Job Results:
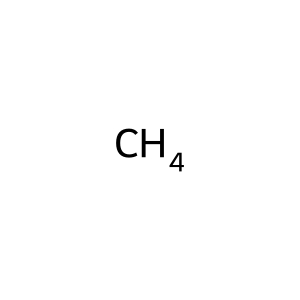
Ligand
Structure
Job ID
9069676bc0da30cae69182dac941f7de
Job name
NA
Time
2026-02-27 11:56:18
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Cytochrome c oxidase subunit 2 | 3VRJ | 3.94 | |
Target general information Gen name MT-CO2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms MTCO2;COXII;COII;COX2 Protein family Cytochrome c oxidase subunit 2 family Biochemical class Immune system Function Copper ion binding.Cytochrome-c oxidase activity. Related diseases Mitochondrial complex IV deficiency (MT-C4D) [MIM:220110]: A disorder of the mitochondrial respiratory chain with heterogeneous clinical manifestations, ranging from isolated myopathy to severe multisystem disease affecting several tissues and organs. Features include hypertrophic cardiomyopathy, hepatomegaly and liver dysfunction, hypotonia, muscle weakness, exercise intolerance, developmental delay, delayed motor development and intellectual disability. Some affected individuals manifest a fatal hypertrophic cardiomyopathy resulting in neonatal death. A subset of patients manifest Leigh syndrome. {ECO:0000269|PubMed:10486321}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02659; DB04464; DB05412 Interacts with Q9NZ94-2; P49281-3 EC number 7.1.1.9 Uniprot keywords 3D-structure; Copper; Disease variant; Electron transport; Magnesium; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Primary mitochondrial disease; Proteomics identification; Reference proteome; Respiratory chain; Translocase; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID C Molecular weight (Da) 21687.9 Length 189 Aromaticity 0.11 Instability index 38 Isoelectric point 5.68 Charge (pH=7) -3.26 2D Binding mode Binding energy (Kcal/mol) -5.37 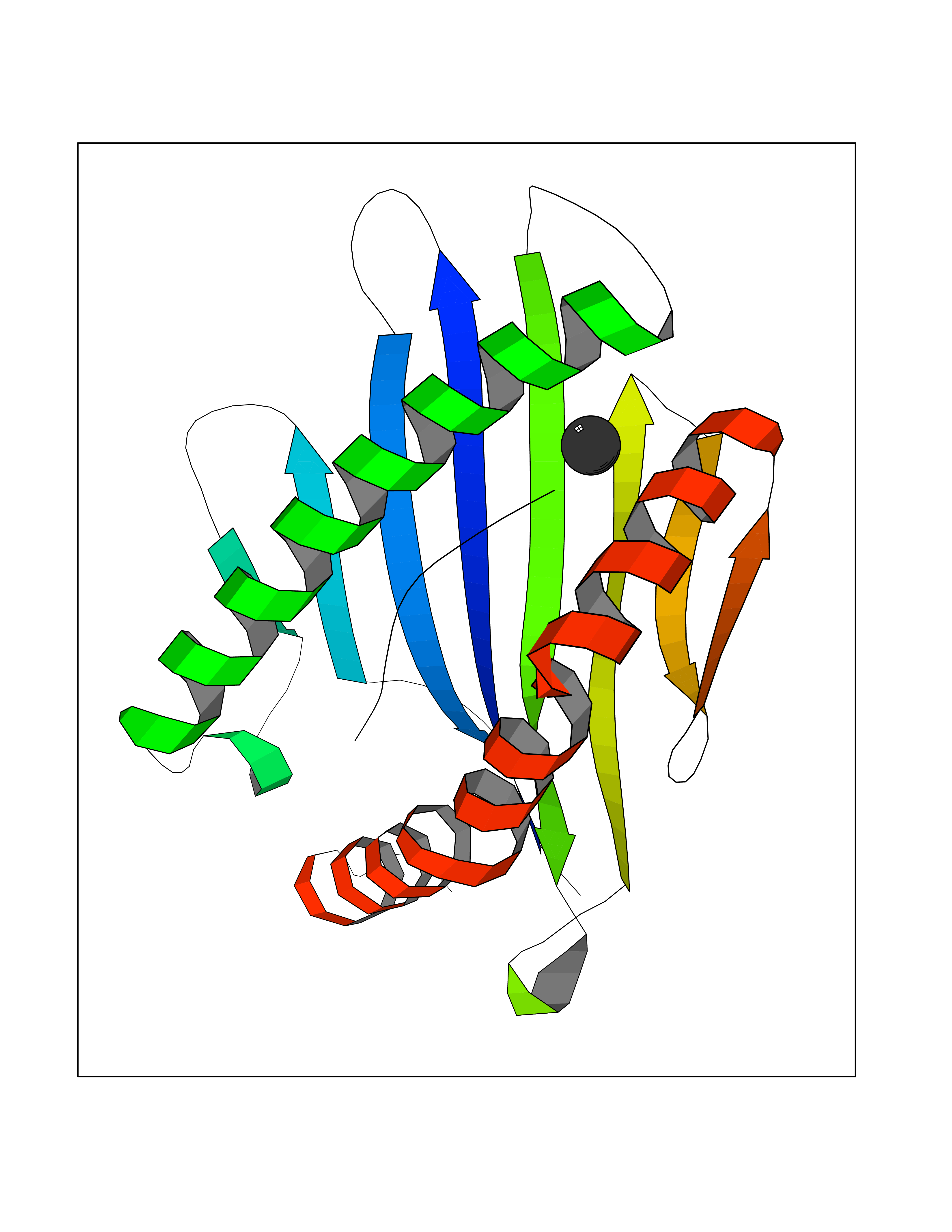 Molscript Map 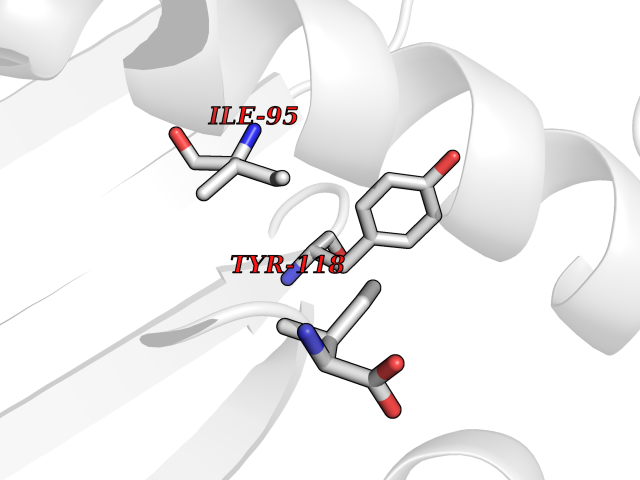 Pymol Map 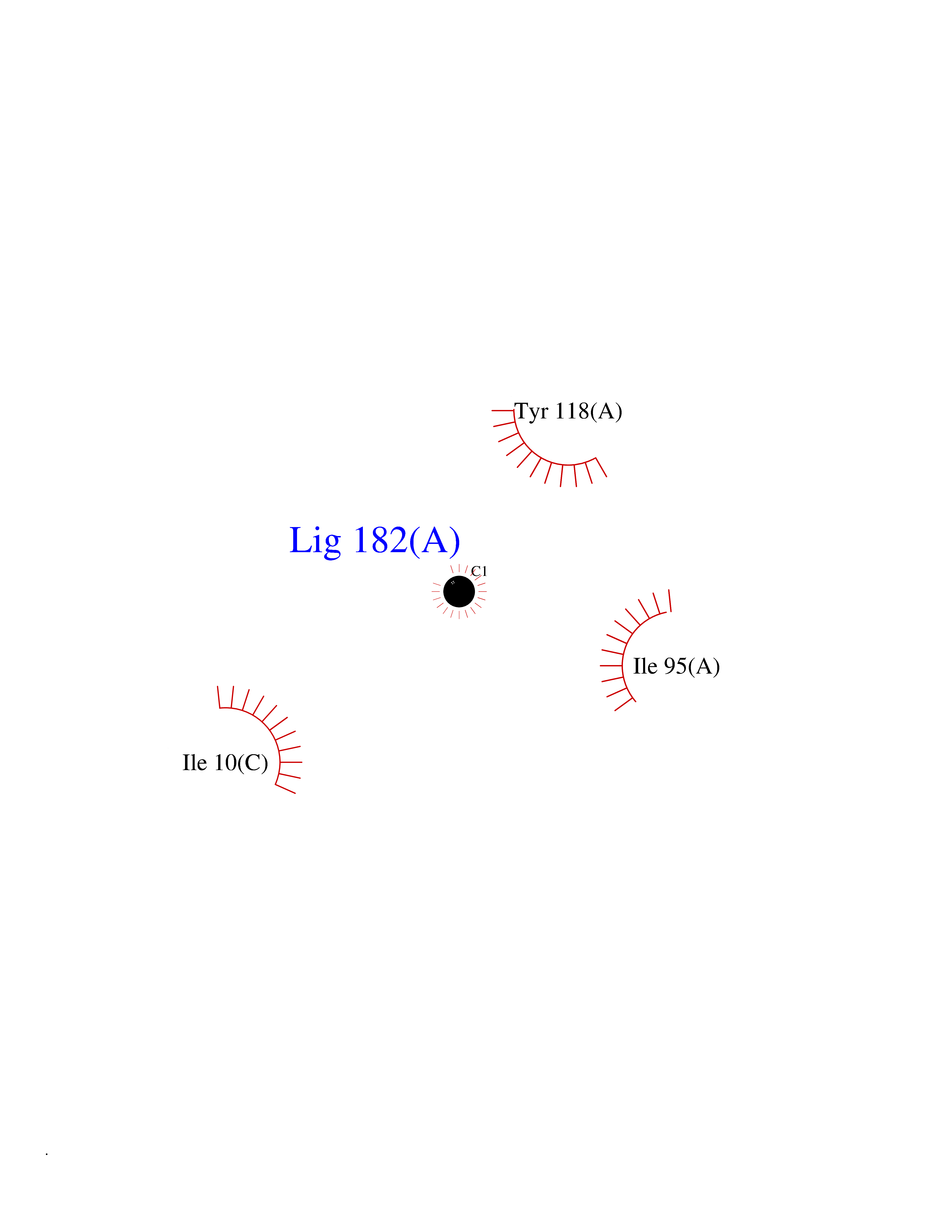 Ligplot Map 3D Binding mode Sequence SHSMRYFYTAMSRPGRGEPRFIAVGYVDDTQFVRFDSDAASPRMAPRAPWIEQEGPEYWDGETRNMKASAQTYRENLRIALRYYNQSEAGSHIIQVMYGCDVGPDGRLLRGHDQSAYDGKDYIALNEDLSSWTAADTAAQITQRKWEAARVAEQLRAYLEGLCVEWLRRYLENGKETLQLTTKLTNTNI Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Vitamin D3 receptor (VDR) | 3B0T | 3.94 | |
Target general information Gen name VDR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin D(3) receptor; Nuclear vitamin D receptor; Nuclear receptor subfamily 1 group I member 1; NR1I1; 1,25-dihydroxyvitamin D3 receptor Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Enters the nucleus upon vitamin D3 binding where it forms heterodimers with the retinoid X receptor/RXR. The VDR-RXR heterodimers bind to specific response elements on DNA and activate the transcription of vitamin D3-responsive target genes. Plays a central role in calcium homeostasis. Nuclear receptor for calcitriol, the active form of vitamin D3 which mediates the action of this vitamin on cells. Related diseases Rickets vitamin D-dependent 2A (VDDR2A) [MIM:277440]: A disorder of vitamin D metabolism resulting in severe rickets, hypocalcemia and secondary hyperparathyroidism. Most patients have total alopecia in addition to rickets. {ECO:0000269|PubMed:1652893, ECO:0000269|PubMed:17970811, ECO:0000269|PubMed:2177843, ECO:0000269|PubMed:2849209, ECO:0000269|PubMed:28698609, ECO:0000269|PubMed:7828346, ECO:0000269|PubMed:8106618, ECO:0000269|PubMed:8381803, ECO:0000269|PubMed:8392085, ECO:0000269|PubMed:8675579, ECO:0000269|PubMed:8961271, ECO:0000269|PubMed:9005998}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07530; DB08742; DB01436; DB04891; DB00146; DB02300; DB00136; DB00169; DB04540; DB05024; DB11672; DB14635; DB01070; DB06410; DB05295; DB06194; DB00153; DB04796; DB03451; DB00910; DB04258; DB11094 Interacts with P35222; Q09472; Q15648; P50222; Q15788; P26045; P19793; Q13573; Q13501; P04637; Q15645; Q9JLI4; P28700; X5D778; Q96HA8; Q01804; Q96S38; P48443 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 28781 Length 254 Aromaticity 0.07 Instability index 47.69 Isoelectric point 6.15 Charge (pH=7) -3.44 2D Binding mode Binding energy (Kcal/mol) -5.37 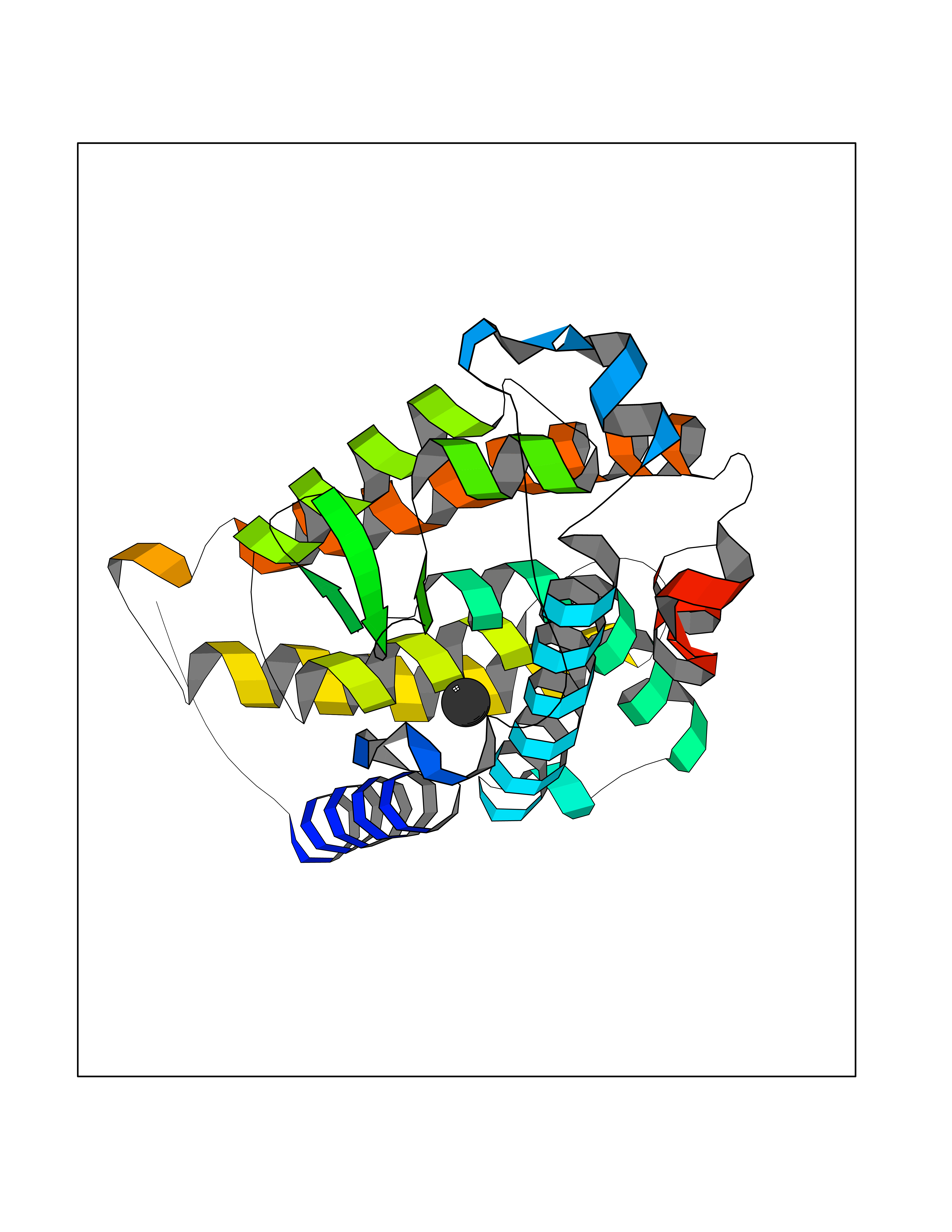 Molscript Map 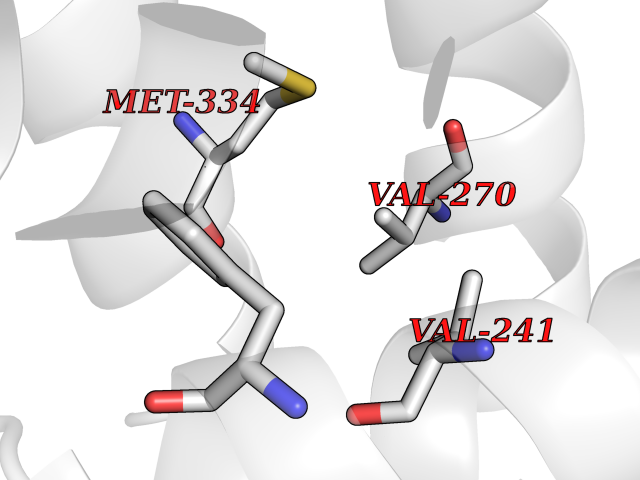 Pymol Map 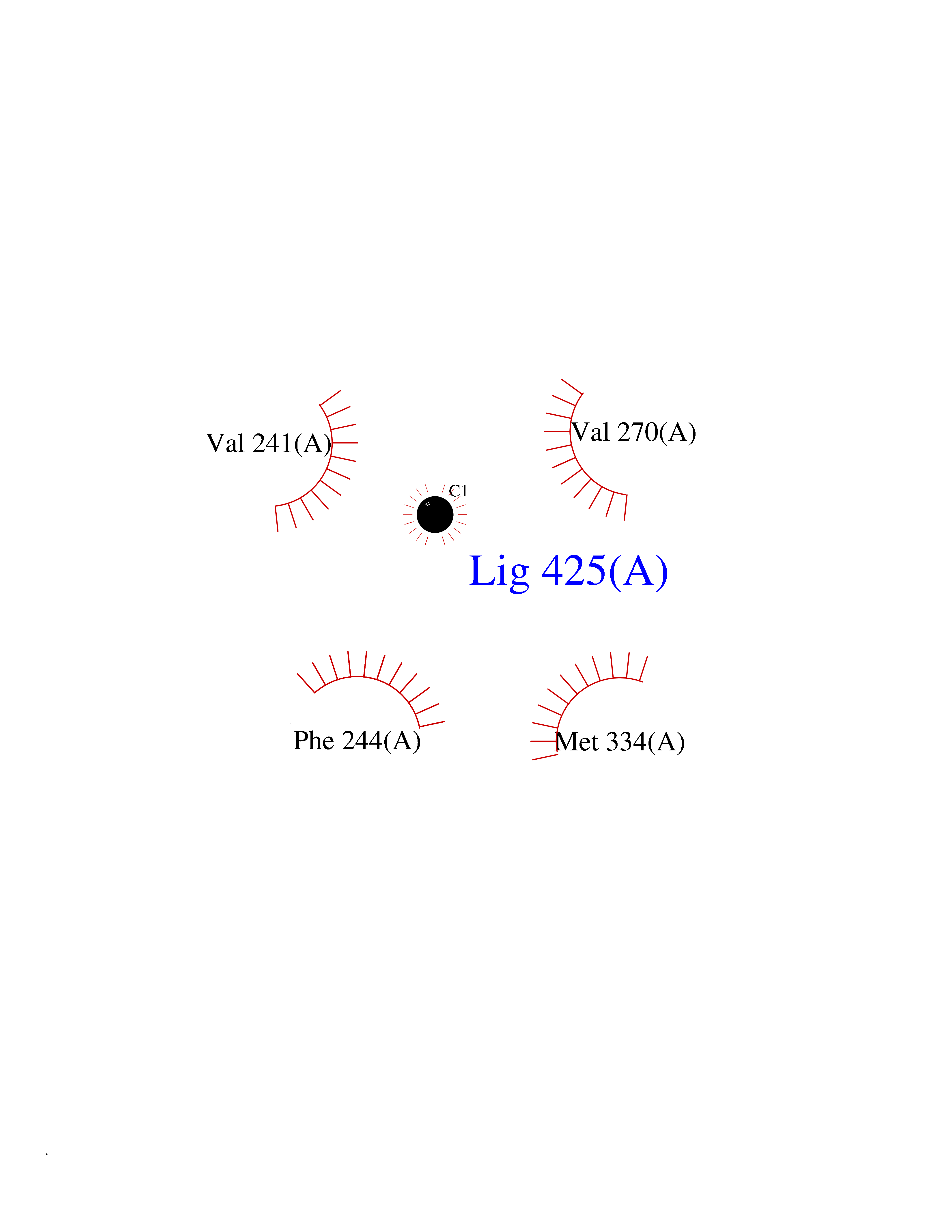 Ligplot Map 3D Binding mode Sequence ALRPKLSEEQQRIIAILLDAHHKTYDPTYSDFCQFRPPVRVNDGGGSVTLELSQLSMLPHLADLVSYSIQKVIGFAKMIPGFRDLTSEDQIVLLKSSAIEVIMLRSNESFTMDDMSWTCGNQDYKYRVSDVTKAGHSLELIEPLIKFQVGLKKLNLHEEEHVLLMAICIVSPDRPGVQDAALIEAIQDRLSNTLQTYIRCRHPPPGSHLLYAKMIQKLADLRSLNEEHSKQYRCLSFQPECSMKLTPLVLEVFG Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Tyrosine aminotransferase | 3DYD | 3.94 | |
Target general information Gen name TAT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class-I pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transferase Function Amino acid binding.L-phenylalanine:2-oxoglutarate aminotransferase activity.L-tyrosine:2-oxoglutarate aminotransferase activity.Pyridoxal phosphate binding. Related diseases Tyrosinemia 2 (TYRSN2) [MIM:276600]: An inborn error of metabolism characterized by elevations of tyrosine in the blood and urine, and oculocutaneous manifestations. Typical features include palmoplantar keratosis, painful corneal ulcers, and intellectual disability. {ECO:0000269|PubMed:1357662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00142; DB00120; DB00114; DB00135 Interacts with P15104; P28799; P28799-2; P17735; Q05086; Q05086-3 EC number 2.6.1.5 Uniprot keywords 3D-structure; Acetylation; Aminotransferase; Disease variant; Intellectual disability; Palmoplantar keratoderma; Phenylalanine catabolism; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase; Tyrosine catabolism Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42209.5 Length 380 Aromaticity 0.08 Instability index 51.79 Isoelectric point 5.29 Charge (pH=7) -10.66 2D Binding mode Binding energy (Kcal/mol) -5.37 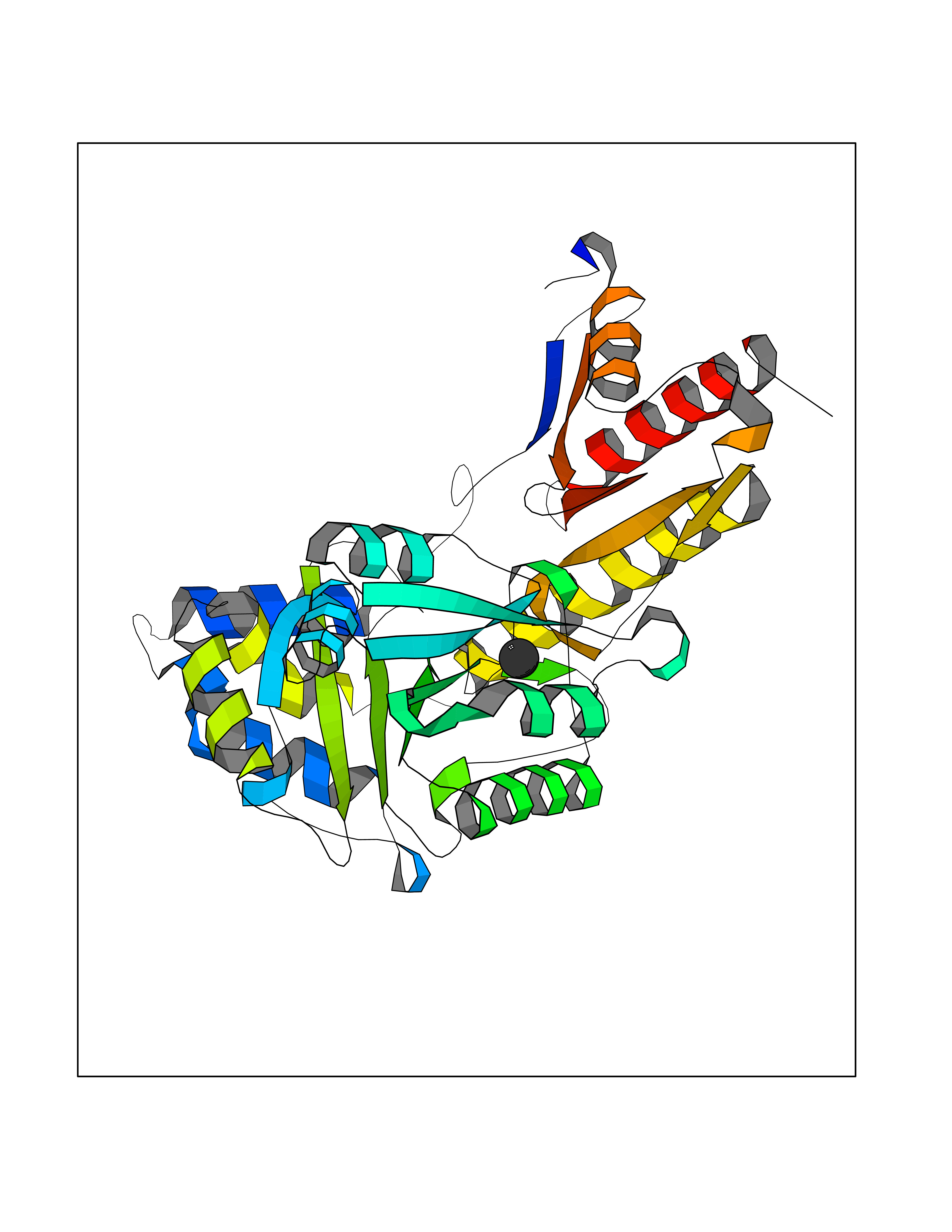 Molscript Map 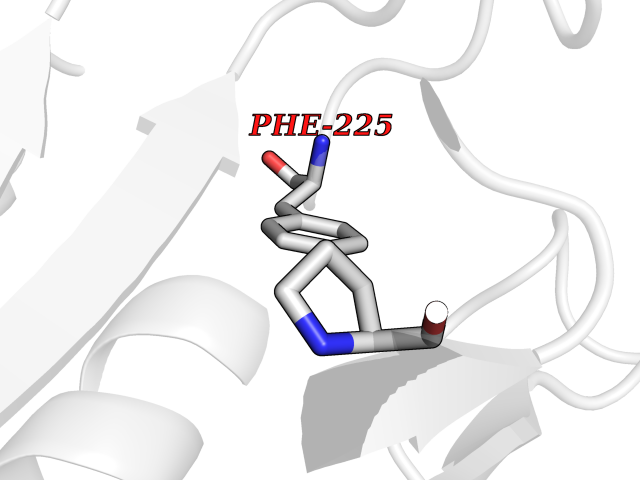 Pymol Map 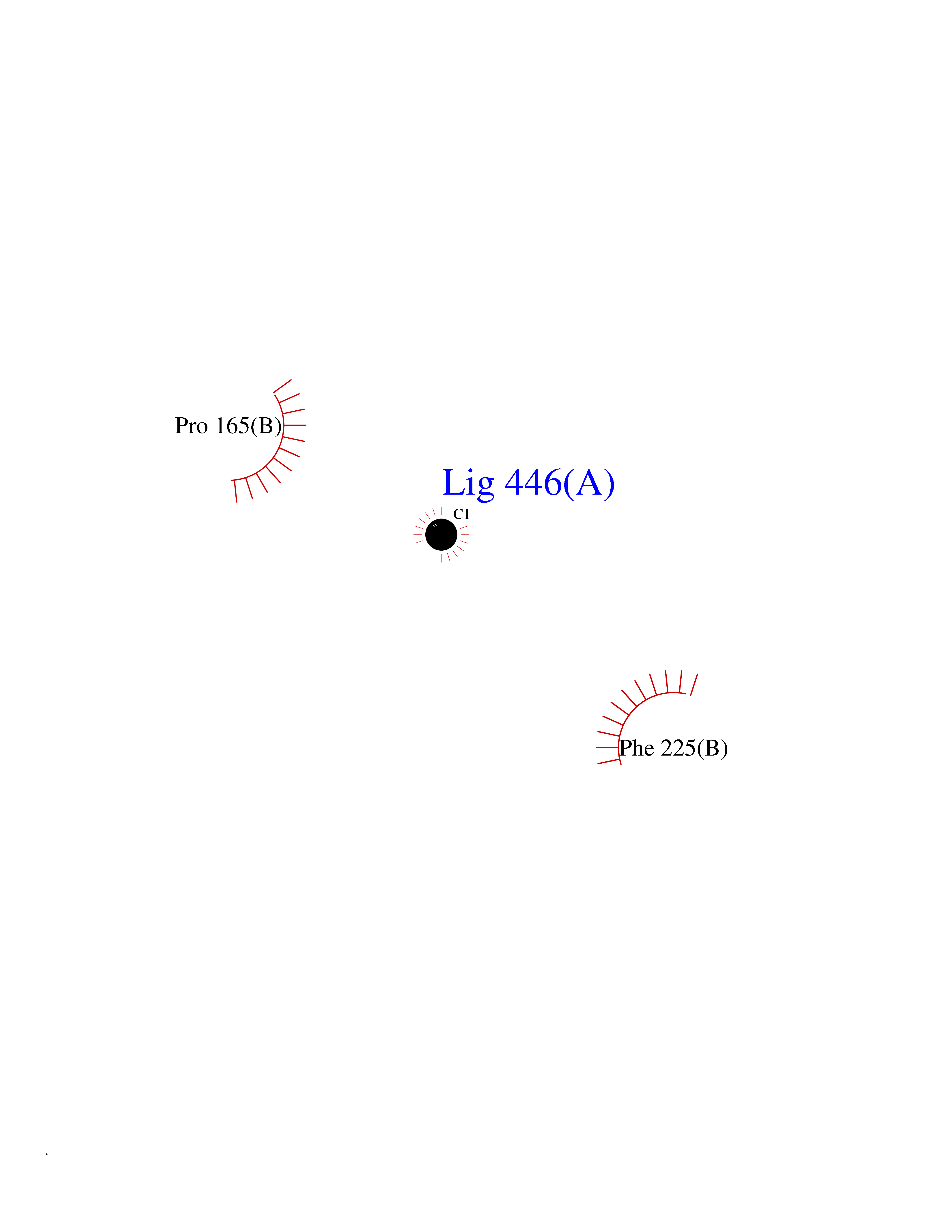 Ligplot Map 3D Binding mode Sequence VKPNPNKTMISLSIGDPTVFGNLPTDPEVTQAMKDALDSGKYNGYAPSIGFLSSREEIASYYHCPEAPLEAKDVILTSGCSQAIDLCLAVLANPGQNILVPRPGFSLYKTLAESMGIEVKLYNLLPEKSWEIDLKQLEYLIDEKTACLIVNNPSNPCGSVFSKRHLQKILAVAARQCVPILADEIYGDMVFSDCKYEPLATLSTDVPILSCGGLAKRWLVPGWRLGWILIHDRRDIFGNEIRDGLVKLSQRILGPCTIVQGALKSILCRTPGEFYHNTLSFLKSNADLCYGALAAIPGLRPVRPSGAMYLMVGIEMEHFPEFENDVEFTERLVAEQSVHCLPATCFEYPNFIRVVITVPEVMMLEACSRIQEFCEQHYHC Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Acetylcholinesterase (AChE) | 4M0E | 3.94 | |
