Job Results:
Ligand
Structure
Job ID
1aab99f222a82120b630ad6d44e2ad5f
Job name
NA
Time
2026-02-27 11:58:02
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Cholesterol 24-hydroxylase (CYP46A1) | 3MDR | 4.56 | |
Target general information Gen name CYP46A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cytochrome P450 46A1; CYP46; Cholesterol 24S-hydroxylase; Cholesterol 24-monooxygenase; CH24H Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function Primarily catalyzes the hydroxylation (with S stereochemistry) at C-24 of cholesterol side chain, triggering cholesterol diffusion out of neurons and its further degradation. By promoting constant cholesterol elimination in neurons, may activate the mevalonate pathway and coordinate the synthesis of new cholesterol and nonsterol isoprenoids involved in synaptic activity and learning. Further hydroxylates cholesterol derivatives and hormone steroids on both the ring and side chain of these molecules, converting them into active oxysterols involved in lipid signaling and biosynthesis. Acts as an epoxidase converting cholesta-5,24-dien-3beta-ol/desmosterol into (24S),25-epoxycholesterol, an abundant lipid ligand of nuclear NR1H2 and NR1H3 receptors shown to promote neurogenesis in developing brain. May also catalyze the oxidative metabolism of xenobiotics, such as clotrimazole. P450 monooxygenase that plays a major role in cholesterol homeostasis in the brain. Related diseases Spinocerebellar ataxia, autosomal recessive, with axonal neuropathy 1 (SCAN1) [MIM:607250]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCAN1 is an autosomal recessive cerebellar ataxia (ARCA) associated with peripheral axonal motor and sensory neuropathy, distal muscular atrophy, pes cavus and steppage gait as seen in Charcot-Marie-Tooth neuropathy. All affected individuals have normal intelligence. {ECO:0000269|PubMed:12244316, ECO:0000269|PubMed:15647511, ECO:0000269|PubMed:15920477, ECO:0000269|PubMed:16141202, ECO:0000269|PubMed:17948061}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.14.14.25 Uniprot keywords 3D-structure; Alternative splicing; Cell projection; Cholesterol metabolism; Endoplasmic reticulum; Heme; Iron; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism; Synapse; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 48977 Length 427 Aromaticity 0.1 Instability index 49.59 Isoelectric point 9.04 Charge (pH=7) 6.25 2D Binding mode Binding energy (Kcal/mol) -6.21 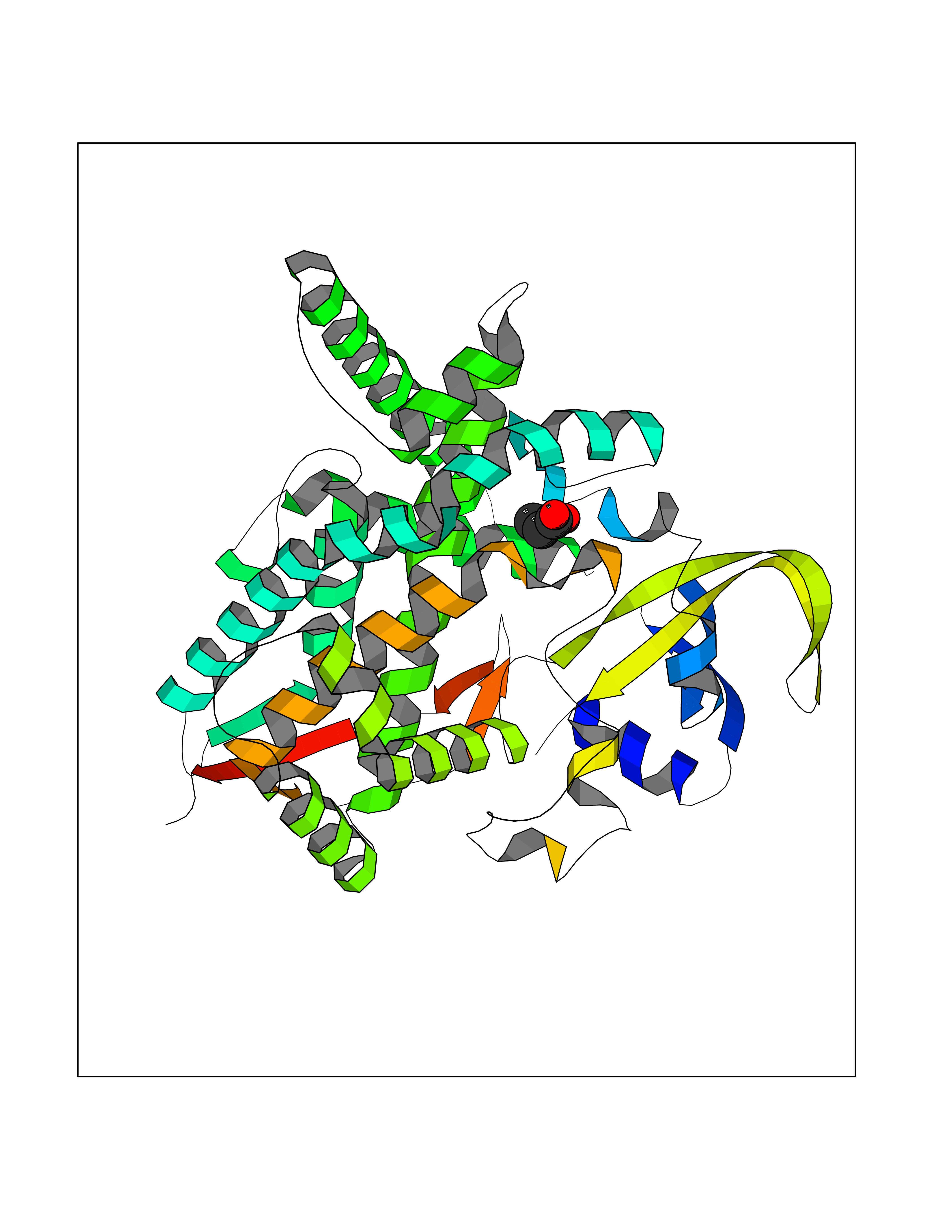 Molscript Map 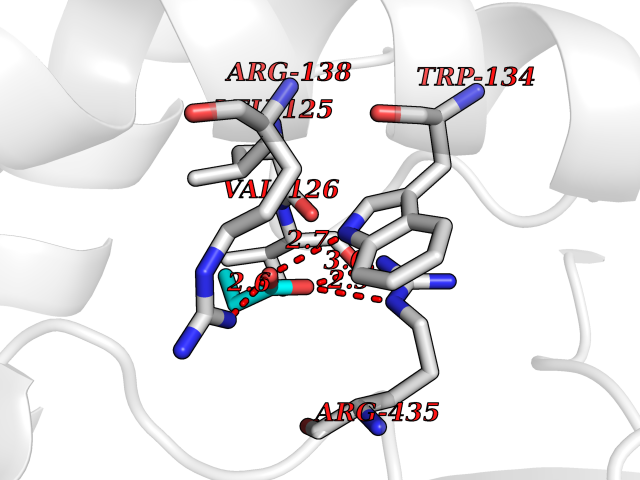 Pymol Map 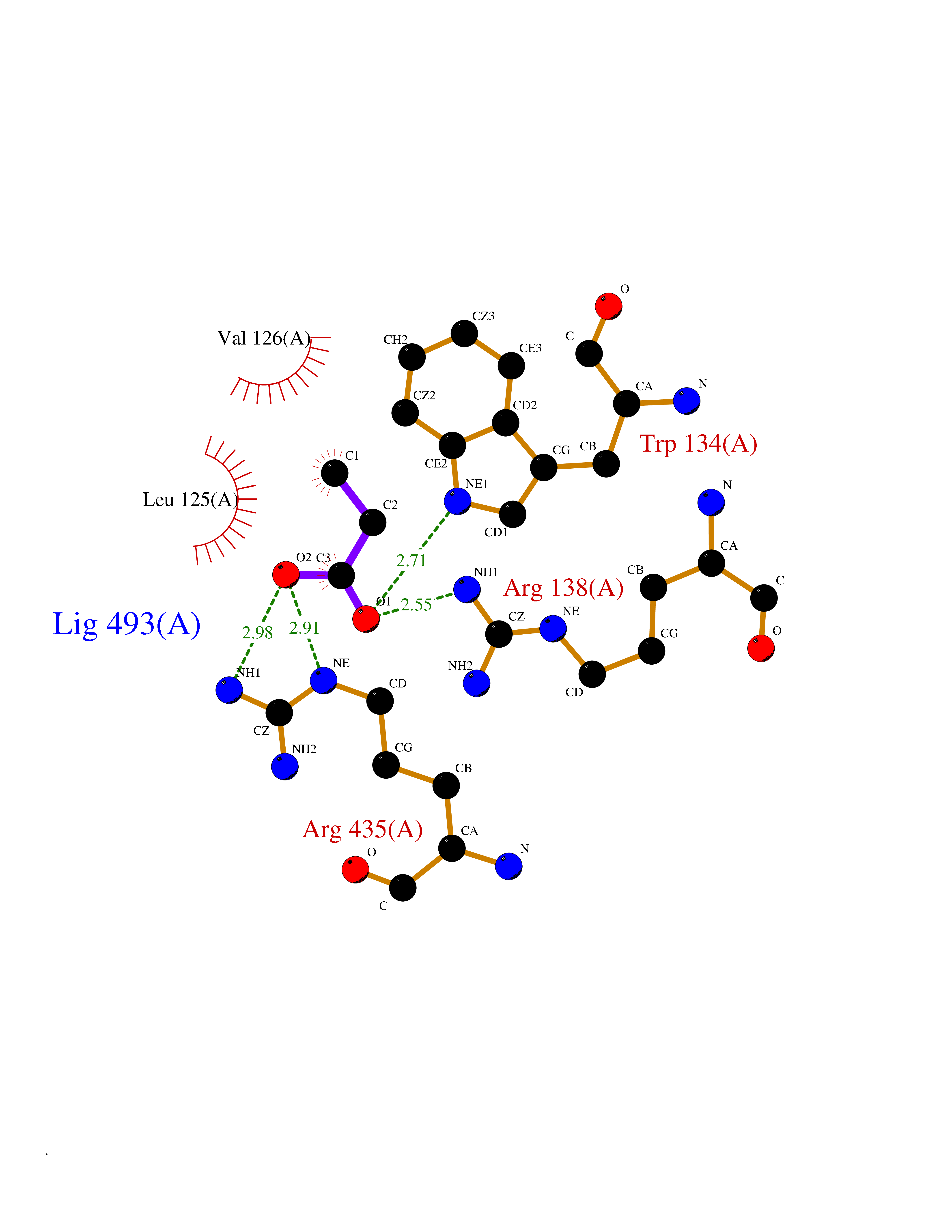 Ligplot Map 3D Binding mode Sequence RVLQDVFLDWAKKYGPVVRVNVFHKTSVIVTSPESVKKFLMSTKYNKDSKMYRALQTVFGERLFGQGLVSECNYERWHKQRRVIDLAFSRSSLVSLMETFNEKAEQLVEILEAKADGQTPVSMQDMLTYTAMDILAKAAFGMETSMLLGAQKPLSQAVKLMLEGITASRNTKRKQLREVRESIRFLRQVGRDWVQRRREALKRGEEVPADILTQILKAEEGAQDDEGLLDNFVTFFIAGHETSANHLAFTVMELSRQPEIVARLQAEVDEVIGSKRYLDFEDLGRLQYLSQVLKESLRLYPPAWGTFRLLEEETLIDGVRVPGNTPLLFSTYVMGRMDTYFEDPLTFNPDRFGPGAPKPRFTYFPFSLGHRSCIGQQFAQMEVKVVMAKLLQRLEFRLVPGQRFGLQEQATLKPLDPVLCTLRPRGW Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Chloride channel protein 7 (ClC-7) | 7CQ5 | 4.56 | |
Target general information Gen name CLCN7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms H(+)/Cl(-) exchange transporter 7; ClC-7; Chloride channel 7 alpha subunit Protein family Chloride channel (TC 2.A.49) family, ClC-7/CLCN7 subfamily Biochemical class Chloride channel Function Slowly voltage-gated channel mediating the exchange of chloride ions against protons. Functions as antiporter and contributes to the acidification of the lysosome lumen. Related diseases Osteopetrosis, autosomal recessive 4 (OPTB4) [MIM:611490]: A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves. {ECO:0000269|PubMed:11207362, ECO:0000269|PubMed:11741829, ECO:0000269|PubMed:14584882, ECO:0000269|PubMed:17033731, ECO:0000269|PubMed:19953639, ECO:0000269|PubMed:26395888, ECO:0000269|PubMed:26477479}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Osteopetrosis, autosomal dominant 2 (OPTA2) [MIM:166600]: A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. OPTA2 is the most common form of osteopetrosis, occurring in adolescence or adulthood. It is characterized by sclerosis, predominantly involving the spine, the pelvis and the skull base. {ECO:0000269|PubMed:11741829, ECO:0000269|PubMed:14584882, ECO:0000269|PubMed:19288050, ECO:0000269|PubMed:19953639, ECO:0000269|PubMed:26395888}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hypopigmentation, organomegaly, and delayed myelination and development (HOD) [MIM:618541]: An autosomal dominant pleiotropic syndrome characterized by skin and hair hypopigmentation, growth and developmental delay, organomegaly including enlarged liver, spleen and kidneys, delayed brain myelination and developmental deficit in motor skills. Skin and liver biopsies show cellular accumulation of large intracellular vacuoles. {ECO:0000269|PubMed:31155284}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q6P5T0; P19397; Q8TB36; O00258; Q53GS7; Q9UGI6-2; Q8TAF8; Q9Y5Y7; P35372-10; Q86WC4; Q9NVC3; Q8N2U9; Q7Z699; Q96MV1; Q7Z7N9 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Antiport; ATP-binding; CBS domain; Chloride; Disease variant; Ion transport; Lysosome; Membrane; Nucleotide-binding; Osteopetrosis; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID C Molecular weight (Da) 75175 Length 681 Aromaticity 0.11 Instability index 38.7 Isoelectric point 8.88 Charge (pH=7) 8.95 2D Binding mode Binding energy (Kcal/mol) -6.21 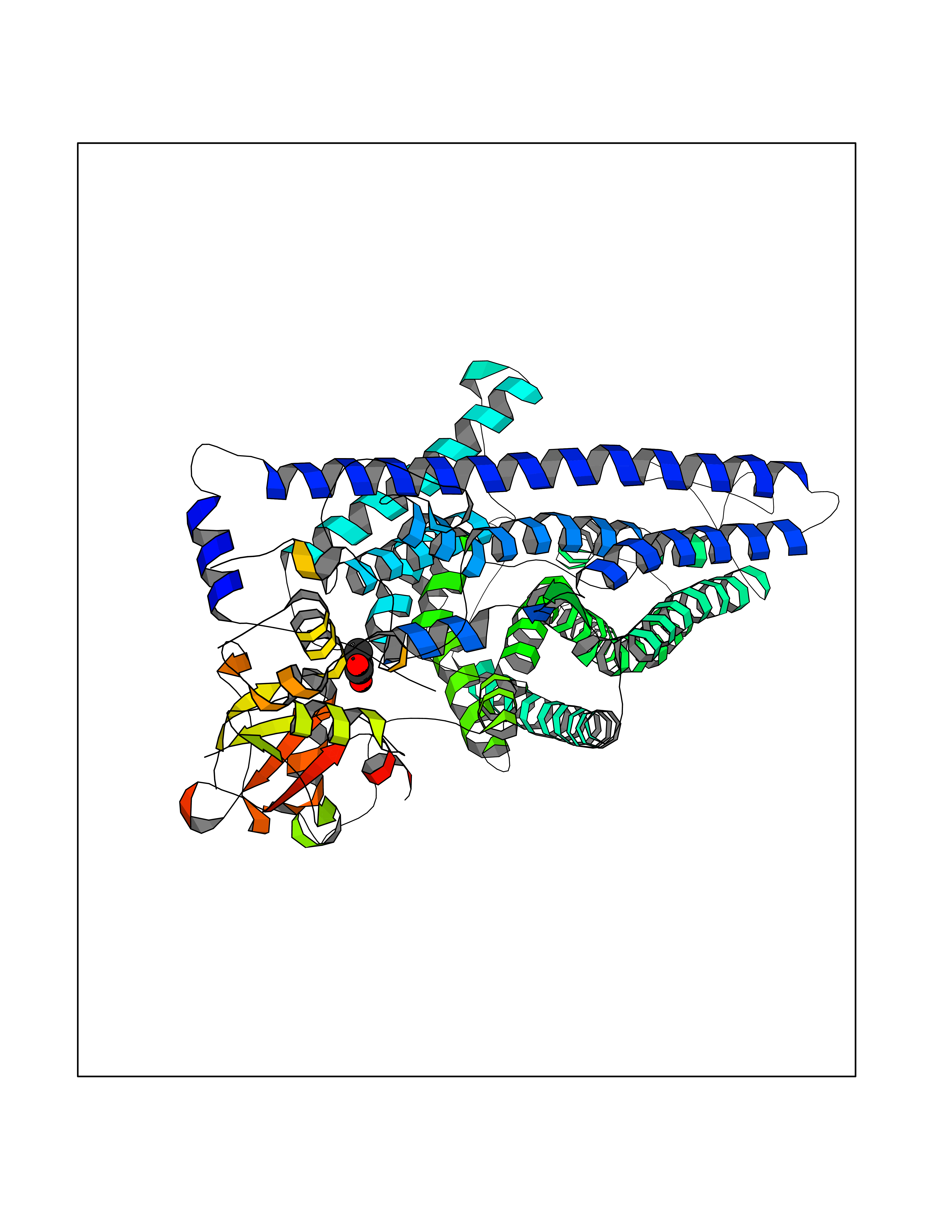 Molscript Map 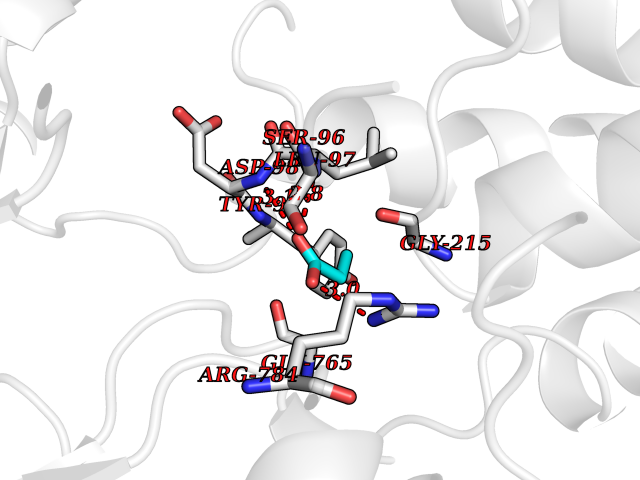 Pymol Map 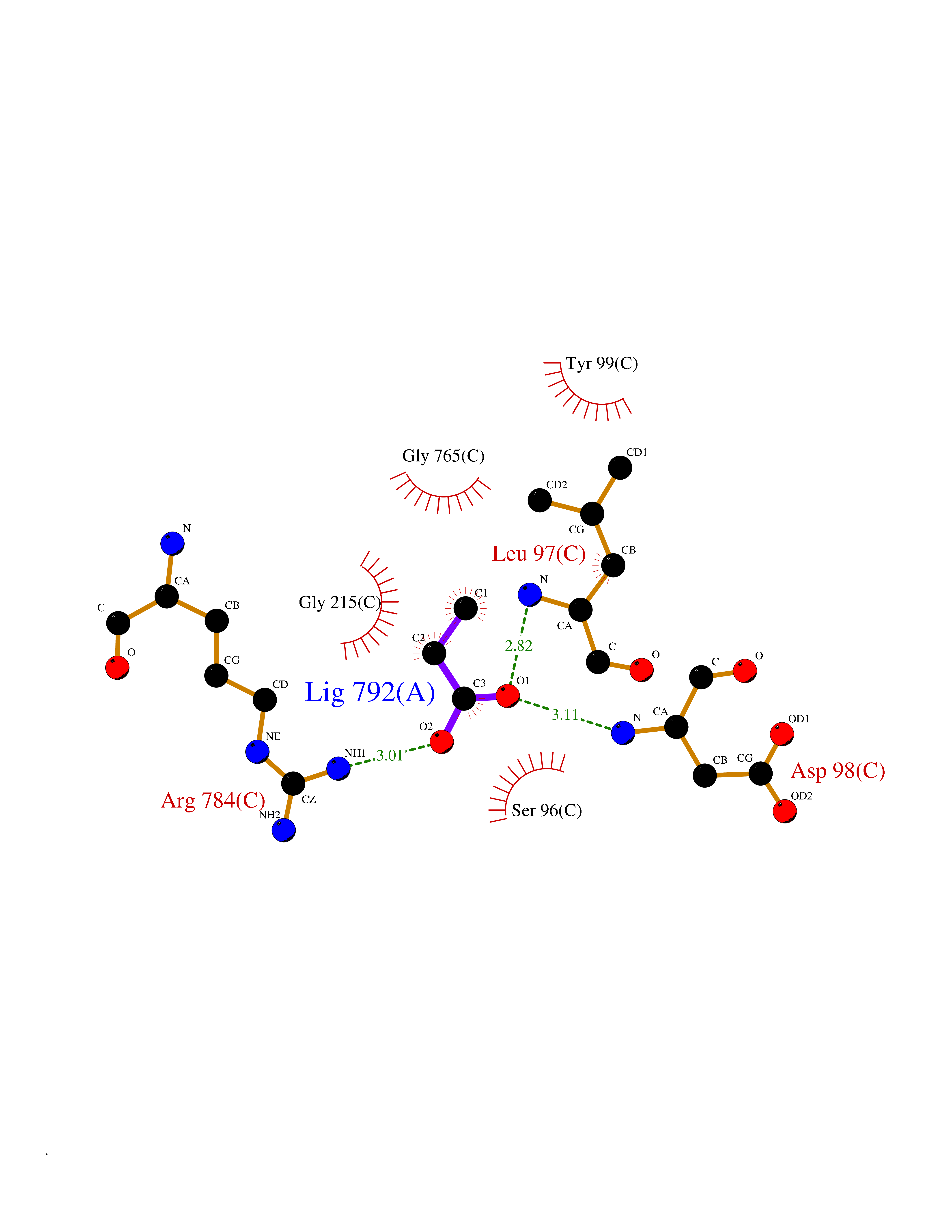 Ligplot Map 3D Binding mode Sequence YESLDYDNSENQLFLEEERRINHTAFRTVEIKRWVICALIGILTGLVACFIDIVVENLAGLKYRVIKGNIDKFTEKGGLSFSLLLWATLNAAFVLVGSVIVAFIEPVAAGSGIPQIKCFLNGVKIPHVVRLKTLVIKVSGVILSVVGGLAVGKEGPMIHSGSVIAAGISQGRSTSLKRDFKIFEYFRRDTEKRDFVSAGAAAGVSAAFGAPVGGVLFSLEEGASFWNQFLTWRIFFASMISTFTLNFVLSIYHGNMWDLSSPGLINFGRFDSEKMAYTIHEIPVFIAMGVVGGVLGAVFNALNYWLTMFRIRYIHRPCLQVIEAVLVAAVTATVAFVLIYSSRDCQPLQGGSMSYPLQLFCADGEYNSMAAAFFNTPEKSVVSLFHDPPGSYNPLTLGLFTLVYFFLACWTYGLTVSAGVFIPSLLIGAAWGRLFGISLSYLTGAAIWADPGKYALMGAAAQLGGIVRMTLSLTVIMMEATSNVTYGFPIMLVLMTAKIVGDVFIEGLYDMHIQLQSVPFLHWEAPVTSHSLTAREVMSTPVTCLRRREKVGVIVDVLSDTASNHNGFPVVEARLQGLILRSQLIVLLKHKVFVRRLRLKDFRDAYPRFPPIQSIHVSQDERECTMDLSEFMNPSPYTVPQEASLPRVFKLFRALGLRHLVVVDNRNQVVGLVTRKDLARY Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Dopamine beta-hydroxylase | 4ZEL | 4.55 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -6.2 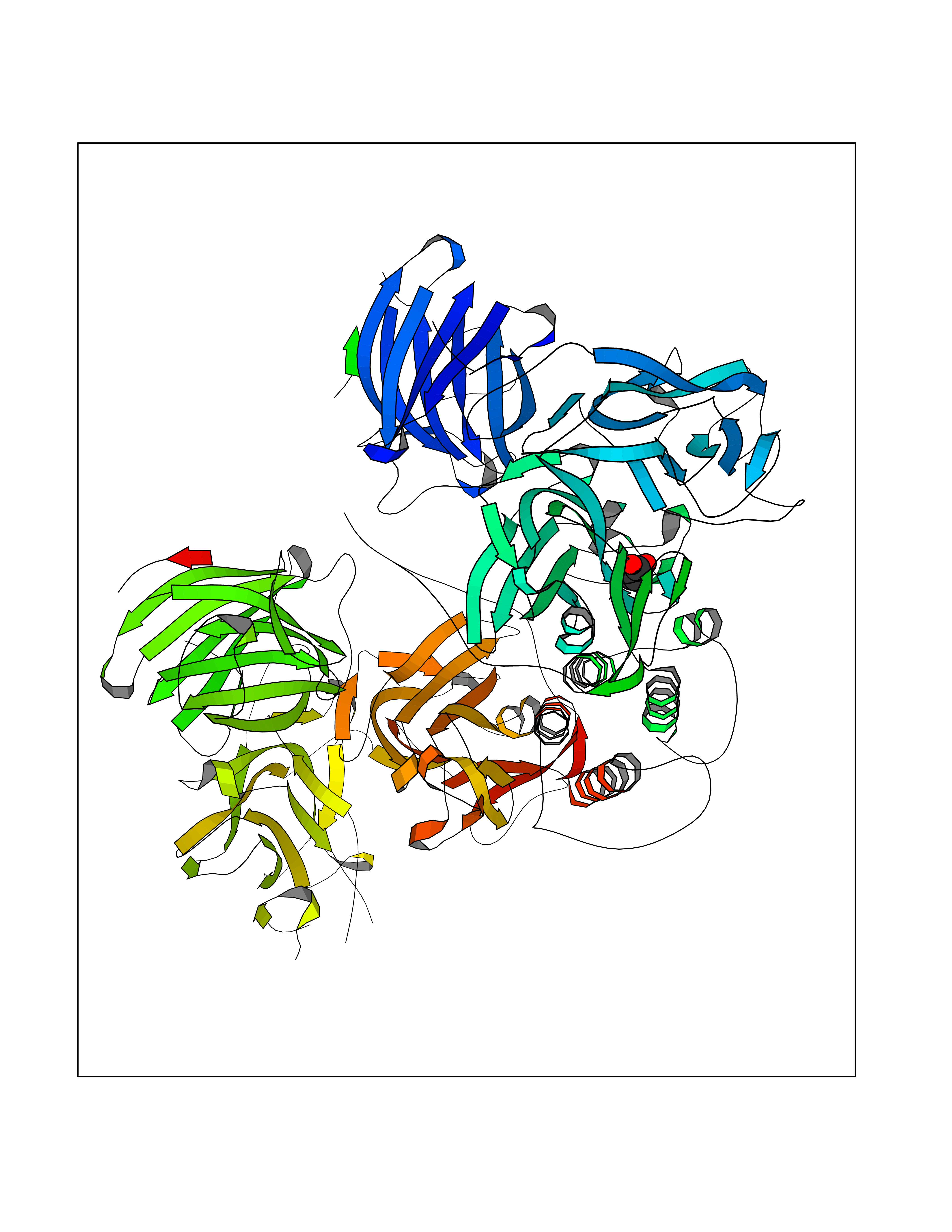 Molscript Map 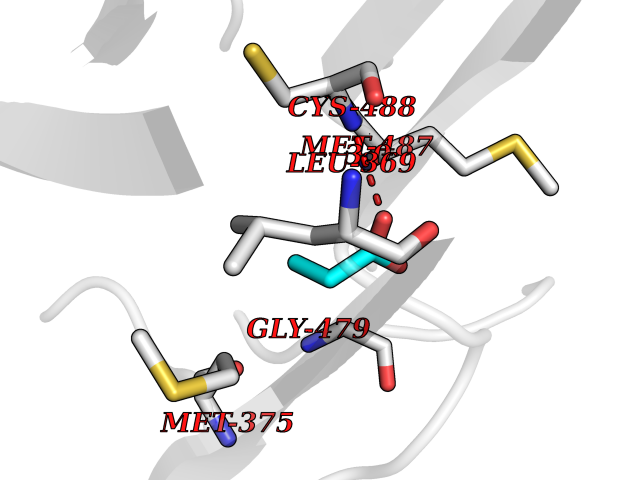 Pymol Map 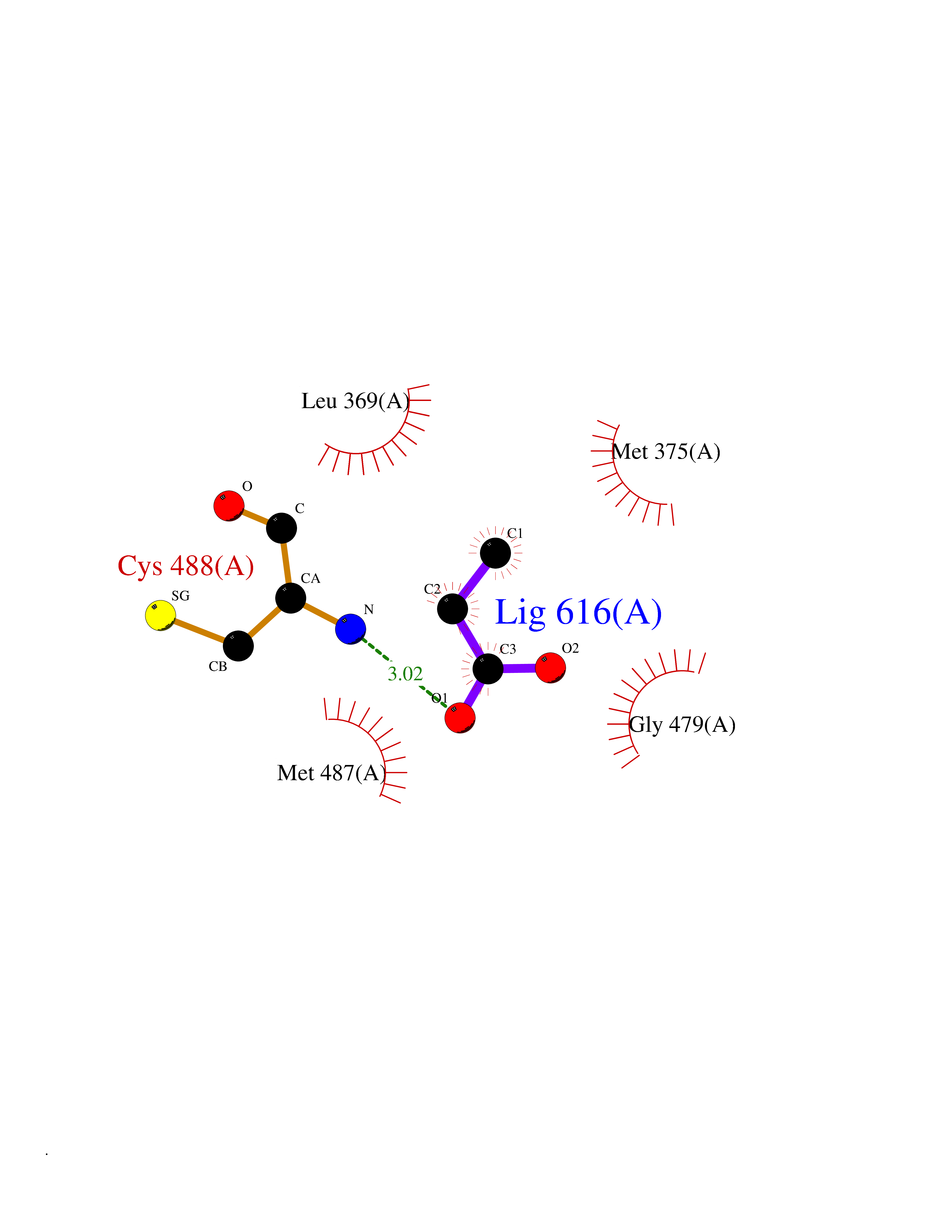 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | D-alanyl-D-alanine carboxypeptidase DacA | 3MZF | 4.55 | |
