Job Results:
Ligand
Structure
Job ID
f5a9e34d4d6e0fad4e04e6dd0d3ae3aa
Job name
NA
Time
2026-01-10 22:50:50
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Matrix metalloproteinase-12 (MMP-12) | 1JK3 | 7.03 | |
Target general information Gen name MMP12 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Macrophage metalloelastase; Macrophage elastase; MME; ME; HME Protein family Peptidase M10A family Biochemical class Peptidase Function Has significant elastolytic activity. Can accept large and small amino acids at the P1' site, but has a preference for leucine. Aromatic or hydrophobic residues are preferred at the P1 site, with small hydrophobic residues (preferably alanine) occupying P3. May be involved in tissue injury and remodeling. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB07026; DB07921; DB04405; DB00551; DB03880; DB07556; DB02118; DB00786; DB07446; DB07683; DB08599; DB08271; DB07922; DB07920; DB05387; DB03367; DB00013 Interacts with NA EC number EC 3.4.24.65 Uniprot keywords 3D-structure; Calcium; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 17435.3 Length 158 Aromaticity 0.13 Instability index 11.56 Isoelectric point 6.52 Charge (pH=7) -1.45 2D Binding mode Binding energy (Kcal/mol) -9.59 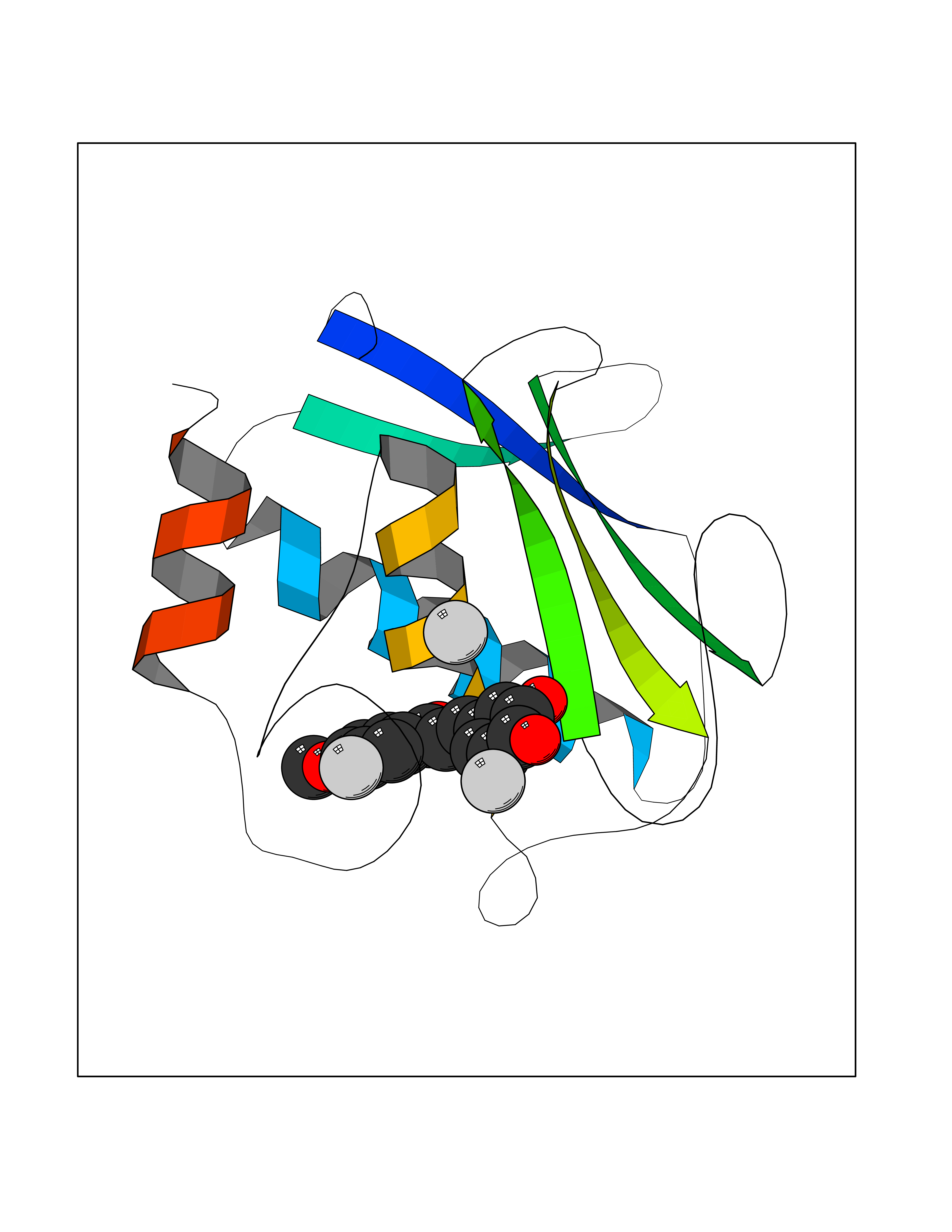 Molscript Map 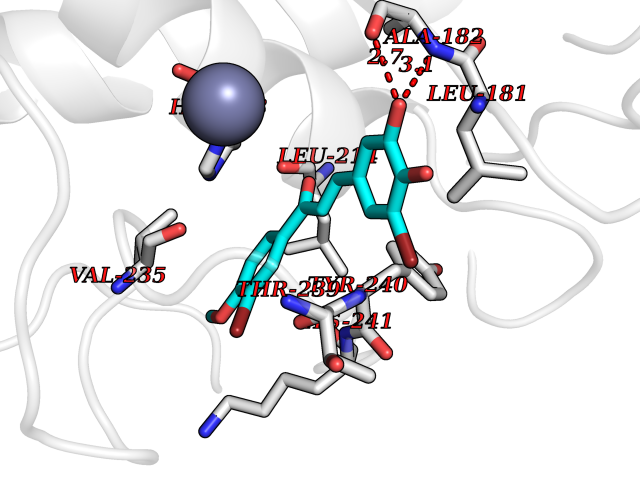 Pymol Map 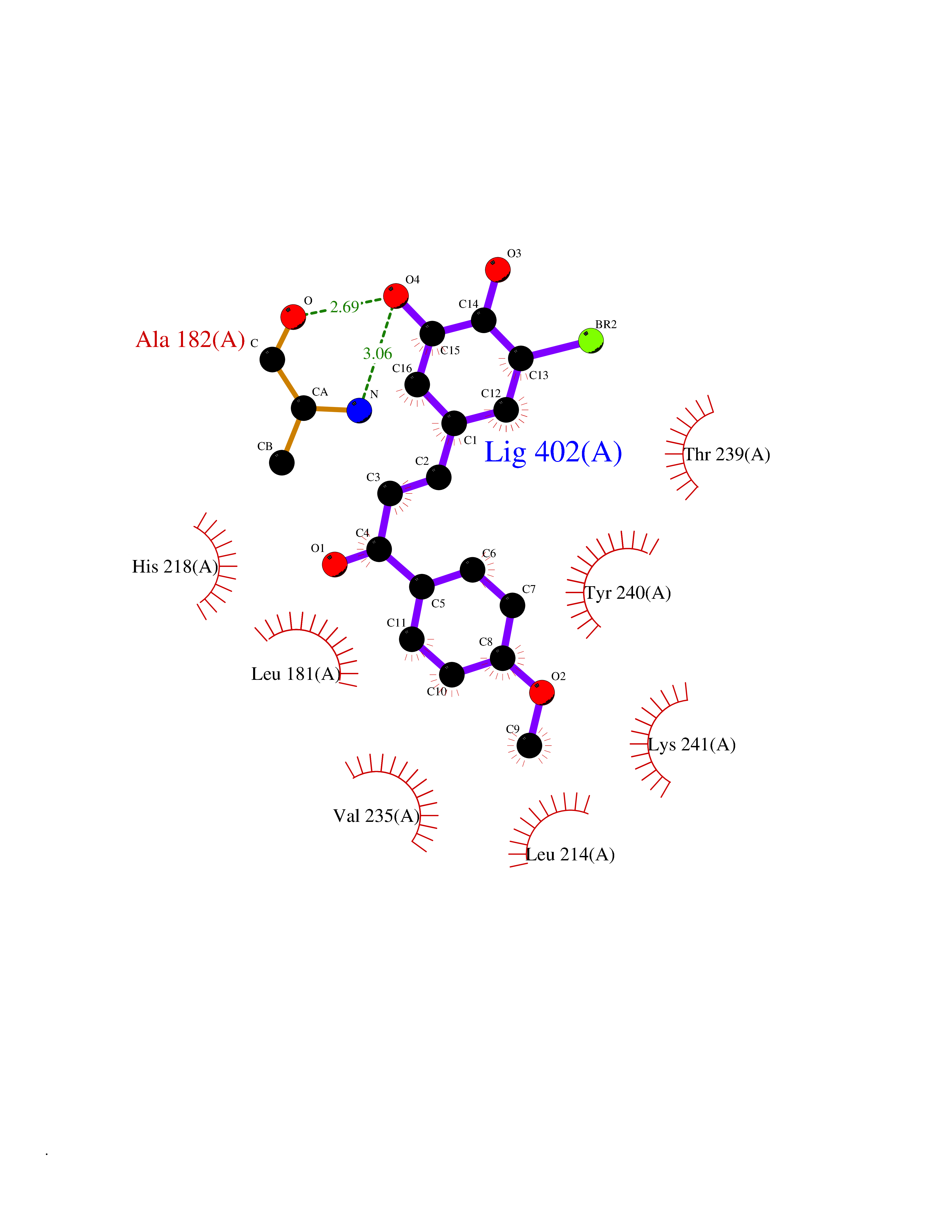 Ligplot Map 3D Binding mode Sequence GPVWRKHYITYRINNYTPDMNREDVDYAIRKAFQVWSNVTPLKFSKINTGMADILVVFARGAHGDFHAFDGKGGILAHAFGPGSGIGGDAHFDEDEFWTTHSGGTNLFLTAVHAIGHSLGLGHSSDPKAVMFPTYKYVDINTFRLSADDIRGIQSLYG Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Vitamin D3 receptor (VDR) | 3B0T | 7.02 | |
Target general information Gen name VDR Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin D(3) receptor; Nuclear vitamin D receptor; Nuclear receptor subfamily 1 group I member 1; NR1I1; 1,25-dihydroxyvitamin D3 receptor Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Enters the nucleus upon vitamin D3 binding where it forms heterodimers with the retinoid X receptor/RXR. The VDR-RXR heterodimers bind to specific response elements on DNA and activate the transcription of vitamin D3-responsive target genes. Plays a central role in calcium homeostasis. Nuclear receptor for calcitriol, the active form of vitamin D3 which mediates the action of this vitamin on cells. Related diseases Rickets vitamin D-dependent 2A (VDDR2A) [MIM:277440]: A disorder of vitamin D metabolism resulting in severe rickets, hypocalcemia and secondary hyperparathyroidism. Most patients have total alopecia in addition to rickets. {ECO:0000269|PubMed:1652893, ECO:0000269|PubMed:17970811, ECO:0000269|PubMed:2177843, ECO:0000269|PubMed:2849209, ECO:0000269|PubMed:28698609, ECO:0000269|PubMed:7828346, ECO:0000269|PubMed:8106618, ECO:0000269|PubMed:8381803, ECO:0000269|PubMed:8392085, ECO:0000269|PubMed:8675579, ECO:0000269|PubMed:8961271, ECO:0000269|PubMed:9005998}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07530; DB08742; DB01436; DB04891; DB00146; DB02300; DB00136; DB00169; DB04540; DB05024; DB11672; DB14635; DB01070; DB06410; DB05295; DB06194; DB00153; DB04796; DB03451; DB00910; DB04258; DB11094 Interacts with P35222; Q09472; Q15648; P50222; Q15788; P26045; P19793; Q13573; Q13501; P04637; Q15645; Q9JLI4; P28700; X5D778; Q96HA8; Q01804; Q96S38; P48443 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 28781 Length 254 Aromaticity 0.07 Instability index 47.69 Isoelectric point 6.15 Charge (pH=7) -3.44 2D Binding mode Binding energy (Kcal/mol) -9.58 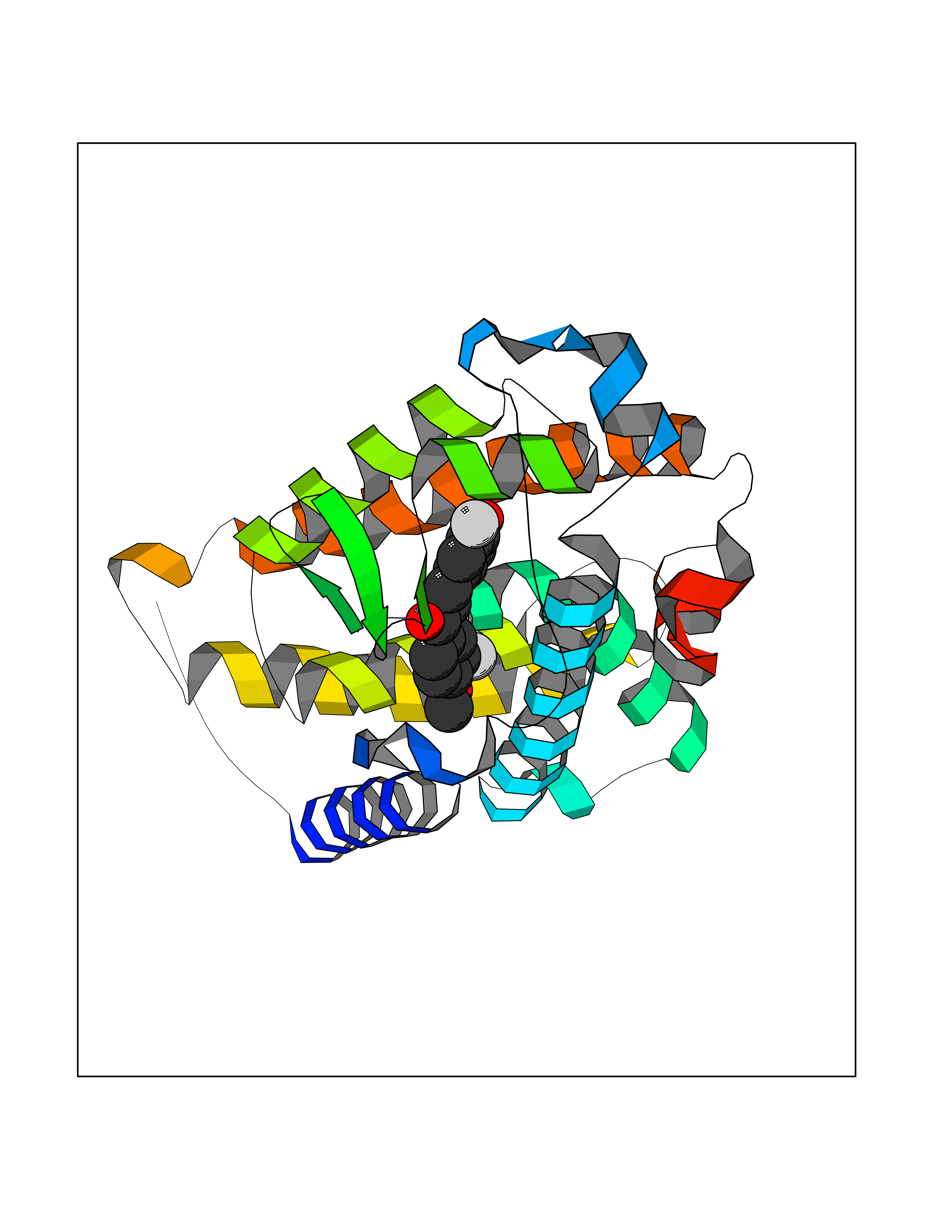 Molscript Map 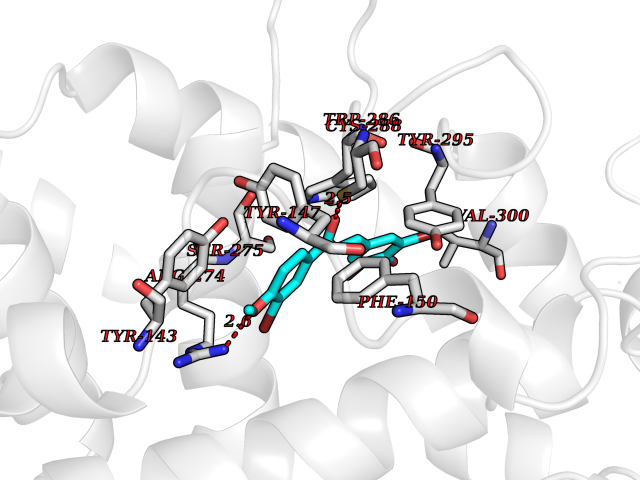 Pymol Map 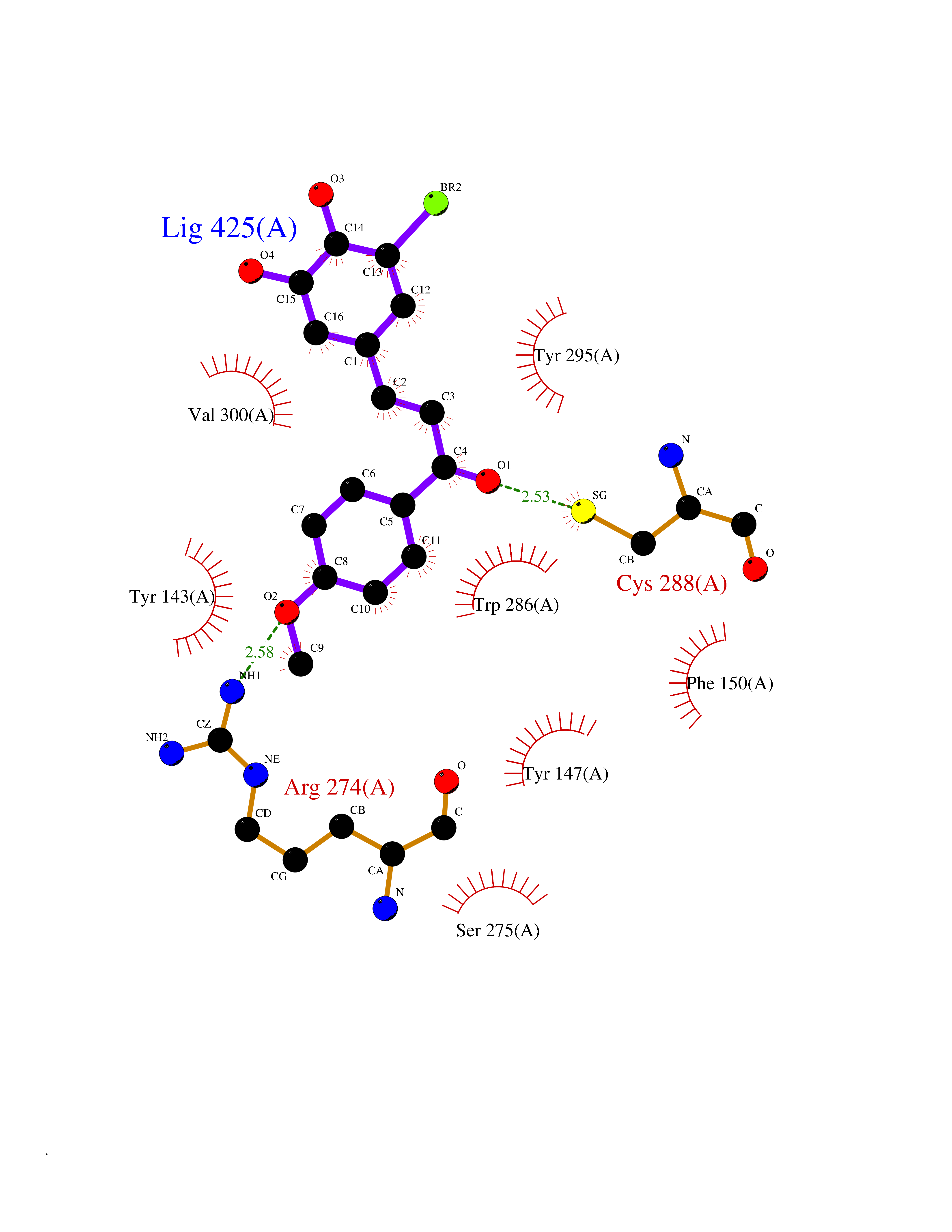 Ligplot Map 3D Binding mode Sequence ALRPKLSEEQQRIIAILLDAHHKTYDPTYSDFCQFRPPVRVNDGGGSVTLELSQLSMLPHLADLVSYSIQKVIGFAKMIPGFRDLTSEDQIVLLKSSAIEVIMLRSNESFTMDDMSWTCGNQDYKYRVSDVTKAGHSLELIEPLIKFQVGLKKLNLHEEEHVLLMAICIVSPDRPGVQDAALIEAIQDRLSNTLQTYIRCRHPPPGSHLLYAKMIQKLADLRSLNEEHSKQYRCLSFQPECSMKLTPLVLEVFG Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Penicillin acylase | 2PVA | 7.02 | |
Target general information Gen name N/A Organism Lysinibacillus sphaericus (Bacillus sphaericus) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase C59 family Biochemical class Hydrolase Function Penicillin amidase activity. Related diseases Usher syndrome 3B (USH3B) [MIM:614504]: A syndrome characterized by progressive vision and hearing loss during early childhood. Some patients have the so-called 'Charles Bonnet syndrome,' involving decreased visual acuity and vivid visual hallucinations. USH is a genetically heterogeneous condition characterized by the association of retinitis pigmentosa with sensorineural deafness. Age at onset and differences in auditory and vestibular function distinguish Usher syndrome type 1 (USH1), Usher syndrome type 2 (USH2) and Usher syndrome type 3 (USH3). USH3 is characterized by postlingual, progressive hearing loss, variable vestibular dysfunction, and onset of retinitis pigmentosa symptoms, including nyctalopia, constriction of the visual fields, and loss of central visual acuity, usually by the second decade of life. {ECO:0000269|PubMed:22279524}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, axonal, 2W (CMT2W) [MIM:616625]: An autosomal dominant, axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. CMT2W patients manifest a peripheral neuropathy mainly affecting the lower limbs and resulting in gait difficulties and distal sensory impairment. Most patients also have upper limb involvement. {ECO:0000269|PubMed:22930593, ECO:0000269|PubMed:26072516, ECO:0000269|PubMed:29235198}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01822; DB03661; DB00417 Interacts with NA EC number 3.5.1.11 Uniprot keywords 3D-structure; Antibiotic resistance; Direct protein sequencing; Hydrolase; Zymogen Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 32972.1 Length 295 Aromaticity 0.11 Instability index 30.94 Isoelectric point 5.65 Charge (pH=7) -4.18 2D Binding mode Binding energy (Kcal/mol) -9.57 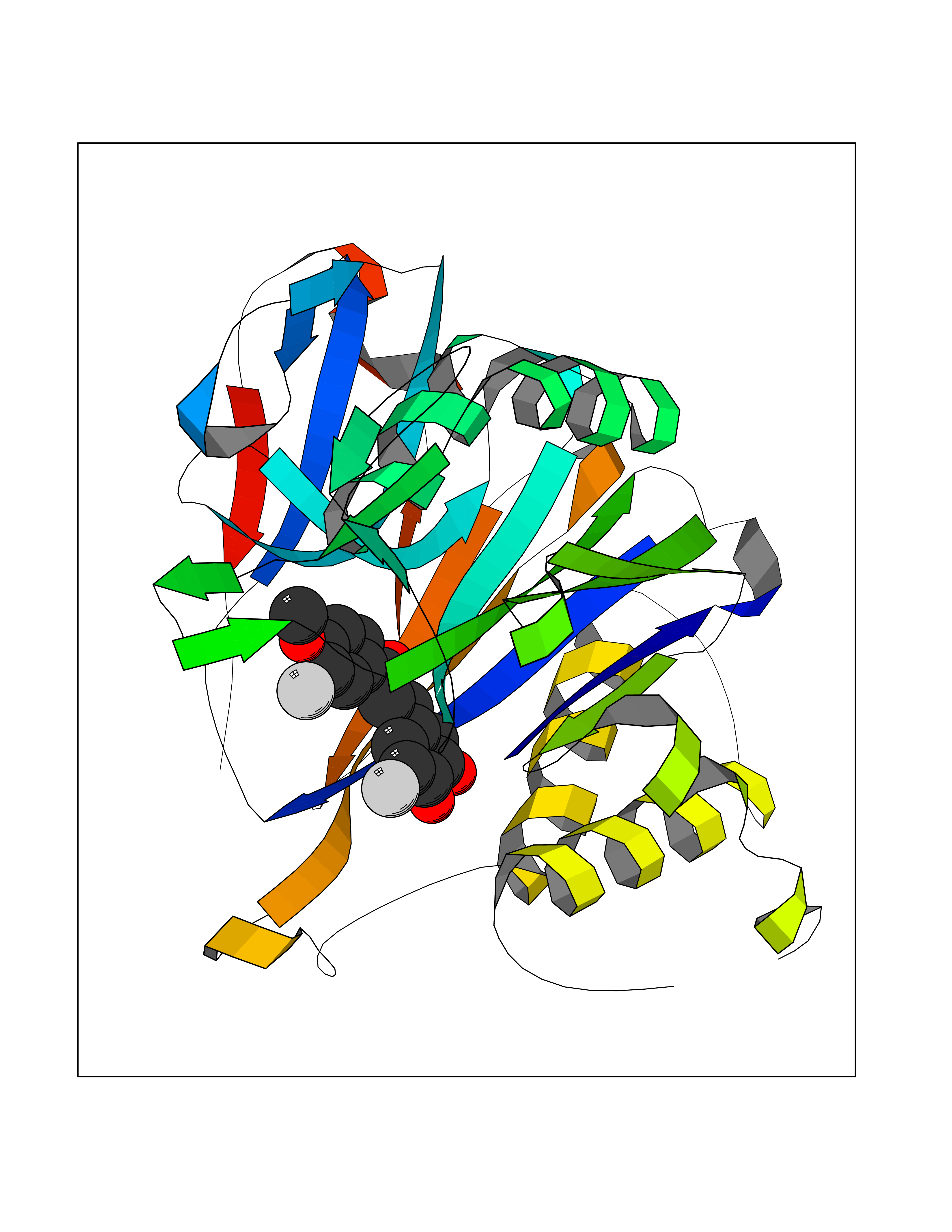 Molscript Map 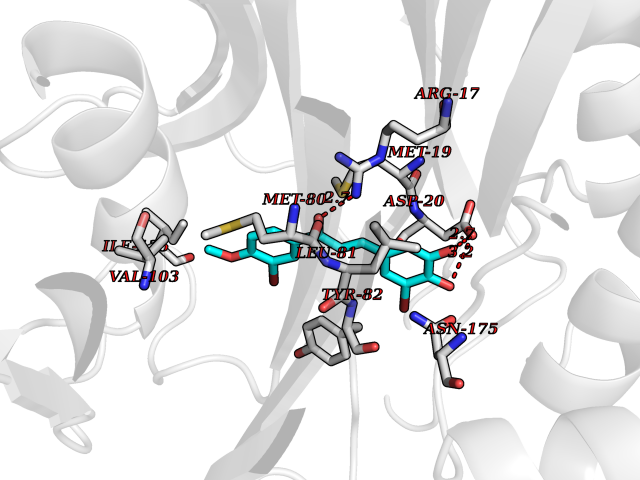 Pymol Map 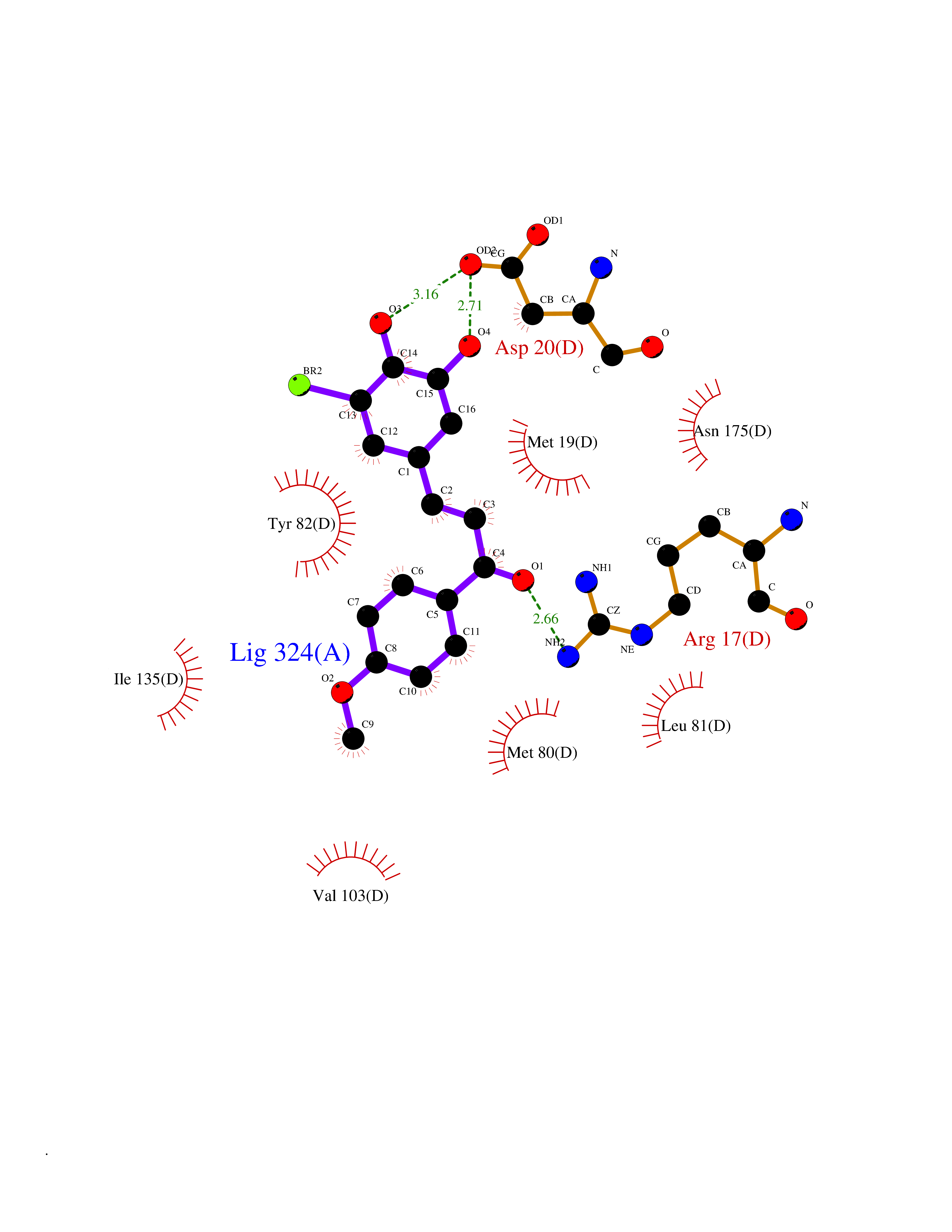 Ligplot Map 3D Binding mode Sequence SSLSIRTTDDKSLFARTMDFTMEPDSKVIIVPRNYGIRLLEKENVVINNSYAFVGMGSTDITSPVLYDGVNEKGLMGAMLYYATFATYADEPKKGTTGINPVYVISQVLGNCVTVDDVIEKLTSYTLLNEANIILGFAPPLHYTFTDASGESIVIEPDKTGITIHRKTIGVMTNSPGYEWHQTNLRAYIGVLPGDFTPSARFLRVAYWKKYTEKAKNETEGVTNLFHILSSVNIPKGVVLTNEGKTDYTIYTSAMCAQSKNYYFKLYDNSRISAVSLMAENLNSQDLITFEWDRK Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | TetR family transcriptional regulator | 2V57 | 7.02 | |
Target general information Gen name lfrR Organism Mycolicibacterium smegmatis (Mycobacterium smegmatis) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Transcription Function DNA binding. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB01123 Interacts with NA EC number NA Uniprot keywords 3D-structure Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 37947.8 Length 348 Aromaticity 0.07 Instability index 36.27 Isoelectric point 5.5 Charge (pH=7) -9.3 2D Binding mode Binding energy (Kcal/mol) -9.57 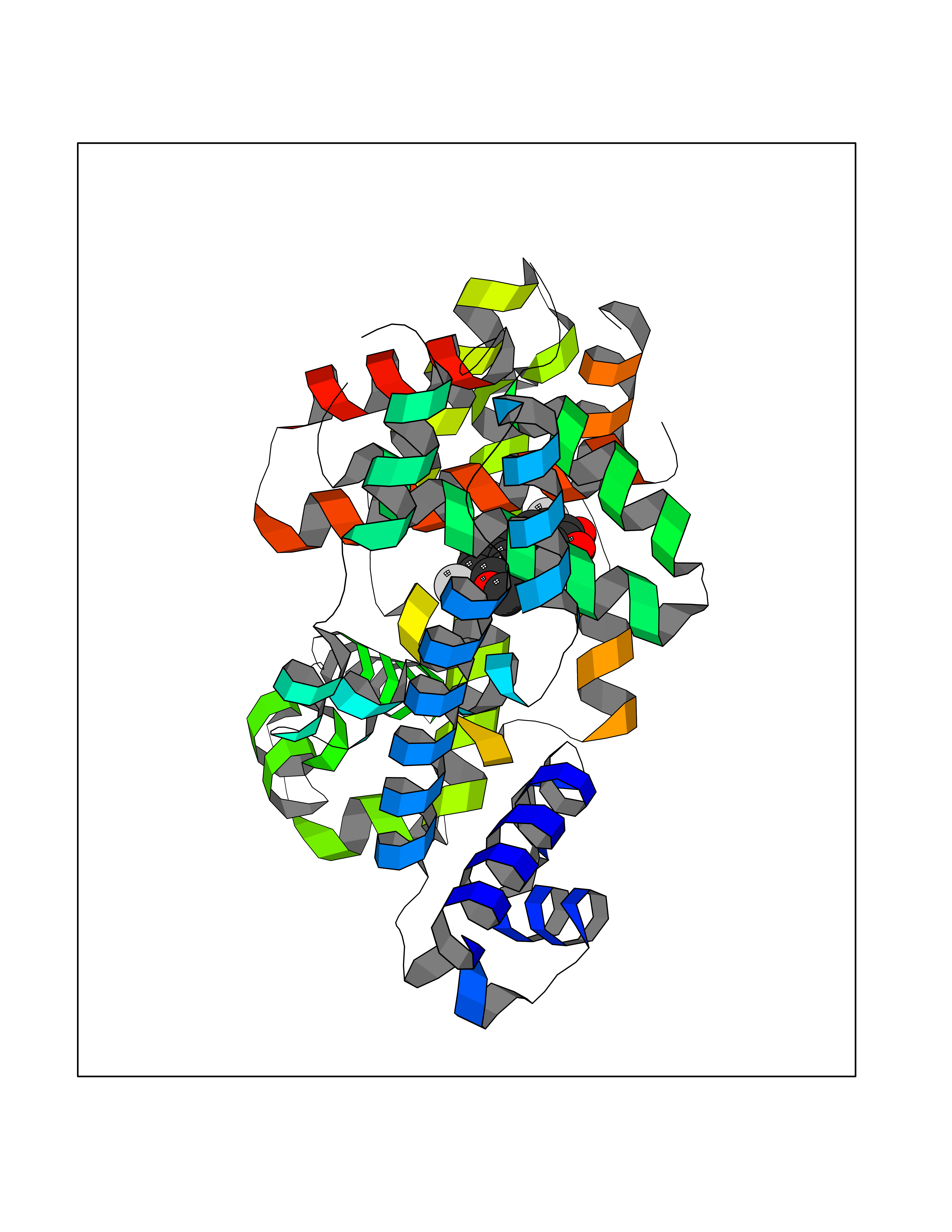 Molscript Map 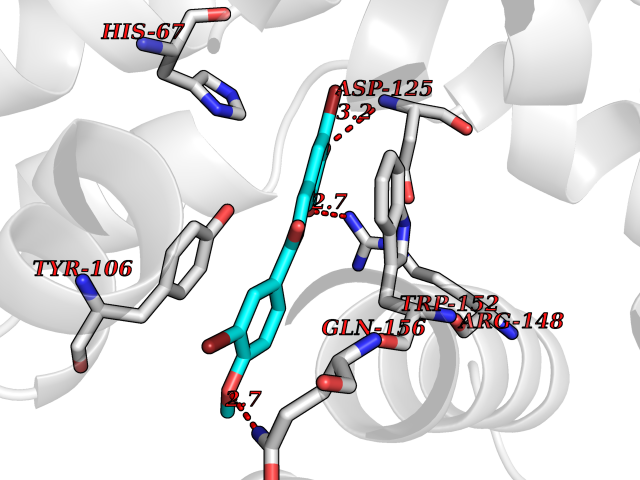 Pymol Map 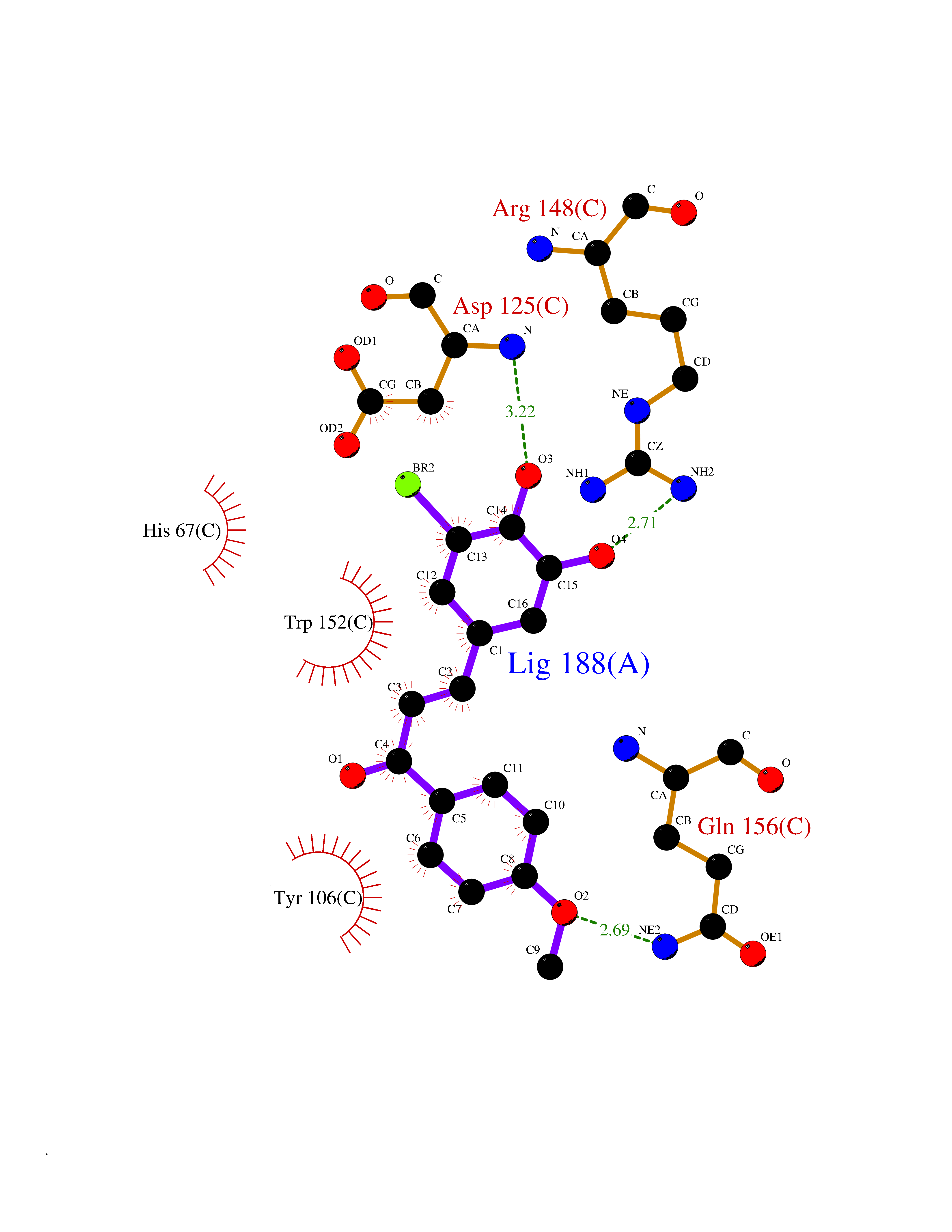 Ligplot Map 3D Binding mode Sequence GARERTRRAILDAAMLVLADHPTAALGDIAAAAGVGRSTVHRYYPERTDLLRALARHVHDLSNAAIERADPTSGPVDAALRRVVESQLDLGPIVLFVYYEPSILADPELAAYFDIGDEAIVEVLNRASTERYPPGWARRVFWALMQAGYEAAKDGMPRHQIVDAIMTSLTSGIITLARERTRRAILDAAMLVLADHPTAALGDIAAAAGVGRSTVHRYYPERTDLLRALARHVHDLSNAAIERADPTSGPVDAALRRVVESQLDLGPIVLFVYYEPSILADPELAAYFDIGDEAIVEVLNRASYPPGWARRVFWALMQAGYEAAKDGMPRHQIVDAIMTSLTSGIITL Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Bacterial DNA ligase (Bact ligA) | 2OWO | 7.02 | |
