Job Results:
Ligand
Structure
Job ID
f5eac8ba4ac90e4335358bfb735bd782
Job name
NA
Time
2025-10-13 17:37:26
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Thiopurine S-methyltransferase | 2BZG | 5.68 | |
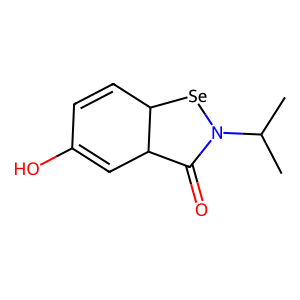
Target general information Gen name TPMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, TPMT family Biochemical class Transferase Function Thiopurine S-methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00993; DB00436; DB01327; DB01033; DB01250; DB01021 Interacts with Q8TAP4-4; Q15047-2; P61981 EC number 2.1.1.67 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 25971.5 Length 229 Aromaticity 0.12 Instability index 32.58 Isoelectric point 6.74 Charge (pH=7) -0.6 2D Binding mode Binding energy (Kcal/mol) -7.75 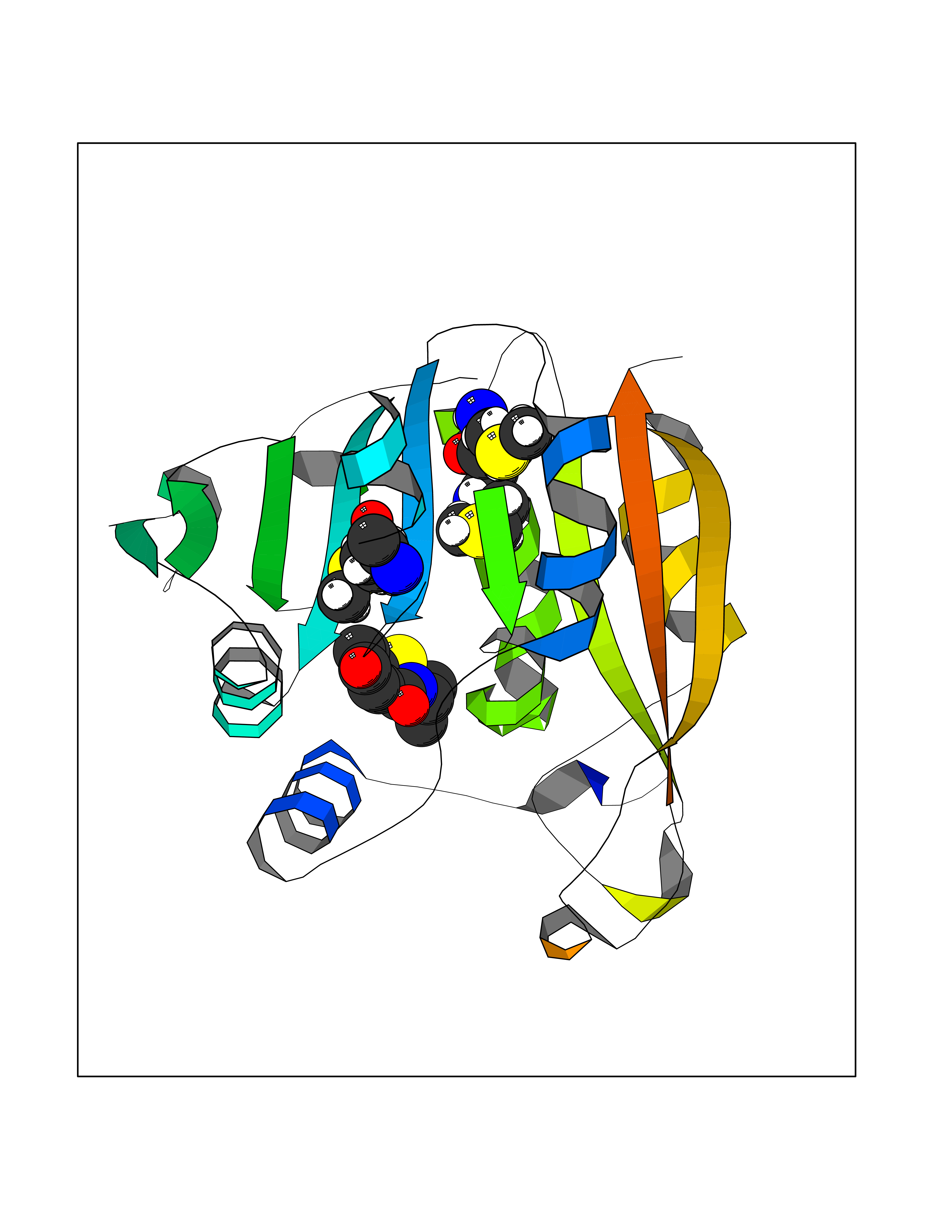 Molscript Map 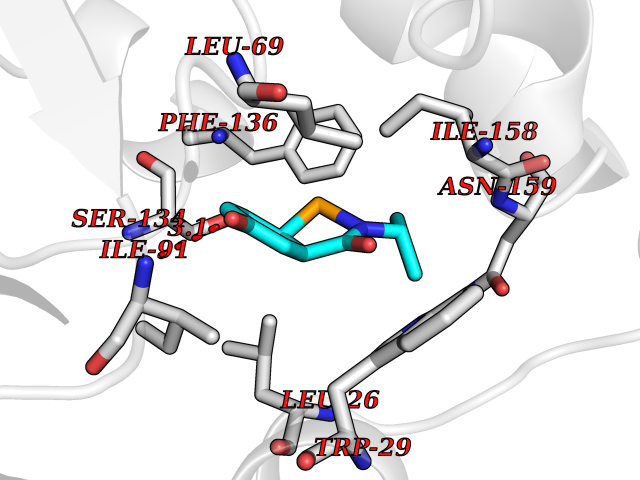 Pymol Map 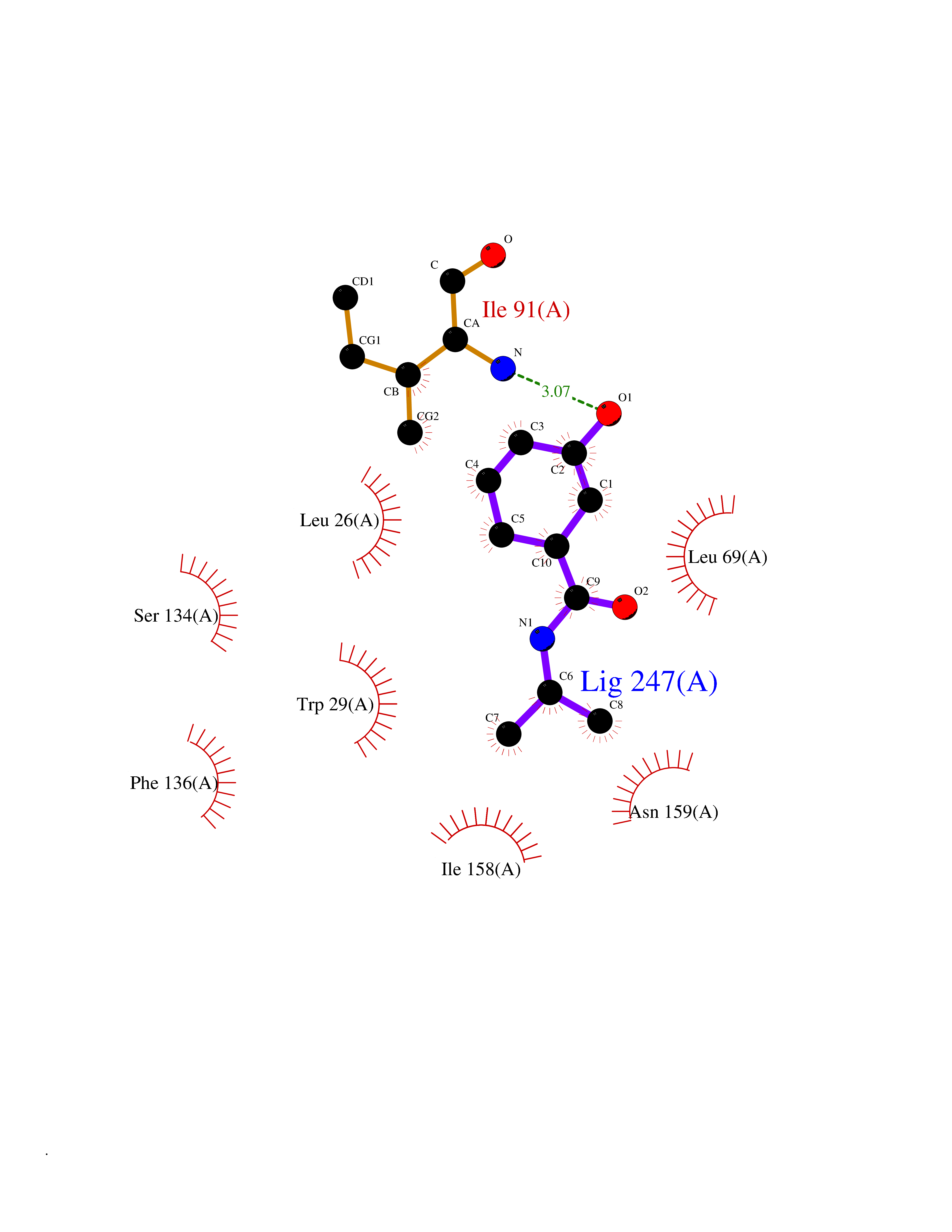 Ligplot Map 3D Binding mode Sequence EVQKNQVLTLEEWQDKWVNGKTAFHQEQGHQLLKKHLDTFLKGKSGLRVFFPLCGKAVEXKWFADRGHSVVGVEISELGIQEFFTEQNLSYSEEPITEIPGTKVFKSSSGNISLYCCSIFDLPRTNIGKFDXIWDRGALVAINPGDRKCYADTXFSLLGKKFQYLLCVLSYDPTKHPGPPFYVPHAEIERLFGKICNIRCLEKVDAFEERHKSWGIDCLFEKLYLLTEK Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | 2-iminobutanoate/2-iminopropanoate deaminase | 1ONI | 5.68 | |
Target general information Gen name RIDA Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms HRSP12 Protein family RutC family Biochemical class Translation Function Deaminase activity.Endoribonuclease activity, producing 3'-phosphomonoesters.Long-chain fatty acid binding.Platinum binding.Protein homodimerization activity.RNA binding.Transition metal ion binding.Xenon atom binding. Related diseases Congenital bile acid synthesis defect 2 (CBAS2) [MIM:235555]: A condition characterized by jaundice, intrahepatic cholestasis and hepatic failure. Patients with this liver disease show absence or low levels of chenodeoxycholic acid and cholic acid in plasma and urine. {ECO:0000269|PubMed:12970144, ECO:0000269|PubMed:15030995, ECO:0000269|PubMed:19175828, ECO:0000269|PubMed:20522910}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q8N9N5-2 EC number 3.5.99.10 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Hydrolase; Lipid metabolism; Mitochondrion; Nucleus; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome; RNA-binding Protein physicochemical properties Chain ID A,B,C,D,E,F,G,H,I Molecular weight (Da) 42624.3 Length 404 Aromaticity 0.07 Instability index 36.76 Isoelectric point 8.99 Charge (pH=7) 5.46 2D Binding mode Binding energy (Kcal/mol) -7.75 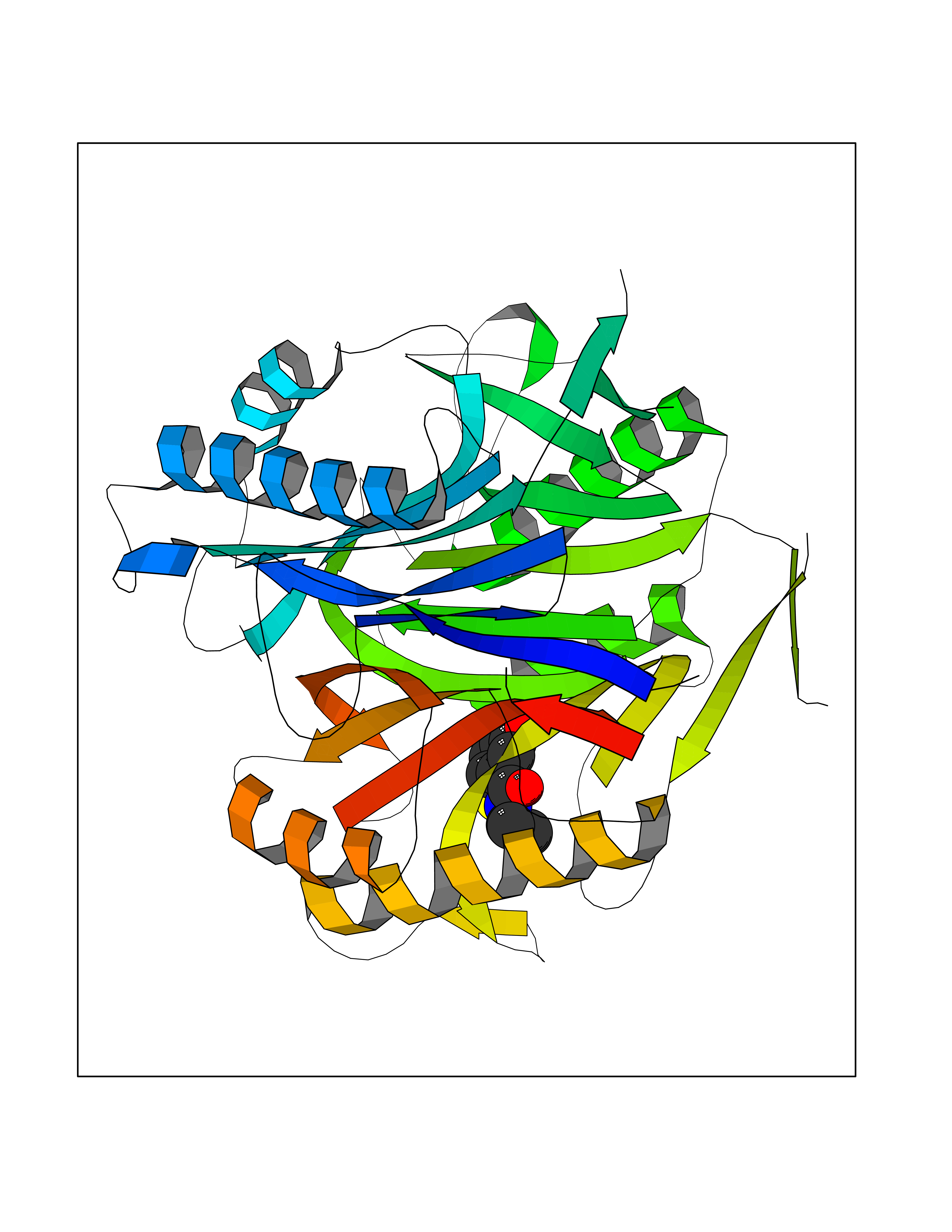 Molscript Map 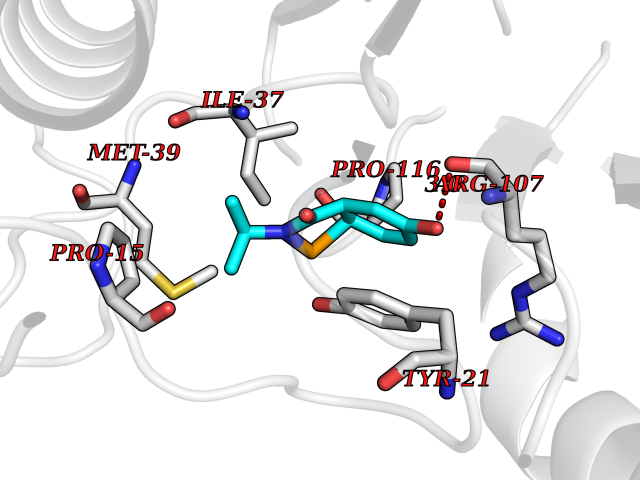 Pymol Map 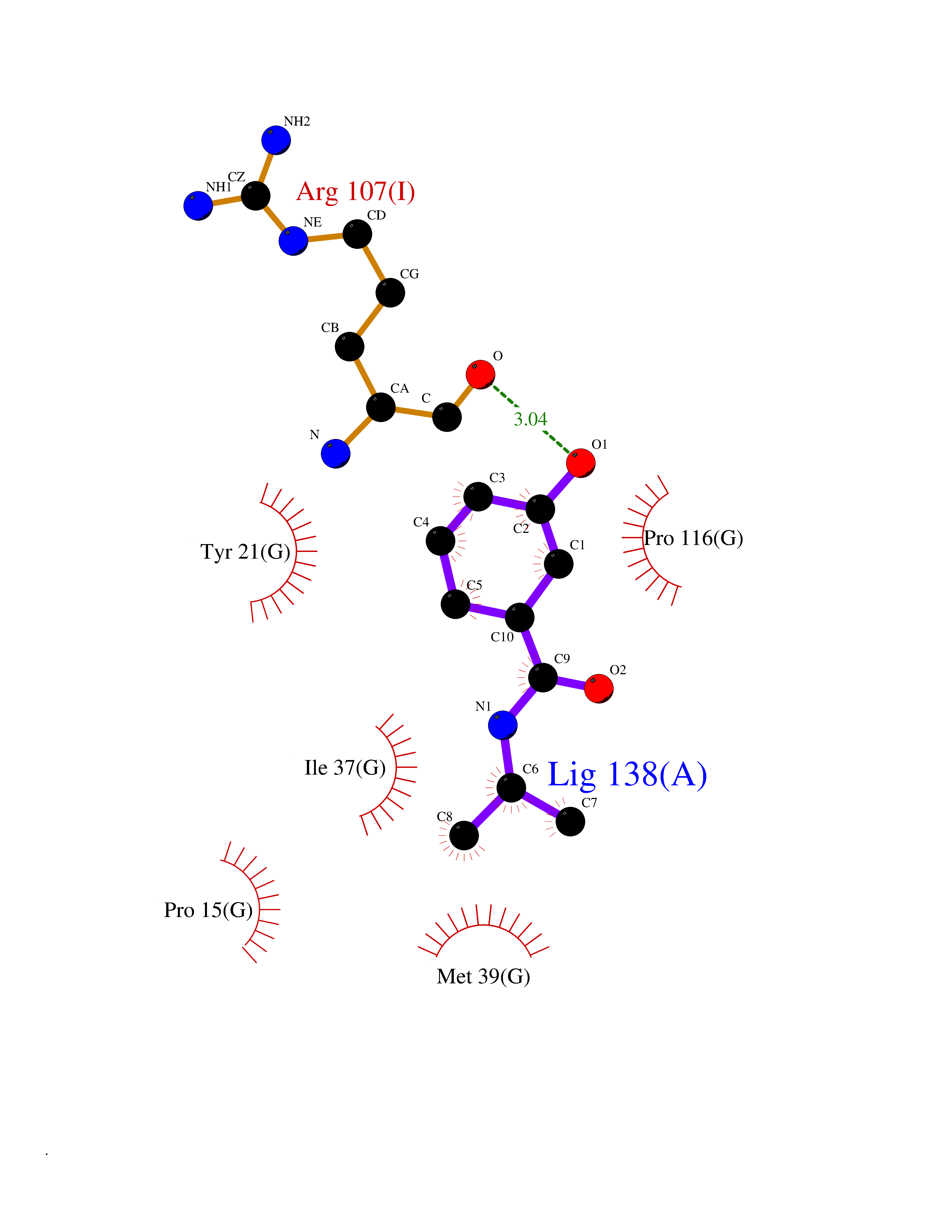 Ligplot Map 3D Binding mode Sequence SSLIRRVISTAKAPGAIGPYSQAVLVDRTIYISGQIGMDPSSGQLVSGGVAEEAKQALKNMGEILKAAGCDFTNVVKTTVLLADINDFNTVNEIYKQYFKSNFPARAAYQVAALPKGSRIEIEAVAIQGPLTTASSSLIRRVISTAKAPGAIGPYSQAVLVDRTIYISGQIGMDPSSGQLVSGGVAEEAKQALKNMGEILKAAGCDFTNVVKTTVLLADINDFNTVNEIYKQYFKSNFPARAAYQVAALPKGSRIEIEAVAIQGPLTTASSSLIRRVISTAKAPGAIGPYSQAVLVDRTIYISGQIGMDPSSGQLVSGGVAEEAKQALKNMGEILKAAGCDFTNVVKTTVLLADINDFNTVNEIYKQYFKSNFPARAAYQVAALPKGSRIEIEAVAIQGPLTTA Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Acetyl-CoA carboxylase (ACC) (EC 6.4.1.2) (Fatty acid synthetase 3) (mRNA transport-defective protein 7) [Includes: Biotin carboxylase (EC 6.3.4.14)] | 1UYS | 5.68 | |
Target general information Gen name ACC1 Organism Saccharomyces cerevisiae (strain ATCC 204508 / S288c) (Baker's yeast) Uniprot ID TTD ID NA Synonyms MTR7;YNR016C;N3175;ABP2;FAS3 Protein family NA Biochemical class NA Function Carries out three functions: biotin carboxyl carrier protein, biotin carboxylase and carboxyltransferase. Involved in the synthesis of very-long-chain fatty acid synthesis which is required to maintain a functional nuclear envelope. Required for acylation and vacuolar membrane association of VAC8 which is necessary to maintain a normal morphology of the vacuole. {ECO:0000269|PubMed:10757783, ECO:0000269|PubMed:12730220, ECO:0000269|PubMed:6103540, ECO:0000269|PubMed:6108218, ECO:0000269|PubMed:8943372}." Related diseases A chromosomal aberration involving NFKB2 is found in a case of B-cell non Hodgkin lymphoma (B-NHL). Translocation t(10;14)(q24;q32) with IGHA1. The resulting oncogene is also called Lyt-10C alpha variant.; DISEASE: A chromosomal aberration involving NFKB2 is found in a cutaneous T-cell leukemia (C-TCL) cell line. This rearrangement produces the p80HT gene which codes for a truncated 80 kDa protein (p80HT).; DISEASE: In B-cell leukemia (B-CLL) cell line, LB40 and EB308, can be found after heterogeneous chromosomal aberrations, such as internal deletions.; DISEASE: Immunodeficiency, common variable, 10 (CVID10) [MIM:615577]: A primary immunodeficiency characterized by childhood-onset of recurrent infections, hypogammaglobulinemia, and decreased numbers of memory and marginal zone B-cells. Some patients may develop autoimmune features and have circulating autoantibodies. An unusual feature is central adrenal insufficiency. {ECO:0000269|PubMed:24140114, ECO:0000269|PubMed:25524009}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q00955 EC number 6.3.4.14; 6.4.1.2 Uniprot keywords 3D-structure; Acetylation; ATP-binding; Biotin; Cytoplasm; Direct protein sequencing; Endoplasmic reticulum; Fatty acid biosynthesis; Fatty acid metabolism; Ligase; Lipid biosynthesis; Lipid metabolism; Manganese; Membrane; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Reference proteome Protein physicochemical properties Chain ID B,C Molecular weight (Da) 145619 Length 1328 Aromaticity 0.1 Instability index 30.31 Isoelectric point 5.32 Charge (pH=7) -26.79 2D Binding mode Binding energy (Kcal/mol) -7.74  Molscript Map 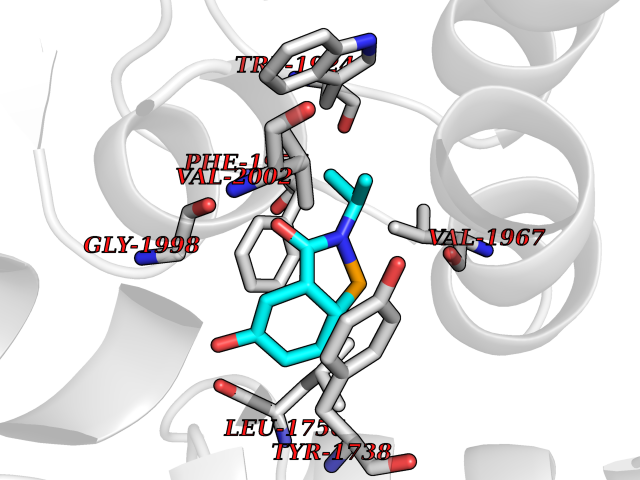 Pymol Map 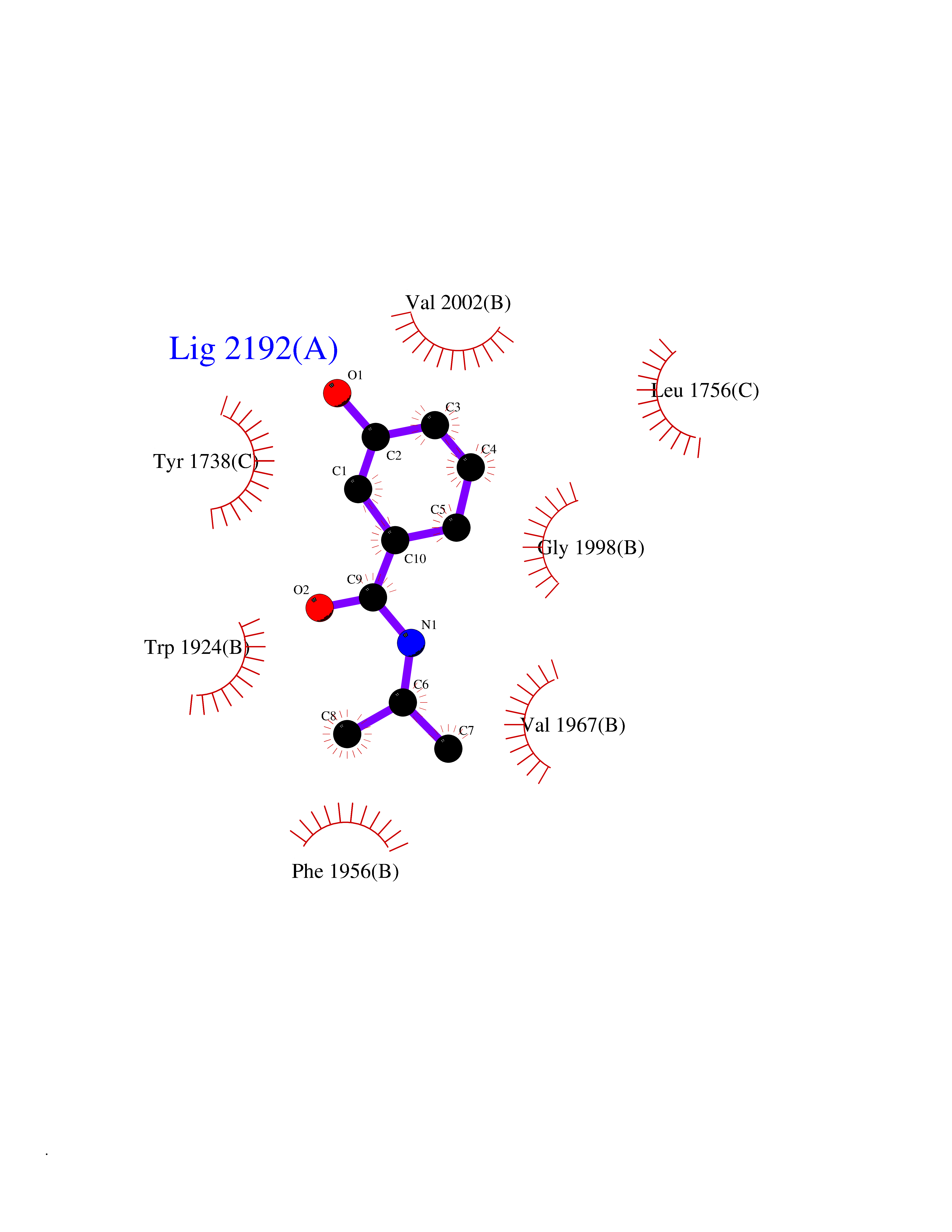 Ligplot Map 3D Binding mode Sequence WLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGLPIATPYPVKEWLQPKRYKAHLXGTTYVYDFPELFRQASSSQWKNFSADVKLTDDFFISNELIEDENGELTEVEREPGANAIGXVAFKITVKTPEYPRGRQFVVVANDITFKIGSFGPQEDEFFNKVTEYARKRGIPRIYLAANSGARIGXAEEIVPLFQVAWNDAANPDKGFQYLYLTSEGXETLKKFDKENSVLTERTVINGEERFVIKTIIGSEDGLGVECLRGSGLIAGATSRAYHDIFTITLVTCRSVGIGAYLVRLGQRAIQVEGQPIILTGAPAINKXLGREVYTSNLQLGGTQIXYNNGVSHLTAVDDLAGVEKIVEWXSYVPAKRNXPVPILETKDTWDRPVDFTPTNDETYDVRWXIEGRETESGFEYGLFDKGSFFETLSGWAKGVVVGRARLGGIPLGVIGVETRTVENLIPADPANPNSAETLIQEPGQVWHPNSAFKTAQAINDFNNGEQLPXXILANWRGFSGNEVLKYGSFIVDALVDYKQPIIIYIPPTGELRGGSWVVVDPTINADQXEXYADVNARAGVLEPQGXVGIKFRREKLLDTXNRLELLPIYGQISLQFADLHDRSSRXVAKGVISKELEWTEARRFFFWRLRRRLNEEYLIKRLSHQVGEASRLEKIARIRSWYPASVDHEDDRQVATWIEENYKTLDDKLKGL Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Riboflavin kinase | 1NB0 | 5.68 | |
Target general information Gen name RFK Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Transferase Function ATP binding.Metal ion binding.Riboflavin kinase activity. Related diseases Glutaric aciduria 1 (GA1) [MIM:231670]: An autosomal recessive metabolic disorder characterized by progressive dystonia and athetosis due to gliosis and neuronal loss in the basal ganglia. {ECO:0000269|PubMed:14707522, ECO:0000269|PubMed:18775954, ECO:0000269|PubMed:24973495, ECO:0000269|PubMed:8541831, ECO:0000269|PubMed:8900227, ECO:0000269|PubMed:8900228, ECO:0000269|PubMed:9600243, ECO:0000269|PubMed:9711871}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03247; DB00140 Interacts with Q9NXG0-2; P19438; P19438-1 EC number 2.7.1.26 Uniprot keywords 3D-structure; ATP-binding; Cytoplasm; Flavoprotein; FMN; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Proteomics identification; Reference proteome; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 16749.9 Length 147 Aromaticity 0.12 Instability index 41.55 Isoelectric point 7.09 Charge (pH=7) 0.12 2D Binding mode Binding energy (Kcal/mol) -7.75 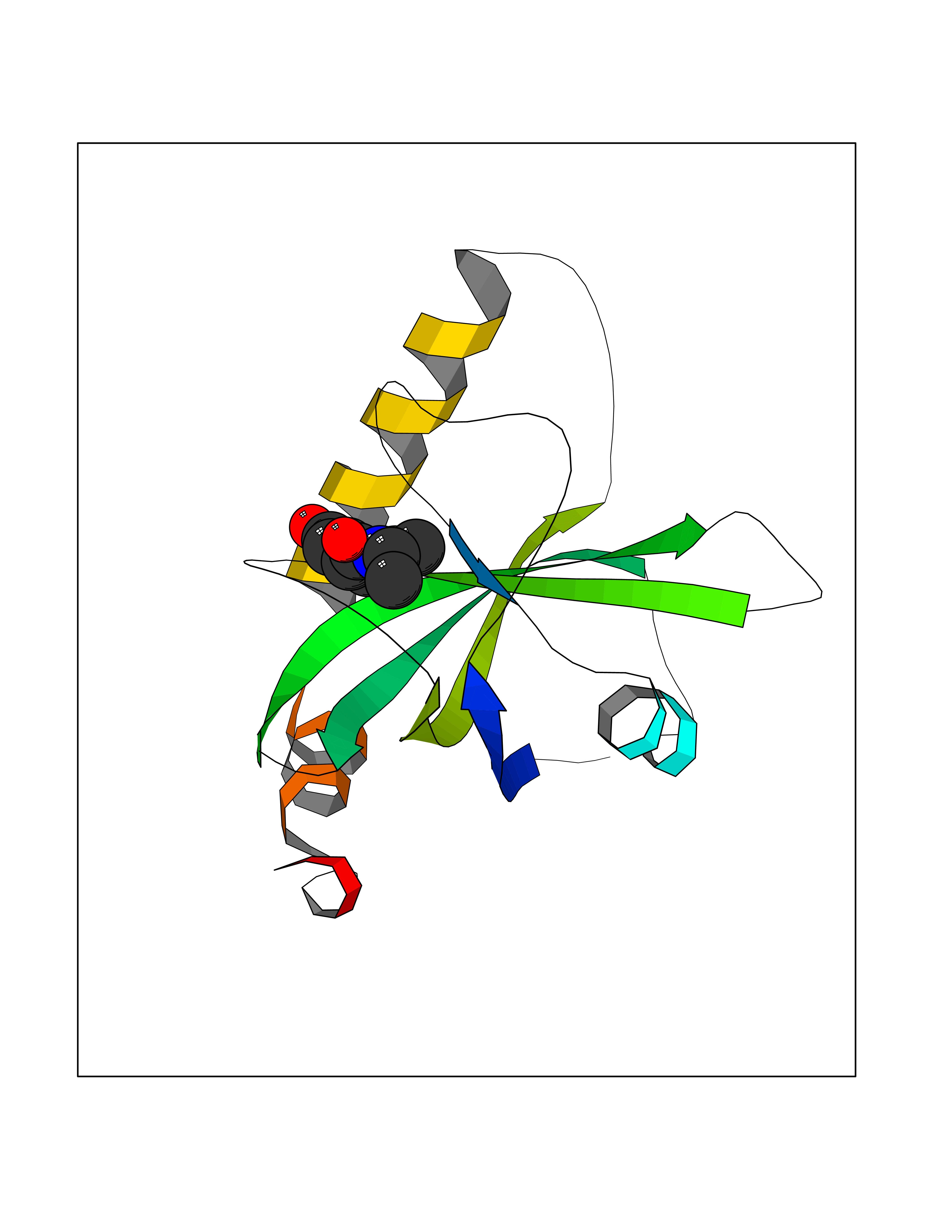 Molscript Map 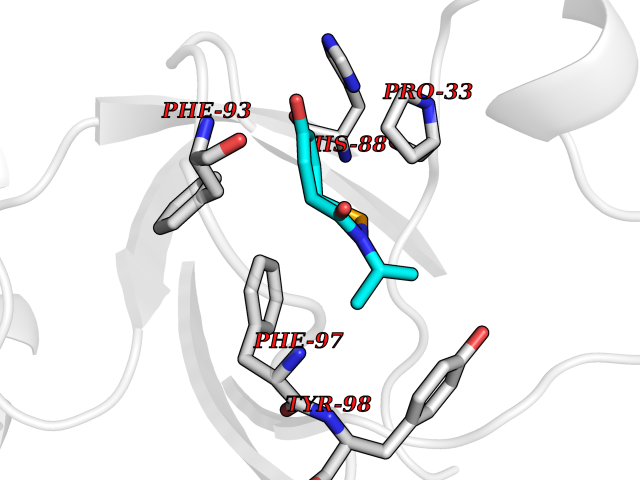 Pymol Map 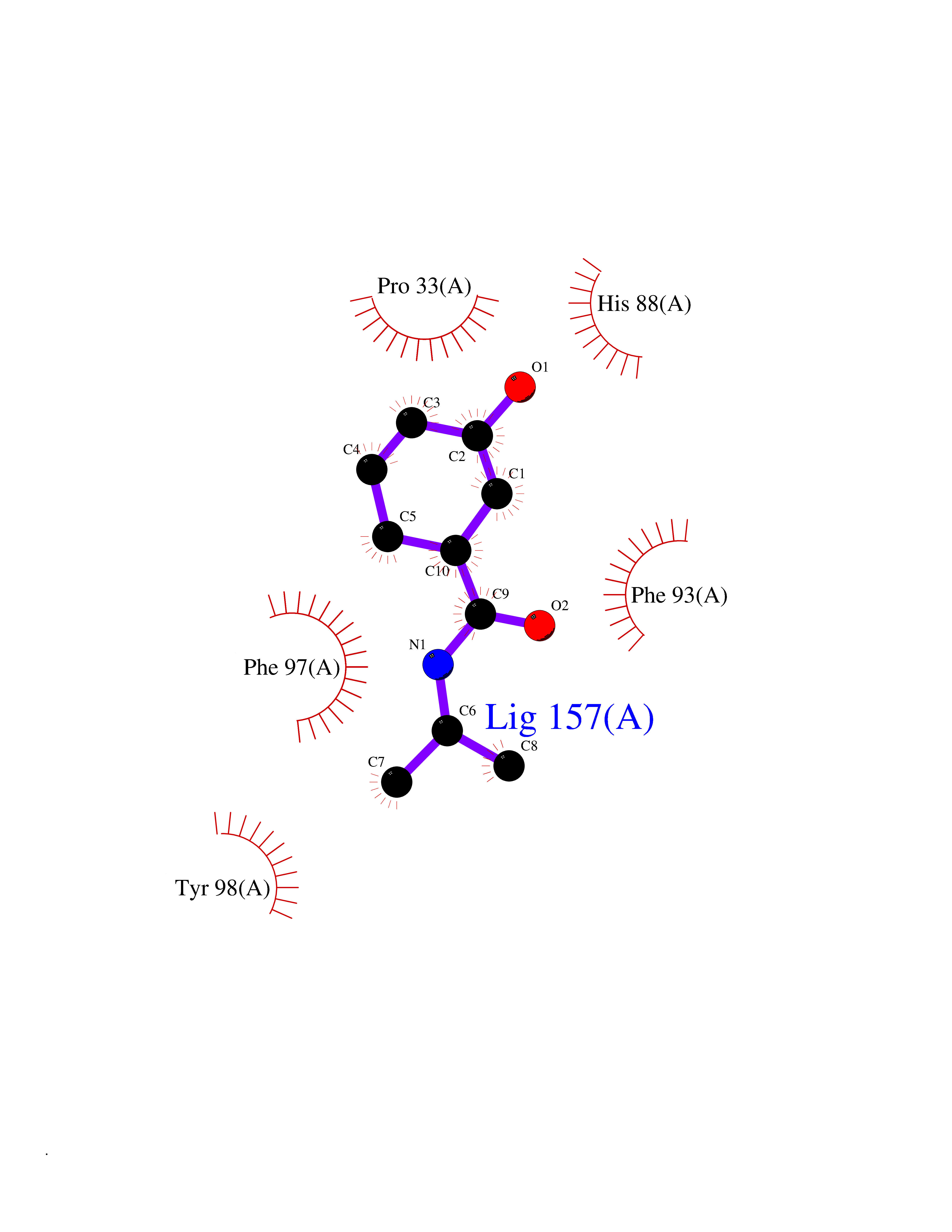 Ligplot Map 3D Binding mode Sequence RHLPYFCRGQVVRGFGRGSKQLGIPTANFPEQVVDNLPADISTGIYYGWASVGSGDVHKMVVSIGWNPYYKNTKKSMETHIMHTFKEDFYGEILNVAIVGYLRPEKNFDSLESLISAIQGDIEEAKKRLELPEYLKIKEDNFFQVSK Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Tryptophan 2,3-dioxygenase (TDO) | 6PYZ | 5.68 | |
