Job Results:
Ligand
Structure
Job ID
5a9d2b2db14ecd17b747d14262c01470
Job name
NA
Time
2025-04-03 16:12:50
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Cytochrome P450 1B1 (CYP1B1) | 3PM0 | 5.41 | |
Target general information Gen name CYP1B1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms CYPIB1 Protein family Cytochrome P450 family Biochemical class Paired donor oxygen oxidoreductase Function In liver microsomes, this enzyme is involved in an NADPH-dependent electron transport pathway. It oxidizes a variety of structurally unrelated compounds, including steroids, fatty acids, retinoid and xenobiotics. Preferentially oxidizes 17beta-estradiol to the carcinogenic 4-hydroxy derivative, and a variety of procarcinogenic compounds to their activated forms, including polycyclic aromatic hydrocarbons. Promotes angiogenesis by removing cellular oxygenation products, thereby decreasing oxidative stress, release of antiangiogenic factor THBS2, then allowing endothelial cells migration, cell adhesion and capillary morphogenesis. These changes are concommitant with the endothelial nitric oxide synthase activity and nitric oxide synthesis. Plays an important role in the regulation of perivascular cell proliferation, migration, and survival through modulation of the intracellular oxidative state and NF-kappa-B expression and/or activity, during angiogenesis. Contributes to oxidative homeostasis and ultrastructural organization and function of trabecular meshwork tissue through modulation of POSTN expression. Cytochromes P450 are a group of heme-thiolate monooxygenases. Related diseases Anterior segment dysgenesis 6 (ASGD6) [MIM:617315]: A form of anterior segment dysgenesis, a group of defects affecting anterior structures of the eye including cornea, iris, lens, trabecular meshwork, and Schlemm canal. Anterior segment dysgeneses result from abnormal migration or differentiation of the neural crest derived mesenchymal cells that give rise to components of the anterior chamber during eye development. Different anterior segment anomalies may exist alone or in combination, including iris hypoplasia, enlarged or reduced corneal diameter, corneal vascularization and opacity, posterior embryotoxon, corectopia, polycoria, abnormal iridocorneal angle, ectopia lentis, and anterior synechiae between the iris and posterior corneal surface. Clinical conditions falling within the phenotypic spectrum of anterior segment dysgeneses include aniridia, Axenfeld anomaly, Reiger anomaly/syndrome, Peters anomaly, and iridogoniodysgenesis. ASGD6 patients predominantly manifest Peters anomaly. Peters anomaly consists of corneal leukoma, defects in the posterior structures of the cornea such as absence of the posterior corneal stroma and Descemet membrane, and a variable degree of iridocorneal and/or keratolenticular adhesions. Over 50% of patients develop glaucoma in childhood. {ECO:0000269|PubMed:11403040}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Glaucoma 3, primary congenital, A (GLC3A) [MIM:231300]: An autosomal recessive form of primary congenital glaucoma (PCG). PCG is characterized by marked increase of intraocular pressure at birth or early childhood, large ocular globes (buphthalmos) and corneal edema. It results from developmental defects of the trabecular meshwork and anterior chamber angle of the eye that prevent adequate drainage of aqueous humor. {ECO:0000269|PubMed:10227395, ECO:0000269|PubMed:10655546, ECO:0000269|PubMed:11184479, ECO:0000269|PubMed:11527932, ECO:0000269|PubMed:11774072, ECO:0000269|PubMed:11980847, ECO:0000269|PubMed:12036985, ECO:0000269|PubMed:12525557, ECO:0000269|PubMed:14635112, ECO:0000269|PubMed:14640114, ECO:0000269|PubMed:15255109, ECO:0000269|PubMed:15342693, ECO:0000269|PubMed:15475877, ECO:0000269|PubMed:16490498, ECO:0000269|PubMed:16688110, ECO:0000269|PubMed:16735994, ECO:0000269|PubMed:16862072, ECO:0000269|PubMed:18470941, ECO:0000269|PubMed:9463332, ECO:0000269|PubMed:9497261}. The disease is caused by variants affecting distinct genetic loci, including the gene represented in this entry.; DISEASE: Glaucoma 1, open angle, A (GLC1A) [MIM:137750]: A form of primary open angle glaucoma (POAG). POAG is characterized by a specific pattern of optic nerve and visual field defects. The angle of the anterior chamber of the eye is open, and usually the intraocular pressure is increased. However, glaucoma can occur at any intraocular pressure. The disease is generally asymptomatic until the late stages, by which time significant and irreversible optic nerve damage has already taken place. {ECO:0000269|PubMed:11774072}. The gene represented in this entry acts as a disease modifier. Digenic mutations in CYP1B1 and MYOC have been found in a family segregating both primary adult-onset and juvenile forms of open angle glaucoma (PubMed:11774072). All affected family members with mutations in both MYOC and CYP1B1 had juvenile glaucoma, whereas those with only the MYOC mutation had the adult-onset form (PubMed:11774072). {ECO:0000269|PubMed:11774072}. Drugs (DrugBank ID) DB02342; DB00613; DB06732; DB00443; DB00121; DB01222; DB00201; DB09061; DB14737; DB01254; DB00694; DB01248; DB00997; DB00470; DB00530; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB00655; DB07776; DB00499; DB01645; DB01381; DB00741; DB01064; DB01026; DB00448; DB14009; DB01065; DB00170; DB00959; DB01204; DB14011; DB03467; DB00338; DB01229; DB14631; DB00635; DB01087; DB00396; DB00818; DB04216; DB02709; DB00675; DB00624; DB13946; DB00277; DB12245; DB11155 Interacts with Q02763 EC number EC 1.14.14.- Uniprot keywords 3D-structure; Disease variant; Endoplasmic reticulum; Fatty acid metabolism; Glaucoma; Heme; Iron; Lipid metabolism; Lyase; Membrane; Metal-binding; Microsome; Mitochondrion; Monooxygenase; Oxidoreductase; Peters anomaly; Proteomics identification; Reference proteome; Steroid metabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 51875.9 Length 459 Aromaticity 0.1 Instability index 34.16 Isoelectric point 8.64 Charge (pH=7) 4.89 2D Binding mode Binding energy (Kcal/mol) -7.39 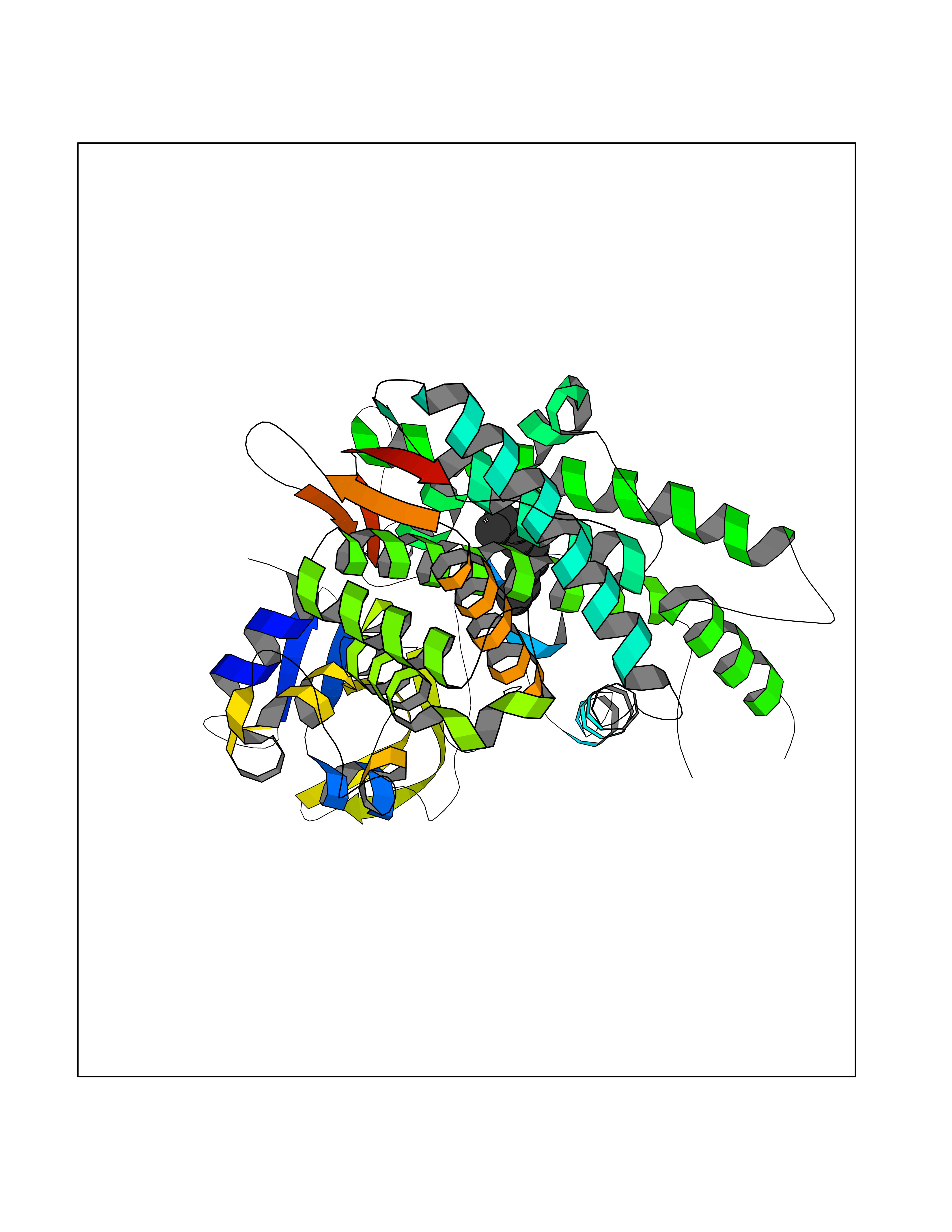 Molscript Map 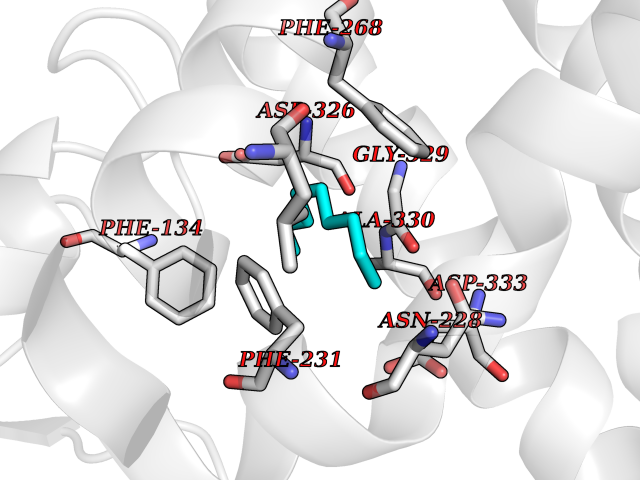 Pymol Map 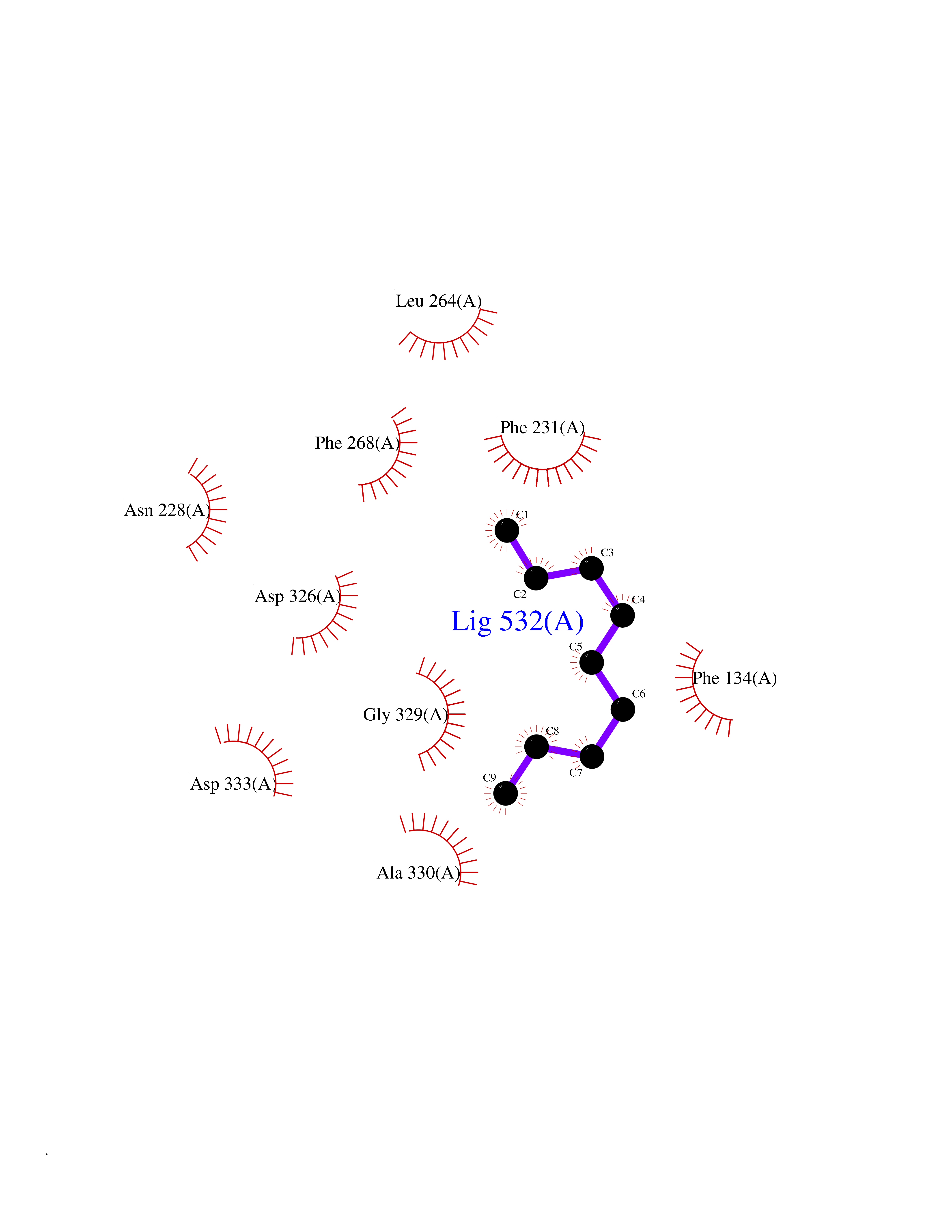 Ligplot Map 3D Binding mode Sequence QAAHLSFARLARRYGDVFQIRLGSCPIVVLNGERAIHQALVQQGSAFADRPSFASFRVVSGGRSMAFGHYSEHWKVQRRAAHSMMRNFFTRQPRSRQVLEGHVLSEARELVALLVRGSADGAFLDPRPLTVVAVANVMSAVCFGCRYSHDDPEFRELLSHNEEFGRTVGAGSLVDVMPWLQYFPNPVRTVFREFEQLNRNFSNFILDKFLRHCESLRPGAAPRDMMDAFILSAEKKAAGDGARLDLENVPATITDIFGASQDTLSTALQWLLLLFTRYPDVQTRVQAELDQVVGRDRLPCMGDQPNLPYVLAFLYEAMRFSSFVPVTIPHATTANTSVLGYHIPKDTVVFVNQWSVNHDPLKWPNPENFDPARFLDKDGLINKDLTSRVMIFSVGKRRCIGEELSKMQLFLFISILAHQCDFRANPNEPAKMNFSYGLTIKPKSFKVNVTLRESMELLD Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Hydroxymethylglutaryl-CoA synthase 2 (HMGCS2) | 2WYA | 5.41 | |