Target general information Gen name ACHE Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms YT; N-ACHE; ARACHE Protein family Type-B carboxylesterase/lipase family Biochemical class Carboxylic ester hydrolase Function Role in neuronal apoptosis. Terminates signal transduction at the neuromuscular junction by rapid hydrolysis of the acetylcholine released into the synaptic cleft. Related diseases Phosphoribosylaminoimidazole carboxylase deficiency (PAICSD) [MIM:619859]: An autosomal recessive inborn error of purine metabolism, clinically characterized by multiple congenital anomalies and early neonatal death. {ECO:0000269|PubMed:31600779}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07846; DB02673; DB04617; DB04614; DB04615; DB07756; DB07701; DB02404; DB03814; DB08615; DB02343; DB02226; DB03005; DB04114; DB03128; DB01122; DB03283; DB00411; DB00122; DB14006; DB01245; DB00944; DB08357; DB08996; DB00449; DB00843; DB01010; DB01364; DB00898; DB00674; DB00483; DB06525; DB04864; DB03348; DB07555; DB00677; DB04924; DB03359; DB00358; DB00940; DB02825; DB02845; DB08167; DB04021; DB00805; DB01805; DB03740; DB04556; DB01400; DB04892; DB00981; DB00733; DB02166; DB00545; DB00863; DB00989; DB00382; DB04616; DB12816; DB01199; DB13503; DB04859 Interacts with Q9Y215; P06733; P63244 EC number EC 3.1.1.7 Uniprot keywords 3D-structure; Alternative splicing; Blood group antigen; Cell membrane; Direct protein sequencing; Disulfide bond; Glycoprotein; GPI-anchor; Hydrolase; Lipoprotein; Membrane; Neurotransmitter degradation; Nucleus; Proteomics identification; Reference proteome; Secreted; Serine esterase; Signal; Synapse Protein physicochemical properties Chain ID A,B Molecular weight (Da) 58804.1 Length 537 Aromaticity 0.11 Instability index 40.85 Isoelectric point 5.73 Charge (pH=7) -8.18 2D Binding mode Binding energy (Kcal/mol) -5.37 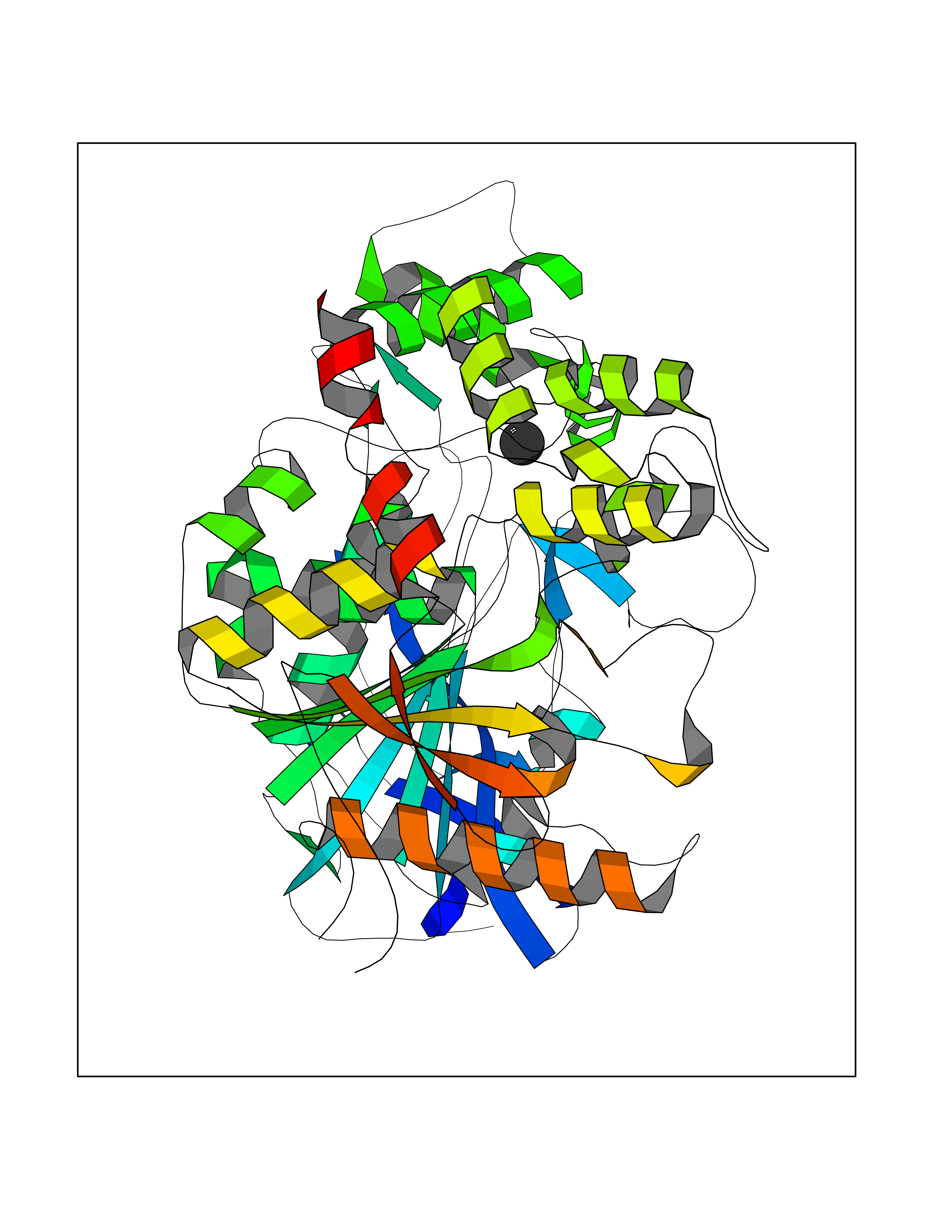 Molscript Map  Pymol Map 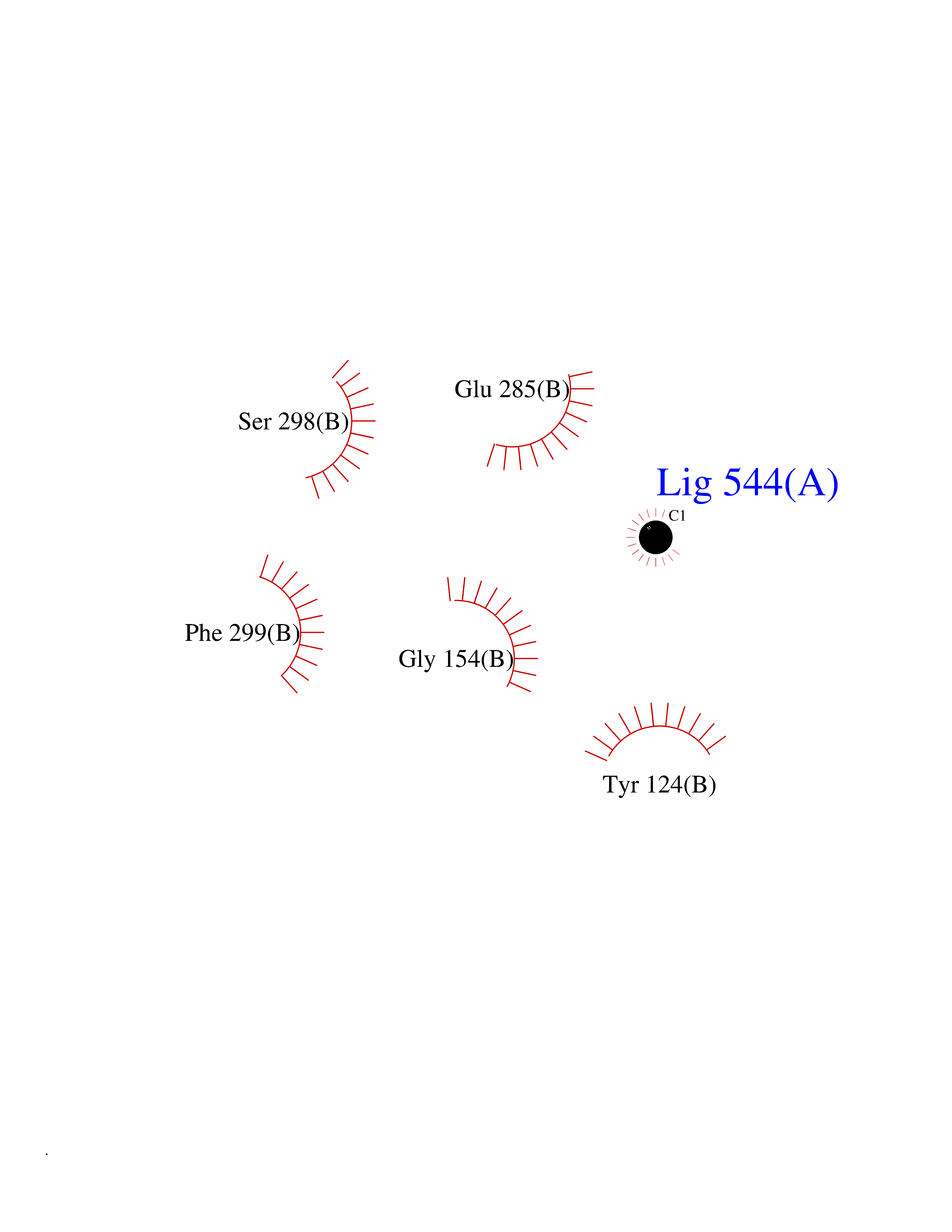 Ligplot Map 3D Binding mode Sequence EDAELLVTVRGGRLRGIRLKTPGGPVSAFLGIPFAEPPMGPRRFLPPEPKQPWSGVVDATTFQSVCYQYVDTLYPGFEGTEMWNPNRELSEDCLYLNVWTPYPRPTSPTPVLVWIYGGGFYSGASSLDVYDGRFLVQAERTVLVSMNYRVGAFGFLALPGSREAPGNVGLLDQRLALQWVQENVAAFGGDPTSVTLFGESAGAASVGMHLLSPPSRGLFHRAVLQSGAPNGPWATVGMGEARRRATQLAHLVGCPPGGTGGNDTELVACLRTRPAQVLVNHEWHVLPQESVFRFSFVPVVDGDFLSDTPEALINAGDFHGLQVLVGVVKDEGSYFLVYGAPGFSKDNESLISRAEFLAGVRVGVPQVSDLAAEAVVLHYTDWLHPEDPARLREALSDVVGDHNVVCPVAQLAGRLAAQGARVYAYVFEHRASTLSWPLWMGVPHGYEIEFIFGIPLDPSRNYTAEEKIFAQRLMRYWANFARTGDPNEPPKAPQWPPYTAGAQQYVSLDLRPLEVRRGLRAQACAFWNRFLPKLLSA Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Serum paraoxonase/arylesterase 1 | 1V04 | 3.94 | |
Target general information Gen name PON1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PON Protein family Paraoxonase family Biochemical class Hydrolase Function Acyl-L-homoserine-lactone lactonohydrolase activity.Aryldialkylphosphatase activity.Arylesterase activity.Calcium ion binding.Phospholipid binding.Protein homodimerization activity. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB01327; DB09130; DB01395; DB14598; DB14600; DB14596; DB00218; DB01085; DB01593; DB14487; DB14533; DB14548 Interacts with NA EC number 3.1.1.2; 3.1.1.81; 3.1.8.1 Uniprot keywords 3D-structure; Calcium; Direct protein sequencing; Disulfide bond; Glycoprotein; HDL; Hydrolase; Metal-binding; Proteomics identification; Reference proteome; Secreted; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 37232.8 Length 332 Aromaticity 0.11 Instability index 35.09 Isoelectric point 5.06 Charge (pH=7) -17.08 2D Binding mode Binding energy (Kcal/mol) -5.37 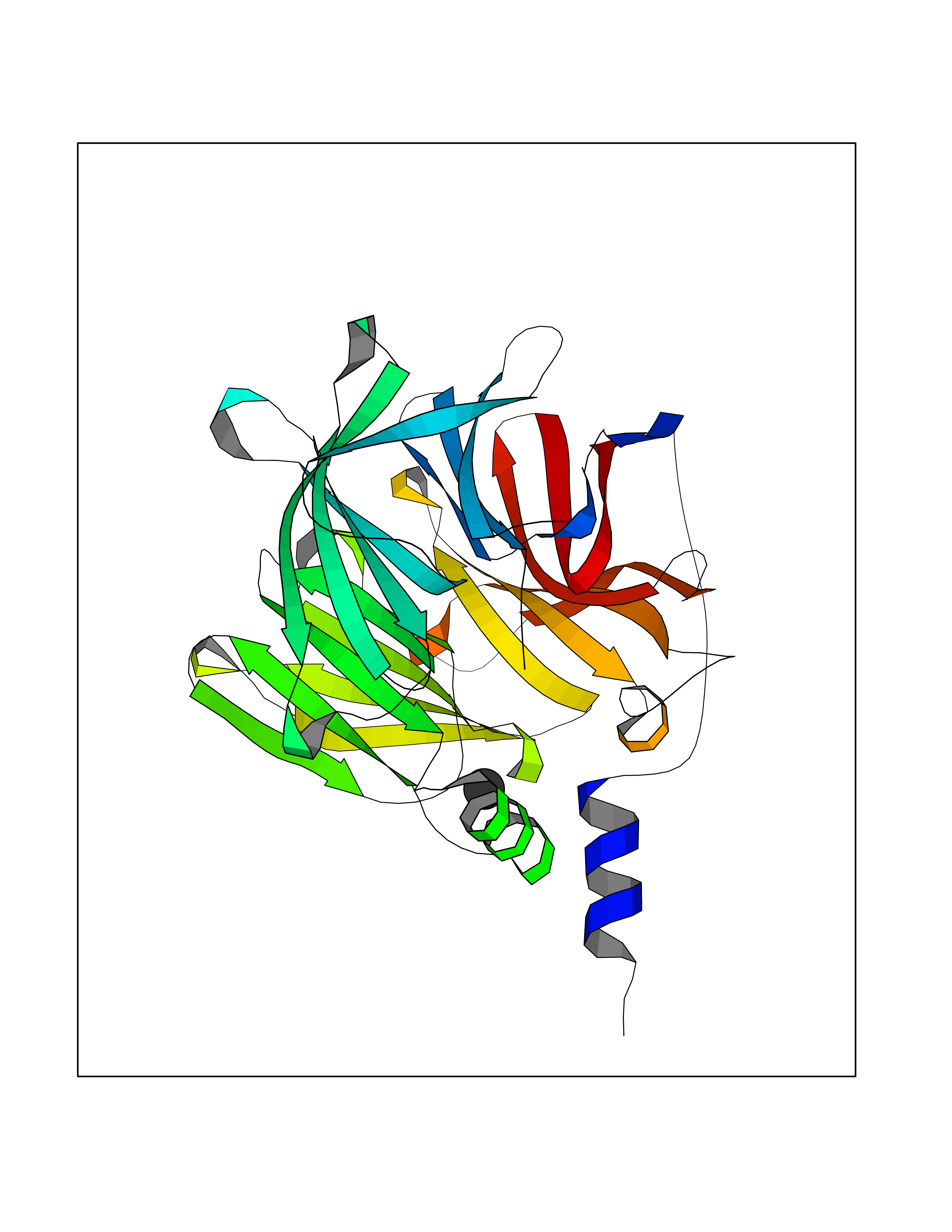 Molscript Map 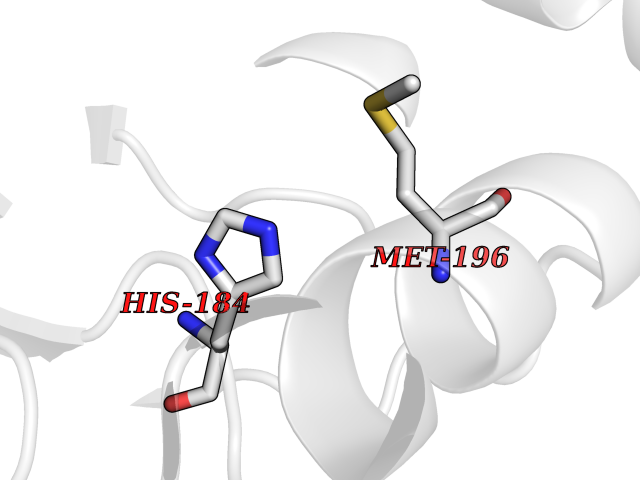 Pymol Map 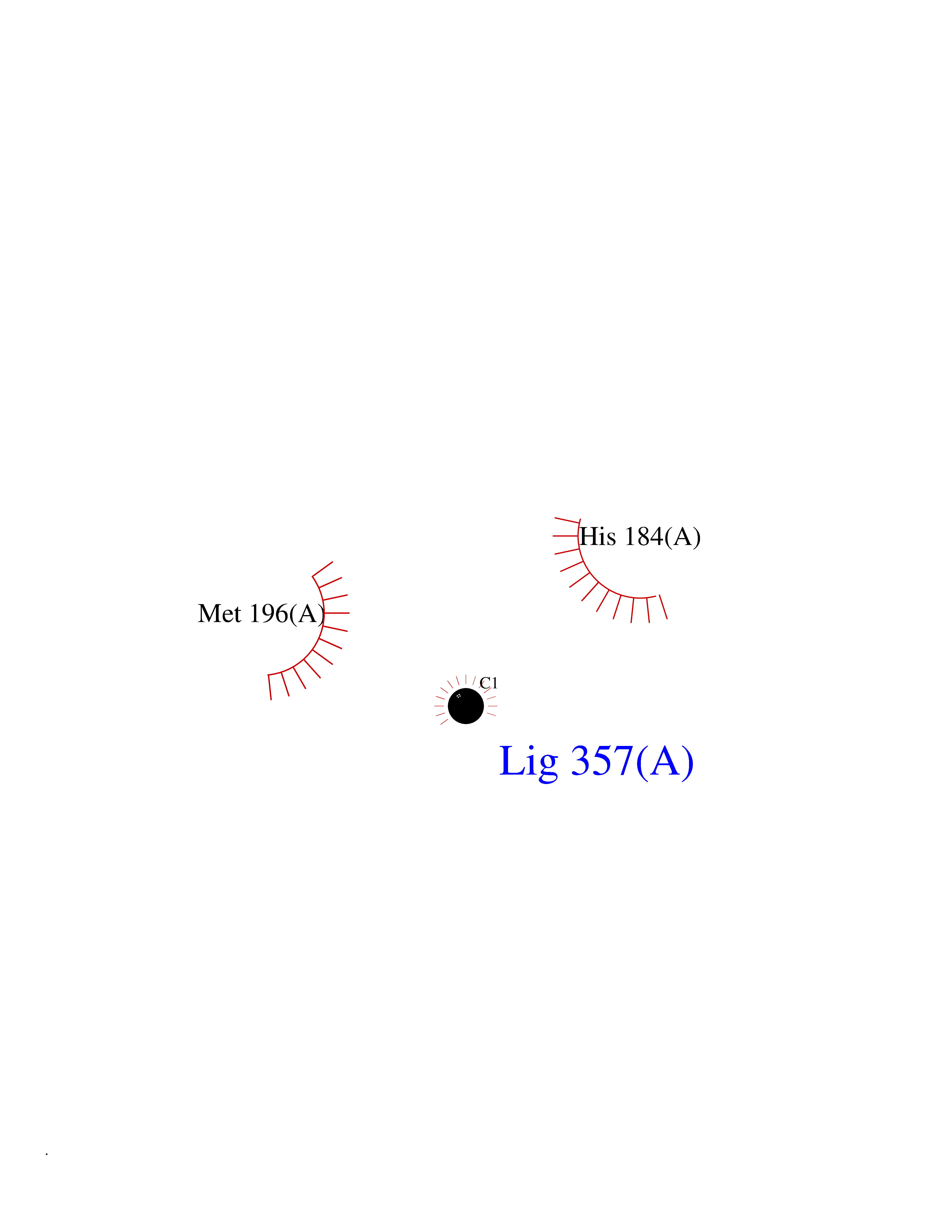 Ligplot Map 3D Binding mode Sequence LFDRQKSSFQTRFNVHREVTPVELPNCNLVKGIDNGSEDLEILPNGLAFISSGLKYDKSGKILLMDLNEKEPAVSELEIIGNTLDISSFNPHGISTFIDDDNTVYLLVVNHPGSSSTVEVFKFQEEEKSLLHLKTIRHKLLPSVNDIVAVGPEHFYATNDHYFIDPYLKSWEMHLGLAWSFVTYYSPNDVRVVAEGFDFANGINISPDGKYVYIAELLAHKIHVYEKHANWTLTPLRVLSFDTLVDNISVDPVTGDLWVGCHPNGMRIFFYDAENPPGSEVLRIQDILSEEPKVTVVYAENGTVLQGSTVAAVYKGKLLIGTVFHKALYCDL Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Camphor 5-monooxygenase | 4L4E | 3.93 | |
Target general information Gen name camC Organism Pseudomonas putida (Arthrobacter siderocapsulatus) Uniprot ID TTD ID NA Synonyms cyp101 Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Camphor 5-monooxygenase activity.Heme binding.Iron ion binding. Related diseases Combined oxidative phosphorylation deficiency 6 (COXPD6) [MIM:300816]: A mitochondrial disease resulting in a neurodegenerative disorder characterized by psychomotor delay, hypotonia, areflexia, muscle weakness and wasting. Some patients manifest prenatal ventriculomegaly and severe postnatal encephalomyopathy. {ECO:0000269|PubMed:20362274, ECO:0000269|PubMed:22019070, ECO:0000269|PubMed:25583628, ECO:0000269|PubMed:26004228, ECO:0000269|PubMed:26173962, ECO:0000269|PubMed:27178839}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, X-linked recessive, 4, with or without cerebellar ataxia (CMTX4) [MIM:310490]: A neuromuscular disorder characterized by progressive sensorimotor axonal neuropathy, distal sensory impairment, difficulty walking due to peripheral neuropathy and/or cerebellar ataxia, and deafness due to auditory neuropathy. Additional features include cognitive impairment, cerebellar atrophy, dysarthria, abnormal extraocular movements, tremor, dysmetria and spasticity. The age at onset ranges from infancy to young