Target general information Gen name TDO2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Tryptophanase; Tryptophan pyrrolase; Tryptophan oxygenase; Tryptamin 2,3-dioxygenase; TRPO; TO Protein family Tryptophan 2,3-dioxygenase family Biochemical class Oxygenase Function Catalyzes the oxidative cleavage of the indole moiety. Heme-dependent dioxygenase that catalyzes the oxidative cleavage of the L-tryptophan (L-Trp) pyrrole ring and converts L-tryptophan to N-formyl-L-kynurenine. Related diseases Hypertryptophanemia (HYPTRP) [MIM:600627]: An autosomal recessive condition characterized by persistent hypertryptophanemia and hyperserotoninemia. {ECO:0000269|PubMed:28285122}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00779; DB00500; DB00150 Interacts with O43865; O95671; P27797; P12830; P36957; O60762; P06730; Q8TBB1; Q9H8S9; Q70IA8; Q8TDX7; Q9NPG2; Q9HAN9; P20393; Q9NRD5; Q8IYS1; O00560; Q9H190; P48775; Q68DK2-5 EC number EC 1.13.11.11 Uniprot keywords 3D-structure; Dioxygenase; Disease variant; Heme; Iron; Metal-binding; Oxidoreductase; Proteomics identification; Reference proteome; Tryptophan catabolism Protein physicochemical properties Chain ID A,B Molecular weight (Da) 83454.8 Length 701 Aromaticity 0.11 Instability index 43.93 Isoelectric point 6.93 Charge (pH=7) -0.48 2D Binding mode Binding energy (Kcal/mol) -7.75  Molscript Map 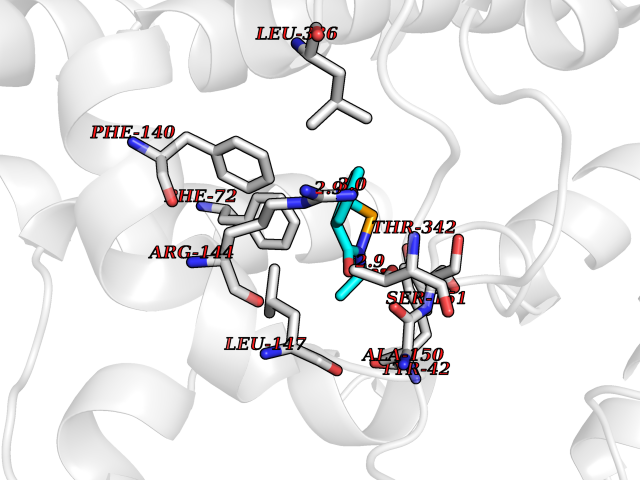 Pymol Map 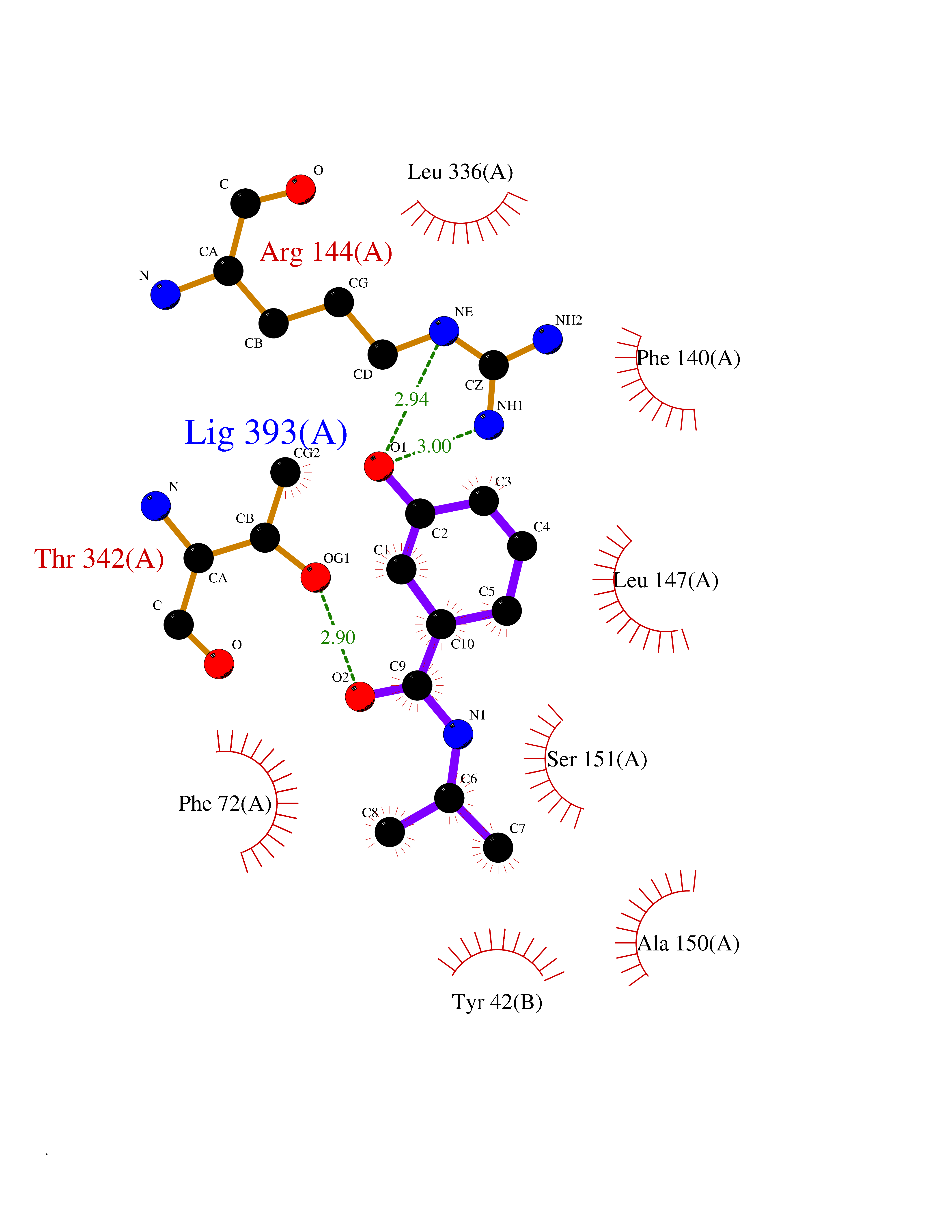 Ligplot Map 3D Binding mode Sequence GLIYGNYLHLEKVLNAQELQSETKGNKIHDEHLFIITHQAYELWFKQILWELDSVREIFQNGHVRDERNMLKVVSRMHRVSVILKLLVQQFSILETMTALDFNDFREYLSPASGFQSLQFRLLENKIGVLQNMRVPYNRRHYRDNFKGEENELLLKSEQEKTLLELVEAWLERTPGLEPHGFNFWGKLEKNITRGLEEEFIRIQAKEESEEKEEQVAEFQKQKEVLLSLFDEKRHEHLLSKGERRLSYRALQGALMIYFYREEPRFQVPFQLLTSLMDIDSLMTKWRYNHVCMVHRMLGSKAGTGGSSGYHYLRSTVSDRYKVFVDLFNLSTYLIPRHWIPKMNPTIHKFLEHGGLIYGNYLHLEKVLNAQELQSETKGNKIHDEHLFIITHQAYELWFKQILWELDSVREIFQNGHVRDERNMLKVVSRMHRVSVILKLLVQQFSILETMTALDFNDFREYLSPASGFQSLQFRLLENKIGVLQNMRVPYYRDNFKGEENELLLKSEQEKTLLELVEAWLERTPGLEPHGFNFWGKLEKNITRGLEEEFIRIQAKEESEEKEEQVAEFQKQKEVLLSLFDEKRHEHLLSKGERRLSYRALQGALMIYFYREEPRFQVPFQLLTSLMDIDSLMTKWRYNHVCMVHRMLGSKAGTGGSSGYHYLRSTVSDRYKVFVDLFNLSTYLIPRHWIPKMNPTIHKFL Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Cardiac phospholamban (PLN) | 7E0Z | 5.68 | |
Target general information Gen name PLN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Phospholamban; PLN; PLB Protein family Phospholamban family Biochemical class NA Function Reversibly inhibits the activity of ATP2A2 in cardiac sarcoplasmic reticulum by decreasing the apparent affinity of the ATPase for Ca(2+). Modulates the contractility of the heart muscle in response to physiological stimuli via its effects on ATP2A2. Modulatescalcium re-uptake during muscle relaxation and plays an important role in calcium homeostasis in the heart muscle. The degree of ATP2A2 inhibition depends on the oligomeric state of PLN. ATP2A2 inhibition is alleviated by PLN phosphorylation. Related diseases Cardiomyopathy, dilated, 1P (CMD1P) [MIM:609909]: A disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death. {ECO:0000269|PubMed:12610310, ECO:0000269|PubMed:16432188, ECO:0000269|PubMed:22137083, ECO:0000269|PubMed:22427649, ECO:0000269|PubMed:22707725}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Cardiomyopathy, familial hypertrophic, 18 (CMH18) [MIM:613874]: A hereditary heart disorder characterized by ventricular hypertrophy, which is usually asymmetric and often involves the interventricular septum. The symptoms include dyspnea, syncope, collapse, palpitations, and chest pain. They can be readily provoked by exercise. The disorder has inter- and intrafamilial variability ranging from benign to malignant forms with high risk of cardiac failure and sudden cardiac death. {ECO:0000269|PubMed:12705874}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q3SXY8; P07307-3; O15342; Q9BXK5; Q13323; P19397; O95471; Q9UHP7-3; Q7Z7G2; O43889-2; Q96BA8; Q09013; Q92838; Q9GZR5; Q5JX71; Q14318; Q8TBE3; P48165; Q8TDT2; O60883; Q8TED1; P31937; Q7Z5P4; P43628; Q5T700; Q8N112; Q9GZY8-5; Q6N075; Q99735; O14880; Q9GZW8; Q9H2K0; P15941-11; Q8TBJ4; O95197-3; Q9NR31; A0A0S2Z4U3; Q9Y3P8; Q15849; Q8IWU4; O95436-2; Q9NQQ7-3; Q9NP94; Q9HBV2; Q9NPE6; Q16623; P32856-2; Q9BVX2; Q7Z7N9; Q6UW68; Q9NWC5; Q96B21; Q4KMG9; Q8N661; O15393-2 EC number NA Uniprot keywords 3D-structure; Acetylation; Cardiomyopathy; Disease variant; Endoplasmic reticulum; Lipoprotein; Membrane; Mitochondrion; Palmitate; Phosphoprotein; Proteomics identification; Reference proteome; Sarcoplasmic reticulum; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 40393 Length 349 Aromaticity 0.13 Instability index 35.1 Isoelectric point 8.98 Charge (pH=7) 6.56 2D Binding mode Binding energy (Kcal/mol) -7.75  Molscript Map 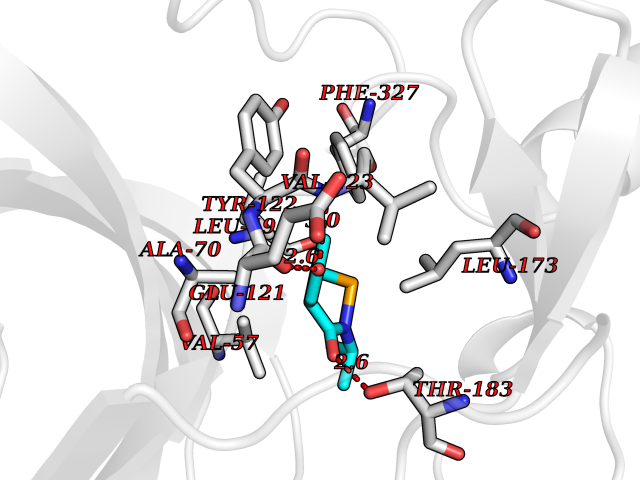 Pymol Map 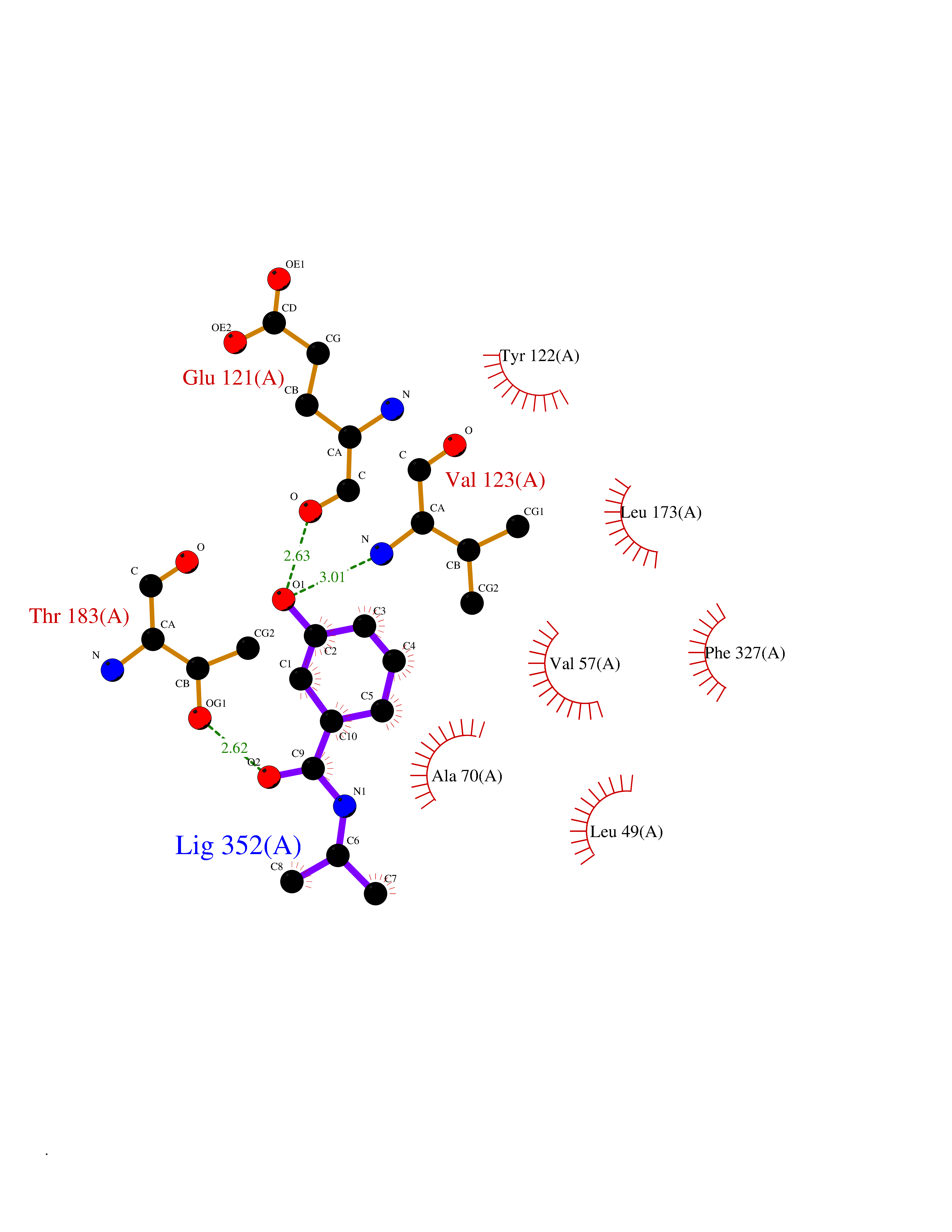 Ligplot Map 3D Binding mode Sequence ESVKEFLAKAKEDFLKKWETPQNTAQLDQFDRIKTLGTGSFGRVMLVKHKESGNHYAMKILDKQKVVKLKQIEHTLNEKRILQAVNFPFLVKLEFSFKDNSNLYMVMEYVAGGEMFSHLRRIGRFSEPHARFYAAQIVLTFEYLHSLDLIYRDLKPENLLIDQQGYIQVTDFGFAKRVKGRTWXLCGTPEYLAPEIILSKGYNKAVDWWALGVLIYEMAAGYPPFFADQPIQIYEKIVSGKVRFPSHFSSDLKDLLRNLLQVDLTKRFGNLKNGVNDIKNHKWFATTDWIAIYQRKVEAPFIPKFKGPGDTSNFDDYEEEEIRVXINEKCGKEFTEFTRSAIRRASTIE Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Transforming protein RhoA (RHOA) | 4XSH | 5.68 | |
Target general information Gen name RHOA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms h12; Rho cDNA clone 12; RHO12; ARHA; ARH12 Protein family Small GTPase superfamily, Rho family Biochemical class Small GTPase Function Involved in a microtubule-dependent signal that is required for the myosin contractile ring formation during cell cycle cytokinesis. Plays an essential role in cleavage furrow formation. Required for the apical junction formation of keratinocyte cell-cell adhesion. Stimulates PKN2 kinase activity. May be an activator of PLCE1. Activated by ARHGEF2, which promotes the exchange of GDP for GTP. Essential for the SPATA13-mediated regulation of cell migration and adhesion assembly and disassembly. The MEMO1-RHOA-DIAPH1 signaling pathway plays an important role in ERBB2-dependent stabilization of microtubules at the cell cortex. It controls the localization of APC and CLASP2 to the cell membrane, via the regulation of GSK3B activity. In turn, membrane-bound APC allows the localization of the MACF1 to the cell membrane, which is required for microtubule capture and stabilization. Regulates a signal transduction pathway linking plasma membrane receptors to the assembly of focal adhesions and actin stress fibers. Involved in a microtubule-dependent signal that is required for the myosin contractile ring formation during cell cycle cytokinesis. Plays an essential role in cleavage furrow formation. Required for the apical junction formation of keratinocyte cell-cell adhesion. May be an activator of PLCE1. Activated by ARHGEF2, which promotes the exchange of GDP for GTP. Essential for the SPATA13-mediated regulation of cell migration and adhesion assembly and disassembly. The MEMO1-RHOA-DIAPH1 signaling pathway plays an important role in ERBB2-dependent stabilization of microtubules at the cell cortex. It controls the localization of APC and CLASP2 to the cell membrane, via the regulation of GSK3B activity. In turn, membrane-bound APC allows the localization of the MACF1 to the cell membrane, which is required for microtubule capture and stabilization. Regulates KCNA2 potassium channel activity by reducing its