Job Results:
Ligand
Structure
Job ID
8fa392a7f47e3bb9cb55af0501d4ab6f
Job name
NA
Time
2025-11-13 18:42:40
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Opioid receptor sigma 1 (OPRS1) | 6DJZ | 7.54 | |
Target general information Gen name SIGMAR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hSigmaR1; Sigma1R; Sigma1-receptor; Sigma non-opioid intracellular receptor 1; Sigma 1-type opioid receptor; SRBP; SR31747-binding protein; SR31747 binding protein 1; SR-BP; SIG-1R; Opioid receptor, s Protein family ERG2 family Biochemical class GPCR rhodopsin Function Involved in the regulation of different receptors it plays a role in BDNF signaling and EGF signaling. Also regulates ion channels like the potassium channel and could modulate neurotransmitter release. Plays a role in calcium signaling through modulation together with ANK2 of the ITP3R-dependent calcium efflux at the endoplasmic reticulum. Plays a role in several other cell functions including proliferation, survival and death. Originally identified for its ability to bind various psychoactive drugs it is involved in learning processes, memory and mood alteration. Necessary for proper mitochondrial axonal transport in motor neurons, in particular the retrograde movement of mitochondria. Plays a role in protecting cells against oxidative stress-induced cell death via its interaction with RNF112. Functions in lipid transport from the endoplasmic reticulum and is involved in a wide array of cellular functions probably through regulation of the biogenesis of lipid microdomains at the plasma membrane. Related diseases Amyotrophic lateral sclerosis 16, juvenile (ALS16) [MIM:614373]: A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. {ECO:0000269|PubMed:21842496}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal recessive 2 (HMNR2) [MIM:605726]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. HMNR2 is characterized by onset of distal muscle weakness and wasting affecting the lower and upper limbs in the first decade. {ECO:0000269|PubMed:26078401, ECO:0000269|PubMed:27629094}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00321; DB09014; DB00907; DB00514; DB01488; DB00574; DB00502; DB00956; DB00704; DB00540; DB06174; DB00652; DB11186; DB03575; DB05316; DB01708; DB00409; DB01104 Interacts with Q92847-1; Q99720-1; O00213-2; P17612; P50454; P37173 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Amyotrophic lateral sclerosis; Cell junction; Cell membrane; Cell projection; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Lipid droplet; Lipid transport; Membrane; Neurodegeneration; Neuropathy; Nucleus; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23901 Length 212 Aromaticity 0.14 Instability index 33.12 Isoelectric point 5.61 Charge (pH=7) -5.6 2D Binding mode Binding energy (Kcal/mol) -10.29 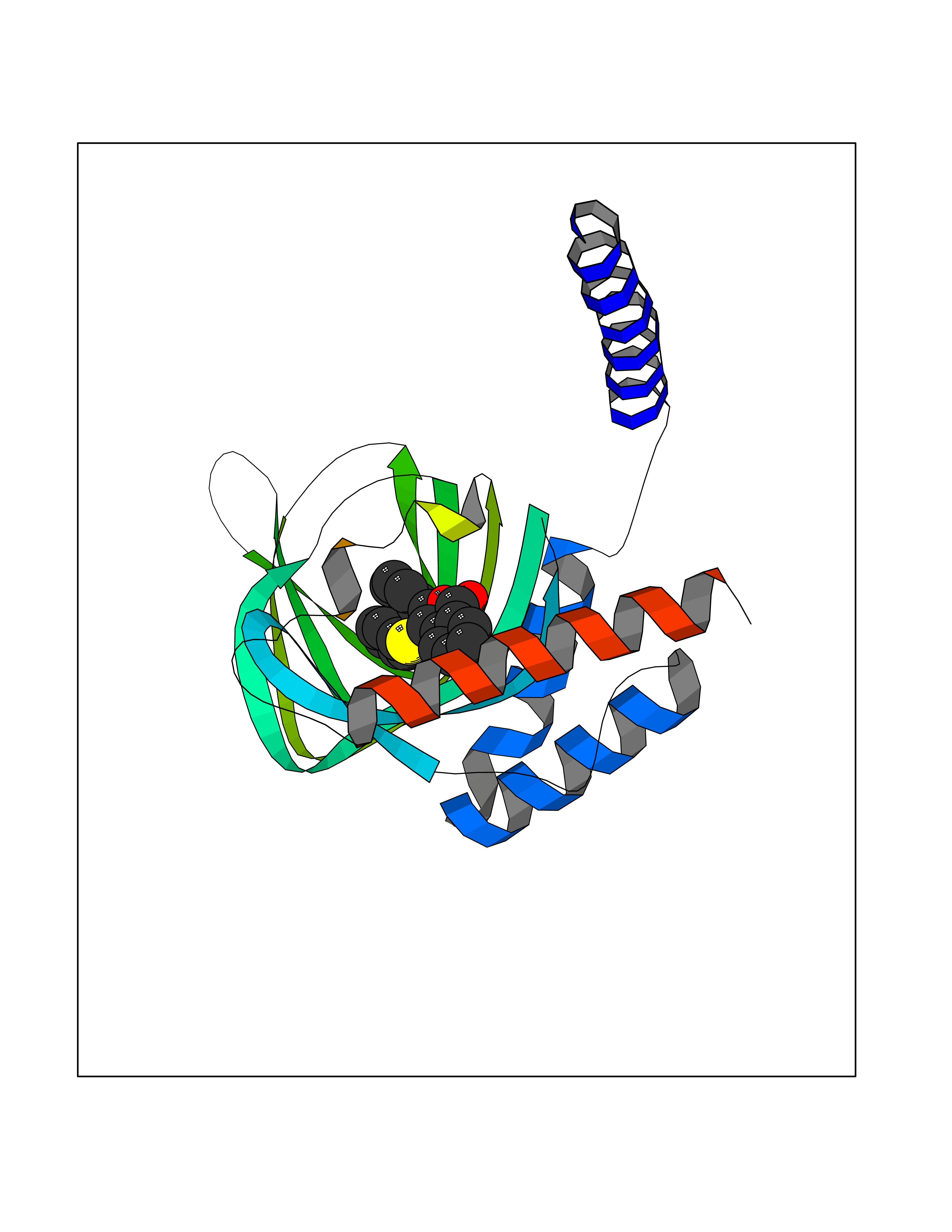 Molscript Map 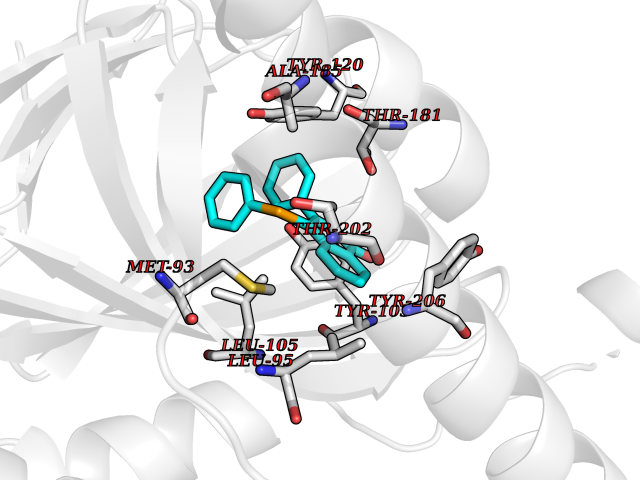 Pymol Map 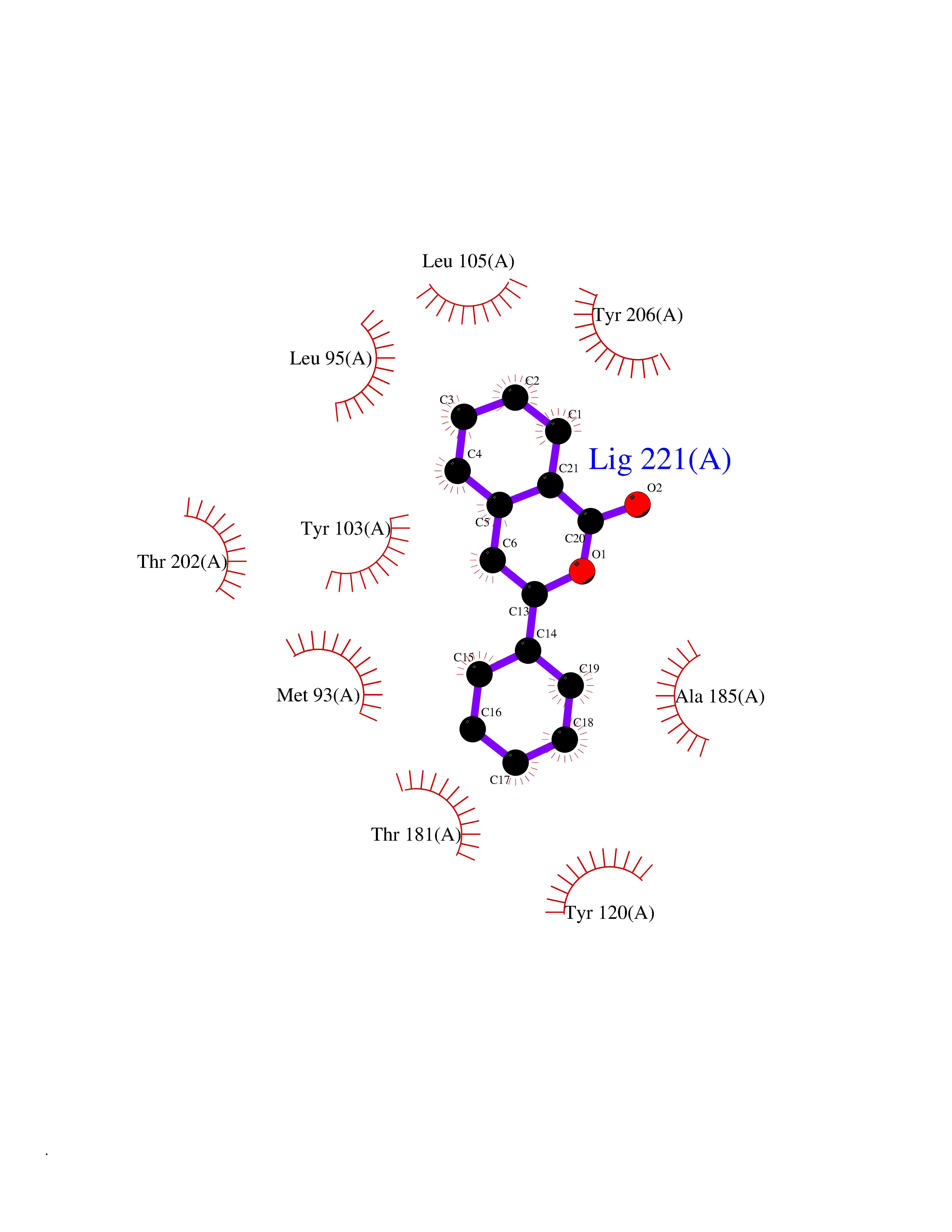 Ligplot Map 3D Binding mode Sequence RWAWAALLLAVAAVLTQVVWLWLGTQSFVFQREEIAQLARQYAGLDHELAFSRLIVELRRLHPGHVLPDEELQWVFVNAGGWMGAMCLLHASLSEYVLLFGTALGSRGHSGRYWAEISDTIISGTFHQWREGTTKSEVFYPGETVVHGPGEATAVEWGPNTWMVEYGRGVIPSTLAFALADTVFSTQDFLTLFYTLRSYARGLRLELTTYLF Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Sphingosine-1-phosphate receptor 2 (S1PR2) | 7T6B | 7.54 | |
Target general information Gen name S1PR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Sphingosine 1-phosphate receptor Edg-5; S1PR2; S1P2; S1P receptor Edg-5; S1P receptor 2; Endothelial differentiation G-protein coupled receptor 5 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for the lysosphingolipid sphingosine 1- phosphate (S1P). S1P is a bioactive lysophospholipid that elicits diverse physiological effect on most types of cells and tissues. When expressed in rat HTC4 hepatoma cells, is capable of mediating S1P-induced cell proliferation and suppression of apoptosis. Related diseases Deafness, autosomal recessive, 68 (DFNB68) [MIM:610419]: A form of non-syndromic sensorineural hearing loss. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. {ECO:0000269|PubMed:26805784}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P16144; Q9JK11-1 EC number NA Uniprot keywords 3D-structure; Cell membrane; Deafness; Disease variant; G-protein coupled receptor; Glycoprotein; Lipoprotein; Membrane; Non-syndromic deafness; Palmitate; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 28917.3 Length 264 Aromaticity 0.11 Instability index 38.95 Isoelectric point 9.11 Charge (pH=7) 9.27 2D Binding mode Binding energy (Kcal/mol) -10.28 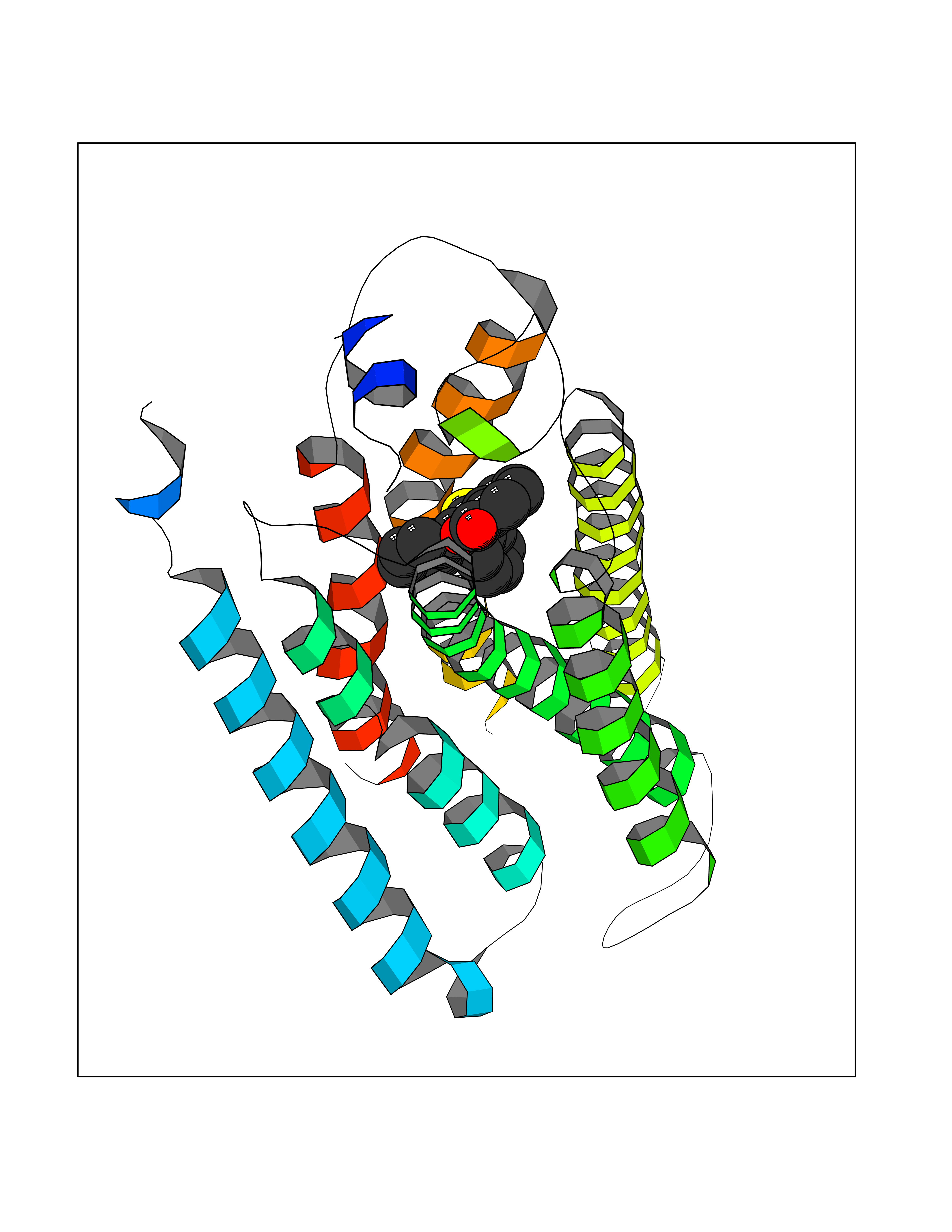 Molscript Map 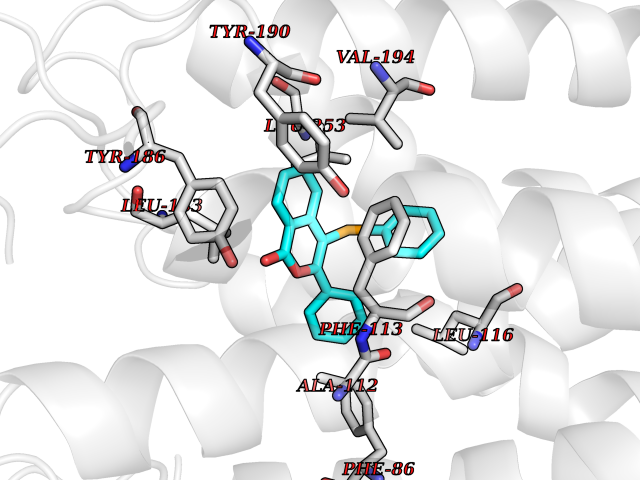 Pymol Map 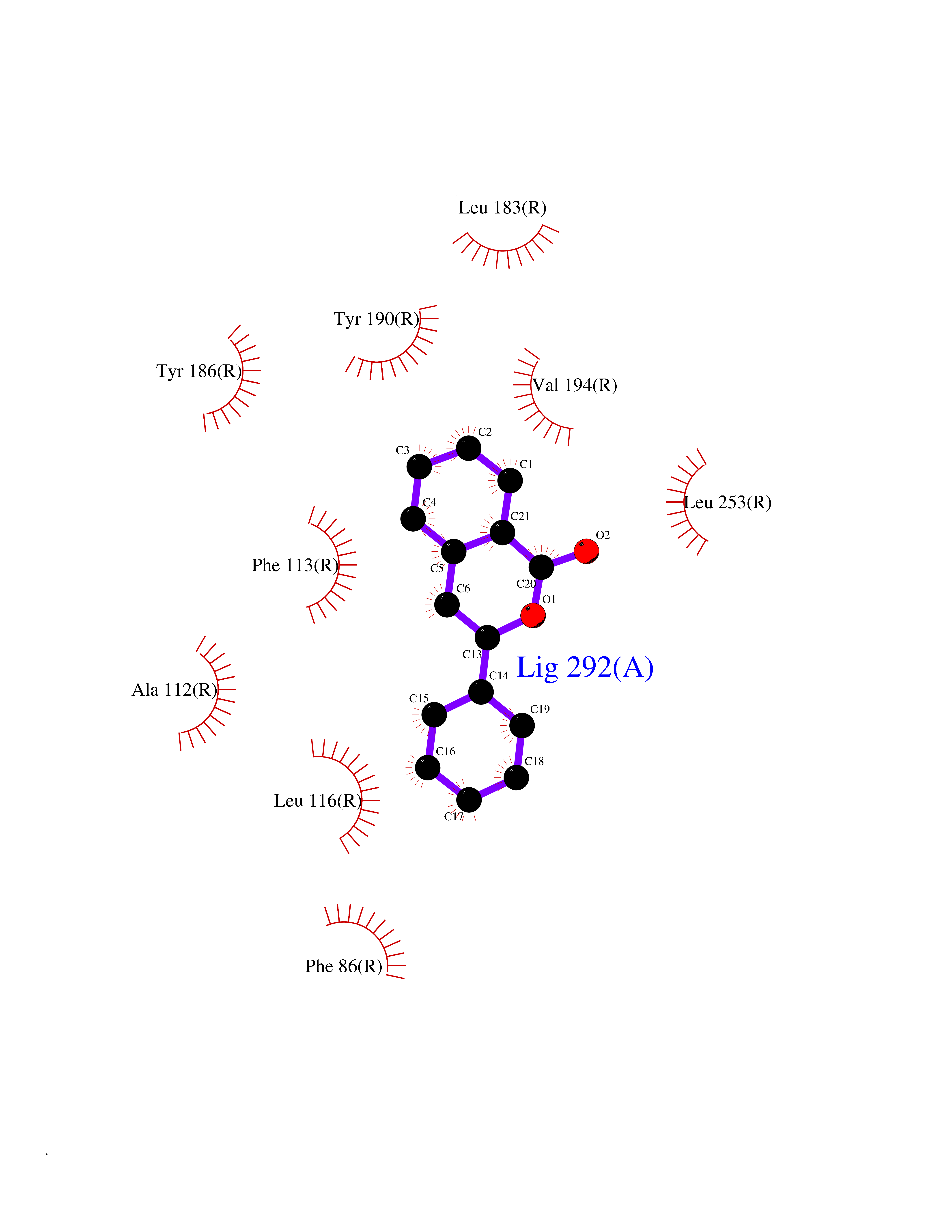 Ligplot Map 3D Binding mode Sequence NKVQEHYNYTKTSRQVASAFIVILCCAIVVENLLVLIAVARNSKFHSAMYLFLGNLAASDLLAGVAFVANTLLSGSVTLRLTPVQWFAREGSAFITLSASVFSLLAIAIERHVAIAKVKLYGSDKSCRMLLLIGASWLISLVLGGLPILGWNCLGHLEACSTVLPLYAKHYVLCVVTIFSIILLAIVALYVRIYCVVRSSQTLALLKTVTIVLGVFIVCWLPAFSILLLDYACPVHSCPILYKAHYFFAVSTLNSLLNPVIYTW Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Guanylate cyclase soluble beta-1 (GUCY1B1) | 7D9R | 7.54 | |
Target general information Gen name GUCY1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Soluble guanylate cyclase small subunit; Guanylate cyclase soluble subunit beta-3; Guanylate cyclase soluble subunit beta-1; GUCY1B3; GUCSB3; GUC1B3; GCS-beta-3; GCS-beta-1 Protein family Adenylyl cyclase class-4/guanylyl cyclase family Biochemical class Phosphorus-oxygen lyase Function Mediates responses to nitric oxide (NO) by catalyzing the biosynthesis of the signaling molecule cGMP. Related diseases Neurodevelopmental disorder with seizures, hypotonia, and brain imaging abnormalities (NEDSHBA) [MIM:618922]: An autosomal recessive neurodevelopmental disorder characterized by global developmental delay, hypotonia, severe to profound intellectual disability, early-onset epilepsy, and microcephaly. Neuroimaging shows cerebral atrophy, thin corpus callosum and hypomyelination in a majority of cases. Death in childhood may occur. {ECO:0000269|PubMed:27435318, ECO:0000269|PubMed:28097321, ECO:0000269|PubMed:32286009, ECO:0000269|PubMed:33476302, ECO:0000269|PubMed:33500274}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09401; DB15456 Interacts with Q02108; Q02108-1 EC number EC 4.6.1.2 Uniprot keywords 3D-structure; Alternative splicing; cGMP biosynthesis; Cytoplasm; Direct protein sequencing; GTP-binding; Heme; Iron; Lyase; Metal-binding; Nucleotide-binding; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A,B