Job Results:
Ligand
Structure
Job ID
7eae9f0de32fdedcd28c1404448b5ea8
Job name
NA
Time
2025-04-07 15:33:07
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | FK506-binding protein 1A (FKBP1A) | 1BKF | 4.27 | |
Target general information Gen name FKBP1A Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Rotamase; Peptidyl-prolyl cis-trans isomerase FKBP1A; PPIase FKBP1A; Immunophillin FKBP; Immunophilin FKBP12; FKBP12; FKBP1; FKBP-1A; FKBP-12; FK-binding protein 12; Calstabin-1; 12 kDa FKBP; 12 kDa F Protein family FKBP-type PPIase family, FKBP1 subfamily Biochemical class Cis-trans-isomerase Function Recruits SMAD7 to ACVR1B which prevents the association of SMAD2 and SMAD3 with the activin receptor complex, thereby blocking the activin signal. May modulate the RYR1 calcium channel activity. PPIases accelerate the folding of proteins. It catalyzes the cis-trans isomerization of proline imidic peptide bonds in oligopeptides. Keeps in an inactive conformation TGFBR1, the TGF-beta type I serine/threonine kinase receptor, preventing TGF-beta receptor activation in absence of ligand. Related diseases WHIM syndrome 1 (WHIMS1) [MIM:193670]: An autosomal dominant immunologic disease characterized by neutropenia, hypogammaglobulinemia and extensive human papillomavirus (HPV) infection. Despite the peripheral neutropenia, bone marrow aspirates from affected individuals contain abundant mature myeloid cells, a condition termed myelokathexis. {ECO:0000269|PubMed:12692554, ECO:0000269|PubMed:15536153}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: CXCR4 mutations play a role in the pathogenesis of Waldenstroem macroglobulinemia (WM) and influence disease presentation and outcome, as well as response to therapy. WM is a B-cell lymphoma characterized by accumulation of malignant lymphoplasmacytic cells in the bone marrow, lymph nodes and spleen, and hypersecretion of monoclonal immunoglobulin M (IgM). Excess IgM production results in serum hyperviscosity, tissue infiltration, and autoimmune-related pathology. {ECO:0000269|PubMed:24366360, ECO:0000269|PubMed:24553177}. Drugs (DrugBank ID) DB08520; DB04012; DB01712; DB04094; DB08597; DB02888; DB01951; DB05814; DB03338; DB03621; DB02311; DB08231; DB00337; DB00864; DB01723 Interacts with P36896; Q9NZD4; P05067; P42345; Q92736; O15105; P36897; PRO_0000037309 [P0C6X7]; PRO_0000037312 [P0C6X7]; P11716 EC number EC 5.2.1.8 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Isomerase; Membrane; Proteomics identification; Reference proteome; Rotamase; Sarcoplasmic reticulum Protein physicochemical properties Chain ID A Molecular weight (Da) 11753.3 Length 107 Aromaticity 0.08 Instability index 43.48 Isoelectric point 8.06 Charge (pH=7) 0.94 2D Binding mode Binding energy (Kcal/mol) -5.82 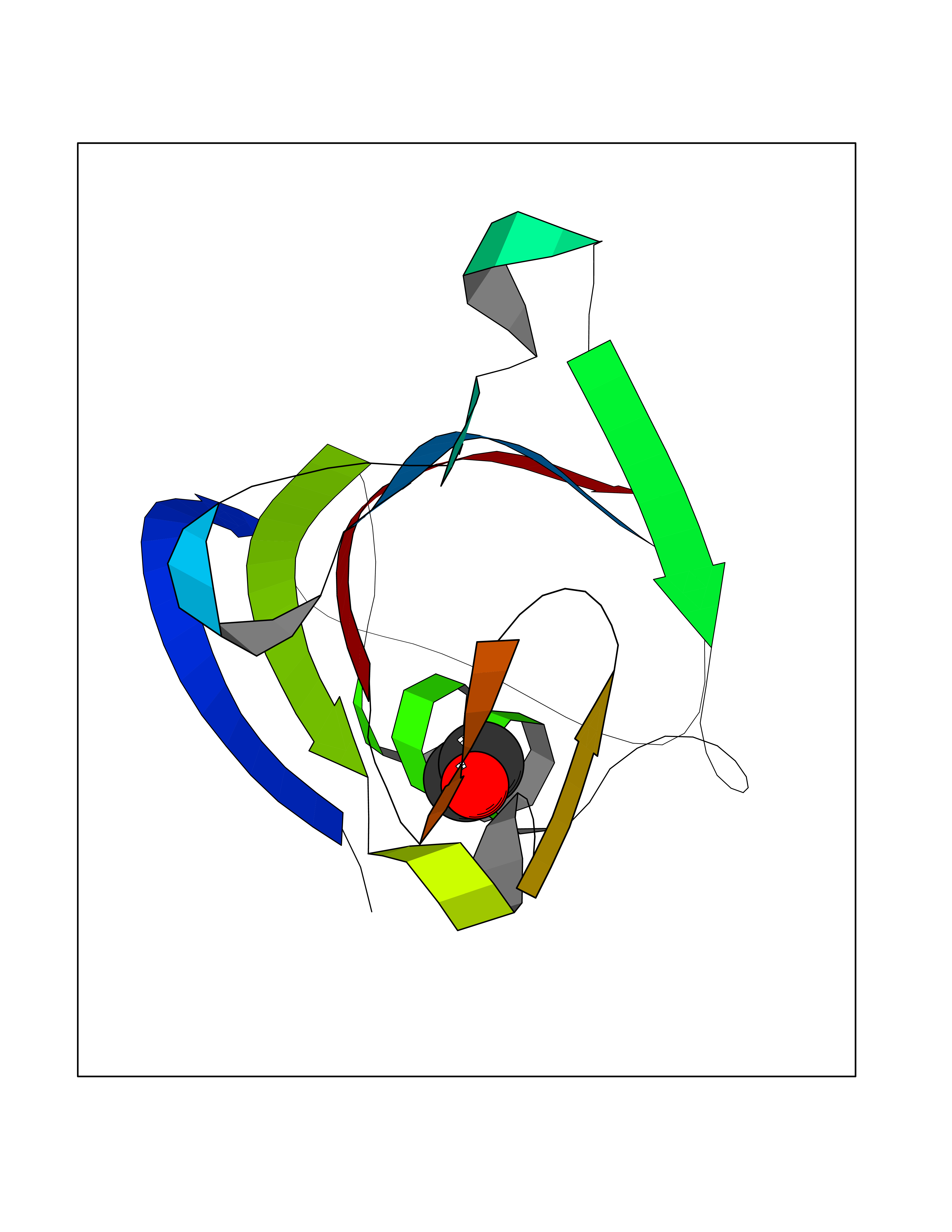 Molscript Map 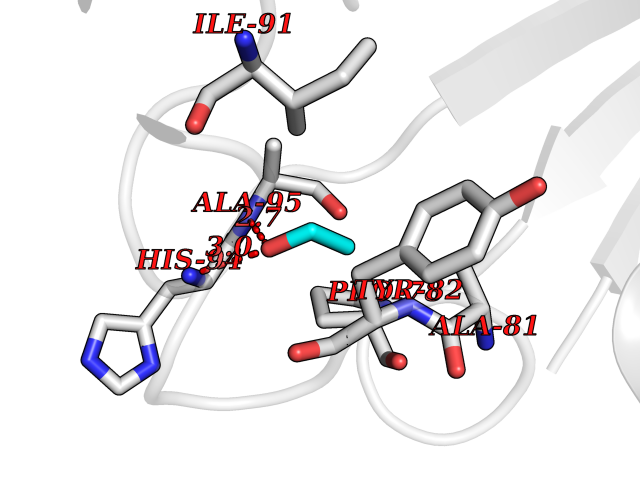 Pymol Map 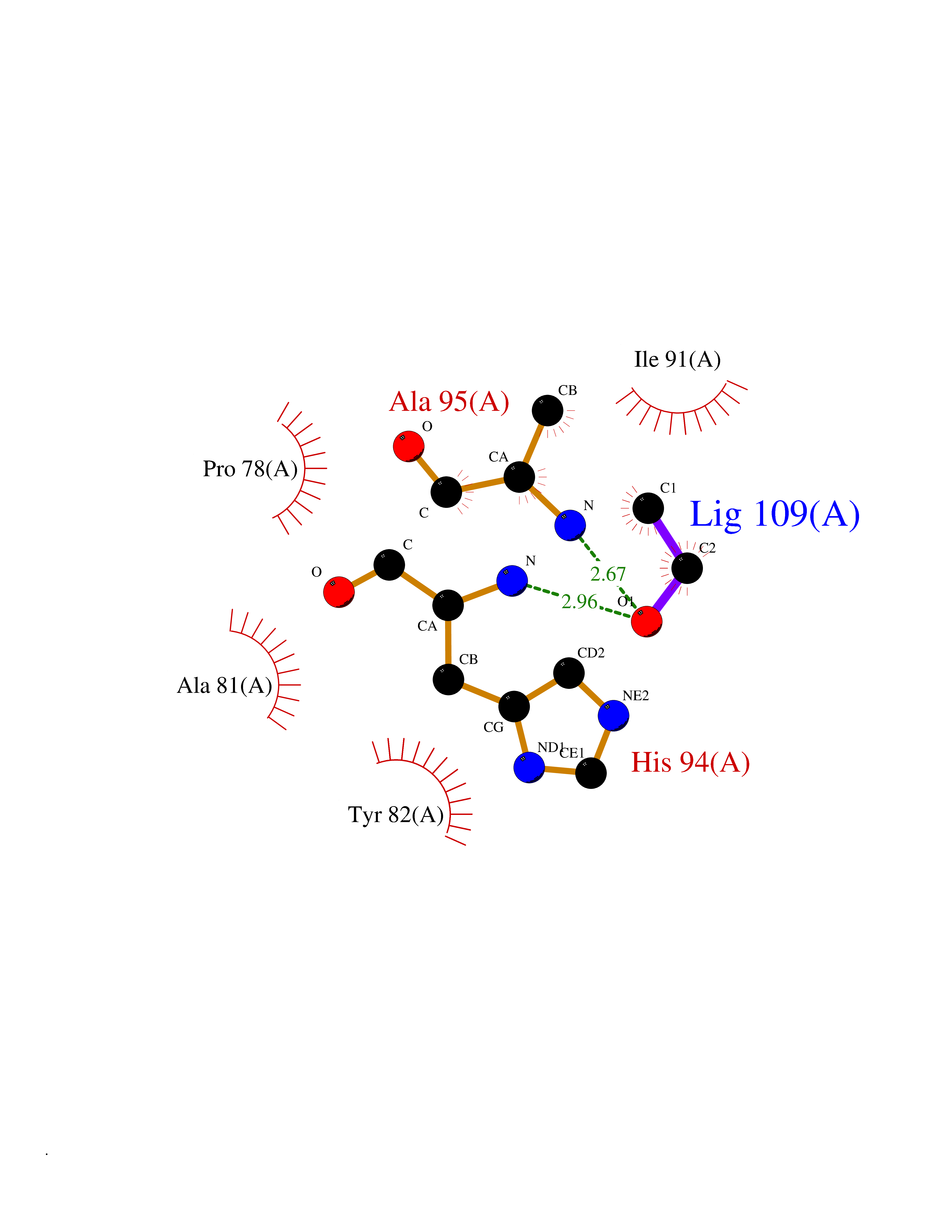 Ligplot Map 3D Binding mode Sequence GVQVETISPGDGRTFPKRGQTCVVHYTGMLEDGKKFDSSRDKNKPFKFMLGKQEVIRGWEEGVAQMSVGQRAKLTISPDYAYGATGVPGIIPPHATLVFDVELLKLE Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | 2-hydroxy-6-oxo-7-methylocta-2,4-dienoate hydrolase | 1UK8 | 4.27 | |
Target general information Gen name cumD Organism Pseudomonas fluorescens Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function Hydrolase activity. Related diseases Intellectual developmental disorder, autosomal dominant 62 (MRD62) [MIM:618793]: An autosomal dominant form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD62 is characterized by mild to moderately impaired intellectual development. {ECO:0000269|PubMed:27479843, ECO:0000269|PubMed:29460436}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03741; DB03793; DB03568; DB02531; DB03750; DB02406; DB03766 Interacts with NA EC number NA Uniprot keywords 3D-structure; Hydrolase Protein physicochemical properties Chain ID A Molecular weight (Da) 30307.9 Length 271 Aromaticity 0.1 Instability index 37.49 Isoelectric point 5.02 Charge (pH=7) -11.58 2D Binding mode Binding energy (Kcal/mol) -5.83  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence NLEIGKSILAAGVLTNYHDVGEGQPVILIHGSGPGVSAYANWRLTIPALSKFYRVIAPDMVGFGFTDRPENYNYSKDSWVDHIIGIMDALEIEKAHIVGNAFGGGLAIATALRYSERVDRMVLMGAAGTRFDVTEGLNAVWGYTPSIENMRNLLDIFAYDRSLVTDELARLRYEASIQPGFQESFSSMFPEPRQRWIDALASSDEDIKTLPNETLIIHGREDQVVPLSSSLRLGELIDRAQLHVFGRCGHWTQIEQTDRFNRLVVEFFNEA Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Pectate lyase | 1R76 | 4.27 | |
Target general information Gen name pelA Organism Niveispirillum irakense (Azospirillum irakense) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Lyase Function Lyase activity. Related diseases A chromosomal aberration involving ALK is found in a form of non-Hodgkin lymphoma. Translocation t(2;5)(p23;q35) with NPM1. The resulting chimeric NPM1-ALK protein homodimerize and the kinase becomes constitutively activated. The constitutively active fusion proteins are responsible for 5-10% of non-Hodgkin lymphomas. {ECO:0000269|PubMed:15938644}.; DISEASE: A chromosomal aberration involving ALK is associated with inflammatory myofibroblastic tumors (IMTs). Translocation t(2;11)(p23;p15) with CARS; translocation t(2;4)(p23;q21) with SEC31A. {ECO:0000269|PubMed:12112524, ECO:0000269|PubMed:16161041}.; DISEASE: A chromosomal aberration involving ALK is associated with anaplastic large-cell lymphoma (ALCL). Translocation t(2;17)(p23;q25) with ALO17. {ECO:0000269|PubMed:12112524}.; DISEASE: Neuroblastoma 3 (NBLST3) [MIM:613014]: A common neoplasm of early childhood arising from embryonic cells that form the primitive neural crest and give rise to the adrenal medulla and the sympathetic nervous system. {ECO:0000269|PubMed:18724359, ECO:0000269|PubMed:18923523, ECO:0000269|PubMed:18923525, ECO:0000269|PubMed:21242967, ECO:0000269|PubMed:22932897}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: The ALK signaling pathway plays an important role in glioblastoma, the most common malignant brain tumor of adults and one of the most lethal cancers. It regulates both glioblastoma migration and growth. {ECO:0000269|PubMed:15908427}.; DISEASE: A chromosomal aberration involving ALK is found in one subject with colorectal cancer. Translocation t(2;2)(p23.1;p23.3). A 5 million base pair tandem duplication generates an in-frame WDCP-ALK gene fusion. {ECO:0000269|PubMed:22327622}.; DISEASE: A chromosomal aberration involving ALK has been identified in a subset of patients with non-small-cell lung carcinoma. This aberration leads to the production of a fusion protein between the N-terminus of EML4 et the C-terminus of ALK. It is unclear whether the fusion protein is caused by a simple inversion within 2p (inv(2)(p21p23)) or whether the chromosome translocation involving 2p is more complex. When tested in a heterologous system, the fusion protein EML4-ALK possesses transforming activity that is dependent on ALK catalytic activity, possibly due to spontaneous dimerization mediated by the EML4 moiety, leading to ALK kinase activation. {ECO:0000269|PubMed:17625570}. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Lyase; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 41907.5 Length 384 Aromaticity 0.08 Instability index 43.72 Isoelectric point 6.11 Charge (pH=7) -3.46 2D Binding mode Binding energy (Kcal/mol) -5.83  Molscript Map 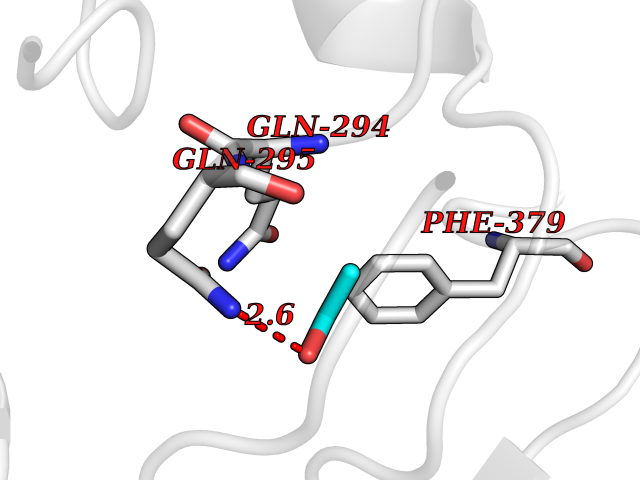 Pymol Map 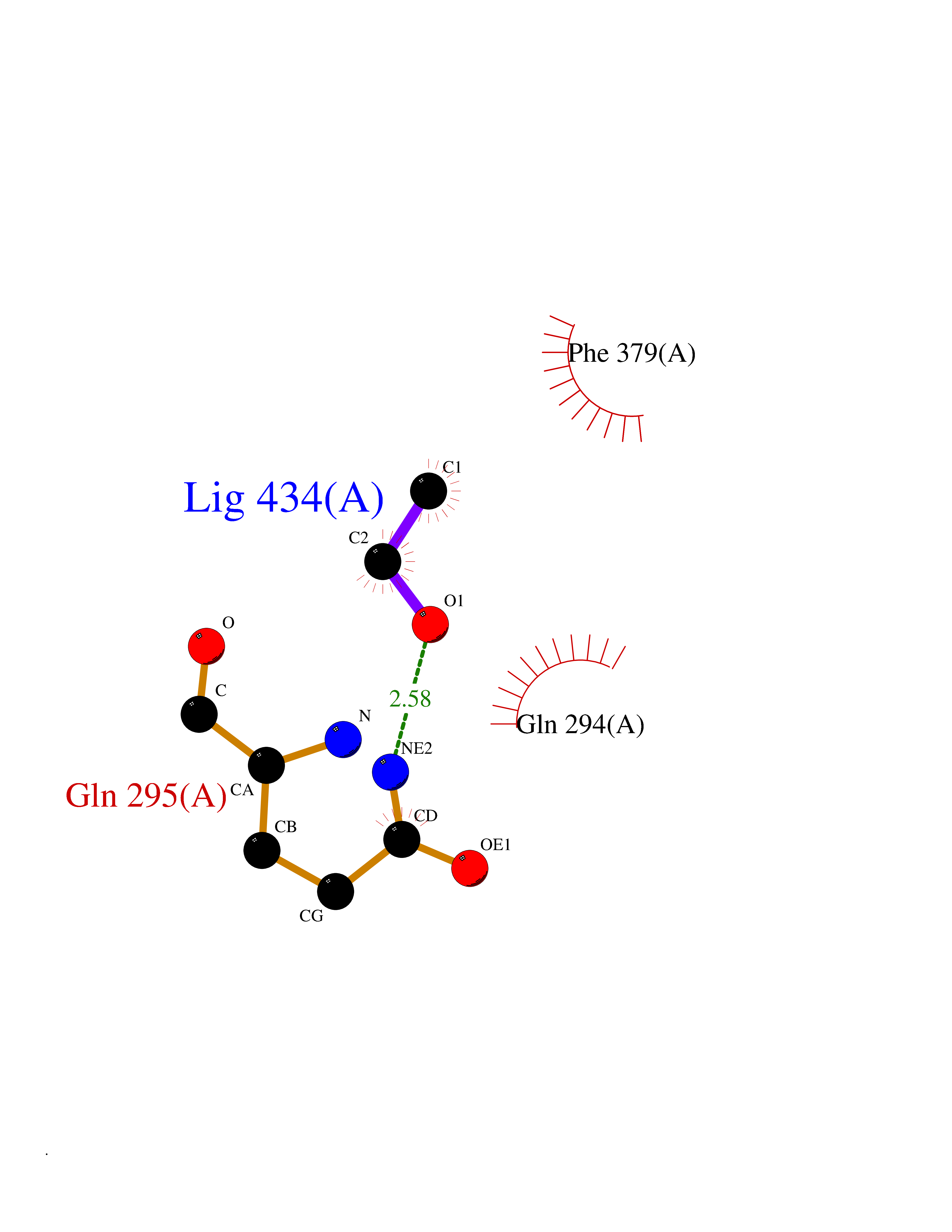 Ligplot Map 3D Binding mode Sequence AVIGMNEAASALTPSRVSSLPDTQRAAWQEYLARSEAQLSRDKASLAAELAPGQPLPPPPAEGKGADTMPLDKPAAWYTSKAARHVADVIVSFQTPAGGWGKNQPRDGALRLPGQHYTGENVAKVKRDRDWHYVGTIDNDATVTEIRFLAQVVSQLAPEEAAPYRDAALKGIEYLLASQFPNGGWPQVWPLEGGYHDAITYNDDALVHVAELLSDIAAGRDGFGFVPPAIRTRALEATNAAIHCIVETQVVQDGKRLGWGQQHDALTLRPTSARNFEPAALSSTESARILLFLMEIEAPSDAVKQAIRGGVAWLNTSVIRDQGAKPLWSRFYSLDGNKPVFGDRDKTIHDDVMGISQERRTGYAWYTTSPQKALSAFTKWEKRS Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | Sodium channel subunit beta-3 | 4L1D | 4.27 | |