Target general information Gen name dacA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b0632;pfv;JW0627 Protein family Peptidase S11 family Biochemical class Hydrolase / antibiotic Function Beta-lactamase activity.Carboxypeptidase activity.Endopeptidase activity.Penicillin binding.Serine-type D-Ala-D-Ala carboxypeptidase activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01602; DB04647; DB00578; DB09319; DB00671; DB00274; DB01329; DB01331; DB09050; DB01147; DB01000 Interacts with NA EC number 3.4.16.4; 3.5.2.6 Uniprot keywords 3D-structure; Carboxypeptidase; Cell inner membrane; Cell membrane; Cell shape; Cell wall biogenesis/degradation; Direct protein sequencing; Hydrolase; Membrane; Peptidoglycan synthesis; Protease; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 38972 Length 355 Aromaticity 0.08 Instability index 21.23 Isoelectric point 7.1 Charge (pH=7) 0.14 2D Binding mode Binding energy (Kcal/mol) -6.21 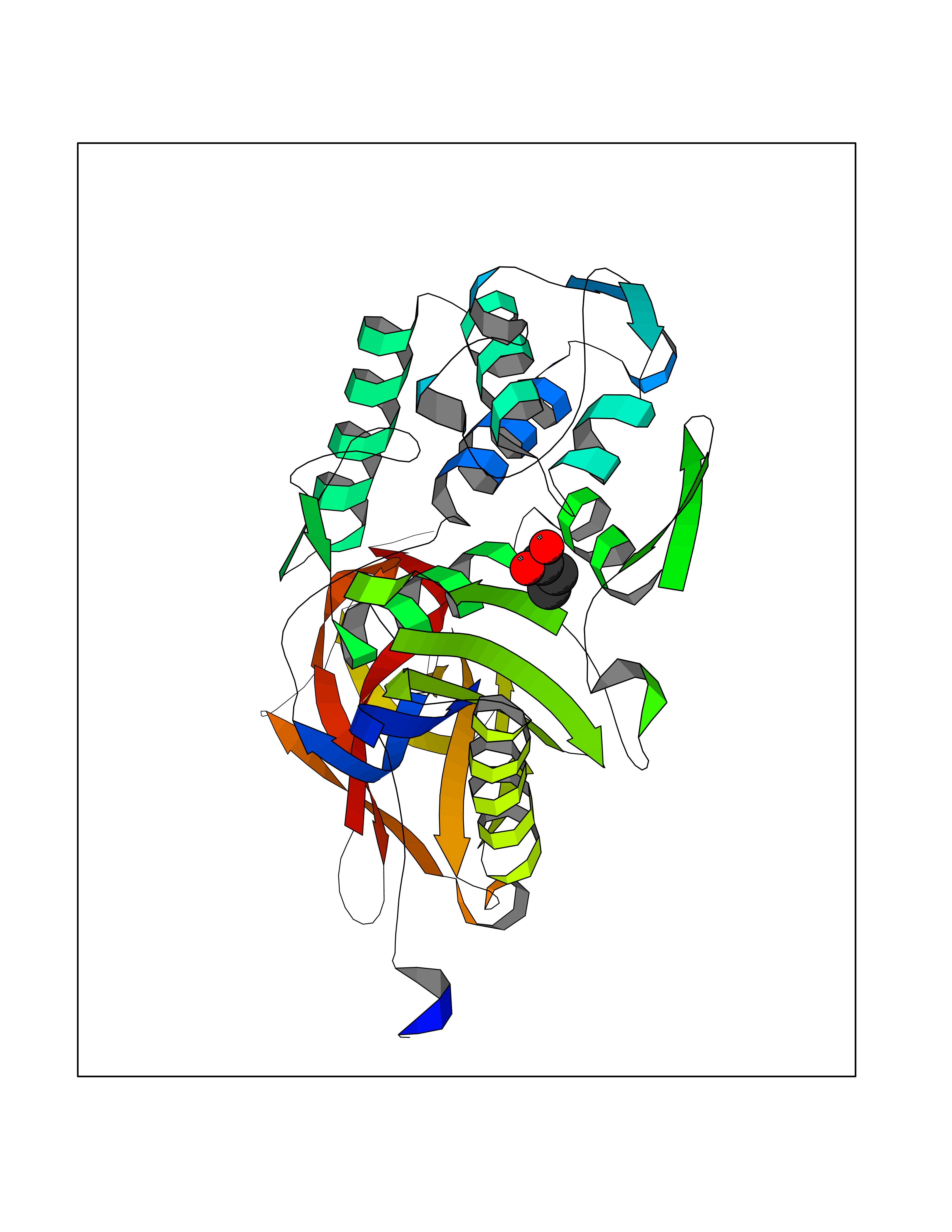 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LNIKTMIPGVPQIDAESYILIDYNSGKVLAEQNADVRRDPASLTKMMTSYVIGQAMKAGKFKETDLVTIGNDAWATGNPVFKGSSLMFLKPGMQVPVSQLIRGINLQSGNDACVAMADFAAGSQDAFVGLMNSYVNALGLKNTHFQTVHGLDADGQYSSARDMALIGQALIRDVPNEYSIYKEKEFTFNGIRQLNRNGLLWDNSLNVDGIKTGHTDKAGYNLVASATEGQMRLISAVMGGRTFKGREAESKKLLTWGFRFFETVNPLKVGKEFASEPVWFGDSDRASLGVDKDVYLTIPRGRMKDLKASYVLNSSELHAPLQKNQVVGTINFQLDGKTIEQRPLVVLQEIPEGNF Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Pyruvate dehydrogenase [ubiquinone] | 3EYA | 4.55 | |
Target general information Gen name poxB Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms b0871;JW0855 Protein family TPP enzyme family Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.Identical protein binding.Lipid binding.Magnesium ion binding.Pyruvate dehydrogenase (quinone) activity.Thiamine pyrophosphate binding. Related diseases Glycogen storage disease 6 (GSD6) [MIM:232700]: A metabolic disorder characterized by mild to moderate hypoglycemia, mild ketosis, growth retardation, and prominent hepatomegaly. Heart and skeletal muscle are not affected. {ECO:0000269|PubMed:9529348}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07003 EC number 1.2.5.1 Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Direct protein sequencing; FAD; Flavoprotein; Lipid-binding; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Oxidoreductase; Pyruvate; Reference proteome; Thiamine pyrophosphate; Ubiquinone Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H,I,J,K,L Molecular weight (Da) 113027 Length 1046 Aromaticity 0.07 Instability index 35.99 Isoelectric point 5.75 Charge (pH=7) -24.38 2D Binding mode Binding energy (Kcal/mol) -6.21  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MKQTVAAYIAKTLESAGVKRIWGVTGDSLNGLSDSLNRMGTIEWMSTRHEEVAAFAAGAEAQLSGELAVCAGSCGPGNLHLINGLFDCHRNHVPVLAIAAHIPSSEIGSGYFQETHPQELFRECSHYCELVSSPEQIPQVLAIAMRKAVLNRGVSVVVLPGDVALKPAPEGATMHWYHAPQPVVTPEEEELRKLAQLLRYSSNIALMCGSGCAGAHKELVEFAGKIKAPIVHALRGKEHVEYDNPYDVGMTGLIGFSSGFHTMMNADTLVLLGTQFPYRAFYPTDAKIIQIDINPASIGAHSKVDMALVGDIKSTLRALLPLVEEKADRKFLDKALEDYRDARKGLDDLAKPSEKAIHPQYLAQQISHFAADDAIFTCDVGTPTVWAARYLKMNGKRRLLGSFNHGSMANAMPQALGAQATEPERQVVAMCGDGGFSMLMGDFLSVVQMKLPVKIVVFNNSVLGFDGTELHDTNFARIAEACGITGIRVEKASEVDEALQRAFSIDGPVLVDVVVAKEELAIPMKQTVAAYIAKTLESAGVKRIWGVTGDSLNGLSDSLNRMGTIEWMSTRHEEVAAFAAGAEAQLSGELAVCAGSCGPGNLHLINGLFDCHRNHVPVLAIAAHIPSSEIGSGYFQETHPQELFRECSHYCELVSSPEQIPQVLAIAMRKAVLNRGVSVVVLPGDVALKPAPEGATMHWYHAPQPVVTPEEEELRKLAQLLRYSSNIALMCGSGCAGAHKELVEFAGKIKAPIVHALRGKEHVEYDNPYDVGMTGLIGFSSGFHTMMNADTLVLLGTQFPYRAFYPTDAKIIQIDINPASIGAHSKVDMALVGDIKSTLRALLPLVEEKADRKFLDKALEDYRDARKGLDDLAKPSEKAIHPQYLAQQISHFAADDAIFTCDVGTPTVWAARYLKMNGKRRLLGSFNHGSMANAMPQALGAQATEPERQVVAMCGDGGFSMLMGDFLSVVQMKLPVKIVVFNNSVLGFVGTELHDTNFARIAEACGITGIRVEKASEVDEALQRAFSIDGPVLVDVVVAKEELAIP Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Adrenergic receptor beta-2 (ADRB2) | 2RH1 | 4.55 | |
Target general information Gen name ADRB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Beta-2 adrenoreceptor; Beta-2 adrenoceptor; Beta-2 adrenergic receptor; B2AR; ADRB2R Protein family G-protein coupled receptor 1 family, Adrenergic receptor subfamily, ADRB2 sub-subfamily Biochemical class GPCR rhodopsin Function The beta-2-adrenergic receptor binds epinephrine with an approximately 30-fold greater affinity than it does norepinephrine. Beta-adrenergic receptors mediate the catecholamine-induced activation of adenylate cyclase through the action of G proteins. Related diseases Cortical dysplasia, complex, with other brain malformations 6 (CDCBM6) [MIM:615771]: A disorder of aberrant neuronal migration and disturbed axonal guidance. Affected individuals have microcephaly, ataxia, and severe delayed psychomotor development. Brain imaging shows variable malformations of cortical development, including white matter streaks, dysmorphic basal ganglia, corpus callosum abnormalities, brainstem and cerebellar hypoplasia, cortical dysplasia, polymicrogyria. {ECO:0000269|PubMed:23246003}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Skin creases, congenital symmetric circumferential, 1 (CSCSC1) [MIM:156610]: An autosomal dominant disease characterized by multiple, symmetric, circumferential rings of folded skin, affecting primarily the limbs. Affected individuals also exhibit intellectual disability, cleft palate, and dysmorphic features. {ECO:0000269|PubMed:26637975}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07543; DB01193; DB00866; DB01118; DB00182; DB01102; DB01274; DB01238; DB09204; DB06216; DB00335; DB01408; DB05590; DB09013; DB00195; DB00217; DB01295; DB00612; DB00901; DB08807; DB06726; DB08808; DB00248; DB00521; DB01136; DB04846; DB01407; DB00785; DB01151; DB11273; DB13345; DB00449; DB11278; DB00841; DB09273; DB06262; DB01363; DB01364; DB00668; DB01049; DB11587; DB01288; DB00983; DB05039; DB00221; DB01064; DB00598; DB01210; DB13139; DB01365; DB13624; DB01214; DB00264; DB01203; DB05849; DB04861; DB00368; DB00540; DB00334; DB09080; DB00816; DB01580; DB00715; DB01359; DB00925; DB00397; DB00960; DB01291; DB01366; DB01182; DB00571; DB06814; DB00852; DB01917; DB11124; DB00867; DB01001; DB00938; DB00489; DB03566; DB00127; DB00871; DB00373; DB00726; DB12248; DB09082; DB09185 