Job Results:
Ligand
Structure
Job ID
5c75d0793a5c7fa3ef7fd4eba45db829
Job name
NA
Time
2026-02-27 11:58:42
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Polybromo-1 (PBRM1) | 4Y03 | 5.04 | |
Target general information Gen name PBRM1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hPB1; Protein polybromo-1; Polybromo-1D; PB1; BRG1-associated factor 180; BAF180 Protein family NA Biochemical class NA Function Required for the stability of the SWI/SNF chromatin remodeling complex SWI/SNF-B (PBAF). Acts as a negative regulator of cell proliferation. Involved in transcriptional activation and repression of select genes by chromatin remodeling (alteration of DNA-nucleosome topology). Related diseases Renal cell carcinoma (RCC) [MIM:144700]: Renal cell carcinoma is a heterogeneous group of sporadic or hereditary carcinoma derived from cells of the proximal renal tubular epithelium. It is subclassified into clear cell renal carcinoma (non-papillary carcinoma), papillary renal cell carcinoma, chromophobe renal cell carcinoma, collecting duct carcinoma with medullary carcinoma of the kidney, and unclassified renal cell carcinoma. Clear cell renal cell carcinoma is the most common subtype. {ECO:0000269|PubMed:21248752}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q68CP9; O95696-1; O95696-2; Q9P2D1; P51532; P04608 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Bromodomain; Chromatin regulator; DNA-binding; Isopeptide bond; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Tumor suppressor; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 13709.7 Length 115 Aromaticity 0.1 Instability index 57.29 Isoelectric point 6.71 Charge (pH=7) -0.35 2D Binding mode Binding energy (Kcal/mol) -6.87 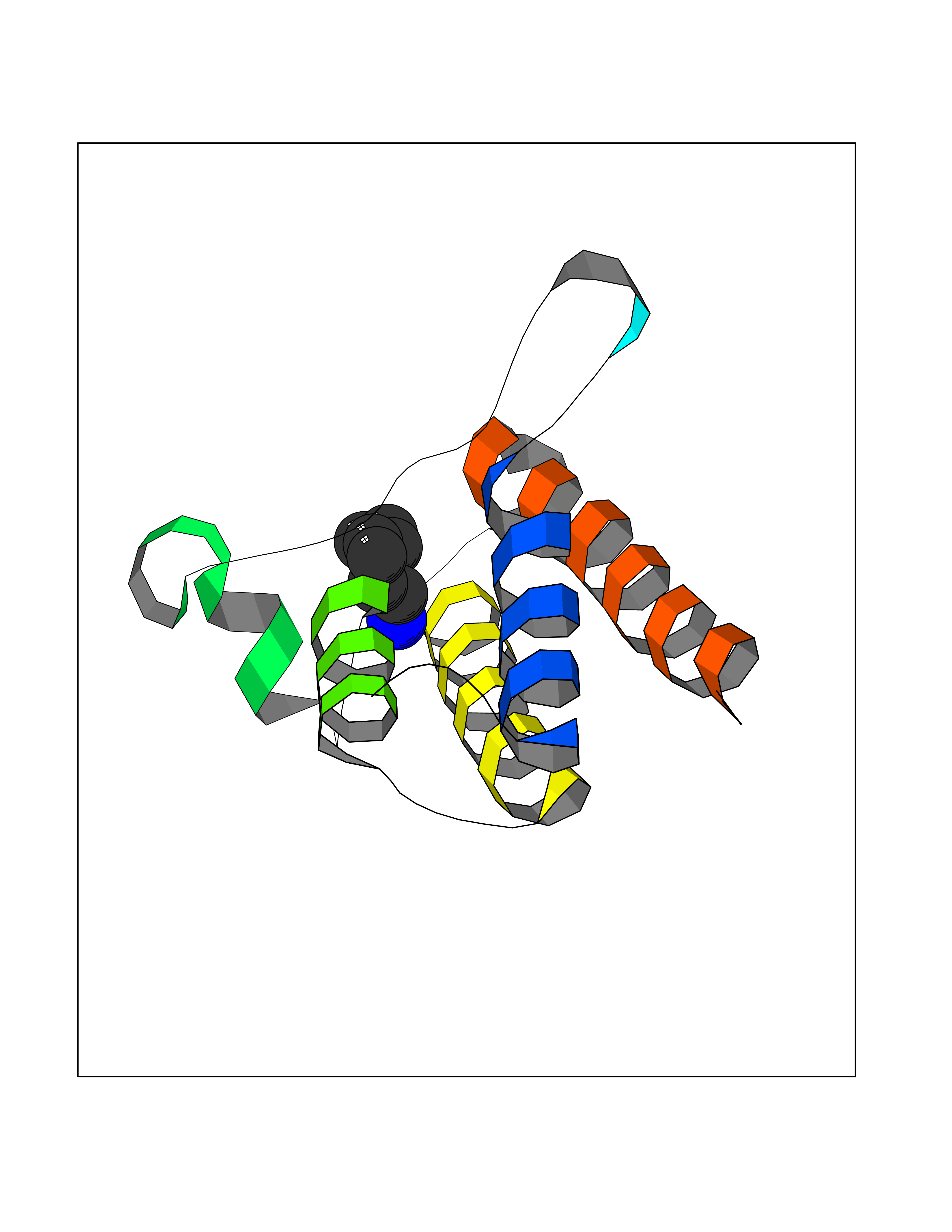 Molscript Map 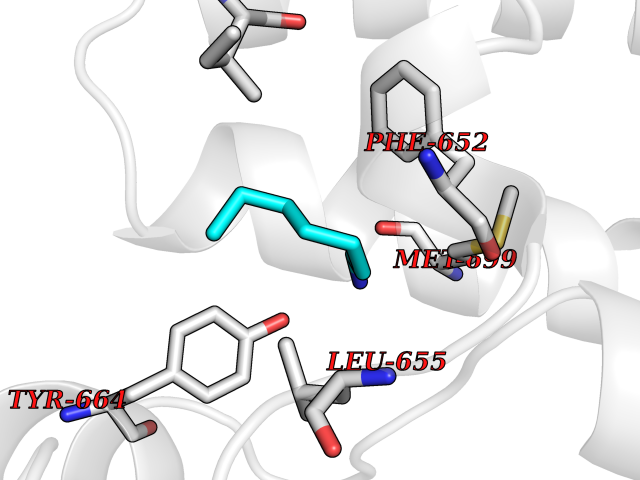 Pymol Map 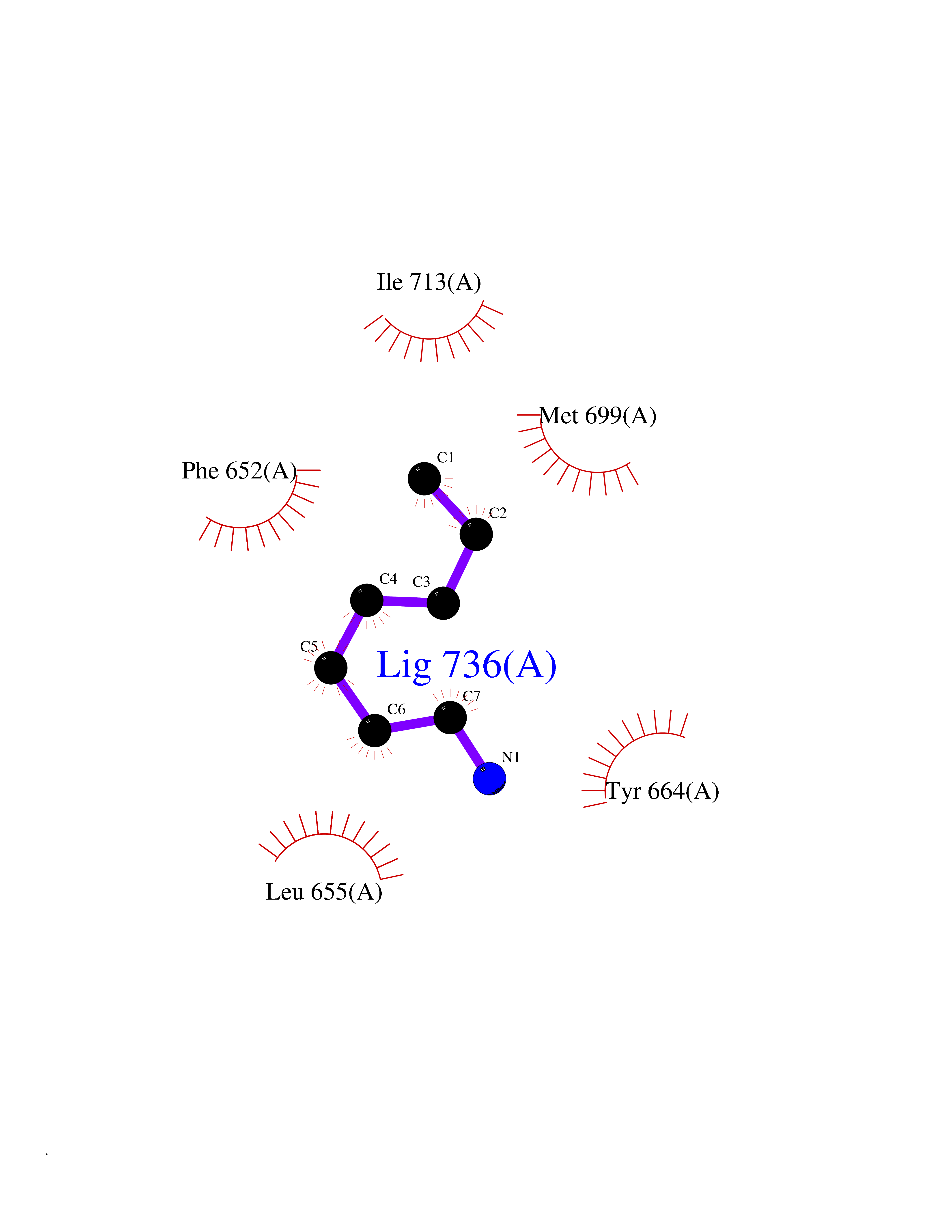 Ligplot Map 3D Binding mode Sequence SKYMTPMQQKLNEVYEAVKNYTDKRGRRLSAIFLRLPSRSELPDYYLTIKKPMDMEKIRSHMMANKYQDIDSMVEDFVMMFNNACTYNEPESLIYKDALVLHKVLLETRRDLEGD Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Histone-lysine N-methyltransferase SMYD3 (SMYD3) | 6P7Z | 5.04 | |
Target general information Gen name SMYD3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SET and MYND domain-containing protein 3; Zinc finger MYND domain-containing protein 1 Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family Biochemical class NA Function Histone methyltransferase. Specifically methylates 'Lys-4' of histone H3, inducing di- and tri-methylation, but not monomethylation . Also methylates 'Lys-5' of histone H4. Plays an important role in transcriptional activation as a member of an RNA polymerase complex. Binds DNA containing 5'-CCCTCC-3' or 5'-GAGGGG-3' sequences. Related diseases Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:17332249}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 6 (NS6) [MIM:613224]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:19966803}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: RAS-associated autoimmune leukoproliferative disorder (RALD) [MIM:614470]: A disorder of apoptosis, characterized by chronic accumulation of non-malignant lymphocytes, defective lymphocyte apoptosis, and an increased risk for the development of hematologic malignancies. {ECO:0000269|PubMed:17517660}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanocytic nevus syndrome, congenital (CMNS) [MIM:137550]: A syndrome characterized by congenital pigmentary skin lesions which can occur at any site and can cover most of the body surface. These lesions may or may not be hairy. Congenital melanocytic nevi are associated with neuromelanosis (the presence of melanin-producing cells within the brain parenchyma or leptomeninges). Less commonly they are associated with malignant melanoma in childhood, both in the skin and the central nervous system. CMNS patients also tend to have a characteristic facial appearance, including wide or prominent forehead, periorbital fullness, small short nose with narrow nasal bridge, round face, full cheeks, prominent premaxilla, and everted lower lip. {ECO:0000269|PubMed:18633438, ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanosis, neurocutaneous (NCMS) [MIM:249400]: A rare congenital disease characterized by the presence of giant or multiple melanocytic nevi on the skin, foci of melanin-producing cells within the brain parenchyma, and infiltration of leptomeninges by abnormal melanin deposits. Neurologic abnormalities include seizures, hydrocephalus, arachnoid cysts, tumors, and syringomyelia. Some patients may develop malignant melanoma. {ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Keratinocytic non-epidermolytic nevus (KNEN) [MIM:162900]: Epidermal nevi of the common, non-organoid and non-epidermolytic type are benign skin lesions and may vary in their extent from a single (usually linear) lesion to widespread and systematized involvement. They may be present at birth or develop early during childhood. {ECO:0000269|PubMed:22499344}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid cancer, non-medullary, 2 (NMTC2) [MIM:188470]: A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms. {ECO:0000269|PubMed:12727991}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9H0L4; Q9H0I2; Q13064; Q7Z3B4; Q16512; Q92529; Q15915; Q9Y2U5 EC number EC 2.1.1.354 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Chromatin regulator; Cytoplasm; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 48694.1 Length 425 Aromaticity 0.08 Instability index 44.91 Isoelectric point 6.86 Charge (pH=7) -0.4 2D Binding mode Binding energy (Kcal/mol) -6.87  Molscript Map 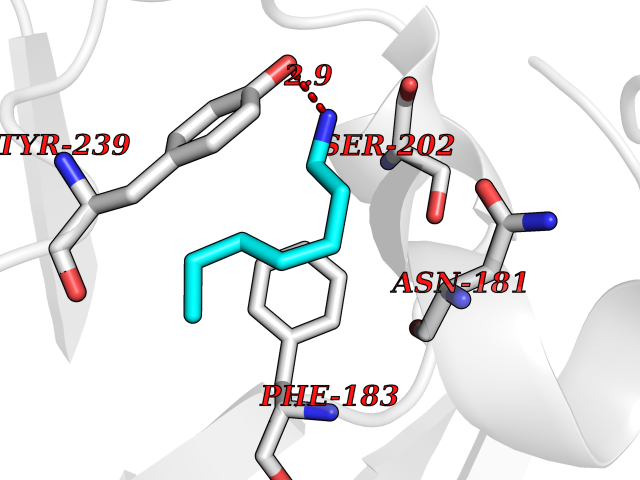 Pymol Map 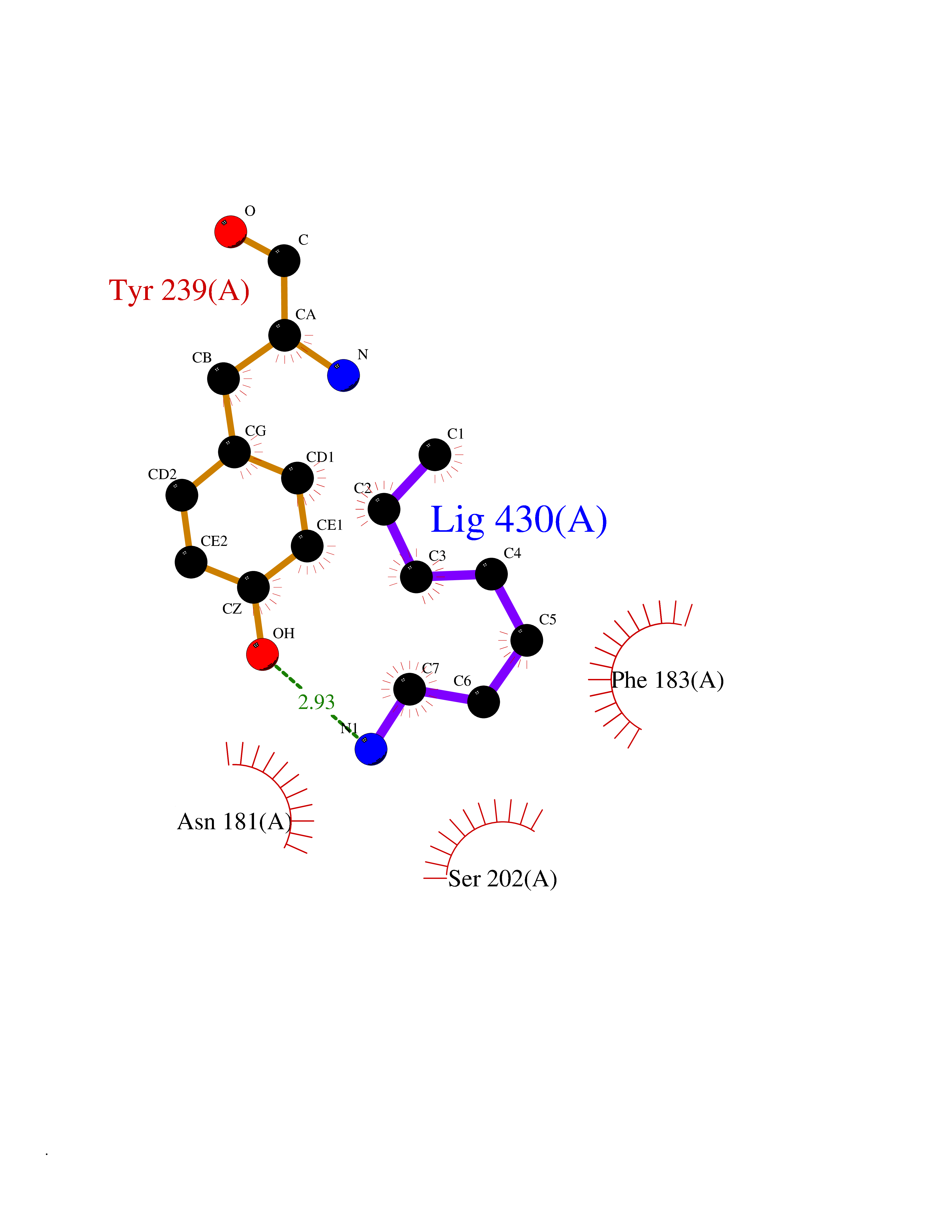 Ligplot Map 3D Binding mode Sequence PLKVEKFATANRGNGLRAVTPLRPGELLFRSDPLAYTVCKGSRGVVCDRCLLGKEKLMRCSQCRVAKYCSAKCQKKAWPDHKRECKCLKSCPRYPPDSVRLLGRVVFKLMDGAPSESEKLYSFYDLESNINKLTEDKKEGLRQLVMTFQHFMREEIQDASQLPPAFDLFEAFAKVICNSFTICNAEMQEVGVGLYPSISLLNHSCDPNCSIVFNGPHLLLRAVRDIEVGEELTICYLDMLMTSEERRKQLRDQYCFECDCFRCQTQDKDADMLTGDEQVWKEVQESLKKIEELKAHWKWEQVLAMCQAIISSNSERLPDINIYQLKVLDCAMDACINLGLLEEALFYGTRTMEPYRIFFPGSHPVRGVQVMKVGKLQLHQGMFPQAMKNLRLAFDIMRVTHGREHSLIEDLILLLEECDANIRAS Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Caspase-7 (CASP7) | 1SHJ | 5.04 | |
Target general information Gen name CASP7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MCH3; ICE-like apoptotic protease 3; ICE-LAP3; CMH-1; CASP-7; Apoptotic protease Mch-3 Protein family Peptidase C14A family Biochemical class Peptidase Function Cleaves and activates sterol regulatory element binding proteins (SREBPs). Proteolytically cleaves poly(ADP-ribose) polymerase (PARP) at a '216-Asp-|-Gly-217' bond. Overexpression promotes programmed cell death. Involved in the activation cascade of caspases responsible for apoptosis execution. Related diseases Pregnancy loss, recurrent, 3 (RPRGL3) [MIM:614391]: A common complication of pregnancy, resulting in spontaneous abortion before the fetus has reached viability. The term includes all miscarriages from the time of conception until 24 weeks of gestation. Recurrent pregnancy loss is defined as 3 or more consecutive spontaneous abortions. {ECO:0000269|PubMed:17339269}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05408; DB03384; DB06255 Interacts with Q13490; P83105; P42858; Q8N4N3-2; P43364; Q16236; Q9GZT8; Q13177; P27986-2; P21673; Q86WV1-2; P17405; P98170 EC number EC 3.4.22.60 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; Apoptosis; Cytoplasm; Hydrolase; Nucleus; Phosphoprotein; Protease; Proteomics identification; Reference proteome; RNA-binding; Secreted; Thiol protease; Ubl conjugation; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 47441.5 Length 417 Aromaticity 0.11 Instability index 20.98 Isoelectric point 8.38 Charge (pH=7) 6.12 2D Binding mode Binding energy (Kcal/mol) -6.88  Molscript Map 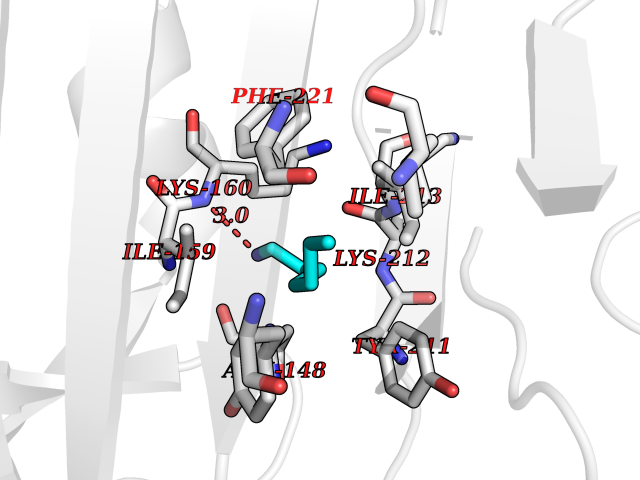 Pymol Map 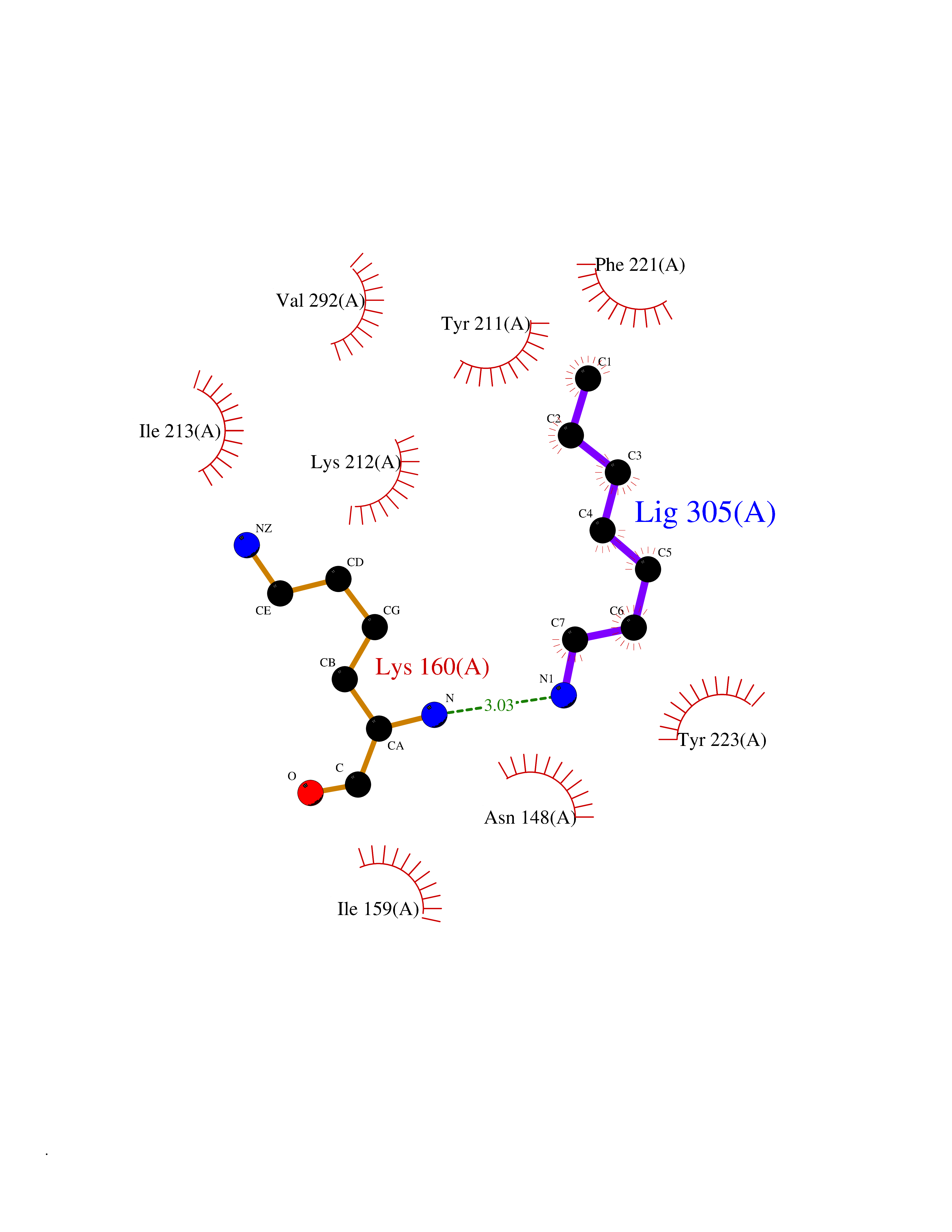 Ligplot Map 3D Binding mode Sequence TYQYNMNFEKLGKCIIINNKNFDKVTGMGVRNGTDKDAEALFKCFRSLGFDVIVYNDCSCAKMQDLLKKASEEDHTNAACFACILLSHGEENVIYGKDGVTPIKDLTAHFRGARCKTLLEKPKLFFIQACRGTEPRYKIPVEADFLFAYSTVRGSWFVQALCSILEEHGKDLEIMQILTRVNDRVARHFKKQIPCVVSMLTKELYFSQVPTYQYNMNFEKLGKCIIINNKNFDKVTGMGVRNGTDKDAEALFKCFRSLGFDVIVYNDCSCAKMQDLLKKASEEDHTNAACFACILLSHGEENVIYGKDGVTPIKDLTAHFRGARCKTLLEKPKLFFIQACRGPRYKIPVEADFLFAYSTVPGSWFVQALCSILEEHGKDLEIMQILTRVNDRVARHFESKQIPCVVSMLTKELYFSQ Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Peroxiredoxin-4 (PRDX4) | 4RQX | 5.04 | |