Target general information Gen name SCN3B Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms KIAA1158 Protein family Sodium channel auxiliary subunit SCN3B (TC 8.A.17) family Biochemical class Membrane protein Function Ion channel binding.Sodium channel regulator activity.Voltage-gated sodium channel activity.Voltage-gated sodium channel activity involved in cardiac muscle cell action potential. Related diseases Brugada syndrome 7 (BRGDA7) [MIM:613120]: A tachyarrhythmia characterized by right bundle branch block and ST segment elevation on an electrocardiogram (ECG). It can cause the ventricles to beat so fast that the blood is prevented from circulating efficiently in the body. When this situation occurs, the individual will faint and may die in a few minutes if the heart is not reset. {ECO:0000269|PubMed:20031595}. The gene represented in this entry may be involved in disease pathogenesis.; DISEASE: Atrial fibrillation, familial, 16 (ATFB16) [MIM:613120]: A familial form of atrial fibrillation, a common sustained cardiac rhythm disturbance. Atrial fibrillation is characterized by disorganized atrial electrical activity and ineffective atrial contraction promoting blood stasis in the atria and reduces ventricular filling. It can result in palpitations, syncope, thromboembolic stroke, and congestive heart failure. {ECO:0000269|PubMed:20558140, ECO:0000269|PubMed:21051419}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05541; DB00907; DB13269; DB13961; DB00776; DB00243; DB00313; DB00909 Interacts with Q15848; Q9NRZ5; Q9NUQ2; Q86W74-2; Q96PS8; Q9H2C2; Q9HD20-3; P27449; Q12981; Q12983; Q6PL45-2; Q4LDR2; P78329; P81534; Q9BUN8; Q6ZPD8; Q9UKR5; Q96D05-2; Q92520; Q9Y3D6; Q14318; Q14802-3; Q8WWP7; Q96F15; Q8TDV0; Q9BZJ8; Q9Y5U9; Q9Y5U4; Q6H9L7; P11215; Q68G75; O95214; Q9UBY5; Q969L2; Q9P0N8; Q6N075; O14880; Q5J8X5; Q9NZG7; Q9P0S3; Q53FV1; Q8NHP8; Q04941; Q96GM1; Q01453; Q02161-2; O76064; Q96GQ5; Q9NTJ5; O00767; Q96IW7; Q8N6R1; Q8WV19; Q9H9B4; Q9BWM7; P22732; Q99726; Q8IWU4; Q96G79; Q6ICL7; Q9H2H9; Q9NVC3; P30825; Q9NRQ5; B2RUZ4; P0DN84; Q13277; O43752; Q9UNK0; Q9UPZ6; O14925; Q9BZW4; P55061; Q9BXJ8; A0PK00; Q5BJH2-2; Q9NV12; Q9Y6G1; Q9NUH8; Q9BTX3; Q8WW34-2; Q9BU79; Q69YG0; P56557; Q9H2L4; Q6PI78; Q8N2M4; Q6ZT21; Q6ZUI0; O60636; A5PKU2; Q9NYZ1; Q9Y5Z9; Q9NZ43; P23763-3; P63027; Q15836; Q9P0L0; O95292; O95070; Q9BSR8; O95159 EC number NA Uniprot keywords 3D-structure; Atrial fibrillation; Brugada syndrome; Cell membrane; Disease variant; Disulfide bond; Glycoprotein; Immunoglobulin domain; Ion channel; Ion transport; Membrane; Proteomics identification; Reference proteome; Signal; Sodium; Sodium channel; Sodium transport; Transmembrane; Transmembrane helix; Transport; Voltage-gated channel Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 38121 Length 332 Aromaticity 0.09 Instability index 42.72 Isoelectric point 6.59 Charge (pH=7) -1.45 2D Binding mode Binding energy (Kcal/mol) -5.83 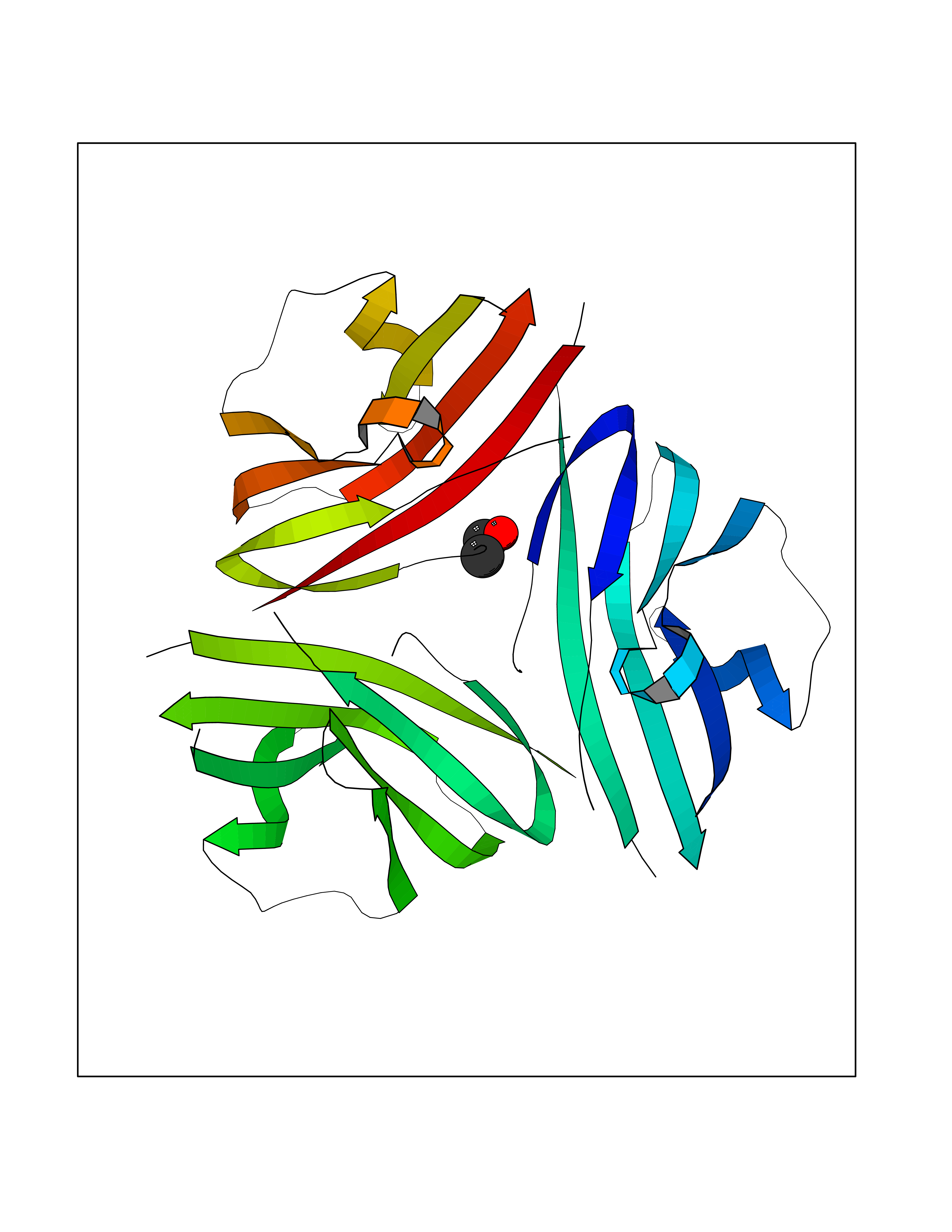 Molscript Map 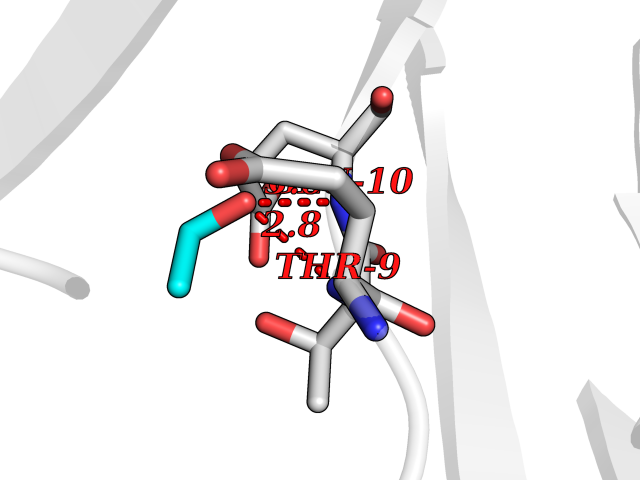 Pymol Map 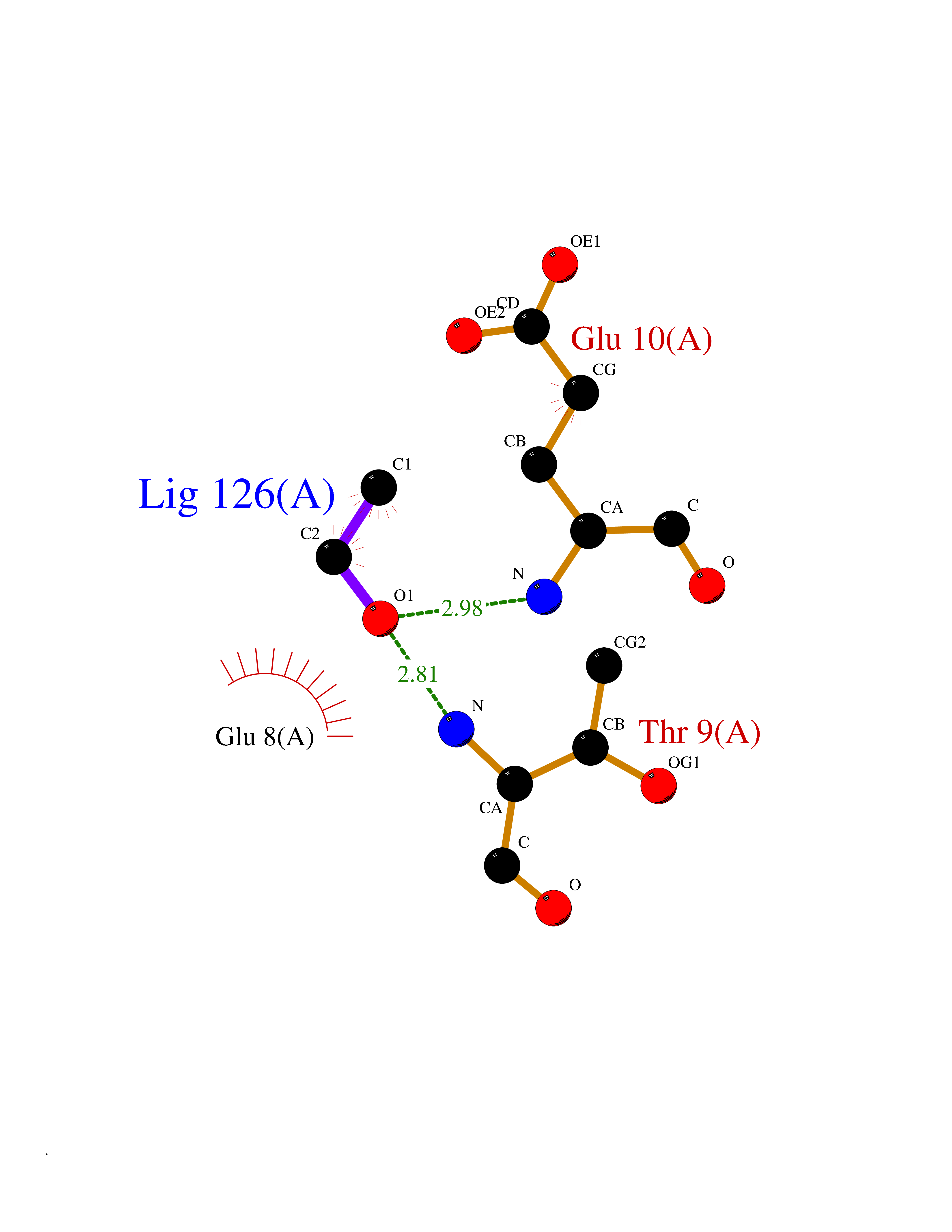 Ligplot Map 3D Binding mode Sequence VCVEVPSETEAVQGNPMKLRCISCMKRATTVVEWFYRPEGGKDFLIYEYRNGHQEVESPFQGRLQWNGSKDLQDVSITVLNVTLNDSGLYTCNVSREFFVKTTRLIPLRVHHHCVEVPSETEAVQGNPMKLRCISCMKATTVVEWFYRPEGGKDFLIYEYRNGHQEVESPFQGRLQWNGSKDLQDVSITVLNVTLNDSGLYTCNVSREFVKTTRLIPLRVHHVCVEVPSETEAVQGNPMKLRCISCMKATTVVEWFYRPEGGKDFLIYEYRNGHQEVESPFQGRLQWNGSKDLQDVSITVLNVTLNDSGLYTCNVSREFVKTTRLIPLRVHH Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Cyclin-dependent kinase 8 (CDK8) | 3RGF | 4.27 | |
Target general information Gen name CDK8 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein kinase K35; Mediator of RNA polymerase II transcription subunit CDK8; Mediator complex subunit CDK8; Cell division protein kinase 8 Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, CDC2/CDKX subfamily Biochemical class Kinase Function Mediator functions as a bridge to convey information from gene-specific regulatory proteins to the basal RNA polymerase II transcription machinery. Mediator is recruited to promoters by direct interactions with regulatory proteins and serves as a scaffold for the assembly of a functional preinitiation complex with RNA polymerase II and the general transcription factors. Phosphorylates the CTD (C-terminal domain) of the large subunit of RNA polymerase II (RNAp II), which may inhibit the formation of a transcription initiation complex. Phosphorylates CCNH leading to down-regulation of the TFIIH complex and transcriptional repression. Recruited through interaction with MAML1 to hyperphosphorylate the intracellular domain of NOTCH, leading to its degradation. Component of the Mediator complex, a coactivator involved in regulated gene transcription of nearly all RNA polymerase II-dependent genes. Related diseases Intellectual developmental disorder with hypotonia and behavioral abnormalities (IDDHBA) [MIM:618748]: An autosomal dominant neurodevelopmental disorder with onset in infancy. IDDHBA is characterized by hypotonia, global developmental delay, learning disability, and behavioral abnormalities, such as autistic features and attention deficit-hyperactivity disorder. Additional variable features may include non-specific facial dysmorphism, congenital heart defects, ocular anomalies, and poor feeding. {ECO:0000269|PubMed:30905399}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03496 Interacts with P24863; Q01094; P02489; Q14204; Q92876 EC number EC 2.7.11.22 Uniprot keywords 3D-structure; Activator; Alternative splicing; ATP-binding; Autism spectrum disorder; Disease variant; Intellectual disability; Kinase; Nucleotide-binding; Nucleus; Proteomics identification; Reference proteome; Repressor; Serine/threonine-protein kinase; Transcription; Transcription regulation; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 37655.2 Length 321 Aromaticity 0.11 Instability index 37.04 Isoelectric point 8.56 Charge (pH=7) 5.13 2D Binding mode Binding energy (Kcal/mol) -5.83 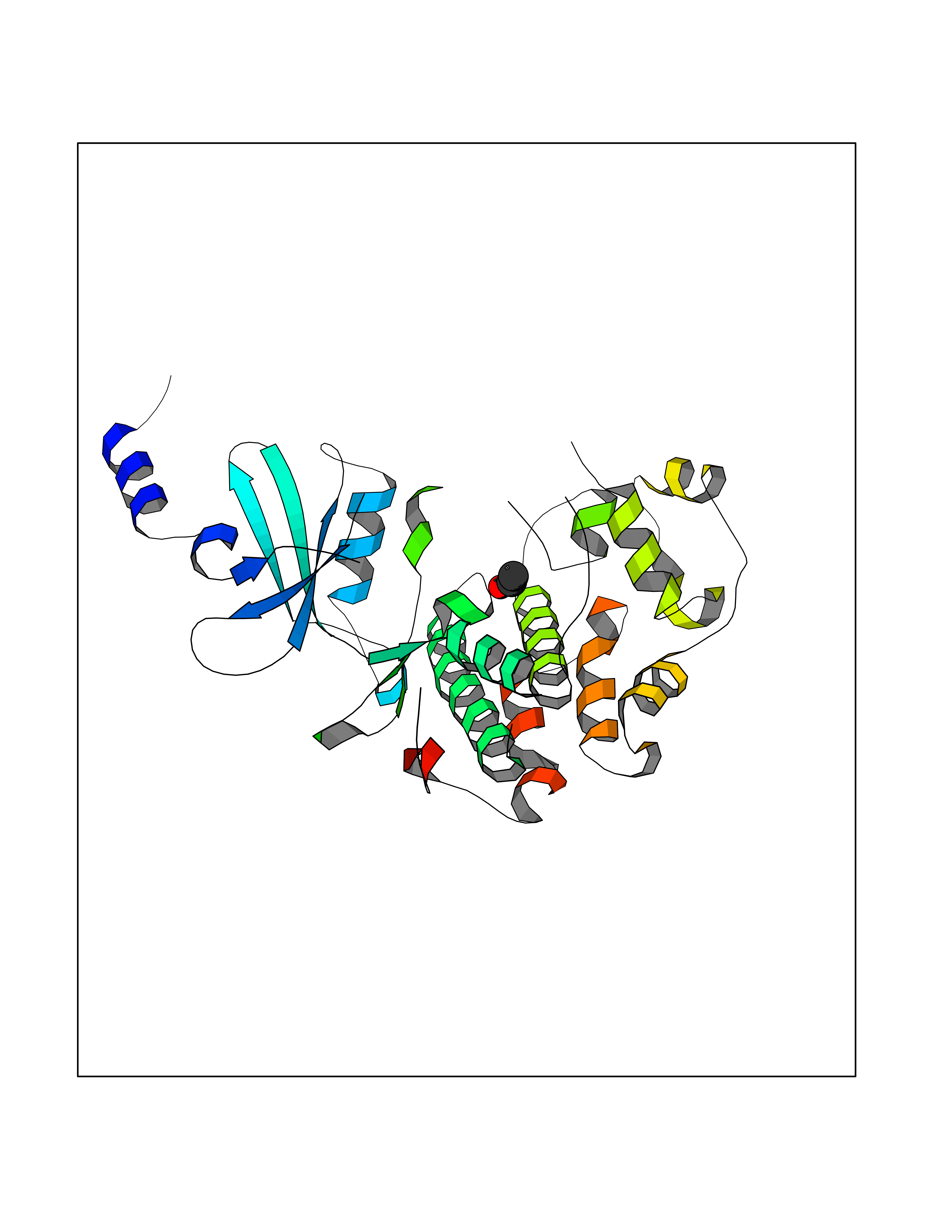 Molscript Map 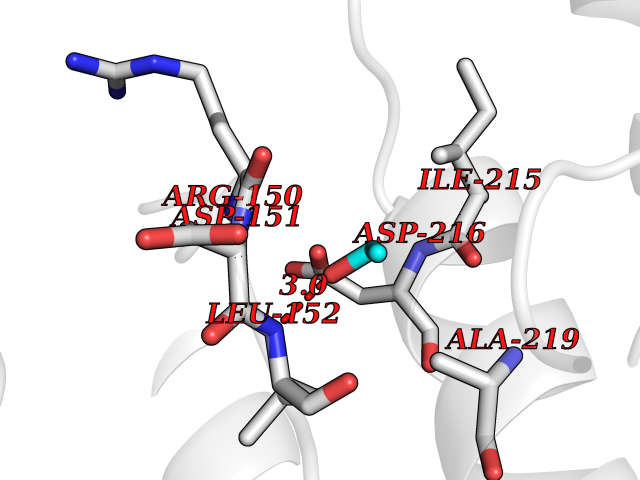 Pymol Map 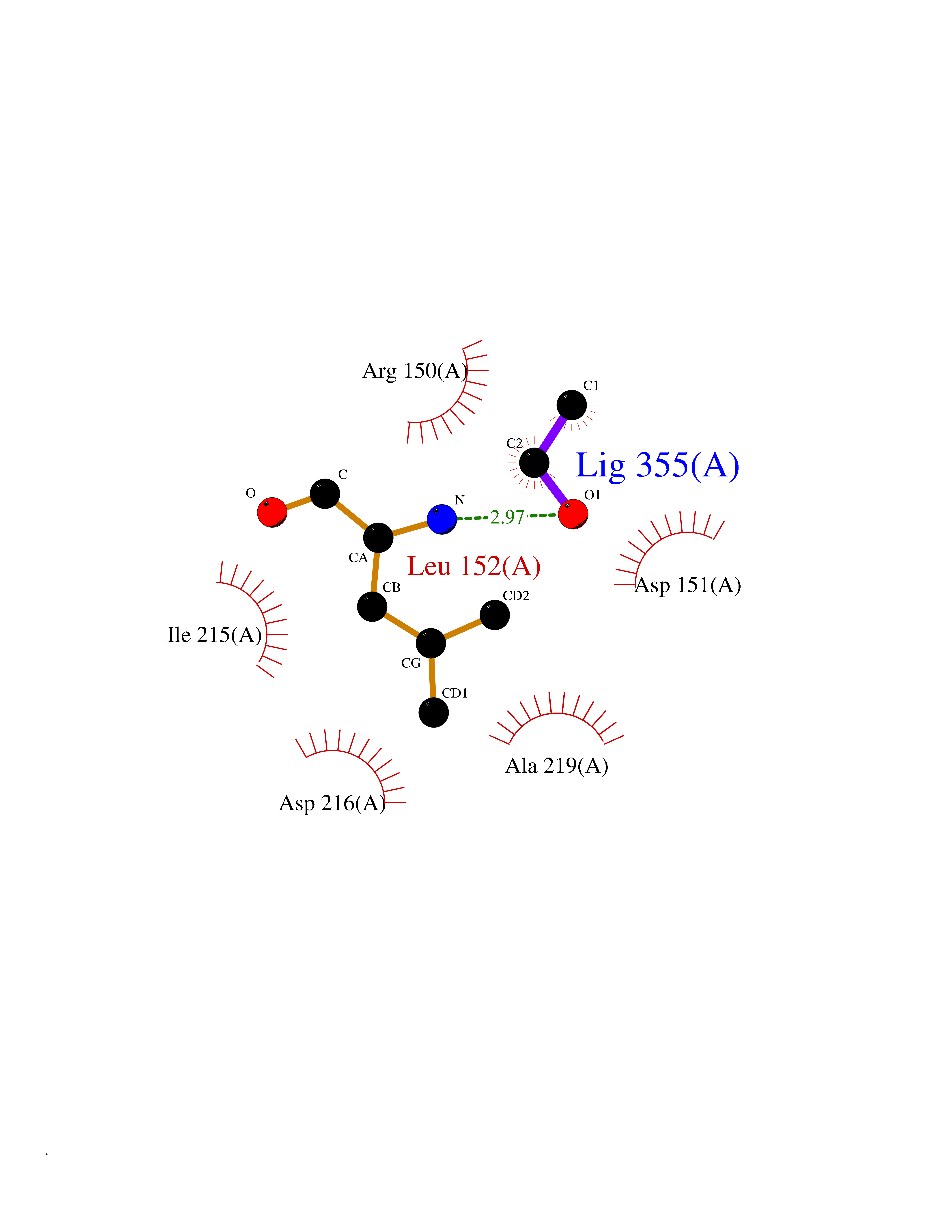 Ligplot Map 3D Binding mode Sequence DKMDYDFKVKLSSERERVEDLFEYEGCKVGHVYKAKRKDGKDDKDYALKQIEGTGISMSACREIALLRELKHPNVISLQKVFLSHADRKVWLLFDYAEHDLWHIIKFHRASKLPRGMVKSLLYQILDGIHYLHANWVLHRDLKPANILVMGEGPERGRVKIADMGFARVTFWYRAPELLLGARHYTKAIDIWAIGCIFAELLTSEPIFHCRQNPYHHDQLDRIFNVMGFPADKDWEDIKKMPEHSTLMKDFRRNTYTNCSLIKYMEKHKVKPDSKAFHLLQKLLTMDPIKRITSEQAMQDPYFLEDPLPTSDVFAGCQIPY Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Lysine-specific demethylase 4E (KDM4E) | 2W2I | 4.27 | |
