Job Results:
Ligand
Structure
Job ID
8e57bce38c3d7c3304510f6b58eb7b68
Job name
NA
Time
2026-02-27 11:59:36
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Cytochrome c oxidase subunit 2 | 3VRJ | 4.75 | |
Target general information Gen name MT-CO2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms MTCO2;COXII;COII;COX2 Protein family Cytochrome c oxidase subunit 2 family Biochemical class Immune system Function Copper ion binding.Cytochrome-c oxidase activity. Related diseases Mitochondrial complex IV deficiency (MT-C4D) [MIM:220110]: A disorder of the mitochondrial respiratory chain with heterogeneous clinical manifestations, ranging from isolated myopathy to severe multisystem disease affecting several tissues and organs. Features include hypertrophic cardiomyopathy, hepatomegaly and liver dysfunction, hypotonia, muscle weakness, exercise intolerance, developmental delay, delayed motor development and intellectual disability. Some affected individuals manifest a fatal hypertrophic cardiomyopathy resulting in neonatal death. A subset of patients manifest Leigh syndrome. {ECO:0000269|PubMed:10486321}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02659; DB04464; DB05412 Interacts with Q9NZ94-2; P49281-3 EC number 7.1.1.9 Uniprot keywords 3D-structure; Copper; Disease variant; Electron transport; Magnesium; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Primary mitochondrial disease; Proteomics identification; Reference proteome; Respiratory chain; Translocase; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID C Molecular weight (Da) 21687.9 Length 189 Aromaticity 0.11 Instability index 38 Isoelectric point 5.68 Charge (pH=7) -3.26 2D Binding mode Binding energy (Kcal/mol) -6.48 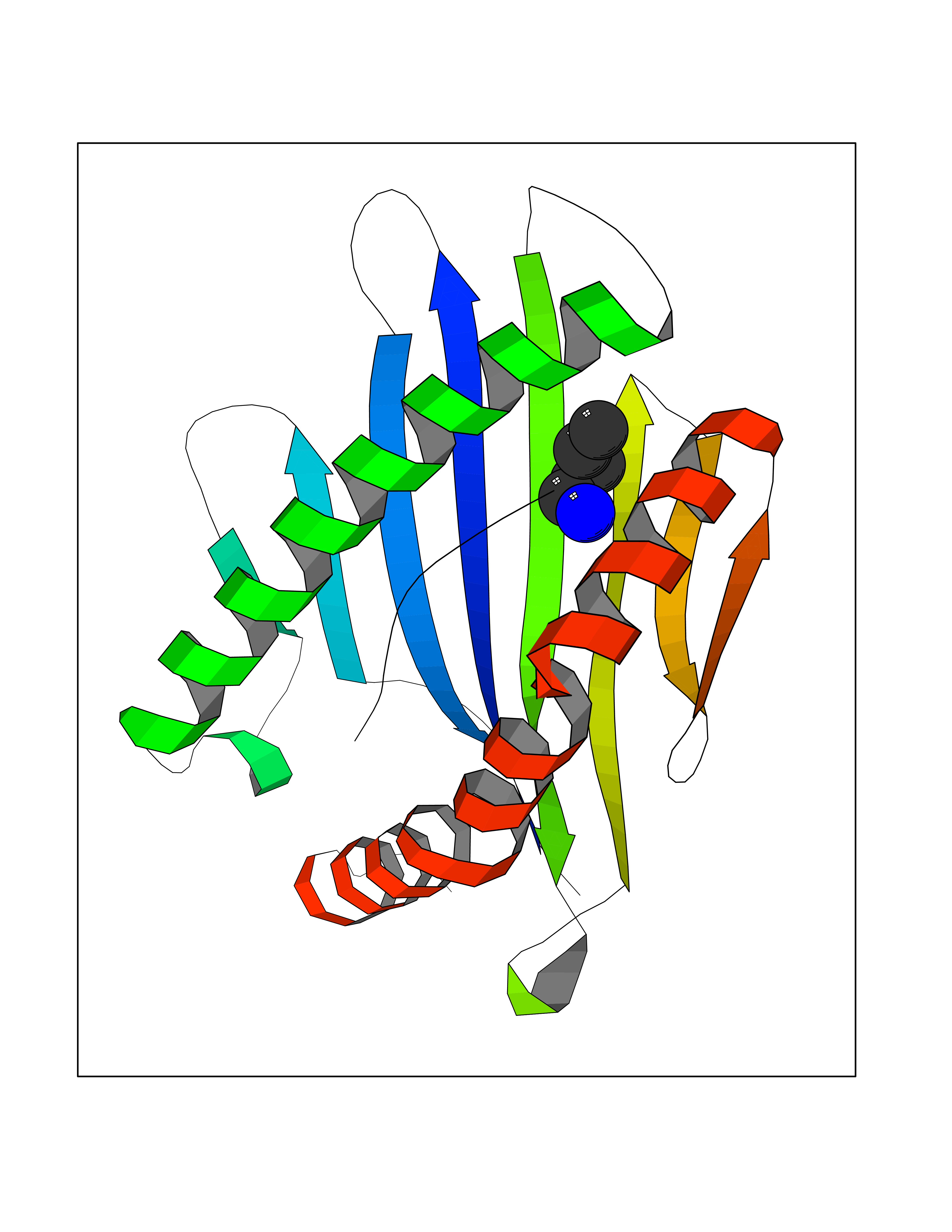 Molscript Map 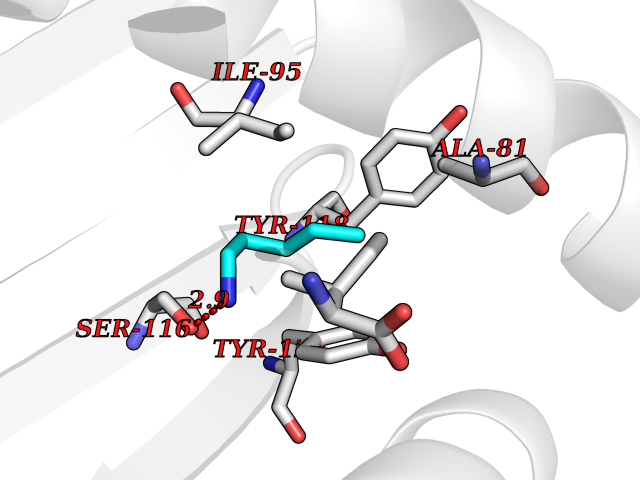 Pymol Map 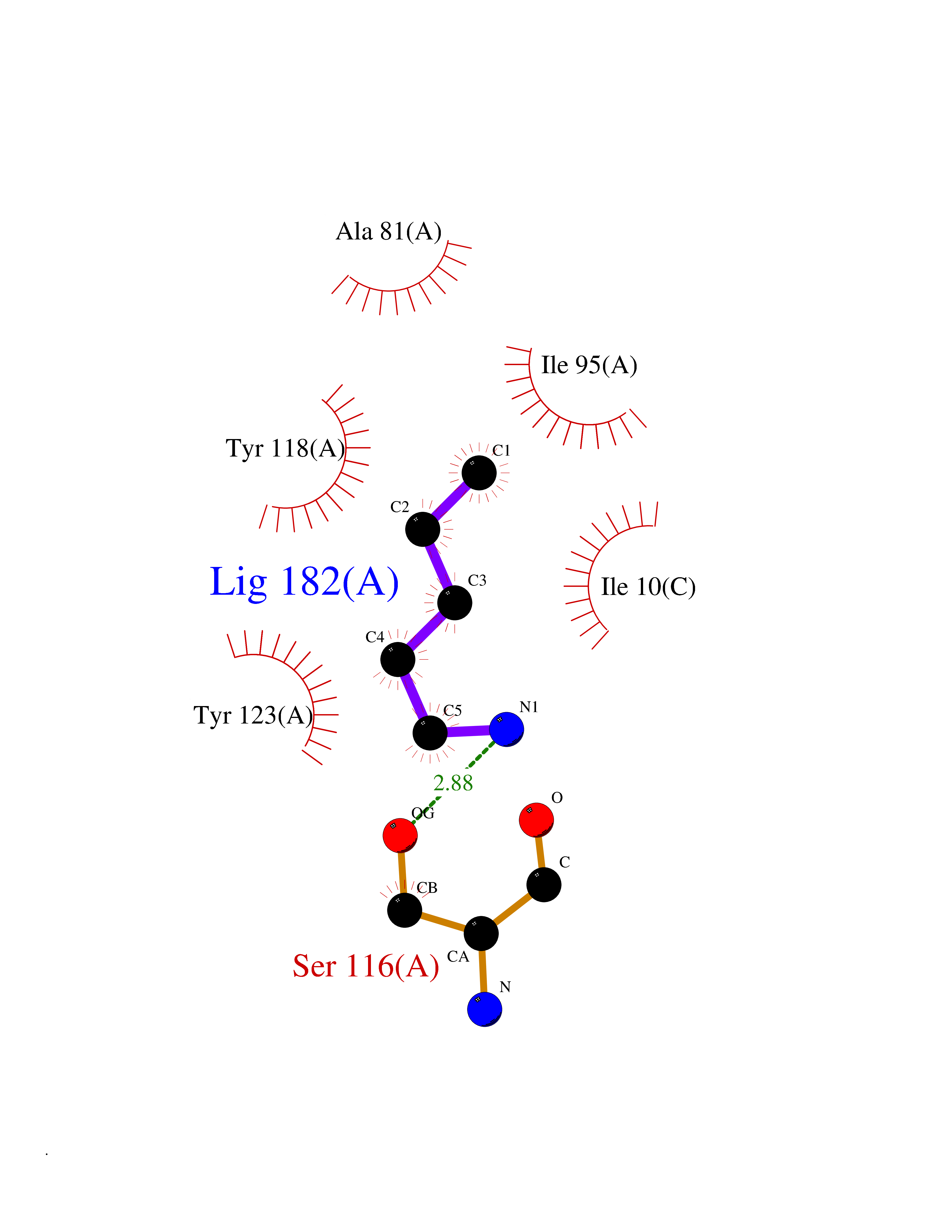 Ligplot Map 3D Binding mode Sequence SHSMRYFYTAMSRPGRGEPRFIAVGYVDDTQFVRFDSDAASPRMAPRAPWIEQEGPEYWDGETRNMKASAQTYRENLRIALRYYNQSEAGSHIIQVMYGCDVGPDGRLLRGHDQSAYDGKDYIALNEDLSSWTAADTAAQITQRKWEAARVAEQLRAYLEGLCVEWLRRYLENGKETLQLTTKLTNTNI Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Natriuretic peptides B | 1YK1 | 4.75 | |
Target general information Gen name NPPB Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Natriuretic peptide family Biochemical class Hormone / growth factor receptor Function Diuretic hormone activity.Hormone activity.Peptide hormone receptor binding.Receptor binding. Related diseases Multiple fibroadenomas of the breast (MFAB) [MIM:615554]: A benign breast disease marked by lobuloalveolar growth with abnormally high proliferation of the epithelium, and characterized by the presence of more than 3 fibroadenomas in one breast. Fibroadenomas are adenomas containing fibrous tissue. {ECO:0000269|PubMed:18779591}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperprolactinemia (HPRL) [MIM:615555]: A disorder characterized by increased levels of prolactin in the blood not associated with gestation or the puerperium. HPRL may result in infertility, hypogonadism, and galactorrhea. {ECO:0000269|PubMed:24195502}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01136; DB06412 Interacts with A8MQ03; P57678; Q6A162; P60411; Q7Z3S9; P25788; Q9UJW9 EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Disulfide bond; Glycoprotein; Hormone; Pharmaceutical; Proteoglycan; Proteomics identification; Reference proteome; Secreted; Signal; Vasoactive; Vasodilator Protein physicochemical properties Chain ID E Molecular weight (Da) 46353.1 Length 415 Aromaticity 0.1 Instability index 37.91 Isoelectric point 5.51 Charge (pH=7) -12.09 2D Binding mode Binding energy (Kcal/mol) -6.48 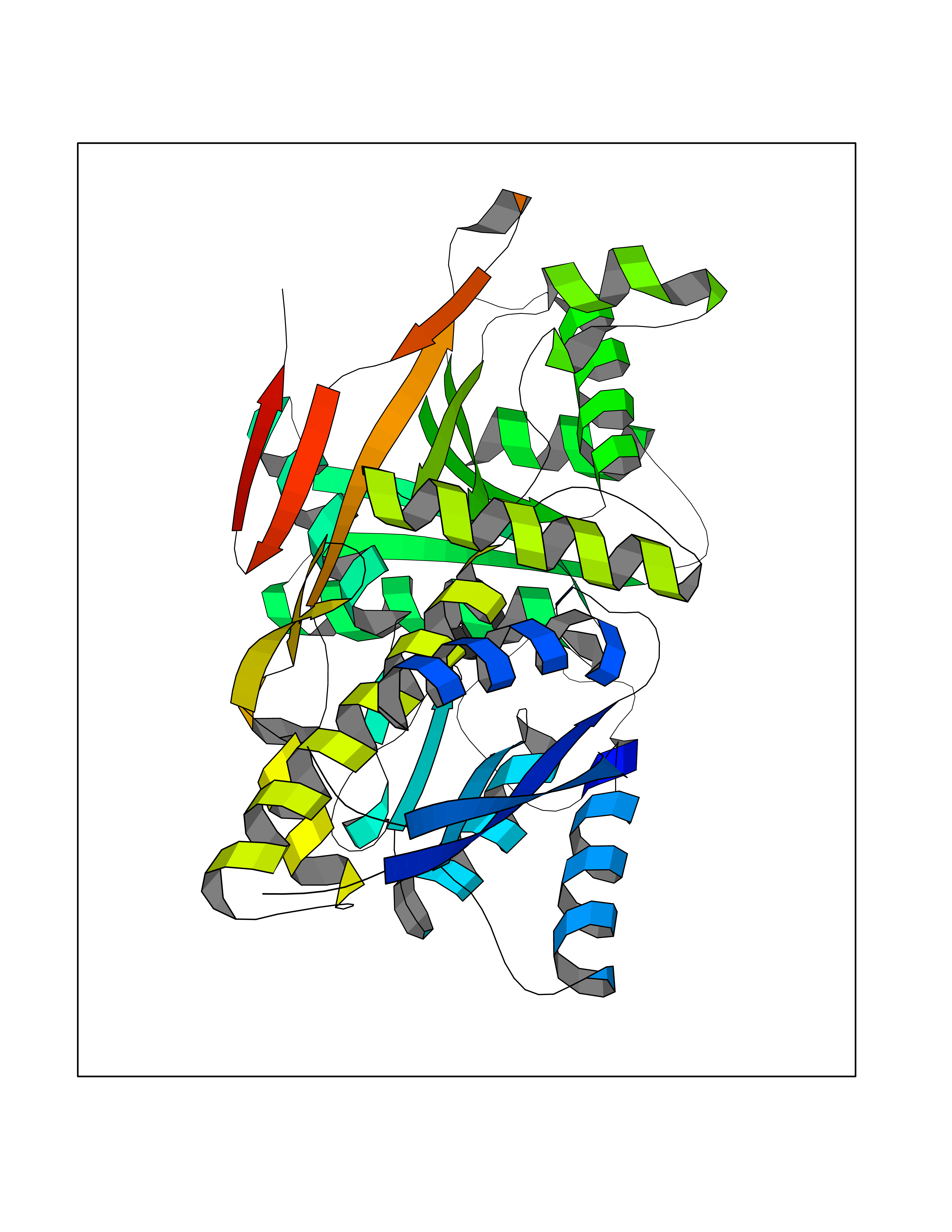 Molscript Map 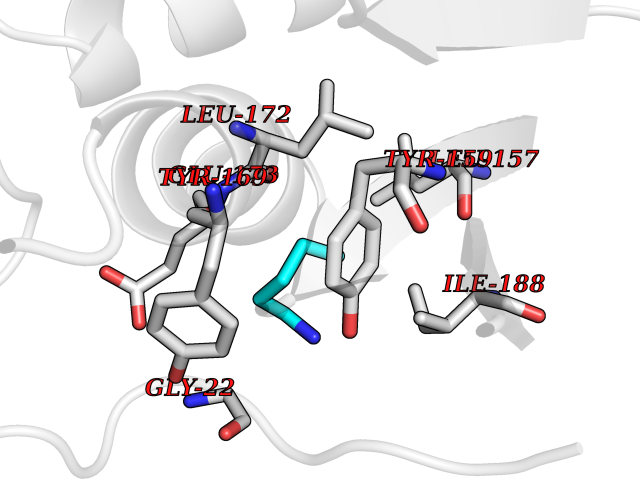 Pymol Map 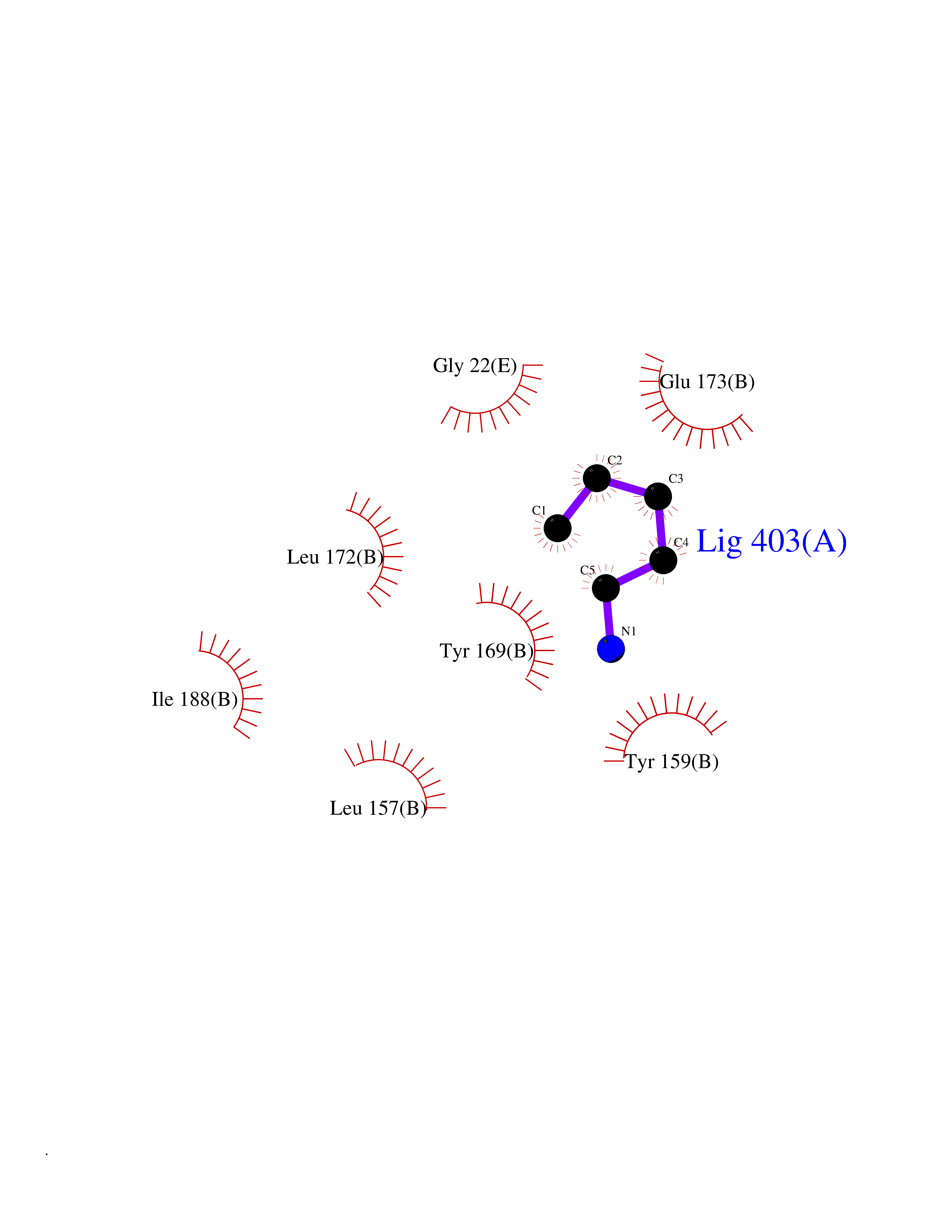 Ligplot Map 3D Binding mode Sequence GCFGRKMDRISSSSGLGCKVLALPPQKIEVLVLLPQDDSYLFSLTRVRPAIEYALRSVEGLLPPGTRFQVAYEDSDCGNRALFSLVDRVAAARGAKPDLILGPVCEYAAAPVARLASHWDLPMLSAGALAAGFQHKDSEYSHLTRVAPAYAKMGEMMLALFRHHHWSRAALVYSDDKLERNCYFTLEGVHEVFQEEGLHTSIYSFDETKDLDLEDIVRNIQASERVVIMCASSDTIRSIMLVAHRHGMTSGDYAFFNIELFNSSSYGDGSWKRGDKHDFEAKQAYSSLQTVTLLRTVKPEFEKFSMEVKSSVEKQGLNMEDYVNMFVEGFHDAILLYVLALHEVLRAGYSKKDGGKIIQQTWNRTFEGIAGQVSIDANGDRYGDFSVIAMTDVEAGTQEVIGDYFGKEGRFEMRP Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | D-amino acid oxidase (DAO) | 3ZNN | 4.75 | |
Target general information Gen name DAO Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Daminoacid oxidase; DAMOX; DAAO Protein family DAMOX/DASOX family Biochemical class CH-NH(2) donor oxidoreductase Function Regulates the level of the neuromodulator D-serine in the brain. Has high activity towards D-DOPA and contributes to dopamine synthesis. Could act as a detoxifying agent which removes D-amino acids accumulated during aging. Acts on a variety of D-amino acids with a preference for those having small hydrophobic side chains followed by those bearing polar, aromatic, and basic groups. Does not act on acidic amino acids. Related diseases Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:12364586}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Amyotrophic lateral sclerosis (ALS) [MIM:105400]: A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. {ECO:0000269|PubMed:20368421, ECO:0000269|PubMed:20538972, ECO:0000269|PubMed:22203986, ECO:0000269|PubMed:23219954, ECO:0000269|PubMed:24138986, ECO:0000269|PubMed:25701391, ECO:0000269|PubMed:37558109, ECO:0000269|PubMed:38035964, ECO:0000269|PubMed:38134563}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07979; DB02838; DB04166; DB03793; DB03225; DB03147; DB03531; DB02988 Interacts with Q9P2K6; O43741 EC number EC 1.4.3.3 Uniprot keywords 3D-structure; Amyotrophic lateral sclerosis; Cell projection; Cytoplasm; Disease variant; FAD; Flavoprotein; Neurodegeneration; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; S-nitrosylation; Schizophrenia; Secreted; Synapse Protein physicochemical properties Chain ID A,B Molecular weight (Da) 38654.6 Length 340 Aromaticity 0.11 Instability index 29.13 Isoelectric point 6.18 Charge (pH=7) -4.45 2D Binding mode Binding energy (Kcal/mol) -6.49 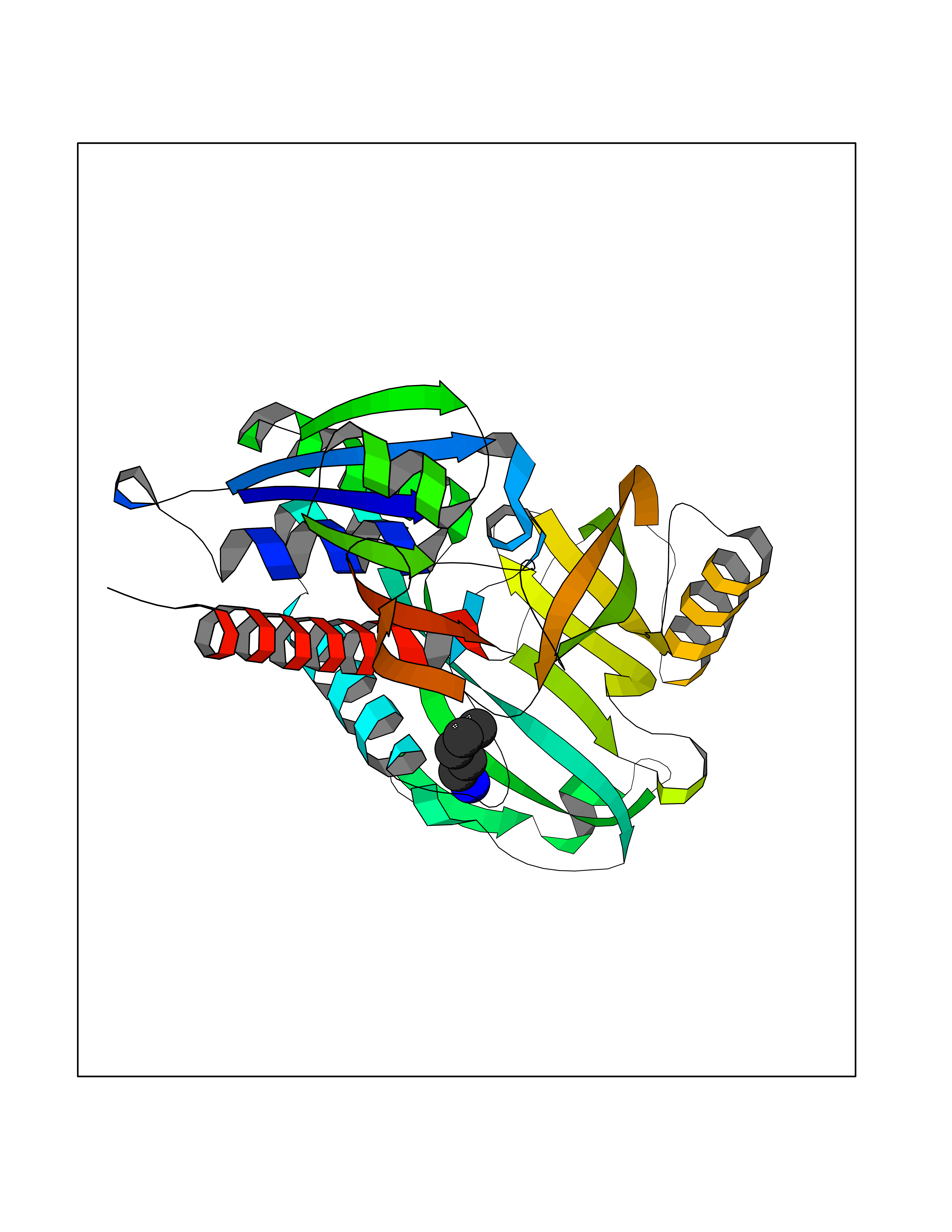 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MRVVVIGAGVIGLSTALCIHERYHSVLQPLDIKVYADRFTPLTTTDVAAGLWQPYLSDPNNPQEADWSQQTFDYLLSHVHSPNAENLGLFLISGYNLFHEAIPDPSWKDTVLGFRKLTPRELDMFPDYGYGWFHTSLILEGKNYLQWLTERLTERGVKFFQRKVESFEEVAREGADVIVNCTGVWAGALQRDPLLQPGRGQIMKVDAPWMKHFILTHDPERGIYNSPYIIPGTQTVTLGGIFQLGNWSELNNIQDHNTIWEGCCRLEPTLKNARIIGERTGFRPVRPQIRLEREQLRTGPSNTEVIHNYGHGGYGLTIHWGCALEAAKLFGRILEEKKLS Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | 2-iminobutanoate/2-iminopropanoate deaminase | 1ONI | 4.75 | |
Target general information Gen name RIDA Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms HRSP12 Protein family RutC family Biochemical class Translation Function Deaminase activity.Endoribonuclease activity, producing 3'-phosphomonoesters.Long-chain fatty acid binding.Platinum binding.Protein homodimerization activity.RNA binding.Transition metal ion binding.Xenon atom binding. Related diseases Congenital bile acid synthesis defect 2 (CBAS2) [MIM:235555]: A condition characterized by jaundice, intrahepatic cholestasis and hepatic failure. Patients with this liver disease show absence or low levels of chenodeoxycholic acid and cholic acid in plasma and urine. {ECO:0000269|PubMed:12970144, ECO:0000269|PubMed:15030995, ECO:0000269|PubMed:19175828, ECO:0000269|PubMed:20522910}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q8N9N5-2 EC number 3.5.99.10 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Hydrolase; Lipid metabolism; Mitochondrion; Nucleus; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; RNA-binding Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H,I Molecular weight (Da) 42624.3 Length 404 Aromaticity 0.07 Instability index 36.76 Isoelectric point 8.99 Charge (pH=7) 5.46 2D Binding mode Binding energy (Kcal/mol) -6.48  Molscript Map  Pymol Map 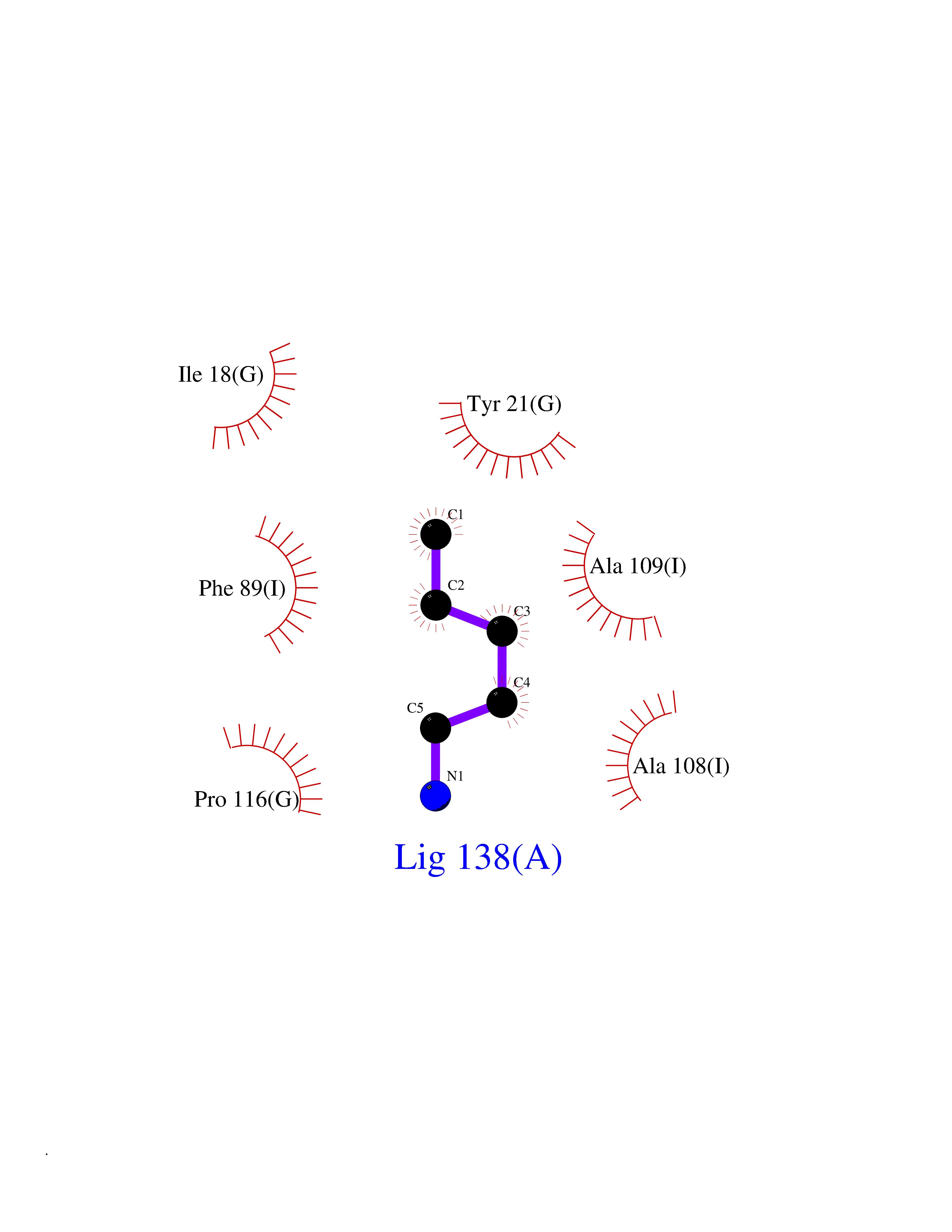 Ligplot Map 3D Binding mode Sequence SSLIRRVISTAKAPGAIGPYSQAVLVDRTIYISGQIGMDPSSGQLVSGGVAEEAKQALKNMGEILKAAGCDFTNVVKTTVLLADINDFNTVNEIYKQYFKSNFPARAAYQVAALPKGSRIEIEAVAIQGPLTTASSSLIRRVISTAKAPGAIGPYSQAVLVDRTIYISGQIGMDPSSGQLVSGGVAEEAKQALKNMGEILKAAGCDFTNVVKTTVLLADINDFNTVNEIYKQYFKSNFPARAAYQVAALPKGSRIEIEAVAIQGPLTTASSSLIRRVISTAKAPGAIGPYSQAVLVDRTIYISGQIGMDPSSGQLVSGGVAEEAKQALKNMGEILKAAGCDFTNVVKTTVLLADINDFNTVNEIYKQYFKSNFPARAAYQVAALPKGSRIEIEAVAIQGPLTTA Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | S-methyl-5'-thioadenosine phosphorylase (MTAP) | 1CB0 | 4.75 | |
