Job Results:
Ligand
Structure
Job ID
e8ee1117e51baec8034629c7ce0a1a1e
Job name
NA
Time
2025-12-29 08:34:20
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Peroxisome proliferator-activated receptor alpha (PPARA) | 3VI8 | 5.91 | |
Target general information Gen name PPARA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Peroxisome proliferater-activated receptor alpha; PPARalpha; PPAR-alpha; PPAR; Nuclear receptor subfamily 1 group C member 1; NR1C1 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Key regulator of lipid metabolism. Activated by the endogenous ligand 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine (16:0/18:1-GPC). Activated by oleylethanolamide, a naturally occurring lipid that regulates satiety. Receptor for peroxisome proliferators such as hypolipidemic drugs and fatty acids. Regulates the peroxisomal beta-oxidation pathway of fatty acids. Functions as transcription activator for the ACOX1 and P450 genes. Transactivation activity requires heterodimerization with RXRA and is antagonized by NR2C2. May be required for the propagation of clock information to metabolic pathways regulated by PER2. Ligand-activated transcription factor. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08915; DB00132; DB01118; DB04557; DB01393; DB04519; DB05416; DB09064; DB09006; DB00636; DB09213; DB03756; DB05187; DB06521; DB01039; DB13873; DB00573; DB13961; DB02266; DB01241; DB07215; DB01050; DB00159; DB07724; DB00328; DB12007; DB03017; DB12961; DB06510; DB08231; DB11605; DB01890; DB04224; DB11133; DB03796; DB02746; DB01708; DB06533; DB04971; DB02709; DB00412; DB09422; DB03193; DB06536; DB00197; DB00313 Interacts with P02768-3; P55212; P45973; P06307; Q3L8U1-3; G5E9A7; P22607; P62993; Q14957; P06396; P42858; Q8WXH2; P13473-2; O75376; Q13133; A0A6Q8PF08; P54725; P62826; Q7Z699; P37173; P55072; P55055-1; Q13133 EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Biological rhythms; DNA-binding; Lipid-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 29322.1 Length 258 Aromaticity 0.09 Instability index 35.53 Isoelectric point 6.09 Charge (pH=7) -3.57 2D Binding mode Binding energy (Kcal/mol) -8.06 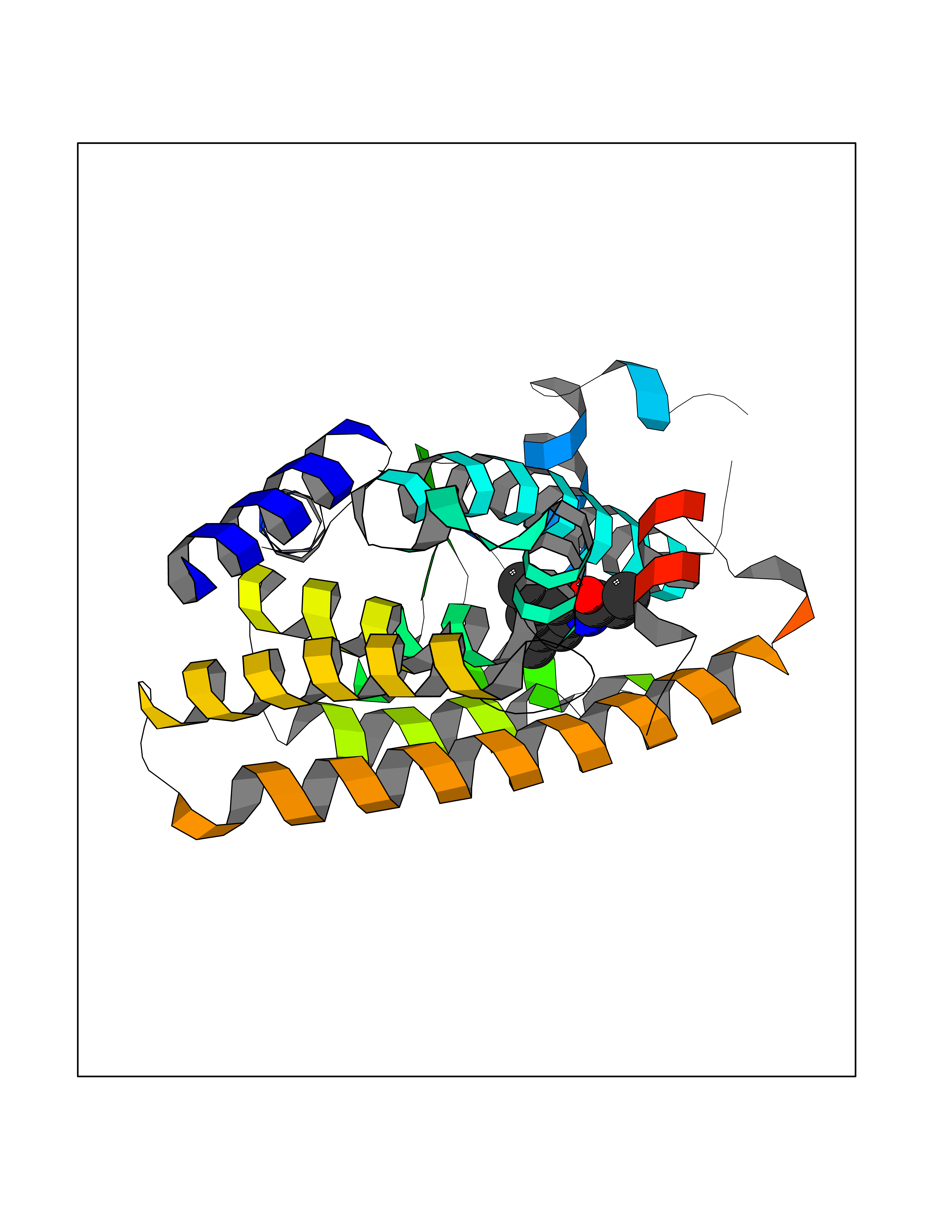 Molscript Map 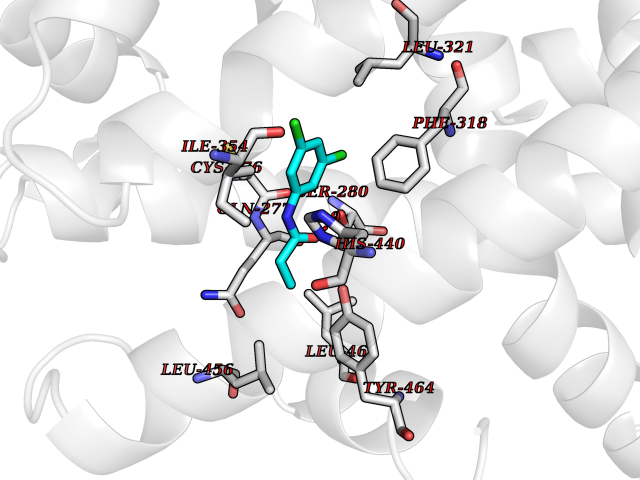 Pymol Map 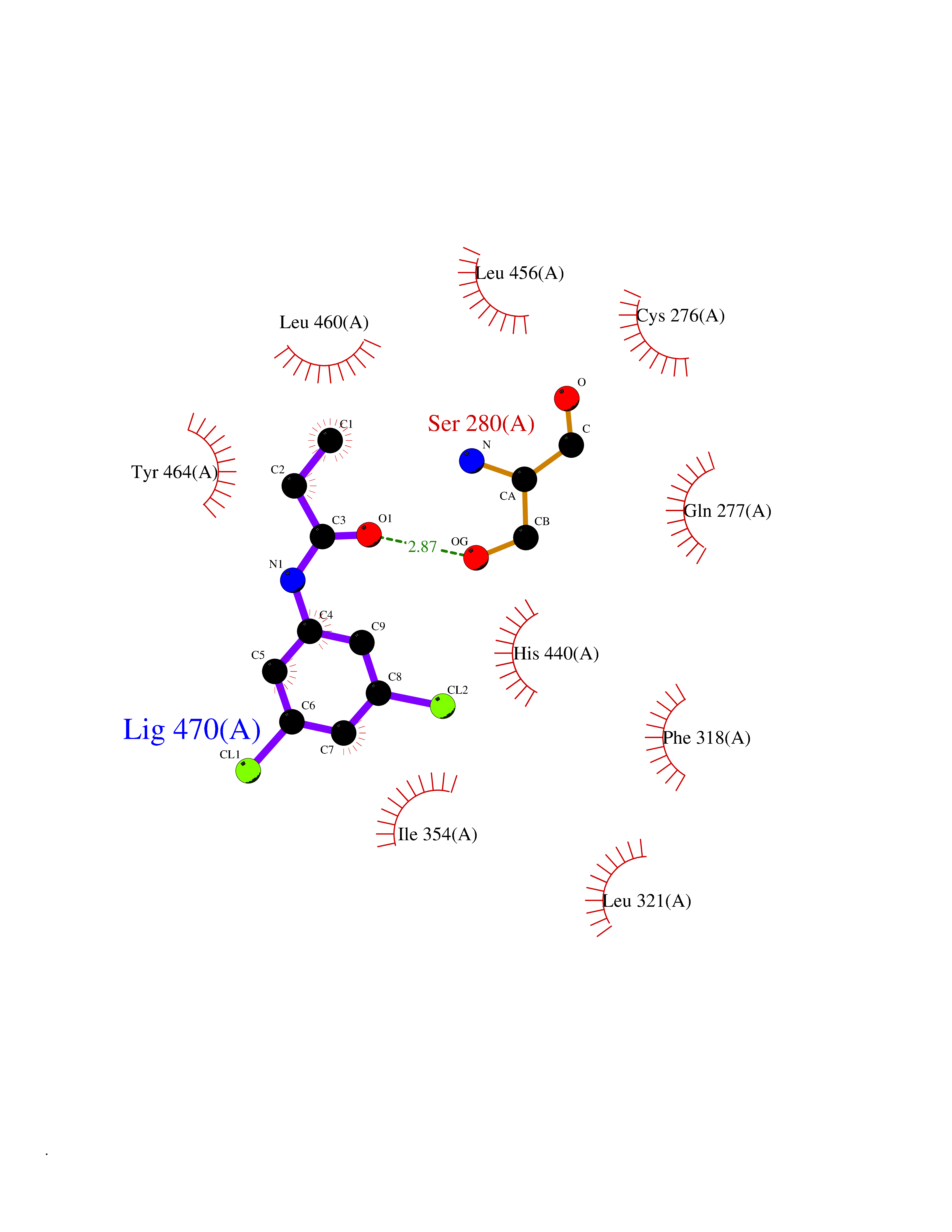 Ligplot Map 3D Binding mode Sequence DLKSLAKRIYEAYLKNFNMNKVKARVILSPFVIHDMETLCMAEKTLVAKLVANGNKEAEVRIFHCCQCTSVETVTELTEFAKAIPGFANLDLNDQVTLLKYGVYEAIFAMLSSVMNKDGMLVAYGNGFITREFLKSLRKPFCDIMEPKFDFAMKFNALELDDSDISLFVAAIICCGDRPGLLNVGHIEKMQEGIVHVLRLHLQSNHPDDIFLFPKLLQKMADLRQLVTEHAQLVQIIKKTESDAALHPLLQEIYRDMY Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Glutamate receptor ionotropic kainate 2 (GRIK2) | 5CMM | 5.91 | |
Target general information Gen name GRIK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Glutamate receptor ionotropic, kainate 2; Glutamate receptor 6; GluR6; GluR-6; GluK2; Excitatory amino acid receptor 4; EAA4 Protein family Glutamate-gated ion channel (TC 1.A.10.1) family, GRIK2 subfamily Biochemical class Glutamate-gated ion channel Function L-glutamate acts as an excitatory neurotransmitter at many synapses in the central nervous system. Binding of the excitatory neurotransmitter L-glutamate induces a conformation change, leading to the opening of the cation channel, and thereby converts the chemical signal to an electrical impulse. The receptor then desensitizes rapidly and enters a transient inactive state, characterized by the presence of bound agonist. May be involved in the transmission of light information from the retina to the hypothalamus. Modulates cell surface expression of NETO2. Ionotropic glutamate receptor. Related diseases Intellectual developmental disorder, autosomal recessive 6 (MRT6) [MIM:611092]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT6 patients display mild to severe intellectual disability and psychomotor development delay in early childhood. Patients do not have neurologic problems, congenital malformations, or facial dysmorphism. Body height, weight, and head circumference are normal. {ECO:0000269|PubMed:17847003}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurodevelopmental disorder with impaired language and ataxia and with or without seizures (NEDLAS) [MIM:619580]: An autosomal dominant disorder characterized by axial hypotonia and global developmental delay. Affected individuals show impaired intellectual development, delayed walking, poor speech, and behavioral abnormalities. Some patients have a more severe phenotype with early-onset seizures resembling epileptic encephalopathy, inability to walk or speak, and hypomyelination on brain imaging. {ECO:0000269|PubMed:28180184, ECO:0000269|PubMed:34375587}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03425; DB01351; DB01352; DB01483; DB00237; DB00241; DB01353; DB01496; DB02852; DB00142; DB01354; DB01355; DB00463; DB00849; DB00312; DB01174; DB00794; DB02999; DB00418; DB00306; DB00599; DB00273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Intellectual disability; Ion channel; Ion transport; Isopeptide bond; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; RNA editing; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 29150.1 Length 257 Aromaticity 0.1 Instability index 35.11 Isoelectric point 5.89 Charge (pH=7) -2.05 2D Binding mode Binding energy (Kcal/mol) -8.06 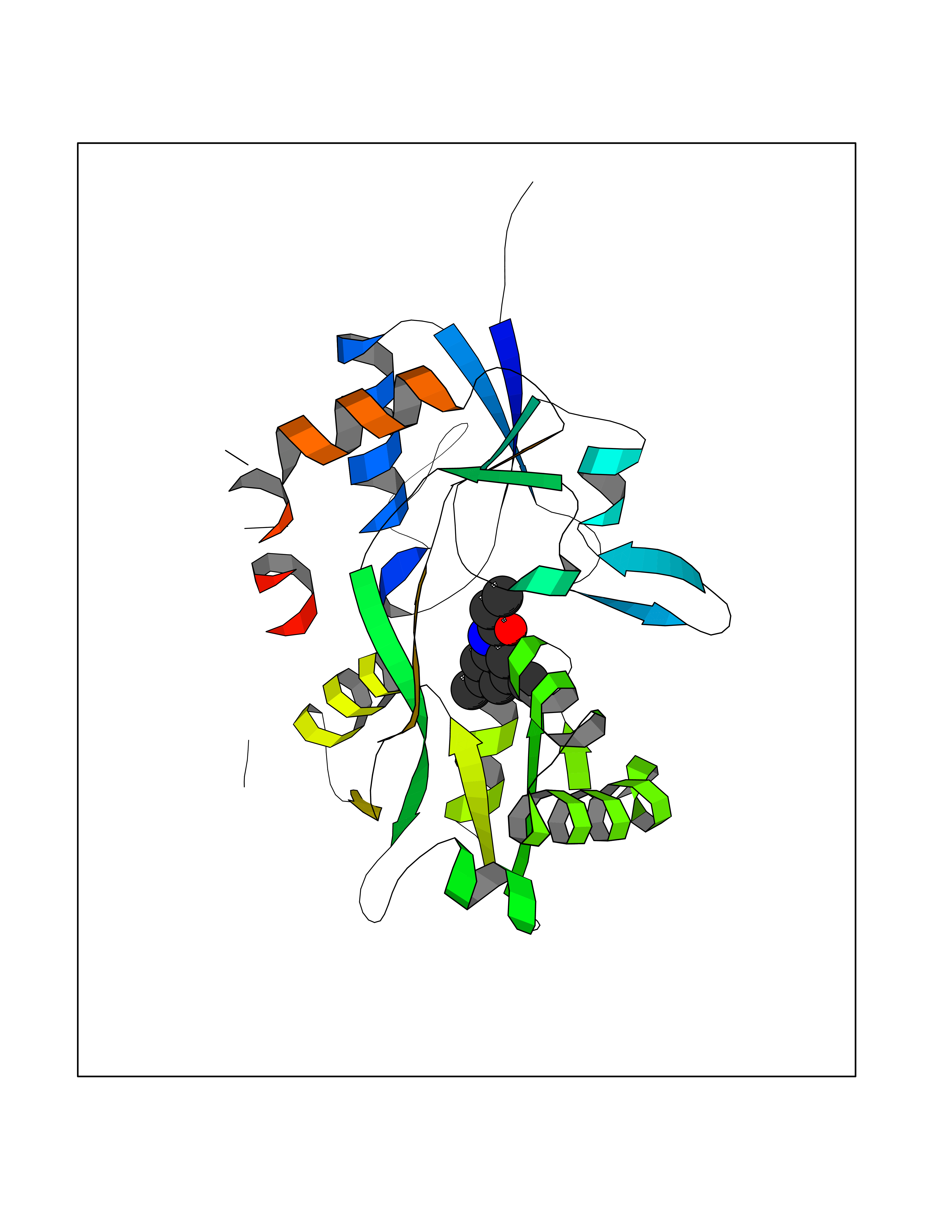 Molscript Map 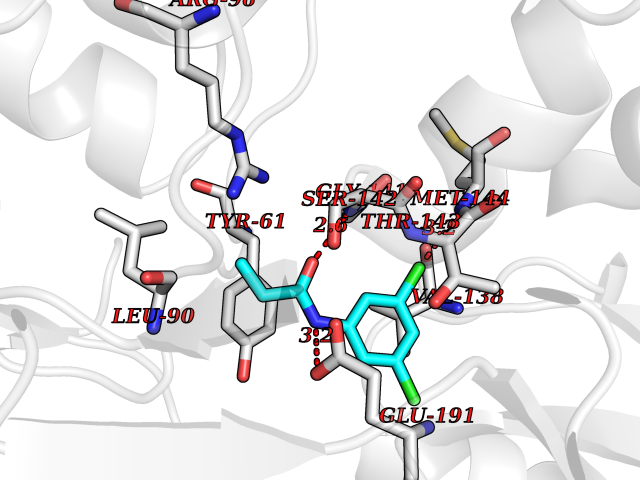 Pymol Map 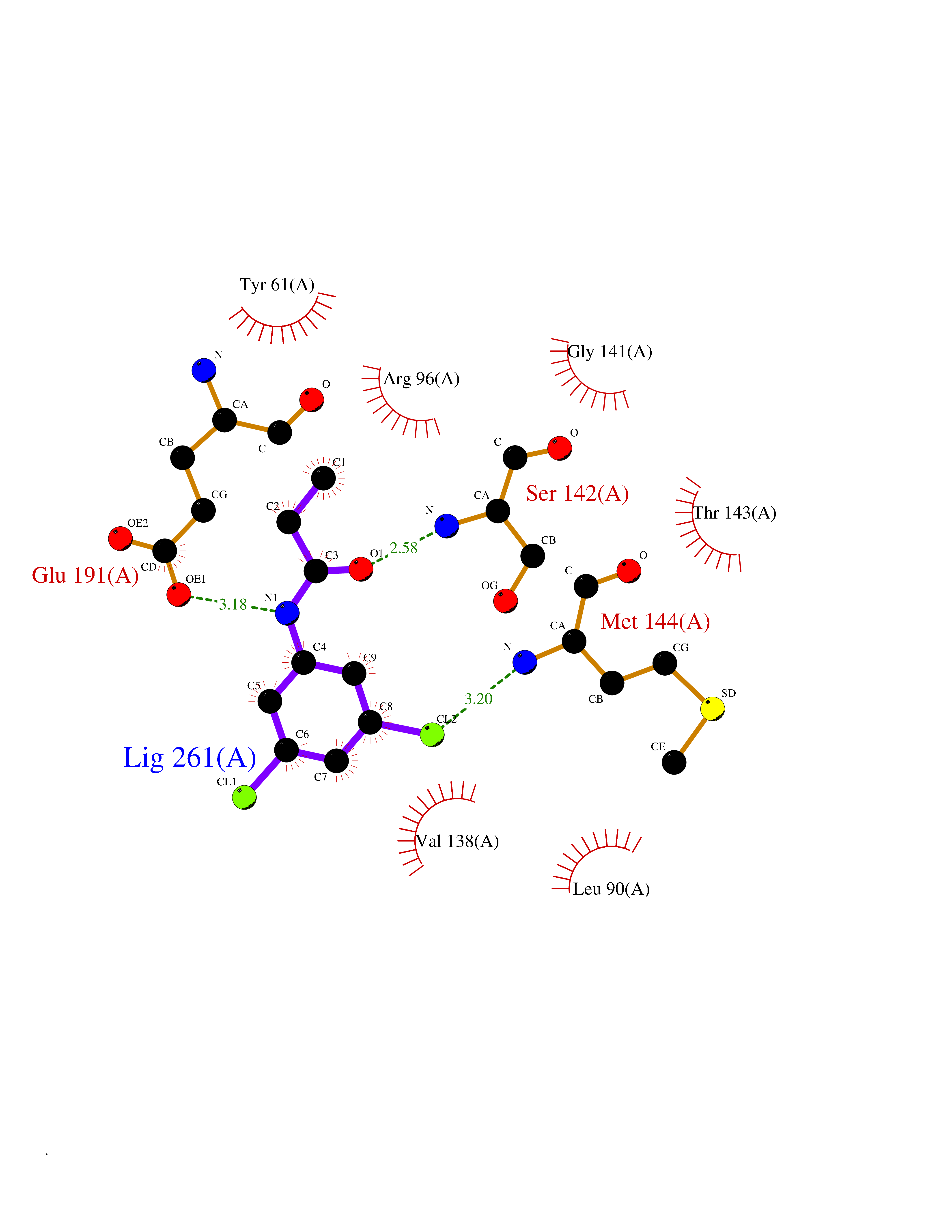 Ligplot Map 3D Binding mode Sequence GSNRSLIVTTILEEPYVLFKKSDKPLYGNDRFEGYCIDLLRELSTILGFTYEIRLVEDGKYGAQDDVNGQWNGMVRELIDHKADLAVAPLTITYVREKVIDFSKPFMTLGISILYRKGTPIDSADDLAKQTKIEYGAVEDGSTMTFFKKSKISTYDKMWAFMSSRRQSVLVKSSEEGIQRVLTSDYALLMESTTIEFVTQRNCNLTQIGGLIDSKGYGVGTPMGSPYRDKITIAILQLQEEGKLHMMKEKWWRGCPE Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Phosphoribosylaminoimidazolecarboxamide formyltransferase (ATIC) | 1P4R | 5.91 | |
Target general information Gen name ATIC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PURH; OK/SW-cl.86; Bifunctional purine biosynthesis protein PURH Protein family PurH family Biochemical class Methyltransferase Function Bifunctional enzyme that catalyzes 2 steps in purine biosynthesis. Related diseases AICA-ribosuria due to ATIC deficiency (AICAR) [MIM:608688]: A neurologically devastating inborn error of purine biosynthesis. Patients excrete massive amounts of AICA-riboside in the urine and accumulate AICA-ribotide and its derivatives in erythrocytes and fibroblasts. Clinical features include profound intellectual disability, epilepsy, dysmorphic features and congenital blindness. AICAR inheritance is autosomal recessive. {ECO:0000269|PubMed:15114530}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02309; DB03442; DB01700; DB01972; DB00563; DB04057; DB00642; DB00116 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Direct protein sequencing; Disease variant; Epilepsy; Hydrolase; Intellectual disability; Multifunctional enzyme; Proteomics