Target general information Gen name KDM4E Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Lysine-specific demethylase 4D-like; KDM4DL; KDM4D-like protein Protein family JHDM3 histone demethylase family Biochemical class Paired donor oxygen oxidoreductase Function Histone demethylase that specifically demethylates 'Lys-9' of histone H3, thereby playing a central role in histone code. Related diseases Defects in KAT2B has been found in a patient with isolated coloboma, a defect of the eye characterized by the absence of ocular structures due to abnormal morphogenesis of the optic cup and stalk, and the fusion of the fetal fissure (optic fissure). Isolated colobomas may be associated with an abnormally small eye (microphthalmia) or small cornea. {ECO:0000269|PubMed:28493397}. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.14.11.- Uniprot keywords 3D-structure; Chromatin regulator; Dioxygenase; Iron; Metal-binding; Nucleus; Oxidoreductase; Reference proteome; Transcription; Transcription regulation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 35131.5 Length 305 Aromaticity 0.14 Instability index 39.34 Isoelectric point 6 Charge (pH=7) -6.1 2D Binding mode Binding energy (Kcal/mol) -5.83 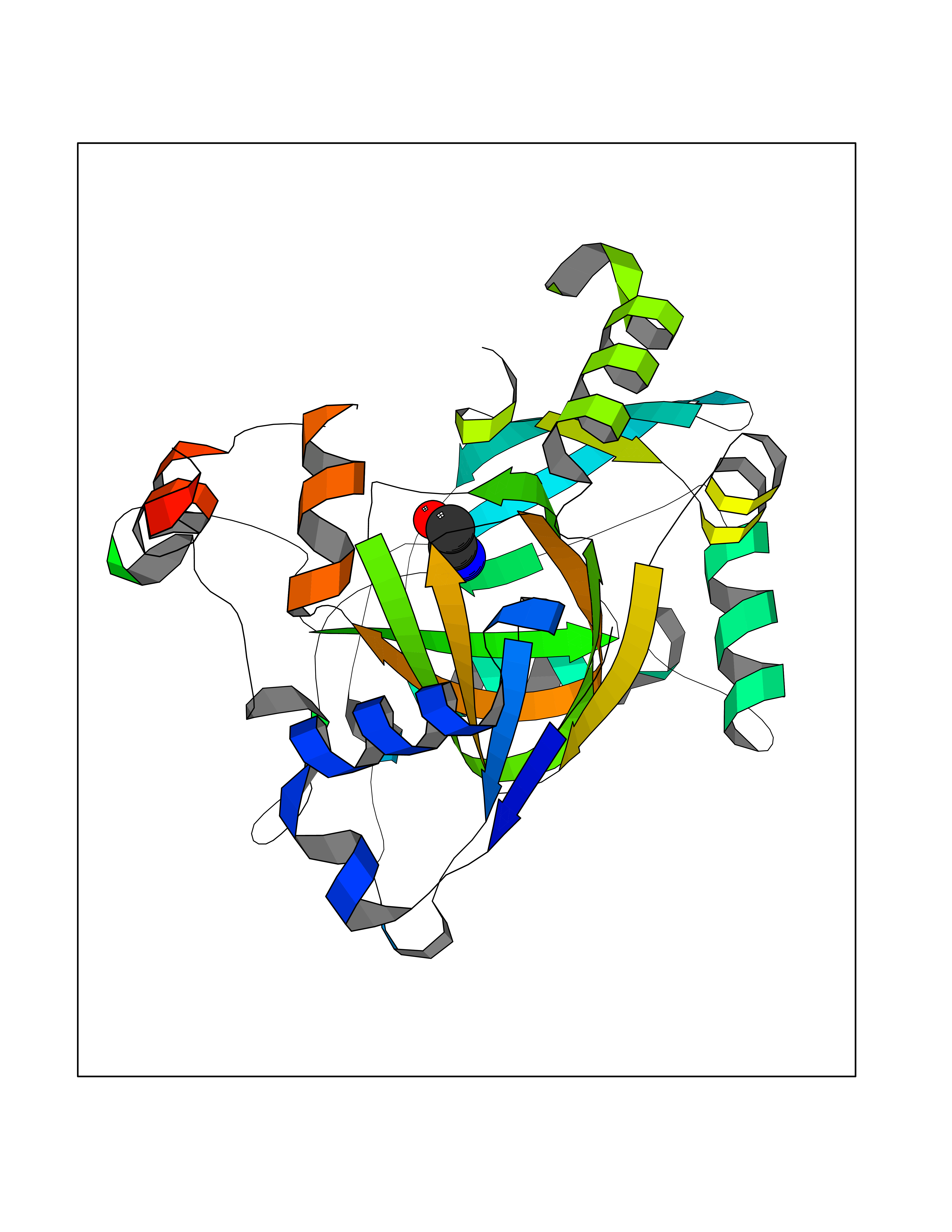 Molscript Map 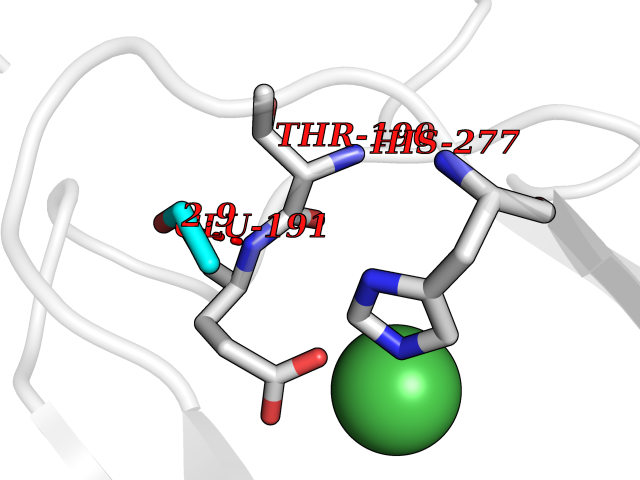 Pymol Map 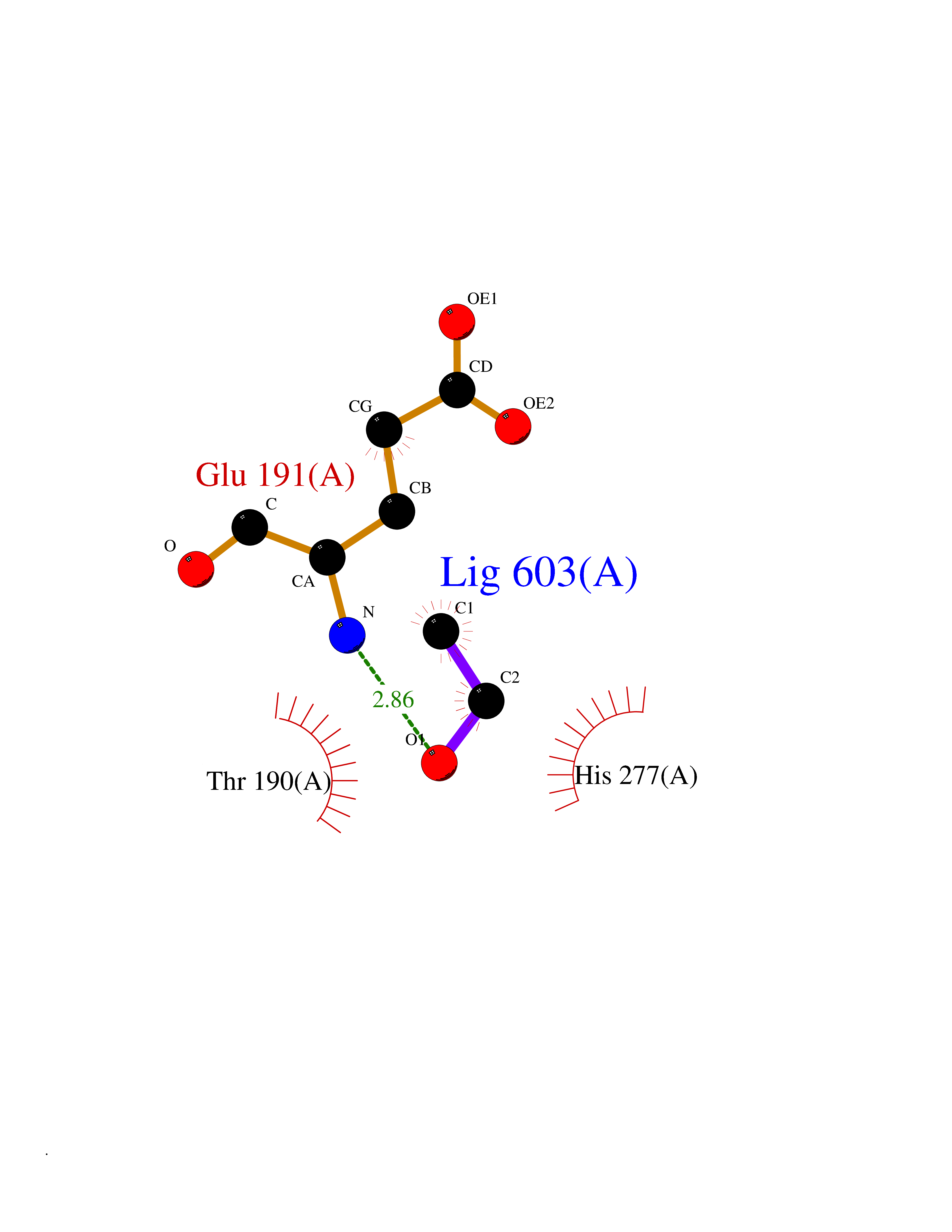 Ligplot Map 3D Binding mode Sequence HTIMTFYPTMEEFADFNTYVAYMESQGAHQAGLAKVIPPKEWKARQMYDDIEDILIATPLQQVTSGQGGVFTQYHKKKKAMRVGQYRRLANSKKYQTPPHQNFADLEQRYWKSHPGNPPIYGADISGSLFEESTKQWNLGHLGTILDLLEQECGVVIEGVNTPYLYFGMWKTTFAWHTEDMDLYSINYLHFGEPKTWYVVPPEHGQHLERLARELFPDISAFLRHKVALISPTVLKENGIPFNCMTQEAGEFMVTFPYGYHAGFNHGFNCAEAINFATPRWIDYGKMAVTFSMDPFVRIVQPESY Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | CaM-kinase IV kinase (CAMKK1) | 6CD6 | 4.27 | |
Target general information Gen name CAMKK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Calcium/calmodulin-dependent protein kinase kinase alpha; Calcium/calmodulin-dependent protein kinase kinase 1; CaMKK alpha; CaMKK 1; CaM-kinase kinase alpha; CaM-kinase kinase 1; CaM-KK alpha; CaM-KK Protein family Protein kinase superfamily, Ser/Thr protein kinase family Biochemical class Kinase Function Calcium/calmodulin-dependent protein kinase that belongs to a proposed calcium-triggered signaling cascade involved in a number of cellular processes. Phosphorylates CAMK1, CAMK1D, CAMK1G and CAMK4. Involved in regulating cell apoptosis. Promotes cell survival by phosphorylating AKT1/PKB that inhibits pro-apoptotic BAD/Bcl2-antagonist of cell death. Related diseases Intellectual developmental disorder with macrocephaly, seizures, and speech delay (IDDMSSD) [MIM:618158]: An autosomal dominant neurodevelopmental disorder characterized by impaired intellectual development, poor speech, postnatal macrocephaly, and seizures. {ECO:0000269|PubMed:30290153}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with O15063; P62258; Q04917; P21333-2; P04792; P42858; O60333-2; P07196; Q99497; Q8N2W9; P60891; Q9Y3C5; P37840; P00441; O76024 EC number EC 2.7.11.17 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Calmodulin-binding; Cytoplasm; Kinase; Methylation; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 29932.3 Length 264 Aromaticity 0.08 Instability index 39.1 Isoelectric point 5.71 Charge (pH=7) -5.63 2D Binding mode Binding energy (Kcal/mol) -5.82 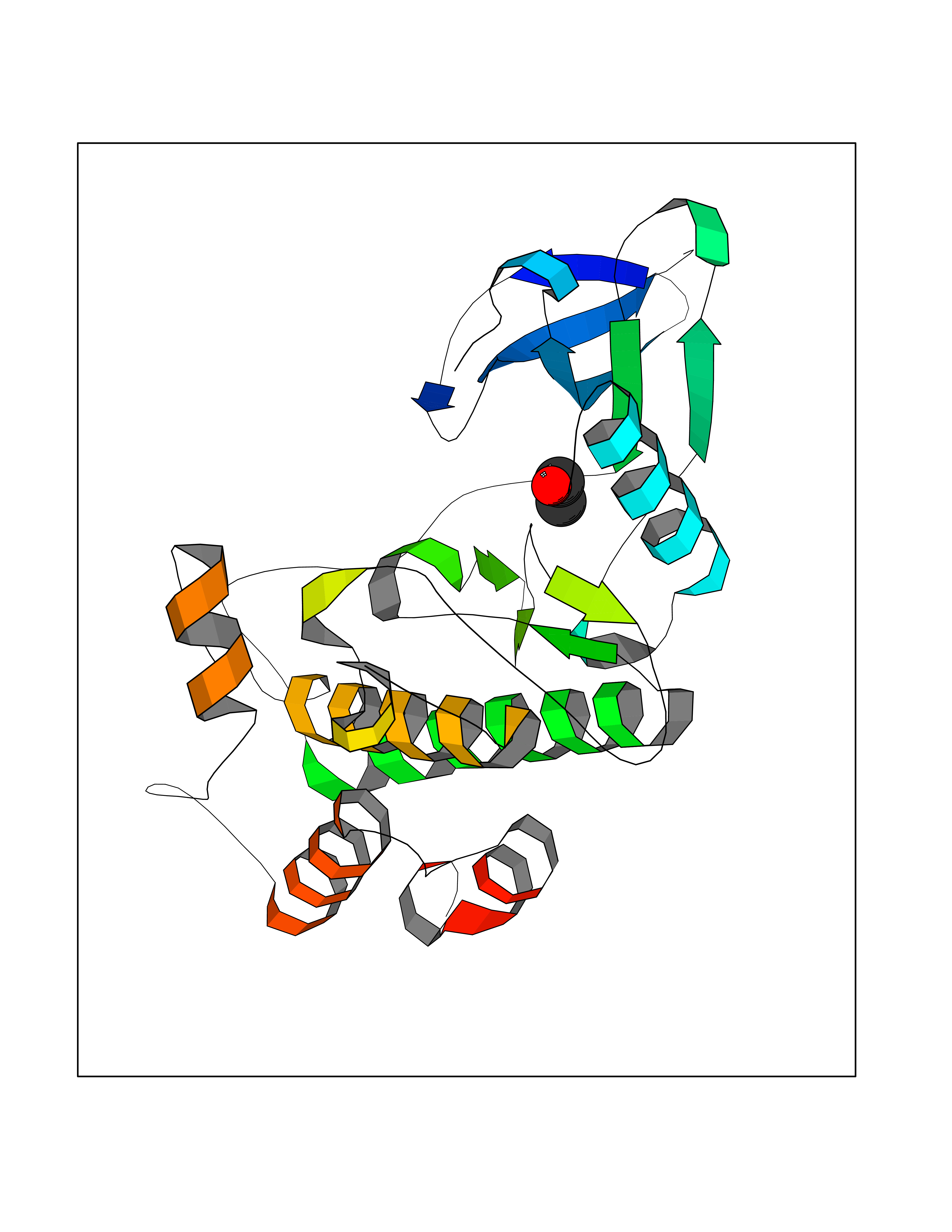 Molscript Map 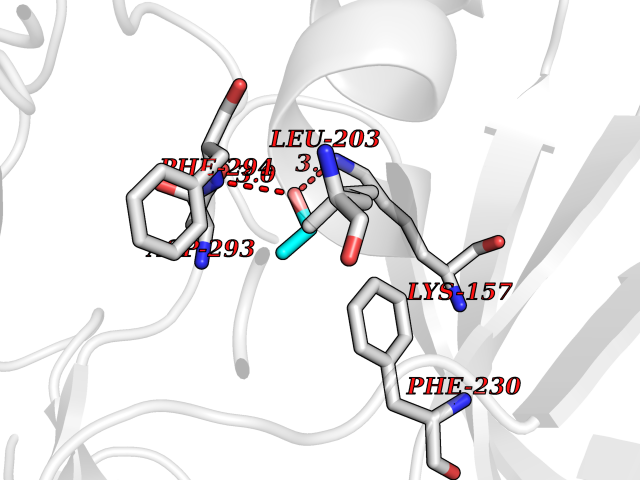 Pymol Map 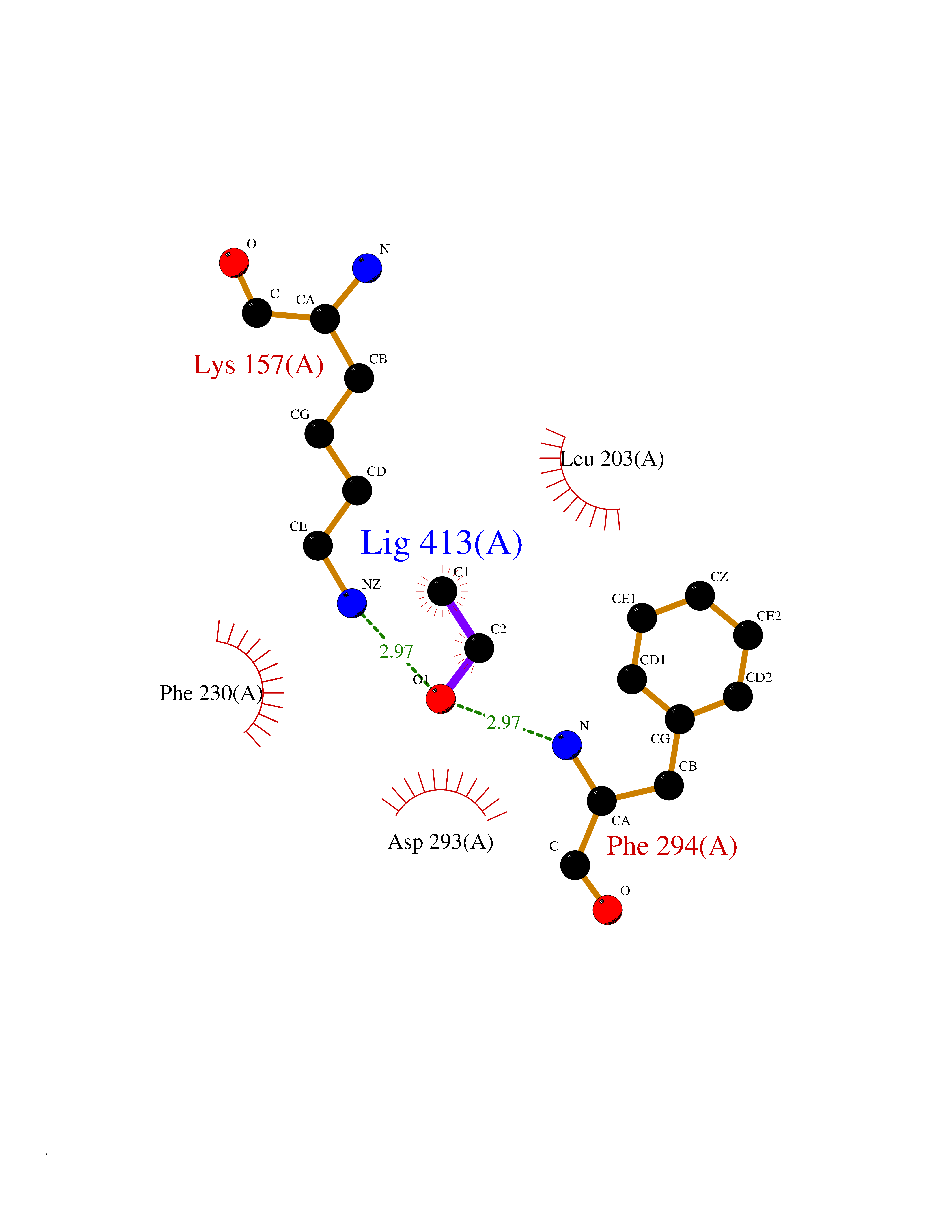 Ligplot Map 3D Binding mode Sequence LNQYKLQSEIGKGAYGVVRLAYNESEDRHYAMKVLSKKKLLKQQLLPLERVYQEIAILKKLDHVNVVKLIEVLDDPAEDNLYLVFDLLRKGPVMEVPCDKPFSEEQARLYLRDVILGLEYLHCQKIVHRDIKPSNLLLGDDGHVKIADFGVSNQFEGNDAQLSSTAGTPAFMAPEAISGQSFSGKALDVWATGVTLYCFVYGKCPFIDDFILALHRKIKNEPVVFPEEPEISEELKDLILKMLDKNPETRIGVPDIKLHPWVTK Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Aspartyl aminopeptidase (DNPEP) | 4DYO | 4.27 | |
Target general information Gen name DNPEP Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms DAP; ASPEP Protein family Peptidase M18 family Biochemical class Peptidase Function Likely to play an important role in intracellular protein and peptide metabolism. Aminopeptidase with specificity towards an acidic amino acid at the N-terminus. Related diseases Cohen-Gibson syndrome (COGIS) [MIM:617561]: An autosomal dominant overgrowth disorder characterized by accelerated osseous maturation, advanced bone age, skeletal abnormalities including flaring of the metaphyses of the long bones, large hands with long fingers and camptodactyly, scoliosis, cervical spine anomalies, dysmorphic facial features, and variable intellectual disability. {ECO:0000269|PubMed:25787343, ECO:0000269|PubMed:27193220, ECO:0000269|PubMed:27868325, ECO:0000269|PubMed:28229514, ECO:0000269|PubMed:28475857}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00142 Interacts with Q9ULA0; Q8TBB1; Q00013; Q9UPN6 EC number EC 3.4.11.21 Uniprot keywords 3D-structure; Acetylation; Alternative initiation; Aminopeptidase; Cytoplasm; Hydrolase; Metal-binding; Metalloprotease; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 50574.6 Length 457 Aromaticity 0.07 Instability index 55.73 Isoelectric point 7.73 Charge (pH=7) 2.3 2D Binding mode Binding energy (Kcal/mol) -5.82 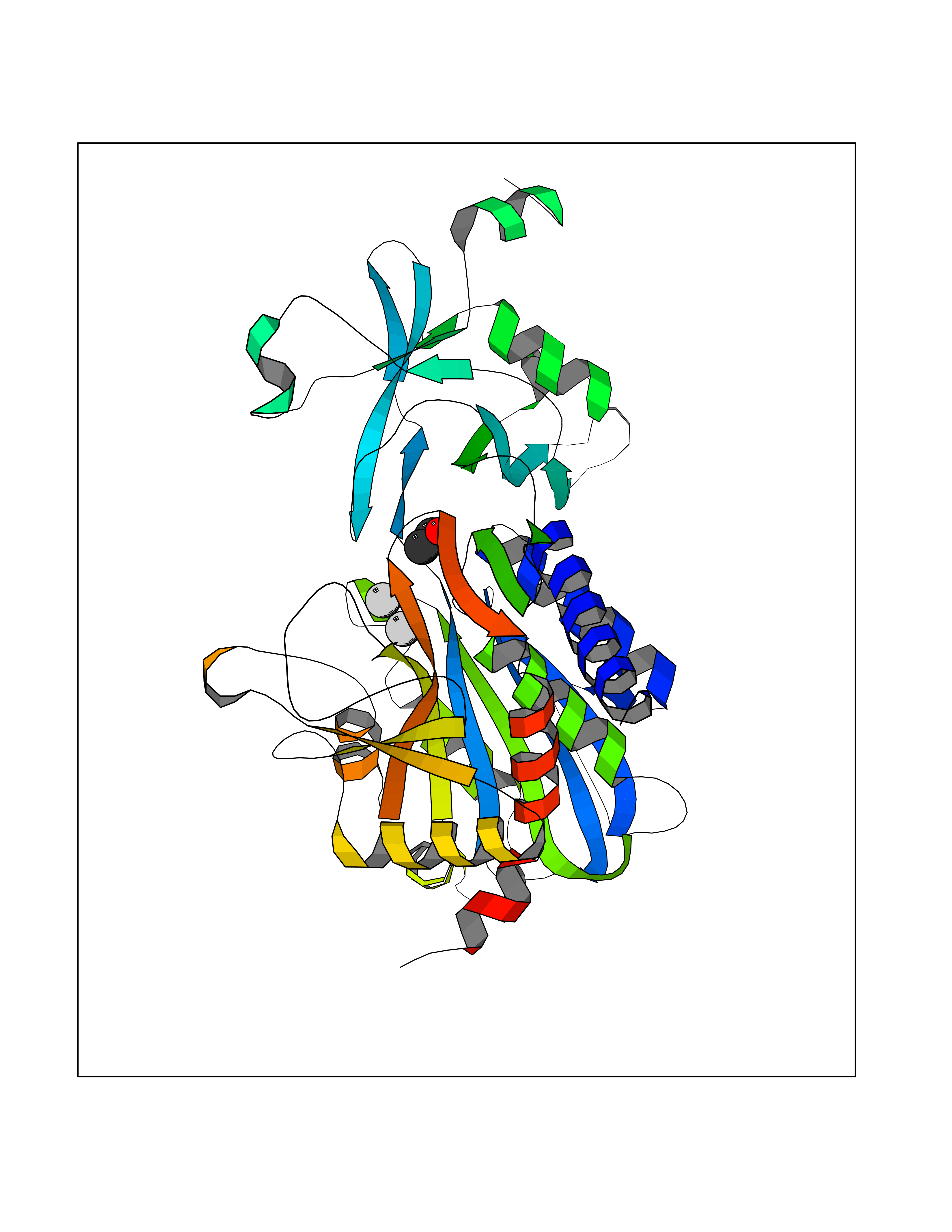 Molscript Map 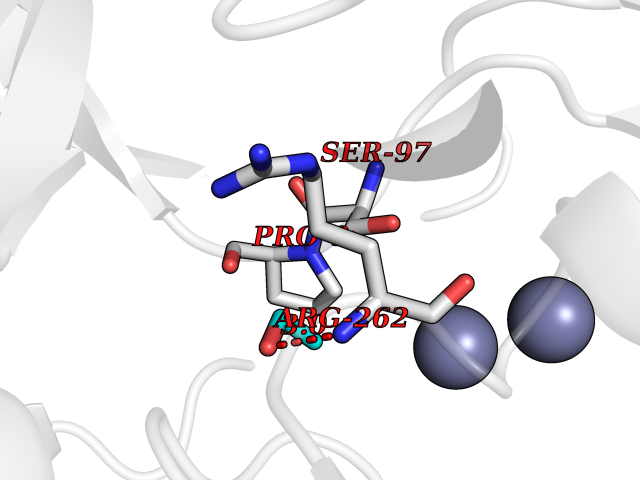 Pymol Map 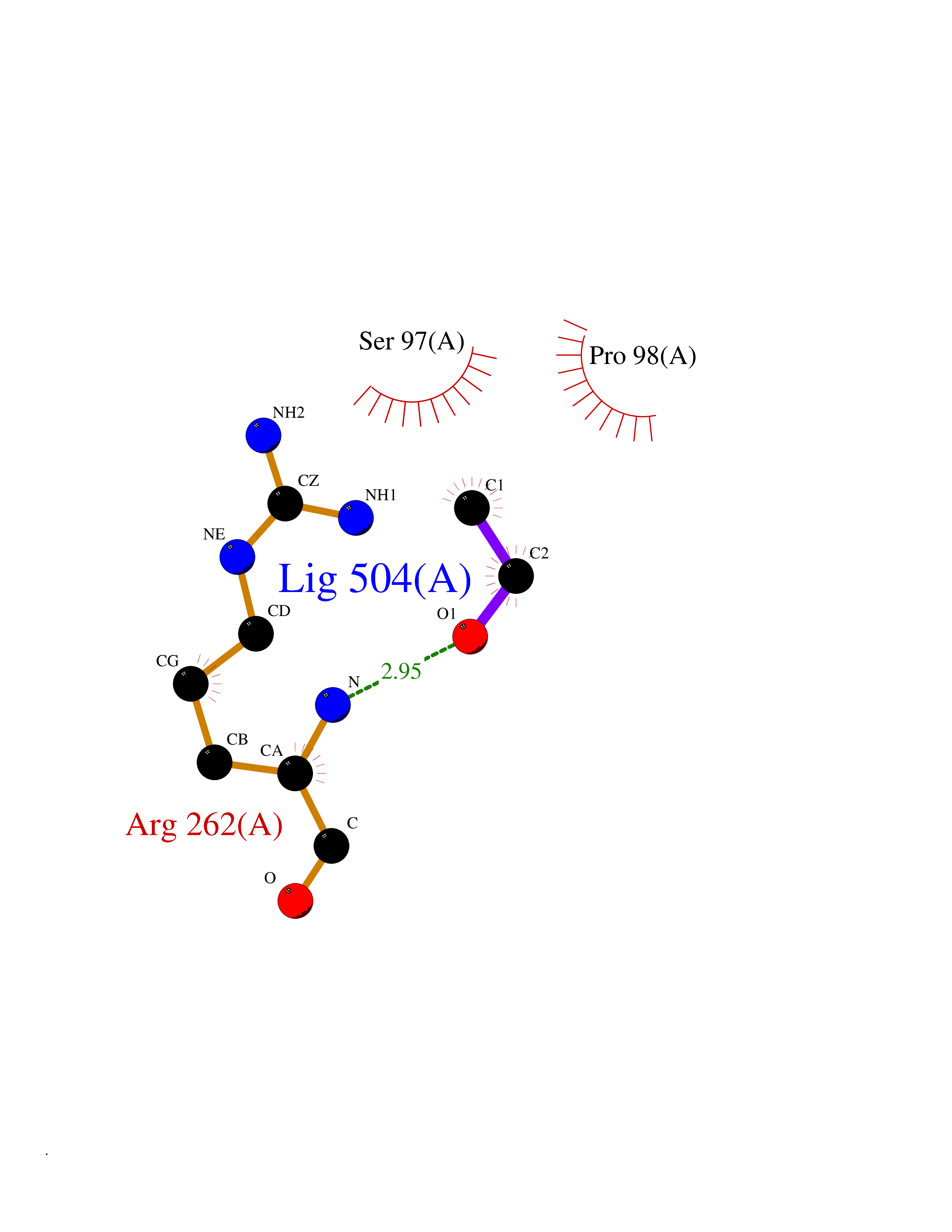 Ligplot Map 3D Binding mode Sequence GKARKEAVQTAAKELLKFVNRSPSPFHAVAECRNRLLQAGFSELKETEKWNIKPESKYFMTRNSSTIIAFAVGGQYVPGNGFSLIGAHTDSPCLRVKRRSRRSQVGFQQVGVETYGGGIWSTWFDRDLTLAGRVIVKCPTSGRLEQQLVHVERPILRIPHLAIHLQRNINENFGPNTEMHLVPILATAIQEELEKGTERHHSVLMSLLCAHLGLSPKDIVEMELCLADTQPAVLGGAYDEFIFAPRLDNLHSCFCALQALIDSCAGPGSLATEPHVRMVTLYDNEEVGSESAQGAQSLLTELVLRRISASCQHPTAFEEAIPKSFMISADMAHAVHPNYLDKHEENHRPLFHKGPVIKVNSKQRYASNAVSEALIREVANKVKVPLQDLMVRNDTPCGTTIGPILASRLGLRVLDLGSPQLAMHSIREMACTTGVLQTLTLFKGFFELFPSLAENLY Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | Mycobacterium Decaprenylphosphoryl-beta-D-ribose oxidase (McyB dprE1) | 4P8N | 4.27 | |
Target general information Gen name McyB dprE1 Organism Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv) Uniprot ID TTD ID Synonyms FAD-dependent decaprenylphosphoryl-beta-D-ribofuranose 2-oxidase; Decaprenylphosphoryl-beta-D-ribose oxidase; Decaprenylphosphoryl-beta-D-ribose 2-epimerase flavoprotein subunit; Decaprenylphosphoryl- Protein family DprE1 family Biochemical class NA Function Component of the DprE1-DprE2 complex that catalyzes the 2-step epimerization of decaprenyl-phospho-ribose (DPR) to decaprenyl-phospho-arabinose (DPA), a key precursor that serves as the arabinose donor required for the synthesis of cell-wall arabinans. DprE1 catalyzes the first step of epimerization, namely FAD-dependent oxidation of the C2' hydroxyl of DPR to yield the keto intermediate decaprenyl-phospho-2'-keto-D-arabinose (DPX). The intermediate DPX is then transferred to DprE2 subunit of the epimerase complex, most probably through a 'substrate channel' at the interface of DprE1-DprE2 complex. Can also use farnesyl-phosphoryl-beta-D-ribofuranose (FPR) as substrate in vitro. Appears to be essential for the growth and survival of M.tuberculosis. Related diseases Neuronopathy, distal hereditary motor, autosomal recessive 8 (HMNR8) [MIM:618912]: An autosomal recessive disorder characterized by motor axonal neuropathy, slowly progressive distal