Target general information Gen name HMGCS2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms HMGCS2; HMG-CoAsynthase; 3-hydroxy-3-methylglutaryl coenzyme A synthase 2 Protein family Thiolase-like superfamily, HMG-CoA synthase family Biochemical class Acyltransferase Function This enzyme condenses acetyl-CoA with acetoacetyl-CoA to form HMG-CoA, which is the substrate for HMG-CoA reductase. Related diseases 3-hydroxy-3-methylglutaryl-CoA synthase-2 deficiency (HMGCS2D) [MIM:605911]: A metabolic disorder characterized by severe hypoketotic hypoglycemia, encephalopathy, and hepatomegaly. {ECO:0000269|PubMed:11228257, ECO:0000269|PubMed:11479731, ECO:0000269|PubMed:12647205, ECO:0000269|PubMed:16601895, ECO:0000269|PubMed:23751782, ECO:0000269|PubMed:25511235, ECO:0000269|PubMed:29597274}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 2.3.3.10 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cholesterol biosynthesis; Cholesterol metabolism; Disease variant; Lipid biosynthesis; Lipid metabolism; Mitochondrion; Phosphoprotein; Proteomics identification; Reference proteome; Steroid biosynthesis; Steroid metabolism; Sterol biosynthesis; Sterol metabolism; Transferase; Transit peptide Protein physicochemical properties Chain ID A,D Molecular weight (Da) 102499 Length 920 Aromaticity 0.1 Instability index 33.48 Isoelectric point 6.72 Charge (pH=7) -1.41 2D Binding mode Binding energy (Kcal/mol) -7.38 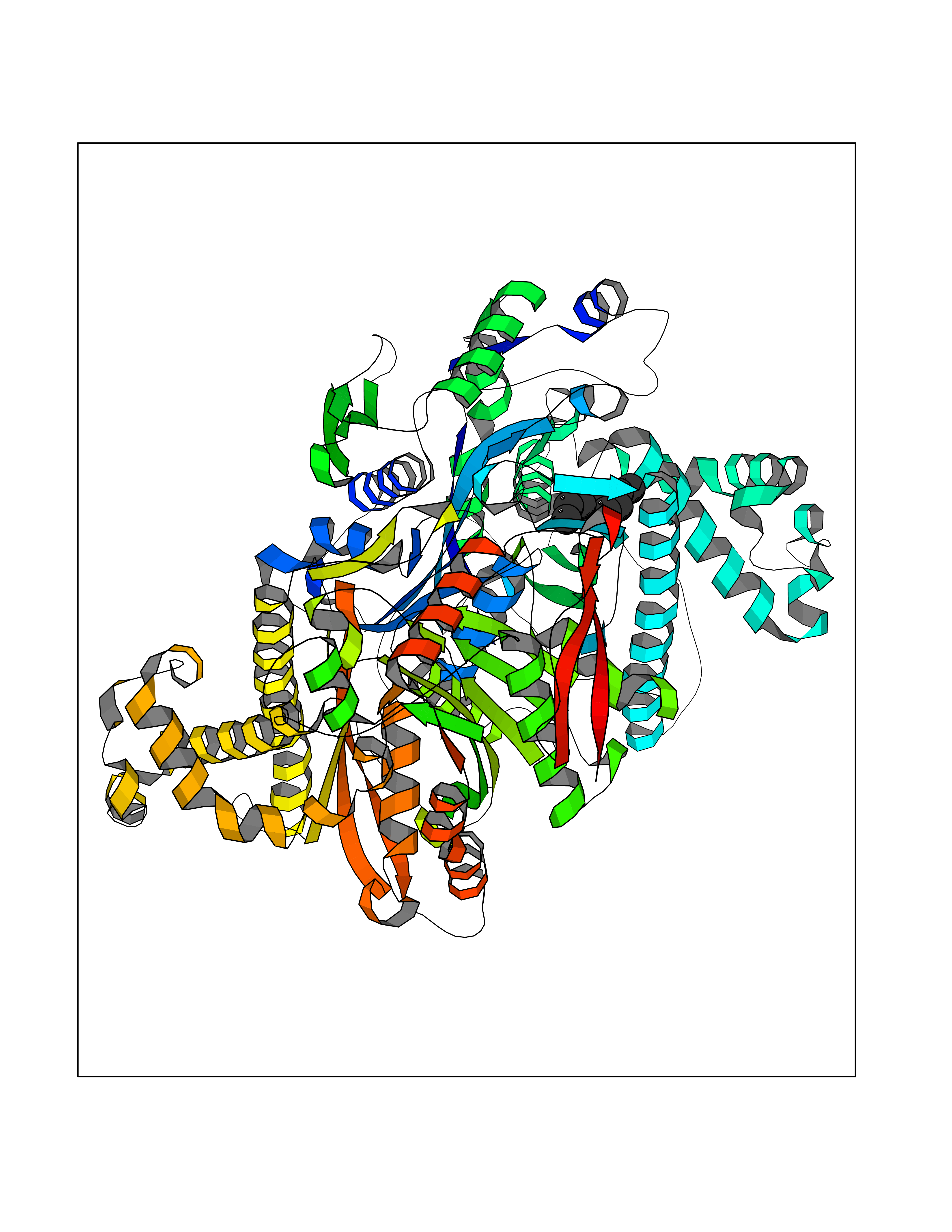 Molscript Map 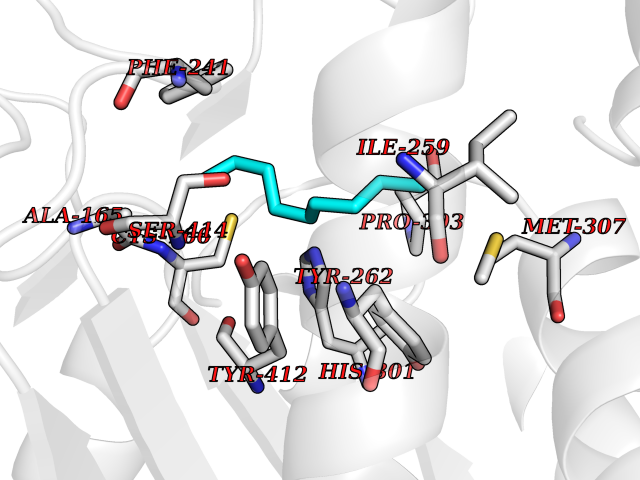 Pymol Map 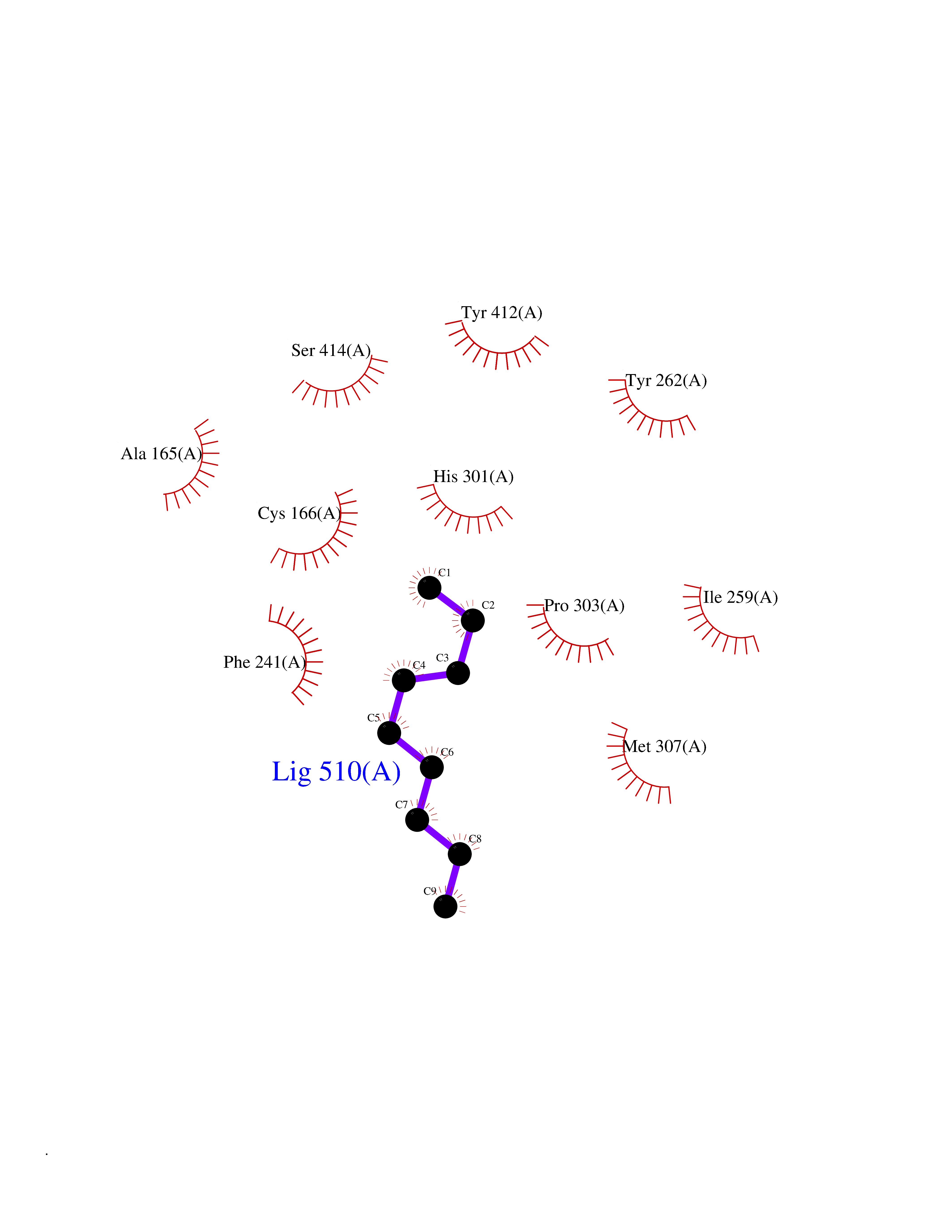 Ligplot Map 3D Binding mode Sequence SMPKDVGILALEVYFPAQYVDQTDLEKYNNVEAGKYTVGLGQTRMGFCSVQEDINSLCLTVVQRLMERIQLPWDSVGRLEVGTETIIDKSKAVKTVLMELFQDSGNTDIEGIDTTNACYGGTASLFNAANWMESSSWDGRYAMVVCGDIAVYPSGNARPTGGAGAVAMLIGPKAPLALERGLRGTHMENVYDFYKPNLASEYPIVDGKLSIQCYLRALDRCYTSYRKKIQNQWKQAGSDRPFTLDDLQYMIFHTPFCKMVQKSLARLMFNDFLSASSDTQTSLYKGLEAFGGLKLEDTYTNKDLDKALLKASQDMFDKKTKASLYLSTHNGNMYTSSLYGCLASLLSHHSAQELAGSRIGAFSYGSGLAASFFSFRVSQDAAPGSPLDKLVSSTSDLPKRLASRKCVSPEEFTEIMNQREQFYHKVNFSPPGDTNSLFPGTWYLERVDEQHRRKYARRPVSMPKDVGILALEVYFPAQYVDQTDLEKYNNVEAGKYTVGLGQTRMGFCSVQEDINSLCLTVVQRLMERIQLPWDSVGRLEVGTETIIDKSKAVKTVLMELFQDSGNTDIEGIDTTNACYGGTASLFNAANWMESSSWDGRYAMVVCGDIAVYPSGNARPTGGAGAVAMLIGPKAPLALERGLRGTHMENVYDFYKPNLASEYPIVDGKLSIQCYLRALDRCYTSYRKKIQNQWKQAGSDRPFTLDDLQYMIFHTPFCKMVQKSLARLMFNDFLSASSDTQTSLYKGLEAFGGLKLEDTYTNKDLDKALLKASQDMFDKKTKASLYLSTHNGNMYTSSLYGCLASLLSHHSAQELAGSRIGAFSYGSGLAASFFSFRVSQDAAPGSPLDKLVSSTSDLPKRLASRKCVSPEEFTEIMNQREQFYHKVNFSPPGDTNSLFPGTWYLERVDEQHRRKYARRPV Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Glucose-dependent insulinotropic receptor (GPR119) | 7XZ6 | 5.41 | |
Target general information Gen name GPR119 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms GPR119; G-protein coupled receptor 119 Protein family G-protein coupled receptor 1 family Biochemical class GPCR rhodopsin Function Receptor for the endogenous fatty-acid ethanolamide oleoylethanolamide (OEA) and lysophosphatidylcholine (LPC). Functions as a glucose-dependent insulinotropic receptor. The activity of this receptor is mediated by G proteins which activate adenylate cyclase. Seems to act through a G(s) mediated pathway. Related diseases Developmental and epileptic encephalopathy 24 (DEE24) [MIM:615871]: A disease characterized by early-onset seizures, intellectual disability of varying degrees, and behavioral disturbances or autistic features in most individuals. {ECO:0000269|PubMed:24747641, ECO:0000269|PubMed:27864847, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Generalized epilepsy with febrile seizures plus 10 (GEFSP10) [MIM:618482]: An autosomal dominant neurologic disorder with incomplete penetrance, characterized by variable types of seizures including absence, tonic-clonic, febrile, focal, and eyelid myoclonia. Some patients have normal neurologic development. Others have mild-to-moderate intellectual disability or autism spectrum disorder. {ECO:0000269|PubMed:29936235, ECO:0000269|PubMed:30351409}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB05166 Interacts with Q12797-6 EC number NA Uniprot keywords 3D-structure; Cell membrane; G-protein coupled receptor; Lipid-binding; Membrane; Receptor; Reference proteome; Transducer; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID R Molecular weight (Da) 32134.1 Length 292 Aromaticity 0.12 Instability index 34.96 Isoelectric point 9.12 Charge (pH=7) 8.03 2D Binding mode Binding energy (Kcal/mol) -7.38 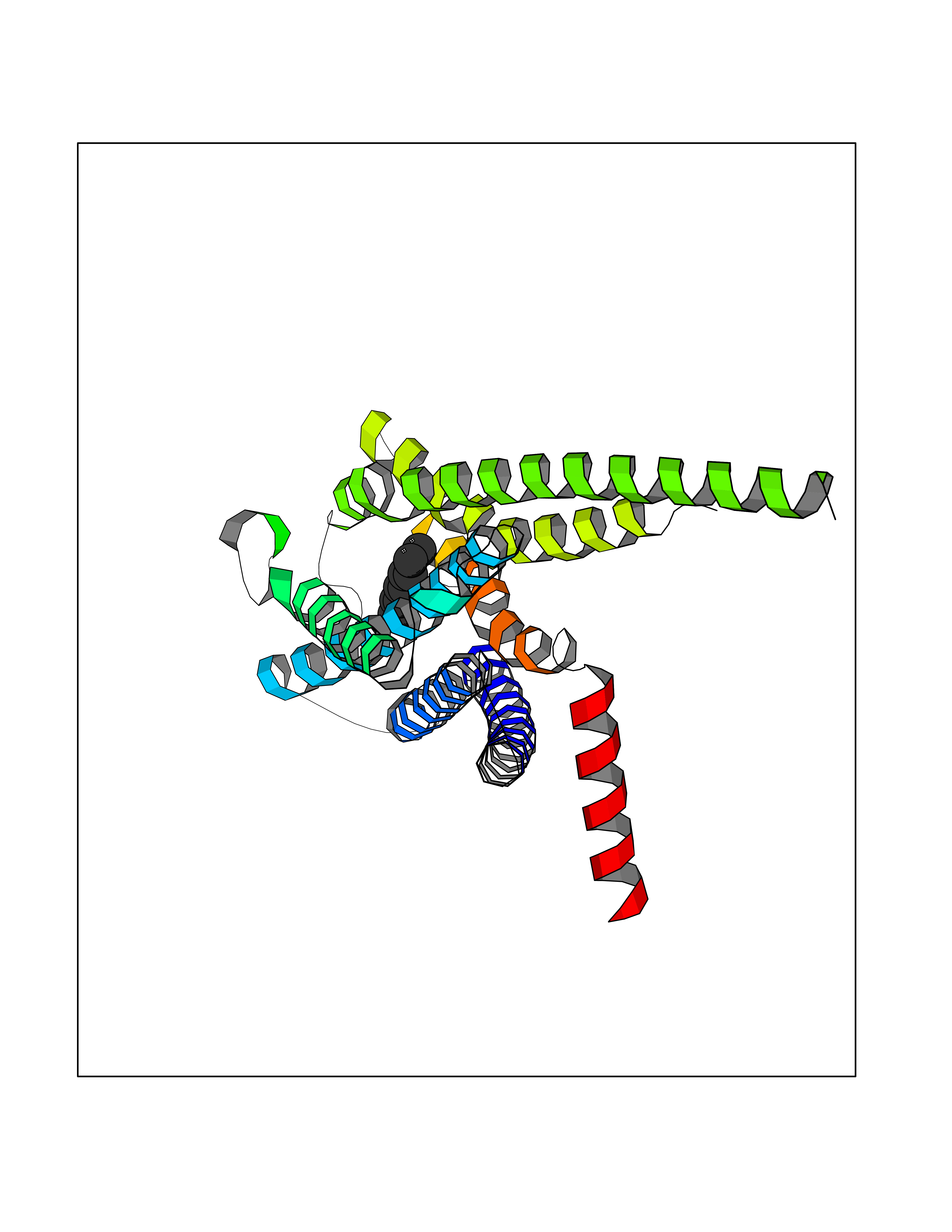 Molscript Map 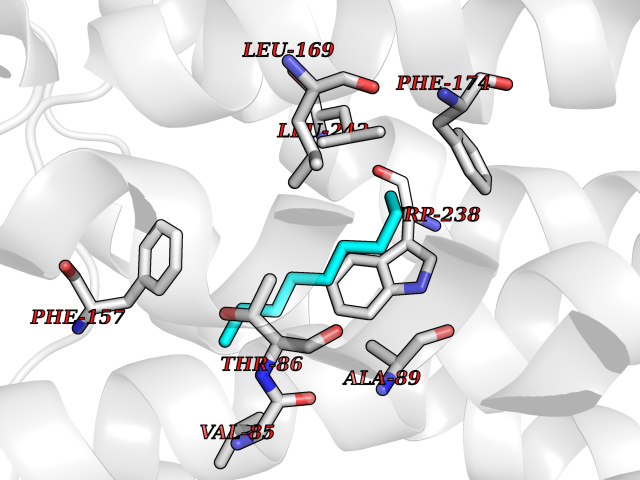 Pymol Map 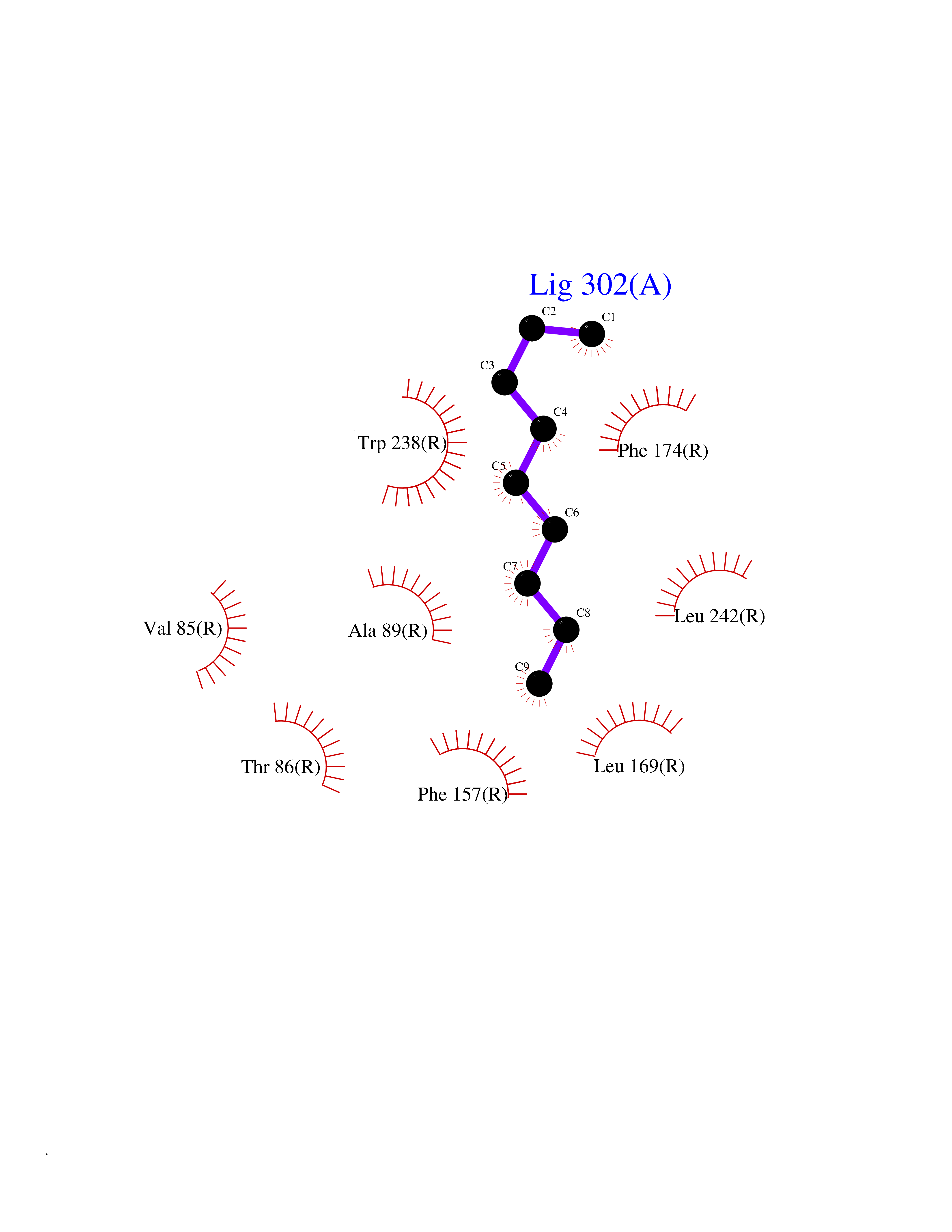 Ligplot Map 3D Binding mode Sequence MESSFSFGVILAVLASLIIATNTLVAVAVLLLIHKNDGVSLCFTLNLAVADTLIGVAISGLLTDQLSSPSRPTQKTLCSLRMAFVTSSAAASVLTVMLITFDRYLAIKQPFRYLKIMSGFVAGACIAGLWLVSYLIGFLPLGIPMFQQTAYKGQCSFFAVFHPHFVLTLSCVGFFPAMLLFVFFYCDMLKIASMHSQQIRKMEHAGAMAGSDFKALRTVSVLIGSFALSWTPFLITGIVQVACQECHLYLVLERYLWLLGVGNSLLNPLIYAYWQKEVRLQLYHMALGVKKV Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | MLK-related kinase (MLTK) | 5HES | 5.41 | |
Target general information Gen name MAP3K20 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms ZAK; Sterile alpha motif- and leucine zipper-containing kinase AZK; Mixed lineage kinase-related kinase; Mitogen-activated protein kinase kinase kinase MLT; Mitogen-activated protein kinase kinase kin Protein family Protein kinase superfamily, STE Ser/Thr protein kinase family, MAP kinase kinase kinase subfamily Biochemical class NA Function Stress-activated component of a protein kinase signal transduction cascade. Regulates the JNK and p38 pathways. Part of a signaling cascade that begins with the activation of the adrenergic receptor ADRA1B and leads to the activation of MAPK14. Pro-apoptotic. Role in regulation of S and G2 cell cycle checkpoint by direct phosphorylation of CHEK2. Involved in limb development. Related diseases Split-foot malformation with mesoaxial polydactyly (SFMMP) [MIM:616890]: An autosomal recessive disorder characterized by a split-foot defect, mesoaxial polydactyly, nail abnormalities of the hands, and sensorineural hearing loss. {ECO:0000269|PubMed:26755636, ECO:0000269|PubMed:32266845}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Myopathy, centronuclear, 6, with fiber-type disproportion (CNM6) [MIM:617760]: A form of centronuclear myopathy, a congenital muscle disorder characterized by progressive muscular weakness and wasting involving mainly limb girdle, trunk, and neck muscles. It may also affect distal muscles. Weakness may be present during childhood or adolescence or may not become evident until the third decade of life. Ptosis is a frequent clinical feature. The most prominent histopathologic features include high frequency of centrally located nuclei in muscle fibers not secondary to regeneration, radial arrangement of sarcoplasmic strands around the central nuclei, and predominance and hypotrophy of type 1 fibers. CNM6 is an autosomal recessive, slowly progressive form with onset in infancy or early childhood. {ECO:0000269|PubMed:27816943, ECO:0000269|PubMed:30237576}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01254; DB12010 Interacts with O75582; P31947; P63104; Q8N184; Q16512; Q6P2D0; Q6ZN57; P13682; Q8N184; Q6AZW8; Q9NQZ8 EC number EC 2.7.11.25 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ATP-binding; Cell cycle; Cytoplasm; Disease variant; Kinase; Magnesium; Metal-binding; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; RNA-binding; rRNA-binding; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 64591.5 Length 566 Aromaticity 0.1 Instability index 52.08 Isoelectric point 5.69 Charge (pH=7) -12.83 2D Binding mode Binding energy (Kcal/mol) -7.38  Molscript Map  Pymol Map 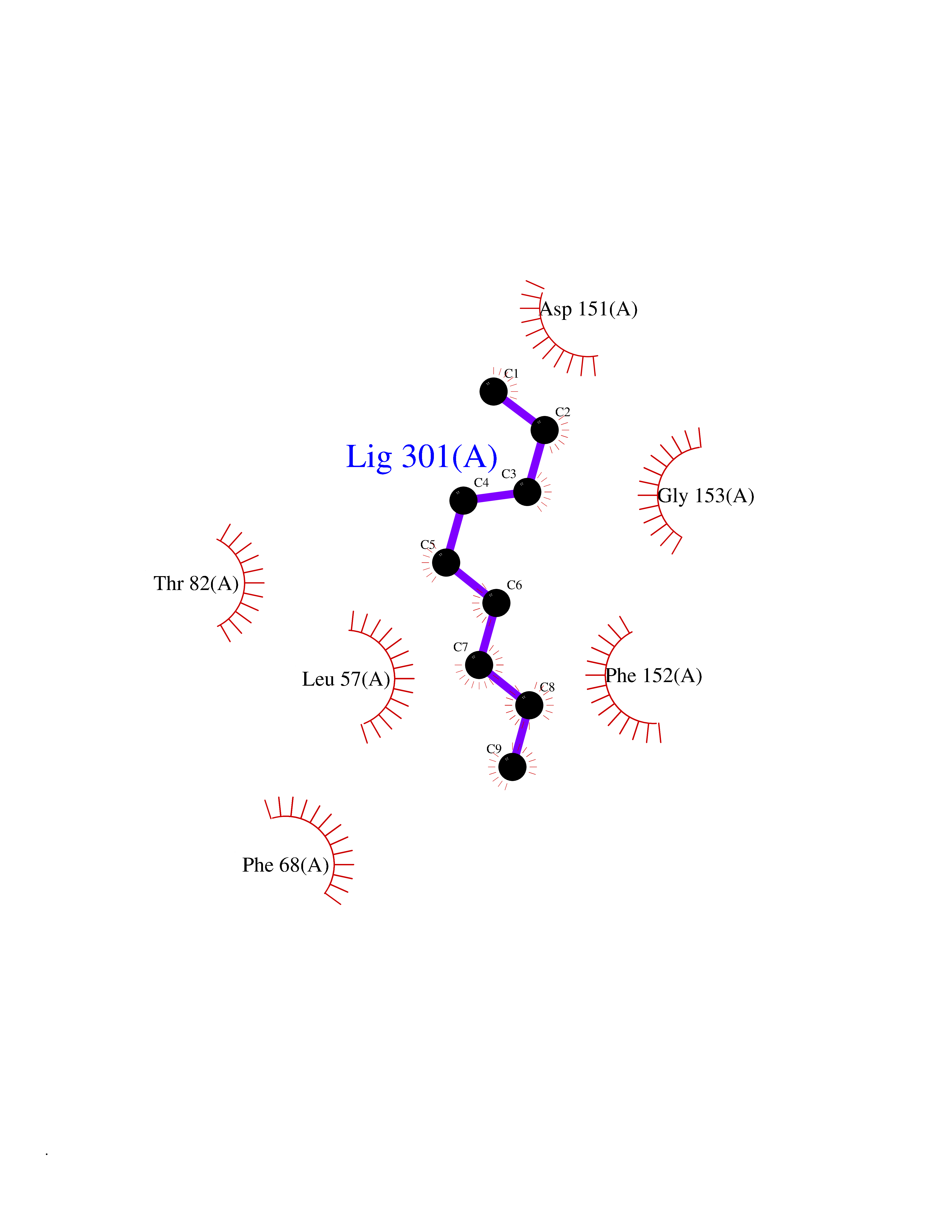 Ligplot Map 3D Binding mode Sequence ASFVQIKFDDLQFFENCGGGSFGSVYRAKWISQDKEVAVKKLLKIEKEAEILSVLSHRNIIQFYGVILEPPNYGIVTEYASLGSLYDYINSNRSEEMDMDHIMTWATDVAKGMHYLHMEAPVKVIHRDLKSRNVVIAADGVLKICDFGASRFHNHXGTFPWMAPEVIQSLPVSETCDTYSYGVVLWEMLTREVPFKGLEGLQVAWLVVEKNERLTIPSSCPRSFAELLHQCWEADAKKRPSFKQIISILESMSNDTSLPDKCNSFLHNKAEWRCEIEATLERLKKLERSFVQIKFDDLQFFENCGGGSFGSVYRAKWISQDKEVAVKKLLKIEKEAEILSVLSHRNIIQFYGVILEPPNYGIVTEYASLGSLYDYINSNRSEEMDMDHIMTWATDVAKGMHYLHMEAPVKVIHRDLKSRNVVIAADGVLKICDFGGTFPWMAPEVIQSLPVSETCDTYSYGVVLWEMLTREVPFKGLEGLQVAWLVVEKNERLTIPSSCPRSFAELLHQCWEADAKKRPSFKQIISILESMSNDTSLPDKCNSFLHNKAEWRCEIEATLERLKKLE Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Thyroid hormone receptor alpha (THRA) | 3ILZ | 5.40 | |
