Job Results:
Ligand
Structure
Job ID
5217c115ac480b5185ad5781ec63f66c
Job name
NA
Time
2026-02-27 11:51:57
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Dihydroorotate dehydrogenase (quinone), mitochondrial | 4CQ8 | 4.56 | |
Target general information Gen name PFF0160c Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID NA Synonyms NA Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class Oxidoreductase Function Dihydroorotate dehydrogenase activity. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42573.5 Length 378 Aromaticity 0.1 Instability index 36.63 Isoelectric point 8.21 Charge (pH=7) 3.17 2D Binding mode Binding energy (Kcal/mol) -6.21 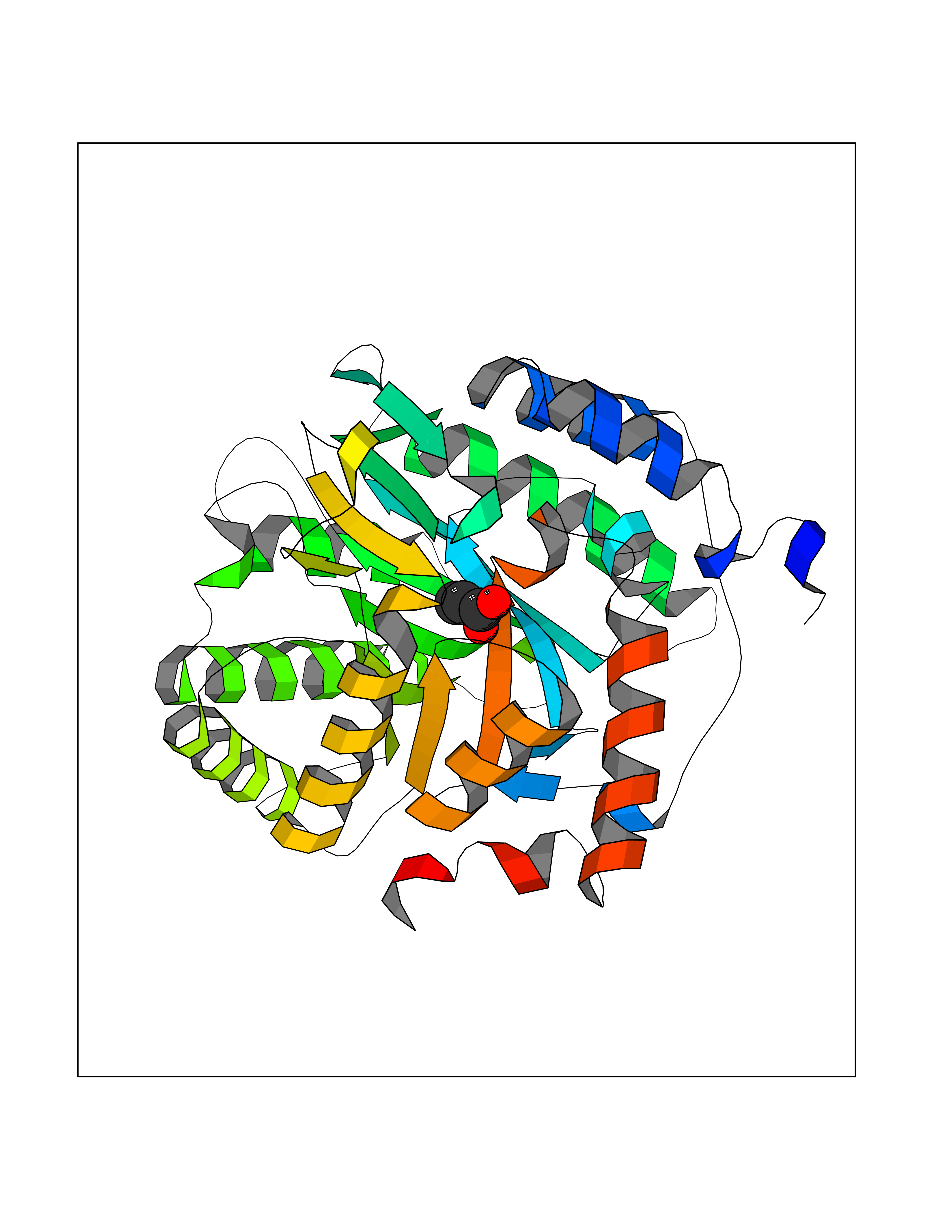 Molscript Map 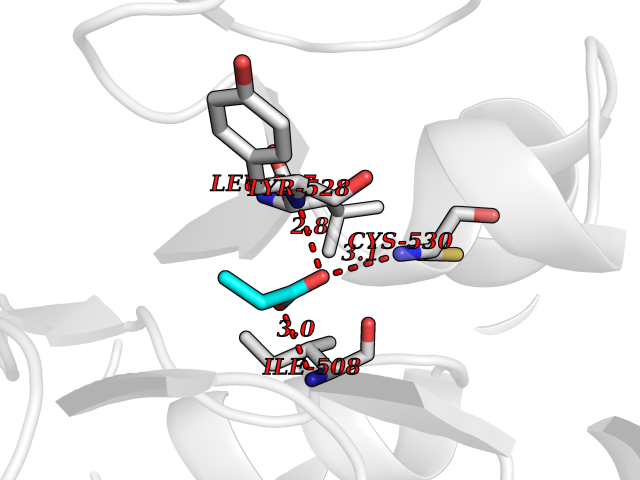 Pymol Map 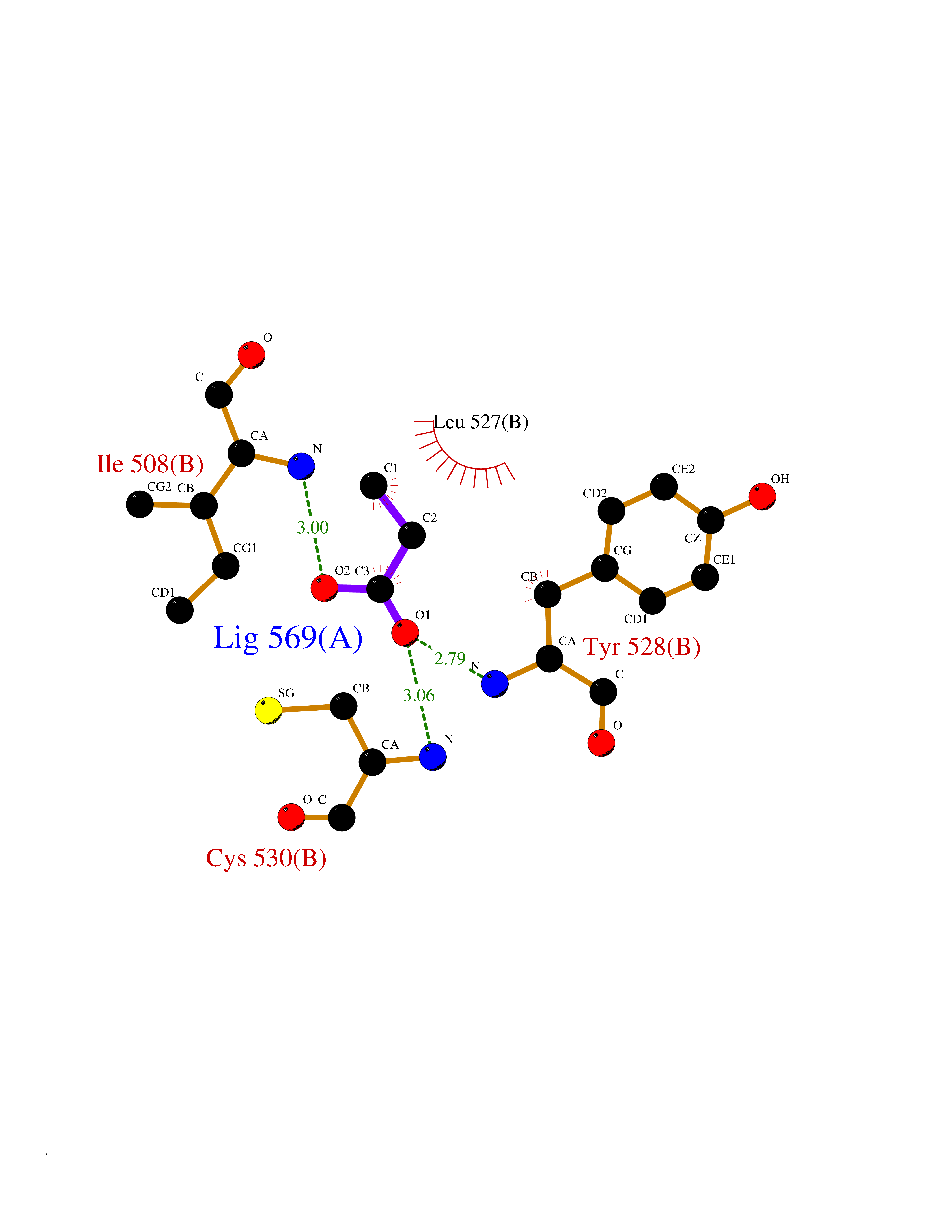 Ligplot Map 3D Binding mode Sequence ADPFESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEKNNFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKHS Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | NDR1 protein kinase (STK38) | 6BXI | 4.56 | |
Target general information Gen name STK38 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Serine/threonine-protein kinase 38; Nuclear Dbf2-related kinase 1; NDR1 Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family Biochemical class Kinase Function Converts MAP3K2 from its phosphorylated form to its non-phosphorylated form and inhibits autophosphorylation of MAP3K2. Negative regulator of MAP3K1/2 signaling. Related diseases Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. {ECO:0000269|PubMed:8955068}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:17332249}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 3 (NS3) [MIM:609942]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:16474405, ECO:0000269|PubMed:16773572, ECO:0000269|PubMed:17056636, ECO:0000269|PubMed:17468812, ECO:0000269|PubMed:19396835, ECO:0000269|PubMed:20949621}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Gastric cancer (GASC) [MIM:613659]: A malignant disease which starts in the stomach, can spread to the esophagus or the small intestine, and can extend through the stomach wall to nearby lymph nodes and organs. It also can metastasize to other parts of the body. The term gastric cancer or gastric carcinoma refers to adenocarcinoma of the stomach that accounts for most of all gastric malignant tumors. Two main histologic types are recognized, diffuse type and intestinal type carcinomas. Diffuse tumors are poorly differentiated infiltrating lesions, resulting in thickening of the stomach. In contrast, intestinal tumors are usually exophytic, often ulcerating, and associated with intestinal metaplasia of the stomach, most often observed in sporadic disease. {ECO:0000269|PubMed:14534542, ECO:0000269|PubMed:3034404, ECO:0000269|PubMed:7773929}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in KRAS are a cause of pylocytic astrocytoma (PA). Pylocytic astrocytomas are neoplasms of the brain and spinal cord derived from glial cells which vary from histologically benign forms to highly anaplastic and malignant tumors. {ECO:0000269|PubMed:16247081}.; DISEASE: Cardiofaciocutaneous syndrome 2 (CFC2) [MIM:615278]: A form of cardiofaciocutaneous syndrome, a multiple congenital anomaly disorder characterized by a distinctive facial appearance, heart defects and intellectual disability. Heart defects include pulmonic stenosis, atrial septal defects and hypertrophic cardiomyopathy. Some affected individuals present with ectodermal abnormalities such as sparse, friable hair, hyperkeratotic skin lesions and a generalized ichthyosis-like condition. Typical facial features are similar to Noonan syndrome. They include high forehead with bitemporal constriction, hypoplastic supraorbital ridges, downslanting palpebral fissures, a depressed nasal bridge, and posteriorly angulated ears with prominent helices. CFC2 patients often do not have the skin abnormalities, such as ichthyosis, hyperkeratosis, and hemangioma observed in CFC1. {ECO:0000269|PubMed:16474404, ECO:0000269|PubMed:16474405, ECO:0000269|PubMed:17056636, ECO:0000269|PubMed:20949621, ECO:0000269|PubMed:21797849}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: KRAS mutations are involved in cancer development. {ECO:0000269|PubMed:14534542, ECO:0000269|PubMed:1553789, ECO:0000269|PubMed:16533793, ECO:0000269|PubMed:24623306, ECO:0000269|PubMed:3034404, ECO:0000269|PubMed:3627975, ECO:0000269|PubMed:6092920, ECO:0000269|PubMed:6695174, ECO:0000269|PubMed:7773929}.; DISEASE: Oculoectodermal syndrome (OES) [MIM:600268]: A syndrome characterized by the association of epibulbar dermoids and aplasia cutis congenita. Affected individuals show multiple, asymmetric, atrophic, non-scarring and hairless regions that may be associated with hamartomas. Ectodermal changes include linear hyperpigmentation that may follow the lines of Blaschko and rarely epidermal nevus-like lesions. Epibulbar dermoids may be uni-or bilateral. Additional ocular anomalies such as skin tags of the upper eyelid, rarely optic nerve or retinal changes, and microphthalmia can be present. The phenotypic expression is highly variable, and various other abnormalities have occasionally been reported including growth failure, lymphedema, cardiovascular defects, as well as neurodevelopmental symptoms like developmental delay, epilepsy, learning difficulties, and behavioral abnormalities. Benign tumor-like lesions such as nonossifying fibromas of the long bones and giant cell granulomas of the jaws have repeatedly been observed and appear to be age-dependent, becoming a common manifestation in individuals aged 5 years or older. {ECO:0000269|PubMed:25808193, ECO:0000269|PubMed:26970110, ECO:0000269|PubMed:30891959}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Schimmelpenning-Feuerstein-Mims syndrome (SFM) [MIM:163200]: A disease characterized by sebaceous nevi, often on the face, associated with variable ipsilateral abnormalities of the central nervous system, ocular anomalies, and skeletal defects. Many oral manifestations have been reported, not only including hypoplastic and malformed teeth, and mucosal papillomatosis, but also ankyloglossia, hemihyperplastic tongue, intraoral nevus, giant cell granuloma, ameloblastoma, bone cysts, follicular cysts, oligodontia, and odontodysplasia. Sebaceous nevi follow the lines of Blaschko and these can continue as linear intraoral lesions, as in mucosal papillomatosis. {ECO:0000269|PubMed:30891959}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with P49407; P32121; Q8N9N5-2; Q03135; P08238; Q9H8S9; Q70IA6; P16333; P30086; P02638 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Chromosome; Cytoplasm; Direct protein sequencing; DNA damage; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 38701.1 Length 333 Aromaticity 0.11 Instability index 38.05 Isoelectric point 5.69 Charge (pH=7) -7.8 2D Binding mode Binding energy (Kcal/mol) -6.21 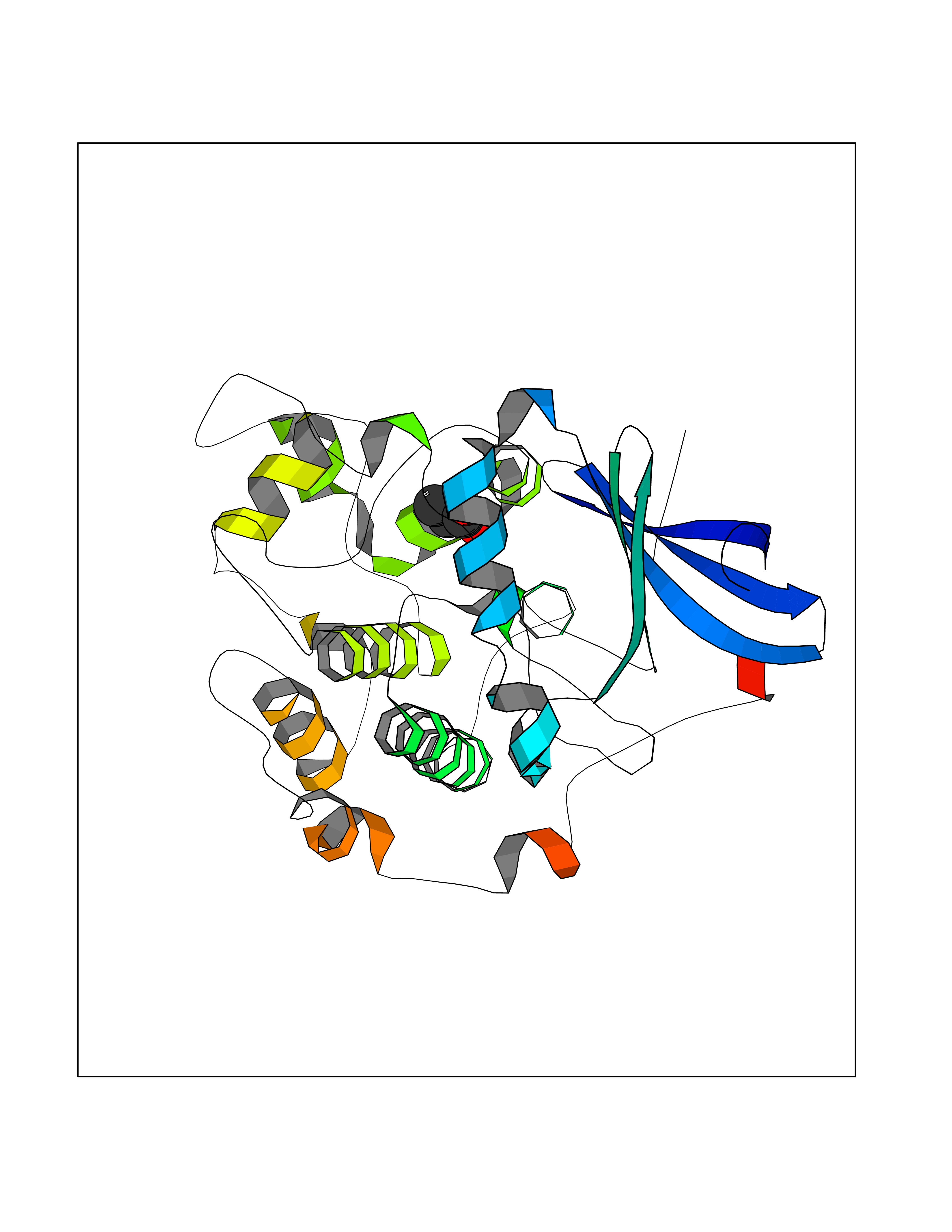 Molscript Map 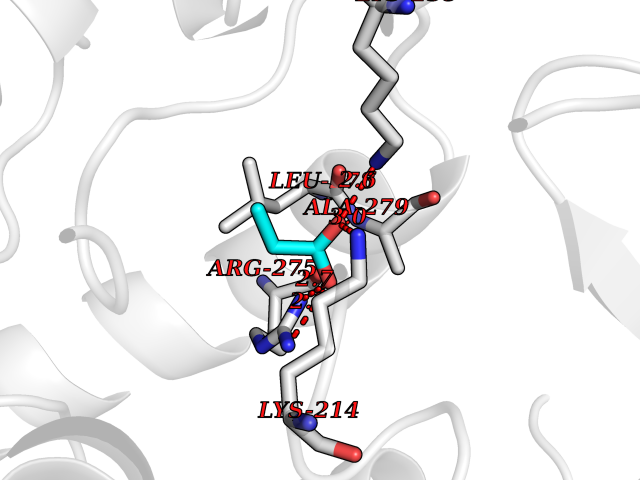 Pymol Map 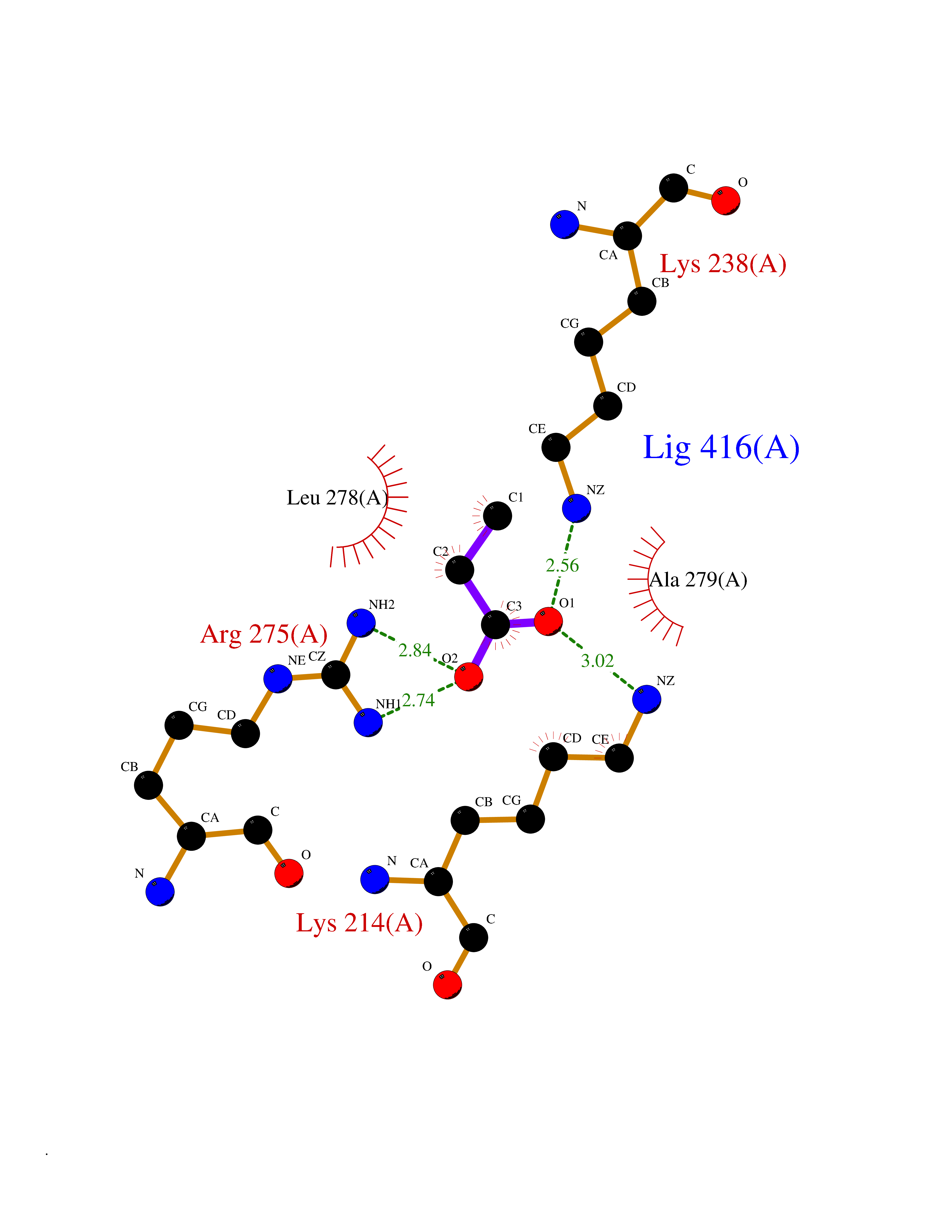 Ligplot Map 3D Binding mode Sequence TRLGLEDFESLKVIGRGAFGEVRLVQKKDTGHVYAMKILRKADMLEKEQVGHIRAERDILVEADSLWVVKMFYSFQDKLNLYLIMEFLPGGDMMTLLMKKDTLTEEETQFYIAETVLAIDSIHQLGFIHRDIKPDNLLLDSKGHVKLSDFGLCTGLKKAHRTEFYRNLNHSLPSDFTFQNMNSKRKAETWKRNRRQLAFSTVGTPDYIAPEVFMQTGYNKLCDWWSLGVIMYEMLIGYPPFCSETPQETYKKVMNWKETLTFPPEVPISEKAKDLILRFCCEWEHRIGAPGVEEIKSNSFFEGVDWEHIRERPAAISIEIKSIDDTSNFDEFP Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | cAMP-dependent protein kinase A type I (PRKAR1A) | 5KJZ | 4.56 | |
Target general information Gen name PRKAR1A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cAMP-dependent protein kinase type I-alpha regulatory subunit; Tissue-specific extinguisher 1; TSE1; PRKAR1; PKR1 Protein family CAMP-dependent kinase regulatory chain family Biochemical class Kinase Function Regulatory subunit of the cAMP-dependent protein kinases involved in cAMP signaling in cells. Related diseases Carney complex 1 (CNC1) [MIM:160980]: CNC is a multiple neoplasia syndrome characterized by spotty skin pigmentation, cardiac and other myxomas, endocrine tumors, and psammomatous melanotic schwannomas. {ECO:0000269|PubMed:15371594, ECO:0000269|PubMed:18241045, ECO:0000269|PubMed:22785148, ECO:0000269|PubMed:23323113, ECO:0000269|PubMed:26405036}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Intracardiac myxoma (INTMYX) [MIM:255960]: Inheritance is autosomal recessive. