Job Results:
Ligand
Structure
Job ID
176b63e19640f4b1f79a8fdfdb32fb77
Job name
NA
Time
2025-10-13 17:39:01
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Potassium voltage-gated channel subfamily D member 3 | 1S1G | 5.77 | |
Target general information Gen name KCND3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Potassium channel family, D (Shal) (TC 1.A.1.2) subfamily, Kv4.3/KCND3 sub-subfamily Biochemical class Transport protein Function A-type (transient outward) potassium channel activity.Ion channel binding.Metal ion binding. Related diseases Spinocerebellar ataxia 19 (SCA19) [MIM:607346]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA19 is a relatively mild, cerebellar ataxic syndrome with cognitive impairment, pyramidal tract involvement, tremor and peripheral neuropathy, and mild atrophy of the cerebellar hemispheres and vermis. {ECO:0000269|PubMed:23280837, ECO:0000269|PubMed:23280838, ECO:0000269|PubMed:28895081}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Brugada syndrome 9 (BRGDA9) [MIM:616399]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:21349352, ECO:0000269|PubMed:22457051}. The gene represented in this entry may be involved in disease pathogenesis. Drugs (DrugBank ID) DB06637; DB00280; DB04855; DB00228; DB00458; DB11633; DB01110; DB01115; DB01069; DB06217 Interacts with Q9NZI2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Brugada syndrome; Cell membrane; Cell projection; Disease variant; Ion channel; Ion transport; Membrane; Metal-binding; Neurodegeneration; Phosphoprotein; Potassium; Potassium channel; Potassium transport; Proteomics identification; Reference proteome; Spinocerebellar ataxia; Transmembrane; Transmembrane helix; Transport; Voltage-gated channel; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 26497.2 Length 217 Aromaticity 0.18 Instability index 36.4 Isoelectric point 4.76 Charge (pH=7) -15.9 2D Binding mode Binding energy (Kcal/mol) -7.87  Molscript Map 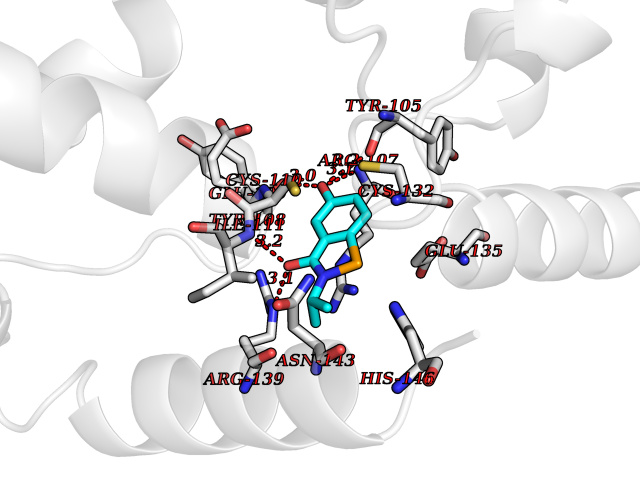 Pymol Map 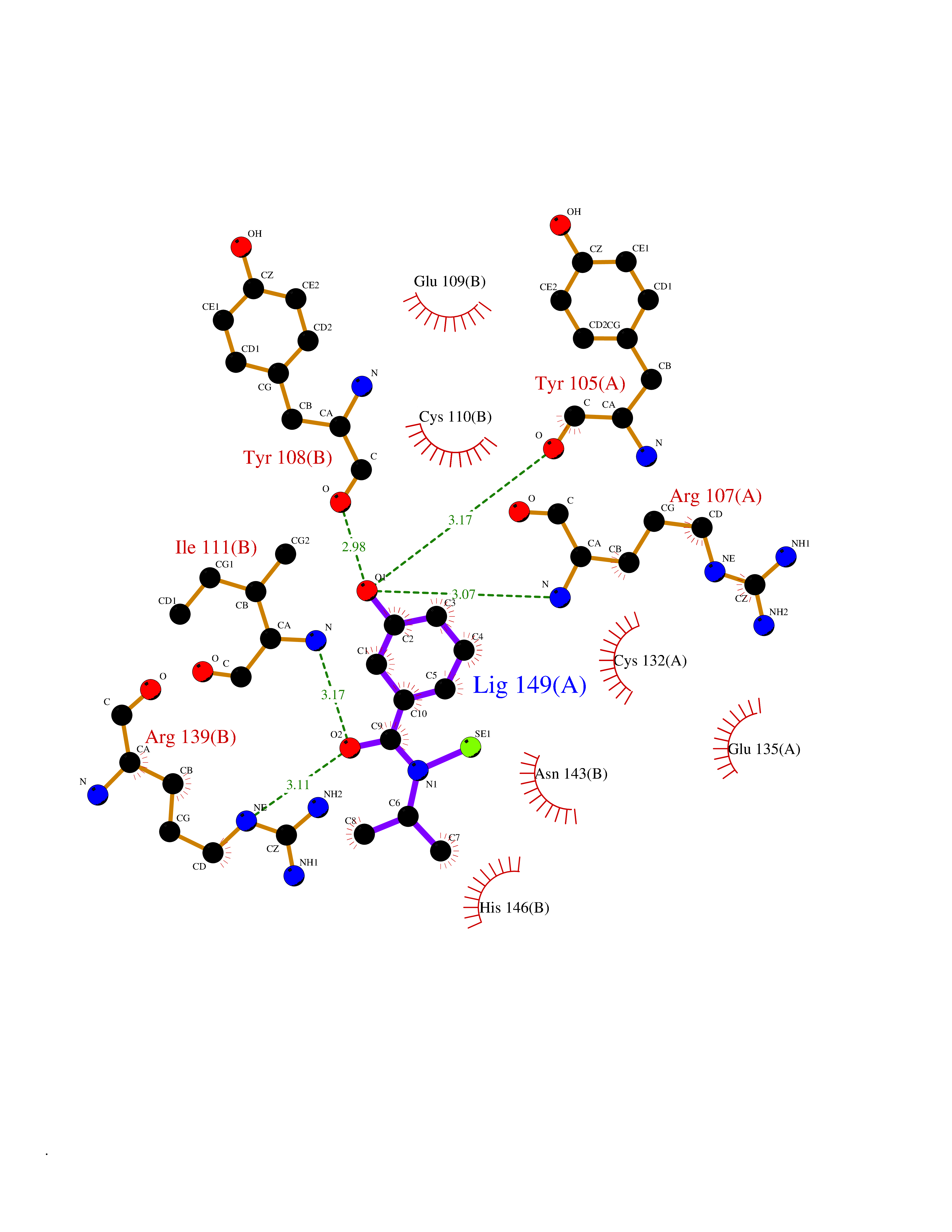 Ligplot Map 3D Binding mode Sequence DELIVLNVSGRRFQTWRTTLERYPDTLLGSTEKEFFFNEDTKEYFFDRDPEVFRCVLNFYRTGKLHYPRYECISAYDDELAFYGILPEIIGDCCYEEYKDRKRENLEQDELIVLNVSGRRFQTWRTTLERYPDTLLGSTEKEFFFNEDTKEYFFDRDPEVFRCVLNFYRTGKLHYPRYECISAYDDELAFYGILPEIIGDCCYEEYKDRKRENLEHH Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Metabotropic glutamate receptor 3 (mGluR3) | 4XAR | 5.77 | |
Target general information Gen name GRM3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms mGLUR3; Group III metabotropic glutamate receptor; GPRC1C Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling inhibits adenylate cyclase activity. G-protein coupled receptor for glutamate. Related diseases Paramyotonia congenita (PMC) [MIM:168300]: An autosomal dominant channelopathy characterized by myotonia, increased by exposure to cold, intermittent flaccid paresis, not necessarily dependent on cold or myotonia, lability of serum potassium, non-progressive nature and lack of atrophy or hypertrophy of muscles. In some patients, myotonia is not increased by cold exposure (paramyotonia without cold paralysis). Patients may have a combination phenotype of PMC and HYPP. {ECO:0000269|PubMed:10369308, ECO:0000269|PubMed:10727489, ECO:0000269|PubMed:1310898, ECO:0000269|PubMed:1316765, ECO:0000269|PubMed:1338909, ECO:0000269|PubMed:15318338, ECO:0000269|PubMed:15790667, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:18166706, ECO:0000269|PubMed:18690054, ECO:0000269|PubMed:19077043, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:8242056, ECO:0000269|PubMed:8308722, ECO:0000269|PubMed:8388676, ECO:0000269|PubMed:8580427}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hypokalemic 2 (HOKPP2) [MIM:613345]: An autosomal dominant disorder manifested by episodic flaccid generalized muscle weakness associated with falls of serum potassium levels. {ECO:0000269|PubMed:10599760, ECO:0000269|PubMed:10851391, ECO:0000269|PubMed:10944223, ECO:0000269|PubMed:11558801, ECO:0000269|PubMed:11591859, ECO:0000269|PubMed:16890191, ECO:0000269|PubMed:17898326, ECO:0000269|PubMed:18162704, ECO:0000269|PubMed:19118277, ECO:0000269|PubMed:20522878, ECO:0000269|PubMed:21043388, ECO:0000269|PubMed:24549961}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hyperkalemic (HYPP) [MIM:170500]: An autosomal dominant channelopathy characterized by episodic flaccid generalized muscle weakness associated with high levels of serum potassium. Concurrence of myotonia is found in HYPP patients. {ECO:0000269|PubMed:1659668, ECO:0000269|PubMed:1659948, ECO:0000269|PubMed:20076800}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis normokalemic (NKPP) [MIM:170500]: A disorder closely related to hyperkalemic periodic paralysis, but marked by a lack of alterations in potassium levels during attacks of muscle weakness. {ECO:0000269|PubMed:15596759, ECO:0000269|PubMed:18046642, ECO:0000269|PubMed:20522878}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myotonia SCN4A-related (MYOSCN4A) [MIM:608390]: A phenotypically highly variable myotonia aggravated by potassium loading, and sometimes by cold. Myotonia is characterized by sustained muscle tensing that prevents muscles from relaxing normally. It causes muscle stiffness that can interfere with movement. In some people the stiffness is very mild, while in other cases it may be severe enough to interfere with walking, running, and other activities of daily life. Myotonia SCN4A-related includes myotonia permanens and myotonia fluctuans. In myotonia permanens, the myotonia is generalized and there is a hypertrophy of the muscle, particularly in the neck and the shoulder. Attacks of severe muscle stiffness of the thoracic muscles may be life threatening due to impaired ventilation. In myotonia fluctuans, the muscle stiffness may fluctuate from day to day, provoked by exercise. {ECO:0000269|PubMed:10218481, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:16832098, ECO:0000269|PubMed:17212350, ECO:0000269|PubMed:17998485, ECO:0000269|PubMed:18203179, ECO:0000269|PubMed:18337100, ECO:0000269|PubMed:19015483, ECO:0000269|PubMed:19347921, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:27653901, ECO:0000269|PubMed:8058156, ECO:0000269|PubMed:9392583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 16 (CMS16) [MIM:614198]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS16 is characterized by fatigable generalized weakness and recurrent attacks of respiratory and bulbar paralysis since birth. The fatigable weakness involves lid-elevator, external ocular, facial, limb and truncal muscles and an decremental response of the compound muscle action potential on repetitive stimulation. {ECO:0000269|PubMed:12766226, ECO:0000269|PubMed:25707578, ECO:0000269|PubMed:26659129}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22A, classic (CMYO22A) [MIM:620351]: A form of congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22A is an autosomal recessive form characterized by fetal hypokinesia, polyhydramnios, and severe neonatal hypotonia associated with respiratory insufficiency. Affected individuals who survive the neonatal