adulthood. {ECO:0000269|PubMed:23217327, ECO:0000269|PubMed:26004228}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, X-linked, 5, with peripheral neuropathy (DFNX5) [MIM:300614]: A form of hearing loss characterized by absent or severely abnormal auditory brainstem response, abnormal middle ear reflexes, abnormal speech discrimination, loss of outer hair cell function, and cochlear nerve hypoplasia. DFNX5 patients manifest auditory neuropathy with childhood onset, associated with distal sensory impairment affecting the peripheral nervous system. {ECO:0000269|PubMed:25986071}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepimetaphyseal dysplasia, X-linked, with hypomyelinating leukodystrophy (SEMDHL) [MIM:300232]: An X-linked recessive developmental disorder characterized by slowly progressive skeletal and neurologic abnormalities, including short stature, large and deformed joints, significant motor impairment, visual defects, and sometimes cognitive deficits. Affected individuals typically have normal early development in the first year or so of life, followed by development regression and the development of symptoms. Brain imaging shows white matter abnormalities consistent with hypomyelinating leukodystrophy. {ECO:0000269|PubMed:28842795}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03836; DB02617; DB02817; DB03627; DB04032; DB03031; DB02125; DB04501; DB01744; DB01663; DB01011; DB01703; DB01826; DB03540; DB02851 Interacts with P00259 EC number 1.14.15.1 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Heme; Iron; Metal-binding; Monooxygenase; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 45446.3 Length 405 Aromaticity 0.08 Instability index 45.33 Isoelectric point 5.23 Charge (pH=7) -16.08 2D Binding mode Binding energy (Kcal/mol) -5.36 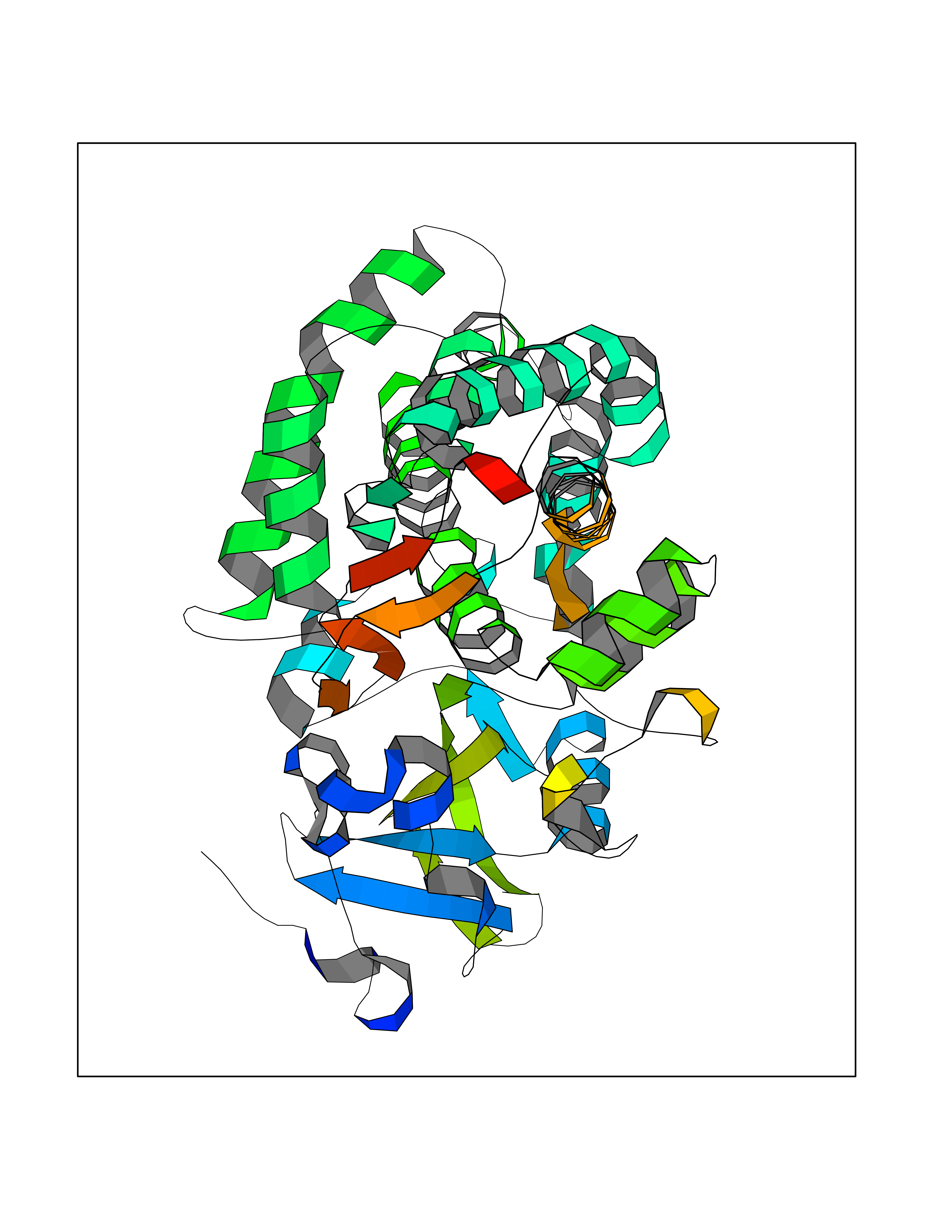 Molscript Map 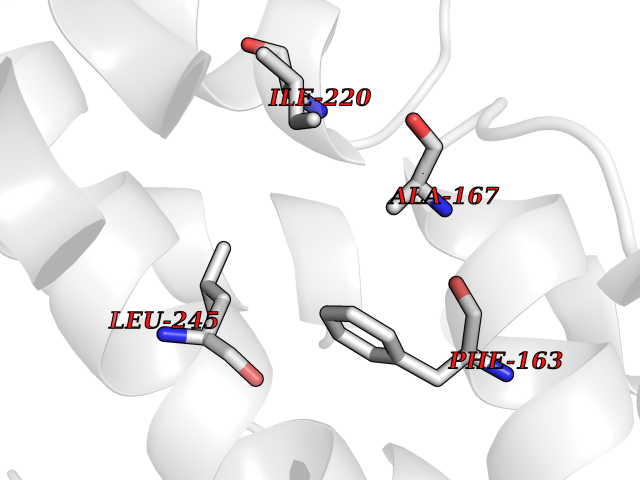 Pymol Map 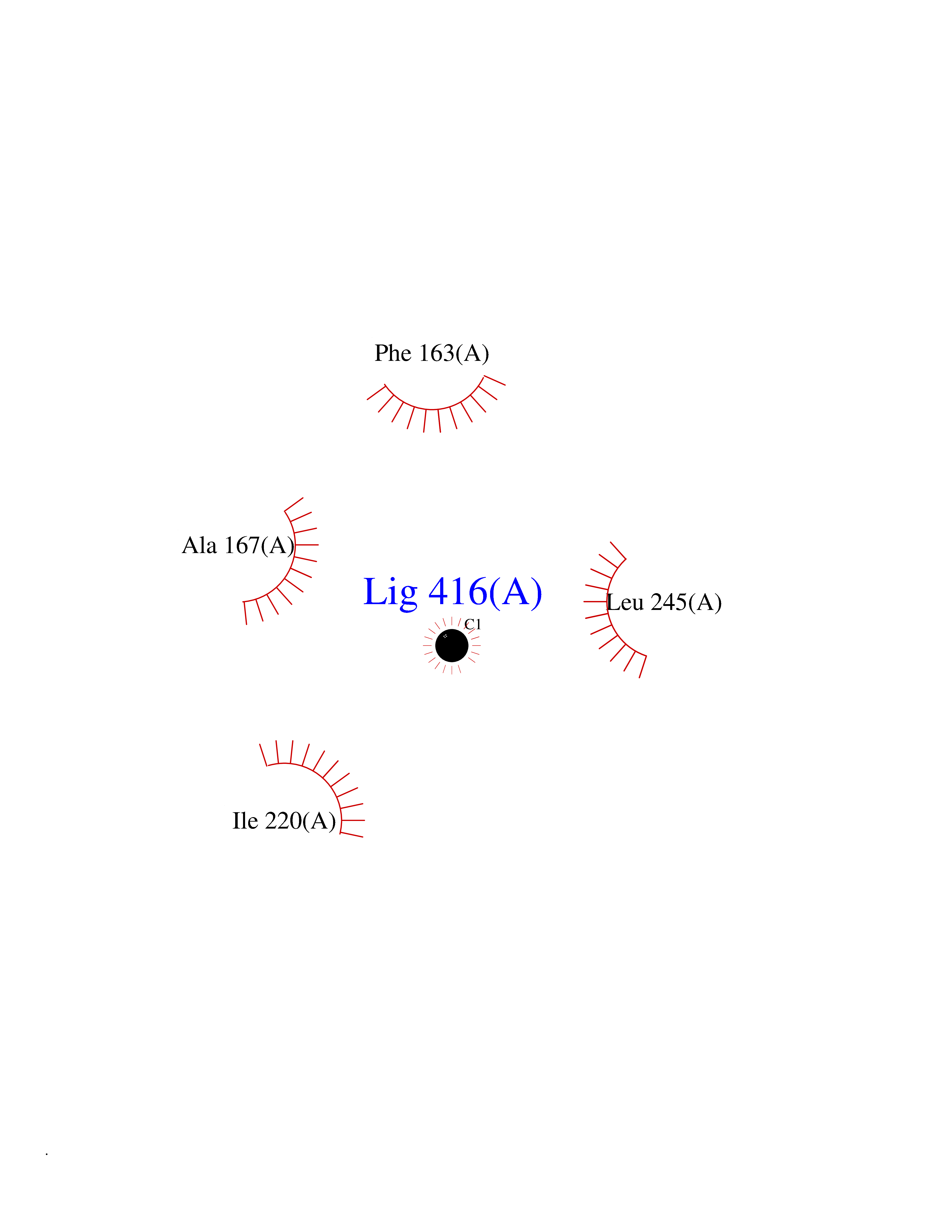 Ligplot Map 3D Binding mode Sequence NLAPLPPHVPEHLVFDFDMYNPSNLSAGVQEAWAVLQESNVPDLVWTRCNGGHWIATRGQLIREAYEDYRHFSSECPFIPREAGEAYDFIPTSMDPPEQRQFRALANQVVGMPVVDKLENRIQELACSLIESLRPQGQCNFTEDYAEPFPIRIFMLLAGLPEEDIPHLGYLTDQMTRPDGSMTFAEAKEALYDYLIPIIEQRRQKPGTDAISIVANGQVNGRPITSDEAKRMCGLLLVGGLDTVVNFLSFSMEFLAKSPEHRQELIERPERIPAACEELLRRFSLVADGRILTSDYEFHGVQLKKGDQILLPQMLSGLDERENAAPMHVDFSRQKVSHTTFGHGSHLCAGQHLARREIIVTLKEWLTRIPDFSIAPGAQIQHKSGIVSGVQALPLVWDPATTKAV Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Chromodomain-helicase-DNA-binding protein 1 | 4O42 | 3.93 | |
Target general information Gen name CHD1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family SNF2/RAD54 helicase family Biochemical class Dna binding protein / viral protein Function ATP binding.ATP-dependent DNA helicase activity.DNA binding.Methylated histone binding. Related diseases Pilarowski-Bjornsson syndrome (PILBOS) [MIM:617682]: An autosomal dominant disorder characterized by developmental delay, speech apraxia, intellectual disability, autism, and facial dysmorphic features. Some patients may have seizures. {ECO:0000269|PubMed:28866611}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O60341-1; B2BUF1; P28799; O76024 EC number 3.6.4.12 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Chromatin regulator; Cytoplasm; Disease variant; DNA-binding; Helicase; Hydrolase; Intellectual disability; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 20969.1 Length 180 Aromaticity 0.12 Instability index 46.35 Isoelectric point 5.88 Charge (pH=7) -2.83 2D Binding mode Binding energy (Kcal/mol) -5.37 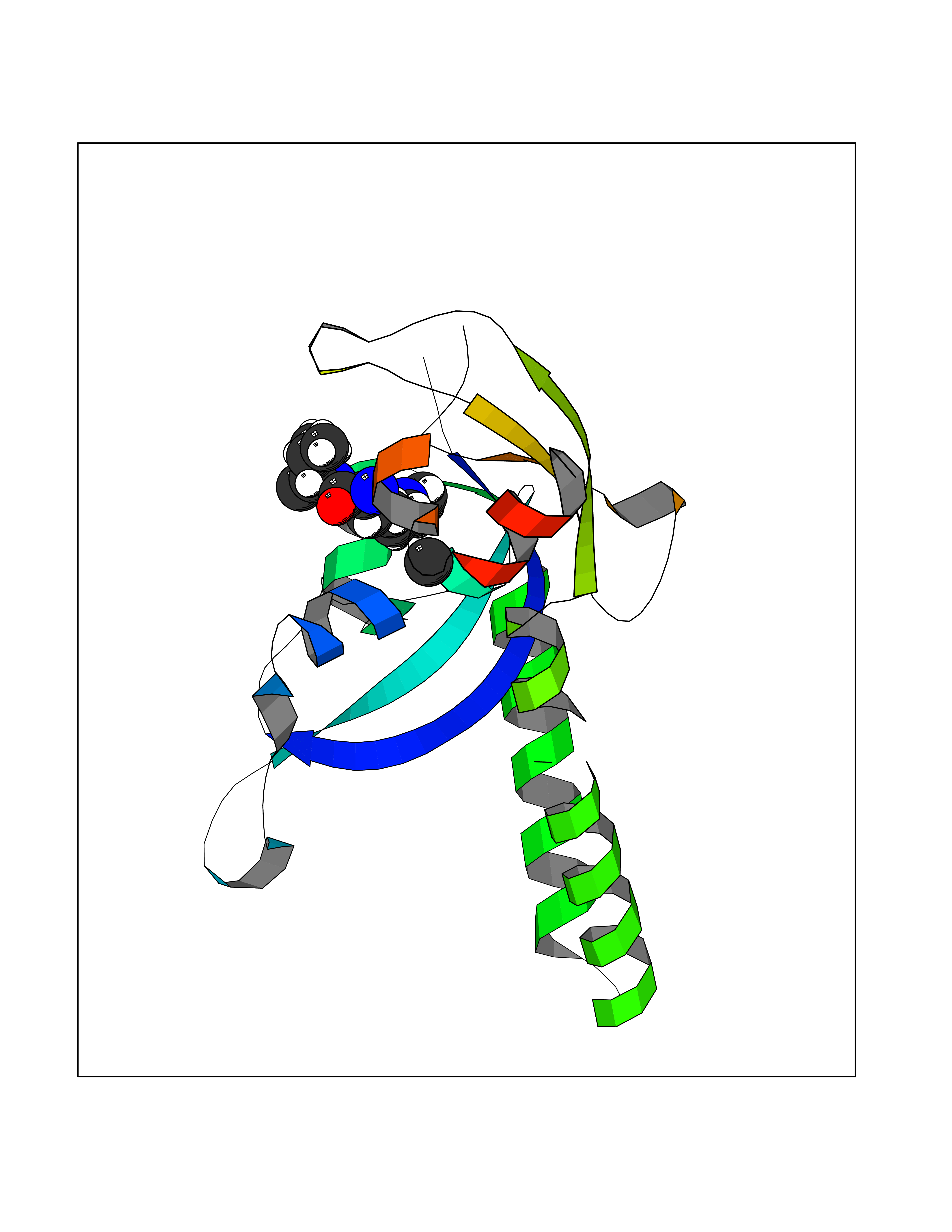 Molscript Map 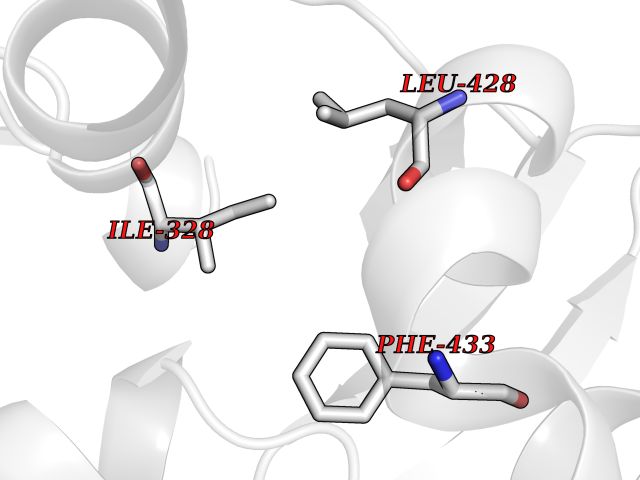 Pymol Map 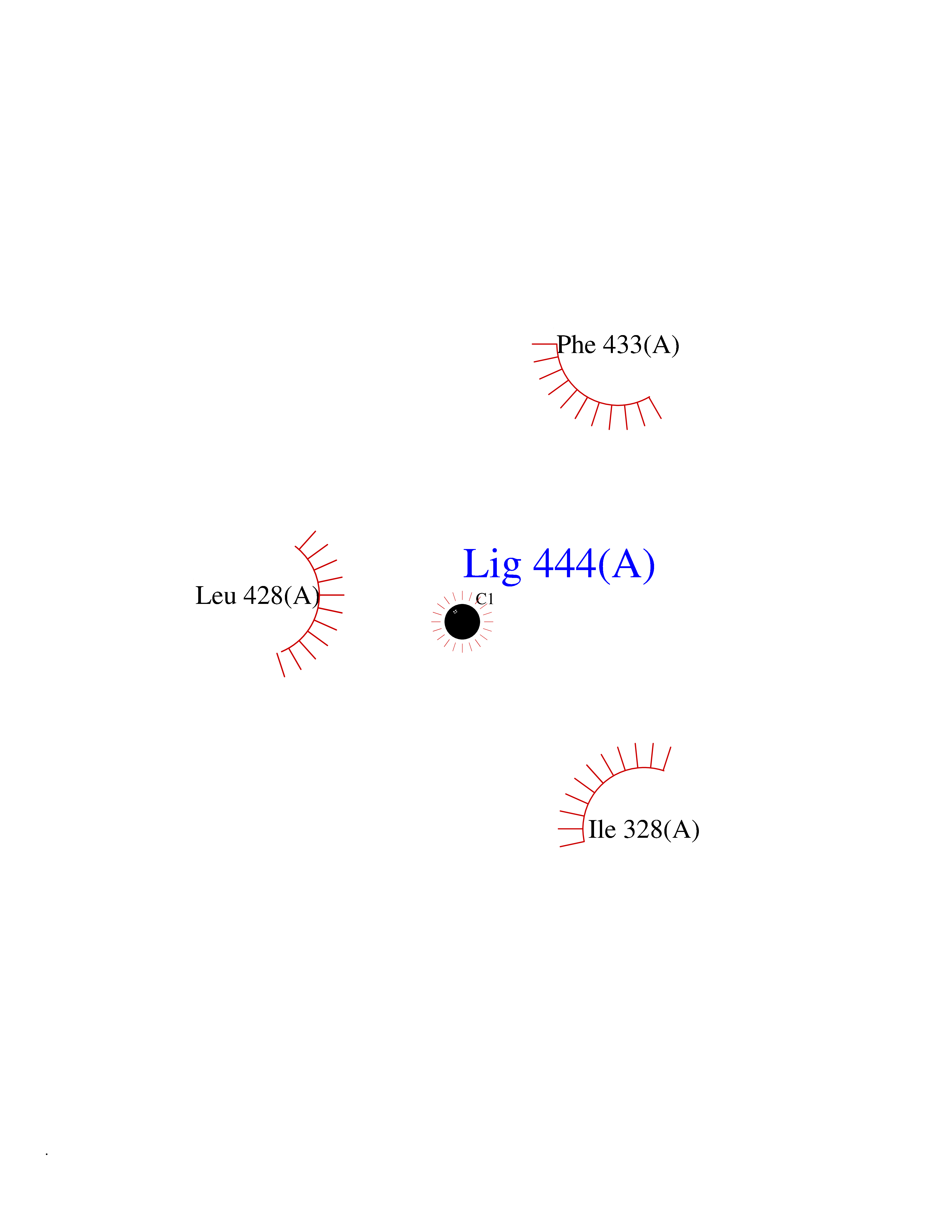 Ligplot Map 3D Binding mode Sequence EFETIERFMDCRIGRKGATGATTTIYAVEADGDPNAGFEKNKEPGEIQYLIKWKGWSHIHNTWETEETLKQQNVRGMKKLDNYKKKDQETKRWLKNASPEDVEYYNCQQELTDDLHKQYQIVERIIAHSNQKSAAGYPDYYCKWQGLPYSECSWEDGALISKKFQACIDEYFSRTARSXV Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Quinone-dependent D-lactate dehydrogenase | 1F0X | 3.93 | |
Target general information Gen name dld Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW2121;b2133 Protein family Quinone-dependent D-lactate dehydrogenase family Biochemical class Oxidoreductase Function (R)-2-hydroxyglutarate dehydrogenase activity.D-lactate dehydrogenase (cytochrome) activity.Electron carrier activity.FAD binding.Flavin adenine dinucleotide binding.NAD binding.Oxidoreductase activity, acting on the CH-OH group of donors, quinone or similar compound as acceptor.Quinone binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147; DB00756 Interacts with NA EC number 1.1.5.12 Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Direct protein sequencing; FAD; Flavoprotein; Membrane; Oxidoreductase; Quinone; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56475.2 Length 502 Aromaticity 0.09 Instability index 32.5 Isoelectric point 5.97 Charge (pH=7) -10.38 2D Binding mode Binding energy (Kcal/mol) -5.36 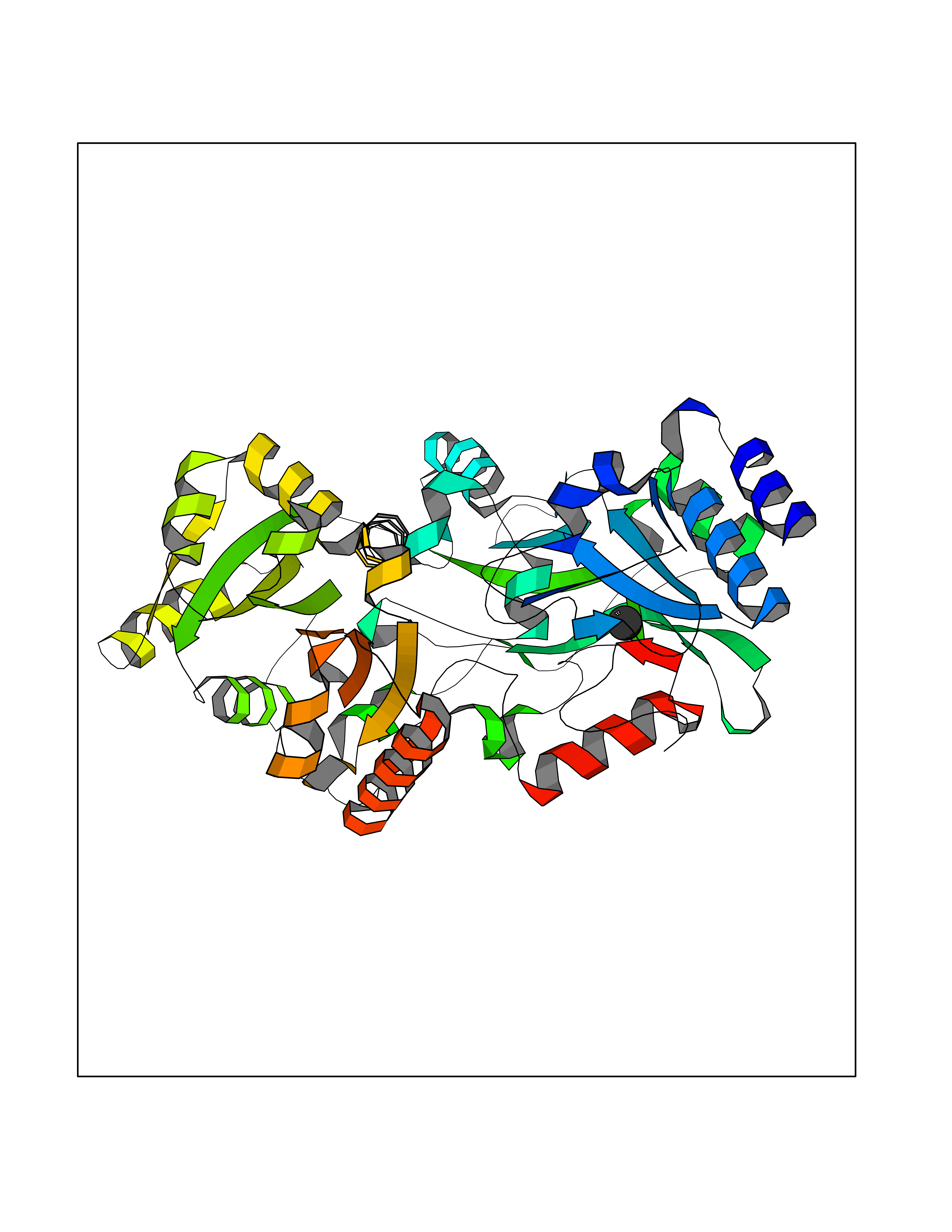 Molscript Map 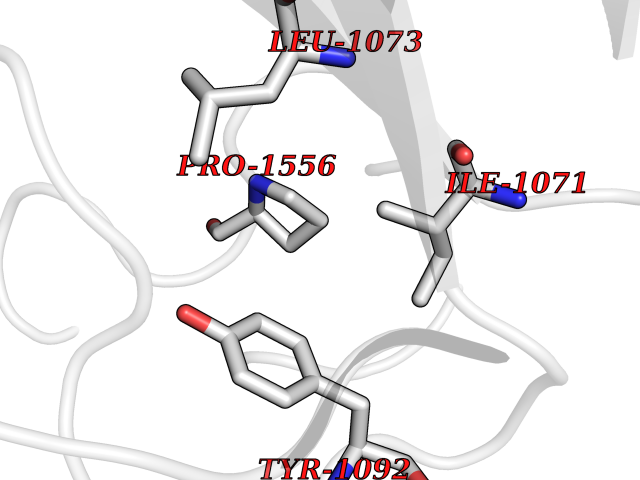 Pymol Map 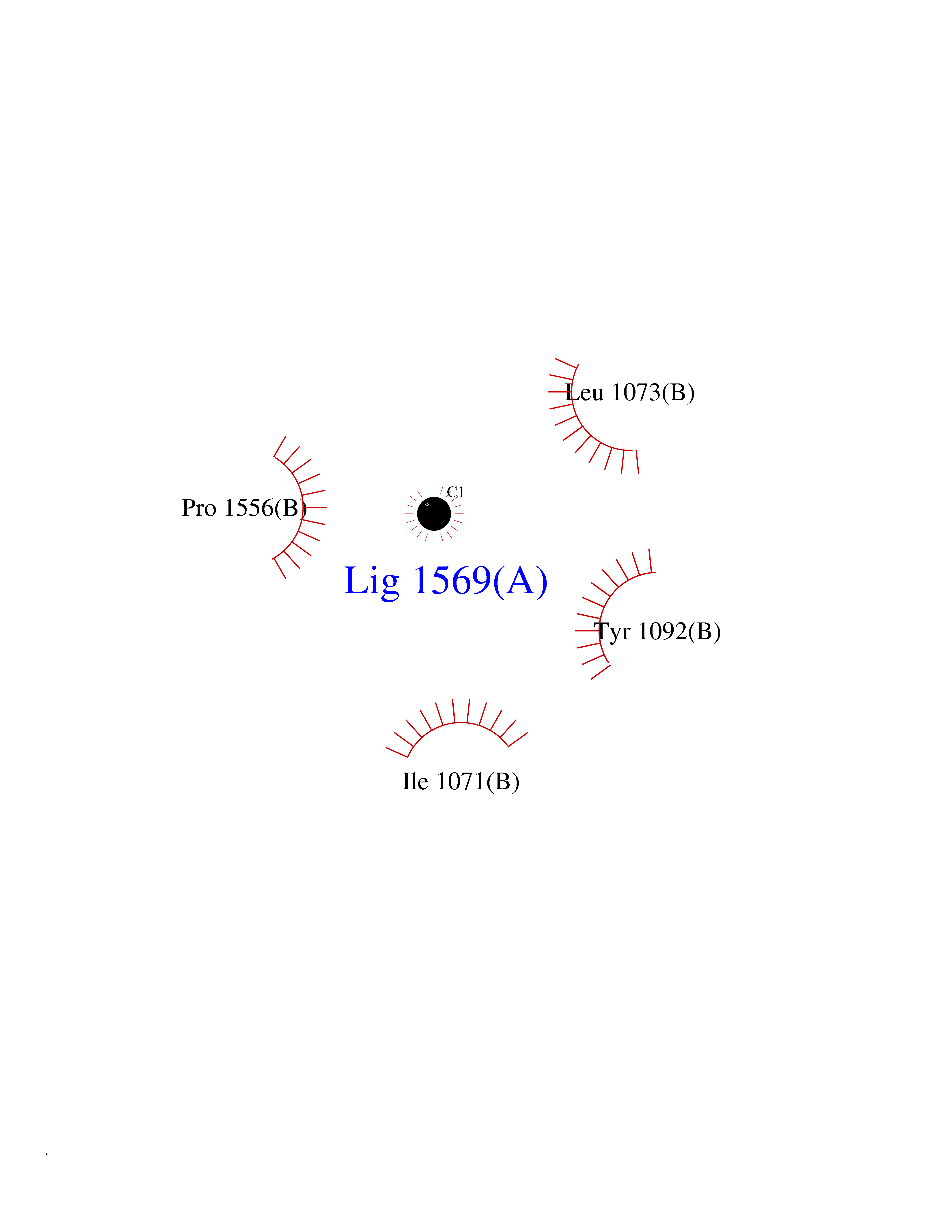 Ligplot Map 3D Binding mode Sequence NKAFLNELARLVGSSHLLTDPAKTARYRKGFRSGQGDALAVVFPGSLLELWRVLKACVTADKIILMQAANTGLTEGSTPNGNDYDRDVVIISTLRLDKLHVLGKGEQVLAYPGTTLYSLEKALKPLGREPHSVIGSSCIGASVIGGICNNSGGSLVQRGPAYTEMSLFARINEDGKLTLVNHLGIDLGETPEQILSKLDDDRIKDDDVRHDGRHAHDYDYVHRVRDIEADTPARYNADPDRLFESSGCAGKLAVFAVRLDTFEAEKNQQVFYIGTNQPEVLTEIRRHILANFENLPVAGEYMHRDIYDIAELPPRMKNWRDKYEHHLLLKMAGDGVGEAKSWLVDYFKQAEGDFFVCTPEEGSKAFLHRFAAAGAAIRYQAVHSDEVEDILALDIALRRNDTEWYEHLPPEIDSQLVHKLYYGHFMCYVFHQDYIVKKGVDVHALKEQMLELLQQRGAQYPAEHNVGHLYKAPETLQKFYRENDPTNSMNPGIGKTSKRKNW Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Glutathione S-transferase P (GSTP1) | 5J41 | 3.93 | |