Target general information Gen name MTAP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Methylthioadenosine phosphorylase; MTAPase; MTA phosphorylase; MSAP; 5'-methylthioadenosine phosphorylase Protein family PNP/MTAP phosphorylase family, MTAP subfamily Biochemical class Glycosyltransferases Function Involved in the breakdown of MTA, a major by-product of polyamine biosynthesis. Responsible for the first step in the methionine salvage pathway after MTA has been generated from S-adenosylmethionine. Has broad substrate specificity with 6-aminopurine nucleosides as preferred substrates. Catalyzes the reversible phosphorylation of S-methyl-5'-thioadenosine (MTA) to adenine and 5-methylthioribose-1-phosphate. Related diseases Diaphyseal medullary stenosis with malignant fibrous histiocytoma (DMSMFH) [MIM:112250]: An autosomal dominant bone dysplasia characterized by pathologic fractures due to abnormal cortical growth and diaphyseal medullary stenosis. The fractures heal poorly, and there is progressive bowing of the lower extremities. Some patients show a limb-girdle myopathy, with muscle weakness and atrophy. Approximately 35% of affected individuals develop an aggressive form of bone sarcoma consistent with malignant fibrous histiocytoma or osteosarcoma. {ECO:0000269|PubMed:22464254}. The disease is caused by variants affecting the gene represented in this entry. DMSMFH causing mutations found in MTAP exon 9 result in exon skipping and dysregulated alternative splicing of all MTAP isoforms (PubMed:22464254). {ECO:0000269|PubMed:22464254}.; DISEASE: Loss of MTAP activity may play a role in human cancer. MTAP loss has been reported in a number of cancers, including osteosarcoma, malignant melanoma and gastric cancer. Drugs (DrugBank ID) DB02158; DB02933; DB02282; DB00173; DB02281 Interacts with Q9H3R5; Q9P0I2 EC number EC 2.4.2.28 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Glycosyltransferase; Nucleus; Proteomics identification; Purine salvage; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 29538.9 Length 268 Aromaticity 0.06 Instability index 40.97 Isoelectric point 7.18 Charge (pH=7) 0.36 2D Binding mode Binding energy (Kcal/mol) -6.48 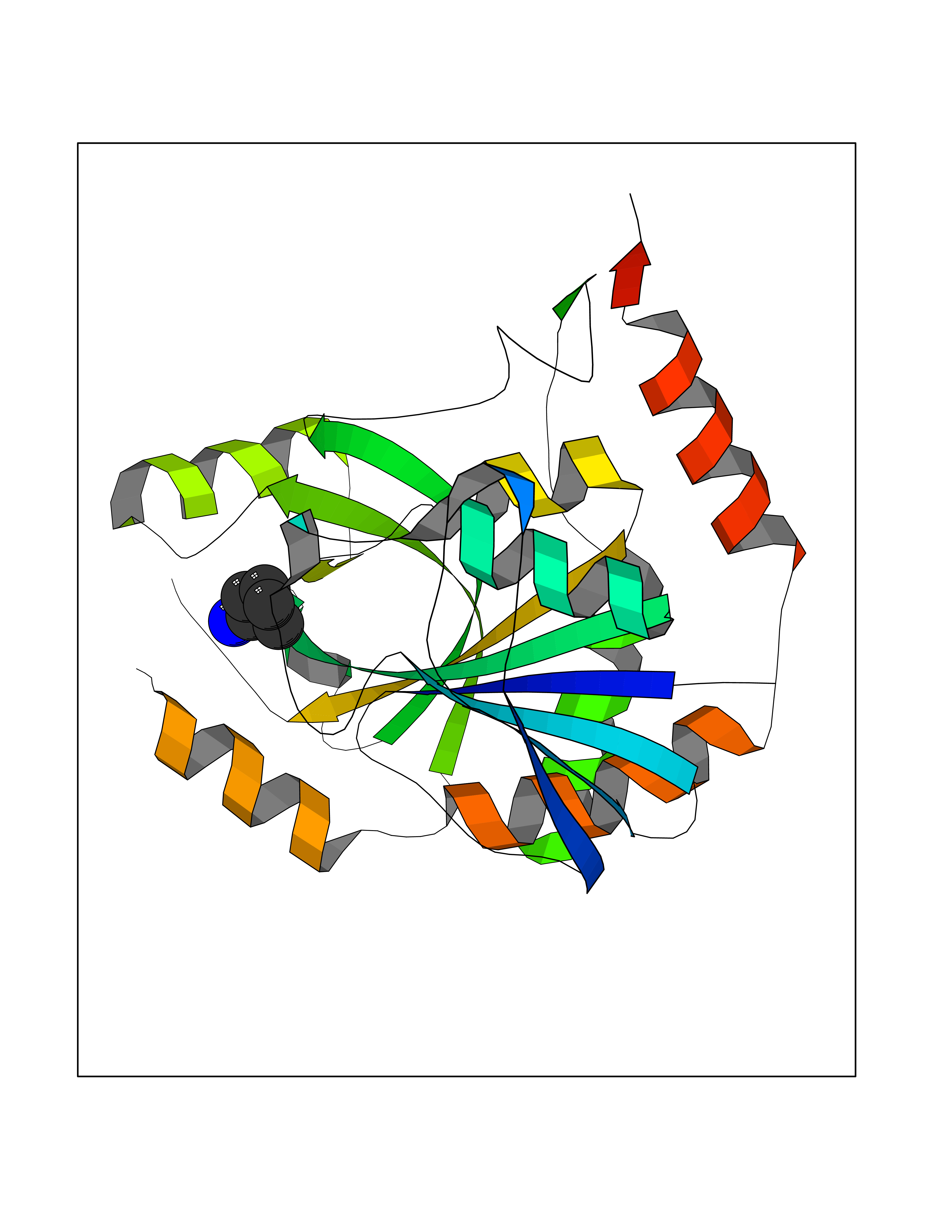 Molscript Map 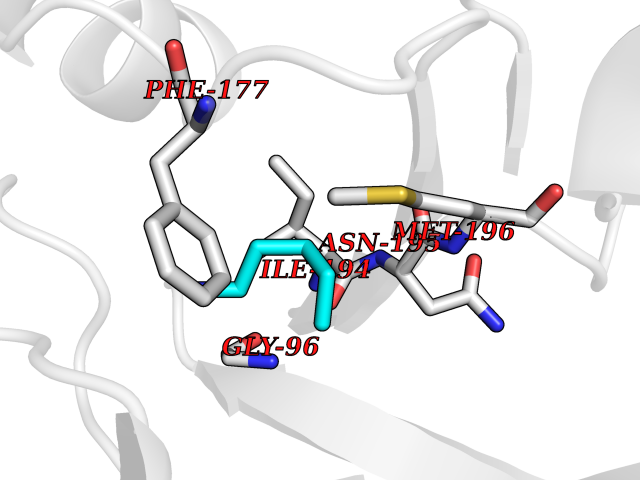 Pymol Map 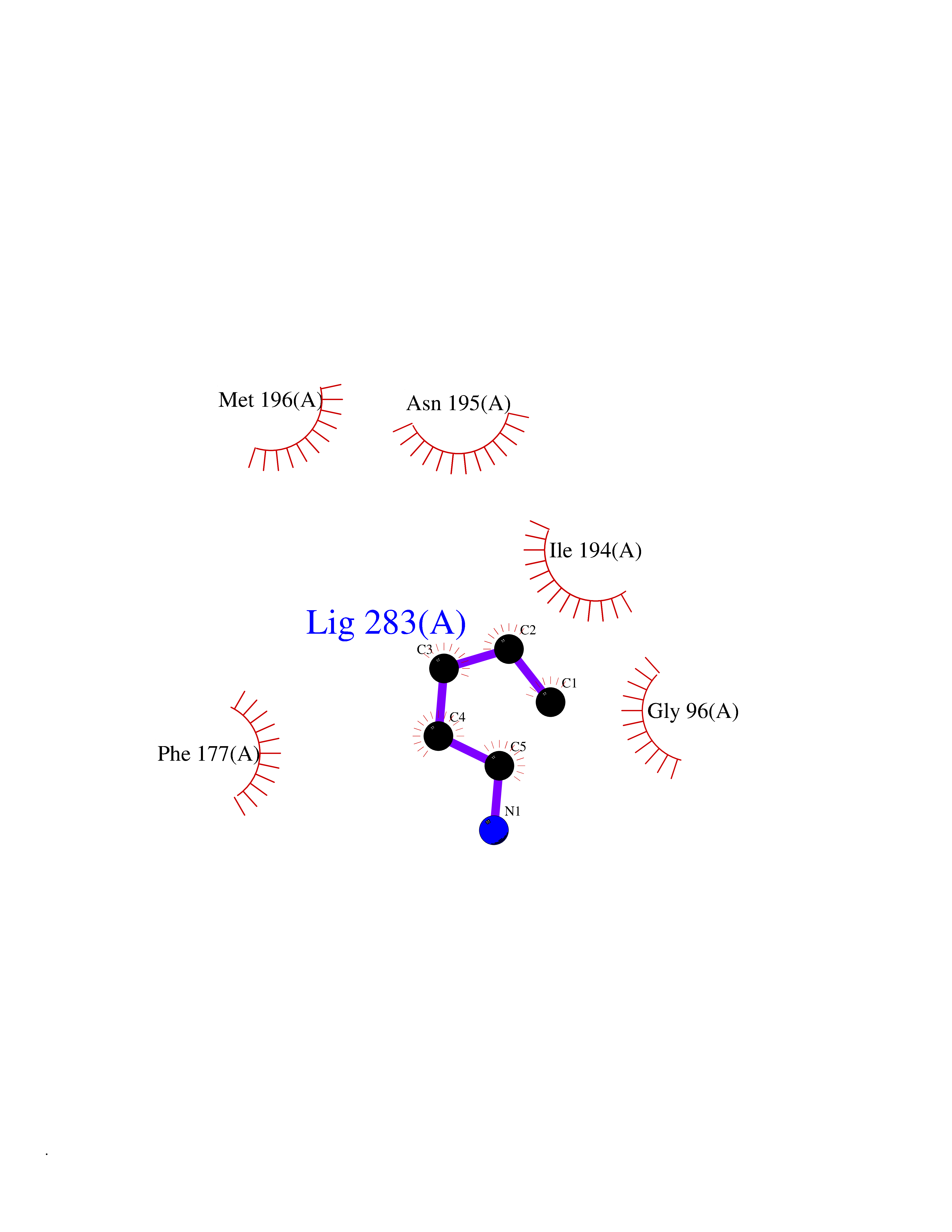 Ligplot Map 3D Binding mode Sequence AVKIGIIGGTGLDDPEILEGRTEKYVDTPFGKPSDALILGKIKNVDCVLLARHGRQHTIMPSKVNYQANIWALKEEGCTHVIVTTACGSLREEIQPGDIVIIDQFIDRTTMRPQSFYDGSHSCARGVCHIPMAEPFCPKTREVLIETAKKLGLRCHSKGTMVTIEGPRFSSRAESFMFRTWGADVINMTTVPEVVLAKEAGICYASIAMATDYDCWAVSVDRVLKTLKENANKAKSLLLTTIPQIGSTEWSETLHNLKNMAQFSVLLP Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Lysine-specific histone demethylase 1B (KDM1B) | 4HSU | 4.75 | |
Target general information Gen name KDM1B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine-specific histone demethylase 2; LSD2; Flavin-containing amine oxidase domain-containing protein 1; C6orf193; AOF1 Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Required for de novo DNA methylation of a subset of imprinted genes during oogenesis. Acts by oxidizing the substrate by FAD to generate the corresponding imine that is subsequently hydrolyzed. Demethylates both mono- and di-methylated 'Lys-4' of histone H3. Has no effect on tri-methylated 'Lys-4', mono-, di- or tri-methylated 'Lys-9', mono-, di- or tri-methylated 'Lys-27', mono-, di- or tri-methylated 'Lys-36' of histone H3, or on mono-, di- or tri-methylated 'Lys-20' of histone H4. Histone demethylase that demethylates 'Lys-4' of histone H3, a specific tag for epigenetic transcriptional activation, thereby acting as a corepressor. Related diseases Angioedema, hereditary, 1 (HAE1) [MIM:106100]: An autosomal dominant disorder characterized by episodic local swelling involving subcutaneous or submucous tissue of the upper respiratory and gastrointestinal tracts, face, extremities, and genitalia. Hereditary angioedema due to C1 esterase inhibitor deficiency is comprised of two clinically indistinguishable forms. In hereditary angioedema type 1, serum levels of C1 esterase inhibitor are decreased, while in type 2, the levels are normal or elevated, but the protein is non-functional. {ECO:0000269|PubMed:12773530, ECO:0000269|PubMed:1363816, ECO:0000269|PubMed:1451784, ECO:0000269|PubMed:14635117, ECO:0000269|PubMed:16409206, ECO:0000269|PubMed:2118657, ECO:0000269|PubMed:2296585, ECO:0000269|PubMed:22994404, ECO:0000269|PubMed:2365061, ECO:0000269|PubMed:24456027, ECO:0000269|PubMed:3178731, ECO:0000269|PubMed:7814636, ECO:0000269|PubMed:7883978, ECO:0000269|PubMed:8172583, ECO:0000269|PubMed:8529136, ECO:0000269|PubMed:8755917, ECO:0000269|Ref.41}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q96L03 EC number EC 1.-.-.- Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Developmental protein; FAD; Flavoprotein; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,C Molecular weight (Da) 85795.5 Length 763 Aromaticity 0.1 Instability index 37.87 Isoelectric point 8.41 Charge (pH=7) 9.16 2D Binding mode Binding energy (Kcal/mol) -6.48 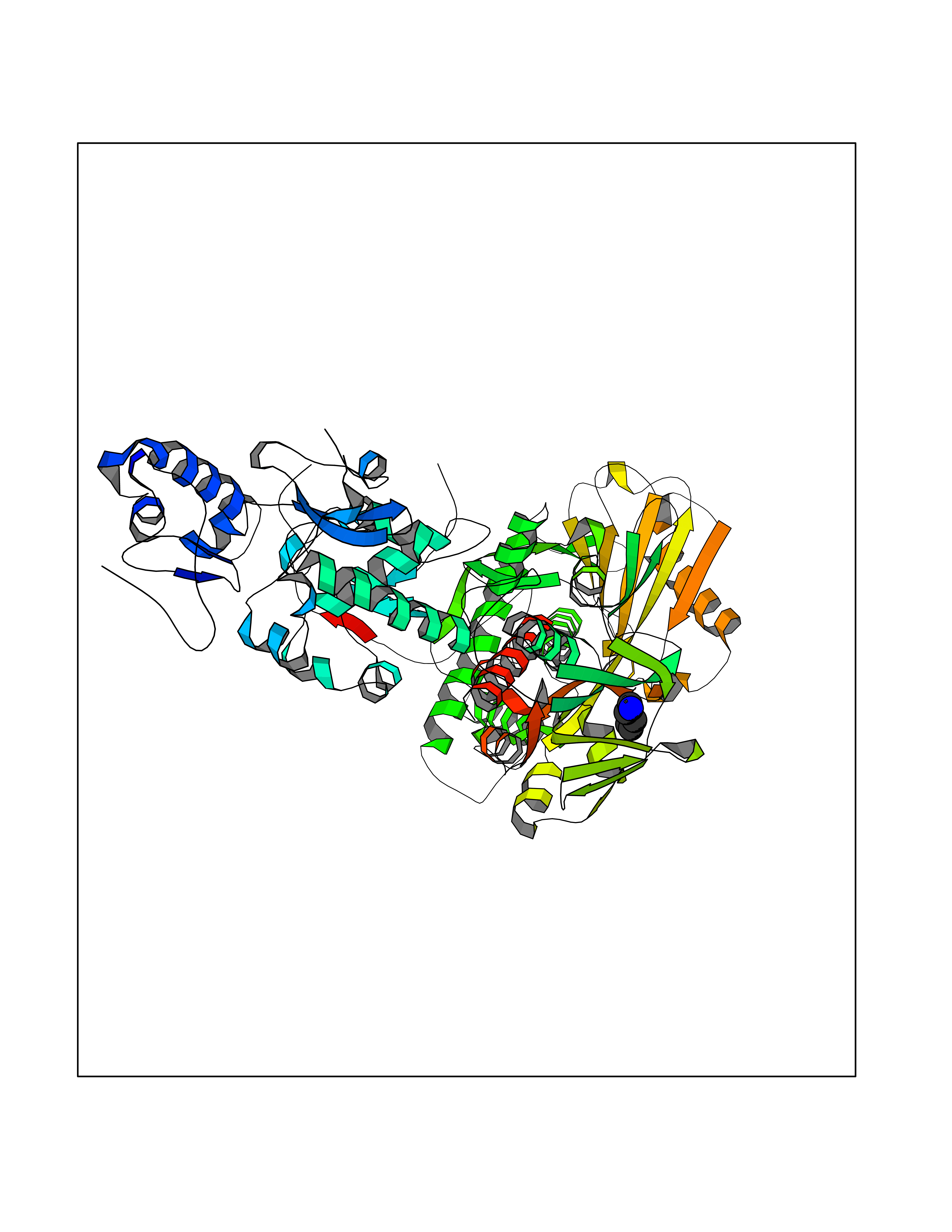 Molscript Map 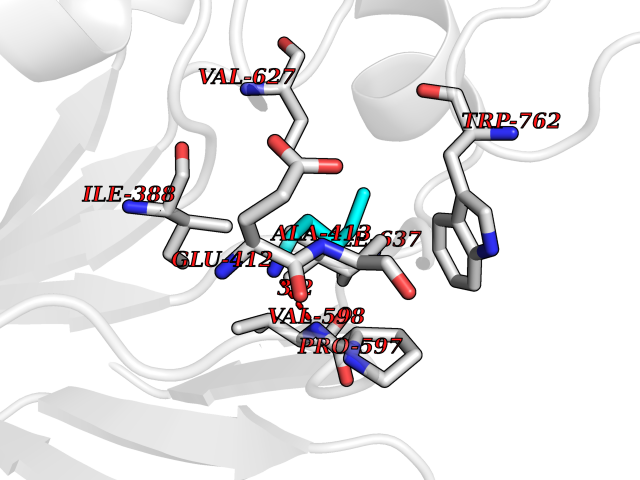 Pymol Map 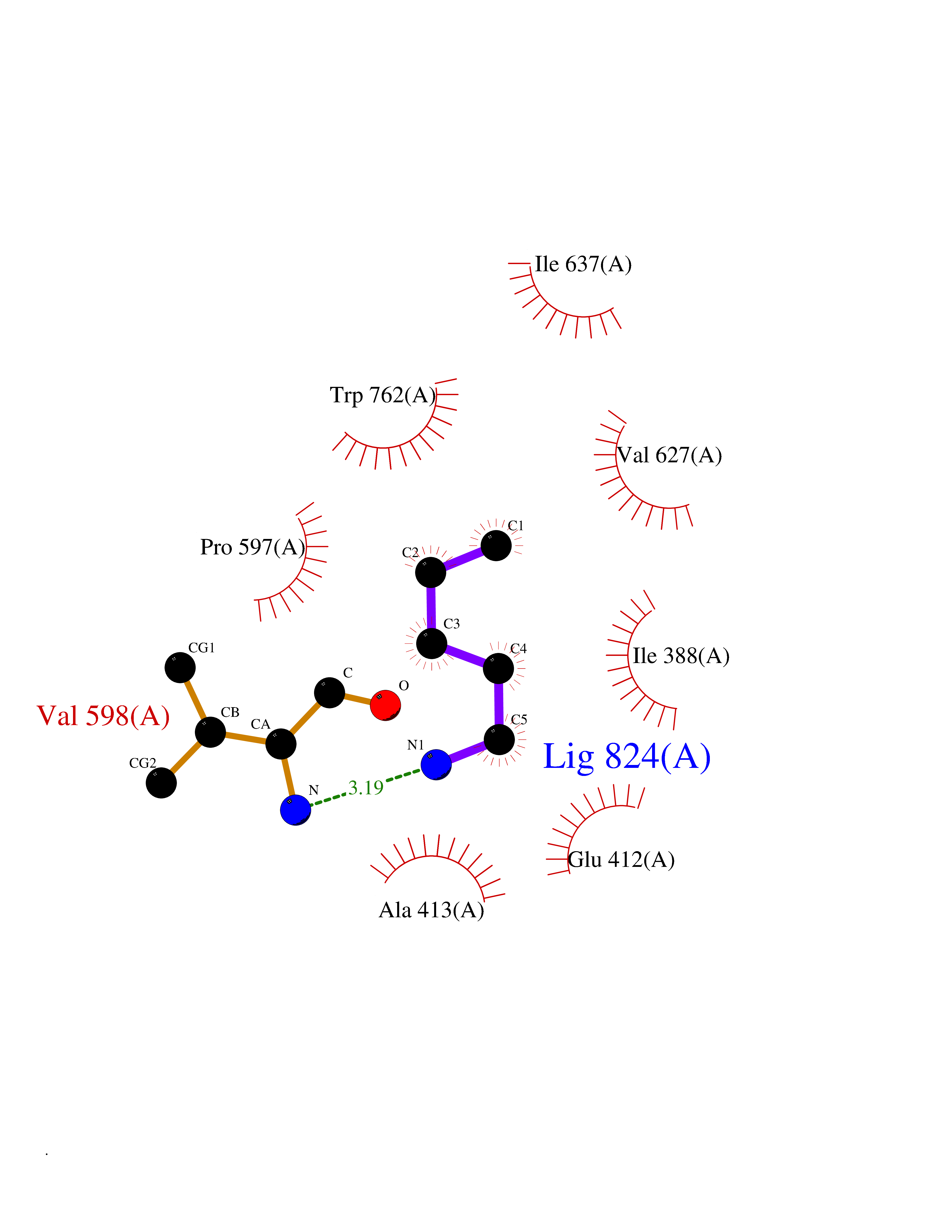 Ligplot Map 3D Binding mode Sequence GSRKCEKAGCTATCPVCFASASERCAKNGYTSRWYHLSCGEHFCNECFDHYYRSHKDGYDKYTTWKKIWTSNGKTEPSPKAFMADQQLPYWVQCTKPECRKWRQLTKEIQLTPQIAKTYRCGMKSDHCSLPEDLRVLEVSNHWWYSMLILPPLLKDSVAAPLLSAYYPDCVGMSPSCTGMNRYFQPFYQPNECGKALCVRPDVMELDELYEFPEYSRDPTMYLALRNLILALWYTNCKEALTPQKCIPHIIVRGLVRIRCVQEVERILYFMTRKGLINTGVLSVGADQYLLPKDYHNKSVIIIGAGPAGLAAARQLHNFGIKVTVLEAKDRIGGRVWDDKSFKGVTVGRGAQIVNGCINNPVALMCEQLGISMHKFGERCDLIQEGGRITDPTIDKRMDFHFNALLDVVSEWRKDKTQLQDVPLGEKIEEIYKAFIKESGIQFSELEGQVLQFHLSNLEYACGSNLHQVSARSWDHNEFFAQFAGDHTLLTPGYSVIIEKLAEGLDIQLKSPVQCIDYSGDEVQVTTTDGTGYSAQKVLVTVPLALLQKGAIQFNPPLSEKKMKAINSLGAGIIEKIALQFPYRFWDSKVQGADFFGHVPPSASKRGLFAVFYDMDPQKKHSVLMSVIAGEAVASVRTLDDKQVLQQCMATLRELFKEQEVPDPTKYFVTRWSTDPWIQMAYSFVKTGGSGEAYDIIAEDIQGTVFFAGEATNRHFPQTVTGAYLSGVREASKIAAFARTMQTARKSTGGKAPRKQLATKAAR Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Tryptophan 2,3-dioxygenase (TDO) | 6PYZ | 4.75 | |
Target general information Gen name TDO2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tryptophanase; Tryptophan pyrrolase; Tryptophan oxygenase; Tryptamin 2,3-dioxygenase; TRPO; TO Protein family Tryptophan 2,3-dioxygenase family Biochemical class Oxygenase Function Catalyzes the oxidative cleavage of the indole moiety. Heme-dependent dioxygenase that catalyzes the oxidative cleavage of the L-tryptophan (L-Trp) pyrrole ring and converts L-tryptophan to N-formyl-L-kynurenine. Related diseases Hypertryptophanemia (HYPTRP) [MIM:600627]: An autosomal recessive condition characterized by persistent hypertryptophanemia and hyperserotoninemia. {ECO:0000269|PubMed:28285122}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00779; DB00500; DB00150 Interacts with O43865; O95671; P27797; P12830; P36957; O60762; P06730; Q8TBB1; Q9H8S9; Q70IA8; Q8TDX7; Q9NPG2; Q9HAN9; P20393; Q9NRD5; Q8IYS1; O00560; Q9H190; P48775; Q68DK2-5 EC number EC 1.13.11.11 Uniprot keywords 3D-structure; Dioxygenase; Disease variant; Heme; Iron; Metal-binding; Oxidoreductase; Proteomics identification; Reference proteome; Tryptophan catabolism Protein physicochemical properties Chain ID A,B Molecular weight (Da) 83454.8 Length 701 Aromaticity 0.11 Instability index 43.93 Isoelectric point 6.93 Charge (pH=7) -0.48 2D Binding mode Binding energy (Kcal/mol) -6.48 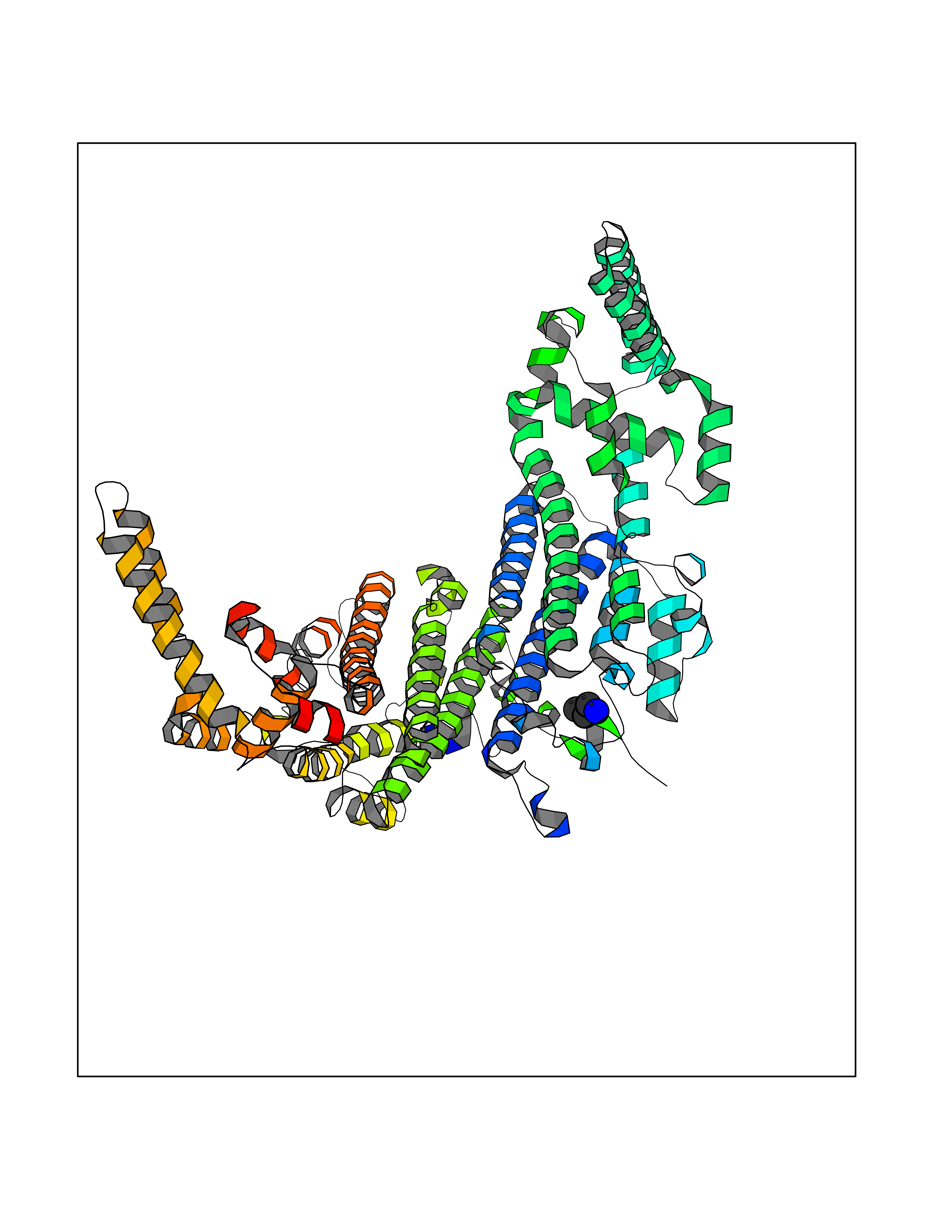 Molscript Map 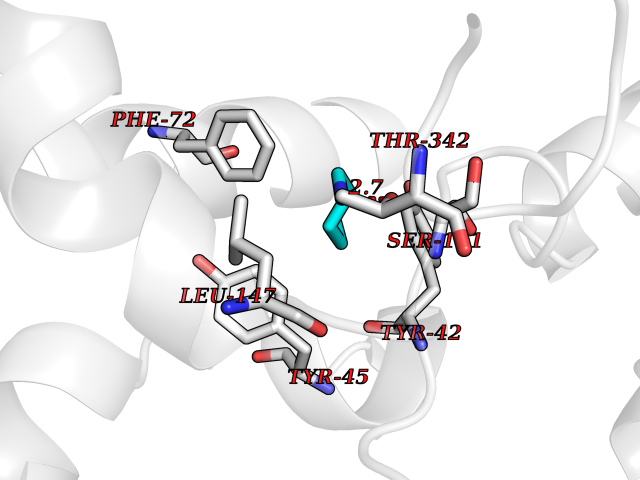 Pymol Map 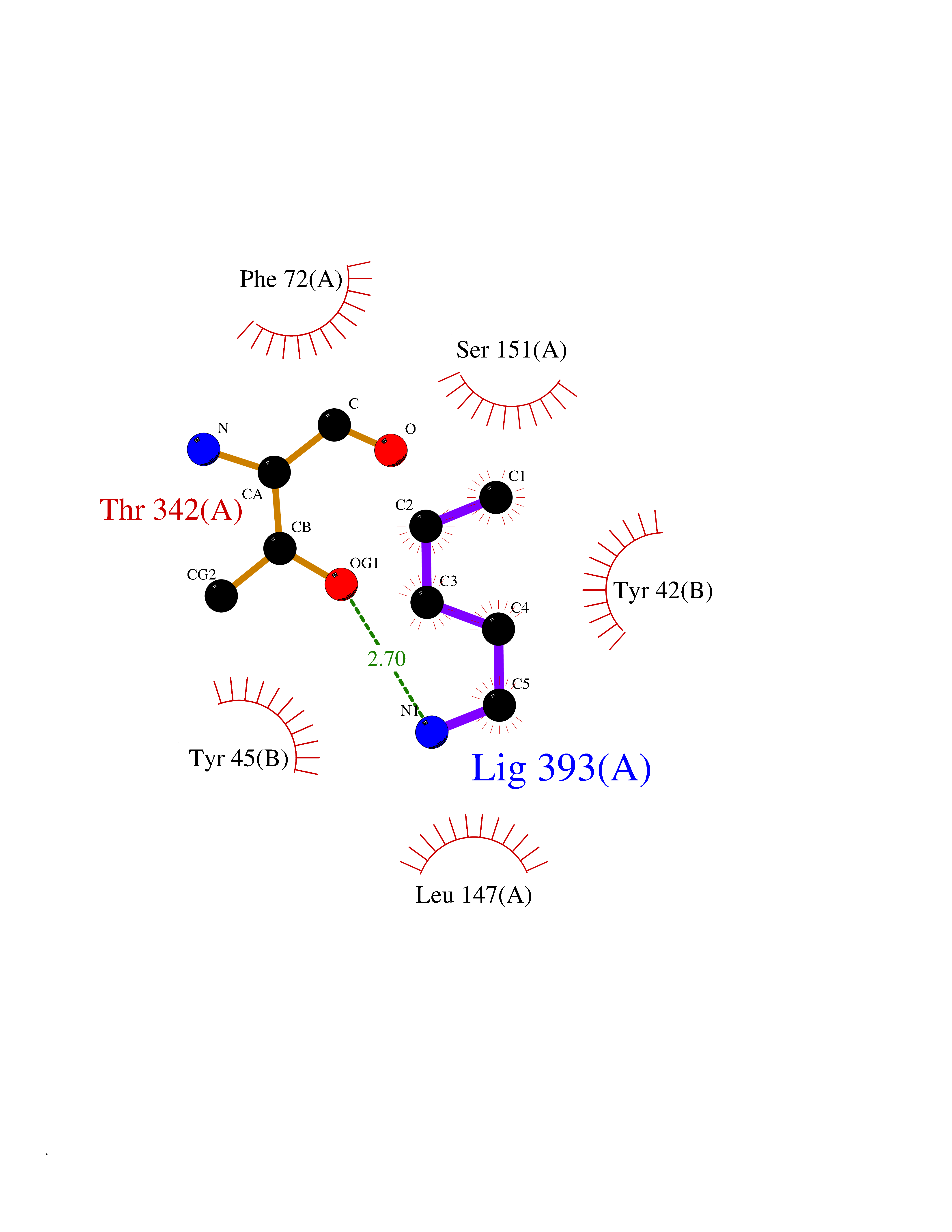 Ligplot Map 3D Binding mode Sequence GLIYGNYLHLEKVLNAQELQSETKGNKIHDEHLFIITHQAYELWFKQILWELDSVREIFQNGHVRDERNMLKVVSRMHRVSVILKLLVQQFSILETMTALDFNDFREYLSPASGFQSLQFRLLENKIGVLQNMRVPYNRRHYRDNFKGEENELLLKSEQEKTLLELVEAWLERTPGLEPHGFNFWGKLEKNITRGLEEEFIRIQAKEESEEKEEQVAEFQKQKEVLLSLFDEKRHEHLLSKGERRLSYRALQGALMIYFYREEPRFQVPFQLLTSLMDIDSLMTKWRYNHVCMVHRMLGSKAGTGGSSGYHYLRSTVSDRYKVFVDLFNLSTYLIPRHWIPKMNPTIHKFLEHGGLIYGNYLHLEKVLNAQELQSETKGNKIHDEHLFIITHQAYELWFKQILWELDSVREIFQNGHVRDERNMLKVVSRMHRVSVILKLLVQQFSILETMTALDFNDFREYLSPASGFQSLQFRLLENKIGVLQNMRVPYYRDNFKGEENELLLKSEQEKTLLELVEAWLERTPGLEPHGFNFWGKLEKNITRGLEEEFIRIQAKEESEEKEEQVAEFQKQKEVLLSLFDEKRHEHLLSKGERRLSYRALQGALMIYFYREEPRFQVPFQLLTSLMDIDSLMTKWRYNHVCMVHRMLGSKAGTGGSSGYHYLRSTVSDRYKVFVDLFNLSTYLIPRHWIPKMNPTIHKFL Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Insulin-like growth factor I receptor (IGF1R) | 3I81 | 4.75 | |