identification; Purine biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 128556 Length 1177 Aromaticity 0.07 Instability index 38.21 Isoelectric point 6.28 Charge (pH=7) -7.98 2D Binding mode Binding energy (Kcal/mol) -8.06 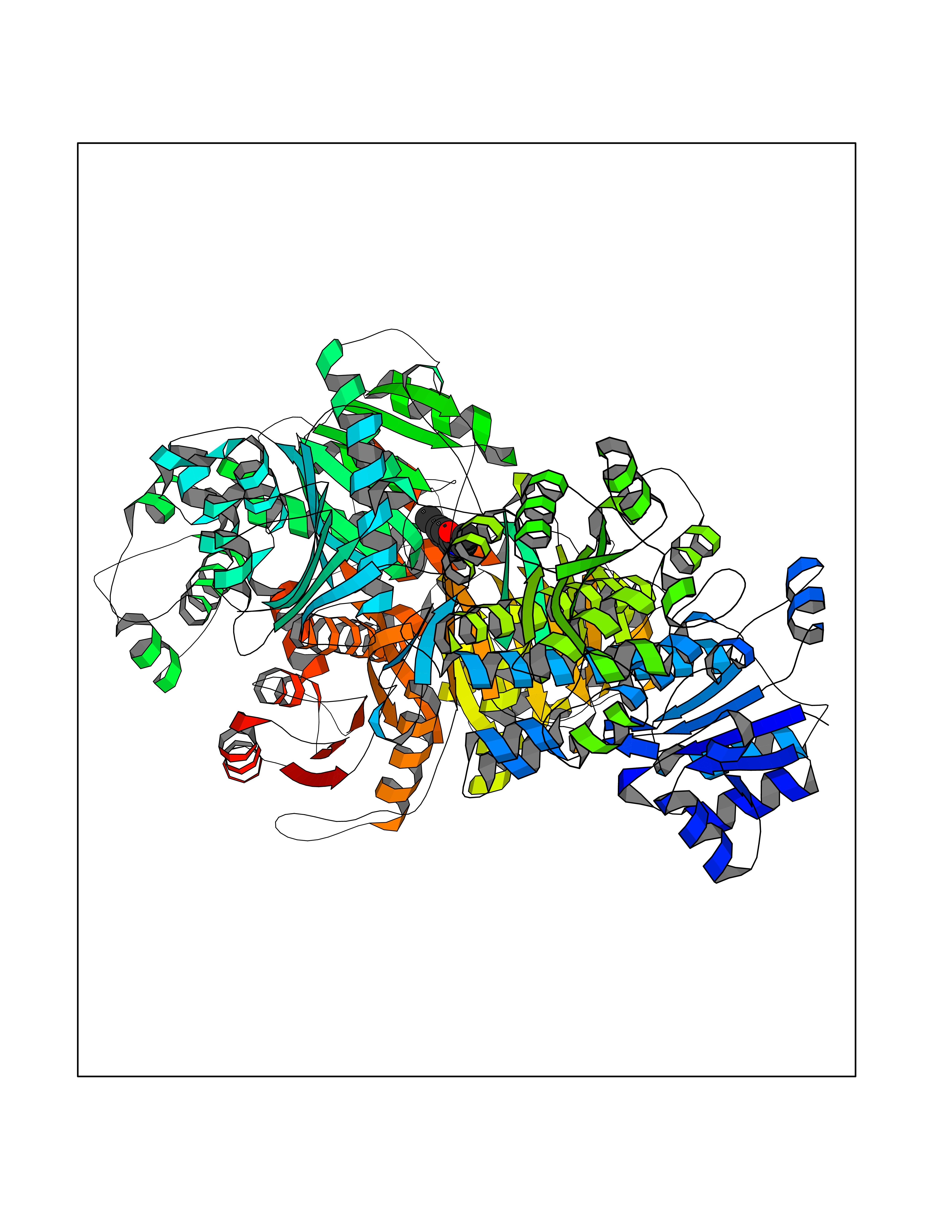 Molscript Map 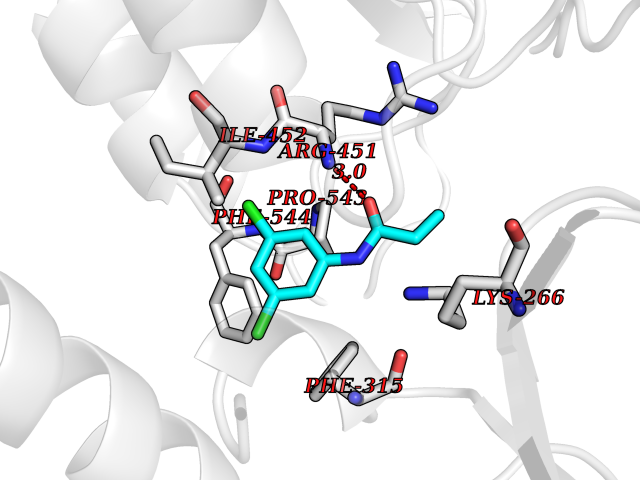 Pymol Map 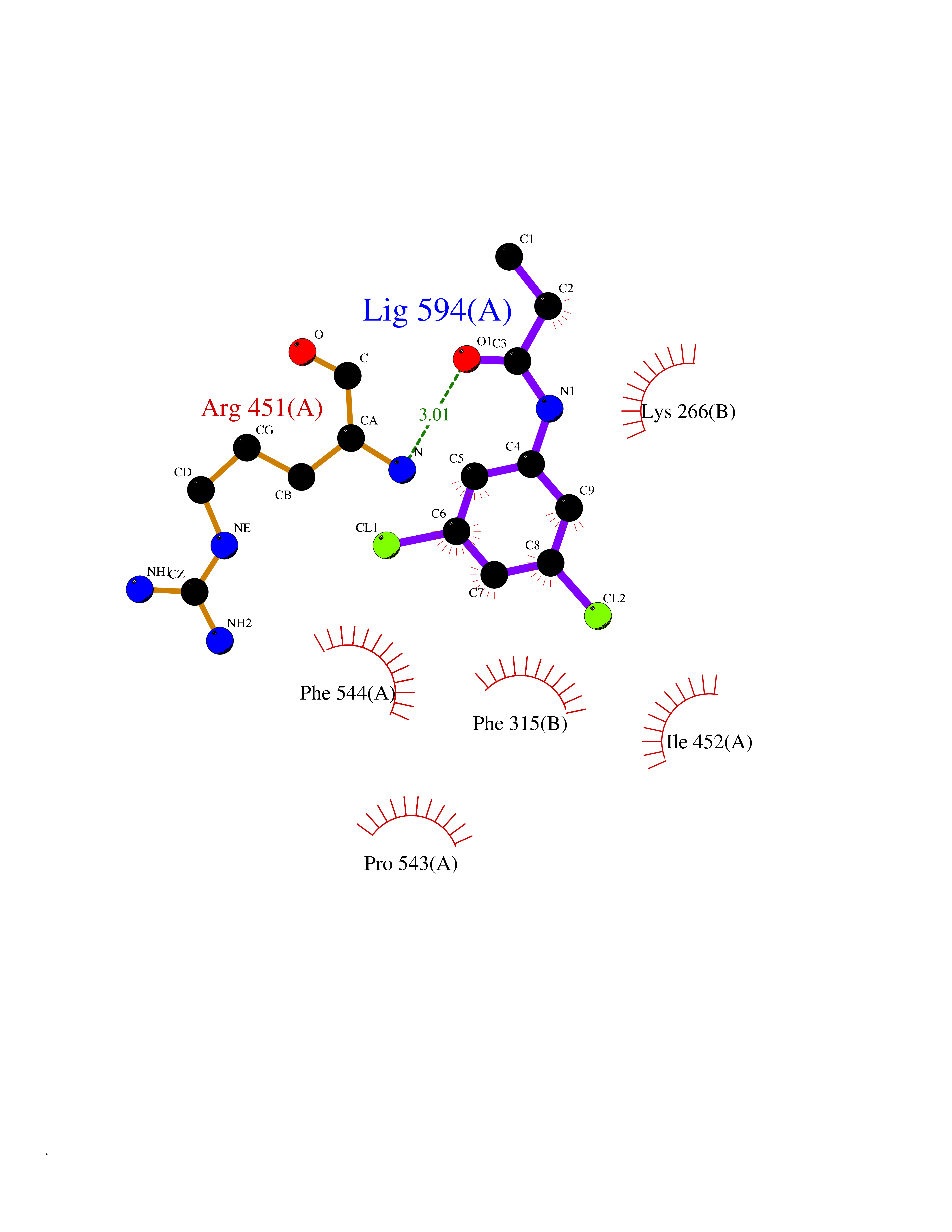 Ligplot Map 3D Binding mode Sequence GQLALFSVSDKTGLVEFARNLTALGLNLVASGGTAKALRDAGLAVRDVSELTGFPEMLGGRVKTLHPAVHAGILARNIPEDNADMARLDFNLIRVVACNLYPFVKTVASPGVTVEEAVEQIDIGGVTLLRAAAKNHARVTVVCEPEDYVVVSTEMQSSESKDTSLETRRQLALKAFTHTAQYDEAISDYFRKQYSKGVSQMPLRYGMNPHQTPAQLYTLQPKLPITVLNGAPGFINLCDALNAWQLVKELKEALGIPAAASFKHVSPAGAAVGIPLSEDEAKVCMVYDLYKTLTPISAAYARARGADRMSSFGDFVALSDVCDVPTAKIISREVSDGIIAPGYEEEALTILSKKKNGNYCVLQMDQSYKPDENEVRTLFGLHLSQKRNNGVVDKSLFSNVVTKNKDLPESALRDLIVATIAVKYTQSNSVCYAKNGQVIGIGAGQQSRIHCTRLAGDKANYWWLRHHPQVLSMKFKTGVKRAEISNAIDQYVTGTIGEDEDLIKWKALFEEVPELLTEAEKKEWVEKLTEVSISSDAFFPFRDNVDRAKRSGVAYIAAPSGSAADKVVIEACDELGIILAHTNLRLFHHQLALFSVSDKTGLVEFARNLTALGLNLVASGGTAKALRDAGLAVRDVSELTGFPEMLGGRVKTLHPAVHAGILARNIPEDNADMARLDFNLIRVVACNLYPFVKTVASPGVTVEEAVEQIDIGGVTLLRAAAKNHARVTVVCEPEDYVVVSTEMQSSESKDTSLETRRQLALKAFTHTAQYDEAISDYFRKQYSKGVSQMPLRYGMNPHQTPAQLYTLQPKLPITVLNGAPGFINLCDALNAWQLVKELKEALGIPAAASFKHVSPAGAAVGIPLSEDEAKVCMVYDLYKTLTPISAAYARARGADRMSSFGDFVALSDVCDVPTAKIISREVSDGIIAPGYEEEALTILSKKKNGNYCVLQMDQSYKPDENEVRTLFGLHLSQKRNNGVVDKSLFSNVVTKNKDLPESALRDLIVATIAVKYTQSNSVCYAKNGQVIGIGAGQQSRIHCTRLAGDKANYWWLRHHPQVLSMKFKTGVKRAEISNAIDQYVTGTIGEDEDLIKWKALFEEVPELLTEAEKKEWVEKLTEVSISSDAFFPFRDNVDRAKRSGVAYIAAPSGSAADKVVIEACDELGIILAHTNLRLFHH Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Hypoxia-inducible factor 2 alpha (HIF-2A) | 5TBM | 5.91 | |
Target general information Gen name EPAS1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms bHLHe73; PASD2; PAS domain-containing protein 2; Member of PAS protein 2; MOP2; Hypoxia-inducible factor 2-alpha; HLF; HIF2A; HIF2-alpha; HIF-2-alpha; HIF-1-alpha-like factor; Endothelial PAS domain-c Protein family NA Biochemical class NA Function Heterodimerizes with ARNT; heterodimer binds to core DNA sequence 5'-TACGTG-3' within the hypoxia response element (HRE) of target gene promoters. Regulates the vascular endothelial growth factor (VEGF) expression and seems to be implicated in the development of blood vessels and the tubular system of lung. May also play a role in the formation of the endothelium that gives rise to the blood brain barrier. Potent activator of the Tie-2 tyrosine kinase expression. Activation requires recruitment of transcriptional coactivators such as CREBBP and probably EP300. Interaction with redox regulatory protein APEX seems to activate CTAD. Transcription factor involved in the induction of oxygen regulated genes. Related diseases Erythrocytosis, familial, 4 (ECYT4) [MIM:611783]: An autosomal dominant disorder characterized by elevated serum hemoglobin and hematocrit, and normal platelet and leukocyte counts. {ECO:0000269|PubMed:18184961, ECO:0000269|PubMed:18378852, ECO:0000269|PubMed:19208626, ECO:0000269|PubMed:22367913}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15463; DB12255 Interacts with P27540; Q96RK4; O00327-8; Q8WYA1-3; Q9GZT9; P60228; O60573; P09467; P61244; Q9BWF3-1; P08047; Q9Y2K6; P40818 EC number NA Uniprot keywords 3D-structure; Activator; Angiogenesis; Congenital erythrocytosis; Developmental protein; Differentiation; Disease variant; DNA-binding; Host-virus interaction; Hydroxylation; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 12249.8 Length 106 Aromaticity 0.11 Instability index 40.77 Isoelectric point 5.25 Charge (pH=7) -5.82 2D Binding mode Binding energy (Kcal/mol) -8.06 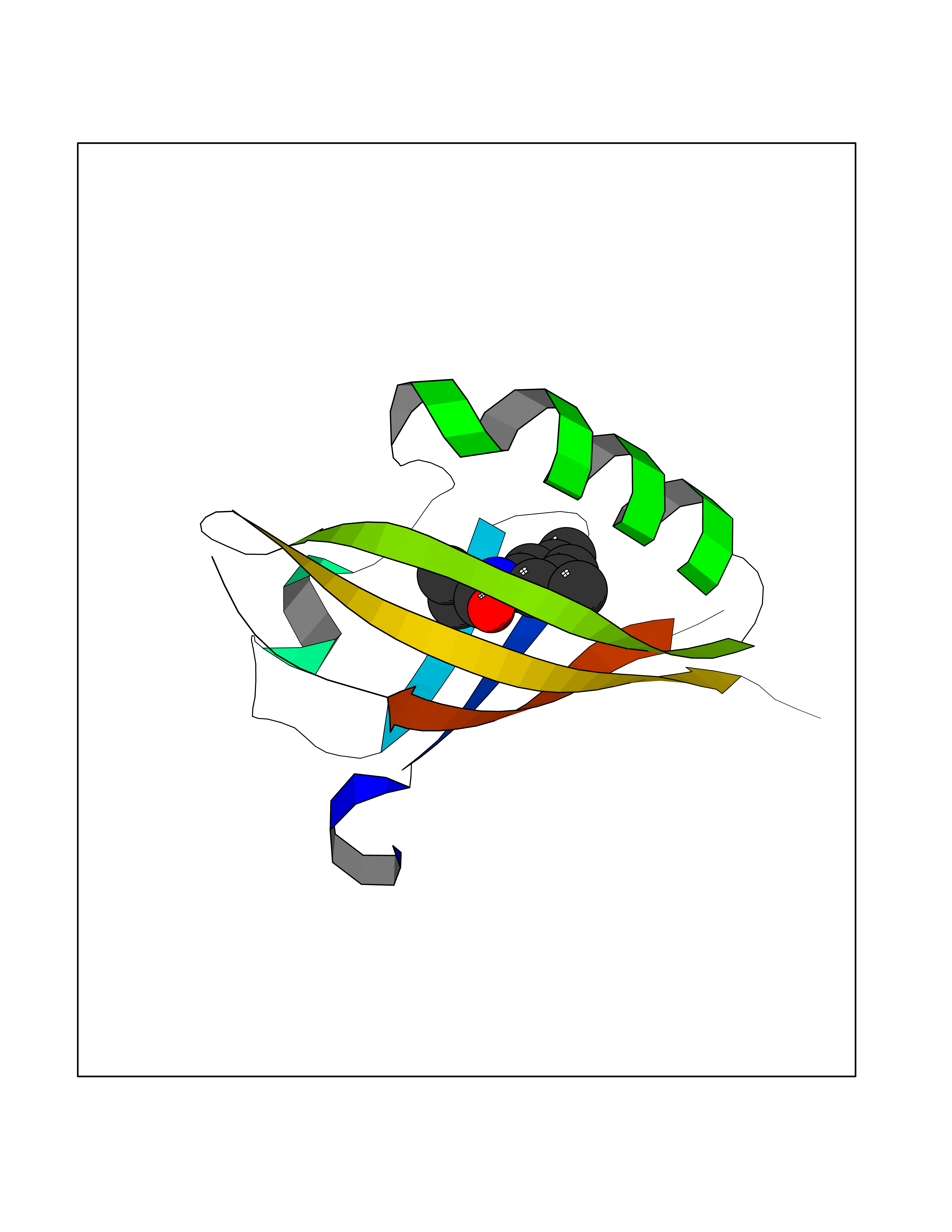 Molscript Map 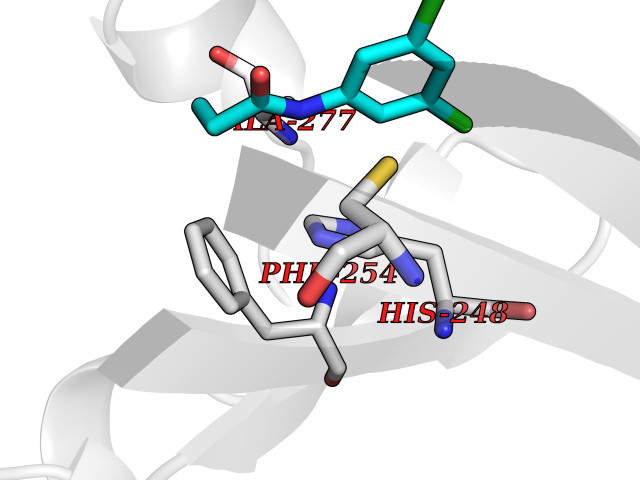 Pymol Map 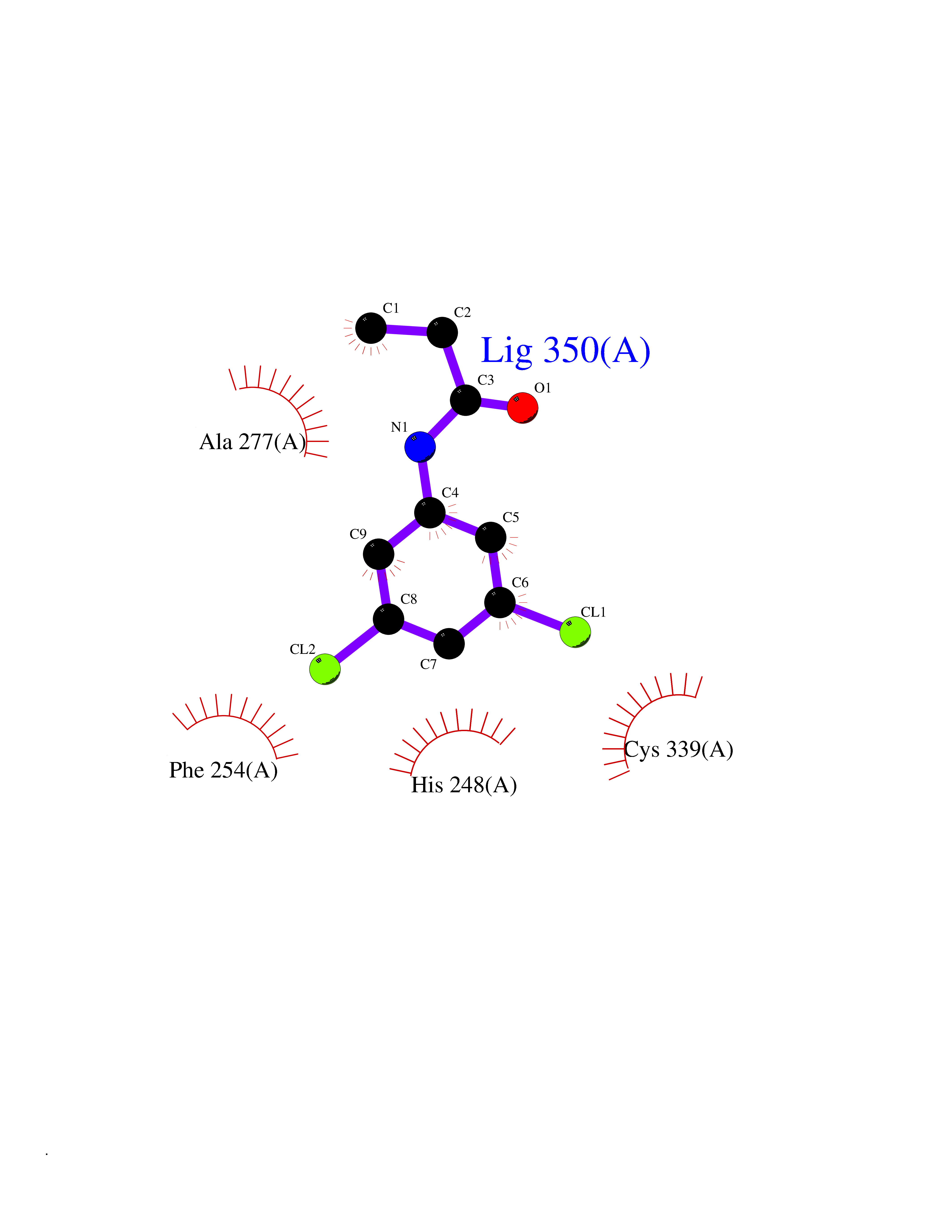 Ligplot Map 3D Binding mode Sequence LDSKTFLSEHSMDMKFTYCDDRITELIGYHPEELLGRSAYEFYHALDSENMTKSHQNLCTKGQVVSGQYRMLAKHGGYVWLETQGTVIYNPPQCIMCVNYVLSEIE Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Receptor-type protein-tyrosine phosphatase zeta (PTPRZ1) | 5H08 | 5.91 | |
Target general information Gen name PTPRZ1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Receptor protein tyrosine phosphatase zeta; R-PTP-zeta; PTPRZ1 Protein family Protein-tyrosine phosphatase family, Receptor class 5 subfamily Biochemical class Phosphoric monoester hydrolase Function Protein tyrosine phosphatase that negatively regulates oligodendrocyte precursor proliferation in the embryonic spinal cord. Required for normal differentiation of the precursor cells into mature, fully myelinating oligodendrocytes. May play a role in protecting oligondendrocytes against apoptosis. May play a role in the establishment of contextual memory, probably via the dephosphorylation of proteins that are part of important signaling cascades. Related diseases Optic atrophy 1 (OPA1) [MIM:165500]: A condition that features progressive visual loss in association with optic atrophy. Atrophy of the optic disk indicates a deficiency in the number of nerve fibers which arise in the retina and converge to form the optic disk, optic nerve, optic chiasm and optic tracts. OPA1 is characterized by an insidious onset of visual impairment in early childhood with moderate to severe loss of visual acuity, temporal optic disk pallor, color vision deficits, and centrocecal scotoma of variable density. {ECO:0000269|PubMed:11017079, ECO:0000269|PubMed:11017080, ECO:0000269|PubMed:11440988, ECO:0000269|PubMed:11440989, ECO:0000269|PubMed:11810270, ECO:0000269|PubMed:12036970, ECO:0000269|PubMed:12566046, ECO:0000269|PubMed:14961560, ECO:0000269|PubMed:15948788, ECO:0000269|PubMed:16513463, ECO:0000269|PubMed:16617242, ECO:0000269|PubMed:18204809, ECO:0000269|PubMed:18360822, ECO:0000269|PubMed:19319978, ECO:0000269|PubMed:19325939, ECO:0000269|PubMed:19969356, ECO:0000269|PubMed:20185555, ECO:0000269|PubMed:22382025, ECO:0000269|PubMed:22857269, ECO:0000269|PubMed:23401657}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Dominant optic atrophy plus syndrome (DOA+) [MIM:125250]: A neurologic disorder characterized most commonly by an insidious onset of visual loss and sensorineural hearing loss in childhood with variable presentation of other clinical manifestations including progressive external ophthalmoplegia, muscle cramps, hyperreflexia, and ataxia. There appears to be a wide range of intermediate phenotypes. {ECO:0000269|PubMed:15531309, ECO:0000269|PubMed:16240368, ECO:0000269|PubMed:18065439, ECO:0000269|PubMed:18158317, ECO:0000269|PubMed:18195150, ECO:0000269|PubMed:20185555, ECO:0000269|PubMed:21112924, ECO:0000269|PubMed:23387428}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Behr syndrome (BEHRS) [MIM:210000]: An autosomal recessive syndrome characterized by optic atrophy beginning in early childhood associated with ataxia, pyramidal signs, spasticity, intellectual disability, and posterior column sensory loss. The ataxia, spasticity, and muscle contractures, mainly of the hip