Target general information Gen name THRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms V-erbA-related protein 7; THRA2; THRA1; Nuclear receptor subfamily 1 group A member 1; NR1A1; ERBA1; EAR7; EAR-7; C-erbA-alpha; C-erbA-1 Protein family Nuclear hormone receptor family, NR1 subfamily Biochemical class Nuclear hormone receptor Function High affinity receptor for thyroid hormones, including triiodothyronine and thyroxine. Isoform Alpha-1: Nuclear hormone receptor that can act as a repressor or activator of transcription. Related diseases Hypothyroidism, congenital, non-goitrous, 6 (CHNG6) [MIM:614450]: A disease characterized by growth retardation, developmental retardation, skeletal dysplasia, borderline low thyroxine levels and high triiodothyronine levels. There is differential sensitivity to thyroid hormone action, with retention of hormone responsiveness in the hypothalamic pituitary axis and liver but skeletal, gastrointestinal, and myocardial resistance. {ECO:0000269|PubMed:22168587, ECO:0000269|PubMed:24969835, ECO:0000269|PubMed:25670821, ECO:0000269|PubMed:26037512}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01118; DB00509; DB04855; DB05035; DB03176; DB00451; DB00279; DB01583; DB05235; DB09100 Interacts with Q9Y2J4; Q9Y2J4-4; O95971; Q8TAP6; Q96JM7; Q15648; Q6FHY5; P31321; Q96A49; O75410-7; Q9JLI4 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Congenital hypothyroidism; Cytoplasm; Disease variant; DNA-binding; Metal-binding; Nucleus; Proteomics identification; Receptor; Reference proteome; Transcription; Transcription regulation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A Molecular weight (Da) 29910.1 Length 267 Aromaticity 0.07 Instability index 52.75 Isoelectric point 5.31 Charge (pH=7) -11.32 2D Binding mode Binding energy (Kcal/mol) -7.36 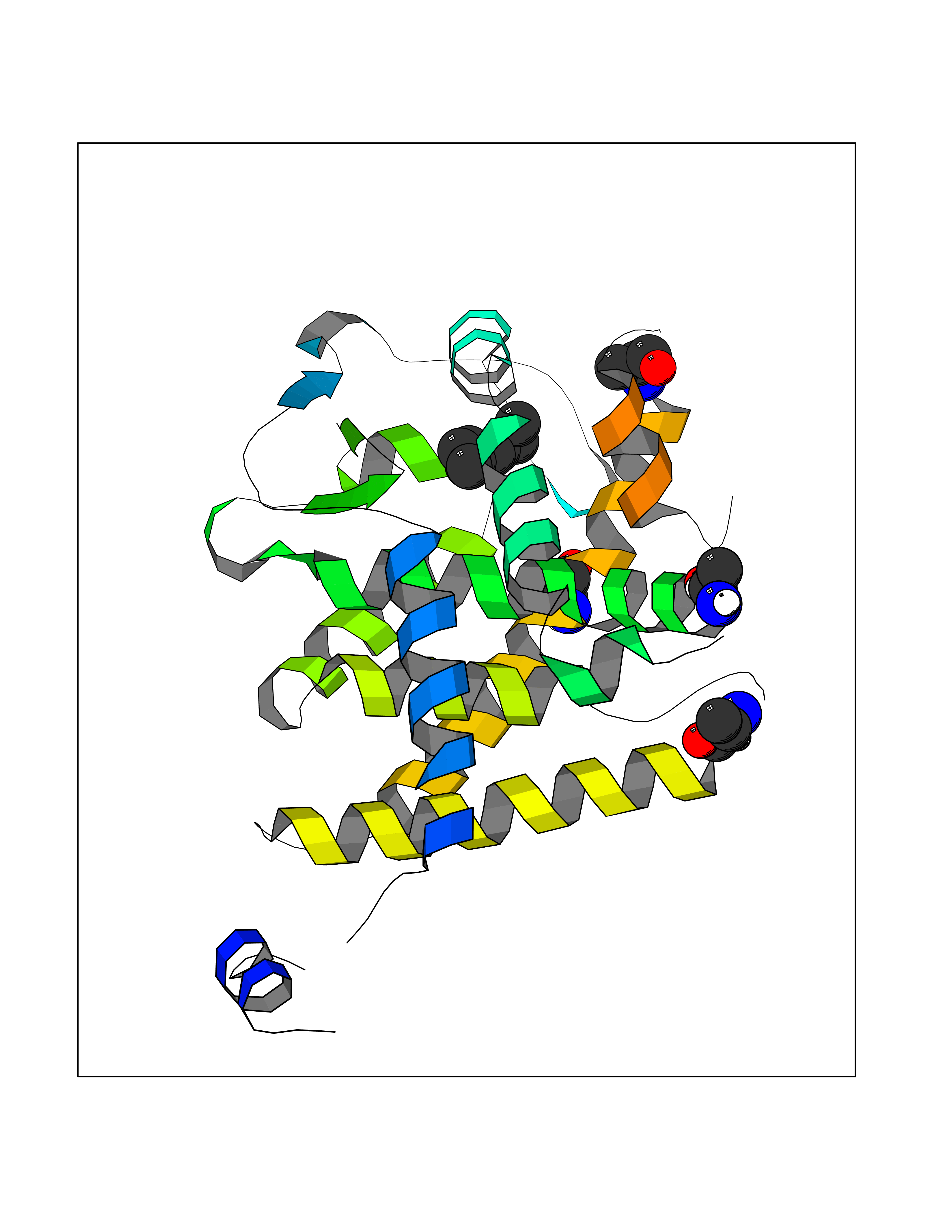 Molscript Map 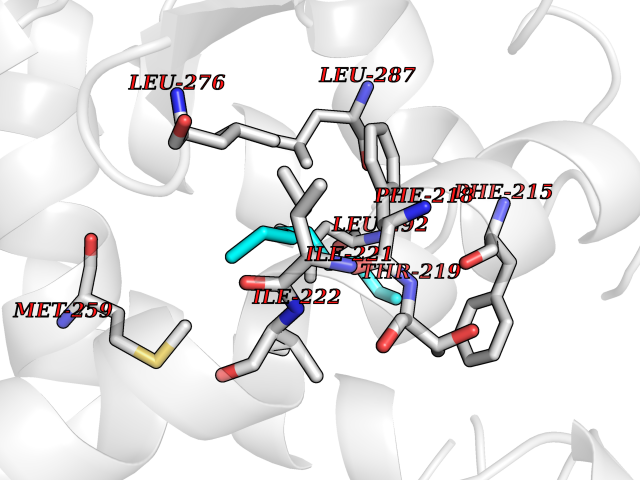 Pymol Map 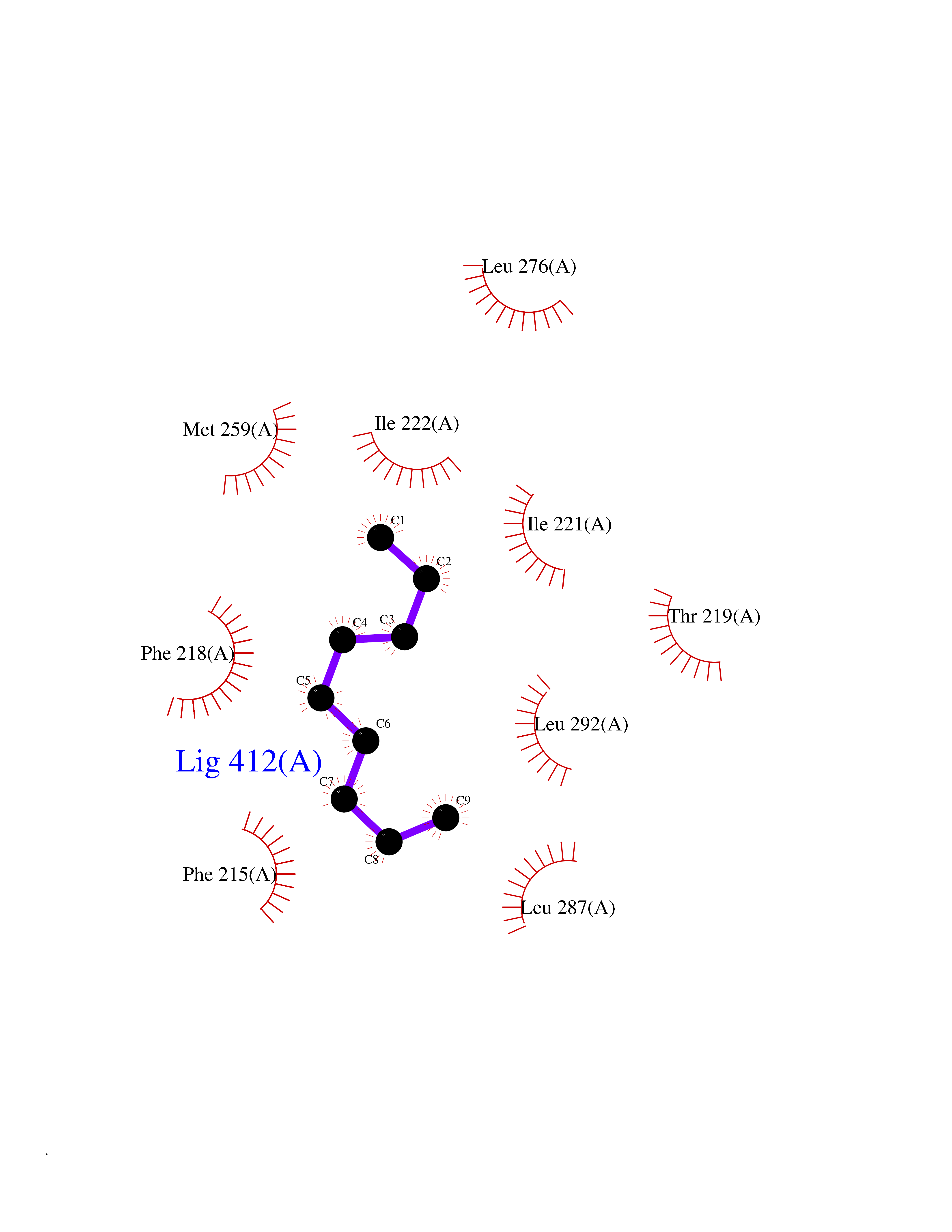 Ligplot Map 3D Binding mode Sequence GSHMEEMIRSLQQRPEPTPEEWDLIHIATEAHRSTNAQGSHWKQRRKFLPDDIGQSPIVSMPDGDKVDLEAFSEFTKIITPAITRVVDFAKKLPMFSELPXEDQIILLKGCCMEIMSLRAAVRYDPESDTLTLSGEMAVKREQLKNGGLGVVSDAIFELGKSLSAFNLDDTEVALLQAVLLMSTDRSGLLXVDKIEKSQEAYLLAFEHYVNHRKHNIPHFWPKLLMKVTDLRMIGAXHASRFLHMKVEXPTELFPPLFLEVFEDQEV Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Angiotensinogenase renin (REN) | 3K1W | 5.40 | |
Target general information Gen name REN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Renin; Angiotensinogenase Protein family Peptidase A1 family Biochemical class Peptidase Function Renin is a highly specific endopeptidase, whose only knownfunction is to generate angiotensin I from angiotensinogen in the plasma, initiating a cascade of reactions that produce an elevation of blood pressure and increased sodium retention by the kidney. Related diseases Renal tubular dysgenesis (RTD) [MIM:267430]: Autosomal recessive severe disorder of renal tubular development characterized by persistent fetal anuria and perinatal death, probably due to pulmonary hypoplasia from early-onset oligohydramnios (the Potter phenotype). {ECO:0000269|PubMed:16116425}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Tubulointerstitial kidney disease, autosomal dominant, 4 (ADTKD4) [MIM:613092]: A form of autosomal dominant tubulointerstitial kidney disease, a genetically heterogeneous disorder characterized by slowly progressive loss of kidney function, bland urinary sediment, hyperuricemia, absent or mildly increased albuminuria, lack of severe hypertension during the early stages, and normal or small kidneys on ultrasound. Renal histology shows variable abnormalities including interstitial fibrosis with tubular atrophy, microcystic dilatation of the tubules, thickening of tubular basement membranes, medullary cysts, and secondary glomerulosclerotic or glomerulocystic changes with abnormal glomerular tufting. There is significant variability, as well as incomplete penetrance. {ECO:0000269|PubMed:19664745}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07113; DB04387; DB03968; DB03736; DB03024; DB02803; DB07244; DB07174; DB08099; DB06967; DB09026; DB03395; DB02296; DB00722; DB00350; DB01844; DB04379; DB06899; DB07059; DB00212; DB05203 Interacts with P01019 EC number EC 3.4.23.15 Uniprot keywords 3D-structure; Alternative splicing; Aspartyl protease; Cleavage on pair of basic residues; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Membrane; Protease; Proteomics identification; Reference proteome; Secreted; Signal; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 36039.5 Length 328 Aromaticity 0.12 Instability index 40.22 Isoelectric point 5.51 Charge (pH=7) -6.69 2D Binding mode Binding energy (Kcal/mol) -7.37  Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence VILTNYMDTQYYGEIGIGTPPQTFKVVFDTGSSNVWVPSSKCSRLYTACVYHKLFDASDSSSYKHNGTELTLRYSTGTVSGFLSQDIITVGGITVTQMFGEVTEMPALPFMLAEFDGVVGMGFIEQAIGRVTPIFDNIISQGVLKEDVFSFYYNRDSSLGGQIVLGGSDPQHYEGNFHYINLIKTGVWQIQMKGVSVGSSTLLCEDGCLALVDTGASYISGSTSSIEKLMEALGAKKRLFDYVVKCNEGPTLPDISFHLGGKEYTLTSADYVFQESYSSKKLCTLAIHAMDIPPPTGPTWALGATFIRKFYTEFDRRNNRIGFALARH Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Nitric-oxide synthase endothelial (NOS3) | 4D1P | 5.40 | |
Target general information Gen name NOS3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Nitric oxide synthase, endothelial; NOSIII; NOS,type III; NOS type III; Endothelial nitric oxide synthase; Endothelial NOS; ENOS; EC-NOS; Constitutive NOS; CNOS Protein family NOS family Biochemical class Paired donor oxygen oxidoreductase Function NO mediates vascular endothelial growth factor (VEGF)-induced angiogenesis in coronary vessels and promotes blood clotting through the activation of platelets. Produces nitric oxide (NO) which is implicated in vascular smooth muscle relaxation through a cGMP-mediated signal transduction pathway. Related diseases Variation Asp-298 in NOS3 may be associated with susceptibility to coronary spasm. {ECO:0000269|PubMed:11740345, ECO:0000269|PubMed:9737779}. Drugs (DrugBank ID) DB07001; DB02048; DB02911; DB02335; DB01997; DB03332; DB04534; DB07244; DB03100; DB03918; DB02207; DB03065; DB00125; DB02994; DB01833; DB00155; DB00997; DB07388; DB03974; DB02077; DB01821; DB09237; DB01110; DB03144; DB03305; DB01686; DB04559; DB02044; DB08019; DB08018; DB02027; DB02979; DB00435; DB04223; DB06154; DB03910; DB02141; DB03963; DB03707; DB02234; DB04018; DB00360; DB02589 Interacts with P60709; P63010-2; Q8N6T3-3; Q9Y575-3; Q96FT7-4; Q5SZD1; Q16543; Q9UNS2; Q8IUI8; P35222; Q05193; O15287; Q08379; Q71DI3; P69905; P61978; Q12891; Q9UKT9; Q9Y2M5; Q14525; Q6DKI2; P43364-2; Q8N6F8; O94851; A4FUJ8; Q8N594; Q8IVI9; Q6X4W1-6; O15381-5; Q9NV79; Q16549; Q5T2W1; O75925; Q96I34; Q6ZMI0-5; P57052; Q9GZR2; Q96D59; Q8N6K7-2; Q9GZS3; Q8IUW3; Q7Z699; Q7Z698; P50502; Q9BR01-2; Q9NVV9; Q86WT6-2; Q9H347; P58304; Q9NZC7-5; Q9UNY5; P14079 EC number EC 1.14.13.39 Uniprot keywords 3D-structure; Alternative splicing; Calcium; Calmodulin-binding; Cell membrane; Cytoplasm; Cytoskeleton; Direct protein sequencing; FAD; Flavoprotein; FMN; Golgi apparatus; Heme; Iron; Lipoprotein; Membrane; Metal-binding; Myristate; NADP; Oxidoreductase; Palmitate; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A,B Molecular weight (Da) 90790.1 Length 803 Aromaticity 0.11 Instability index 50.67 Isoelectric point 6.03 Charge (pH=7) -9.56 2D Binding mode Binding energy (Kcal/mol) -7.36  Molscript Map  Pymol Map 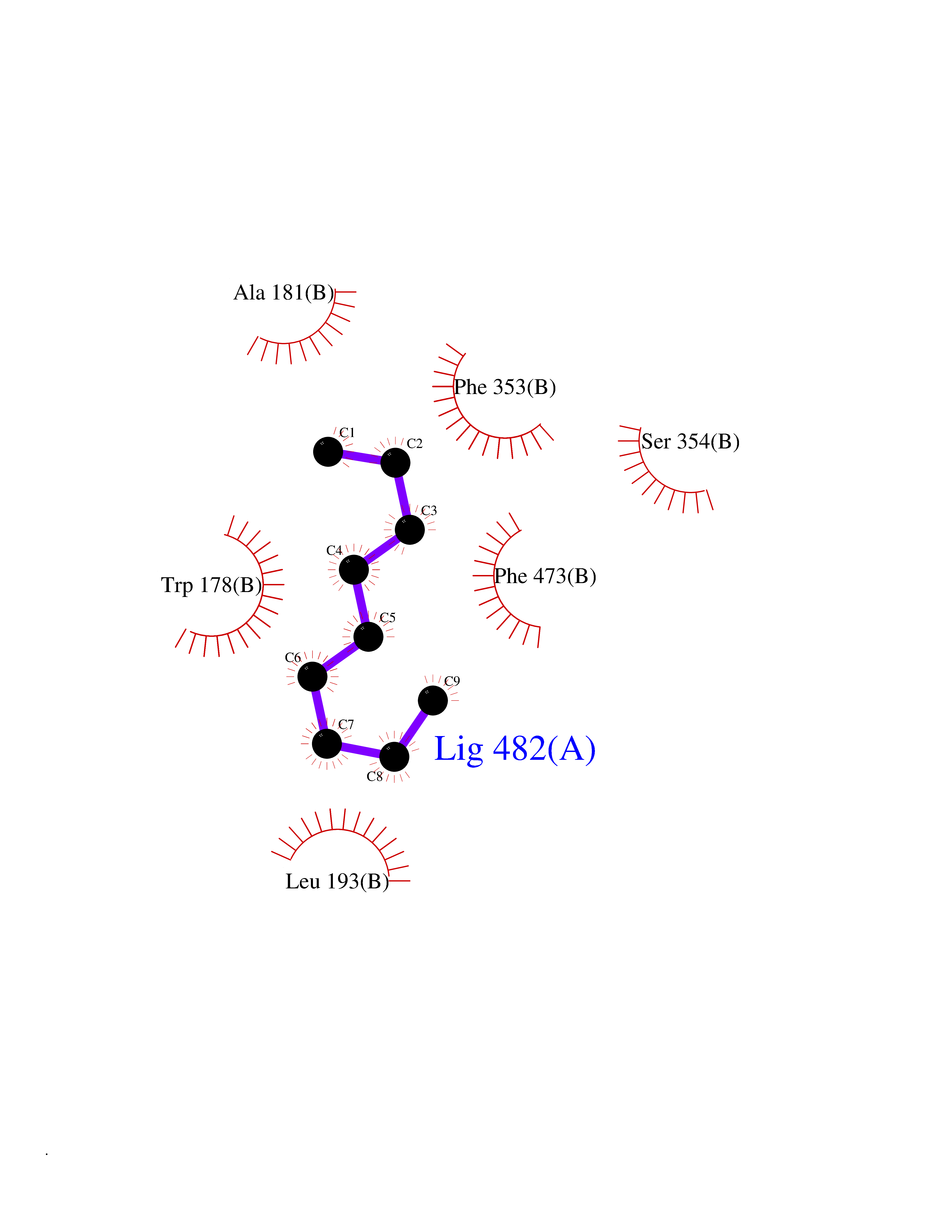 Ligplot Map 3D Binding mode Sequence FPRVKNWEVGSITYDTLSAQAQQDGPCTPRRCLGSLVFPAPEQLLSQARDFINQYYSSIKRSGSQAHEQRLQEVEAEVAATGTYQLRESELVFGAKQAWRNAPRCVGRIQWGKLQVFDARDCRSAQEMFTYICNHIKYATNRGNLRSAITVFPQRCPGRGDFRIWNSQLVRYAGYRQQDGSVRGDPANVEITELCIQHGWTPGNGRFDVLPLLLQAPDEPPELFLLPPELVLEVPLEHPTLEWFAALGLRWYALPAVSNMLLEIGGLEFPAAPFSGWYMSTEIGTRNLCDPHRYNILEDVAVCMDLDTRTTSSLWKDKAAVEINVAVLHSYQLAKVTIVDHHAATASFMKHLENEQKARGGCPADWAWIVPPISGSLTPVFHQEMVNYFLSPAFRYQPDPWKFPRVKNWEVGSITYDTLSAQAQQDGPCTPRRCLGSLVFPAPEQLLSQARDFINQYYSSIKRSGSQAHEQRLQEVEAEVAATGTYQLRESELVFGAKQAWRNAPRCVGRIQWGKLQVFDARDCRSAQEMFTYICNHIKYATNRGNLRSAITVFPQRCPGRGDFRIWNSQLVRYAGYRQQDGSVRGDPANVEITELCIQHGWTPGNGRFDVLPLLLQAPDEPPELFLLPPELVLEVPLEHPTLEWFAALGLRWYALPAVSNMLLEIGGLEFPAAPFSGWYMSTEIGTRNLCDPHRYNILEDVAVCMDLDTRTTSSLWKDKAAVEINVAVLHSYQLAKVTIVDHHAATASFMKHLENEQKARGGCPADWAWIVPPISGSLTPVFHQEMVNYFLSPAFRYQPDPW Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Thiopurine S-methyltransferase | 2BZG | 5.40 | |
