Job Results:
Ligand
Structure
Job ID
bb998bd8d55e750bf9fe315d9dd71eb1
Job name
NA
Time
2025-12-26 14:34:43
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Matrix metalloproteinase-16 (MMP-16) | 1RM8 | 5.72 | |
Target general information Gen name MMP16 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Membrane-type-3 matrix metalloproteinase; Membrane-type matrix metalloproteinase 3; MTMMP3; MT3MMP; MT3-MMP; MT-MMP 3; MMPX2; MMP-X2; C8orf57 Protein family Peptidase M10A family Biochemical class Peptidase Function Activates progelatinase A. Involved in the matrix remodeling of blood vessels. Isoform short cleaves fibronectin and also collagen type III, but at lower rate. It has no effect on type I, II, IV and V collagen. However, upon interaction with CSPG4, it may be involved in degradation and invasion of type I collagen by melanoma cells. Endopeptidase that degrades various components of the extracellular matrix, such as collagen type III and fibronectin. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03880; DB00786 Interacts with NA EC number EC 3.4.24.- Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Cleavage on pair of basic residues; Collagen degradation; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Membrane; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 18853.6 Length 169 Aromaticity 0.12 Instability index 33.65 Isoelectric point 4.88 Charge (pH=7) -12.42 2D Binding mode Binding energy (Kcal/mol) -7.8  Molscript Map 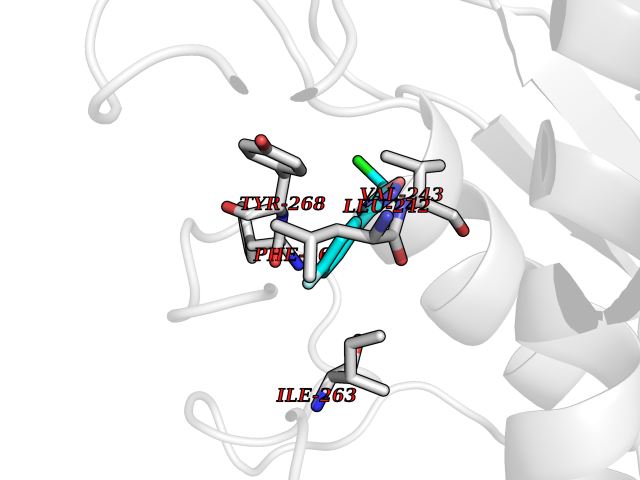 Pymol Map 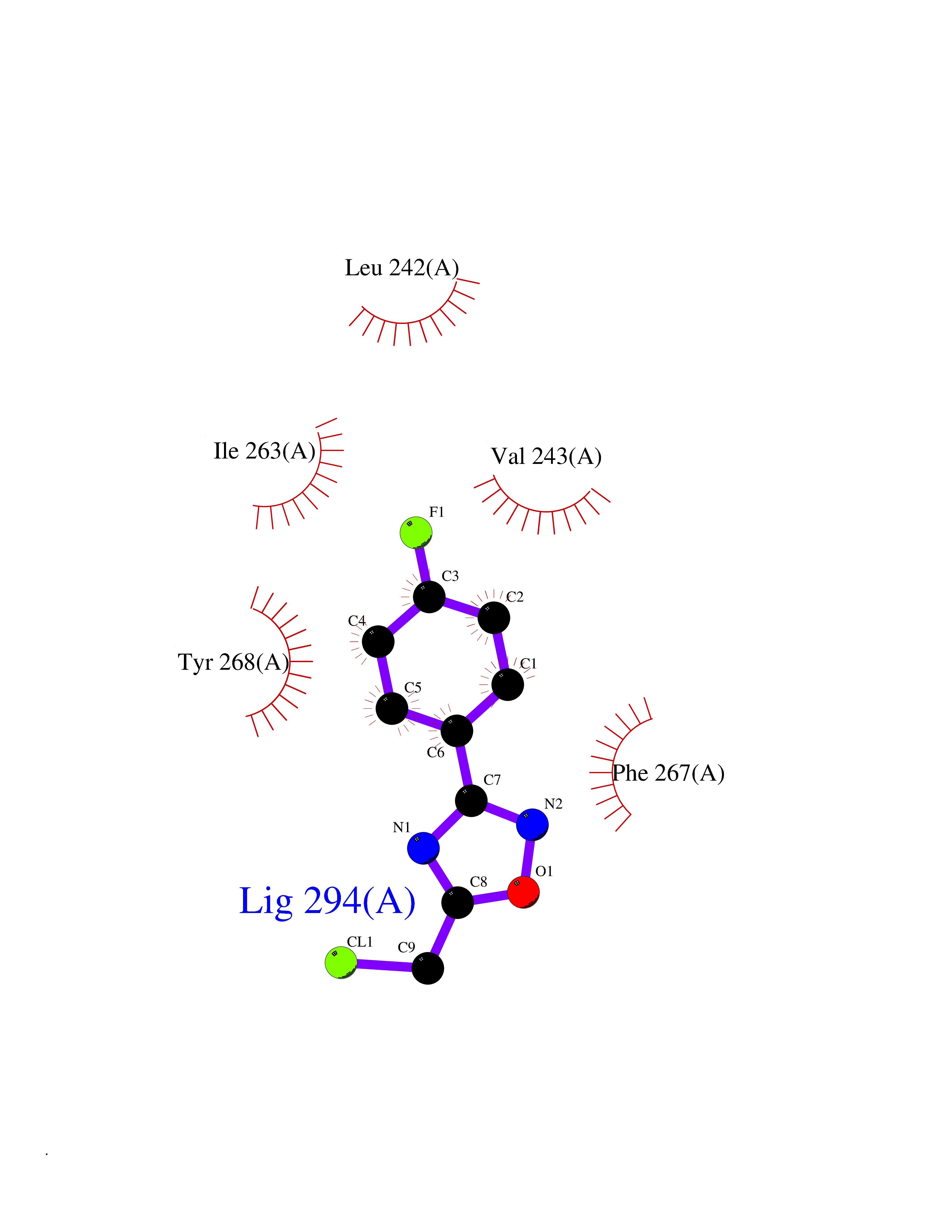 Ligplot Map 3D Binding mode Sequence GQKWQHKHITYSIKNVTPKVGDPETRKAIRRAFDVWQNVTPLTFEEVPYSELENGKRDVDITIIFASGFHGDSSPFDGEGGFLAHAYFPGPGIGGDTHFDSDEPWTLGNPNHDGNDLFLVAVHELGHALGLEHSNDPTAIMAPFYQYMETDNFKLPNDDLQGIQKIYGP Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Tyrosine-protein kinase BTK (ATK) | 4RFZ | 5.72 | |
Target general information Gen name BTK Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Bruton's tyrosine kinase; Bruton tyrosine kinase; BPK; B-cell progenitor kinase; B cell progenitor kinase; Agammaglobulinemia tyrosine kinase; Agammaglobulinaemia tyrosine kinase; AGMX1 Protein family Protein kinase superfamily, Tyr protein kinase family, TEC subfamily Biochemical class Kinase Function Binding of antigen to the B-cell antigen receptor (BCR) triggers signaling that ultimately leads to B-cell activation. After BCR engagement and activation at the plasma membrane, phosphorylates PLCG2 at several sites, igniting the downstream signaling pathway through calcium mobilization, followed by activation of the protein kinase C (PKC) family members. PLCG2 phosphorylation is performed in close cooperation with the adapter protein B-cell linker protein BLNK. BTK acts as a platform to bring together a diverse array of signaling proteins and is implicated in cytokine receptor signaling pathways. Plays an important role in the function of immune cells of innate as well as adaptive immunity, as a component of the Toll-like receptors (TLR) pathway. The TLR pathway acts as a primary surveillance system for the detection of pathogens and are crucial to the activation of host defense. Especially, is a critical molecule in regulating TLR9 activation in splenic B-cells. Within the TLR pathway, induces tyrosine phosphorylation of TIRAP which leads to TIRAP degradation. BTK plays also a critical role in transcription regulation. Induces the activity of NF-kappa-B, which is involved in regulating the expression of hundreds of genes. BTK is involved on the signaling pathway linking TLR8 and TLR9 to NF-kappa-B. Transiently phosphorylates transcription factor GTF2I on tyrosine residues in response to BCR. GTF2I then translocates to the nucleus to bind regulatory enhancer elements to modulate gene expression. ARID3A and NFAT are other transcriptional target of BTK. BTK is required for the formation of functional ARID3A DNA-binding complexes. There is however no evidence that BTK itself binds directly to DNA. BTK has a dual role in the regulation of apoptosis. Non-receptor tyrosine kinase indispensable for B lymphocyte development, differentiation and signaling. Related diseases X-linked agammaglobulinemia (XLA) [MIM:300755]: Humoral immunodeficiency disease which results in developmental defects in the maturation pathway of B-cells. Affected boys have normal levels of pre-B-cells in their bone marrow but virtually no circulating mature B-lymphocytes. This results in a lack of immunoglobulins of all classes and leads to recurrent bacterial infections like otitis, conjunctivitis, dermatitis, sinusitis in the first few years of life, or even some patients present overwhelming sepsis or meningitis, resulting in death in a few hours. Treatment in most cases is by infusion of intravenous immunoglobulin. {ECO:0000269|PubMed:10220140, ECO:0000269|PubMed:10612838, ECO:0000269|PubMed:10678660, ECO:0000269|PubMed:7627183, ECO:0000269|PubMed:7633420, ECO:0000269|PubMed:7633429, ECO:0000269|PubMed:7711734, ECO:0000269|PubMed:7809124, ECO:0000269|PubMed:7849006, ECO:0000269|PubMed:7849697, ECO:0000269|PubMed:7849721, ECO:0000269|PubMed:7880320, ECO:0000269|PubMed:7897635, ECO:0000269|PubMed:8013627, ECO:0000269|PubMed:8162018, ECO:0000269|PubMed:8162056, ECO:0000269|PubMed:8594569, ECO:0000269|PubMed:8634718, ECO:0000269|PubMed:8695804, ECO:0000269|PubMed:8723128, ECO:0000269|PubMed:8834236, ECO:0000269|PubMed:9016530, ECO:0000269|PubMed:9260159, ECO:0000269|PubMed:9280283, ECO:0000269|PubMed:9445504, ECO:0000269|PubMed:9545398}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 3, with agammaglobulinemia (IGHD3) [MIM:307200]: An X-linked recessive disorder characterized by growth hormone deficiency, short stature, delayed bone age, agammaglobulinemia with markedly reduced numbers of B cells, and good response to treatment with growth hormone. {ECO:0000269|PubMed:8013627}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15327; DB11703; DB15347; DB01254; DB15170; DB14785; DB12010; DB09053; DB01863; DB17472; DB14924; DB11764; DB15227; DB16657; DB05204; DB15035 Interacts with Q13444; Q99856; Q8WV28; Q06187; P78347; P08238; Q9BVA0; P21145; P50222; Q04759; O60239; P42768 EC number EC 2.7.10.2 Uniprot keywords 3D-structure; Acetylation; Adaptive immunity; Alternative promoter usage; Apoptosis; ATP-binding; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Dwarfism; Immunity; Innate immunity; Kinase; Lipid-binding; Membrane; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; SH2 domain; SH3 domain; Transcription; Transcription regulation; Transferase; Tyrosine-protein kinase; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 30491.7 Length 263 Aromaticity 0.12 Instability index 45.93 Isoelectric point 5.39 Charge (pH=7) -7.65 2D Binding mode Binding energy (Kcal/mol) -7.81 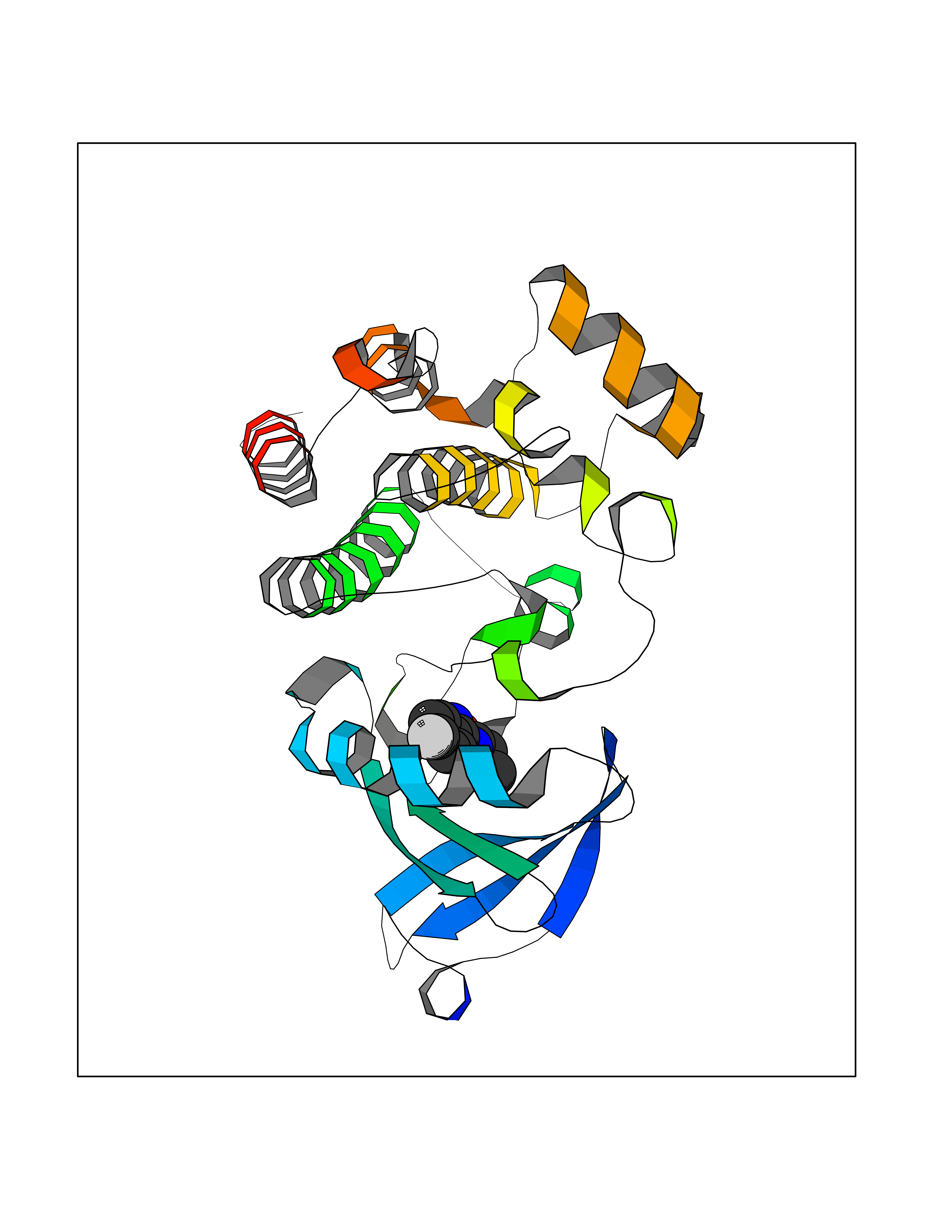 Molscript Map 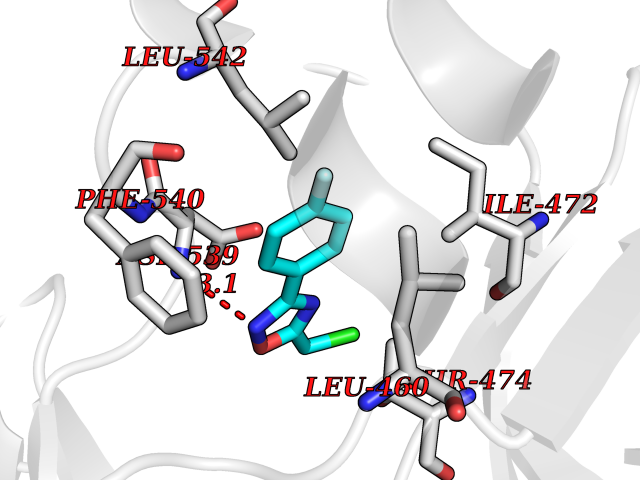 Pymol Map 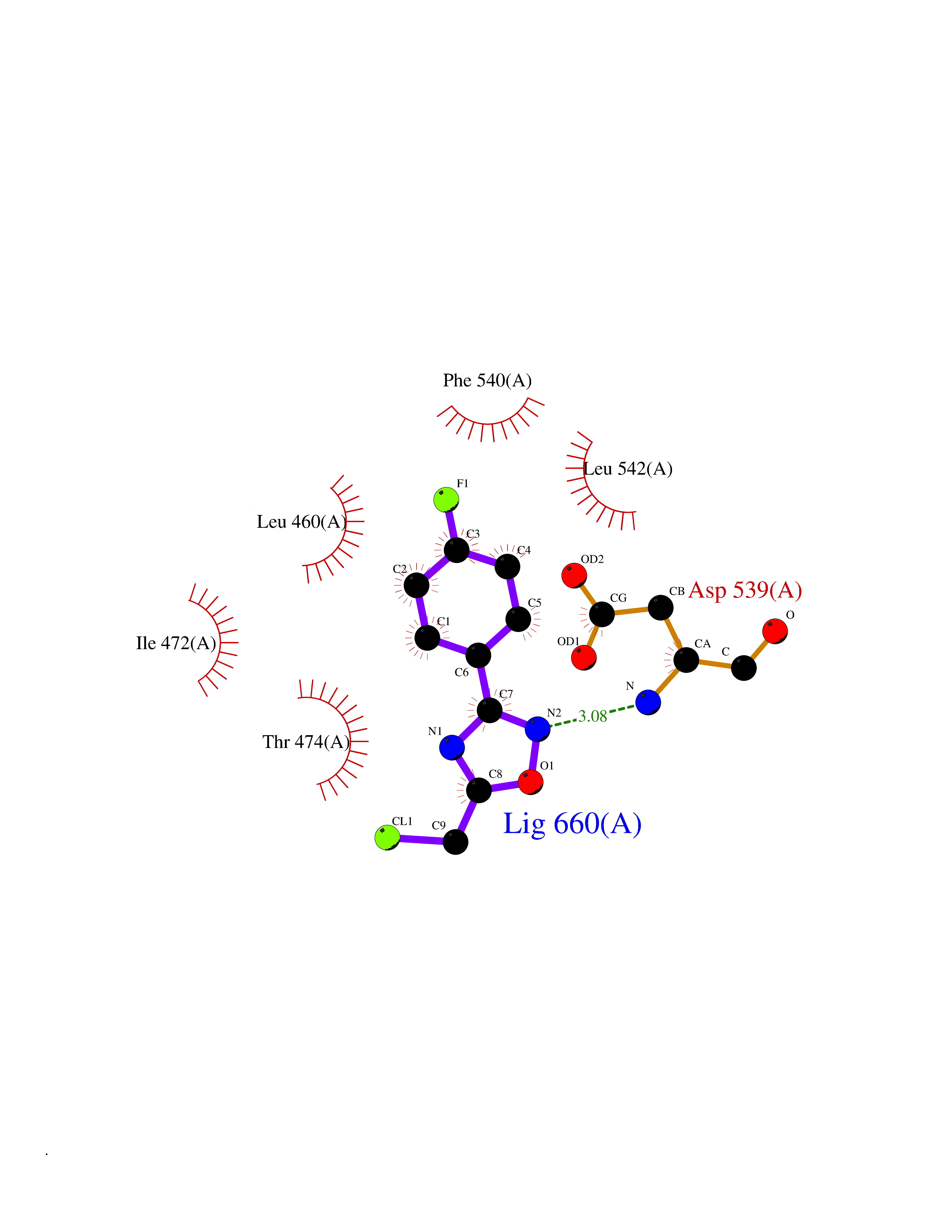 Ligplot Map 3D Binding mode Sequence EIDPKDLTFLKELGTGQFGVVKYGKWRGQYDVAIKMIKEGSMSEDEFIEEAKVMMNLSHEKLVQLYGVCTKQRPIFIITEYMANGCLLNYLREARHAFQTQQLLEMCKDVCEAMEYLESKQFLHRDLAARNCLVNDQGVVKVSDFGLSRYVLDDEYTSSVGSKFPVRWSPPEVLMYSKFSSKSDIWAFGVLMWEIYSLGKMPYERFTNSETAEHIAQGLRLYRPHLASAAVYTIMYSCWHEKADERPTFKILLSNILDVMDEE Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Peptostreptococcal albumin-binding protein | 2VDB | 5.72 | |