adductors, hamstrings, and soleus, are progressive and become more prominent in the second decade. {ECO:0000269|PubMed:21636302, ECO:0000269|PubMed:25012220, ECO:0000269|PubMed:25146916}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mitochondrial DNA depletion syndrome 14, cardioencephalomyopathic type (MTDPS14) [MIM:616896]: An autosomal recessive mitochondrial disorder characterized by lethal infantile encephalopathy, hypertrophic cardiomyopathy and optic atrophy. Skeletal muscle biopsies show significant mtDNA depletion and abnormal mitochondria. {ECO:0000269|PubMed:26561570}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9UM73; Q12860 EC number EC 3.1.3.48 Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; Glycoprotein; Hydrolase; Membrane; Phosphoprotein; Protein phosphatase; Proteoglycan; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 32208.1 Length 282 Aromaticity 0.11 Instability index 38.98 Isoelectric point 7.35 Charge (pH=7) 0.89 2D Binding mode Binding energy (Kcal/mol) -8.07 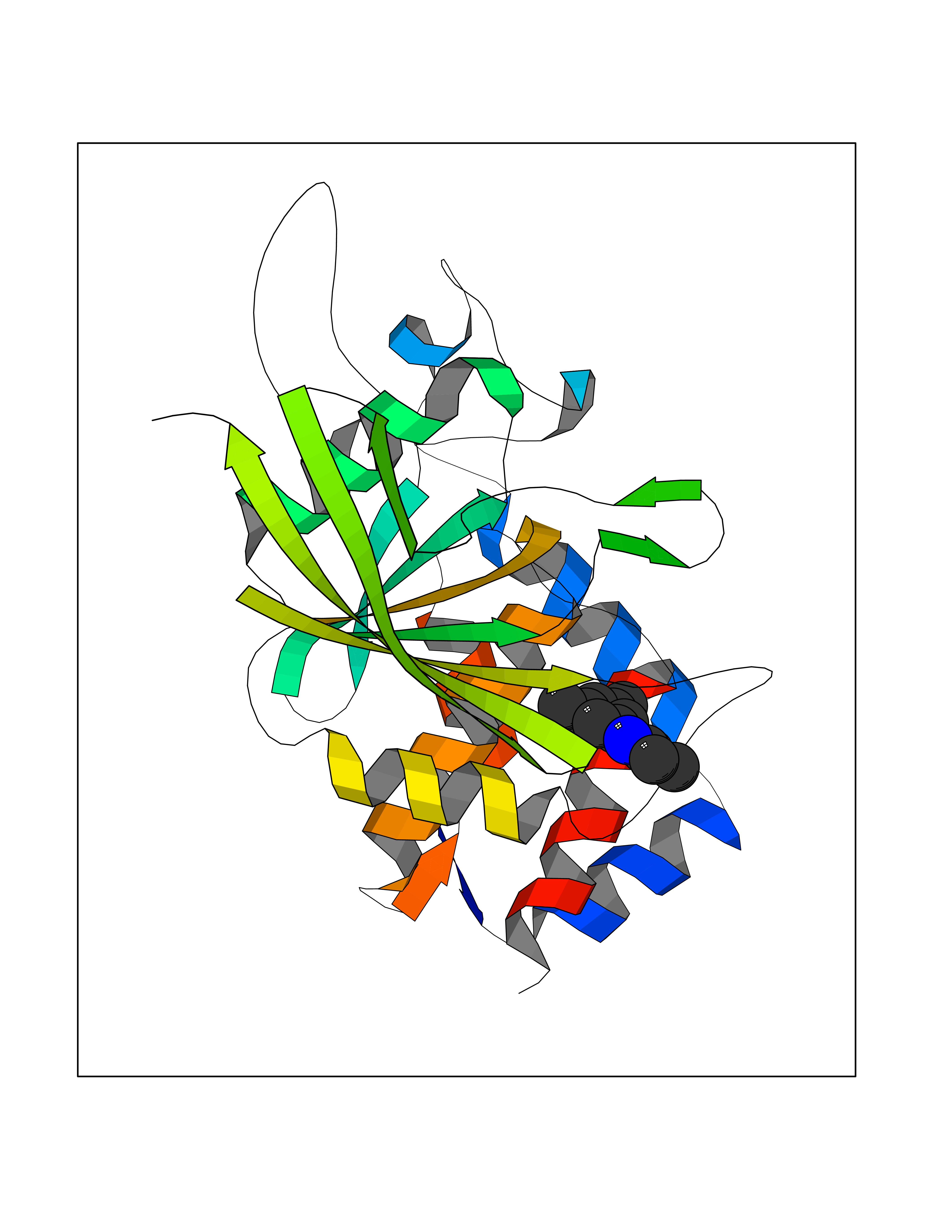 Molscript Map 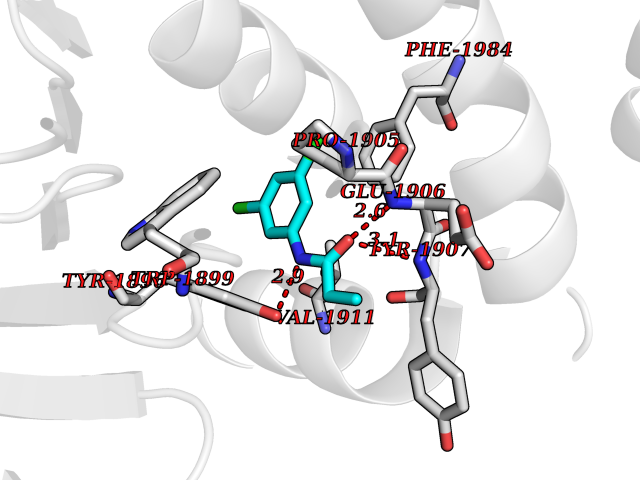 Pymol Map 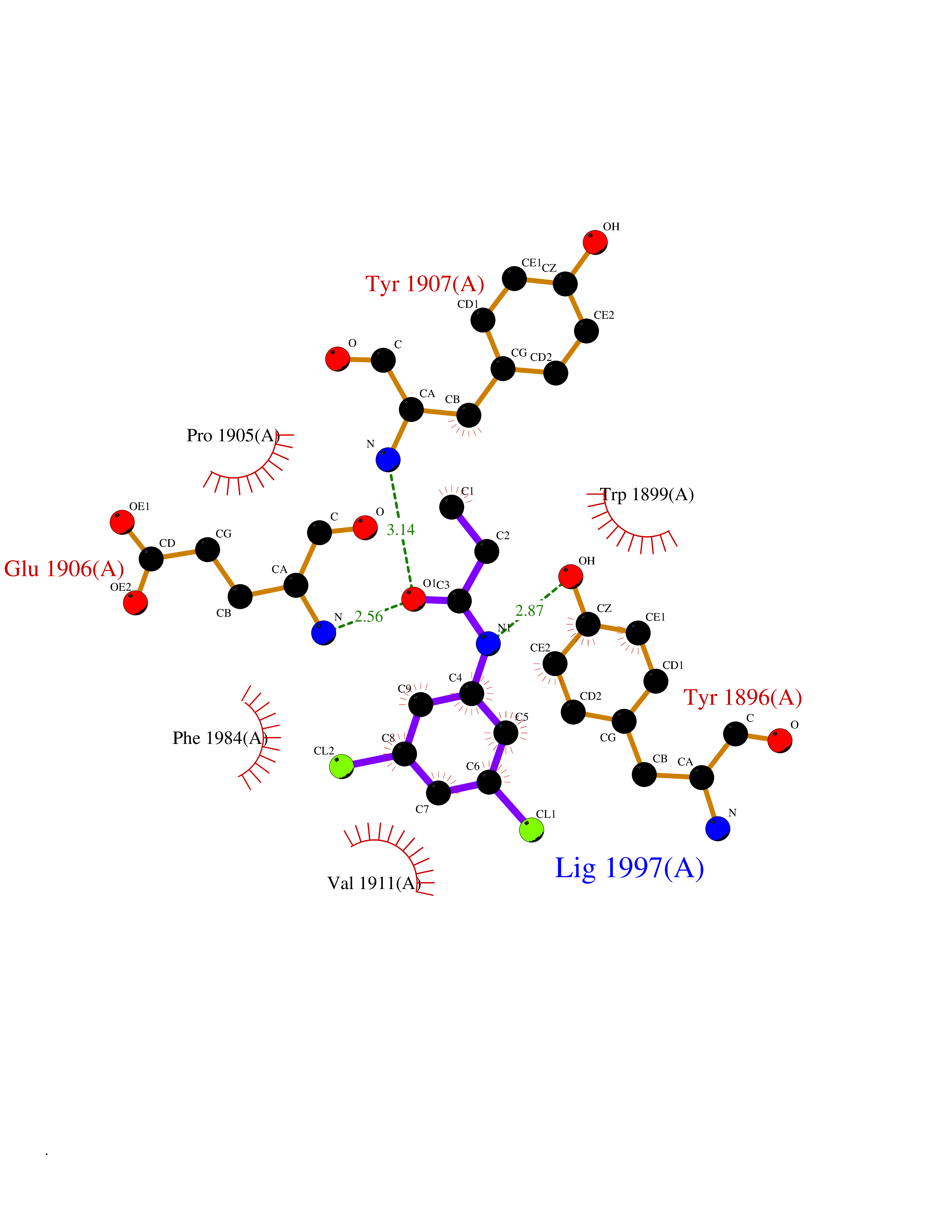 Ligplot Map 3D Binding mode Sequence GPAIPIKHFPKHVADLHASSGFTEEFEEVQSCTVDLGITADSSNHPDNKHKNRYINIVAYDHSRVKLAQLAEKDGKLTDYINANYVDGYNRPKAYIAAQGPLKSTAEDFWRMIWEHNVEVIVMITNLVEKGRRKCDQYWPADGSEEYGNFLVTQKSVQVLAYYTVRNFTLRNTKIRVVTQYHYTQWPDMGVPEYSLPVLTFVRKAAYAKRHAVGPVVVHCSAGVGRTGTYIVLDSMLQQIQHEGTVNIFGFLKHIRSQRNYLVQTEEQYVFIHDTLVEAILS Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Dual-specificity tyrosine-phosphorylation regulated kinase 3 (DYRK3) | 5Y86 | 5.91 | |
Target general information Gen name DYRK3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Regulatory erythroid kinase; REDK; Dual specificity tyrosine-phosphorylation-regulated kinase 3 Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, MNB/DYRK subfamily Biochemical class Kinase Function Dual-specificity tyrosine-regulated kinases (DYRKs) autophosphorylate a critical tyrosine residue in their activation loop and phosphorylate their substrate on serine and threonine residues. Acts as a central dissolvase of membraneless organelles during the G2-to-M transition, after the nuclear-envelope breakdown: acts by mediating phosphorylation of multiple serine and threonine residues in unstructured domains of proteins, such as SRRM1 and PCM1. Does not mediate disassembly of all membraneless organelles: disassembly of P-body and nucleolus is not regulated by DYRK3. Dissolution of membraneless organelles at the onset of mitosis is also required to release mitotic regulators, such as ZNF207, from liquid-unmixed organelles where they are sequestered and keep them dissolved during mitosis. Regulates mTORC1 by mediating the dissolution of stress granules: during stressful conditions, DYRK3 partitions from the cytosol to the stress granule, together with mTORC1 components, which prevents mTORC1 signaling. When stress signals are gone, the kinase activity of DYRK3 is required for the dissolution of stress granule and mTORC1 relocation to the cytosol: acts by mediating the phosphorylation of the mTORC1 inhibitor AKT1S1, allowing full reactivation of mTORC1 signaling. Also acts as a negative regulator of EPO-dependent erythropoiesis: may place an upper limit on red cell production during stress erythropoiesis. Inhibits cell death due to cytokine withdrawal in hematopoietic progenitor cells. Promotes cell survival upon genotoxic stress through phosphorylation of SIRT1: this in turn inhibits p53/TP53 activity and apoptosis. Dual-specificity protein kinase that promotes disassembly of several types of membraneless organelles during mitosis, such as stress granules, nuclear speckles and pericentriolar material. Related diseases Defects in MELK are associated with some cancers, such as brain or breast cancers. Expression is dramatically increased in aggressive undifferentiated tumors, correlating with poor patient outcome in breast and brain cancers, suggesting a role in tumor-initiating cells and proliferation via its function in cell proliferation regulation. Drugs (DrugBank ID) NA Interacts with Q9H8Y8 EC number EC 2.7.12.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell cycle; Cell division; Cytoplasm; Cytoskeleton; Kinase; Magnesium; Metal-binding; Mitosis; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 44821.5 Length 395 Aromaticity 0.1 Instability index 49.38 Isoelectric point 9.52 Charge (pH=7) 21.08 2D Binding mode Binding energy (Kcal/mol) -8.06  Molscript Map 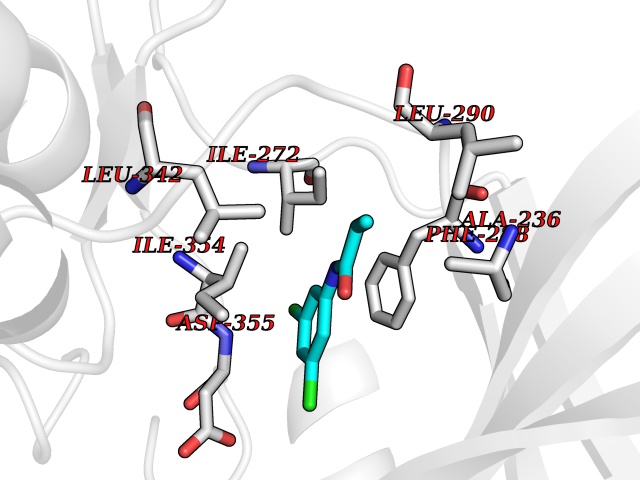 Pymol Map 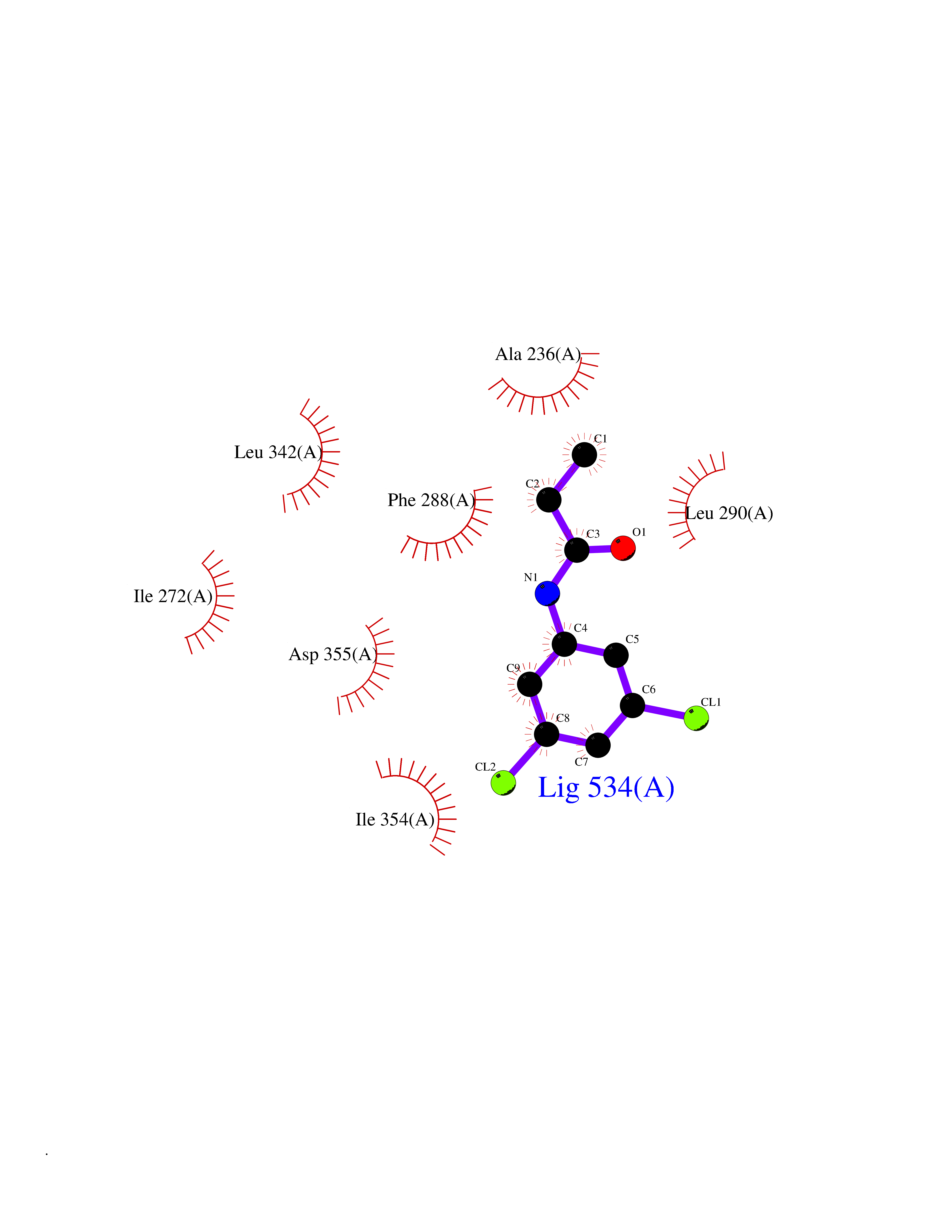 Ligplot Map 3D Binding mode Sequence VVPLTPEQALKQYKHHLTAYEKLEIINYPEIYFVGPNAKKRHGVIGGPNNGGYDDADGAYIHVPRDHLAYRYEVLKIIGKGSFGQVARVYDHKLRQYVALKMVRNEKRFHRQAAEEIRILEHLKKQDKTGSMNVIHMLESFTFRNHVCMAFELLSIDLYELIKKNKFQGFSVQLVRKFAQSILQSLDALHKNKIIHCDLKPENILLKHHGRSXTKVIDFGSSCFEYQKLYTXIQSRFYRAPEIILGSRYSTPIDIWSFGCILAELLTGQPLFPGEDEGDQLACMMELLGMPPPKLLEQSKRAKYFINXKGIPRYCSVTTQADGRVVLVGGRSRRGKKRGPPGSKDWGTALKGCDDYLFIEFLKRCLHWDPSARLXPAQALRHPWISKSVPRPLTT Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Retinoic acid receptor beta (RARB) | 4DM6 | 5.90 | |
Target general information Gen name RARB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-epsilon; RAR-beta; Nuclear receptor subfamily 1 group B member 2; NR1B2; HBV-activated protein; HAP Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RXR/RAR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence or presence of hormone ligand, acts mainly as an activator of gene expression due to weak binding to corepressors. In concert with RARG, required for skeletal growth, matrix homeostasis and growth plate function. Receptor for retinoic acid. Related diseases Microphthalmia, syndromic, 12 (MCOPS12) [MIM:615524]: A form of microphthalmia, a disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues (anophthalmia). In many cases, microphthalmia/anophthalmia occurs in association with syndromes that include non-ocular abnormalities. MCOPS12 patients manifest variable features, including diaphragmatic hernia, pulmonary hypoplasia, and cardiac abnormalities. {ECO:0000269|PubMed:24075189, ECO:0000269|PubMed:27120018}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00459; DB00210; DB00523; DB02877; DB00926; DB05785; DB04942; DB00799; DB00755; DB12808 Interacts with O95273; P50222; Q9UBK2; P62195; P28702; P28702-3; P48443; P03255 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Microphthalmia; Nucleus; Phosphoprotein; Proto-oncogene; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 25904.1 Length 229 Aromaticity 0.06 Instability index 44.34 Isoelectric point 7.55 Charge (pH=7) 0.73 2D Binding mode Binding energy (Kcal/mol) -8.04  Molscript Map 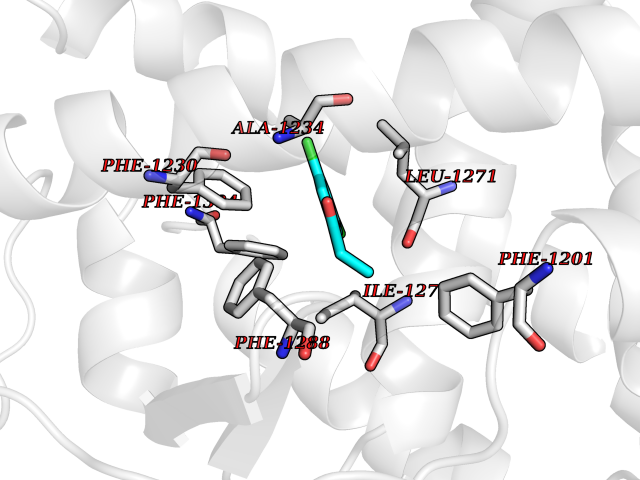 Pymol Map 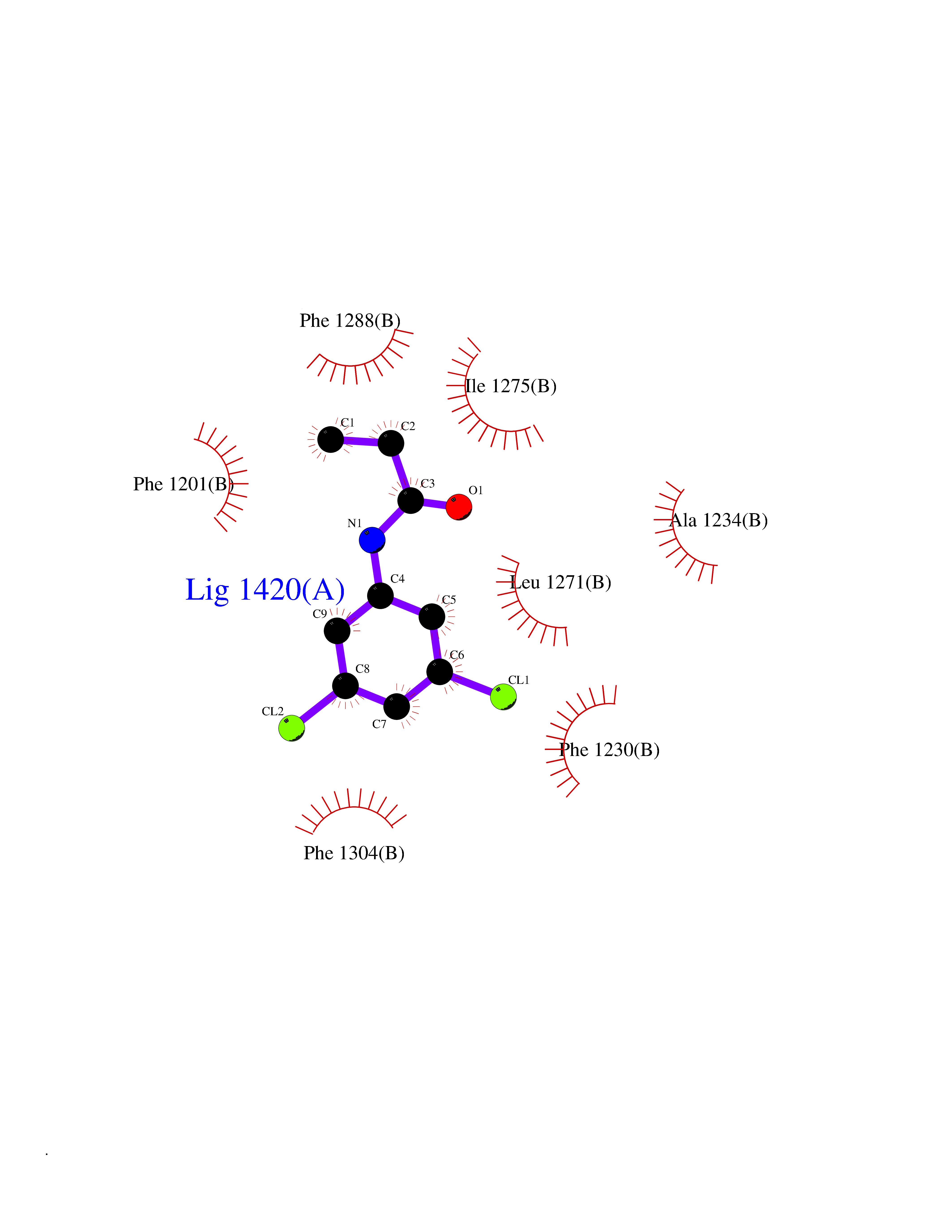 Ligplot Map 3D Binding mode Sequence TEKIRKAHQETFPSLCQLGKYTTNSSADHRVRLDLGLWDKFSELATKCIIKIVEFAKRLPGFTGLTIADQITLLKAACLDILILRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFTFANQLLPLEMDDTETGLLSAICLICGDRQDLEEPTKVDKLQEPLLEALKIYIRKRRPSKPHMFPKILMKITDLRSISAKGAERVITLKMEIPGSMPPLIQEMLEN Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Aspartate aminotransferase, mitochondrial | 5AX8 | 5.90 | |