muscle weakness mainly affecting the lower limbs, difficulty walking, and increased serum sorbitol. Additional variable features are distal sensory impairment, upper limb tremor, scoliosis, and mild hearing loss. {ECO:0000269|PubMed:32367058, ECO:0000269|PubMed:33314640, ECO:0000269|PubMed:33397963}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 1.1.98.3 Uniprot keywords 3D-structure; Antibiotic resistance; Cell wall biogenesis/degradation; FAD; Flavoprotein; Oxidoreductase; Periplasm; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 49442.7 Length 454 Aromaticity 0.09 Instability index 28.24 Isoelectric point 7.29 Charge (pH=7) 0.64 2D Binding mode Binding energy (Kcal/mol) -5.83  Molscript Map 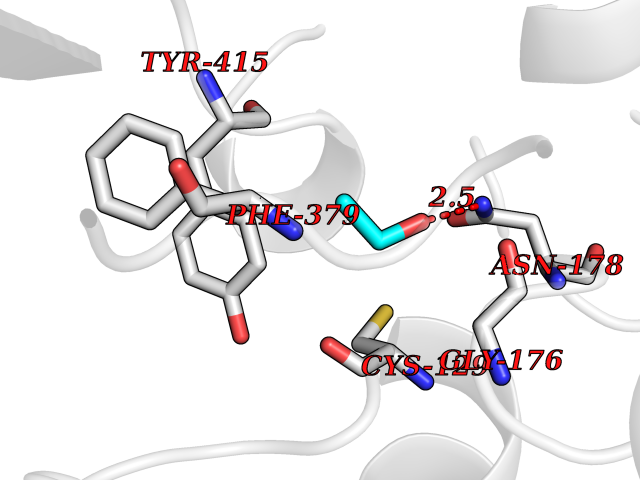 Pymol Map 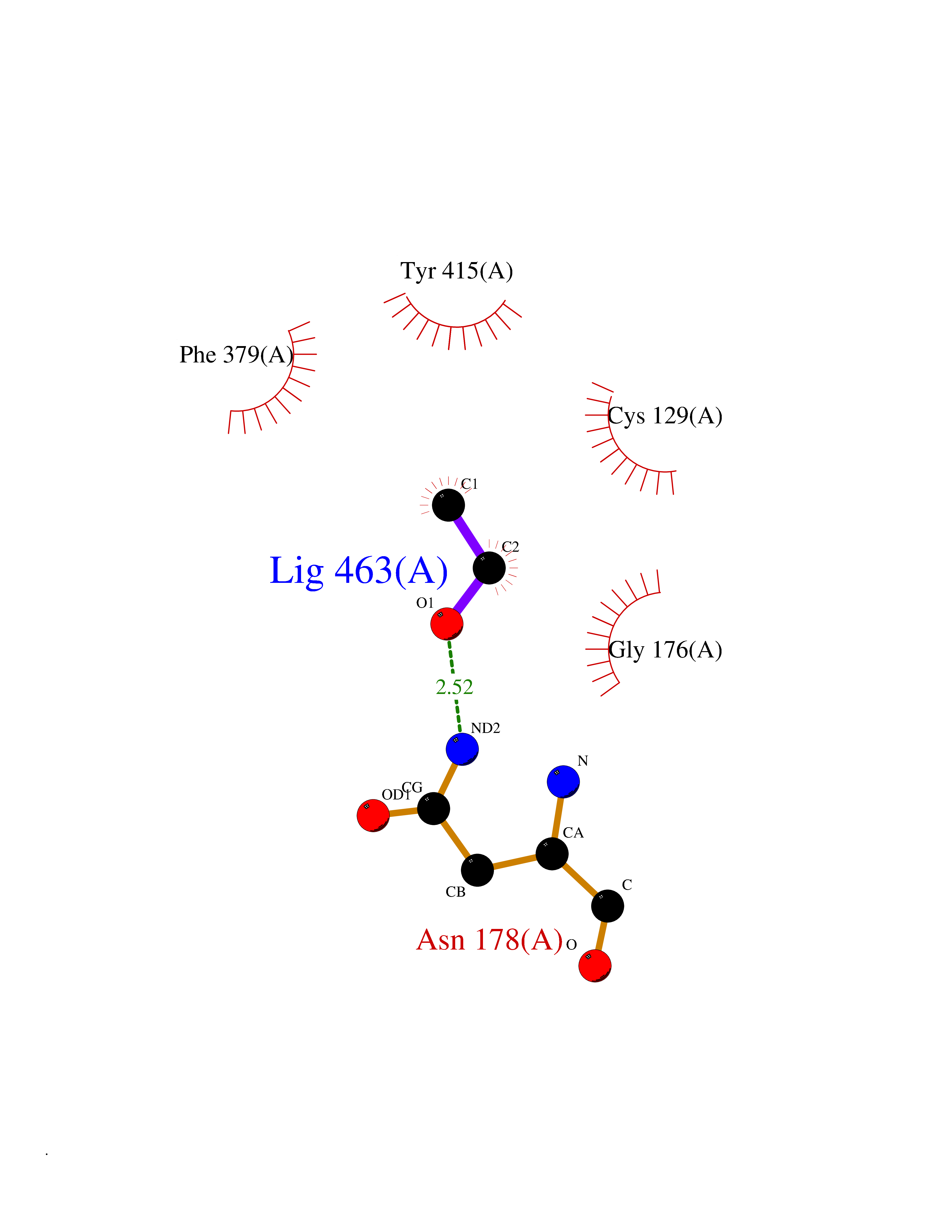 Ligplot Map 3D Binding mode Sequence VGATTTATRLTGWGRTAPSVANVLRTPDAEMIVKAVARVAESGGGRGAIARGLGRSYGDNAQNGGGLVIDMTPLNTIHSIDADTKLVDIDAGVNLDQLMKAALPFGLWVPVLPGTRQVTVGGAIACDIHGKNHHSAGSFGNHVRSMDLLTADGEIRHLTPTGEDAELFWATVGGNGLTGIIMRATIEMTPTSTAYFIADGDVTASLDETIALHSDGSEARYTYSSAWFDAISAPPKLGRAAVSRGRLATVEQLPAKLRSEPLKFDAPQLLTLPDVFPNGLANKYTFGPIGELWYRKSGTYRGKVQNLTQFYHPLDMFGEWNRAGFLQYQFVIPTEAVDEFKKIIGVIQASGHYSFLNVFKLFGPRNQAPLSFPIPGWNICVDFPIKDGLGKFVSELDRRVLEFGGRLYTAKDSRTTAETFHAMYPRVDEWISVRRKVDPLRVFASDMARRLELL Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Extracellular signal-regulated kinase 2 (ERK2) | 4N0S | 4.27 | |
Target general information Gen name MAPK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms PRKM2; PRKM1; P42-MAPK; P42 Mitogen-activated protein kinase; Mitogen-activated protein kinase 2; Mitogen-activated protein kinase 1; MAPK 2; MAPK 1; MAP kinase isoform p42; MAP kinase 2; MAP kinase 1 Protein family Protein kinase superfamily, CMGC Ser/Thr protein kinase family, MAP kinase subfamily Biochemical class Kinase Function MAPK1/ERK2 and MAPK3/ERK1 are the 2 MAPKs which play an important role in the MAPK/ERK cascade. They participate also in a signaling cascade initiated by activated KIT and KITLG/SCF. Depending on the cellular context, the MAPK/ERK cascade mediates diverse biological functions such as cell growth, adhesion, survival and differentiation through the regulation of transcription, translation, cytoskeletal rearrangements. The MAPK/ERK cascade plays also a role in initiation and regulation of meiosis, mitosis, and postmitotic functions in differentiated cells by phosphorylating a number of transcription factors. About 160 substrates have already been discovered for ERKs. Many of these substrates are localized in the nucleus, and seem to participate in the regulation of transcription upon stimulation. However, other substrates are found in the cytosol as well as in other cellular organelles, and those are responsible for processes such as translation, mitosis and apoptosis. Moreover, the MAPK/ERK cascade is also involved in the regulation of the endosomal dynamics, including lysosome processing and endosome cycling through the perinuclear recycling compartment (PNRC); as well as in the fragmentation of the Golgi apparatus during mitosis. The substrates include transcription factors (such as ATF2, BCL6, ELK1, ERF, FOS, HSF4 or SPZ1), cytoskeletal elements (such as CANX, CTTN, GJA1, MAP2, MAPT, PXN, SORBS3 or STMN1), regulators of apoptosis (such as BAD, BTG2, CASP9, DAPK1, IER3, MCL1 or PPARG), regulators of translation (such as EIF4EBP1) and a variety of other signaling-related molecules (like ARHGEF2, DCC, FRS2 or GRB10). Protein kinases (such as RAF1, RPS6KA1/RSK1, RPS6KA3/RSK2, RPS6KA2/RSK3, RPS6KA6/RSK4, SYK, MKNK1/MNK1, MKNK2/MNK2, RPS6KA5/MSK1, RPS6KA4/MSK2, MAPKAPK3 or MAPKAPK5) and phosphatases (such as DUSP1, DUSP4, DUSP6 or DUSP16) are other substrates which enable the propagation the MAPK/ERK signal to additional cytosolic and nuclear targets, thereby extending the specificity of the cascade. Mediates phosphorylation of TPR in respons to EGF stimulation. May play a role in the spindle assembly checkpoint. Phosphorylates PML and promotes its interaction with PIN1, leading to PML degradation. Phosphorylates CDK2AP2. Serine/threonine kinase which acts as an essential component of the MAP kinase signal transduction pathway. Related diseases Noonan syndrome 13 (NS13) [MIM:619087]: A form of Noonan syndrome, a disease characterized by short stature, facial dysmorphic features such as hypertelorism, a downward eyeslant and low-set posteriorly rotated ears, and a high incidence of congenital heart defects and hypertrophic cardiomyopathy. Other features can include a short neck with webbing or redundancy of skin, deafness, motor delay, variable intellectual deficits, multiple skeletal defects, cryptorchidism, and bleeding diathesis. Individuals with Noonan syndrome are at risk of juvenile myelomonocytic leukemia, a myeloproliferative disorder characterized by excessive production of myelomonocytic cells. NS13 inheritance is autosomal dominant. There is considerable variability in severity. {ECO:0000269|PubMed:32721402}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07788; DB07264; DB08521; DB07794; DB08513; DB00945; DB01169; DB02587; DB07905; DB01017; DB06877; DB07010; DB02116; DB06641; DB02482; DB02733; DB04338; DB06195; DB11120; DB13930 Interacts with P05067; P53004; P15882; Q7L5N1; P28562; Q13115; Q16690; Q16828; Q16829; Q99956; Q9BRK4; Q02750; P28482; Q16539-3; Q99750; Q9BUB5; P35548; Q15121; Q9UPG8; P62487; P35813; Q6NYC8; P25786; A2A3K4; P35236; Q12913; Q15256; Q15256-5; O76064; Q15418; Q15349; P51812; P29353-2; P61764; Q08117-2; Q9Y296; B2RXF5; A0A384NQ31; Q9U1H0; Q05922; P02687; Q8VSP9; P0A2M9; Q69559 EC number EC 2.7.11.24 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Apoptosis; ATP-binding; Cell cycle; Cell junction; Cytoplasm; Cytoskeleton; Direct protein sequencing; Disease variant; DNA-binding; Host-virus interaction; Kinase; Membrane; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; Serine/threonine-protein kinase; Transcription; Transcription regulation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 40154.7 Length 352 Aromaticity 0.1 Instability index 40.3 Isoelectric point 6.38 Charge (pH=7) -3.06 2D Binding mode Binding energy (Kcal/mol) -5.82 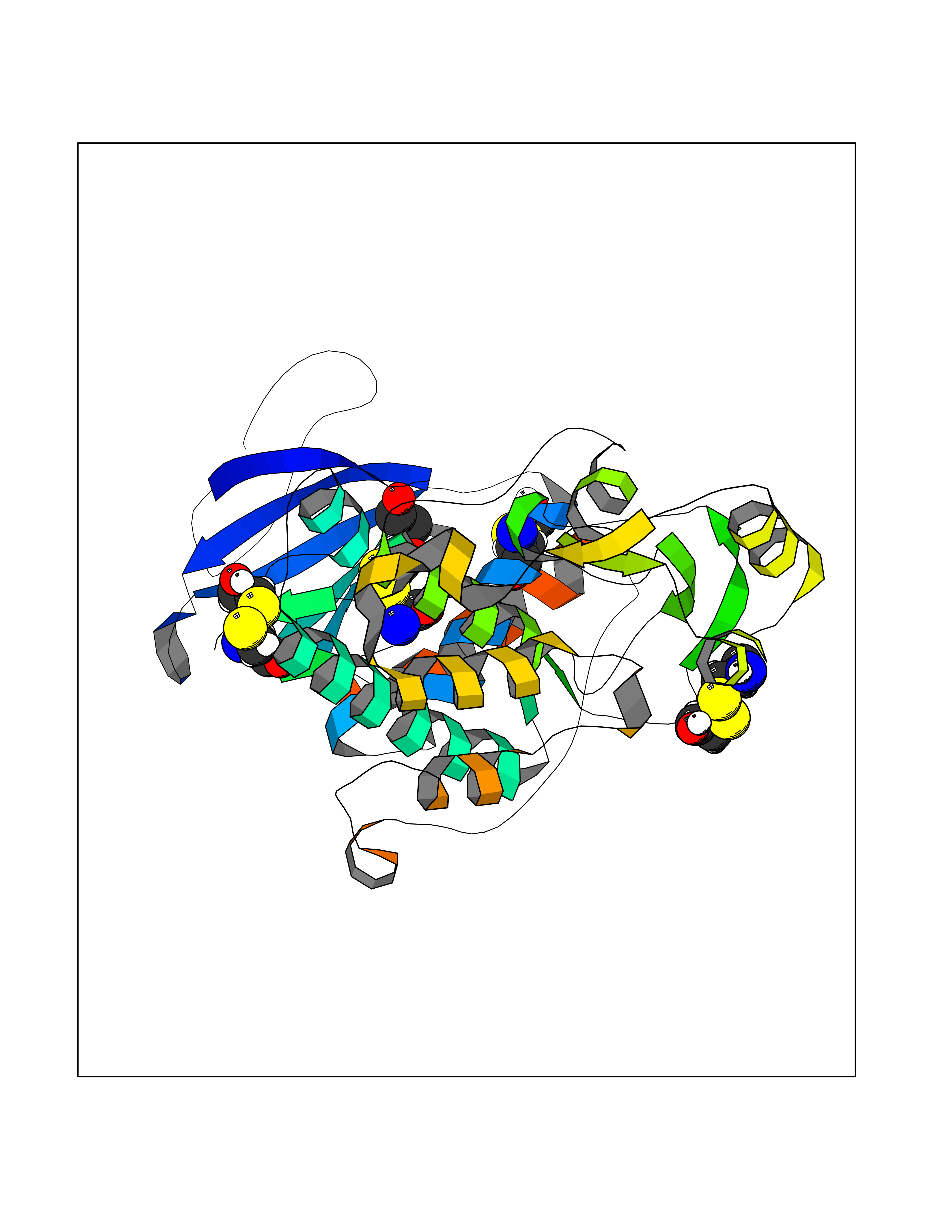 Molscript Map 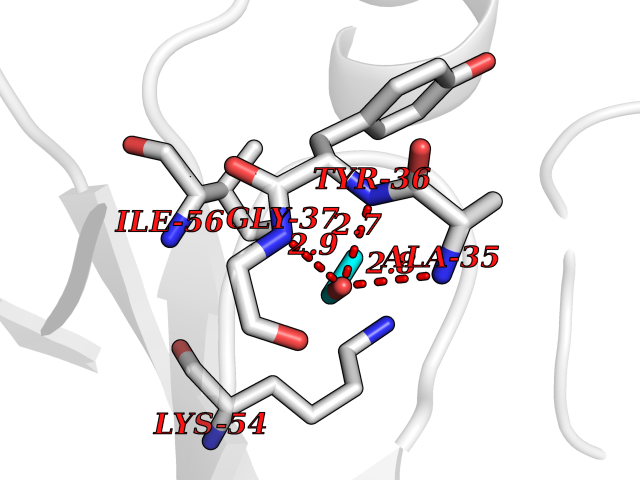 Pymol Map 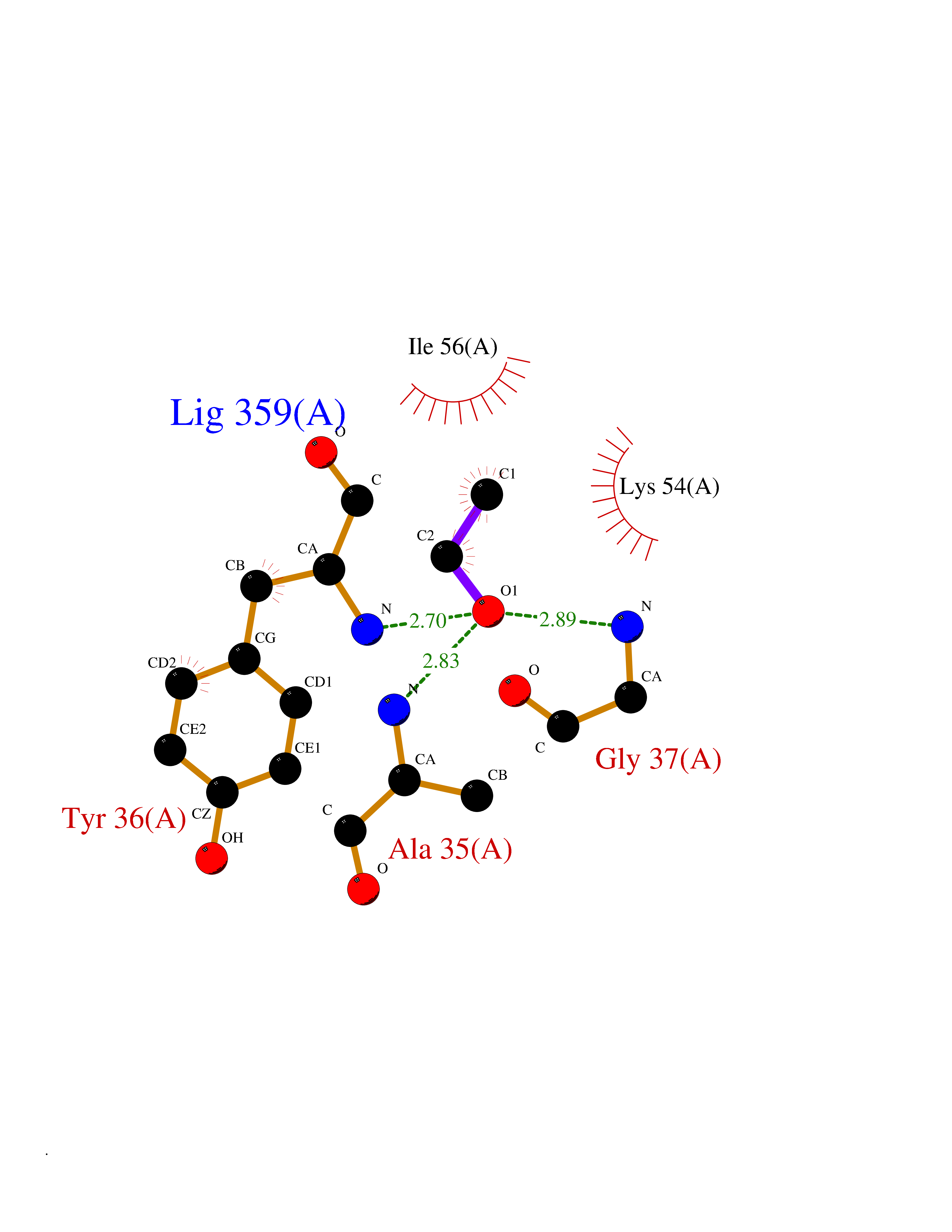 Ligplot Map 3D Binding mode Sequence AAGAGPEMVRGQVFDVGPRYTNLSYIGEGAYGMVCSAYDNVNKVRVAIKKISPFEHQTYXQRTLREIKILLRFRHENIIGINDIIRAPTIEQMKDVYIVQDLMETDLYKLLKTQHLSNDHICYFLYQILRGLKYIHSANVLHRDLKPSNLLLNTTXDLKIXDFGLARVADPDHDHTGFLTEYVATRWYRAPEIMLNSKGYTKSIDIWSVGCILAEMLSNRPIFPGKHYLDQLNHILGILGSPSQEDLNXIINLKARNYLLSLPHKNKVPWNRLFPNADSKALDLLDKMLTFNPHKRIEVEQALAHPYLEQYYDPSDEPIAEAPFKFDMELDDLPKEKLKELIFEETARFQPG Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | IL-1 receptor-associated kinase 1 (IRAK1) | 6BFN | 4.27 | |
