Job Results:
Ligand
Structure
Job ID
a9ea1a8efdae070d2ad25867c2a2c1f3
Job name
NA
Time
2026-02-24 16:12:06
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Gamma-aminobutyric acid type B receptor subunit 1 | 4MS4 | 5.60 | |
Target general information Gen name GABBR1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms GPRC3B;GPR51 Protein family G-protein coupled receptor 3 family, GABA-B receptor subfamily Biochemical class Signaling protein / antagonist Function G-protein coupled GABA receptor activity. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08891; DB08892; DB00181; DB00363; DB02530; DB05010; DB09072 Interacts with Q9UBS5; Q9UBS5-2; P46459; Q86UR5 EC number NA Uniprot keywords 3D-structure; Cell membrane; Coiled coil; Direct protein sequencing; Disease variant; Disulfide bond; Epilepsy; G-protein coupled receptor; Glycoprotein; Intellectual disability; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 46502.1 Length 408 Aromaticity 0.12 Instability index 50.05 Isoelectric point 5.78 Charge (pH=7) -5.62 2D Binding mode Binding energy (Kcal/mol) -7.64 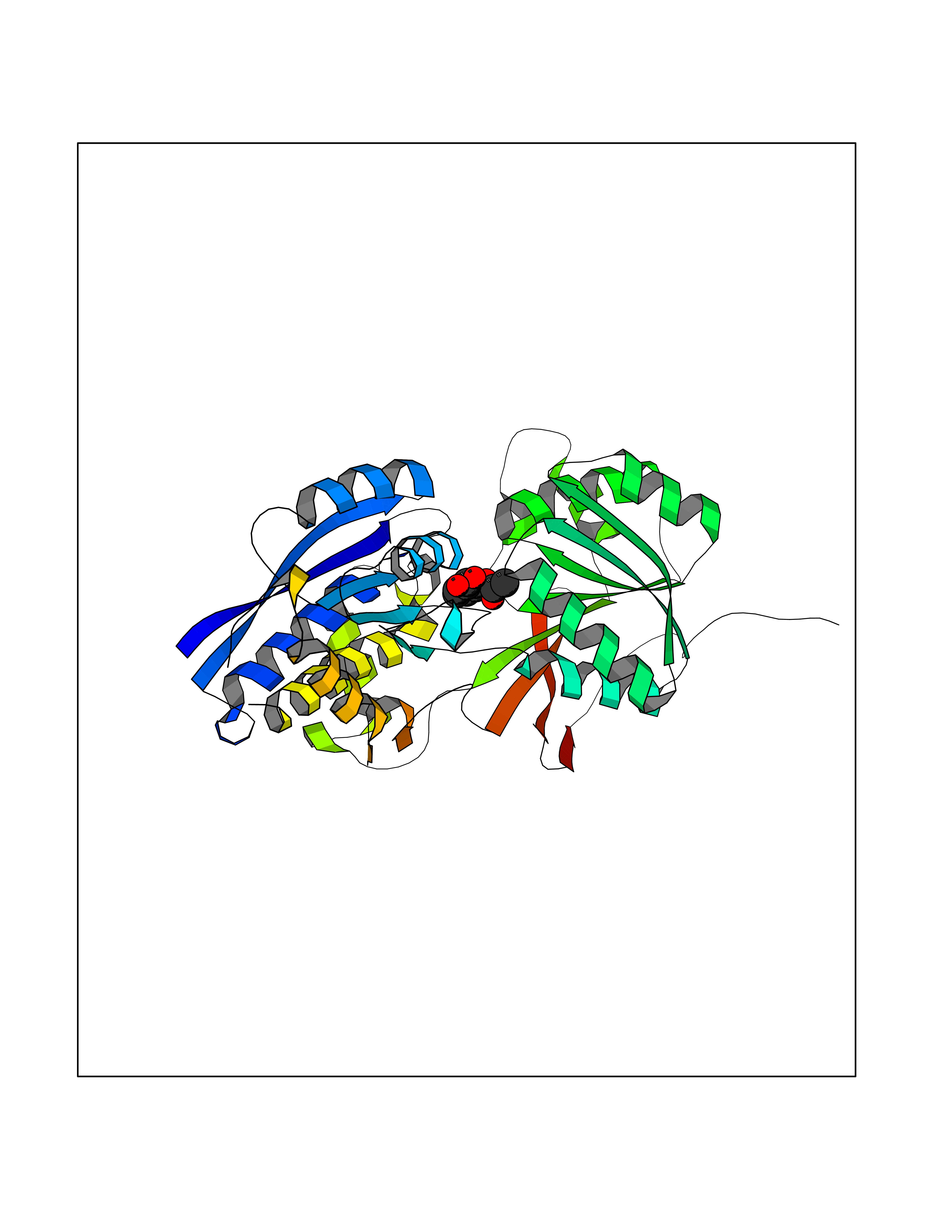 Molscript Map 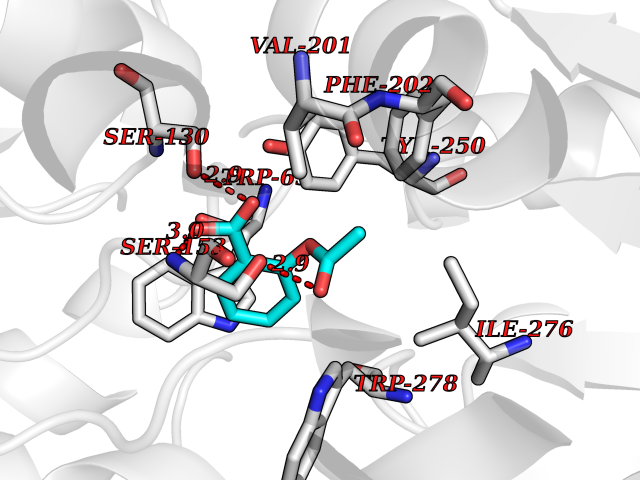 Pymol Map 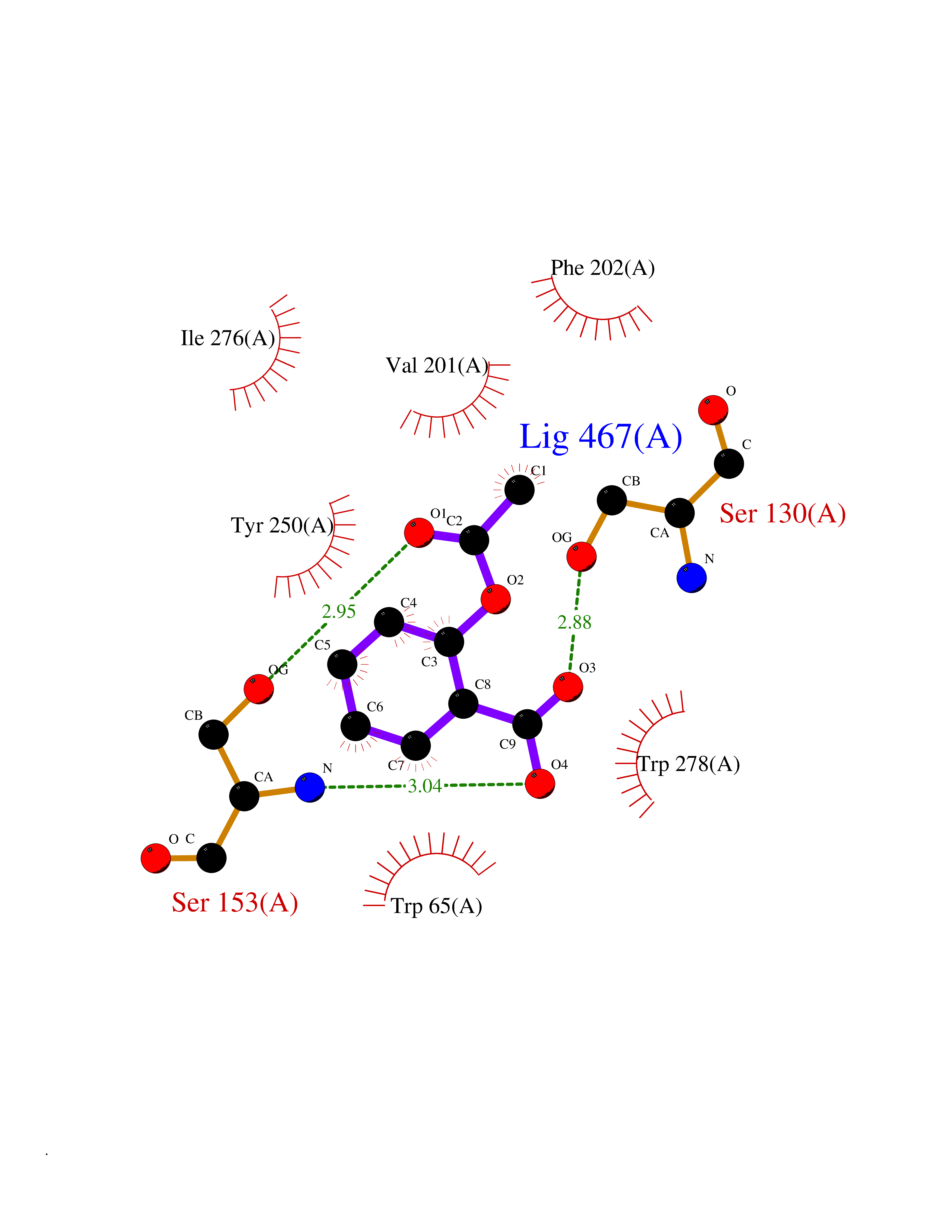 Ligplot Map 3D Binding mode Sequence RRAVYIGALFPMSGGWPGGQACQPAVEMALEDVNSRRDILPDYELKLIHHDSKCDPGQATKYLYELLYNDPIKIILMPGCSSVSTLVAEAARMWNLIVLSYGSSSPALSNRQRFPTFFRTHPSATLHNPTRVKLFEKWGWKKIATIQQTTEVFTSTLDDLEERVKEAGIEITFRQSFFSDPAVPVKNLKRQDARIIVGLFYETEARKVFCEVYKERLFGKKYVWFLIGWYADNWFKIYDPSINCTVDEMTEAVEGHITTEIVMLNPANTRSISNMTSQEFVEKLTKRLKRHPEETGGFQEAPLAYDAIWALALALNKTSRLEDFNYNNQTITDQIYRAMNSSSFEGVSGHVVFDASGSRMAWTLIEQLQGGSYKKIGYYDSTKDDLSWSKTDKWIGGSPPADDYKDDD Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Cystathionine gamma-lyase (CTH) | 3COG | 5.60 | |
Target general information Gen name CTH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Gamma-cystathionase; Cysteine-protein sulfhydrase Protein family Trans-sulfuration enzymes family Biochemical class NA Function Catalyzes the last step in the trans-sulfuration pathway from methionine to cysteine. Has broad substrate specificity. Converts cystathionine to cysteine, ammonia and 2-oxobutanoate. Converts two cysteine molecules to lanthionine and hydrogen sulfide. Can also accept homocysteine as substrate. Specificity depends on the levels of the endogenous substrates. Generates the endogenous signaling molecule hydrogen sulfide (H2S), and so contributes to the regulation of blood pressure. Acts as a cysteine-protein sulfhydrase by mediating sulfhydration of target proteins: sulfhydration consists of converting -SH groups into -SSH on specific cysteine residues of target proteins such as GAPDH, PTPN1 and NF-kappa-B subunit RELA, thereby regulating their function. Related diseases Cystathioninuria (CSTNU) [MIM:219500]: Autosomal recessive phenotype characterized by abnormal accumulation of plasma cystathionine, leading to increased urinary excretion. {ECO:0000269|PubMed:12574942, ECO:0000269|PubMed:18476726}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02328; DB03928; DB00151; DB04217; DB00114 Interacts with P32929; Q96NT3; Q96NT3-2; Q96HA8; Q6P9E2 EC number EC 4.4.1.1 Uniprot keywords 3D-structure; Alternative splicing; Amino-acid biosynthesis; Calmodulin-binding; Cysteine biosynthesis; Cytoplasm; Disease variant; Lipid metabolism; Lyase; Proteomics identification; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 86026 Length 782 Aromaticity 0.08 Instability index 32.4 Isoelectric point 6.27 Charge (pH=7) -9.46 2D Binding mode Binding energy (Kcal/mol) -7.64 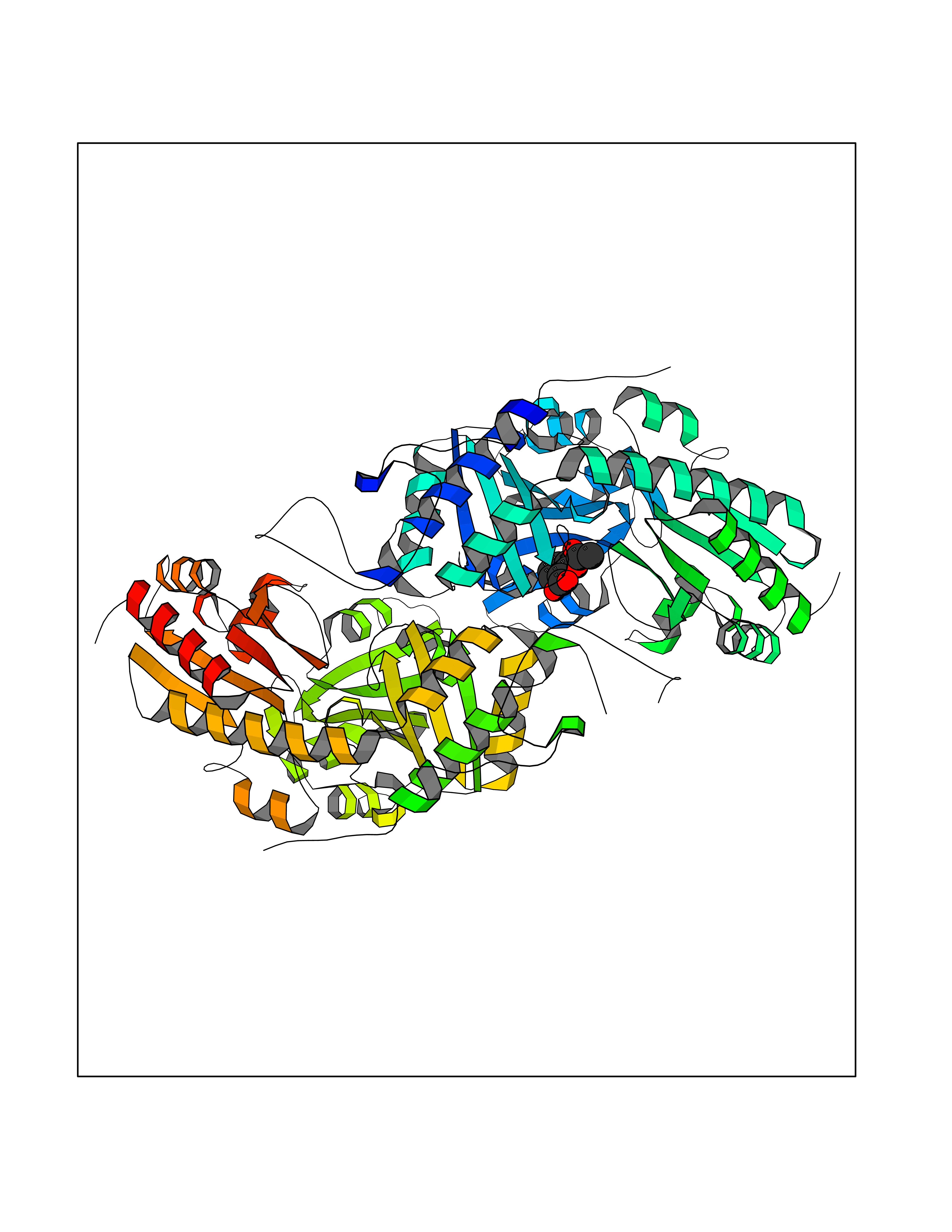 Molscript Map 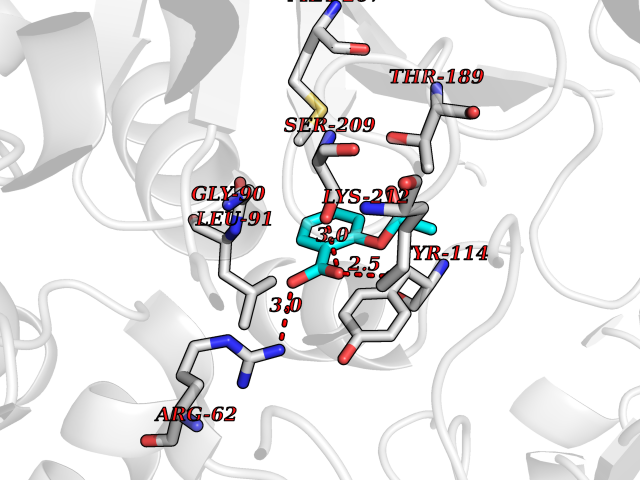 Pymol Map 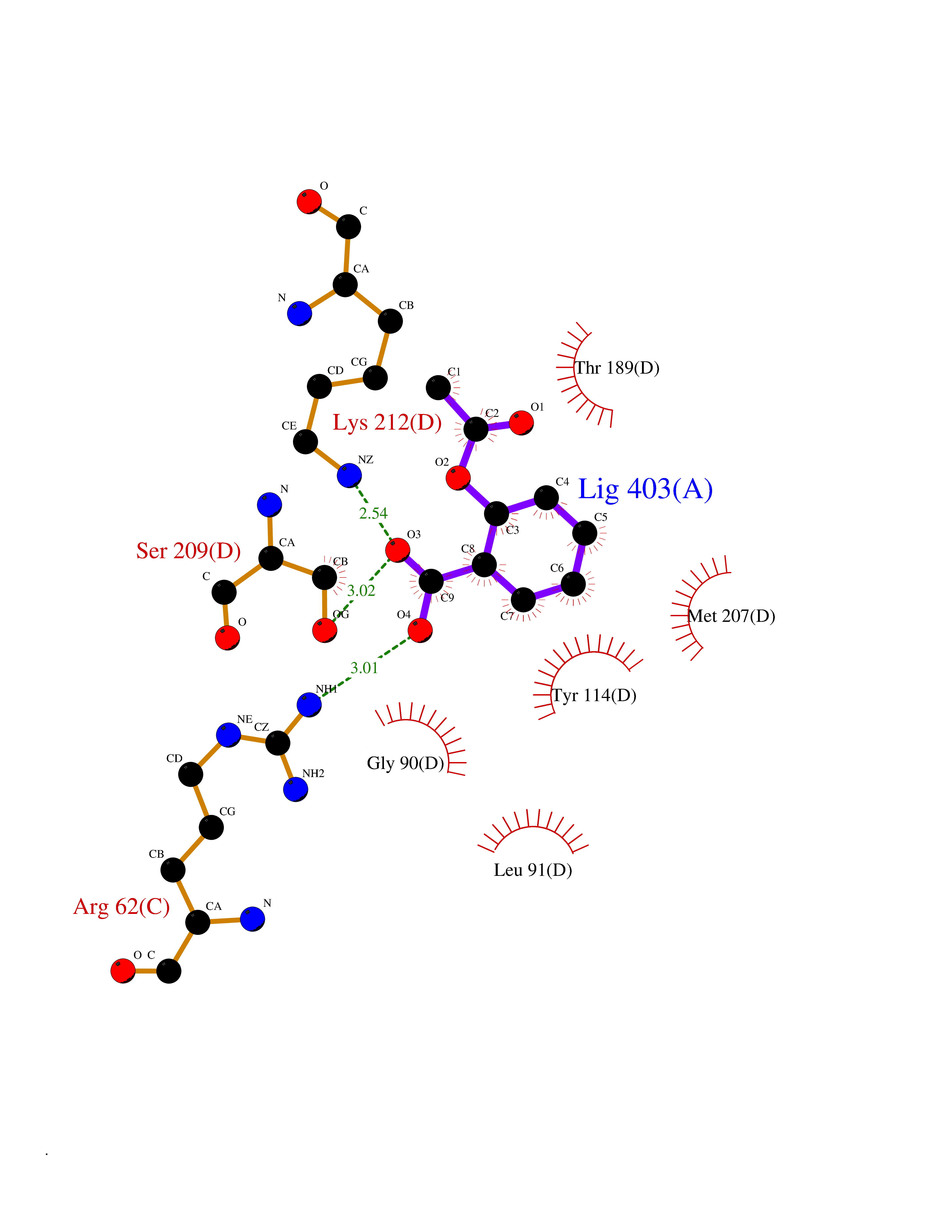 Ligplot Map 3D Binding mode Sequence GFLPHFQHFATQAIHVGQDPEQWTSRAVVPPISLSTTFKQGAPGQHSGFEYSRSGNPTRNCLEKAVAALDGAKYCLAFASGLAATVTITHLLKAGDQIICMDDVYGGTNRYFRQVASEFGLKISFVDCSKIKLLEAAITPETKLVWIETPTNPTQKVIDIEGCAHIVHKHGDIILVVDNTFMSPYFQRPLALGADISMYSATKYMNGHSDVVMGLVSVNCESLHNRLRFLQNSLGAVPSPIDCYLCNRGLKTLHVRMEKHFKNGMAVAQFLESNPWVEKVIYPGLPSHPQHELVKRQCTGCTGMVTFYIKGTLQHAEIFLKNLKLFTLAESLGGFESLAELPAIMTHASVLKNDRDVLGISDTLIRLSVGLEDEEDLLEDLDQALKAAHPPSGFLPHFQHFATQAIHVGQDPEQWTSRAVVPPISLSTTFKQGAPGQGFEYSRSGNPTRNCLEKAVAALDGAKYCLAFASGLAATVTITHLLKAGDQIICMDDVYGGTNRYFRQVASEFGLKISFVDCSKIKLLEAAITPETKLVWIETPTNPTQKVIDIEGCAHIVHKHGDIILVVDNTFMSPYFQRPLALGADISMYSATKYMNGHSDVVMGLVSVNCESLHNRLRFLQNSLGAVPSPIDCYLCNRGLKTLHVRMEKHFKNGMAVAQFLESNPWVEKVIYPGLPSHPQHELVKRQCTGCTGMVTFYIKGTLQHAEIFLKNLKLFTLAESLGGFESLAELPAIMTHASVLKNDRDVLGISDTLIRLSVGLEDEEDLLEDLDQALKAAHPPS Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Pyruvate oxidase | 2EZ9 | 5.60 | |
Target general information Gen name pox5 Organism Lactiplantibacillus plantarum (strain ATCC BAA-793 / NCIMB 8826 / WCFS1) (Lactobacillus plantarum) Uniprot ID TTD ID NA Synonyms lp_3589 Protein family TPP enzyme family Biochemical class Oxidoreductase Function Magnesium ion binding.Pyruvate oxidase activity.Thiamine pyrophosphate binding. Related diseases Telangiectasia, hereditary hemorrhagic, 2 (HHT2) [MIM:600376]: A multisystemic vascular dysplasia leading to dilation of permanent blood vessels and arteriovenous malformations of skin, mucosa, and viscera. The disease is characterized by recurrent epistaxis and gastro-intestinal hemorrhage. Visceral involvement includes arteriovenous malformations of the lung, liver, and brain. {ECO:0000269|PubMed:10694922, ECO:0000269|PubMed:10767348, ECO:0000269|PubMed:11170071, ECO:0000269|PubMed:11484689, ECO:0000269|PubMed:14684682, ECO:0000269|PubMed:15024723, ECO:0000269|PubMed:15712270, ECO:0000269|PubMed:16525724, ECO:0000269|PubMed:16752392, ECO:0000269|PubMed:20414677, ECO:0000269|PubMed:26176610, ECO:0000269|PubMed:8640225, ECO:0000269|PubMed:9245985}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01987; DB03147 Interacts with NA EC number 1.2.3.3 Uniprot keywords 3D-structure; FAD; Flavoprotein; Magnesium; Metal-binding; Oxidoreductase; Reference proteome; Thiamine pyrophosphate Protein physicochemical properties Chain ID A,B Molecular weight (Da) 64137.9 Length 585 Aromaticity 0.08 Instability index 28.31 Isoelectric point 5.17 Charge (pH=7) -18.41 2D Binding mode Binding energy (Kcal/mol) -7.63 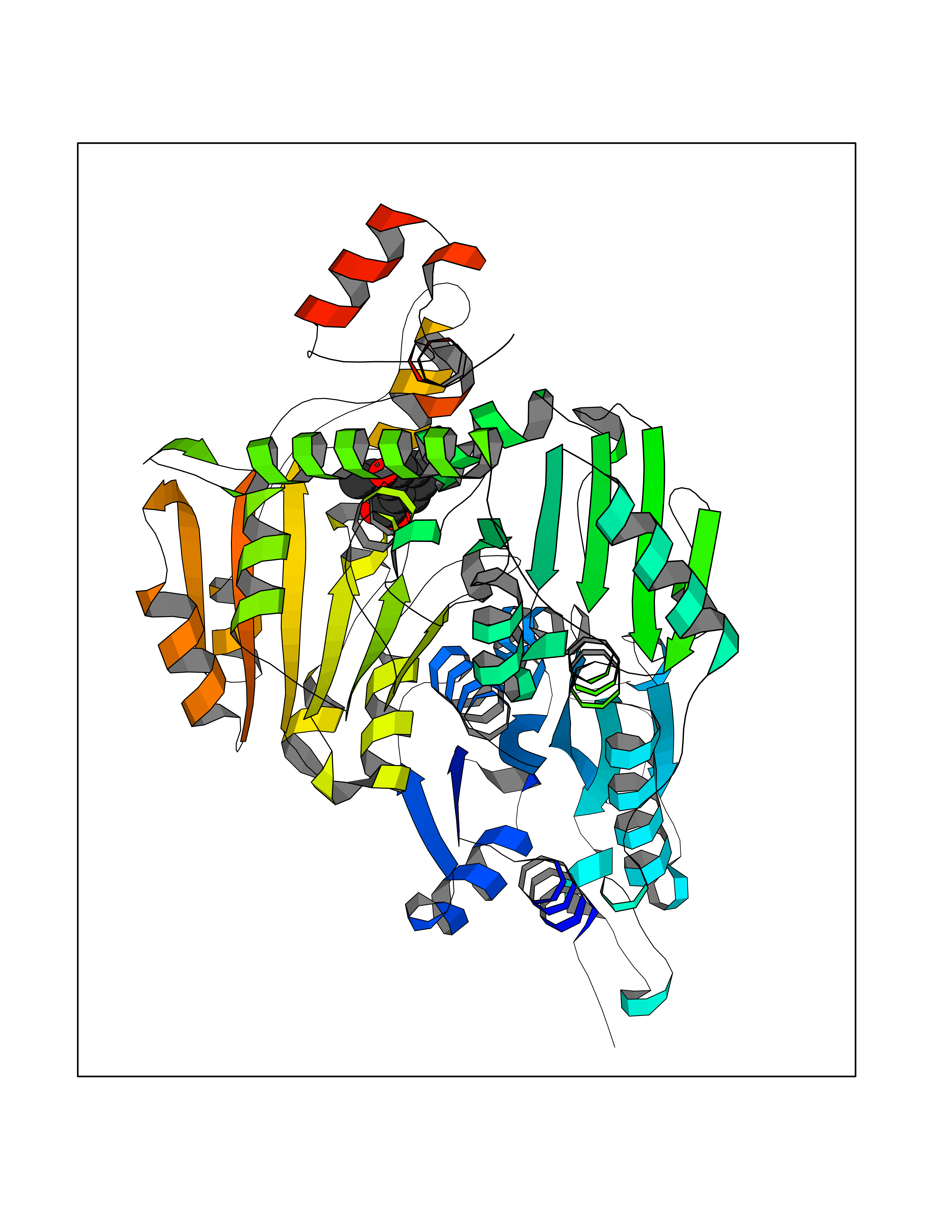 Molscript Map 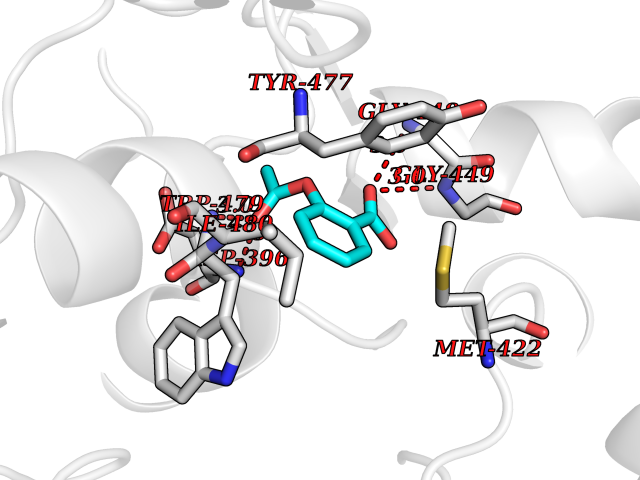 Pymol Map 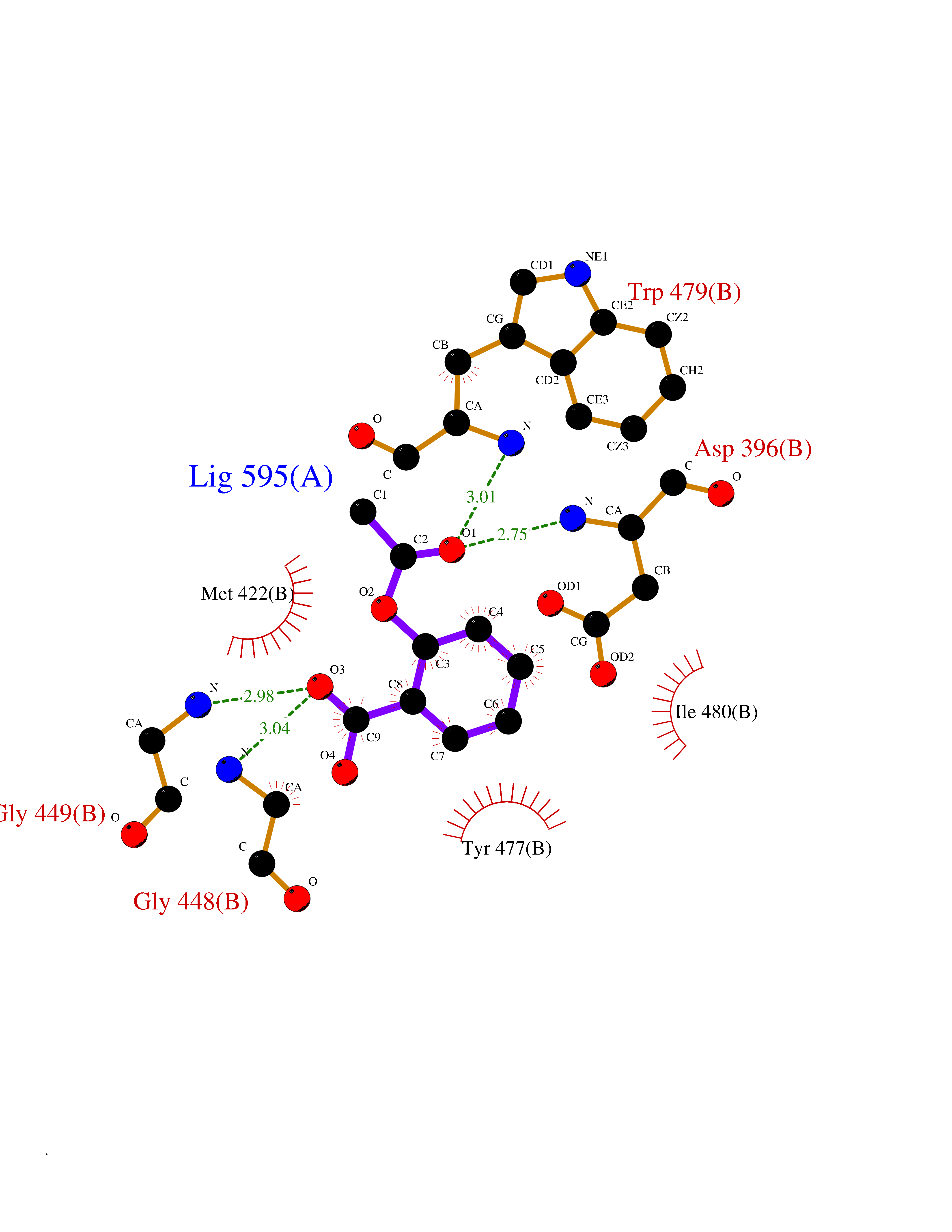 Ligplot Map 3D Binding mode Sequence TNILAGAAVIKVLEAWGVDHLYGIPGGSINSIMDALSAERDRIHYIQVRHEEVGAMAAAADAKLTGKIGVCFGSAGPGGTHLMNGLYDAREDHVPVLALIGQFGTTGMNMDTFQEMNENPIYADVADYNVTAVNAATLPHVIDEAIRRAYAHQGVAVVQIPVDLPWQQIPAEDWYASANSYQTPLLPEPDVQAVTRLTQTLLAAERPLIYYGIGARKAGKELEQLSKTLKIPLMSTYPAKGIVADRYPAYLGSANRVAQKPANEALAQADVVLFVGNNYPFAEVSKAFKNTRYFLQIDIDPAKLGKRHKTDIAVLADAQKTLAAILAQVSERESTPWWQANLANVKNWRAYLASLEDKQEGPLQAYQVLRAVNKIAEPDAIYSIDVGDINLNANRHLKLTPSNRHITSNLFATMGVGIPGAIAAKLNYPERQVFNLAGDGGASMTMQDLATQVQYHLPVINVVFTNCQYGWIKDEQEDTNQNDFIGVEFNDIDFSKIADGVHMQAFRVNKIEQLPDVFEQAKAIAQHEPVLIDAVITGDRPLPAEKLRLDSAMSSAADIEAFKQRYEAQDLQPLSTYLKQFGLDD Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Monoglyceride lipase (MAGL) | 3PE6 | 5.60 | |