Target general information Gen name Bact ligA Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms ligA; Polydeoxyribonucleotide synthase [NAD+]; NAD+-dependent DNA ligase Protein family NAD-dependent DNA ligase family, LigA subfamily Biochemical class Phosphoric ester ligase Function DNA ligase that catalyzes the formation of phosphodiester linkages between 5'-phosphoryl and 3'-hydroxyl groups in double-stranded DNA using NAD as a coenzyme and as the energy source for the reaction. It is essential for DNA replication and repair of damaged DNA. Related diseases Dystonia 1, torsion, autosomal dominant (DYT1) [MIM:128100]: A primary torsion dystonia, and the most common and severe form. Dystonia is defined by the presence of sustained involuntary muscle contractions, often leading to abnormal postures. Dystonia type 1 is characterized by involuntary, repetitive, sustained muscle contractions or postures involving one or more sites of the body, in the absence of other neurological symptoms. Typically, symptoms develop first in an arm or leg in middle to late childhood and progress in approximately 30% of patients to other body regions (generalized dystonia) within about five years. 'Torsion' refers to the twisting nature of body movements observed in DYT1, often affecting the trunk. Distribution and severity of symptoms vary widely between affected individuals, ranging from mild focal dystonia to severe generalized dystonia, even within families. {ECO:0000269|PubMed:14970196, ECO:0000269|PubMed:15505207, ECO:0000269|PubMed:16361107, ECO:0000269|PubMed:17428918, ECO:0000269|PubMed:18167355, ECO:0000269|PubMed:18477710, ECO:0000269|PubMed:18827015, ECO:0000269|PubMed:19955557, ECO:0000269|PubMed:20169475, ECO:0000269|PubMed:21102408, ECO:0000269|PubMed:24930953, ECO:0000269|PubMed:27490483, ECO:0000269|PubMed:9288096}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Arthrogryposis multiplex congenita 5 (AMC5) [MIM:618947]: A form of arthrogryposis multiplex congenita, a developmental condition characterized by multiple joint contractures resulting from reduced or absent fetal movements. AMC5 is an autosomal recessive form characterized by severe congenital contractures, developmental delay, strabismus and tremor. {ECO:0000269|PubMed:28516161, ECO:0000269|PubMed:29053766, ECO:0000269|PubMed:30244176}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07813 EC number EC 6.5.1.2 Uniprot keywords 3D-structure; Direct protein sequencing; DNA damage; DNA repair; DNA replication; Ligase; Magnesium; Metal-binding; NAD; Reference proteome; Zinc Protein physicochemical properties Chain ID D,A,C Molecular weight (Da) 66922.6 Length 612 Aromaticity 0.06 Instability index 37.23 Isoelectric point 5.48 Charge (pH=7) -16.8 2D Binding mode Binding energy (Kcal/mol) -9.58 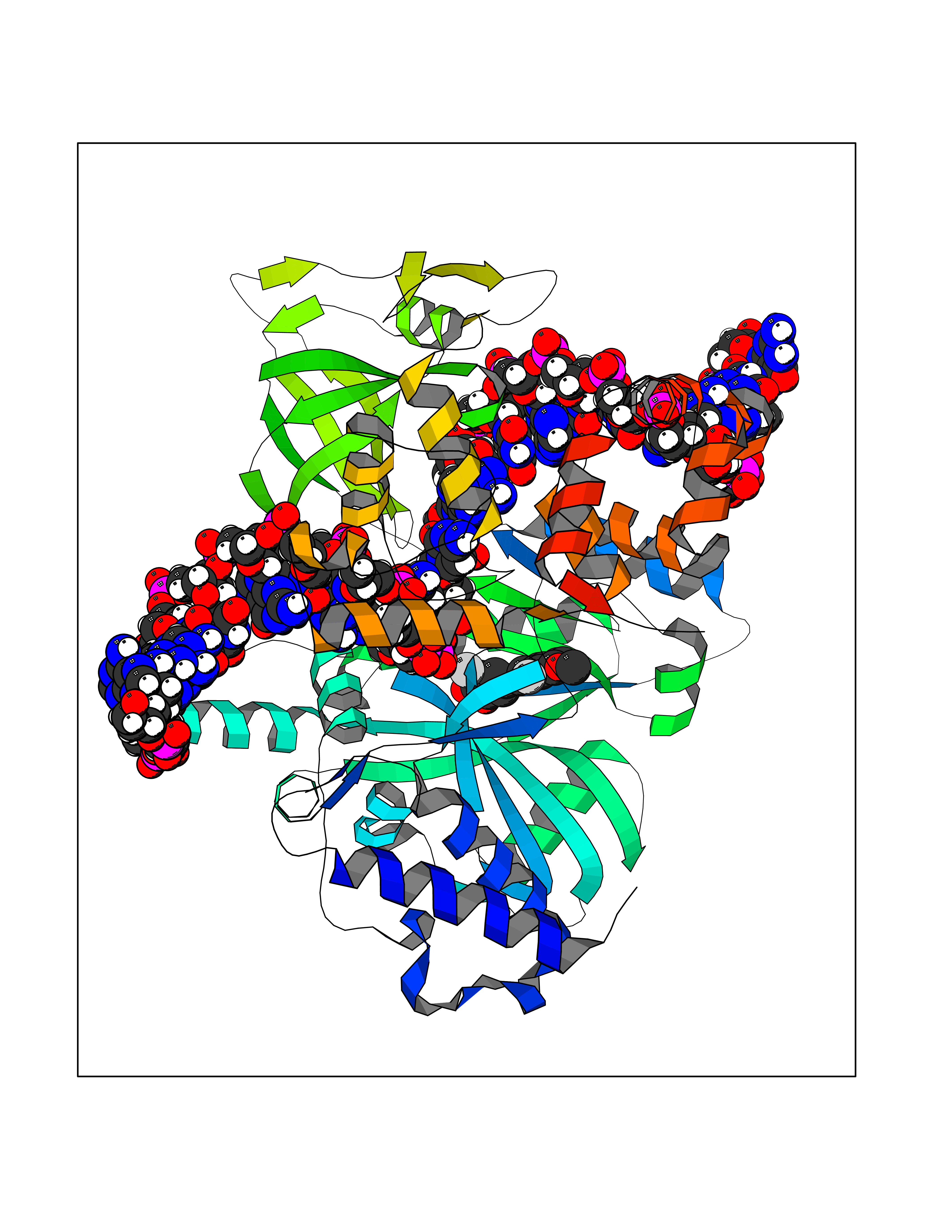 Molscript Map 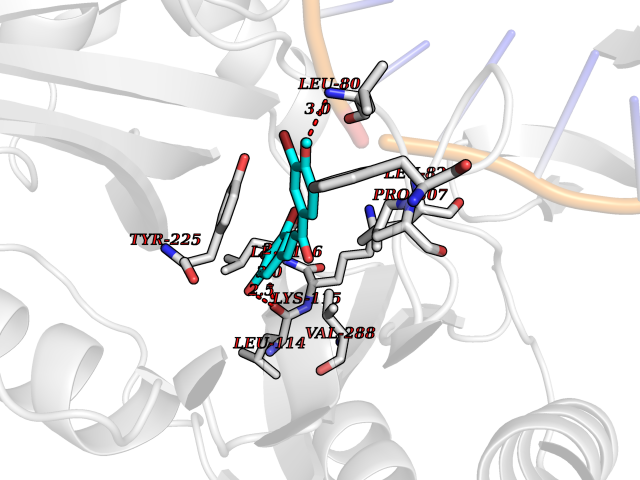 Pymol Map 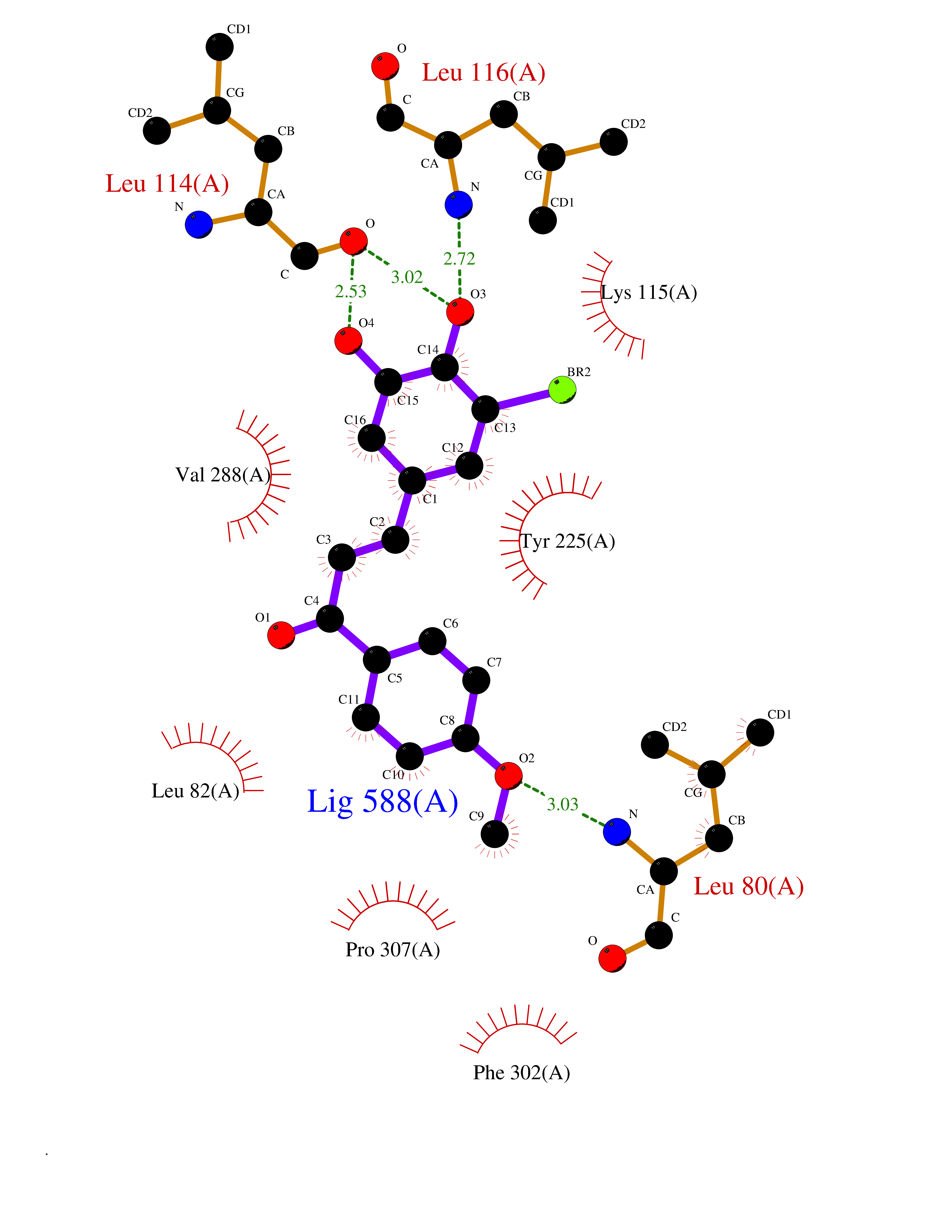 Ligplot Map 3D Binding mode Sequence ACAATTGCGACXCCACTATCGGAATGMESIEQQLTELRTTLRHHEYLYHVMDAPEIPDAEYDRLMRELRELETKHPELITPDSPTQRVGAAPLAAFSQIRHEVPMLSLDNVFDEESFLAFNKRVQDRLKNNEKVTWCCELKLDGLAVSILYENGVLVSAATRGDGTTGEDITSNVRTIRAIPLKLHGENIPARLEVRGEVFLPQAGFEKINEDARRTGGKVFANPRNAAAGSLRQLDPRITAKRPLTFFCYGVGVLEGGELPDTHLGRLLQFKKWGLPVSDRVTLCESAEEVLAFYHKVEEDRPTLGFDIDGVVIKVNSLAQQEQLGFVARAPRWAVAFKFPAQEQMTFVRDVEFQVGRTGAITPVARLEPVHVAGVLVSNATLHNADEIERLGLRIGDKVVIRRAGDVIPQVVNVVLSERPEDTREVVFPTHCPVCGSDVERVEGEAVARCTGGLICGAQRKESLKHFVSRRAMDVDGMGDKIIDQLVEKEYVHTPADLFKLTAGKLTGLERMGPKSAQNVVNALEKAKETTFARFLYALGIREVGEATAAGLAAYFGTLEALEAASIEELQKVPDVGIVVASHVHNFFAEESNRNVISELLAEGVHWPAP Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | "15-cis-phytoene desaturase, chloroplastic/chromoplastic (EC 1.3.5.5) (Phytoene dehydrogenase) (Phytoene desaturase)" | 5MOG | 7.01 | |
Target general information Gen name PDS1 Organism Oryza sativa subsp. indica (Rice) Uniprot ID TTD ID NA Synonyms PDS;OsI_010044 Protein family Carotenoid/retinoid oxidoreductase family Biochemical class NA Function Converts phytoene into zeta-carotene via the intermediary of phytofluene by the symmetrical introduction of two double bonds at the C-11 and C-11' positions of phytoene with a concomitant isomerization of two neighboring double bonds at the C9 and C9' positions from trans to cis. Active with decylplastoquinone (DPQ) as substrate (PubMed:26147209, PubMed:29176862). Also active with other benzoquinones, which are strongly preferred over naphthoquinones as substrates (PubMed:26147209). {ECO:0000269|PubMed:26147209, ECO:0000269|PubMed:29176862}." Related diseases NA Drugs (DrugBank ID) NA Interacts with NA EC number 1.3.5.5 Uniprot keywords 3D-structure; Carotenoid biosynthesis; Chloroplast; Chromoplast; Direct protein sequencing; FAD; Flavoprotein; Membrane; Oxidoreductase; Plastid; Reference proteome; Transit peptide Protein physicochemical properties Chain ID E Molecular weight (Da) 52485.1 Length 466 Aromaticity 0.1 Instability index 45.53 Isoelectric point 5.93 Charge (pH=7) -5.81 2D Binding mode Binding energy (Kcal/mol) -9.56 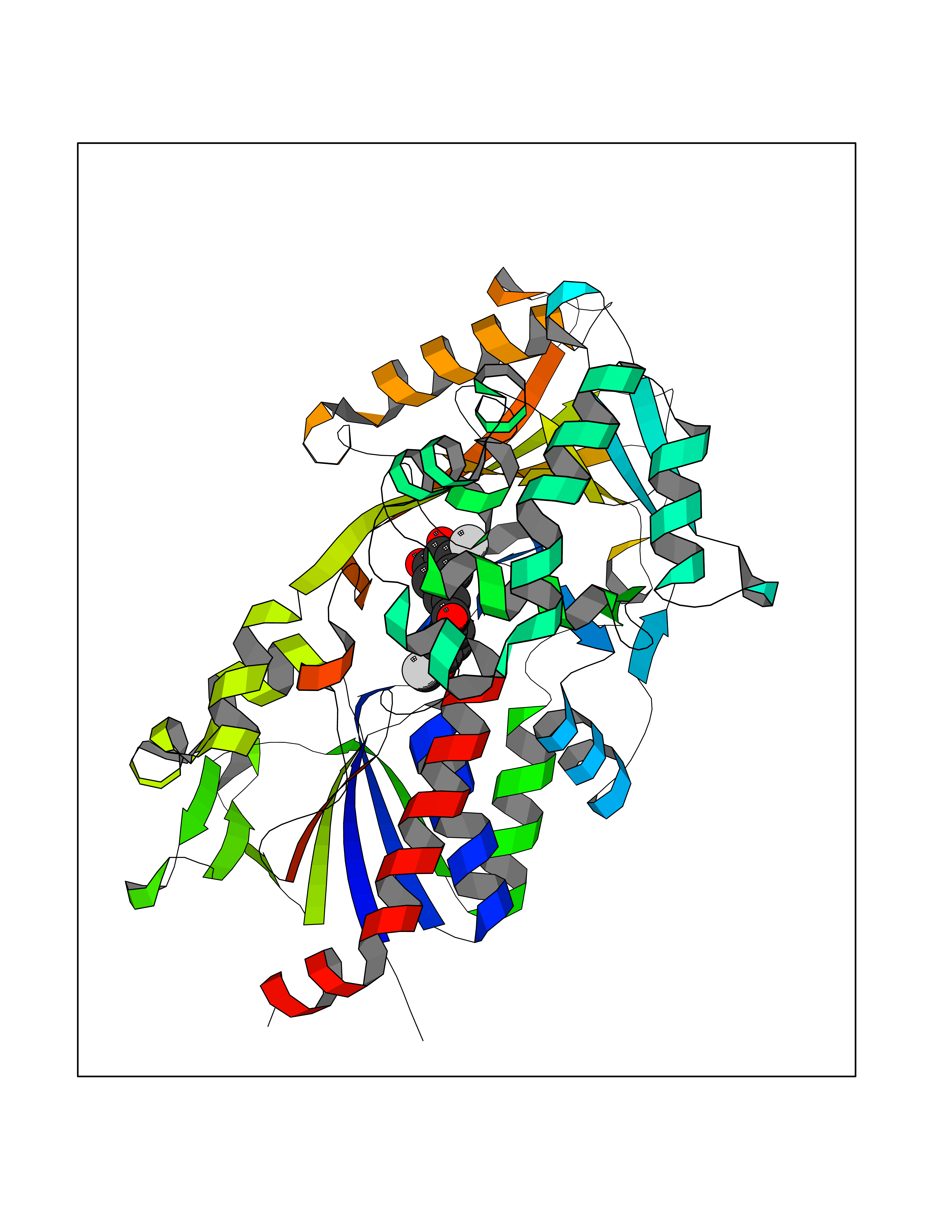 Molscript Map 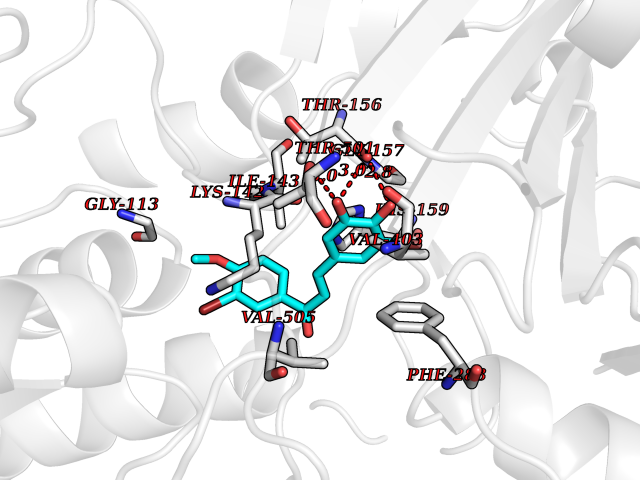 Pymol Map 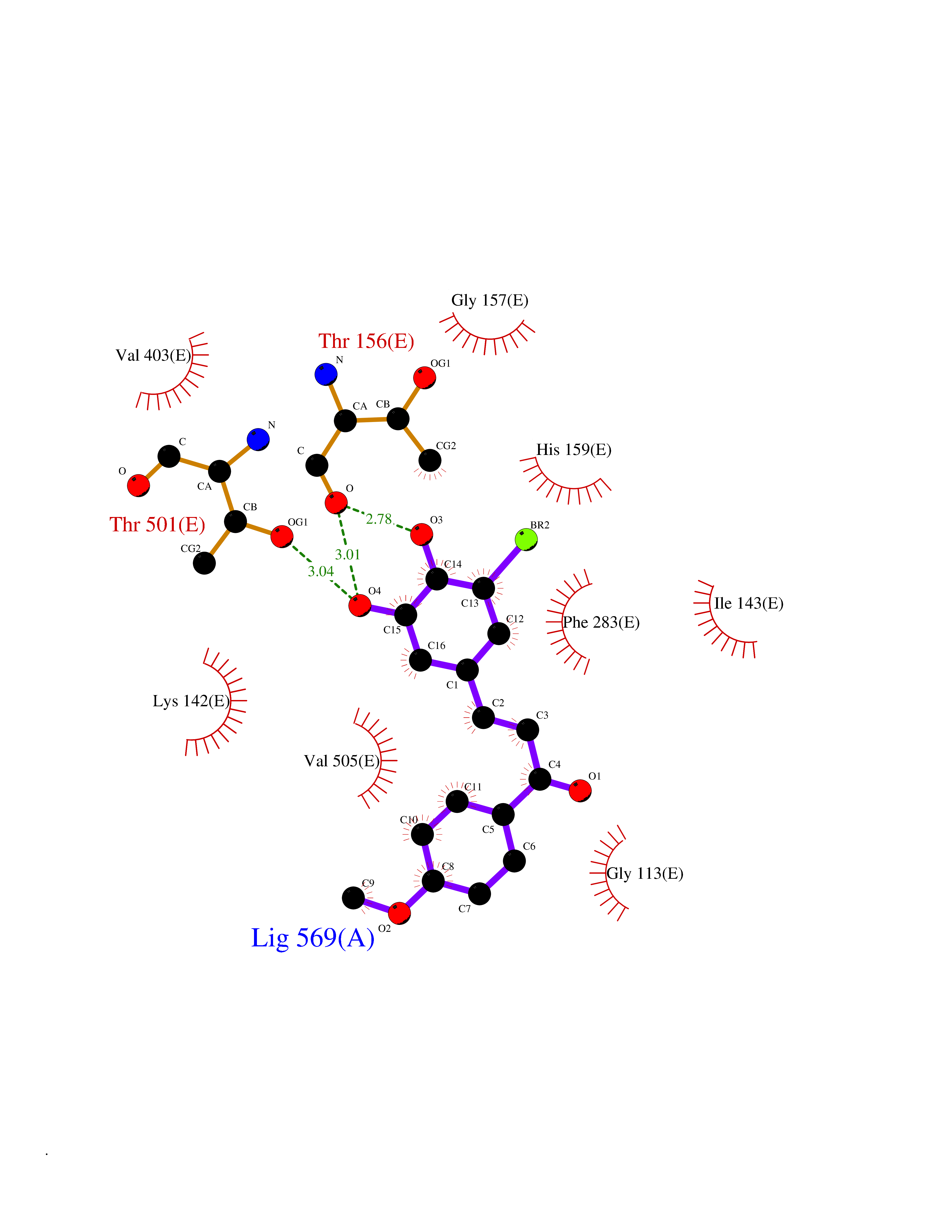 Ligplot Map 3D Binding mode Sequence TKPLQVVIAGAGLAGLSTAKYLADAGHKPILLEARDVLGGKIAAWKDEDGDWYETGLHIFFGAYPNIQNLFGELGINDRLQWKEHSMIFAMPNKPGEFSRFDFPETLPAPLNGIWAILRNNEMLTWPEKVKFALGLLPAMVGGQAYVEAQDGFTVSEWMKKQGVPDRVNDEVFIAMSKALNFINPDELSMQCILIALNRFLQEKHGSKMAFLDGNPPERLCMPIVDHVRSLGGEVRLNSRIQKIELNPDGTVKHFALTDGTQITGDAYVFATPVDILKLLVPQEWKEISYFKKLEKLVGVPVINVHIWFDRKLKNTYDHLLFSRSSLLSVYADMSVTCKEYYDPNRSMLELVFAPAEEWVGRSDTEIIEATMQELAKLFPDEIAADQSKAKILKYHVVKTPRSVYKTIPDCEPCRPLQRSPIEGFYLAGDYTKQKYLASMEGAVLSGKLCAQSVVEDYKMLSRRSL Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Pyruvate kinase M2 (PKM) | 3GR4 | 7.01 | |
Target general information Gen name PKM Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p58; Tumor M2-PK; Thyroid hormone-binding protein 1; THBP1; Pyruvate kinase muscle isozyme; Pyruvate kinase isozymes M1/M2; Pyruvate kinase PKM; Pyruvate kinase 2/3; PKM2; PK3; PK2; Opa-interacting pr Protein family Pyruvate kinase family Biochemical class Kinase Function Stimulates POU5F1-mediated transcriptional activation. Plays a general role in caspase independent cell death of tumor cells. The ratio between the highly active tetrameric form and nearly inactive dimeric form determines whether glucose carbons are channeled to biosynthetic processes or used for glycolytic ATP production. The transition between the 2 forms contributes to the control of glycolysis and is important for tumor cell proliferation and survival. Promotes in a STAT1-dependent manner, the expression of the immune checkpoint protein CD274 in ARNTL/BMAL1-deficient macrophages. Glycolytic enzyme that catalyzes the transfer of a phosphoryl group from phosphoenolpyruvate (PEP) to ADP, generating ATP. Related diseases Congenital sucrase-isomaltase deficiency (CSID) [MIM:222900]: Autosomal