Target general information Gen name GOT2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms KYAT4 Protein family Class-I pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transferase Function 4-hydroxyglutamate transaminase activity.Amino acid binding.Enzyme binding.Kynurenine-oxoglutarate transaminase activity.L-aspartate:2-oxoglutarate aminotransferase activity.L-phenylalanine:2-oxoglutarate aminotransferase activity.Phospholipid binding.Protein homodimerization activity.Pyridoxal phosphate binding.RNA binding. Related diseases Developmental and epileptic encephalopathy 82 (DEE82) [MIM:618721]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE82 is an autosomal recessive metabolic encephalopathy characterized by epilepsy from the first year of life, global developmental delay, hypotonia and feeding difficulties apparent soon after birth, and intellectual and motor disabilities. Other features include poor overall growth, progressive microcephaly and biochemical abnormalities, including increased serum lactate and ammonia. {ECO:0000269|PubMed:31422819}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02783; DB00128; DB00151; DB00142; DB00114 Interacts with NA EC number 2.6.1.1; 2.6.1.7 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminotransferase; Cell membrane; Direct protein sequencing; Disease variant; Epilepsy; Lipid transport; Membrane; Methylation; Mitochondrion; Nitration; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase; Transit peptide; Transport Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 44694.8 Length 401 Aromaticity 0.1 Instability index 26.15 Isoelectric point 8.98 Charge (pH=7) 8.22 2D Binding mode Binding energy (Kcal/mol) -8.05 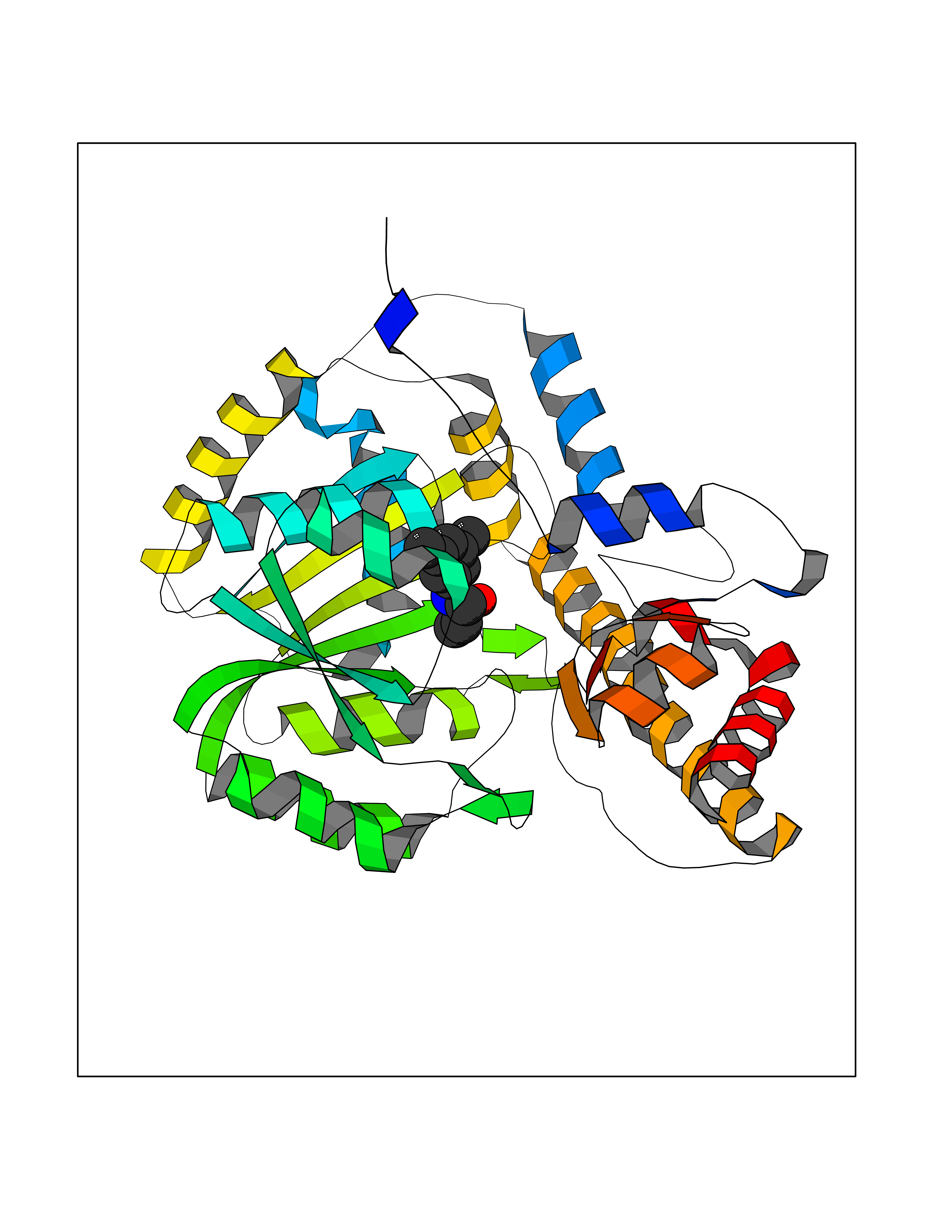 Molscript Map 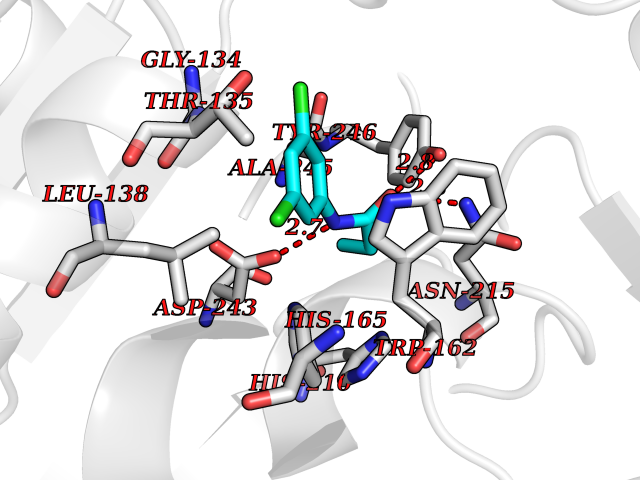 Pymol Map 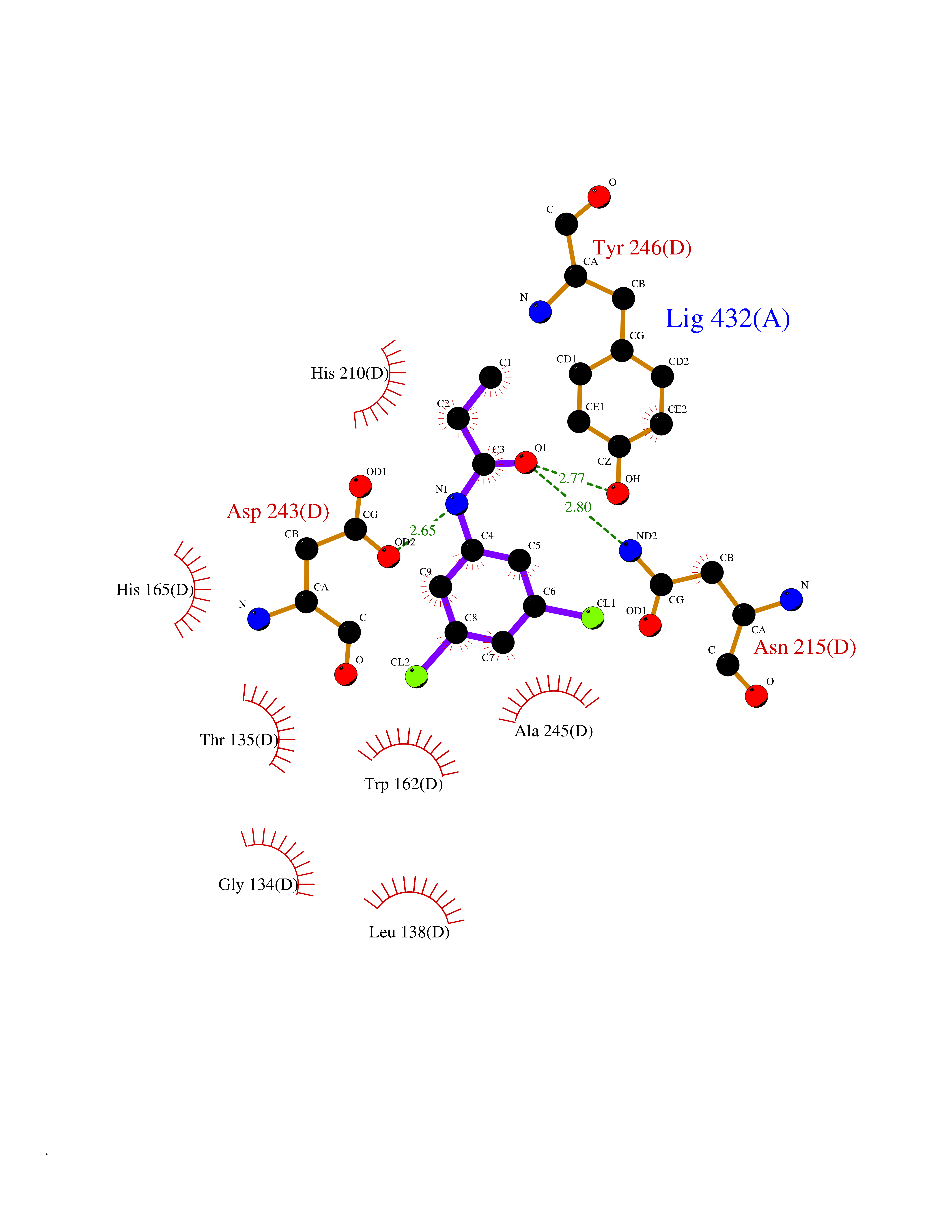 Ligplot Map 3D Binding mode Sequence SSWWTHVEMGPPDPILGVTEAFKRDTNSKKMNLGVGAYRDDNGKPYVLPSVRKAEAQIAAKNLDKEYLPIGGLAEFCKASAELALGENSEVLKSGRFVTVQTISGTGALRIGASFLQRFFKFSRDVFLPKPTWGNHTPIFRDAGMQLQGYRYYDPKTCGFDFTGAVEDISKIPEQSVLLLHACAHNPTGVDPRPEQWKEIATVVKKRNLFAFFDMAYQGFASGDGDKDAWAVRHFIEQGINVCLCQSYAKNMGLYGERVGAFTMVCKDADEAKRVESQLKILIRPMYSNPPLNGARIAAAILNTPDLRKQWLQEVKGMADRIIGMRTQLVSNLKKEGSTHNWQHITDQIGMFCFTGLKPEQVERLIKEFSIYMTKDGRISVAGVTSSNVGYLAHAIHQVTK Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Histone-lysine N-methyltransferase KMT5B (KMT5B) | 3S8P | 5.90 | |
Target general information Gen name KMT5B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine N-methyltransferase 5B; Lysine-specific methyltransferase 5B; Suppressor of variegation 4-20 homolog 1; Su(var)4-20 homolog 1; Suv4-20h1; [histone H4]-N-methyl-L-lysine20 N-methyltransferase KM Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, Suvar4-20 subfamily Biochemical class NA Function Histone methyltransferase that specifically methylates monomethylated 'Lys-20' (H4K20me1) and dimethylated 'Lys-20' (H4K20me2) of histone H4 to produce respectively dimethylated 'Lys-20' (H4K20me2) and trimethylated 'Lys-20' (H4K20me3) and thus regulates transcription and maintenance of genome integrity. In vitro also methylates unmodified 'Lys-20' (H4K20me0) of histone H4 and nucleosomes. H4 'Lys-20' trimethylation represents a specific tag for epigenetic transcriptional repression. Mainly functions in pericentric heterochromatin regions, thereby playing a central role in the establishment of constitutive heterochromatin in these regions. KMT5B is targeted to histone H3 via its interaction with RB1 family proteins (RB1, RBL1 and RBL2) (By similarity). Plays a role in myogenesis by regulating the expression of target genes, such as EID3. Facilitates TP53BP1 foci formation upon DNA damage and proficient non-homologous end-joining (NHEJ)-directed DNA repair by catalyzing the di- and trimethylation of 'Lys-20' of histone H4. May play a role in class switch reconbination by catalyzing the di- and trimethylation of 'Lys-20' of histone H4 (By similarity). Related diseases Intellectual developmental disorder, autosomal dominant 51 (MRD51) [MIM:617788]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. {ECO:0000269|PubMed:28191889, ECO:0000269|PubMed:29276005}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9H2G4; Q61026 EC number EC 2.1.1.361 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Disease variant; Intellectual disability; Isopeptide bond; Metal-binding; Methyltransferase; Myogenesis; Nucleus; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 26230.2 Length 233 Aromaticity 0.11 Instability index 42.29 Isoelectric point 5.64 Charge (pH=7) -6.07 2D Binding mode Binding energy (Kcal/mol) -8.05 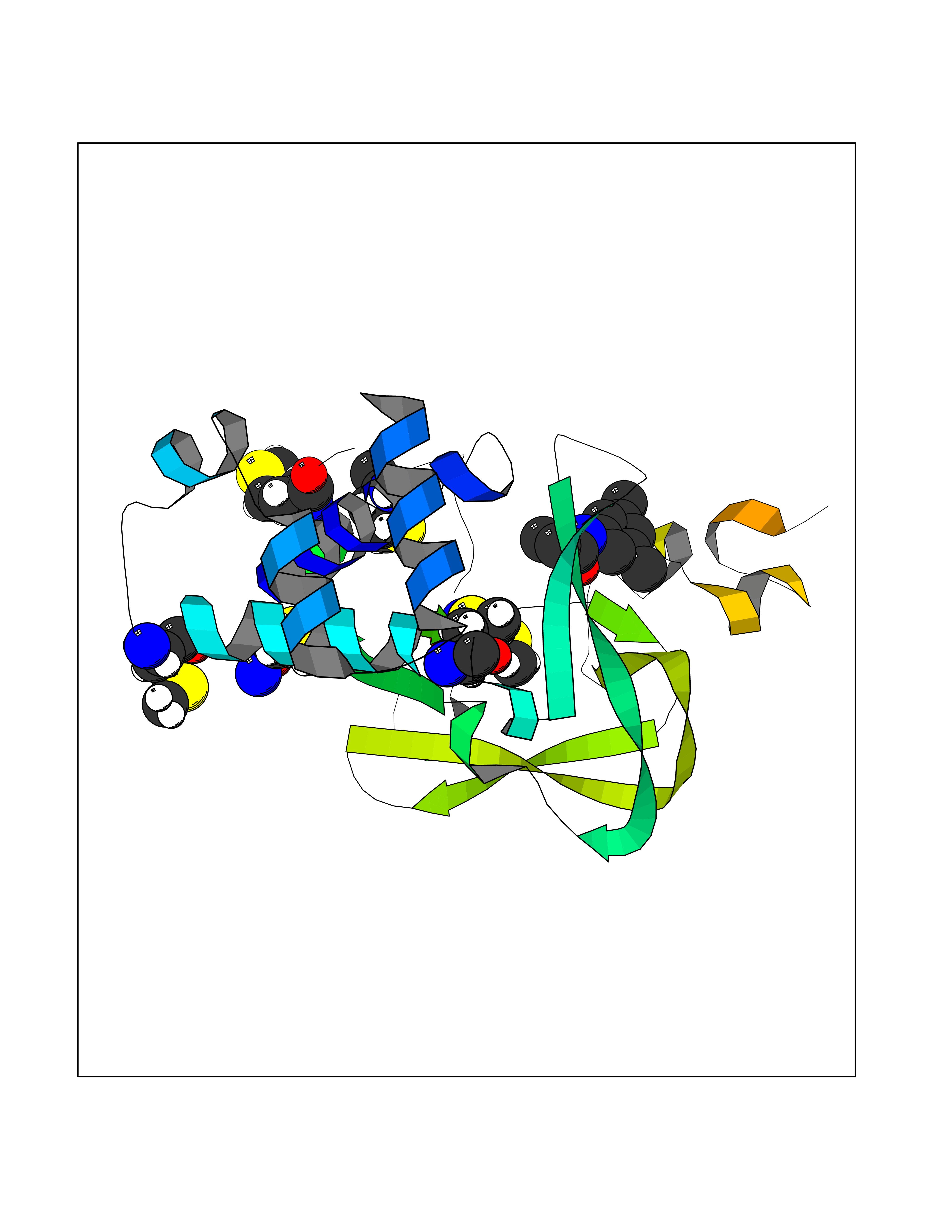 Molscript Map 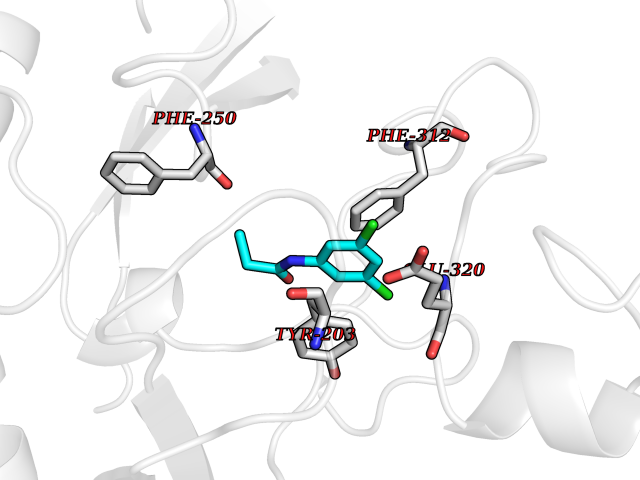 Pymol Map 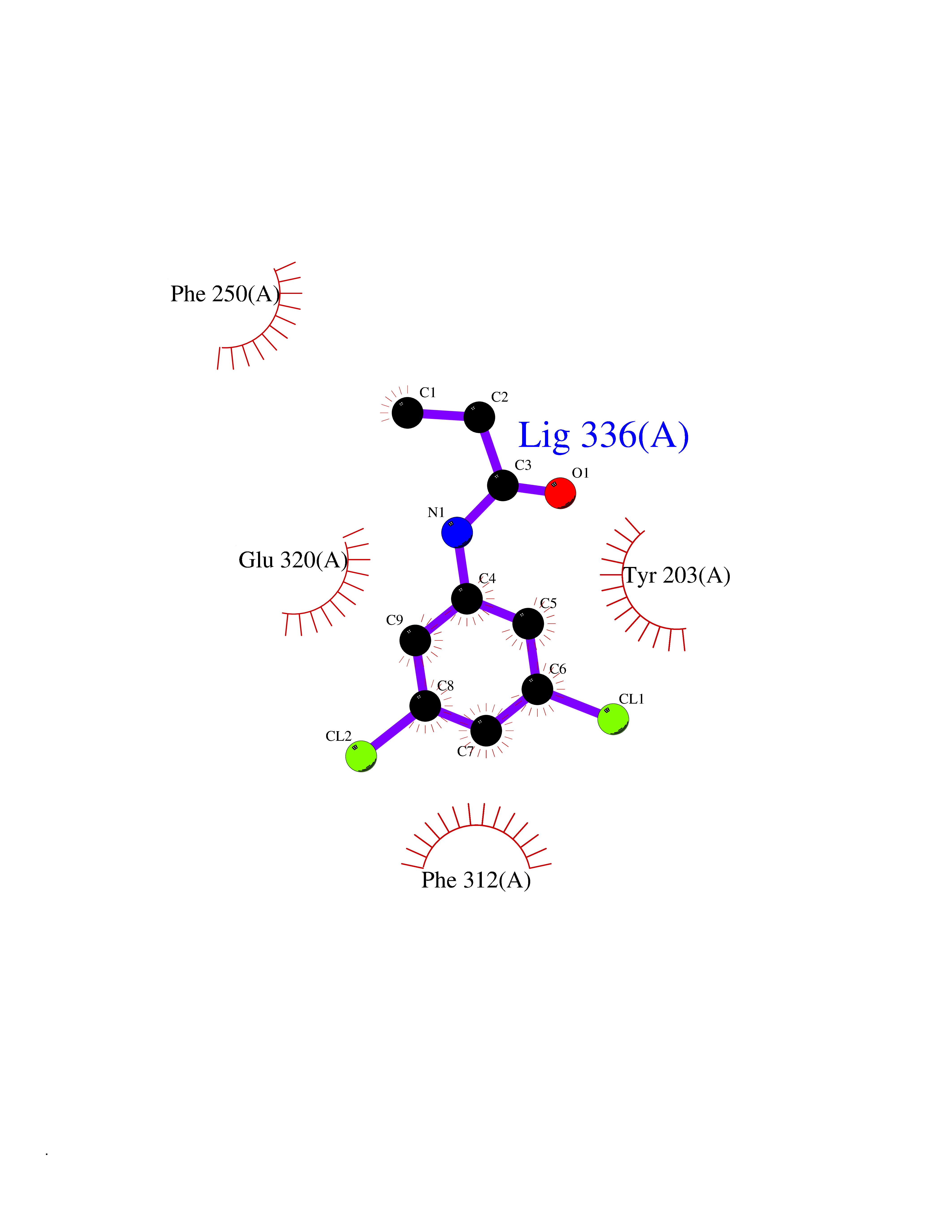 Ligplot Map 3D Binding mode Sequence XSAKELCENDDLATSLVLDPYLGFQTHKXNTRQEELKEVIERFKKDEHLEKAFKCLTSGEWARHYFLNKNKXQEKLFKEHVFIYLRXFATDSGFEILPCNRYSSEQNGAKIVATKEWKRNDKIELLVGCIAELSEIEENXLLRHGENDFSVXYSTRKNCAQLWLGPAAFINHDCRPNCKFVSTGRDTACVKALRDIEPGEEISCYYGDGFFGENNEFCECYTCERRGTGAFKS Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Pseudomonas Methionine gamma-lyase (Pseudo mdeA) | 1PG8 | 5.90 | |
Target general information Gen name Pseudo mdeA Organism Pseudomonas putida (Arthrobacter siderocapsulatus) Uniprot ID TTD ID Synonyms Pseudo MGL; L-methionine gamma-lyase; L-methioninase; Homocysteine desulfhydrase Protein family Trans-sulfuration enzymes family, L-methionine gamma-lyase subfamily Biochemical class Carbon-sulfur lyases Function Catalyzes the alpha,gamma-elimination of L-methionine to produce methanethiol, 2-oxobutanoate and ammonia. Is involved in L-methionine catabolism. In fact, shows a multicatalytic function since it also catalyzes gamma-replacement of L-methionine with thiol compounds, alpha,gamma-elimination and gamma-replacement reactions of L-homocysteine and its S-substituted derivatives, O-substituted-L-homoserines and DL-selenomethionine, and, to a lesser extent, alpha,beta-elimination and beta-replacement reactions of L-cysteine, S-methyl-L-cysteine, and O-acetyl-L-serine. Also catalyzes deamination and gamma-addition reactions of L-vinylglycine. Thus, the enzyme is able to cleave C-S, C-Se, and C-O bonds of sulfur, selenium, and oxygen amino acids, respectively. Related diseases Lecithin-cholesterol acyltransferase deficiency (LCATD) [MIM:245900]: A disorder of lipoprotein metabolism characterized by inadequate esterification of plasmatic cholesterol. Two clinical forms are recognized: complete LCAT deficiency and fish-eye disease. LCATD is generally referred to the complete form which is associated with absence of both alpha and beta LCAT activities resulting in esterification anomalies involving both HDL (alpha-LCAT activity) and LDL (beta-LCAT