Job Results:
Ligand
Structure
Job ID
8adf9aabe9fa12b13024fa8d07ca9b9b
Job name
NA
Time
2026-01-21 12:38:59
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Dihydroorotate dehydrogenase (DHODH) | 4OQV | 6.47 | |
Target general information Gen name DHODH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dihydroorotate oxidase; Dihydroorotate dehydrogenase (quinone), mitochondrial; DHOdehase; DHODH Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Postaxial acrofacial dysostosis (POADS) [MIM:263750]: POADS is characterized by severe micrognathia, cleft lip and/or palate, hypoplasia or aplasia of the posterior elements of the limbs, coloboma of the eyelids and supernumerary nipples. POADS is a very rare disorder: only 2 multiplex families, each consisting of 2 affected siblings born to unaffected, nonconsanguineous parents, have been described among a total of around 30 reported cases. {ECO:0000269|PubMed:19915526}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07559; DB07561; DB08172; DB08169; DB07443; DB07978; DB07975; DB04281; DB08249; DB07977; DB07976; DB04583; DB08008; DB01117; DB03523; DB03480; DB02613; DB04147; DB03247; DB01097; DB06481; DB08006; DB02262; DB05125; DB08880; DB07646 Interacts with Q6ZMZ0; P49638 EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Disease variant; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Proteomics identification; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 38341.4 Length 353 Aromaticity 0.05 Instability index 39.27 Isoelectric point 9.28 Charge (pH=7) 5.52 2D Binding mode Binding energy (Kcal/mol) -8.82  Molscript Map 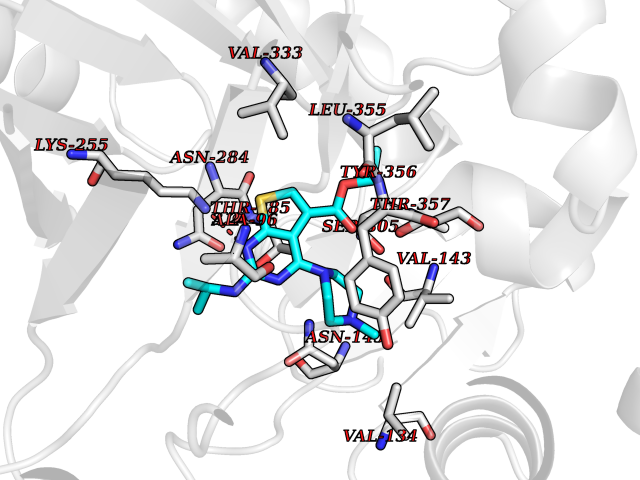 Pymol Map 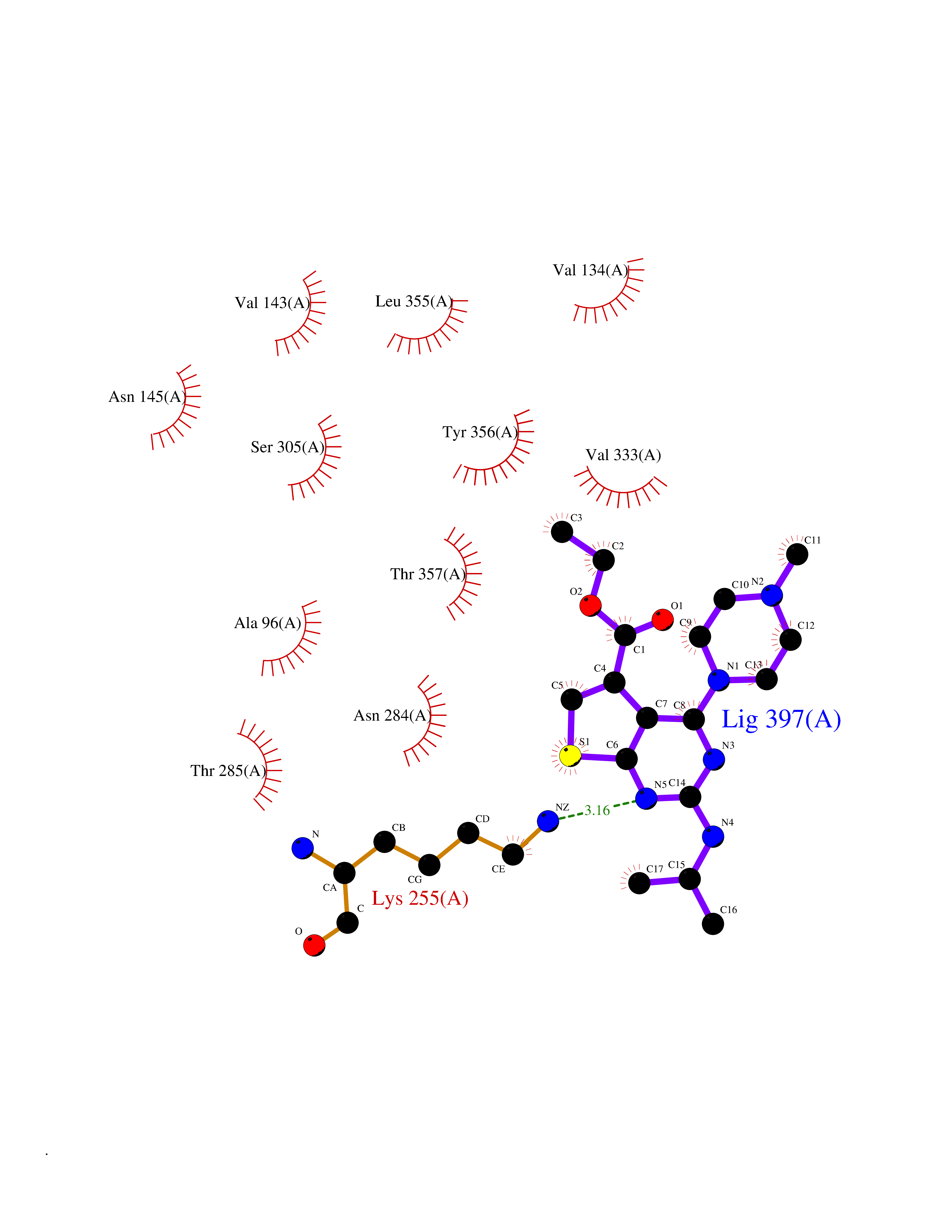 Ligplot Map 3D Binding mode Sequence DERFYAEHLMPTLQGLLDPESAHRLAVRFTSLGLLPRARFQDSDMLEVRVLGHKFRNPVGIAAGFDKHGEAVDGLYKMGFGFVEIGSVTPKPQEGNPRPRVFRLPEDQAVINRYGFNSHGLSVVEHRLRARQQKQAKLTEDGLPLGVNLGKNKTSVDAAEDYAEGVRVLGPLADYLVVNVSSPGKAELRRLLTKVLQERDGLRRVHRPAVLVKIAPDLTSQDKEDIASVVKELGIDGLIVTNTTVSRPAGLQGALRSETGGLSGKPLRDLSTQTIREMYALTQGRVPIIGVGGVSSGQDALEKIRAGASLVQLYTALTFWGPPVVGKVKRELEALLKEQGFGGVTDAIGADHR Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Cannabinoid receptor 1 (CB1) | 5U09 | 6.46 | |
Target general information Gen name CNR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Cannabinoid CB1 receptor; CNR; CB-R; CANN6 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Mediates many cannabinoid-induced effects, acting, among others, on food intake, memory loss, gastrointestinal motility, catalepsy, ambulatory activity, anxiety, chronic pain. Signaling typically involves reduction in cyclic AMP. In the hypothalamus, may have a dual effect on mitochondrial respiration depending upon the agonist dose and possibly upon the cell type. Increases respiration at low doses, while decreases respiration at high doses. At high doses, CNR1 signal transduction involves G-protein alpha-i protein activation and subsequent inhibition of mitochondrial soluble adenylate cyclase, decrease in cyclic AMP concentration, inhibition of protein kinase A (PKA)-dependent phosphorylation of specific subunits of the mitochondrial electron transport system, including NDUFS2. In the hypothalamus, inhibits leptin-induced reactive oxygen species (ROS) formation and mediates cannabinoid-induced increase in SREBF1 and FASN gene expression. In response to cannabinoids, drives the release of orexigenic beta-endorphin, but not that of melanocyte-stimulating hormone alpha/alpha-MSH, from hypothalamic POMC neurons, hence promoting food intake. In the hippocampus, regulates cellular respiration and energy production in response to cannabinoids. Involved in cannabinoid-dependent depolarization-induced suppression of inhibition (DSI), a process in which depolarization of CA1 postsynaptic pyramidal neurons mobilizes eCBs, which retrogradely activate presynaptic CB1 receptors, transiently decreasing GABAergic inhibitory neurotransmission. Also reduces excitatory synaptic transmission. In superior cervical ganglions and cerebral vascular smooth muscle cells, inhibits voltage-gated Ca(2+) channels in a constitutive, as well as agonist-dependent manner. In cerebral vascular smooth muscle cells, cannabinoid-induced inhibition of voltage-gated Ca(2+) channels leads to vasodilation and decreased vascular tone. Induces leptin production in adipocytes and reduces LRP2-mediated leptin clearance in the kidney, hence participating in hyperleptinemia. In adipose tissue, CNR1 signaling leads to increased expression of SREBF1, ACACA and FASN genes. In the liver, activation by endocannabinoids leads to increased de novo lipogenesis and reduced fatty acid catabolism, associated with increased expression of SREBF1/SREBP-1, GCK, ACACA, ACACB and FASN genes. May also affect de novo cholesterol synthesis and HDL-cholesteryl ether uptake. Peripherally modulates energy metabolism. In high carbohydrate diet-induced obesity, may decrease the expression of mitochondrial dihydrolipoyl dehydrogenase/DLD in striated muscles, as well as that of selected glucose/ pyruvate metabolic enzymes, hence affecting energy expenditure through mitochondrial metabolism. In response to cannabinoid anandamide, elicits a proinflammatory response in macrophages, which involves NLRP3 inflammasome activation and IL1B and IL18 secretion. In macrophages infiltrating pancreatic islets, this process may participate in the progression of type-2 diabetes and associated loss of pancreatic beta-cells. G-protein coupled receptor for endogenous cannabinoids (eCBs), including N-arachidonoylethanolamide (also called anandamide or AEA) and 2-arachidonoylglycerol (2-AG), as well as phytocannabinoids, such as delta(9)-tetrahydrocannabinol (THC). Related diseases Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:18177726}. The protein represented in this entry may be involved in disease pathogenesis. May contribute to the development of diet-induced obesity and several obesity-associated features, such as dyslipidemia and liver steatosis, regulating peripheral lipogenesis, energy expenditure and feeding behavior. CNR1 inverse agonists have been shown to reduce body weight and improve metabolic abnormalities in obese subjects, although adverse neuropsychiatric effects, including anxiety, irritability, and depressed mood, halted their therapeutic development (PubMed:18177726). In obese mice, peripherally restricted CNR1 inverse agonists have been shown to normalize metabolic abnormalities, including insulin resistance and fatty liver, and to reverse leptin resistance. {ECO:0000269|PubMed:18177726}.; DISEASE: Dysfunction of the endogenous cannabinoid system including CNR1 has been implicated in the pathogenesis of a number of central nervous system disorders, including Huntington disease, Parkinson disease, and Alzheimer disease (PubMed:32549916). In post-mortem brains from Huntington disease patients, a progressive CNR1 loss has been observed in the caudate nucleus, putamen, and substantia nigra pars reticulata, and altered expression and abnormal endocannabinoid levels precede motor symptoms in a disease mouse model (PubMed:10828533, PubMed:19524019, PubMed:8255419). In Parkinson disease, low CNR1 expression in mid-superior frontal gyrus and mid-cingulate cortex has been associated with poor mind, poor executive functioning and poor episode memory, while patients with more severe visuospatial dysfunction showed decreased receptor availability in the precuneus, mid-cingulate, supplementary motor cortex, inferior orbitofrontal gyrus and thalamus (PubMed:31342135). In an animal model for Alzheimer disease, CNR1 heterozygous deletion has been associated with decreased levels of postsynaptic density protein 95 (DLG4/PSD95) and accelerated memory impairment, suggesting synaptic dysfunction and a crucial role for CNR1 in the progression of disease symptoms (PubMed:10828533, PubMed:19524019, PubMed:30096288, PubMed:31342135, PubMed:8255419). {ECO:0000269|PubMed:10828533, ECO:0000269|PubMed:19524019, ECO:0000269|PubMed:30096288, ECO:0000269|PubMed:31342135, ECO:0000269|PubMed:32549916, ECO:0000269|PubMed:8255419}. Drugs (DrugBank ID) DB05750; DB09061; DB00470; DB14009; DB00486; DB14011; DB11745; DB09288; DB02955; DB06155; DB05077; DB11755; DB05201 Interacts with P29274; P21554 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Cell projection; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Neurodegeneration; Obesity; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 32070.3 Length 282 Aromaticity 0.13 Instability index 40.15 Isoelectric point 9.16 Charge (pH=7) 9.36 2D Binding mode Binding energy (Kcal/mol) -8.81 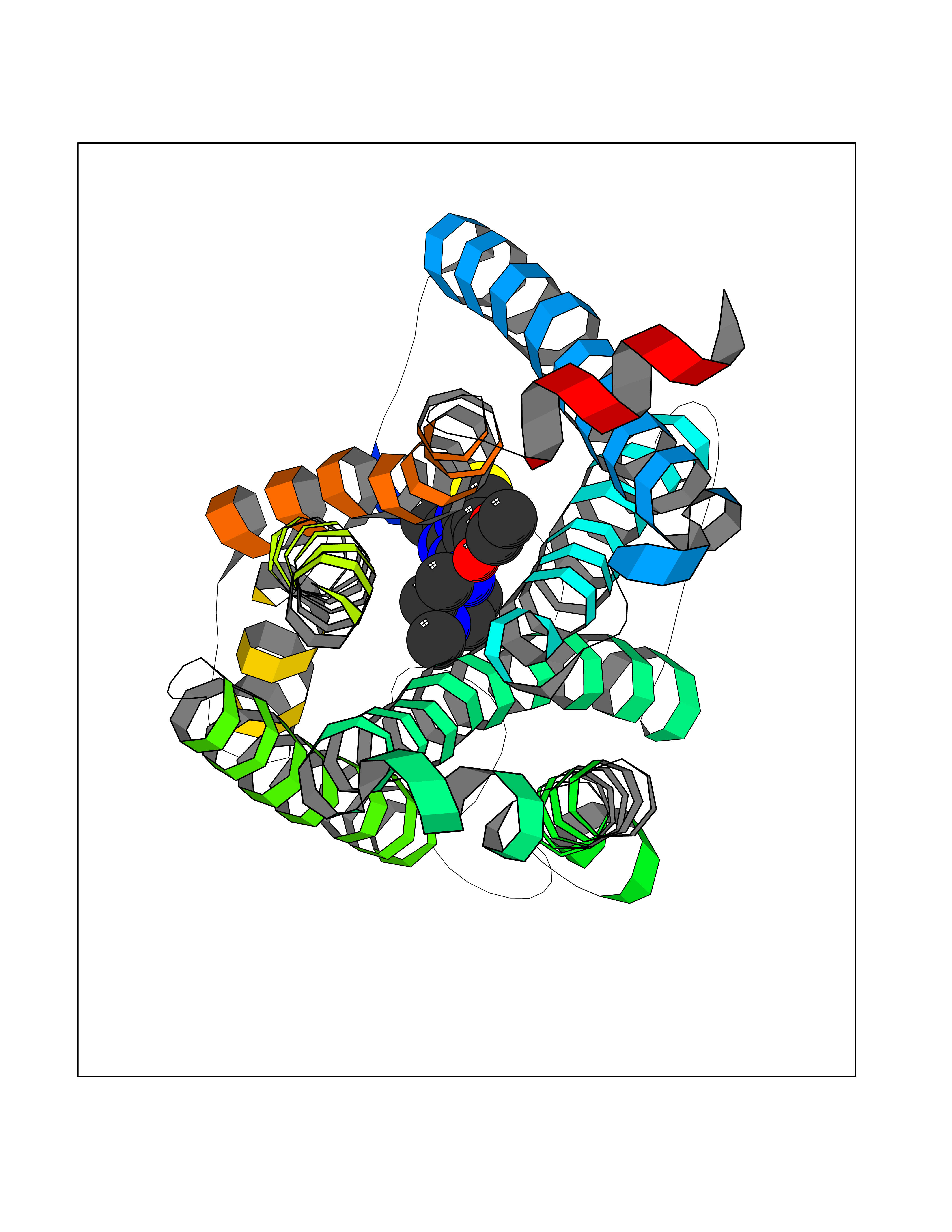 Molscript Map 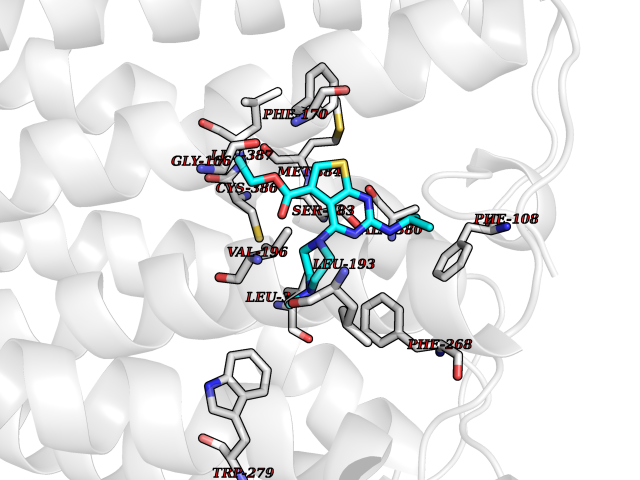 Pymol Map 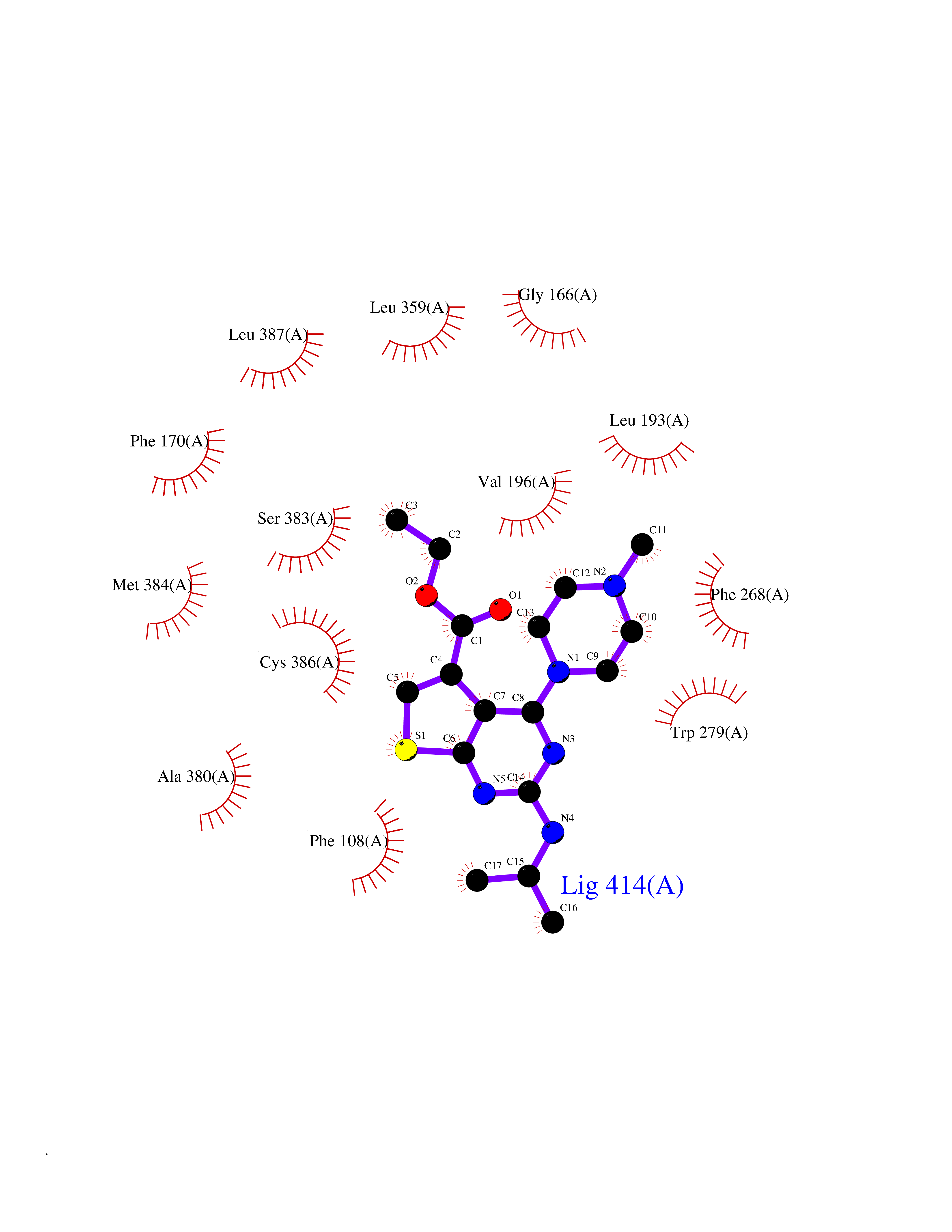 Ligplot Map 3D Binding mode Sequence ENFMDIECFMVLNPSQQLAIAVLSLTLGTFTVLENLLVLCVILHSRSLRCRPSYHFIGSLAVADLLGSVIFVYSFIDFHVFHRKDSRNVFLFKLGGVTASFTASVGSLFLAAIDRYISIHRPLAYKRIVTRPKAVVAFCLMWTIAIVIAVLPLLGWNCEKLQSVCSDIFPHIDETYLMFWIGVTSVLLLFIVYAYMYILWKADQARMDIRLAKTLVLILVVLIICWGPLLAIMVYDVFGKMNKLIKTVFAFCSMLCLLNSTVNPIIYALRSKDLRHAFRSMF Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Ribonucleoside-diphosphate reductase large subunit | 2WGH | 6.46 | |
