Job Results:
Ligand
Structure
Job ID
9d5454904e548acd822dcc3f8261b50f
Job name
NA
Time
2026-02-27 11:53:48
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Kynurenine oxoglutarate transaminase II (AADAT) | 2XH1 | 3.94 | |
Target general information Gen name AADAT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Kynurenineoxoglutarate transaminase II; Kynurenineoxoglutarate transaminase 2; Kynurenineoxoglutarate aminotransferase II; Kynurenine/alphaaminoadipate aminotransferase, mitochondrial; Kynurenine amin Protein family Class-I pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transaminase Function Transaminase with broad substrate specificity. Has transaminase activity towards aminoadipate, kynurenine, methionine and glutamate. Shows activity also towards tryptophan, aspartate and hydroxykynurenine. Accepts a variety of oxo-acids as amino- group acceptors, with a preference for 2-oxoglutarate, 2- oxocaproic acid, phenylpyruvate and alpha-oxo-gamma-methiol butyric acid. Can also use glyoxylate as amino-group acceptor (in vitro). Related diseases Epilepsy, nocturnal frontal lobe, 3 (ENFL3) [MIM:605375]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:11062464, ECO:0000269|PubMed:11104662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00142; DB00114 Interacts with NA EC number EC 2.6.1.39 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminotransferase; Mitochondrion; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase; Transit peptide Protein physicochemical properties Chain ID A,B Molecular weight (Da) 91792.6 Length 824 Aromaticity 0.09 Instability index 47.71 Isoelectric point 5.96 Charge (pH=7) -8.73 2D Binding mode Binding energy (Kcal/mol) -5.37  Molscript Map 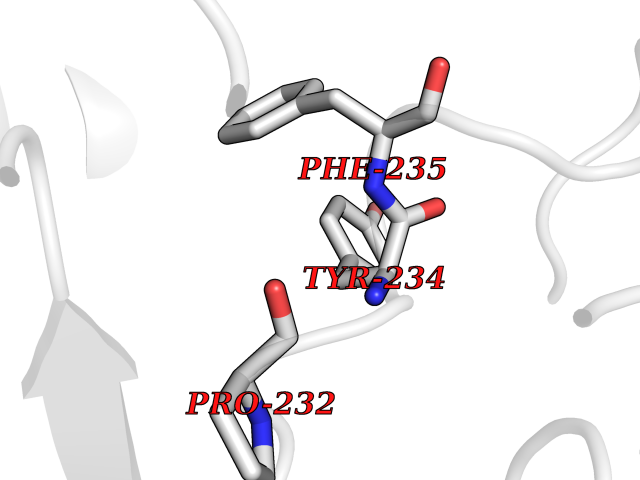 Pymol Map 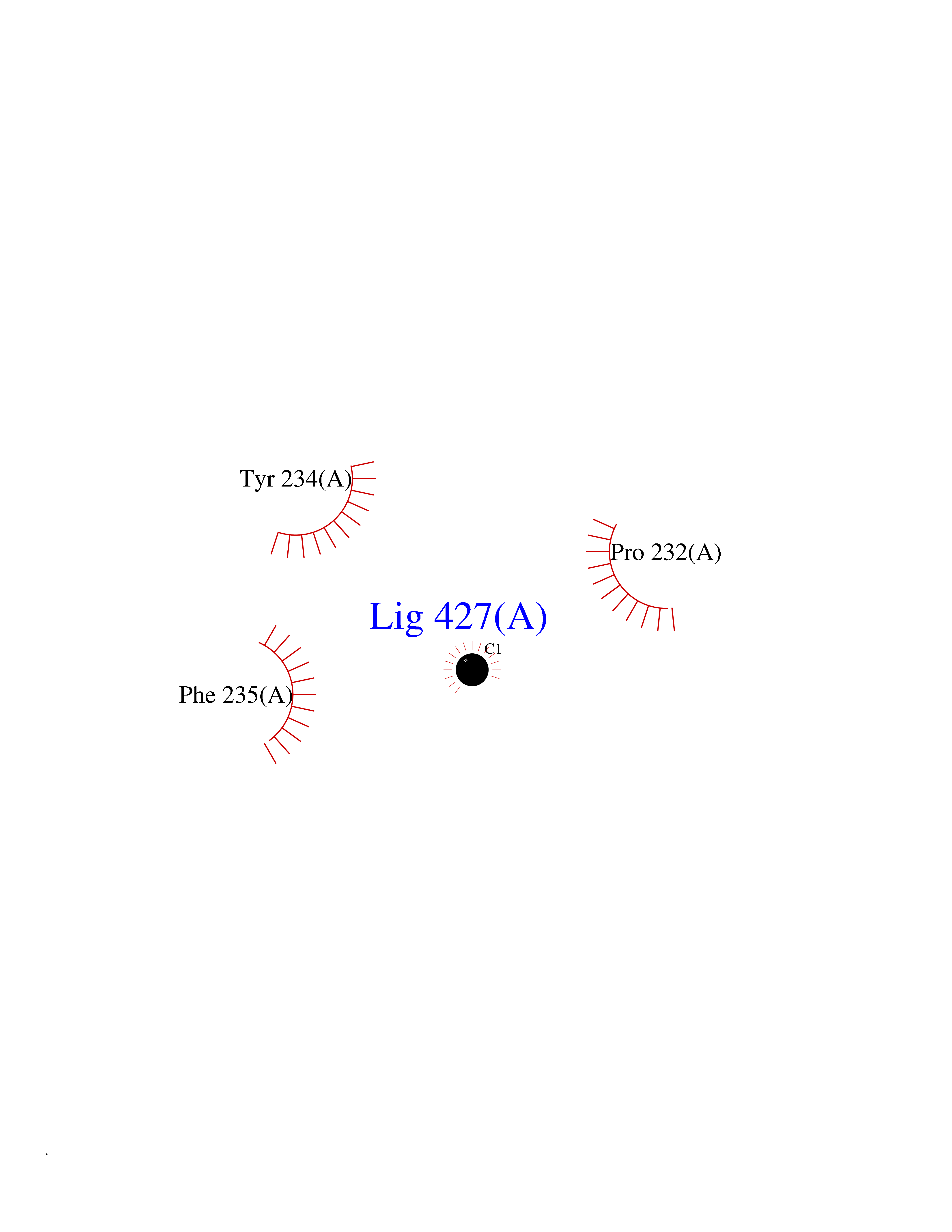 Ligplot Map 3D Binding mode Sequence ENYARFITAASAARNPSPIMISLAGGLPNPNMFPFKTAVITVENGKTIQFGEEMMKRALQYSPSAGIPELLSWLKQLQIKLHNPPTIHYPPSQGQMDLCVTSGSQQGLCKVFEMIINPGDNVLLDEPAYSGTLQSLHPLGCNIINVASDESGIVPDSLRDILSRWKPEDAKNPQKNTPKFLYTVPNGNNPTGNSLTSERKKEIYELARKYDFLIIEDDPYYFLQFNKFRVPTFLSMDVDGRVIRADSFSKIISSGLRIGFLTGPKPLIERVILHIQVSTLHPSTFNQLMISQLLHEWGEEGFMAHVDRVIDFYSNQKDAILAAADKWLTGLAEWHVPAAGMFLWIKVKGINDVKELIEEKAVKMGVLMLPGNAFYVDSSAPSPYLRASFSSASPEQMDVAFQVLAQLIKESLENYARFITAASAARNPSPIMISLAGGLPNPNMFPFKTAVITVENGKTIQFGEEMMKRALQYSPSAGIPELLSWLKQLQIKLHNPPTIHYPPSQGQMDLCVTSGSQQGLCKVFEMIINPGDNVLLDEPAYSGTLQSLHPLGCNIINVASDESGIVPDSLRDILSRWKPEDAKNPQKNTPKFLYTVPNGNNPTGNSLTSERKKEIYELARKYDFLIIEDDPYYFLQFNKFRVPTFLSMDVDGRVIRADSFSKIISSGLRIGFLTGPKPLIERVILHIQVSTLHPSTFNQLMISQLLHEWGEEGFMAHVDRVIDFYSNQKDAILAAADKWLTGLAEWHVPAAGMFLWIKVKGINDVKELIEEKAVKMGVLMLPGNAFYVDSSAPSPYLRASFSSASPEQMDVAFQVLAQLIKESL Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Neuronal acetylcholine receptor beta-2 (CHRNB2) | 6CNJ | 3.94 | |
Target general information Gen name CHRNB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nicotinic acetylcholine receptor beta2; Nicotinic acetylcholine receptor beta 2-subunit protein; CHRNB2; Beta-2 nAChR; Alpha-4/beta-2 nicotinic receptor Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Beta-2/CHRNB2 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane permeable to sodiun ions. Related diseases Epilepsy, nocturnal frontal lobe, 3 (ENFL3) [MIM:605375]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:11062464, ECO:0000269|PubMed:11104662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00572; DB00237; DB00565; DB09028; DB01245; DB00514; DB07720; DB00898; DB00753; DB00657; DB00333; DB00184; DB00981; DB05458; DB05855; DB05740; DB00747; DB00202; DB01273 Interacts with P43681-1; P30532 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 84601.2 Length 728 Aromaticity 0.13 Instability index 39.72 Isoelectric point 5.86 Charge (pH=7) -9.84 2D Binding mode Binding energy (Kcal/mol) -5.37  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ETRAHAEERLLKKLFSGYNKWSRPVANISDVVLVRFGLSIAQLIDVDEKNQMMTTNVWVKQEWHDYKLRWDPADYENVTSIRIPSELIWRPDIVLYNNADGDFAVTHLTKAHLFHDGRVQWTPPAIYKSSCSIDVTFFPFDQQNCTMKFGSWTYDKAKIDLVNMHSRVDQLDFWESGEWVIVDAVGTYNTRKYECCAEIYPDITYAFVIRRLPLFYTINLIIPCLLISCLTVLVFYLPSECGEKITLCISVLLSLTVFLLLITEIIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHHRSPRTHTMPTWVRRVFLDIVPRLLLMKRFERSVKEDWKYVAMVIDRIFLWMFIIVCLLGTVGLFLPPWDTEERLVEHLLDPSRYNKLIRPATNGSELVTVQLMVSLAQLISVHEREQIMTTNVWLTQEWEDYRLTWKPEEFDNMKKVRLPSKHIWLPDVVLYNNADGMYEVSFYSNAVVSYDGSIFWLPPAIYKSACKIEVKHFPFDQQNCTMKFRSWTYDRTEIDLVLKSEVASLDDFTPSGEWDIVALPGRRNENPDDSTYVDITYDFIIRRKPLFYTINLIIPCVLITSLAILVFYLPSDCGEKMTLCISVLLALTVFLLLISKIVPPTSLDVPLVGKYLMFTMVLVTFSIVTSVCVLNVHHRSPTTHTMAPWVKVVFLEKLPALLFMQQSVSEDWKYVAMVIDRLFLWIFVFVCVFGTIGMF Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Hydroxymethylglutaryl-CoA synthase 2 (HMGCS2) | 2WYA | 3.94 | |
Target general information Gen name HMGCS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HMGCS2; HMG-CoAsynthase; 3-hydroxy-3-methylglutaryl coenzyme A synthase 2 Protein family Thiolase-like superfamily, HMG-CoA synthase family Biochemical class Acyltransferase Function This enzyme condenses acetyl-CoA with acetoacetyl-CoA to form HMG-CoA, which is the substrate for HMG-CoA reductase. Related diseases 3-hydroxy-3-methylglutaryl-CoA synthase-2 deficiency (HMGCS2D) [MIM:605911]: A metabolic disorder characterized by severe hypoketotic hypoglycemia, encephalopathy, and hepatomegaly. {ECO:0000269|PubMed:11228257, ECO:0000269|PubMed:11479731, ECO:0000269|PubMed:12647205, ECO:0000269|PubMed:16601895, ECO:0000269|PubMed:23751782, ECO:0000269|PubMed:25511235, ECO:0000269|PubMed:29597274}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 2.3.3.10 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Disease variant; Lipid biosynthesis; Lipid metabolism; Mitochondrion; Phosphoprotein; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transferase; Transit peptide Protein physicochemical properties Chain ID A,D Molecular weight (Da) 102499 Length 920 Aromaticity 0.1 Instability index 33.48 Isoelectric point 6.72 Charge (pH=7) -1.41 2D Binding mode