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Primary pigmented nodular adrenocortical disease 1 (PPNAD1) [MIM:610489]: A rare bilateral adrenal defect causing ACTH-independent Cushing syndrome. Macroscopic appearance of the adrenals is characteristic with small pigmented micronodules observed in the cortex. Clinical manifestations of Cushing syndrome include facial and truncal obesity, abdominal striae, muscular weakness, osteoporosis, arterial hypertension, diabetes. PPNAD1 is most often diagnosed in patients with Carney complex, a multiple neoplasia syndrome. However it can also be observed in patients without other manifestations. {ECO:0000269|PubMed:12213893}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Acrodysostosis 1, with or without hormone resistance (ACRDYS1) [MIM:101800]: A form of skeletal dysplasia characterized by short stature, severe brachydactyly, facial dysostosis, and nasal hypoplasia. Affected individuals often have advanced bone age and obesity. Laboratory studies show resistance to multiple hormones, including parathyroid, thyrotropin, calcitonin, growth hormone-releasing hormone, and gonadotropin. However, not all patients show endocrine abnormalities. {ECO:0000269|PubMed:21651393, ECO:0000269|PubMed:22464250, ECO:0000269|PubMed:22464252, ECO:0000269|PubMed:22723333, ECO:0000269|PubMed:23043190, ECO:0000269|PubMed:23425300, ECO:0000269|PubMed:26405036}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01790; DB02527; DB02315; DB05798 Interacts with Q9GZX7; P24588; O43687-2; Q9BSF0; Q9H6J7-2; Q86Y01; P0C7A2-2; Q9H0R8; Q9H8W4; P17612; P31321; P51817; P35250; Q86UC2; Q01105; Q8N0X7; O96006; P03259-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; cAMP; cAMP-binding; Cell membrane; Cushing syndrome; Direct protein sequencing; Disease variant; Disulfide bond; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Repeat Protein physicochemical properties Chain ID A Molecular weight (Da) 17007.4 Length 149 Aromaticity 0.08 Instability index 49.36 Isoelectric point 6.36 Charge (pH=7) -0.52 2D Binding mode Binding energy (Kcal/mol) -6.21 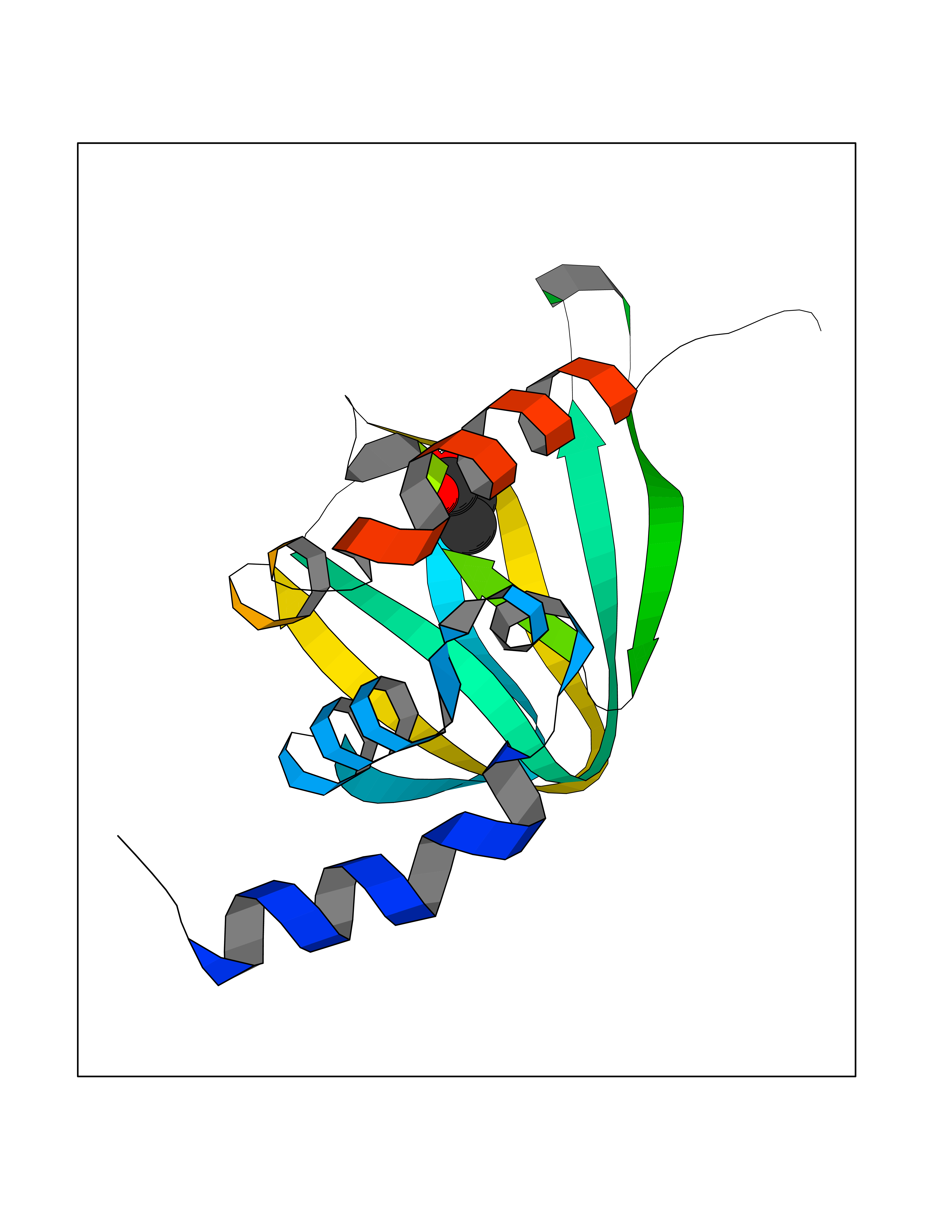 Molscript Map 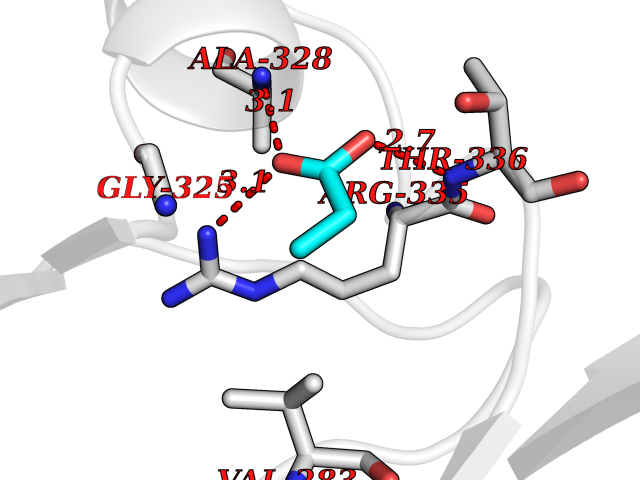 Pymol Map 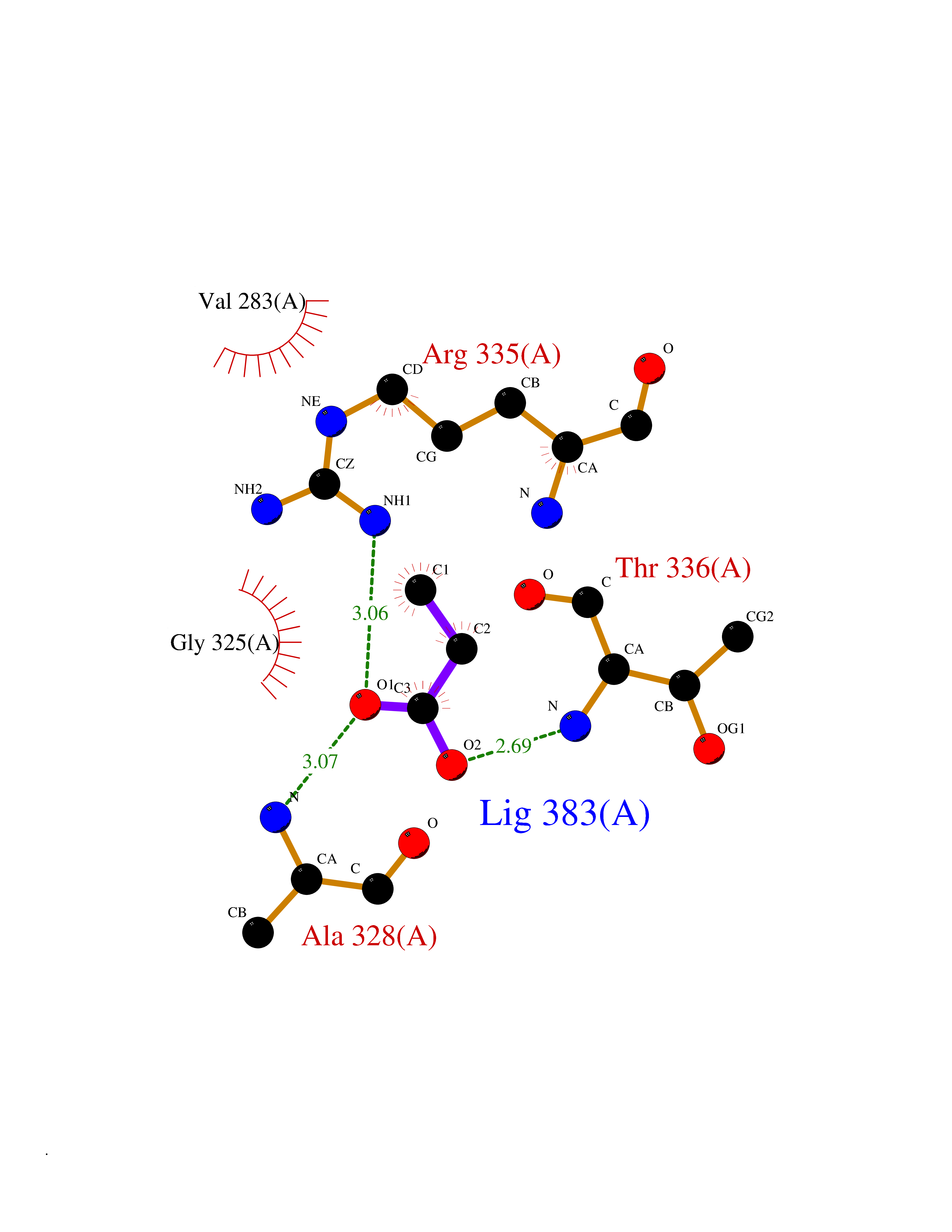 Ligplot Map 3D Binding mode Sequence SILMGSTLRKRKMYEEFLSKVSILESLDKWERLTVADALEPVQFEDGQKIVVQGEPGDEFFIILEGSAAVLQRRSENEEFVEVRRLGPSDYFGEIALLMNRPRTATVVARGPLKCVKLDRPRFERVLGPCSDILKRNIQQYNSFVSLSV Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | NAPE-hydrolyzing phospholipase D (NAPE-PLD) | 4QN9 | 4.56 | |
Target general information Gen name NAPEPLD Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NAPE-PLD; N-acyl-phosphatidylethanolamine-hydrolyzing phospholipase D; N-acyl phosphatidylethanolamine phospholipase D; C7orf18 Protein family NAPE-PLD family Biochemical class NA Function Hydrolyzes N-acyl-phosphatidylethanolamines (NAPEs) to produce N-acylethanolamines (NAEs) and phosphatidic acid. Responsible for the generation of these bioactive fatty acid ethanolamides (FAEs), including anandamide (N-arachidonoylethanolamine), the ligand of cannabinoid and vanilloid receptors. As a regulator of lipid metabolism in the adipose tissue, mediates the crosstalk between adipocytes, gut microbiota and immune cells to control body temperature and weight. In particular, regulates energy homeostasis by promoting cold-induced brown or beige adipocyte differentiation program to generate heat from fatty acids and glucose (By similarity). Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539269}. The gene represented in this entry is involved in disease pathogenesis. H3F3A mutations affecting residues involved in post-translational modifications of histone H3.3 are recurrent in malignant, aggressive gliomas including glioblastoma multiforme (GBM) and diffuse intrinsic pontine glioma (DIPG) (PubMed:22286061, PubMed:22286216). The mechanism through which mutations lead to tumorigenesis involves altered histones methylation, impaired regulation of Polycomb repressive complex 2 (PRC2) activity, and aberrant epigenetic regulation of gene expression (PubMed:23539183, PubMed:23539269, PubMed:23603901). {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539183, ECO:0000269|PubMed:23539269, ECO:0000269|PubMed:23603901}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 1 (BRYLIB1) [MIM:619720]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB1 is caused by variants in H3-3A. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 2 (BRYLIB2) [MIM:619721]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB2 is caused by variants in H3-3B. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: H3F3A and H3F3B mutations affecting residues involved in post-translational modifications of histone H3.3 are implicated in the pathogenesis of some bone and cartilage neoplasms. Mutations have been found with high prevalence in chondroblastoma and giant cell tumors of bone, and with low frequency in osteosarcoma, conventional chondrosarcoma and clear cell chondrosarcoma. Chondroblastoma samples frequently carry a H3F3B mutation affecting residue Lys-37 (H3K36), although H3F3A is mutated in some cases. Most giant cell tumors of bone harbor H3F3A mutations affecting residue Gly-35 (H3G34). {ECO:0000269|PubMed:24162739}. Drugs (DrugBank ID) DB14009 Interacts with Q6IQ20 EC number EC 3.1.4.54 Uniprot keywords 3D-structure; Acetylation; Endosome; Golgi apparatus; Hydrolase; Lipid degradation; Lipid metabolism; Membrane; Metal-binding; Nucleus; Phospholipid degradation; Phospholipid metabolism; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 74256.5 Length 643 Aromaticity 0.13 Instability index 48.34 Isoelectric point 5.65 Charge (pH=7) -17.61 2D Binding mode Binding energy (Kcal/mol) -6.22 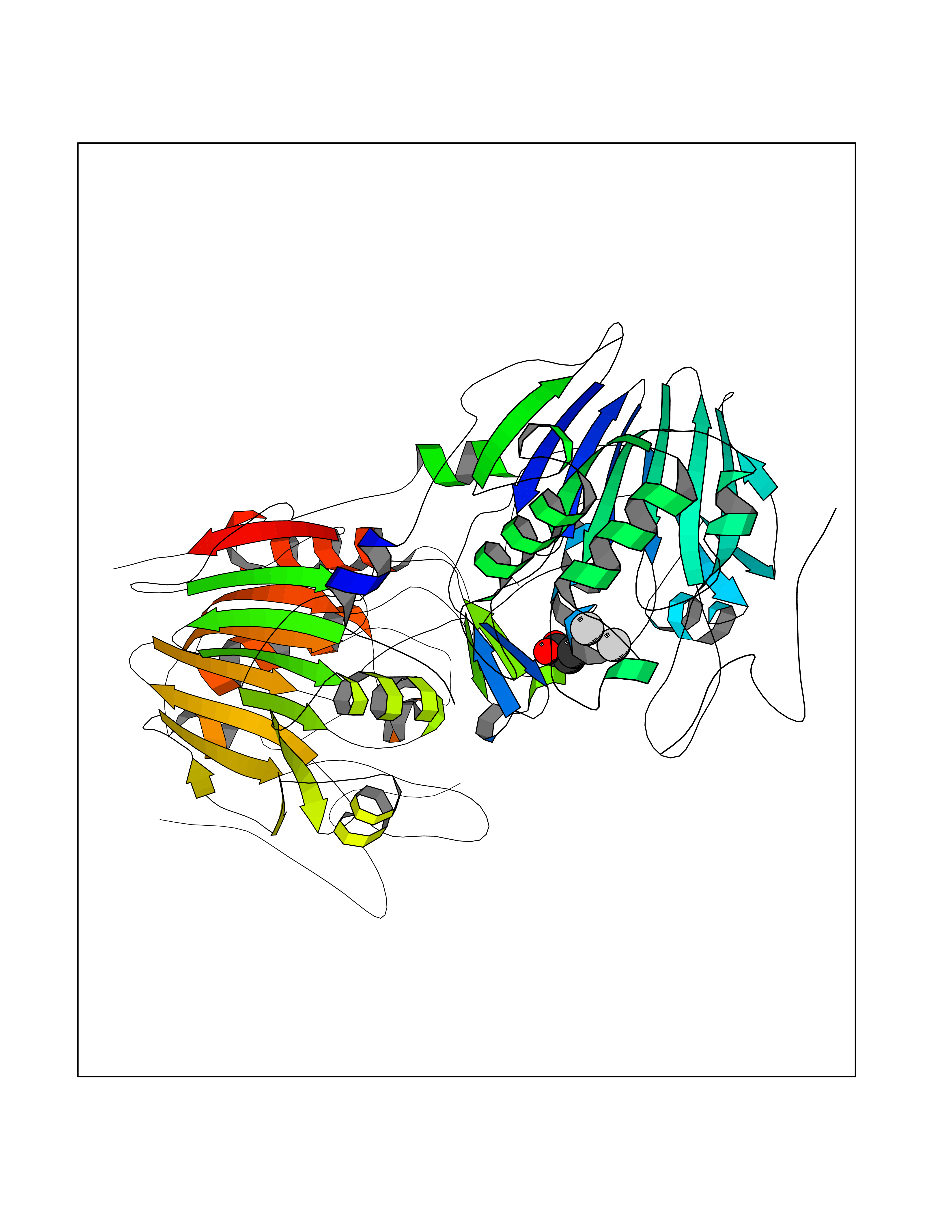 Molscript Map 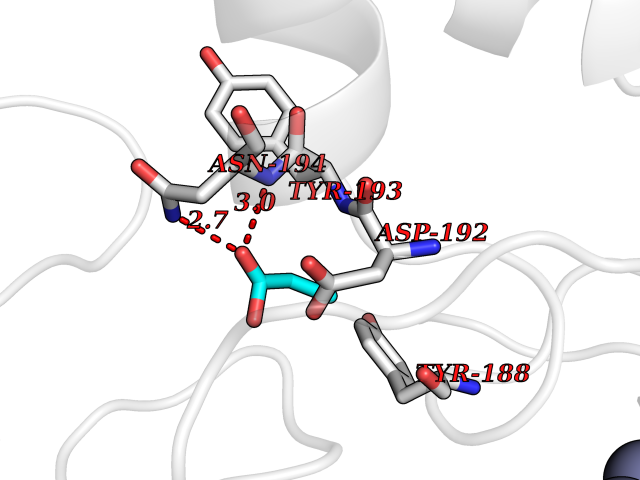 Pymol Map 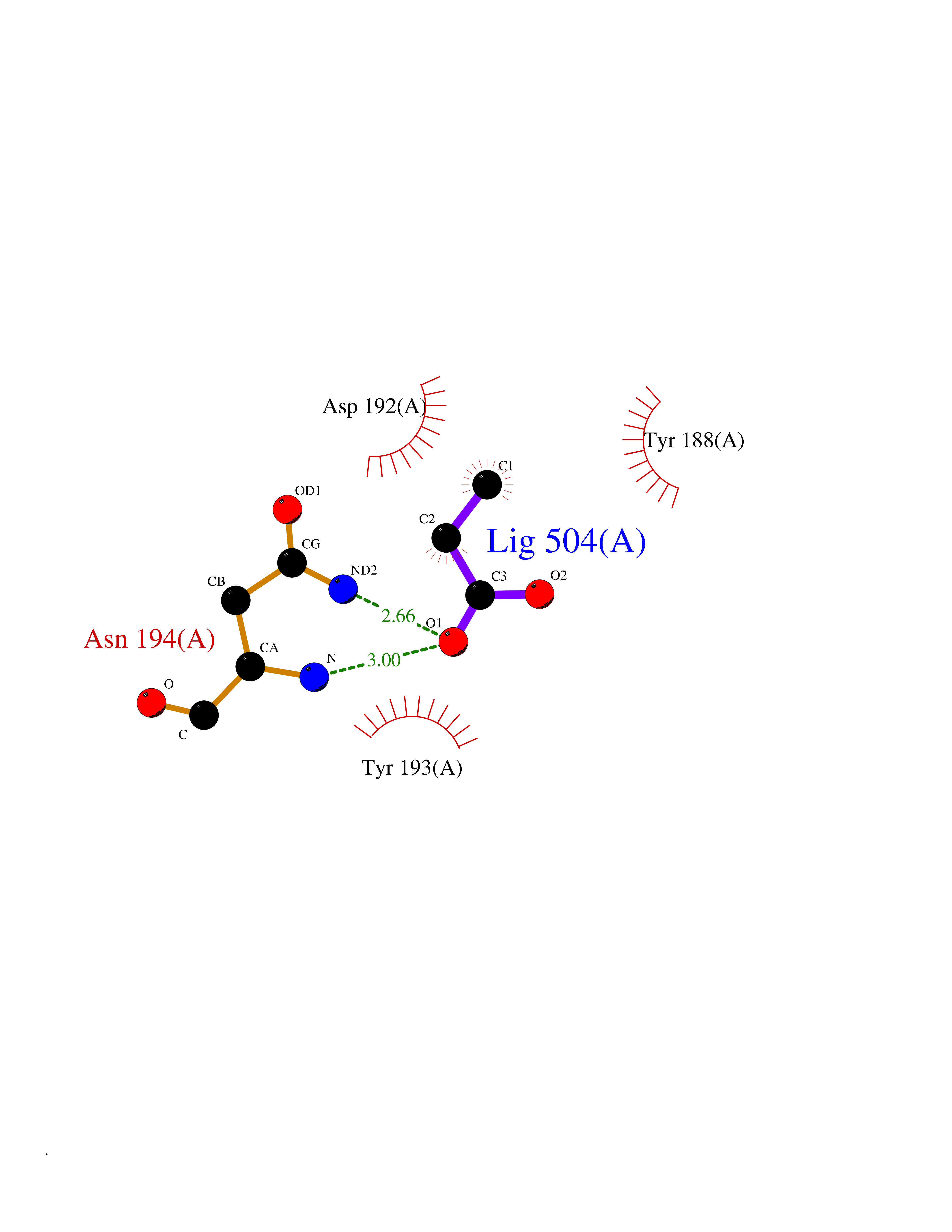 Ligplot Map 3D Binding mode Sequence SKKGKDGRFVNPWPTWKNPSIPNSSVPSSKEELDKELPVLKPYFITNPEEAGVREAGLRVTWLGHATVMVEMDELIFLTDPIFSSRASPSQYMGPKRFRRSPCTISELPPIDAVLISHNHYDHLDYNSVIALNERFGNELRWFVPLGLLDWMQKCGCENVIELDWWEENCVPGHDKVTFVFTPSQHWCKRTLMDDNKVLWGSWSVLGPWNRFFFAGDTGYCPAFEEIGKRFGPFDLAAIPIGAYEPRWFMKYQHVDPEEAVRIHTDVQTKKSMAIHWGTFALANEHYLEPPVKLNEALERYGLNAEDFFVLKHGESRYLNNSKKGKDGRFVNPWPTWKNPSIPNSSVPSSKEELDKELPVLKPYFITNPEEAGVREAGLRVTWLGHATVMVEMDELIFLTDPIFSSRASPSQYMGPKRFRRSPCTISELPPIDAVLISHNHYDHLDYNSVIALNERFGNELRWFVPLGLLDWMQKCGCENVIELDWWEENCVPGHDKVTFVFTPSQHWCKRTLMDDNKVLWGSWSVLGPWNRFFFAGDTGYCPAFEEIGKRFGPFDLAAIPIGAYEPRWFMKYQHVDPEEAVRIHTDVQTKKSMAIHWGTFALANEHYLEPPVKLNEALERYGLNAEDFFVLKHGESRYLNND Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Phosphatidylethanolamine-binding protein 1 (PEBP1) | 2QYQ | 4.56 | |
Target general information Gen name PEBP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Raf kinase inhibitor protein; RKIP; Prostatic-binding protein; PEBP-1; PEBP; PBP; Neuropolypeptide h3; Hippocampal cholinergic neurostimulating peptide; HCNPpp; HCNP Protein family Phosphatidylethanolamine-binding protein family Biochemical class Phosphatidylethanolamine-binding protein family Function Binds ATP, opioids and phosphatidylethanolamine. Has lower affinity for phosphatidylinositol and phosphatidylcholine. Serine protease inhibitor which inhibits thrombin, neuropsin and chymotrypsin but not trypsin, tissue type plasminogen activator and elastase. Inhibits the kinase activity of RAF1 by inhibiting its activation and by dissociating the RAF1/MEK complex and acting as a competitive inhibitor of MEK phosphorylation. Related diseases Retinitis pigmentosa 49 (RP49) [MIM:613756]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:15570217, ECO:0000269|PubMed:7479749}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09130; DB09568 Interacts with P16050; Q9NRD5; P04049; Q15208; Q9NS68; Q9JLL3 EC number NA Uniprot keywords 3D-structure; ATP-binding; Cytoplasm; Direct protein sequencing; Disulfide bond; Lipid-binding; Nucleotide-binding; Phosphoprotein; Protease inhibitor; Proteomics identification; Reference proteome; Serine protease inhibitor Protein physicochemical properties Chain ID A Molecular weight (Da) 20928.3 Length 186 Aromaticity 0.1 Instability index 24.05 Isoelectric point 6.59 Charge (pH=7) -0.98 2D Binding mode Binding energy (Kcal/mol) -6.22  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MPVDLSKWSGPLSLQEVDEQPQHPLHVTYAGAAVDELGKVLTPTQVKNRPTSISWDGLDSGKLYTLVLTDPDAPSRKDPKYREWHHFLVVNMKGNDISSGTVLSDYVGSGPPKGTGLHRYVWLVYEQDRPLKCDEPILSNRSGDHRGKFKVASFRKKYELRAPVAGTCYQAEWDDYVPKLYEQLSG Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Glutathione S-transferase LANCL1 (LANCL1) | 3E73 | 4.56 | |
Target general information Gen name LANCL1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p40; LanC-like protein 1; GPR69A; 40 kDa erythrocyte membrane protein Protein family LanC-like protein family Biochemical class NA Function Functions as glutathione transferase. Catalyzes conjugation of the glutathione (GSH) to artificial substrates 1-chloro-2,4-dinitrobenzene (CDNB) and p-nitrophenyl acetate. Mitigates neuronal oxidative stress during normal postnatal development and in response to oxidative stresses probably through GSH antioxidant defense mechanism (By similarity). May play a role in EPS8 signaling. Binds glutathione. Related diseases Spermatogenic failure 5 (SPGF5) [MIM:243060]: An infertility disorder caused by spermatogenesis defects. Semen from affected men show close to 100% morphologically abnormal multiflagellar spermatozoa with low motility, oversized irregular heads, and abnormal midpiece and acrosome. {ECO:0000269|PubMed:17435757, ECO:0000269|PubMed:21733974}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9UHR4; P42858; Q08509 EC number EC 2.5.1.18 Uniprot keywords 3D-structure; Acetylation; Cell membrane; Cytoplasm; Direct protein sequencing; Membrane; Metal-binding; Proteomics identification; Reference proteome; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 46005.4 Length 405 Aromaticity 0.14 Instability index 33.7 Isoelectric point 7.13 Charge (pH=7) 0.4 2D Binding mode Binding energy (Kcal/mol) -6.22 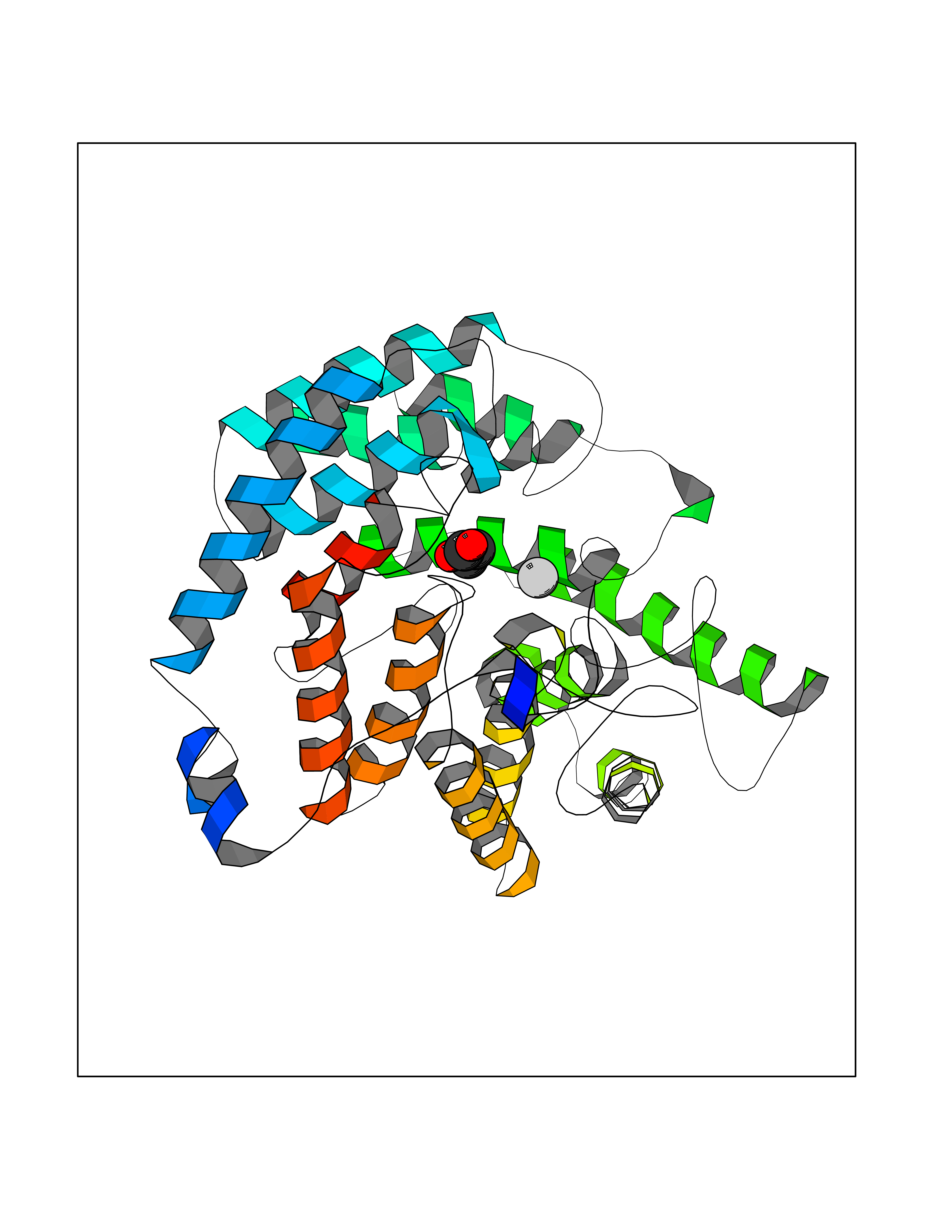 Molscript Map 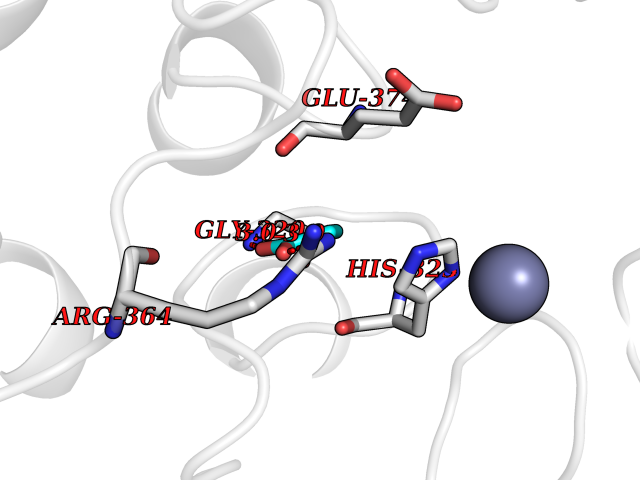 Pymol Map 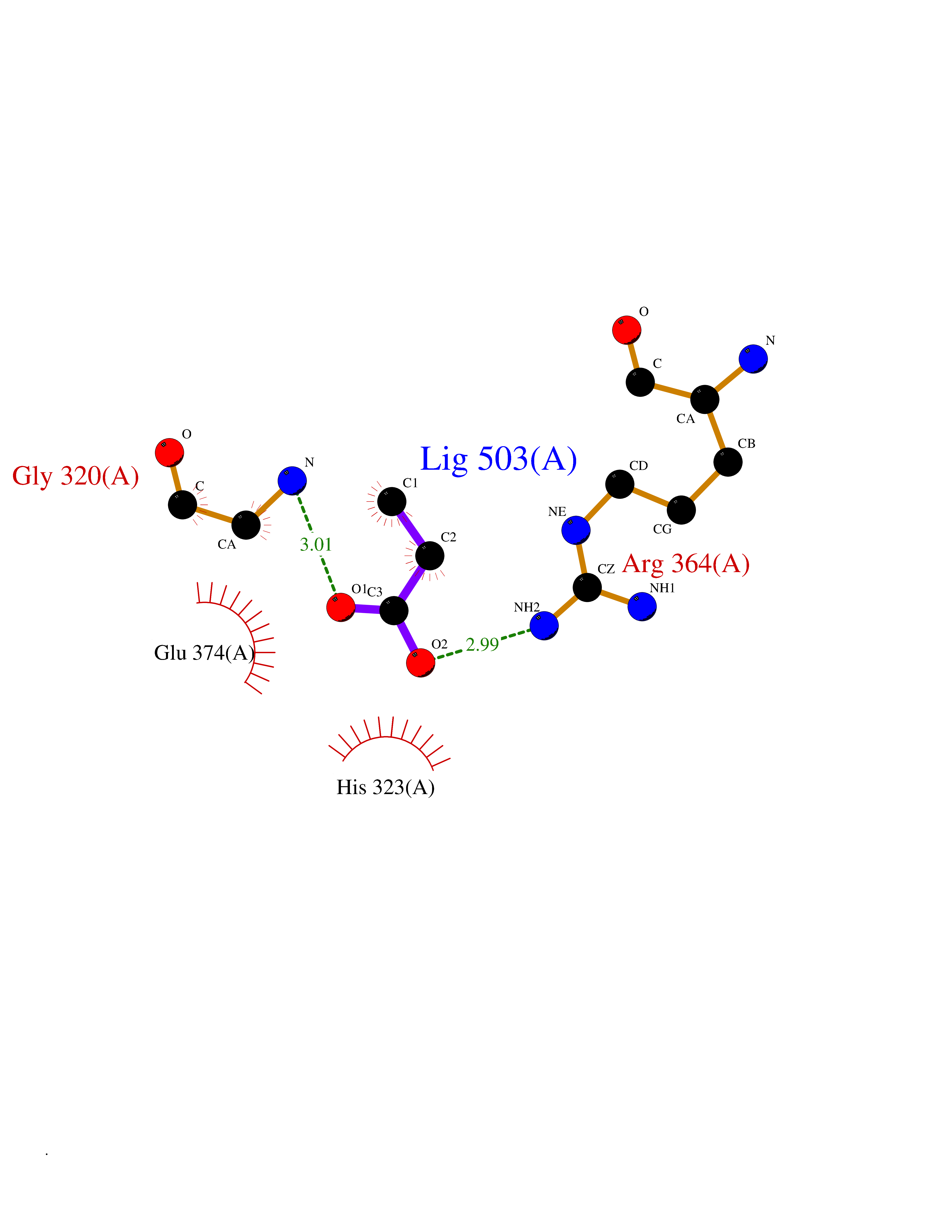 Ligplot Map 3D Binding mode Sequence SMDIEFMAQRAFPNPYADYNKSLAEGYFDAAGRLTPEFSQRLTNKIRELLQQMERGLKSADPRDGTGYTGWAGIAVLYLHLYDVFGDPAYLQLAHGYVKQSLNCLTKRSITFLCGDAGPLAVAAVLYHKMNNEKQAEDCITRLIHLNKIDPHAPNEMLYGRIGYIYALLFVNKNFGVEKIPQSHIQQICETILTSGENLARKRNFTAKSPLMYEWYQEYYVGAAHGLAGIYYYLMQPSLQVSQGKLHSLVKPSVDYVCQLKFPSGNYPPCIGDNRDLLVHWCHGAPGVIYMLIQAYKVFREEKYLCDAYQCADVIWQYGLLKKGYGLCHGSAGNAYAFLTLYNLTQDMKYLYRACKFAEWCLEYGEHGCRTPDTPFSLFEGMAGTIYFLADLLVPTKARFPAFEL Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Non-heme chloroperoxidase | 1A8U | 4.55 | |