Target general information Gen name RRM1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms RR1 Protein family Ribonucleoside diphosphate reductase large chain family Biochemical class Oxidoreductase Function ATP binding.Identical protein binding.Ribonucleoside-diphosphate reductase activity, thioredoxin disulfide as acceptor. Related diseases Progressive external ophthalmoplegia with mitochondrial DNA deletions, autosomal recessive 6 (PEOB6) [MIM:620647]: A form of progressive external ophthalmoplegia, a mitochondrial myopathy characterized by progressive paralysis of the levator palpebrae, orbicularis oculi, and extraocular muscles. Ragged red fibers are seen on muscle biopsy. {ECO:0000269|PubMed:35617047}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00242; DB00631; DB01073; DB05420; DB00441; DB01005; DB05003; DB01280; DB06433 Interacts with P23921; P31350; Q8N720 EC number 1.17.4.1 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; ATP-binding; Cytoplasm; Deoxyribonucleotide synthesis; Disulfide bond; Nucleotide-binding; Oxidoreductase; Phosphoprotein; Primary mitochondrial disease; Progressive external ophthalmoplegia; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 147968 Length 1301 Aromaticity 0.1 Instability index 43.58 Isoelectric point 7.14 Charge (pH=7) 0.86 2D Binding mode Binding energy (Kcal/mol) -8.81 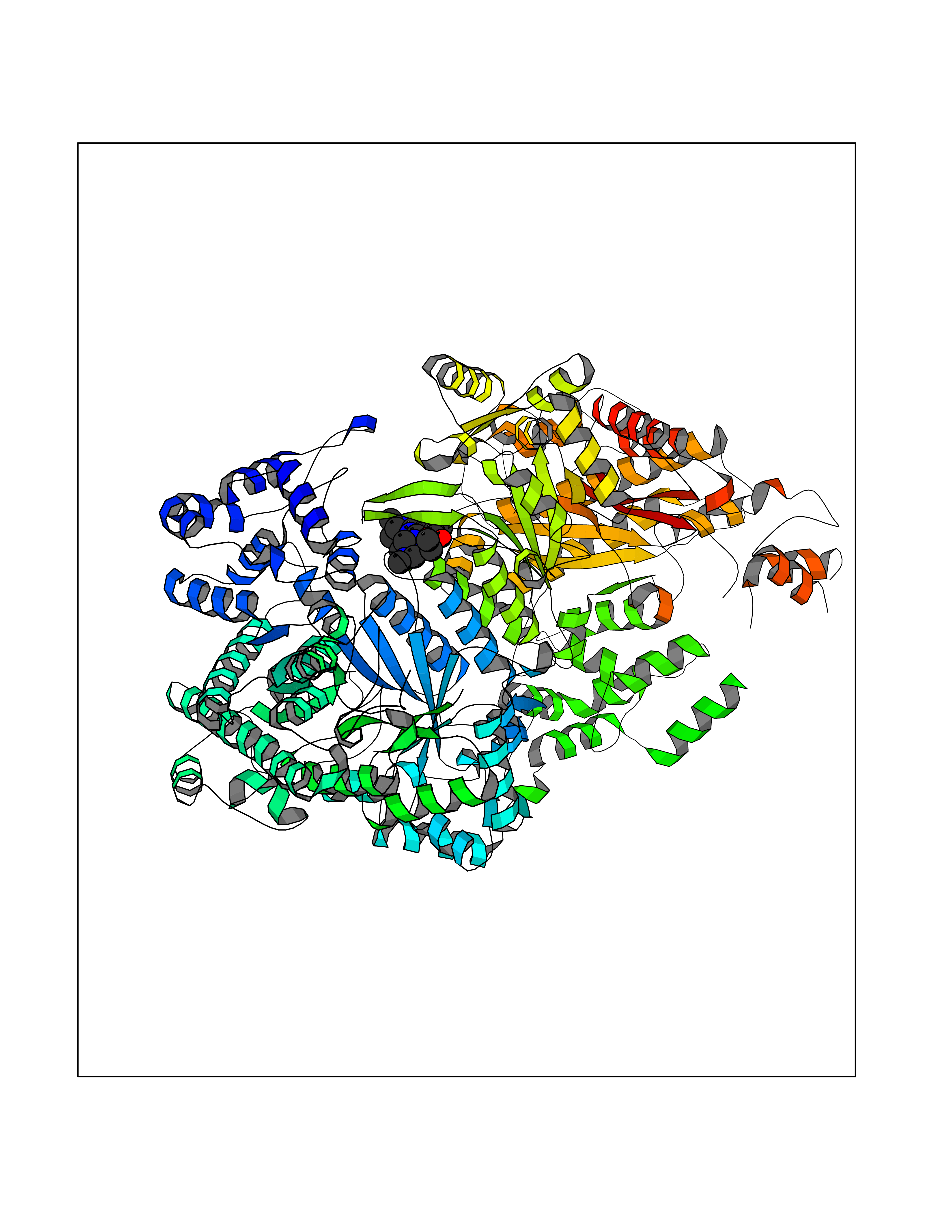 Molscript Map 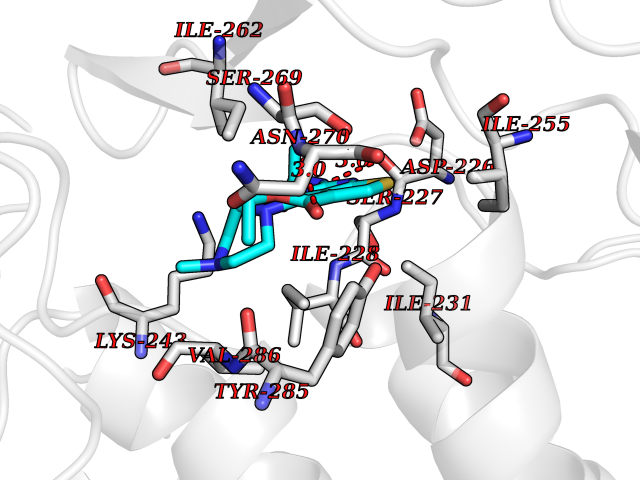 Pymol Map  Ligplot Map 3D Binding mode Sequence LAARIAVSNLHKETKKVFSDVMEDLYNYINPHNGKHSPMVAKSTLDIVLANKDRLNSAIIYDRDFSYNYFGFKTLERSYLLKINGKVAERPQHMLMRVSVGIHKEDIDAAIETYNLLSERWFTHASPTLFNAGTNRPQLSSCFLLSMKDDSIEGIYDTLKQCALISKSAGGIGVAVSCIRATGSYIAGTNGNSNGLVPMLRVYNNTARYVDQGGNKRPGAFAIYLEPWHLDIFEFLDLKKNTGKEEQRARDLFFALWIPDLFMKRVETNQDWSLMCPNECPGLDEVWGEEFEKLYASYEKQGRVRKVVKAQQLWYAIIESQTETGTPYMLYKDSCNRKSNQQNLGTIKCSNLCTEIVEYTSKDEVAVCNLASLALNMYVTSEHTYDFKKLAEVTKVVVRNLNKIIDINYYPVPEACLSNKRHRPIGIGVQGLADAFILMRYPFESAEAQLLNKQIFETIYYGALEASCDLAKEQGPYETYEGSPVSKGILQYDMWNVTPTDLWDWKVLKEKIAKYGIRNSLLIAPMPTASTAQILGNNESIEPYTSNIYQIVNPHLLKDLTERGLGSIQSIPEIPDDLKQLYKTVWEISQKTVLKMAAERGAFIDQSQSLNIHIAEPNYGKLTSMHFYGWKQGLKTGMYYLRTRAARIAVSNLHKETKKVFSDVMEDLYNYINPHNGKHSPMVAKSTLDIVLANKDRLNSAIIYDRDFSYNYFGFKTLERSYLLKINGKVAERPQHMLMRVSVGIHKEDIDAAIETYNLLSERWFTHASPTLFNAGTNRPQLSSCFLLSMKDDSIEGIYDTLKQCALISKSAGGIGVAVSCIRATGSYIAGTNGNSNGLVPMLRVYNNTARYVDQGGNKRPGAFAIYLEPWHLDIFEFLDLKKNTGKEEQRARDLFFALWIPDLFMKRVETNQDWSLMCPNECPGLDEVWGEEFEKLYASYEKQGRVRKVVKAQQLWYAIIESQTETGTPYMLYKDSCNRKSNQQNLGTIKCSNLCTEIVEYTSKDEVAVCNLASLALNMYVTSEHTYDFKKLAEVTKVVVRNLNKIIDINYYPVPEACLSNKRHRPIGIGVQGLADAFILMRYPFESAEAQLLNKQIFETIYYGALEASCDLAKEQGPYETYEGSPVSKGILQYDMWNVTPTDLWDWKVLKEKIAKYGIRNSLLIAPMPTASTAQILGNNESIEPYTSNIYQIVNPHLLKDLTERGLWEEMKNQIIACNGSIQSIPEIPDDLKQLYKTVWEISQKTVLKMAAERGAFIDQSQSLNIHIAEPNYGKLTSMHFYGWKQGLKTGMYYLRTRAH Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Dipeptidyl peptidase 8 (DPP-8) | 6EOP | 6.46 | |
Target general information Gen name DPP8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Prolyl dipeptidase DPP8; MSTP141; MSTP135; MSTP097; Dipeptidyl peptidase VIII; Dipeptidyl peptidase IV-related protein 1; DPRP1; DPRP-1; DPP VIII; DP8 Protein family Peptidase S9B family, DPPIV subfamily Biochemical class Peptidase Function Dipeptidyl peptidase that cleaves off N-terminal dipeptides from proteins having a Pro or Ala residue at position 2. Related diseases Orotic aciduria 1 (ORAC1) [MIM:258900]: A disorder of pyrimidine metabolism resulting in megaloblastic anemia and orotic acid crystalluria that is frequently associated with some degree of physical and intellectual disability. A minority of cases have additional features, particularly congenital malformations and immune deficiencies. {ECO:0000269|PubMed:9042911}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 3.4.14.5 Uniprot keywords 3D-structure; Alternative splicing; Aminopeptidase; Apoptosis; Cytoplasm; Hydrolase; Protease; Proteomics identification; Reference proteome; Serine protease Protein physicochemical properties Chain ID A,D Molecular weight (Da) 97764.9 Length 849 Aromaticity 0.12 Instability index 47.71 Isoelectric point 5.69 Charge (pH=7) -21.66 2D Binding mode Binding energy (Kcal/mol) -8.81  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence LEPFYVERYSWSQLKKLLADTRKYHGYMMAKAPHDFMFVKRNDPDGPHSDRIYYLAMSNRENTLFYSEIPKTINRAAVLMLSWKPLLDLFQYSREEELLRERKRIGTVGIASYDYHQGSGTFLFQAGSGIYHVKDGGPQGFTQQPLRPNLVETSCPNIRMDPKLCPADPDWIAFIHSNDIWISNIVTREERRLTYVHNELANMEEDARSAGVATFVLQEEFDRYSGYWWCPKAETTPSGGKILRILYEENDESEVEIIHVTSPMLETRRADSFRYPKTGTANPKVTFKMSEIMIDAEGRIIDVIDKELIQPFEILFEGVEYIARAGWTPEGKYAWSILLDRSQTRLQIVLISPELFIPVEDDVMERQRLIESVPDSVTPLIIYEETTDIWINIHDIFHVFPQSHEEEIEFIFASECKTGFRHLYKITSILKESKYKRSSGGLPAPSDFKCPIKEEIAITSGEWEVLGRHGSNIQVDEVRRLVYFEGTKDSPLEHHLYVVSYVNPGEVTRLTDRGYSHSCCISQHCDFFISKYSNQKNPHCVSLYKLSSPEDDPTCKTKEFWATILDSAGPLPDYTPPEIFSFESTTGFTLYGMLYKPHDLQPGKKYPTVLFIYGGPQVQLVNNRFKGVKYFRLNTLASLGYVVVVIDNRGSXHRGLKFEGAFKYKMGQIEIDDQVEGLQYLASRYDFIDLDRVGIHGWSYGGYLSLMALMQRSDIFRVAIAGAPVTLWIFYDTGYTERYMGHPDQNEQGYYLGSVAMQAEKFPSEPNRLLLLHGFLDENVHFAHTSILLSFLVRAGKPYDLQIYPQERHSIRVPESGEHYELHLLHYLQENLGSRIAALKVSLRFLYEG Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Protein arginine methyltransferase 5 (PRMT5) | 7MXC | 6.46 | |
Target general information Gen name PRMT5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Shk1 kinase-binding protein 1 homolog; SKB1Hs; SKB1 homolog; SKB1; Protein arginine N-methyltransferase 5; Jak-binding protein 1; JBP1; IBP72; Histone-arginine N-methyltransferase PRMT5; HRMT1L5; 72 k Protein family Class I-like SAM-binding methyltransferase superfamily, Protein arginine N-methyltransferase family Biochemical class Methyltransferase Function Specifically mediates the symmetrical dimethylation of arginine residues in the small nuclear ribonucleoproteins Sm D1 (SNRPD1) and Sm D3 (SNRPD3); such methylation being required for the assembly and biogenesis of snRNP core particles. Methylates SUPT5H and may regulate its transcriptional elongation properties. Mono- and dimethylates arginine residues of myelin basic protein (MBP) in vitro. May play a role in cytokine-activated transduction pathways. Negatively regulates cyclin E1 promoter activity and cellular proliferation. Methylates histone H2A and H4 'Arg-3' during germ cell development. Methylates histone H3 'Arg-8', which may repress transcription. Methylates the Piwi proteins (PIWIL1, PIWIL2 and PIWIL4), methylation of Piwi proteins being required for the interaction with Tudor domain-containing proteins and subsequent localization to the meiotic nuage. Methylates RPS10. Attenuates EGF signaling through the MAPK1/MAPK3 pathway acting at 2 levels. First, monomethylates EGFR; this enhances EGFR 'Tyr-1197' phosphorylation and PTPN6 recruitment, eventually leading to reduced SOS1 phosphorylation. Second, methylates RAF1 and probably BRAF, hence destabilizing these 2 signaling proteins and reducing their catalytic activity. Required for induction of E-selectin and VCAM-1, on the endothelial cells surface at sites of inflammation. Methylates HOXA9. Methylates and regulates SRGAP2 which is involved in cell migration and differentiation. Acts as a transcriptional corepressor in CRY1-mediated repression of the core circadian component PER1 by regulating the H4R3 dimethylation at the PER1 promoter. Methylates GM130/GOLGA2, regulating Golgi ribbon formation. Methylates H4R3 in genes involved in glioblastomagenesis in a CHTOP- and/or TET1-dependent manner. Symmetrically methylates POLR2A, a modification that allows the recruitment to POLR2A of proteins including SMN1/SMN2 and SETX. This is required for resolving RNA-DNA hybrids created by RNA polymerase II, that form R-loop in transcription terminal regions, an important step in proper transcription termination. Along with LYAR, binds the promoter of gamma-globin HBG1/HBG2 and represses its expression. Symmetrically methylates NCL. Methylates TP53; methylation might possibly affect TP53 target gene specificity. Arginine methyltransferase that can both catalyze the formation of omega-N monomethylarginine (MMA) and symmetrical dimethylarginine (sDMA), with a preference for the formation of MMA. Related diseases Epilepsy, nocturnal frontal lobe, 3 (ENFL3) [MIM:605375]: An autosomal dominant focal epilepsy characterized by nocturnal seizures with hyperkinetic automatisms and poorly organized stereotyped