activity). It causes a typical triad of diffuse corneal opacities, target cell hemolytic anemia, and proteinuria with renal failure. {ECO:0000269|PubMed:11423760, ECO:0000269|PubMed:12957688, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:16051254, ECO:0000269|PubMed:16216249, ECO:0000269|PubMed:1681161, ECO:0000269|PubMed:1859405, ECO:0000269|PubMed:2370048, ECO:0000269|PubMed:7607641, ECO:0000269|PubMed:7711728, ECO:0000269|PubMed:8318557, ECO:0000269|PubMed:8432868, ECO:0000269|PubMed:8807342, ECO:0000269|PubMed:9007616, ECO:0000269|PubMed:9741700}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fish-eye disease (FED) [MIM:136120]: A disorder of lipoprotein metabolism due to partial lecithin-cholesterol acyltransferase deficiency that affects only alpha-LCAT activity. FED is characterized by low plasma HDL and corneal opacities due to accumulation of cholesterol deposits in the cornea ('fish-eye'). {ECO:0000269|PubMed:1516702, ECO:0000269|PubMed:1571050, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:1737840, ECO:0000269|PubMed:21901787, ECO:0000269|PubMed:8620346, ECO:0000269|PubMed:9261271}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04083 Interacts with NA EC number EC 4.4.1.11 Uniprot keywords 3D-structure; Direct protein sequencing; Lyase; Pyridoxal phosphate Protein physicochemical properties Chain ID A,C Molecular weight (Da) 85234.4 Length 796 Aromaticity 0.07 Instability index 37.1 Isoelectric point 6.21 Charge (pH=7) -11.34 2D Binding mode Binding energy (Kcal/mol) -8.04  Molscript Map 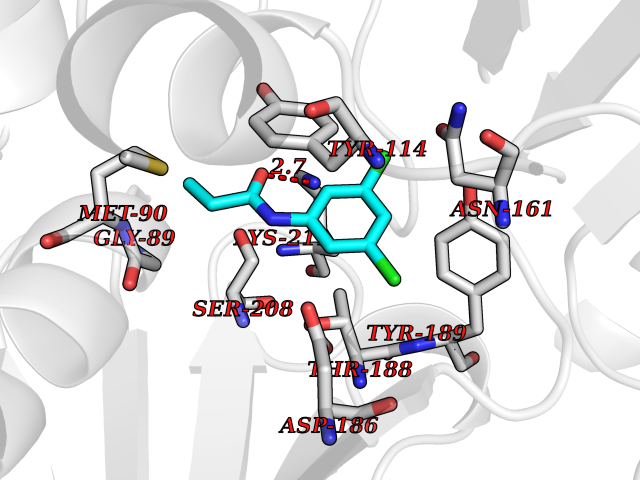 Pymol Map 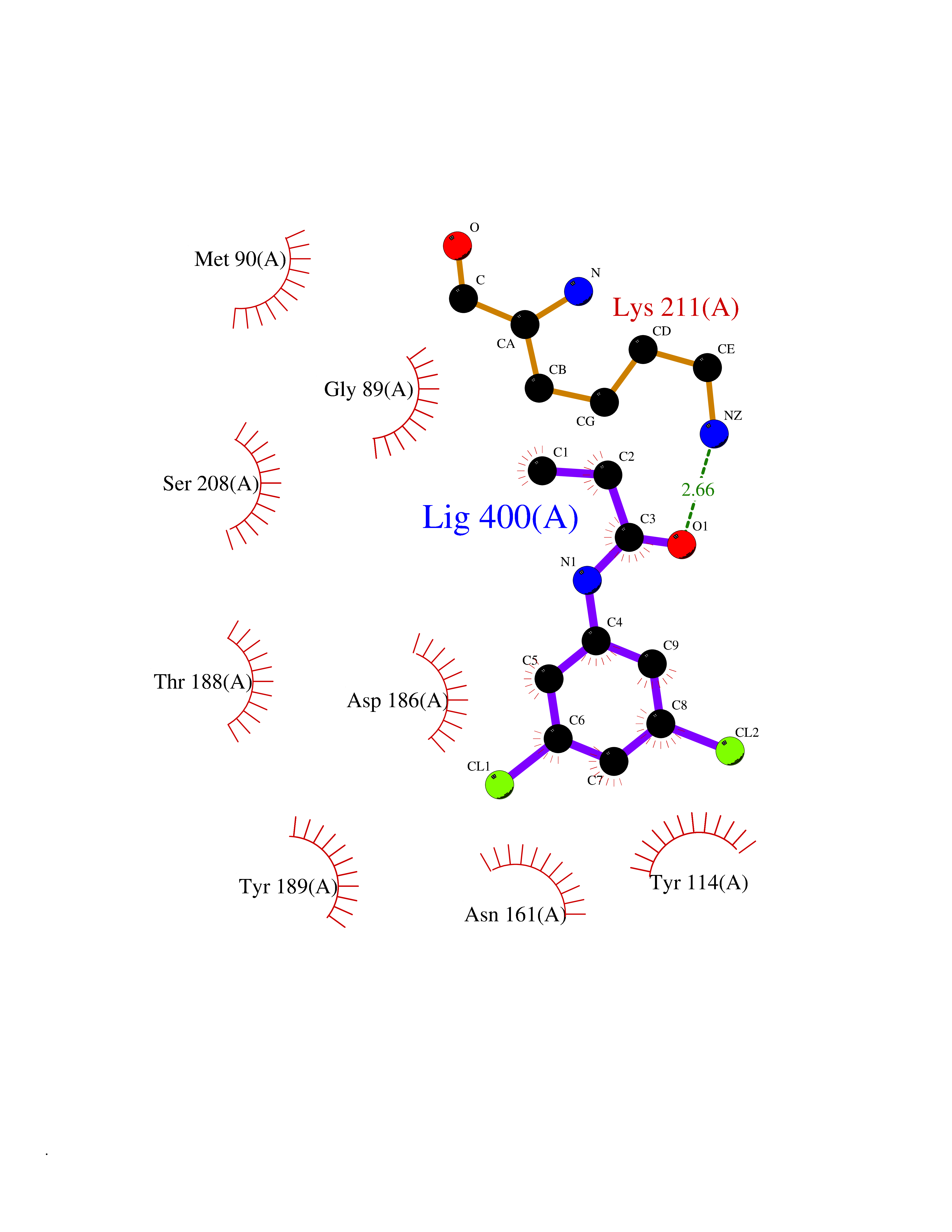 Ligplot Map 3D Binding mode Sequence MHGSNKLPGFATRAIHHGYDPQDHGGALVPPVYQTATFTFPTVEYGAACFAGEQAGHFYSRISNPTLNLLEARMASLEGGEAGLALASGMGAITSTLWTLLRPGDEVLLGNTLYGCTFAFLHHGIGEFGVKLRHVDMADLQALEAAMTPATRVIYFESPANPNMHMADIAGVAKIARKHGATVVVDNTYCTPYLQRPLELGADLVVHSATKYLSGHGDITAGIVVGSQALVDRIRLQGLKDMTGAVLSPHDAALLMRGIKTLNLRMDRHCANAQVLAEFLARQPQVELIHYPGLASFPQYTLARQQMSQPGGMIAFELKGGIGAGRRFMNALQLFSRAVSLGDAESLAQHPASMTHSSYTPEERAHYGISEGLVRLSVGLEDIDDLLADVQQALKASAMHGSNKLPGFATRAIHHGYDPQDHGGALVPPVYQTATFTFPTVEYGAACFAGEQAGHFYSRISNPTLNLLEARMASLEGGEAGLALASGMGAITSTLWTLLRPGDEVLLGNTLYGCTFAFLHHGIGEFGVKLRHVDMADLQALEAAMTPATRVIYFESPANPNMHMADIAGVAKIARKHGATVVVDNTYCTPYLQRPLELGADLVVHSATKYLSGHGDITAGIVVGSQALVDRIRLQGLKDMTGAVLSPHDAALLMRGIKTLNLRMDRHCANAQVLAEFLARQPQVELIHYPGLASFPQYTLARQQMSQPGGMIAFELKGGIGAGRRFMNALQLFSRAVSLGDAESLAQHPASMTHSSYTPEERAHYGISEGLVRLSVGLEDIDDLLADVQQALKASA Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Caspase-6 (CASP6) | 4NBL | 5.90 | |
Target general information Gen name CASP6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MCH2; Caspase-6 subunit p18; Caspase-6 subunit p11; CASP-6; Apoptotic protease Mch-2 Protein family Peptidase C14A family Biochemical class Peptidase Function Cleaves poly(ADP-ribose) polymerase in vitro, as well as lamins. Overexpression promotes programmed cell death. Involved in the activation cascade of caspases responsible for apoptosis execution. Related diseases Growth hormone deficiency, isolated, 1A (IGHD1A) [MIM:262400]: An autosomal recessive, severe deficiency of growth hormone leading to dwarfism. Patients often develop antibodies to administered growth hormone. {ECO:0000269|PubMed:8364549}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 1B (IGHD1B) [MIM:612781]: An autosomal recessive deficiency of growth hormone leading to short stature. Patients have low but detectable levels of growth hormone, significantly retarded bone age, and a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:12655557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Kowarski syndrome (KWKS) [MIM:262650]: A syndrome clinically characterized by short stature associated with bioinactive growth hormone, normal or slightly increased growth hormone secretion, pathologically low insulin-like growth factor 1 levels, and normal catch-up growth on growth hormone replacement therapy. {ECO:0000269|PubMed:17519310, ECO:0000269|PubMed:8552145, ECO:0000269|PubMed:9276733}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 2 (IGHD2) [MIM:173100]: An autosomal dominant deficiency of growth hormone leading to short stature. Clinical severity is variable. Patients have a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:11502836, ECO:0000269|PubMed:9152628}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9Y614; Q6DHV7-2; Q6UY14-3; Q96MA6; Q5T2L2; Q96Q83-2; Q9Y303-2; Q9NU02; P09525; P06727; Q8WW27; Q66PJ3-4; Q6XD76; P18848; Q9H0Y0; Q14032; P54687-4; P06276; Q9NSI6-4; Q96Q07-2; Q9H0W9-3; Q9NQ89; Q13901; Q3SXR2; Q8N1A6; P17655; P20807-4; P42574; P55212; O00257-3; P24863; Q9NNX6-10; Q9UJX2; P42773; O95674; Q494V2-2; Q8WUX9; Q9Y3D0; Q8N365; Q3SX64; Q99966; P09496-2; Q6PJW8-3; Q96BR5; P02458-1; Q9UGL9; Q9UKG9-2; P26998; P35222; Q53TN4; P61962; O60479; Q96EY1-3; Q92782-2; Q9BPU6; A0AVK6; Q658K8; O00303; Q13347; O00472; O00423; Q6NXG1-3; Q49AJ0-4; Q8N128-2; Q8IZU1; Q6ZNL6; Q9NSA1; Q06547-3; Q49A26-4; Q9HAV0; Q6NXT2; Q9BT25; Q9NRZ9-6; Q96EW2-2; P42858; Q8N6M8-2; Q92613; P0C870; Q9UK76; Q8N5Z5; Q8TBB5-2; Q9UH77; Q8N4N3-2; Q5JUW0-3; Q8N1A0; P13473-2; Q6DKI2; Q9H2C1; Q8N0U6; Q9Y234; Q8TBB1; Q1L5Z9; Q96JB6; Q16609; Q8IYG6; P0DP58-2; Q969L2; P27338; A6NJ78-4; Q96C03-3; Q8N5J2-3; A0A0A0MR05; P34949-2; Q9BV20; Q6IN84-2; A2RUH7; P01106; Q9H7X0; Q15742-2; Q9UJ70-2; Q8NDH3-5; Q96HA8; P36639-4; Q8NFH4; Q8NFH3; Q7Z3B4; Q6N063-2; Q6GQQ9-2; Q9H8K7; Q99447; P27815-4; O15534; Q9BUL5; Q00169; P48739; P61925; Q58EX7-2; O60664; Q14181; P0DPB6; P36954; Q07869; O60927; Q6ZMI0-5; P54619; Q8NCQ7-2; P41222; P29074; Q8WUD1-2; Q5R372-9; Q9HD47-3; Q09028; Q04206; P47804-3; Q15382; Q06587; Q8N5U6; P62701; Q66K80; Q01826; O15126; P22307-3; Q9BRK5; Q9NTN9-3; P01011; Q15393; Q9NR46; Q9BZQ2; O60902-3; Q86US8; P37840; Q96H20; Q13573; Q7Z6I5; Q496A3; Q9C004; Q5W111-2; Q96BD6; Q92797-2; O60506-4; O15273; Q86WV5; Q96A09; P54274-2; P22735; O43548; Q9NQ88; Q9UIK5-2; Q53NU3; P04637; Q12888; P36406; Q86WT6-2; Q13885; P49459; Q9P1Q0-4; Q9NX94; Q8NA23-2; Q9BQA1; O00755; O95070; O43829; Q8IWT0-2; Q53FD0-2; Q05CR2; Q96JL9-2; Q96LX8; Q3KNS6-3; Q5JTY5; A0A384MDV8; B7Z3E8; Q86V28 EC number EC 3.4.22.59 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; Autocatalytic cleavage; Cytoplasm; Hydrolase; Lipoprotein; Nucleus; Palmitate; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Thiol protease; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 57170 Length 500 Aromaticity 0.12 Instability index 31.33 Isoelectric point 8.05 Charge (pH=7) 4.52 2D Binding mode Binding energy (Kcal/mol) -8.05  Molscript Map 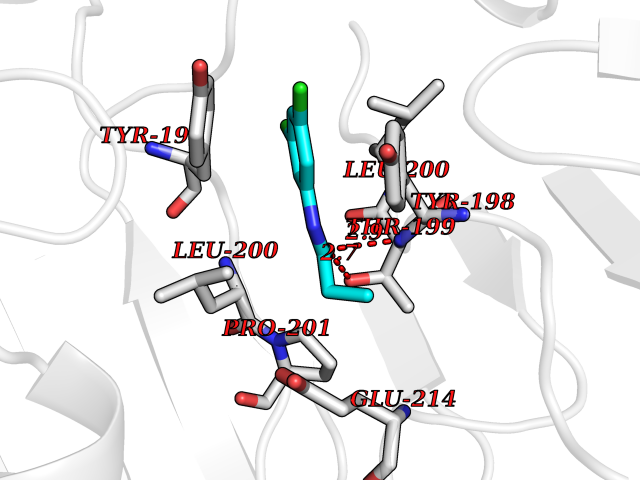 Pymol Map 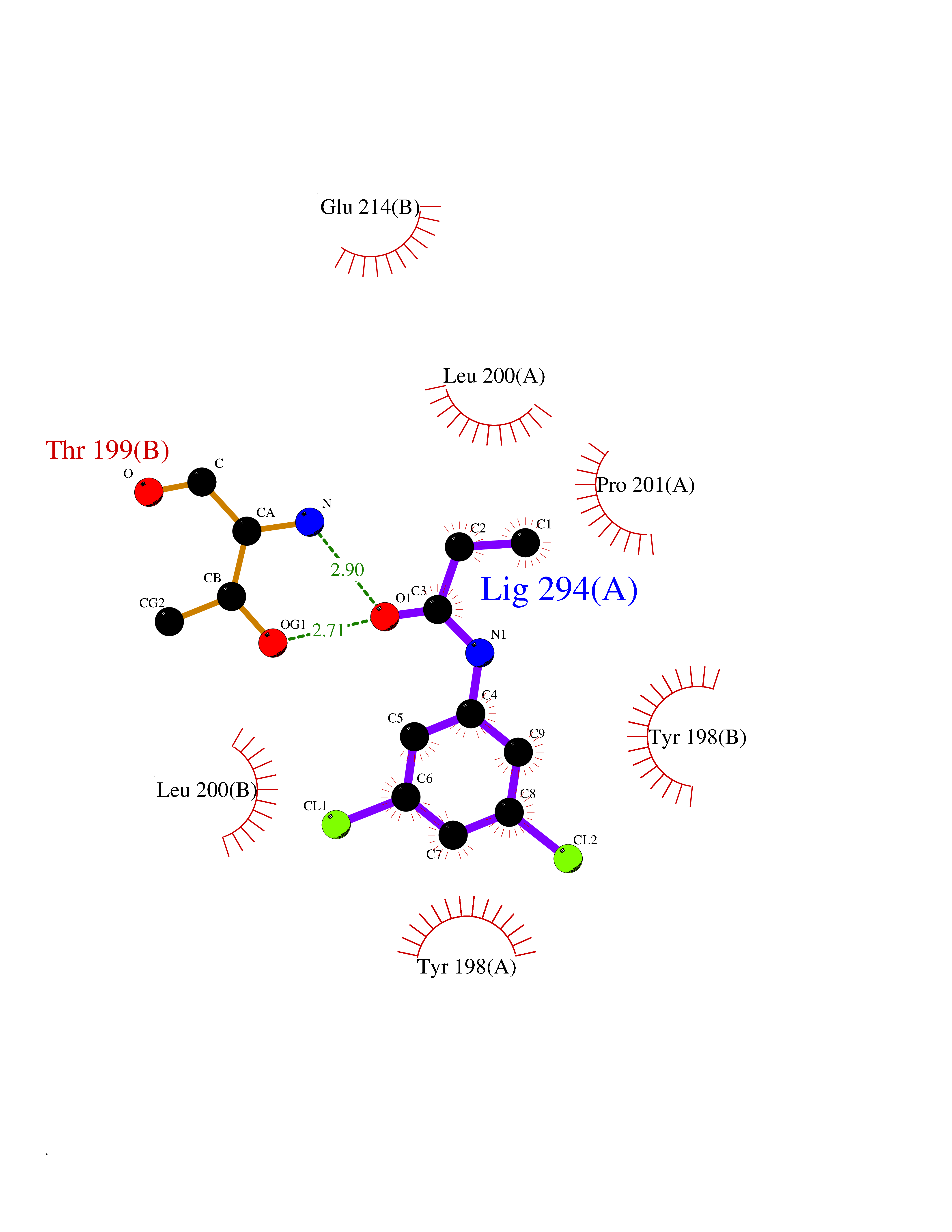 Ligplot Map 3D Binding mode Sequence AFYKREMFDPAEKYKMDHRRRGIALIFNHERFFWHLTLPERRGTCADRDNLTRRFSDLGFEVKCFNDLKAEELLLKIHEVSTVSHADADCFVCVFLSHGEGNHIYAYDAKIEIQTLTGLFKGDKCHSLVGKPKIFIIQAARGNQHDVPVIPDTNITEVDAASVYTLPAGADFLMCYSVAEGYYSHRETVNGSWYIQDLCEMLGKYGSSLEFTELLTLVNRKVSQRRVDFCKDPSAIGKKQVPCFASMLTKKLHFFPKSMFDPAEKYKMDHRRRGIALIFNHERFFWHLTLPERRGTCADRDNLTRRFSDLGFEVKCFNDLKAEELLLKIHEVSTVSHADADCFVCVFLSHGEGNHIYAYDAKIEIQTLTGLFKGDKCHSLVGKPKIFIIQAARGNTNITEVDAASVYTLPAGADFLMCYSVAEGYYSHRETVNGSWYIQDLCEMLGKYGSSLEFTELLTLVNRKVSQRRVDFCKDPSAIGKKQVPCFASMLTKKLHFFPK Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | C-C chemokine receptor type 9 (CCR9) | 5LWE | 5.90 | |
