILRLSGGGDWHIAYVLLYGPRRV Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | IL-1 receptor-associated kinase 1 (IRAK1) | 6BFN | 5.19 | |
Target general information Gen name IRAK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Interleukin-1 receptor-associated kinase 1; IRAK-1; IRAK Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family, Pelle subfamily Biochemical class Kinase Function Involved in Toll-like receptor (TLR) and IL-1R signaling pathways. Is rapidly recruited by MYD88 to the receptor-signaling complex upon TLR activation. Association with MYD88 leads to IRAK1 phosphorylation by IRAK4 and subsequent autophosphorylation and kinase activation. Phosphorylates E3 ubiquitin ligases Pellino proteins (PELI1, PELI2 and PELI3) to promote pellino-mediated polyubiquitination of IRAK1. Then, the ubiquitin-binding domain of IKBKG/NEMO binds to polyubiquitinated IRAK1 bringing together the IRAK1-MAP3K7/TAK1-TRAF6 complex and the NEMO-IKKA-IKKB complex. In turn, MAP3K7/TAK1 activates IKKs (CHUK/IKKA and IKBKB/IKKB) leading to NF-kappa-B nuclear translocation and activation. Alternatively, phosphorylates TIRAP to promote its ubiquitination and subsequent degradation. Phosphorylates the interferon regulatory factor 7 (IRF7) to induce its activation and translocation to the nucleus, resulting in transcriptional activation of type I IFN genes, which drive the cell in an antiviral state. When sumoylated, translocates to the nucleus and phosphorylates STAT3. Serine/threonine-protein kinase that plays a critical role in initiating innate immune response against foreign pathogens. Related diseases Anemia, non-spherocytic hemolytic, due to G6PD deficiency (NSHA) [MIM:300908]: A disease characterized by G6PD deficiency, acute hemolytic anemia, fatigue, back pain, and jaundice. In most patients, the disease is triggered by an exogenous agent, such as some drugs, food, or infection. Increased unconjugated bilirubin, lactate dehydrogenase, and reticulocytosis are markers of the disorder. Although G6PD deficiency can be life-threatening, most patients are asymptomatic throughout their life. {ECO:0000269|PubMed:12524354, ECO:0000269|PubMed:1303180, ECO:0000269|PubMed:1303182, ECO:0000269|PubMed:1536798, ECO:0000269|PubMed:1611091, ECO:0000269|PubMed:1889820, ECO:0000269|PubMed:1945893, ECO:0000269|PubMed:20007901, ECO:0000269|PubMed:26479991, ECO:0000269|PubMed:2836867, ECO:0000269|PubMed:2912069, ECO:0000269|PubMed:30988594, ECO:0000269|PubMed:38066190, ECO:0000269|PubMed:7858267, ECO:0000269|PubMed:7959695, ECO:0000269|PubMed:8193373, ECO:0000269|PubMed:8490627, ECO:0000269|PubMed:8533762, ECO:0000269|PubMed:8733135, ECO:0000269|PubMed:9452072}. The disease is caused by variants affecting the gene represented in this entry. Deficiency of G6PD is associated with hemolytic anemia in two different situations. First, in areas in which malaria has been endemic, G6PD-deficiency alleles have reached high frequencies (1% to 50%) and deficient individuals, though essentially asymptomatic in the steady state, have a high risk of acute hemolytic attacks. Secondly, sporadic cases of G6PD