Target general information Gen name cpo Organism Kitasatospora aureofaciens (Streptomyces aureofaciens) Uniprot ID TTD ID NA Synonyms cpoT Protein family AB hydrolase superfamily, Bacterial non-heme haloperoxidase / perhydrolase family Biochemical class Haloperoxidase Function Chloride peroxidase activity. Related diseases Hypervalinemia and hyperleucine-isoleucinemia (HVLI) [MIM:618850]: An autosomal recessive metabolic disorder characterized by highly elevated plasma concentrations of valine and leucine/isoleucine. Affected individuals suffer from headache and mild memory impairment. {ECO:0000269|PubMed:25653144}. The disease is caused by variants affecting the gene represented in this entry. A patient with hypervalinemia and hyperleucine-isoleucinemia was identified as compound heterozygote for Gln-170 (inherited from his father) and Lys-264 (inherited from his mother), both variants reduced the catalytic activity of the enzyme. After treatment with vitamin B6, a precursor of pyridoxal 5'-phosphate, a BCAT2 cofactor, the blood levels of branched chain amino acids, especially valine, were decreased and brain lesions were improved. {ECO:0000269|PubMed:25653144}. Drugs (DrugBank ID) DB03793 Interacts with NA EC number 1.11.1.- Uniprot keywords 3D-structure; Chloride; Oxidoreductase; Peroxidase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 60428.4 Length 554 Aromaticity 0.13 Instability index 31.26 Isoelectric point 4.65 Charge (pH=7) -32.72 2D Binding mode Binding energy (Kcal/mol) -6.21 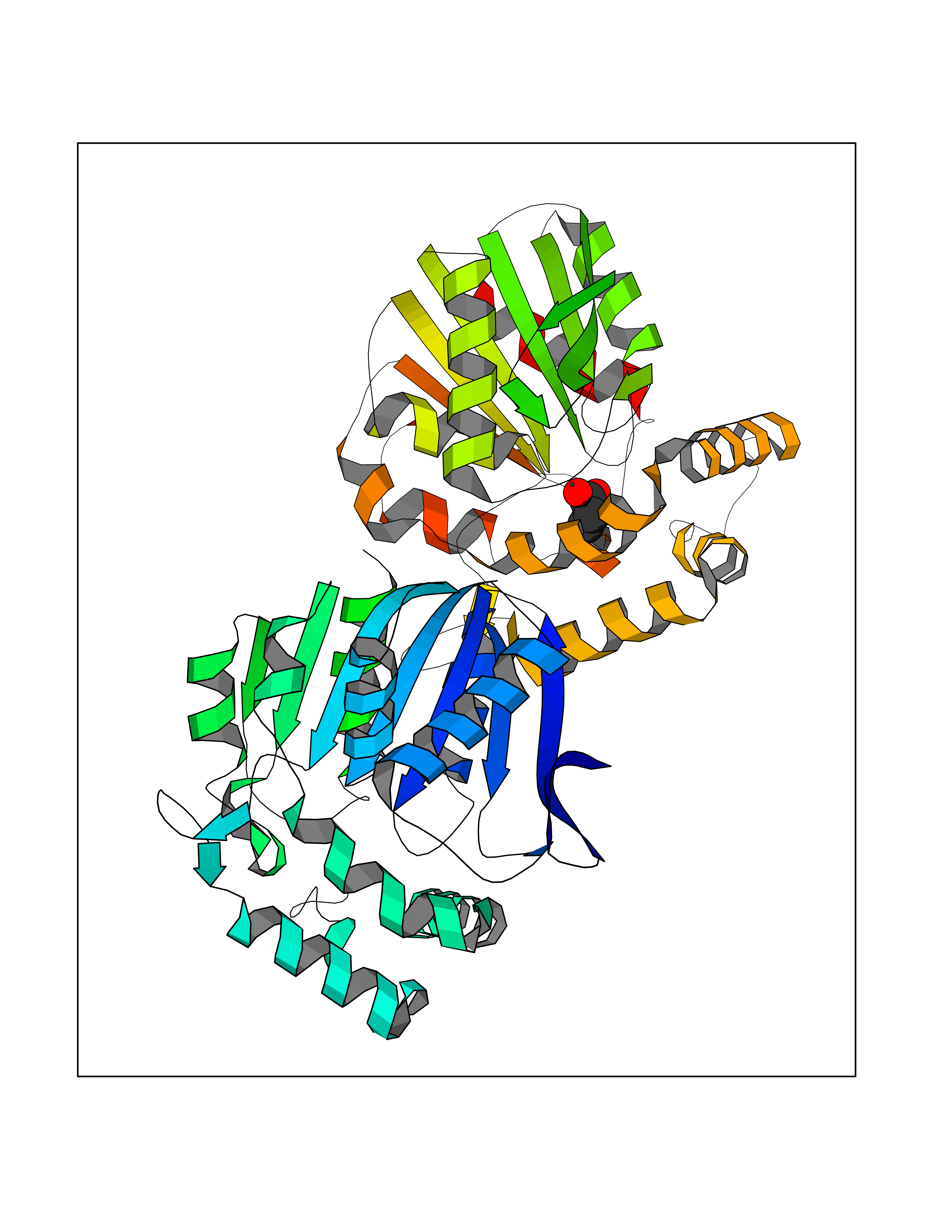 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence PFITVGQENSTSIDLYYEDHGAGQPVVLIHGFPLSGHSWERQSAALLDAGYRVITYDRRGFGQSSQPTTGYDYDTFAADLNTVLETLDLQDAVLVGFSMGTGEVARYVSSYGTARIAKVAFLASLEPFLLKTDDNPDGAAPKEFFDGIVAAVKADRYAFYTGFFNDFYNLDENLGTRISEEAVRNSWNTAASGGFFAAAAAPTTWYTDFRADIPRIDVPALILHGTGDRTLPIENTARVFHKALPSAEYVEVEGAPHGLLWTHAEEVNTALLAFLAKPFITVGQENSTSIDLYYEDHGAGQPVVLIHGFPLSGHSWERQSAALLDAGYRVITYDRRGFGQSSQPTTGYDYDTFAADLNTVLETLDLQDAVLVGFSMGTGEVARYVSSYGTARIAKVAFLASLEPFLLKTDDNPDGAAPKEFFDGIVAAVKADRYAFYTGFFNDFYNLDENLGTRISEEAVRNSWNTAASGGFFAAAAAPTTWYTDFRADIPRIDVPALILHGTGDRTLPIENTARVFHKALPSAEYVEVEGAPHGLLWTHAEEVNTALLAFLAK Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Type 2 DNA topoisomerase 6 subunit B (EC 5.6.2.2) (Type II DNA topoisomerase VI subunit B) (TopoVI-B) | 2HKJ | 4.55 | |
Target general information Gen name top6B Organism Saccharolobus shibatae (strain ATCC 51178 / DSM 5389 / JCM 8931 / NBRC 15437 / B12) (Sulfolobus shibatae) Uniprot ID TTD ID NA Synonyms J5U23_02508 Protein family TOP6B family Biochemical class NA Function Relaxes both positive and negative supercoils and exhibits a strong decatenase and unknotting activity; it cannot introduce DNA supercoils (PubMed:7961685). ATP is absolutely required for DNA cleavage; the nonhydrolyzable analog AMP-PNP generates nicked or linear products from a supercoiled dsDNA substrate. Generates staggered two-nucleotide long 5' overhangs. The enzyme is covalently attached transiently to the 5'-ends of the cleaved strands (PubMed:11485995). {ECO:0000269|PubMed:11485995, ECO:0000269|PubMed:7961685}." Related diseases Neuropathy, hereditary motor and sensory, 6C, with optic atrophy (HMSN6C) [MIM:618511]: An autosomal recessive neurologic disorder characterized by childhood onset of axonal, sensorimotor polyneuropathy affecting mainly the lower limbs, and adult-onset optic atrophy. Clinical features include progressive distal muscle weakness and atrophy, significant standing and walking difficulties, areflexia, neurogenic pain and progressive visual impairment. {ECO:0000269|PubMed:31187503}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O05208; O05207 EC number 5.6.2.2 Uniprot keywords 3D-structure; ATP-binding; Direct protein sequencing; DNA-binding; Isomerase; Nucleotide-binding; Topoisomerase Protein physicochemical properties Chain ID A Molecular weight (Da) 52553.8 Length 461 Aromaticity 0.09 Instability index 38.32 Isoelectric point 8.62 Charge (pH=7) 4.35 2D Binding mode Binding energy (Kcal/mol) -6.2  Molscript Map  Pymol Map 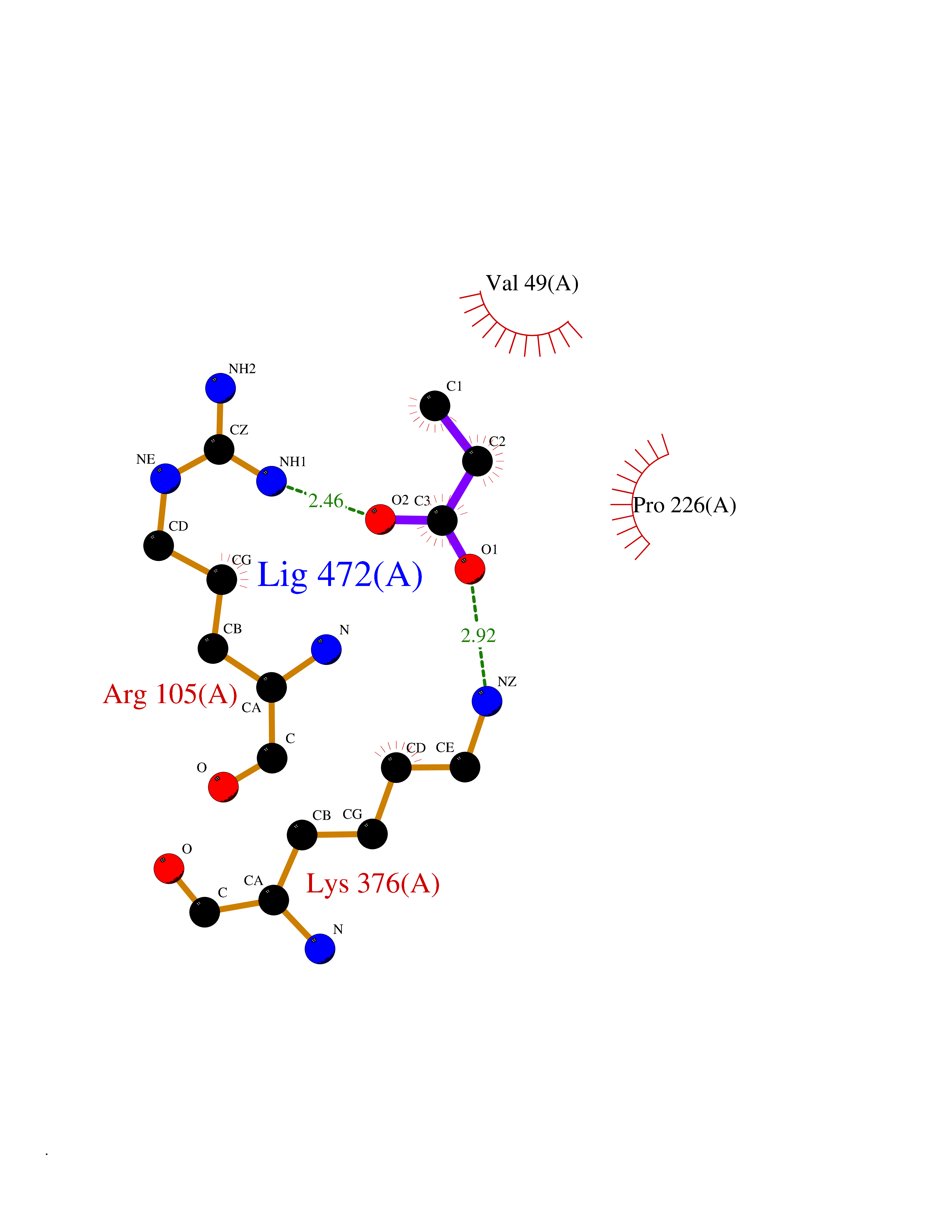 Ligplot Map 3D Binding mode Sequence LSPAEFFKRNPELAGFPNPARALYQTVRELIENSLDATDVHGILPNIKITIDLIDDARQIYKVNVVDNGIGIPPQEVPNAFGRVLYSSKYVNRQTRGMYGLGVKAAVLYSQMHQDKPIEIETSPVNSKRIYTFKLKIDINKNEPIIVERGSVENTRGFHGTSVAISIPGDWPKAKSRIYEYIKRTYIITPYAEFIFKDPEGNVTYYPRLTNKIPKPPQEVKPHPYGVDREEIKILINNLKRDYTIKEFLVNEFQSIGDTTADKILELAGLKPNKKVKNLTEEEITRLVETFKKYEDFRSPSADSLSVIGEDLIELGLKKIFNPDFAASITRKPKAYQGHPFIVEAGVAFGGSIPVGEEPIVLRYANKIPLIYDEKSDVIWKVVEELDWKRYGIESDQYQMVVMVHLCSTKIPYKSAGKESIAEVEDIEKEIKNALMEVARKLKQYLSEKRKEQEAKKKLLA Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Adrenergic receptor beta-2 (ADRB2) | 2RH1 | 4.55 | |
Target general information Gen name ADRB2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Beta-2 adrenoreceptor; Beta-2 adrenoceptor; Beta-2 adrenergic receptor; B2AR; ADRB2R Protein family G-protein coupled receptor 1 family, Adrenergic receptor subfamily, ADRB2 sub-subfamily Biochemical class GPCR rhodopsin Function The beta-2-adrenergic receptor binds epinephrine with an approximately 30-fold greater affinity than it does norepinephrine. Beta-adrenergic receptors mediate the catecholamine-induced activation of adenylate cyclase through the action of G proteins. Related diseases Cortical dysplasia, complex, with other brain malformations 6 (CDCBM6) [MIM:615771]: A disorder of aberrant neuronal migration and disturbed axonal guidance. Affected individuals have microcephaly, ataxia, and severe delayed psychomotor development. Brain imaging shows variable malformations of cortical development, including white matter streaks, dysmorphic basal ganglia, corpus callosum abnormalities, brainstem and cerebellar hypoplasia, cortical dysplasia, polymicrogyria. {ECO:0000269|PubMed:23246003}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Skin creases, congenital symmetric circumferential, 1 (CSCSC1) [MIM:156610]: An autosomal dominant disease characterized by multiple, symmetric, circumferential rings of folded skin, affecting primarily the limbs. Affected individuals also exhibit intellectual disability, cleft palate, and dysmorphic features. {ECO:0000269|PubMed:26637975}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07543; DB01193; DB00866; DB01118; DB00182; DB01102; DB01274; DB01238; DB09204; DB06216; DB00335; DB01408; DB05590; DB09013; DB00195; DB00217; DB01295; DB00612; DB00901; DB08807; DB06726; DB08808; DB00248; DB00521; DB01136; DB04846; DB01407; DB00785; DB01151; DB11273; DB13345; DB00449; DB11278; DB00841; DB09273; DB06262; DB01363; DB01364; DB00668; DB01049; DB11587; DB01288; DB00983; DB05039; DB00221; DB01064; DB00598; DB01210; DB13139; DB01365; DB13624; DB01214; DB00264; DB01203; DB05849; DB04861; DB00368; DB00540; DB00334; DB09080; DB00816; DB01580; DB00715; DB01359; DB00925; DB00397; DB00960; DB01291; DB01366; DB01182; DB00571; DB06814; DB00852; DB01917; DB11124; DB00867; DB01001; DB00938; DB00489; DB03566; DB00127; DB00871; DB00373; DB00726; DB12248; DB09082; DB09185 Interacts with P30542; P07550; P32121; Q96B67; Q9UII2; Q9ULD4-2; Q9NSI6-4; Q5M9N0-2; A0AVK6; Q658K8; O00472; Q15910-2; Q15486; P61978; Q5TCQ9; Q99685; O14745; Q9NR21-5; Q8WVD3; Q9H0X6; Q13573; P12931; Q5T0J7-2; Q8N0U2 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; Endosome; G-protein coupled receptor; Glycoprotein; Golgi apparatus; Hydroxylation; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32266.1 Length 282 Aromaticity 0.15 Instability index 36.1 Isoelectric point 8.02 Charge (pH=7) 2.1 2D Binding mode Binding energy (Kcal/mol) -6.2 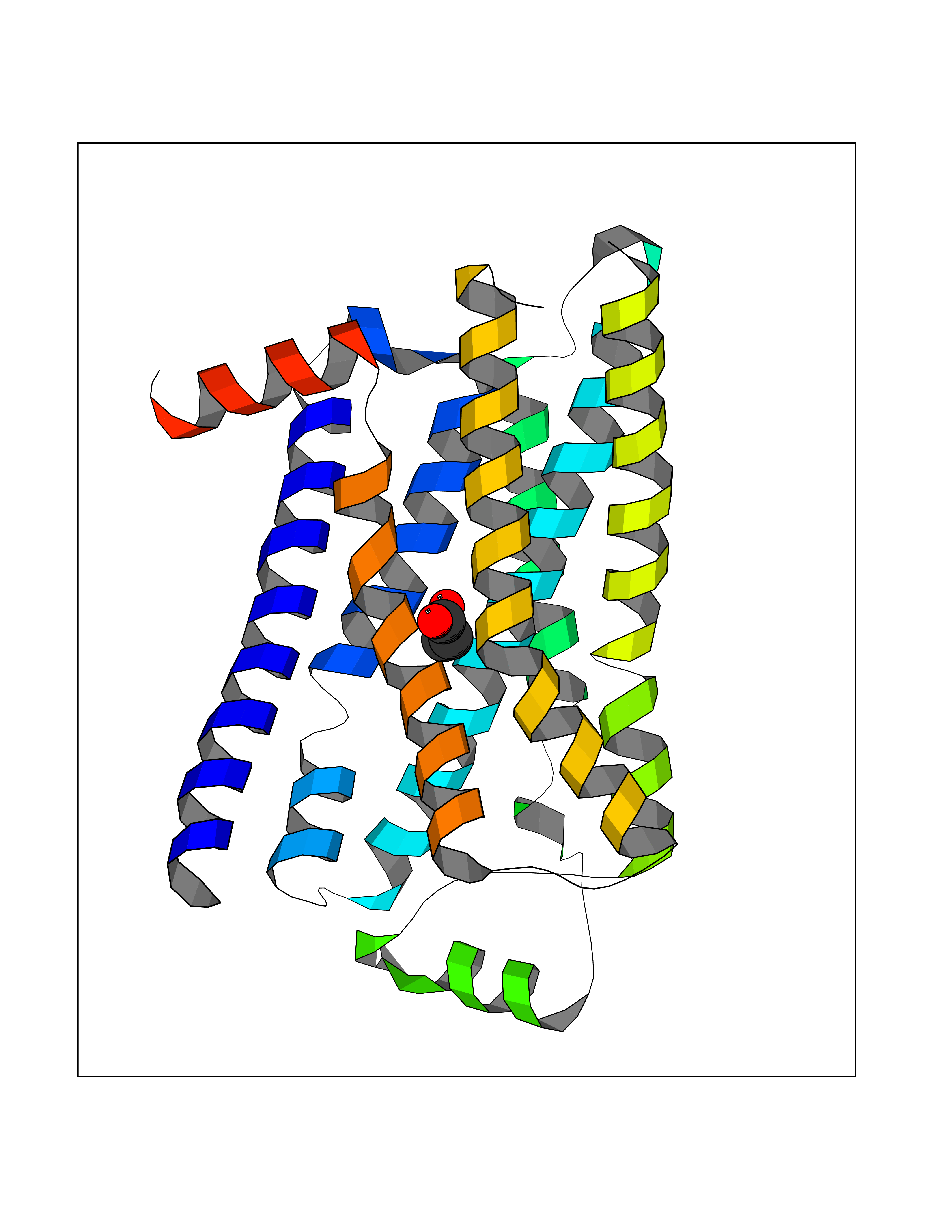 Molscript Map 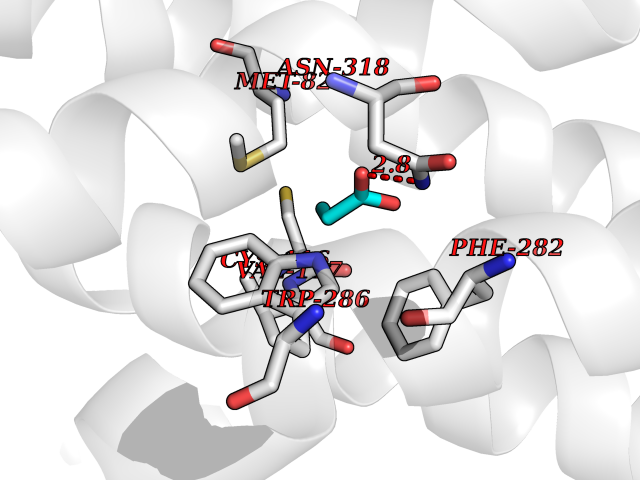 Pymol Map 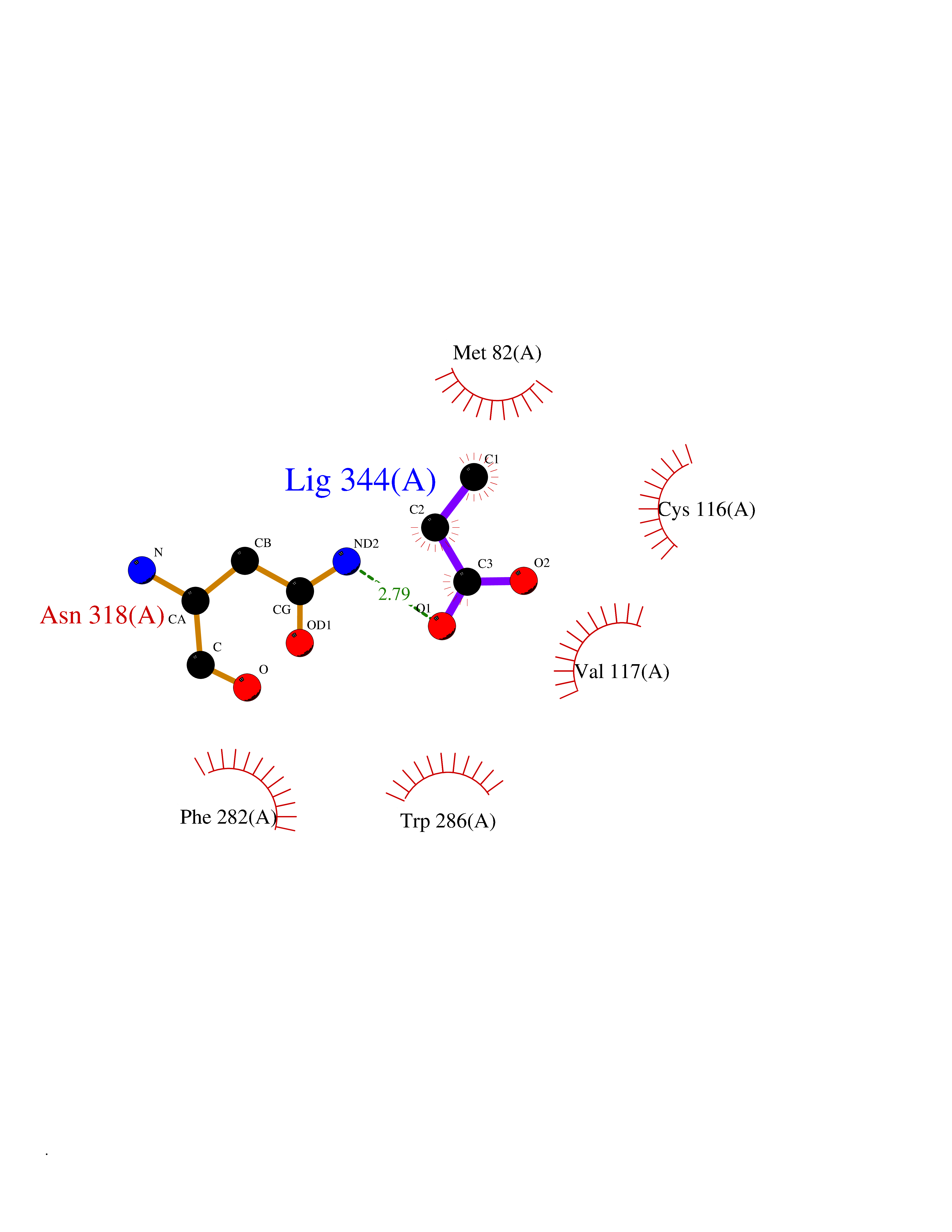 Ligplot Map 3D Binding mode Sequence DEVWVVGMGIVMSLIVLAIVFGNVLVITAIAKFERLQTVTNYFITSLACADLVMGLAVVPFGAAHILMKMWTFGNFWCEFWTSIDVLCVTASIETLCVIAVDRYFAITSPFKYQSLLTKNKARVIILMVWIVSGLTSFLPIQMHWYRATHQEAINCYAEETCCDFFTNQAYAIASSIVSFYVPLVIMVFVYSRVFQEAKRQLKFCLKEHKALKTLGIIMGTFTLCWLPFFIVNIVHVIQDNLIRKEVYILLNWIGYVNSGFNPLIYCRSPDFRIAFQELLCL Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Acetylcholinesterase (AChE) | 4M0E | 4.55 | |