Target general information Gen name IRAK1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Interleukin-1 receptor-associated kinase 1; IRAK-1; IRAK Protein family Protein kinase superfamily, TKL Ser/Thr protein kinase family, Pelle subfamily Biochemical class Kinase Function Involved in Toll-like receptor (TLR) and IL-1R signaling pathways. Is rapidly recruited by MYD88 to the receptor-signaling complex upon TLR activation. Association with MYD88 leads to IRAK1 phosphorylation by IRAK4 and subsequent autophosphorylation and kinase activation. Phosphorylates E3 ubiquitin ligases Pellino proteins (PELI1, PELI2 and PELI3) to promote pellino-mediated polyubiquitination of IRAK1. Then, the ubiquitin-binding domain of IKBKG/NEMO binds to polyubiquitinated IRAK1 bringing together the IRAK1-MAP3K7/TAK1-TRAF6 complex and the NEMO-IKKA-IKKB complex. In turn, MAP3K7/TAK1 activates IKKs (CHUK/IKKA and IKBKB/IKKB) leading to NF-kappa-B nuclear translocation and activation. Alternatively, phosphorylates TIRAP to promote its ubiquitination and subsequent degradation. Phosphorylates the interferon regulatory factor 7 (IRF7) to induce its activation and translocation to the nucleus, resulting in transcriptional activation of type I IFN genes, which drive the cell in an antiviral state. When sumoylated, translocates to the nucleus and phosphorylates STAT3. Serine/threonine-protein kinase that plays a critical role in initiating innate immune response against foreign pathogens. Related diseases Anemia, non-spherocytic hemolytic, due to G6PD deficiency (NSHA) [MIM:300908]: A disease characterized by G6PD deficiency, acute hemolytic anemia, fatigue, back pain, and jaundice. In most patients, the disease is triggered by an exogenous agent, such as some drugs, food, or infection. Increased unconjugated bilirubin, lactate dehydrogenase, and reticulocytosis are markers of the disorder. Although G6PD deficiency can be life-threatening, most patients are asymptomatic throughout their life. {ECO:0000269|PubMed:12524354, ECO:0000269|PubMed:1303180, ECO:0000269|PubMed:1303182, ECO:0000269|PubMed:1536798, ECO:0000269|PubMed:1611091, ECO:0000269|PubMed:1889820, ECO:0000269|PubMed:1945893, ECO:0000269|PubMed:20007901, ECO:0000269|PubMed:26479991, ECO:0000269|PubMed:2836867, ECO:0000269|PubMed:2912069, ECO:0000269|PubMed:30988594, ECO:0000269|PubMed:38066190, ECO:0000269|PubMed:7858267, ECO:0000269|PubMed:7959695, ECO:0000269|PubMed:8193373, ECO:0000269|PubMed:8490627, ECO:0000269|PubMed:8533762, ECO:0000269|PubMed:8733135, ECO:0000269|PubMed:9452072}. The disease is caused by variants affecting the gene represented in this entry. Deficiency of G6PD is associated with hemolytic anemia in two different situations. First, in areas in which malaria has been endemic, G6PD-deficiency alleles have reached high frequencies (1% to 50%) and deficient individuals, though essentially asymptomatic in the steady state, have a high risk of acute hemolytic attacks. Secondly, sporadic cases of G6PD deficiency occur at a very low frequencies, and they usually present a more severe phenotype. Several types of NSHA are recognized. Class-I variants are associated with severe NSHA; class-II have an activity <10% of normal; class-III have an activity of 10% to 60% of normal; class-IV have near normal activity. Drugs (DrugBank ID) DB12010 Interacts with Q15306; Q92985; Q99836; Q96FA3; Q9HAT8; Q8N2H9-2; Q13526; Q86WV6; P58753; Q9Y4K3; Q8VCW4; Q5D1E7 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cytoplasm; Direct protein sequencing; Host-virus interaction; Immunity; Innate immunity; Isopeptide bond; Kinase; Lipid droplet; Magnesium; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 33681.4 Length 301 Aromaticity 0.09 Instability index 39.86 Isoelectric point 8.6 Charge (pH=7) 5.09 2D Binding mode Binding energy (Kcal/mol) -5.82 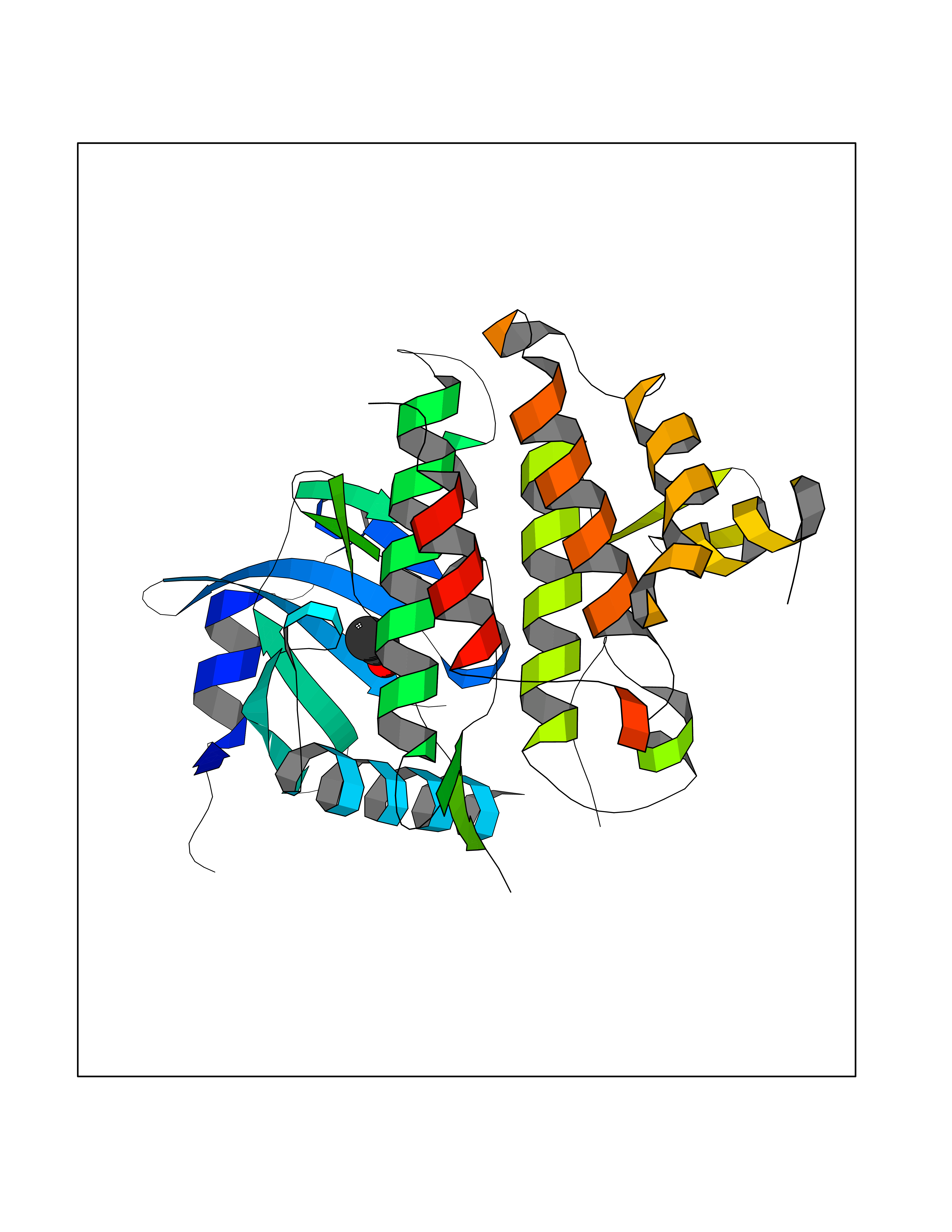 Molscript Map 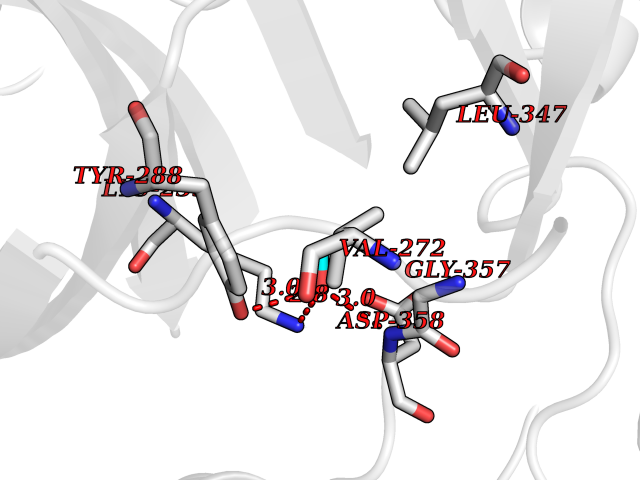 Pymol Map 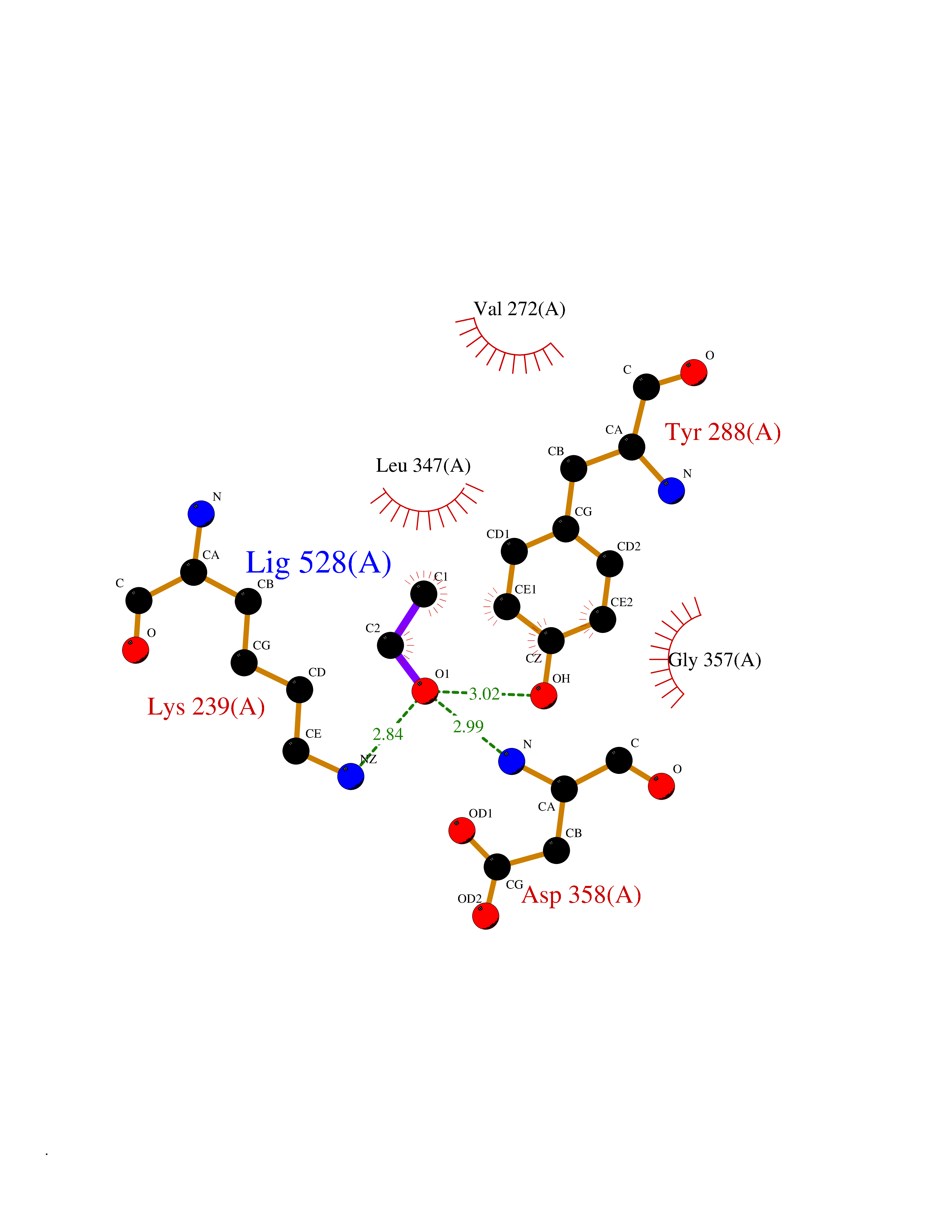 Ligplot Map 3D Binding mode Sequence SRPFPFCWPLCEISRGTHNFSEELKIGEGGFGCVYRAVMRNTVYAVKRLKEWTAVKQSFLTEVEQLSRFRHPNIVDFAGYCAQNGFYCLVYGFLPNGSLEDRLHCQTQACPPLSWPQRLDILLGTARAIQFLHQDSPSLIHGDIKSSNVLLDERLTPKLGDFGLARFSRTVRGTLAYLPEEYIKTGRLAVDTDTFSFGVVVLETLAGQRAVKTHGARTKYLKDLVEEEAEEAGVAAADAWAAPIAMQIYKKHLDPRPGPCPPELGLGLGQLACCCLHRRAKRRPPMTQVYERLEKLQAVVA Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Diamine oxidase (AOC1) | 3HII | 4.27 | |
Target general information Gen name AOC1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Kidney amine oxidase; KAO; Histaminase; Amiloride-binding protein; AOC1; ABP Protein family Copper/topaquinone oxidase family Biochemical class CH-NH(2) donor oxidoreductase Function Catalyzes the degradation of compounds such as putrescine, histamine, spermine, and spermidine, substances involved in allergic and immune responses, cell proliferation, tissue differentiation, tumor formation, and possibly apoptosis. Placental DAO is thought to play a role in the regulation of the female reproductive function. Related diseases Lichtenstein-Knorr syndrome (LIKNS) [MIM:616291]: An autosomal recessive neurologic disorder characterized by progressive cerebellar ataxia and severe progressive sensorineural hearing loss. {ECO:0000269|PubMed:25205112, ECO:0000269|PubMed:30237576}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00594; DB01373; DB09130; DB03608; DB05383 Interacts with Q15038; O75593; Q8IUC2; Q96HA8; Q7Z3K3; Q6ZRY4; Q01085-2; O43711; Q96K80 EC number EC 1.4.3.22 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Copper; Direct protein sequencing; Disulfide bond; Glycoprotein; Heparin-binding; Membrane; Metal-binding; Oxidoreductase; Proteomics identification; Reference proteome; Secreted; Signal; TPQ Protein physicochemical properties Chain ID A,B Molecular weight (Da) 162607 Length 1425 Aromaticity 0.13 Instability index 45.72 Isoelectric point 6.76 Charge (pH=7) -4.07 2D Binding mode Binding energy (Kcal/mol) -5.83  Molscript Map 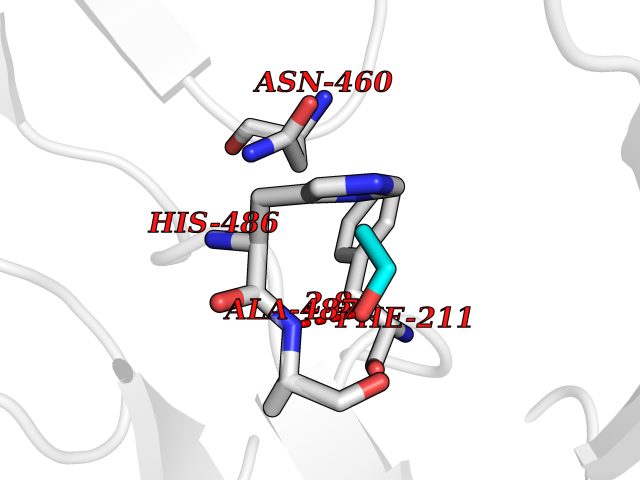 Pymol Map 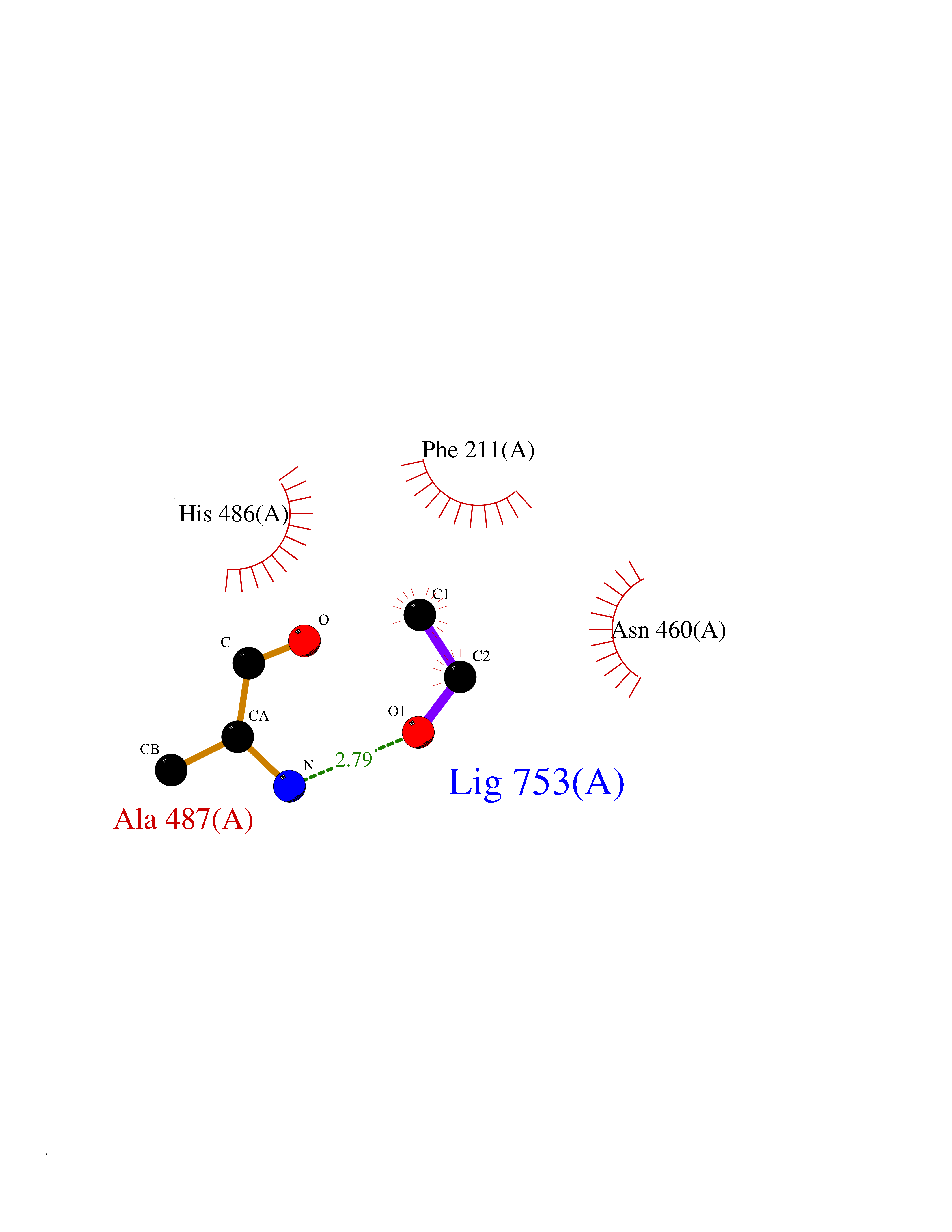 Ligplot Map 3D Binding mode Sequence PRKAGVFSDLSNQELKAVHSFLWSKKELRLQPSSTTTMAKNTVFLIEMLLPKKYHVLRFLDKGERHPVREARAVIFFGDQEHPNVTEFAVGPLPGPCYMRALSPRPGYQSSWASRPISTAEYALLYHTLQEATKPLHQFFLNTTGFSFQDCHDRCLAFTDVAPRGVASGQRRSWLIIQRYVEGYFLHPTGLELLVDHGSTDAGHWAVEQVWYNGKFYGSPEELARKYADGEVDVVVLEDPLEPPLFSSHKPRGDFPSPIHVSGPRLVQPHGPRFRLEGNAVLYGGWSFAFRLRSSSGLQVLNVHFGGERIAYEVSVQEAVALYGGHTPAGMQTKYLDVGWGLGSVTHELAPGIDCPETATFLDTFHYYDADDPVHYPRALCLFEMPTGVPLRRHFNSNFKGGFNFYAGLKGQVLVLRTTSTVYNXDYIWDFIFYPNGVMEAKMHATGYVHATFYTPEGLRHGTRLHTHLIGNIHTHLVHYRVDLDVAGTKNSFQTLQMKLENITNPWSPRHRVVQPTLEQTQYSWERQAAFRFKRKLPKYLLFTSPQENPWGHKRSYRLQIHSMADQVLPPGWQEEQAITWARYPLAVTKYRESELCSSSIYHQNDPWDPPVVFEQFLHNNENIENEDLVAWVTVGFLHIPHSEDIPNTATPGNSVGFLLRPFNFFPEDPSLASRDTVIVWPRDNGPNYVQRWIPEDRDCSMPPPFSYNGTYRPVRKAGVFSDLSNQELKAVHSFLWSKKELRLQPSSTTTMAKNTVFLIEMLLPKKYHVLRFLDKGERHPVREARAVIFFGDQEHPNVTEFAVGPLPGPCYMRALSPRPGYQSSWASRPISTAEYALLYHTLQEATKPLHQFFLNTTGFSFQDCHDRCLAFTDVAPRGVASGQRRSWLIIQRYVEGYFLHPTGLELLVDHGSTDAGHWAVEQVWYNGKFYGSPEELARKYADGEVDVVVLEPPLFSSHKPRGDFPSPIHVSGPRLVQPHGPRFRLEGNAVLYGGWSFAFRLRSSSGLQVLNVHFGGERIAYEVSVQEAVALYGGHTPAGMQTKYLDVGWGLGSVTHELAPGIDCPETATFLDTFHYYDADDPVHYPRALCLFEMPTGVPLRRHFNSNFKGGFNFYAGLKGQVLVLRTTSTVYNXDYIWDFIFYPNGVMEAKMHATGYVHATFYTPEGLRHGTRLHTHLIGNIHTHLVHYRVDLDVAGTKNSFQTLQMKLENITNPWSPRHRVVQPTLEQTQYSWERQAAFRFKRKLPKYLLFTSPQENPWGHKRSYRLQIHSMADQVLPPGWQEEQAITWARYPLAVTKYRESELCSSSIYHQNDPWDPPVVFEQFLHNNENIENEDLVAWVTVGFLHIPHSEDIPNTATPGNSVGFLLRPFNFFPEDPSLASRDTVIVWPRDNGPNYVQRWIPEDRDCSMPPPFSYNGTYRPV Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Melanoma derived growth regulator (MIA) | 5IXB | 4.27 | |