Target general information Gen name TPMT Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms NA Protein family Class I-like SAM-binding methyltransferase superfamily, TPMT family Biochemical class Transferase Function Thiopurine S-methyltransferase activity. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00993; DB00436; DB01327; DB01033; DB01250; DB01021 Interacts with Q8TAP4-4; Q15047-2; P61981 EC number 2.1.1.67 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Methyltransferase; Phosphoprotein; Proteomics identification; Reference proteome; S-adenosyl-L-methionine; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 25971.5 Length 229 Aromaticity 0.12 Instability index 32.58 Isoelectric point 6.74 Charge (pH=7) -0.6 2D Binding mode Binding energy (Kcal/mol) -7.36 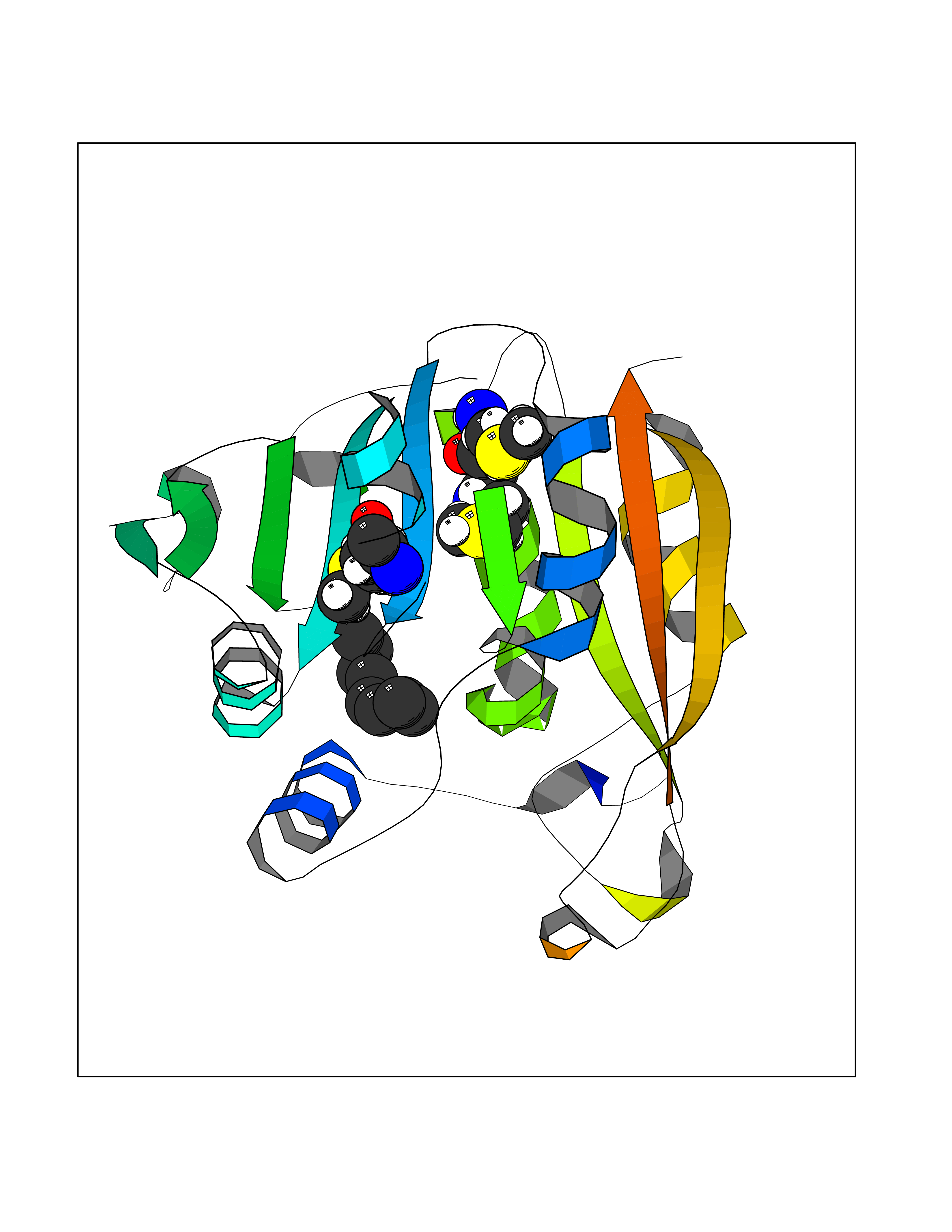 Molscript Map 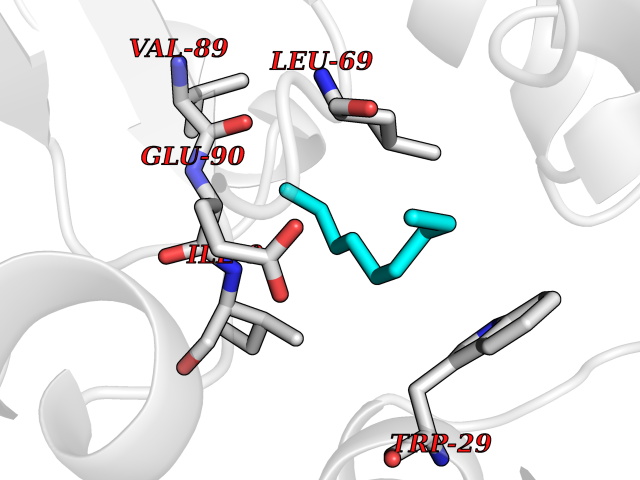 Pymol Map 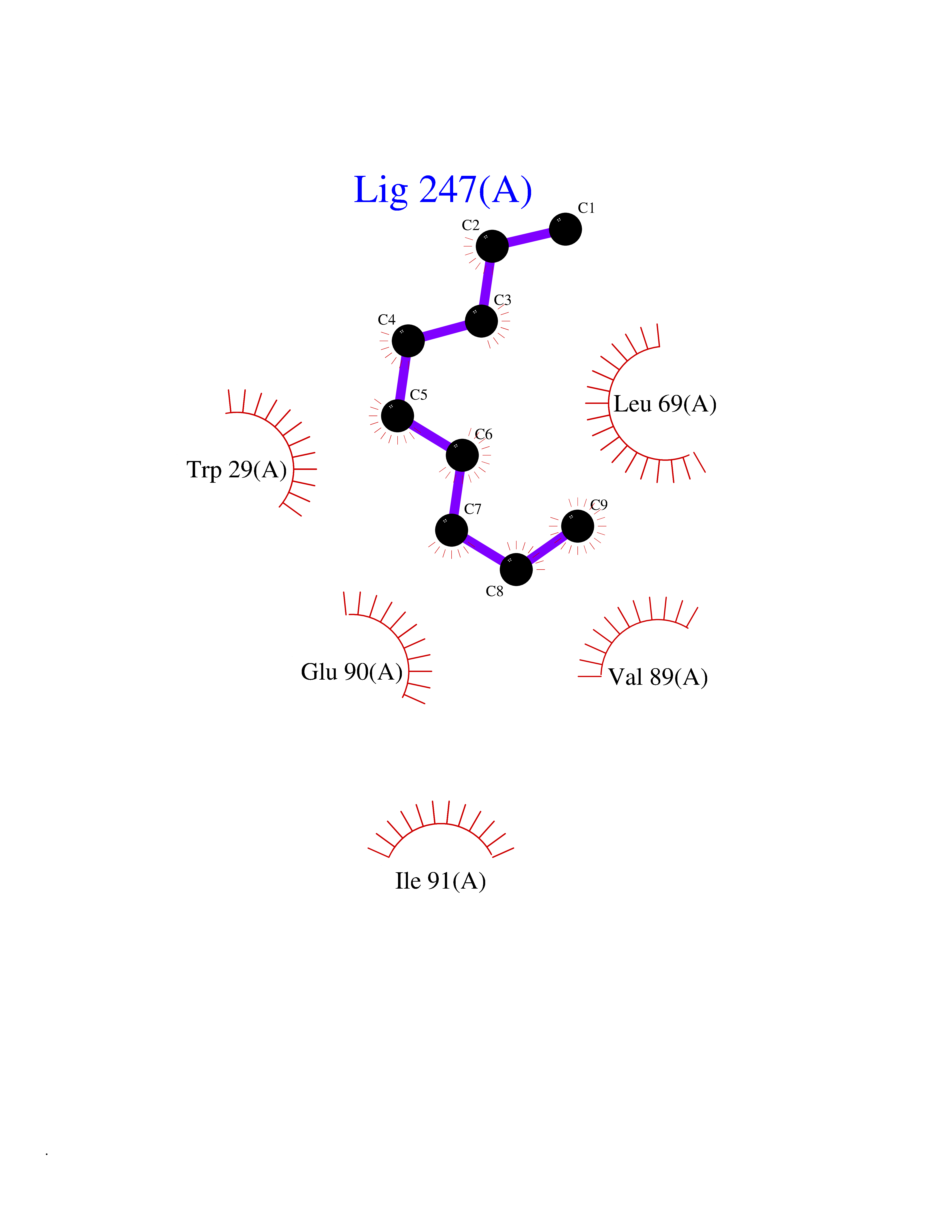 Ligplot Map 3D Binding mode Sequence EVQKNQVLTLEEWQDKWVNGKTAFHQEQGHQLLKKHLDTFLKGKSGLRVFFPLCGKAVEXKWFADRGHSVVGVEISELGIQEFFTEQNLSYSEEPITEIPGTKVFKSSSGNISLYCCSIFDLPRTNIGKFDXIWDRGALVAINPGDRKCYADTXFSLLGKKFQYLLCVLSYDPTKHPGPPFYVPHAEIERLFGKICNIRCLEKVDAFEERHKSWGIDCLFEKLYLLTEK Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | SEC14-like protein 3 | 4UYB | 5.40 | |
Target general information Gen name SEC14L3 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms TAP2 Protein family NA Biochemical class Transport protein Function Lipid binding.Transporter activity. Related diseases Chondrodysplasia with platyspondyly, distinctive brachydactyly, hydrocephaly, and microphthalmia (CDP-PBHM) [MIM:300863]: A disease characterized by chondrodysplasia, severe platyspondyly, hydrocephaly, and facial features with microphthalmia. Bone abnormalities include a distinctive metaphyseal cupping of the metacarpals, metatarsals, and phalanges. Affected females show a milder phenotype with small stature, sometimes associated with body asymmetry and mild intellectual disability. {ECO:0000269|PubMed:20181727}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB14003; DB14001; DB14002; DB11635; DB11251; DB00163 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Lipid-binding; Proteomics identification; Reference proteome; Transport Protein physicochemical properties Chain ID A Molecular weight (Da) 46148.7 Length 401 Aromaticity 0.1 Instability index 45.19 Isoelectric point 5.79 Charge (pH=7) -5.94 2D Binding mode Binding energy (Kcal/mol) -7.37 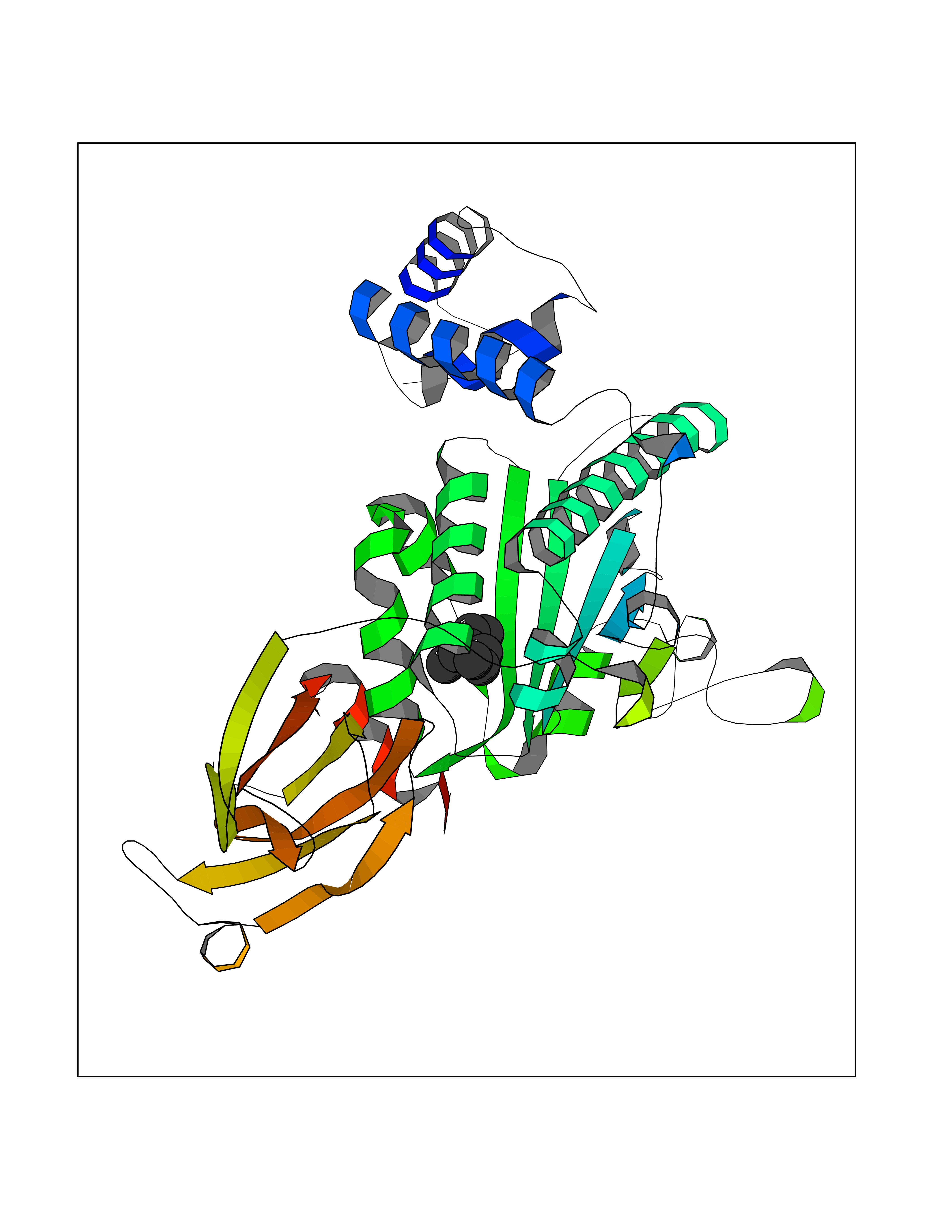 Molscript Map 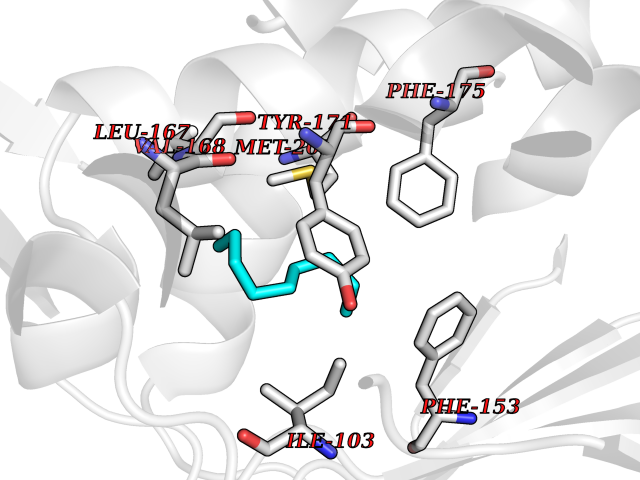 Pymol Map 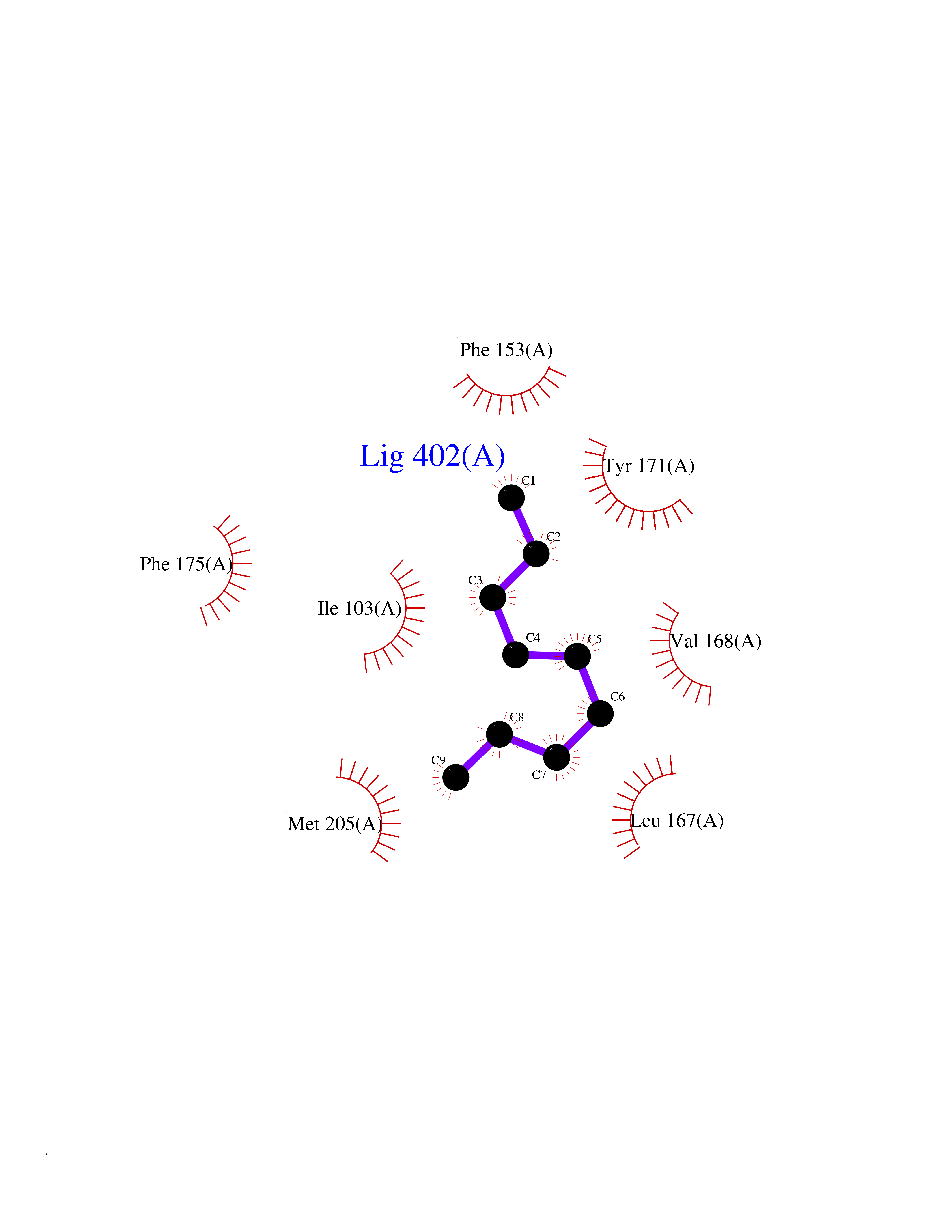 Ligplot Map 3D Binding mode Sequence SMSGRVGDLSPKQAETLAKFRENVQDVLPALPNPDDYFLLRWLRARNFDLQKSEALLRKYMEFRKTMDIDHILDWQPPEVIQKYMPGGLCGYDRDGCPVWYDIIGPLDPKGLLFSVTKQDLLKTKMRDCERILHECDLQTERLGKKIETIVMIFDCEGLGLKHFWKPLVEVYQEFFGLLEENYPETLKFMLIVKATKLFPVGYNLMKPFLSEDTRRKIIVLGNNWKEGLLKLISPEELPAQFGGTLTDPDGNPKCLTKINYGGEIPKSMYVRDQVKTQYEHSVQINRGSSHQVEYEILFPGCVLRWQFSSDGADIGFGVFLKTKMGERQRAGEMTEVLPSQRYNAHMVPEDGNLTCSEAGVYVLRFDNTYSFVHAKKVSFTVEVLLPDEGMQKYDKELTPV Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Monoglyceride lipase (MAGL) | 3PE6 | 5.40 | |
Target general information Gen name MGLL Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Monoacylglycerol lipase; MGL; Lysophospholipaselike; Lysophospholipase-like; Lysophospholipase homolog; HUK5; HU-K5 Protein family AB hydrolase superfamily, Monoacylglycerol lipase family Biochemical class Carboxylic ester hydrolase Function Hydrolyzes the endocannabinoid 2-arachidonoylglycerol, and thereby contributes to the regulation of endocannabinoid signaling, nociperception and perception of pain. Regulates the levels of fatty acids that serve as signaling molecules and promote cancer cell migration, invasion and tumor growth. Converts monoacylglycerides to free fatty acids and glycerol. Related diseases Systemic lupus erythematosus 9 (SLEB9) [MIM:610927]: A chronic, relapsing, inflammatory, and often febrile multisystemic disorder of connective tissue, characterized principally by involvement of the skin, joints, kidneys and serosal membranes. It is of unknown etiology, but is thought to represent a failure of the regulatory mechanisms of the autoimmune system. The disease is marked by a wide range of system dysfunctions, an elevated erythrocyte sedimentation rate, and the formation of LE cells in the blood or bone marrow. {ECO:0000269|PubMed:17360460}. Disease susceptibility is associated with variants affecting the gene represented in this entry.; DISEASE: Immunodeficiency, common variable, 7 (CVID7) [MIM:614699]: A primary immunodeficiency characterized by antibody deficiency, hypogammaglobulinemia, recurrent bacterial infections and an inability to mount an antibody response to antigen. The defect results from a failure of B-cell differentiation and impaired secretion of immunoglobulins; the numbers of circulating B-cells is usually in the normal range, but can be low. {ECO:0000269|PubMed:22035880}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P07550; P37235 EC number EC 3.1.1.23 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Lipid biosynthesis; Lipid degradation; Lipid metabolism; Membrane; Nitration; Phosphoprotein; Proteomics identification; Reference proteome; Serine esterase Protein physicochemical properties Chain ID A Molecular weight (Da) 31808.4 Length 289 Aromaticity 0.08 Instability index 29.7 Isoelectric point 6.73 Charge (pH=7) -0.91 2D Binding mode Binding energy (Kcal/mol) -7.37 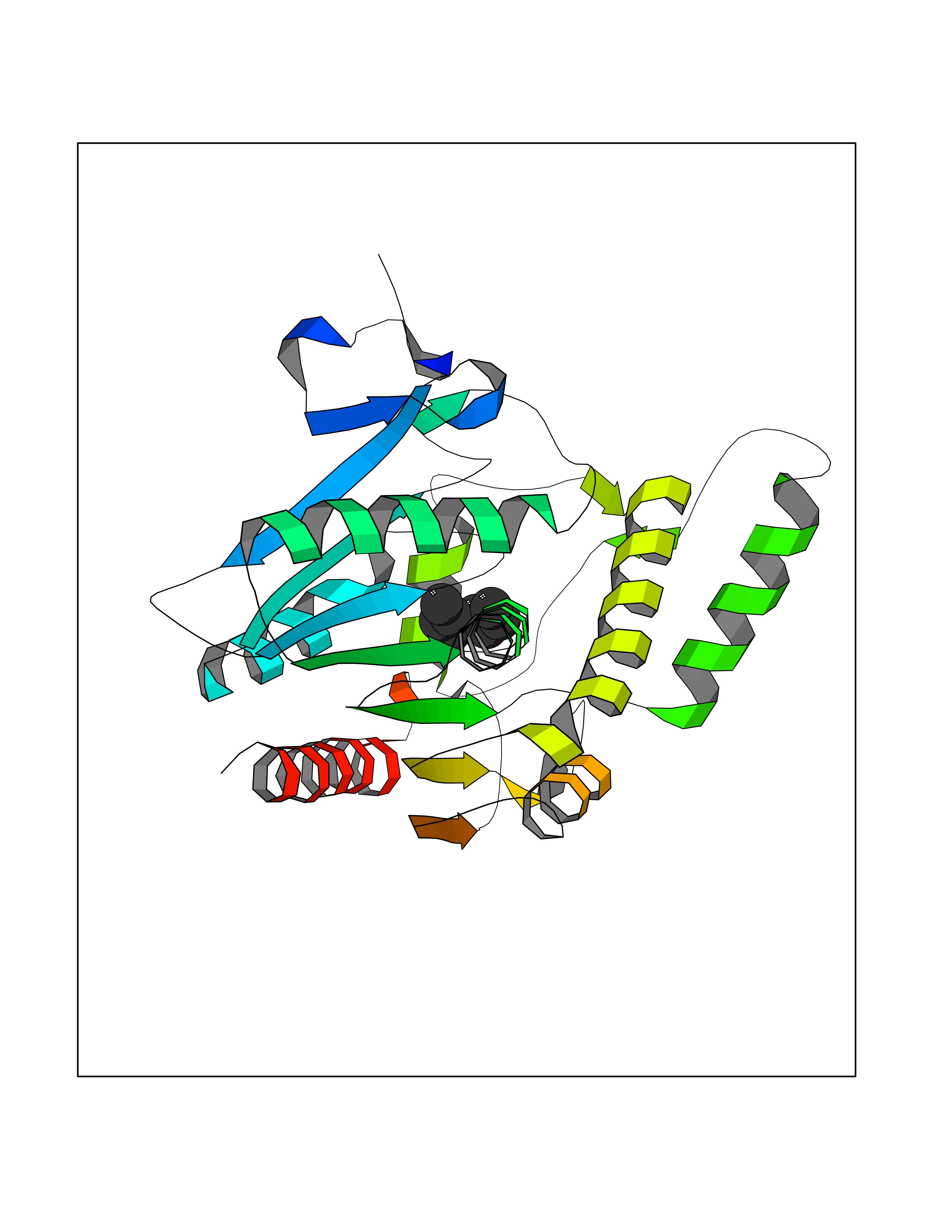 Molscript Map 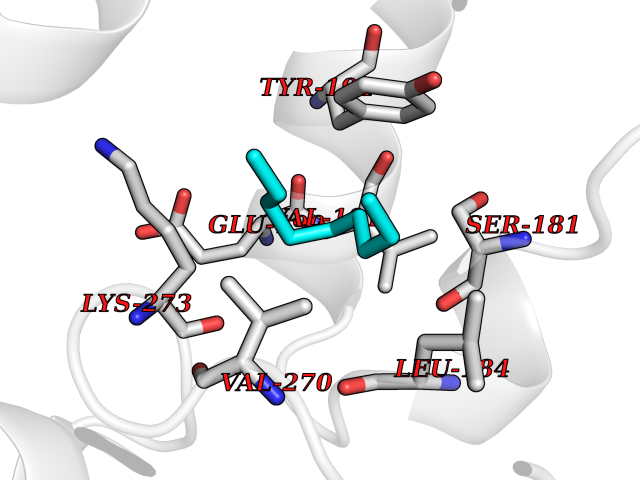 Pymol Map 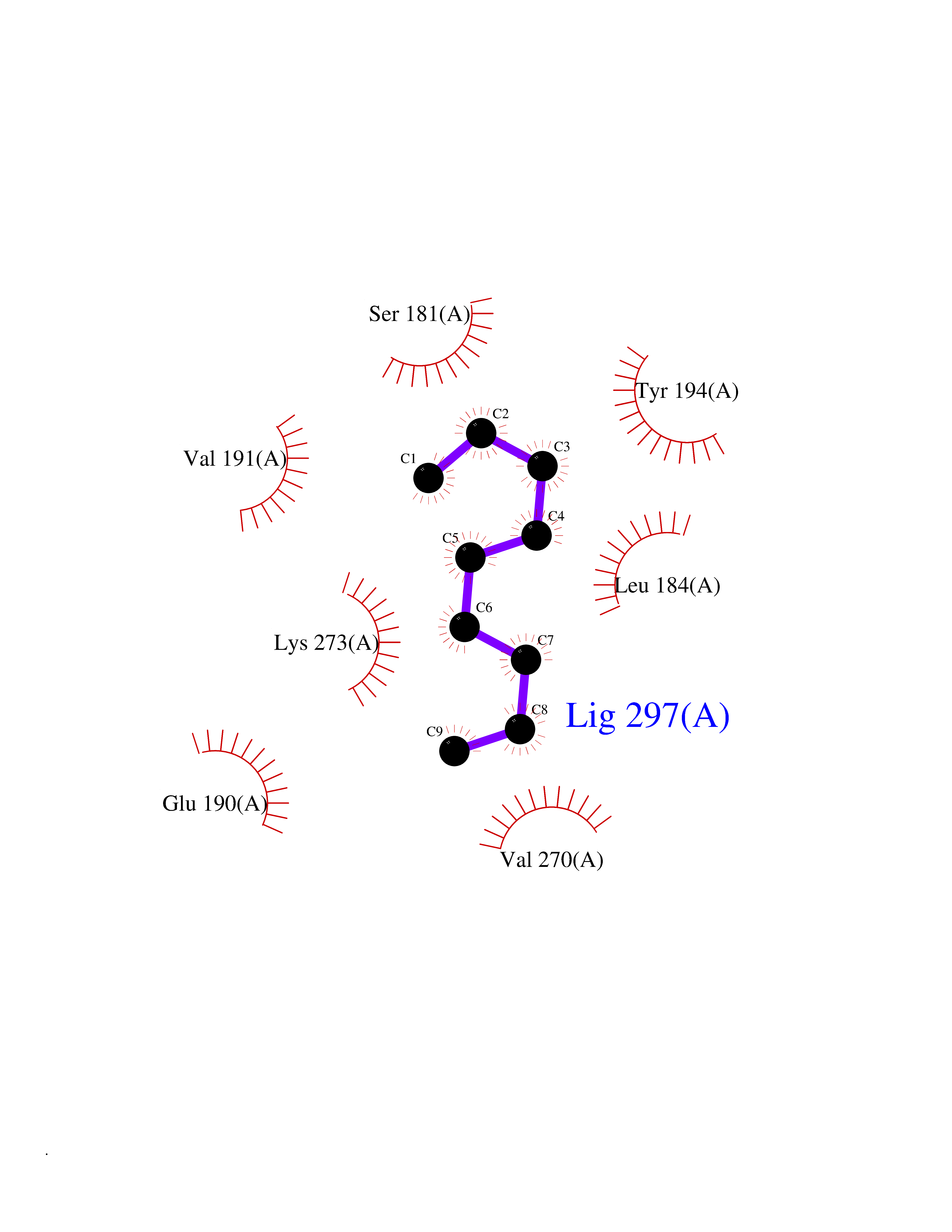 Ligplot Map 3D Binding mode Sequence PRRTPQSIPYQDLPHLVNADGQYLFCRYWAPTGTPKALIFVSHGAGEHSGRYEELARMLMGLDLLVFAHDHVGHGQSEGERMVVSDFHVFVRDVLQHVDSMQKDYPGLPVFLLGHSMGGAIAILTAAERPGHFAGMVLISPLVLANPESATTFKVLAAKVLNSVLPNLSSGPIDSSVLSRNKTEVDIYNSDPLICRAGLKVCFGIQLLNAVSRVERALPKLTVPFLLLQGSADRLCDSKGAYLLMELAKSQDKTLKIYEGAYHVLHKELPEVTNSVFHEINMWVSQRTA Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | BUB1 mitotic checkpoint serine/threonine kinase (BUB1) | 6F7B | 5.40 | |