Interacts with P30542; P07550; P32121; Q96B67; Q9UII2; Q9ULD4-2; Q9NSI6-4; Q5M9N0-2; A0AVK6; Q658K8; O00472; Q15910-2; Q15486; P61978; Q5TCQ9; Q99685; O14745; Q9NR21-5; Q8WVD3; Q9H0X6; Q13573; P12931; Q5T0J7-2; Q8N0U2 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; Endosome; G-protein coupled receptor; Glycoprotein; Golgi apparatus; Hydroxylation; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32266.1 Length 282 Aromaticity 0.15 Instability index 36.1 Isoelectric point 8.02 Charge (pH=7) 2.1 2D Binding mode Binding energy (Kcal/mol) -6.21  Molscript Map 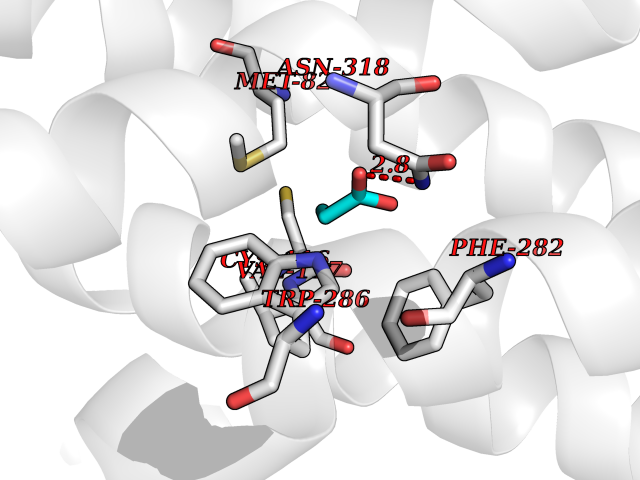 Pymol Map 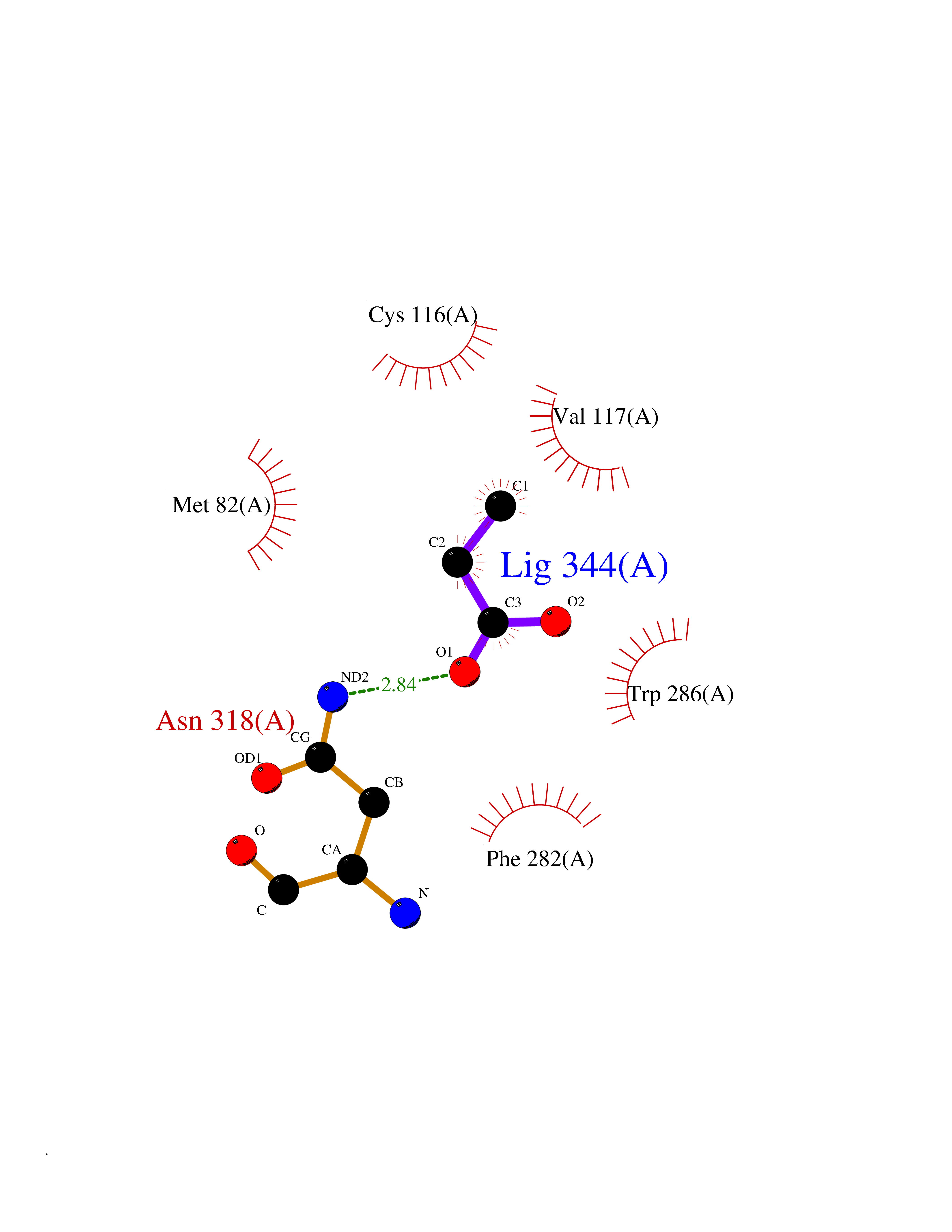 Ligplot Map 3D Binding mode Sequence DEVWVVGMGIVMSLIVLAIVFGNVLVITAIAKFERLQTVTNYFITSLACADLVMGLAVVPFGAAHILMKMWTFGNFWCEFWTSIDVLCVTASIETLCVIAVDRYFAITSPFKYQSLLTKNKARVIILMVWIVSGLTSFLPIQMHWYRATHQEAINCYAEETCCDFFTNQAYAIASSIVSFYVPLVIMVFVYSRVFQEAKRQLKFCLKEHKALKTLGIIMGTFTLCWLPFFIVNIVHVIQDNLIRKEVYILLNWIGYVNSGFNPLIYCRSPDFRIAFQELLCL Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Acetylcholinesterase (AChE) | 4M0E | 4.55 | |
Target general information Gen name ACHE Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms YT; N-ACHE; ARACHE Protein family Type-B carboxylesterase/lipase family Biochemical class Carboxylic ester hydrolase Function Role in neuronal apoptosis. Terminates signal transduction at the neuromuscular junction by rapid hydrolysis of the acetylcholine released into the synaptic cleft. Related diseases Phosphoribosylaminoimidazole carboxylase deficiency (PAICSD) [MIM:619859]: An autosomal recessive inborn error of purine metabolism, clinically characterized by multiple congenital anomalies and early neonatal death. {ECO:0000269|PubMed:31600779}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07846; DB02673; DB04617; DB04614; DB04615; DB07756; DB07701; DB02404; DB03814; DB08615; DB02343; DB02226; DB03005; DB04114; DB03128; DB01122; DB03283; DB00411; DB00122; DB14006; DB01245; DB00944; DB08357; DB08996; DB00449; DB00843; DB01010; DB01364; DB00898; DB00674; DB00483; DB06525; DB04864; DB03348; DB07555; DB00677; DB04924; DB03359; DB00358; DB00940; DB02825; DB02845; DB08167; DB04021; DB00805; DB01805; DB03740; DB04556; DB01400; DB04892; DB00981; DB00733; DB02166; DB00545; DB00863; DB00989; DB00382; DB04616; DB12816; DB01199; DB13503; DB04859 Interacts with Q9Y215; P06733; P63244 EC number EC 3.1.1.7 Uniprot keywords 3D-structure; Alternative splicing; Blood group antigen; Cell membrane; Direct protein sequencing; Disulfide bond; Glycoprotein; GPI-anchor; Hydrolase; Lipoprotein; Membrane; Neurotransmitter degradation; Nucleus; Proteomics identification; Reference proteome; Secreted; Serine esterase; Signal; Synapse Protein physicochemical properties Chain ID A,B Molecular weight (Da) 58804.1 Length 537 Aromaticity 0.11 Instability index 40.85 Isoelectric point 5.73 Charge (pH=7) -8.18 2D Binding mode Binding energy (Kcal/mol) -6.2 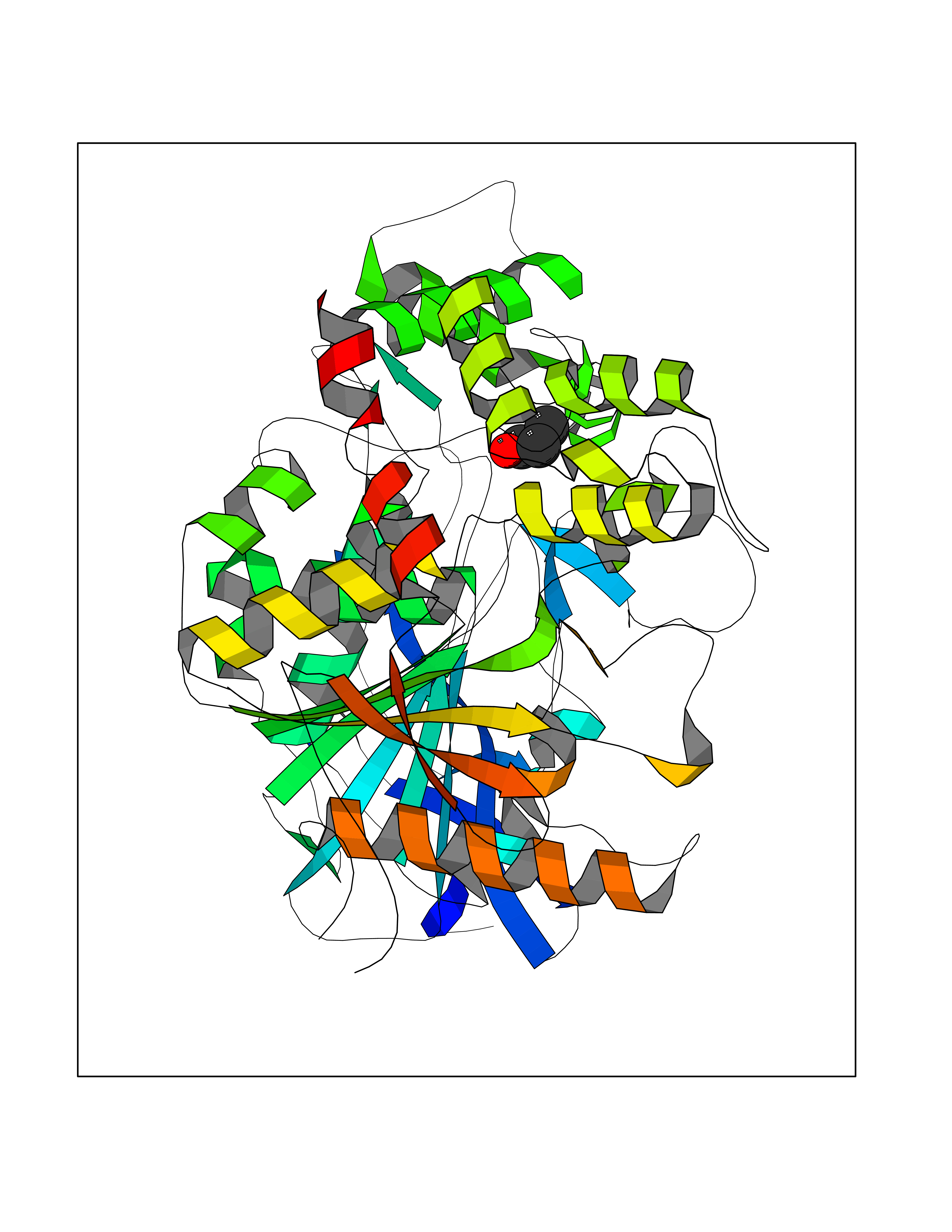 Molscript Map 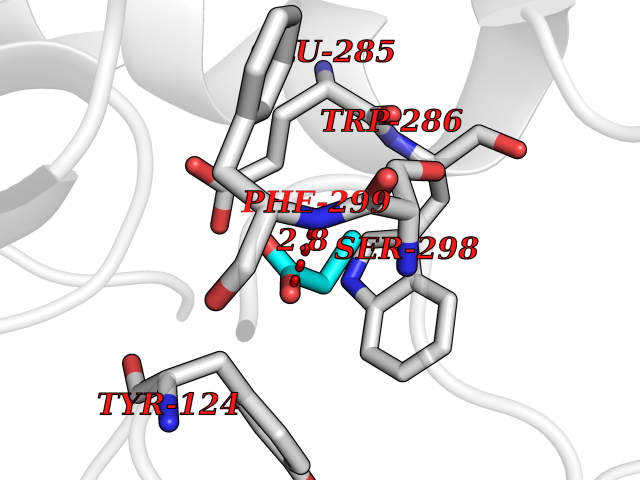 Pymol Map 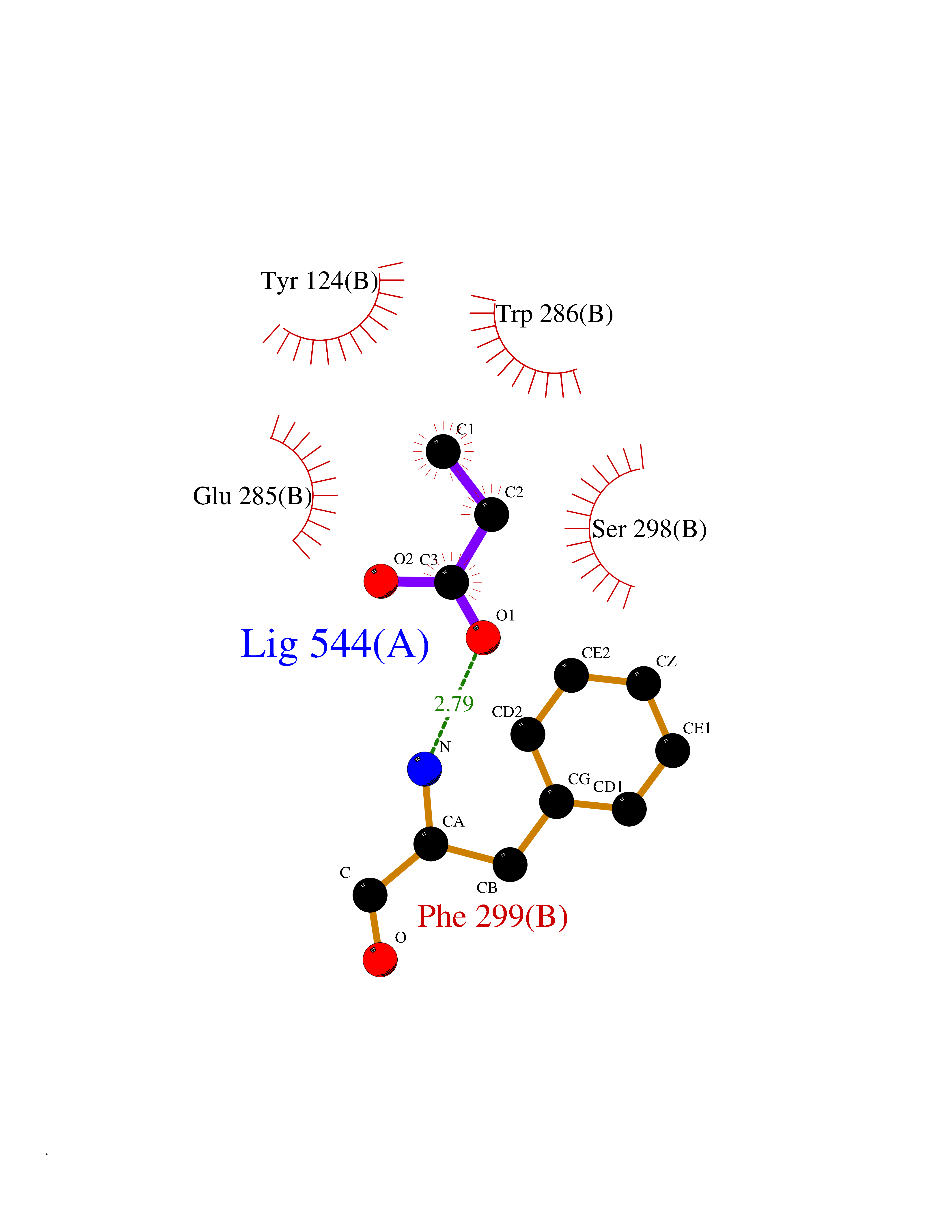 Ligplot Map 3D Binding mode Sequence EDAELLVTVRGGRLRGIRLKTPGGPVSAFLGIPFAEPPMGPRRFLPPEPKQPWSGVVDATTFQSVCYQYVDTLYPGFEGTEMWNPNRELSEDCLYLNVWTPYPRPTSPTPVLVWIYGGGFYSGASSLDVYDGRFLVQAERTVLVSMNYRVGAFGFLALPGSREAPGNVGLLDQRLALQWVQENVAAFGGDPTSVTLFGESAGAASVGMHLLSPPSRGLFHRAVLQSGAPNGPWATVGMGEARRRATQLAHLVGCPPGGTGGNDTELVACLRTRPAQVLVNHEWHVLPQESVFRFSFVPVVDGDFLSDTPEALINAGDFHGLQVLVGVVKDEGSYFLVYGAPGFSKDNESLISRAEFLAGVRVGVPQVSDLAAEAVVLHYTDWLHPEDPARLREALSDVVGDHNVVCPVAQLAGRLAAQGARVYAYVFEHRASTLSWPLWMGVPHGYEIEFIFGIPLDPSRNYTAEEKIFAQRLMRYWANFARTGDPNEPPKAPQWPPYTAGAQQYVSLDLRPLEVRRGLRAQACAFWNRFLPKLLSA Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Multifunctional protein ADE2 | 2H31 | 4.55 | |
Target general information Gen name PAICS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ADE2;PAIS;AIRC Protein family SAICAR synthetase family; AIR carboxylase family, Class II subfamily Biochemical class Ligase Function ATP binding.Cadherin binding.Identical protein binding.Phosphoribosylaminoimidazole carboxylase activity.Phosphoribosylaminoimidazolesuccinocarboxamide synthase activity. Related diseases Phosphoribosylaminoimidazole carboxylase deficiency (PAICSD) [MIM:619859]: An autosomal recessive inborn error of purine metabolism, clinically characterized by multiple congenital anomalies and early neonatal death. {ECO:0000269|PubMed:31600779}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00128 Interacts with Q16543; P51116; Q969R5; Q8TBB1; Q6ZVK8; P22234; O75928-2; C9JJ79; Q9Y4B4; P78317; Q7KZS0; Q9UBW7 EC number 4.1.1.21; 6.3.2.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Decarboxylase; Direct protein sequencing; Disease variant; Ligase; Lyase; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Proteomics identification; Purine biosynthesis; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 41862.7 Length 386 Aromaticity 0.08 Instability index 34.99 Isoelectric point 7.02 Charge (pH=7) 0.04 2D Binding mode Binding energy (Kcal/mol) -6.2 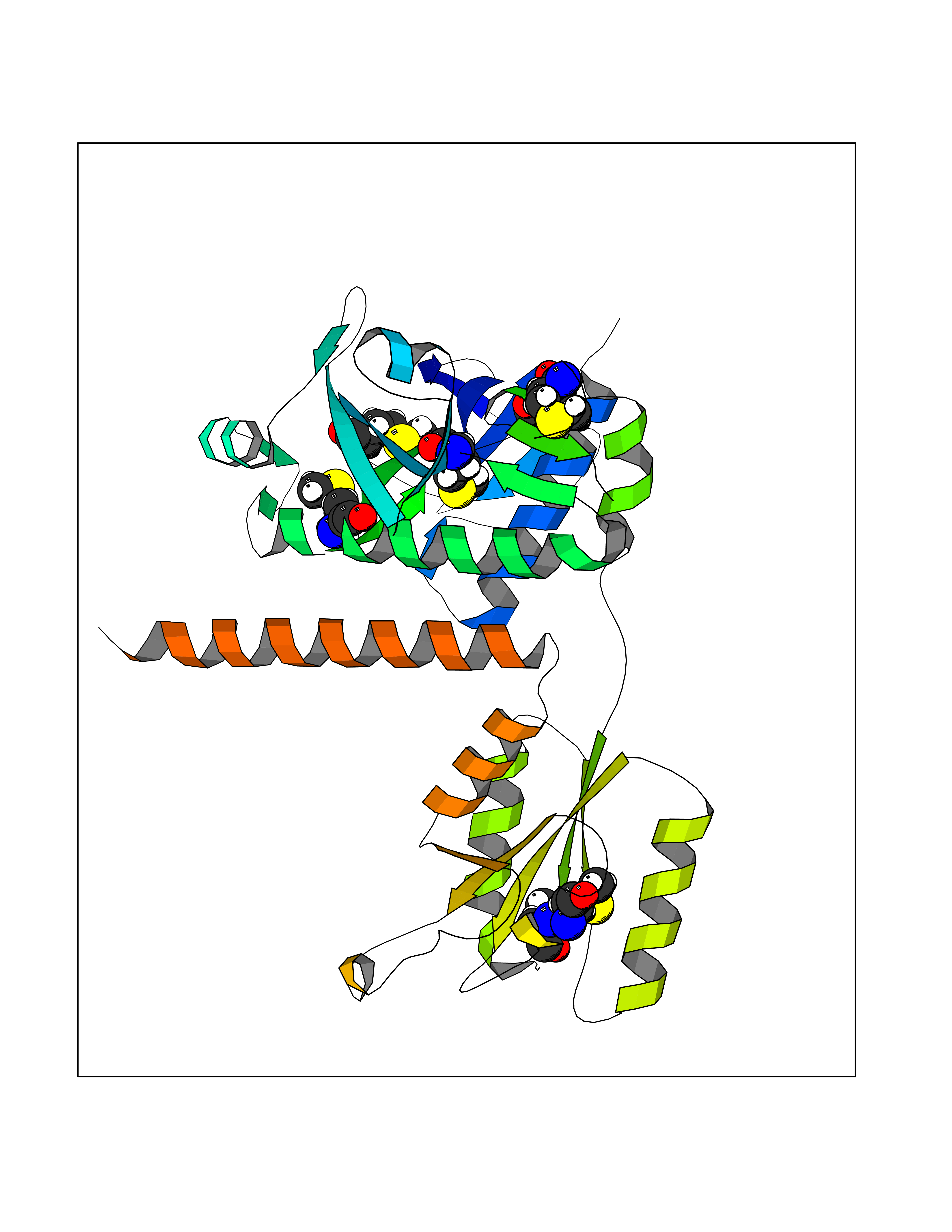 Molscript Map 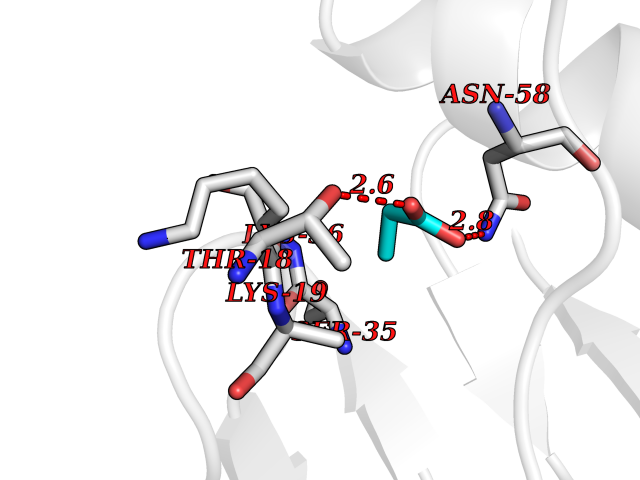 Pymol Map 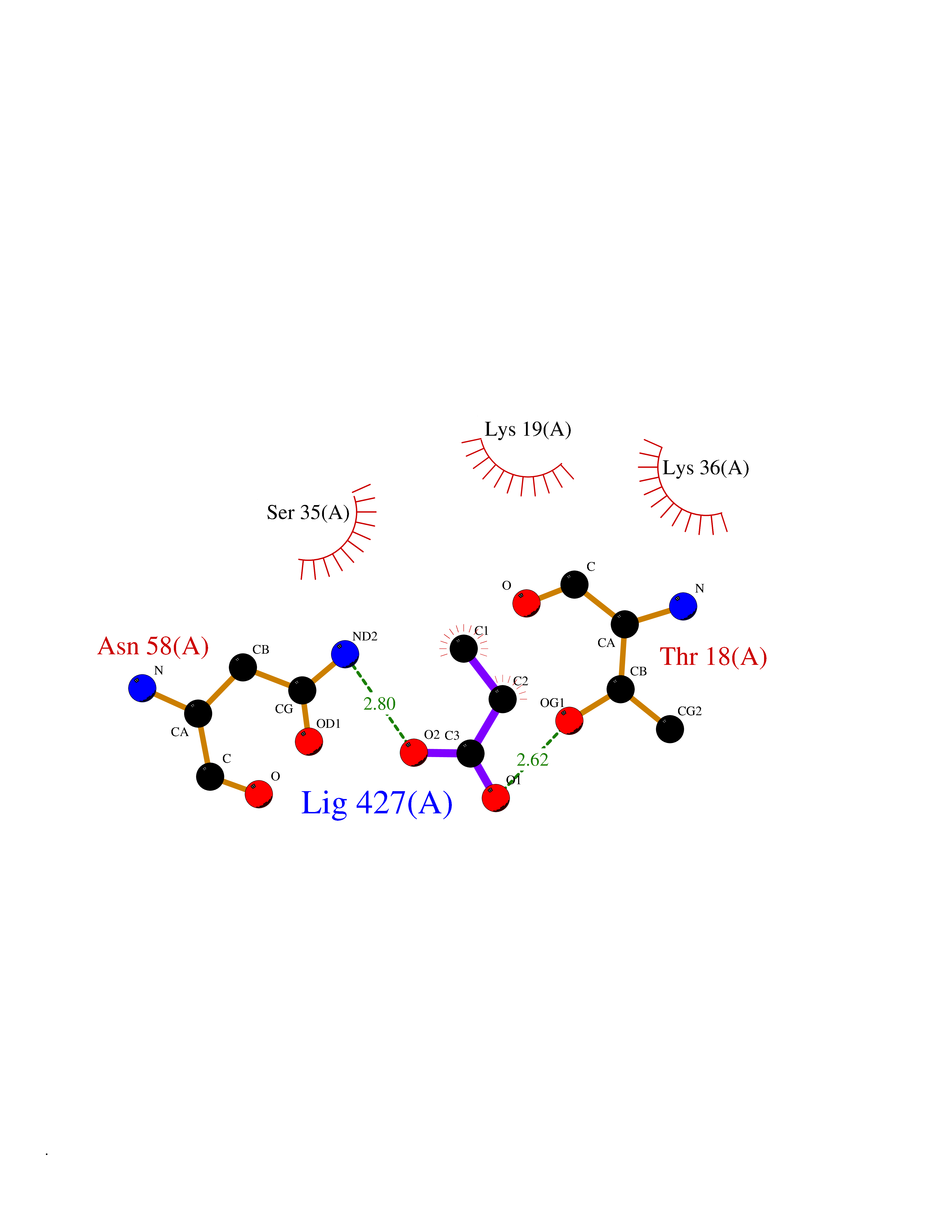 Ligplot Map 3D Binding mode Sequence LNIGKKLYEGKTKEVYELLDSPGKVLLQSKGKAAISNKITSCIFQLLQEAGIKTAFTRKCGETAFIAPQCEXIPIEWVCRRIATGSFLKRNPGVKEGYKFYPPKVELFFKDDANNDPQWSEEQLIAAKFCFAGLLIGQTEVDIXSHATQAIFEILEKSWLPQNCTLVDXKIEFGVDVTTKEIVLADVIDNDSWRLWPSGPEGLQXVKKNFEWVAERVELLLKSESQCRVVVLXGSTSDLGHCEKIKKACGNFGIPCELRVTSAHKGPDETLRIKAEYEGDGIPTVFVAVAGRSNGLGPVXSGNTAYPVISCPPLTPDWGVQDVWSSLRLPSGLGCSTVLSPEGSAQFAAQIFGLSNHLVWSKLRASILNTWISLKQADKKIRECNL Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Acetyl-CoA carboxylase (ACC) (EC 6.4.1.2) (Fatty acid synthetase 3) (mRNA transport-defective protein 7) [Includes: Biotin carboxylase (EC 6.3.4.14)] | 1UYS | 4.55 | |