deficiency occur at a very low frequencies, and they usually present a more severe phenotype. Several types of NSHA are recognized. Class-I variants are associated with severe NSHA; class-II have an activity <10% of normal; class-III have an activity of 10% to 60% of normal; class-IV have near normal activity. Drugs (DrugBank ID) DB12010 Interacts with Q15306; Q92985; Q99836; Q96FA3; Q9HAT8; Q8N2H9-2; Q13526; Q86WV6; P58753; Q9Y4K3; Q8VCW4; Q5D1E7 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cytoplasm; Direct protein sequencing; Host-virus interaction; Immunity; Innate immunity; Isopeptide bond; Kinase; Lipid droplet; Magnesium; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 33681.4 Length 301 Aromaticity 0.09 Instability index 39.86 Isoelectric point 8.6 Charge (pH=7) 5.09 2D Binding mode Binding energy (Kcal/mol) -7.08  Molscript Map 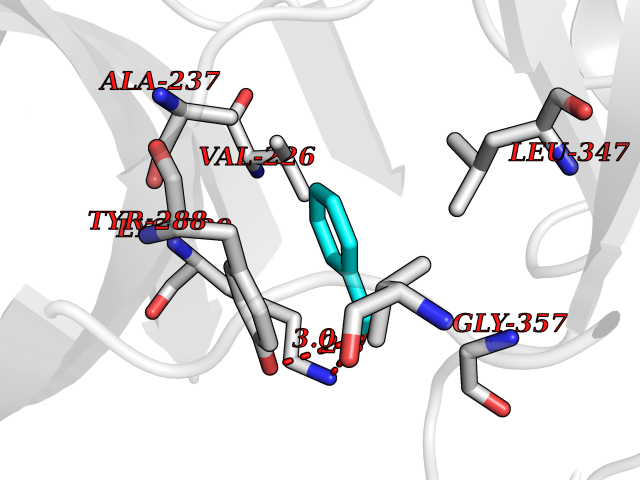 Pymol Map 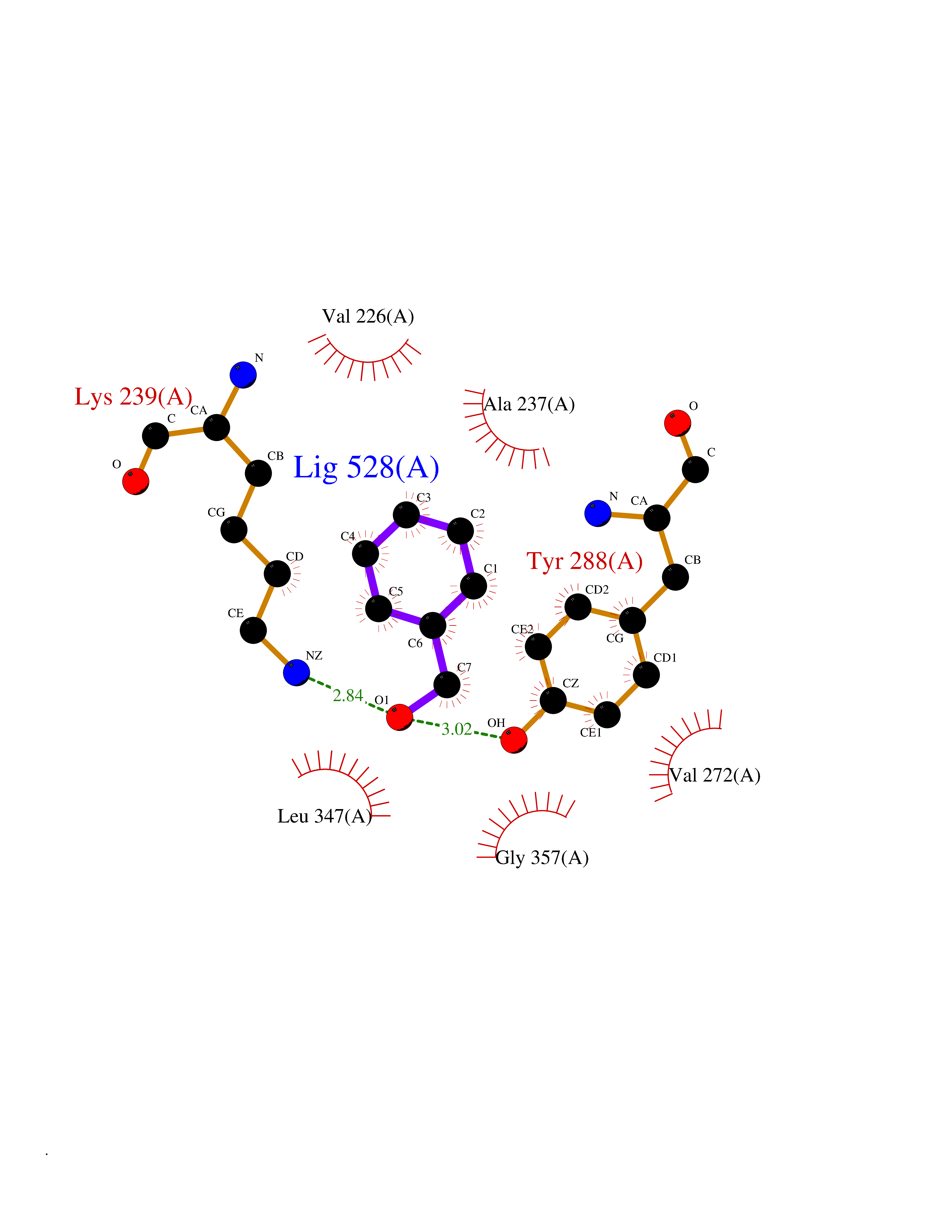 Ligplot Map 3D Binding mode Sequence SRPFPFCWPLCEISRGTHNFSEELKIGEGGFGCVYRAVMRNTVYAVKRLKEWTAVKQSFLTEVEQLSRFRHPNIVDFAGYCAQNGFYCLVYGFLPNGSLEDRLHCQTQACPPLSWPQRLDILLGTARAIQFLHQDSPSLIHGDIKSSNVLLDERLTPKLGDFGLARFSRTVRGTLAYLPEEYIKTGRLAVDTDTFSFGVVVLETLAGQRAVKTHGARTKYLKDLVEEEAEEAGVAAADAWAAPIAMQIYKKHLDPRPGPCPPELGLGLGQLACCCLHRRAKRRPPMTQVYERLEKLQAVVA Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Programmed cell death 1 ligand 1 (PD-L1) | 5J89 | 5.19 | |
Target general information Gen name CD274 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hPD-L1; Programmed death ligand 1; PDL1; PDCD1LG1; PDCD1L1; PDCD1 ligand 1; B7H1; B7-H1; B7 homolog 1 Protein family Immunoglobulin superfamily, BTN/MOG family Biochemical class Immunoglobulin Function As a ligand for the inhibitory receptor PDCD1/PD-1, modulates the activation threshold of T-cells and limits T-cell effector response. Through a yet unknown activating receptor, may costimulate T-cell subsets that predominantly produce interleukin-10 (IL10). Plays a critical role in induction and maintenance of immune tolerance to self. Related diseases Truncation of the 3'-untranslated (3'-UTR) region of CD274 transcripts leads to elevated expression of CD274 in multiple cancers including T-cell leukemia, diffuse large B-cell lymphoma and stomach adenocarcinoma (PubMed:27281199). Disruption of 3'-UTR region is caused by structural variants that stabilize CD274 transcripts, leading to overexpression (PubMed:27281199). Increased expression in tumors promotes immune evasion and tumor cell growth by allowing malignant cells to escape destruction by the immune system (PubMed:27281199). {ECO:0000269|PubMed:27281199}. Drugs (DrugBank ID) DB15773; DB11595; DB15771; DB11945; DB15772; DB14776; DB15770; DB11714; DB15769; DB09035; DB09037; DB00203; DB00313 Interacts with P33681; Q8IZR5; Q9NX76; Q15116; Q15116 EC number NA Uniprot keywords 3D-structure; Adaptive immunity; Alternative splicing; Cell membrane; Disulfide bond; Endosome; Glycoprotein; Immunity; Immunoglobulin domain; Membrane; Nucleus; Proteomics identification; Receptor; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID C,D Molecular weight (Da) 28335.2 