Target general information Gen name ACHE Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms YT; N-ACHE; ARACHE Protein family Type-B carboxylesterase/lipase family Biochemical class Carboxylic ester hydrolase Function Role in neuronal apoptosis. Terminates signal transduction at the neuromuscular junction by rapid hydrolysis of the acetylcholine released into the synaptic cleft. Related diseases Phosphoribosylaminoimidazole carboxylase deficiency (PAICSD) [MIM:619859]: An autosomal recessive inborn error of purine metabolism, clinically characterized by multiple congenital anomalies and early neonatal death. {ECO:0000269|PubMed:31600779}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07846; DB02673; DB04617; DB04614; DB04615; DB07756; DB07701; DB02404; DB03814; DB08615; DB02343; DB02226; DB03005; DB04114; DB03128; DB01122; DB03283; DB00411; DB00122; DB14006; DB01245; DB00944; DB08357; DB08996; DB00449; DB00843; DB01010; DB01364; DB00898; DB00674; DB00483; DB06525; DB04864; DB03348; DB07555; DB00677; DB04924; DB03359; DB00358; DB00940; DB02825; DB02845; DB08167; DB04021; DB00805; DB01805; DB03740; DB04556; DB01400; DB04892; DB00981; DB00733; DB02166; DB00545; DB00863; DB00989; DB00382; DB04616; DB12816; DB01199; DB13503; DB04859 Interacts with Q9Y215; P06733; P63244 EC number EC 3.1.1.7 Uniprot keywords 3D-structure; Alternative splicing; Blood group antigen; Cell membrane; Direct protein sequencing; Disulfide bond; Glycoprotein; GPI-anchor; Hydrolase; Lipoprotein; Membrane; Neurotransmitter degradation; Nucleus; Proteomics identification; Reference proteome; Secreted; Serine esterase; Signal; Synapse Protein physicochemical properties Chain ID A,B Molecular weight (Da) 58804.1 Length 537 Aromaticity 0.11 Instability index 40.85 Isoelectric point 5.73 Charge (pH=7) -8.18 2D Binding mode Binding energy (Kcal/mol) -6.2 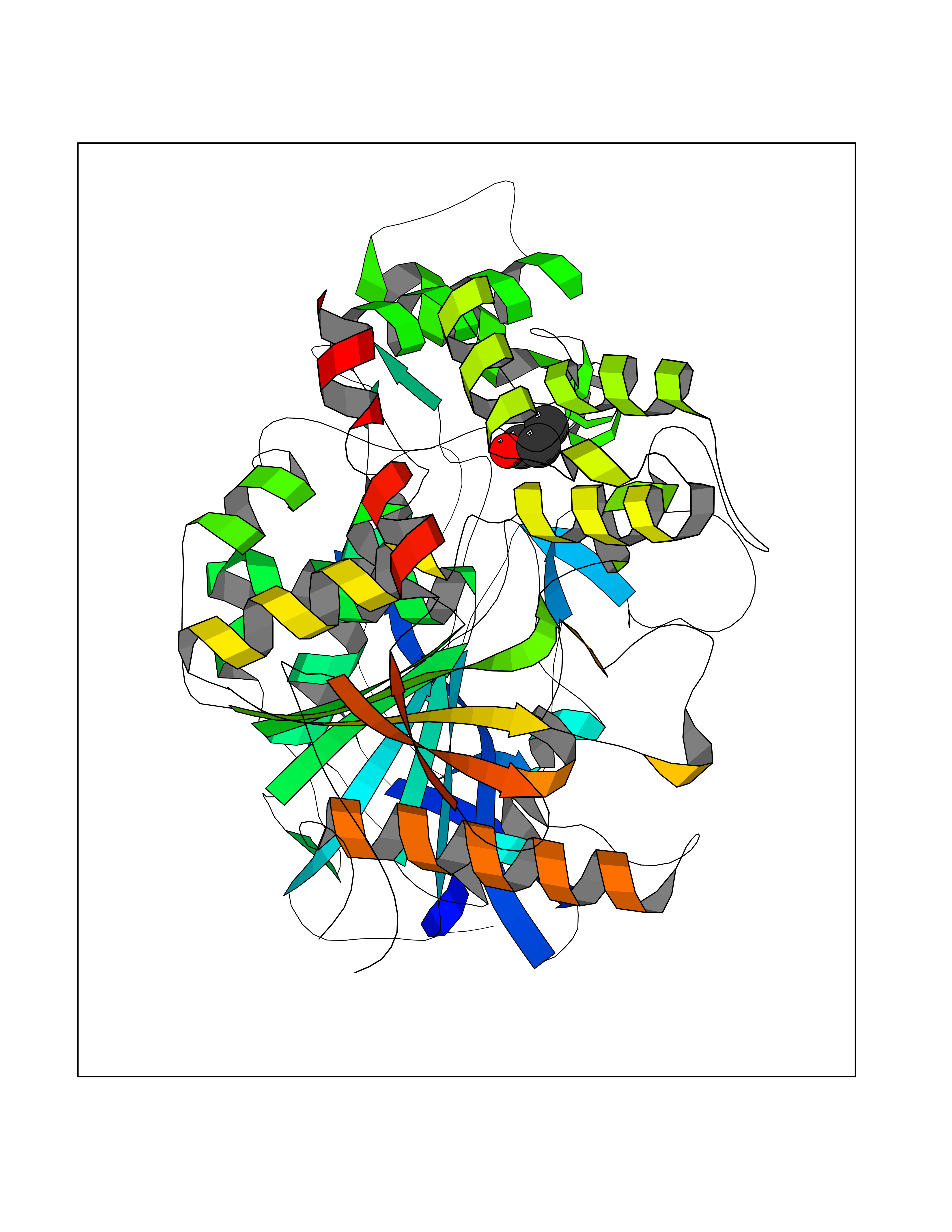 Molscript Map 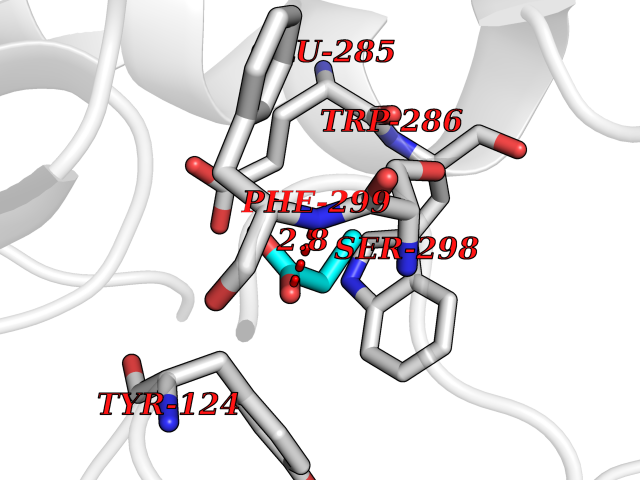 Pymol Map 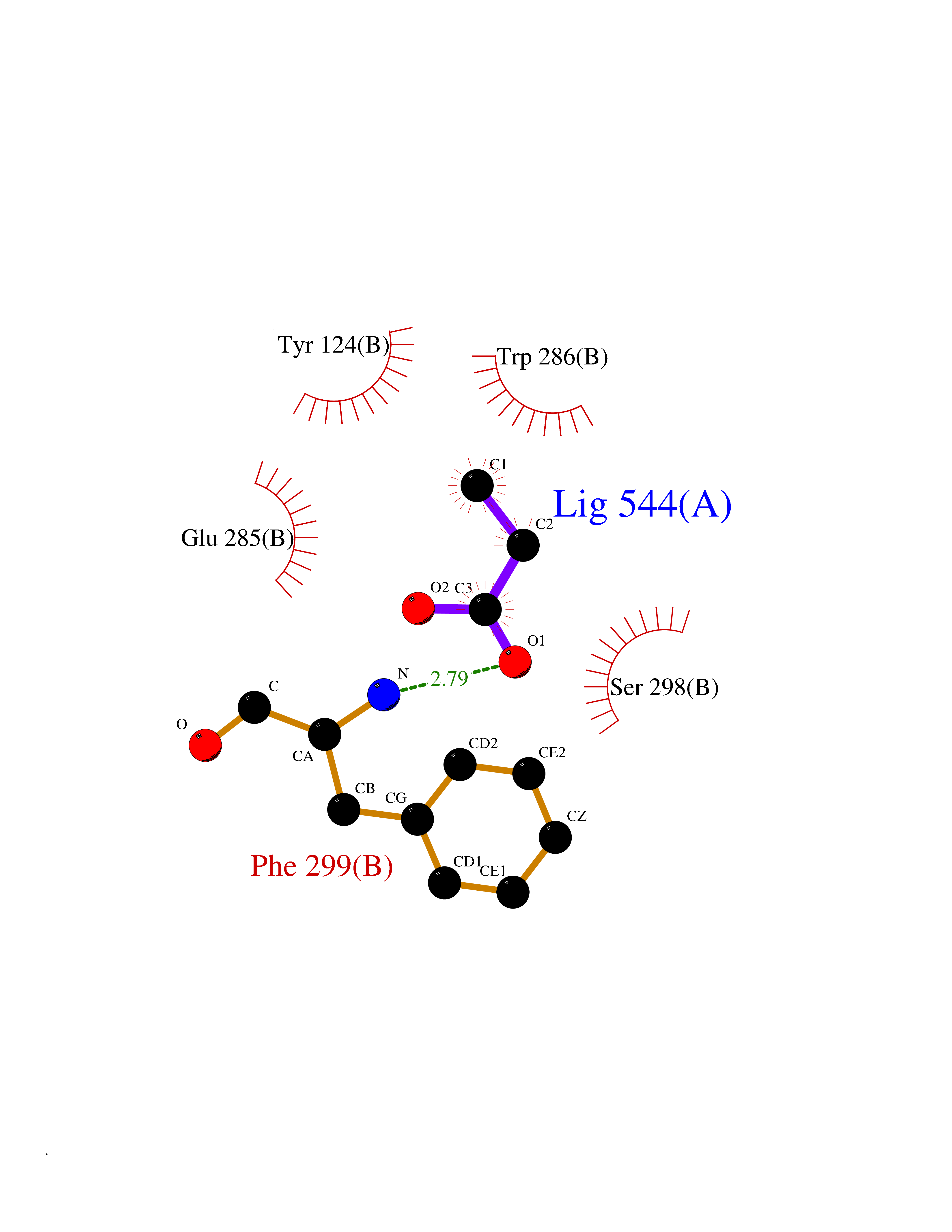 Ligplot Map 3D Binding mode Sequence EDAELLVTVRGGRLRGIRLKTPGGPVSAFLGIPFAEPPMGPRRFLPPEPKQPWSGVVDATTFQSVCYQYVDTLYPGFEGTEMWNPNRELSEDCLYLNVWTPYPRPTSPTPVLVWIYGGGFYSGASSLDVYDGRFLVQAERTVLVSMNYRVGAFGFLALPGSREAPGNVGLLDQRLALQWVQENVAAFGGDPTSVTLFGESAGAASVGMHLLSPPSRGLFHRAVLQSGAPNGPWATVGMGEARRRATQLAHLVGCPPGGTGGNDTELVACLRTRPAQVLVNHEWHVLPQESVFRFSFVPVVDGDFLSDTPEALINAGDFHGLQVLVGVVKDEGSYFLVYGAPGFSKDNESLISRAEFLAGVRVGVPQVSDLAAEAVVLHYTDWLHPEDPARLREALSDVVGDHNVVCPVAQLAGRLAAQGARVYAYVFEHRASTLSWPLWMGVPHGYEIEFIFGIPLDPSRNYTAEEKIFAQRLMRYWANFARTGDPNEPPKAPQWPPYTAGAQQYVSLDLRPLEVRRGLRAQACAFWNRFLPKLLSA Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Multifunctional protein ADE2 | 2H31 | 4.55 | |
Target general information Gen name PAICS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ADE2;PAIS;AIRC Protein family SAICAR synthetase family; AIR carboxylase family, Class II subfamily Biochemical class Ligase Function ATP binding.Cadherin binding.Identical protein binding.Phosphoribosylaminoimidazole carboxylase activity.Phosphoribosylaminoimidazolesuccinocarboxamide synthase activity. Related diseases Phosphoribosylaminoimidazole carboxylase deficiency (PAICSD) [MIM:619859]: An autosomal recessive inborn error of purine metabolism, clinically characterized by multiple congenital anomalies and early neonatal death. {ECO:0000269|PubMed:31600779}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00128 Interacts with Q16543; P51116; Q969R5; Q8TBB1; Q6ZVK8; P22234; O75928-2; C9JJ79; Q9Y4B4; P78317; Q7KZS0; Q9UBW7 EC number 4.1.1.21; 6.3.2.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Decarboxylase; Direct protein sequencing; Disease variant; Ligase; Lyase; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Proteomics identification; Purine biosynthesis; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 41862.7 Length 386 Aromaticity 0.08 Instability index 34.99 Isoelectric point 7.02 Charge (pH=7) 0.04 2D Binding mode Binding energy (Kcal/mol) -6.2 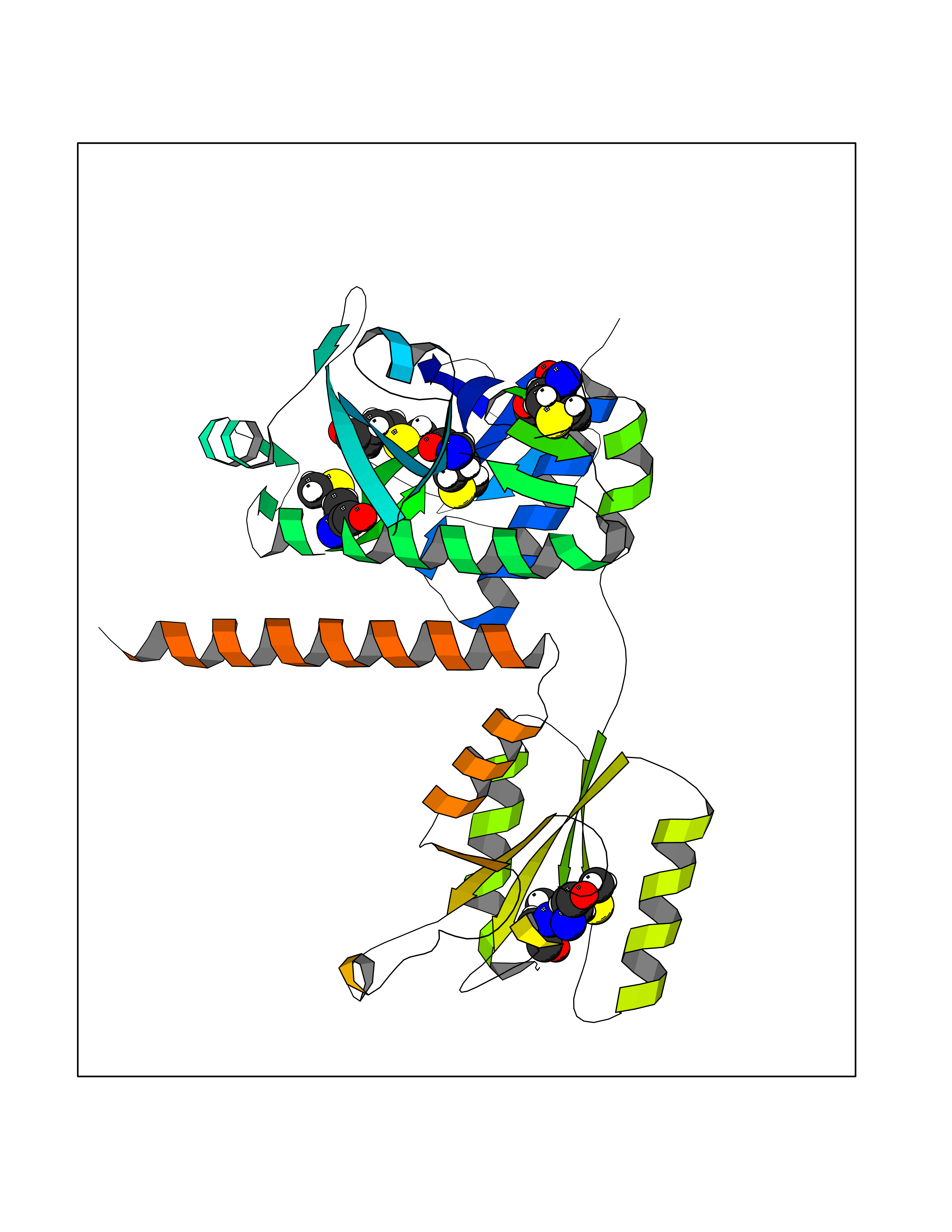 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LNIGKKLYEGKTKEVYELLDSPGKVLLQSKGKAAISNKITSCIFQLLQEAGIKTAFTRKCGETAFIAPQCEXIPIEWVCRRIATGSFLKRNPGVKEGYKFYPPKVELFFKDDANNDPQWSEEQLIAAKFCFAGLLIGQTEVDIXSHATQAIFEILEKSWLPQNCTLVDXKIEFGVDVTTKEIVLADVIDNDSWRLWPSGPEGLQXVKKNFEWVAERVELLLKSESQCRVVVLXGSTSDLGHCEKIKKACGNFGIPCELRVTSAHKGPDETLRIKAEYEGDGIPTVFVAVAGRSNGLGPVXSGNTAYPVISCPPLTPDWGVQDVWSSLRLPSGLGCSTVLSPEGSAQFAAQIFGLSNHLVWSKLRASILNTWISLKQADKKIRECNL Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | RET M918T mutant (RET M918T) | 4CKI | 4.55 | |
Target general information Gen name RET Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RET51 M918T mutant; Proto-oncogene tyrosine-protein kinase receptor Ret M918T mutant; Proto-oncogene c-Ret M918T mutant; PTC M918T mutant; Cadherin family member 12 M918T mutant; CDHR16 M918T mutant; Protein family Protein kinase superfamily, Tyr protein kinase family Biochemical class Kinase Function Phosphorylates PTK2/FAK1. Regulates both cell death/survival balance and positional information. Required for the molecular mechanisms orchestration during intestine organogenesis; involved in the development of enteric nervous system and renal organogenesis during embryonic life, and promotes the formation of Peyer's patch-like structures, a major component of the gut-associated lymphoid tissue. Modulates cell adhesion via its cleavage by caspase in sympathetic neurons and mediates cell migration in an integrin (e. g. ITGB1 and ITGB3)-dependent manner. Involved in the development of the neural crest. Active in the absence of ligand, triggering apoptosis through a mechanism that requires receptor intracellular caspase cleavage. Acts as a dependence receptor; in the presence of the ligand GDNF in somatotrophs (within pituitary), promotes survival and down regulates growth hormone (GH) production, but triggers apoptosis in absence of GDNF. Regulates nociceptor survival and size. Triggers the differentiation of rapidly adapting (RA) mechanoreceptors. Mediator of several diseases such as neuroendocrine cancers; these diseases are characterized by aberrant integrins-regulated cell migration. Mediates, through interaction with GDF15-receptor GFRAL, GDF15-induced cell-signaling in the brainstem which induces inhibition of food-intake. Activates MAPK- and AKT-signaling pathways. Isoform 1 in complex with GFRAL induces higher activation of MAPK-signaling pathway than isoform 2 in complex with GFRAL. Receptor tyrosine-protein kinase involved in numerous cellular mechanisms including cell proliferation, neuronal navigation, cell migration, and cell differentiation upon binding with glial cell derived neurotrophic factor family ligands. Related diseases Hirschsprung disease 1 (HSCR1) [MIM:142623]: A disorder of neural crest development characterized by absence of enteric ganglia along a variable length of the intestine. It is the most common cause of congenital intestinal obstruction. Early symptoms range from complete acute neonatal obstruction, characterized by vomiting, abdominal distention and failure to pass stool, to chronic constipation in the older child. {ECO:0000269|PubMed:10090908, ECO:0000269|PubMed:10484767, ECO:0000269|PubMed:10618407, ECO:0000269|PubMed:22174939, ECO:0000269|PubMed:7581377, ECO:0000269|PubMed:7633441, ECO:0000269|PubMed:7704557, ECO:0000269|PubMed:7881414, ECO:0000269|PubMed:8114938, ECO:0000269|PubMed:8114939, ECO:0000269|PubMed:9043870, ECO:0000269|PubMed:9090527, ECO:0000269|PubMed:9094028, ECO:0000269|PubMed:9259198, ECO:0000269|PubMed:9384613, ECO:0000269|PubMed:9497256, ECO:0000269|Ref.70}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Medullary thyroid carcinoma (MTC) [MIM:155240]: Rare tumor derived from the C cells of the thyroid. Three hereditary forms are known, that are transmitted in an autosomal dominant fashion: (a) multiple neoplasia type 2A (MEN2A), (b) multiple neoplasia type IIB (MEN2B) and (c) familial MTC (FMTC), which occurs in 25-30% of MTC cases and where MTC is the only clinical manifestation. {ECO:0000269|PubMed:10323403, ECO:0000269|PubMed:10826520, ECO:0000269|PubMed:11692159, ECO:0000269|PubMed:24560924, ECO:0000269|PubMed:29908090, ECO:0000269|PubMed:31118272, ECO:0000269|PubMed:7784092, ECO:0000269|PubMed:7845675, ECO:0000269|PubMed:7849720, ECO:0000269|PubMed:7874109, ECO:0000269|PubMed:7881414, ECO:0000269|PubMed:7915165, ECO:0000269|PubMed:8103403, ECO:0000269|PubMed:8557249, ECO:0000269|PubMed:8625130, ECO:0000269|PubMed:8807338, ECO:0000269|PubMed:9223675, ECO:0000269|PubMed:9259198, ECO:0000269|PubMed:9398735, ECO:0000269|PubMed:9452077, ECO:0000269|PubMed:9506724, ECO:0000269|PubMed:9621513, ECO:0000269|PubMed:9677065}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Multiple neoplasia 2B (MEN2B) [MIM:162300]: Uncommon inherited cancer syndrome characterized by predisposition to MTC and phaeochromocytoma which is associated with marfanoid habitus, mucosal neuromas, skeletal and ophthalmic abnormalities, and ganglioneuromas of the intestine tract. Then the disease