Target general information Gen name pab Organism Finegoldia magna (Peptostreptococcus magnus) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Protein binding Function Binds serum albumin. Related diseases LTC4 synthase deficiency is associated with a neurometabolic developmental disorder characterized by muscular hypotonia, psychomotor retardation, failure to thrive, and microcephaly. {ECO:0000269|PubMed:10896305, ECO:0000269|PubMed:9820300}. Drugs (DrugBank ID) DB03600; DB00788 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell wall; Peptidoglycan-anchor; Secreted; Signal Protein physicochemical properties Chain ID B Molecular weight (Da) 21751.6 Length 189 Aromaticity 0.1 Instability index 49.17 Isoelectric point 5.44 Charge (pH=7) -6.7 2D Binding mode Binding energy (Kcal/mol) -7.8 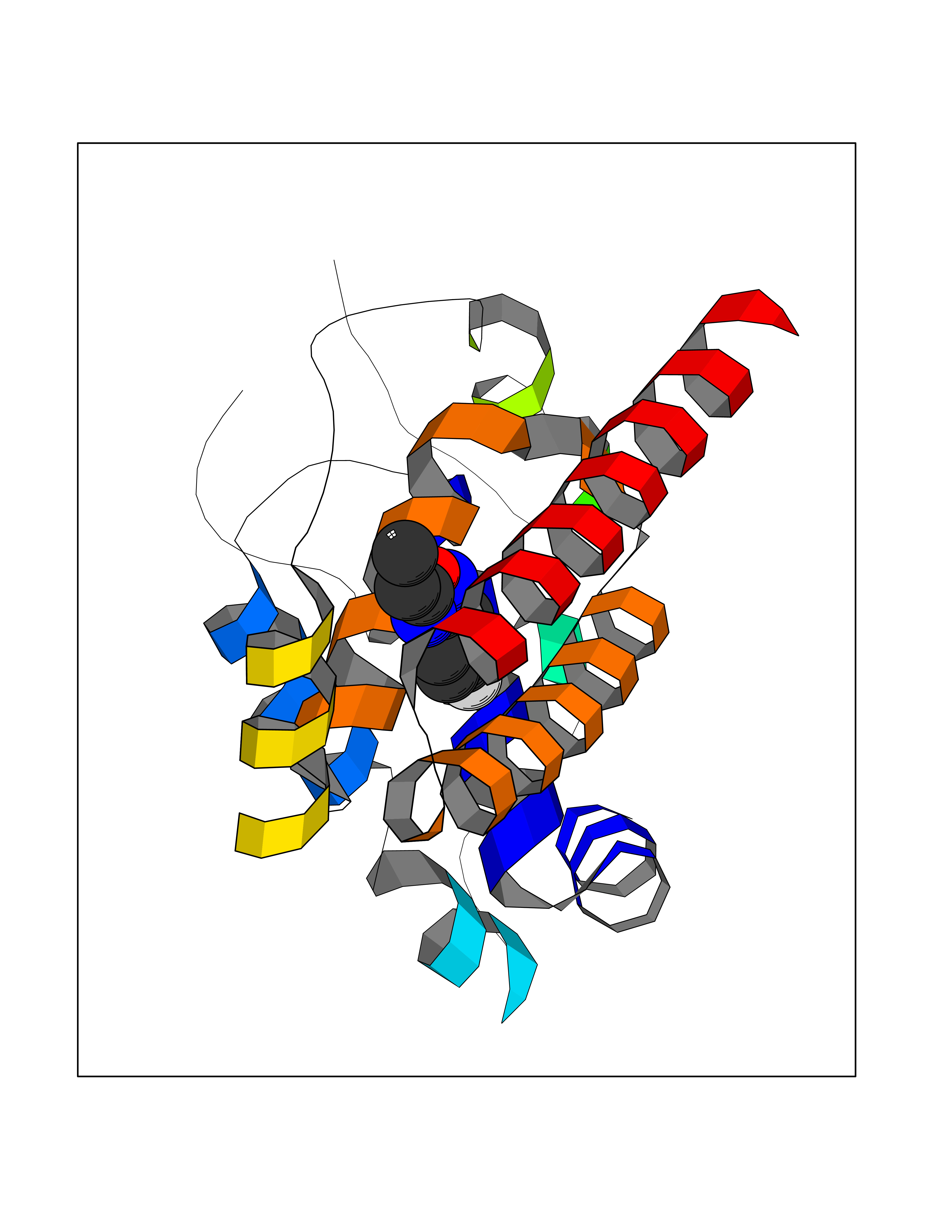 Molscript Map 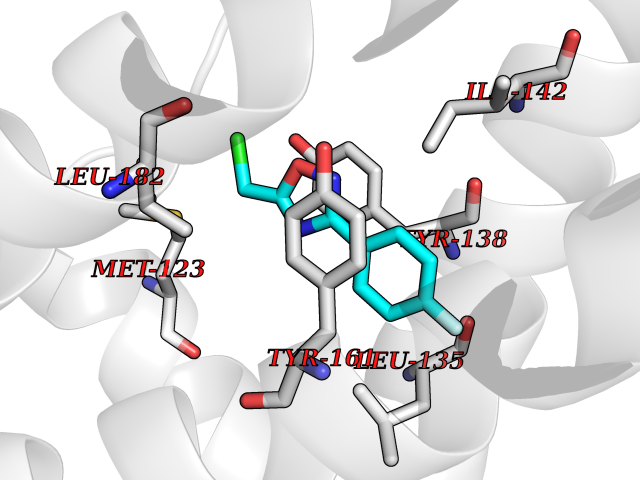 Pymol Map 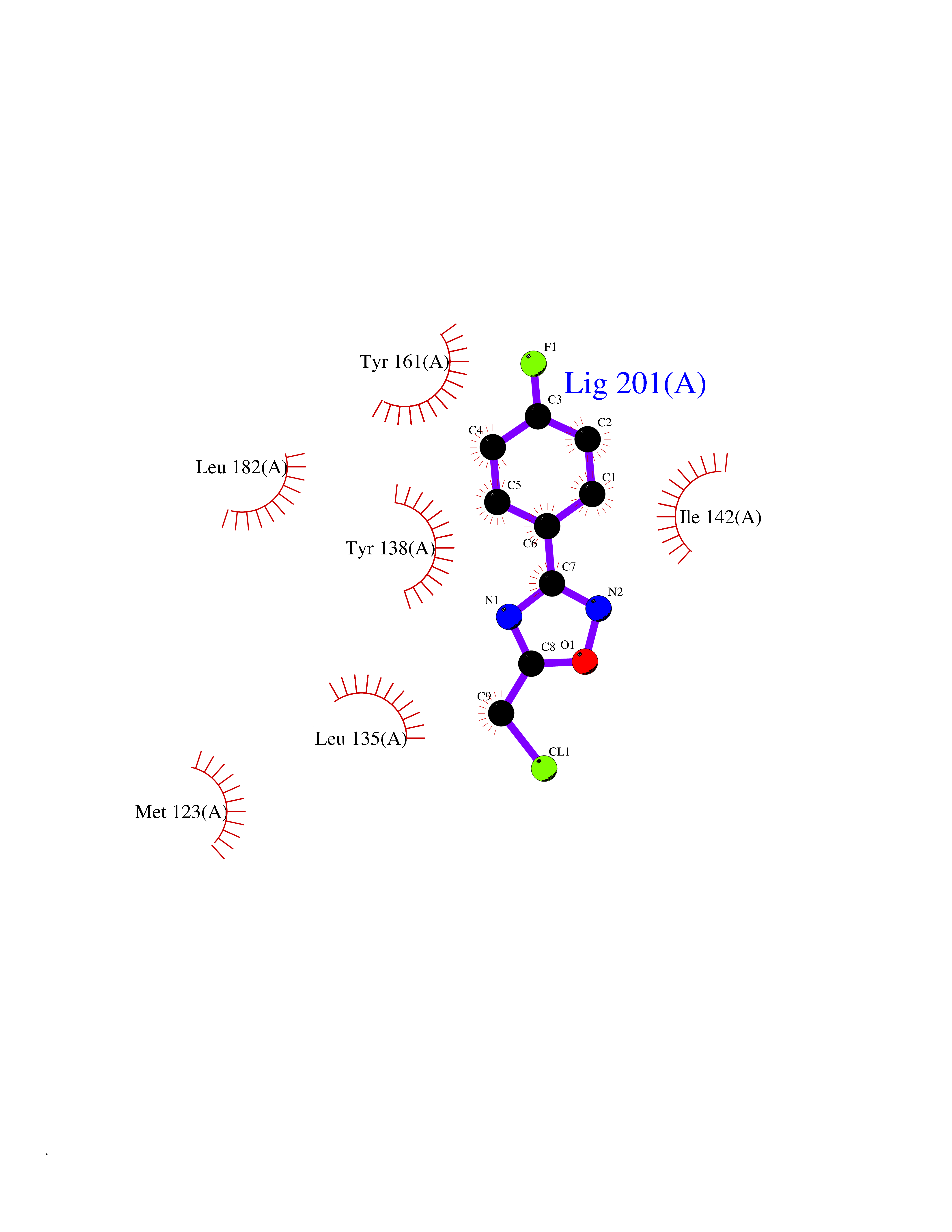 Ligplot Map 3D Binding mode Sequence EVAHRFKDLGEENFKALVLIAFAQYLQQCPFEDHVKLVNEVTEFAKTCVADESAENCDKSLHTLFGDKLCTVATLEMADCCAKQEPERNECFLQHKDDNPNLPRLVRPEVDVMCTAFHDNEETFLKKYLYEIARRHPYFYAPELLFFAKRYKAAFTECCQAADKAACLLPKLDELRDEGKASSAKQRLK Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Histone-lysine N-methyltransferase KMT5B (KMT5B) | 3S8P | 5.72 | |
Target general information Gen name KMT5B Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine N-methyltransferase 5B; Lysine-specific methyltransferase 5B; Suppressor of variegation 4-20 homolog 1; Su(var)4-20 homolog 1; Suv4-20h1; [histone H4]-N-methyl-L-lysine20 N-methyltransferase KM Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, Suvar4-20 subfamily Biochemical class NA Function Histone methyltransferase that specifically methylates monomethylated 'Lys-20' (H4K20me1) and dimethylated 'Lys-20' (H4K20me2) of histone H4 to produce respectively dimethylated 'Lys-20' (H4K20me2) and trimethylated 'Lys-20' (H4K20me3) and thus regulates transcription and maintenance of genome integrity. In vitro also methylates unmodified 'Lys-20' (H4K20me0) of histone H4 and nucleosomes. H4 'Lys-20' trimethylation represents a specific tag for epigenetic transcriptional repression. Mainly functions in pericentric heterochromatin regions, thereby playing a central role in the establishment of constitutive heterochromatin in these regions. KMT5B is targeted to histone H3 via its interaction with RB1 family proteins (RB1, RBL1 and RBL2) (By similarity). Plays a role in myogenesis by regulating the expression of target genes, such as EID3. Facilitates TP53BP1 foci formation upon DNA damage and proficient non-homologous end-joining (NHEJ)-directed DNA repair by catalyzing the di- and trimethylation of 'Lys-20' of histone H4. May play a role in class switch reconbination by catalyzing the di- and trimethylation of 'Lys-20' of histone H4 (By similarity). Related diseases Intellectual developmental disorder, autosomal dominant 51 (MRD51) [MIM:617788]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. {ECO:0000269|PubMed:28191889, ECO:0000269|PubMed:29276005}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9H2G4; Q61026 EC number EC 2.1.1.361 Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Chromosome; Disease variant; Intellectual disability; Isopeptide bond; Metal-binding; Methyltransferase; Myogenesis; Nucleus; Proteomics identification; Reference proteome; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 26230.2 Length 233 Aromaticity 0.11 Instability index 42.29 Isoelectric point 5.64 Charge (pH=7) -6.07 2D Binding mode Binding energy (Kcal/mol) -7.8  Molscript Map  Pymol Map 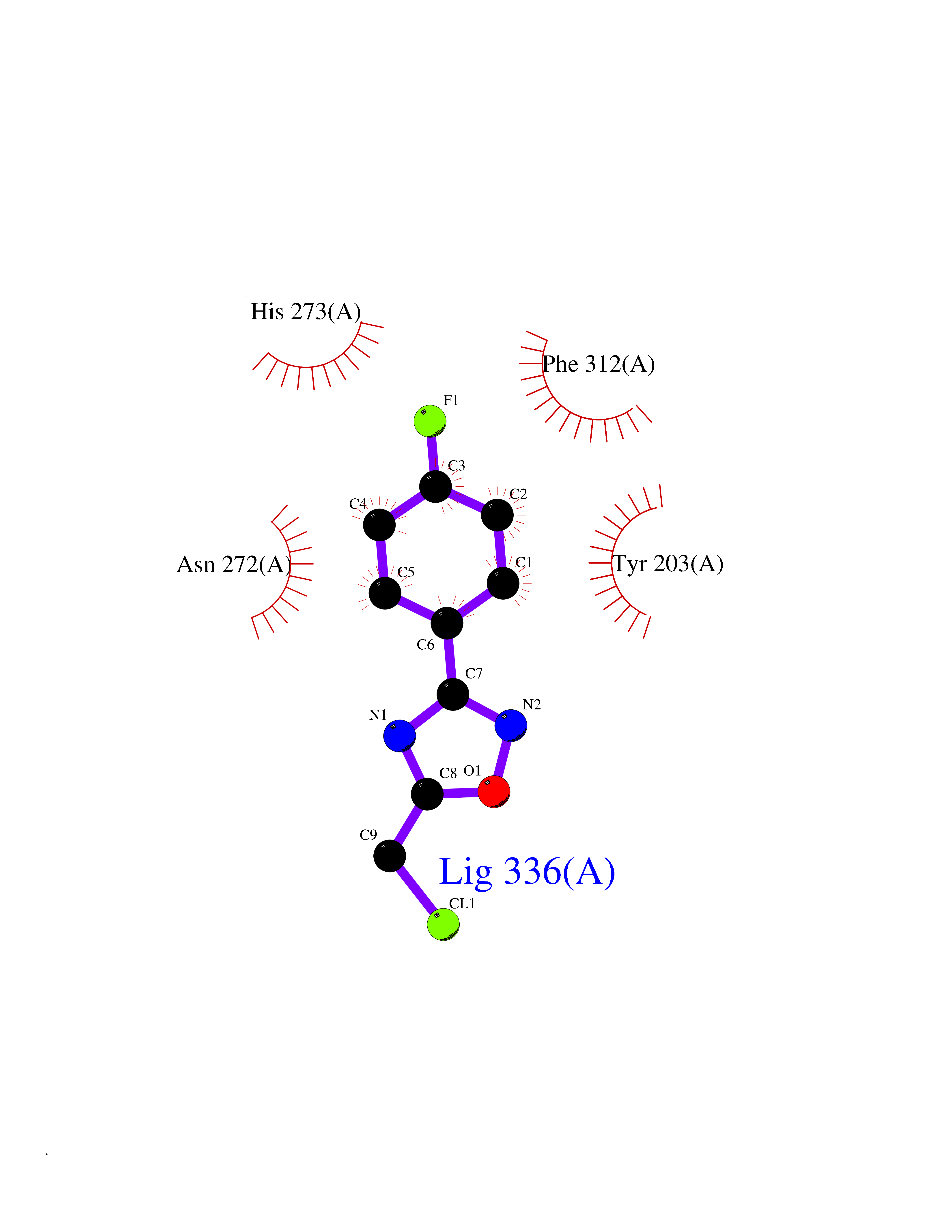 Ligplot Map 3D Binding mode Sequence XSAKELCENDDLATSLVLDPYLGFQTHKXNTRQEELKEVIERFKKDEHLEKAFKCLTSGEWARHYFLNKNKXQEKLFKEHVFIYLRXFATDSGFEILPCNRYSSEQNGAKIVATKEWKRNDKIELLVGCIAELSEIEENXLLRHGENDFSVXYSTRKNCAQLWLGPAAFINHDCRPNCKFVSTGRDTACVKALRDIEPGEEISCYYGDGFFGENNEFCECYTCERRGTGAFKS Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Matrix metalloproteinase-13 (MMP-13) | 2OW9 | 5.72 | |
Target general information Gen name MMP13 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Matrix metalloproteinase 13; Collagenase-3; Collagenase 3 Protein family Peptidase M10A family Biochemical class Peptidase Function Cleaves triple helical collagens, including type I, type II and type III collagen, but has the highest activity with soluble type II collagen. Can also degrade collagen type IV, type XIV and type X. May also function by activating or degrading key regulatory proteins, such as TGFB1 and CTGF. Plays a role in wound healing, tissue remodeling, cartilage degradation, bone development, bone mineralization and ossification. Required for normal embryonic bone development and ossification. Plays a role in the healing of bone fractures via endochondral ossification. Plays a role in wound healing, probably by a mechanism that involves proteolytic activation of TGFB1 and degradation of CTGF. Plays a role in keratinocyte migration during wound healing. May play a role in cell migration and in tumor cell invasion. Plays a role in the degradation of extracellular matrix proteins including fibrillar collagen, fibronectin, TNC and ACAN. Related diseases Spondyloepimetaphyseal dysplasia, Missouri type (SEMDM) [MIM:602111]: A bone disease characterized by moderate to severe metaphyseal changes, mild epiphyseal involvement, rhizomelic shortening of the lower limbs with bowing of the femora and/or tibiae, coxa vara, genu varum and pear-shaped vertebrae in childhood. Epimetaphyseal changes improve with age. {ECO:0000269|PubMed:16167086}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Metaphyseal anadysplasia 1 (MANDP1) [MIM:602111]: A bone development disorder characterized by skeletal anomalies that resolve spontaneously with age. Clinical characteristics are evident from the first months of life and include slight shortness of stature and a mild varus deformity of the legs. Patients attain a normal stature in adolescence and show improvement or complete resolution of varus deformity of the legs and rhizomelic micromelia. {ECO:0000269|PubMed:19615667}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Metaphyseal dysplasia, Spahr type (MDST) [MIM:250400]: An autosomal recessive, rare disease characterized by moderate short stature, mild genua vara, and radiographic signs of metaphyseal dysplasia, but no biochemical signs of rickets. {ECO:0000269|PubMed:24648384, ECO:0000269|PubMed:24781753}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02049; DB01996; DB03033; DB07827; DB08388; DB08561; DB08490; DB06423; DB02697; DB00786; DB04759; DB04760; DB04761; DB07013; DB02071 Interacts with NA EC number EC 3.4.24.- Uniprot keywords 3D-structure; Calcium; Collagen degradation; Direct protein sequencing; Disease variant; Disulfide bond; Dwarfism; Extracellular matrix; Glycoprotein; Hydrolase; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 18757.8 Length 167 Aromaticity 0.15 Instability index 9.97 Isoelectric point 5.47 Charge (pH=7) -7.44 2D Binding mode Binding energy (Kcal/mol) -7.8  Molscript Map 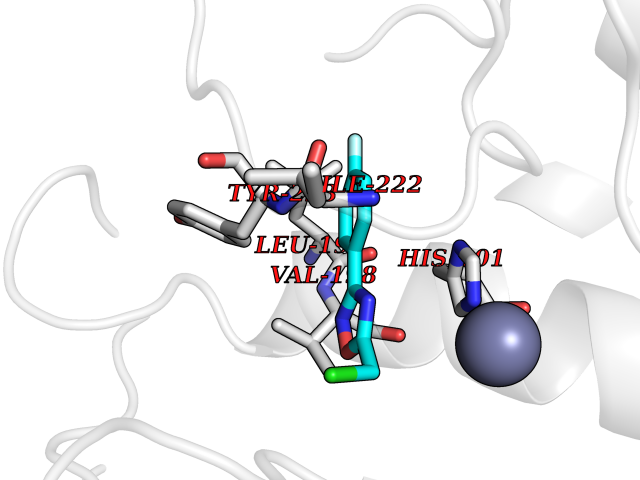 Pymol Map 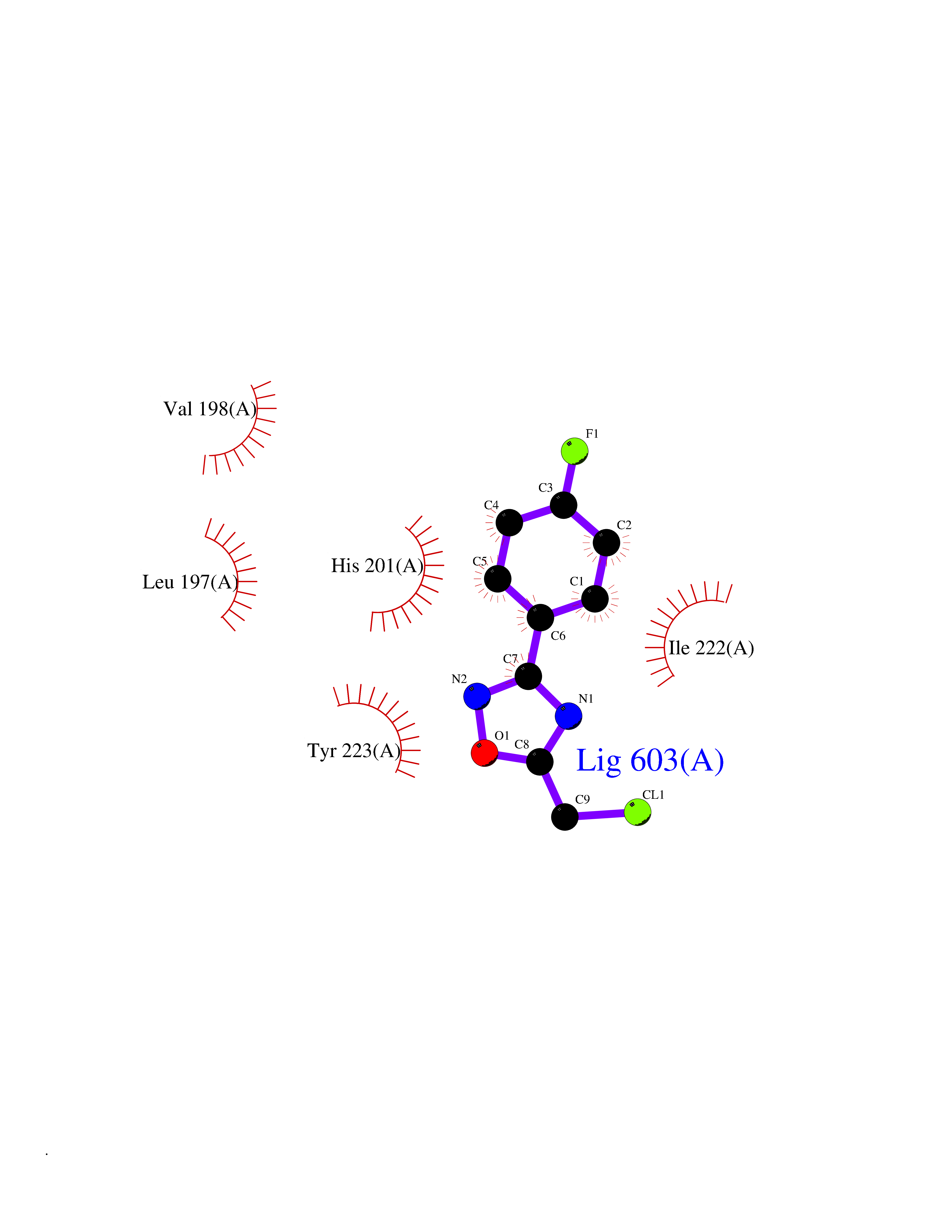 Ligplot Map 3D Binding mode Sequence YNVFPRTLKWSKMNLTYRIVNYTPDMTHSEVEKAFKKAFKVWSDVTPLNFTRLHDGIADIMISFGIKEHGDFYPFDGPSGLLAHAFPPGPNYGGDAHFDDDETWTSSSKGYNLFLVAAHEFGHSLGLDHSKDPGALMFPIYTYTGKSHFMLPDDDVQGIQSLYGPGD Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Retinoic acid receptor gamma (RARG) | 1FCY | 5.71 | |