recessive intestinal disorder that is clinically characterized by fermentative diarrhea, abdominal pain, and cramps upon ingestion of sugar. The symptoms are the consequence of absent or drastically reduced enzymatic activities of sucrase and isomaltase. The prevalence of CSID is 0.02 % in individuals of European descent and appears to be much higher in Greenland, Alaskan, and Canadian native people. CSID arises due to post-translational perturbations in the intracellular transport, polarized sorting, aberrant processing, and defective function of SI. {ECO:0000269|PubMed:10903344, ECO:0000269|PubMed:11340066, ECO:0000269|PubMed:14724820, ECO:0000269|PubMed:16329100, ECO:0000269|PubMed:8609217}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07697; DB07692; DB02726; DB07628; DB00787; DB11638; DB09130; DB08951; DB01733; DB11263; DB00119 Interacts with P49407; P32121; Q96IK1-2; P35222; P53355; P22607; P42858; P04049; Q8N488; Q7Z699; Q9BSI4; Q9UMX0; Q9Y649; Q9WMX2; P35222; P53355; Q9H6Z9; P68431; Q16665; P27361 EC number EC 2.7.1.40 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; ATP-binding; Cytoplasm; Direct protein sequencing; Glycolysis; Hydroxylation; Isopeptide bond; Kinase; Magnesium; Metal-binding; Methylation; Nucleotide-binding; Nucleus; Phosphoprotein; Potassium; Proteomics identification; Pyruvate; Reference proteome; S-nitrosylation; Transferase; Translation regulation; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 112053 Length 1024 Aromaticity 0.05 Instability index 27.06 Isoelectric point 7.34 Charge (pH=7) 1.66 2D Binding mode Binding energy (Kcal/mol) -9.57 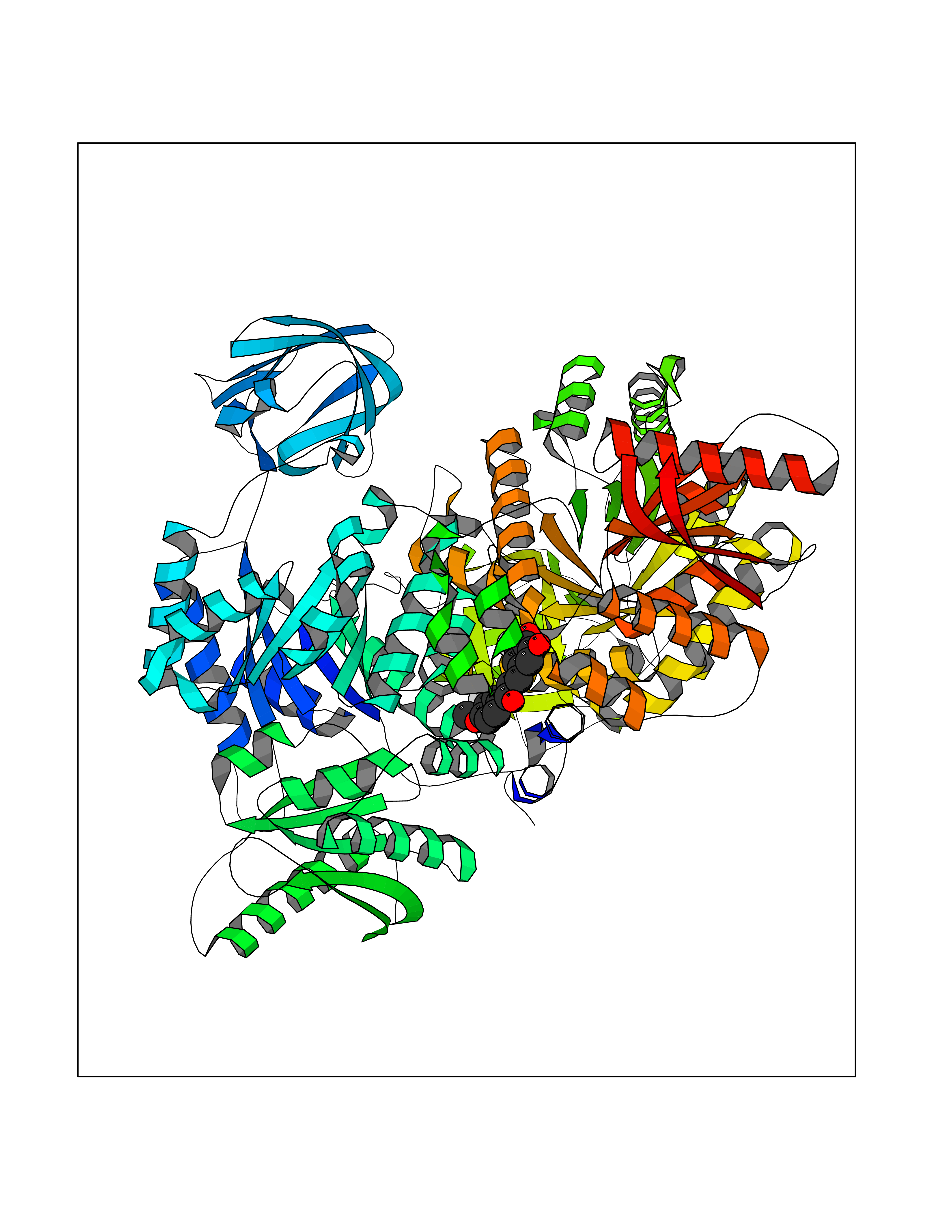 Molscript Map 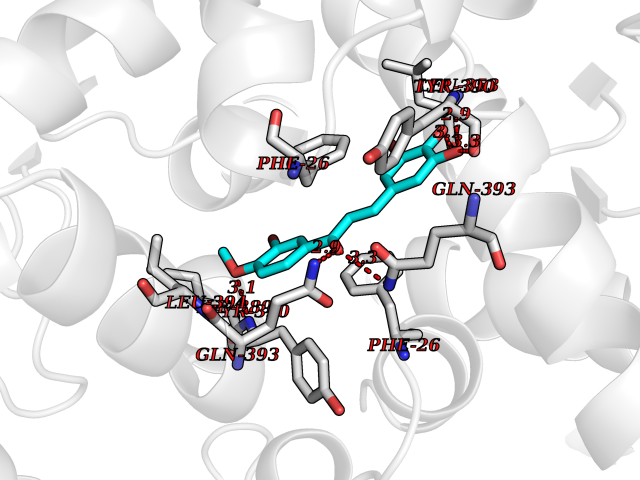 Pymol Map 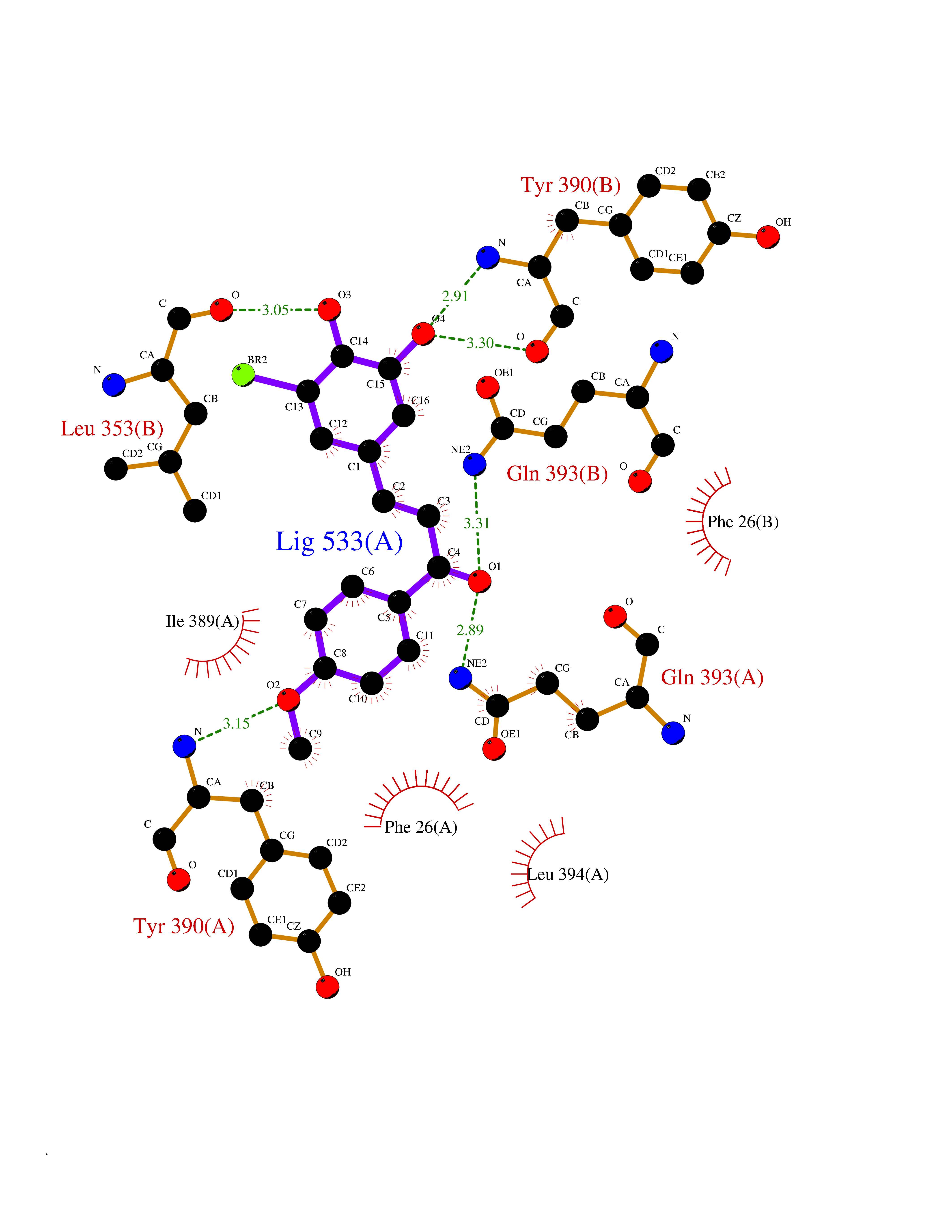 Ligplot Map 3D Binding mode Sequence IQTQQLHAAMADTFLEHMCRLDIDSPPITARNTGIICTIGPASRSVETLKEMIKSGMNVARLNFSHGTHEYHAETIKNVRTATESFASDPILYRPVAVALDTKGPEIRTGLIKGSGTAEVELKKGATLKITLDNAYMEKCDENILWLDYKNICKVVEVGSKIYVDDGLISLQVKQKGADFLVTEVENGGSLGSKKGVNLPGAAVDLPAVSEKDIQDLKFGVEQDVDMVFASFIRKASDVHEVRKVLGEKGKNIKIISKIENHEGVRRFDEILEASDGIMVARGDLGIEIPAEKVFLAQKMMIGRCNRAGKPVICATQMLESMIKKPRPTRAEGSDVANAVLDGADCIMLSGETAKGDYPLEAVRMQHLIAREAEAAIYHLQLFEELRRLAPITSDPTEATAVGAVEASFKCCSGAIIVLTKSGRSAHQVARYRPRAPIIAVTRNPQTARQAHLYRGIFPVLCKDPVQEAWAEDVDLRVNFAMNVGKARGFFKKGDVVIVLTGWRPGSGFTNTMRVVPVPIQTQQLHAAMADTFLEHMCRLDIDSPPITARNTGIICTIGPASRSVETLKEMIKSGMNVARLNFSHGTHEYHAETIKNVRTATESFASDPILYRPVAVALDTKGPEIRTGLIKEVEATLKITLDNAYMEKCDENILWLDYKNICKVVEVGSKIYVDDGLISLQVDFLVTEVENGGSLGSKKGVNLPGAAVDLPAVSEKDIQDLKFGVEQDVDMVFASFIRKASDVHEVRKVLGEKGKNIKIISKIENHEGVRRFDEILEASDGIMVARGDLGIEIPAEKVFLAQKMMIGRCNRAGKPVICATQMLESMIKKPRPTRAEGSDVANAVLDGADCIMLSGETAKGDYPLEAVRMQHLIAREAEAAIYHLQLFEELRRLAPITSDPTEATAVGAVEASFKCCSGAIIVLTKSGRSAHQVARYRPRAPIIAVTRNPQTARQAHLYRGIFPVLCKDPVQEAWAEDVDLRVNFAMNVGKARGFFKKGDVVIVLTGWRPGSGFTNTMRVVPVP Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Cholestenol delta-isomerase (EBP) | 6OHU | 7.01 | |
Target general information Gen name EBP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Emopamilbinding protein; EBP; Delta(8)Delta(7) sterol isomerase; D8D7 sterol isomerase; 3betahydroxysteroidDelta(8),Delta(7)isomerase Protein family EBP family Biochemical class Intramolecular oxidoreductases Function Catalyzes the conversion of Delta(8)-sterols to their corresponding Delta(7)-isomers. Related diseases Chondrodysplasia punctata 2, X-linked dominant (CDPX2) [MIM:302960]: A clinically and genetically heterogeneous disorder characterized by punctiform calcification of the bones. The key clinical features of CDPX2 are chondrodysplasia punctata, linear ichthyosis, cataracts and short stature. CDPX2 is a rare disorder of defective cholesterol biosynthesis, biochemically characterized by an increased amount of 8-dehydrocholesterol and cholest-8(9)-en-3-beta-ol in the plasma and tissues. {ECO:0000269|PubMed:10391218, ECO:0000269|PubMed:10391219, ECO:0000269|PubMed:10942423, ECO:0000269|PubMed:11493318, ECO:0000269|PubMed:18176751, ECO:0000269|PubMed:25814754}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: MEND syndrome (MEND) [MIM:300960]: An X-linked recessive disorder associated with a defect in sterol biosynthesis. Disease manifestations and severity are highly variable. Clinical features include intellectual disability, short stature, scoliosis, digital abnormalities, cataracts, and dermatologic abnormalities. {ECO:0000269|PubMed:12503101, ECO:0000269|PubMed:20949533, ECO:0000269|PubMed:24459067, ECO:0000269|PubMed:24700572}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00675 Interacts with O95870; Q86W74-2; Q13520; Q3SXY8; Q8N6S5; Q9H2C2; Q9HD20-3; O95393; Q12983; Q8WVV5; P06681; O14523; Q8WVX3-2; P01031; Q6UWT4; Q9P0B6; Q8NHW4; P25942; Q07108; P60033; O14735; P23141-3; Q9H9P2; Q8NHS1; O43889-2; Q96BA8; P49447; O43169; P78329; P81534; Q9H1M4; Q96LL9; Q15125; Q08426; Q9BV81; P54849; Q9UKR5; Q9Y282; Q7L5A8; Q5JX71; Q96IV6; Q9UGM5; Q9Y3D6; Q14318; Q9BWH2; Q14802-3; Q9H0Q3; Q8WWP7; Q96F15; P29033; O95452; O14653; Q8TDT2; P02724; P30519; Q7Z5P4; Q9Y5U9; Q9Y5U4; Q8N5M9; Q5T700; Q68G75; Q7L5N7; Q7Z4F1; Q96AG4; Q16873; Q6ZSS7; P50281; Q5J8X5; Q9UHE5; O95167; Q9NX14; Q99519; Q92982; Q9NZG7; Q16617; Q8IXM6; Q2M2E3; Q9P0S3; Q53FV1; Q8N138; Q7RTS5; Q9Y342; Q04941; Q8IY26; Q01453; P54315; P43378; P15151; Q8N8N0; Q5QGT7; Q96GQ5; Q9NTJ5; Q969E2; O75396; Q9Y6X1; Q8N6R1; Q9BWM7; Q8TD22; Q8IWU4; Q96G79; Q2M3R5; Q9NVC3; P08195-4; Q96JW4; Q6P1K1; Q0VAQ4; Q9NRQ5; B2RUZ4; Q9BZL3; Q6UX34; Q86Y82; P61266; Q13190; O43752; O15400; Q9UNK0; O43759-2; P57105; Q8N2H4; Q96BZ9; P07204; O14925; Q96CP7; Q96MV1; P55061; Q9NV29; P17152; Q9BXJ8; A0PK00; Q9NV12; Q9BVK8; Q9NUH8; Q9P0S9; Q9NRX6; Q8N511; Q969S6; Q9BTX3; A2RU14; Q9H0R3; Q8NBD8; Q8WW34-2; Q9NWH2; Q9BU79; Q8TBM7; Q69YG0; Q9NW97; Q9H2L4; Q6PI78; Q8N2M4; Q8N661; Q5BJF2; Q9NSU2-1; A0AVG3; Q5TGU0; A5PKU2; Q9Y385; Q9Y5Z9; Q53HI1; Q9H1C4; Q9NZ43; P23763-3; P63027; Q15836; O75379; Q9P0L0; O95292; O95070; Q9Y548; Q9BSR8; Q96EC8; Q6UX98; O95159 EC number EC 5.3.3.5 Uniprot keywords 3D-structure; Acetylation; Cataract; Cholesterol biosynthesis; Cholesterol metabolism; Cytoplasmic vesicle; Disease variant; Dwarfism; Endoplasmic reticulum; Ichthyosis; Isomerase; Lipid biosynthesis; Lipid metabolism; Membrane; Nucleus; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 24012.7 Length 206 Aromaticity 0.17 Instability index 42.52 Isoelectric point 6.19 Charge (pH=7) -3.19 2D Binding mode Binding energy (Kcal/mol) -9.56 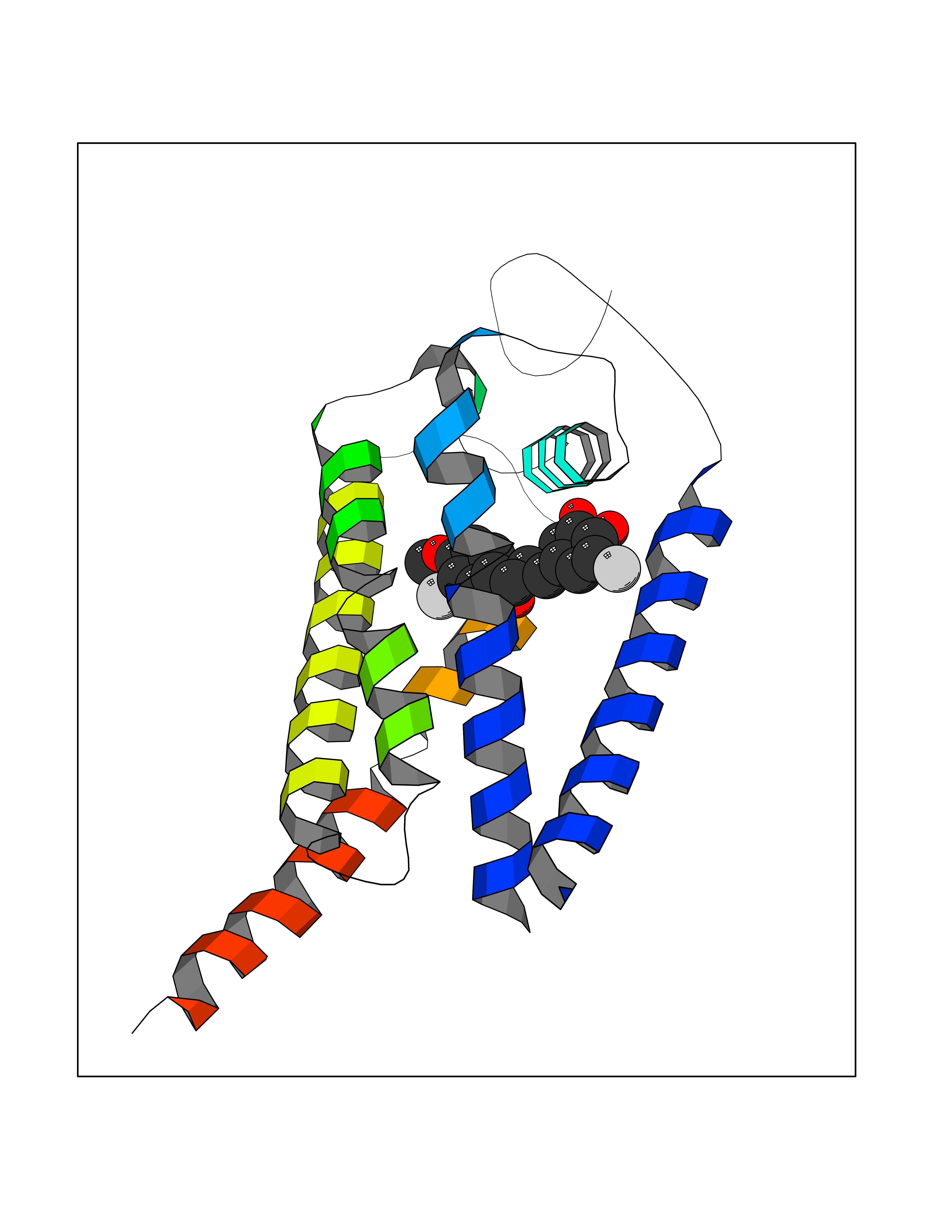 Molscript Map 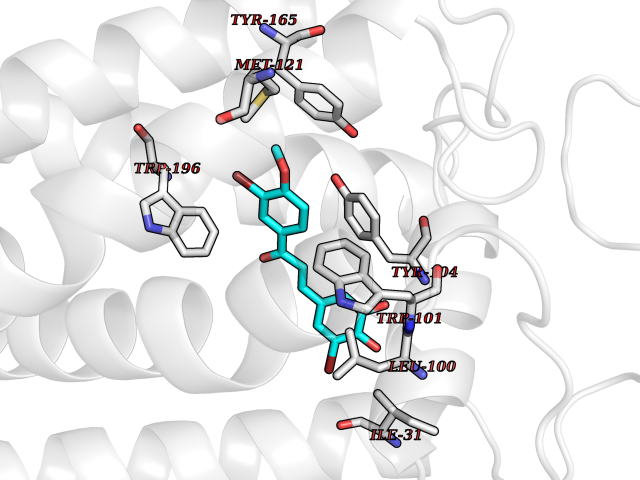 Pymol Map 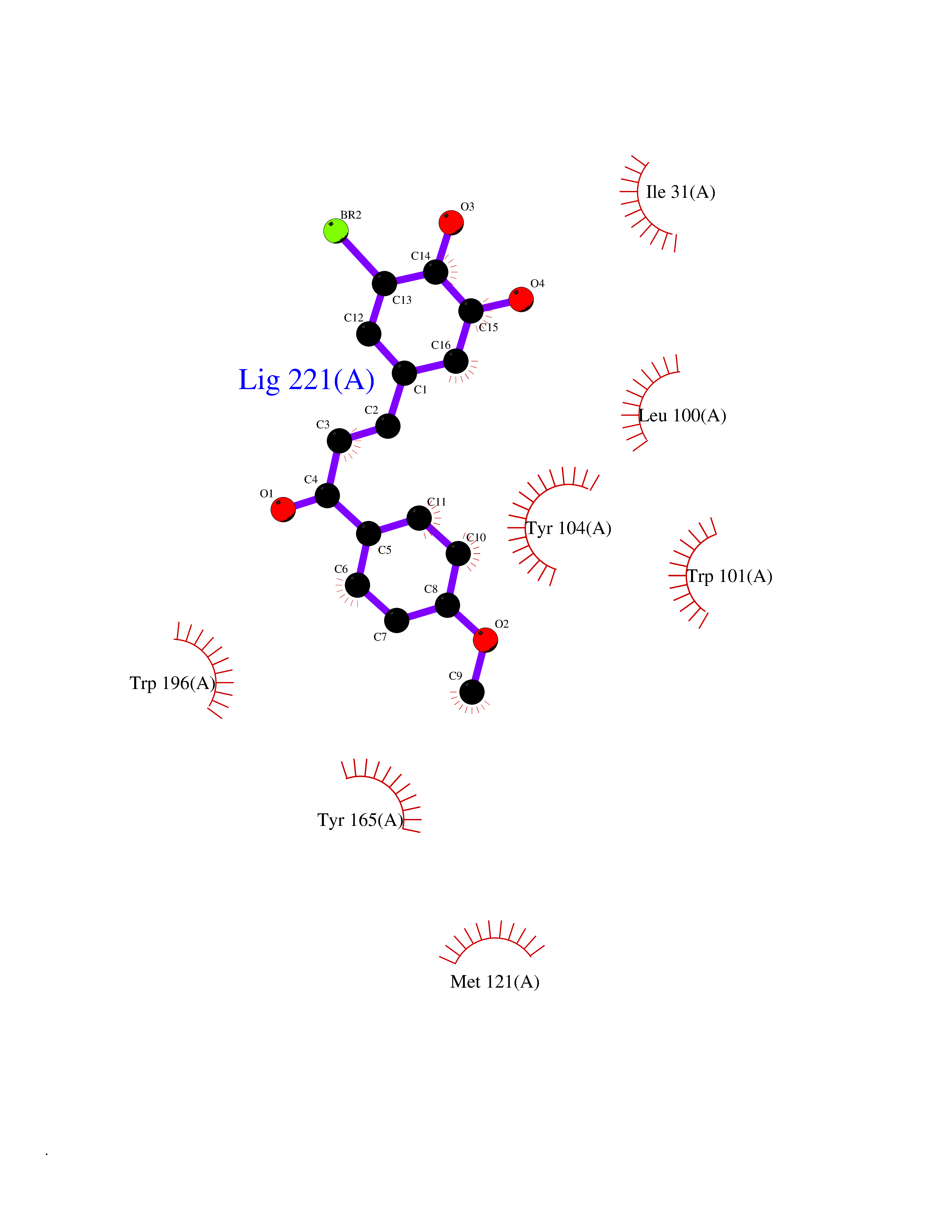 Ligplot Map 3D Binding mode Sequence PLHPYWPQHLRLDNFVPNDRPTWHILAGLFSVTGVLVVTTWLLSGRTWRRLSLCWFAVCGFIHLVIEGWFVLYYEDLLGDQAFLSQLWKEYAKGDSRYILGDNFTVCMETITACLWGPLSLWVVIAFLRQHPLRFILQLVVSVGQIYGDVLYFLTEHRDGFQHGELGHPLYFWFYFVFMNALWLVLPGVLVLDAVKHLTHAQSTLD Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Sodium/glucose cotransporter 2 (SGLT2) | 7VSI | 7.01 | |
