Job Results:
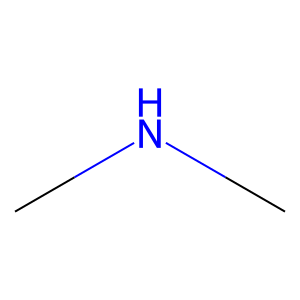
Ligand
Structure
Job ID
3c17296c67deee94355bca3744afb8a3
Job name
NA
Time
2025-02-13 15:25:56
| Rank | Target | PDB ID |
AirScore
|
Detail
|
|---|---|---|---|---|
| 61 | Ornithine delta-aminotransferase (OAT) | 2OAT | 4.00 | |
Target general information Gen name OAT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Ornithine--oxo-acid aminotransferase; Ornithine aminotransferase, mitochondrial Protein family Class-III pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transaminase Function Catalyzes the transfer of the delta-amino group from L-ornithine. Related diseases Hyperornithinemia with gyrate atrophy of choroid and retina (HOGA) [MIM:258870]: A disorder clinically characterized by a triad of progressive chorioretinal degeneration, early cataract formation, and type II muscle fiber atrophy. Characteristic chorioretinal atrophy with progressive constriction of the visual fields leads to blindness at the latest during the sixth decade of life. Patients generally have normal intelligence. {ECO:0000269|PubMed:1612597, ECO:0000269|PubMed:1737786, ECO:0000269|PubMed:23076989, ECO:0000269|PubMed:2793865, ECO:0000269|PubMed:3375240, ECO:0000269|PubMed:7668253, ECO:0000269|PubMed:7887415}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02821; DB02054; DB00129; DB00114 