period have delayed motor development, difficulty walking, proximal muscle weakness of the upper and lower limbs, facial and neck muscle weakness, easy fatigability, and mild limb contractures or foot deformities. {ECO:0000269|PubMed:26700687, ECO:0000269|PubMed:28262468, ECO:0000269|PubMed:36090556}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22B, severe fetal (CMYO22B) [MIM:620369]: A severe congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22B is an autosomal recessive form characterized by onset in utero. Affected individuals show fetal akinesia, and develop fetal hydrops with pulmonary hypoplasia, severe joint contractures, and generalized muscle hypoplasia. Death occurs in utero or soon after birth. {ECO:0000269|PubMed:26700687}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05096 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50355.5 Length 445 Aromaticity 0.11 Instability index 38.26 Isoelectric point 6.52 Charge (pH=7) -1.53 2D Binding mode Binding energy (Kcal/mol) -7.87  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence RREIKIEGDLVLGGLFPINEKGTGTEECGRINEDRGIQRLEAMLFAIDEINKDDYLLPGVKLGVHILDTCSRDTYALEQSLEFVRASLLLIAGVIGGSYSSVSIQVANLLRLFQIPQISYASTSAKLSDKSRYDYFARTVPPDFYQAKAMAEILRFFNWTYVSTVASEGDYGETGIEAFEQEARLRNISIATAEKVGRSNIRKSYDSVIRELLQKPNARVVVLFMRSDDSRELIAAASRANASFTWVASDGWGAQESIIKGSEHVAYGAITLELASQPVRQFDRYFQSLNPYNNHRNPWFRDFWEQKFQCSLRVCDKHLAIDSSNYEQESKIMFVVNAVYAMAHALHKMQRTLCPNTTKLCDAMKILDGKKLYKDYLLKINFTAPDADSIVKFDTFGDGMGRYNVFNFQNVGGKYSYLKVGHWAETLSLDVNSIHWSRNSVPTSE Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Acyloxyacyl hydrolase (neutrophil) | 5W7C | 5.77 | |
Target general information Gen name AOAH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Acyloxyacyl hydrolase Protein family NA Biochemical class NA Function Removes the secondary (acyloxyacyl-linked) fatty acyl chains from the lipid A region of bacterial lipopolysaccharides. By breaking down LPS, terminates the host response to bacterial infection and prevents prolonged and damaging inflammatory responses (By similarity). In peritoneal macrophages, seems to be important for recovery from a state of immune tolerance following infection by Gram-negative bacteria (By similarity). Related diseases Major depressive disorder (MDD) [MIM:608516]: A common psychiatric disorder. It is a complex trait characterized by one or more major depressive episodes without a history of manic, mixed, or hypomanic episodes. A major depressive episode is characterized by at least 2 weeks during which there is a new onset or clear worsening of either depressed mood or loss of interest or pleasure in nearly all activities. Four additional symptoms must also be present including changes in appetite, weight, sleep, and psychomotor activity; decreased energy; feelings of worthlessness or guilt; difficulty thinking, concentrating, or making decisions; or recurrent thoughts of death or suicidal ideation, plans, or attempts. The episode must be accompanied by distress or impairment in social, occupational, or other important areas of functioning. {ECO:0000269|PubMed:15229186}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q15700 EC number EC 3.1.1.77 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cytoplasmic vesicle; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Lipid metabolism; Metal-binding; Proteomics identification; Reference proteome; Secreted; Signal; Zymogen Protein physicochemical properties Chain ID C Molecular weight (Da) 47779.7 Length 420 Aromaticity 0.1 Instability index 43.45 Isoelectric point 7.72 Charge (pH=7) 2.1 2D Binding mode Binding energy (Kcal/mol) -7.87  Molscript Map 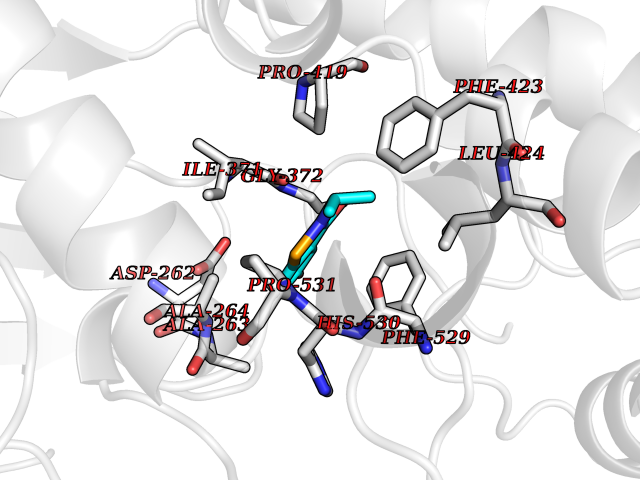 Pymol Map 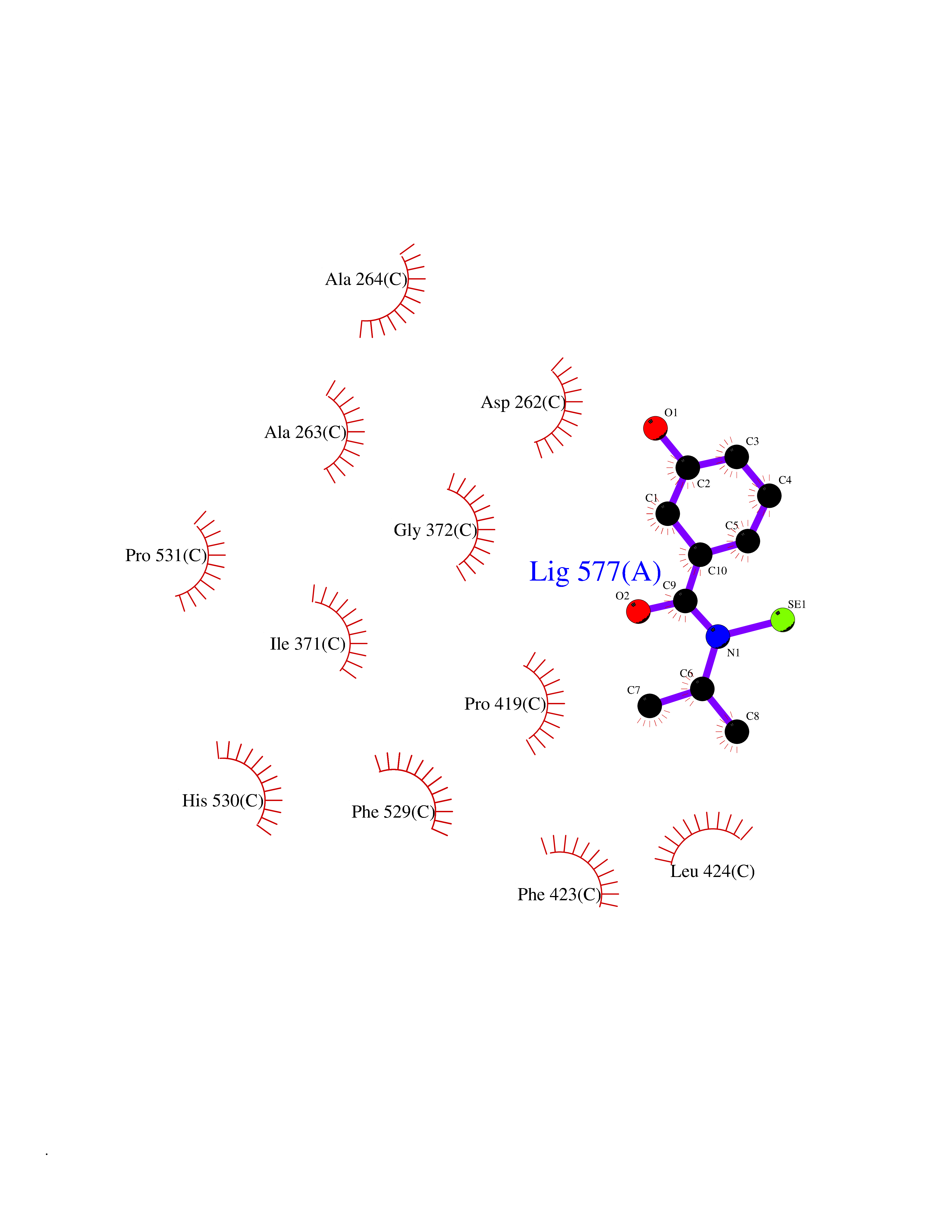 Ligplot Map 3D Binding mode Sequence GSDICSLPVLAKICQKIKLAMEQSVPFKDVDSDKYSVFPTLRGYHWRGRDCNDSDESVYPGRRPNNWDVHQDSNCNGIWGVDPKDGVPYEKKFCEGSQPRGIILLGDAAGAHFHISPEWITASQMSLNSFINLPTALTNELDWPQLSGATGFLDSTVGIKEKSIYLRLWKRNHCNHRDYQNISRNGASSRNLKKFIESLSRNKVLDYPAIVIYAMIGNDVCSGKSDPVPAMTTPEKLYSNVMQTLKHLNSHLPNGSHVILYGLPDGTFLWDNLHNRYHPLGQLNKDMTYAQLYSFLNCLQVSPCHGWMSSNKTLRTLTSERAEQLSNTLKKIAASEKFTNFNLFYMDFAFHEIIQEWQKRGGQPWQLIEPVDGFHPNEVALLLLADHFWKKVQLQWPQILGKENPFNPQIKQVFGDQGGH Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Lysine N-methyltransferase 3B (NSD1) | 3OOI | 5.77 | |
Target general information Gen name NSD1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nuclear receptor-binding SET domain-containing protein 1; NR-binding SET domain-containing protein; NR-binding SET domain containing protein; KMT3B; Hypothetical protein FLJ22263 similar to nuclear re Protein family Class V-like SAM-binding methyltransferase superfamily Biochemical class Methyltransferase Function Preferentially methylates 'Lys-36' of histone H3 and 'Lys-20' of histone H4 (in vitro). Transcriptional intermediary factor capable of both negatively or positively influencing transcription, depending on the cellular context. Histone methyltransferase. Related diseases Sotos syndrome (SOTOS) [MIM:117550]: An autosomal dominant, childhood overgrowth syndrome characterized by pre- and postnatal overgrowth, developmental delay, intellectual disability, advanced bone age, and abnormal craniofacial morphology including macrodolichocephaly with frontal bossing, frontoparietal sparseness of hair, apparent hypertelorism, downslanting palpebral fissures, and facial flushing. Common oral findings include: premature eruption of teeth; high, arched palate; pointed chin and, more rarely, prognathism. {ECO:0000269|PubMed:11896389, ECO:0000269|PubMed:12464997, ECO:0000269|PubMed:12807965, ECO:0000269|PubMed:14997421}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Beckwith-Wiedemann syndrome (BWS) [MIM:130650]: A disorder characterized by anterior abdominal wall defects including exomphalos (omphalocele), pre- and postnatal overgrowth, and macroglossia. Additional less frequent complications include specific developmental defects and a predisposition to embryonal tumors. {ECO:0000269|PubMed:14997421}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: A chromosomal aberration involving NSD1 is found in childhood acute myeloid leukemia. Translocation t(5;11)(q35;p15.5) with NUP98.; DISEASE: A chromosomal aberration involving NSD1 is found in an adult form of myelodysplastic syndrome (MDS). Insertion of NUP98 into NSD1 generates a NUP98-NSD1 fusion product. {ECO:0000269|PubMed:15382262}. Drugs (DrugBank ID) NA Interacts with Q13283; Q16778; Q04206; O95994; Q86Z20; A8MQ03; Q3LI66; Q99750 EC number EC 2.1.1.43 Uniprot keywords 3D-structure; Activator; Alternative splicing; Chromatin regulator; Chromosomal rearrangement; Chromosome; Disease variant; Isopeptide bond; Metal-binding; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Proto-oncogene; Reference proteome; Repeat; Repressor; S-adenosyl-L-methionine; Transcription; Transcription regulation; Transferase; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 26487.9 Length 232 Aromaticity 0.08 Instability index 37.29 Isoelectric point 8.1 Charge (pH=7) 2.69 2D Binding mode Binding energy (Kcal/mol) -7.87  Molscript Map 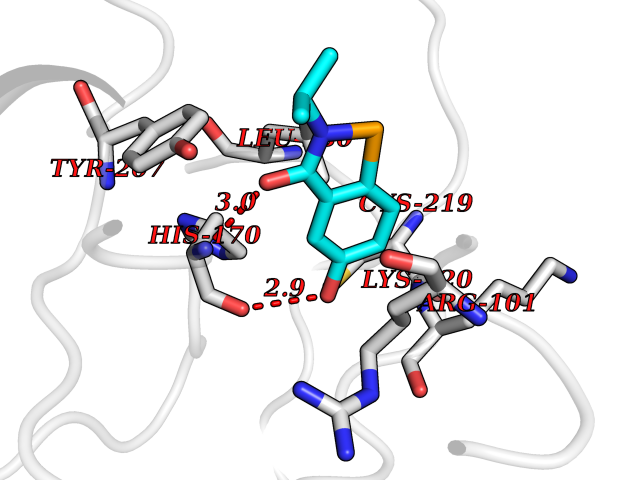 Pymol Map 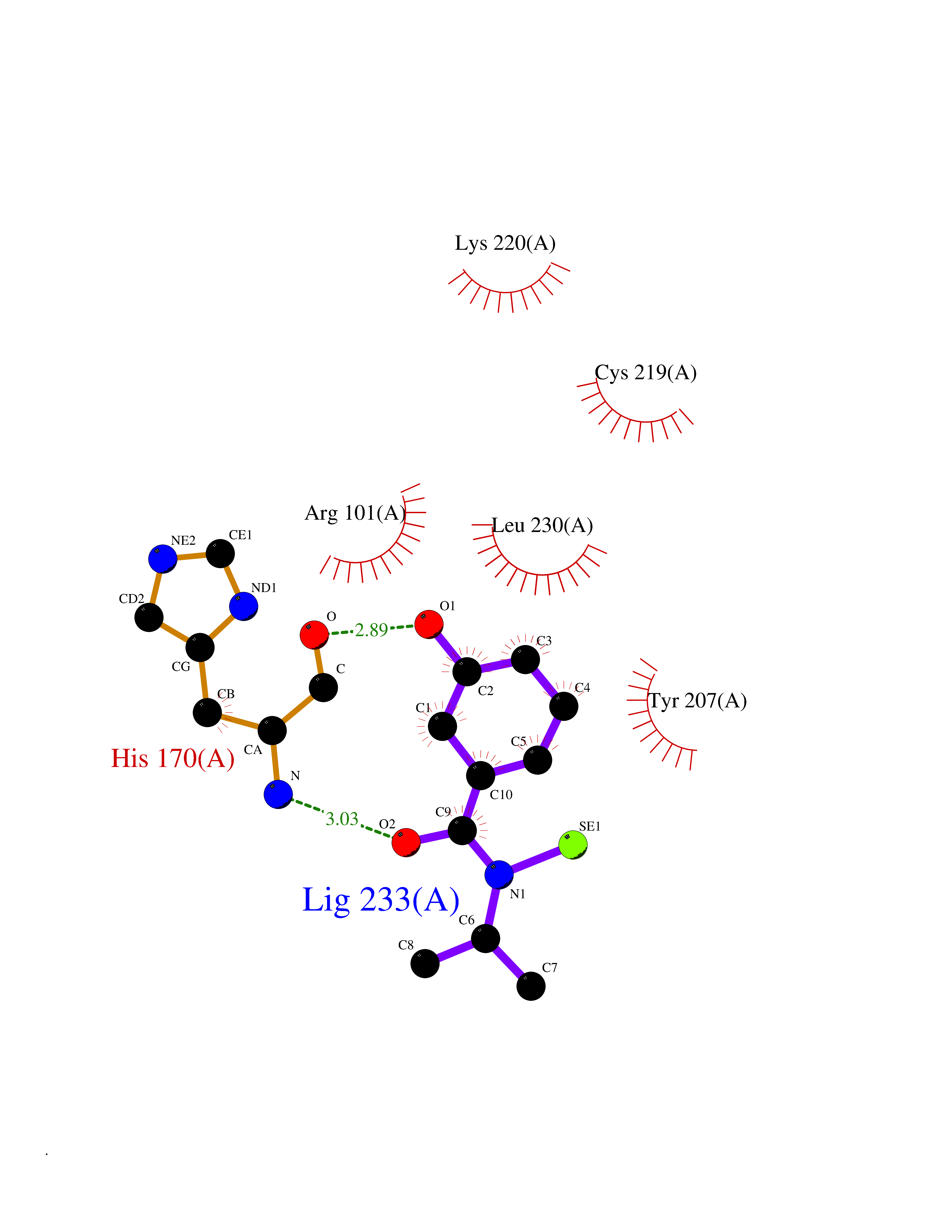 Ligplot Map 3D Binding mode Sequence SKELRQLQEDRKNDKKPPPYKHIKVNRPIGRVQIFTADLSEIPRCNCKATDENPCGIDSECINRMLLYECHPTVCPAGGRCQNQCFSKRQYPEVEIFRTLQRGWGLRTKTDIKKGEFVNEYVGELIDEEECRARIRYAQEHDITNFYMLTLDKDRIIDAGPKGNYARFMNHCCQPNCETQKWSVNGDTRVGLFALSDIKAGTELTFNYNLECLGNGKTVCKCGAPNCSGFLG Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Zinc finger protein Helios (IKZF2) | 7LPS | 5.77 | |
