Job Results:
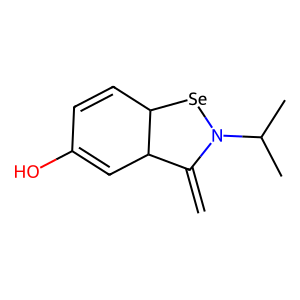
Ligand
Structure
Job ID
6035497cf04470e01d5d7dbbed13b512
Job name
NA
Time
2025-10-13 17:35:32
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Thyroid hormone receptor alpha (THRA) | 3ILZ | 5.73 | |
Target general information Gen name THRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms V-erbA-related protein 7; THRA2; THRA1; Nuclear receptor subfamily 1 group A member 1; NR1A1; ERBA1; EAR7; EAR-7; C-erbA-alpha; C-erbA-1 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function High affinity receptor for thyroid hormones, including triiodothyronine and thyroxine. Isoform Alpha-1: Nuclear hormone receptor that can act as a repressor or activator of transcription. Related diseases Hypothyroidism, congenital, non-goitrous, 6 (CHNG6) [MIM:614450]: A disease characterized by growth retardation, developmental retardation, skeletal dysplasia, borderline low thyroxine levels and high triiodothyronine levels. There is differential sensitivity to thyroid hormone action, with retention of hormone responsiveness in the hypothalamic pituitary axis and liver but skeletal, gastrointestinal, and myocardial resistance. {ECO:0000269|PubMed:22168587, ECO:0000269|PubMed:24969835, ECO:0000269|PubMed:25670821, ECO:0000269|PubMed:26037512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01118; DB00509; DB04855; DB05035; DB03176; DB00451; DB00279; DB01583; DB05235; DB09100 Interacts with Q9Y2J4; Q9Y2J4-4; O95971; Q8TAP6; Q96JM7; Q15648; Q6FHY5; P31321; Q96A49; O75410-7; Q9JLI4 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Congenital hypothyroidism; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 29910.1 Length 267 Aromaticity 0.07 Instability index 52.75 Isoelectric point 5.31 Charge (pH=7) -11.32 2D Binding mode Binding energy (Kcal/mol) -7.81  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GSHMEEMIRSLQQRPEPTPEEWDLIHIATEAHRSTNAQGSHWKQRRKFLPDDIGQSPIVSMPDGDKVDLEAFSEFTKIITPAITRVVDFAKKLPMFSELPXEDQIILLKGCCMEIMSLRAAVRYDPESDTLTLSGEMAVKREQLKNGGLGVVSDAIFELGKSLSAFNLDDTEVALLQAVLLMSTDRSGLLXVDKIEKSQEAYLLAFEHYVNHRKHNIPHFWPKLLMKVTDLRMIGAXHASRFLHMKVEXPTELFPPLFLEVFEDQEV Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Glutamate receptor ionotropic kainate 2 (GRIK2) | 5CMM | 5.73 | |
Target general information Gen name GRIK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Glutamate receptor ionotropic, kainate 2; Glutamate receptor 6; GluR6; GluR-6; GluK2; Excitatory amino acid receptor 4; EAA4 Protein family Glutamate-gated ion channel (TC 1.A.10.1) family, GRIK2 subfamily Biochemical class Glutamate-gated ion channel Function L-glutamate acts as an excitatory neurotransmitter at many synapses in the central nervous system. Binding of the excitatory neurotransmitter L-glutamate induces a conformation change, leading to the opening of the cation channel, and thereby converts the chemical signal to an electrical impulse. The receptor then desensitizes rapidly and enters a transient inactive state, characterized by the presence of bound agonist. May be involved in the transmission of light information from the retina to the hypothalamus. Modulates cell surface expression of NETO2. Ionotropic glutamate receptor. Related diseases Intellectual developmental disorder, autosomal recessive 6 (MRT6) [MIM:611092]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT6 patients display mild to severe intellectual disability and psychomotor development delay in early childhood. Patients do not have neurologic problems, congenital malformations, or facial dysmorphism. Body height, weight, and head circumference are normal. {ECO:0000269|PubMed:17847003}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurodevelopmental disorder with impaired language and ataxia and with or without seizures (NEDLAS) [MIM:619580]: An autosomal dominant disorder characterized by axial hypotonia and global developmental delay. Affected individuals show impaired intellectual development, delayed walking, poor speech, and behavioral abnormalities. Some patients have a more severe phenotype with early-onset seizures resembling epileptic encephalopathy, inability to walk or speak, and hypomyelination on brain imaging. {ECO:0000269|PubMed:28180184, ECO:0000269|PubMed:34375587}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03425; DB01351; DB01352; DB01483; DB00237; DB00241; DB01353; DB01496; DB02852; DB00142; DB01354; DB01355; DB00463; DB00849; DB00312; DB01174; DB00794; DB02999; DB00418; DB00306; DB00599; DB00273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Epilepsy; Glycoprotein; Intellectual disability; Ion channel; Ion transport; Isopeptide bond; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; RNA editing; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 29150.1 Length 257 Aromaticity 0.1 Instability index 35.11 Isoelectric point 5.89 Charge (pH=7) -2.05 2D Binding mode Binding energy (Kcal/mol) -7.81  Molscript Map 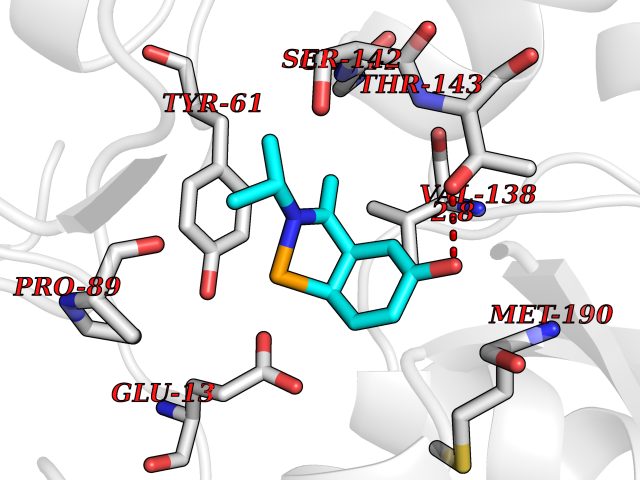 Pymol Map 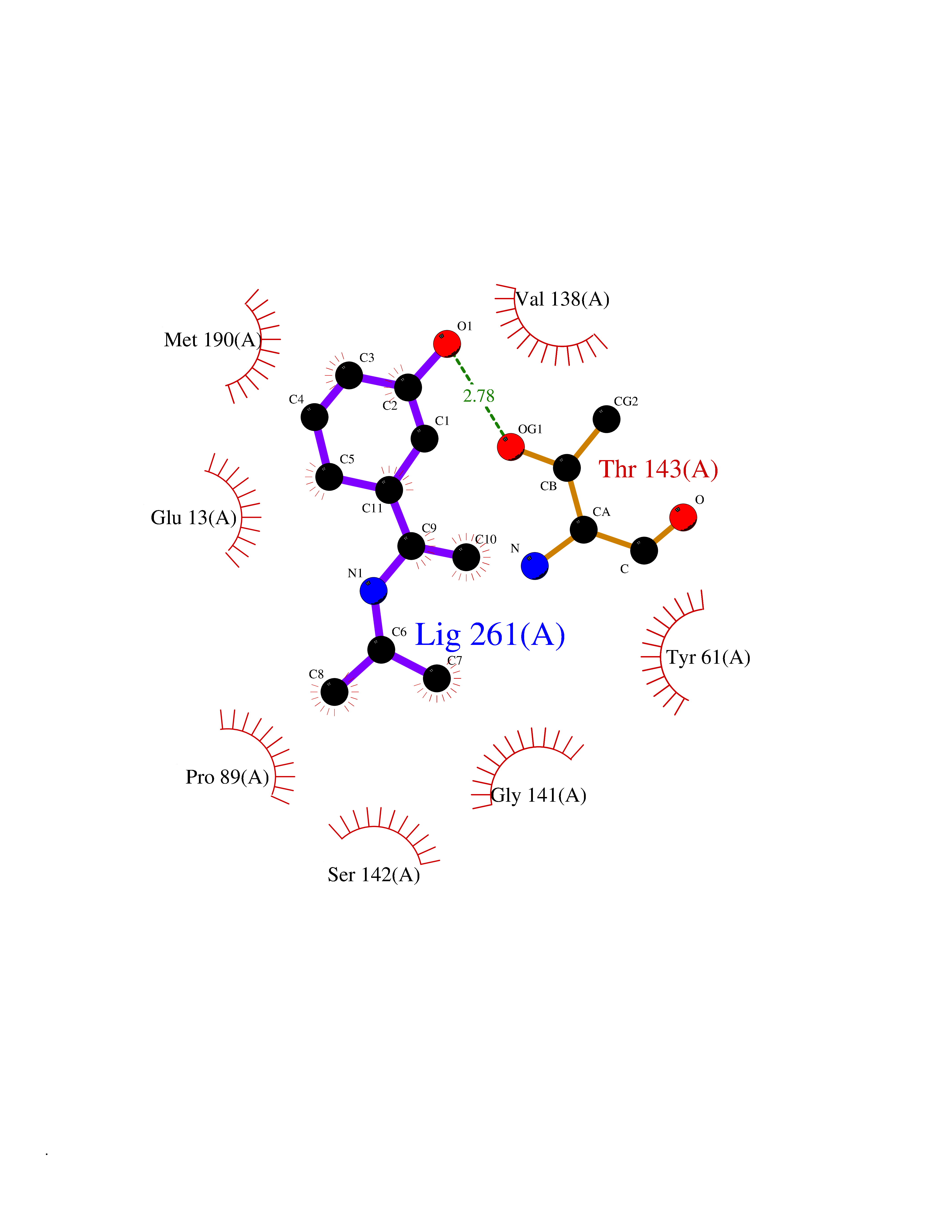 Ligplot Map 3D Binding mode Sequence GSNRSLIVTTILEEPYVLFKKSDKPLYGNDRFEGYCIDLLRELSTILGFTYEIRLVEDGKYGAQDDVNGQWNGMVRELIDHKADLAVAPLTITYVREKVIDFSKPFMTLGISILYRKGTPIDSADDLAKQTKIEYGAVEDGSTMTFFKKSKISTYDKMWAFMSSRRQSVLVKSSEEGIQRVLTSDYALLMESTTIEFVTQRNCNLTQIGGLIDSKGYGVGTPMGSPYRDKITIAILQLQEEGKLHMMKEKWWRGCPE Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Lysine-specific demethylase 7A (KDM7A) | 3KVB | 5.73 | |
Target general information Gen name KDM7A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine-specific demethylase 7; KIAA1718; KDM7; JmjC domain-containing histone demethylation protein 1D; JHDM1D Protein family JHDM1 histone demethylase family, JHDM1D subfamily Biochemical class NA Function Histone demethylase required for brain development. Specifically demethylates dimethylated 'Lys-9' and 'Lys-27' (H3K9me2 and H3K27me2, respectively) of histone H3 and monomethylated histone H4 'Lys-20' residue (H4K20Me1), thereby playing a central role in histone code. Specifically binds trimethylated 'Lys-4' of histone H3 (H3K4me3), affecting histone demethylase specificity: in presence of H3K4me3, it has no demethylase activity toward H3K9me2, while it has high activity toward H3K27me2. Demethylates H3K9me2 in absence of H3K4me3. Has activity toward H4K20Me1 only when nucleosome is used as a substrate and when not histone octamer is used as substrate. Related diseases MUC1/CA 15-3 is used as a serological clinical marker of breast cancer to monitor response to breast cancer treatment and disease recurrence (PubMed:20816948). Decreased levels over time may be indicative of a positive response to treatment. Conversely, increased levels may indicate disease progression. At an early stage disease, only 21% of patients exhibit high MUC1/CA 15-3 levels, that is why CA 15-3 is not a useful screening test. Most antibodies target the highly immunodominant core peptide domain of 20 amino acid (APDTRPAPGSTAPPAHGVTS) tandem repeats. Some antibodies recognize glycosylated epitopes. {ECO:0000269|PubMed:20816948}.; DISEASE: Tubulointerstitial kidney disease, autosomal dominant, 2 (ADTKD2) [MIM:174000]: A form of autosomal dominant tubulointerstitial kidney disease, a genetically heterogeneous disorder characterized by slowly progressive loss of kidney function, bland urinary sediment, hyperuricemia, absent or mildly increased albuminuria, lack of severe hypertension during the early stages, and normal or small kidneys on ultrasound. Renal histology shows variable abnormalities including interstitial fibrosis with tubular atrophy, microcystic dilatation of the tubules, thickening of tubular basement membranes, medullary cysts, and secondary glomerulosclerotic or glomerulocystic changes with abnormal glomerular tufting. There is significant variability, as well as incomplete penetrance. {ECO:0000269|PubMed:23396133}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.14.11.- Uniprot keywords 3D-structure; Alternative splicing; Chromatin regulator; Dioxygenase; Iron; Metal-binding; Neurogenesis; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 42365.5 Length 366 Aromaticity 0.12 Instability index 41.95 Isoelectric point 6.3 Charge (pH=7) -3.09 2D Binding mode Binding energy (Kcal/mol) -7.82  Molscript Map 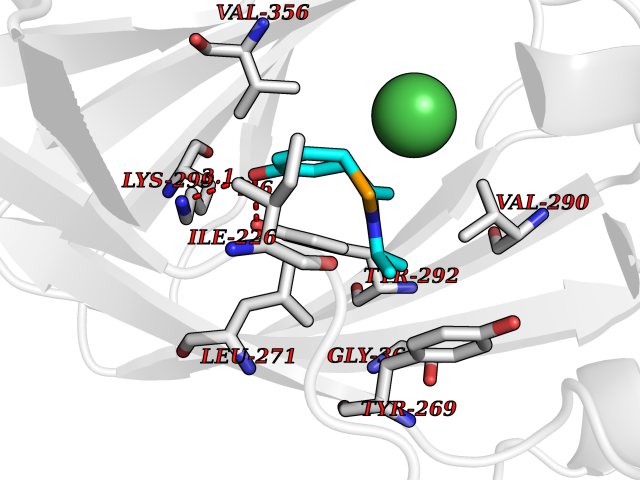 Pymol Map 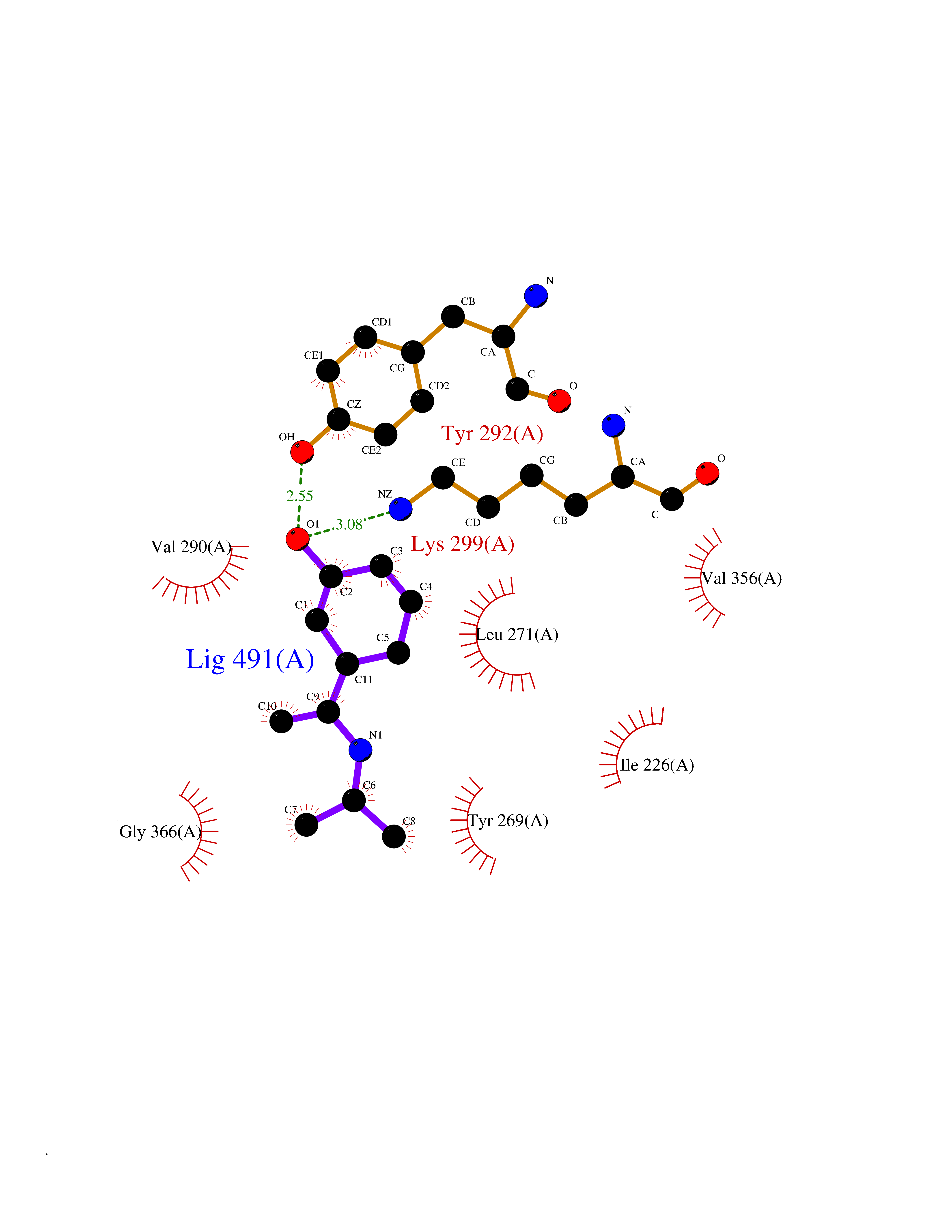 Ligplot Map 3D Binding mode Sequence PVQAGTRTFIKELRSRVFPSADEIIIKMHGSQLTQRYLEKHGFDVPIMVPKLDDLGLRLPSPTFSVMDVERYVGGDKVIDVIDVARQADSKMTLHNYVKYFMNPNRPKVLNVISLEFSDTKMSELVEVPDIAKKLSWVENYWPDDSVFPKPFVQKYCLMGVQDSYTDFHIDFGGTSVWYHVLWGEKIFYLIKPTDENLARYESWSSSVTQSEVFFGDKVDKCYKCVVKQGHTLFVPTGWIHAVLTSQDCMAFGGNFLHNLNIGMQLRCYEMEKRLKTPDLFKFPFFEAICWFVAKNLLETLKELREDGFQPQTYLVQGVKALHTALKLWMKKELVSEHAFEIPDNVRPGHLIKELSKVIRAIEEEN Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Acetylcholine receptor subunit alpha | 4ZJS | 5.72 | |