Target general information Gen name BUB1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms hBUB1; Mitotic checkpoint serine/threonine-protein kinase BUB1; BUB1L; BUB1A Protein family Protein kinase superfamily, Ser/Thr protein kinase family, BUB1 subfamily Biochemical class Kinase Function Has a key role in the assembly of checkpoint proteins at the kinetochore, being required for the subsequent localization of CENPF, BUB1B, CENPE and MAD2L1. Required for the kinetochore localization of PLK1. Required for centromeric enrichment of AUKRB in prometaphase. Plays an important role in defining SGO1 localization and thereby affects sister chromatid cohesion. Acts as a substrate for anaphase-promoting complex or cyclosome (APC/C) in complex with its activator CDH1 (APC/C-Cdh1). Necessary for ensuring proper chromosome segregation and binding to BUB3 is essential for this function. Can regulate chromosome segregation in a kinetochore-independent manner. Can phosphorylate BUB3. The BUB1-BUB3 complex plays a role in the inhibition of APC/C when spindle-assembly checkpoint is activated and inhibits the ubiquitin ligase activity of APC/C by phosphorylating its activator CDC20. This complex can also phosphorylate MAD1L1. Kinase activity is essential for inhibition of APC/CCDC20 and for chromosome alignment but does not play a major role in the spindle-assembly checkpoint activity. Mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. Serine/threonine-protein kinase that performs 2 crucial functions during mitosis: it is essential for spindle-assembly checkpoint signaling and for correct chromosome alignment. Related diseases Microcephaly 30, primary, autosomal recessive (MCPH30) [MIM:620183]: A form of microcephaly, a disease defined as a head circumference more than 3 standard deviations below the age, sex and ethnically matched mean. Brain weight is markedly reduced and the cerebral cortex is disproportionately small. MCPH30 is characterized by small head, poor overall growth, and global developmental delay with variably impaired intellectual development. Affected individuals may also have variable congenital anomalies, including atrial septal defect, dysmorphic facial features, tracheal stenosis, and anomalies of the skin and teeth. {ECO:0000269|PubMed:35044816}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with O95376; O60566; O43684; P46108; Q8NG31; Q8NG31-2; Q9GZQ8; P03070 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; Apoptosis; ATP-binding; Cell cycle; Cell division; Centromere; Chromosome; Chromosome partition; Host-virus interaction; Intellectual disability; Kinase; Kinetochore; Mitosis; Nucleotide-binding; Nucleus; Phosphoprotein; Primary microcephaly; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 39564.8 Length 343 Aromaticity 0.12 Instability index 31.17 Isoelectric point 8.49 Charge (pH=7) 4.7 2D Binding mode Binding energy (Kcal/mol) -7.36 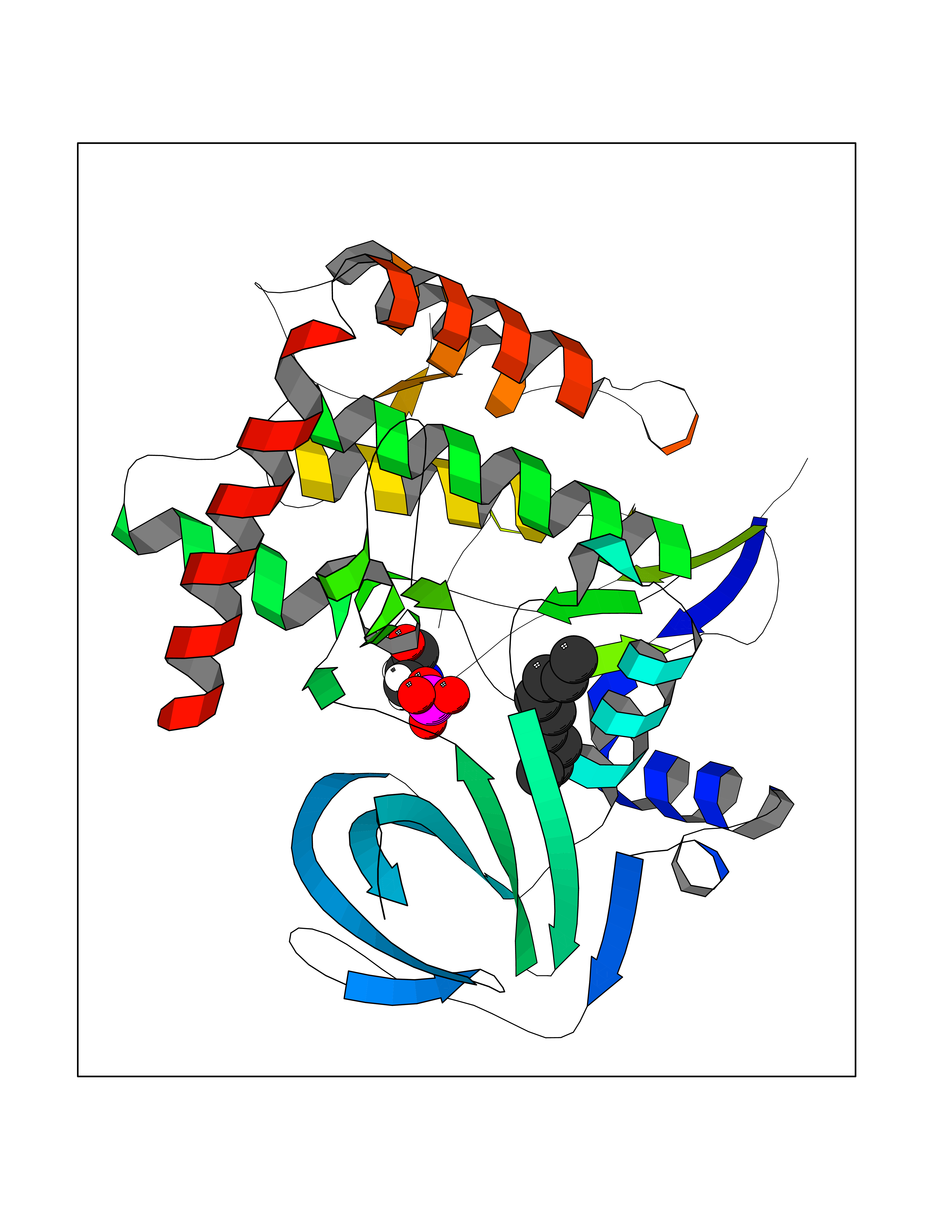 Molscript Map 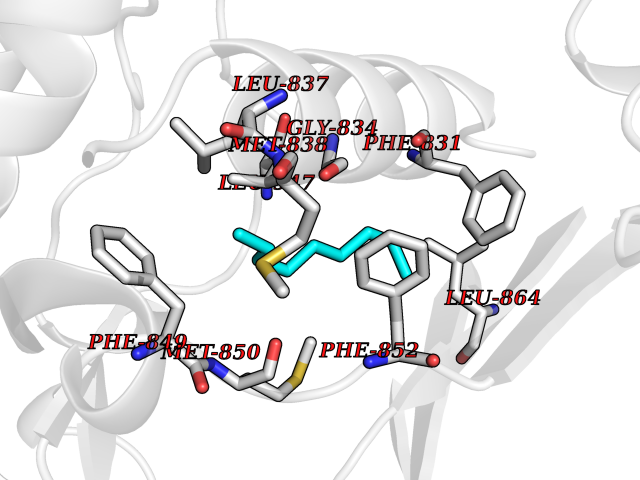 Pymol Map 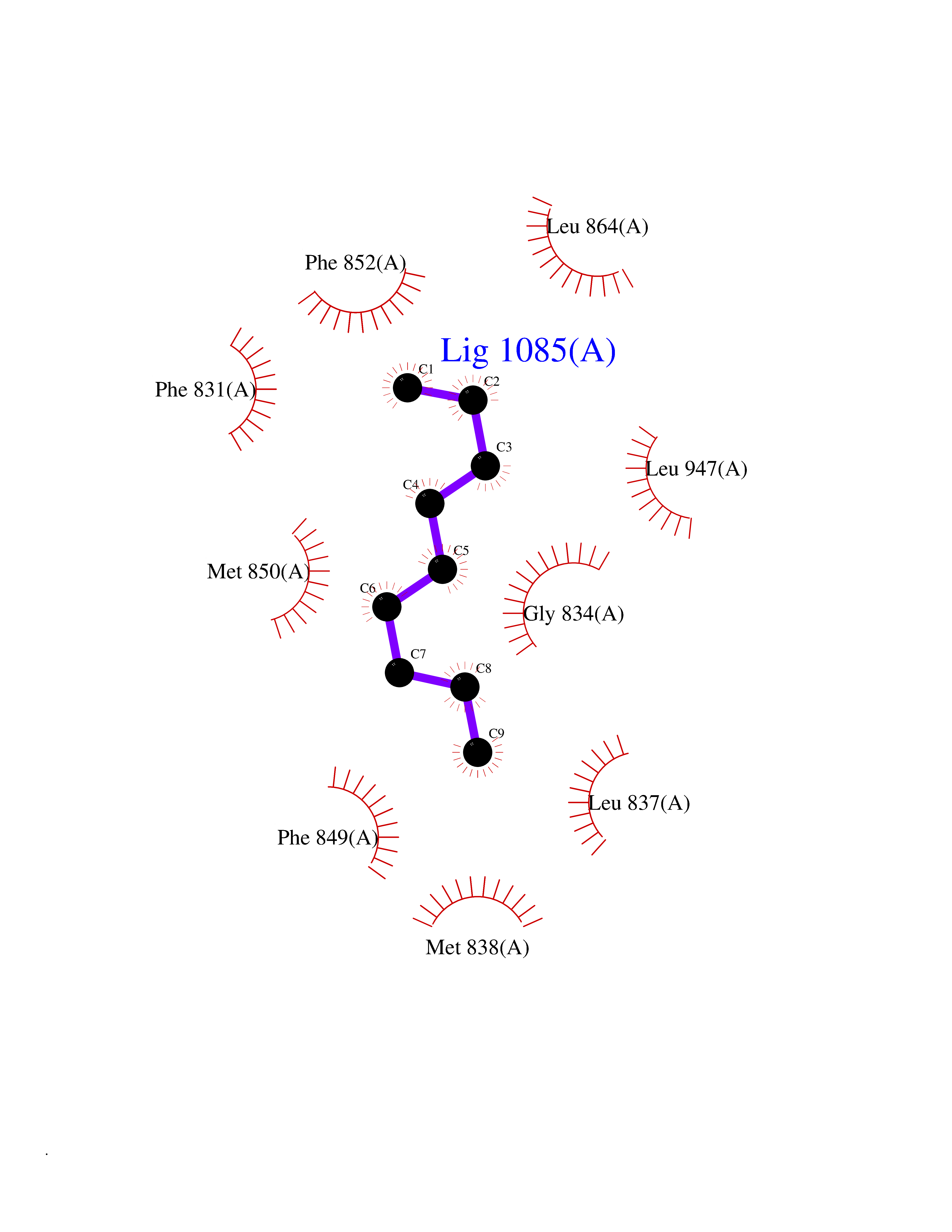 Ligplot Map 3D Binding mode Sequence APNFIVGNPWDDKLIFKLLSGLSKPVSSYPNTFEWQCKLPAIKPKTEFQLGSKLVYVHHLLGEGAFAQVYEATQKNKQKFVLKVQKPANPWEFYIGTQLMERLKPSMQHMFMKFYSAHLFQNGSVLVGELYSYGTLLNAINLYKNTPEKVMPQGLVISFAMRMLYMIEQVHDCEIIHGDIKPDNFILGNGFLEQDDEDDLSAGLALIDLGQSIDMKLFPKGTIFTAKCETXGFQCVEMLSNKPWNYQIDYFGVAATVYCMLFGTYMKVKNEECKPEGLFRRLPHLDMWNEFFHVMLNIPDCHHLPSLDLLRQKLKKVFQQHYTNKIRALRNRLIVLLLECKRS Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | p53-binding protein Mdm4 (MDM4) | 6Q9Y | 5.40 | |
Target general information Gen name MDM4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein Mdmx; Mdm2-like p53-binding protein; Double minute 4 protein Protein family MDM2/MDM4 family Biochemical class MDM2/MDM4 family Function Inhibits p53/TP53- and TP73/p73-mediated cell cycle arrest and apoptosis by binding its transcriptional activation domain. Inhibits degradation of MDM2. Can reverse MDM2-targeted degradation of TP53 while maintaining suppression of TP53 transactivation and apoptotic functions. Related diseases Bone marrow failure syndrome 6 (BMFS6) [MIM:618849]: A form of bone marrow failure syndrome, a heterogeneous group of life-threatening disorders characterized by hematopoietic defects in association with a range of variable extra-hematopoietic manifestations. BMFS6 is an autosomal dominant form characterized by intermittent neutropenia, lymphopenia, or anemia associated with hypocellular bone marrow, and increased susceptibility to cancer. {ECO:0000269|PubMed:32300648}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q9NX04; P10415; Q7Z479; O95971; P48729; Q00987; Q13064; P41227; P06400; Q9Y4L5; P23297; P29034; P33763; P04271; P31947; P04637; P62837; Q93009; O14972; P61964; P62258; P61981; P63104; Q9BRR0; A0A0S2Z6X0; Q3YBA8; P03255-2 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Disease variant; Metal-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation; Zinc; Zinc-finger Protein physicochemical properties Chain ID A,B Molecular weight (Da) 19722 Length 173 Aromaticity 0.08 Instability index 50.78 Isoelectric point 8.48 Charge (pH=7) 2.27 2D Binding mode Binding energy (Kcal/mol) -7.36 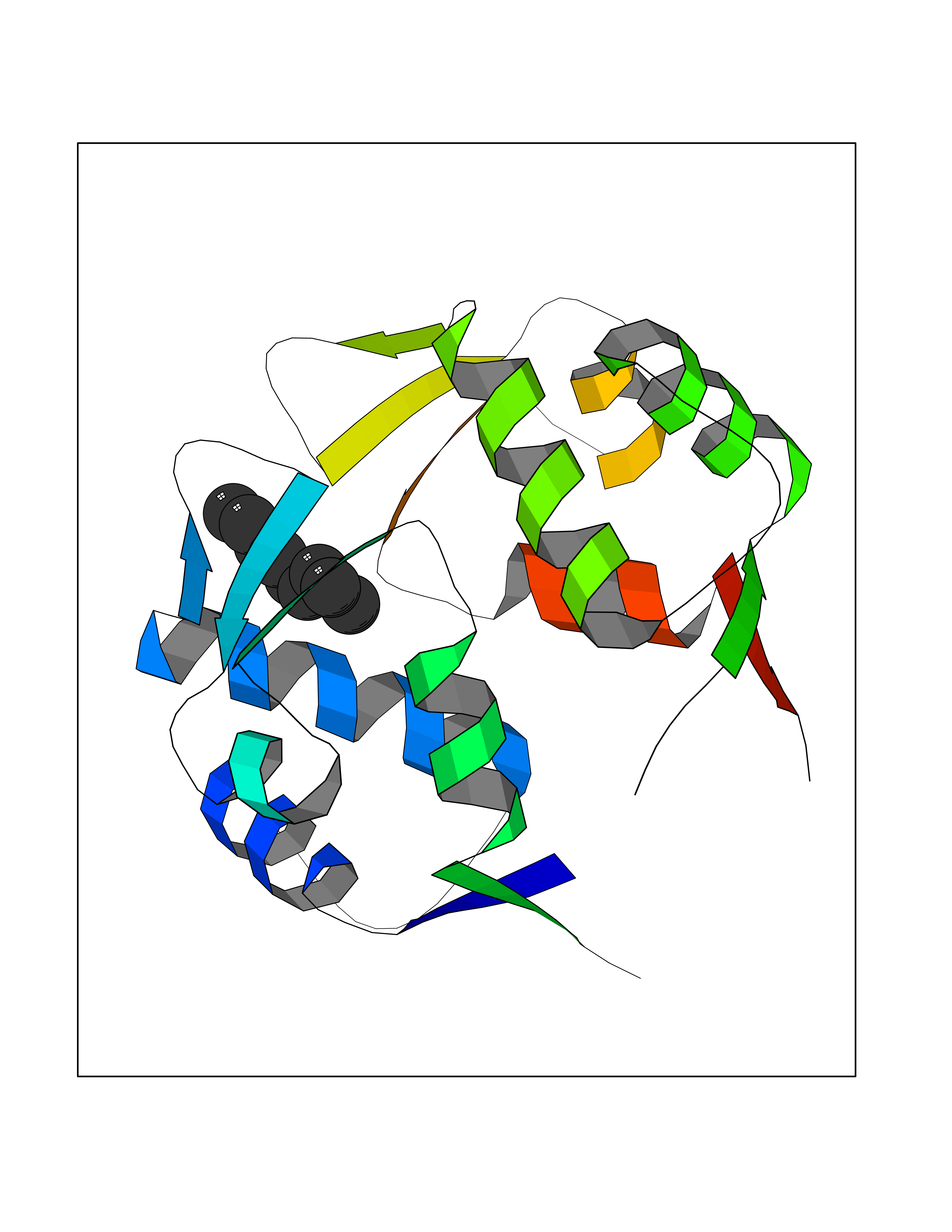 Molscript Map 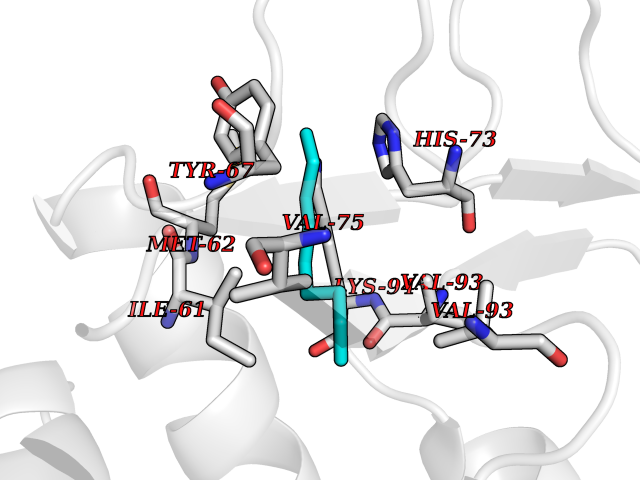 Pymol Map 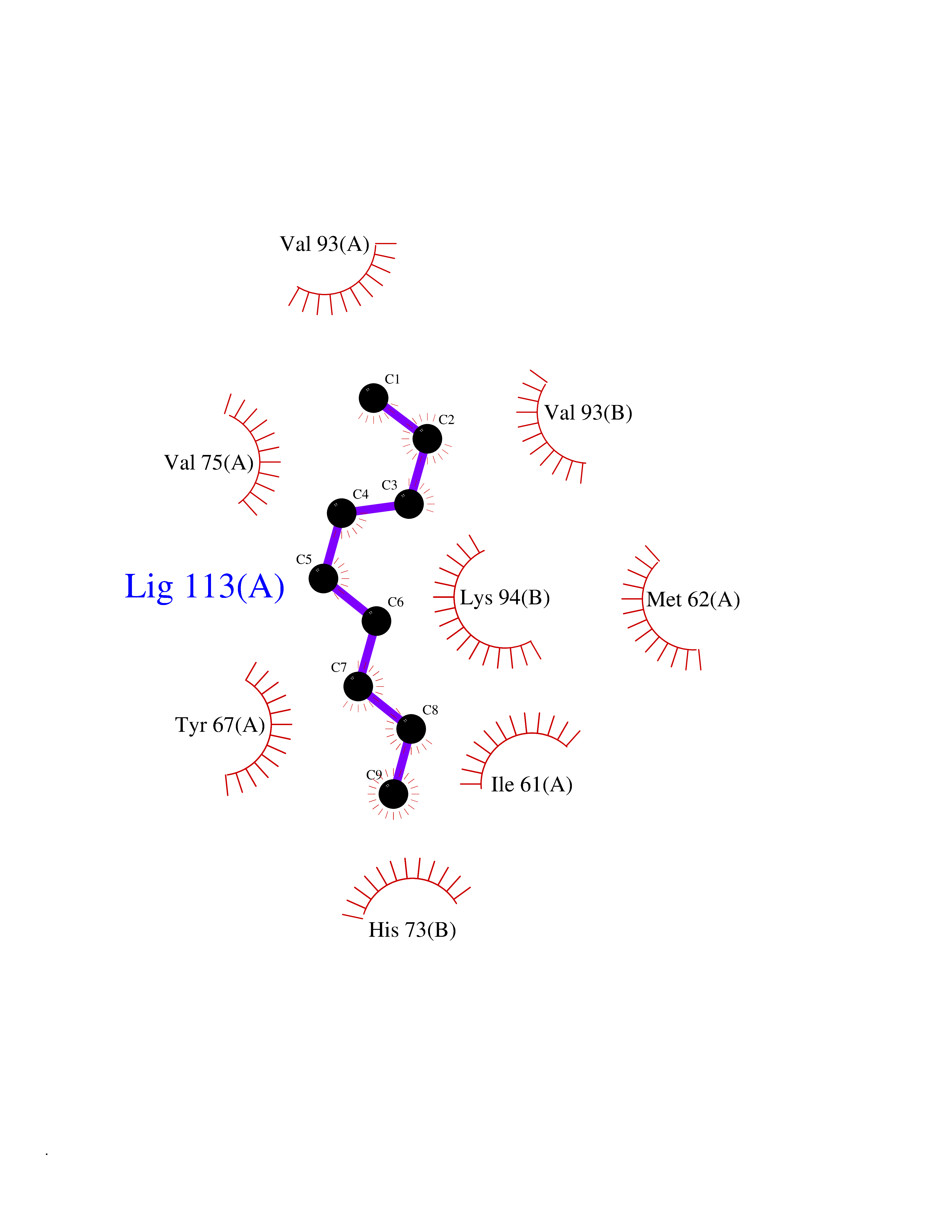 Ligplot Map 3D Binding mode Sequence QVRPKLPLLKILHAAGAQGEMFTVKEVMHYLGQYIMVKQLYDQQEQHMVYCGGDLLGELLGRQSFSVKDPSPLYDMLRKNLVTLAQINQVRPKLPLLKILHAAGAQGEMFTVKEVMHYLGQYIMVKQLYDQQEQHMVYCGGDLLGELLGRQSFSVKDPSPLYDMLRKNLVTLA Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Toxoplasma Calcium-dependent protein kinase 1 (Tg CDPK1) | 6BFA | 5.40 | |
Target general information Gen name Tg CDPK1 Organism Toxoplasma gondii (strain ATCC 50861 / VEG) Uniprot ID TTD ID Synonyms CAM kinase, CDPK family TgCDPK1 Protein family Protein kinase superfamily, Ser/Thr protein kinase family, CDPK subfamily Biochemical class NA Function Provides the mechanistic link between calcium signalling and motility, differentiation and invasion. Required for the microneme secretion at the apical complex and parasite proliferation. Related diseases Factor XI deficiency (FA11D) [MIM:612416]: A hemorrhagic disease characterized by reduced levels and activity of factor XI resulting in moderate bleeding symptoms, usually occurring after trauma or surgery. Patients usually do not present spontaneous bleeding but women can present with menorrhagia. Hemorrhages are usually moderate. {ECO:0000269|PubMed:10027710, ECO:0000269|PubMed:10606881, ECO:0000269|PubMed:11895778, ECO:0000269|PubMed:15026311, ECO:0000269|PubMed:15180874, ECO:0000269|PubMed:1547342, ECO:0000269|PubMed:15953011, ECO:0000269|PubMed:16607084, ECO:0000269|PubMed:18005151, ECO:0000269|PubMed:21457405, ECO:0000269|PubMed:21668437, ECO:0000269|PubMed:21999818, ECO:0000269|PubMed:22016685, ECO:0000269|PubMed:22159456, ECO:0000269|PubMed:22322133, ECO:0000269|PubMed:25158988, ECO:0000269|PubMed:2813350, ECO:0000269|PubMed:7669672, ECO:0000269|PubMed:7888672, ECO:0000269|PubMed:9401068, ECO:0000269|PubMed:9787168}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; ATP-binding; Calcium; Kinase; Metal-binding; Nucleotide-binding; Serine/threonine-protein kinase; Transferase Protein physicochemical properties Chain ID A Molecular weight (Da) 53029.9 Length 464 Aromaticity 0.09 Instability index 29.74 Isoelectric point 5.61 Charge (pH=7) -8.87 2D Binding mode Binding energy (Kcal/mol) -7.36 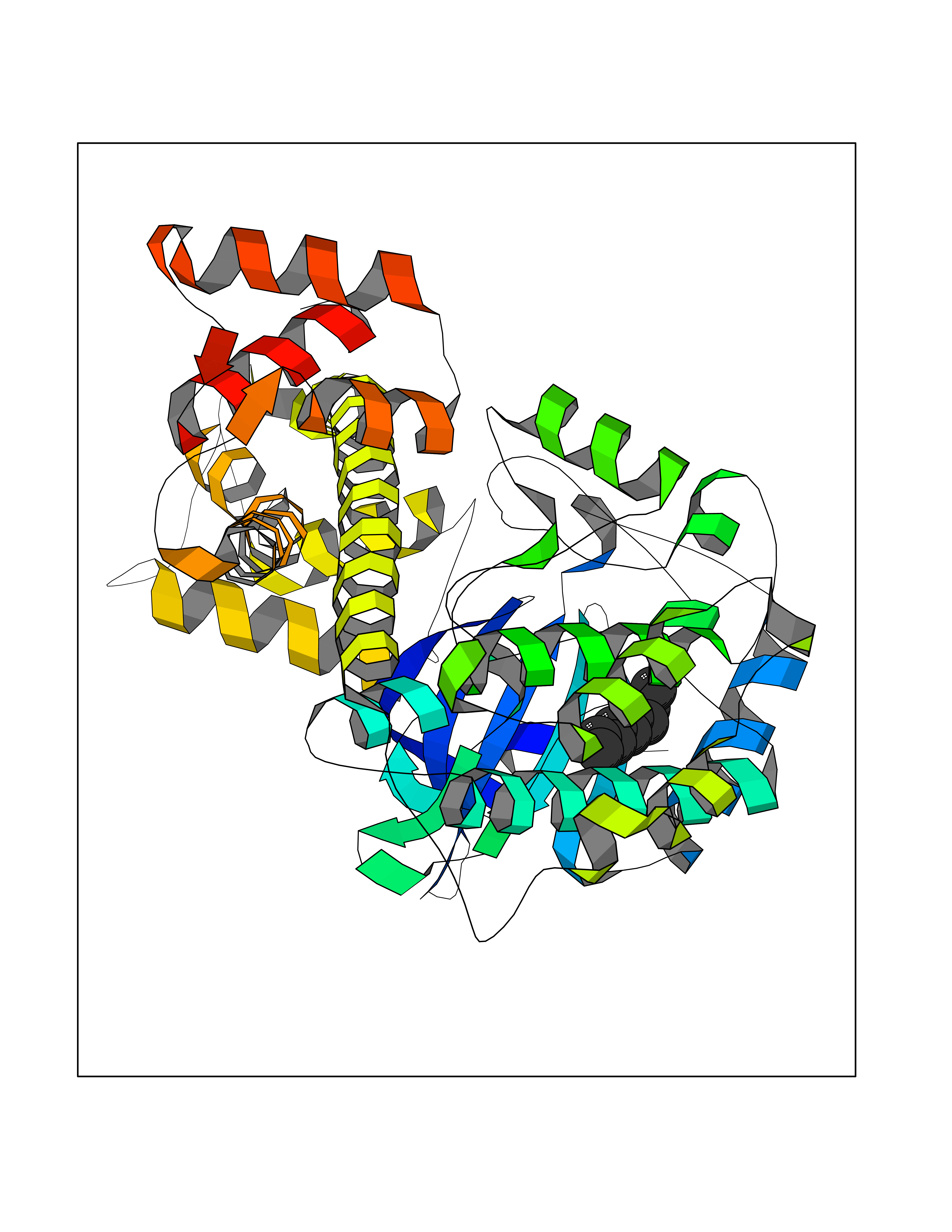 Molscript Map 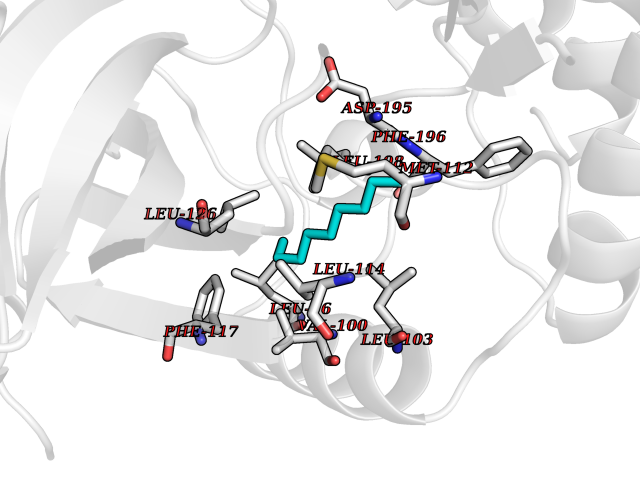 Pymol Map 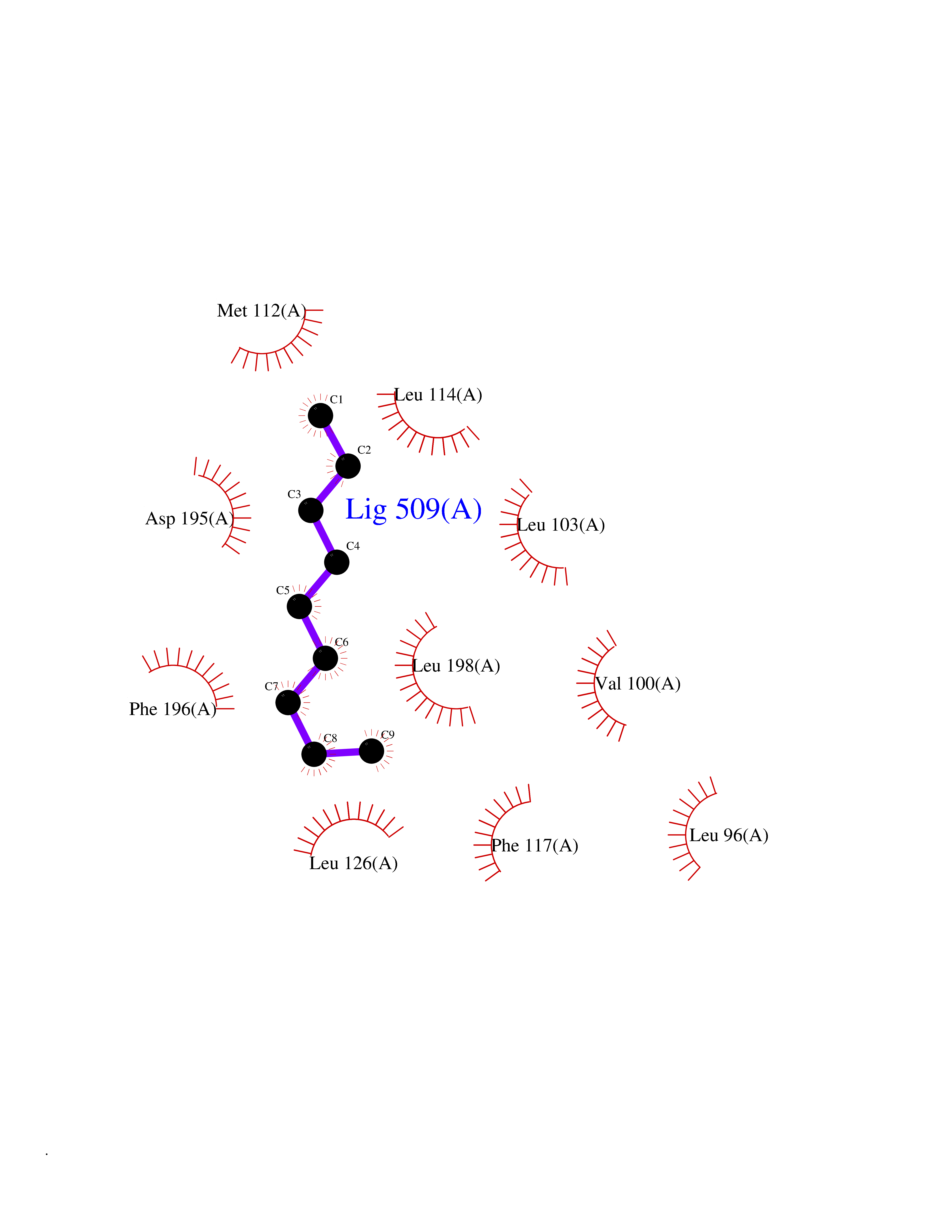 Ligplot Map 3D Binding mode Sequence TAIFSDRYKGQRVLGKGSFGEVILCKDKITGQECAVKVISKRQVKQKTDKESLLREVQLLKQLDHPNIMKLYEFFEDKGYFYLVGEVYTGGELFDEIISRKRFSEVDAARIIRQVLSGITYMHKNKIVHRDLKPENLLLESKSKDANIRIIDFGLSTHFEASKKMKDKIGTAYYIAPEVLHGTYDEKCDVWSTGVILYILLSGCPPFNGANEYDILKKVEKGKYTFELPQWKKVSESAKDLIRKMLTYVPSMRISARDALDHEWIQTYTKEQISVDVPSLDNAILNIRQFQGTQKLAQAALLYMGSKLTSQDETKELTAIFHKMDKNGDGQLDRAELIEGYKELMRMKGQDASMLDASAVEHEVDQVLDAVDFDKNGYIEYSEFVTVAMDRKTLLSRERLERAFRMFDSDNSGKISSTELATIFGVSDVDSETWKSVLSEVDKNNDGEVDFDEFQQMLLKLCGN Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Histone-lysine N-methyltransferase EHMT2 (EHMT2) | 5VSC | 5.40 | |