movements. {ECO:0000269|PubMed:11062464, ECO:0000269|PubMed:11104662}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P01019; Q9NX04; Q8WUW1; Q08289; P78371; Q16543; Q8N8U2; P54105; P21964-2; Q9NQ92; Q16526; Q9Y6K1; Q01094; Q08426; P38919; Q14241; O15197-2; Q6ZV65; P01100; O95995; P62993; Q8TE85; Q9BX10; P62805; P31269; Q00613; Q63ZY3; P03952; Q8TBB1; P06858; Q86UQ8-1; Q96HA8; Q8WVJ2; P24928; O14744; Q86U06; Q9BRS2; O75044; Q96RU7; P31930; P40337-2; Q9BQA1; P63104; Q96E35; Q91XC0; P03418; Q6ZV65-2 EC number EC 2.1.1.320 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Biological rhythms; Chromatin regulator; Chromosome; Cytoplasm; Direct protein sequencing; Golgi apparatus; Methyltransferase; Nucleus; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 71188.3 Length 621 Aromaticity 0.11 Instability index 44.6 Isoelectric point 5.95 Charge (pH=7) -9.68 2D Binding mode Binding energy (Kcal/mol) -8.81 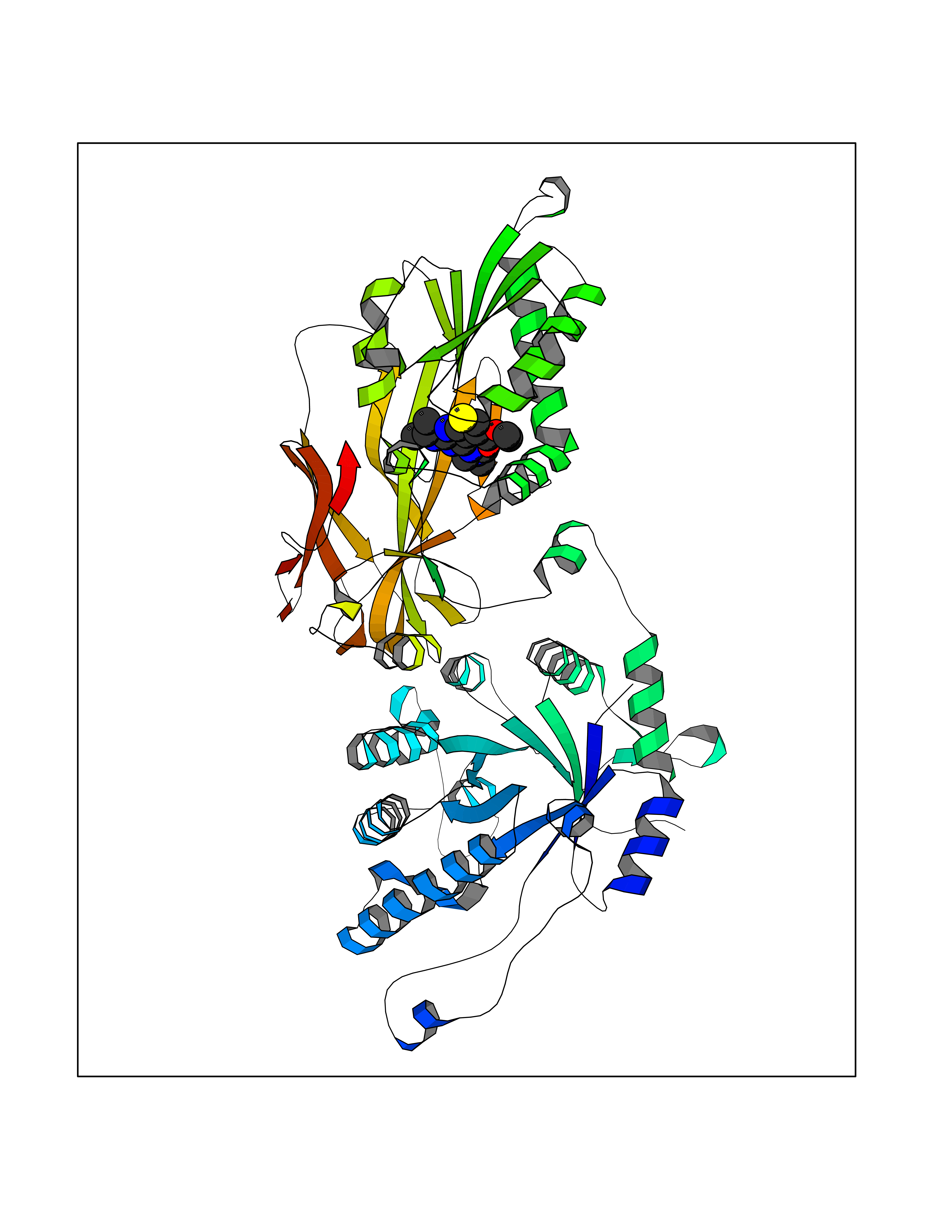 Molscript Map 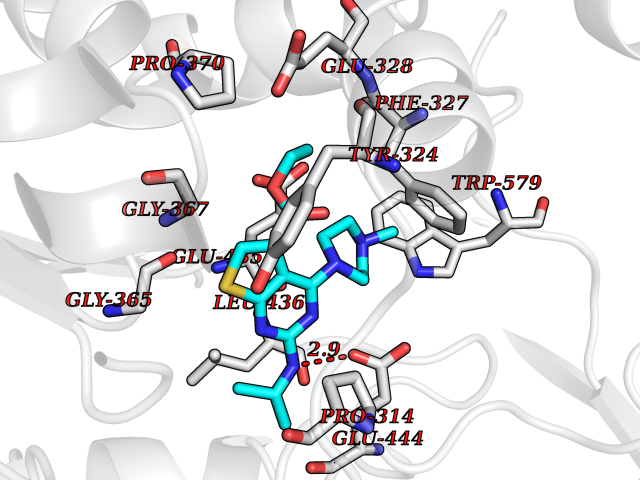 Pymol Map 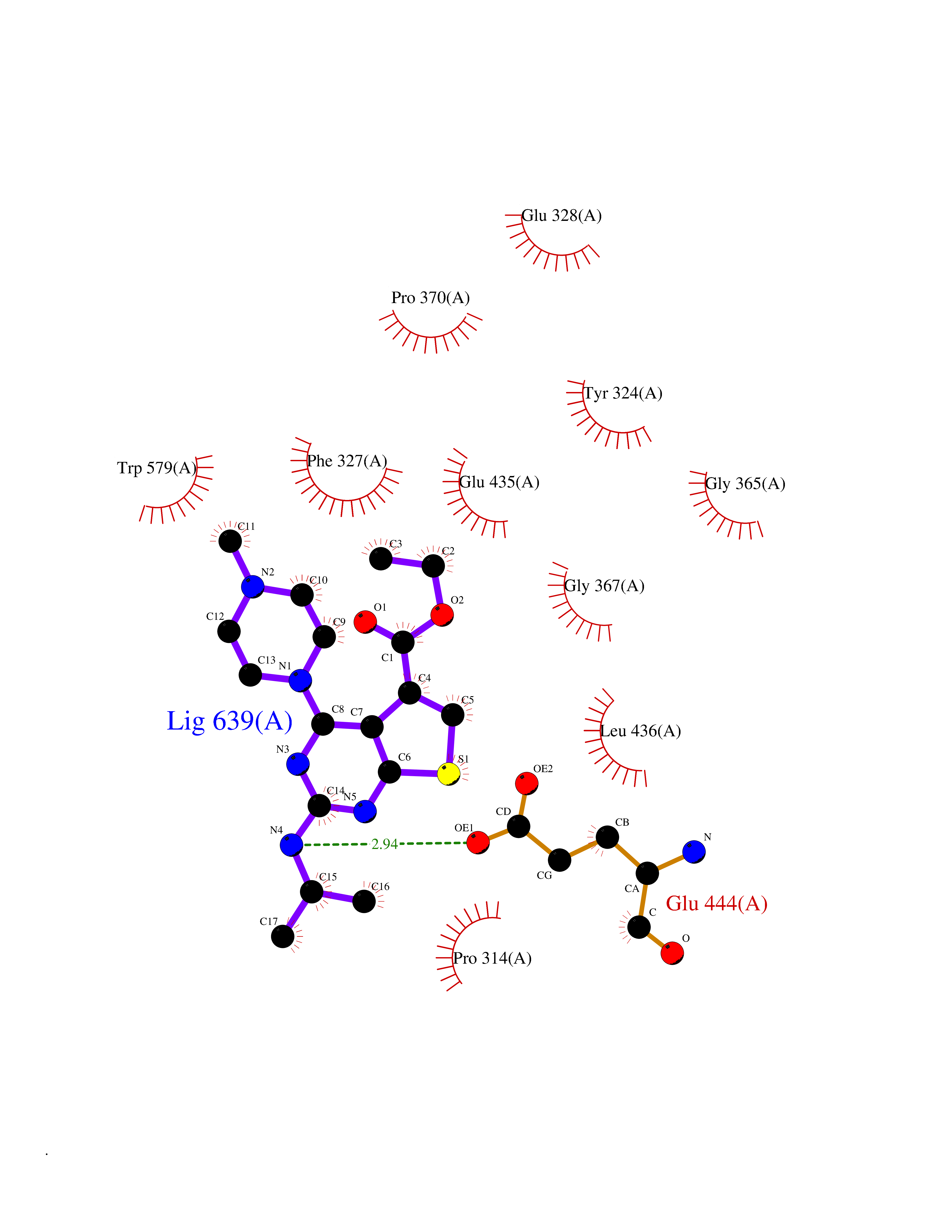 Ligplot Map 3D Binding mode Sequence RVSSGRDLNCVPEIADTLGAVAKQGFDFLCMPVFHPRFKREFIQEPAKNRPGPQTRSDLLLSGRDWNTLIVGKLSPWIRPDSKVEKIRRNSEAAMLQELNFGAYLGLPAFLLPLNQEDNTNLARVLTNHIHTGHHSSMFWMRVPLVAPEDLRDDIIENAPTTHTEEYSGEEKTWMWWHNFRTLCDYSKRIAVALEIGADLPSNHVIDRWLGEPIKAAILPTSIFLTNKKGFPVLSKMHQRLIFRLLKLEVQFIITGTNHHSCSYLQYLEYLSQNRPPPNAYELFAKGYEDYLQSPLQPLMDNLESQTYEVFEKDPIKYSQYQQAIYKCLLDRVPEEEKDTNVQVLMVLGAGRGPLVNASLRAAKQADRRIKLYAVEKNPNAVVTLENWQFEEWGSQVTVVSSDMREWVAPEKADIIVSELLGSFADNELSPECLDGAQHFLKDDGVSIPGEYTSFLAPISSSKLYNEVRACREKDRDPEAQFEMPYVVRLHNFHQLSAPQPCFTFSHPNRDPMIDNNRYCTLEFPVEVNTVLHGFAGYFETVLYQDITLSIRPETHSPGMFSWFPILFPIKQPITVREGQTICVRFWRCSNSKKVWYEWAVTAPVCSAIHNPTGRSYTIGL Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Glycogen phosphorylase, liver form | 3DDS | 6.45 | |
Target general information Gen name PYGL Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Glycogen phosphorylase family Biochemical class Transferase Function AMP binding.ATP binding.Bile acid binding.Drug binding.Glucose binding.Glycogen phosphorylase activity.Linear malto-oligosaccharide phosphorylase activity.Protein homodimerization activity.Purine nucleobase binding.Pyridoxal phosphate binding.SHG alpha-glucan phosphorylase activity.Vitamin binding. Related diseases Glycogen storage disease 6 (GSD6) [MIM:232700]: A metabolic disorder characterized by mild to moderate hypoglycemia, mild ketosis, growth retardation, and prominent hepatomegaly. Heart and skeletal muscle are not affected. {ECO:0000269|PubMed:9529348}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P11216; P11217 EC number 2.4.1.1 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; Carbohydrate metabolism; Cytoplasm; Disease variant; Glycogen metabolism; Glycogen storage disease; Glycosyltransferase; Nucleotide-binding; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89481.9 Length 778 Aromaticity 0.11 Instability index 34.96 Isoelectric point 6.49 Charge (pH=7) -3.54 2D Binding mode Binding energy (Kcal/mol) -8.8 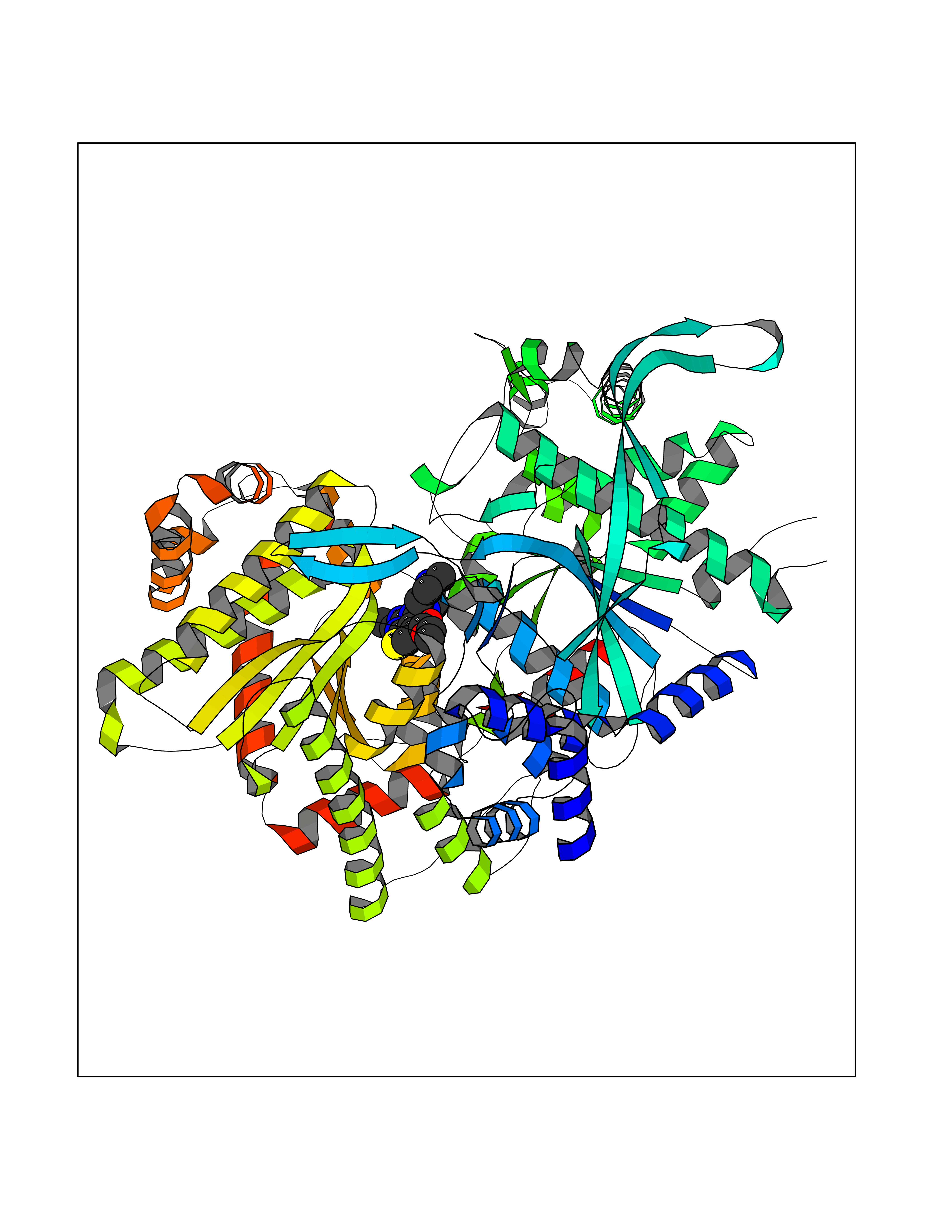 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence NVAELKKSFNRHLHFTLVKDRNVATTRDYYFALAHTVRDHLVGRWIRTQQHYYDKCPKRVYYLSLEFYMGRTLQNTMINLGLQNACDEAIYQLGLDIEELEEIEEDAGLGNGGLGRLAACFLDSMATLGLAAYGYGIRYEYGIFNQKIRDGWQVEEADDWLRYGNPWEKSRPEFMLPVHFYGKVEHTNTGTKWIDTQVVLALPYDTPVPGYMNNTVNTMRLWSENISRVLYPNDNFFEGKELRLKQEYFVVAATLQDIIRRFKASKFGSTGTVFDAFPDQVAIQLNDTHPALAIPELMRIFVDIEKLPWSKAWELTQKTFAYTNHTVLPEALERWPVDLVEKLLPRHLEIIYEINQKHLDRIVALFPKDVDRLRRMSLIEEEGSKRINMAHLCIVGSHAVNGVAKIHSDIVKTKVFKDFSELEPDKFQNKTNGITPRRWLLLCNPGLAELIAEKIGEDYVKDLSQLTKLHSFLGDDVFLRELAKVKQENKLKFSQFLETEYKVKINPSSMFDVQVKRIHEYKRQLLNCLHVITMYNRIKKDPKKLFVPRTVIIGGKAAPGYHMAKMIIKLITSVADVVNNDPMVGSKLKVIFLENYRVSLAEKVIPATDLSEQISTAGTEASGTGNMKFMLNGALTIGTMDGANVEMAEEAGEENLFIFGMRIDDVAALDKKGYEAKEYYEALPELKLVIDQIDNGFFSPKQPDLFKDIINMLFYHDRFKVFADYEAYVKCQDKVSQLYMNPKAWNTMVLKNIAASGKFSSDRTIKEYAQNIWNVEPS Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Cholesterol oxidase | 1COY | 6.45 | |
Target general information Gen name choB Organism Brevibacterium sterolicum Uniprot ID TTD ID NA Synonyms NA Protein family GMC oxidoreductase family Biochemical class Oxidoreductase(oxygen receptor) Function Cholesterol oxidase activity.Flavin adenine dinucleotide binding.Steroid delta-isomerase activity. Related diseases Achondroplasia (ACH) [MIM:100800]: A frequent form of short-limb dwarfism. It is characterized by a long, narrow trunk, short extremities, particularly in the proximal (rhizomelic) segments, a large head with frontal bossing, hypoplasia of the midface and a trident configuration of the hands. ACH is an autosomal dominant disease. {ECO:0000269|PubMed:10611230, ECO:0000269|PubMed:12297284, ECO:0000269|PubMed:7758520, ECO:0000269|PubMed:7847369, ECO:0000269|PubMed:8078586, ECO:0000269|PubMed:8599935}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Crouzon syndrome with acanthosis nigricans (CAN) [MIM:612247]: Classic Crouzon disease which is caused by mutations in the FGFR2 gene is characterized by craniosynostosis (premature fusion of the skull sutures), and facial hypoplasia. Crouzon syndrome with acanthosis nigricans (a skin disorder characterized by pigmentation anomalies), CAN, is considered to be an independent disorder from classic Crouzon syndrome. CAN is characterized by additional more severe physical manifestation, such as Chiari malformation, hydrocephalus, and atresia or stenosis of the choanas, and is caused by a specific mutation (Ala-391 to Glu) in the transmembrane domain of FGFR3. It is proposed to have an autosomal dominant mode of inheritance. {ECO:0000269|PubMed:17935505, ECO:0000269|PubMed:7493034}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thanatophoric dysplasia 1 (TD1) [MIM:187600]: A neonatal lethal skeletal dysplasia. Affected individuals manifest severe shortening of the limbs with macrocephaly, narrow thorax, short ribs, and curved femurs. {ECO:0000269|PubMed:10360402, ECO:0000269|PubMed:10671061, ECO:0000269|PubMed:7773297, ECO:0000269|PubMed:8589699, ECO:0000269|PubMed:8845844, ECO:0000269|PubMed:9790257}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thanatophoric dysplasia 2 (TD2) [MIM:187601]: A neonatal lethal skeletal dysplasia causing severe shortening of the limbs, narrow thorax and short ribs. Patients with thanatophoric dysplasia type 2 have straight femurs and cloverleaf skull. {ECO:0000269|PubMed:12297284, ECO:0000269|PubMed:7773297, ECO:0000269|PubMed:8754806}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hypochondroplasia (HCH) [MIM:146000]: Autosomal dominant disease and is characterized by disproportionate short stature. It resembles achondroplasia, but with a less severe phenotype. {ECO:0000269|PubMed:10215410, ECO:0000269|PubMed:10777366, ECO:0000269|PubMed:11055896, ECO:0000269|PubMed:12707965, ECO:0000269|PubMed:7670477, ECO:0000269|PubMed:9452043}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Bladder cancer (BLC) [MIM:109800]: A malignancy originating in tissues of the urinary bladder. It often presents with multiple tumors appearing at different times and at different sites in the bladder. Most bladder cancers are transitional cell carcinomas that begin in cells that normally make up the inner lining of the bladder. Other types of bladder cancer include squamous cell carcinoma (cancer that begins in thin, flat cells) and adenocarcinoma (cancer that begins in cells that make and release mucus and other fluids). Bladder cancer is a complex disorder with both genetic and environmental influences. {ECO:0000269|PubMed:10471491, ECO:0000269|PubMed:11314002}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Somatic mutations can constitutively activate FGFR3.; DISEASE: Cervical cancer (CERCA) [MIM:603956]: A malignant neoplasm of the cervix, typically originating from a dysplastic or premalignant lesion previously present at the active squamocolumnar junction. The transformation from mild dysplastic to invasive carcinoma generally occurs slowly within several years, although the rate of this process varies widely. Carcinoma in situ is particularly known to precede invasive cervical cancer in most cases. Cervical cancer is strongly associated with infection by oncogenic types of human papillomavirus. {ECO:0000269|PubMed:10471491}. The gene represented in this entry is involved in disease pathogenesis.; DISEASE: Camptodactyly, tall stature, and hearing loss syndrome (CATSHLS) [MIM:610474]: An autosomal dominant syndrome characterized by permanent and irreducible flexion of one or more fingers of the hand and/or feet, tall stature, scoliosis and/or a pectus excavatum, and hearing loss. Affected individuals have developmental delay and/or intellectual disability, and several of these have microcephaly. Radiographic findings included tall vertebral bodies with irregular borders and broad femoral metaphyses with long tubular shafts. On audiological exam, each tested member have bilateral sensorineural hearing loss and absent otoacoustic emissions. The hearing loss was congenital or developed in early infancy, progressed variably in early childhood, and range from