progresses rapidly with the development of metastatic MTC and a pheochromocytome in 50% of cases. {ECO:0000269|PubMed:24560924, ECO:0000269|PubMed:7906417, ECO:0000269|PubMed:7906866, ECO:0000269|PubMed:7911697, ECO:0000269|PubMed:8595427, ECO:0000269|PubMed:8807338, ECO:0000269|PubMed:9294615, ECO:0000269|PubMed:9360560}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pheochromocytoma (PCC) [MIM:171300]: A catecholamine-producing tumor of chromaffin tissue of the adrenal medulla or sympathetic paraganglia. The cardinal symptom, reflecting the increased secretion of epinephrine and norepinephrine, is hypertension, which may be persistent or intermittent. {ECO:0000269|PubMed:12000816}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Multiple neoplasia 2A (MEN2A) [MIM:171400]: The most frequent form of medullary thyroid cancer (MTC). It is an inherited cancer syndrome characterized by MTC, phaeochromocytoma and/or hyperparathyroidism. {ECO:0000269|PubMed:10522989, ECO:0000269|PubMed:7860065, ECO:0000269|PubMed:7874109, ECO:0000269|PubMed:7881414, ECO:0000269|PubMed:7915165, ECO:0000269|PubMed:8099202, ECO:0000269|PubMed:8103403, ECO:0000269|PubMed:8626834, ECO:0000269|PubMed:8807338, ECO:0000269|PubMed:9097963, ECO:0000269|PubMed:9384613, ECO:0000269|PubMed:9452064}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Various chromosomal aberrations involving RET are known. Some of them have been found in papillary thyroid carcinomas (PTCs) (PubMed:10439047, PubMed:10980597, PubMed:12787916, PubMed:2406025). Inversion inv(10)(q11.2;q21) generates the RET/CCDC6 (PTC1) oncogene (PubMed:2406025). Inversion inv(10)(q11.2;q11.2) generates the RET/NCOA4 (PTC3) oncogene. Translocation t(10;14)(q11;q32) with GOLGA5 generates the RET/GOLGA5 (PTC5) oncogene (PubMed:2734021). Translocation t(8;10)(p21.3;q11.2) with PCM1 generates the PCM1/RET fusion (PubMed:10980597). Translocation t(6;10)(p21.3;q11.2) with TRIM27/RFP generates the Delta RFP/RET oncogene (PubMed:12787916). Translocation t(1;10)(p13;q11) with TRIM33 generates the TRIM33/RET (PTC7) oncogene (PubMed:10439047). Translocation t(7;10)(q32;q11) with TRIM24/TIF1 generates the TRIM24/RET (PTC6) oncogene (PubMed:10439047). Translocation t(6;10)(p21.3;q11.2) with TRIM27/RFP generates the TRIM27/RET oncogene (PubMed:3037315). {ECO:0000269|PubMed:10439047, ECO:0000269|PubMed:10980597, ECO:0000269|PubMed:12787916, ECO:0000269|PubMed:2406025, ECO:0000269|PubMed:2734021, ECO:0000269|PubMed:3037315}.; DISEASE: Mutations in RET have been detected in patients with renal agenesis suggesting a possible involvement of this gene in disease pathogenesis. {ECO:0000269|PubMed:18252215}. Drugs (DrugBank ID) DB01809; DB12742; DB08875; DB12147; DB12010; DB00619; DB09078; DB08901; DB15822; DB08896; DB15685; DB00398; DB05294; DB05014 Interacts with Q9UM73; P22455; Q7Z3S9; P40763; Q9Y490; Q62985; Q7Z3S9 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell adhesion; Cell membrane; Chromosomal rearrangement; Disease variant; Disulfide bond; Endosome; Glycoprotein; Hirschsprung disease; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 33472.5 Length 295 Aromaticity 0.09 Instability index 43.68 Isoelectric point 9.07 Charge (pH=7) 6.3 2D Binding mode Binding energy (Kcal/mol) -6.21  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GPLSLSVDAFKILEDPKWEFPRKNLVLGKTLGEGEFGKVVKATAFHLKGRAGYTTVAVKMLKENASPSELRDLLSEFNVLKQVNHPHVIKLYGACSQDGPLLLIVEYAKYGSLRGFLRESRKVDERALTMGDLISFAWQISQGMQYLAEMKLVHRDLAARNILVAEGRKMKISDFGLSRDVXEEDSXVKRSQGRIPVKWTAIESLFDHIYTTQSDVWSFGVLLWEIVTLGGNPYPGIPPERLFNLLKTGHRMERPDNCSEEMYRLMLQCWKQEPDKRPVFADISKDLEKMMVKRR Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Hyperpolarization cyclic nucleotide-gated channel 4 (HCN4) | 3OTF | 4.55 | |
Target general information Gen name HCN4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channel 4 Protein family Potassium channel HCN family Biochemical class Voltage-gated ion channel Function Contributes to the native pacemaker currents in heart (If) that regulate the rhythm of heart beat. May contribute to the native pacemaker currents in neurons (Ih). May mediate responses to sour stimuli. Hyperpolarization-activated ion channel with very slow activation and inactivation exhibiting weak selectivity for potassium over sodium ions. Related diseases Sick sinus syndrome 2 (SSS2) [MIM:163800]: The term 'sick sinus syndrome' encompasses a variety of conditions caused by sinus node dysfunction. The most common clinical manifestations are syncope, presyncope, dizziness, and fatigue. Electrocardiogram typically shows sinus bradycardia, sinus arrest, and/or sinoatrial block. Episodes of atrial tachycardias coexisting with sinus bradycardia ('tachycardia-bradycardia syndrome') are also common in this disorder. SSS occurs most often in the elderly associated with underlying heart disease or previous cardiac surgery, but can also occur in the fetus, infant, or child without heart disease or other contributing factors. SSS2 onset is in utero or at birth. {ECO:0000269|PubMed:15123648, ECO:0000269|PubMed:16407510, ECO:0000269|PubMed:20662977, ECO:0000269|PubMed:23103389}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 8 (BRGDA8) [MIM:613123]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:19165230}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Epilepsy, idiopathic generalized 18 (EIG18) [MIM:619521]: An autosomal dominant form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. EIG18 is characterized by onset of myoclonic seizures in infancy. Although the seizures remit, some patients may have later speech or cognitive impairment. {ECO:0000269|PubMed:30127718}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O60741; Q9Y3Q4 EC number NA Uniprot keywords 3D-structure; Brugada syndrome; cAMP; cAMP-binding; Cell membrane; Disease variant; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Nucleotide-binding; Phosphoprotein; Potassium; Potassium channel; Potassium transport; Proteomics identification; Reference proteome; Sodium; Sodium channel; Sodium transport; Transmembrane; Transmembrane helix; Transport; Voltage-gated channel Protein physicochemical properties Chain ID A Molecular weight (Da) 23211.2 Length 197 Aromaticity 0.12 Instability index 42.69 Isoelectric point 8.67 Charge (pH=7) 3.11 2D Binding mode Binding energy (Kcal/mol) -6.2  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence DSSRRQYQEKYKQVEQYMSFHKLPPDTRQRIHDYYEHRYQGKMFDEESILGELSEPLREEIINFNCRKLVASMPLFANADPNFVTSMLTKLRFEVFQPGDYIIREGTIGKKMYFIQHGVVSVLTKGNKETKLADGSYFGEICLLTRGRRTASVRADTYCRLYSLSVDNFNEVLEEYPMMRRAFETVALDRLDRIGKK Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Cholesterol 24-hydroxylase (CYP46A1) | 3MDR | 4.55 | |
Target general information Gen name CYP46A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cytochrome P450 46A1; CYP46; Cholesterol 24S-hydroxylase; Cholesterol 24-monooxygenase; CH24H Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function Primarily catalyzes the hydroxylation (with S stereochemistry) at C-24 of cholesterol side chain, triggering cholesterol diffusion out of neurons and its further degradation. By promoting constant cholesterol elimination in neurons, may activate the mevalonate pathway and coordinate the synthesis of new cholesterol and nonsterol isoprenoids involved in synaptic activity and learning. Further hydroxylates cholesterol derivatives and hormone steroids on both the ring and side chain of these molecules, converting them into active oxysterols involved in lipid signaling and biosynthesis. Acts as an epoxidase converting cholesta-5,24-dien-3beta-ol/desmosterol into (24S),25-epoxycholesterol, an abundant lipid ligand of nuclear NR1H2 and NR1H3 receptors shown to promote neurogenesis in developing brain. May also catalyze the oxidative metabolism of xenobiotics, such as clotrimazole. P450 monooxygenase that plays a major role in cholesterol homeostasis in the brain. Related diseases Spinocerebellar ataxia, autosomal recessive, with axonal neuropathy 1 (SCAN1) [MIM:607250]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCAN1 is an autosomal recessive cerebellar ataxia (ARCA) associated with peripheral axonal motor and sensory neuropathy, distal muscular atrophy, pes cavus and steppage gait as seen in Charcot-Marie-Tooth neuropathy. All affected individuals have normal intelligence. {ECO:0000269|PubMed:12244316, ECO:0000269|PubMed:15647511, ECO:0000269|PubMed:15920477, ECO:0000269|PubMed:16141202, ECO:0000269|PubMed:17948061}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.14.14.25 Uniprot keywords 3D-structure; Alternative splicing; Cell projection; Cholesterol metabolism; Endoplasmic reticulum; Heme; Iron; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism; Synapse; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 48977 Length 427 Aromaticity 0.1 Instability index 49.59 Isoelectric point 9.04 Charge (pH=7) 6.25 2D Binding mode Binding energy (Kcal/mol) -6.21 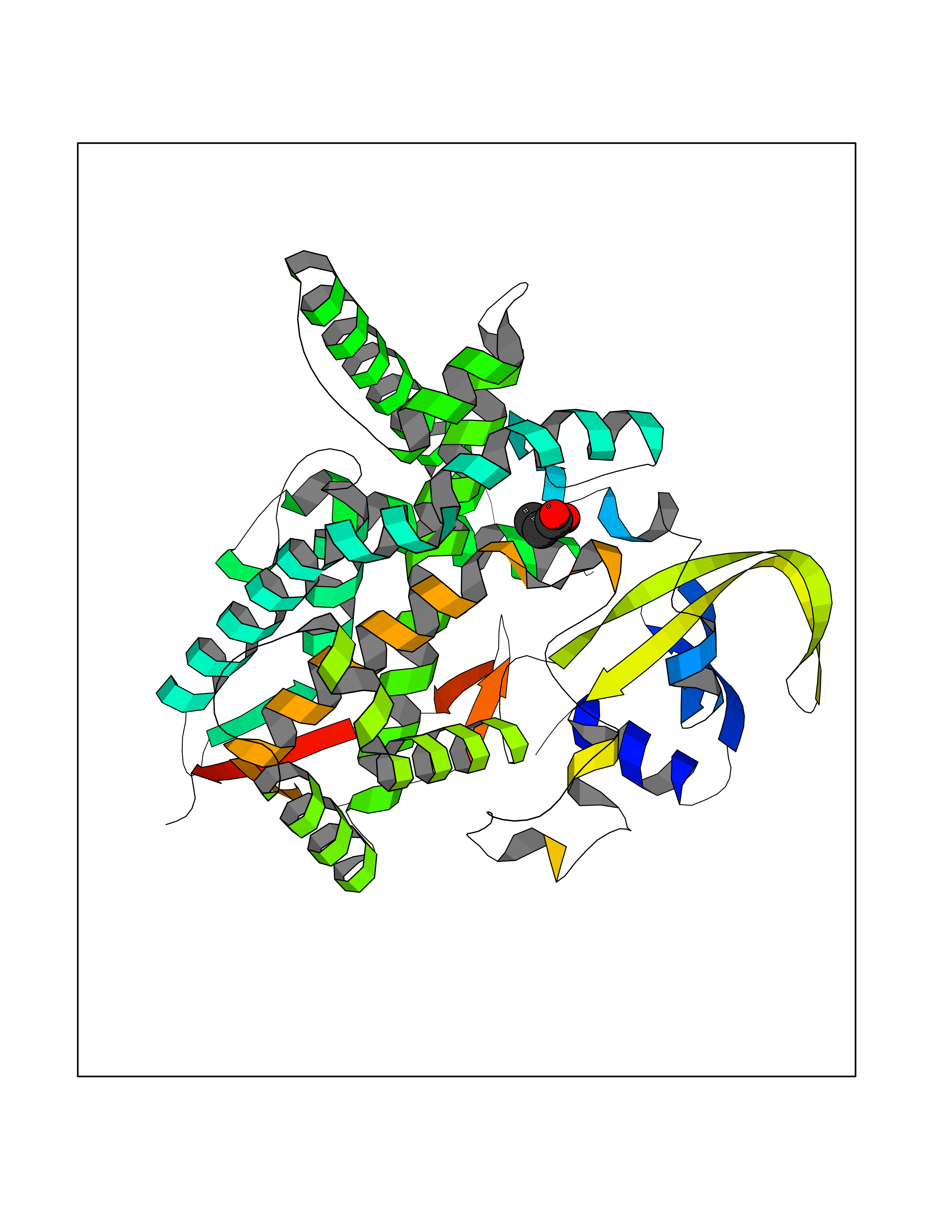 Molscript Map 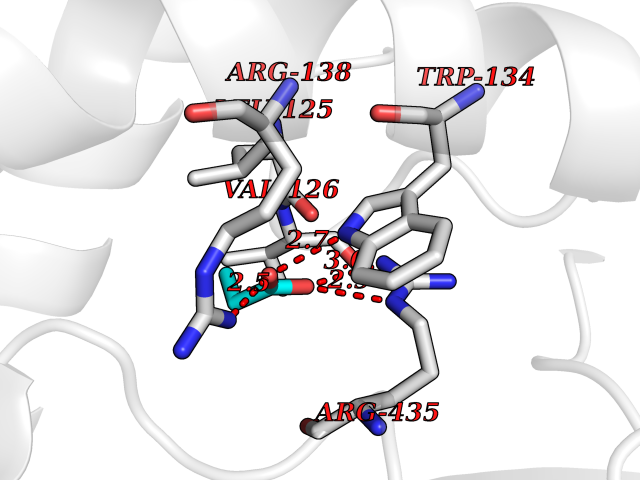 Pymol Map 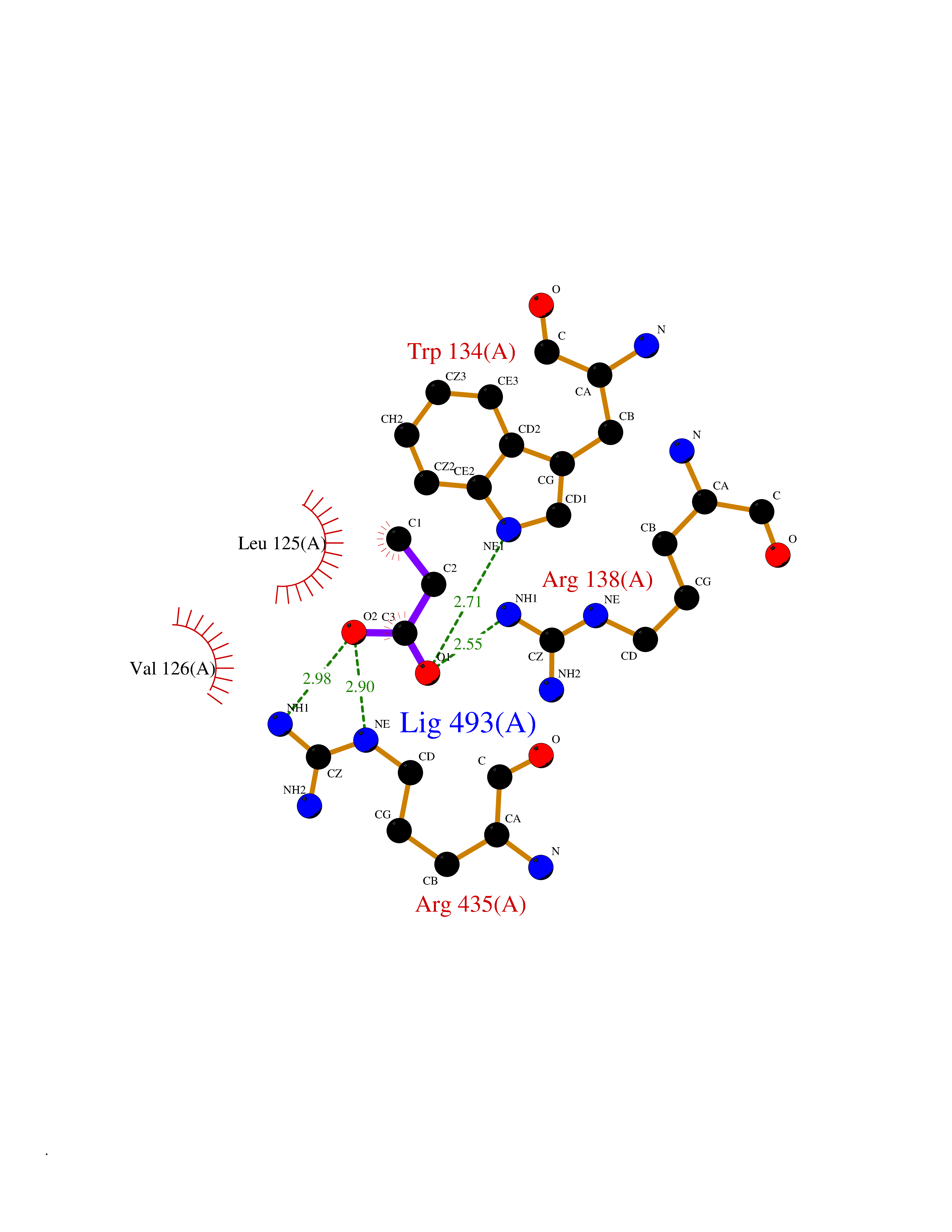 Ligplot Map 3D Binding mode Sequence RVLQDVFLDWAKKYGPVVRVNVFHKTSVIVTSPESVKKFLMSTKYNKDSKMYRALQTVFGERLFGQGLVSECNYERWHKQRRVIDLAFSRSSLVSLMETFNEKAEQLVEILEAKADGQTPVSMQDMLTYTAMDILAKAAFGMETSMLLGAQKPLSQAVKLMLEGITASRNTKRKQLREVRESIRFLRQVGRDWVQRRREALKRGEEVPADILTQILKAEEGAQDDEGLLDNFVTFFIAGHETSANHLAFTVMELSRQPEIVARLQAEVDEVIGSKRYLDFEDLGRLQYLSQVLKESLRLYPPAWGTFRLLEEETLIDGVRVPGNTPLLFSTYVMGRMDTYFEDPLTFNPDRFGPGAPKPRFTYFPFSLGHRSCIGQQFAQMEVKVVMAKLLQRLEFRLVPGQRFGLQEQATLKPLDPVLCTLRPRGW Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Zinc finger-containing ubiquitin peptidase 1 (ZUP1) | 6EI1 | 4.55 | |