Target general information Gen name PRDX4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Thioredoxindependent peroxide reductase A0372; Thioredoxin peroxidase AO372; PrxIV; Peroxiredoxin4; Peroxiredoxin IV; PRDX4; Antioxidant enzyme AOE372; AOE372 Protein family Peroxiredoxin family, AhpC/Prx1 subfamily Biochemical class Peroxidases Function Probably involved in redox regulation of the cell. Regulates the activation of NF-kappa-B in the cytosol by a modulation of I-kappa-B-alpha phosphorylation. Related diseases May be involved in T-cell exhaustion associated with chronic viral infections such as with human immunodeficiency virus (HIV) and hepatitic C virus (HCV). {ECO:0000269|PubMed:19001139, ECO:0000269|PubMed:19587053}.; DISEASE: T-cell lymphoma, subcutaneous panniculitis-like (SPTCL) [MIM:618398]: An uncommon form of T-cell non-Hodgkin lymphoma, in which cytotoxic CD8+ T-cells infiltrate subcutaneous adipose tissue, and rimming adipocytes in a lace-like pattern. Affected individuals typically present with multiple subcutaneous nodules, systemic B-cell symptoms, and, in a subset of cases, autoimmune disorders, most commonly systemic lupus erythematosus. A subset of patients develop hemophagocytic lymphohistiocytosis. SPTCL transmission pattern is consistent with autosomal recessive inheritance with incomplete penetrance. {ECO:0000269|PubMed:30374066, ECO:0000269|PubMed:30792187, ECO:0000269|Ref.2}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P54253; P18428; P07237; P30101; Q15084; Q06830; P21731; Q8NBS9 EC number EC 1.11.1.15 Uniprot keywords 3D-structure; Antioxidant; Cytoplasm; Direct protein sequencing; Disulfide bond; Endoplasmic reticulum; Oxidoreductase; Peroxidase; Proteomics identification; Redox-active center; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 18846.2 Length 166 Aromaticity 0.12 Instability index 30.43 Isoelectric point 5.54 Charge (pH=7) -3.89 2D Binding mode Binding energy (Kcal/mol) -6.88  Molscript Map 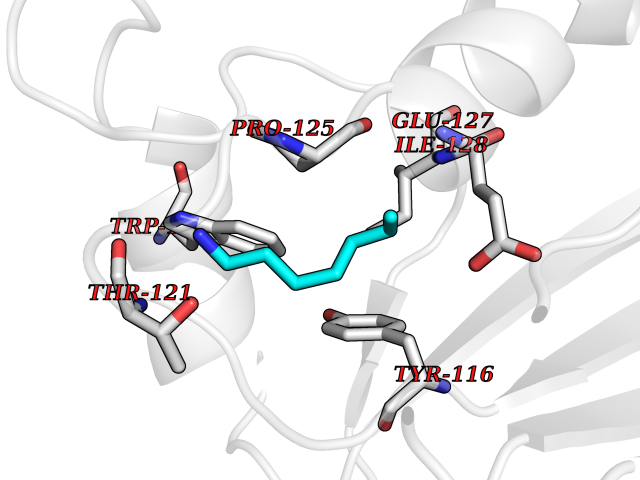 Pymol Map 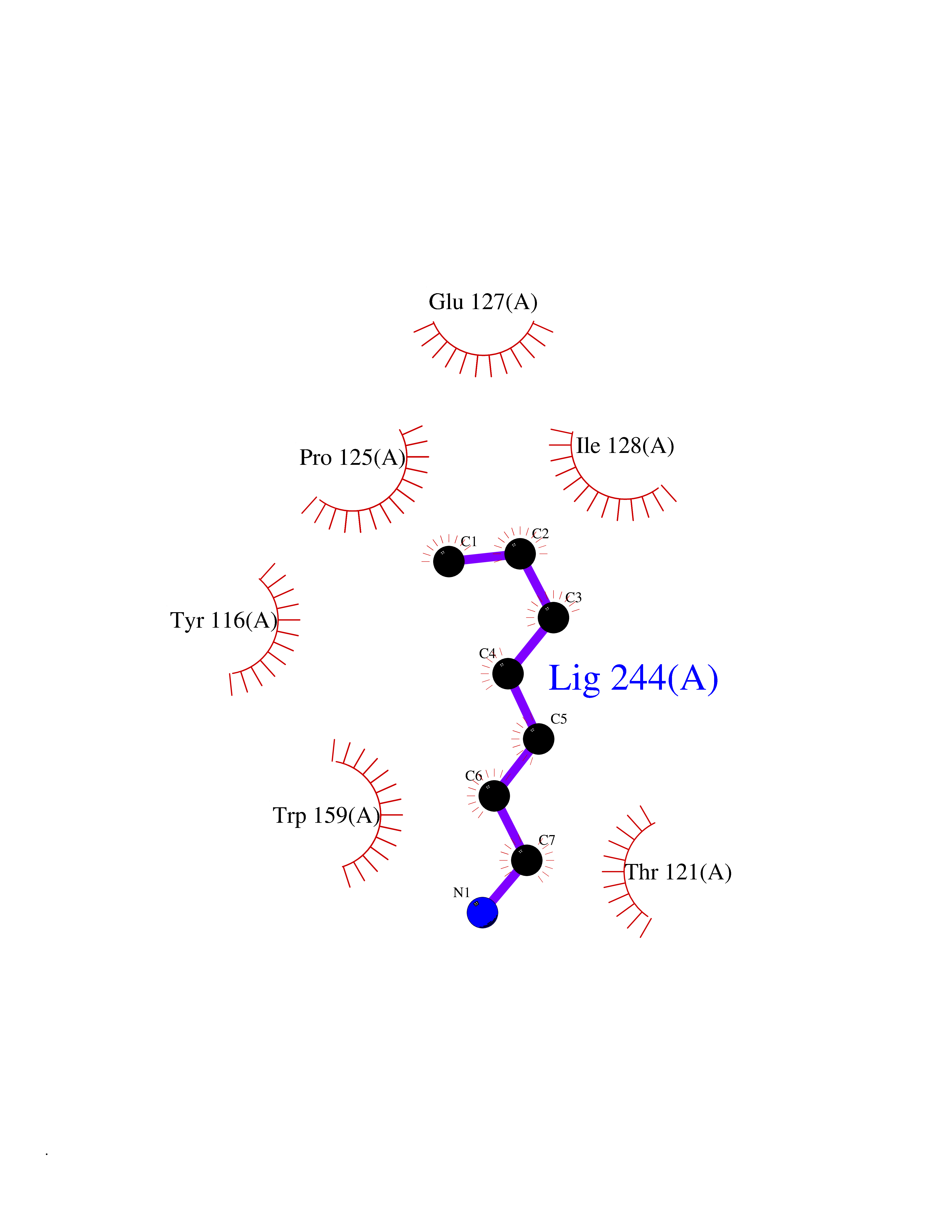 Ligplot Map 3D Binding mode Sequence GTAKISKPAPYWEGTAVIDGEFKELKLTDYRGKYLVFFFYPLDFTFVCPTEIIAFGDRLEEFRSINTEVVACSVDSQFTHLAWINTPRRQGGLGPIRIPLLSDLTHQISKDYGVYLEDSGHTLRGLFIIDDKGILRQITLNDLPVGRSVDETLRLVQAFQYTDKHG Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Cytochrome P450 1B1 (CYP1B1) | 3PM0 | 5.04 | |
Target general information Gen name CYP1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CYPIB1 Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function In liver microsomes, this enzyme is involved in an NADPH-dependent electron transport pathway. It oxidizes a variety of structurally unrelated compounds, including steroids, fatty acids, retinoid and xenobiotics. Preferentially oxidizes 17beta-estradiol to the carcinogenic 4-hydroxy derivative, and a variety of procarcinogenic compounds to their activated forms, including polycyclic aromatic hydrocarbons. Promotes angiogenesis by removing cellular oxygenation products, thereby decreasing oxidative stress, release of antiangiogenic factor THBS2, then allowing endothelial cells migration, cell adhesion and capillary morphogenesis. These changes are concommitant with the endothelial nitric oxide synthase activity and nitric oxide synthesis. Plays an important role in the regulation of perivascular cell proliferation, migration, and survival through modulation of the intracellular oxidative state and NF-kappa-B expression and/or activity, during angiogenesis. Contributes to oxidative homeostasis and ultrastructural organization and function of trabecular meshwork tissue through modulation of POSTN expression. Cytochromes P450 are a group of heme-thiolate monooxygenases. Related diseases Anterior segment dysgenesis 6 (ASGD6) [MIM:617315]: A form of anterior segment dysgenesis, a group of defects affecting anterior structures of the eye including cornea, iris, lens, trabecular meshwork, and Schlemm canal. Anterior segment dysgeneses result from abnormal migration or differentiation of the neural crest derived mesenchymal cells that give rise to components of the anterior chamber during eye development. Different anterior segment anomalies may exist alone or in combination, including iris hypoplasia, enlarged or reduced corneal diameter, corneal vascularization and opacity, posterior embryotoxon, corectopia, polycoria, abnormal iridocorneal angle, ectopia lentis, and anterior synechiae between the iris and posterior corneal surface. Clinical conditions falling within the phenotypic spectrum of anterior segment dysgeneses include aniridia, Axenfeld anomaly, Reiger anomaly/syndrome, Peters anomaly, and iridogoniodysgenesis. ASGD6 patients predominantly manifest Peters anomaly. Peters anomaly consists of corneal leukoma, defects in the posterior structures of the cornea such as absence of the posterior corneal stroma and Descemet membrane, and a variable degree of iridocorneal and/or keratolenticular adhesions. Over 50% of patients develop glaucoma in childhood. {ECO:0000269|PubMed:11403040}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glaucoma 3, primary congenital, A (GLC3A) [MIM:231300]: An autosomal recessive form of primary congenital glaucoma (PCG). PCG is characterized by marked increase of intraocular pressure at birth or early childhood, large ocular globes (buphthalmos) and corneal edema. It results from developmental defects of the trabecular meshwork and anterior chamber angle of the eye that prevent adequate drainage of aqueous humor. {ECO:0000269|PubMed:10227395, ECO:0000269|PubMed:10655546, ECO:0000269|PubMed:11184479, ECO:0000269|PubMed:11527932, ECO:0000269|PubMed:11774072, ECO:0000269|PubMed:11980847, ECO:0000269|PubMed:12036985, ECO:0000269|PubMed:12525557, ECO:0000269|PubMed:14635112, ECO:0000269|PubMed:14640114, ECO:0000269|PubMed:15255109, ECO:0000269|PubMed:15342693, ECO:0000269|PubMed:15475877, ECO:0000269|PubMed:16490498, ECO:0000269|PubMed:16688110, ECO:0000269|PubMed:16735994, ECO:0000269|PubMed:16862072, ECO:0000269|PubMed:18470941, ECO:0000269|PubMed:9463332, ECO:0000269|PubMed:9497261}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Glaucoma 1, open angle, A (GLC1A) [MIM:137750]: A form of primary open angle glaucoma (POAG). POAG is characterized by a specific pattern of optic nerve and visual field defects. The angle of the anterior chamber of the eye is open, and usually the intraocular pressure is increased. However, glaucoma can occur at any intraocular pressure. The disease is generally asymptomatic until the late stages, by which time significant and irreversible optic nerve damage has already taken place. {ECO:0000269|PubMed:11774072}. The gene represented in this entry acts as a disease modifier. Digenic mutations in CYP1B1 and MYOC have been found in a family segregating both primary adult-onset and juvenile forms of open angle glaucoma (PubMed:11774072). All affected family members with mutations in both MYOC and CYP1B1 had juvenile glaucoma, whereas those with only the MYOC mutation had the adult-onset form (PubMed:11774072). {ECO:0000269|PubMed:11774072}. Drugs (DrugBank ID) DB02342; DB00613; DB06732; DB00443; DB00121; DB01222; DB00201; DB09061; DB14737; DB01254; DB00694; DB01248; DB00997; DB00470; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB07776; DB00499; DB01645; DB01381; DB00741; DB01064; DB01026; DB00448; DB14009; DB01065; DB00170; DB00959; DB01204; DB14011; DB03467; DB00338; DB01229; DB14631; DB00635; DB01087; DB00396; DB00818; DB04216; DB02709; DB00675; DB00624; DB13946; DB00277; DB12245; DB11155 Interacts with Q02763 EC number EC 1.14.14.