Interacts with P05067 EC number EC 2.6.1.13 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Aminotransferase; Direct protein sequencing; Disease variant; Mitochondrion; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase; Transit peptide Protein physicochemical properties Chain ID A,B,C Molecular weight (Da) 44807.9 Length 404 Aromaticity 0.09 Instability index 26.67 Isoelectric point 5.72 Charge (pH=7) -6.54 2D Binding mode Binding energy (Kcal/mol) -5.45 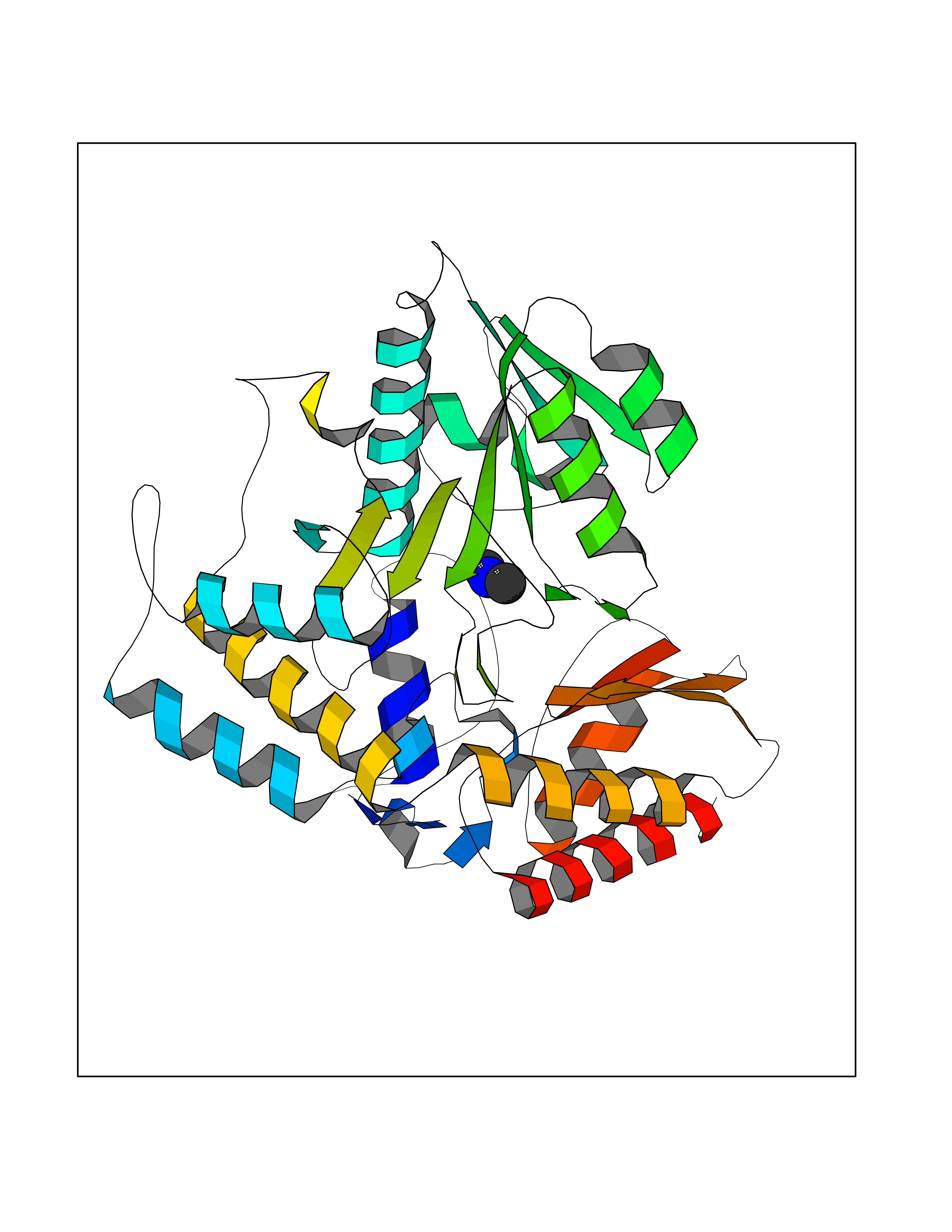 Molscript Map 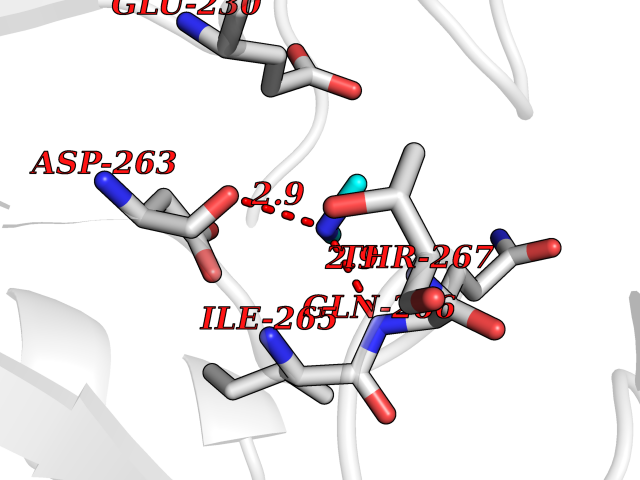 Pymol Map 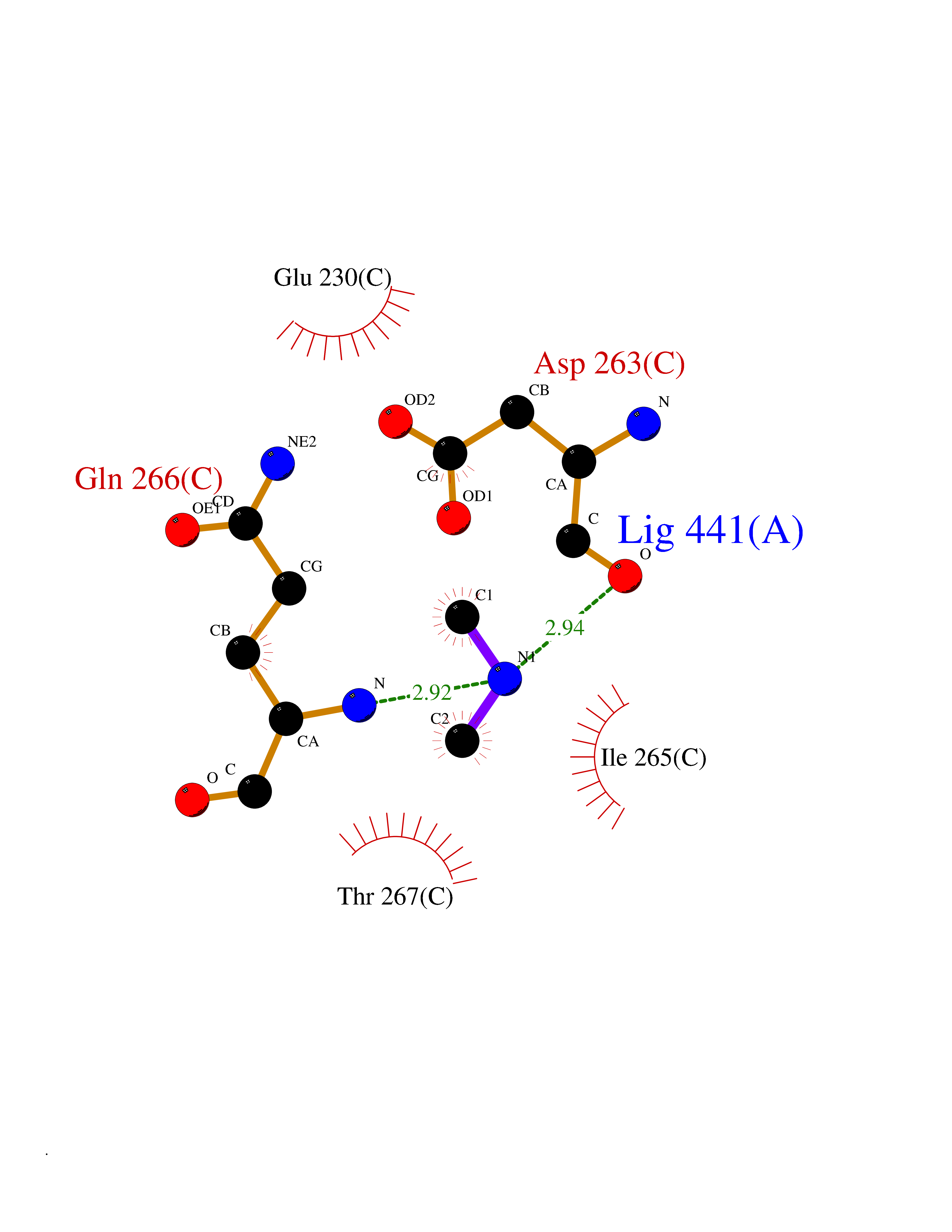 Ligplot Map 3D Binding mode Sequence GPPTSDDIFEREYKYGAHNYHPLPVALERGKGIYLWDVEGRKYFDFLSSYSAVNQGHCHPKIVNALKSQVDKLTLTSRAFYNNVLGEYEEYITKLFNYHKVLPMNTGVEAGETACKLARKWGYTVKGIQKYKAKIVFAAGNFWGRTLSAISSSTDPTSYDGFGPFMPGFDIIPYNDLPALERALQDPNVAAFMVEPIQGEAGVVVPDPGYLMGVRELCTRHQVLFIADEIQTGLARTGRWLAVDYENVRPDIVLLGKALSGGLYPVSAVLCDDDIMLTIKPGEHGSTYGGNPLGCRVAIAALEVLEEENLAENADKLGIILRNELMKLPSDVVTAVRGKGLLNAIVIKETKDWDAWKVCLRLRDNGLLAKPTHGDIIRFAPPLVIKEDELRESIEIINKTILSF Hydrogen bonds contact Hydrophobic contact | ||||
| 62 | Fumarate reductase flavoprotein subunit | 1KF6 | 4.00 | |