Target general information Gen name GSTP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GSTP11; GSTP1-1; GST3; GST classpi; GST class-pi; FAEES3 Protein family GST superfamily, Pi family Biochemical class Alkyl aryl transferase Function Regulates negatively CDK5 activity via p25/p35 translocation to prevent neurodegeneration. Conjugation of reduced glutathione to a wide number of exogenous and endogenous hydrophobic electrophiles. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01834; DB03814; DB00316; DB14001; DB00321; DB01008; DB04972; DB00958; DB00291; DB02633; DB00515; DB01242; DB00363; DB11672; DB14635; DB14002; DB03619; DB11831; DB00903; DB00773; DB06246; DB05460; DB00143; DB03310; DB03003; DB13014; DB00526; DB14924; DB08370; DB03686; DB04132; DB01915; DB07849; DB00197; DB00163 Interacts with Q6UY14-3; Q92624; Q9BWT7; A8MQ03; Q9NRD0; Q5TD97; P49639; Q15323; Q14525; O76011; P78385; Q07627; Q8IUG1; P60409; P60411; Q9BYR8; Q9BYR6; Q3LI66; Q9P2M1; Q5JR59-3; Q7Z3S9; P0DPK4; P22735; Q12933; Q8N720 EC number EC 2.5.1.18 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Lipid metabolism; Mitochondrion; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 46333.6 Length 417 Aromaticity 0.1 Instability index 30.67 Isoelectric point 5.44 Charge (pH=7) -5.73 2D Binding mode Binding energy (Kcal/mol) -5.36  Molscript Map 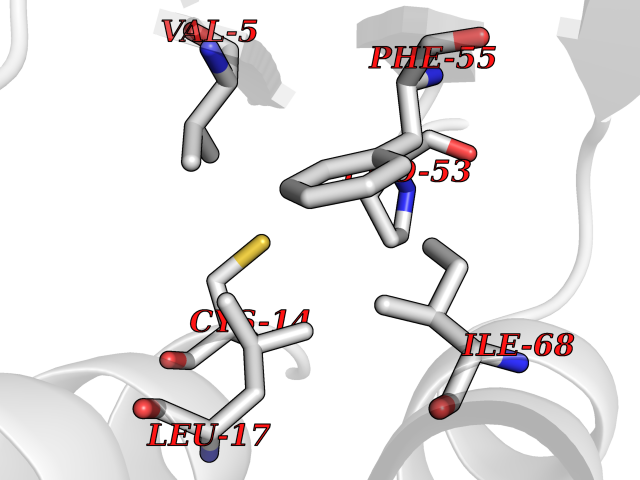 Pymol Map 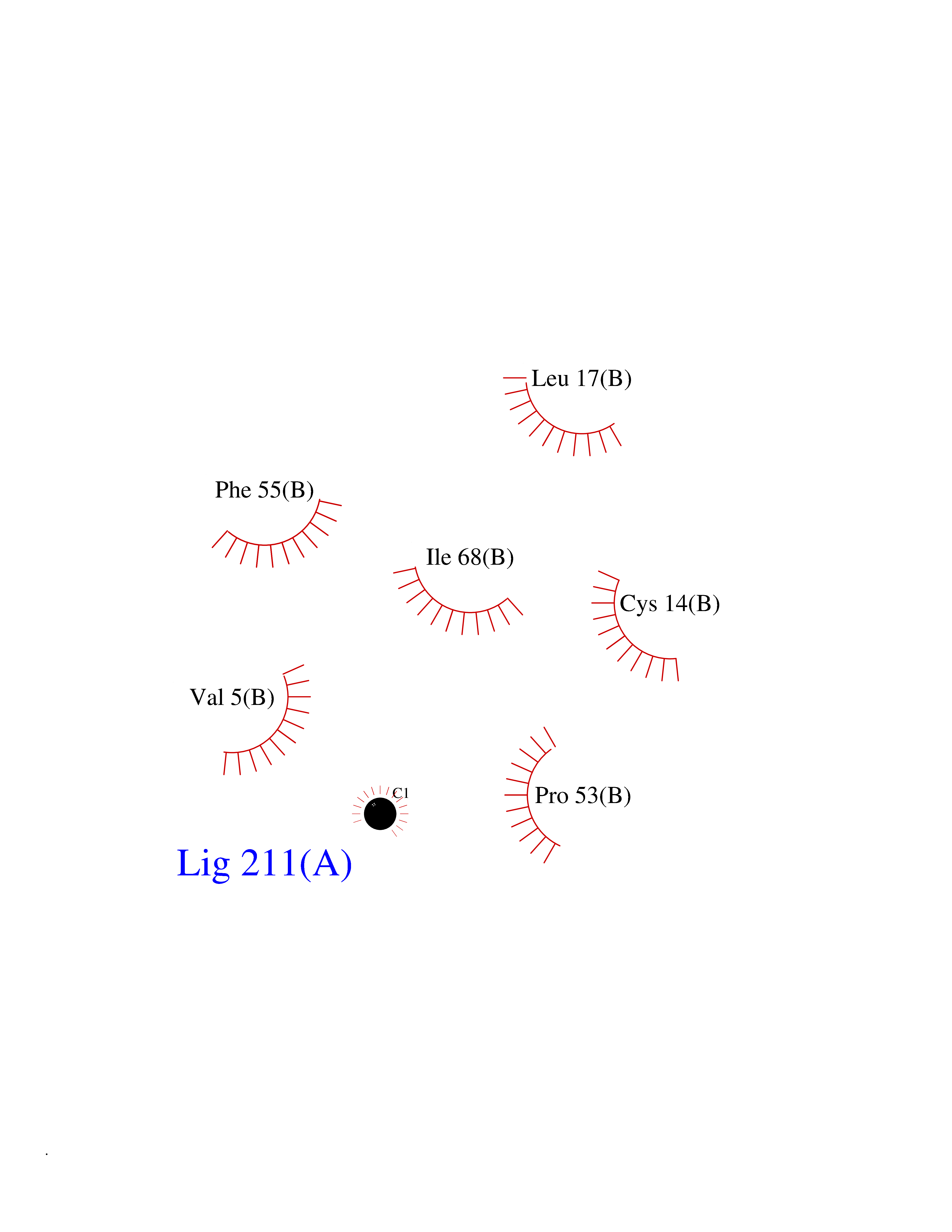 Ligplot Map 3D Binding mode Sequence PPYTVVYFPVRGRCAALRMLLADQGQSWKEEVVTVETWQEGSLKASCLYGQLPKFQDGDLTLYQSNTILRHLGRTLGLYGKDQQEAALVDMVNDGVEDLRCKYISLIYTNYEAGKDDYVKALPGQLKPFETLLSQNQGGKTFIVGDQISFADYNLLDLLLIHEVLAPGCLDAFPLLSAYVGRLSARPKLKAFLASPEYVNLPINGNGKQPYTVVYFPVRGRCAALRMLLADQGQSWKEEVVTVETWQEGSLKASCLYGQLPKFQDGDLTLYQSNTILRHLGRTLGLYGKDQQEAALVDMVNDGVEDLRCKYISLIYTNYEAGKDDYVKALPGQLKPFETLLSQNQGGKTFIVGDQISFADYNLLDLLLIHEVLAPGCLDAFPLLSAYVGRLSARPKLKAFLASPEYVNLPINGNGKQ Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Medium-chain specific acyl-CoA dehydrogenase, mitochondrial | 4P13 | 3.93 | |
Target general information Gen name ACADM Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Acyl-CoA dehydrogenase family Biochemical class Oxidoreductase Function Acyl-CoA dehydrogenase activity.Flavin adenine dinucleotide binding.Identical protein binding.Medium-chain-acyl-CoA dehydrogenase activity. Related diseases Acyl-CoA dehydrogenase medium-chain deficiency (ACADMD) [MIM:201450]: An inborn error of mitochondrial fatty acid beta-oxidation which causes fasting hypoglycemia, hepatic dysfunction and encephalopathy, often resulting in death in infancy. {ECO:0000269|PubMed:10767181, ECO:0000269|PubMed:11349232, ECO:0000269|PubMed:11409868, ECO:0000269|PubMed:11486912, ECO:0000269|PubMed:1363805, ECO:0000269|PubMed:1671131, ECO:0000269|PubMed:1684086, ECO:0000269|PubMed:1902818, ECO:0000269|PubMed:2251268, ECO:0000269|PubMed:2393404, ECO:0000269|PubMed:2394825, ECO:0000269|PubMed:7603790, ECO:0000269|PubMed:7929823, ECO:0000269|PubMed:8198141, ECO:0000269|PubMed:9158144, ECO:0000269|PubMed:9882619}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03415; DB03147; DB02910 Interacts with PRO_0000000502 [P11310] EC number 1.3.8.7 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Disease variant; FAD; Fatty acid metabolism; Flavoprotein; Lipid metabolism; Mitochondrion; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Transit peptide Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 85080.3 Length 773 Aromaticity 0.09 Instability index 30.55 Isoelectric point 5.71 Charge (pH=7) -7.7 2D Binding mode Binding energy (Kcal/mol) -5.36 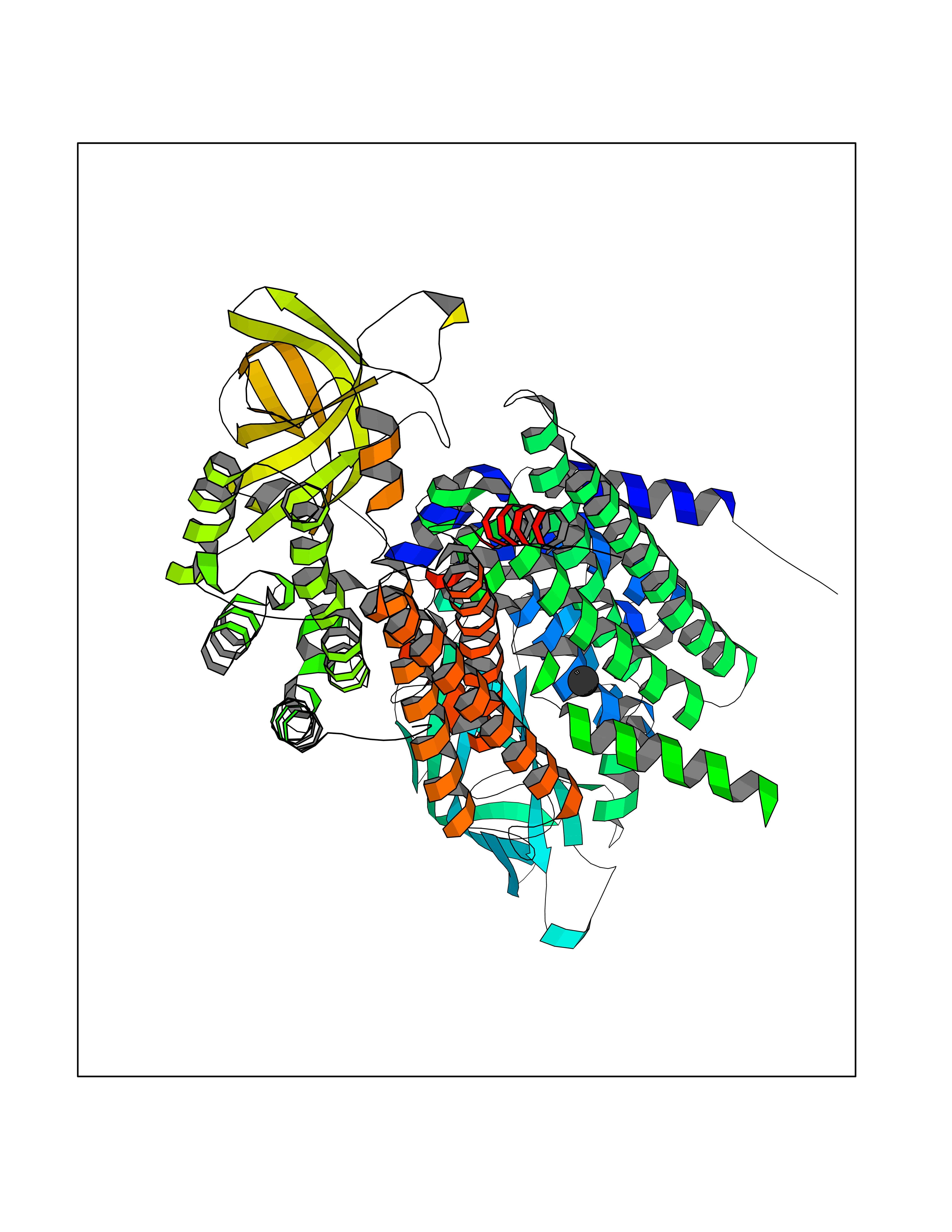 Molscript Map 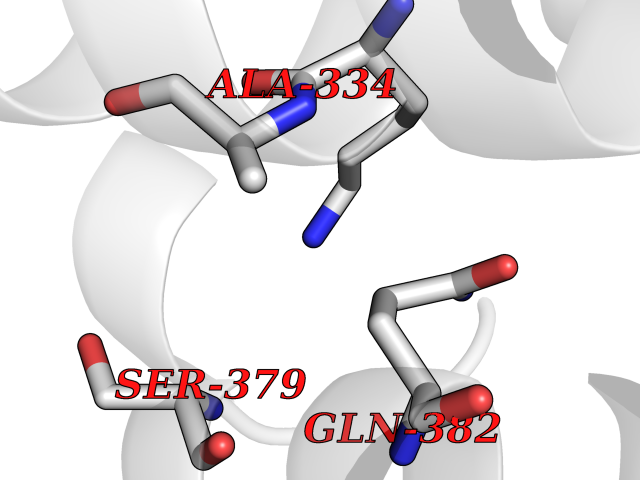 Pymol Map 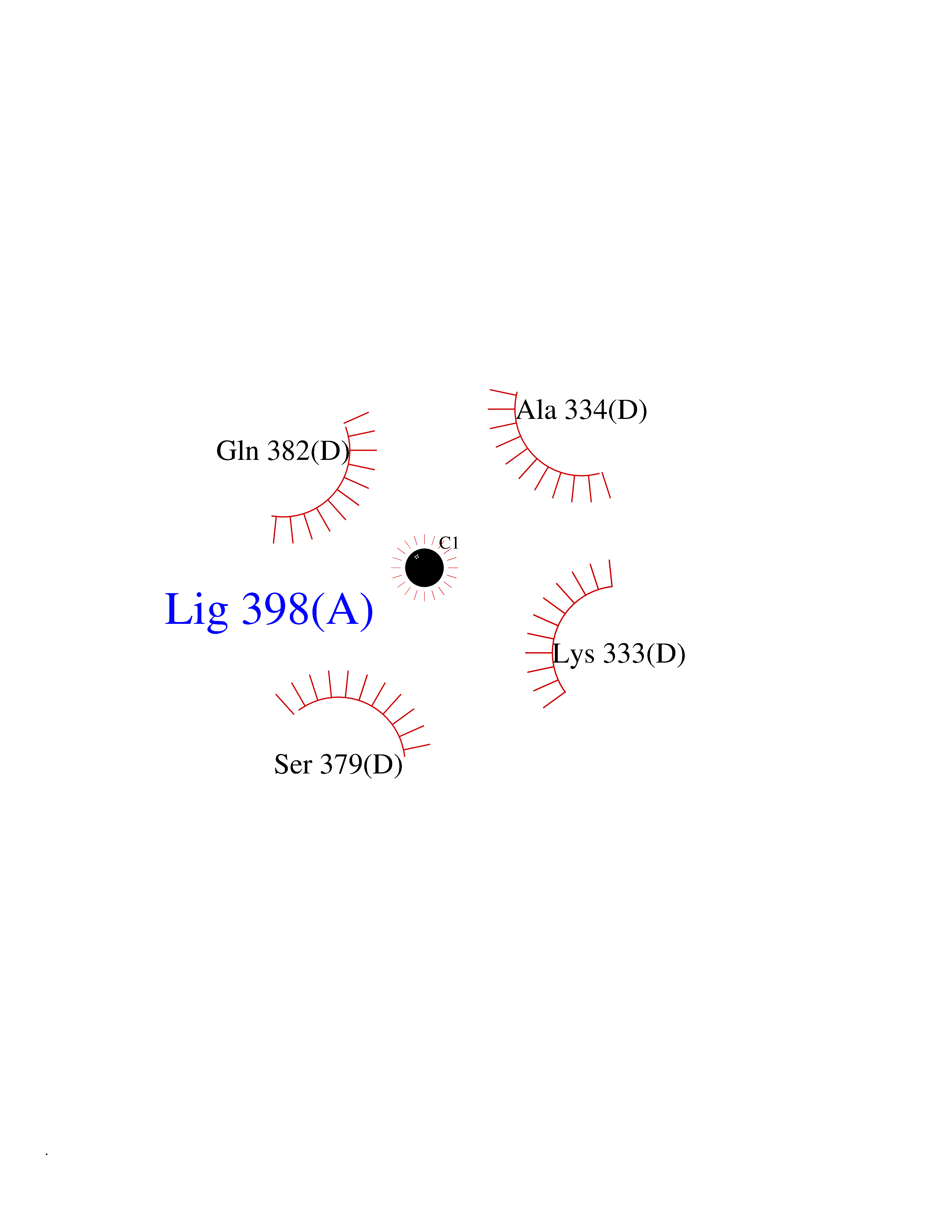 Ligplot Map 3D Binding mode Sequence LGFSFEFTEQQKEFQATARKFAREEIIPVAAEYDKTGEYPVPLIRRAWELGLMNTHIPENCGGLGLGTFDACLISEELAYGCTGVQTAIEGNSLGQMPIIIAGNDQQKKKYLGRMTEEPLMCAYCVTEPGAGSDVAGIKTKAEKKGDEYIINGQKMWITNGGKANWYFLLARSDPDPKAPANKAFTGFIVEADTPGIQIGRKELNMGQRCSDTRGIVFEDVKVPKENVLIGDGAGFKVAMGAFDKTRPVVAAGAVGLAQRALDEATKYALERKTFGKLLVEHQAISFMLAEMAMEVELARMSYQRAAWEVDSGRRNTYYASIAKAFAGDIANQLATDAVQILGGNGFNTEYPVEKLMRDAKIYQIYEGTSQIQRLIVAREHIDKYKLGFSFEFTEQQKEFQATARKFAREEIIPVAAEYDKTGEYPVPLIRRAWELGLMNTHIPENCGGLGLGTFDACLISEELAYGCTGVQTAIEGNSLGQMPIIIAGNDQQKKKYLGRMTEEPLMCAYCVTEPGAGSDVAGIKTKAEKKGDEYIINGQKMWITNGGKANWYFLLARSDPDPKAPANKAFTGFIVEADTPGIQIGRKELNMGQRCSDTRGIVFEDVKVPKENVLIGDGAGFKVAMGAFDKTRPVVAAGAVGLAQRALDEATKYALERKTFGKLLVEHQAISFMLAEMAMEVELARMSYQRAAWEVDSGRRNTYYASIAKAFAGDIANQLATDAVQILGGNGFNTEYPVEKLMRDAKIYQIYEGTSQIQRLIVAREHIDKYKN Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Argininosuccinate synthase | 2NZ2 | 3.93 | |
Target general information Gen name ASS1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ASS Protein family Argininosuccinate synthase family, Type 1 subfamily Biochemical class Ligase Function Amino acid binding.Argininosuccinate synthase activity.ATP binding.Identical protein binding.RNA binding. Related diseases Citrullinemia 1 (CTLN1) [MIM:215700]: The classic form of citrullinemia, an autosomal recessive disease characterized primarily by elevated serum and urine citrulline levels. Ammonia intoxication is another manifestation. It is a disorder of the urea cycle, usually manifesting in the first few days of life. Affected infants appear normal at birth, but as ammonia builds up in the body they present symptoms such as lethargy, poor feeding, vomiting, seizures and loss of consciousness. Less commonly, a milder form can develop later in childhood or adulthood. {ECO:0000269|PubMed:11708871, ECO:0000269|PubMed:11941481, ECO:0000269|PubMed:12815590, ECO:0000269|PubMed:14680976, ECO:0000269|PubMed:15863597, ECO:0000269|PubMed:16475226, ECO:0000269|PubMed:18473344, ECO:0000269|PubMed:19006241, ECO:0000269|PubMed:1943692, ECO:0000269|PubMed:2358466, ECO:0000269|PubMed:23611581, ECO:0000269|PubMed:24889030, ECO:0000269|PubMed:25179242, ECO:0000269|PubMed:27287393, ECO:0000269|PubMed:28111830, ECO:0000269|PubMed:7977368}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00125; DB00128; DB00171; DB00155 Interacts with P10398; P00966; Q9HBL8; Q9NVM4 EC number 6.3.4.5 Uniprot keywords 3D-structure; Acetylation; Amino-acid biosynthesis; Arginine biosynthesis; ATP-binding; Cytoplasm; Direct protein sequencing; Disease variant; Ligase; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Urea cycle Protein physicochemical properties Chain ID A Molecular weight (Da) 41098.8 Length 364 Aromaticity 0.1 Instability index 36.56 Isoelectric point 8.4 Charge (pH=7) 3.55 2D Binding mode Binding energy (Kcal/mol) -5.36  Molscript Map 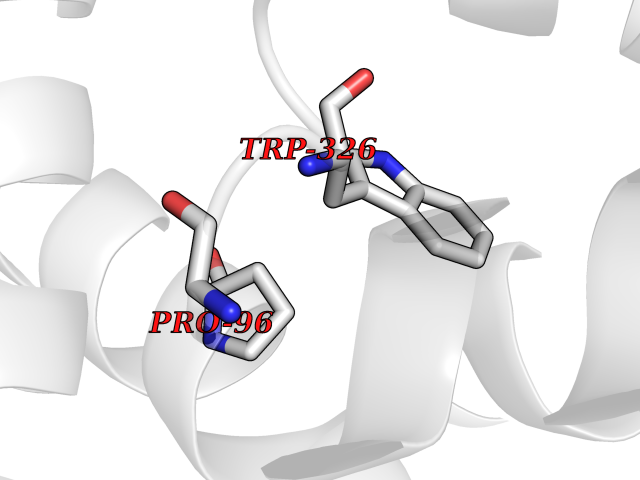 Pymol Map 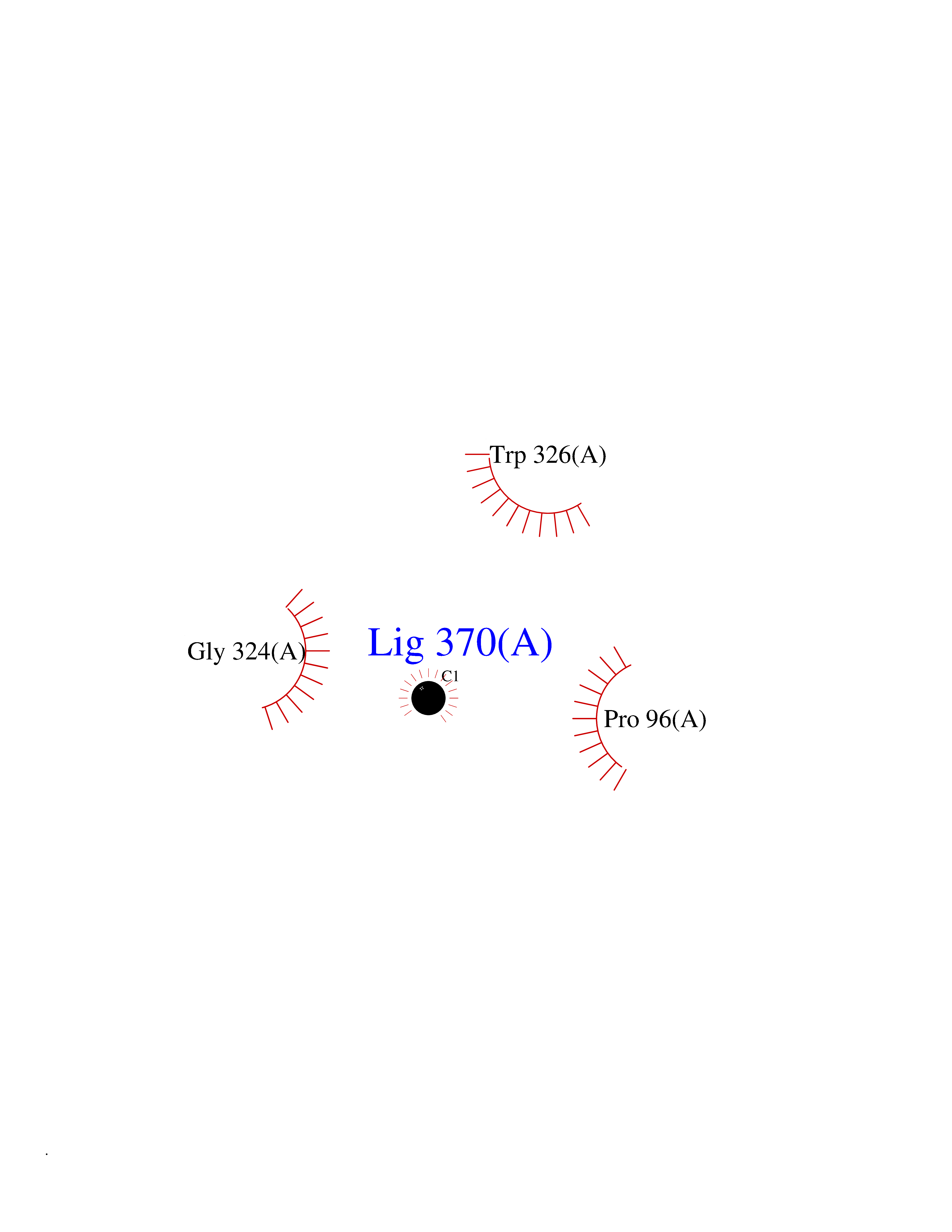 Ligplot Map 3D Binding mode Sequence KGSVVLAYSGGLDTSCILVWLKEQGYDVIAYLANIGQKEDFEEARKKALKLGAKKVFIEDVSREFVEEFIWPAIQSSALYEDRYLLGTSLARPCIARKQVEIAQREGAKYVSHGATGKGNDQVRFELSCYSLAPQIKVIAPWRMPEFYNRFKRNDLMEYAKQHGIPIPVTPKNPWSMDENLMHISYEAGILENPKNQAPPGLYTKTQDPAKAPNTPDILEIEFKKGVPVKVTNVKDGTTHQTSLELFMYLNEVAGKHGVGRIDIVENRFIGMKSRGIYETPAGTILYHAHLDIEAFTMDREVRKIKQGLGLKFAELVYTGFWHSPECEFVRHCIAKSQERVEGKVQVSVLKGQVYILGRESPLS Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Dopamine beta-hydroxylase | 4ZEL | 3.93 | |