Binding energy (Kcal/mol) -5.37  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SMPKDVGILALEVYFPAQYVDQTDLEKYNNVEAGKYTVGLGQTRMGFCSVQEDINSLCLTVVQRLMERIQLPWDSVGRLEVGTETIIDKSKAVKTVLMELFQDSGNTDIEGIDTTNACYGGTASLFNAANWMESSSWDGRYAMVVCGDIAVYPSGNARPTGGAGAVAMLIGPKAPLALERGLRGTHMENVYDFYKPNLASEYPIVDGKLSIQCYLRALDRCYTSYRKKIQNQWKQAGSDRPFTLDDLQYMIFHTPFCKMVQKSLARLMFNDFLSASSDTQTSLYKGLEAFGGLKLEDTYTNKDLDKALLKASQDMFDKKTKASLYLSTHNGNMYTSSLYGCLASLLSHHSAQELAGSRIGAFSYGSGLAASFFSFRVSQDAAPGSPLDKLVSSTSDLPKRLASRKCVSPEEFTEIMNQREQFYHKVNFSPPGDTNSLFPGTWYLERVDEQHRRKYARRPVSMPKDVGILALEVYFPAQYVDQTDLEKYNNVEAGKYTVGLGQTRMGFCSVQEDINSLCLTVVQRLMERIQLPWDSVGRLEVGTETIIDKSKAVKTVLMELFQDSGNTDIEGIDTTNACYGGTASLFNAANWMESSSWDGRYAMVVCGDIAVYPSGNARPTGGAGAVAMLIGPKAPLALERGLRGTHMENVYDFYKPNLASEYPIVDGKLSIQCYLRALDRCYTSYRKKIQNQWKQAGSDRPFTLDDLQYMIFHTPFCKMVQKSLARLMFNDFLSASSDTQTSLYKGLEAFGGLKLEDTYTNKDLDKALLKASQDMFDKKTKASLYLSTHNGNMYTSSLYGCLASLLSHHSAQELAGSRIGAFSYGSGLAASFFSFRVSQDAAPGSPLDKLVSSTSDLPKRLASRKCVSPEEFTEIMNQREQFYHKVNFSPPGDTNSLFPGTWYLERVDEQHRRKYARRPV Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Two pore calcium channel protein 2 (TPC2) | 6NQ2 | 3.94 | |
Target general information Gen name TPCN2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Voltage-dependent calcium channel protein TPC2; TPC2 Protein family Calcium channel alpha-1 subunit (TC 1.A.1.11) family, Two pore calcium channel subfamily Biochemical class NA Function Nicotinic acid adenine dinucleotide phosphate (NAADP) receptor that may function as one of the major voltage-gated Ca(2+) channels (VDCC) across the lysosomal membrane. May be involved in smooth muscle contraction. Related diseases Spermatogenic failure 5 (SPGF5) [MIM:243060]: An infertility disorder caused by spermatogenesis defects. Semen from affected men show close to 100% morphologically abnormal multiflagellar spermatozoa with low motility, oversized irregular heads, and abnormal midpiece and acrosome. {ECO:0000269|PubMed:17435757, ECO:0000269|PubMed:21733974}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O00165; P42345; Q9ULQ1; Q8NHX9 EC number NA Uniprot keywords 3D-structure; Calcium; Calcium channel; Calcium transport; Endosome; Glycoprotein; Ion channel; Ion transport; Lysosome; Membrane; Proteomics identification; Reference proteome; Repeat; Transmembrane; Transmembrane helix; Transport; Voltage-gated channel Protein physicochemical properties Chain ID A Molecular weight (Da) 71128.5 Length 621 Aromaticity 0.14 Instability index 35.3 Isoelectric point 8.84 Charge (pH=7) 8.43 2D Binding mode Binding energy (Kcal/mol) -5.37  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence AARWDLCIDQAVVFIEDAIQYRSINHRVDASSMWLYRRYYSNVCQRTLSFTIFLILFLAFIETPSSLTSTADVRYRAAPWEPPCGLTESVEVLCLLVFAADLSVKGYLFGWAHFQKNLWLLGYLVVLVVSLVDWTVSLSLVCHEPLRIRRLLRPFFLLQNSSMMKKTLKCIRWSLPEMASVGLLLAIHLCLFTMFGMLLFAGLTYFQNLPESLTSLLVLLTTANNPDVMIPAYSKNRAYAIFFIVFTVIGSLFLMNLLTAIIYSQFRGYLMKSLQTSLFRRRLGTRAAFEVLSSMVGAVGVKPQNLLQVLQKVQLDSSHKQAMMEKVRSYGSVLLSAEEFQKLFNELDRSVVKEHPPRPEYQSPFLQSAQFLFGHYYFDYLGNLIALANLVSICVFLVLDADVLPAERDDFILGILNCVFIVYYLLEMLLKVFALGLRGYLSYPSNVFDGLLTVVLLVLEISTLAVYRLSLWDMTRMLNMLIVFRFLRIIPSMKPMAVVASTVLGLVQNMRAFGGILVVVYYVFAIIGINLFRGVIVALSAPCGSFEQLEYWANNFDDFAAALVTLWNLMVVNNWQVFLDAYRRYSGPWSKIYFVLWWLVSSVIWVNLFLALILENFLHKW Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Melatonin receptor type 1A (MTNR1A) | 7DB6 | 3.94 | |
Target general information Gen name MTNR1A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mel1a receptor; Mel1AR; Mel-1A-R Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Likely to mediate the reproductive and circadian actions of melatonin. The activity of this receptor is mediated by pertussis toxin sensitive G proteins that inhibit adenylate cyclase activity. High affinity receptor for melatonin. Related diseases Spermatogenic failure 5 (SPGF5) [MIM:243060]: An infertility disorder caused by spermatogenesis defects. Semen from affected men show close to 100% morphologically abnormal multiflagellar spermatozoa with low motility, oversized irregular heads, and abnormal midpiece and acrosome. {ECO:0000269|PubMed:17435757, ECO:0000269|PubMed:21733974}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06594; DB01065; DB00980; DB02709; DB09071 Interacts with P27797; A8MQ03; Q8IUG1; P49286; O76081; P57088 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID D Molecular weight (Da) 31301 Length 276 Aromaticity 0.15 Instability index 37.33 Isoelectric point 9.22 Charge (pH=7) 9.92 2D Binding mode Binding energy (Kcal/mol) -5.38  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence RPSWLASALACVLIFTIVVDILGNLLVILSVYRNKKLRNAGNIFVVSLAVADLVVAIYPYPLVLMSIFNNGWNLGYLHCQVSGFLMGLSVIGSIFNITGIAINRYCYICHSLKYDKLYSSKNSLCYVLLIWLLTLAAVLPNLRAGTLQYDPRIYSCTFAQSVSSAYTIAVVVFHFLVPMIIVIFCYLRIWILVLQVRQRVPQDFRNFVTMFVVFVLFAICWAPLNFIGLAVASDPASMVPRIPEWLFVASYYMAYFNSCLNAIIYGLLNQNFRKEY Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Interferon gamma | 1FYH | 3.93 | |
Target general information Gen name IFNG Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Type II (or gamma) interferon family Biochemical class Immune system Function Cytokine activity.Interferon-gamma receptor binding. Related diseases Aplastic anemia (AA) [MIM:609135]: A form of anemia in which the bone marrow fails to produce adequate numbers of peripheral blood elements. It is characterized by peripheral pancytopenia and marrow hypoplasia. {ECO:0000269|PubMed:15327519}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Immunodeficiency 69 (IMD69) [MIM:618963]: A form of Mendelian susceptibility to mycobacterial disease, a rare condition caused by impairment of interferon-gamma mediated immunity. It is characterized by predisposition to illness caused by moderately virulent mycobacterial species, such as Bacillus Calmette-Guerin (BCG) vaccine, environmental non-tuberculous mycobacteria, and by the more virulent Mycobacterium tuberculosis. Other microorganisms rarely cause severe clinical disease in individuals with susceptibility to mycobacterial