Target general information Gen name IGF1R Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Type 1 insulin-like growth factor receptor; Insulin-like growth factor 1 receptor; IGF-IR; IGF-I receptor; IGF-1R; IGF-1 receptor; CD221 antigen; CD221 Protein family Protein kinase superfamily, Tyr protein kinase family, Insulin receptor subfamily Biochemical class Kinase Function Binds IGF1 with high affinity and IGF2 and insulin (INS) with a lower affinity. The activated IGF1R is involved in cell growth and survival control. IGF1R is crucial for tumor transformation and survival of malignant cell. Ligand binding activates the receptor kinase, leading to receptor autophosphorylation, and tyrosines phosphorylation of multiple substrates, that function as signaling adapter proteins including, the insulin-receptor substrates (IRS1/2), Shc and 14-3-3 proteins. Phosphorylation of IRSs proteins lead to the activation of two main signaling pathways: the PI3K-AKT/PKB pathway and the Ras-MAPK pathway. The result of activating the MAPK pathway is increased cellular proliferation, whereas activating the PI3K pathway inhibits apoptosis and stimulates protein synthesis. Phosphorylated IRS1 can activate the 85 kDa regulatory subunit of PI3K (PIK3R1), leading to activation of several downstream substrates, including protein AKT/PKB. AKT phosphorylation, in turn, enhances protein synthesis through mTOR activation and triggers the antiapoptotic effects of IGFIR through phosphorylation and inactivation of BAD. In parallel to PI3K-driven signaling, recruitment of Grb2/SOS by phosphorylated IRS1 or Shc leads to recruitment of Ras and activation of the ras-MAPK pathway. In addition to these two main signaling pathways IGF1R signals also through the Janus kinase/signal transducer and activator of transcription pathway (JAK/STAT). Phosphorylation of JAK proteins can lead to phosphorylation/activation of signal transducers and activators of transcription (STAT) proteins. In particular activation of STAT3, may be essential for the transforming activity of IGF1R. The JAK/STAT pathway activates gene transcription and may be responsible for the transforming activity. JNK kinases can also be activated by the IGF1R. IGF1 exerts inhibiting activities on JNK activation via phosphorylation and inhibition of MAP3K5/ASK1, which is able to directly associate with the IGF1R. Receptor tyrosine kinase which mediates actions of insulin-like growth factor 1 (IGF1). Related diseases Insulin-like growth factor 1 resistance (IGF1RES) [MIM:270450]: A disorder characterized by intrauterine growth retardation, poor postnatal growth and increased plasma IGF1 levels. {ECO:0000269|PubMed:14657428, ECO:0000269|PubMed:15928254, ECO:0000269|PubMed:25040157}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07156; DB07474; DB05023; DB15399; DB12267; DB12250; DB11840; DB11564; DB01306; DB09456; DB09564; DB01307; DB00047; DB01309; DB00030; DB00046; DB11567; DB00071; DB11568; DB16637; DB06075; DB01277; DB14751; DB08804; DB04395; DB05897; DB09098; DB06343; DB05184 Interacts with Q9NZN5; PRO_0000004724 [P49913]; P41240; P35222; P05019; P08069; P06213; P05556; Q9Y2W7; Q9UJU2; P27986; O00459; P35813; P18031; Q06124; P29350; P29353-2; Q01995; P62258; Q64010; P01317 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; ATP-binding; Cell membrane; Cleavage on pair of basic residues; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Host-virus interaction; Isopeptide bond; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 33985.7 Length 296 Aromaticity 0.1 Instability index 46.37 Isoelectric point 4.84 Charge (pH=7) -13.94 2D Binding mode Binding energy (Kcal/mol) -6.48 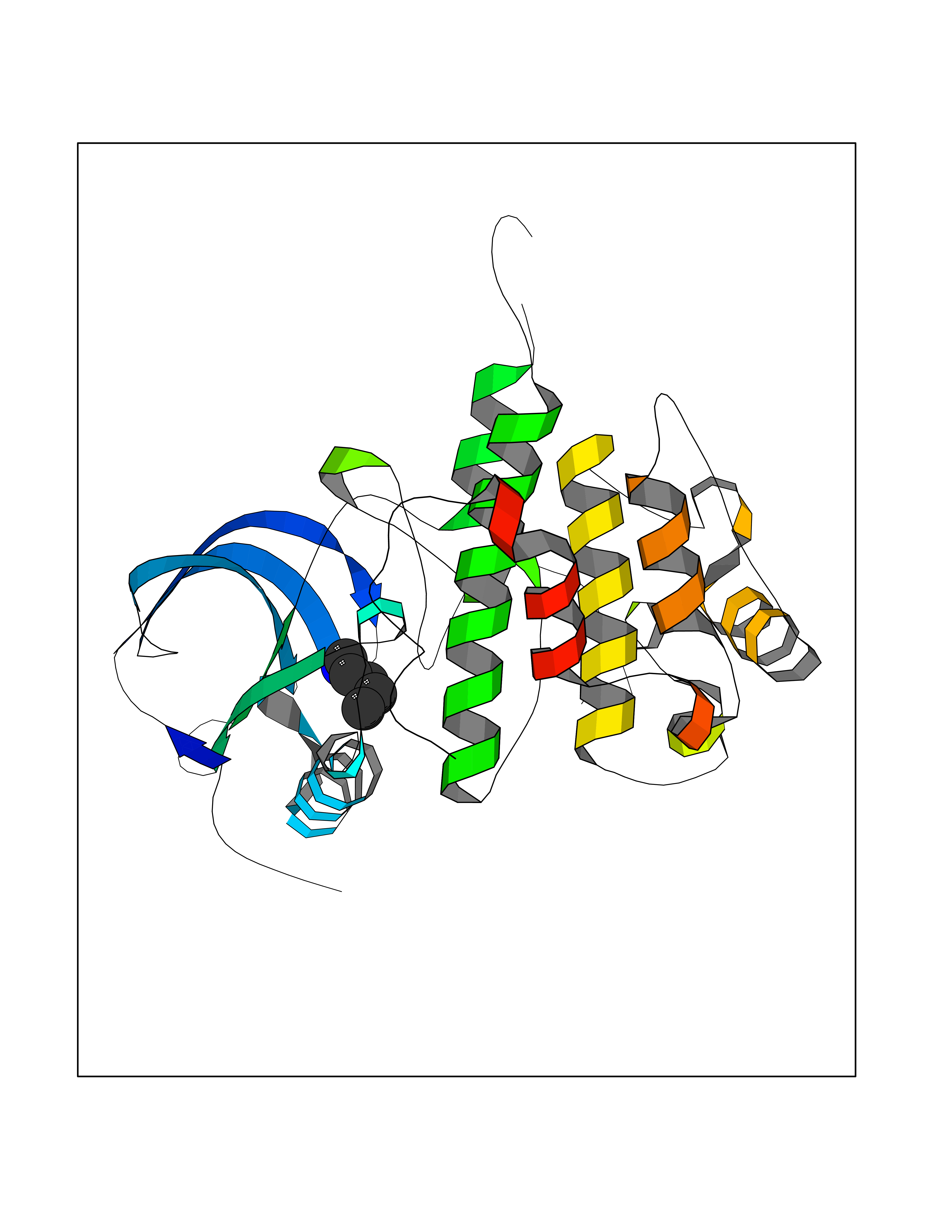 Molscript Map 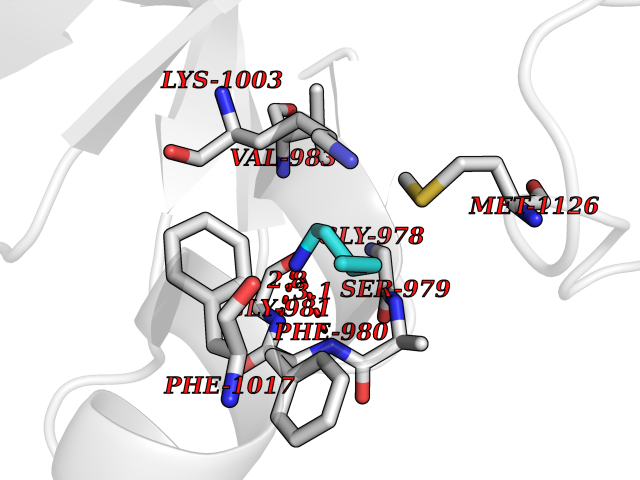 Pymol Map 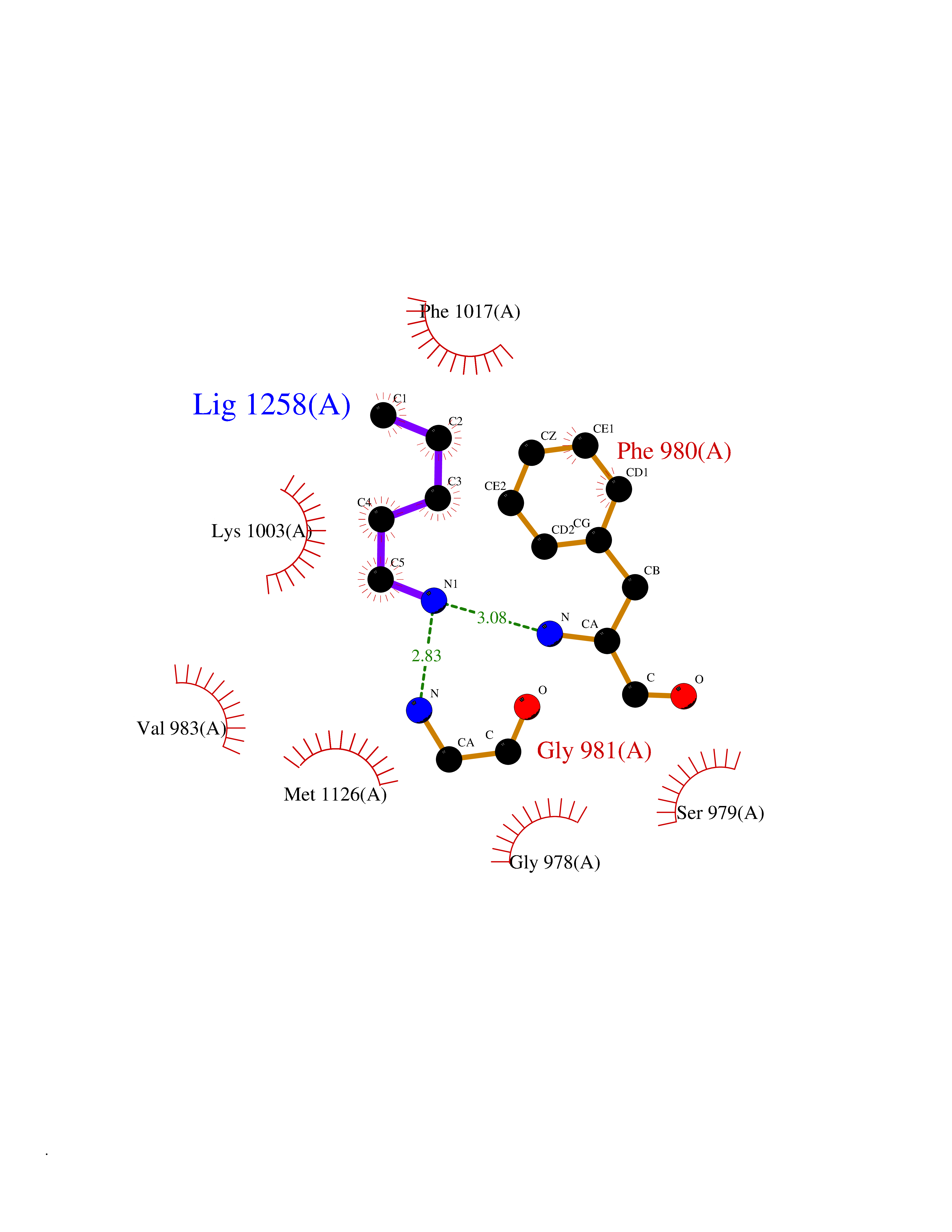 Ligplot Map 3D Binding mode Sequence DVYVPDEWEVAREKITMSRELGQGSFGMVYEGVAKGVVKDEPETRVAIKTVNEAASMRERIEFLNEASVMKEFNCHHVVRLLGVVSQGQPTLVIMELMTRGDLKSYLRSLRPEENNPVLAPPSLSKMIQMAGEIADGMAYLNANKFVHRDLAARNCMVAEDFTVKIGDFGMTRDIYETDYYRLLPVRWMSPESLKDGVFTTYSDVWSFGVVLWEIATLAEQPYQGLSNEQVLRFVMEGGLLDKPDNCPDMLFELMRMCWQYNPKMRPSFLEIISSIKEEMEPGFREVSFYYSEENK Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Beta-galactosidase (GLB1) | 3THD | 4.75 | |
Target general information Gen name GLB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lactase; Elastin receptor 1; ELNR1; Acid beta-galactosidase Protein family Glycosyl hydrolase 35 family Biochemical class NA Function Isoform 1: Cleaves beta-linked terminal galactosyl residues from gangliosides, glycoproteins, and glycosaminoglycans. Related diseases GM1-gangliosidosis 1 (GM1G1) [MIM:230500]: An autosomal recessive lysosomal storage disease marked by the accumulation of GM1 gangliosides, glycoproteins and keratan sulfate primarily in neurons of the central nervous system. GM1-gangliosidosis type 1 is characterized by onset within the first three months of life, central nervous system degeneration, coarse facial features, hepatosplenomegaly, skeletal dysmorphology reminiscent of Hurler syndrome, and rapidly progressive psychomotor deterioration. Urinary oligosaccharide levels are high. It leads to death usually between the first and second year of life. {ECO:0000269|PubMed:10338095, ECO:0000269|PubMed:10737981, ECO:0000269|PubMed:10839995, ECO:0000269|PubMed:1487238, ECO:0000269|PubMed:15365997, ECO:0000269|PubMed:15714521, ECO:0000269|PubMed:15791924, ECO:0000269|PubMed:16538002, ECO:0000269|PubMed:16941474, ECO:0000269|PubMed:17309651, ECO:0000269|PubMed:17664528, ECO:0000269|PubMed:1907800, ECO:0000269|PubMed:1909089, ECO:0000269|PubMed:1928092, ECO:0000269|PubMed:19472408, ECO:0000269|PubMed:24737316, ECO:0000269|PubMed:25936995, ECO:0000269|PubMed:8213816, ECO:0000269|Ref.28, ECO:0000269|Ref.31}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: GM1-gangliosidosis 2 (GM1G2) [MIM:230600]: A gangliosidosis characterized by onset between ages 1 and 5. The main symptom is locomotor ataxia, ultimately leading to a state of decerebration with epileptic seizures. Patients do not display the skeletal changes associated with the infantile form, but they nonetheless excrete elevated amounts of beta-linked galactose-terminal oligosaccharides. Inheritance is autosomal recessive. {ECO:0000269|PubMed:10737981, ECO:0000269|PubMed:12644936, ECO:0000269|PubMed:15714521, ECO:0000269|PubMed:16941474, ECO:0000269|PubMed:17309651, ECO:0000269|PubMed:1907800, ECO:0000269|PubMed:1909089, ECO:0000269|PubMed:19472408, ECO:0000269|PubMed:24737316, ECO:0000269|PubMed:25936995, ECO:0000269|PubMed:8213816}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: GM1-gangliosidosis 3 (GM1G3) [MIM:230650]: A gangliosidosis with a variable phenotype. Patients show mild skeletal abnormalities, dysarthria, gait disturbance, dystonia and visual impairment. Visceromegaly is absent. Intellectual deficit can initially be mild or absent but progresses over time. Inheritance is autosomal