Length 249 Aromaticity 0.1 Instability index 35.39 Isoelectric point 6.15 Charge (pH=7) -3.43 2D Binding mode Binding energy (Kcal/mol) -7.08 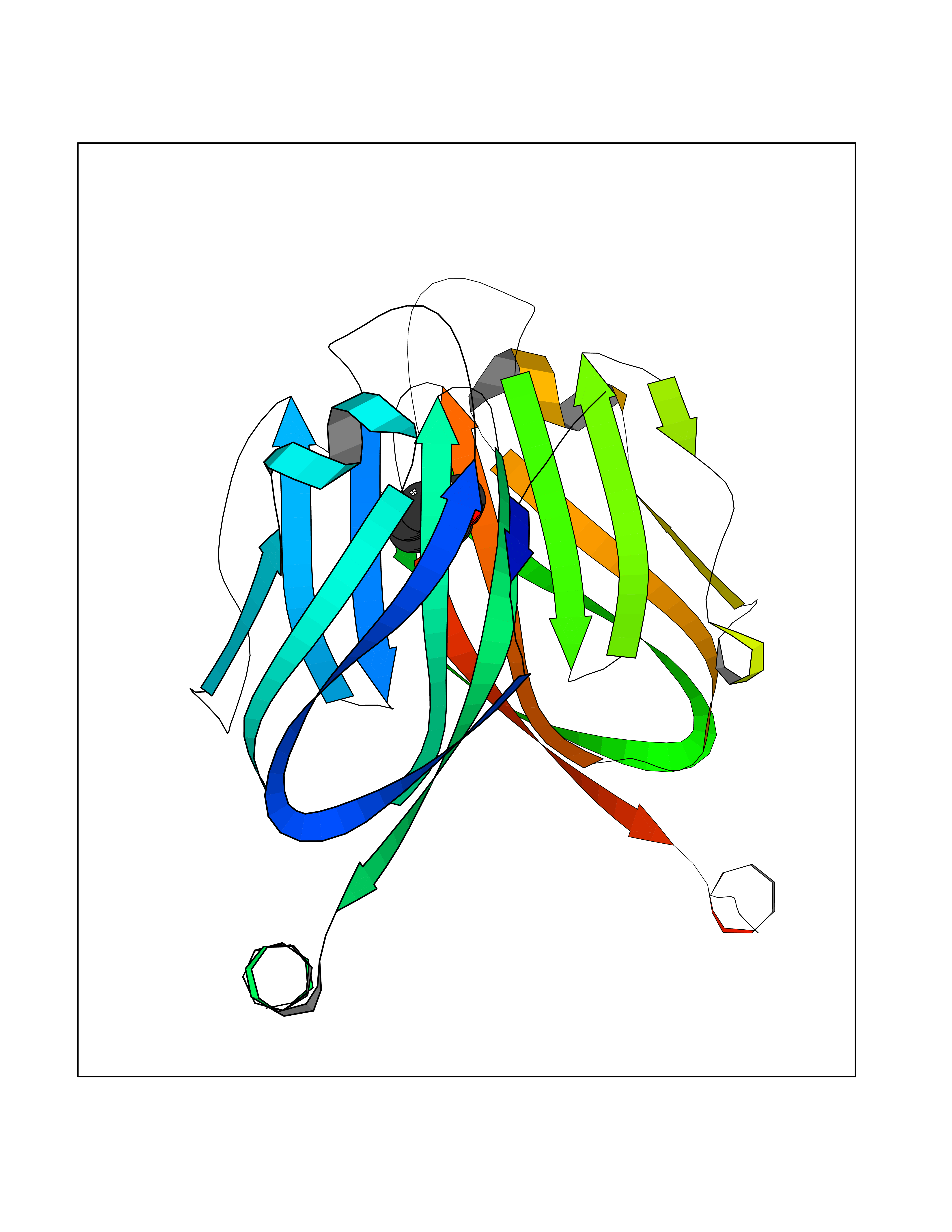 Molscript Map 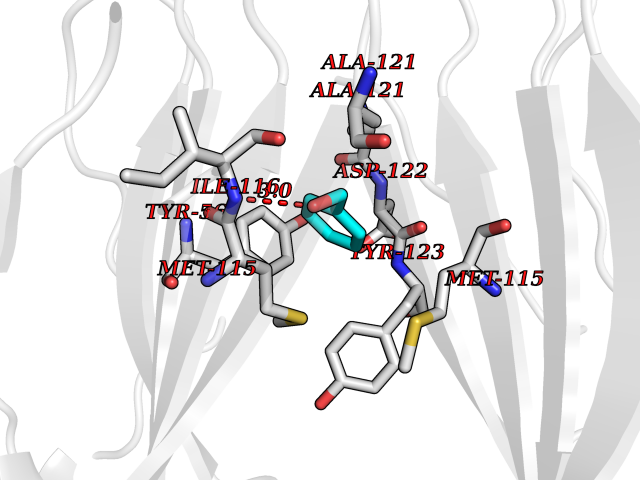 Pymol Map 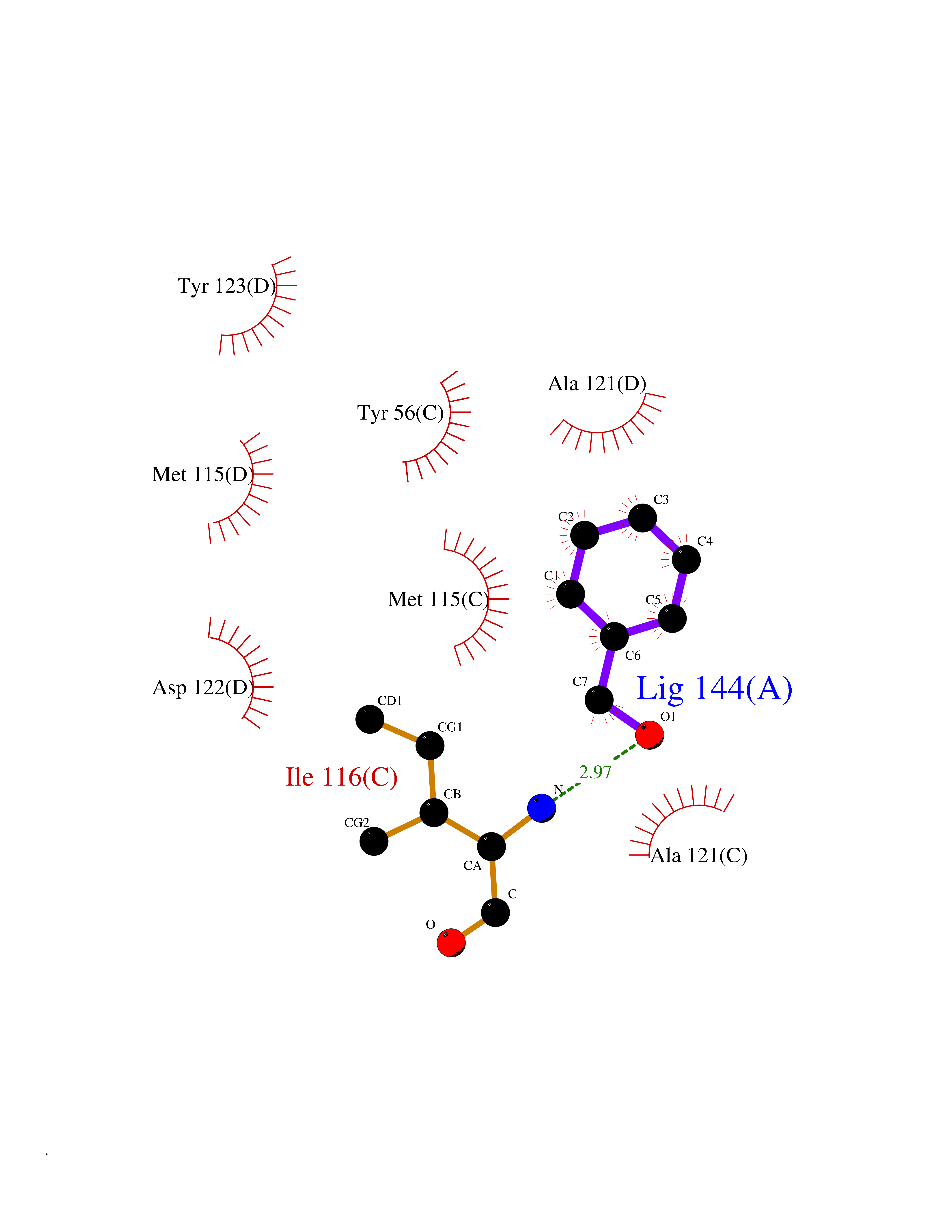 Ligplot Map 3D Binding mode Sequence AFTVTVPKDLYVVEYGSNMTIECKFPVEKQLDLAALIVYWEMEDKNIIQFVHGEEDLKVQHSSYRQRARLLKDQLSLGNAALQITDVKLQDAGVYRCMISYGGADYKRITVKVNAPYAAALEHHHAFTVTVPKDLYVVEYGSNMTIECKFPVEKQLDLAALIVYWEMEDKNIIQFVHGEEDLKVQHSSYRQRARLLKDQLSLGNAALQITDVKLQDAGVYRCMISYGGADYKRITVKVNAPYAAALEHH Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | 5'-methylthioadenosine/S-adenosylhomocysteine nucleosidase | 4WKC | 5.19 | |
Target general information Gen name mtnN Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms pfs;b0159;yadA;JW0155;mtn Protein family PNP/UDP phosphorylase family, MtnN subfamily Biochemical class hydrolase / hydrolase inhibitor Function Adenosylhomocysteine nucleosidase activity.Identical protein binding.Methylthioadenosine nucleosidase activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02158; DB08606; DB02933; DB00173; DB02281 Interacts with P0AF12 EC number 3.2.2.9 Uniprot keywords 3D-structure; Amino-acid biosynthesis; Hydrolase; Methionine biosynthesis; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 24353.7 Length 232 Aromaticity 0.05 Instability index 22.1 Isoelectric point 5.09 Charge (pH=7) -9.9 2D Binding mode Binding energy (Kcal/mol) -7.07  Molscript Map 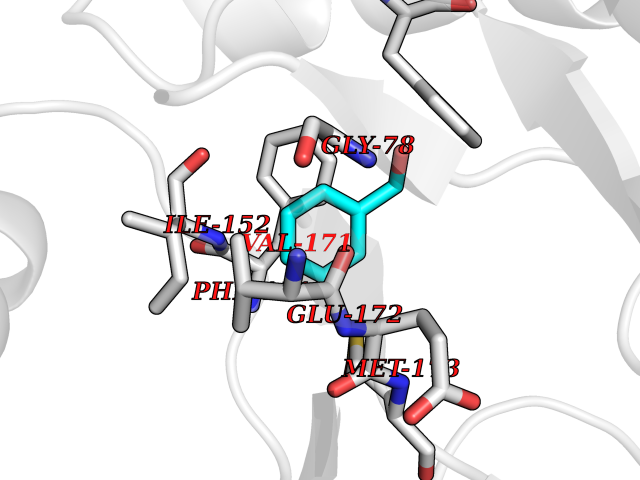 Pymol Map 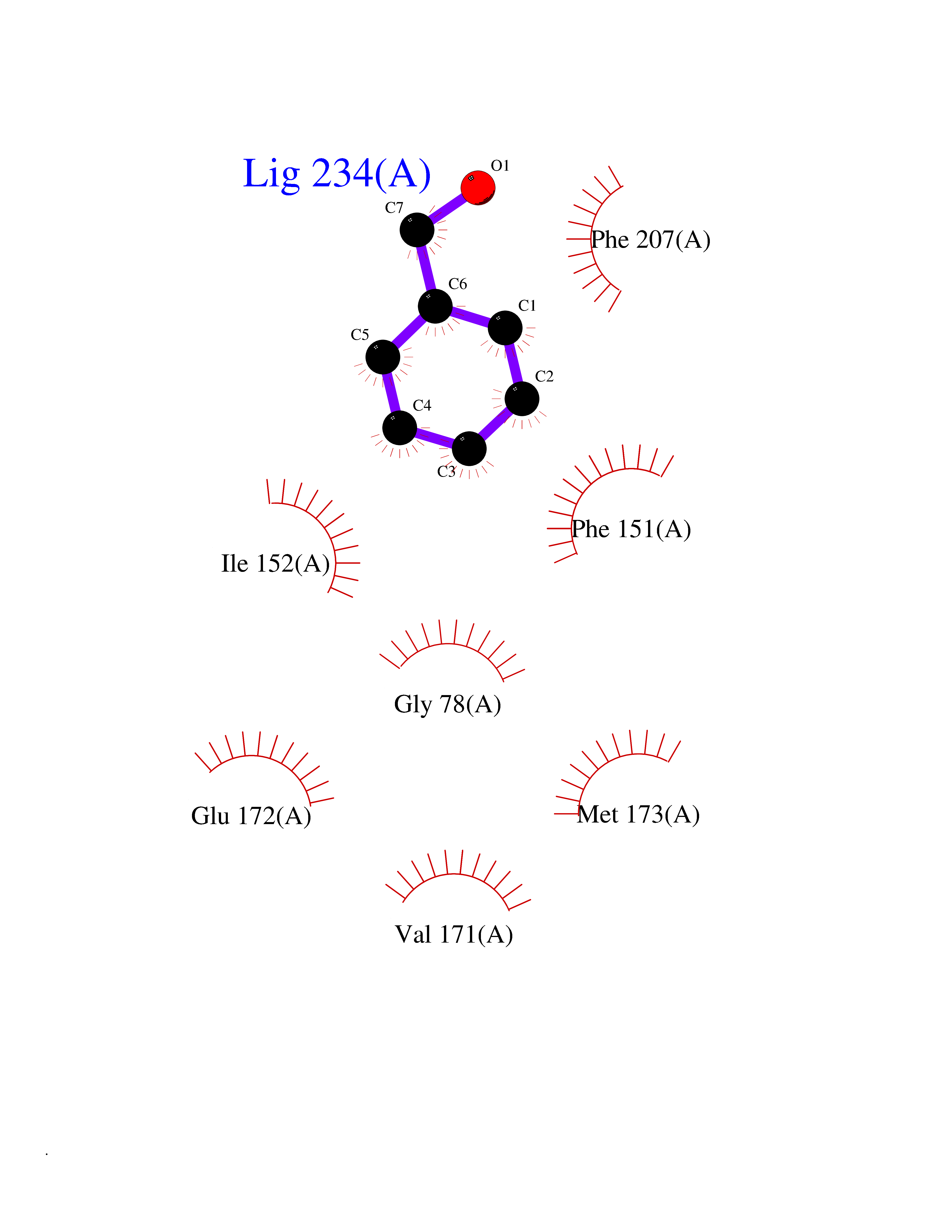 Ligplot Map 3D Binding mode Sequence MKIGIIGAMEEEVTLLRDKIENRQTISLGGCEIYTGQLNGTEVALLKSGIGKVAAALGATLLLEHCKPDVIINTGSAGGLAPTLKVGDIVVSDEARYHDADVTAFGYEYGQLPGCPAGFKADDKLIAAAEACIAELNLNAVRGLIVSGDAFINGSVGLAKIRHNFPQAIAVEMEATAIAHVCHNFNVPFVVVRAISDVADQQSHLSFDEFLAVAAKQSSLMVESLVQKLAHG Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Plasmepsin-2 | 2BJU | 5.19 | |
Target general information Gen name N/A Organism Plasmodium falciparum (isolate HB3) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase A1 family Biochemical class Hydrolase Function Aspartic-type endopeptidase activity. Related diseases Short/branched-chain acyl-CoA dehydrogenase deficiency (SBCADD) [MIM:610006]: Autosomal recessive disorder and consists of a defect in catabolism of L-isoleucine which is characterized by an increase of 2-methylbutyrylglycine and 2-methylbutyrylcarnitine in blood and urine. Affected individuals have seizures and psychomotor delay as the main clinical features. {ECO:0000269|PubMed:10832746, ECO:0000269|PubMed:11013134, ECO:0000269|PubMed:16317551}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04378; DB04373; DB11638; DB01218; DB02505; DB03063 Interacts with NA EC number 3.4.23.39 Uniprot keywords 3D-structure; Aspartyl protease; Direct protein sequencing; Disulfide bond; Hydrolase; Membrane; Protease; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Vacuole; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 36923.5 Length 329 Aromaticity 0.13 Instability index 44.31 Isoelectric point 4.67 Charge (pH=7) -17.94 2D Binding