Target general information Gen name DBH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Copper type II ascorbate-dependent monooxygenase family Biochemical class Oxidoreductase Function Catalytic activity.Copper ion binding.Dopamine beta-monooxygenase activity.L-ascorbic acid binding. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB06774; DB09130; DB05394; DB00822; DB00988; DB00968; DB00550 Interacts with P00352; P63010-2; Q04656; Q8WUW1; Q9UNS2; Q71DI3; P61978; Q9Y2M5; Q92876; P08727; Q14693; P0DPK4; Q6GQQ9-2; P27986-2; Q9ULX5; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q86WT6-2 EC number 1.14.17.1 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Copper; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Membrane; Metal-binding; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal-anchor; Transmembrane; Transmembrane helix; Vitamin C Protein physicochemical properties Chain ID A,B Molecular weight (Da) 123694 Length 1094 Aromaticity 0.1 Instability index 51.85 Isoelectric point 5.84 Charge (pH=7) -24.5 2D Binding mode Binding energy (Kcal/mol) -5.36 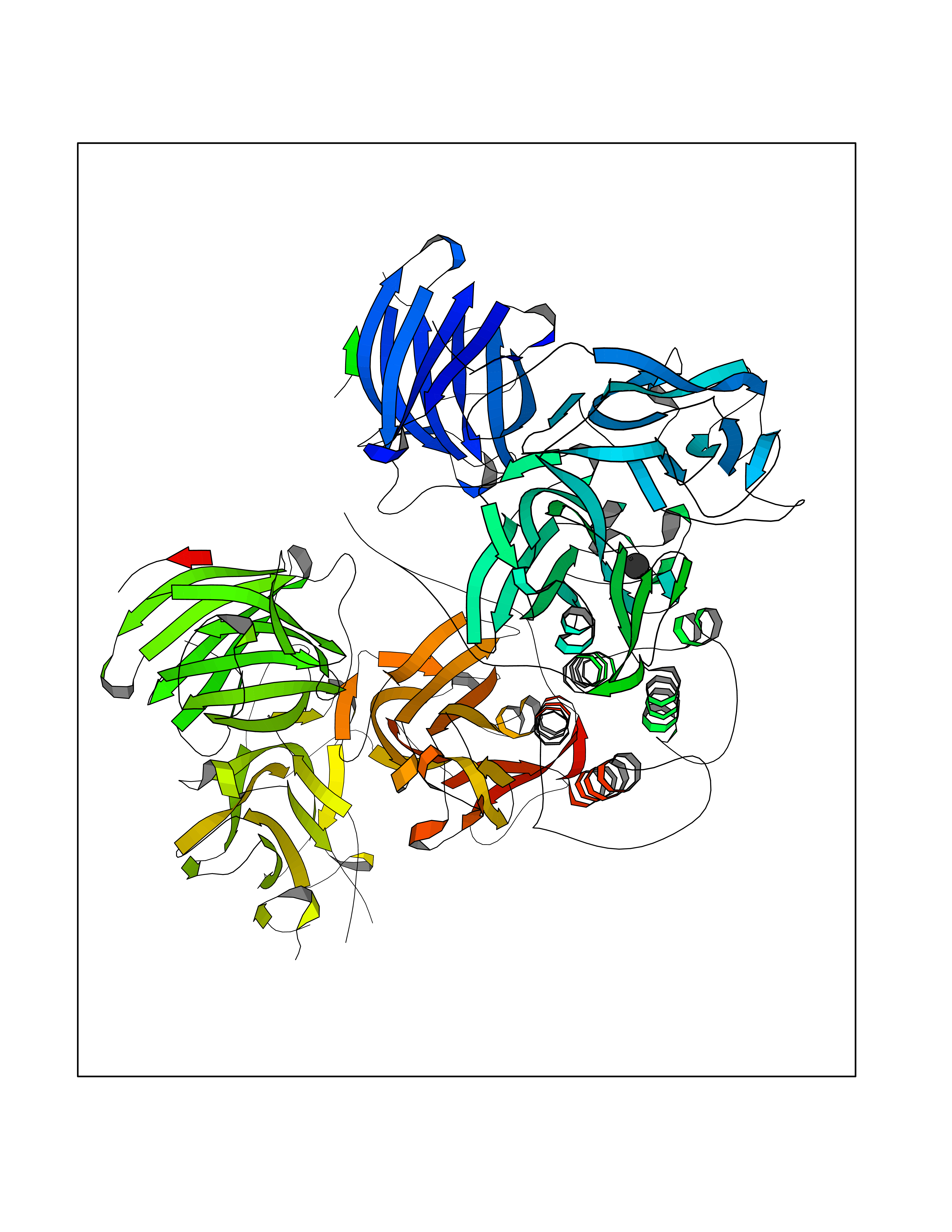 Molscript Map 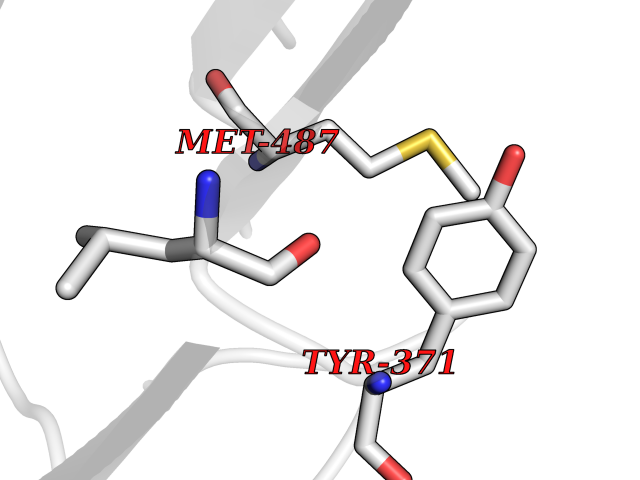 Pymol Map 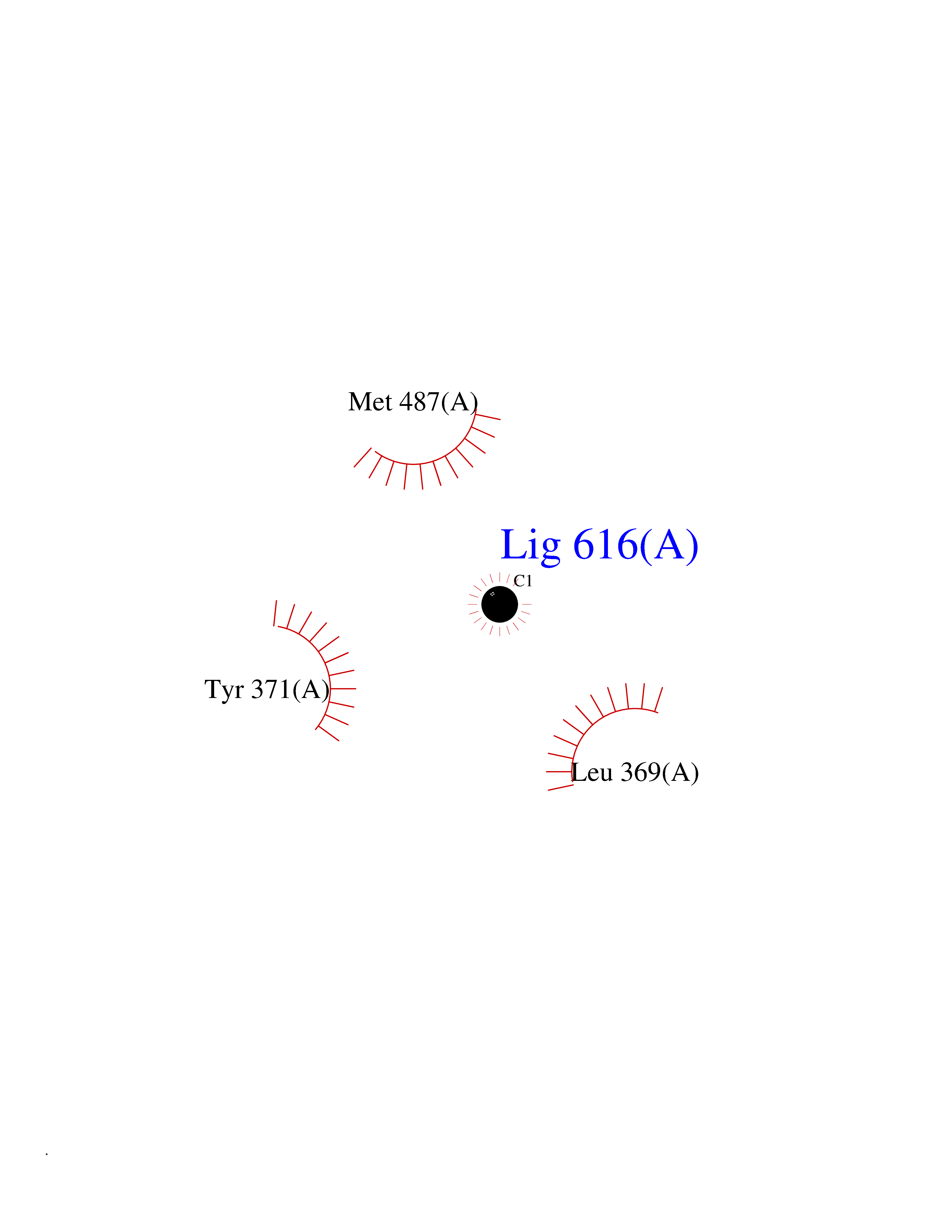 Ligplot Map 3D Binding mode Sequence PLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLWTDGDAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEMDSVPHFSGPCDSKMKPDRLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTVVSPLPYHIPLDPEGSLELSWNVSYTQEAIHFQLLVRRLKAGVLFGMSDRGELENADLVVLAYFADAWSDQKGQIHLDPQQDYQLLQVQRTPEGLTLLFKRPFGTCDPKDYLIEDGTVHLVYGILEEPFRSLEAINGSGLQMGLQRVQLLKPNIPEPELPSDACTMEVQAPNIQIPSQETTYWCYIKELPKGFSRHHIIKYEPIVTKGNEALVHHMEVFQCAPEVPHFSGPCDSKMLNYCRHVLAAWALGAKAFYYPEEAGLAFGGPGSSRYLRLEVHYHNPLVIEGRNDSSGIRLYYTAKLRRFNAGIMELGLVYTPVMAIPPRETAFILTGYCTDKCTQLALPPSGIHIFASQLHTHLTGRKVVTVLVRDGREWEIVNQDNHYSPHFQEIRMLKKVVSVHPGDVLITSCTYNTEDRELATVGGFGILEEMCVNYVHYYPQTQLELCKSAVDAGFLQKYFHLINRFNNEDVCTCPQASVSQQFTSVPWNSFNRDVLKALYSFAPISMHCNKSSAVRFQGEWNLQPLPKVISTLEEPTPQCVVSIGG Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Vitamin K-dependent protein C (PROC) | 1LQV | 3.93 | |
Target general information Gen name PROC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin K-dependent protein C light chain; Vitamin K-dependent protein C heavy chain; PROC; Blood coagulation factor XIV; Autoprothrombin IIA; Anticoagulant protein C; Activation peptide Protein family Peptidase S1 family Biochemical class Peptidase Function Protein C is avitamin K-dependent serine protease that regulates blood coagulation by inactivating factors Va and VIIIa in the presence of calcium ions and phospholipids. Related diseases Thrombophilia due to protein C deficiency, autosomal dominant (THPH3) [MIM:176860]: A hemostatic disorder characterized by impaired regulation of blood coagulation and a tendency to recurrent venous thrombosis. Individuals with decreased amounts of protein C are classically referred to as having type I protein C deficiency and those with normal amounts of a functionally defective protein as having type II deficiency. {ECO:0000269|PubMed:1301959, ECO:0000269|PubMed:1347706, ECO:0000269|PubMed:1511989, ECO:0000269|PubMed:1868249, ECO:0000269|PubMed:2437584, ECO:0000269|PubMed:25618265, ECO:0000269|PubMed:25748729, ECO:0000269|PubMed:2602169, ECO:0000269|PubMed:7792728, ECO:0000269|PubMed:7865674, ECO:0000269|PubMed:8292730, ECO:0000269|PubMed:8398832, ECO:0000269|PubMed:8499568, ECO:0000269|PubMed:8560401, ECO:0000269|PubMed:8829639, ECO:0000269|PubMed:9798967}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thrombophilia due to protein C deficiency, autosomal recessive (THPH4) [MIM:612304]: A hemostatic disorder characterized by impaired regulation of blood coagulation and a tendency to recurrent venous thrombosis. It results in a thrombotic condition that can manifest as a severe neonatal disorder or as a milder disorder with late-onset thrombophilia. The severe form leads to neonatal death through massive neonatal venous thrombosis. Often associated with ecchymotic skin lesions which can turn necrotic called purpura fulminans, this disorder is very rare. {ECO:0000269|PubMed:1511988, ECO:0000269|PubMed:1593215, ECO:0000269|PubMed:1611081, ECO:0000269|PubMed:25618265, ECO:0000269|PubMed:7841323, ECO:0000269|PubMed:7841324, ECO:0000269|PubMed:7878626}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB13192; DB00025; DB09131; DB09332; DB13998; DB00170; DB13999; DB13149; DB00464; DB14738 Interacts with A8MQ03; P51511 EC number EC 3.4.21.69 Uniprot keywords 3D-structure; Alternative splicing; Blood coagulation; Calcium; Cleavage on pair of basic residues; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Endoplasmic reticulum; Gamma-carboxyglutamic acid; Glycoprotein; Golgi apparatus; Hemostasis; Hydrolase; Hydroxylation; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Thrombophilia; Zymogen Protein physicochemical properties Chain ID C,D Molecular weight (Da) 45326 Length 411 Aromaticity 0.12 Instability index 40.68 Isoelectric point 7.07 Charge (pH=7) 0.29 2D Binding mode Binding energy (Kcal/mol) -5.35 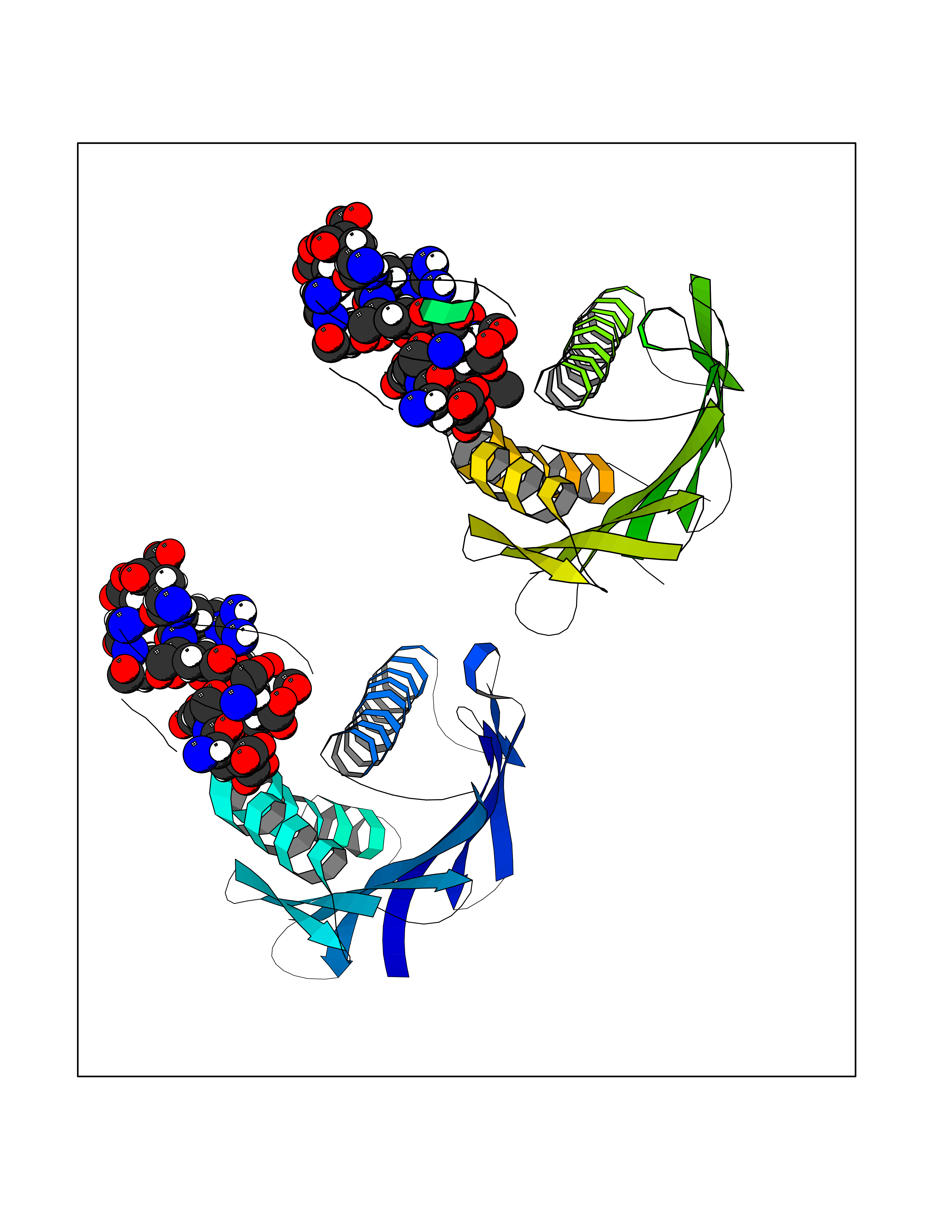 Molscript Map 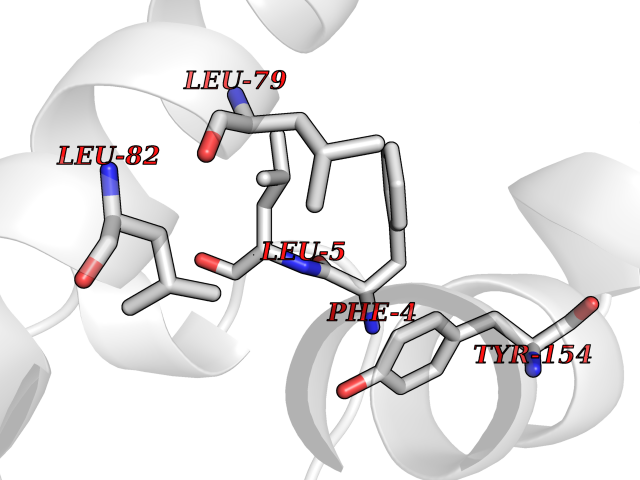 Pymol Map 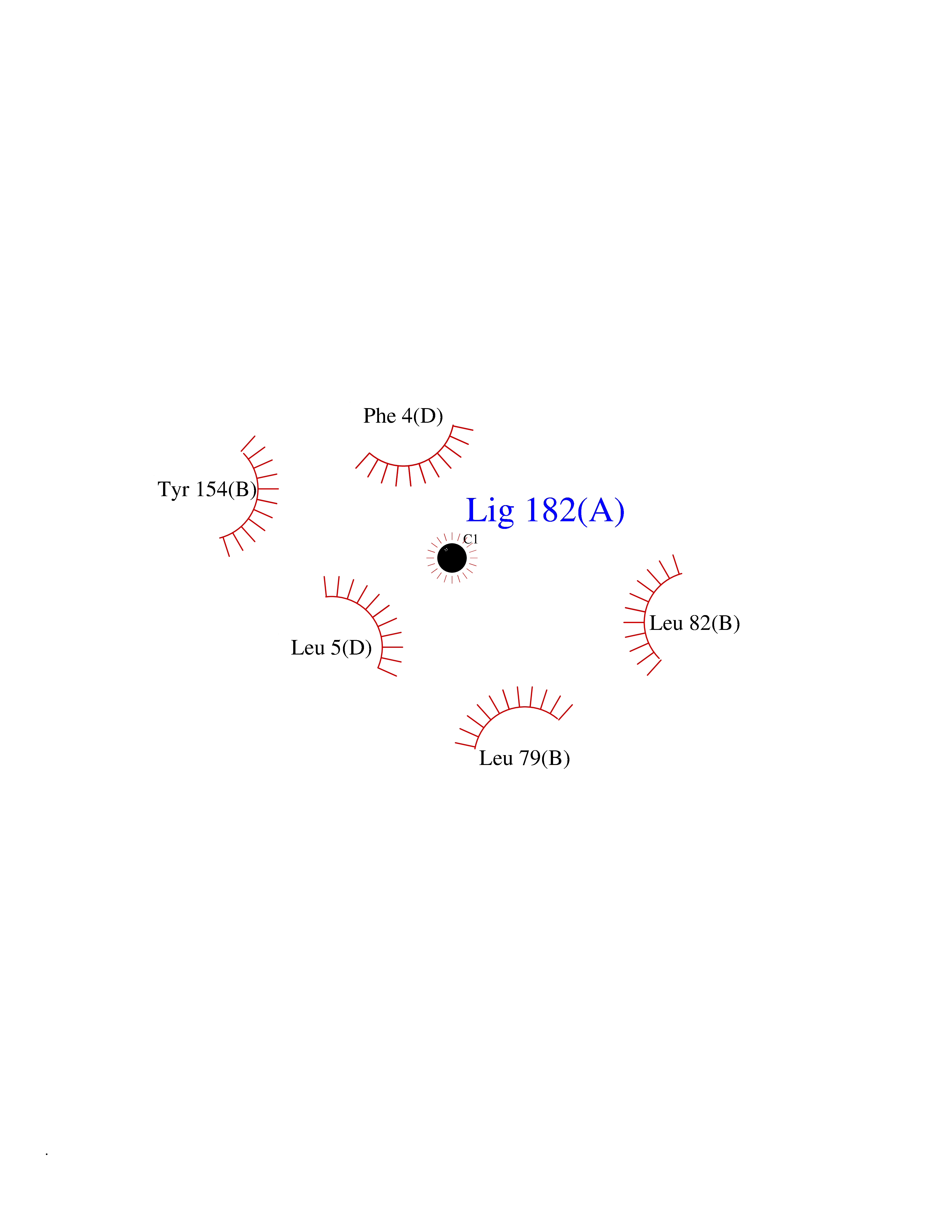 Ligplot Map 3D Binding mode Sequence GLQRLHMLQISYFRDPYHVWYQGNASLGGHLTHVLEGPDTNTTIIQLQPLQEPESWARTQSGLQSYLLQFHGLVRLVHQERTLAFPLTIRCFLGCELPPEGSRAHVFFEVAVNGSSFVSFRPERALWQADTQVTSGVVTFTLQQLNAYNRTRYELREFLEDTCVQYVQKHISANSFLXXLRHSSLXRXCIXXICDFXXAKXIFQNANSFLXXLRHSSLXRXCIXXICDFXXAKXIFQNLQRLHMLQISYFRDPYHVWYQGNASLGGHLTHVLEGPDTNTTIIQLQPLQEPESWARTQSGLQSYLLQFHGLVRLVHQERTLAFPLTIRCFLGCELPPEGSRAHVFFEVAVNGSSFVSFRPERALWQADTQVTSGVVTFTLQQLNAYNRTRYELREFLEDTCVQYVQKHISAE Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Aldose reductase (AKR1B1) | 1US0 | 3.93 | |