Target general information Gen name frdA Organism Escherichia coli (strain K12) Uniprot ID TTD ID NA Synonyms JW4115;b4154 Protein family FAD-dependent oxidoreductase 2 family, FRD/SDH subfamily Biochemical class Oxidoreductase Function Electron carrier activity.FAD binding.Fumarate reductase (menaquinone).Succinate dehydrogenase activity. Related diseases Glycogen storage disease 11 (GSD11) [MIM:612933]: A metabolic disorder that results in exertional myoglobinuria, pain, cramps and easy fatigue. {ECO:0000269|PubMed:2334430}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07490; DB07918; DB00730 Interacts with P0AC47; P0ACB4; P76111 EC number 1.3.5.1 Uniprot keywords 3D-structure; Cell inner membrane; Cell membrane; Direct protein sequencing; Electron transport; FAD; Flavoprotein; Membrane; Nucleotide-binding; Oxidoreductase; Reference proteome; Transport Protein physicochemical properties Chain ID A,M Molecular weight (Da) 90370.7 Length 820 Aromaticity 0.08 Instability index 28.88 Isoelectric point 5.86 Charge (pH=7) -16.21 2D Binding mode Binding energy (Kcal/mol) -5.45 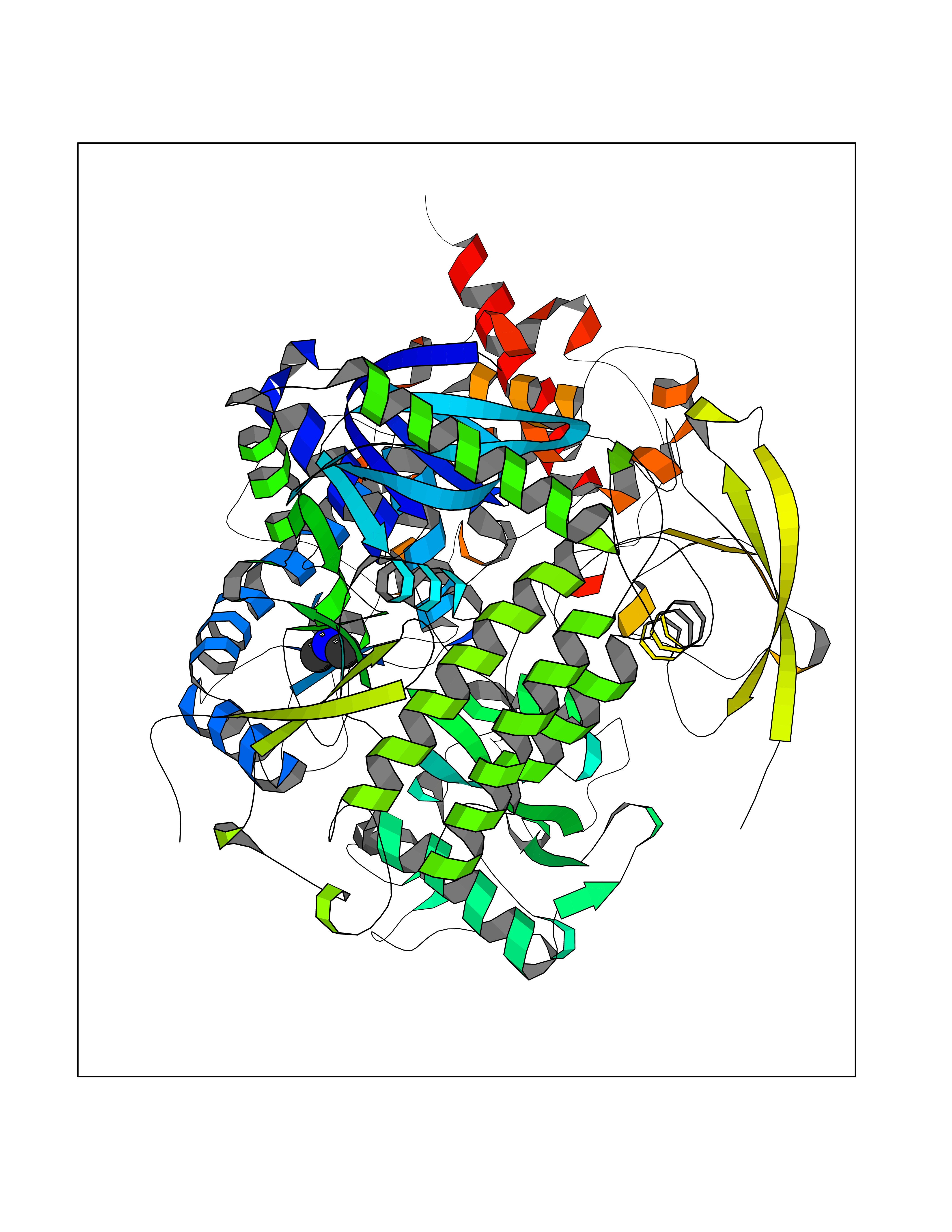 Molscript Map 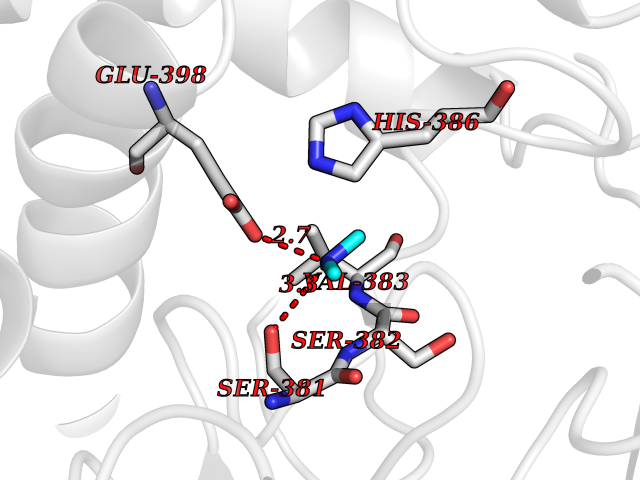 Pymol Map 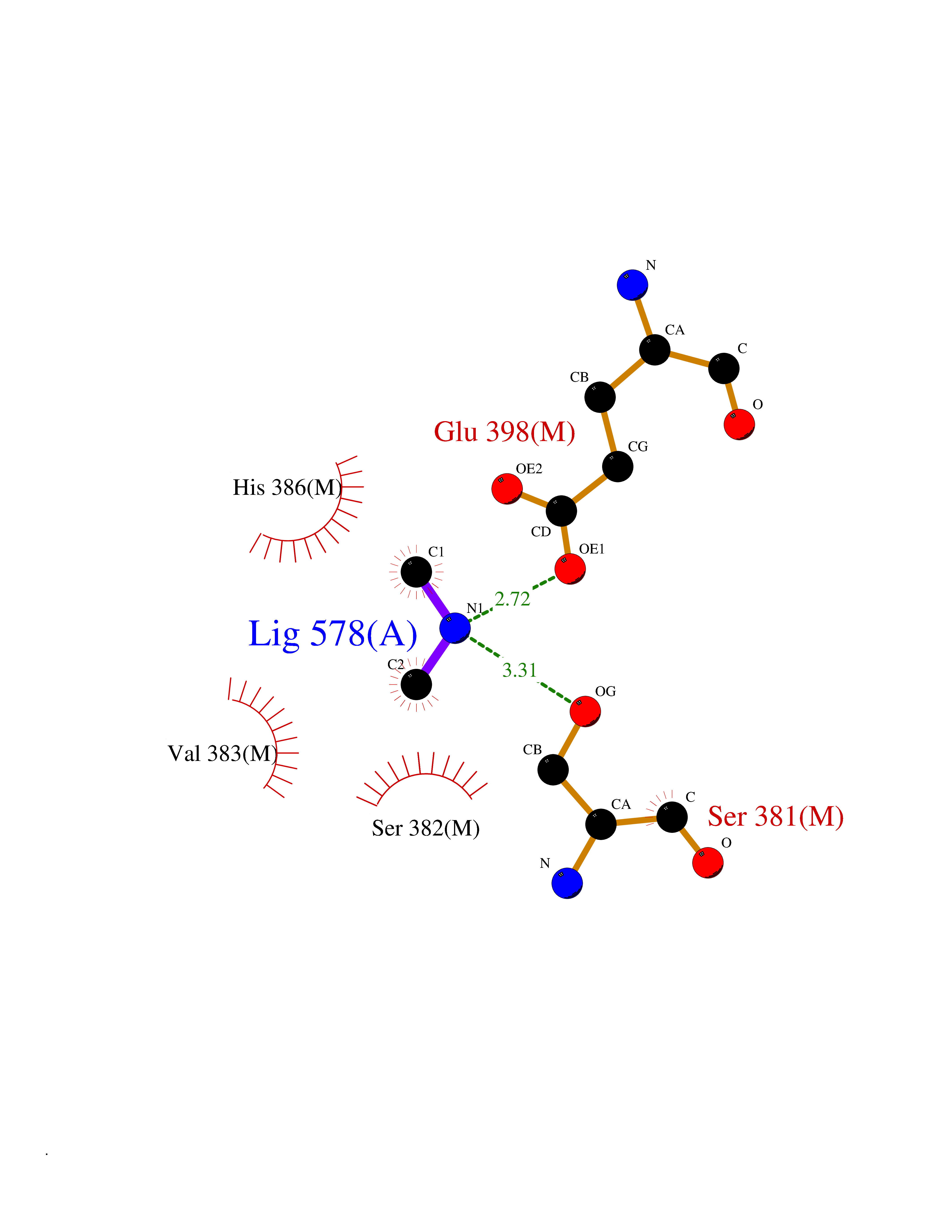 Ligplot Map 3D Binding mode Sequence