mode Binding energy (Kcal/mol) -7.08 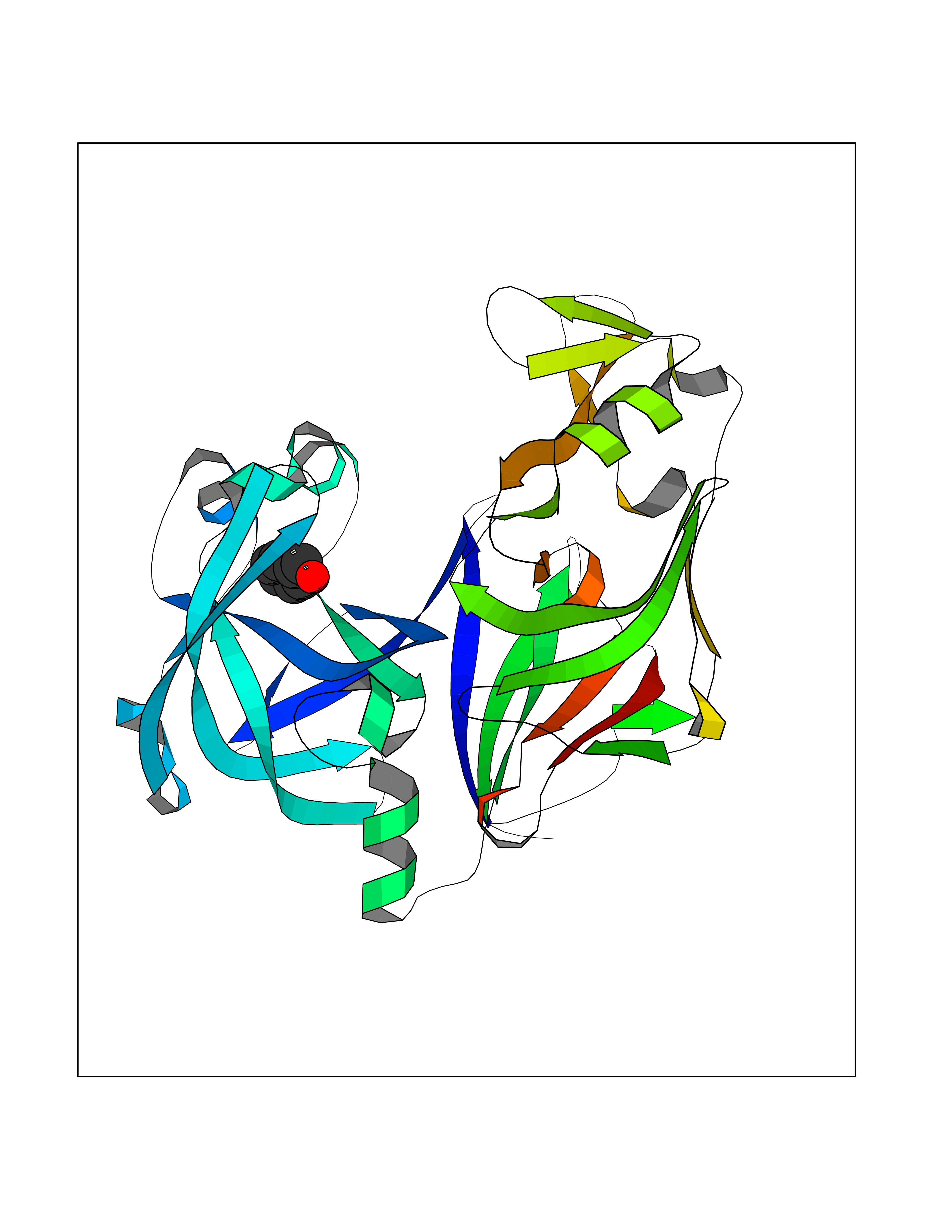 Molscript Map 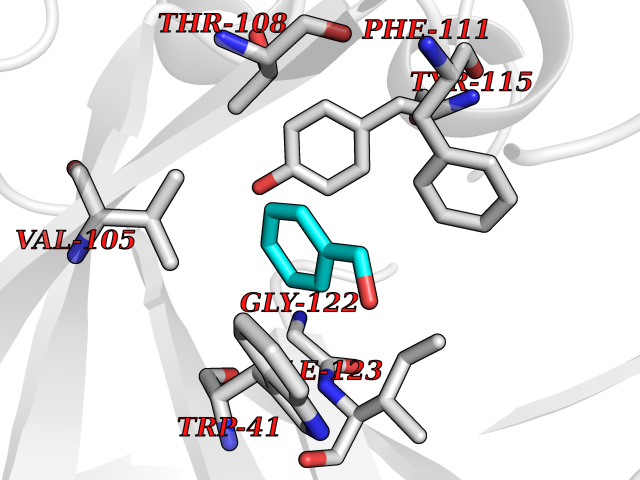 Pymol Map 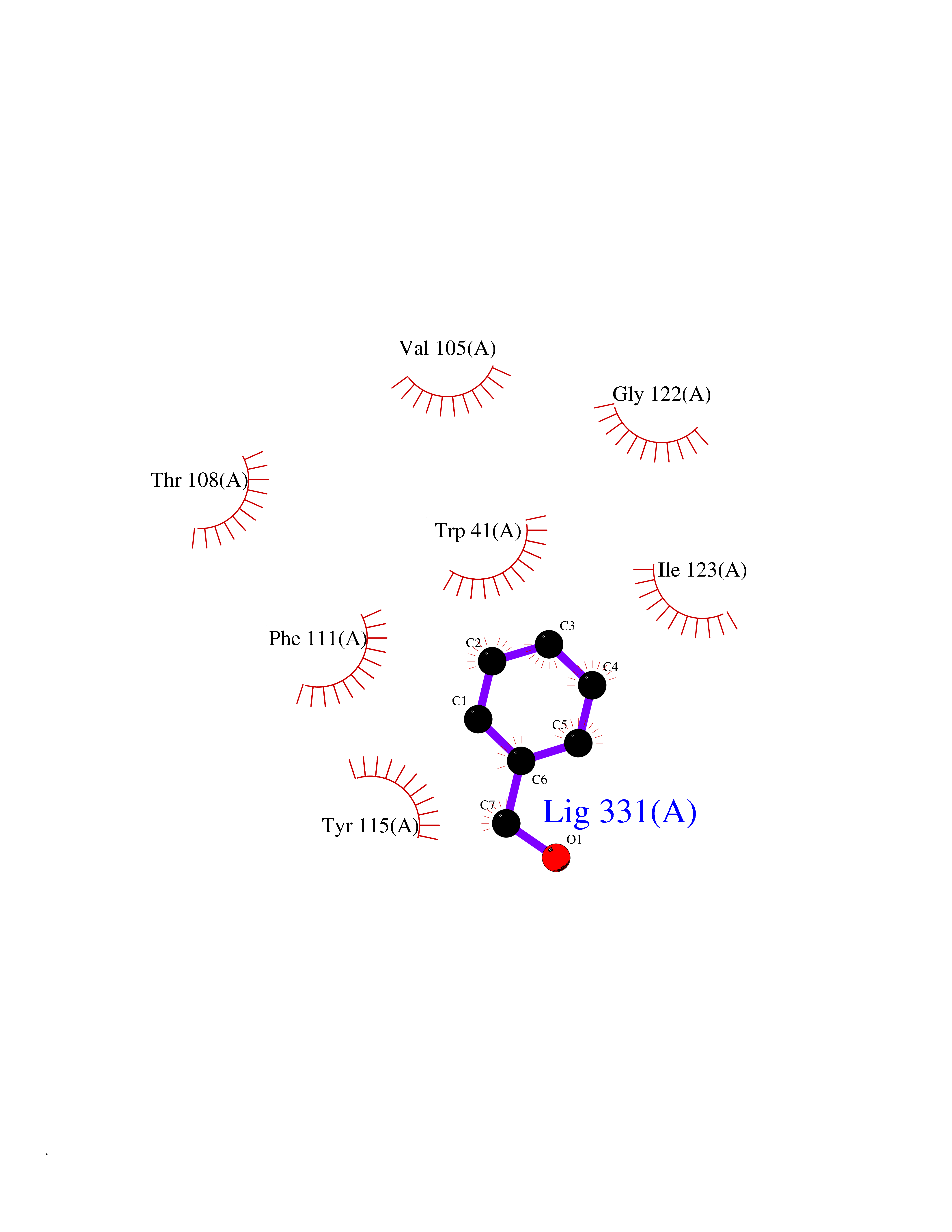 Ligplot Map 3D Binding mode Sequence SSNDNIELVDFQNIMFYGDAEVGDNQQPFTFILDTGSANLWVPSVKCTTAGCLTKHLYDSSKSRTYEKDGTKVEMNYVSGTVSGFFSKDLVTVGNLSLPYKFIEVIDTNGFEPTYTASTFDGILGLGWKDLSIGSVDPIVVELKNQNKIENALFTFYLPVHDKHTGFLTIGGIEERFYEGPLTYEKLNHDLYWQITLDAHVGNIMLEKANCIVDSGTSAITVPTDFLNKMLQNLDVIKVPFLPFYVTLCNNSKLPTFEFTSENGKYTLEPEYYLQHIEDVGPGLCMLNIIGLDFPVPTFILGDPFMRKYFTVFDYDNHSVGIALAKKNL Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Acetylcholine receptor subunit alpha | 4ZJS | 5.18 | |
Target general information Gen name CHRNA1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ACHRA;CHNRA Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-1/CHRNA1 sub-subfamily Biochemical class Immune system Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ion channel activity.Ligand-gated ion channel activity. Related diseases Multiple pterygium syndrome, lethal type (LMPS) [MIM:253290]: Multiple pterygia are found infrequently in children with arthrogryposis and in fetuses with fetal akinesia syndrome. In lethal multiple pterygium syndrome there is intrauterine growth retardation, multiple pterygia, and flexion contractures causing severe arthrogryposis and fetal akinesia. Subcutaneous edema can be severe, causing fetal hydrops with cystic hygroma and lung hypoplasia. Oligohydramnios and facial anomalies are frequent. {ECO:0000269|PubMed:18252226}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: The alpha subunit is the main focus for antibody binding in myasthenia gravis. Myasthenia gravis is characterized by sporadic muscular fatigability and weakness, occurring chiefly in muscles innervated by cranial nerves, and characteristically improved by cholinesterase-inhibiting drugs.; DISEASE: Myasthenic syndrome, congenital, 1A, slow-channel (CMS1A) [MIM:601462]: A common congenital myasthenic syndrome. Congenital myasthenic syndromes are characterized by muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS1A is a slow-channel myasthenic syndrome. It is caused by kinetic abnormalities of the AChR, resulting in prolonged AChR channel opening episodes, prolonged endplate currents, and depolarization block. This is associated with calcium overload, which may contribute to subsequent degeneration of the endplate and postsynaptic membrane. {ECO:0000269|PubMed:16685696, ECO:0000269|PubMed:7619526, ECO:0000269|PubMed:8872460, ECO:0000269|PubMed:9158151, ECO:0000269|PubMed:9221765}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 1B, fast-channel (CMS1B) [MIM:608930]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS1B is a fast-channel myasthenic syndrome. It is caused by kinetic abnormalities of the AChR, resulting in brief opening and activity of the channel, with a rapid decay in endplate current, failure to achieve threshold depolarization of the endplate and consequent failure to fire an action potential. {ECO:0000269|PubMed:10195214, ECO:0000269|PubMed:12588888, ECO:0000269|PubMed:15079006}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08838; DB00565; DB00555 