infections. Clinical outcome severity depends on the degree of impairment of interferon-gamma mediated immunity. IMD69 is an autosomal recessive disorder manifesting with fever, hepatosplenomegaly, leukocytosis, and thrombocytosis during the acute infection. {ECO:0000269|PubMed:32163377}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14724; DB05111; DB10770; DB10772; DB01296; DB01250; DB05110 Interacts with P15260; Q66793 EC number NA Uniprot keywords 3D-structure; Antiviral defense; Cleavage on pair of basic residues; Cytokine; Direct protein sequencing; Glycoprotein; Growth regulation; Pharmaceutical; Proteomics identification; Pyrrolidone carboxylic acid; Reference proteome; Secreted; Signal Protein physicochemical properties Chain ID A,D Molecular weight (Da) 28390 Length 242 Aromaticity 0.12 Instability index 18.95 Isoelectric point 8.42 Charge (pH=7) 1.71 2D Binding mode Binding energy (Kcal/mol) -5.36 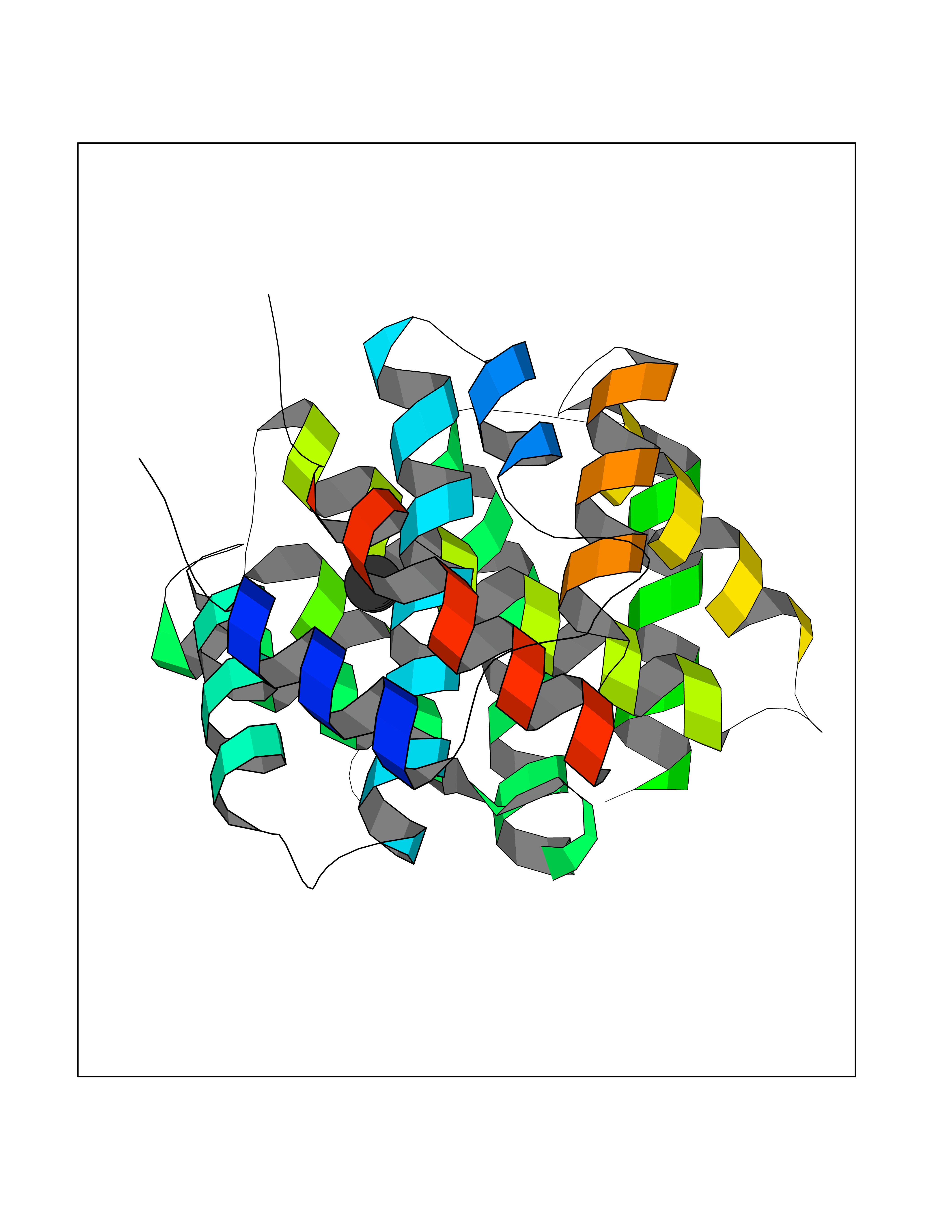 Molscript Map 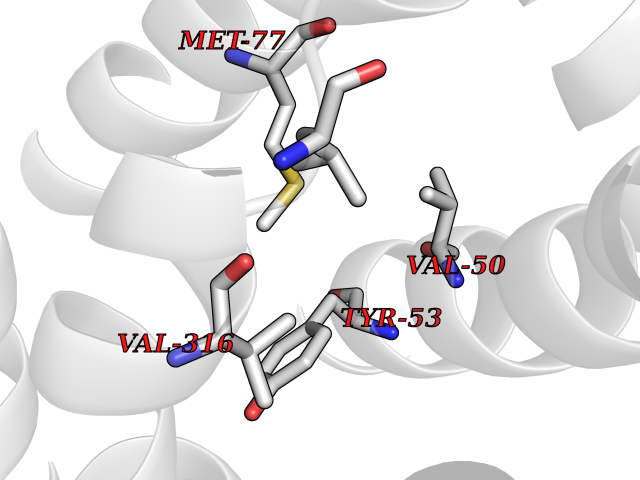 Pymol Map 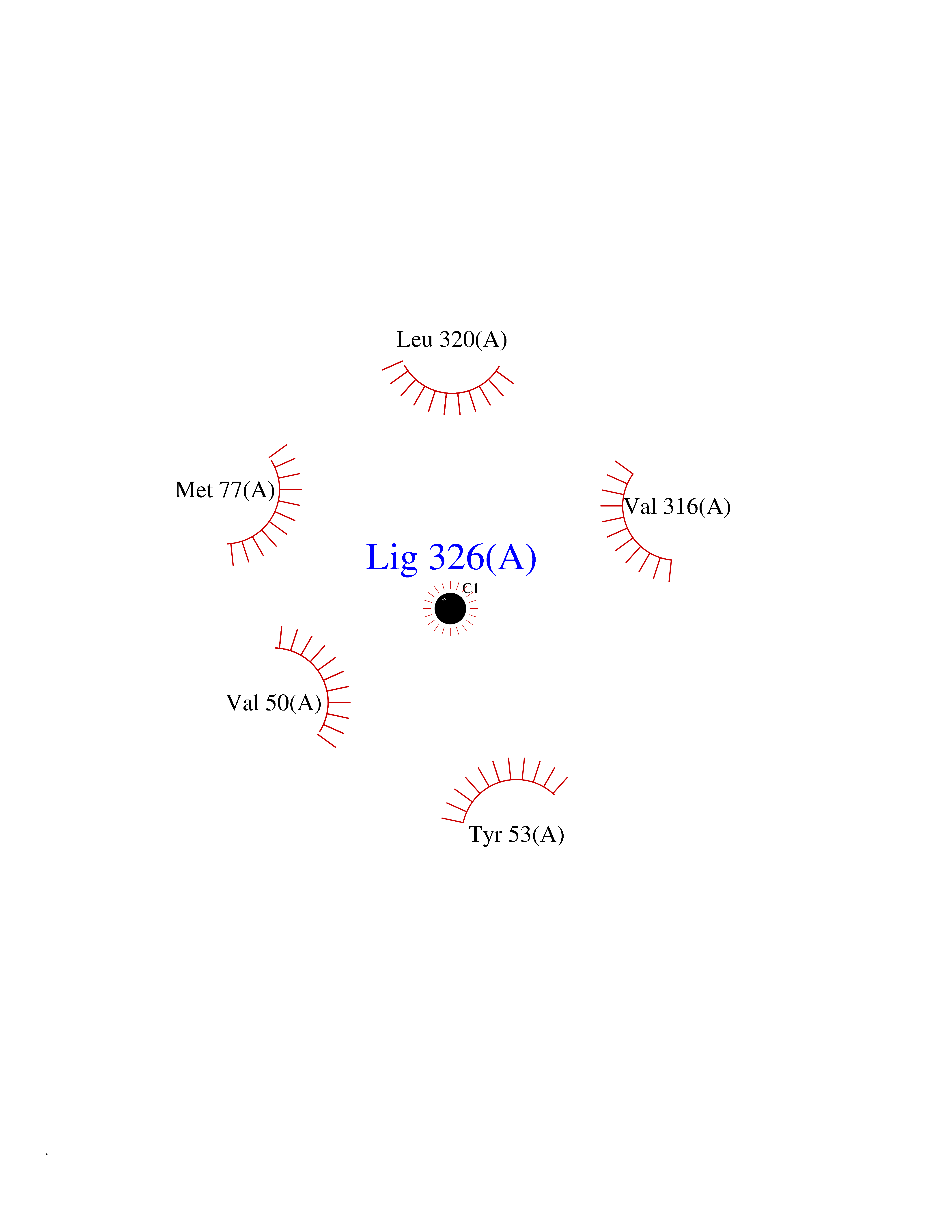 Ligplot Map 3D Binding mode Sequence MQDPYVKEAENLKKYFNAGHSDVADNGTLFLGILKNWKEESDRKIMQSQIVSFYFKLFKNFKDDQSIQKSVETIKEDMNVKFFNSNKKKRDDFEKLTNYSVTDLNVQRKAIDELIQVMAELGANVSGEFVKEAENLKKYFNDNGTLFLGILKNWKEESDRKIMQSQIVSFYFKLFKNFKDDQSIQKSVETIKEDMNVKFFNSNKKKRDDFEKLTNYSVTDLNVQRKAIHELIQVMAELSPAA Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | DNA polymerase alpha catalytic p180 (POLA1) | 4QCL | 3.93 | |
Target general information Gen name POLA1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms POLA; DNA polymerase alpha catalytic subunit p180; DNA polymerase alpha catalytic subunit Protein family DNA polymerase type-B family Biochemical class Kinase Function During the S phase of the cell cycle, the DNA polymerase alpha complex (composed of a catalytic subunit POLA1/p180, a regulatory subunit POLA2/p70 and two primase subunits PRIM1/p49 and PRIM2/p58) is recruited to DNA at the replicative forks via direct interactions with MCM10 and WDHD1. The primase subunit of the polymerase alpha complex initiates DNA synthesis by oligomerising short RNA primers on both leading and lagging strands. These primers are initially extended by the polymerase alpha catalytic subunit and subsequently transferred to polymerase delta and polymerase epsilon for processive synthesis on the lagging and leading strand, respectively. The reason this transfer occurs is because the polymerase alpha has limited processivity and lacks intrinsic 3' exonuclease activity for proofreading error, and therefore is not well suited for replicating long complexes. In the cytosol, responsible for a substantial proportion of the physiological concentration of cytosolic RNA:DNA hybrids, which are necessary to prevent spontaneous activation of type I interferon responses. Plays an essential role in the initiation of DNA replication. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00242; DB00631; DB01073; DB01280 Interacts with P27694; P10193; P03070 EC number EC 2.7.7.7 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Disease variant; DNA replication; DNA-binding; DNA-directed DNA polymerase; Dwarfism; Host-virus interaction; Intellectual disability; Metal-binding; Nucleotidyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 24505.6 Length 212 Aromaticity 0.09 Instability index 43.56 Isoelectric point 9.02 Charge (pH=7) 4.85 2D Binding mode Binding energy (Kcal/mol) -5.35 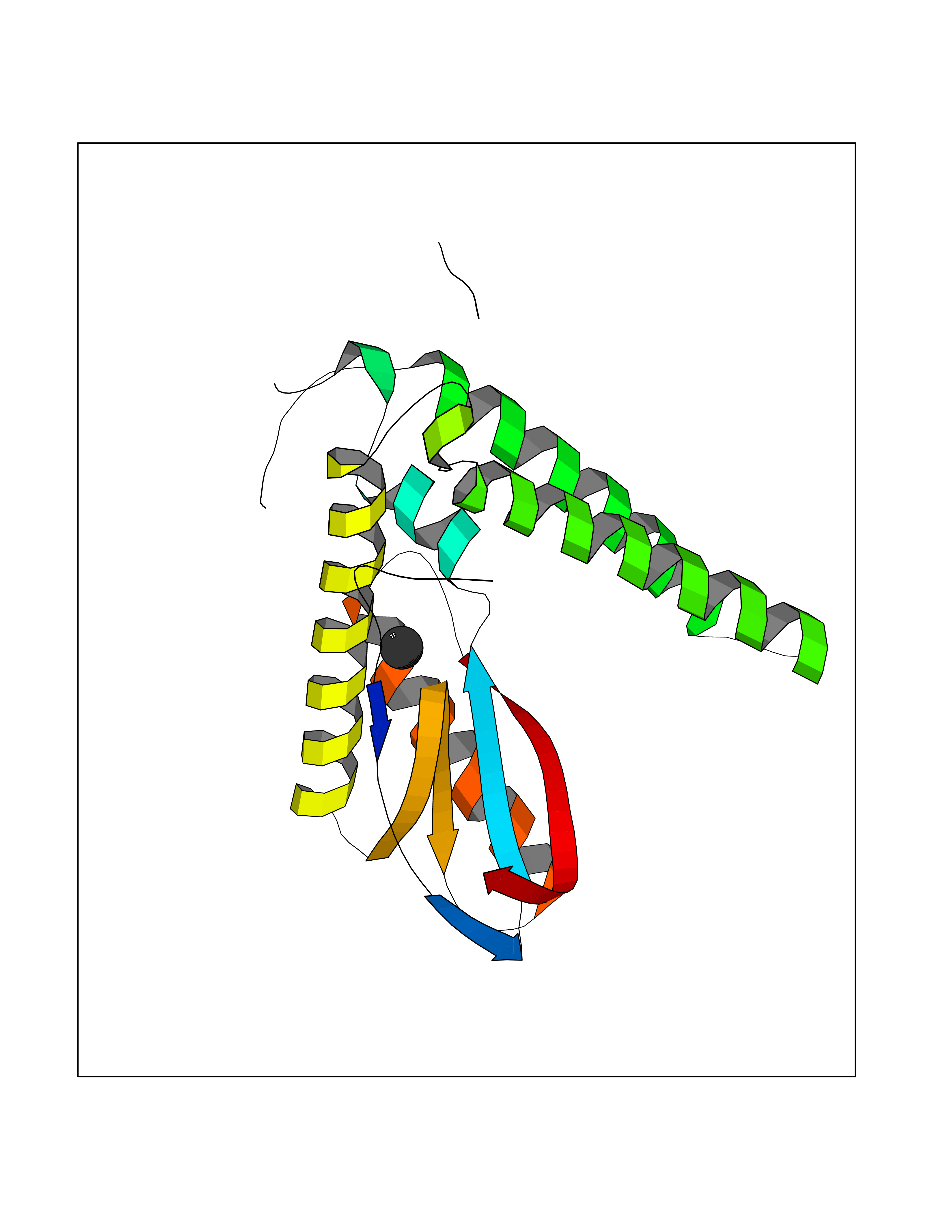 Molscript Map 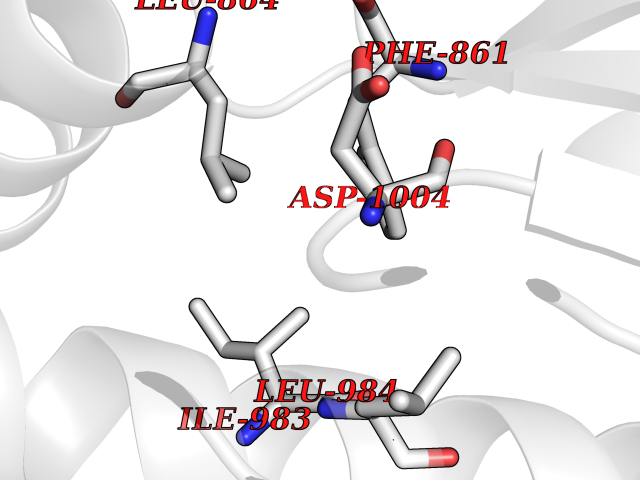 Pymol Map 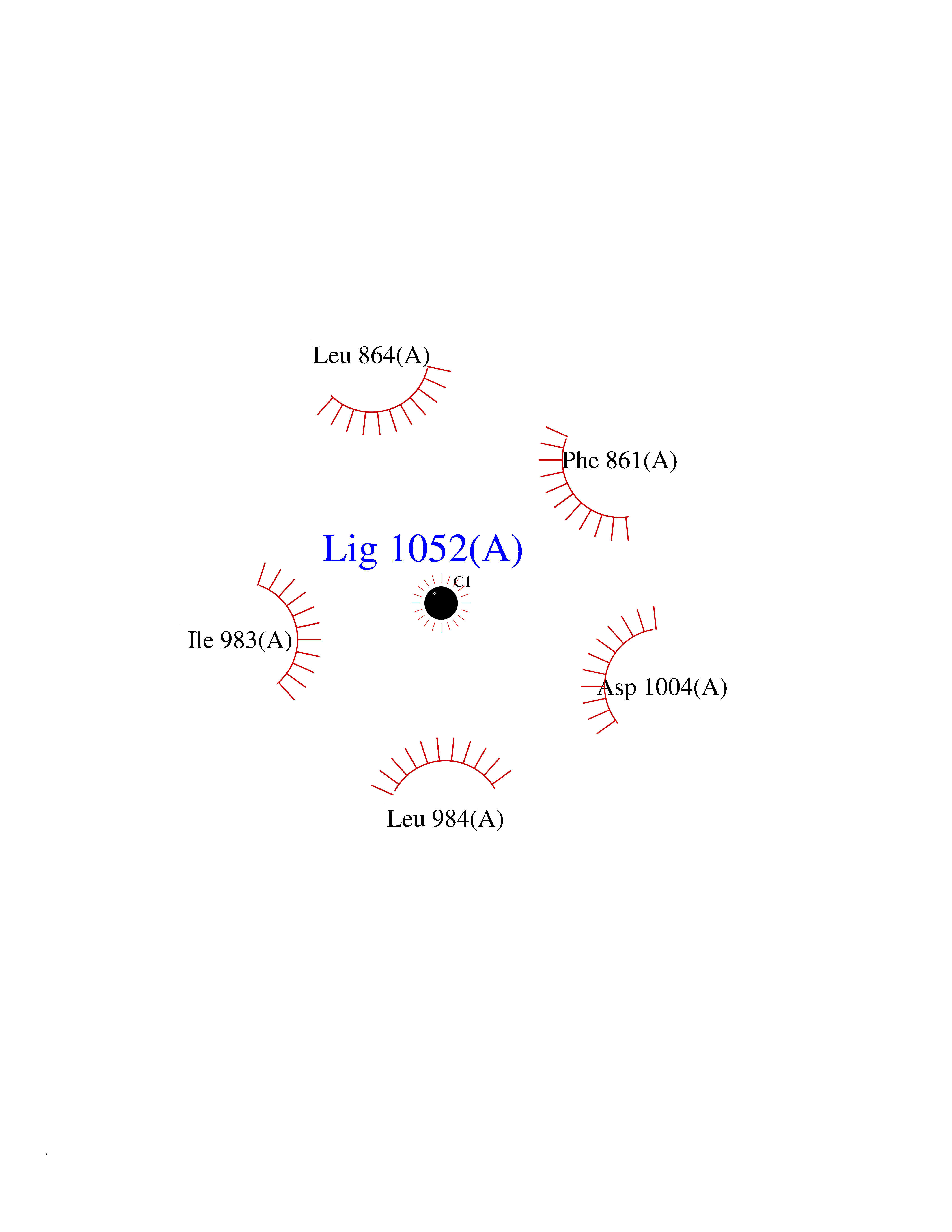 Ligplot Map 3D Binding mode Sequence YIVPDKQIRKKAAYAGGLVLDPKVGFYDKFILLLDFNSLYPSIIQEFNICFTTVQRVEQIPELPDPSLEMGILPREIRKLVERRKQVKQLMKQQDLNPDLILQYDIRQKALKLTANSMYGCLGFSYSRFYAKPLAALVTYKGREILMHTKEMVQKMNLEVIYGDTDSIMINTNSTNLEEVFKLGNKVKSEVNKLYKLLEIDIDGVFKSLLLL Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Bacterial Dihydropteroate synthetase (Bact folP) | 1AJ0 | 3.93 | |
Target general information Gen name Bact folP Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms folP; H2Pte synthase; Dihydropteroate synthase; Dihydropteroate pyrophosphorylase; DHPS Protein family DHPS family Biochemical class Alkyl aryl transferase Function Dhps catalyzes the formation of the immediate precursor of folic acid. It is implicated in resistance to sulfonamide. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14033; DB00634; DB01298; DB01581; DB06821; DB01582; DB00576; DB01015; DB00259; DB06729; DB00263 Interacts with NA EC number EC 2.5.1.15 Uniprot keywords 3D-structure; Direct protein sequencing; Folate biosynthesis; Magnesium; Metal-binding; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 30614.8 Length 282 Aromaticity 0.05 Instability index 32.55 Isoelectric point 5.68 Charge (pH=7) -8.5 2D Binding mode Binding energy (Kcal/mol) -5.35 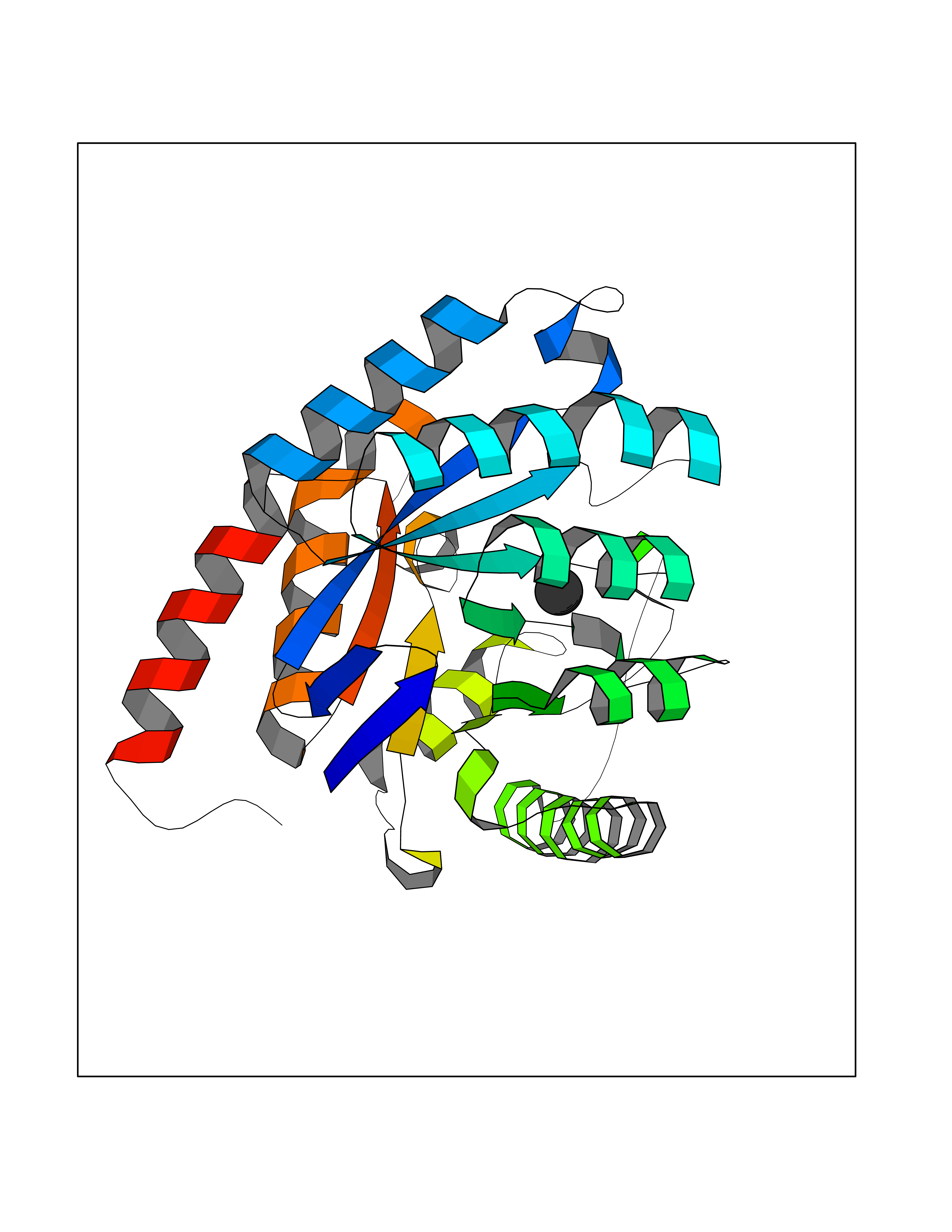 Molscript Map 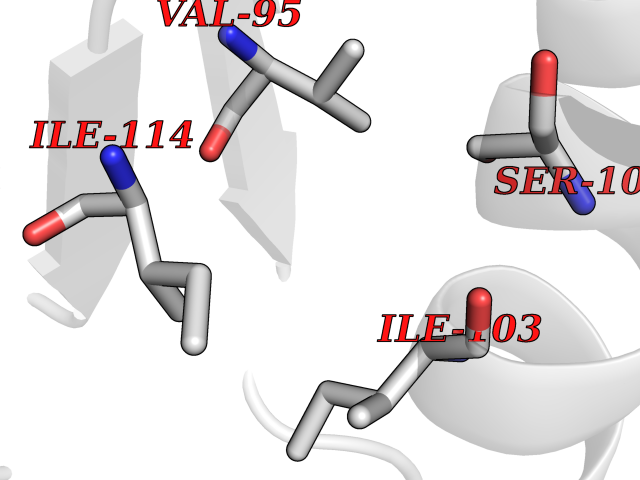 Pymol Map  Ligplot Map 3D Binding mode Sequence MKLFAQGTSLDLSHPHVMGILNVTPDSFSDGGTHNSLIDAVKHANLMINAGATIIDVGGESTRPGAAEVSVEEELQRVIPVVEAIAQRFEVWISVDTSKPEVIRESAKVGAHIINDIRSLSEPGALEAAAETGLPVCLMHMQGNPKTMQEAPKYDDVFAEVNRYFIEQIARCEQAGIAKEKLLLDPGFGFGKNLSHNYSLLARLAEFHHFNLPLLVGMSRKSMIGQLLNVGPSERLSGSLACAVIAAMQGAHIIRVHDVKETVEAMRVVEATLSAKENKRYE Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Medium-chain specific acyl-CoA dehydrogenase, mitochondrial | 4P13 | 3.93 | |