Target general information Gen name MGLL Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Monoacylglycerol lipase; MGL; Lysophospholipaselike; Lysophospholipase-like; Lysophospholipase homolog; HUK5; HU-K5 Protein family AB hydrolase superfamily, Monoacylglycerol lipase family Biochemical class Carboxylic ester hydrolase Function Hydrolyzes the endocannabinoid 2-arachidonoylglycerol, and thereby contributes to the regulation of endocannabinoid signaling, nociperception and perception of pain. Regulates the levels of fatty acids that serve as signaling molecules and promote cancer cell migration, invasion and tumor growth. Converts monoacylglycerides to free fatty acids and glycerol. Related diseases Systemic lupus erythematosus 9 (SLEB9) [MIM:610927]: A chronic, relapsing, inflammatory, and often febrile multisystemic disorder of connective tissue, characterized principally by involvement of the skin, joints, kidneys and serosal membranes. It is of unknown etiology, but is thought to represent a failure of the regulatory mechanisms of the autoimmune system. The disease is marked by a wide range of system dysfunctions, an elevated erythrocyte sedimentation rate, and the formation of LE cells in the blood or bone marrow. {ECO:0000269|PubMed:17360460}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Immunodeficiency, common variable, 7 (CVID7) [MIM:614699]: A primary immunodeficiency characterized by antibody deficiency, hypogammaglobulinemia, recurrent bacterial infections and an inability to mount an antibody response to antigen. The defect results from a failure of B-cell differentiation and impaired secretion of immunoglobulins; the numbers of circulating B-cells is usually in the normal range, but can be low. {ECO:0000269|PubMed:22035880}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07550; P37235 EC number EC 3.1.1.23 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Lipid biosynthesis; Lipid degradation; Lipid metabolism; Membrane; Nitration; Phosphoprotein; Proteomics identification; Reference proteome; Serine esterase Protein physicochemical properties Chain ID A Molecular weight (Da) 31808.4 Length 289 Aromaticity 0.08 Instability index 29.7 Isoelectric point 6.73 Charge (pH=7) -0.91 2D Binding mode Binding energy (Kcal/mol) -7.63 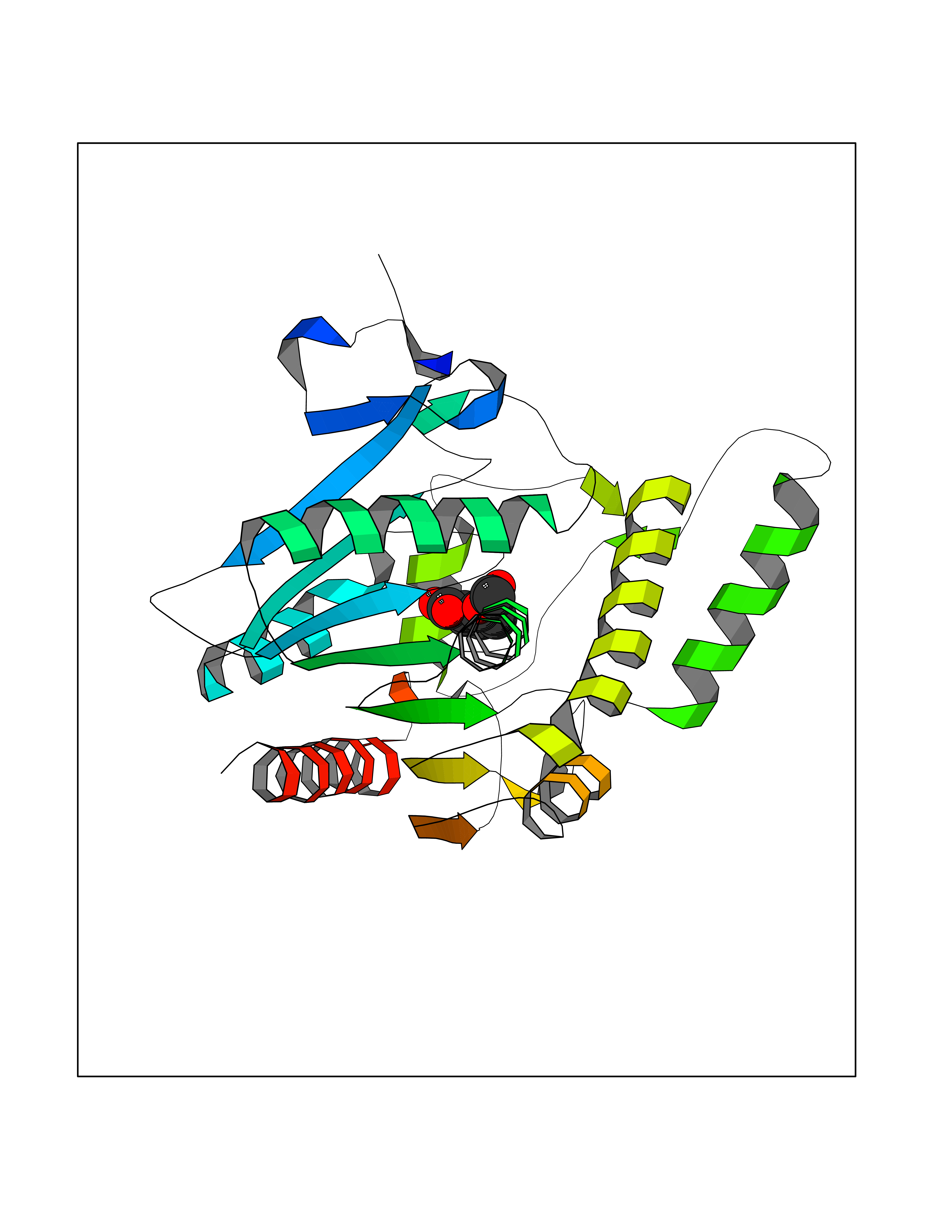 Molscript Map 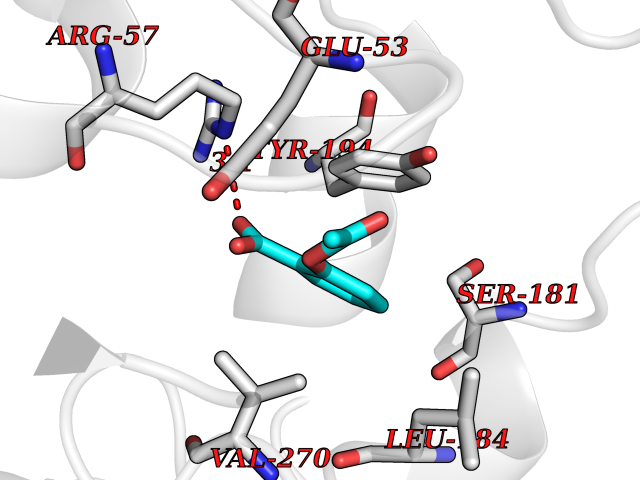 Pymol Map 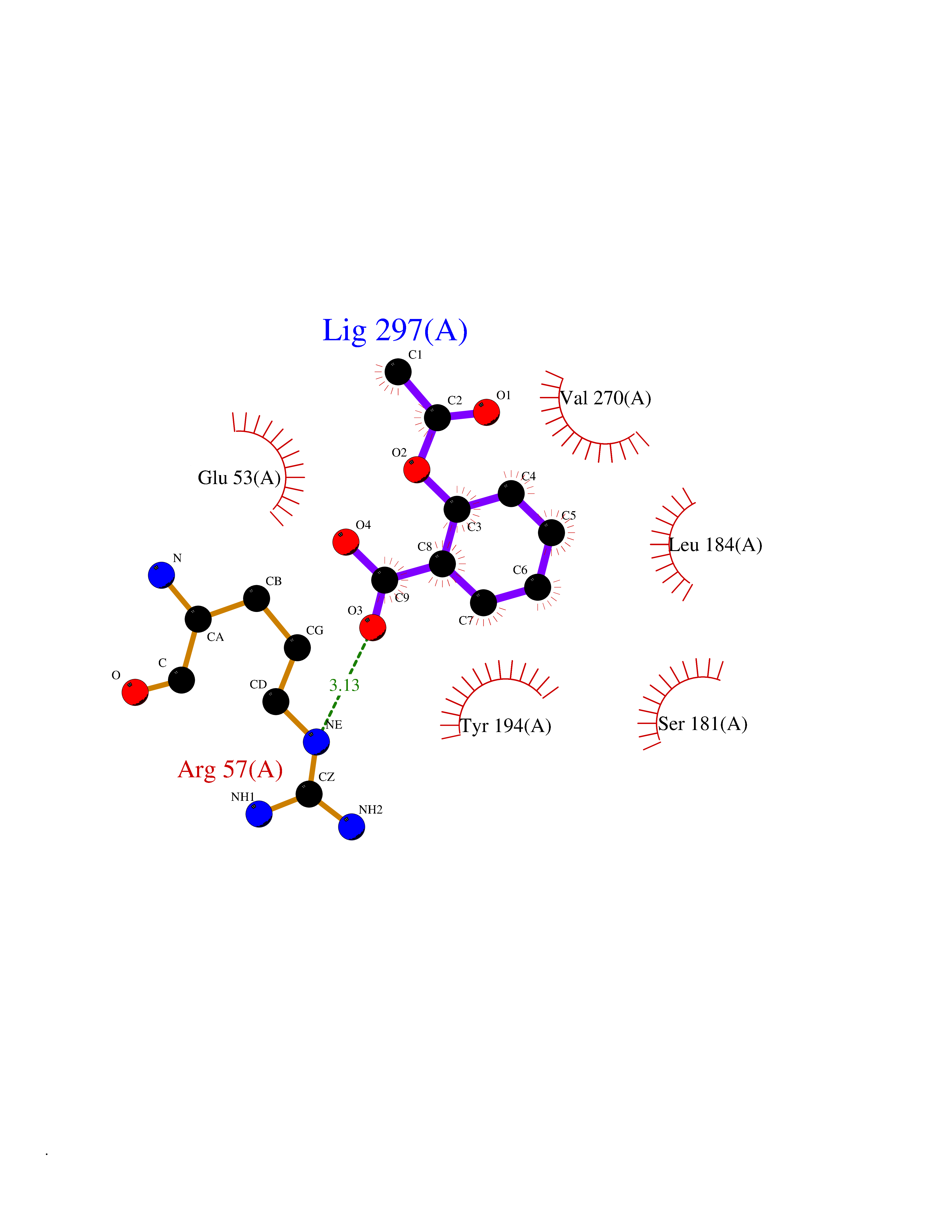 Ligplot Map 3D Binding mode Sequence PRRTPQSIPYQDLPHLVNADGQYLFCRYWAPTGTPKALIFVSHGAGEHSGRYEELARMLMGLDLLVFAHDHVGHGQSEGERMVVSDFHVFVRDVLQHVDSMQKDYPGLPVFLLGHSMGGAIAILTAAERPGHFAGMVLISPLVLANPESATTFKVLAAKVLNSVLPNLSSGPIDSSVLSRNKTEVDIYNSDPLICRAGLKVCFGIQLLNAVSRVERALPKLTVPFLLLQGSADRLCDSKGAYLLMELAKSQDKTLKIYEGAYHVLHKELPEVTNSVFHEINMWVSQRTA Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | IL-1 receptor-associated kinase 1 (IRAK1) | 6BFN | 5.60 | |
Target general information Gen name IRAK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Interleukin-1 receptor-associated kinase 1; IRAK-1; IRAK Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family, Pelle subfamily Biochemical class Kinase Function Involved in Toll-like receptor (TLR) and IL-1R signaling pathways. Is rapidly recruited by MYD88 to the receptor-signaling complex upon TLR activation. Association with MYD88 leads to IRAK1 phosphorylation by IRAK4 and subsequent autophosphorylation and kinase activation. Phosphorylates E3 ubiquitin ligases Pellino proteins (PELI1, PELI2 and PELI3) to promote pellino-mediated polyubiquitination of IRAK1. Then, the ubiquitin-binding domain of IKBKG/NEMO binds to polyubiquitinated IRAK1 bringing together the IRAK1-MAP3K7/TAK1-TRAF6 complex and the NEMO-IKKA-IKKB complex. In turn, MAP3K7/TAK1 activates IKKs (CHUK/IKKA and IKBKB/IKKB) leading to NF-kappa-B nuclear translocation and activation. Alternatively, phosphorylates TIRAP to promote its ubiquitination and subsequent degradation. Phosphorylates the interferon regulatory factor 7 (IRF7) to induce its activation and translocation to the nucleus, resulting in transcriptional activation of type I IFN genes, which drive the cell in an antiviral state. When sumoylated, translocates to the nucleus and phosphorylates STAT3. Serine/threonine-protein kinase that plays a critical role in initiating innate immune response against foreign pathogens. Related diseases Anemia, non-spherocytic hemolytic, due to G6PD deficiency (NSHA) [MIM:300908]: A disease characterized by G6PD deficiency, acute hemolytic anemia, fatigue, back pain, and jaundice. In most patients, the disease is triggered by an exogenous agent, such as some drugs, food, or infection. Increased unconjugated bilirubin, lactate dehydrogenase, and reticulocytosis are markers of the disorder. Although G6PD deficiency can be life-threatening, most patients are asymptomatic throughout their life. {ECO:0000269|PubMed:12524354, ECO:0000269|PubMed:1303180, ECO:0000269|PubMed:1303182, ECO:0000269|PubMed:1536798, ECO:0000269|PubMed:1611091, ECO:0000269|PubMed:1889820, ECO:0000269|PubMed:1945893, ECO:0000269|PubMed:20007901, ECO:0000269|PubMed:26479991, ECO:0000269|PubMed:2836867, ECO:0000269|PubMed:2912069, ECO:0000269|PubMed:30988594, ECO:0000269|PubMed:38066190, ECO:0000269|PubMed:7858267, ECO:0000269|PubMed:7959695, ECO:0000269|PubMed:8193373, ECO:0000269|PubMed:8490627, ECO:0000269|PubMed:8533762, ECO:0000269|PubMed:8733135, ECO:0000269|PubMed:9452072}. The disease is caused by variants affecting the gene represented in this entry. Deficiency of G6PD is associated with hemolytic anemia in two different situations. First, in areas in which malaria has been endemic, G6PD-deficiency alleles have reached high frequencies (1% to 50%) and deficient individuals, though essentially asymptomatic in the steady state, have a high risk of acute hemolytic attacks. Secondly, sporadic cases of G6PD deficiency occur at a very low frequencies, and they usually present a more severe phenotype. Several types of NSHA are recognized. Class-I variants are associated with severe NSHA; class-II have an activity <10% of normal; class-III have an activity of 10% to 60% of normal; class-IV have near normal activity. Drugs (DrugBank ID) DB12010 Interacts with Q15306; Q92985; Q99836; Q96FA3; Q9HAT8; Q8N2H9-2; Q13526; Q86WV6; P58753; Q9Y4K3; Q8VCW4; Q5D1E7 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cytoplasm; Direct protein sequencing; Host-virus interaction; Immunity; Innate immunity; Isopeptide bond; Kinase; Lipid droplet; Magnesium; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 33681.4 Length 301 Aromaticity 0.09 Instability index 39.86 Isoelectric point 8.6 Charge (pH=7) 5.09 2D Binding mode Binding energy (Kcal/mol) -7.64 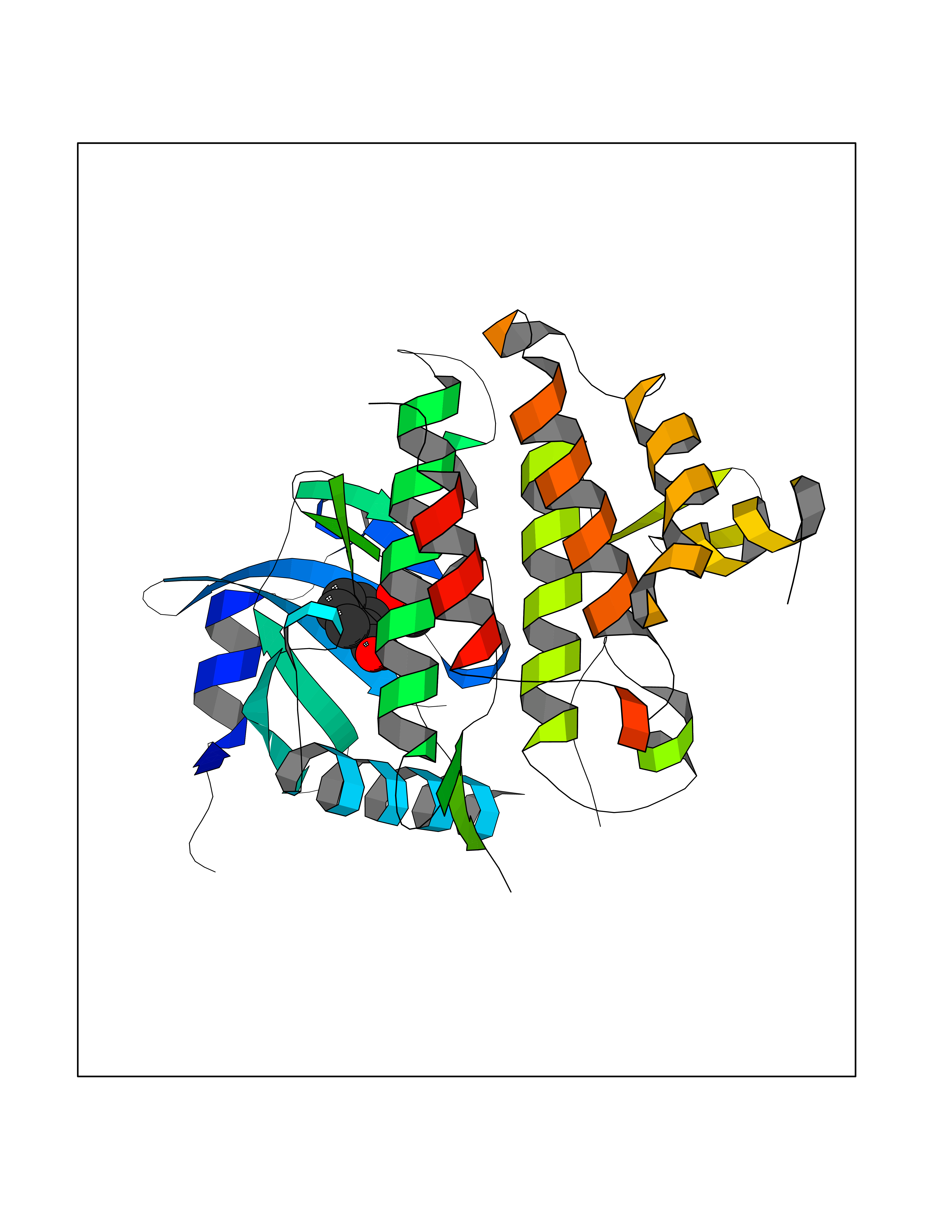 Molscript Map 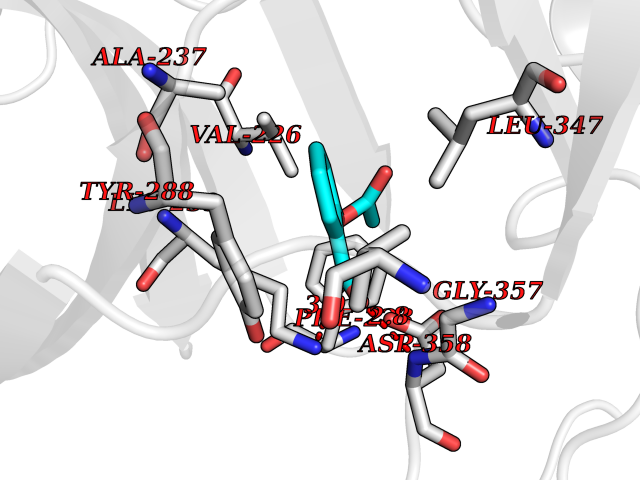 Pymol Map 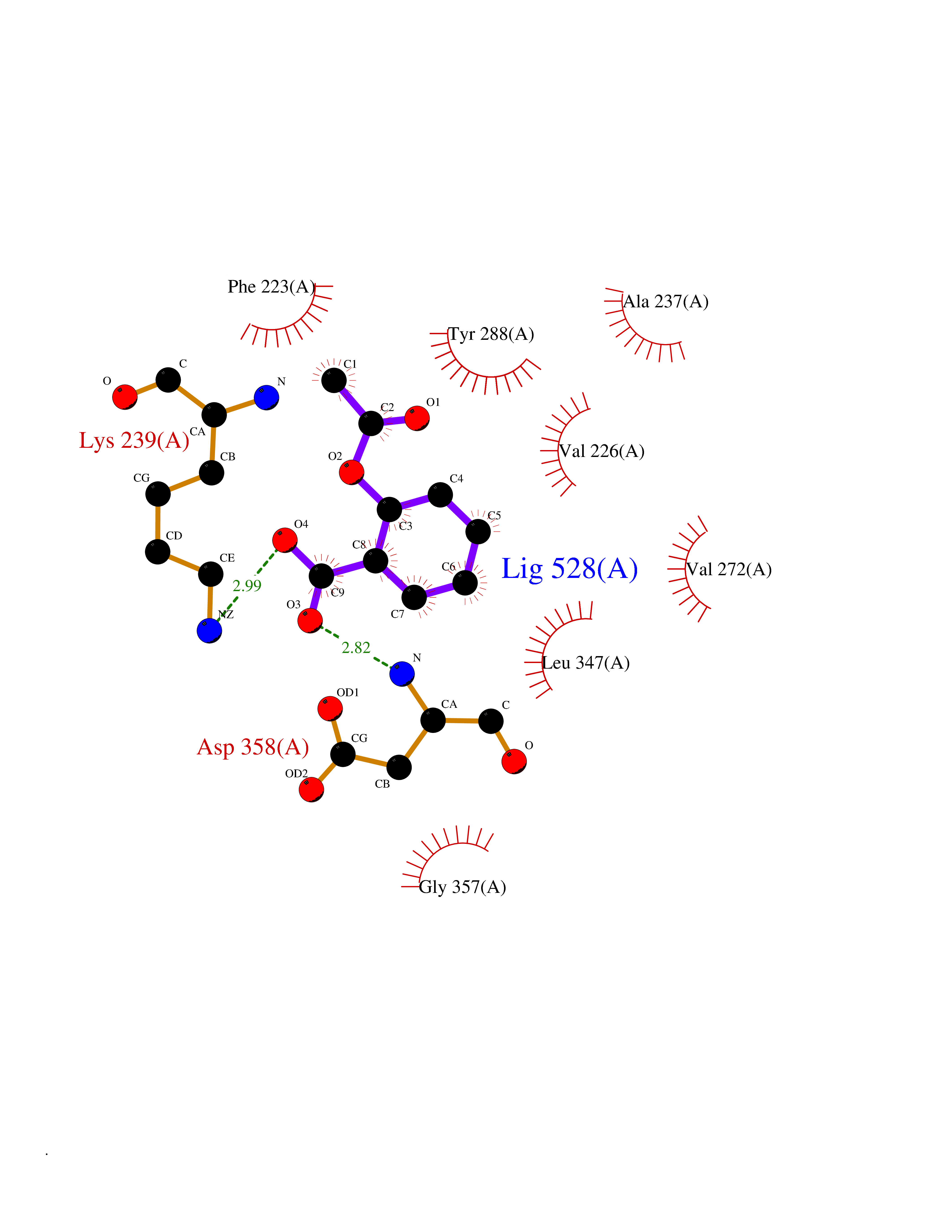 Ligplot Map 3D Binding mode Sequence SRPFPFCWPLCEISRGTHNFSEELKIGEGGFGCVYRAVMRNTVYAVKRLKEWTAVKQSFLTEVEQLSRFRHPNIVDFAGYCAQNGFYCLVYGFLPNGSLEDRLHCQTQACPPLSWPQRLDILLGTARAIQFLHQDSPSLIHGDIKSSNVLLDERLTPKLGDFGLARFSRTVRGTLAYLPEEYIKTGRLAVDTDTFSFGVVVLETLAGQRAVKTHGARTKYLKDLVEEEAEEAGVAAADAWAAPIAMQIYKKHLDPRPGPCPPELGLGLGQLACCCLHRRAKRRPPMTQVYERLEKLQAVVA Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Wnt-7a protein (WNT7A) | 4UZQ | 5.60 | |