Target general information Gen name IKZF2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ikaros family zinc finger protein 2 Protein family Ikaros C2H2-type zinc-finger protein family Biochemical class NA Function Associates with Ikaros at centromeric heterochromatin. Related diseases Developmental and epileptic encephalopathy 25, with amelogenesis imperfecta (DEE25) [MIM:615905]: An autosomal recessive disease characterized by subclinical seizures appearing in the first days of life, evolving to severe epileptic disease. Affected individuals have profound or severe delayed development with lack of speech, and most patients do not acquire the ability to sit. Additional variable features include axial hypotonia, peripheral hypertonia, and abnormal involuntary movements such as dystonia and choreoathetosis. Dental abnormalities, including delayed eruption, hypodontia, tooth hypoplasia, yellow discoloration, thin enamel, and enamel chipping are observed in most patients. {ECO:0000269|PubMed:24995870, ECO:0000269|PubMed:26384929, ECO:0000269|PubMed:30054523}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P29972; P56545; P56545-3; Q17RB8; P09022; Q8N8B7-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Activator; Alternative splicing; DNA-binding; Isopeptide bond; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID B,C Molecular weight (Da) 47006.6 Length 410 Aromaticity 0.09 Instability index 44.28 Isoelectric point 7.23 Charge (pH=7) 0.69 2D Binding mode Binding energy (Kcal/mol) -7.86  Molscript Map  Pymol Map 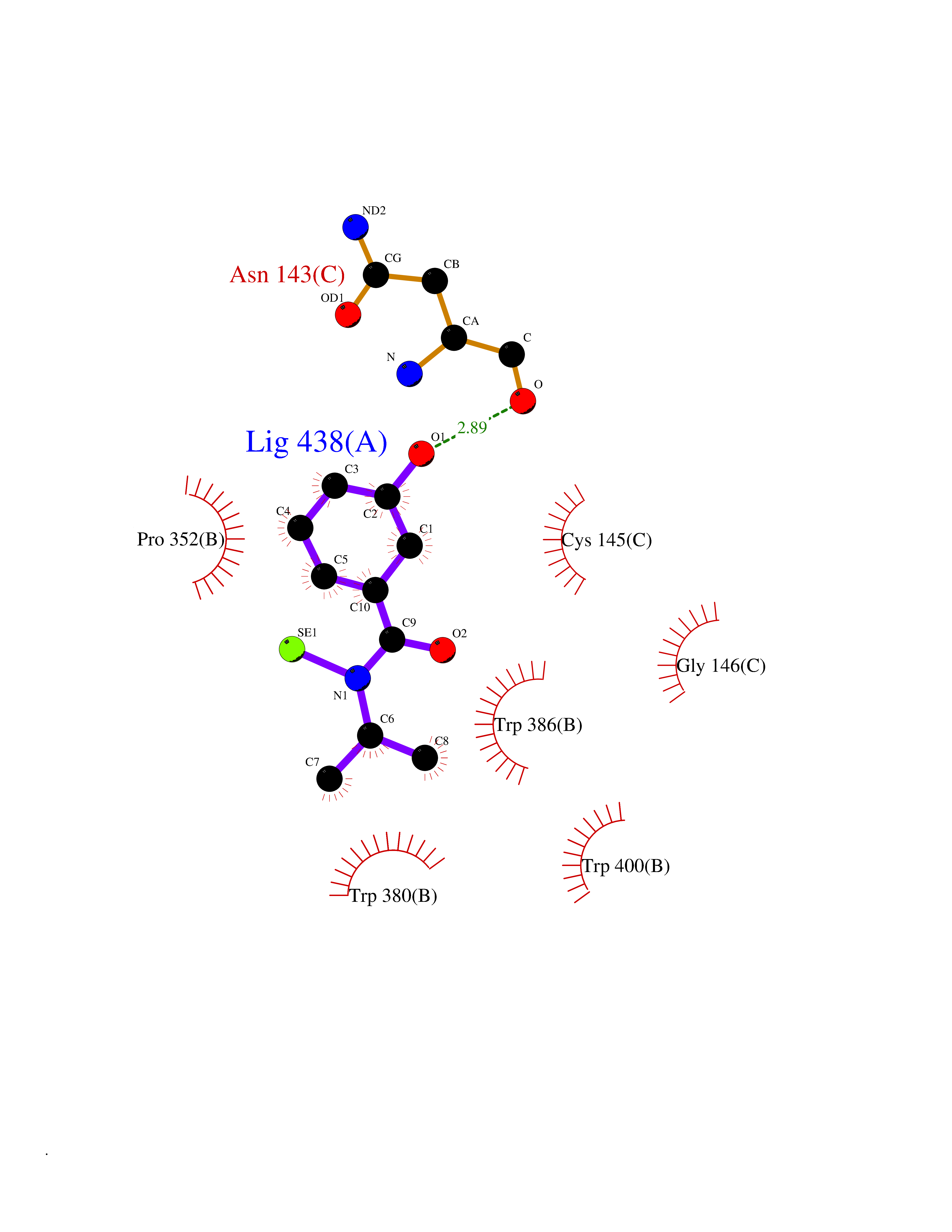 Ligplot Map 3D Binding mode Sequence INFDTSLPTSHTYLGADMEEFHGRTLHDDDSCQVIPVLPQVMMILIPGQTLPLQLFHPQEVSMVRNLIQKDRTFAVLAYSNVQEREAQFGTTAEIYAYREEQDFGIEIVKVKAIGRQRFKVLELRTQSDGIQQAKVQILPECVLPSTMSAVQLESLNKCQIFPCSYKWWQKYQKRKFHCANLTSWPRWLYSLYDAETLMDRIKKQLREWDENLKDDSLPSNPIDFSYRVAACLPIDDVLRIQLLKIGSAIQRLRCELDIMNKCTSLCCKQCQETEITTKNEIFSLSLCGPMAAYVNPHGYVHETLTVYKACNLNLIGRPSTEHSWFPGYAWTVAQCKICASHIGWKFTATKKDMSPQKFWGLTRSALLPTIPDTEDEISPDGERPFHCNQCGASFTQKGNLLRHIKLHSG Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Fungal Scytalone dehydratase (Fung SDH1) | 3STD | 5.77 | |
Target general information Gen name Fung SDH1 Organism Pyricularia oryzae (strain 70-15 / ATCC MYA-4617 / FGSC 8958) (Rice blast fungus) (Magnaporthe oryzae) Uniprot ID TTD ID Synonyms SDH1 Protein family Scytalone dehydratase family Biochemical class Alpha-carbonic anhydrase Function Catalyzes two steps in melanin biosynthesis. From scytalone they are two dehydration steps and one reduction step to yield melanin. Related diseases CODAS syndrome (CODASS) [MIM:600373]: A rare syndrome characterized by the combination of cerebral, ocular, dental, auricular, and skeletal features. These include developmental delay, craniofacial anomalies, cataracts, ptosis, median nasal groove, delayed tooth eruption, hearing loss, short stature, delayed epiphyseal ossification, metaphyseal hip dysplasia, and vertebral coronal clefts. {ECO:0000269|PubMed:25574826, ECO:0000269|PubMed:25808063}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 4.2.1.94 Uniprot keywords 3D-structure; Calcium; Direct protein sequencing; Endosome; Lyase; Melanin biosynthesis; Metal-binding; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 19102.4 Length 162 Aromaticity 0.14 Instability index 31.72 Isoelectric point 5.87 Charge (pH=7) -3.7 2D Binding mode Binding energy (Kcal/mol) -7.87 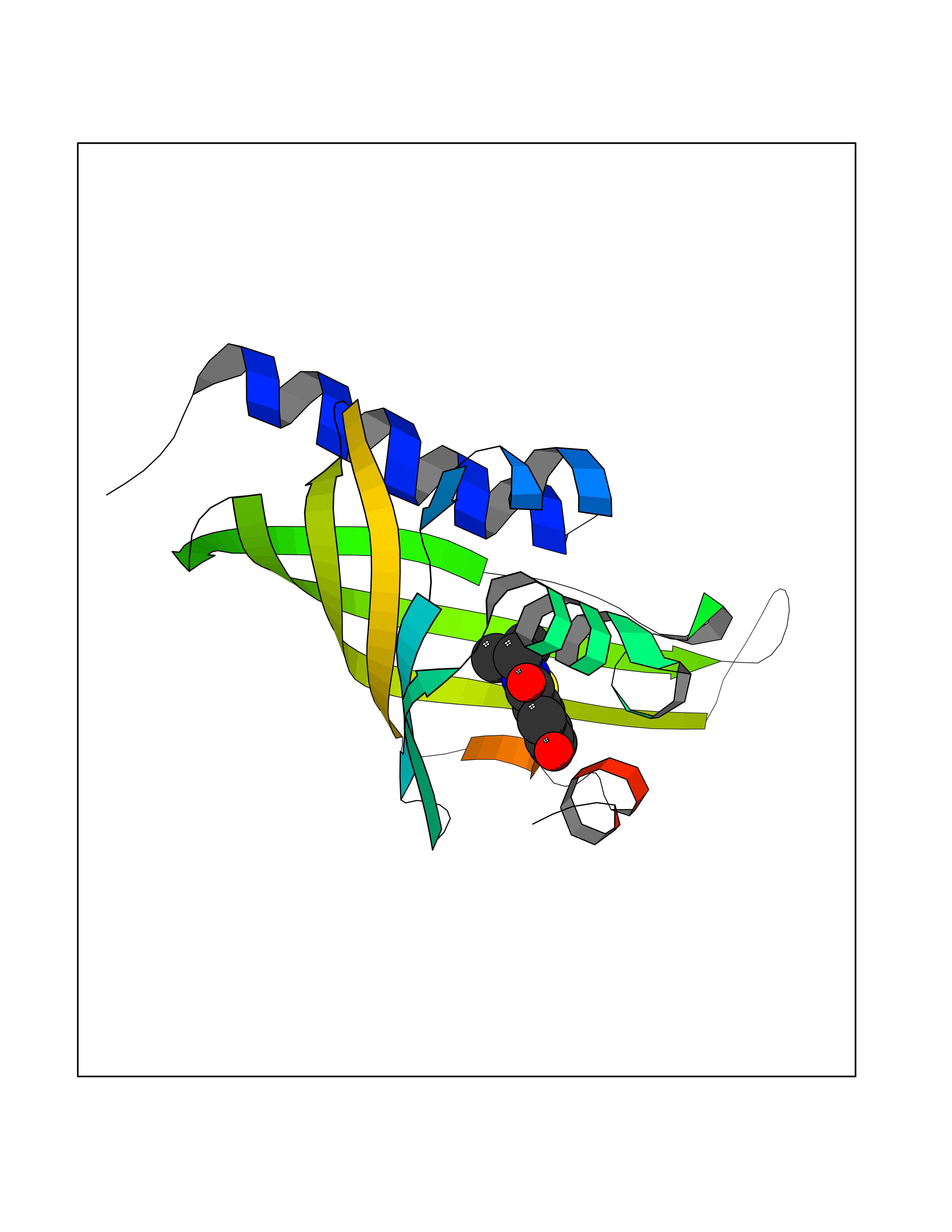 Molscript Map 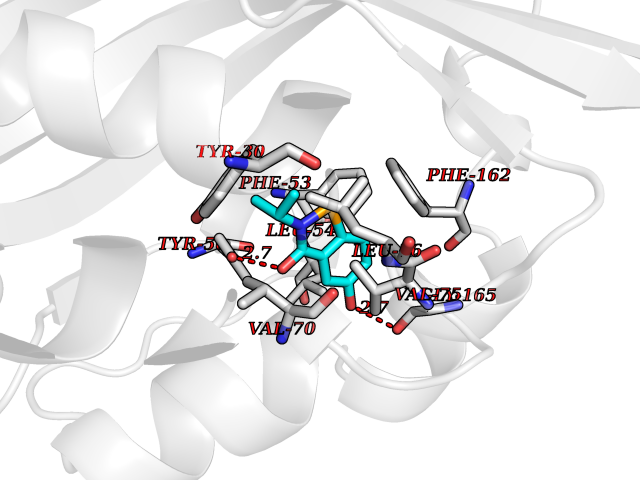 Pymol Map 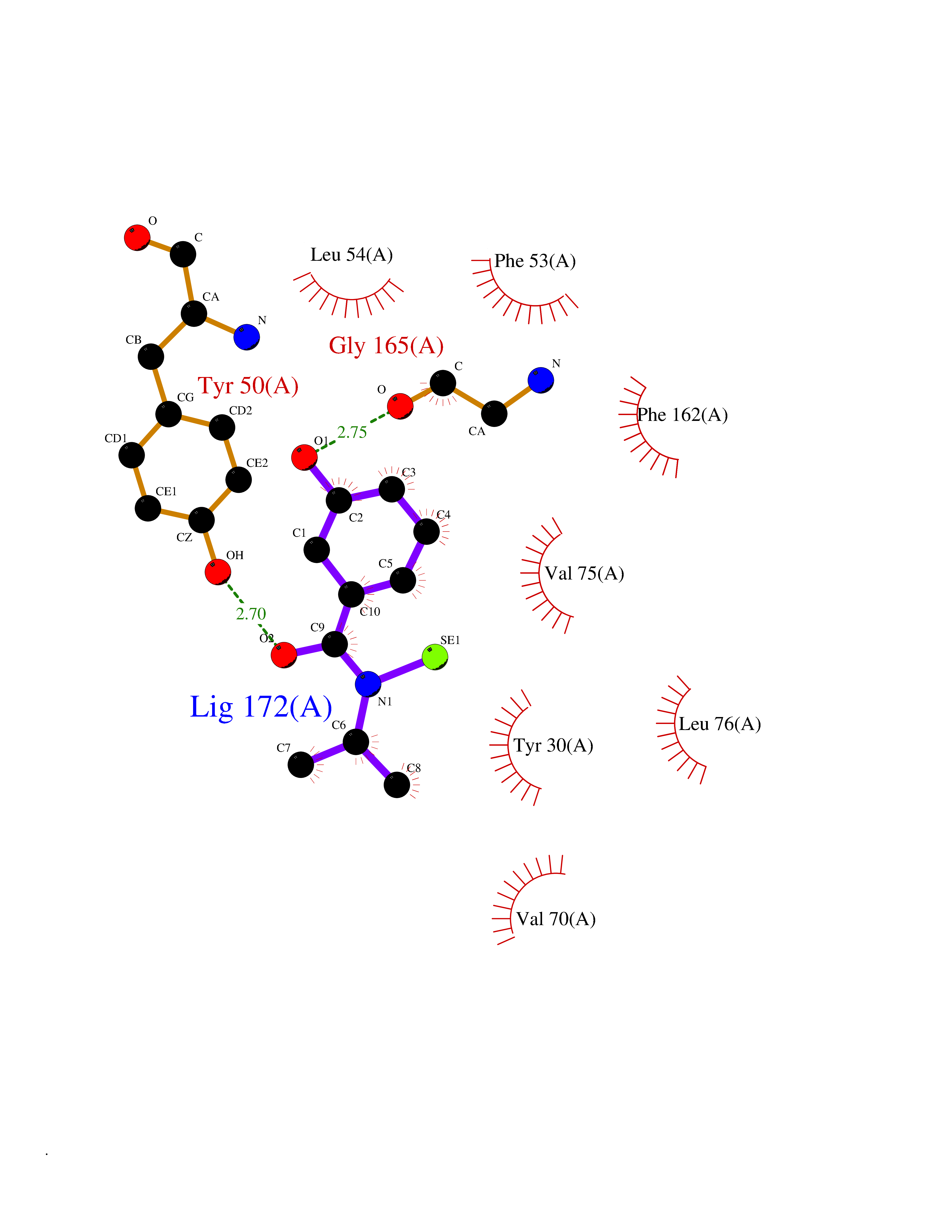 Ligplot Map 3D Binding mode Sequence GEITFSDYLGLMTCVYEWADSYDSKDWDRLRKVIAPTLRIDYRSFLDKLWEAMPAEEFVGMVSSKQVLGDPTLRTQHFIGGTRWEKVSEDEVIGYHQLRVPHQRYKDTTMKEVTMKGHAHSANLHWYKKIDGVWKFAGLKPDIRWGEFDFDRIFEDGRETFG Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | MAPK signal-integrating kinase 1 (MKNK1) | 5WVD | 5.77 | |
Target general information Gen name MKNK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Mnk1; MAP kinase signal-integrating kinase 1 Protein family Protein kinase superfamily, CAMK Ser/Thr protein kinase family Biochemical class Protein kinase superfamily. CAMK Ser/Thr protein kinase family Function May play a role in the response to environmental stress and cytokines. Appears to regulate translation by phosphorylating EIF4E, thus increasing the affinity of this protein for the 7-methylguanosine-containing mRNA cap. Related diseases Defects in MELK are associated with some cancers, such as brain or breast cancers. Expression is dramatically increased in aggressive undifferentiated tumors, correlating with poor patient outcome in breast and brain cancers, suggesting a role in tumor-initiating cells and proliferation via its function in cell proliferation regulation. Drugs (DrugBank ID) DB12010 Interacts with P54253; Q03060-25; P42858; P28482; Q16539; Q96CV9 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cytoplasm; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Translation regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 27536.2 Length 241 Aromaticity 0.11 Instability index 50.42 Isoelectric point 6.02 Charge (pH=7) -3.43 2D Binding mode Binding energy (Kcal/mol) -7.87 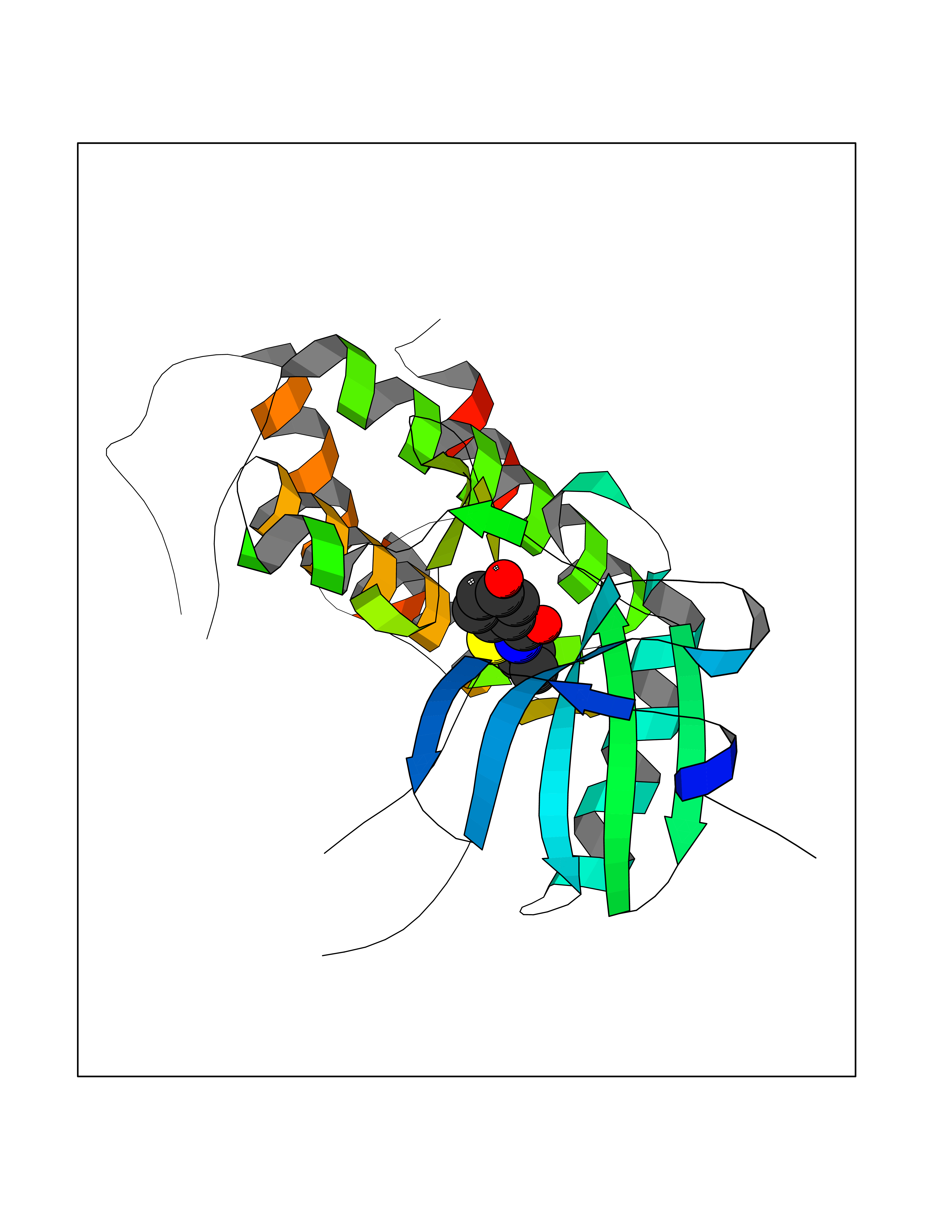 Molscript Map 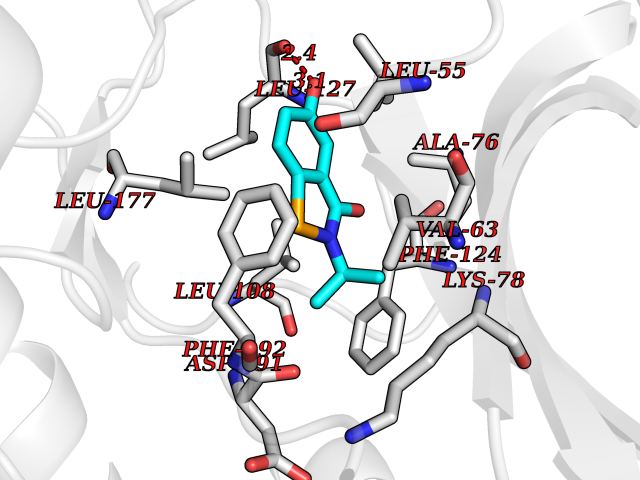 Pymol Map 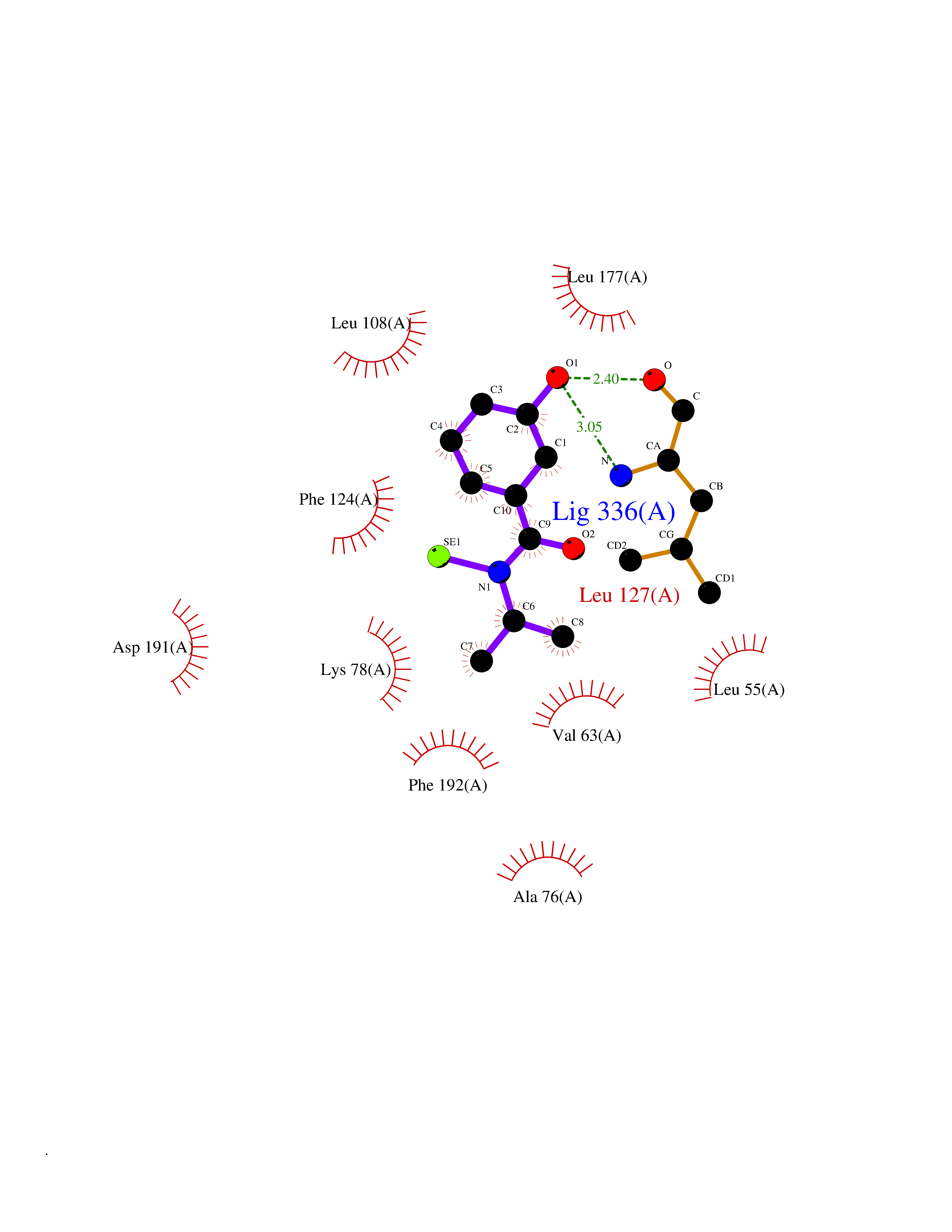 Ligplot Map 3D Binding mode Sequence PGKFEDMYKLTSELLGEGAYAKVQGAVSLQNGKEYAVKIIEKQAGHSRSRVFREVETLYQCQGNKNILELIEFFEDDTRFYLVFEKLQGGSILAHIQKQKHFNEREASRVVRDVAAALDFLHTKGIAHRDLKPENILCESPEKVSPVKICDFDLGSGYMAPEVVEVFTDQATFYDKRCDLWSLGVVLYIMLSGYPPFKYEFPDKDWAHISSEAKDLISKLLVRDAKQRLSAAQVLQHPWVQ Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Neprilysin | 1R1H | 5.76 | |