Target general information Gen name ACC1 Organism Saccharomyces cerevisiae (strain ATCC 204508 / S288c) (Baker's yeast) Uniprot ID TTD ID NA Synonyms MTR7;YNR016C;N3175;ABP2;FAS3 Protein family NA Biochemical class NA Function Carries out three functions: biotin carboxyl carrier protein, biotin carboxylase and carboxyltransferase. Involved in the synthesis of very-long-chain fatty acid synthesis which is required to maintain a functional nuclear envelope. Required for acylation and vacuolar membrane association of VAC8 which is necessary to maintain a normal morphology of the vacuole. {ECO:0000269|PubMed:10757783, ECO:0000269|PubMed:12730220, ECO:0000269|PubMed:6103540, ECO:0000269|PubMed:6108218, ECO:0000269|PubMed:8943372}." Related diseases A chromosomal aberration involving NFKB2 is found in a case of B-cell non Hodgkin lymphoma (B-NHL). Translocation t(10;14)(q24;q32) with IGHA1. The resulting oncogene is also called Lyt-10C alpha variant.; DISEASE: A chromosomal aberration involving NFKB2 is found in a cutaneous T-cell leukemia (C-TCL) cell line. This rearrangement produces the p80HT gene which codes for a truncated 80 kDa protein (p80HT).; DISEASE: In B-cell leukemia (B-CLL) cell line, LB40 and EB308, can be found after heterogeneous chromosomal aberrations, such as internal deletions.; DISEASE: Immunodeficiency, common variable, 10 (CVID10) [MIM:615577]: A primary immunodeficiency characterized by childhood-onset of recurrent infections, hypogammaglobulinemia, and decreased numbers of memory and marginal zone B-cells. Some patients may develop autoimmune features and have circulating autoantibodies. An unusual feature is central adrenal insufficiency. {ECO:0000269|PubMed:24140114, ECO:0000269|PubMed:25524009}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q00955 EC number 6.3.4.14; 6.4.1.2 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Biotin; Cytoplasm; Direct protein sequencing; Endoplasmic reticulum; Fatty acid biosynthesis; Fatty acid metabolism; Ligase; Lipid biosynthesis; Lipid metabolism; Manganese; Membrane; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Reference proteome Protein physicochemical properties Chain ID B,C Molecular weight (Da) 145619 Length 1328 Aromaticity 0.1 Instability index 30.31 Isoelectric point 5.32 Charge (pH=7) -26.79 2D Binding mode Binding energy (Kcal/mol) -6.21  Molscript Map 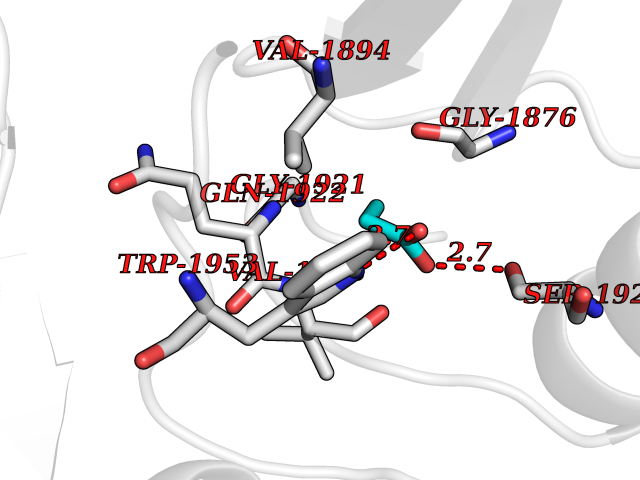 Pymol Map 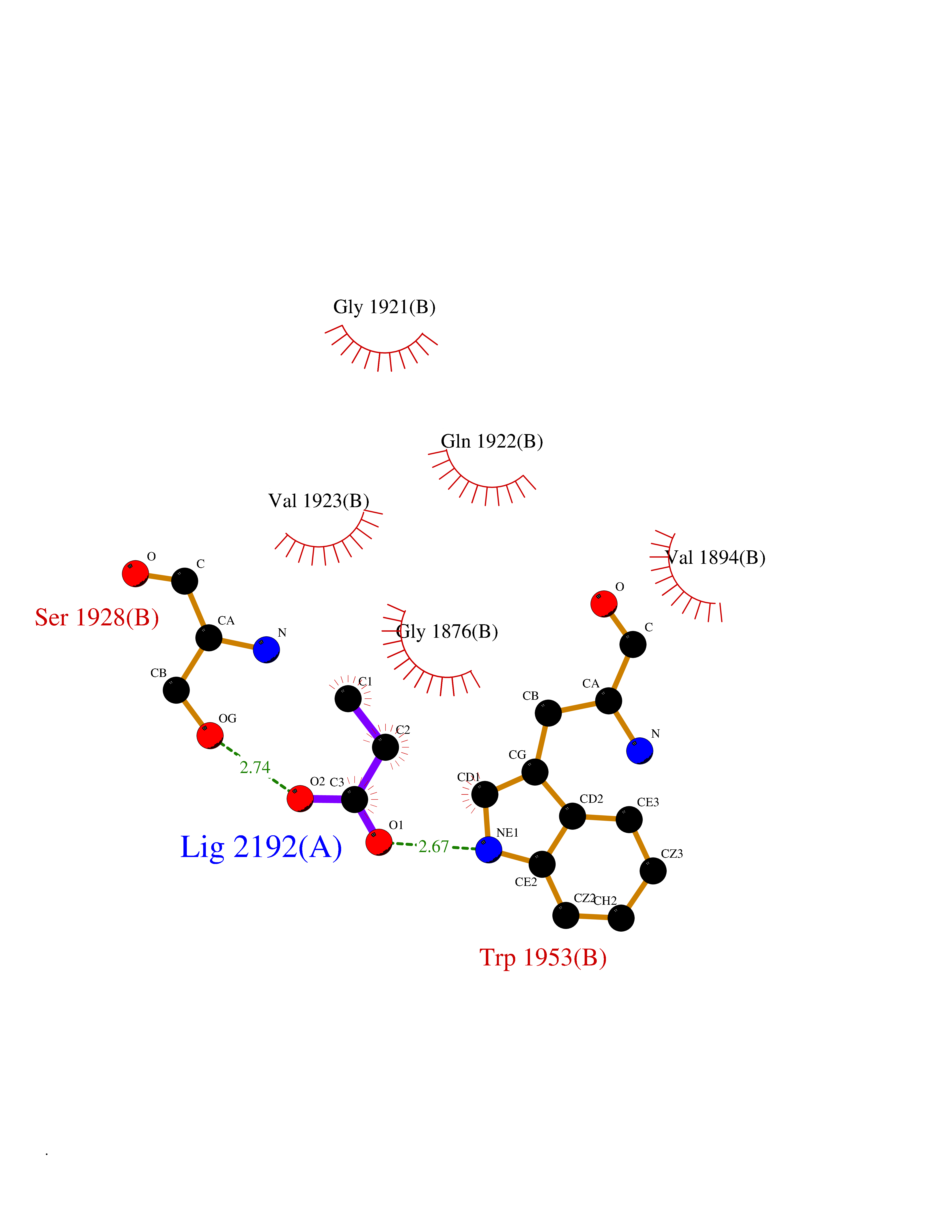 Ligplot Map 3D Binding mode Sequence WLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGLPIATPYPVKEWLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGL Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Cytochrome c | 3ZOO | 4.55 | |
Target general information Gen name CYCS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CYC Protein family Cytochrome c family Biochemical class Oxidoreductase Function Electron transporter, transferring electrons from CoQH2-cytochrome c reductase complex and cytochrome c oxidase complex activity.Heme binding.Metal ion binding. Related diseases Thrombocytopenia 4 (THC4) [MIM:612004]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. {ECO:0000269|PubMed:18345000}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11638; DB03317; DB03366; DB01017; DB02110; DB03977; DB03934; DB04249 Interacts with O14727; P05067; Q6XD76; Q9NSI6-4; Q3SXR2; Q96BR5; Q9UKG9-2; O00303; Q8IZU1; Q3SYB3; P06241; Q8N5Z5; Q6A162; Q1L5Z9; P02750; Q8IYG6; Q6FHY5; A0A0A0MR05; Q9BUL5; Q6ZMI0-5; Q66K80; Q9NTN9-3; P37840; Q13573; Q92797-2; O43829; Q9FKS5 EC number NA Uniprot keywords 3D-structure; Acetylation; Apoptosis; Direct protein sequencing; Disease variant; Electron transport; Heme; Iron; Metal-binding; Mitochondrion; Phosphoprotein; Proteomics identification; Reference proteome; Respiratory chain; Transport Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 11601.4 Length 104 Aromaticity 0.09 Instability index 12.21 Isoelectric point 9.61 Charge (pH=7) 9.01 2D Binding mode Binding energy (Kcal/mol) -6.2  Molscript Map 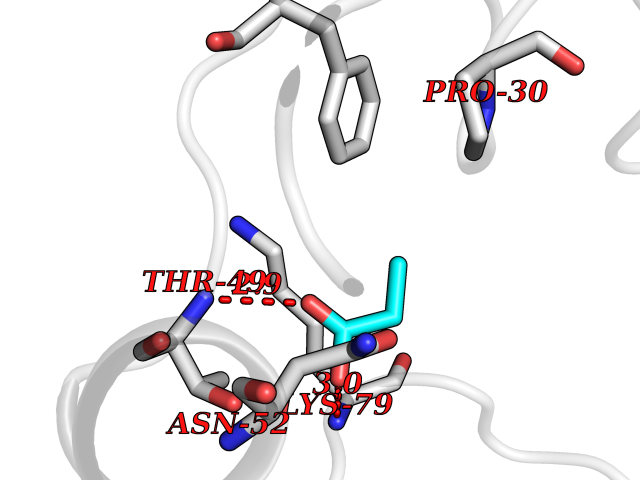 Pymol Map 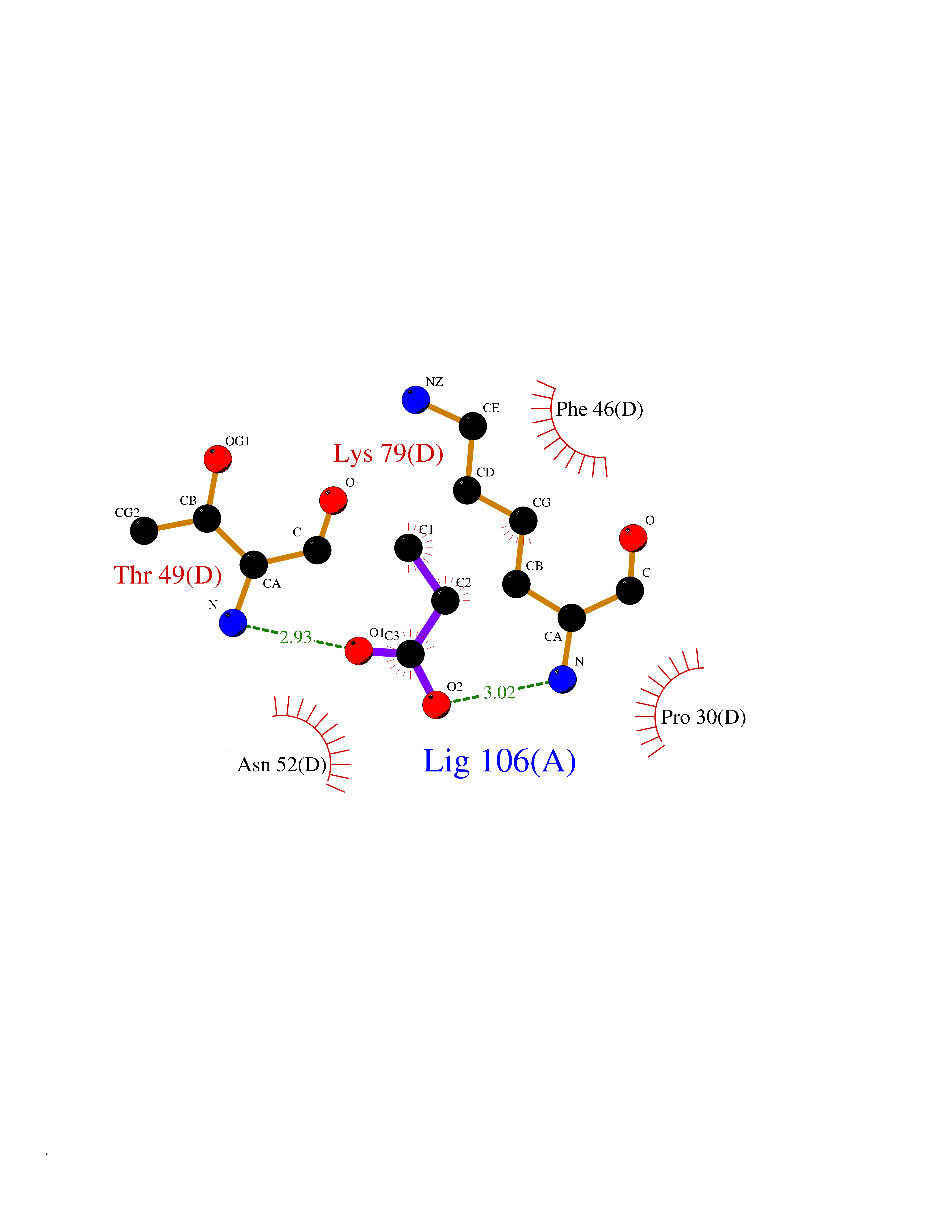 Ligplot Map 3D Binding mode Sequence GDVEKGKKIFIMKCSQCHTVEKGGKHKTGPNLHGLFGRKTGQAPGFSYTAANKNKGIIWGEDTLMEYLENPKKYIPGTKMIFVGIKKKEERADLIAYLKKATNE Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Carbapenem-hydrolyzing beta-lactamase KPC | 3RXW | 4.55 | |
Target general information Gen name bla Organism Klebsiella pneumoniae Uniprot ID TTD ID NA Synonyms kpc;kpc1 Protein family Class-A beta-lactamase family Biochemical class Hydrolase / hydrolase inhibitor Function Beta-lactamase activity. Related diseases Cornelia de Lange syndrome 5 (CDLS5) [MIM:300882]: A form of Cornelia de Lange syndrome, a clinically heterogeneous developmental disorder associated with malformations affecting multiple systems. It is characterized by facial dysmorphisms, abnormal hands and feet, growth delay, cognitive retardation, hirsutism, gastroesophageal dysfunction and cardiac, ophthalmologic and genitourinary anomalies. {ECO:0000269|PubMed:22885700, ECO:0000269|PubMed:22889856}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09060; DB12107 Interacts with NA EC number 3.5.2.6 Uniprot keywords 3D-structure; Antibiotic resistance; Hydrolase; Plasmid; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 27965.2 Length 264 Aromaticity 0.08 Instability index 26.27 Isoelectric point 6.14 Charge (pH=7) -1.42 2D Binding mode Binding energy (Kcal/mol) -6.21 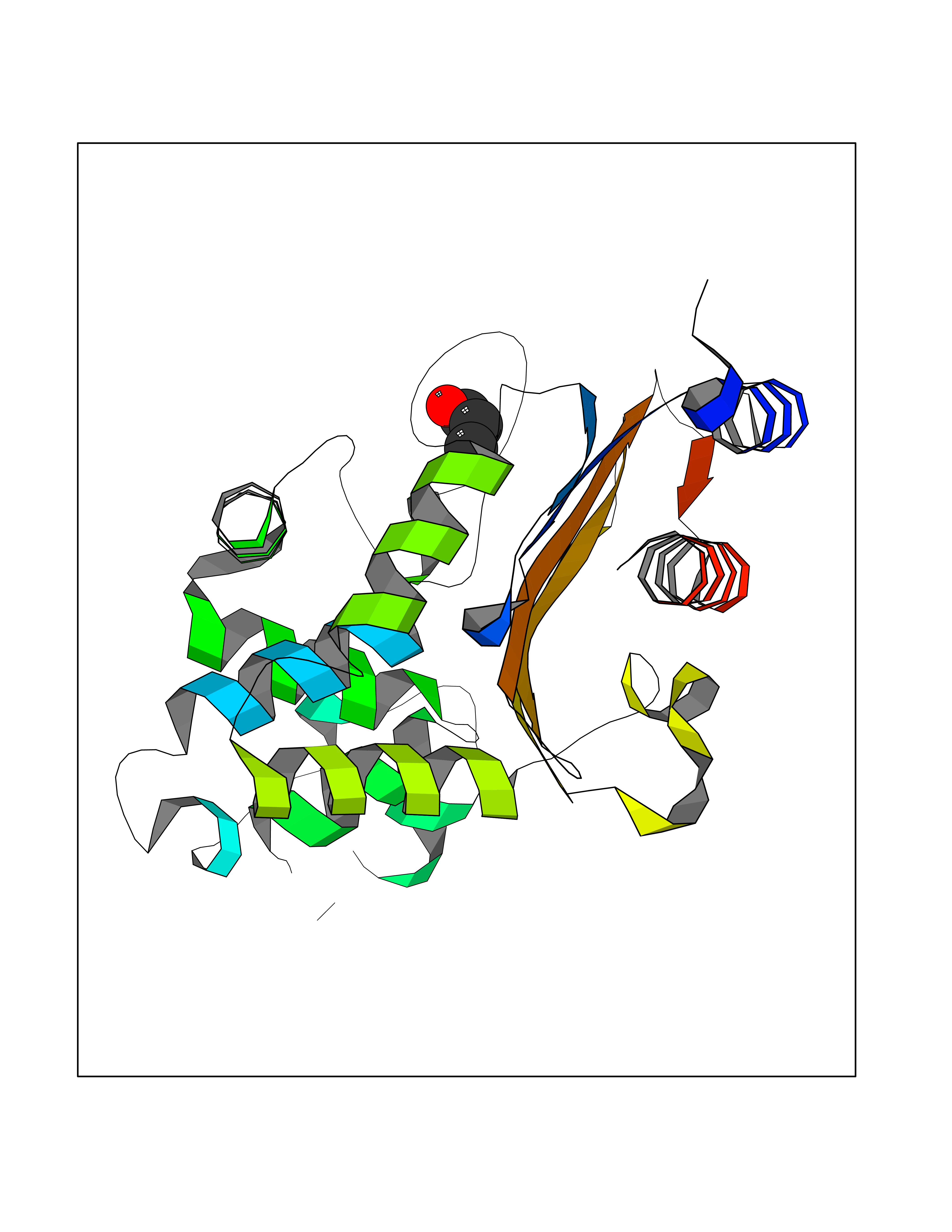 Molscript Map 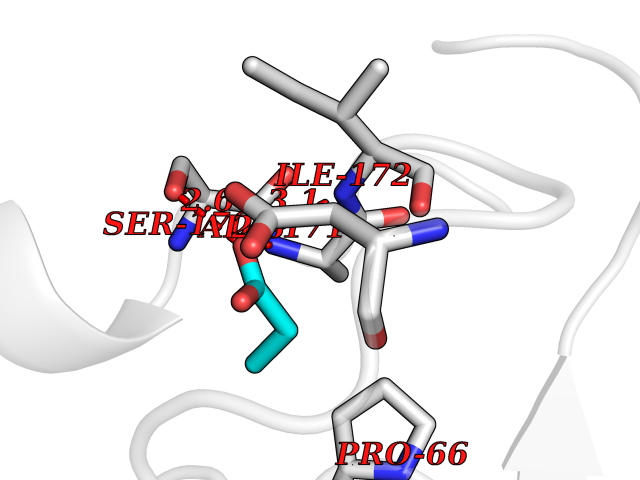 Pymol Map  Ligplot Map 3D Binding mode Sequence TNLVAEPFAKLEQDFGGSIGVYAMDTGSGATVSYRAEERFPLCSSFKGFLAAAVLARSQQQAGLLDTPIRYGKNALVPWSPISEKYLTTGMTVAELSAAAVQYSDNAAANLLLKELGGPAGLTAFMRSIGDTTFRLDRWELELNSAIPGDARDTSSPRAVTESLQKLTLGSALAAPQRQQFVDWLKGNTTGNHRIRAAVPADWAVGDKTGTCGVYGTANDYAVVWPTGRAPIVLAVYTRAPNKDDKHSEAVIAAAARLALEGLG Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Nischarin | 3P0C | 4.55 | |