Target general information Gen name AKR1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aldehyde reductase; AKR1B1 Protein family Aldo/keto reductase family Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the NADPH-dependent reduction of a wide variety of carbonyl-containing compounds to their corresponding alcohols with a broad range of catalytic efficiencies. Related diseases Glutamine deficiency, congenital (GLND) [MIM:610015]: An autosomal recessive disorder characterized by variable brain malformations, encephalopathy, severe developmental delay, seizures, and decreased glutamine levels in bodily fluids. Death in early infancy may occur. {ECO:0000269|PubMed:16267323, ECO:0000269|PubMed:26711351, ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 116 (DEE116) [MIM:620806]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE116 is autosomal dominant form characterized by severe developmental delay, seizures, and white matter abnormalities. {ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry. DEE116 is caused by variants that disrupt the canonical translation start codon in GLUL resulting in initiation of translation at Met-18 (PubMed:38579670). The resulting protein is enzymatically competent but insensitive to negative feedback regulation via glutamine-induced degradation. {ECO:0000269|PubMed:38579670}. Drugs (DrugBank ID) DB07028; DB07030; DB07450; DB02101; DB08449; DB08000; DB07139; DB07498; DB02007; DB02020; DB11859; DB02994; DB04272; DB07187; DB00694; DB00997; DB06246; DB01039; DB02021; DB16707; DB00143; DB02834; DB08084; DB01689; DB07063; DB06077; DB02518; DB00157; DB03461; DB05383; DB05533; DB05327; DB02712; DB00605; DB02383; DB02132; DB08772; DB07093; DB07999; DB08098 Interacts with Q9BUY7 EC number EC 1.1.1.300 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Lipid metabolism; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 35447.6 Length 313 Aromaticity 0.09 Instability index 36.41 Isoelectric point 7.1 Charge (pH=7) 0.26 2D Binding mode Binding energy (Kcal/mol) -5.36 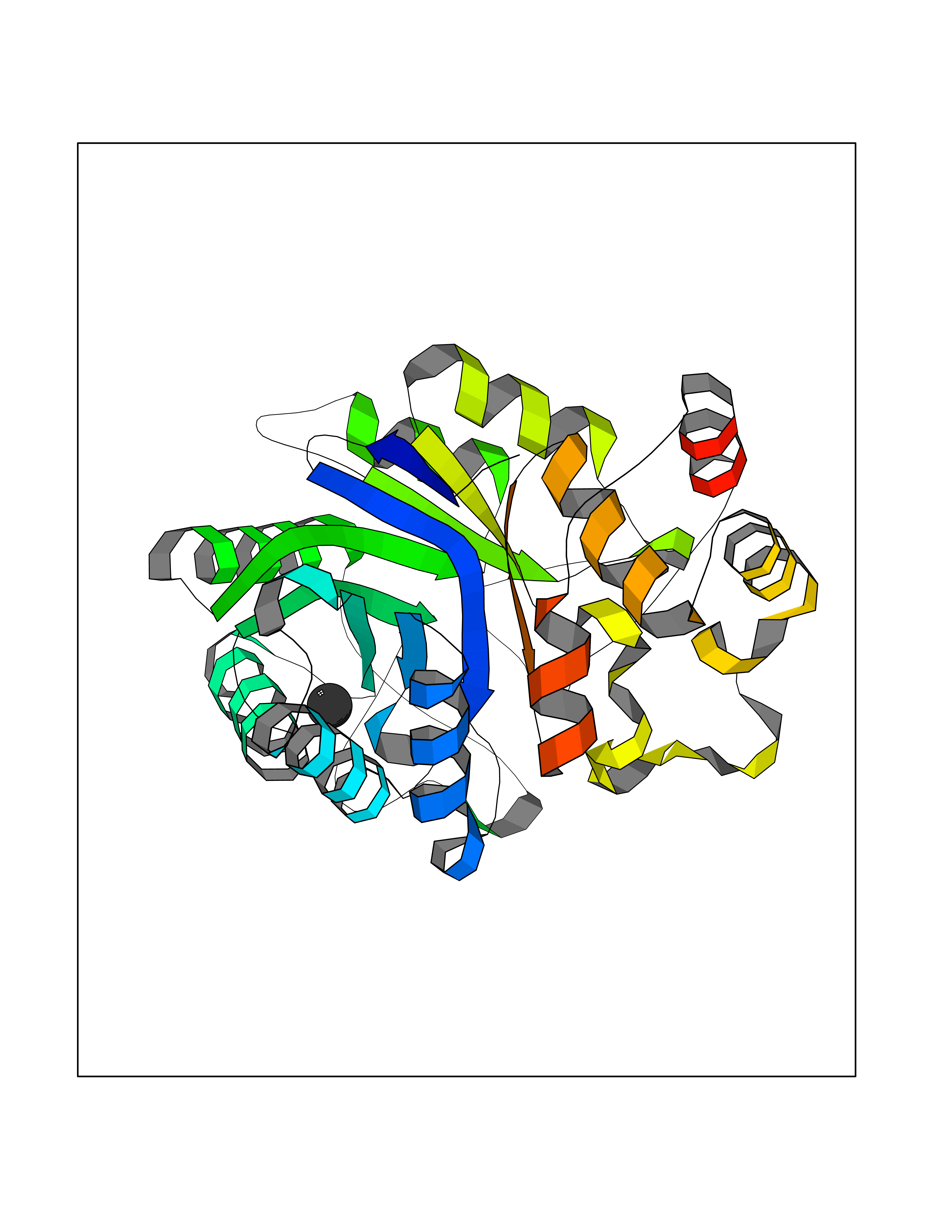 Molscript Map 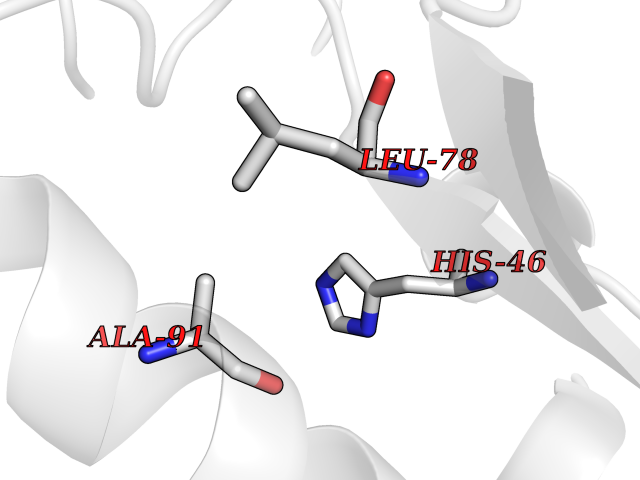 Pymol Map 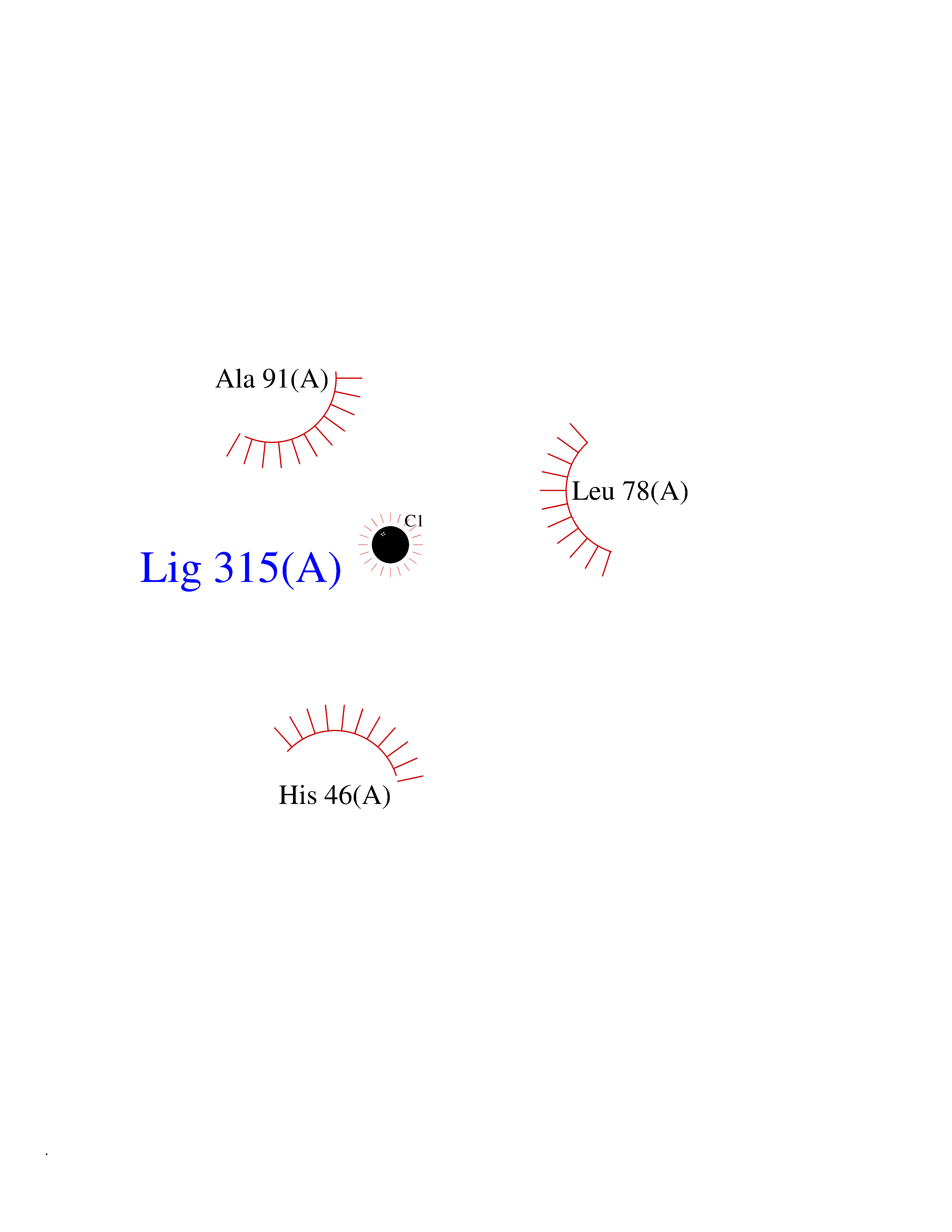 Ligplot Map 3D Binding mode Sequence MASRILLNNGAKMPILGLGTWKSPPGQVTEAVKVAIDVGYRHIDCAHVYQNENEVGVAIQEKLREQVVKREELFIVSKLWCTYHEKGLVKGACQKTLSDLKLDYLDLYLIHWPTGFKPGKEFFPLDESGNVVPSDTNILDTWAAMEELVDEGLVKAIGISNFNHLQVEMILNKPGLKYKPAVNQIECHPYLTQEKLIQYCQSKGIVVTAYSPLGSPDRPWAKPEDPSLLEDPRIKAIAAKHNKTTAQVLIRFPMQRNLVVIPKSVTPERIAENFKVFDFELSSQDMTTLLSYNRNWRVCALLSCTSHKDYPFH Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | DNA polymerase alpha catalytic p180 (POLA1) | 4QCL | 3.93 | |
Target general information Gen name POLA1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms POLA; DNA polymerase alpha catalytic subunit p180; DNA polymerase alpha catalytic subunit Protein family DNA polymerase type-B family Biochemical class Kinase Function During the S phase of the cell cycle, the DNA polymerase alpha complex (composed of a catalytic subunit POLA1/p180, a regulatory subunit POLA2/p70 and two primase subunits PRIM1/p49 and PRIM2/p58) is recruited to DNA at the replicative forks via direct interactions with MCM10 and WDHD1. The primase subunit of the polymerase alpha complex initiates DNA synthesis by oligomerising short RNA primers on both leading and lagging strands. These primers are initially extended by the polymerase alpha catalytic subunit and subsequently transferred to polymerase delta and polymerase epsilon for processive synthesis on the lagging and leading strand, respectively. The reason this transfer occurs is because the polymerase alpha has limited processivity and lacks intrinsic 3' exonuclease activity for proofreading error, and therefore is not well suited for replicating long complexes. In the cytosol, responsible for a substantial proportion of the physiological concentration of cytosolic RNA:DNA hybrids, which are necessary to prevent spontaneous activation of type I interferon responses. Plays an essential role in the initiation of DNA replication. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00242; DB00631; DB01073; DB01280 Interacts with P27694; P10193; P03070 EC number EC 2.7.7.7 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Disease variant; DNA replication; DNA-binding; DNA-directed DNA polymerase; Dwarfism; Host-virus interaction; Intellectual disability; Metal-binding; Nucleotidyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 24505.6 Length 212 Aromaticity 0.09 Instability index 43.56 Isoelectric point 9.02 Charge (pH=7) 4.85 2D Binding mode Binding energy (Kcal/mol) -5.35 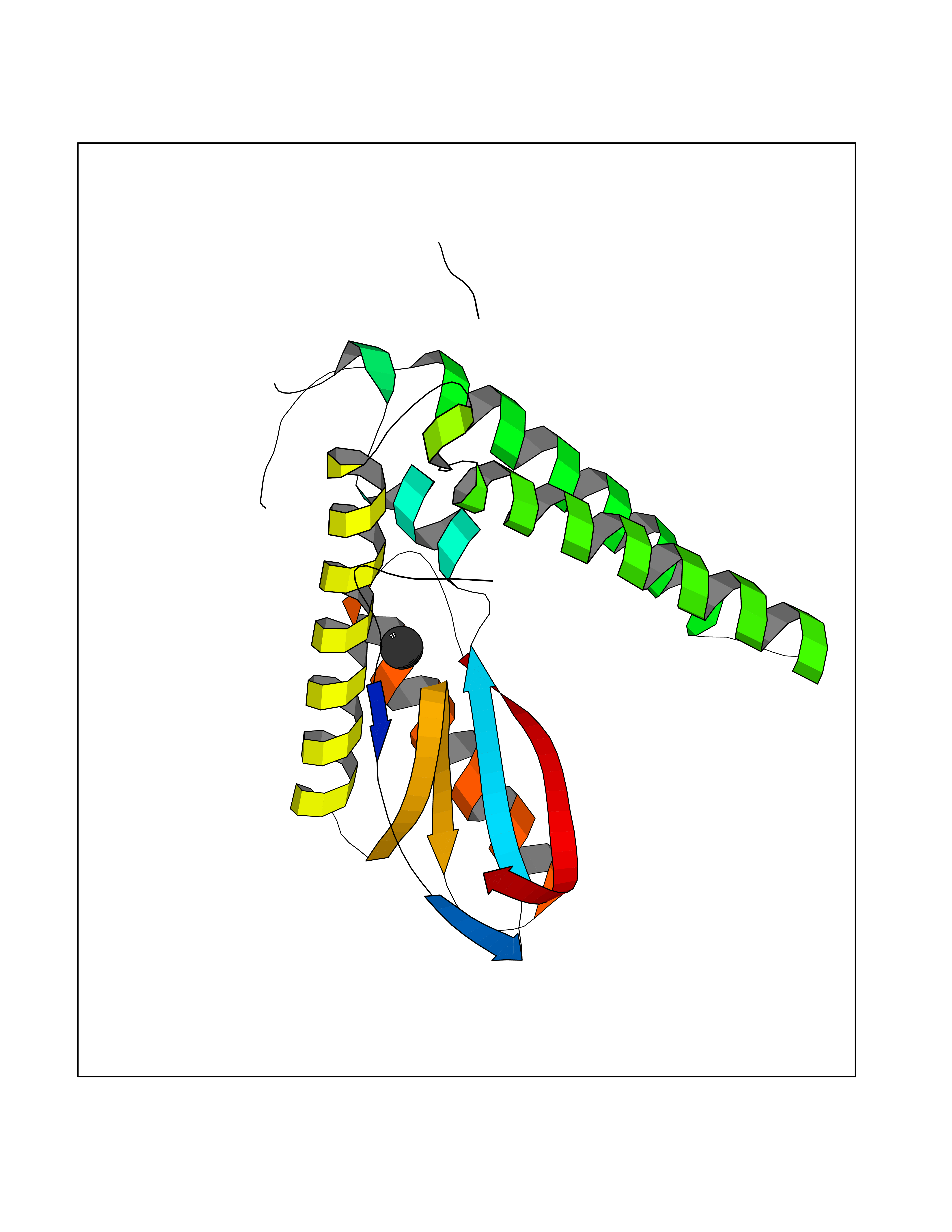 Molscript Map  Pymol Map 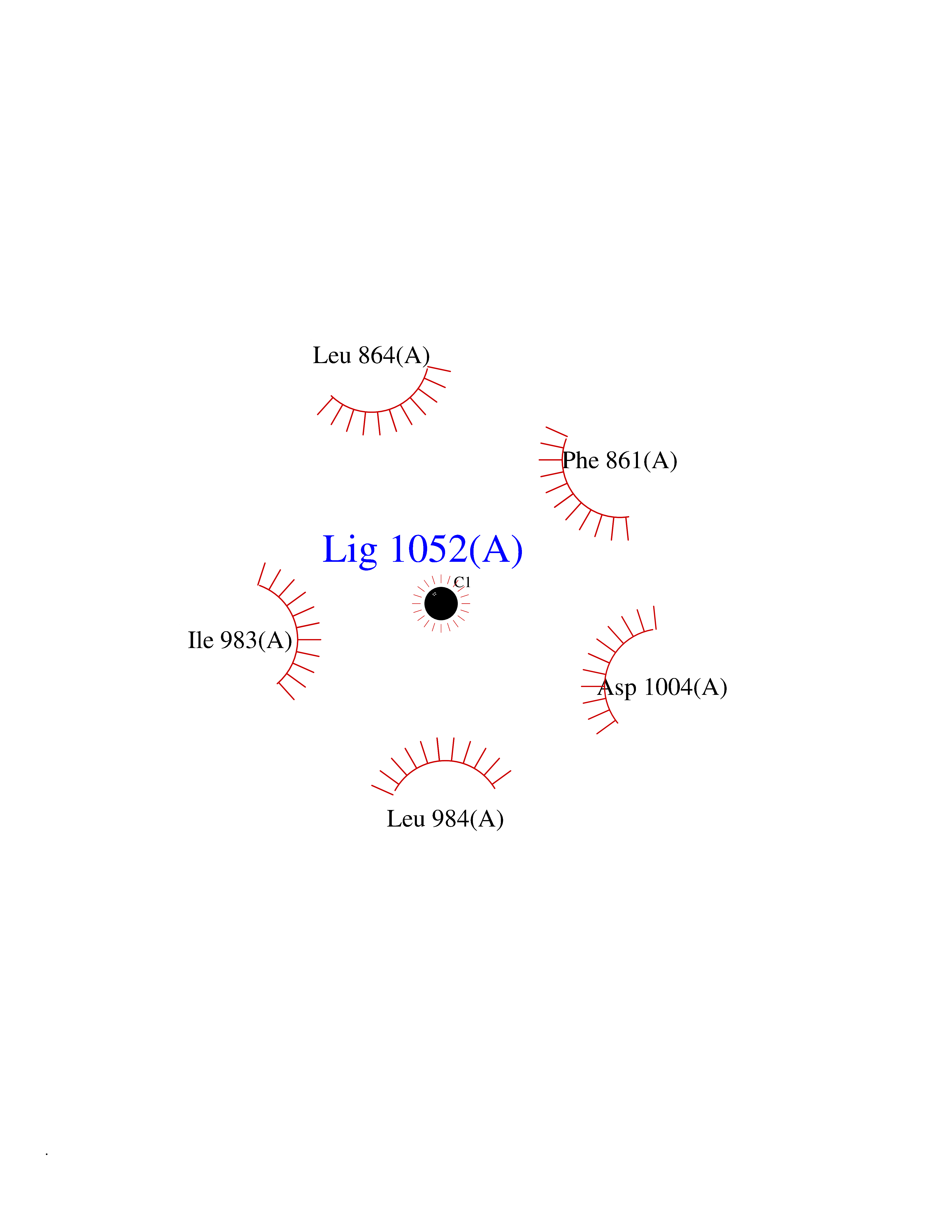 Ligplot Map 3D Binding mode Sequence YIVPDKQIRKKAAYAGGLVLDPKVGFYDKFILLLDFNSLYPSIIQEFNICFTTVQRVEQIPELPDPSLEMGILPREIRKLVERRKQVKQLMKQQDLNPDLILQYDIRQKALKLTANSMYGCLGFSYSRFYAKPLAALVTYKGREILMHTKEMVQKMNLEVIYGDTDSIMINTNSTNLEEVFKLGNKVKSEVNKLYKLLEIDIDGVFKSLLLL Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | Cytochrome P450 1A2 | 2HI4 | 3.93 | |