Target general information Gen name CHRNA1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ACHRA;CHNRA Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-1/CHRNA1 sub-subfamily Biochemical class Immune system Function Acetylcholine binding.Acetylcholine-gated cation-selective channel activity.Acetylcholine receptor activity.Ion channel activity.Ligand-gated ion channel activity. Related diseases Multiple pterygium syndrome, lethal type (LMPS) [MIM:253290]: Multiple pterygia are found infrequently in children with arthrogryposis and in fetuses with fetal akinesia syndrome. In lethal multiple pterygium syndrome there is intrauterine growth retardation, multiple pterygia, and flexion contractures causing severe arthrogryposis and fetal akinesia. Subcutaneous edema can be severe, causing fetal hydrops with cystic hygroma and lung hypoplasia. Oligohydramnios and facial anomalies are frequent. {ECO:0000269|PubMed:18252226}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: The alpha subunit is the main focus for antibody binding in myasthenia gravis. Myasthenia gravis is characterized by sporadic muscular fatigability and weakness, occurring chiefly in muscles innervated by cranial nerves, and characteristically improved by cholinesterase-inhibiting drugs.; DISEASE: Myasthenic syndrome, congenital, 1A, slow-channel (CMS1A) [MIM:601462]: A common congenital myasthenic syndrome. Congenital myasthenic syndromes are characterized by muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS1A is a slow-channel myasthenic syndrome. It is caused by kinetic abnormalities of the AChR, resulting in prolonged AChR channel opening episodes, prolonged endplate currents, and depolarization block. This is associated with calcium overload, which may contribute to subsequent degeneration of the endplate and postsynaptic membrane. {ECO:0000269|PubMed:16685696, ECO:0000269|PubMed:7619526, ECO:0000269|PubMed:8872460, ECO:0000269|PubMed:9158151, ECO:0000269|PubMed:9221765}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myasthenic syndrome, congenital, 1B, fast-channel (CMS1B) [MIM:608930]: A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS1B is a fast-channel myasthenic syndrome. It is caused by kinetic abnormalities of the AChR, resulting in brief opening and activity of the channel, with a rapid decay in endplate current, failure to achieve threshold depolarization of the endplate and consequent failure to fire an action potential. {ECO:0000269|PubMed:10195214, ECO:0000269|PubMed:12588888, ECO:0000269|PubMed:15079006}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08838; DB00565; DB00555 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Congenital myasthenic syndrome; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A,B,C,D,E Molecular weight (Da) 46717.8 Length 411 Aromaticity 0.11 Instability index 38.02 Isoelectric point 4.77 Charge (pH=7) -22.31 2D Binding mode Binding energy (Kcal/mol) -7.8  Molscript Map 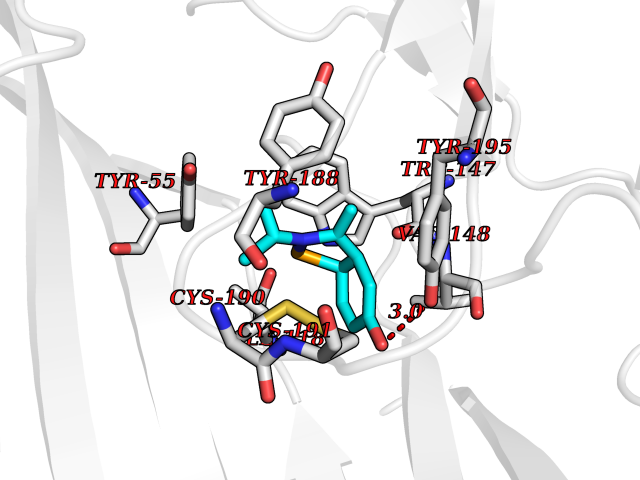 Pymol Map  Ligplot Map 3D Binding mode Sequence EHETRLVAKLFKDYSSVVRPVEDHRQVVEVTLGFTLQDIVKADSSTNEVDLVYYEQQRWVDYNLKWNPDDYGGVKKIHIPAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQTRQVQHYSCCPEPYIDVNLVVKFREEHETRLVAKLFKDYSSVVRPVEDHRQVVEVTLGFTLQDIVKADSSTNEVDLVYYEQQRWVDYNLKWNPDDYGGVKKIHIPAADIWTPDITAYSSTRPVQVLSPQIAVVTHDGSVMFIPAQRLSFMCDPTGVDSEEGATCAVKFGSWVYSGFEIDLKTDTDQVDLSSYYASSKYEILSATQTRQVQHYSCCPEPYIDVNLVVKFRER Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Muscarinic acetylcholine receptor M4 (CHRM4) | 5DSG | 5.72 | |
Target general information Gen name CHRM4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms M4 receptor; CHRM4 Protein family G-protein coupled receptor 1 family, Muscarinic acetylcholine receptor subfamily, CHRM4 sub-subfamily Biochemical class GPCR rhodopsin Function The muscarinic acetylcholine receptor mediates various cellular responses, including inhibition of adenylate cyclase, breakdown of phosphoinositides and modulation of potassium channels through the action of G proteins. Primary transducing effect is inhibition of adenylate cyclase. Related diseases Leukodystrophy, hypomyelinating, 15 (HLD15) [MIM:617951]: An autosomal recessive disorder characterized by hypomyelinating leukodystrophy with thinning of the corpus callosum. Clinical features include motor and cognitive impairment appearing in the first or second decade of life, dystonia, ataxia, spasticity, and dysphagia. Most patients develop severe optic atrophy, and some have hearing loss. {ECO:0000269|PubMed:29576217}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03128; DB08897; DB05752; DB00321; DB00543; DB01238; DB14185; DB00572; DB00767; DB01019; DB00835; DB00411; DB01239; DB00568; DB00363; DB00496; DB01151; DB09167; DB01142; DB00366; DB09194; DB06702; DB00986; DB06787; DB11181; DB00725; DB00424; DB00458; DB01625; DB01221; DB00408; DB00934; DB00454; DB06709; DB00940; DB01403; DB00340; DB00622; DB00540; DB00334; DB00715; DB01085; DB00387; DB01069; DB00777; DB12278; DB01224; DB11855; DB13581; DB00747; DB01591; DB00342; DB11235; DB01409; DB01036; DB00376; DB09089; DB00726; DB00809; DB09076; DB09185; DB00246 Interacts with NA EC number NA Uniprot keywords 3D-structure; Cell membrane; Disulfide bond; G-protein coupled receptor; Glycoprotein; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Synapse; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 32545.4 Length 287 Aromaticity 0.14 Instability index 30.63 Isoelectric point 9.34 Charge (pH=7) 11.91 2D Binding mode Binding energy (Kcal/mol) -7.8  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence HNRYETVEMVFIATVTGSLSLVTVVGNILVMLSIKVNRQLQTVNNYFLFSLACADLIIGAFSMNLYTVYIIKGYWPLGAVVCDLWLALDYVVSNASVMNLLIISFDRYFCVTKPLTYPARRTTKMAGLMIAAAWVLSFVLWAPAILFWQFVVGKRTVPDNQCFIQFLSNPAVTFGTAIAAFYLPVVIMTVLYIHISLASRSRVQMAARERKVTRTIFAILLAFILTWTPYNVMVLVNTFCQSCIPDTVWSIGYWLCYVNSTINPACYALCNATFKKTFRHLLLCQYR Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Guanidinoacetate N-methyltransferase | 3ORH | 5.72 | |
Target general information Gen name GAMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, RMT2 methyltransferase family Biochemical class Transferase Function Guanidinoacetate N-methyltransferase activity.Methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 2 (CCDS2) [MIM:612736]: An autosomal recessive disorder characterized by developmental delay and regression, intellectual disability, severe disturbance of expressive and cognitive speech, intractable seizures, movement disturbances, severe depletion of creatine and phosphocreatine in the brain, and accumulation of guanidinoacetic acid in brain and body fluids. {ECO:0000269|PubMed:12468279, ECO:0000269|PubMed:15108290, ECO:0000269|PubMed:15651030, ECO:0000269|PubMed:16293431, ECO:0000269|PubMed:16855203, ECO:0000269|PubMed:17101918, ECO:0000269|PubMed:17466557, ECO:0000269|PubMed:19388150, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:24415674, ECO:0000269|PubMed:8651275}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00148; DB02751; DB00536; DB13191; DB01752 Interacts with O95363; Q969Q5; Q9HCM9-2 EC number 2.1.1.2 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Disease variant; Methyltransferase; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 24656 Length 219 Aromaticity 0.11 Instability index 46.5 Isoelectric point 5.91 Charge (pH=7) -4.34 2D Binding mode Binding energy (Kcal/mol) -7.8  Molscript Map 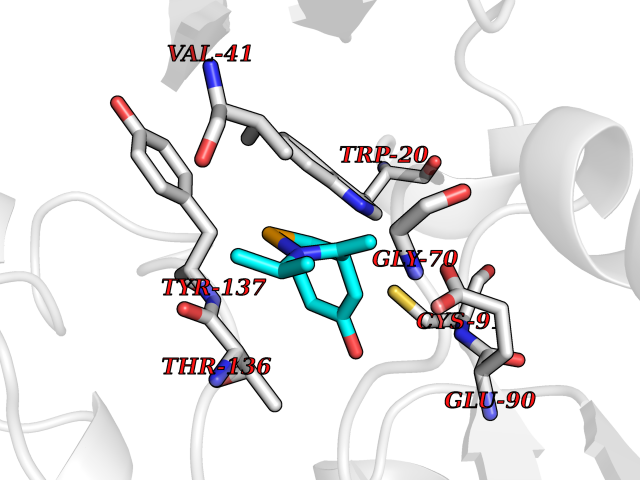 Pymol Map 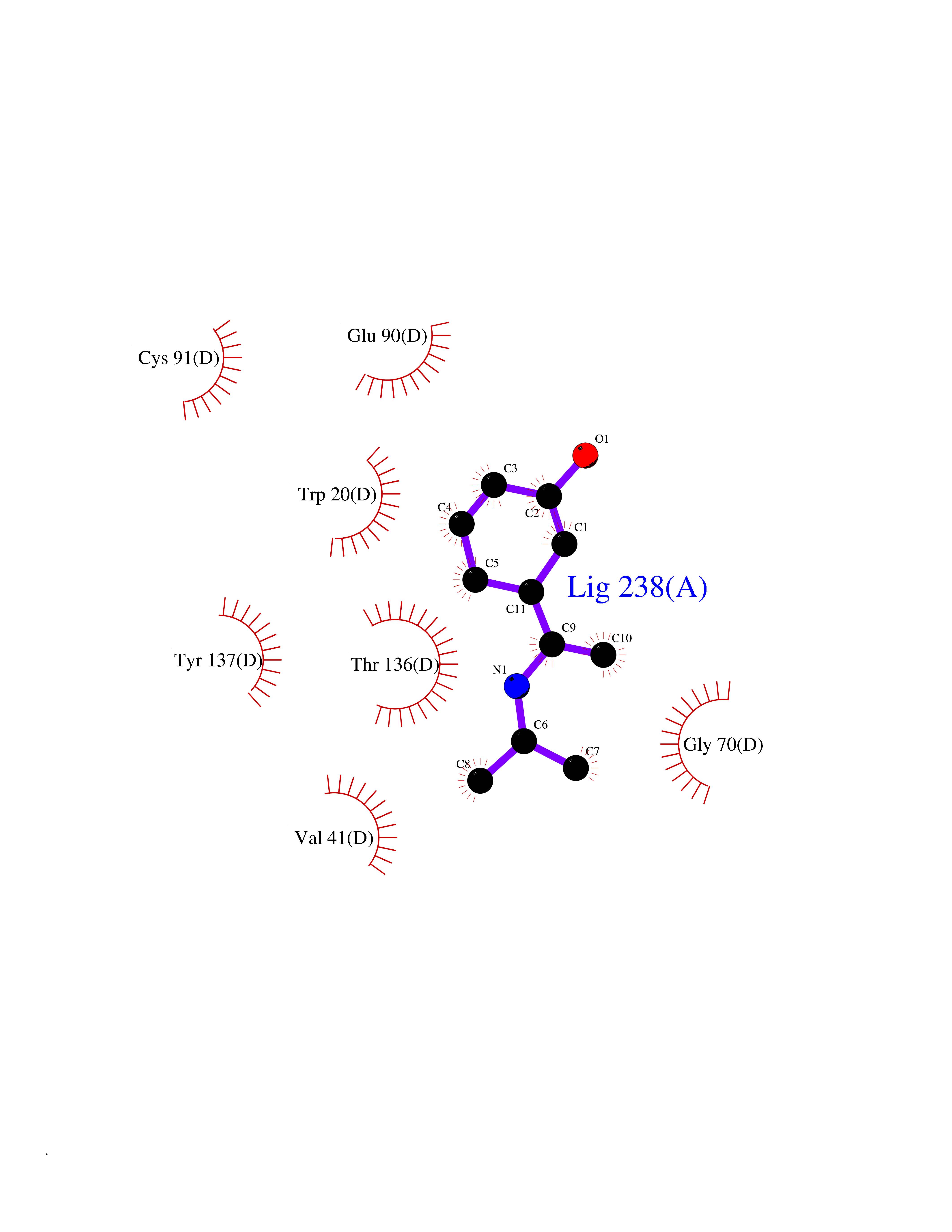 Ligplot Map 3D Binding mode Sequence PAWGAAPAAYDAADTHLRILGKPVMERWETPYMHALAAAASSKGGRVLEVGFGMAIAASKVQEAPIDEHWIIECNDGVFQRLRDWAPRQTHKVIPLKGLWEDVAPTLPDGHFDGILYDTYPLSEETWHTHQFNFIKNHAFRLLKPGGVLTYCNLTSWGELMKSKYSDITIMFEETQVPALLEAGFRRENIRTEVMALVPPADCRYYAFPQMITPLVTKG Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Pyruvate dehydrogenase kinase 1 (PDHK1) | 2Q8G | 5.72 | |