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Congenital myasthenic syndrome; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46717.8 Length 411 Aromaticity 0.11 Instability index 38.02 Isoelectric point 4.77 Charge (pH=7) -22.31 2D Binding mode Binding energy (Kcal/mol) -7.07 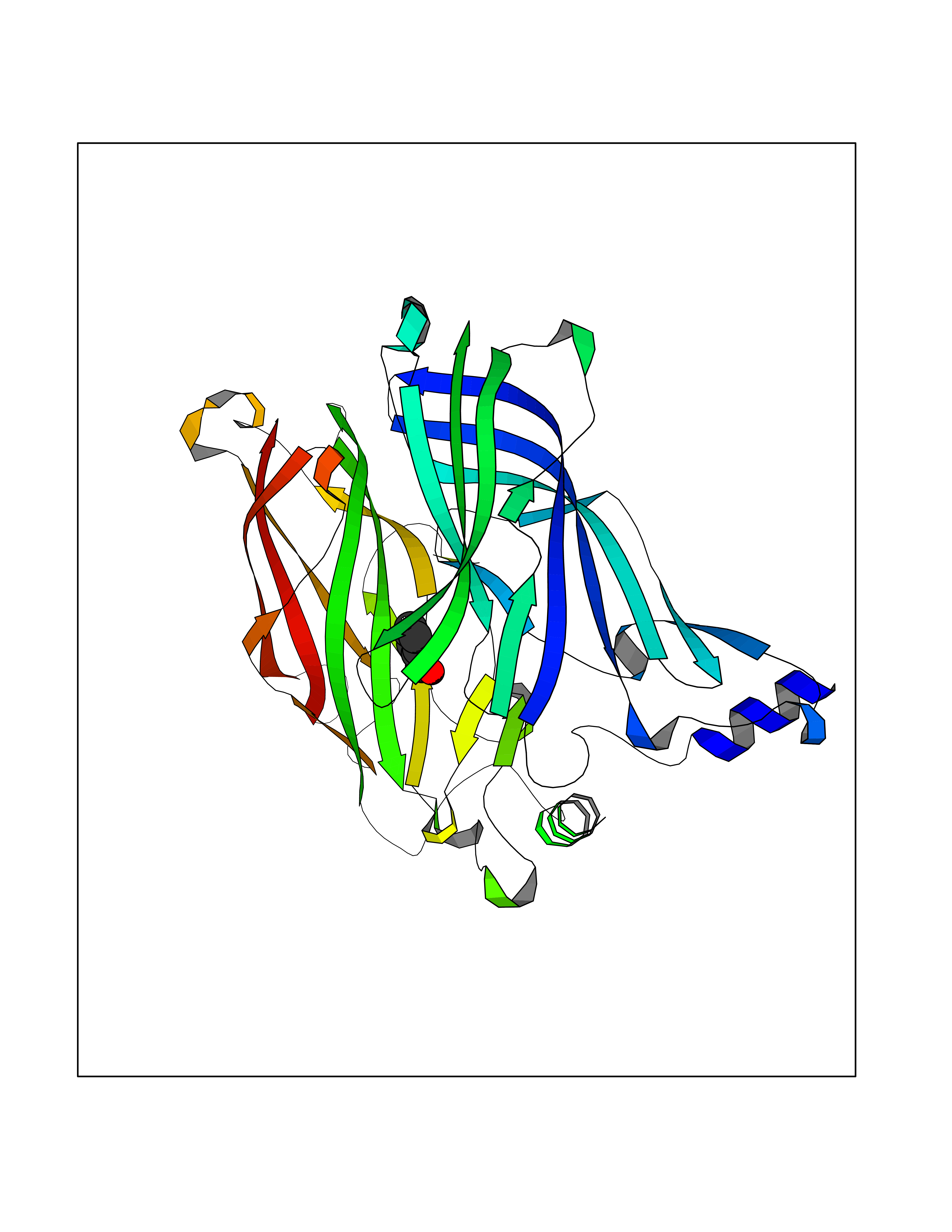 Molscript Map 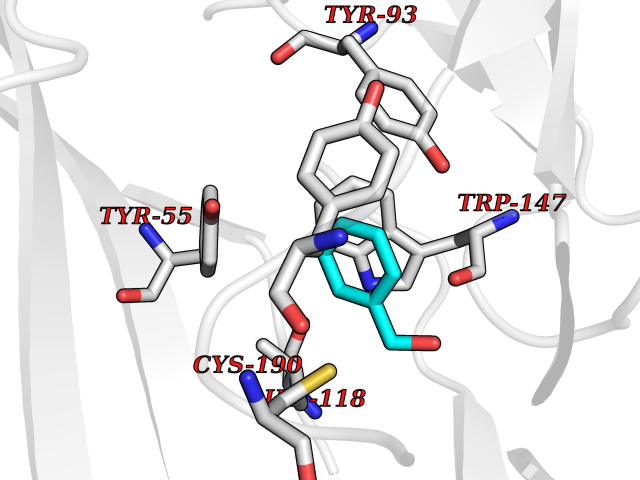 Pymol Map 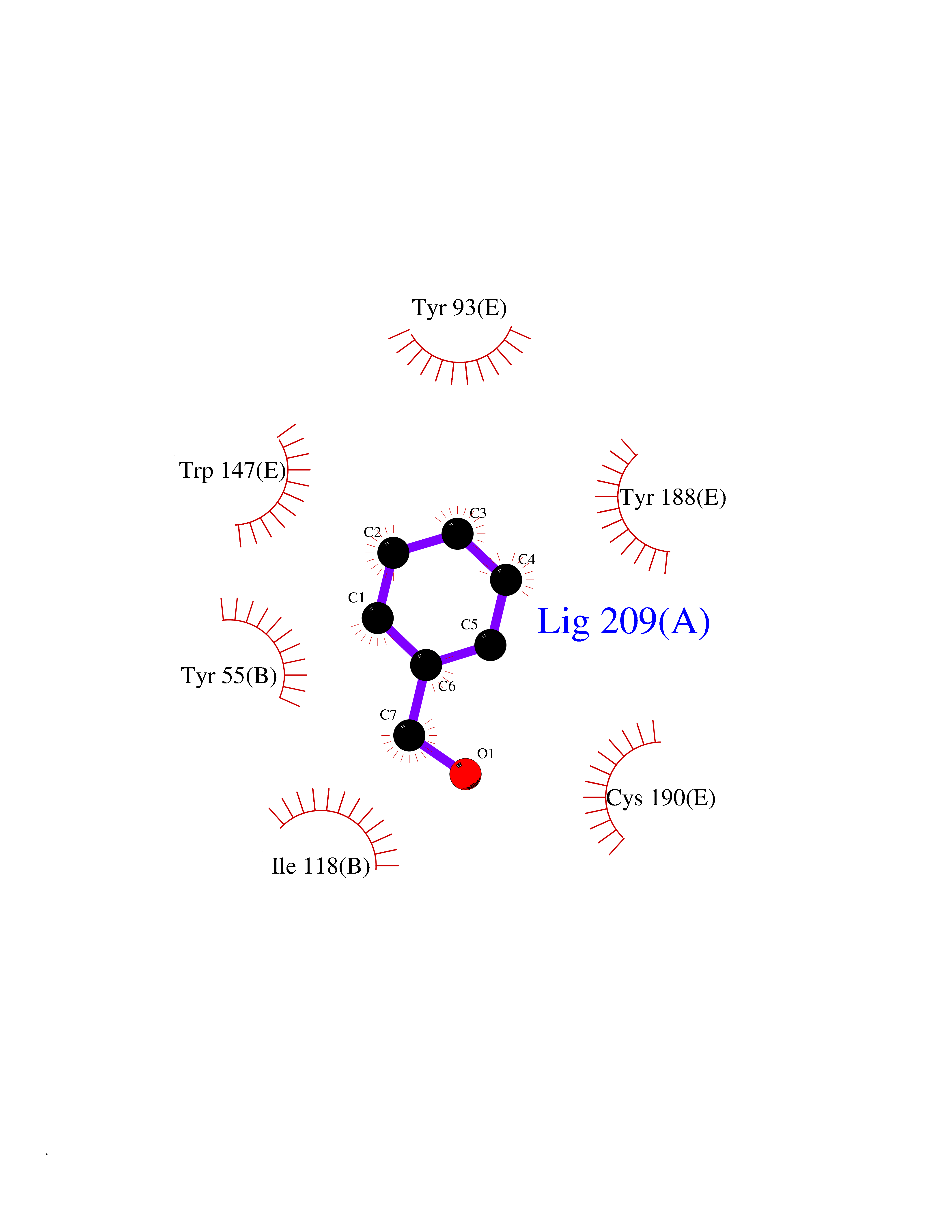 Ligplot Map 3D Binding mode Sequence EHETRLVAKLFKDYSSVVRPVEDHRQVVEVTLGFTLQDIVKADSSTNEVDLVYYEQQRWVDYNLKWNPDDYGGVKKIHIPAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQTRQVQHYSCCPEPYIDVNLVVKFREEHETRLVAKLFKDYSSVVRPVEDHRQVVEVTLGFTLQDIVKADSSTNEVDLVYYEQQRWVDYNLKWNPDDYGGVKKIHIPAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQTRQVQHYSCCPEPYIDVNLVVKFRER Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | Cyclin-dependent kinase 4 | 2W96 | 5.18 | |
Target general information Gen name CDK4 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, CDC2/CDKX subfamily Biochemical class Cell cycle Function ATP binding.Cyclin binding.Cyclin-dependent protein serine/threonine kinase activity.Cyclin-dependent protein serine/threonine kinase regulator activity.Protein complex binding. Related diseases Melanoma, cutaneous malignant 3 (CMM3) [MIM:609048]: A malignant neoplasm of melanocytes, arising de novo or from a pre-existing benign nevus, which occurs most often in the skin but may also involve other sites. {ECO:0000269|PubMed:7652577, ECO:0000269|PubMed:8528263, ECO:0000269|PubMed:9311594, ECO:0000269|PubMed:9425228}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12001; DB03496; DB12010; DB09073; DB02733; DB11730; DB15442 Interacts with Q9UH17; P24385; P30279; P30281; Q16543; P50613; P38936; P46527; P49918; P42771; P42772; P42773; P55273; Q9UJC3; P08238; Q9UKT9; Q0VD86; P01106; Q9ULD0; P28749; Q08999; P09936; Q8N720 EC number 2.7.11.22 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cell cycle; Cell division; Cytoplasm; Disease variant; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID B Molecular weight (Da) 30138.4 Length 267 Aromaticity 0.09 Instability index 36.2 Isoelectric point 5.78 Charge (pH=7) -5.83 2D Binding mode Binding energy (Kcal/mol) -7.07  Molscript Map  Pymol Map 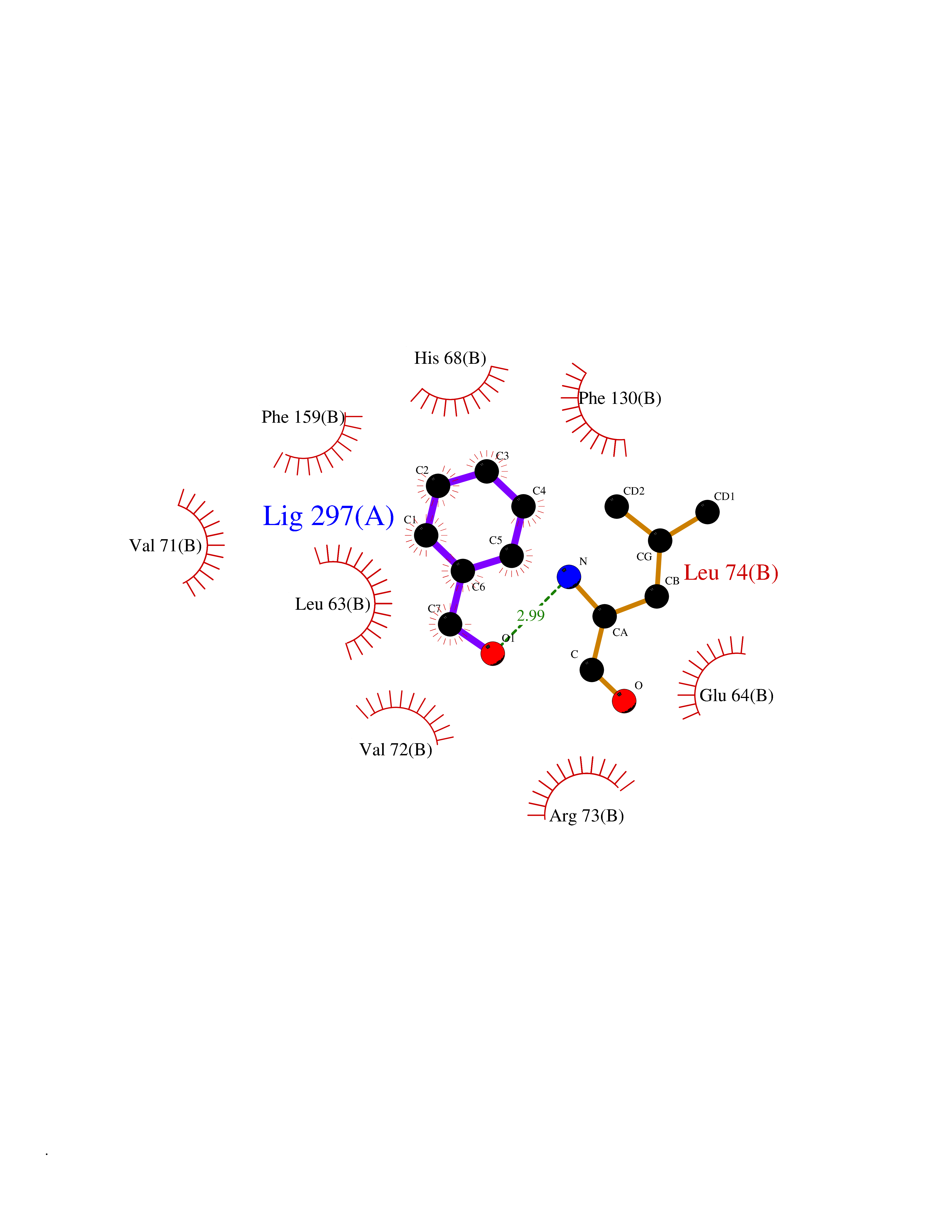 Ligplot Map 3D Binding mode Sequence SRYEPVAEIGVGAYGTVYKARDPHSGHFVALKSVRVPNGEEGLPISTVREVALLRRLEAFEHPNVVRLMDVCATSRTDREIKVTLVFEHVDQDLRTYLDKAPPPGLPAETIKDLMRQFLRGLDFLHANCIVHRDLKPENILVTSGGTVKLADFGLARIYSYQMALDPVVVTLWYRAPEVLLQSTYATPVDMWSVGCIFAEMFRRKPLFCGNSEADQLGKIFDLIGLPPEDDWVPEMEESGAQLLLEMLTFNPHKRISAFRALQHSYL Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Tyrosine 3-monooxygenase (TH) | 2XSN | 5.18 | |