Molecular weight (Da) 121177 Length 1071 Aromaticity 0.09 Instability index 42.35 Isoelectric point 6.18 Charge (pH=7) -11.86 2D Binding mode Binding energy (Kcal/mol) -10.28 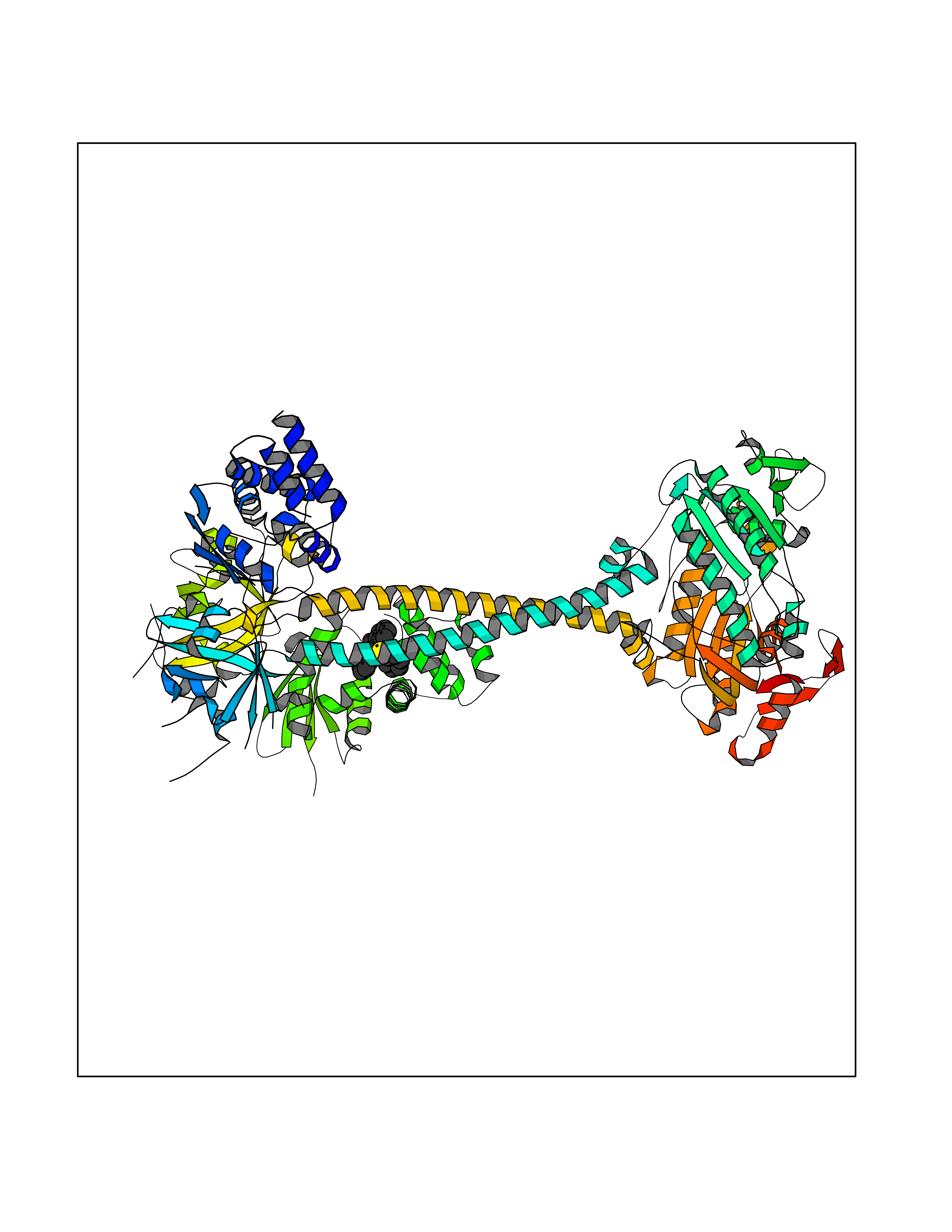 Molscript Map 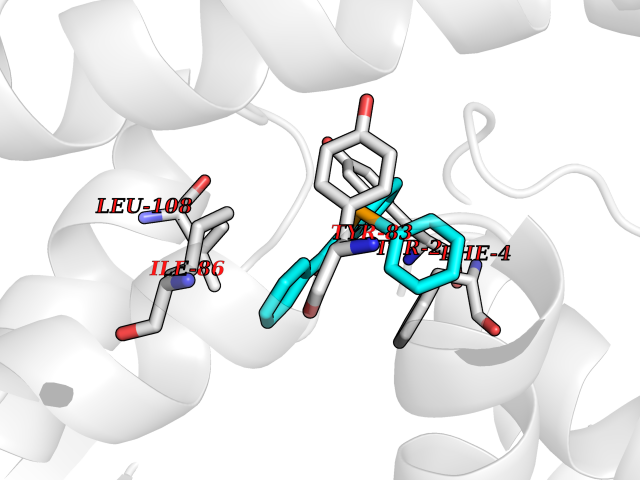 Pymol Map 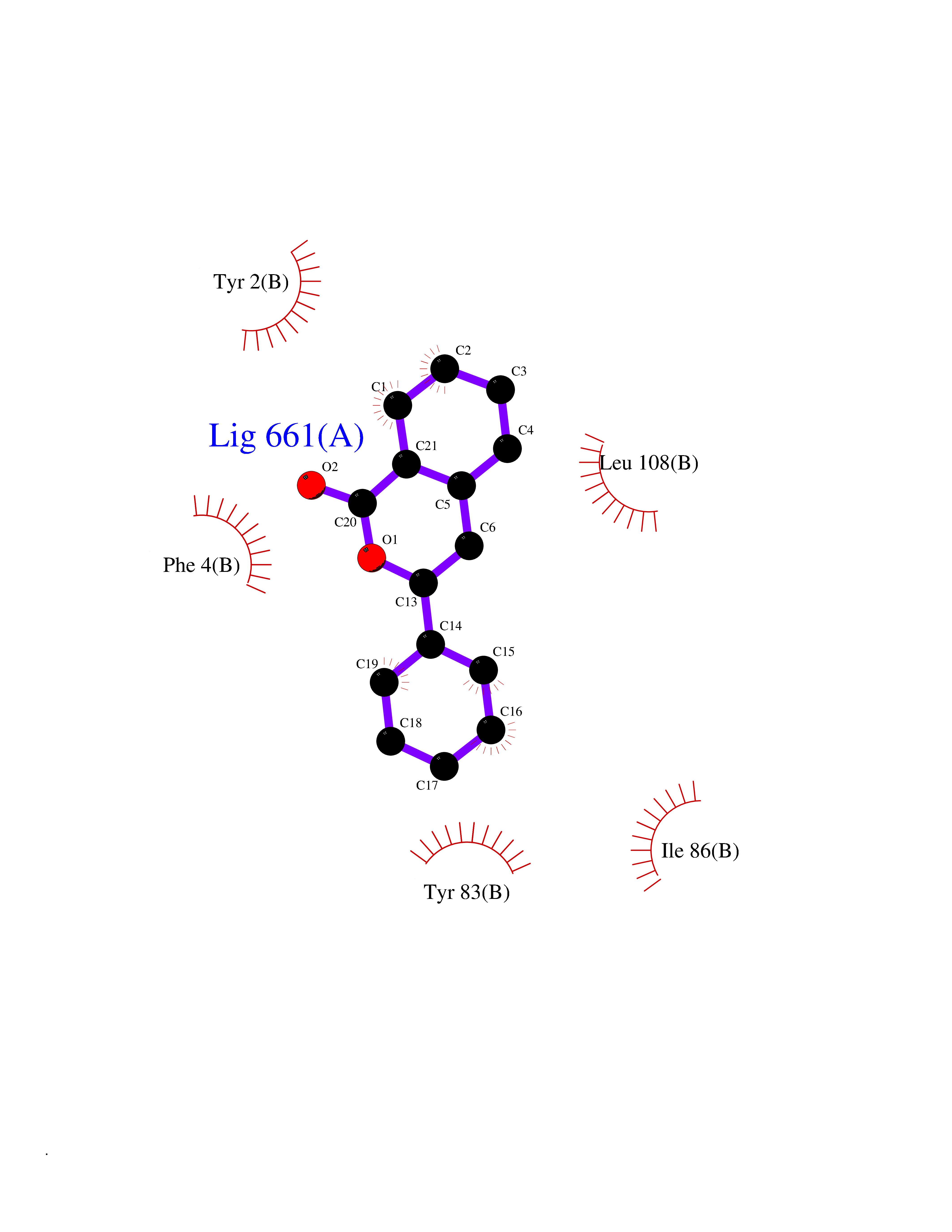 Ligplot Map 3D Binding mode Sequence SRVYLHTLAESICKLIFPEFERLNVALQRTLAKHKFEKTIAEQAVAAGVPVEVIKESLGEEVFKICYEEDENILGVVGGTLKDFLNSFSTLLKQASILCLLHVYYFTTSLILPGIIKAAAHVLYETEVEVSLYLLYSVHSLVIPTSLFCKTFPFHFMFDKDMTILQFGNGIRRLMNRRDKPNFEEYFEILTPKINQTFSGIMTMLNMQFVVRVRRVMDLKGQMIYIVESSAILFLGSPCVLYLSDIPIHNALRDVVLIGEQARAQDGLKKRLGKLKATLEQAHQALEEEKKKTVDLLCSIFPCEVAQQLWQGQVVQAKKFSNVTMLFSDIVGFTAICSQCSPLQVITMLNALYTRFDQQCGELDVYKVETIGDAYCVAGGLHKESDTHAVQIALMAVKMMELSDEVMSPHGEPIKMRIGLHSGSVFAGVVGVKMPRYCLFGNNVTLANKFESCSVPRKINVSPTTYRLLKDCPGFVFTPRSREELPPNFPSEIPGICHFLDAMYGFVNHALELLVIRNYGPEVWEDIKKEAQLDEEGQFLVRIIYDDSKTYDLVAAASKVLNLNAGEILQMFGKMFFVFCQESGYDTILRVLGSNVREFLQNLDALHDHLATIYPGMRAPSFRCTDAEKGKGLILHYYSEREGLQDIVIGIIKTVAQQIHGTEIDMKVIQQRNEECDHTQFLIEEKEESRISPYTFCKAFPFHIIFDRDLVVTQCGNAIYRVLPQLQPGNCSLLSVFSLVRPHIDISFHGILSHINTVFVLRSKEGLLDSCLRLKGQMIYLPEADSILFLCSPSVMNLDDLTRRGLYLSDIPLHDATRDLVLLGEQFREEYKLTQELEILTDRLQLTLRALEDEKKKTDTLLYSVLPPSVANELRHKRPVPAKRYDNVTILFSGIVGFNAFCSKHAGAMKIVNLLNDLYTRFDTLTDSRKNPFVYKVETVGDKYMTVSGLPEPCIHHARSICHLALDMMEIAGQVQVDGESVQITIGIHTGEVVTGVIGQRMPRYCLFGNTVNLTSRTETTGEKGKINVSEYTYRCLMSPENSDPQFHLEHRGPVSMKGKKEPMQVWFLSR Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Folate receptor beta (FOLR2) | 4KN0 | 7.53 | |
Target general information Gen name FOLR2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Placental folate-binding protein; Folate receptor, fetal/placental; Folate receptor type-beta; Folate receptor 2; FR-beta; FOLR2 Protein family Folate receptor family Biochemical class Folate receptor Function Binds to folate and reduced folic acid derivatives and mediates delivery of 5-methyltetrahydrofolate and folate analogs into the interior of cells. Has high affinity for folate and folic acid analogs at neutral pH. Exposure to slightly acidic pH after receptor endocytosis triggers a conformation change that strongly reduces its affinity for folates and mediates their release. Related diseases Acute hepatic porphyria (AHEPP) [MIM:612740]: A form of porphyria. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. AHP is characterized by attacks of gastrointestinal disturbances, abdominal colic, paralyses and peripheral neuropathy. Most attacks are precipitated by drugs, alcohol, caloric deprivation, infections, or endocrine factors. {ECO:0000269|PubMed:10706561, ECO:0000269|PubMed:1309003, ECO:0000269|PubMed:1569184, ECO:0000269|PubMed:17236137, ECO:0000269|PubMed:2063868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00158; DB00563; DB05168 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Direct protein sequencing; Disulfide bond; Folate-binding; Glycoprotein; GPI-anchor; Lipoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Secreted; Signal; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23841.6 Length 205 Aromaticity 0.12 Instability index 56.78 Isoelectric point 7.92 Charge (pH=7) 2.58 2D Binding mode Binding energy (Kcal/mol) -10.27 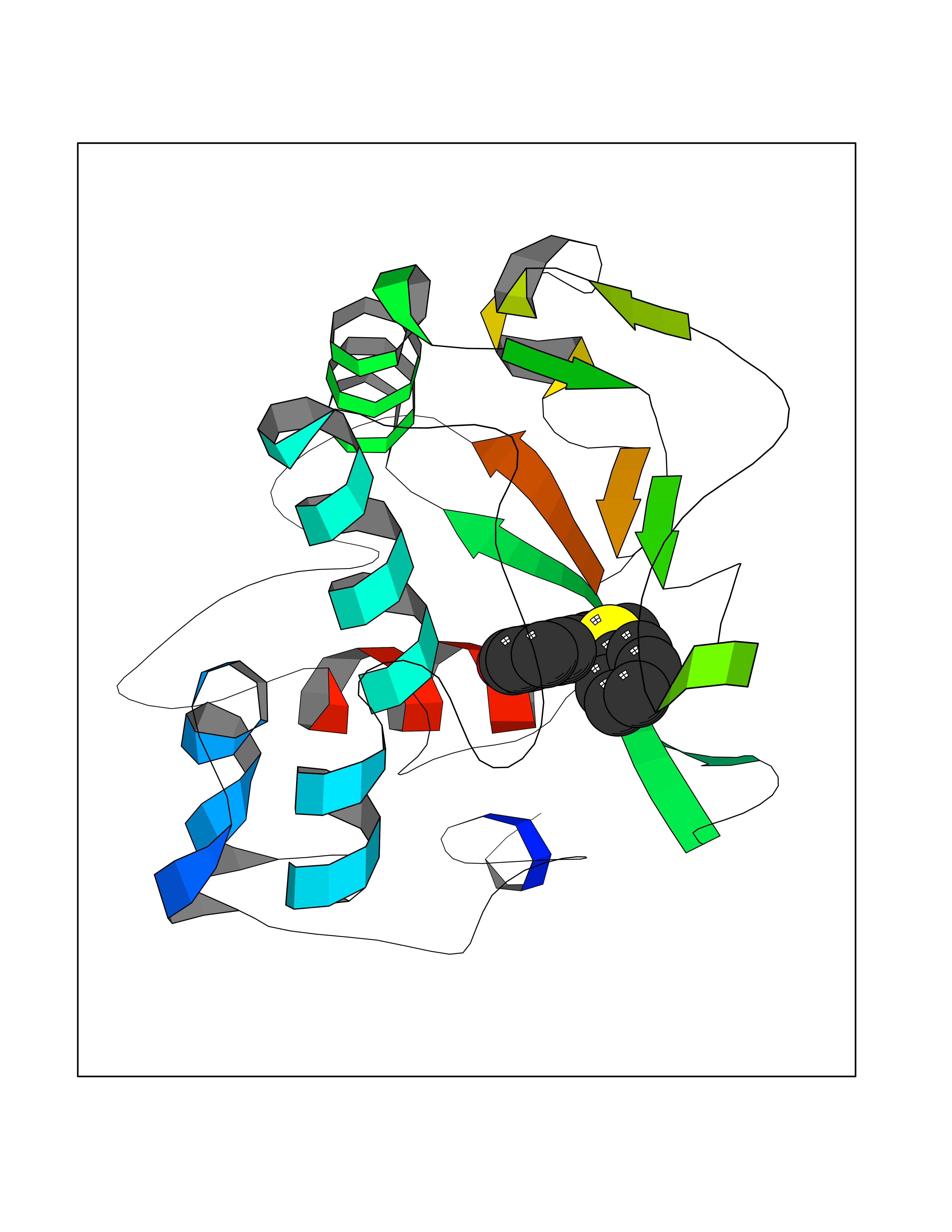 Molscript Map 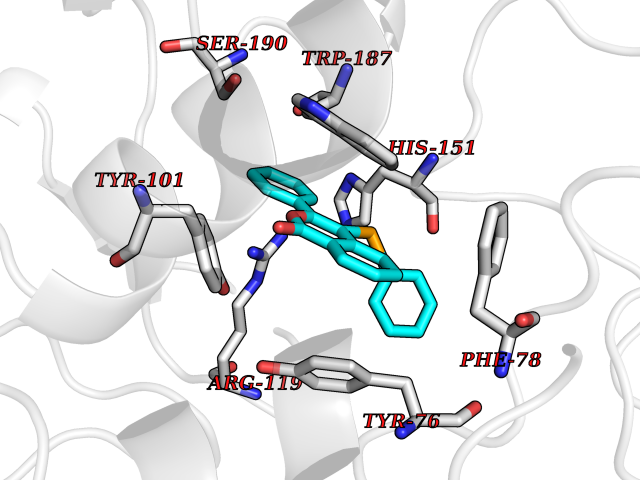 Pymol Map 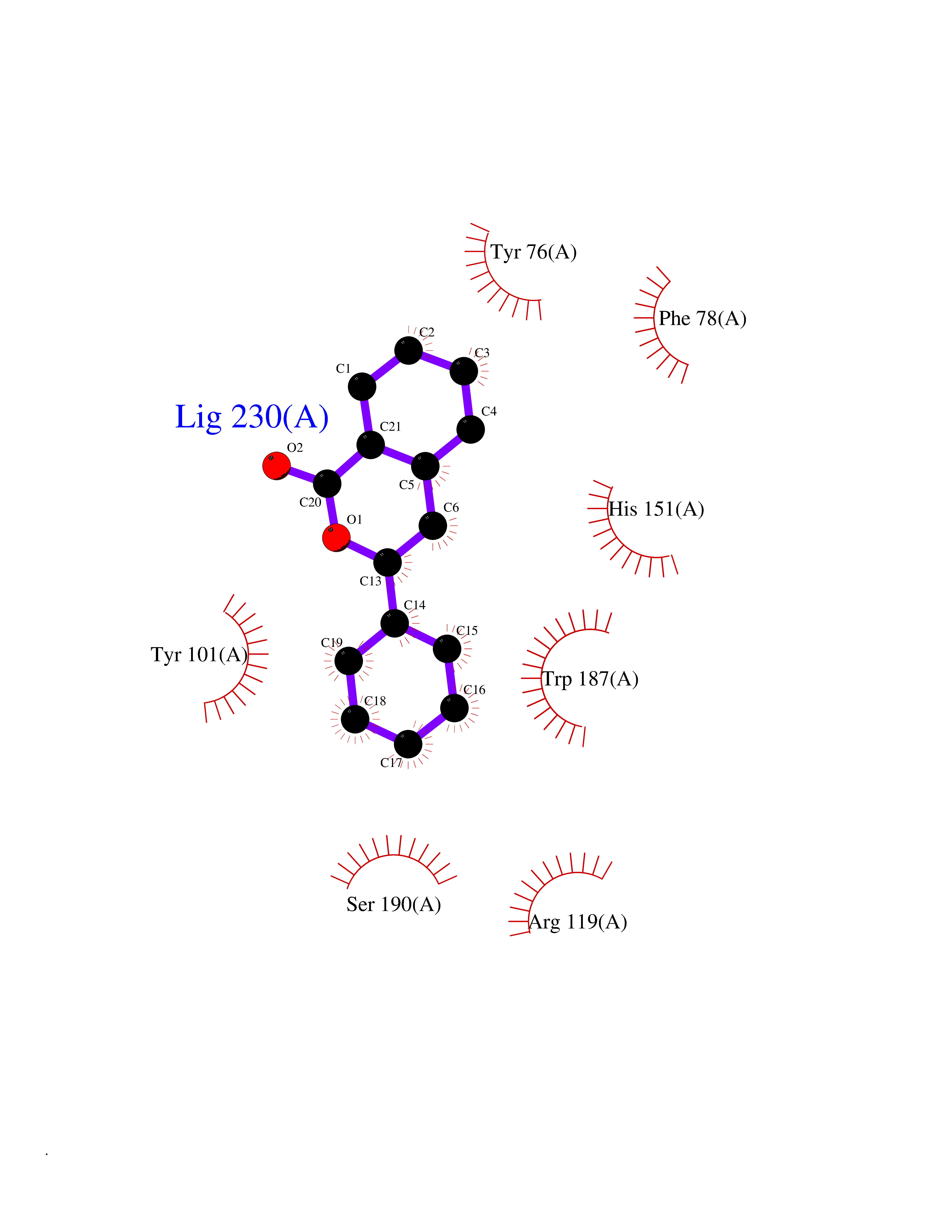 Ligplot Map 3D Binding mode Sequence RTDLLNVCMDAKHHKTKPGPEDKLHDQCSPWKKNACCTASTSQELHKDTSRLYNFNWDHCGKMEPACKRHFIQDTCLYECSPNLGPWIQQVNQSWRKERFLDVPLCKEDCQRWWEDCHTSHTCKSNWHRGWDWTSGVNKCPAGALCRTFESYFPTPAALCEGLWSHSYKVSNYSRGSGRCIQMWFDSAQGNPNEEVARFYAAAMH Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | SEC14-like protein 2 | 4OMJ | 7.53 | |
Target general information Gen name SEC14L2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms KIAA1658;C22orf6;KIAA1186 Protein family NA Biochemical class Transport protein Function Phospholipid binding.Transporter activity.Vitamin E binding. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB14001; DB14002; DB11635; DB11251; DB00163 Interacts with NA EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Cytoplasm; Lipid-binding; Nucleus; Proteomics identification; Reference proteome; Transcription; Transcription regulation; Transport Protein physicochemical properties Chain ID A,B Molecular weight (Da) 31533.3 Length 274 Aromaticity 0.1 Instability index 49.26 Isoelectric point 8.34 Charge (pH=7) 2.81 2D Binding mode Binding energy (Kcal/mol) -10.26 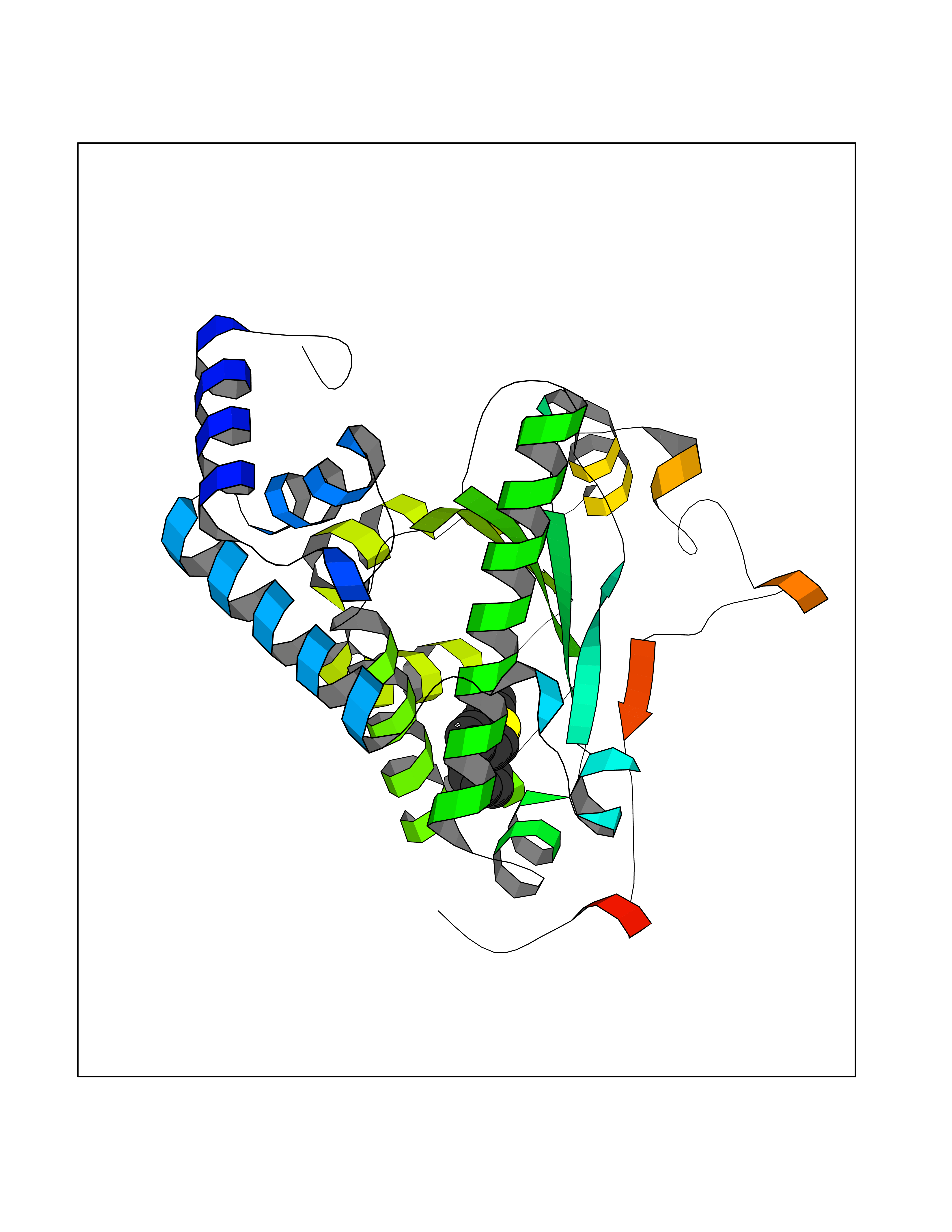 Molscript Map 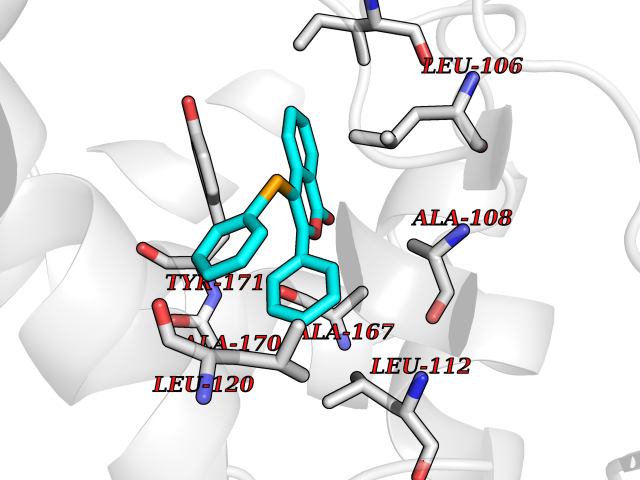 Pymol Map 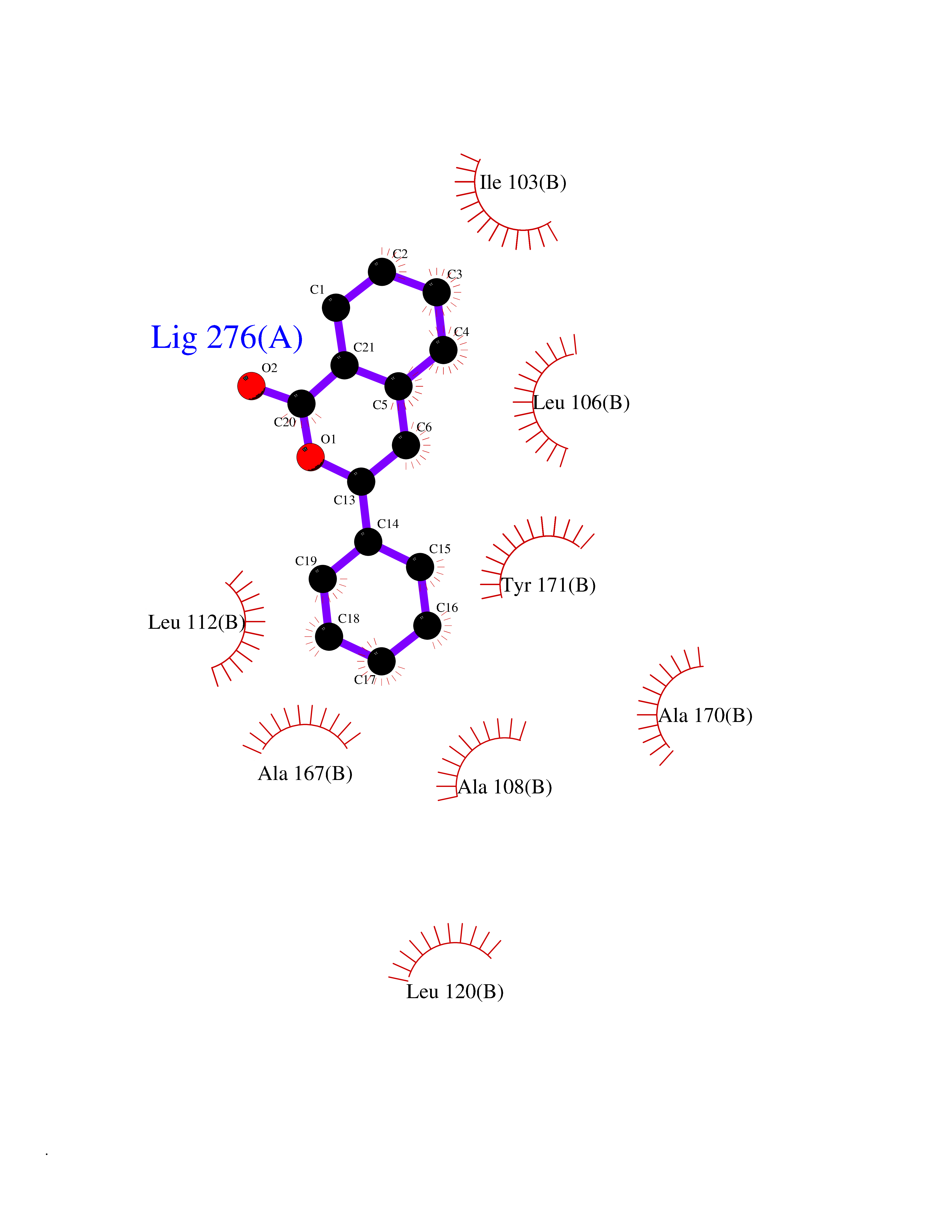 Ligplot Map 3D Binding mode Sequence MSGRVGDLSPRQKEALAKFRENVQDVLPALPNPDDYFLLRWLRARSFDLQKSEAMLRKHVEFRKQKDIDNIISWQPPEVIQQYLSGGMCGYDLDGCPVWYDIIGPLDAKGLLFSASKQDLLRTKMRECELLLQECAHQTTKLGRKVETITIIYDCEGLGLKHLWKPAVEAYGEFLCMFEENYPETLKRLFVVKAPKLFPVAYNLIKPFLSEDTRKKIMVLGANWKEVLLKHISPDQVPVEYGGTMTDPDGNPKCKSKINYGGDIPRKYYVRDQV Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Histidine decarboxylase (HDC) | 4E1O | 7.53 | |
Target general information Gen name HDC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Human histidine decarboxylase Protein family Group II decarboxylase family Biochemical class Carbon-carbon lyase Function Catalyzes the biosynthesis of histamine from histidine. Related diseases Corticosterone methyloxidase 1 deficiency (CMO-1 deficiency) [MIM:203400]: Autosomal recessive disorder of aldosterone biosynthesis. There are two biochemically different forms of selective aldosterone deficiency be termed corticosterone methyloxidase (CMO) deficiency type 1 and type 2. In CMO-1 deficiency, aldosterone is undetectable in plasma, while its immediate precursor, 18-hydroxycorticosterone, is low or normal. {ECO:0000269|PubMed:11238478, ECO:0000269|PubMed:8439335, ECO:0000269|PubMed:9177280}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Corticosterone methyloxidase 2 deficiency (CMO-2 deficiency) [MIM:610600]: Autosomal recessive disorder of aldosterone biosynthesis. In CMO-2 deficiency, aldosterone can be low or normal, but at the expense of increased secretion of 18-hydroxycorticosterone. Consequently, patients have a greatly increased ratio of 18-hydroxycorticosterone to aldosterone and a low ratio of corticosterone to 18-hydroxycorticosterone in serum. {ECO:0000269|PubMed:12788848, ECO:0000269|PubMed:1346492, ECO:0000269|PubMed:1594605, ECO:0000269|PubMed:9625333, ECO:0000269|PubMed:9814506}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00117; DB00114 Interacts with Q86UW9 