Target general information Gen name SLC5A2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Solute carrier family 5 member 2; Na(+)/glucose cotransporter 2; Low affinity sodium-glucose cotransporter Protein family Sodium:solute symporter (SSF) (TC 2.A.21) family Biochemical class Solute:sodium symporter Function Has a Na(+) to glucose coupling ratio of 1:1. Sodium-dependent glucose transporter. Related diseases Renal glucosuria (GLYS) [MIM:233100]: A disorder characterized by persistent isolated glucosuria, normal fasting serum glucose concentration, decreased renal tubular resorption of glucose from the urine, and absence of any other signs of tubular dysfunction. {ECO:0000269|PubMed:14614622}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12236; DB08907; DB01914; DB06292; DB09038; DB11827; DB12713 Interacts with O14556; Q13113 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Ion transport; Membrane; Metal-binding; Proteomics identification; Reference proteome; Sodium; Sodium transport; Sugar transport; Symport; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 63858.9 Length 586 Aromaticity 0.12 Instability index 39.46 Isoelectric point 8.62 Charge (pH=7) 7.41 2D Binding mode Binding energy (Kcal/mol) -9.57 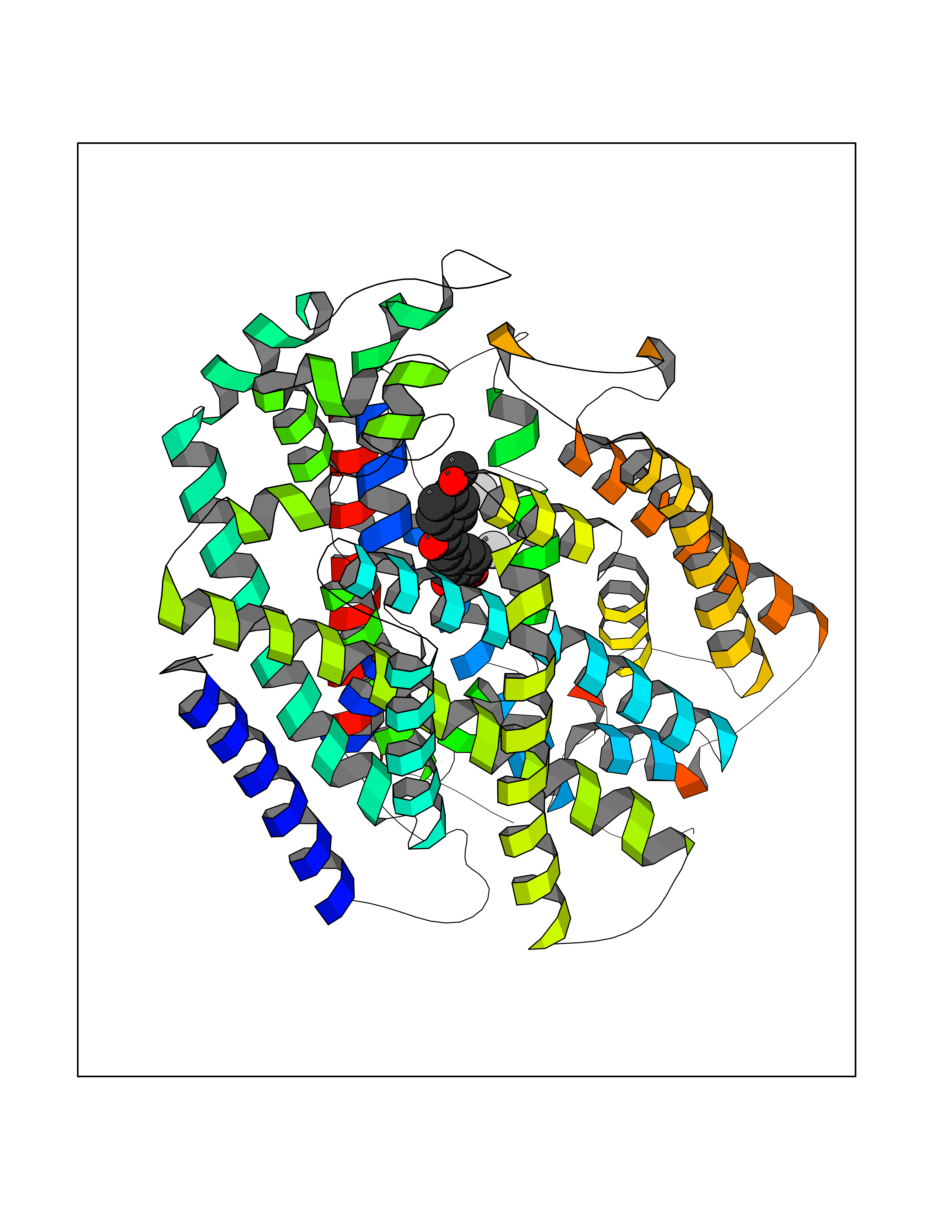 Molscript Map 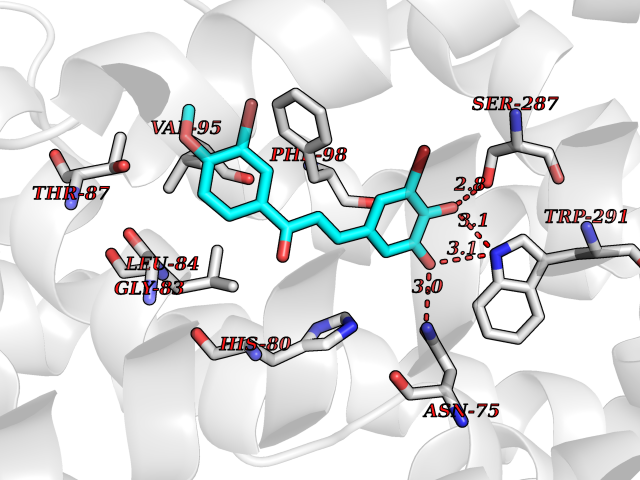 Pymol Map 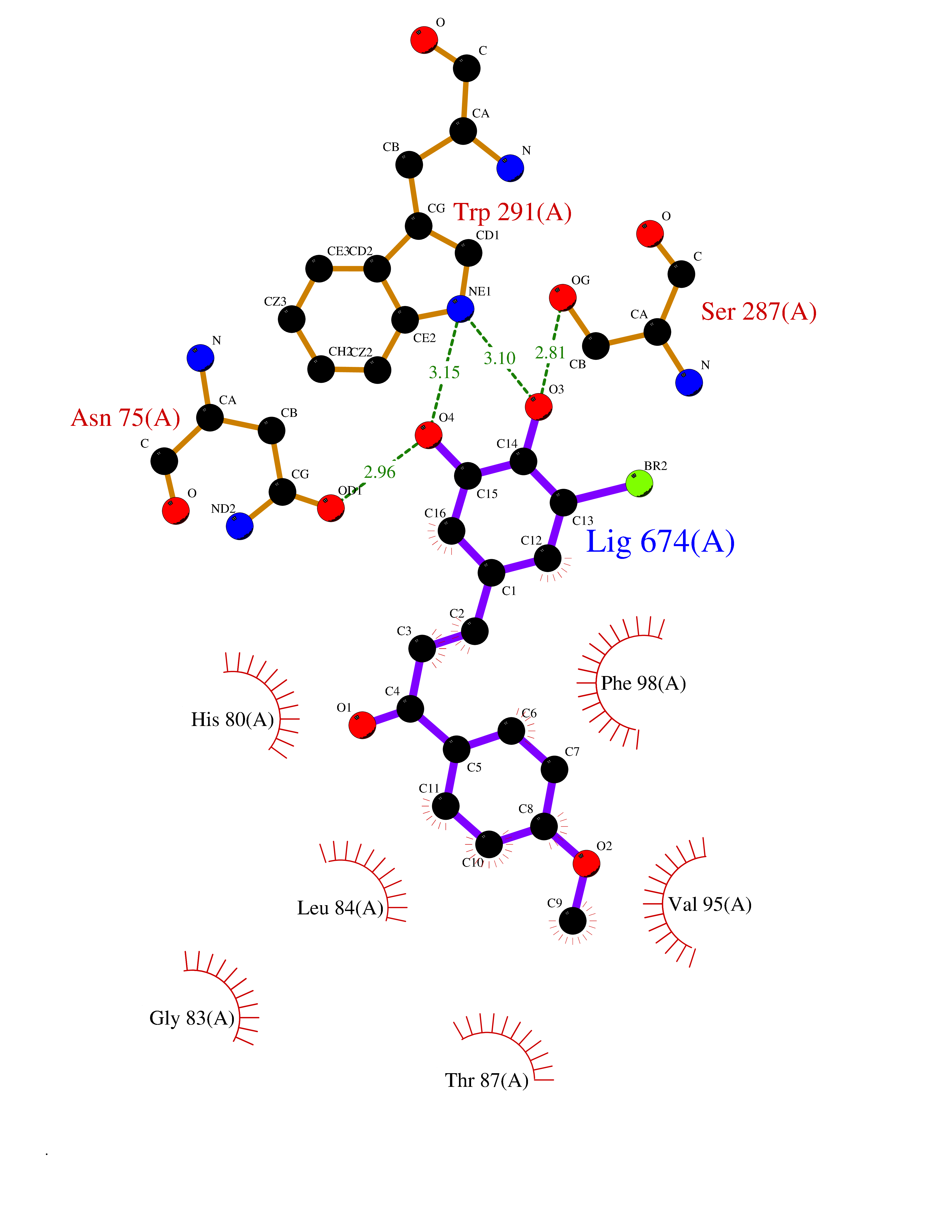 Ligplot Map 3D Binding mode Sequence DNPADILVIAAYFLLVIGVGLWSMCRTNRGTVGGYFLAGRSMVWWPVGASLFASNIGSGHFVGLAGTGAASGLAVAGFEWNALFVVLLLGWLFAPVYLTAGVITMPQYLRKRFGGRRIRLYLSVLSLFLYIFTKISVDMFSGAVFIQQALGWNIYASVIALLGITMIYTVTGGLAALMYTDTVQTFVILGGACILMGYAFHEVGGYSGLFDKYLGAATSLTVSEDPAVGNISSFCYRPRPDSYHLLRHPVTGDLPWPALLLGLTIVSGWYWCSDQVIVQRCLAGKSLTHIKAGCILCGYLKLTPMFLMVMPGMISRILYPDEVACVVPEVCRRVCGTEVGCSNIAYPRLVVKLMPNGLRGLMLAVMLAALMSSLASIFNSSSTLFTMDIYTRLRPRAGDRELLLVGRLWVVFIVVVSVAWLPVVQAAQGGQLFDYIQAVSSYLAPPVSAVFVLALFVPRVNEQGAFWGLIGGLLMGLARLIPEFSFGSGSCVQPSACPAFLCGVHYLYFAIVLFFCSGLLTLTVSLCTAPIPRKHLHRLVFSLRHSKEEREDLDEDISEDPSWARVVNLNALLMMAVAVFLWGFYA Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Vitamin K-dependent protein C (PROC) | 1LQV | 7.00 | |
Target general information Gen name PROC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin K-dependent protein C light chain; Vitamin K-dependent protein C heavy chain; PROC; Blood coagulation factor XIV; Autoprothrombin IIA; Anticoagulant protein C; Activation peptide Protein family Peptidase S1 family Biochemical class Peptidase Function Protein C is avitamin K-dependent serine protease that regulates blood coagulation by inactivating factors Va and VIIIa in the presence of calcium ions and phospholipids. Related diseases Thrombophilia due to protein C deficiency, autosomal dominant (THPH3) [MIM:176860]: A hemostatic disorder characterized by impaired regulation of blood coagulation and a tendency to recurrent venous thrombosis. Individuals with decreased amounts of protein C are classically referred to as having type I protein C deficiency and those with normal amounts of a functionally defective protein as having type II deficiency. {ECO:0000269|PubMed:1301959, ECO:0000269|PubMed:1347706, ECO:0000269|PubMed:1511989, ECO:0000269|PubMed:1868249, ECO:0000269|PubMed:2437584, ECO:0000269|PubMed:25618265, ECO:0000269|PubMed:25748729, ECO:0000269|PubMed:2602169, ECO:0000269|PubMed:7792728, ECO:0000269|PubMed:7865674, ECO:0000269|PubMed:8292730, ECO:0000269|PubMed:8398832, ECO:0000269|PubMed:8499568, ECO:0000269|PubMed:8560401, ECO:0000269|PubMed:8829639, ECO:0000269|PubMed:9798967}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thrombophilia due to protein C deficiency, autosomal recessive (THPH4) [MIM:612304]: A hemostatic disorder characterized by impaired regulation of blood coagulation and a tendency to recurrent venous thrombosis. It results in a thrombotic condition that can manifest as a severe neonatal disorder or as a milder disorder with late-onset thrombophilia. The severe form leads to neonatal death through massive neonatal venous thrombosis. Often associated with ecchymotic skin lesions which can turn necrotic called purpura fulminans, this disorder is very rare. {ECO:0000269|PubMed:1511988, ECO:0000269|PubMed:1593215, ECO:0000269|PubMed:1611081, ECO:0000269|PubMed:25618265, ECO:0000269|PubMed:7841323, ECO:0000269|PubMed:7841324, ECO:0000269|PubMed:7878626}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB13192; DB00025; DB09131; DB09332; DB13998; DB00170; DB13999; DB13149; DB00464; DB14738 Interacts with A8MQ03; P51511 EC number EC 3.4.21.69 Uniprot keywords 3D-structure; Alternative splicing; Blood coagulation; Calcium; Cleavage on pair of basic residues; Direct protein sequencing; Disease variant; Disulfide bond; EGF-like domain; Endoplasmic reticulum; Gamma-carboxyglutamic acid; Glycoprotein; Golgi apparatus; Hemostasis; Hydrolase; Hydroxylation; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Serine protease; Signal; Thrombophilia; Zymogen Protein physicochemical properties Chain ID C,D Molecular weight (Da) 45326 Length 411 Aromaticity 0.12 Instability index 40.68 Isoelectric point 7.07 Charge (pH=7) 0.29 2D Binding mode Binding energy (Kcal/mol) -9.55  Molscript Map 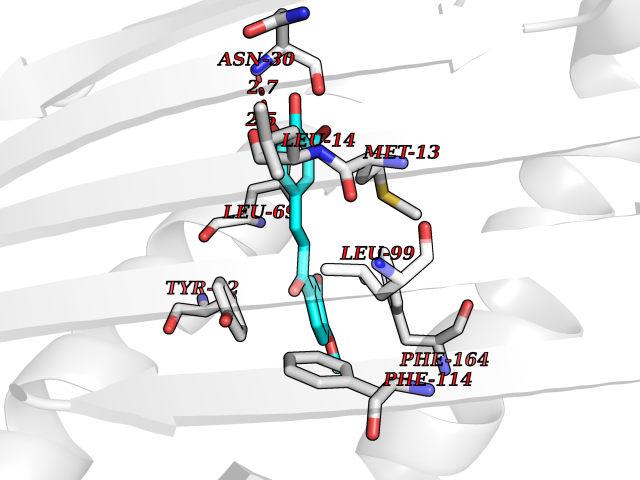 Pymol Map 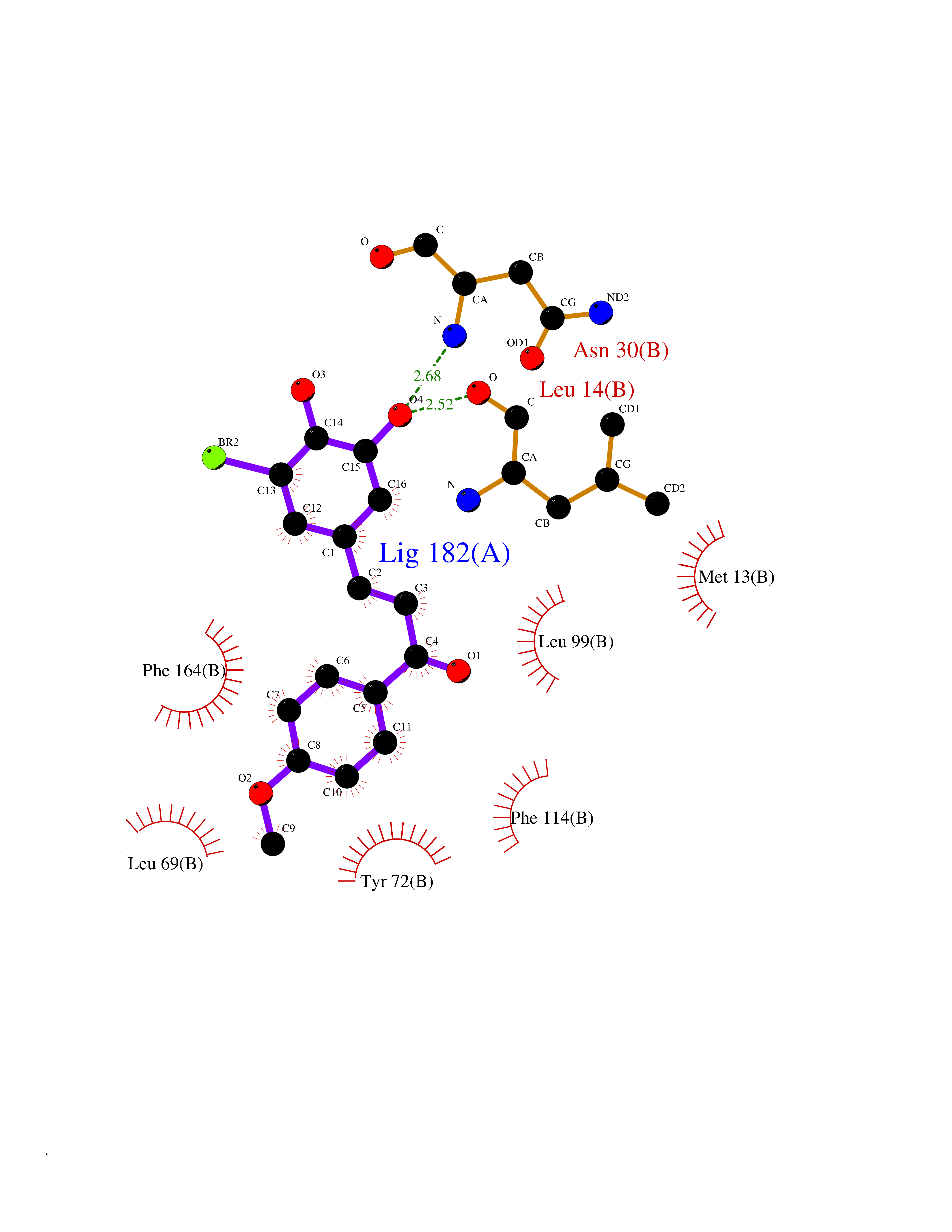 Ligplot Map 3D Binding mode Sequence GLQRLHMLQISYFRDPYHVWYQGNASLGGHLTHVLEGPDTNTTIIQLQPLQEPESWARTQSGLQSYLLQFHGLVRLVHQERTLAFPLTIRCFLGCELPPEGSRAHVFFEVAVNGSSFVSFRPERALWQADTQVTSGVVTFTLQQLNAYNRTRYELREFLEDTCVQYVQKHISANSFLXXLRHSSLXRXCIXXICDFXXAKXIFQNANSFLXXLRHSSLXRXCIXXICDFXXAKXIFQNLQRLHMLQISYFRDPYHVWYQGNASLGGHLTHVLEGPDTNTTIIQLQPLQEPESWARTQSGLQSYLLQFHGLVRLVHQERTLAFPLTIRCFLGCELPPEGSRAHVFFEVAVNGSSFVSFRPERALWQADTQVTSGVVTFTLQQLNAYNRTRYELREFLEDTCVQYVQKHISAE Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Cytochrome c oxidase subunit 2 | 3VRJ | 7.00 | |
Target general information Gen name MT-CO2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms MTCO2;COXII;COII;COX2 Protein family Cytochrome c oxidase subunit 2 family Biochemical class Immune system Function Copper ion binding.Cytochrome-c oxidase activity. Related diseases Mitochondrial complex IV deficiency (MT-C4D) [MIM:220110]: A disorder of the mitochondrial respiratory chain with heterogeneous clinical manifestations, ranging from isolated myopathy to severe multisystem disease affecting several tissues and organs. Features include hypertrophic cardiomyopathy, hepatomegaly and liver dysfunction, hypotonia, muscle weakness, exercise intolerance, developmental delay, delayed motor development and intellectual disability. Some affected individuals manifest a fatal hypertrophic cardiomyopathy resulting in neonatal death. A subset of patients manifest Leigh syndrome. {ECO:0000269|PubMed:10486321}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02659; DB04464; DB05412 Interacts with Q9NZ94-2; P49281-3 EC number 7.1.1.9 Uniprot keywords 3D-structure; Copper; Disease variant; Electron transport; Magnesium; Membrane; Metal-binding; Mitochondrion; Mitochondrion inner membrane; Primary mitochondrial disease; Proteomics identification; Reference proteome; Respiratory chain; Translocase; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID C Molecular weight (Da) 21687.9 Length 189 Aromaticity 0.11 Instability index 38 Isoelectric point 5.68 Charge (pH=7) -3.26 2D Binding mode Binding energy (Kcal/mol) -9.55  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence SHSMRYFYTAMSRPGRGEPRFIAVGYVDDTQFVRFDSDAASPRMAPRAPWIEQEGPEYWDGETRNMKASAQTYRENLRIALRYYNQSEAGSHIIQVMYGCDVGPDGRLLRGHDQSAYDGKDYIALNEDLSSWTAADTAAQITQRKWEAARVAEQLRAYLEGLCVEWLRRYLENGKETLQLTTKLTNTNI Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Monoamine oxidase type B (MAO-B) | 2V5Z | 7.00 | |