Target general information Gen name RARG Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-gamma; Nuclear receptor subfamily 1 group B member 3; NR1B3 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Receptor for retinoic acid. Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RAR/RXR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence of ligand, acts mainly as an activator of gene expression due to weak binding to corepressors. Required for limb bud development. In concert with RARA or RARB, required for skeletal growth, matrix homeostasis and growth plate function (By similarity). Related diseases Cystic fibrosis (CF) [MIM:219700]: A common generalized disorder of the exocrine glands which impairs clearance of secretions in a variety of organs. It is characterized by the triad of chronic bronchopulmonary disease (with recurrent respiratory infections), pancreatic insufficiency (which leads to malabsorption and growth retardation) and elevated sweat electrolytes. It is the most common genetic disease in Caucasians, with a prevalence of about 1 in 2'000 live births. Inheritance is autosomal recessive. {ECO:0000269|PubMed:10094564, ECO:0000269|PubMed:10869121, ECO:0000269|PubMed:10923036, ECO:0000269|PubMed:11242048, ECO:0000269|PubMed:12167682, ECO:0000269|PubMed:12394343, ECO:0000269|PubMed:12529365, ECO:0000269|PubMed:1284466, ECO:0000269|PubMed:1284468, ECO:0000269|PubMed:1284529, ECO:0000269|PubMed:1284530, ECO:0000269|PubMed:1284548, ECO:0000269|PubMed:1379210, ECO:0000269|PubMed:15528182, ECO:0000269|PubMed:15716351, ECO:0000269|PubMed:16822950, ECO:0000269|PubMed:1695717, ECO:0000269|PubMed:1699669, ECO:0000269|PubMed:17098864, ECO:0000269|PubMed:1710600, ECO:0000269|PubMed:1712898, ECO:0000269|PubMed:17182731, ECO:0000269|PubMed:20008117, ECO:0000269|PubMed:20150177, ECO:0000269|PubMed:20691141, ECO:0000269|PubMed:21884936, ECO:0000269|PubMed:2236053, ECO:0000269|PubMed:23818989, ECO:0000269|PubMed:25330774, ECO:0000269|PubMed:26846474, ECO:0000269|PubMed:27241308, ECO:0000269|PubMed:28001373, ECO:0000269|PubMed:28067262, ECO:0000269|PubMed:28087700, ECO:0000269|PubMed:32026723, ECO:0000269|PubMed:33572515, ECO:0000269|PubMed:7504969, ECO:0000269|PubMed:7505694, ECO:0000269|PubMed:7505767, ECO:0000269|PubMed:7508414, ECO:0000269|PubMed:7513296, ECO:0000269|PubMed:7517264, ECO:0000269|PubMed:7520022, ECO:0000269|PubMed:7522211, ECO:0000269|PubMed:7524909, ECO:0000269|PubMed:7524913, ECO:0000269|PubMed:7525450, ECO:0000269|PubMed:7537150, ECO:0000269|PubMed:7541273, ECO:0000269|PubMed:7541510, ECO:0000269|PubMed:7543567, ECO:0000269|PubMed:7544319, ECO:0000269|PubMed:7581407, ECO:0000269|PubMed:7606851, ECO:0000269|PubMed:7680525, ECO:0000269|PubMed:7683628, ECO:0000269|PubMed:7683954, ECO:0000269|PubMed:8081395, ECO:0000269|PubMed:8406518, ECO:0000269|PubMed:8522333, ECO:0000269|PubMed:8723693, ECO:0000269|PubMed:8723695, ECO:0000269|PubMed:8800923, ECO:0000269|PubMed:8829633, ECO:0000269|PubMed:8910473, ECO:0000269|PubMed:8956039, ECO:0000269|PubMed:9101301, ECO:0000269|PubMed:9222768, ECO:0000269|PubMed:9375855, ECO:0000269|PubMed:9401006, ECO:0000269|PubMed:9443874, ECO:0000269|PubMed:9452048, ECO:0000269|PubMed:9452054, ECO:0000269|PubMed:9452073, ECO:0000269|PubMed:9482579, ECO:0000269|PubMed:9507391, ECO:0000269|PubMed:9521595, ECO:0000269|PubMed:9554753, ECO:0000269|PubMed:9736778, ECO:0000269|PubMed:9804160, ECO:0000269|PubMed:9921909}. The disease is caused by variants affecting the gene represented in this entry. There is some evidence that the functional defect caused by the most common variant Phe-508 DEL can be corrected by the binding to the snake phospholipase A2 crotoxin basic subunit CB. This toxin both disrupts the Phe-508 DEL-cytokeratin 8 complex, allowing for the escape from degradation, and increases the chloride channel current (PubMed:27241308). {ECO:0000269|PubMed:27241308}.; DISEASE: Congenital bilateral absence of the vas deferens (CBAVD) [MIM:277180]: An autosomal recessive disease characterized by vas deferens aplasia resulting in azoospermia and male infertility. CBAVD may occur in isolation or as a manifestation of cystic fibrosis. {ECO:0000269|PubMed:10066035, ECO:0000269|PubMed:10651488, ECO:0000269|PubMed:17329263, ECO:0000269|PubMed:7529962, ECO:0000269|PubMed:7539342, ECO:0000269|PubMed:9067761, ECO:0000269|PubMed:9736778, ECO:0000269|Ref.117}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07294; DB07031; DB00459; DB00210; DB00523; DB02466; DB03466; DB02741; DB03279; DB00926; DB00982; DB05785; DB05467; DB02258; DB00799; DB00755; DB12808 Interacts with Q96RK4; P13349; P31321; P28702; P48443; O60504-2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; DNA-binding; Isopeptide bond; Metal-binding; Methylation; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 26574.9 Length 236 Aromaticity 0.06 Instability index 49.98 Isoelectric point 5.76 Charge (pH=7) -2.95 2D Binding mode Binding energy (Kcal/mol) -7.78 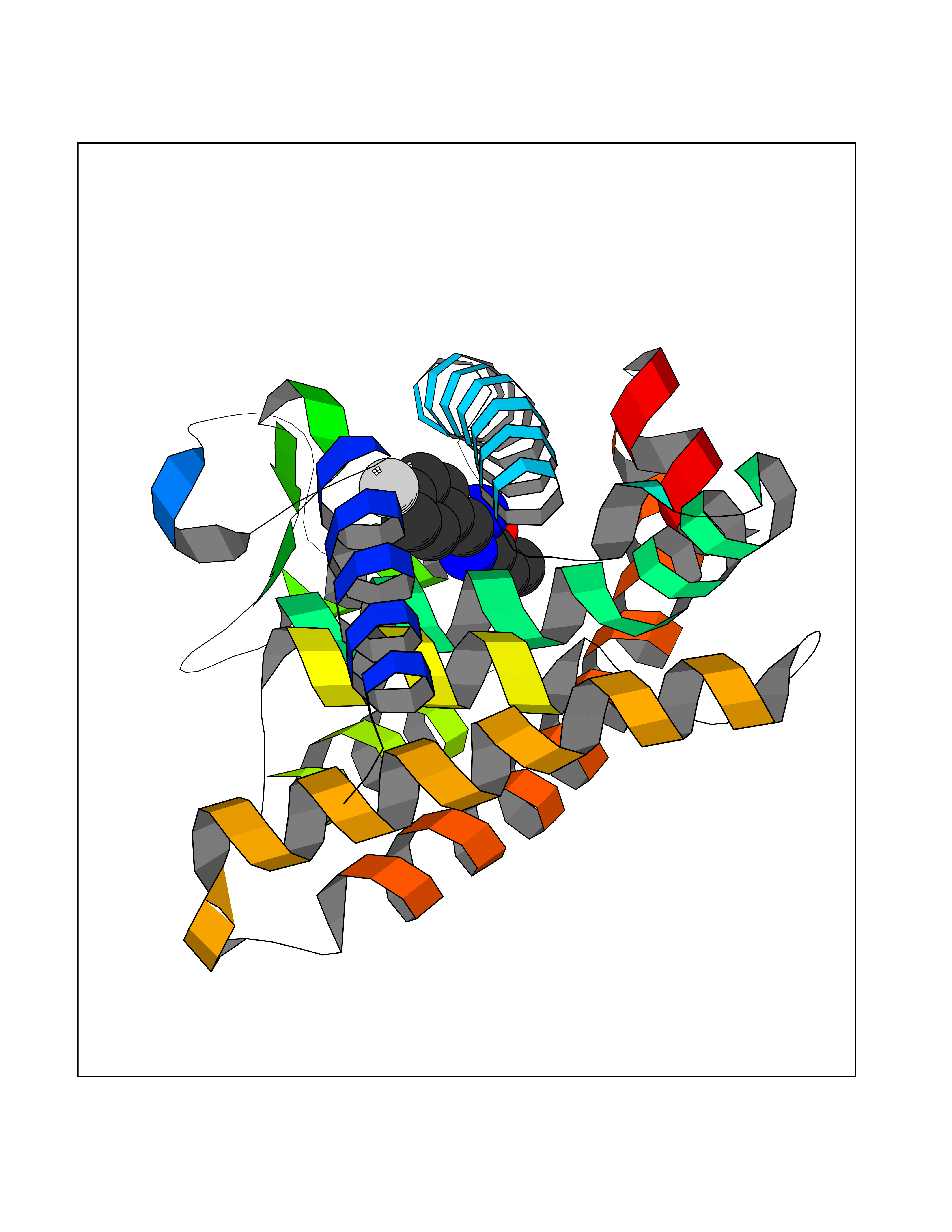 Molscript Map 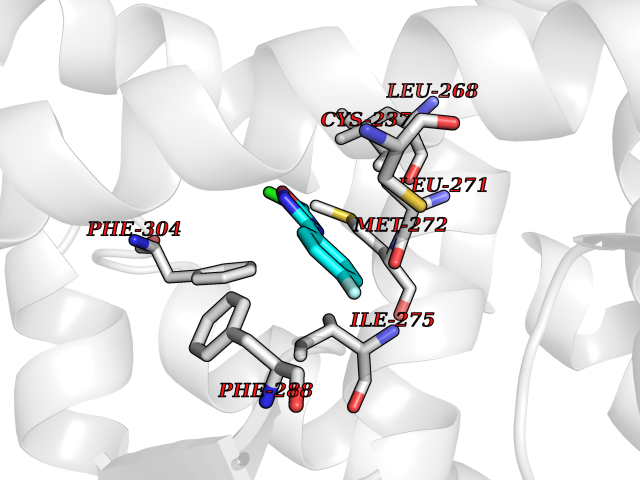 Pymol Map 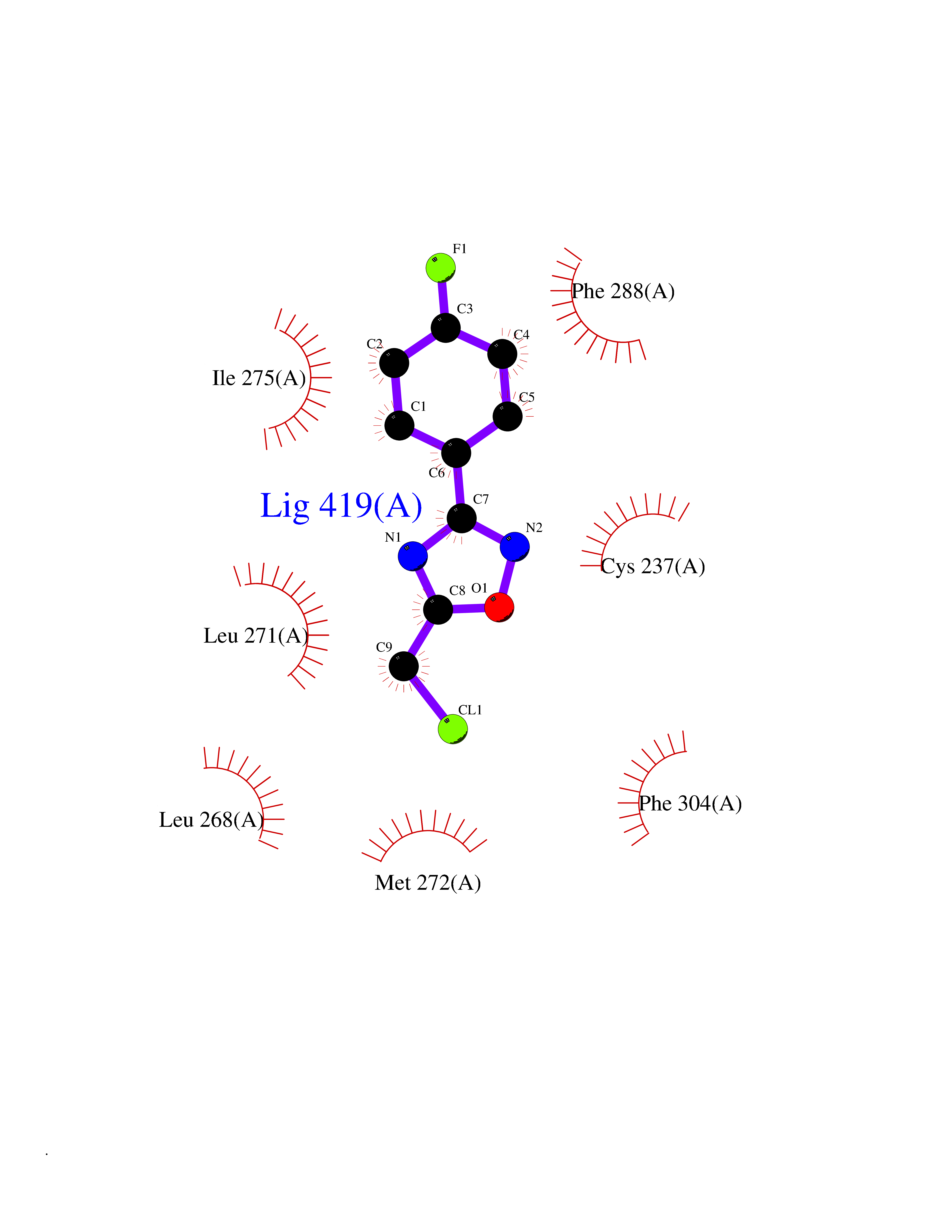 Ligplot Map 3D Binding mode Sequence ASPQLEELITKVSKAHQETFPSLCQLGKYTTNSSADHRVQLDLGLWDKFSELATKCIIKIVEFAKRLPGFTGLSIADQITLLKAACLDILMLRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFAFAGQLLPLEMDDTETGLLSAICLICGDRMDLEEPEKVDKLQEPLLEALRLYARRRRPSQPYMFPRMLMKITDLRGISTKGAERAITLKMEIPGPMPPLIREMLE Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Retinoic acid receptor beta (RARB) | 4DM6 | 5.71 | |
Target general information Gen name RARB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-epsilon; RAR-beta; Nuclear receptor subfamily 1 group B member 2; NR1B2; HBV-activated protein; HAP Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RXR/RAR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence or presence of hormone ligand, acts mainly as an activator of gene expression due to weak binding to corepressors. In concert with RARG, required for skeletal growth, matrix homeostasis and growth plate function. Receptor for retinoic acid. Related diseases Microphthalmia, syndromic, 12 (MCOPS12) [MIM:615524]: A form of microphthalmia, a disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues (anophthalmia). In many cases, microphthalmia/anophthalmia occurs in association with syndromes that include non-ocular abnormalities. MCOPS12 patients manifest variable features, including diaphragmatic hernia, pulmonary hypoplasia, and cardiac abnormalities. {ECO:0000269|PubMed:24075189, ECO:0000269|PubMed:27120018}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00459; DB00210; DB00523; DB02877; DB00926; DB05785; DB04942; DB00799; DB00755; DB12808 Interacts with O95273; P50222; Q9UBK2; P62195; P28702; P28702-3; P48443; P03255 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Microphthalmia; Nucleus; Phosphoprotein; Proto-oncogene; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 25904.1 Length 229 Aromaticity 0.06 Instability index 44.34 Isoelectric point 7.55 Charge (pH=7) 0.73 2D Binding mode Binding energy (Kcal/mol) -7.79  Molscript Map 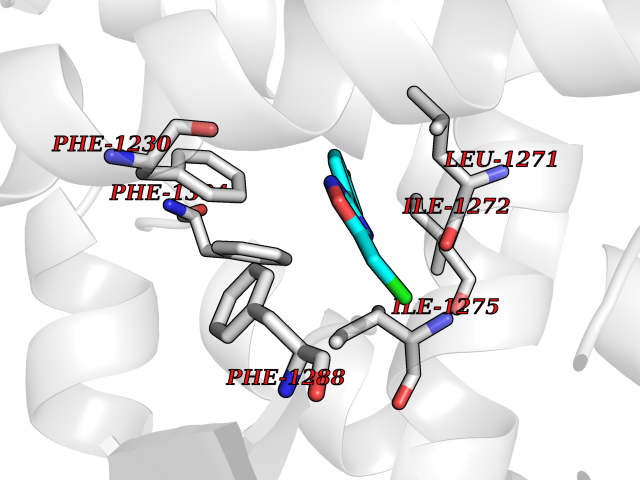 Pymol Map 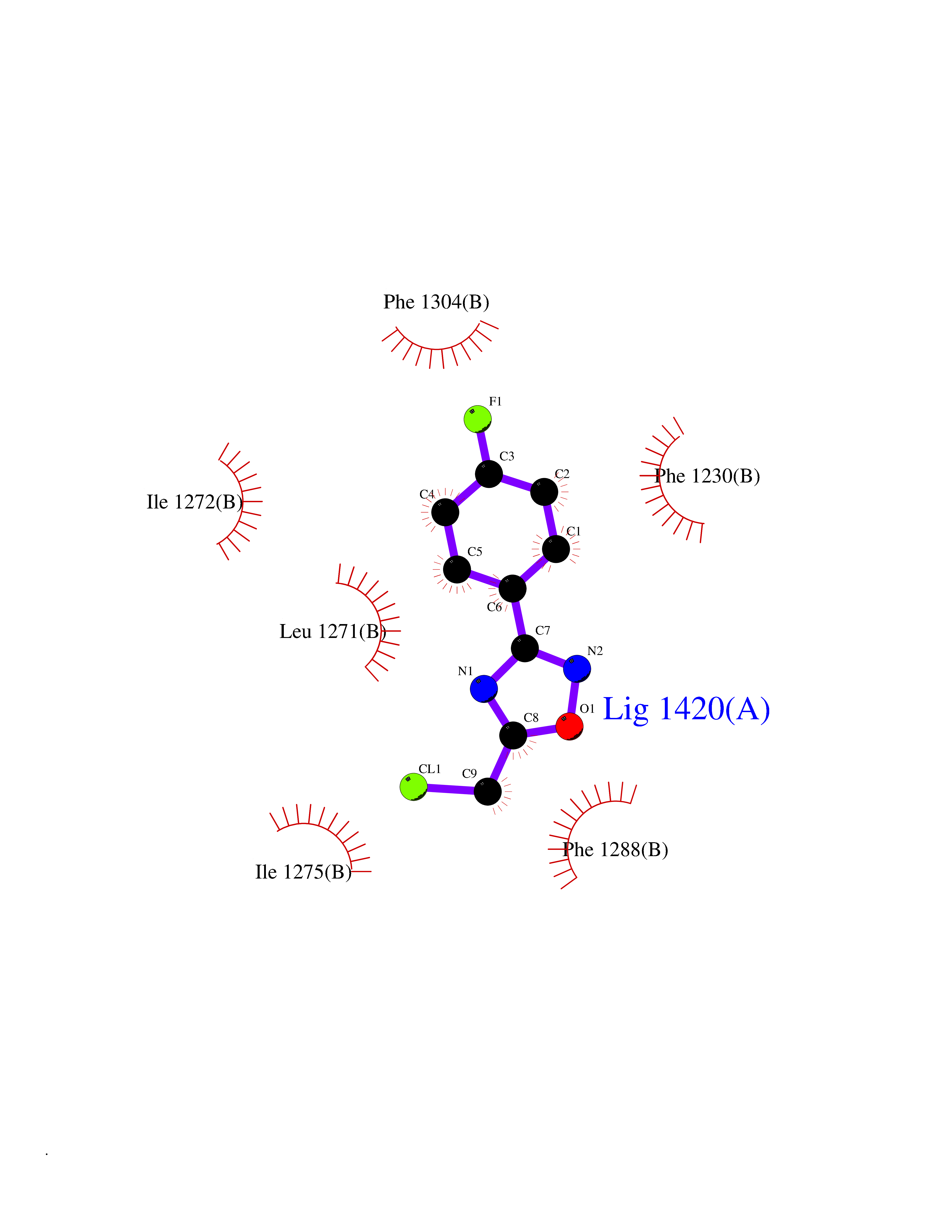 Ligplot Map 3D Binding mode Sequence TEKIRKAHQETFPSLCQLGKYTTNSSADHRVRLDLGLWDKFSELATKCIIKIVEFAKRLPGFTGLTIADQITLLKAACLDILILRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFTFANQLLPLEMDDTETGLLSAICLICGDRQDLEEPTKVDKLQEPLLEALKIYIRKRRPSKPHMFPKILMKITDLRSISAKGAERVITLKMEIPGSMPPLIQEMLEN Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Leucine carboxyl methyltransferase 1 | 3IEI | 5.71 | |