EC number EC 4.1.1.22 Uniprot keywords 3D-structure; Alternative splicing; Catecholamine biosynthesis; Decarboxylase; Lyase; Proteomics identification; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A,B,C,D,E,F Molecular weight (Da) 107706 Length 956 Aromaticity 0.1 Instability index 55.17 Isoelectric point 6.23 Charge (pH=7) -9.63 2D Binding mode Binding energy (Kcal/mol) -10.27  Molscript Map 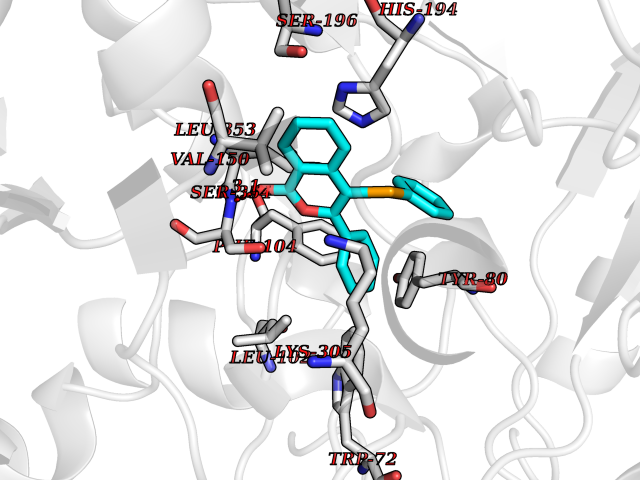 Pymol Map 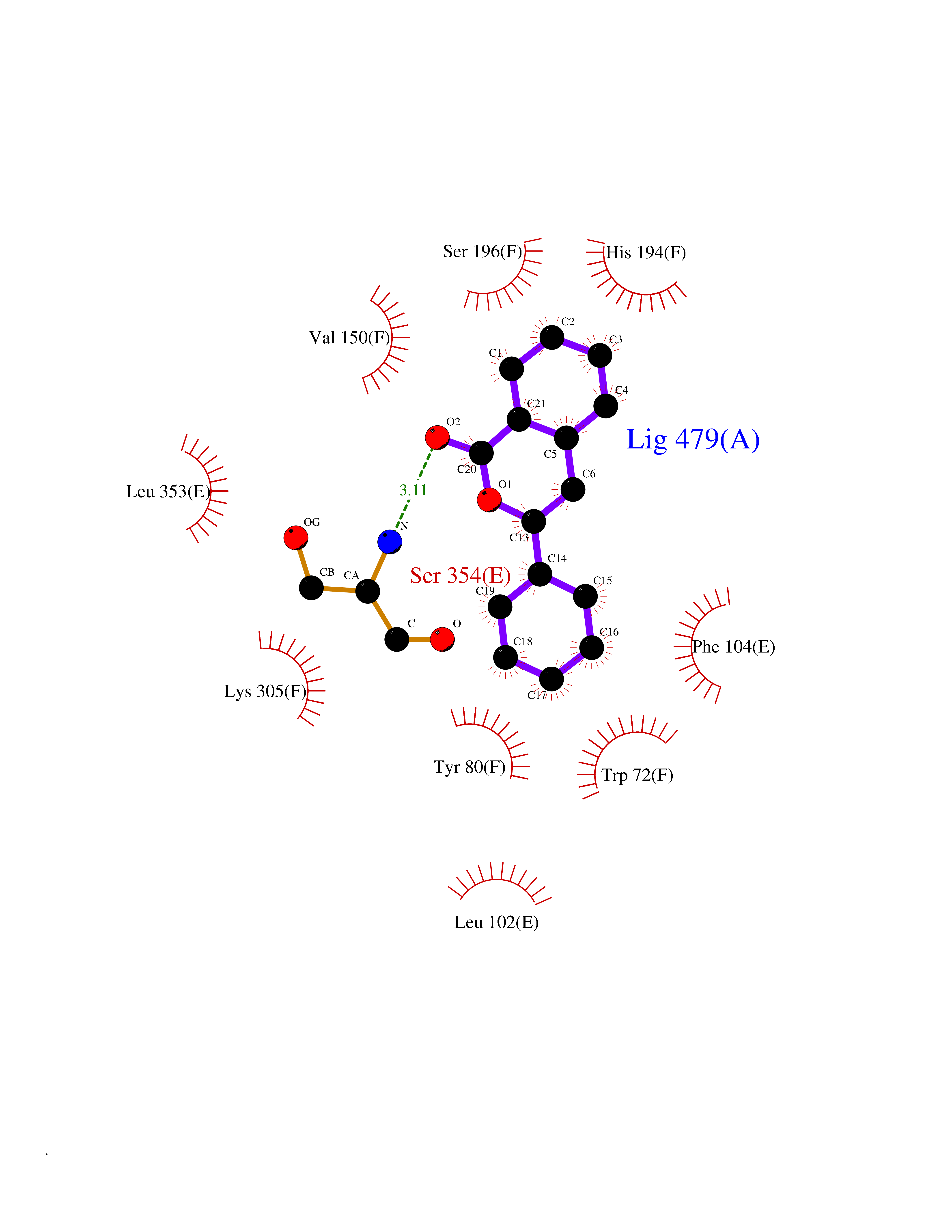 Ligplot Map 3D Binding mode Sequence GSMEPEEYRERGREMVDYICQYLSTVRERRVTPDVQPGYLRAQLPESAPEDPDSWDSIFGDIERIIMPGVVHWQSPHMHAYYPALTSWPSLLGDMLADAINCLGFTWASSPACTELEMNVMDWLAKMLGLPEHFLHHHPSSQGGGVLQSTVSESTLIALLAARKNKILEMKTSEPDADESSLNARLVAYASDQAHSSVEKAGLISLVKMKFLPVDDNFSLRGEALQKAIEEDKQRGLVPVFVCATLGTTGVCAFDXLSELGPICAREGLWLHIDAAYAGTAFLCPEFRGFLKGIEYADSFTFNPSKWMMVHFDCTGFWVKDKYKLQQTFSVNPIYLRHANSGVATDFMHWQIPLSRRFRSVKLWFVIRSFGVKNLQAHVRHGTEMAKYFESLVRNDPSFEIPAKRHLGLVVFRLKGPNSLTENVLKEIAKAGRLFLIPATIQDKLIIRFTVTSQFTTRDDILRDWNLIRDAATLILSQGSMEPEEYRERGREMVDYICQYLSTVRERRVTPDVQPGYLRAQLPESAPEDPDSWDSIFGDIERIIMPGVVHWQSPHMHAYYPALTSWPSLLGDMLADAINCLGFTWASSPACTELEMNVMDWLAKMLGLPEHFLHHHPSSQGGGVLQSTVSESTLIALLAARKNKILEMKTSEPDADESSLNARLVAYASDQAHSSVEKAGLISLVKMKFLPVDDNFSLRGEALQKAIEEDKQRGLVPVFVCATLGTTGVCAFDXLSELGPICAREGLWLHIDAAYAGTAFLCPEFRGFLKGIEYADSFTFNPSKWMMVHFDCTGFWVKDKYKLQQTFSVNPIYLRHANSGVATDFMHWQIPLSRRFRSVKLWFVIRSFGVKNLQAHVRHGTEMAKYFESLVRNDPSFEIPAKRHLGLVVFRLKGPNSLTENVLKEIAKAGRLFLIPATIQDKLIIRFTVTSQFTTRDDILRDWNLIRDAATLILSQ Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Bacterial Flavohemoglobin (Bact hmp) | 1GVH | 7.53 | |
Target general information Gen name Bact hmp Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms Nitric oxide dioxygenase; NOD; NO oxygenase; Hemoglobin-like protein; HMP; Ferrisiderophore reductase B; Dihydropteridine reductase Protein family Globin family, Two-domain flavohemoproteins subfamily; Flavoprotein pyridine nucleotide cytochrome reductase family Biochemical class Paired donor oxygen oxidoreductase Function Various electron acceptors arealso reduced by HMP in vitro, including dihydropterine, ferrisiderophores, ferric citrate, cytochrome c, nitrite, S-nitrosoglutathione, and alkylhydroperoxides. However, it is unknown if these reactions are of any biological significance in vivo. Related diseases Ovarian dysgenesis 1 (ODG1) [MIM:233300]: An autosomal recessive disease characterized by primary amenorrhea, variable development of secondary sex characteristics, poorly developed streak ovaries, and high serum levels of follicle-stimulating hormone (FSH) and luteinizing hormone (LH). {ECO:0000269|PubMed:10551778, ECO:0000269|PubMed:11889179, ECO:0000269|PubMed:12571157, ECO:0000269|PubMed:12915623, ECO:0000269|PubMed:7553856, ECO:0000269|PubMed:9769327, ECO:0000269|PubMed:9851774}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Ovarian hyperstimulation syndrome (OHSS) [MIM:608115]: Disorder which occurs either spontaneously or most often as an iatrogenic complication of ovarian stimulation treatments for in vitro fertilization. The clinical manifestations vary from abdominal distention and discomfort to potentially life-threatening, massive ovarian enlargement and capillary leak with fluid sequestration. Pathologic features of this syndrome include the presence of multiple serous and hemorrhagic follicular cysts lined by luteinized cells, a condition called hyperreactio luteinalis. {ECO:0000269|PubMed:12930927, ECO:0000269|PubMed:12930928, ECO:0000269|PubMed:15080154, ECO:0000269|PubMed:16278261, ECO:0000269|PubMed:17721928, ECO:0000269|PubMed:24058690, ECO:0000269|PubMed:25581598}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number EC 1.14.12.17 Uniprot keywords 3D-structure; Cytoplasm; Detoxification; Direct protein sequencing; FAD; Flavoprotein; Heme; Iron; Metal-binding; NAD; NADP; Oxidoreductase; Oxygen transport; Reference proteome; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 43867.1 Length 396 Aromaticity 0.1 Instability index 28.85 Isoelectric point 5.48 Charge (pH=7) -12.32 2D Binding mode Binding energy (Kcal/mol) -10.28  Molscript Map 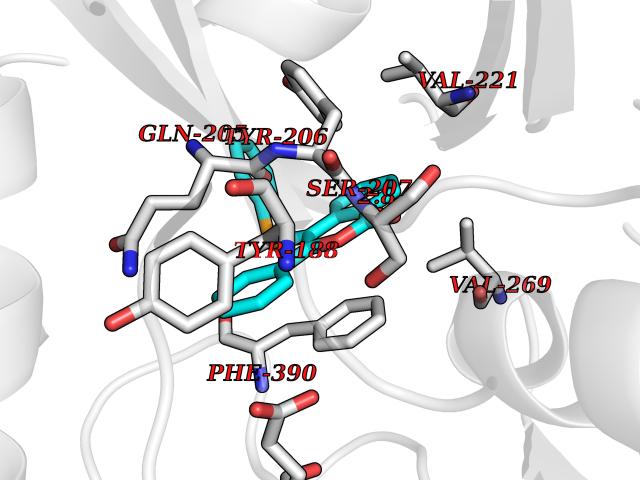 Pymol Map 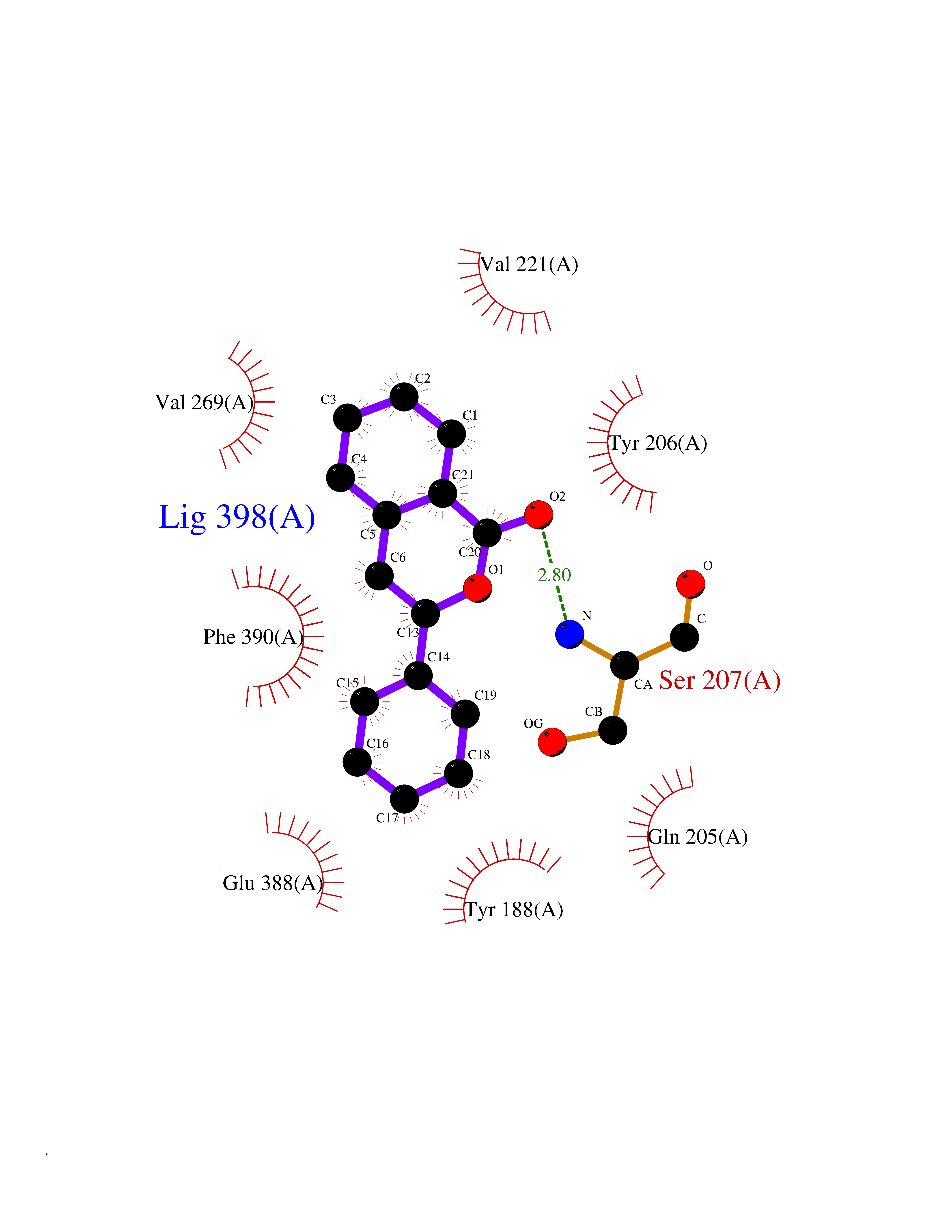 Ligplot Map 3D Binding mode Sequence MLDAQTIATVKATIPLLVETGPKLTAHFYDRMFTHNPELKEIFNMSNQRNGDQREALFNAIAAYASNIENLPALLPAVEKIAQKHTSFQIKPEQYNIVGEHLLATLDEMFSPGQEVLDAWGKAYGVLANVFINREAEIYNENASKAGGWEGTRDFRIVAKTPRSALITSFELEPVDGGAVAEYRPGQYLGVWLKPEGFPHQEIRQYSLTRKPDGKGYRIAVKREEGGQVSNWLHNHANVGDVVKLVAPAGDFFMAVADDTPVTLISAGVGQTPMLAMLDTLAKAGHTAQVNWFHAAENGDVHAFADEVKELGQSLPRFTAHTWYRQPSEADRAKGQFDSEGLMDLSKLEGAFSDPTMQFYLCGPVGFMQFTAKQLVDLGVKQENIHYECFGPHKVL Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Glutamate receptor ionotropic NMDA 2A (NMDAR2A) | 5KCJ | 7.53 | |
Target general information Gen name GRIN2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms NR2A; NMDA receptor NR2A; N-methyl D-aspartate receptor subtype 2A; HNR2A; Glutamate receptor ionotropic, NMDA 2A; Glutamate [NMDA] receptor subunit epsilon-1; GluN2A Protein family Glutamate-gated ion channel (TC 1.A.10.1) family, NR1/GRIN1 subfamily Biochemical class Glutamate-gated ion channel Function Channel activation requires binding of the neurotransmitter glutamate to the epsilon subunit, glycine binding to the zeta subunit, plus membrane depolarization to eliminate channel inhibition by Mg(2+). Sensitivity to glutamate and channel kinetics depend on the subunit composition; channels containing GRIN1 and GRIN2A have higher sensitivity to glutamate and faster kinetics than channels formed by GRIN1 and GRIN2B. Contributes to the slow phase of excitatory postsynaptic current, long-term synaptic potentiation, and learning. Component of NMDA receptor complexes that function as heterotetrameric, ligand-gated ion channels with high calcium permeability and voltage-dependent sensitivity to magnesium. Related diseases Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal dominant (NDHMSD) [MIM:614254]: An autosomal dominant neurodevelopmental disorder characterized by severe intellectual disability and developmental delay, absent speech, muscular hypotonia, dyskinesia, and hyperkinetic movements. Cortical blindness, cerebral atrophy, and seizures are present in some patients. {ECO:0000269|PubMed:21376300, ECO:0000269|PubMed:25167861, ECO:0000269|PubMed:25864721, ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28095420, ECO:0000269|PubMed:28228639, ECO:0000269|PubMed:28389307, ECO:0000269|PubMed:38538865}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurodevelopmental disorder with or without hyperkinetic movements and seizures, autosomal recessive (NDHMSR) [MIM:617820]: An autosomal recessive neurodevelopmental disorder characterized by severe intellectual disability and psychomotor developmental delay, involuntary and stereotypic movements, spasticity, and inability to walk without support. Intractable seizures manifest in some patients. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:28051072}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 101 (DEE101) [MIM:619814]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE101 is an autosomal recessive, severe form characterized by onset of seizures in early infancy. Death in infancy may occur. {ECO:0000269|PubMed:27164704, ECO:0000269|PubMed:34611970}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01931; DB00659; DB06151; DB08838; DB01238; DB00289; DB05824; DB04620; DB03929; DB00647; DB00843; DB00228; DB11823; DB13146; DB06741; DB00142; DB00874; DB08954; DB06738; DB09409; DB09481; DB01043; DB00454; DB00333; DB04896; DB01173; DB00312; DB01174; DB01708; DB00418; DB00193 Interacts with P05067; P35637; Q12879-1; Q13224; Q62936 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Intellectual disability; Ion channel; Ion transport; Ligand-gated ion channel; Magnesium; Membrane; Metal-binding; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Zinc Protein physicochemical properties Chain ID B Molecular weight (Da) 53395.6 Length 469 Aromaticity 0.11 Instability index 29.84 Isoelectric point 8.72 Charge (pH=7) 5.65 2D Binding mode Binding energy (Kcal/mol) -10.27 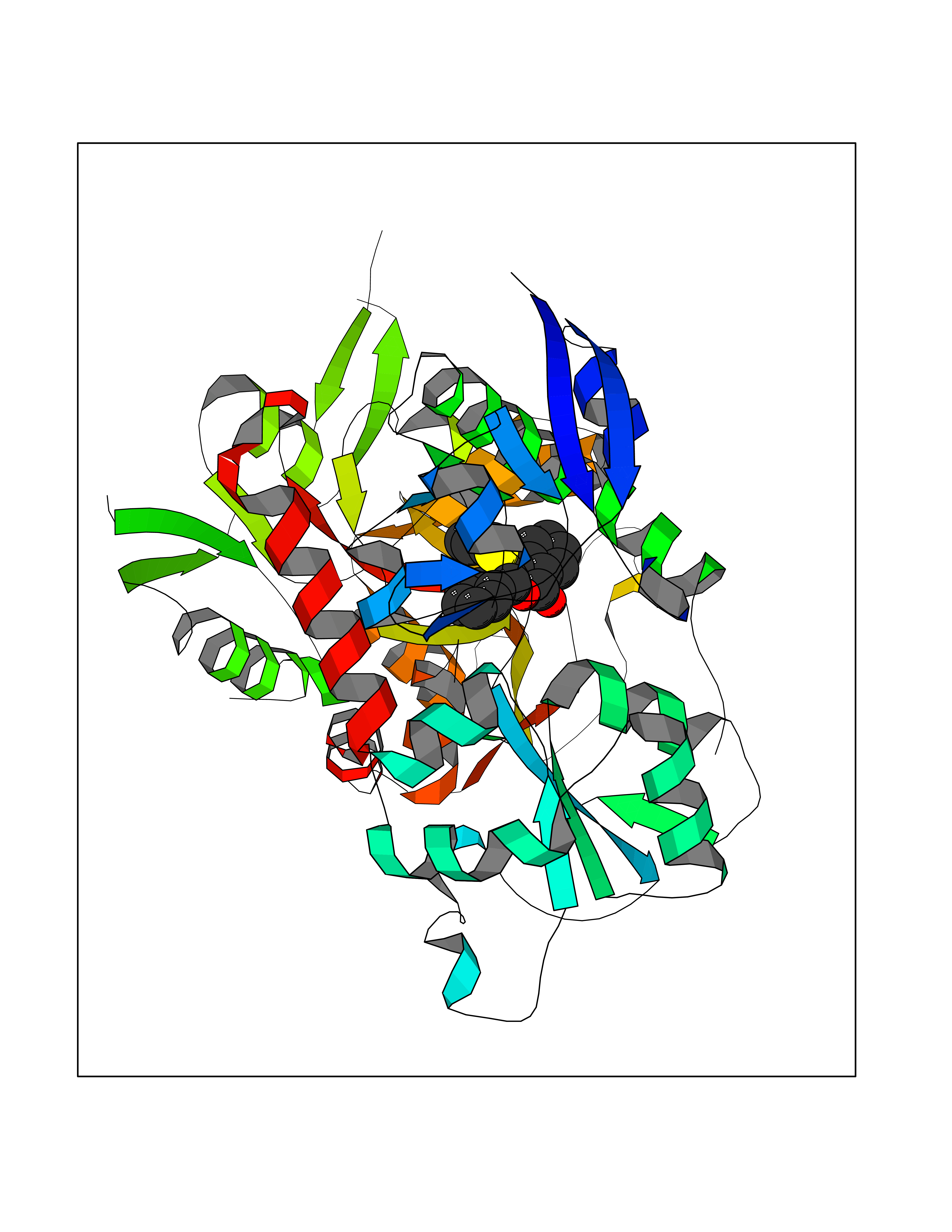 Molscript Map 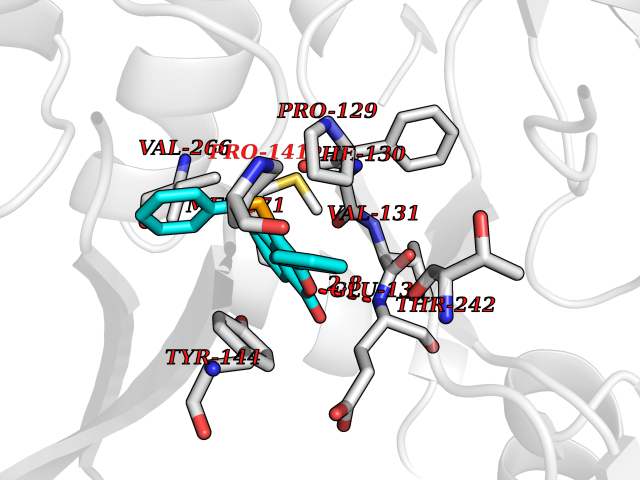 Pymol Map 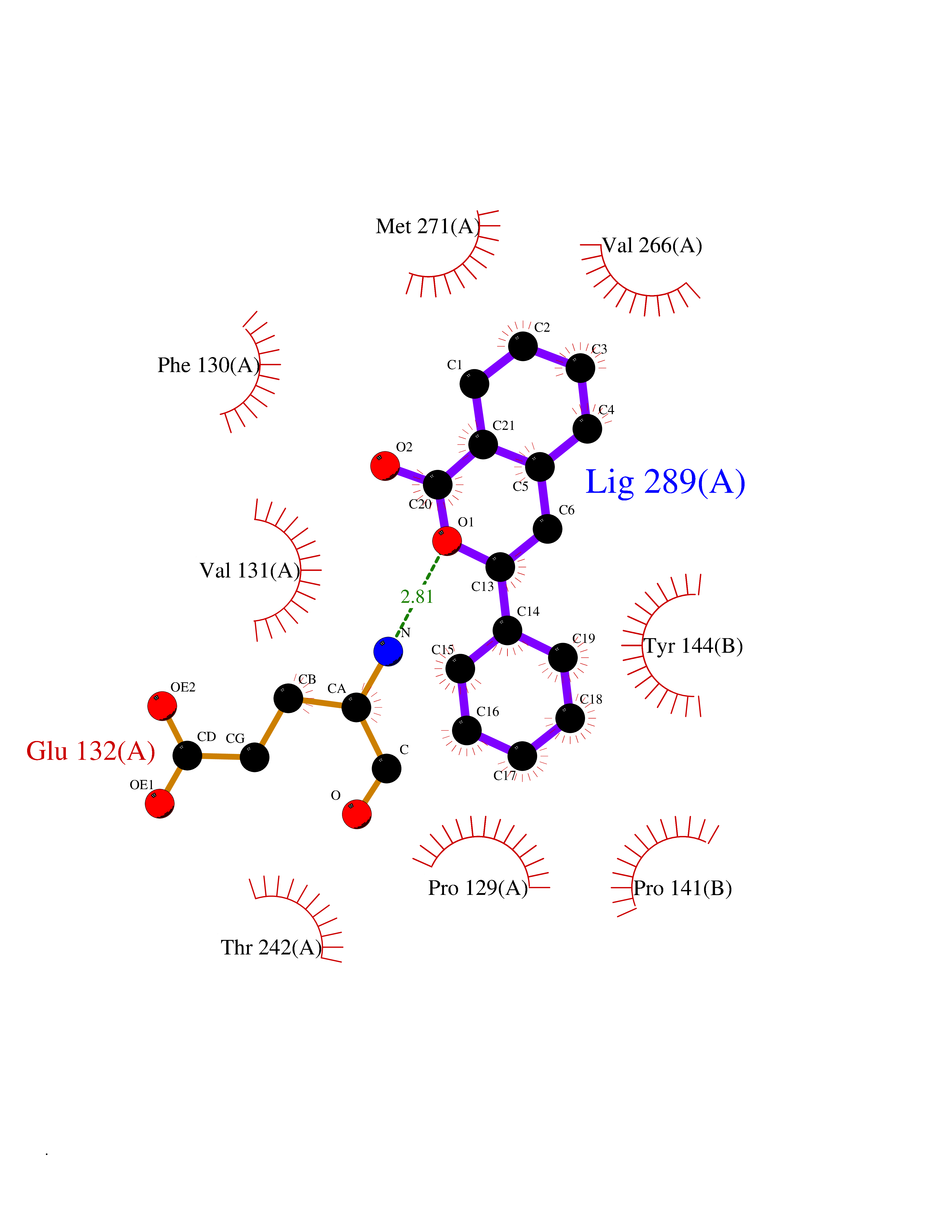 Ligplot Map 3D Binding mode Sequence DNHLSIVTLEEAPFVILKKLSRTVKFTYDLYLVTNGKHGKKVNNVWNGMIGEVVYQRAVMAVGSLTINEERSEVVDFSVPFVETGISVMVSRGTQVTGLSDKKFQRPHDYSPPFRFGTVPNGSTERNIRNNYPYMHQYMTKFNQKGVEDALVSLKTGKLDAFIYDAAVLNYKAGRDEGCKLVTIGSGYIFATTGYGIALQKGSPWKRQIDLALLQFVGDGEMEELETLWLTGICTRLKIVTIHQEPFVYYGFCIDLLIKLARTMNFTYEVHLVADGKFGTQERVNKKEWNGMMGELLSGQADMIVAPLTINNERAQYIEFSKPFKYQGLTILVKKGTRITGINDPRLRNPSDKFIYATVKQSSVDIYFRRQVELSTMYRHMEKHNYESAAEAIQAVRDNKLHAFIWDSAVLEFEASQKCDLVTTGELFFRSGFGIGMRKDSPWKQNVSLSILKSHENGFMEDLDKTWVR Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | LIM domain kinase-2 (LIMK-2) | 7QHG | 7.53 | |
Target general information Gen name LIMK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms LIMK-2; LIM domain kinase 2 Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family Biochemical class Kinase Function Displays serine/threonine-specific phosphorylation of myelin basic protein and histone (MBP) in vitro. Related diseases Intellectual developmental disorder, autosomal recessive 80, with variant lissencephaly (MRT80) [MIM:620653]: An autosomal recessive disorder characterized by global developmental delay, mildly to moderately impaired intellectual development, attention deficit-hyperactivity disorder, hypotonia, seizure, poor social skills, and autistic traits. Brain imaging shows fronto-temporal lissencephaly and pachygyria. {ECO:0000269|PubMed:37880421}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11718; DB12010 Interacts with Q16543; P08238; Q96C90; P62258 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cytoplasm; Cytoskeleton; Kinase; LIM domain; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Serine/threonine-protein kinase; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 32109.2 Length 283 Aromaticity 0.1 Instability index 27.28 Isoelectric point 6.01 Charge (pH=7) -3.93 2D Binding mode Binding energy (Kcal/mol) -10.28  Molscript Map 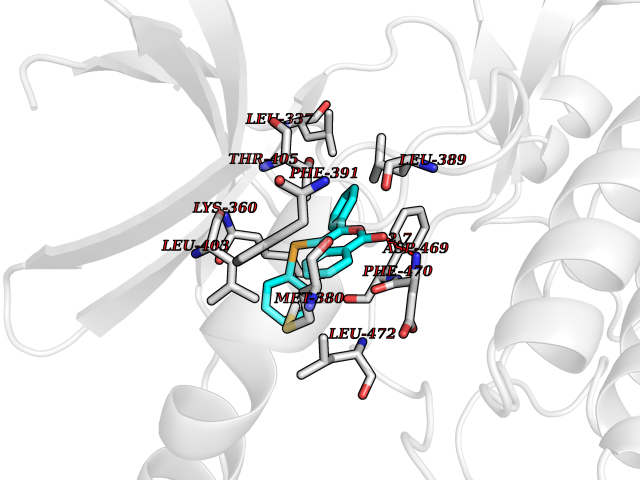 Pymol Map 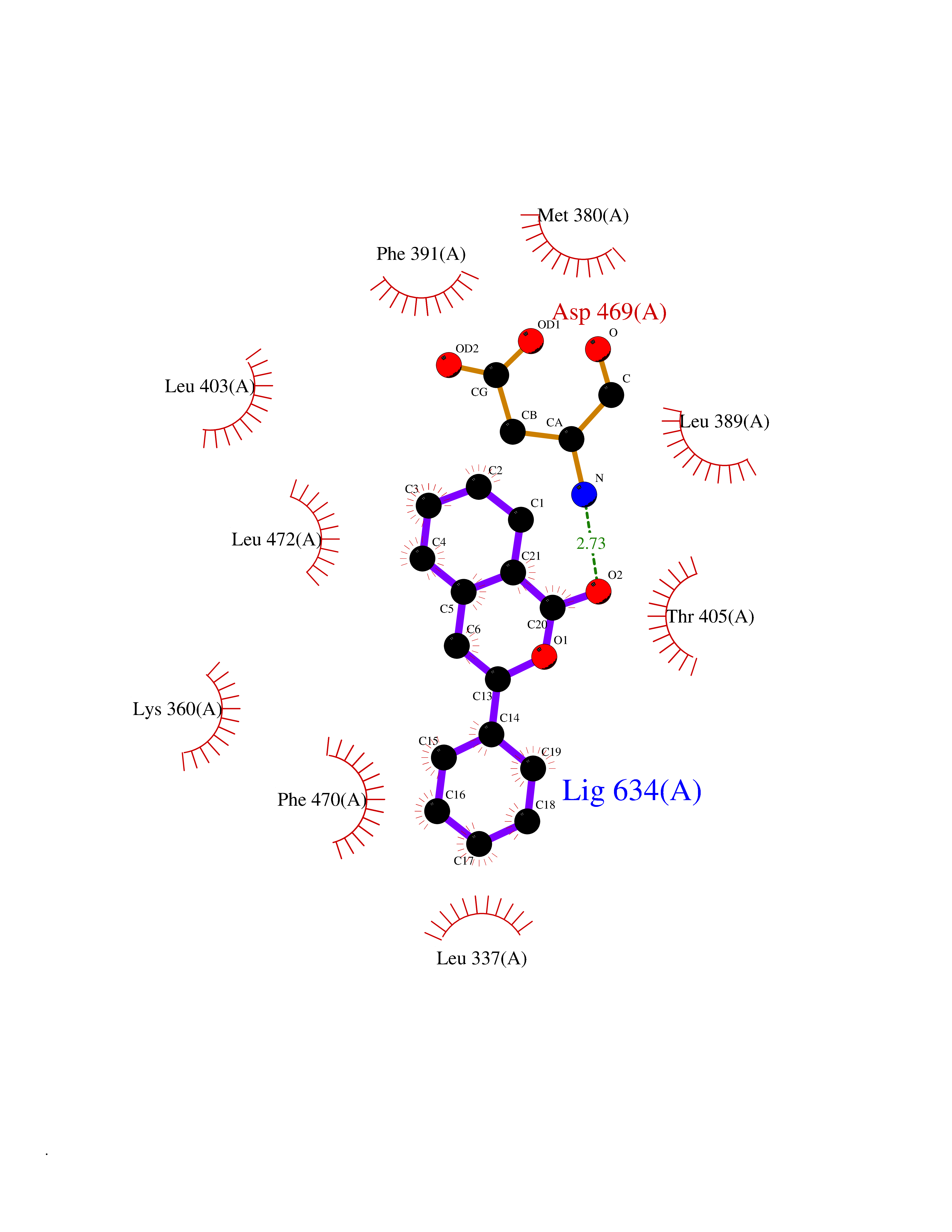 Ligplot Map 3D Binding mode Sequence MDLIHGEVLGKGFFGQAIKVTHKATGKVMVMKELIRCDEETQKTFLTEVKVMRSLDHPNVLKFIGVLYKDKKLNLLTEYIEGGTLKDFLRSMDPFPWQQKVRFAKGIASGMAYLHSMCIIHRDLNSHNCLIKLDKTVVVADFGLSRLIVDRKKRYTVVGNPYWMAPEMLNGKSYDETVDIFSFGIVLCEIIGQVYADPDCLPRTLDFGLNVKLFWEKFVPTDCPPAFFPLAAICCRLEPESRPAFSKLEDSFEALSLYLGELGIPLPAELEELDHTVSMQYGL Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | NAD-dependent deacetylase sirtuin-2 (SIRT2) | 4RMJ | 7.53 | |
Target general information Gen name SIRT2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SIR2like protein 2; SIR2L2; SIR2L; SIR2-like protein 2; Regulatory protein SIR2 homolog 2; NADdependent protein deacetylase sirtuin2; NAD-dependent protein deacetylase sirtuin-2 Protein family Sirtuin family, Class I subfamily Biochemical class Carbon-nitrogen hydrolase Function Participates in the modulation of multiple and diverse biological processes such as cell cycle control, genomic integrity, microtubule dynamics, cell differentiation, metabolic networks, and autophagy. Plays a major role in the control of cell cycle progression and genomic stability. Functions in the antephase checkpoint preventing precocious mitotic entry in response to microtubule stress agents, and hence allowing proper inheritance of chromosomes. Positively regulates the anaphase promoting complex/cyclosome (APC/C) ubiquitin ligase complex activity by deacetylating CDC20 and FZR1, then allowing progression through mitosis. Associates both with chromatin at transcriptional start sites (TSSs) and enhancers of active genes. Plays a role in cell cycle and chromatin compaction through epigenetic modulation of the regulation of histone H4 'Lys-20' methylation (H4K20me1) during early mitosis. Specifically deacetylates histone H4 at 'Lys-16' (H4K16ac) between the G2/M transition and metaphase enabling H4K20me1 deposition by KMT5A leading to ulterior levels of H4K20me2 and H4K20me3 deposition throughout cell cycle, and mitotic S-phase progression. Deacetylates KMT5A modulating KMT5A chromatin localization during the mitotic stress response. Deacetylates also histone H3 at 'Lys-57' (H3K56ac) during the mitotic G2/M transition. Upon bacterium Listeria monocytogenes infection, deacetylates 'Lys-18' of histone H3 in a receptor tyrosine kinase MET- and PI3K/Akt-dependent manner, thereby inhibiting transcriptional activity and promoting late stages of listeria infection. During oocyte meiosis progression, may deacetylate histone H4 at 'Lys-16' (H4K16ac) and alpha-tubulin, regulating spindle assembly and chromosome alignment by influencing microtubule dynamics and kinetochore function. Deacetylates histone H4 at 'Lys-16' (H4K16ac) at the VEGFA promoter and thereby contributes to regulate expression of VEGFA, a key regulator of angiogenesis. Deacetylates alpha-tubulin at 'Lys-40' and hence controls