Target general information Gen name ACADM Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Acyl-CoA dehydrogenase family Biochemical class Oxidoreductase Function Acyl-CoA dehydrogenase activity.Flavin adenine dinucleotide binding.Identical protein binding.Medium-chain-acyl-CoA dehydrogenase activity. Related diseases Acyl-CoA dehydrogenase medium-chain deficiency (ACADMD) [MIM:201450]: An inborn error of mitochondrial fatty acid beta-oxidation which causes fasting hypoglycemia, hepatic dysfunction and encephalopathy, often resulting in death in infancy. {ECO:0000269|PubMed:10767181, ECO:0000269|PubMed:11349232, ECO:0000269|PubMed:11409868, ECO:0000269|PubMed:11486912, ECO:0000269|PubMed:1363805, ECO:0000269|PubMed:1671131, ECO:0000269|PubMed:1684086, ECO:0000269|PubMed:1902818, ECO:0000269|PubMed:2251268, ECO:0000269|PubMed:2393404, ECO:0000269|PubMed:2394825, ECO:0000269|PubMed:7603790, ECO:0000269|PubMed:7929823, ECO:0000269|PubMed:8198141, ECO:0000269|PubMed:9158144, ECO:0000269|PubMed:9882619}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03415; DB03147; DB02910 Interacts with PRO_0000000502 [P11310] EC number 1.3.8.7 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Disease variant; FAD; Fatty acid metabolism; Flavoprotein; Lipid metabolism; Mitochondrion; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Transit peptide Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 85080.3 Length 773 Aromaticity 0.09 Instability index 30.55 Isoelectric point 5.71 Charge (pH=7) -7.7 2D Binding mode Binding energy (Kcal/mol) -5.36  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LGFSFEFTEQQKEFQATARKFAREEIIPVAAEYDKTGEYPVPLIRRAWELGLMNTHIPENCGGLGLGTFDACLISEELAYGCTGVQTAIEGNSLGQMPIIIAGNDQQKKKYLGRMTEEPLMCAYCVTEPGAGSDVAGIKTKAEKKGDEYIINGQKMWITNGGKANWYFLLARSDPDPKAPANKAFTGFIVEADTPGIQIGRKELNMGQRCSDTRGIVFEDVKVPKENVLIGDGAGFKVAMGAFDKTRPVVAAGAVGLAQRALDEATKYALERKTFGKLLVEHQAISFMLAEMAMEVELARMSYQRAAWEVDSGRRNTYYASIAKAFAGDIANQLATDAVQILGGNGFNTEYPVEKLMRDAKIYQIYEGTSQIQRLIVAREHIDKYKLGFSFEFTEQQKEFQATARKFAREEIIPVAAEYDKTGEYPVPLIRRAWELGLMNTHIPENCGGLGLGTFDACLISEELAYGCTGVQTAIEGNSLGQMPIIIAGNDQQKKKYLGRMTEEPLMCAYCVTEPGAGSDVAGIKTKAEKKGDEYIINGQKMWITNGGKANWYFLLARSDPDPKAPANKAFTGFIVEADTPGIQIGRKELNMGQRCSDTRGIVFEDVKVPKENVLIGDGAGFKVAMGAFDKTRPVVAAGAVGLAQRALDEATKYALERKTFGKLLVEHQAISFMLAEMAMEVELARMSYQRAAWEVDSGRRNTYYASIAKAFAGDIANQLATDAVQILGGNGFNTEYPVEKLMRDAKIYQIYEGTSQIQRLIVAREHIDKYKN Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Macrophage colony-stimulating factor 1 receptor (CSF1R) | 2I1M | 3.93 | |
Target general information Gen name CSF1R Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Proto-oncogene c-Fms; M-CSF-R; FMS; CSF-1R; CSF-1-R; CSF-1 receptor; CD115 Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Promotes the release of proinflammatory chemokines in response to IL34 and CSF1, and thereby plays an important role in innate immunity and in inflammatory processes. Plays an important role in the regulation of osteoclast proliferation and differentiation, the regulation of bone resorption, and is required for normal bone and tooth development. Required for normal male and female fertility, and for normal development of milk ducts and acinar structures in the mammary gland during pregnancy. Promotes reorganization of the actin cytoskeleton, regulates formation of membrane ruffles, cell adhesion and cell migration, and promotes cancer cell invasion. Activates several signaling pathways in response to ligand binding. Phosphorylates PIK3R1, PLCG2, GRB2, SLA2 and CBL. Activation of PLCG2 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate, that then lead to the activation of protein kinase C family members, especially PRKCD. Phosphorylation of PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, leads to activation of the AKT1 signaling pathway. Activated CSF1R also mediates activation of the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1, and of the SRC family kinases SRC, FYN and YES1. Activated CSF1R transmits signals both via proteins that directly interact with phosphorylated tyrosine residues in its intracellular domain, or via adapter proteins, such as GRB2. Promotes activation of STAT family members STAT3, STAT5A and/or STAT5B. Promotes tyrosine phosphorylation of SHC1 and INPP5D/SHIP-1. Receptor signaling is down-regulated by protein phosphatases, such as INPP5D/SHIP-1, that dephosphorylate the receptor and its downstream effectors, and by rapid internalization of the activated receptor. Tyrosine-protein kinase that acts as cell-surface receptor for CSF1 and IL34 and plays an essential role in the regulation of survival, proliferation and differentiation of hematopoietic precursor cells, especially mononuclear phagocytes, such as macrophages and monocytes. Related diseases Aberrant expression of CSF1 or CSF1R can promote cancer cell proliferation, invasion and formation of metastases. Overexpression of CSF1 or CSF1R is observed in a significant percentage of breast, ovarian, prostate, and endometrial cancers.; DISEASE: Aberrant expression of CSF1 or CSF1R may play a role in inflammatory diseases, such as rheumatoid arthritis, glomerulonephritis, atherosclerosis, and allograft rejection.; DISEASE: Leukoencephalopathy, hereditary diffuse, with spheroids 1 (HDLS1) [MIM:221820]: An autosomal dominant adult-onset rapidly progressive neurodegenerative disorder characterized by variable behavioral, cognitive, and motor changes. Patients often die of dementia within 6 years of onset. Brain imaging shows patchy abnormalities in the cerebral white matter, predominantly affecting the frontal and parietal lobes. {ECO:0000269|PubMed:22197934, ECO:0000269|PubMed:23408870, ECO:0000269|PubMed:24336230, ECO:0000269|PubMed:24532199}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brain abnormalities, neurodegeneration, and dysosteosclerosis (BANDDOS) [MIM:618476]: An autosomal recessive disease with variable manifestations. Main features are brain malformations with calcifying leukoencephalopathy, progressive neurodegeneration, and bone sclerotic features. The age at onset ranges from infancy to early adulthood. Neurologic features include loss of previous motor and language skills, cognitive impairment, spasticity, and focal seizures. Brain imaging shows periventricular white matter abnormalities and calcifications, large cisterna magna or Dandy-Walker malformation, and sometimes agenesis of the corpus callosum. {ECO:0000269|PubMed:30982608, ECO:0000269|PubMed:30982609}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07167; DB07202; DB12147; DB12010; DB00619; DB06080; DB12978; DB01268 