Target general information Gen name PDK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Pyruvate dehydrogenase kinase isoform 1; Pyruvate dehydrogenase (acetyl-transferring) kinase isozyme 1, mitochondrial; PDHK1; PDH kinase 1 Protein family PDK/BCKDK protein kinase family Biochemical class Kinase Function Kinase that plays a key role in regulation of glucose and fatty acid metabolism and homeostasis via phosphorylation of the pyruvate dehydrogenase subunits PDHA1 and PDHA2. This inhibits pyruvate dehydrogenase activity, and thereby regulates metabolite flux through the tricarboxylic acid cycle, down-regulates aerobic respiration and inhibits the formation of acetyl-coenzyme A from pyruvate. Plays an important role in cellular responses to hypoxia and is important for cell proliferation under hypoxia. Protects cells against apoptosis in response to hypoxia and oxidative stress. Related diseases TP53 is found in increased amounts in a wide variety of transformed cells. TP53 is frequently mutated or inactivated in about 60% of cancers. TP53 defects are found in Barrett metaplasia a condition in which the normally stratified squamous epithelium of the lower esophagus is replaced by a metaplastic columnar epithelium. The condition develops as a complication in approximately 10% of patients with chronic gastroesophageal reflux disease and predisposes to the development of esophageal adenocarcinoma.; DISEASE: Esophageal cancer (ESCR) [MIM:133239]: A malignancy of the esophagus. The most common types are esophageal squamous cell carcinoma and adenocarcinoma. Cancer of the esophagus remains a devastating disease because it is usually not detected until it has progressed to an advanced incurable stage. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Li-Fraumeni syndrome (LFS) [MIM:151623]: An autosomal dominant familial cancer syndrome that in its classic form is defined by the existence of a proband affected by a sarcoma before 45 years with a first degree relative affected by any tumor before 45 years and another first degree relative with any tumor before 45 years or a sarcoma at any age. Other clinical definitions for LFS have been proposed and called Li-Fraumeni like syndrome (LFL). In these families affected relatives develop a diverse set of malignancies at unusually early ages. Four types of cancers account for 80% of tumors occurring in TP53 germline mutation carriers: breast cancers, soft tissue and bone sarcomas, brain tumors (astrocytomas) and adrenocortical carcinomas. Less frequent tumors include choroid plexus carcinoma or papilloma before the age of 15, rhabdomyosarcoma before the age of 5, leukemia, Wilms tumor, malignant phyllodes tumor, colorectal and gastric cancers. {ECO:0000269|PubMed:10484981, ECO:0000269|PubMed:1565144, ECO:0000269|PubMed:1737852, ECO:0000269|PubMed:1933902, ECO:0000269|PubMed:1978757, ECO:0000269|PubMed:2259385, ECO:0000269|PubMed:36108750, ECO:0000269|PubMed:7887414, ECO:0000269|PubMed:8825920, ECO:0000269|PubMed:9452042}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Squamous cell carcinoma of the head and neck (HNSCC) [MIM:275355]: A non-melanoma skin cancer affecting the head and neck. The hallmark of cutaneous SCC is malignant transformation of normal epidermal keratinocytes. The gene represented in this entry is involved in disease pathogenesis.; DISEASE: Lung cancer (LNCR) [MIM:211980]: A common malignancy affecting tissues of the lung. The most common form of lung cancer is non-small cell lung cancer (NSCLC) that can be divided into 3 major histologic subtypes: squamous cell carcinoma, adenocarcinoma, and large cell lung cancer. NSCLC is often diagnosed at an advanced stage and has a poor prognosis. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Papilloma of choroid plexus (CPP) [MIM:260500]: A benign tumor of neuroectodermal origin that generally occurs in childhood, but has also been reported in adults. Although generally found within the ventricular system, choroid plexus papillomas can arise ectopically in the brain parenchyma or disseminate throughout the neuraxis. Patients present with signs and symptoms of increased intracranial pressure including headache, hydrocephalus, papilledema, nausea, vomiting, cranial nerve deficits, gait impairment, and seizures. {ECO:0000269|PubMed:12085209}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Adrenocortical carcinoma (ADCC) [MIM:202300]: A malignant neoplasm of the adrenal cortex and a rare childhood tumor. It occurs with increased frequency in patients with Beckwith-Wiedemann syndrome and Li-Fraumeni syndrome. {ECO:0000269|PubMed:11481490}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Basal cell carcinoma 7 (BCC7) [MIM:614740]: A common malignant skin neoplasm that typically appears on hair-bearing skin, most commonly on sun-exposed areas. It is slow growing and rarely metastasizes, but has potentialities for local invasion and destruction. It usually develops as a flat, firm, pale area that is small, raised, pink or red, translucent, shiny, and waxy, and the area may bleed following minor injury. Tumor size can vary from a few millimeters to several centimeters in diameter. {ECO:0000269|PubMed:21946351}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Bone marrow failure syndrome 5 (BMFS5) [MIM:618165]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFS5 is an autosomal dominant form characterized by infantile onset of severe red cell anemia requiring transfusion. Additional features include hypogammaglobulinemia, poor growth with microcephaly, developmental delay, and seizures. {ECO:0000269|PubMed:30146126}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07403; DB08809 Interacts with P05067; P08559; Q16513; P31749-1; P31751-1 EC number EC 2.7.11.2 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Kinase; Mitochondrion; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Transit peptide Protein physicochemical properties Chain ID A Molecular weight (Da) 42249.9 Length 368 Aromaticity 0.11 Instability index 49.91 Isoelectric point 6.83 Charge (pH=7) -0.46 2D Binding mode Binding energy (Kcal/mol) -7.81  Molscript Map 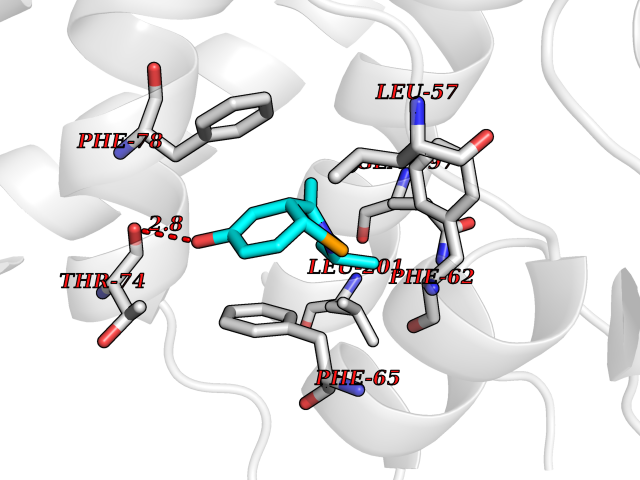 Pymol Map 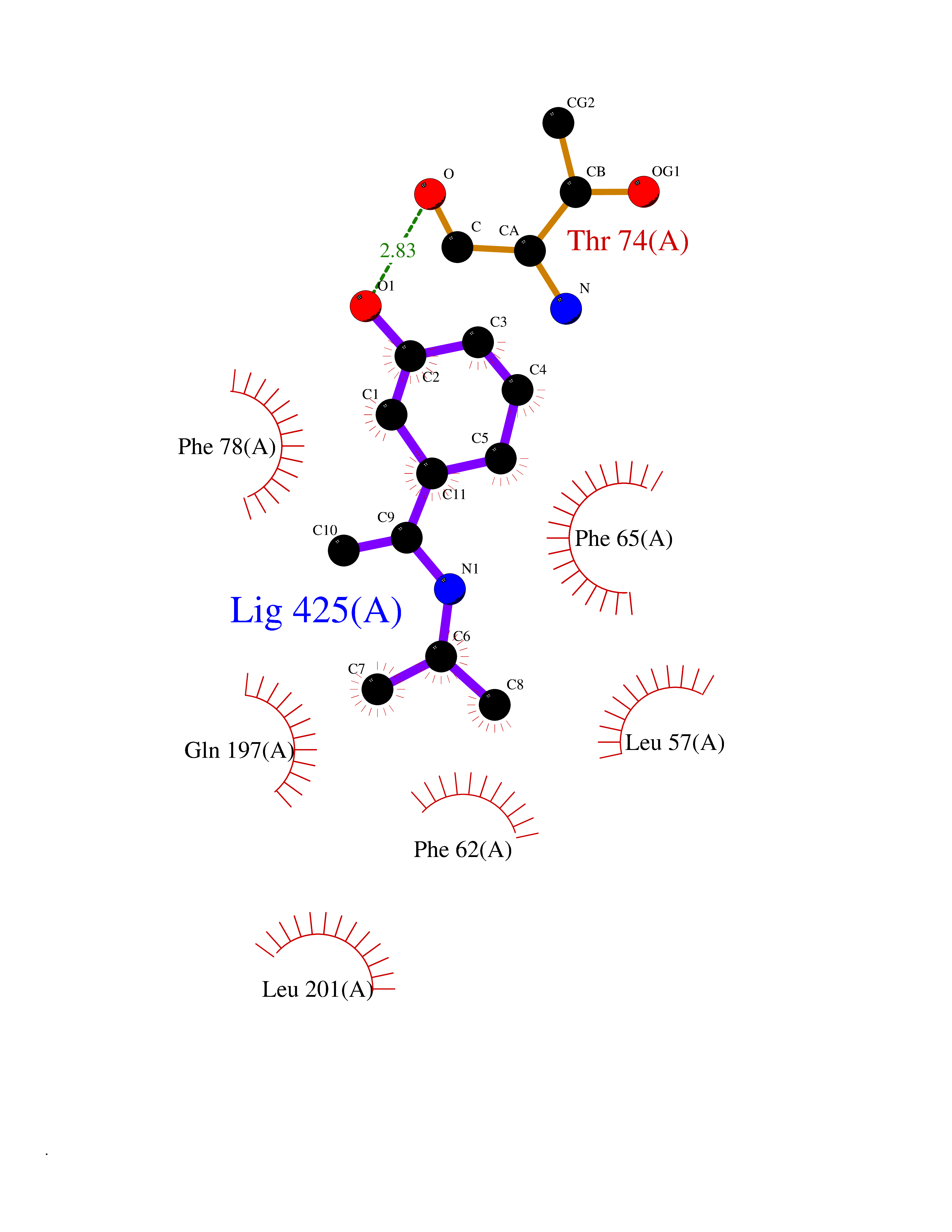 Ligplot Map 3D Binding mode Sequence GVPGQVDFYARFSPSPLSMKQFLDFGSVNACEKTSFMFLRQELPVRLANIMKEISLLPDNLLRTPSVQLVQSWYIQSLQELLDFKDKSAEDAKAIYDFTDTVIRIRNRHNDVIPTMAQGVIEYKESFDPVTSQNVQYFLDRFYMSRISIRMLLNQHSLLFGKHIGSINPNCNVLEVIKDGYENARRLCDLYYINSPELELEELNAKSPGQPIQVVYVPSHLYHMVFELFKNAMRATMEHHANRGVYPPIQVHVTLGNEDLTVKMSDRGGGVPLRKIDRLFNYMYSTAPRPRVETSRAVPLAGFGYGLPISRLYAQYFQGDLKLYSLEGYGTDAVIYIKALSTDSIERLPVYNKAAWKHYNTNDDWCVP Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Hypoxia-inducible factor 2 alpha (HIF-2A) | 5TBM | 5.72 | |