neuronal motility, oligodendroglial cell arbor projection processes and proliferation of non-neuronal cells. Phosphorylation at Ser-368 by a G1/S-specific cyclin E-CDK2 complex inactivates SIRT2-mediated alpha-tubulin deacetylation, negatively regulating cell adhesion, cell migration and neurite outgrowth during neuronal differentiation. Deacetylates PARD3 and participates in the regulation of Schwann cell peripheral myelination formation during early postnatal development and during postinjury remyelination. Involved in several cellular metabolic pathways. Plays a role in the regulation of blood glucose homeostasis by deacetylating and stabilizing phosphoenolpyruvate carboxykinase PCK1 activity in response to low nutrient availability. Acts as a key regulator in the pentose phosphate pathway (PPP) by deacetylating and activating the glucose-6-phosphate G6PD enzyme, and therefore, stimulates the production of cytosolic NADPH to counteract oxidative damage. Maintains energy homeostasis in response to nutrient deprivation as well as energy expenditure by inhibiting adipogenesis and promoting lipolysis. Attenuates adipocyte differentiation by deacetylating and promoting FOXO1 interaction to PPARG and subsequent repression of PPARG-dependent transcriptional activity. Plays a role in the regulation of lysosome-mediated degradation of protein aggregates by autophagy in neuronal cells. Deacetylates FOXO1 in response to oxidative stress or serum deprivation, thereby negatively regulating FOXO1-mediated autophagy. Deacetylates a broad range of transcription factors and co-regulators regulating target gene expression. Deacetylates transcriptional factor FOXO3 stimulating the ubiquitin ligase SCF(SKP2)-mediated FOXO3 ubiquitination and degradation. Deacetylates HIF1A and therefore promotes HIF1A degradation and inhibition of HIF1A transcriptional activity in tumor cells in response to hypoxia. Deacetylates RELA in the cytoplasm inhibiting NF-kappaB-dependent transcription activation upon TNF-alpha stimulation. Inhibits transcriptional activation by deacetylating p53/TP53 and EP300. Deacetylates also EIF5A. Functions as a negative regulator on oxidative stress-tolerance in response to anoxia-reoxygenation conditions. Plays a role as tumor suppressor. NAD-dependent protein deacetylase, which deacetylates internal lysines on histone and alpha-tubulin as well as many other proteins such as key transcription factors. Related diseases Deafness, autosomal recessive, 39 (DFNB39) [MIM:608265]: A form of profound prelingual sensorineural hearing loss. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. {ECO:0000269|PubMed:19576567}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15493 Interacts with O60566; O60729; P11413; Q92831; Q04206; Q9BYB0; Q12834; Q9UM11 EC number EC 3.5.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Autophagy; Cell cycle; Cell division; Cell membrane; Cell projection; Chromosome; Cytoplasm; Cytoskeleton; Differentiation; Immunity; Innate immunity; Meiosis; Membrane; Metal-binding; Microtubule; Mitosis; NAD; Neurodegeneration; Neurogenesis; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 34093.1 Length 301 Aromaticity 0.1 Instability index 45.2 Isoelectric point 5.59 Charge (pH=7) -6.94 2D Binding mode Binding energy (Kcal/mol) -10.27 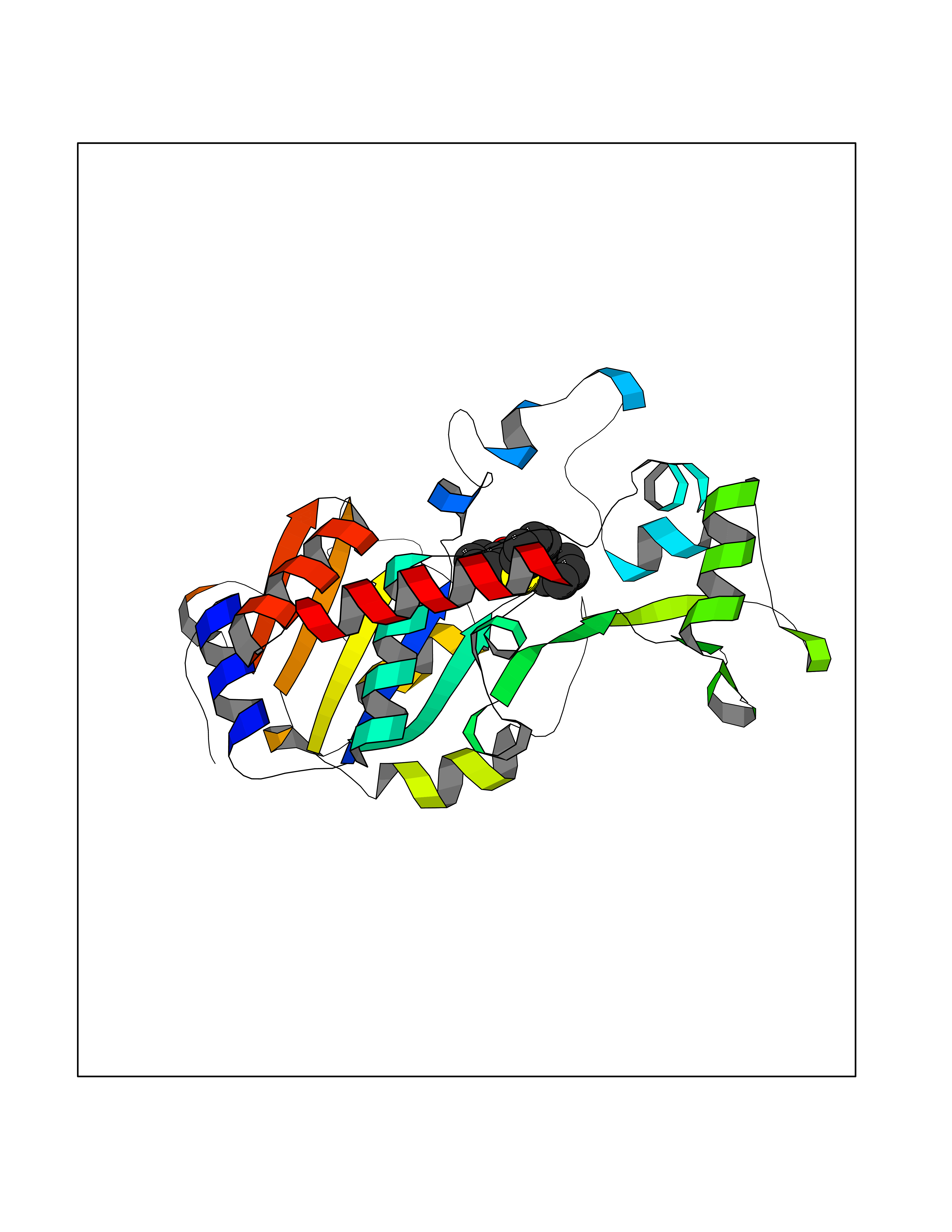 Molscript Map 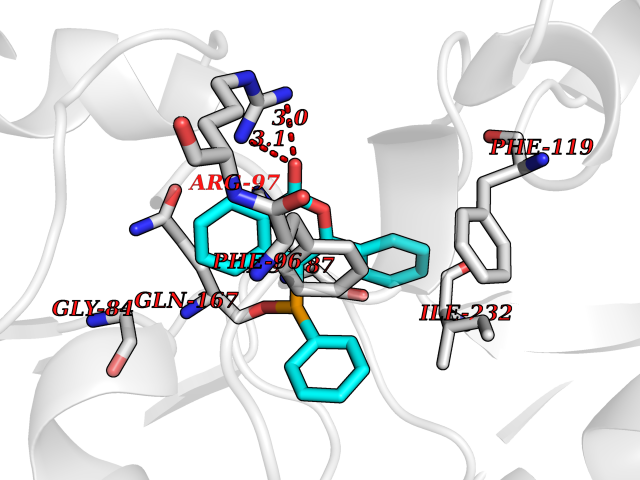 Pymol Map 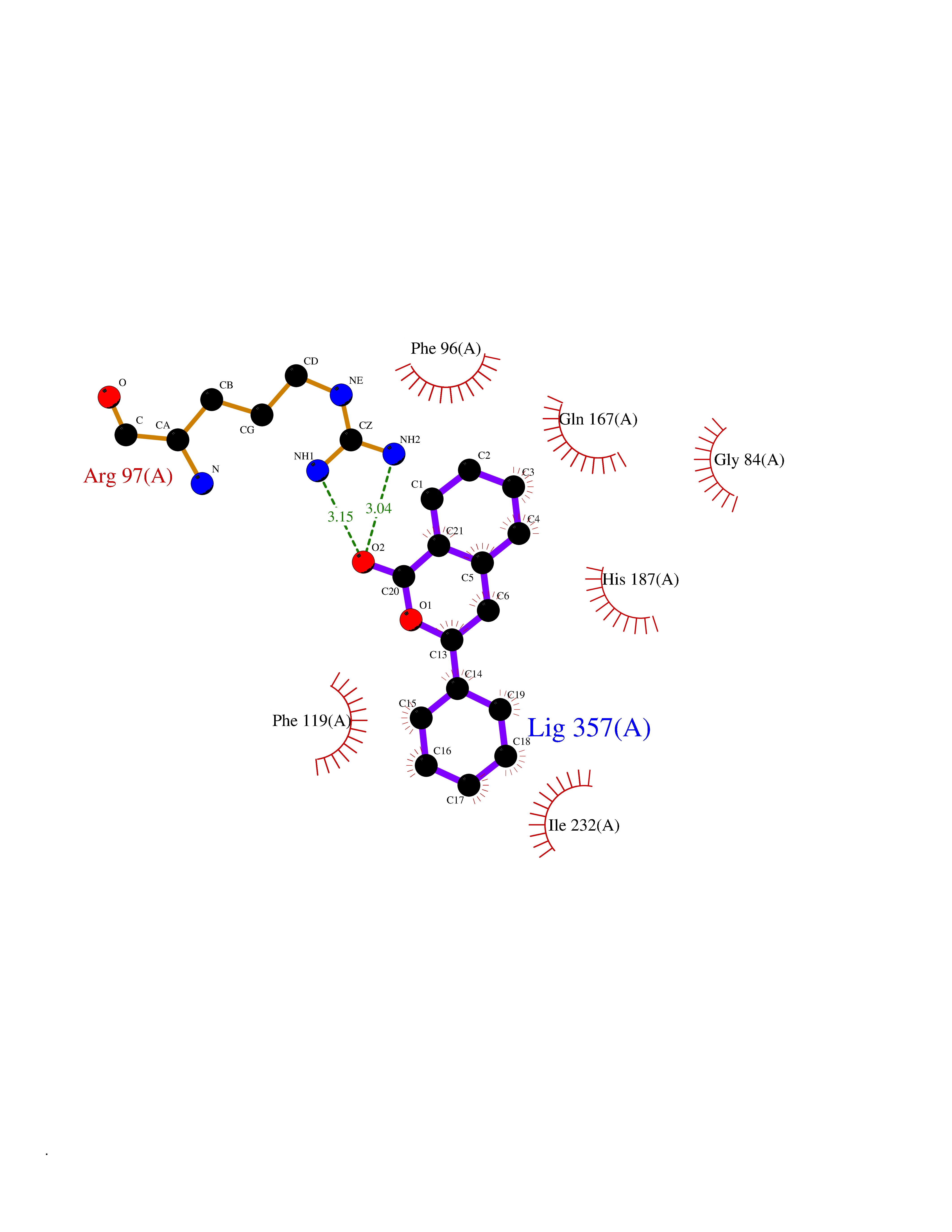 Ligplot Map 3D Binding mode Sequence MERLLDELTLEGVARYMQSERCRRVICLVGAGISTSAGIPDFRSPSTGLYDNLEKYHLPYPEAIFEISYFKKHPEPFFALAKELYPGQFKPTICHYFMRLLKDKGLLLRCYTQNIDTLERIAGLEQEDLVEAHGTFYTSHCVSASCRHEYPLSWMKEKIFSEVTPKCEDCQSLVKPDIVFFGESLPARFFSCMQSDFLKVDLLLVMGTSLQVQPFASLISKAPLSTPRLLINKEKAGQSDPFLGMIMGLGGGMDFDSKKAYRDVAWLGECDQGCLALAELLGWKKELEDLVRREHASIDAQ Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Ecdysone receptor (20-hydroxy-ecdysone receptor) (EcRH) (Ecdysteroid receptor) (Nuclear receptor subfamily 1 group H member 1) | 3IXP | 7.52 | |
Target general information Gen name NA Organism Helicoverpa armigera (Cotton bollworm) (Heliothis armigera) Uniprot ID TTD ID NA Synonyms NA Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class NA Function NA Related diseases NA Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; DNA-binding; Metal-binding; Nucleus; Receptor; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,D Molecular weight (Da) 52799.1 Length 462 Aromaticity 0.09 Instability index 51.97 Isoelectric point 8.09 Charge (pH=7) 2.37 2D Binding mode Binding energy (Kcal/mol) -10.25 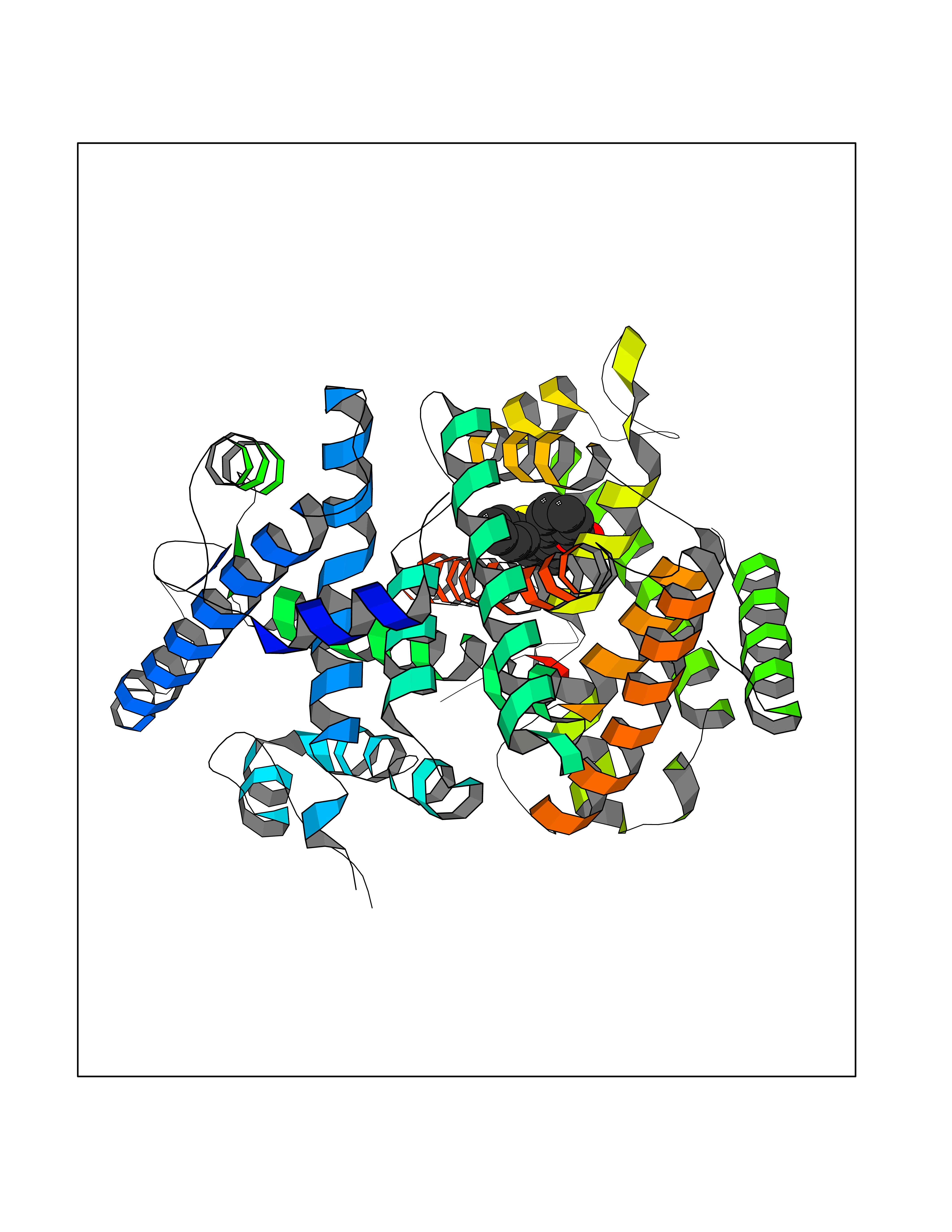 Molscript Map 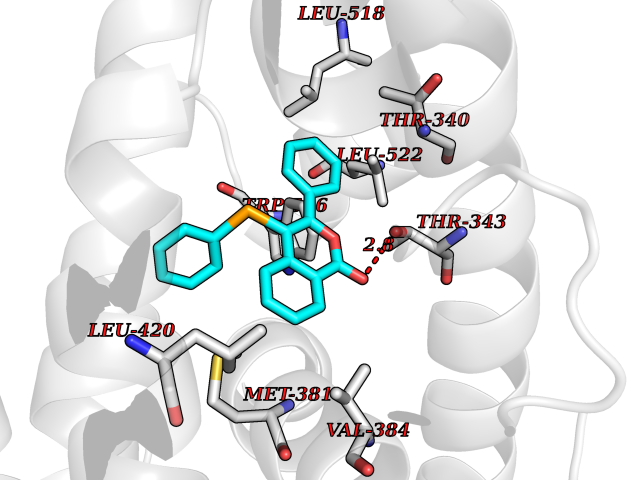 Pymol Map 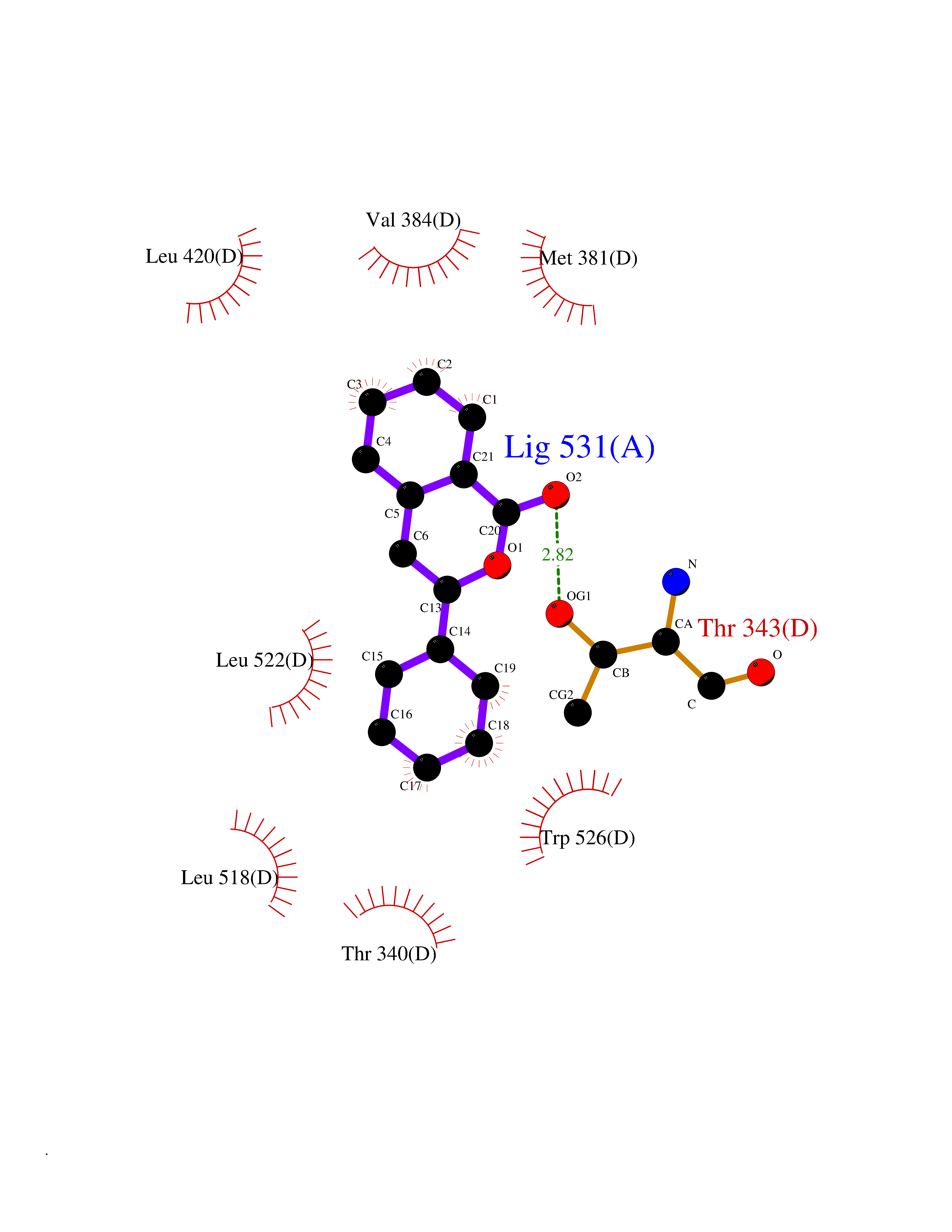 Ligplot Map 3D Binding mode Sequence QELSIERLLEMESLVADPSEEFQFLRVGPDSNVPPKFRAPVSSLCQIGNKQIAALVVWARDIPHFSQLEMEDQILLIKGSWNELLLFAIAWRSMEFLTSPPQLMCLMPGMTLHRNSALQAGVGQIFDRVLSELSLKMRTLRVDQAEYVALKAIILLNPDVKGLKNRQEVEVLREKMFLCLDEYCRRSRSSEEGRFAALLLRLPALRSISLKSFEHLFFFHLVADTSIAGYIRDALRNHAPPIVPPLTANQKSLIARLVYYQEGYEQMPFRQITEMTILTVQLIVEFAKGLPGFSKISQSDQITLLKACSSEVMMLRVARRYDAATDSVLFANNQAYTRDNYRKAGMAYVIEDLLHFCRCMYSMMMDNVHYALLTAIVIFSDRPGLEQPSLVEEIQRYYLNTLRVYILNQNSASPRSAVIFGKILGILTEIRTLGMQNSNMCISLKLKNRKLPPFLEEIWDVA Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Adenosine A2a receptor (ADORA2A) | 5IU4 | 7.52 | |
Target general information Gen name ADORA2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Adenosine receptor A2a; ADORA2; A2a Adenosine receptor; A(2A) adenosine receptor Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function The activity of this receptor is mediated by G proteins which activate adenylyl cyclase. Receptor for adenosine. Related diseases Intellectual developmental disorder, autosomal recessive 59 (MRT59) [MIM:617323]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. {ECO:0000269|PubMed:26416544}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08770; DB14132; DB00640; DB05009; DB05191; DB04853; DB00201; DB04932; DB09273; DB00651; DB00824; DB11757; DB17080; DB00555; DB00358; DB00683; DB01303; DB00806; DB06213; DB01412; DB00277 Interacts with P30542; P29274; P29275; O15155; P21554; Q99418; O15354; Q7Z6G3; O43759-2; Q13107; Q5T9L3-1; P31424-1 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32419.4 Length 296 Aromaticity 0.12 Instability index 39.04 Isoelectric point 8.77 Charge (pH=7) 7.19 2D Binding mode Binding energy (Kcal/mol) -10.26  Molscript Map 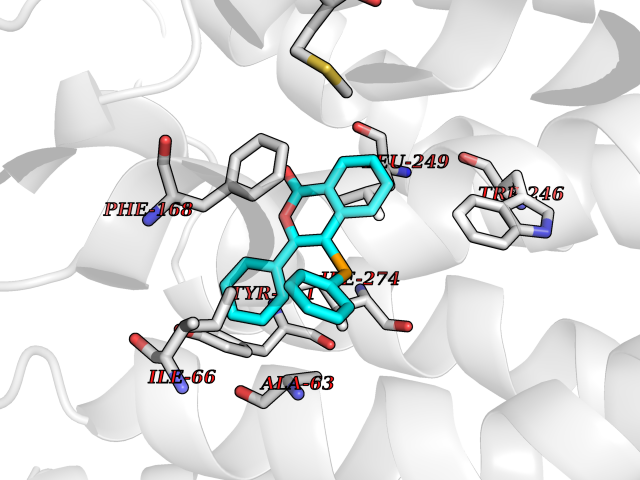 Pymol Map 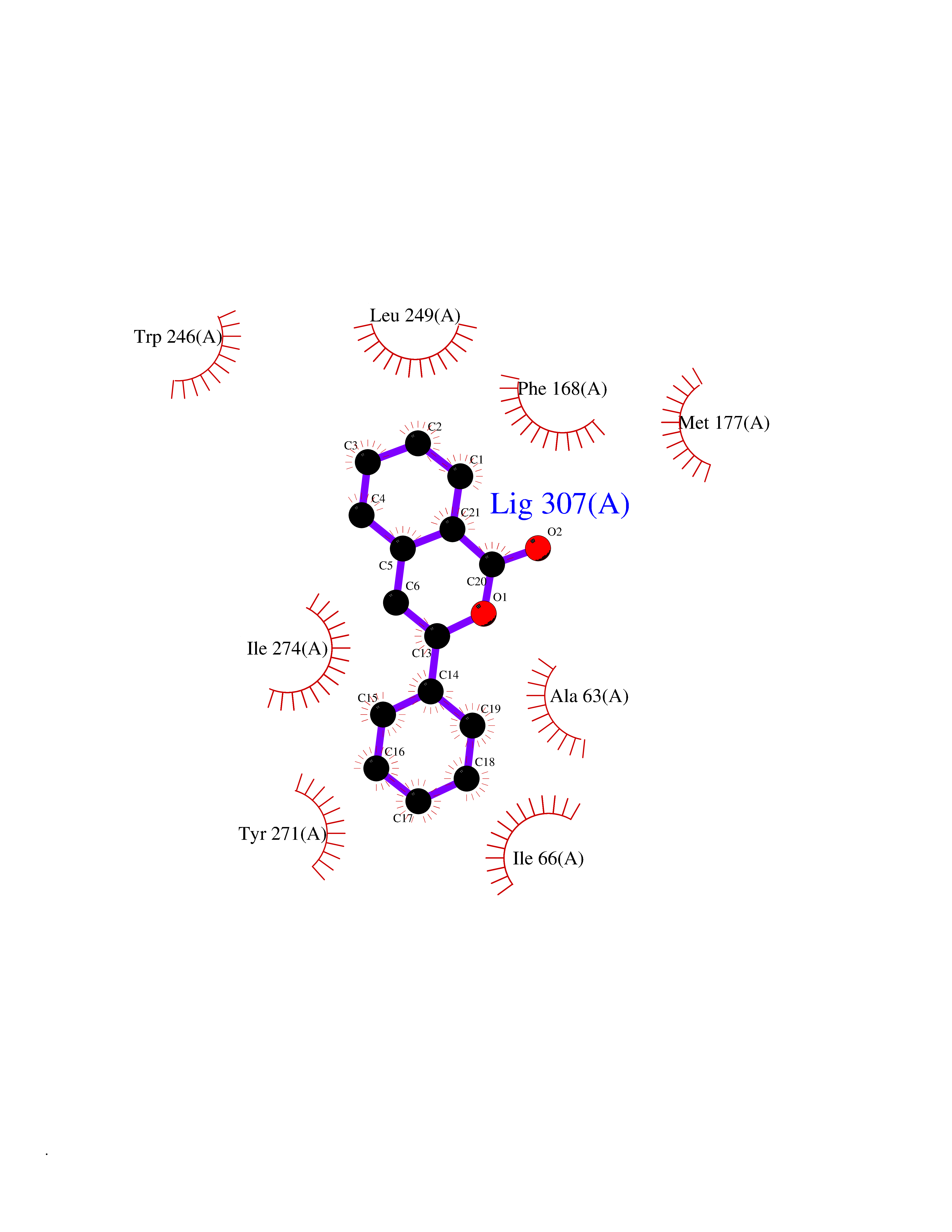 Ligplot Map 3D Binding mode Sequence APPIMGSSVYITVELAIAVLAILGNVLVCWAVWLNSNLQNVTNYFVVSLAAADILVGVLAIPFAITISTGFCAACHGCLFIACFVLVLAQSSIFSLLAIAIDRYIAIAIPLRYNGLVTGTRAAGIIAICWVLSFAIGLTPMLGWNNCGQPKEGKAHSQGCGEGQVACLFEDVVPMNYMVYFNFFACVLVPLLLMLGVYLRIFAAARRQLERARSTLQKEVHAAKSAAIIAGLFALCWLPLHIINCFTFFCPDCSHAPLWLMYLAIVLAHTNSVVNPFIYAYRIREFRQTFRKIIRS Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Adenosine A2a receptor (ADORA2A) | 5IU4 | 7.52 | |