Target general information Gen name LCMT1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CGI-68;LCMT Protein family Methyltransferase superfamily, LCMT family Biochemical class Transferase Function Protein C-terminal carboxyl O-methyltransferase activity.Protein C-terminal leucine carboxyl O-methyltransferase activity.S-adenosylmethionine-dependent methyltransferase activity. Related diseases Neurodevelopmental disorder, mitochondrial, with abnormal movements and lactic acidosis, with or without seizures (NEMMLAS) [MIM:617710]: An autosomal recessive, mitochondrial disorder with a broad phenotypic spectrum ranging from severe neonatal lactic acidosis, encephalomyopathy and early death to an attenuated course with milder manifestations. Clinical features include delayed psychomotor development, intellectual disability, hypotonia, dystonia, ataxia, and spasticity. Severe combined respiratory chain deficiency may be found in severely affected individuals. {ECO:0000269|PubMed:28236339, ECO:0000269|PubMed:28650581, ECO:0000269|PubMed:28905505, ECO:0000269|PubMed:30920170, ECO:0000269|PubMed:35074316}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Parkinsonism-dystonia 3, childhood-onset (PKDYS3) [MIM:619738]: An autosomal recessive neurodegenerative disorder with onset in infancy or early childhood. Affected individuals present with progressive movement abnormalities, including parkinsonism with tremor, dystonia, myoclonus ataxia, and hyperkinetic movements such as ballismus. The parkinsonism features may be responsive to treatment with levodopa, although many patients develop levodopa-induced dyskinesia. Some patients may have mild cognitive impairment or psychiatric disturbances. {ECO:0000269|PubMed:29120065, ECO:0000269|PubMed:31970218, ECO:0000269|PubMed:34890876}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00149 Interacts with P51116 EC number 2.1.1.233 Uniprot keywords 3D-structure; Alternative splicing; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H Molecular weight (Da) 35803 Length 310 Aromaticity 0.08 Instability index 42.77 Isoelectric point 6.13 Charge (pH=7) -3.58 2D Binding mode Binding energy (Kcal/mol) -7.79  Molscript Map 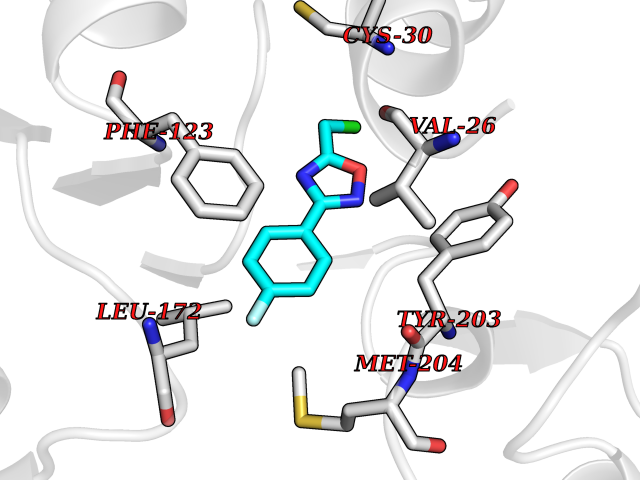 Pymol Map 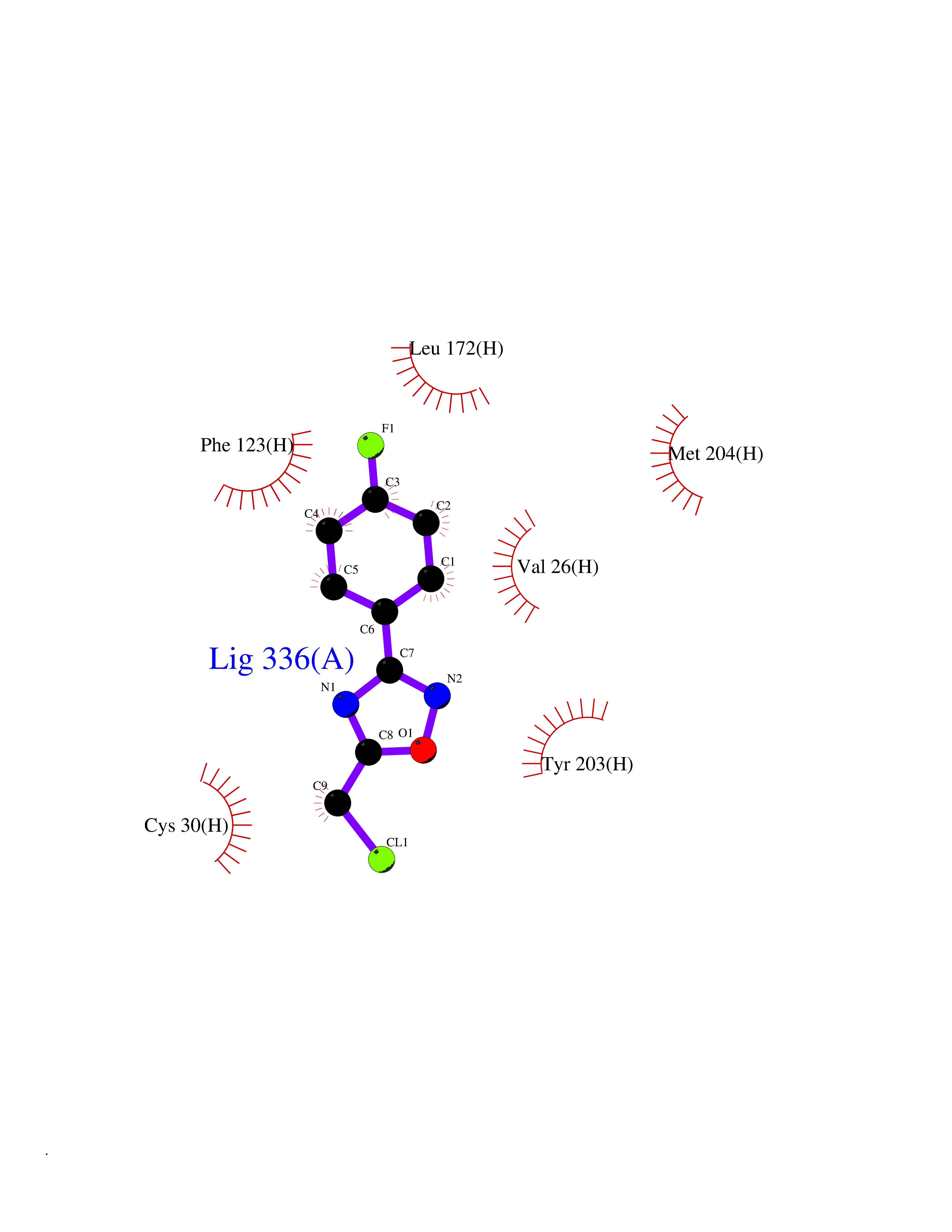 Ligplot Map 3D Binding mode Sequence GVRGTCEDASLCKRFAVSIGYWHDPYIQHFVRLSKERKAPEINRGYFARVHGVSQLIKAFLRKTECHCQIVNLGAGMDTTFWRLKDEDLLSSKYFEVDFPMIVTRKLHSIKCKPPLSSPILELHSEDTLQMDGHILDSKRYAVIGADLRDLSELEEKLKKCNMNTQLPTLLIAECVLVYMTPEQSANLLKWAANSFERAMFINYEQVNMGDRFGQIMIENLRRRQCDLAGVETCKSLESQKERLLSNGWETASAVDMMELYNRLPRAEVSRIESLEFLDEMELLEQLMRHYCLCWATKGGNELGLKEITY Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Pseudomonas Methionine gamma-lyase (Pseudo mdeA) | 1PG8 | 5.71 | |
Target general information Gen name Pseudo mdeA Organism Pseudomonas putida (Arthrobacter siderocapsulatus) Uniprot ID TTD ID Synonyms Pseudo MGL; L-methionine gamma-lyase; L-methioninase; Homocysteine desulfhydrase Protein family Trans-sulfuration enzymes family, L-methionine gamma-lyase subfamily Biochemical class Carbon-sulfur lyases Function Catalyzes the alpha,gamma-elimination of L-methionine to produce methanethiol, 2-oxobutanoate and ammonia. Is involved in L-methionine catabolism. In fact, shows a multicatalytic function since it also catalyzes gamma-replacement of L-methionine with thiol compounds, alpha,gamma-elimination and gamma-replacement reactions of L-homocysteine and its S-substituted derivatives, O-substituted-L-homoserines and DL-selenomethionine, and, to a lesser extent, alpha,beta-elimination and beta-replacement reactions of L-cysteine, S-methyl-L-cysteine, and O-acetyl-L-serine. Also catalyzes deamination and gamma-addition reactions of L-vinylglycine. Thus, the enzyme is able to cleave C-S, C-Se, and C-O bonds of sulfur, selenium, and oxygen amino acids, respectively. Related diseases Lecithin-cholesterol acyltransferase deficiency (LCATD) [MIM:245900]: A disorder of lipoprotein metabolism characterized by inadequate esterification of plasmatic cholesterol. Two clinical forms are recognized: complete LCAT deficiency and fish-eye disease. LCATD is generally referred to the complete form which is associated with absence of both alpha and beta LCAT activities resulting in esterification anomalies involving both HDL (alpha-LCAT activity) and LDL (beta-LCAT activity). It causes a typical triad of diffuse corneal opacities, target cell hemolytic anemia, and proteinuria with renal failure. {ECO:0000269|PubMed:11423760, ECO:0000269|PubMed:12957688, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:16051254, ECO:0000269|PubMed:16216249, ECO:0000269|PubMed:1681161, ECO:0000269|PubMed:1859405, ECO:0000269|PubMed:2370048, ECO:0000269|PubMed:7607641, ECO:0000269|PubMed:7711728, ECO:0000269|PubMed:8318557, ECO:0000269|PubMed:8432868, ECO:0000269|PubMed:8807342, ECO:0000269|PubMed:9007616, ECO:0000269|PubMed:9741700}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fish-eye disease (FED) [MIM:136120]: A disorder of lipoprotein metabolism due to partial lecithin-cholesterol acyltransferase deficiency that affects only alpha-LCAT activity. FED is characterized by low plasma HDL and corneal opacities due to accumulation of cholesterol deposits in the cornea ('fish-eye'). {ECO:0000269|PubMed:1516702, ECO:0000269|PubMed:1571050, ECO:0000269|PubMed:15994445, ECO:0000269|PubMed:1737840, ECO:0000269|PubMed:21901787, ECO:0000269|PubMed:8620346, ECO:0000269|PubMed:9261271}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04083 Interacts with NA EC number EC 4.4.1.11 Uniprot keywords 3D-structure; Direct protein sequencing; Lyase; Pyridoxal phosphate Protein physicochemical properties Chain ID A,C Molecular weight (Da) 85234.4 Length 796 Aromaticity 0.07 Instability index 37.1 Isoelectric point 6.21 Charge (pH=7) -11.34 2D Binding mode Binding energy (Kcal/mol) -7.79 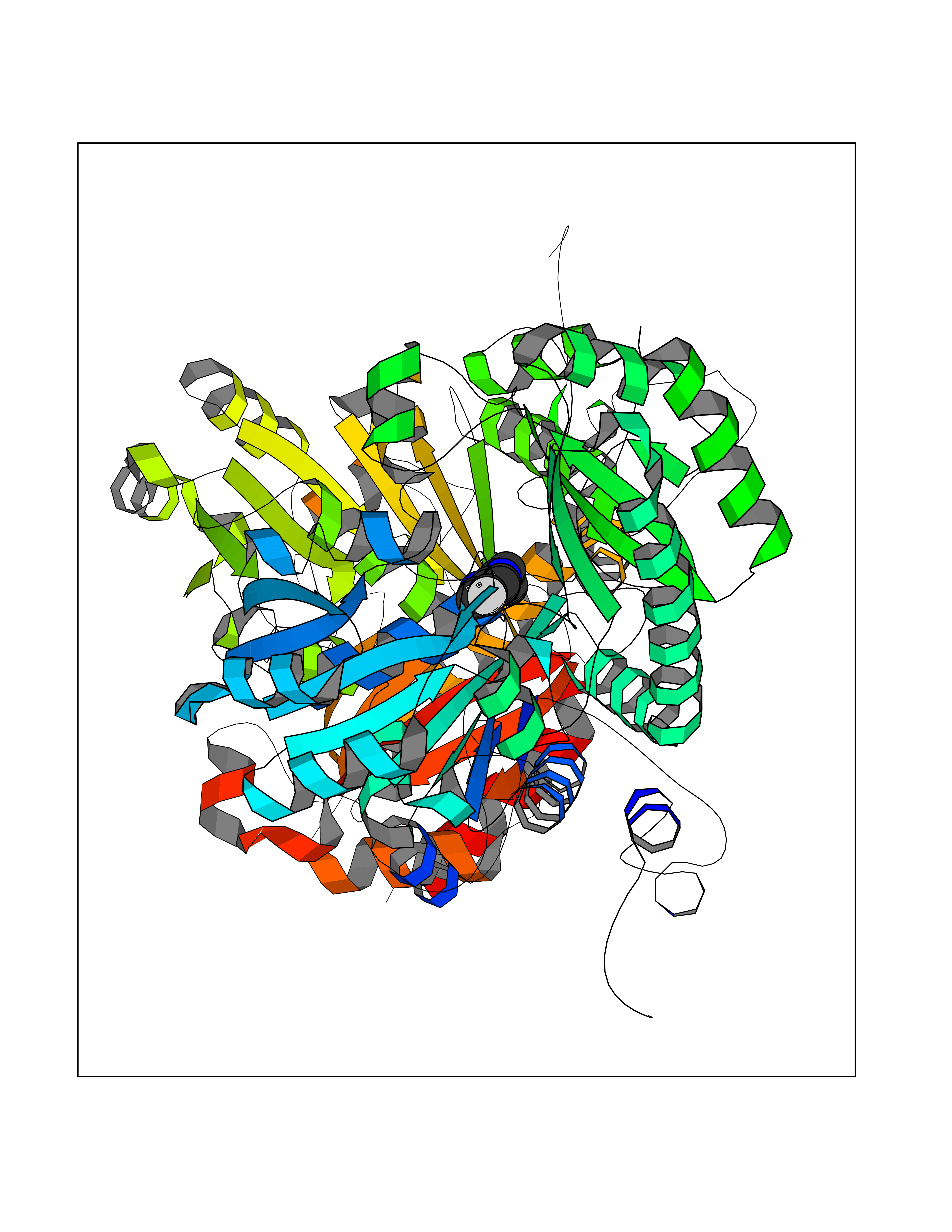 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MHGSNKLPGFATRAIHHGYDPQDHGGALVPPVYQTATFTFPTVEYGAACFAGEQAGHFYSRISNPTLNLLEARMASLEGGEAGLALASGMGAITSTLWTLLRPGDEVLLGNTLYGCTFAFLHHGIGEFGVKLRHVDMADLQALEAAMTPATRVIYFESPANPNMHMADIAGVAKIARKHGATVVVDNTYCTPYLQRPLELGADLVVHSATKYLSGHGDITAGIVVGSQALVDRIRLQGLKDMTGAVLSPHDAALLMRGIKTLNLRMDRHCANAQVLAEFLARQPQVELIHYPGLASFPQYTLARQQMSQPGGMIAFELKGGIGAGRRFMNALQLFSRAVSLGDAESLAQHPASMTHSSYTPEERAHYGISEGLVRLSVGLEDIDDLLADVQQALKASAMHGSNKLPGFATRAIHHGYDPQDHGGALVPPVYQTATFTFPTVEYGAACFAGEQAGHFYSRISNPTLNLLEARMASLEGGEAGLALASGMGAITSTLWTLLRPGDEVLLGNTLYGCTFAFLHHGIGEFGVKLRHVDMADLQALEAAMTPATRVIYFESPANPNMHMADIAGVAKIARKHGATVVVDNTYCTPYLQRPLELGADLVVHSATKYLSGHGDITAGIVVGSQALVDRIRLQGLKDMTGAVLSPHDAALLMRGIKTLNLRMDRHCANAQVLAEFLARQPQVELIHYPGLASFPQYTLARQQMSQPGGMIAFELKGGIGAGRRFMNALQLFSRAVSLGDAESLAQHPASMTHSSYTPEERAHYGISEGLVRLSVGLEDIDDLLADVQQALKASA Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Staphylococcus Enoyl ACP reductase (Stap-coc fabI) | 6YUR | 5.71 | |
Target general information Gen name Stap-coc fabI Organism Staphylococcus aureus (strain NCTC 8325 / PS 47) Uniprot ID TTD ID Synonyms NADPHdependent enoylACP reductase; Enoyl[acylcarrierprotein] reductase [NADPH] FabI Protein family Short-chain dehydrogenases/reductases (SDR) family, FabI subfamily Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the reduction of a carbon-carbon double bond in an enoyl moiety that is covalently linked to an acyl carrier protein (ACP). It has a preference for a long chain (C12) substrate compared to the shorter (C4) acyl group. Involved in the elongation cycle of fatty acid which are used in the lipid metabolism. Related diseases Amyloidosis, hereditary systemic 4, Finnish type (AMYLD4) [MIM:105120]: A form of hereditary systemic amyloidosis, a disorder characterized by amyloid deposition in multiple tissues resulting in a wide clinical spectrum. AMYLD4 is due to gelsolin amyloid deposition and is typically characterized by cranial neuropathy and lattice corneal dystrophy. Most patients have modest involvement of internal organs, but severe systemic disease can develop in some individuals causing peripheral polyneuropathy, amyloid cardiomyopathy, and nephrotic syndrome leading to renal failure. AMYLD4 is usually inherited in an autosomal dominant pattern. However, homozygotes with a more severe phenotype have also been reported. {ECO:0000269|PubMed:1338910, ECO:0000269|PubMed:19666512, ECO:0000269|PubMed:2176481}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; NAD; NADP; Oxidoreductase; Reference proteome Protein physicochemical properties Chain ID A,C Molecular weight (Da) 55674.6 Length 510 Aromaticity 0.07 Instability index 33.86 Isoelectric point 5.64 Charge (pH=7) -9.3 2D Binding mode Binding energy (Kcal/mol) -7.79  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence VNLENKTYVIMGIANKRSIAFGVAKVLDQLGAKLVFTYRKERSRKELEKLLEQLNQPEAHLYQIDVQSDEEVINGFEQIGKDVGNIDGVYHSIAFANMEDLRGRFSETSREGFLLAQDISSYSLTIVAHEAKKLMPEGGSIVATTYLGGEFAVQNYNVMGVAKASLEANVKYLALDLGPDNIRVNAISAGPIRTLSAKGVGGFNTILKEIEERAPLKRNVDQVEVGKTAAYLLSDLSSGVTGENIHVDSGFHAIKVNLENKTYVIMGIANKRSIAFGVAKVLDQLGAKLVFTYRKERSRKELEKLLEQLNQPEAHLYQIDVQSDEEVINGFEQIGKDVGNIDGVYHSIAFANMEDLRGRFSETSREGFLLAQDISSYSLTIVAHEAKKLMPEGGSIVATTYLGGEFAVQNYNVMGVAKASLEANVKYLALDLGPDNIRVNAISAGPIRTLSAKGVGGFNTILKEIEERAPLKRNVDQVEVGKTAAYLLSDLSSGVTGENIHVDSGFHAIK Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Euchromatic histone-lysine N-methyltransferase 1 (EHMT1) | 5TTG | 5.71 | |