Target general information Gen name WNT7A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein Wnt-7a Protein family Wnt family Biochemical class NA Function Plays an important role in embryonic development, including dorsal versus ventral patterning during limb development, skeleton development and urogenital tract development. Required for central nervous system (CNS) angiogenesis and blood-brain barrier regulation. Required for normal, sexually dimorphic development of the Mullerian ducts, and for normal fertility in both sexes. Required for normal neural stem cell proliferation in the hippocampus dentate gyrus. Required for normal progress through the cell cycle in neural progenitor cells, for self-renewal of neural stem cells, and for normal neuronal differentiation and maturation. Promotes formation of synapses via its interaction with FZD5. Ligand for members of the frizzled family of seven transmembrane receptors that functions in the canonical Wnt/beta-catenin signaling pathway. Related diseases Limb pelvis hypoplasia aplasia syndrome (LPHAS) [MIM:276820]: A syndrome of severe deficiency of the extremities due to hypo- or aplasia of one or more long bones of one or more limbs. Pelvic manifestations include hip dislocation, hypoplastic iliac bone and aplastic pubic bones. Thoracic deformity, unusual facies and genitourinary anomalies can be present. {ECO:0000269|PubMed:16826533, ECO:0000269|PubMed:17431918, ECO:0000269|PubMed:20949531, ECO:0000269|PubMed:21271649, ECO:0000269|PubMed:27638328}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fuhrmann syndrome (FUHRS) [MIM:228930]: Distinct limb-malformation disorder characterized also by various degrees of limb aplasia/hypoplasia and joint dysplasia. {ECO:0000269|PubMed:16826533}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P55212; P22607; P06396; P13473-2; Q9UMX0; Q9Y5W5; Q5T9L3; Q9Z0J1 EC number NA Uniprot keywords 3D-structure; Developmental protein; Disease variant; Disulfide bond; Extracellular matrix; Glycoprotein; Lipoprotein; Proteomics identification; Reference proteome; Secreted; Signal; Wnt signaling pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 40475.5 Length 356 Aromaticity 0.1 Instability index 50.49 Isoelectric point 7.67 Charge (pH=7) 1.62 2D Binding mode Binding energy (Kcal/mol) -7.64 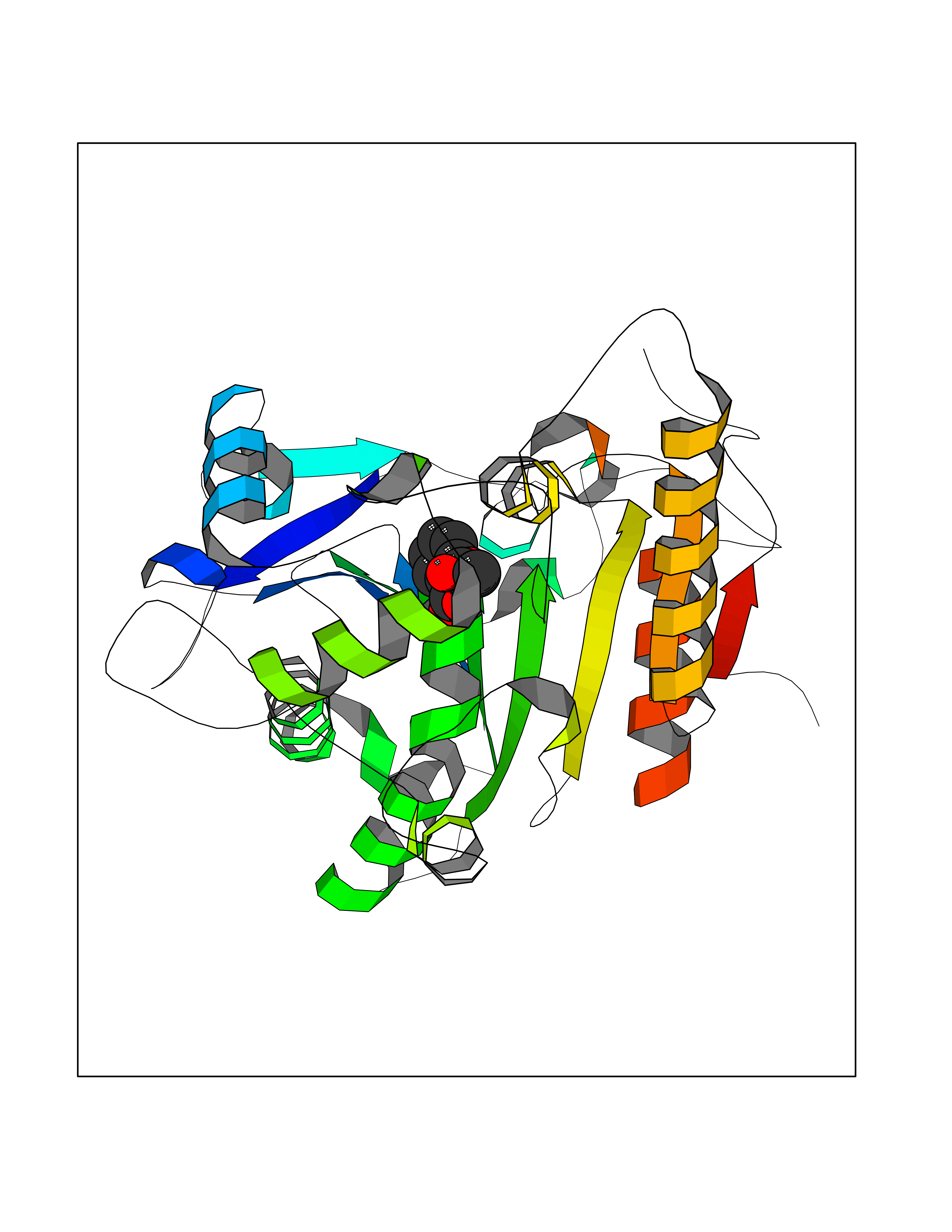 Molscript Map 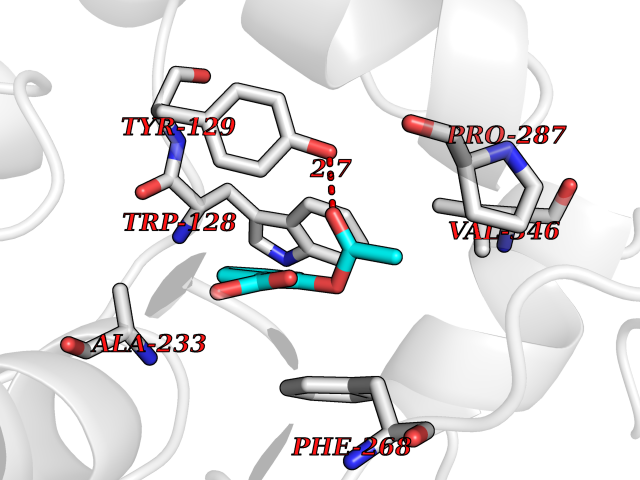 Pymol Map 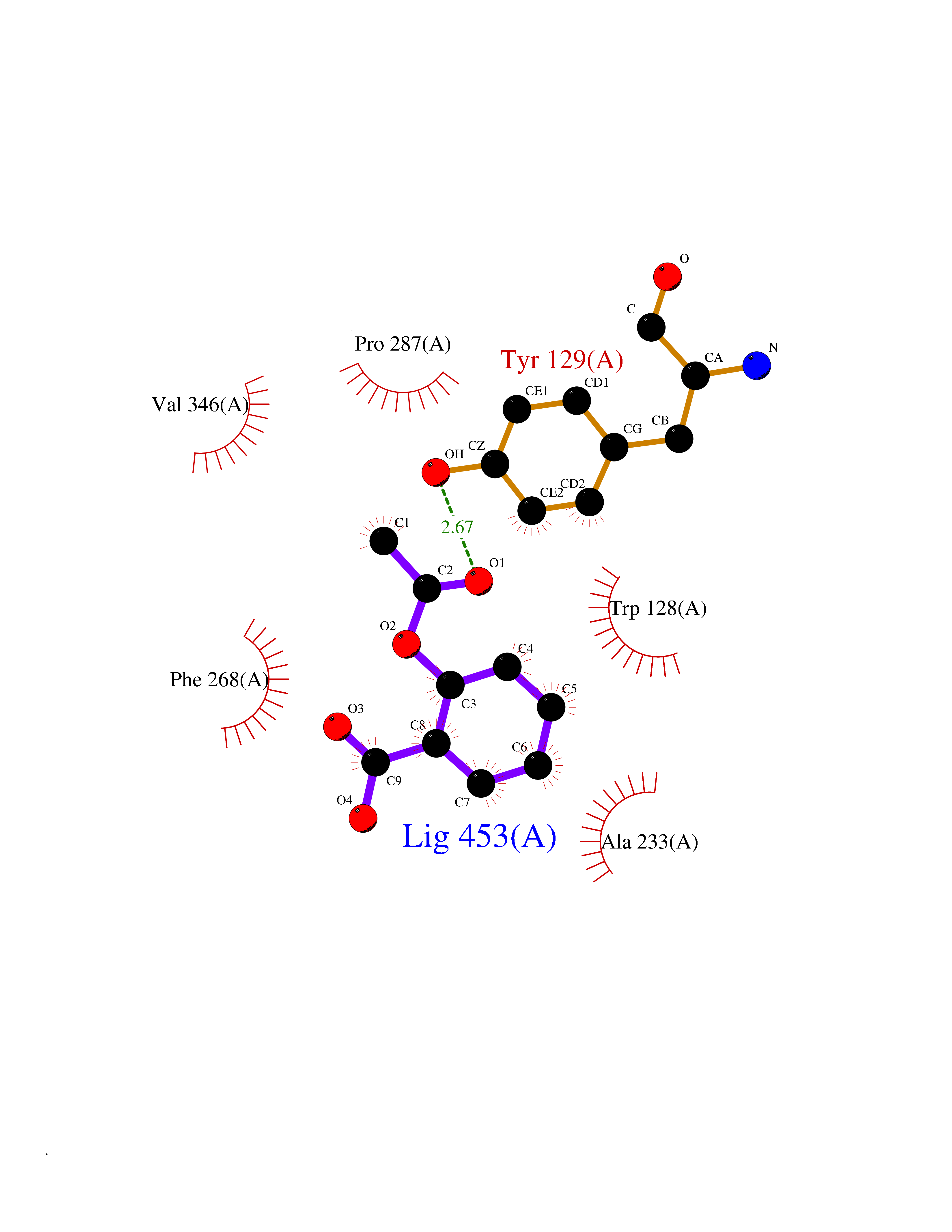 Ligplot Map 3D Binding mode Sequence EDLRLHLLLNTSVTCNDGSPAGYYLKESRGSRRWLLFLEGGWYCFNRENCDSRYDTMRRLMSSRDWPRTRTGTGILSSQPEENPYWWNANMVFIPYCSSDVWSGASSKSEKNEYAFMGALIIQEVVRELLGRGLSGAKVLLLAGSAAGGTGVLLNVDRVAEQLEKLGYPAIQVRGLADSGWFLDNKQYRHTDCVDTITCAPTEAIRRGIRYWNGVVPERCRRQFQEGEEWNCFFGYKVYPTLRSPVFVVQWLFDEAQLTVDNVHLTGQPVQEGLRLYIQNLGRELRHTLKDVPASFAPACLSHEIIIRSHWTDVQVKGTSLPRALHCWDRSLHKGCPVHLVDSCPWPHCNPSCPTS Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Phosphatidylethanolamine-binding protein 1 (PEBP1) | 2QYQ | 5.60 | |
Target general information Gen name PEBP1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Raf kinase inhibitor protein; RKIP; Prostatic-binding protein; PEBP-1; PEBP; PBP; Neuropolypeptide h3; Hippocampal cholinergic neurostimulating peptide; HCNPpp; HCNP Protein family Phosphatidylethanolamine-binding protein family Biochemical class Phosphatidylethanolamine-binding protein family Function Binds ATP, opioids and phosphatidylethanolamine. Has lower affinity for phosphatidylinositol and phosphatidylcholine. Serine protease inhibitor which inhibits thrombin, neuropsin and chymotrypsin but not trypsin, tissue type plasminogen activator and elastase. Inhibits the kinase activity of RAF1 by inhibiting its activation and by dissociating the RAF1/MEK complex and acting as a competitive inhibitor of MEK phosphorylation. Related diseases Retinitis pigmentosa 49 (RP49) [MIM:613756]: A retinal dystrophy belonging to the group of pigmentary retinopathies. Retinitis pigmentosa is characterized by retinal pigment deposits visible on fundus examination and primary loss of rod photoreceptor cells followed by secondary loss of cone photoreceptors. Patients typically have night vision blindness and loss of midperipheral visual field. As their condition progresses, they lose their far peripheral visual field and eventually central vision as well. {ECO:0000269|PubMed:15570217, ECO:0000269|PubMed:7479749}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09130; DB09568 Interacts with P16050; Q9NRD5; P04049; Q15208; Q9NS68; Q9JLL3 EC number NA Uniprot keywords 3D-structure; ATP-binding; Cytoplasm; Direct protein sequencing; Disulfide bond; Lipid-binding; Nucleotide-binding; Phosphoprotein; Protease inhibitor; Proteomics identification; Reference proteome; Serine protease inhibitor Protein physicochemical properties Chain ID A Molecular weight (Da) 20928.3 Length 186 Aromaticity 0.1 Instability index 24.05 Isoelectric point 6.59 Charge (pH=7) -0.98 2D Binding mode Binding energy (Kcal/mol) -7.64 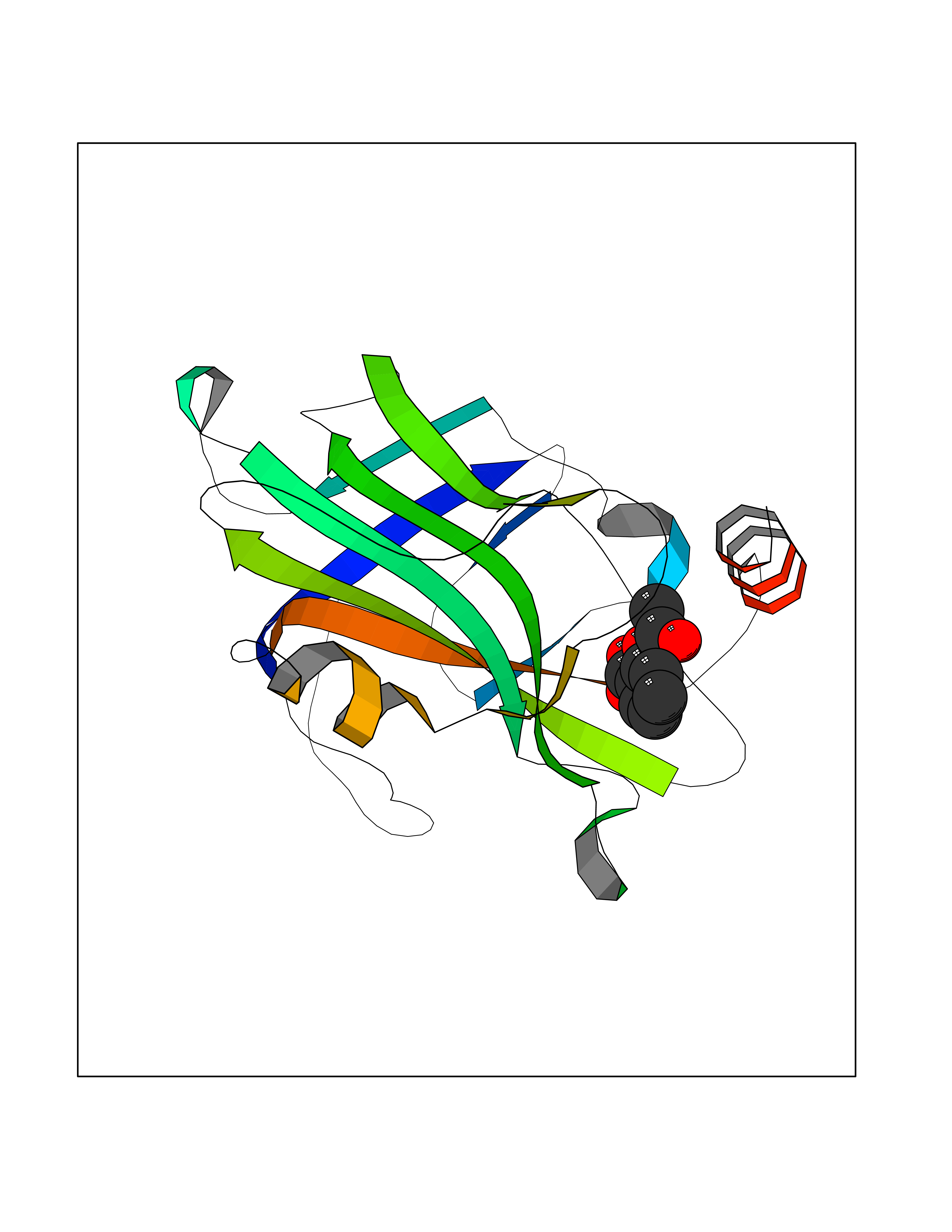 Molscript Map 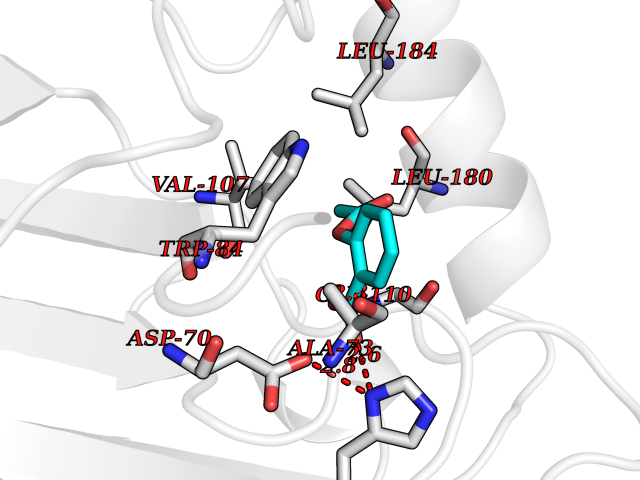 Pymol Map 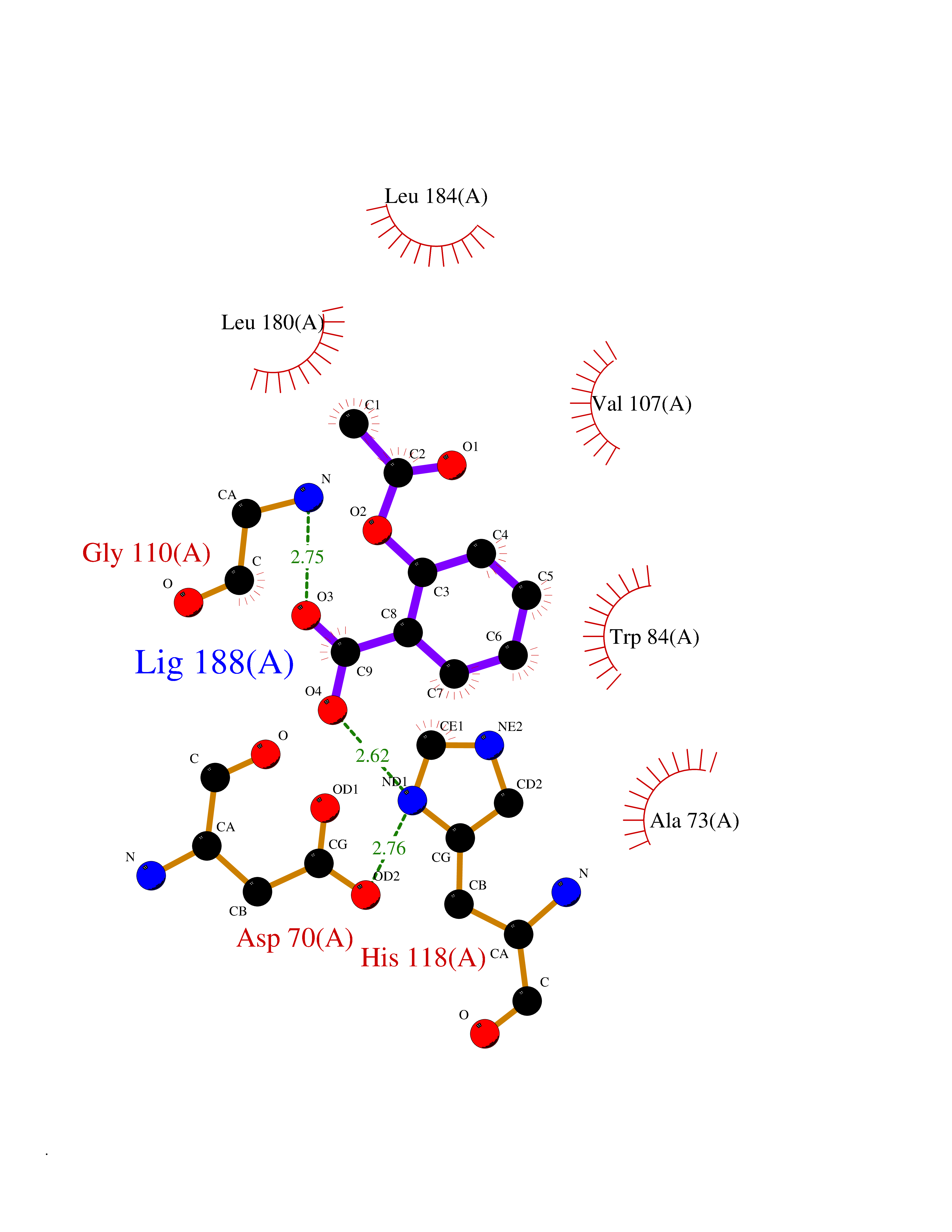 Ligplot Map 3D Binding mode Sequence MPVDLSKWSGPLSLQEVDEQPQHPLHVTYAGAAVDELGKVLTPTQVKNRPTSISWDGLDSGKLYTLVLTDPDAPSRKDPKYREWHHFLVVNMKGNDISSGTVLSDYVGSGPPKGTGLHRYVWLVYEQDRPLKCDEPILSNRSGDHRGKFKVASFRKKYELRAPVAGTCYQAEWDDYVPKLYEQLSG Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Fatty acid-binding protein, intestinal | 3AKM | 5.59 | |