Target general information Gen name ADORA2A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Adenosine receptor A2a; ADORA2; A2a Adenosine receptor; A(2A) adenosine receptor Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function The activity of this receptor is mediated by G proteins which activate adenylyl cyclase. Receptor for adenosine. Related diseases Intellectual developmental disorder, autosomal recessive 59 (MRT59) [MIM:617323]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. {ECO:0000269|PubMed:26416544}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08770; DB14132; DB00640; DB05009; DB05191; DB04853; DB00201; DB04932; DB09273; DB00651; DB00824; DB11757; DB17080; DB00555; DB00358; DB00683; DB01303; DB00806; DB06213; DB01412; DB00277 Interacts with P30542; P29274; P29275; O15155; P21554; Q99418; O15354; Q7Z6G3; O43759-2; Q13107; Q5T9L3-1; P31424-1 EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32419.4 Length 296 Aromaticity 0.12 Instability index 39.04 Isoelectric point 8.77 Charge (pH=7) 7.19 2D Binding mode Binding energy (Kcal/mol) -10.26 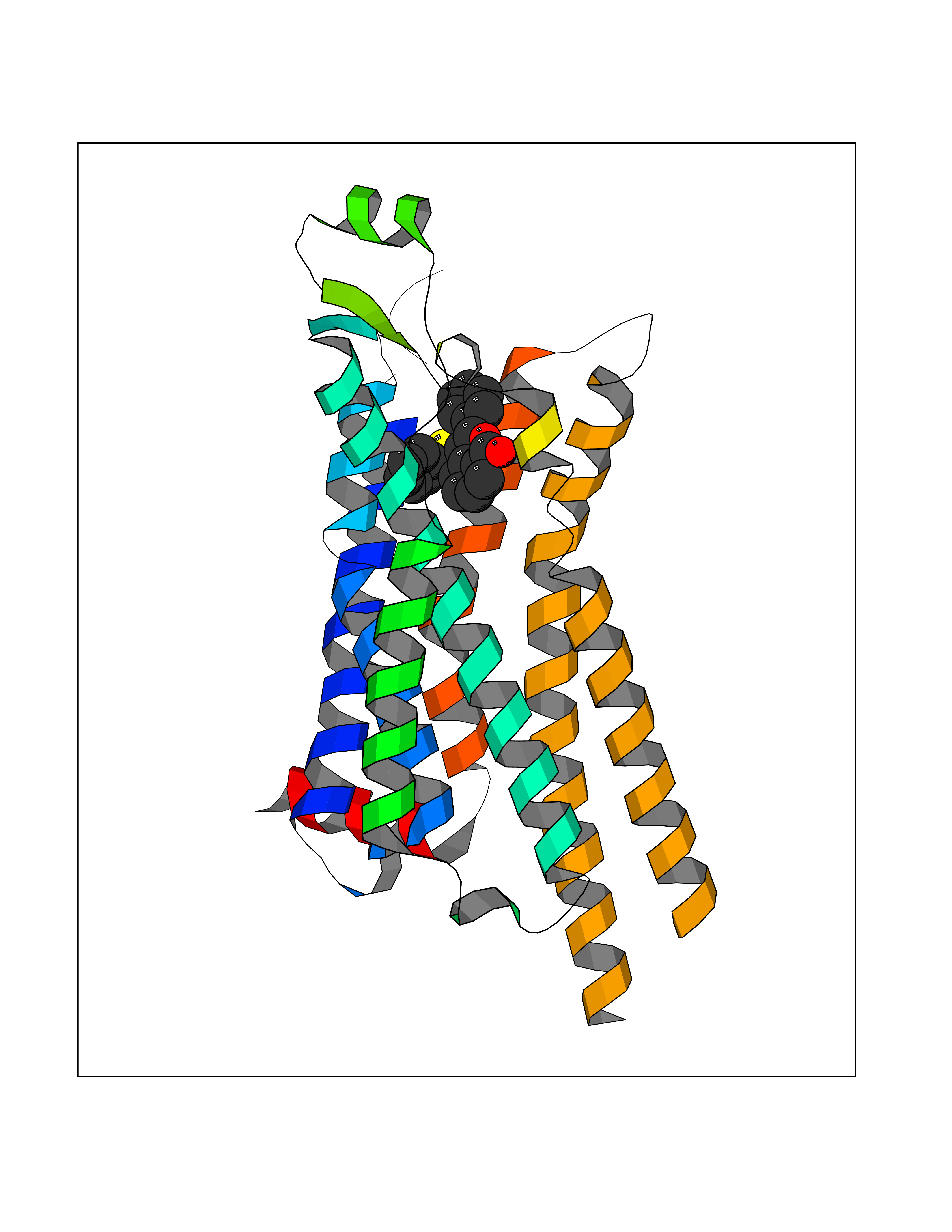 Molscript Map 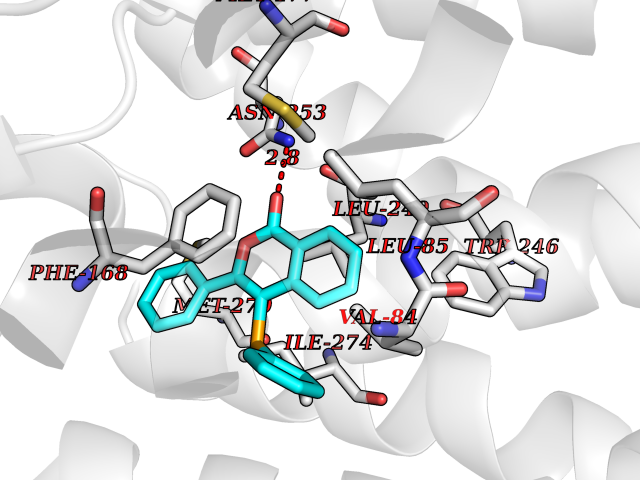 Pymol Map 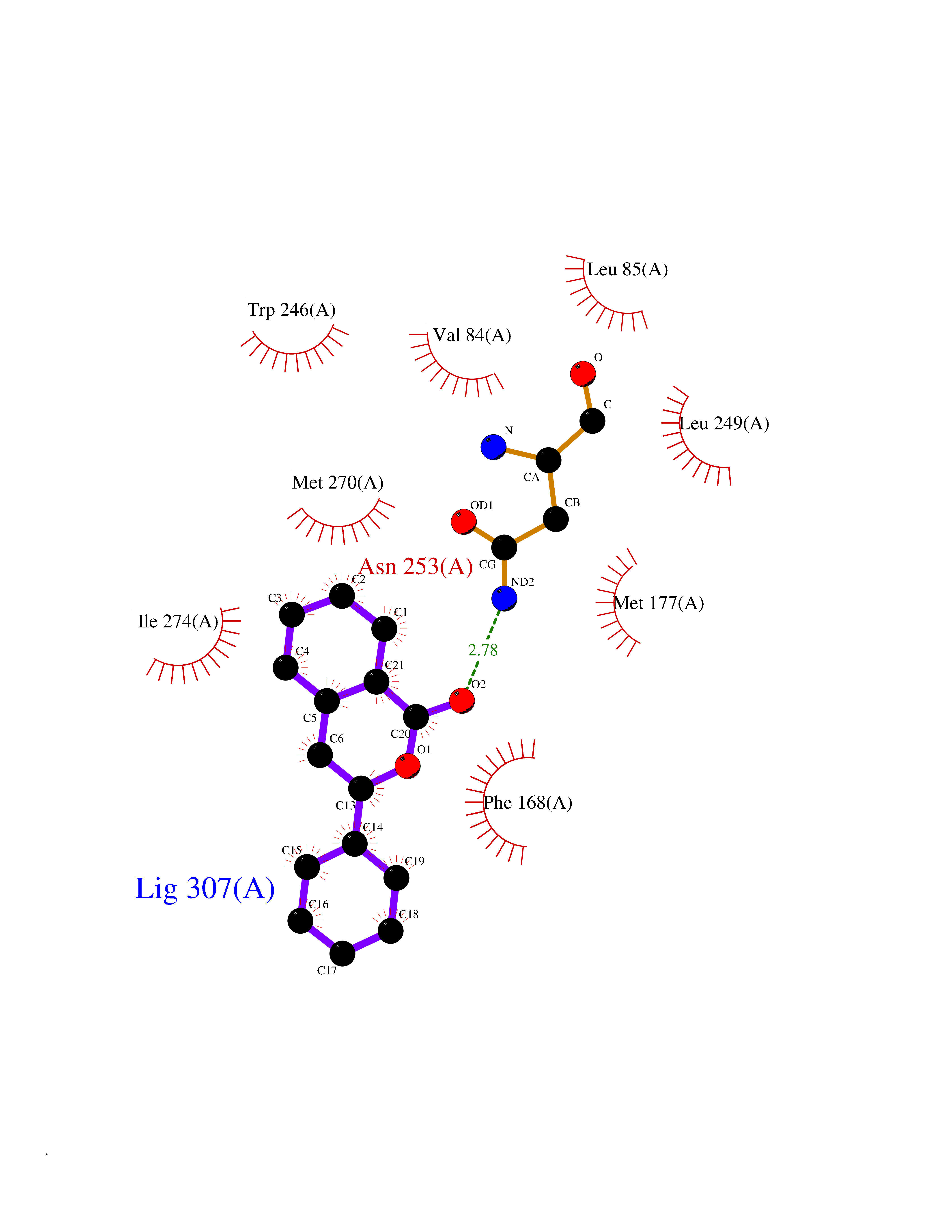 Ligplot Map 3D Binding mode Sequence APPIMGSSVYITVELAIAVLAILGNVLVCWAVWLNSNLQNVTNYFVVSLAAADILVGVLAIPFAITISTGFCAACHGCLFIACFVLVLAQSSIFSLLAIAIDRYIAIAIPLRYNGLVTGTRAAGIIAICWVLSFAIGLTPMLGWNNCGQPKEGKAHSQGCGEGQVACLFEDVVPMNYMVYFNFFACVLVPLLLMLGVYLRIFAAARRQLERARSTLQKEVHAAKSAAIIAGLFALCWLPLHIINCFTFFCPDCSHAPLWLMYLAIVLAHTNSVVNPFIYAYRIREFRQTFRKIIRS Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Monomeric sarcosine oxidase | 2GF3 | 7.52 | |
Target general information Gen name soxA Organism Bacillus sp. (strain B-0618) Uniprot ID TTD ID NA Synonyms sox Protein family MSOX/MTOX family, MSOX subfamily Biochemical class Oxidoreductase Function Sarcosine oxidase activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB03098; DB01918; DB03517; DB03147; DB03366; DB02083; DB02543 Interacts with NA EC number 1.5.3.1 Uniprot keywords 3D-structure; Cytoplasm; Direct protein sequencing; FAD; Flavoprotein; Oxidoreductase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42606.4 Length 385 Aromaticity 0.1 Instability index 26.97 Isoelectric point 5.27 Charge (pH=7) -17.18 2D Binding mode Binding energy (Kcal/mol) -10.26  Molscript Map 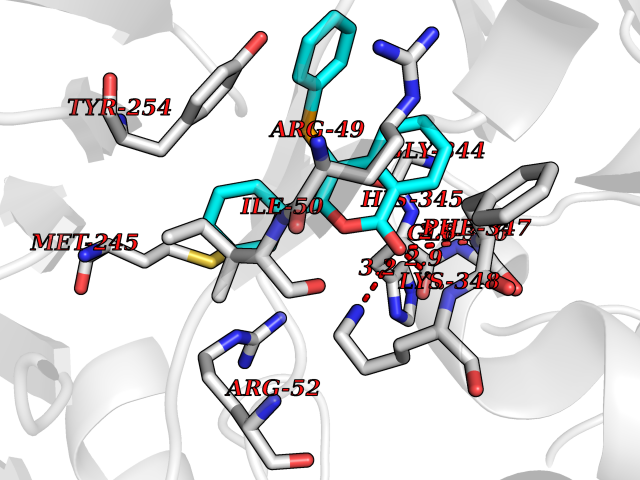 Pymol Map 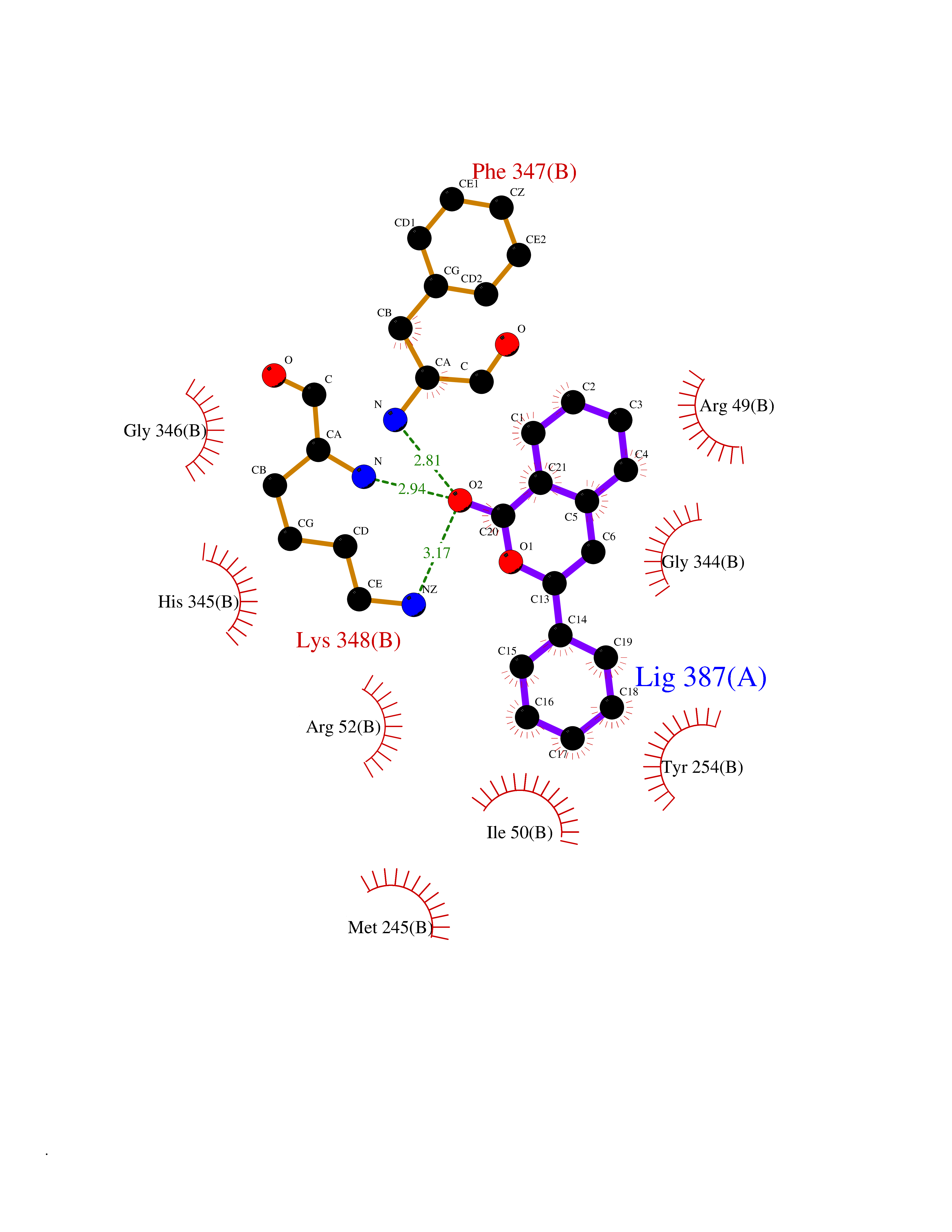 Ligplot Map 3D Binding mode Sequence STHFDVIVVGAGSMGMAAGYQLAKQGVKTLLVDAFDPPHTNGSHHGDTRIIRHAYGEGREYVPLALRSQELWYELEKETHHKIFTKTGVLVFGPKGESAFVAETMEAAKEHSLTVDLLEGDEINKRWPGITVPENYNAIFEPNSGVLFSENCIRAYRELAEARGAKVLTHTRVEDFDISPDSVKIETANGSYTADKLIVSMGAWNSKLLSKLNLDIPLQPYRQVVGFFESDESKYSNDIDFPGFMVEVPNGIYYGFPSFGGCGLKLGYHTFGQKIDPDTINREFGVYPEDESNLRAFLEEYMPGANGELKRGAVCMYTKTLDEHFIIDLHPEHSNVVIAAGFSGHGFKFSSGVGEVLSQLALTGKTEHDISIFSINRPALKESLQ Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Metabotropic glutamate receptor 5 (mGluR5) | 4OO9 | 7.52 | |
Target general information Gen name GRM5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MGLUR5; GPRC1E Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function G-protein coupled receptor for glutamate. Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling activates a phosphatidylinositol-calcium second messenger system and generates a calcium-activated chloride current. Plays an important role in the regulation of synaptic plasticity and the modulation of the neural network activity. Related diseases Charcot-Marie-Tooth disease, axonal, 2D (CMT2D) [MIM:601472]: A dominant axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:12690580, ECO:0000269|PubMed:17035524, ECO:0000269|PubMed:17101916, ECO:0000269|PubMed:17663003, ECO:0000269|PubMed:20169446, ECO:0000269|PubMed:24604904, ECO:0000269|PubMed:25168514, ECO:0000269|PubMed:26244500, ECO:0000269|PubMed:26503042, ECO:0000269|PubMed:31173493}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal dominant 5 (HMND5) [MIM:600794]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. {ECO:0000269|PubMed:12690580, ECO:0000269|PubMed:17035524, ECO:0000269|PubMed:23279345, ECO:0000269|PubMed:24627108, ECO:0000269|PubMed:26503042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinal muscular atrophy, infantile, James type (SMAJI) [MIM:619042]: An autosomal dominant form of spinal muscular atrophy, a group of neuromuscular disorders characterized by degeneration of the anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. SMAJI is a severe disease characterized by hypotonia manifesting in the first weeks or months of life, delayed motor development, motor regression, and muscle weakness and atrophy primarily affecting distal muscles. Additional variable features include feeding difficulties, poor overall growth, foot deformities, kyphosis, hyperlordosis, scoliosis, vocal cord dysfunction, and respiratory insufficiency. {ECO:0000269|PubMed:32181591}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00659; DB05070; DB12733; DB06201 Interacts with P41594; Q7Z6G3 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Methylation; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 27065.4 Length 247 Aromaticity 0.13 Instability index 42.92 Isoelectric point 9.24 Charge (pH=7) 11.34 2D Binding mode Binding energy (Kcal/mol) -10.26 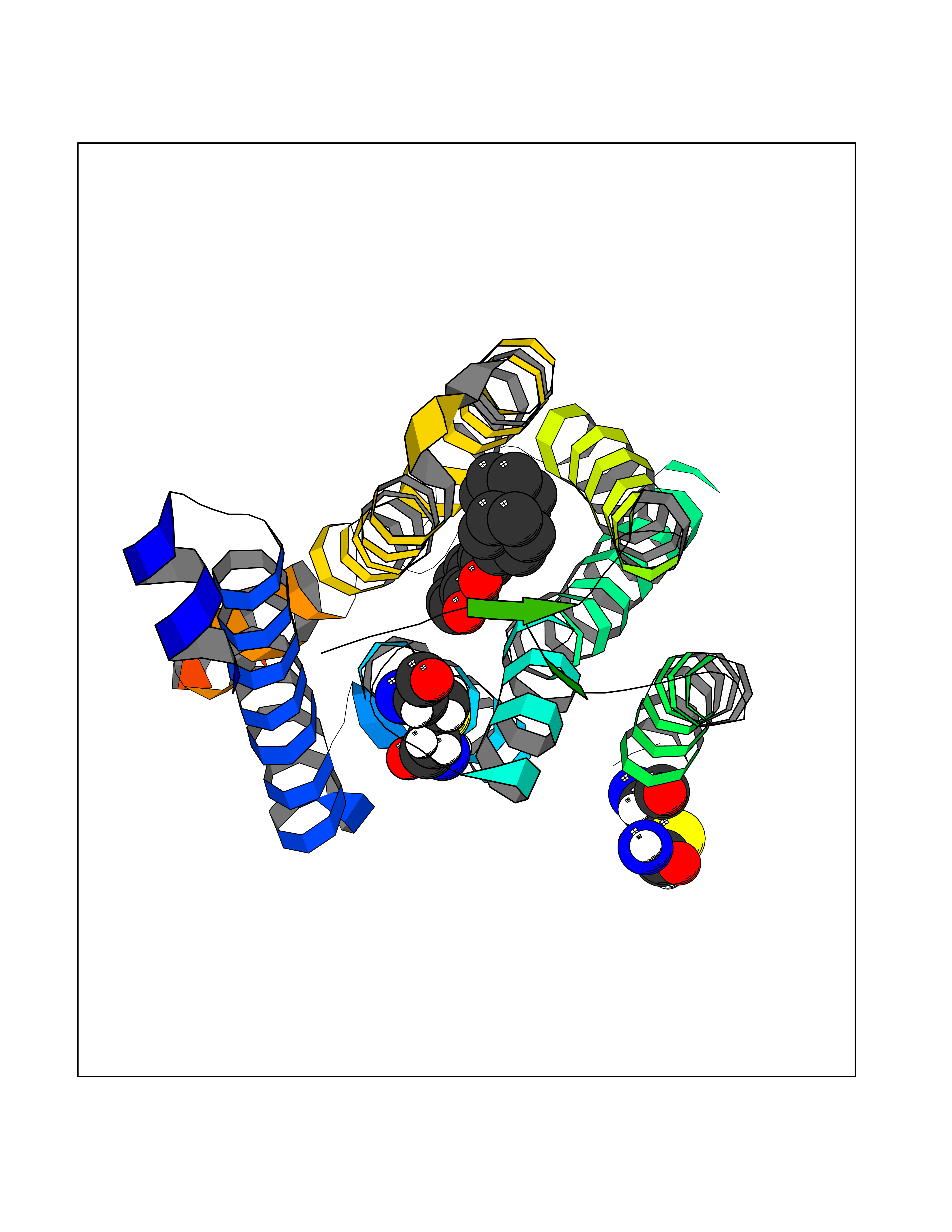 Molscript Map 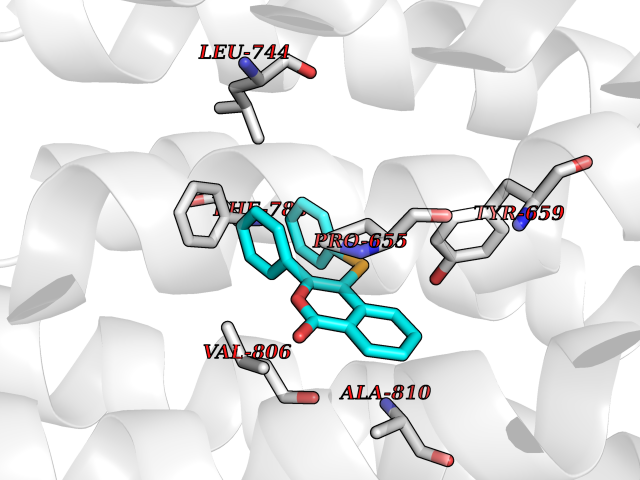 Pymol Map 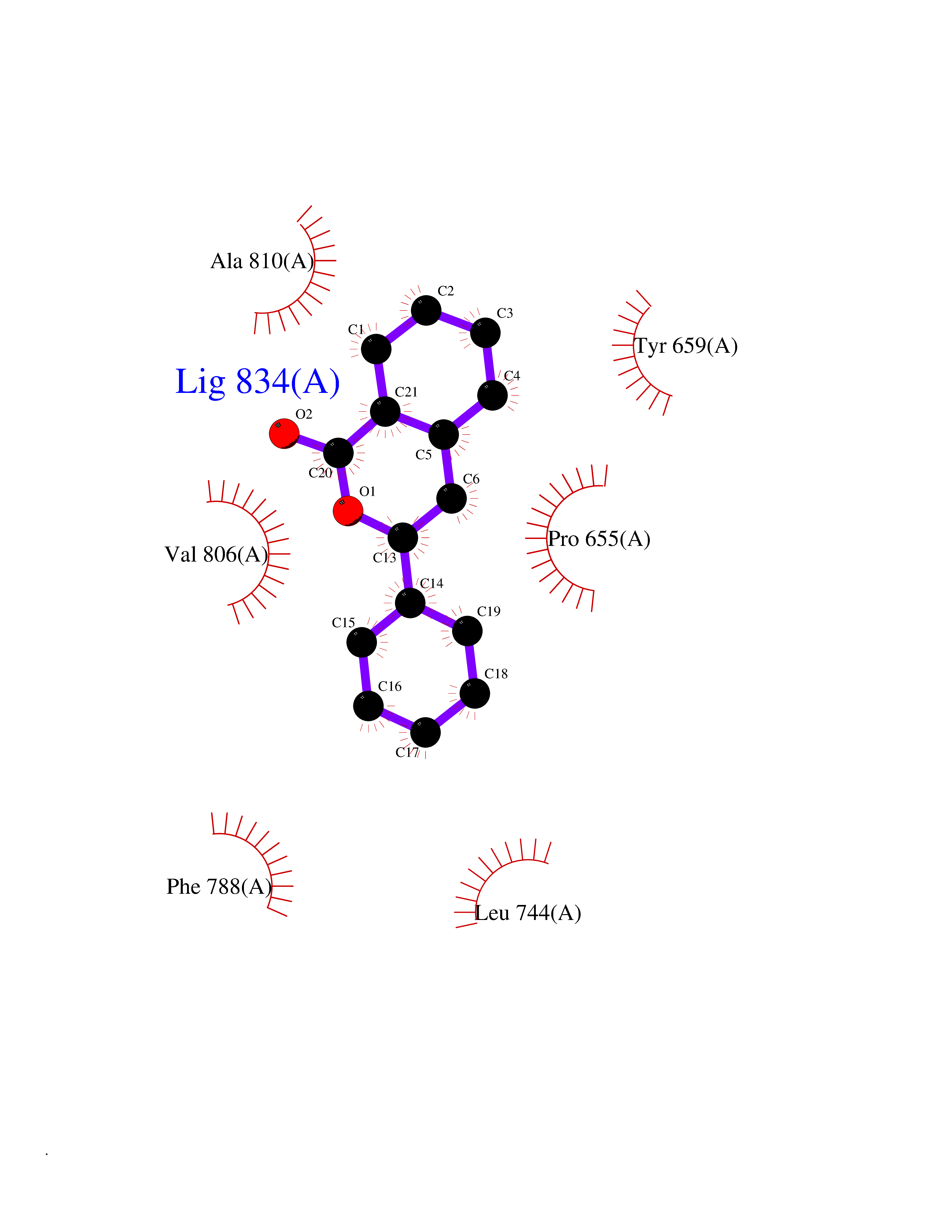 Ligplot Map 3D Binding mode Sequence SPVQYLRWGDPAPIAAVVFACLGLLATLFVTVVFIIYRDTPVVKSSSRELCYIILAGICLGYLCTFXLIAKPKQIYCYLQRIGIGLSPAMSYSALVTKTYRAARILAMSKKSAXAQLVIAFILICIQLGIIVALFIMEPPDIMVYLICNTTNLGVVAPLGYNGLLILACTFYAFKTRNVPANFNEAKYIAFTMYTTCIIWLAFVPIYFGSNYKIITMCFSVSLSATVALGCMFVPKVYIILAKPERN Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | N-acylethanolamine-hydrolyzing acidamidase (NAAA) | 6DXX | 7.52 | |