Target general information Gen name EHMT1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms EHMT1 Protein family Class V-like SAM-binding methyltransferase superfamily Biochemical class NA Function Histone methyltransferase that specifically mono- and dimethylates 'Lys-9' of histone H3 (H3K9me1 and H3K9me2, respectively) in euchromatin. H3K9me represents a specific tag for epigenetic transcriptional repression by recruiting HP1 proteins to methylated histones. Also weakly methylates 'Lys-27' of histone H3 (H3K27me). Also required for DNA methylation, the histone methyltransferase activity is not required for DNA methylation, suggesting that these 2 activities function independently. Probably targeted to histone H3 by different DNA-binding proteins like E2F6, MGA, MAX and/or DP1. During G0 phase, it probably contributes to silencing of MYC- and E2F-responsive genes, suggesting a role in G0/G1 transition in cell cycle. In addition to the histone methyltransferase activity, also methylates non-histone proteins: mediates dimethylation of 'Lys-373' of p53/TP53. Related diseases Kleefstra syndrome 1 (KLEFS1) [MIM:610253]: A form of Kleefstra syndrome, an autosomal dominant disease characterized by variable intellectual disability, psychomotor developmental delay, seizures, behavioral abnormalities, and facial dysmorphisms. KLEFS1 patients additionally manifest brachy(micro)cephaly, congenital heart defects, and urogenital defects. {ECO:0000269|PubMed:16826528, ECO:0000269|PubMed:19264732}. The disease is caused by variants affecting the gene represented in this entry. The syndrome can be either caused by intragenic EHMT1 mutations leading to haploinsufficiency of the EHMT1 gene or by a submicroscopic 9q34.3 deletion. Although it is not known if and to what extent other genes in the 9q34.3 region contribute to the syndrome observed in deletion cases, EHMT1 seems to be the major determinant of the core disease phenotype (PubMed:19264732). {ECO:0000269|PubMed:16826528, ECO:0000269|PubMed:19264732}. Drugs (DrugBank ID) NA Interacts with Q99549; Q04206; Q04207 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ANK repeat; Chromatin regulator; Chromosome; Disease variant; Intellectual disability; Isopeptide bond; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; S-adenosyl-L-methionine; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 30066.9 Length 260 Aromaticity 0.11 Instability index 49.19 Isoelectric point 5.73 Charge (pH=7) -4.88 2D Binding mode Binding energy (Kcal/mol) -7.79  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence VERIVSRDIARGYERIPIPCVNAVDSEPCPSNYKYVSQNCVTSPMNIDRNITHLQYCVCIDDCSSSNCMCGQLSMRCWYDKDGRLLPEFNMAEPPLIFECNHACSCWRNCRNRVVQNGLRARLQLYRTRDMGWGVRSLQDIPPGTFVCEYVGELISDSEADVREEDSYLFDLDNDGEVYCIDARFYGNVSRFINHHCEPNLVPVRVFMAHQDLRFPRIAFFSTRLIEAGEQLGFDYGERFWDIKGKLFSCRCGSPKCRHS Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Histone-lysine N-methyltransferase EHMT2 (EHMT2) | 5VSC | 5.71 | |
Target general information Gen name EHMT2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein G9a; NG36; Lysine N-methyltransferase 1C; KMT1C; Histone H3-K9 methyltransferase 3; HLA-B-associated transcript 8; H3-K9-HMTase 3; G9A; Euchromatic histone-lysine N-methyltransferase 2; C6orf3 Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, Suvar3-9 subfamily Biochemical class Methyltransferase Function H3K9me represents a specific tag for epigenetic transcriptional repression by recruiting HP1 proteins to methylated histones. Also mediates monomethylation of 'Lys-56' of histone H3 (H3K56me1) in G1 phase, leading to promote interaction between histone H3 and PCNA and regulating DNA replication. Also weakly methylates 'Lys-27' of histone H3 (H3K27me). Also required for DNA methylation, the histone methyltransferase activity is not required for DNA methylation, suggesting that these 2 activities function independently. Probably targeted to histone H3 by different DNA-binding proteins like E2F6, MGA, MAX and/or DP1. May also methylate histone H1. In addition to the histone methyltransferase activity, also methylates non-histone proteins: mediates dimethylation of 'Lys-373' of p53/TP53. Also methylates CDYL, WIZ, ACIN1, DNMT1, HDAC1, ERCC6, KLF12 and itself. Histone methyltransferase that specifically mono- and dimethylates 'Lys-9' of histone H3 (H3K9me1 and H3K9me2, respectively) in euchromatin. Related diseases Pseudohypoaldosteronism 2C (PHA2C) [MIM:614492]: An autosomal dominant disorder characterized by severe hypertension, hyperkalemia, hyperchloremia, mild hyperchloremic metabolic acidosis in some cases, and correction of physiologic abnormalities by thiazide diuretics. {ECO:0000269|PubMed:11498583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuropathy, hereditary sensory and autonomic, 2A (HSAN2A) [MIM:201300]: A form of hereditary sensory and autonomic neuropathy, a genetically and clinically heterogeneous group of disorders characterized by degeneration of dorsal root and autonomic ganglion cells, and by sensory and/or autonomic abnormalities. HSAN2A is an autosomal recessive disorder characterized by impairment of pain, temperature and touch sensation, onset of symptoms in infancy or early childhood, occurrence of distal extremity pathologies (paronychia, whitlows, ulcers, and Charcot joints), frequent amputations, sensory loss that affects all modalities of sensation (lower and upper limbs and perhaps the trunk as well), absence or diminution of tendon reflexes (usually in all limbs), minimal autonomic dysfunction, absence of sensory nerve action potentials, and virtual absence of myelinated fibers with decreased numbers of unmyelinated fibers in sural nerves. {ECO:0000269|PubMed:15060842, ECO:0000269|PubMed:15911806, ECO:0000269|PubMed:18521183}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q6VMQ6-2; Q6P1J9; Q9UBC3; P38919; Q9UM22; P23771; Q99684; Q13547; Q96JB3; Q92831; O60341-1; Q9Y4X4; P57682; Q13330; O94776; Q9BTC8; P20592; Q9BSU3; Q99801-1; O60568; Q9NQX1; Q5JSZ5; Q7Z3Z2; Q9P2R6; Q14119; Q96GT9; O60315; Q9NWS9-2; Q96JM2; A0A0S2Z5X4; Q96BV0; Q96EG3; Q07120; O60341-1 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ANK repeat; Chromatin regulator; Chromosome; Isopeptide bond; Metal-binding; Methylation; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; S-adenosyl-L-methionine; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 31010.9 Length 269 Aromaticity 0.1 Instability index 47.49 Isoelectric point 5.16 Charge (pH=7) -9.31 2D Binding mode Binding energy (Kcal/mol) -7.79 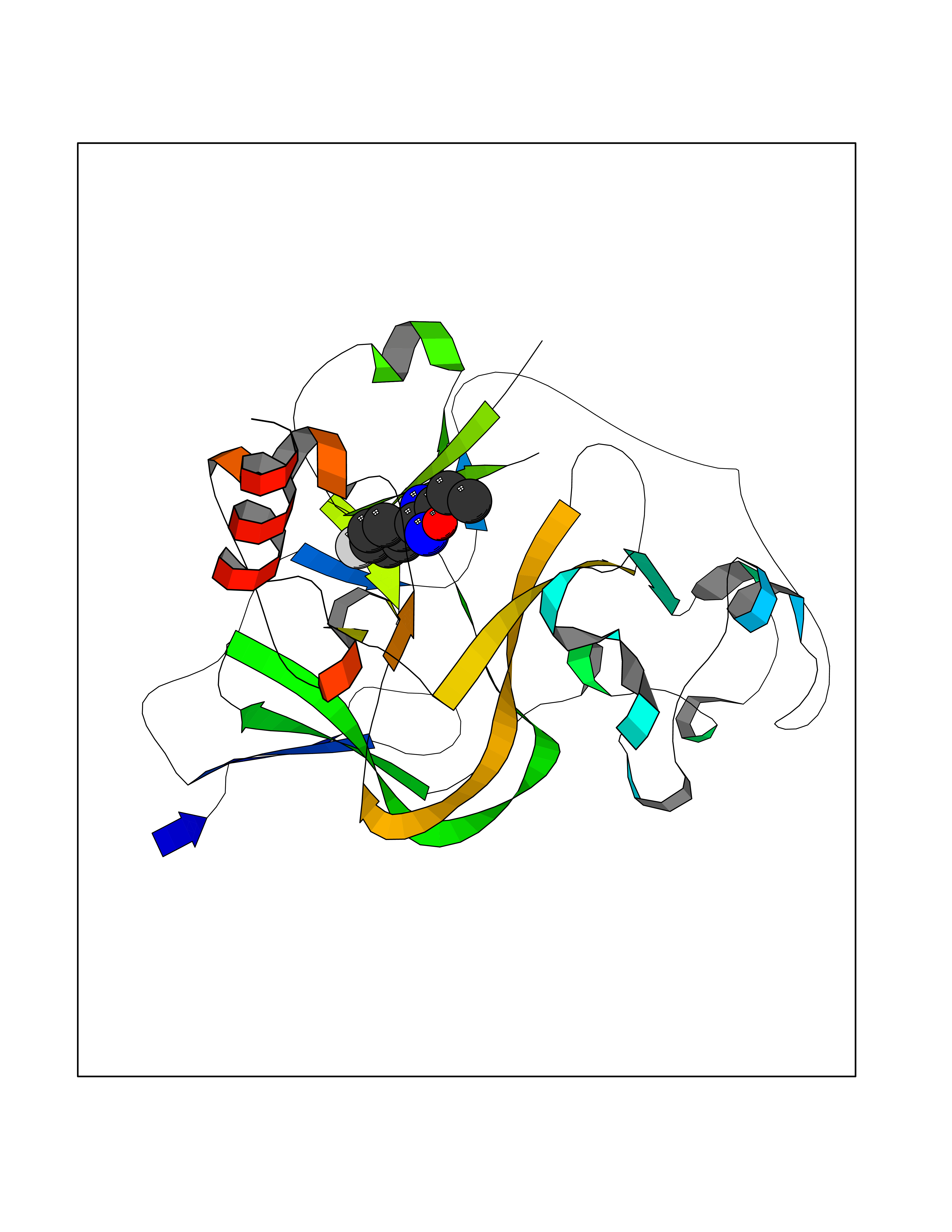 Molscript Map 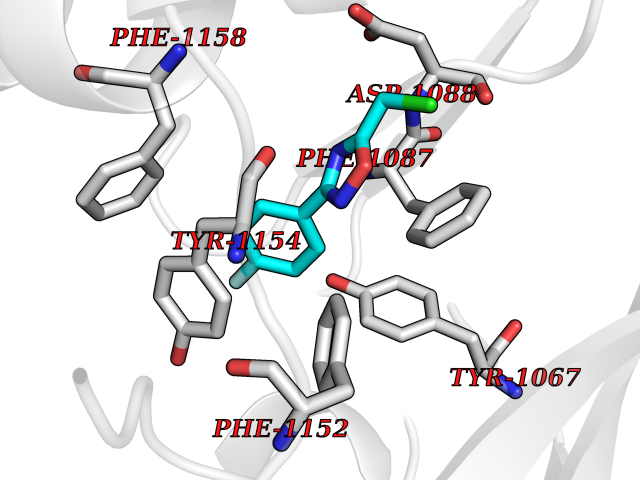 Pymol Map 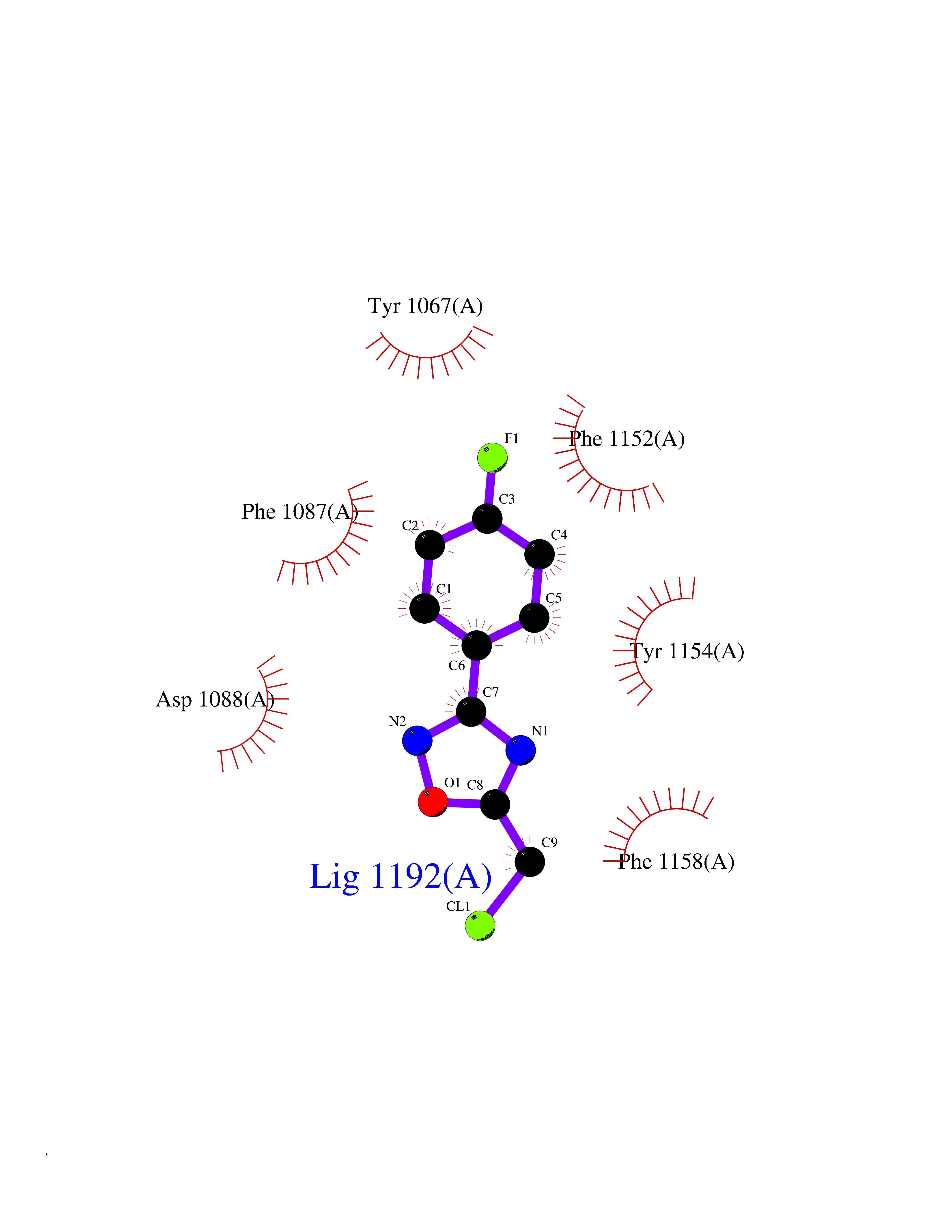 Ligplot Map 3D Binding mode Sequence TEKIICRDVARGYENVPIPCVNGVDGEPCPEDYKYISENCETSTMNIDRNITHLQHCTCVDDCSSSNCLCGQLSIRCWYDKDGRLLQEFNKIEPPLIFECNQACSCWRNCKNRVVQSGIKVRLQLYRTAKMGWGVRALQTIPQGTFICEYVGELISDAEADVREDDSYLFDLDEVYCIDARYYGNISRFINHLCDPNIIPVRVFMLHQDLRFPRIAFFSSRDIRTGEELGFDYGDRFWDIKSKYFTCQCGSEKCKHSAEAIALEQSRLA Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Phenylethanolamine N-methyltransferase (PNMT) | 2G72 | 5.71 | |