location at the cell surface in response to CHRM1 activation; promotes KCNA2 endocytosis. Regulates a signal transduction pathway linking plasma membrane receptors to the assembly of focal adhesions and actin stress fibers. Related diseases Ectodermal dysplasia with facial dysmorphism and acral, ocular, and brain anomalies (EDFAOB) [MIM:618727]: A neuroectodermal syndrome characterized by linear hypopigmentation, alopecia, apparently asymptomatic leukoencephalopathy, and facial, ocular, dental and acral anomalies. Patients show no intellectual or neurologic impairment. {ECO:0000269|PubMed:31570889}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04315 Interacts with Q15109; Q7Z6G8-3; P05067; Q07960; P52565; O15085; Q9NZN5; Q8IW93; Q92974; Q12774; P46527; Q9Y4D1; O60610; Q9UKT9; P19338; Q9Y4F9; Q9Y4F9-2; Q96MK2; Q13464; Q9BST9; Q15796; Q9HCE7-2; Q15654; O08808; Q6PDM6; Q9Z0S9; Q8C6B2; A0A0F6B1Q8; Q9FD10 EC number EC 3.6.5.2 Uniprot keywords 3D-structure; ADP-ribosylation; Cell cycle; Cell division; Cell membrane; Cell projection; Cytoplasm; Cytoskeleton; Direct protein sequencing; Disease variant; Ectodermal dysplasia; Glycoprotein; GTP-binding; Host-virus interaction; Hydrolase; Isopeptide bond; Lipoprotein; Magnesium; Membrane; Methylation; Nucleotide-binding; Nucleus; Phosphoprotein; Prenylation; Proteomics identification; Proto-oncogene; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID B,A Molecular weight (Da) 43736.4 Length 382 Aromaticity 0.09 Instability index 35.31 Isoelectric point 8.29 Charge (pH=7) 3.14 2D Binding mode Binding energy (Kcal/mol) -7.75  Molscript Map 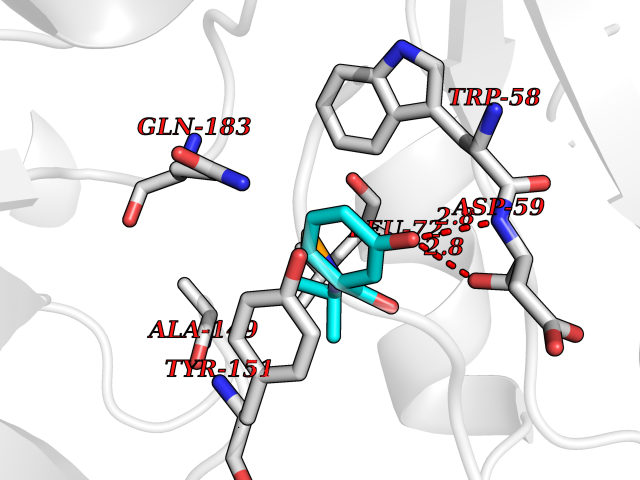 Pymol Map 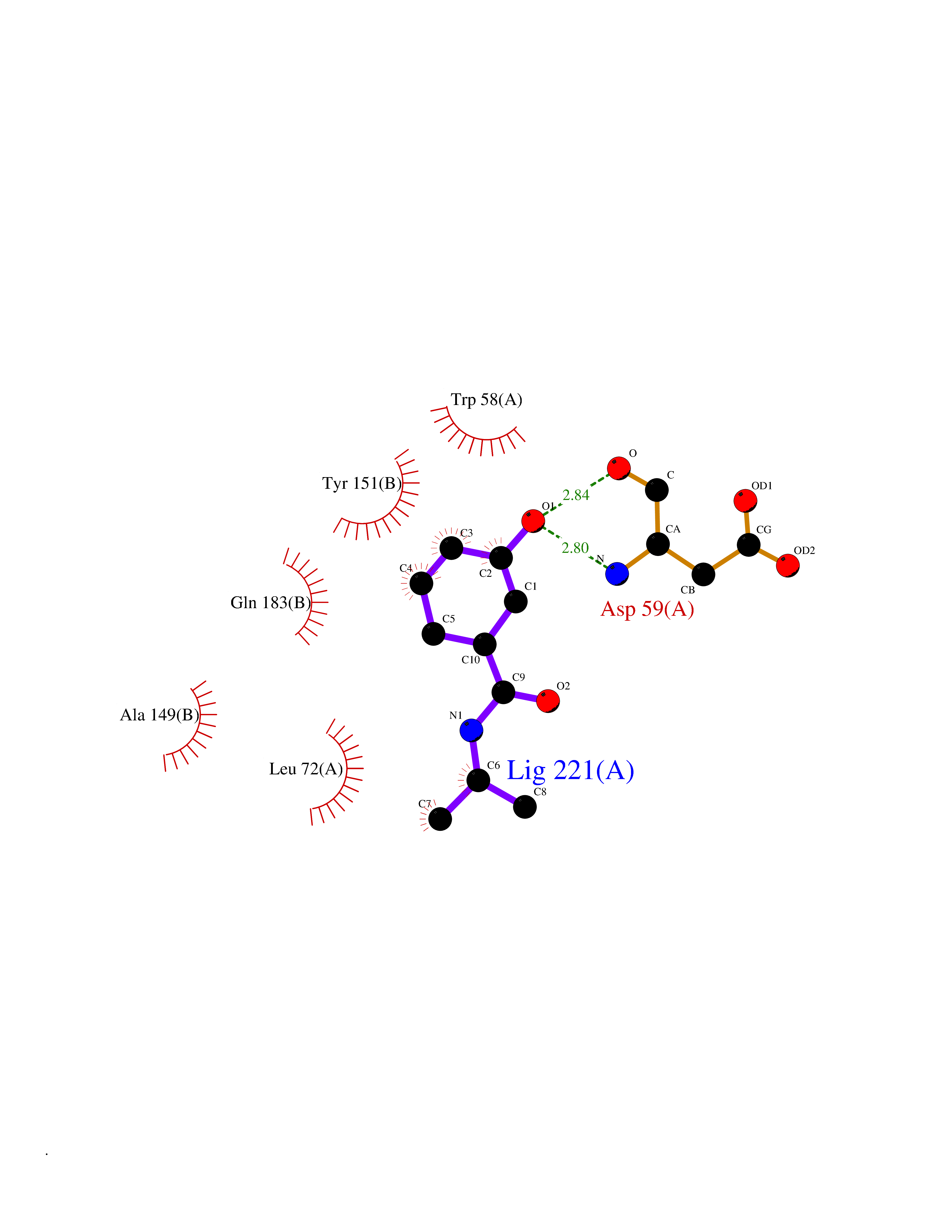 Ligplot Map 3D Binding mode Sequence AIRKKLVIVGDGACGKTCLLIVNSKDQFPEVYVPTVFENYVADIEVDGKQVELALWDTAGQEDYDRLRPLSYPDTDVILMCFSIDSPDSLENIPEKWTPEVKHFCPNVPIILVGNKKDLRNDEHTRRELAKMKQEPVKPEEGRDMANRIGAFGYMECSAKTKDGVREVFEMATRAALKYKLCTNKEEADAWGKKQFNKWSKEEKSAIRDYTKNARPYNEFLRMHAGKLDSDPTMKKKIESLDKALNRKEAKVNDNIKVYRGDDAWIFGKEYDNSIIKNGKVDREKFKEIQKKFQGKTTTEFGYISTSILIDAGYAKTRPVMTEFKVGSGTHGAYMNSDDLTAYPGQYELLLPRNTVYKIEKIYIAIDNNTQKEQIKVEATIK Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Matrix metalloproteinase-10 (MMP-10) | 1Q3A | 5.67 | |
Target general information Gen name MMP10 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Transin-2; Stromelysin-2; STMY2; SL-2 Protein family Peptidase M10A family Biochemical class Peptidase Function Activates procollagenase. Can degrade fibronectin, gelatins of type I, III, IV, and V; weakly collagens III, IV, and V. Related diseases Orthostatic hypotension 1 (ORTHYP1) [MIM:223360]: A form of orthostatic hypotension due to congenital dopamine beta-hydroxylase deficiency. Orthostatic hypotension, also known as postural hypotension, is a finding defined as a 20-mm Hg decrease in systolic pressure or a 10-mm Hg decrease in diastolic pressure occurring 3 minutes after a person has risen from supine to standing. Symptoms include dizziness, blurred vision, and sometimes syncope. ORTHYP1 is an autosomal recessive condition apparent from infancy or early childhood and characterized by low plasma and urinary levels of norepinephrine and epinephrine, and episodic hypoglycemia. {ECO:0000269|PubMed:11857564}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00786; DB08271 Interacts with NA EC number EC 3.4.24.22 Uniprot keywords 3D-structure; Calcium; Collagen degradation; Disulfide bond; Extracellular matrix; Hydrolase; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Zinc; Zymogen Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 52822 Length 471 Aromaticity 0.12 Instability index 21.13 Isoelectric point 4.83 Charge (pH=7) -35.32 2D Binding mode Binding energy (Kcal/mol) -7.74  Molscript Map  Pymol Map 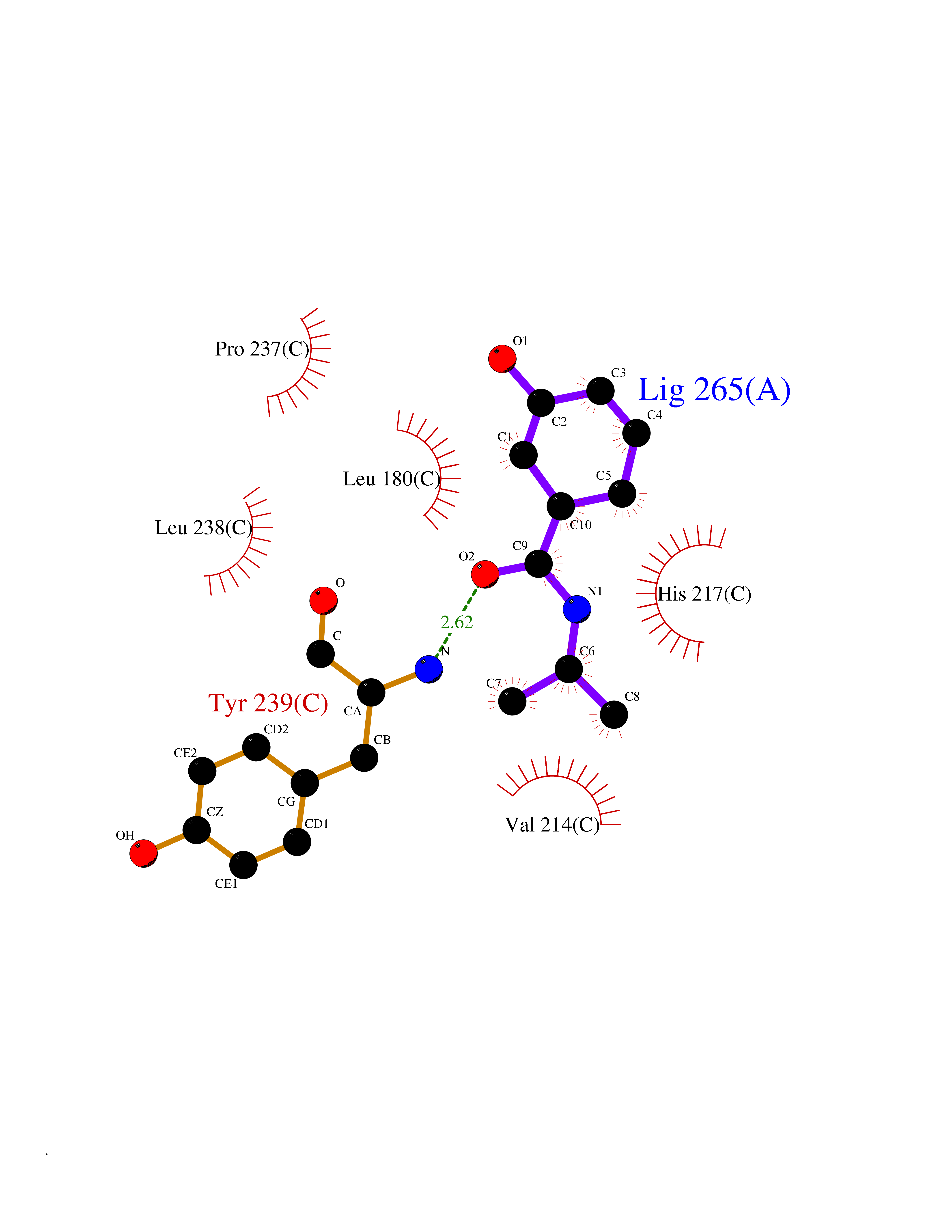 Ligplot Map 3D Binding mode Sequence MPKWRKTHLTYRIVNYTPDLPRDAVDSAIEKALKVWEEVTPLTFSRLYEGEADIMISFAVKEHGDNYSFDGPGHSLAHAYPPGPGLYGDIHFDDDEKWTEDASGTNLFLVAAHELGHSLGLFHSANTEALMYPLYNSLAQFRLSQDDVNGIQSLYGPKWRKTHLTYRIVNYTPDLPRDAVDSAIEKALKVWEEVTPLTFSRLYEGEADIMISFAVKEHGDNYSFDGPGHSLAHAYPPGPGLYGDIHFDDDEKWTEDASGTNLFLVAAHELGHSLGLFHSANTEALMYPLYNSLAQFRLSQDDVNGIQSLYGGMPKWRKTHLTYRIVNYTPDLPRDAVDSAIEKALKVWEEVTPLTFSRLYEGEADIMISFAVKEHGDNYSFDGPGHSLAHAYPPGPGLYGDIHFDDDEKWTEDASGTNLFLVAAHELGHSLGLFHSANTEALMYPLYNSFTELAQFRLSQDDVNGIQSLYG Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Tyrosine-protein kinase Kit (KIT) | 1T46 | 5.67 | |
Target general information Gen name KIT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog; p145 c-kit; Proto-oncogene tyrosine-protein kinase Kit; Proto-oncogene c-Kit; Piebald trait protein; PBT; Mast/stem cell growth factor re Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function In response to KITLG/SCF binding, KIT can activate several signaling pathways. Phosphorylates PIK3R1, PLCG1, SH2B2/APS and CBL. Activates the AKT1 signaling pathway by phosphorylation of PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase. Activated KIT also transmits signals via GRB2 and activation of RAS, RAF1 and the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1. Promotes activation of STAT family members STAT1, STAT3, STAT5A and STAT5B. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate. KIT signaling is modulated by protein phosphatases, and by rapid internalization and degradation of the receptor. Activated KIT promotes phosphorylation of the protein phosphatases PTPN6/SHP-1 and PTPRU, and of the transcription factors STAT1, STAT3, STAT5A and STAT5B. Promotes phosphorylation of PIK3R1, CBL, CRK (isoform Crk-II), LYN, MAPK1/ERK2 and/or MAPK3/ERK1, PLCG1, SRC and SHC1. Tyrosine-protein kinase that acts as cell-surface receptor for the cytokine KITLG/SCF and plays an essential role in the regulation of cell survival and proliferation, hematopoiesis, stem cell maintenance, gametogenesis, mast cell development, migration and function, and in melanogenesis. Related diseases Piebald trait (PBT) [MIM:172800]: Autosomal dominant genetic developmental abnormality of pigmentation characterized by congenital patches of white skin and hair that lack melanocytes. {ECO:0000269|PubMed:11074500, ECO:0000269|PubMed:1370874, ECO:0000269|PubMed:1376329, ECO:0000269|PubMed:1717985, ECO:0000269|PubMed:7687267, ECO:0000269|PubMed:8680409, ECO:0000269|PubMed:9450866, ECO:0000269|PubMed:9699740}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Gastrointestinal stromal tumor (GIST) [MIM:606764]: Common mesenchymal neoplasms arising in the gastrointestinal tract, most often in the stomach. They are histologically, immunohistochemically, and genetically different from typical leiomyomas, leiomyosarcomas, and schwannomas. Most GISTs are composed of a fairly uniform population of spindle-shaped cells. Some tumors are dominated by epithelioid cells or contain a mixture of spindle and epithelioid morphologies. Primary GISTs in the gastrointestinal tract commonly metastasize in the omentum and mesenteries, often as multiple nodules. However, primary tumors may also occur outside of the gastrointestinal tract, in other intra-abdominal locations, especially in the omentum and mesentery. {ECO:0000269|PubMed:11505412, ECO:0000269|PubMed:15824741, ECO:0000269|PubMed:9438854, ECO:0000269|PubMed:9697690}. The gene represented in this entry is involved in disease pathogenesis.; DISEASE: Testicular germ cell tumor (TGCT) [MIM:273300]: A common malignancy in males representing 95% of all testicular neoplasms. TGCTs have various pathologic subtypes including: unclassified intratubular germ cell neoplasia, seminoma (including cases with syncytiotrophoblastic cells), spermatocytic seminoma, embryonal carcinoma, yolk sac tumor, choriocarcinoma, and teratoma. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Leukemia, acute myelogenous (AML) [MIM:601626]: A subtype of acute leukemia, a cancer of the white blood cells. AML is a malignant disease of bone marrow characterized by maturational arrest of hematopoietic precursors at an early stage of development. Clonal expansion of myeloid blasts occurs in bone marrow, blood, and other tissue. Myelogenous leukemias develop from changes in cells that normally produce neutrophils, basophils, eosinophils and monocytes. The gene represented in this entry is involved in disease pathogenesis. Somatic mutations that lead to constitutive activation of KIT are detected in AML patients. These mutations fall into two classes, the most common being in-frame internal tandem duplications of variable length in the juxtamembrane region that disrupt the normal regulation of the kinase activity. Likewise, point mutations in the kinase domain can result in a constitutively activated kinase.; DISEASE: Mastocytosis, cutaneous (MASTC) [MIM:154800]: A form of mastocytosis, a heterogeneous group of disorders associated with abnormal proliferation and accumulation of mast cells in various tissues, especially in the skin and hematopoietic organs. MASTC is an autosomal dominant form characterized by macules, papules, nodules, or diffuse infiltration of the skin, often associated with localized hyperpigmentation. Gentle rubbing of the lesions induces histamine release from mechanically activated mast cells, causing local wheals, erythema, and often pruritus, a phenomenon termed Darier sign. {ECO:0000269|PubMed:15173254, ECO:0000269|PubMed:19865100, ECO:0000269|PubMed:21689725, ECO:0000269|PubMed:24289326, ECO:0000269|PubMed:9990072}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Mastocytosis, systemic (MASTSYS) [MIM:154800]: A severe form of mastocytosis characterized by abnormal proliferation and accumulation of mast cells in several organs, resulting in a systemic disease that may affect bone, gastrointestinal tract, lymphatics, spleen, and liver. In some cases, it is associated with a clonal hematologic non-mast-cell lineage disease, such as a myelodysplastic or myeloproliferative disorder. It can also lead to mast cell leukemia, which carries a high risk of mortality. {ECO:0000269|PubMed:9990072}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12742; DB09103; DB15233; DB01254; DB12147; DB12010; DB00619; DB09078; DB06080; DB06595; DB04868; DB05913; DB06589; DB12978; DB01962; DB08901; DB08896; DB14840; DB00398; DB01268; DB11800; DB05146 Interacts with P00519; P42684; O75815; P51451; Q8WV28; P46108; P07332; P09769; O75791; P62993; Q14451; P08631; Q96JZ2; P21583; P06239; P07948; P16333; O43639; P27986; O00459; Q92569; P19174; P16885; Q13882; Q06124; Q92729; P20936; Q9UQQ2; O14796; Q9NP31; Q8N5H7; P78314; Q15464; P29353; P98077; Q92529; Q9H6Q3; O14508; O14543; O14544; P12931; Q9ULZ2; Q9HBL0; Q63HR2; Q68CZ2; P42681; P07947; P43403; Q8VBX6; P35235 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cytoplasm; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Immunoglobulin domain; Kinase; Magnesium; Membrane; Metal-binding; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 33575.6 Length 297 Aromaticity 0.11 Instability index 45.37 Isoelectric point 8.37 Charge (pH=7) 3.24 2D Binding mode Binding energy (Kcal/mol) -7.73  Molscript Map 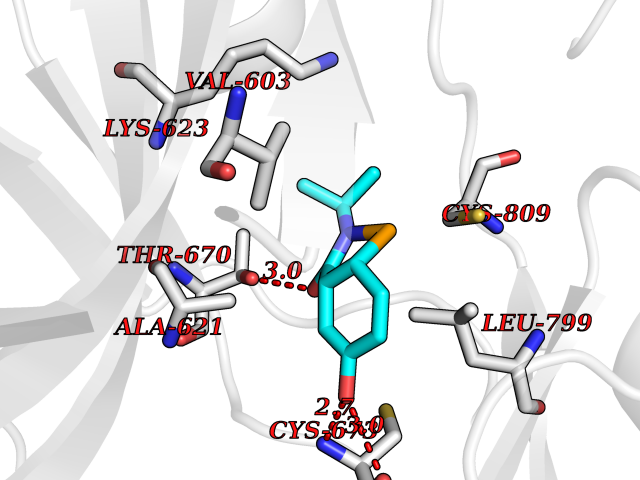 Pymol Map 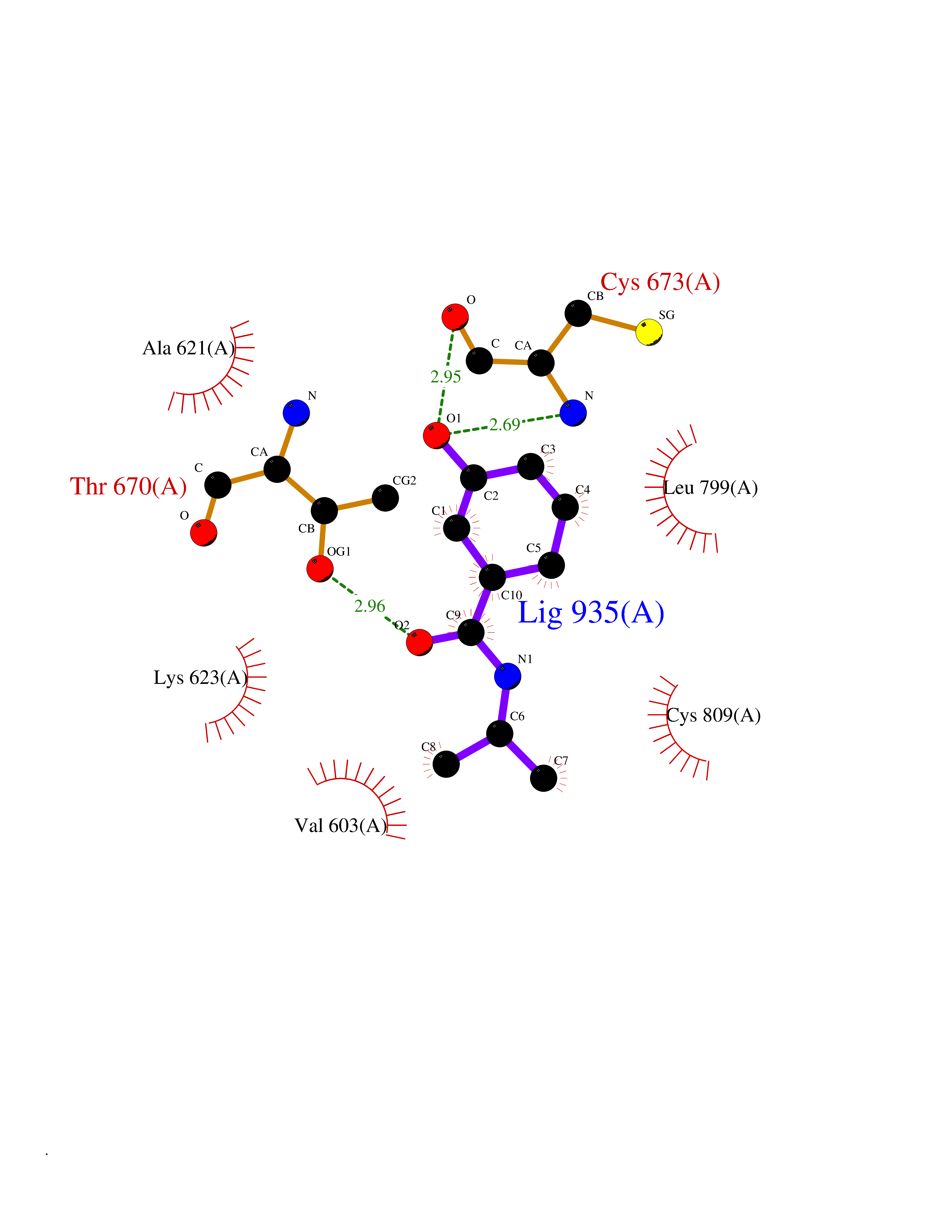 Ligplot Map 3D Binding mode Sequence GNNYVYIDPTQLPYDHKWEFPRNRLSFGKTLGAGAFGKVVEATAYGLIKSDAAMTVAVKMLKPSAHLTEREALMSELKVLSYLGNHMNIVNLLGACTIGGPTLVITEYCCYGDLLNFLRRKRDSFLALDLEDLLSFSYQVAKGMAFLASKNCIHRDLAARNILLTHGRITKICDFGLARDIKNDSNYVVKGNARLPVKWMAPESIFNCVYTFESDVWSYGIFLWELFSLGSSPYPGMPVDSKFYKMIKEGFRMLSPEHAPAEMYDIMKTCWDADPLKRPTFKQIVQLIEKQISESTN Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Cytochrome P450 1A2 | 2HI4 | 5.67 | |
Target general information Gen name CYP1A2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Cytochrome P450 family Biochemical class Oxidoreductase Function Aromatase activity.Caffeine oxidase activity.Demethylase activity.Electron carrier activity.Enzyme binding.Heme binding.Iron ion binding.Monooxygenase activity.Oxidoreductase activity.Oxidoreductase activity, acting on paired donors, with incorporation or reduction of molecular oxygen, reduced flavin or flavoprotein as one donor, and incorporation of one atom of oxygen.Oxygen binding. Related diseases Myeloperoxidase deficiency (MPOD) [MIM:254600]: A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. {ECO:0000269|PubMed:37198333, ECO:0000269|PubMed:7904599, ECO:0000269|PubMed:8142659, ECO:0000269|PubMed:8621627, ECO:0000269|PubMed:9354683, ECO:0000269|PubMed:9637725}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08496; DB01667; DB14132; DB04356; DB02489; DB11932; DB12001; DB05812; DB13573; DB01418; DB00316; DB15568; DB06594; DB00518; DB05396; DB00969; DB07453; DB01424; DB01223; DB01118; DB00321; DB00261; DB01217; DB01435; DB06605; DB05676; DB06413; DB06216; DB01072; DB15011; DB06442; DB06626; DB00993; DB00972; DB13203; DB05015; DB16703; DB06769; DB01086; DB06770; DB06771; DB06732; DB00195; DB04889; DB11967; DB13975; DB00188; DB12151; DB01558; DB14018; DB13812; DB00201; DB09061; DB14737; DB11791; DB06774; DB00564; DB06016; DB01136; DB12814; DB00477; DB00356; DB01166; DB00501; DB01012; DB00568; DB00827; DB00537; DB00215; DB12499; DB14025; DB00349; DB01242; DB00575; DB00758; DB00363; DB00286; DB11672; DB14635; DB00924; DB08912; DB00851; DB06292; DB01254; DB01609; DB01151; DB16650; DB12161; DB01191; DB00633; DB11994; DB00586; DB11511; DB12945; DB00280; DB01184; DB09167; DB05928; DB01142; DB09273; DB00470; DB00476; DB00625; DB15444; DB06210; DB13874; DB11718; DB00467; DB11404; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB04574; DB13592; DB00330; DB00898; DB00977; DB00773; DB01628; DB00927; DB04854; DB01482; DB00574; DB12265; DB15669; DB01195; DB08972; DB04841; DB00544; DB00472; DB00499; DB00176; DB01320; DB00998; DB14029; DB06160; DB01044; DB01241; DB01155; DB01645; DB01381; DB00986; DB00365; DB00400; DB05708; DB00629; DB00502; DB01094; DB14999; DB04076; DB11737; DB00619; DB00458; DB11564; DB01306; DB09456; DB09564; DB01307; DB00047; DB01309; DB00030; DB00046; DB11567; DB00071; DB11568; DB05258; DB00034; DB00105; DB15131; DB00011; DB00018; DB00069; DB00060; DB00068; DB00033; DB00951; DB11757; DB09570; DB01026; DB01097; DB16217; DB09078; DB01002; DB05667; DB00281; DB12406; DB09198; DB04948; DB00978; DB06448; DB16220; DB01601; DB00455; DB04871; DB06077; DB01283; DB00772; DB00934; DB06234; DB14009; DB00784; DB01065; DB00170; DB00454; DB00532; DB00333; DB00763; DB00553; DB01028; DB09241; DB01233; DB00379; DB06148; DB01388; DB06595; DB00370; DB16236; DB00745; DB11763; DB00218; DB06510; DB14011; DB00461; DB00607; DB00779; DB00788; DB06600; DB00238; DB06803; DB00184; DB01115; DB11793; DB00435; DB05115; DB00717; DB01059; DB00540; DB05990; DB01165; DB00334; DB16267; DB00338; DB00904; DB11632; DB11443; DB01173; DB11837; DB09330; DB01303; DB11697; DB00377; DB00715; DB06589; DB11774; DB00487; DB00008; DB00022; DB09122; DB13634; DB00806; DB11198; DB08883; DB00850; DB03783; DB01174; DB00388; DB00252; DB11450; DB01100; DB13823; DB04951; DB17472; DB11642; DB08910; DB15822; DB01058; DB01087; DB00794; DB00420; DB09288; DB01182; DB06479; DB00818; DB00571; DB13449; DB11892; DB04216; DB00908; DB00468; DB01129; DB00980; DB09290; DB00863; DB01367; DB00409; DB02709; DB13174; DB01045; DB11753; DB00740; DB14924; DB00503; DB00533; DB01656; DB15119; DB00268; DB00296; DB00412; DB00817; DB12332; DB13772; DB06654; DB11491; DB00418; DB01037; DB11689; DB06290; DB13261; DB15093; DB00052; DB00398; DB01208; DB09118; DB00428; DB06820; DB00382; DB00675; DB06083; DB09071; DB05488; DB09256; DB01079; DB01405; DB00857; DB08880; DB11712; DB01412; DB00277; DB00730; DB01623; DB00208; DB06137; DB00697; DB01056; DB06264; DB00752; DB00384; DB12245; DB00831; DB15442; DB00440; DB00685; DB08867; DB14989; DB13609; DB06235; DB00313; DB08881; DB00661; DB09185; DB12026; DB00682; DB02134; DB00549; DB00744; DB00315; DB00425; DB09225; DB09120 