Target general information Gen name ZUP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Zinc finger with UFM1-specific peptidase domain protein; ZUFSP; Lys-63-specific deubiquitinase ZUFSP; DUB; C6orf113 Protein family Peptidase C78 family, ZUFSP subfamily Biochemical class Peptidase Function Shows only weak activity against 'Lys-11' and 'Lys-48'-linked chains. Plays an important role in genome stability pathways, functioning to prevent spontaneous DNA damage and also promote cellular survival in response to exogenous DNA damage. Modulates the ubiquitination status of replication protein A (RPA) complex proteins in response to replication stress. Deubiquitinase with endodeubiquitinase activity that specifically interacts with and cleaves 'Lys-63'-linked long polyubiquitin chains. Related diseases WHIM syndrome 2 (WHIMS2) [MIM:619407]: An autosomal recessive form of WHIM syndrome, a primary immunodeficiency disorder characterized by warts, hypogammaglobulinemia, infections, and myelokathexis. Myelokathexis is a unique form of non-cyclic severe congenital neutropenia caused by accumulation of mature and degenerating neutrophils in the bone marrow. Monocytopenia and lymphopenia, especially B lymphopenia, also commonly occur. There is significant phenotypic variation among patients, such that some individuals may have an incomplete form of the disorder in which one or more of the classic tetrad features are not present. {ECO:0000269|PubMed:24777453}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q92619; P50281; Q8WVC2 EC number EC 3.4.19.12 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Hydrolase; Metal-binding; Nucleus; Proteomics identification; Reference proteome; Repeat; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 46930.3 Length 410 Aromaticity 0.07 Instability index 58.67 Isoelectric point 9 Charge (pH=7) 9.7 2D Binding mode Binding energy (Kcal/mol) -6.2 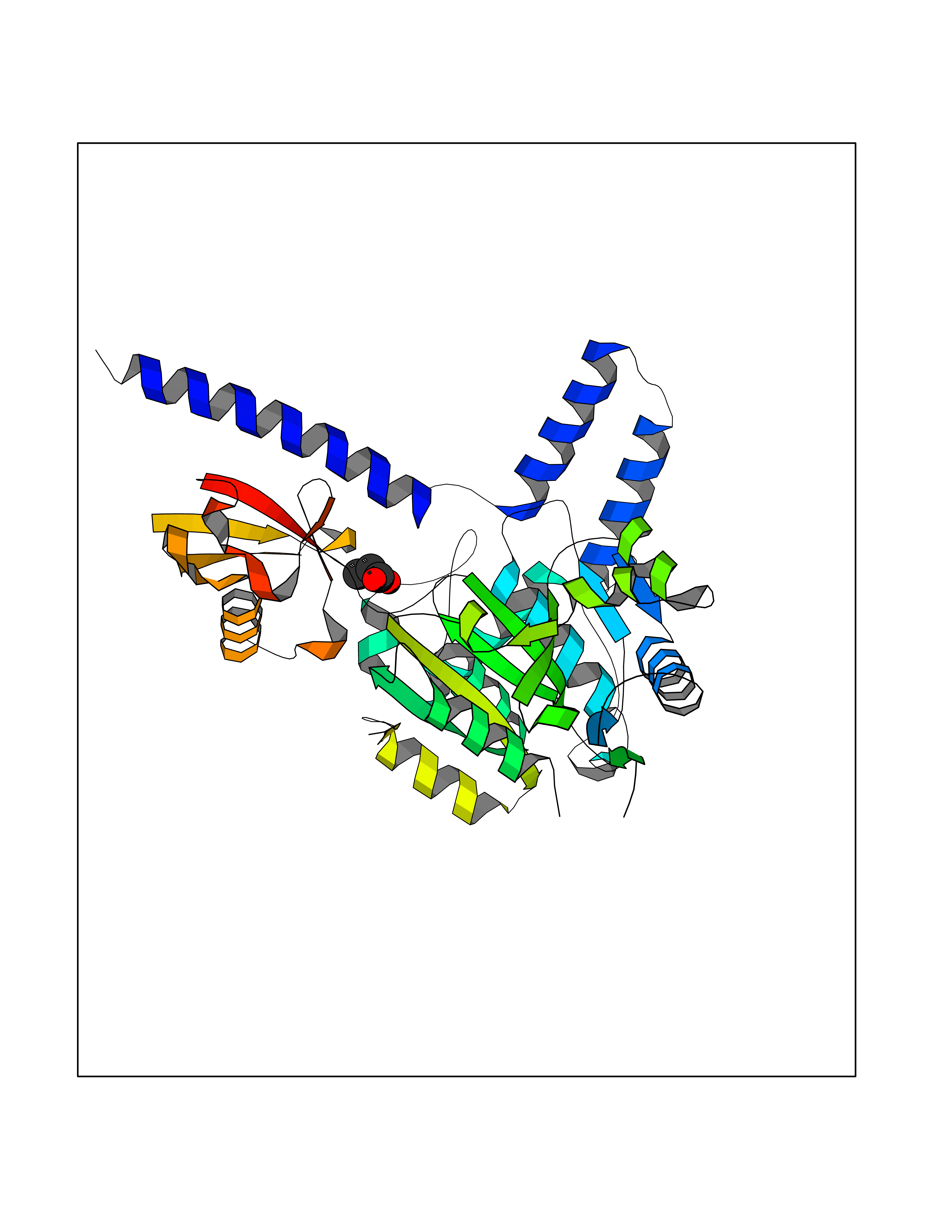 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LQQEEDRKRRSEESRQEIEEFQKLQRQYGLDNSGGYKQQQLRNMEIEVNRGRMPPSEFHRRKADMMESLALGFDDGKTKTSGIIEALHRYYQNAATDVRRVWLSSVVDHFHSSLGDKGWGCGYRNFQMLLSSLLQNDAYNDCLKGMLIPCIPKIQSMIEDAWKEGFDPQGASQLNNRLQGTKAWIGACEVYILLTSLRVKCHIVDFHKSTGPLGTHPRLFEWILNYYSSSPKVVCTSKPPIYLQHQGHSRTVIGIEEKKNRTLCLLILDPGCPSREMQKLLKQDIEASSLKQLRKSMGNLKHKQYQILAVEGALSLEEKLARRQASQVFTAEKIPMQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRG Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | Debrisoquine 4-hydroxylase (CYP2D6) | 4WNV | 4.55 | |
Target general information Gen name CYP2D6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms P450-DB1; Cytochrome P450-DB1; Cytochrome P450 2D6; CYPIID6; CYP2DL1 Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function It is involved in the metabolism of drugs such as antiarrhythmics, adrenoceptor antagonists, and tricyclic antidepressants. Responsible for the metabolism of many drugs and environmental chemicals that it oxidizes. Related diseases A chromosomal aberration involving BCL2 has been found in chronic lymphatic leukemia. Translocation t(14;18)(q32;q21) with immunoglobulin gene regions. BCL2 mutations found in non-Hodgkin lymphomas carrying the chromosomal translocation could be attributed to the Ig somatic hypermutation mechanism resulting in nucleotide transitions. {ECO:0000269|PubMed:2875799, ECO:0000269|PubMed:3285301}. Drugs (DrugBank ID) DB01562; DB01472; DB14010; DB12001; DB05812; DB01193; DB00316; DB15568; DB00918; DB06203; DB00866; DB01424; DB01118; DB00321; DB00381; DB00613; DB00543; DB00182; DB00701; DB11785; DB01435; DB01429; DB01274; DB01238; DB14185; DB09204; DB11638; DB06216; DB00637; DB11586; DB00335; DB00289; DB01076; DB00972; DB04957; DB09013; DB16703; DB01086; DB06770; DB01244; DB15982; DB00195; DB01295; DB12236; DB01128; DB04889; DB00810; DB13975; DB08807; DB00188; DB09128; DB12151; DB12752; DB06726; DB00297; DB08808; DB00921; DB01156; DB00490; DB09173; DB00201; DB09061; DB14737; DB06016; DB00521; DB01136; DB00482; DB04846; DB00439; DB00185; DB00608; DB01114; DB00477; DB00356; DB01410; DB01166; DB00501; DB01012; DB00568; DB00604; DB00215; DB12499; DB00283; DB04920; DB14025; DB00349; DB00845; DB01242; DB00575; DB13508; DB00257; DB00363; DB09065; DB05239; DB00907; DB00318; DB11672; DB14635; DB00924; DB00091; DB11963; DB06292; DB04884; DB00496; DB01264; DB09183; DB04840; DB00705; DB06512; DB01151; DB06700; DB16650; DB12161; DB13679; DB09555; DB01191; DB00633; DB01576; DB00514; DB00647; DB11994; DB01551; DB00343; DB01093; DB01075; DB00757; DB01184; DB00843; DB09167; DB00590; DB01142; DB00997; DB00470; DB04855; DB00476; DB00625; DB11979; DB00216; DB15444; DB09039; DB13874; DB01228; DB06735; DB11718; DB00494; DB13757; DB00751; DB00530; DB13443; DB01175; DB06678; DB00187; DB00330; DB01466; DB01628; DB01590; DB12500; DB01023; DB00574; DB06702; DB12265; DB01195; DB04841; DB00472; DB00623; DB01095; DB00176; DB00983; DB02703; DB15149; DB00674; DB05087; DB00317; DB08909; DB00986; DB01218; DB00502; DB00956; DB01611; DB00557; DB09053; DB01177; DB04946; DB00619; DB00458; DB08952; DB00224; DB06370; DB13293; DB04818; DB16200; DB11633; DB06636; DB00951; DB11757; DB00602; DB09570; DB01026; DB00598; DB12212; DB00448; DB11732; DB16217; DB09078; DB00528; DB12070; DB09351; DB01210; DB08918; DB00281; DB04948; DB01206; DB00836; DB01601; DB00455; DB04871; DB09195; DB06708; DB04829; DB09238; DB00934; DB14921; DB00737; DB14009; DB09224; DB00170; DB00454; DB00532; DB13530; DB06691; DB01071; DB00933; DB01577; DB00333; DB00763; DB01403; DB01028; DB09241; DB01214; DB01233; DB00264; DB00379; DB06148; DB01388; DB01110; DB00211; DB01454; DB06595; DB00834; DB00805; DB08893; DB00370; DB12523; DB01171; DB00745; DB14011; DB09049; DB00731; DB04861; DB01149; DB00220; DB09048; DB00238; DB00627; DB00622; DB00699; DB02701; DB00184; DB01115; DB04868; DB12005; DB00540; DB00334; DB14881; DB00338; DB00904; DB11130; DB04911; DB01173; DB11837; DB04938; DB01096; DB01580; DB01062; DB00497; DB06412; DB01192; DB01267; DB00377; DB06603; DB00715; DB06589; DB00022; DB01359; DB00738; DB01074; DB08922; DB00850; DB03783; DB00780; DB00914; DB00252; DB05316; DB01100; DB00960; DB00592; DB01621; DB04951; DB17472; DB11642; DB08901; DB01297; DB15822; DB01087; DB01035; DB00433; DB00396; DB01131; DB00420; DB01069; DB09288; DB01182; DB00571; DB04216; DB01224; DB00908; DB00468; DB01129; DB00863; DB00243; DB00234; DB14761; DB00409; DB06506; DB02709; DB11855; DB13174; DB11753; DB08864; DB14840; DB00734; DB12693; DB00503; DB00953; DB09291; DB15119; DB00412; DB05271; DB12332; DB11614; DB06654; DB01232; DB01037; DB06144; DB01104; DB00203; DB00641; DB01591; DB00398; DB12713; DB00489; DB06727; DB01323; DB09118; DB06820; DB06729; DB06608; DB11770; DB00675; DB00706; DB06204; DB06083; DB01079; DB12095; DB06287; DB00857; DB00342; DB13775; DB04905; DB04844; DB11712; DB00277; DB00679; DB01623; DB00208; DB00373; DB01409; DB00932; DB06137; DB01036; DB05109; DB00193; DB00752; DB00656; DB12245; DB00726; DB00792; DB00209; DB15328; DB09076; DB13609; DB15091; DB11915; DB00862; DB08881; DB00285; DB00661; DB06217; DB06684; DB09185; DB00570; DB00361; DB11739; DB09068; DB01392; DB00549; DB15688; DB00425; DB01624 Interacts with NA EC number EC 1.14.14.- Uniprot keywords 3D-structure; Alternative splicing; Cholesterol metabolism; Endoplasmic reticulum; Fatty acid metabolism; Heme; Iron; Lipid metabolism; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 51898.1 Length 464 Aromaticity 0.09 Instability index 43.83 Isoelectric point 6.76 Charge (pH=7) -0.99 2D Binding mode Binding energy (Kcal/mol) -6.21  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GKLPPGPLPLPGLGNLLFQNTPYCFDQLRRRFGDVFSLQLAWTPVVVLNGLAAVREALVTHGEDTADRPPVPITQILGFGPRSQGVFLARYGPAWREQRRFSVSTLRNLGLGKKSLEQWVTEEAACLCAAFANHSGRPFRPNGLLDKAVSNVIASLTCGRRFEYDDPRFLRLLDLAQEGLKEESGFLREVLNAVPVLLHIPALAGKVLRFQKAFLTQLDELLTEHRMTWDPAQPPRDLTEAFLAEMEKAKGNPESSFNDENLRIVVADLFSAGMVTTSTTLAWGLLLMILHPDVQRRVQQEIDDVIGQVRRPEMGDQAHMPYTTAVIHEVQRFGDIVPLGVTHMTSRDIEVQGFRIPKGTTLITNLSSVLKDEAVWEKPFRFHPEHFLDAQGHFVKPEAFLPFSAGRRACLGEPLARMELFLFFTSLLQHFSFSVPTGQPRPSHHGVFAFLVSPSPYELCAVPR Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Torsin-1A (TOR1A) | 5J1S | 4.55 | |