Target general information Gen name PNMT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PNMTase; PENT; Noradrenaline N-methyltransferase Protein family Class I-like SAM-binding methyltransferase superfamily, NNMT/PNMT/TEMT family Biochemical class NA Function Converts noradrenaline to adrenaline. Related diseases A chromosomal aberration involving TRIM24/TIF1 is found in papillary thyroid carcinomas (PTCs). Translocation t(7;10)(q32;q11) with RET. The translocation generates the TRIM24/RET (PTC6) oncogene. {ECO:0000269|PubMed:10439047}. Drugs (DrugBank ID) DB08129; DB08128; DB07739; DB07798; DB07747; DB03468; DB08550; DB03824; DB04273; DB07906; DB07597; DB09571; DB00968; DB08631; DB01752; DB08654 Interacts with Q9P2G9-2; Q8TBB1 EC number EC 2.1.1.28 Uniprot keywords 3D-structure; Catecholamine biosynthesis; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 29198.9 Length 264 Aromaticity 0.09 Instability index 54.33 Isoelectric point 5.91 Charge (pH=7) -3.69 2D Binding mode Binding energy (Kcal/mol) -7.79  Molscript Map 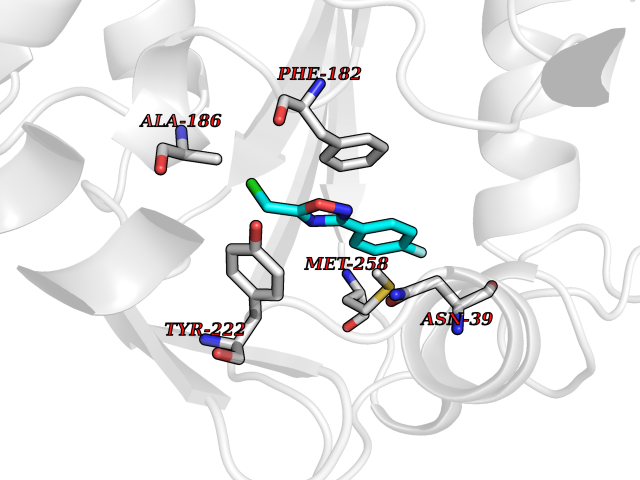 Pymol Map 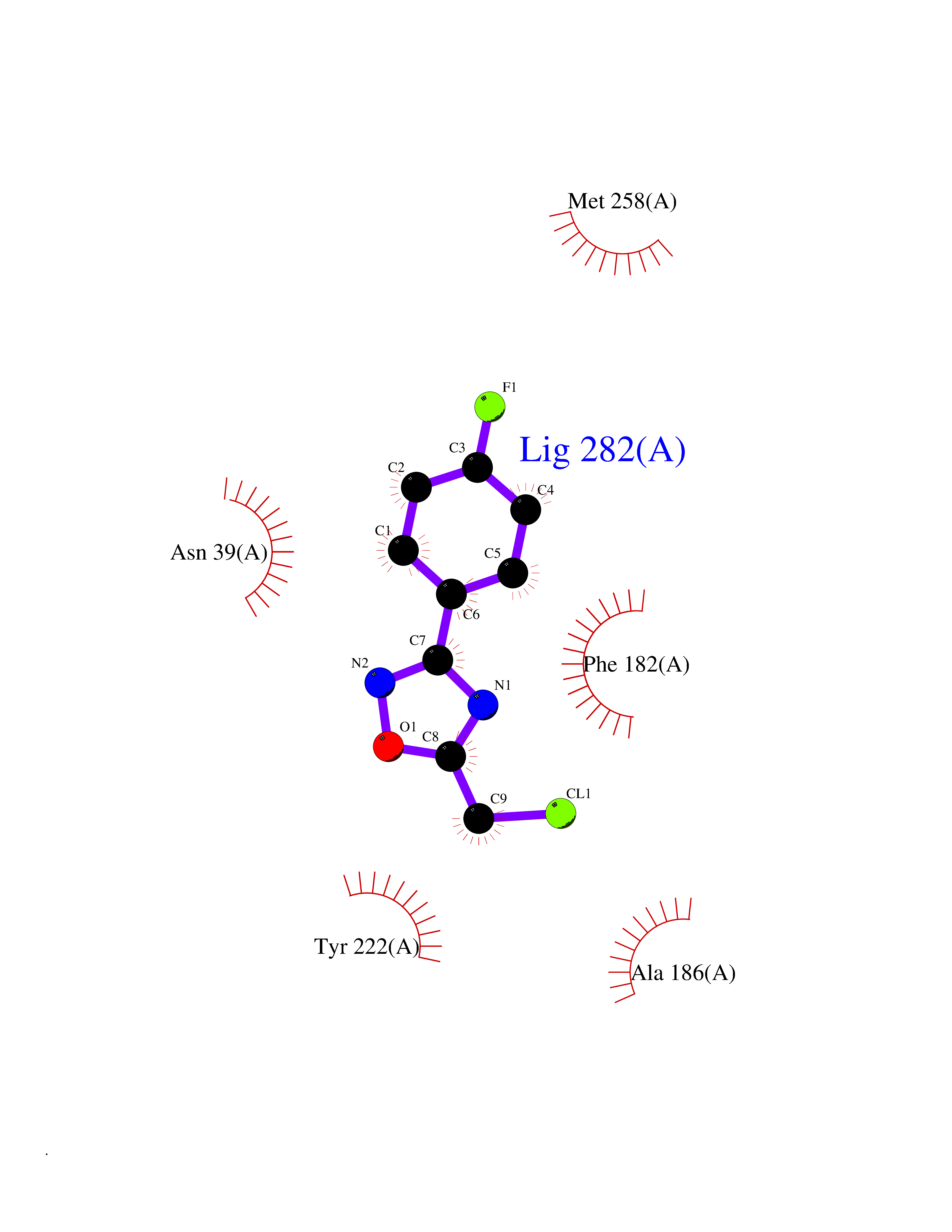 Ligplot Map 3D Binding mode Sequence APGQAAVASAYQRFEPRAYLRNNYAPPRGDLCNPNGVGPWKLRCLAQTFATGEVSGRTLIDIGSGPTVYQLLSACSHFEDITMTDFLEVNRQELGRWLQEEPGAFNWSMYSQHACLIEGKGECWQDKERQLRARVKRVLPIDVHQPQPLGAGSPAPLPADALVSAFCLEAVSPDLASFQRALDHITTLLRPGGHLLLIGALEESWYLAGEARLTVVPVSEEEVREALVRSGYKVRDLRTYIMPAHLQTGVDDVKGVFFAWAQKV Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Corynebacterium Pup-protein ligase (Cory pafA) | 4BJR | 5.71 | |
Target general information Gen name Cory pafA Organism Corynebacterium glutamicum (strain ATCC 13032 / DSM 20300 / JCM 1318 / BCRC 11384 / CCUG 27702 / LMG 3730 / NBRC 12168 / NCIMB 10025 / NRRL B-2784 / 534) Uniprot ID TTD ID Synonyms Pup-conjugating enzyme; Pup--protein ligase; Proteasome accessory factor A Protein family Pup ligase/Pup deamidase family, Pup-conjugating enzyme subfamily Biochemical class NA Function Catalyzes the covalent attachment of the prokaryotic ubiquitin-like protein modifier Pup to the proteasomal substrate proteins, thereby targeting them for proteasomal degradation. This tagging system is termed pupylation. The ligation reaction involves the side-chain carboxylate of the C-terminal glutamate of Pup and the side-chain amino group of a substrate lysine. Related diseases Anemia, non-spherocytic hemolytic, due to G6PD deficiency (NSHA) [MIM:300908]: A disease characterized by G6PD deficiency, acute hemolytic anemia, fatigue, back pain, and jaundice. In most patients, the disease is triggered by an exogenous agent, such as some drugs, food, or infection. Increased unconjugated bilirubin, lactate dehydrogenase, and reticulocytosis are markers of the disorder. Although G6PD deficiency can be life-threatening, most patients are asymptomatic throughout their life. {ECO:0000269|PubMed:12524354, ECO:0000269|PubMed:1303180, ECO:0000269|PubMed:1303182, ECO:0000269|PubMed:1536798, ECO:0000269|PubMed:1611091, ECO:0000269|PubMed:1889820, ECO:0000269|PubMed:1945893, ECO:0000269|PubMed:20007901, ECO:0000269|PubMed:26479991, ECO:0000269|PubMed:2836867, ECO:0000269|PubMed:2912069, ECO:0000269|PubMed:30988594, ECO:0000269|PubMed:38066190, ECO:0000269|PubMed:7858267, ECO:0000269|PubMed:7959695, ECO:0000269|PubMed:8193373, ECO:0000269|PubMed:8490627, ECO:0000269|PubMed:8533762, ECO:0000269|PubMed:8733135, ECO:0000269|PubMed:9452072}. The disease is caused by variants affecting the gene represented in this entry. Deficiency of G6PD is associated with hemolytic anemia in two different situations. First, in areas in which malaria has been endemic, G6PD-deficiency alleles have reached high frequencies (1% to 50%) and deficient individuals, though essentially asymptomatic in the steady state, have a high risk of acute hemolytic attacks. Secondly, sporadic cases of G6PD deficiency occur at a very low frequencies, and they usually present a more severe phenotype. Several types of NSHA are recognized. Class-I variants are associated with severe NSHA; class-II have an activity <10% of normal; class-III have an activity of 10% to 60% of normal; class-IV have near normal activity. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; ATP-binding; Ligase; Magnesium; Metal-binding; Nucleotide-binding; Reference proteome; Ubl conjugation pathway Protein physicochemical properties Chain ID A,B Molecular weight (Da) 110572 Length 994 Aromaticity 0.07 Instability index 42.33 Isoelectric point 5.26 Charge (pH=7) -34.19 2D Binding mode Binding energy (Kcal/mol) -7.79 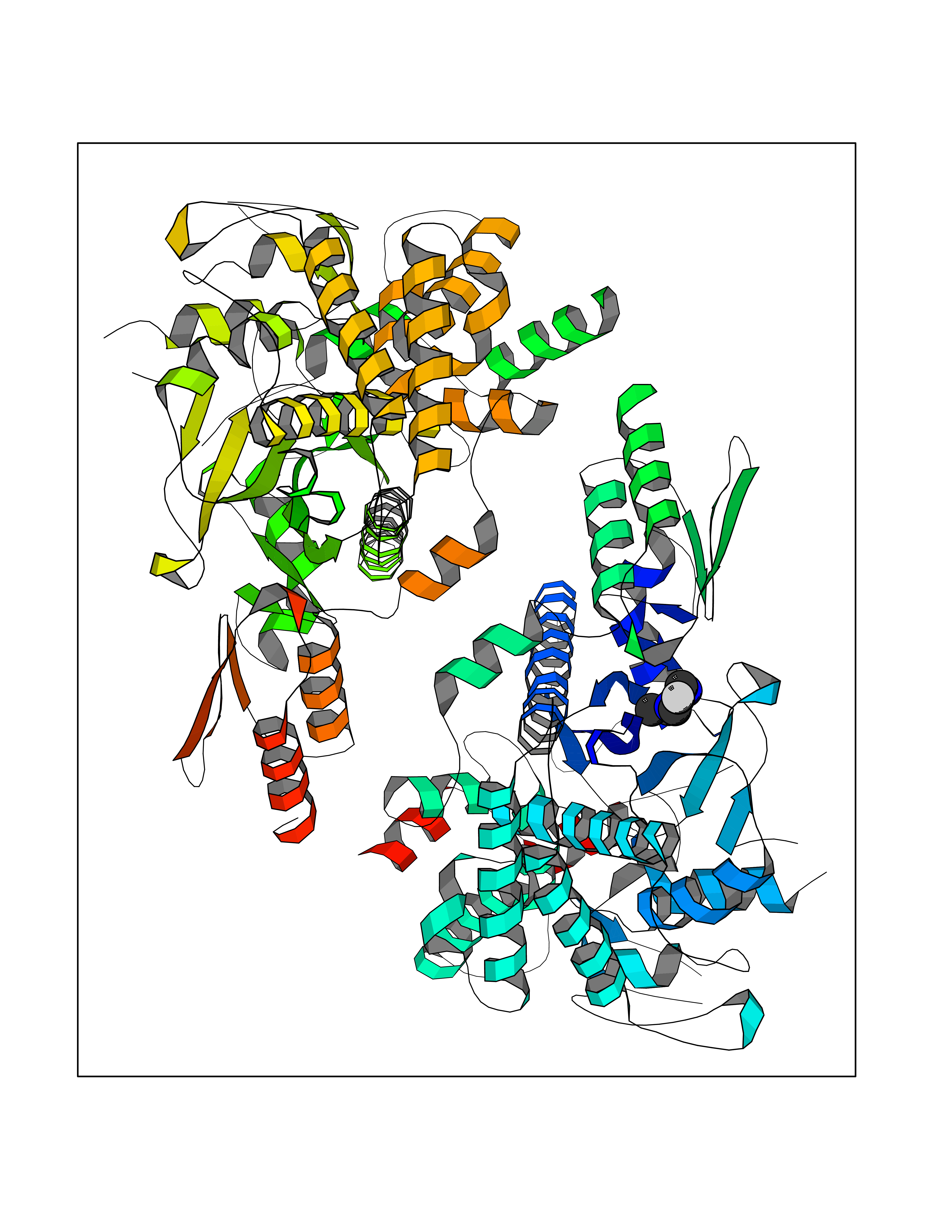 Molscript Map 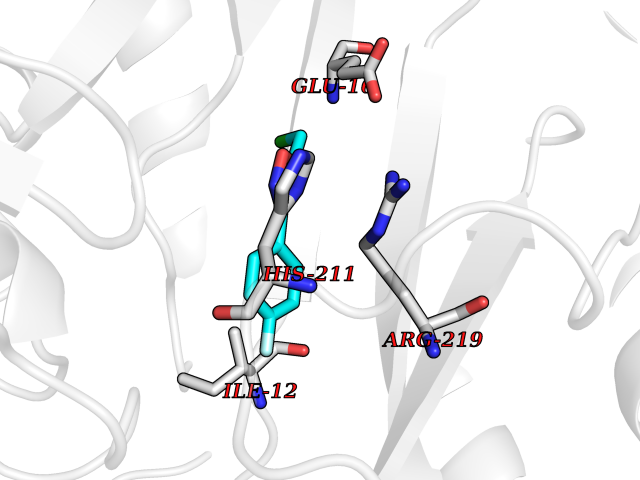 Pymol Map 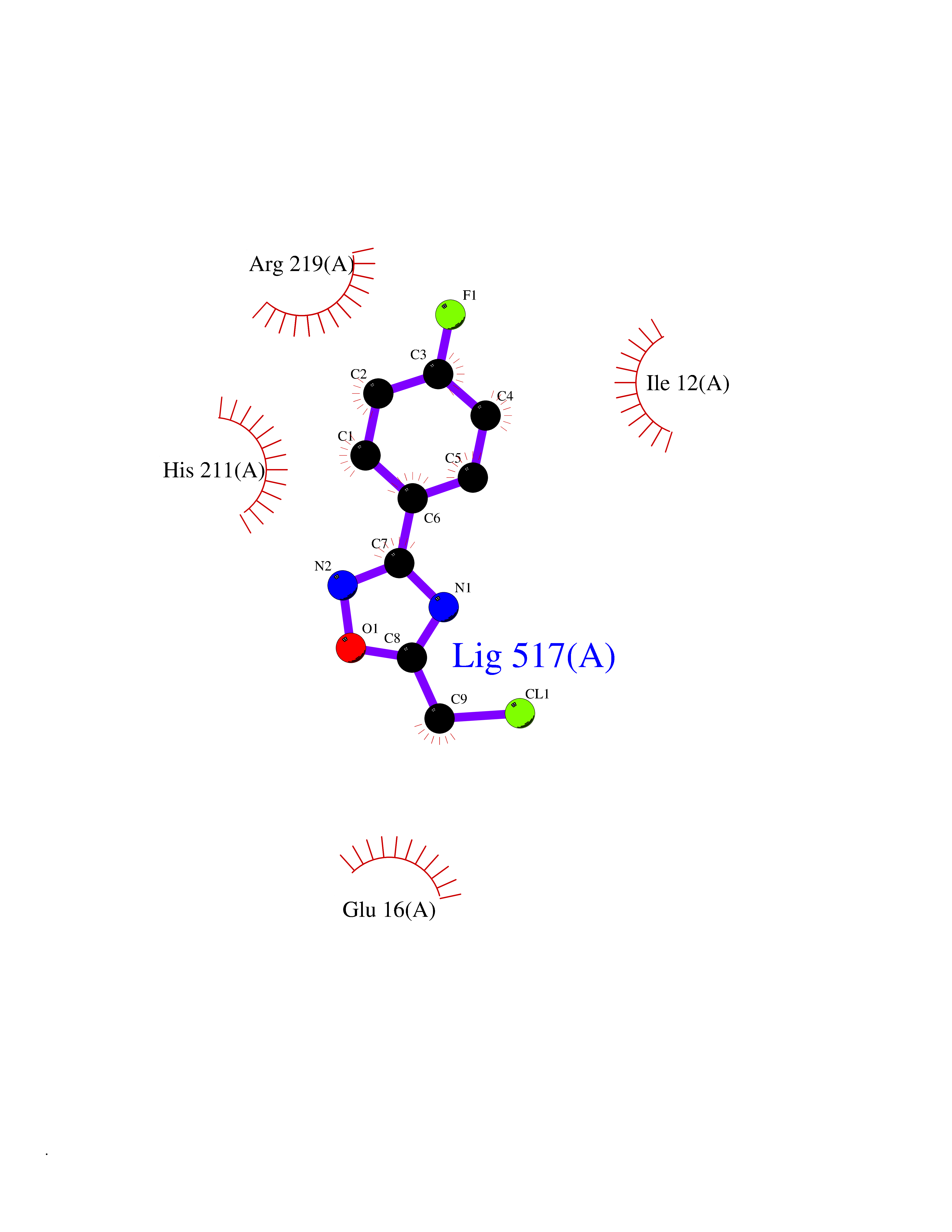 Ligplot Map 3D Binding mode Sequence TVESALTRRIMGIETEYGLTFVDGDSKKLRPDEIARRMFRPIVEKYSSSNIFIPNGSRLYLNVGSHPEYATAECDNLTQLINFEKAGDVIADRMAVDAEESLAKEDIAGQVYLFKNNVDSVGNSYGCHENYLVGRSMPLKALGKRLMPFLITRQLICGAGRIHHPNPSFPLGYCISQRSDHVWEGVSSASRPIINTRDEPHADSHSYRRLHVIVGDANMAEPSIALKVGSTLLVLEMIEADFGLPSLELANDIASIREISRDATGSTLLSLKDGTTMTALQIQQVVFEHASKWLEQRPEPEFSGTSNTEMARVLDLWGRMLKAIESGDFSEVDTEIDWVIKKKLIDRFIQRGNLGLDDPKLAQVDLTYHDIRPGRGLFSVLQSRGMIKRWTTDEAILAAVDTAPDTTRAHLRGRILKAADTLGVPVTVDWMRHKVNRPEPQSVELGDPFSAVNSEVDQLIEYMTVHAGSASGTSLLDEIDGLLENNAEEFVRSYVQKGGETVESALTRRIMGIETEYGLTFVDGDSKKLRPDEIARRMFRPIVEKYSSSNIFIPNGSRLYLNVGSHPEYATAECDNLTQLINFEKAGDVIADRMAVDAEESLAKEDIAGQVYLFKNNVDSVGNSYGCHENYLVGRSMPLKALGKRLMPFLITRQLICGAGRIHHPNPSFPLGYCISQRSDHVWEGVSSASRPIINTRDEPHADSHSYRRLHVIVGDANMAEPSIALKVGSTLLVLEMIEADFGLPSLELANDIASIREISRDATGSTLLSLKDGTTMTALQIQQVVFEHASKWLEQRPEPEFSGTSNTEMARVLDLWGRMLKAIESGDFSEVDTEIDWVIKKKLIDRFIQRGNLGLDDPKLAQVDLTYHDIRPGRGLFSVLQSRGMIKRWTTDEAILAAVDTAPDTTRAHLRGRILKAADTLGVPVTVDWMRHKVNRPEPQSVELGDPFSAVNSEVDQLIEYMTVHASLLDEIDGLLENNAEEFVRSYVQKGGE Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Optic atrophy protein 1 (OPA1) | 6JTG | 5.71 | |
Target general information Gen name OPA1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms OPA1; Dynaminlike 120 kDa protein, mitochondrial; Dynaminlike 120 kDa protein, form S1 Protein family TRAFAC class dynamin-like GTPase superfamily, Dynamin/Fzo/YdjA family Biochemical class Carbon-carbon hydrolase Function Dynamin-like 120 kDa protein, form S1: Inactive form produced by cleavage at S1 position by OMA1 following stress conditions that induce loss of mitochondrial membrane potential, leading to negative regulation of mitochondrial fusion. Related diseases Optic atrophy 1 (OPA1) [MIM:165500]: A condition that features progressive visual loss in association with optic atrophy. Atrophy of the optic disk indicates a deficiency in the number of nerve fibers which arise in the retina and converge to form the optic disk, optic nerve, optic chiasm and optic tracts. OPA1 is characterized by an insidious onset of visual impairment in early childhood with moderate to severe loss of visual acuity, temporal optic disk pallor, color vision deficits, and centrocecal scotoma of variable density. {ECO:0000269|PubMed:11017079, ECO:0000269|PubMed:11017080, ECO:0000269|PubMed:11440988, ECO:0000269|PubMed:11440989, ECO:0000269|PubMed:11810270, ECO:0000269|PubMed:12036970, ECO:0000269|PubMed:12566046, ECO:0000269|PubMed:14961560, ECO:0000269|PubMed:15948788, ECO:0000269|PubMed:16513463, ECO:0000269|PubMed:16617242, ECO:0000269|PubMed:18204809, ECO:0000269|PubMed:18360822, ECO:0000269|PubMed:19319978, ECO:0000269|PubMed:19325939, ECO:0000269|PubMed:19969356, ECO:0000269|PubMed:20185555, ECO:0000269|PubMed:22382025, ECO:0000269|PubMed:22857269, ECO:0000269|PubMed:23401657}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Dominant optic atrophy plus syndrome (DOA+) [MIM:125250]: A neurologic disorder characterized most commonly