Target general information Gen name TH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tyrosine 3-hydroxylase; TH Protein family Biopterin-dependent aromatic amino acid hydroxylase family Biochemical class Paired donor oxygen oxidoreductase Function Plays an important role in the physiology of adrenergic neurones. Related diseases Segawa syndrome autosomal recessive (ARSEGS) [MIM:605407]: A form of DOPA-responsive dystonia presenting in infancy or early childhood. Dystonia is defined by the presence of sustained involuntary muscle contractions, often leading to abnormal postures. Some cases present with parkinsonian symptoms in infancy. Unlike all other forms of dystonia, it is an eminently treatable condition, due to a favorable response to L-DOPA. {ECO:0000269|PubMed:10585338, ECO:0000269|PubMed:11196107, ECO:0000269|PubMed:11246459, ECO:0000269|PubMed:15505183, ECO:0000269|PubMed:15747353, ECO:0000269|PubMed:16049992, ECO:0000269|PubMed:17696123, ECO:0000269|PubMed:18058633, ECO:0000269|PubMed:18554280, ECO:0000269|PubMed:19491146, ECO:0000269|PubMed:20056467, ECO:0000269|PubMed:20430833, ECO:0000269|PubMed:21940685, ECO:0000269|PubMed:22264700, ECO:0000269|PubMed:22815559, ECO:0000269|PubMed:23762320, ECO:0000269|PubMed:23939262, ECO:0000269|PubMed:24753243, ECO:0000269|PubMed:7814018, ECO:0000269|PubMed:8528210, ECO:0000269|PubMed:8817341, ECO:0000269|PubMed:9613851, ECO:0000269|PubMed:9703425}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: May play a role in the pathogenesis of Parkinson disease (PD). A genome-wide copy number variation analysis has identified a 34 kilobase deletion over the TH gene in a PD patient but not in any controls. {ECO:0000269|PubMed:20809526}. Drugs (DrugBank ID) DB03552; DB04400; DB00765; DB00120; DB00360; DB00135 Interacts with P29762; P61978-2; Q99750; P08651-5; O75928-2; Q9UHX1-2; P0DJD3-2; P07101-3; Q9UJ04; C9J7I0; Q5MCW4 EC number EC 1.14.16.2 Uniprot keywords 3D-structure; Alternative splicing; Catecholamine biosynthesis; Cell projection; Cytoplasm; Cytoplasmic vesicle; Disease variant; Dystonia; Iron; Metal-binding; Monooxygenase; Neurotransmitter biosynthesis; Nucleus; Oxidoreductase; Parkinson disease; Parkinsonism; Phosphoprotein; Proteomics identification; Reference proteome; Synapse Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 34997 Length 306 Aromaticity 0.12 Instability index 42.59 Isoelectric point 5.32 Charge (pH=7) -12.31 2D Binding mode Binding energy (Kcal/mol) -7.07  Molscript Map 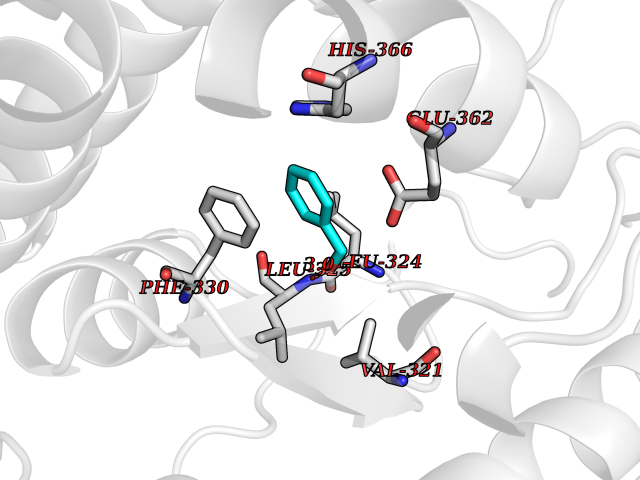 Pymol Map 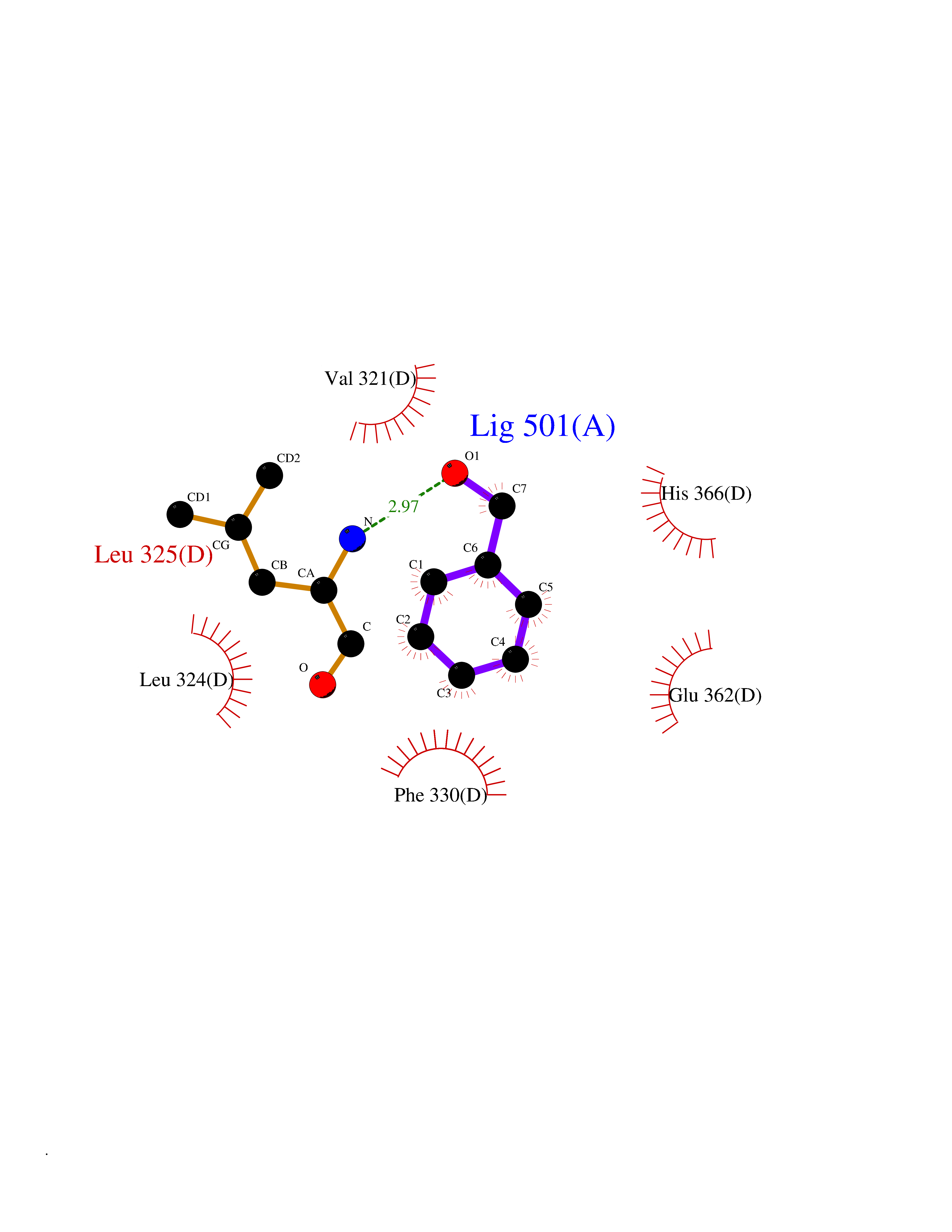 Ligplot Map 3D Binding mode Sequence VPWFPRKVSELDKCHHLVTKFDPDLDLDHPGFSDQVYRQRRKLIAEIAFQYRHGDPIPRVEYTAEEIATWKEVYTTLKGLYATHACGEHLEAFALLERFSGYREDNIPQLEDVSRFLKERTGFQLRPVAGLLSARDFLASLAFRVFQCTQYIRHASSPMHSPEPDCCHELLGHVPMLADRTFAQFSQDIGLASLGASDEEIEKLSTLYWFTVEFGLCKQNGEVKAYGAGLLSSYGELLHCLSEEPEIRAFDPEAAAVQPYQDQTYQSVYFVSESFSDAKDKLRSYASRIQRPFSVKFDPYTLAIDV Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Ribonucleoside-diphosphate reductase subunit M2 | 3OLJ | 5.18 | |