Target general information Gen name MAOB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MAO-B; Amine oxidase [flavin-containing] B Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. MAOB preferentially degrades benzylamine and phenylethylamine. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB08176; DB02211; DB08516; DB08480; DB01472; DB04307; DB07512; DB07513; DB00915; DB00182; DB06698; DB04889; DB00215; DB09130; DB04147; DB00988; DB01363; DB00668; DB01175; DB02509; DB03147; DB14914; DB00614; DB04818; DB02095; DB01247; DB00601; DB01577; DB01442; DB01171; DB08082; DB02643; DB04677; DB03894; DB08804; DB04820; DB00184; DB04821; DB12612; DB01626; DB00780; DB00191; DB00388; DB01132; DB00721; DB01168; DB01367; DB09363; DB06654; DB01037; DB01104; DB14569; DB09042; DB00752; DB16446; DB09185; DB04832; DB00909 Interacts with P55212; P28329-3; Q8NI60; Q5RI15; Q92915-2; P22607; Q53GS7; P06396; P01112; O14901; P13473-2; P21397; Q9BVL2; O75400-2; P62826; Q6NTF9-3; Q9Y371; Q7Z699; Q9UMX0; Q9Y649 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; FAD; Flavoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Oxidoreductase; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56019.9 Length 494 Aromaticity 0.09 Instability index 34.81 Isoelectric point 6.51 Charge (pH=7) -2.2 2D Binding mode Binding energy (Kcal/mol) -9.54  Molscript Map  Pymol Map 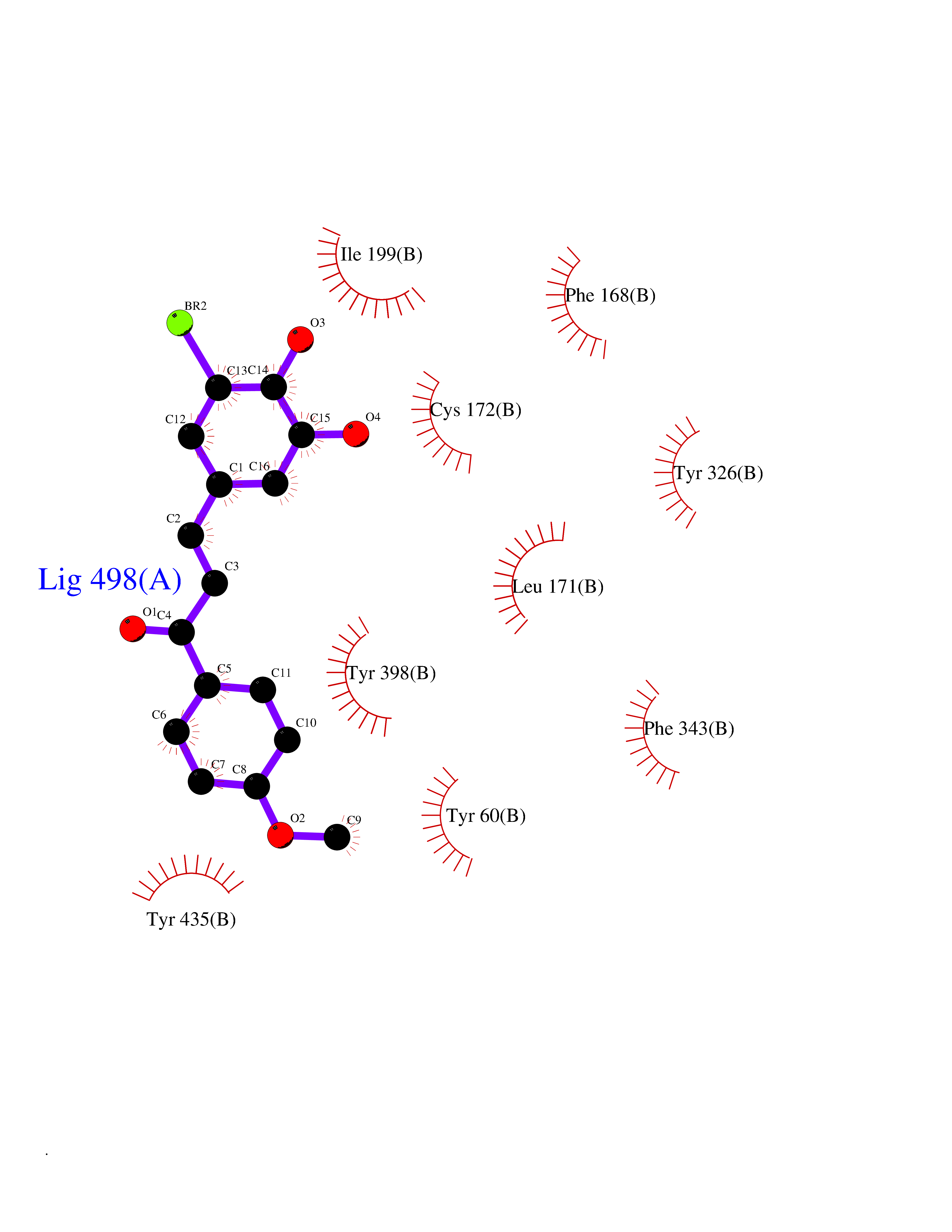 Ligplot Map 3D Binding mode Sequence NKCDVVVVGGGISGMAAAKLLHDSGLNVVVLEARDRVGGRTYTLRNQKVKYVDLGGSYVGPTQNRILRLAKELGLETYKVNEVERLIHHVKGKSYPFRGPFPPVWNPITYLDHNNFWRTMDDMGREIPSDAPWKAPLAEEWDNMTMKELLDKLCWTESAKQLATLFVNLCVTAETHEVSALWFLWYVKQCGGTTRIISTTNGGQERKFVGGSGQVSERIMDLLGDRVKLERPVIYIDQTRENVLVETLNHEMYEAKYVISAIPPTLGMKIHFNPPLPMMRNQMITRVPLGSVIKCIVYYKEPFWRKKDYCGTMIIDGEEAPVAYTLDDTKPEGNYAAIMGFILAHKARKLARLTKEERLKKLCELYAKVLGSLEALEPVHYEEKNWCEEQYSGGCYTTYFPPGILTQYGRVLRQPVDRIYFAGTETATHWSGYMEGAVEAGERAAREILHAMGKIPEDEIWQSEPESVDVPAQPITTTFLERHLPSVPGLLRLI Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Matrix metalloproteinase-12 (MMP-12) | 1Y93 | 7.00 | |
Target general information Gen name MMP12 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Macrophage metalloelastase; Macrophage elastase; MME; ME; HME Protein family Peptidase M10A family Biochemical class Peptidase Function Has significant elastolytic activity. Can accept large and small amino acids at the P1' site, but has a preference for leucine. Aromatic or hydrophobic residues are preferred at the P1 site, with small hydrophobic residues (preferably alanine) occupying P3. May be involved in tissue injury and remodeling. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB07026; DB07921; DB04405; DB00551; DB03880; DB07556; DB02118; DB00786; DB07446; DB07683; DB08599; DB08271; DB07922; DB07920; DB05387; DB03367; DB00013 Interacts with NA EC number EC 3.4.24.65 Uniprot keywords 3D-structure; Calcium; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 17461.3 Length 158 Aromaticity 0.13 Instability index 13.25 Isoelectric point 6.11 Charge (pH=7) -3.44 2D Binding mode Binding energy (Kcal/mol) -9.54  Molscript Map 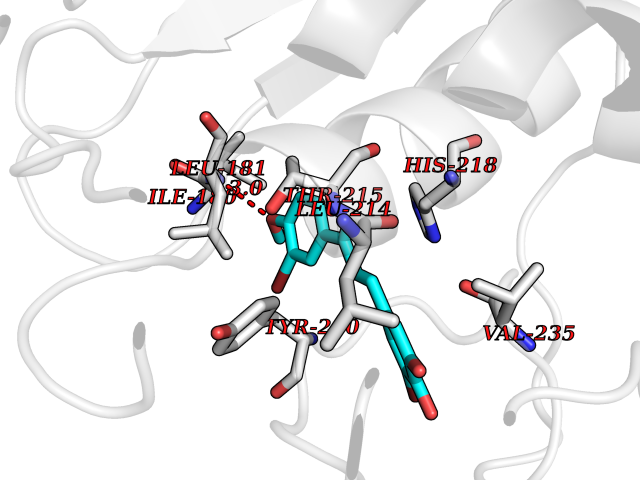 Pymol Map 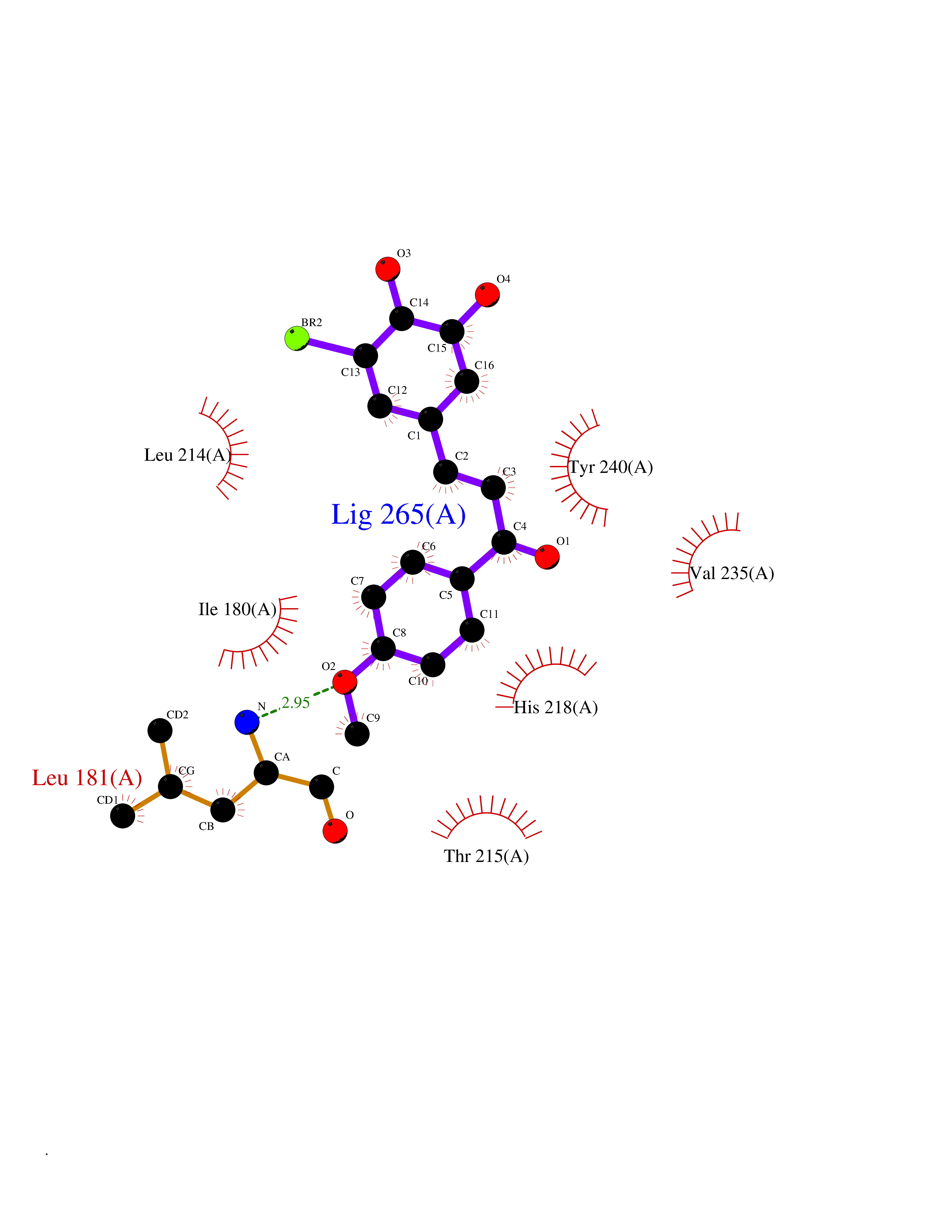 Ligplot Map 3D Binding mode Sequence GPVWRKHYITYRINNYTPDMNREDVDYAIRKAFQVWSNVTPLKFSKINTGMADILVVFARGAHGDDHAFDGKGGILAHAFGPGSGIGGDAHFDEDEFWTTHSGGTNLFLTAVHEIGHSLGLGHSSDPKAVMFPTYKYVDINTFRLSADDIRGIQSLYG Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Histone acetyltransferase p300 (EP300) | 5LKX | 7.00 | |
Target general information Gen name EP300 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms p300 HAT; Protein propionyltransferase p300; P300; Histone crotonyltransferase p300; Histone butyryltransferase p300; E1Aassociated protein p300; E1A-associated protein p300 Protein family NA Biochemical class Acyltransferase Function Acetylates all four core histones in nucleosomes. Histone acetylation gives an epigenetic tag for transcriptional activation. Mediates cAMP-gene regulation by binding specifically to phosphorylated CREB protein. Mediates acetylation of histone H3 at 'Lys-122' (H3K122ac), a modification that localizes at the surface of the histone octamer and stimulates transcription, possibly by promoting nucleosome instability. Mediates acetylation of histone H3 at 'Lys-27' (H3K27ac). Also functions as acetyltransferase for non-histone targets, such as ALX1, HDAC1, PRMT1 or SIRT2. Acetylates 'Lys-131' of ALX1 and acts as its coactivator. Acetylates SIRT2 and is proposed to indirectly increase the transcriptional activity of TP53 through acetylation and subsequent attenuation of SIRT2 deacetylase function. Acetylates HDAC1 leading to its inactivation and modulation of transcription. Acts as a TFAP2A-mediated transcriptional coactivator in presence of CITED2. Plays a role as a coactivator of NEUROD1-dependent transcription of the secretin and p21 genes and controls terminal differentiation of cells in the intestinal epithelium. Promotes cardiac myocyte enlargement. Can also mediate transcriptional repression. Acetylates FOXO1 and enhances its transcriptional activity. Acetylates BCL6 wich disrupts its ability to recruit histone deacetylases and hinders its transcriptional repressor activity. Participates in CLOCK or NPAS2-regulated rhythmic gene transcription; exhibits a circadian association with CLOCK or NPAS2, correlating with increase in PER1/2 mRNA and histone H3 acetylation on the PER1/2 promoter. Acetylates MTA1 at 'Lys-626' which is essential for its transcriptional coactivator activity. Acetylates XBP1 isoform 2; acetylation increases protein stability of XBP1 isoform 2 and enhances its transcriptional activity. Acetylates PCNA; acetylation promotes removal of chromatin-bound PCNA and its degradation during nucleotide excision repair (NER). Acetylates MEF2D. Acetylates and stabilizes ZBTB7B protein by antagonizing ubiquitin conjugation and degragation, this mechanism may be involved in CD4/CD8 lineage differentiation. In addition to protein acetyltransferase, can use different acyl-CoA substrates, such as (2E)-butenoyl-CoA (crotonyl-CoA), butanoyl-CoA (butyryl-CoA) or propanoyl-CoA (propionyl-CoA), and is able to mediate protein crotonylation, butyrylation or propionylation, respectively. Acts as a histone crotonyltransferase; crotonylation marks active promoters and enhancers and confers resistance to transcriptional repressors. Histone crotonyltransferase activity is dependent on the concentration of (2E)-butenoyl-CoA (crotonyl-CoA) substrate and such activity is weak when (E)-but-2-enoyl-CoA (crotonyl-CoA) concentration is low. Also acts as a histone butyryltransferase; butyrylation marks active promoters. Functions as a transcriptional coactivator for SMAD4 in the TGF-beta signaling pathway. Acetylates PCK1 and promotes PCK1 anaplerotic activity. Functions as histone acetyltransferase and regulates transcription via chromatin remodeling. Related diseases Defects in EP300 may play a role in epithelial cancer.; DISEASE: Chromosomal aberrations involving EP300 may be a cause of acute myeloid leukemias. Translocation t(8;22)(p11;q13) with KAT6A.; DISEASE: Rubinstein-Taybi syndrome 2 (RSTS2) [MIM:613684]: A disorder characterized by craniofacial abnormalities, postnatal growth deficiency, broad thumbs, broad big toes, intellectual disability and a propensity for development of malignancies. Some individuals with RSTS2 have less severe mental impairment, more severe microcephaly, and a greater degree of changes