recessive. {ECO:0000269|PubMed:11511921, ECO:0000269|PubMed:15714521, ECO:0000269|PubMed:15986423, ECO:0000269|PubMed:16941474, ECO:0000269|PubMed:17309651, ECO:0000269|PubMed:17664528, ECO:0000269|PubMed:1907800, ECO:0000269|PubMed:1909089, ECO:0000269|PubMed:19472408, ECO:0000269|PubMed:24737316, ECO:0000269|PubMed:25936995, ECO:0000269|PubMed:8198123, ECO:0000269|Ref.28, ECO:0000269|Ref.30}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mucopolysaccharidosis 4B (MPS4B) [MIM:253010]: A form of mucopolysaccharidosis type 4, an autosomal recessive lysosomal storage disease characterized by intracellular accumulation of keratan sulfate and chondroitin-6-sulfate. Key clinical features include short stature, skeletal dysplasia, dental anomalies, and corneal clouding. Intelligence is normal and there is no direct central nervous system involvement, although the skeletal changes may result in neurologic complications. There is variable severity, but patients with the severe phenotype usually do not survive past the second or third decade of life. {ECO:0000269|PubMed:11511921, ECO:0000269|PubMed:12393180, ECO:0000269|PubMed:16538002, ECO:0000269|PubMed:16941474, ECO:0000269|PubMed:17664528, ECO:0000269|PubMed:1928092, ECO:0000269|PubMed:19472408, ECO:0000269|PubMed:7586649}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04465 Interacts with Q8NBJ4; Q3KNW5; Q9BRI3; P30825 EC number EC 3.2.1.23 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Direct protein sequencing; Disease variant; Disulfide bond; Gangliosidosis; Glycoprotein; Glycosidase; Hydrolase; Lysosome; Mucopolysaccharidosis; Proteomics identification; Reference proteome; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 67980.6 Length 605 Aromaticity 0.13 Instability index 40.91 Isoelectric point 5.81 Charge (pH=7) -9.05 2D Binding mode Binding energy (Kcal/mol) -6.48 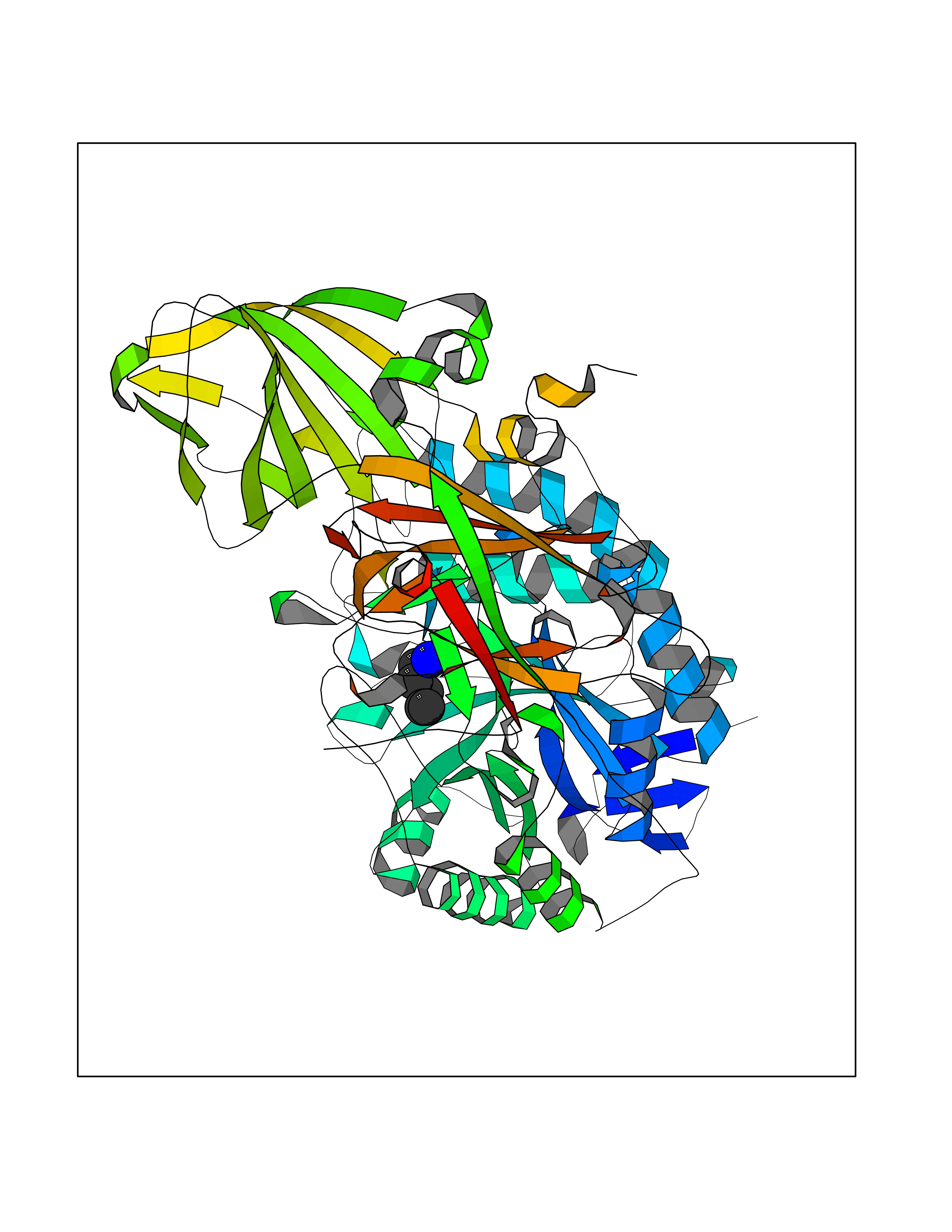 Molscript Map 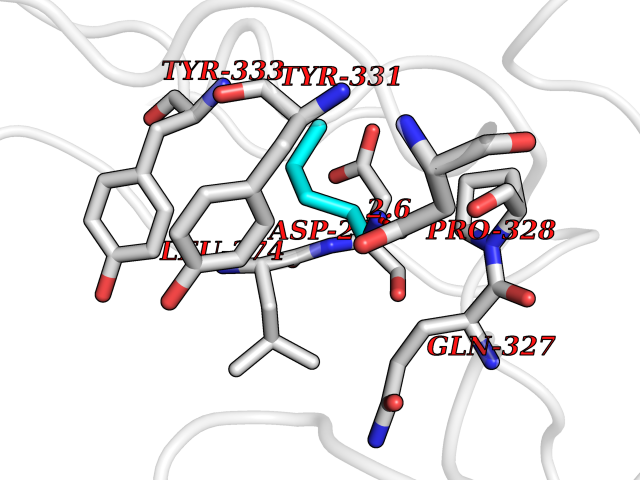 Pymol Map 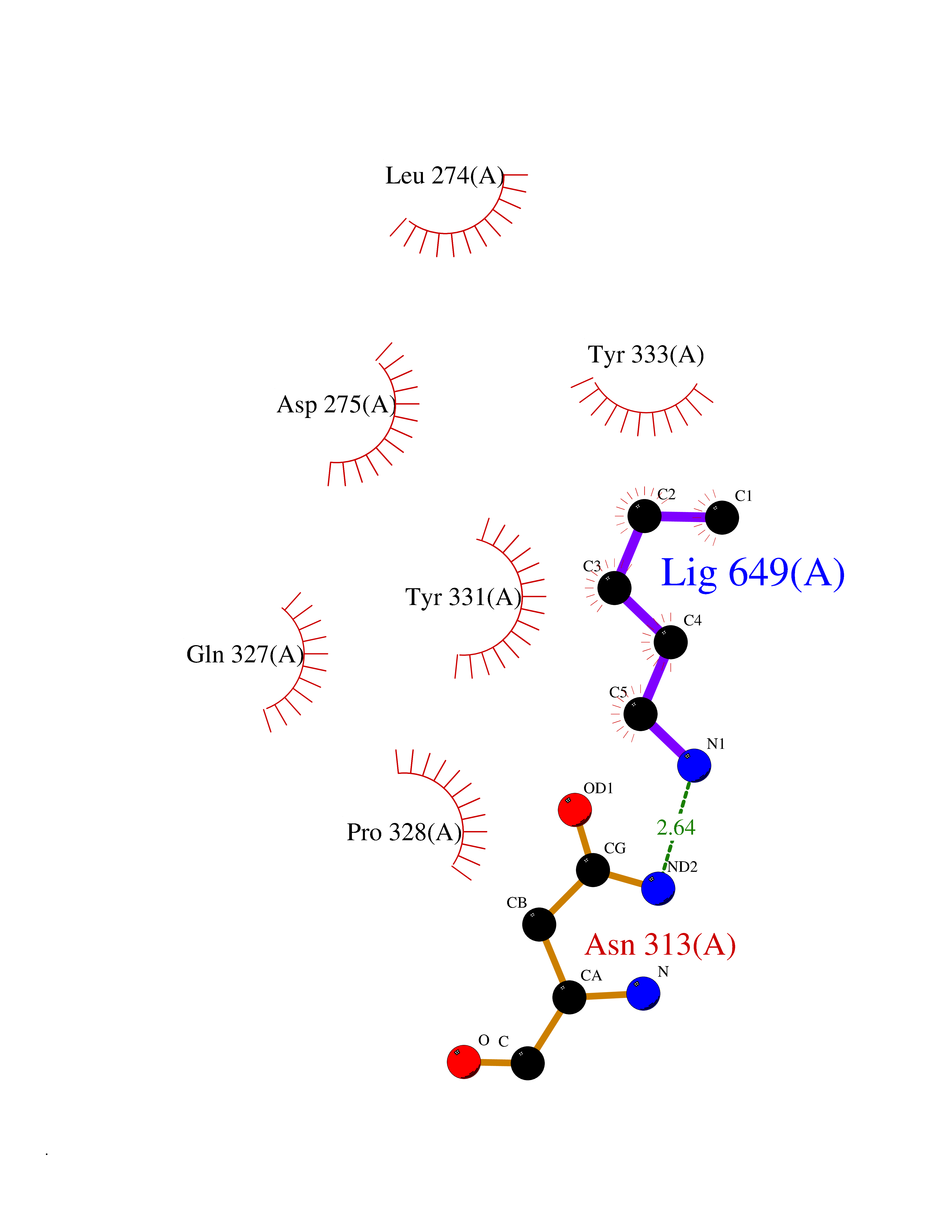 Ligplot Map 3D Binding mode Sequence QRMFEIDYSRDSFLKDGQPFRYISGSIHYSRVPRFYWKDRLLKMKMAGLNAIQTYVPWNFHEPWPGQYQFSEDHDVEYFLRLAHELGLLVILRPGPYICAEWEMGGLPAWLLEKESILLRSSDPDYLAAVDKWLGVLLPKMKPLLYQNGGPVITVQVENEYGSYFACDFDYLRFLQKRFRHHLGDDVVLFTTDGAHKTFLKCGALQGLYTTVDFGTGSNITDAFLSQRKCEPKGPLINSEFYTGWLDHWGQPHSTIKTEAVASSLYDILARGASVNLYMFIGGTNFAYWNGANSPYAAQPTSYDYDAPLSEAGDLTEKYFALRNIIQKFEKVPEGPIPPSTPKFAYGKVTLEKLKTVGAALDILCPSGPIKSLYPLTFIQVKQHYGFVLYRTTLPQDCSNPAPLSSPLNGVHDRAYVAVDGIPQGVLERNNVITLNITGKAGATLDLLVENMGRVNYGAYINDFKGLVSNLTLSSNILTDWTIFPLDTEDAVRSHLGGWGHRNYTLPAFYMGNFSIPSGIPDLPQDTFIQFPGWTKGQVWINGFNLGRYWPARGPQLTLFVPQHILMTSAPNTITVLELEWAPCSSDDPELCAVTFVDRPVIGSS Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Neutrophil gelatinase-associated lipocalin (LCN2) | 5NKN | 4.75 | |
Target general information Gen name LCN2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p25; Siderocalin LCN2; Oncogene 24p3; NGAL; Lipocalin-2; LCN2; 25 kDa alpha-2-microglobulin-related subunit of MMP-9 Protein family Calycin superfamily, Lipocalin family Biochemical class Calycin family Function Iron-trafficking protein involved in multiple processes such as apoptosis, innate immunity and renal development. Binds iron through association with 2,5-dihydroxybenzoic acid (2,5- DHBA), a siderophore that shares structural similarities withbacterial enterobactin, and delivers or removes iron from the cell, depending on the context. Iron-bound form (holo-24p3) is internalized following binding to the SLC22A17 (24p3R) receptor, leading to release of iron and subsequent increase of intracellular iron concentration. In contrast, association of the iron-free form (apo-24p3) with the SLC22A17 (24p3R) receptor is followed by association with an intracellular siderophore, iron chelation and iron transfer to the extracellular medium, thereby reducing intracellular iron concentration. Involved in apoptosis due to interleukin-3 (IL3) deprivation: iron-loaded form increases intracellular iron concentration without promoting apoptosis, while iron-free form decreases intracellular iron levels, inducing expression of the proapoptotic protein BCL2L11/BIM, resulting in apoptosis. Involved in innate immunity, possibly by sequestrating iron, leading to limit bacterial growth. . Related diseases Pseudovaginal perineoscrotal hypospadias (PPSH) [MIM:264600]: A form of male pseudohermaphroditism in which 46,XY males show ambiguous genitalia at birth, including perineal hypospadias and a blind perineal pouch, and develop masculinization at puberty. The name of the disorder stems from the finding of a blind-ending perineal opening resembling a vagina and a severely hypospadiac penis with the urethra opening onto the perineum. {ECO:0000269|PubMed:10718838, ECO:0000269|PubMed:10898110, ECO:0000269|PubMed:10999800, ECO:0000269|PubMed:12843198, ECO:0000269|PubMed:15064320, ECO:0000269|PubMed:1522235, ECO:0000269|PubMed:15528927, ECO:0000269|PubMed:15770495, ECO:0000269|PubMed:16098368, ECO:0000269|PubMed:16181229, ECO:0000269|PubMed:7554313, ECO:0000269|PubMed:8626825, ECO:0000269|PubMed:8768837, ECO:0000269|PubMed:9208814, ECO:0000269|PubMed:9745434, ECO:0000269|PubMed:9843052}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02710; DB01672; DB01926; DB04043; DB01631; DB04476 Interacts with P49419-2; Q9NXW9; Q8WXI3; Q12797-6; Q9BXY8; Q96LC9; P49069; P24863; Q9UKJ5; Q9H1P6; Q9H6B4; O14595; Q08426; Q6NZ36-4; B3EWG5; Q7Z4H3; Q6ISS4; Q5TA76; P80188; Q9UIQ6-2; Q9Y6Y9; Q96JG8; Q8IXL7-2; Q969H8; Q969S2; Q17RF5; P07237; P13667; Q96FA3; Q9NRD5; Q13526; Q9UGP5-2; Q12837; P54646; Q86Y79; O60895; Q9BWG6; P60059; O43765; Q96EQ0; Q8IYX1; Q9UL33-2; P20396; O43715; Q13049; Q99816; Q5W5X9-3; Q99757; P57075-2; Q969M7; Q9UMX0; Q9UHD9; P15692-12; Q14119; Q9Y6T4; Q9H0D6; O96006; A0A1U9X8X8 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Glycoprotein; Immunity; Innate immunity; Ion transport; Iron; Iron transport; Proteomics identification; Pyrrolidone carboxylic acid; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 19748.4 Length 172 Aromaticity 0.13 Instability index 30.73 Isoelectric point 7.71 Charge (pH=7) 0.72 2D Binding mode Binding energy (Kcal/mol) -6.48  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SDLIPAPPLSKVPLQQNFQDNQFHGKWYVVGVAGNGFLREDKDPIKMAATIYELKEDKSYNVTFQKFPMKKCQYMTDTLVPGSQPGEFTLGNIKSEPGYTSWLVRVVSTNYNQHAMVFFKAVQQNREDFFITLYGRTKELTSELKENFIRFSKSLGLPENHIVFPVPIDQCI Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Neuronal acetylcholine receptor beta-2 (CHRNB2) | 6CNJ | 4.75 | |
Target general information Gen name CHRNB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nicotinic acetylcholine receptor beta2; Nicotinic acetylcholine receptor beta 2-subunit protein; CHRNB2; Beta-2 nAChR; Alpha-4/beta-2 nicotinic receptor Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Beta-2/CHRNB2 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane permeable to sodiun ions. Related diseases Epilepsy, nocturnal frontal lobe, 3 (ENFL3) [MIM:605375]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:11062464, ECO:0000269|PubMed:11104662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00572; DB00237; DB00565; DB09028; DB01245; DB00514; DB07720; DB00898; DB00753; DB00657; DB00333; DB00184; DB00981; DB05458; DB05855; DB05740; DB00747; DB00202; DB01273 Interacts with P43681-1; P30532 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84601.2 Length 728 Aromaticity 0.13 Instability index 39.72 Isoelectric point 5.86 Charge (pH=7) -9.84 2D Binding mode Binding energy (Kcal/mol) -6.47  Molscript Map 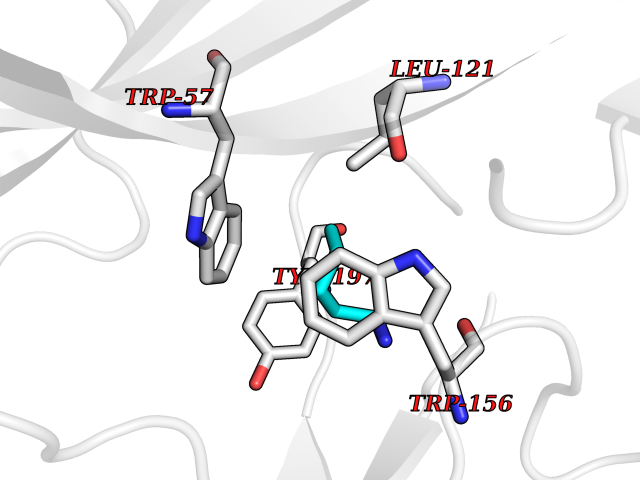 Pymol Map 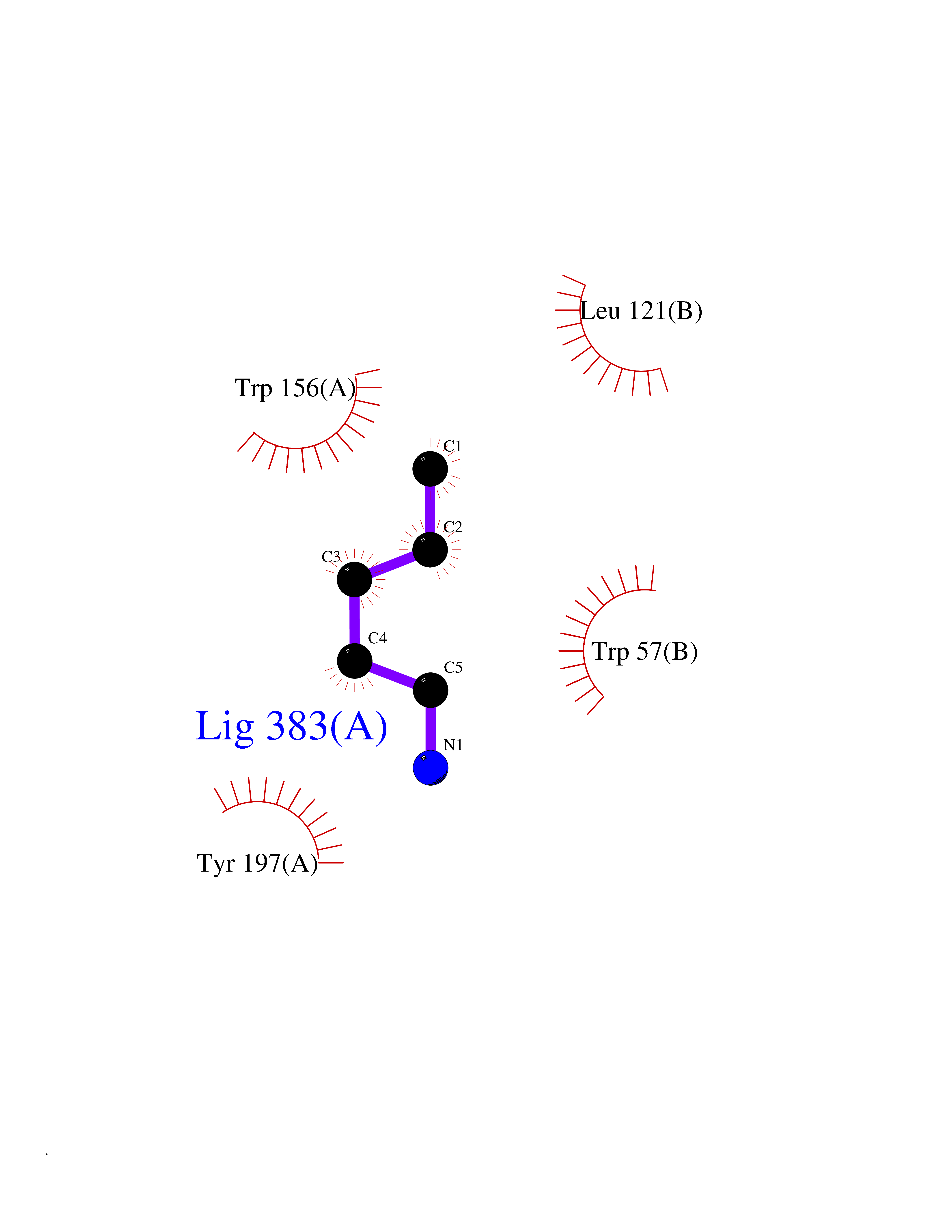 Ligplot Map 3D Binding mode Sequence ETRAHAEERLLKKLFSGYNKWSRPVANISDVVLVRFGLSIAQLIDVDEKNQMMTTNVWVKQEWHDYKLRWDPADYENVTSIRIPSELIWRPDIVLYNNADGDFAVTHLTKAHLFHDGRVQWTPPAIYKSSCSIDVTFFPFDQQNCTMKFGSWTYDKAKIDLVNMHSRVDQLDFWESGEWVIVDAVGTYNTRKYECCAEIYPDITYAFVIRRLPLFYTINLIIPCLLISCLTVLVFYLPSECGEKITLCISVLLSLTVFLLLITEIIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHHRSPRTHTMPTWVRRVFLDIVPRLLLMKRFERSVKEDWKYVAMVIDRIFLWMFIIVCLLGTVGLFLPPWDTEERLVEHLLDPSRYNKLIRPATNGSELVTVQLMVSLAQLISVHEREQIMTTNVWLTQEWEDYRLTWKPEEFDNMKKVRLPSKHIWLPDVVLYNNADGMYEVSFYSNAVVSYDGSIFWLPPAIYKSACKIEVKHFPFDQQNCTMKFRSWTYDRTEIDLVLKSEVASLDDFTPSGEWDIVALPGRRNENPDDSTYVDITYDFIIRRKPLFYTINLIIPCVLITSLAILVFYLPSDCGEKMTLCISVLLALTVFLLLISKIVPPTSLDVPLVGKYLMFTMVLVTFSIVTSVCVLNVHHRSPTTHTMAPWVKVVFLEKLPALLFMQQSVSEDWKYVAMVIDRLFLWIFVFVCVFGTIGMF Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Neprilysin | 1R1H | 4.74 | |