Target general information Gen name MME Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms EPN Protein family Peptidase M13 family Biochemical class Hydrolase Function Endopeptidase activity.Exopeptidase activity.Metalloendopeptidase activity.Metallopeptidase activity.Peptide binding.Zinc ion binding. Related diseases Charcot-Marie-Tooth disease, axonal, 2T (CMT2T) [MIM:617017]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:26991897, ECO:0000269|PubMed:27588448}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinocerebellar ataxia 43 (SCA43) [MIM:617018]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA43 is a slowly progressive, autosomal dominant form. {ECO:0000269|PubMed:27583304}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08575; DB02597; DB00616; DB11623; DB05796; DB06655; DB02558; DB02062; DB00886; DB02557; DB09292; DB13928; DB08626 Interacts with P05067; P21926; Q06787-7; P08107; P04792 EC number 3.4.24.11 Uniprot keywords 3D-structure; Cell membrane; Charcot-Marie-Tooth disease; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lipoprotein; Membrane; Metal-binding; Metalloprotease; Myristate; Neurodegeneration; Neuropathy; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Spinocerebellar ataxia; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 79435.8 Length 696 Aromaticity 0.11 Instability index 37.5 Isoelectric point 5.53 Charge (pH=7) -11.46 2D Binding mode Binding energy (Kcal/mol) -7.85  Molscript Map 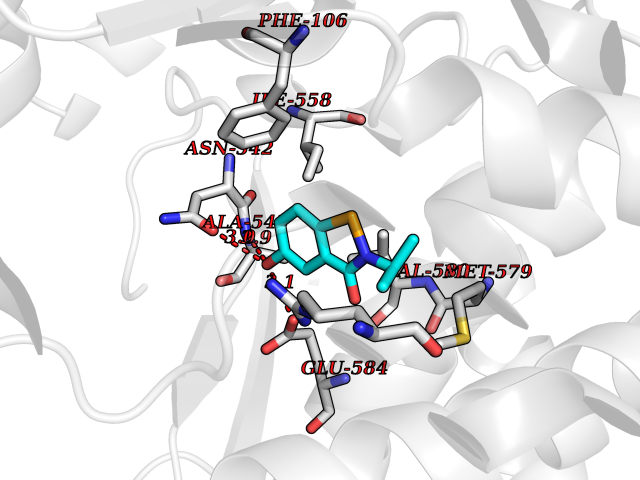 Pymol Map 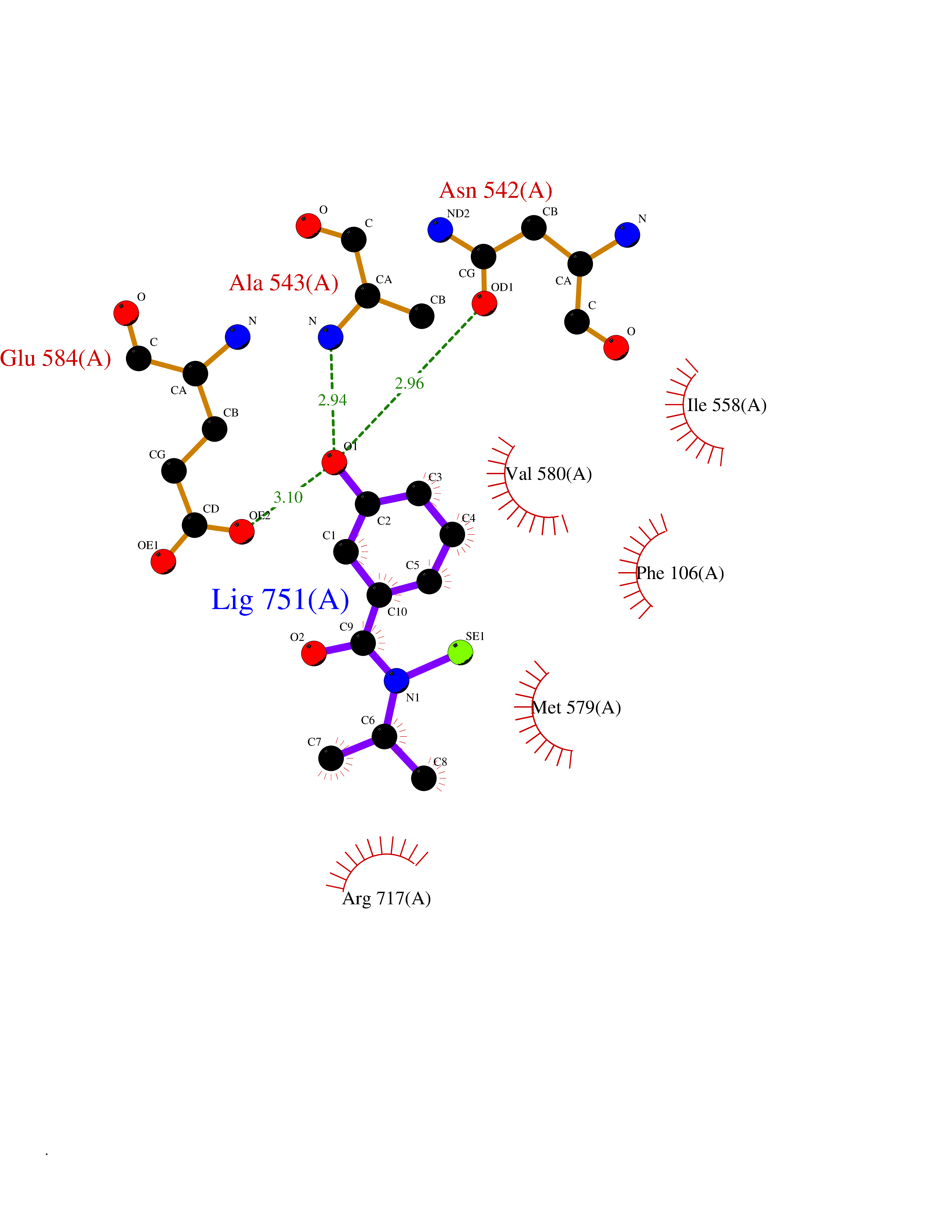 Ligplot Map 3D Binding mode Sequence GICKSSDCIKSAARLIQNMDATTEPCTDFFKYACGGWLKRNVIPETSSRYGNFDILRDELEVVLKDVLQEPKTEDIVAVQKAKALYRSCINESAIDSRGGEPLLKLLPDIYGWPVATENWEQKYGASWTAEKAIAQLNSKYGKKVLINLFVGTDDKNSVNHVIHIDQPRLGLPSRDYYECTGIYKEACTAYVDFMISVARLIRQEERLPIDENQLALEMNKVMELEKEIANATAKPEDRNDPMLLYNKMTLAQIQNNFSLEINGKPFSWLNFTNEIMSTVNISITNEEDVVVYAPEYLTKLKPILTKYSARDLQNLMSWRFIMDLVSSLSRTYKESRNAFRKALYGTTSETATWRRCANYVNGNMENAVGRLYVEAAFAGESKHVVEDLIAQIREVFIQTLDDLTWMDAETKKRAEEKALAIKERIGYPDDIVSNDNKLNNEYLELNYKEDEYFENIIQNLKFSQSKQLKKLREKVDKDEWISGAAVVNAFYSSGRNQIVFPAGILQPPFFSAQQSNSLNYGGIGMVIGHEITHGFDDNGRNFNKDGDLVDWWTQQSASNFKEQSQCMVYQYGNFSWDLAGGQHLNGINTLGENIADNGGLGQAYRAYQNYIKKNGEEKLLPGLDLNHKQLFFLNFAQVWCGTYRPEYAVNSIKTDVHSPGNFRIIGTLQNSAEFSEAFHCRKNSYMNPEKKCRVW Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Monoamine oxidase type B (MAO-B) | 2V5Z | 5.76 | |
Target general information Gen name MAOB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MAO-B; Amine oxidase [flavin-containing] B Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. MAOB preferentially degrades benzylamine and phenylethylamine. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB08176; DB02211; DB08516; DB08480; DB01472; DB04307; DB07512; DB07513; DB00915; DB00182; DB06698; DB04889; DB00215; DB09130; DB04147; DB00988; DB01363; DB00668; DB01175; DB02509; DB03147; DB14914; DB00614; DB04818; DB02095; DB01247; DB00601; DB01577; DB01442; DB01171; DB08082; DB02643; DB04677; DB03894; DB08804; DB04820; DB00184; DB04821; DB12612; DB01626; DB00780; DB00191; DB00388; DB01132; DB00721; DB01168; DB01367; DB09363; DB06654; DB01037; DB01104; DB14569; DB09042; DB00752; DB16446; DB09185; DB04832; DB00909 Interacts with P55212; P28329-3; Q8NI60; Q5RI15; Q92915-2; P22607; Q53GS7; P06396; P01112; O14901; P13473-2; P21397; Q9BVL2; O75400-2; P62826; Q6NTF9-3; Q9Y371; Q7Z699; Q9UMX0; Q9Y649 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Direct protein sequencing; FAD; Flavoprotein; Membrane; Mitochondrion; Mitochondrion outer membrane; Oxidoreductase; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 56019.9 Length 494 Aromaticity 0.09 Instability index 34.81 Isoelectric point 6.51 Charge (pH=7) -2.2 2D Binding mode Binding energy (Kcal/mol) -7.85 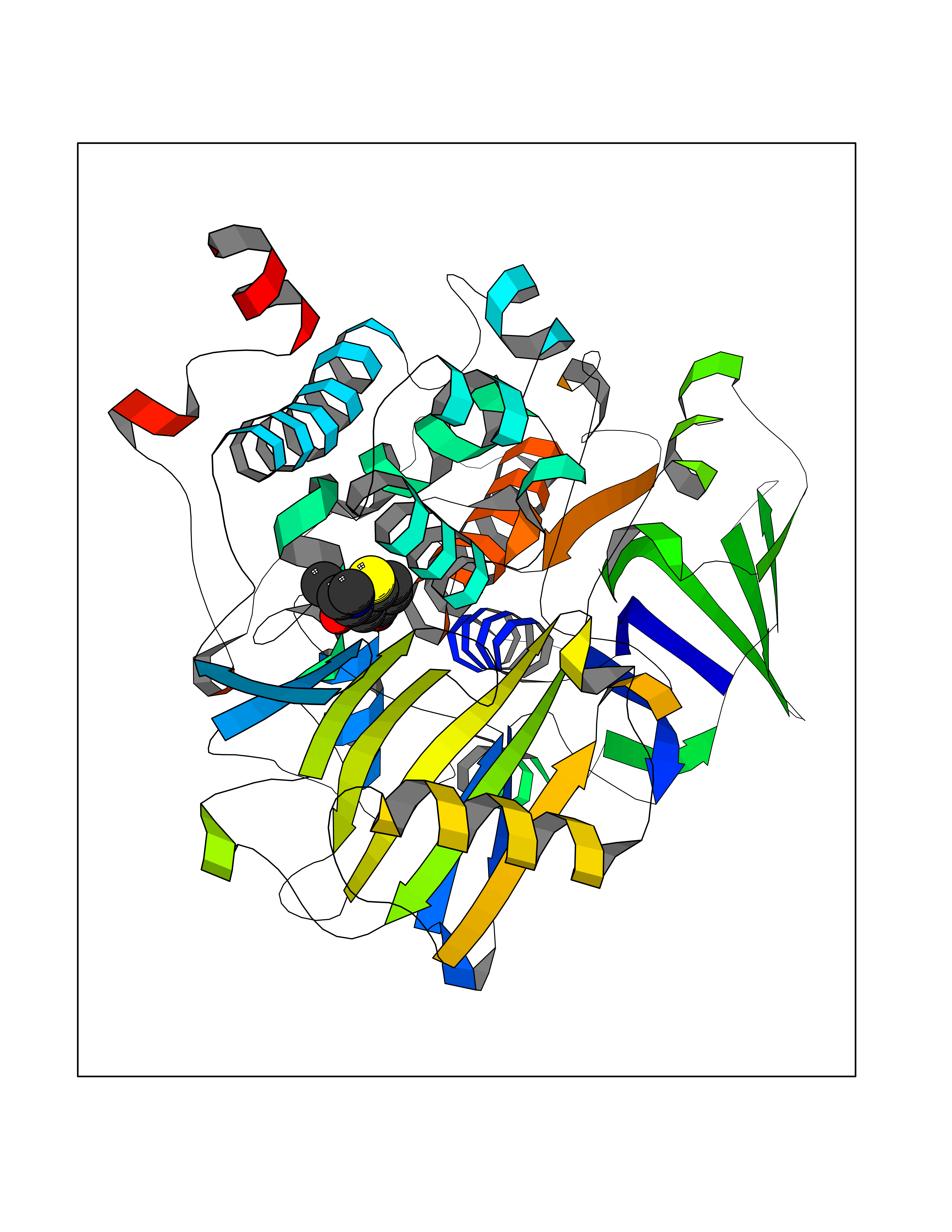 Molscript Map 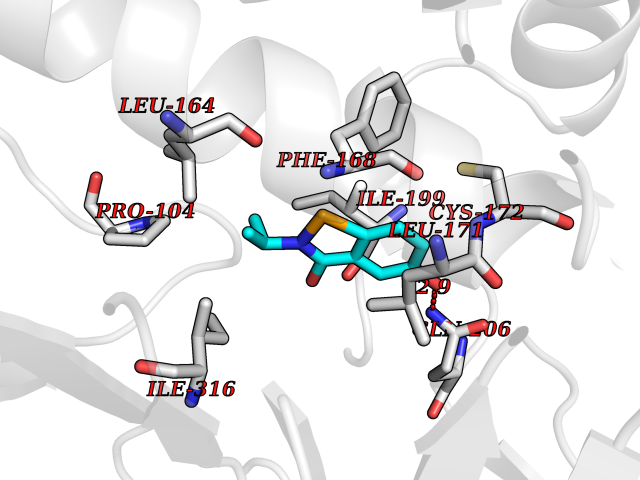 Pymol Map 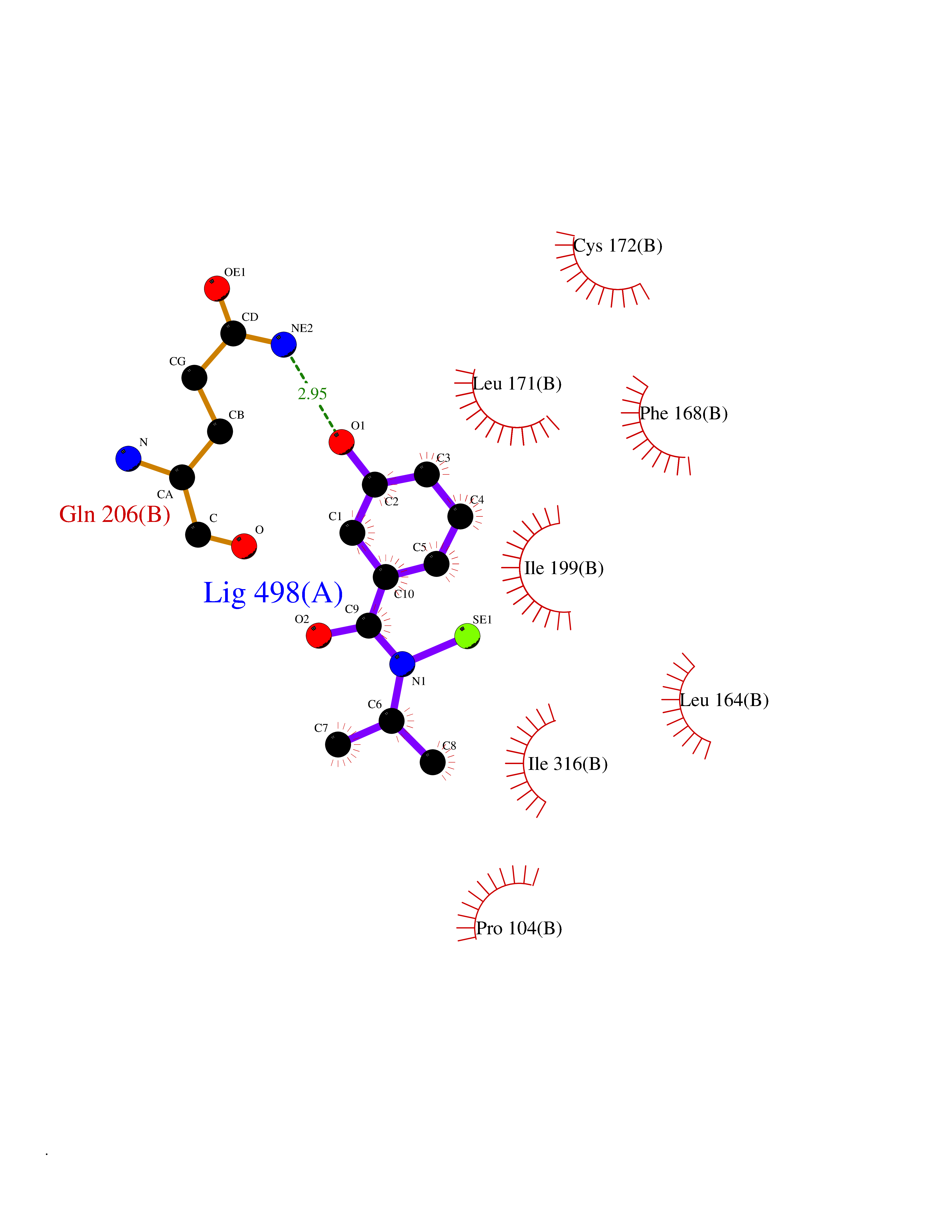 Ligplot Map 3D Binding mode Sequence NKCDVVVVGGGISGMAAAKLLHDSGLNVVVLEARDRVGGRTYTLRNQKVKYVDLGGSYVGPTQNRILRLAKELGLETYKVNEVERLIHHVKGKSYPFRGPFPPVWNPITYLDHNNFWRTMDDMGREIPSDAPWKAPLAEEWDNMTMKELLDKLCWTESAKQLATLFVNLCVTAETHEVSALWFLWYVKQCGGTTRIISTTNGGQERKFVGGSGQVSERIMDLLGDRVKLERPVIYIDQTRENVLVETLNHEMYEAKYVISAIPPTLGMKIHFNPPLPMMRNQMITRVPLGSVIKCIVYYKEPFWRKKDYCGTMIIDGEEAPVAYTLDDTKPEGNYAAIMGFILAHKARKLARLTKEERLKKLCELYAKVLGSLEALEPVHYEEKNWCEEQYSGGCYTTYFPPGILTQYGRVLRQPVDRIYFAGTETATHWSGYMEGAVEAGERAAREILHAMGKIPEDEIWQSEPESVDVPAQPITTTFLERHLPSVPGLLRLI Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Tankyrase-2 (TNKS-2) | 3U9H | 5.76 | |
Target general information Gen name TNKS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tankyrase-related protein; Tankyrase-like protein; Tankyrase II; TRF1-interacting ankyrin-related ADP-ribose polymerase 2; TNKL; TANK2; Protein poly-ADP-ribosyltransferase tankyrase-2; Poly [ADP-ribos Protein family ARTD/PARP family Biochemical class Glycosyltransferases Function Acts as an activator of the Wnt signaling pathway by mediating poly-ADP-ribosylation of AXIN1 and AXIN2, 2 key components of the beta-catenin destruction complex: poly-ADP-ribosylated target proteins are recognized by RNF146, which mediates their ubiquitination and subsequent degradation. Also mediates poly-ADP-ribosylation of BLZF1 and CASC3, followed by recruitment of RNF146 and subsequent ubiquitination. Mediates poly-ADP-ribosylation of TERF1, thereby contributing to the regulation of telomere length. Stimulates 26S proteasome activity. Poly-ADP-ribosyltransferase involved in various processes such as Wnt signaling pathway, telomere length and vesicle trafficking. Related diseases Intellectual developmental disorder with macrocephaly, seizures, and speech delay (IDDMSSD) [MIM:618158]: An autosomal dominant neurodevelopmental disorder characterized by impaired intellectual development, poor speech, postnatal macrocephaly, and seizures. {ECO:0000269|PubMed:30290153}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O15084; Q7Z6K5-1; O15169; Q9NWV8; P11274; Q13698; Q9NRI5; Q6V0I7; Q9NWT6; P14652; Q9UIQ6; Q14980; Q9BZL4; Q92698; P78314; O43815; P54274; Q9C0C2; Q9UHP3; Q06649 EC number EC 2.4.2.30 Uniprot keywords 3D-structure; ADP-ribosylation; ANK repeat; Chromosome; Cytoplasm; Glycosyltransferase; Golgi apparatus; Hydroxylation; Membrane; Metal-binding; NAD; Nucleotidyltransferase; Nucleus; Proteomics identification; Reference proteome; Repeat; Telomere; Transferase; Ubl conjugation; Wnt signaling pathway; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 23695.5 Length 208 Aromaticity 0.11 Instability index 47.61 Isoelectric point 8.28 Charge (pH=7) 2.88 2D Binding mode Binding energy (Kcal/mol) -7.86  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GTILIDLSPDDKEFQSVEEEMQSTVREHRDGGHAGGIFNRYNILKIQKVCNKKLWERYTHRRKEVSEENHNHANERMLFHGSPFVNAIIHKGFDERHAYIGGMFGAGIYFAENSSKSNQYVYGIGGGTGCPVHKDRSCYICHRQLLFCRVTLGKSFLQFSAMAHSPPGHHSVTGRPSVNGLALAEYVIYRGEQAYPEYLITYQIMRPE Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Beta-arrestin-1 (ARRB1) | 6TKO | 5.76 | |
Target general information Gen name ARRB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Non-visual arrestin-2; Betaarrestin1; Arrestin beta1; Arrestin beta-1; ARR1 Protein family Arrestin family Biochemical class Arrestin protein Function During homologous desensitization, beta-arrestins bind to the GPRK-phosphorylated receptor and sterically preclude its coupling to the cognate G-protein; the binding appears to require additional receptor determinants exposed only in the active receptor conformation. The beta-arrestins target many receptors for internalization by acting as endocytic adapters (CLASPs, clathrin-associated sorting proteins) and recruiting the GPRCs to the adapter protein 2 complex 2 (AP-2) in clathrin-coated pits (CCPs). However, the extent of beta-arrestin involvement appears to vary significantly depending on the receptor, agonist and cell type. Internalized arrestin-receptor complexes traffic to intracellular endosomes, where they remain uncoupled from G-proteins. Two different modes of arrestin-mediated internalization occur. Class A receptors, like ADRB2, OPRM1, ENDRA, D1AR and ADRA1B dissociate from beta-arrestin at or near the plasma membrane