Interacts with P09603; Q15375; P29323; Q6ZMJ4-1 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Immunity; Immunoglobulin domain; Inflammatory response; Innate immunity; Kinase; Membrane; Neurodegeneration; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 35082.9 Length 311 Aromaticity 0.11 Instability index 44.6 Isoelectric point 8.13 Charge (pH=7) 2.42 2D Binding mode Binding energy (Kcal/mol) -5.36 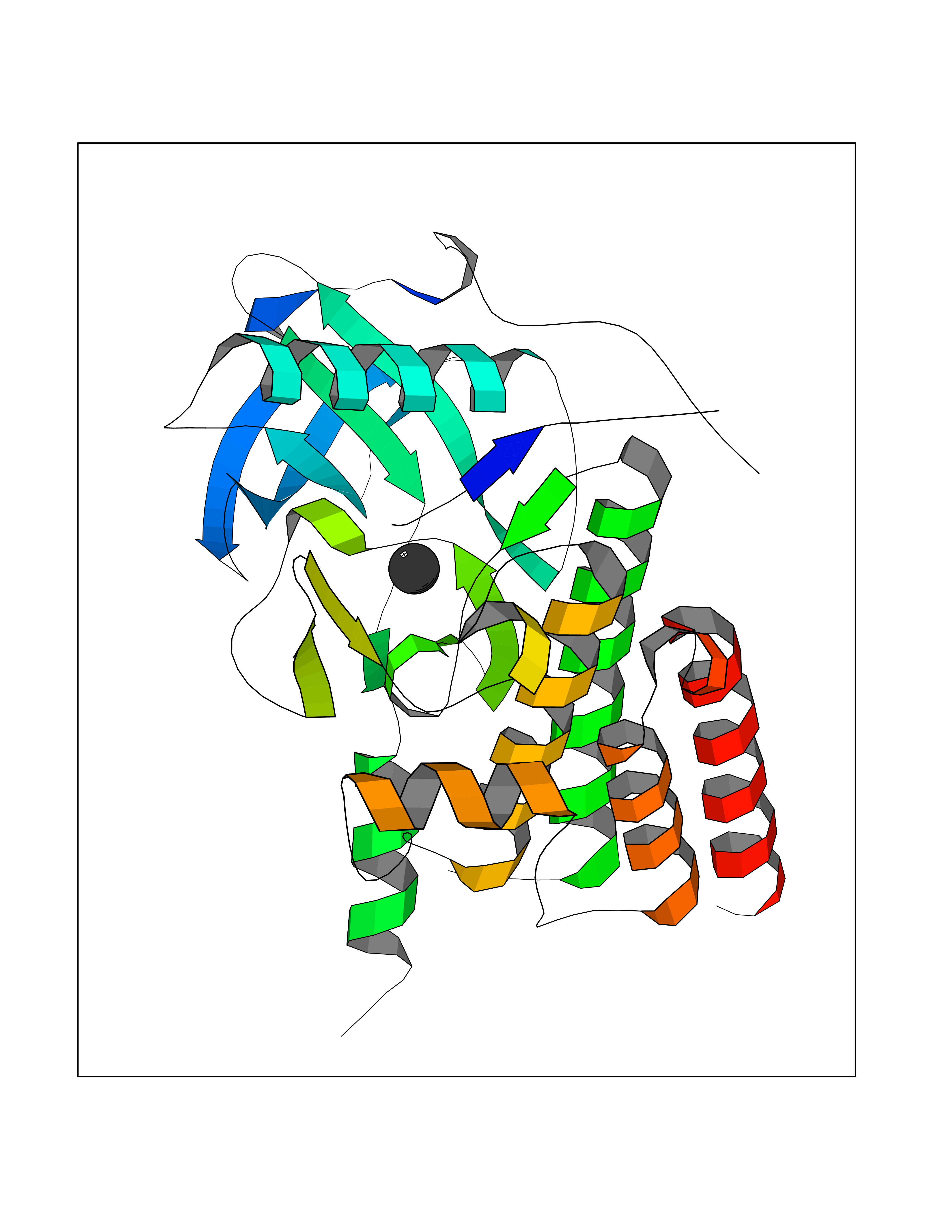 Molscript Map 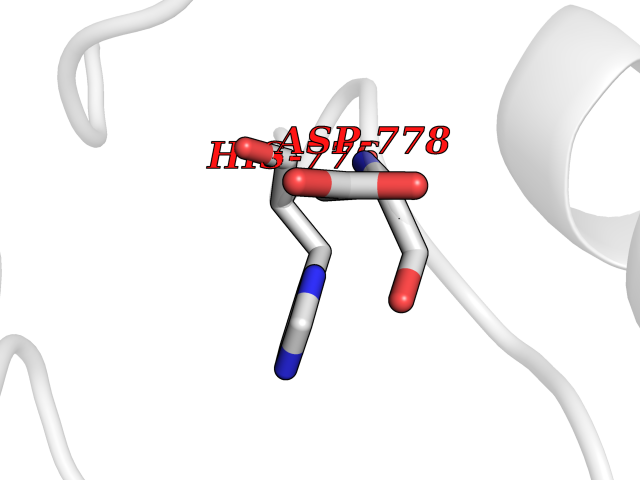 Pymol Map 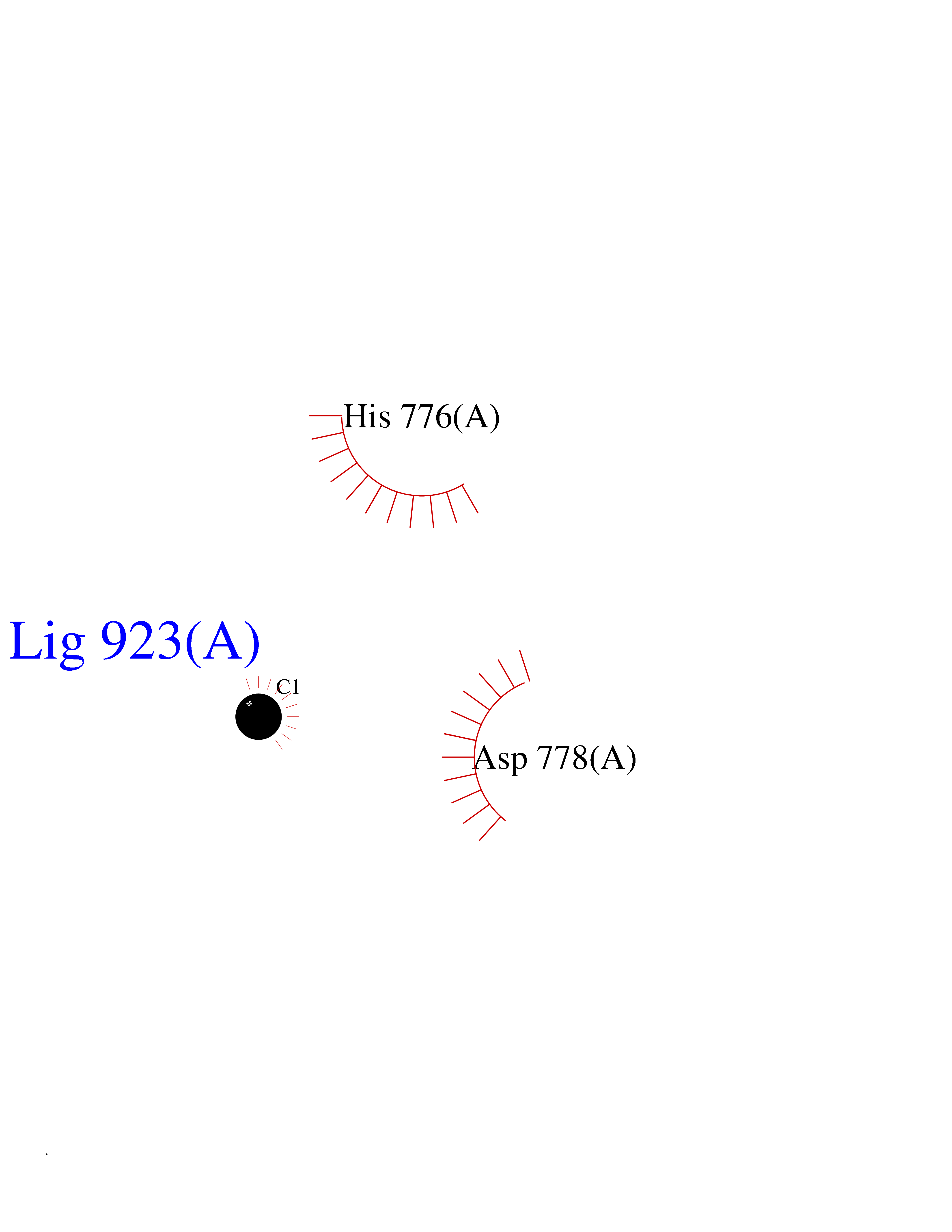 Ligplot Map 3D Binding mode Sequence QVRWKIIESYNSYTFIDPTQLPYNEKWEFPRNNLQFGKTLGAGAFGKVVEATAFGLGKEDAVLKVAVKMLKSTAHADEKEALMSELKIMSHLGQHENIVNLLGACTHGGPVLVITEYCCYGDLLNFLRRKSRVLETDSTASTRDLLHFSSQVAQGMAFLASKNCIHRDVAARNVLLTNGHVAKIGDFGLARDIMNDSNYIVKGNARLPVKWMAPESIFDCVYTVQSDVWSYGILLWEIFSLGLNPYPGILVNSKFYKLVKDGYQMAQPAFAPKNIYSIMQACWALEPTHRPTFQQICSFLQEQAQEDRRER Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Penicillin-binding protein 2B | 2WAD | 3.93 | |
Target general information Gen name penA Organism Streptococcus pneumoniae (strain ATCC BAA-255 / R6) Uniprot ID TTD ID NA Synonyms spr1517;pbp2b Protein family Transpeptidase family Biochemical class Peptide binding protein Function Penicillin binding. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01163; DB00415; DB08795; DB01140; DB00456; DB01066; DB00493; DB01331; DB01212; DB00567; DB03313; DB00485; DB00739; DB01603; DB00607; DB00713; DB00319 Interacts with NA EC number NA Uniprot keywords 3D-structure; Antibiotic resistance; Cell membrane; Cell shape; Cell wall biogenesis/degradation; Membrane; Peptidoglycan synthesis; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 65444.4 Length 607 Aromaticity 0.08 Instability index 30.15 Isoelectric point 4.95 Charge (pH=7) -20.68 2D Binding mode Binding energy (Kcal/mol) -5.36 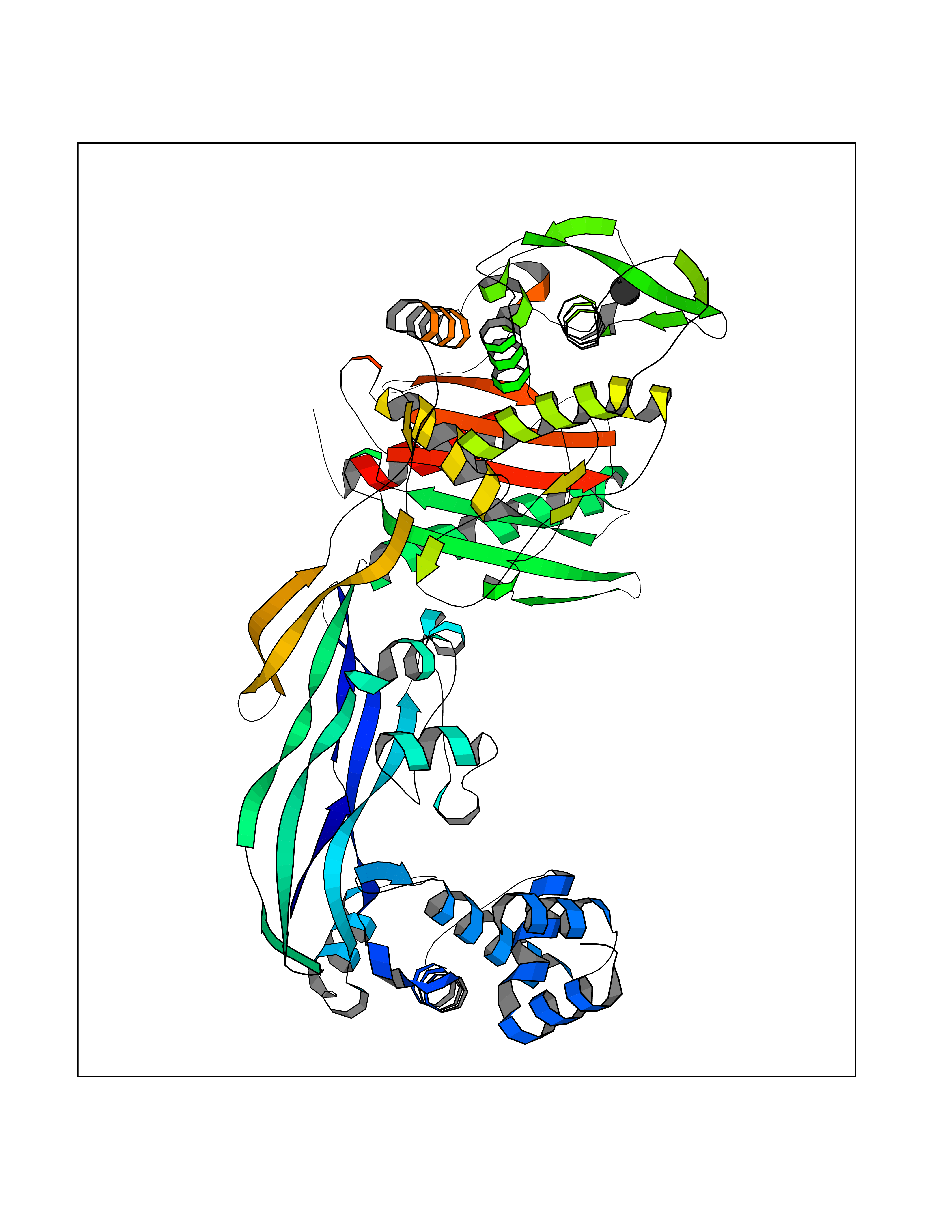 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SQTKVTTSSARGEIYDASGKPLVENTLKQVVSFTRSNKMTATDLKEIAKKLLTYVSISSPNLTERQLADYYLADPEIYKKTVEALPSESELYNNAVDSVPTSQLNYTEDEKKEIYLFSQLNAVGNFATGTIATDPLNDSQVAVIASISKEMPGISISTSWDRKILETSLSSIVGSVSSEKAGLPAEEAESYLKKGYSLNDRVGTSYLEKQYEEVLQGKRPVKEIHLDKHGDMESVENIEEGSKGKNIKLTIDLAFQDSVDALLKSYFNSELGNGGAKYSEGVYAVALNPQTGAVLSMSGLKHDLKTGELTPDSLGTVTNVFVPGSVVKAATISSGWENGVLSGNQTLTDQPIVFQGSAPIYSWYKLAYGSFPITAVEALEYSSNAYVVQTALGIMGQTYQPNMFVGTSNLESAMGKLRSTFGEYGLGSATGIDLPDESTGLVPKEYNFANFITNAFGQFDNYTPMQLAQYVATIANNGVRLAPHIVEGIYDNNDKGGLGELIQAIDTKEINKVNISESDMAILHQGFYQVSHGTSPLTTGRAFSDGATVSISGKTGTNTNAVAYAPTENPQIAVAVVFPHNTNLTKNVGPAIARDIINLYNQHHPMN Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | D-amino acid oxidase (DAO) | 3ZNN | 3.93 | |
Target general information Gen name DAO Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Daminoacid oxidase; DAMOX; DAAO Protein family DAMOX/DASOX family Biochemical class CH-NH(2) donor oxidoreductase Function Regulates the level of the neuromodulator D-serine in the brain. Has high activity towards D-DOPA and contributes to dopamine synthesis. Could act as a detoxifying agent which removes D-amino acids accumulated during aging. Acts on a variety of D-amino acids with a preference for those having small hydrophobic side chains followed by those bearing polar, aromatic, and basic groups. Does not act on acidic amino acids. Related diseases Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:12364586}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Amyotrophic lateral sclerosis (ALS) [MIM:105400]: A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. {ECO:0000269|PubMed:20368421, ECO:0000269|PubMed:20538972, ECO:0000269|PubMed:22203986, ECO:0000269|PubMed:23219954, ECO:0000269|PubMed:24138986, ECO:0000269|PubMed:25701391, ECO:0000269|PubMed:37558109, ECO:0000269|PubMed:38035964, ECO:0000269|PubMed:38134563}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07979; DB02838; DB04166; DB03793; DB03225; DB03147; DB03531; DB02988 Interacts with Q9P2K6; O43741 EC number EC 1.4.3.3 Uniprot keywords 3D-structure; Amyotrophic lateral sclerosis; Cell projection; Cytoplasm; Disease variant; FAD; Flavoprotein; Neurodegeneration; Oxidoreductase; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; S-nitrosylation; Schizophrenia; Secreted; Synapse Protein physicochemical properties Chain ID A,B Molecular weight (Da) 38654.6 Length 340 Aromaticity 0.11 Instability index 29.13 Isoelectric point 6.18 Charge (pH=7) -4.45 2D Binding mode Binding energy (Kcal/mol) -5.36  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MRVVVIGAGVIGLSTALCIHERYHSVLQPLDIKVYADRFTPLTTTDVAAGLWQPYLSDPNNPQEADWSQQTFDYLLSHVHSPNAENLGLFLISGYNLFHEAIPDPSWKDTVLGFRKLTPRELDMFPDYGYGWFHTSLILEGKNYLQWLTERLTERGVKFFQRKVESFEEVAREGADVIVNCTGVWAGALQRDPLLQPGRGQIMKVDAPWMKHFILTHDPERGIYNSPYIIPGTQTVTLGGIFQLGNWSELNNIQDHNTIWEGCCRLEPTLKNARIIGERTGFRPVRPQIRLEREQLRTGPSNTEVIHNYGHGGYGLTIHWGCALEAAKLFGRILEEKKLS Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Argininosuccinate synthase | 2NZ2 | 3.93 | |
Target general information Gen name ASS1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ASS Protein family Argininosuccinate synthase family, Type 1 subfamily Biochemical class Ligase Function Amino acid binding.Argininosuccinate synthase activity.ATP binding.Identical protein binding.RNA binding. Related diseases Citrullinemia 1 (CTLN1) [MIM:215700]: The classic form of citrullinemia, an autosomal recessive disease characterized primarily by elevated serum and urine citrulline levels. Ammonia intoxication is another manifestation. It is a disorder of the urea cycle, usually manifesting in the first few days of life. Affected infants appear normal at birth, but as ammonia builds up in the body they present symptoms such as lethargy, poor feeding, vomiting, seizures and loss of consciousness. Less commonly, a milder form can develop later in childhood or