Target general information Gen name CYP1A2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Aromatase activity.Caffeine oxidase activity.Demethylase activity.Electron carrier activity.Enzyme binding.Heme binding.Iron ion binding.Monooxygenase activity.Oxidoreductase activity.Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, reduced flavin or flavoprotein as one donor, and incorporation of one atom of oxygen.Oxygen binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08496; DB01667; DB14132; DB04356; DB02489; DB11932; DB12001; DB05812; DB13573; DB01418; DB00316; DB15568; DB06594; DB00518; DB05396; DB00969; DB07453; DB01424; DB01223; DB01118; DB00321; DB00261; DB01217; DB01435; DB06605; DB05676; DB06413; DB06216; DB01072; DB15011; DB06442; DB06626; DB00993; DB00972; DB13203; DB05015; DB16703; DB06769; DB01086; DB06770; DB06771; DB06732; DB00195; DB04889; DB11967; DB13975; DB00188; DB12151; DB01558; DB14018; DB13812; DB00201; DB09061; DB14737; DB11791; DB06774; DB00564; DB06016; DB01136; DB12814; DB00477; DB00356; DB01166; DB00501; DB01012; DB00568; DB00827; DB00537; DB00215; DB12499; DB14025; DB00349; DB01242; DB00575; DB00758; DB00363; DB00286; DB11672; DB14635; DB00924; DB08912; DB00851; DB06292; DB01254; DB01609; DB01151; DB16650; DB12161; DB01191; DB00633; DB11994; DB00586; DB11511; DB12945; DB00280; DB01184; DB09167; DB05928; DB01142; DB09273; DB00470; DB00476; DB00625; DB15444; DB06210; DB13874; DB11718; DB00467; DB11404; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB04574; DB13592; DB00330; DB00898; DB00977; DB00773; DB01628; DB00927; DB04854; DB01482; DB00574; DB12265; DB15669; DB01195; DB08972; DB04841; DB00544; DB00472; DB00499; DB00176; DB01320; DB00998; DB14029; DB06160; DB01044; DB01241; DB01155; DB01645; DB01381; DB00986; DB00365; DB00400; DB05708; DB00629; DB00502; DB01094; DB14999; DB04076; DB11737; DB00619; DB00458; DB11564; DB01306; DB09456; DB09564; DB01307; DB00047; DB01309; DB00030; DB00046; DB11567; DB00071; DB11568; DB05258; DB00034; DB00105; DB15131; DB00011; DB00018; DB00069; DB00060; DB00068; DB00033; DB00951; DB11757; DB09570; DB01026; DB01097; DB16217; DB09078; DB01002; DB05667; DB00281; DB12406; DB09198; DB04948; DB00978; DB06448; DB16220; DB01601; DB00455; DB04871; DB06077; DB01283; DB00772; DB00934; DB06234; DB14009; DB00784; DB01065; DB00170; DB00454; DB00532; DB00333; DB00763; DB00553; DB01028; DB09241; DB01233; DB00379; DB06148; DB01388; DB06595; DB00370; DB16236; DB00745; DB11763; DB00218; DB06510; DB14011; DB00461; DB00607; DB00779; DB00788; DB06600; DB00238; DB06803; DB00184; DB01115; DB11793; DB00435; DB05115; DB00717; DB01059; DB00540; DB05990; DB01165; DB00334; DB16267; DB00338; DB00904; DB11632; DB11443; DB01173; DB11837; DB09330; DB01303; DB11697; DB00377; DB00715; DB06589; DB11774; DB00487; DB00008; DB00022; DB09122; DB13634; DB00806; DB11198; DB08883; DB00850; DB03783; DB01174; DB00388; DB00252; DB11450; DB01100; DB13823; DB04951; DB17472; DB11642; DB08910; DB15822; DB01058; DB01087; DB00794; DB00420; DB09288; DB01182; DB06479; DB00818; DB00571; DB13449; DB11892; DB04216; DB00908; DB00468; DB01129; DB00980; DB09290; DB00863; DB01367; DB00409; DB02709; DB13174; DB01045; DB11753; DB00740; DB14924; DB00503; DB00533; DB01656; DB15119; DB00268; DB00296; DB00412; DB00817; DB12332; DB13772; DB06654; DB11491; DB00418; DB01037; DB11689; DB06290; DB13261; DB15093; DB00052; DB00398; DB01208; DB09118; DB00428; DB06820; DB00382; DB00675; DB06083; DB09071; DB05488; DB09256; DB01079; DB01405; DB00857; DB08880; DB11712; DB01412; DB00277; DB00730; DB01623; DB00208; DB06137; DB00697; DB01056; DB06264; DB00752; DB00384; DB12245; DB00831; DB15442; DB00440; DB00685; DB08867; DB14989; DB13609; DB06235; DB00313; DB08881; DB00661; DB09185; DB12026; DB00682; DB02134; DB00549; DB00744; DB00315; DB00425; DB09225; DB09120 Interacts with O95870 EC number 1.14.14.1; 4.2.1.152 Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Fatty acid metabolism; Glycoprotein; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54475 Length 480 Aromaticity 0.1 Instability index 40.43 Isoelectric point 9.16 Charge (pH=7) 9.89 2D Binding mode Binding energy (Kcal/mol) -5.36  Molscript Map 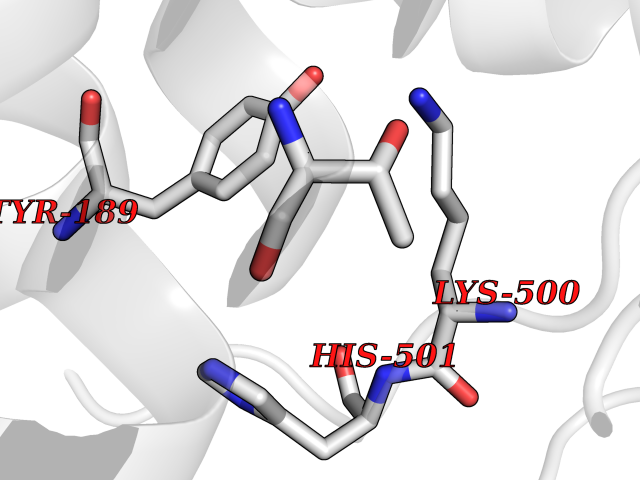 Pymol Map 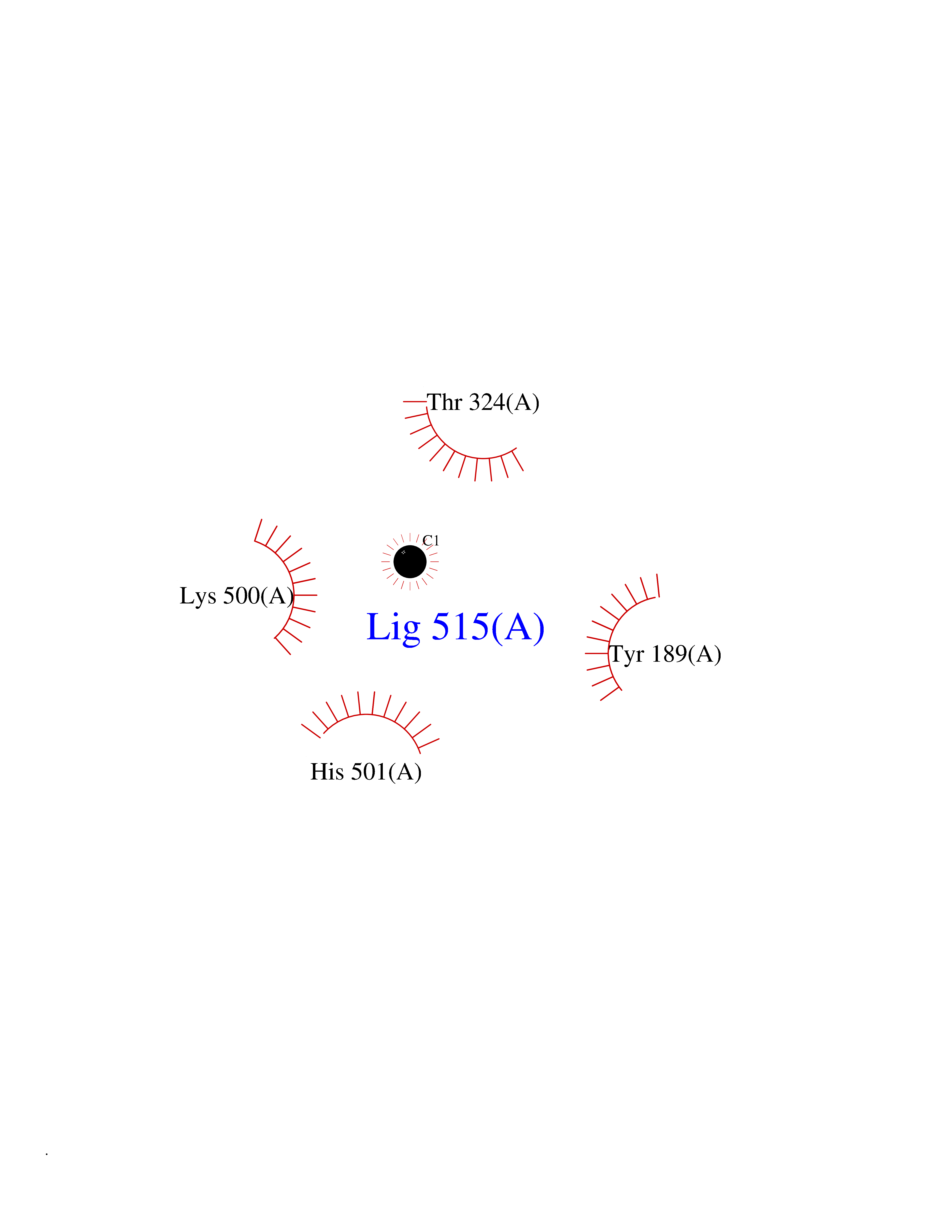 Ligplot Map 3D Binding mode Sequence RVPKGLKSPPEPWGWPLLGHVLTLGKNPHLALSRMSQRYGDVLQIRIGSTPVLVLSRLDTIRQALVRQGDDFKGRPDLYTSTLITDGQSLTFSTDSGPVWAARRRLAQNALNTFSIASDPASSSSCYLEEHVSKEAKALISRLQELMAGPGHFDPYNQVVVSVANVIGAMCFGQHFPESSDEMLSLVKNTHEFVETASSGNPLDFFPILRYLPNPALQRFKAFNQRFLWFLQKTVQEHYQDFDKNSVRDITGALFKHSKKGPRASGNLIPQEKIVNLVNDIFGAGFDTVTTAISWSLMYLVTKPEIQRKIQKELDTVIGRERRPRLSDRPQLPYLEAFILETFRHSSFLPFTIPHSTTRDTTLNGFYIPKKCCVFVNQWQVNHDPELWEDPSEFRPERFLTADGTAINKPLSEKMMLFGMGKRRCIGEVLAKWEIFLFLAILLQQLEFSVPPGVKVDLTPIYGLTMKHARCEHVQARRFS Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Penicillin-binding protein 2B | 2WAD | 3.93 | |
Target general information Gen name penA Organism Streptococcus pneumoniae (strain ATCC BAA-255 / R6) Uniprot ID TTD ID NA Synonyms spr1517;pbp2b Protein family Transpeptidase family Biochemical class Peptide binding protein Function Penicillin binding. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01163; DB00415; DB08795; DB01140; DB00456; DB01066; DB00493; DB01331; DB01212; DB00567; DB03313; DB00485; DB00739; DB01603; DB00607; DB00713; DB00319 Interacts with NA EC number NA Uniprot keywords 3D-structure; Antibiotic resistance; Cell membrane; Cell shape; Cell wall biogenesis/degradation; Membrane; Peptidoglycan synthesis; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 65444.4 Length 607 Aromaticity 0.08 Instability index 30.15 Isoelectric point 4.95 Charge (pH=7) -20.68 2D Binding mode Binding energy (Kcal/mol) -5.36 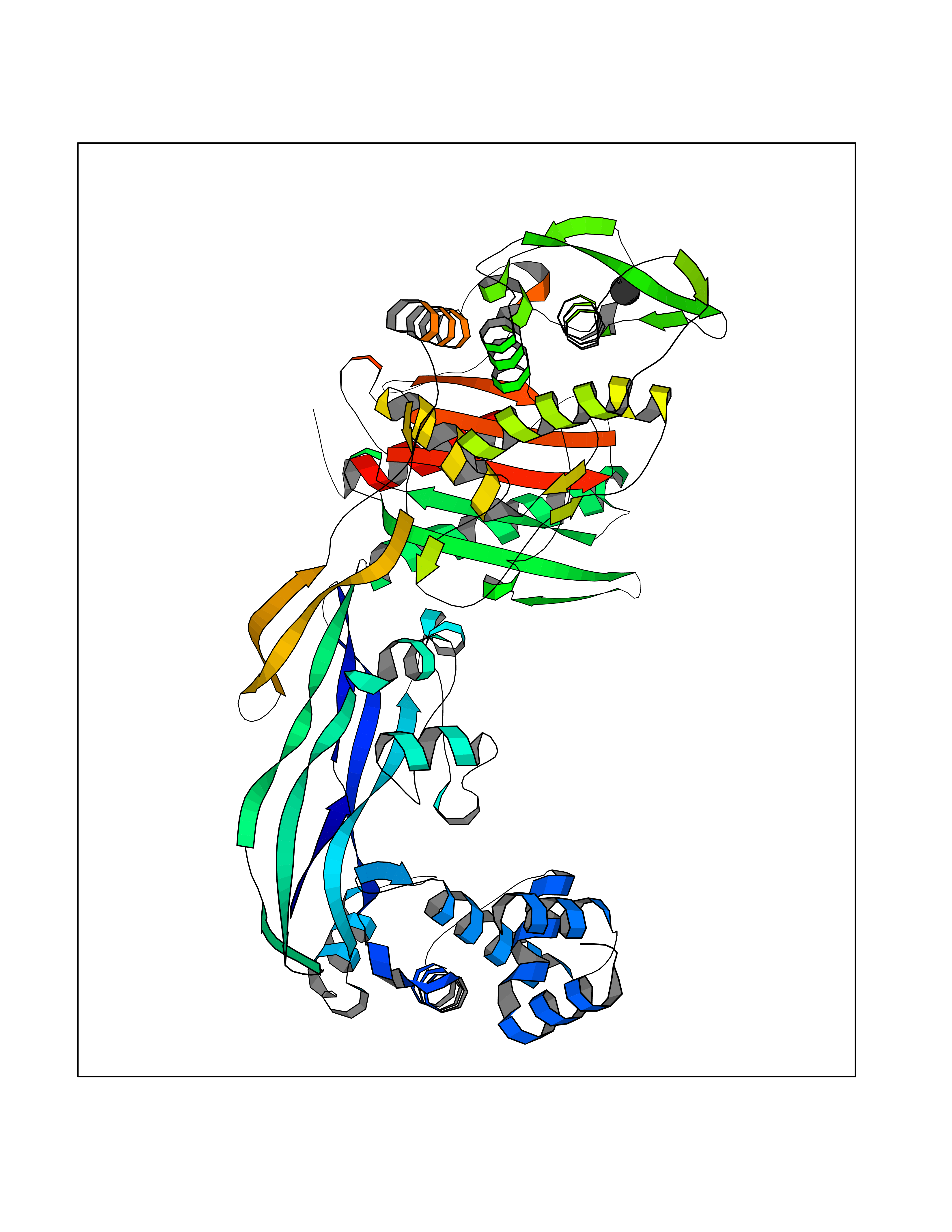 Molscript Map 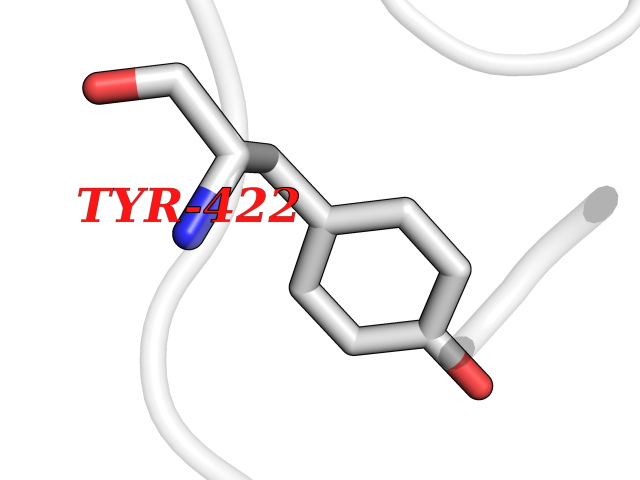 Pymol Map  Ligplot Map 3D Binding mode Sequence SQTKVTTSSARGEIYDASGKPLVENTLKQVVSFTRSNKMTATDLKEIAKKLLTYVSISSPNLTERQLADYYLADPEIYKKTVEALPSESELYNNAVDSVPTSQLNYTEDEKKEIYLFSQLNAVGNFATGTIATDPLNDSQVAVIASISKEMPGISISTSWDRKILETSLSSIVGSVSSEKAGLPAEEAESYLKKGYSLNDRVGTSYLEKQYEEVLQGKRPVKEIHLDKHGDMESVENIEEGSKGKNIKLTIDLAFQDSVDALLKSYFNSELGNGGAKYSEGVYAVALNPQTGAVLSMSGLKHDLKTGELTPDSLGTVTNVFVPGSVVKAATISSGWENGVLSGNQTLTDQPIVFQGSAPIYSWYKLAYGSFPITAVEALEYSSNAYVVQTALGIMGQTYQPNMFVGTSNLESAMGKLRSTFGEYGLGSATGIDLPDESTGLVPKEYNFANFITNAFGQFDNYTPMQLAQYVATIANNGVRLAPHIVEGIYDNNDKGGLGELIQAIDTKEINKVNISESDMAILHQGFYQVSHGTSPLTTGRAFSDGATVSISGKTGTNTNAVAYAPTENPQIAVAVVFPHNTNLTKNVGPAIARDIINLYNQHHPMN Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Argininosuccinate lyase | 1K62 | 3.93 | |