Target general information Gen name RRM2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms RR2 Protein family Ribonucleoside diphosphate reductase small chain family Biochemical class Oxidoreductase Function Metal ion binding.Ribonucleoside-diphosphate reductase activity, thioredoxin disulfide as acceptor. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00242; DB05260; DB05801; DB05003; DB05428 Interacts with P41002; Q9UM11; P23921; O00560 EC number 1.17.4.1 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Deoxyribonucleotide synthesis; Iron; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 33579.4 Length 286 Aromaticity 0.14 Instability index 43.7 Isoelectric point 5.12 Charge (pH=7) -12.86 2D Binding mode Binding energy (Kcal/mol) -7.06 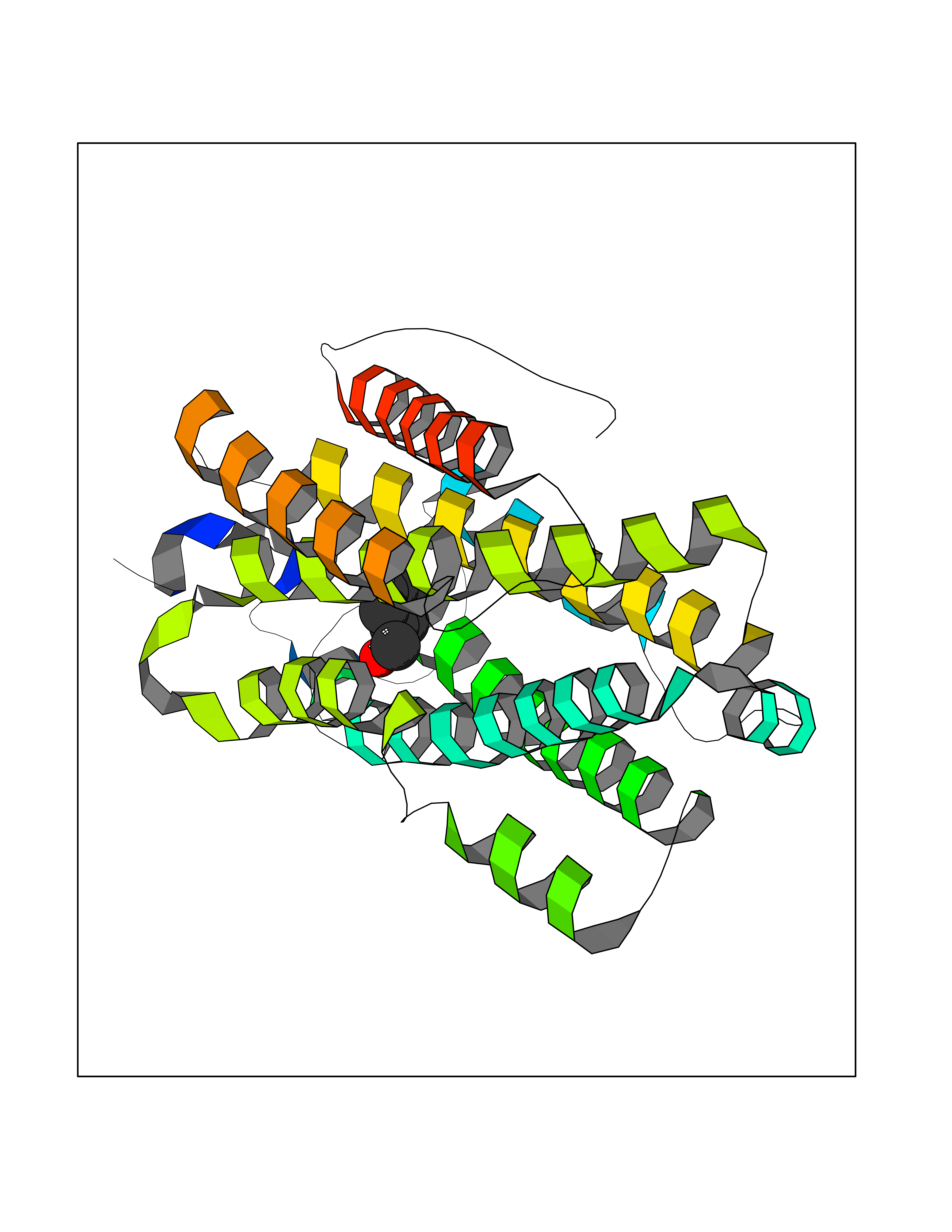 Molscript Map 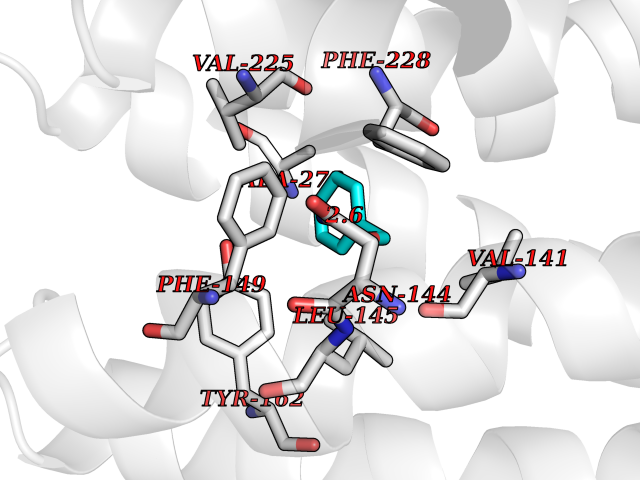 Pymol Map 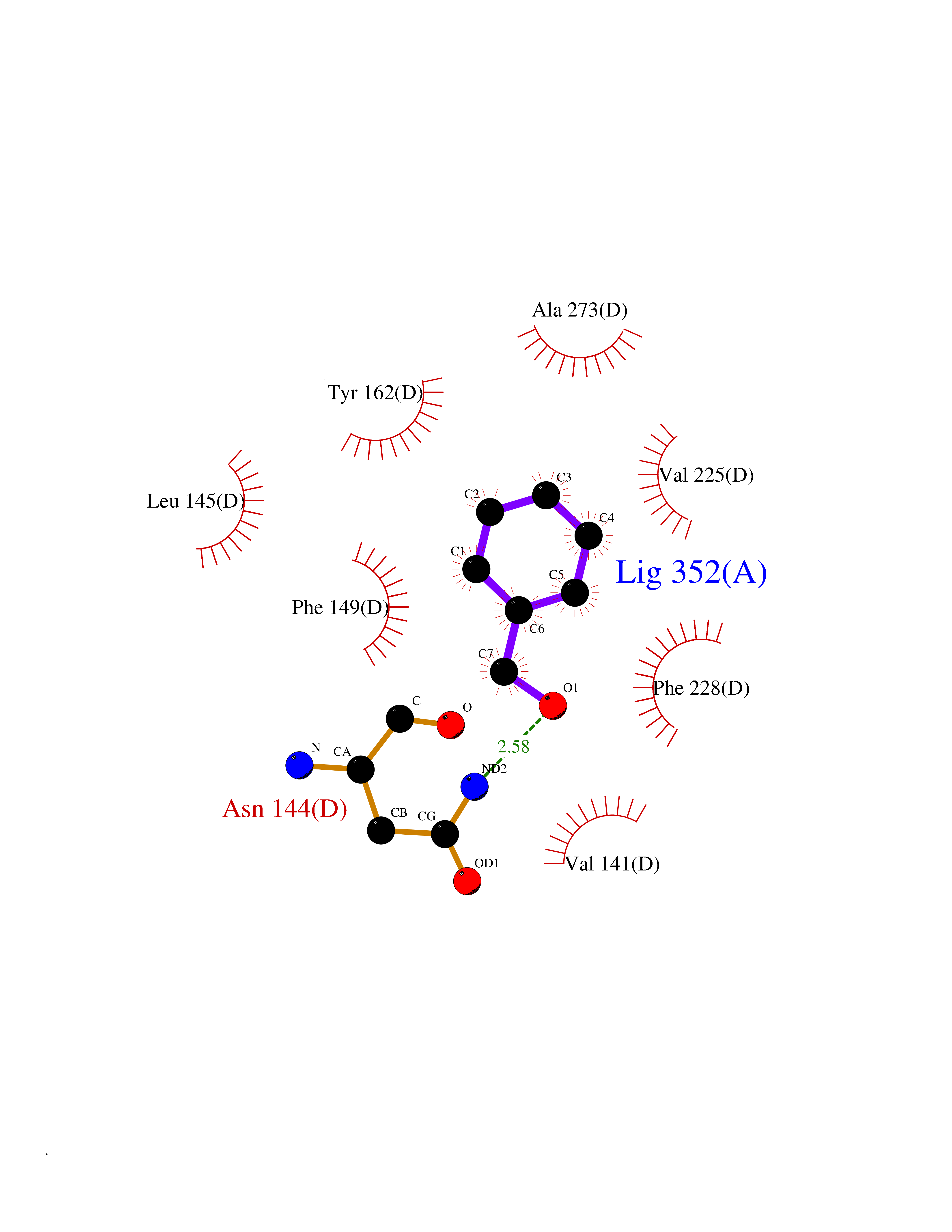 Ligplot Map 3D Binding mode Sequence MGVEDEPLLRENPRRFVIFPIEYHDIWQMYKKAEASFWTAEEVDLSKDIQHWESLKPEERYFISHVLAFFAASDGIVNENLVERFSQEVQITEARCFYGFQIAMENIHSEMYSLLIDTYIKDPKEREFLFNAIETMPCVKKKADWALRWIGDKEATYGERVVAFAAVEGIFFSGSFASIFWLKKRGLMPGLTFSNELISRDEGLHCDFACLMFKHLVHKPSEERVREIIINAVRIEQEFLTEALPVKLIGMNCTLMKQYIEFVADRLMLELGFSKVFRVENPFDFM Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Cocaine esterase | 3I2K | 5.18 | |
Target general information Gen name cocE Organism Rhodococcus sp. (strain MB1 Bresler) Uniprot ID TTD ID NA Synonyms NA Protein family CocE/NonD hydrolase family Biochemical class Hydrolase Function Carboxylic ester hydrolase activity.Dipeptidyl-peptidase activity. Related diseases Thiamine metabolism dysfunction syndrome 5, episodic encephalopathy type (THMD5) [MIM:614458]: An autosomal recessive metabolic disorder due to an inborn error of thiamine metabolism. The phenotype is highly variable, but in general, affected individuals have onset in early childhood of acute encephalopathic episodes associated with increased serum and CSF lactate. These episodes result in progressive neurologic dysfunction manifest as gait disturbances, ataxia, dystonia, and spasticity, which in some cases may result in loss of ability to walk. Cognitive function is usually preserved, although mildly delayed development has been reported. These episodes are usually associated with infection and metabolic decompensation. Some patients may have recovery of some neurologic deficits. {ECO:0000269|PubMed:22152682}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03793; DB01795 Interacts with NA EC number 3.1.1.84 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Hydrolase; Serine esterase Protein physicochemical properties Chain ID A Molecular weight (Da) 62127.9 Length 574 Aromaticity 0.09 Instability index 26.62 Isoelectric point 4.56 Charge (pH=7) -33.24 2D Binding mode Binding energy (Kcal/mol) -7.06 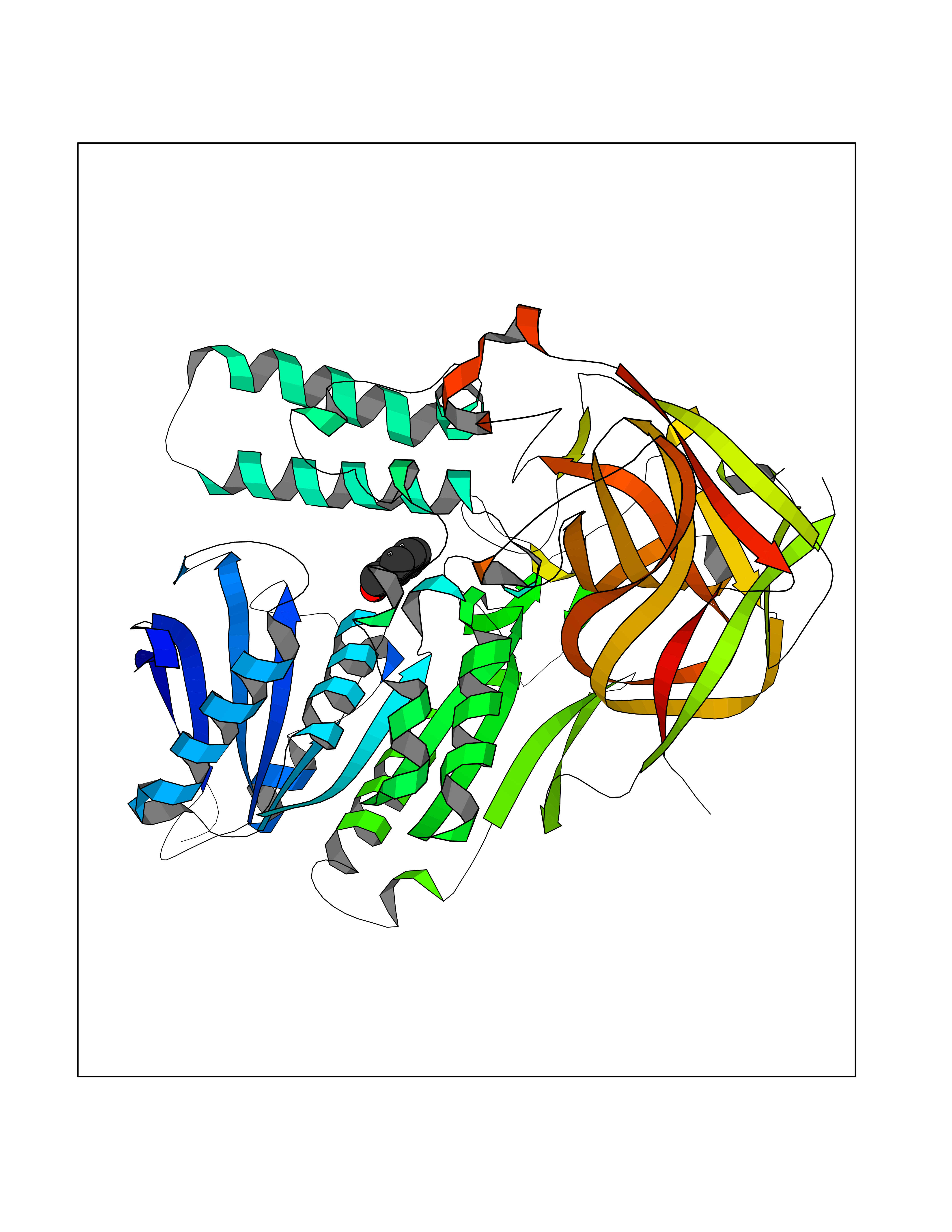 Molscript Map 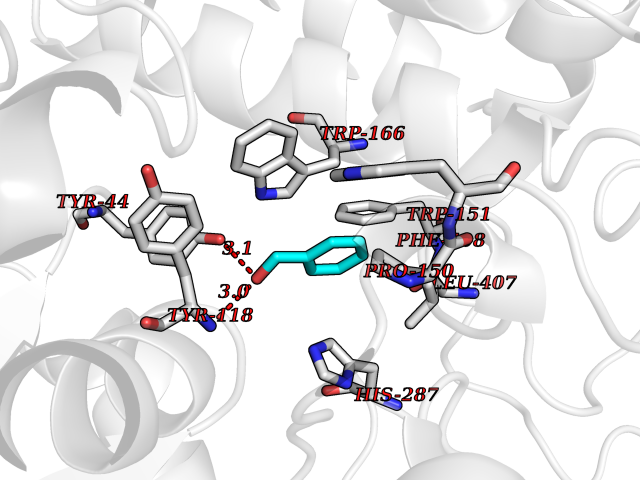 Pymol Map 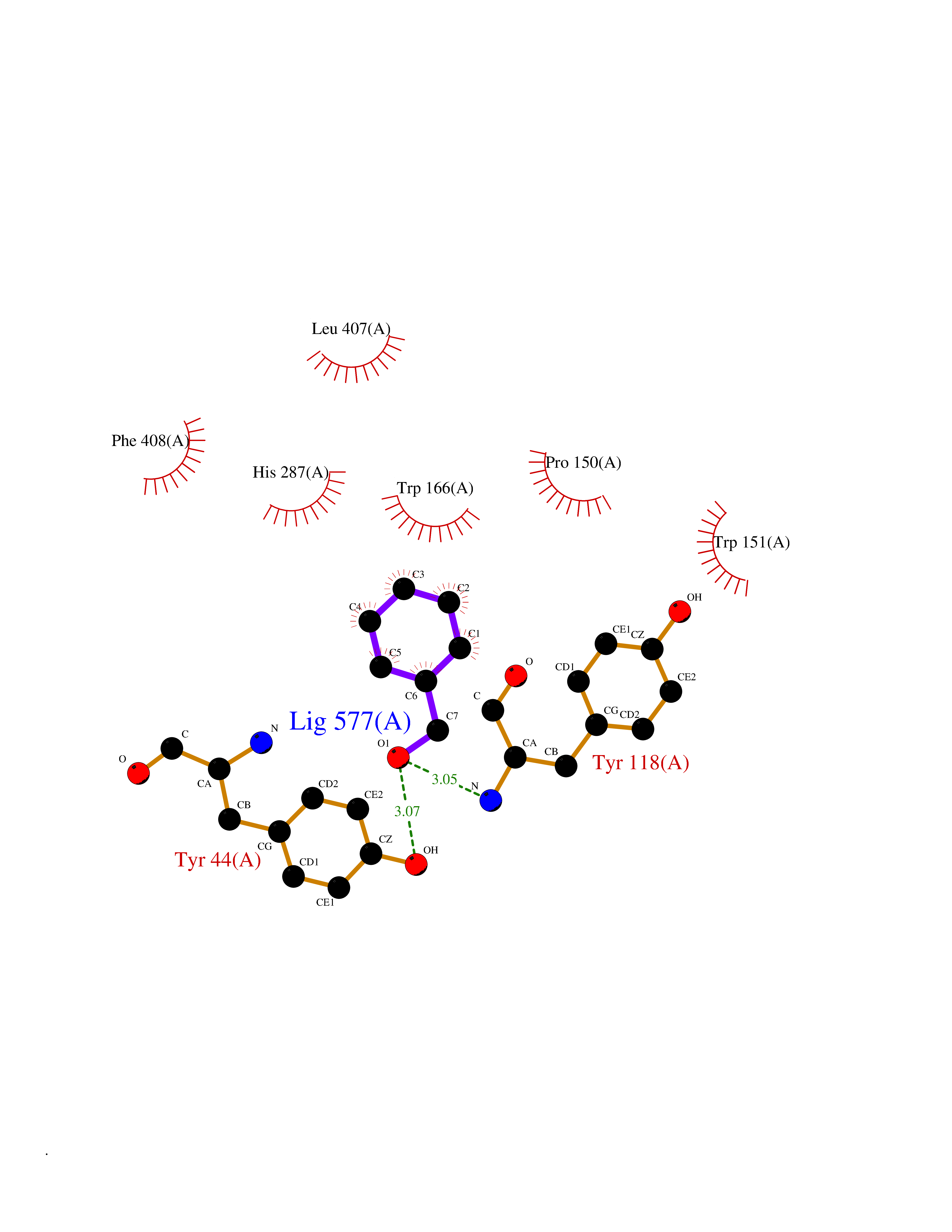 Ligplot Map 3D Binding mode Sequence VDGNYSVASNVMVPMRDGVRLAVDLYRPDADGPVPVLLVRNPYDKFDVFAWSTQSTNWLEFVRDGYAVVIQDTRGLFASEGEFVPHVDDEADAEDTLSWILEQAWCDGNVGMFGVSYLGVTQWQAAVSGVGGLKAIAPSMASADLYRAPWYGPGGALSVEALLGWSALIGTGLITSRSDARPEDAADFVQLAAILNDVAGAASVTPLAEQPLLGRLIPWVIDQVVDHPDNDESWQSISLFERLGGLATPALITAGWYDGFVGESLRTFVAVKDNADARLVVGPWSHSNLTGRNADRKFGIAATYPIQEATTMHKAFFDRHLRGETDALAGVPKVRLFVMGIDEWRDETDWPLPDTAYTPFYLGGSGAANTSTGGGTLSTSISGTESADTYLYDPADPVPSLGGTLLFHNGDNGPADQRPIHDRDDVLCYSTEVLTDPVEVTGTVSARLFVSSSAVDTDFTAKLVDVFPDGRAIALCDGIVRMRYRETLVNPTLIEAGEIYEVAIDMLATSNVFLPGHRIMVQVSSSNFPKYDRNSNTGGVIAREQLEEMCTAVNRIHRGPEHPSHIVLPIIKRK Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Euchromatic histone-lysine N-methyltransferase 1 (EHMT1) | 5TTG | 5.18 | |