adulthood. {ECO:0000269|PubMed:11708871, ECO:0000269|PubMed:11941481, ECO:0000269|PubMed:12815590, ECO:0000269|PubMed:14680976, ECO:0000269|PubMed:15863597, ECO:0000269|PubMed:16475226, ECO:0000269|PubMed:18473344, ECO:0000269|PubMed:19006241, ECO:0000269|PubMed:1943692, ECO:0000269|PubMed:2358466, ECO:0000269|PubMed:23611581, ECO:0000269|PubMed:24889030, ECO:0000269|PubMed:25179242, ECO:0000269|PubMed:27287393, ECO:0000269|PubMed:28111830, ECO:0000269|PubMed:7977368}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00125; DB00128; DB00171; DB00155 Interacts with P10398; P00966; Q9HBL8; Q9NVM4 EC number 6.3.4.5 Uniprot keywords 3D-structure; Acetylation; Amino-acid biosynthesis; Arginine biosynthesis; ATP-binding; Cytoplasm; Direct protein sequencing; Disease variant; Ligase; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Urea cycle Protein physicochemical properties Chain ID A Molecular weight (Da) 41098.8 Length 364 Aromaticity 0.1 Instability index 36.56 Isoelectric point 8.4 Charge (pH=7) 3.55 2D Binding mode Binding energy (Kcal/mol) -5.36  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence KGSVVLAYSGGLDTSCILVWLKEQGYDVIAYLANIGQKEDFEEARKKALKLGAKKVFIEDVSREFVEEFIWPAIQSSALYEDRYLLGTSLARPCIARKQVEIAQREGAKYVSHGATGKGNDQVRFELSCYSLAPQIKVIAPWRMPEFYNRFKRNDLMEYAKQHGIPIPVTPKNPWSMDENLMHISYEAGILENPKNQAPPGLYTKTQDPAKAPNTPDILEIEFKKGVPVKVTNVKDGTTHQTSLELFMYLNEVAGKHGVGRIDIVENRFIGMKSRGIYETPAGTILYHAHLDIEAFTMDREVRKIKQGLGLKFAELVYTGFWHSPECEFVRHCIAKSQERVEGKVQVSVLKGQVYILGRESPLS Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Camphor 5-monooxygenase | 4L4E | 3.93 | |
Target general information Gen name camC Organism Pseudomonas putida (Arthrobacter siderocapsulatus) Uniprot ID TTD ID NA Synonyms cyp101 Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Camphor 5-monooxygenase activity.Heme binding.Iron ion binding. Related diseases Combined oxidative phosphorylation deficiency 6 (COXPD6) [MIM:300816]: A mitochondrial disease resulting in a neurodegenerative disorder characterized by psychomotor delay, hypotonia, areflexia, muscle weakness and wasting. Some patients manifest prenatal ventriculomegaly and severe postnatal encephalomyopathy. {ECO:0000269|PubMed:20362274, ECO:0000269|PubMed:22019070, ECO:0000269|PubMed:25583628, ECO:0000269|PubMed:26004228, ECO:0000269|PubMed:26173962, ECO:0000269|PubMed:27178839}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, X-linked recessive, 4, with or without cerebellar ataxia (CMTX4) [MIM:310490]: A neuromuscular disorder characterized by progressive sensorimotor axonal neuropathy, distal sensory impairment, difficulty walking due to peripheral neuropathy and/or cerebellar ataxia, and deafness due to auditory neuropathy. Additional features include cognitive impairment, cerebellar atrophy, dysarthria, abnormal extraocular movements, tremor, dysmetria and spasticity. The age at onset ranges from infancy to young adulthood. {ECO:0000269|PubMed:23217327, ECO:0000269|PubMed:26004228}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Deafness, X-linked, 5, with peripheral neuropathy (DFNX5) [MIM:300614]: A form of hearing loss characterized by absent or severely abnormal auditory brainstem response, abnormal middle ear reflexes, abnormal speech discrimination, loss of outer hair cell function, and cochlear nerve hypoplasia. DFNX5 patients manifest auditory neuropathy with childhood onset, associated with distal sensory impairment affecting the peripheral nervous system. {ECO:0000269|PubMed:25986071}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spondyloepimetaphyseal dysplasia, X-linked, with hypomyelinating leukodystrophy (SEMDHL) [MIM:300232]: An X-linked recessive developmental disorder characterized by slowly progressive skeletal and neurologic abnormalities, including short stature, large and deformed joints, significant motor impairment, visual defects, and sometimes cognitive deficits. Affected individuals typically have normal early development in the first year or so of life, followed by development regression and the development of symptoms. Brain imaging shows white matter abnormalities consistent with hypomyelinating leukodystrophy. {ECO:0000269|PubMed:28842795}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03836; DB02617; DB02817; DB03627; DB04032; DB03031; DB02125; DB04501; DB01744; DB01663; DB01011; DB01703; DB01826; DB03540; DB02851 Interacts with P00259 EC number 1.14.15.1 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Heme; Iron; Metal-binding; Monooxygenase; Oxidoreductase Protein physicochemical properties Chain ID A Molecular weight (Da) 45446.3 Length 405 Aromaticity 0.08 Instability index 45.33 Isoelectric point 5.23 Charge (pH=7) -16.08 2D Binding mode Binding energy (Kcal/mol) -5.36 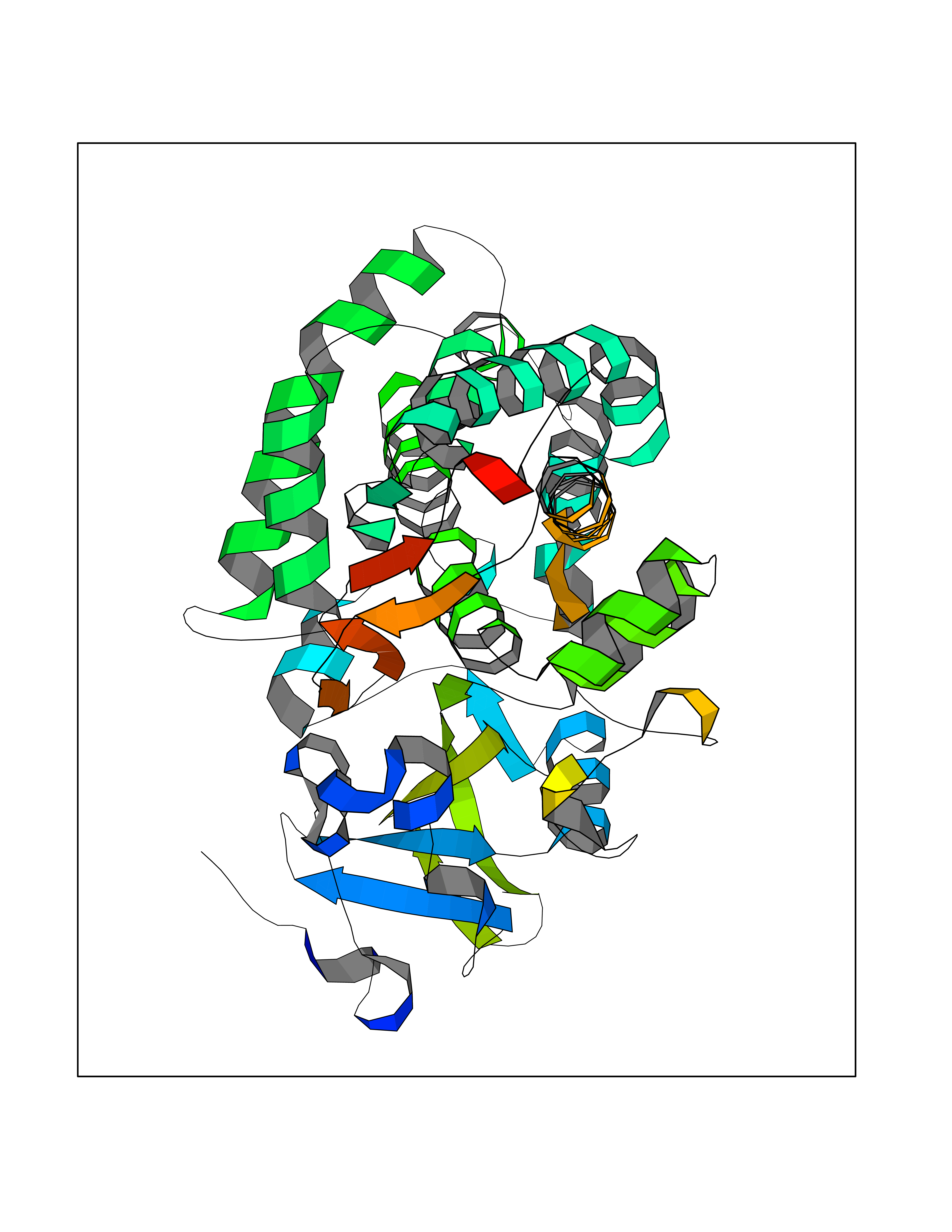 Molscript Map 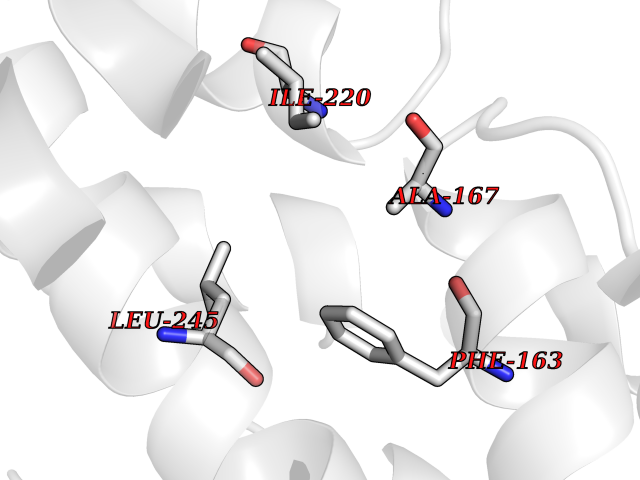 Pymol Map 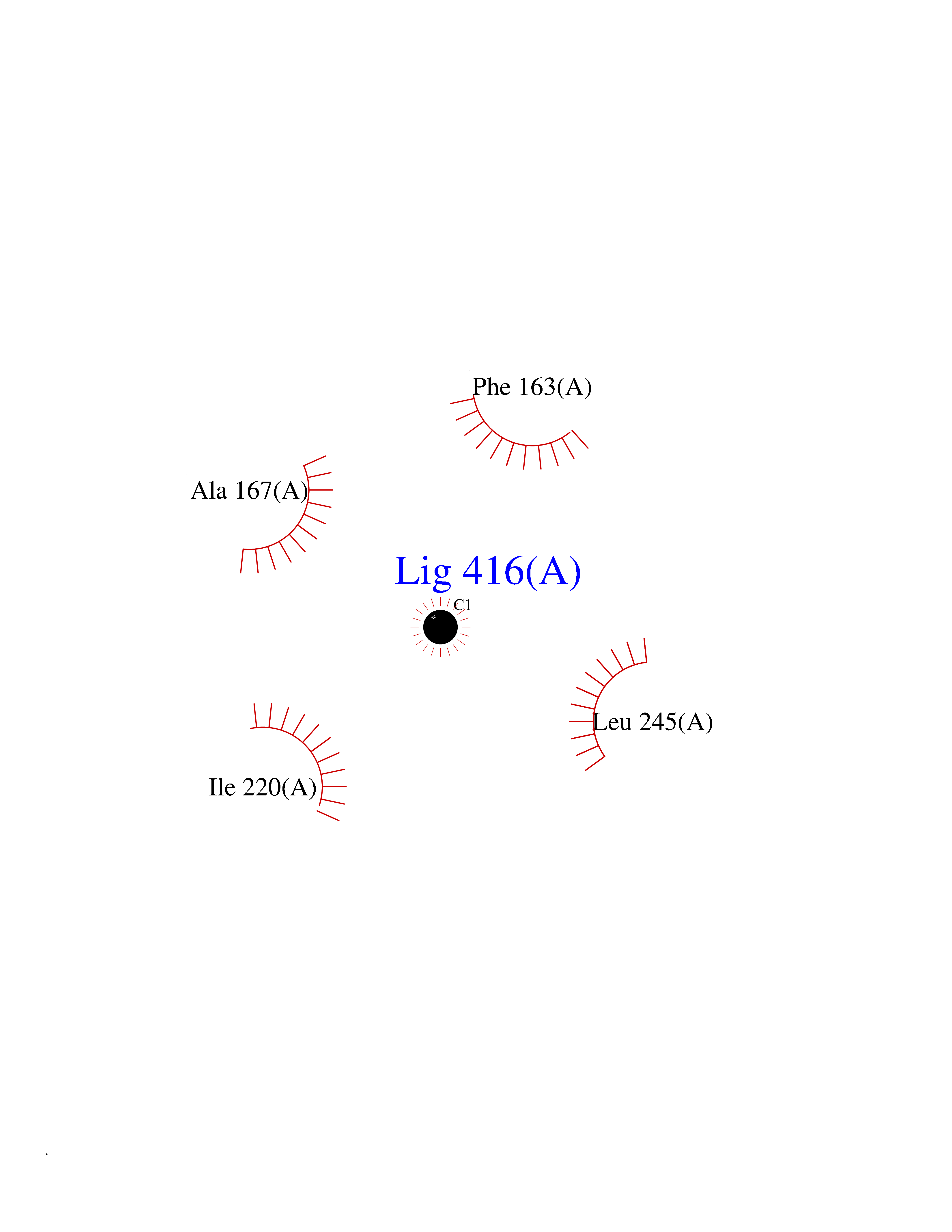 Ligplot Map 3D Binding mode Sequence NLAPLPPHVPEHLVFDFDMYNPSNLSAGVQEAWAVLQESNVPDLVWTRCNGGHWIATRGQLIREAYEDYRHFSSECPFIPREAGEAYDFIPTSMDPPEQRQFRALANQVVGMPVVDKLENRIQELACSLIESLRPQGQCNFTEDYAEPFPIRIFMLLAGLPEEDIPHLGYLTDQMTRPDGSMTFAEAKEALYDYLIPIIEQRRQKPGTDAISIVANGQVNGRPITSDEAKRMCGLLLVGGLDTVVNFLSFSMEFLAKSPEHRQELIERPERIPAACEELLRRFSLVADGRILTSDYEFHGVQLKKGDQILLPQMLSGLDERENAAPMHVDFSRQKVSHTTFGHGSHLCAGQHLARREIIVTLKEWLTRIPDFSIAPGAQIQHKSGIVSGVQALPLVWDPATTKAV Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Pyridoxal kinase (PDXK) | 3KEU | 3.93 | |