Target general information Gen name MME Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms EPN Protein family Peptidase M13 family Biochemical class Hydrolase Function Endopeptidase activity.Exopeptidase activity.Metalloendopeptidase activity.Metallopeptidase activity.Peptide binding.Zinc ion binding. Related diseases Charcot-Marie-Tooth disease, axonal, 2T (CMT2T) [MIM:617017]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:26991897, ECO:0000269|PubMed:27588448}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinocerebellar ataxia 43 (SCA43) [MIM:617018]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA43 is a slowly progressive, autosomal dominant form. {ECO:0000269|PubMed:27583304}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08575; DB02597; DB00616; DB11623; DB05796; DB06655; DB02558; DB02062; DB00886; DB02557; DB09292; DB13928; DB08626 Interacts with P05067; P21926; Q06787-7; P08107; P04792 EC number 3.4.24.11 Uniprot keywords 3D-structure; Cell membrane; Charcot-Marie-Tooth disease; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lipoprotein; Membrane; Metal-binding; Metalloprotease; Myristate; Neurodegeneration; Neuropathy; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Spinocerebellar ataxia; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 79435.8 Length 696 Aromaticity 0.11 Instability index 37.5 Isoelectric point 5.53 Charge (pH=7) -11.46 2D Binding mode Binding energy (Kcal/mol) -6.47  Molscript Map 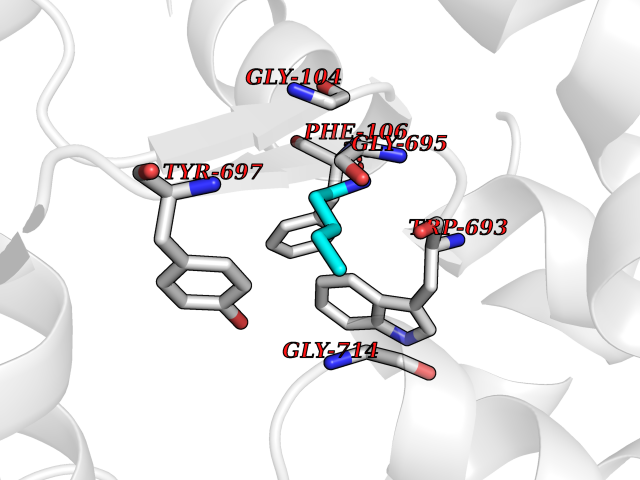 Pymol Map 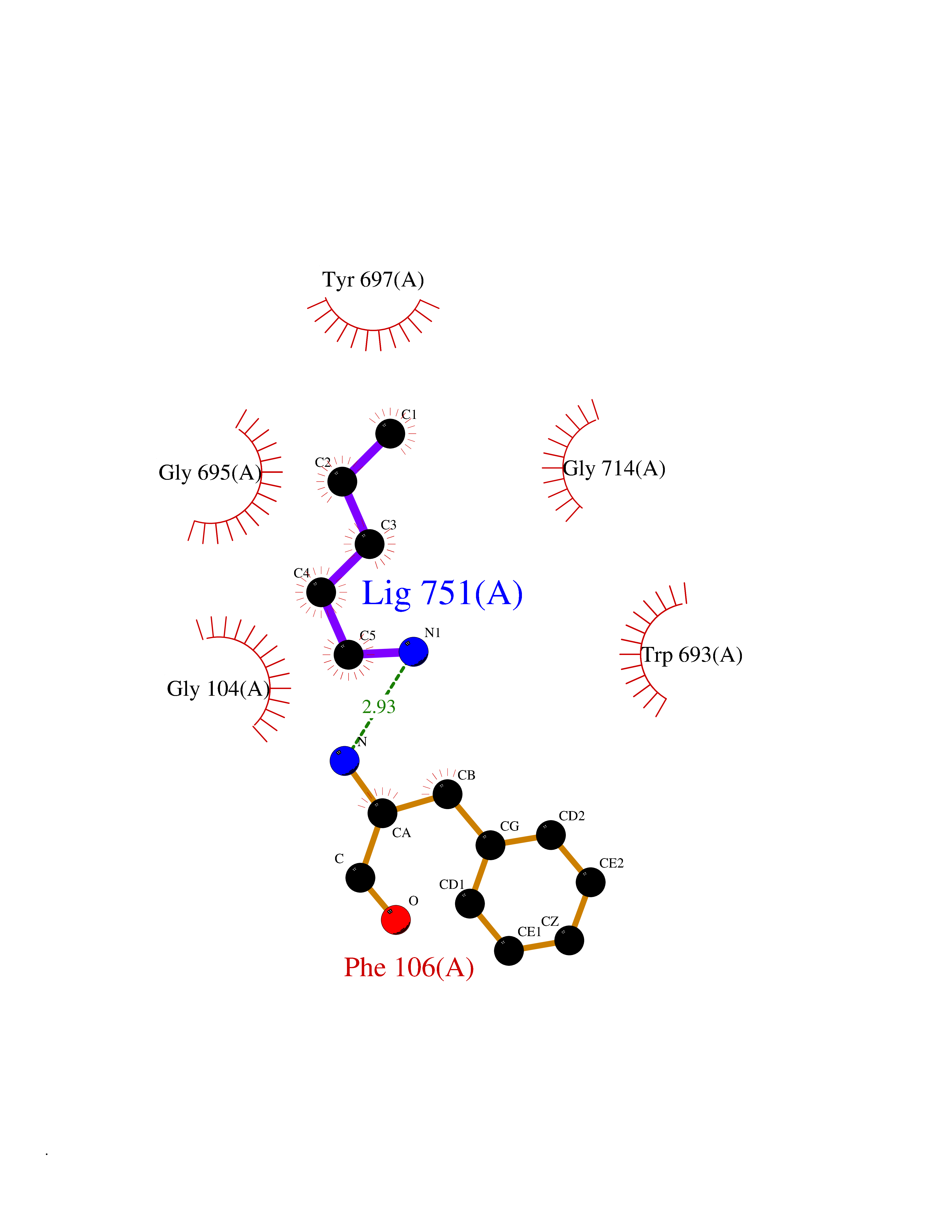 Ligplot Map 3D Binding mode Sequence GICKSSDCIKSAARLIQNMDATTEPCTDFFKYACGGWLKRNVIPETSSRYGNFDILRDELEVVLKDVLQEPKTEDIVAVQKAKALYRSCINESAIDSRGGEPLLKLLPDIYGWPVATENWEQKYGASWTAEKAIAQLNSKYGKKVLINLFVGTDDKNSVNHVIHIDQPRLGLPSRDYYECTGIYKEACTAYVDFMISVARLIRQEERLPIDENQLALEMNKVMELEKEIANATAKPEDRNDPMLLYNKMTLAQIQNNFSLEINGKPFSWLNFTNEIMSTVNISITNEEDVVVYAPEYLTKLKPILTKYSARDLQNLMSWRFIMDLVSSLSRTYKESRNAFRKALYGTTSETATWRRCANYVNGNMENAVGRLYVEAAFAGESKHVVEDLIAQIREVFIQTLDDLTWMDAETKKRAEEKALAIKERIGYPDDIVSNDNKLNNEYLELNYKEDEYFENIIQNLKFSQSKQLKKLREKVDKDEWISGAAVVNAFYSSGRNQIVFPAGILQPPFFSAQQSNSLNYGGIGMVIGHEITHGFDDNGRNFNKDGDLVDWWTQQSASNFKEQSQCMVYQYGNFSWDLAGGQHLNGINTLGENIADNGGLGQAYRAYQNYIKKNGEEKLLPGLDLNHKQLFFLNFAQVWCGTYRPEYAVNSIKTDVHSPGNFRIIGTLQNSAEFSEAFHCRKNSYMNPEKKCRVW Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Fibrinogen gamma chain | 1DUG | 4.74 | |
Target general information Gen name FGG Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PRO2061 Protein family NA Biochemical class transferase Function Cell adhesion molecule binding.Metal ion binding.Protein binding, bridging.Protein homodimerization activity.Receptor binding.Structural molecule activity. Related diseases Congenital afibrinogenemia (CAFBN) [MIM:202400]: Rare autosomal recessive disorder is characterized by bleeding that varies from mild to severe and by complete absence or extremely low levels of plasma and platelet fibrinogen. {ECO:0000269|PubMed:25427968}. The disease is caused by variants affecting the gene represented in this entry. Patients with congenital fibrinogen abnormalities can manifest different clinical pictures. Some cases are clinically silent, some show a tendency toward bleeding and some show a predisposition for thrombosis with or without bleeding.; DISEASE: Dysfibrinogenemia, congenital (DYSFIBRIN) [MIM:616004]: A disorder characterized by qualitative abnormalities (dysfibrinogenemia) of the circulating fibrinogen. Affected individuals are frequently asymptomatic, but some patients have bleeding diathesis, thromboembolic complications, or both. In some cases, dysfibrinogenemia is associated with low circulating fibrinogen levels (hypodysfibrinogenemia). {ECO:0000269|PubMed:15632207, ECO:0000269|PubMed:2257302, ECO:0000269|PubMed:2976995, ECO:0000269|PubMed:3708159}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00009; DB11571; DB00364; DB11300; DB11572 Interacts with P75358 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Blood coagulation; Calcium; Coiled coil; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hemostasis; Isopeptide bond; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Secreted; Signal; Sulfation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 54170 Length 468 Aromaticity 0.12 Instability index 35.94 Isoelectric point 6.08 Charge (pH=7) -7.16 2D Binding mode Binding energy (Kcal/mol) -6.47 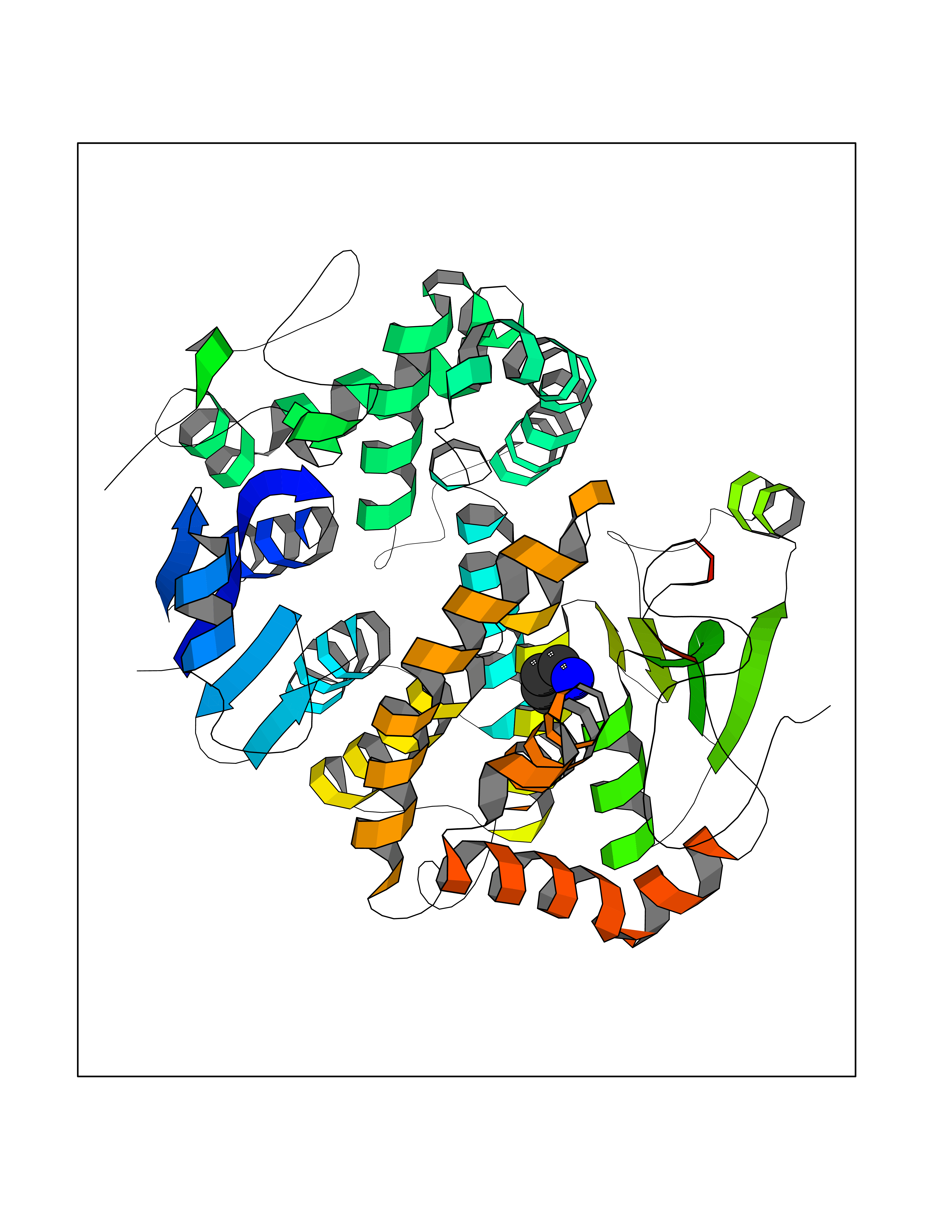 Molscript Map 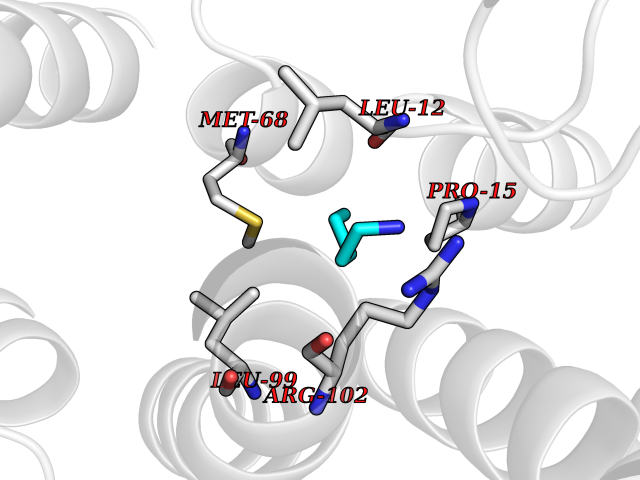 Pymol Map 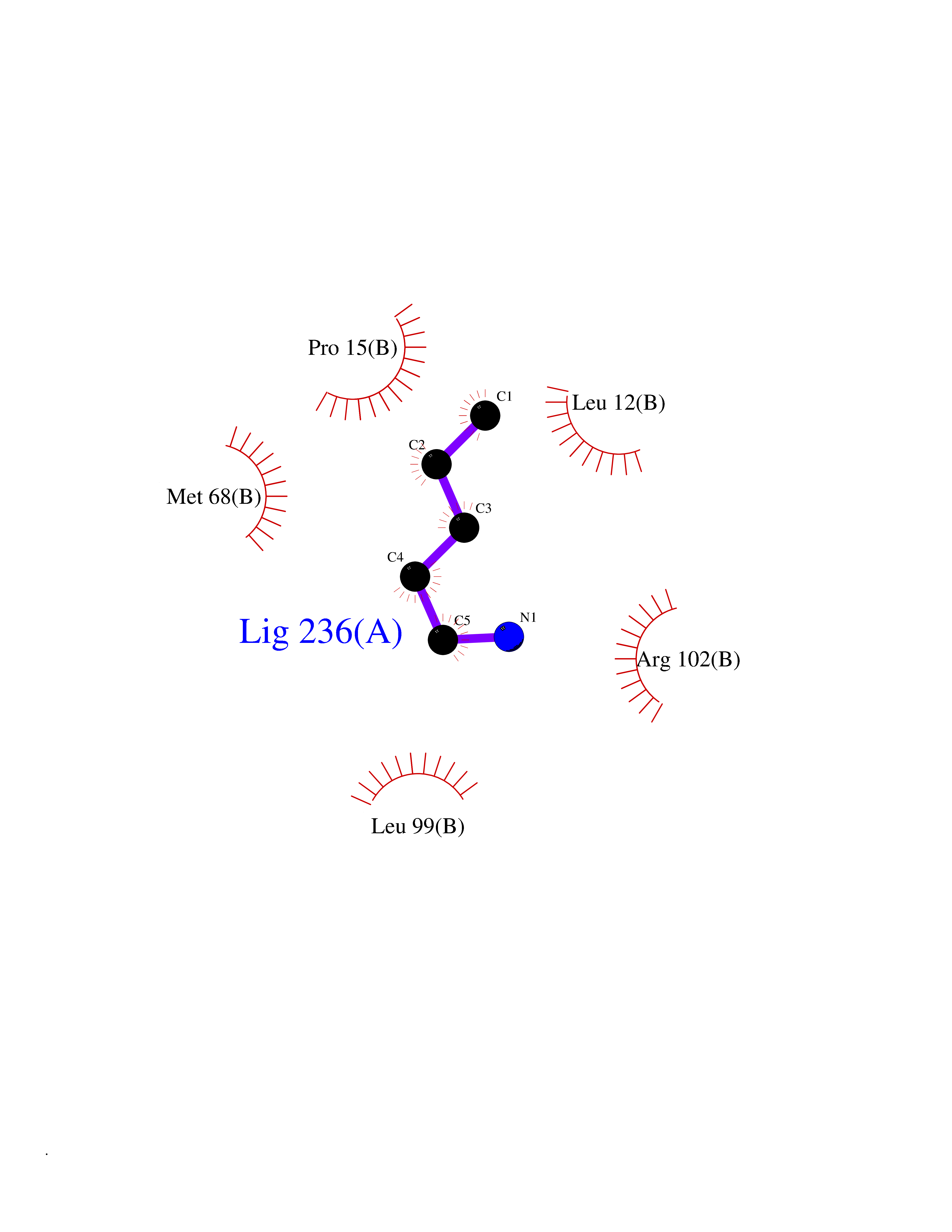 Ligplot Map 3D Binding mode Sequence SPILGYWKIKGLVQPTRLLLEYLEEKYEEHLYERDEGDKWRNKKFELGLEFPNLPYYIDGDVKLTQSMAIIRYIADKHNMLGGCPKERAEISMLEGAVLDIRYGVSRIAYSKDFETLKVDFLSKLPEMLKMFEDRLCHKTYLNGDHVTHPDFMLYDALDVVLYMDPMCLDAFPKLVCFKKRIEAIPQIDKYLKSSKYIAWPLQGWQATFGGGDHPPKSDPQQHHLGGAKQAGDVSPILGYWKIKGLVQPTRLLLEYLEEKYEEHLYERDEGDKWRNKKFELGLEFPNLPYYIDGDVKLTQSMAIIRYIADKHNMLGGCPKERAEISMLEGAVLDIRYGVSRIAYSKDFETLKVDFLSKLPEMLKMFEDRLCHKTYLNGDHVTHPDFMLYDALDVVLYMDPMCLDAFPKLVCFKKRIEAIPQIDKYLKSSKYIAWPLQGWQATFGGGDHPPKSDPQQHHLGGAKQAGDV Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Tyrosine-protein kinase ABL1 (ABL) | 5HU9 | 4.74 | |
Target general information Gen name ABL1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p150; Proto-oncogene tyrosine-protein kinase ABL1; Proto-oncogene c-Abl; JTK7; C-ABL; Abl; Abelson tyrosine-protein kinase 1; Abelson murine leukemia viral oncogene homolog 1 Protein family Protein kinase superfamily, Tyr protein kinase family, ABL subfamily Biochemical class Kinase Function Coordinates actin remodeling through tyrosine phosphorylation of proteins controlling cytoskeleton dynamics like WASF3 (involved in branch formation); ANXA1 (involved in membrane anchoring); DBN1, DBNL, CTTN, RAPH1 and ENAH (involved in signaling); or MAPT and PXN (microtubule-binding proteins). Phosphorylation of WASF3 is critical for the stimulation of lamellipodia formation and cell migration. Involved in the regulation of cell adhesion and motility through phosphorylation of key regulators of these processes such as BCAR1, CRK, CRKL, DOK1, EFS or NEDD9. Phosphorylates multiple receptor tyrosine kinases and more particularly promotes endocytosis of EGFR, facilitates the formation of neuromuscular synapses through MUSK, inhibits PDGFRB-mediated chemotaxis and modulates the endocytosis of activated B-cell receptor complexes. Other substrates which are involved in endocytosis regulation are the caveolin (CAV1) and RIN1. Moreover, ABL1 regulates the CBL family of ubiquitin ligases that drive receptor down-regulation and actin remodeling. Phosphorylation of CBL leads to increased EGFR stability. Involved in late-stage autophagy by regulating positively the trafficking and function of lysosomal components. ABL1 targets to mitochondria in response to oxidative stress and thereby mediates mitochondrial dysfunction and cell death. In response to oxidative stress, phosphorylates serine/threonine kinase PRKD2 at 'Tyr-717'. ABL1 is also translocated in the nucleus where it has DNA-binding activity and is involved in DNA-damage response and apoptosis. Many substrates are known mediators of DNA repair: DDB1, DDB2, ERCC3, ERCC6, RAD9A, RAD51, RAD52 or WRN. Activates the proapoptotic pathway when the DNA damage is too severe to be repaired. Phosphorylates TP73, a primary regulator for this type of damage-induced apoptosis. Phosphorylates the caspase CASP9 on 'Tyr-153' and regulates its processing in the apoptotic response to DNA damage. Phosphorylates PSMA7 that leads to an inhibition of proteasomal activity and cell cycle transition blocks. ABL1 acts also as a regulator of multiple pathological