MQTFQADLAIVGAGGAGLRAAIAAAQANPNAKIALISKVYPMRSHTVAAEGGSAAVAQDHDSFEYHFHDTVAGGDWLCEQDVVDYFVHHCPTEMTQLELWGCPWSRRPDGSVNVRRFGGMKIERTWFAADKTGFHMLHTLFQTSLQFPQIQRFDEHFVLDILVDDGHVRGLVAMNMMEGTLVQIRANAVVMATGGAGRVYRYNTNGGIVTGDGMGMALSHGVPLRDMEFVQYHPTGLPGSGILMTEGCRGEGGILVNKNGYRYLQDYGMGPETPLGEPKNKYMELGPRDKVSQAFWHEWRKGNTISTPRGDVVYLDLRHLGEKKLHERLPFICELAKAYVGVDPVKEPIPVRPTAHYTMGGIETDQNCETRIKGLFAVGECSSVGLHGANRLGSNSLAELVVFGRLAGEQATERAATAGNGNEAAIEAQAAGVEQRLKDLVNQDGGENWAKIRDEMGLAMEEGCGIYRTPELMQKTIDKLAELQERFKRVRITDTSSVFNTDLLYTIELGHGLNVAECMAHSAMARKESRGAHQRLDEGCTERDDVNFLKHTLAFRDADGTTRLEYSDVKITTLPPAAEMKNLKIEVVRYNPEVDTAPHSAFYEVPYDATTSLLDALGYIKDNLAPDLSYRWSCRMAICGSCGMMVNNVPKLACKTFLRDYTDGMKVEALANFPIERDLVVDMTHFIESLEAIKPYIIGNSRTADQGTNIQTPAQMAKYHQFSGCINCGLCYAACPQFGLNPEFIGPAAITLAHRYNEDSRDHGKKERMAQLNSQNGVWSCTFVGYCSEVCPKHVDPAAAIQQGKVESSKDFLIATLKPR Hydrogen bonds contact Hydrophobic contact | ||||
| 63 | Neprilysin | 1R1H | 4.00 | |
Target general information Gen name MME Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms EPN Protein family Peptidase M13 family Biochemical class Hydrolase Function Endopeptidase activity.Exopeptidase activity.Metalloendopeptidase activity.Metallopeptidase activity.Peptide binding.Zinc ion binding. Related diseases Charcot-Marie-Tooth disease, axonal, 2T (CMT2T) [MIM:617017]: An axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. {ECO:0000269|PubMed:26991897, ECO:0000269|PubMed:27588448}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Spinocerebellar ataxia 43 (SCA43) [MIM:617018]: A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA43 is a slowly progressive, autosomal dominant form. {ECO:0000269|PubMed:27583304}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08575; DB02597; DB00616; DB11623; DB05796; DB06655; DB02558; DB02062; DB00886; DB02557; DB09292; DB13928; DB08626 Interacts with P05067; P21926; Q06787-7; P08107; P04792 EC number 3.4.24.11 Uniprot keywords 3D-structure; Cell membrane; Charcot-Marie-Tooth disease; Disease variant; Disulfide bond; Glycoprotein; Hydrolase; Lipoprotein; Membrane; Metal-binding; Metalloprotease; Myristate; Neurodegeneration; Neuropathy; Phosphoprotein; Protease; Proteomics identification; Reference proteome; Signal-anchor; Spinocerebellar ataxia; Transmembrane; Transmembrane helix; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 79435.8 Length 696 Aromaticity 0.11 Instability index 37.5 Isoelectric point 5.53 Charge (pH=7) -11.46 2D Binding mode Binding energy (Kcal/mol) -5.45 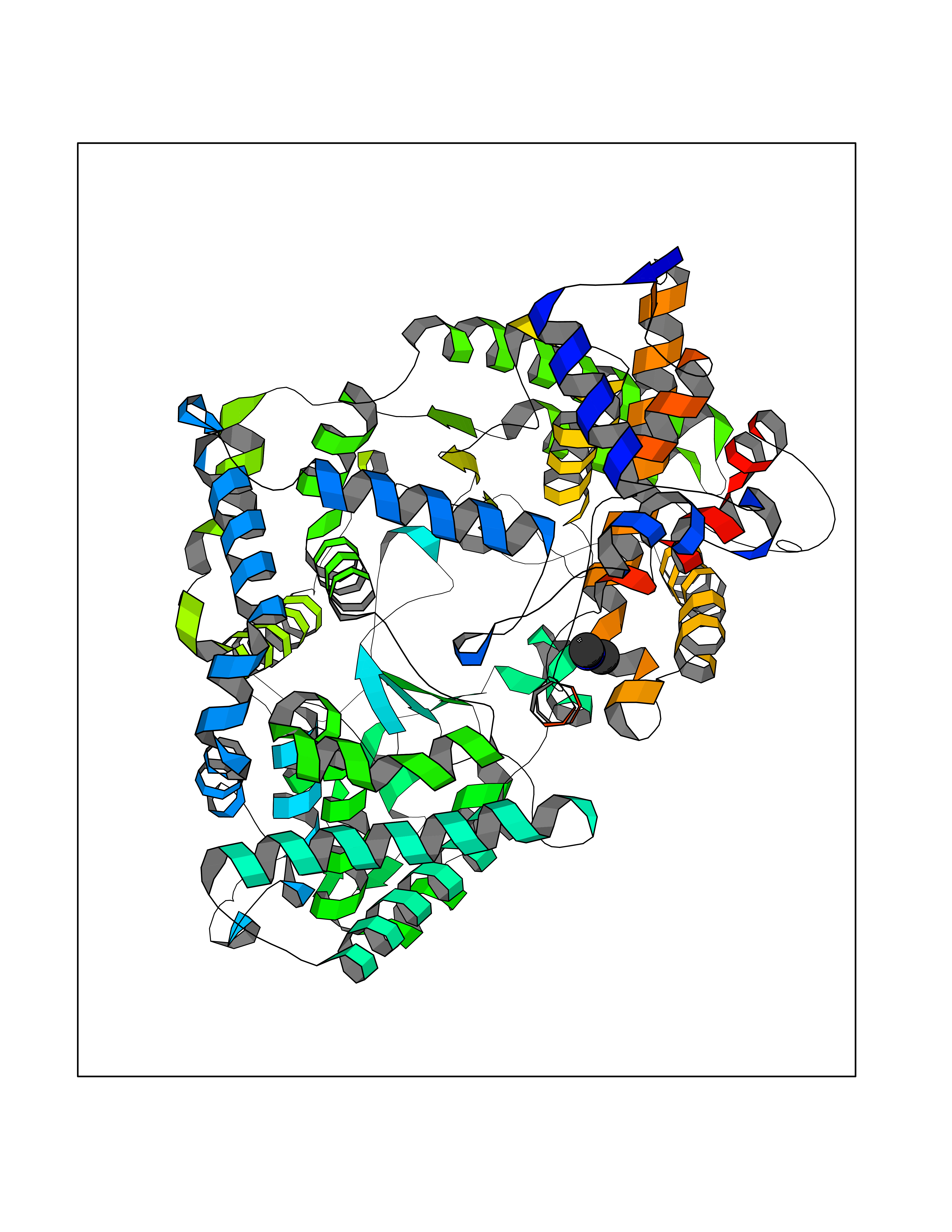 Molscript Map 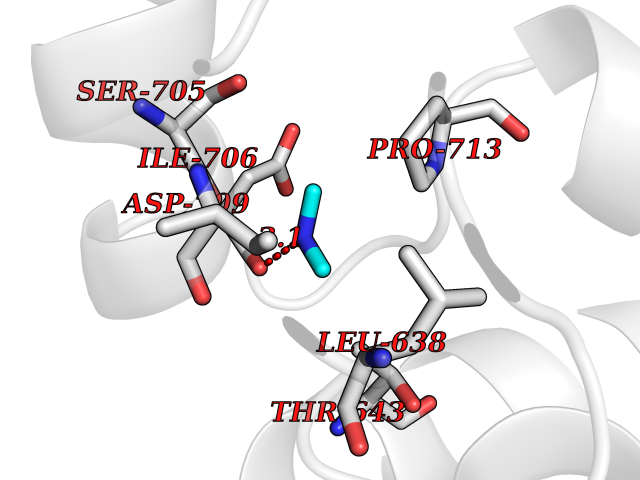 Pymol Map 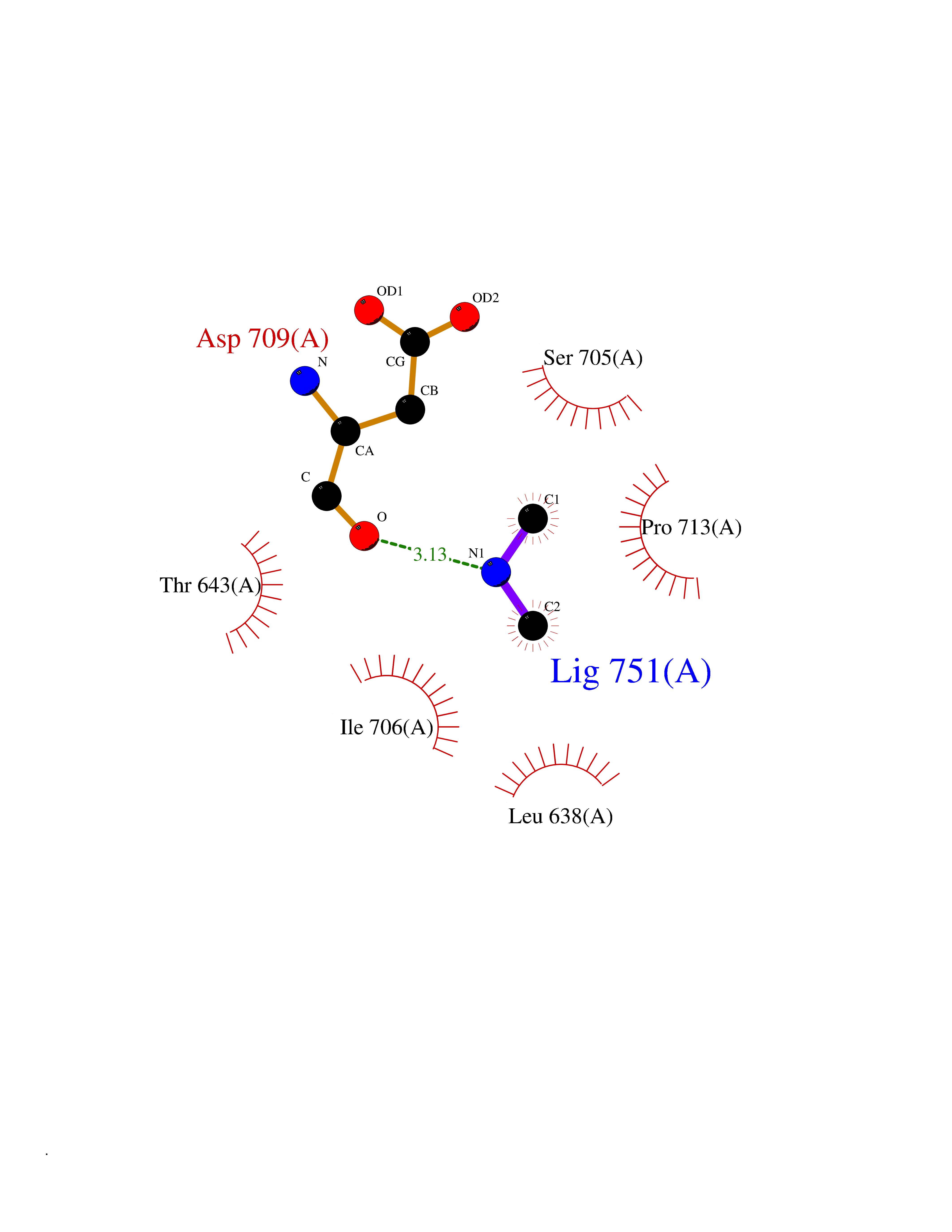 Ligplot Map 3D Binding mode Sequence