Target general information Gen name CCR9 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Gprotein coupled receptor 28; GPR96; GPR28; GPR-9-6; G-protein coupled receptor 28; CDw199; CCR-9; CCCKR9; CC-CKR-9; CC chemokine receptor type 9; CC CKR9; C-C CKR-9 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Subsequently transduces a signal by increasing the intracellular calcium ions level. Receptor for chemokine SCYA25/TECK. Related diseases Split-foot malformation with mesoaxial polydactyly (SFMMP) [MIM:616890]: An autosomal recessive disorder characterized by a split-foot defect, mesoaxial polydactyly, nail abnormalities of the hands, and sensorineural hearing loss. {ECO:0000269|PubMed:26755636, ECO:0000269|PubMed:32266845}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myopathy, centronuclear, 6, with fiber-type disproportion (CNM6) [MIM:617760]: A form of centronuclear myopathy, a congenital muscle disorder characterized by progressive muscular weakness and wasting involving mainly limb girdle, trunk, and neck muscles. It may also affect distal muscles. Weakness may be present during childhood or adolescence or may not become evident until the third decade of life. Ptosis is a frequent clinical feature. The most prominent histopathologic features include high frequency of centrally located nuclei in muscle fibers not secondary to regeneration, radial arrangement of sarcoplasmic strands around the central nuclei, and predominance and hypotrophy of type 1 fibers. CNM6 is an autosomal recessive, slowly progressive form with onset in infancy or early childhood. {ECO:0000269|PubMed:27816943, ECO:0000269|PubMed:30237576}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15250 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Host-virus interaction; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 31137.4 Length 275 Aromaticity 0.13 Instability index 25.98 Isoelectric point 9.31 Charge (pH=7) 15.22 2D Binding mode Binding energy (Kcal/mol) -8.05 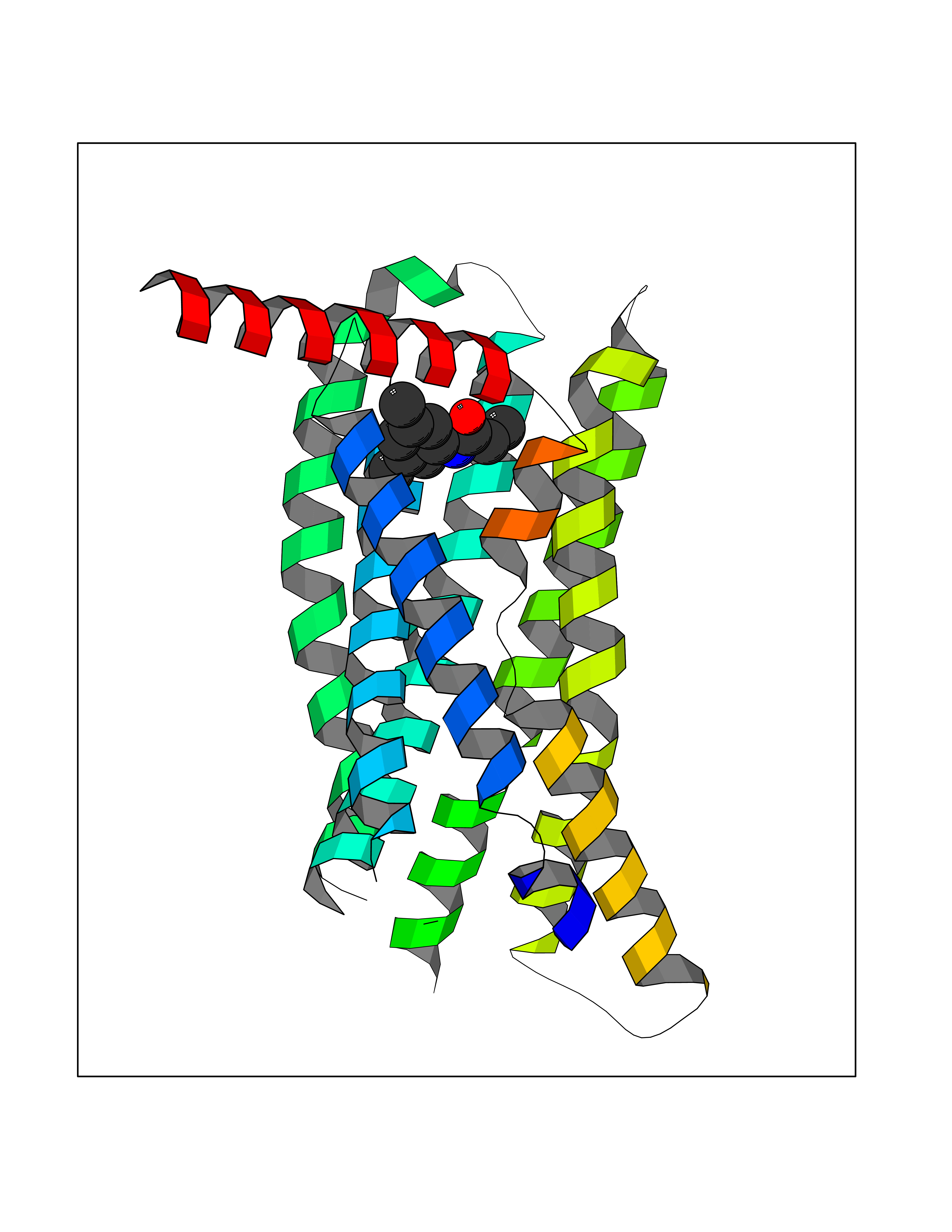 Molscript Map 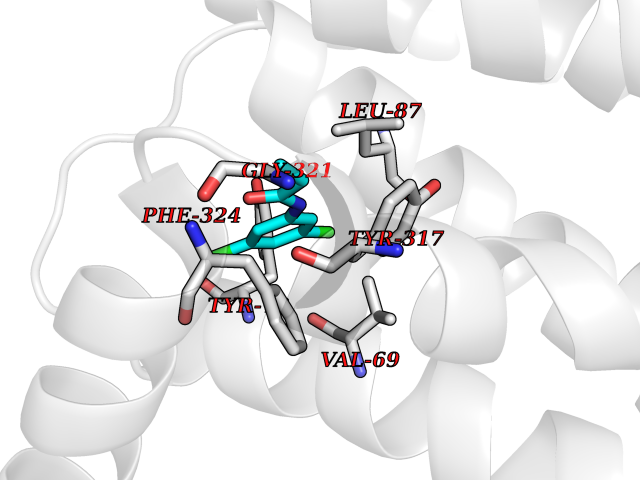 Pymol Map 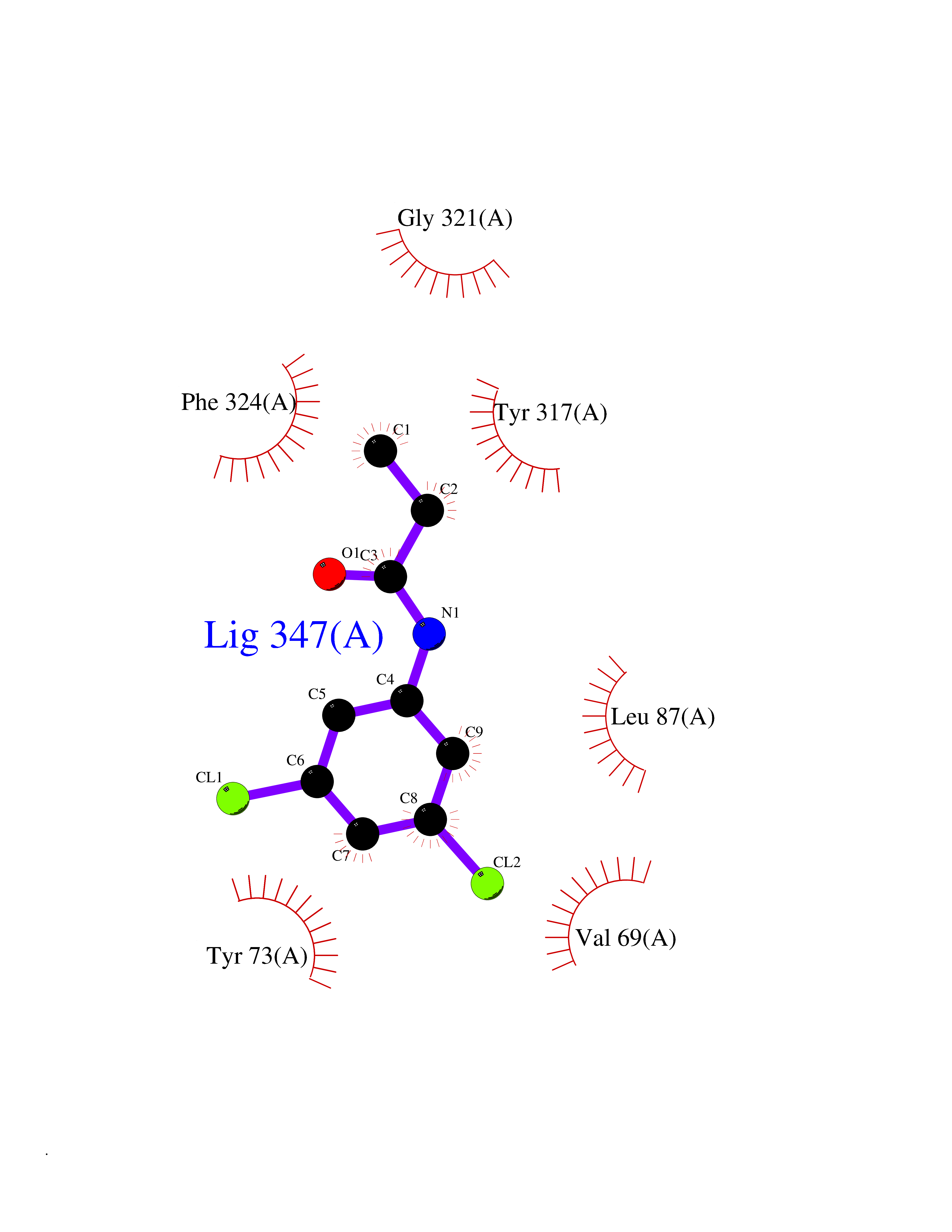 Ligplot Map 3D Binding mode Sequence RQFASHFLPPLYWLVFIVGALGNSLVILVYWYCARAKTATDMFLLNLAIADLLFLVTLPFWAIATFMCKVVNSMYKMNFYSCVLLIMCICVDRYIAIAQAMRAHTWREKRLLYSKMVCFTIWVLAAALCIPEILYCTTKLKSAVLALKVILGFFLPFVVMACCYTIIIHTLIQAKKSSKHKALKATITVLTVFVLSQFPYNCILLVQTIDAYAMFISNCAVSTAIDICFQVTQAIAFFHSCLNPVLYVFVGERFRRDLVKTLKNLGAISQAAAHH Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Quinone reductase 1 (NQO1) | 1D4A | 5.89 | |
Target general information Gen name NQO1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Qui reductase 1; QR1; Phylloquinone reductase; Phylloqui reductase; NMOR1; NAD(P)H:quinone oxidoreductase 1; NAD(P)H dehydrogenase [quinone] 1; Menadione reductase; DTD; DT-diaphorase 1; DT-diaphorase Protein family NAD(P)H dehydrogenase (quinone) family Biochemical class NADH/NADPH oxidoreductase Function The enzyme apparently serves as a quinone reductase in connection with conjugation reactions of hydroquinons involved in detoxification pathways as well as in biosynthetic processes such as the vitamin K-dependent gamma-carboxylation of glutamate residues in prothrombin synthesis. Related diseases Congenital disorder of glycosylation 2D (CDG2D) [MIM:607091]: A multisystem disorder caused by a defect in glycoprotein biosynthesis and characterized by under-glycosylated serum glycoproteins. Congenital disorders of glycosylation result in a wide variety of clinical features, such as defects in the nervous system development, psychomotor retardation, dysmorphic features, hypotonia, coagulation disorders, and immunodeficiency. The broad spectrum of features reflects the critical role of N-glycoproteins during embryonic development, differentiation, and maintenance of cell functions. {ECO:0000269|PubMed:11901181, ECO:0000269|PubMed:30653653, ECO:0000269|PubMed:32157688}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Combined low LDL and fibrinogen (CLDLFIB) [MIM:620364]: An autosomal recessive condition characterized by low plasma LDL-cholesterol and fibrinogen levels, and associated with a decreased risk of coronary artery disease. {ECO:0000269|PubMed:34855475}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07385; DB02395; DB03626; DB14001; DB04392; DB09061; DB00958; DB02633; DB00515; DB14002; DB00266; DB00997; DB01927; DB02400; DB03147; DB00170; DB00526; DB00252; DB04090; DB00163 Interacts with P07902; Q9UK53; P15559 EC number EC 1.6.5.2 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; FAD; Flavoprotein; Isopeptide bond; NAD; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 61454.1 Length 546 Aromaticity 0.12 Instability index 36.74 Isoelectric point 8.99 Charge (pH=7) 8.99 2D Binding mode Binding energy (Kcal/mol) -8.04 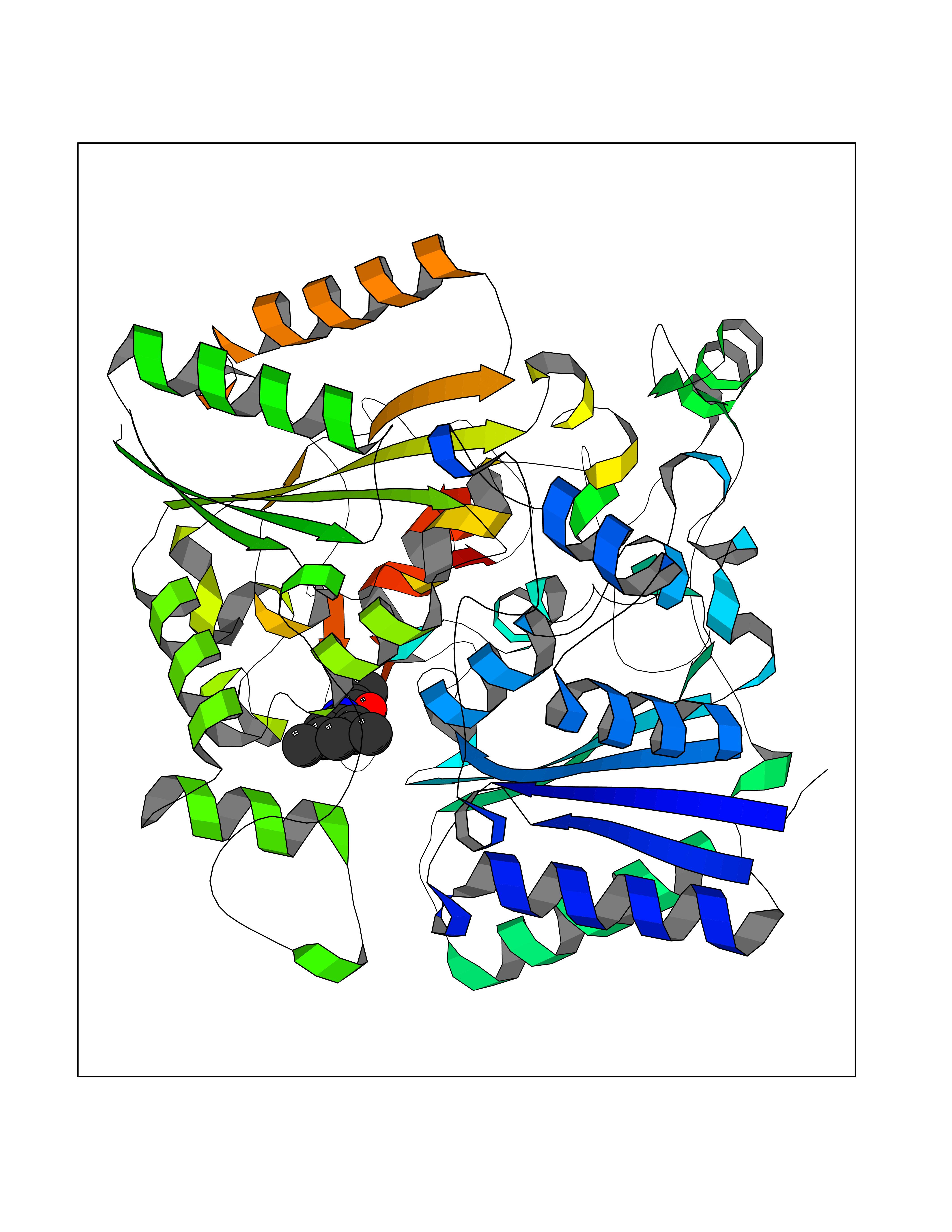 Molscript Map 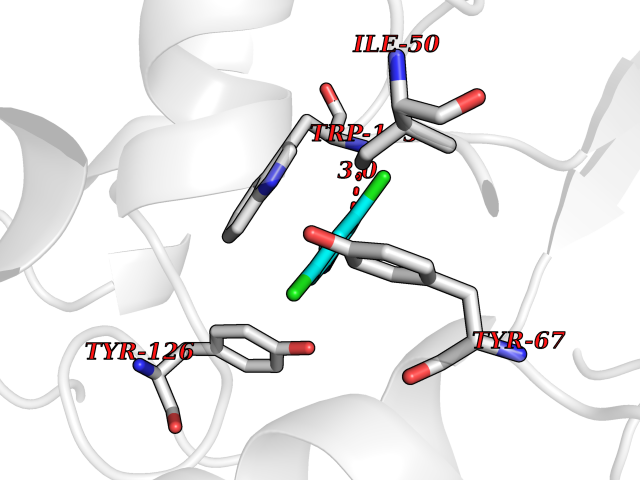 Pymol Map 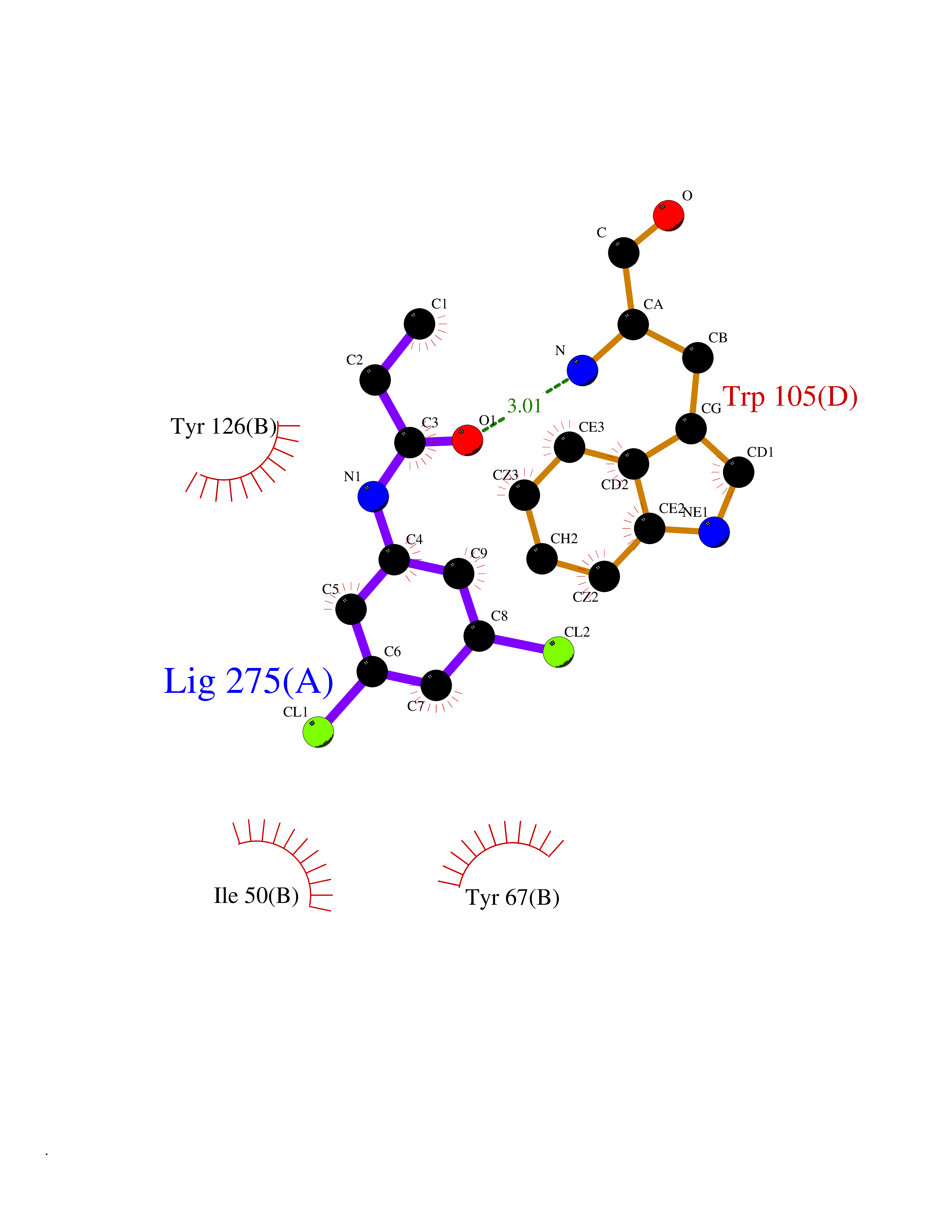 Ligplot Map 3D Binding mode Sequence VGRRALIVLAHSERTSFNYAMKEAAAAALKKKGWEVVESDLYAMNFNPIISRKDITGKLKDPANFQYPAESVLAYKEGHLSPDIVAEQKKLEAADLVIFQFPLQWFGVPAILKGWFERVFIGEFAYTYAAMYDKGPFRSKKAVLSITTGGSGSMYSLQGIHGDMNVILWPIQSGILHFCGFQVLEPQLTYSIGHTPADARIQILEGWKKRLENIWDETPLYFAPSSLFDLNFQAGFLMKKEVQDEEKNKKFGLSVGHHLGKSIPTDNQIKARKVGRRALIVLAHSERTSFNYAMKEAAAAALKKKGWEVVESDLYAMNFNPIISRKDITGKLKDPANFQYPAESVLAYKEGHLSPDIVAEQKKLEAADLVIFQFPLQWFGVPAILKGWFERVFIGEFAYTYAAMYDKGPFRSKKAVLSITTGGSGSMYSLQGIHGDMNVILWPIQSGILHFCGFQVLEPQLTYSIGHTPADARIQILEGWKKRLENIWDETPLYFAPSSLFDLNFQAGFLMKKEVQDEEKNKKFGLSVGHHLGKSIPTDNQIKARK Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Mitochondrial aldehyde dehydrogenase (ALDH2) | 1O04 | 5.89 | |