Target general information Gen name EHMT2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein G9a; NG36; Lysine N-methyltransferase 1C; KMT1C; Histone H3-K9 methyltransferase 3; HLA-B-associated transcript 8; H3-K9-HMTase 3; G9A; Euchromatic histone-lysine N-methyltransferase 2; C6orf3 Protein family Class V-like SAM-binding methyltransferase superfamily, Histone-lysine methyltransferase family, Suvar3-9 subfamily Biochemical class Methyltransferase Function H3K9me represents a specific tag for epigenetic transcriptional repression by recruiting HP1 proteins to methylated histones. Also mediates monomethylation of 'Lys-56' of histone H3 (H3K56me1) in G1 phase, leading to promote interaction between histone H3 and PCNA and regulating DNA replication. Also weakly methylates 'Lys-27' of histone H3 (H3K27me). Also required for DNA methylation, the histone methyltransferase activity is not required for DNA methylation, suggesting that these 2 activities function independently. Probably targeted to histone H3 by different DNA-binding proteins like E2F6, MGA, MAX and/or DP1. May also methylate histone H1. In addition to the histone methyltransferase activity, also methylates non-histone proteins: mediates dimethylation of 'Lys-373' of p53/TP53. Also methylates CDYL, WIZ, ACIN1, DNMT1, HDAC1, ERCC6, KLF12 and itself. Histone methyltransferase that specifically mono- and dimethylates 'Lys-9' of histone H3 (H3K9me1 and H3K9me2, respectively) in euchromatin. Related diseases Pseudohypoaldosteronism 2C (PHA2C) [MIM:614492]: An autosomal dominant disorder characterized by severe hypertension, hyperkalemia, hyperchloremia, mild hyperchloremic metabolic acidosis in some cases, and correction of physiologic abnormalities by thiazide diuretics. {ECO:0000269|PubMed:11498583}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neuropathy, hereditary sensory and autonomic, 2A (HSAN2A) [MIM:201300]: A form of hereditary sensory and autonomic neuropathy, a genetically and clinically heterogeneous group of disorders characterized by degeneration of dorsal root and autonomic ganglion cells, and by sensory and/or autonomic abnormalities. HSAN2A is an autosomal recessive disorder characterized by impairment of pain, temperature and touch sensation, onset of symptoms in infancy or early childhood, occurrence of distal extremity pathologies (paronychia, whitlows, ulcers, and Charcot joints), frequent amputations, sensory loss that affects all modalities of sensation (lower and upper limbs and perhaps the trunk as well), absence or diminution of tendon reflexes (usually in all limbs), minimal autonomic dysfunction, absence of sensory nerve action potentials, and virtual absence of myelinated fibers with decreased numbers of unmyelinated fibers in sural nerves. {ECO:0000269|PubMed:15060842, ECO:0000269|PubMed:15911806, ECO:0000269|PubMed:18521183}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with Q6VMQ6-2; Q6P1J9; Q9UBC3; P38919; Q9UM22; P23771; Q99684; Q13547; Q96JB3; Q92831; O60341-1; Q9Y4X4; P57682; Q13330; O94776; Q9BTC8; P20592; Q9BSU3; Q99801-1; O60568; Q9NQX1; Q5JSZ5; Q7Z3Z2; Q9P2R6; Q14119; Q96GT9; O60315; Q9NWS9-2; Q96JM2; A0A0S2Z5X4; Q96BV0; Q96EG3; Q07120; O60341-1 EC number EC 2.1.1.- Uniprot keywords 3D-structure; Acetylation; Alternative splicing; ANK repeat; Chromatin regulator; Chromosome; Isopeptide bond; Metal-binding; Methylation; Methyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; S-adenosyl-L-methionine; Transferase; Ubl conjugation; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 31010.9 Length 269 Aromaticity 0.1 Instability index 47.49 Isoelectric point 5.16 Charge (pH=7) -9.31 2D Binding mode Binding energy (Kcal/mol) -7.37 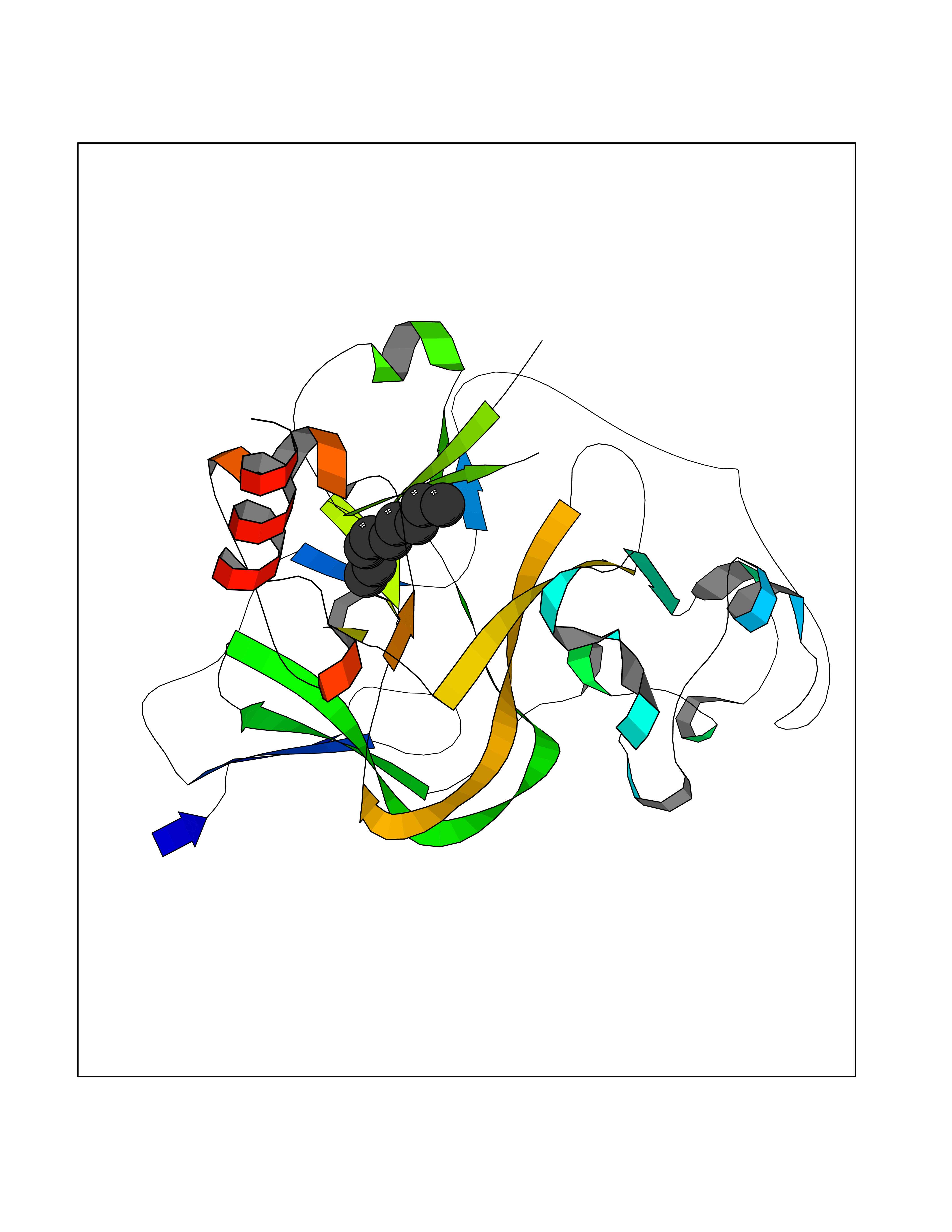 Molscript Map 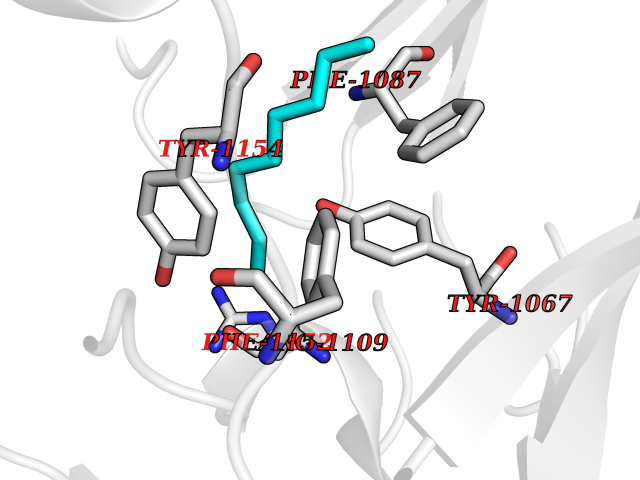 Pymol Map 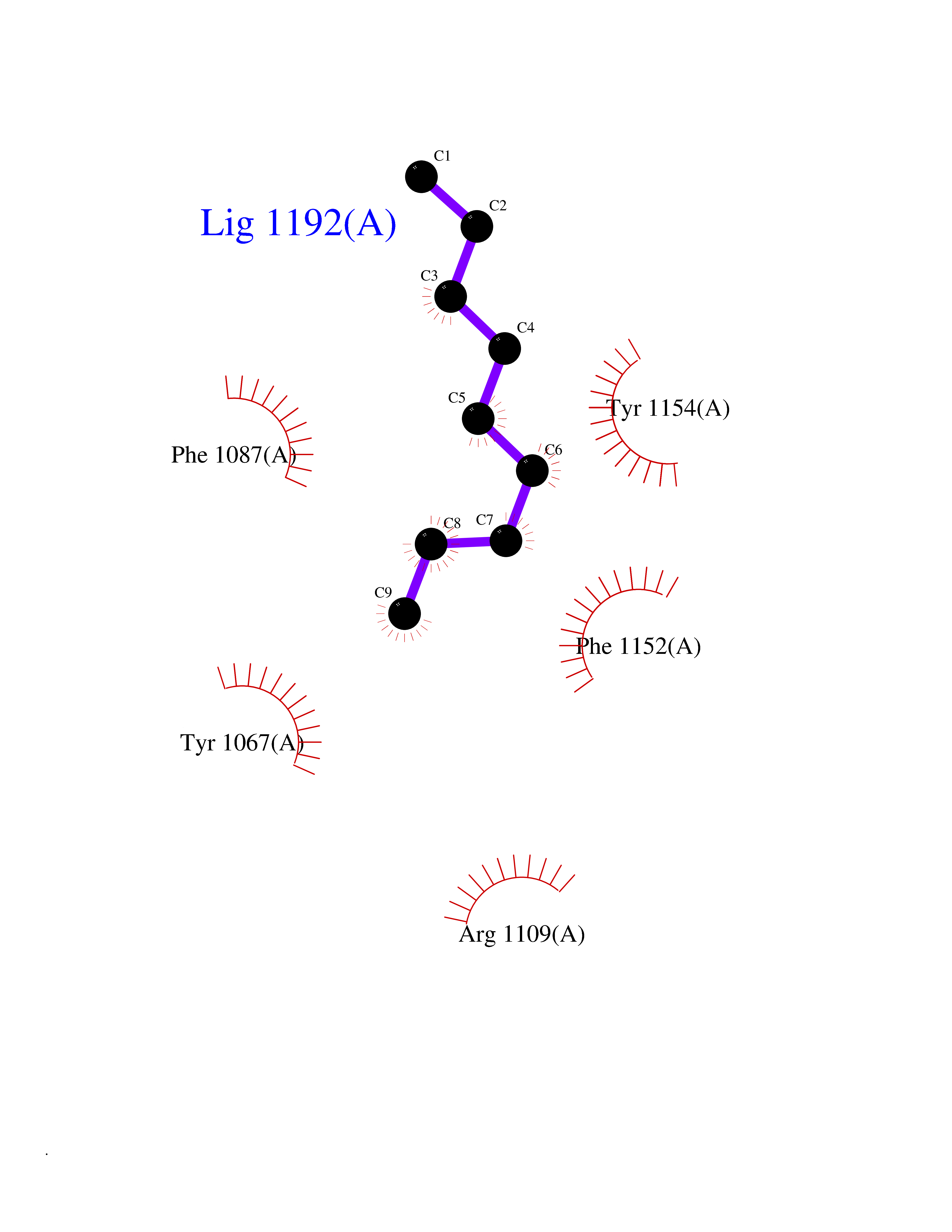 Ligplot Map 3D Binding mode Sequence TEKIICRDVARGYENVPIPCVNGVDGEPCPEDYKYISENCETSTMNIDRNITHLQHCTCVDDCSSSNCLCGQLSIRCWYDKDGRLLQEFNKIEPPLIFECNQACSCWRNCKNRVVQSGIKVRLQLYRTAKMGWGVRALQTIPQGTFICEYVGELISDAEADVREDDSYLFDLDEVYCIDARYYGNISRFINHLCDPNIIPVRVFMLHQDLRFPRIAFFSSRDIRTGEELGFDYGDRFWDIKSKYFTCQCGSEKCKHSAEAIALEQSRLA Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Neuronal acetylcholine receptor alpha-3/beta-4 (CHRNA3/B4) | 6PV7 | 5.40 | |
Target general information Gen name CHRNA3-CHRNB4 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Neuronal acetylcholine receptor Protein family Ligand-gated ion channel (TC 1.A.9) family, Acetylcholine receptor (TC 1.A.9.1) subfamily, Alpha-3/CHRNA3 sub-subfamily Biochemical class Neurotransmitter receptor Function A type of nicotinic acetylcholine receptor, consisting of 3 and 4 subunits. Related diseases Bladder dysfunction, autonomic, with impaired pupillary reflex and secondary CAKUT (BAIPRCK) [MIM:191800]: An autosomal recessive disease characterized by impaired innervation and autonomic dysfunction of the urinary bladder, hydronephrosis, vesicoureteral reflux, small kidneys, recurrent urinary tract infections, and progressive renal insufficiency. Additional autonomic features are impaired pupillary reflex and orthostatic hypotension. The disease manifests in utero or early childhood. {ECO:0000269|PubMed:31708116}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00915; DB01156; DB00237; DB00565; DB09028; DB00514; DB07720; DB00898; DB00472; DB05710; DB01227; DB00848; DB00333; DB00184; DB01090; DB00202; DB01273 Interacts with NA EC number NA Uniprot keywords 3D-structure; Alternative splicing; Cell membrane; Disease variant; Disulfide bond; Endoplasmic reticulum; Glycoprotein; Golgi apparatus; Ion channel; Ion transport; Ligand-gated ion channel; Membrane; Phosphoprotein; Postsynaptic cell membrane; Proteomics identification; Receptor; Reference proteome; Signal; Synapse; Transmembrane; Transmembrane helix; Transport; Ubl conjugation Protein physicochemical properties Chain ID A,B Molecular weight (Da) 89661.2 Length 775 Aromaticity 0.12 Instability index 35.11 Isoelectric point 6.07 Charge (pH=7) -5.67 2D Binding mode Binding energy (Kcal/mol) -7.37  Molscript Map 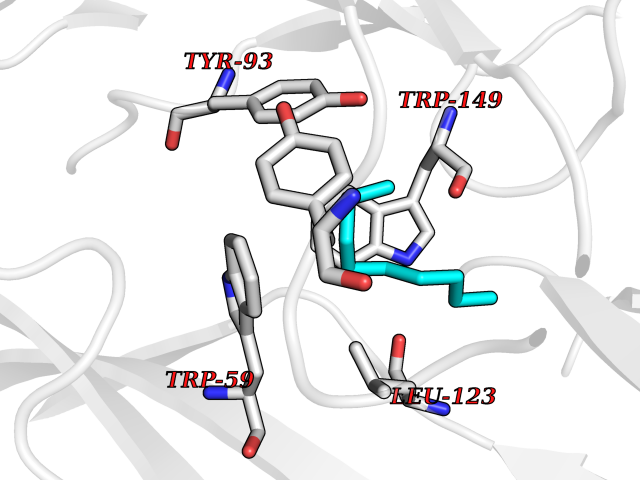 Pymol Map 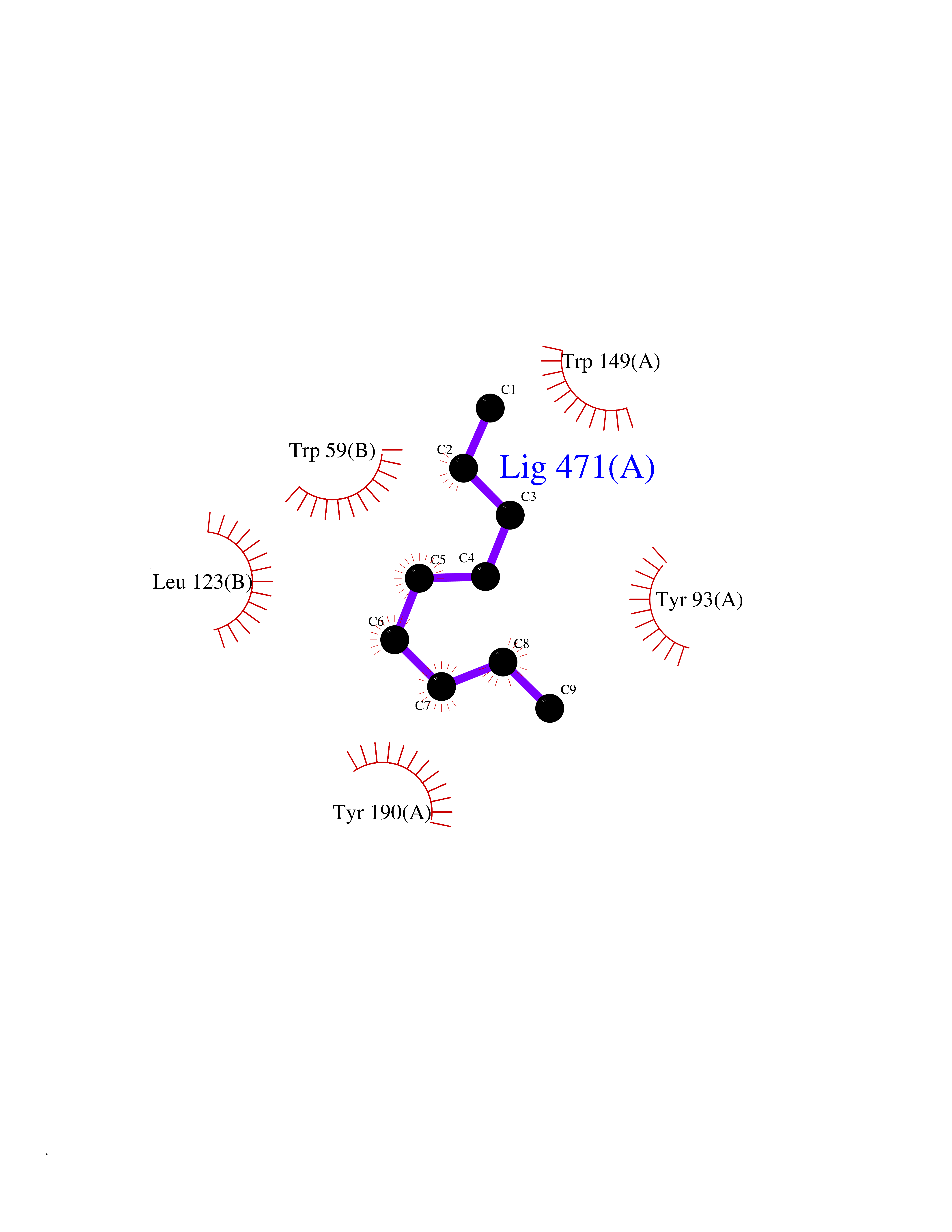 Ligplot Map 3D Binding mode Sequence SEAEHRLFERLFEDYNEIIRPVANVSDPVIIHFEVSMSQLVKVDEVNQIMETNLWLKQIWNDYKLKWNPSDYGGAEFMRVPAQKIWKPDIVLYNNAVGDFQVDDKTKALLKYTGEVTWIPPAIFKSSCKIDVTYFPFDYQNCTMKFGSWSYDKAKIDLVLIGSSMNLKDYWESGEWAIIKAPGYKHDIKYNCCEEIYPDITYSLYIRRLPLFYTINLIIPCLLISFLTVLVFYLPSDCGEKVTLCISVLLSLTVFLLVITETIPSTSLVIPLIGEYLLFTMIFVTLSIVITVFVLNVHYRTPTTHTMPSWVKTVFLNLLPRVMFMTRIKEAIQSVKYIAENMKAQNEAKEIQDDWKYVAMVIDRIFLWVFTLVCILGTAGLFLQPLMRVANAEEKLMDDLLNKTRYNNLIRPATSSSQLISIKLQLSLAQLISVNEREQIMTTNVWLKQEWTDYRLTWNSSRYEGVNILRIPAKRIWLPDIVLYNNADGTYEVSVYTNLIVRSNGSVLWLPPAIYKSACKIEVKYFPFDQQNCTLKFRSWTYDHTEIDMVLMTPTASMDDFTPSGEWDIVALPGRRTVNPQDPSYVDVTYDFIIKRKPLFYTINLIIPCVLTTLLAILVFYLPSDCGEKMTLCISVLLALTFFLLLISKIVPPTSLDVPLIGKYLMFTMVLVTFSIVTSVCVLNVHHRSPSTHTMAPWVKRCFLHKLPTFLFMKRRQDVQEALEGVSFIAQHMKNDDEDQSVVEDWKYVAMVVDRLFLWVFMFVCVLGTVGLFLP Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | Fatty acid-binding protein, intestinal | 3AKM | 5.39 | |