in facial bone structure than RSTS1 patients. {ECO:0000269|PubMed:15706485}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Menke-Hennekam syndrome 2 (MKHK2) [MIM:618333]: A form of Menke-Hennekam syndrome, a congenital autosomal dominant disease characterized by developmental delay, growth retardation, and craniofacial dysmorphism. Patients have intellectual disability of variable severity, speech delay, autistic behavior, short stature and microcephaly. Main facial characteristics include short palpebral fissures, telecanthi, depressed nasal ridge, short nose, anteverted nares, short columella and long philtrum. {ECO:0000269|PubMed:29460469}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NXW9; P27695; Q9UBL3; Q8WXX7; Q9NPI1; P24941; Q99967; P61201; P16220-1; P17844; Q01844; P35637; Q00403; Q16665; Q9H2X6; Q92831; P55209; O60934; P20265; Q96KQ4; Q8WUF5; Q13761; Q96EB6; Q13309; O95863; P42226; Q9UL17; P56279; P05549; P04637; Q13625; O15350; P11473; P67809; K4P3M7; P03122; P06422; P06790; Q61221; Q9QXM1; P04608; P03070; P03255; P03255-2; P03259 EC number EC 2.3.1.48 Uniprot keywords 3D-structure; Acetylation; Acyltransferase; Biological rhythms; Bromodomain; Cell cycle; Chromosomal rearrangement; Chromosome; Citrullination; Cytoplasm; Direct protein sequencing; Disease variant; Host-virus interaction; Intellectual disability; Isopeptide bond; Metal-binding; Methylation; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 64477.2 Length 554 Aromaticity 0.12 Instability index 45.78 Isoelectric point 7.01 Charge (pH=7) 0.05 2D Binding mode Binding energy (Kcal/mol) -9.55  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence KKIFKPEELRQALMPTLEALYRQDPESLPFRQPVDPQLLGIPDYFDIVKSPMDLSTIKRKLDTGQYQEPWQYVDDIWLMFNNAWLYNRKTSRVYKYCSKLSEVFEQEIDPVMQSLGYCCGRKLEFSPQTLCCYGKQLCTIPRDATYYSYQNRYHFCEKCFNEIQGESVSLGQTTINKEQFSKRKNDTLDPELFVECTECGRKMHQICVLHHEIIWPAGFVCDGCLKKSARTRKENKFSAKRLPSTRLGTFLENRVNDFLRRQNHPESGEVTVRVVHASDKTVEVKPGMKARFVDSGEMAESFPYRTKALFAFEEIDGVDLCFFGMHVQEYGSDCPPPNQRRVYISYLDSVHFFRPKCLRTAVYHEILIGYLEYVKKLGYTTGHIWACPPSEGDDYIFHCHPPDQKIPKPKRLQEWFKKMLDKAVSERIVHDYKDIFKQATEDRLTSAKELPYFEGDFWPNVLEESIKESGGSGSQKLYATMEKHKEVFFVIRLIAGPAANSLPPIVDPDPLIPCDLMDGRDAFLTLARDKHLEFSSLRRAQWSTMCMLVELHTQ Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Plasma retinol-binding protein (RBP4) | 5NU7 | 7.00 | |
Target general information Gen name RBP4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Retinol-binding protein 4; RBP4; RBP; Plasma retinol-binding protein(1-176); PRBP Protein family Calycin superfamily, Lipocalin family Biochemical class Calycin family Function Delivers retinol from the liver stores to the peripheral tissues. In plasma, the RBP-retinol complex interacts with transthyretin, this prevents its loss by filtration through the kidney glomeruli. Related diseases Retinal dystrophy, iris coloboma, and comedogenic acne syndrome (RDCCAS) [MIM:615147]: A disease characterized by retinal degeneration, ocular colobomas involving both the anterior and posterior segment, impaired night vision and loss of visual acuity. Additional characteristic features include developmental abnormalities and severe acne. {ECO:0000269|PubMed:10232633, ECO:0000269|PubMed:23189188, ECO:0000269|PubMed:9888420}. The disease is caused by variants affecting the gene represented in this entry. Loss of functional RBP4 protein results in serum retinol deficiency. Lack of normal levels of retinol impairs the visual cycle leading to night blindness at early stages; prolonged deficiency may lead to retinal degeneration. Additionally, retinol deficiency may result in dry skin, increased susceptibility to infection and acne (PubMed:23189188). {ECO:0000269|PubMed:23189188}.; DISEASE: Microphthalmia/Coloboma 10 (MCOPCB10) [MIM:616428]: A disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues. Ocular abnormalities like opacities of the cornea and lens, scaring of the retina and choroid, and other abnormalities may also be present. Ocular colobomas are a set of malformations resulting from abnormal morphogenesis of the optic cup and stalk, and the fusion of the fetal fissure (optic fissure). {ECO:0000269|PubMed:25910211}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06985; DB06755; DB05076; DB03917; DB00755; DB00162 Interacts with Q9UBX0; P02766; O55245 EC number NA Uniprot keywords 3D-structure; Direct protein sequencing; Disease variant; Disulfide bond; Methylation; Microphthalmia; Proteomics identification; Reference proteome; Retinol-binding; Secreted; Sensory transduction; Signal; Transport; Vision; Vitamin A Protein physicochemical properties Chain ID A Molecular weight (Da) 20030.2 Length 174 Aromaticity 0.13 Instability index 28.54 Isoelectric point 5.24 Charge (pH=7) -4.02 2D Binding mode Binding energy (Kcal/mol) -9.54  Molscript Map 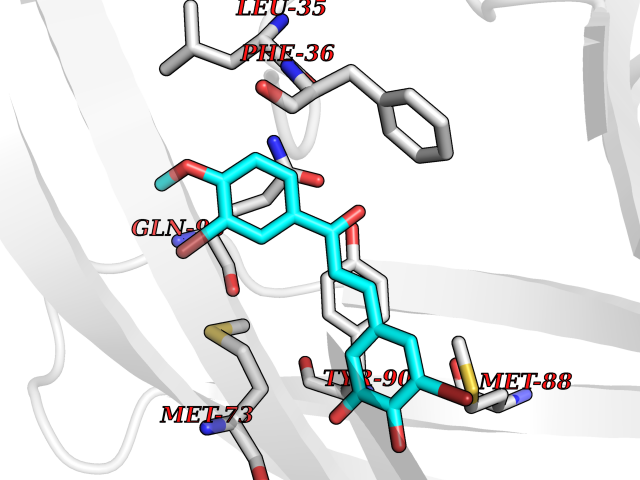 Pymol Map 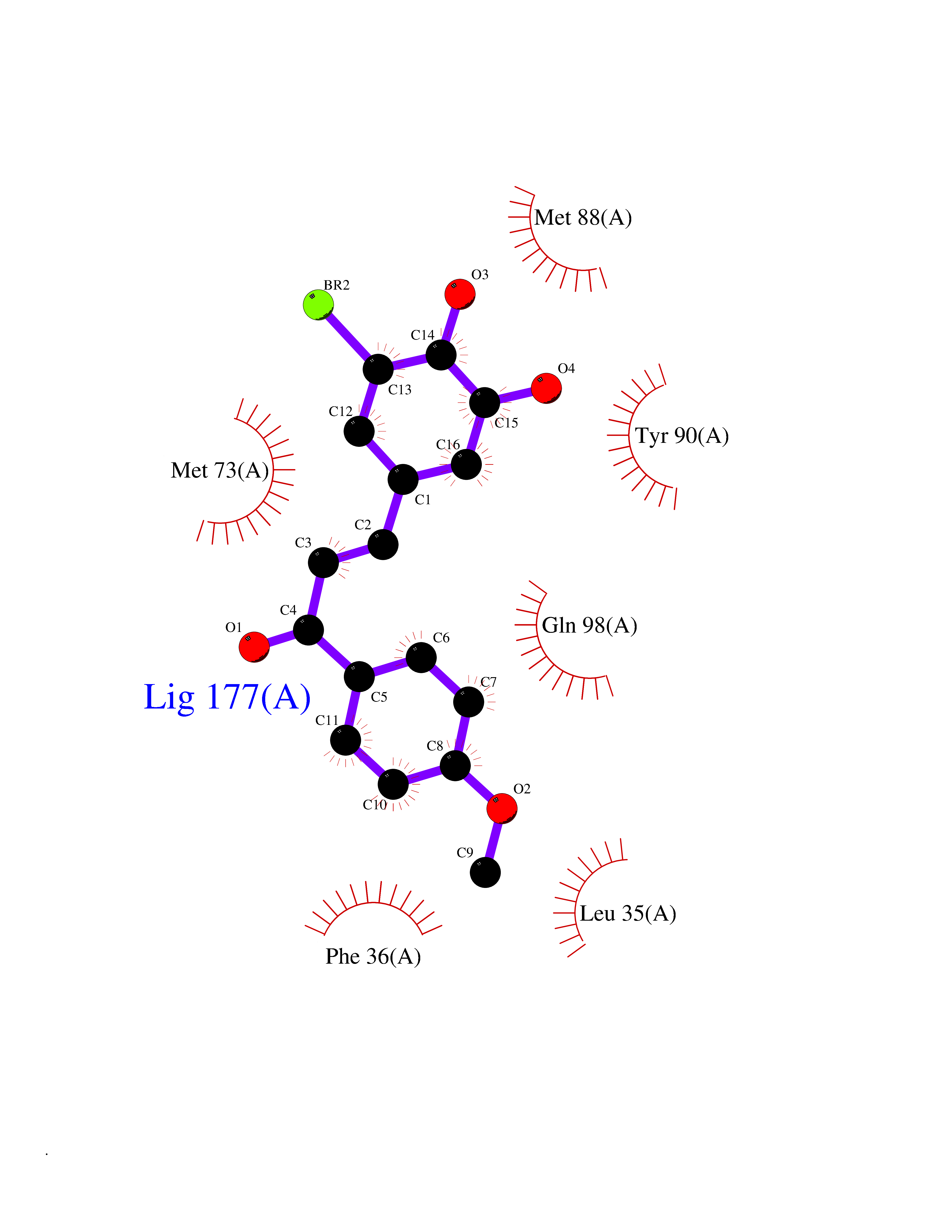 Ligplot Map 3D Binding mode Sequence ERDCRVSSFRVKENFDKARFSGTWYAMAKKDPEGLFLQDNIVAEFSVDETGQMSATAKGRVRLLNNWDVCADMVGTFTDTEDPAKFKMKYWGVASFLQKGNDDHWIVDTDYDTYAVQYSCRLLNLDGTCADSYSFVFSRDPNGLPPEAQKIVRQRQEELCLARQYRLIVHNGYC Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | Protein-serine O-palmitoleoyltransferase porcupine (PORCN) | 7URD | 7.00 | |
Target general information Gen name PORCN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein-cysteine N-palmitoyltransferase porcupine; Protein MG61; PORCN Protein family Membrane-bound acyltransferase family, Porcupine subfamily Biochemical class Acyltransferase Function protein-cysteine N-palmitoyltransferase that modulates the processingof Wnt proteins by mediating serine palmitoylation of Wnt family members. Related diseases Focal dermal hypoplasia (FODH) [MIM:305600]: A rare congenital ectomesodermal disorder characterized by a combination of skin defects, skeletal abnormalities, and ocular anomalies. Affected individuals have patchy dermal hypoplasia, often in a distribution pattern following the Blaschko lines, and areas of subcutaneous fat herniation or deposition of fat into the dermis. In addition, sparse and brittle hair, hypoplastic nails and papillomas have been described. Skeletal abnormalities usually comprise syndactyly, ectrodactyly, and brachydactyly, and in some cases osteopathia striata has been seen. Patients frequently have ocular anomalies, including microphthalmia/ anophthalmia, coloboma, pigmentary and vascularization defects of the retina. Dental abnormalities are often present. {ECO:0000269|PubMed:17546030, ECO:0000269|PubMed:17546031, ECO:0000269|PubMed:18325042, ECO:0000269|PubMed:19277062, ECO:0000269|PubMed:19309688, ECO:0000269|PubMed:19586929, ECO:0000269|PubMed:19863546, ECO:0000269|PubMed:21472892}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P55056; Q8N350-4 EC number EC 2.3.1.250 Uniprot keywords 3D-structure; Acyltransferase; Alternative splicing; Disease variant; Endoplasmic reticulum; Lipoprotein; Membrane; Palmitate; Proteomics identification; Reference proteome; Transferase; Transmembrane; Transmembrane helix; Wnt signaling pathway Protein physicochemical properties Chain ID A Molecular weight (Da) 49013.3 Length 432 Aromaticity 0.15 Instability index 36.17 Isoelectric point 9.03 Charge (pH=7) 11.23 2D Binding mode Binding energy (Kcal/mol) -9.54 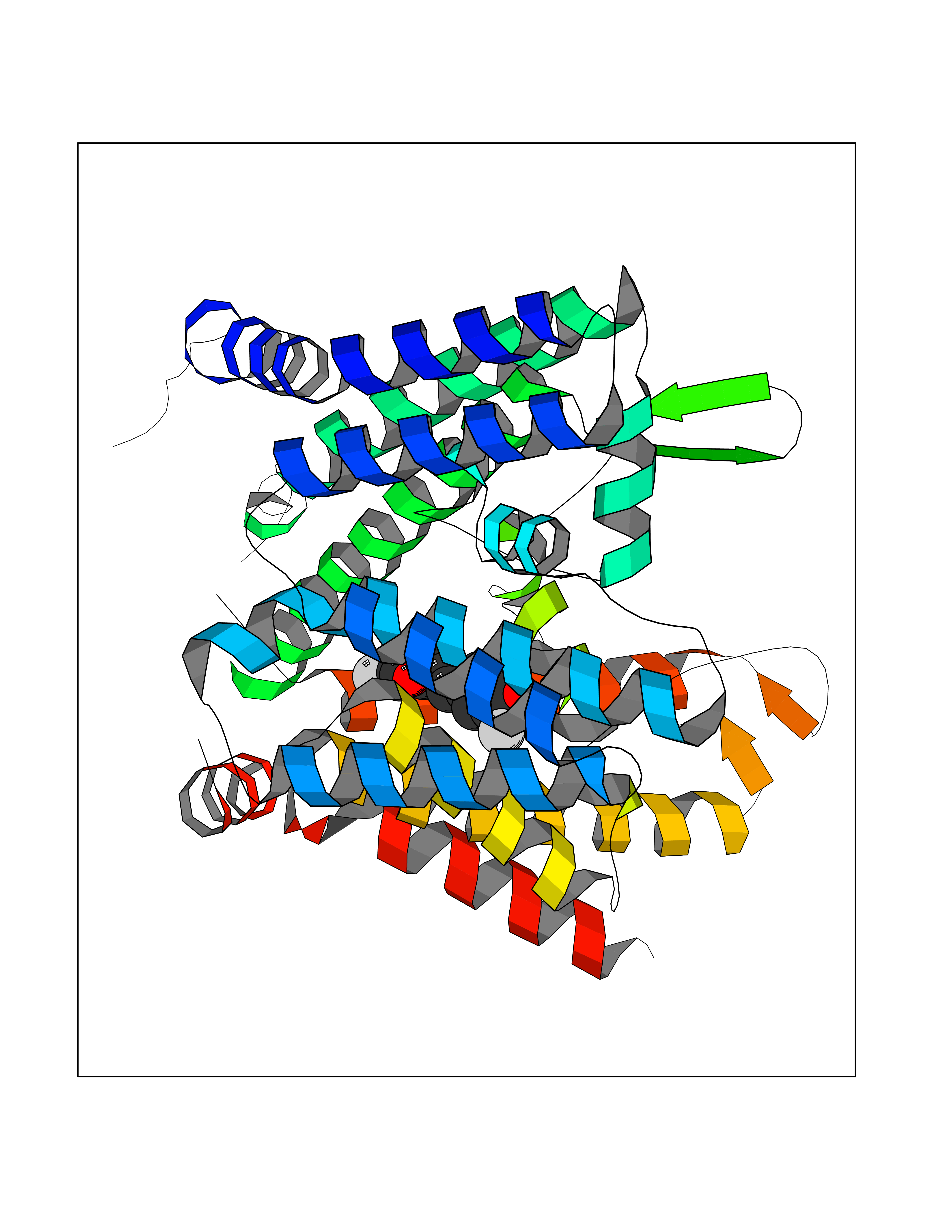 Molscript Map 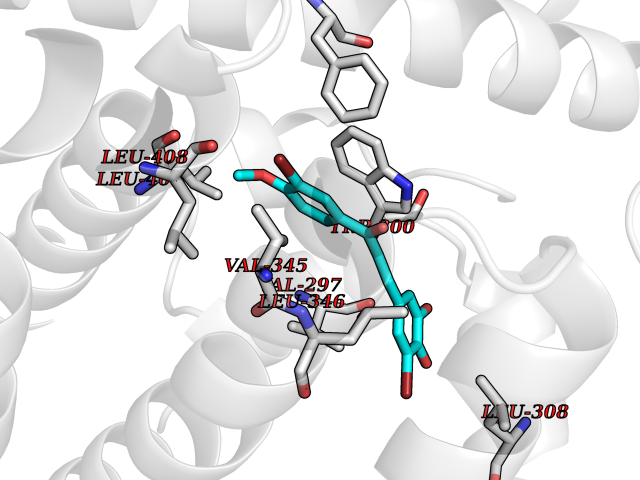 Pymol Map 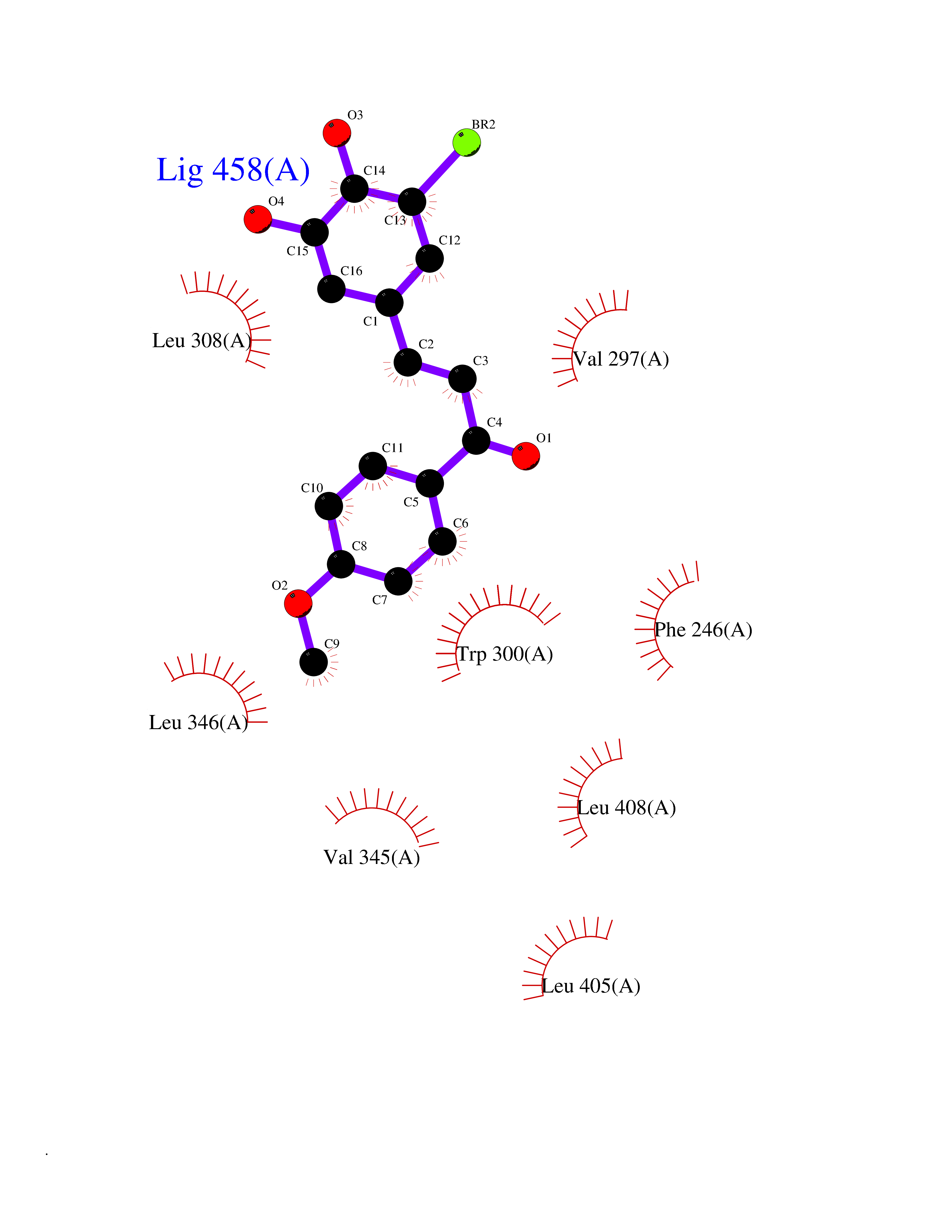 Ligplot Map 3D Binding mode Sequence FSRQEFFQQLLQGCLLPTAQQGLDQIWLLLAICLACRLLWRLGLPSYLKHASTVAGGFFSLYHFFQLHMVWVVLLSLLCYLVLFLCRHSSHRGVFLSVTILIYLLMGEMHMVDTVTWHKMRGAQMIVAMKAVSLGFDLDRGEVGTVPSPVEFMGYLYFVGTIVFGPWISFHSYLQAVQGRPLSCRWLQKVARSLALALLCLVLSTCVGPYLFPYFIPLNARWLRAYESAVSFHFSNYFVGFLSEATATLAGAGFTEEKDHLEWDLTVSKPLNVELPRSMVEVVTSWNLPMSYWLNNYVFKNALRLGTFSAVLVTYAASALLHGFSFHLAAVLLSLAFITYVEHVLRKRLARILSACVLSKRCPPDCSHQHRLGLGVRALNLLFGALAIFHLAYLGSLFDVYGMAYTVHKWSELSWASHWVTFGCWIFYRLIG Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Leucine carboxyl methyltransferase 1 | 3IEI | 6.99 | |