Target general information Gen name MIA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Melanoma-derived growth regulatory protein; Melanoma inhibitory activity protein Protein family MIA/OTOR family Biochemical class NA Function Elicits growth inhibition on melanoma cells in vitro as well as some other neuroectodermal tumors, including gliomas. Related diseases Hypertriglyceridemia, transient infantile (HTGTI) [MIM:614480]: An autosomal recessive disorder characterized by onset of moderate to severe transient hypertriglyceridemia in infancy that normalizes with age. The hypertriglyceridemia is associated with hepatomegaly, moderately elevated transaminases, persistent fatty liver, and the development of hepatic fibrosis. {ECO:0000269|PubMed:22226083, ECO:0000269|PubMed:24549054}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Direct protein sequencing; Disulfide bond; Growth factor; Proteomics identification; Reference proteome; Secreted; SH3 domain; Signal Protein physicochemical properties Chain ID A Molecular weight (Da) 11955.6 Length 105 Aromaticity 0.12 Instability index 26.45 Isoelectric point 8.7 Charge (pH=7) 2.64 2D Binding mode Binding energy (Kcal/mol) -5.82 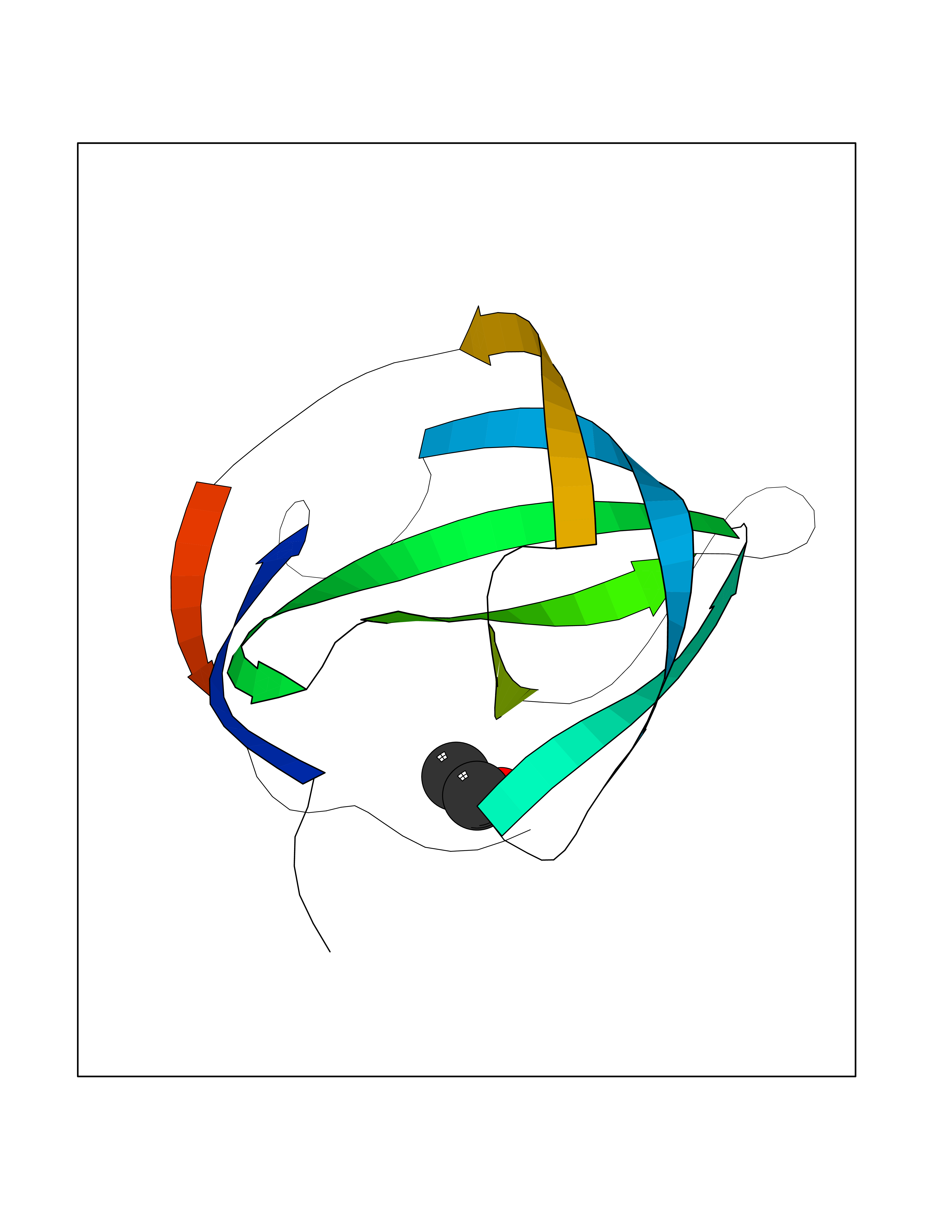 Molscript Map 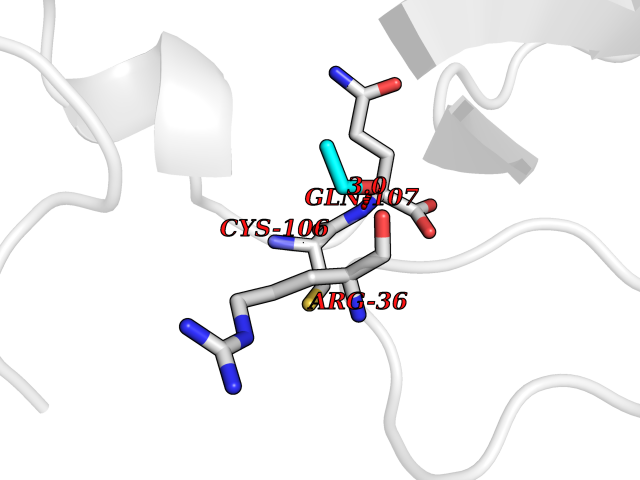 Pymol Map 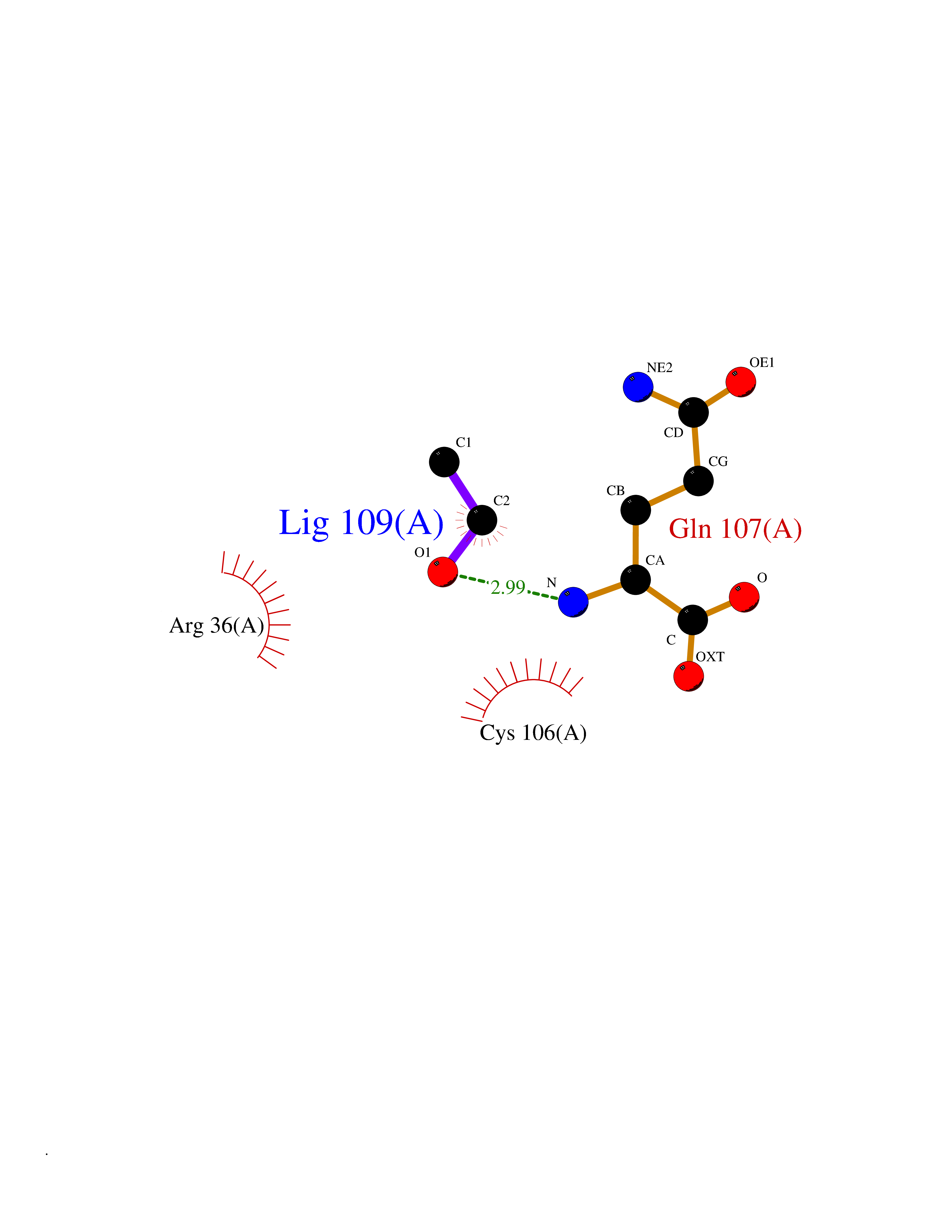 Ligplot Map 3D Binding mode Sequence MPKLADRKLCADQECSHPISMAVALQDYMAPDCRFLTIHRGQVVYVFSKLKGRGRLFWGGSVQGDYYGDLAARLGYFPSSIVREDQTLKPGKVDVKTDKWDFYCQ Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | N-acylethanolamine-hydrolyzing acidamidase (NAAA) | 6DXX | 4.27 | |
Target general information Gen name NAAA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nacylsphingosine amidohydrolaselike; Nacylethanolaminehydrolyzing acid amidase subunit beta; NAAA; Acid ceramidaselike protein; ASAHlike protein Protein family Acid ceramidase family Biochemical class Carbon-nitrogen hydrolase Function Degrades bioactive fatty acid amides to their corresponding acids, with the following preference: N- palmitoylethanolamine > N-myristoylethanolamine > N- lauroylethanolamine = N-stearoylethanolamine > N- arachidonoylethanolamine > N-oleoylethanolamine. Also exhibits weak hydrolytic activity against the ceramides N- lauroylsphingosine and N-palmitoylsphingosine. Related diseases Hypertriglyceridemia, transient infantile (HTGTI) [MIM:614480]: An autosomal recessive disorder characterized by onset of moderate to severe transient hypertriglyceridemia in infancy that normalizes with age. The hypertriglyceridemia is associated with hepatomegaly, moderately elevated transaminases, persistent fatty liver, and the development of hepatic fibrosis. {ECO:0000269|PubMed:22226083, ECO:0000269|PubMed:24549054}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB09061; DB14009; DB14011 Interacts with NA EC number EC 3.5.1.- Uniprot keywords 3D-structure; Alternative splicing; Autocatalytic cleavage; Direct protein sequencing; Disulfide bond; Fatty acid metabolism; Glycoprotein; Hydrolase; Lipid degradation; Lipid metabolism; Lysosome; Membrane; Proteomics identification; Reference proteome; Signal; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 36877.8 Length 328 Aromaticity 0.11 Instability index 44.37 Isoelectric point 7.72 Charge (pH=7) 1.08 2D Binding mode Binding energy (Kcal/mol) -5.83 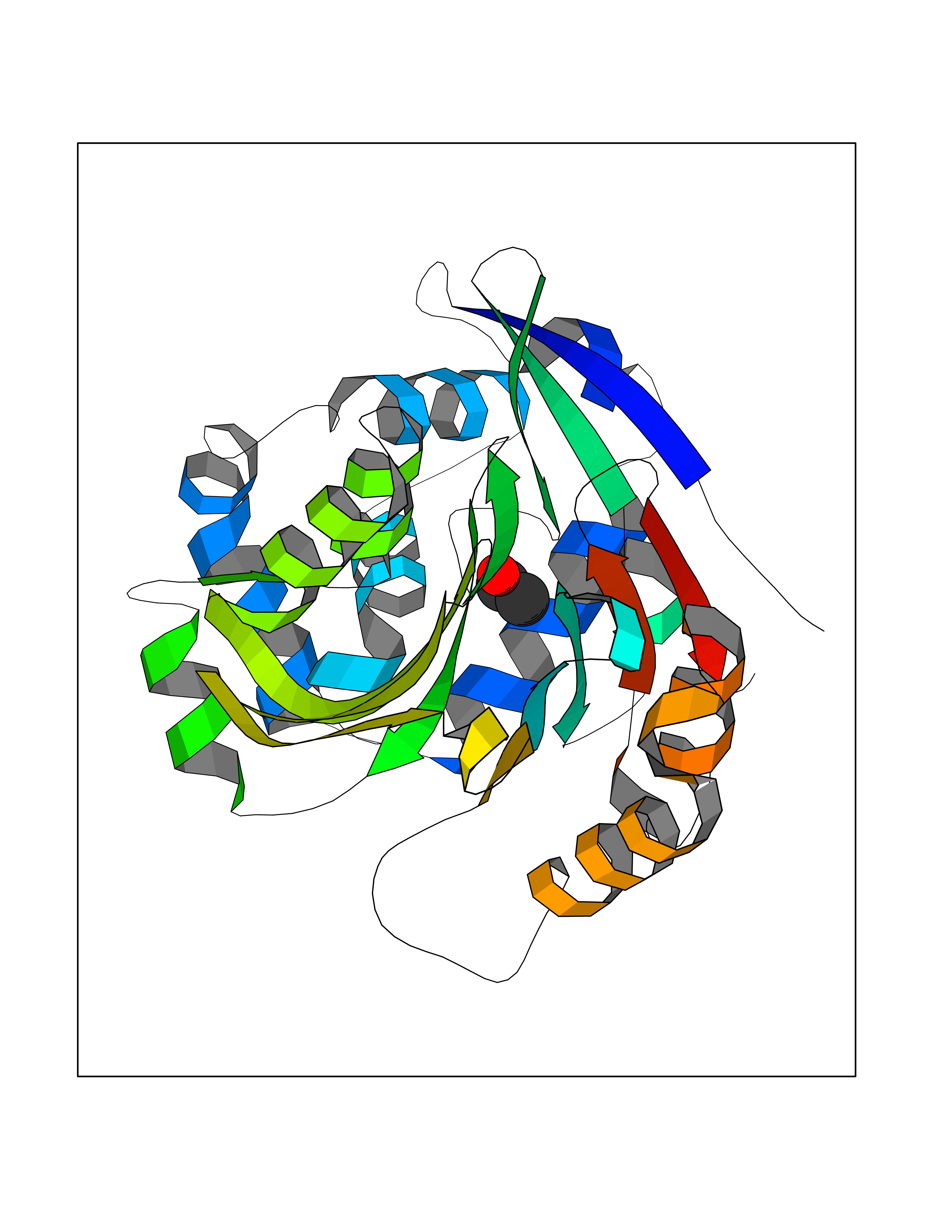 Molscript Map 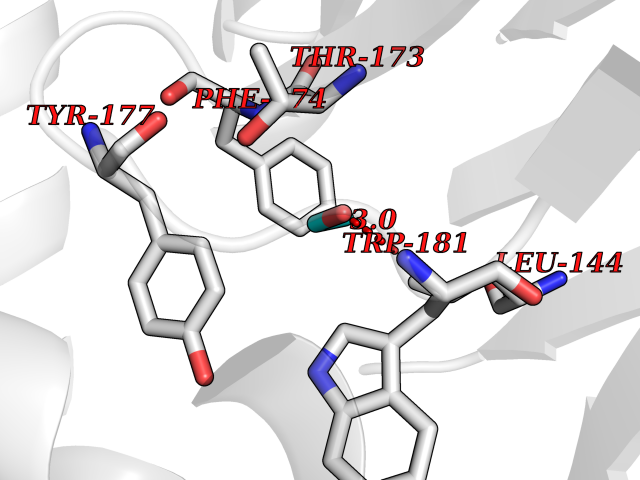 Pymol Map 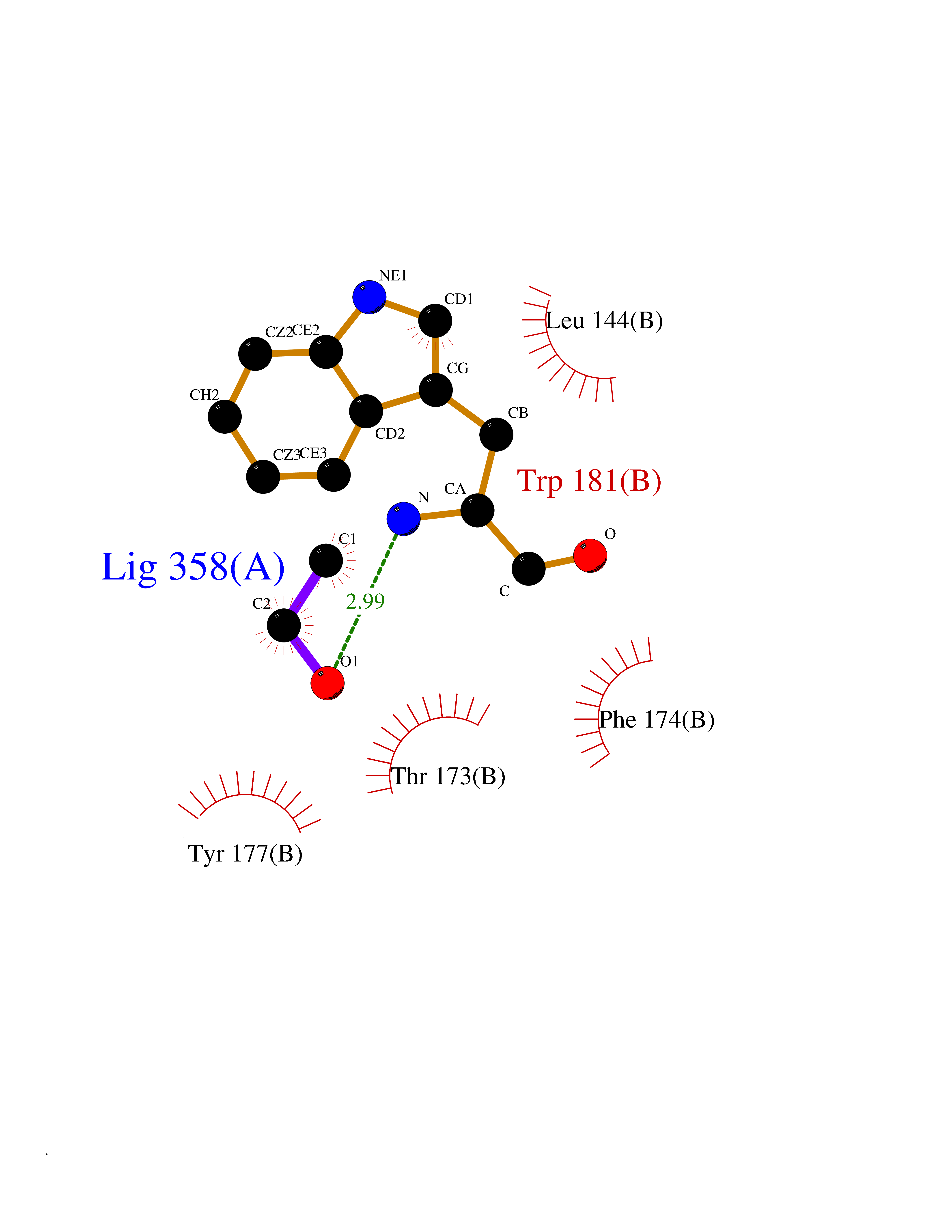 Ligplot Map 3D Binding mode Sequence SPPAAPRFNVSLDSVPELRWLPVLRHYDLDLVRAAMAQVIGDRVPKWVHVLIGKVVLELERFLPQPFTGEIRGMCDFMNLSLADCLLVNLAYESSVFCTSIVAQDSRGHIYHGRNLDYPFGNVLRKLTVDVQFLKNGQIAFTGTTFIGYVGLWTGQSPHKFTVSGDERDKGWWWENAIAALFRRHIPVSWLIRATLSESENFEAAVGKLAKTPLIADVYYIVGGTSPREGVVITRNRDGPADIWPLDPLNGAWFRVETNYDHWKPAPKEDDRRTSAIKALNATGQANLSLEALFQILSVVPVYNNFTIYTTVMSAGSPDKYMTRIRNP Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Cathepsin G (CTSG) | 1KYN | 4.27 | |
Target general information Gen name CTSG Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CG Protein family Peptidase S1 family Biochemical class Peptidase Function Cleaves complement C3. Has antibacterial activity against the Gram-negative bacterium P. aeruginosa, antibacterial activity is inhibited by LPS from P. aeruginosa, Z-Gly-Leu-Phe-CH2Cl and phenylmethylsulfonyl fluoride. Serine protease with trypsin- and chymotrypsin-like specificity. Related diseases Lethal congenital contracture syndrome 2 (LCCS2) [MIM:607598]: A form of lethal congenital contracture syndrome, an autosomal recessive disorder characterized by degeneration of anterior horn neurons, extreme skeletal muscle atrophy, and congenital non-progressive joint contractures (arthrogryposis). The contractures can involve the upper or lower limbs and/or the vertebral column, leading to various degrees of flexion or extension limitations evident at birth. LCCS2 patients manifest craniofacial/ocular findings, lack of hydrops, multiple pterygia, and fractures, as well as a normal duration of pregnancy and a unique feature of a markedly distended urinary bladder (neurogenic bladder defect). The phenotype suggests a spinal cord neuropathic etiology. {ECO:0000269|PubMed:17701904}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Erythroleukemia, familial (FERLK) [MIM:133180]: An autosomal dominant myeloproliferative disorder characterized by neoplastic proliferation of erythroblastic and myeloblastic elements with atypical erythroblasts and myeloblasts in the peripheral blood. Disease penetrance is incomplete. {ECO:0000269|PubMed:27416908}. Disease susceptibility may be associated with variants affecting the gene represented in this entry.; DISEASE: Visceral neuropathy, familial, 1, autosomal recessive (VSCN1) [MIM:243180]: An autosomal recessive disorder characterized by intestinal dysmotility due to aganglionosis (Hirschsprung disease), hypoganglionosis, and/or chronic intestinal pseudoobstruction. Additional variable features are progressive peripheral neuropathy, arthrogryposis, hypoplasia or aplasia of the olfactory bulb and of the external auditory canals, microtia or anotia, and facial dysmorphism. Some patients present structural cardiac anomalies and arthrogryposis with multiple pterygia. {ECO:0000269|PubMed:33497358}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04016; DB02360 Interacts with A8MQ03; Q15323; Q7Z3S9; Q9NRD5 EC number EC 3.4.21.20 Uniprot keywords 3D-structure; Antibiotic; Antimicrobial; Cell membrane; Chemotaxis; Cytoplasm; Direct protein sequencing; Disulfide bond; Glycoprotein; Hydrolase; Lysosome; Membrane; Nucleus; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 25353.8 Length 223 Aromaticity 0.06 Instability index 66.1 Isoelectric point 11.51 Charge (pH=7) 23.24 2D Binding mode Binding energy (Kcal/mol) -5.83 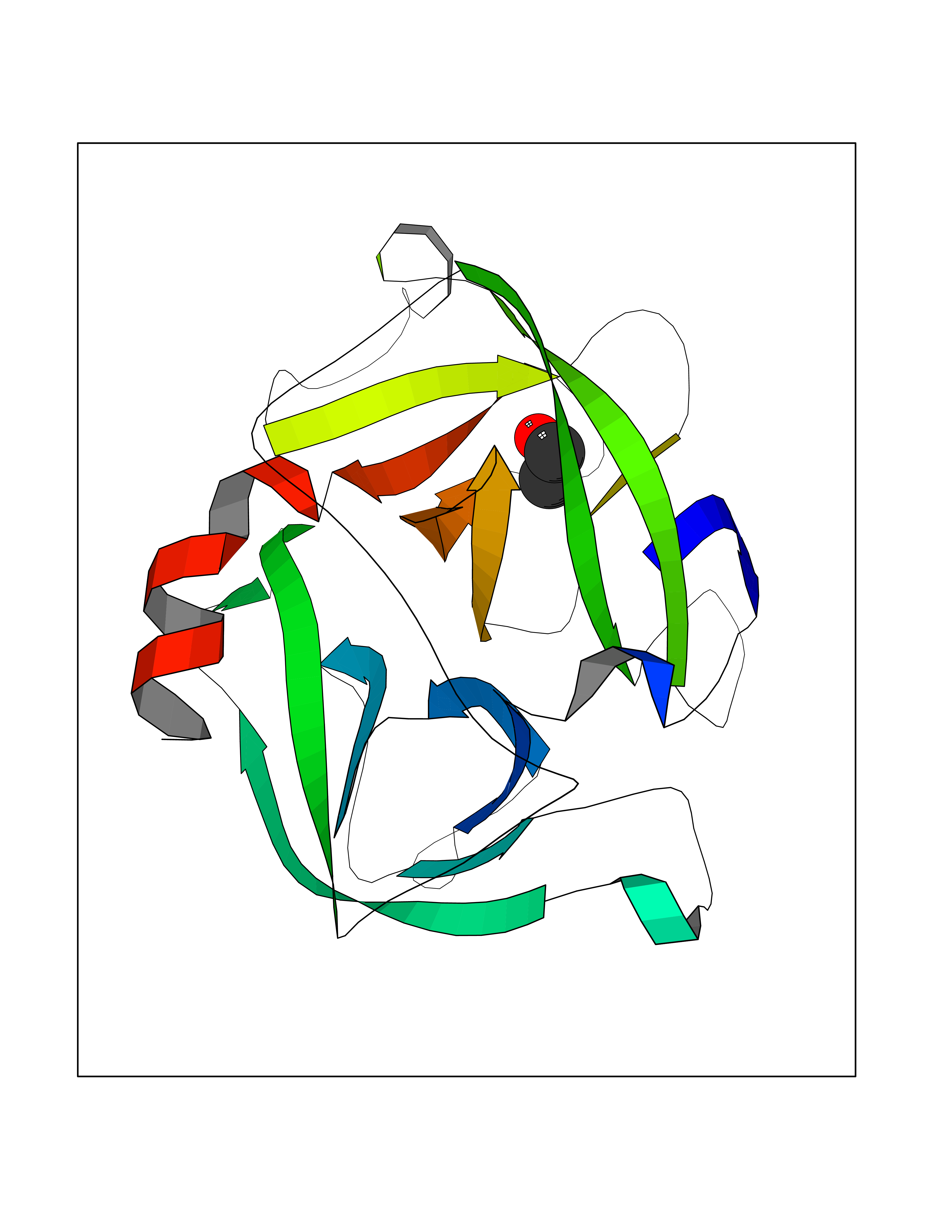 Molscript Map  Pymol Map 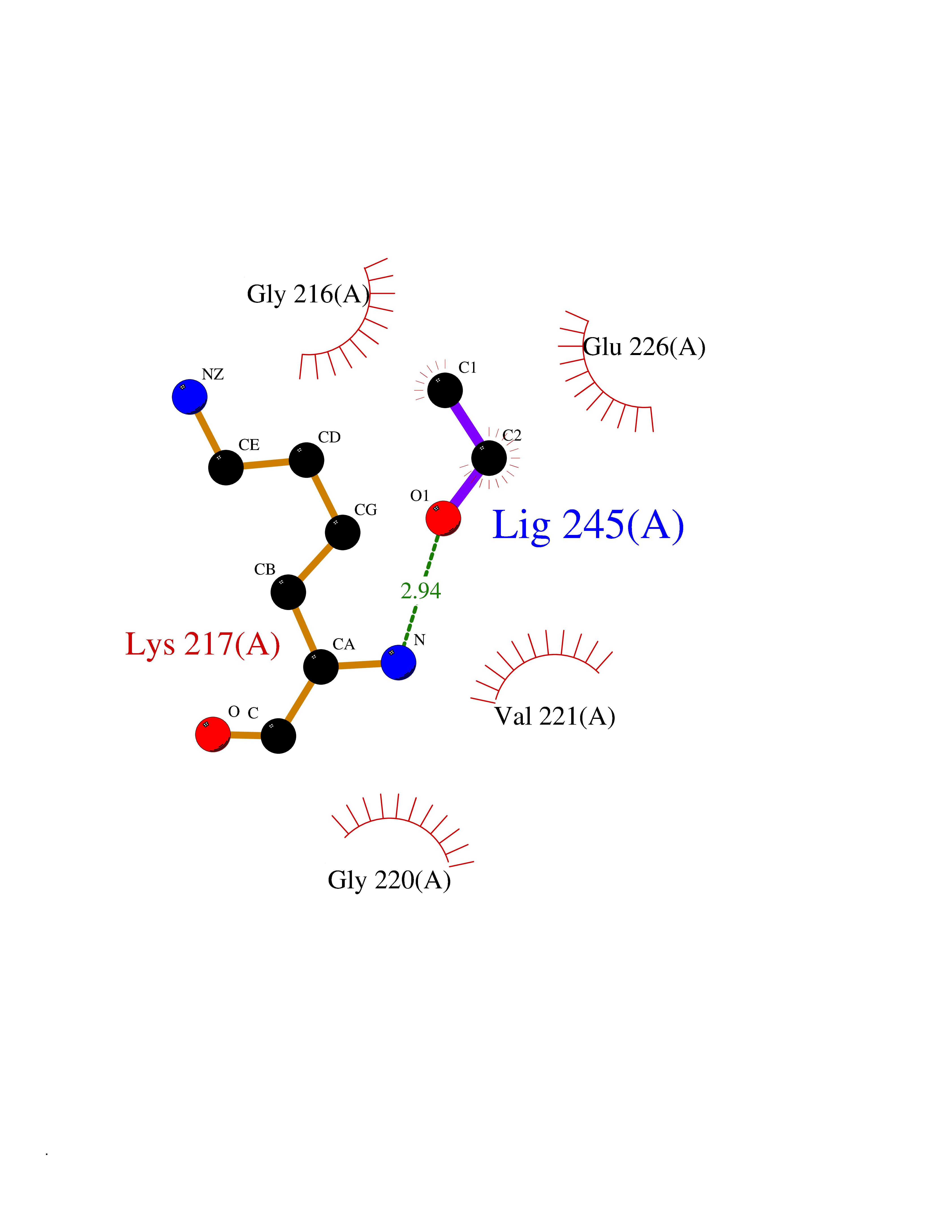 Ligplot Map 3D Binding mode Sequence IIGGRESRPHSRPYMAYLQIQSPAGQSRCGGFLVREDFVLTAAHCWGSNINVTLGAHNIQRRENTQQHITARRAIRHPQYNQRTIQNDIMLLQLSRRVRRNRNVNPVALPRAQEGLRPGTLCTVAGWGRVSMRRGTDTLREVQLRVQRDRQCLRIFGSYDPRRQICVGDRRERKAAFKGDSGGPLLCNNVAHGIVSYGKSSGVPPEVFTRVSSFLPWIRTTMR Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | Ribosomal protein S6 kinase alpha-6 (RSK6) | 6G77 | 4.27 | |