Target general information Gen name NISCH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms KIAA0975;IRAS Protein family NA Biochemical class Signaling protein Function Identical protein binding.Integrin binding.Phosphatidylinositol binding. Related diseases Dyskinesia, limb and orofacial, infantile-onset (IOLOD) [MIM:616921]: An autosomal recessive, early-onset hyperkinetic movement disorder characterized by axial hypotonia, dyskinesia of the limbs and trunk, orofacial dyskinesia, drooling, and dysarthria. The severity of the hyperkinesis is variable. {ECO:0000269|PubMed:27058446}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Striatal degeneration, autosomal dominant 2 (ADSD2) [MIM:616922]: An autosomal dominant disorder characterized by striatal degeneration and dysfunction of basal ganglia, resulting in hyperkinesis. {ECO:0000269|PubMed:27058447}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08838; DB09242; DB15133; DB00697 Interacts with Q9Y2I1; P51151; P61107; P05714 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Apoptosis; Cell membrane; Coiled coil; Cytoplasm; Endosome; Leucine-rich repeat; Membrane; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Repeat Protein physicochemical properties Chain ID A,B Molecular weight (Da) 25709.3 Length 219 Aromaticity 0.11 Instability index 13.52 Isoelectric point 8.57 Charge (pH=7) 3.01 2D Binding mode Binding energy (Kcal/mol) -6.21  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence NLYFQSMEARVVGSELVDTYTVYIIQVTDGSHEWTVKHRYSDFHDLHEKLVAERKIDKNLLPPKKIIGKNSRSLVEKREKDLEVYLQKLLAAFPGVTPRVLAHFLHFHFYESMEARVVGSELVDTYTVYIIQVTDGSHEWTVKHRYSDFHDLHEKLVAERKIDKNLLPPKKIIGKNSRSLVEKREKDLEVYLQKLLAAFPGVTPRVLAHFLHFHFYEIN Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Valacyclovir hydrolase (BPHL) | 2OCI | 4.55 | |
Target general information Gen name BPHL Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms VACVase; MCNAA; DJ40E16.6.1; Breast epithelial mucin-associated antigen; Bph-rp; Biphenyl hydrolase-like protein (Serine hydrolase) (Breast epithelial mucin-associated antigen, MCNAA, Bph-rp), variant Protein family AB hydrolase superfamily, Lipase family Biochemical class Carboxylic ester hydrolase Function Serine hydrolase that catalyzes the hydrolytic activation of amino acid ester prodrugs of nucleoside analogs such as valacyclovir and valganciclovir. Activates valacyclovir to acyclovir. May play a role in detoxification processes. It is a specific alpha-amino acid ester hydrolase that prefers small, hydrophobic, and aromatic side chains and does not have a stringent requirement for the leaving group other than preferring aprimary alcohol. Related diseases Lecithin-cholesterol acyltransferase deficiency (LCATD) [MIM:245900]: A disorder of lipoprotein metabolism characterized by inadequate esterification of plasmatic cholesterol. Two clinical forms are recognized: complete LCAT deficiency and fish-eye disease. LCATD is generally referred to the complete form which is associated with absence of both alpha and beta LCAT activities resulting in esterification anomalies involving both HDL (alpha-LCAT activity) and LDL (beta-LCAT activity). It causes a typical triad of diffuse corneal opacities, target cell hemolytic anemia, and proteinuria with renal failure. {ECO:0000269|PubMed:11423760, ECO:0000269|PubMed:12957688, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:16051254, ECO:0000269|PubMed:16216249, ECO:0000269|PubMed:1681161, ECO:0000269|PubMed:1859405, ECO:0000269|PubMed:2370048, ECO:0000269|PubMed:7607641, ECO:0000269|PubMed:7711728, ECO:0000269|PubMed:8318557, ECO:0000269|PubMed:8432868, ECO:0000269|PubMed:8807342, ECO:0000269|PubMed:9007616, ECO:0000269|PubMed:9741700}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fish-eye disease (FED) [MIM:136120]: A disorder of lipoprotein metabolism due to partial lecithin-cholesterol acyltransferase deficiency that affects only alpha-LCAT activity. FED is characterized by low plasma HDL and corneal opacities due to accumulation of cholesterol deposits in the cornea ('fish-eye'). {ECO:0000269|PubMed:1516702, ECO:0000269|PubMed:1571050, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:1737840, ECO:0000269|PubMed:21901787, ECO:0000269|PubMed:8620346, ECO:0000269|PubMed:9261271}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03380 Interacts with P62508-3 EC number EC 3.1.-.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Hydrolase; Proteomics identification; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 28823.7 Length 254 Aromaticity 0.11 Instability index 25.18 Isoelectric point 8.31 Charge (pH=7) 2.59 2D Binding mode Binding energy (Kcal/mol) -6.21 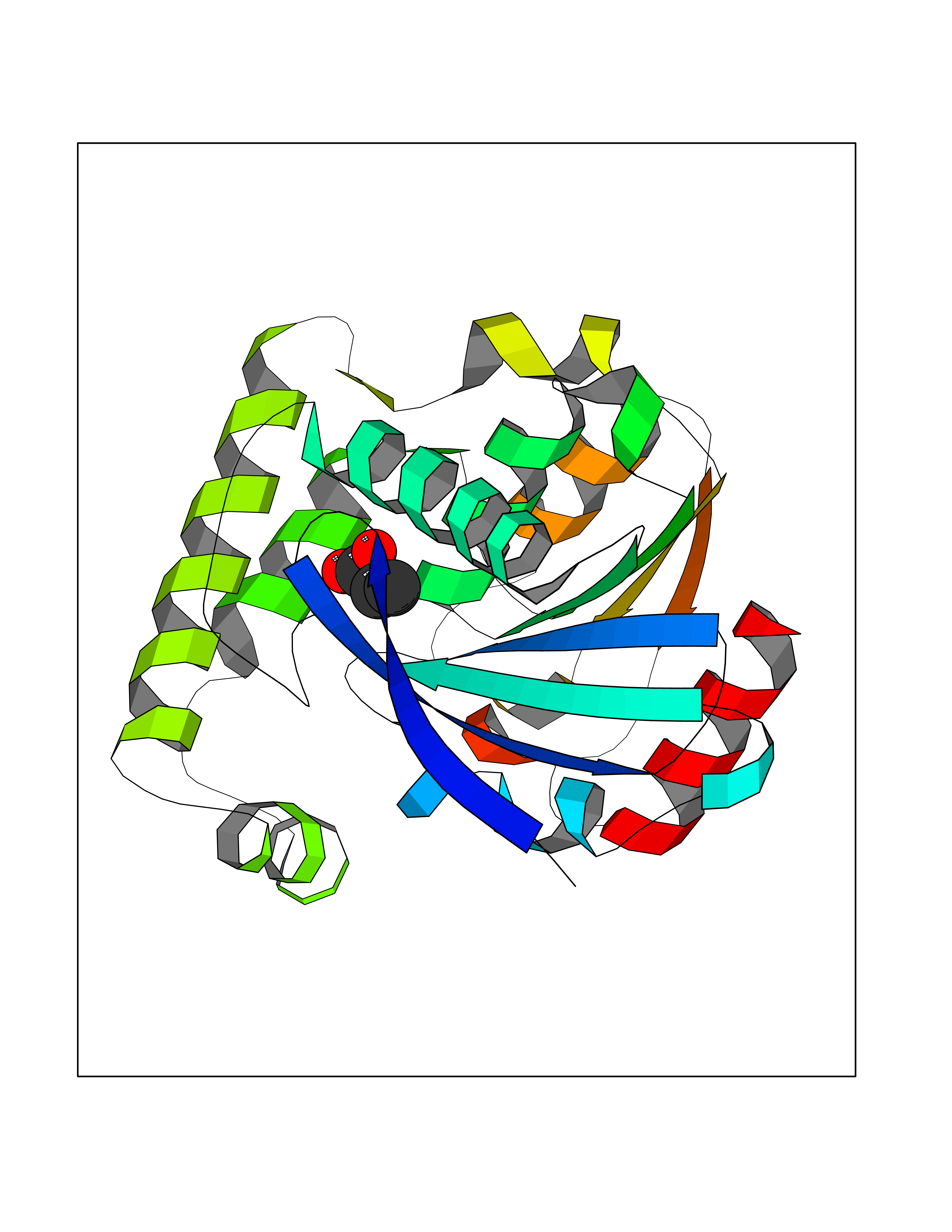 Molscript Map 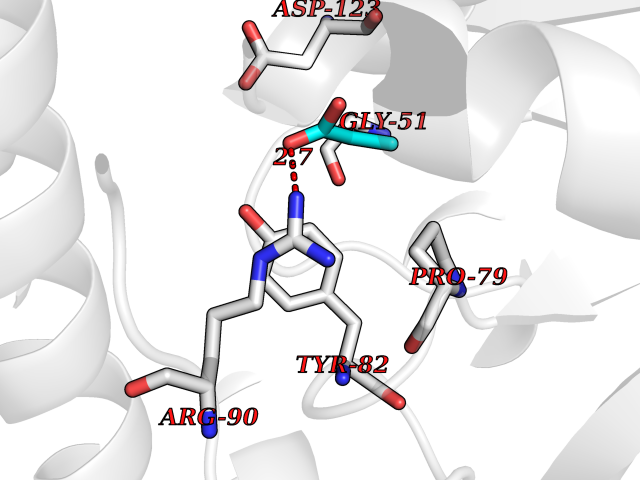 Pymol Map 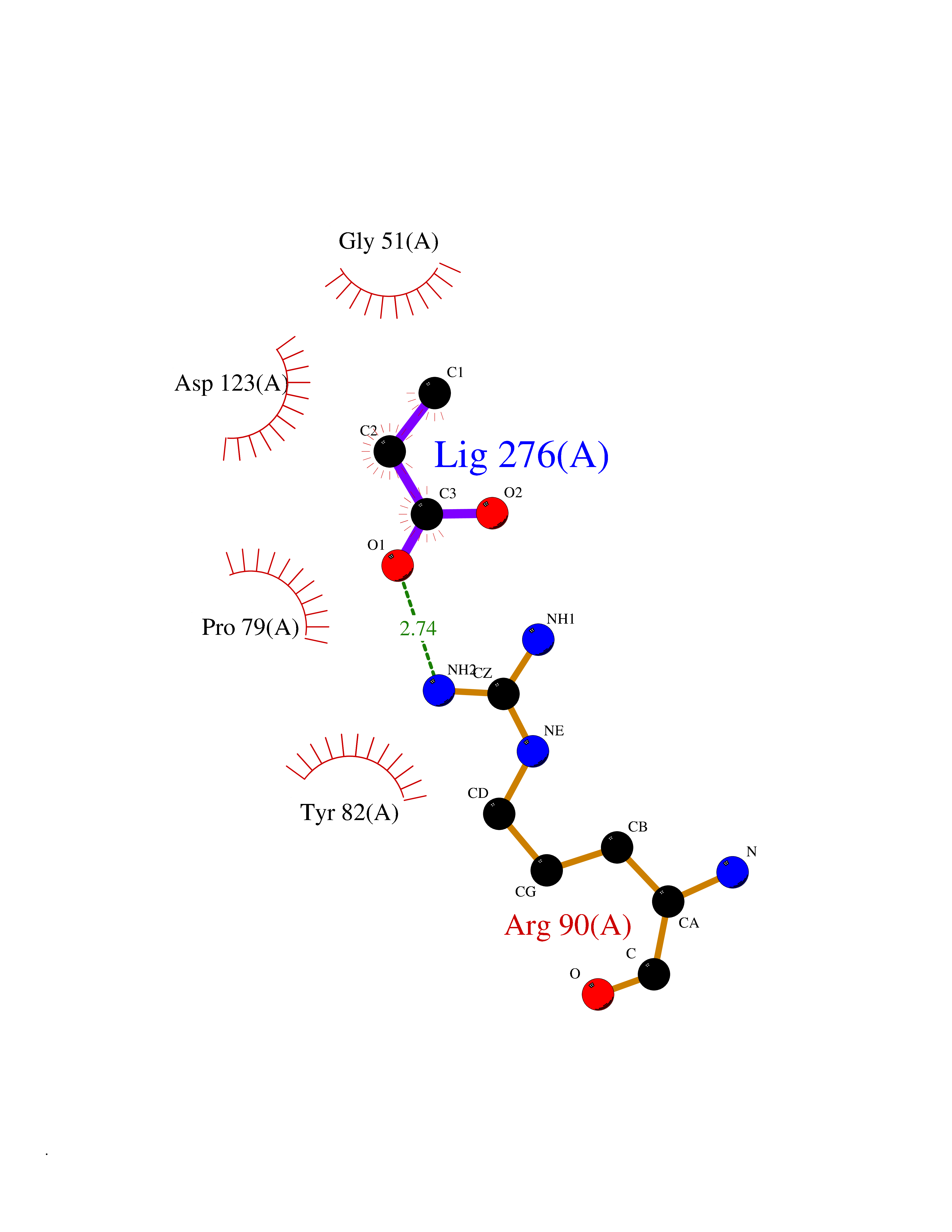 Ligplot Map 3D Binding mode Sequence SVTSAKVAVNGVQLHYQQTGEGDHAVLLLPGMLGSGETDFGPQLKNLNKKLFTVVAWDPRGYGHSRPPDRDFPADFFERDAKDAVDLMKALKFKKVSLLGWSDGGITALIAAAKYPSYIHKMVIWGANAYVTDEDSMIYEGIRDVSKWSERTRKPLEALYGYDYFARTCEKWVDGIRQFKHLPDGNICRHLLPRVQCPALIVHGEKDPLVPRFHADFIHKHVKGSRLHLMPEGKHNLHLRFADEFNKLAEDFLQ Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Alpha-1-antitrypsin (SERPINA1) | 5NBU | 4.55 | |
Target general information Gen name SERPINA1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SERPINA1; PRO0684/PRO2209; Alpha1-proteinase; Alpha-1-antiproteinase; Alpha-1 protease inhibitor Protein family Serpin family Biochemical class Serpin protein Function Inhibitor of serine proteases. Its primary target is elastase, but it also has a moderate affinity for plasmin and thrombin. Related diseases Alpha-1-antitrypsin deficiency (A1ATD) [MIM:613490]: A disorder whose most common manifestation is emphysema, which becomes evident by the third to fourth decade. A less common manifestation of the deficiency is liver disease, which occurs in children and adults, and may result in cirrhosis and liver failure. Environmental factors, particularly cigarette smoking, greatly increase the risk of emphysema at an earlier age. {ECO:0000269|PubMed:1905728, ECO:0000269|PubMed:2227940, ECO:0000269|PubMed:2390072}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01998; DB09130; DB00080; DB03345; DB14007; DB05961; DB05481; DB01593; DB14487; DB14533; DB14548 Interacts with Q9Y282; Q8N7X4; P01009; P43307; O15393; P00772; P71213; P00760 EC number NA Uniprot keywords 3D-structure; Acute phase; Alternative splicing; Blood coagulation; Direct protein sequencing; Endoplasmic reticulum; Extracellular matrix; Glycoprotein; Hemostasis; Phosphoprotein; Protease inhibitor; Proteomics identification; Reference proteome; Secreted; Serine protease inhibitor; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 41542.2 Length 370 Aromaticity 0.09 Instability index 30.24 Isoelectric point 5.56 Charge (pH=7) -9.66 2D Binding mode Binding energy (Kcal/mol) -6.2 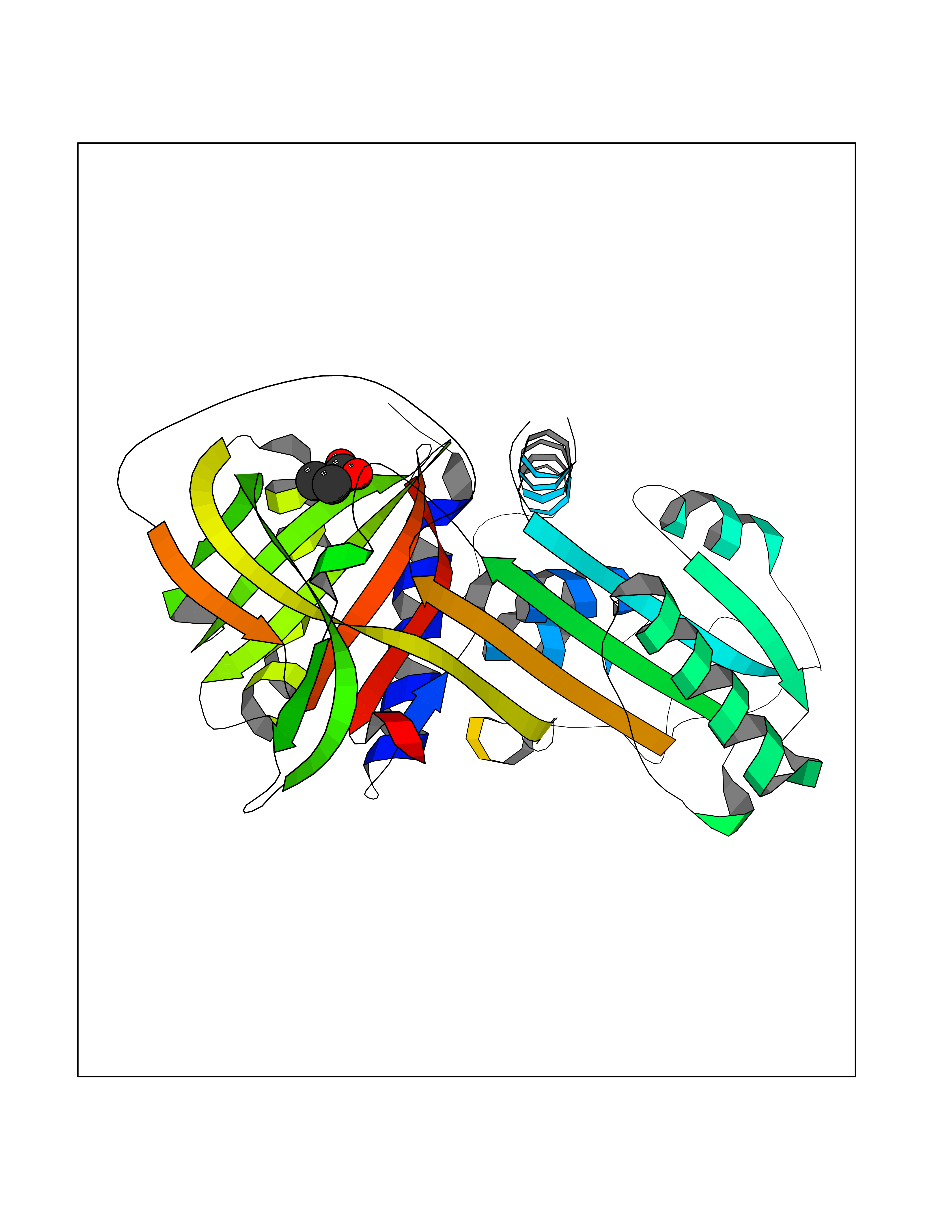 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence TFNKITPNLAEFAFSLYRQLAHQSNSTNILFSPVSIAAAFAMLSLGAKGDTHDEILEGLNFNLTEIPEAQIHEGFQELLRTLNQSQLQLTTGNGLFLSEGLKLVDKFLEDVKKLYHSEAFTVNFGDTEEAKKQINDYVEKGTQGKIVDLVKELDRDTVFALVNYIFFKGKWERPFEVKDTEEEDFHVDQVTTVKVPMMKRLGMFNIQHSKKLSSWVLLMKYLGNATAIFFLPDEGKLQHLENELTHDIITKFLENEDRRSASLHLPKLSITGTYDLKSVLGQLGITKVFSNGADLSGVTEEAPLKLSKAVHKAVLTIDEKGTEAAGAMFLEAIPMSIPPEVKFNKPFVFLIIEQNTKAPLFMGRVVNPTQ Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Sphingosine-1-phosphate receptor 2 (S1PR2) | 7T6B | 4.55 | |