and undergo rapid recycling. Class B receptors, like AVPR2, AGTR1, NTSR1, TRHR and TACR1 internalize as a complex with arrestin and traffic with it to endosomal vesicles, presumably as desensitized receptors, for extended periods of time. Receptor resensitization then requires that receptor-bound arrestin is removed so that the receptor can be dephosphorylated and returned to the plasma membrane. Involved in internalization of P2RY4 and UTP-stimulated internalization of P2RY2. Involved in phosphorylation-dependent internalization of OPRD1 ands subsequent recycling. Involved in the degradation of cAMP by recruiting cAMP phosphodiesterases to ligand-activated receptors. Beta-arrestins function as multivalent adapter proteins that can switch the GPCR from a G-protein signaling mode that transmits short-lived signals from the plasma membrane via small molecule second messengers and ion channels to a beta-arrestin signaling mode that transmits a distinct set of signals that are initiated as the receptor internalizes and transits the intracellular compartment. Acts as signaling scaffold for MAPK pathways such as MAPK1/3 (ERK1/2). ERK1/2 activated by the beta-arrestin scaffold is largely excluded from the nucleus and confined to cytoplasmic locations such as endocytic vesicles, also called beta-arrestin signalosomes. Recruits c-Src/SRC to ADRB2 resulting in ERK activation. GPCRs for which the beta-arrestin-mediated signaling relies on both ARRB1 and ARRB2 (codependent regulation) include ADRB2, F2RL1 and PTH1R. For some GPCRs the beta-arrestin-mediated signaling relies on either ARRB1 or ARRB2 and is inhibited by the other respective beta-arrestin form (reciprocal regulation). Inhibits ERK1/2 signaling in AGTR1- and AVPR2-mediated activation (reciprocal regulation). Is required for SP-stimulated endocytosis of NK1R and recruits c-Src/SRC to internalized NK1R resulting in ERK1/2 activation, which is required for the antiapoptotic effects of SP. Is involved in proteinase-activated F2RL1-mediated ERK activity. Acts as signaling scaffold for the AKT1 pathway. Is involved in alpha-thrombin-stimulated AKT1 signaling. Is involved in IGF1-stimulated AKT1 signaling leading to increased protection from apoptosis. Involved in activation of the p38 MAPK signaling pathway and in actin bundle formation. Involved in F2RL1-mediated cytoskeletal rearrangement and chemotaxis. Involved in AGTR1-mediated stress fiber formation by acting together with GNAQ to activate RHOA. Appears to function as signaling scaffold involved in regulation of MIP-1-beta-stimulated CCR5-dependent chemotaxis. Involved in attenuation of NF-kappa-B-dependent transcription in response to GPCR or cytokine stimulation by interacting with and stabilizing CHUK. May serve as nuclear messenger for GPCRs. Involved in OPRD1-stimulated transcriptional regulation by translocating to CDKN1B and FOS promoter regions and recruiting EP300 resulting in acetylation of histone H4. Involved in regulation of LEF1 transcriptional activity via interaction with DVL1 and/or DVL2 Also involved in regulation of receptors other than GPCRs. Involved in Toll-like receptor and IL-1 receptor signaling through the interaction with TRAF6 which prevents TRAF6 autoubiquitination and oligomerization required for activation of NF-kappa-B and JUN. Binds phosphoinositides. Binds inositolhexakisphosphate (InsP6). Involved in IL8-mediated granule release in neutrophils. Required for atypical chemokine receptor ACKR2-induced RAC1-LIMK1-PAK1-dependent phosphorylation of cofilin (CFL1) and for the up-regulation of ACKR2 from endosomal compartment to cell membrane, increasing its efficiency in chemokine uptake and degradation. Involved in the internalization of the atypical chemokine receptor ACKR3. Negatively regulates the NOTCH signaling pathway by mediating the ubiquitination and degradation of NOTCH1 by ITCH. Participates to the recruitment of the ubiquitin-protein ligase to the receptor. Functions in regulating agonist-mediated G-protein coupled receptor (GPCR) signaling by mediating both receptor desensitization and resensitization processes. Related diseases Intellectual developmental disorder with dysmorphic facies and ptosis (IDDDFP) [MIM:617333]: An autosomal dominant neurodevelopmental disorder characterized by delayed psychomotor development, intellectual disability, delayed language, and facial dysmorphisms, most notably ptosis. Additional features may include poor growth, hypotonia, and seizures. {ECO:0000269|PubMed:27939639, ECO:0000269|PubMed:27939640}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P63010-2; O15169; P0DP25; P20963; P25101; P50148; Q5JWF2; Q14749; P06396; Q16665; P11142; Q99683; P53779; P45984; Q00987; P19338; Q14978; P14618; P14859-6; P35813; O75688; Q13523; P06702; P12931; Q15208; Q13428; P04637; P27348; P25490; O43298; O95218; Q7DB77 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Cell projection; Coated pit; Cytoplasm; Cytoplasmic vesicle; Membrane; Nucleus; Phosphoprotein; Protein transport; Proteomics identification; Reference proteome; Signal transduction inhibitor; Transcription; Transcription regulation; Transport; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32455.6 Length 293 Aromaticity 0.12 Instability index 28.99 Isoelectric point 9.12 Charge (pH=7) 9.86 2D Binding mode Binding energy (Kcal/mol) -7.86  Molscript Map 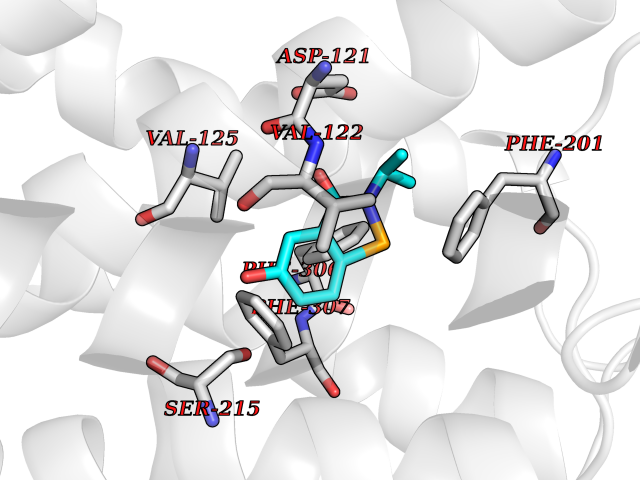 Pymol Map 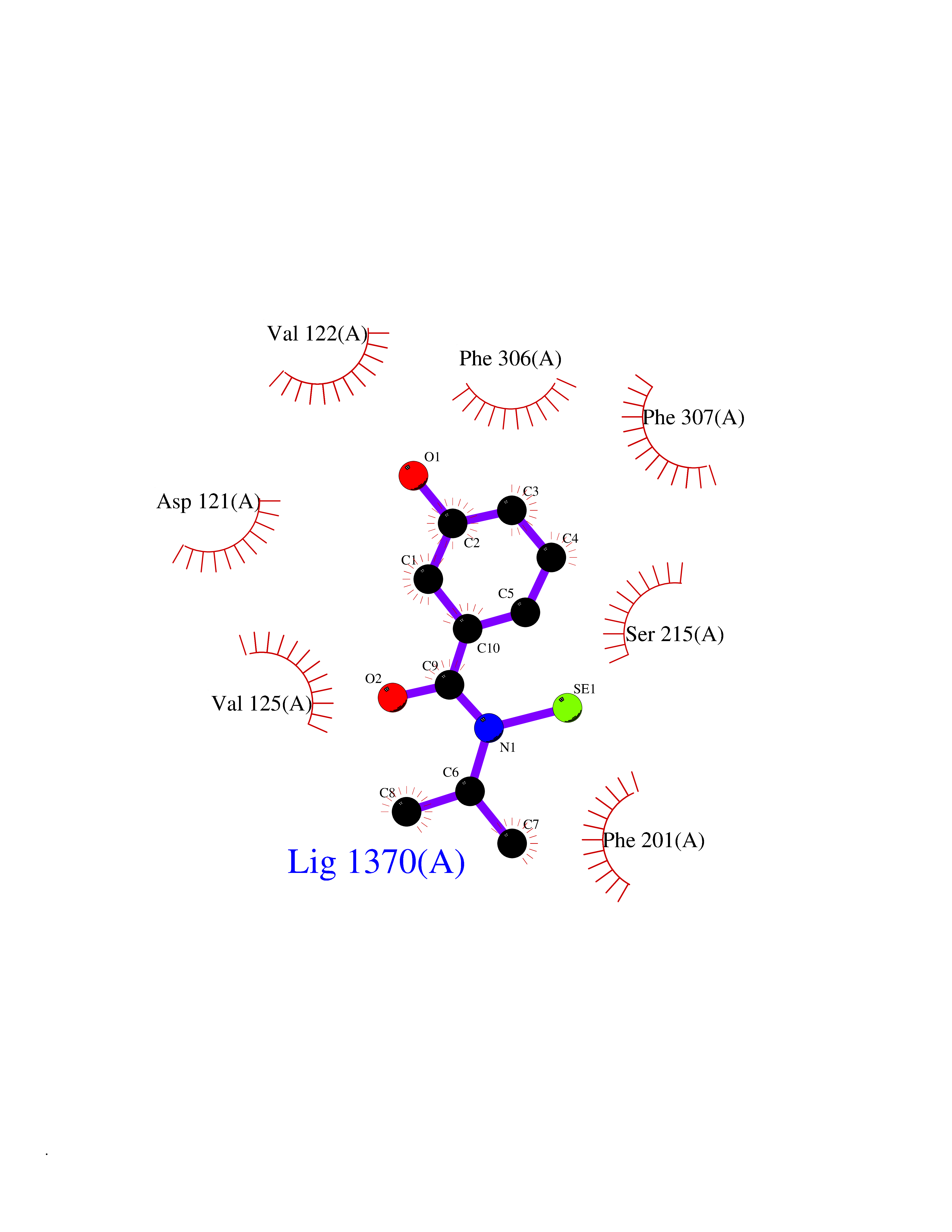 Ligplot Map 3D Binding mode Sequence AGCSLLMALVVLLIVAGNVLVIAAIGRTQRLQTLTNLFITSLACADLVVGLLVVPFGATLVCRGTWLWGSFLCELWTSLDVLCVTASIWTLCVIAIDRYLAITSPFRYQSLMTRARAKVIICTVWAISALVSFLPIMMHWWRDEDPQALKCYQDPGCCDFVTNRAYAIASSIISFYIPLLIMIFVYLRVYREAKEQIRKIDVMAMREHKALKTLGIIMGVFTLCWLPFFLVNIVNVFNRDLVPKWLFVAFNWLGYANSAMNPIIYCRSPDFRKAFKRLLAEXAXXAXXXLAKD Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Schistosoma Purine nucleoside phosphorylase (sch PNP) | 3F8W | 5.76 | |
Target general information Gen name sch PNP Organism Schistosoma mansoni (Blood fluke) Uniprot ID TTD ID Synonyms sch Purine nucleoside phosphorylase; sch Inosine-guanosine phosphorylase Protein family PNP/MTAP phosphorylase family Biochemical class NA Function The purine nucleoside phosphorylases catalyze the phosphorolytic breakdown of the N-glycosidic bond in the beta-(deoxy)ribonucleoside molecules, with the formation of the corresponding free purine bases and pentose-1-phosphate. Related diseases Hemolytic anemia, non-spherocytic, due to glucose phosphate isomerase deficiency (HA-GPID) [MIM:613470]: A form of anemia in which there is no abnormal hemoglobin or spherocytosis. It is caused by glucose phosphate isomerase deficiency. {ECO:0000269|PubMed:28803808, ECO:0000269|PubMed:7989588, ECO:0000269|PubMed:8499925, ECO:0000269|PubMed:8822952, ECO:0000269|PubMed:8822954, ECO:0000269|PubMed:9446754, ECO:0000269|PubMed:9856489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 2.4.2.1 Uniprot keywords 3D-structure; Glycosyltransferase; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 61140.6 Length 564 Aromaticity 0.06 Instability index 29.8 Isoelectric point 7.6 Charge (pH=7) 1.82 2D Binding mode Binding energy (Kcal/mol) -7.86 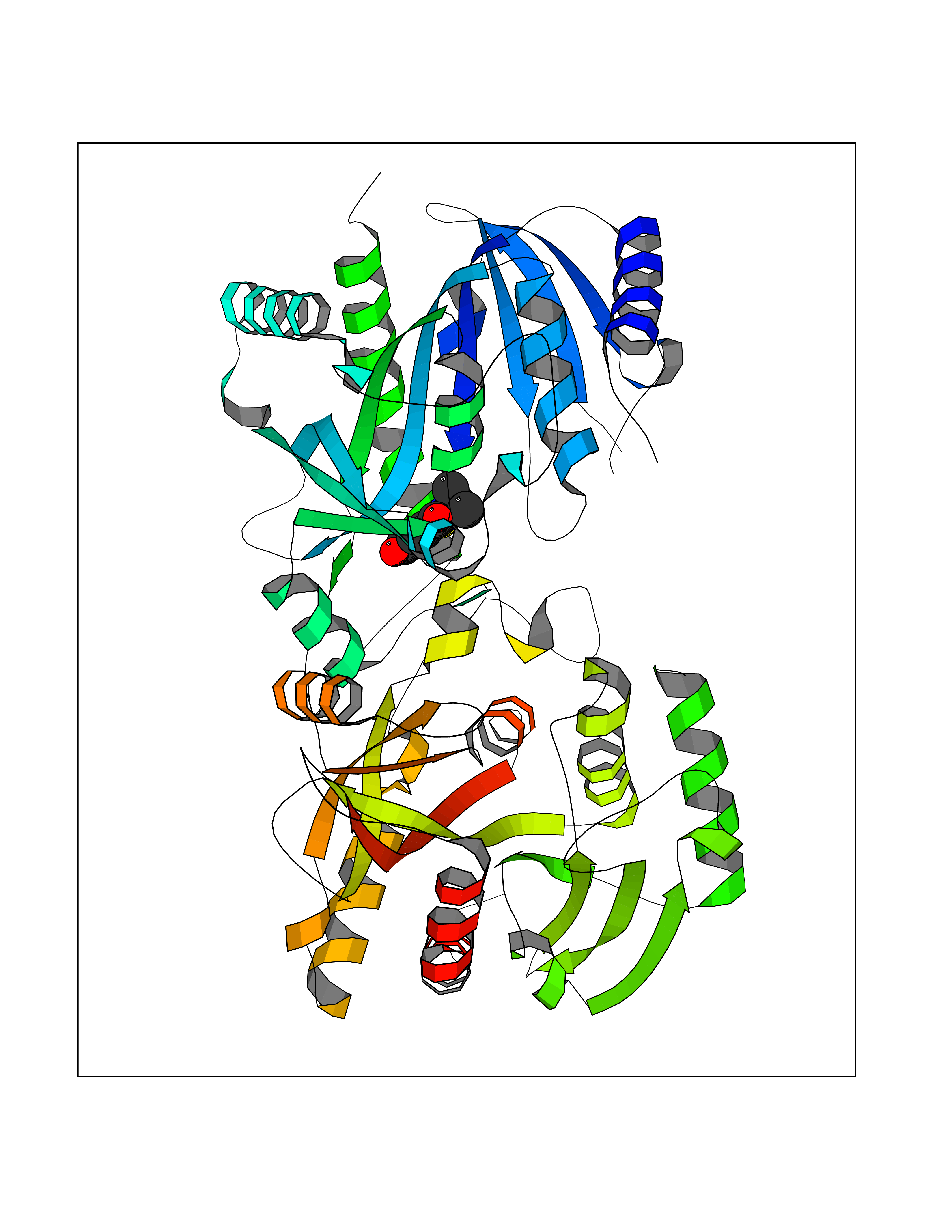 Molscript Map 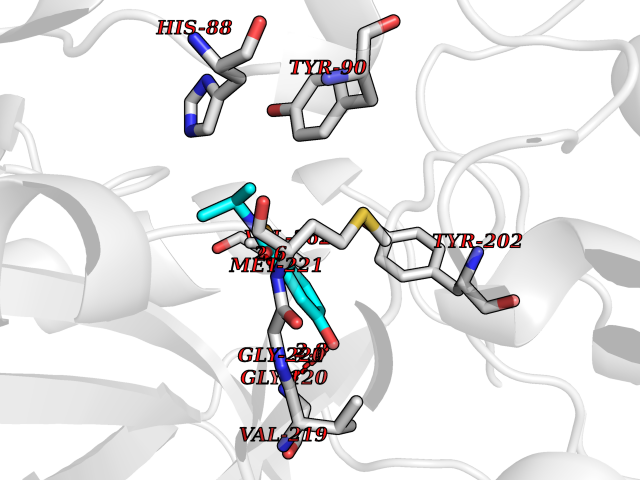 Pymol Map 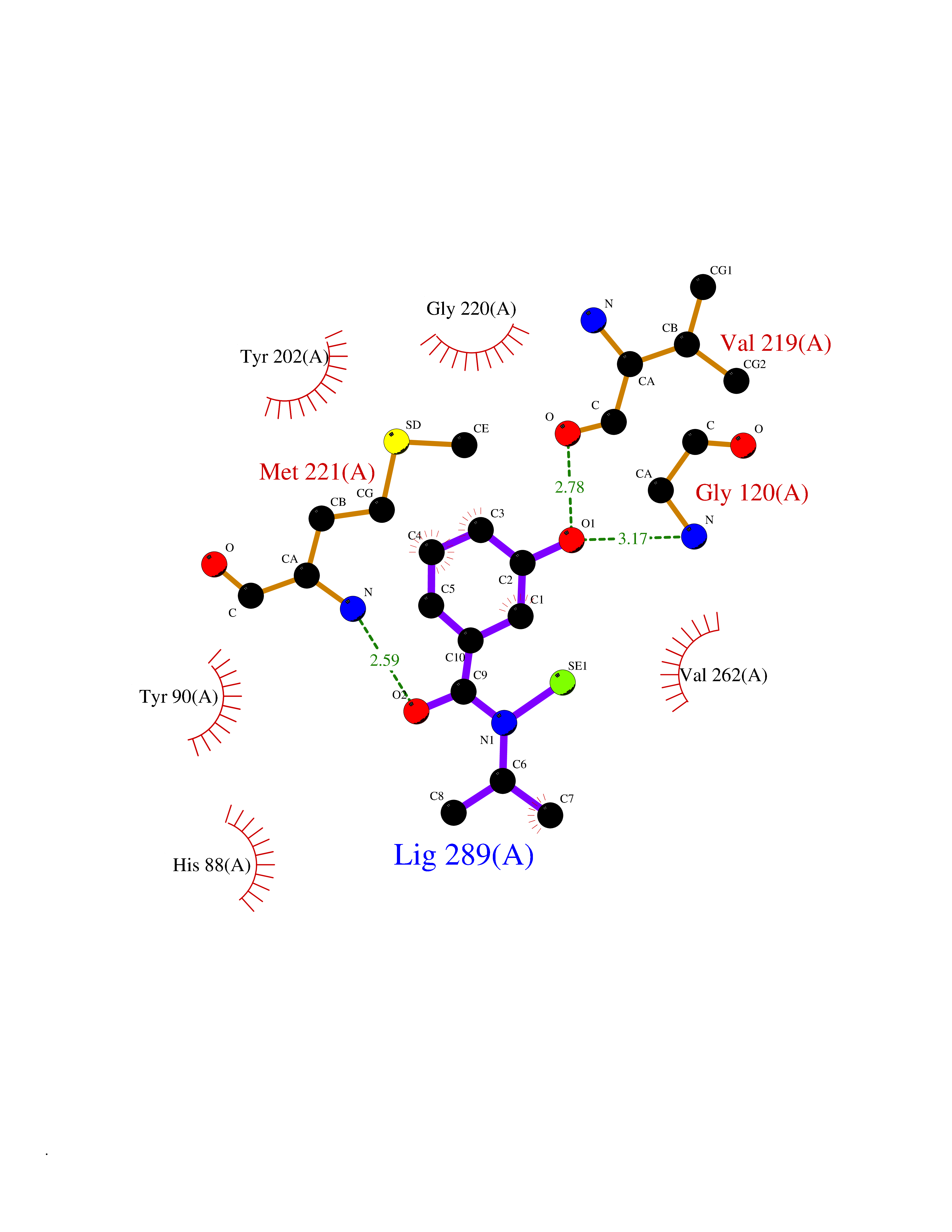 Ligplot Map 3D Binding mode Sequence SVTANIENVKKVAHHIQKLTSIVPEIGIICGSGLGKLADGVKDKITIPYTKIPNFPQTSHSGNLIFGTLSGRKVVVMQGRFHMYEGYSNDTVALPIRVMKLLGVKILMVSNAAGGLNRSLKLGDFVILKDHIYLPGLGLNNILVGPNQEAFGTRFPALSNAYDRDLRKLAVQVAEENGFGNLVHQGVYVMNGGPCYETPAECTMLLNMGCDVVGMSTIPEVVIARHCGIQVFAVSLVTNISVLDVESDLKPNHEEVLATGAQRAELMQSWFEKIIEKLPKSVTANIENVKKVAHHIQKLTSIVPEIGIICGSGLGKLADGVKDKITIPYTKIPNFPQTSVVGHSGNLIFGTLSGRKVVVMQGRFHMYEGYSNDTVALPIRVMKLLGVKILMVSNAAGGLNRSLKLGDFVILKDHIYLPGLGLNNILVGPNQEAFGTRFPALSNAYDRDLRKLAVQVAEENGFGNLVHQGVYVMNGGPCYETPAECTMLLNMGCDVVGMSTIPEVVIARHCGIQVFAVSLVTNISVLDVESDLKPNHEEVLATGAQRAELMQSWFEKIIEKLPKD Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Hyperpolarization cyclic nucleotide-gated channel 2 (HCN2) | 3U10 | 5.76 | |