Target general information Gen name FABP2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms FABPI Protein family Calycin superfamily, Fatty-acid binding protein (FABP) family Biochemical class Transport protein Function Fatty acid binding.Transporter activity. Related diseases Usher syndrome 3B (USH3B) [MIM:614504]: A syndrome characterized by progressive vision and hearing loss during early childhood. Some patients have the so-called 'Charles Bonnet syndrome,' involving decreased visual acuity and vivid visual hallucinations. USH is a genetically heterogeneous condition characterized by the association of retinitis pigmentosa with sensorineural deafness. Age at onset and differences in auditory and vestibular function distinguish Usher syndrome type 1 (USH1), Usher syndrome type 2 (USH2) and Usher syndrome type 3 (USH3). USH3 is characterized by postlingual, progressive hearing loss, variable vestibular dysfunction, and onset of retinitis pigmentosa symptoms, including nyctalopia, constriction of the visual fields, and loss of central visual acuity, usually by the second decade of life. {ECO:0000269|PubMed:22279524}. The disease may be caused by variants affecting the gene represented in this entry.; DISEASE: Charcot-Marie-Tooth disease, axonal, 2W (CMT2W) [MIM:616625]: An autosomal dominant, axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. CMT2W patients manifest a peripheral neuropathy mainly affecting the lower limbs and resulting in gait difficulties and distal sensory impairment. Most patients also have upper limb involvement. {ECO:0000269|PubMed:22930593, ECO:0000269|PubMed:26072516, ECO:0000269|PubMed:29235198}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB04557; DB09213; DB00783; DB13952; DB13953; DB13954; DB13955; DB13956; DB01050; DB08231; DB03796; DB01138 Interacts with O95994; Q9NYB0 EC number NA Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Lipid-binding; Proteomics identification; Reference proteome; Transport Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 15075.9 Length 131 Aromaticity 0.11 Instability index 32.01 Isoelectric point 6.88 Charge (pH=7) -0.09 2D Binding mode Binding energy (Kcal/mol) -7.35  Molscript Map 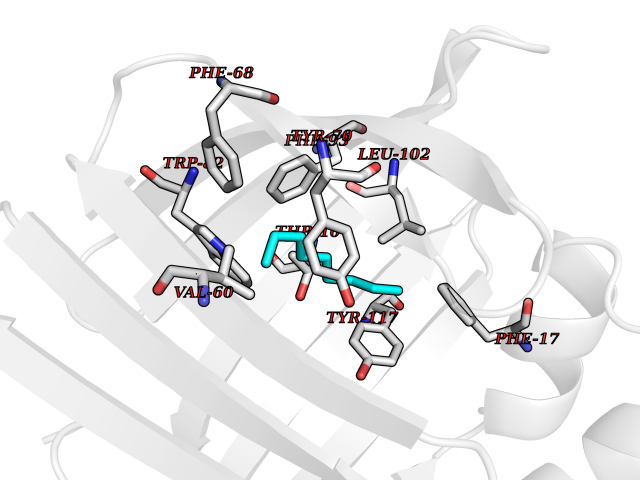 Pymol Map 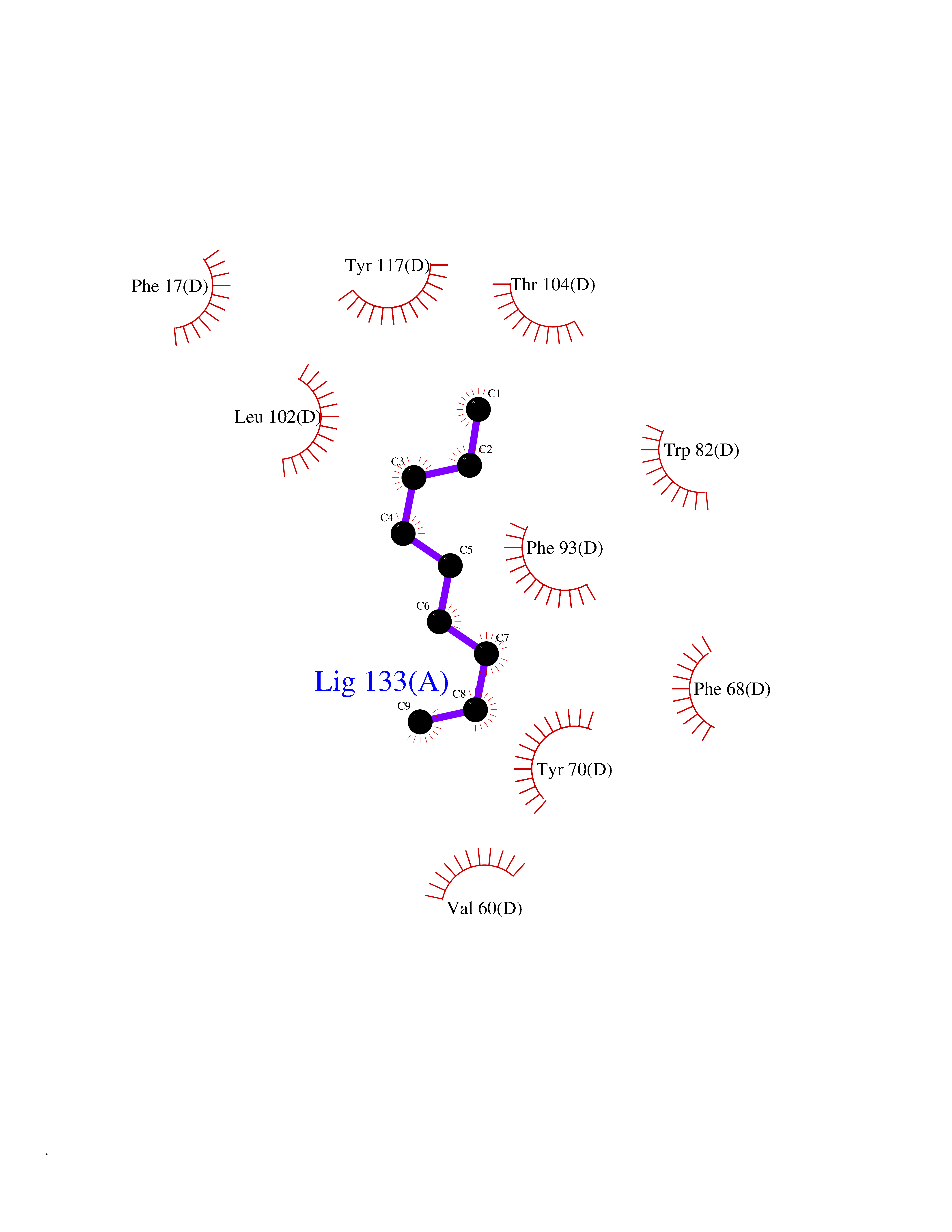 Ligplot Map 3D Binding mode Sequence AFDSTWKVDRSENYDKFMEKMGVNIVKRKLAAHDNLKLTITQEGNKFTVKESSAFRNIEVVFELGVTFNYNLADGTELRGTWSLEGNKLIGKFKRTDNGNELNTVREIIGDELVQTYVYEGVEAKRIFKKD Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | Platelet-derived growth factor receptor alpha (PDGFRA) | 5K5X | 5.39 | |
Target general information Gen name PDGFRA Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms RHEPDGFRA; Platelet-derived growth factor receptor 2; Platelet-derived growth factor alpha receptor; PDGFR2; PDGFR-alpha; PDGFR-2; PDGF-R-alpha; CD140a antigen; CD140a; CD140 antigen-like family membe Protein family Protein kinase superfamily, Tyr protein kinase family, CSF-1/PDGF receptor subfamily Biochemical class Kinase Function Depending on the context, promotes or inhibits cell proliferation and cell migration. Plays an important role in the differentiation of bone marrow-derived mesenchymal stem cells. Required for normal skeleton development and cephalic closure during embryonic development. Required for normal development of the mucosa lining the gastrointestinal tract, and for recruitment of mesenchymal cells and normal development of intestinal villi. Plays a role in cell migration and chemotaxis in wound healing. Plays a role in platelet activation, secretion of agonists from platelet granules, and in thrombin-induced platelet aggregation. Binding of its cognate ligands - homodimeric PDGFA, homodimeric PDGFB, heterodimers formed by PDGFA and PDGFB or homodimeric PDGFC -leads to the activation of several signaling cascades; the response depends on the nature of the bound ligand and is modulated by the formation of heterodimers between PDGFRA and PDGFRB. Phosphorylates PIK3R1, PLCG1, and PTPN11. Activation of PLCG1 leads to the production of the cellular signaling molecules diacylglycerol and inositol 1,4,5-trisphosphate, mobilization of cytosolic Ca(2+) and the activation of protein kinase C. Phosphorylates PIK3R1, the regulatory subunit of phosphatidylinositol 3-kinase, and thereby mediates activation of the AKT1 signaling pathway. Mediates activation of HRAS and of the MAP kinases MAPK1/ERK2 and/or MAPK3/ERK1. Promotes activation of STAT family members STAT1, STAT3 and STAT5A and/or STAT5B. Receptor signaling is down-regulated by protein phosphatases that dephosphorylate the receptor and its down-stream effectors, and by rapid internalization of the activated receptor. Tyrosine-protein kinase that acts as a cell-surface receptor for PDGFA, PDGFB and PDGFC and plays an essential role in the regulation of embryonic development, cell proliferation, survival and chemotaxis. Related diseases A chromosomal aberration involving PDGFRA is found in some cases of hypereosinophilic syndrome. Interstitial chromosomal deletion del(4)(q12q12) causes the fusion of FIP1L1 and PDGFRA (FIP1L1-PDGFRA). Mutations that cause overexpression and/or constitutive activation of PDGFRA may be a cause of hypereosinophilic syndrome. {ECO:0000269|PubMed:12808148}.; DISEASE: Gastrointestinal stromal tumor (GIST) [MIM:606764]: Common mesenchymal neoplasms arising in the gastrointestinal tract, most often in the stomach. They are histologically, immunohistochemically, and genetically different from typical leiomyomas, leiomyosarcomas, and schwannomas. Most GISTs are composed of a fairly uniform population of spindle-shaped cells. Some tumors are dominated by epithelioid cells or contain a mixture of spindle and epithelioid morphologies. Primary GISTs in the gastrointestinal tract commonly metastasize in the omentum and mesenteries, often as multiple nodules. However, primary tumors may also occur outside of the gastrointestinal tract, in other intra-abdominal locations, especially in the omentum and mesentery. {ECO:0000269|PubMed:12522257, ECO:0000269|PubMed:15928335}. The gene represented in this entry may be involved in disease pathogenesis. Mutations causing PDGFRA constitutive activation have been found in gastrointestinal stromal tumors lacking KIT mutations (PubMed:12522257). {ECO:0000269|PubMed:12522257}.; DISEASE: GIST-plus syndrome (GISTPS) [MIM:175510]: A disorder characterized by multiple mesenchymal tumors of the gastrointestinal tract, including gastrointestinal stromal tumor, inflammatory fibroid polyps, and fibroid tumors. Additional features are coarse facies and skin, broad hands and feet, and premature tooth loss. GISTPS is an autosomal dominant disease with incomplete penetrance. Gastrointestinal stromal tumor and inflammatory fibroid polyps may also occur in isolation. {ECO:0000269|PubMed:14699510, ECO:0000269|PubMed:17087943, ECO:0000269|PubMed:25975287}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12742; DB00102; DB12147; DB10772; DB12010; DB00619; DB09078; DB06595; DB09079; DB06043; DB06589; DB08901; DB08896; DB14840; DB01268; DB11800; DB05146 Interacts with P46108; P46109; P00533; Q8N6L0; P04085; P01127; Q9NRA1; P31947; P62258; Q9NRA1-1; A8T7D5; P05067; Q8IY26 EC number EC 2.7.10.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cell membrane; Cell projection; Chemotaxis; Developmental protein; Disease variant; Disulfide bond; Glycoprotein; Golgi apparatus; Host-virus interaction; Immunoglobulin domain; Kinase; Membrane; Nucleotide-binding; Phosphoprotein; Proteomics identification; Proto-oncogene; Receptor; Reference proteome; Repeat; Signal; Transferase; Transmembrane; Transmembrane helix; Tyrosine-protein kinase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 39294.8 Length 345 Aromaticity 0.11 Instability index 46.74 Isoelectric point 6.6 Charge (pH=7) -1.38 2D Binding mode Binding energy (Kcal/mol) -7.35 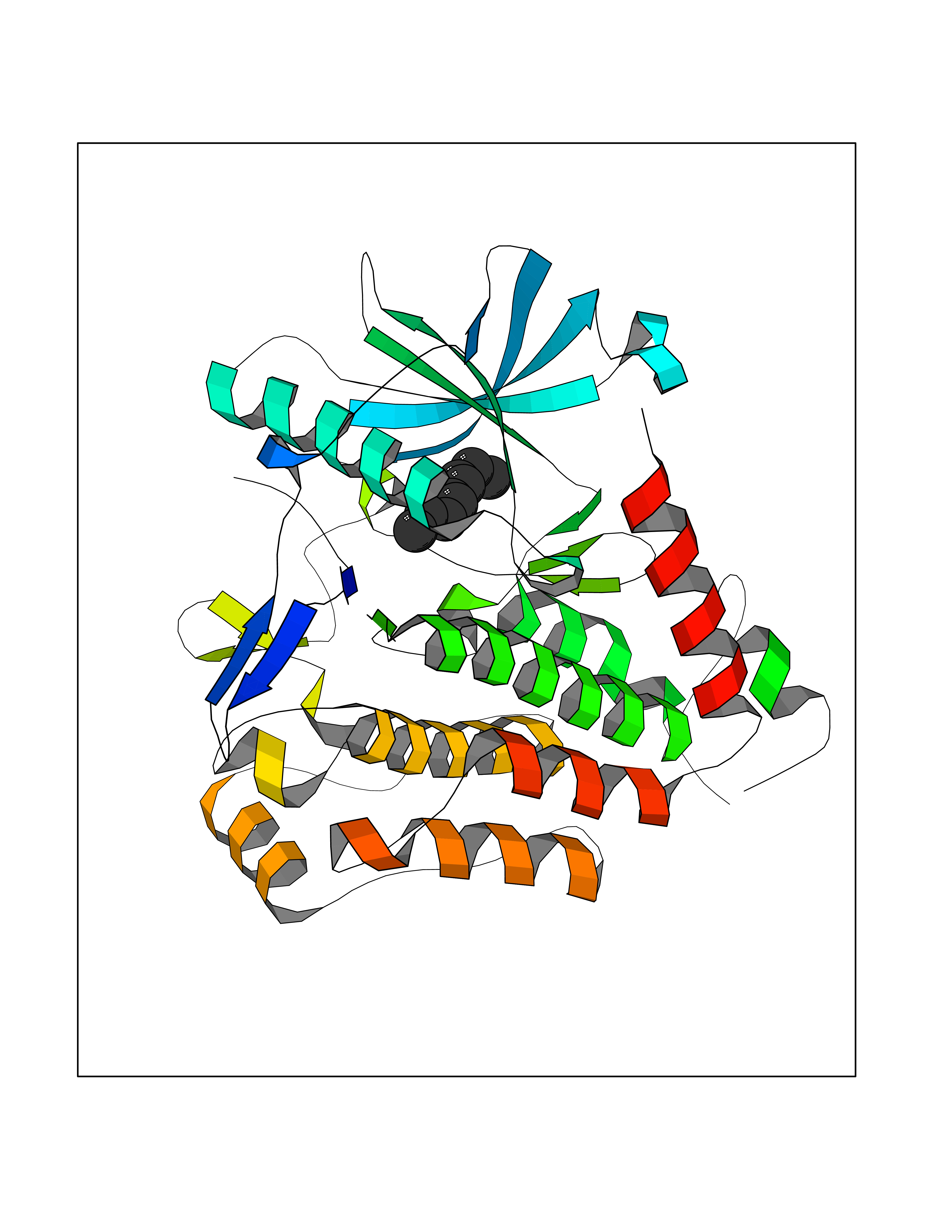 Molscript Map 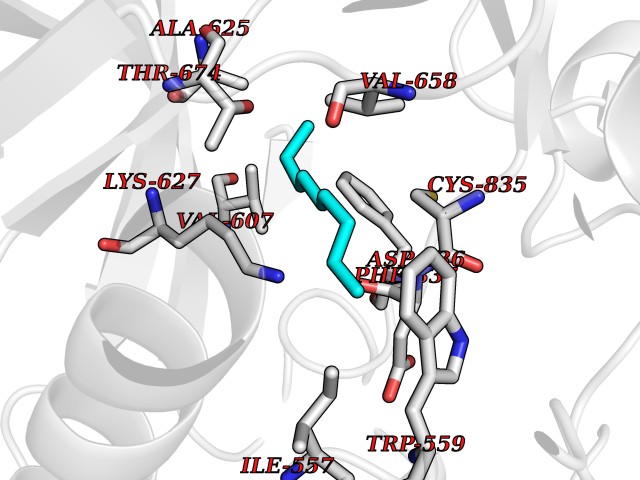 Pymol Map 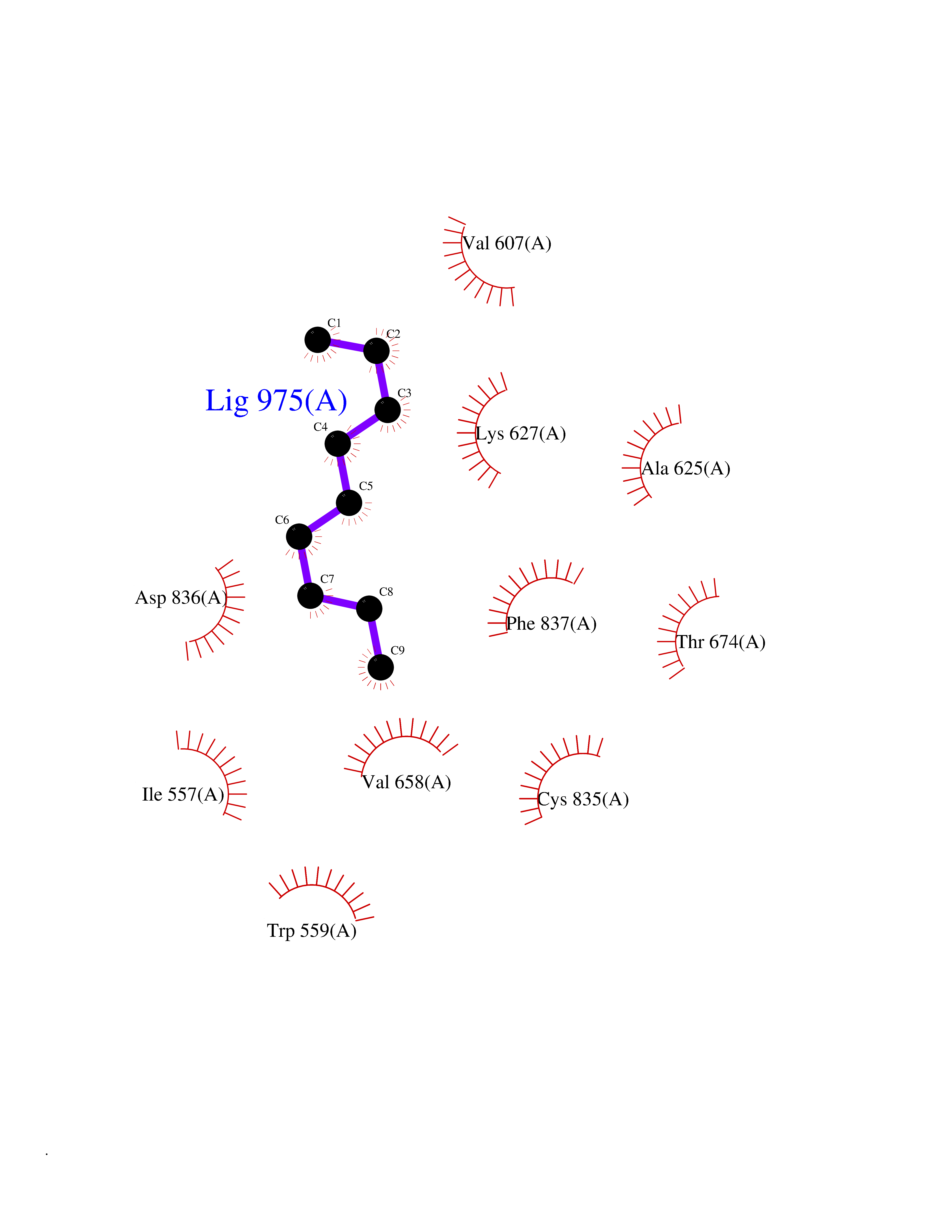 Ligplot Map 3D Binding mode Sequence RYEIRWRVIESISPDGHEYIYVDPMQLPYDSRWEFPRDGLVLGRVLGSGAFGKVVEGTAYGLSRSQPVMKVAVKMLKPTARSSEKQALMSELKIMTHLGPHLNIVNLLGACTKSGPIYIITEYCFYGDLVNYLHKNRDSFLSHSMLDSEVKNLLSDDNSEGLTLLDLLSFTYQVARGMEFLASKNCVHRDLAARNVLLAQGKIVKICDFGLARDIMHDSNYVSKGSTFLPVKWMAPESIFDNLYTTLSDVWSYGILLWEIFSLGGTPYPGMMVDSTFYNKIKSGYRMAKPDHATSEVYEIMVKCWNSEPEKRPSFYHLSEIVENLLPGQYKKSYEKIHLDFLKSD Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Gamma-butyrobetaine dioxygenase | 4C5W | 5.39 | |