Target general information Gen name FABP2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms FABPI Protein family Calycin superfamily, Fatty-acid binding protein (FABP) family Biochemical class Transport protein Function Fatty acid binding.Transporter activity. Related diseases Usher syndrome 3B (USH3B) [MIM:614504]: A syndrome characterized by progressive vision and hearing loss during early childhood. Some patients have the so-called 'Charles Bonnet syndrome,' involving decreased visual acuity and vivid visual hallucinations. USH is a genetically heterogeneous condition characterized by the association of retinitis pigmentosa with sensorineural deafness. Age at onset and differences in auditory and vestibular function distinguish Usher syndrome type 1 (USH1), Usher syndrome type 2 (USH2) and Usher syndrome type 3 (USH3). USH3 is characterized by postlingual, progressive hearing loss, variable vestibular dysfunction, and onset of retinitis pigmentosa symptoms, including nyctalopia, constriction of the visual fields, and loss of central visual acuity, usually by the second decade of life. {ECO:0000269|PubMed:22279524}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, axonal, 2W (CMT2W) [MIM:616625]: An autosomal dominant, axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. CMT2W patients manifest a peripheral neuropathy mainly affecting the lower limbs and resulting in gait difficulties and distal sensory impairment. Most patients also have upper limb involvement. {ECO:0000269|PubMed:22930593, ECO:0000269|PubMed:26072516, ECO:0000269|PubMed:29235198}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04557; DB09213; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB01050; DB08231; DB03796; DB01138 Interacts with O95994; Q9NYB0 EC number NA Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Lipid-binding; Proteomics identification; Reference proteome; Transport Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 15075.9 Length 131 Aromaticity 0.11 Instability index 32.01 Isoelectric point 6.88 Charge (pH=7) -0.09 2D Binding mode Binding energy (Kcal/mol) -7.62  Molscript Map 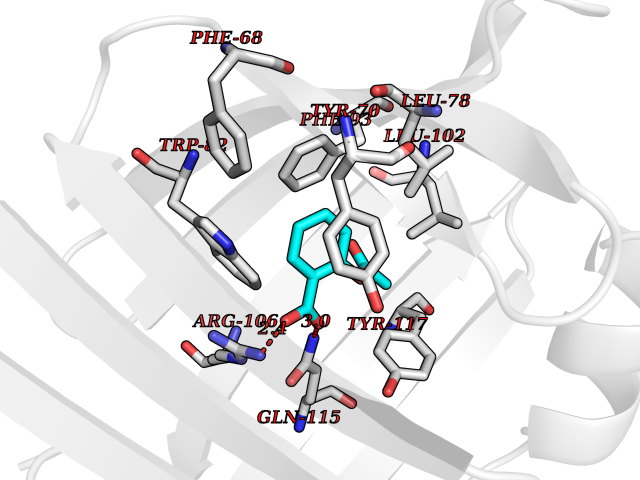 Pymol Map 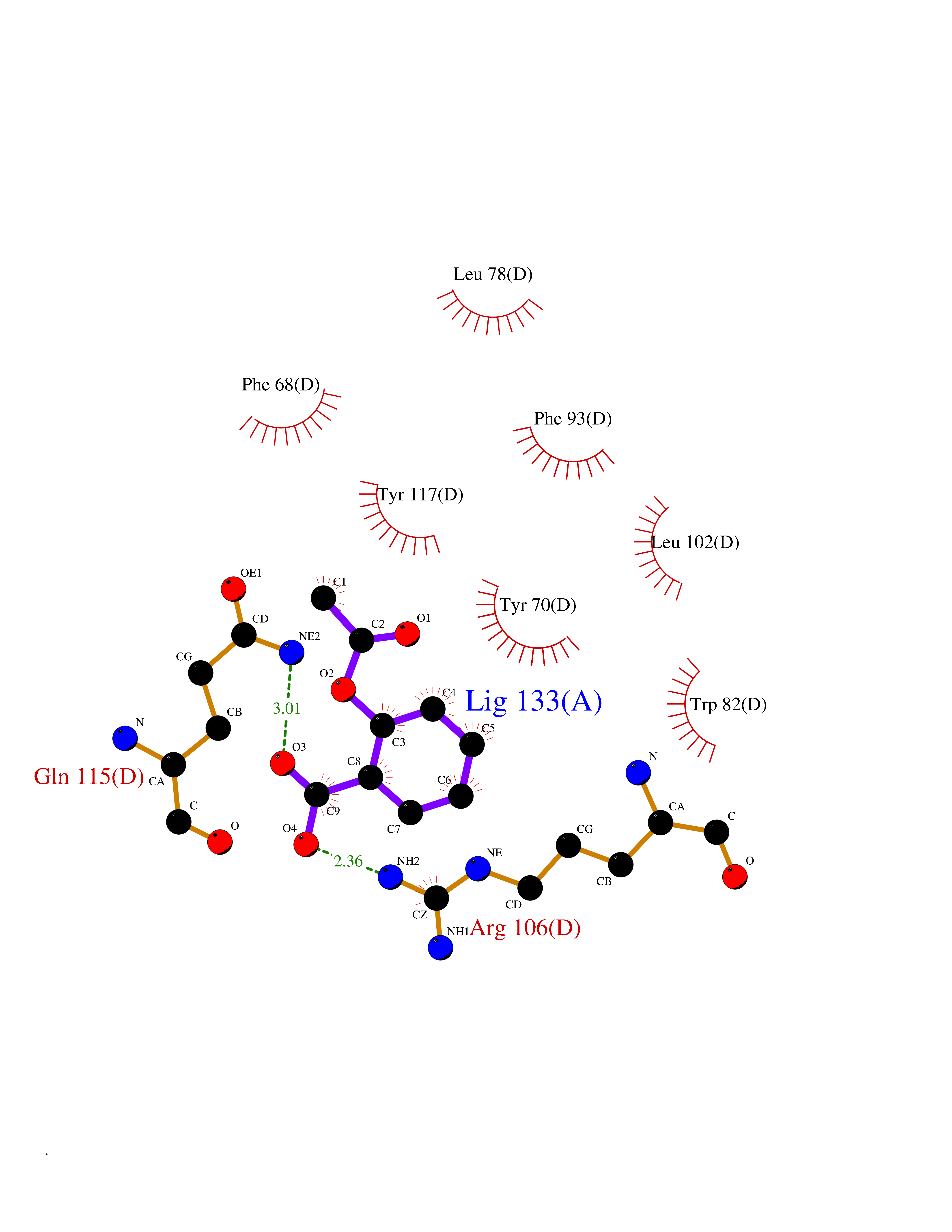 Ligplot Map 3D Binding mode Sequence AFDSTWKVDRSENYDKFMEKMGVNIVKRKLAAHDNLKLTITQEGNKFTVKESSAFRNIEVVFELGVTFNYNLADGTELRGTWSLEGNKLIGKFKRTDNGNELNTVREIIGDELVQTYVYEGVEAKRIFKKD Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Acetyl-CoA carboxylase (ACC) (EC 6.4.1.2) (Fatty acid synthetase 3) (mRNA transport-defective protein 7) [Includes: Biotin carboxylase (EC 6.3.4.14)] | 1UYS | 5.59 | |
Target general information Gen name ACC1 Organism Saccharomyces cerevisiae (strain ATCC 204508 / S288c) (Baker's yeast) Uniprot ID TTD ID NA Synonyms MTR7;YNR016C;N3175;ABP2;FAS3 Protein family NA Biochemical class NA Function Carries out three functions: biotin carboxyl carrier protein, biotin carboxylase and carboxyltransferase. Involved in the synthesis of very-long-chain fatty acid synthesis which is required to maintain a functional nuclear envelope. Required for acylation and vacuolar membrane association of VAC8 which is necessary to maintain a normal morphology of the vacuole. {ECO:0000269|PubMed:10757783, ECO:0000269|PubMed:12730220, ECO:0000269|PubMed:6103540, ECO:0000269|PubMed:6108218, ECO:0000269|PubMed:8943372}." Related diseases A chromosomal aberration involving NFKB2 is found in a case of B-cell non Hodgkin lymphoma (B-NHL). Translocation t(10;14)(q24;q32) with IGHA1. The resulting oncogene is also called Lyt-10C alpha variant.; DISEASE: A chromosomal aberration involving NFKB2 is found in a cutaneous T-cell leukemia (C-TCL) cell line. This rearrangement produces the p80HT gene which codes for a truncated 80 kDa protein (p80HT).; DISEASE: In B-cell leukemia (B-CLL) cell line, LB40 and EB308, can be found after heterogeneous chromosomal aberrations, such as internal deletions.; DISEASE: Immunodeficiency, common variable, 10 (CVID10) [MIM:615577]: A primary immunodeficiency characterized by childhood-onset of recurrent infections, hypogammaglobulinemia, and decreased numbers of memory and marginal zone B-cells. Some patients may develop autoimmune features and have circulating autoantibodies. An unusual feature is central adrenal insufficiency. {ECO:0000269|PubMed:24140114, ECO:0000269|PubMed:25524009}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q00955 EC number 6.3.4.14; 6.4.1.2 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Biotin; Cytoplasm; Direct protein sequencing; Endoplasmic reticulum; Fatty acid biosynthesis; Fatty acid metabolism; Ligase; Lipid biosynthesis; Lipid metabolism; Manganese; Membrane; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Reference proteome Protein physicochemical properties Chain ID B,C Molecular weight (Da) 145619 Length 1328 Aromaticity 0.1 Instability index 30.31 Isoelectric point 5.32 Charge (pH=7) -26.79 2D Binding mode Binding energy (Kcal/mol) -7.63  Molscript Map  Pymol Map 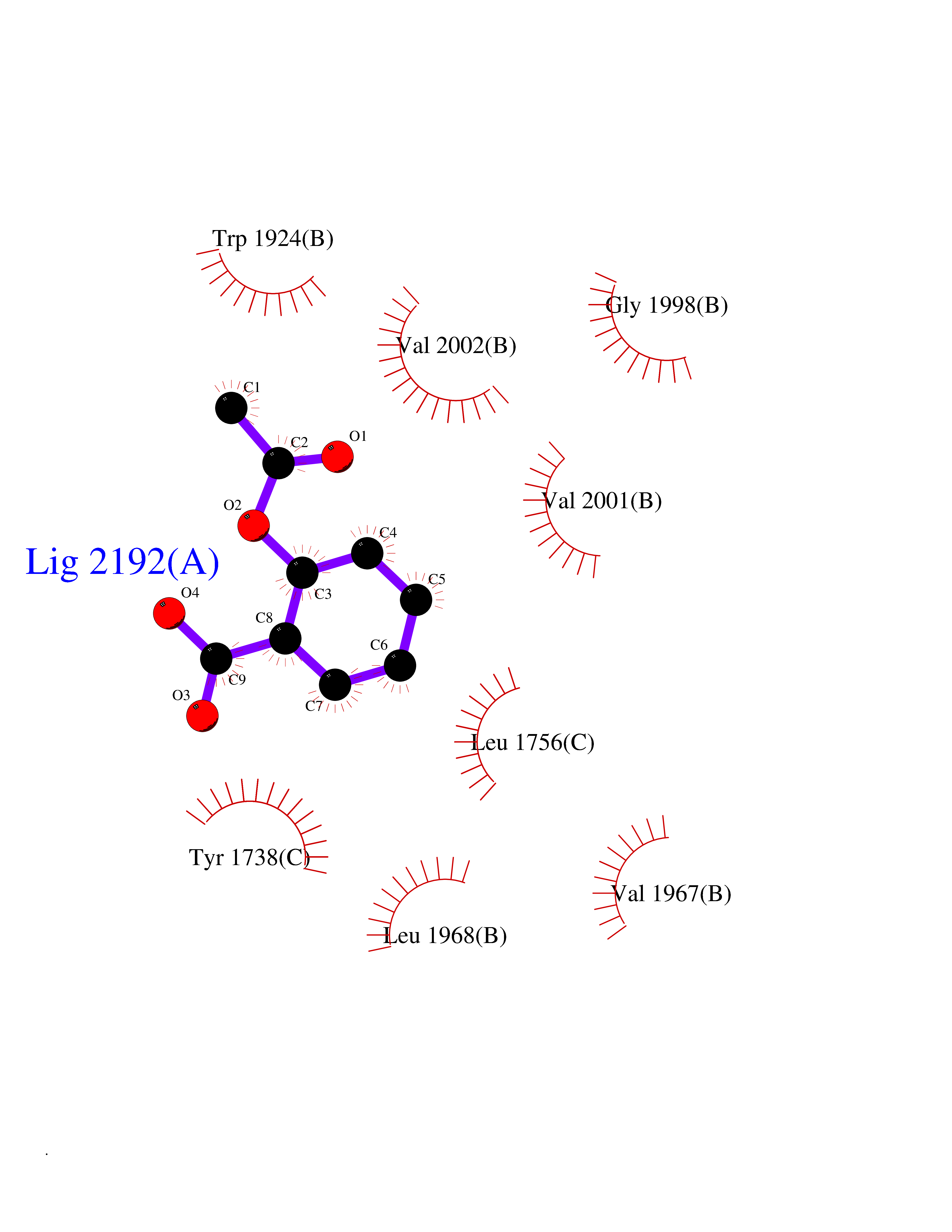 Ligplot Map 3D Binding mode Sequence WLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGLPIATPYPVKEWLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGL Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Isobutyryl-CoA dehydrogenase, mitochondrial | 1RX0 | 5.59 | |
Target general information Gen name ACAD8 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms IBD;ARC42 Protein family Acyl-CoA dehydrogenase family Biochemical class Oxidoreductase Function Acyl-CoA dehydrogenase activity.Flavin adenine dinucleotide binding. Related diseases Isobutyryl-CoA dehydrogenase deficiency (IBDD) [MIM:611283]: An autosomal recessive metabolic disorder characterized by plasma carnitine deficiency and elevated C4-acylcarnitine. Patients manifest variable clinical features including failure to thrive, seizures, anemia, muscular hypotonia and developmental delay. Some patients may be asymptomatic. {ECO:0000269|PubMed:12359132, ECO:0000269|PubMed:15505379, ECO:0000269|PubMed:16857760}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147; DB01675 Interacts with Q8TAG5 EC number 1.3.8.5 Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; Branched-chain amino acid catabolism; Direct protein sequencing; Disease variant; FAD; Flavoprotein; Mitochondrion; Oxidoreductase; Proteomics identification; Reference proteome; Transcription; Transcription regulation; Transit peptide Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 41689.3 Length 384 Aromaticity 0.08 Instability index 40.81 Isoelectric point 6.39 Charge (pH=7) -2.06 2D Binding mode Binding energy (Kcal/mol) -7.63  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence TSCIDPSMGLNEEQKEFQKVAFDFAAREMAPNMAEWDQKELFPVDVMRKAAQLGFGGVYIQTDVGGSGLSRLDTSVIFEALATGCTSTTAYISIHNMCAWMIDSFGNEEQRHKFCPPLCTMEKFASYCLTEPGSGSDAASLLTSAKKQGDHYILNGSKAFISGAGESDIYVVMCRTGGPGPKGISCIVVEKGTPGLSFGKKEKKVGWNSQPTRAVIFEDCAVPVANRIGSEGQGFLIAVRGLNGGRINIASCSLGAAHASVILTRDHLNVRKQFGEPLASNQYLQFTLADMATRLVAARLMVRNAAVALQEERKDAVALCSMAKLFATDECFAICNQALQMHGGYGYLKDYAVQQYVRDSRVHQILEGSNEVMRILISRSLLQE Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Neuronal acetylcholine receptor alpha-2 (CHRNA2) | 5FJV | 5.59 | |