Target general information Gen name S1PR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Sphingosine 1-phosphate receptor Edg-5; S1PR2; S1P2; S1P receptor Edg-5; S1P receptor 2; Endothelial differentiation G-protein coupled receptor 5 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for the lysosphingolipid sphingosine 1- phosphate (S1P). S1P is a bioactive lysophospholipid that elicits diverse physiological effect on most types of cells and tissues. When expressed in rat HTC4 hepatoma cells, is capable of mediating S1P-induced cell proliferation and suppression of apoptosis. Related diseases Deafness, autosomal recessive, 68 (DFNB68) [MIM:610419]: A form of non-syndromic sensorineural hearing loss. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. {ECO:0000269|PubMed:26805784}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P16144; Q9JK11-1 EC number NA Uniprot keywords 3D-structure; Cell membrane; Deafness; Disease variant; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Non-syndromic deafness; Palmitate; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 28917.3 Length 264 Aromaticity 0.11 Instability index 38.95 Isoelectric point 9.11 Charge (pH=7) 9.27 2D Binding mode Binding energy (Kcal/mol) -6.2  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence NKVQEHYNYTKTSRQVASAFIVILCCAIVVENLLVLIAVARNSKFHSAMYLFLGNLAASDLLAGVAFVANTLLSGSVTLRLTPVQWFAREGSAFITLSASVFSLLAIAIERHVAIAKVKLYGSDKSCRMLLLIGASWLISLVLGGLPILGWNCLGHLEACSTVLPLYAKHYVLCVVTIFSIILLAIVALYVRIYCVVRSSQTLALLKTVTIVLGVFIVCWLPAFSILLLDYACPVHSCPILYKAHYFFAVSTLNSLLNPVIYTW Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | Hyperpolarization cyclic nucleotide-gated channel 2 (HCN2) | 3U10 | 4.55 | |
Target general information Gen name HCN2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channel 2; Brain cyclic nucleotide-gated channel 2; BCNG2; BCNG-2 Protein family Potassium channel HCN family Biochemical class Voltage-gated ion channel Function Contributes to the native pacemaker currents in heart (If) and in neurons (Ih). Can also transport ammonium in the distal nephron. Produces a large instantaneous current. Modulated by intracellular chloride ions and pH; acidic pH shifts the activation to more negative voltages. Hyperpolarization-activated ion channel exhibiting weak selectivity for potassium over sodium ions. Related diseases Epilepsy, idiopathic generalized 17 (EIG17) [MIM:602477]: A form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. Both autosomal dominant and autosomal recessive EIG17 inheritance have been reported. {ECO:0000269|PubMed:22131395, ECO:0000269|PubMed:29064616}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Febrile seizures, familial, 2 (FEB2) [MIM:602477]: Seizures associated with febrile episodes in childhood without any evidence of intracranial infection or defined pathologic or traumatic cause. It is a common condition, affecting 2-5% of children aged 3 months to 5 years. The majority are simple febrile seizures (generally defined as generalized onset, single seizures with a duration of less than 30 minutes). Complex febrile seizures are characterized by focal onset, duration greater than 30 minutes, and/or more than one seizure in a 24 hour period. The likelihood of developing epilepsy following simple febrile seizures is low. Complex febrile seizures are associated with a moderately increased incidence of epilepsy. FEB2 transmission pattern is consistent with autosomal dominant inheritance. {ECO:0000269|PubMed:24324597}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02527; DB02315; DB09083 Interacts with Q9UL51; Q4ACU6-1 EC number NA Uniprot keywords 3D-structure; Ammonia transport; cAMP; cAMP-binding; Cell membrane; Disease variant; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Lipoprotein; Membrane; Methylation; Nucleotide-binding; Palmitate; Phosphoprotein; Potassium; Potassium channel; Potassium transport; Proteomics identification; Reference proteome; Sodium; Sodium channel; Sodium transport; Transmembrane; Transmembrane helix; Transport; Voltage-gated channel Protein physicochemical properties Chain ID A Molecular weight (Da) 23672.9 Length 202 Aromaticity 0.12 Instability index 38.05 Isoelectric point 8.85 Charge (pH=7) 4.11 2D Binding mode Binding energy (Kcal/mol) -6.2  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence DSSRRQYQEKYKQVEQYMSFHKLPADFRQKIHDYYEHRYQGKMFDEDSILGELNGPLREEIVNFNCRKLVASMPLFANADPNFVTAMLTKLKFEVFQPGDYIIREGTIGKKMYFIQHGVVSVLTKGNKEMKLSDGSYFGEICLLTRGRRTASVRADTYCRLYSLSVDNFNEVLEEYPMMRRAFETVAIDRLDRIGKKNSILL Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Endoplasmic reticulum chaperone BiP (HSPA5) | 6ASY | 4.55 | |
Target general information Gen name HSPA5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Immunoglobulin heavy chainbinding protein; Immunoglobulin heavy chain-binding protein; Heat shock protein family A member 5; Heat shock protein 70 family protein 5; Heat shock 70 kDa protein 5; HSP70 Protein family Heat shock protein 70 family Biochemical class Acid anhydride hydrolase Function Involved in the correct folding of proteins and degradation of misfolded proteins via its interaction with DNAJC10/ERdj5, probably to facilitate the release of DNAJC10/ERdj5 from its substrate. Acts as a key repressor of the ERN1/IRE1-mediated unfolded protein response (UPR). In the unstressed endoplasmic reticulum, recruited by DNAJB9/ERdj4 to the luminal region of ERN1/IRE1, leading to disrupt the dimerization of ERN1/IRE1, thereby inactivating ERN1/IRE1. Accumulation of misfolded protein in the endoplasmic reticulum causes release of HSPA5/BiP from ERN1/IRE1, allowing homodimerization and subsequent activation of ERN1/IRE1. Plays an auxiliary role in post-translational transport of small presecretory proteins across endoplasmic reticulum (ER). May function as an allosteric modulator for SEC61 channel-forming translocon complex, likely cooperating with SEC62 to enable the productive insertion of these precursors into SEC61 channel. Appears to specifically regulate translocation of precursors having inhibitory residues in their mature region that weaken channel gating. Endoplasmic reticulum chaperone that plays a key