Target general information Gen name NAAA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nacylsphingosine amidohydrolaselike; Nacylethanolaminehydrolyzing acid amidase subunit beta; NAAA; Acid ceramidaselike protein; ASAHlike protein Protein family Acid ceramidase family Biochemical class Carbon-nitrogen hydrolase Function Degrades bioactive fatty acid amides to their corresponding acids, with the following preference: N- palmitoylethanolamine > N-myristoylethanolamine > N- lauroylethanolamine = N-stearoylethanolamine > N- arachidonoylethanolamine > N-oleoylethanolamine. Also exhibits weak hydrolytic activity against the ceramides N- lauroylsphingosine and N-palmitoylsphingosine. Related diseases Hypertriglyceridemia, transient infantile (HTGTI) [MIM:614480]: An autosomal recessive disorder characterized by onset of moderate to severe transient hypertriglyceridemia in infancy that normalizes with age. The hypertriglyceridemia is associated with hepatomegaly, moderately elevated transaminases, persistent fatty liver, and the development of hepatic fibrosis. {ECO:0000269|PubMed:22226083, ECO:0000269|PubMed:24549054}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09061; DB14009; DB14011 Interacts with NA EC number EC 3.5.1.- Uniprot keywords 3D-structure; Alternative splicing; Autocatalytic cleavage; Direct protein sequencing; Disulfide bond; Fatty acid metabolism; Glycoprotein; Hydrolase; Lipid degradation; Lipid metabolism; Lysosome; Membrane; Proteomics identification; Reference proteome; Signal; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 36877.8 Length 328 Aromaticity 0.11 Instability index 44.37 Isoelectric point 7.72 Charge (pH=7) 1.08 2D Binding mode Binding energy (Kcal/mol) -10.26  Molscript Map 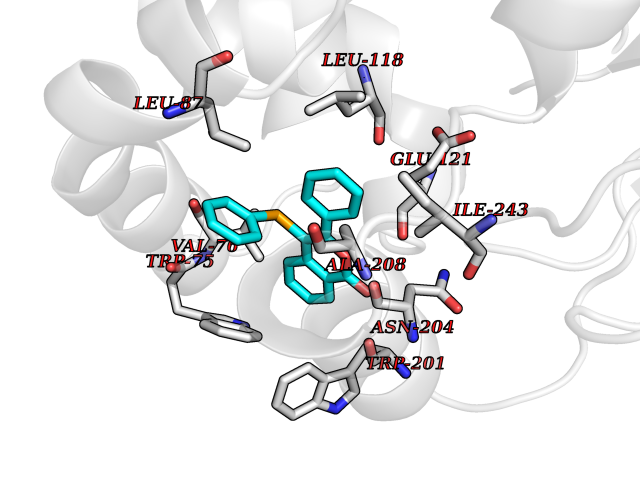 Pymol Map 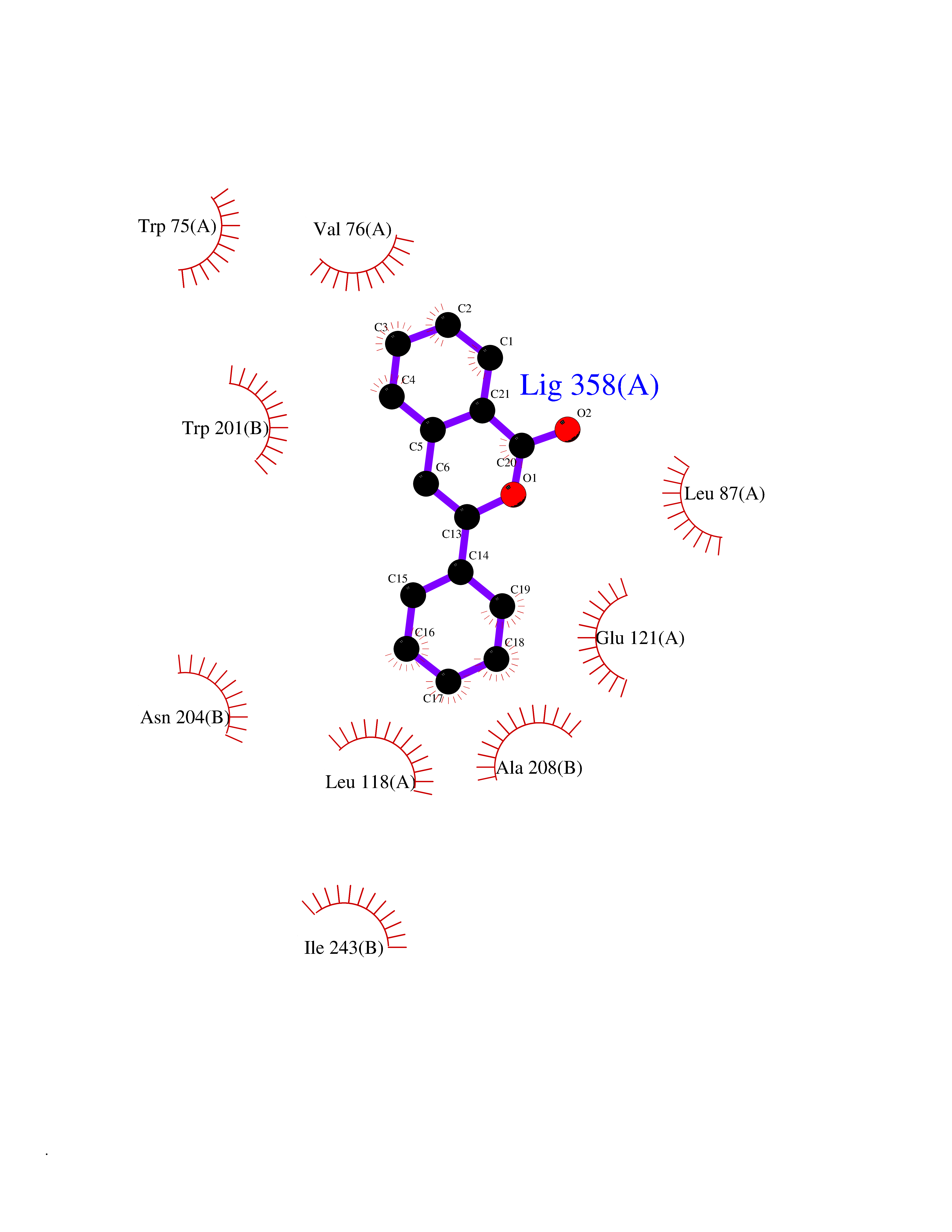 Ligplot Map 3D Binding mode Sequence SPPAAPRFNVSLDSVPELRWLPVLRHYDLDLVRAAMAQVIGDRVPKWVHVLIGKVVLELERFLPQPFTGEIRGMCDFMNLSLADCLLVNLAYESSVFCTSIVAQDSRGHIYHGRNLDYPFGNVLRKLTVDVQFLKNGQIAFTGTTFIGYVGLWTGQSPHKFTVSGDERDKGWWWENAIAALFRRHIPVSWLIRATLSESENFEAAVGKLAKTPLIADVYYIVGGTSPREGVVITRNRDGPADIWPLDPLNGAWFRVETNYDHWKPAPKEDDRRTSAIKALNATGQANLSLEALFQILSVVPVYNNFTIYTTVMSAGSPDKYMTRIRNP Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Vitamin K epoxide reductase complex 1 (VKORC1) | 6WV3 | 7.51 | |
Target general information Gen name VKORC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Vitamin K1 2,3-epoxide reductase subunit 1; VKORC1; VKOR; UNQ308/PRO351; MSTP576; MSTP134 Protein family VKOR family Biochemical class Short-chain dehydrogenases reductase Function Involved invitamin K metabolism. Catalytic subunit of the vitamin K epoxide reductase (VKOR) complex which reduces inactive vitamin K 2,3-epoxide to active vitamin K. Vitamin K is required for the gamma-carboxylation of various proteins, including clotting factors, and is required for normal blood coagulation, but also for normal bone development. Related diseases Combined deficiency of vitamin K-dependent clotting factors 2 (VKCFD2) [MIM:607473]: VKCFD leads to a bleeding tendency that is usually reversed by oral administration of vitamin K. {ECO:0000269|PubMed:14765194, ECO:0000269|PubMed:16270630}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Coumarin resistance (CMRES) [MIM:122700]: A condition characterized by partial or complete resistance to warfarin or other 4-hydroxycoumarin derivatives. These drugs are used as anti-coagulants for the prevention of thromboembolic diseases in subjects with deep vein thrombosis, atrial fibrillation, or mechanical heart valve replacement. {ECO:0000269|PubMed:14765194, ECO:0000269|PubMed:20946155}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01418; DB00266; DB09332; DB00170; DB00498; DB00946; DB01022; DB00682 Interacts with Q13323; Q7Z7G2; Q96BA8; Q9Y282; Q5JX71; Q96KR6; Q5T7V8; Q8TDT2; Q9NQG1; P15941-11; Q96TC7; Q9NR31; A0A0S2Z4U3; Q8TBB6; O15393-2; Q19QW4 EC number EC 1.17.4.4 Uniprot keywords 3D-structure; Alternative splicing; Disease variant; Disulfide bond; Endoplasmic reticulum; Membrane; Oxidoreductase; Proteomics identification; Quinone; Redox-active center; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 42656.4 Length 381 Aromaticity 0.1 Instability index 32.12 Isoelectric point 7.73 Charge (pH=7) 1.93 2D Binding mode Binding energy (Kcal/mol) -10.25  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence KGEELFTGVVPILVELDGDVNGHKFSVRGEGEGDATNGKLTLKFICTTGKLPVPWPTLVTTLXVQCFSRYPDHMKRHDFFKSAMPEGYVQERTISFKDDGTYKTRAEVKFEGDTLVNRIELKGIDFKEDGNILGHKLEYNSTWGSPGWVRLALCLTGLVLSLYALHVKAARARDRDYRALCDVGTAISCSRVFSSRWGRGFGLVEHVLGQDSILNQSNSIFGCIFYTLQLLLGCLRTRWASVLMLLSSLVSLAGSVYLAWILFFVLYDFCIVCITTYAINVSLMWLSFRKVQENSHNVYITADKQKNGIKANFKIRHNVEDGSVQLADHYQQNTPIGDGPVLLPDNHYLSTQSVLSKDPNEKRDHMVLLEFVTAAGITHHH Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Cholestenol delta-isomerase (EBP) | 6OHU | 7.51 | |
Target general information Gen name EBP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Emopamilbinding protein; EBP; Delta(8)Delta(7) sterol isomerase; D8D7 sterol isomerase; 3betahydroxysteroidDelta(8),Delta(7)isomerase Protein family EBP family Biochemical class Intramolecular oxidoreductases Function Catalyzes the conversion of Delta(8)-sterols to their corresponding Delta(7)-isomers. Related diseases Chondrodysplasia punctata 2, X-linked dominant (CDPX2) [MIM:302960]: A clinically and genetically heterogeneous disorder characterized by punctiform calcification of the bones. The key clinical features of CDPX2 are chondrodysplasia punctata, linear ichthyosis, cataracts and short stature. CDPX2 is a rare disorder of defective cholesterol biosynthesis, biochemically characterized by an increased amount of 8-dehydrocholesterol and cholest-8(9)-en-3-beta-ol in the plasma and tissues. {ECO:0000269|PubMed:10391218, ECO:0000269|PubMed:10391219, ECO:0000269|PubMed:10942423, ECO:0000269|PubMed:11493318, ECO:0000269|PubMed:18176751, ECO:0000269|PubMed:25814754}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: MEND syndrome (MEND) [MIM:300960]: An X-linked recessive disorder associated with a defect in sterol biosynthesis. Disease manifestations and severity are highly variable. Clinical features include intellectual disability, short stature, scoliosis, digital abnormalities, cataracts, and dermatologic abnormalities. {ECO:0000269|PubMed:12503101, ECO:0000269|PubMed:20949533, ECO:0000269|PubMed:24459067, ECO:0000269|PubMed:24700572}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00675 Interacts with