Target general information Gen name HCN2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channel 2; Brain cyclic nucleotide-gated channel 2; BCNG2; BCNG-2 Protein family Potassium channel HCN family Biochemical class Voltage-gated ion channel Function Contributes to the native pacemaker currents in heart (If) and in neurons (Ih). Can also transport ammonium in the distal nephron. Produces a large instantaneous current. Modulated by intracellular chloride ions and pH; acidic pH shifts the activation to more negative voltages. Hyperpolarization-activated ion channel exhibiting weak selectivity for potassium over sodium ions. Related diseases Epilepsy, idiopathic generalized 17 (EIG17) [MIM:602477]: A form of idiopathic generalized epilepsy, a disorder characterized by recurring generalized seizures in the absence of detectable brain lesions and/or metabolic abnormalities. Generalized seizures arise diffusely and simultaneously from both hemispheres of the brain. Seizure types include juvenile myoclonic seizures, absence seizures, and generalized tonic-clonic seizures. Both autosomal dominant and autosomal recessive EIG17 inheritance have been reported. {ECO:0000269|PubMed:22131395, ECO:0000269|PubMed:29064616}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Febrile seizures, familial, 2 (FEB2) [MIM:602477]: Seizures associated with febrile episodes in childhood without any evidence of intracranial infection or defined pathologic or traumatic cause. It is a common condition, affecting 2-5% of children aged 3 months to 5 years. The majority are simple febrile seizures (generally defined as generalized onset, single seizures with a duration of less than 30 minutes). Complex febrile seizures are characterized by focal onset, duration greater than 30 minutes, and/or more than one seizure in a 24 hour period. The likelihood of developing epilepsy following simple febrile seizures is low. Complex febrile seizures are associated with a moderately increased incidence of epilepsy. FEB2 transmission pattern is consistent with autosomal dominant inheritance. {ECO:0000269|PubMed:24324597}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02527; DB02315; DB09083 Interacts with Q9UL51; Q4ACU6-1 EC number NA Uniprot keywords 3D-structure; Ammonia transport; cAMP; cAMP-binding; Cell membrane; Disease variant; Epilepsy; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Lipoprotein; Membrane; Methylation; Nucleotide-binding; Palmitate; Phosphoprotein; Potassium; Potassium channel; Potassium transport; Proteomics identification; Reference proteome; Sodium; Sodium channel; Sodium transport; Transmembrane; Transmembrane helix; Transport; Voltage-gated channel Protein physicochemical properties Chain ID A Molecular weight (Da) 23672.9 Length 202 Aromaticity 0.12 Instability index 38.05 Isoelectric point 8.85 Charge (pH=7) 4.11 2D Binding mode Binding energy (Kcal/mol) -7.85 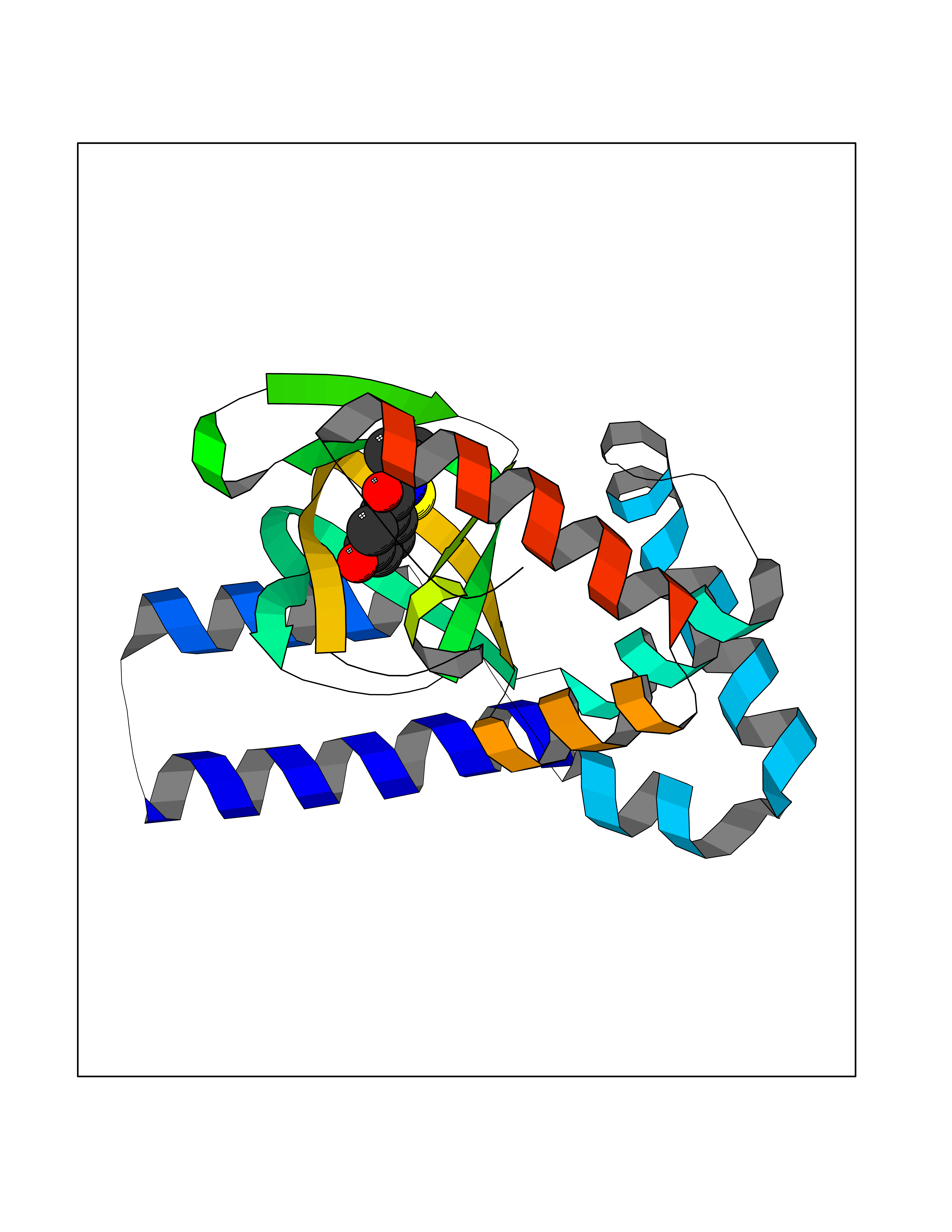 Molscript Map 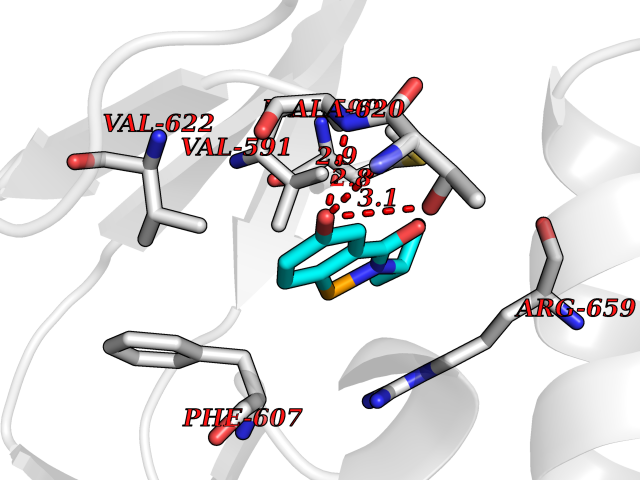 Pymol Map 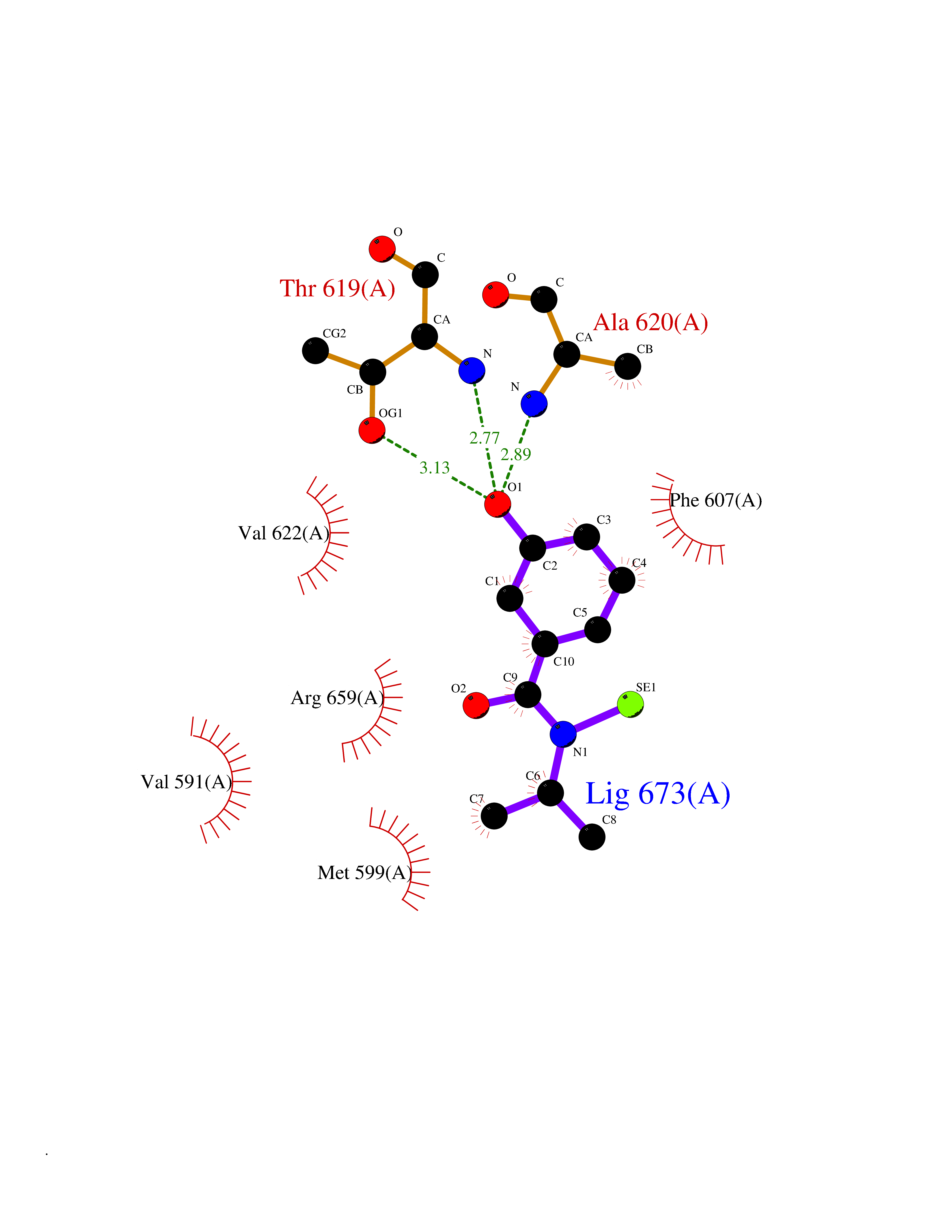 Ligplot Map 3D Binding mode Sequence DSSRRQYQEKYKQVEQYMSFHKLPADFRQKIHDYYEHRYQGKMFDEDSILGELNGPLREEIVNFNCRKLVASMPLFANADPNFVTAMLTKLKFEVFQPGDYIIREGTIGKKMYFIQHGVVSVLTKGNKEMKLSDGSYFGEICLLTRGRRTASVRADTYCRLYSLSVDNFNEVLEEYPMMRRAFETVAIDRLDRIGKKNSILL Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Caspase-7 (CASP7) | 1SHJ | 5.76 | |
Target general information Gen name CASP7 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MCH3; ICE-like apoptotic protease 3; ICE-LAP3; CMH-1; CASP-7; Apoptotic protease Mch-3 Protein family Peptidase C14A family Biochemical class Peptidase Function Cleaves and activates sterol regulatory element binding proteins (SREBPs). Proteolytically cleaves poly(ADP-ribose) polymerase (PARP) at a '216-Asp-|-Gly-217' bond. Overexpression promotes programmed cell death. Involved in the activation cascade of caspases responsible for apoptosis execution. Related diseases Pregnancy loss, recurrent, 3 (RPRGL3) [MIM:614391]: A common complication of pregnancy, resulting in spontaneous abortion before the fetus has reached viability. The term includes all miscarriages from the time of conception until 24 weeks of gestation. Recurrent pregnancy loss is defined as 3 or more consecutive spontaneous abortions. {ECO:0000269|PubMed:17339269}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05408; DB03384; DB06255 Interacts with Q13490; P83105; P42858; Q8N4N3-2; P43364; Q16236; Q9GZT8; Q13177; P27986-2; P21673; Q86WV1-2; P17405; P98170 EC number EC 3.4.22.60 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; Apoptosis; Cytoplasm; Hydrolase; Nucleus; Phosphoprotein; Protease; Proteomics identification; Reference proteome; RNA-binding; Secreted; Thiol protease; Ubl conjugation; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 47441.5 Length 417 Aromaticity 0.11 Instability index 20.98 Isoelectric point 8.38 Charge (pH=7) 6.12 2D Binding mode Binding energy (Kcal/mol) -7.86 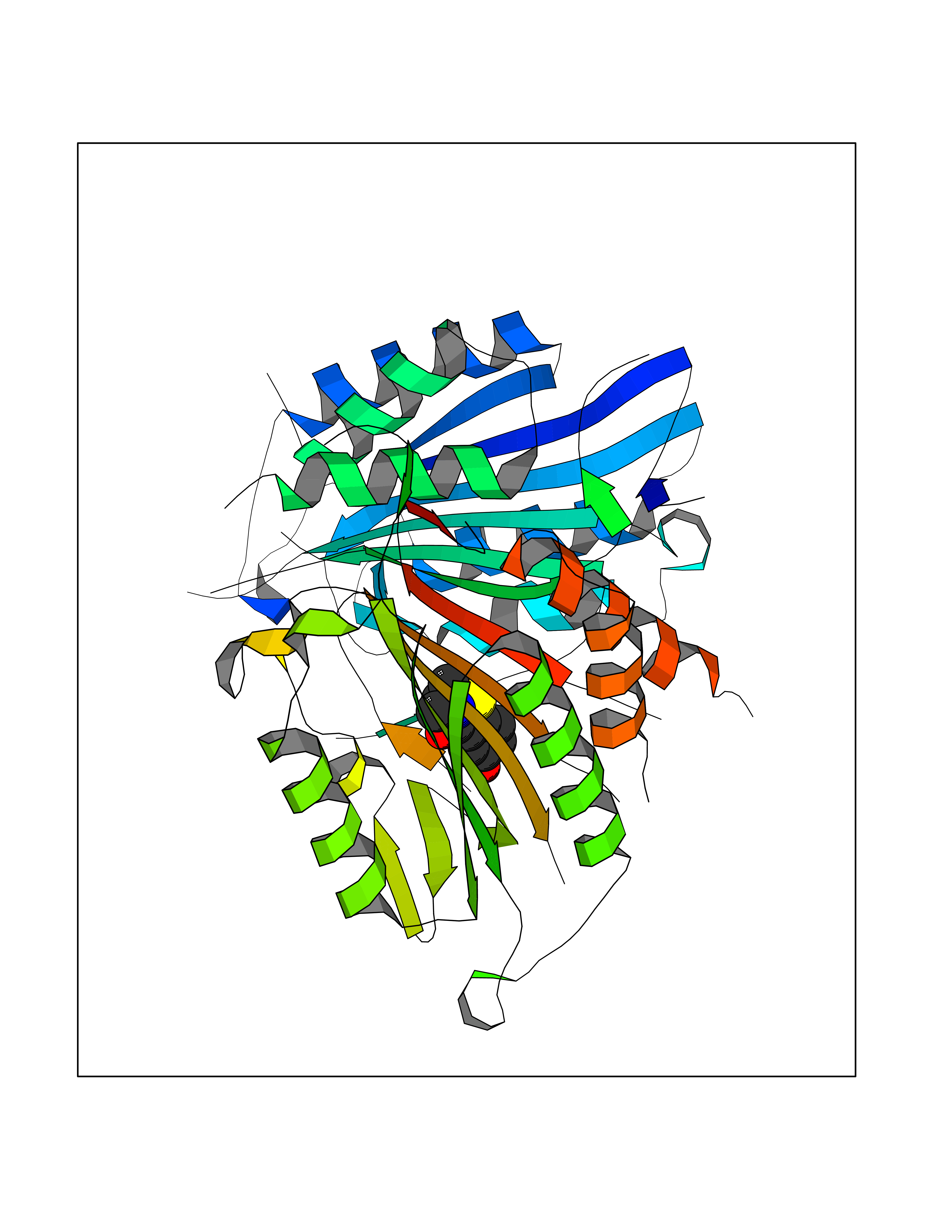 Molscript Map 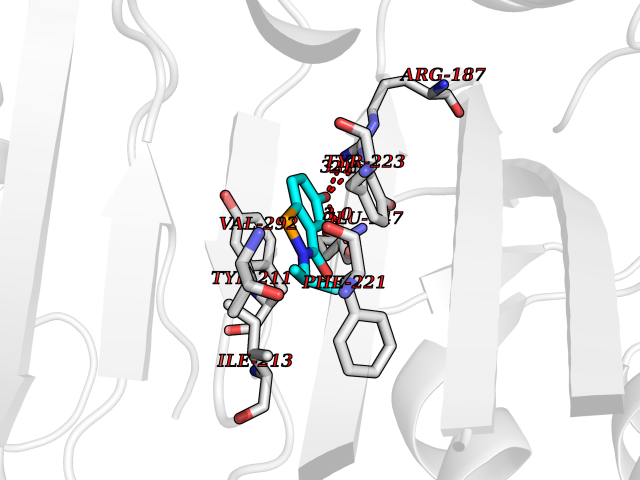 Pymol Map 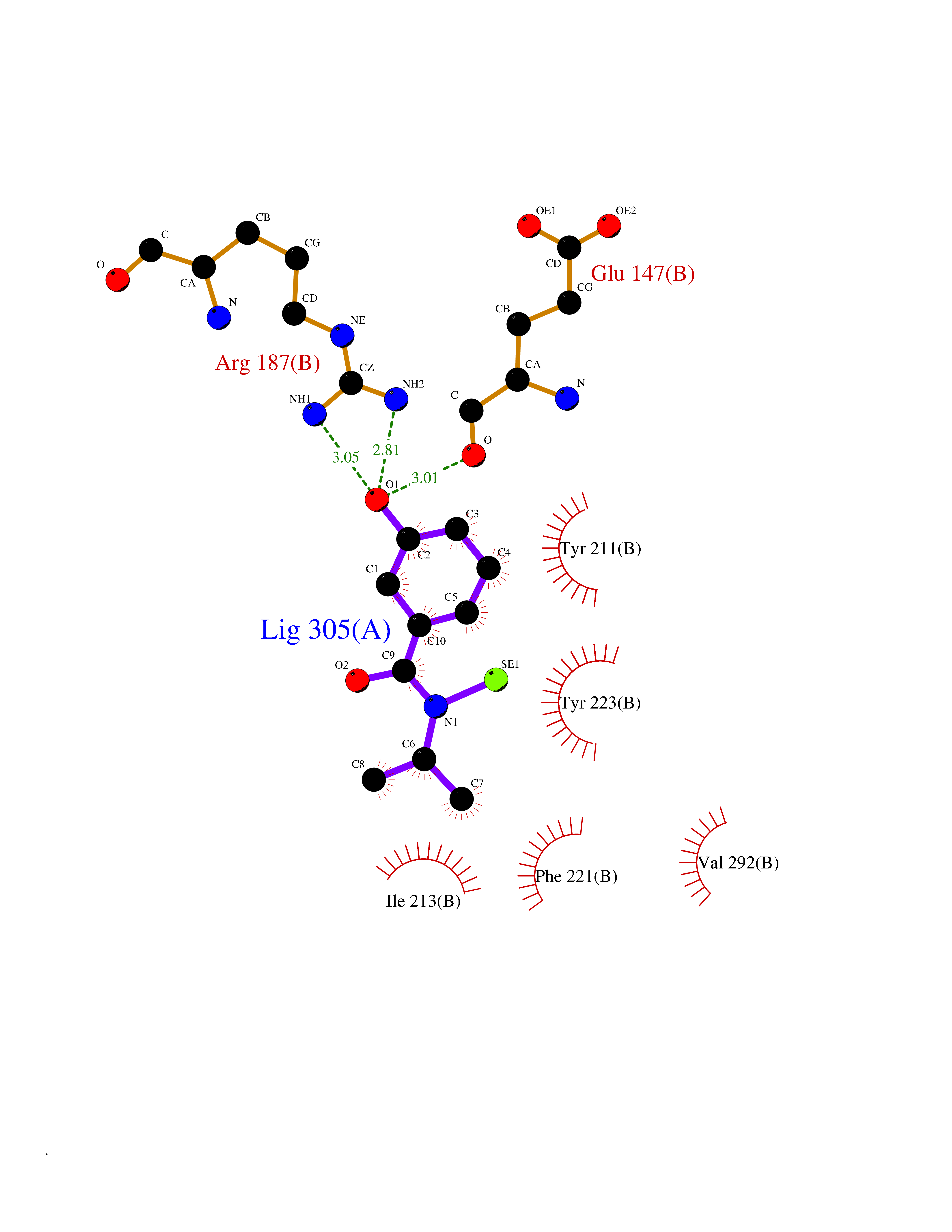 Ligplot Map 3D Binding mode Sequence TYQYNMNFEKLGKCIIINNKNFDKVTGMGVRNGTDKDAEALFKCFRSLGFDVIVYNDCSCAKMQDLLKKASEEDHTNAACFACILLSHGEENVIYGKDGVTPIKDLTAHFRGARCKTLLEKPKLFFIQACRGTEPRYKIPVEADFLFAYSTVRGSWFVQALCSILEEHGKDLEIMQILTRVNDRVARHFKKQIPCVVSMLTKELYFSQVPTYQYNMNFEKLGKCIIINNKNFDKVTGMGVRNGTDKDAEALFKCFRSLGFDVIVYNDCSCAKMQDLLKKASEEDHTNAACFACILLSHGEENVIYGKDGVTPIKDLTAHFRGARCKTLLEKPKLFFIQACRGPRYKIPVEADFLFAYSTVPGSWFVQALCSILEEHGKDLEIMQILTRVNDRVARHFESKQIPCVVSMLTKELYFSQ Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Bromodomain-containing protein 2 (BRD2) | 4A9J | 5.76 | |