by an insidious onset of visual loss and sensorineural hearing loss in childhood with variable presentation of other clinical manifestations including progressive external ophthalmoplegia, muscle cramps, hyperreflexia, and ataxia. There appears to be a wide range of intermediate phenotypes. {ECO:0000269|PubMed:15531309, ECO:0000269|PubMed:16240368, ECO:0000269|PubMed:18065439, ECO:0000269|PubMed:18158317, ECO:0000269|PubMed:18195150, ECO:0000269|PubMed:20185555, ECO:0000269|PubMed:21112924, ECO:0000269|PubMed:23387428}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Behr syndrome (BEHRS) [MIM:210000]: An autosomal recessive syndrome characterized by optic atrophy beginning in early childhood associated with ataxia, pyramidal signs, spasticity, intellectual disability, and posterior column sensory loss. The ataxia, spasticity, and muscle contractures, mainly of the hip adductors, hamstrings, and soleus, are progressive and become more prominent in the second decade. {ECO:0000269|PubMed:21636302, ECO:0000269|PubMed:25012220, ECO:0000269|PubMed:25146916}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mitochondrial DNA depletion syndrome 14, cardioencephalomyopathic type (MTDPS14) [MIM:616896]: An autosomal recessive mitochondrial disorder characterized by lethal infantile encephalopathy, hypertrophic cardiomyopathy and optic atrophy. Skeletal muscle biopsies show significant mtDNA depletion and abnormal mitochondria. {ECO:0000269|PubMed:26561570}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q12983; Q5S007; Q9NTG7 EC number EC 3.6.5.5 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Apoptosis; Cardiomyopathy; Coiled coil; Deafness; Disease variant; Disulfide bond; GTP-binding; Hydrolase; Lipid-binding; Membrane; Mitochondrion; Mitochondrion inner membrane; Neurodegeneration; Nucleotide-binding; Primary mitochondrial disease; Proteomics identification; Reference proteome; Sensory transduction; Transit peptide; Transmembrane; Transmembrane helix; Vision Protein physicochemical properties Chain ID A Molecular weight (Da) 36858.7 Length 330 Aromaticity 0.06 Instability index 41.24 Isoelectric point 5.51 Charge (pH=7) -7.89 2D Binding mode Binding energy (Kcal/mol) -7.79 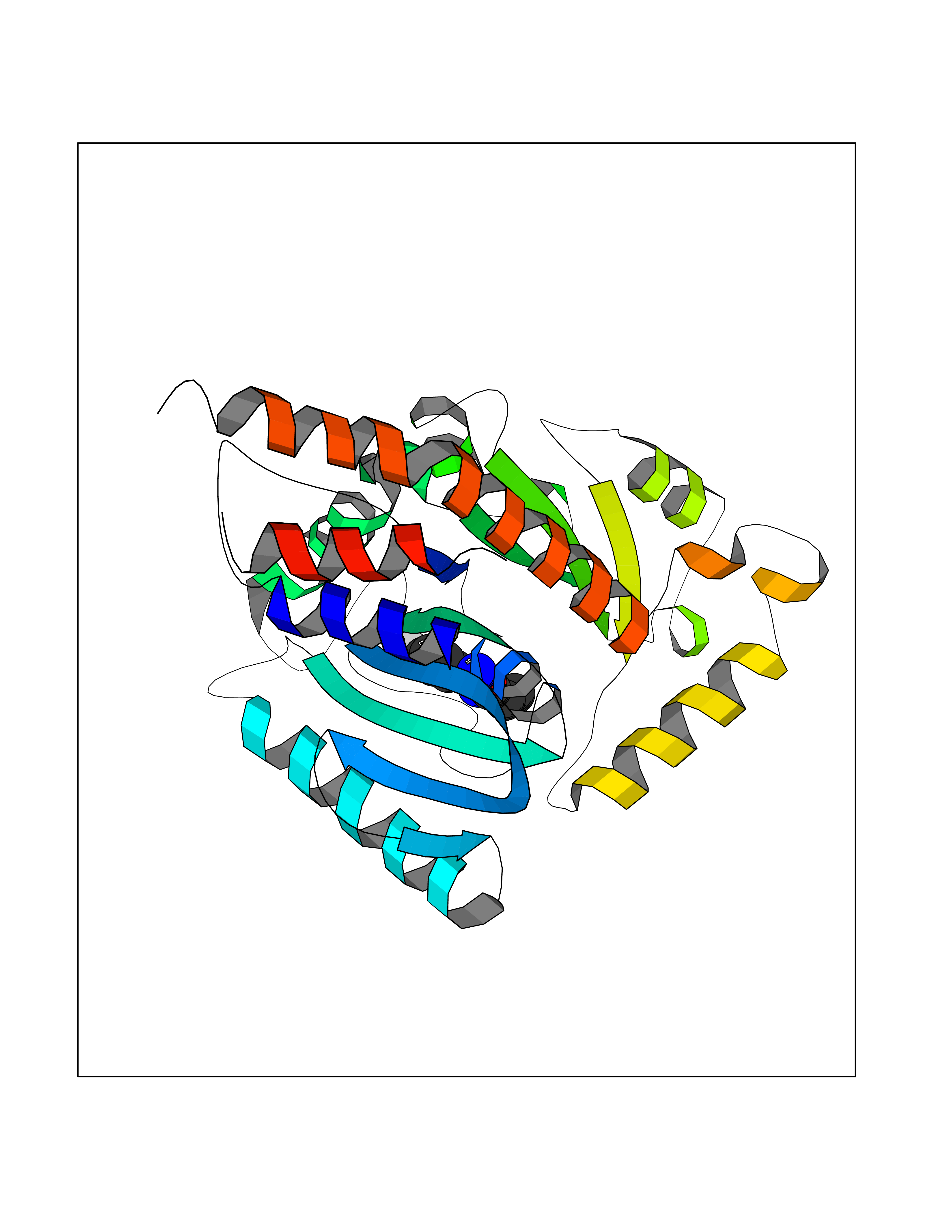 Molscript Map 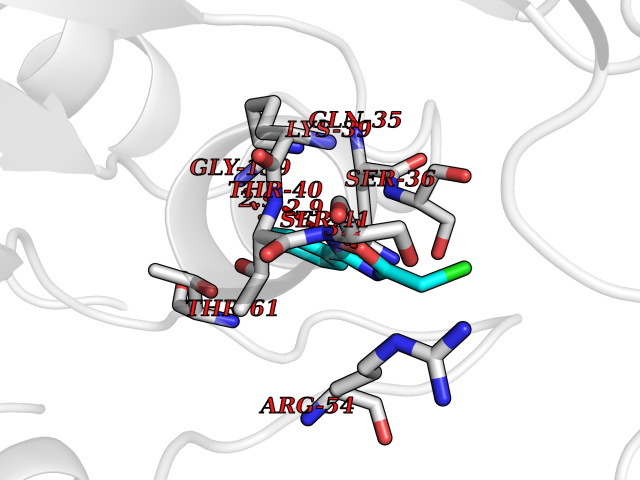 Pymol Map 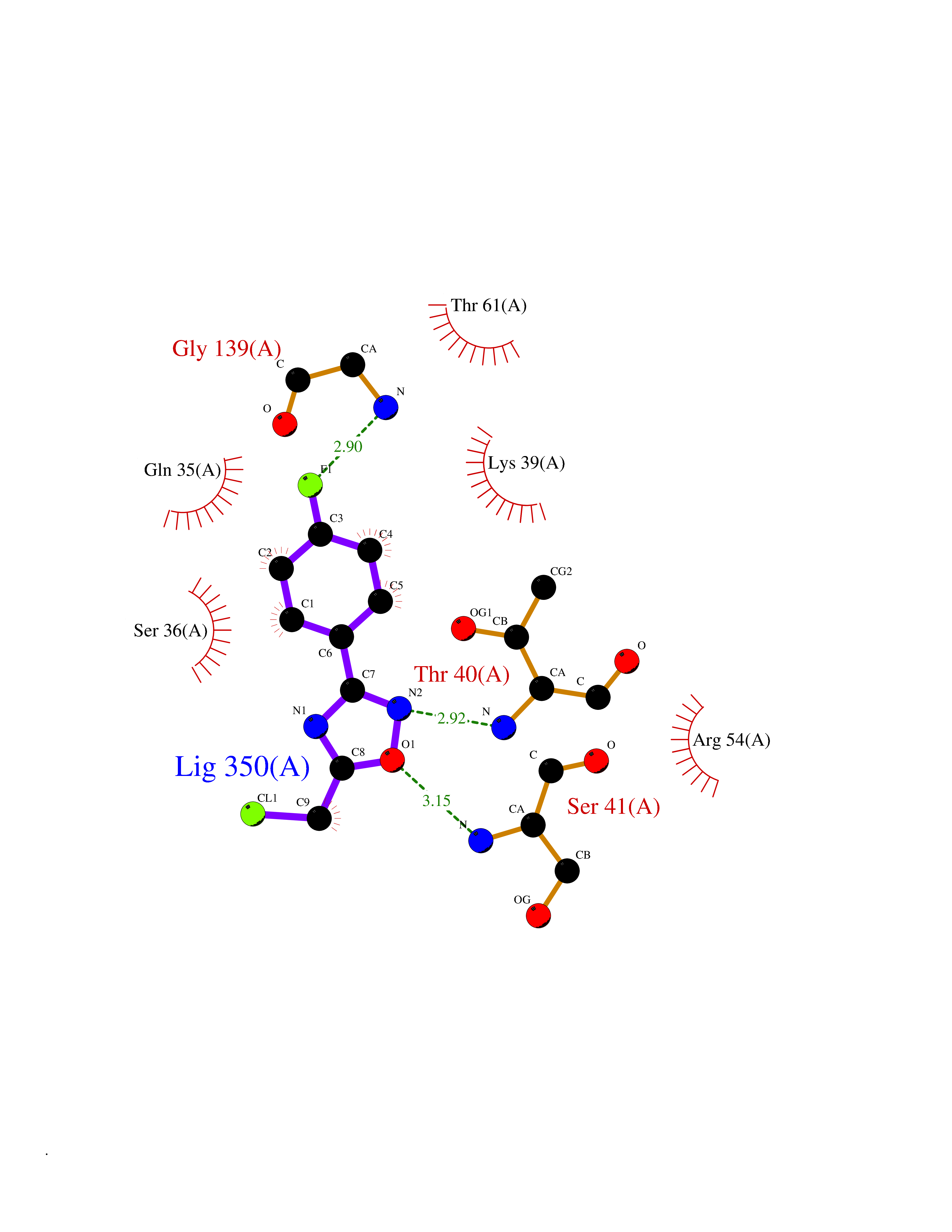 Ligplot Map 3D Binding mode Sequence SLIDMYSEVLDVLSDYDASYNTQDHLPRVVVVGDQSAGKTSVLEMIAQARIFPRGSGEMMTRSPVKVTLSEGPHHVALFKDSSREFDLTKEEDLAALRHEIELRMRKNVKEGCTVSPETISLNVKGPGLQRMVLVDLPGVINTVTSGMAPDTKETIFSISKAYMQNPNAIILCIQDGSVDAERSIVTDLVSQMDPHGRRTIFVLTKVDLAEKNVASPSRIQQIIEGKLFPMKALGYFAVVTGKGNSSESIEAIREYEEEFFQNSKLLKTSMLKAHQVTTRNLSLAVSDCFWKMVRESVEQQADSFKATRFNLEVREIQEKLDAFIEALHQ Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | Receptor-interacting protein 1 (RIPK1) | 5TX5 | 5.71 | |
Target general information Gen name RIPK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Receptor-interacting serine/threonine-protein kinase 1; RIP1; RIP-1; RIP; Cell death protein RIP Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family Biochemical class Kinase Function Upon activation of TNFR1 by the TNF-alpha family cytokines, TRADD and TRAF2 are recruited to the receptor. Phosphorylates DAB2IP at 'Ser-728' in a TNF-alpha-dependent manner, and thereby activates the MAP3K5-JNK apoptotic cascade. Ubiquitination by TRAF2 via 'Lys-63'-link chains acts as a critical enhancer of communication with downstream signal transducers in the mitogen-activated protein kinase pathway and the NF-kappa-B pathway, which in turn mediate downstream events including the activation of genes encoding inflammatory molecules. Polyubiquitinated protein binds to IKBKG/NEMO, the regulatory subunit of the IKK complex, a critical event for NF-kappa-B activation. Interaction with other cellular RHIM-containing adapters initiates gene activation and cell death. RIPK1 and RIPK3 association, in particular, forms a necrosis-inducing complex. Serine-threonine kinase which transduces inflammatory and cell-death signals (programmed necrosis) following death receptors ligation, activation of pathogen recognition receptors (PRRs), and DNA damage. Related diseases Immunodeficiency 57 with autoinflammation (IMD57) [MIM:618108]: An autosomal recessive primary immunodeficiency characterized by lymphopenia and recurrent viral, bacterial, and fungal infections. Patients exhibit early-onset inflammatory bowel disease involving the upper and lower gastrointestinal tract, and develop progressive polyarthritis. {ECO:0000269|PubMed:30026316}. The disease is caused by variants affecting the gene represented in this entry. RIPK1-deficient immune cells from IMD57 patients have impaired proinflammatory signaling leading to dysregulated cytokine secretion and are prone to necroptosis. {ECO:0000269|PubMed:30026316}.; DISEASE: Autoinflammation with episodic fever and lymphadenopathy (AIEFL) [MIM:618852]: An autosomal dominant immunologic disorder characterized by early onset of recurrent episodes of unexplained fever, lymphadenopathy, hepatosplenomegaly, and increased levels of inflammatory cytokines and chemokines in patient serum. {ECO:0000269|PubMed:31827280, ECO:0000269|PubMed:31827281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with P04083; Q13490; Q13489; Q92851; Q14790; Q8IVM0; P48729; Q13158; Q9Y6K9; Q96AB6; Q9ULZ3; Q13546; Q9Y572; P19438; Q13077; Q12933; Q13114; Q13107; B7UI21; PRO_0000449629 [P0DTD1]; U5TQE9 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cell membrane; Cytoplasm; Disease variant; Glycoprotein; Host-virus interaction; Inflammatory response; Isopeptide bond; Kinase; Membrane; Necrosis; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 29554.2 Length 259 Aromaticity 0.08 Instability index 48.26 Isoelectric point 6.29 Charge (pH=7) -2.52 2D Binding mode Binding energy (Kcal/mol) -7.79 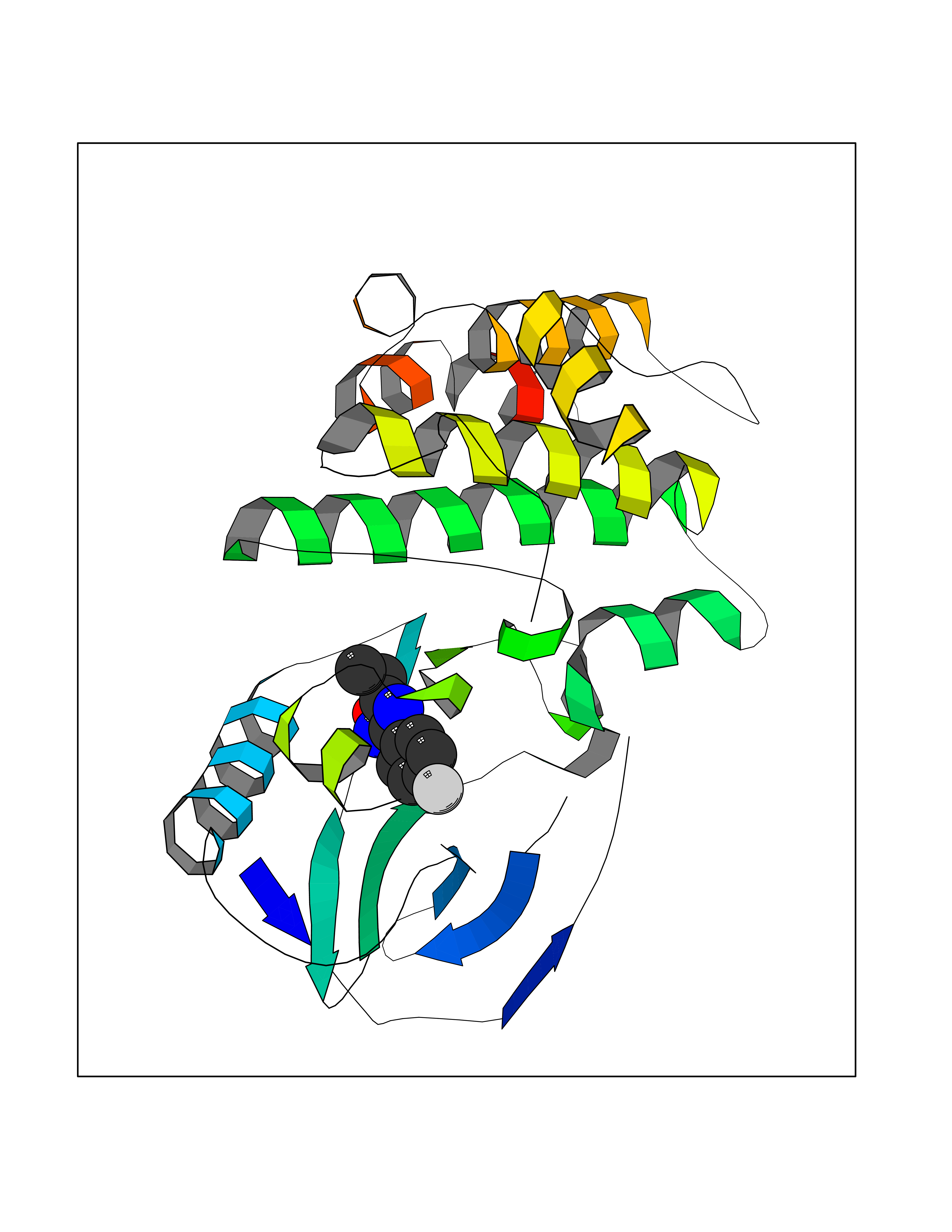 Molscript Map 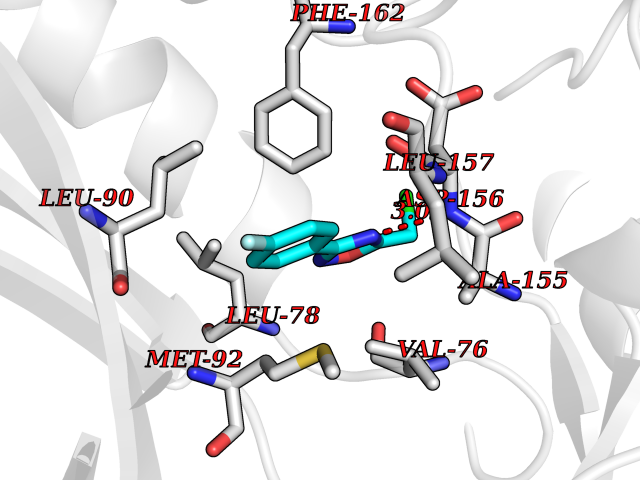 Pymol Map 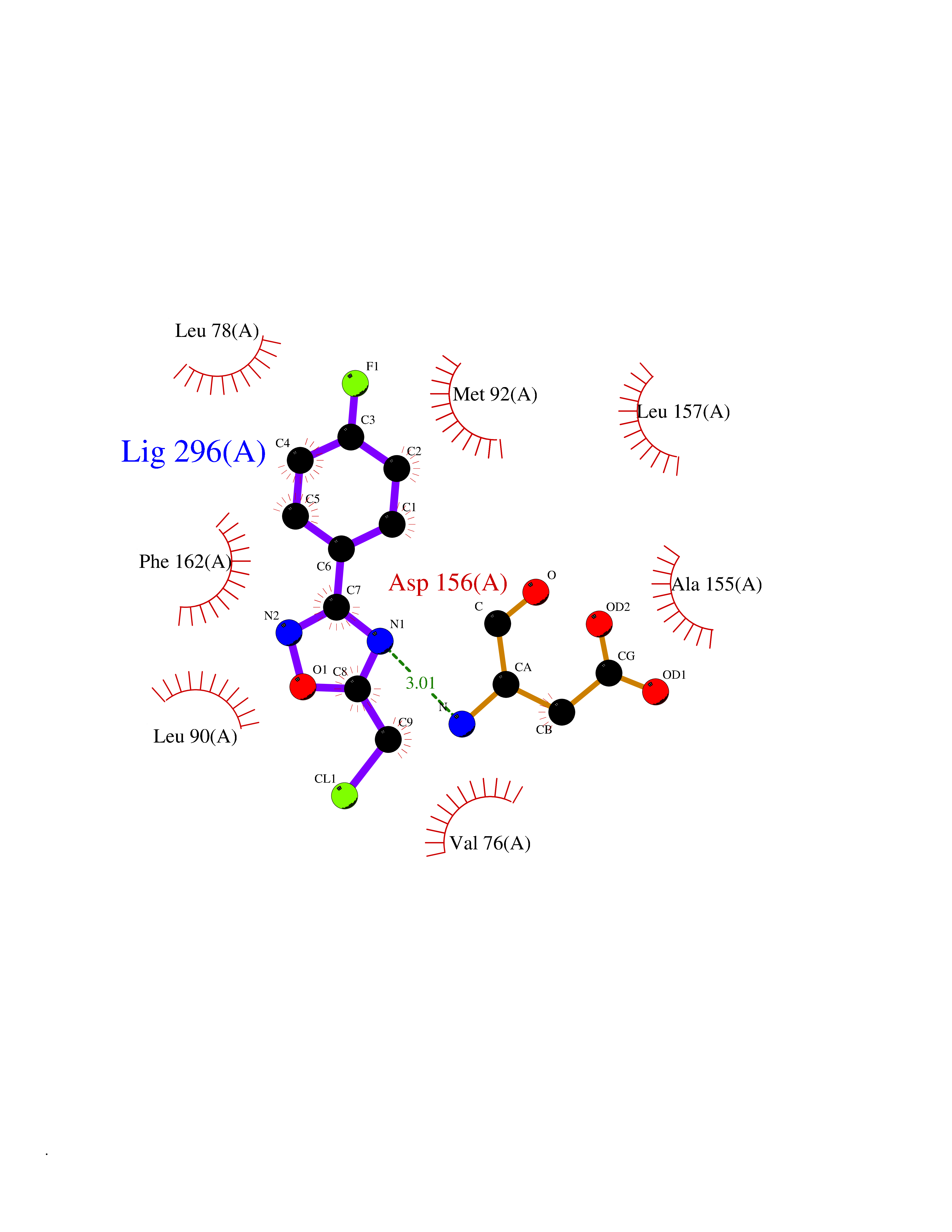 Ligplot Map 3D Binding mode Sequence IKMKSSDFLESAELDSGGKVSLAFHRTQGLMIMKTVYKGPNCIEHNEALLEEAKMMNRLRHSRVVKLLGVIIEEGKYSLVMEYMEKGNLMHVLKAEMSTPLSVKGRIILEIIEGMAYLHGKGVIHKDLKPENILVDNDFHIKIADLGLASFKMWSKLNGTLYYMAPEHLNDVNAKPTEKSDVYSFAVVLWAIFANKEPYQQLIMAIKSGNRPDVDDITEYCPREIISLMKLCWEANPEARPTFPGIEEKFRPFYLSQLE Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Matrix metalloproteinase-10 (MMP-10) | 1Q3A | 5.70 | |
Target general information Gen name MMP10 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Transin-2; Stromelysin-2; STMY2; SL-2 Protein family Peptidase M10A family Biochemical class Peptidase Function Activates procollagenase. Can degrade fibronectin, gelatins of type I, III, IV, and V; weakly collagens III, IV, and V. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00786; DB08271 Interacts with NA EC number EC 3.4.24.22 Uniprot keywords 3D-structure; Calcium; Collagen degradation; Disulfide bond; Extracellular matrix; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 52822 Length 471 Aromaticity 0.12 Instability index 21.13 Isoelectric point 4.83 Charge (pH=7) -35.32 2D Binding mode Binding energy (Kcal/mol) -7.77  Molscript Map 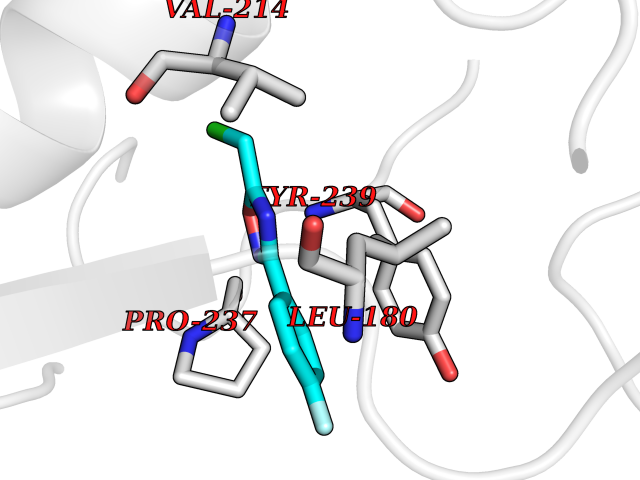 Pymol Map 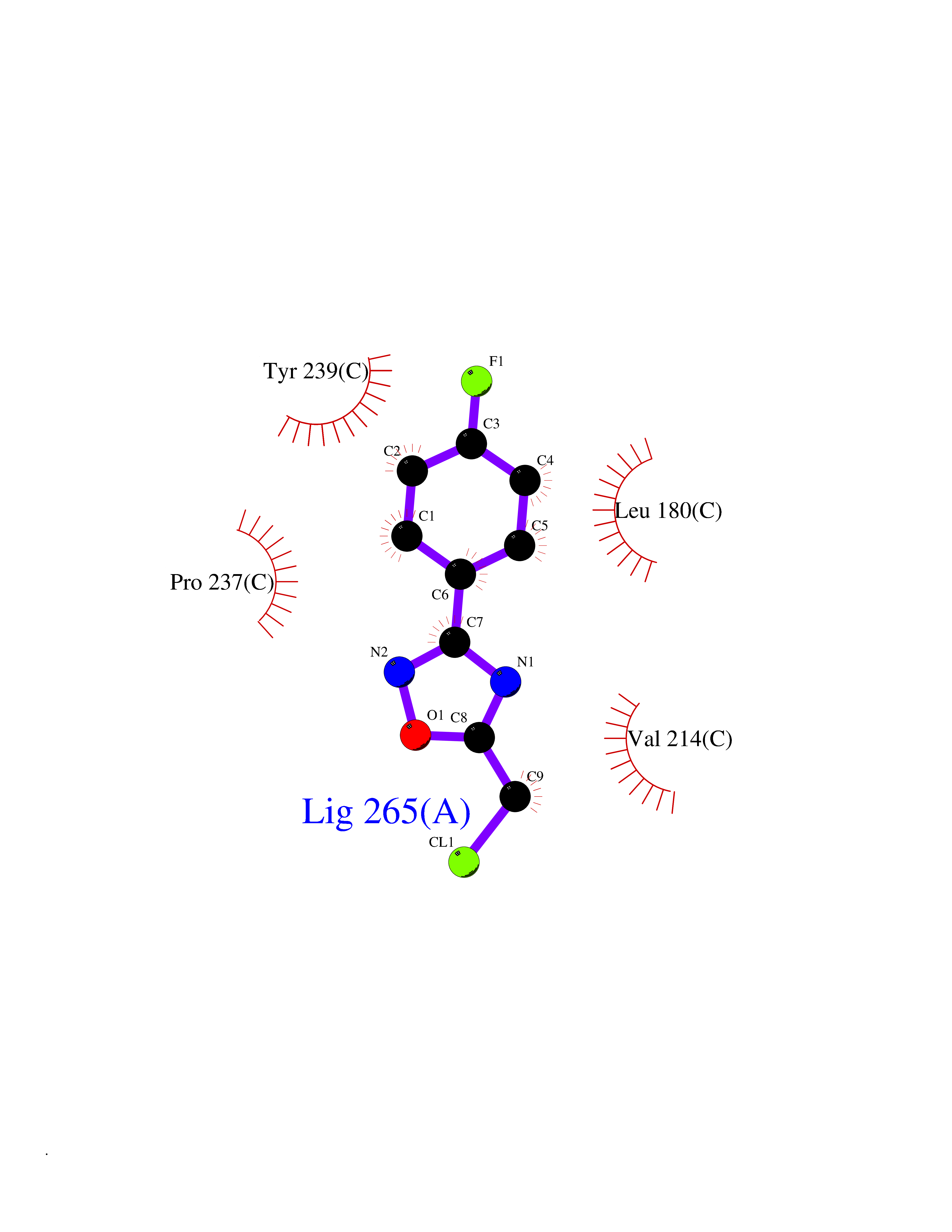 Ligplot Map 3D Binding mode Sequence MPKWRKTHLTYRIVNYTPDLPRDAVDSAIEKALKVWEEVTPLTFSRLYEGEADIMISFAVKEHGDNYSFDGPGHSLAHAYPPGPGLYGDIHFDDDEKWTEDASGTNLFLVAAHELGHSLGLFHSANTEALMYPLYNSLAQFRLSQDDVNGIQSLYGPKWRKTHLTYRIVNYTPDLPRDAVDSAIEKALKVWEEVTPLTFSRLYEGEADIMISFAVKEHGDNYSFDGPGHSLAHAYPPGPGLYGDIHFDDDEKWTEDASGTNLFLVAAHELGHSLGLFHSANTEALMYPLYNSLAQFRLSQDDVNGIQSLYGGMPKWRKTHLTYRIVNYTPDLPRDAVDSAIEKALKVWEEVTPLTFSRLYEGEADIMISFAVKEHGDNYSFDGPGHSLAHAYPPGPGLYGDIHFDDDEKWTEDASGTNLFLVAAHELGHSLGLFHSANTEALMYPLYNSFTELAQFRLSQDDVNGIQSLYG Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | 3-oxoacyl-[acyl-carrier-protein] synthase 2 | 3I8P | 5.70 | |