Target general information Gen name CHRNA2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CHRNA2 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-2/CHRNA2 sub-subfamily Biochemical class Neurotransmitter receptor Function After binding acetylcholine, the AChR responds by an extensive change in conformation that affects all subunits and leads to opening of an ion-conducting channel across the plasma membrane. Related diseases Epilepsy, nocturnal frontal lobe, 4 (ENFL4) [MIM:610353]: An autosomal dominant focal epilepsy characterized by nocturnal seizures associated with fear sensation, tongue movements, and nocturnal wandering, closely resembling nightmares and sleep walking. {ECO:0000269|PubMed:16826524}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Seizures, benign familial infantile, 6 (BFIS6) [MIM:610353]: A form of benign familial infantile epilepsy, a neurologic disorder characterized by afebrile seizures occurring in clusters during the first year of life, without neurologic sequelae. BFIS6 inheritance is autosomal dominant. {ECO:0000269|PubMed:25847220}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00732; DB00237; DB00411; DB00565; DB01245; DB00514; DB01135; DB07720; DB00898; DB00472; DB00483; DB08960; DB00657; DB01336; DB00416; DB01226; DB00184; DB01337; DB01338; DB00721; DB00728; DB05740; DB00202; DB01199; DB01339 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 48323.4 Length 413 Aromaticity 0.15 Instability index 32 Isoelectric point 5.69 Charge (pH=7) -6.58 2D Binding mode Binding energy (Kcal/mol) -7.63  Molscript Map 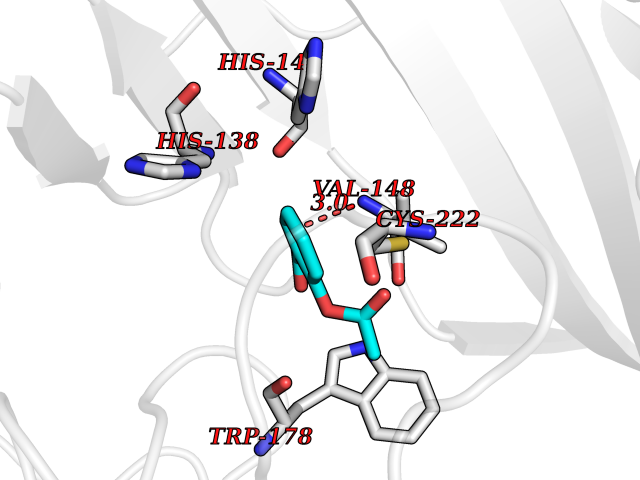 Pymol Map 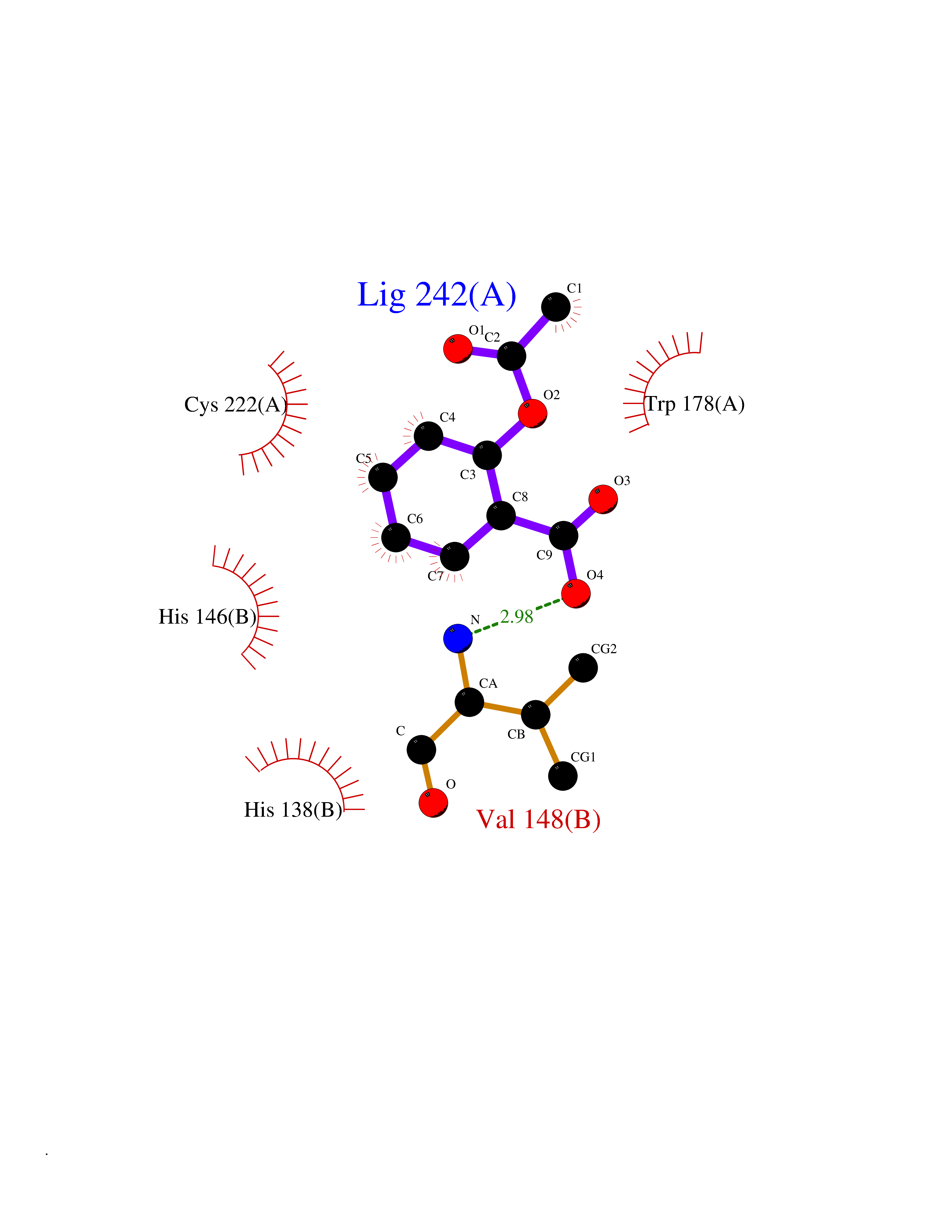 Ligplot Map 3D Binding mode Sequence DRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLPEDRLFKHLFRGYNRWARPVPNTSDVVIVRFGLSIAQLIDVDEKNQMMTTNVWLKQEWSDYKLRWNPTDFGNITSLRVPSEMIWIPDIVLYNNADGEFAVTHMTKAHLFSTGTVHWVPPAIYKSSCSIDVTFFPFDQQNCKMKFGSWTYDKAKIDLEQMEQTVDLKDYWESGEWAIVNATGTYNSKKYDCCAEIYPDVTYAFVIRRLP Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Mutated Histone H3.3 (H3F3A) | 4GUS | 5.59 | |
Target general information Gen name H3F3A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PP781; Histone H3.3; H3F3; H3.3B; H3.3A Protein family Histone H3 family Biochemical class NA Function Variant histone H3 which replaces conventional H3 in a wide range of nucleosomes in active genes. Constitutes the predominant form of histone H3 in non-dividing cells and is incorporated into chromatin independently of DNA synthesis. Deposited at sites of nucleosomal displacement throughout transcribed genes, suggesting that it represents an epigenetic imprint of transcriptionally active chromatin. Nucleosomes wrap and compact DNA into chromatin, limiting DNA accessibility to the cellular machineries which require DNA as a template. Histones thereby play a central role in transcription regulation, DNA repair, DNA replication and chromosomal stability. DNA accessibility is regulated via a complex set of post-translational modifications of histones, also called histone code, and nucleosome remodeling. Related diseases Glioma (GLM) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539269}. The gene represented in this entry is involved in disease pathogenesis. H3F3A mutations affecting residues involved in post-translational modifications of histone H3.3 are recurrent in malignant, aggressive gliomas including glioblastoma multiforme (GBM) and diffuse intrinsic pontine glioma (DIPG) (PubMed:22286061, PubMed:22286216). The mechanism through which mutations lead to tumorigenesis involves altered histones methylation, impaired regulation of Polycomb repressive complex 2 (PRC2) activity, and aberrant epigenetic regulation of gene expression (PubMed:23539183, PubMed:23539269, PubMed:23603901). {ECO:0000269|PubMed:22286061, ECO:0000269|PubMed:22286216, ECO:0000269|PubMed:23539183, ECO:0000269|PubMed:23539269, ECO:0000269|PubMed:23603901}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 1 (BRYLIB1) [MIM:619720]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB1 is caused by variants in H3-3A. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: Bryant-Li-Bhoj neurodevelopmental syndrome 2 (BRYLIB2) [MIM:619721]: An autosomal dominant disorder predominantly characterized by global developmental delay, impaired intellectual development, poor or absent speech, and delayed motor milestones. Clinical manifestations are highly variable, including abnormal head shape, dysmorphic facial features, oculomotor abnormalities, feeding problems, and non-specific brain imaging abnormalities. Additional features may include hearing loss, seizures, short stature, and mild skeletal defects. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}. The disease is caused by variants affecting the gene represented in this entry. BRYLIB2 is caused by variants in H3-3B. {ECO:0000269|PubMed:33268356, ECO:0000269|PubMed:34876591}.; DISEASE: H3F3A and H3F3B mutations affecting residues involved in post-translational modifications of histone H3.3 are implicated in the pathogenesis of some bone and cartilage neoplasms. Mutations have been found with high prevalence in chondroblastoma and giant cell tumors of bone, and with low frequency in osteosarcoma, conventional chondrosarcoma and clear cell chondrosarcoma. Chondroblastoma samples frequently carry a H3F3B mutation affecting residue Lys-37 (H3K36), although H3F3A is mutated in some cases. Most giant cell tumors of bone harbor H3F3A mutations affecting residue Gly-35 (H3G34). {ECO:0000269|PubMed:24162739}. Drugs (DrugBank ID) NA Interacts with Q9NVP2; P45973; Q13111; Q9UER7; Q9UER7-1; Q9Y6K1; P62805; P49321-2; Q8IZL8; Q5VWG9; Q9VK33; Q8R5C8 EC number NA Uniprot keywords 3D-structure; Acetylation; ADP-ribosylation; Chromosome; Citrullination; Direct protein sequencing; Disease variant; DNA-binding; Hydroxylation; Intellectual disability; Lipoprotein; Methylation; Nucleosome core; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A,C Molecular weight (Da) 86148.9 Length 766 Aromaticity 0.1 Instability index 37.57 Isoelectric point 8.25 Charge (pH=7) 7.16 2D Binding mode Binding energy (Kcal/mol) -7.63 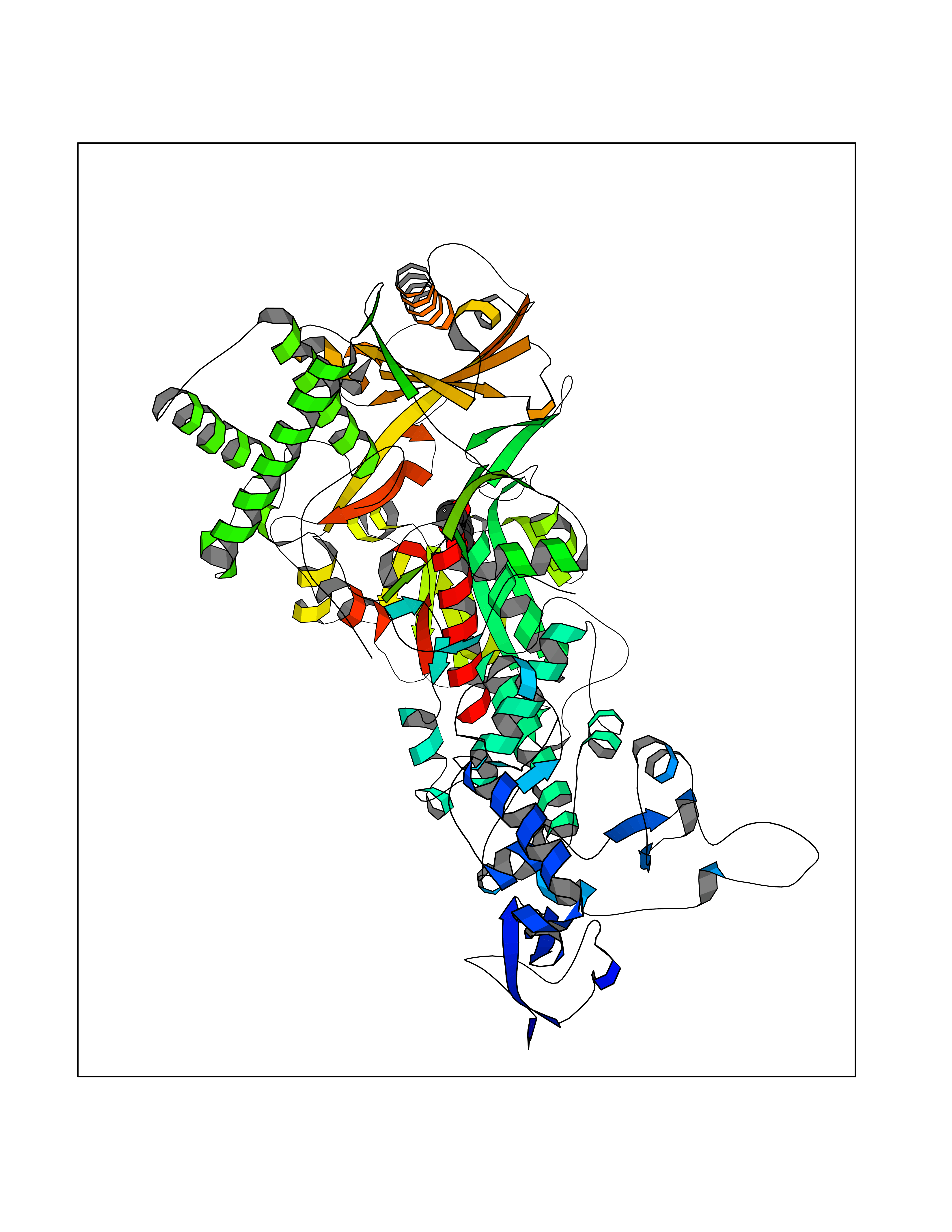 Molscript Map  Pymol Map 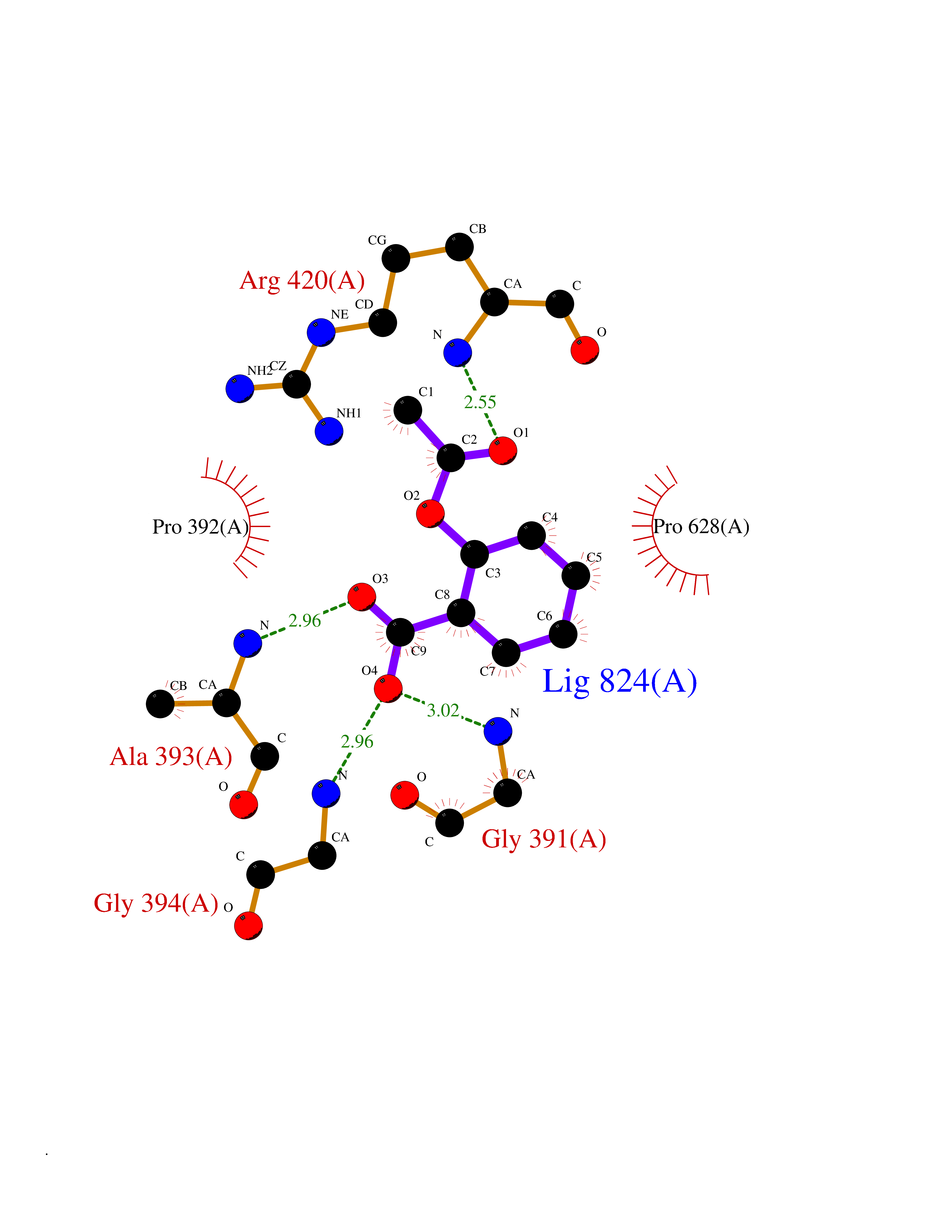 Ligplot Map 3D Binding mode Sequence GSRKCEKAGCTATCPVCFASASERCAKNGYTSRWYHLSCGEHFCNECFDHYYRSHKDGYDKYTTWKKIWTSNGKTEPSPKAFMADQQLPYWVQCTKPECRKWRQLTKEIQLTPQIAKTYRCGMKPNTAIKPETSDHCSLPEDLRVLEVSNHWWYSMLILPPLLKDSVAAPLLSAYYPDCVGMSPSCTGMNRYFQPFYQPNECGKALCVRPDVMELDELYEFPEYSRDPTMYLALRNLILALWYTNCKEALTPQKCIPHIIVRGLVRIRCVQEVERILYFMTRKGLINTGVLSVGADQYLLPKDYHNKSVIIIGAGPAGLAAARQLHNFGIKVTVLEAKDRIGGRVWDDKSFKGVTVGRGAQIVNGCINNPVALMCEQLGISMHKFGERCDLIQEGGRITDPTIDKRMDFHFNALLDVVSEWRKDKTQLQDVPLGEKIEEIYKAFIKESGIQFSELEGQVLQFHLSNLEYACGSNLHQVSARSWDHNEFFAQFAGDHTLLTPGYSVIIEKLAEGLDIQLKSPVQCIDYSGDEVQVTTTDGTGYSAQKVLVTVPLALLQKGAIQFNPPLSEKKMKAINSLGAGIIEKIALQFPYRFWDSKVQGADFFGHVPPSASKRGLFAVFYDMDPQKKHSVLMSVIAGEAVASVRTLDDKQVLQQCMATLRELFKEQEVPDPTKYFVTRWSTDPWIQMAYSFVKTGGSGEAYDIIAEDIQGTVFFAGEATNRHFPQTVTGAYLSGVREASKIAAFARTMQTARKSTGGKAPRKQL Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Tyrosine-protein kinase Kit (KIT) | 1T46 | 5.58 | |