Target general information Gen name RPS6KA6 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms pp90RSK4; p90RSK6; p90-RSK 6; S6K-alpha-6; Ribosomal S6 kinase 4; RSK4; RSK-4; 90 kDa ribosomal protein S6 kinase 6 Protein family Protein kinase superfamily, AGC Ser/Thr protein kinase family, S6 kinase subfamily Biochemical class NA Function Constitutively active serine/threonine-protein kinase that exhibits growth-factor-independent kinase activity and that may participate in p53/TP53-dependent cell growth arrest signaling and play an inhibitory role during embryogenesis. Related diseases Symptomatic deficiency in lactate transport (SDLT) [MIM:245340]: Deficiency of lactate transporter may result in an acidic intracellular environment created by muscle activity with consequent degeneration of muscle and release of myoglobin and creatine kinase. This defect might compromise extreme performance in otherwise healthy individuals. {ECO:0000269|PubMed:10590411}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hyperinsulinemic hypoglycemia, familial, 7 (HHF7) [MIM:610021]: A form of hyperinsulinemic hypoglycemia, a clinically and genetically heterogeneous disorder characterized by inappropriate insulin secretion from the pancreatic beta-cells in the presence of low blood glucose levels. HHF7 features include exercise-induced hyperinsulinism, loss of consciousness due to hypoglycemia, and hypoglycemic seizures. HHF7 inheritance is autosomal dominant. {ECO:0000269|PubMed:17701893}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Monocarboxylate transporter 1 deficiency (MCT1D) [MIM:616095]: A metabolic disorder characterized by recurrent ketoacidosis, a pathologic state due to ketone formation exceeding ketone utilization. The clinical consequences of ketoacidosis are vomiting, osmotic diuresis, dehydration, and Kussmaul breathing. The condition may progress to decreased consciousness and, ultimately, death. {ECO:0000269|PubMed:25390740}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12010 Interacts with Q7Z698 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cytoplasm; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 33406.4 Length 291 Aromaticity 0.11 Instability index 38.08 Isoelectric point 8.4 Charge (pH=7) 2.55 2D Binding mode Binding energy (Kcal/mol) -5.82  Molscript Map  Pymol Map 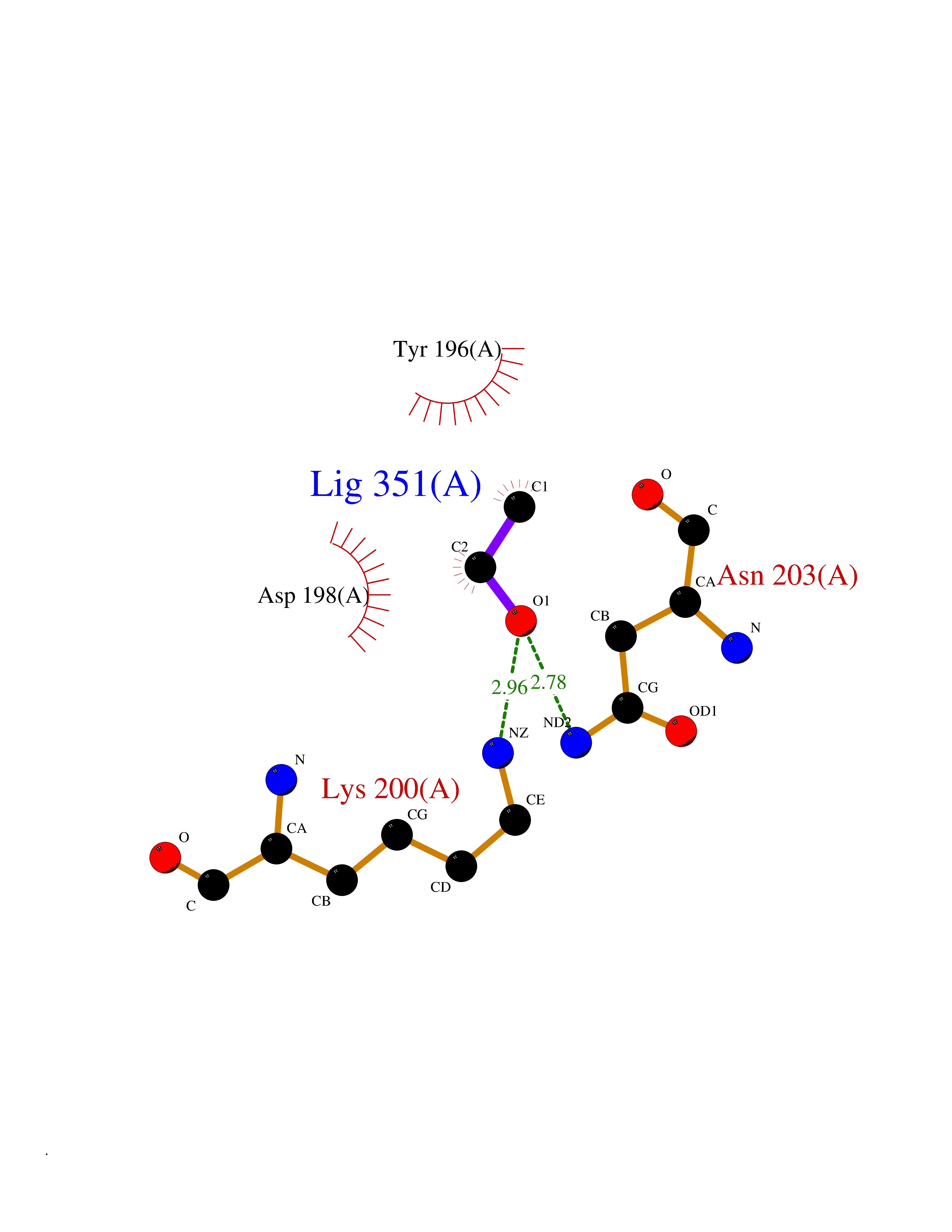 Ligplot Map 3D Binding mode Sequence VVKEIPITHHVKEGYEKADPAQFELLKVLGQGSFGKVFLVRKKTGPDAGQLYAMKVLKKASLKVRDDILVEVNHPFIVKLHYAFQTEGKLYLILDFLRGGDVFTRLSKEVLFTEEDVKFYLAELALALDHLHQLGIVYRDLKPENILLDEIGHIKLTDFGLSKESVDQEKKAYSFCGTVEYMAPEVVNRRGHSQSADWWSYGVLMFEMLTGTLPFQGKDRNETMNMILKAKLGMPQFLSAEAQSLLRMLFKRNPANRLGSEGVEEIKRHLFFANIDWDKLYKREVQPPFKP Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Argininosuccinate lyase | 1K62 | 4.26 | |
Target general information Gen name ASL Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Lyase 1 family, Argininosuccinate lyase subfamily Biochemical class Lyase Function Argininosuccinate lyase activity.Identical protein binding. Related diseases Argininosuccinic aciduria (ARGINSA) [MIM:207900]: An autosomal recessive disorder of the urea cycle. The disease is characterized by mental and physical retardation, liver enlargement, skin lesions, dry and brittle hair showing trichorrhexis nodosa microscopically and fluorescing red, convulsions, and episodic unconsciousness. {ECO:0000269|PubMed:11747432, ECO:0000269|PubMed:11747433, ECO:0000269|PubMed:12408190, ECO:0000269|PubMed:1705937, ECO:0000269|PubMed:17326097, ECO:0000269|PubMed:19703900, ECO:0000269|PubMed:22081021, ECO:0000269|PubMed:2263616, ECO:0000269|PubMed:24166829, ECO:0000269|PubMed:9045711}. The disease is caused by variants affecting the gene represented in this entry. The phenotype heterogeneity among patients is associated with interallelic complementation resulting in either complete loss of activity or partial regeneration of functional active sites in the heterotetrameric mutant protein. {ECO:0000269|PubMed:11747433}. Drugs (DrugBank ID) DB03814; DB00125; DB02267 Interacts with P04424; Q9BTE3-2; Q96HA8; O75382 EC number 4.3.2.1 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Amino-acid biosynthesis; Arginine biosynthesis; Disease variant; Lyase; Proteomics identification; Reference proteome; Urea cycle Protein physicochemical properties Chain ID A,B Molecular weight (Da) 51364.1 Length 459 Aromaticity 0.08 Instability index 35.82 Isoelectric point 6.66 Charge (pH=7) -1.25 2D Binding mode Binding energy (Kcal/mol) -5.81 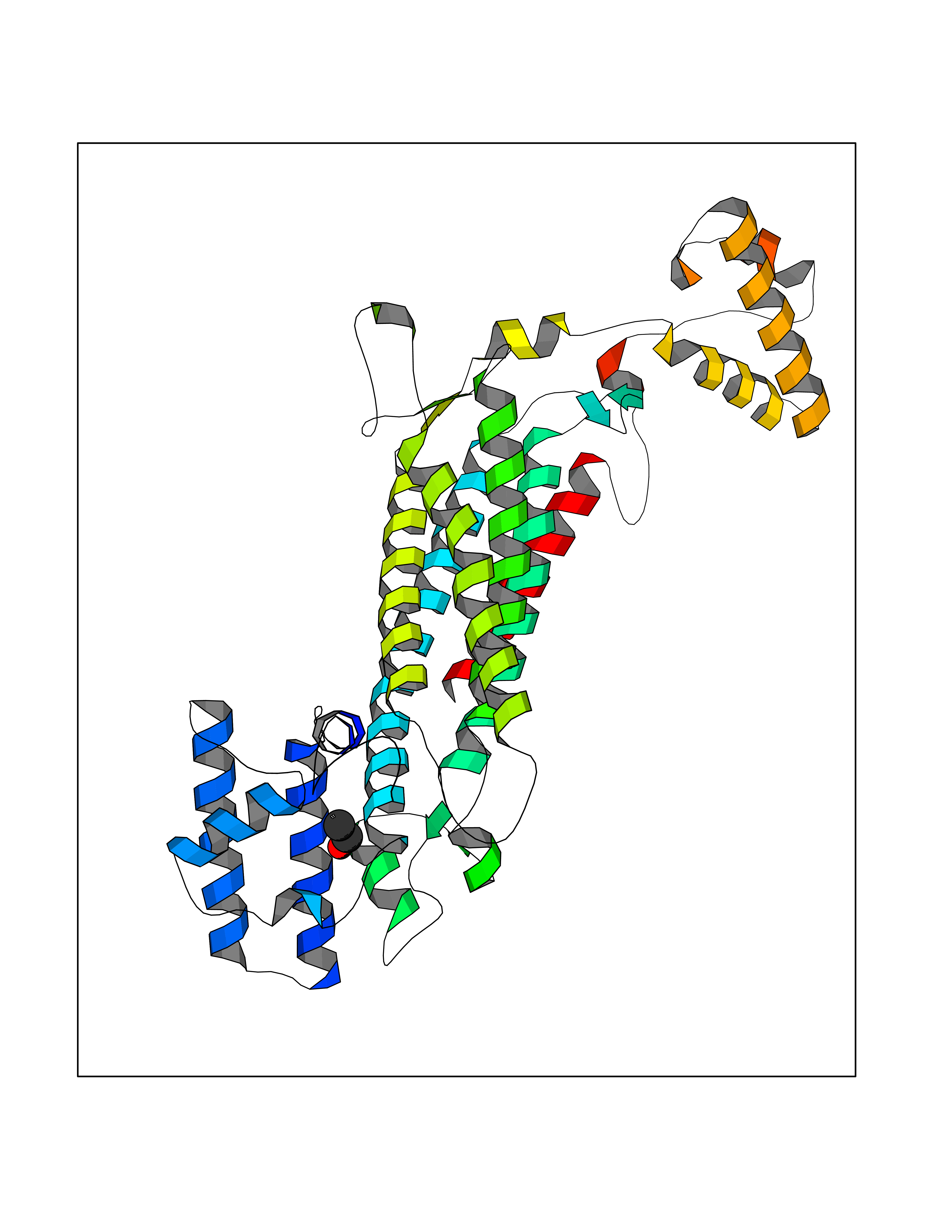 Molscript Map 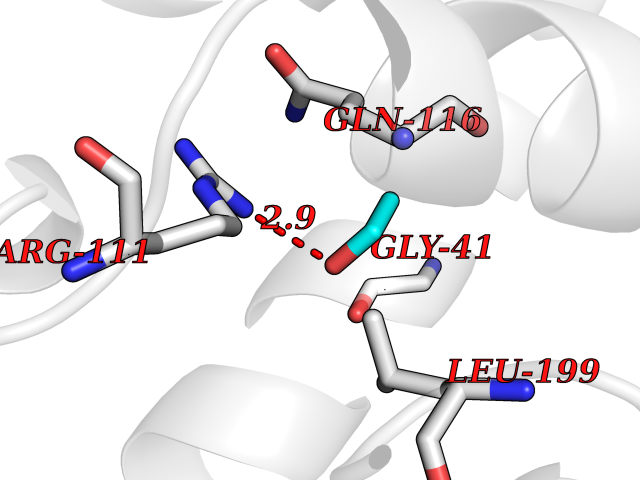 Pymol Map 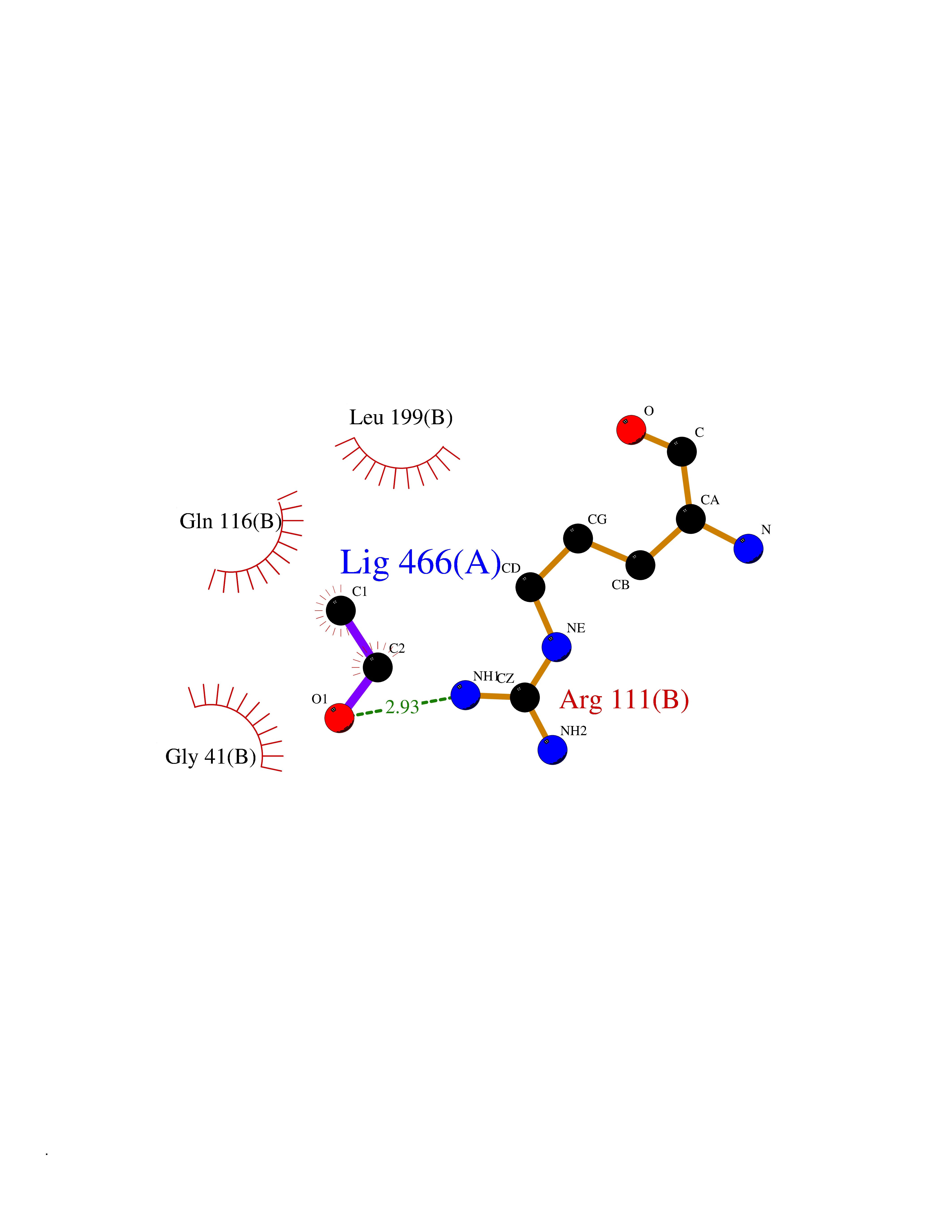 Ligplot Map 3D Binding mode Sequence GKLWGGRFVGAVDPIMEKFNASIAYDRHLWEVDVQGSKAYSRGLEKAGLLTKAEMDQILHGLDKVAEEWAQGTFKLNSNDEDIHTANERRLKELIGATAGKLHTGRSRNDQVVTDLRLWMRQTCSTLSGLLWELIRTMVDRAEAERDVLFPGYTHLQRAQPIRWSHWILSHAVALTRDSERLLEVRKRINVLPLGSGAIAGNPLGVDRELLRAELNFGAITLNSMDATSERDFVAEFLFWRSLCMTHLSRMAEDLILYCTKEFSFVQLSDAYSTGSSLMPRKKNPDSLELIRSKAGRVFGRCAGLLMTLKGLPSTYNKDLQEDKEAVFEVSDTMSAVLQVATGVISTLQIHQENMGQALSPDMLATDLAYYLVRKGMPFRQAHEASGKAVFMAETKGVALNQLSLQELQTISPLFSGDVICVWDYRHSVEQYGALGGTARSSVDWQIRQVRALLQAQQA Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Bifunctional methylenetetrahydrofolate dehydrogenase/cyclohydrolase, mitochondrial | 5TC4 | 4.26 | |
Target general information Gen name MTHFD2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NMDMC Protein family Tetrahydrofolate dehydrogenase/cyclohydrolase family Biochemical class Oxidoreductase Function Magnesium ion binding.Methenyltetrahydrofolate cyclohydrolase activity.Methylenetetrahydrofolate dehydrogenase (NAD+) activity.Methylenetetrahydrofolate dehydrogenase (NADP+) activity.Phosphate ion binding. Related diseases Acute hepatic porphyria (AHEPP) [MIM:612740]: A form of porphyria. Porphyrias are inherited defects in the biosynthesis of heme, resulting in the accumulation and increased excretion of porphyrins or porphyrin precursors. They are classified as erythropoietic or hepatic, depending on whether the enzyme deficiency occurs in red blood cells or in the liver. AHP is characterized by attacks of gastrointestinal disturbances, abdominal colic, paralyses and peripheral neuropathy. Most attacks are precipitated by drugs, alcohol, caloric deprivation, infections, or endocrine factors. {ECO:0000269|PubMed:10706561, ECO:0000269|PubMed:1309003, ECO:0000269|PubMed:1569184, ECO:0000269|PubMed:17236137, ECO:0000269|PubMed:2063868}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00157; DB00116 Interacts with Q9UJ70-2 EC number 1.5.1.15; 3.5.4.9 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Hydrolase; Isopeptide bond; Magnesium; Mitochondrion; Multifunctional enzyme; NAD; NADP; One-carbon metabolism; Oxidoreductase; Proteomics identification; Reference proteome; Transit peptide; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 31600.3 Length 292 Aromaticity 0.03 Instability index 27.9 Isoelectric point 8 Charge (pH=7) 1.64 2D Binding mode Binding energy (Kcal/mol) -5.81 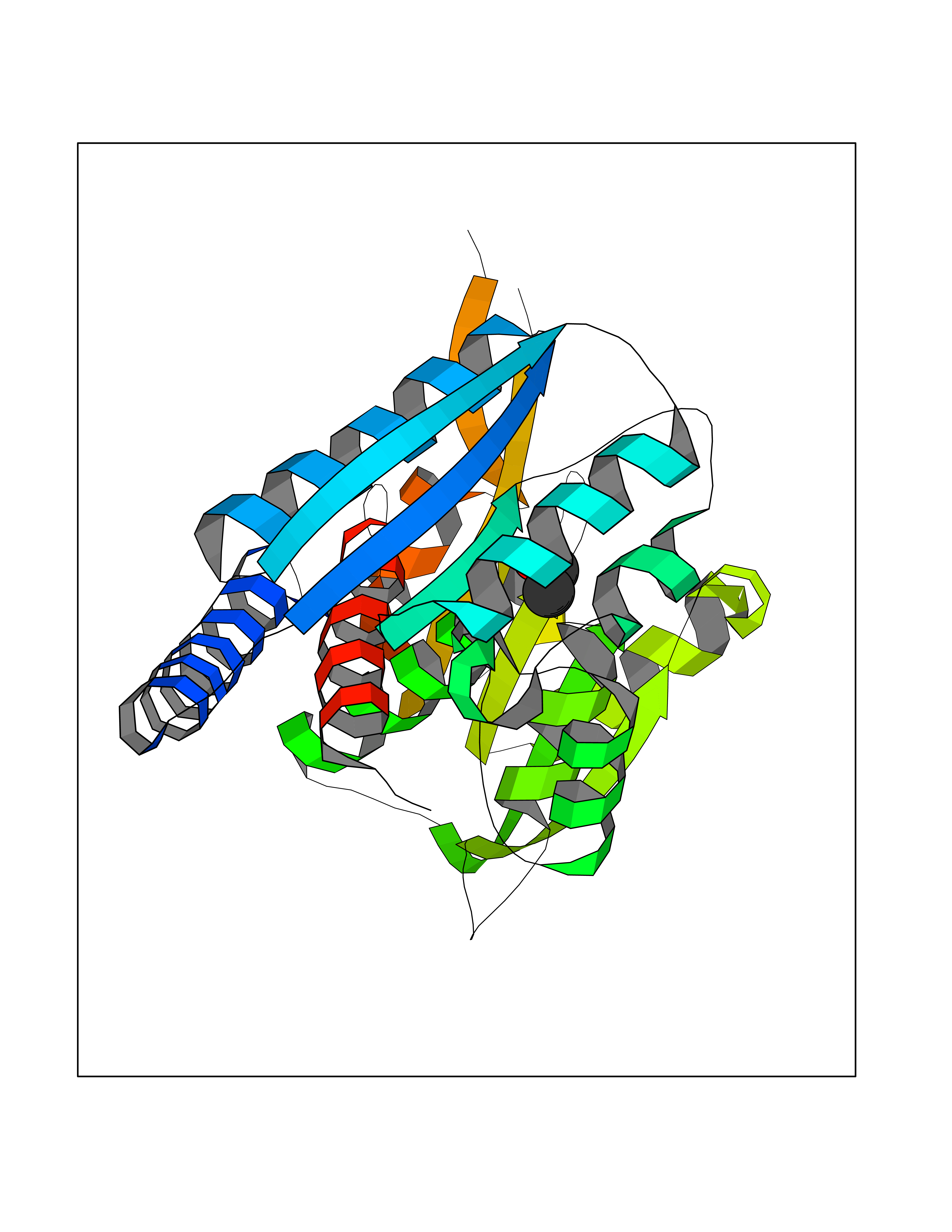 Molscript Map 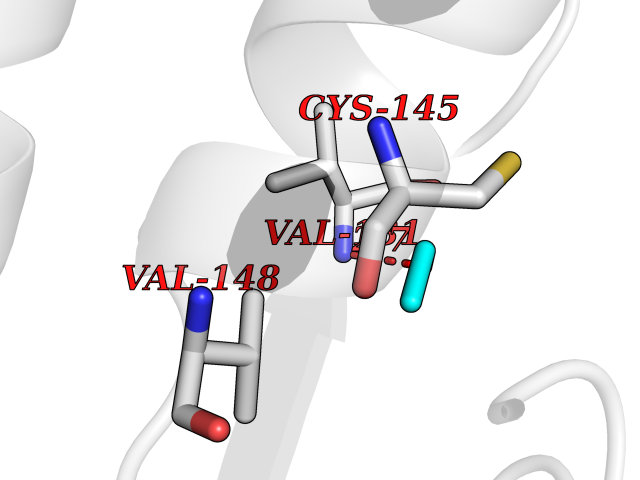 Pymol Map 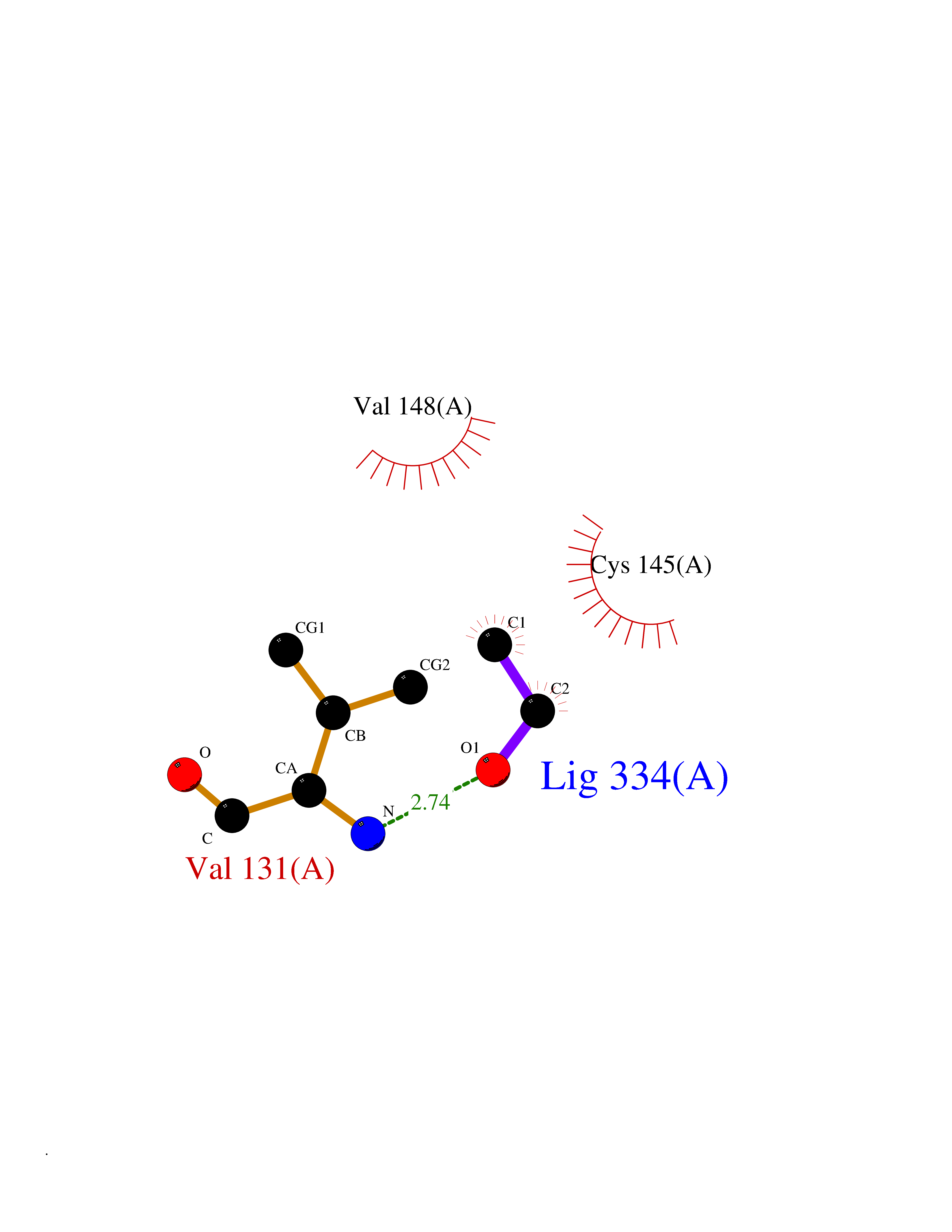 Ligplot Map 3D Binding mode Sequence EAVVISGRKLAQQIKQEVRQEVEEWVASGNKRPHLSVILVGENPASHSYVLNKTRAAAVVGINSETIMKPASISEEELLNLINKLNNDDNVDGLLVQLPLPEHIDERRICNAVSPDKDVDGFHVINVGRMCLDQYSMLPATPWGVWEIIKRTGIPTLGKNVVVAGRSKNVGMPIAMLLHTDGAHERPGGDATVTISHRYTPKEQLKKHTILADIVISAAGIPNLITADMIKEGAAVIDVGINRVHKPKLVGDVDFEGVRQKAGYITPVPGGVGPMTVAMLMKNTIIAAKKVL Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Steroid 17-alpha-monooxygenase (S17AH) | 3SWZ | 4.26 | |