Interacts with O95870 EC number 1.14.14.1; 4.2.1.152 Uniprot keywords 3D-structure; Direct protein sequencing; Endoplasmic reticulum; Fatty acid metabolism; Glycoprotein; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Proteomics identification; Reference proteome; Steroid metabolism; Sterol metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 54475 Length 480 Aromaticity 0.1 Instability index 40.43 Isoelectric point 9.16 Charge (pH=7) 9.89 2D Binding mode Binding energy (Kcal/mol) -7.74 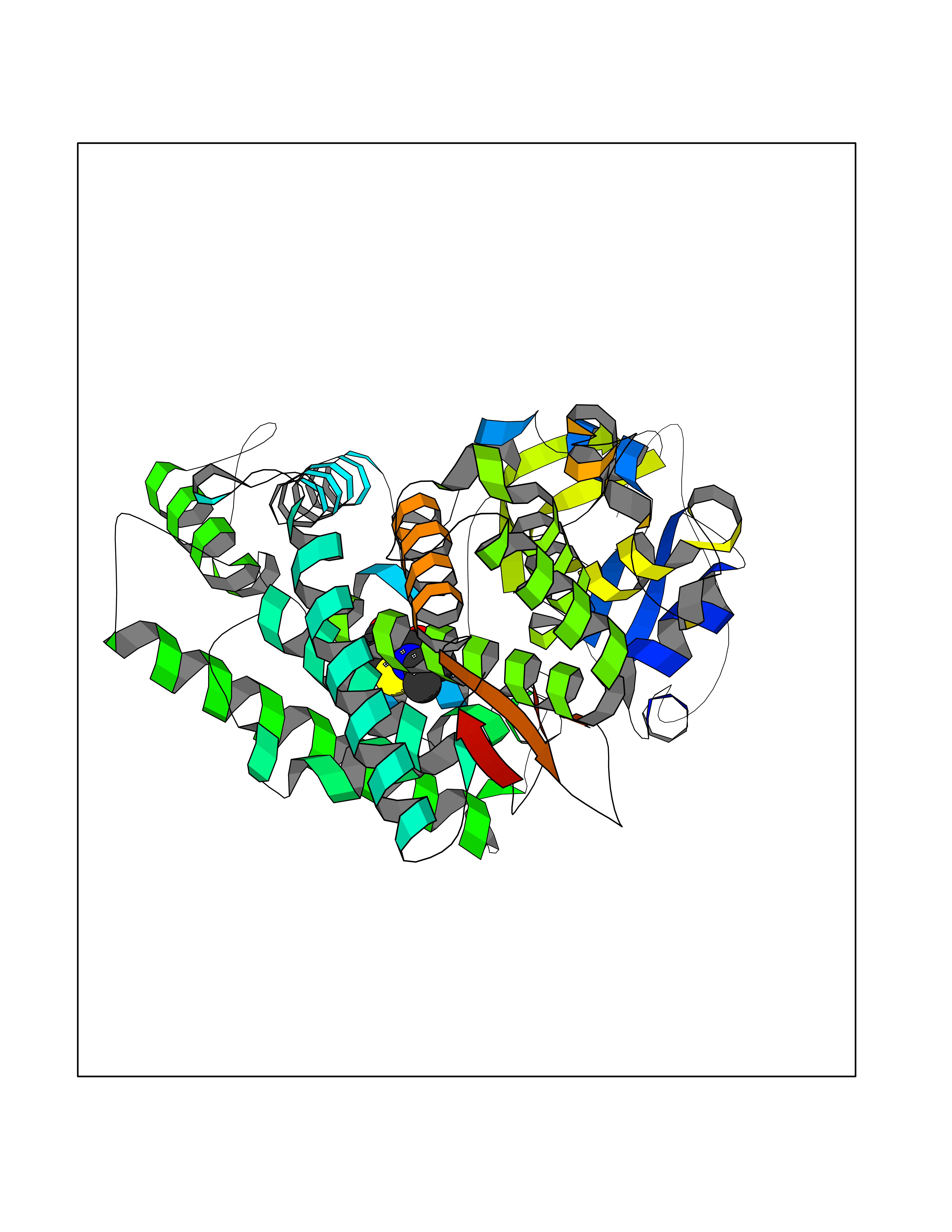 Molscript Map 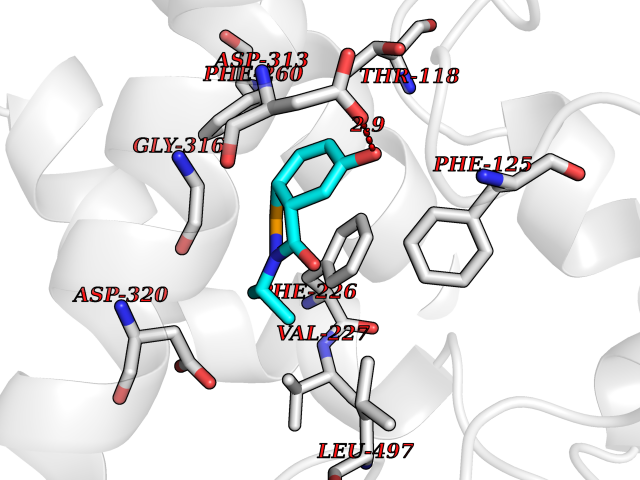 Pymol Map 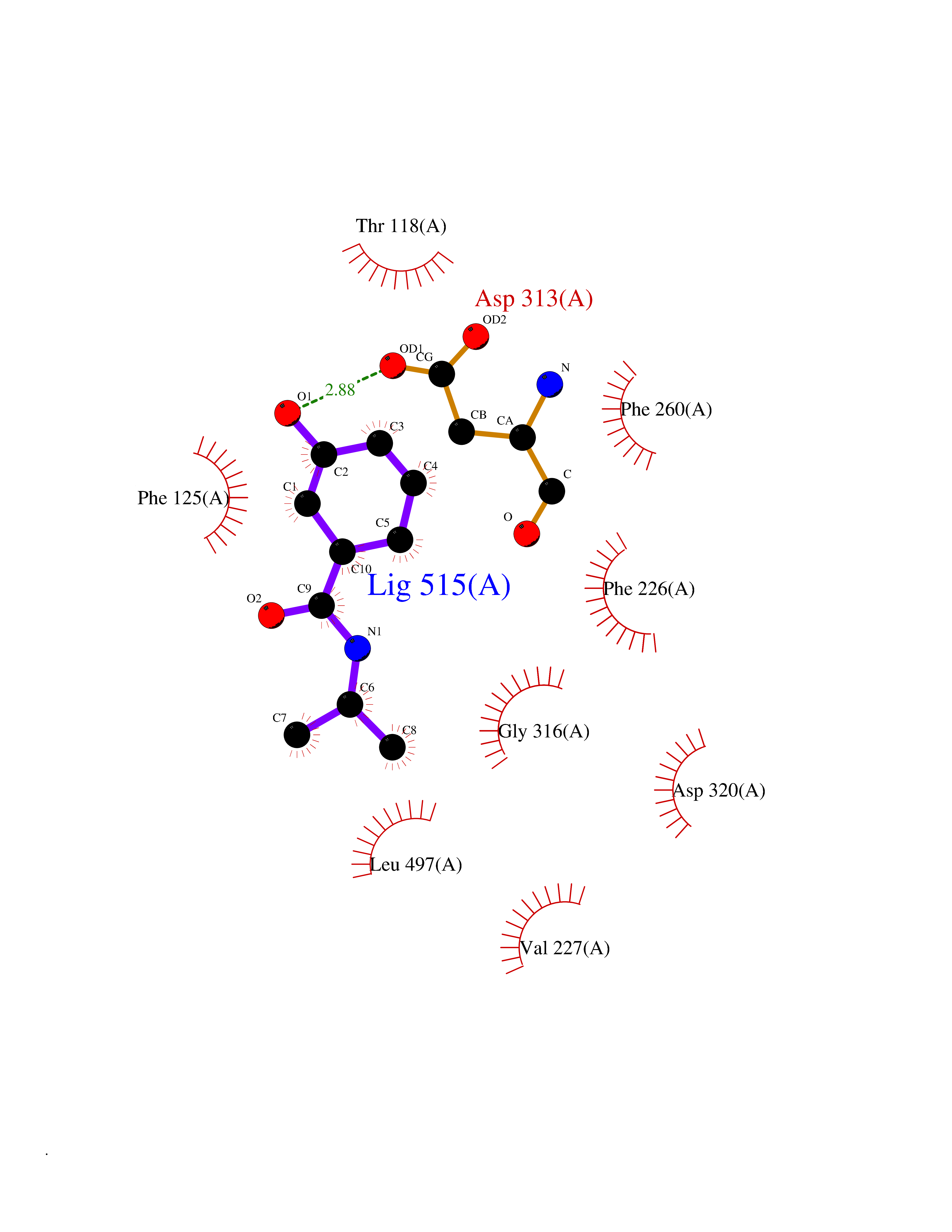 Ligplot Map 3D Binding mode Sequence RVPKGLKSPPEPWGWPLLGHVLTLGKNPHLALSRMSQRYGDVLQIRIGSTPVLVLSRLDTIRQALVRQGDDFKGRPDLYTSTLITDGQSLTFSTDSGPVWAARRRLAQNALNTFSIASDPASSSSCYLEEHVSKEAKALISRLQELMAGPGHFDPYNQVVVSVANVIGAMCFGQHFPESSDEMLSLVKNTHEFVETASSGNPLDFFPILRYLPNPALQRFKAFNQRFLWFLQKTVQEHYQDFDKNSVRDITGALFKHSKKGPRASGNLIPQEKIVNLVNDIFGAGFDTVTTAISWSLMYLVTKPEIQRKIQKELDTVIGRERRPRLSDRPQLPYLEAFILETFRHSSFLPFTIPHSTTRDTTLNGFYIPKKCCVFVNQWQVNHDPELWEDPSEFRPERFLTADGTAINKPLSEKMMLFGMGKRRCIGEVLAKWEIFLFLAILLQQLEFSVPPGVKVDLTPIYGLTMKHARCEHVQARRFS Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Plasmepsin-2 | 2BJU | 5.67 | |
Target general information Gen name N/A Organism Plasmodium falciparum (isolate HB3) Uniprot ID TTD ID NA Synonyms NA Protein family Peptidase A1 family Biochemical class Hydrolase Function Aspartic-type endopeptidase activity. Related diseases Short/branched-chain acyl-CoA dehydrogenase deficiency (SBCADD) [MIM:610006]: Autosomal recessive disorder and consists of a defect in catabolism of L-isoleucine which is characterized by an increase of 2-methylbutyrylglycine and 2-methylbutyrylcarnitine in blood and urine. Affected individuals have seizures and psychomotor delay as the main clinical features. {ECO:0000269|PubMed:10832746, ECO:0000269|PubMed:11013134, ECO:0000269|PubMed:16317551}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04378; DB04373; DB11638; DB01218; DB02505; DB03063 Interacts with NA EC number 3.4.23.39 Uniprot keywords 3D-structure; Aspartyl protease; Direct protein sequencing; Disulfide bond; Hydrolase; Membrane; Protease; Reference proteome; Signal-anchor; Transmembrane; Transmembrane helix; Vacuole; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 36923.5 Length 329 Aromaticity 0.13 Instability index 44.31 Isoelectric point 4.67 Charge (pH=7) -17.94 2D Binding mode Binding energy (Kcal/mol) -7.74 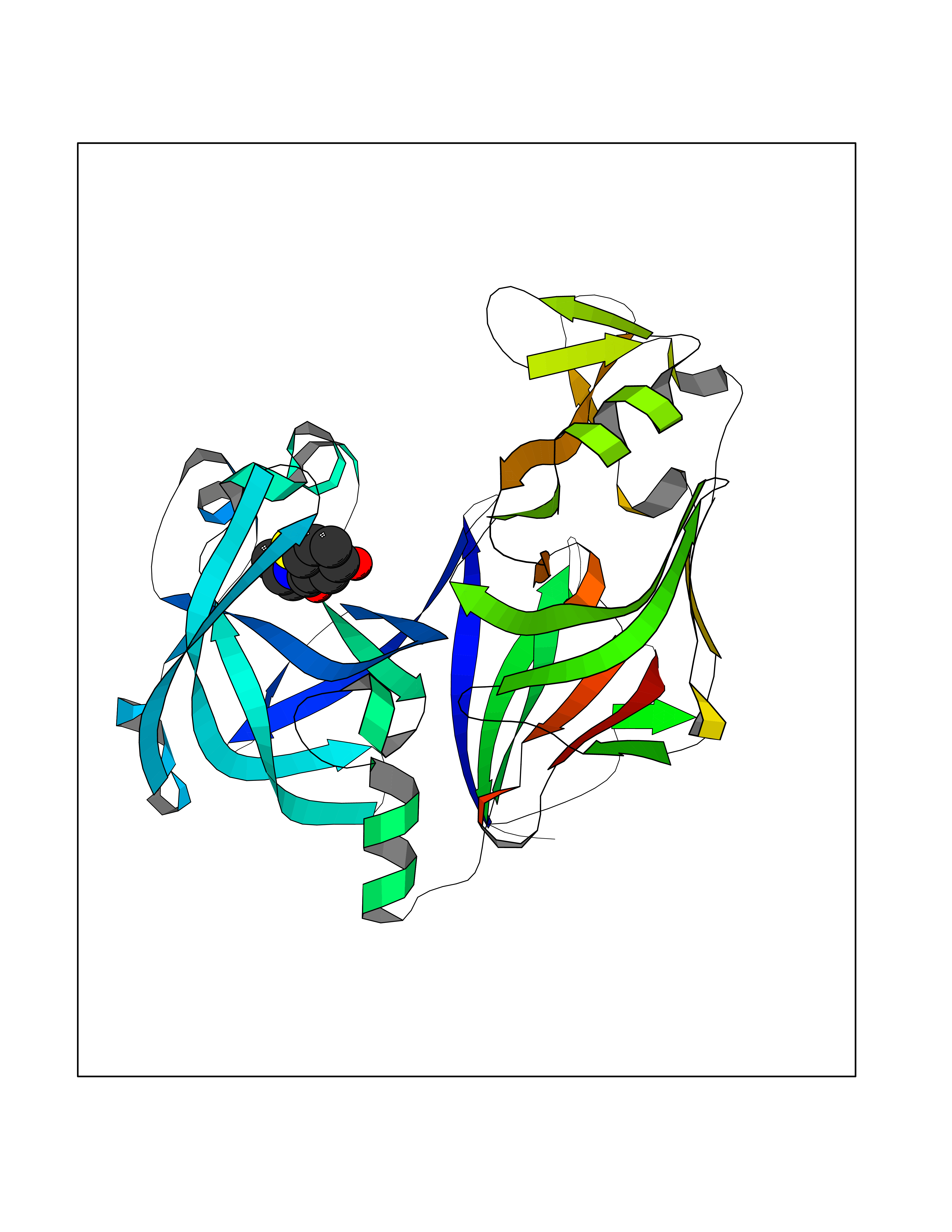 Molscript Map 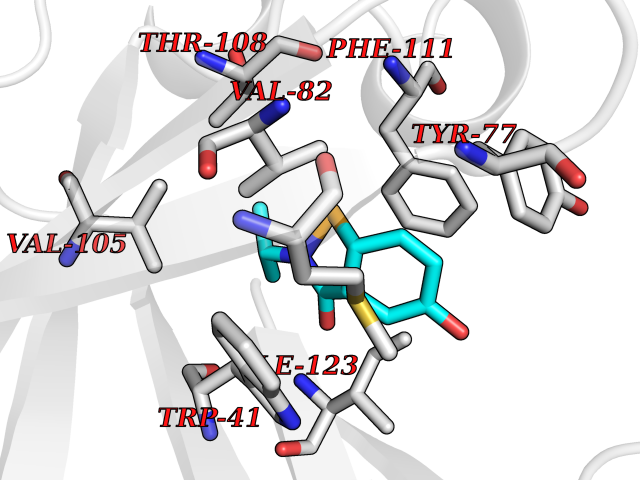 Pymol Map 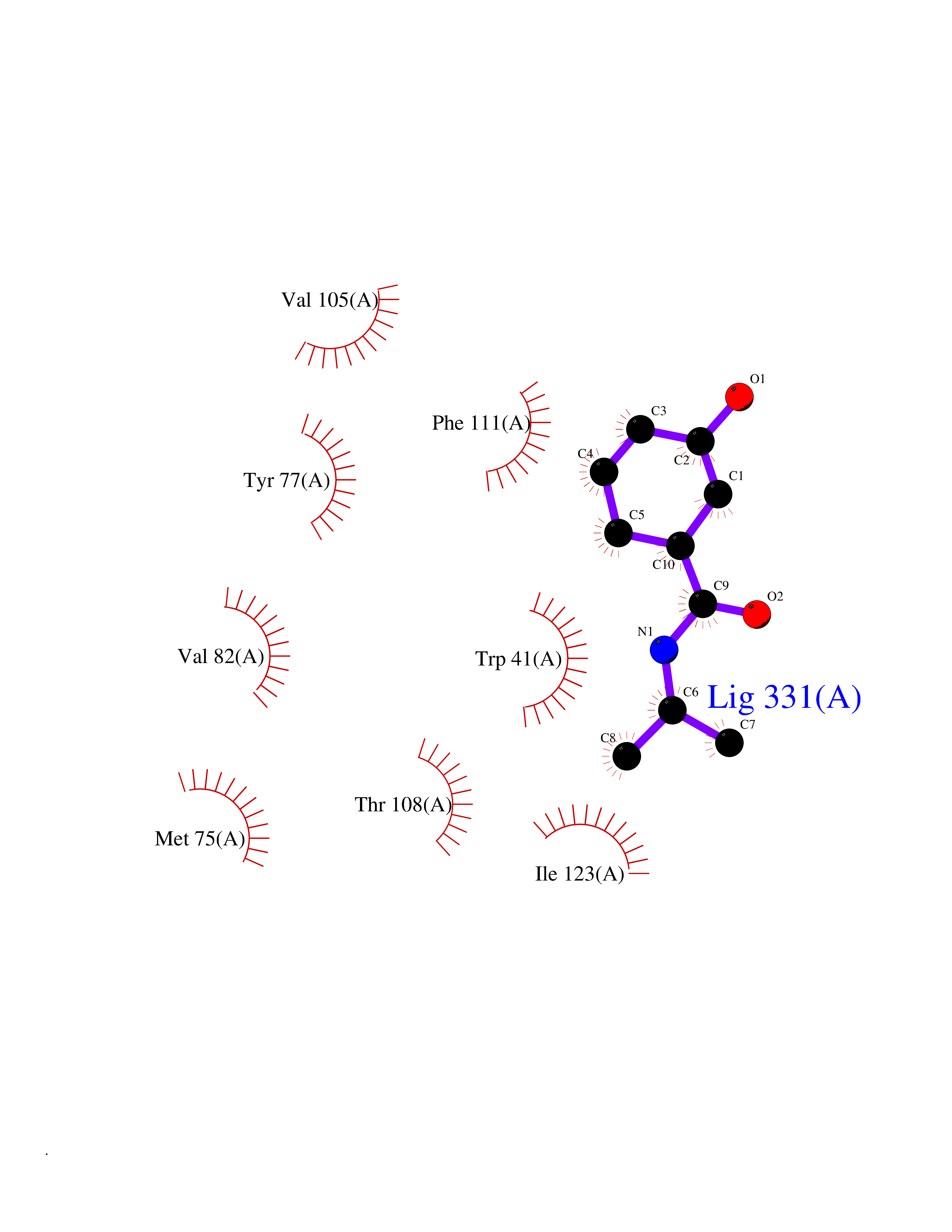 Ligplot Map 3D Binding mode Sequence SSNDNIELVDFQNIMFYGDAEVGDNQQPFTFILDTGSANLWVPSVKCTTAGCLTKHLYDSSKSRTYEKDGTKVEMNYVSGTVSGFFSKDLVTVGNLSLPYKFIEVIDTNGFEPTYTASTFDGILGLGWKDLSIGSVDPIVVELKNQNKIENALFTFYLPVHDKHTGFLTIGGIEERFYEGPLTYEKLNHDLYWQITLDAHVGNIMLEKANCIVDSGTSAITVPTDFLNKMLQNLDVIKVPFLPFYVTLCNNSKLPTFEFTSENGKYTLEPEYYLQHIEDVGPGLCMLNIIGLDFPVPTFILGDPFMRKYFTVFDYDNHSVGIALAKKNL Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Plasmodium Dihydroorotate dehydrogenase (Malaria DHOdehase) | 1TV5 | 5.67 | |
Target general information Gen name Malaria DHOdehase Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID Synonyms PFF0160c; Mitochondrially bound dihydroorotate-ubiqui oxidoreductase; Dihydroorotate oxidase of Plasmodium falciparum; Dihydroorotate dehydrogenase of Plasmodium falciparum; DHOdehase of Plasmodium fa Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class CH-CH donor oxidoreductase Function Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number EC 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 41846.8 Length 371 Aromaticity 0.1 Instability index 37.25 Isoelectric point 8.21 Charge (pH=7) 3.13 2D Binding mode Binding energy (Kcal/mol) -7.73  Molscript Map 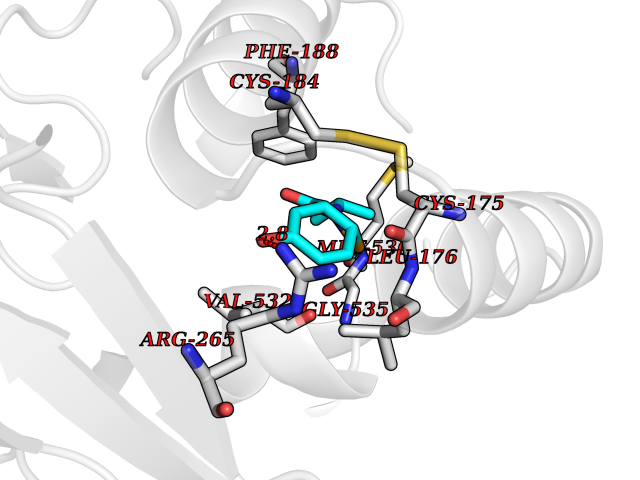 Pymol Map 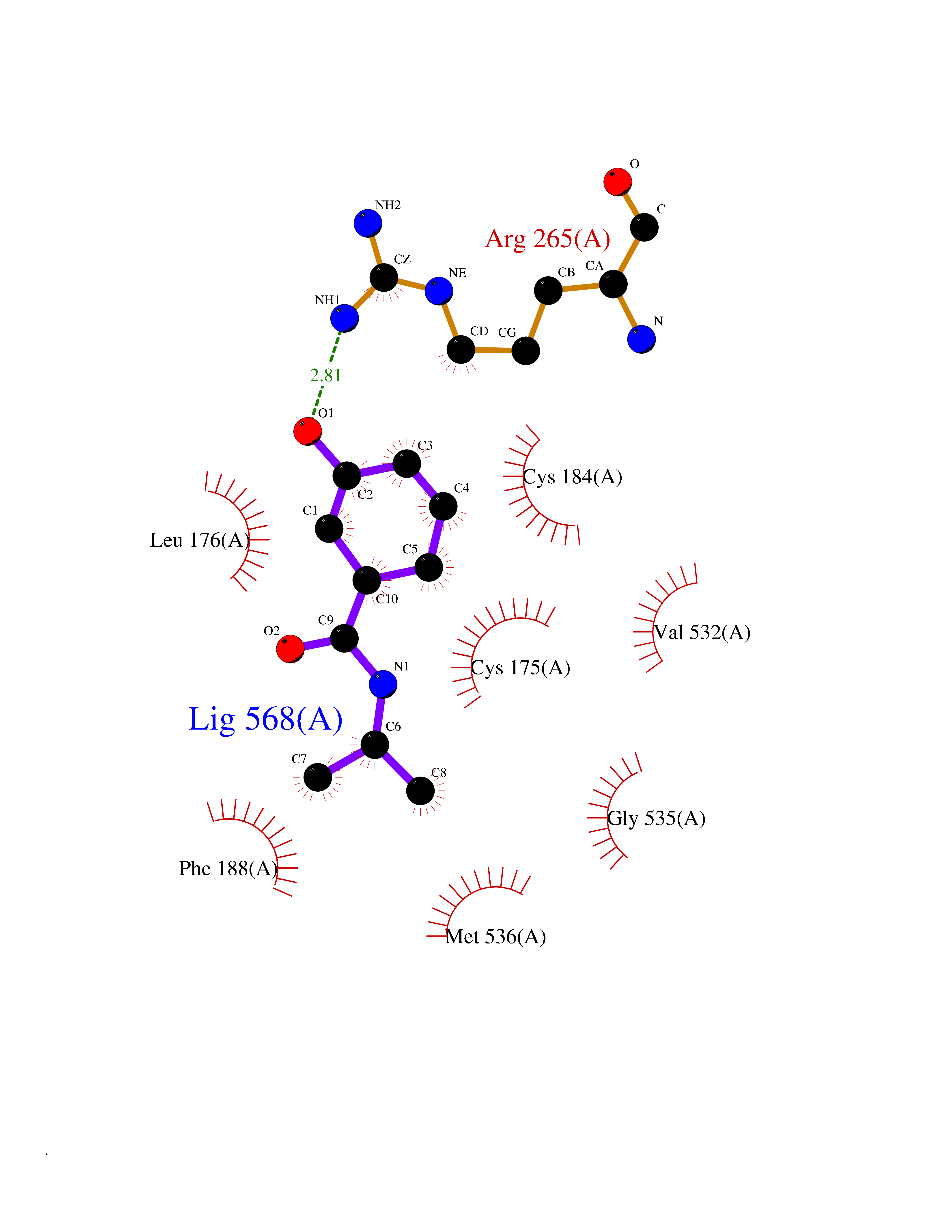 Ligplot Map 3D Binding mode Sequence FESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKH Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Metabotropic glutamate receptor 3 (mGluR3) | 4XAR | 5.67 | |
Target general information Gen name GRM3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms mGLUR3; Group III metabotropic glutamate receptor; GPRC1C Protein family G-protein coupled receptor 3 family Biochemical class GPCR glutamate Function Ligand binding causes a conformation change that triggers signaling via guanine nucleotide-binding proteins (G proteins) and modulates the activity of down-stream effectors. Signaling inhibits adenylate cyclase activity. G-protein coupled receptor for glutamate. Related diseases Paramyotonia congenita (PMC) [MIM:168300]: An autosomal dominant channelopathy characterized by myotonia, increased by exposure to cold, intermittent flaccid paresis, not necessarily dependent on cold or myotonia, lability of serum potassium, non-progressive nature and lack of atrophy or hypertrophy of muscles. In some patients, myotonia is not increased by cold exposure (paramyotonia without cold paralysis). Patients may have a combination phenotype of PMC and HYPP. {ECO:0000269|PubMed:10369308, ECO:0000269|PubMed:10727489, ECO:0000269|PubMed:1310898, ECO:0000269|PubMed:1316765, ECO:0000269|PubMed:1338909, ECO:0000269|PubMed:15318338, ECO:0000269|PubMed:15790667, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:18166706, ECO:0000269|PubMed:18690054, ECO:0000269|PubMed:19077043, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:8242056, ECO:0000269|PubMed:8308722, ECO:0000269|PubMed:8388676, ECO:0000269|PubMed:8580427}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hypokalemic 2 (HOKPP2) [MIM:613345]: An autosomal dominant disorder manifested by episodic flaccid generalized muscle weakness associated with falls of serum potassium levels. {ECO:0000269|PubMed:10599760, ECO:0000269|PubMed:10851391, ECO:0000269|PubMed:10944223, ECO:0000269|PubMed:11558801, ECO:0000269|PubMed:11591859, ECO:0000269|PubMed:16890191, ECO:0000269|PubMed:17898326, ECO:0000269|PubMed:18162704, ECO:0000269|PubMed:19118277, ECO:0000269|PubMed:20522878, ECO:0000269|PubMed:21043388, ECO:0000269|PubMed:24549961}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis hyperkalemic (HYPP) [MIM:170500]: An autosomal dominant channelopathy characterized by episodic flaccid generalized muscle weakness associated with high levels of serum potassium. Concurrence of myotonia is found in HYPP patients. {ECO:0000269|PubMed:1659668, ECO:0000269|PubMed:1659948, ECO:0000269|PubMed:20076800}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Periodic paralysis normokalemic (NKPP) [MIM:170500]: A disorder closely related to hyperkalemic periodic paralysis, but marked by a lack of alterations in potassium levels during attacks of muscle weakness. {ECO:0000269|PubMed:15596759, ECO:0000269|PubMed:18046642, ECO:0000269|PubMed:20522878}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myotonia SCN4A-related (MYOSCN4A) [MIM:608390]: A phenotypically highly variable myotonia aggravated by potassium loading, and sometimes by cold. Myotonia is characterized by sustained muscle tensing that prevents muscles from relaxing normally. It causes muscle stiffness that can interfere with movement. In some people the stiffness is very mild, while in other cases it may be severe enough to interfere with walking, running, and other activities of daily life. Myotonia SCN4A-related includes myotonia permanens and myotonia fluctuans. In myotonia permanens, the myotonia is generalized and there is a hypertrophy of the muscle, particularly in the neck and the shoulder. Attacks of severe muscle stiffness of the thoracic muscles may be life threatening due to impaired ventilation. In myotonia fluctuans, the muscle stiffness may fluctuate from day to day, provoked by