Target general information Gen name KIT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog; p145 c-kit; Proto-oncogene tyrosine-protein kinase Kit; Proto-oncogene c-Kit; Piebald trait protein; PBT; Mast/stem cell growth factor re Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function In response to KITLG/SCF binding, KIT can activate several signaling pathways. Phosphorylates PIK3R1, PLCG1, SH2B2/APS and CBL. Activates the AKT1 signaling pathway by phosphorylation of PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase. Activated KIT also transmits signals via GRB2 and activation of RAS, RAF1 and the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1. Promotes activation of STAT family members STAT1, STAT3, STAT5A and STAT5B. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate. KIT signaling is modulated by protein phosphatases, and by rapid internalization and degradation of the receptor. Activated KIT promotes phosphorylation of the protein phosphatases PTPN6/SHP-1 and PTPRU, and of the transcription factors STAT1, STAT3, STAT5A and STAT5B. Promotes phosphorylation of PIK3R1, CBL, CRK (isoform Crk-II), LYN, MAPK1/ERK2 and/or MAPK3/ERK1, PLCG1, SRC and SHC1. Tyrosine-protein kinase that acts as cell-surface receptor for the cytokine KITLG/SCF and plays an essential role in the regulation of cell survival and proliferation, hematopoiesis, stem cell maintenance, gametogenesis, mast cell development, migration and function, and in melanogenesis. Related diseases Piebald trait (PBT) [MIM:172800]: Autosomal dominant genetic developmental abnormality of pigmentation characterized by congenital patches of white skin and hair that lack melanocytes. {ECO:0000269|PubMed:11074500, ECO:0000269|PubMed:1370874, ECO:0000269|PubMed:1376329, ECO:0000269|PubMed:1717985, ECO:0000269|PubMed:7687267, ECO:0000269|PubMed:8680409, ECO:0000269|PubMed:9450866, ECO:0000269|PubMed:9699740}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Gastrointestinal stromal tumor (GIST) [MIM:606764]: Common mesenchymal neoplasms arising in the gastrointestinal tract, most often in the stomach. They are histologically, immunohistochemically, and genetically different from typical leiomyomas, leiomyosarcomas, and schwannomas. Most GISTs are composed of a fairly uniform population of spindle-shaped cells. Some tumors are dominated by epithelioid cells or contain a mixture of spindle and epithelioid morphologies. Primary GISTs in the gastrointestinal tract commonly metastasize in the omentum and mesenteries, often as multiple nodules. However, primary tumors may also occur outside of the gastrointestinal tract, in other intra-abdominal locations, especially in the omentum and mesentery. {ECO:0000269|PubMed:11505412, ECO:0000269|PubMed:15824741, ECO:0000269|PubMed:9438854, ECO:0000269|PubMed:9697690}. The gene represented in this entry is involved in disease pathogenesis.; DISEASE: Testicular germ cell tumor (TGCT) [MIM:273300]: A common malignancy in males representing 95% of all testicular neoplasms. TGCTs have various pathologic subtypes including: unclassified intratubular germ cell neoplasia, seminoma (including cases with syncytiotrophoblastic cells), spermatocytic seminoma, embryonal carcinoma, yolk sac tumor, choriocarcinoma, and teratoma. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. The gene represented in this entry is involved in disease pathogenesis. Somatic mutations that lead to constitutive activation of KIT are detected in AML patients. These mutations fall into two classes, the most common being in-frame internal tandem duplications of variable length in the juxtamembrane region that disrupt the normal regulation of the kinase activity. Likewise, point mutations in the kinase domain can result in a constitutively activated kinase.; DISEASE: Mastocytosis, cutaneous (MASTC) [MIM:154800]: A form of mastocytosis, a heterogeneous group of disorders associated with abnormal proliferation and accumulation of mast cells in various tissues, especially in the skin and hematopoietic organs. MASTC is an autosomal dominant form characterized by macules, papules, nodules, or diffuse infiltration of the skin, often associated with localized hyperpigmentation. Gentle rubbing of the lesions induces histamine release from mechanically activated mast cells, causing local wheals, erythema, and often pruritus, a phenomenon termed Darier sign. {ECO:0000269|PubMed:15173254, ECO:0000269|PubMed:19865100, ECO:0000269|PubMed:21689725, ECO:0000269|PubMed:24289326, ECO:0000269|PubMed:9990072}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mastocytosis, systemic (MASTSYS) [MIM:154800]: A severe form of mastocytosis characterized by abnormal proliferation and accumulation of mast cells in several organs, resulting in a systemic disease that may affect bone, gastrointestinal tract, lymphatics, spleen, and liver. In some cases, it is associated with a clonal hematologic non-mast-cell lineage disease, such as a myelodysplastic or myeloproliferative disorder. It can also lead to mast cell leukemia, which carries a high risk of mortality. {ECO:0000269|PubMed:9990072}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12742; DB09103; DB15233; DB01254; DB12147; DB12010; DB00619; DB09078; DB06080; DB06595; DB04868; DB05913; DB06589; DB12978; DB01962; DB08901; DB08896; DB14840; DB00398; DB01268; DB11800; DB05146 Interacts with P00519; P42684; O75815; P51451; Q8WV28; P46108; P07332; P09769; O75791; P62993; Q14451; P08631; Q96JZ2; P21583; P06239; P07948; P16333; O43639; P27986; O00459; Q92569; P19174; P16885; Q13882; Q06124; Q92729; P20936; Q9UQQ2; O14796; Q9NP31; Q8N5H7; P78314; Q15464; P29353; P98077; Q92529; Q9H6Q3; O14508; O14543; O14544; P12931; Q9ULZ2; Q9HBL0; Q63HR2; Q68CZ2; P42681; P07947; P43403; Q8VBX6; P35235 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Immunoglobulin domain; Kinase; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 33575.6 Length 297 Aromaticity 0.11 Instability index 45.37 Isoelectric point 8.37 Charge (pH=7) 3.24 2D Binding mode Binding energy (Kcal/mol) -7.61  Molscript Map 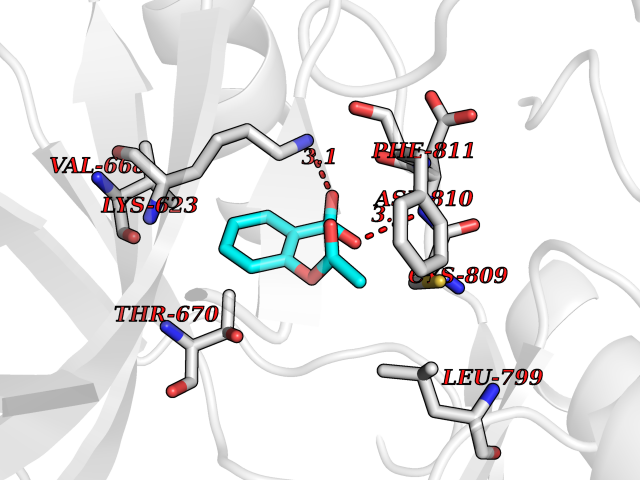 Pymol Map 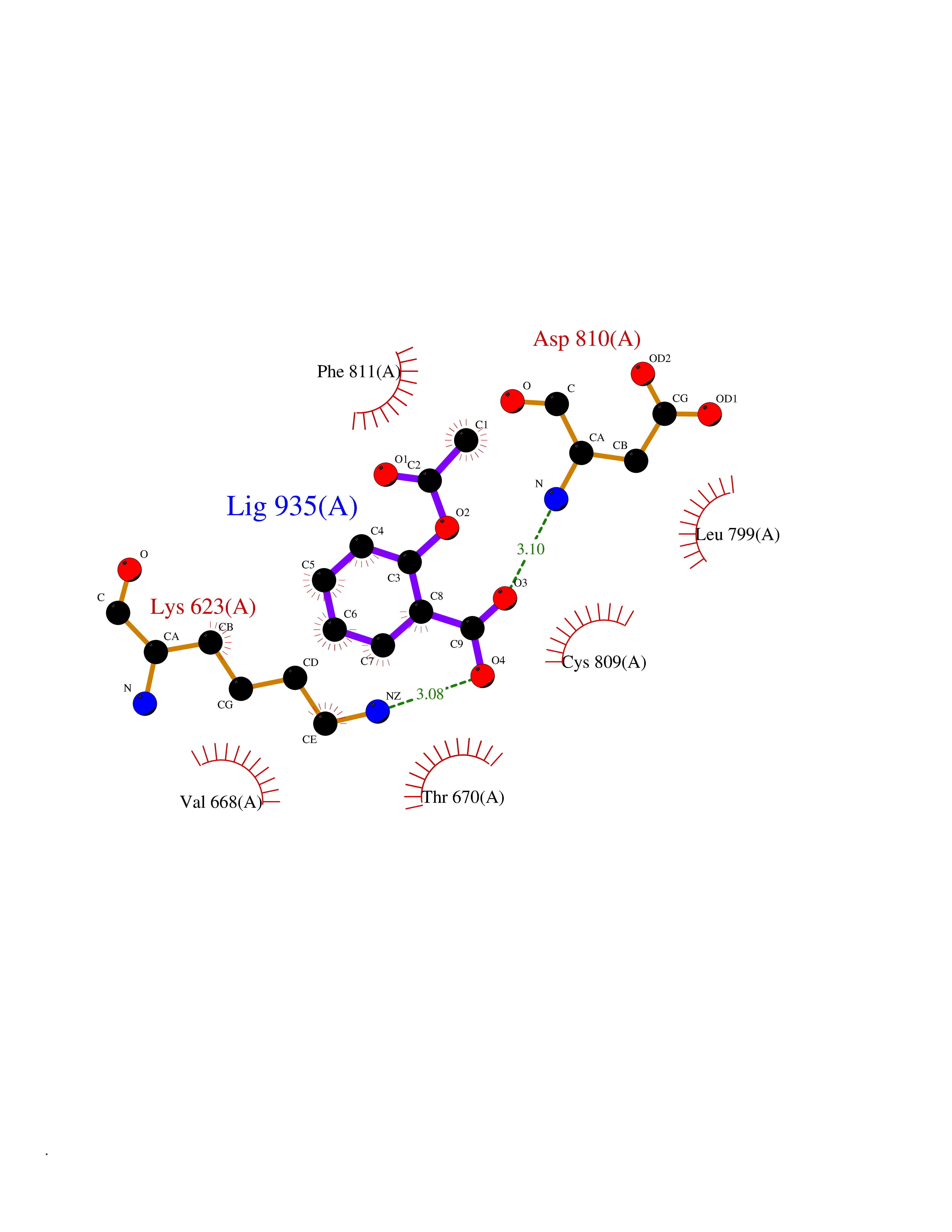 Ligplot Map 3D Binding mode Sequence GNNYVYIDPTQLPYDHKWEFPRNRLSFGKTLGAGAFGKVVEATAYGLIKSDAAMTVAVKMLKPSAHLTEREALMSELKVLSYLGNHMNIVNLLGACTIGGPTLVITEYCCYGDLLNFLRRKRDSFLALDLEDLLSFSYQVAKGMAFLASKNCIHRDLAARNILLTHGRITKICDFGLARDIKNDSNYVVKGNARLPVKWMAPESIFNCVYTFESDVWSYGIFLWELFSLGSSPYPGMPVDSKFYKMIKEGFRMLSPEHAPAEMYDIMKTCWDADPLKRPTFKQIVQLIEKQISESTN Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Gamma-butyrobetaine dioxygenase | 4C5W | 5.58 | |