Target general information Gen name fabF Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1081;b1095;fabJ Protein family Thiolase-like superfamily, Beta-ketoacyl-ACP synthases family Biochemical class Transferase Function 3-oxoacyl-[acyl-carrier-protein] synthase activity.Beta-ketoacyl-acyl-carrier-protein synthase II activity. Related diseases Pigmentary disorder, reticulate, with systemic manifestations, X-linked (PDR) [MIM:301220]: An X-linked recessive disorder characterized by recurrent infections and sterile inflammation in various organs. Diffuse skin hyperpigmentation with a distinctive reticulate pattern is universally evident by early childhood. This is later followed in many patients by hypohidrosis, corneal inflammation and scarring, enterocolitis that resembles inflammatory bowel disease, and recurrent urethral strictures. Melanin and amyloid deposition is present in the dermis. Affected males also have a characteristic facies with frontally upswept hair and flared eyebrows. Female carriers have only restricted pigmentary changes along Blaschko's lines. {ECO:0000269|PubMed:27019227}. The disease is caused by variants affecting the gene represented in this entry. XLPDR is caused by a recurrent intronic mutation that results in missplicing and reduced POLA1 expression. This leads to a decrease in cytosolic RNA:DNA hybrids and constitutive activation of type I interferon responses, but has no effect on cell replication. {ECO:0000269|PubMed:27019227}.; DISEASE: Van Esch-O'Driscoll syndrome (VEODS) [MIM:301030]: An X-linked recessive syndrome characterized by different degrees of intellectual disability, moderate to severe short stature, microcephaly, hypogonadism, and variable congenital malformations. {ECO:0000269|PubMed:31006512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08366; DB01034; DB03017; DB08407 Interacts with P0A6Y8 EC number 2.3.1.179 Uniprot keywords 3D-structure; Acyltransferase; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Lipid biosynthesis; Lipid metabolism; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 42794.9 Length 411 Aromaticity 0.06 Instability index 31.41 Isoelectric point 5.72 Charge (pH=7) -7.34 2D Binding mode Binding energy (Kcal/mol) -7.77  Molscript Map 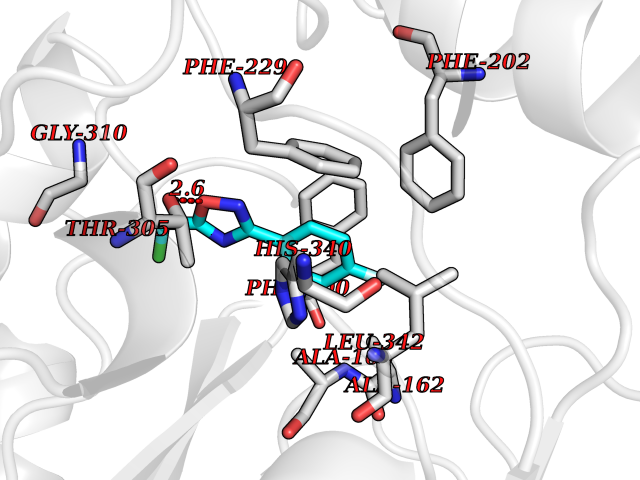 Pymol Map 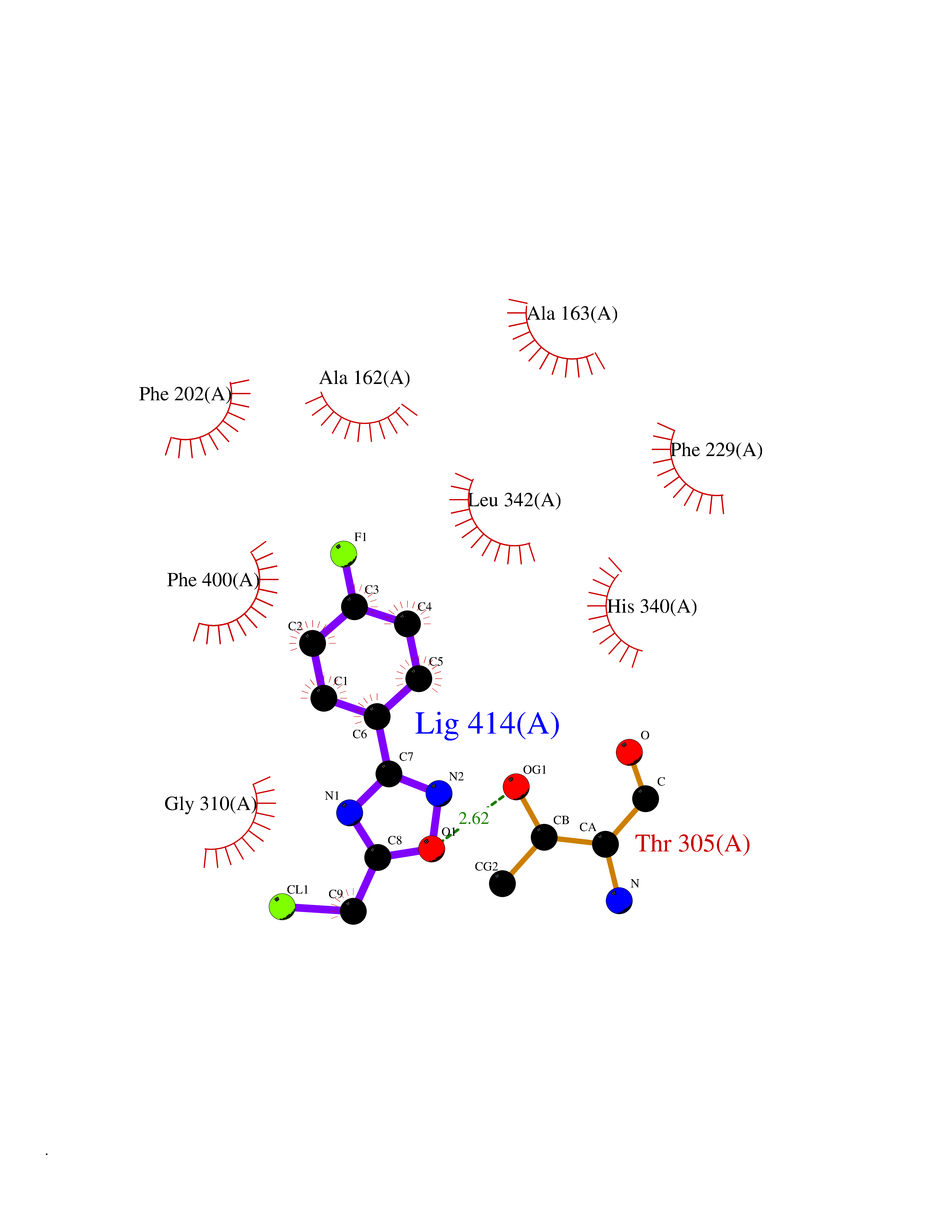 Ligplot Map 3D Binding mode Sequence KRRVVVTGLGMLSPVGNTVESTWKALLAGQSGISLIDHFDTSAYATKFAGLVKDFNCEDIISRKEQRKMDAFIQYGIVAGVQAMQDSGLEITEENATRIGAAIGSGIGGLGLIEENHTSLMNGGPRKISPFFVPSTIVNMVAGHLTIMYGLRGPSISIATAATSGVHNIGHAARIIAYGDADVMVAGGAEKASTPLGVGGFGAARALSTRNDNPQAASRPWDKERDGFVLGDGAGMLVLEEYEHAKKRGAKIYAELVGFGMSSDAYHMTSPPENGAGAALAMANALRDAGIEASQIGYVNAHGTSTPAGDKAEAQAVKTIFGEAASRVLVSSTKSMTGHLLGAAGAVESIYSILALRDQAVPPTINLDNPDEGCDLDFVPHEARQVSGMEYTLCNSFGFGGTNGSLIFKKI Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Bacterial Flavohemoglobin (Bact hmp) | 1GVH | 5.70 | |
Target general information Gen name Bact hmp Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms Nitric oxide dioxygenase; NOD; NO oxygenase; Hemoglobin-like protein; HMP; Ferrisiderophore reductase B; Dihydropteridine reductase Protein family Globin family, Two-domain flavohemoproteins subfamily; Flavoprotein pyridine nucleotide cytochrome reductase family Biochemical class Paired donor oxygen oxidoreductase Function Various electron acceptors arealso reduced by HMP in vitro, including dihydropterine, ferrisiderophores, ferric citrate, cytochrome c, nitrite, S-nitrosoglutathione, and alkylhydroperoxides. However, it is unknown if these reactions are of any biological significance in vivo. Related diseases Ovarian dysgenesis 1 (ODG1) [MIM:233300]: An autosomal recessive disease characterized by primary amenorrhea, variable development of secondary sex characteristics, poorly developed streak ovaries, and high serum levels of follicle-stimulating hormone (FSH) and luteinizing hormone (LH). {ECO:0000269|PubMed:10551778, ECO:0000269|PubMed:11889179, ECO:0000269|PubMed:12571157, ECO:0000269|PubMed:12915623, ECO:0000269|PubMed:7553856, ECO:0000269|PubMed:9769327, ECO:0000269|PubMed:9851774}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ovarian hyperstimulation syndrome (OHSS) [MIM:608115]: Disorder which occurs either spontaneously or most often as an iatrogenic complication of ovarian stimulation treatments for in vitro fertilization. The clinical manifestations vary from abdominal distention and discomfort to potentially life-threatening, massive ovarian enlargement and capillary leak with fluid sequestration. Pathologic features of this syndrome include the presence of multiple serous and hemorrhagic follicular cysts lined by luteinized cells, a condition called hyperreactio luteinalis. {ECO:0000269|PubMed:12930927, ECO:0000269|PubMed:12930928, ECO:0000269|PubMed:15080154, ECO:0000269|PubMed:16278261, ECO:0000269|PubMed:17721928, ECO:0000269|PubMed:24058690, ECO:0000269|PubMed:25581598}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number EC 1.14.12.17 Uniprot keywords 3D-structure; Cytoplasm; Detoxification; Direct protein sequencing; FAD; Flavoprotein; Heme; Iron; Metal-binding; NAD; NADP; Oxidoreductase; Oxygen transport; Reference proteome; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 43867.1 Length 396 Aromaticity 0.1 Instability index 28.85 Isoelectric point 5.48 Charge (pH=7) -12.32 2D Binding mode Binding energy (Kcal/mol) -7.78 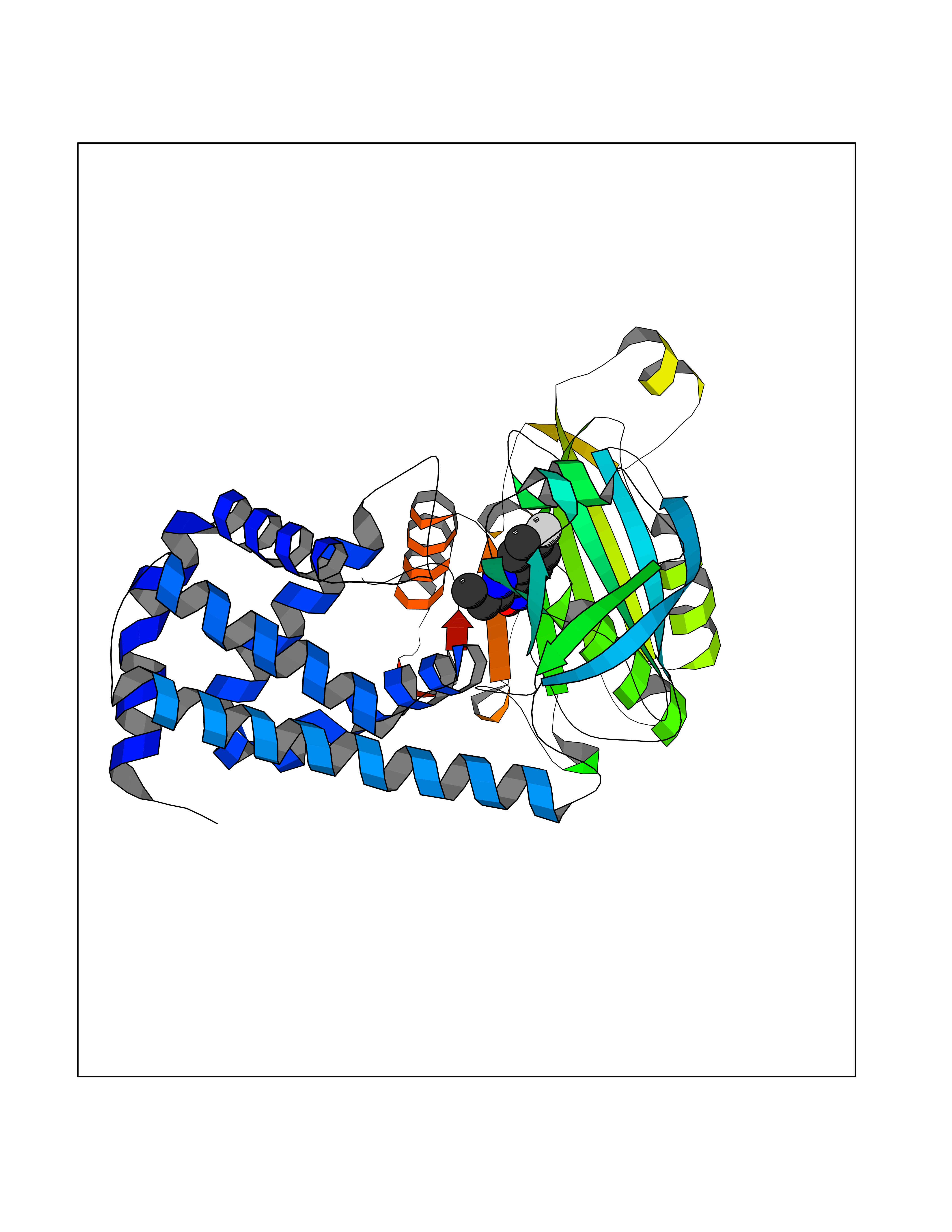 Molscript Map 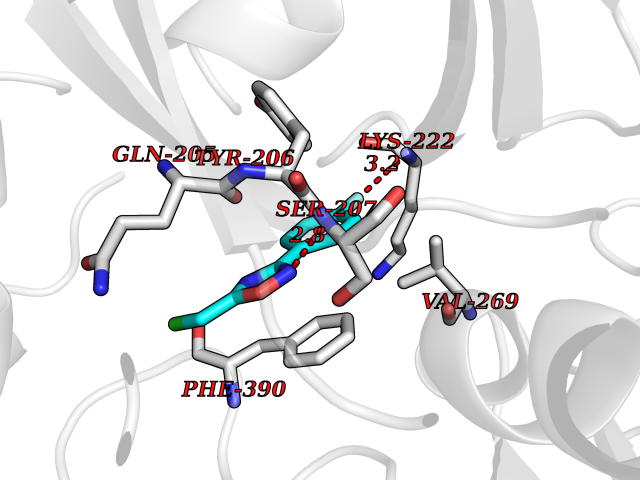 Pymol Map 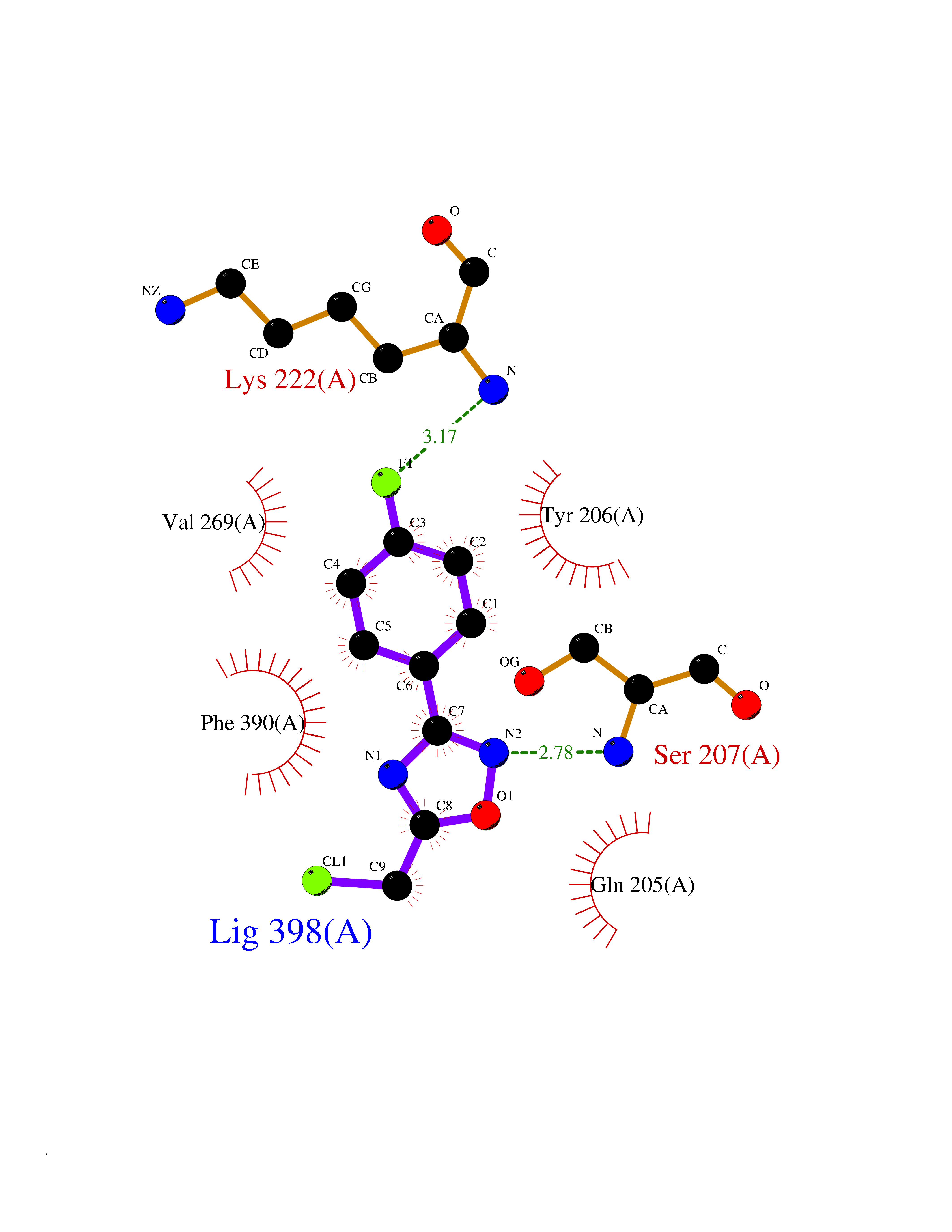 Ligplot Map 3D Binding mode Sequence MLDAQTIATVKATIPLLVETGPKLTAHFYDRMFTHNPELKEIFNMSNQRNGDQREALFNAIAAYASNIENLPALLPAVEKIAQKHTSFQIKPEQYNIVGEHLLATLDEMFSPGQEVLDAWGKAYGVLANVFINREAEIYNENASKAGGWEGTRDFRIVAKTPRSALITSFELEPVDGGAVAEYRPGQYLGVWLKPEGFPHQEIRQYSLTRKPDGKGYRIAVKREEGGQVSNWLHNHANVGDVVKLVAPAGDFFMAVADDTPVTLISAGVGQTPMLAMLDTLAKAGHTAQVNWFHAAENGDVHAFADEVKELGQSLPRFTAHTWYRQPSEADRAKGQFDSEGLMDLSKLEGAFSDPTMQFYLCGPVGFMQFTAKQLVDLGVKQENIHYECFGPHKVL Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Spermine synthase | 3C6K | 5.70 | |
Target general information Gen name SMS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Spermidine/spermine synthase family Biochemical class Transferase Function Spermine synthase activity. Related diseases Intellectual developmental disorder, X-linked, syndromic, Snyder-Robinson type (MRXSSR) [MIM:309583]: An X-linked intellectual disability syndrome characterized by a collection of clinical features including facial asymmetry, marfanoid habitus, hypertonia, osteoporosis and unsteady gait. {ECO:0000269|PubMed:14508504, ECO:0000269|PubMed:18550699, ECO:0000269|PubMed:19206178, ECO:0000269|PubMed:22612257, ECO:0000269|PubMed:23696453, ECO:0000269|PubMed:23897707}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00127 Interacts with NA EC number 2.5.1.22 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; Intellectual disability; Phosphoprotein; Polyamine biosynthesis; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27177.1 Length 238 Aromaticity 0.11 Instability index 41.31 Isoelectric point 4.82 Charge (pH=7) -9.19 2D Binding mode Binding energy (Kcal/mol) -7.77 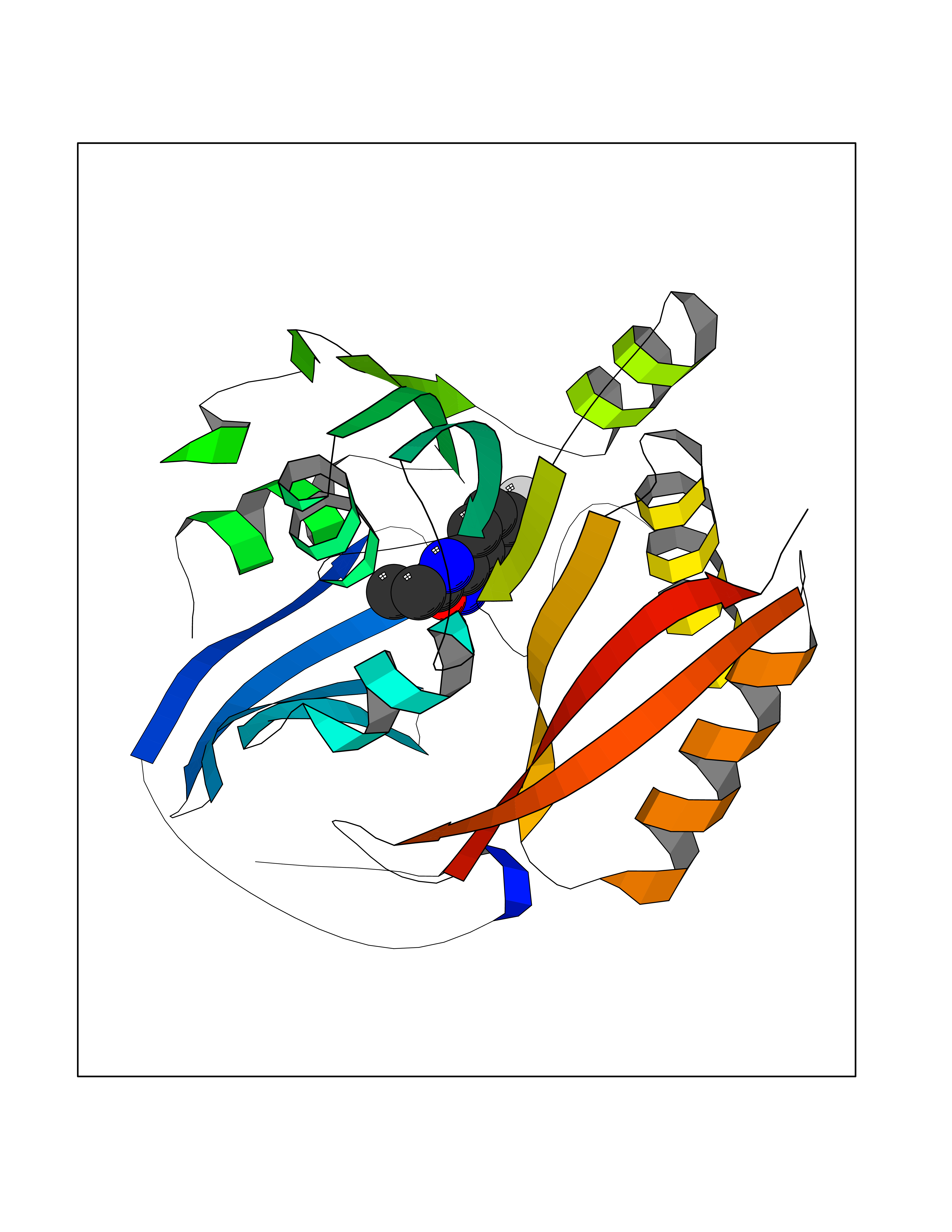 Molscript Map 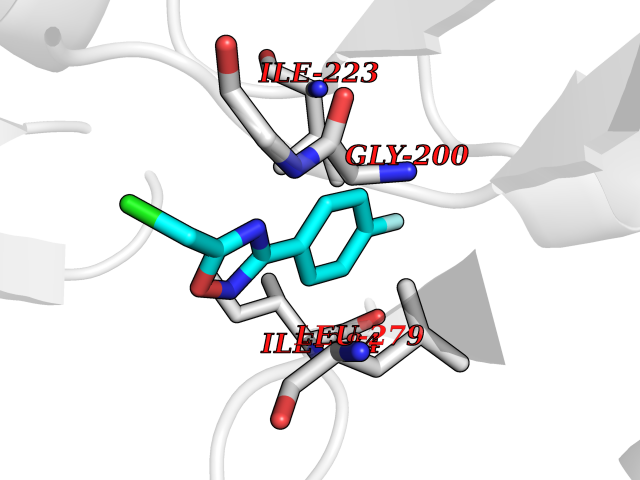 Pymol Map 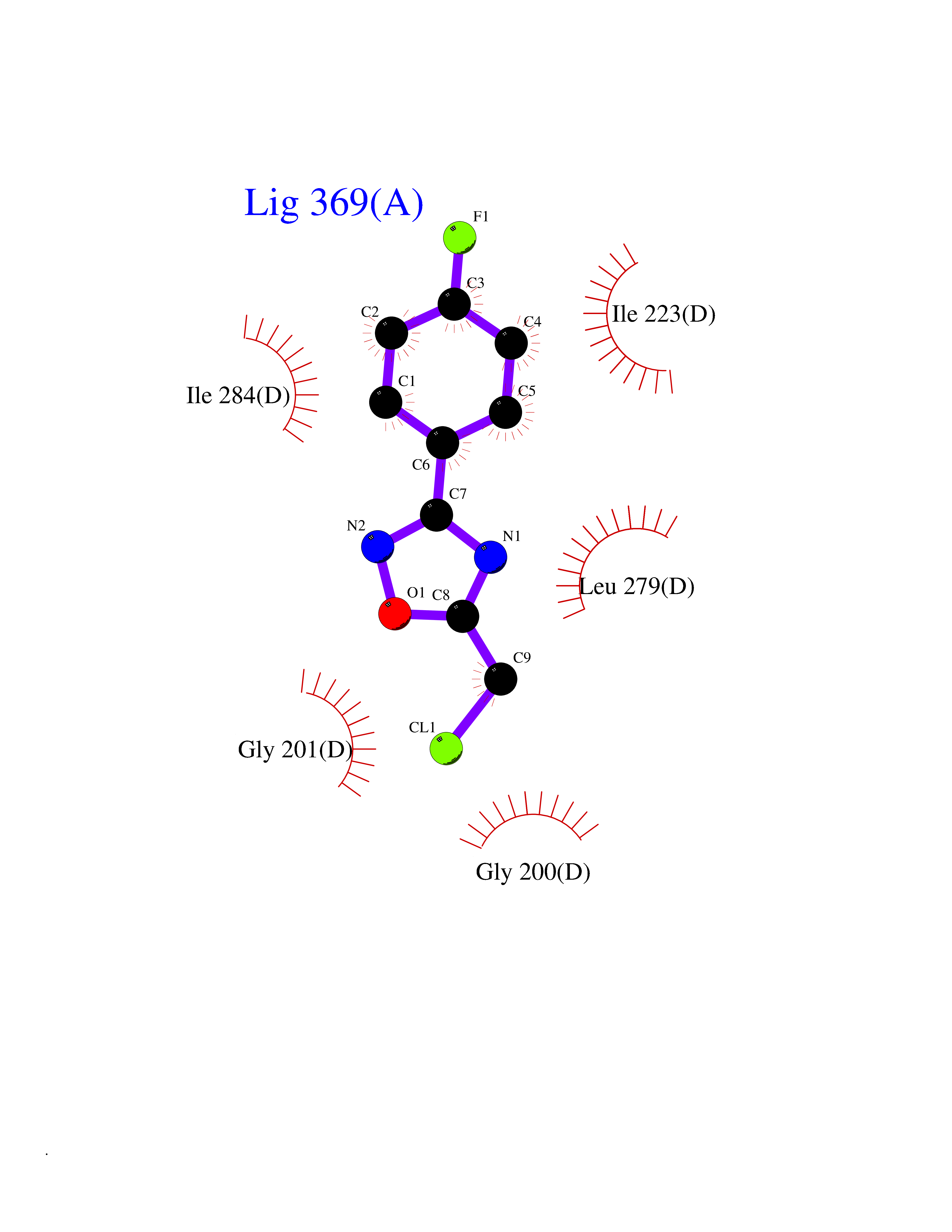 Ligplot Map 3D Binding mode Sequence RYWPTADGRLVEYDIDEVVYDEDSPYQNIKILHSKQFGNILILSGDVNLAESDLAYTRAIMGSGKEDYTGKDVLILGGGDGGILCEIVKLKPKMVTMVEIDQMVIDGCKKYMRKDVLDNLKGDCYQVLIEDCIPVLKRYAKEGREFDYVINDLTAVPISTSPSTWEFLRLILDLSMKVLKQDGKYFTQGNCVNLTEALSLYEEQLGRLYCPVEFSKEIVCVPSYLELWVFYTVWKKAK Hydrogen bonds contact Hydrophobic contact | ||||