Target general information Gen name CYP17A1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Steroid 17-alpha-hydroxylase/17,20 lyase; P450c17; P450-C17; P450 17; CYPXVII; CYP17A1; CYP 17; 17 alpha-Hydroxylase/C17-20-lyase Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function Conversion of pregnenolone and progesterone to their 17- alpha-hydroxylated products and subsequently to dehydroepiandrosterone (DHEA) and androstenedione. Catalyzes both the 17-alpha-hydroxylation and the 17,20-lyase reaction. Involved in sexual development during fetal life and at puberty. Related diseases Adrenal hyperplasia 5 (AH5) [MIM:202110]: A form of congenital adrenal hyperplasia, a common recessive disease due to defective synthesis of cortisol. Congenital adrenal hyperplasia is characterized by androgen excess leading to ambiguous genitalia in affected females, rapid somatic growth during childhood in both sexes with premature closure of the epiphyses and short adult stature. Four clinical types: 'salt wasting' (SW, the most severe type), 'simple virilizing' (SV, less severely affected patients), with normal aldosterone biosynthesis, 'non-classic form' or late-onset (NC or LOAH) and 'cryptic' (asymptomatic). {ECO:0000269|PubMed:10720067, ECO:0000269|PubMed:11549685, ECO:0000269|PubMed:11836339, ECO:0000269|PubMed:12466376, ECO:0000269|PubMed:14671162, ECO:0000269|PubMed:1515452, ECO:0000269|PubMed:1714904, ECO:0000269|PubMed:1740503, ECO:0000269|PubMed:19793597, ECO:0000269|PubMed:24140098, ECO:0000269|PubMed:24498484, ECO:0000269|PubMed:25650406, ECO:0000269|PubMed:2808364, ECO:0000269|PubMed:8027220, ECO:0000269|PubMed:8245018, ECO:0000269|PubMed:8345056, ECO:0000269|PubMed:8396144, ECO:0000269|PubMed:8550762, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05812; DB04630; DB01424; DB09061; DB00882; DB01234; DB14649; DB01026; DB05667; DB14009; DB14011; DB00157; DB01708; DB00396; DB02901 Interacts with NA EC number EC 1.14.14.19 Uniprot keywords 3D-structure; Congenital adrenal hyperplasia; Disease variant; Endoplasmic reticulum; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Monooxygenase; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Steroidogenesis Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 51385.8 Length 453 Aromaticity 0.08 Instability index 32.67 Isoelectric point 8.48 Charge (pH=7) 4.08 2D Binding mode Binding energy (Kcal/mol) -5.81  Molscript Map 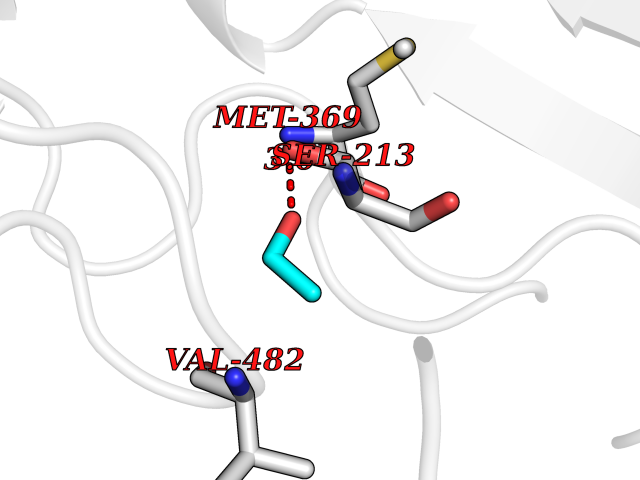 Pymol Map 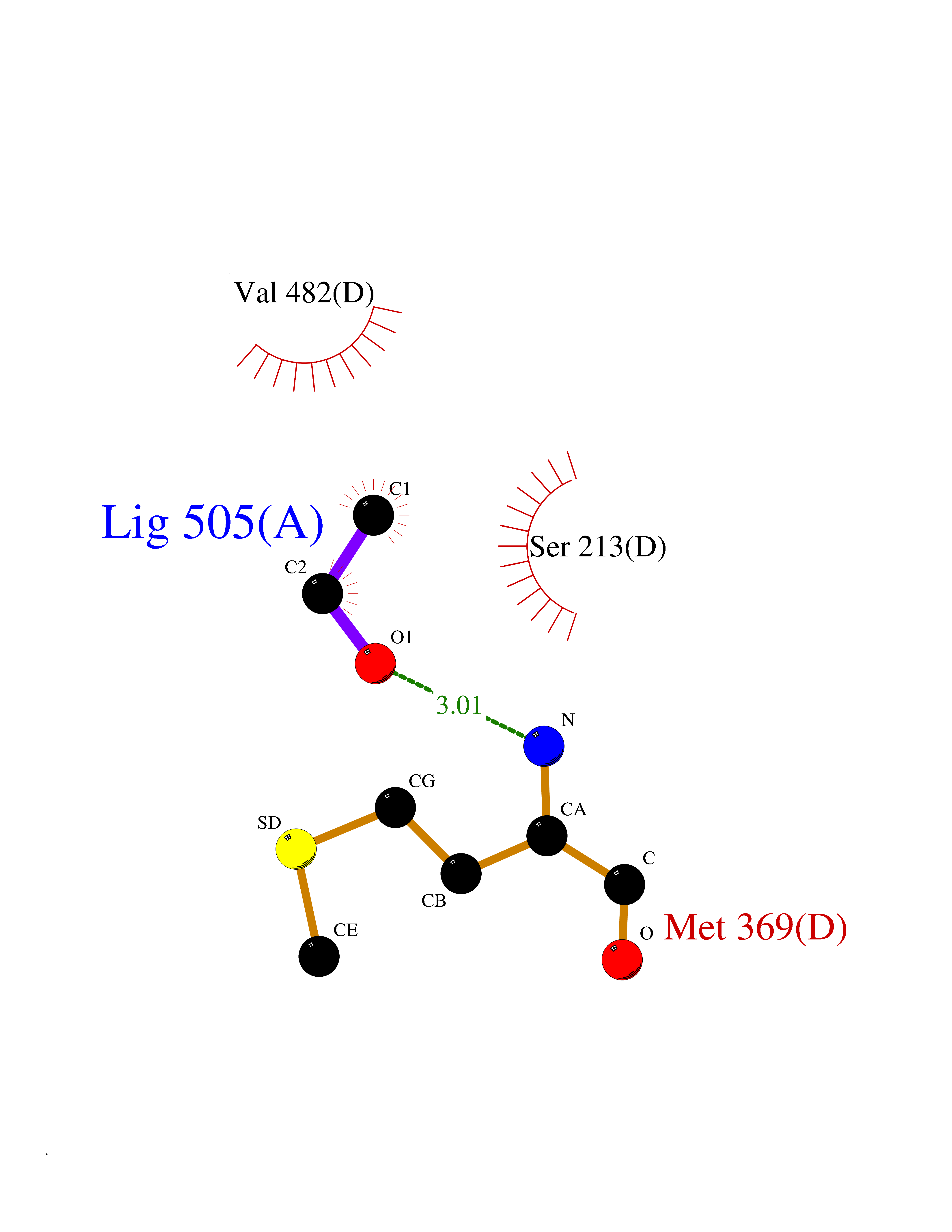 Ligplot Map 3D Binding mode Sequence LPRHGHMHNNFFKLQKKYGPIYSVRMGTKTTVIVGHHQLAKEVLIKKGKDFSGRPQMATLDIASNNRKGIAFADSGAHWQLHRRLAMATFALFKDGDQKLEKIICQEISTLCDMLATHNGQSIDISFPVFVAVTNVISLICFNTSYKNGDPELNVIQNYNEGIIDNLSKDSLVDLVPWLKIFPNKTLEKLKSHVKIRNDLLNKILENYKEKFRSDSITNMLDTLMQAKMNSDDSELLSDNHILTTIGDIFGAGVETTTSVVKWTLAFLLHNPQVKKKLYEEIDQNVGFSRTPTISDRNRLLLLEATIREVLRLRPVAPMLIPHKANVDSSIGEFAVDKGTEVIINLWALHHNEKEWHQPDQFMPERFLNPAGTQLISPSVSYLPFGAGPRSCIGEILARQELFLIMAWLLQRFDLEVPDDGQLPSLEGIPKVVFLIDSFKVKIKVRQAWREAQ Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Histone deacetylase 3 (HDAC3) | 4A69 | 4.26 | |
Target general information Gen name HDAC3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SMAP45; RPD32; RPD3-2; HD3 Protein family Histone deacetylase family, HD type 1 subfamily Biochemical class Carbon-nitrogen hydrolase Function Gives a tag for epigenetic repression and plays an important role in transcriptional regulation, cell cycle progression and developmental events. Histone deacetylases act via the formation of large multiprotein complexes. Participates in the BCL6 transcriptional repressor activity by deacetylating the H3 'Lys-27' (H3K27) on enhancer elements, antagonizing EP300 acetyltransferase activity and repressing proximal gene expression. Probably participates in the regulation of transcription through its binding to the zinc-finger transcription factor YY1; increases YY1 repression activity. Required to repress transcription of the POU1F1 transcription factor. Acts as a molecular chaperone for shuttling phosphorylated NR2C1 to PML bodies for sumoylation. Contributes, together with XBP1 isoform 1, to the activation of NFE2L2-mediated HMOX1 transcription factor gene expression in a PI(3)K/mTORC2/Akt-dependent signaling pathway leading to endothelial cell (EC) survival under disturbed flow/oxidative stress. Regulates both the transcriptional activation and repression phases of the circadian clock in a deacetylase activity-independent manner. During the activation phase, promotes the accumulation of ubiquitinated ARNTL/BMAL1 at the E-boxes and during the repression phase, blocks FBXL3-mediated CRY1/2 ubiquitination and promotes the interaction of CRY1 and ARNTL/BMAL1. The NCOR1-HDAC3 complex regulates the circadian expression of the core clock gene ARTNL/BMAL1 and the genes involved in lipid metabolism in the liver. Responsible for the deacetylation of lysine residues on the N-terminal part of the core histones (H2A, H2B, H3 and H4), and some other non-histone substrates. Related diseases Cocoon syndrome (COCOS) [MIM:613630]: A lethal syndrome characterized by multiple fetal malformations including defective face and seemingly absent limbs, which are bound to the trunk and encased under the skin. {ECO:0000269|PubMed:20961246}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Bartsocas-Papas syndrome 2 (BPS2) [MIM:619339]: An autosomal recessive, severe form of popliteal pterygium syndrome. Popliteal pterygia syndromes have considerable variability in severity and in the associated phenotypic features but they are all characterized by cutaneous webbing across one or more major joints, cleft lip and/or palate, syndactyly, and genital malformations. {ECO:0000269|PubMed:25691407}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12565; DB05015; DB01262; DB11841; DB12645; DB11830; DB06603; DB06819; DB05223; DB03766; DB12847; DB06176; DB00313; DB02546 Interacts with O43823; Q9ULX6; Q9UKG1; P24385; P23771; Q13227; Q13547; O60341; Q969R5; P43356; Q9UIS9; P01106; O75376; Q9Y618; Q15466; P48552; P00558; P00558-1; P60510; Q15022; O09106; P08393; Q60974; Q8CBD1; P12504 EC number EC 3.5.1.98 Uniprot keywords 3D-structure; Alternative splicing; Biological rhythms; Chromatin regulator; Chromosome; Cytoplasm; Host-virus interaction; Hydrolase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repressor; Transcription; Transcription regulation; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 50376.9 Length 437 Aromaticity 0.14 Instability index 29.95 Isoelectric point 6.1 Charge (pH=7) -6.84 2D Binding mode Binding energy (Kcal/mol) -5.81  Molscript Map 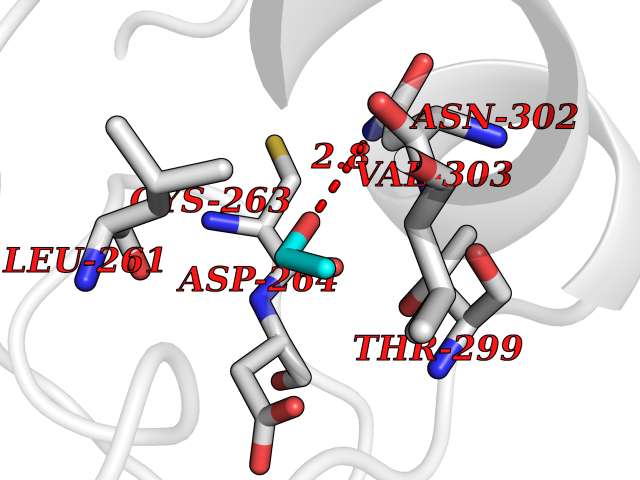 Pymol Map 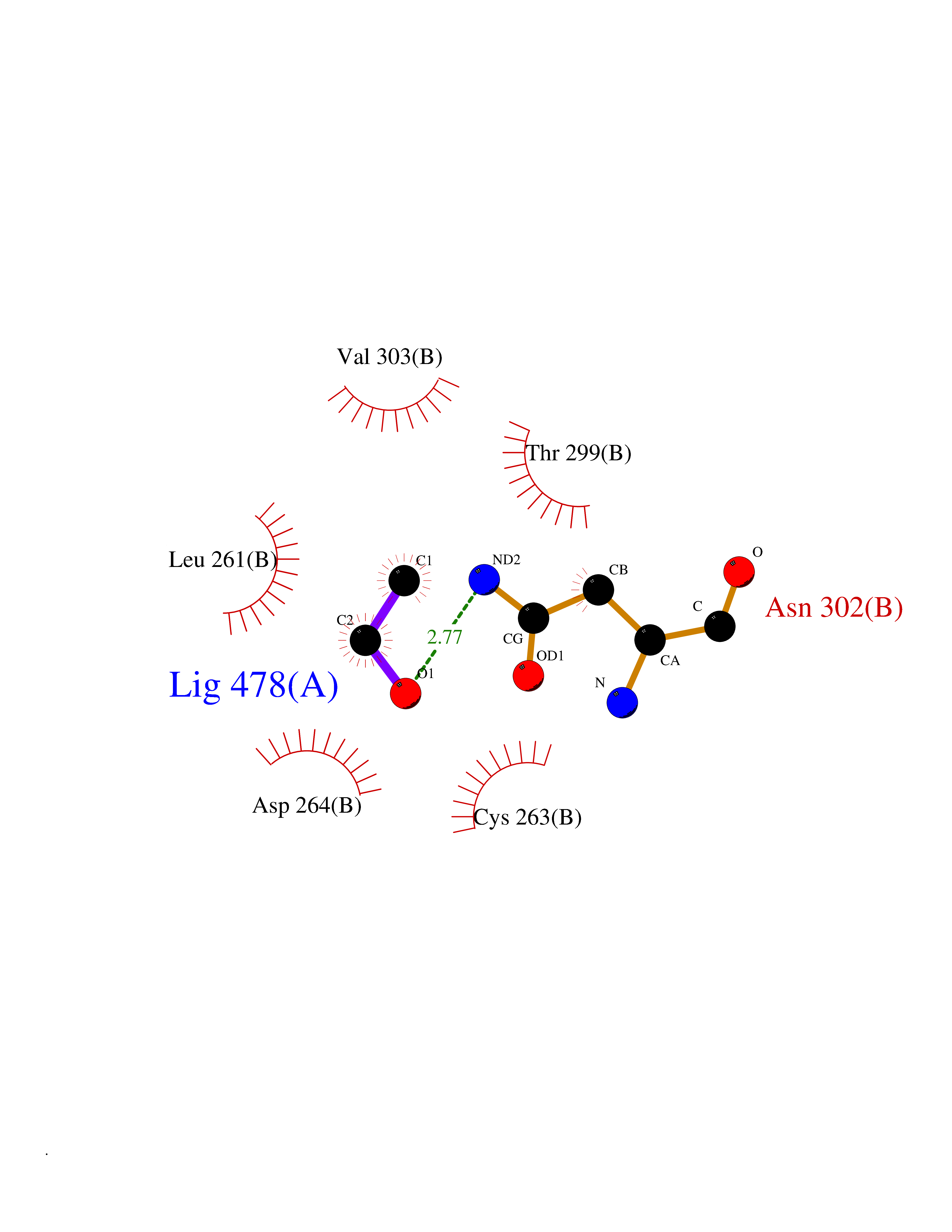 Ligplot Map 3D Binding mode Sequence KFINMNGLMADPMKVYKDRQVMNMWSEQEKETFREKFMQHPKNFGLIASFLERKTVAECVLYYYLTKKAKTVAYFYDPDVGNFHYGAGHPMKPHRLALTHSLVLHYGLYKKMIVFKPYQASQHDMCRFHSEDYIDFLQRVSPTNMQGFTKSLNAFNVGDDCPVFPGLFEFCSRYTGASLQGATQLNNKICDIAINWAGGLHHAKKFEASGFCYVNDIVIGILELLKYHPRVLYIDIDIHHGDGVQEAFYLTDRVMTVSFHKYGNYFFPGTGDMYEVGAESGRYYCLNVPLRDGIDDQSYKHLFQPVINQVVDFYQPTCIVLQCGADSLGCDRLGCFNLSIRGHGECVEYVKSFNIPLLVLGGGGYTVRNVARCWTYETSLLVEEAISEELPYSEYFEYFAPDFTLHPDVSTRIENQNSRQYLDQIRQTIFENLKMLN Hydrogen bonds contact Hydrophobic contact | ||||