Target general information Gen name PDXK Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Pyridoxine kinase; PDXK Protein family Pyridoxine kinase family Biochemical class Kinase Function Required for synthesisof pyridoxal-5-phosphate from vitamin B6. Related diseases Neuropathy, hereditary motor and sensory, 6C, with optic atrophy (HMSN6C) [MIM:618511]: An autosomal recessive neurologic disorder characterized by childhood onset of axonal, sensorimotor polyneuropathy affecting mainly the lower limbs, and adult-onset optic atrophy. Clinical features include progressive distal muscle weakness and atrophy, significant standing and walking difficulties, areflexia, neurogenic pain and progressive visual impairment. {ECO:0000269|PubMed:31187503}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04776; DB03909; DB04770; DB00147; DB00165 Interacts with NA EC number EC 2.7.1.35 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Charcot-Marie-Tooth disease; Cobalt; Cytoplasm; Disease variant; Kinase; Magnesium; Manganese; Metal-binding; Neurodegeneration; Neuropathy; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Sodium; Transferase; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 34370.1 Length 305 Aromaticity 0.07 Instability index 39.92 Isoelectric point 6.16 Charge (pH=7) -3.37 2D Binding mode Binding energy (Kcal/mol) -5.36 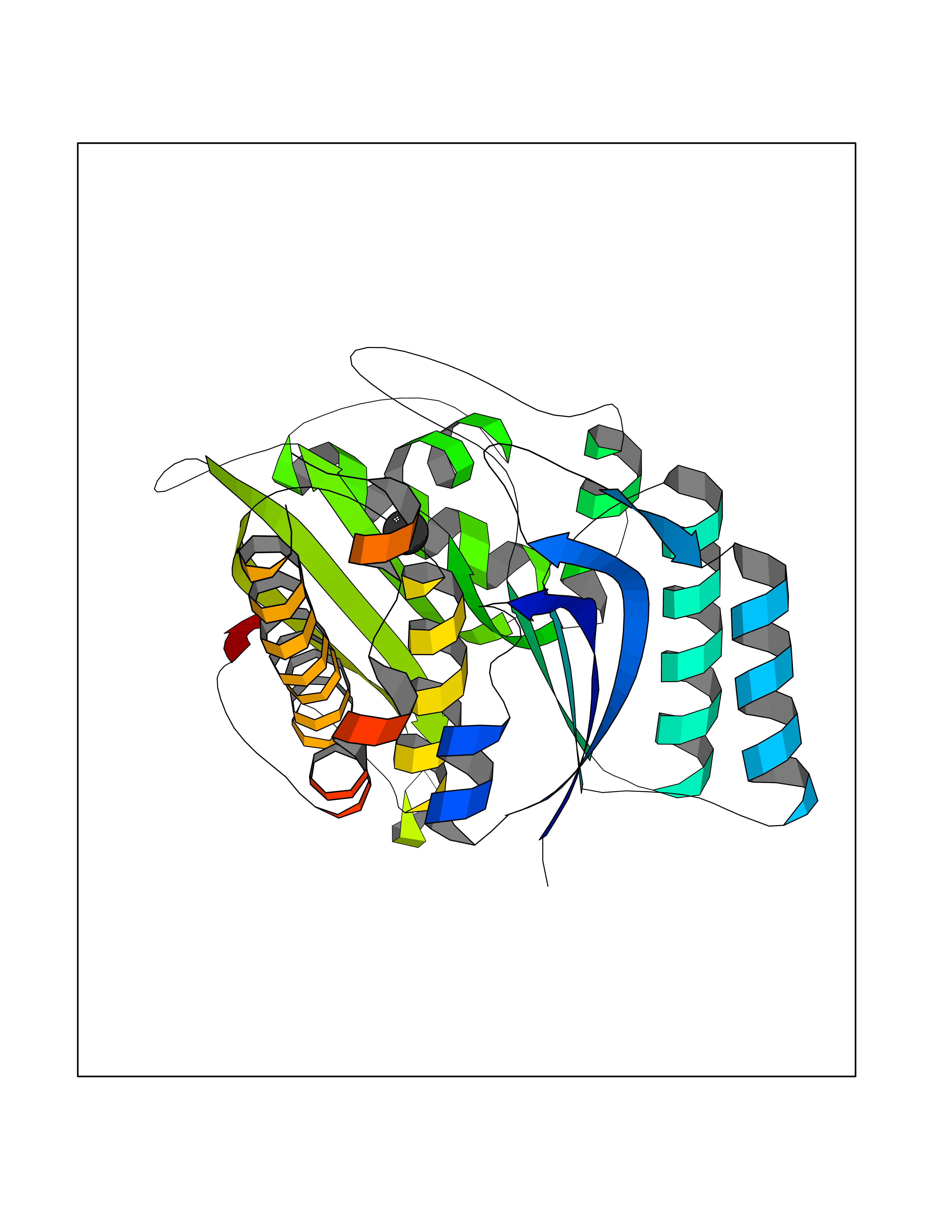 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ECRVLSIQSHVIRGYVGNRAATFPLQVLGFEIDAVNSVQFSNHTGYAHWKGQVLNSDELQELYEGLRLNNMNKYDYVLTGYTRDKSFLAMVVDIVQELKQQNPRLVYVCDPVLGDKWDGEGSMYVPEDLLPVYKEKVVPLADIITPNQFEAELLSGRKIHSQEEALRVMDMLHSMGPDTVVITSSDLPSPQGSNYLIVLGSQRRRNPAGSVVMERIRMDIRKVDAVFVGTGDLFAAMLLAWTHKHPNNLKVACEKTVSTLHHVLQRTIQCAKAQARPSPMQLELRMVQSKRDIEDPEIVVQATVL Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | Retinoic acid receptor beta (RARB) | 4DM6 | 3.93 | |
Target general information Gen name RARB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-epsilon; RAR-beta; Nuclear receptor subfamily 1 group B member 2; NR1B2; HBV-activated protein; HAP Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RXR/RAR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence or presence of hormone ligand, acts mainly as an activator of gene expression due to weak binding to corepressors. In concert with RARG, required for skeletal growth, matrix homeostasis and growth plate function. Receptor for retinoic acid. Related diseases Microphthalmia, syndromic, 12 (MCOPS12) [MIM:615524]: A form of microphthalmia, a disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues (anophthalmia). In many cases, microphthalmia/anophthalmia occurs in association with syndromes that include non-ocular abnormalities. MCOPS12 patients manifest variable features, including diaphragmatic hernia, pulmonary hypoplasia, and cardiac abnormalities. {ECO:0000269|PubMed:24075189, ECO:0000269|PubMed:27120018}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00459; DB00210; DB00523; DB02877; DB00926; DB05785; DB04942; DB00799; DB00755; DB12808 Interacts with O95273; P50222; Q9UBK2; P62195; P28702; P28702-3; P48443; P03255 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Microphthalmia; Nucleus; Phosphoprotein; Proto-oncogene; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 25904.1 Length 229 Aromaticity 0.06 Instability index 44.34 Isoelectric point 7.55 Charge (pH=7) 0.73 2D Binding mode Binding energy (Kcal/mol) -5.35  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence TEKIRKAHQETFPSLCQLGKYTTNSSADHRVRLDLGLWDKFSELATKCIIKIVEFAKRLPGFTGLTIADQITLLKAACLDILILRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFTFANQLLPLEMDDTETGLLSAICLICGDRQDLEEPTKVDKLQEPLLEALKIYIRKRRPSKPHMFPKILMKITDLRSISAKGAERVITLKMEIPGSMPPLIQEMLEN Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Chromodomain-helicase-DNA-binding protein 1 | 4O42 | 3.93 | |
Target general information Gen name CHD1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family SNF2/RAD54 helicase family Biochemical class Dna binding protein / viral protein Function ATP binding.ATP-dependent DNA helicase activity.DNA binding.Methylated histone binding. Related diseases Pilarowski-Bjornsson syndrome (PILBOS) [MIM:617682]: An autosomal dominant disorder characterized by developmental delay, speech apraxia, intellectual disability, autism, and facial dysmorphic features. Some patients may have seizures. {ECO:0000269|PubMed:28866611}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O60341-1; B2BUF1; P28799; O76024 EC number 3.6.4.12 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Chromatin regulator; Cytoplasm; Disease variant; DNA-binding; Helicase; Hydrolase; Intellectual disability; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 20969.1 Length 180 Aromaticity 0.12 Instability index 46.35 Isoelectric point 5.88 Charge (pH=7) -2.83 2D Binding mode Binding energy (Kcal/mol) -5.37  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence EFETIERFMDCRIGRKGATGATTTIYAVEADGDPNAGFEKNKEPGEIQYLIKWKGWSHIHNTWETEETLKQQNVRGMKKLDNYKKKDQETKRWLKNASPEDVEYYNCQQELTDDLHKQYQIVERIIAHSNQKSAAGYPDYYCKWQGLPYSECSWEDGALISKKFQACIDEYFSRTARSXV Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Farnesyl pyrophosphate synthase | 4NUA | 3.93 | |
Target general information Gen name FDPS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms KIAA1293;FPS Protein family FPP/GGPP synthase family Biochemical class Transferase Function Dimethylallyltranstransferase activity.Geranyltranstransferase activity.Metal ion binding.RNA binding. Related diseases Porokeratosis 9, multiple types (POROK9) [MIM:616631]: A form of porokeratosis, a disorder of faulty keratinization characterized by one or more atrophic patches surrounded by a distinctive hyperkeratotic ridgelike border called the cornoid lamella. The keratotic lesions can progress to overt cutaneous neoplasms, typically squamous cell carcinomas. Multiple clinical variants of porokeratosis are recognized, including porokeratosis of Mibelli, linear porokeratosis, disseminated superficial actinic porokeratosis, palmoplantar porokeratosis, and punctate porokeratosis. Different clinical presentations can be observed among members of the same family. Individuals expressing more than one variant have also been reported. {ECO:0000269|PubMed:26202976}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00630; DB01785; DB07780; DB02552; DB07841; DB00710; DB06255; DB04714; DB02508; DB06548; DB00282; DB00884; DB00399 Interacts with O95870; P54253; Q6ZMZ0; Q9BRI3; Q8WWF3 EC number 2.5.1.1; 2.5.1.10 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Cytoplasm; Disease variant; Host-virus interaction; Hydroxylation; Isoprene biosynthesis; Lipid biosynthesis; Lipid metabolism; Magnesium; Metal-binding; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 38722.9 Length 338 Aromaticity 0.12 Instability index 45.6 Isoelectric point 5.1 Charge (pH=7) -9.86 2D Binding mode Binding energy (Kcal/mol) -5.37 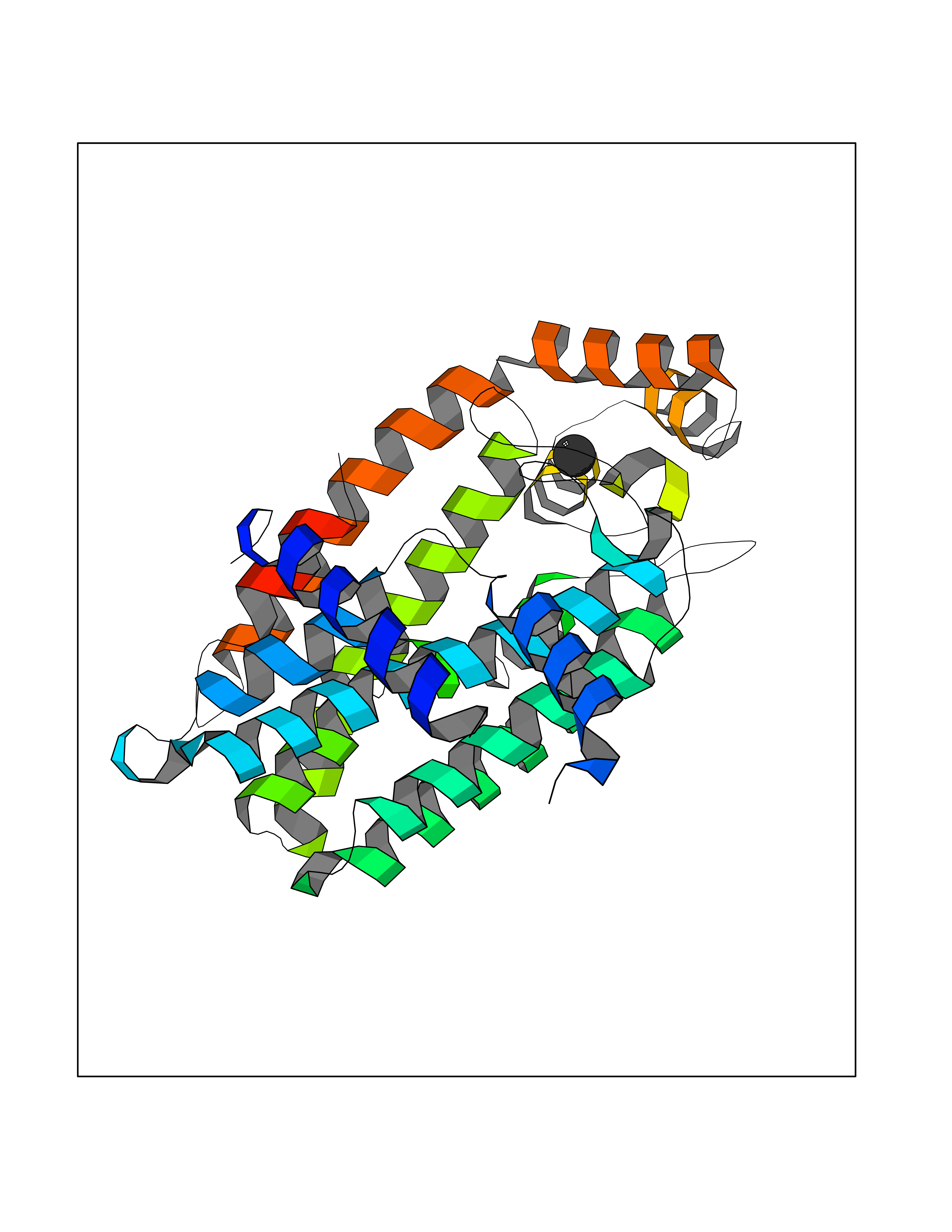 Molscript Map 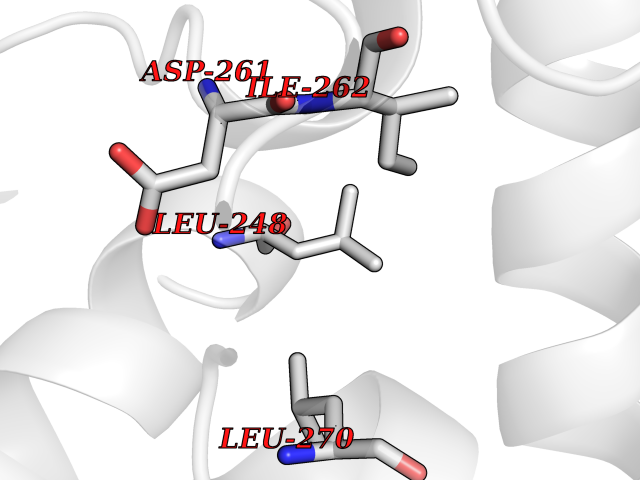 Pymol Map 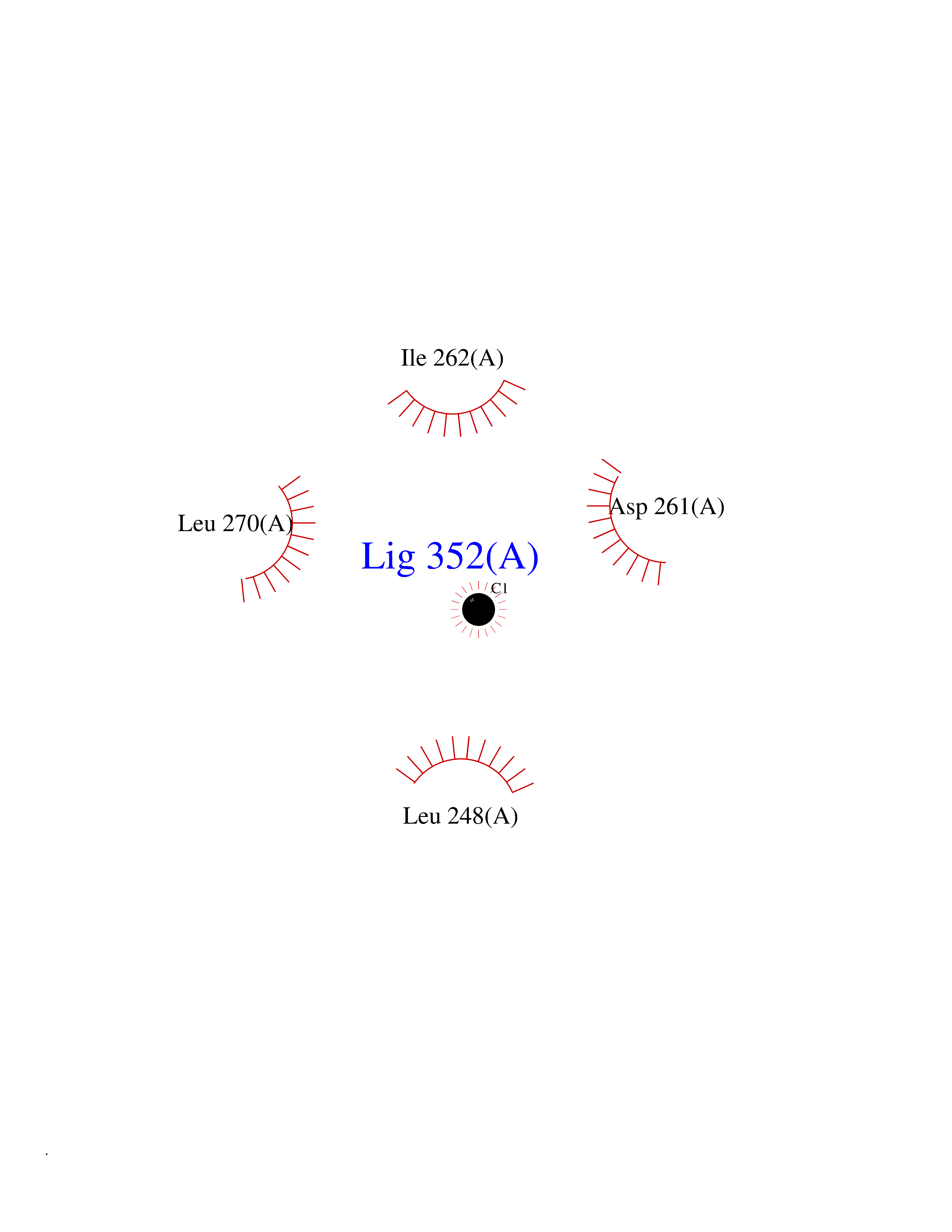 Ligplot Map 3D Binding mode Sequence AYAQEKQDFVQHFSQIVRVLTEHPEIGDAIARLKEVLEYNTIGGKYNRGLTVVVAFRELVEPRKQDADSLQRAWTVGWCVELLQAFFLVADDIMDSSLTRRGQICWYQKPGVGLDAINDANLLEACIYRLLKLYCREQPYYLNLIELFLQSSYQTEIGQTLDLLTAPQGNVDLVRFTEKRYKSIVKYKTAFYSFYLPIAAAMYMAGIDGEKEHANAKKILLEMGEFAQIQDDYLDLFGDPSVTGKIGTDIQDNKCSWLVVQCLQRATPEQYQILKENYGQKEAEKVARVKALYEELDLPAVFLQYEEDSYSHIMALIEQYAAPLPPAVFLGLARKIYK Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Ornithine decarboxylase (ODC1) | 2OO0 | 3.93 | |