mild to severe. Computed tomography and magnetic resonance imaging reveal that the brain, middle ear, and inner ear are structurally normal. {ECO:0000269|PubMed:17033969}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Multiple myeloma (MM) [MIM:254500]: A malignant tumor of plasma cells usually arising in the bone marrow and characterized by diffuse involvement of the skeletal system, hyperglobulinemia, Bence-Jones proteinuria and anemia. Complications of multiple myeloma are bone pain, hypercalcemia, renal failure and spinal cord compression. The aberrant antibodies that are produced lead to impaired humoral immunity and patients have a high prevalence of infection. Amyloidosis may develop in some patients. Multiple myeloma is part of a spectrum of diseases ranging from monoclonal gammopathy of unknown significance (MGUS) to plasma cell leukemia. {ECO:0000269|PubMed:11529856, ECO:0000269|PubMed:9207791}. The gene represented in this entry may be involved in disease pathogenesis. A chromosomal aberration involving FGFR3 is found in multiple myeloma. Translocation t(4;14)(p16.3;q32.3) with the IgH locus.; DISEASE: Lacrimo-auriculo-dento-digital syndrome 2 (LADD2) [MIM:620192]: A form of lacrimo-auriculo-dento-digital syndrome, an autosomal dominant disease characterized by aplastic/hypoplastic lacrimal and salivary glands and ducts, cup-shaped ears, hearing loss, hypodontia and enamel hypoplasia, and distal limb segments anomalies. In addition to these cardinal features, facial dysmorphism, malformations of the kidney and respiratory system and abnormal genitalia have been reported. Craniosynostosis and severe syndactyly are not observed. {ECO:0000269|PubMed:16501574}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Keratinocytic non-epidermolytic nevus (KNEN) [MIM:162900]: Epidermal nevi of the common, non-organoid and non-epidermolytic type are benign skin lesions and may vary in their extent from a single (usually linear) lesion to widespread and systematized involvement. They may be present at birth or develop early during childhood. {ECO:0000269|PubMed:16841094}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Muenke syndrome (MNKS) [MIM:602849]: A condition characterized by premature closure of coronal suture of skull during development (coronal craniosynostosis), which affects the shape of the head and face. It may be uni- or bilateral. When bilateral, it is characterized by a skull with a small antero-posterior diameter (brachycephaly), often with a decrease in the depth of the orbits and hypoplasia of the maxillae. Unilateral closure of the coronal sutures leads to flattening of the orbit on the involved side (plagiocephaly). The intellect is normal. In addition to coronal craniosynostosis some affected individuals show skeletal abnormalities of hands and feet, sensorineural hearing loss, intellectual disability and respiratory insufficiency. {ECO:0000269|PubMed:11746040, ECO:0000269|PubMed:9042914, ECO:0000269|PubMed:9950359}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Keratosis, seborrheic (KERSEB) [MIM:182000]: A common benign skin tumor. Seborrheic keratoses usually begin with the appearance of one or more sharply defined, light brown, flat macules. The lesions may be sparse or numerous. As they initially grow, they develop a velvety to finely verrucous surface, followed by an uneven warty surface with multiple plugged follicles and a dull or lackluster appearance. {ECO:0000269|PubMed:15772091}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Testicular germ cell tumor (TGCT) [MIM:273300]: A common malignancy in males representing 95% of all testicular neoplasms. TGCTs have various pathologic subtypes including: unclassified intratubular germ cell neoplasia, seminoma (including cases with syncytiotrophoblastic cells), spermatocytic seminoma, embryonal carcinoma, yolk sac tumor, choriocarcinoma, and teratoma. {ECO:0000269|PubMed:19855393}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Achondroplasia, severe, with developmental delay and acanthosis nigricans (SADDAN) [MIM:616482]: A severe form of achondroplasia associated with developmental delay and acanthosis nigricans. Patients manifest short-limb dwarfism, with a long, narrow trunk, short extremities, particularly in the proximal (rhizomelic) segments, a large head with frontal bossing, hypoplasia of the midface and a trident configuration of the hands. Acanthosis nigricans is a skin condition characterized by brown-pigmented, velvety verrucosities in body folds and creases. {ECO:0000269|PubMed:10053006}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147; DB01708 Interacts with NA EC number 1.1.3.6; 5.3.3.1 Uniprot keywords 3D-structure; Cholesterol metabolism; Direct protein sequencing; FAD; Flavoprotein; Isomerase; Lipid metabolism; Oxidoreductase; Secreted; Signal; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54295.8 Length 501 Aromaticity 0.1 Instability index 19.97 Isoelectric point 8.88 Charge (pH=7) 5.13 2D Binding mode Binding energy (Kcal/mol) -8.8  Molscript Map  Pymol Map 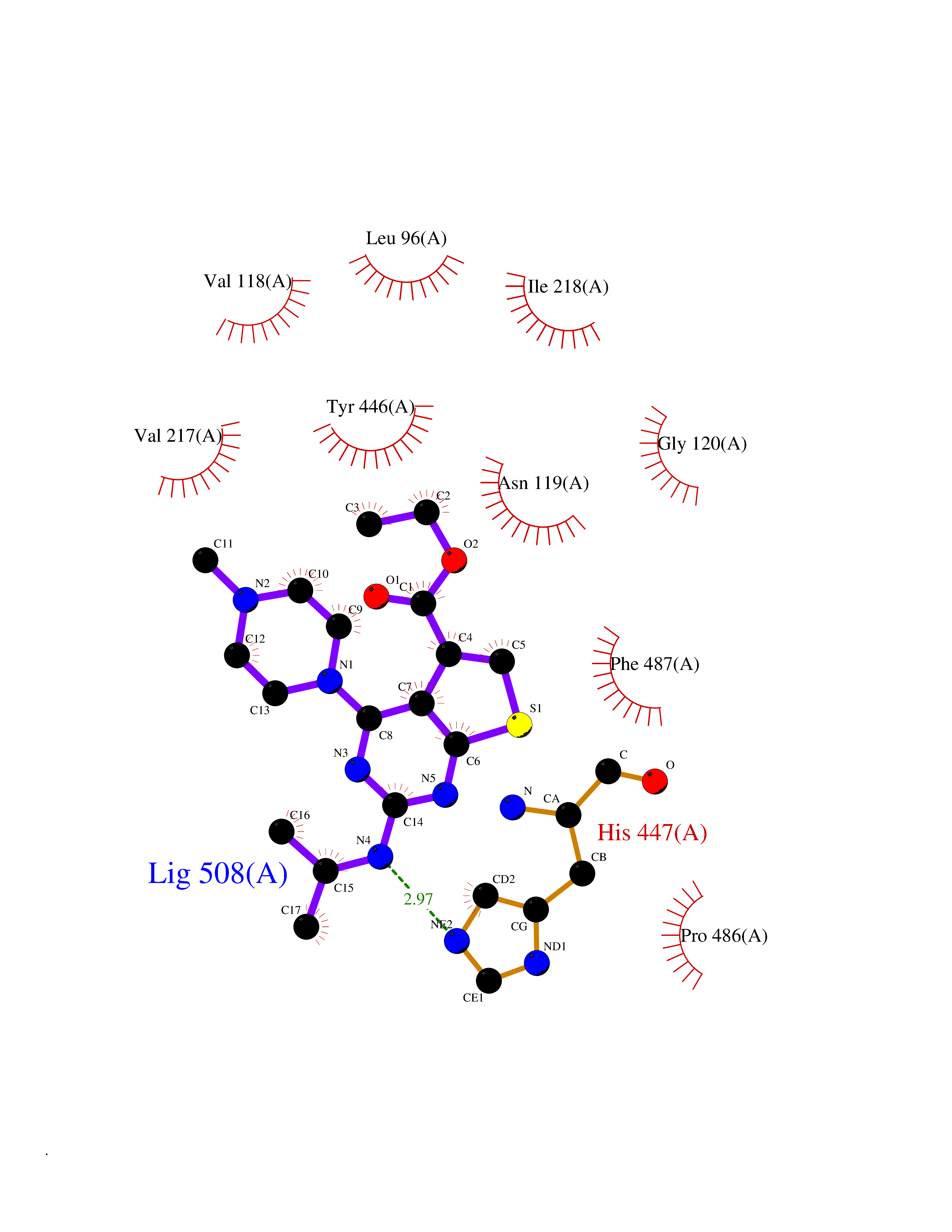 Ligplot Map 3D Binding mode Sequence RTLADGDRVPALVIGSGYGGAVAALRLTQAGIPTQIVEMGRSWDTPGSDGKIFCGMLNPDKRSMWLADKTDQPVSNFMGFGINKSIDRYVGVLDSERFSGIKVYQGRGVGGGSLVNGGMAVTPKRNYFEEILPSVDSNEMYNKYFPRANTGLGVNNIDQAWFESTEWYKFARTGRKTAQRSGFTTAFVPNVYDFEYMKKEAAGQVTKSGLGGEVIYGNNAGKKSLDKTYLAQAAATGKLTITTLHRVTKVAPATGSGYSVTMEQIDEQGNVVATKVVTADRVFFAAGSVGTSKLLVSMKAQGHLPNLSSQVGEGWGNNGNIMVGRANHMWDATGSKQATIPTMGIDNWADPTAPIFAEIAPLPAGLETYVSLYLAITKNPERARFQFNSGTGKVDLTWAQSQNQKGIDMAKKVFDKINQKEGTIYRTDLFYYKTWGDDFTYHPLGGVLLNKATDNFGRLPEYPGLYVVDGSLVPGNVGVNPFVTITALAERNMDKIISSDI Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Sphingosine-1-phosphate receptor 1 (S1PR1) | 7EW0 | 6.45 | |
Target general information Gen name S1PR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Sphingosine 1-phosphate receptor Edg-1; S1P1; S1P receptor Edg-1; S1P receptor 1; Endothelial differentiation G-protein coupled receptor 1; CHEDG1; CD363 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Signaling leads to the activation of RAC1, SRC, PTK2/FAK1 and MAP kinases. Plays an important role in cell migration, probably via its role in the reorganization of the actin cytoskeleton and the formation of lamellipodia in response to stimuli that increase the activity of the sphingosine kinase SPHK1. Required for normal chemotaxis toward sphingosine 1-phosphate. Required for normal embryonic heart development and normal cardiac morphogenesis. Plays an important role in the regulation of sprouting angiogenesis and vascular maturation. Inhibits sprouting angiogenesis to prevent excessive sprouting during blood vessel development. Required for normal egress of mature T-cells from the thymus into the blood stream and into peripheral lymphoid organs. Plays a role in the migration of osteoclast precursor cells, the regulation of bone mineralization and bone homeostasis. Plays a role in responses to oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine by pulmonary endothelial cells and in the protection against ventilator-induced lung injury. G-protein coupled receptor for the bioactive lysosphingolipid sphingosine 1-phosphate (S1P) that seems to be coupled to the G(i) subclass of heteromeric G proteins. Related diseases Wolcott-Rallison syndrome (WRS) [MIM:226980]: A rare autosomal recessive disorder, characterized by permanent neonatal or early infancy insulin-dependent diabetes and, at a later age, epiphyseal dysplasia, osteoporosis, growth retardation and other multisystem manifestations, such as hepatic and renal dysfunctions, intellectual disability and cardiovascular abnormalities. {ECO:0000269|PubMed:10932183, ECO:0000269|PubMed:12086964, ECO:0000269|PubMed:12960215, ECO:0000269|PubMed:16813601, ECO:0000269|PubMed:24168455, ECO:0000269|PubMed:24194294, ECO:0000269|PubMed:27145240, ECO:0000269|PubMed:28220546, ECO:0000269|PubMed:30906465, ECO:0000269|PubMed:30922274, ECO:0000269|PubMed:32216767, ECO:0000269|PubMed:34123975}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14766; DB08868; DB12612; DB12016; DB12371 Interacts with Q07108 EC number NA Uniprot keywords 3D-structure; Acetylation; Angiogenesis; Cell membrane; Chemotaxis; Disulfide bond; Endosome; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID D Molecular weight (Da) 32418.9 Length 284 Aromaticity 0.13 Instability index 39.5 Isoelectric point 9.71 Charge (pH=7) 19.09 2D Binding mode Binding energy (Kcal/mol) -8.8  Molscript Map 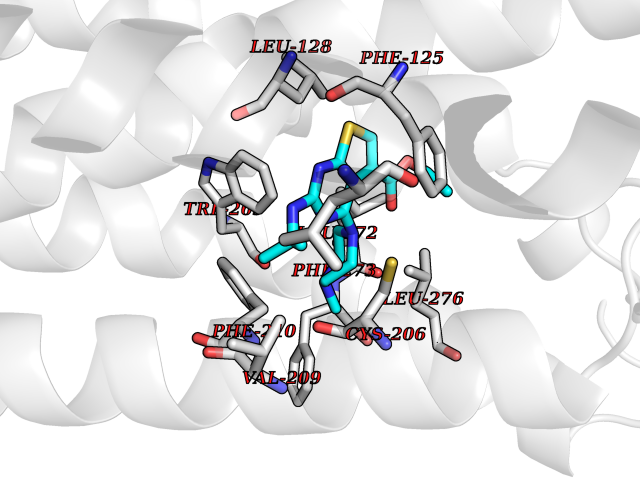 Pymol Map 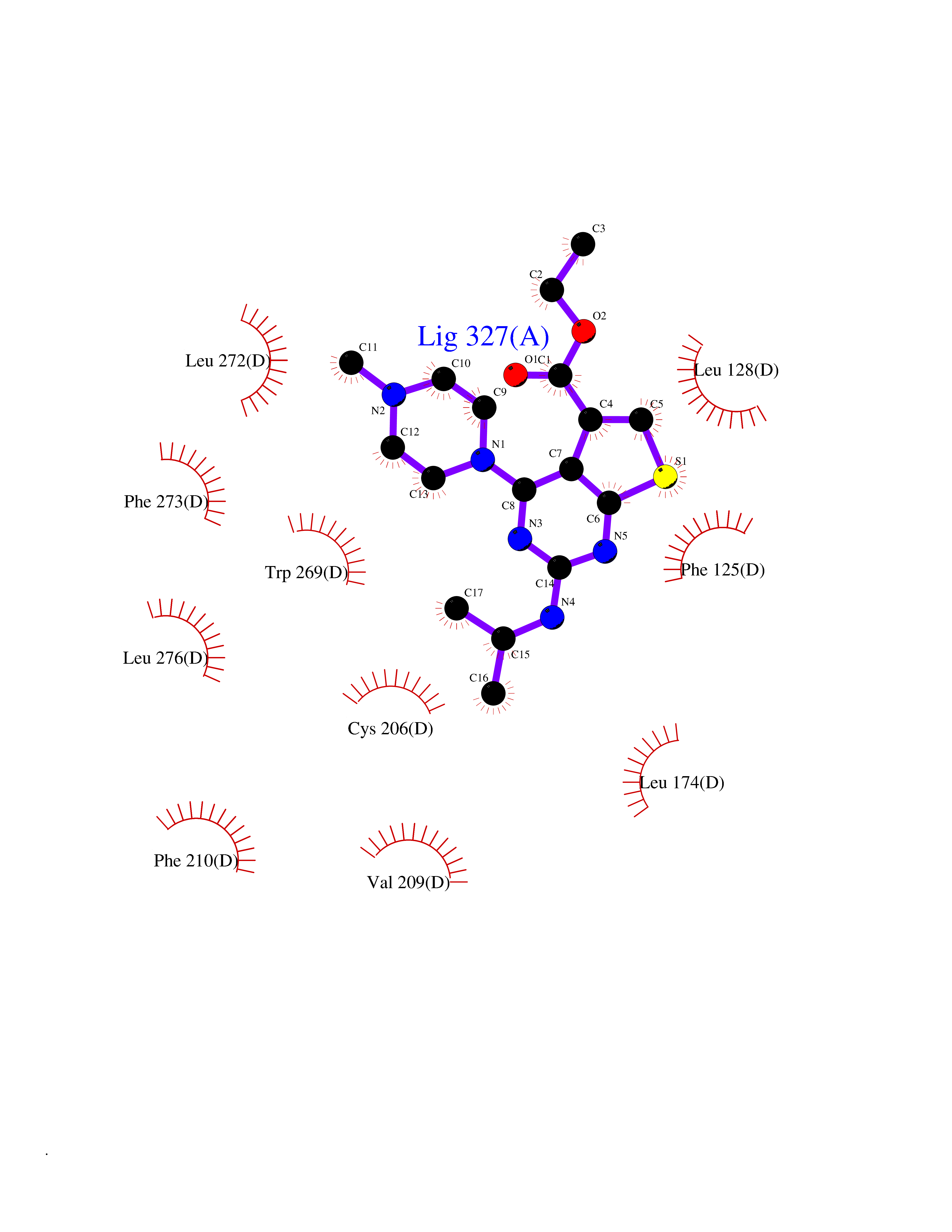 Ligplot Map 3D Binding mode Sequence YDIIVRHYNYTGKLTSVVFILICCFIILENIFVLLTIWKTKKFHRPMYYFIGNLALSDLLAGVAYTANLLLSGATTYKLTPAQWFLREGSMFVALSASVFSLLAIAIERYITMLKMKLHNGSNNFRLFLLISACWVISLILGGLPIMGWNCISALSSCSTVLPLYHKHYILFCTTVFTLLLLSIVILYCRIYSLVRTRSRRLTFRKSEKSLALLKTVIIVLSVFIACWAPLFILLLLDVGCKVKTCDILFRAEYFLVLAVLNSGTNPIIYTLTNKEMRRAFIRI Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Dihydrolipoyl dehydrogenase | 1LVL | 6.44 | |