Target general information Gen name ALDH2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aldehyde dehydrogenase, mitochondrial; ALDM; ALDHI; ALDH-E2; ALDH class 2 Protein family Aldehyde dehydrogenase family Biochemical class Aldehyde/oxo donor oxidoreductase Function Second enzyme of the major oxidative pathway of alcohol metabolism. Catalyzes the chemical transformation from acetaldehyde to acetic acid. Additionally, functions as a protector against oxidative stress. Related diseases AMED syndrome, digenic (AMEDS) [MIM:619151]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. AMEDS is an autosomal recessive, digenic form characterized by childhood onset of bone marrow failure resulting in aplastic anemia, in association with global developmental delay, intellectual disability, and poor overall growth with short stature. {ECO:0000269|PubMed:33355142}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry. AMEDS patients carry ADH5 biallelic variants and homozygous or heterozygous ALDH2 variant p.Glu504Lys, affecting protein activity. Cellular and animal studies demonstrate that the simultaneous loss of ALDH2 and ADH5 activities leads to an increase of cellular formaldehyde sensitivity and multisystem abnormalities including hematopoietic failure. {ECO:0000269|PubMed:33355142}. Drugs (DrugBank ID) DB01612; DB06770; DB04381; DB02115; DB00822; DB00536; DB00157; DB00435; DB00727; DB09117; DB06154; DB06207 Interacts with NA EC number EC 1.2.1.3 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Disease variant; Dwarfism; Intellectual disability; Mitochondrion; NAD; Oxidoreductase; Proteomics identification; Reference proteome; Transit peptide; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H Molecular weight (Da) 50223.5 Length 462 Aromaticity 0.1 Instability index 33.68 Isoelectric point 5.29 Charge (pH=7) -7.87 2D Binding mode Binding energy (Kcal/mol) -8.03 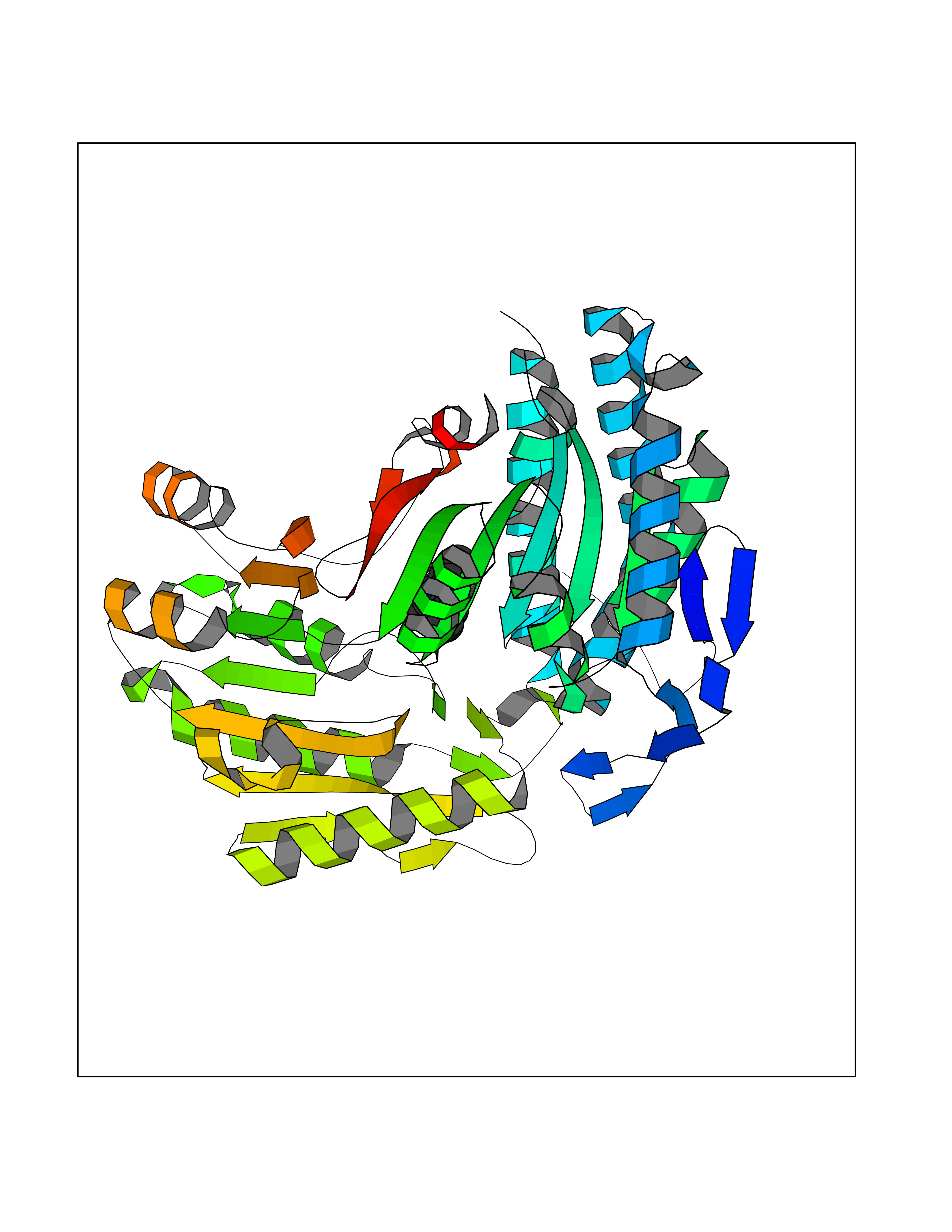 Molscript Map 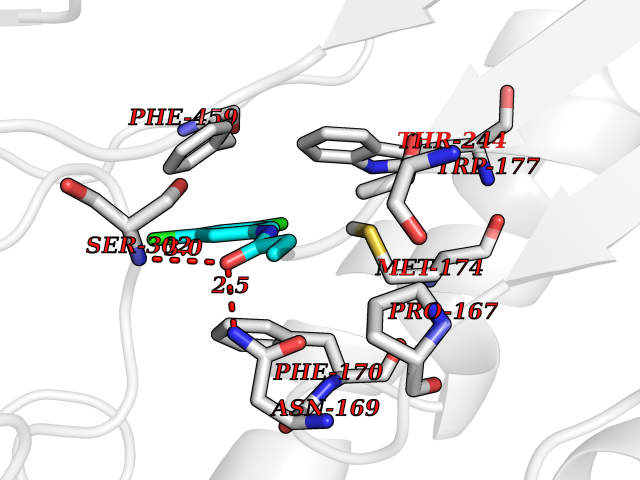 Pymol Map 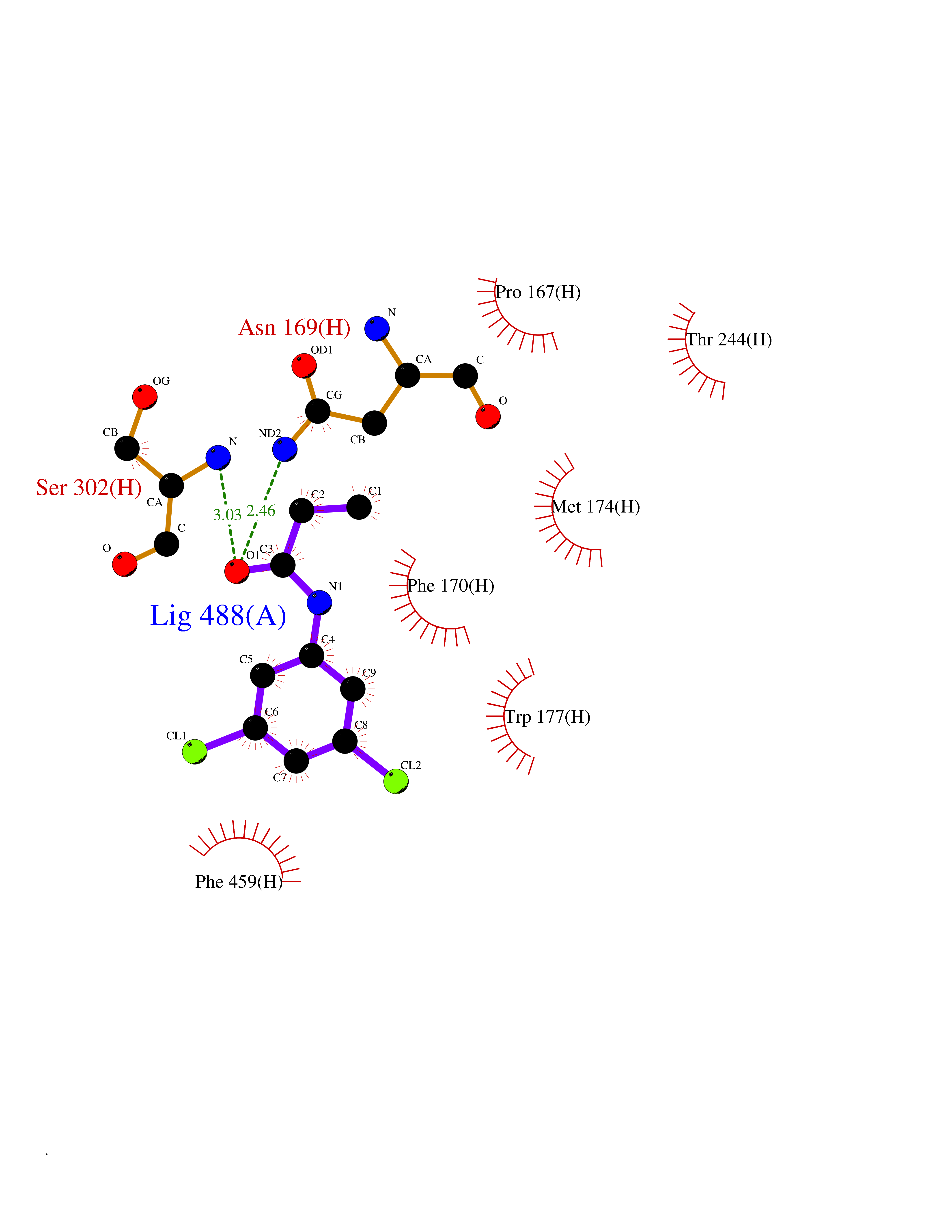 Ligplot Map 3D Binding mode Sequence AVPAPNQQPEVFCNQIFINNEWHDAVSRKTFPTVNPSTGEVICQVAEGDKEDVDKAVKAARAAFQLGSPWRRMDASHRGRLLNRLADLIERDRTYLAALETLDNGKPYVISYLVDLDMVLKCLRYYAGWADKEPVGVCGQIIPWNFPLLMQAWKLGPALATGNVVVMKVAEQTPLTALYVANLIKEAGFPPGVVNIVPGFGPTAGAAIASHEDVDKVAFTGSTEIGRVIQVAAGSSNLKRVTLELGGKSPNIIMSDADMDWAVEQAHFALFFNQGQCSCAGSRTFVQEDIYDEFVERSVARAKSRVVGNPFDSKTEQGPQVDETQFKKILGYINTGKQEGAKLLCGGGIAADRGYFIQPTVFGDVQDGMTIAKEEIFGPVMQILKFKTIEEVVGRANNSTYGLAAAVFTKDLDKANYLSQALQAGTVWVNCYDVFGAQSPFGGYKMSGSGRELGEYGLQAYT Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Neuronal acetylcholine receptor subunit alpha-3 | 4ZK4 | 5.89 | |
Target general information Gen name CHRNA3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NACHRA3 Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Acetylcholine binding protein Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ligand-gated ion channel activity.Serotonin-gated cation-selective channel activity. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46391.5 Length 408 Aromaticity 0.12 Instability index 30.23 Isoelectric point 4.6 Charge (pH=7) -22.73 2D Binding mode Binding energy (Kcal/mol) -8.03 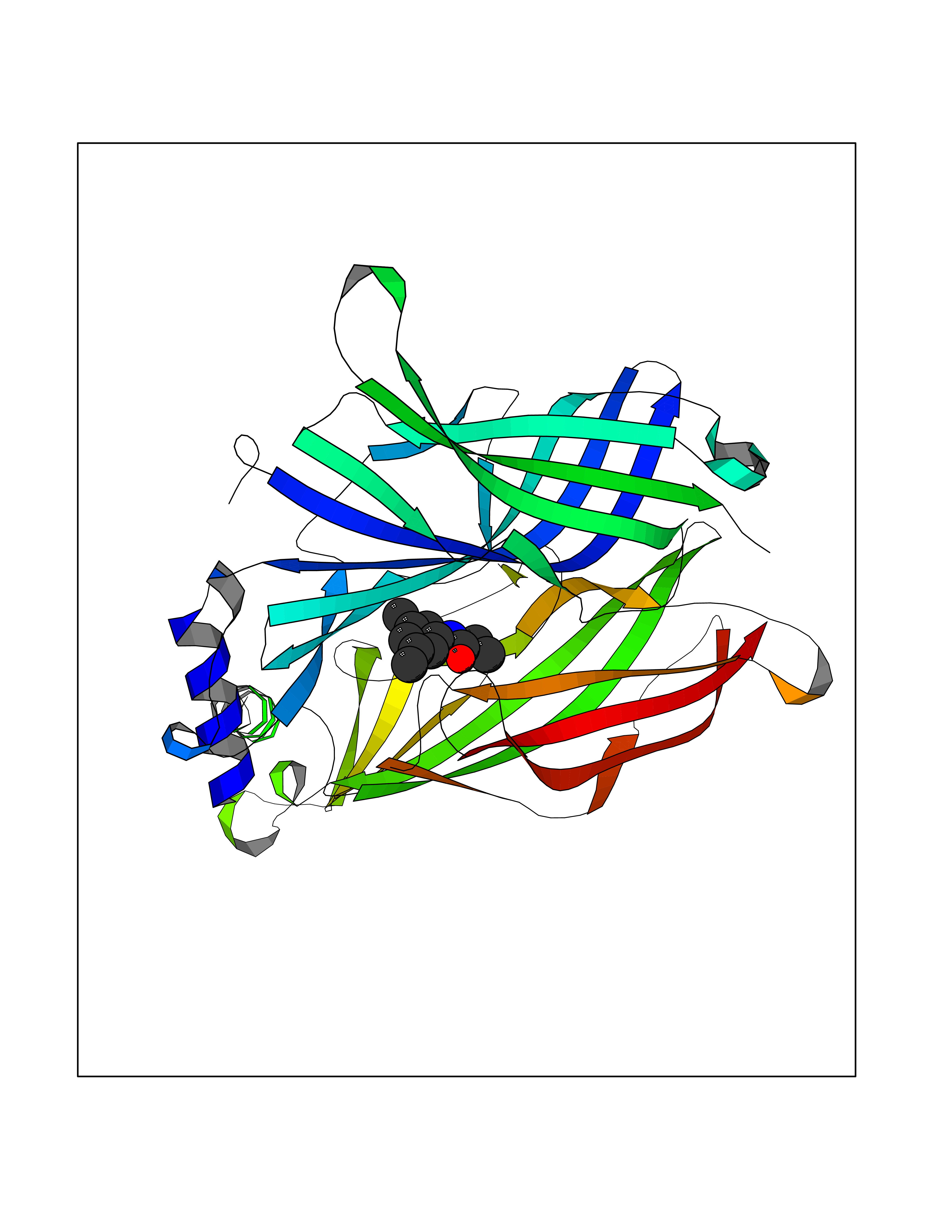 Molscript Map 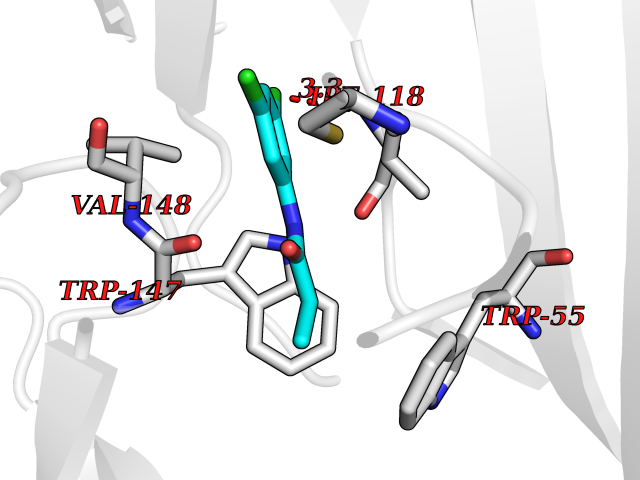 Pymol Map 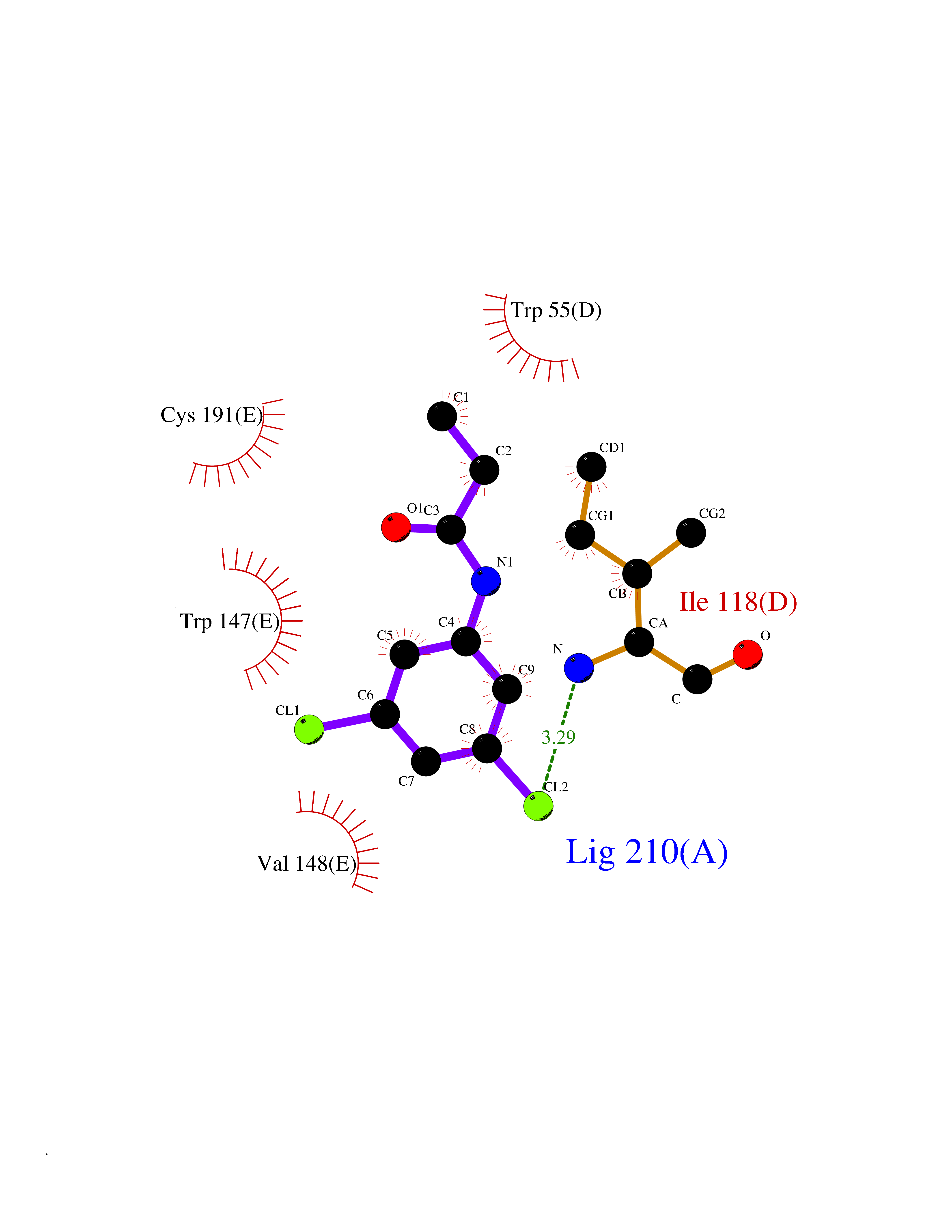 Ligplot Map 3D Binding mode Sequence LHSQANLMRLKSDLFYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRERRLHSQANLMRLKSDLFNRYPGPTKDDPLTVTLGFTLQDIVKADSSTNEVDLVYWEQQRWKLNSLMWDPNEYGNITDFRTSAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQYKHDIKYNCCEEIYPDVVLVVKFRE Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | Matrix metalloproteinase-12 (MMP-12) | 1Y93 | 5.89 | |
Target general information Gen name MMP12 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Macrophage metalloelastase; Macrophage elastase; MME; ME; HME Protein family Peptidase M10A family Biochemical class Peptidase Function Has significant elastolytic activity. Can accept large and small amino acids at the P1' site, but has a preference for leucine. Aromatic or hydrophobic residues are preferred at the P1 site, with small hydrophobic residues (preferably alanine) occupying P3. May be involved in tissue injury and remodeling. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB07026; DB07921; DB04405; DB00551; DB03880; DB07556; DB02118; DB00786; DB07446; DB07683; DB08599; DB08271; DB07922; DB07920; DB05387; DB03367; DB00013 Interacts with NA EC number EC 3.4.24.65 Uniprot keywords 3D-structure; Calcium; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 17461.3 Length 158 Aromaticity 0.13 Instability index 13.25 Isoelectric point 6.11 Charge (pH=7) -3.44 2D Binding mode Binding energy (Kcal/mol) -8.04 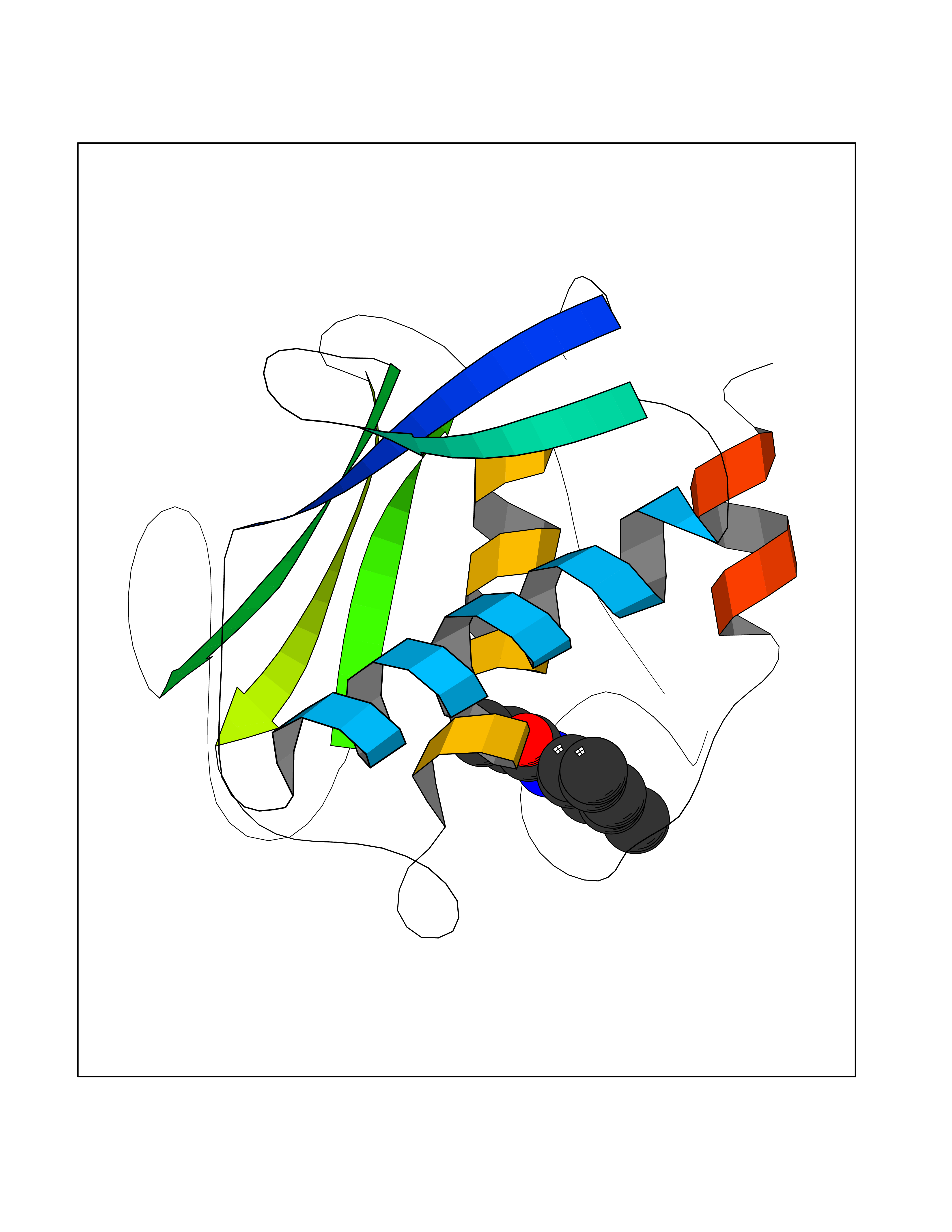 Molscript Map 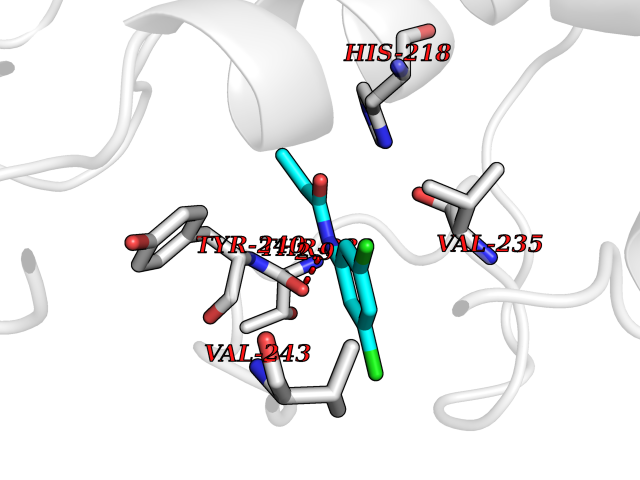 Pymol Map 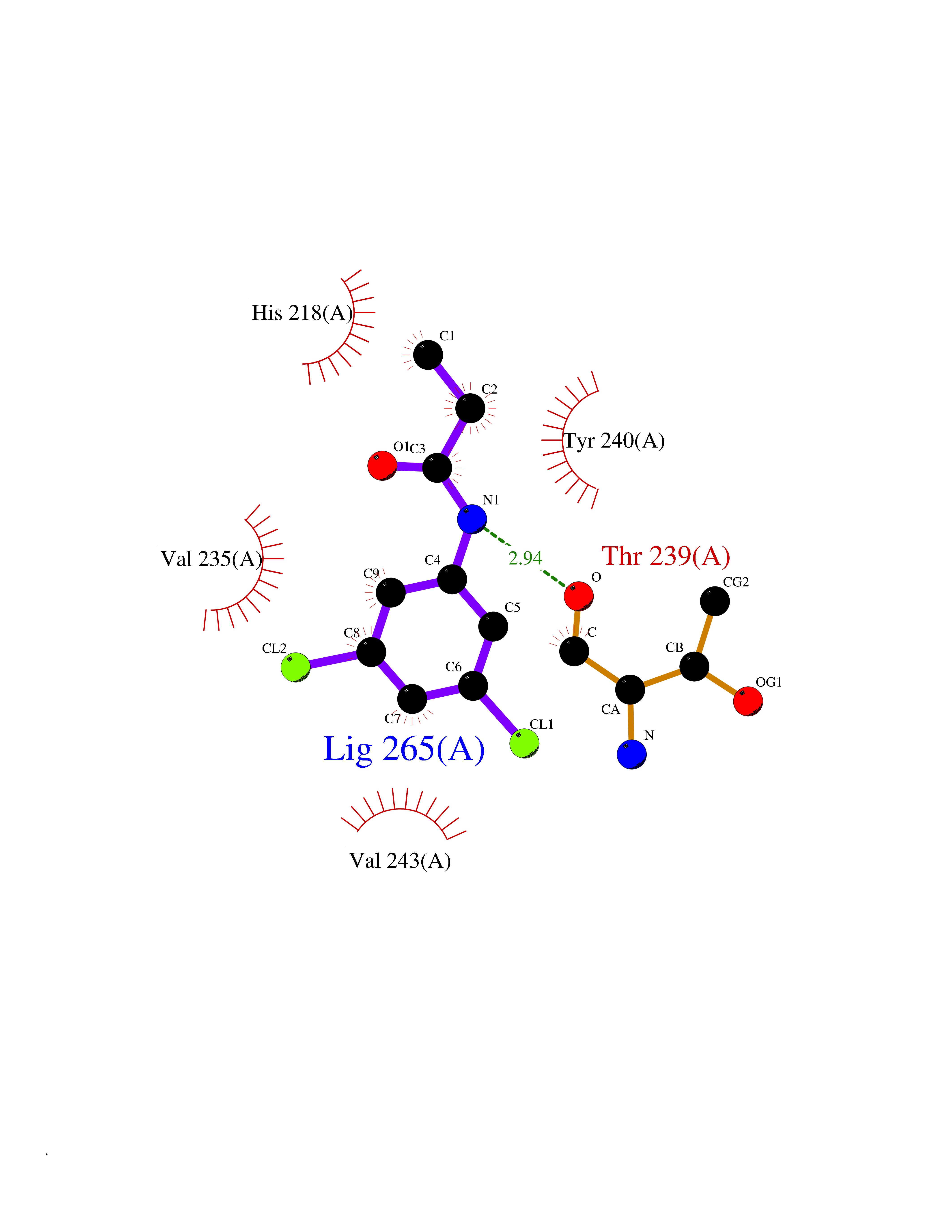 Ligplot Map 3D Binding mode Sequence GPVWRKHYITYRINNYTPDMNREDVDYAIRKAFQVWSNVTPLKFSKINTGMADILVVFARGAHGDDHAFDGKGGILAHAFGPGSGIGGDAHFDEDEFWTTHSGGTNLFLTAVHEIGHSLGLGHSSDPKAVMFPTYKYVDINTFRLSADDIRGIQSLYG Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Adenosine kinase (ADK) | 1BX4 | 5.89 | |
Target general information Gen name ADK Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Adenosine 5'-phosphotransferase; AK Protein family Carbohydrate kinase PfkB family Biochemical class Kinase Function Serves as a potential regulator of concentrations of extracellular adenosine and intracellular adenine nucleotides. ATP dependent phosphorylation of adenosine and other related nucleoside analogs to monophosphate derivatives. Related diseases Hypermethioninemia due to adenosine kinase deficiency (HMAKD) [MIM:614300]: A metabolic disorder characterized by global developmental delay, early-onset seizures, mild dysmorphic features, and characteristic biochemical anomalies, including persistent hypermethioninemia with increased levels of S-adenosylmethionine and S-adenosylhomocysteine. Homocysteine levels are typically normal. {ECO:0000269|PubMed:21963049}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07280; DB07173; DB01048; DB00640; DB00131; DB00171; DB00811 Interacts with NA EC number EC 2.7.1.20 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cytoplasm; Direct protein sequencing; Disease variant; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Purine salvage; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 38383.4 Length 342 Aromaticity 0.09 Instability index 37.64 Isoelectric point 6.24 Charge (pH=7) -3.41 2D Binding mode Binding energy (Kcal/mol) -8.04 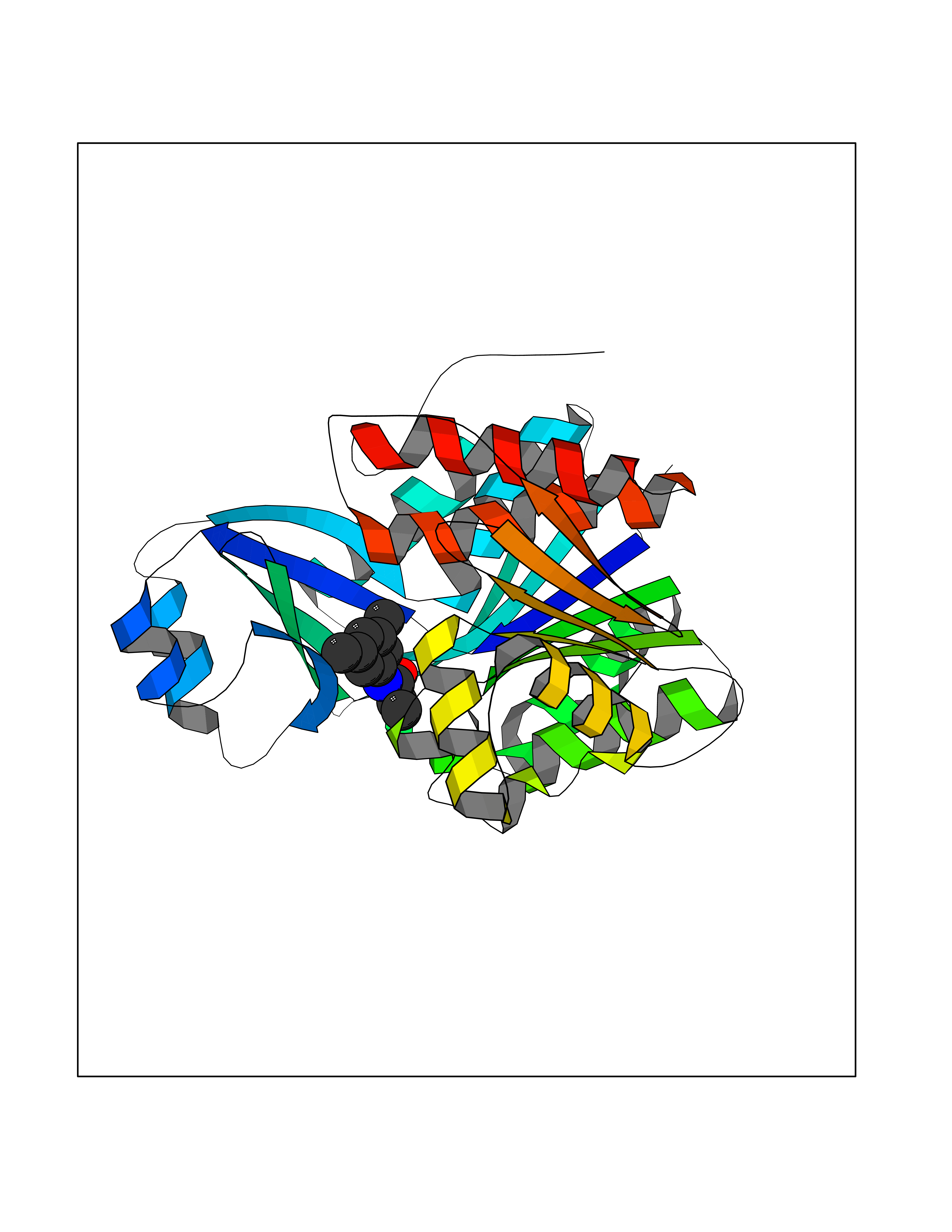 Molscript Map 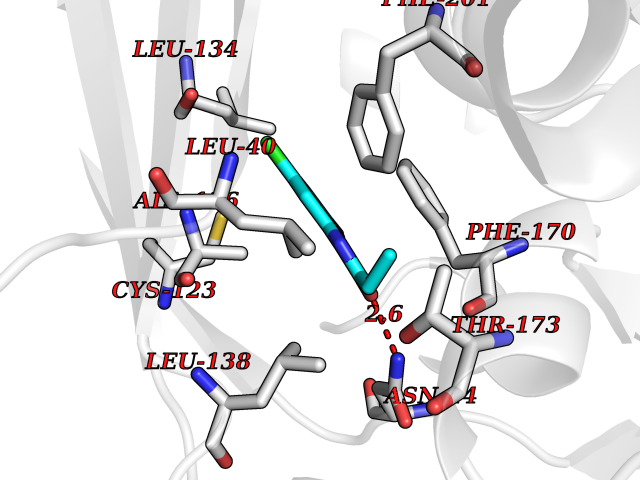 Pymol Map 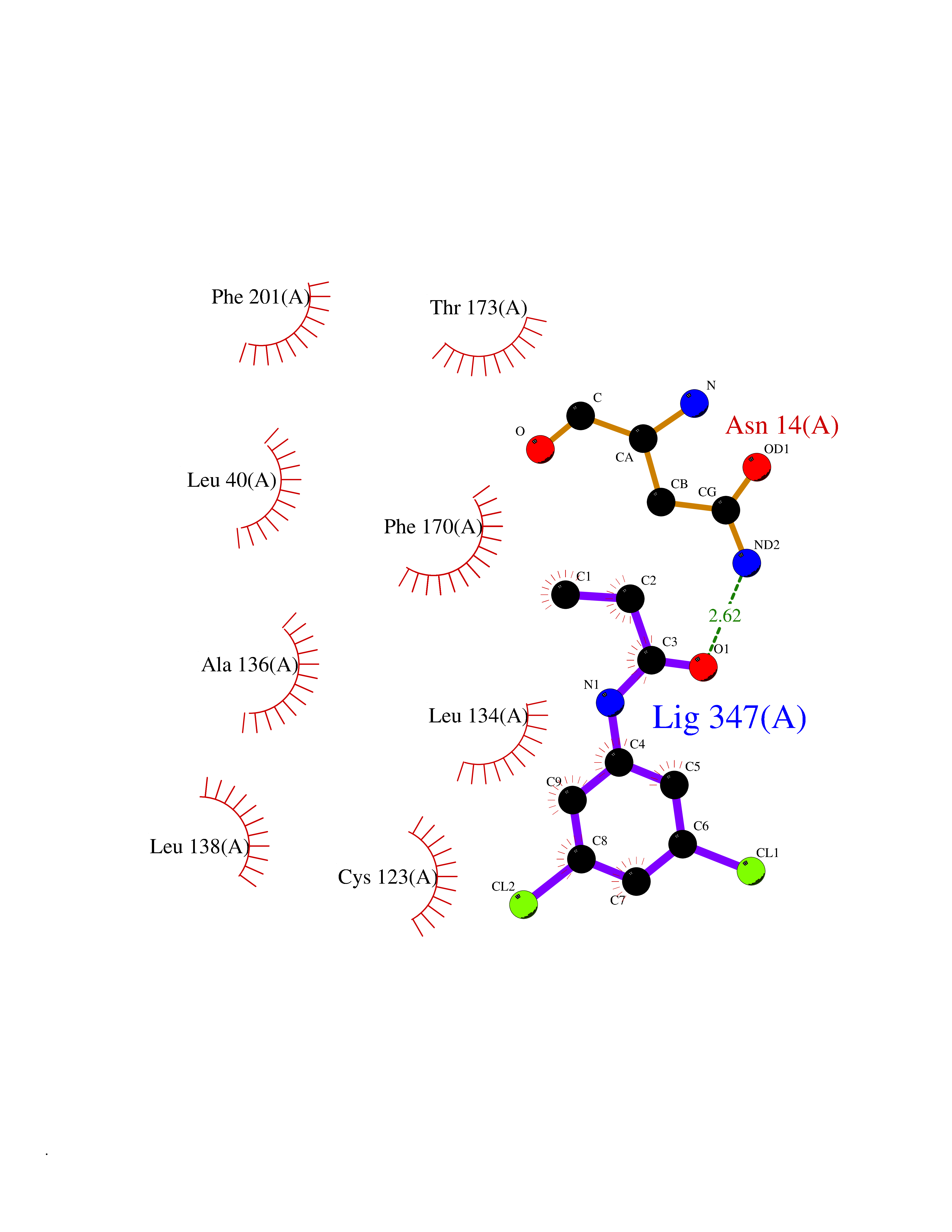 Ligplot Map 3D Binding mode Sequence VRENILFGMGNPLLDISAVVDKDFLDKYSLKPNDQILAEDKHKELFDELVKKFKVEYHAGGSTQNSIKVAQWMIQQPHKAATFFGCIGIDKFGEILKRKAAEAHVDAHYYEQNEQPTGTCAACITGDNRSLIANLAAANCYKKEKHLDLEKNWMLVEKARVCYIAGFFLTVSPESVLKVAHHASENNRIFTLNLSAPFISQFYKESLMKVMPYVDILFGNETEAATFAREQGFETKDIKEIAKKTQALPKMNSKRQRIVIFTQGRDDTIMATESEVTAFAVLDQDQKEIIDTNGAGDAFVGGFLSQLVSDKPLTECIRAGHYAASIIIRRTGCTFPEKPDFH Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Acetyl-CoA carboxylase (ACC) (EC 6.4.1.2) (Fatty acid synthetase 3) (mRNA transport-defective protein 7) [Includes: Biotin carboxylase (EC 6.3.4.14)] | 1UYS | 5.89 | |