Target general information Gen name ODC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ODC Protein family Orn/Lys/Arg decarboxylase class-II family Biochemical class Carbon-carbon lyase Function Polyamines are essential for cell proliferation and are implicated in cellular processes, ranging from DNA replication to apoptosis. Catalyzes the first and rate-limiting step of polyamine biosynthesis that converts ornithine into putrescine, which is the precursor for the polyamines, spermidine and spermine. Related diseases Bachmann-Bupp syndrome (BABS) [MIM:619075]: An autosomal dominant disorder characterized by global developmental delay, alopecia, absolute or relative macrocephaly, and facial dysmorphism. Neuroimaging shows white matter abnormalities, prominent Virchow-Robin spaces, periventricular cysts, and abnormalities of the corpus callosum. {ECO:0000269|PubMed:30239107, ECO:0000269|PubMed:30475435}. The disease is caused by variants affecting the gene represented in this entry. BABS is due to truncating variants that lead to a gain of function. This phenomenon apparently results from truncation proximal to or involving the C-terminal region of ODC1 protein, distal enough to allow escape from nonsense-mediated decay. A gain of function is corroborated by elevated plasma levels of N-acetylputrescine, with otherwise normal polyamine levels, in affected individuals. {ECO:0000269|PubMed:30475435}. Drugs (DrugBank ID) DB06243; DB04263; DB03856; DB04083; DB02824; DB01917; DB00114; DB02209; DB00203; DB00127; DB00313 Interacts with Q9H8Y8; Q92993; Q9UMX2; Q9UMX2-2 EC number EC 4.1.1.17 Uniprot keywords 3D-structure; Decarboxylase; Disease variant; Hypotrichosis; Lyase; Phosphoprotein; Polyamine biosynthesis; Proteomics identification; Pyridoxal phosphate; Reference proteome; S-nitrosylation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 45682.9 Length 410 Aromaticity 0.11 Instability index 40.93 Isoelectric point 5.61 Charge (pH=7) -6.68 2D Binding mode Binding energy (Kcal/mol) -5.37 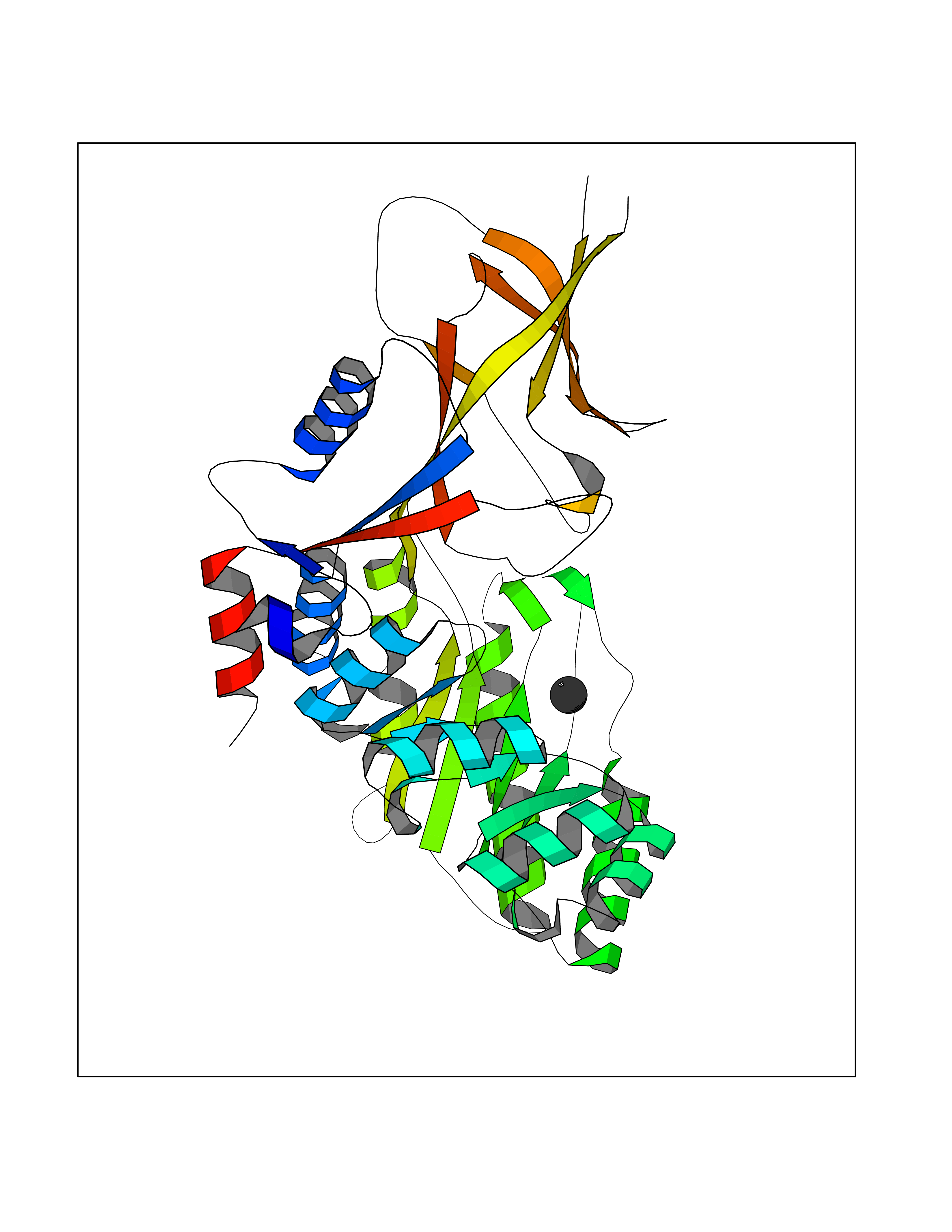 Molscript Map 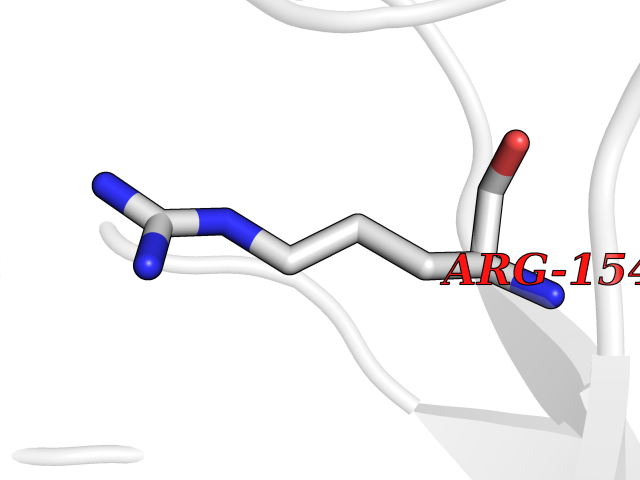 Pymol Map  Ligplot Map 3D Binding mode Sequence LMNNFGNEEFDCHFLDEGFTAKDILDQKINEVSSSDDKDAFYVADLGDILKKHLRWLKALPRVTPFYAVKCNDSKAIVKTLAATGTGFDCASKTEIQLVQSLGVPPERIIYANPCKQVSQIKYAANNGVQMMTFDSEVELMKVARAHPKAKLVLRIATDDSKAVCRLSVKFGATLRTSRLLLERAKELNIDVVGVSFHVGSGCTDPETFVQAISDARCVFDMGAEVGFSMYLLDIGGGFPGSEDVKLKFEEITGVINPALDKYFPSDSGVRIIAEPGRYYVASAFTLAVNIIAKKIVLEQTFMYYVNDGVYGSFNCILYDHAHVKPLLQKRPKPDEKYYSSSIWGPTCDGLDRIVERCDLPEMHVGDWMLFENMGAYTVAAASTFNGFQRPTIYYVMSGPAWQLMQQFQN Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Aldose reductase (AKR1B1) | 1US0 | 3.93 | |
Target general information Gen name AKR1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aldehyde reductase; AKR1B1 Protein family Aldo/keto reductase family Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the NADPH-dependent reduction of a wide variety of carbonyl-containing compounds to their corresponding alcohols with a broad range of catalytic efficiencies. Related diseases Glutamine deficiency, congenital (GLND) [MIM:610015]: An autosomal recessive disorder characterized by variable brain malformations, encephalopathy, severe developmental delay, seizures, and decreased glutamine levels in bodily fluids. Death in early infancy may occur. {ECO:0000269|PubMed:16267323, ECO:0000269|PubMed:26711351, ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 116 (DEE116) [MIM:620806]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE116 is autosomal dominant form characterized by severe developmental delay, seizures, and white matter abnormalities. {ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry. DEE116 is caused by variants that disrupt the canonical translation start codon in GLUL resulting in initiation of translation at Met-18 (PubMed:38579670). The resulting protein is enzymatically competent but insensitive to negative feedback regulation via glutamine-induced degradation. {ECO:0000269|PubMed:38579670}. Drugs (DrugBank ID) DB07028; DB07030; DB07450; DB02101; DB08449; DB08000; DB07139; DB07498; DB02007; DB02020; DB11859; DB02994; DB04272; DB07187; DB00694; DB00997; DB06246; DB01039; DB02021; DB16707; DB00143; DB02834; DB08084; DB01689; DB07063; DB06077; DB02518; DB00157; DB03461; DB05383; DB05533; DB05327; DB02712; DB00605; DB02383; DB02132; DB08772; DB07093; DB07999; DB08098 Interacts with Q9BUY7 EC number EC 1.1.1.300 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Lipid metabolism; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 35447.6 Length 313 Aromaticity 0.09 Instability index 36.41 Isoelectric point 7.1 Charge (pH=7) 0.26 2D Binding mode Binding energy (Kcal/mol) -5.36 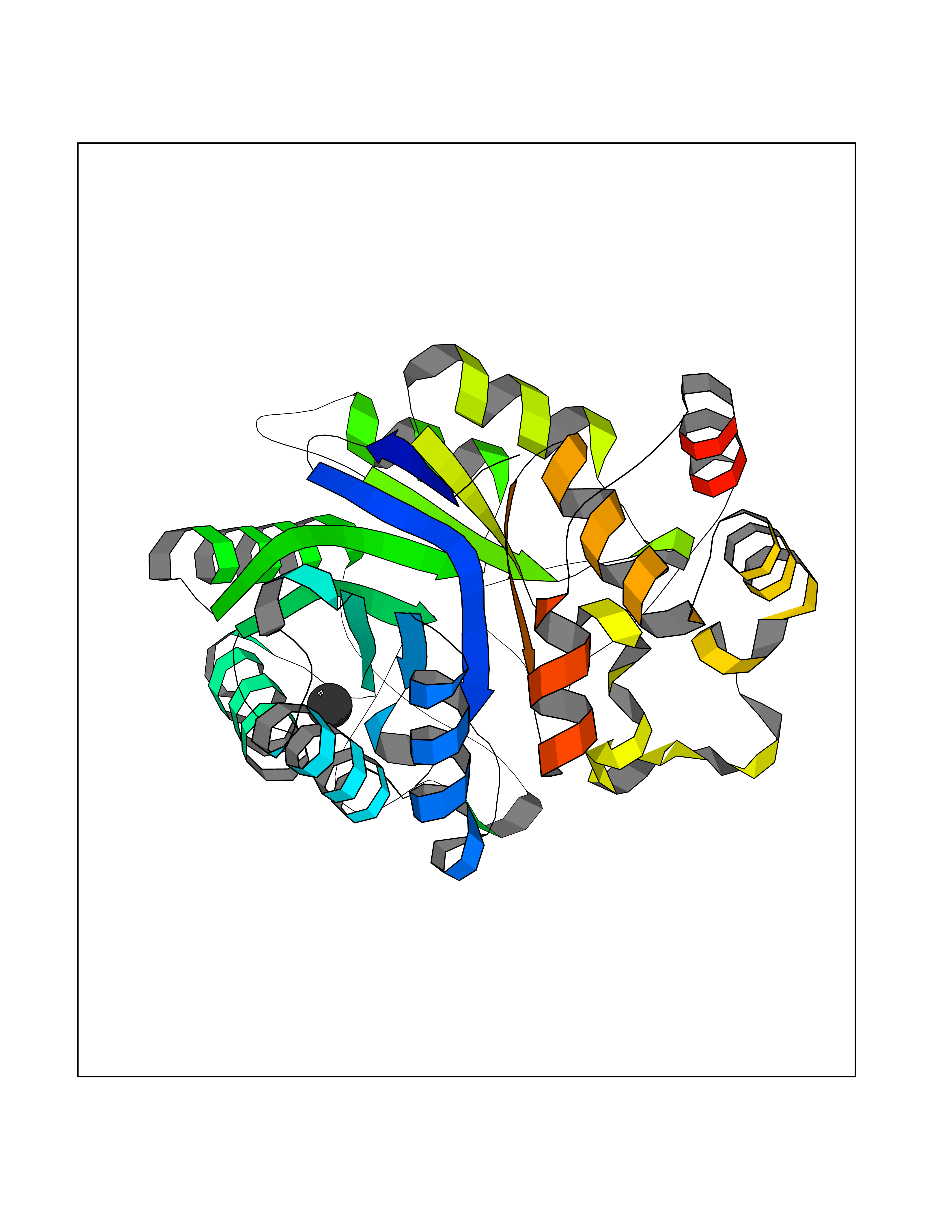 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MASRILLNNGAKMPILGLGTWKSPPGQVTEAVKVAIDVGYRHIDCAHVYQNENEVGVAIQEKLREQVVKREELFIVSKLWCTYHEKGLVKGACQKTLSDLKLDYLDLYLIHWPTGFKPGKEFFPLDESGNVVPSDTNILDTWAAMEELVDEGLVKAIGISNFNHLQVEMILNKPGLKYKPAVNQIECHPYLTQEKLIQYCQSKGIVVTAYSPLGSPDRPWAKPEDPSLLEDPRIKAIAAKHNKTTAQVLIRFPMQRNLVVIPKSVTPERIAENFKVFDFELSSQDMTTLLSYNRNWRVCALLSCTSHKDYPFH Hydrogen bonds contact Hydrophobic contact | ||||