- Uniprot keywords 3D-structure; Disease variant; Endoplasmic reticulum; Fatty acid metabolism; Glaucoma; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Mitochondrion; Monooxygenase; Oxidoreductase; Peters anomaly; Proteomics identification; Reference proteome; Steroid metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 51875.9 Length 459 Aromaticity 0.1 Instability index 34.16 Isoelectric point 8.64 Charge (pH=7) 4.89 2D Binding mode Binding energy (Kcal/mol) -6.88 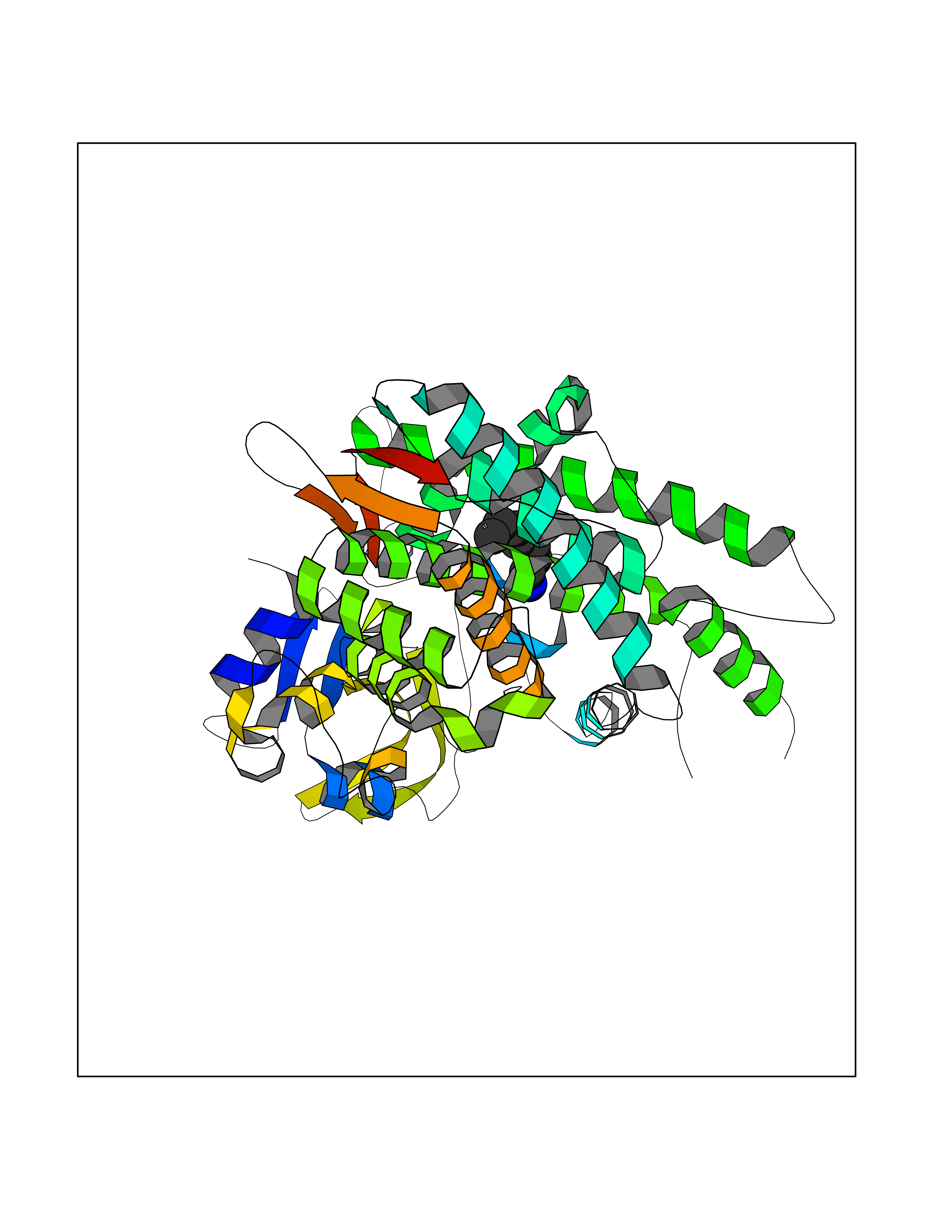 Molscript Map 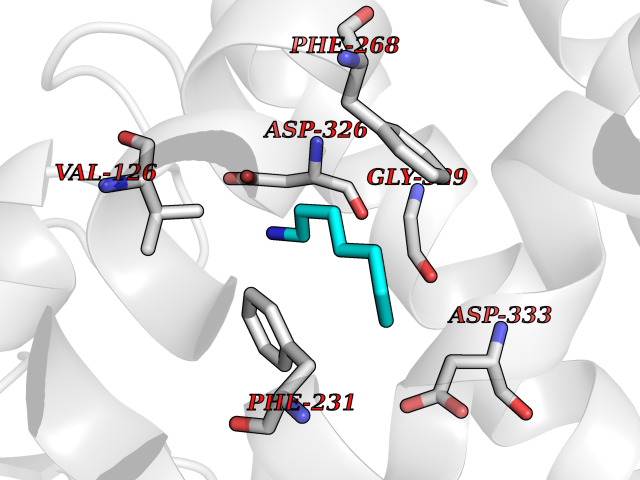 Pymol Map 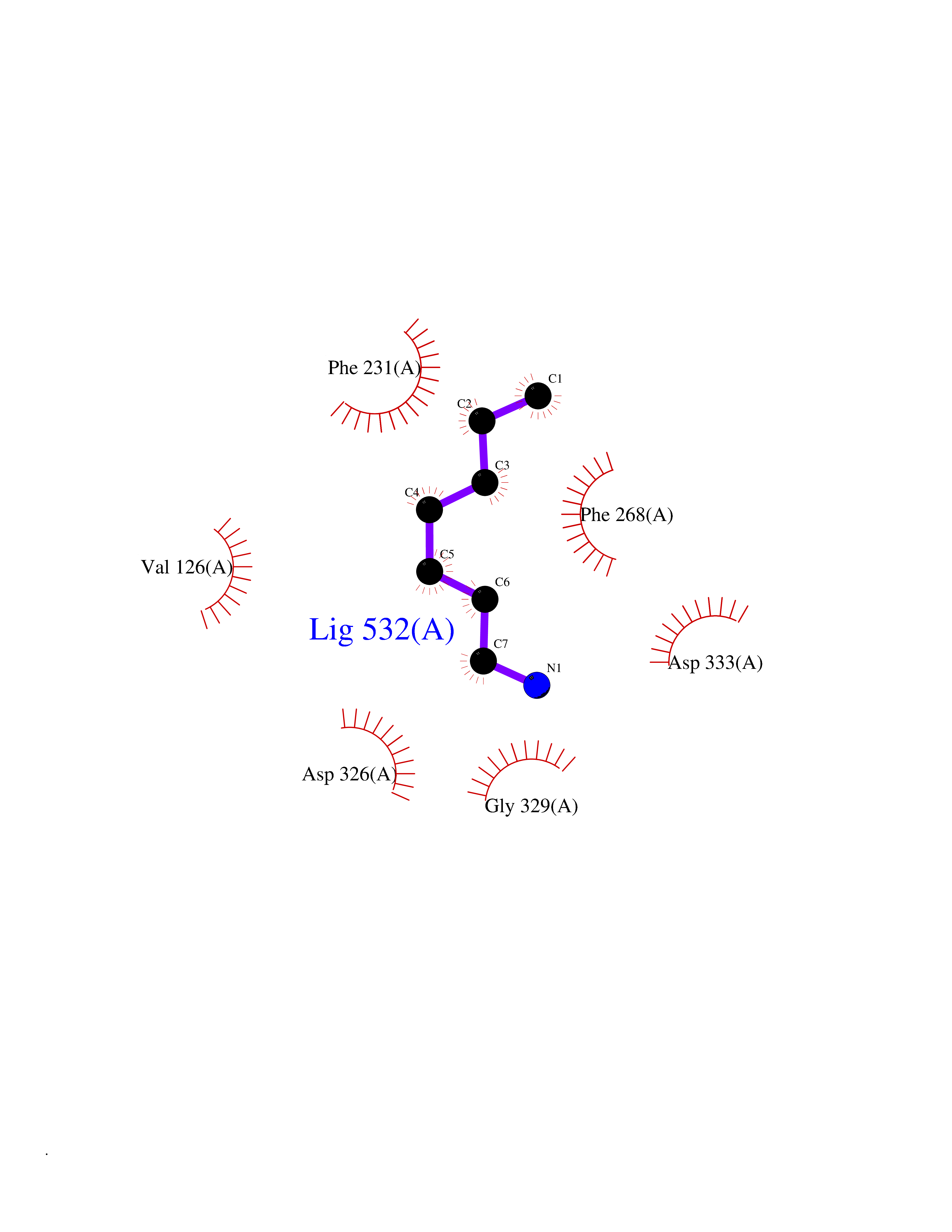 Ligplot Map 3D Binding mode Sequence QAAHLSFARLARRYGDVFQIRLGSCPIVVLNGERAIHQALVQQGSAFADRPSFASFRVVSGGRSMAFGHYSEHWKVQRRAAHSMMRNFFTRQPRSRQVLEGHVLSEARELVALLVRGSADGAFLDPRPLTVVAVANVMSAVCFGCRYSHDDPEFRELLSHNEEFGRTVGAGSLVDVMPWLQYFPNPVRTVFREFEQLNRNFSNFILDKFLRHCESLRPGAAPRDMMDAFILSAEKKAAGDGARLDLENVPATITDIFGASQDTLSTALQWLLLLFTRYPDVQTRVQAELDQVVGRDRLPCMGDQPNLPYVLAFLYEAMRFSSFVPVTIPHATTANTSVLGYHIPKDTVVFVNQWSVNHDPLKWPNPENFDPARFLDKDGLINKDLTSRVMIFSVGKRRCIGEELSKMQLFLFISILAHQCDFRANPNEPAKMNFSYGLTIKPKSFKVNVTLRESMELLD Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Cytochrome c oxidase subunit 2 | 3VRJ | 5.03 | |
Target general information Gen name MT-CO2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms MTCO2;COXII;COII;COX2 Protein family Cytochrome c oxidase subunit 2 family Biochemical class Immune system Function Copper ion binding.Cytochrome-c oxidase activity. Related diseases Mitochondrial complex IV deficiency (MT-C4D) [MIM:220110]: A disorder of the mitochondrial respiratory chain with heterogeneous clinical manifestations, ranging from isolated myopathy to severe multisystem disease affecting several tissues and organs. Features include hypertrophic cardiomyopathy, hepatomegaly and liver dysfunction, hypotonia, muscle weakness, exercise intolerance, developmental delay, delayed motor development and intellectual disability. Some affected individuals manifest a fatal hypertrophic cardiomyopathy resulting in neonatal death. A subset of patients manifest Leigh syndrome. {ECO:0000269|PubMed:10486321}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02659; DB04464; DB05412 Interacts with Q9NZ94-2; P49281-3 EC number 7.1.1.9 Uniprot keywords 3D-structure; Copper; Disease variant; Electron transport; Magnesium; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Primary mitochondrial disease; Proteomics identification; Reference proteome; Respiratory chain; Translocase; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID C Molecular weight (Da) 21687.9 Length 189 Aromaticity 0.11 Instability index 38 Isoelectric point 5.68 Charge (pH=7) -3.26 2D Binding mode Binding energy (Kcal/mol) -6.87 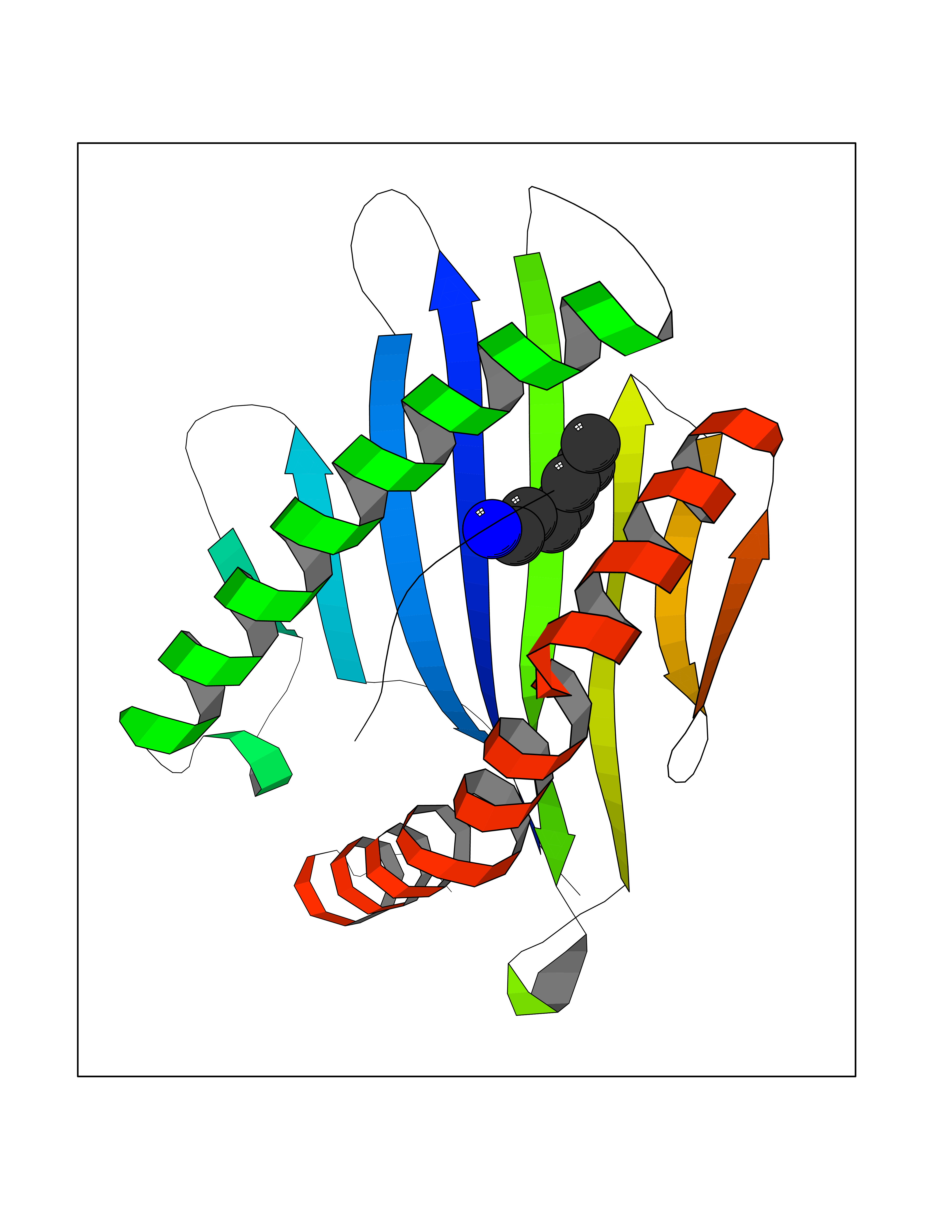 Molscript Map 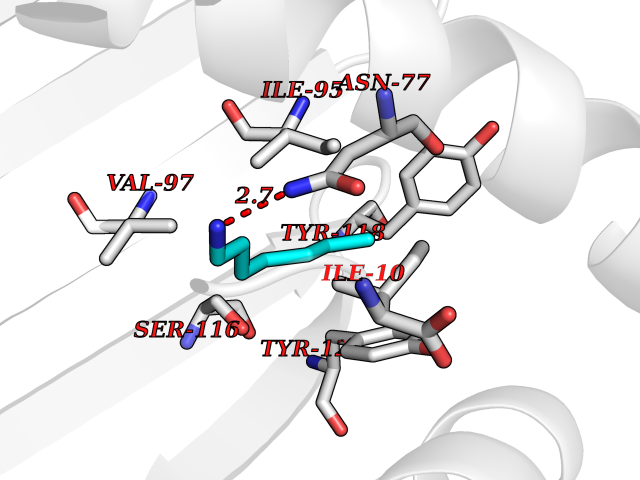 Pymol Map 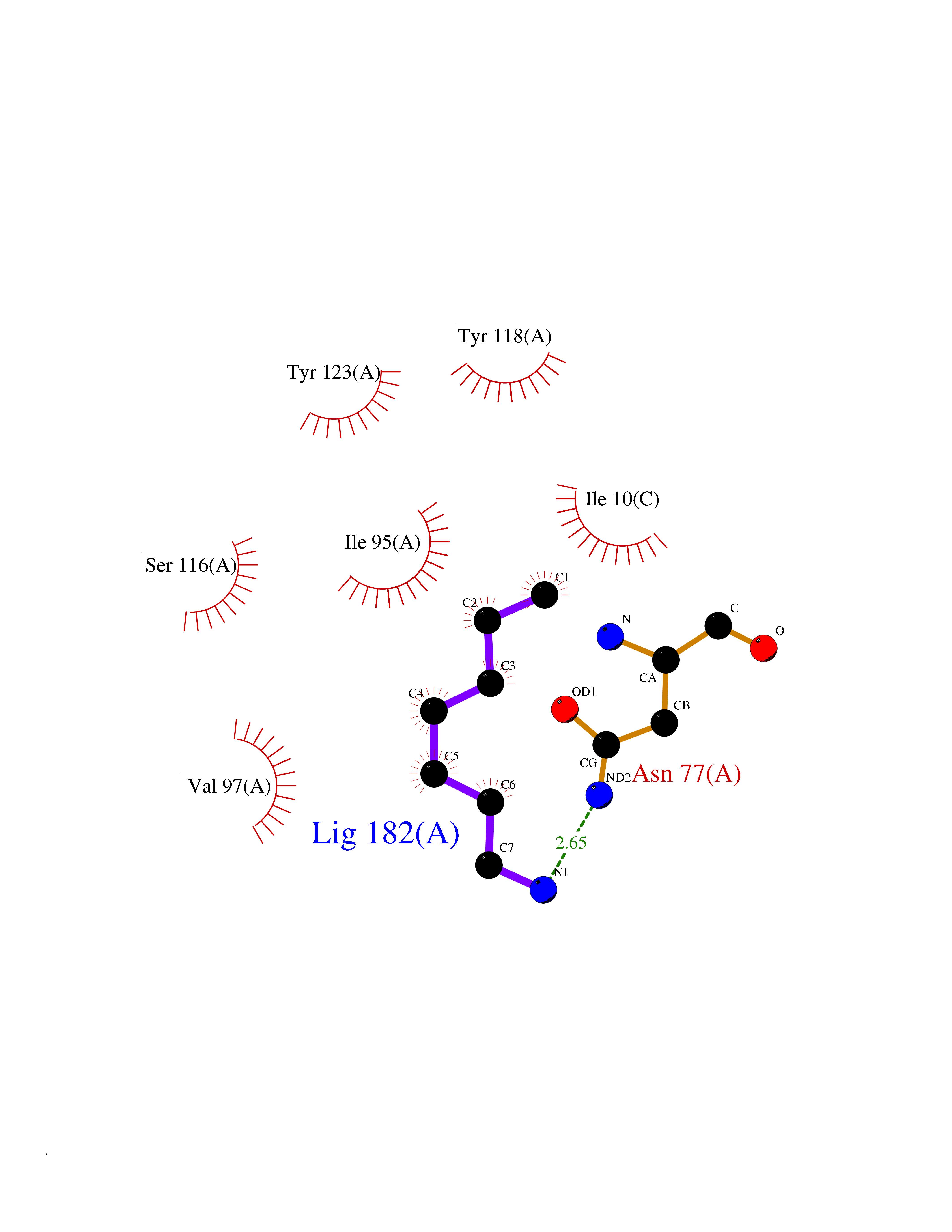 Ligplot Map 3D Binding mode Sequence SHSMRYFYTAMSRPGRGEPRFIAVGYVDDTQFVRFDSDAASPRMAPRAPWIEQEGPEYWDGETRNMKASAQTYRENLRIALRYYNQSEAGSHIIQVMYGCDVGPDGRLLRGHDQSAYDGKDYIALNEDLSSWTAADTAAQITQRKWEAARVAEQLRAYLEGLCVEWLRRYLENGKETLQLTTKLTNTNI Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Medium-chain specific acyl-CoA dehydrogenase, mitochondrial | 4P13 | 5.03 | |
Target general information Gen name ACADM Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Acyl-CoA dehydrogenase family Biochemical class Oxidoreductase Function Acyl-CoA dehydrogenase activity.Flavin adenine dinucleotide binding.Identical protein binding.Medium-chain-acyl-CoA dehydrogenase activity. Related diseases Acyl-CoA dehydrogenase medium-chain deficiency (ACADMD) [MIM:201450]: An inborn error of mitochondrial fatty acid beta-oxidation which causes fasting hypoglycemia, hepatic dysfunction and encephalopathy, often resulting in death in infancy. {ECO:0000269|PubMed:10767181, ECO:0000269|PubMed:11349232, ECO:0000269|PubMed:11409868, ECO:0000269|PubMed:11486912, ECO:0000269|PubMed:1363805, ECO:0000269|PubMed:1671131, ECO:0000269|PubMed:1684086, ECO:0000269|PubMed:1902818, ECO:0000269|PubMed:2251268, ECO:0000269|PubMed:2393404, ECO:0000269|PubMed:2394825, ECO:0000269|PubMed:7603790, ECO:0000269|PubMed:7929823, ECO:0000269|PubMed:8198141, ECO:0000269|PubMed:9158144, ECO:0000269|PubMed:9882619}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03415; DB03147; DB02910 Interacts with PRO_0000000502 [P11310] EC number 1.3.8.7 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Disease variant; FAD; Fatty acid metabolism; Flavoprotein; Lipid metabolism; Mitochondrion; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Transit peptide Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 85080.3 Length 773 Aromaticity 0.09 Instability index 30.55 Isoelectric point 5.71 Charge (pH=7) -7.7 2D Binding mode Binding energy (Kcal/mol) -6.87 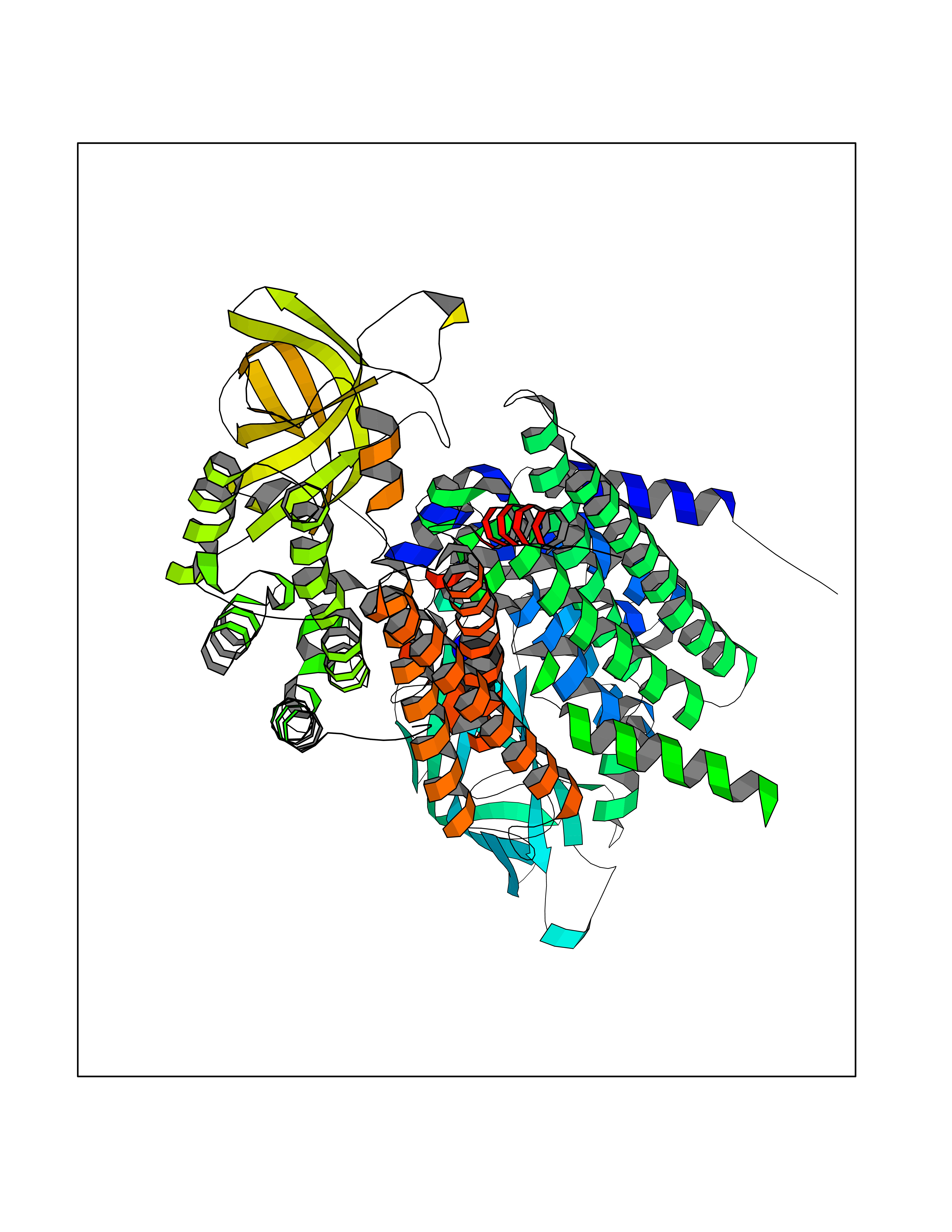 Molscript Map 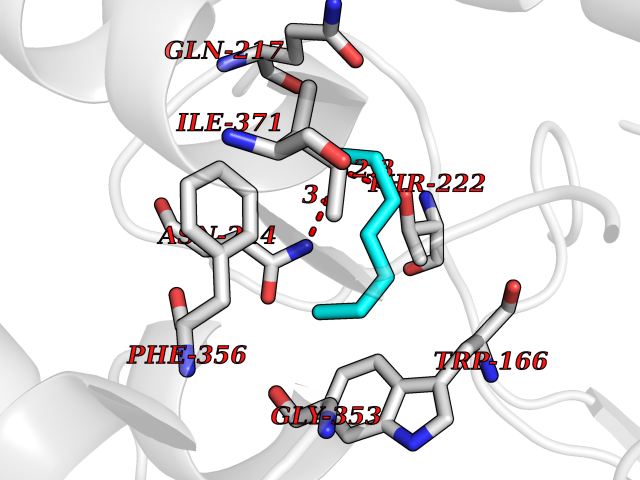 Pymol Map 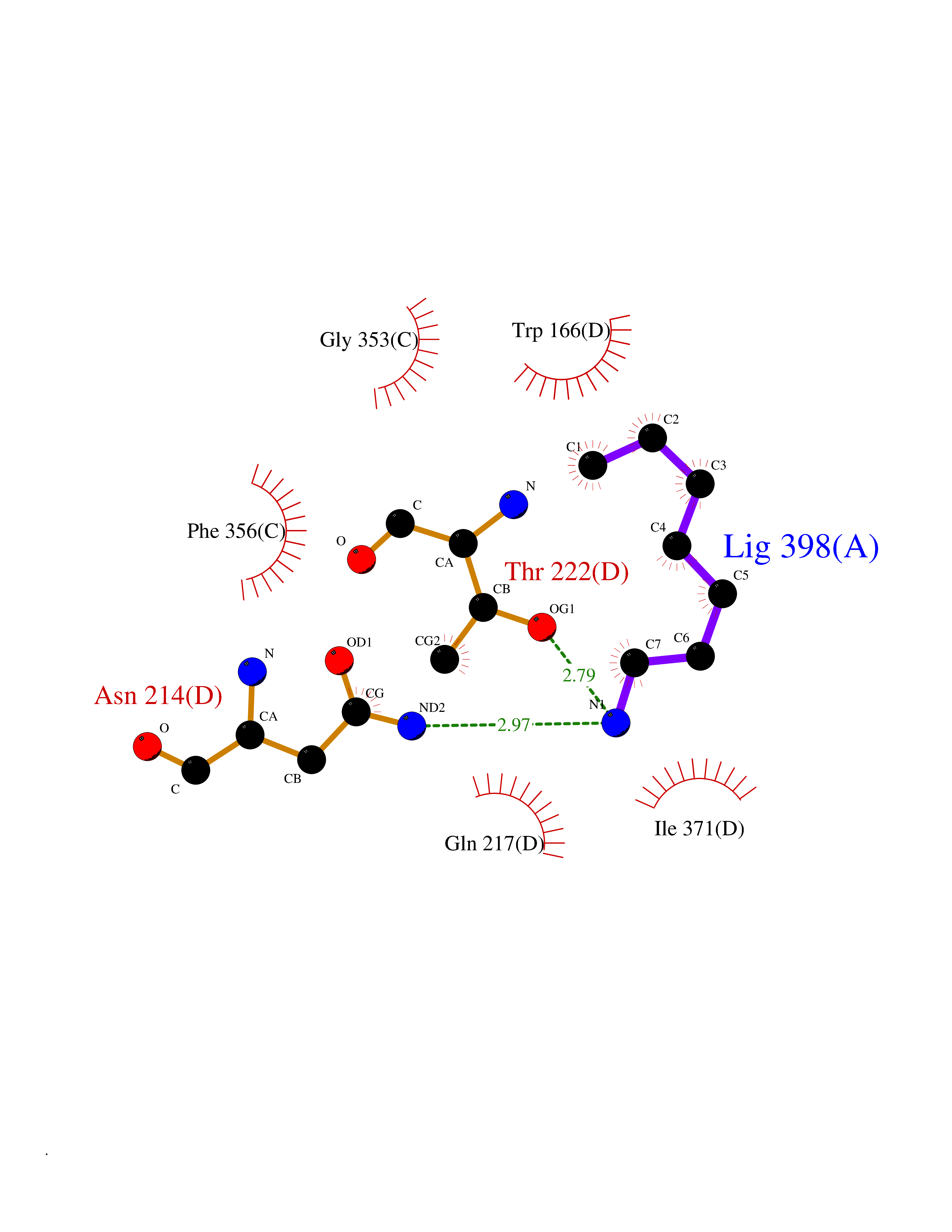 Ligplot Map 3D Binding mode Sequence LGFSFEFTEQQKEFQATARKFAREEIIPVAAEYDKTGEYPVPLIRRAWELGLMNTHIPENCGGLGLGTFDACLISEELAYGCTGVQTAIEGNSLGQMPIIIAGNDQQKKKYLGRMTEEPLMCAYCVTEPGAGSDVAGIKTKAEKKGDEYIINGQKMWITNGGKANWYFLLARSDPDPKAPANKAFTGFIVEADTPGIQIGRKELNMGQRCSDTRGIVFEDVKVPKENVLIGDGAGFKVAMGAFDKTRPVVAAGAVGLAQRALDEATKYALERKTFGKLLVEHQAISFMLAEMAMEVELARMSYQRAAWEVDSGRRNTYYASIAKAFAGDIANQLATDAVQILGGNGFNTEYPVEKLMRDAKIYQIYEGTSQIQRLIVAREHIDKYKLGFSFEFTEQQKEFQATARKFAREEIIPVAAEYDKTGEYPVPLIRRAWELGLMNTHIPENCGGLGLGTFDACLISEELAYGCTGVQTAIEGNSLGQMPIIIAGNDQQKKKYLGRMTEEPLMCAYCVTEPGAGSDVAGIKTKAEKKGDEYIINGQKMWITNGGKANWYFLLARSDPDPKAPANKAFTGFIVEADTPGIQIGRKELNMGQRCSDTRGIVFEDVKVPKENVLIGDGAGFKVAMGAFDKTRPVVAAGAVGLAQRALDEATKYALERKTFGKLLVEHQAISFMLAEMAMEVELARMSYQRAAWEVDSGRRNTYYASIAKAFAGDIANQLATDAVQILGGNGFNTEYPVEKLMRDAKIYQIYEGTSQIQRLIVAREHIDKYKN Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Diamine oxidase (AOC1) | 3HIG | 5.03 | |