Target general information Gen name lpdV Organism Pseudomonas putida (Arthrobacter siderocapsulatus) Uniprot ID TTD ID NA Synonyms NA Protein family Class-I pyridine nucleotide-disulfide oxidoreductase family Biochemical class Oxidoreductase Function Dihydrolipoyl dehydrogenase activity.Electron carrier activity.Flavin adenine dinucleotide binding. Related diseases Factor VII deficiency (FA7D) [MIM:227500]: A hemorrhagic disease with variable presentation. The clinical picture can be very severe, with the early occurrence of intracerebral hemorrhages or repeated hemarthroses, or, in contrast, moderate with cutaneous-mucosal hemorrhages (epistaxis, menorrhagia) or hemorrhages provoked by a surgical intervention. Finally, numerous subjects are completely asymptomatic despite very low factor VII levels. {ECO:0000269|PubMed:10862079, ECO:0000269|PubMed:11091194, ECO:0000269|PubMed:11129332, ECO:0000269|PubMed:12472587, ECO:0000269|PubMed:14717781, ECO:0000269|PubMed:1634227, ECO:0000269|PubMed:18976247, ECO:0000269|PubMed:19432927, ECO:0000269|PubMed:19751712, ECO:0000269|PubMed:2070047, ECO:0000269|PubMed:21206266, ECO:0000269|PubMed:21372693, ECO:0000269|PubMed:26761581, ECO:0000269|PubMed:7974346, ECO:0000269|PubMed:7981691, ECO:0000269|PubMed:8043443, ECO:0000269|PubMed:8204879, ECO:0000269|PubMed:8242057, ECO:0000269|PubMed:8364544, ECO:0000269|PubMed:8652821, ECO:0000269|PubMed:8844208, ECO:0000269|PubMed:8883260, ECO:0000269|PubMed:8940045, ECO:0000269|PubMed:9414278, ECO:0000269|PubMed:9452082, ECO:0000269|PubMed:9576180}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.8.1.4 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Disulfide bond; FAD; Flavoprotein; NAD; Oxidoreductase; Redox-active center Protein physicochemical properties Chain ID A Molecular weight (Da) 48026.9 Length 458 Aromaticity 0.04 Instability index 36.03 Isoelectric point 6.33 Charge (pH=7) -3.95 2D Binding mode Binding energy (Kcal/mol) -8.78 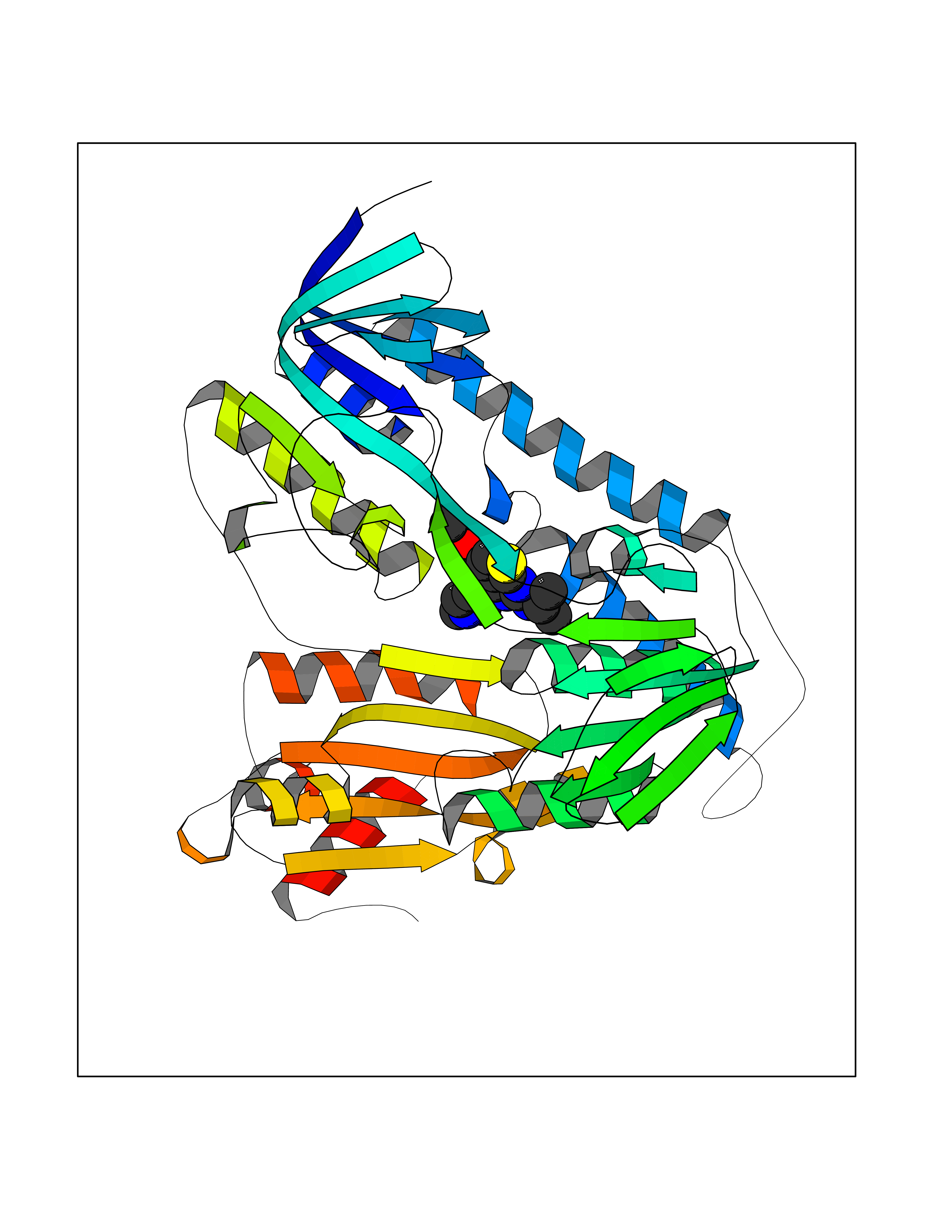 Molscript Map 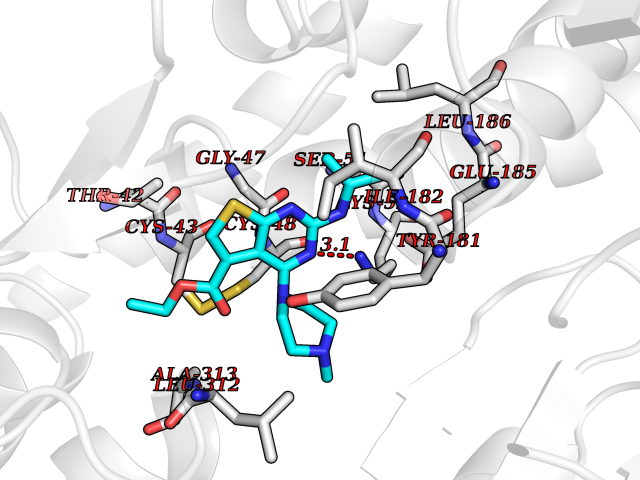 Pymol Map 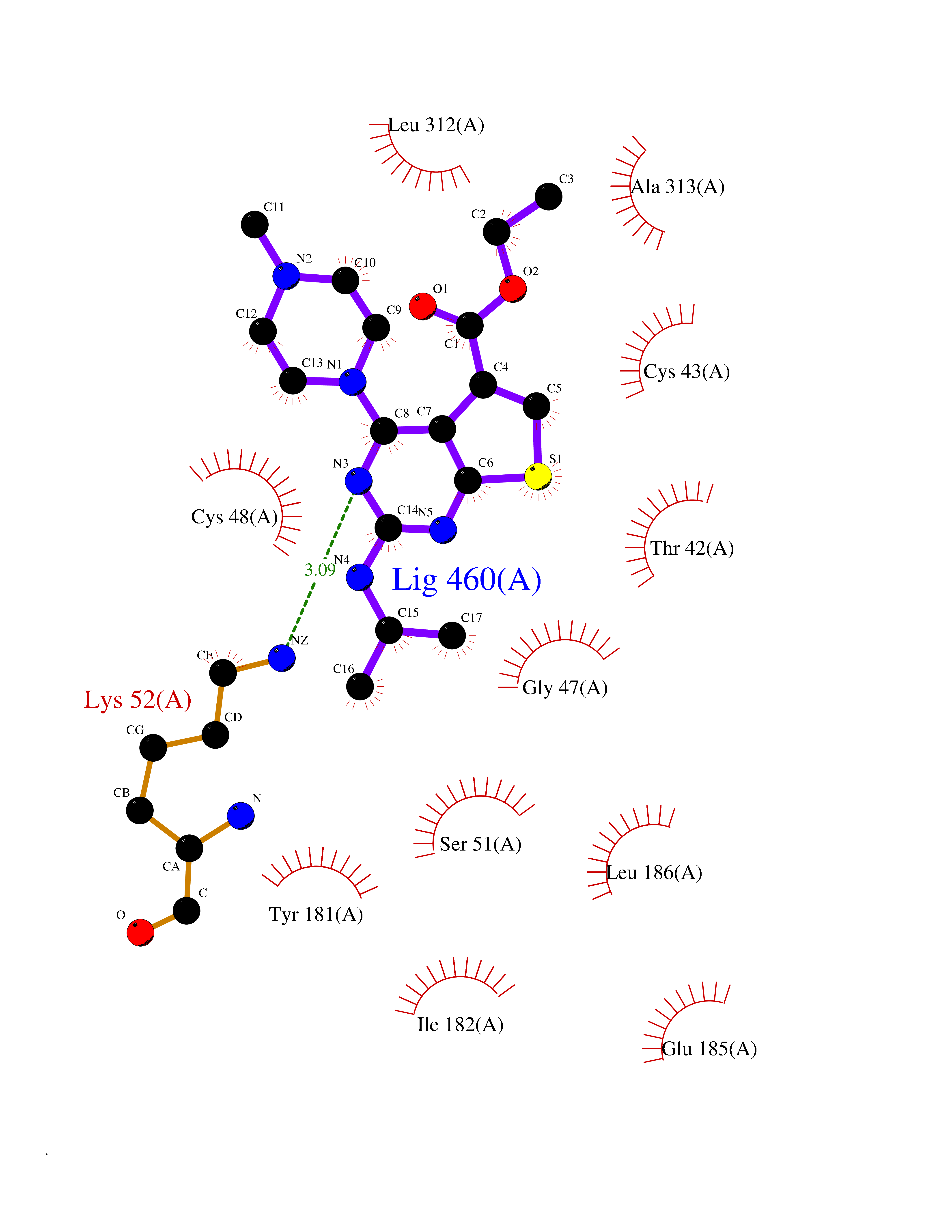 Ligplot Map 3D Binding mode Sequence QQTIQTTLLIIGGGPGGYVAAIRAGQLGIPTVLVEGQALGGTCLNIGCIPSKALIHVAEQFHQASRFTEPSPLGISVASPRLDIGQSVAWKDGIVDRLTTGVAALLKKHGVKVVHGWAKVLDGKQVEVDGQRIQCEHLLLATGSSSVELPMLPLGGPVISSTEALAPKALPQHLVVVGGGYIGLELGIAYRKLGAQVSVVEARERILPTYDSELTAPVAESLKKLGIALHLGHSVEGYENGCLLANDGKGGQLRLEADRVLVAVGRRPRTKGFNLECLDLKMNGAAIAIDERCQTSMHNVWAIGDVAGEPMLAHRAMAQGEMVAEIIAGKARRFEPAAIAAVCFTDPEVVVVGKTPEQASQQGLDCIVAQFPFAANGRAMSLESKSGFVRVVARRDNHLILGWQAVGVAVSELSTAFAQSLEMGACLEDVAGTIHAHPTLGEAVQEAALRALGHALHI Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | 2-oxopropyl-CoM reductase, carboxylating | 1MO9 | 6.44 | |
Target general information Gen name xecC Organism Xanthobacter autotrophicus (strain ATCC BAA-1158 / Py2) Uniprot ID TTD ID NA Synonyms Xaut_4867 Protein family Class-I pyridine nucleotide-disulfide oxidoreductase family Biochemical class Oxidoreductase Function 2-oxopropyl-CoM reductase (carboxylating) activity.Flavin adenine dinucleotide binding. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB03163; DB03147 Interacts with NA EC number 1.8.1.5 Uniprot keywords 3D-structure; Disulfide bond; FAD; Flavoprotein; NADP; Oxidoreductase; Plasmid; Redox-active center; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 114413 Length 1044 Aromaticity 0.08 Instability index 25.66 Isoelectric point 5.68 Charge (pH=7) -21.74 2D Binding mode Binding energy (Kcal/mol) -8.78 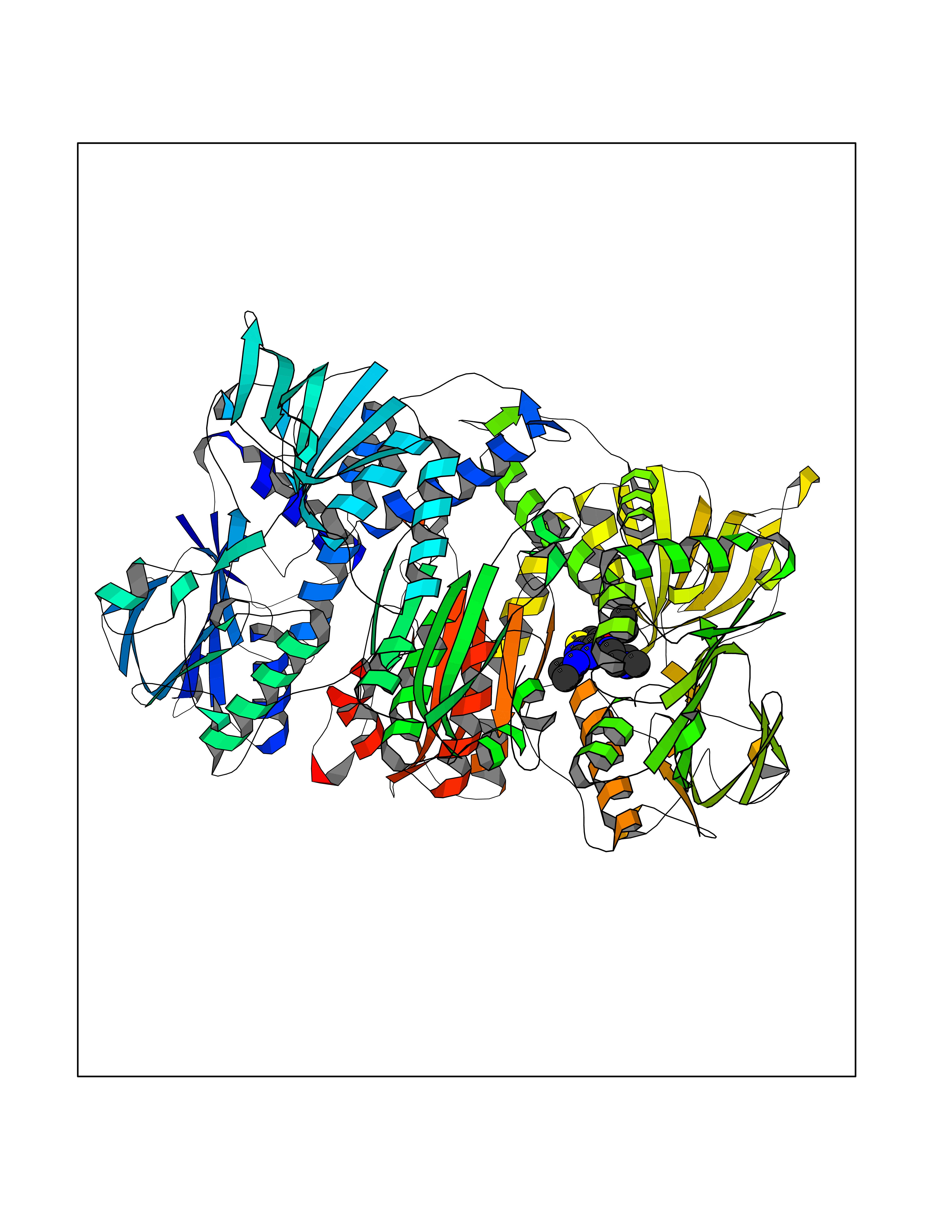 Molscript Map 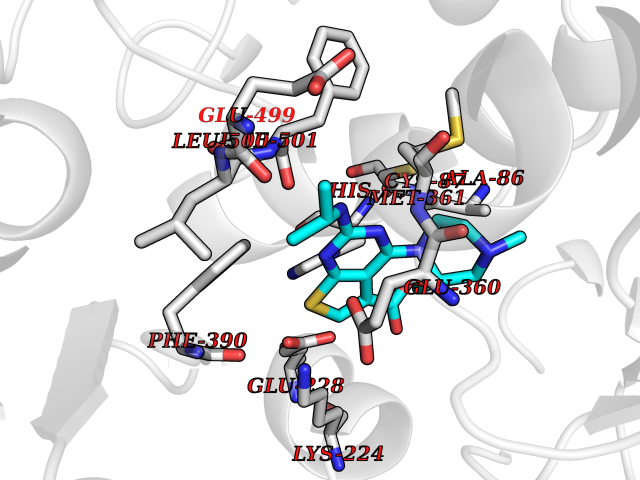 Pymol Map 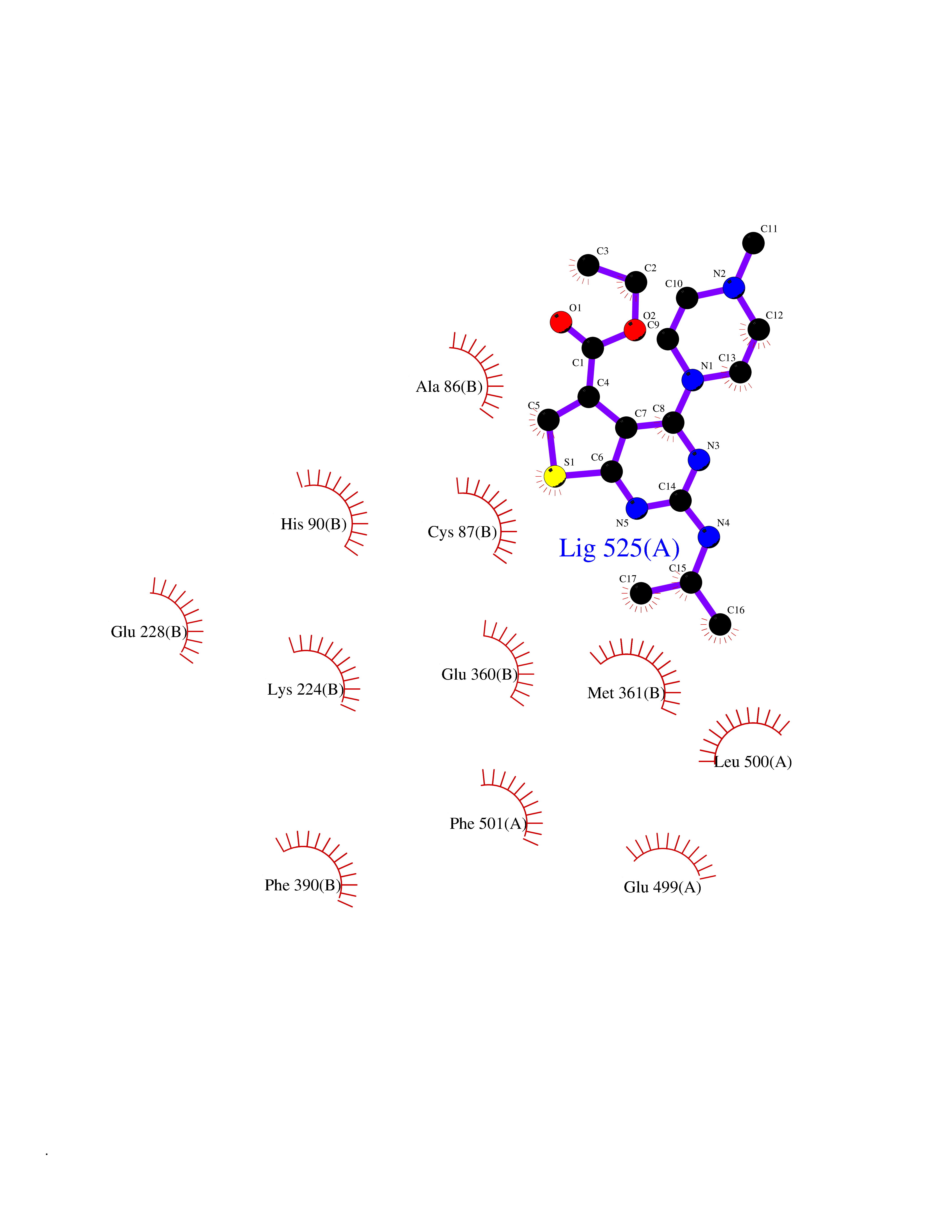 Ligplot Map 3D Binding mode Sequence KVWNARNDHLTINQWATRIDEILEAPDGGEVIYNVDENDPREYDAIFIGGGAAGRFGSAYLRAMGGRQLIVDRWPFLGGSCPHNACVPHHLFSDCAAELMLARTFSGQYWFPDMTEKVVGIKEVVDLFRAGRNGPHGIMNFQSKEQLNLEYILNCPAKVIDNHTVEAAGKVFKAKNLILAVGAGPGTLDVPGVNAKGVFDHATLVEELDYEPGSTVVVVGGSKTAVEYGCFFNATGRRTVMLVRTEPLKLIKDNETRAYVLDRMKEQGMEIISGSNVTRIEEDANGRVQAVVAMTPNGEMRIETDFVFLGLGEQPRSAELAKILGLDLGPKGEVLVNEYLQTSVPNVYAVGDLIGGPMEMFKARKSGCYAARNVMGEKISYTPKNYPDFLHTHYEVSFLGMGEEEARAAGHEIVTIKMPPDTENGLNVALPASDRTMLYAFGKGTAHMSGFQKIVIDAKTRKVLGAHHVGYGAKDAFQYLNVLIKQGLTVDELGDMDELFLNPTHFIQLSRLRAGSKNLVSLKVWNARNDHLTINQWATRIDEILEAPDGGEVIYNVDENDPREYDAIFIGGGAAGRFGSAYLRAMGGRQLIVDRWPFLGGSCPHNACVPHHLFSDCAAELMLARTFSGQYWFPDMTEKVVGIKEVVDLFRAGRNGPHGIMNFQSKEQLNLEYILNCPAKVIDNHTVEAAGKVFKAKNLILAVGAGPGTLDVPGVNAKGVFDHATLVEELDYEPGSTVVVVGGSKTAVEYGCFFNATGRRTVMLVRTEPLKLIKDNETRAYVLDRMKEQGMEIISGSNVTRIEEDANGRVQAVVAMTPNGEMRIETDFVFLGLGEQPRSAELAKILGLDLGPKGEVLVNEYLQTSVPNVYAVGDLIGGPMEMFKARKSGCYAARNVMGEKISYTPKNYPDFLHTHYEVSFLGMGEEEARAAGHEIVTIKMPPDTENGLNVALPASDRTMLYAFGKGTAHMSGFQKIVIDAKTRKVLGAHHVGYGAKDAFQYLNVLIKQGLTVDELGDMDELFLNPTHFIQLSRLRAGSKNLVSL Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Adrenergic receptor alpha-2A (ADRA2A) | 7EJ8 | 6.44 | |
Target general information Gen name ADRA2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Alpha-2AAR; Alpha-2A adrenoreceptor; Alpha-2A adrenoceptor; Alpha-2A adrenergic receptor; Alpha-2 adrenergic receptor subtype C10; ADRAR; ADRA2R Protein family G-protein coupled receptor 1 family, Adrenergic receptor subfamily, ADRA2A sub-subfamily Biochemical class GPCR rhodopsin Function The rank order of potency for agonists of this receptor is oxymetazoline > clonidine > epinephrine > norepinephrine > phenylephrine > dopamine > p-synephrine > p-tyramine > serotonin = p-octopamine. For antagonists, the rank order is yohimbine > phentolamine = mianserine > chlorpromazine = spiperone = prazosin > propanolol > alprenolol = pindolol. Alpha-2 adrenergic receptors mediate the catecholamine-induced inhibition of adenylate cyclase through the action of G proteins. Related diseases Lipodystrophy, familial partial, 8 (FPLD8) [MIM:620679]: An autosomal dominant form of partial lipodystrophy, a disorder characterized by abnormal subcutaneous fat distribution. FPLD8 patients show selective loss of subcutaneous adipose tissue from the limbs, beginning around 13 to 15 years of age, and abnormal accumulation of subcutaneous adipose tissue in the dorsal neck and face, as well as in the posterior thoracic and abdominal regions. The disorder is associated with metabolic abnormalities, including diabetes mellitus and hyperlipidemia. {ECO:0000269|PubMed:27376152}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01472; DB00321; DB00543; DB00182; DB00714; DB00964; DB09229; DB01238; DB14185; DB06216; DB00865; DB00217; DB00484; DB01200; DB00248; DB01136; DB04846; DB00477; DB09202; DB00575; DB00363; DB01151; DB00633; DB01576; DB11273; DB13345; DB00320; DB00449; DB11278; DB09167; DB04855; DB06262; DB01363; DB05492; DB00751; DB00668; DB01049; DB00696; DB01175; DB06678; DB09194; DB00800; DB06623; DB00629; DB01018; DB00502; DB11577; DB00555; DB06707; DB00589; DB04948; DB09195; DB00408; DB08815; DB00934; DB01365; DB01577; DB01403; DB00968; DB06148; DB00370; DB09205; DB09242; DB06711; DB01149; DB00368; DB00540; DB06229; DB00935; DB01267; DB00715; DB01186; DB01608; DB00925; DB00692; DB00397; DB09286; DB09244; DB06153; DB00413; DB00457; DB00433; DB01069; DB00852; DB01224; DB11124; DB11738; DB00268; DB09304; DB06764; DB13025; DB00697; DB00797; DB00193; DB00656; DB00726; DB11477; DB06694; DB01392; DB00246; DB01624 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Direct protein sequencing; Disulfide bond; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Methylation; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 30303.9 Length 263 Aromaticity 0.16 Instability index 35.08 Isoelectric point 9.66 Charge (pH=7) 16.82 2D Binding mode Binding energy (Kcal/mol) -8.78 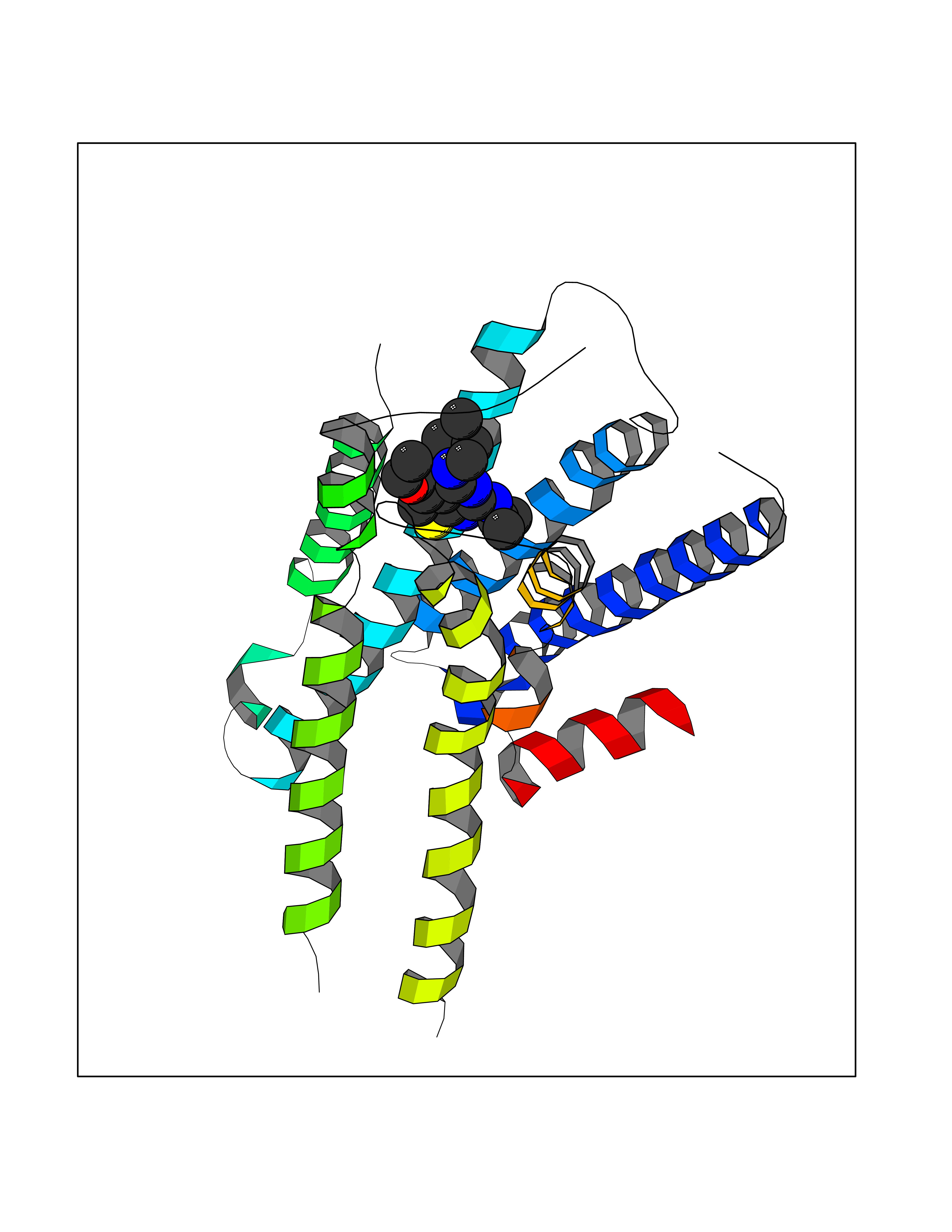 Molscript Map 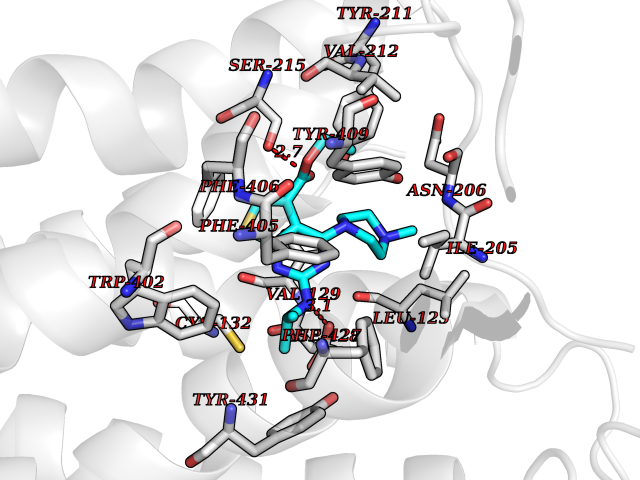 Pymol Map 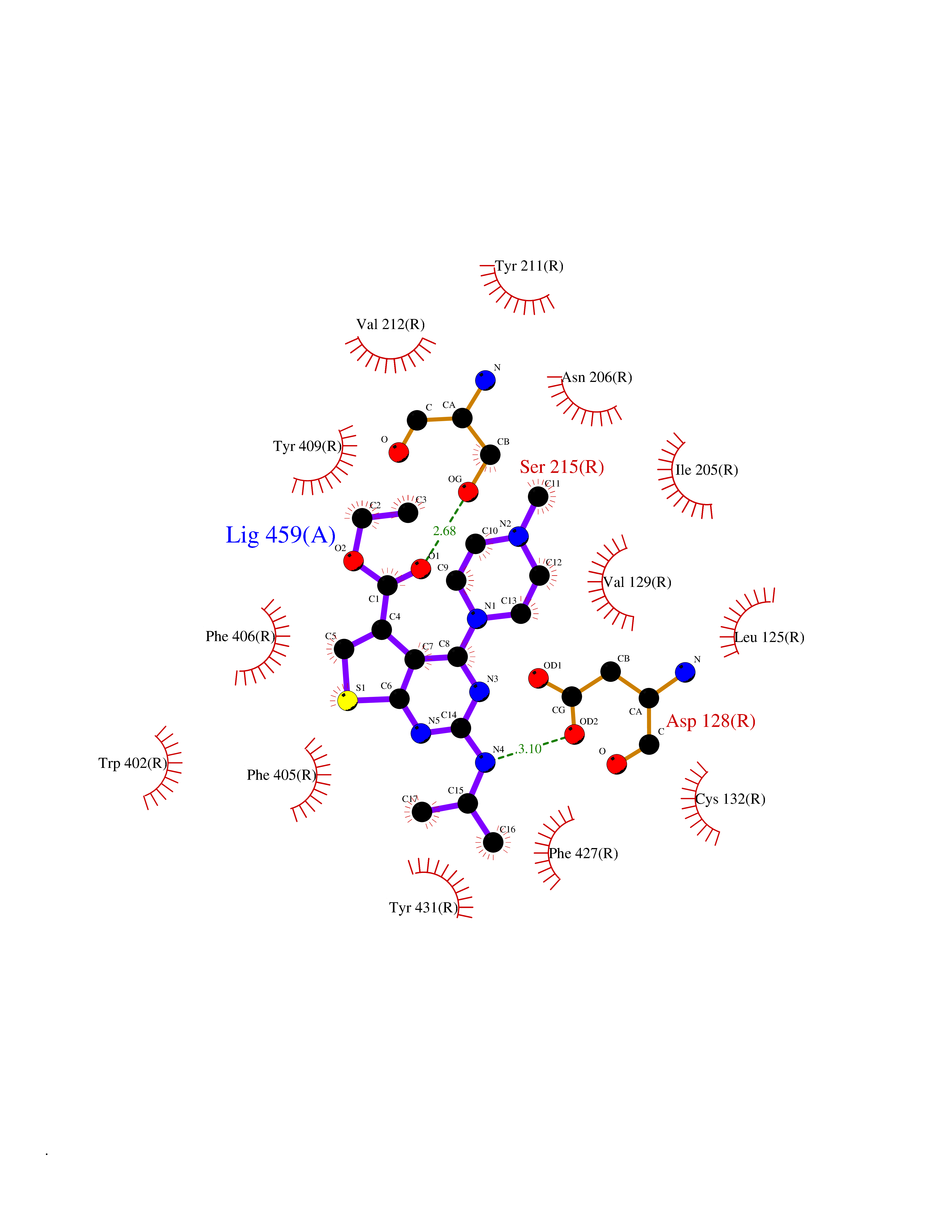 Ligplot Map 3D Binding mode Sequence YSLQVTLTLVCLAGLLMLLTVFGNVLVIIAVFTSRALKAPQNLFLVSLASADILVATLVIPFSLANEVMGYWYFGKAWCEIYLALDVLFCTSSIVHLCAISLDRYWSITQAIEYNLKRTPRRIKAIIITVWVISAVISFPPRCEINDQKWYVISSCIGSFFAPCLIMILVYVRIYQIAKRRTRRGRQNREKRFTFVLAVVIGVFVVCWFPFFFTYTLTAVGCSVPRTLFKFFFWFGYCNSSLNPVIYTIFNHDFRRAFKKILC Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Sodium/glucose cotransporter 2 (SGLT2) | 7VSI | 6.44 | |