role in protein folding and quality control in the endoplasmic reticulum lumen. Related diseases Autoantigen in rheumatoid arthritis. {ECO:0000269|PubMed:11160188}. Drugs (DrugBank ID) DB00945; DB00025; DB09130; DB13998; DB13999 Interacts with Q9BYF1; P05067; P18850; Q9ULD4-2; Q6E0U4; Q9UBS4; Q9UBS3; P49184; Q92874; Q9NZJ5; P04626; O75460; O75460-1; P62993; Q15486; P14625; Q9Y4L1; P01721; O95868; Q9Y328; Q96IZ0; Q15084; P04049; P61619; P36955; Q9H173; Q9UHI5; Q13573; Q6URK8; Q6PF05; Q13404; Q9NYU1; P09544; Q6T424; Q9QXT0; Q91YW3; Q9Z2B5; Q62627; K0BRG7; P0DTC2 EC number EC 3.6.4.10 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Chaperone; Cytoplasm; Direct protein sequencing; Endoplasmic reticulum; Host-virus interaction; Hydrolase; Isopeptide bond; Methylation; Nitration; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Signal; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 67147.3 Length 606 Aromaticity 0.06 Instability index 32.09 Isoelectric point 5.22 Charge (pH=7) -15.9 2D Binding mode Binding energy (Kcal/mol) -6.21 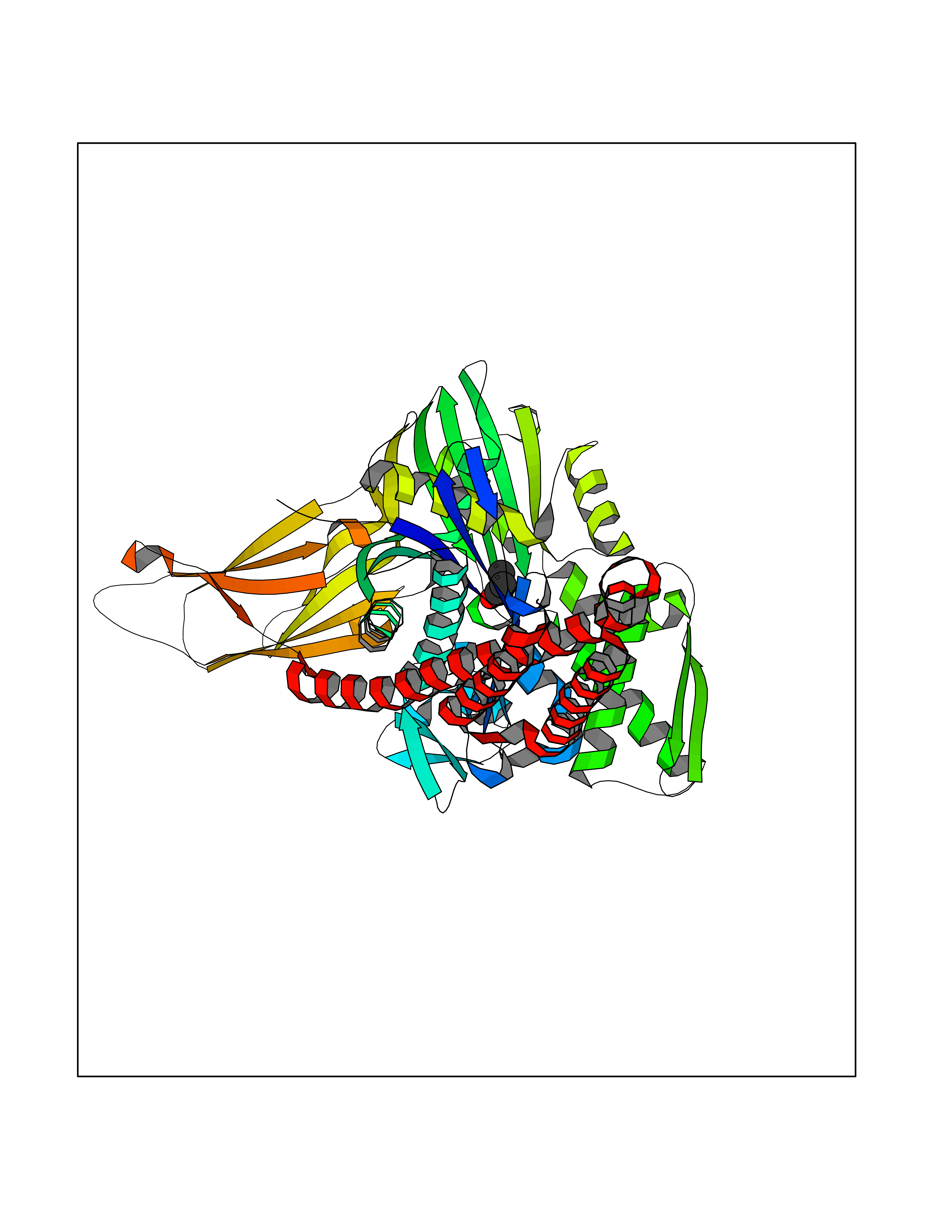 Molscript Map 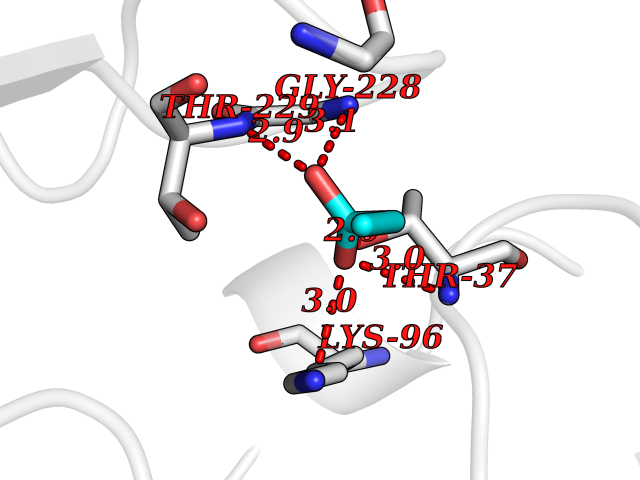 Pymol Map 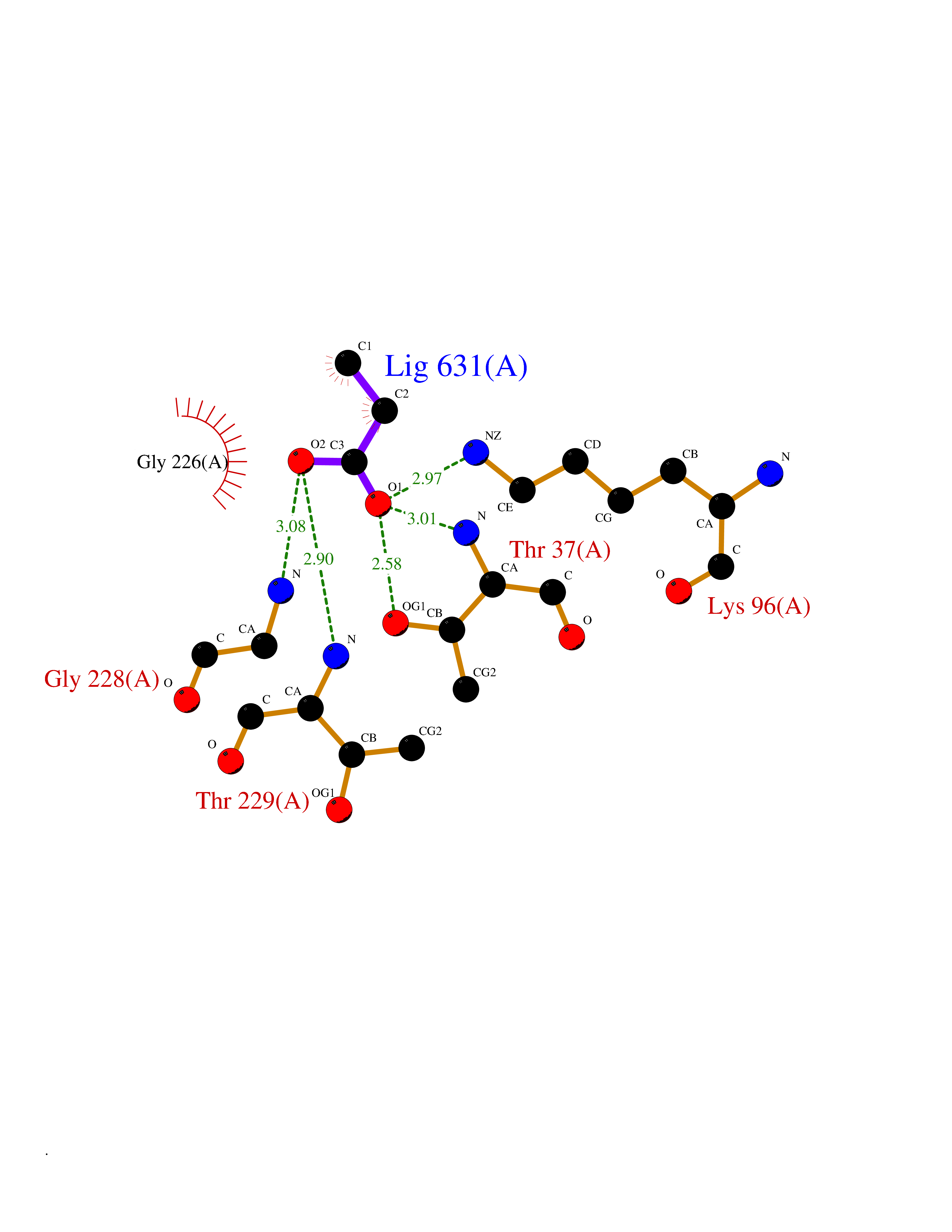 Ligplot Map 3D Binding mode Sequence SEDVGTVVGIDLGTTYSCVGVFKNGRVEIIANDQGNRITPSYVAFTPEGERLIGDAAKNQLTSNPENTVFDAKRLIGRTWNDPSVQQDIKFLPFKVVEKKTKPYIQVDIGGGQTKTFAPEEISAMVLTKMKETAEAYLGKKVTHAVVTVPAYFNDAQRQATKDAGTIAGLNVMRIINEPTAAAIAYGLDKREGEKNILVFDLGGGTFDVSLLTIDNGVFEVVATNGDTHLGGEDFDQRVMEHFIKLYKKKTGKDVRKDNRAVQKLRREVEKAKRALSSQHQARIEIESFYEGEDFSETLTRAKFEELNMDLFRSTMKPVQKVLEDSDLKKSDIDEIVLVGGSTRIPKIQQLVKEFFNGKEPSRGINPDEAVAYGAAVQAGVLSGDQDTGDLVLLDVCPLTLGIETVGGVMTKLIPRNTVVPTKKSQIFSVGGTVTIKVYEGERPLTKDNHLLGTFDLTGIPPAPRGVPQIEVTFEIDVNGILRVTAEDKGTGNKNKITITNDQNRLTPEEIERMVNDAEKFAEEDKKLKERIDTRNELESYAYSLKNQIGDKEKLGGKLSSEDKETMEKAVEEKIEWLESHQDADIEDFKAKKKELEEIVQPIISK Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | RAC-beta serine/threonine-protein kinase (AKT2) | 3D0E | 4.55 | |
Target general information Gen name AKT2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAC-PK-beta; Protein kinase B beta; Protein kinase Akt-2; PKB beta Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family, RAC subfamily Biochemical class Kinase Function AKT2 is one of 3 closely related serine/threonine-protein kinases (AKT1, AKT2 and AKT3) called the AKT kinase, and which regulate many processes including metabolism, proliferation, cell survival, growth and angiogenesis. This is mediated through serine and/or threonine phosphorylation of a range of downstream substrates. Over 100 substrate candidates have been reported so far, but for most of them, no isoform specificity has been reported. AKT is responsible of the regulation of glucose uptake by mediating insulin-induced translocation of the SLC2A4/GLUT4 glucose transporter to the cell surface. Phosphorylation of PTPN1 at 'Ser-50' negatively modulates its phosphatase activity preventing dephosphorylation of the insulin receptor and the attenuation of insulin signaling. Phosphorylation of TBC1D4 triggers the binding of this effector to inhibitory 14-3-3 proteins, which is required for insulin-stimulated glucose transport. AKT regulates also the storage of glucose in the form of glycogen by phosphorylating GSK3A at 'Ser-21' and GSK3B at 'Ser-9', resulting in inhibition of its kinase activity. Phosphorylation of GSK3 isoforms by AKT is also thought to be one mechanism by which cell proliferation is driven. AKT regulates also cell survival via the phosphorylation of MAP3K5 (apoptosis signal-related kinase). Phosphorylation of 'Ser-83' decreases MAP3K5 kinase activity stimulated by oxidative stress and thereby prevents apoptosis. AKT mediates insulin-stimulated protein synthesis by phosphorylating TSC2 at 'Ser-939' and 'Thr-1462', thereby activating mTORC1 signaling and leading to both phosphorylation of 4E-BP1 and in activation of RPS6KB1. AKT is involved in the phosphorylation of members of the FOXO factors (Forkhead family of transcription factors), leading to binding of 14-3-3 proteins and cytoplasmic localization. In particular, FOXO1 is phosphorylated at 'Thr-24', 'Ser-256' and 'Ser-319'. FOXO3 and FOXO4 are phosphorylated on equivalent sites. AKT has an important role in the regulation of NF-kappa-B-dependent gene transcription and positively regulates the activity of CREB1 (cyclic AMP (cAMP)-response element binding protein). The phosphorylation of CREB1 induces the binding of accessory proteins that are necessary for the transcription of pro-survival genes such as BCL2 and MCL1. AKT phosphorylates 'Ser-454' on ATP citrate lyase (ACLY), thereby potentially regulating ACLY activity and fatty acid synthesis. Activates the 3B isoform of cyclic nucleotide phosphodiesterase (PDE3B) via phosphorylation of 'Ser-273', resulting in reduced cyclic AMP levels and inhibition of lipolysis. Phosphorylates PIKFYVE on 'Ser-318', which results in increased PI(3)P-5 activity. The Rho GTPase-activating protein DLC1 is another substrate and its phosphorylation is implicated in the regulation cell proliferation and cell growth. AKT plays a role as key modulator of the AKT-mTOR signaling pathway controlling the tempo of the process of newborn neurons integration during adult neurogenesis, including correct neuron positioning, dendritic development and synapse formation. Signals downstream of phosphatidylinositol 3-kinase (PI(3)K) to mediate the effects of various growth factors such as platelet-derived growth factor (PDGF), epidermal growth factor (EGF), insulin and insulin-like growth factor I (IGF-I). AKT mediates the antiapoptotic effects of IGF-I. Essential for the SPATA13-mediated regulation of cell migration and adhesion assembly and disassembly. May be involved in the regulation of the placental development. Related diseases Defects in AKT2 are a cause of susceptibility to breast cancer (BC). AKT2 promotes metastasis of tumor cells without affecting the latency of tumor development. May play a role in glioblastoma cell survival (PubMed:20167810). {ECO:0000269|PubMed:20167810}.; DISEASE: Type 2 diabetes mellitus (T2D) [MIM:125853]: A multifactorial disorder of glucose homeostasis caused by a lack of sensitivity to insulin. Affected individuals usually have an obese body habitus and manifestations of a metabolic syndrome characterized by diabetes, insulin resistance, hypertension and hypertriglyceridemia. The disease