signaling cascades during infection. Several known tyrosine-phosphorylated microbial proteins have been identified as ABL1 substrates. This is the case of A36R of Vaccinia virus, Tir (translocated intimin receptor) of pathogenic E. coli and possibly Citrobacter, CagA (cytotoxin-associated gene A) of H. pylori, or AnkA (ankyrin repeat-containing protein A) of A. phagocytophilum. Pathogens can highjack ABL1 kinase signaling to reorganize the host actin cytoskeleton for multiple purposes, like facilitating intracellular movement and host cell exit. Finally, functions as its own regulator through autocatalytic activity as well as through phosphorylation of its inhibitor, ABI1. Regulates T-cell differentiation in a TBX21-dependent manner. Phosphorylates TBX21 on tyrosine residues leading to an enhancement of its transcriptional activator activity. Non-receptor tyrosine-protein kinase that plays a role in many key processes linked to cell growth and survival such as cytoskeleton remodeling in response to extracellular stimuli, cell motility and adhesion, receptor endocytosis, autophagy, DNA damage response and apoptosis. Related diseases Leukemia, chronic myeloid (CML) [MIM:608232]: A clonal myeloproliferative disorder of a pluripotent stem cell with a specific cytogenetic abnormality, the Philadelphia chromosome (Ph), involving myeloid, erythroid, megakaryocytic, B-lymphoid, and sometimes T-lymphoid cells, but not marrow fibroblasts. The gene represented in this entry is involved in disease pathogenesis.; DISEASE: A chromosomal aberration involving ABL1 has been found in patients with chronic myeloid leukemia. Translocation t(9;22)(q34;q11) with BCR. The translocation produces a BCR-ABL found also in acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL). {ECO:0000269|PubMed:3021337}.; DISEASE: A chromosomal aberration involving ABL1 is found in a form of acute lymphoblastic leukemia (PubMed:15361874). Translocation t(9;9)(q34;q34) with NUP214 (PubMed:15361874). {ECO:0000269|PubMed:15361874}.; DISEASE: Congenital heart defects and skeletal malformations syndrome (CHDSKM) [MIM:617602]: An autosomal dominant disorder characterized by congenital heart disease with atrial and ventricular septal defects, variable skeletal abnormalities, and failure to thrive. Skeletal defects include pectus excavatum, scoliosis, and finger contractures. Some patient exhibit joint laxity. {ECO:0000269|PubMed:28288113}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08043; DB08583; DB07831; DB08350; DB12597; DB00171; DB06616; DB12267; DB01254; DB12010; DB00619; DB13749; DB08231; DB03878; DB04868; DB08339; DB08901; DB12323; DB08896; DB14989; DB05184 Interacts with Q8IZP0; Q9NYB9; O14672; P10275; Q13315; Q4KMG0; P46108; P46109; P35222; P00533; P04626; Q03468; Q14315; P36888; P05107; P10721; Q38SD2; Q92918; Q7Z434; O43196; P15941; P15941-12; P16333; O43900; Q13905; Q86UR5; Q13671; P31947; Q15464; O75751; P37840; Q9BX66; O60504-2; Q07890; P12931; P51692; Q9Y4G6; P11387; P04637; P15498; Q9Y6W5; P62258; P61981; P63104; O35158; P37840; P48165; Q15323; P37840 EC number EC 2.7.10.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Apoptosis; ATP-binding; Autophagy; Cell adhesion; Chromosomal rearrangement; Cytoplasm; Cytoskeleton; Disease variant; DNA damage; DNA repair; DNA-binding; Endocytosis; Kinase; Lipoprotein; Magnesium; Manganese; Membrane; Metal-binding; Mitochondrion; Myristate; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Reference proteome; SH2 domain; SH3 domain; Transferase; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 31264.6 Length 270 Aromaticity 0.12 Instability index 37.99 Isoelectric point 5.42 Charge (pH=7) -7.67 2D Binding mode Binding energy (Kcal/mol) -6.46 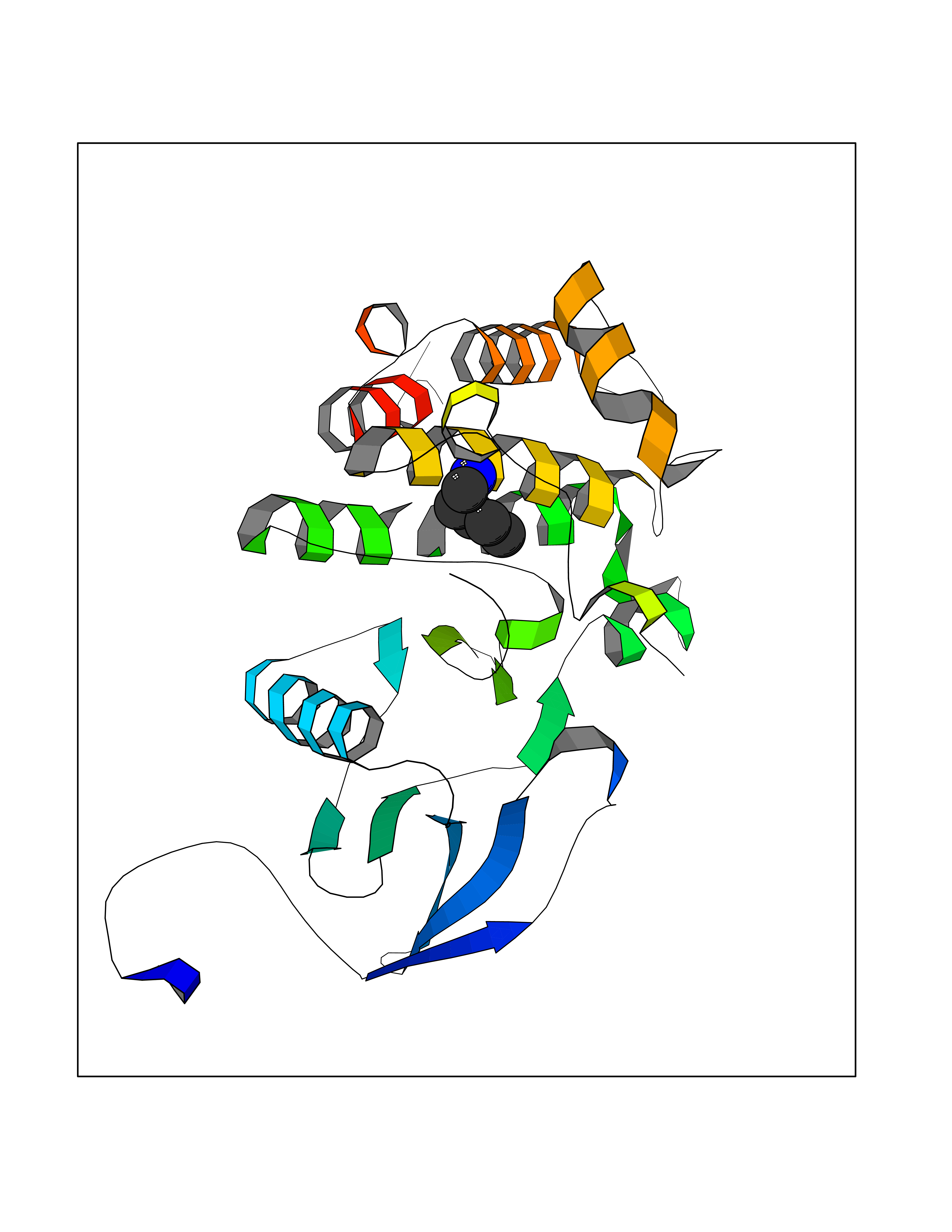 Molscript Map 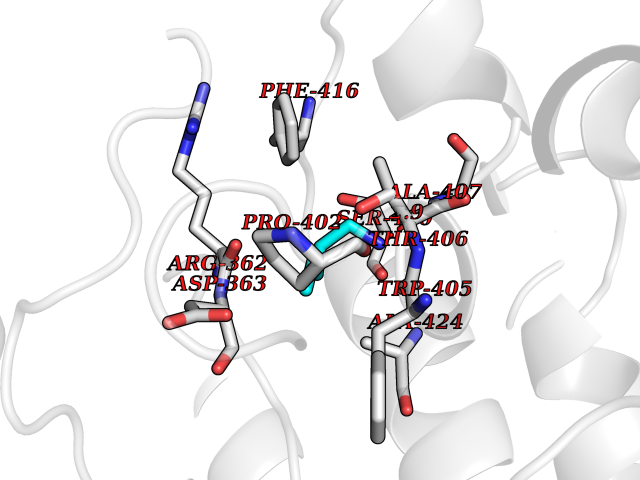 Pymol Map 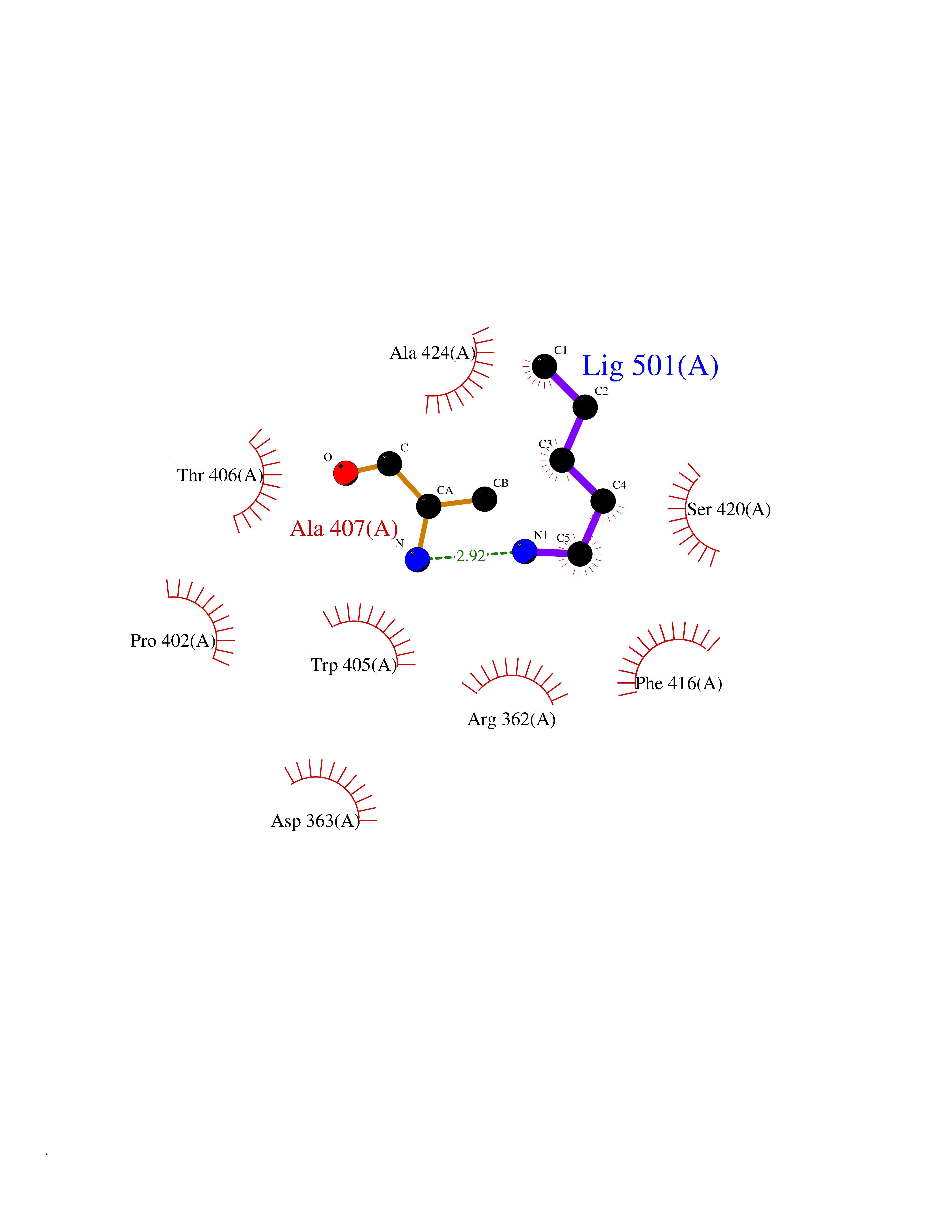 Ligplot Map 3D Binding mode Sequence AMGSSPNYDKWEMERTDITMKHKLGGGQYGEVYEGVWKKYSLTVAVKTLKEDTMEVEEFLKEAAVMKEIKHPNLVQLLGVCTREPPFYIITEFMTYGNLLDYLRECNRQEVNAVVLLYMATQISSAMEYLEKKNFIHRDLAARNCLVGENHLVKVADFGLSRLMTAHAGAKFPIKWTAPESLAYNKFSIKSDVWAFGVLLWEIATYGMSPYPGIDLSQVYELLEKDYRMERPEGCPEKVYELMRACWQWNPSDRPSFAEIHQAFETMFQE Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Glycine--tRNA ligase | 2ZT5 | 4.74 | |
Target general information Gen name GARS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms GARS Protein family Class-II aminoacyl-tRNA synthetase family Biochemical class Ligase Function ATP binding.Bis(5'-nucleosyl)-tetraphosphatase (asymmetrical) activity.Glycine-tRNA ligase activity.Identical protein binding.Protein dimerization activity. Related diseases Charcot-Marie-Tooth disease, axonal, 2D (CMT2D) [MIM:601472]: A dominant axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:12690580, ECO:0000269|PubMed:17035524, ECO:0000269|PubMed:17101916, ECO:0000269|PubMed:17663003, ECO:0000269|PubMed:20169446, ECO:0000269|PubMed:24604904, ECO:0000269|PubMed:25168514, ECO:0000269|PubMed:26244500, ECO:0000269|PubMed:26503042, ECO:0000269|PubMed:31173493}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal dominant 5 (HMND5) [MIM:600794]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. {ECO:0000269|PubMed:12690580, ECO:0000269|PubMed:17035524, ECO:0000269|PubMed:23279345, ECO:0000269|PubMed:24627108, ECO:0000269|PubMed:26503042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinal muscular atrophy, infantile, James type (SMAJI) [MIM:619042]: An autosomal dominant form of spinal muscular atrophy, a group of neuromuscular disorders characterized by degeneration of the anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. SMAJI is a severe disease characterized by hypotonia manifesting in the first weeks or months of life, delayed motor development, motor regression, and muscle weakness and atrophy primarily affecting distal muscles. Additional variable features include feeding difficulties, poor overall growth, foot deformities, kyphosis, hyperlordosis, scoliosis, vocal cord dysfunction, and respiratory insufficiency. {ECO:0000269|PubMed:32181591}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00145 Interacts with Q16520; Q9UNS2; P41250; Q99612; Q9BR81; Q8WXH5; Q99932-2; Q9CZD3 EC number 2.7.7.-; 6.1.1.14 Uniprot keywords 3D-structure; Acetylation; Alternative initiation; Aminoacyl-tRNA synthetase; ATP-binding; Cell projection; Charcot-Marie-Tooth disease; Cytoplasm; Disease variant; Hydrolase; Ligase; Mitochondrion; Neurodegeneration; Neuropathy; Nucleotide-binding; Phosphoprotein; Protein biosynthesis; Proteomics identification; Reference proteome; Secreted; Transferase; Transit peptide Protein physicochemical properties Chain ID A Molecular weight (Da) 43254.1 Length 381 Aromaticity 0.11 Instability index 47.89 Isoelectric point 6.6 Charge (pH=7) -1.4 2D Binding mode Binding energy (Kcal/mol) -6.46 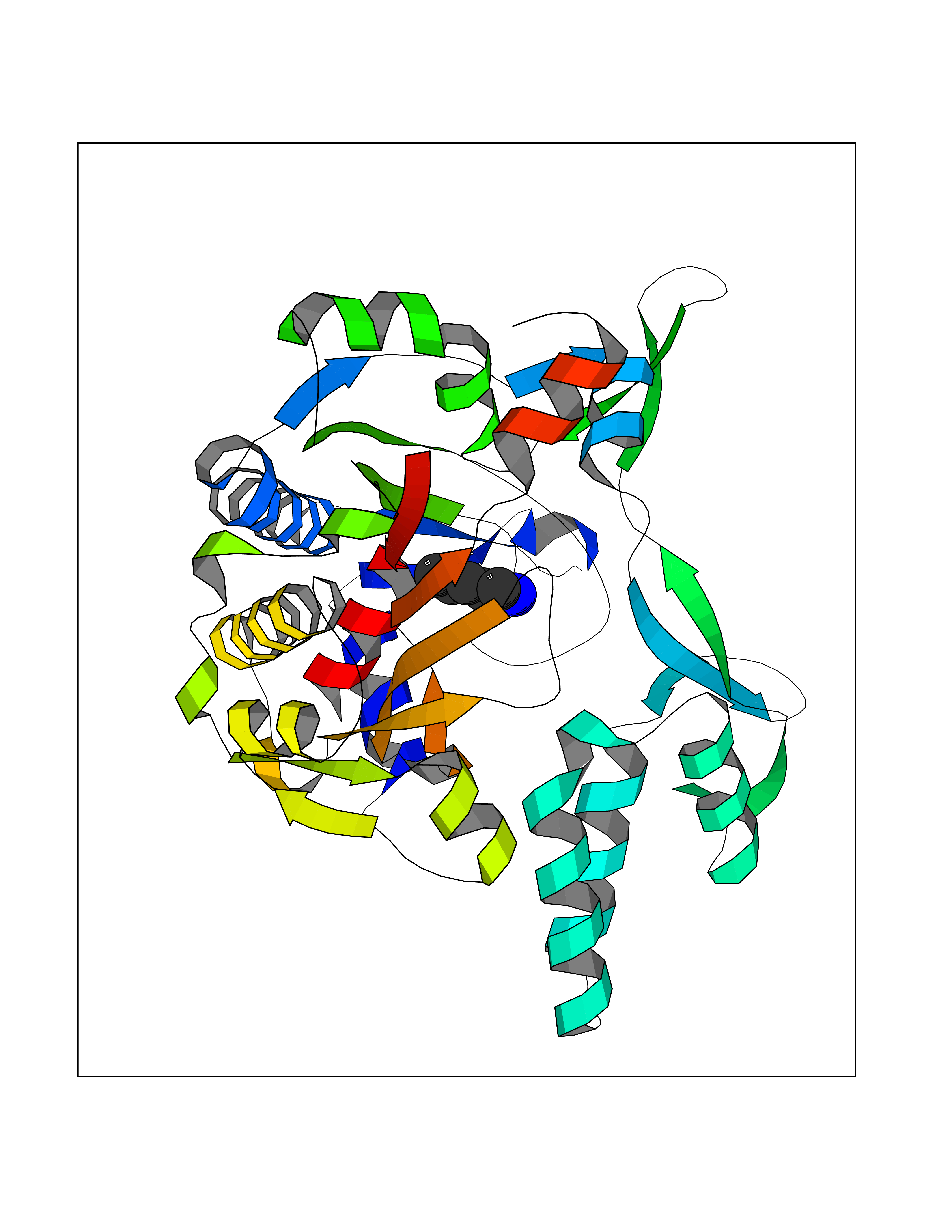 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence IVDRAKMEDTLKRRFFYDQAFAIYGGVSGLYDFGPVGCALKNNIIQTWRQHFIQEEQILEIDCTMLTPEPVLKTSGHVDKFADFMVKDVKNGECFRADHLLKAHLQKLMSDKKCSVEKKSEMESVLAQLDNYGQQELADLFVNYNVKSPITGNDLSPPVSFNLMFKTFIGPGGNMPGYLRPETAQGIFLNFKRLLEFNQGKLPFAAAQIGNSFRNEISPRSGLIRVREFTMAEIEHFVDPSEKDHPKFQNVADLHLYLYSAKAQVSGQSARKMRLGDAVEQGVINNTVLGYFIGRIYLYLTKVGISPDKLRFRQHMENEMAHYACDCWDAESKTSYGWIEIVGCADRSCYDLSCHARATKVPPNVIEPSFGLGRIMYTVFE Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | 2-hydroxy-6-oxo-7-methylocta-2,4-dienoate hydrolase | 1UK8 | 4.74 | |
Target general information Gen name cumD Organism Pseudomonas fluorescens Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function Hydrolase activity. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03741; DB03793; DB03568; DB02531; DB03750; DB02406; DB03766 Interacts with NA EC number NA Uniprot keywords 3D-structure; Hydrolase Protein physicochemical properties Chain ID A Molecular weight (Da) 30307.9 Length 271 Aromaticity 0.1 Instability index 37.49 Isoelectric point 5.02 Charge (pH=7) -11.58 2D Binding mode Binding energy (Kcal/mol) -6.46  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence NLEIGKSILAAGVLTNYHDVGEGQPVILIHGSGPGVSAYANWRLTIPALSKFYRVIAPDMVGFGFTDRPENYNYSKDSWVDHIIGIMDALEIEKAHIVGNAFGGGLAIATALRYSERVDRMVLMGAAGTRFDVTEGLNAVWGYTPSIENMRNLLDIFAYDRSLVTDELARLRYEASIQPGFQESFSSMFPEPRQRWIDALASSDEDIKTLPNETLIIHGREDQVVPLSSSLRLGELIDRAQLHVFGRCGHWTQIEQTDRFNRLVVEFFNEA Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | "Succinate dehydrogenase [ubiquinone] iron-sulfur subunit, mitochondrial (EC 1.3.5.1) (Iron-sulfur subunit of complex II) (Ip) (Malate dehydrogenase [quinone] iron-sulfur subunit) (EC 1.1.5.-)" | 1ZOY | 4.74 | |