Target general information Gen name SLC5A2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Solute carrier family 5 member 2; Na(+)/glucose cotransporter 2; Low affinity sodium-glucose cotransporter Protein family Sodium:solute symporter (SSF) (TC 2.A.21) family Biochemical class Solute:sodium symporter Function Has a Na(+) to glucose coupling ratio of 1:1. Sodium-dependent glucose transporter. Related diseases Renal glucosuria (GLYS) [MIM:233100]: A disorder characterized by persistent isolated glucosuria, normal fasting serum glucose concentration, decreased renal tubular resorption of glucose from the urine, and absence of any other signs of tubular dysfunction. {ECO:0000269|PubMed:14614622}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12236; DB08907; DB01914; DB06292; DB09038; DB11827; DB12713 Interacts with O14556; Q13113 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Ion transport; Membrane; Metal-binding; Proteomics identification; Reference proteome; Sodium; Sodium transport; Sugar transport; Symport; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 63858.9 Length 586 Aromaticity 0.12 Instability index 39.46 Isoelectric point 8.62 Charge (pH=7) 7.41 2D Binding mode Binding energy (Kcal/mol) -8.78  Molscript Map 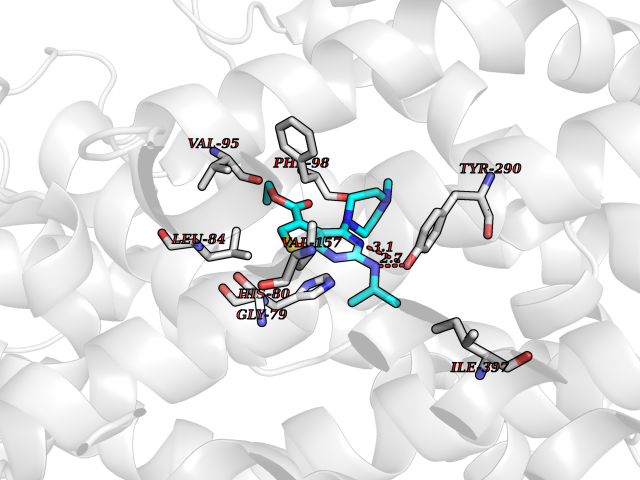 Pymol Map 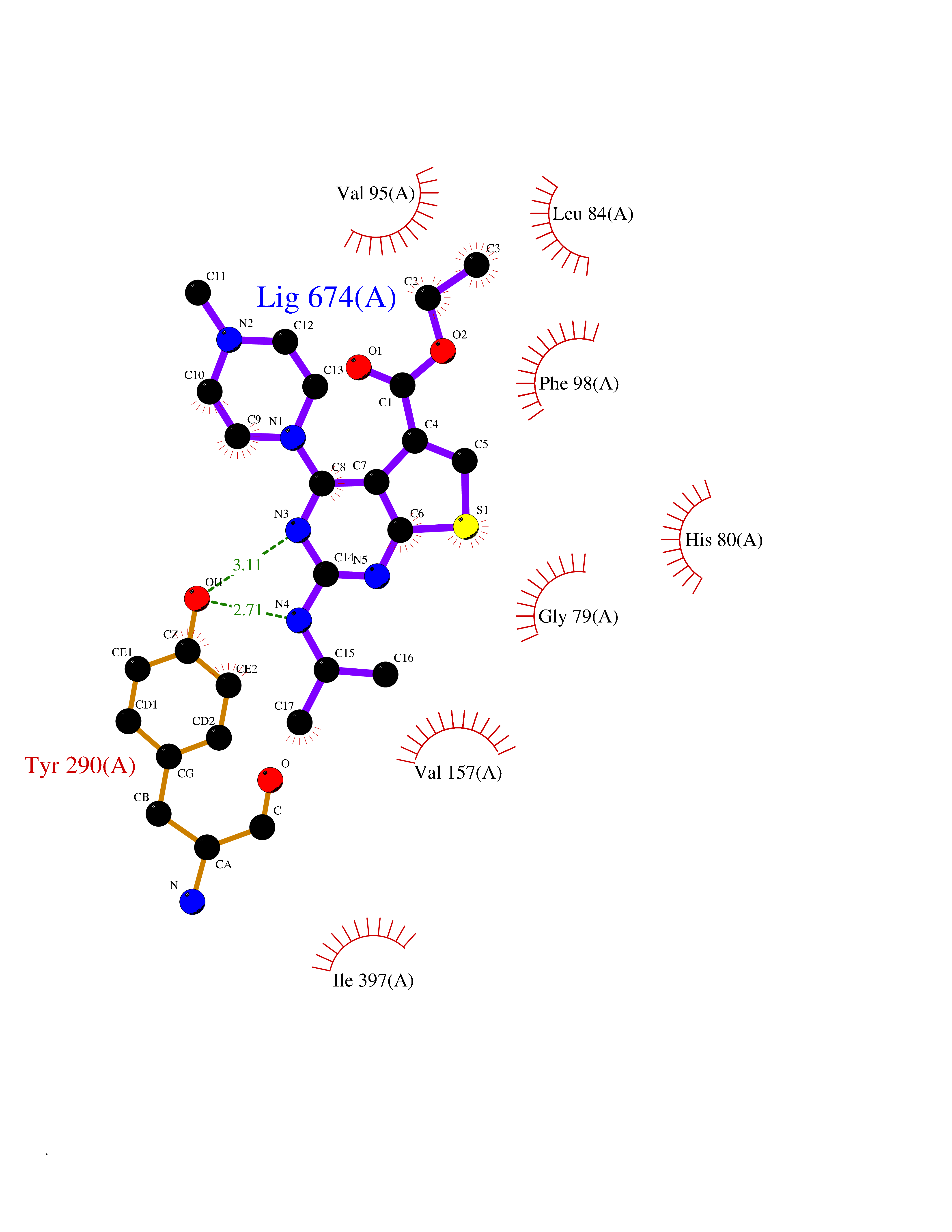 Ligplot Map 3D Binding mode Sequence DNPADILVIAAYFLLVIGVGLWSMCRTNRGTVGGYFLAGRSMVWWPVGASLFASNIGSGHFVGLAGTGAASGLAVAGFEWNALFVVLLLGWLFAPVYLTAGVITMPQYLRKRFGGRRIRLYLSVLSLFLYIFTKISVDMFSGAVFIQQALGWNIYASVIALLGITMIYTVTGGLAALMYTDTVQTFVILGGACILMGYAFHEVGGYSGLFDKYLGAATSLTVSEDPAVGNISSFCYRPRPDSYHLLRHPVTGDLPWPALLLGLTIVSGWYWCSDQVIVQRCLAGKSLTHIKAGCILCGYLKLTPMFLMVMPGMISRILYPDEVACVVPEVCRRVCGTEVGCSNIAYPRLVVKLMPNGLRGLMLAVMLAALMSSLASIFNSSSTLFTMDIYTRLRPRAGDRELLLVGRLWVVFIVVVSVAWLPVVQAAQGGQLFDYIQAVSSYLAPPVSAVFVLALFVPRVNEQGAFWGLIGGLLMGLARLIPEFSFGSGSCVQPSACPAFLCGVHYLYFAIVLFFCSGLLTLTVSLCTAPIPRKHLHRLVFSLRHSKEEREDLDEDISEDPSWARVVNLNALLMMAVAVFLWGFYA Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Thymidine phosphorylase (TYMP) | 2J0F | 6.44 | |
Target general information Gen name TYMP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms TdRPase; TYMP; TP; Platelet-derived endothelial cell growth factor; PDECGF; PD-ECGF; Gliostatin Protein family Thymidine/pyrimidine-nucleoside phosphorylase family Biochemical class Pentosyltransferase Function Catalyzes the reversible phosphorolysis of thymidine. The produced molecules are then utilized as carbon and energy sources or in the rescue of pyrimidine bases for nucleotide synthesis. Related diseases Mitochondrial DNA depletion syndrome 1, MNGIE type (MTDPS1) [MIM:603041]: A multisystem disease associated with mitochondrial dysfunction. It is clinically characterized by onset between the second and fifth decades of life, ptosis, progressive external ophthalmoplegia, gastrointestinal dysmotility (often pseudoobstruction), diffuse leukoencephalopathy, cachexia, peripheral neuropathy, and myopathy. {ECO:0000269|PubMed:12177387, ECO:0000269|PubMed:9924029}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01101; DB00369; DB00322; DB00544; DB06433; DB09343; DB00432 Interacts with Q14696; Q9H0C1 EC number EC 2.4.2.4 Uniprot keywords 3D-structure; Alternative splicing; Angiogenesis; Chemotaxis; Developmental protein; Differentiation; Direct protein sequencing; Disease variant; Glycosyltransferase; Growth factor; Neuropathy; Phosphoprotein; Primary mitochondrial disease; Progressive external ophthalmoplegia; Proteomics identification; Reference proteome; Repeat; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 46508.2 Length 446 Aromaticity 0.03 Instability index 37.49 Isoelectric point 5.81 Charge (pH=7) -4.7 2D Binding mode Binding energy (Kcal/mol) -8.79 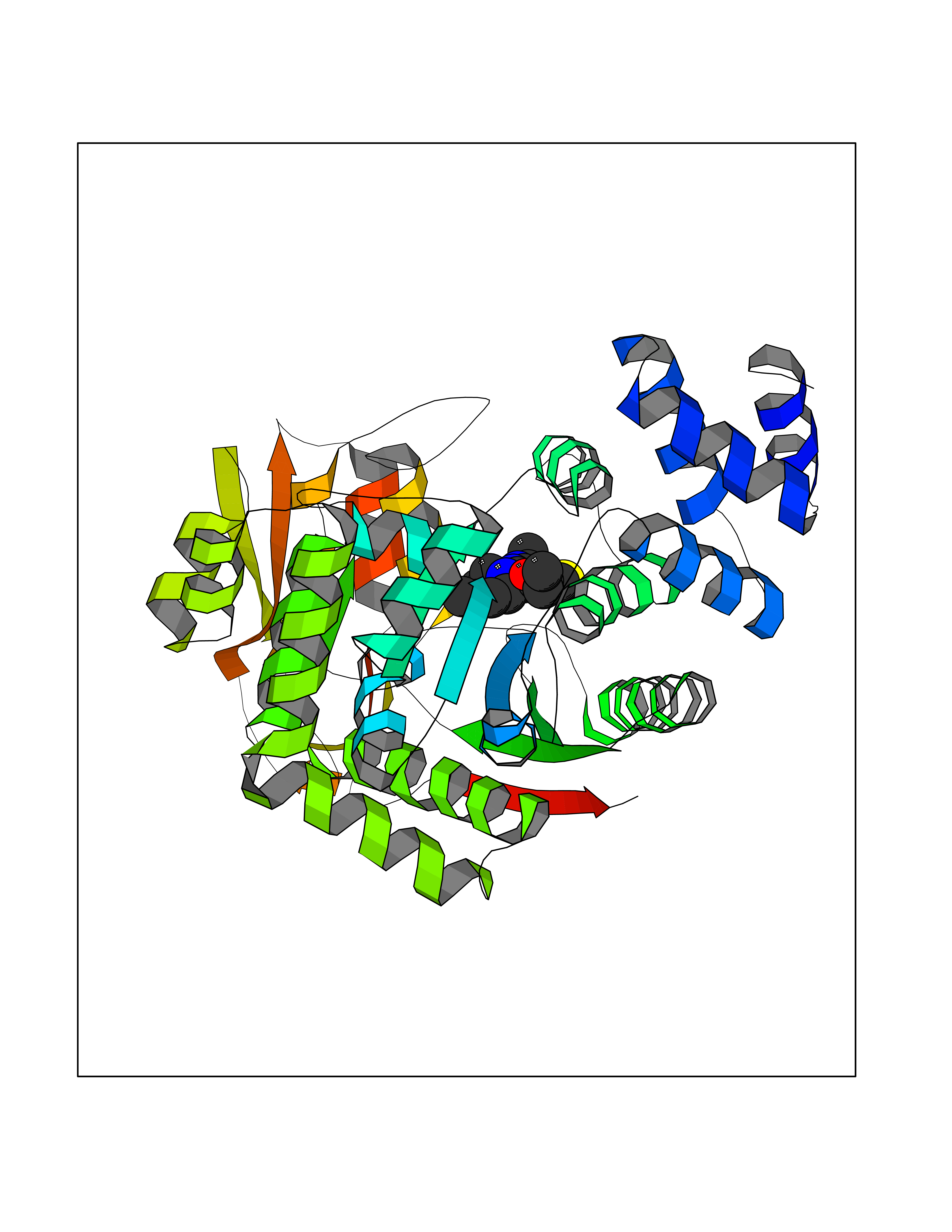 Molscript Map 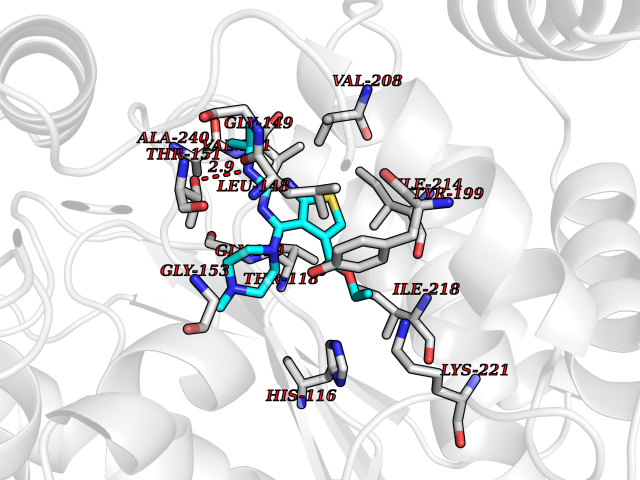 Pymol Map 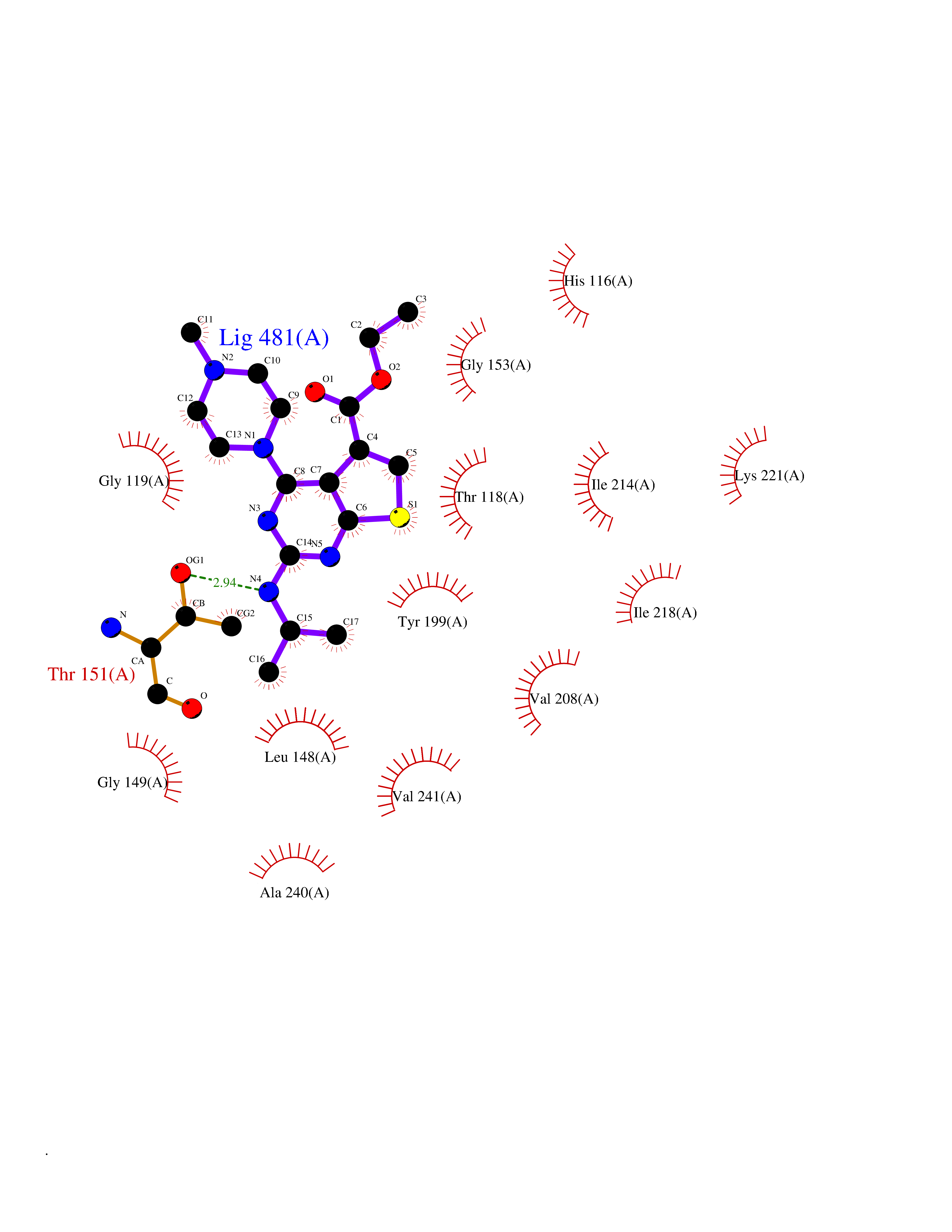 Ligplot Map 3D Binding mode Sequence KQLPELIRMKRDGGRLSEADIRGFVAAVVNGSAQGAQIGAMLMAIRLRGMDLEETSVLTQALAQSGQQLEWPEAWRQQLVDKHSTGGVGDKVSLVLAPALAACGCKVPMISGRGLGHTGGTLDKLESIPGFNVIQSPEQMQVLLDQAGCCIVGQSEQLVPADGILYAARDVTATVDSLPLITASILSKKLVEGLSALVVDVKFGAGAVFPNQEQARELAKTLVGVGASLGLRVAAALTAMDKPLGRCVGHALEVEEALLCMDGAGPPDLRDLVTTLGGALLWLSGHAGTQAQGAARVAAALDDGSALGRFERMLAAQGVDPGLARALCSGSPAERRQLLPRAREQEELLAPADGTVELVRALPLALVLHELGAGRSRAGEPLRLGVGAELLVDVGQRLRRGTPWLRVHRDGPALSGPQSRALQEALVLSDRAPFAAPLPFAELVLP Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | GTPase HRas (HRAS) | 7L0F | 6.44 | |
Target general information Gen name HRAS Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p21ras; cHras; c-H-ras; Transforming protein p21; HaRas; Ha-Ras; H-Ras-1; GTPase HRas, Nterminally processed Protein family Small GTPase superfamily, Ras family Biochemical class Small GTPase Function Ras proteins bind GDP/GTP and possess intrinsic GTPase activity. Involved in the activation of Ras protein signal transduction. Related diseases Costello syndrome (CSTLO) [MIM:218040]: A rare condition characterized by prenatally increased growth, postnatal growth deficiency, intellectual disability, distinctive facial appearance, cardiovascular abnormalities (typically pulmonic stenosis, hypertrophic cardiomyopathy and/or atrial tachycardia), tumor predisposition, skin and musculoskeletal abnormalities. {ECO:0000269|PubMed:16170316, ECO:0000269|PubMed:16329078, ECO:0000269|PubMed:16443854, ECO:0000269|PubMed:17054105, ECO:0000269|PubMed:18039947, ECO:0000269|PubMed:18247425, ECO:0000269|PubMed:19995790}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy with excess of muscle spindles (CMEMS) [MIM:218040]: Variant of Costello syndrome. {ECO:0000269|PubMed:17412879}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid cancer, non-medullary, 2 (NMTC2) [MIM:188470]: A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms. {ECO:0000269|PubMed:12727991}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Mutations which change positions 12, 13 or 61 activate the potential of HRAS to transform cultured cells and are implicated in a variety of human tumors. {ECO:0000269|PubMed:3670300}.; DISEASE: Bladder cancer (BLC) [MIM:109800]: A malignancy originating in tissues of the urinary bladder. It often presents with multiple tumors appearing at different times and at different sites in the bladder. Most bladder cancers are transitional cell carcinomas that begin in cells that normally make up the inner lining of the bladder. Other types of bladder cancer include squamous cell carcinoma (cancer that begins in thin, flat cells) and adenocarcinoma (cancer that begins in cells that make and release mucus and other fluids). Bladder cancer is a complex disorder with both genetic and environmental influences. {ECO:0000269|PubMed:6298635, ECO:0000269|PubMed:6844927}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Schimmelpenning-Feuerstein-Mims syndrome (SFM) [MIM:163200]: A disease characterized by sebaceous nevi, often on the face, associated with variable ipsilateral abnormalities of the central nervous system, ocular anomalies, and skeletal defects. Many oral manifestations have been reported, not only including hypoplastic and malformed teeth, and mucosal papillomatosis, but also ankyloglossia, hemihyperplastic tongue, intraoral nevus, giant cell granuloma, ameloblastoma, bone cysts, follicular cysts, oligodontia, and odontodysplasia. Sebaceous nevi follow the lines of Blaschko and these can continue as linear intraoral lesions, as in mucosal papillomatosis. {ECO:0000269|PubMed:22683711}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04315; DB04137; DB02210; DB08751; DB03226; DB15588 Interacts with Q99996-3; P53677-2; P10398; Q9NXL2-1; Q9UII2; Q9H7T9; Q00994; Q9H2G9; P15056; Q7Z569; Q5PSV4; Q9ULD4-2; Q96LL4; Q96HB5; Q49A88-3; Q96GN5-2; P24941; O95674; Q9H3R5; Q9Y4F5-3; Q86XR8; Q494V2-2; Q8WUX9; Q14117; Q9Y6W6; O14641; A0AVK6; Q8NB25; Q8IZU1; O94868-3; P15407; P15408; P52655; Q96CS2; Q9BT25; Q8IV36; O43248; Q53GQ0; P10809; Q8NDH6-2; Q8IY31-2; Q8NA54; Q13352; P28290-2; Q9BVG8-5; Q2M2Z5; Q6P597; P57682; Q9UH77; P08727; Q14525; Q14847-2; Q96LR2; P27338; Q99558; Q96EZ8; Q8TAC0; Q5JXC2; Q8NEH6; Q9Y605; Q96HT8; Q9GZM8; P21359; Q8N5V2; Q6PHZ7; Q9BZ95-3; A5D8V7; O43482; Q9BR81; O15534; Q9BUL5; O00329; O00329-2; Q9UPR0; Q96I34; Q15435-3; P04049; P11233; Q15311; Q12967; Q9NS23-2; Q9NS23-4; Q8WWW0; Q8TBY0; Q9P2K3-2; Q9NZL6; O15211; Q8IXN7; Q13671; Q13671-1; Q8WVD3; Q9BY12-3; Q13435; Q12824; Q13573; Q07889; Q86W54-2; Q92783-2; O75886; Q13586; Q8N4C7; O75528; P54274-2; Q9BXU0; Q5T0J7-2; Q5T1C6; Q8IUR5-4; P36406; Q86WT6-2; Q99598; Q6PF05; Q9UGJ1-2; Q9Y5Z9; P22415; Q495M9; Q9H270; Q8NEZ2; P19544-6; O43829; Q9C0F3; Q7Z637; Q86V28; P42337; Q9Z0S9; Q9EQZ6; P27671; Q5EBH1; Q5EBH1-1; P52306-5 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Glycoprotein; Golgi apparatus; GTP-binding; Hydrolase; Isopeptide bond; Lipoprotein; Membrane; Methylation; Nucleotide-binding; Nucleus; Palmitate; Prenylation; Proteomics identification; Proto-oncogene; Reference proteome; S-nitrosylation; Ubl conjugation Protein physicochemical properties Chain ID E,F Molecular weight (Da) 28737.2 Length 259 Aromaticity 0.1 Instability index 30.69 Isoelectric point 5.64 Charge (pH=7) -4.15 2D Binding mode Binding energy (Kcal/mol) -8.79 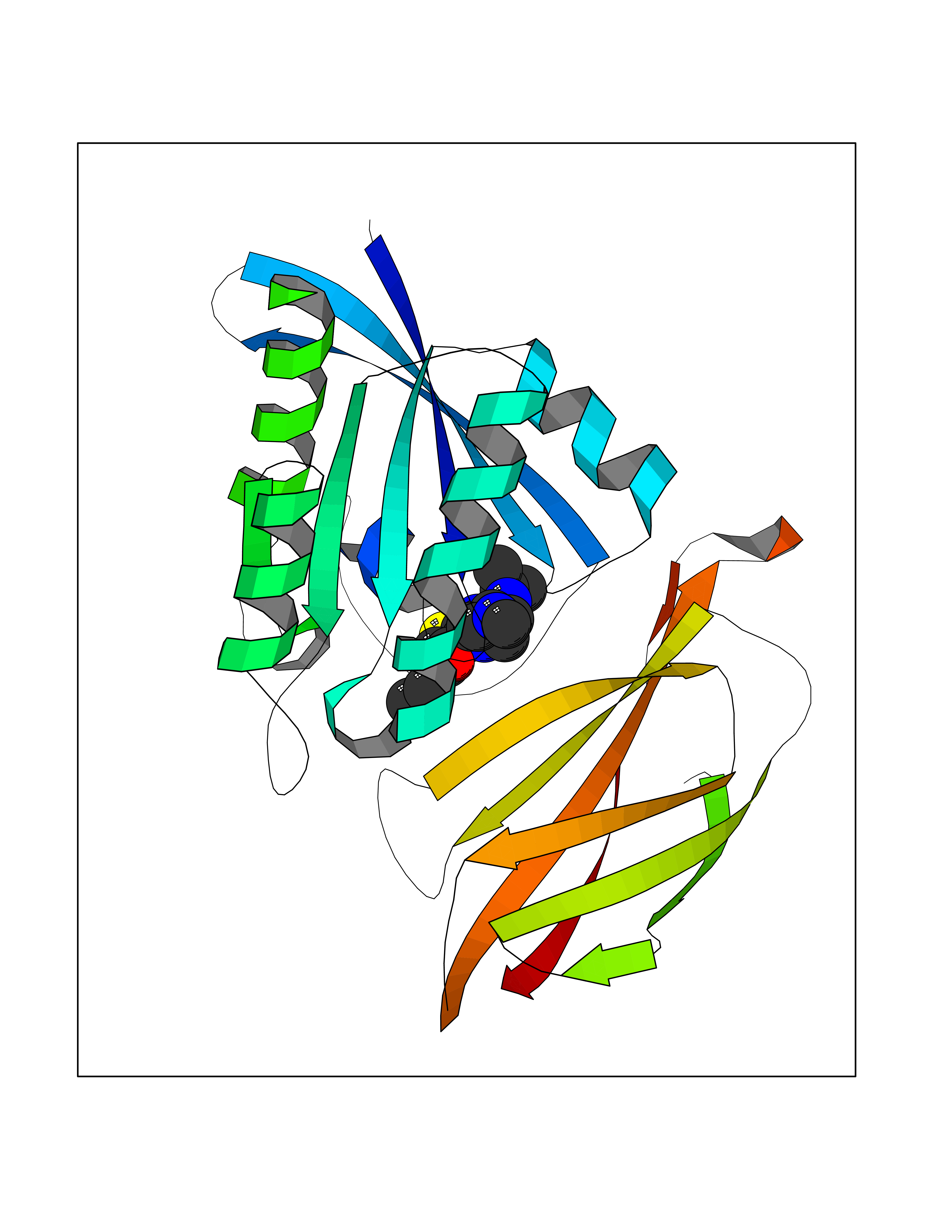 Molscript Map 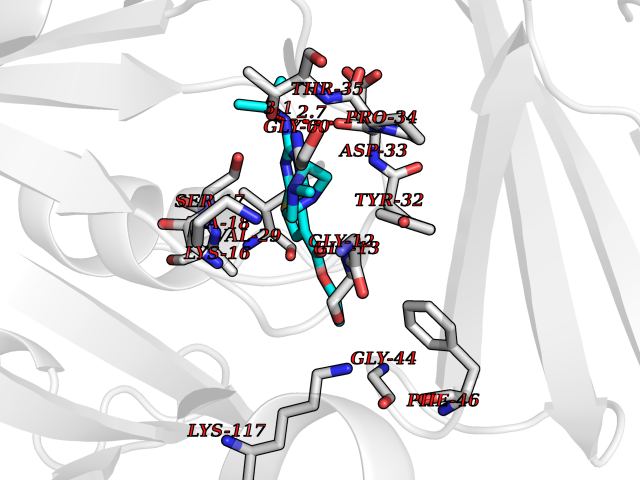 Pymol Map 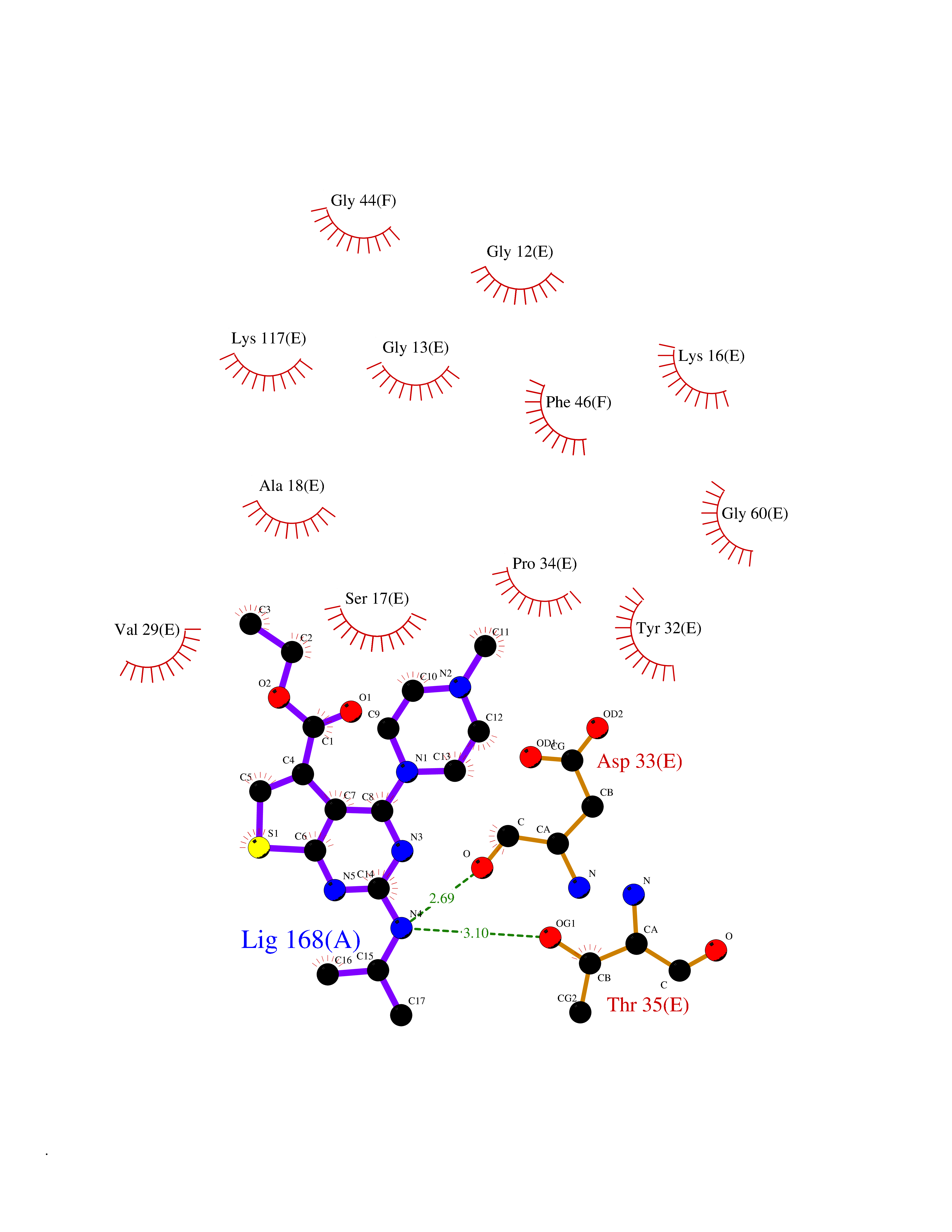 Ligplot Map 3D Binding mode Sequence MTEYKLVVVGAGGVGKSALTIQLIQNHFVDEYDPTIEDSYRKQVVIDGETCLLDILDTAGQEEYSAMRDQYMRTGEGFLCVFAINNTKSFEDIHQYREQIKRVKDSDDVPMVLVGNKCDLAARTVESRQAQDLARSYGIPYIETSAKTRQGVEDAFYTLVREIRQHSVPTKLEVVAATPTSLLISWDAPAVTVFFYIIAYGETGHGVGAFQAFRVPGSKSTATISGLKPGVDYTITVYARGYSKQGPYKPSPISINYRT Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | GABA(A) receptor gamma-2 (GABRG2) | 6X3X | 6.43 | |