Target general information Gen name LCMT1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CGI-68;LCMT Protein family Methyltransferase superfamily, LCMT family Biochemical class Transferase Function Protein C-terminal carboxyl O-methyltransferase activity.Protein C-terminal leucine carboxyl O-methyltransferase activity.S-adenosylmethionine-dependent methyltransferase activity. Related diseases Neurodevelopmental disorder, mitochondrial, with abnormal movements and lactic acidosis, with or without seizures (NEMMLAS) [MIM:617710]: An autosomal recessive, mitochondrial disorder with a broad phenotypic spectrum ranging from severe neonatal lactic acidosis, encephalomyopathy and early death to an attenuated course with milder manifestations. Clinical features include delayed psychomotor development, intellectual disability, hypotonia, dystonia, ataxia, and spasticity. Severe combined respiratory chain deficiency may be found in severely affected individuals. {ECO:0000269|PubMed:28236339, ECO:0000269|PubMed:28650581, ECO:0000269|PubMed:28905505, ECO:0000269|PubMed:30920170, ECO:0000269|PubMed:35074316}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Parkinsonism-dystonia 3, childhood-onset (PKDYS3) [MIM:619738]: An autosomal recessive neurodegenerative disorder with onset in infancy or early childhood. Affected individuals present with progressive movement abnormalities, including parkinsonism with tremor, dystonia, myoclonus ataxia, and hyperkinetic movements such as ballismus. The parkinsonism features may be responsive to treatment with levodopa, although many patients develop levodopa-induced dyskinesia. Some patients may have mild cognitive impairment or psychiatric disturbances. {ECO:0000269|PubMed:29120065, ECO:0000269|PubMed:31970218, ECO:0000269|PubMed:34890876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00149 Interacts with P51116 EC number 2.1.1.233 Uniprot keywords 3D-structure; Alternative splicing; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H Molecular weight (Da) 35803 Length 310 Aromaticity 0.08 Instability index 42.77 Isoelectric point 6.13 Charge (pH=7) -3.58 2D Binding mode Binding energy (Kcal/mol) -9.53  Molscript Map 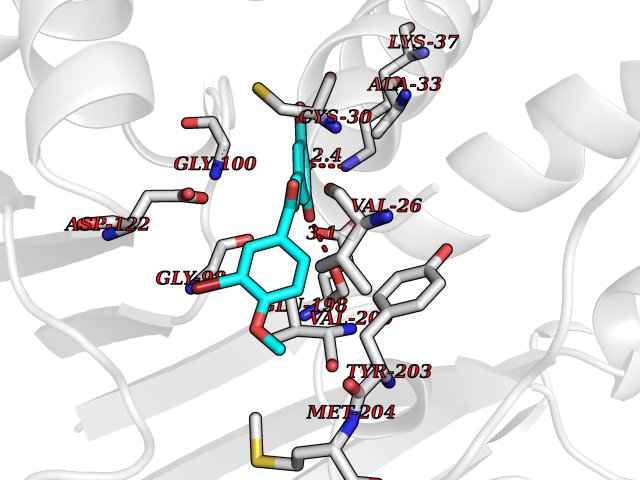 Pymol Map 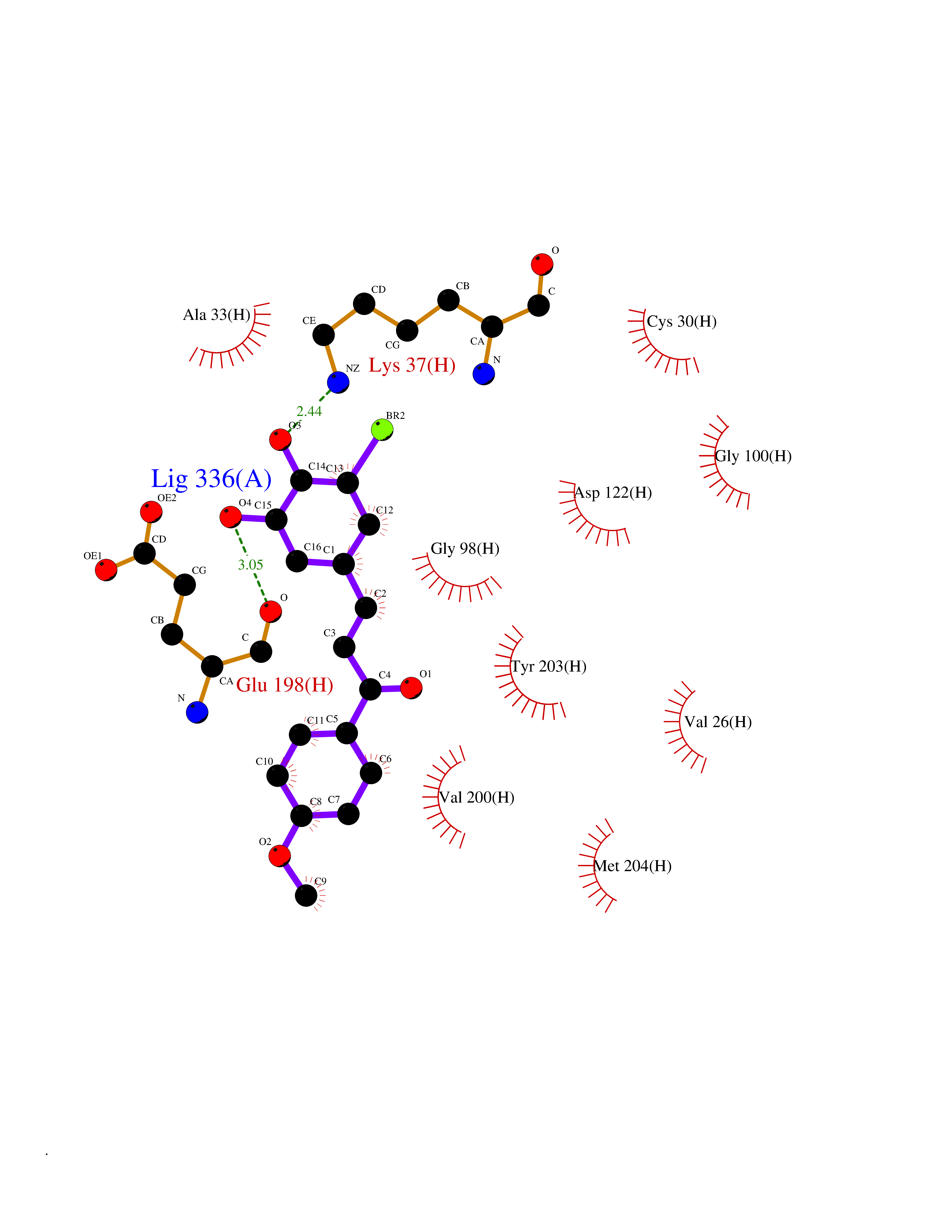 Ligplot Map 3D Binding mode Sequence GVRGTCEDASLCKRFAVSIGYWHDPYIQHFVRLSKERKAPEINRGYFARVHGVSQLIKAFLRKTECHCQIVNLGAGMDTTFWRLKDEDLLSSKYFEVDFPMIVTRKLHSIKCKPPLSSPILELHSEDTLQMDGHILDSKRYAVIGADLRDLSELEEKLKKCNMNTQLPTLLIAECVLVYMTPEQSANLLKWAANSFERAMFINYEQVNMGDRFGQIMIENLRRRQCDLAGVETCKSLESQKERLLSNGWETASAVDMMELYNRLPRAEVSRIESLEFLDEMELLEQLMRHYCLCWATKGGNELGLKEITY Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Tankyrase-2 (TNKS-2) | 3U9H | 6.99 | |
Target general information Gen name TNKS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tankyrase-related protein; Tankyrase-like protein; Tankyrase II; TRF1-interacting ankyrin-related ADP-ribose polymerase 2; TNKL; TANK2; Protein poly-ADP-ribosyltransferase tankyrase-2; Poly [ADP-ribos Protein family ARTD/PARP family Biochemical class Glycosyltransferases Function Acts as an activator of the Wnt signaling pathway by mediating poly-ADP-ribosylation of AXIN1 and AXIN2, 2 key components of the beta-catenin destruction complex: poly-ADP-ribosylated target proteins are recognized by RNF146, which mediates their ubiquitination and subsequent degradation. Also mediates poly-ADP-ribosylation of BLZF1 and CASC3, followed by recruitment of RNF146 and subsequent ubiquitination. Mediates poly-ADP-ribosylation of TERF1, thereby contributing to the regulation of telomere length. Stimulates 26S proteasome activity. Poly-ADP-ribosyltransferase involved in various processes such as Wnt signaling pathway, telomere length and vesicle trafficking. Related diseases Intellectual developmental disorder with macrocephaly, seizures, and speech delay (IDDMSSD) [MIM:618158]: An autosomal dominant neurodevelopmental disorder characterized by impaired intellectual development, poor speech, postnatal macrocephaly, and seizures. {ECO:0000269|PubMed:30290153}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O15084; Q7Z6K5-1; O15169; Q9NWV8; P11274; Q13698; Q9NRI5; Q6V0I7; Q9NWT6; P14652; Q9UIQ6; Q14980; Q9BZL4; Q92698; P78314; O43815; P54274; Q9C0C2; Q9UHP3; Q06649 EC number EC 2.4.2.30 Uniprot keywords 3D-structure; ADP-ribosylation; ANK repeat; Chromosome; Cytoplasm; Glycosyltransferase; Golgi apparatus; Hydroxylation; Membrane; Metal-binding; NAD; Nucleotidyltransferase; Nucleus; Proteomics identification; Reference proteome; Repeat; Telomere; Transferase; Ubl conjugation; Wnt signaling pathway; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 23695.5 Length 208 Aromaticity 0.11 Instability index 47.61 Isoelectric point 8.28 Charge (pH=7) 2.88 2D Binding mode Binding energy (Kcal/mol) -9.53  Molscript Map 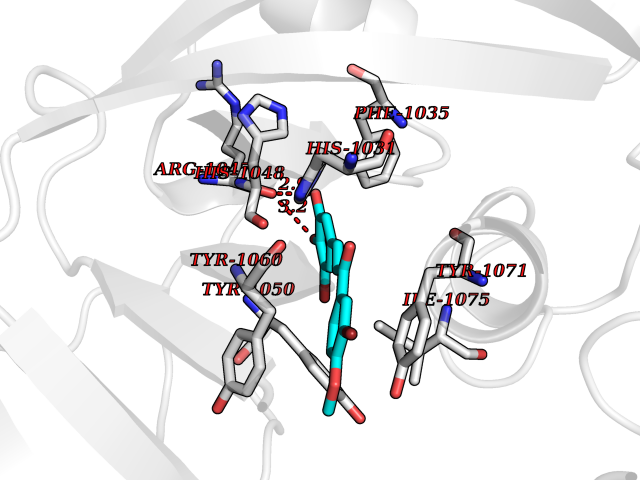 Pymol Map 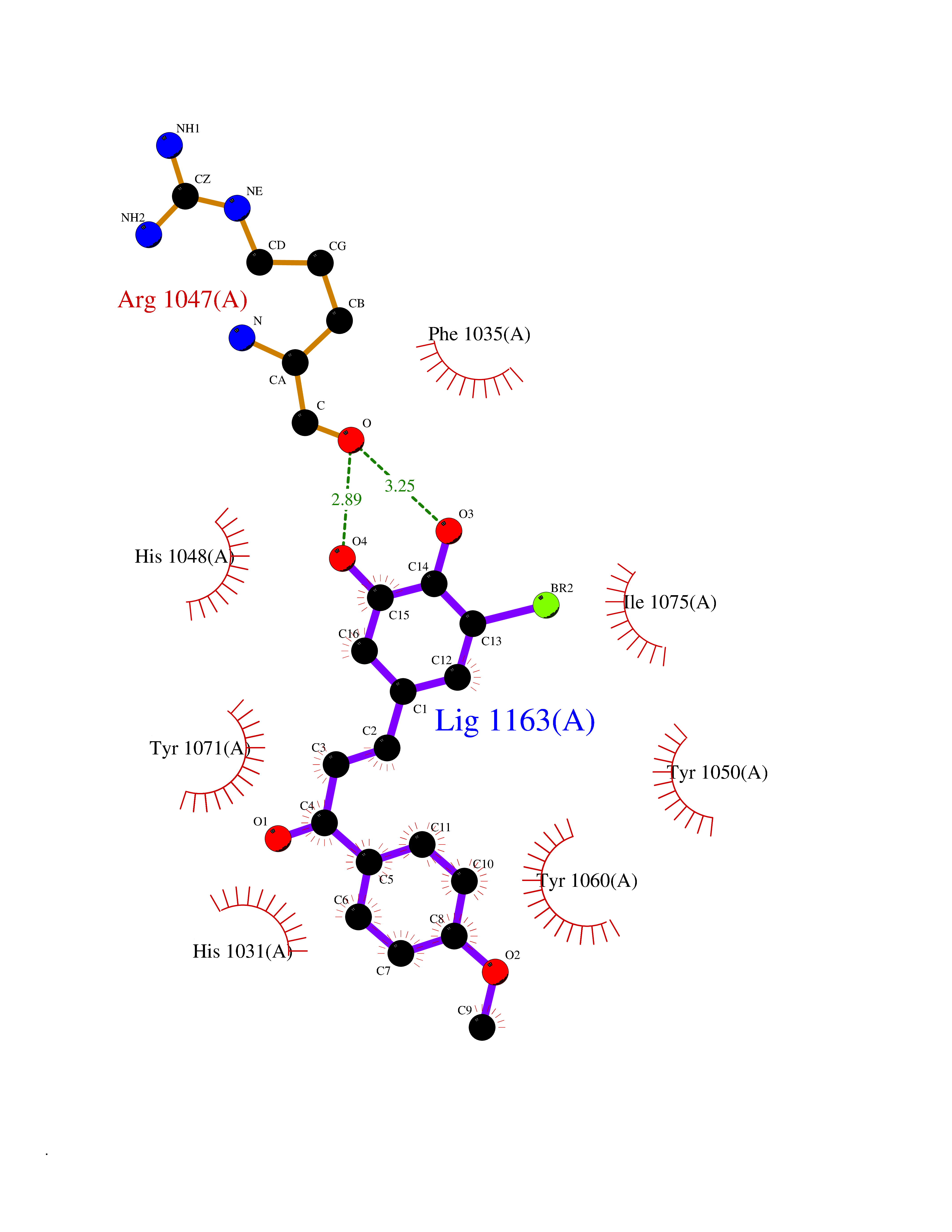 Ligplot Map 3D Binding mode Sequence GTILIDLSPDDKEFQSVEEEMQSTVREHRDGGHAGGIFNRYNILKIQKVCNKKLWERYTHRRKEVSEENHNHANERMLFHGSPFVNAIIHKGFDERHAYIGGMFGAGIYFAENSSKSNQYVYGIGGGTGCPVHKDRSCYICHRQLLFCRVTLGKSFLQFSAMAHSPPGHHSVTGRPSVNGLALAEYVIYRGEQAYPEYLITYQIMRPE Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Beta-arrestin-1 (ARRB1) | 6TKO | 6.99 | |
Target general information Gen name ARRB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Non-visual arrestin-2; Betaarrestin1; Arrestin beta1; Arrestin beta-1; ARR1 Protein family Arrestin family Biochemical class Arrestin protein Function During homologous desensitization, beta-arrestins bind to the GPRK-phosphorylated receptor and sterically preclude its coupling to the cognate G-protein; the binding appears to require additional receptor determinants exposed only in the active receptor conformation. The beta-arrestins target many receptors for internalization by acting as endocytic adapters (CLASPs, clathrin-associated sorting proteins) and recruiting the GPRCs to the adapter protein 2 complex 2 (AP-2) in clathrin-coated pits (CCPs). However, the extent of beta-arrestin involvement appears to vary significantly depending on the receptor, agonist and cell type. Internalized arrestin-receptor complexes traffic to intracellular endosomes, where they remain uncoupled from G-proteins. Two different modes of arrestin-mediated internalization occur. Class A receptors, like ADRB2, OPRM1, ENDRA, D1AR and ADRA1B dissociate from beta-arrestin at or near the plasma membrane and undergo rapid recycling. Class B receptors, like AVPR2, AGTR1, NTSR1, TRHR and TACR1 internalize as a complex with arrestin and traffic with it to endosomal vesicles, presumably as desensitized receptors, for extended periods of time. Receptor resensitization then requires that