Target general information Gen name BBOX1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms BBH;BBOX Protein family Gamma-BBH/TMLD family Biochemical class Oxidoreductase Function Gamma-butyrobetaine dioxygenase activity.Identical protein binding.Iron ion binding.Zinc ion binding. Related diseases Neurodevelopmental disorder with poor language and loss of hand skills (NDPLHS) [MIM:617903]: An autosomal dominant disorder characterized by psychomotor developmental stagnation or regression. NDPLHS manifest in the first years of life as loss of purposeful hand movements, loss of language, and intellectual disability. {ECO:0000269|PubMed:26740508, ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Developmental and epileptic encephalopathy 59 (DEE59) [MIM:617904]: A form of epileptic encephalopathy, a heterogeneous group of severe early-onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. DEE59 is an autosomal dominant condition characterized by onset of refractory seizures in early infancy. {ECO:0000269|PubMed:28856709, ECO:0000269|PubMed:29100083, ECO:0000269|PubMed:29369404}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00126 Interacts with O75936; A0MZ66-7 EC number 1.14.11.1 Uniprot keywords 3D-structure; Carnitine biosynthesis; Cytoplasm; Dioxygenase; Iron; Metal-binding; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 31642.5 Length 275 Aromaticity 0.12 Instability index 35.68 Isoelectric point 6.33 Charge (pH=7) -2.46 2D Binding mode Binding energy (Kcal/mol) -7.35 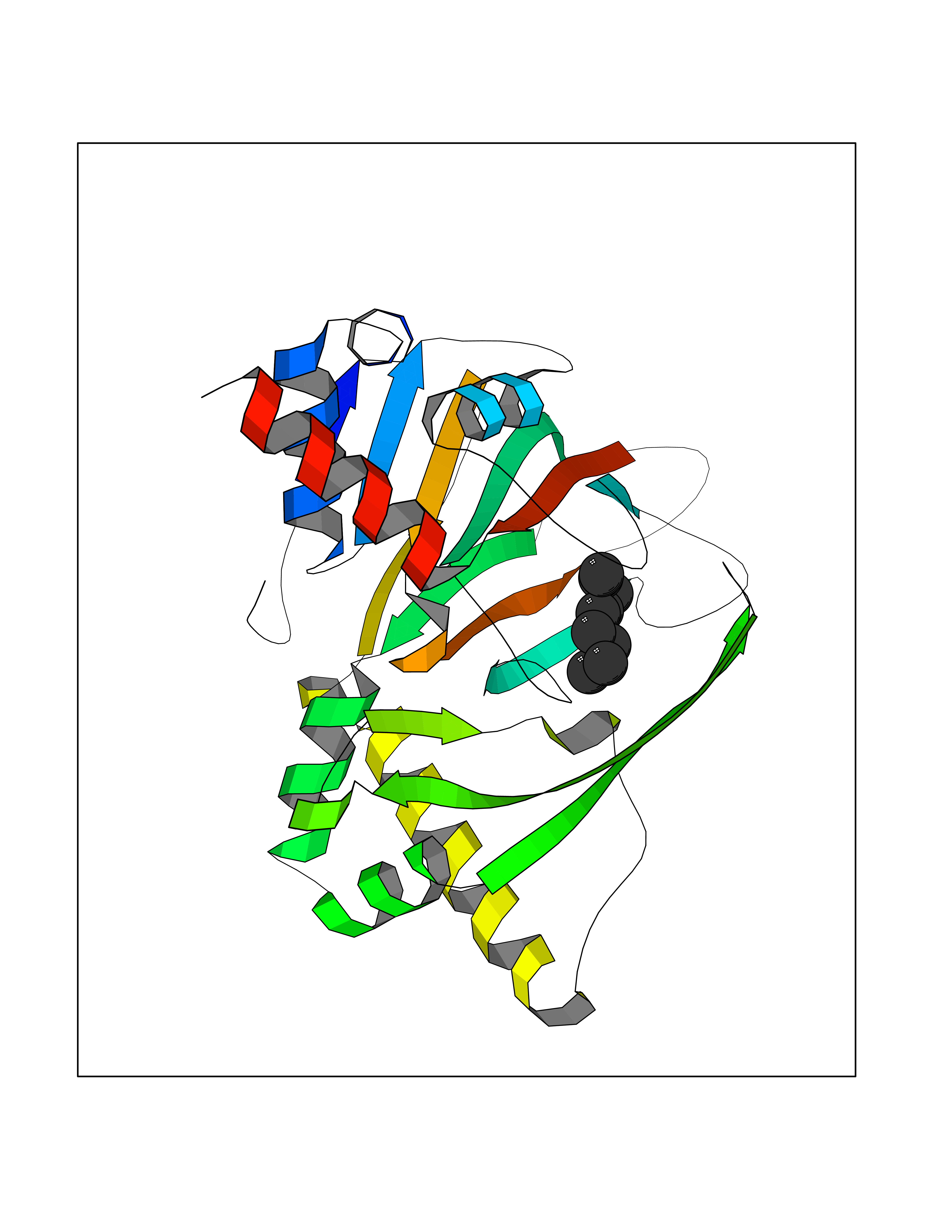 Molscript Map 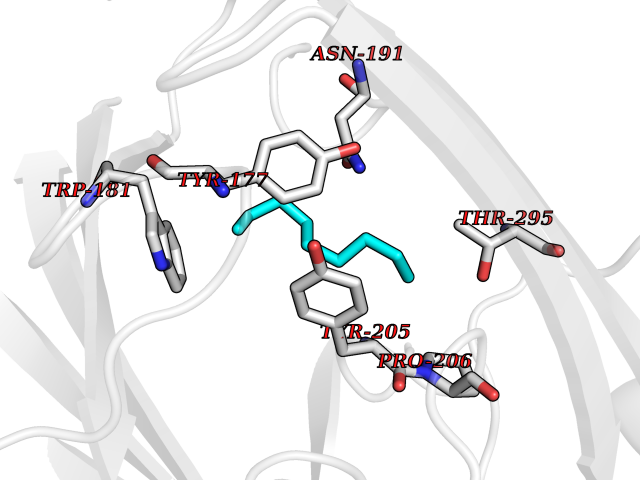 Pymol Map 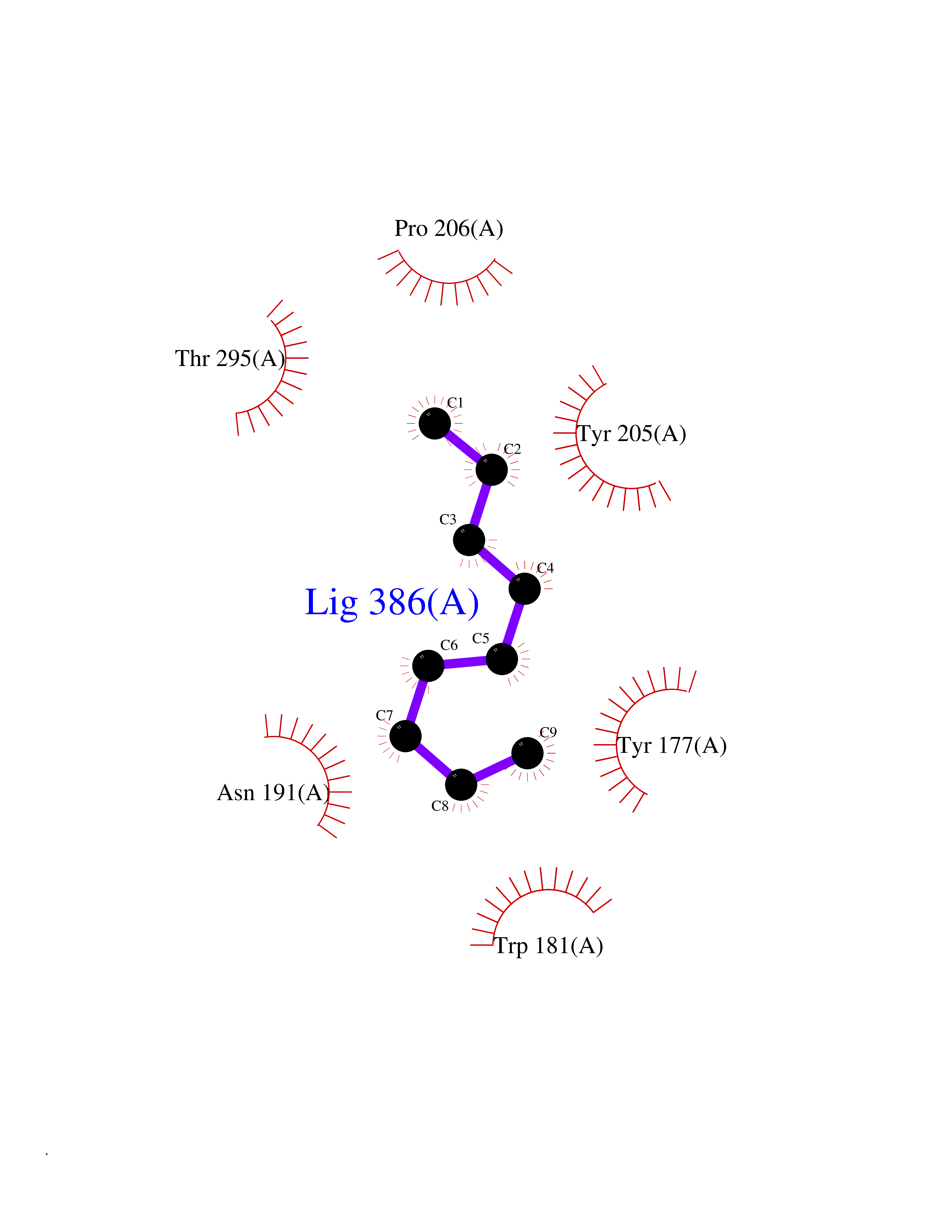 Ligplot Map 3D Binding mode Sequence FPECQYWGSELQLPTLDFEDVLRYDEHAYKWLSTLKKVGIVRLTGASDKPGEVSKLGKRMGFLYLTFYGHTWQVQDKIDANNVAYTTGKLSFHTDYPALHHPPGVQLLHCIKQTVTGGDSEIVDGFNVCQKLKKNNPQAFQILSSTFVDFTDIGVDYCDFSVQSKHKIIELDDKGQVVRINFNNATRDTIFDVPVERVQPFYAALKEFVDLMNSKESKFTFKMNPGDVITFDNWRLLHGRRSYEAGTEISRHLEGAYADWDVVMSRLRILRQRVE Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Fatty acid synthase (FASN) | 3TJM | 5.39 | |
Target general information Gen name FASN Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Yeast fatty acid synthase; Fatty-acyl-CoA synthase; Fatty acyl-CoA synthetase enzyme; FAS Protein family NA Biochemical class Acyltransferase Function Fatty acid synthetase catalyzes the formation of long-chain fatty acids from acetyl-CoA, malonyl-CoA and NADPH. This multifunctional protein has 7 catalytic activities as an acyl carrier protein. Related diseases Glycine encephalopathy 2 (GCE2) [MIM:620398]: A form of glycine encephalopathy, a metabolic disorder characterized by a high concentration of glycine in the body fluids. Affected individuals typically have severe neurological symptoms, including seizure, lethargy, and muscular hypotonia soon after birth. Most of them die within the neonatal period. Atypical cases have later disease onset and less severely affected psychomotor development. {ECO:0000269|PubMed:10873393, ECO:0000269|PubMed:11286506, ECO:0000269|PubMed:16051266, ECO:0000269|PubMed:26371980, ECO:0000269|PubMed:28244183, ECO:0000269|PubMed:8005589, ECO:0000269|PubMed:9600239, ECO:0000269|PubMed:9621520}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01034; DB01083 Interacts with Q15848; Q16665; P42858; Q8IV20; Q8TBB1; PRO_0000045603 [Q99IB8] EC number EC 2.3.1.85 Uniprot keywords 3D-structure; Acetylation; Cytoplasm; Direct protein sequencing; Fatty acid biosynthesis; Fatty acid metabolism; Hydrolase; Isopeptide bond; Lipid biosynthesis; Lipid metabolism; Lyase; Multifunctional enzyme; NAD; NADP; Oxidoreductase; Phosphopantetheine; Phosphoprotein; Proteomics identification; Pyridoxal phosphate; Reference proteome; S-nitrosylation; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 30174.9 Length 275 Aromaticity 0.09 Instability index 43.28 Isoelectric point 5.92 Charge (pH=7) -5.4 2D Binding mode Binding energy (Kcal/mol) -7.35 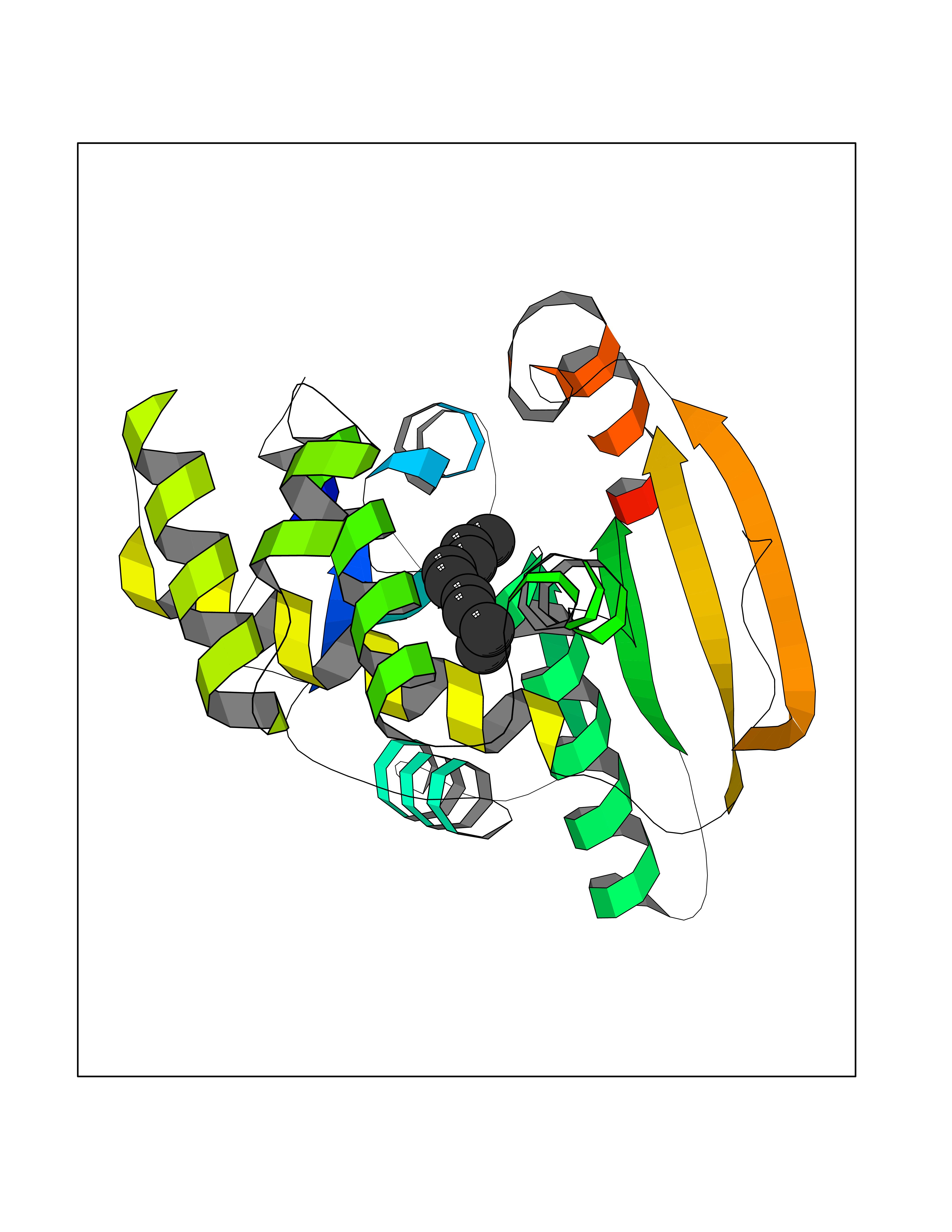 Molscript Map 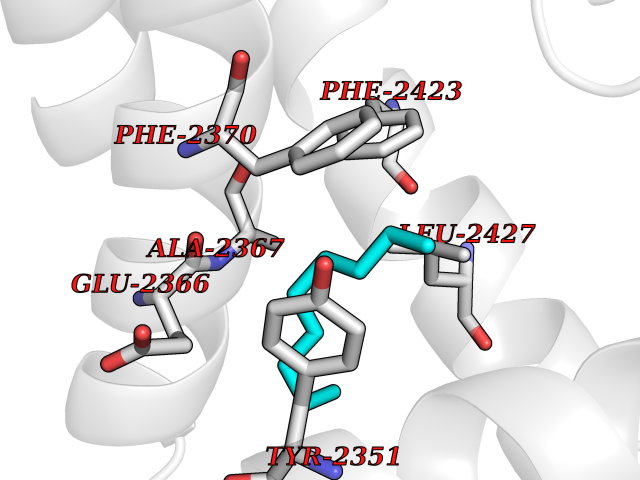 Pymol Map 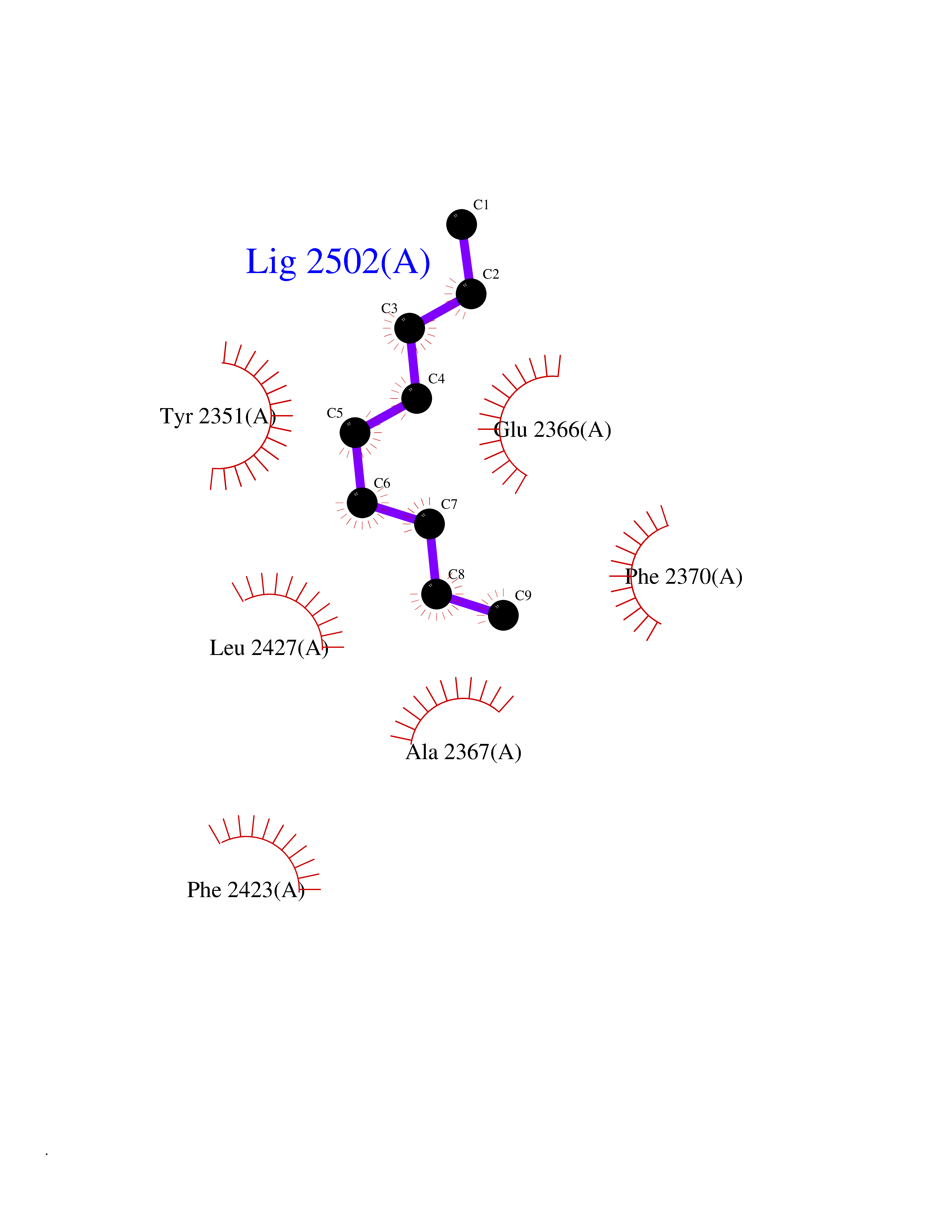 Ligplot Map 3D Binding mode Sequence NLRSLLVNPEGPTLMRLNSVQSSERPLFLVHPIEGSTTVFHSLASRLSIPTYGLQCTRAAPLDSIHSLAAYYIDCIRQVQPEGPYRVAGYSYGACVAFEMCSQLQAQQSPAPTHNSLFLFDGSPTYVLAYTGSYRAKLTPGCEAEAETEAICFFVQQFTDMEHNRVLEALLPLKGLEERVAAAVDLIIKSHQGLDRQELSFAARSFYYKLRAAEQYTPKAKYHGNVMLLRAAAGADYNLSQVCDGKVSVHVIEGDHATLLEGSGLESIISIIHSS Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Matrix metalloproteinase-16 (MMP-16) | 1RM8 | 5.39 | |
Target general information Gen name MMP16 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Membrane-type-3 matrix metalloproteinase; Membrane-type matrix metalloproteinase 3; MTMMP3; MT3MMP; MT3-MMP; MT-MMP 3; MMPX2; MMP-X2; C8orf57 Protein family Peptidase M10A family Biochemical class Peptidase Function Activates progelatinase A. Involved in the matrix remodeling of blood vessels. Isoform short cleaves fibronectin and also collagen type III, but at lower rate. It has no effect on type I, II, IV and V collagen. However, upon interaction with CSPG4, it may be involved in degradation and invasion of type I collagen by melanoma cells. Endopeptidase that degrades various components of the extracellular matrix, such as collagen type III and fibronectin. Related diseases Cerebral creatine deficiency syndrome 3 (CCDS3) [MIM:612718]: An autosomal recessive disorder characterized by developmental delay/regression, intellectual disability, severe disturbance of expressive and cognitive speech, and severe depletion of creatine/phosphocreatine in the brain. Most patients develop a myopathy characterized by muscle weakness and atrophy later in life. {ECO:0000269|PubMed:11555793, ECO:0000269|PubMed:20682460, ECO:0000269|PubMed:22386973, ECO:0000269|PubMed:23660394, ECO:0000269|PubMed:23770102, ECO:0000269|PubMed:26490222, ECO:0000269|PubMed:27233232}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Fanconi renotubular syndrome 1 (FRTS1) [MIM:134600]: A form of Fanconi renotubular syndrome, a disease due to a generalized dysfunction of the proximal kidney tubule resulting in decreased solute and water reabsorption. Patients have polydipsia and polyuria with phosphaturia, glycosuria and aminoaciduria. They may develop hypophosphatemic rickets or osteomalacia, acidosis and a tendency toward dehydration. Some eventually develop renal insufficiency. FRTS1 inheritance is autosomal dominant. {ECO:0000269|PubMed:29654216}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03880; DB00786 Interacts with NA EC number EC 3.4.24.- Uniprot keywords 3D-structure; Alternative splicing; Calcium; Cell membrane; Cleavage on pair of basic residues; Collagen degradation; Disulfide bond; Extracellular matrix; Glycoprotein; Hydrolase; Membrane; Metal-binding; Metalloprotease; Protease; Proteomics identification; Reference proteome; Repeat; Secreted; Signal; Transmembrane; Transmembrane helix; Zinc; Zymogen Protein physicochemical properties Chain ID A Molecular weight (Da) 18853.6 Length 169 Aromaticity 0.12 Instability index 33.65 Isoelectric point 4.88 Charge (pH=7) -12.42 2D Binding mode Binding energy (Kcal/mol) -7.35 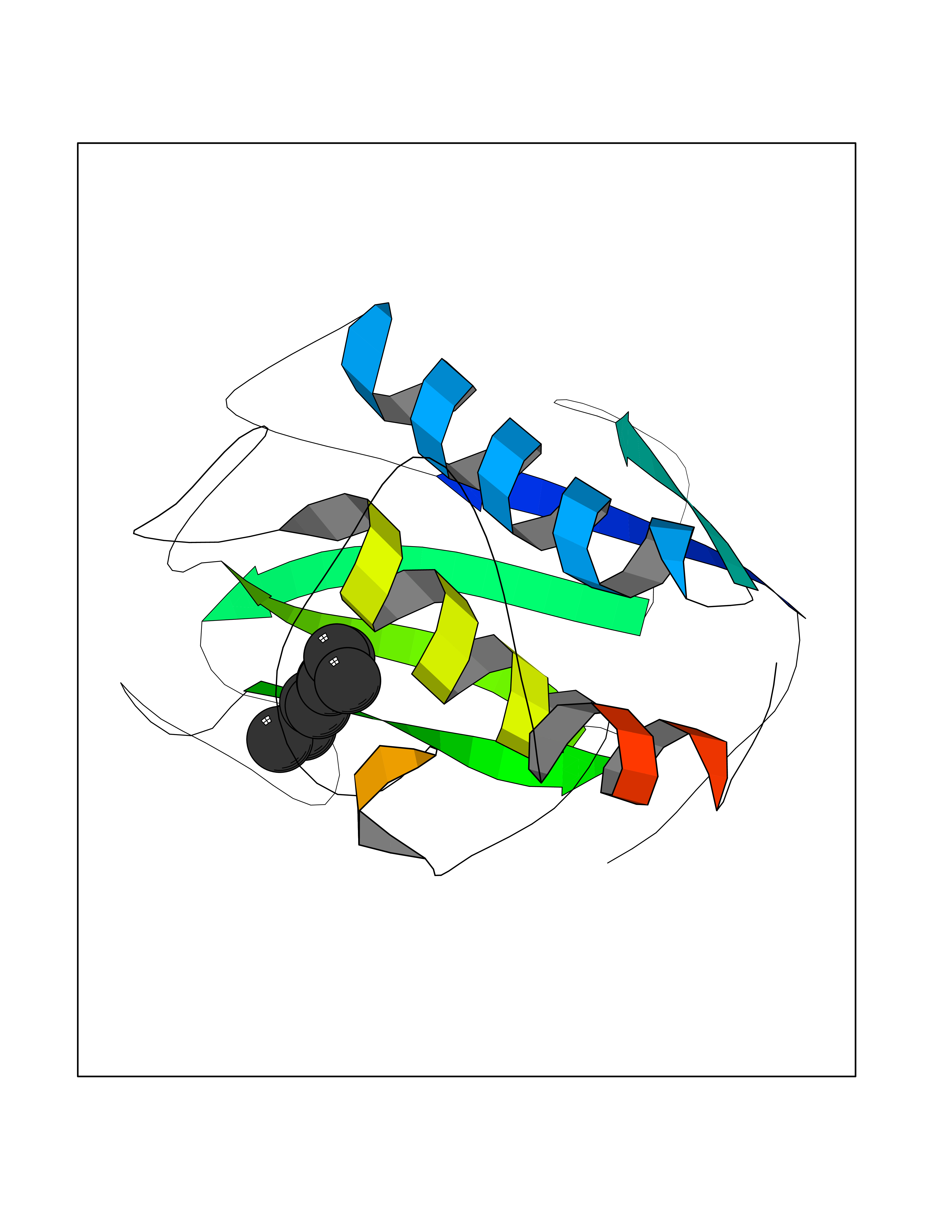 Molscript Map 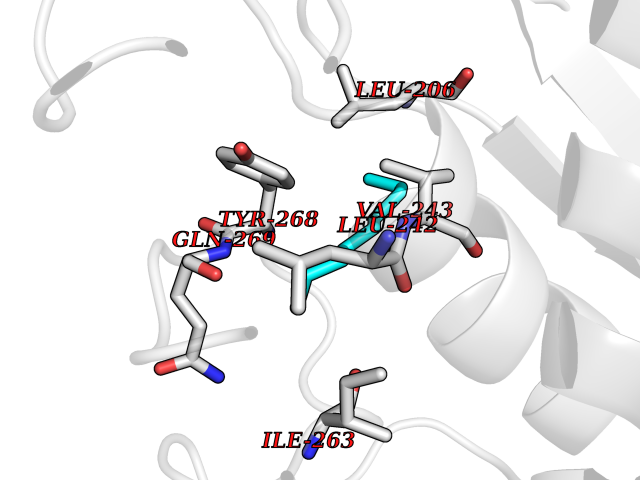 Pymol Map 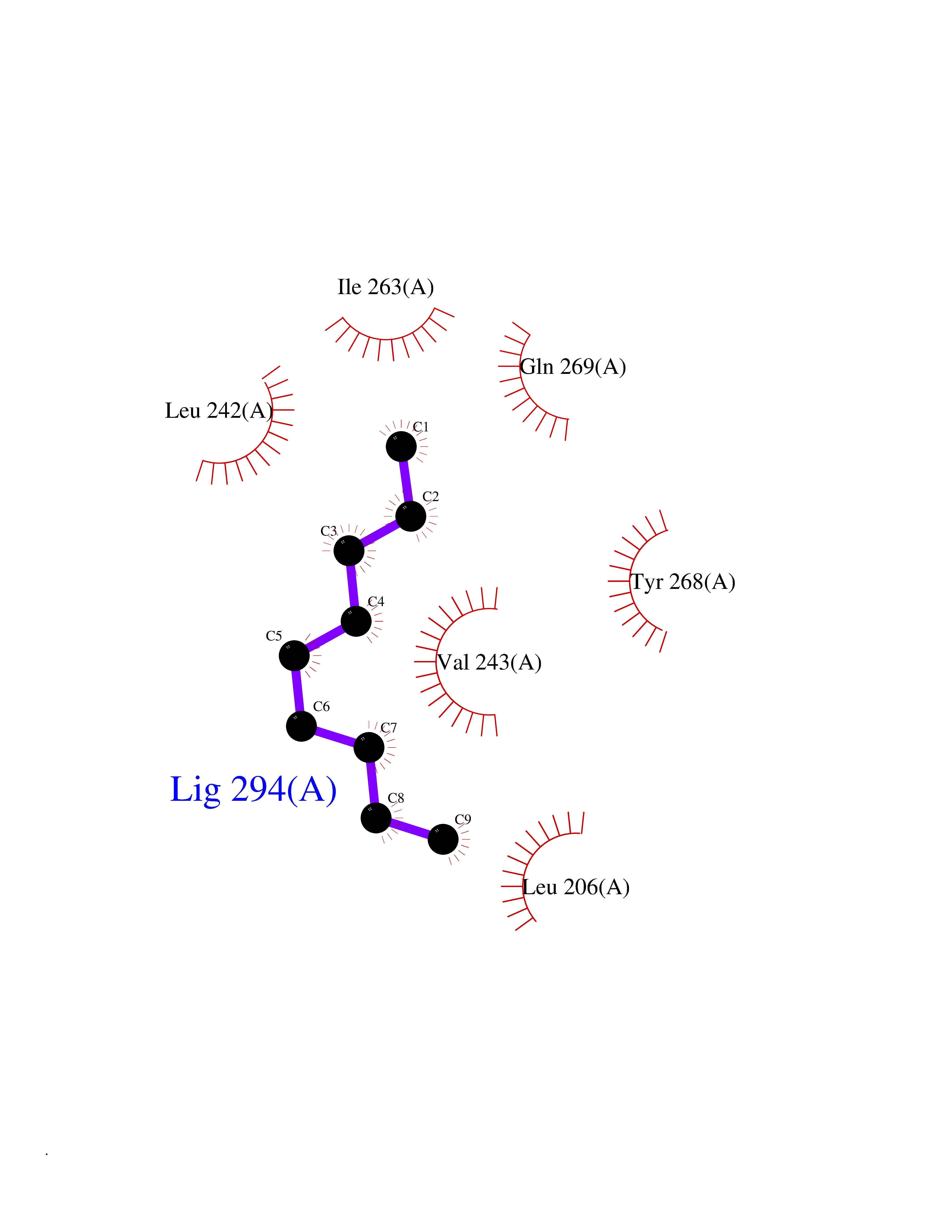 Ligplot Map 3D Binding mode Sequence GQKWQHKHITYSIKNVTPKVGDPETRKAIRRAFDVWQNVTPLTFEEVPYSELENGKRDVDITIIFASGFHGDSSPFDGEGGFLAHAYFPGPGIGGDTHFDSDEPWTLGNPNHDGNDLFLVAVHELGHALGLEHSNDPTAIMAPFYQYMETDNFKLPNDDLQGIQKIYGP Hydrogen bonds contact Hydrophobic contact | ||||