Target general information Gen name EPAS1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms bHLHe73; PASD2; PAS domain-containing protein 2; Member of PAS protein 2; MOP2; Hypoxia-inducible factor 2-alpha; HLF; HIF2A; HIF2-alpha; HIF-2-alpha; HIF-1-alpha-like factor; Endothelial PAS domain-c Protein family NA Biochemical class NA Function Heterodimerizes with ARNT; heterodimer binds to core DNA sequence 5'-TACGTG-3' within the hypoxia response element (HRE) of target gene promoters. Regulates the vascular endothelial growth factor (VEGF) expression and seems to be implicated in the development of blood vessels and the tubular system of lung. May also play a role in the formation of the endothelium that gives rise to the blood brain barrier. Potent activator of the Tie-2 tyrosine kinase expression. Activation requires recruitment of transcriptional coactivators such as CREBBP and probably EP300. Interaction with redox regulatory protein APEX seems to activate CTAD. Transcription factor involved in the induction of oxygen regulated genes. Related diseases Erythrocytosis, familial, 4 (ECYT4) [MIM:611783]: An autosomal dominant disorder characterized by elevated serum hemoglobin and hematocrit, and normal platelet and leukocyte counts. {ECO:0000269|PubMed:18184961, ECO:0000269|PubMed:18378852, ECO:0000269|PubMed:19208626, ECO:0000269|PubMed:22367913}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB15463; DB12255 Interacts with P27540; Q96RK4; O00327-8; Q8WYA1-3; Q9GZT9; P60228; O60573; P09467; P61244; Q9BWF3-1; P08047; Q9Y2K6; P40818 EC number NA Uniprot keywords 3D-structure; Activator; Angiogenesis; Congenital erythrocytosis; Developmental protein; Differentiation; Disease variant; DNA-binding; Host-virus interaction; Hydroxylation; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 12249.8 Length 106 Aromaticity 0.11 Instability index 40.77 Isoelectric point 5.25 Charge (pH=7) -5.82 2D Binding mode Binding energy (Kcal/mol) -7.81 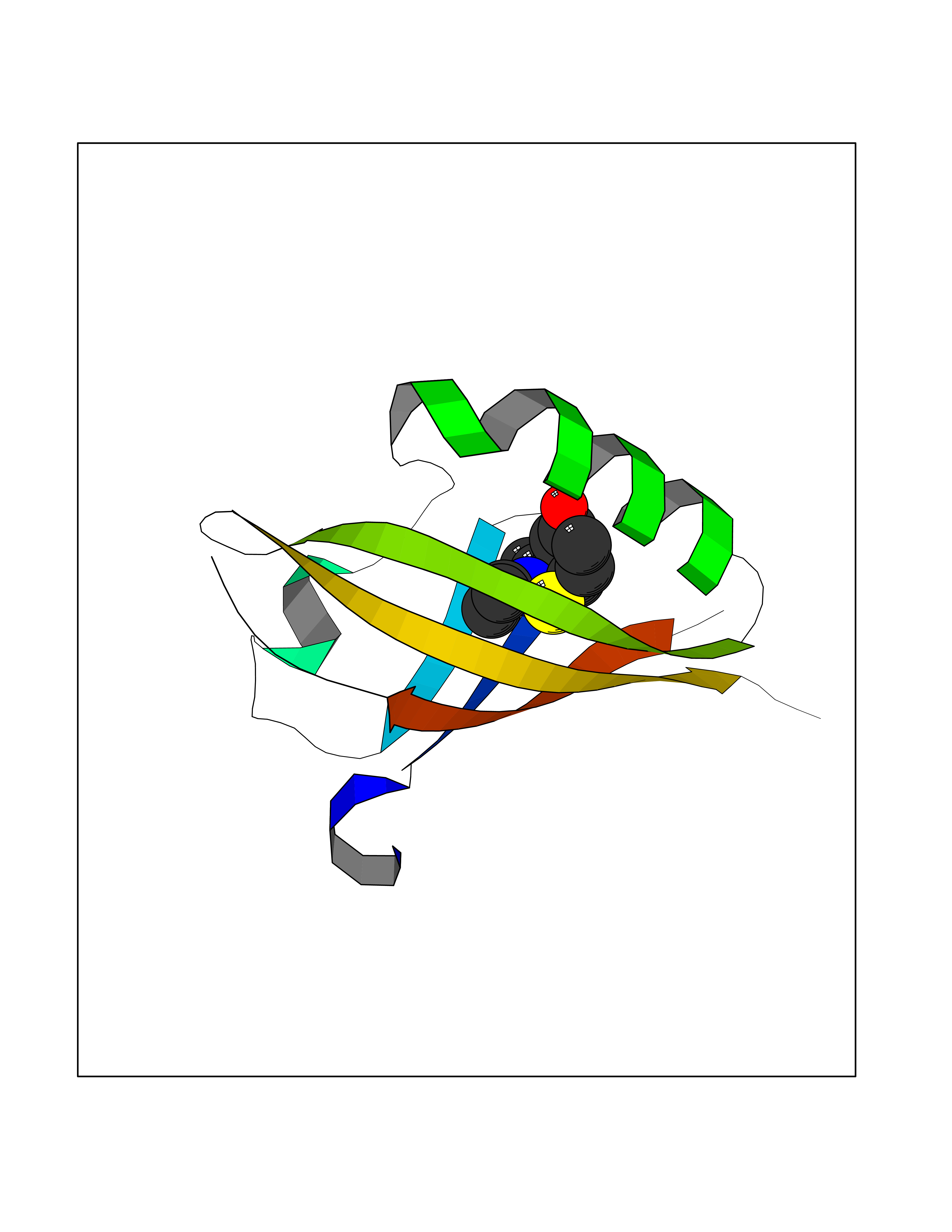 Molscript Map 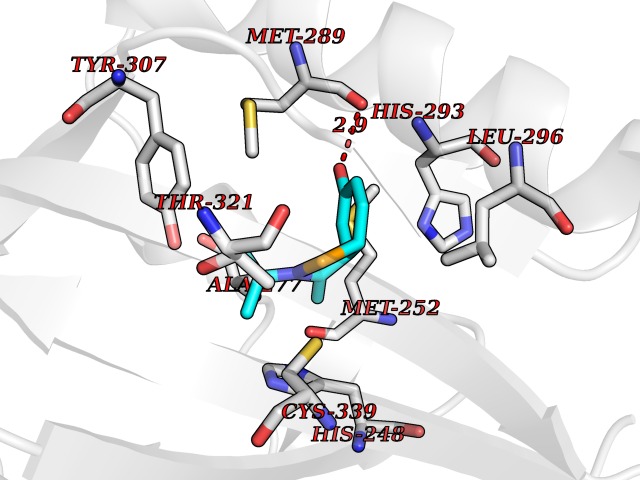 Pymol Map 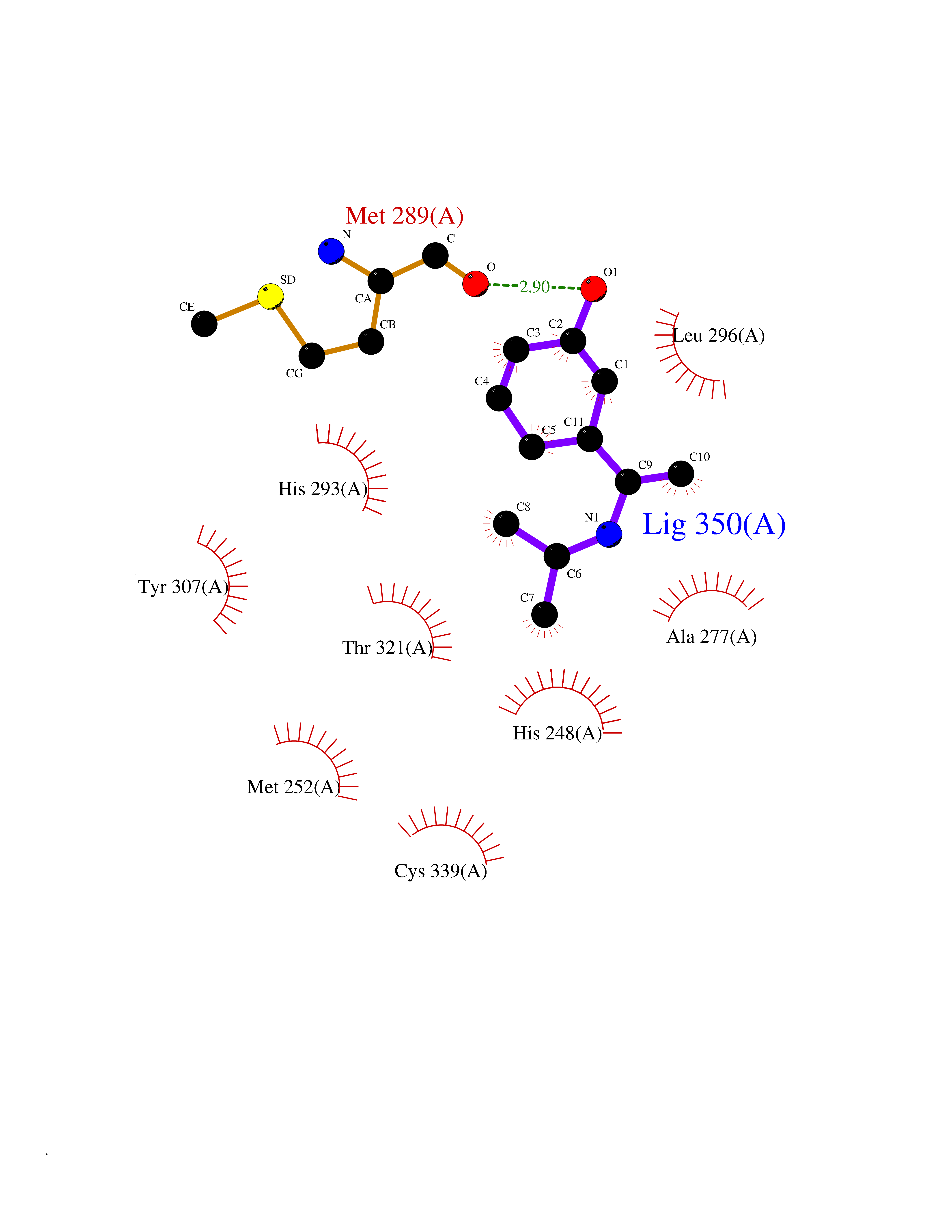 Ligplot Map 3D Binding mode Sequence LDSKTFLSEHSMDMKFTYCDDRITELIGYHPEELLGRSAYEFYHALDSENMTKSHQNLCTKGQVVSGQYRMLAKHGGYVWLETQGTVIYNPPQCIMCVNYVLSEIE Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Protein kinase G2 (PRKG2) | 5BV6 | 5.72 | |
Target general information Gen name PRKG2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cGMP-dependent protein kinase II; cGMP-dependent protein kinase 2; cGKII; cGK2; cGK 2; PRKGR2 Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family, cGMP subfamily Biochemical class NA Function Crucial regulator of intestinal secretion and bone growth (By similarity). Phosphorylates and activates CFTR on the plasma membrane. Plays a key role in intestinal secretion by regulating cGMP-dependent translocation of CFTR in jejunum (By similarity). Acts downstream of NMDAR to activate the plasma membrane accumulation of GRIA1/GLUR1 in synapse and increase synaptic plasticity. Phosphorylates GRIA1/GLUR1 at Ser-863 (By similarity). Acts as regulator of gene expression and activator of the extracellular signal-regulated kinases MAPK3/ERK1 and MAPK1/ERK2 in mechanically stimulated osteoblasts. Under fluid shear stress, mediates ERK activation and subsequent induction of FOS, FOSL1/FRA1, FOSL2/FRA2 and FOSB that play a key role in the osteoblast anabolic response to mechanical stimulation (By similarity). Related diseases Spondylometaphyseal dysplasia, Pagnamenta type (SMDP) [MIM:619638]: A form of spondylometaphyseal dysplasia, a group of short stature disorders distinguished by abnormalities in the vertebrae and the metaphyses of the tubular bones. SMDP is an autosomal recessive form characterized by short stature and mild platyspondyly with no disproportion between the limbs. Mild metaphyseal changes are present. {ECO:0000269|PubMed:34782440}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Acromesomelic dysplasia 4 (AMD4) [MIM:619636]: A form of acromesomelic dysplasia, a skeletal disorder characterized by short stature, very short limbs and hand/foot malformations. The severity of limb abnormalities increases from proximal to distal with profoundly affected hands and feet showing brachydactyly and/or rudimentary fingers (knob-like fingers). AMD4 radiographic hallmarks include mild to moderate platyspondyly, moderate brachydactyly, iliac flaring, and metaphyseal alterations of the long bones that progressively increase with age. AMD4 inheritance is autosomal recessive. {ECO:0000269|PubMed:33106379, ECO:0000269|PubMed:34782440}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with NA EC number EC 2.7.11.12 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; cGMP; cGMP-binding; Coiled coil; Disease variant; Dwarfism; Kinase; Lipoprotein; Membrane; Myristate; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 17227.1 Length 150 Aromaticity 0.09 Instability index 43.7 Isoelectric point 4.88 Charge (pH=7) -8.41 2D Binding mode Binding energy (Kcal/mol) -7.81 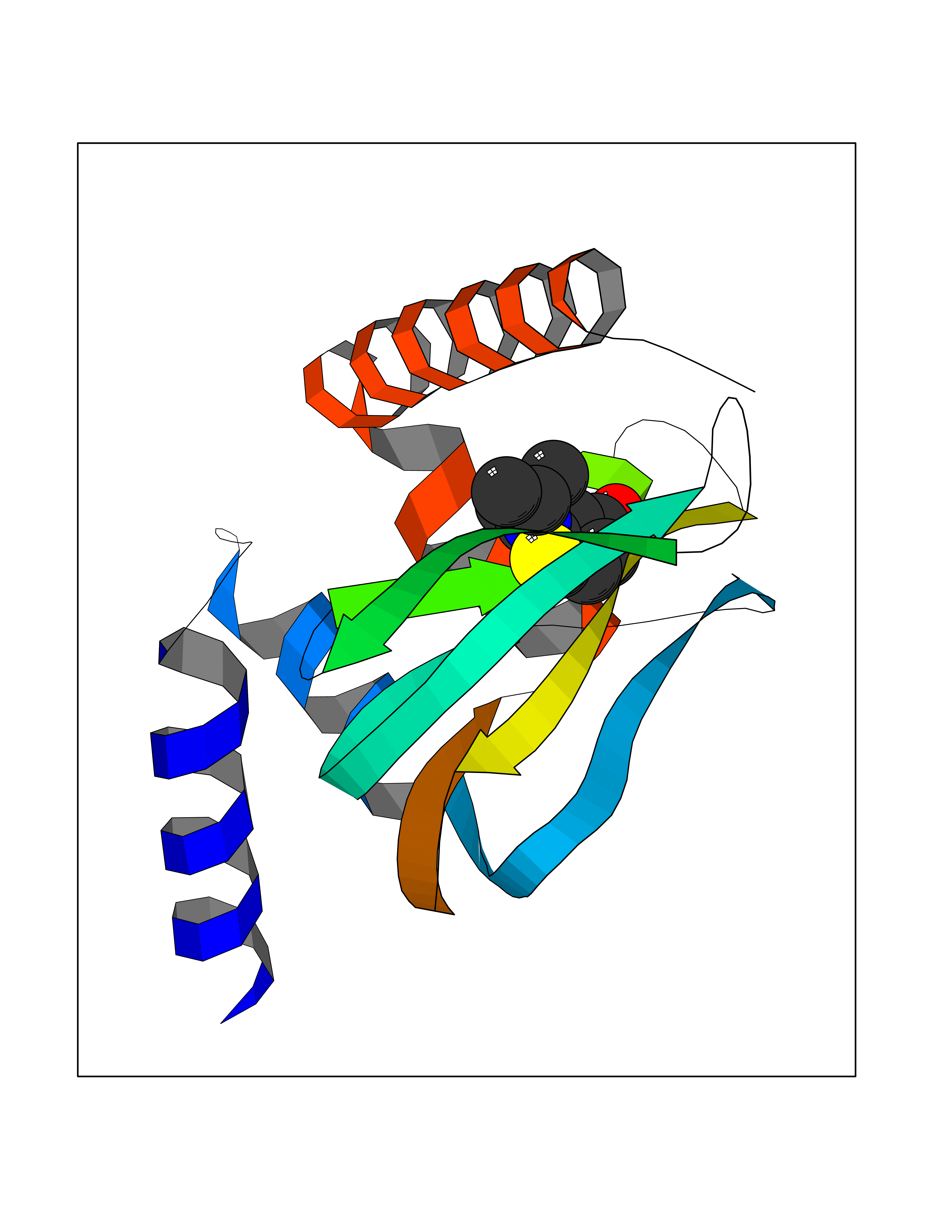 Molscript Map 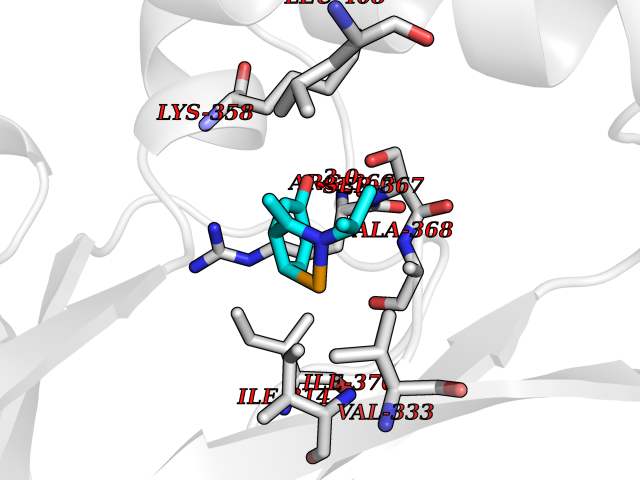 Pymol Map 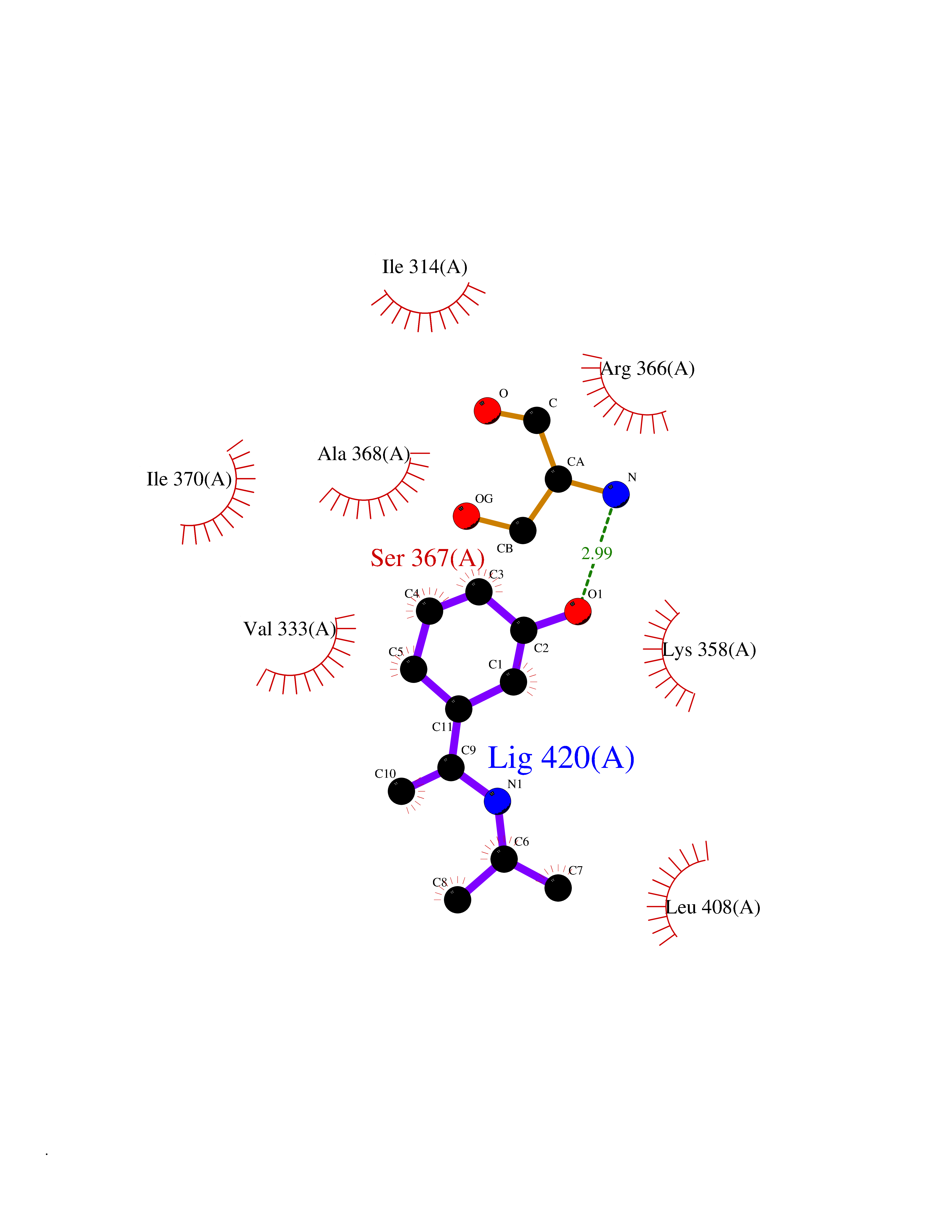 Ligplot Map 3D Binding mode Sequence TAQARDEQYRNFLRSVSLLKNLPEDKLTKIIDCLEVEYYDKGDYIIREGEEGSTFFILAKGKVKVTQSTEGHDQPQLIKTLQKGEYFGEKALISDDVRSANIIAEENDVACLVIDRETFNQTVGTFEELQKYLEGYVANLNRDDEKRHAK Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Soluble epoxide hydrolase (EPHX2) | 1ZD3 | 5.72 | |
Target general information Gen name EPHX2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Bifunctional epoxide hydrolase 2 Protein family AB hydrolase superfamily, Epoxide hydrolase family Biochemical class Ether bond hydrolase Function Bifunctional enzyme. The C-terminal domain has epoxide hydrolase activity and acts on epoxides (alkene oxides, oxiranes) and arene oxides. Plays a role in xenobiotic metabolism by degrading potentially toxic epoxides (By similarity). Also determines steady-state levels of physiological mediators. The N-terminal domain has lipid phosphatase activity, with the highest activity towards threo-9,10-phosphonooxy-hydroxy-octadecanoic acid, followed by erythro-9,10-phosphonooxy-hydroxy-octadecanoic acid, 12-phosphonooxy-octadec-9Z-enoic acid and 12-phosphonooxy-octadec-9E-enoic acid. Related diseases Leukemia, juvenile myelomonocytic (JMML) [MIM:607785]: An aggressive pediatric myelodysplastic syndrome/myeloproliferative disorder characterized by malignant transformation in the hematopoietic stem cell compartment with proliferation of differentiated progeny. Patients have splenomegaly, enlarged lymph nodes, rashes, and hemorrhages. {ECO:0000269|PubMed:17332249}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Noonan syndrome 6 (NS6) [MIM:613224]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. {ECO:0000269|PubMed:19966803}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: RAS-associated autoimmune leukoproliferative disorder (RALD) [MIM:614470]: A disorder of apoptosis, characterized by chronic accumulation of non-malignant lymphocytes, defective lymphocyte apoptosis, and an increased risk for the development of hematologic malignancies. {ECO:0000269|PubMed:17517660}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanocytic nevus syndrome, congenital (CMNS) [MIM:137550]: A syndrome characterized by congenital pigmentary skin lesions which can occur at any site and can cover most of the body surface. These lesions may or may not be hairy. Congenital melanocytic nevi are associated with neuromelanosis (the presence of melanin-producing cells within the brain parenchyma or leptomeninges). Less commonly they are associated with malignant melanoma in childhood, both in the skin and the central nervous system. CMNS patients also tend to have a characteristic facial appearance, including wide or prominent forehead, periorbital fullness, small short nose with narrow nasal bridge, round face, full cheeks, prominent premaxilla, and everted lower lip. {ECO:0000269|PubMed:18633438, ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Melanosis, neurocutaneous (NCMS) [MIM:249400]: A rare congenital disease