receptor-bound arrestin is removed so that the receptor can be dephosphorylated and returned to the plasma membrane. Involved in internalization of P2RY4 and UTP-stimulated internalization of P2RY2. Involved in phosphorylation-dependent internalization of OPRD1 ands subsequent recycling. Involved in the degradation of cAMP by recruiting cAMP phosphodiesterases to ligand-activated receptors. Beta-arrestins function as multivalent adapter proteins that can switch the GPCR from a G-protein signaling mode that transmits short-lived signals from the plasma membrane via small molecule second messengers and ion channels to a beta-arrestin signaling mode that transmits a distinct set of signals that are initiated as the receptor internalizes and transits the intracellular compartment. Acts as signaling scaffold for MAPK pathways such as MAPK1/3 (ERK1/2). ERK1/2 activated by the beta-arrestin scaffold is largely excluded from the nucleus and confined to cytoplasmic locations such as endocytic vesicles, also called beta-arrestin signalosomes. Recruits c-Src/SRC to ADRB2 resulting in ERK activation. GPCRs for which the beta-arrestin-mediated signaling relies on both ARRB1 and ARRB2 (codependent regulation) include ADRB2, F2RL1 and PTH1R. For some GPCRs the beta-arrestin-mediated signaling relies on either ARRB1 or ARRB2 and is inhibited by the other respective beta-arrestin form (reciprocal regulation). Inhibits ERK1/2 signaling in AGTR1- and AVPR2-mediated activation (reciprocal regulation). Is required for SP-stimulated endocytosis of NK1R and recruits c-Src/SRC to internalized NK1R resulting in ERK1/2 activation, which is required for the antiapoptotic effects of SP. Is involved in proteinase-activated F2RL1-mediated ERK activity. Acts as signaling scaffold for the AKT1 pathway. Is involved in alpha-thrombin-stimulated AKT1 signaling. Is involved in IGF1-stimulated AKT1 signaling leading to increased protection from apoptosis. Involved in activation of the p38 MAPK signaling pathway and in actin bundle formation. Involved in F2RL1-mediated cytoskeletal rearrangement and chemotaxis. Involved in AGTR1-mediated stress fiber formation by acting together with GNAQ to activate RHOA. Appears to function as signaling scaffold involved in regulation of MIP-1-beta-stimulated CCR5-dependent chemotaxis. Involved in attenuation of NF-kappa-B-dependent transcription in response to GPCR or cytokine stimulation by interacting with and stabilizing CHUK. May serve as nuclear messenger for GPCRs. Involved in OPRD1-stimulated transcriptional regulation by translocating to CDKN1B and FOS promoter regions and recruiting EP300 resulting in acetylation of histone H4. Involved in regulation of LEF1 transcriptional activity via interaction with DVL1 and/or DVL2 Also involved in regulation of receptors other than GPCRs. Involved in Toll-like receptor and IL-1 receptor signaling through the interaction with TRAF6 which prevents TRAF6 autoubiquitination and oligomerization required for activation of NF-kappa-B and JUN. Binds phosphoinositides. Binds inositolhexakisphosphate (InsP6). Involved in IL8-mediated granule release in neutrophils. Required for atypical chemokine receptor ACKR2-induced RAC1-LIMK1-PAK1-dependent phosphorylation of cofilin (CFL1) and for the up-regulation of ACKR2 from endosomal compartment to cell membrane, increasing its efficiency in chemokine uptake and degradation. Involved in the internalization of the atypical chemokine receptor ACKR3. Negatively regulates the NOTCH signaling pathway by mediating the ubiquitination and degradation of NOTCH1 by ITCH. Participates to the recruitment of the ubiquitin-protein ligase to the receptor. Functions in regulating agonist-mediated G-protein coupled receptor (GPCR) signaling by mediating both receptor desensitization and resensitization processes. Related diseases Intellectual developmental disorder with dysmorphic facies and ptosis (IDDDFP) [MIM:617333]: An autosomal dominant neurodevelopmental disorder characterized by delayed psychomotor development, intellectual disability, delayed language, and facial dysmorphisms, most notably ptosis. Additional features may include poor growth, hypotonia, and seizures. {ECO:0000269|PubMed:27939639, ECO:0000269|PubMed:27939640}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P63010-2; O15169; P0DP25; P20963; P25101; P50148; Q5JWF2; Q14749; P06396; Q16665; P11142; Q99683; P53779; P45984; Q00987; P19338; Q14978; P14618; P14859-6; P35813; O75688; Q13523; P06702; P12931; Q15208; Q13428; P04637; P27348; P25490; O43298; O95218; Q7DB77 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Cell projection; Coated pit; Cytoplasm; Cytoplasmic vesicle; Membrane; Nucleus; Phosphoprotein; Protein transport; Proteomics identification; Reference proteome; Signal transduction inhibitor; Transcription; Transcription regulation; Transport; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32455.6 Length 293 Aromaticity 0.12 Instability index 28.99 Isoelectric point 9.12 Charge (pH=7) 9.86 2D Binding mode Binding energy (Kcal/mol) -9.54  Molscript Map 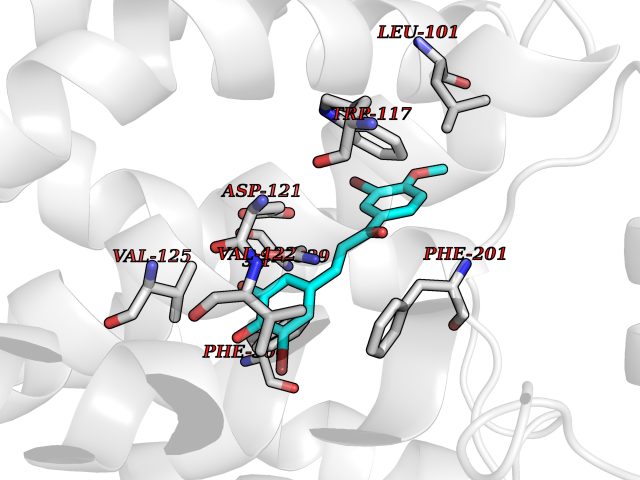 Pymol Map 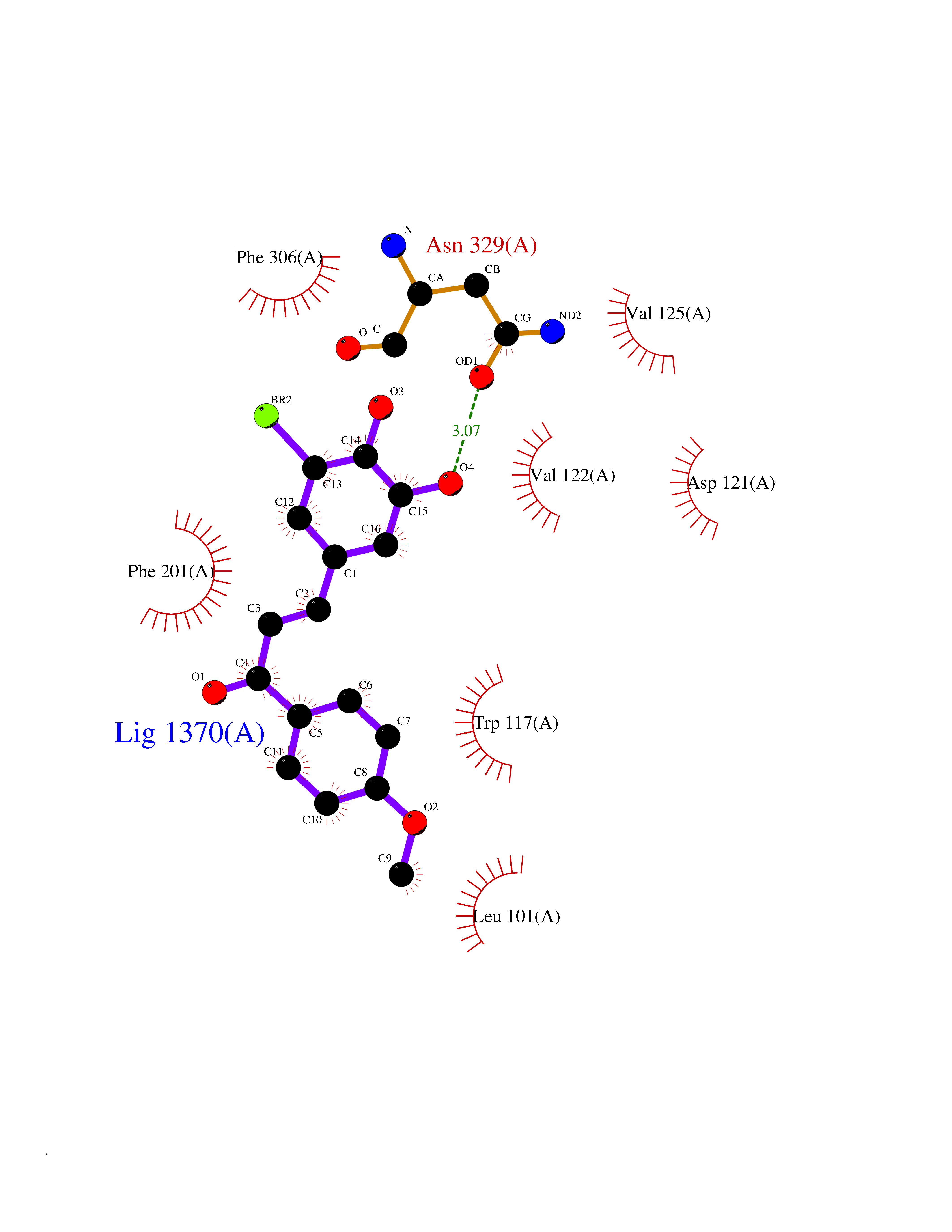 Ligplot Map 3D Binding mode Sequence AGCSLLMALVVLLIVAGNVLVIAAIGRTQRLQTLTNLFITSLACADLVVGLLVVPFGATLVCRGTWLWGSFLCELWTSLDVLCVTASIWTLCVIAIDRYLAITSPFRYQSLMTRARAKVIICTVWAISALVSFLPIMMHWWRDEDPQALKCYQDPGCCDFVTNRAYAIASSIISFYIPLLIMIFVYLRVYREAKEQIRKIDVMAMREHKALKTLGIIMGVFTLCWLPFFLVNIVNVFNRDLVPKWLFVAFNWLGYANSAMNPIIYCRSPDFRKAFKRLLAEXAXXAXXXLAKD Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Melatonin receptor type 1B (MTNR1B) | 6ME9 | 6.99 | |
Target general information Gen name MTNR1B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mel1b receptor; Mel1b melatonin receptor; Mel-1B-R Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Likely to mediate the reproductive and circadian actions of melatonin. The activity of this receptor is mediated by pertussis toxin sensitive G proteins that inhibit adenylate cyclase activity. High affinity receptor for melatonin. Related diseases Insulin-like growth factor 1 resistance (IGF1RES) [MIM:270450]: A disorder characterized by intrauterine growth retardation, poor postnatal growth and increased plasma IGF1 levels. {ECO:0000269|PubMed:14657428, ECO:0000269|PubMed:15928254, ECO:0000269|PubMed:25040157}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB06594; DB01065; DB00980; DB02709; DB09071; DB15133 Interacts with P28335; P48039; O76081; Q14669 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50184.9 Length 448 Aromaticity 0.11 Instability index 37.2 Isoelectric point 5.72 Charge (pH=7) -5.68 2D Binding mode Binding energy (Kcal/mol) -9.53 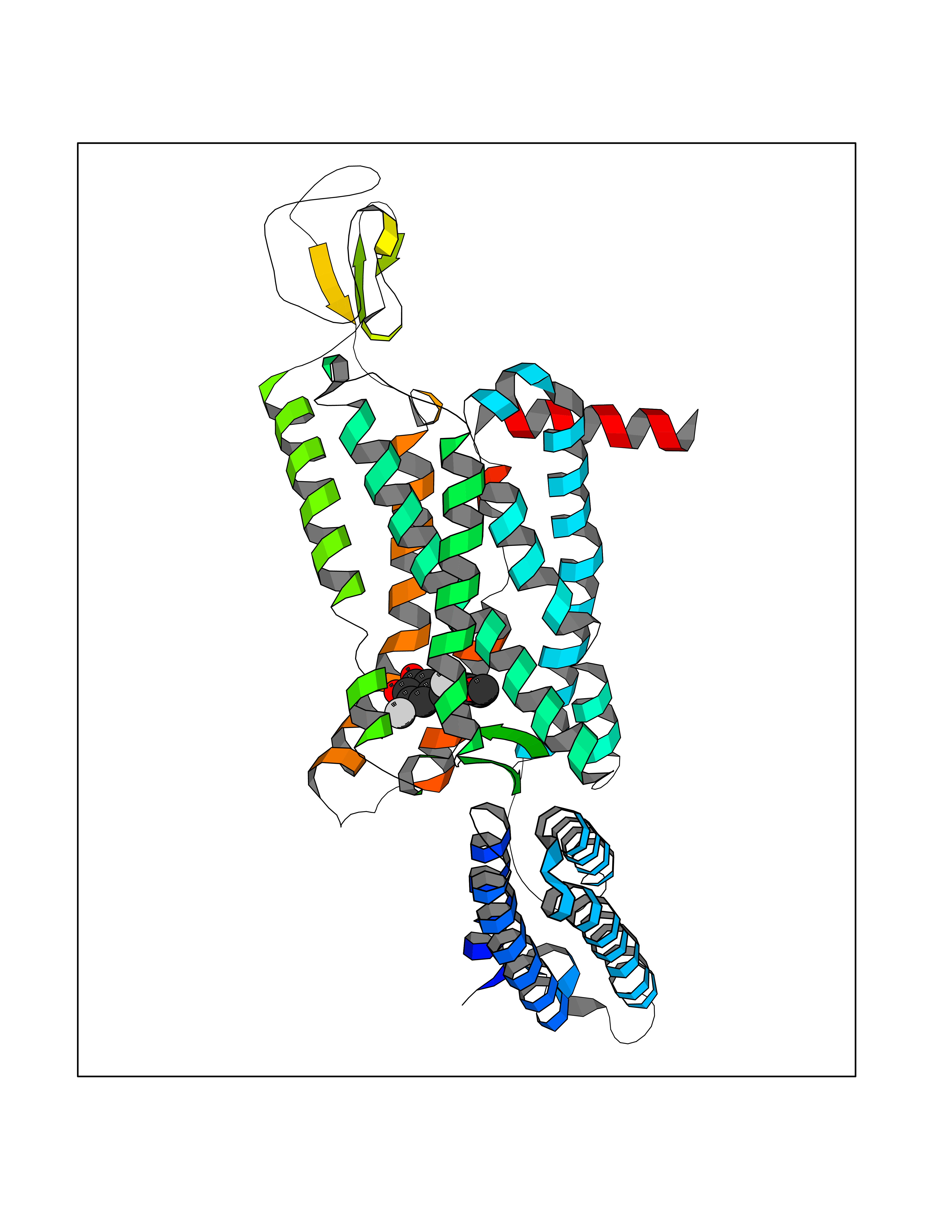 Molscript Map 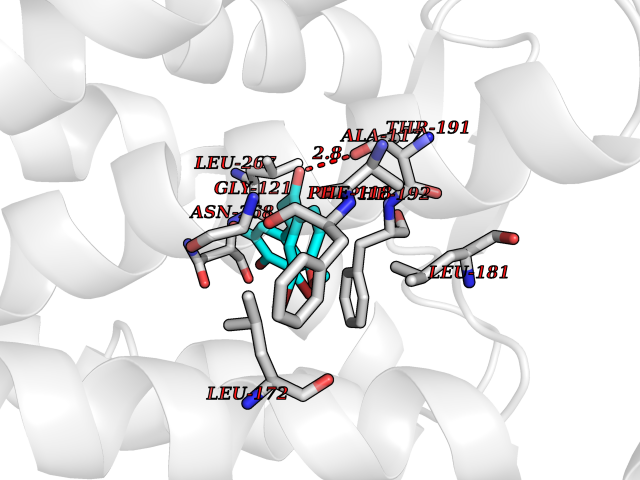 Pymol Map 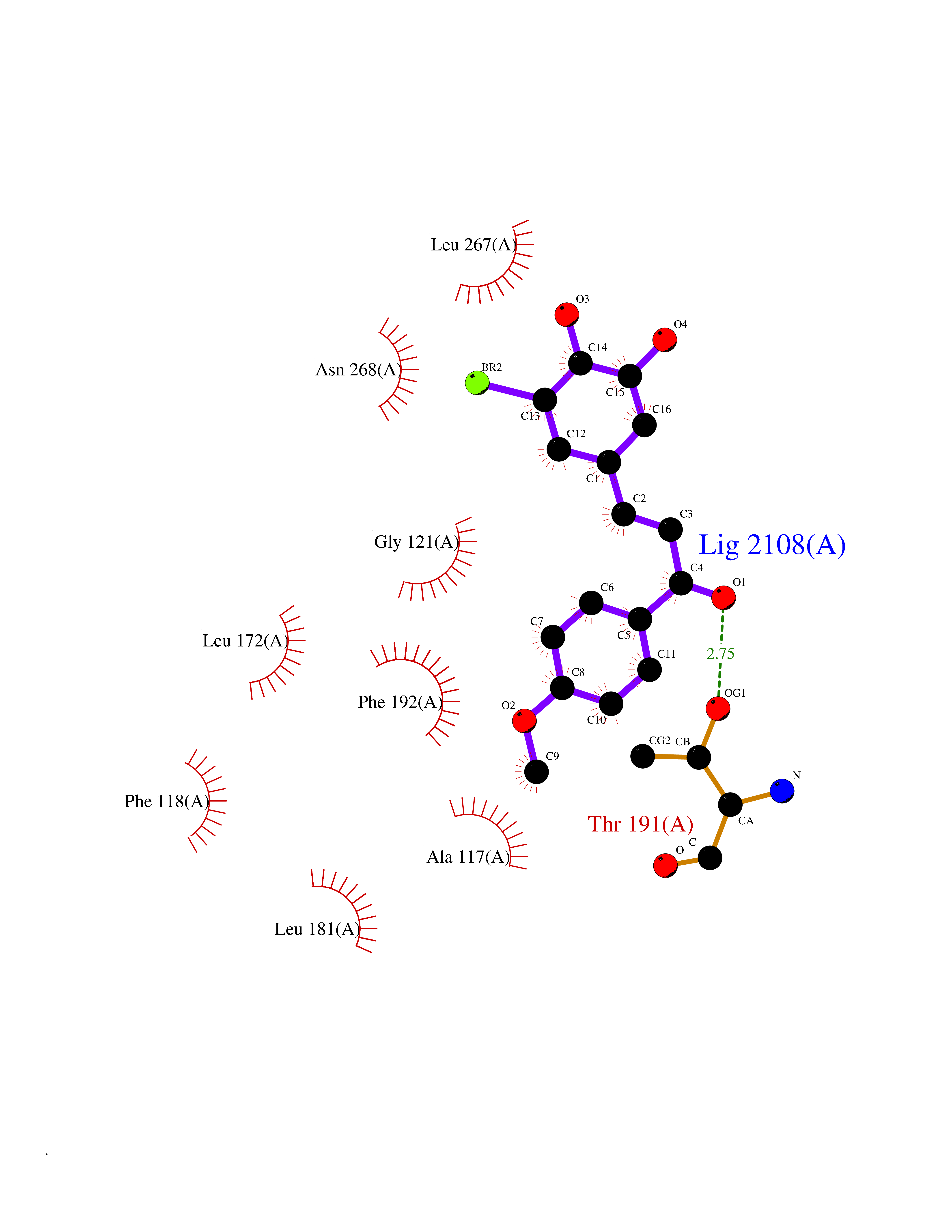 Ligplot Map 3D Binding mode Sequence ADLEDNWETLNDNLKVIEKADNAAQVKDALTKMRAAALDAQKATPPKLEDKSPDSPEMKDFRHGFDILVGQIDDALKLANEGKVKEAQAAAEQLKTTRNAYIQKYLGDGARPSWVAPALSAVLIVTTAVDVVGNLLVILSVLRNRKLRNAGNLFLVSLALANLVVAFYPYPLILVAIFYDGWAFGEEHCKASAFVMGLSVIGSVWNITAIAIDRYLYICHSMAYHRIYRRWHTPLHICLIWLLTVVALLPNFFVGSLEYDPRIYSCTFIQTASTQYTAAVVVIHFLLPIAVVSFCYLRIWVLVLQARMKKYTCTVCGYIYNPEDGDPDNGVNPGTDFKDIPDDWVCPLCGVGKDQFEEVECLKPSDLRSFLTMFVVFVIFAICFAPLNCIGLAVAINPQEMAPQIPEGLFVTSYLLAYFNSCLNPIVYGLLDQNFRREYKRILLALWN Hydrogen bonds contact Hydrophobic contact | ||||