exercise. {ECO:0000269|PubMed:10218481, ECO:0000269|PubMed:16786525, ECO:0000269|PubMed:16832098, ECO:0000269|PubMed:17212350, ECO:0000269|PubMed:17998485, ECO:0000269|PubMed:18203179, ECO:0000269|PubMed:18337100, ECO:0000269|PubMed:19015483, ECO:0000269|PubMed:19347921, ECO:0000269|PubMed:20076800, ECO:0000269|PubMed:27653901, ECO:0000269|PubMed:8058156, ECO:0000269|PubMed:9392583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 16 (CMS16) [MIM:614198]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS16 is characterized by fatigable generalized weakness and recurrent attacks of respiratory and bulbar paralysis since birth. The fatigable weakness involves lid-elevator, external ocular, facial, limb and truncal muscles and an decremental response of the compound muscle action potential on repetitive stimulation. {ECO:0000269|PubMed:12766226, ECO:0000269|PubMed:25707578, ECO:0000269|PubMed:26659129}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22A, classic (CMYO22A) [MIM:620351]: A form of congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22A is an autosomal recessive form characterized by fetal hypokinesia, polyhydramnios, and severe neonatal hypotonia associated with respiratory insufficiency. Affected individuals who survive the neonatal period have delayed motor development, difficulty walking, proximal muscle weakness of the upper and lower limbs, facial and neck muscle weakness, easy fatigability, and mild limb contractures or foot deformities. {ECO:0000269|PubMed:26700687, ECO:0000269|PubMed:28262468, ECO:0000269|PubMed:36090556}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Congenital myopathy 22B, severe fetal (CMYO22B) [MIM:620369]: A severe congenital myopathy, a clinically and genetically heterogeneous group of muscle disorders characterized by hypotonia and muscle weakness apparent at birth, and specific pathological features on muscle biopsy. CMYO22B is an autosomal recessive form characterized by onset in utero. Affected individuals show fetal akinesia, and develop fetal hydrops with pulmonary hypoplasia, severe joint contractures, and generalized muscle hypoplasia. Death occurs in utero or soon after birth. {ECO:0000269|PubMed:26700687}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05096 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Proteomics identification; Receptor; Reference proteome; Signal; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 50355.5 Length 445 Aromaticity 0.11 Instability index 38.26 Isoelectric point 6.52 Charge (pH=7) -1.53 2D Binding mode Binding energy (Kcal/mol) -7.73  Molscript Map 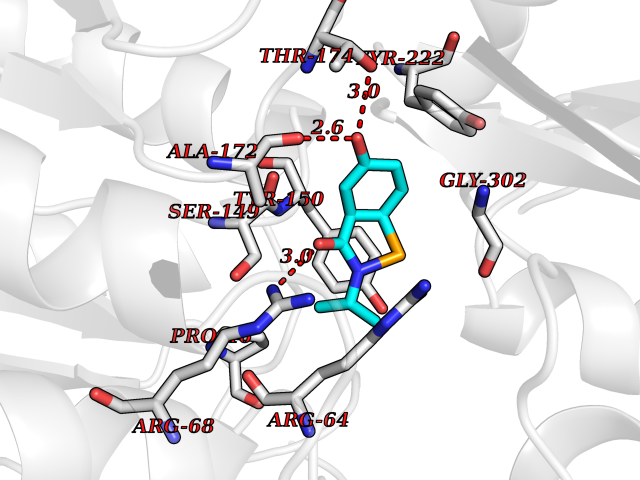 Pymol Map 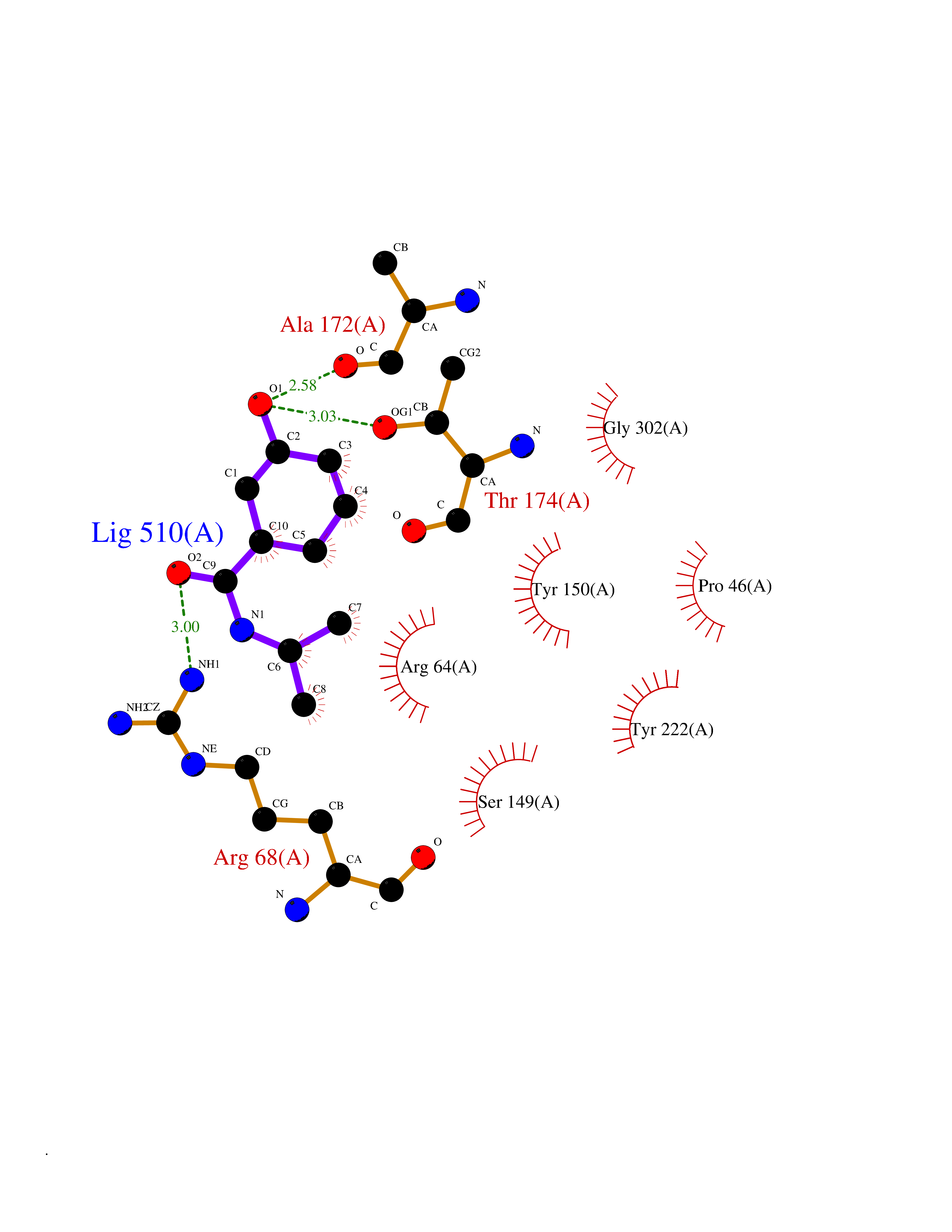 Ligplot Map 3D Binding mode Sequence RREIKIEGDLVLGGLFPINEKGTGTEECGRINEDRGIQRLEAMLFAIDEINKDDYLLPGVKLGVHILDTCSRDTYALEQSLEFVRASLLLIAGVIGGSYSSVSIQVANLLRLFQIPQISYASTSAKLSDKSRYDYFARTVPPDFYQAKAMAEILRFFNWTYVSTVASEGDYGETGIEAFEQEARLRNISIATAEKVGRSNIRKSYDSVIRELLQKPNARVVVLFMRSDDSRELIAAASRANASFTWVASDGWGAQESIIKGSEHVAYGAITLELASQPVRQFDRYFQSLNPYNNHRNPWFRDFWEQKFQCSLRVCDKHLAIDSSNYEQESKIMFVVNAVYAMAHALHKMQRTLCPNTTKLCDAMKILDGKKLYKDYLLKINFTAPDADSIVKFDTFGDGMGRYNVFNFQNVGGKYSYLKVGHWAETLSLDVNSIHWSRNSVPTSE Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Caspase-6 (CASP6) | 4NBL | 5.67 | |
Target general information Gen name CASP6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MCH2; Caspase-6 subunit p18; Caspase-6 subunit p11; CASP-6; Apoptotic protease Mch-2 Protein family Peptidase C14A family Biochemical class Peptidase Function Cleaves poly(ADP-ribose) polymerase in vitro, as well as lamins. Overexpression promotes programmed cell death. Involved in the activation cascade of caspases responsible for apoptosis execution. Related diseases Growth hormone deficiency, isolated, 1A (IGHD1A) [MIM:262400]: An autosomal recessive, severe deficiency of growth hormone leading to dwarfism. Patients often develop antibodies to administered growth hormone. {ECO:0000269|PubMed:8364549}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 1B (IGHD1B) [MIM:612781]: An autosomal recessive deficiency of growth hormone leading to short stature. Patients have low but detectable levels of growth hormone, significantly retarded bone age, and a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:12655557}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Kowarski syndrome (KWKS) [MIM:262650]: A syndrome clinically characterized by short stature associated with bioinactive growth hormone, normal or slightly increased growth hormone secretion, pathologically low insulin-like growth factor 1 levels, and normal catch-up growth on growth hormone replacement therapy. {ECO:0000269|PubMed:17519310, ECO:0000269|PubMed:8552145, ECO:0000269|PubMed:9276733}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Growth hormone deficiency, isolated, 2 (IGHD2) [MIM:173100]: An autosomal dominant deficiency of growth hormone leading to short stature. Clinical severity is variable. Patients have a positive response and immunologic tolerance to growth hormone therapy. {ECO:0000269|PubMed:11502836, ECO:0000269|PubMed:9152628}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9Y614; Q6DHV7-2; Q6UY14-3; Q96MA6; Q5T2L2; Q96Q83-2; Q9Y303-2; Q9NU02; P09525; P06727; Q8WW27; Q66PJ3-4; Q6XD76; P18848; Q9H0Y0; Q14032; P54687-4; P06276; Q9NSI6-4; Q96Q07-2; Q9H0W9-3; Q9NQ89; Q13901; Q3SXR2; Q8N1A6; P17655; P20807-4; P42574; P55212; O00257-3; P24863; Q9NNX6-10; Q9UJX2; P42773; O95674; Q494V2-2; Q8WUX9; Q9Y3D0; Q8N365; Q3SX64; Q99966; P09496-2; Q6PJW8-3; Q96BR5; P02458-1; Q9UGL9; Q9UKG9-2; P26998; P35222; Q53TN4; P61962; O60479; Q96EY1-3; Q92782-2; Q9BPU6; A0AVK6; Q658K8; O00303; Q13347; O00472; O00423; Q6NXG1-3; Q49AJ0-4; Q8N128-2; Q8IZU1; Q6ZNL6; Q9NSA1; Q06547-3; Q49A26-4; Q9HAV0; Q6NXT2; Q9BT25; Q9NRZ9-6; Q96EW2-2; P42858; Q8N6M8-2; Q92613; P0C870; Q9UK76; Q8N5Z5; Q8TBB5-2; Q9UH77; Q8N4N3-2; Q5JUW0-3; Q8N1A0; P13473-2; Q6DKI2; Q9H2C1; Q8N0U6; Q9Y234; Q8TBB1; Q1L5Z9; Q96JB6; Q16609; Q8IYG6; P0DP58-2; Q969L2; P27338; A6NJ78-4; Q96C03-3; Q8N5J2-3; A0A0A0MR05; P34949-2; Q9BV20; Q6IN84-2; A2RUH7; P01106; Q9H7X0; Q15742-2; Q9UJ70-2; Q8NDH3-5; Q96HA8; P36639-4; Q8NFH4; Q8NFH3; Q7Z3B4; Q6N063-2; Q6GQQ9-2; Q9H8K7; Q99447; P27815-4; O15534; Q9BUL5; Q00169; P48739; P61925; Q58EX7-2; O60664; Q14181; P0DPB6; P36954; Q07869; O60927; Q6ZMI0-5; P54619; Q8NCQ7-2; P41222; P29074; Q8WUD1-2; Q5R372-9; Q9HD47-3; Q09028; Q04206; P47804-3; Q15382; Q06587; Q8N5U6; P62701; Q66K80; Q01826; O15126; P22307-3; Q9BRK5; Q9NTN9-3; P01011; Q15393; Q9NR46; Q9BZQ2; O60902-3; Q86US8; P37840; Q96H20; Q13573; Q7Z6I5; Q496A3; Q9C004; Q5W111-2; Q96BD6; Q92797-2; O60506-4; O15273; Q86WV5; Q96A09; P54274-2; P22735; O43548; Q9NQ88; Q9UIK5-2; Q53NU3; P04637; Q12888; P36406; Q86WT6-2; Q13885; P49459; Q9P1Q0-4; Q9NX94; Q8NA23-2; Q9BQA1; O00755; O95070; O43829; Q8IWT0-2; Q53FD0-2; Q05CR2; Q96JL9-2; Q96LX8; Q3KNS6-3; Q5JTY5; A0A384MDV8; B7Z3E8; Q86V28 EC number EC 3.4.22.59 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; Autocatalytic cleavage; Cytoplasm; Hydrolase; Lipoprotein; Nucleus; Palmitate; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Thiol protease; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 57170 Length 500 Aromaticity 0.12 Instability index 31.33 Isoelectric point 8.05 Charge (pH=7) 4.52 2D Binding mode Binding energy (Kcal/mol) -7.73 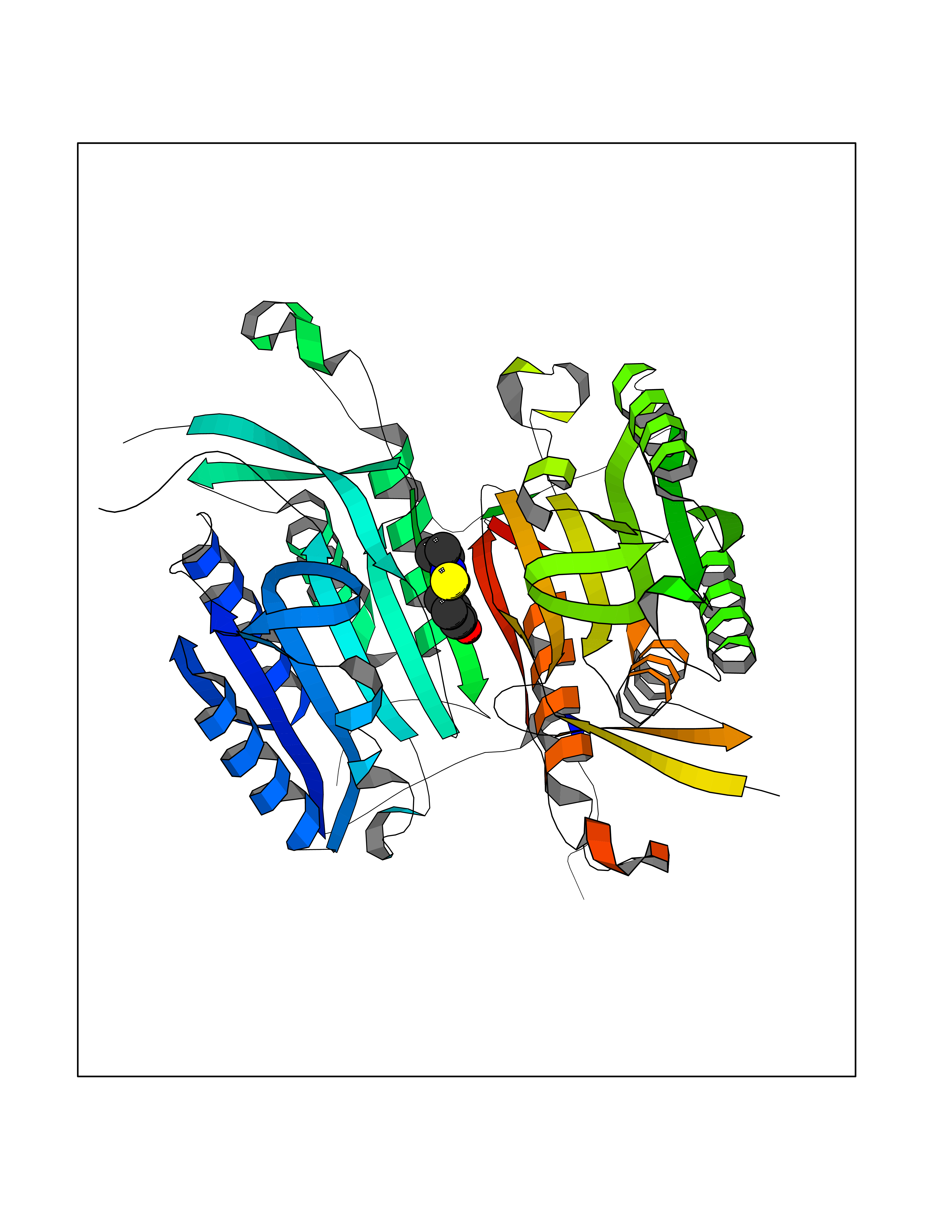 Molscript Map 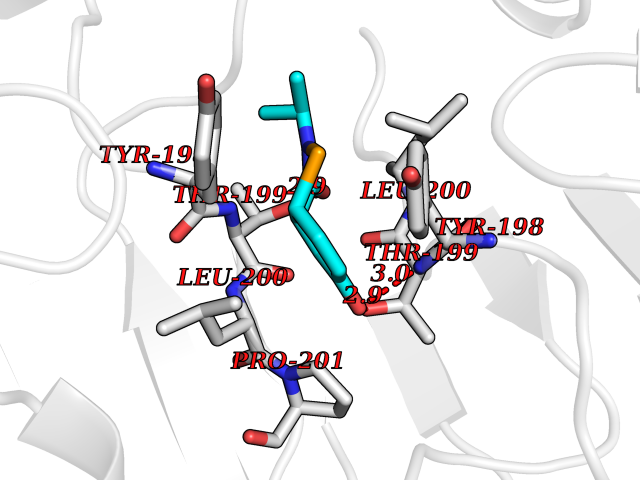 Pymol Map 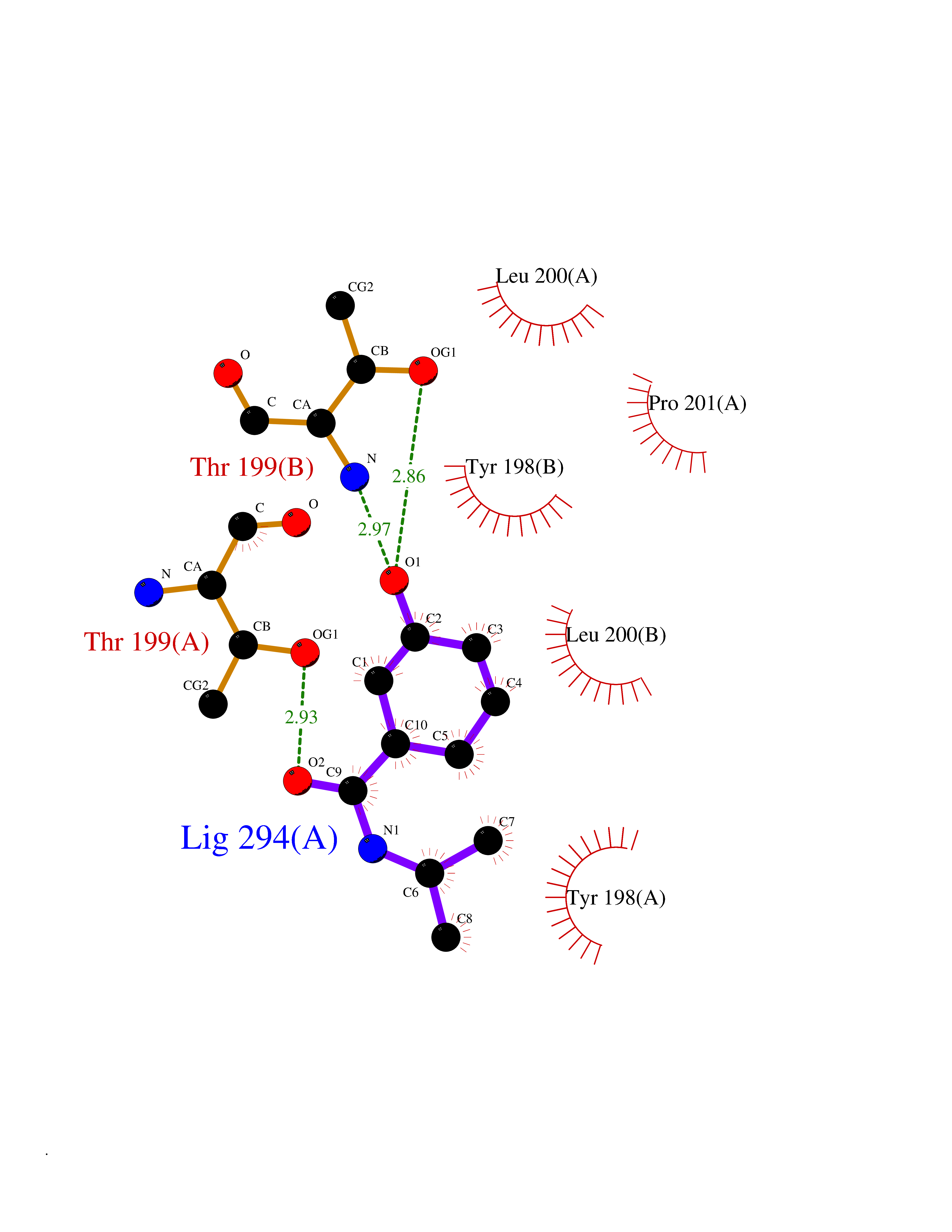 Ligplot Map 3D Binding mode Sequence AFYKREMFDPAEKYKMDHRRRGIALIFNHERFFWHLTLPERRGTCADRDNLTRRFSDLGFEVKCFNDLKAEELLLKIHEVSTVSHADADCFVCVFLSHGEGNHIYAYDAKIEIQTLTGLFKGDKCHSLVGKPKIFIIQAARGNQHDVPVIPDTNITEVDAASVYTLPAGADFLMCYSVAEGYYSHRETVNGSWYIQDLCEMLGKYGSSLEFTELLTLVNRKVSQRRVDFCKDPSAIGKKQVPCFASMLTKKLHFFPKSMFDPAEKYKMDHRRRGIALIFNHERFFWHLTLPERRGTCADRDNLTRRFSDLGFEVKCFNDLKAEELLLKIHEVSTVSHADADCFVCVFLSHGEGNHIYAYDAKIEIQTLTGLFKGDKCHSLVGKPKIFIIQAARGNTNITEVDAASVYTLPAGADFLMCYSVAEGYYSHRETVNGSWYIQDLCEMLGKYGSSLEFTELLTLVNRKVSQRRVDFCKDPSAIGKKQVPCFASMLTKKLHFFPK Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Protein kinase G2 (PRKG2) | 5BV6 | 5.67 | |