Target general information Gen name GABRG2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Gamma-aminobutyric acid receptor subunit gamma-2; GABA(A) receptor subunit gamma-2 Protein family Ligand-gated ion channel (TC 1.A.9) family, Gamma-aminobutyric acid receptor (TC 1.A.9.5) subfamily, GABRG2 sub-subfamily Biochemical class Neurotransmitter receptor Function Plays an important role in the formation of functional inhibitory GABAergic synapses in addition to mediating synaptic inhibition as a GABA-gated ion channel. The gamma2 subunit is necessary but not sufficient for a rapid formation of active synaptic contacts and the synaptogenic effect of this subunit is influenced by the type of alpha and beta subunits present in the receptor pentamer. The alpha1/beta2/gamma2 receptor and the alpha1/beta3/gamma2 receptor exhibit synaptogenic activity. The alpha2/beta2/gamma2 receptor exhibits synatogenic activity whereas the alpha2/beta3/gamma2 receptor shows very little or no synaptogenic activity. Functions also as histamine receptor and mediates cellular responses to histamine. Ligand-gated chloride channel which is a component of the heteropentameric receptor for GABA, the major inhibitory neurotransmitter in the brain. Related diseases Developmental and epileptic encephalopathy 74 (DEE74) [MIM:618396]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE74 is an autosomal dominant form with onset in the first year of life. {ECO:0000269|PubMed:27864268}. The gene represented in this entry is involved in disease pathogenesis.; DISEASE: Epilepsy, childhood absence 2 (ECA2) [MIM:607681]: A subtype of idiopathic generalized epilepsy characterized by an onset at age 6-7 years, frequent absence seizures (several per day) and bilateral, synchronous, symmetric 3-Hz spike waves on EEG. Tonic-clonic seizures often develop in adolescence. Some individuals manifest febrile seizures. Absence seizures may either remit or persist into adulthood. {ECO:0000269|PubMed:11326275}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Febrile seizures, familial, 8 (FEB8) [MIM:607681]: Seizures associated with febrile episodes in childhood without any evidence of intracranial infection or defined pathologic or traumatic cause. It is a common condition, affecting 2-5% of children aged 3 months to 5 years. The majority are simple febrile seizures (generally defined as generalized onset, single seizures with a duration of less than 30 minutes). Complex febrile seizures are characterized by focal onset, duration greater than 30 minutes, and/or more than one seizure in a 24 hour period. The likelihood of developing epilepsy following simple febrile seizures is low. Complex febrile seizures are associated with a moderately increased incidence of epilepsy. {ECO:0000269|PubMed:16924025}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Generalized epilepsy with febrile seizures plus 3 (GEFSP3) [MIM:607681]: A rare autosomal dominant, familial condition with incomplete penetrance and large intrafamilial variability. Patients display febrile seizures persisting sometimes beyond the age of 6 years and/or a variety of afebrile seizure types. This disease combines febrile seizures, generalized seizures often precipitated by fever at age 6 years or more, and partial seizures, with a variable degree of severity. {ECO:0000269|PubMed:11326274, ECO:0000269|PubMed:23708187}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12537; DB00546; DB06579; DB00404; DB00543; DB11901; DB14719; DB11859; DB01558; DB09017; DB00237; DB00241; DB01489; DB00395; DB00475; DB14715; DB01594; DB00349; DB01068; DB00628; DB01559; DB01553; DB01511; DB01189; DB00829; DB13837; DB00228; DB01215; DB00402; DB00189; DB01545; DB09166; DB00292; DB01567; DB01205; DB01544; DB00690; DB05087; DB01381; DB01437; DB00801; DB01159; DB00753; DB01587; DB00555; DB13643; DB00186; DB13872; DB13437; DB00603; DB01043; DB00371; DB00463; DB01028; DB01107; DB15489; DB00683; DB12458; DB01595; DB14028; DB00334; DB00842; DB14672; DB00312; DB00252; DB13335; DB01708; DB01588; DB00794; DB00818; DB01589; DB12404; DB01236; DB09118; DB00306; DB01956; DB00231; DB11582; DB00897; DB00425; DB00909; DB15490 Interacts with P51513; O95166 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Cell projection; Chloride; Chloride channel; Cytoplasmic vesicle; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Ion channel; Ion transport; Lipoprotein; Membrane; Palmitate; Postsynaptic cell membrane; Proteomics identification; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 77460 Length 672 Aromaticity 0.14 Instability index 35.95 Isoelectric point 8.75 Charge (pH=7) 7.31 2D Binding mode Binding energy (Kcal/mol) -8.77  Molscript Map 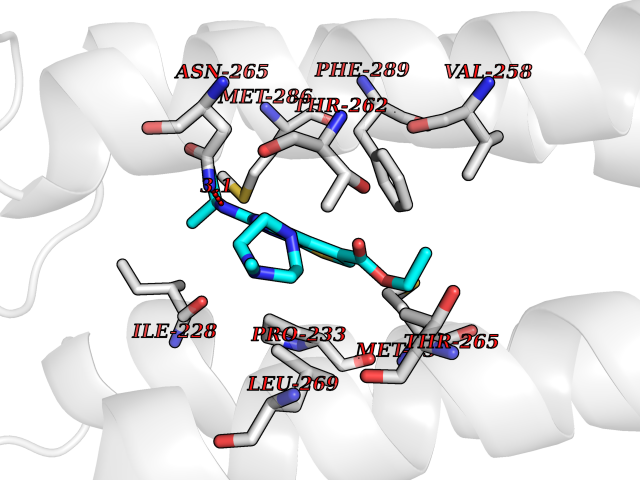 Pymol Map 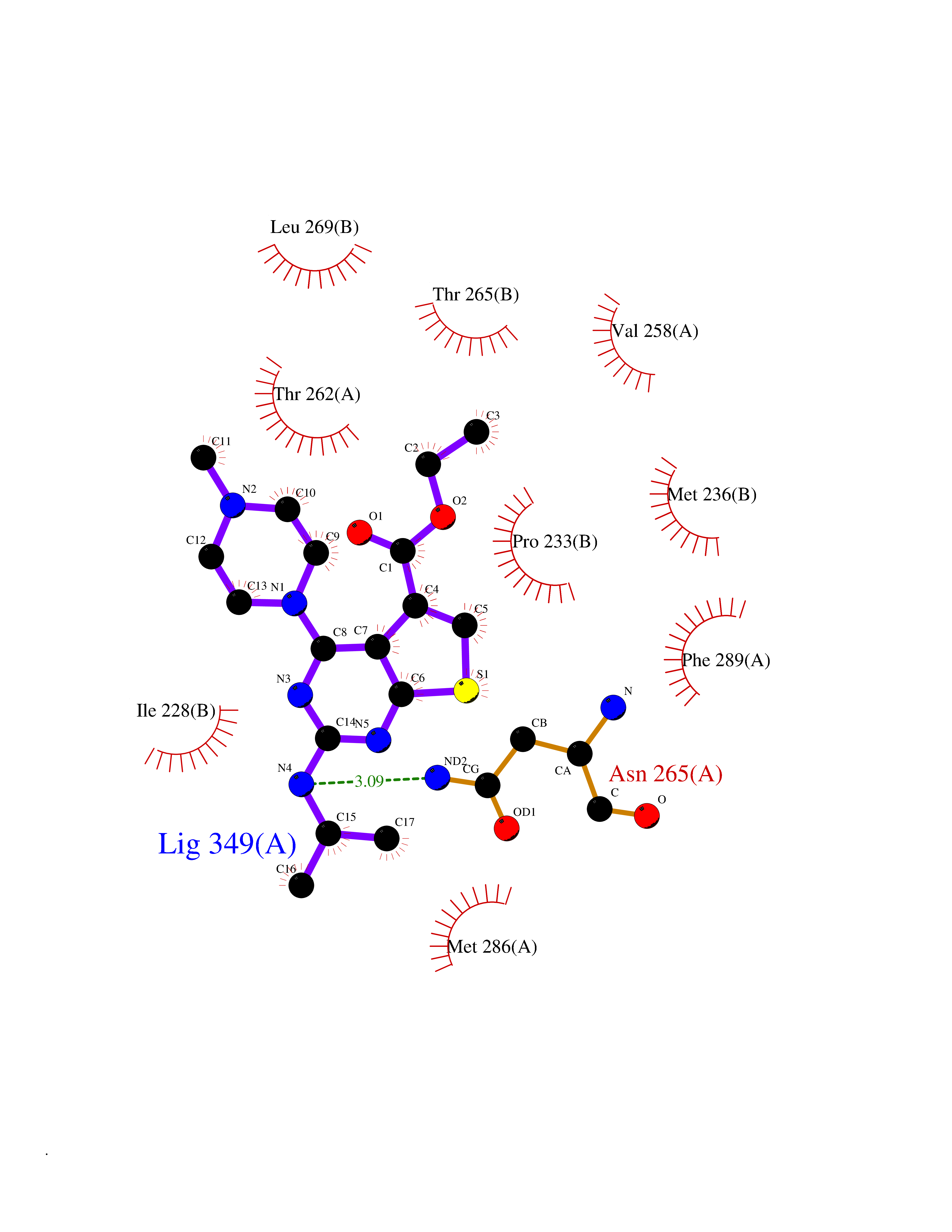 Ligplot Map 3D Binding mode Sequence SNMSLVKETVDRLLKGYDIRLRPDFGGPPVAVGMNIDIASIDMVSEVNMDYTLTMYFQQAWRDKRLSYNVIPLNLTLDNRVADQLWVPDTYFLNDKKSFVHGVTVKNRMIRLHPDGTVLYGLRITTTAACMMDLRRYPLDEQNCTLEIESYGYTTDDIEFYWRGDDNAVTGVTKIELPQFSIVDYKLITKKVVFSTGSYPRLSLSFKLKRNIGYFILQTYMPSILITILSWVSFWINYDASAARVALGITTVLTMTTINTHLRETLPKIPYVKAIDMYLMGCFVFVFMALLEYALVNYIFFSQPARAAAIDRWSRIFFPVVFSFFNIVYWLYYVDNTTVFTRILDRLLDGYDNRLRPGLGERVTEVKTDIFVTSFGPVSDHDMEYTIDVFFRQSWKDERLKFKGPMTVLRLNNLMASKIWTPDTFFHNGKKSVAHNMTMPNKLLRITEDGTLLYTMRLTVRAECPMHLEDFPMDAHACPLKFGSYAYTRAEVVYEWTREPARSVVVAEDGSRLNQYDLLGQTVDSGIVQSSTGEYVVMTTHFHLKRKIGYFVIQTYLPCIMTVILSQVSFWLNRESVPARTVFGVTTVLTMTTLSISARNSLPKVAYATAMDWFIAVCYAFVFSALIEFATVNYFTKSQPARAAKIDRLSRIAFPLLFGIFNLVYWATYLNR Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | CDC-like kinase 1 (CLK1) | 6KHD | 6.43 | |
Target general information Gen name CLK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dual specificity protein kinase CLK1; CLK; CDClike kinase 1 Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, Lammer subfamily Biochemical class Kinase Function Phosphorylates serine- and arginine-rich (SR) proteins of the spliceosomal complex and may be a constituent of a network of regulatory mechanisms that enable SR proteins to control RNA splicing. Phosphorylates: SRSF1, SRSF3 and PTPN1. Regulates the alternative splicing of tissue factor (F3) pre-mRNA in endothelial cells and adenovirus E1A pre-mRNA. Dual specificity kinase acting on both serine/threonine and tyrosine-containing substrates. Related diseases Ischemic stroke (ISCHSTR) [MIM:601367]: A stroke is an acute neurologic event leading to death of neural tissue of the brain and resulting in loss of motor, sensory and/or cognitive function. Ischemic strokes, resulting from vascular occlusion, is considered to be a highly complex disease consisting of a group of heterogeneous disorders with multiple genetic and environmental risk factors. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Genetic variations in ALOX5AP may be associated with susceptibility to myocardial infarction. Involvement in myocardial infarction is however unclear: according to some authors (PubMed:14770184), a 4-SNP haplotype in ALOX5AP confers risk of myocardial infarction, while according to other (PubMed:17304054) ALOX5AP is not implicated in this condition. {ECO:0000269|PubMed:14770184, ECO:0000269|PubMed:17304054}. Drugs (DrugBank ID) DB06376; DB04367; DB08691; DB12010 Interacts with P60409; O76083; Q8N2M8; P49760; Q92997; P60371; P60409; Q8TBB1; O76083-2; O14492-2; A7MD48; Q07955; O75494; P84103; Q16629; P62995 EC number EC 2.7.12.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Kinase; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase Protein physicochemical properties Chain ID A Molecular weight (Da) 37557.7 Length 322 Aromaticity 0.1 Instability index 37.24 Isoelectric point 6.38 Charge (pH=7) -4.53 2D Binding mode Binding energy (Kcal/mol) -8.78 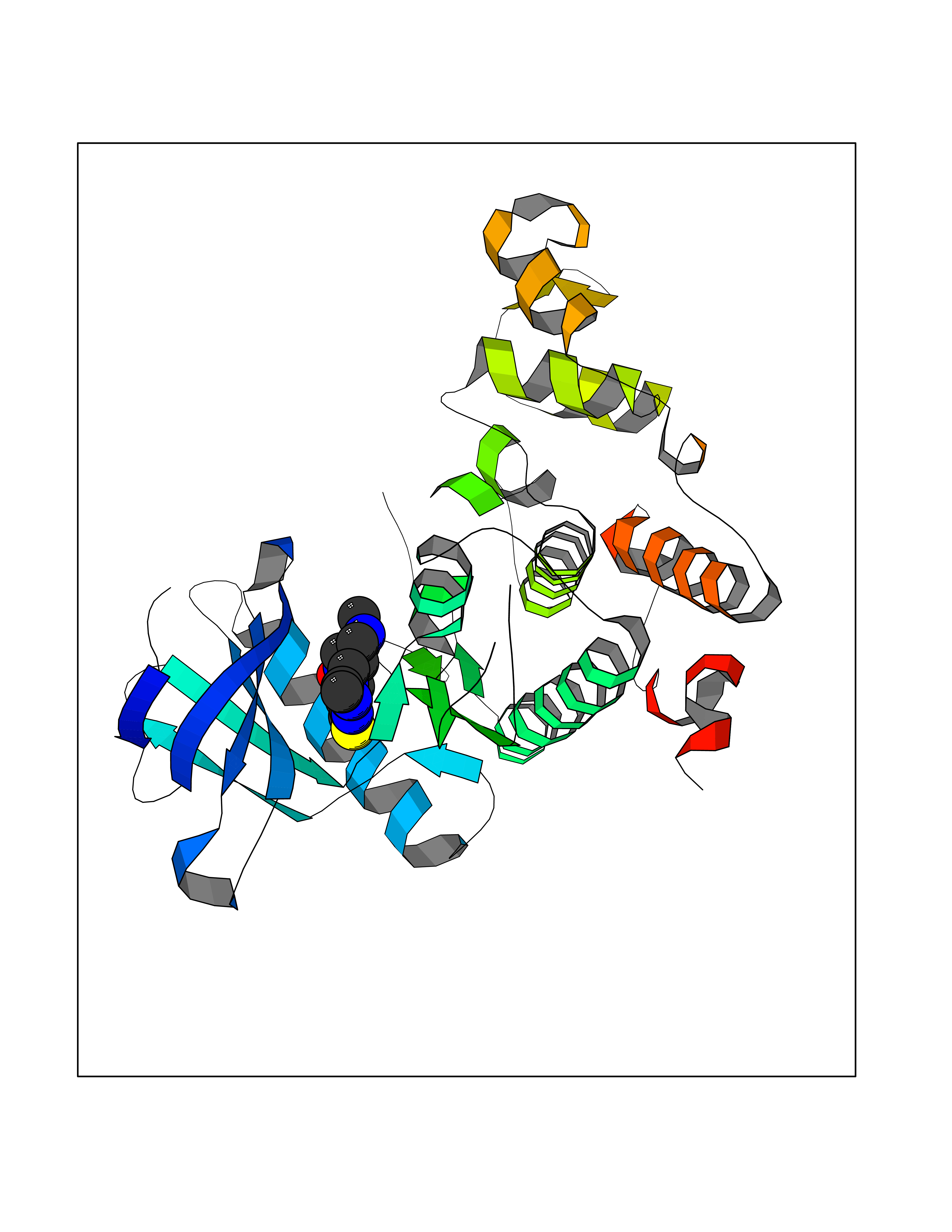 Molscript Map 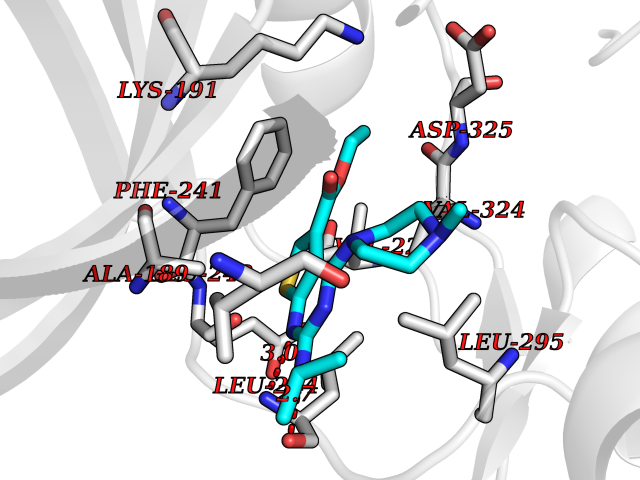 Pymol Map 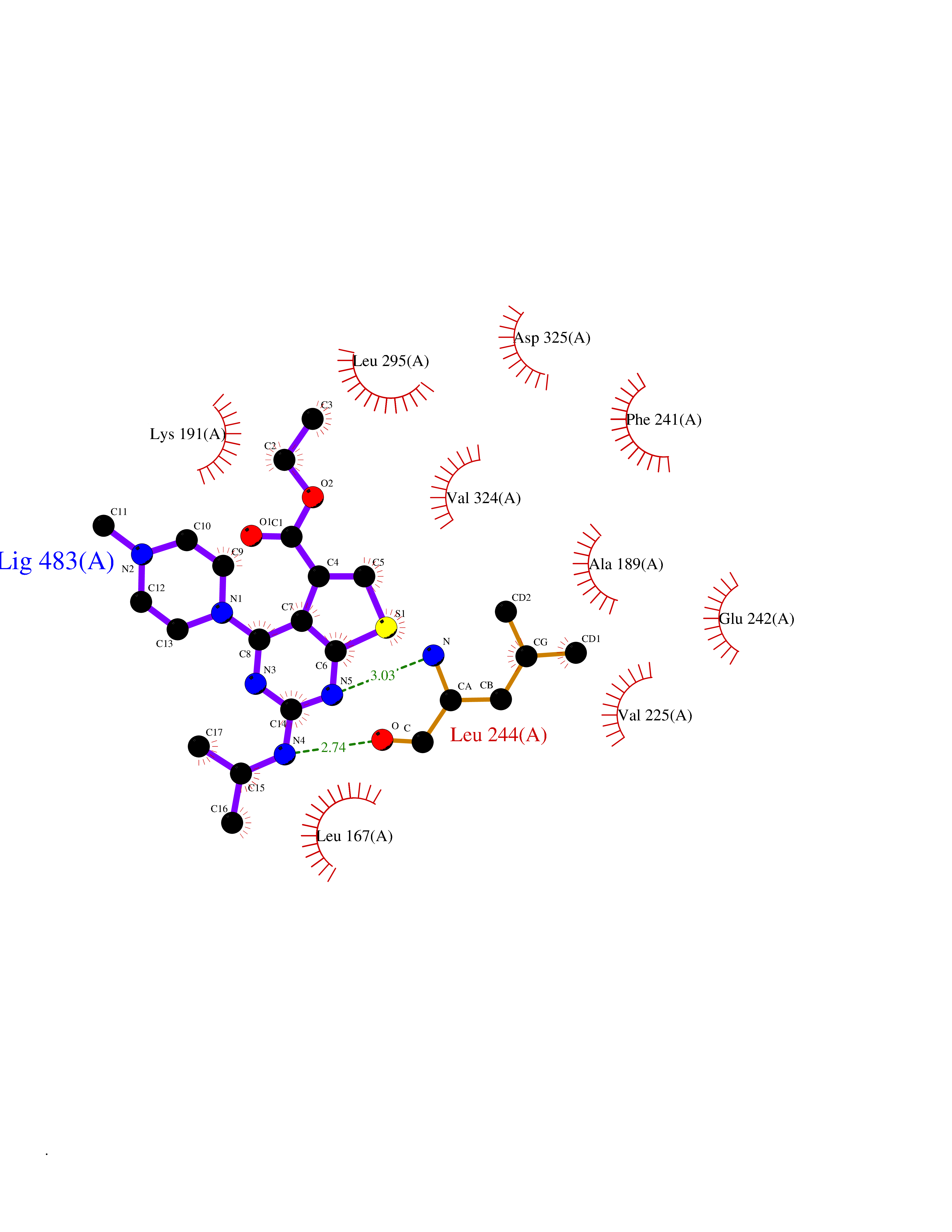 Ligplot Map 3D Binding mode Sequence LICQSGDVLSARYEIVDTLGEGAFGKVVECIDHKAGGRHVAVKIVKNVDRYCEAARSEIQVLEHLNTTDPNSTFRCVQMLEWFEHHGHICIVFELLGLSTYDFIKENGFLPFRLDHIRKMAYQICKSVNFLHSNKLTHTDLKPENILFVQSDYTEERTLINPDIKVVDFGSATYDDEHHSTLVRHYRAPEVILALGWSQPCDVWSIGCILIEYYLGFTVFPTHDSKEHLAMMERILGPLPKHMIQKTRKRKYFHHDRLDWDEHSSAGRYVSRACKPLKEFMLSQDVEHERLFDLIQKMLEYDPAKRITLREALKHPFFDLLK Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | N-acylethanolamine-hydrolyzing acidamidase (NAAA) | 6DXX | 6.43 | |