Target general information Gen name BBOX1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms BBH;BBOX Protein family Gamma-BBH/TMLD family Biochemical class Oxidoreductase Function Gamma-butyrobetaine dioxygenase activity.Identical protein binding.Iron ion binding.Zinc ion binding. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126 Interacts with O75936; A0MZ66-7 EC number 1.14.11.1 Uniprot keywords 3D-structure; Carnitine biosynthesis; Cytoplasm; Dioxygenase; Iron; Metal-binding; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 31642.5 Length 275 Aromaticity 0.12 Instability index 35.68 Isoelectric point 6.33 Charge (pH=7) -2.46 2D Binding mode Binding energy (Kcal/mol) -7.61 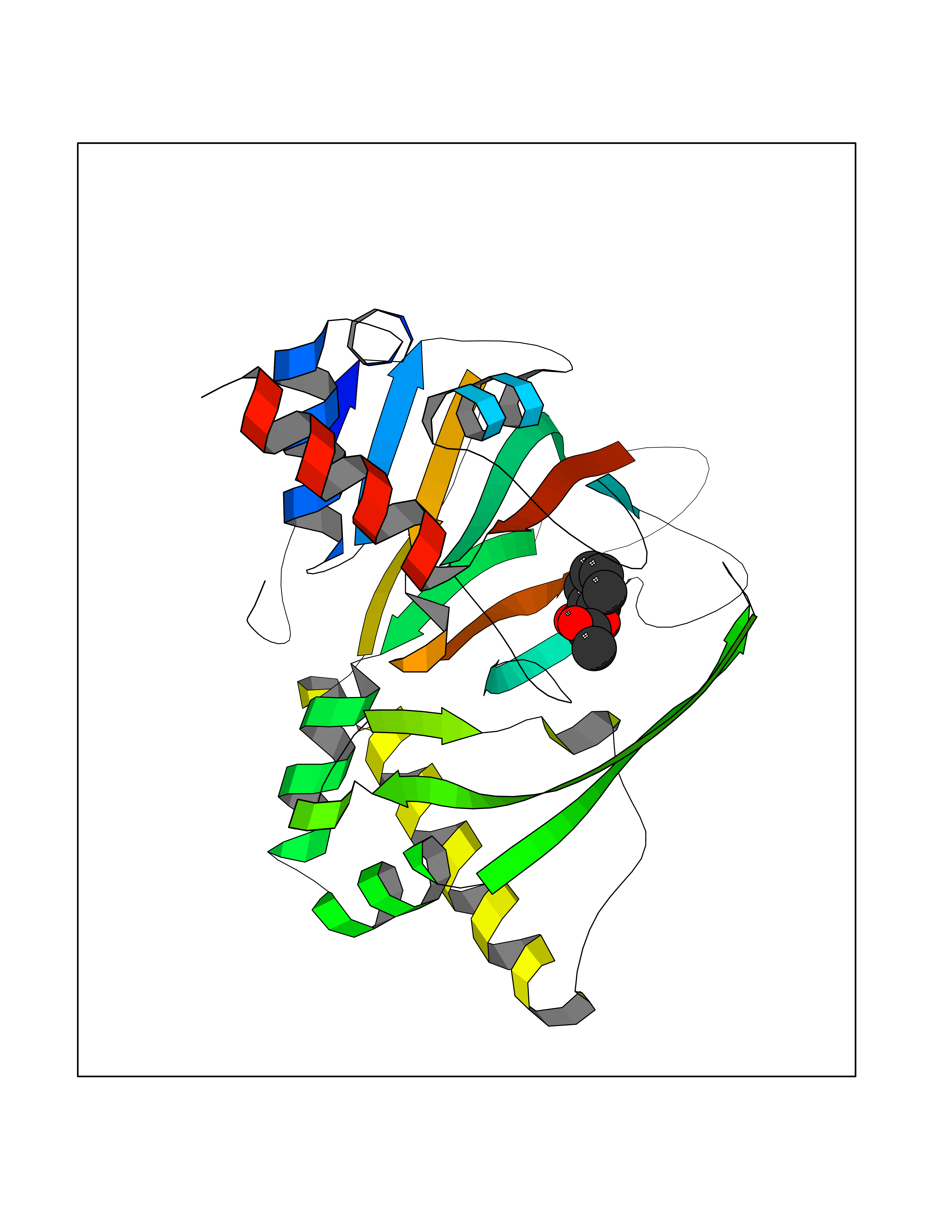 Molscript Map 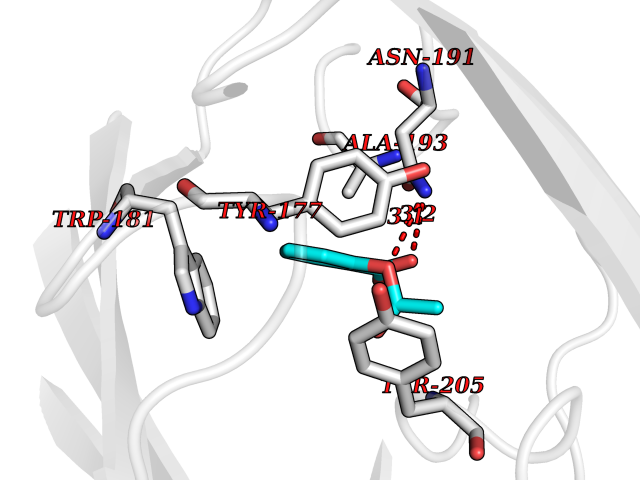 Pymol Map 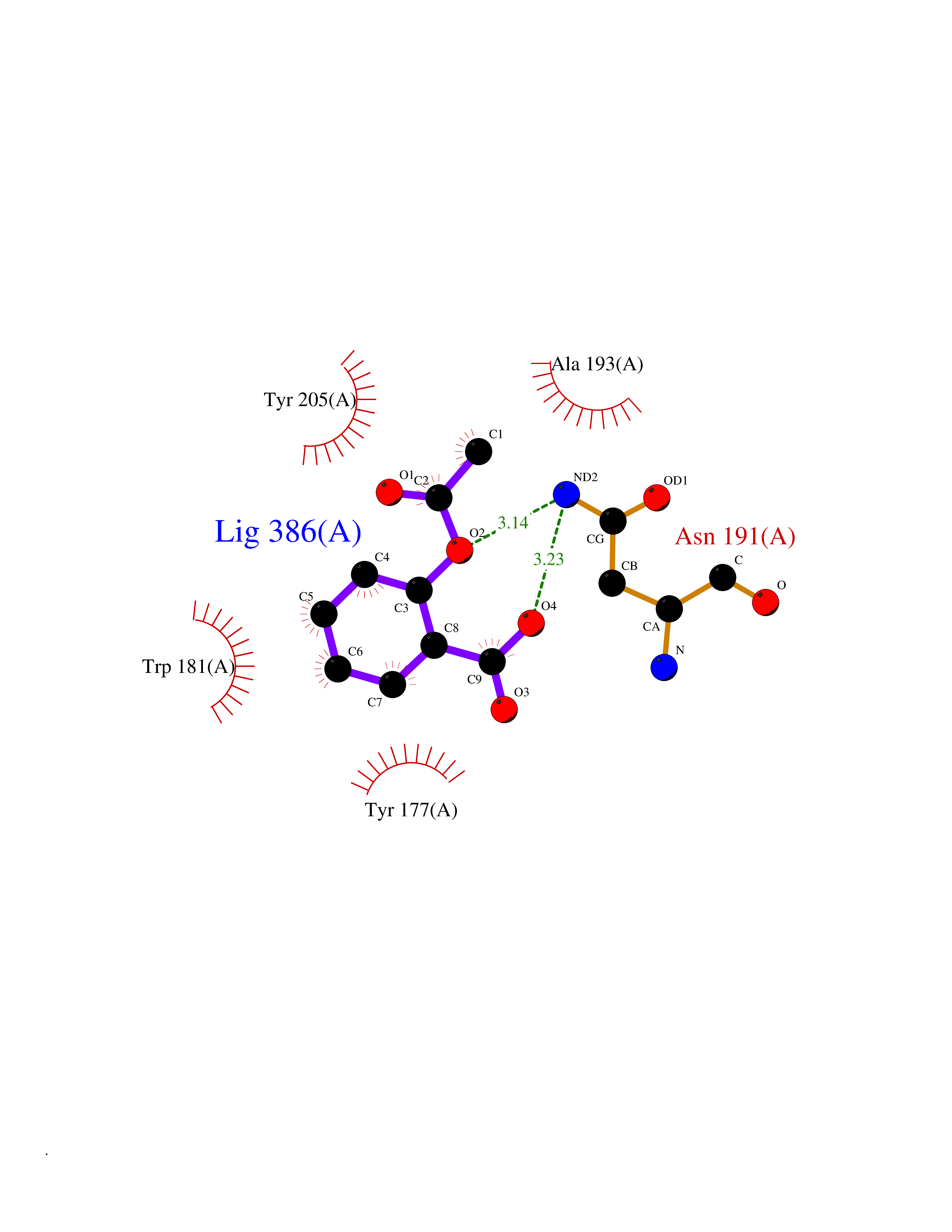 Ligplot Map 3D Binding mode Sequence FPECQYWGSELQLPTLDFEDVLRYDEHAYKWLSTLKKVGIVRLTGASDKPGEVSKLGKRMGFLYLTFYGHTWQVQDKIDANNVAYTTGKLSFHTDYPALHHPPGVQLLHCIKQTVTGGDSEIVDGFNVCQKLKKNNPQAFQILSSTFVDFTDIGVDYCDFSVQSKHKIIELDDKGQVVRINFNNATRDTIFDVPVERVQPFYAALKEFVDLMNSKESKFTFKMNPGDVITFDNWRLLHGRRSYEAGTEISRHLEGAYADWDVVMSRLRILRQRVE Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Short/branched chain specific acyl-CoA dehydrogenase, mitochondrial | 2JIF | 5.58 | |
Target general information Gen name ACADSB Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Acyl-CoA dehydrogenase family Biochemical class Oxidoreductase Function Acyl-CoA dehydrogenase activity.Flavin adenine dinucleotide binding. Related diseases Short/branched-chain acyl-CoA dehydrogenase deficiency (SBCADD) [MIM:610006]: Autosomal recessive disorder and consists of a defect in catabolism of L-isoleucine which is characterized by an increase of 2-methylbutyrylglycine and 2-methylbutyrylcarnitine in blood and urine. Affected individuals have seizures and psychomotor delay as the main clinical features. {ECO:0000269|PubMed:10832746, ECO:0000269|PubMed:11013134, ECO:0000269|PubMed:16317551}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00167; DB00313 Interacts with NA EC number 1.3.8.5 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Disease variant; FAD; Fatty acid metabolism; Flavoprotein; Lipid metabolism; Mitochondrion; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Transit peptide Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 41740.2 Length 381 Aromaticity 0.09 Instability index 31.31 Isoelectric point 5.63 Charge (pH=7) -6.59 2D Binding mode Binding energy (Kcal/mol) -7.61 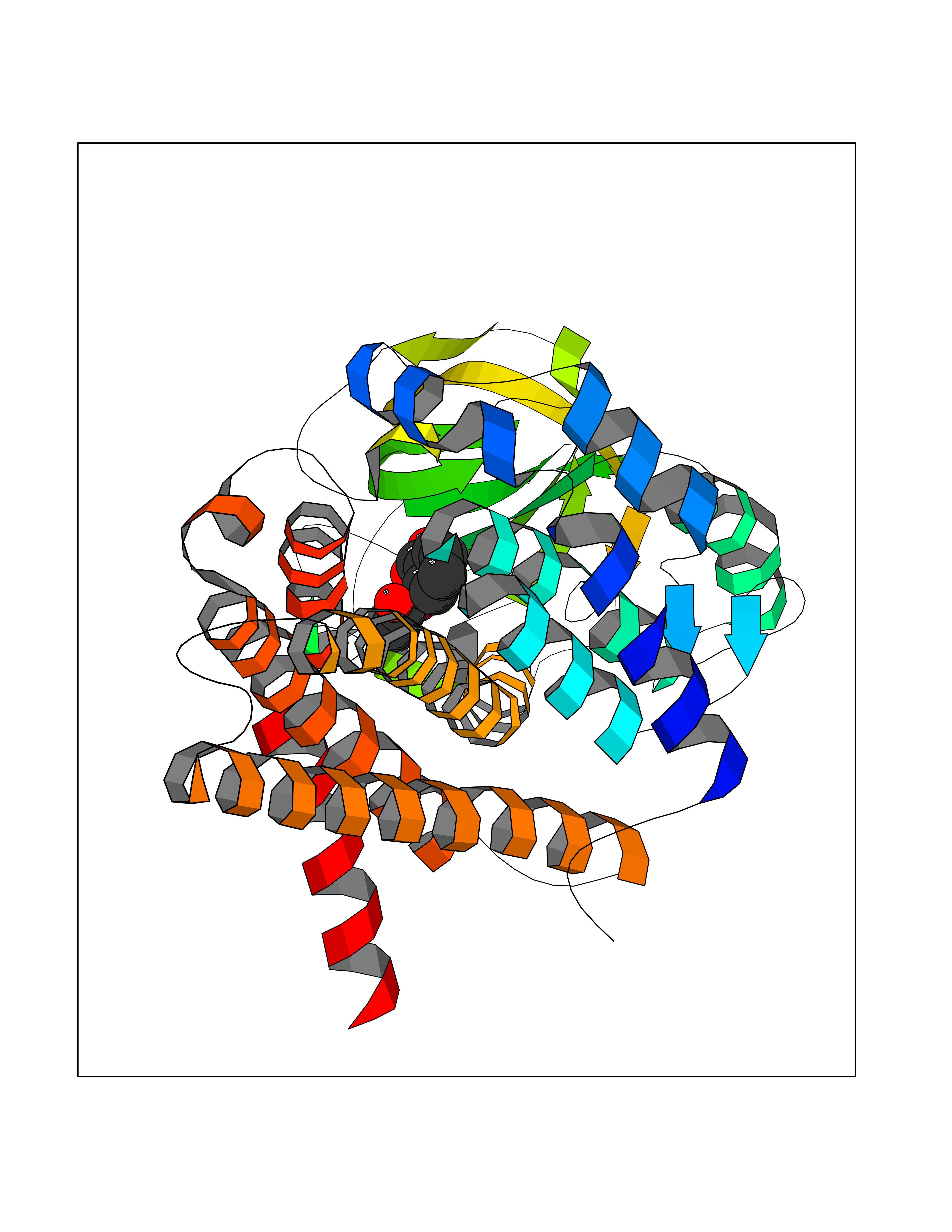 Molscript Map 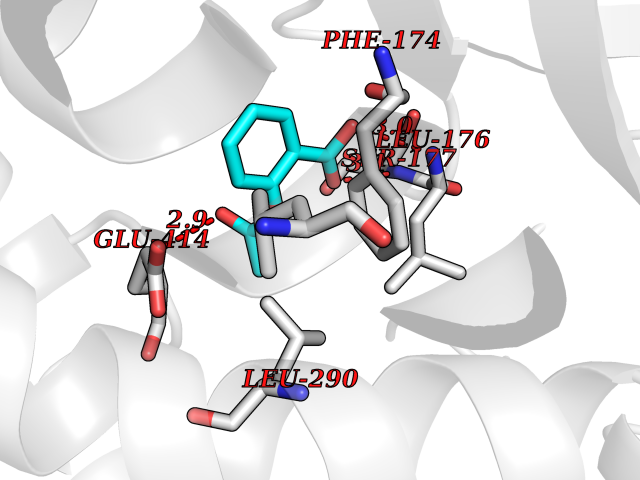 Pymol Map 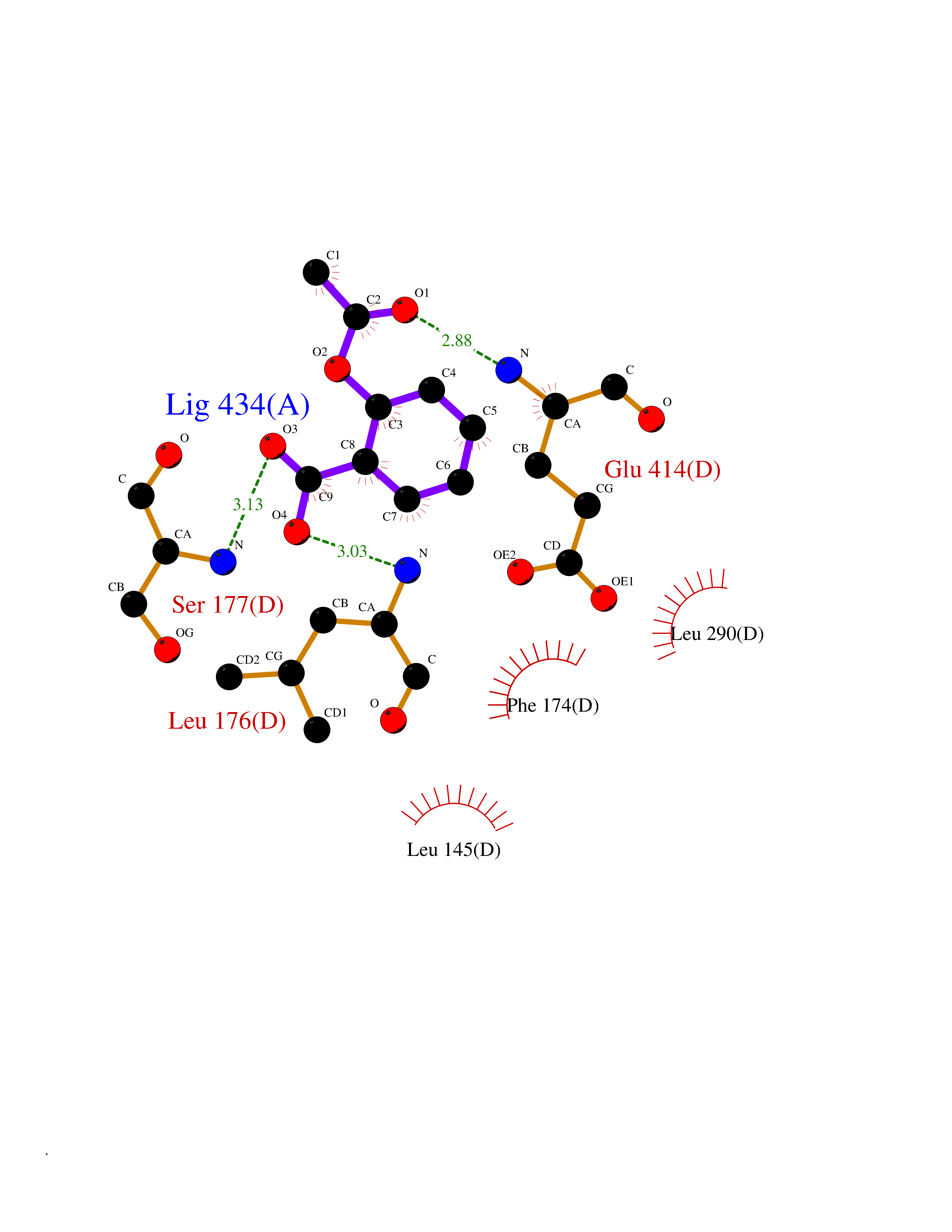 Ligplot Map 3D Binding mode Sequence APLQTFTDEEMMIKSSVKKFAQEQIAPLVSTMDENSKMEKSVIQGLFQQGLMGIEVDPEYGGTGASFLSTVLVIEELAKVDASVAVFCEIQNTLINTLIRKHGTEEQKATYLPQLTTEKVGSFCLSEAGAGSDSFALKTRADKEGDYYVLNGSKMWISSAEHAGLFLVMANVDPTIGYKGITSFLVDRDTPGLHIGKPENKLGLRASSTCPLTFENVKVPEANILGQIGHGYKYAIGSLNEGRIGIAAQMLGLAQGCFDYTIPYIKERIQFGKRLFDFQGLQHQVAHVATQLEAARLLTYNAARLLEAGKPFIKEASMAKYYASEIAGQTTSKCIEWMGGVGYTKDYPVEKYFRDAKIGTIYEGASNIQLNTIAKHIDAEY Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | 2-oxoglutarate dehydrogenase, mitochondrial | 3ERY | 5.58 | |
Target general information Gen name OGDH Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Alpha-ketoglutarate dehydrogenase family Biochemical class Immune system Function Chaperone binding.Heat shock protein binding.Metal ion binding.Oxoglutarate dehydrogenase (NAD+) activity.Oxoglutarate dehydrogenase (succinyl-transferring) activity.Thiamine pyrophosphate binding. Related diseases Postaxial acrofacial dysostosis (POADS) [MIM:263750]: POADS is characterized by severe micrognathia, cleft lip and/or palate, hypoplasia or aplasia of the posterior elements of the limbs, coloboma of the eyelids and supernumerary nipples. POADS is a very rare disorder: only 2 multiplex families, each consisting of 2 affected siblings born to unaffected, nonconsanguineous parents, have been described among a total of around 30 reported cases. {ECO:0000269|PubMed:19915526}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00157; DB00313; DB09092 Interacts with P54253; P42858 EC number 1.2.4.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Calcium; Glycolysis; Isopeptide bond; Magnesium; Metal-binding; Mitochondrion; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Thiamine pyrophosphate; Transit peptide; Ubl conjugation Protein physicochemical properties Chain ID P,Q Molecular weight (Da) 18311.1 Length 158 Aromaticity 0.15 Instability index 37.26 Isoelectric point 5.64 Charge (pH=7) -2.98 2D Binding mode Binding energy (Kcal/mol) -7.6  Molscript Map 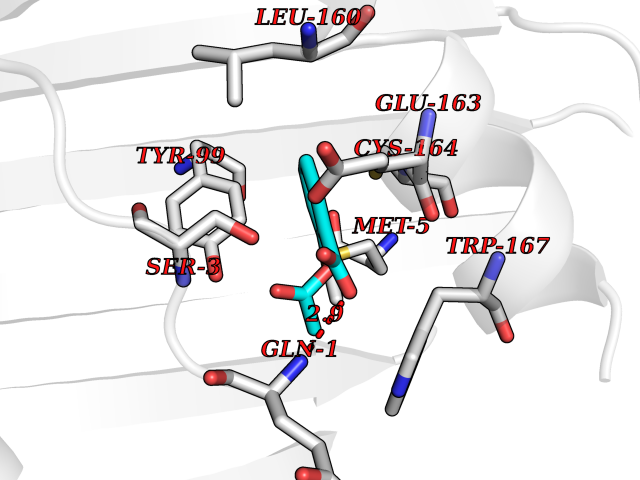 Pymol Map 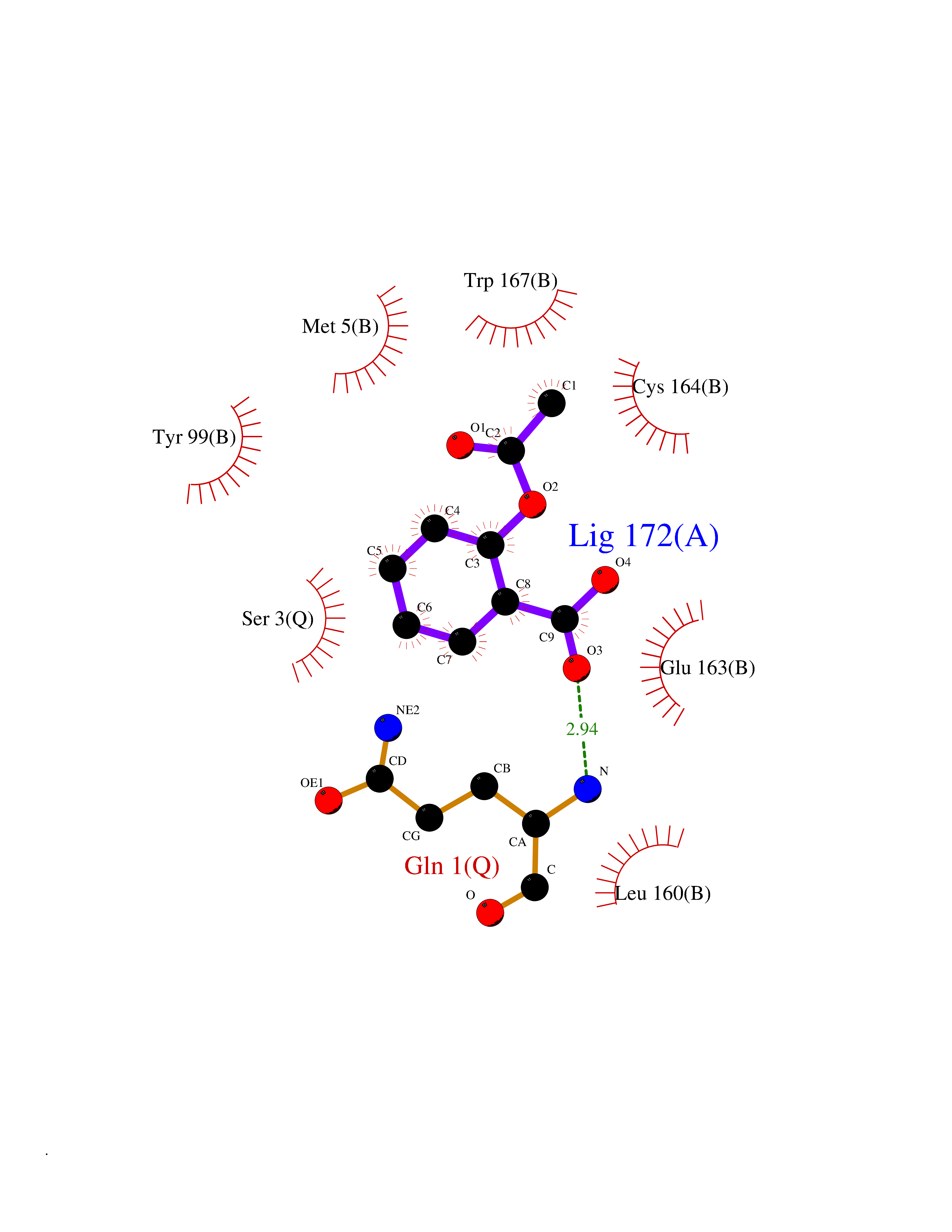 Ligplot Map 3D Binding mode Sequence QLSPFPFDLGPHSMRYYETATSRRGLGEPRYTSVGYVDDKEFVRFDSDARITQVAKGQEQWFRVNLRTLLGYYNQSAGGTHTLQRMYGCDVGSDGRLLRGYEQFAYDGCDYIALNEDLRTWTAADMAAQITRRKWEQAGAAEYYRAYLEGECVEWLHR Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Plasmodium Dihydroorotate dehydrogenase (Malaria DHOdehase) | 1TV5 | 5.58 | |
Target general information Gen name Malaria DHOdehase Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID Synonyms PFF0160c; Mitochondrially bound dihydroorotate-ubiqui oxidoreductase; Dihydroorotate oxidase of Plasmodium falciparum; Dihydroorotate dehydrogenase of Plasmodium falciparum; DHOdehase of Plasmodium fa Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 41846.8 Length 371 Aromaticity 0.1 Instability index 37.25 Isoelectric point 8.21 Charge (pH=7) 3.13 2D Binding mode Binding energy (Kcal/mol) -7.62 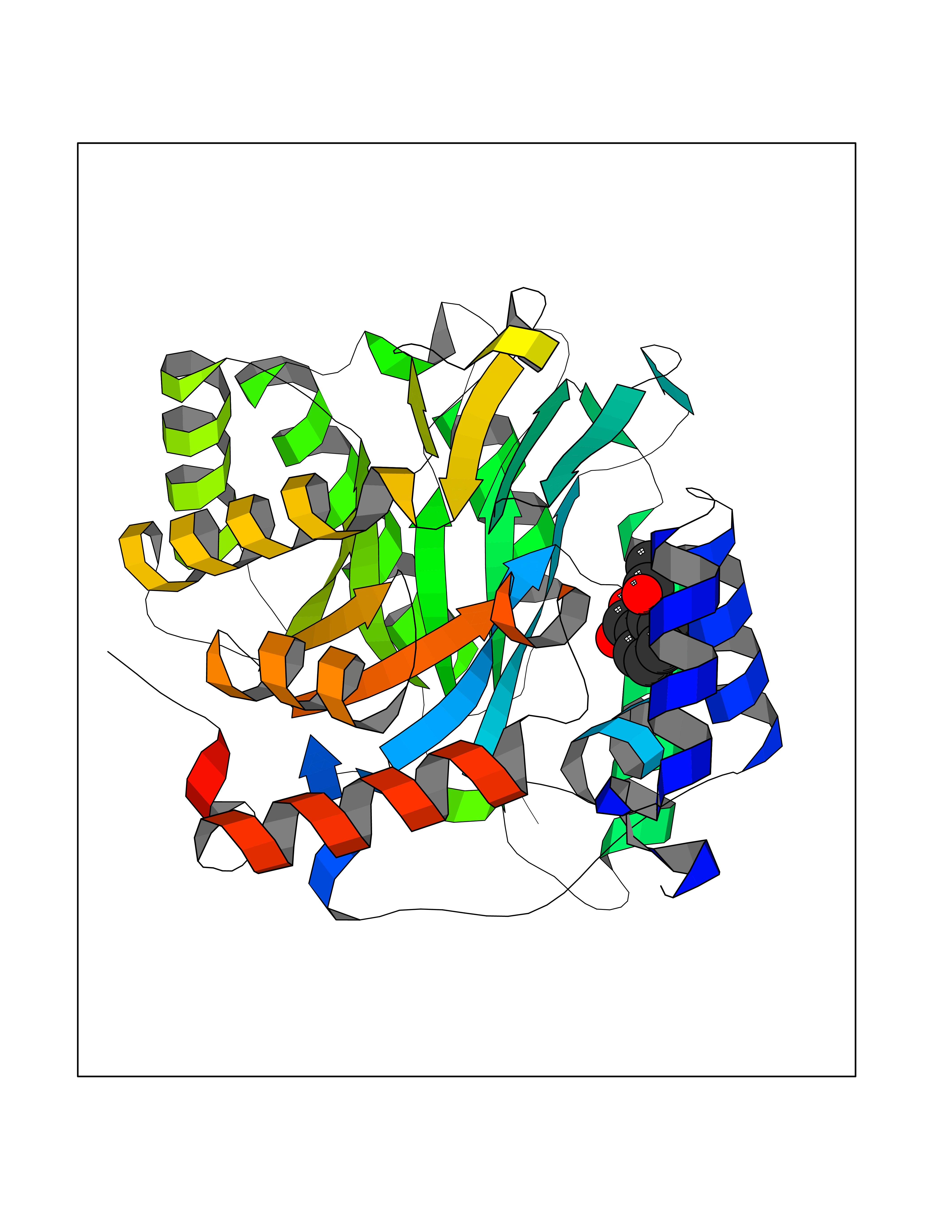 Molscript Map 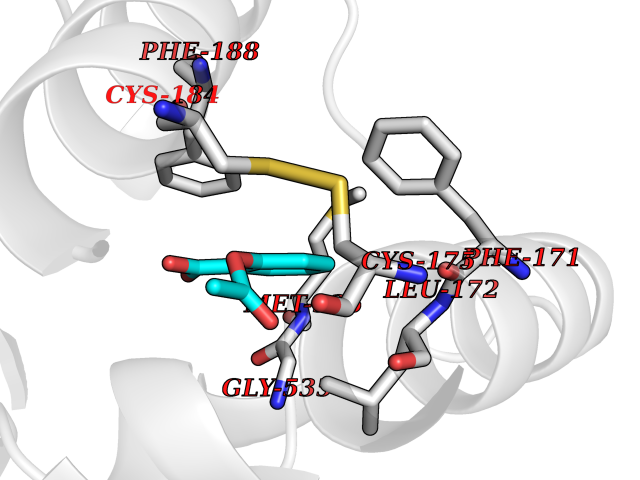 Pymol Map 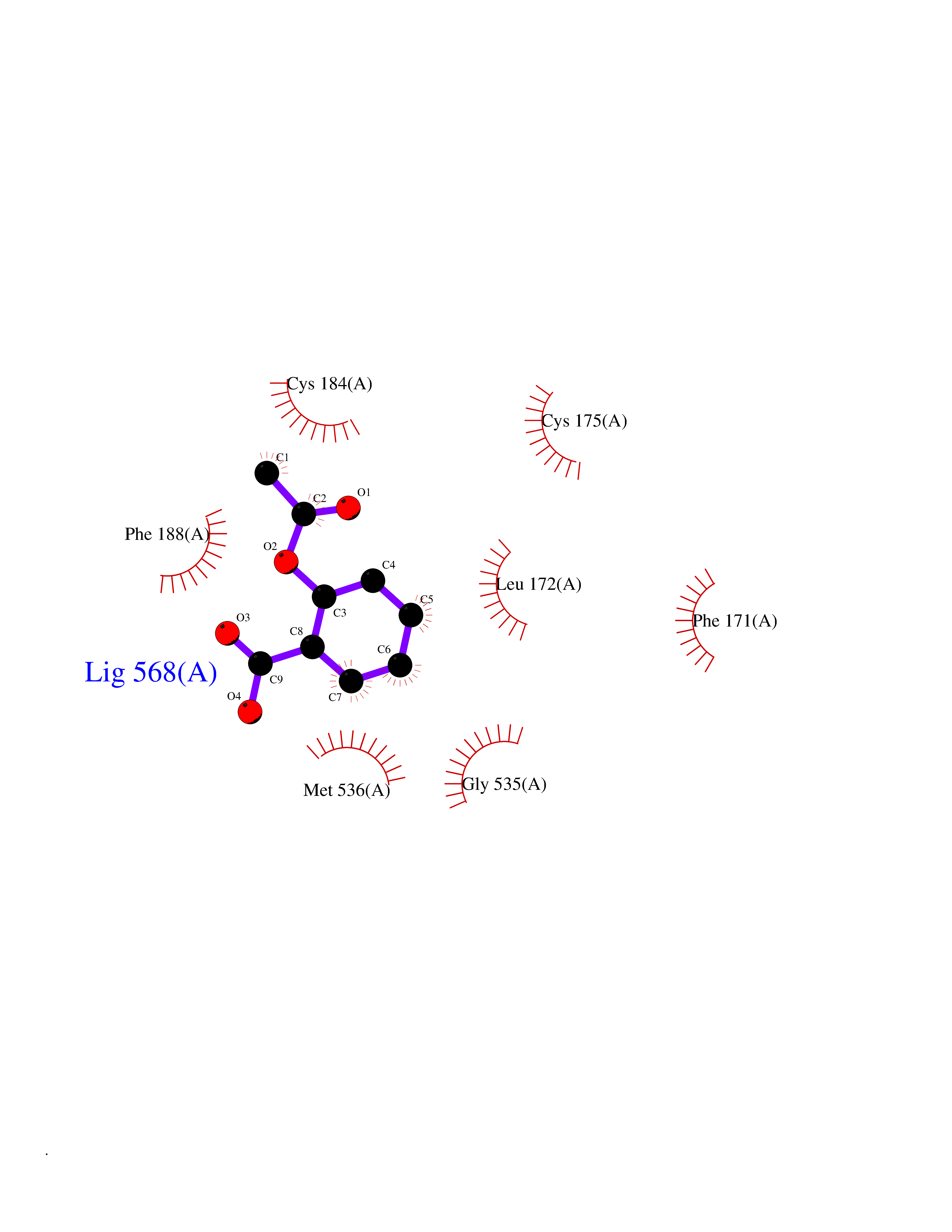 Ligplot Map 3D Binding mode Sequence FESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKH Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Tryptophan 2,3-dioxygenase (TDO) | 6PYZ | 5.58 | |