Target general information Gen name SDHB Organism Sus scrofa (Pig) Uniprot ID TTD ID NA Synonyms NA Protein family Succinate dehydrogenase/fumarate reductase iron-sulfur protein family Biochemical class NA Function Iron-sulfur protein (IP) subunit of the succinate dehydrogenase complex (mitochondrial respiratory chain complex II), responsible for transferring electrons from succinate to ubiquinone (coenzyme Q) (PubMed:15989954). SDH also oxidizes malate to the non-canonical enol form of oxaloacetate, enol-oxaloacetate. Enol-oxaloacetate, which is a potent inhibitor of the succinate dehydrogenase activity, is further isomerized into keto-oxaloacetate (By similarity). {ECO:0000250|UniProtKB:Q3T189, ECO:0000269|PubMed:15989954}." Related diseases A chromosomal aberration involving NFKB2 is found in a case of B-cell non Hodgkin lymphoma (B-NHL). Translocation t(10;14)(q24;q32) with IGHA1. The resulting oncogene is also called Lyt-10C alpha variant.; DISEASE: A chromosomal aberration involving NFKB2 is found in a cutaneous T-cell leukemia (C-TCL) cell line. This rearrangement produces the p80HT gene which codes for a truncated 80 kDa protein (p80HT).; DISEASE: In B-cell leukemia (B-CLL) cell line, LB40 and EB308, can be found after heterogeneous chromosomal aberrations, such as internal deletions.; DISEASE: Immunodeficiency, common variable, 10 (CVID10) [MIM:615577]: A primary immunodeficiency characterized by childhood-onset of recurrent infections, hypogammaglobulinemia, and decreased numbers of memory and marginal zone B-cells. Some patients may develop autoimmune features and have circulating autoantibodies. An unusual feature is central adrenal insufficiency. {ECO:0000269|PubMed:24140114, ECO:0000269|PubMed:25524009}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 1.1.5.-; 1.3.5.1 Uniprot keywords 2Fe-2S; 3D-structure; 3Fe-4S; 4Fe-4S; Acetylation; Electron transport; Iron; Iron-sulfur; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Reference proteome; Transit peptide; Transport; Tricarboxylic acid cycle Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 120840 Length 1092 Aromaticity 0.09 Instability index 39.34 Isoelectric point 7.64 Charge (pH=7) 4.11 2D Binding mode Binding energy (Kcal/mol) -6.46  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence STQYPVVDHEFDAVVVGAGGAGLRAAFGLSEAGFNTACVTKLFPTRSHTVAAQGGINAALGNMEEDNWRWHFYDTVKGSDWLGDQDAIHYMTEQAPASVVELENYGMPFSRTEDGKIYQRAFGGQSLKFGKGGQAHRCCCVADRTGHSLLHTLYGRSLRYDTSYFVEYFALDLLMENGECRGVIALCIEDGSIHRIRARNTVVATGGYGRTYFSCTSAHTSTGDGTAMVTRAGLPCQDLEFVQFHPTGIYGAGCLITEGCRGEGGILINSQGERFMERYAPVAKDLASRDVVSRSMTLEIREGRGCGPEKDHVYLQLHHLPPEQLAVRLPGISETAMIFAGVDVTKEPIPVLPTVHYNMGGIPTNYKGQVLRHVNGQDQVVPGLYACGEAACASVHGANRLGANSLLDLVVFGRACALSIAESCRPGDKVPSIKPNAGEESVMNLDKLRFANGTIRTSELRLSMQKSMQSHAAVFRVGSVLQEGCEKILRLYGDLQHLKTFDRGMVWNTDLVETLELQNLMLCALQTIYGAEARKESRGAHAREDFKERVDEYDYSKPIQGQQKKPFQEHWRKHTLSYVDVKTGKVSLEYRPVIDKTLNEADCATVPPAIRSYSSKAASLHWTGERVVSVLLLGLLPAAYLNPCSAMDYSLAAALTLHGHWGIGQVVTDYVRGDALQKAAKAGLLALSAFTFAGLCYFNYHDVGICKAVAMLWKLTTAKEEMERFWNKNLGSNRPLSPHITIYRWSLPMAMSICHRGTGIALSAGVSLFGLSALLLPGNFESHLELVKSLCLGPTLIYTAKFGIVFPLMYHTWNGIRHLIWDLGKGLTIPQLTQSGVVVLILTVLSSVGLAAMPRIKKFAIYRWDPDKTGDKPHMQTYEIDLNNCGPMVLDALIKIKNEIDSTLTFRRSCREGICGSCAMNINGGNTLACTRRIDTNLDKVSKIYPLPHMYVIKDLVPDLSNFYAQYKSIEPYLKKKDESQEGKQQYLQSIEEREKLDGLYECILCACCSTSCPSYWWNGDKYLGPAVLMQAYRWMIDSRDDFTEERLAKLQDPFSLYRCHTIMNCTGTCPKGLNPGKAIAEIKKMMATYKE Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Histone deacetylase 8 (HDAC8) | 5BWZ | 4.74 | |
Target general information Gen name HDAC8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Histone deacetylase-8; HDACL1; HD8; CDA07 Protein family Histone deacetylase family, HD type 1 subfamily Biochemical class Carbon-nitrogen hydrolase Function Gives a tag for epigenetic repression and plays an important role in transcriptional regulation, cell cycle progression and developmental events. Histone deacetylases act via the formation of large multiprotein complexes. Also involved in the deacetylation of cohesin complex protein SMC3 regulating release of cohesin complexes from chromatin. May play a role in smooth muscle cell contractility. Responsible for the deacetylation of lysine residues on the N-terminal part of the core histones (H2A, H2B, H3 and H4). Related diseases Cornelia de Lange syndrome 5 (CDLS5) [MIM:300882]: A form of Cornelia de Lange syndrome, a clinically heterogeneous developmental disorder associated with malformations affecting multiple systems. It is characterized by facial dysmorphisms, abnormal hands and feet, growth delay, cognitive retardation, hirsutism, gastroesophageal dysfunction and cardiac, ophthalmologic and genitourinary anomalies. {ECO:0000269|PubMed:22885700, ECO:0000269|PubMed:22889856}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07350; DB02565; DB07586; DB12565; DB05015; DB08168; DB01262; DB11841; DB14490; DB14491; DB14488; DB14501; DB14489; DB12645; DB01592; DB02917; DB06603; DB06819; DB03766; DB12847; DB06176; DB04297; DB00313; DB02546; DB01593; DB14487; DB14533; DB14548 Interacts with NA EC number EC 3.5.1.98 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Cytoplasm; Disease variant; Hydrolase; Intellectual disability; Metal-binding; Nucleus; Obesity; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; Transcription; Transcription regulation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 39018.4 Length 351 Aromaticity 0.11 Instability index 38.57 Isoelectric point 6.06 Charge (pH=7) -5.26 2D Binding mode Binding energy (Kcal/mol) -6.47 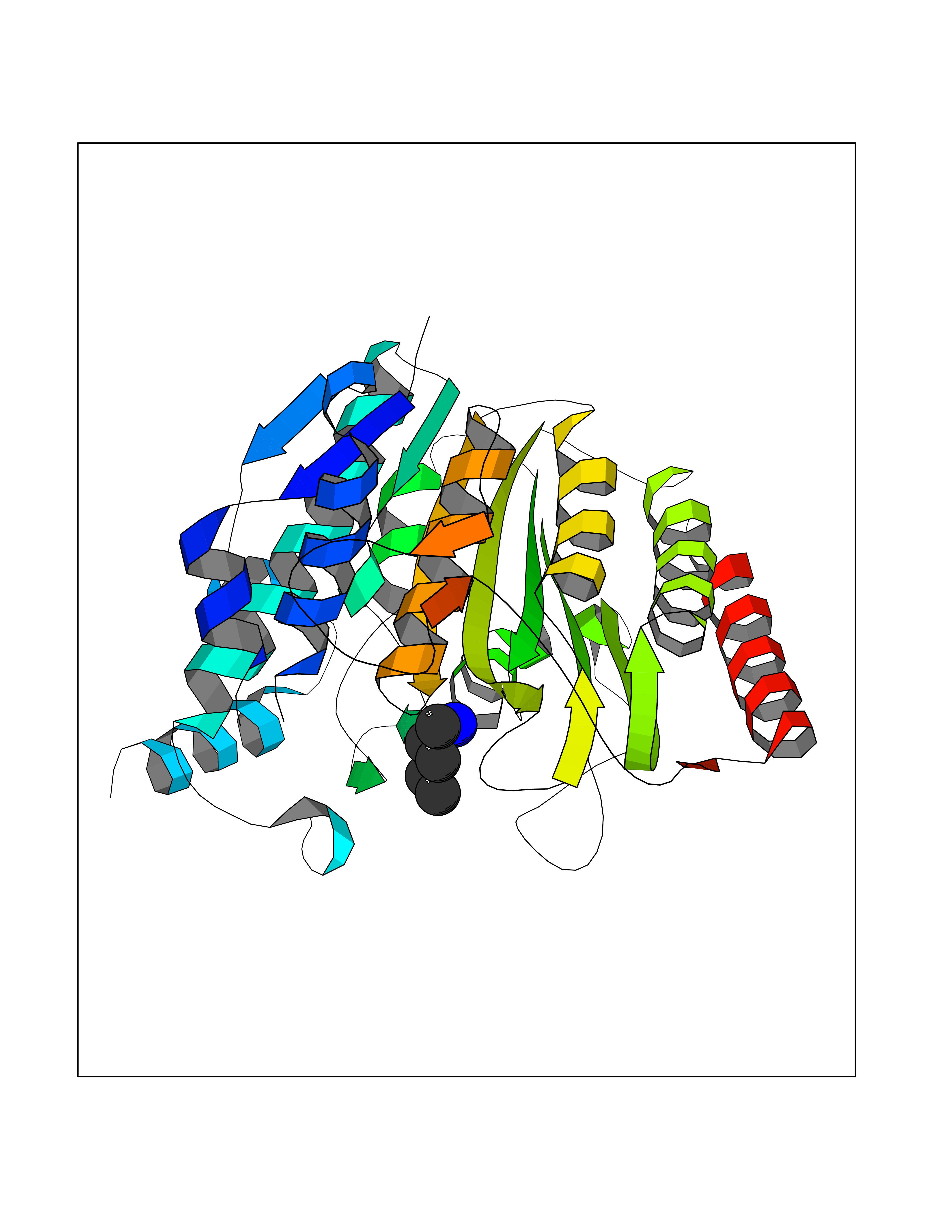 Molscript Map 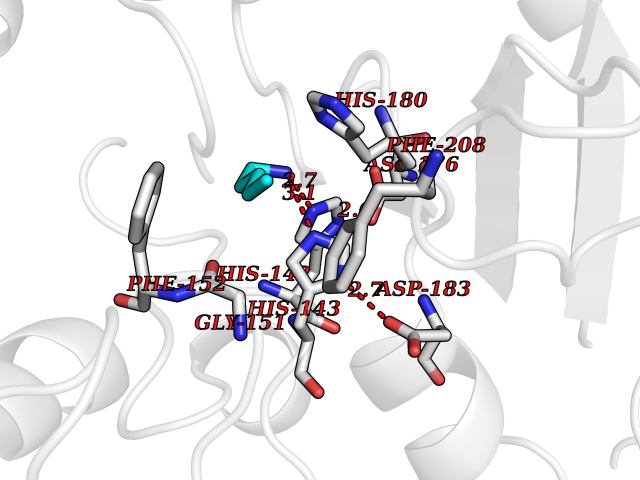 Pymol Map 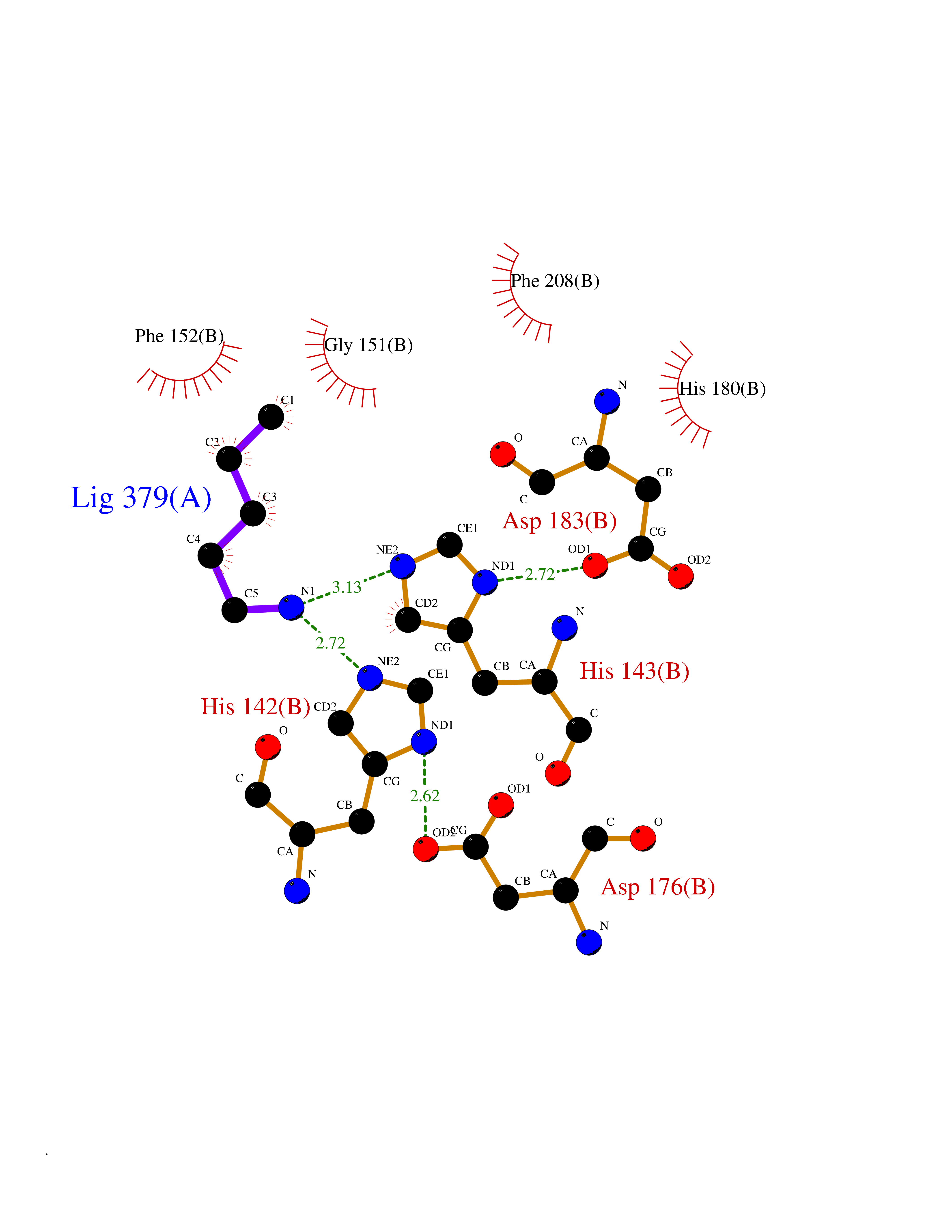 Ligplot Map 3D Binding mode Sequence LVPVYIYSPEYVSMCDSLPKRAEMVHSLIEAYALHKQMRIVKPKVASMEEMATFHTDAYLQHLQKVSQEYGLGYDCPATEGIFDYAAAIGGATITAAQCLIDGMCKVAINWSGGWHHAKKDEASGFCYLNDAVLGILRLRRKFERILYVDLDLHHGDGVEDAFSFTSKVMTVSLHKFSPGFFPGTGDVSDVGLGKGRYYSVNVPIQDGIQDEKYYQICESVLKEVYQAFNPKAVVLQLGADTIAGDPMCSFNMTPVGIGKCLKYILQWQLATLILGGGGYNLANTARCWTYLTGVILGKTLSSEIPDHEFFTAYGPDYVLEITPSCRPDRNEPHRIQQILNYIKGNLKHVV Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Calcium-binding mitochondrial carrier protein Aralar2 | 4P5W | 4.74 | |
Target general information Gen name SLC25A13 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Mitochondrial carrier (TC 2.A.29) family Biochemical class Transport protein Function Acidic amino acid transmembrane transporter activity.Calcium ion binding.L-aspartate transmembrane transporter activity.L-glutamate transmembrane transporter activity.Transporter activity. Related diseases Citrullinemia 2 (CTLN2) [MIM:603471]: A form of citrullinemia, an autosomal recessive disease characterized primarily by elevated serum and urine citrulline levels. Ammonia intoxication is another manifestation. Citrullinemia type 2 is characterized by neuropsychiatric symptoms including abnormal behaviors, loss of memory, seizures and coma. Death can result from brain edema. Onset is sudden and usually between the ages of 20 and 50 years. {ECO:0000269|PubMed:10369257, ECO:0000269|PubMed:10610724}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Cholestasis, neonatal intrahepatic, caused by citrin deficiency (NICCD) [MIM:605814]: A form of citrullinemia type 2 with neonatal onset, characterized by suppression of the bile flow, hepatic fibrosis, low birth weight, growth retardation, hypoproteinemia, variable liver dysfunction. Neonatal intrahepatic cholestasis due to citrin deficiency