GICKSSDCIKSAARLIQNMDATTEPCTDFFKYACGGWLKRNVIPETSSRYGNFDILRDELEVVLKDVLQEPKTEDIVAVQKAKALYRSCINESAIDSRGGEPLLKLLPDIYGWPVATENWEQKYGASWTAEKAIAQLNSKYGKKVLINLFVGTDDKNSVNHVIHIDQPRLGLPSRDYYECTGIYKEACTAYVDFMISVARLIRQEERLPIDENQLALEMNKVMELEKEIANATAKPEDRNDPMLLYNKMTLAQIQNNFSLEINGKPFSWLNFTNEIMSTVNISITNEEDVVVYAPEYLTKLKPILTKYSARDLQNLMSWRFIMDLVSSLSRTYKESRNAFRKALYGTTSETATWRRCANYVNGNMENAVGRLYVEAAFAGESKHVVEDLIAQIREVFIQTLDDLTWMDAETKKRAEEKALAIKERIGYPDDIVSNDNKLNNEYLELNYKEDEYFENIIQNLKFSQSKQLKKLREKVDKDEWISGAAVVNAFYSSGRNQIVFPAGILQPPFFSAQQSNSLNYGGIGMVIGHEITHGFDDNGRNFNKDGDLVDWWTQQSASNFKEQSQCMVYQYGNFSWDLAGGQHLNGINTLGENIADNGGLGQAYRAYQNYIKKNGEEKLLPGLDLNHKQLFFLNFAQVWCGTYRPEYAVNSIKTDVHSPGNFRIIGTLQNSAEFSEAFHCRKNSYMNPEKKCRVW Hydrogen bonds contact Hydrophobic contact | ||||
| 64 | S-adenosylmethionine decarboxylase proenzyme (AMD1) | 1JL0 | 4.00 | |
Target general information Gen name AMD1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms SamDC; S-adenosylmethioninedecarboxylase; AdoMetDC; AMD Protein family Eukaryotic AdoMetDC family Biochemical class Carbon-carbon lyase Function Promotes maintenance and self-renewal of embryonic stem cells, by maintaining spermine levels. Essential for biosynthesis of the polyamines spermidine and spermine. Related diseases Niemann-Pick disease A (NPDA) [MIM:257200]: An early-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Niemann-Pick disease type A is a primarily neurodegenerative disorder characterized by onset within the first year of life, intellectual disability, digestive