Target general information Gen name AOC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Kidney amine oxidase; KAO; Histaminase; Amiloride-binding protein; AOC1; ABP Protein family Copper/topaquinone oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the degradation of compounds such as putrescine, histamine, spermine, and spermidine, substances involved in allergic and immune responses, cell proliferation, tissue differentiation, tumor formation, and possibly apoptosis. Placental DAO is thought to play a role in the regulation of the female reproductive function. Related diseases Lichtenstein-Knorr syndrome (LIKNS) [MIM:616291]: An autosomal recessive neurologic disorder characterized by progressive cerebellar ataxia and severe progressive sensorineural hearing loss. {ECO:0000269|PubMed:25205112, ECO:0000269|PubMed:30237576}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00594; DB01373; DB09130; DB03608; DB05383 Interacts with Q15038; O75593; Q8IUC2; Q96HA8; Q7Z3K3; Q6ZRY4; Q01085-2; O43711; Q96K80 EC number EC 1.4.3.22 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Copper; Direct protein sequencing; Disulfide bond; Glycoprotein; Heparin-binding; Membrane; Metal-binding; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal; TPQ Protein physicochemical properties Chain ID A,B Molecular weight (Da) 69037.3 Length 607 Aromaticity 0.13 Instability index 43.33 Isoelectric point 6.52 Charge (pH=7) -4.61 2D Binding mode Binding energy (Kcal/mol) -6.85 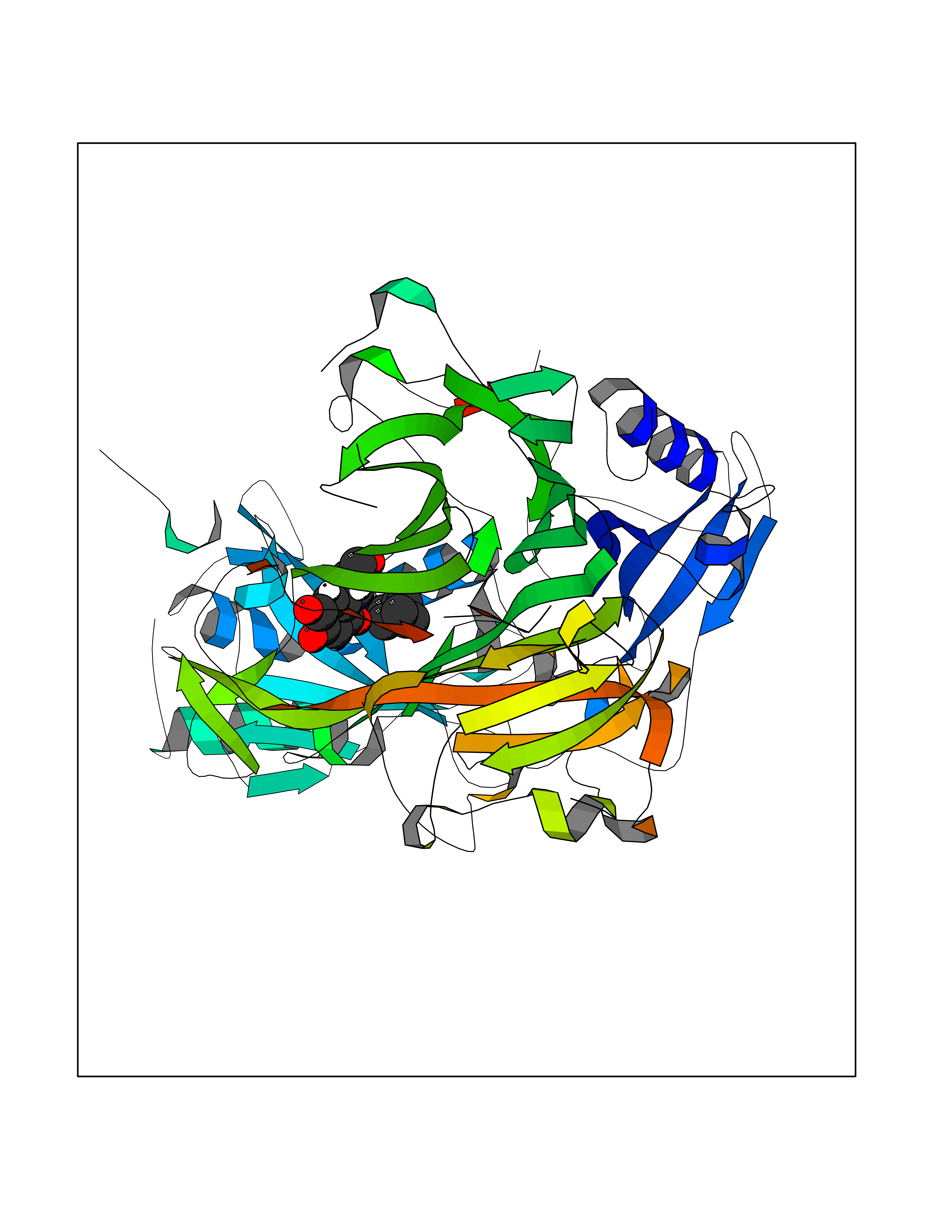 Molscript Map 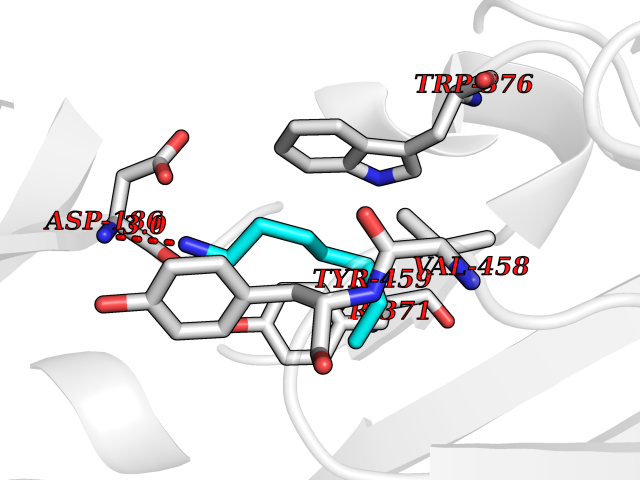 Pymol Map  Ligplot Map 3D Binding mode Sequence RKAGVFSDLSNQELKAVHSFLWSKKELRLQPSSTTTMAKNTVFLIEMLLPKKYHVLRFLDKGERHPVREARAVIFFGDQEHPNVTEFAVGPLPGPCYMRALSPRPGYQSSWASRPISTAEYALLYHTLQEATKPLHQFFLNTTGFSFQDCHDRCLAFTDVAPRGVASGQRRSWLIIQRYVEGYFLHPTGLELLVDHGSTDAGHWAVEQVWYNGKFYGSPEELARKYADGEVDVVVLEPPLFSSLVQPHGPRFRLEGNAVLYGGWSFAFRLRSSSGLQVLNVHFGGERIAYEVSVQEAVALYGGHTPAGMQTKYLDVGWGLGSVTHELAPGIDCPETATFLDTFHYYDADDPVHYPRALCLFEMPTGVPLKGQVLVLRTTSTVYNXDYIWDFIFYPNGVMEAKMHATGYVHATFYTPEGLRHGTRLHTHLIGNIHTHLVHYRVDLDVAGTKNSFQTLQYSWERQAAFRFKRKLPKYLLFTSPQENPWGHKRSYRLQIHSMADQVLPPGWQEEQAITWARYPLAVTKYRESELCSSSIYHQNDPWDPPVVFEQFLHNNENIENEDLVAWVTVGFLHIPHSEDIPNTATPGNSVGFLLRPFNFNGTYRPV Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Fatty acid synthase (FASN) | 3TJM | 5.03 | |
Target general information Gen name FASN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Yeast fatty acid synthase; Fatty-acyl-CoA synthase; Fatty acyl-CoA synthetase enzyme; FAS Protein family NA Biochemical class Acyltransferase Function Fatty acid synthetase catalyzes the formation of long-chain fatty acids from acetyl-CoA, malonyl-CoA and NADPH. This multifunctional protein has 7 catalytic activities as an acyl carrier protein. Related diseases Glycine encephalopathy 2 (GCE2) [MIM:620398]: A form of glycine encephalopathy, a metabolic disorder characterized by a high concentration of glycine in the body fluids. Affected individuals typically have severe neurological symptoms, including seizure, lethargy, and muscular hypotonia soon after birth. Most of them die within the neonatal period. Atypical cases have later disease onset and less severely affected psychomotor development. {ECO:0000269|PubMed:10873393, ECO:0000269|PubMed:11286506, ECO:0000269|PubMed:16051266, ECO:0000269|PubMed:26371980, ECO:0000269|PubMed:28244183, ECO:0000269|PubMed:8005589, ECO:0000269|PubMed:9600239, ECO:0000269|PubMed:9621520}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01034; DB01083 Interacts with Q15848; Q16665; P42858; Q8IV20; Q8TBB1; PRO_0000045603 [Q99IB8] EC number EC 2.3.1.85 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Isopeptide bond; Lipid biosynthesis; Lipid metabolism; Lyase; Multifunctional enzyme; NAD; NADP; Oxidoreductase; Phosphopantetheine; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; S-nitrosylation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30174.9 Length 275 Aromaticity 0.09 Instability index 43.28 Isoelectric point 5.92 Charge (pH=7) -5.4 2D Binding mode Binding energy (Kcal/mol) -6.86  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence NLRSLLVNPEGPTLMRLNSVQSSERPLFLVHPIEGSTTVFHSLASRLSIPTYGLQCTRAAPLDSIHSLAAYYIDCIRQVQPEGPYRVAGYSYGACVAFEMCSQLQAQQSPAPTHNSLFLFDGSPTYVLAYTGSYRAKLTPGCEAEAETEAICFFVQQFTDMEHNRVLEALLPLKGLEERVAAAVDLIIKSHQGLDRQELSFAARSFYYKLRAAEQYTPKAKYHGNVMLLRAAAGADYNLSQVCDGKVSVHVIEGDHATLLEGSGLESIISIIHSS Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | S-methyl-5'-thioadenosine phosphorylase (MTAP) | 1CB0 | 5.03 | |
Target general information Gen name MTAP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Methylthioadenosine phosphorylase; MTAPase; MTA phosphorylase; MSAP; 5'-methylthioadenosine phosphorylase Protein family PNP/MTAP phosphorylase family, MTAP subfamily Biochemical class Glycosyltransferases Function Involved in the breakdown of MTA, a major by-product of polyamine biosynthesis. Responsible for the first step in the methionine salvage pathway after MTA has been generated from S-adenosylmethionine. Has broad substrate specificity with 6-aminopurine nucleosides as preferred substrates. Catalyzes the reversible phosphorylation of S-methyl-5'-thioadenosine (MTA) to adenine and 5-methylthioribose-1-phosphate. Related diseases Diaphyseal medullary stenosis with malignant fibrous histiocytoma (DMSMFH) [MIM:112250]: An autosomal dominant bone dysplasia characterized by pathologic fractures due to abnormal cortical growth and diaphyseal medullary stenosis. The fractures heal poorly, and there is progressive bowing of the lower extremities. Some patients show a limb-girdle myopathy, with muscle weakness and atrophy. Approximately 35% of affected individuals develop an aggressive form of bone sarcoma consistent with malignant fibrous histiocytoma or osteosarcoma. {ECO:0000269|PubMed:22464254}. The disease is caused by variants affecting the gene represented in this entry. DMSMFH causing mutations found in MTAP exon 9 result in exon skipping and dysregulated alternative splicing of all MTAP isoforms (PubMed:22464254). {ECO:0000269|PubMed:22464254}.; DISEASE: Loss of MTAP activity may play a role in human cancer. MTAP loss has been reported in a number of cancers, including osteosarcoma, malignant melanoma and gastric cancer. Drugs (DrugBank ID) DB02158; DB02933; DB02282; DB00173; DB02281 Interacts with Q9H3R5; Q9P0I2 EC number EC 2.4.2.28 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Glycosyltransferase; Nucleus; Proteomics identification; Purine salvage; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 29538.9 Length 268 Aromaticity 0.06 Instability index 40.97 Isoelectric point 7.18 Charge (pH=7) 0.36 2D Binding mode Binding energy (Kcal/mol) -6.86 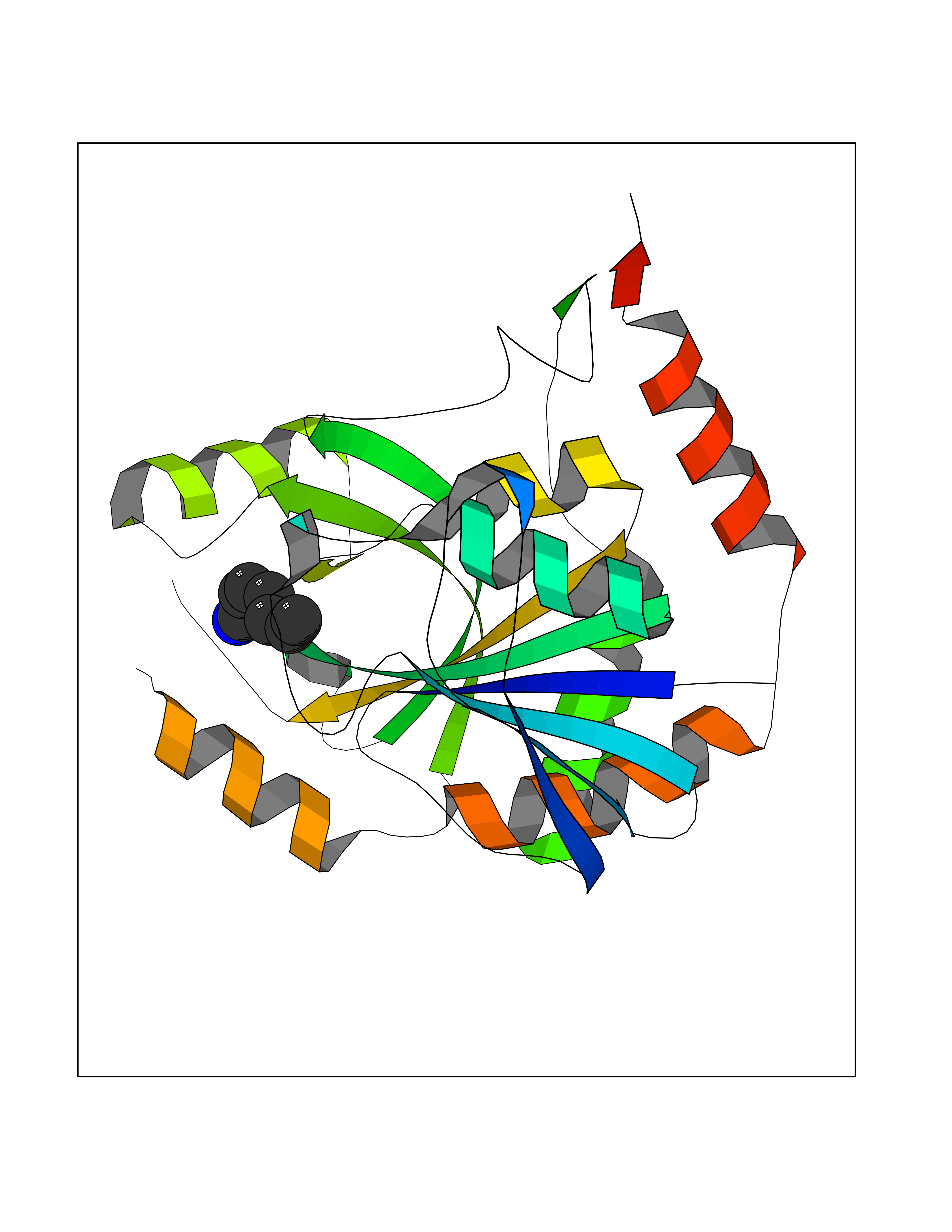 Molscript Map 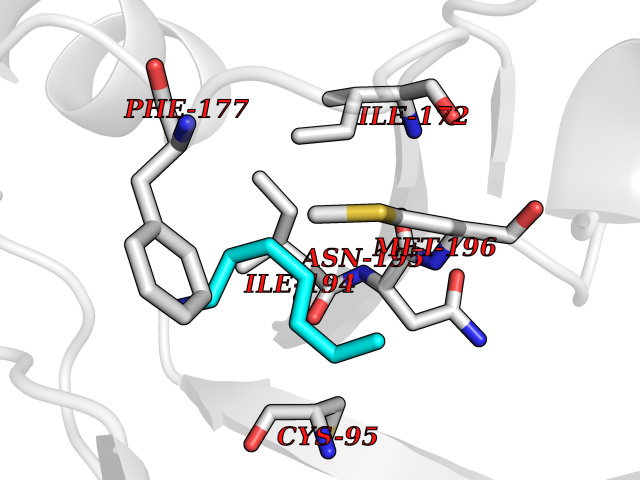 Pymol Map 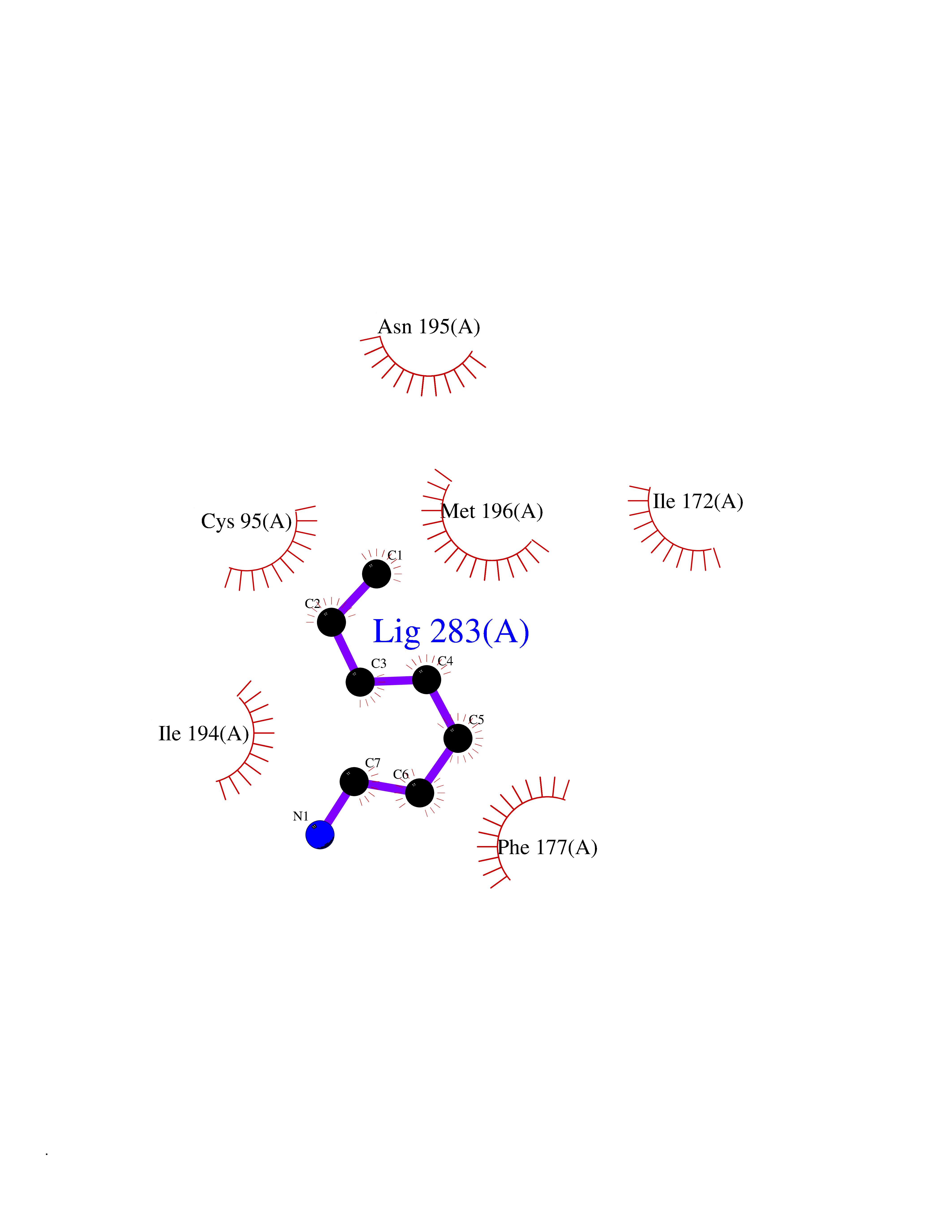 Ligplot Map 3D Binding mode Sequence AVKIGIIGGTGLDDPEILEGRTEKYVDTPFGKPSDALILGKIKNVDCVLLARHGRQHTIMPSKVNYQANIWALKEEGCTHVIVTTACGSLREEIQPGDIVIIDQFIDRTTMRPQSFYDGSHSCARGVCHIPMAEPFCPKTREVLIETAKKLGLRCHSKGTMVTIEGPRFSSRAESFMFRTWGADVINMTTVPEVVLAKEAGICYASIAMATDYDCWAVSVDRVLKTLKENANKAKSLLLTTIPQIGSTEWSETLHNLKNMAQFSVLLP Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Macrophage-stimulating protein receptor (RON) | 3PLS | 5.03 | |