characterized by the presence of giant or multiple melanocytic nevi on the skin, foci of melanin-producing cells within the brain parenchyma, and infiltration of leptomeninges by abnormal melanin deposits. Neurologic abnormalities include seizures, hydrocephalus, arachnoid cysts, tumors, and syringomyelia. Some patients may develop malignant melanoma. {ECO:0000269|PubMed:23392294}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Keratinocytic non-epidermolytic nevus (KNEN) [MIM:162900]: Epidermal nevi of the common, non-organoid and non-epidermolytic type are benign skin lesions and may vary in their extent from a single (usually linear) lesion to widespread and systematized involvement. They may be present at birth or develop early during childhood. {ECO:0000269|PubMed:22499344}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thyroid cancer, non-medullary, 2 (NMTC2) [MIM:188470]: A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms. {ECO:0000269|PubMed:12727991}. Disease susceptibility is associated with variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08257; DB08258; DB08259; DB06345; DB12610; DB08256; DB02029; DB04213; DB03677 Interacts with NA EC number NA Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aromatic hydrocarbons catabolism; Cytoplasm; Detoxification; Direct protein sequencing; Hydrolase; Lipid metabolism; Lipoprotein; Magnesium; Metal-binding; Multifunctional enzyme; Peroxisome; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 61744.9 Length 547 Aromaticity 0.09 Instability index 43.97 Isoelectric point 5.81 Charge (pH=7) -7.76 2D Binding mode Binding energy (Kcal/mol) -7.8  Molscript Map 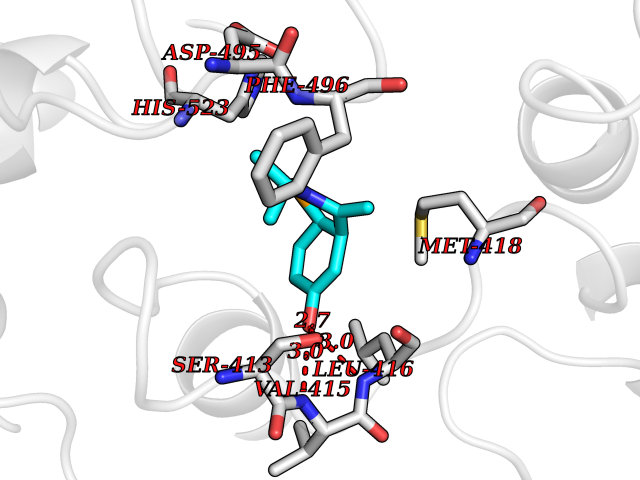 Pymol Map 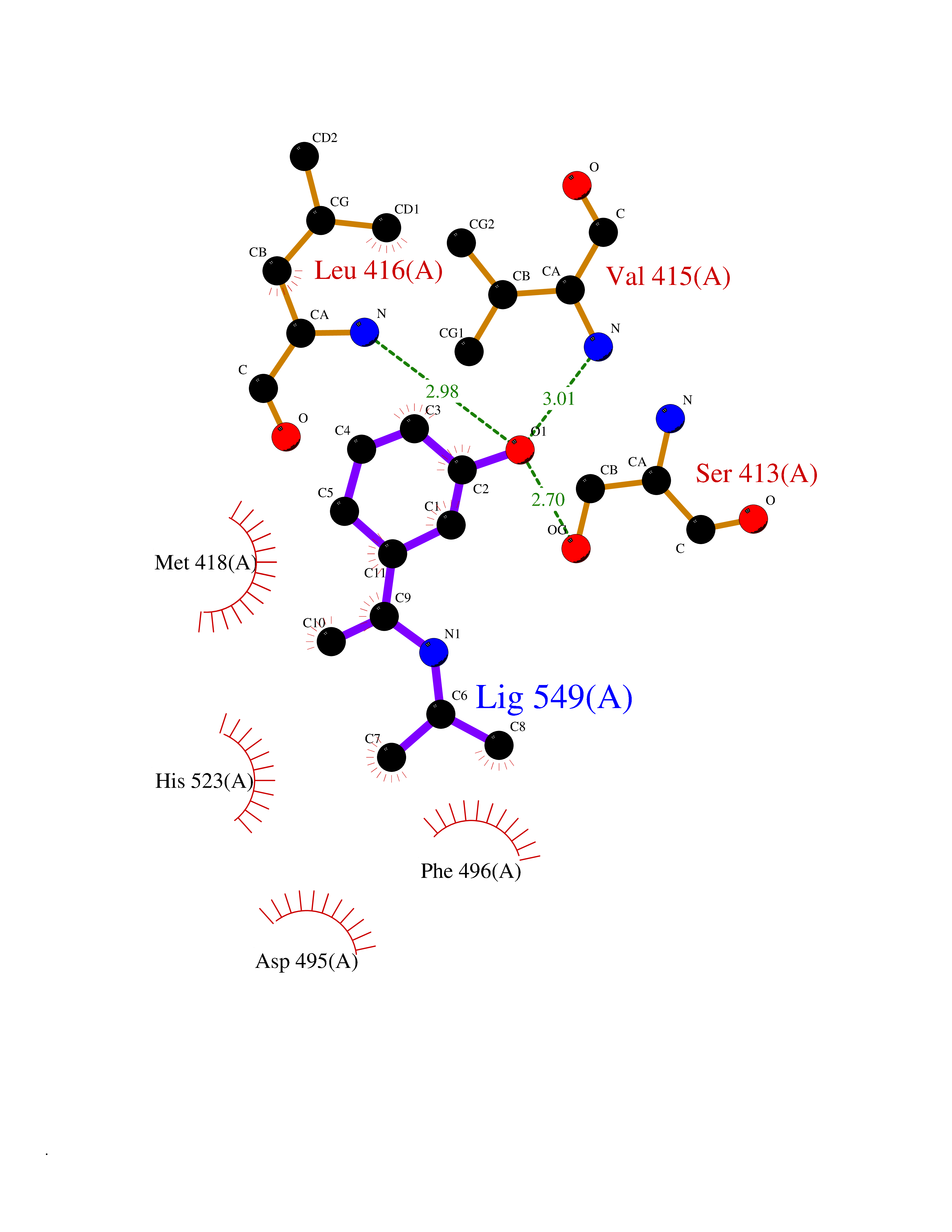 Ligplot Map 3D Binding mode Sequence TLRAAVFDLDGVLALPAVFGVLGRTEEALALPRGLLNDAFQKGGPEGATTRLMKGEITLSQWIPLMEENCRKCSETAKVCLPKNFSIKEIFDKAISARKINRPMLQAALMLRKKGFTTAILTNTWLDDRAERDGLAQLMCELKMHFDFLIESCQVGMVKPEPQIYKFLLDTLKASPSEVVFLDDIGANLKPARDLGMVTILVQDTDTALKELEKVTGIQLLNTPAPLPTSCNPSDMSHGYVTVKPRVRLHFVELGSGPAVCLCHGFPESWYSWRYQIPALAQAGYRVLAMDMKGYGESSAPPEIEEYCMEVLCKEMVTFLDKLGLSQAVFIGHDWGGMLVWYMALFYPERVRAVASLNTPFIPANPNMSPLESIKANPVFDYQLYFQEPGVAEAELEQNLSRTFKSLFRASDESVLSMHKVCEAGGLFVNSPEEPSLSRMVTEEEIQFYVQQFKKSGFRGPLNWYRNMERNWKWACKSLGRKILIPALMVTAEKDFVLVPQMSQHMEDWIPHLKRGHIEDCGHWTQMDKPTEVNQILIKWLDSDARN Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Cyclin-dependent kinase 13 (CDK13) | 5EFQ | 5.72 | |
Target general information Gen name CDK13 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CDC2-related protein kinase 5; Cell division cycle 2-like protein kinase 5; Cell division protein kinase 13; hCDK13; Cholinesterase-related cell division controller Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, CDC2/CDKX subfamily Biochemical class Kinase Function Cyclin-dependent kinase which displays CTD kinase activity and is required for RNA splicing. Has CTD kinase activity by hyperphosphorylating the C-terminal heptapeptide repeat domain (CTD) of the largest RNA polymerase II subunit RPB1, thereby acting as a key regulator of transcription elongation. Required for RNA splicing, probably by phosphorylating SRSF1/SF2. Required during hematopoiesis. In case of infection by HIV-1 virus, interacts with HIV-1 Tat protein acetylated at 'Lys-50' and 'Lys-51', thereby increasing HIV-1 mRNA splicing and promoting the production of the doubly spliced HIV-1 protein Nef. Related diseases Congenital heart defects, dysmorphic facial features, and intellectual developmental disorder (CHDFIDD) [MIM:617360]: An autosomal dominant syndrome characterized by atrial and/or ventricular septal congenital heart defects, facial dysmorphism with hypertelorism, upslanted palpebral fissures, epicanthal folds, ptosis, strabismus, posteriorly rotated ears, thin upper lip, and small mouth. Patients manifest global developmental delay, delayed walking and speech acquisition, and intellectual disability. Some patients have mild microcephaly, a small cerebral cortex, and agenesis of corpus callosum. More variable features include clinodactyly and/or camptodactyly of the fingers, hypotonia, and joint hypermobility. {ECO:0000269|PubMed:27479907, ECO:0000269|PubMed:28807008}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O75909; Q16543; Q07021 EC number EC 2.7.11.22 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Disease variant; Host-virus interaction; Intellectual disability; Isopeptide bond; Kinase; mRNA processing; mRNA splicing; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; RNA editing; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 37413.1 Length 322 Aromaticity 0.1 Instability index 46.11 Isoelectric point 7.64 Charge (pH=7) 1.32 2D Binding mode Binding energy (Kcal/mol) -7.8  Molscript Map  Pymol Map 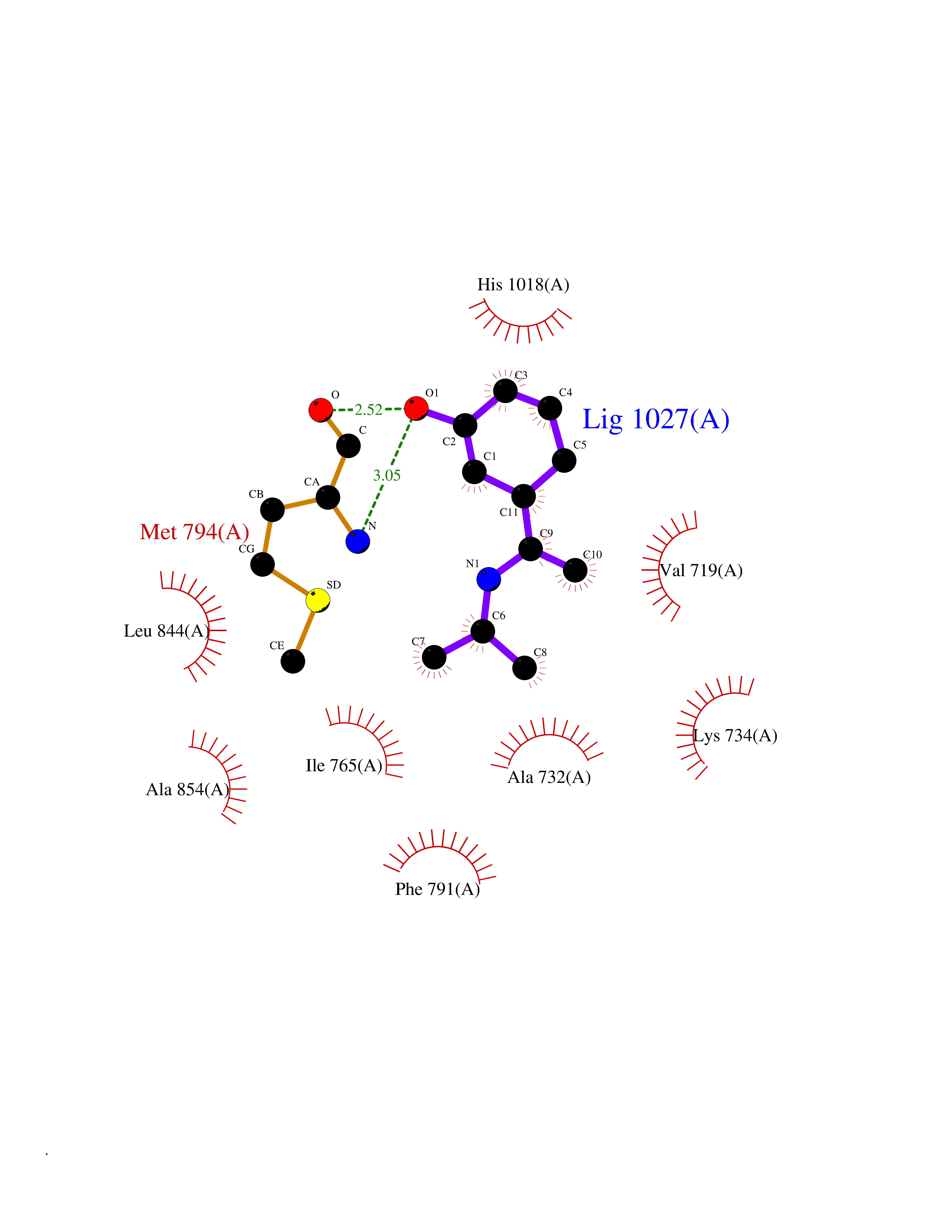 Ligplot Map 3D Binding mode Sequence IDWGKRCVDKFDIIGIIGEGTYGQVYKARDKDTGEMVALKKVRLDNEKEGFPITAIREIKILRQLTHQSIINMKEIVTDKEGAFYLVFEYMDHDLMGLLESGLVHFNENHIKSFMRQLMEGLDYCHKKNFLHRDIKCSNILLNNRGQIKLADFGLARLYSSEESRPYXNKVITLWYRPPELLLGEERYTPAIDVWSCGCILGELFTKKPIFQANQELAQLELISRICGSPCPAVWPDVIKLPYFNTMKPKKQYRRKLREEFVFIPAAALDLFDYMLALDPSKRCTAEQALQCEFLRDVEPSKMPPPDLPLWQDCHELWSKKR Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Protein kinase G1 (PRKG1) | 6BG2 | 5.72 | |
Target general information Gen name PRKG1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms cGMP-dependent protein kinase I; cGMP-dependent protein kinase 1; cGKI; cGK1; cGK 1; PRKGR1B; PRKGR1A; PRKG1B Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family, cGMP subfamily Biochemical class Kinase Function GMP binding activates PRKG1, which phosphorylates serines and threonines on many cellular proteins. Numerous protein targets for PRKG1 phosphorylation are implicated in modulating cellular calcium, but the contribution of each of these targets may vary substantially among cell types. Proteins that are phosphorylated by PRKG1 regulate platelet activation and adhesion, smooth muscle contraction, cardiac function, gene expression, feedback of the NO-signaling pathway, and other processes involved in several aspects of the CNS like axon guidance, hippocampal and cerebellar learning, circadian rhythm and nociception. Smooth muscle relaxation is mediated through lowering of intracellular free calcium, by desensitization of contractile proteins to calcium, and by decrease in the contractile state of smooth muscle or in platelet activation. Regulates intracellular calcium levels via several pathways: phosphorylates MRVI1/IRAG and inhibits IP3-induced Ca(2+) release from intracellular stores, phosphorylation of KCNMA1 (BKCa) channels decreases intracellular Ca(2+) levels, which leads to increased opening of this channel. PRKG1 phosphorylates the canonical transient receptor potential channel (TRPC) family which inactivates the associated inward calcium current. Another mode of action of NO/cGMP/PKGI signaling involves PKGI-mediated inactivation of the Ras homolog gene family member A (RhoA). Phosphorylation of RHOA by PRKG1 blocks the action of this protein in myriad processes: regulation of RHOA translocation; decreasing contraction; controlling vesicle trafficking, reduction of myosin light chain phosphorylation resulting in vasorelaxation. Activation of PRKG1 by NO signaling alters also gene expression in a number of tissues. In smooth muscle cells, increased cGMP and PRKG1 activity influence expression of smooth muscle-specific contractile proteins, levels of proteins in the NO/cGMP signaling pathway, down-regulation of the matrix proteins osteopontin and thrombospondin-1 to limit smooth muscle cell migration and phenotype. Regulates vasodilator-stimulated phosphoprotein (VASP) functions in platelets and smooth muscle. Serine/threonine protein kinase that acts as key mediator of the nitric oxide (NO)/cGMP signaling pathway. Related diseases Aortic aneurysm, familial thoracic 8 (AAT8) [MIM:615436]: A disease characterized by permanent dilation of the thoracic aorta usually due to degenerative changes in the aortic wall. It is primarily associated with a characteristic histologic appearance known as 'medial necrosis' or 'Erdheim cystic medial necrosis' in which there is degeneration and fragmentation of elastic fibers, loss of smooth muscle cells, and an accumulation of basophilic ground substance. {ECO:0000269|PubMed:23910461}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q13873; Q86V42; P25791; O76074; O15015; O94989; P25791-3; Q6FHY5; Q13976-2; Q9NYW8 EC number EC 2.7.11.12 Uniprot keywords 3D-structure; Acetylation; Allosteric enzyme; Alternative splicing; Aortic aneurysm; ATP-binding; cGMP; cGMP-binding; Coiled coil; Cytoplasm; Disease variant; Disulfide bond; Kinase; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID C Molecular weight (Da) 38032 Length 335 Aromaticity 0.12 Instability index 38.26 Isoelectric point 6.23 Charge (pH=7) -2.54 2D Binding mode Binding energy (Kcal/mol) -7.8  Molscript Map 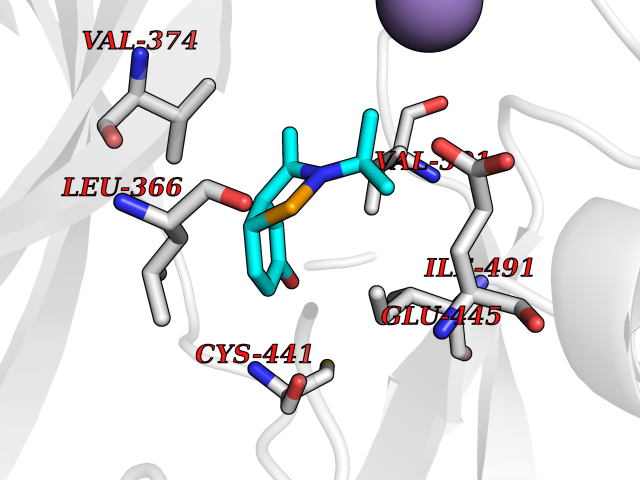 Pymol Map 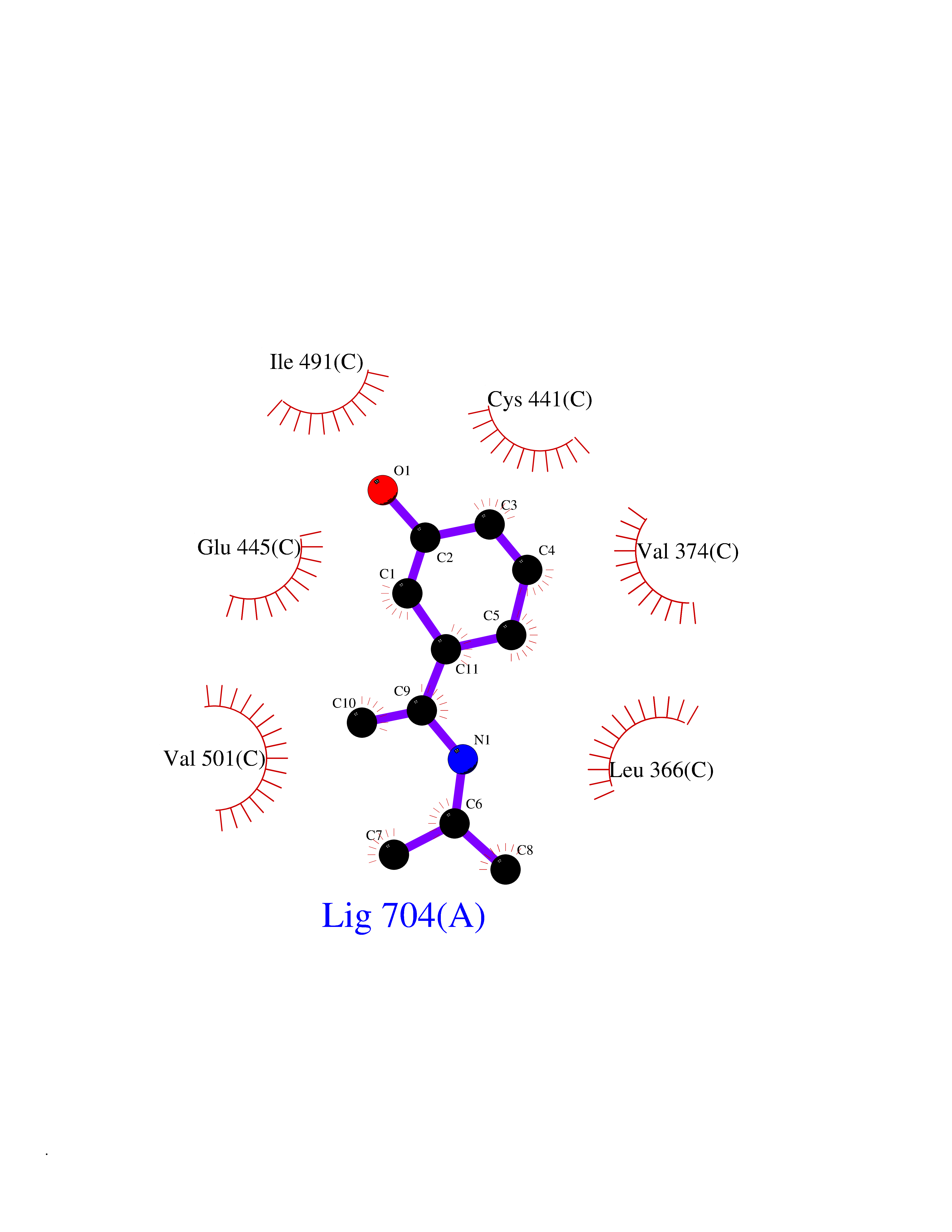 Ligplot Map 3D Binding mode Sequence EDAEAKAKYEAEAAFFANLKLSDFNIIDTLGVGGFGRVELVQLKSEESKTFAMKILKKRHIVDTRQQEHIRSEKQIMQGAHSDFIVRLYRTFKDSKYLYMLMEACLGGELWTILRDRGSFEDSTTRFYTACVVEAFAYLHSKGIIYRDLKPENLILDHRGYAKLVDFGFAKKIGFGKKTWXFCGTPEYVAPEIILNKGHDISADYWSLGILMYELLTGSPPFSGPDPMKTYNIILRGIDMIEFPKKIAKNAANLIKKLCRDNPSERLGNLKNGVKDIQKHKWFEGFNWEGLRKGTLTPPIIPSVASPTDTSNFDSFPEDNDEPPPDDNSGWDIDF Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Receptor-interacting protein 1 (RIPK1) | 5TX5 | 5.72 | |
Target general information Gen name RIPK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Receptor-interacting serine/threonine-protein kinase 1; RIP1; RIP-1; RIP; Cell death protein RIP Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family Biochemical class Kinase Function Upon activation of TNFR1 by the TNF-alpha family cytokines, TRADD and TRAF2 are recruited to the receptor. Phosphorylates DAB2IP at 'Ser-728' in a TNF-alpha-dependent manner, and thereby activates the MAP3K5-JNK apoptotic cascade. Ubiquitination by TRAF2 via 'Lys-63'-link chains acts as a critical enhancer of communication with downstream signal transducers in the mitogen-activated protein kinase pathway and the NF-kappa-B pathway, which in turn mediate downstream events including the activation of genes encoding inflammatory molecules. Polyubiquitinated protein binds to IKBKG/NEMO, the regulatory subunit of the IKK complex, a critical event for NF-kappa-B activation. Interaction with other cellular RHIM-containing adapters initiates gene activation and cell death. RIPK1 and RIPK3 association, in particular, forms a necrosis-inducing complex. Serine-threonine kinase which transduces inflammatory and cell-death signals (programmed necrosis) following death receptors ligation, activation of pathogen recognition receptors (PRRs), and DNA damage. Related diseases Immunodeficiency 57 with autoinflammation (IMD57) [MIM:618108]: An autosomal recessive primary immunodeficiency characterized by lymphopenia and recurrent viral, bacterial, and fungal infections. Patients exhibit early-onset inflammatory bowel disease involving the upper and lower gastrointestinal tract, and develop progressive polyarthritis. {ECO:0000269|PubMed:30026316}. The disease is caused by variants affecting the gene represented in this entry. RIPK1-deficient immune cells from IMD57 patients have impaired proinflammatory signaling leading to dysregulated cytokine secretion and are prone to necroptosis. {ECO:0000269|PubMed:30026316}.; DISEASE: Autoinflammation with episodic fever and lymphadenopathy (AIEFL) [MIM:618852]: An autosomal dominant immunologic disorder characterized by early onset of recurrent episodes of unexplained fever, lymphadenopathy, hepatosplenomegaly, and increased levels of inflammatory cytokines and chemokines in patient serum. {ECO:0000269|PubMed:31827280, ECO:0000269|PubMed:31827281}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with P04083; Q13490; Q13489; Q92851; Q14790; Q8IVM0; P48729; Q13158; Q9Y6K9; Q96AB6; Q9ULZ3; Q13546; Q9Y572; P19438; Q13077; Q12933; Q13114; Q13107; B7UI21; PRO_0000449629 [P0DTD1]; U5TQE9 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cell membrane; Cytoplasm; Disease variant; Glycoprotein; Host-virus interaction; Inflammatory response; Isopeptide bond; Kinase; Membrane; Necrosis; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 29554.2 Length 259 Aromaticity 0.08 Instability index 48.26 Isoelectric point 6.29 Charge (pH=7) -2.52 2D Binding mode Binding energy (Kcal/mol) -7.8 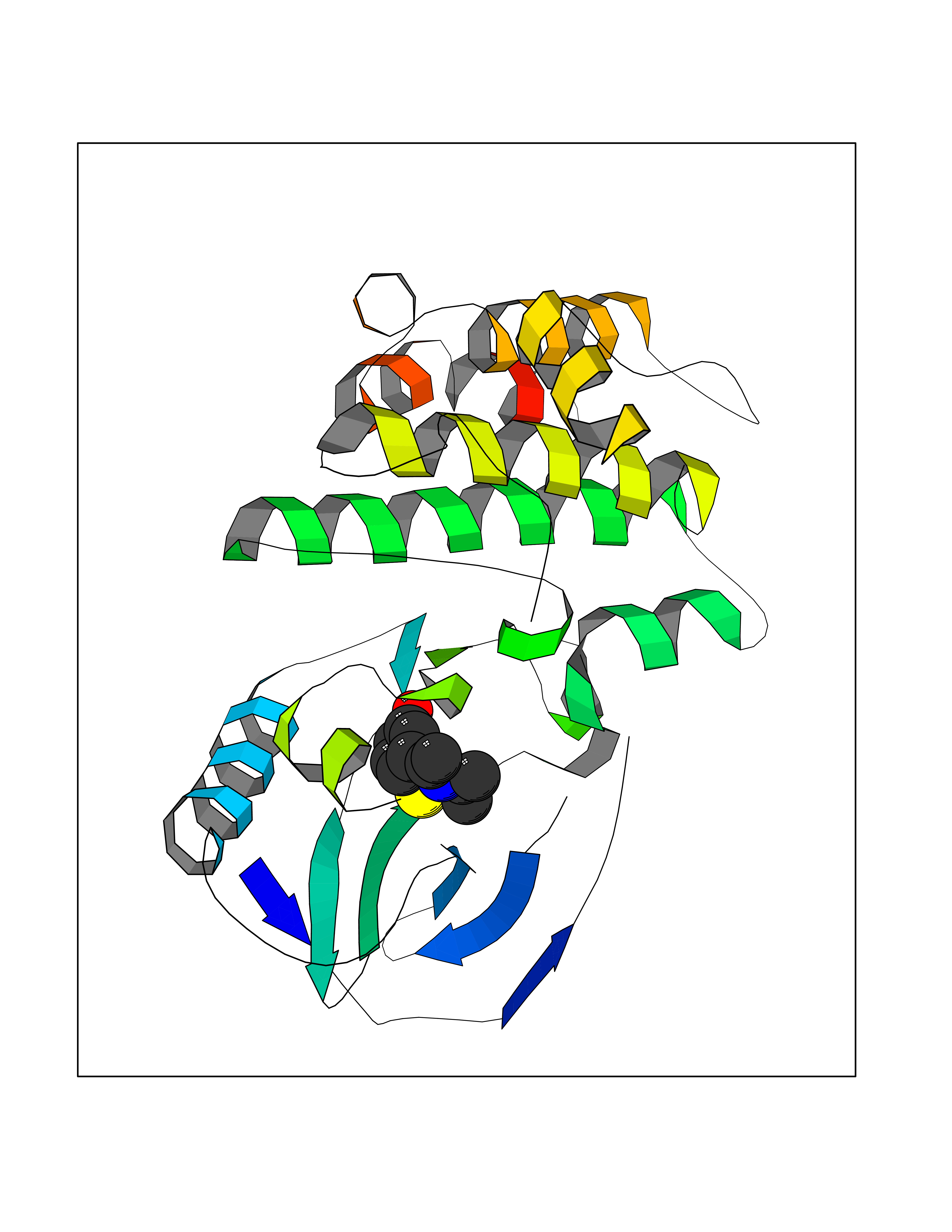 Molscript Map 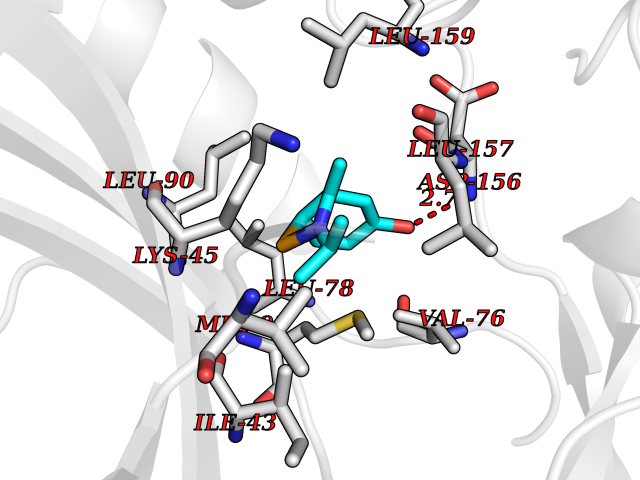 Pymol Map 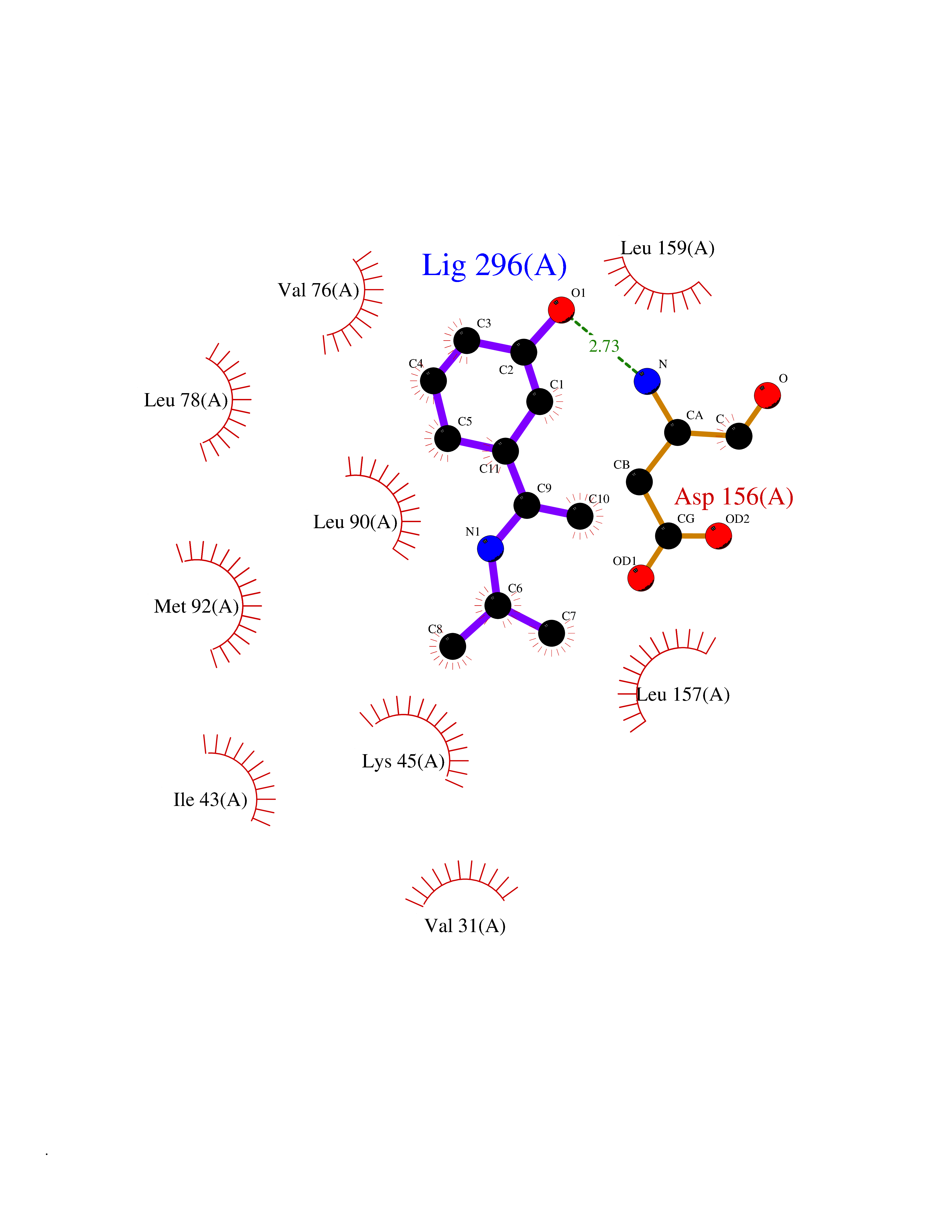 Ligplot Map 3D Binding mode Sequence IKMKSSDFLESAELDSGGKVSLAFHRTQGLMIMKTVYKGPNCIEHNEALLEEAKMMNRLRHSRVVKLLGVIIEEGKYSLVMEYMEKGNLMHVLKAEMSTPLSVKGRIILEIIEGMAYLHGKGVIHKDLKPENILVDNDFHIKIADLGLASFKMWSKLNGTLYYMAPEHLNDVNAKPTEKSDVYSFAVVLWAIFANKEPYQQLIMAIKSGNRPDVDDITEYCPREIISLMKLCWEANPEARPTFPGIEEKFRPFYLSQLE Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Aldose reductase (AKR1B1) | 1US0 | 5.71 | |
Target general information Gen name AKR1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Aldehyde reductase; AKR1B1 Protein family Aldo/keto reductase family Biochemical class Short-chain dehydrogenases reductase Function Catalyzes the NADPH-dependent reduction of a wide variety of carbonyl-containing compounds to their corresponding alcohols with a broad range of catalytic efficiencies. Related diseases Glutamine deficiency, congenital (GLND) [MIM:610015]: An autosomal recessive disorder characterized by variable brain malformations, encephalopathy, severe developmental delay, seizures, and decreased glutamine levels in bodily fluids. Death in early infancy may occur. {ECO:0000269|PubMed:16267323, ECO:0000269|PubMed:26711351, ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 116 (DEE116) [MIM:620806]: A form of epileptic encephalopathy, a heterogeneous group of early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE116 is autosomal dominant form characterized by severe developmental delay, seizures, and white matter abnormalities. {ECO:0000269|PubMed:38579670}. The disease is caused by variants affecting the gene represented in this entry. DEE116 is caused by variants that disrupt the canonical translation start codon in GLUL resulting in initiation of translation at Met-18 (PubMed:38579670). The resulting protein is enzymatically competent but insensitive to negative feedback regulation via glutamine-induced degradation. {ECO:0000269|PubMed:38579670}. Drugs (DrugBank ID) DB07028; DB07030; DB07450; DB02101; DB08449; DB08000; DB07139; DB07498; DB02007; DB02020; DB11859; DB02994; DB04272; DB07187; DB00694; DB00997; DB06246; DB01039; DB02021; DB16707; DB00143; DB02834; DB08084; DB01689; DB07063; DB06077; DB02518; DB00157; DB03461; DB05383; DB05533; DB05327; DB02712; DB00605; DB02383; DB02132; DB08772; DB07093; DB07999; DB08098 Interacts with Q9BUY7 EC number EC 1.1.1.300 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Lipid metabolism; NADP; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 35447.6 Length 313 Aromaticity 0.09 Instability index 36.41 Isoelectric point 7.1 Charge (pH=7) 0.26 2D Binding mode Binding energy (Kcal/mol) -7.79 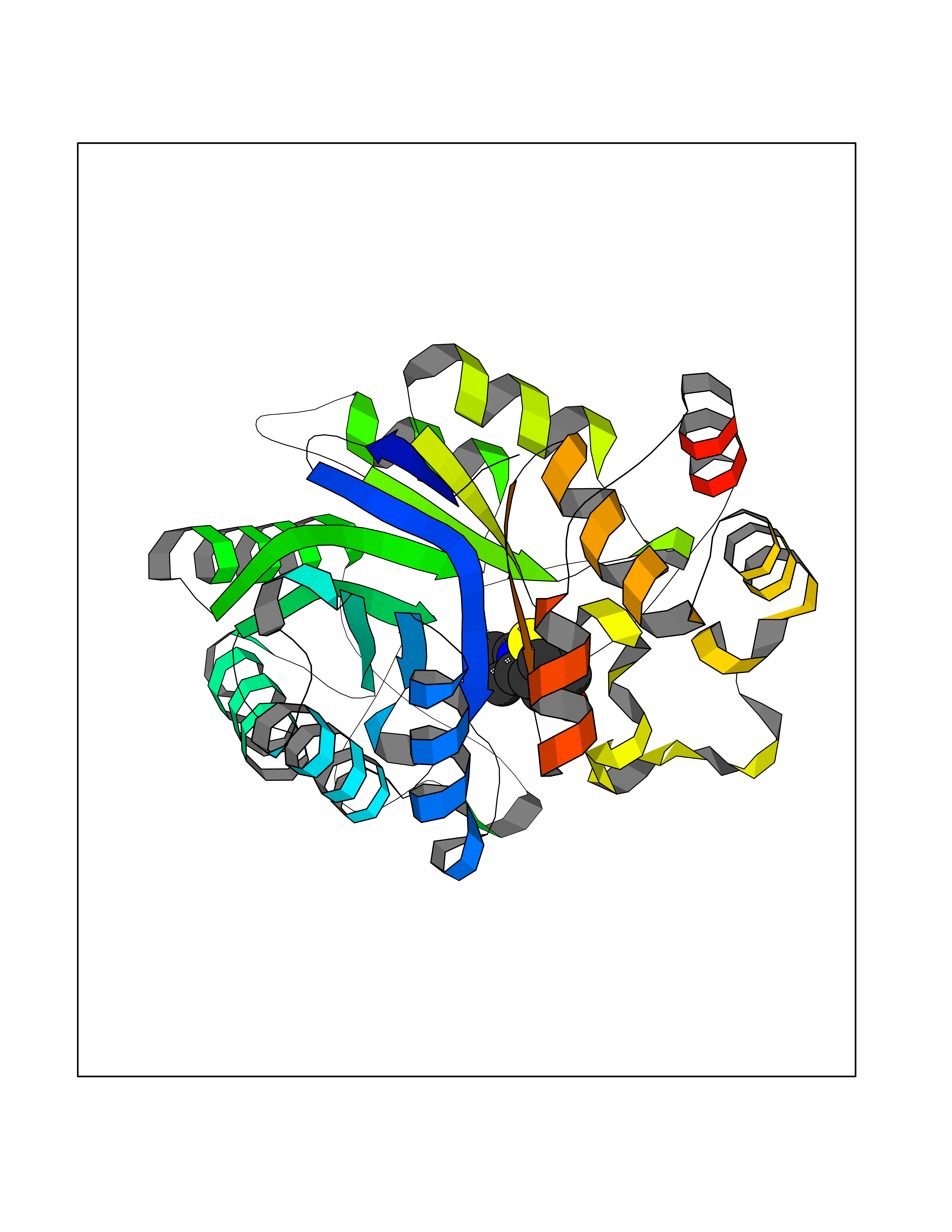 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence MASRILLNNGAKMPILGLGTWKSPPGQVTEAVKVAIDVGYRHIDCAHVYQNENEVGVAIQEKLREQVVKREELFIVSKLWCTYHEKGLVKGACQKTLSDLKLDYLDLYLIHWPTGFKPGKEFFPLDESGNVVPSDTNILDTWAAMEELVDEGLVKAIGISNFNHLQVEMILNKPGLKYKPAVNQIECHPYLTQEKLIQYCQSKGIVVTAYSPLGSPDRPWAKPEDPSLLEDPRIKAIAAKHNKTTAQVLIRFPMQRNLVVIPKSVTPERIAENFKVFDFELSSQDMTTLLSYNRNWRVCALLSCTSHKDYPFH Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Monoamine oxidase type A (MAO-A) | 2Z5Y | 5.71 | |