Target general information Gen name PRKG2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cGMP-dependent protein kinase II; cGMP-dependent protein kinase 2; cGKII; cGK2; cGK 2; PRKGR2 Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family, cGMP subfamily Biochemical class NA Function Crucial regulator of intestinal secretion and bone growth (By similarity). Phosphorylates and activates CFTR on the plasma membrane. Plays a key role in intestinal secretion by regulating cGMP-dependent translocation of CFTR in jejunum (By similarity). Acts downstream of NMDAR to activate the plasma membrane accumulation of GRIA1/GLUR1 in synapse and increase synaptic plasticity. Phosphorylates GRIA1/GLUR1 at Ser-863 (By similarity). Acts as regulator of gene expression and activator of the extracellular signal-regulated kinases MAPK3/ERK1 and MAPK1/ERK2 in mechanically stimulated osteoblasts. Under fluid shear stress, mediates ERK activation and subsequent induction of FOS, FOSL1/FRA1, FOSL2/FRA2 and FOSB that play a key role in the osteoblast anabolic response to mechanical stimulation (By similarity). Related diseases Spondylometaphyseal dysplasia, Pagnamenta type (SMDP) [MIM:619638]: A form of spondylometaphyseal dysplasia, a group of short stature disorders distinguished by abnormalities in the vertebrae and the metaphyses of the tubular bones. SMDP is an autosomal recessive form characterized by short stature and mild platyspondyly with no disproportion between the limbs. Mild metaphyseal changes are present. {ECO:0000269|PubMed:34782440}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Acromesomelic dysplasia 4 (AMD4) [MIM:619636]: A form of acromesomelic dysplasia, a skeletal disorder characterized by short stature, very short limbs and hand/foot malformations. The severity of limb abnormalities increases from proximal to distal with profoundly affected hands and feet showing brachydactyly and/or rudimentary fingers (knob-like fingers). AMD4 radiographic hallmarks include mild to moderate platyspondyly, moderate brachydactyly, iliac flaring, and metaphyseal alterations of the long bones that progressively increase with age. AMD4 inheritance is autosomal recessive. {ECO:0000269|PubMed:33106379, ECO:0000269|PubMed:34782440}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with NA EC number EC 2.7.11.12 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; cGMP; cGMP-binding; Coiled coil; Disease variant; Dwarfism; Kinase; Lipoprotein; Membrane; Myristate; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 17227.1 Length 150 Aromaticity 0.09 Instability index 43.7 Isoelectric point 4.88 Charge (pH=7) -8.41 2D Binding mode Binding energy (Kcal/mol) -7.74 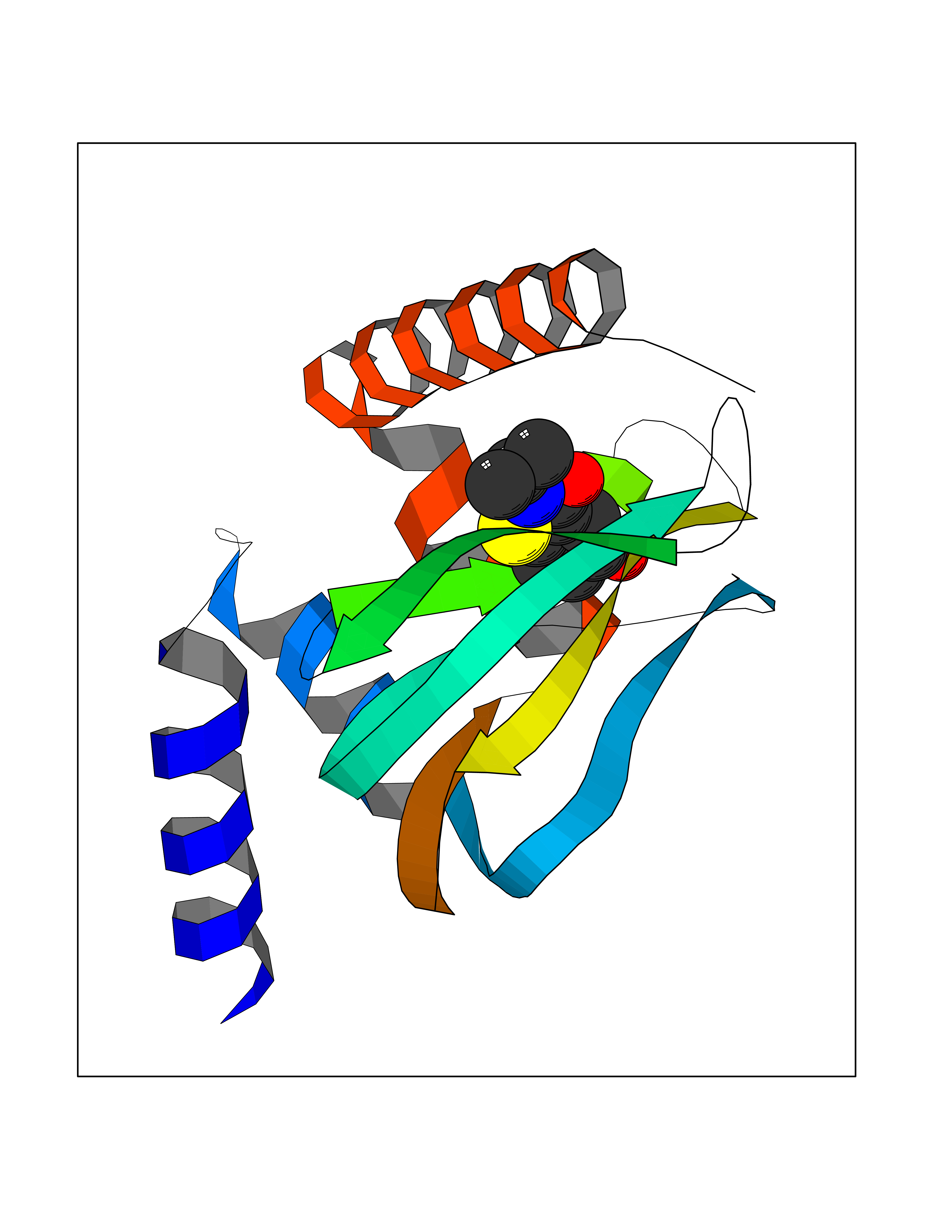 Molscript Map 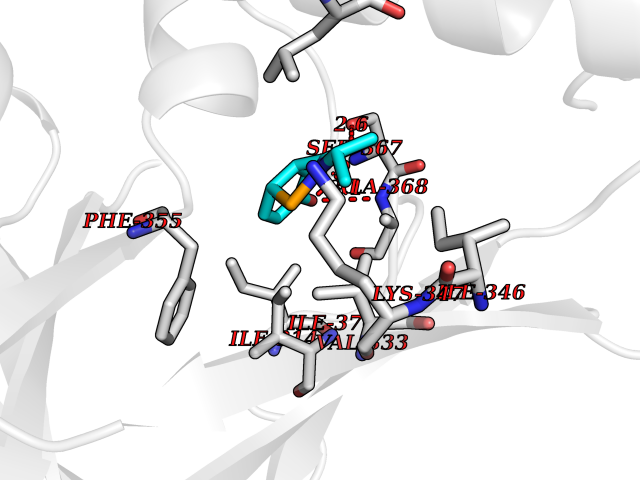 Pymol Map 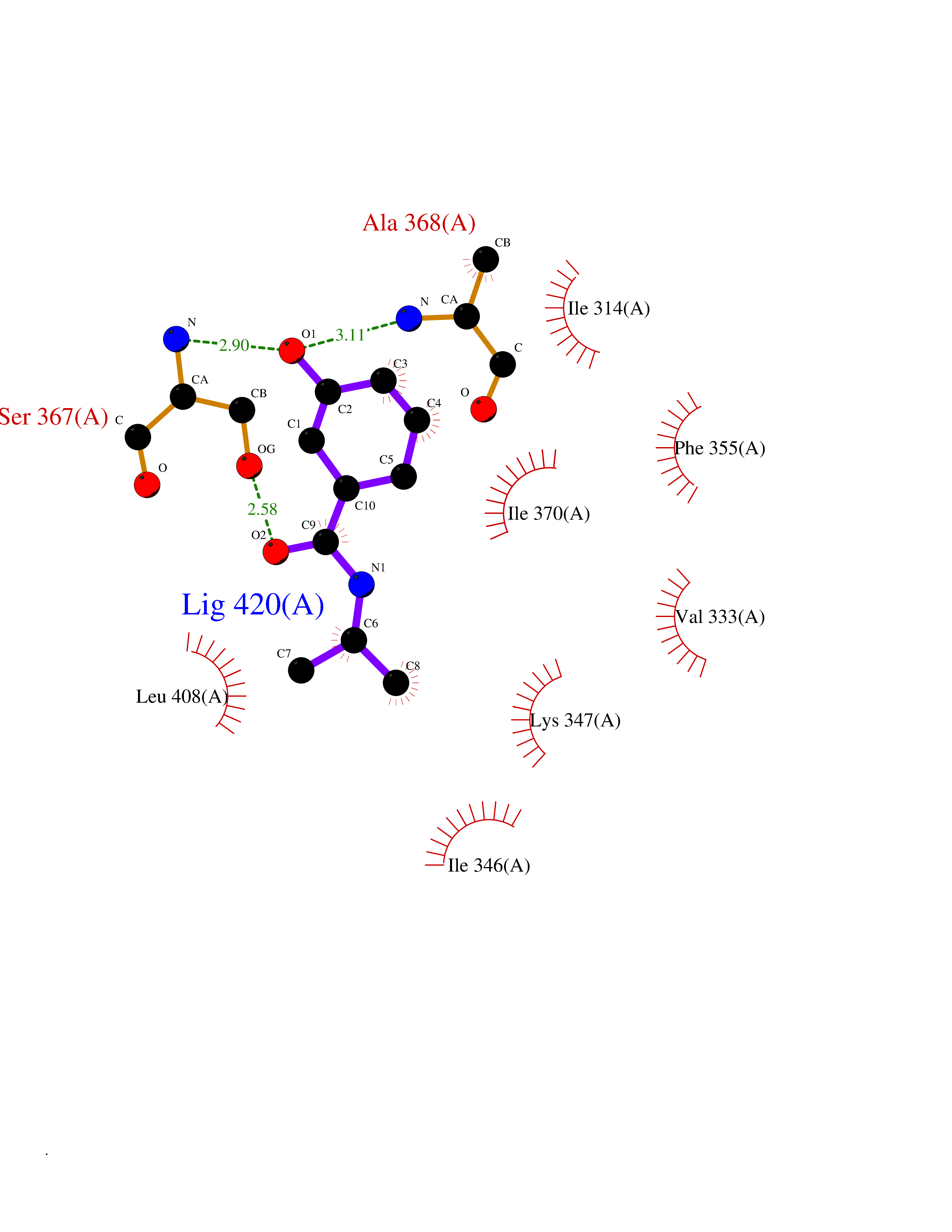 Ligplot Map 3D Binding mode Sequence TAQARDEQYRNFLRSVSLLKNLPEDKLTKIIDCLEVEYYDKGDYIIREGEEGSTFFILAKGKVKVTQSTEGHDQPQLIKTLQKGEYFGEKALISDDVRSANIIAEENDVACLVIDRETFNQTVGTFEELQKYLEGYVANLNRDDEKRHAK Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | Dual-specificity tyrosine-phosphorylation regulated kinase 2 (DYRK2) | 6HDR | 5.67 | |
Target general information Gen name DYRK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Dual specificity tyrosine-phosphorylation-regulated kinase 2 Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, MNB/DYRK subfamily Biochemical class Kinase Function Functions in part via its role in ubiquitin-dependent proteasomal protein degradation. Functions downstream of ATM and phosphorylates p53/TP53 at 'Ser-46', and thereby contributes to the induction of apoptosis in response to DNA damage. Phosphorylates NFATC1, and thereby inhibits its accumulation in the nucleus and its transcription factor activity. Phosphorylates EIF2B5 at 'Ser-544', enabling its subsequent phosphorylation and inhibition by GSK3B. Likewise, phosphorylation of NFATC1, CRMP2/DPYSL2 and CRMP4/DPYSL3 promotes their subsequent phosphorylation by GSK3B. May play a general role in the priming of GSK3 substrates. Inactivates GYS1 by phosphorylation at 'Ser-641', and potentially also a second phosphorylation site, thus regulating glycogen synthesis. Mediates EDVP E3 ligase complex formation and is required for the phosphorylation and subsequent degradation of KATNA1. Phosphorylates TERT at 'Ser-457', promoting TERT ubiquitination by the EDVP complex. Phosphorylates SIAH2, and thereby increases its ubiquitin ligase activity. Promotes the proteasomal degradation of MYC and JUN, and thereby regulates progress through the mitotic cell cycle and cell proliferation. Promotes proteasomal degradation of GLI2 and GLI3, and thereby plays a role in smoothened and sonic hedgehog signaling. Plays a role in cytoskeleton organization and neurite outgrowth via its phosphorylation of DCX and DPYSL2. Phosphorylates CRMP2/DPYSL2, CRMP4/DPYSL3, DCX, EIF2B5, EIF4EBP1, GLI2, GLI3, GYS1, JUN, MDM2, MYC, NFATC1, p53/TP53, TAU/MAPT and KATNA1. Can phosphorylate histone H1, histone H3 and histone H2B (in vitro). Can phosphorylate CARHSP1 (in vitro). Serine/threonine-protein kinase involved in the regulation of the mitotic cell cycle, cell proliferation, apoptosis, organization of the cytoskeleton and neurite outgrowth. Related diseases Bone marrow failure and diabetes mellitus syndrome (BMFDMS) [MIM:620044]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFDMS is an autosomal recessive form characterized by various degrees of bone marrow failure, ranging from dyserythropoiesis to bone marrow aplasia, with onset in infancy or early childhood, and non-autoimmune insulin-dependent diabetes mellitus appearing in the first or second decades. Many patients show pigmentary skin abnormalities and short stature. {ECO:0000269|PubMed:28073829, ECO:0000269|PubMed:35611808, ECO:0000269|PubMed:35931051}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NR20; Q13422; Q9BQD3; Q9BRK4; P23497; O43379; P62258; Q96C00 EC number EC 2.7.12.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cytoplasm; Kinase; Magnesium; Manganese; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Tyrosine-protein kinase; Ubl conjugation; Ubl conjugation pathway Protein physicochemical properties Chain ID A Molecular weight (Da) 46422.1 Length 407 Aromaticity 0.09 Instability index 44.91 Isoelectric point 9.09 Charge (pH=7) 12.37 2D Binding mode Binding energy (Kcal/mol) -7.73 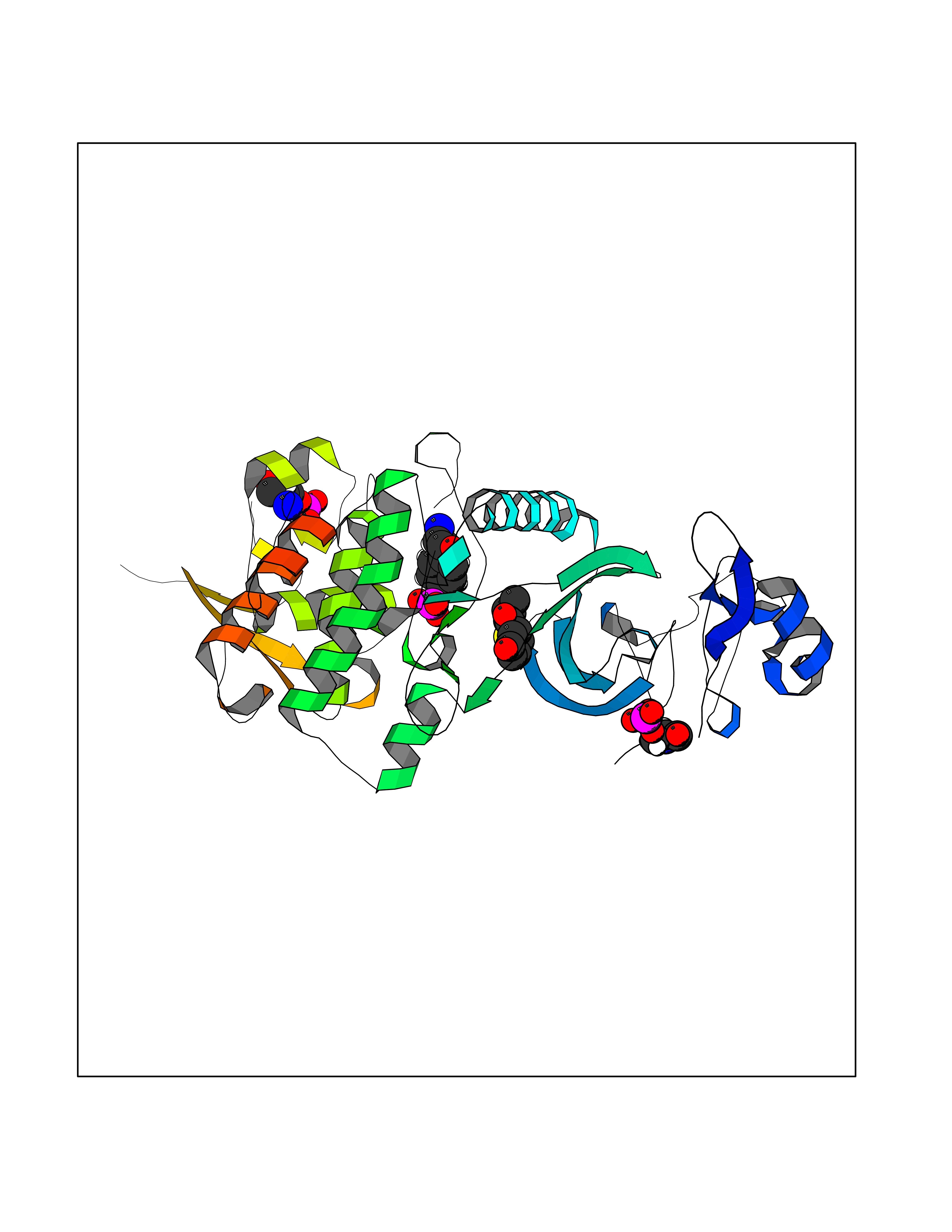 Molscript Map 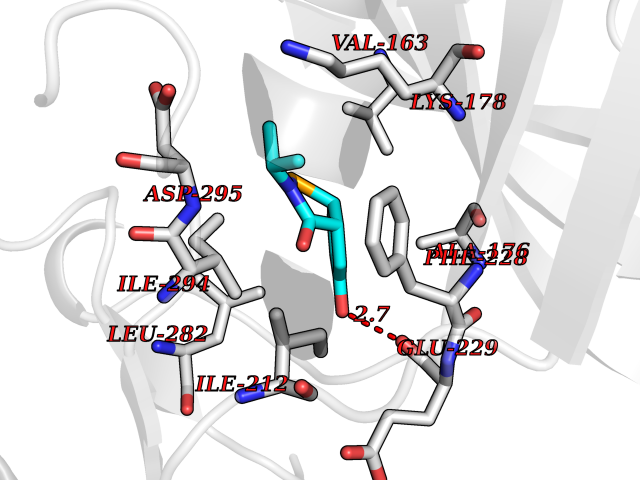 Pymol Map 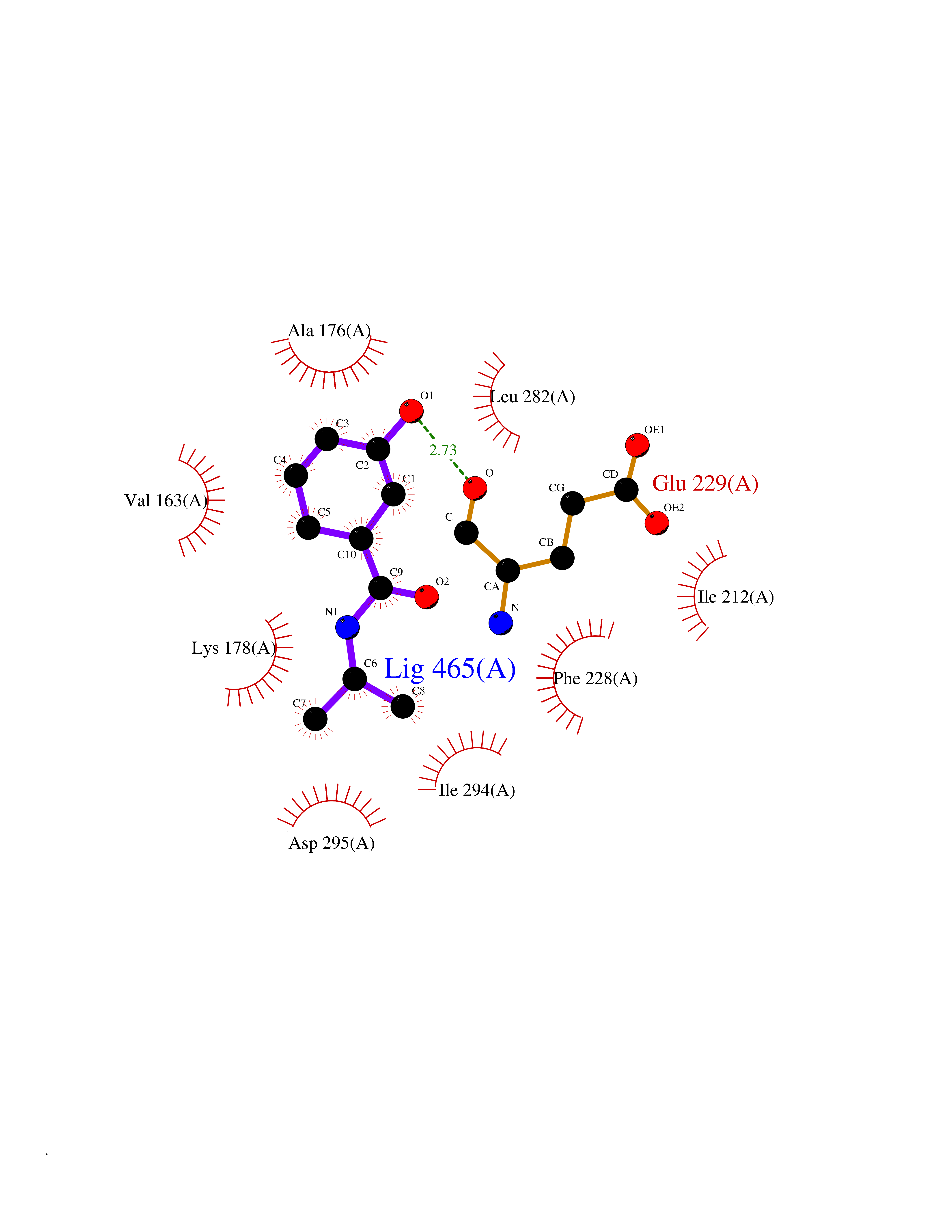 Ligplot Map 3D Binding mode Sequence HHHSXGVDLGTENLYFQSMGKVKATPMTPEQAMKQYMQKLTAFEHHEIFSYPEIYFLGLNAKKRQGMTGGPNNGGYDDDQGSYVQVPHDHVAYRYEVLKVIGKGSFGQVVKAYDHKVHQHVALKMVRNEKRFHRQAAEEIRILEHLRKQDKDNTMNVIHMLENFTFRNHICMTFELLSMNLYELIKKNKFQGFSLPLVRKFAHSILQCLDALHKNRIIHCDLKPENILLKQQGRSGIKVIDFGSSCYEHQRVYTXIQSRFYRAPEVILGARYGMPIDMWSLGCILAELLTGYPLLPGEDEGDQLACMIELLGMPSQKLLDASKRAKNFVSXKGYPRYCTVTTLSDVVLNGGRSRRGKLRGPPESREWGNALKGCDDPLFLDFLKQCLEWDPAVRMTPGQALRHPWLR Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Glucose-dependent insulinotropic receptor (GPR119) | 7XZ6 | 5.67 | |