Target general information Gen name MST1R Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p185Ron; Proteintyrosine kinase 8; Macrophagestimulating protein receptor beta chain; Macrophagestimulating protein receptor; MST1R; MSP receptor; CDw136; CD136 Protein family Protein kinase superfamily, Tyr protein kinase family Biochemical class Kinase Function Receptor tyrosine kinase that transduces signals from the extracellular matrix into the cytoplasm by binding to MST1 ligand. Regulates many physiological processes including cell survival, migration and differentiation. Ligand binding at the cell surface induces autophosphorylation of RON on its intracellular domain that provides docking sites for downstream signaling molecules. Following activation by ligand, interacts with the PI3-kinase subunit PIK3R1, PLCG1 or the adapter GAB1. Recruitment of these downstream effectors by RON leads to the activation of several signaling cascades including the RAS-ERK, PI3 kinase-AKT, or PLCgamma-PKC. RON signaling activates the wound healing response by promoting epithelial cell migration, proliferation as well as survival at the wound site. Plays also a role in the innate immune response by regulating the migration and phagocytic activity of macrophages. Alternatively, RON can also promote signals such as cell migration and proliferation in response to growth factors other than MST1 ligand. Related diseases Nasopharyngeal carcinoma, 3 (NPCA3) [MIM:617075]: A form of nasopharyngeal carcinoma, a malignant neoplasm that originates in the nasopharyngeal epithelium and includes 4 subtypes: keratinizing squamous cell, non-keratinizing, basaloid squamous cell, and papillary adenocarcinoma. {ECO:0000269|PubMed:26951679}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08865; DB12010 Interacts with P26927; O43157; O15031; Q9ULL4 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cleavage on pair of basic residues; Disease variant; Disulfide bond; Glycoprotein; Immunity; Innate immunity; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 34204.1 Length 298 Aromaticity 0.09 Instability index 38.81 Isoelectric point 6.37 Charge (pH=7) -3.11 2D Binding mode Binding energy (Kcal/mol) -6.86 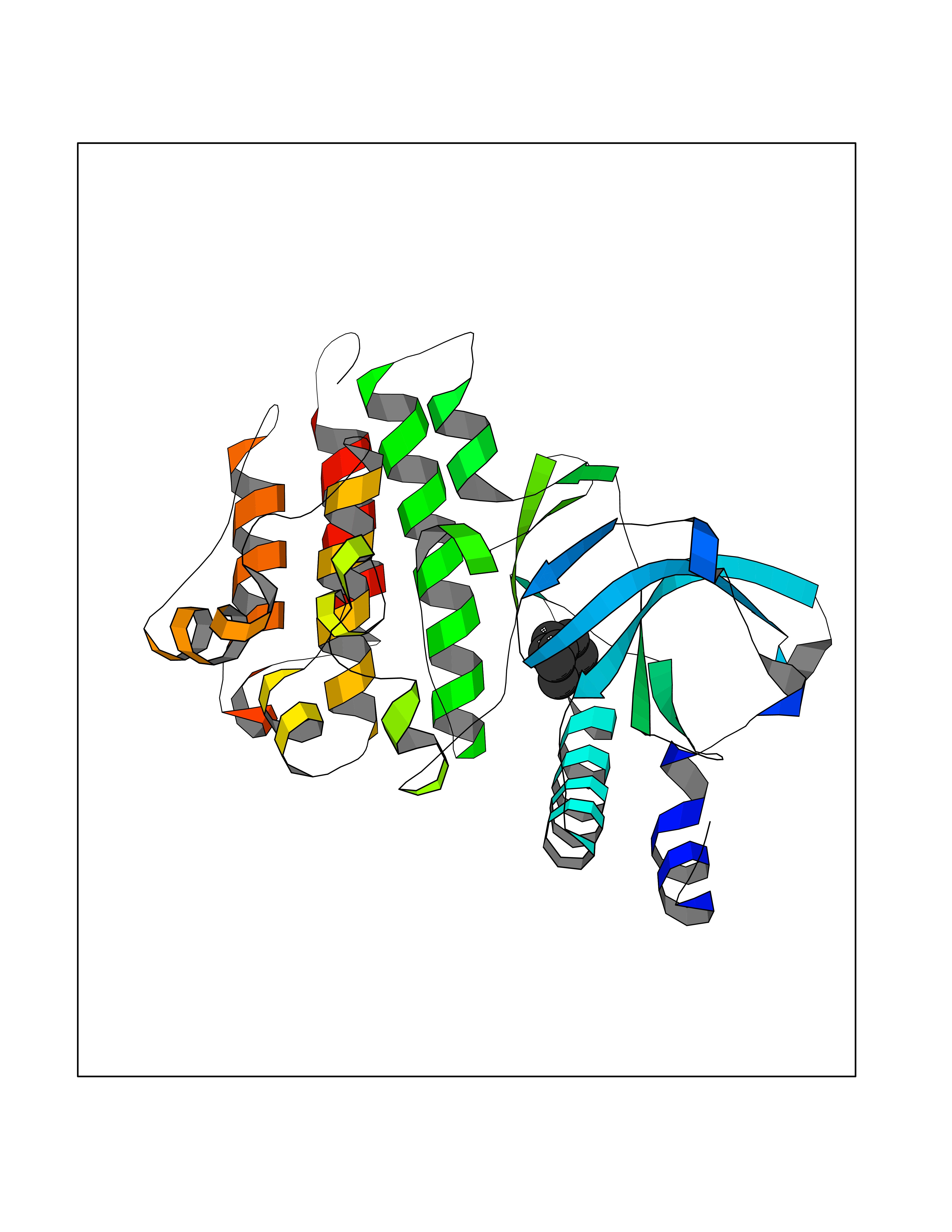 Molscript Map 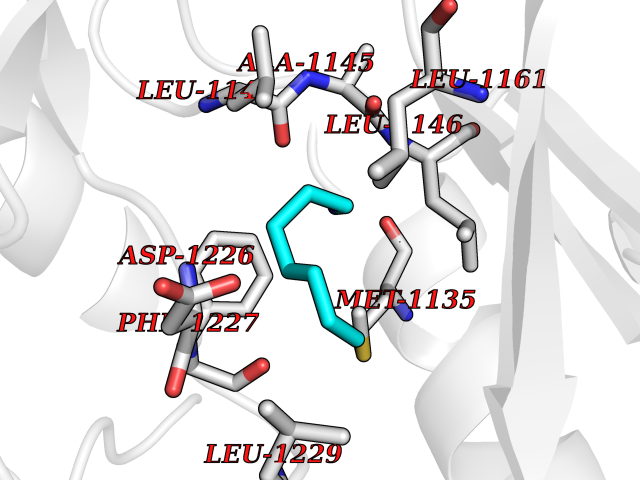 Pymol Map 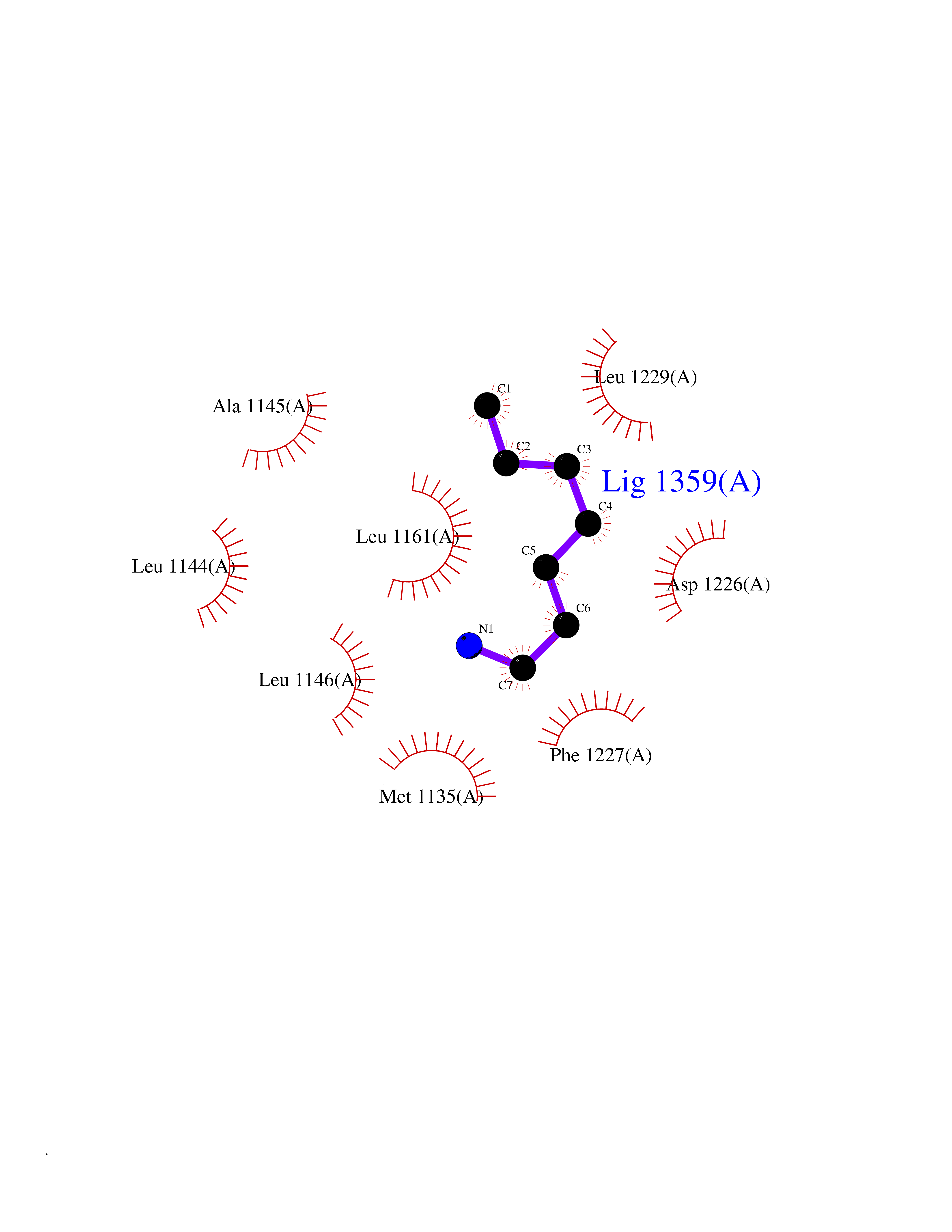 Ligplot Map 3D Binding mode Sequence RDLDSALLAEVKDVLIPHERVVTHSDRVIGKGHFGVVYHGEYIDQAQNRIQCAIKSLSRITEMQQVEAFLREGLLMRGLNHPNVLALIGIMLPPEGLPHVLLPYMCHGDLLQFIRSPQRNPTVKDLISFGLQVARGMEYLAEQKFVHRDLAARNCMLDESFTVKVADFGLARDILDREYYSVQQHRHARLPVKWTALESLQTYRFTTKSDVWSFGVLLWELLTRGAPPYRHIDPFDLTHFLAQGRRLPQPEYCPDSLYQVMQQCWEADPAVRPTFRVLVGEVEQIVSALLGDHYVQLP Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | PRKR-like endoplasmic reticulum kinase (PERK) | 4G31 | 5.03 | |
Target general information Gen name EIF2AK3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PEK Protein family Protein kinase superfamily, Ser/Thr protein kinase family, GCN2 subfamily Biochemical class Kinase Function Converts phosphorylated eIF-2-alpha/EIF2S1 either in a global protein synthesis inhibitor, leading to a reduced overall utilization of amino acids, or to a translation initiation activator of specific mRNAs, such as the transcriptional activator ATF4, and hence allowing ATF4-mediated reprogramming of amino acid biosynthetic gene expression to alleviate nutrient depletion. Serves as a critical effector of unfolded protein response (UPR)-induced G1 growth arrest due to the loss of cyclin-D1 (CCND1). Involved in control of mitochondrial morphology and function. Metabolic-stress sensing protein kinase that phosphorylates the alpha subunit of eukaryotic translation initiation factor 2 (eIF-2-alpha/EIF2S1) on 'Ser-52' during the unfolded protein response (UPR) and in response to low amino acid availability. Related diseases Wolcott-Rallison syndrome (WRS) [MIM:226980]: A rare autosomal recessive disorder, characterized by permanent neonatal or early infancy insulin-dependent diabetes and, at a later age, epiphyseal dysplasia, osteoporosis, growth retardation and other multisystem manifestations, such as hepatic and renal dysfunctions, intellectual disability and cardiovascular abnormalities. {ECO:0000269|PubMed:10932183, ECO:0000269|PubMed:12086964, ECO:0000269|PubMed:12960215, ECO:0000269|PubMed:16813601, ECO:0000269|PubMed:24168455, ECO:0000269|PubMed:24194294, ECO:0000269|PubMed:27145240, ECO:0000269|PubMed:28220546, ECO:0000269|PubMed:30906465, ECO:0000269|PubMed:30922274, ECO:0000269|PubMed:32216767, ECO:0000269|PubMed:34123975}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NZJ5; P11021 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; ADP-ribosylation; ATP-binding; Diabetes mellitus; Disease variant; Endoplasmic reticulum; Glycoprotein; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Signal; Stress response; Transferase; Translation regulation; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Unfolded protein response Protein physicochemical properties Chain ID A Molecular weight (Da) 29033.5 Length 248 Aromaticity 0.1 Instability index 43.71 Isoelectric point 7.75 Charge (pH=7) 1.27 2D Binding mode Binding energy (Kcal/mol) -6.86 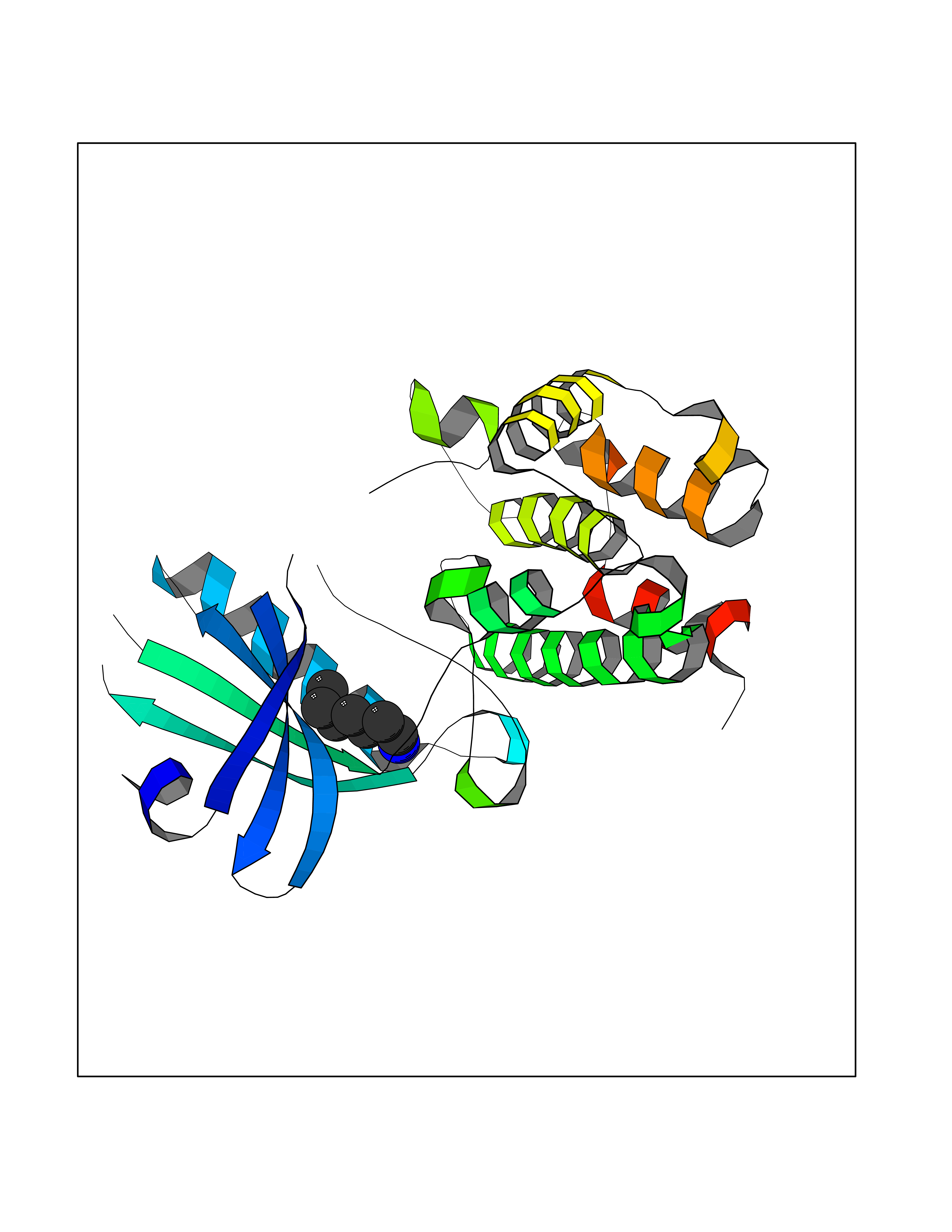 Molscript Map 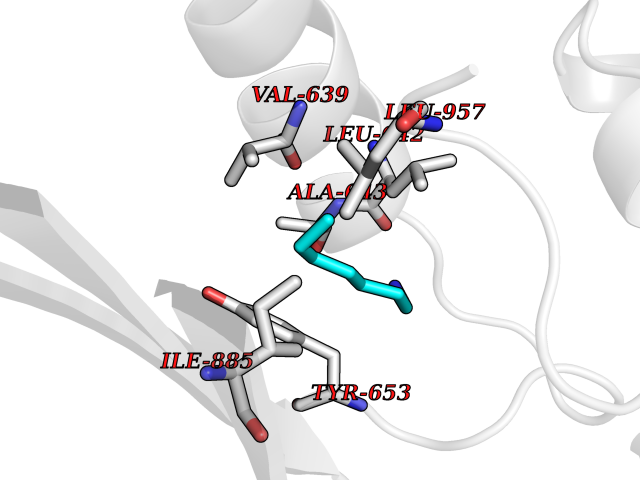 Pymol Map 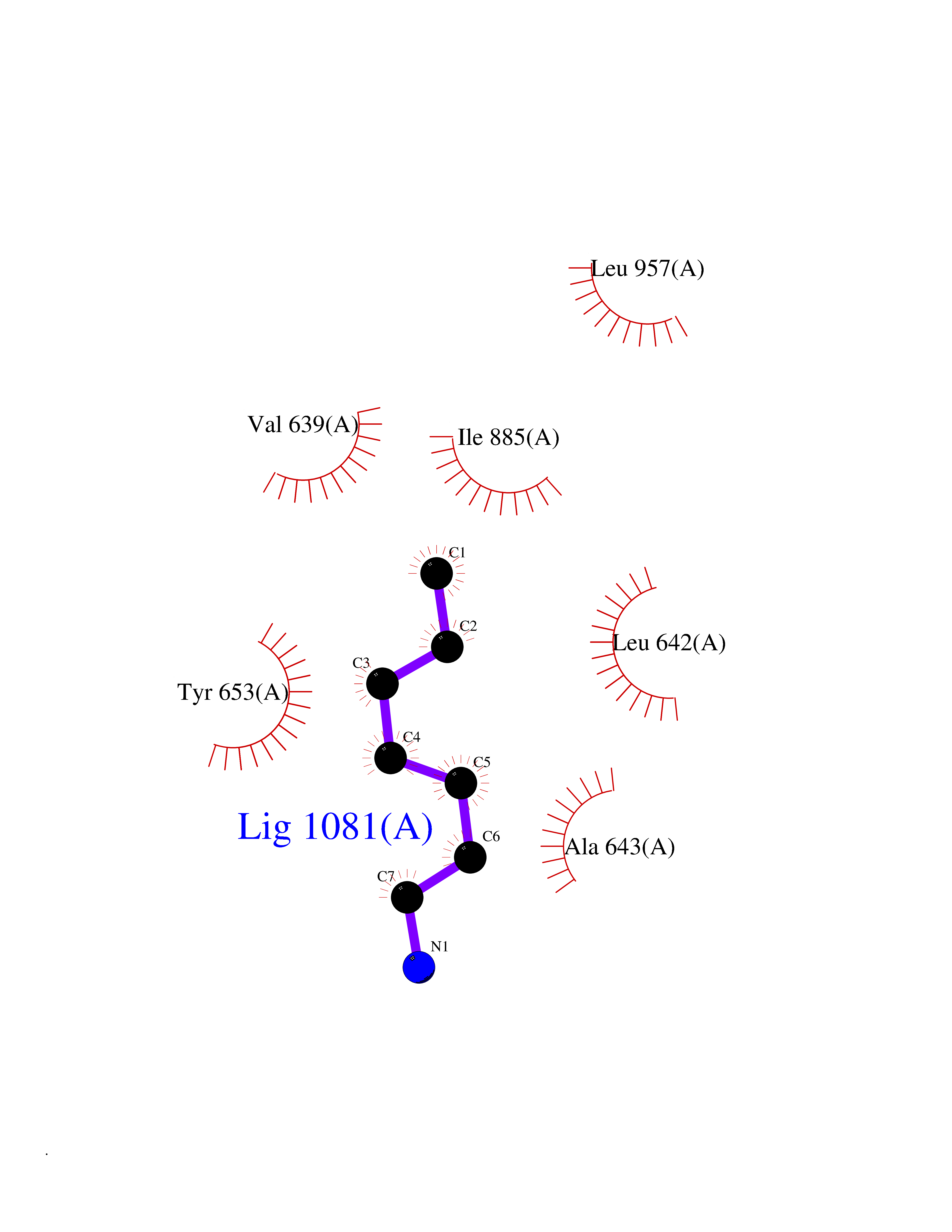 Ligplot Map 3D Binding mode Sequence GRYLTDFEPIQCLGRGGVVFEAKNKVDDCNYAIKRIRLPNRELAREKVMREVKALAKLEHPGIVRYFNAWLEKNKVYLYIQMQLCRKENLKDWMNGRCTIEERERSVCLHIFLQIAEAVEFLHSKGLMHRDLKPSNIFFTMDDVVKVGDFGLVGTKLYMSPEQIHGNSYSHKVDIFSLGLILFELLYPFSTQMERVRTLTDVRNLKFPPLFTQKYPCEYVMVQDMLSPSPMERPEAINIIENAVFEDL Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Dual-specificity tyrosine-phosphorylation regulated kinase 2 (DYRK2) | 6HDR | 5.03 | |
Target general information Gen name DYRK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dual specificity tyrosine-phosphorylation-regulated kinase 2 Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, MNB/DYRK subfamily Biochemical class Kinase Function Functions in part via its role in ubiquitin-dependent proteasomal protein degradation. Functions downstream of ATM and phosphorylates p53/TP53 at 'Ser-46', and thereby contributes to the induction of apoptosis in response to DNA damage. Phosphorylates NFATC1, and thereby inhibits its accumulation in the nucleus and its transcription factor activity. Phosphorylates EIF2B5 at 'Ser-544', enabling its subsequent phosphorylation and inhibition by GSK3B. Likewise, phosphorylation of NFATC1, CRMP2/DPYSL2 and CRMP4/DPYSL3 promotes their subsequent phosphorylation by GSK3B. May play a general role in the priming of GSK3 substrates. Inactivates GYS1 by phosphorylation at 'Ser-641', and potentially also a second phosphorylation site, thus regulating glycogen synthesis. Mediates EDVP E3 ligase complex formation and is required for the phosphorylation and subsequent degradation of KATNA1. Phosphorylates TERT at 'Ser-457', promoting TERT ubiquitination by the EDVP complex. Phosphorylates SIAH2, and thereby increases its ubiquitin ligase activity. Promotes the proteasomal degradation of MYC and JUN, and thereby regulates progress through the mitotic cell cycle and cell proliferation. Promotes proteasomal degradation of GLI2 and GLI3, and thereby plays a role in smoothened and sonic hedgehog signaling. Plays a role in cytoskeleton organization and neurite outgrowth via its phosphorylation of DCX and DPYSL2. Phosphorylates CRMP2/DPYSL2, CRMP4/DPYSL3, DCX, EIF2B5, EIF4EBP1, GLI2, GLI3, GYS1, JUN, MDM2, MYC, NFATC1, p53/TP53, TAU/MAPT and KATNA1. Can phosphorylate histone H1, histone H3 and histone H2B (in vitro). Can phosphorylate CARHSP1 (in vitro). Serine/threonine-protein kinase involved in the regulation of the mitotic cell cycle, cell proliferation, apoptosis, organization of the cytoskeleton and neurite outgrowth. Related diseases Bone marrow failure and diabetes mellitus syndrome (BMFDMS) [MIM:620044]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFDMS is an autosomal recessive form characterized by various degrees of bone marrow failure, ranging from dyserythropoiesis to bone marrow aplasia, with onset in infancy or early childhood, and non-autoimmune insulin-dependent diabetes mellitus appearing in the first or second decades. Many patients show pigmentary skin abnormalities and short stature. {ECO:0000269|PubMed:28073829, ECO:0000269|PubMed:35611808, ECO:0000269|PubMed:35931051}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NR20; Q13422; Q9BQD3; Q9BRK4; P23497; O43379; P62258; Q96C00 EC number EC 2.7.12.