Target general information Gen name MAOA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Monoamine oxidase A; Amine oxidase [flavin-containing] A Protein family Flavin monoamine oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function MAOA preferentially oxidizes biogenic amines such as 5-hydroxytryptamine (5-HT), norepinephrine and epinephrine. Catalyzes the oxidative deamination of biogenic and xenobiotic amines and has important functions in the metabolism of neuroactive and vasoactive amines in the central nervous system and peripheral tissues. Related diseases Brunner syndrome (BRNRS) [MIM:300615]: A form of X-linked non-dysmorphic mild intellectual disability. Male patients are affected by borderline intellectual deficit and exhibit abnormal behavior, including disturbed regulation of impulsive aggression. Obligate female carriers have normal intelligence and behavior. {ECO:0000269|PubMed:8211186}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01472; DB00918; DB00182; DB06698; DB04889; DB13876; DB01445; DB06774; DB00215; DB04017; DB09130; DB05205; DB07641; DB00988; DB01363; DB00668; DB12329; DB01175; DB03147; DB14914; DB00614; DB01381; DB07919; DB04818; DB01247; DB00601; DB01577; DB00805; DB01442; DB01171; DB08804; DB00952; DB04820; DB00184; DB04821; DB06412; DB01626; DB00780; DB00191; DB00388; DB00397; DB09244; DB04850; DB00721; DB01168; DB00571; DB00852; DB09363; DB00140; DB00953; DB06654; DB01037; DB01104; DB00669; DB14569; DB09042; DB00624; DB13943; DB13944; DB13946; DB09245; DB00752; DB15328; DB09185; DB04832; DB00315; DB00909 Interacts with P27338 EC number EC 1.4.3.4 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Catecholamine metabolism; Direct protein sequencing; Disease variant; FAD; Flavoprotein; Intellectual disability; Membrane; Mitochondrion; Mitochondrion outer membrane; Neurotransmitter degradation; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A Molecular weight (Da) 58195.3 Length 513 Aromaticity 0.11 Instability index 34.97 Isoelectric point 7.98 Charge (pH=7) 2.87 2D Binding mode Binding energy (Kcal/mol) -7.78  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence HMFDVVVIGGGISGLSAAKLLTEYGVSVLVLEARDRVGGRTYTIRNEHVDYVDVGGAYVGPTQNRILRLSKELGIETYKVNVSERLVQYVKGKTYPFRAAFPPVWNPIAYLDYNNLWRTIDNMGKEIPTDAPWEAQHADKWDKMTMKELIDKICWTKTARRFAYLFVNINVTSEPHEVSALWFLWYVKQCGGTTRIFSVTNGGQERKFVGGSGQVSERIMDLLGDQVKLNHPVTHVDQSSDNIIIETLNHEHYECKYVINAIPPTLTAKIHFRPELPAERNQLIQRLPMGAVIKCMMYYKEAFWKKKDYCGCMIIEDEDAPISITLDDTKPDGSLPAIMGFILARKADRLAKLHKEIRKKKICELYAKVLGSQEALHPVHYEEKNWCEEQYSGGCYTAYFPPGIMTQYGRVIRQPVGRIFFAGTETATKWSGYMEGAVEAGERAAREVLNGLGKVTEKDIWVQEPESKDVPAVEITHTFWERNLPSVSGLLKIIGFSTSVTALGFVLYKYKLL Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | Thiopurine S-methyltransferase | 2BZG | 5.71 | |
Target general information Gen name TPMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, TPMT family Biochemical class Transferase Function Thiopurine S-methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00993; DB00436; DB01327; DB01033; DB01250; DB01021 Interacts with Q8TAP4-4; Q15047-2; P61981 EC number 2.1.1.67 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 25971.5 Length 229 Aromaticity 0.12 Instability index 32.58 Isoelectric point 6.74 Charge (pH=7) -0.6 2D Binding mode Binding energy (Kcal/mol) -7.79  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence EVQKNQVLTLEEWQDKWVNGKTAFHQEQGHQLLKKHLDTFLKGKSGLRVFFPLCGKAVEXKWFADRGHSVVGVEISELGIQEFFTEQNLSYSEEPITEIPGTKVFKSSSGNISLYCCSIFDLPRTNIGKFDXIWDRGALVAINPGDRKCYADTXFSLLGKKFQYLLCVLSYDPTKHPGPPFYVPHAEIERLFGKICNIRCLEKVDAFEERHKSWGIDCLFEKLYLLTEK Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Cytochrome c | 3ZOO | 5.71 | |
Target general information Gen name CYCS Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms CYC Protein family Cytochrome c family Biochemical class Oxidoreductase Function Electron transporter, transferring electrons from CoQH2-cytochrome c reductase complex and cytochrome c oxidase complex activity.Heme binding.Metal ion binding. Related diseases Thrombocytopenia 4 (THC4) [MIM:612004]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. {ECO:0000269|PubMed:18345000}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB11638; DB03317; DB03366; DB01017; DB02110; DB03977; DB03934; DB04249 Interacts with O14727; P05067; Q6XD76; Q9NSI6-4; Q3SXR2; Q96BR5; Q9UKG9-2; O00303; Q8IZU1; Q3SYB3; P06241; Q8N5Z5; Q6A162; Q1L5Z9; P02750; Q8IYG6; Q6FHY5; A0A0A0MR05; Q9BUL5; Q6ZMI0-5; Q66K80; Q9NTN9-3; P37840; Q13573; Q92797-2; O43829; Q9FKS5 EC number NA Uniprot keywords 3D-structure; Acetylation; Apoptosis; Direct protein sequencing; Disease variant; Electron transport; Heme; Iron; Metal-binding; Mitochondrion; Phosphoprotein; Proteomics identification; Reference proteome; Respiratory chain; Transport Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 11601.4 Length 104 Aromaticity 0.09 Instability index 12.21 Isoelectric point 9.61 Charge (pH=7) 9.01 2D Binding mode Binding energy (Kcal/mol) -7.79  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence GDVEKGKKIFIMKCSQCHTVEKGGKHKTGPNLHGLFGRKTGQAPGFSYTAANKNKGIIWGEDTLMEYLENPKKYIPGTKMIFVGIKKKEERADLIAYLKKATNE Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | HIF-prolyl hydroxylase 2 (HPH-2) | 6ZBO | 5.71 | |
Target general information Gen name EGLN1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SM-20; Prolyl hydroxylase domain-containing protein 2; PHD2; Hypoxia-inducible factor prolyl hydroxylase 2; HPH-2; HIF-PH2; Egl nine homolog 1; C1orf12 Protein family NA Biochemical class Paired donor oxygen oxidoreductase Function Cellular oxygen sensor that catalyzes, under normoxic conditions, the post-translational formation of 4-hydroxyproline in hypoxia-inducible factor (HIF) alpha proteins. Hydroxylates a specific proline found in each of the oxygen-dependent degradation (ODD) domains (N-terminal, NODD, and C-terminal, CODD) of HIF1A. Also hydroxylates HIF2A. Has a preference for the CODD site for both HIF1A and HIF1B. Hydroxylated HIFs are then targeted for proteasomal degradation via the von Hippel-Lindau ubiquitination complex. Under hypoxic conditions, the hydroxylation reaction is attenuated allowing HIFs to escape degradation resulting in their translocation to the nucleus, heterodimerization with HIF1B, and increased expression of hypoxy-inducible genes. EGLN1 is the most important isozyme under normoxia and, through regulating the stability of HIF1, involved in various hypoxia-influenced processes such as angiogenesis in retinal and cardiac functionality. Target proteins are preferentially recognized via a LXXLAP motif. Related diseases Erythrocytosis, familial, 3 (ECYT3) [MIM:609820]: An autosomal dominant disorder characterized by elevated serum hemoglobin and hematocrit, and normal serum erythropoietin levels. {ECO:0000269|PubMed:16407130, ECO:0000269|PubMed:17579185}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126; DB11682; DB14490; DB14491; DB14488; DB14501; DB14489; DB08687; DB01592; DB07112; DB04847; DB12255 Interacts with Q99814; Q14318; Q16665; Q13438; PRO_0000037551 [Q9WMX2] EC number EC 1.14.11.29 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Congenital erythrocytosis; Cytoplasm; Dioxygenase; Disease variant; Iron; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; S-nitrosylation; Vitamin C; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,D Molecular weight (Da) 45717.8 Length 406 Aromaticity 0.11 Instability index 24.41 Isoelectric point 7.59 Charge (pH=7) 1.54 2D Binding mode Binding energy (Kcal/mol) -7.8  Molscript Map 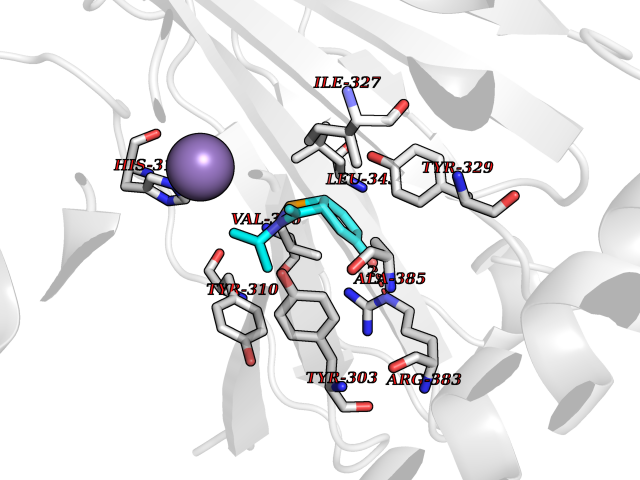 Pymol Map 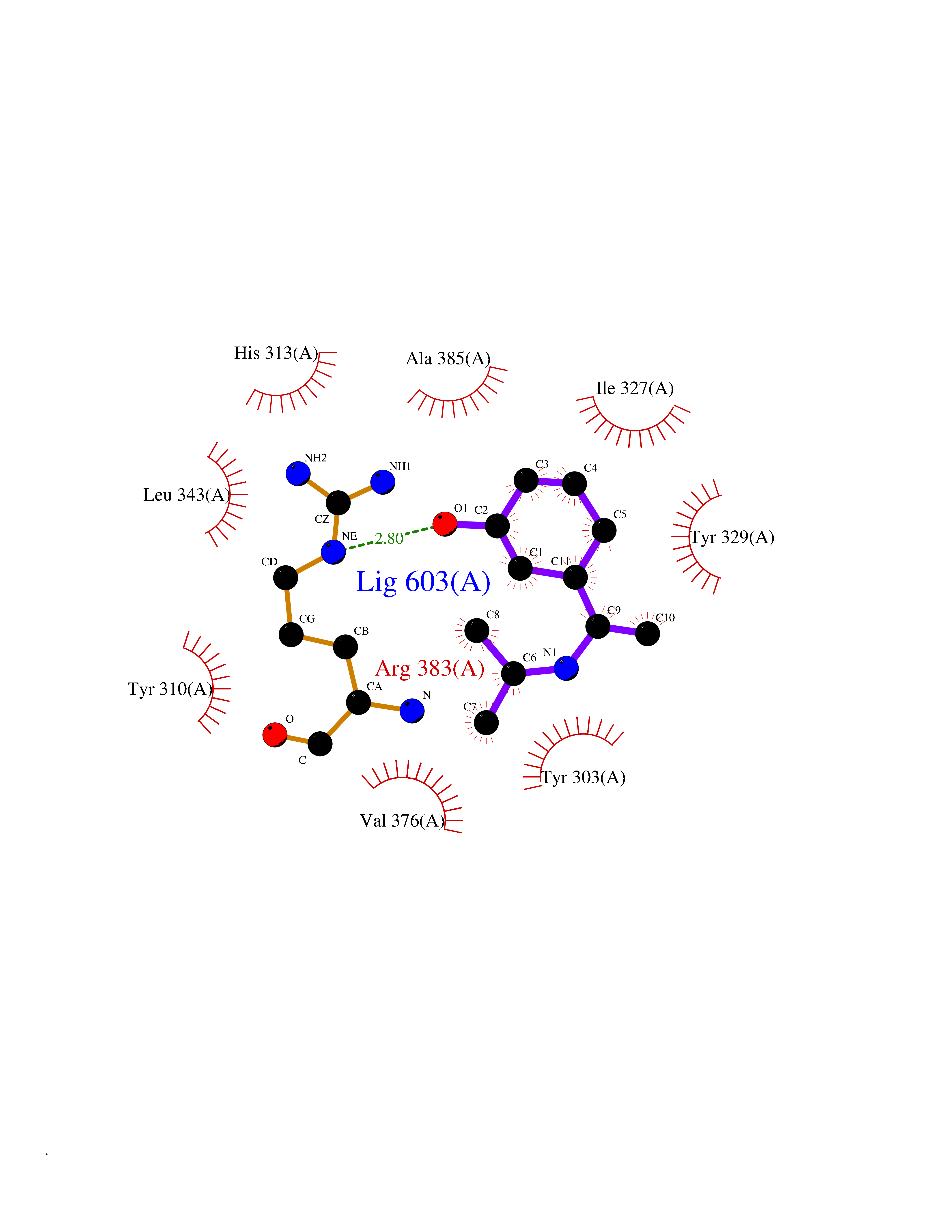 Ligplot Map 3D Binding mode Sequence LPALKLALEYIVPCMNKHGICVVDDFLGKETGQQIGDEVRALHDTGKFTGDKITWIEGKEPGCETIGLLMSSMDDLIRHCNGKLGSYKINGRTKAMVACYPGNGTGYVRHVDNPNGDGRCVTCIYYLNKDWDAKVSGGILRIFPEGKAQFADIEPKFDRLLFFWSDRRNPHEVQPAYATRYAITVWYFDADERARAKVKYLTGELPALKLALEYIVPCMNKHGICVVDDFLGKETGQQIGDEVRALHDTGKFTGDKITWIEGKEPGCETIGLLMSSMDDLIRHCNGKLGSYKINGRTKAMVACYPGNGTGYVRHVDNPNGDGRCVTCIYYLNKDWDAKVSGGILRIFPEGKAQFADIEPKFDRLLFFWSDRRNPHEVQPAYATRYAITVWYFDADERARAKVKYLT Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Opioid receptor sigma 1 (OPRS1) | 6DJZ | 5.71 | |