Target general information Gen name ACC1 Organism Saccharomyces cerevisiae (strain ATCC 204508 / S288c) (Baker's yeast) Uniprot ID TTD ID NA Synonyms MTR7;YNR016C;N3175;ABP2;FAS3 Protein family NA Biochemical class NA Function Carries out three functions: biotin carboxyl carrier protein, biotin carboxylase and carboxyltransferase. Involved in the synthesis of very-long-chain fatty acid synthesis which is required to maintain a functional nuclear envelope. Required for acylation and vacuolar membrane association of VAC8 which is necessary to maintain a normal morphology of the vacuole. {ECO:0000269|PubMed:10757783, ECO:0000269|PubMed:12730220, ECO:0000269|PubMed:6103540, ECO:0000269|PubMed:6108218, ECO:0000269|PubMed:8943372}." Related diseases A chromosomal aberration involving NFKB2 is found in a case of B-cell non Hodgkin lymphoma (B-NHL). Translocation t(10;14)(q24;q32) with IGHA1. The resulting oncogene is also called Lyt-10C alpha variant.; DISEASE: A chromosomal aberration involving NFKB2 is found in a cutaneous T-cell leukemia (C-TCL) cell line. This rearrangement produces the p80HT gene which codes for a truncated 80 kDa protein (p80HT).; DISEASE: In B-cell leukemia (B-CLL) cell line, LB40 and EB308, can be found after heterogeneous chromosomal aberrations, such as internal deletions.; DISEASE: Immunodeficiency, common variable, 10 (CVID10) [MIM:615577]: A primary immunodeficiency characterized by childhood-onset of recurrent infections, hypogammaglobulinemia, and decreased numbers of memory and marginal zone B-cells. Some patients may develop autoimmune features and have circulating autoantibodies. An unusual feature is central adrenal insufficiency. {ECO:0000269|PubMed:24140114, ECO:0000269|PubMed:25524009}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q00955 EC number 6.3.4.14; 6.4.1.2 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Biotin; Cytoplasm; Direct protein sequencing; Endoplasmic reticulum; Fatty acid biosynthesis; Fatty acid metabolism; Ligase; Lipid biosynthesis; Lipid metabolism; Manganese; Membrane; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Reference proteome Protein physicochemical properties Chain ID B,C Molecular weight (Da) 145619 Length 1328 Aromaticity 0.1 Instability index 30.31 Isoelectric point 5.32 Charge (pH=7) -26.79 2D Binding mode Binding energy (Kcal/mol) -8.04  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence WLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGLPIATPYPVKEWLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGL Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Cocaine esterase | 3I2K | 5.89 | |
Target general information Gen name cocE Organism Rhodococcus sp. (strain MB1 Bresler) Uniprot ID TTD ID NA Synonyms NA Protein family CocE/NonD hydrolase family Biochemical class Hydrolase Function Carboxylic ester hydrolase activity.Dipeptidyl-peptidase activity. Related diseases Thiamine metabolism dysfunction syndrome 5, episodic encephalopathy type (THMD5) [MIM:614458]: An autosomal recessive metabolic disorder due to an inborn error of thiamine metabolism. The phenotype is highly variable, but in general, affected individuals have onset in early childhood of acute encephalopathic episodes associated with increased serum and CSF lactate. These episodes result in progressive neurologic dysfunction manifest as gait disturbances, ataxia, dystonia, and spasticity, which in some cases may result in loss of ability to walk. Cognitive function is usually preserved, although mildly delayed development has been reported. These episodes are usually associated with infection and metabolic decompensation. Some patients may have recovery of some neurologic deficits. {ECO:0000269|PubMed:22152682}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03793; DB01795 Interacts with NA EC number 3.1.1.84 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; Hydrolase; Serine esterase Protein physicochemical properties Chain ID A Molecular weight (Da) 62127.9 Length 574 Aromaticity 0.09 Instability index 26.62 Isoelectric point 4.56 Charge (pH=7) -33.24 2D Binding mode Binding energy (Kcal/mol) -8.04  Molscript Map 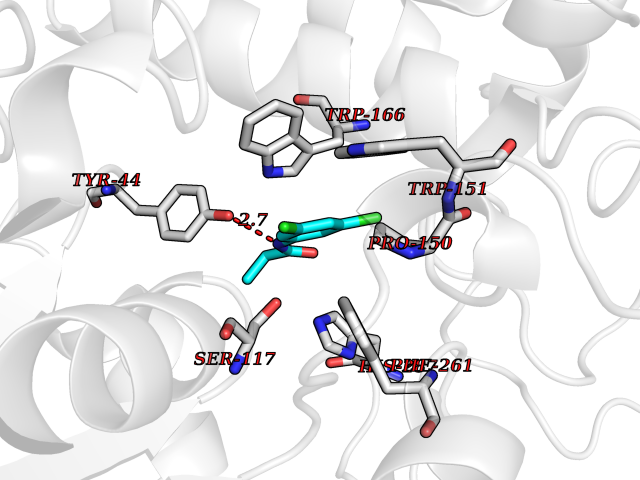 Pymol Map 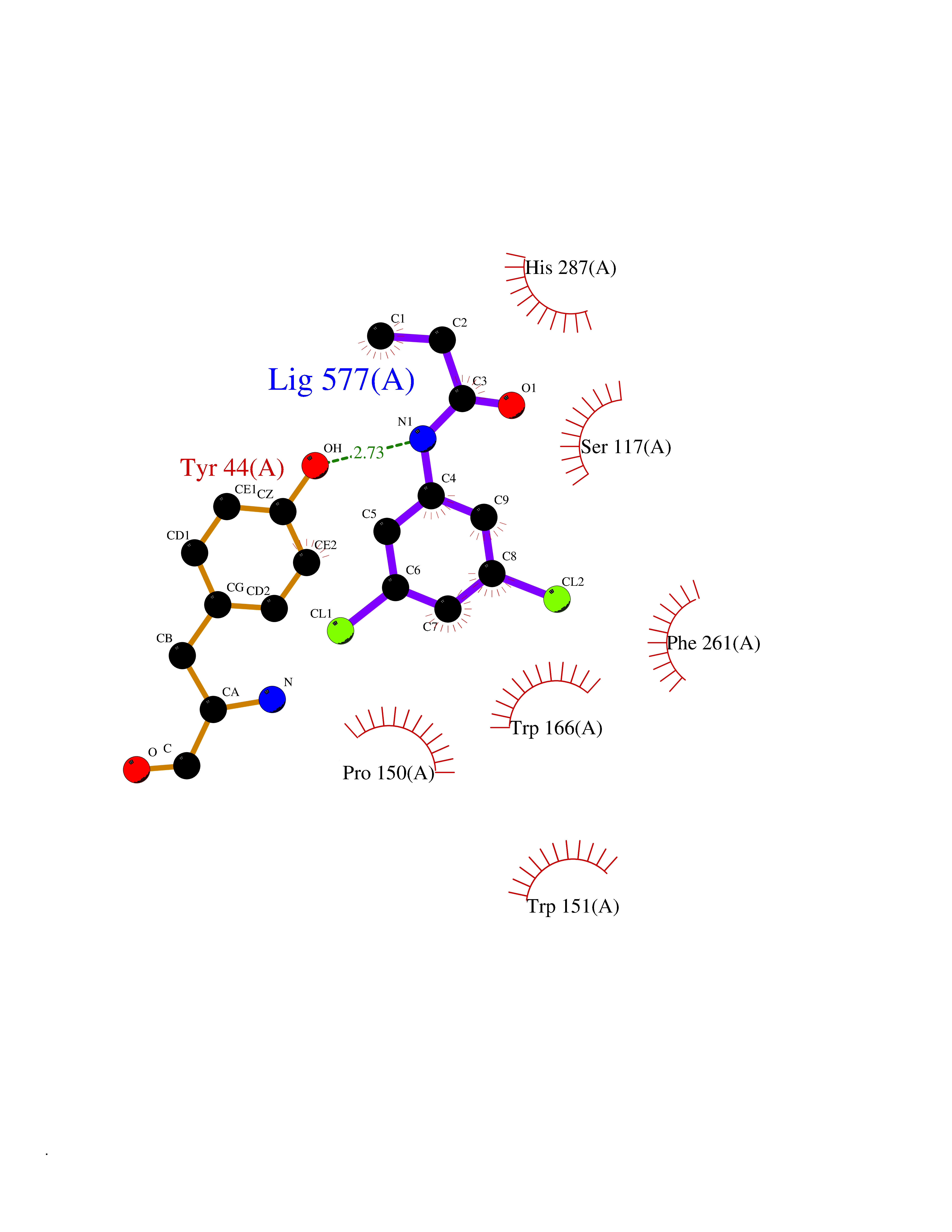 Ligplot Map 3D Binding mode Sequence VDGNYSVASNVMVPMRDGVRLAVDLYRPDADGPVPVLLVRNPYDKFDVFAWSTQSTNWLEFVRDGYAVVIQDTRGLFASEGEFVPHVDDEADAEDTLSWILEQAWCDGNVGMFGVSYLGVTQWQAAVSGVGGLKAIAPSMASADLYRAPWYGPGGALSVEALLGWSALIGTGLITSRSDARPEDAADFVQLAAILNDVAGAASVTPLAEQPLLGRLIPWVIDQVVDHPDNDESWQSISLFERLGGLATPALITAGWYDGFVGESLRTFVAVKDNADARLVVGPWSHSNLTGRNADRKFGIAATYPIQEATTMHKAFFDRHLRGETDALAGVPKVRLFVMGIDEWRDETDWPLPDTAYTPFYLGGSGAANTSTGGGTLSTSISGTESADTYLYDPADPVPSLGGTLLFHNGDNGPADQRPIHDRDDVLCYSTEVLTDPVEVTGTVSARLFVSSSAVDTDFTAKLVDVFPDGRAIALCDGIVRMRYRETLVNPTLIEAGEIYEVAIDMLATSNVFLPGHRIMVQVSSSNFPKYDRNSNTGGVIAREQLEEMCTAVNRIHRGPEHPSHIVLPIIKRK Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Monoglyceride lipase (MAGL) | 3PE6 | 5.89 | |
Target general information Gen name MGLL Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Monoacylglycerol lipase; MGL; Lysophospholipaselike; Lysophospholipase-like; Lysophospholipase homolog; HUK5; HU-K5 Protein family AB hydrolase superfamily, Monoacylglycerol lipase family Biochemical class Carboxylic ester hydrolase Function Hydrolyzes the endocannabinoid 2-arachidonoylglycerol, and thereby contributes to the regulation of endocannabinoid signaling, nociperception and perception of pain. Regulates the levels of fatty acids that serve as signaling molecules and promote cancer cell migration, invasion and tumor growth. Converts monoacylglycerides to free fatty acids and glycerol. Related diseases Systemic lupus erythematosus 9 (SLEB9) [MIM:610927]: A chronic, relapsing, inflammatory, and often febrile multisystemic disorder of connective tissue, characterized principally by involvement of the skin, joints, kidneys and serosal membranes. It is of unknown etiology, but is thought to represent a failure of the regulatory mechanisms of the autoimmune system. The disease is marked by a wide range of system dysfunctions, an elevated erythrocyte sedimentation rate, and the formation of LE cells in the blood or bone marrow. {ECO:0000269|PubMed:17360460}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Immunodeficiency, common variable, 7 (CVID7) [MIM:614699]: A primary immunodeficiency characterized by antibody deficiency, hypogammaglobulinemia, recurrent bacterial infections and an inability to mount an antibody response to antigen. The defect results from a failure of B-cell differentiation and impaired secretion of immunoglobulins; the numbers of circulating B-cells is usually in the normal range, but can be low. {ECO:0000269|PubMed:22035880}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07550; P37235 EC number EC 3.1.1.23 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Lipid biosynthesis; Lipid degradation; Lipid metabolism; Membrane; Nitration; Phosphoprotein; Proteomics identification; Reference proteome; Serine esterase Protein physicochemical properties Chain ID A Molecular weight (Da) 31808.4 Length 289 Aromaticity 0.08 Instability index 29.7 Isoelectric point 6.73 Charge (pH=7) -0.91 2D Binding mode Binding energy (Kcal/mol) -8.03 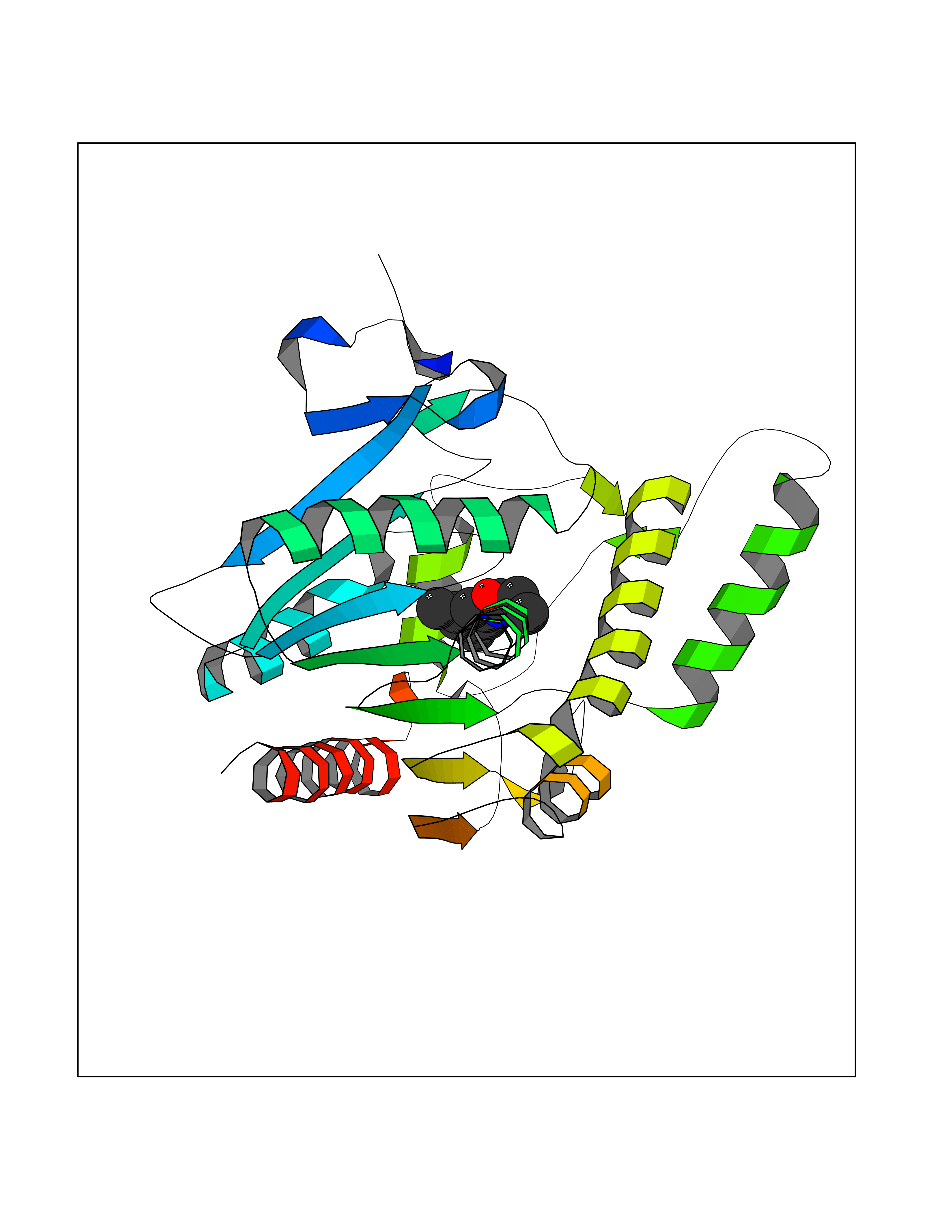 Molscript Map 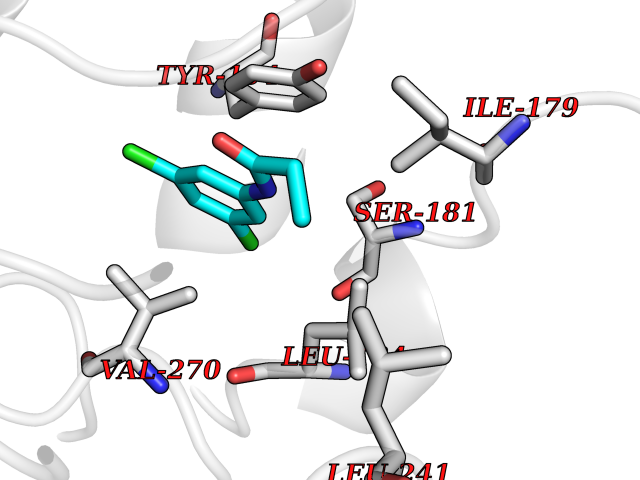 Pymol Map  Ligplot Map 3D Binding mode Sequence PRRTPQSIPYQDLPHLVNADGQYLFCRYWAPTGTPKALIFVSHGAGEHSGRYEELARMLMGLDLLVFAHDHVGHGQSEGERMVVSDFHVFVRDVLQHVDSMQKDYPGLPVFLLGHSMGGAIAILTAAERPGHFAGMVLISPLVLANPESATTFKVLAAKVLNSVLPNLSSGPIDSSVLSRNKTEVDIYNSDPLICRAGLKVCFGIQLLNAVSRVERALPKLTVPFLLLQGSADRLCDSKGAYLLMELAKSQDKTLKIYEGAYHVLHKELPEVTNSVFHEINMWVSQRTA Hydrogen bonds contact Hydrophobic contact | ||||