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cytoplasm; Kinase; Magnesium; Manganese; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID A Molecular weight (Da) 46422.1 Length 407 Aromaticity 0.09 Instability index 44.91 Isoelectric point 9.09 Charge (pH=7) 12.37 2D Binding mode Binding energy (Kcal/mol) -6.87 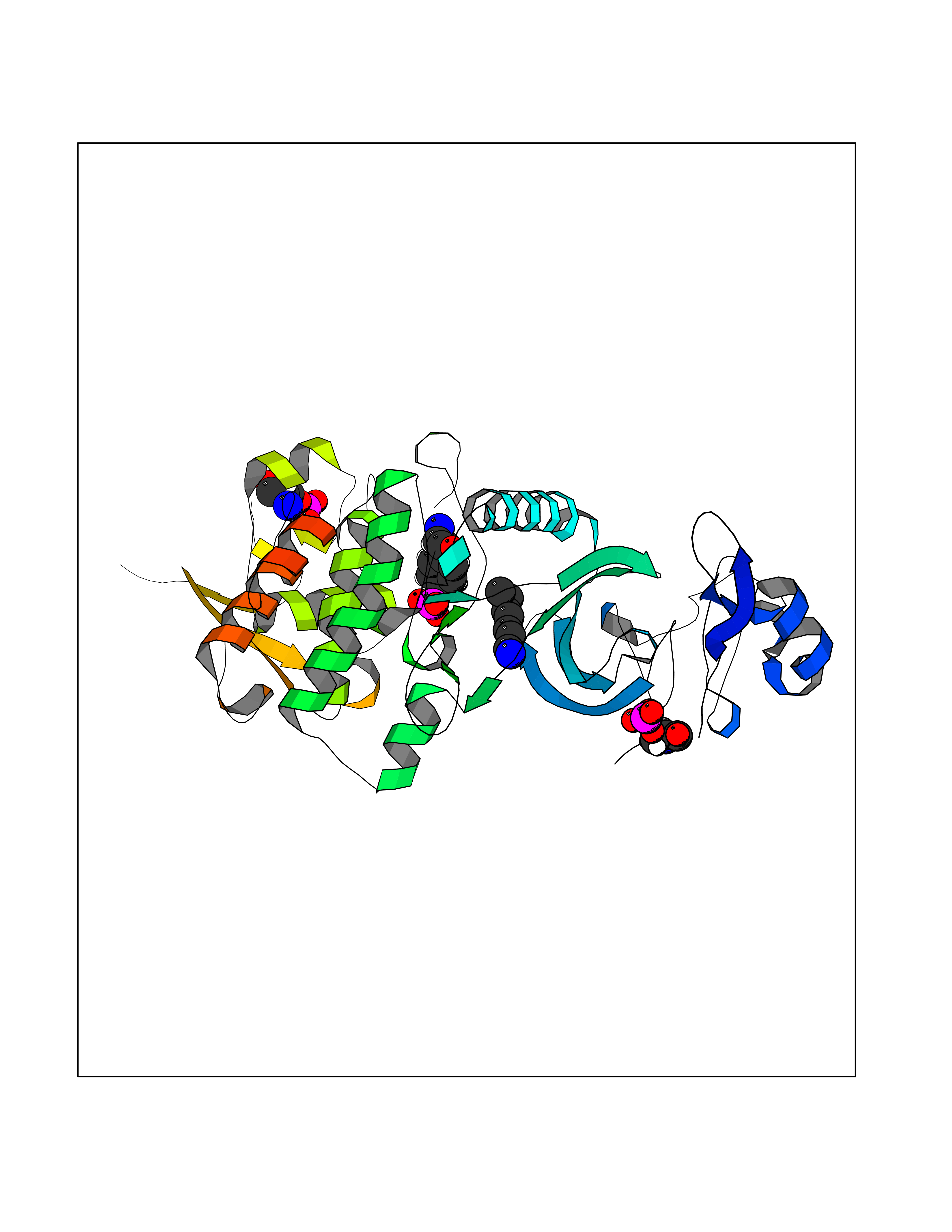 Molscript Map 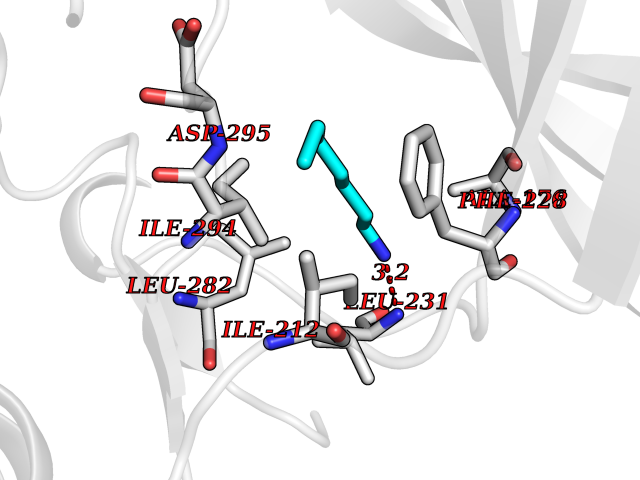 Pymol Map 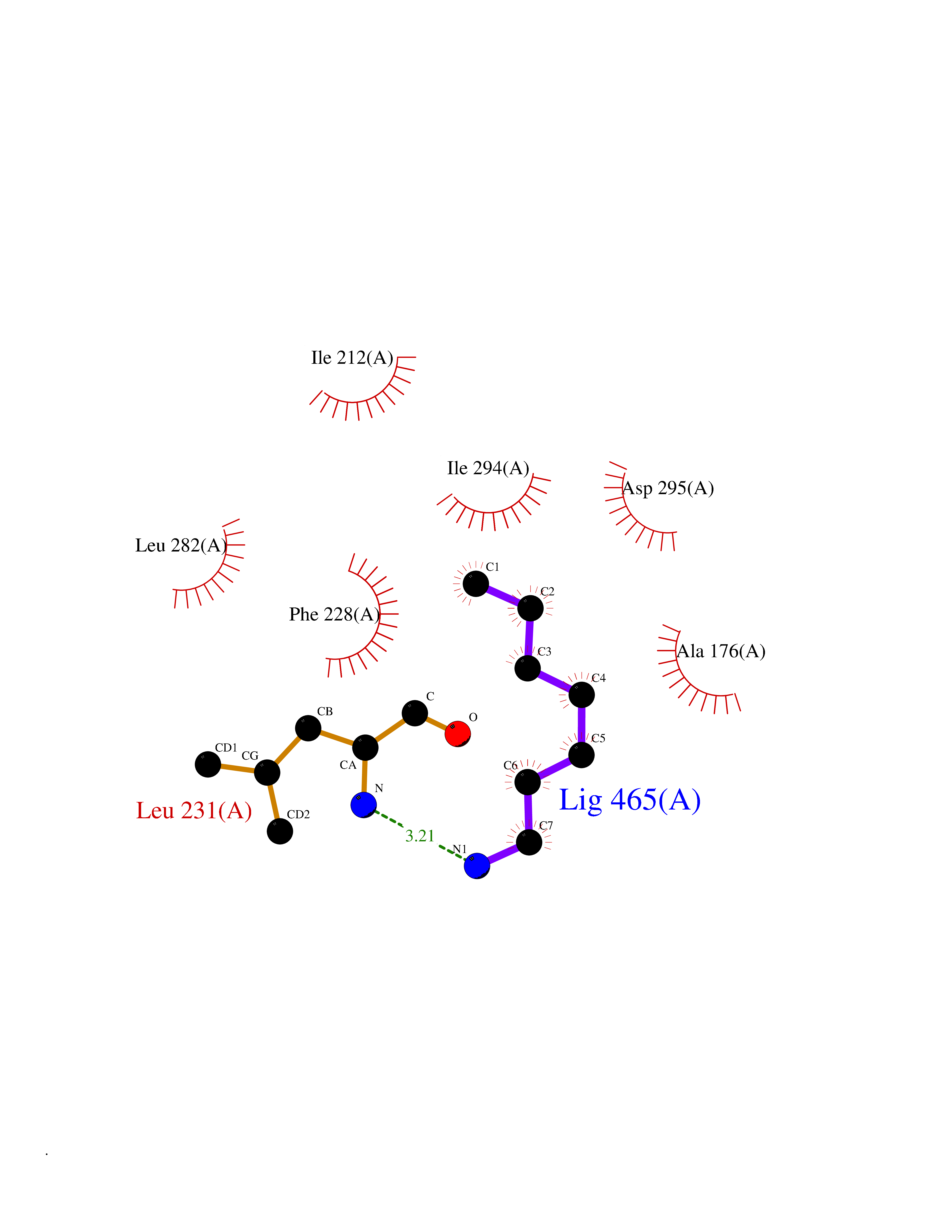 Ligplot Map 3D Binding mode Sequence HHHSXGVDLGTENLYFQSMGKVKATPMTPEQAMKQYMQKLTAFEHHEIFSYPEIYFLGLNAKKRQGMTGGPNNGGYDDDQGSYVQVPHDHVAYRYEVLKVIGKGSFGQVVKAYDHKVHQHVALKMVRNEKRFHRQAAEEIRILEHLRKQDKDNTMNVIHMLENFTFRNHICMTFELLSMNLYELIKKNKFQGFSLPLVRKFAHSILQCLDALHKNRIIHCDLKPENILLKQQGRSGIKVIDFGSSCYEHQRVYTXIQSRFYRAPEVILGARYGMPIDMWSLGCILAELLTGYPLLPGEDEGDQLACMIELLGMPSQKLLDASKRAKNFVSXKGYPRYCTVTTLSDVVLNGGRSRRGKLRGPPESREWGNALKGCDDPLFLDFLKQCLEWDPAVRMTPGQALRHPWLR Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Histone-lysine N-methyltransferase EHMT2 (EHMT2) | 5VSC | 5.03 | |
Target general information Gen name EHMT2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein G9a; NG36; Lysine N-methyltransferase 1C; KMT1C; Histone H3-K9 methyltransferase 3; HLA-B-associated transcript 8; H3-K9-HMTase 3; G9A; Euchromatic histone-lysine N-methyltransferase 2; C6orf3 Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, Suvar3-9 subfamily Biochemical class Methyltransferase Function H3K9me represents a specific tag for epigenetic transcriptional repression by recruiting HP1 proteins to methylated histones. Also mediates monomethylation of 'Lys-56' of histone H3 (H3K56me1) in G1 phase, leading to promote interaction between histone H3 and PCNA and regulating DNA replication. Also weakly methylates 'Lys-27' of histone H3 (H3K27me). Also required for DNA methylation, the histone methyltransferase activity is not required for DNA methylation, suggesting that these 2 activities function independently. Probably targeted to histone H3 by different DNA-binding proteins like E2F6, MGA, MAX and/or DP1. May also methylate histone H1. In addition to the histone methyltransferase activity, also methylates non-histone proteins: mediates dimethylation of 'Lys-373' of p53/TP53. Also methylates CDYL, WIZ, ACIN1, DNMT1, HDAC1, ERCC6, KLF12 and itself. Histone methyltransferase that specifically mono- and dimethylates 'Lys-9' of histone H3 (H3K9me1 and H3K9me2, respectively) in euchromatin. Related diseases Pseudohypoaldosteronism 2C (PHA2C) [MIM:614492]: An autosomal dominant disorder characterized by severe hypertension, hyperkalemia, hyperchloremia, mild hyperchloremic metabolic acidosis in some cases, and correction of physiologic abnormalities by thiazide diuretics. {ECO:0000269|PubMed:11498583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuropathy, hereditary sensory and autonomic, 2A (HSAN2A) [MIM:201300]: A form of hereditary sensory and autonomic neuropathy, a genetically and clinically heterogeneous group of disorders characterized by degeneration of dorsal root and autonomic ganglion cells, and by sensory and/or autonomic abnormalities. HSAN2A is an autosomal recessive disorder characterized by impairment of pain, temperature and touch sensation, onset of symptoms in infancy or early childhood, occurrence of distal extremity pathologies (paronychia, whitlows, ulcers, and Charcot joints), frequent amputations, sensory loss that affects all modalities of sensation (lower and upper limbs and perhaps the trunk as well), absence or diminution of tendon reflexes (usually in all limbs), minimal autonomic dysfunction, absence of sensory nerve action potentials, and virtual absence of myelinated fibers with decreased numbers of unmyelinated fibers in sural nerves. {ECO:0000269|PubMed:15060842, ECO:0000269|PubMed:15911806, ECO:0000269|PubMed:18521183}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q6VMQ6-2; Q6P1J9; Q9UBC3; P38919; Q9UM22; P23771; Q99684; Q13547; Q96JB3; Q92831; O60341-1; Q9Y4X4; P57682; Q13330; O94776; Q9BTC8; P20592; Q9BSU3; Q99801-1; O60568; Q9NQX1; Q5JSZ5; Q7Z3Z2; Q9P2R6; Q14119; Q96GT9; O60315; Q9NWS9-2; Q96JM2; A0A0S2Z5X4; Q96BV0; Q96EG3; Q07120; O60341-1 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ANK repeat; Chromatin regulator; Chromosome; Isopeptide bond; Metal-binding; Methylation; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; S-adenosyl-L-methionine; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 31010.9 Length 269 Aromaticity 0.1 Instability index 47.49 Isoelectric point 5.16 Charge (pH=7) -9.31 2D Binding mode Binding energy (Kcal/mol) -6.86 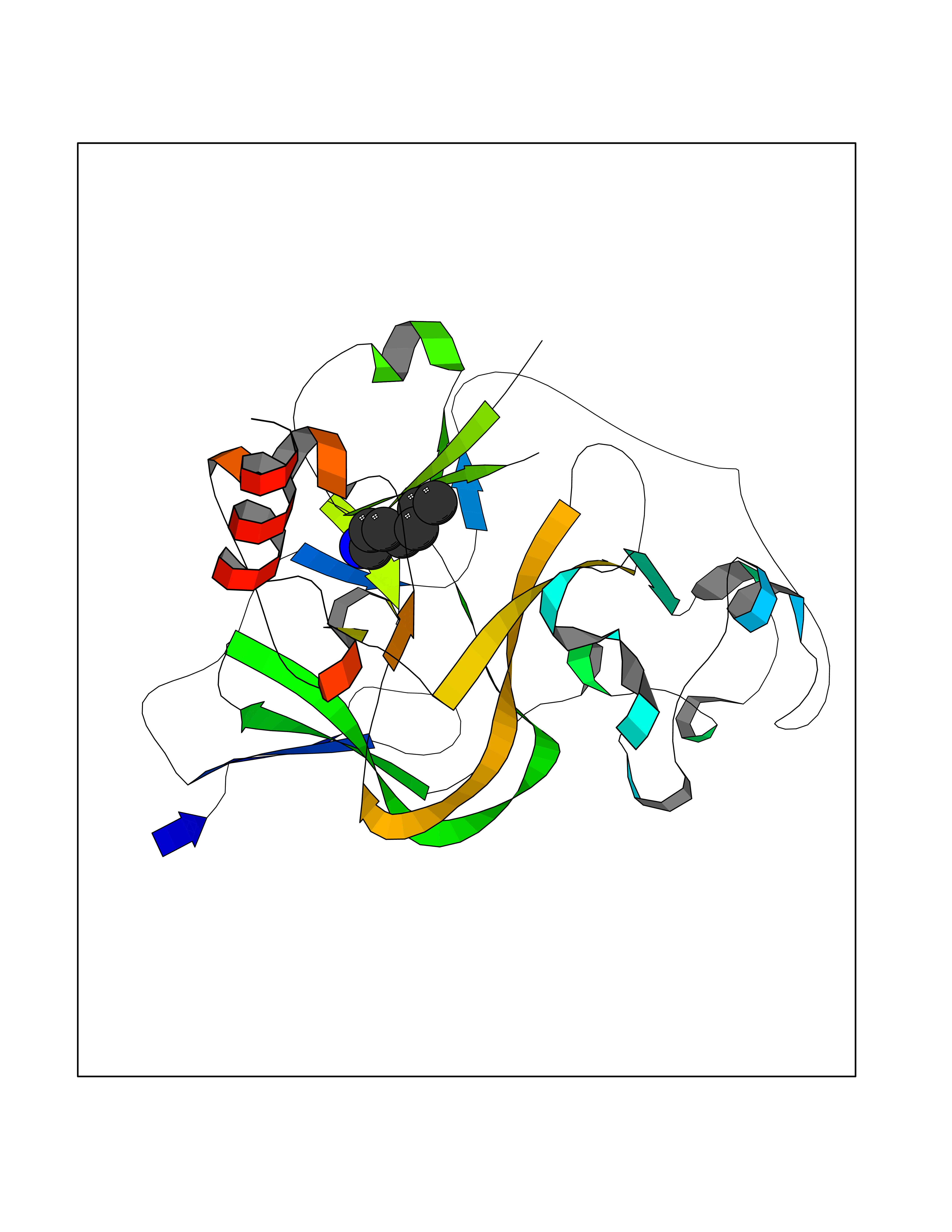 Molscript Map 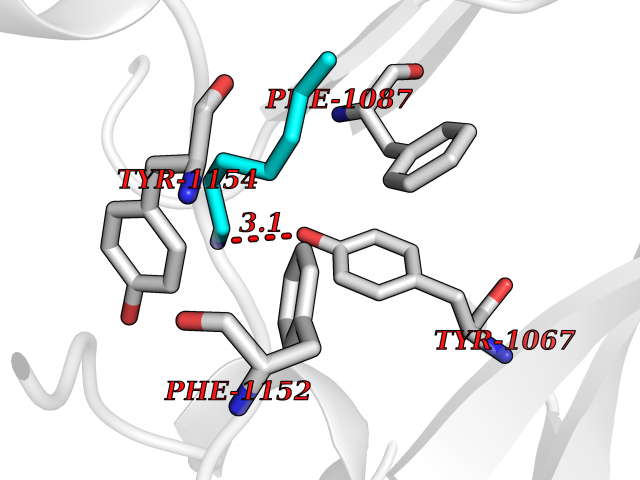 Pymol Map 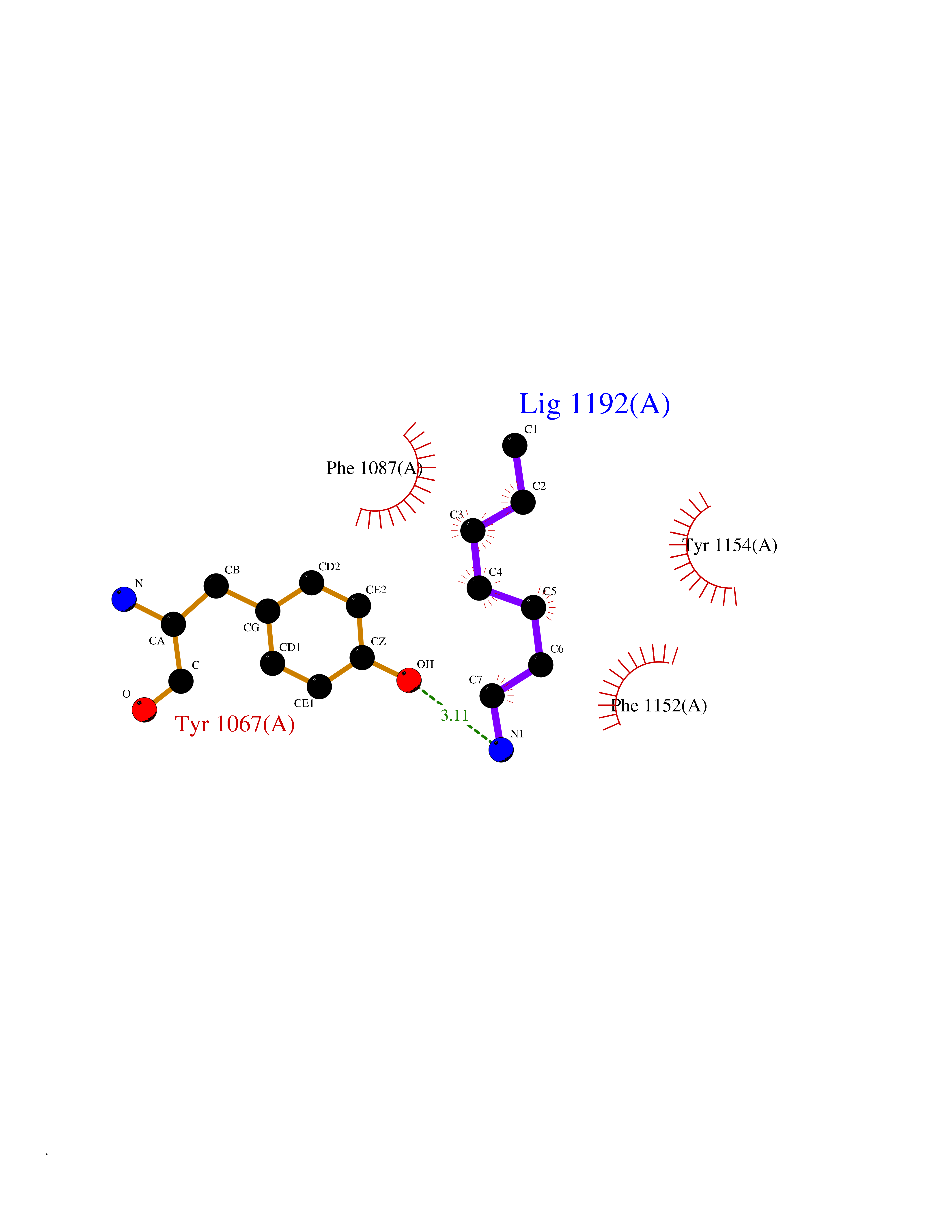 Ligplot Map 3D Binding mode Sequence TEKIICRDVARGYENVPIPCVNGVDGEPCPEDYKYISENCETSTMNIDRNITHLQHCTCVDDCSSSNCLCGQLSIRCWYDKDGRLLQEFNKIEPPLIFECNQACSCWRNCKNRVVQSGIKVRLQLYRTAKMGWGVRALQTIPQGTFICEYVGELISDAEADVREDDSYLFDLDEVYCIDARYYGNISRFINHLCDPNIIPVRVFMLHQDLRFPRIAFFSSRDIRTGEELGFDYGDRFWDIKSKYFTCQCGSEKCKHSAEAIALEQSRLA Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Fascin (FSCN1) | 6I18 | 5.03 | |
Target general information Gen name FSCN1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Singed-like protein; SNL; HSN; FAN1; 55 kDa actin-bundling protein; 55 kDa actin bundling protein Protein family Fascin family Biochemical class NA Function Plays a role in the organization of actin filament bundles and the formation of microspikes, membrane ruffles, and stress fibers. Important for the formation of a diverse set of cell protrusions, such as filopodia, and for cell motility and migration. Organizes filamentous actin into bundles with a minimum of 4. 1:1 actin/fascin ratio. Related diseases Familial adenomatous polyposis 2 (FAP2) [MIM:608456]: A condition characterized by the development of multiple colorectal adenomatous polyps, benign neoplasms derived from glandular epithelium. Some affected individuals may develop colorectal carcinoma. {ECO:0000269|PubMed:11818965, ECO:0000269|PubMed:12606733, ECO:0000269|PubMed:12853198, ECO:0000269|PubMed:15366000, ECO:0000269|PubMed:16134147, ECO:0000269|PubMed:16287072, ECO:0000269|PubMed:16557584, ECO:0000269|PubMed:16941501, ECO:0000269|PubMed:18091433, ECO:0000269|PubMed:18515411, ECO:0000269|PubMed:19953527, ECO:0000269|PubMed:20418187, ECO:0000269|PubMed:20848659, ECO:0000269|PubMed:25820570, ECO:0000269|PubMed:26694661}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Gastric cancer (GASC) [MIM:613659]: A malignant disease which starts in the stomach, can spread to the esophagus or the small intestine, and can extend through the stomach wall to nearby lymph nodes and organs. It also can metastasize to other parts of the body. The term gastric cancer or gastric carcinoma refers to adenocarcinoma of the stomach that accounts for most of all gastric malignant tumors. Two main histologic types are recognized, diffuse type and intestinal type carcinomas. Diffuse tumors are poorly differentiated infiltrating lesions, resulting in thickening of the stomach. In contrast, intestinal tumors are usually exophytic, often ulcerating, and associated with intestinal metaplasia of the stomach, most often observed in sporadic disease. {ECO:0000269|PubMed:15273732, ECO:0000269|PubMed:25820570}. The gene represented in this entry may be involved in disease