Target general information Gen name BRD2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Really interesting new gene 3 protein; RING3; O27.1.1; KIAA9001 Protein family BET family Biochemical class Bromodomain Function Binds hyperacetylated chromatin and plays a role in the regulation of transcription, probably by chromatin remodeling. Regulates transcription of the CCND1 gene. Plays a role in nucleosome assembly. May play a role in spermatogenesis or folliculogenesis. Related diseases Lethal congenital contracture syndrome 2 (LCCS2) [MIM:607598]: A form of lethal congenital contracture syndrome, an autosomal recessive disorder characterized by degeneration of anterior horn neurons, extreme skeletal muscle atrophy, and congenital non-progressive joint contractures (arthrogryposis). The contractures can involve the upper or lower limbs and/or the vertebral column, leading to various degrees of flexion or extension limitations evident at birth. LCCS2 patients manifest craniofacial/ocular findings, lack of hydrops, multiple pterygia, and fractures, as well as a normal duration of pregnancy and a unique feature of a markedly distended urinary bladder (neurogenic bladder defect). The phenotype suggests a spinal cord neuropathic etiology. {ECO:0000269|PubMed:17701904}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythroleukemia, familial (FERLK) [MIM:133180]: An autosomal dominant myeloproliferative disorder characterized by neoplastic proliferation of erythroblastic and myeloblastic elements with atypical erythroblasts and myeloblasts in the peripheral blood. Disease penetrance is incomplete. {ECO:0000269|PubMed:27416908}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Visceral neuropathy, familial, 1, autosomal recessive (VSCN1) [MIM:243180]: An autosomal recessive disorder characterized by intestinal dysmotility due to aganglionosis (Hirschsprung disease), hypoganglionosis, and/or chronic intestinal pseudoobstruction. Additional variable features are progressive peripheral neuropathy, arthrogryposis, hypoplasia or aplasia of the olfactory bulb and of the external auditory canals, microtia or anotia, and facial dysmorphism. Some patients present structural cardiac anomalies and arthrogryposis with multiple pterygia. {ECO:0000269|PubMed:33497358}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P62805; Q13761 EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Bromodomain; Chromatin regulator; Chromosome; Host-virus interaction; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation Protein physicochemical properties Chain ID A Molecular weight (Da) 13375.4 Length 112 Aromaticity 0.12 Instability index 51.4 Isoelectric point 7.92 Charge (pH=7) 0.99 2D Binding mode Binding energy (Kcal/mol) -7.85 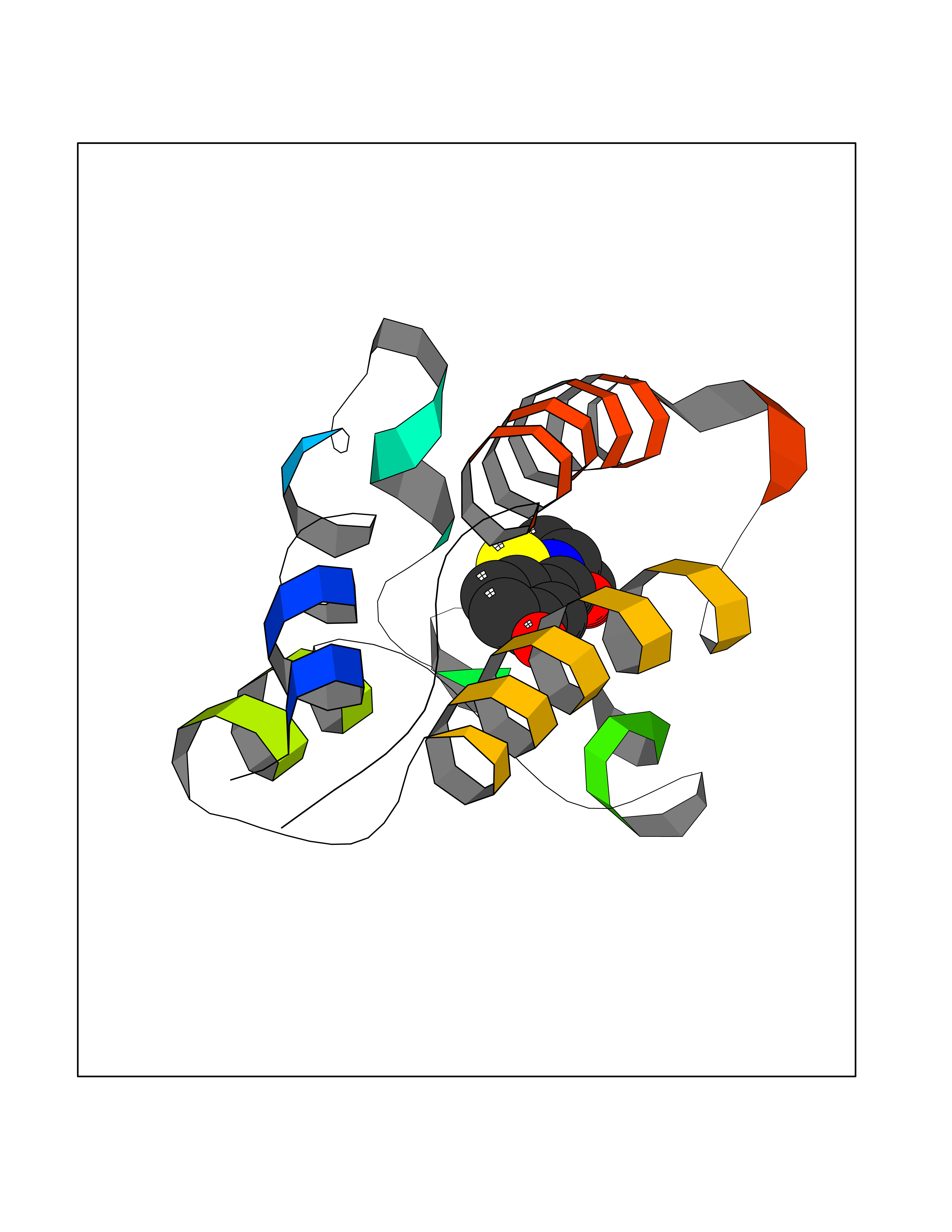 Molscript Map 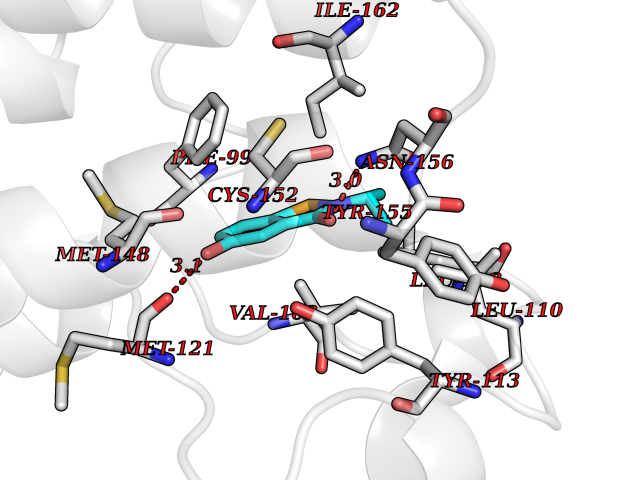 Pymol Map 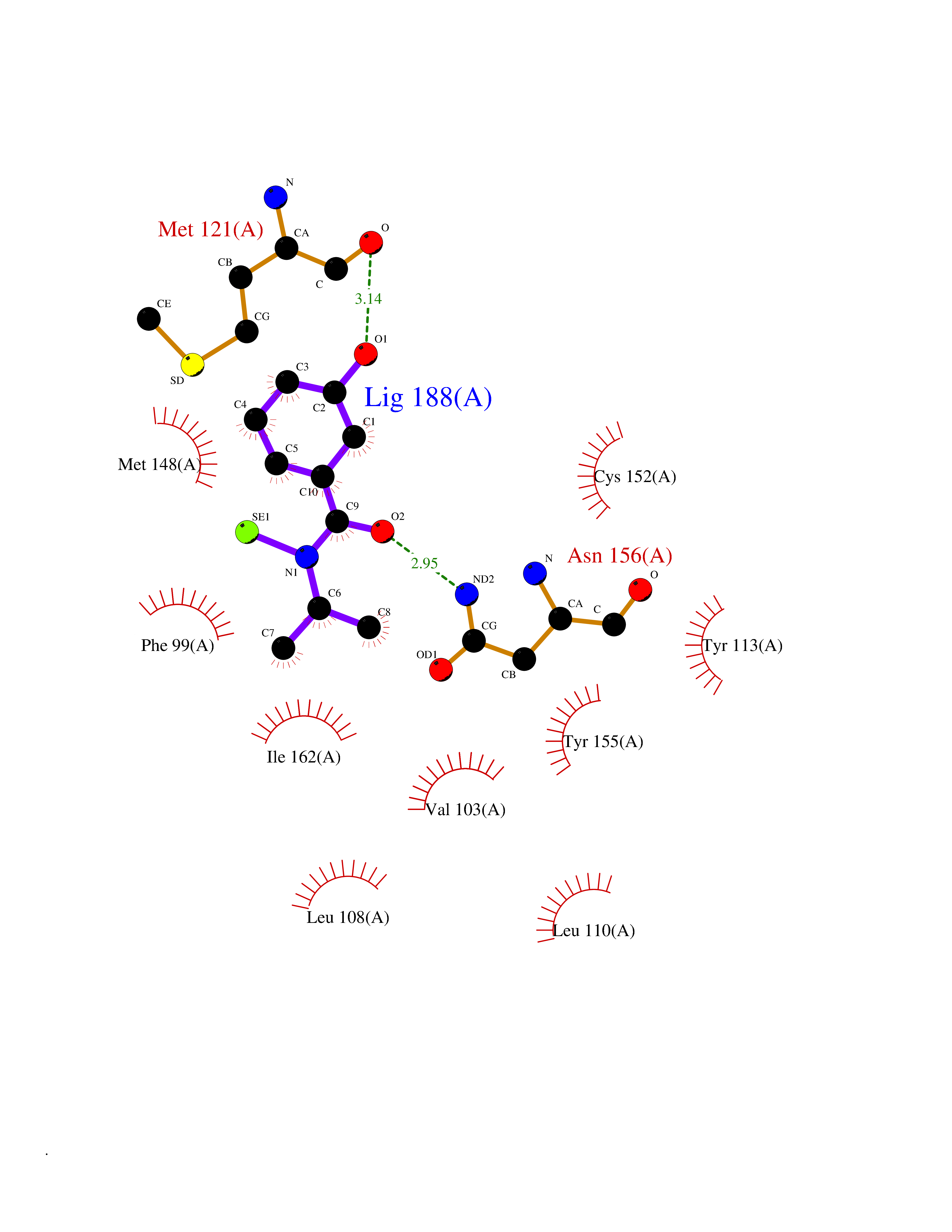 Ligplot Map 3D Binding mode Sequence VTNQLQYLHKVVMKALWKHQFAWPFRQPVDAVKLGLPDYHKIIKQPMDMGTIKRRLENNYYWAASECMQDFNTMFTNCYIYNKPTDDIVLMAQTLEKIFLQKVASMPQEEQE Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | Glucose-dependent insulinotropic receptor (GPR119) | 7XZ6 | 5.76 | |
Target general information Gen name GPR119 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GPR119; G-protein coupled receptor 119 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for the endogenous fatty-acid ethanolamide oleoylethanolamide (OEA) and lysophosphatidylcholine (LPC). Functions as a glucose-dependent insulinotropic receptor. The activity of this receptor is mediated by G proteins which activate adenylate cyclase. Seems to act through a G(s) mediated pathway. Related diseases Developmental and epileptic encephalopathy 24 (DEE24) [MIM:615871]: A disease characterized by early-onset seizures, intellectual disability of varying degrees, and behavioral disturbances or autistic features in most individuals. {ECO:0000269|PubMed:24747641, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Generalized epilepsy with febrile seizures plus 10 (GEFSP10) [MIM:618482]: An autosomal dominant neurologic disorder with incomplete penetrance, characterized by variable types of seizures including absence, tonic-clonic, febrile, focal, and eyelid myoclonia. Some patients have normal neurologic development. Others have mild-to-moderate intellectual disability or autism spectrum disorder. {ECO:0000269|PubMed:29936235, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05166 Interacts with Q12797-6 EC number NA Uniprot keywords 3D-structure; Cell membrane; G-protein coupled receptor; Lipid-binding; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32134.1 Length 292 Aromaticity 0.12 Instability index 34.96 Isoelectric point 9.12 Charge (pH=7) 8.03 2D Binding mode Binding energy (Kcal/mol) -7.86  Molscript Map 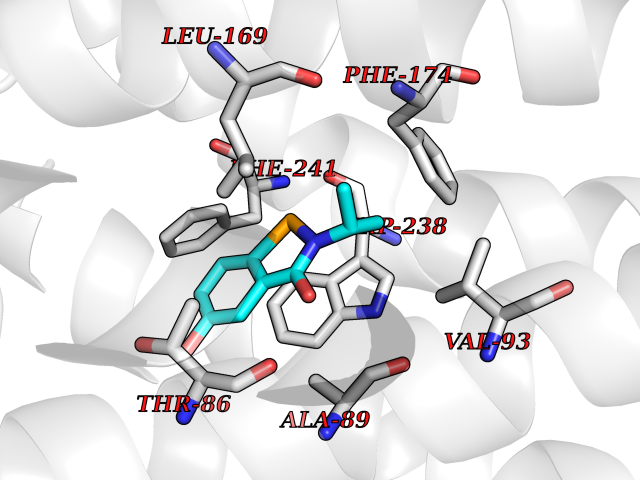 Pymol Map 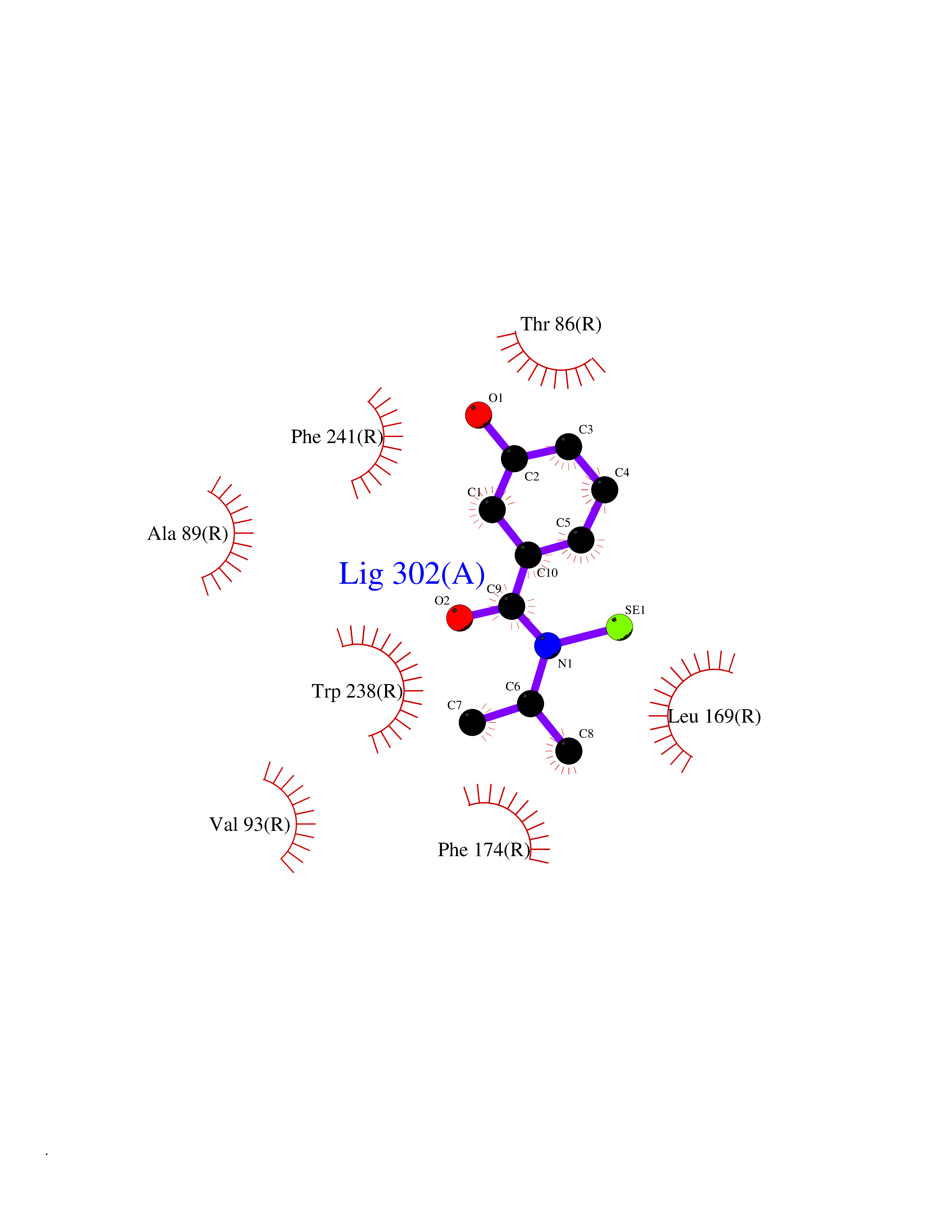 Ligplot Map 3D Binding mode Sequence MESSFSFGVILAVLASLIIATNTLVAVAVLLLIHKNDGVSLCFTLNLAVADTLIGVAISGLLTDQLSSPSRPTQKTLCSLRMAFVTSSAAASVLTVMLITFDRYLAIKQPFRYLKIMSGFVAGACIAGLWLVSYLIGFLPLGIPMFQQTAYKGQCSFFAVFHPHFVLTLSCVGFFPAMLLFVFFYCDMLKIASMHSQQIRKMEHAGAMAGSDFKALRTVSVLIGSFALSWTPFLITGIVQVACQECHLYLVLERYLWLLGVGNSLLNPLIYAYWQKEVRLQLYHMALGVKKV Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Mineralocorticoid receptor (MR) | 4PF3 | 5.75 | |
Target general information Gen name NR3C2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nuclear receptor subfamily 3 group C member 2; Mineralocorticoid receptor; MLR; MCR; Inner ear mineralocorticoid receptor; Delta Protein family Nuclear hormone receptor family, NR3 subfamily Biochemical class Nuclear hormone receptor Function Binds to mineralocorticoid response elements (MRE) and transactivates target genes. The effect of MC is to increase ion and water transport and thus raise extracellular fluid volume and blood pressure and lower potassium levels. Receptor for both mineralocorticoids (MC) such as aldosterone and glucocorticoids (GC) such as corticosterone or cortisol. Related diseases Pseudohypoaldosteronism 1, autosomal dominant (PHA1A) [MIM:177735]: A salt wasting disease resulting from target organ unresponsiveness to mineralocorticoids. PHA1A is a mild form characterized by target organ defects confined to kidney. Patients may present with neonatal renal salt wasting with hyperkalaemic acidosis despite high aldosterone levels. These patients improve with age and usually become asymptomatic without treatment. {ECO:0000269|PubMed:11134129, ECO:0000269|PubMed:12788847, ECO:0000269|PubMed:16954160, ECO:0000269|PubMed:16972228}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Early-onset hypertension with severe exacerbation in pregnancy (EOHSEP) [MIM:605115]: Inheritance is autosomal dominant. The disease is characterized by the onset of severe hypertension before the age of 20, and by suppression of aldosterone secretion. {ECO:0000269|PubMed:10884226, ECO:0000269|PubMed:15908963, ECO:0000269|PubMed:15967794}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04630; DB01013; DB04652; DB06780; DB01134; DB01395; DB00700; DB01023; DB16165; DB00687; DB13867; DB08906; DB00588; DB02998; DB00393; DB00396; DB00421; DB02901; DB13951; DB00624; DB13943; DB13944; DB15114 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Endoplasmic reticulum; Lipid-binding; Membrane; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Receptor; Reference proteome; Steroid-binding; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 29012.4 Length 249 Aromaticity 0.12 Instability index 51.27 Isoelectric point 6.3 Charge (pH=7) -2.08 2D Binding mode Binding energy (Kcal/mol) -7.85 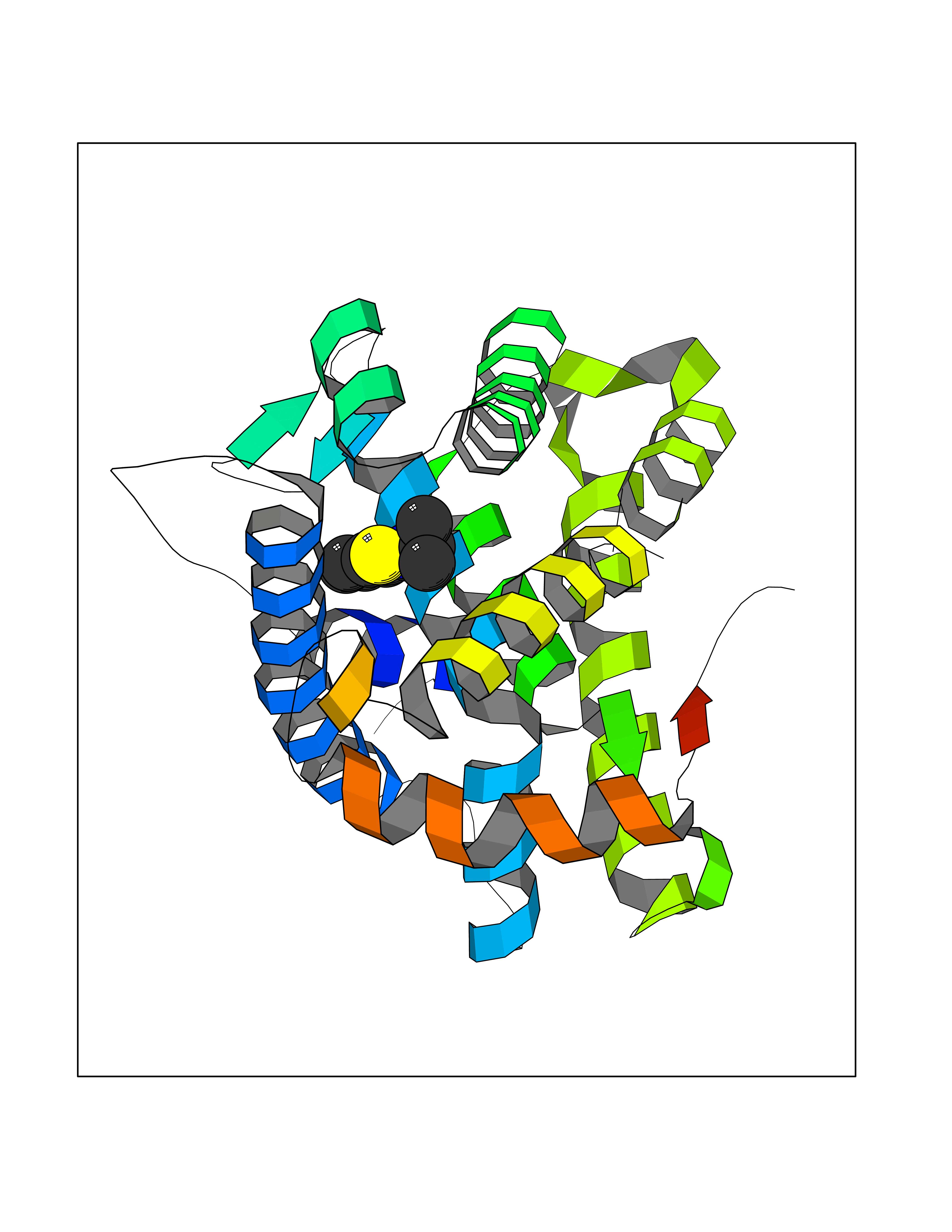 Molscript Map 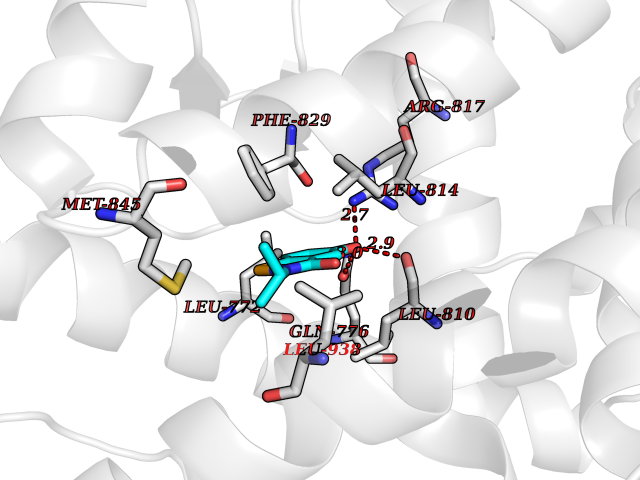 Pymol Map 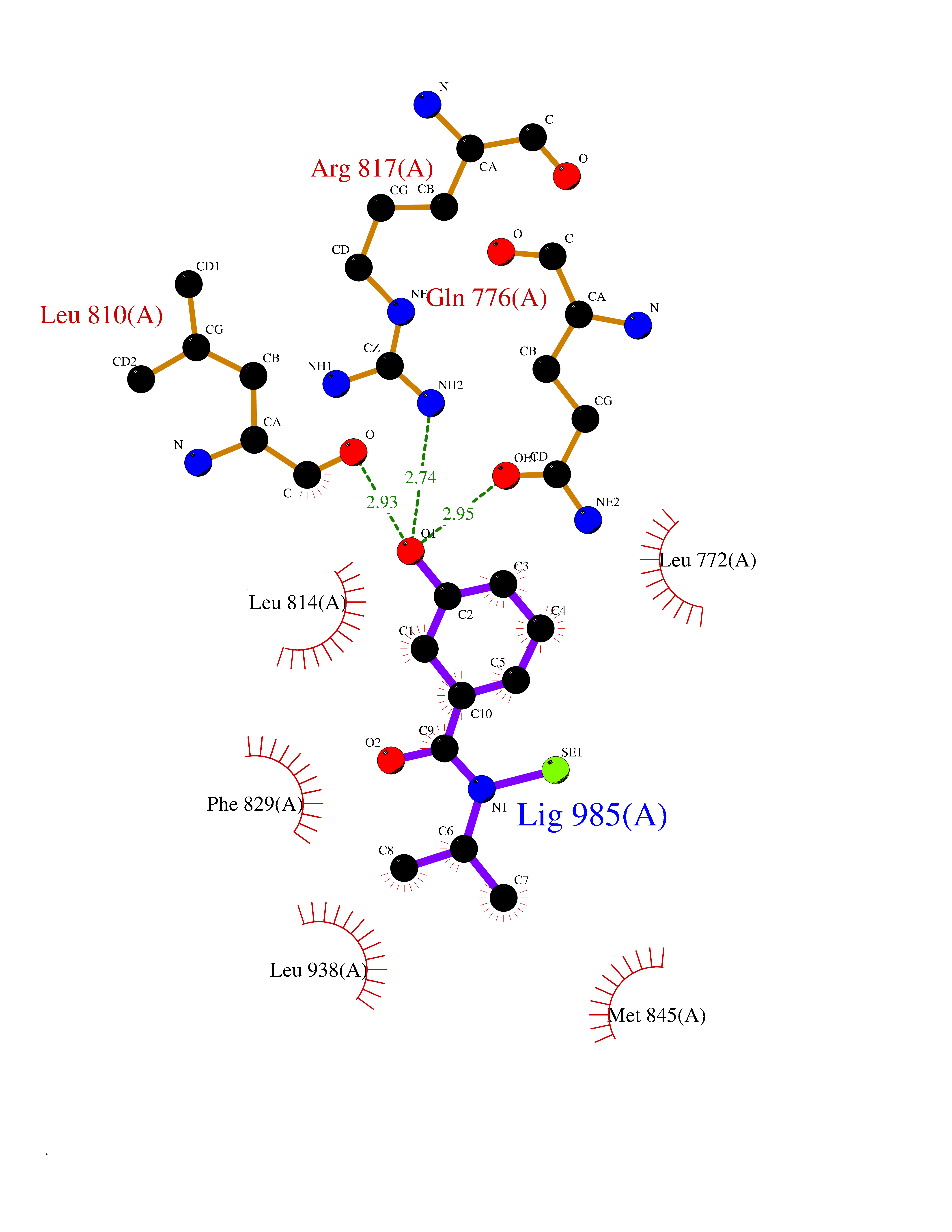 Ligplot Map 3D Binding mode Sequence TPSPVMVLENIEPEIVYAGYDSSKPDTAENLLSTLNRLAGKQMIQVVKWAKVLPGFKNLPLEDQITLIQYSWMSLLSFALSWRSYKHTNSQFLYFAPDLVFNEEKMHQSAMYELCQGMHQISLQFVRLQLTFEEYTIMKVLLLLSTIPKDGLKSQAAFEEMRTNYIKELRKMVTKCPNNSGQSWQRFYQLTKLLDSMHDLVSDLLEFCFYTFRESHALKVEFPAMLVEIISDQLPKVESGNVKPLYFHR Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Guanidinoacetate N-methyltransferase | 3ORH | 5.75 | |