is generally not severe and symptoms disappear by one year of age with an appropriate diet. Years or even decades later, however, some individuals develop the characteristic features of citrullinemia type 2 with neuropsychiatric symptoms. {ECO:0000269|PubMed:11793471}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00128 Interacts with O75746 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Calcium; Disease variant; Intrahepatic cholestasis; Membrane; Metal-binding; Methylation; Mitochondrion; Mitochondrion inner membrane; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34724.3 Length 306 Aromaticity 0.12 Instability index 28.83 Isoelectric point 5.55 Charge (pH=7) -7.5 2D Binding mode Binding energy (Kcal/mol) -6.46 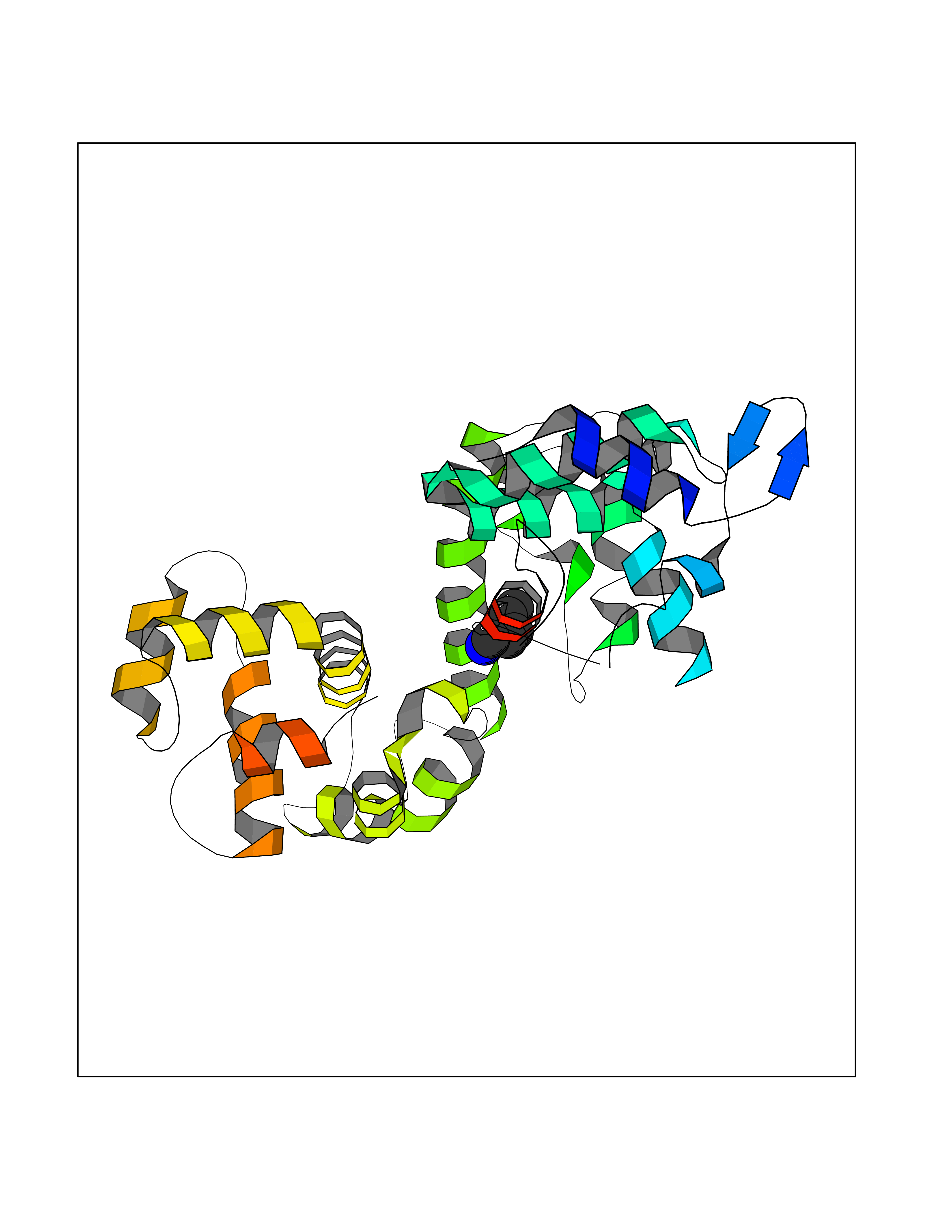 Molscript Map 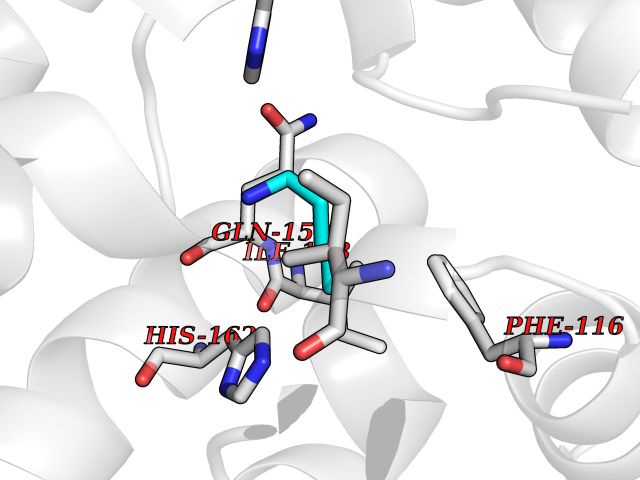 Pymol Map 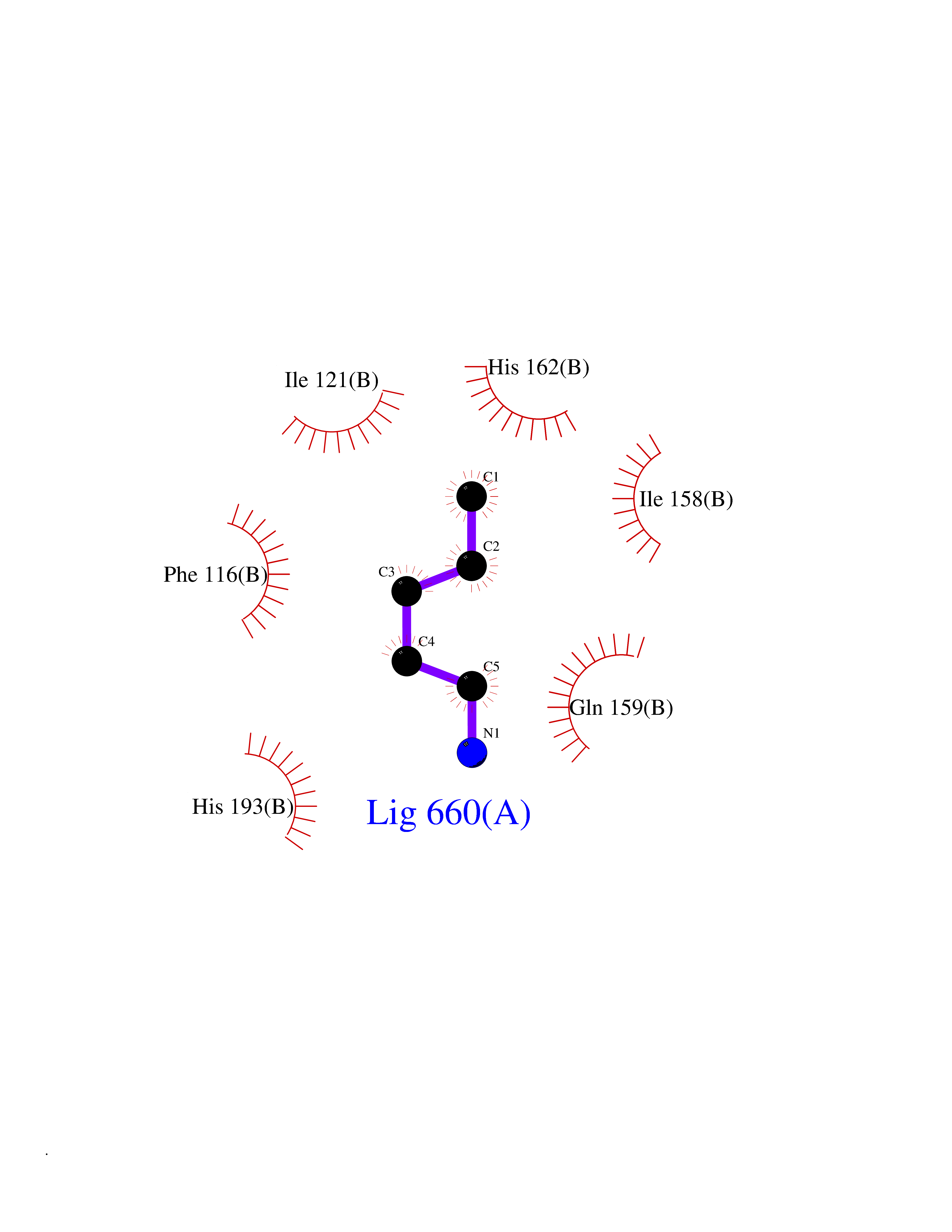 Ligplot Map 3D Binding mode Sequence RADPAELRTIFLKYASIEKNGEFFMSPNDFVTRYLNINPKTVELLSGVVDQTKDGLISFQEFVAFESVLCAPDALFMVAFQLFDKAGKGEVTFEDVKQVFGQTTIHQHIPFNWDSEFVQLHFGKERKRHLTYAEFTQFLLEIQLEHAKQAFVQRDNARTGRVTAIDFRDIMVTIRPHVLTPFVEECLVAAAGGTTSHQVSFSYFNGFNSLLNNMELIRKIYSTLAGTRKDVEVTKEEFVLAAQKFGQVTPMEVDILFQLADLYEPRGRMTLADIERIAPPNPDHVGGYKLAVATFAGIENKFGLYL Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Caterpiller protein 1.1 (NLRP3) | 7PZC | 4.74 | |
Target general information Gen name NLRP3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PYRIN-containing APAF1-like protein 1; PYPAF1; NALP3; NACHT, LRR and PYD domains-containing protein 3; Cryopyrin; Cold-induced autoinflammatory syndrome 1 protein; CLR1.1; CIAS1; C1orf7; Angiotensin/v Protein family NLRP family Biochemical class NA Function In response to pathogens and other damage-associated signals, initiates the formation of the inflammasome polymeric complex, made of NLRP3, PYCARD and CASP1 (and possibly CASP4 and CASP5). Recruitment of proCASP1 to the inflammasome promotes its activation and CASP1-catalyzed IL1B and IL18 maturation and secretion in the extracellular milieu. Activation of NLRP3 inflammasome is also required for HMGB1 secretion. The active cytokines and HMGB1 stimulate inflammatory responses. Inflammasomes can also induce pyroptosis, an inflammatory form of programmed cell death. Under resting conditions, NLRP3 is autoinhibited. NLRP3 activation stimuli include extracellular ATP, reactive oxygen species, K(+) efflux, crystals of monosodium urate or cholesterol, amyloid-beta fibers, environmental or industrial particles and nanoparticles, cytosolic dsRNA, etc. However, it is unclear what constitutes the direct NLRP3 activator. Activation in presence of cytosolic dsRNA is mediated by DHX33. Independently of inflammasome activation, regulates the differentiation of T helper 2 (Th2) cells and has a role in Th2 cell-dependent asthma and tumor growth. During Th2 differentiation, required for optimal IRF4 binding to IL4 promoter and for IRF4-dependent IL4 transcription. Binds to the consensus DNA sequence 5'-GRRGGNRGAG-3'. May also participate in the transcription of IL5, IL13, GATA3, CCR3, CCR4 and MAF. As the sensor component of the NLRP3 inflammasome, plays a crucial role in innate immunity and inflammation. Related diseases Familial cold autoinflammatory syndrome 1 (FCAS1) [MIM:120100]: A rare autosomal dominant systemic inflammatory disease characterized by recurrent episodes of maculopapular rash associated with arthralgias, myalgias, fever and chills, swelling of the extremities, and conjunctivitis after generalized exposure to cold. Rarely, some patients may also develop late-onset renal amyloidosis. {ECO:0000269|PubMed:11687797, ECO:0000269|PubMed:11992256, ECO:0000269|PubMed:12355493, ECO:0000269|PubMed:12522564, ECO:0000269|PubMed:15593220, ECO:0000269|PubMed:17284928, ECO:0000269|PubMed:24952504}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Muckle-Wells syndrome (MWS) [MIM:191900]: A hereditary periodic fever syndrome characterized by fever, chronic recurrent urticaria, arthralgias, progressive sensorineural deafness, and reactive renal amyloidosis. The disease may be severe if generalized reactive amyloidosis occurs. {ECO:0000269|PubMed:11687797, ECO:0000269|PubMed:11992256, ECO:0000269|PubMed:12355493, ECO:0000269|PubMed:15593220, ECO:0000269|PubMed:24952504}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Chronic infantile neurologic cutaneous and articular syndrome (CINCA) [MIM:607115]: Rare congenital inflammatory disorder characterized by a triad of neonatal onset of cutaneous symptoms, chronic meningitis, and joint manifestations with recurrent fever and inflammation. {ECO:0000269|PubMed:12032915, ECO:0000269|PubMed:12483741, ECO:0000269|PubMed:14630794, ECO:0000269|PubMed:15231984, ECO:0000269|PubMed:15334500, ECO:0000269|PubMed:15593220, ECO:0000269|PubMed:24952504, ECO:0000269|PubMed:31086329}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Keratoendothelitis fugax hereditaria (KEFH) [MIM:148200]: An autosomal dominant corneal disease that periodically, and fleetingly, affects the corneal endothelium, stroma, and vision, eventually leading to central corneal stromal opacities in some patients. The disease is characterized by unilateral attacks of ocular pain, pericorneal injection, and photophobia. The acute symptoms vanish in 1-2 days but vision remains blurry for several weeks. The attacks start at the age of 3-12 years and can affect either eye. They generally decrease in frequency and get milder with age. {ECO:0000269|PubMed:29366613, ECO:0000269|PubMed:35559676}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, autosomal dominant, 34, with or without inflammation (DFNA34) [MIM:617772]: A form of sensorineural hearing loss. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. DFNA34 is a postlingual, slowly progressive form with variable severity and variable additional features. Some DFNA34 patients have autoinflammatory manifestations. {ECO:0000269|PubMed:28847925}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P27797; P36957; P19525; Q7Z434; Q8TDX7; Q96P20; Q9ULZ3; Q80H93; P0DTC9; Q9ES74; Q8TDX7-1 EC number NA Uniprot keywords 3D-structure; Activator; ADP-ribosylation; Alternative splicing; Amyloidosis; ATP-binding; Cytoplasm; Cytoskeleton; Deafness; Disease variant; Disulfide bond; Endoplasmic reticulum; Golgi apparatus; Hydrolase; Immunity; Inflammasome; Inflammatory response; Innate immunity; Isopeptide bond; Leucine-rich repeat; Lipoprotein; Membrane; Mitochondrion; Non-syndromic deafness; Nucleotide-binding; Nucleus; Palmitate; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Secreted; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 115171 Length 1010 Aromaticity 0.08 Instability index 45.92 Isoelectric point 6.17 Charge (pH=7) -11.22 2D Binding mode Binding energy (Kcal/mol) -6.47 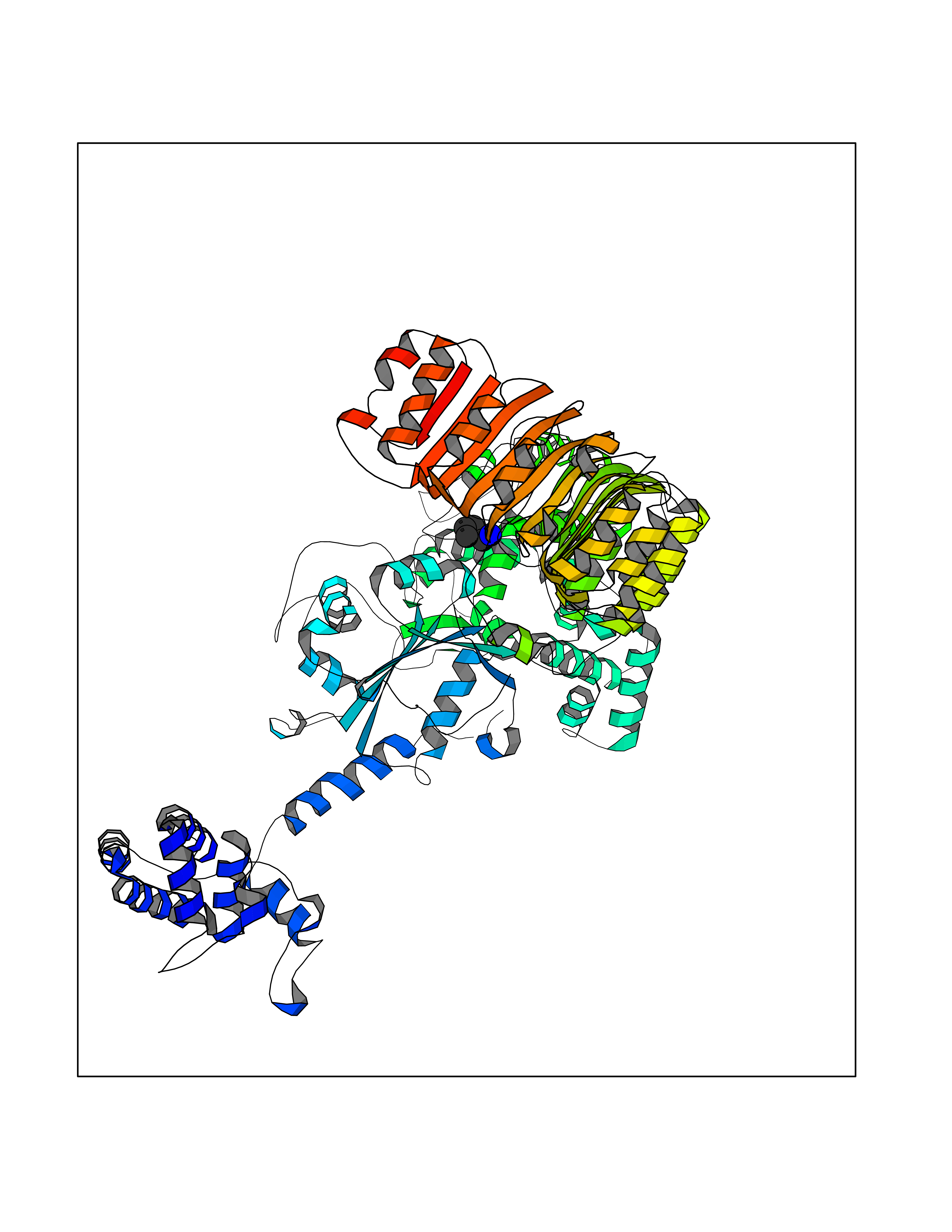 Molscript Map 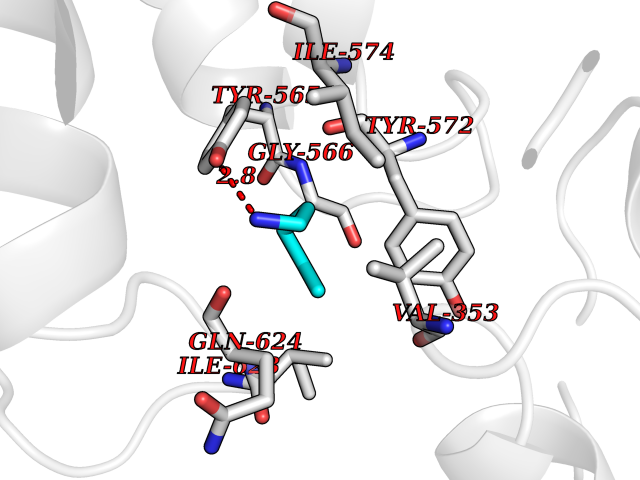 Pymol Map 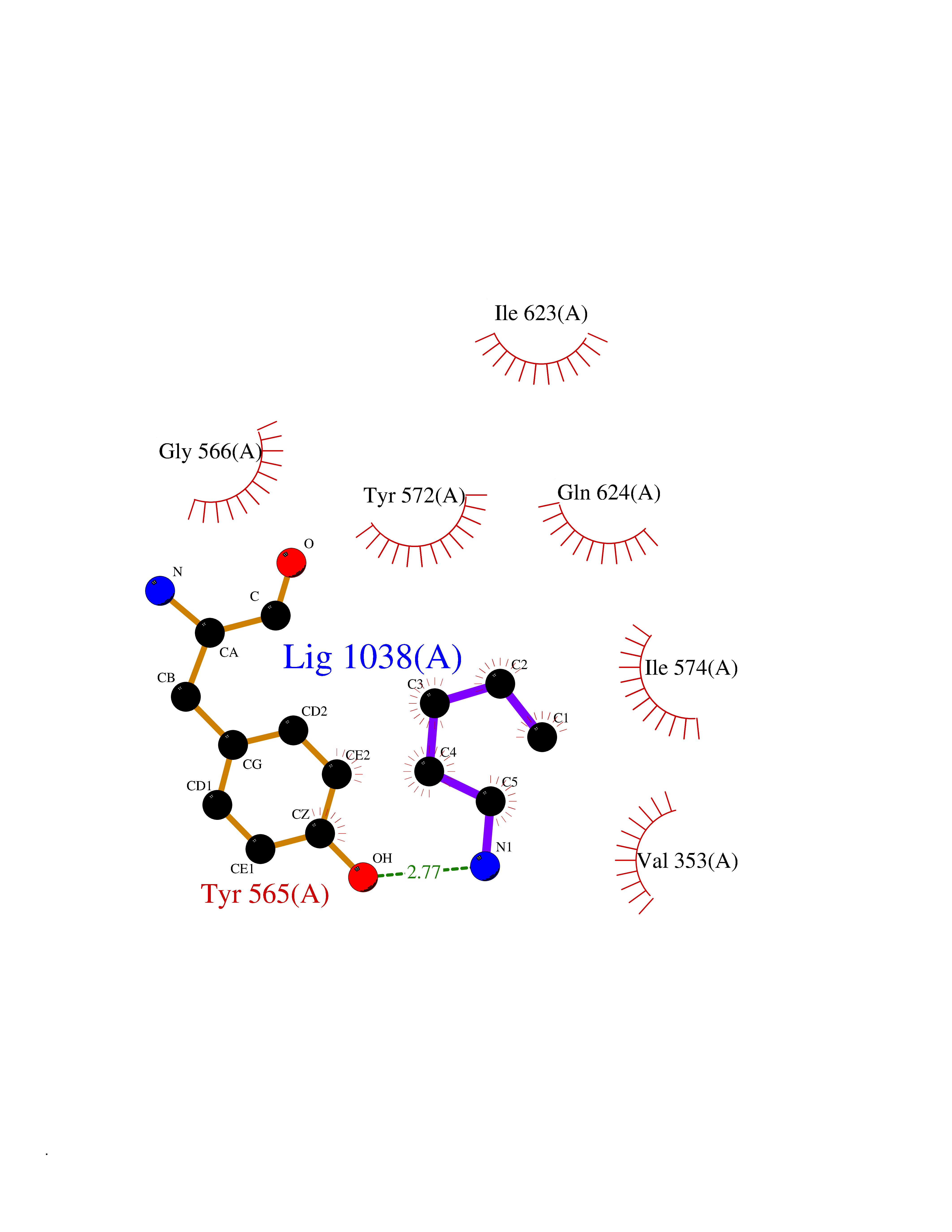 Ligplot Map 3D Binding mode Sequence MASTRCKLARYLEDLEDVDLKKFKMHLEDYPPQKGCIPLPRGQTEKADHVDLATLMIDFNGEEKAWAMAVWIFAAINRRDLYEKAKRDEPKWGSDNARVSNPTVICQEDSIEEEWMGLLEYLSRISICKMKKDYRKKYRKYVRSRFQCIEESVSLNKRYTRLRLIKEHRSQSPVSPIKMELLFDPDDEHSEPVHTVVFQGAAGIGKTILARKMMLDWASGTLYQDRFDYLFYIHCREVSLVTQRSLGDLIMSCCPDPNPPIHKIVRKPSRILFLMDGFDELQGAFDEHIGPLCTDWQKAERGDILLSSLIRKKLLPEASLLITTRPVALEKLQHLLDHPRHVEILGFSEAKRKEYFFKYFSDEAQARAAFSLIQENEVLFTMCFIPLVCWIVCTGLKQQMESGKSLAQTSKTTTAVYVFFLSSLLQPRGGSQEHGLCAHLWGLCSLAADGIWNQKILFEESDLRNHGLQKADVSAFLRMNLFQKEVDCEKFYSFIHMTFQEFFAAMYYLLEEEKEGRTNVPGSRLKLPSRDVTVLLENYGKFEKGYLIFVVRFLFGLVNQERTSYLEKKLSCKISQQIRLELLKWIEVKAKAKKLQIQPSQLELFYCLYEMQEEDFVQRAMDYFPKIEINLSTRMDHMVSSFCIENCHRVESLSLGFLHNMPKEEEEEEKEGRHLDMVQCVLPSSSHAACSHGLVNSHLTSSFCRGLFSVLSTSQSLTELDLSDNSLGDPGMRVLCETLQHPGCNIRRLWLGRCGLSHECCFDISLVLSSNQKLVELDLSDNALGDFGIRLLCVGLKHLLCNLKKLWLVSCCLTSACCQDLASVLSTSHSLTRLYVGENALGDSGVAILCEKAKNPQCNLQKLGLVNSGLTSVCCSALSSVLSTNQNLTHLYLRGNTLGDKGIKLLCEGLLHPDCKLQVLELDNCNLTSHCCWDLSTLLTSSQSLRKLSLGNNDLGDLGVMMFCEVLKQQSCLLQNLGLSEMYFNYETKSALETLQEEKPELTVVFEPSW Hydrogen bonds contact Hydrophobic contact | ||||