Target general information Gen name ASL Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Lyase 1 family, Argininosuccinate lyase subfamily Biochemical class Lyase Function Argininosuccinate lyase activity.Identical protein binding. Related diseases Argininosuccinic aciduria (ARGINSA) [MIM:207900]: An autosomal recessive disorder of the urea cycle. The disease is characterized by mental and physical retardation, liver enlargement, skin lesions, dry and brittle hair showing trichorrhexis nodosa microscopically and fluorescing red, convulsions, and episodic unconsciousness. {ECO:0000269|PubMed:11747432, ECO:0000269|PubMed:11747433, ECO:0000269|PubMed:12408190, ECO:0000269|PubMed:1705937, ECO:0000269|PubMed:17326097, ECO:0000269|PubMed:19703900, ECO:0000269|PubMed:22081021, ECO:0000269|PubMed:2263616, ECO:0000269|PubMed:24166829, ECO:0000269|PubMed:9045711}. The disease is caused by variants affecting the gene represented in this entry. The phenotype heterogeneity among patients is associated with interallelic complementation resulting in either complete loss of activity or partial regeneration of functional active sites in the heterotetrameric mutant protein. {ECO:0000269|PubMed:11747433}. Drugs (DrugBank ID) DB03814; DB00125; DB02267 Interacts with P04424; Q9BTE3-2; Q96HA8; O75382 EC number 4.3.2.1 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Amino-acid biosynthesis; Arginine biosynthesis; Disease variant; Lyase; Proteomics identification; Reference proteome; Urea cycle Protein physicochemical properties Chain ID A,B Molecular weight (Da) 51364.1 Length 459 Aromaticity 0.08 Instability index 35.82 Isoelectric point 6.66 Charge (pH=7) -1.25 2D Binding mode Binding energy (Kcal/mol) -5.36 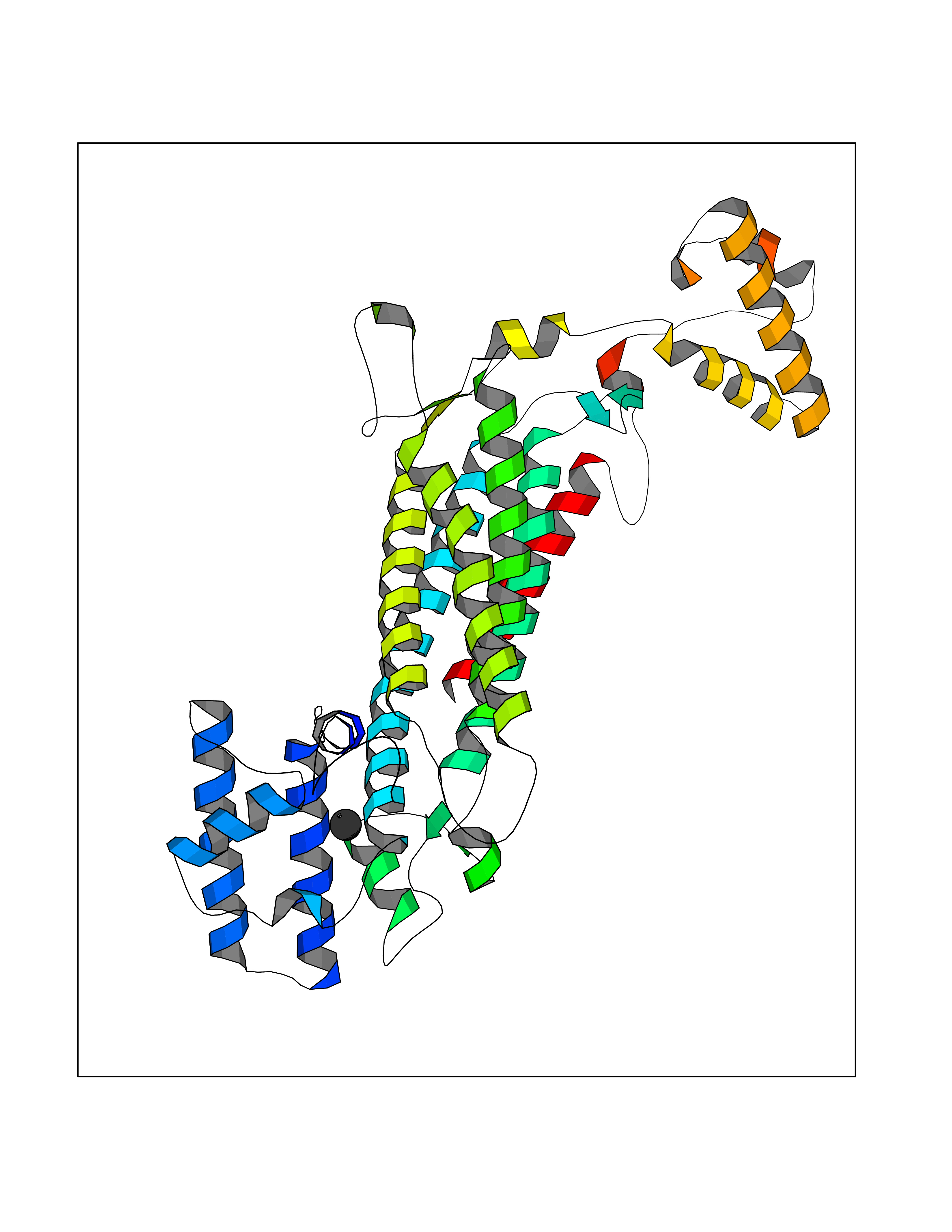 Molscript Map 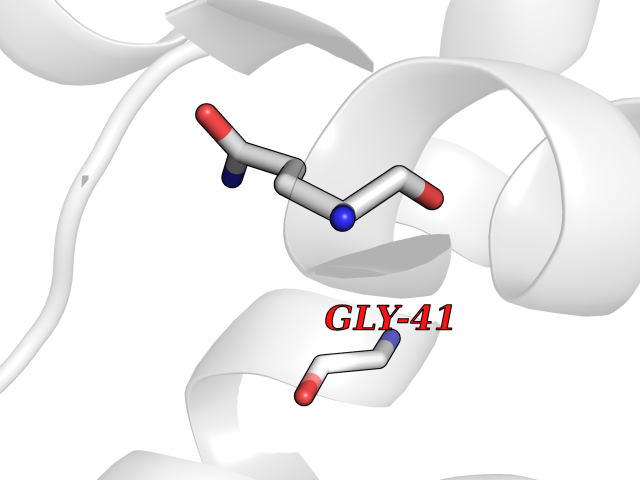 Pymol Map 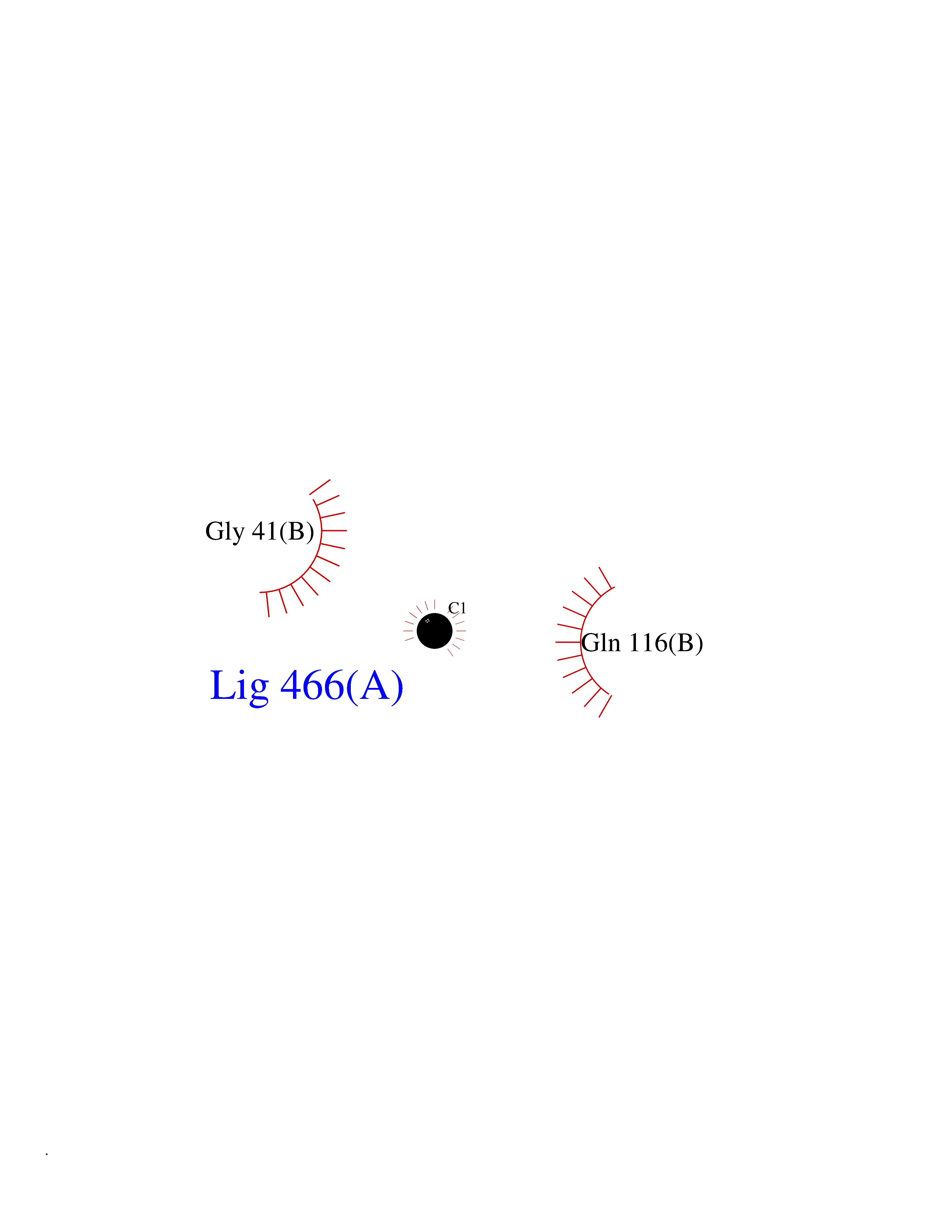 Ligplot Map 3D Binding mode Sequence GKLWGGRFVGAVDPIMEKFNASIAYDRHLWEVDVQGSKAYSRGLEKAGLLTKAEMDQILHGLDKVAEEWAQGTFKLNSNDEDIHTANERRLKELIGATAGKLHTGRSRNDQVVTDLRLWMRQTCSTLSGLLWELIRTMVDRAEAERDVLFPGYTHLQRAQPIRWSHWILSHAVALTRDSERLLEVRKRINVLPLGSGAIAGNPLGVDRELLRAELNFGAITLNSMDATSERDFVAEFLFWRSLCMTHLSRMAEDLILYCTKEFSFVQLSDAYSTGSSLMPRKKNPDSLELIRSKAGRVFGRCAGLLMTLKGLPSTYNKDLQEDKEAVFEVSDTMSAVLQVATGVISTLQIHQENMGQALSPDMLATDLAYYLVRKGMPFRQAHEASGKAVFMAETKGVALNQLSLQELQTISPLFSGDVICVWDYRHSVEQYGALGGTARSSVDWQIRQVRALLQAQQA Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Adenosine deaminase (EC 3.5.4.4) (S-methyl-5'-thioadenosine deaminase) (EC 3.5.4.31) | 3EWD | 3.93 | |
Target general information Gen name ADA Organism Plasmodium vivax (strain Salvador I) Uniprot ID TTD ID NA Synonyms PVX_111245 Protein family Metallo-dependent hydrolases superfamily, Adenosine and AMP deaminases family Biochemical class NA Function Catalyzes the hydrolytic deamination of adenosine to produce inosine (PubMed:19728741). Unlike mammalian adenosine deaminases, also catalyzes the deamination of 5'-methylthioadenosine (MTA), a by-product of polyamine biosynthesis, to produce 5'-methylthioinosine (MTI) (PubMed:19728741). Plays an essential role in the purine salvage pathway which allows the parasite to use host cell purines for the synthesis of nucleic acids (PubMed:19728741). {ECO:0000269|PubMed:19728741}." Related diseases NA Drugs (DrugBank ID) NA Interacts with NA EC number 3.5.4.31; 3.5.4.4 Uniprot keywords 3D-structure; Hydrolase; Metal-binding; Purine salvage; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 41950.8 Length 364 Aromaticity 0.11 Instability index 27.19 Isoelectric point 5.49 Charge (pH=7) -13.73 2D Binding mode Binding energy (Kcal/mol) -5.36  Molscript Map  Pymol Map 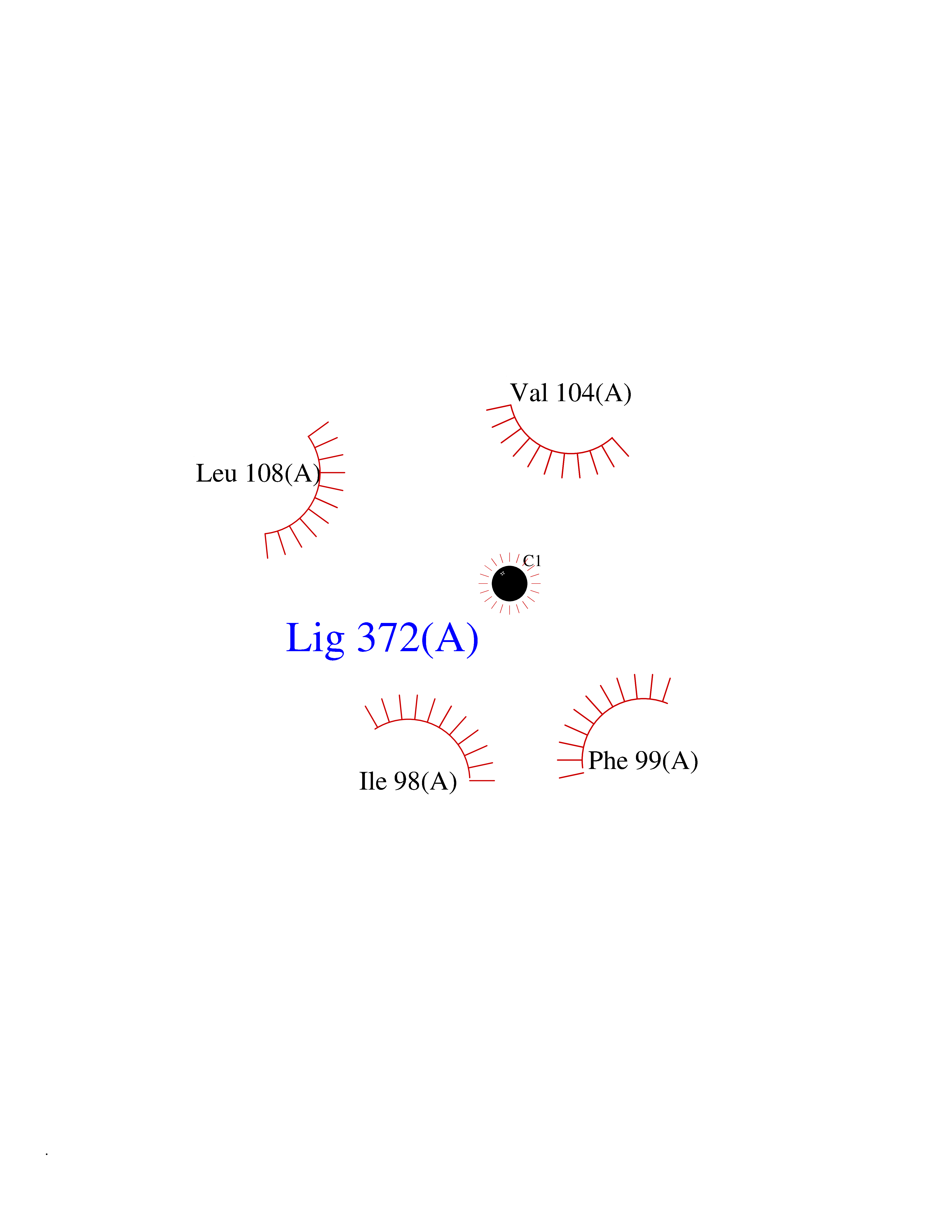 Ligplot Map 3D Binding mode Sequence PIDFLKKEELKNIDLSQMSKKERYKIWKRIPKCELHCHLDLCFSADFFVSCIRKYNLQPNLSDEEVLDYYLFAKGGKSLGEFVEKAIKVADIFHDYEVIEDLAKHAVFNKYKEGVVLMEFRYSPTFVAFKYNLDIELIHQAIVKGIKEVVELLDHKIHVALMCIGTGHEAANIKASADFCLKHKADFVGFDHGGHEVDLKEYKEIFDYVRESGVPLSVHAGEDVTLPNLNTLYSAIQVLKVERIGHGIRVAESQELIDMVKEKNILLEVCPISNVLLKNAKSMDTHPIRQLYDAGVKVSVNSDDPGMFLTNINDDYEELYTHLNFTLEDFMKMNEWALEKSFMDSNIKDKIKNLYFKGEFEAYV Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | 72 kDa type IV collagenase | 3AYU | 3.93 | |
Target general information Gen name MMP2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CLG4A Protein family Peptidase M10A family Biochemical class Hydrolase / hydrolase inhibitor Function Metalloendopeptidase activity.Metallopeptidase activity.Serine-type endopeptidase activity.Zinc ion binding. Related diseases Multicentric osteolysis, nodulosis, and arthropathy (MONA) [MIM:259600]: An autosomal recessive syndrome characterized by severe multicentric osteolysis with predominant involvement of the hands and feet. Additional features include coarse face, corneal opacities, patches of thickened, hyperpigmented skin, hypertrichosis and gum hypertrophy. {ECO:0000269|PubMed:11431697, ECO:0000269|PubMed:15691365, ECO:0000269|PubMed:16542393}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01197; DB06423; DB04866; DB00786; DB05387; DB12843; DB01630 Interacts with P05067; PRO_0000000092 [P05067]; PRO_0000000093 [P05067]; P20908; Q8NBP7; Q04864-2; Q8IX30; P16035 EC number 3.4.24.24 Uniprot keywords 3D-structure; Alternative splicing; Angiogenesis; Autocatalytic cleavage; Calcium; Collagen degradation; Cytoplasm; Direct protein sequencing; Disease variant; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Membrane; Metal-binding; Metalloprotease; Mitochondrion; Nucleus; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 19623.6 Length 176 Aromaticity 0.14 Instability index 15.48 Isoelectric point 4.84 Charge (pH=7) -10.61 2D Binding mode Binding energy (Kcal/mol) -5.35 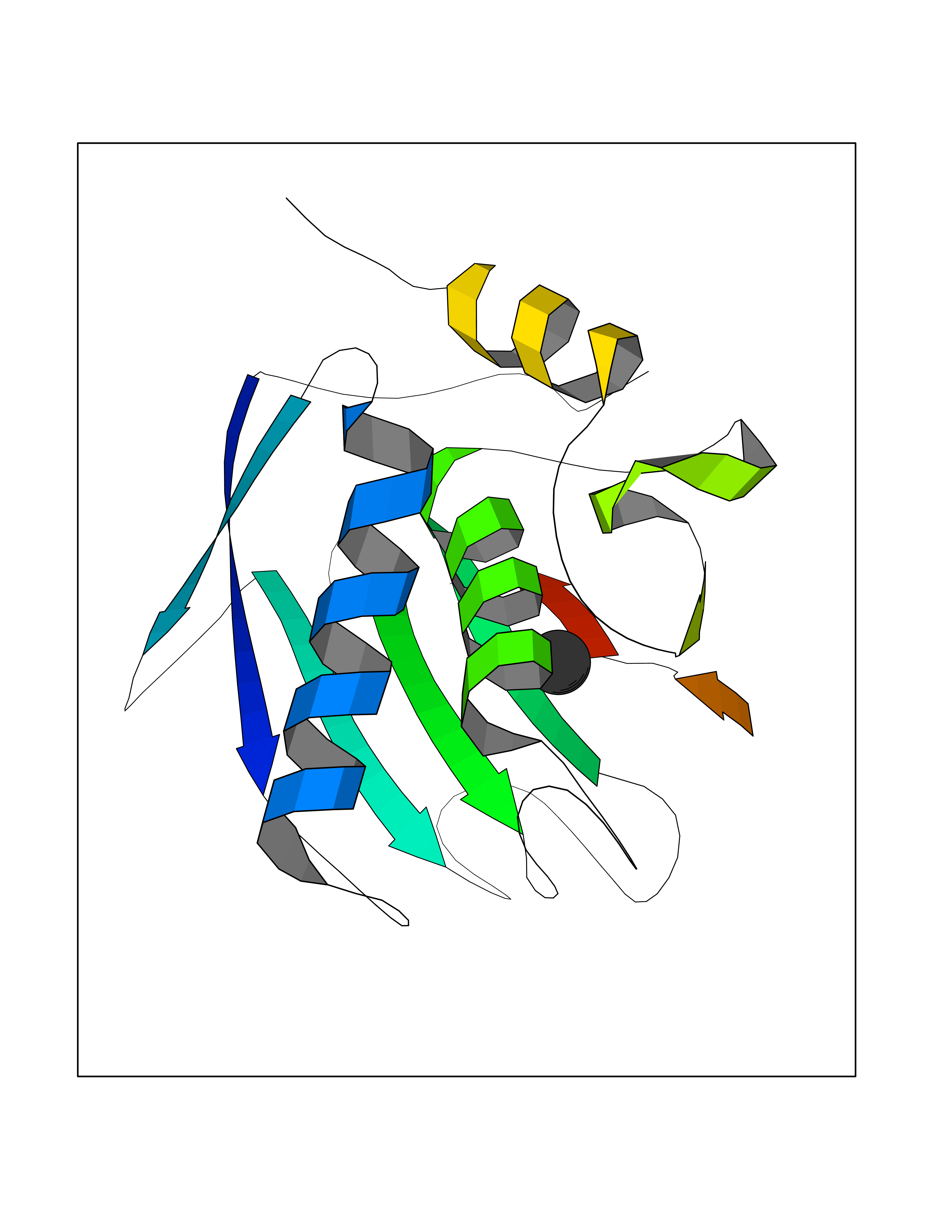 Molscript Map 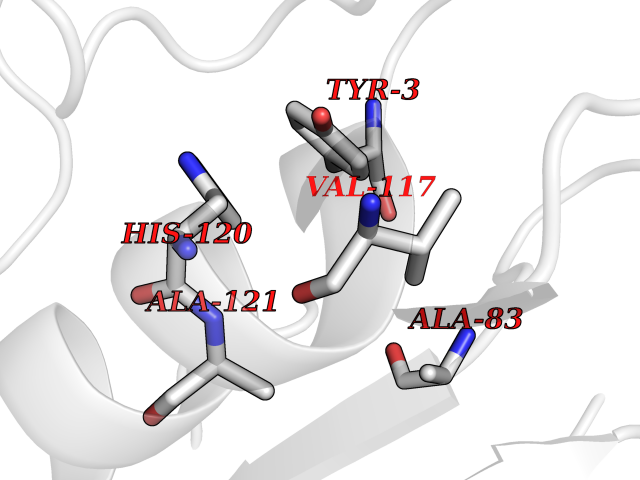 Pymol Map 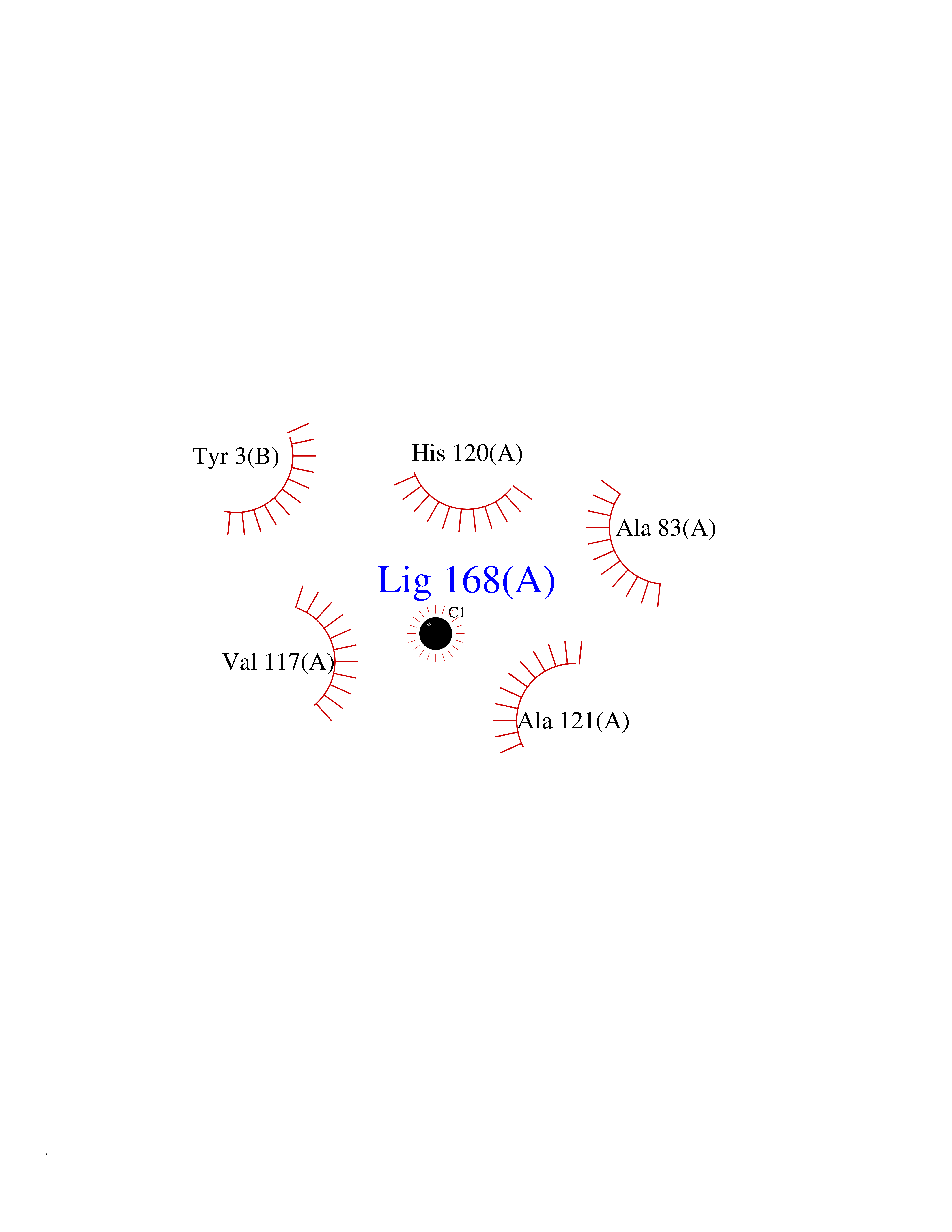 Ligplot Map 3D Binding mode Sequence YNFFPRKPKWDKNQITYRIIGYTPDLDPETVDDAFARAFQVWSDVTPLRFSRIHDGEADIMINFGRWEHGDGYPFDGKDGLLAHAFAPGTGVGGDSHFDDDELWTLGKGVGYSLFLVAAHAFGHAMGLEHSQDPGALMAPIYTYTKNFRLSQDDIKGIQELYGASPISYGNDALMP Hydrogen bonds contact Hydrophobic contact | ||||