Target general information Gen name TOR1A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Torsin family 1 member A; Torsin ATPase-1A; TORA; TA; Dystonia 1 protein; DYT1; DQ2 Protein family ClpA/ClpB family, Torsin subfamily Biochemical class Acid anhydride hydrolase Function Involved in the regulation of synaptic vesicle recycling, controls STON2 protein stability in collaboration with the COP9 signalosome complex (CSN). In the nucleus, may link the cytoskeleton with the nuclear envelope, this mechanism seems to be crucial for the control of nuclear polarity, cell movement and, specifically in neurons, nuclear envelope integrity. Participates in the cellular trafficking and may regulate the subcellular location of multipass membrane proteins such as the dopamine transporter SLC6A3, leading to the modulation of dopamine neurotransmission. In the endoplasmic reticulum, plays a role in the quality control of protein folding by increasing clearance of misfolded proteins such as SGCE variants or holding them in an intermediate state for proper refolding. May have a redundant function with TOR1B in non-neural tissues. Protein with chaperone functions important for the control of protein folding, processing, stability and localization as well as for the reduction of misfolded protein aggregates. Related diseases Dystonia 1, torsion, autosomal dominant (DYT1) [MIM:128100]: A primary torsion dystonia, and the most common and severe form. Dystonia is defined by the presence of sustained involuntary muscle contractions, often leading to abnormal postures. Dystonia type 1 is characterized by involuntary, repetitive, sustained muscle contractions or postures involving one or more sites of the body, in the absence of other neurological symptoms. Typically, symptoms develop first in an arm or leg in middle to late childhood and progress in approximately 30% of patients to other body regions (generalized dystonia) within about five years. 'Torsion' refers to the twisting nature of body movements observed in DYT1, often affecting the trunk. Distribution and severity of symptoms vary widely between affected individuals, ranging from mild focal dystonia to severe generalized dystonia, even within families. {ECO:0000269|PubMed:14970196, ECO:0000269|PubMed:15505207, ECO:0000269|PubMed:16361107, ECO:0000269|PubMed:17428918, ECO:0000269|PubMed:18167355, ECO:0000269|PubMed:18477710, ECO:0000269|PubMed:18827015, ECO:0000269|PubMed:19955557, ECO:0000269|PubMed:20169475, ECO:0000269|PubMed:21102408, ECO:0000269|PubMed:24930953, ECO:0000269|PubMed:27490483, ECO:0000269|PubMed:9288096}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Arthrogryposis multiplex congenita 5 (AMC5) [MIM:618947]: A form of arthrogryposis multiplex congenita, a developmental condition characterized by multiple joint contractures resulting from reduced or absent fetal movements. AMC5 is an autosomal recessive form characterized by severe congenital contractures, developmental delay, strabismus and tremor. {ECO:0000269|PubMed:28516161, ECO:0000269|PubMed:29053766, ECO:0000269|PubMed:30244176}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q6PCB6; P05067; Q96FT7-4; Q9UNS2; P22692; P60409; Q99750; Q5JTV8; Q8NFQ8; Q6PCB6; P63010-2; P05067; Q9UII2; Q9UJX2; Q53EZ4; Q96FZ7; Q92478; P35222; Q9NR90-2; Q9UHI6; Q14154; Q9H147; O75616; Q9UI08-2; Q6PIV2; Q9H4A5; P0C0S5; Q6DN90-2; Q8NA54; Q92993; Q8IUB9; A2RU56; Q8TDB4; P41218; Q9Y605; O43196-2; O00746; Q96CV9-2; Q99497; Q13113; P27986-2; P12273; O75626-3; P57729; Q96QF0-7; Q9BY12-3; Q15019-3; Q9NQ40; Q12824; Q05C28; Q7Z6I5; O75558; P21980-2; Q9BUZ4; Q9NX07-2; Q5JTY5 EC number EC 3.6.4.- Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell projection; Chaperone; Cytoplasm; Cytoplasmic vesicle; Cytoskeleton; Disease variant; Dystonia; Endoplasmic reticulum; Glycoprotein; Hydrolase; Membrane; Nucleotide-binding; Nucleus; Proteomics identification; Reference proteome; Signal; Synapse Protein physicochemical properties Chain ID A,B Molecular weight (Da) 58238.4 Length 512 Aromaticity 0.12 Instability index 39.75 Isoelectric point 5.81 Charge (pH=7) -11.04 2D Binding mode Binding energy (Kcal/mol) -6.21  Molscript Map 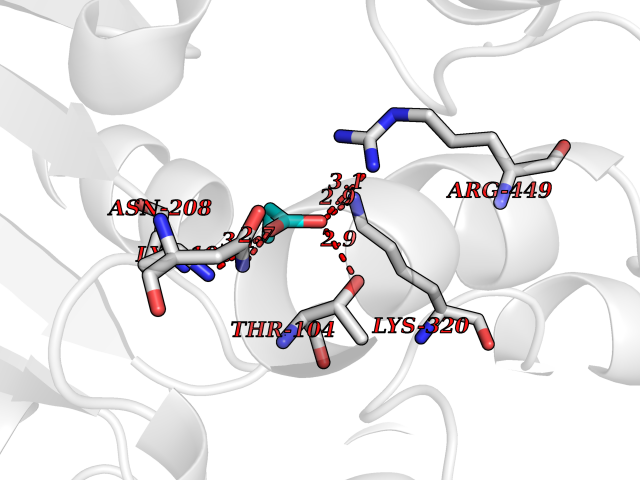 Pymol Map 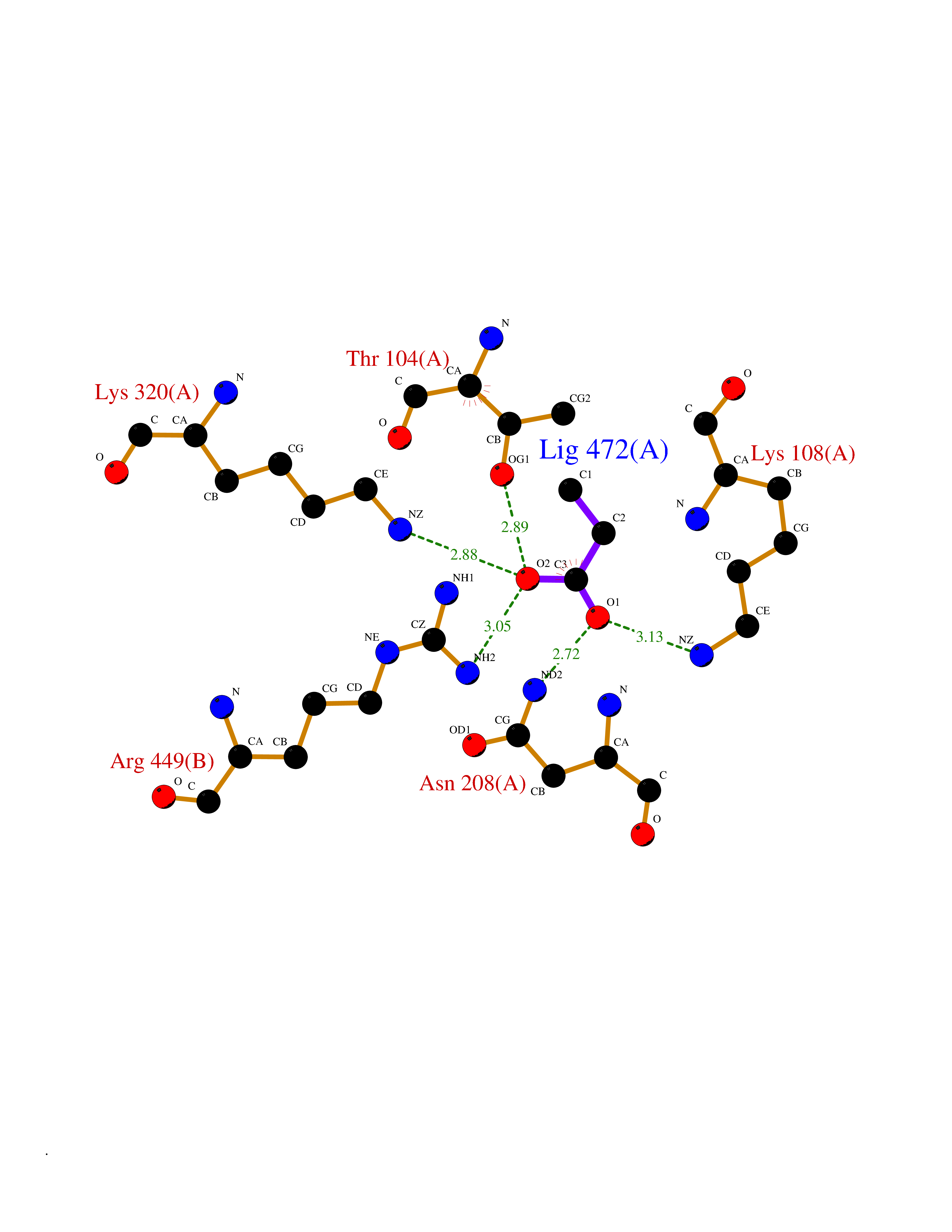 Ligplot Map 3D Binding mode Sequence SLSREALQKDLDDNLFGQHLAKKIILNAVFGFINNPKPKKPLTLSLHGWTGTGKNFVSKIIAENIYEGGLNSDYVHLFVATLHFPHASNITLYKDQLQLWIRGNVSACARSIFIFDQMDKMHAGLIDAIKPFLDYYDLVDGVSYQKAMFIFLSNAGAERITDVALDFWRSGKQREDIKLKDIEHALSVSVFNNKNSGFWHSSLIDRNLIDYFVPFLPLEYKHLKMCIRVEMQSRGYEIDEDIVSRVAEEMTFFPKEERVFSDKGCKTVFTKLDYYYDNSYYSSPAQQVPKNPALEAFLAQFSQLEDKFPGQSSFLWQRGRKFLQKHLNASNPTEPATIIFTAAREGRETLKCLSHHVADAYTSSQKVSPIQIDGAGRTWQDSDTVKLLVDLELSYGFENGQKAAVVHHFESFPAGSTLIFYKYCDHENAAFKDVALVLTVLLEEETLEASVGPRETEEKVRDLLWAKFTNSDTPTSFNHMDSDKLSGLWSRISHLVLPVQPVSSIEEQGCLF Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Hyperpolarization cyclic nucleotide-gated channel 2 (HCN2) | 3U10 | 4.55 | |
Target general information Gen name HCN2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channel 2; Brain cyclic nucleotide-gated channel 2; BCNG2; BCNG-2 Protein family Potassium channel HCN family Biochemical class Voltage-gated ion channel Function Contributes to the native pacemaker currents in heart (If) and in neurons (Ih). Can also transport ammonium in the distal nephron. Produces a large instantaneous current. Modulated by intracellular chloride ions and pH; acidic pH shifts the activation to more negative voltages. Hyperpolarization-activated ion channel exhibiting weak selectivity for potassium over sodium ions. Related diseases Epilepsy, idiopathic generalized 17 (EIG17) [MIM:602477]: A form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. Both autosomal dominant and autosomal recessive EIG17 inheritance have been reported. {ECO:0000269|PubMed:22131395, ECO:0000269|PubMed:29064616}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Febrile seizures, familial, 2 (FEB2) [MIM:602477]: Seizures associated with febrile episodes in childhood without any evidence of intracranial infection or defined pathologic or traumatic cause. It is a common condition, affecting 2-5% of children aged 3 months to 5 years. The majority are simple febrile seizures (generally defined as generalized onset, single seizures with a duration of less than 30 minutes). Complex febrile seizures are characterized by focal onset, duration greater than 30 minutes, and/or more than one seizure in a 24 hour period. The likelihood of developing epilepsy following simple febrile seizures is low. Complex febrile seizures are associated with a moderately increased incidence of epilepsy. FEB2 transmission pattern is consistent with autosomal dominant inheritance. {ECO:0000269|PubMed:24324597}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02527; DB02315; DB09083 Interacts with Q9UL51; Q4ACU6-1 EC number NA Uniprot keywords 3D-structure; Ammonia transport; cAMP; cAMP-binding; Cell membrane; Disease variant; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Lipoprotein; Membrane; Methylation; Nucleotide-binding; Palmitate; Phosphoprotein; Potassium; Potassium channel; Potassium transport; Proteomics identification; Reference proteome; Sodium; Sodium channel; Sodium transport; Transmembrane; Transmembrane helix; Transport; Voltage-gated channel Protein physicochemical properties Chain ID A Molecular weight (Da) 23672.9 Length 202 Aromaticity 0.12 Instability index 38.05 Isoelectric point 8.85 Charge (pH=7) 4.11 2D Binding mode Binding energy (Kcal/mol) -6.2  Molscript Map 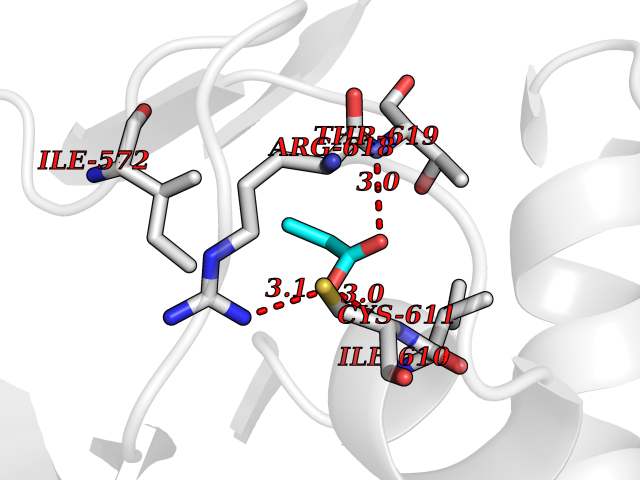 Pymol Map 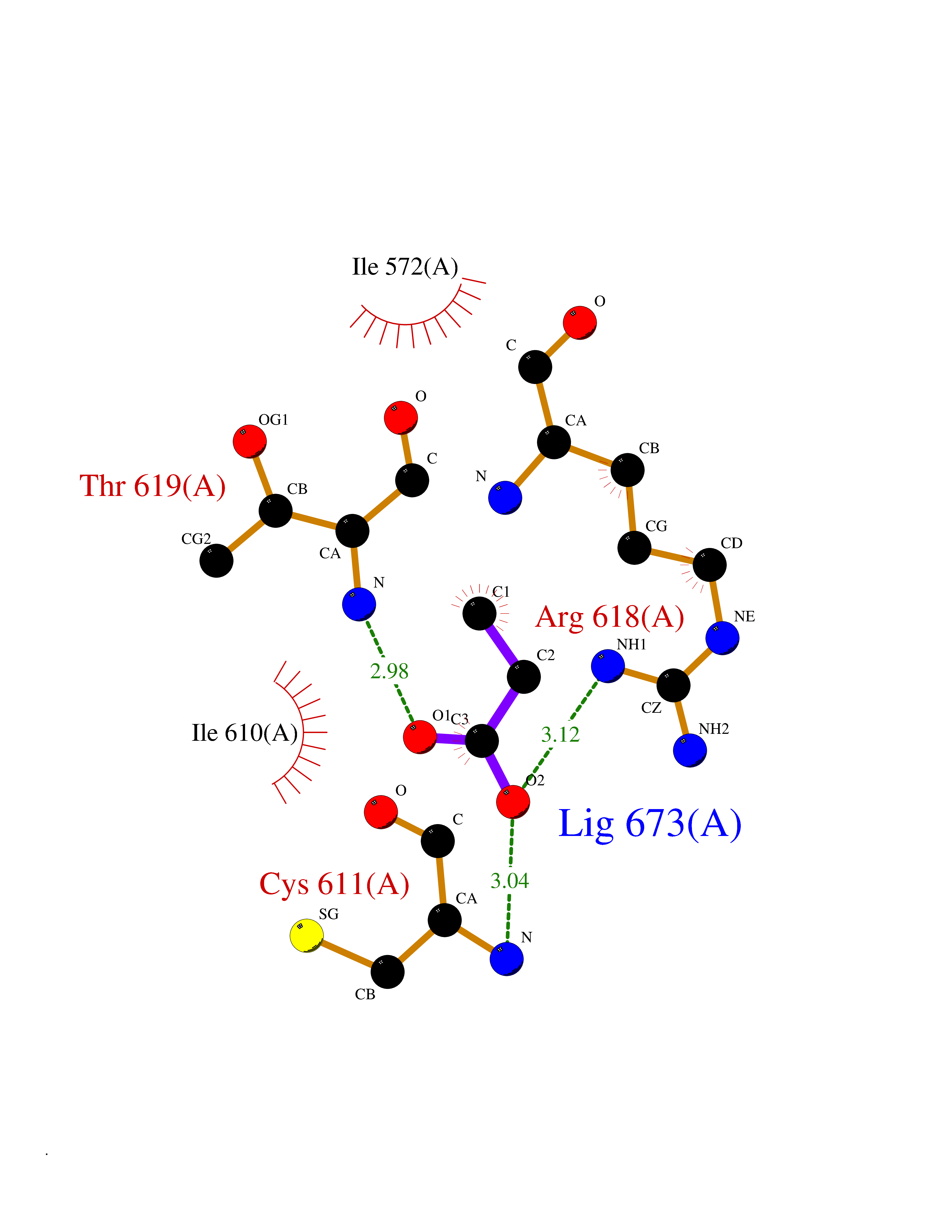 Ligplot Map 3D Binding mode Sequence DSSRRQYQEKYKQVEQYMSFHKLPADFRQKIHDYYEHRYQGKMFDEDSILGELNGPLREEIVNFNCRKLVASMPLFANADPNFVTAMLTKLKFEVFQPGDYIIREGTIGKKMYFIQHGVVSVLTKGNKEMKLSDGSYFGEICLLTRGRRTASVRADTYCRLYSLSVDNFNEVLEEYPMMRRAFETVAIDRLDRIGKKNSILL Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Endoplasmic reticulum chaperone BiP (HSPA5) | 6ASY | 4.55 | |
Target general information Gen name HSPA5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Immunoglobulin heavy chainbinding protein; Immunoglobulin heavy chain-binding protein; Heat shock protein family A member 5; Heat shock protein 70 family protein 5; Heat shock 70 kDa protein 5; HSP70 Protein family Heat shock protein 70 family Biochemical class Acid anhydride hydrolase Function Involved in the correct folding of proteins and degradation of misfolded proteins via its interaction with DNAJC10/ERdj5, probably to facilitate the release of DNAJC10/ERdj5 from its substrate. Acts as a key repressor of the ERN1/IRE1-mediated unfolded protein response (UPR). In the unstressed endoplasmic reticulum, recruited by DNAJB9/ERdj4 to the luminal region of ERN1/IRE1, leading to disrupt the dimerization of ERN1/IRE1, thereby inactivating ERN1/IRE1. Accumulation of misfolded protein in the endoplasmic reticulum causes release of HSPA5/BiP from ERN1/IRE1, allowing homodimerization and subsequent activation of ERN1/IRE1. Plays an auxiliary role in post-translational transport of small presecretory proteins across endoplasmic reticulum (ER). May function as an allosteric modulator for SEC61 channel-forming translocon complex, likely cooperating with SEC62 to enable the productive insertion of these precursors into SEC61 channel. Appears to specifically regulate translocation of precursors having inhibitory residues in their mature region that weaken channel gating. Endoplasmic reticulum chaperone that plays a key role in protein