Target general information Gen name TDO2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tryptophanase; Tryptophan pyrrolase; Tryptophan oxygenase; Tryptamin 2,3-dioxygenase; TRPO; TO Protein family Tryptophan 2,3-dioxygenase family Biochemical class Oxygenase Function Catalyzes the oxidative cleavage of the indole moiety. Heme-dependent dioxygenase that catalyzes the oxidative cleavage of the L-tryptophan (L-Trp) pyrrole ring and converts L-tryptophan to N-formyl-L-kynurenine. Related diseases Hypertryptophanemia (HYPTRP) [MIM:600627]: An autosomal recessive condition characterized by persistent hypertryptophanemia and hyperserotoninemia. {ECO:0000269|PubMed:28285122}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00779; DB00500; DB00150 Interacts with O43865; O95671; P27797; P12830; P36957; O60762; P06730; Q8TBB1; Q9H8S9; Q70IA8; Q8TDX7; Q9NPG2; Q9HAN9; P20393; Q9NRD5; Q8IYS1; O00560; Q9H190; P48775; Q68DK2-5 EC number EC 1.13.11.11 Uniprot keywords 3D-structure; Dioxygenase; Disease variant; Heme; Iron; Metal-binding; Oxidoreductase; Proteomics identification; Reference proteome; Tryptophan catabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 42122.8 Length 353 Aromaticity 0.11 Instability index 45.07 Isoelectric point 7.06 Charge (pH=7) 0.23 2D Binding mode Binding energy (Kcal/mol) -7.61 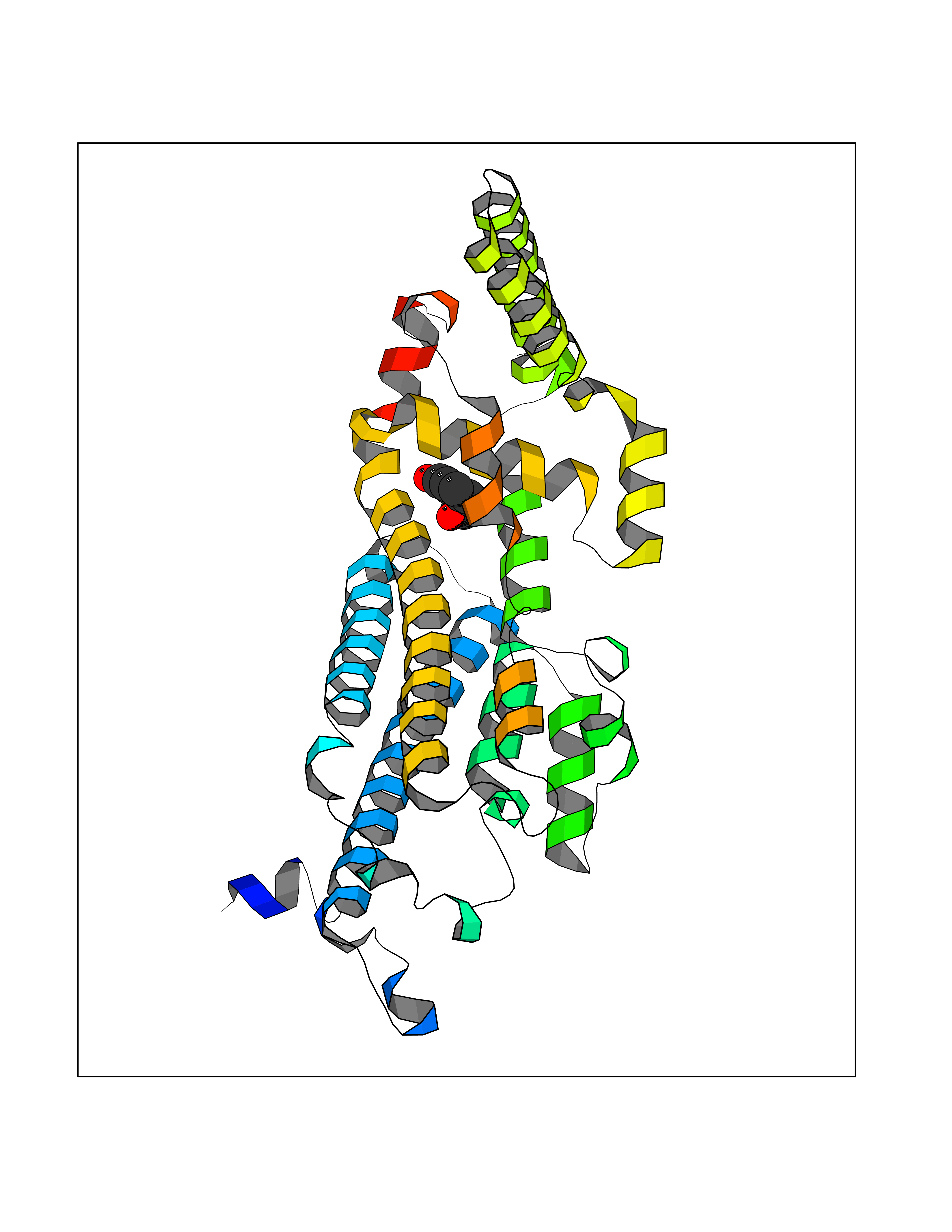 Molscript Map 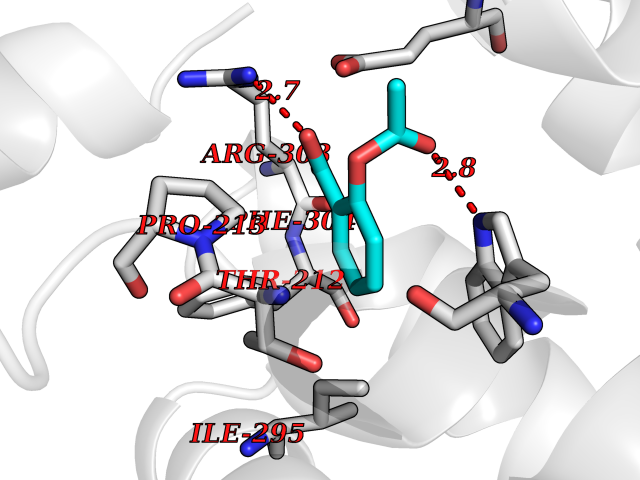 Pymol Map 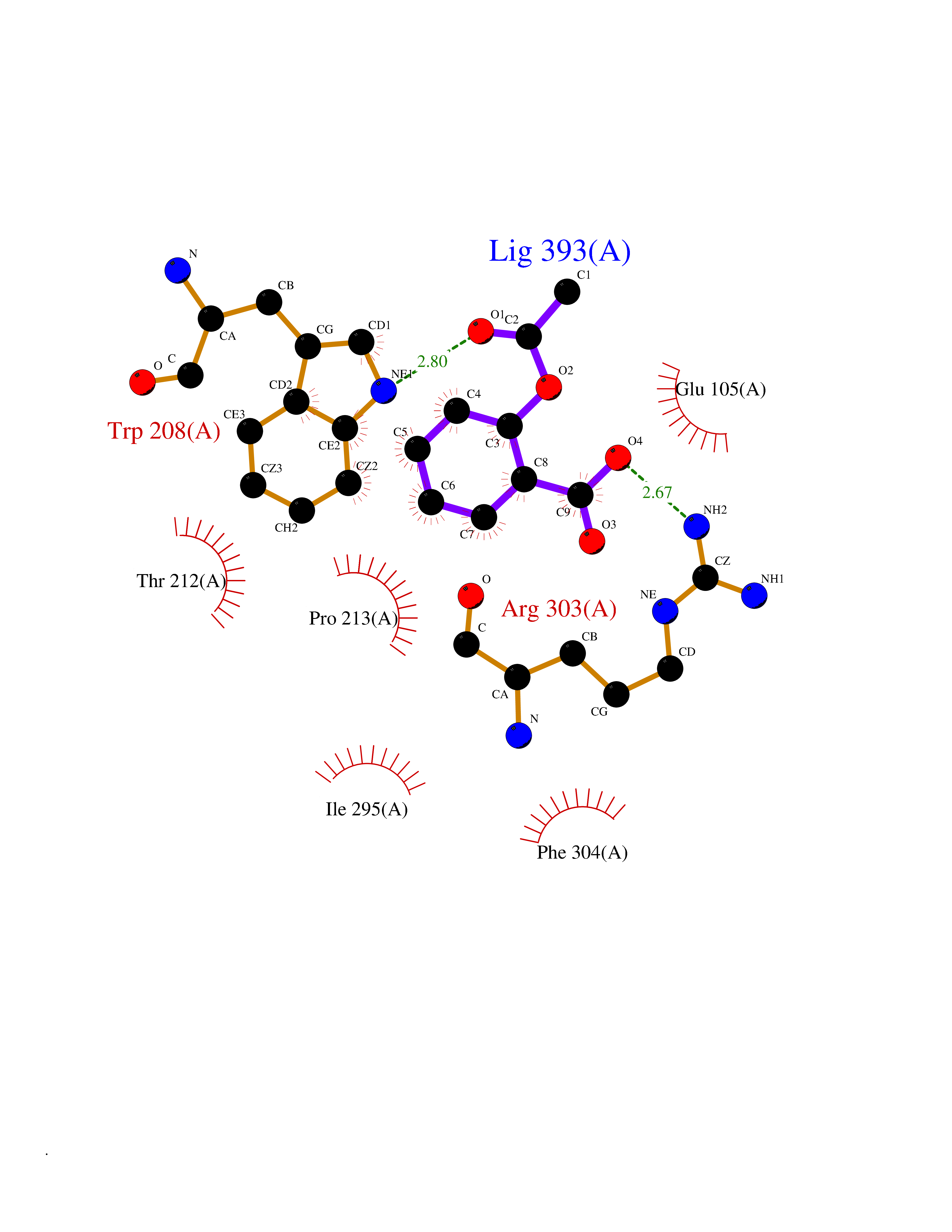 Ligplot Map 3D Binding mode Sequence GLIYGNYLHLEKVLNAQELQSETKGNKIHDEHLFIITHQAYELWFKQILWELDSVREIFQNGHVRDERNMLKVVSRMHRVSVILKLLVQQFSILETMTALDFNDFREYLSPASGFQSLQFRLLENKIGVLQNMRVPYNRRHYRDNFKGEENELLLKSEQEKTLLELVEAWLERTPGLEPHGFNFWGKLEKNITRGLEEEFIRIQAKEESEEKEEQVAEFQKQKEVLLSLFDEKRHEHLLSKGERRLSYRALQGALMIYFYREEPRFQVPFQLLTSLMDIDSLMTKWRYNHVCMVHRMLGSKAGTGGSSGYHYLRSTVSDRYKVFVDLFNLSTYLIPRHWIPKMNPTIHKFLEH Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Hepatitis B virus Capsid protein (HBV C) | 7PZL | 5.58 | |
Target general information Gen name HBV C Organism Hepatitis B virus genotype D subtype ayw (isolate France/Tiollais/1979) (HBV-D) Uniprot ID TTD ID Synonyms Core antigen; Core protein; HBcAg; p21.5 Protein family Orthohepadnavirus core antigen family Biochemical class NA Function Self assembles to form an icosahedral capsid. Most capsid appear to be large particles with an icosahedral symmetry of T=4 and consist of 240 copies of capsid protein, though a fraction forms smaller T=3 particles consisting of 180 capsid proteins. Entering capsid are transported along microtubules to the nucleus. Phosphorylation of the capsid is thought to induce exposure of nuclear localization signal in the C-terminal portion of the capsid protein that allows binding to the nuclear pore complex via the importin (karyopherin-) alpha and beta. Capsids are imported in intact form through the nuclear pore into the nuclear basket, where it probably binds NUP153. Only capsids that contain the mature viral genome can release the viral DNA and capsid protein into the nucleoplasm. Immature capsids get stucked in the basket. Capsids encapsulate the pre-genomic RNA and the P protein. Pre-genomic RNA is reverse transcribed into DNA while the capsid is still in the cytoplasm. The capsid can then either be directed to the nucleus, providing more genome for transcription, or bud through the endoplasmic reticulum to provide new virions. Related diseases Oocyte/zygote/embryo maturation arrest 14 (OZEMA14) [MIM:620276]: An autosomal recessive female infertility disorder characterized by oocyte maturation arrest, fertilization failure, and/or early embryonic arrest. {ECO:0000269|PubMed:32666501, ECO:0000269|PubMed:33683667, ECO:0000269|PubMed:33898437, ECO:0000269|PubMed:34218387}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative initiation; Capsid protein; Cytoplasmic inwards viral transport; DNA-binding; Host cytoplasm; Host-virus interaction; Microtubular inwards viral transport; Phosphoprotein; Reference proteome; Repeat; RNA-binding; T=4 icosahedral capsid protein; Viral penetration into host nucleus; Virion; Virus entry into host cell Protein physicochemical properties Chain ID A,B Molecular weight (Da) 32388.9 Length 286 Aromaticity 0.12 Instability index 42.05 Isoelectric point 5.06 Charge (pH=7) -11.81 2D Binding mode Binding energy (Kcal/mol) -7.62 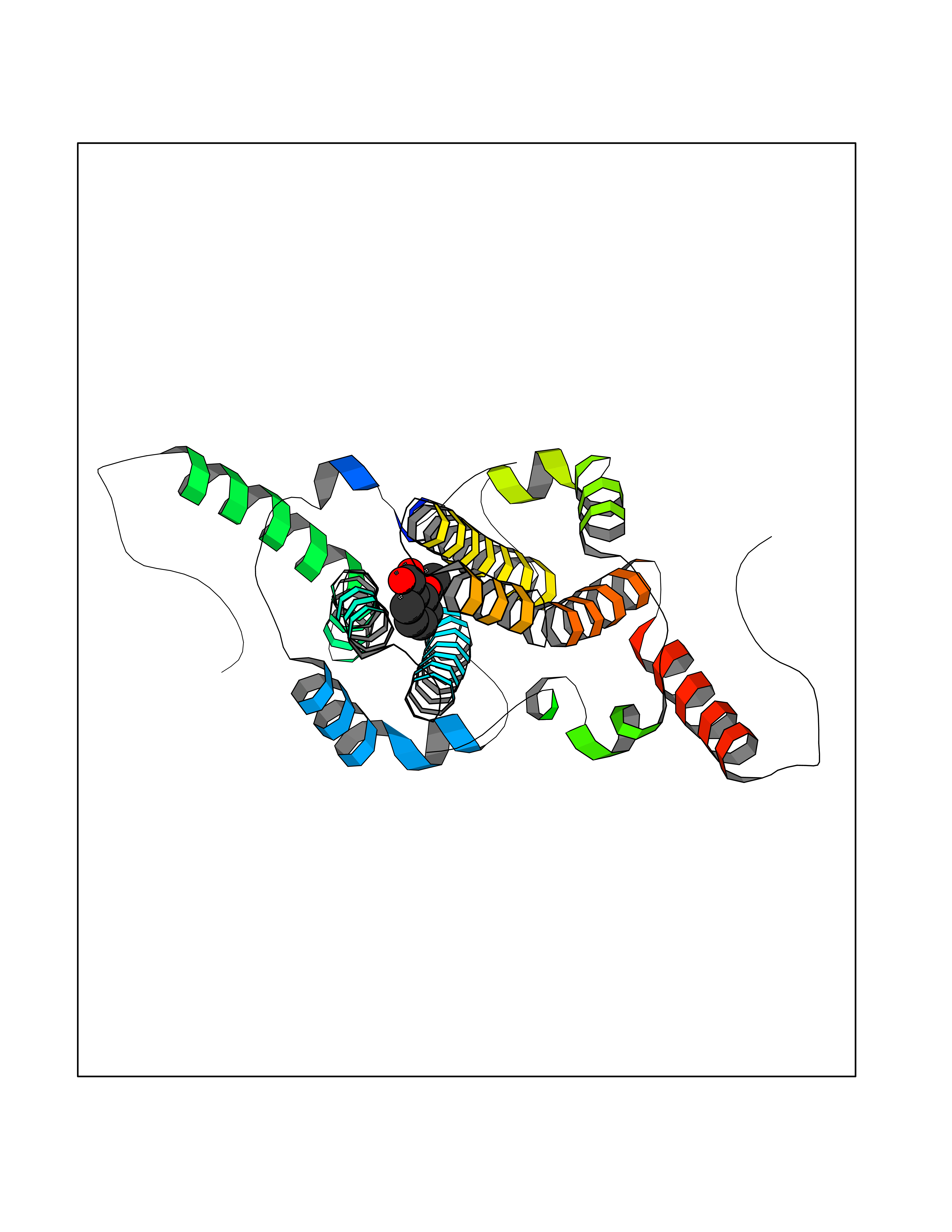 Molscript Map 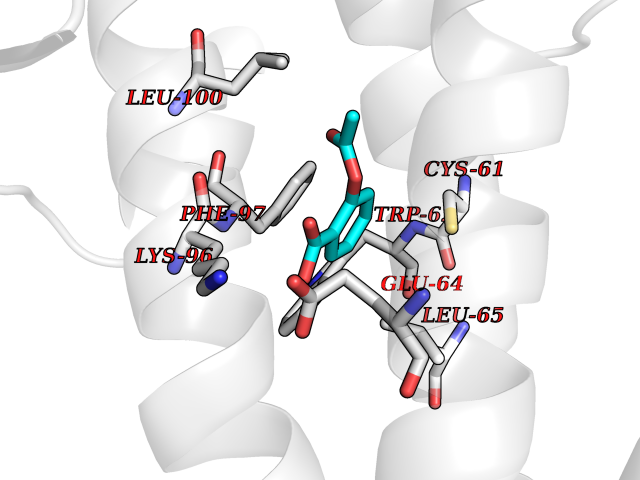 Pymol Map 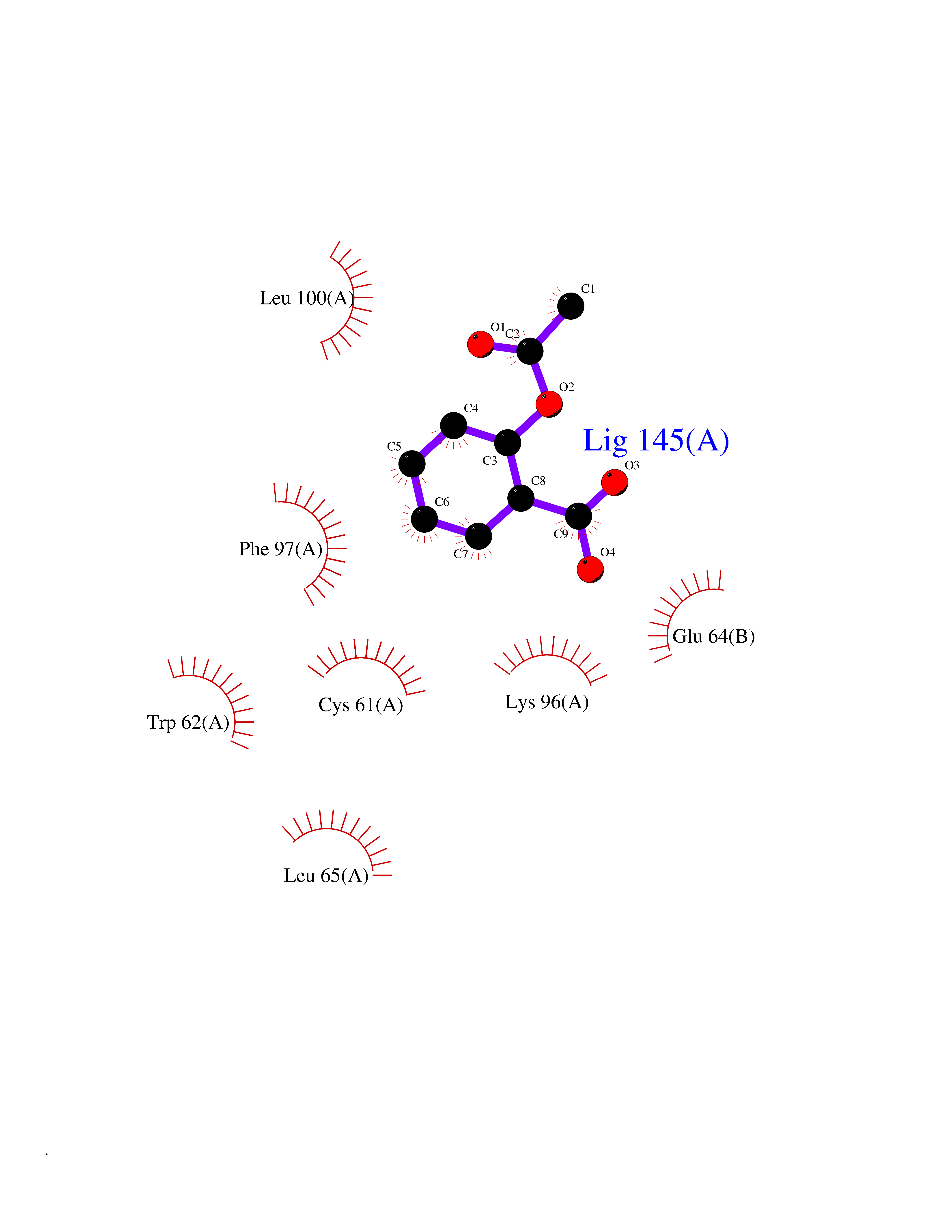 Ligplot Map 3D Binding mode Sequence MDIDPYKEFGATVELLSFLPSDFFPSVRDLLDTASALYREALESPEHCSPHHTALRQAIVCWGELMTLATWVGVNLEDPASRDLVVSYVNTNMGLKFRQLLWFHISCLTFGRETVIEYLVSFGVWIRTPPAYRPPNAPILSTLMDIDPYKEFGATVELLSFLPSDFFPSVRDLLDTASALYREALESPEHCSPHHTALRQAIVCWGELMTLATWVGVNLEDPASRDLVVSYVNTNMGLKFRQLLWFHISCLTFGRETVIEYLVSFGVWIRTPPAYRPPNAPILSTL Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Leukotriene A-4 hydrolase (LTA4H) | 3U9W | 5.57 | |
Target general information Gen name LTA4H Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Leukotriene A4 hydrolase; Leukotriene A(4)Leukotriene A-4 hydrolase hydrolase; Leukotriene A(4) hydrolase; LTA4; LTA-H; LTA-4hydrolase; LTA-4 hydrolase Protein family Peptidase M1 family Biochemical class Ether bond hydrolase Function Has also aminopeptidase activity. Epoxide hydrolase that catalyzes the final step in the biosynthesis of the proinflammatory mediator leukotriene B4. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07102; DB06917; DB07258; DB07094; DB07259; DB02352; DB07292; DB07104; DB06828; DB08466; DB01197; DB05177; DB03366; DB08040; DB06851; DB02062; DB07099; DB07260; DB07196; DB11781; DB03424; DB07237 Interacts with Q9BSI4 EC number EC 3.3.2.6 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Hydrolase; Leukotriene biosynthesis; Lipid metabolism; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 68927 Length 608 Aromaticity 0.1 Instability index 38.84 Isoelectric point 5.87 Charge (pH=7) -9.86 2D Binding mode Binding energy (Kcal/mol) -7.6 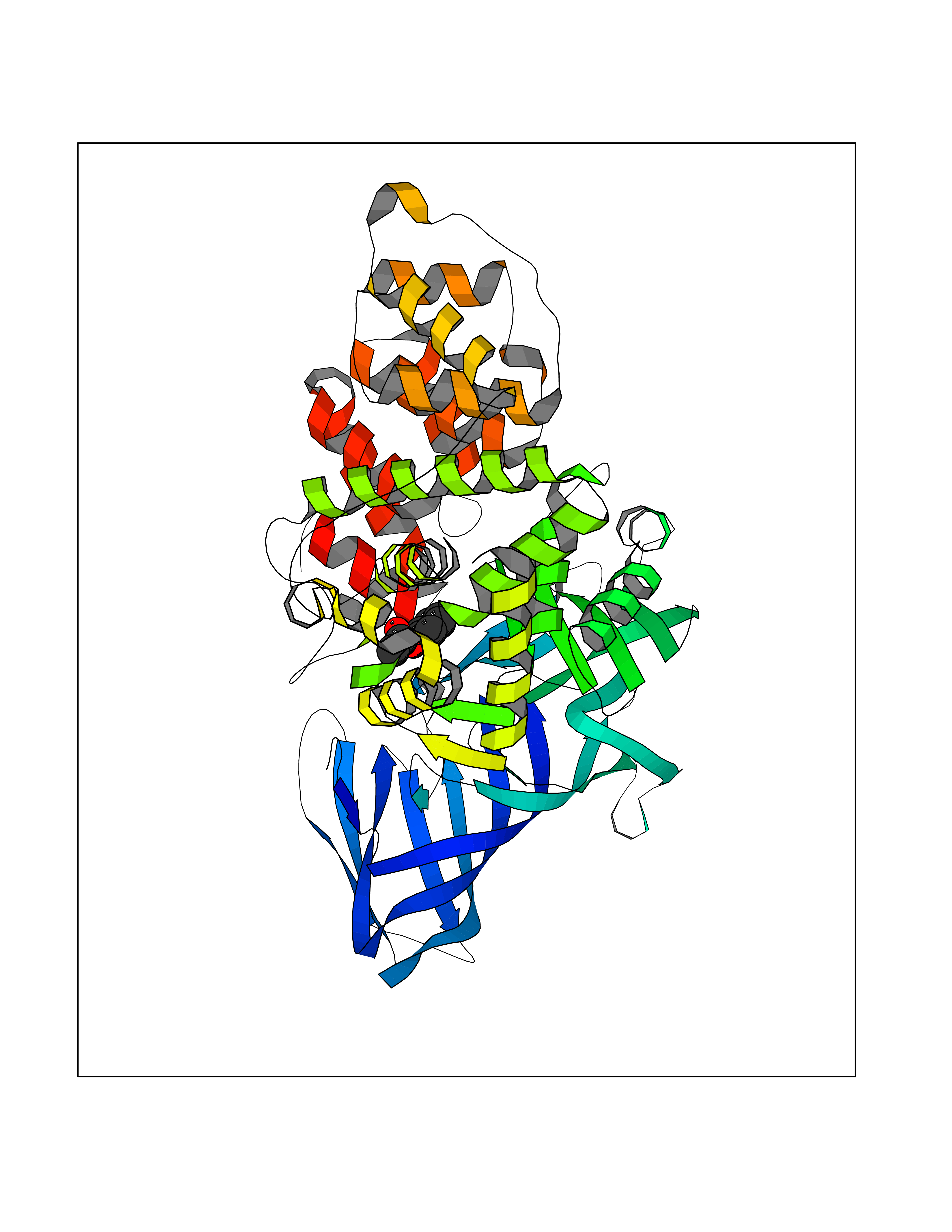 Molscript Map 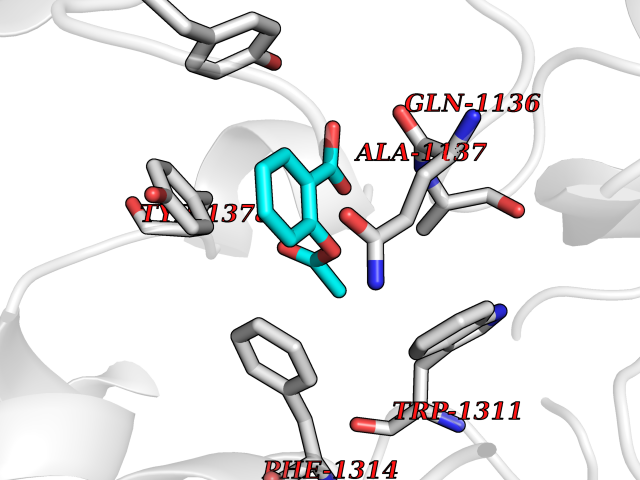 Pymol Map 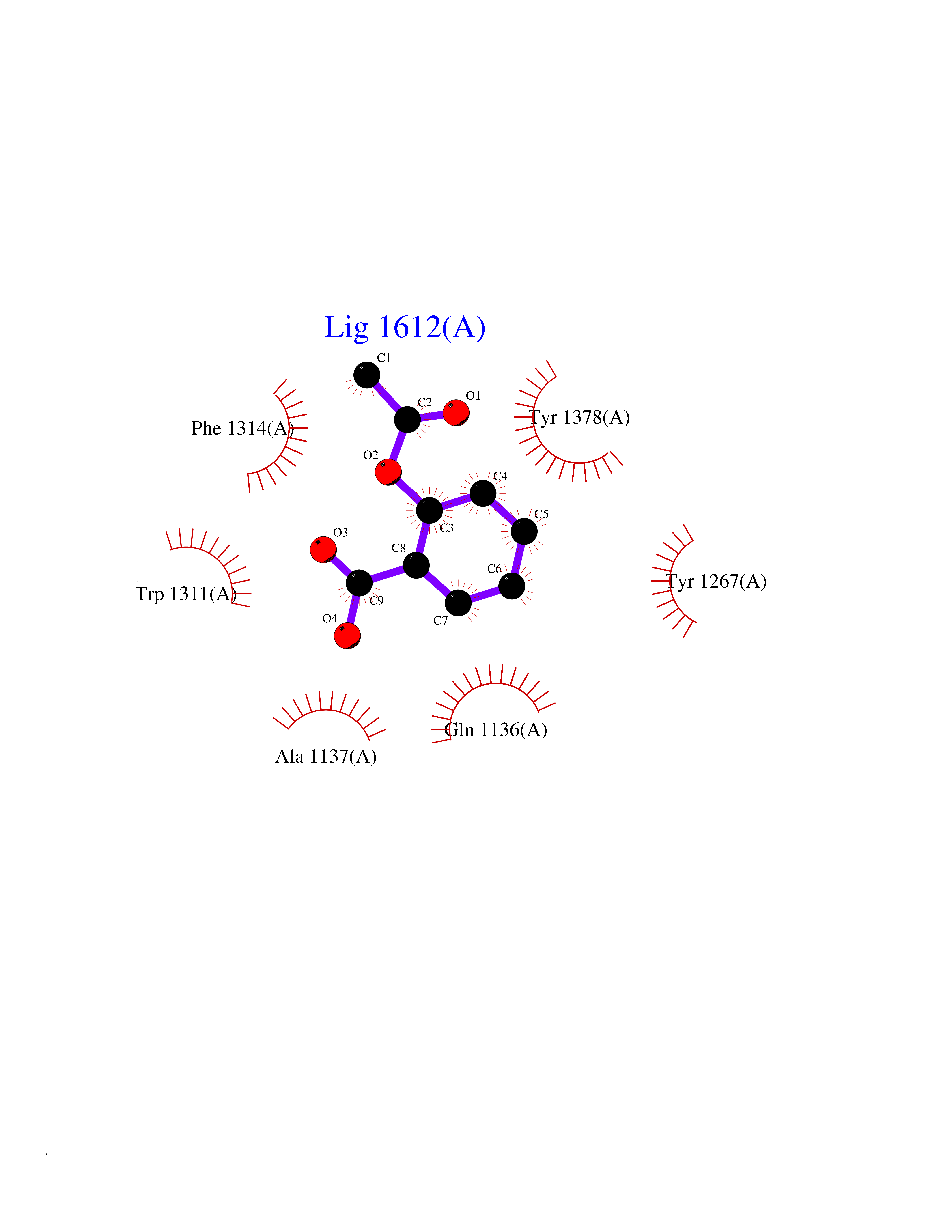 Ligplot Map 3D Binding mode Sequence IVDTCSLASPASVCRTKHLHLRCSVDFTRRTLTGTAALTVQSQEDNLRSLVLDTKDLTIEKVVINGQEVKYALGERQSYKGSPMEISLPIALSKNQEIVIEISFETSPKSSALQWLTPEQTSGKEHPYLFSQCQAIHCRAILPCQDTPSVKLTYTAEVSVPKELVALMSAIRDGETPDPEDPSRKIYKFIQKVPIPCYLIALVVGALESRQIGPRTLVWSEKEQVEKSAYEFSETESMLKIAEDLGGPYVWGQYDLLVLPPSFPYGGMENPCLTFVTPTLLAGDKSLSNVIAHEISHSWTGNLVTNKTWDHFWLNEGHTVYLERHICGRLFGEKFRHFNALGGWGELQNSVKTFGETHPFTKLVVDLTDIDPDVAYSSVPYEKGFALLFYLEQLLGGPEIFLGFLKAYVEKFSYKSITTDDWKDFLYSYFKDKVDVLNQVDWNAWLYSPGLPPIKPNYDMTLTNACIALSQRWITAKEDDLNSFNATDLKDLSSHQLNEFLAQTLQRAPLPLGHIKRMQEVYNFNAINNSEIRFRWLRLCIQSKWEDAIPLALKMATEQGRMKFTRPLFKDLAAFDKSHDQAVRTYQEHKASMHPVTAMLVGKDLKVD Hydrogen bonds contact Hydrophobic contact | ||||