Target general information Gen name GPR119 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GPR119; G-protein coupled receptor 119 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for the endogenous fatty-acid ethanolamide oleoylethanolamide (OEA) and lysophosphatidylcholine (LPC). Functions as a glucose-dependent insulinotropic receptor. The activity of this receptor is mediated by G proteins which activate adenylate cyclase. Seems to act through a G(s) mediated pathway. Related diseases Developmental and epileptic encephalopathy 24 (DEE24) [MIM:615871]: A disease characterized by early-onset seizures, intellectual disability of varying degrees, and behavioral disturbances or autistic features in most individuals. {ECO:0000269|PubMed:24747641, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Generalized epilepsy with febrile seizures plus 10 (GEFSP10) [MIM:618482]: An autosomal dominant neurologic disorder with incomplete penetrance, characterized by variable types of seizures including absence, tonic-clonic, febrile, focal, and eyelid myoclonia. Some patients have normal neurologic development. Others have mild-to-moderate intellectual disability or autism spectrum disorder. {ECO:0000269|PubMed:29936235, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05166 Interacts with Q12797-6 EC number NA Uniprot keywords 3D-structure; Cell membrane; G-protein coupled receptor; Lipid-binding; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32134.1 Length 292 Aromaticity 0.12 Instability index 34.96 Isoelectric point 9.12 Charge (pH=7) 8.03 2D Binding mode Binding energy (Kcal/mol) -7.74  Molscript Map 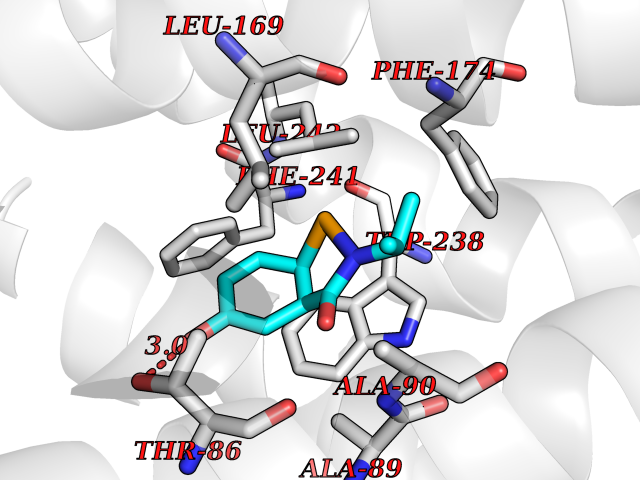 Pymol Map 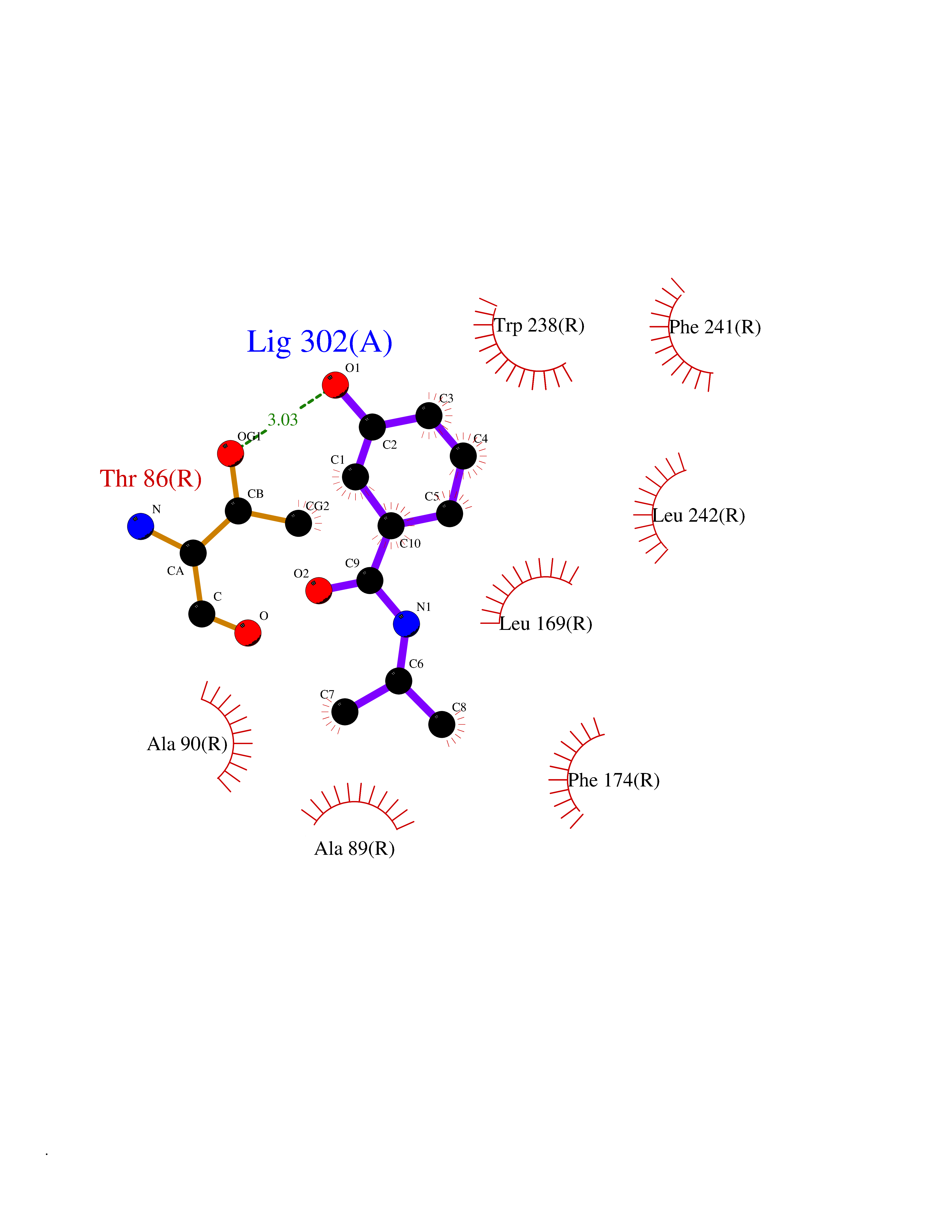 Ligplot Map 3D Binding mode Sequence MESSFSFGVILAVLASLIIATNTLVAVAVLLLIHKNDGVSLCFTLNLAVADTLIGVAISGLLTDQLSSPSRPTQKTLCSLRMAFVTSSAAASVLTVMLITFDRYLAIKQPFRYLKIMSGFVAGACIAGLWLVSYLIGFLPLGIPMFQQTAYKGQCSFFAVFHPHFVLTLSCVGFFPAMLLFVFFYCDMLKIASMHSQQIRKMEHAGAMAGSDFKALRTVSVLIGSFALSWTPFLITGIVQVACQECHLYLVLERYLWLLGVGNSLLNPLIYAYWQKEVRLQLYHMALGVKKV Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Retinoic acid receptor beta (RARB) | 4DM6 | 5.66 | |
Target general information Gen name RARB Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RAR-epsilon; RAR-beta; Nuclear receptor subfamily 1 group B member 2; NR1B2; HBV-activated protein; HAP Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RXR/RAR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence or presence of hormone ligand, acts mainly as an activator of gene expression due to weak binding to corepressors. In concert with RARG, required for skeletal growth, matrix homeostasis and growth plate function. Receptor for retinoic acid. Related diseases Microphthalmia, syndromic, 12 (MCOPS12) [MIM:615524]: A form of microphthalmia, a disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues (anophthalmia). In many cases, microphthalmia/anophthalmia occurs in association with syndromes that include non-ocular abnormalities. MCOPS12 patients manifest variable features, including diaphragmatic hernia, pulmonary hypoplasia, and cardiac abnormalities. {ECO:0000269|PubMed:24075189, ECO:0000269|PubMed:27120018}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00459; DB00210; DB00523; DB02877; DB00926; DB05785; DB04942; DB00799; DB00755; DB12808 Interacts with O95273; P50222; Q9UBK2; P62195; P28702; P28702-3; P48443; P03255 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Microphthalmia; Nucleus; Phosphoprotein; Proto-oncogene; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 25904.1 Length 229 Aromaticity 0.06 Instability index 44.34 Isoelectric point 7.55 Charge (pH=7) 0.73 2D Binding mode Binding energy (Kcal/mol) -7.72  Molscript Map 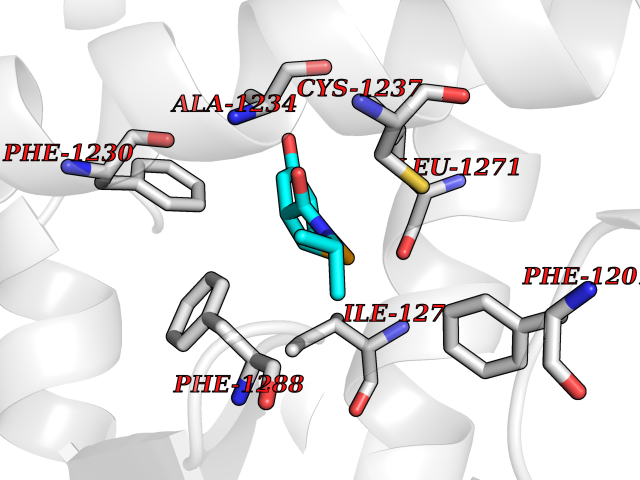 Pymol Map 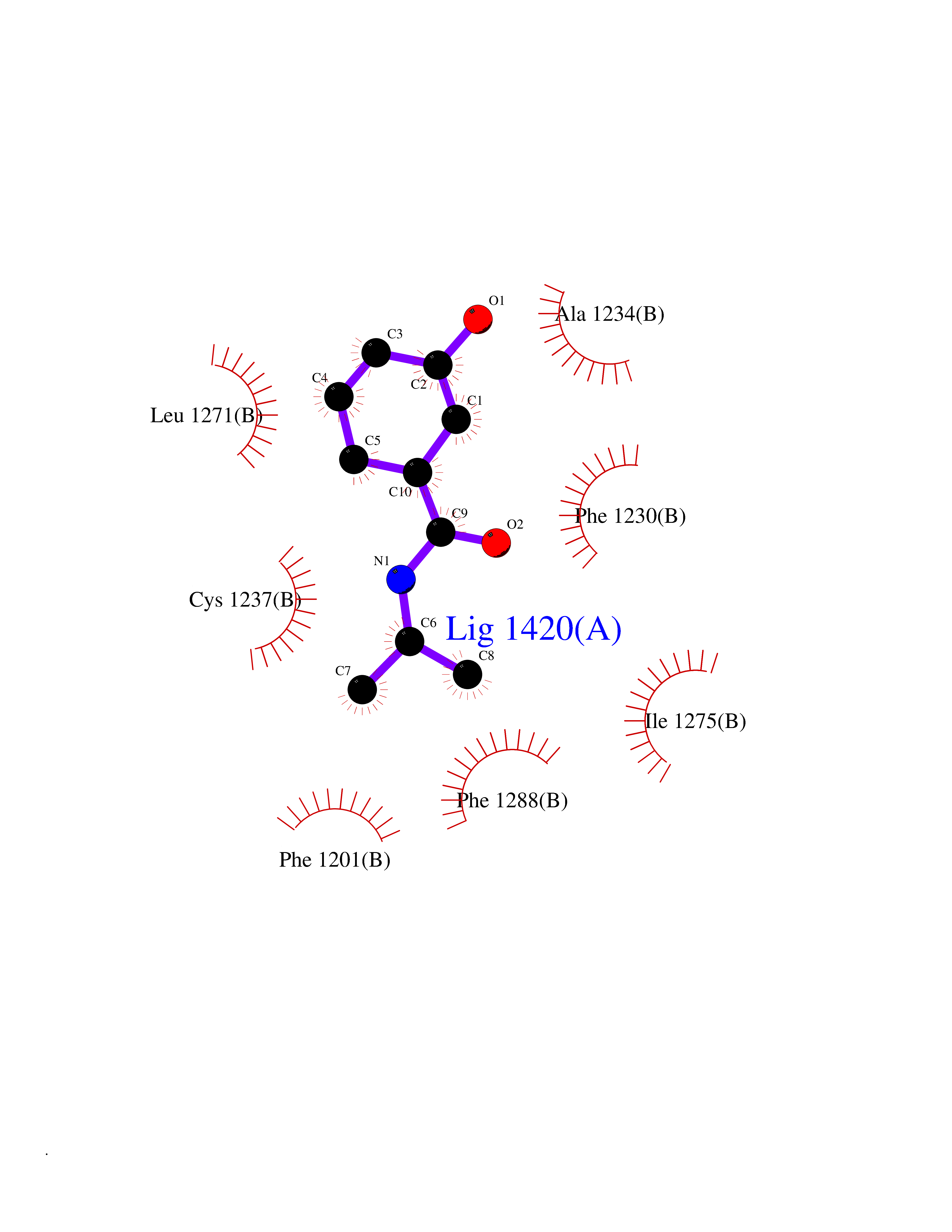 Ligplot Map 3D Binding mode Sequence TEKIRKAHQETFPSLCQLGKYTTNSSADHRVRLDLGLWDKFSELATKCIIKIVEFAKRLPGFTGLTIADQITLLKAACLDILILRICTRYTPEQDTMTFSDGLTLNRTQMHNAGFGPLTDLVFTFANQLLPLEMDDTETGLLSAICLICGDRQDLEEPTKVDKLQEPLLEALKIYIRKRRPSKPHMFPKILMKITDLRSISAKGAERVITLKMEIPGSMPPLIQEMLEN Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | "Periplasmic trehalase (EC 3.2.1.28) (Alpha,alpha-trehalase) (Alpha,alpha-trehalose glucohydrolase) (Tre37A)" | 2JG0 | 5.66 | |
Target general information Gen name treA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW1186;osmA;b1197 Protein family Glycosyl hydrolase 37 family Biochemical class NA Function Provides the cells with the ability to utilize trehalose at high osmolarity by splitting it into glucose molecules that can subsequently be taken up by the phosphotransferase-mediated uptake system. Related diseases SRC kinase activity has been shown to be increased in several tumor tissues and tumor cell lines such as colon carcinoma cells. {ECO:0000269|PubMed:2498394, ECO:0000269|PubMed:3093483}.; DISEASE: Thrombocytopenia 6 (THC6) [MIM:616937]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC6 is an autosomal dominant form. Affected individuals may also have bone abnormalities and an increased risk for myelofibrosis. {ECO:0000269|PubMed:26936507}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number 3.2.1.28 Uniprot keywords 3D-structure; Direct protein sequencing; Glycosidase; Hydrolase; Periplasm; Reference proteome; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 57508.9 Length 507 Aromaticity 0.11 Instability index 48.32 Isoelectric point 5.48 Charge (pH=7) -10.13 2D Binding mode Binding energy (Kcal/mol) -7.72  Molscript Map 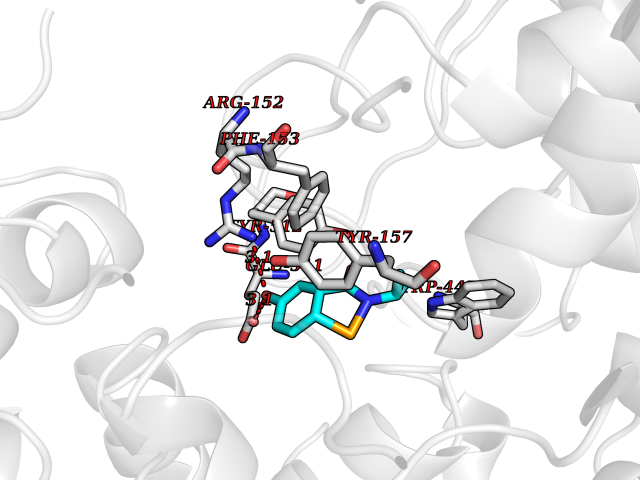 Pymol Map 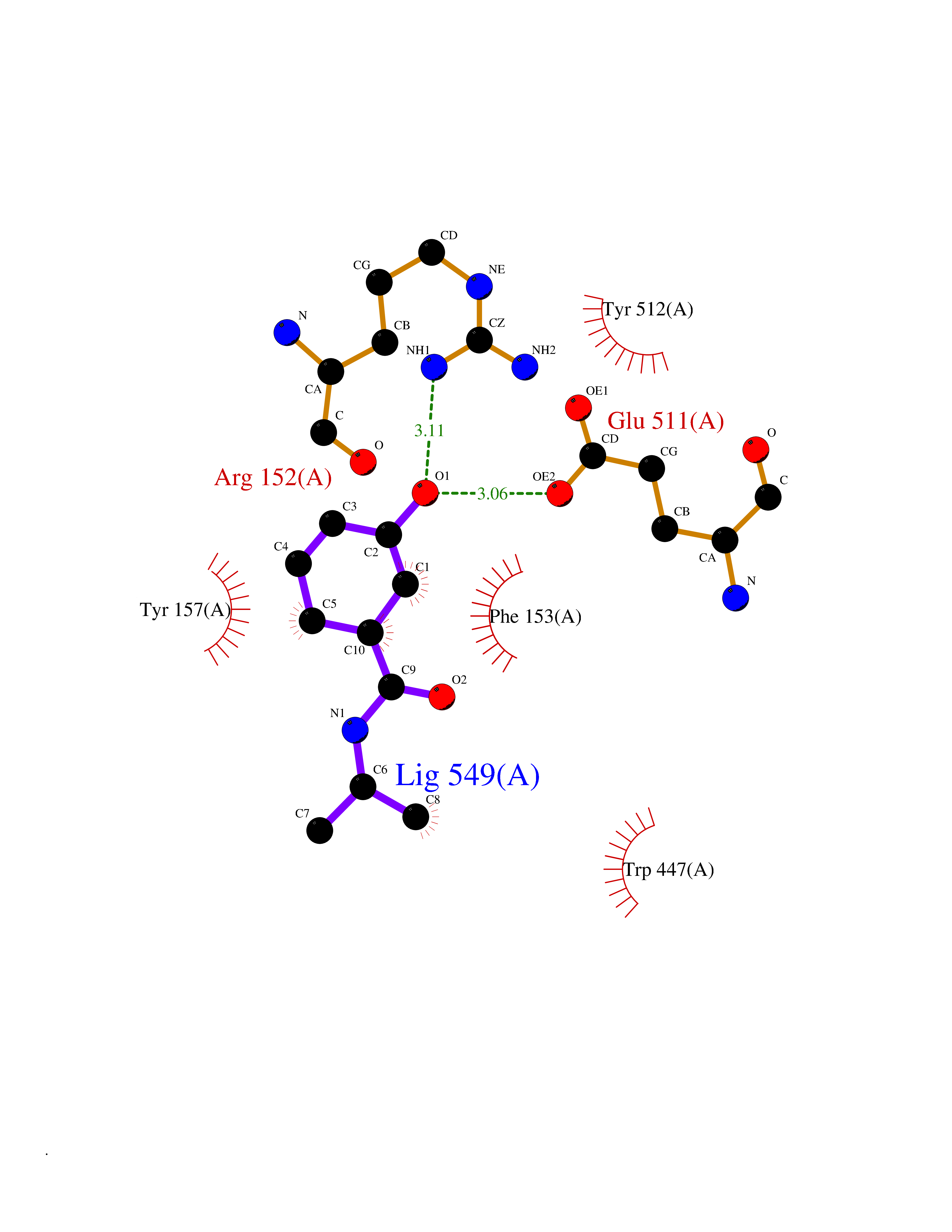 Ligplot Map 3D Binding mode Sequence PQPPDILLGPLFNDVQNAKLFPDQKTFADAVPNSDPLMILADYRMQQNQSGFDLRHFVNVNFTLPKYVPPEGQSLREHIDGLWPVLTRSTENTEKWDSLLPLPEPYVVPGGRFREVYYWDSYFTMLGLAESGHWDKVADMVANFAHEIDTYGHIPNGNRSYYLSRSQPPFFALMVELLAQHEGDAALKQYLPQMQKEYAYWMDGVENLQAGQQEKRVVKLQDGTLLNRYWDDRDTPRPESWVEDIATAKSNPNRPATEIYRDLRSAAASGWDFSSRWMDNPQQLNTLRTTSIVPVDLNSLMFKMEKILARASKAAGDNAMANQYETLANARQKGIEKYLWNDQQGWYADYDLKSHKVRNQLTAAALFPLYVNAAAKDRANKMATATKTHLLQPGGLNTTSVKSGQQWDAPNGWAPLQWVATEGLQNYGQKEVAMDISWHFLTNVQHTYDREKKLVEKYDVSTTGTGGGGGEYPLQDGFGWTNGVTLKMLDLICPKEQPCDNVPATRP Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Glycinamide ribonucleotide formyltransferase (GART) | 4ZZ1 | 5.66 | |
Target general information Gen name GART Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Trifunctional purine biosynthetic protein adenosine-3; PRGS; PGFT Protein family GARS family; AIR synthase family; GART family Biochemical class Carbon-nitrogen ligase Function A trifunctional polypeptide. Has Phosphoribosylamineglycine ligase, Phosphoribosylglycinamide formyltransferase, AIR synthetase (FGAM cyclase) activity which is required for de novo purine biosynthesis. Related diseases Schizophrenia (SCZD) [MIM:181500]: A complex, multifactorial psychotic disorder or group of disorders characterized by disturbances in the form and content of thought (e.g. delusions, hallucinations), in mood (e.g. inappropriate affect), in sense of self and relationship to the external world (e.g. loss of ego boundaries, withdrawal), and in behavior (e.g bizarre or apparently purposeless behavior). Although it affects emotions, it is distinguished from mood disorders in which such disturbances are primary. Similarly, there may be mild impairment of cognitive function, and it is distinguished from the dementias in which disturbed cognitive function is considered primary. Some patients manifest schizophrenic as well as bipolar disorder symptoms and are often given the diagnosis of schizoaffective disorder. {ECO:0000269|PubMed:15645182}. Disease susceptibility may be associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02236; DB03546; DB00642 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Ligase; Magnesium; Manganese; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Proteomics identification; Purine biosynthesis; Reference proteome; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 21438.4 Length 200 Aromaticity 0.05 Instability index 33.54 Isoelectric point 6.23 Charge (pH=7) -1.77 2D Binding mode Binding energy (Kcal/mol) -7.72 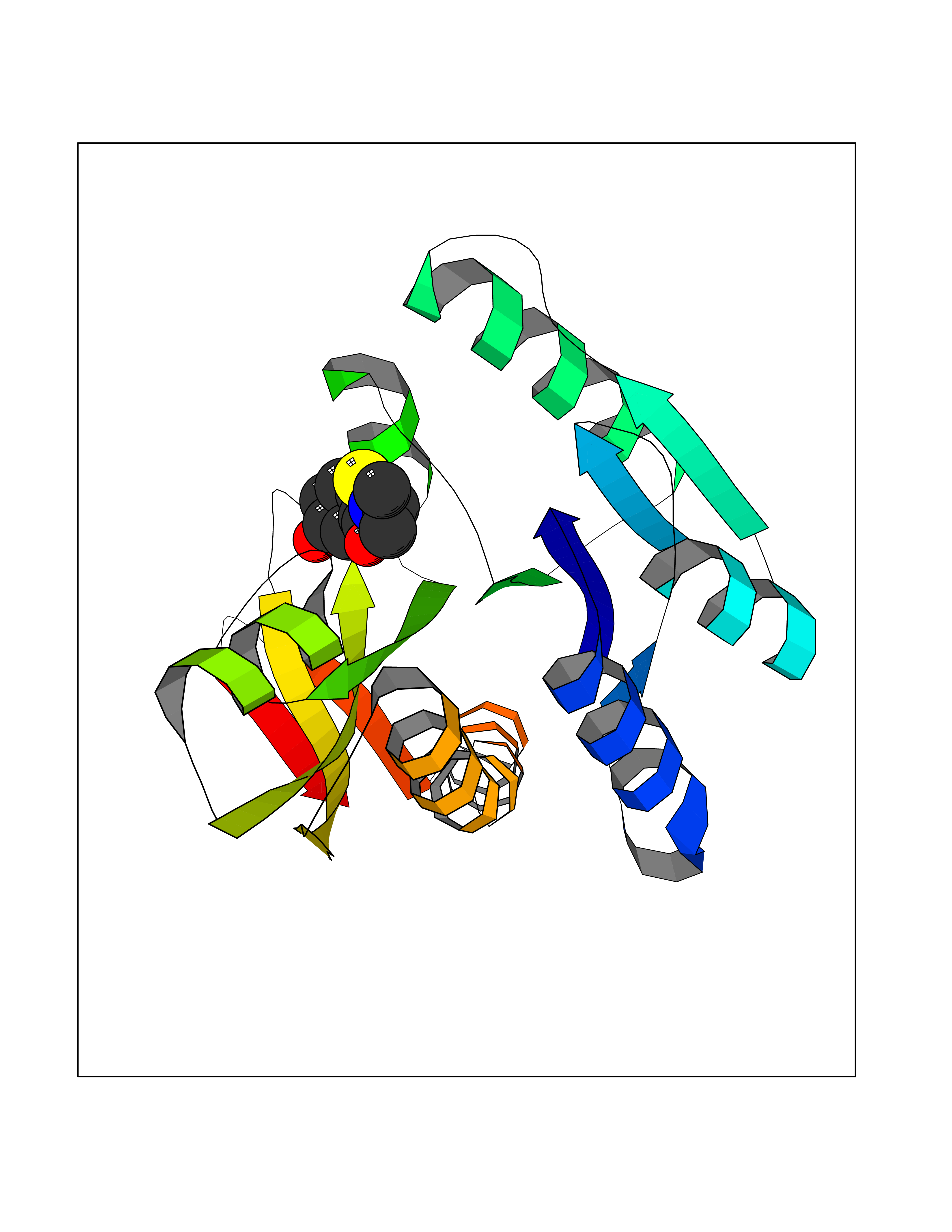 Molscript Map  Pymol Map 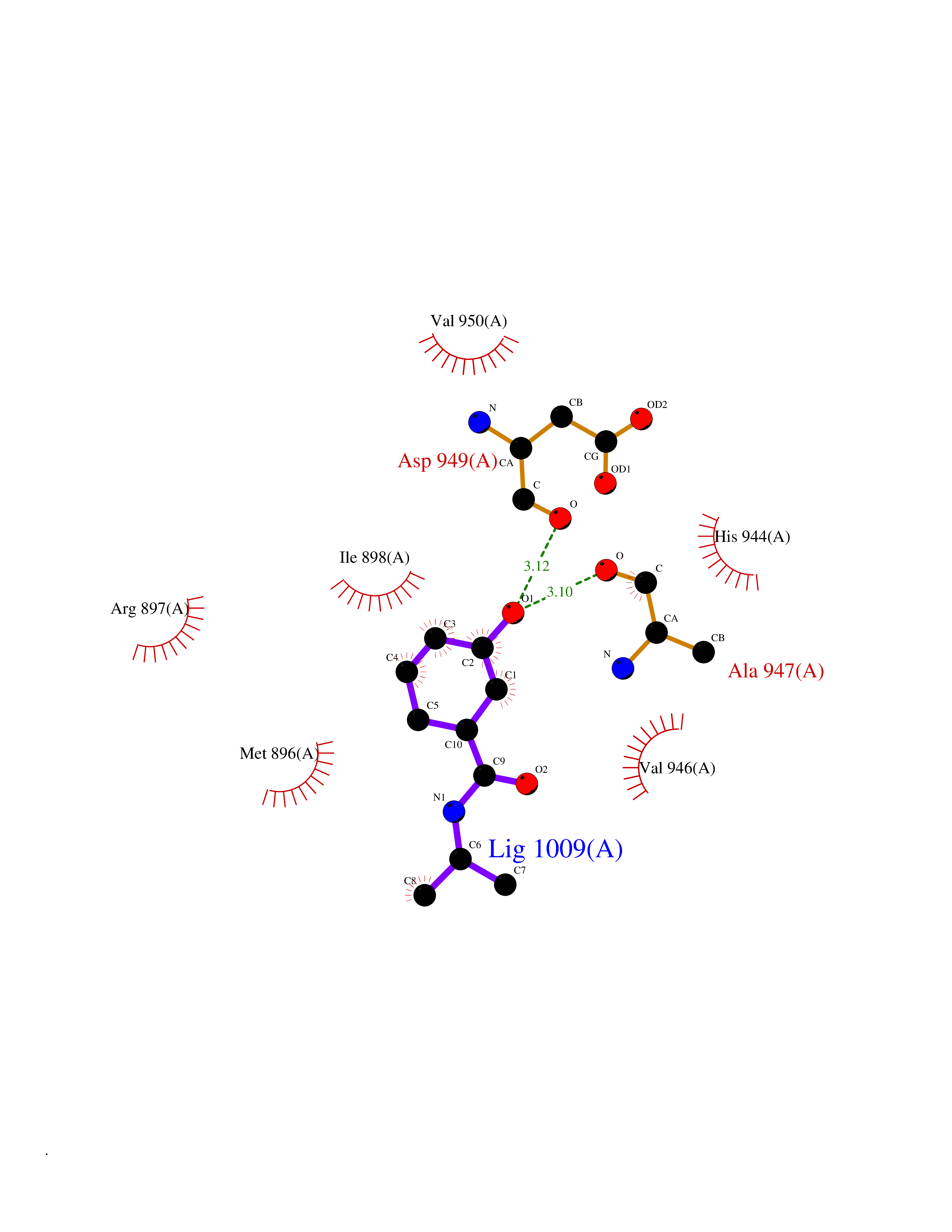 Ligplot Map 3D Binding mode Sequence ARVAVLISGTGSNLQALIDSTREPNSSAQIDIVISNKAAVAGLDKAERAGIPTRVINHKLYKNRVEFDSAIDLVLEEFSIDIVCLAGFMRILSGPFVQKWNGKMLNIHPSLLPSFKGSNAHEQALETGVTVTGCTVHFVAEDVDAGQIILQEAVPVKRGDTVATLSERVKLAEHKIFPAALQLVASGTVQLGENGKICWV Hydrogen bonds contact Hydrophobic contact | ||||