Target general information Gen name EHMT1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms EHMT1 Protein family Class V-like SAM-binding methyltransferase superfamily Biochemical class NA Function Histone methyltransferase that specifically mono- and dimethylates 'Lys-9' of histone H3 (H3K9me1 and H3K9me2, respectively) in euchromatin. H3K9me represents a specific tag for epigenetic transcriptional repression by recruiting HP1 proteins to methylated histones. Also weakly methylates 'Lys-27' of histone H3 (H3K27me). Also required for DNA methylation, the histone methyltransferase activity is not required for DNA methylation, suggesting that these 2 activities function independently. Probably targeted to histone H3 by different DNA-binding proteins like E2F6, MGA, MAX and/or DP1. During G0 phase, it probably contributes to silencing of MYC- and E2F-responsive genes, suggesting a role in G0/G1 transition in cell cycle. In addition to the histone methyltransferase activity, also methylates non-histone proteins: mediates dimethylation of 'Lys-373' of p53/TP53. Related diseases Kleefstra syndrome 1 (KLEFS1) [MIM:610253]: A form of Kleefstra syndrome, an autosomal dominant disease characterized by variable intellectual disability, psychomotor developmental delay, seizures, behavioral abnormalities, and facial dysmorphisms. KLEFS1 patients additionally manifest brachy(micro)cephaly, congenital heart defects, and urogenital defects. {ECO:0000269|PubMed:16826528, ECO:0000269|PubMed:19264732}. The disease is caused by variants affecting the gene represented in this entry. The syndrome can be either caused by intragenic EHMT1 mutations leading to haploinsufficiency of the EHMT1 gene or by a submicroscopic 9q34.3 deletion. Although it is not known if and to what extent other genes in the 9q34.3 region contribute to the syndrome observed in deletion cases, EHMT1 seems to be the major determinant of the core disease phenotype (PubMed:19264732). {ECO:0000269|PubMed:16826528, ECO:0000269|PubMed:19264732}. Drugs (DrugBank ID) NA Interacts with Q99549; Q04206; Q04207 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ANK repeat; Chromatin regulator; Chromosome; Disease variant; Intellectual disability; Isopeptide bond; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; S-adenosyl-L-methionine; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 30066.9 Length 260 Aromaticity 0.11 Instability index 49.19 Isoelectric point 5.73 Charge (pH=7) -4.88 2D Binding mode Binding energy (Kcal/mol) -7.06  Molscript Map 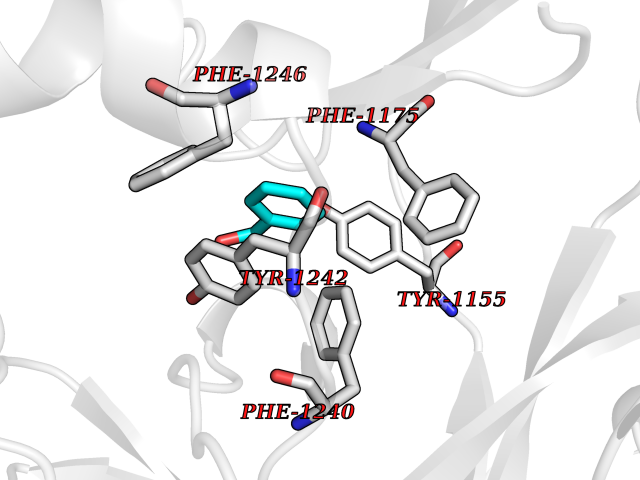 Pymol Map 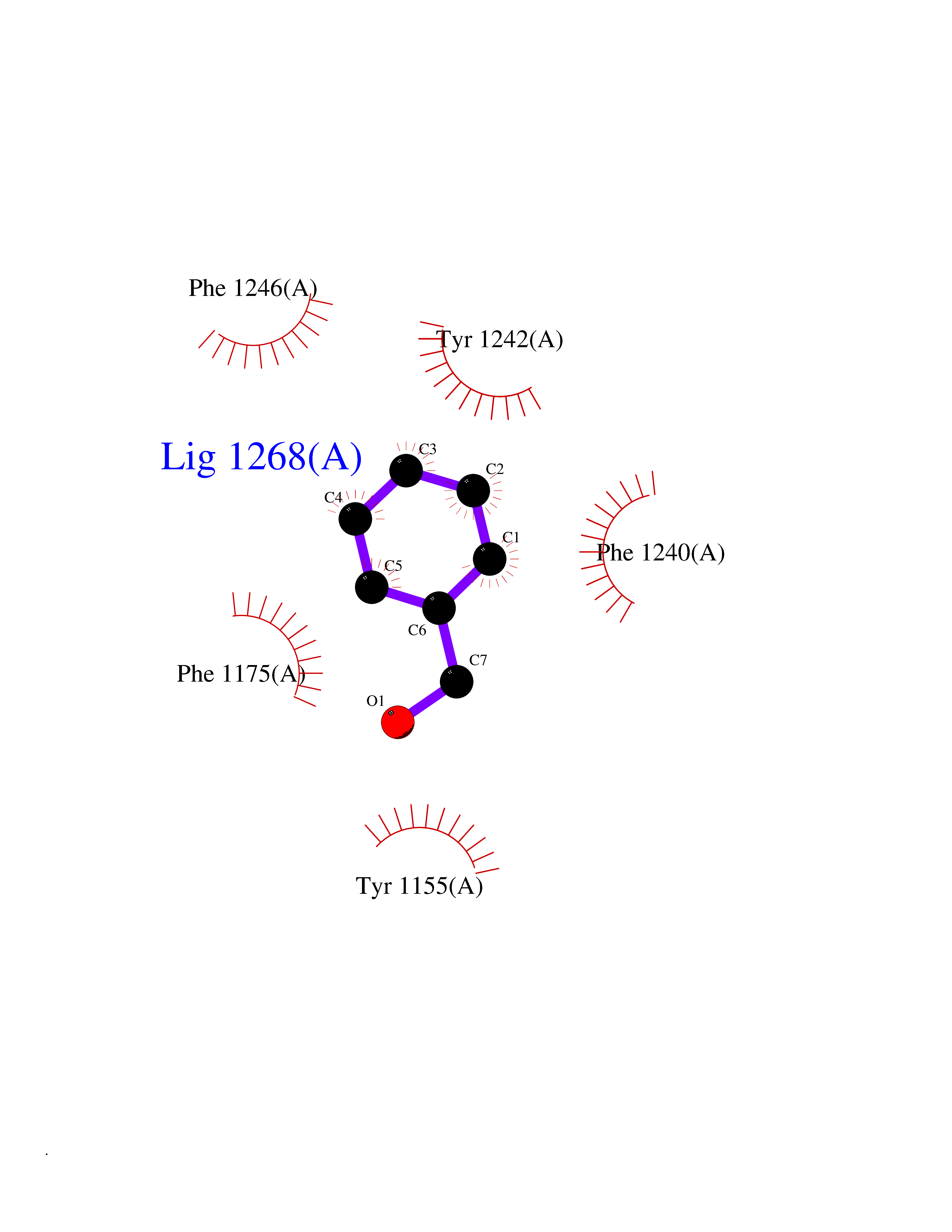 Ligplot Map 3D Binding mode Sequence VERIVSRDIARGYERIPIPCVNAVDSEPCPSNYKYVSQNCVTSPMNIDRNITHLQYCVCIDDCSSSNCMCGQLSMRCWYDKDGRLLPEFNMAEPPLIFECNHACSCWRNCRNRVVQNGLRARLQLYRTRDMGWGVRSLQDIPPGTFVCEYVGELISDSEADVREEDSYLFDLDNDGEVYCIDARFYGNVSRFINHHCEPNLVPVRVFMAHQDLRFPRIAFFSTRLIEAGEQLGFDYGERFWDIKGKLFSCRCGSPKCRHS Hydrogen bonds contact Hydrophobic contact | ||||