Target general information Gen name NAAA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nacylsphingosine amidohydrolaselike; Nacylethanolaminehydrolyzing acid amidase subunit beta; NAAA; Acid ceramidaselike protein; ASAHlike protein Protein family Acid ceramidase family Biochemical class Carbon-nitrogen hydrolase Function Degrades bioactive fatty acid amides to their corresponding acids, with the following preference: N- palmitoylethanolamine > N-myristoylethanolamine > N- lauroylethanolamine = N-stearoylethanolamine > N- arachidonoylethanolamine > N-oleoylethanolamine. Also exhibits weak hydrolytic activity against the ceramides N- lauroylsphingosine and N-palmitoylsphingosine. Related diseases Hypertriglyceridemia, transient infantile (HTGTI) [MIM:614480]: An autosomal recessive disorder characterized by onset of moderate to severe transient hypertriglyceridemia in infancy that normalizes with age. The hypertriglyceridemia is associated with hepatomegaly, moderately elevated transaminases, persistent fatty liver, and the development of hepatic fibrosis. {ECO:0000269|PubMed:22226083, ECO:0000269|PubMed:24549054}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09061; DB14009; DB14011 Interacts with NA EC number EC 3.5.1.- Uniprot keywords 3D-structure; Alternative splicing; Autocatalytic cleavage; Direct protein sequencing; Disulfide bond; Fatty acid metabolism; Glycoprotein; Hydrolase; Lipid degradation; Lipid metabolism; Lysosome; Membrane; Proteomics identification; Reference proteome; Signal; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 36877.8 Length 328 Aromaticity 0.11 Instability index 44.37 Isoelectric point 7.72 Charge (pH=7) 1.08 2D Binding mode Binding energy (Kcal/mol) -8.77 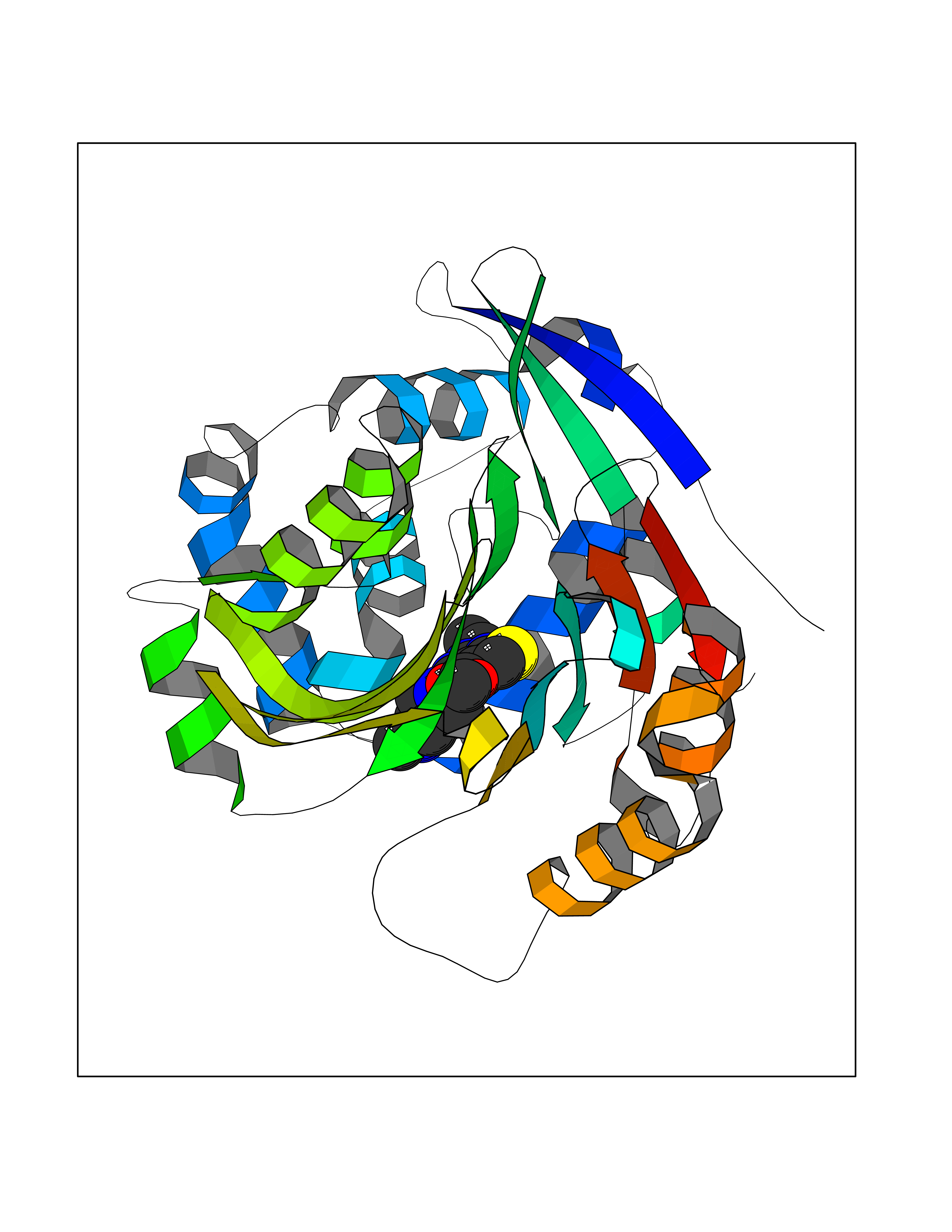 Molscript Map 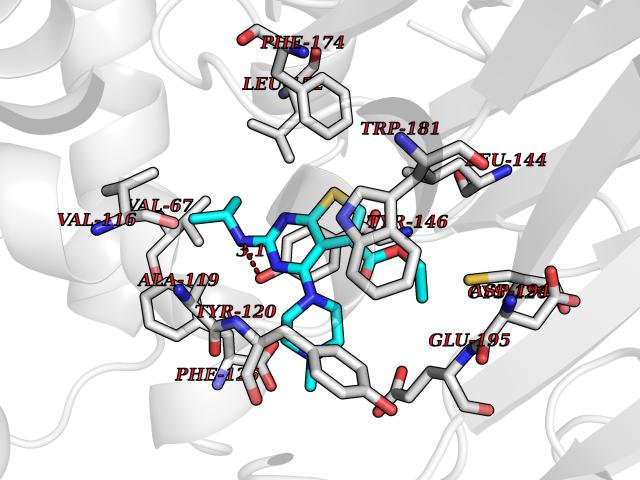 Pymol Map 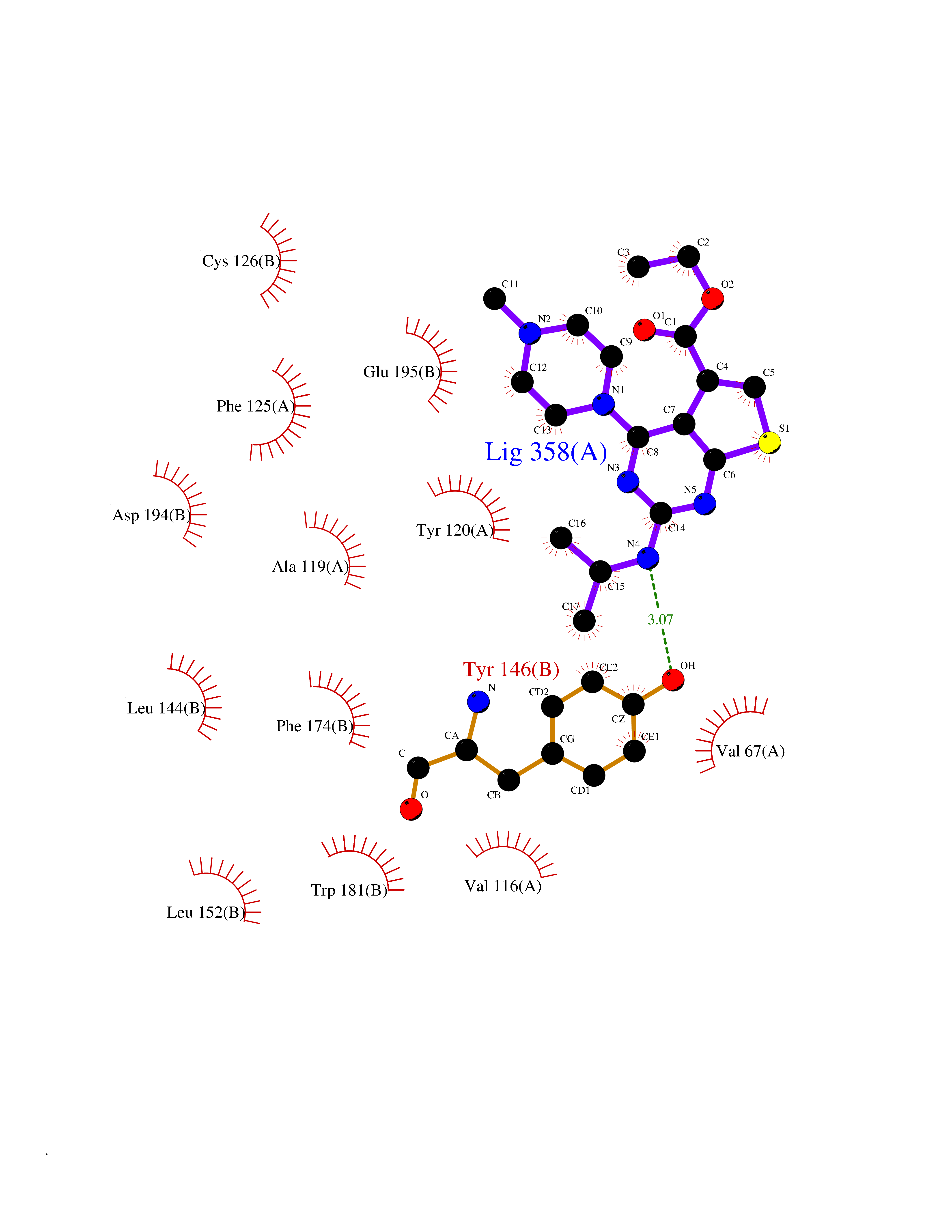 Ligplot Map 3D Binding mode Sequence SPPAAPRFNVSLDSVPELRWLPVLRHYDLDLVRAAMAQVIGDRVPKWVHVLIGKVVLELERFLPQPFTGEIRGMCDFMNLSLADCLLVNLAYESSVFCTSIVAQDSRGHIYHGRNLDYPFGNVLRKLTVDVQFLKNGQIAFTGTTFIGYVGLWTGQSPHKFTVSGDERDKGWWWENAIAALFRRHIPVSWLIRATLSESENFEAAVGKLAKTPLIADVYYIVGGTSPREGVVITRNRDGPADIWPLDPLNGAWFRVETNYDHWKPAPKEDDRRTSAIKALNATGQANLSLEALFQILSVVPVYNNFTIYTTVMSAGSPDKYMTRIRNP Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Progesterone receptor (PGR) | 1SQN | 6.42 | |
Target general information Gen name PGR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PR; Nuclear receptor subfamily 3 group C member 3; NR3C3 Protein family Nuclear hormone receptor family, NR3 subfamily Biochemical class Nuclear hormone receptor Function Depending on the isoform, progesterone receptor functions as transcriptional activator or repressor. The steroid hormones and their receptors are involved in the regulation of eukaryotic gene expression and affect cellular proliferation and differentiation in target tissues. Related diseases Butyrylcholinesterase deficiency (BCHED) [MIM:617936]: An autosomal recessive metabolic condition characterized by increased sensitivity to certain anesthetic drugs, including the muscle relaxants succinylcholine or mivacurium. BCHED results in slower hydrolysis of these drugs and, consequently, a prolonged neuromuscular block, leading to apnea. The duration of the prolonged apnea varies significantly depending on the extent of the enzyme deficiency. {ECO:0000269|PubMed:10404729, ECO:0000269|PubMed:11928765, ECO:0000269|PubMed:12881446, ECO:0000269|PubMed:1306123, ECO:0000269|PubMed:1349196, ECO:0000269|PubMed:1415224, ECO:0000269|PubMed:15563885, ECO:0000269|PubMed:15781196, ECO:0000269|PubMed:1611188, ECO:0000269|PubMed:16788378, ECO:0000269|PubMed:17700357, ECO:0000269|PubMed:18075469, ECO:0000269|PubMed:18300943, ECO:0000269|PubMed:25054547, ECO:0000269|PubMed:25264279, ECO:0000269|PubMed:2915989, ECO:0000269|PubMed:7634491, ECO:0000269|PubMed:8554068, ECO:0000269|PubMed:8680411, ECO:0000269|PubMed:9110359, ECO:0000269|PubMed:9191541, ECO:0000269|PubMed:9388484, ECO:0000269|PubMed:9543549, ECO:0000269|PubMed:9694584}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01431; DB06680; DB01406; DB12941; DB13857; DB00304; DB09123; DB01395; DB00378; DB11219; DB00823; DB00294; DB13867; DB08906; DB00588; DB06730; DB11619; DB11064; DB06789; DB00367; DB00431; DB09124; DB00603; DB00351; DB02998; DB00834; DB00648; DB00764; DB14512; DB06713; DB00717; DB00957; DB09389; DB01428; DB02746; DB00396; DB14583; DB00421; DB04787; DB05253; DB08867 Interacts with Q9H467; P03372; P06401; P40763; P03372 EC number NA Uniprot keywords 3D-structure; Alternative promoter usage; Alternative splicing; Cytoplasm; Direct protein sequencing; DNA-binding; Isopeptide bond; Lipid-binding; Lipoprotein; Membrane; Metal-binding; Mitochondrion; Mitochondrion outer membrane; Nucleus; Palmitate; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Steroid-binding; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 28853.6 Length 250 Aromaticity 0.09 Instability index 54.82 Isoelectric point 8.4 Charge (pH=7) 2.28 2D Binding mode Binding energy (Kcal/mol) -8.75 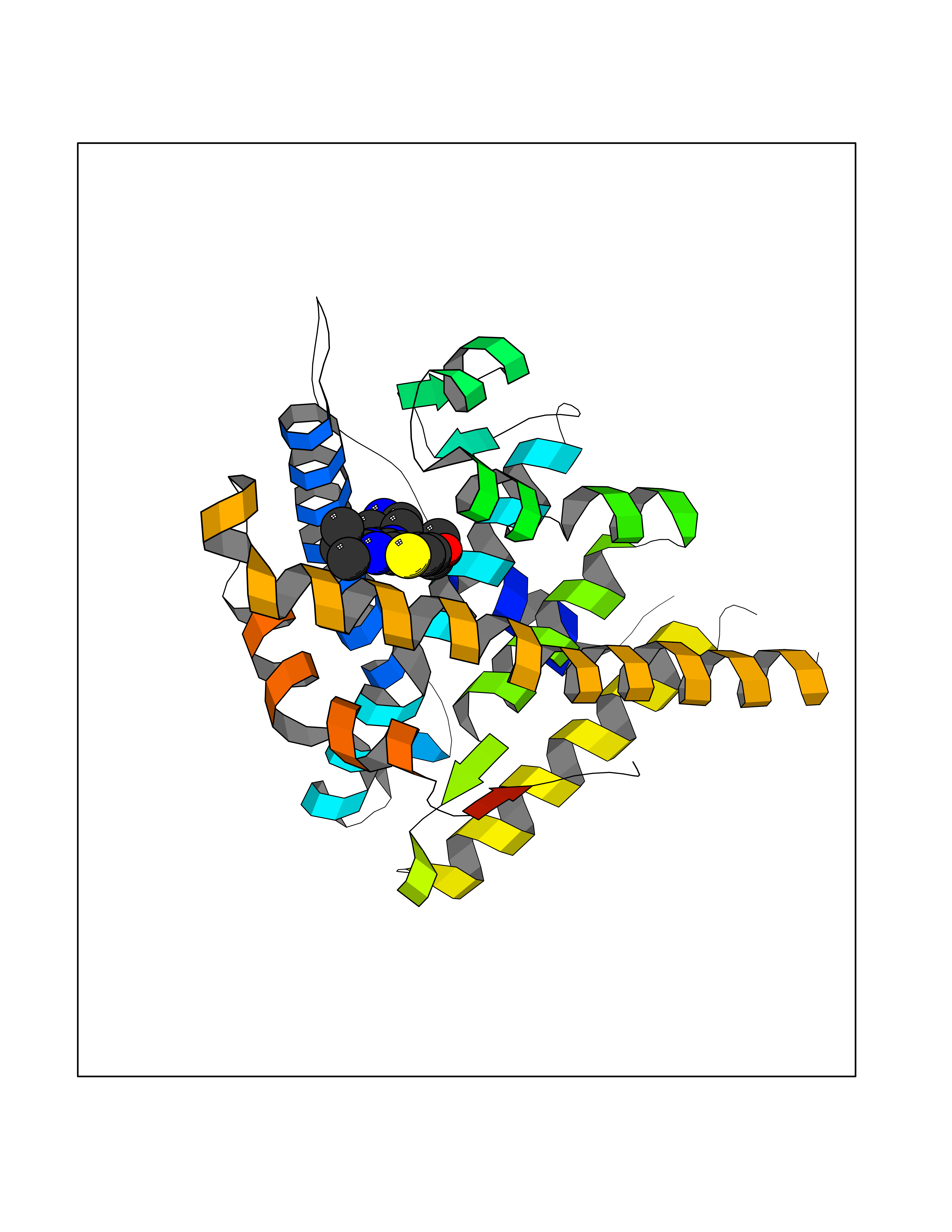 Molscript Map 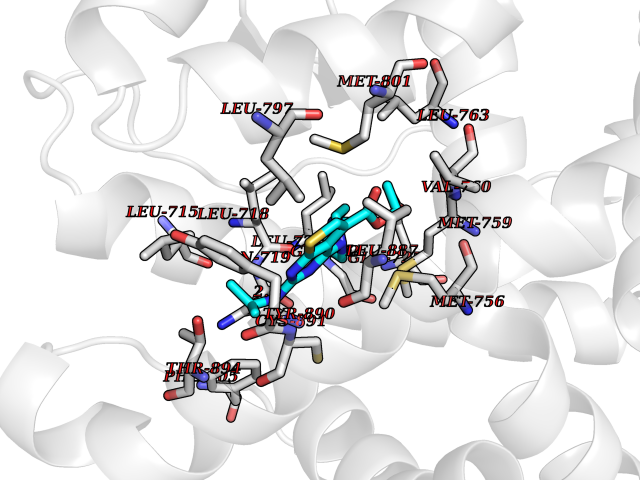 Pymol Map 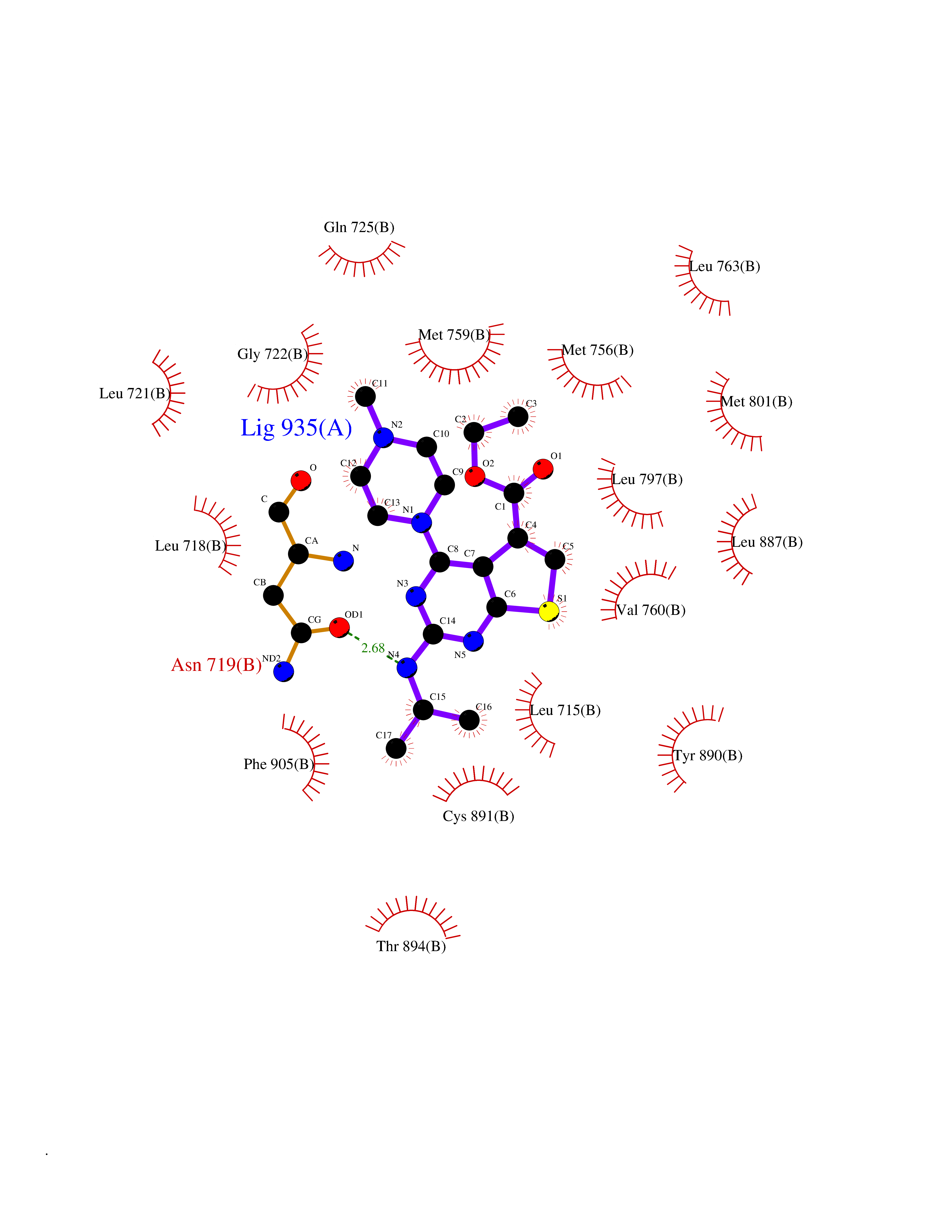 Ligplot Map 3D Binding mode Sequence LIPPLINLLMSIEPDVIYAGHDNTKPDTSSSLLTSLNQLGERQLLSVVKWSKSLPGFRNLHIDDQITLIQYSWMSLMVFGLGWRSYKHVSGQMLYFAPDLILNEQRMKESSFYSLCLTMWQIPQEFVKLQVSQEEFLCMKVLLLLNTIPLEGLRSQTQFEEMRSSYIRELIKAIGLRQGVVSSSQRFYQLTKLLDNLHDLVKQLHLYCLNTFIQSRALSVEFPEMMSEVIAAQLPKILAGMVKPLLFHKK Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Pyruvate synthase | 2C42 | 6.42 | |
Target general information Gen name por Organism Desulfocurvibacter africanus (Desulfovibrio africanus) Uniprot ID TTD ID NA Synonyms NA Protein family Pyruvate:ferredoxin/flavodoxin oxidoreductase family Biochemical class Oxidoreductase Function 4 iron, 4 sulfur cluster binding.Iron ion binding.Pyruvate synthase activity.Thiamine pyrophosphate binding. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02410; DB01987; DB00507 Interacts with NA EC number 1.2.7.1 Uniprot keywords 3D-structure; 4Fe-4S; Calcium; Cytoplasm; Direct protein sequencing; Disulfide bond; Electron transport; Iron; Iron-sulfur; Magnesium; Metal-binding; Oxidoreductase; Pyruvate; Thiamine pyrophosphate; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 115569 Length 1065 Aromaticity 0.09 Instability index 31.51 Isoelectric point 6.32 Charge (pH=7) -5.62 2D Binding mode Binding energy (Kcal/mol) -8.75  Molscript Map 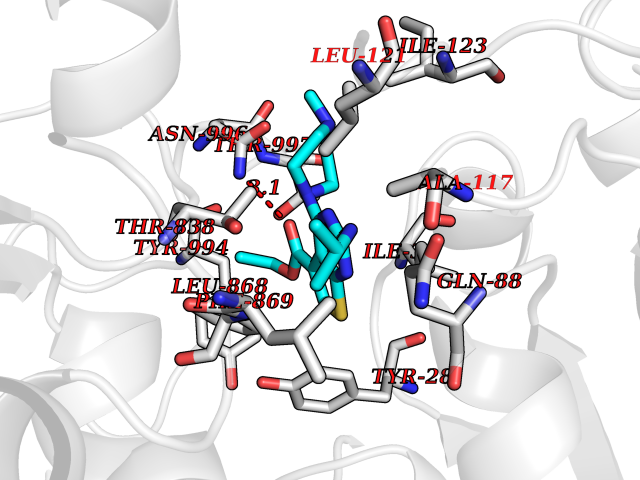 Pymol Map 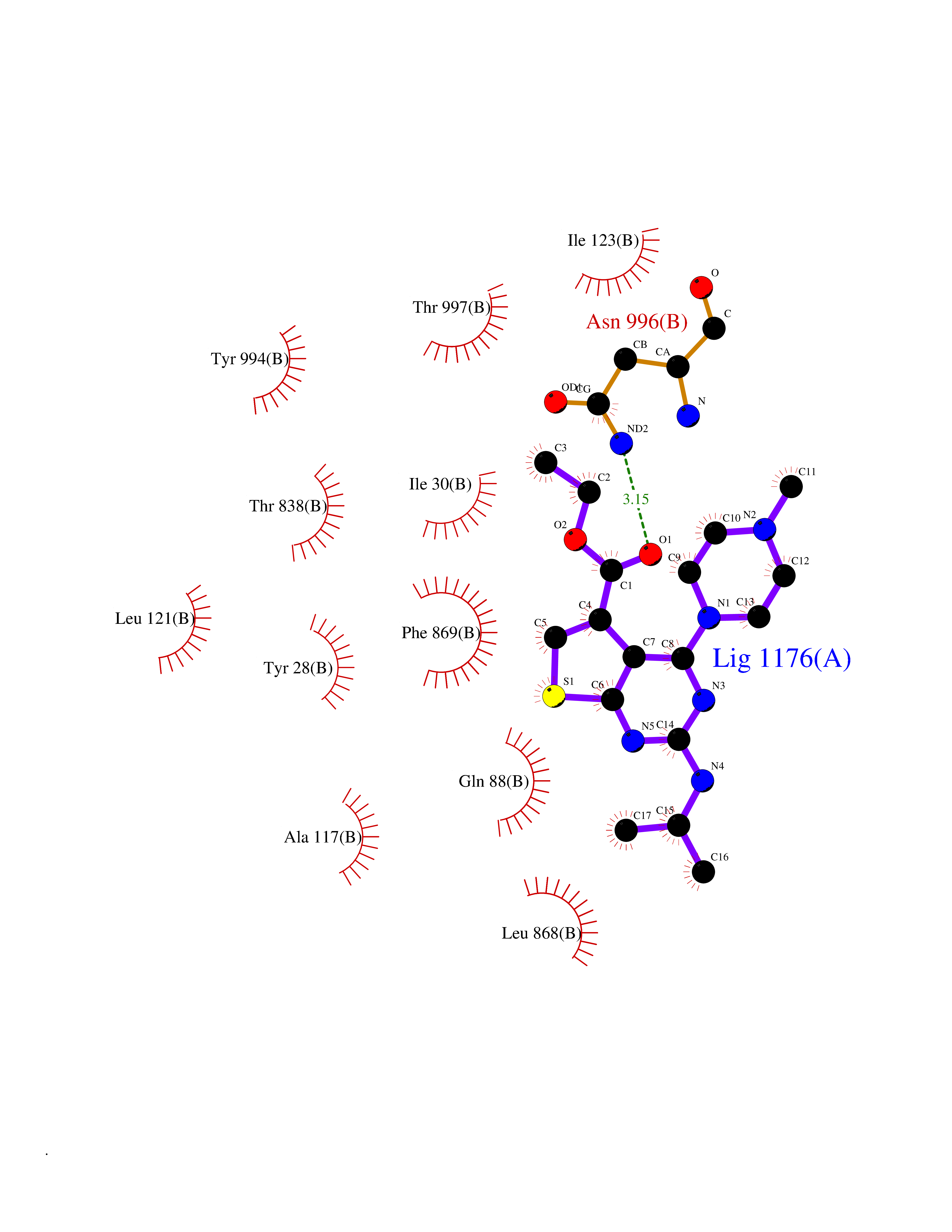 Ligplot Map 3D Binding mode Sequence GKKMMTTDGNTATAHVAYAMSEVAAIYPITPSSTMGEEADDWAAQGRKNIFGQTLTIREMQSEAGAAGAVHGALAAGALTTTFTASQGLLLMIPNMYKISGELLPGVFHVTARAIAAHALSIFGDHQDIYAARQTGFAMLASSSVQEAHDMALVAHLAAIESNVPFMHFFDGFRTSHEIQKIEVLDYADMASLVNQKALAEFRAKSPGIVAEYMQKVASLTGRSYKLFDYVGAPDAERVIVSMGSSCETIEEVINHLAAKGEKIGLIKVRLYRPFVSEAFFAALPASAKVITVLDRTKEPGAPGDPLYLDVCSAFVERGEAMPKILAGRYGLGSKEFSPAMVKSVYDNMSGAKKNHFTVGIEDDVTGTSLPVDNAFADTTPKGTIQCQFWGLGADGTVGANKQAIKIIGDNTDLFAQGYFSYDSKKSGGITISHLRFGEKPIQSTYLVNRADYVACHNPAYVGIYDILEGIKDGGTFVLNSPWSSLEDMDKHLPSGIKRTIANKKLKFYNIDAVKIATDVGLGGRINMIMQTAFFKLAGVLPFEKAVDLLKKSIHKAYGKKGEKIVKMNTDAVDQAVTSLQEFKYPDSWKDAPAETKAEPMTNEFFKNVVKPILTQQGDKLPVSAFEADGRFPLGTSQFEKRGVAINVPQWVPENCIQCNQCAFVCPHSAILPVLAKEEELVGAPANFTALEAKGKELKGYKFRIQINTLDCMGCGNCADICPPKEKALVMQPLDTQRDAQVPNLEYAARIPVKSEVLPRDSLKGSQFQEPLMEFSGACSGCGETPYVRVITQLFGERMFIANATGCSSIWGASAPSMPYKTNRLGQGPAWGNSLFEDAAEYGFGMSVWIFGGDGWAYDIGYGGLDHVLASGEDVNVFVMDTEVYSNTGGQSSKATPTGAVAKFAAAGKRTGKKDLARMVMTYGYVYVATVSMGYSKQQFLKVLKEAESFPGPSLVIAYATCINQGLRKGMGKSQDVMNTAVKSGYWPLFRYDPRLAAQGKNPFQLDSKAPDGSVEEFLMAQNRFAVLDRSFPEDAKRLRAQVAHELDVRFKELEHMAATNIFES Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Riboflavin kinase | 1NB0 | 6.42 | |
Target general information Gen name RFK Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Transferase Function ATP binding.Metal ion binding.Riboflavin kinase activity. Related diseases Glutaric aciduria 1 (GA1) [MIM:231670]: An autosomal recessive metabolic disorder characterized by progressive dystonia and athetosis due to gliosis and neuronal loss in the basal ganglia. {ECO:0000269|PubMed:14707522, ECO:0000269|PubMed:18775954, ECO:0000269|PubMed:24973495, ECO:0000269|PubMed:8541831, ECO:0000269|PubMed:8900227, ECO:0000269|PubMed:8900228, ECO:0000269|PubMed:9600243, ECO:0000269|PubMed:9711871}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03247; DB00140 Interacts with Q9NXG0-2; P19438; P19438-1 EC number 2.7.1.26 Uniprot keywords 3D-structure; ATP-binding; Cytoplasm; Flavoprotein; FMN; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Proteomics identification; Reference proteome; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 16749.9 Length 147 Aromaticity 0.12 Instability index 41.55 Isoelectric point 7.09 Charge (pH=7) 0.12 2D Binding mode Binding energy (Kcal/mol) -8.76  Molscript Map 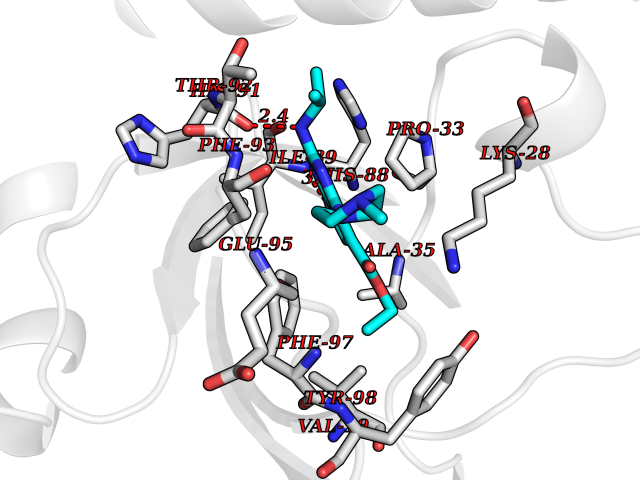 Pymol Map 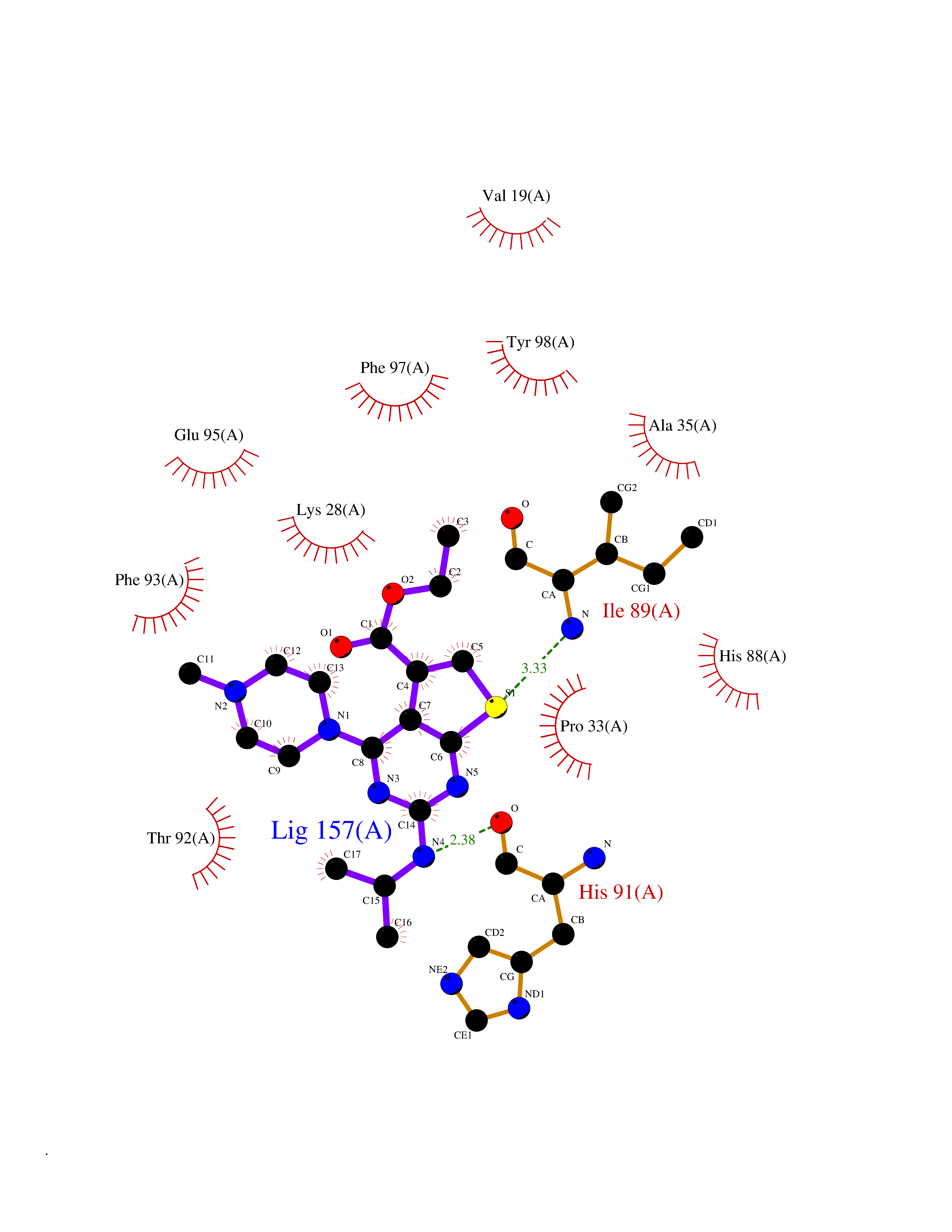 Ligplot Map 3D Binding mode Sequence RHLPYFCRGQVVRGFGRGSKQLGIPTANFPEQVVDNLPADISTGIYYGWASVGSGDVHKMVVSIGWNPYYKNTKKSMETHIMHTFKEDFYGEILNVAIVGYLRPEKNFDSLESLISAIQGDIEEAKKRLELPEYLKIKEDNFFQVSK Hydrogen bonds contact Hydrophobic contact | ||||