Target general information Gen name GAMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, RMT2 methyltransferase family Biochemical class Transferase Function Guanidinoacetate N-methyltransferase activity.Methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 2 (CCDS2) [MIM:612736]: An autosomal recessive disorder characterized by developmental delay and regression, intellectual disability, severe disturbance of expressive and cognitive speech, intractable seizures, movement disturbances, severe depletion of creatine and phosphocreatine in the brain, and accumulation of guanidinoacetic acid in brain and body fluids. {ECO:0000269|PubMed:12468279, ECO:0000269|PubMed:15108290, ECO:0000269|PubMed:15651030, ECO:0000269|PubMed:16293431, ECO:0000269|PubMed:16855203, ECO:0000269|PubMed:17101918, ECO:0000269|PubMed:17466557, ECO:0000269|PubMed:19388150, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:24415674, ECO:0000269|PubMed:8651275}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00148; DB02751; DB00536; DB13191; DB01752 Interacts with O95363; Q969Q5; Q9HCM9-2 EC number 2.1.1.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Disease variant; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 24656 Length 219 Aromaticity 0.11 Instability index 46.5 Isoelectric point 5.91 Charge (pH=7) -4.34 2D Binding mode Binding energy (Kcal/mol) -7.84 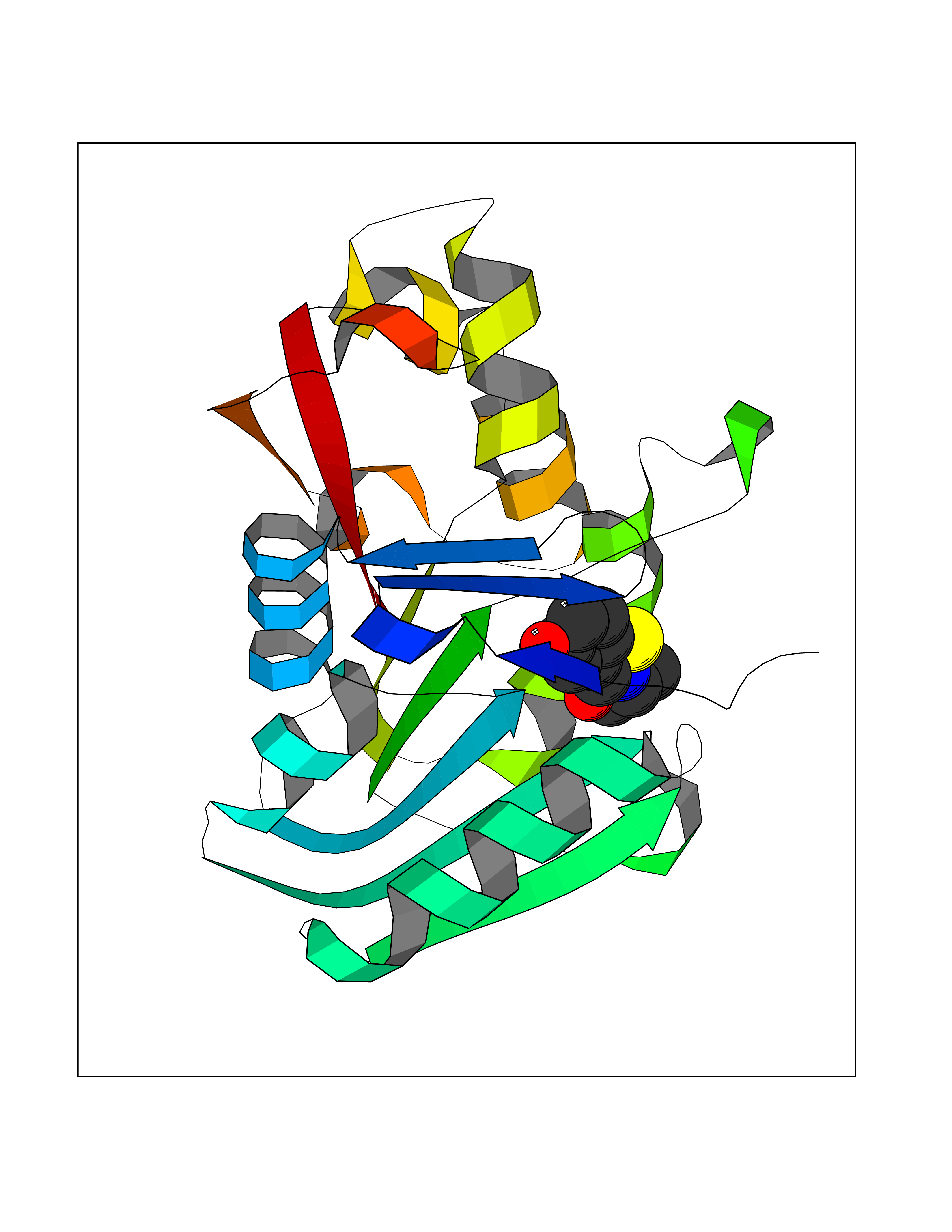 Molscript Map 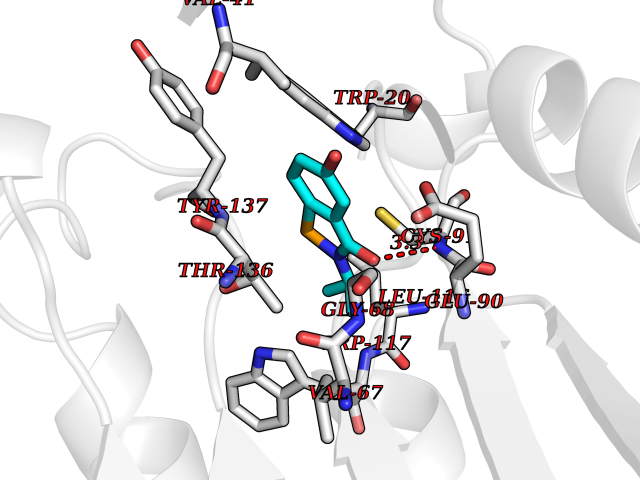 Pymol Map 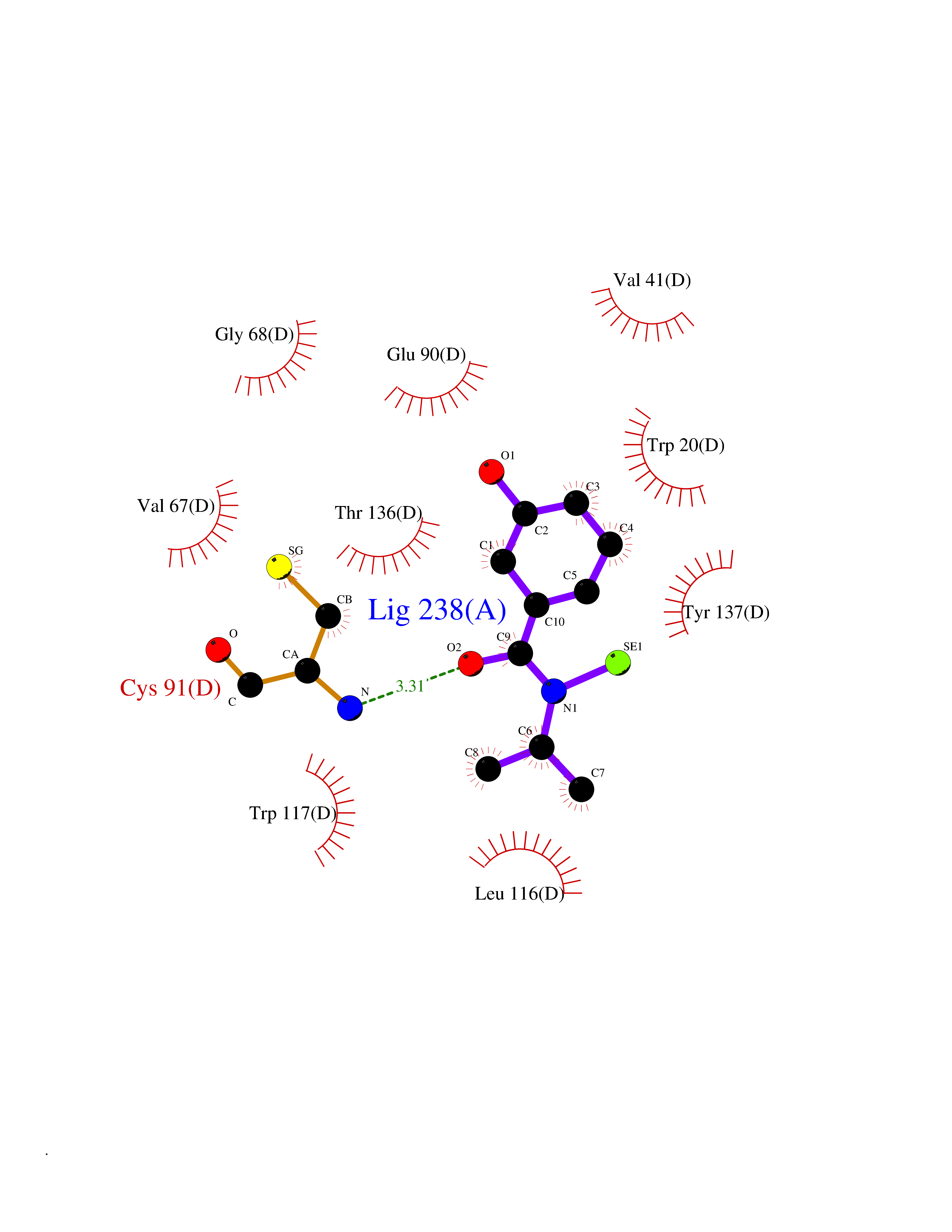 Ligplot Map 3D Binding mode Sequence PAWGAAPAAYDAADTHLRILGKPVMERWETPYMHALAAAASSKGGRVLEVGFGMAIAASKVQEAPIDEHWIIECNDGVFQRLRDWAPRQTHKVIPLKGLWEDVAPTLPDGHFDGILYDTYPLSEETWHTHQFNFIKNHAFRLLKPGGVLTYCNLTSWGELMKSKYSDITIMFEETQVPALLEAGFRRENIRTEVMALVPPADCRYYAFPQMITPLVTKG Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Peroxisome proliferator-activated receptor alpha (PPARA) | 3VI8 | 5.75 | |
Target general information Gen name PPARA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Peroxisome proliferater-activated receptor alpha; PPARalpha; PPAR-alpha; PPAR; Nuclear receptor subfamily 1 group C member 1; NR1C1 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Key regulator of lipid metabolism. Activated by the endogenous ligand 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine (16:0/18:1-GPC). Activated by oleylethanolamide, a naturally occurring lipid that regulates satiety. Receptor for peroxisome proliferators such as hypolipidemic drugs and fatty acids. Regulates the peroxisomal beta-oxidation pathway of fatty acids. Functions as transcription activator for the ACOX1 and P450 genes. Transactivation activity requires heterodimerization with RXRA and is antagonized by NR2C2. May be required for the propagation of clock information to metabolic pathways regulated by PER2. Ligand-activated transcription factor. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08915; DB00132; DB01118; DB04557; DB01393; DB04519; DB05416; DB09064; DB09006; DB00636; DB09213; DB03756; DB05187; DB06521; DB01039; DB13873; DB00573; DB13961; DB02266; DB01241; DB07215; DB01050; DB00159; DB07724; DB00328; DB12007; DB03017; DB12961; DB06510; DB08231; DB11605; DB01890; DB04224; DB11133; DB03796; DB02746; DB01708; DB06533; DB04971; DB02709; DB00412; DB09422; DB03193; DB06536; DB00197; DB00313 Interacts with P02768-3; P55212; P45973; P06307; Q3L8U1-3; G5E9A7; P22607; P62993; Q14957; P06396; P42858; Q8WXH2; P13473-2; O75376; Q13133; A0A6Q8PF08; P54725; P62826; Q7Z699; P37173; P55072; P55055-1; Q13133 EC number NA Uniprot keywords 3D-structure; Activator; Alternative splicing; Biological rhythms; DNA-binding; Lipid-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 29322.1 Length 258 Aromaticity 0.09 Instability index 35.53 Isoelectric point 6.09 Charge (pH=7) -3.57 2D Binding mode Binding energy (Kcal/mol) -7.85 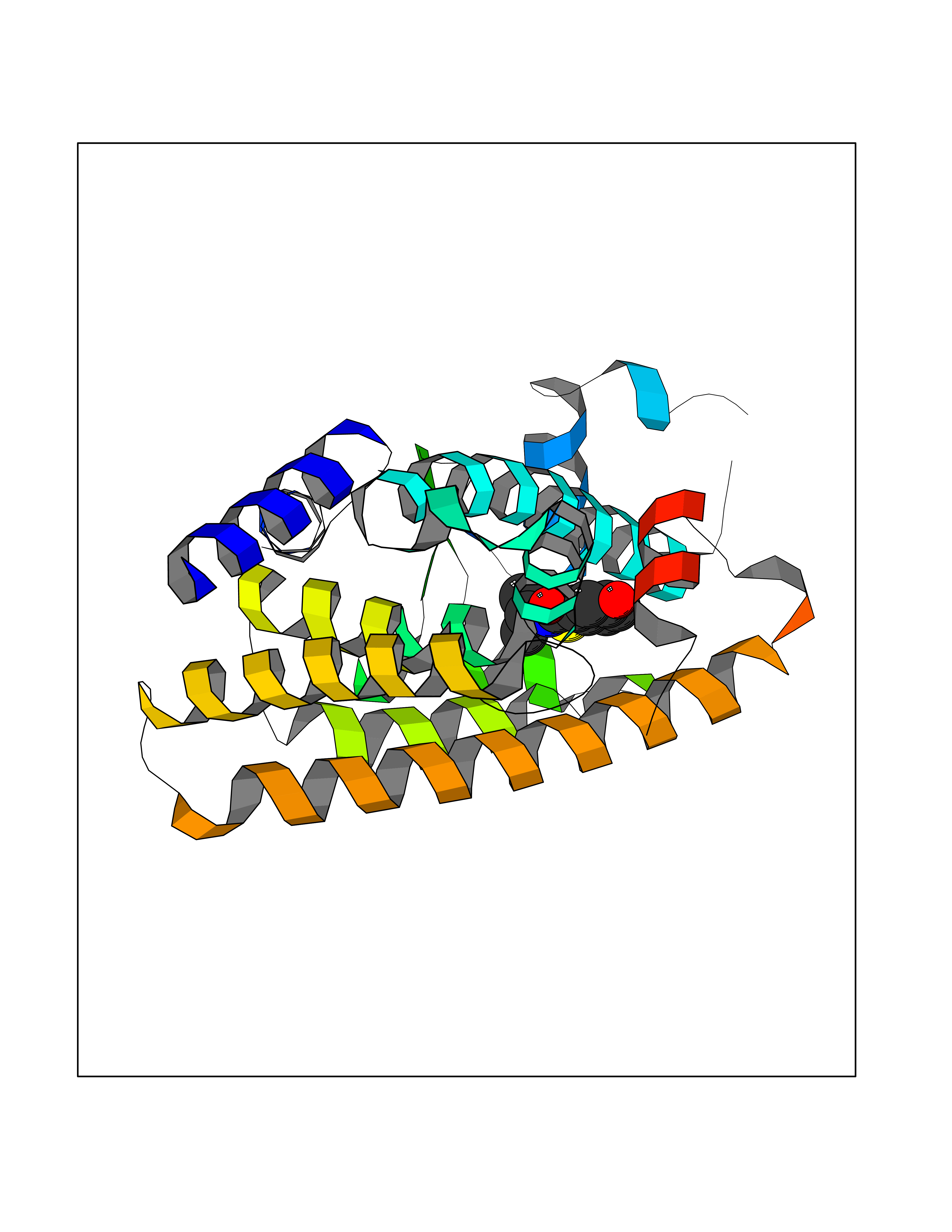 Molscript Map 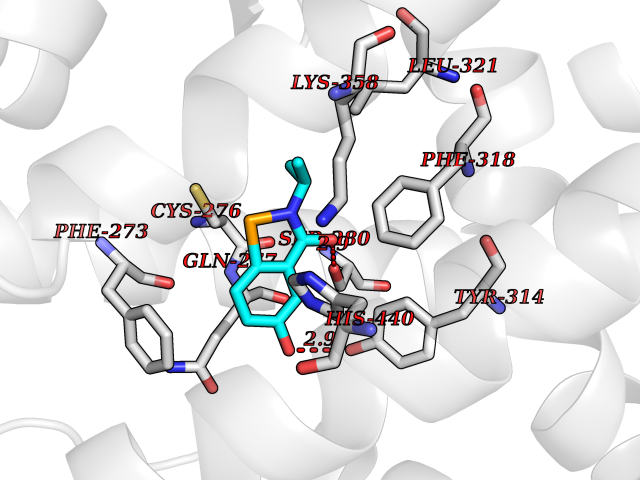 Pymol Map 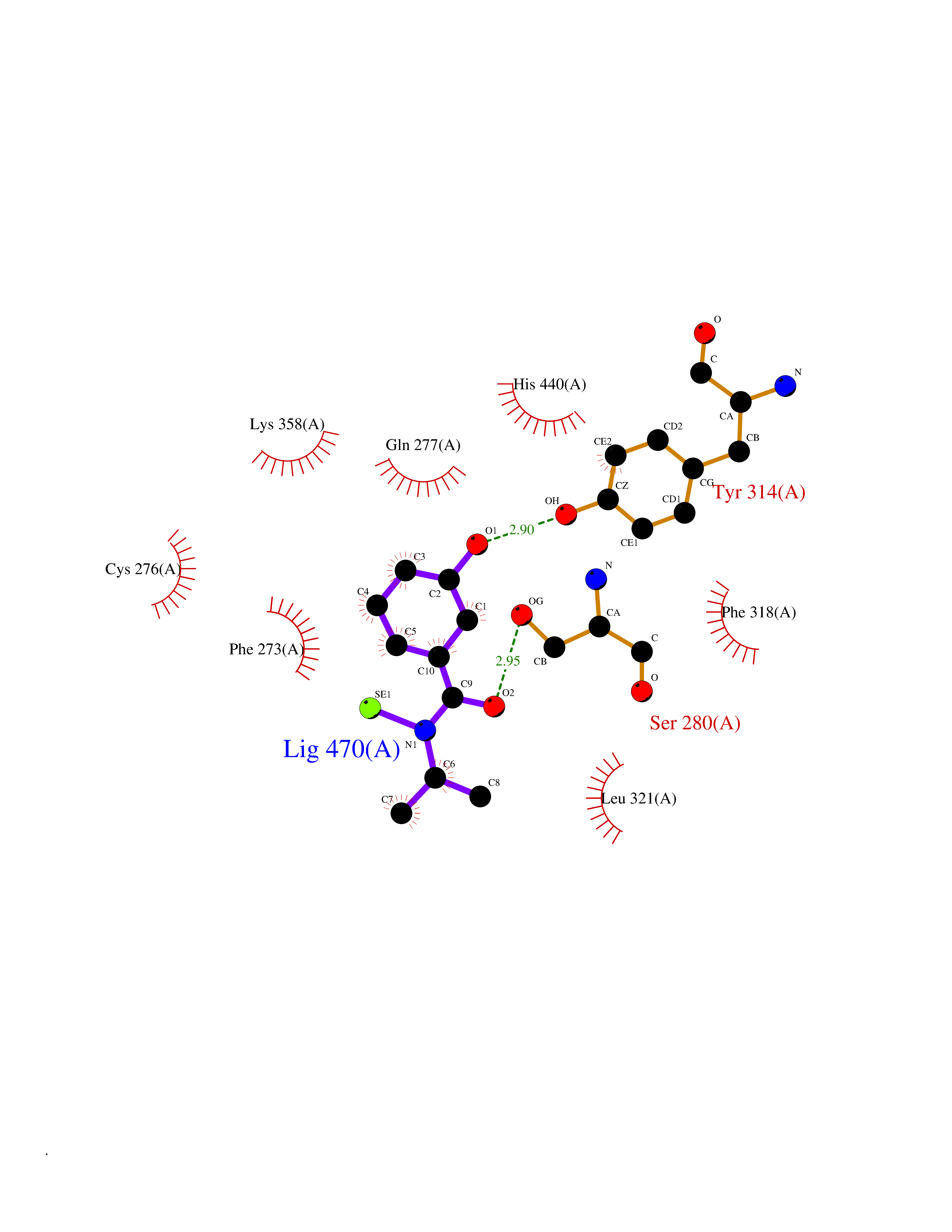 Ligplot Map 3D Binding mode Sequence DLKSLAKRIYEAYLKNFNMNKVKARVILSPFVIHDMETLCMAEKTLVAKLVANGNKEAEVRIFHCCQCTSVETVTELTEFAKAIPGFANLDLNDQVTLLKYGVYEAIFAMLSSVMNKDGMLVAYGNGFITREFLKSLRKPFCDIMEPKFDFAMKFNALELDDSDISLFVAAIICCGDRPGLLNVGHIEKMQEGIVHVLRLHLQSNHPDDIFLFPKLLQKMADLRQLVTEHAQLVQIIKKTESDAALHPLLQEIYRDMY Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | N-acylethanolamine-hydrolyzing acidamidase (NAAA) | 6DXX | 5.75 | |
Target general information Gen name NAAA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nacylsphingosine amidohydrolaselike; Nacylethanolaminehydrolyzing acid amidase subunit beta; NAAA; Acid ceramidaselike protein; ASAHlike protein Protein family Acid ceramidase family Biochemical class Carbon-nitrogen hydrolase Function Degrades bioactive fatty acid amides to their corresponding acids, with the following preference: N- palmitoylethanolamine > N-myristoylethanolamine > N- lauroylethanolamine = N-stearoylethanolamine > N- arachidonoylethanolamine > N-oleoylethanolamine. Also exhibits weak hydrolytic activity against the ceramides N- lauroylsphingosine and N-palmitoylsphingosine. Related diseases Hypertriglyceridemia, transient infantile (HTGTI) [MIM:614480]: An autosomal recessive disorder characterized by onset of moderate to severe transient hypertriglyceridemia in infancy that normalizes with age. The hypertriglyceridemia is associated with hepatomegaly, moderately elevated transaminases, persistent fatty liver, and the development of hepatic fibrosis. {ECO:0000269|PubMed:22226083, ECO:0000269|PubMed:24549054}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09061; DB14009; DB14011 Interacts with NA EC number EC 3.5.1.- Uniprot keywords 3D-structure; Alternative splicing; Autocatalytic cleavage; Direct protein sequencing; Disulfide bond; Fatty acid metabolism; Glycoprotein; Hydrolase; Lipid degradation; Lipid metabolism; Lysosome; Membrane; Proteomics identification; Reference proteome; Signal; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 36877.8 Length 328 Aromaticity 0.11 Instability index 44.37 Isoelectric point 7.72 Charge (pH=7) 1.08 2D Binding mode Binding energy (Kcal/mol) -7.84 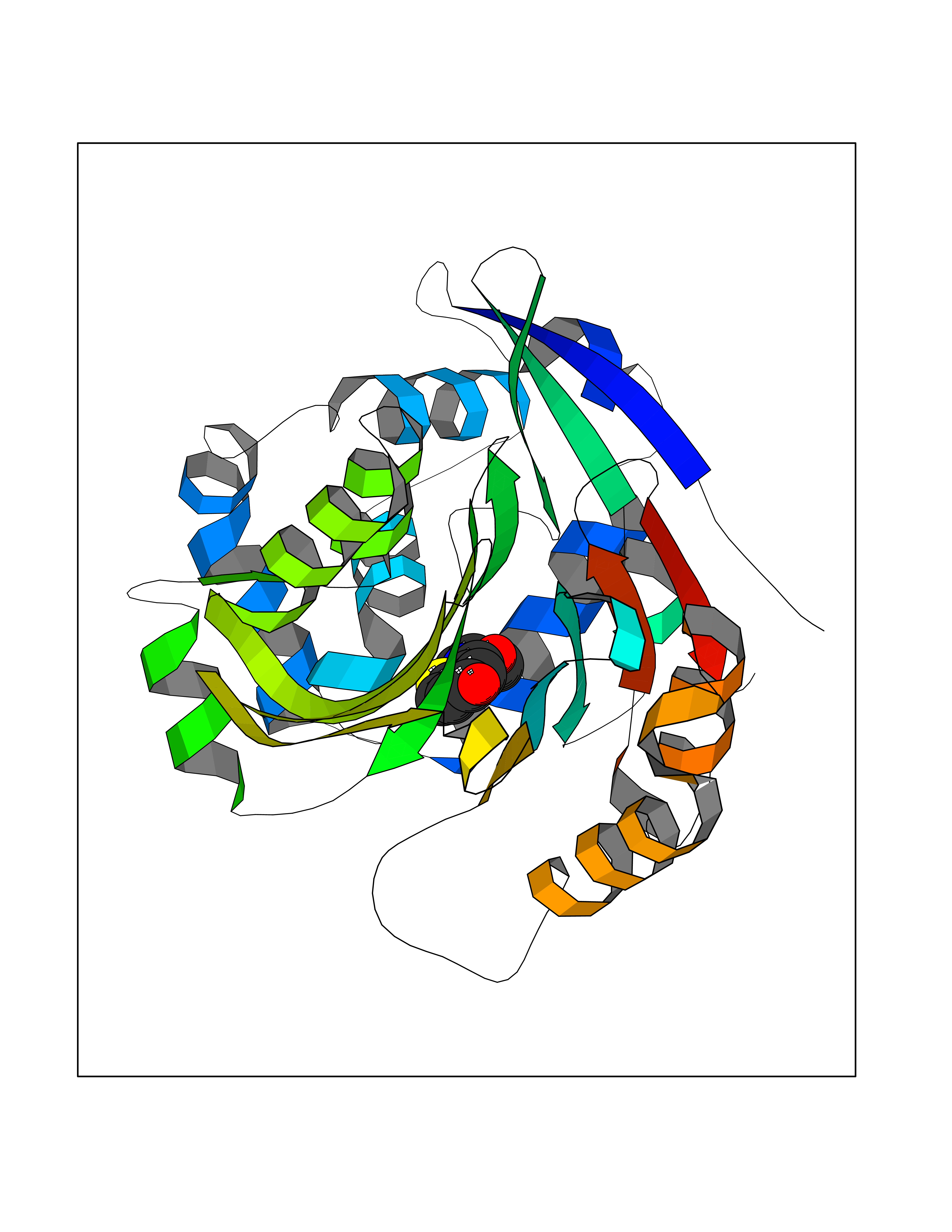 Molscript Map 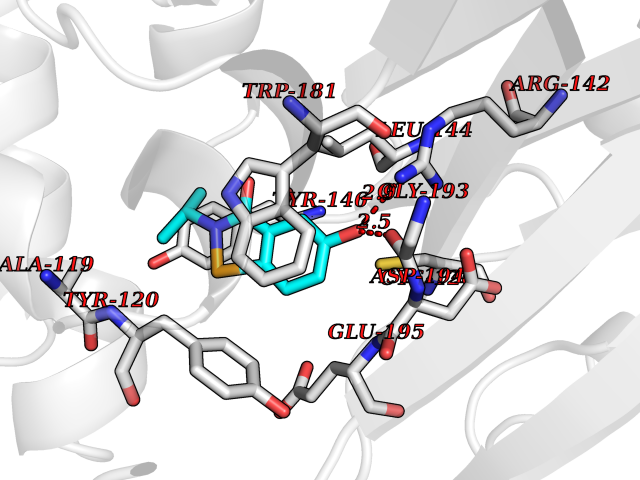 Pymol Map 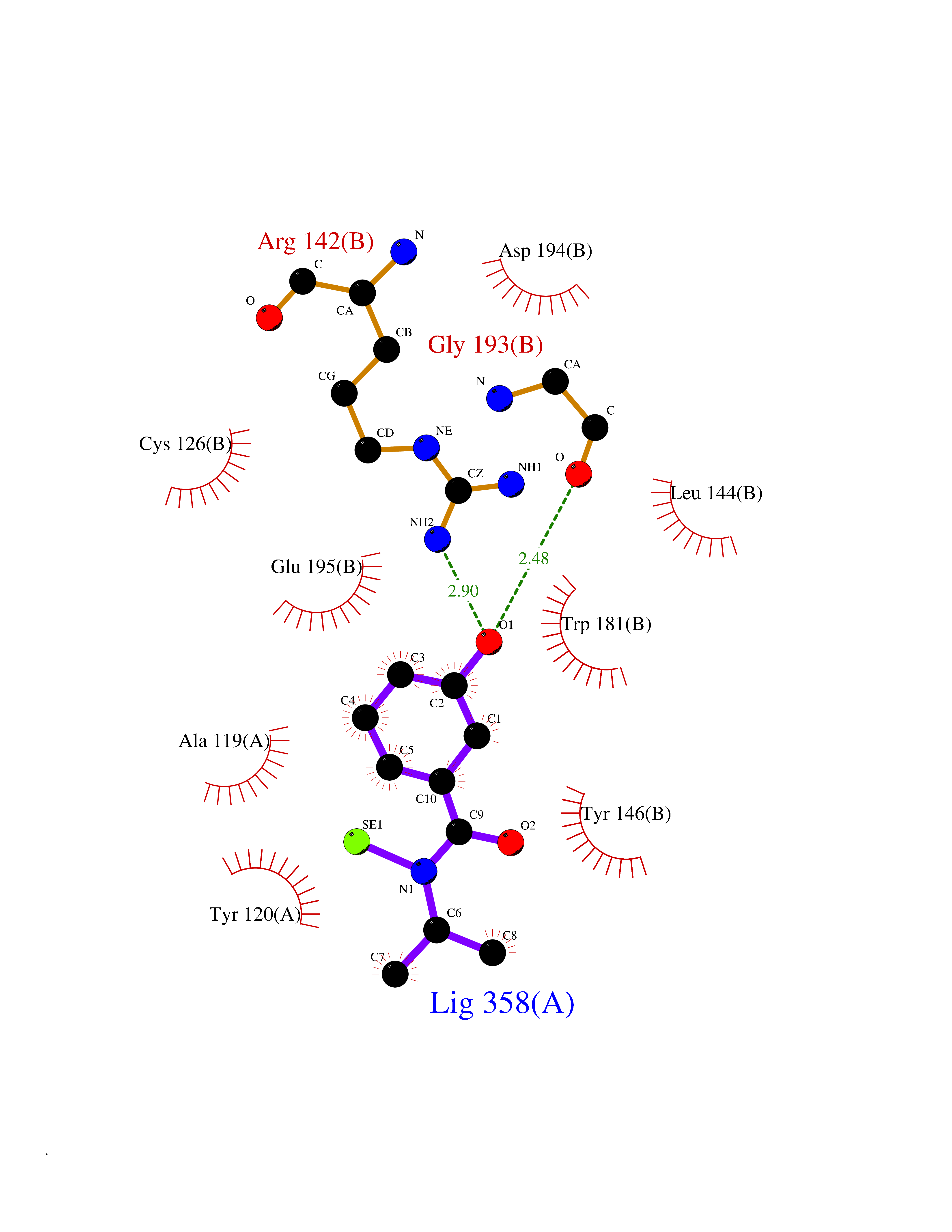 Ligplot Map 3D Binding mode Sequence SPPAAPRFNVSLDSVPELRWLPVLRHYDLDLVRAAMAQVIGDRVPKWVHVLIGKVVLELERFLPQPFTGEIRGMCDFMNLSLADCLLVNLAYESSVFCTSIVAQDSRGHIYHGRNLDYPFGNVLRKLTVDVQFLKNGQIAFTGTTFIGYVGLWTGQSPHKFTVSGDERDKGWWWENAIAALFRRHIPVSWLIRATLSESENFEAAVGKLAKTPLIADVYYIVGGTSPREGVVITRNRDGPADIWPLDPLNGAWFRVETNYDHWKPAPKEDDRRTSAIKALNATGQANLSLEALFQILSVVPVYNNFTIYTTVMSAGSPDKYMTRIRNP Hydrogen bonds contact Hydrophobic contact | ||||