O95870; Q86W74-2; Q13520; Q3SXY8; Q8N6S5; Q9H2C2; Q9HD20-3; O95393; Q12983; Q8WVV5; P06681; O14523; Q8WVX3-2; P01031; Q6UWT4; Q9P0B6; Q8NHW4; P25942; Q07108; P60033; O14735; P23141-3; Q9H9P2; Q8NHS1; O43889-2; Q96BA8; P49447; O43169; P78329; P81534; Q9H1M4; Q96LL9; Q15125; Q08426; Q9BV81; P54849; Q9UKR5; Q9Y282; Q7L5A8; Q5JX71; Q96IV6; Q9UGM5; Q9Y3D6; Q14318; Q9BWH2; Q14802-3; Q9H0Q3; Q8WWP7; Q96F15; P29033; O95452; O14653; Q8TDT2; P02724; P30519; Q7Z5P4; Q9Y5U9; Q9Y5U4; Q8N5M9; Q5T700; Q68G75; Q7L5N7; Q7Z4F1; Q96AG4; Q16873; Q6ZSS7; P50281; Q5J8X5; Q9UHE5; O95167; Q9NX14; Q99519; Q92982; Q9NZG7; Q16617; Q8IXM6; Q2M2E3; Q9P0S3; Q53FV1; Q8N138; Q7RTS5; Q9Y342; Q04941; Q8IY26; Q01453; P54315; P43378; P15151; Q8N8N0; Q5QGT7; Q96GQ5; Q9NTJ5; Q969E2; O75396; Q9Y6X1; Q8N6R1; Q9BWM7; Q8TD22; Q8IWU4; Q96G79; Q2M3R5; Q9NVC3; P08195-4; Q96JW4; Q6P1K1; Q0VAQ4; Q9NRQ5; B2RUZ4; Q9BZL3; Q6UX34; Q86Y82; P61266; Q13190; O43752; O15400; Q9UNK0; O43759-2; P57105; Q8N2H4; Q96BZ9; P07204; O14925; Q96CP7; Q96MV1; P55061; Q9NV29; P17152; Q9BXJ8; A0PK00; Q9NV12; Q9BVK8; Q9NUH8; Q9P0S9; Q9NRX6; Q8N511; Q969S6; Q9BTX3; A2RU14; Q9H0R3; Q8NBD8; Q8WW34-2; Q9NWH2; Q9BU79; Q8TBM7; Q69YG0; Q9NW97; Q9H2L4; Q6PI78; Q8N2M4; Q8N661; Q5BJF2; Q9NSU2-1; A0AVG3; Q5TGU0; A5PKU2; Q9Y385; Q9Y5Z9; Q53HI1; Q9H1C4; Q9NZ43; P23763-3; P63027; Q15836; O75379; Q9P0L0; O95292; O95070; Q9Y548; Q9BSR8; Q96EC8; Q6UX98; O95159 EC number EC 5.3.3.5 Uniprot keywords 3D-structure; Acetylation; Cataract; Cholesterol biosynthesis; Cholesterol metabolism; Cytoplasmic vesicle; Disease variant; Dwarfism; Endoplasmic reticulum; Ichthyosis; Isomerase; Lipid biosynthesis; Lipid metabolism; Membrane; Nucleus; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 24012.7 Length 206 Aromaticity 0.17 Instability index 42.52 Isoelectric point 6.19 Charge (pH=7) -3.19 2D Binding mode Binding energy (Kcal/mol) -10.24  Molscript Map 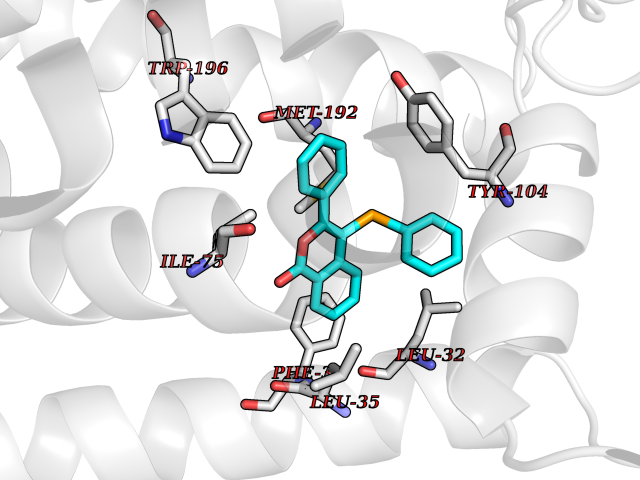 Pymol Map 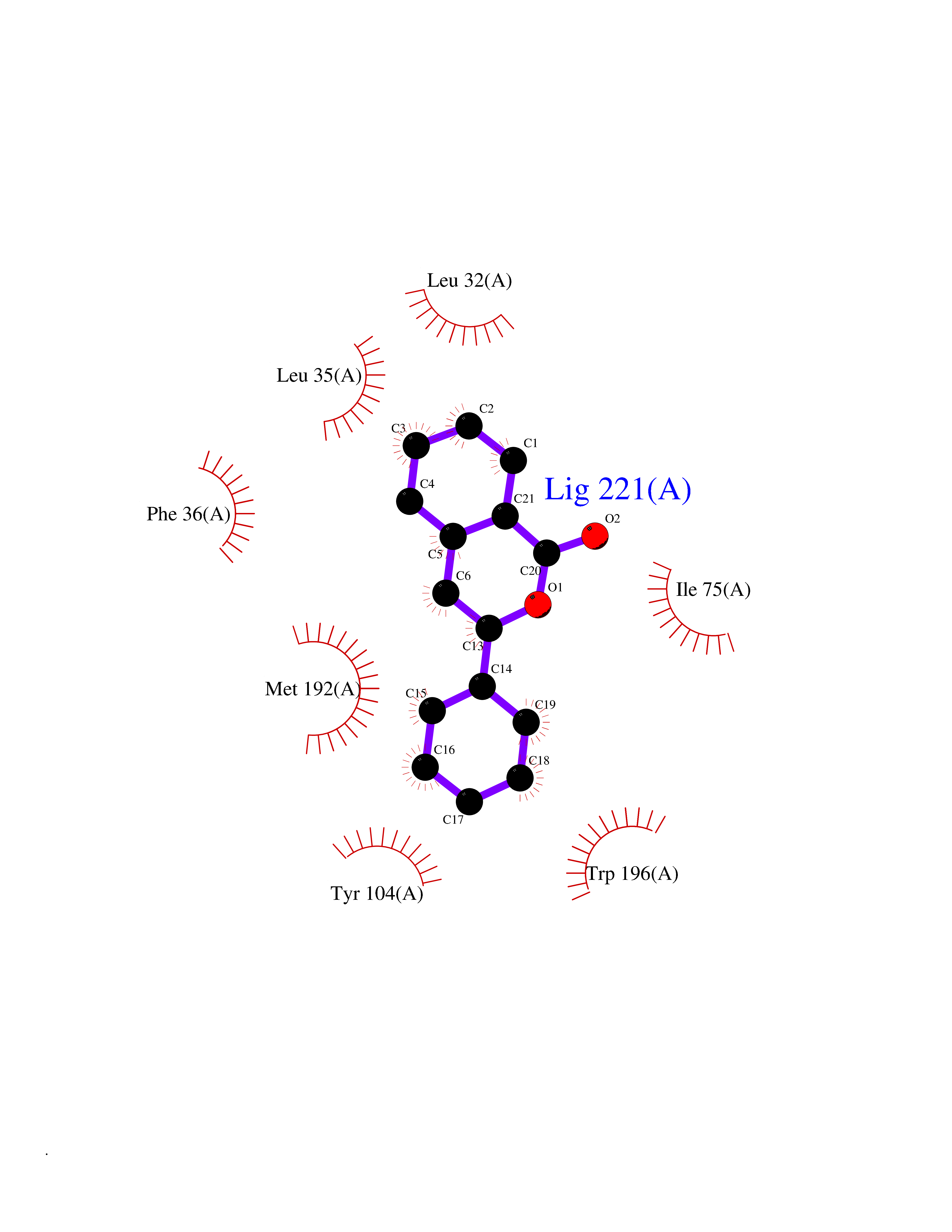 Ligplot Map 3D Binding mode Sequence PLHPYWPQHLRLDNFVPNDRPTWHILAGLFSVTGVLVVTTWLLSGRTWRRLSLCWFAVCGFIHLVIEGWFVLYYEDLLGDQAFLSQLWKEYAKGDSRYILGDNFTVCMETITACLWGPLSLWVVIAFLRQHPLRFILQLVVSVGQIYGDVLYFLTEHRDGFQHGELGHPLYFWFYFVFMNALWLVLPGVLVLDAVKHLTHAQSTLD Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Putidaredoxin reductase | 1Q1R | 7.50 | |
Target general information Gen name camA Organism Pseudomonas putida (Arthrobacter siderocapsulatus) Uniprot ID TTD ID NA Synonyms NA Protein family FAD-dependent oxidoreductase family Biochemical class Oxidoreductase Function Flavin adenine dinucleotide binding.Oxidoreductase activity. Related diseases Multiple fibroadenomas of the breast (MFAB) [MIM:615554]: A benign breast disease marked by lobuloalveolar growth with abnormally high proliferation of the epithelium, and characterized by the presence of more than 3 fibroadenomas in one breast. Fibroadenomas are adenomas containing fibrous tissue. {ECO:0000269|PubMed:18779591}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperprolactinemia (HPRL) [MIM:615555]: A disorder characterized by increased levels of prolactin in the blood not associated with gestation or the puerperium. HPRL may result in infertility, hypogonadism, and galactorrhea. {ECO:0000269|PubMed:24195502}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03147 Interacts with NA EC number 1.18.1.5 Uniprot keywords 3D-structure; FAD; Flavoprotein; NAD; Oxidoreductase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 45089 Length 417 Aromaticity 0.06 Instability index 32.36 Isoelectric point 5.41 Charge (pH=7) -8.61 2D Binding mode Binding energy (Kcal/mol) -10.24 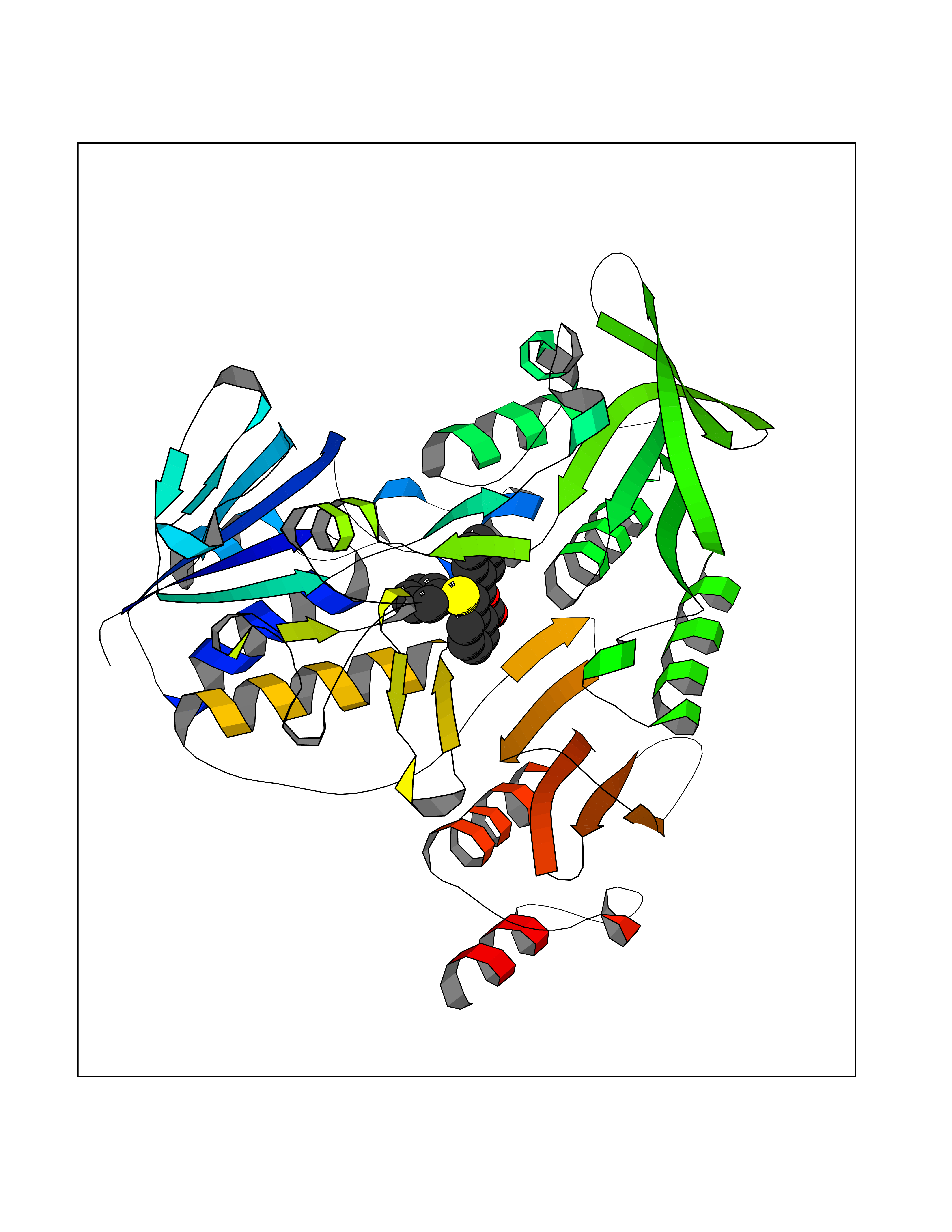 Molscript Map 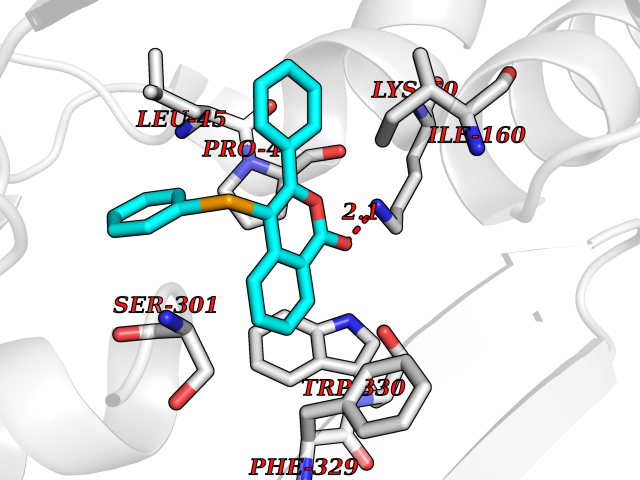 Pymol Map 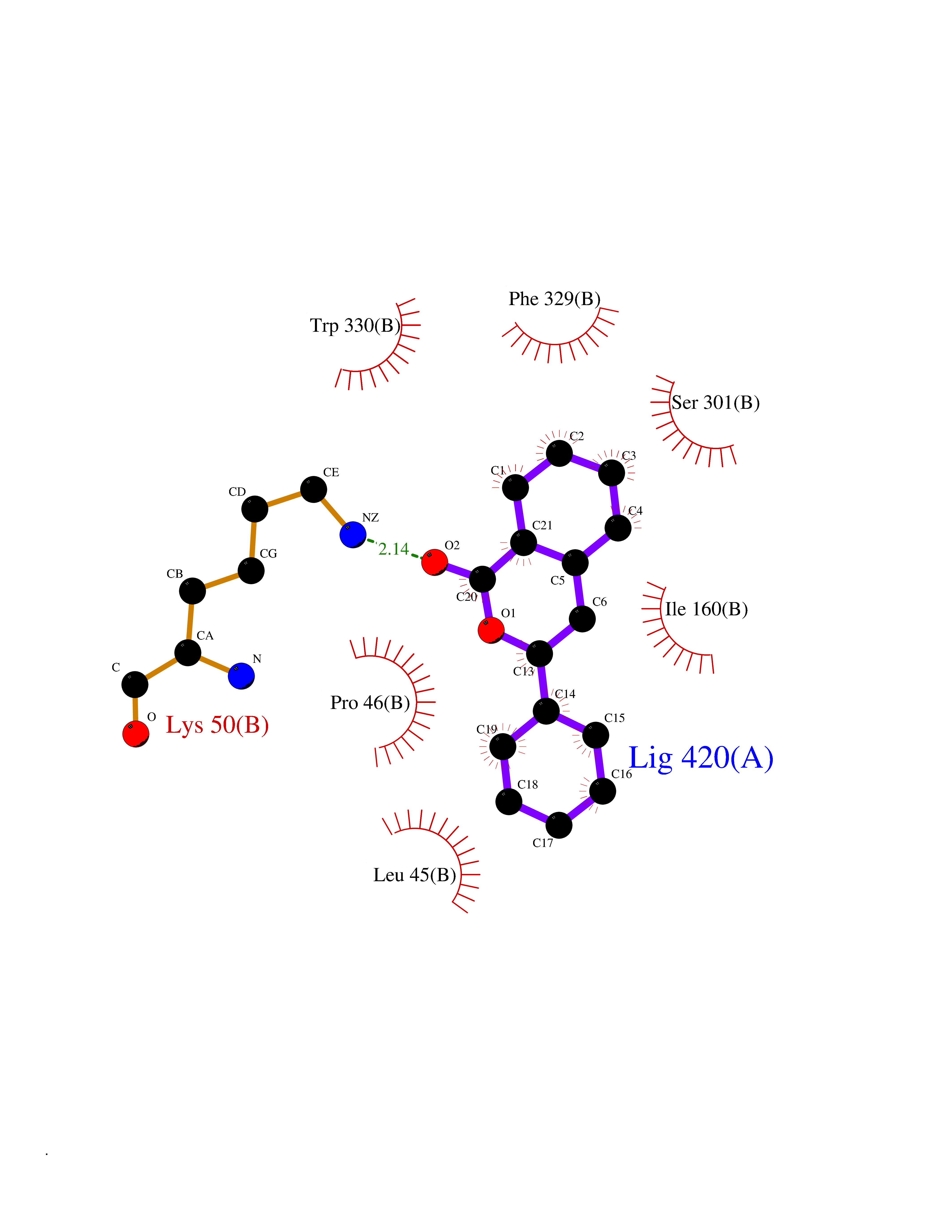 Ligplot Map 3D Binding mode Sequence NANDNVVIVGTGLAGVEVAFGLRASGWEGNIRLVGDATVIPHHLPPLSKAYLAGKATAESLYLRTPDAYAAQNIQLLGGTQVTAINRDRQQVILSDGRALDYDRLVLATGGRPRPLPVASGAVGKANNFRYLRTLEDAECIRRQLIADNRLVVIGGGYIGLEVAATAIKANMHVTLLDTAARVLERVTAPPVSAFYEHLHREAGVDIRTGTQVCGFEMSTDQQKVTAVLCEDGTRLPADLVIAGIGLIPNCELASAAGLQVDNGIVINEHMQTSDPLIMAVGDCARFHSQLYDRWVRIESVPNALEQARKIAAILCGKVPRDEAAPWFWSDQYEIGLKMVGLSEGYDRIIVRGSLAQPDFSVFYLQGDRVLAVDTVNRPVEFNQSKQIITDRLPVEPNLLGDESVPLKEIIAAAKAE Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Nicotinamide N-methyltransferase | 2IIP | 7.50 | |
Target general information Gen name NNMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, NNMT/PNMT/TEMT family Biochemical class Transferase Function Nicotinamide N-methyltransferase activity.Pyridine N-methyltransferase activity. Related diseases Defects in PPARG can lead to type 2 insulin-resistant diabetes and hyptertension. PPARG mutations may be associated with colon cancer. {ECO:0000269|PubMed:10394368}.; DISEASE: Obesity (OBESITY) [MIM:601665]: A condition characterized by an increase of body weight beyond the limitation of skeletal and physical requirements, as the result of excessive accumulation of body fat. {ECO:0000269|PubMed:9753710}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Lipodystrophy, familial partial, 3 (FPLD3) [MIM:604367]: A form of lipodystrophy characterized by marked loss of subcutaneous fat from the extremities. Facial adipose tissue may be increased, decreased or normal. Affected individuals show an increased preponderance of insulin resistance, diabetes mellitus and dyslipidemia. {ECO:0000269|PubMed:11788685, ECO:0000269|PubMed:12453919}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glioma 1 (GLM1) [MIM:137800]: Gliomas are benign or malignant central nervous system neoplasms derived from glial cells. They comprise astrocytomas and glioblastoma multiforme that are derived from astrocytes, oligodendrogliomas derived from oligodendrocytes and ependymomas derived from ependymocytes. {ECO:0000269|PubMed:10851250}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Polymorphic PPARG alleles have been found to be significantly over-represented among a cohort of American patients with sporadic glioblastoma multiforme suggesting a possible contribution to disease susceptibility. Drugs (DrugBank ID) DB00627 Interacts with NA EC number 2.1.1.1 Uniprot keywords 3D-structure; Acetylation; Citrullination; Cytoplasm; Direct protein sequencing; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 27886.8 Length 251 Aromaticity 0.1 Instability index 40.66 Isoelectric point 5.23 Charge (pH=7) -5.11 2D Binding mode Binding energy (Kcal/mol) -10.24  Molscript Map 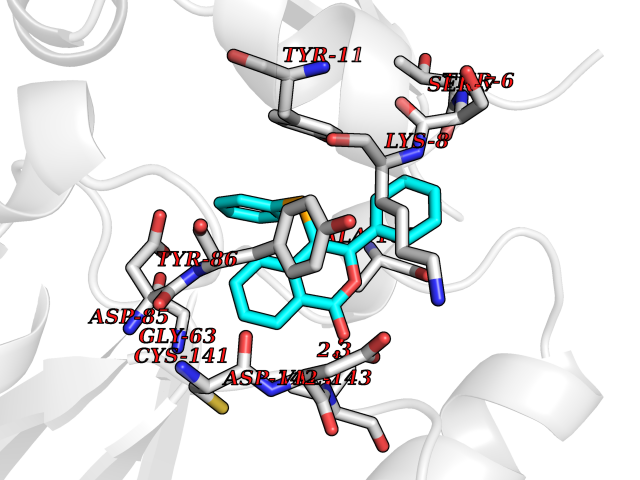 Pymol Map 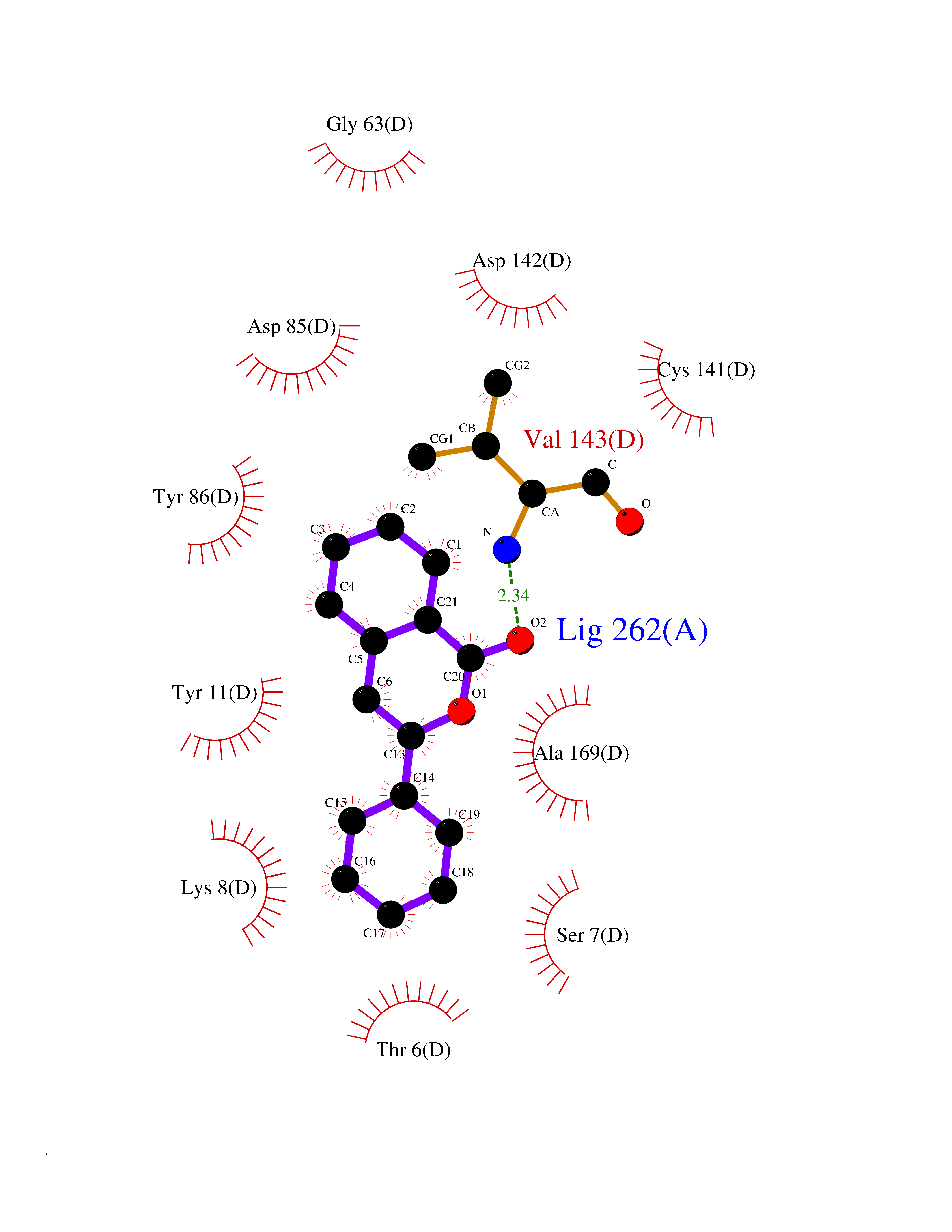 Ligplot Map 3D Binding mode Sequence GFTSKDTYLSHFNPRDYLEKYYSAESQILKHLLKNLFKIFCLDGVKGDLLIDIGSGPTIYQLLSACESFKEIVVTDYSDQNLQELEKWLKAAPAAFDWSPVVTYVCDLEGNRVKGPEKEEKLRQAVKQVLKCDVTQSQPLGAVPLPPADCVLSTLCLDAACPDLPTYCRALRNLGSLLKPGGFLVIMDALKSSYYMIGEQKFSSLPLGREAVEAAVKEAGYTIEWFEVISQSYSSTMANNEGLFSLVARKL Hydrogen bonds contact Hydrophobic contact | ||||