pathogenesis. Somatic mutations contribute to the development of a sub-set of sporadic gastric cancers in carriers of Helicobacter pylori (PubMed:15273732). {ECO:0000269|PubMed:15273732}. Drugs (DrugBank ID) NA Interacts with Q9BXW9; P40692; Q9ULL4; P63104 EC number NA Uniprot keywords 3D-structure; Acetylation; Actin-binding; Cell junction; Cell projection; Cytoplasm; Cytoskeleton; Direct protein sequencing; Isopeptide bond; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 53682.4 Length 484 Aromaticity 0.1 Instability index 34.21 Isoelectric point 7.24 Charge (pH=7) 0.65 2D Binding mode Binding energy (Kcal/mol) -6.86 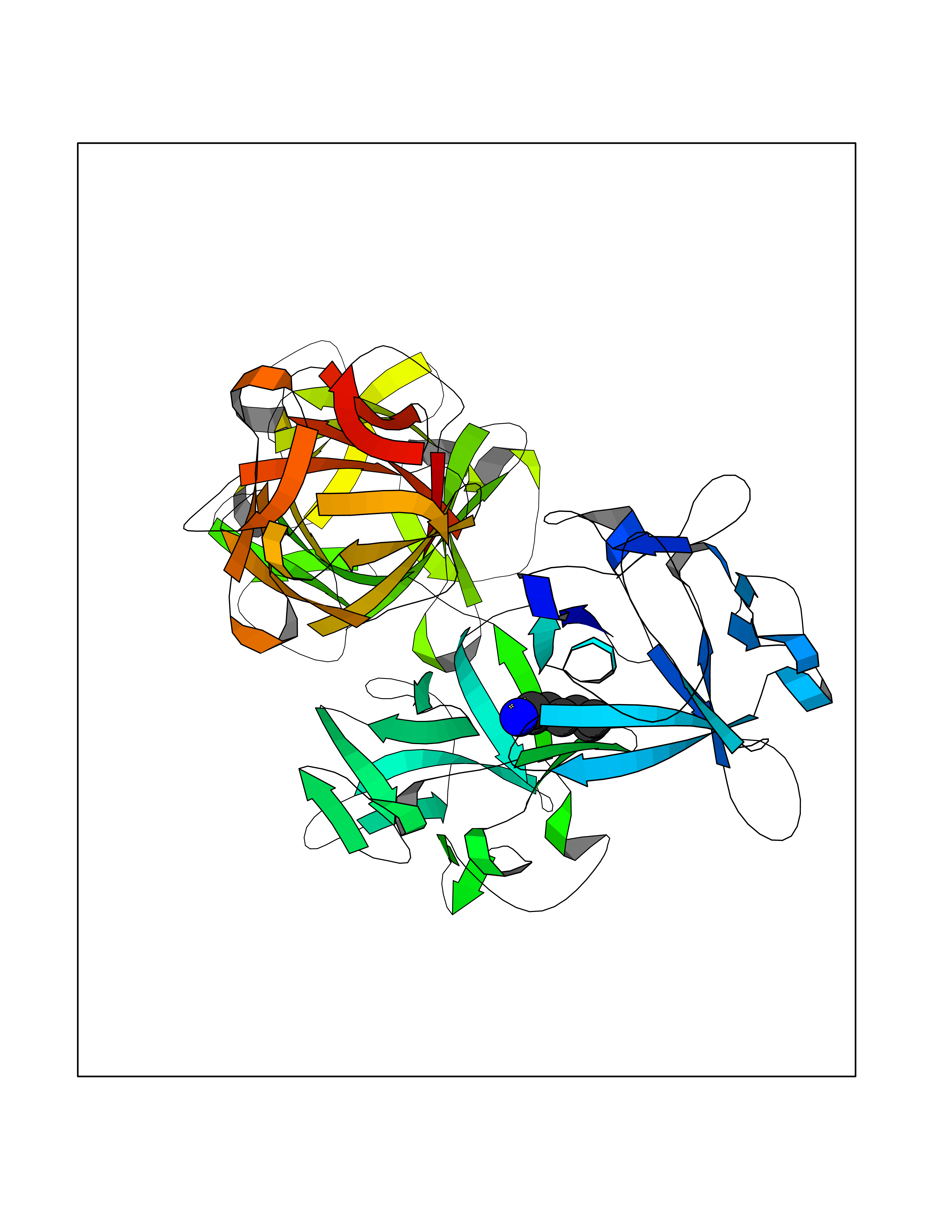 Molscript Map 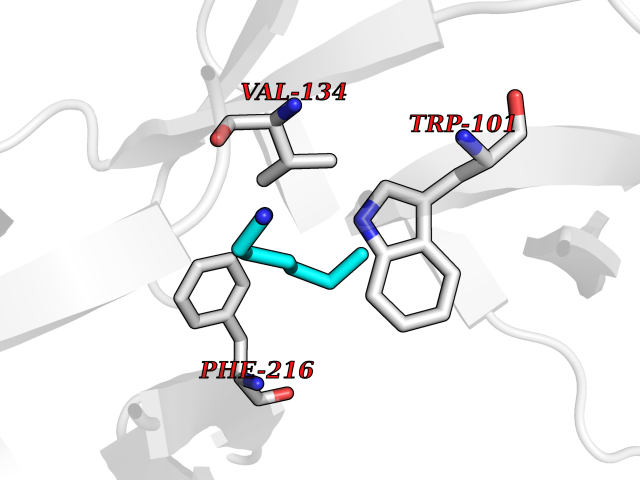 Pymol Map 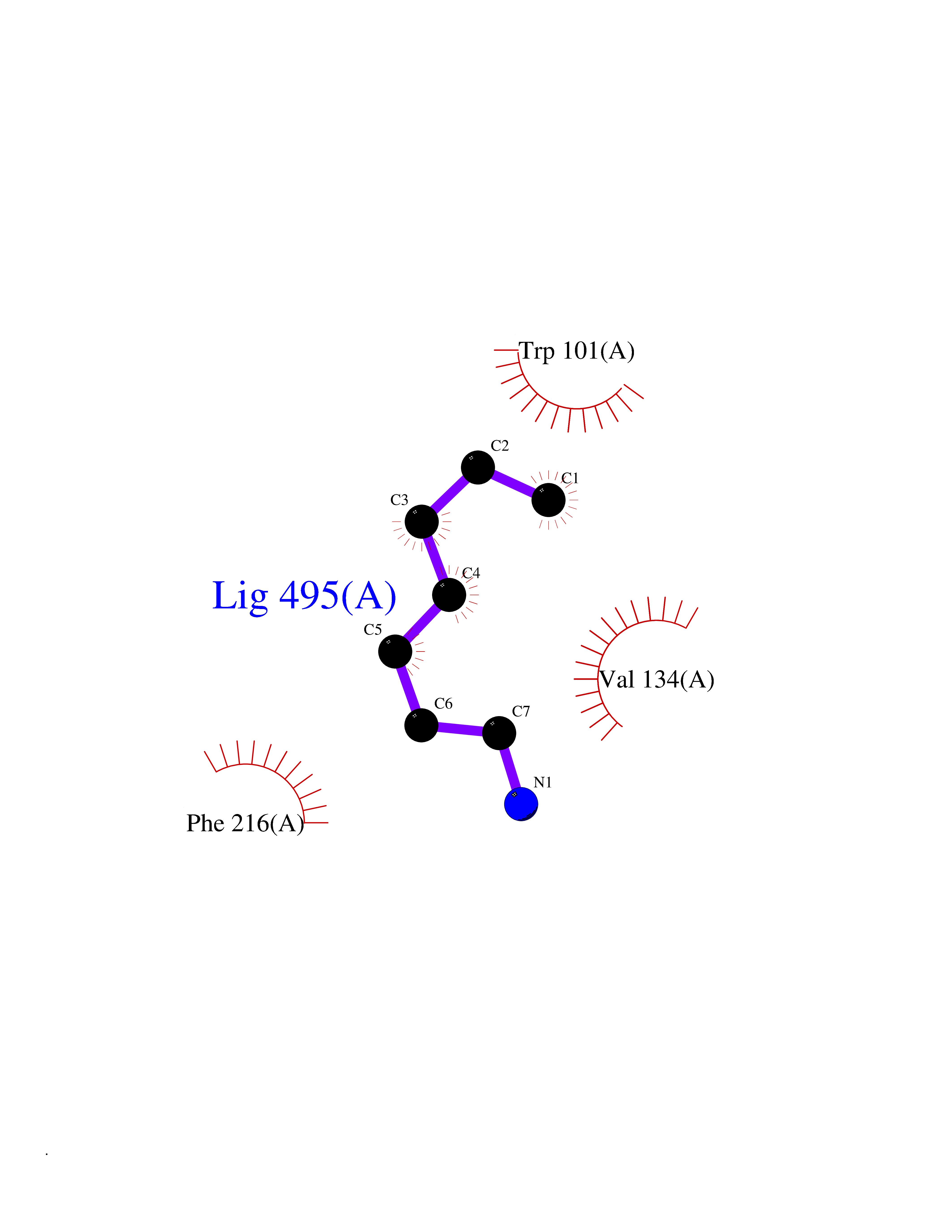 Ligplot Map 3D Binding mode Sequence VQIQFGLINCGNKYLTAEAFGFKVNASASSLKKKQIWTLEQPPDEAGSAAVCLRSHLGRYLAADKDGNVTCEREVPGPDCRFLIVAHDDGRWSLQSEAHRRYFGGTEDRLSCFAQTVSPAEKWSVHIAMHPQVNIYSVTRKRYAHLSARPADEIAVDRDVPWGVDSLITLAFQDQRYSVQTADHRFLRHDGRLVARPEPATGYTLEFRSGKVAFRDCEGRYLAPSGPSGTLKAGKATKVGKDELFALEQSCAQVVLQAANERNVSTRQGMDLSANQDEETDQETFQLEIDRDTKKCAFRTHTGKYWTLTATGGVQSTASSKNASCYFDIEWRDRRITLRASNGKFVTSKKNGQLAASVETAGDSELFLMKLINRPIIVFRGEHGFIGCRKVTGTLDANRSSYDVFQLEFNDGAYNIKDSTGKYWTVGSDSAVTSSGDTPVDFFFEFCDYNKVAIKVGGRYLKGDHAGVLKASAETVDPASLWEY Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | Aggrecanase-1 (ADAMTS4) | 2RJP | 5.03 | |
Target general information Gen name ADAMTS4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aggrecanase 1; ADMP-1; ADAMTS4; ADAM-TS4; ADAM-TS 4; A disintegrin and metalloproteinase with thrombospondin motifs 4 Protein family NA Biochemical class Peptidase Function Cleaves aggrecan, a cartilage proteoglycan, and may be involved in its turnover. May play an important role in the destruction of aggrecan in arthritic diseases. Could also be a critical factor in the exacerbation of neurodegeneration in Alzheimer disease. Cleaves aggrecan at the '392-Glu-|-Ala-393' site. Related diseases Familial male precocious puberty (FMPP) [MIM:176410]: In FMPP the receptor is constitutively activated. {ECO:0000269|PubMed:11134146, ECO:0000269|PubMed:11391350, ECO:0000269|PubMed:7629248, ECO:0000269|PubMed:7692306, ECO:0000269|PubMed:7714085, ECO:0000269|PubMed:7757065, ECO:0000269|PubMed:8281137, ECO:0000269|PubMed:8829636, ECO:0000269|PubMed:8929952, ECO:0000269|PubMed:9467560, ECO:0000269|PubMed:9661624}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Luteinizing hormone resistance (LHR) [MIM:238320]: An autosomal recessive disorder characterized by unresponsiveness to luteinizing hormone, defective sexual development in males, and defective follicular development and ovulation, amenorrhea and infertility in females. Two forms of the disorder have been defined in males. Type 1 is a severe form characterized by complete 46,XY male pseudohermaphroditism, low testosterone and high luteinizing hormone levels, total lack of responsiveness to luteinizing and chorionic gonadotropin hormones, lack of breast development, and absent development of secondary male sex characteristics. Type 2, a milder form, displays a broader range of phenotypic expression ranging from micropenis to severe hypospadias. {ECO:0000269|PubMed:12050206, ECO:0000269|PubMed:15372531, ECO:0000269|PubMed:15472221, ECO:0000269|PubMed:19551906, ECO:0000269|PubMed:7719343, ECO:0000269|PubMed:8559204, ECO:0000269|PubMed:9215288, ECO:0000269|PubMed:9514160, ECO:0000269|PubMed:9626144, ECO:0000269|PubMed:9626653}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06822 Interacts with NA EC number EC 3.4.24.82 Uniprot keywords 3D-structure; Alternative splicing; Cleavage on pair of basic residues; Direct protein sequencing; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 31309.4 Length 291 Aromaticity 0.07 Instability index 42.35 Isoelectric point 5.97 Charge (pH=7) -7.02 2D Binding mode Binding energy (Kcal/mol) -6.86 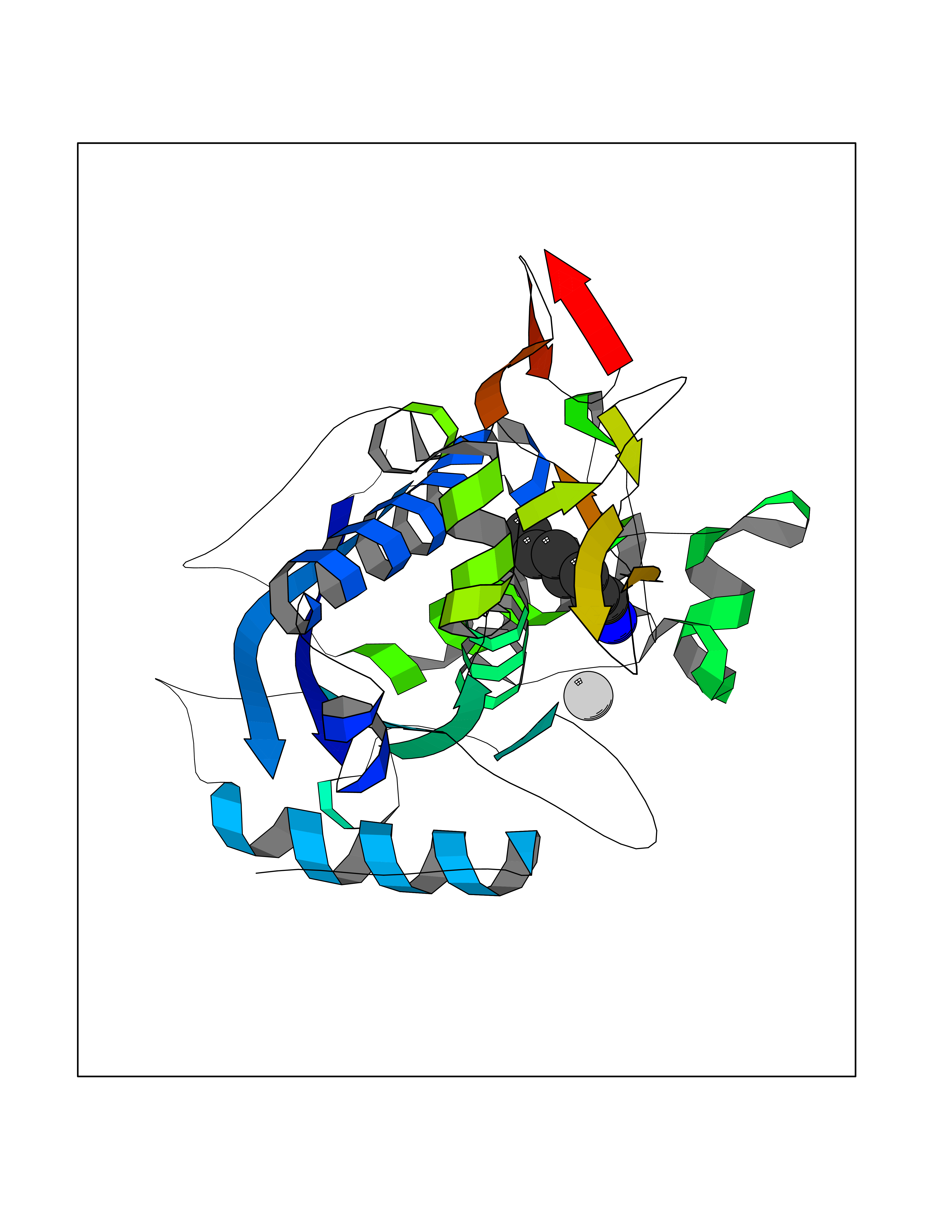 Molscript Map 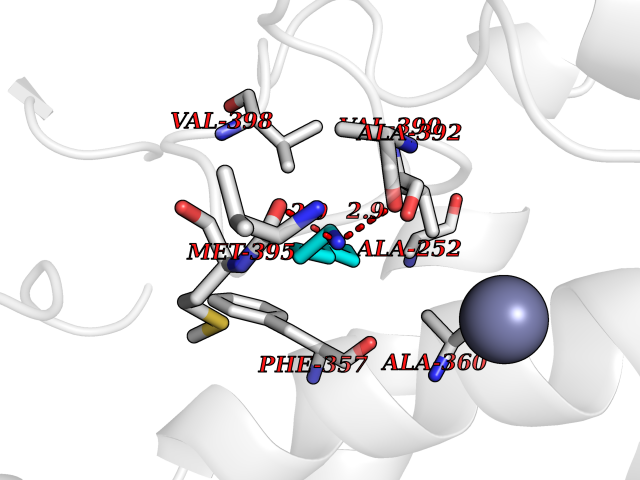 Pymol Map 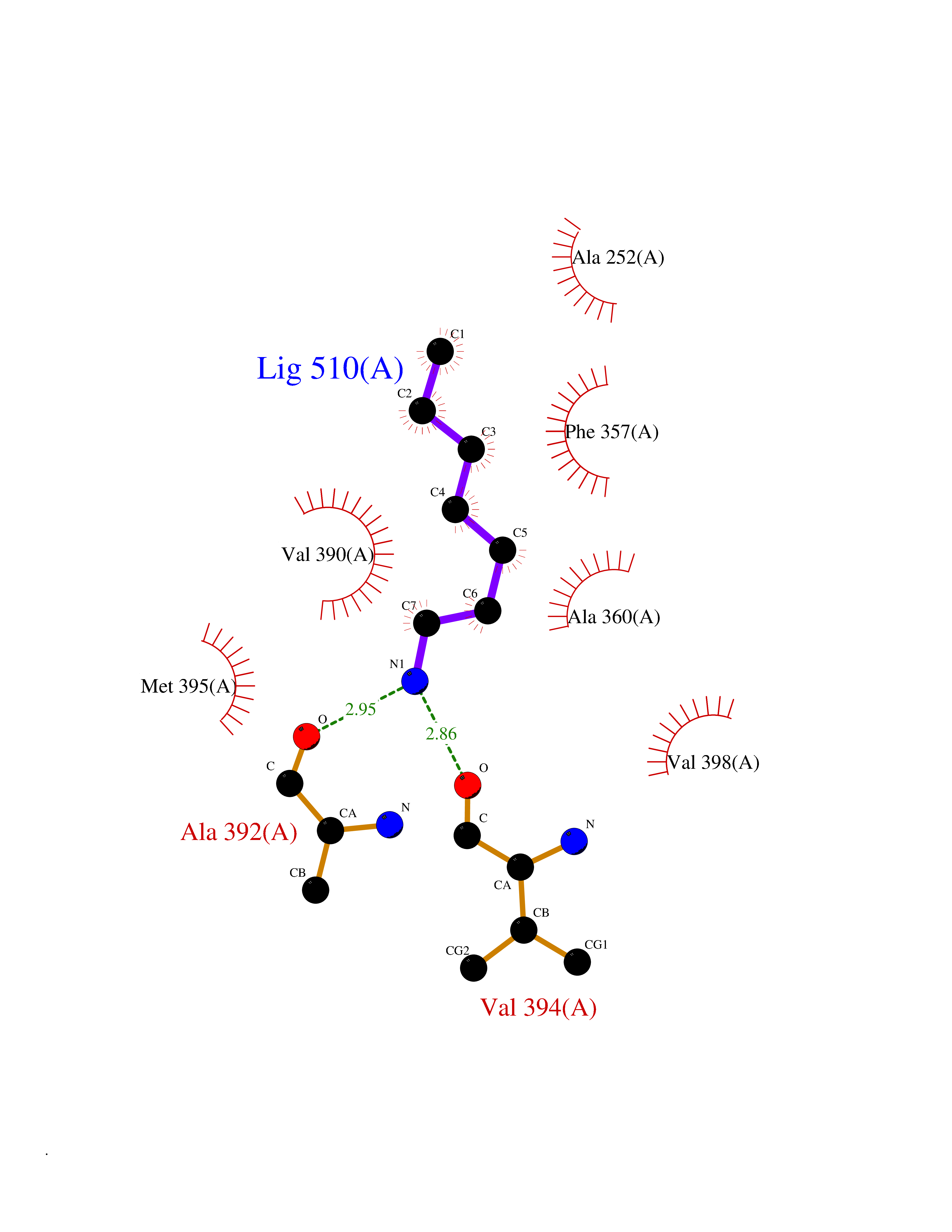 Ligplot Map 3D Binding mode Sequence ASLSRFVETLVVADDKMAAFHGAGLKRYLLTVMAAAAKAFKHPSIRNPVSLVVTRLVILEGPQVGPSAAQTLRSFCAWQRGLNTPEDSDPDHFDTAILFTRQDLCGVSTCDTLGMADVGTVCDPARSCAIVEDDGLQSAFTAAHQLGHVFNMLHDNSKPCISLNGPLSTSRHVMAPVMAHVDPEEPWSPCSARFITDFLDNGYGHCLLDKPEAPLHLPVTFPGKDYDADRQCQLTFGPDSRHCPQLPPPCAALWCSGHLNGHAMCQTKHSPWADGTPCGPAQACMGGRCLH Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Sucrase-isomaltase, intestinal | 3LPP | 5.02 | |
Target general information Gen name SI Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Glycosyl hydrolase 31 family Biochemical class Hydrolase Function Alpha-1,4-glucosidase activity.Carbohydrate binding.Oligo-1,6-glucosidase activity.Sucrose alpha-glucosidase activity. Related diseases Congenital sucrase-isomaltase deficiency (CSID) [MIM:222900]: Autosomal recessive intestinal disorder that is clinically characterized by fermentative diarrhea, abdominal pain, and cramps upon ingestion of sugar. The symptoms are the consequence of absent or drastically reduced enzymatic activities of sucrase and isomaltase. The prevalence of CSID is 0.02 % in individuals of European descent and appears to be much higher in Greenland, Alaskan, and Canadian native people. CSID arises due to post-translational perturbations in the intracellular transport, polarized sorting, aberrant processing, and defective function of SI. {ECO:0000269|PubMed:10903344, ECO:0000269|PubMed:11340066, ECO:0000269|PubMed:14724820, ECO:0000269|PubMed:16329100, ECO:0000269|PubMed:8609217}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00284; DB00747 Interacts with O43451 EC number 3.2.1.10; 3.2.1.48 Uniprot keywords 3D-structure; Cell membrane; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Glycosidase; Hydrolase; Membrane; Multifunctional enzyme; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Signal-anchor; Sulfation; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 97633.9 Length 853 Aromaticity 0.13 Instability index 32 Isoelectric point 5.23 Charge (pH=7) -23.62 2D Binding mode Binding energy (Kcal/mol) -6.84  Molscript Map 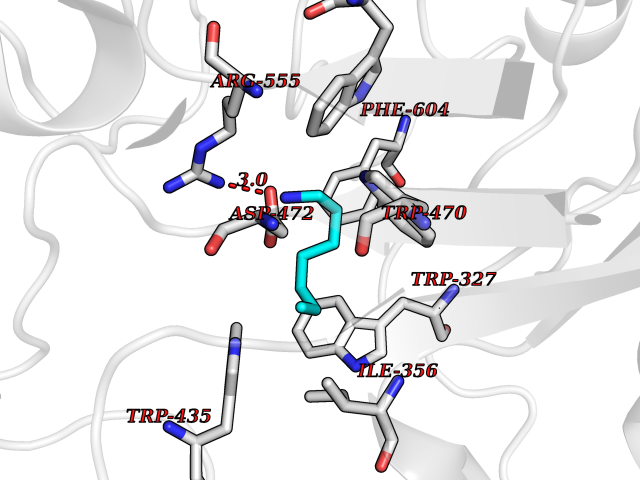 Pymol Map 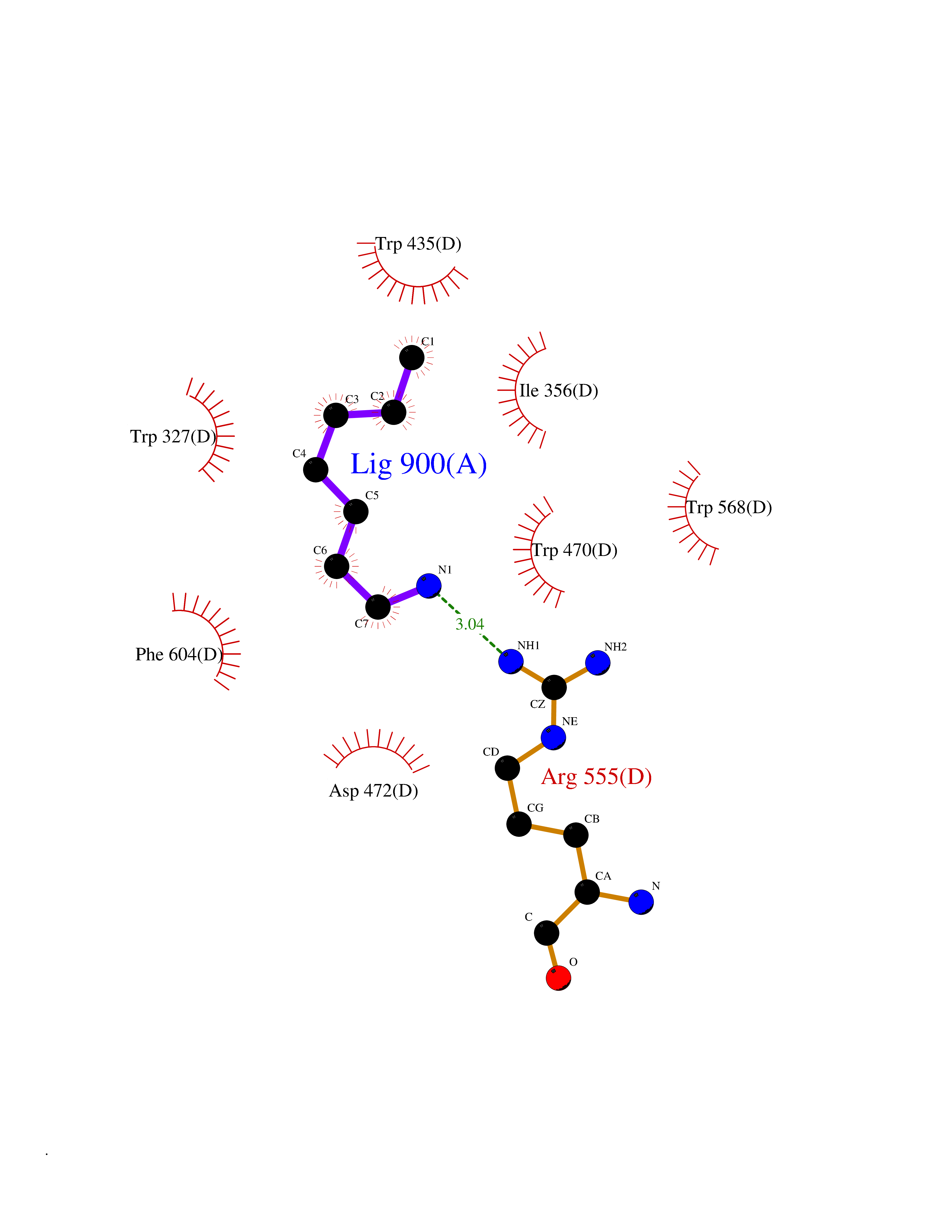 Ligplot Map 3D Binding mode Sequence LNDPVNVRINCIPEQFPTEGICAQRGCCWRPWNDSLIPWCFFVDNHGYNVQDMTTTSIGVEAKLNRIPSPTLFGNDINSVLFTTQNQTPNRFRFKITDPNNRRYEVPHQYVKEFTGPTVSDTLYDVKVAQNPFSIQVIRKSNGKTLFDTSIGPLVYSDQYLQISARLPSDYIYGIGEQVHKRFRHDLSWKTWPIFTRDQLPGDNNNNLYGHQTFFMCIEDTSGKSFGVFLMNSNAMEIFIQPTPIVTYRVTGGILDFYILLGDTPEQVVQQYQQLVGLPAMPAYWNLGFQLSRWNYKSLDVVKEVVRRNREAGIPFDTQVTDIDYMEDKKDFTYDQVAFNGLPQFVQDLHDHGQKYVIILDPAISIGRRYATYERGNTQHVWINESDGSTPIIGEVWPGLTVYPDFTNPNCIDWWANECSIFHQEVQYDGLWIDMNEVSSFIQGSTKGCNVNKLNYPPFTPDILDKLMYSKTICMDAVQNWGKQYDVHSLYGYSMAIATEQAVQKVFPNKRSFILTRSTFAGSGRHAAHWLGDNTASWEQMEWSITGMLEFSLFGIPLVGADICGFVAETTEELCRRWMQLGAFYPFSRNHNSDGYEHQDPAFFGQNSLLVKSSRQYLTIRYTLLPFLYTLFYKAHVFGETVARPVLHEFYEDTNSWIEDTEFLWGPALLITPVLKQGADTVSAYIPDAIWYDYESGAKRPWRKQRVDMYLPADKIGLHLRGGYIIPIQEPDVTTTASRKNPLGLIVALGENNTAKGDFFWDDGETKDTIQNGNYILYTFSVSNNTLDIVCTHSSYQEGTTLAFQTVKILGLTDSVTEVRVAEHSNFTYDASNQVLLIADLKLNLGRNFSVQW Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Serine--pyruvate aminotransferase | 5HHY | 5.02 | |