folding and quality control in the endoplasmic reticulum lumen. Related diseases Autoantigen in rheumatoid arthritis. {ECO:0000269|PubMed:11160188}. Drugs (DrugBank ID) DB00945; DB00025; DB09130; DB13998; DB13999 Interacts with Q9BYF1; P05067; P18850; Q9ULD4-2; Q6E0U4; Q9UBS4; Q9UBS3; P49184; Q92874; Q9NZJ5; P04626; O75460; O75460-1; P62993; Q15486; P14625; Q9Y4L1; P01721; O95868; Q9Y328; Q96IZ0; Q15084; P04049; P61619; P36955; Q9H173; Q9UHI5; Q13573; Q6URK8; Q6PF05; Q13404; Q9NYU1; P09544; Q6T424; Q9QXT0; Q91YW3; Q9Z2B5; Q62627; K0BRG7; P0DTC2 EC number EC 3.6.4.10 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Chaperone; Cytoplasm; Direct protein sequencing; Endoplasmic reticulum; Host-virus interaction; Hydrolase; Isopeptide bond; Methylation; Nitration; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Signal; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 67147.3 Length 606 Aromaticity 0.06 Instability index 32.09 Isoelectric point 5.22 Charge (pH=7) -15.9 2D Binding mode Binding energy (Kcal/mol) -6.2  Molscript Map 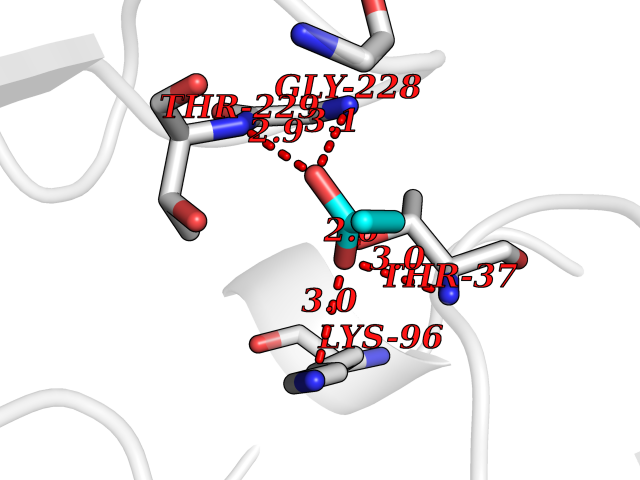 Pymol Map  Ligplot Map 3D Binding mode Sequence SEDVGTVVGIDLGTTYSCVGVFKNGRVEIIANDQGNRITPSYVAFTPEGERLIGDAAKNQLTSNPENTVFDAKRLIGRTWNDPSVQQDIKFLPFKVVEKKTKPYIQVDIGGGQTKTFAPEEISAMVLTKMKETAEAYLGKKVTHAVVTVPAYFNDAQRQATKDAGTIAGLNVMRIINEPTAAAIAYGLDKREGEKNILVFDLGGGTFDVSLLTIDNGVFEVVATNGDTHLGGEDFDQRVMEHFIKLYKKKTGKDVRKDNRAVQKLRREVEKAKRALSSQHQARIEIESFYEGEDFSETLTRAKFEELNMDLFRSTMKPVQKVLEDSDLKKSDIDEIVLVGGSTRIPKIQQLVKEFFNGKEPSRGINPDEAVAYGAAVQAGVLSGDQDTGDLVLLDVCPLTLGIETVGGVMTKLIPRNTVVPTKKSQIFSVGGTVTIKVYEGERPLTKDNHLLGTFDLTGIPPAPRGVPQIEVTFEIDVNGILRVTAEDKGTGNKNKITITNDQNRLTPEEIERMVNDAEKFAEEDKKLKERIDTRNELESYAYSLKNQIGDKEKLGGKLSSEDKETMEKAVEEKIEWLESHQDADIEDFKAKKKELEEIVQPIISK Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | DNA-dependent protein kinase catalytic (PRKDC) | 7SGL | 4.55 | |
Target general information Gen name PRKDC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p460; HYRC1; HYRC; DNPK1; DNA-dependent protein kinase catalytic subunit; DNA-PKcs; DNA-PK catalytic subunit Protein family PI3/PI4-kinase family Biochemical class Kinase Function Serine/threonine-protein kinase that acts as a molecular sensor for DNA damage. Involved in DNA non-homologous end joining (NHEJ) required for double-strand break (DSB) repair and V(D)J recombination. Must be bound to DNA to express its catalytic properties. Promotes processing of hairpin DNA structures in V(D)J recombination by activation of the hairpin endonuclease artemis (DCLRE1C). The assembly of the DNA-PK complex at DNA ends is also required for the NHEJ ligation step. Required to protect and align broken ends of DNA. May also act as a scaffold protein to aid the localization of DNA repair proteins to the site of damage. Found at the ends of chromosomes, suggesting a further role in the maintenance of telomeric stability and the prevention of chromosomal end fusion. Also involved in modulation of transcription. Recognizes the substrate consensus sequence [ST]-Q. Phosphorylates 'Ser-139' of histone variant H2AX/H2AFX, thereby regulating DNA damage response mechanism. Phosphorylates DCLRE1C, c-Abl/ABL1, histone H1, HSPCA, c-jun/JUN, p53/TP53, PARP1, POU2F1, DHX9, FH, SRF, XRCC1, XRCC1, XRCC4, XRCC5, XRCC6, WRN, MYC and RFA2. Can phosphorylate C1D not only in the presence of linear DNA but also in the presence of supercoiled DNA. Ability to phosphorylate p53/TP53 in the presence of supercoiled DNA is dependent on C1D. Contributes to the determination of the circadian period length by antagonizing phosphorylation of CRY1 'Ser-588' and increasing CRY1 protein stability, most likely through an indirect mechanism (By similarity). Plays a role in the regulation of DNA virus-mediated innate immune response by assembling into the HDP-RNP complex, a complex that serves as a platform for IRF3 phosphorylation and subsequent innate immune response activation through the cGAS-STING pathway. Related diseases Immunodeficiency 26 with or without neurologic abnormalities (IMD26) [MIM:615966]: A form of severe combined immunodeficiency characterized by reduced or absent T and B cells, recurrent candidiasis, and lower respiratory tract infections. Some patients show dysmorphic features, severe growth failure, microcephaly, seizures, and impaired neurological functions. {ECO:0000269|PubMed:19075392, ECO:0000269|PubMed:23722905}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00201; DB05210 Interacts with O43918; P10275; Q96SD1; P14921; P50549; P09629; Q9BPZ7; Q96RI1-2; P10276; P17947; P13010; P12956; P25490 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Biological rhythms; Disease variant; DNA damage; DNA recombination; DNA repair; DNA-binding; Immunity; Innate immunity; Kinase; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Ribosome biogenesis; SCID; Serine/threonine-protein kinase; TPR repeat; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 437836 Length 3852 Aromaticity 0.09 Instability index 47.88 Isoelectric point 6.63 Charge (pH=7) -10.35 2D Binding mode Binding energy (Kcal/mol) -6.21  Molscript Map 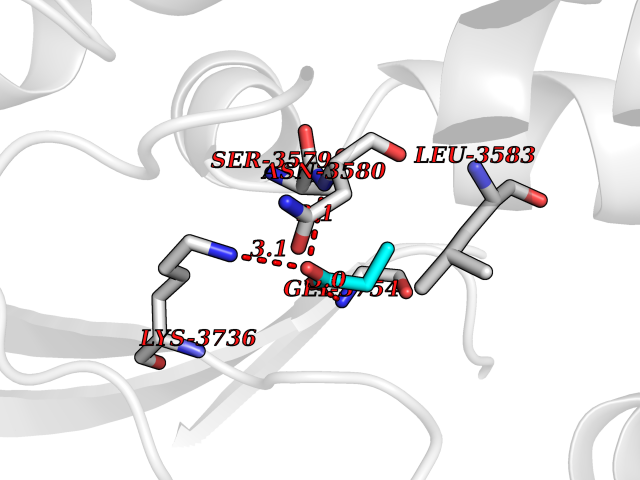 Pymol Map 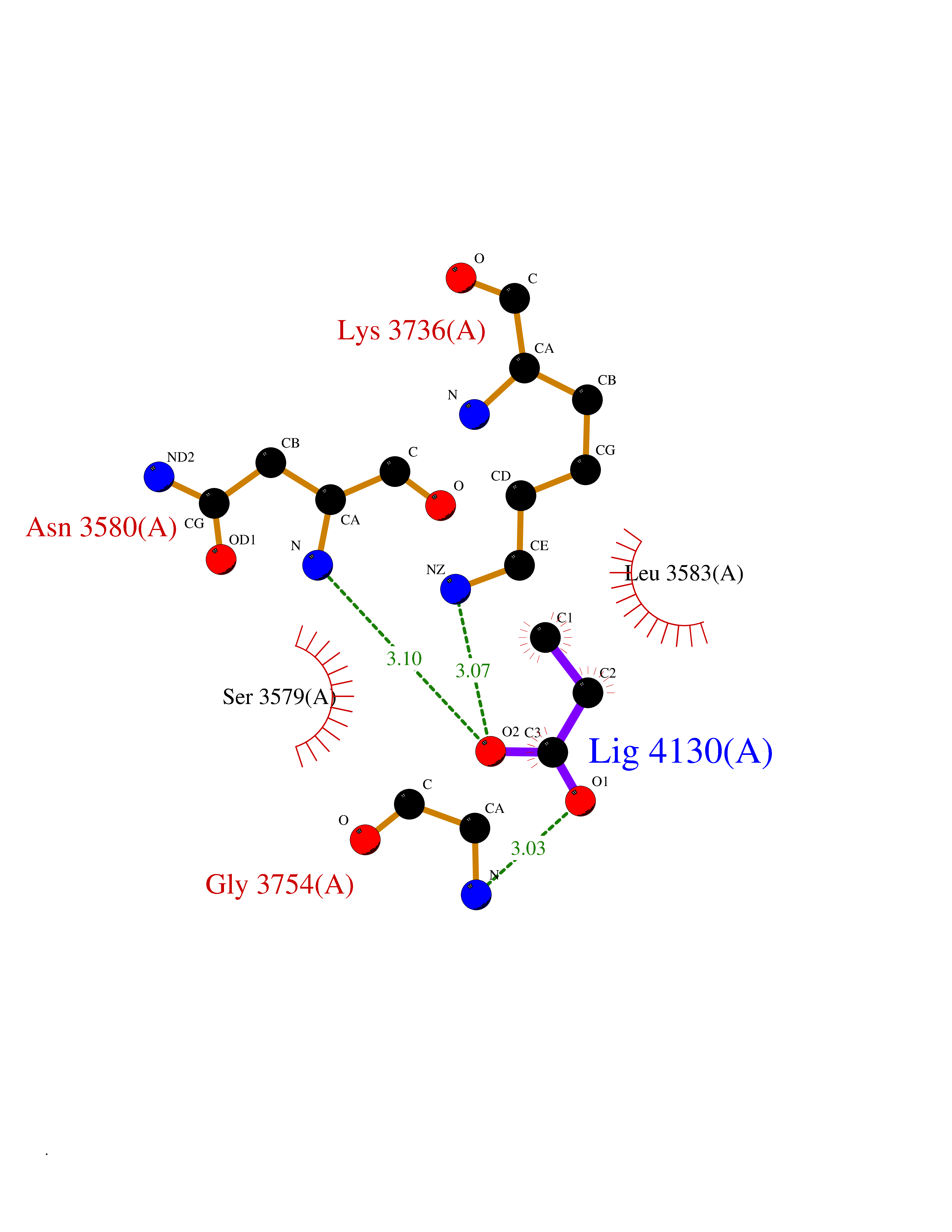 Ligplot Map 3D Binding mode Sequence MAGSGAGVRCSLLRLQETLSAADRCGAALAGHQLIRGLGQECVLSSSPAVLALQTSLVFSRDFGLLVFVRKSLNSIEFRECREEILKFLCIFLEKMGQKIAPYSVEIKNTCTSVYTKDRAAKCKIPALDLLIKLLQTFRSSRLMDEFKIGELFSKFYGELALKKKIPDTVLEKVYELLGLLGEVHPSEMINNAENLFRAFLGELKTQMTSAVREPKLPVLAGCLKGLSSLLCNFTKSMEEDPQTSREIFNFVLKAIRPQIDLKRYAVPSAGLRLFALHASQFSTCLLDNYVSLFEVLLKWCAHTNVELKKAALSALESFLKQVSNMVAKNAEMHKNKLQYFMEQFYGIIRNVDSNNKELSIAIRGYGLFAGPCKVINAKDVDFMYVELIQRCKQMFLTQTDTGDDRVYQMPSFLQSVASVLLYLDTVPEVYTPVLEHLVVMQIDSFPQYSPKMQLVCCRAIVKVFLALAAKGPVLRNCISTVVHQGLIRICSKPVVLPKGPESESEDHRASGEVRTGKWKVPTYKDYVDLFRHLLSSDQMMDSILADEAFFSVNSSSESLNHLLYDEFVKSVLKIVEKLDLTLEIDPAANLHPAKPKDFSAFINLVEFCREILPEKQAEFFEPWVYSFSYELILQSTRLPLISGFYKLLSITVRNAKKIKYFEGVSDPEKYSCFALFVKFGKEVAVKMKQYKDELLASCLTFLLSLPHNIIELDVRAYVPALQMAFKLGLSYTPLAEVGLNALEEWSIYIDRHVMQPYYKDILPCLDGYLKTNWEVSALSRAAAISLEEIRIRVVQMLGSLGGQINKNLLTVTSSDEMMKSYVAWDREKRLSFAVPFREMKPVIFLDVFLPRVTELALTASDRQTKVAACELLHSMVMFMLGKATQMPEGGQGAPPMYQLYKRTFPVLLRLACDVDQVTRQLYEPLVMQLIHWFTNNKKFESQDTVALLEAILDGIVDPVDSTLRDFCGRCIREFLKWSIKQITPQQQEKSPVNTKSLFKRLYSLALHPNAFKRLGASLAFNNIYREFREEESLVEQFVFEALVIYMESLALAHADEKSLGTIQQCCDAIDHLCRIIEKKHVSLNKAKKRRLPRGFPPSASLCLLDLVKWLLAHCGRPQTECRHKSIELFYKFVPLLPGNRSPNLWLKDVLKEEGVSFLINTFEGGGCGQPSGILAQPTLLYLRGPFSLQATLCWLDLLLAALECYNTFIGERTVGALQVLGTEAQSSLLKAVAFFLESIAMHDIIAAEKCFGTGAAGNRTSPQEGERYNYSKCTVVVRIMEFTTTLLNTSPEGWKLLKKDLCNTHLMRVLVQTLCEPASIGFNIGDVQVMAHLPDVCVNLMKALKMSPYKDILETHLREKITAQSIEELCAVNLYGPDAQVDRSRLAAVVSACKQLHRAGLLHNILPSQSTDLHHSVGTELLSLVYKGIAPGDERQCLPSLDLSCKQLASGLLELAFAFGGLCERLVSLLLNPAVLSTSVIHFSHGEYFYSLFSETINTELLKNLDLAVLELMQSSVDNTKMVSAVLNGMLDQSFRERANQKHQGLKLATTILQHWKKCDSWWAKDSPLETKMAVLALLAKILQIDSSVSFNTSHGSFPEVFTTYISLLADTKLDLHLKGQAVTLLPFFTSLTGGSLEELRRVLEQLIVAHFPMQSREFPPGTPRFNNYVDCMKKFLDALELSQSPMLLELMTEVLCREQQHVMEELFQSSFRRIARRGSCVTQVGLLESVYEMFRKDDPRLSFTRQSFVDRSLLTLLWHCSLDALREFFSTIVVDAIDVLKSRFTKLNESTFDTQITKKMGYYKILDVMYSRLPKDDVHAKESKINQVFHGSCITEGNELTKTLIKLCYDAFTENMAGENQLLERRRLYHCAAYNCAISVICCVFNELKFYQGFLFSEKPEKNLLIFENLIDLKRRYNFPVEVEVPMERKKKYIEIRKEAREAANGDSDGPSYMSSLSYLADSTLSEEMSQFDFSTGVQSYSYSSQLEMDELNRHECMAPLTALVKHMHRSPRDLPSWMKFLHGKLGNPIVPLNIRLFLAKLVINTEEVFRPYAKHWLSPLLQLAASENNGGEGIHYMVVEIVATILSWTGLATPTGVPKDEVLANRLLNFLMKHVFHPKRAVFRHNLEIIKTLVECWKDCLSIPYRLIFEKFSGKDPNSKDNSVGIQLLGIVMANDLPPYDPQCGIQSSEYFQALVNNMSFVRYKEVYAAAAEVLGLILRYVMERKNILEESLCELVAKQLKQHQNTMEDKFIVCLNKVTKSFPPLADRFMNAVFFLLPKFHGVLKTLCLEVVLCRVEGMTELYFQLKSKDFVQVMRHRDDERQKVCLDIIYKMMPKLKPVELRELLNPVVEFVSHPSTTCREQMYNILMWIHDNYRDPESETDNDSQEIFKLAKDVLIQGLIDENPGLQLIIRNFWSHETRLPSNTLDRLLALNSLYSPKIEVHFLSLATNFLLEMTSMSPDYPNPMFEHPLSECEFQEYTIDSDWRFRSTVLTPMFVEXQASQGPVAGQIRAXQQQHDFXLXQTQVVLYRSYRHGDLPDIQIKHSSLITPLQAVAQRDPIIAKQLFSSLFSGILKEMDKFKTLSEKNNITQKLLQDFNRFLNTTFSFFPPFVSCIQDISCQHAALLSLDPAAVSAGCLASLQQPVGIRLLEEALLRLLPALPPDVLRWVELAKLYRSIGEYDVLRGIFTSEIGTKQITQSALLAEARSDYSEAAKQYDEALNKQDWVDGEPTEAEKDFWELASLDCYNHLAEWKSLEYCSTASIDSENPPDLNKIWSEPFYQETYLPYMIRSKLKLLLQGEADQSLLTFIDKAMHGELQKAILELHYSQELSLLYLLQDDVDRAKYYIQNGIQSFMQNYSSIDVLLHQSRLTKLQSVQALTEIQEFISFISKQGNLSSQVPLKRLLNTWTNRYPDAKMDPMNIWDDIITNRCFFLSKIEEKLTEDISSLIRSCKFSMKMKMIDSARKQNNFSLAMKLLKELHKESKTRDDWLVSWVQSYCRLSHCRSRSQGCSEQVLTVLKTVSLLDENNVSSYLSKNILAFRDQNILLGTTYRIIANALSSEPACLAEIEEDKARRILELSGSSSEDSEKVIAGLYQRAFQHLSEAVQAAEEEAQPPSWSCGPAAGVIDAYMTLADFCDQQLRKEEENASVIDSAELQAYPALVVEKMLKALKLNSNEARLKFPRLLQIIERYPEETLSLMTKEISSVPCWQFISWISHMVALLDKDQAVAVQHSVEEITDNYPQAIVYPFIISSESYSFKDTSTGHKNKEFVARIKSKLDQGGVIQDFINALDQLSNPELLFKDWSNDVRAELAKTPVNKKNIEKMYERMYAALGDPKAPGLGAFRRKFIQTFGKEFDKHFGKGGSKLLRMKLSDFNDITNMLLLKMNKDSKPPGNLKECSPWMSDFKVEFLRNELEIPGQYDGRGKPLPEYHVRIAGFDERVTVMASLRRPKRIIIRGHDEREHPFLVKGGEDLRQDQRVEQLFQVMNGILAQDSACSQRALQLRTYSVVPMTSRLGLIEWLENTVTLKDLLLNTMSQEEKAAYLSDPRAPPCEYKDWLTKMSGKHDVGAYMLMYKGANRTETVTSFRKRESKVPADLLKRAFVRMSTSPEAFLALRSHFASSHALICISHWILGIGDRHLNNFMVAMETGGVIGIDFGHAFGSATQFLPVPELMPFRLTRQFINLMLPMKETGLMYSIMVHALRAFRSDPGLLTNTMDVFVKEPSFDWKNFEQKMLKKGGSWIQEINVAEKNWYPRQKICYAKRKLAGANPAVITCDELLLGHEKAPAFRDYVAVARGSKDHNIRAQEPESGLSEETQVKCLMDQATDPNILGRTWEGWEPWM Hydrogen bonds contact Hydrophobic contact | ||||