Target general information Gen name SIGMAR1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hSigmaR1; Sigma1R; Sigma1-receptor; Sigma non-opioid intracellular receptor 1; Sigma 1-type opioid receptor; SRBP; SR31747-binding protein; SR31747 binding protein 1; SR-BP; SIG-1R; Opioid receptor, s Protein family ERG2 family Biochemical class GPCR rhodopsin Function Involved in the regulation of different receptors it plays a role in BDNF signaling and EGF signaling. Also regulates ion channels like the potassium channel and could modulate neurotransmitter release. Plays a role in calcium signaling through modulation together with ANK2 of the ITP3R-dependent calcium efflux at the endoplasmic reticulum. Plays a role in several other cell functions including proliferation, survival and death. Originally identified for its ability to bind various psychoactive drugs it is involved in learning processes, memory and mood alteration. Necessary for proper mitochondrial axonal transport in motor neurons, in particular the retrograde movement of mitochondria. Plays a role in protecting cells against oxidative stress-induced cell death via its interaction with RNF112. Functions in lipid transport from the endoplasmic reticulum and is involved in a wide array of cellular functions probably through regulation of the biogenesis of lipid microdomains at the plasma membrane. Related diseases Amyotrophic lateral sclerosis 16, juvenile (ALS16) [MIM:614373]: A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. {ECO:0000269|PubMed:21842496}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuronopathy, distal hereditary motor, autosomal recessive 2 (HMNR2) [MIM:605726]: A form of distal hereditary motor neuronopathy, a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs. HMNR2 is characterized by onset of distal muscle weakness and wasting affecting the lower and upper limbs in the first decade. {ECO:0000269|PubMed:26078401, ECO:0000269|PubMed:27629094}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00321; DB09014; DB00907; DB00514; DB01488; DB00574; DB00502; DB00956; DB00704; DB00540; DB06174; DB00652; DB11186; DB03575; DB05316; DB01708; DB00409; DB01104 Interacts with Q92847-1; Q99720-1; O00213-2; P17612; P50454; P37173 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Amyotrophic lateral sclerosis; Cell junction; Cell membrane; Cell projection; Cytoplasmic vesicle; Direct protein sequencing; Disease variant; Endoplasmic reticulum; Lipid droplet; Lipid transport; Membrane; Neurodegeneration; Neuropathy; Nucleus; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Synapse; Transmembrane; Transmembrane helix; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 23901 Length 212 Aromaticity 0.14 Instability index 33.12 Isoelectric point 5.61 Charge (pH=7) -5.6 2D Binding mode Binding energy (Kcal/mol) -7.79 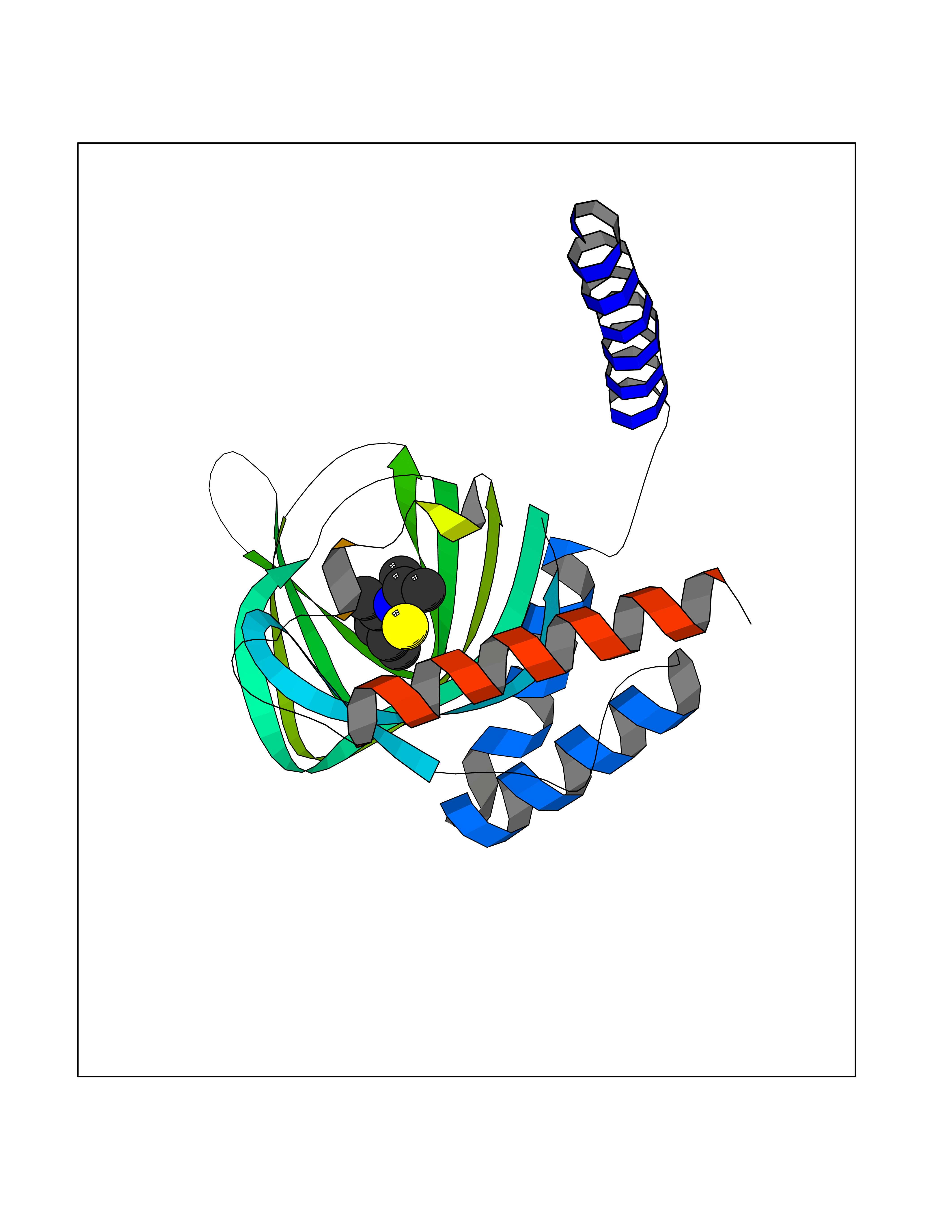 Molscript Map 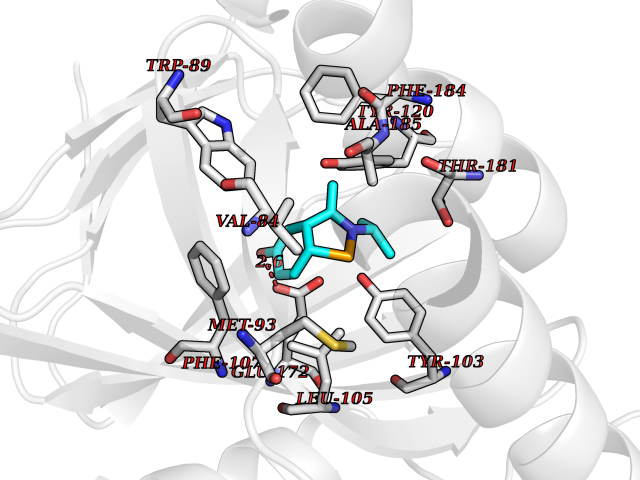 Pymol Map 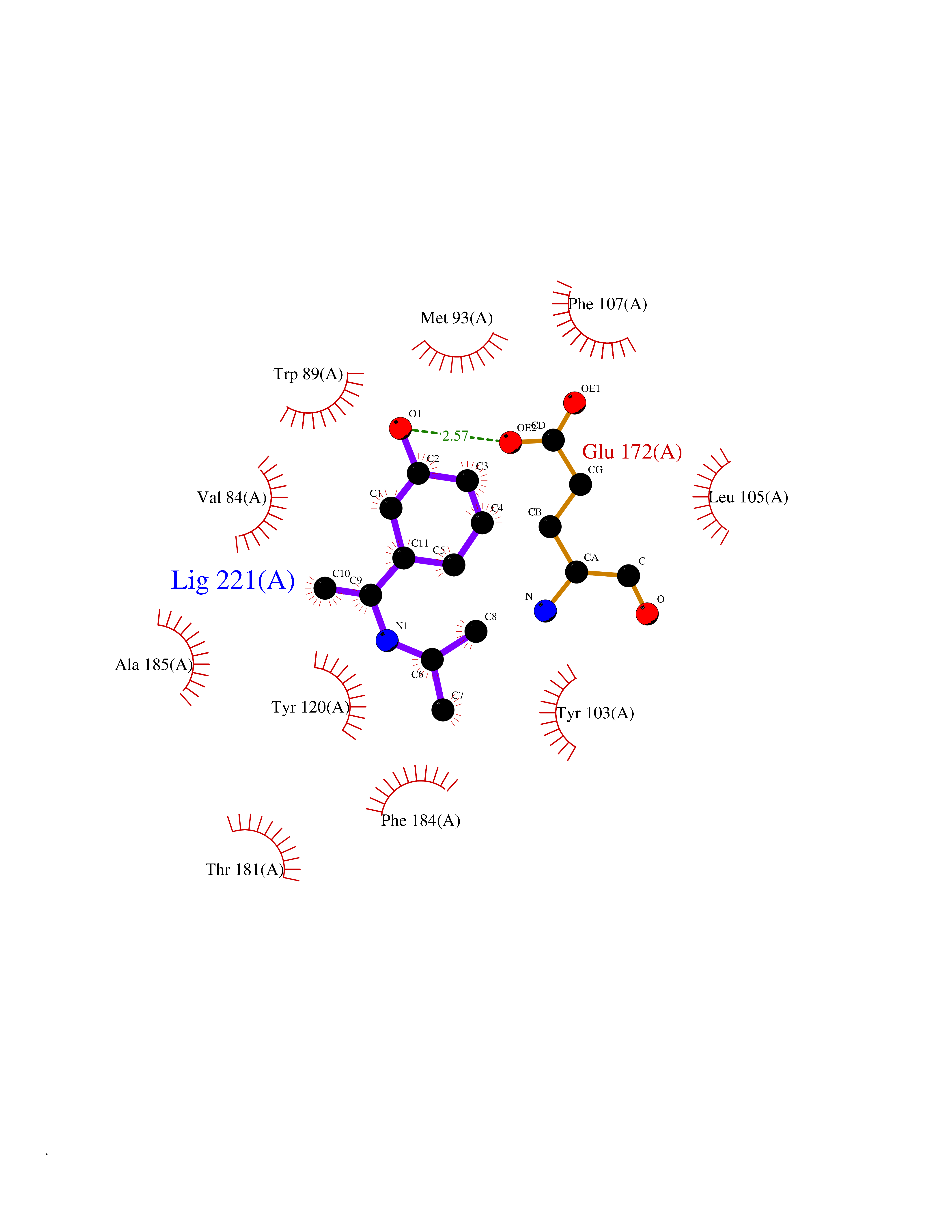 Ligplot Map 3D Binding mode Sequence RWAWAALLLAVAAVLTQVVWLWLGTQSFVFQREEIAQLARQYAGLDHELAFSRLIVELRRLHPGHVLPDEELQWVFVNAGGWMGAMCLLHASLSEYVLLFGTALGSRGHSGRYWAEISDTIISGTFHQWREGTTKSEVFYPGETVVHGPGEATAVEWGPNTWMVEYGRGVIPSTLAFALADTVFSTQDFLTLFYTLRSYARGLRLELTTYLF Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | IL-1 receptor-associated kinase 1 (IRAK1) | 6BFN | 5.71 | |
Target general information Gen name IRAK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Interleukin-1 receptor-associated kinase 1; IRAK-1; IRAK Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family, Pelle subfamily Biochemical class Kinase Function Involved in Toll-like receptor (TLR) and IL-1R signaling pathways. Is rapidly recruited by MYD88 to the receptor-signaling complex upon TLR activation. Association with MYD88 leads to IRAK1 phosphorylation by IRAK4 and subsequent autophosphorylation and kinase activation. Phosphorylates E3 ubiquitin ligases Pellino proteins (PELI1, PELI2 and PELI3) to promote pellino-mediated polyubiquitination of IRAK1. Then, the ubiquitin-binding domain of IKBKG/NEMO binds to polyubiquitinated IRAK1 bringing together the IRAK1-MAP3K7/TAK1-TRAF6 complex and the NEMO-IKKA-IKKB complex. In turn, MAP3K7/TAK1 activates IKKs (CHUK/IKKA and IKBKB/IKKB) leading to NF-kappa-B nuclear translocation and activation. Alternatively, phosphorylates TIRAP to promote its ubiquitination and subsequent degradation. Phosphorylates the interferon regulatory factor 7 (IRF7) to induce its activation and translocation to the nucleus, resulting in transcriptional activation of type I IFN genes, which drive the cell in an antiviral state. When sumoylated, translocates to the nucleus and phosphorylates STAT3. Serine/threonine-protein kinase that plays a critical role in initiating innate immune response against foreign pathogens. Related diseases Anemia, non-spherocytic hemolytic, due to G6PD deficiency (NSHA) [MIM:300908]: A disease characterized by G6PD deficiency, acute hemolytic anemia, fatigue, back pain, and jaundice. In most patients, the disease is triggered by an exogenous agent, such as some drugs, food, or infection. Increased unconjugated bilirubin, lactate dehydrogenase, and reticulocytosis are markers of the disorder. Although G6PD deficiency can be life-threatening, most patients are asymptomatic throughout their life. {ECO:0000269|PubMed:12524354, ECO:0000269|PubMed:1303180, ECO:0000269|PubMed:1303182, ECO:0000269|PubMed:1536798, ECO:0000269|PubMed:1611091, ECO:0000269|PubMed:1889820, ECO:0000269|PubMed:1945893, ECO:0000269|PubMed:20007901, ECO:0000269|PubMed:26479991, ECO:0000269|PubMed:2836867, ECO:0000269|PubMed:2912069, ECO:0000269|PubMed:30988594, ECO:0000269|PubMed:38066190, ECO:0000269|PubMed:7858267, ECO:0000269|PubMed:7959695, ECO:0000269|PubMed:8193373, ECO:0000269|PubMed:8490627, ECO:0000269|PubMed:8533762, ECO:0000269|PubMed:8733135, ECO:0000269|PubMed:9452072}. The disease is caused by variants affecting the gene represented in this entry. Deficiency of G6PD is associated with hemolytic anemia in two different situations. First, in areas in which malaria has been endemic, G6PD-deficiency alleles have reached high frequencies (1% to 50%) and deficient individuals, though essentially asymptomatic in the steady state, have a high risk of acute hemolytic attacks. Secondly, sporadic cases of G6PD deficiency occur at a very low frequencies, and they usually present a more severe phenotype. Several types of NSHA are recognized. Class-I variants are associated with severe NSHA; class-II have an activity <10% of normal; class-III have an activity of 10% to 60% of normal; class-IV have near normal activity. Drugs (DrugBank ID) DB12010 Interacts with Q15306; Q92985; Q99836; Q96FA3; Q9HAT8; Q8N2H9-2; Q13526; Q86WV6; P58753; Q9Y4K3; Q8VCW4; Q5D1E7 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cytoplasm; Direct protein sequencing; Host-virus interaction; Immunity; Innate immunity; Isopeptide bond; Kinase; Lipid droplet; Magnesium; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 33681.4 Length 301 Aromaticity 0.09 Instability index 39.86 Isoelectric point 8.6 Charge (pH=7) 5.09 2D Binding mode Binding energy (Kcal/mol) -7.79  Molscript Map 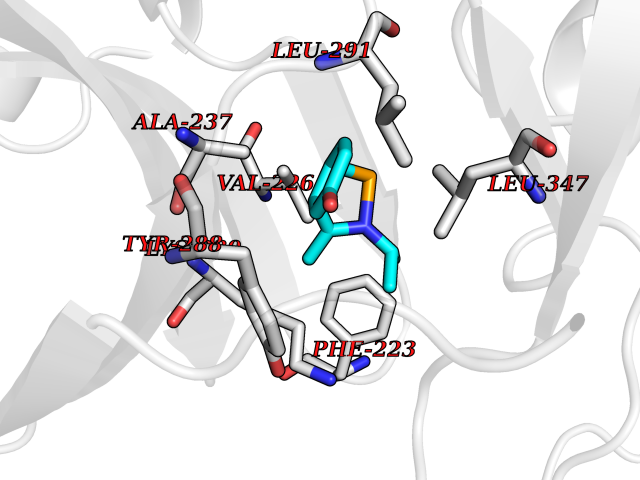 Pymol Map 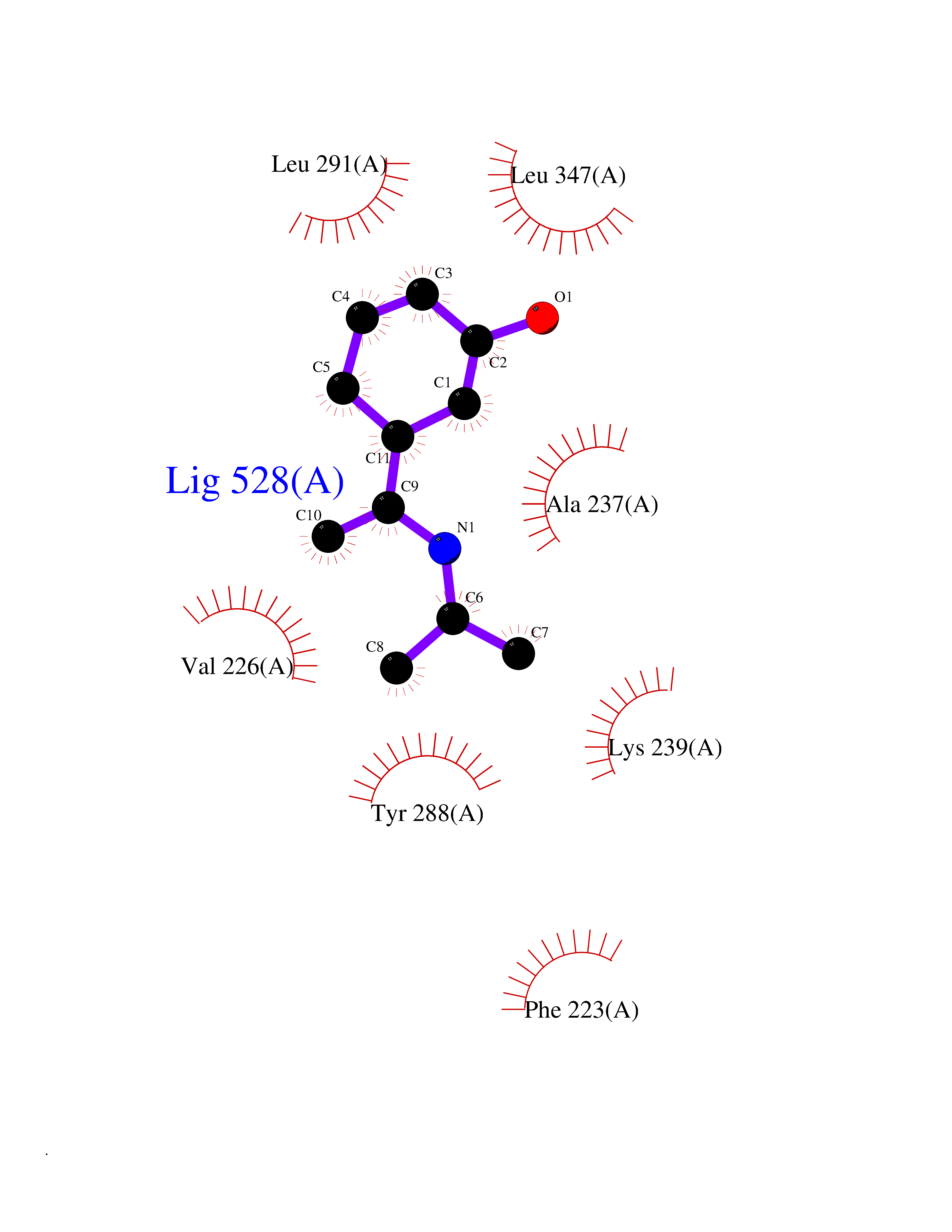 Ligplot Map 3D Binding mode Sequence SRPFPFCWPLCEISRGTHNFSEELKIGEGGFGCVYRAVMRNTVYAVKRLKEWTAVKQSFLTEVEQLSRFRHPNIVDFAGYCAQNGFYCLVYGFLPNGSLEDRLHCQTQACPPLSWPQRLDILLGTARAIQFLHQDSPSLIHGDIKSSNVLLDERLTPKLGDFGLARFSRTVRGTLAYLPEEYIKTGRLAVDTDTFSFGVVVLETLAGQRAVKTHGARTKYLKDLVEEEAEEAGVAAADAWAAPIAMQIYKKHLDPRPGPCPPELGLGLGQLACCCLHRRAKRRPPMTQVYERLEKLQAVVA Hydrogen bonds contact Hydrophobic contact | ||||