Target general information Gen name AGXT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms AGT1;SPAT Protein family Class-V pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transferase Function Alanine-glyoxylate transaminase activity.Amino acid binding.Identical protein binding.Protein homodimerization activity.Protein self-association.Pyridoxal phosphate binding.Receptor binding.Serine-pyruvate transaminase activity.Transaminase activity. Related diseases Hyperoxaluria primary 1 (HP1) [MIM:259900]: An inborn error of glyoxylate metabolism characterized by increased excretion of oxalate and glycolate, and progressive tissue accumulation of insoluble calcium oxalate. Affected individuals are at risk for nephrolithiasis, nephrocalcinosis and early onset end-stage renal disease. {ECO:0000269|PubMed:10394939, ECO:0000269|PubMed:10453743, ECO:0000269|PubMed:10541294, ECO:0000269|PubMed:10862087, ECO:0000269|PubMed:10960483, ECO:0000269|PubMed:12559847, ECO:0000269|PubMed:12777626, ECO:0000269|PubMed:1301173, ECO:0000269|PubMed:1349575, ECO:0000269|PubMed:15253729, ECO:0000269|PubMed:15849466, ECO:0000269|PubMed:15961946, ECO:0000269|PubMed:15963748, ECO:0000269|PubMed:16971151, ECO:0000269|PubMed:1703535, ECO:0000269|PubMed:17495019, ECO:0000269|PubMed:2039493, ECO:0000269|PubMed:23229545, ECO:0000269|PubMed:24055001, ECO:0000269|PubMed:24934730, ECO:0000269|PubMed:26149463, ECO:0000269|PubMed:8101040, ECO:0000269|PubMed:9192270, ECO:0000269|PubMed:9604803}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08060; DB00160; DB02079; DB00145; DB04083; DB00114; DB00133 Interacts with Q5BKX5-3; P21549; P0C7T5; Q9NR55; Q9H5F2; Q9UKJ5; A8MQ03; O43281-2; A1KXE4-2; Q5TD97; P49356; P53539; P49639; Q15323; O76011; O76014; Q6A162; Q07627; P60328; Q9BYR8; Q3LI67; Q8IUC2; Q9BYQ4; O60711; Q99750; Q5VZ52; P0DPK4; O43482; P50542-1; O15496; Q6ZR37; Q9NQX0; Q8HWS3; Q5W111-2; Q8WVR3; Q6DKK2 EC number 2.6.1.44; 2.6.1.51 Uniprot keywords 3D-structure; Acetylation; Aminotransferase; Disease variant; Peroxisome; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84537 Length 771 Aromaticity 0.07 Instability index 36.81 Isoelectric point 8.42 Charge (pH=7) 6.81 2D Binding mode Binding energy (Kcal/mol) -6.84 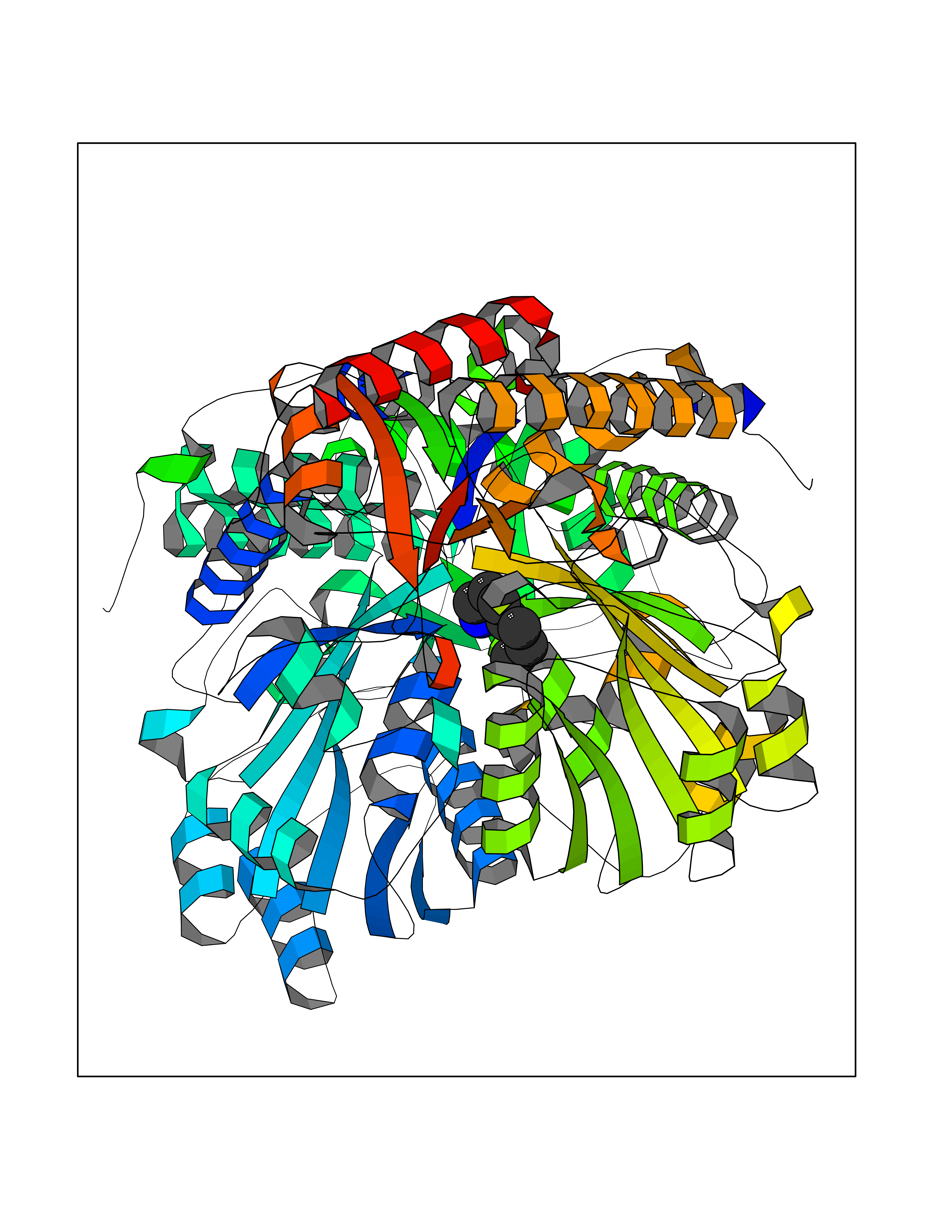 Molscript Map 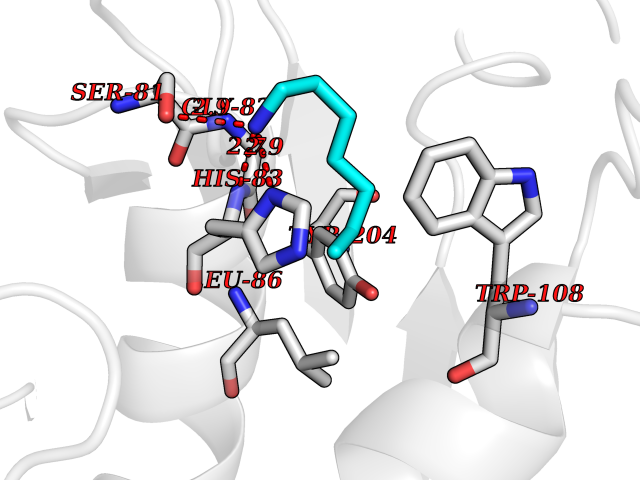 Pymol Map 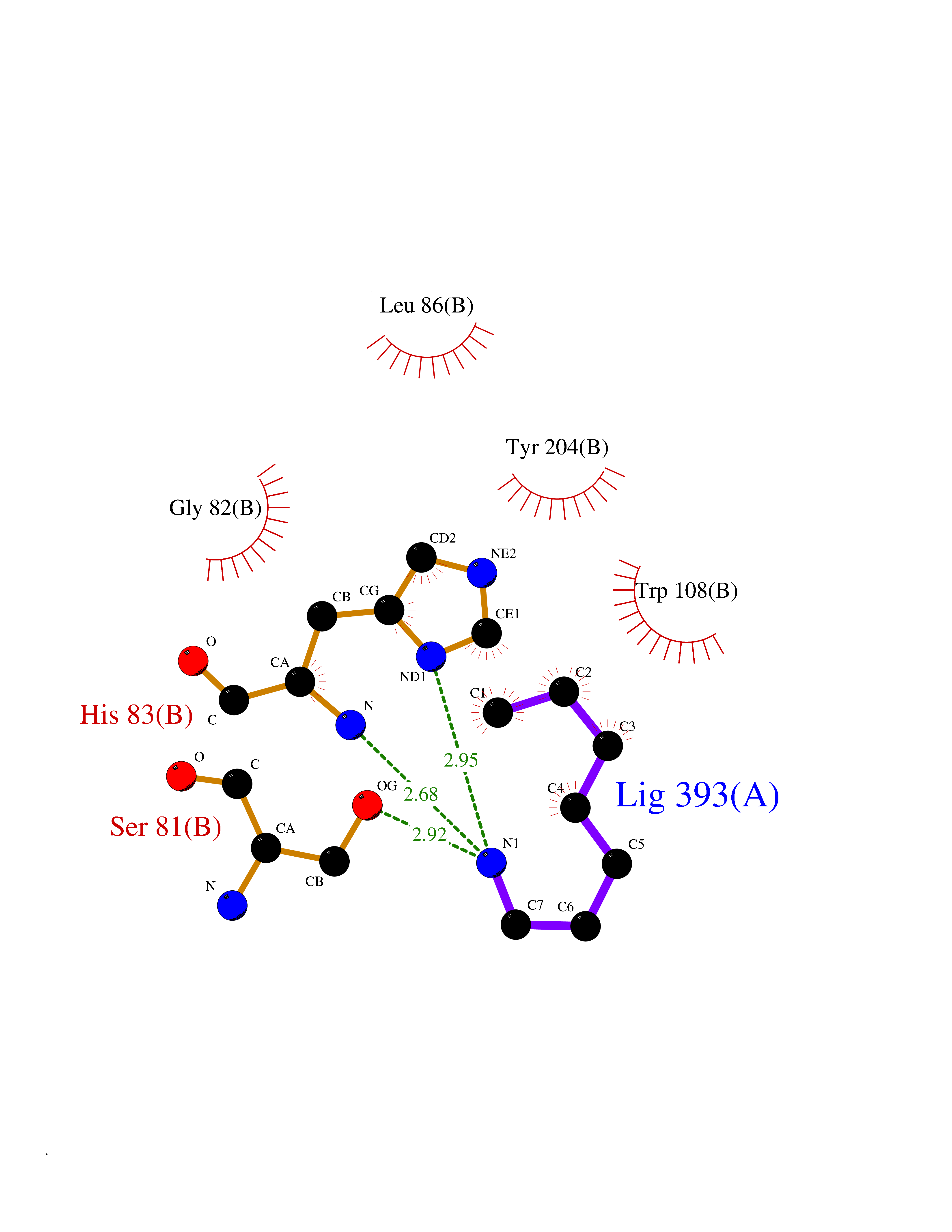 Ligplot Map 3D Binding mode Sequence LLVTPPKALLKPLSIPNQLLLGPGPSNLPPRIMAAGGLQMIGSMSKDMYQIMDEIKEGIQYVFQTRNPLTLVISGSGHCALEAALVNVLEPGDSFLVGANGIWGQRAVDIGERIGARVHPMTKDPGGHYTLQEVEEGLAQHKPVLLFLTHGESSTGVLQPLDGFGELCHRYKCLLLVDSVASLGGTPLYMDRQGIDILYSGSQKALNAPPGTSLISFSDKAKKKMYSRKTKPFSFYLDIKWLANFWGCDDQPRMYHHTIPVISLYSLRESLALIAEQGLENSWRQHREAAAYLHGRLQALGLQLFVKDPALRLPTVTTVAVPAGYDWRDIVSYVIDHFDIEIMGGLGPSTGKVLRIGLLGCNATRENVDRVTEALRAALQHCPKKLLVTPPKALLKPLSIPNQLLLGPGPSNLPPRIMAAGGLQMIGSMSKDMYQIMDEIKEGIQYVFQTRNPLTLVISGSGHCALEAALVNVLEPGDSFLVGANGIWGQRAVDIGERIGARVHPMTKDPGGHYTLQEVEEGLAQHKPVLLFLTHGESSTGVLQPLDGFGELCHRYKCLLLVDSVASLGGTPLYMDRQGIDILYSGSQKALNAPPGTSLISFSDKAKKKMYSRKTKPFSFYLDIKWLANFWGCDDQPRMYHHTIPVISLYSLRESLALIAEQGLENSWRQHREAAAYLHGRLQALGLQLFVKDPALRLPTVTTVAVPAGYDWRDIVSYVIDHFDIEIMGGLGPSTGKVLRIGLLGCNATRENVDRVTEALRAALQHCPKKK Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Plasmepsin-2 | 2BJU | 5.02 | |
Target general information Gen name N/A Organism Plasmodium falciparum (isolate HB3) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase A1 family Biochemical class Hydrolase Function Aspartic-type endopeptidase activity. Related diseases Short/branched-chain acyl-CoA dehydrogenase deficiency (SBCADD) [MIM:610006]: Autosomal recessive disorder and consists of a defect in catabolism of L-isoleucine which is characterized by an increase of 2-methylbutyrylglycine and 2-methylbutyrylcarnitine in blood and urine. Affected individuals have seizures and psychomotor delay as the main clinical features. {ECO:0000269|PubMed:10832746, ECO:0000269|PubMed:11013134, ECO:0000269|PubMed:16317551}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04378; DB04373; DB11638; DB01218; DB02505; DB03063 Interacts with NA EC number 3.4.23.39 Uniprot keywords 3D-structure; Aspartyl protease; Direct protein sequencing; Disulfide bond; Hydrolase; Membrane; Protease; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Vacuole; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 36923.5 Length 329 Aromaticity 0.13 Instability index 44.31 Isoelectric point 4.67 Charge (pH=7) -17.94 2D Binding mode Binding energy (Kcal/mol) -6.84 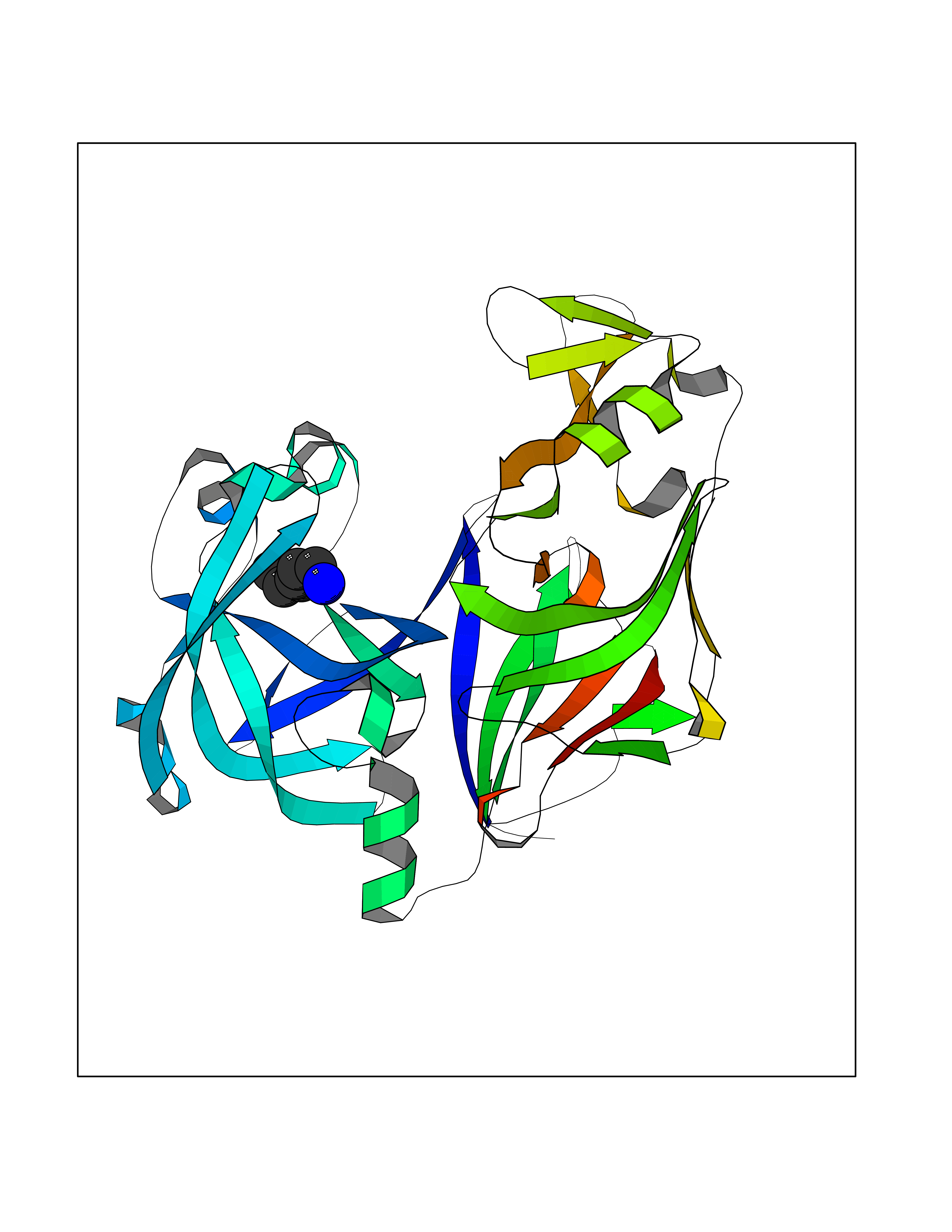 Molscript Map 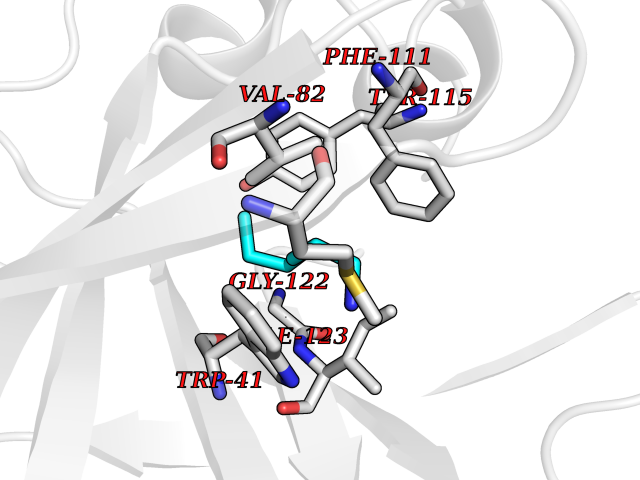 Pymol Map 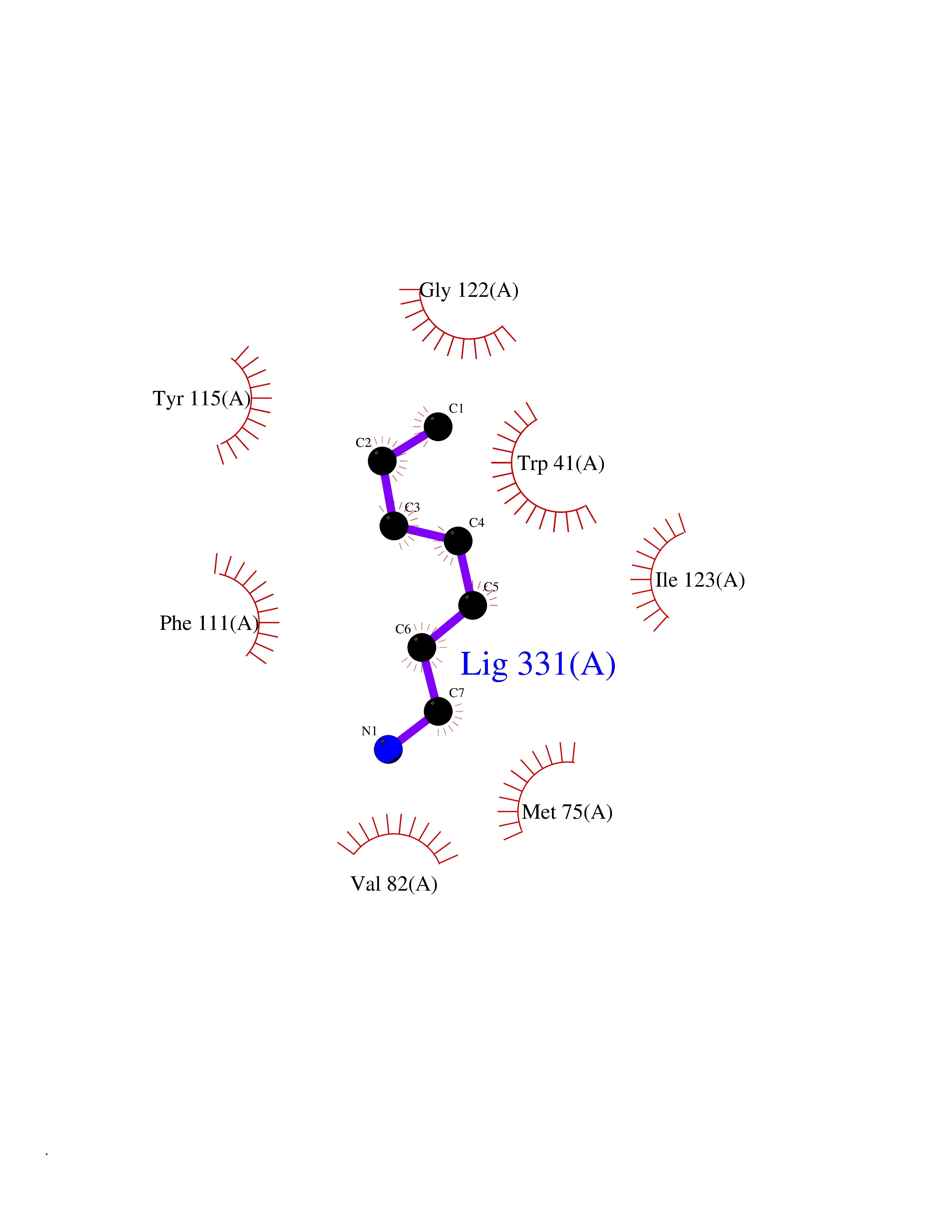 Ligplot Map 3D Binding mode Sequence SSNDNIELVDFQNIMFYGDAEVGDNQQPFTFILDTGSANLWVPSVKCTTAGCLTKHLYDSSKSRTYEKDGTKVEMNYVSGTVSGFFSKDLVTVGNLSLPYKFIEVIDTNGFEPTYTASTFDGILGLGWKDLSIGSVDPIVVELKNQNKIENALFTFYLPVHDKHTGFLTIGGIEERFYEGPLTYEKLNHDLYWQITLDAHVGNIMLEKANCIVDSGTSAITVPTDFLNKMLQNLDVIKVPFLPFYVTLCNNSKLPTFEFTSENGKYTLEPEYYLQHIEDVGPGLCMLNIIGLDFPVPTFILGDPFMRKYFTVFDYDNHSVGIALAKKNL Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Acetyl-CoA carboxylase (ACC) (EC 6.4.1.2) (Fatty acid synthetase 3) (mRNA transport-defective protein 7) [Includes: Biotin carboxylase (EC 6.3.4.14)] | 1UYS | 5.02 | |
Target general information Gen name ACC1 Organism Saccharomyces cerevisiae (strain ATCC 204508 / S288c) (Baker's yeast) Uniprot ID TTD ID NA Synonyms MTR7;YNR016C;N3175;ABP2;FAS3 Protein family NA Biochemical class NA Function Carries out three functions: biotin carboxyl carrier protein, biotin carboxylase and carboxyltransferase. Involved in the synthesis of very-long-chain fatty acid synthesis which is required to maintain a functional nuclear envelope. Required for acylation and vacuolar membrane association of VAC8 which is necessary to maintain a normal morphology of the vacuole. {ECO:0000269|PubMed:10757783, ECO:0000269|PubMed:12730220, ECO:0000269|PubMed:6103540, ECO:0000269|PubMed:6108218, ECO:0000269|PubMed:8943372}." Related diseases A chromosomal aberration involving NFKB2 is found in a case of B-cell non Hodgkin lymphoma (B-NHL). Translocation t(10;14)(q24;q32) with IGHA1. The resulting oncogene is also called Lyt-10C alpha variant.; DISEASE: A chromosomal aberration involving NFKB2 is found in a cutaneous T-cell leukemia (C-TCL) cell line. This rearrangement produces the p80HT gene which codes for a truncated 80 kDa protein (p80HT).; DISEASE: In B-cell leukemia (B-CLL) cell line, LB40 and EB308, can be found after heterogeneous chromosomal aberrations, such as internal deletions.; DISEASE: Immunodeficiency, common variable, 10 (CVID10) [MIM:615577]: A primary immunodeficiency characterized by childhood-onset of recurrent infections, hypogammaglobulinemia, and decreased numbers of memory and marginal zone B-cells. Some patients may develop autoimmune features and have circulating autoantibodies. An unusual feature is central adrenal insufficiency. {ECO:0000269|PubMed:24140114, ECO:0000269|PubMed:25524009}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q00955 EC number 6.3.4.14; 6.4.1.2 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Biotin; Cytoplasm; Direct protein sequencing; Endoplasmic reticulum; Fatty acid biosynthesis; Fatty acid metabolism; Ligase; Lipid biosynthesis; Lipid metabolism; Manganese; Membrane; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Reference proteome Protein physicochemical properties Chain ID B,C Molecular weight (Da) 145619 Length 1328 Aromaticity 0.1 Instability index 30.31 Isoelectric point 5.32 Charge (pH=7) -26.79 2D Binding mode Binding energy (Kcal/mol) -6.84  Molscript Map 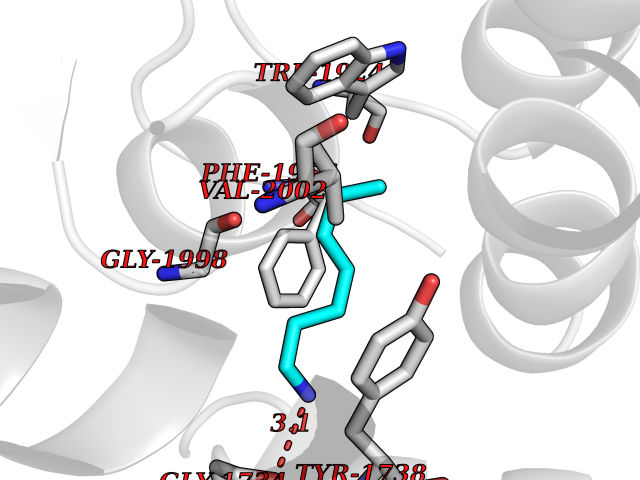 Pymol Map 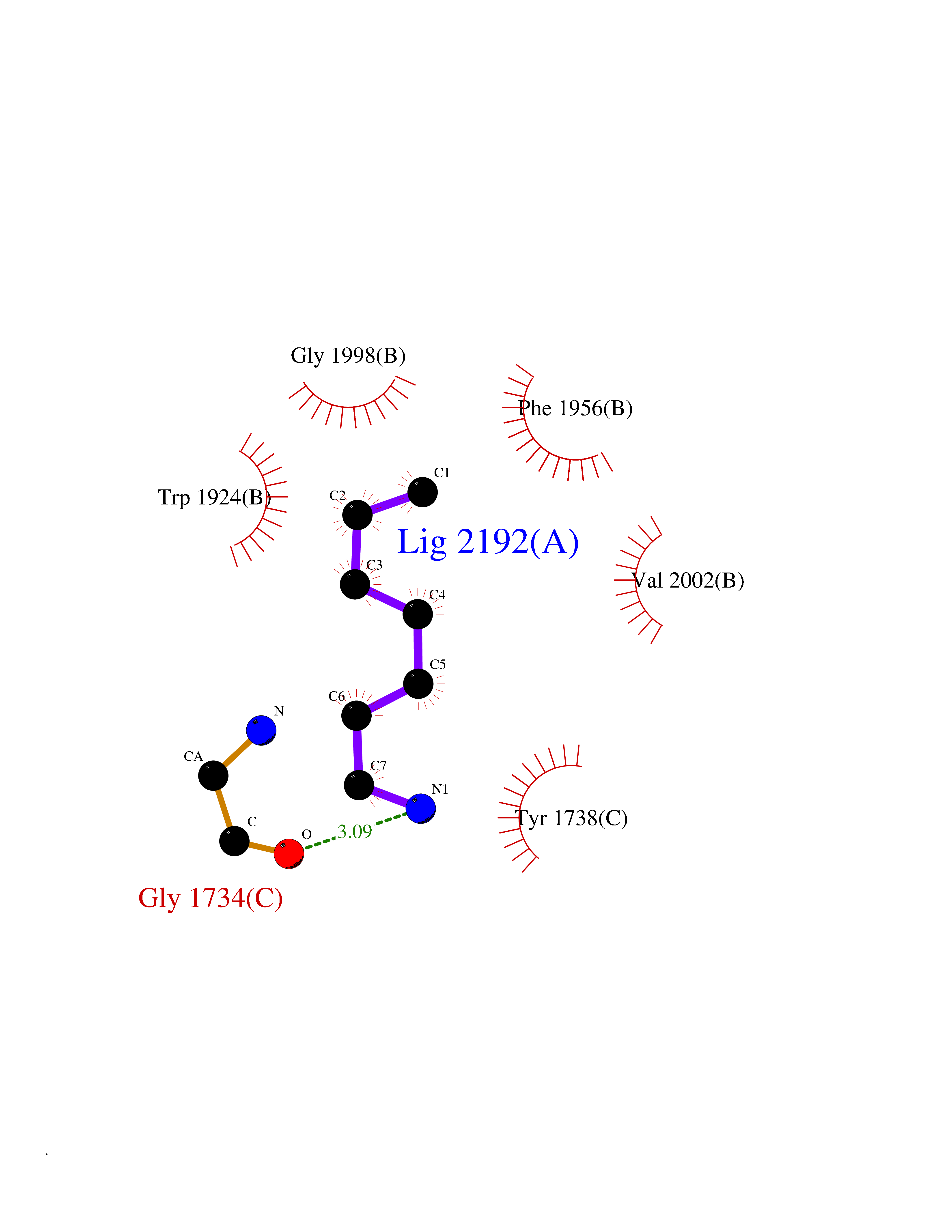 Ligplot Map 3D Binding mode Sequence WLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGLPIATPYPVKEWLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGL Hydrogen bonds contact Hydrophobic contact | ||||