results in long-term complications that affect the eyes, kidneys, nerves, and blood vessels. {ECO:0000269|PubMed:15166380, ECO:0000269|PubMed:19164855}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Hypoinsulinemic hypoglycemia with hemihypertrophy (HIHGHH) [MIM:240900]: A disorder characterized by hypoglycemia, low insulin levels, low serum levels of ketone bodies and branched-chain amino acids, left-sided hemihypertrophy, neonatal macrosomia, reduced consciousness and hypoglycemic seizures. {ECO:0000269|PubMed:21979934}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08073; DB07859; DB12218; DB07947; DB07812 Interacts with P31749; P49841; P08238; Q6FHY5; Q9NRD5; Q04864-2; O60504; P53804; Q9C0C9; P08670; Q15118-1 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Apoptosis; ATP-binding; Carbohydrate metabolism; Cell membrane; Cytoplasm; Developmental protein; Diabetes mellitus; Disease variant; Disulfide bond; Endosome; Glucose metabolism; Glycogen biosynthesis; Glycogen metabolism; Glycoprotein; Kinase; Manganese; Membrane; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Reference proteome; Serine/threonine-protein kinase; Sugar transport; Transferase; Translation regulation; Transport; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 37380.5 Length 324 Aromaticity 0.12 Instability index 29.68 Isoelectric point 6.19 Charge (pH=7) -3.43 2D Binding mode Binding energy (Kcal/mol) -6.2 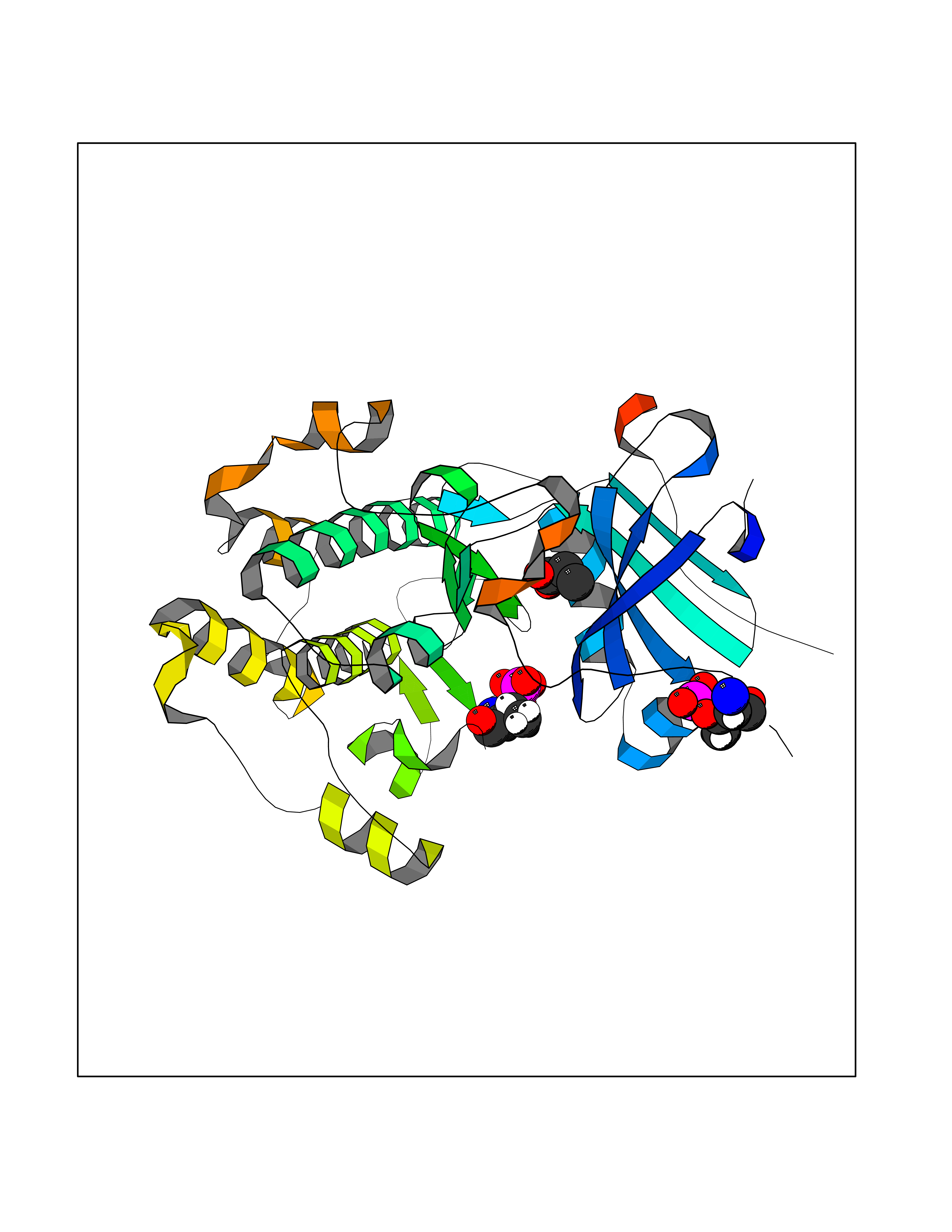 Molscript Map 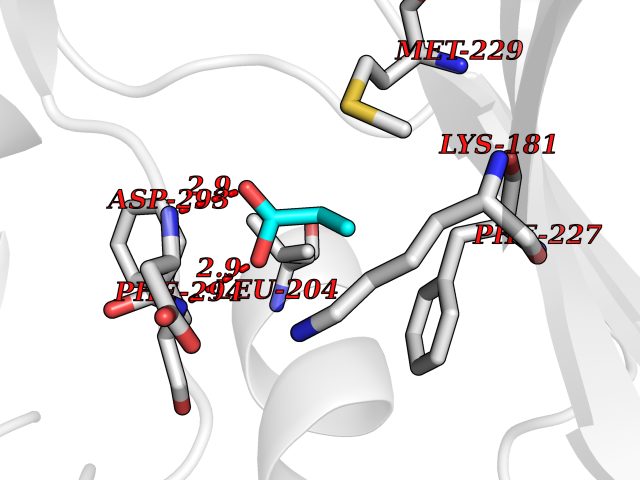 Pymol Map 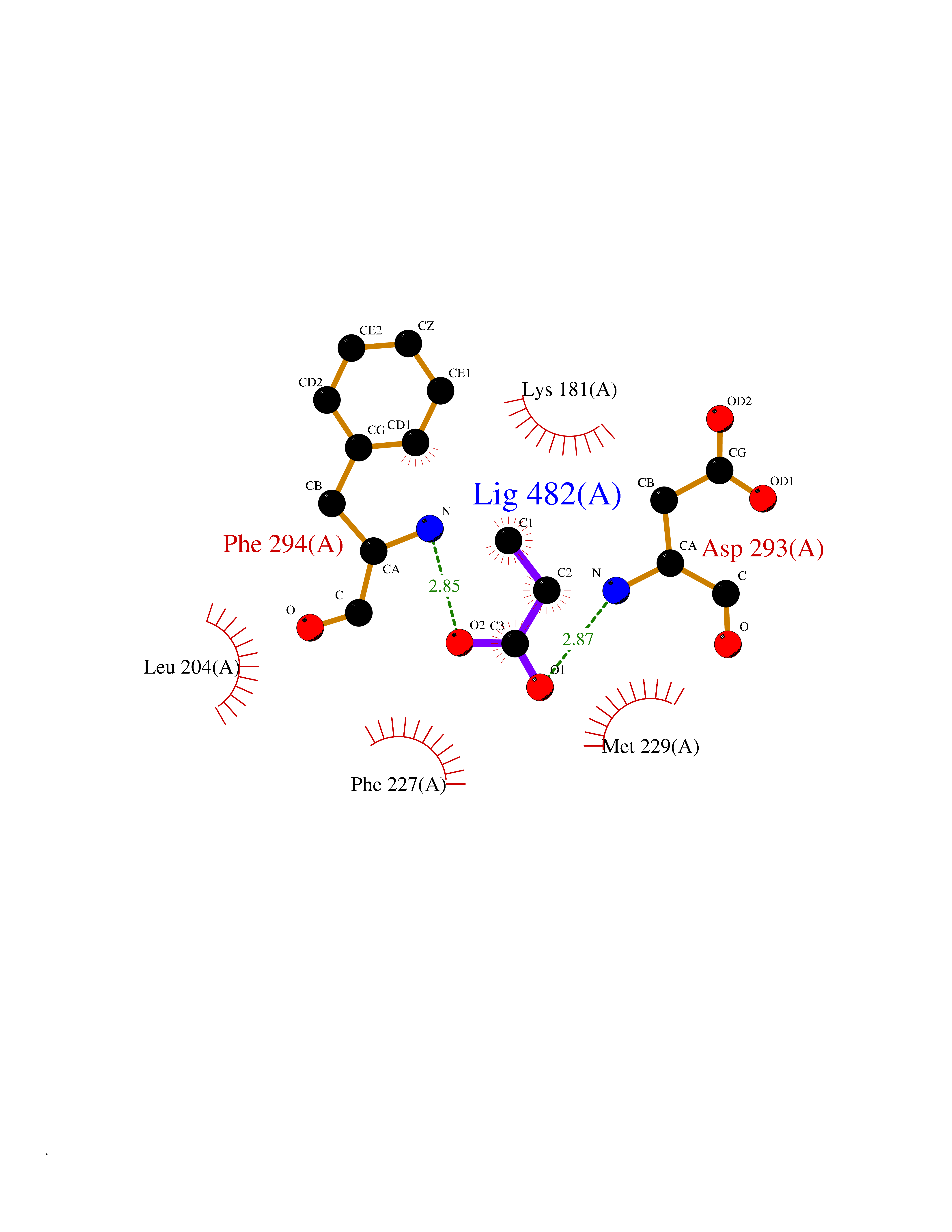 Ligplot Map 3D Binding mode Sequence KVTMNDFDYLKLLGKGTFGKVILVREKATGRYYAMKILRKEVIIAKDEVAHTVTESRVLQNTRHPFLTALKYAFQTHDRLCFVMEYANGGELFFHLSRERVFTEERARFYGAEIVSALEYLHSRDVVYRDIKLENLMLDKDGHIKITDFGLCKEGISDGATMKXFCGTPEYLAPEVLEDNDYGRAVDWWGLGVVMYEMMCGRLPFYNQDHERLFELILMEEIRFPRTLSPEAKSLLAGLLKKDPKQRLGGGPSDAKEVMEHRFFLSINWQDVVQKKLLPPFKPQVTSEVDTRYFDDEFTAQSITIXPPDQRTHFPQFDYSASIR Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Glycogen phosphorylase, liver form | 3DDS | 4.54 | |
Target general information Gen name PYGL Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Glycogen phosphorylase family Biochemical class Transferase Function AMP binding.ATP binding.Bile acid binding.Drug binding.Glucose binding.Glycogen phosphorylase activity.Linear malto-oligosaccharide phosphorylase activity.Protein homodimerization activity.Purine nucleobase binding.Pyridoxal phosphate binding.SHG alpha-glucan phosphorylase activity.Vitamin binding. Related diseases Glycogen storage disease 6 (GSD6) [MIM:232700]: A metabolic disorder characterized by mild to moderate hypoglycemia, mild ketosis, growth retardation, and prominent hepatomegaly. Heart and skeletal muscle are not affected. {ECO:0000269|PubMed:9529348}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P11216; P11217 EC number 2.4.1.1 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; Carbohydrate metabolism; Cytoplasm; Disease variant; Glycogen metabolism; Glycogen storage disease; Glycosyltransferase; Nucleotide-binding; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89481.9 Length 778 Aromaticity 0.11 Instability index 34.96 Isoelectric point 6.49 Charge (pH=7) -3.54 2D Binding mode Binding energy (Kcal/mol) -6.2  Molscript Map 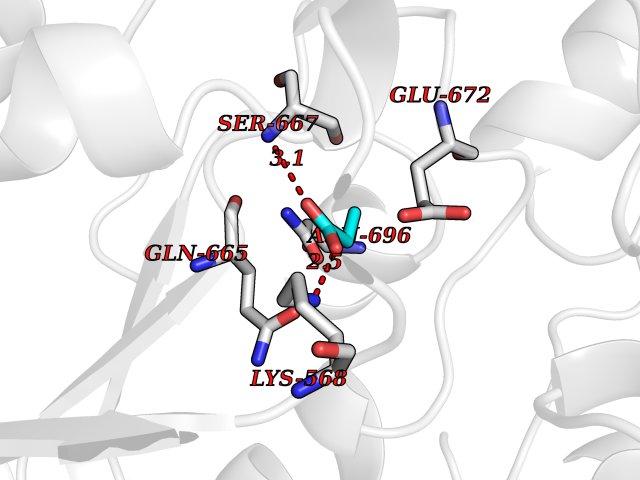 Pymol Map 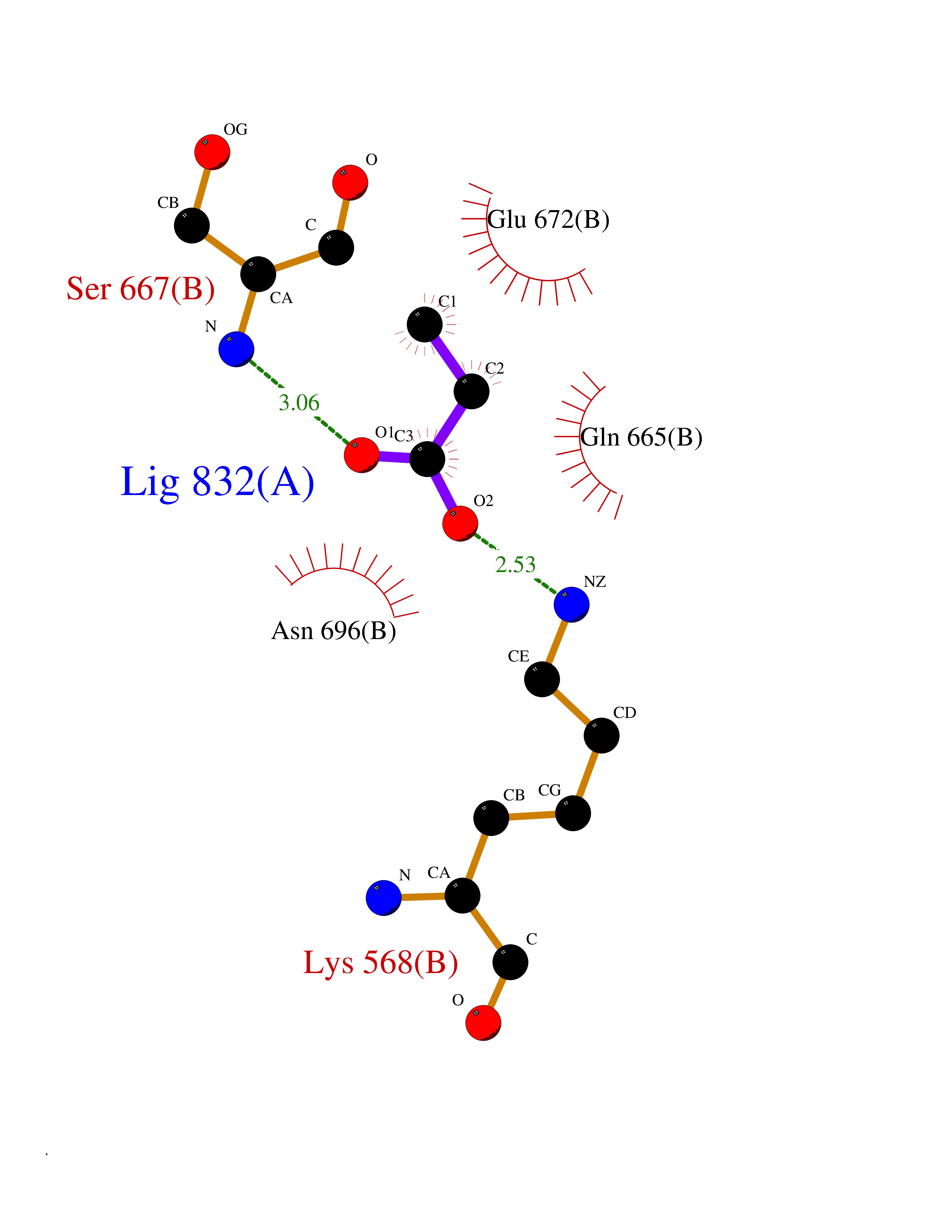 Ligplot Map 3D Binding mode Sequence NVAELKKSFNRHLHFTLVKDRNVATTRDYYFALAHTVRDHLVGRWIRTQQHYYDKCPKRVYYLSLEFYMGRTLQNTMINLGLQNACDEAIYQLGLDIEELEEIEEDAGLGNGGLGRLAACFLDSMATLGLAAYGYGIRYEYGIFNQKIRDGWQVEEADDWLRYGNPWEKSRPEFMLPVHFYGKVEHTNTGTKWIDTQVVLALPYDTPVPGYMNNTVNTMRLWSENISRVLYPNDNFFEGKELRLKQEYFVVAATLQDIIRRFKASKFGSTGTVFDAFPDQVAIQLNDTHPALAIPELMRIFVDIEKLPWSKAWELTQKTFAYTNHTVLPEALERWPVDLVEKLLPRHLEIIYEINQKHLDRIVALFPKDVDRLRRMSLIEEEGSKRINMAHLCIVGSHAVNGVAKIHSDIVKTKVFKDFSELEPDKFQNKTNGITPRRWLLLCNPGLAELIAEKIGEDYVKDLSQLTKLHSFLGDDVFLRELAKVKQENKLKFSQFLETEYKVKINPSSMFDVQVKRIHEYKRQLLNCLHVITMYNRIKKDPKKLFVPRTVIIGGKAAPGYHMAKMIIKLITSVADVVNNDPMVGSKLKVIFLENYRVSLAEKVIPATDLSEQISTAGTEASGTGNMKFMLNGALTIGTMDGANVEMAEEAGEENLFIFGMRIDDVAALDKKGYEAKEYYEALPELKLVIDQIDNGFFSPKQPDLFKDIINMLFYHDRFKVFADYEAYVKCQDKVSQLYMNPKAWNTMVLKNIAASGKFSSDRTIKEYAQNIWNVEPS Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Carbonic anhydrase 3 | 3UYQ | 4.54 | |
Target general information Gen name CA3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Alpha-carbonic anhydrase family Biochemical class Lyase Function Carbonate dehydratase activity.Nickel cation binding.Phosphatase activity.Zinc ion binding. Related diseases Cortical dysplasia, complex, with other brain malformations 6 (CDCBM6) [MIM:615771]: A disorder of aberrant neuronal migration and disturbed axonal guidance. Affected individuals have microcephaly, ataxia, and severe delayed psychomotor development. Brain imaging shows variable malformations of cortical development, including white matter streaks, dysmorphic basal ganglia, corpus callosum abnormalities, brainstem and cerebellar hypoplasia, cortical dysplasia, polymicrogyria. {ECO:0000269|PubMed:23246003}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Skin creases, congenital symmetric circumferential, 1 (CSCSC1) [MIM:156610]: An autosomal dominant disease characterized by multiple, symmetric, circumferential rings of folded skin, affecting primarily the limbs. Affected individuals also exhibit intellectual disability, cleft palate, and dysmorphic features. {ECO:0000269|PubMed:26637975}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00819; DB00562; DB01194; DB00482; DB00606; DB01144; DB00869; DB08846; DB00311; DB00703; DB12418; DB00391; DB00273; DB00580; DB00909 Interacts with P37235; Q9BS40 EC number 4.2.1.1 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Glutathionylation; Lyase; Metal-binding; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 29416.7 Length 259 Aromaticity 0.11 Instability index 38.2 Isoelectric point 6.71 Charge (pH=7) -1.07 2D Binding mode Binding energy (Kcal/mol) -6.19  Molscript Map 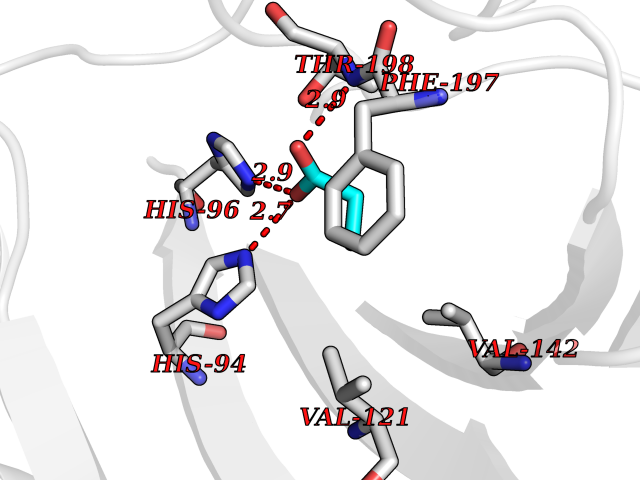 Pymol Map 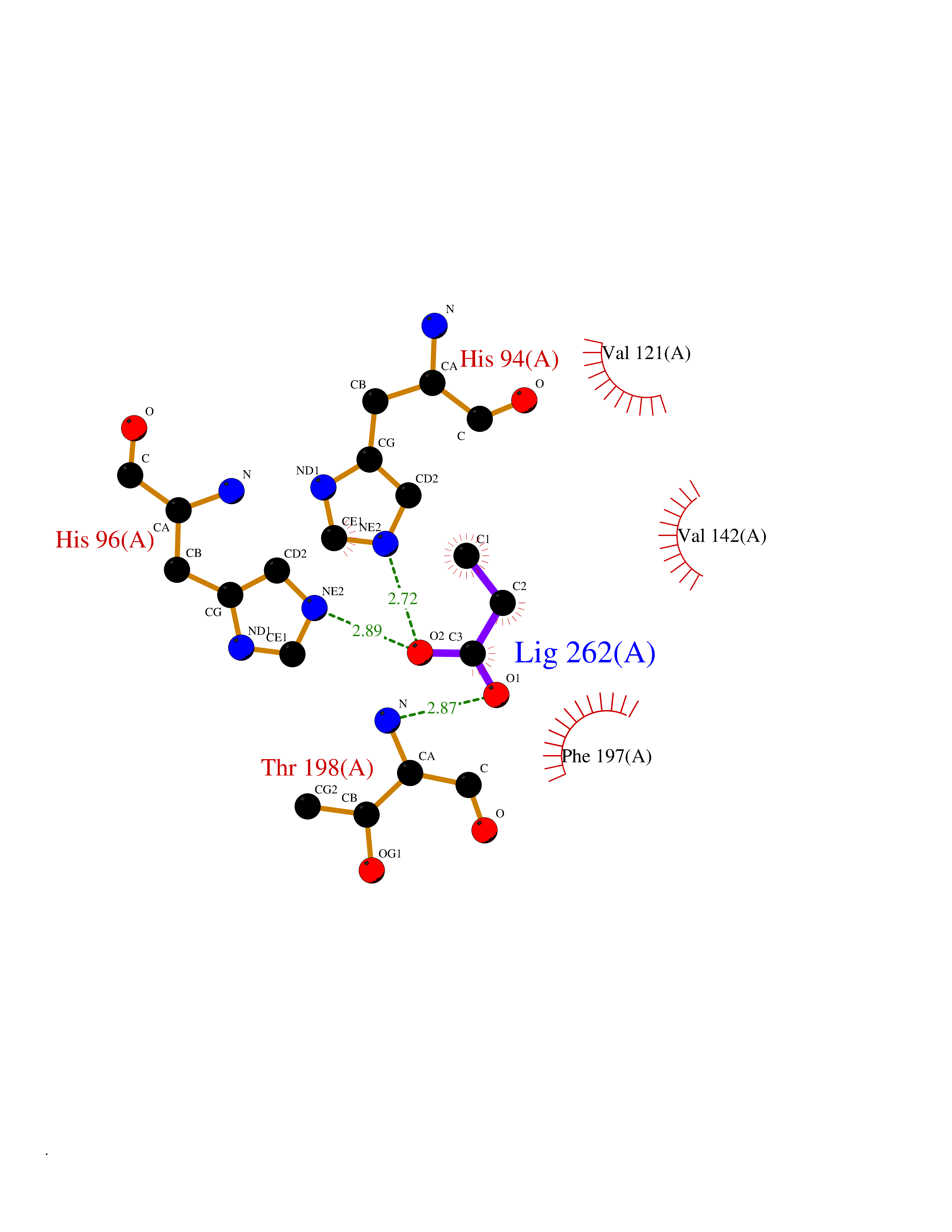 Ligplot Map 3D Binding mode Sequence AKEWGYASHNGPDHWHELFPNAKGENQSPIELHTKDIRHDPSLQPWSVSYDGGSAKTILNNGHTCRVVFDDTYDRSMLRGGPLPGPYRLRQFHLHWGSSDDHGSEHTVDGVKYAAELHLVHWNPKYNTFKEALKQRDGIAVIGIFLKIGHENGEFQIFLDALDKIKTKGKEAPFTKFDPSSLFPASRDYWTYQGSFTTPPCEECIVWLLLKEPMTVSSDQMAKLRSLLSSAENEPPVPLVSNWRPPQPINNRVVRASFK Hydrogen bonds contact Hydrophobic contact | ||||