disorders, failure to thrive, major hepatosplenomegaly, and severe neurologic symptoms. The severe neurological disorders and pulmonary infections lead to an early death, often around the age of four. Clinical features are variable. A phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. {ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1391960, ECO:0000269|PubMed:15221801, ECO:0000269|PubMed:15877209, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:1718266, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:2023926, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430884, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:8680412, ECO:0000269|PubMed:8693491, ECO:0000269|PubMed:9266408, ECO:0000269|PubMed:9660788}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Niemann-Pick disease B (NPDB) [MIM:607616]: A late-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Clinical signs involve only visceral organs. The most constant sign is hepatosplenomegaly which can be associated with pulmonary symptoms. Patients remain free of neurologic manifestations. However, a phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. In Niemann-Pick disease type B, onset of the first symptoms occurs in early childhood and patients can survive into adulthood. {ECO:0000269|PubMed:12369017, ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1301192, ECO:0000269|PubMed:15241805, ECO:0000269|PubMed:16010684, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:16472269, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:1885770, ECO:0000269|PubMed:19050888, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:21098024, ECO:0000269|PubMed:21621718, ECO:0000269|PubMed:22613662, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430512, ECO:0000269|PubMed:25920558, ECO:0000269|PubMed:26084044, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:27659707, ECO:0000269|PubMed:8051942, ECO:0000269|PubMed:8664904}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB08163; DB00118; DB01917 Interacts with P17707; Q96A98; Q8WY91 EC number EC 4.1.1.50 Uniprot keywords 3D-structure; Alternative splicing; Autocatalytic cleavage; Decarboxylase; Direct protein sequencing; Lyase; Phosphoprotein; Polyamine biosynthesis; Proteomics identification; Pyruvate; Reference proteome; S-adenosyl-L-methionine; Schiff base; Spermidine biosynthesis; Zymogen Protein physicochemical properties Chain ID A,B Molecular weight (Da) 35790.5 Length 311 Aromaticity 0.14 Instability index 39.47 Isoelectric point 6.03 Charge (pH=7) -2.01 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map 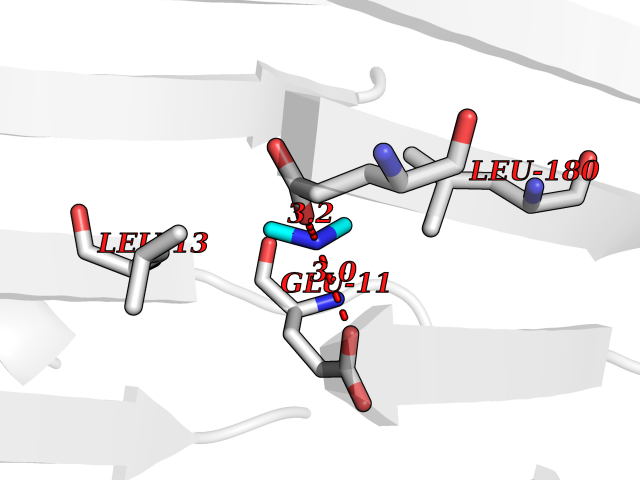 Pymol Map 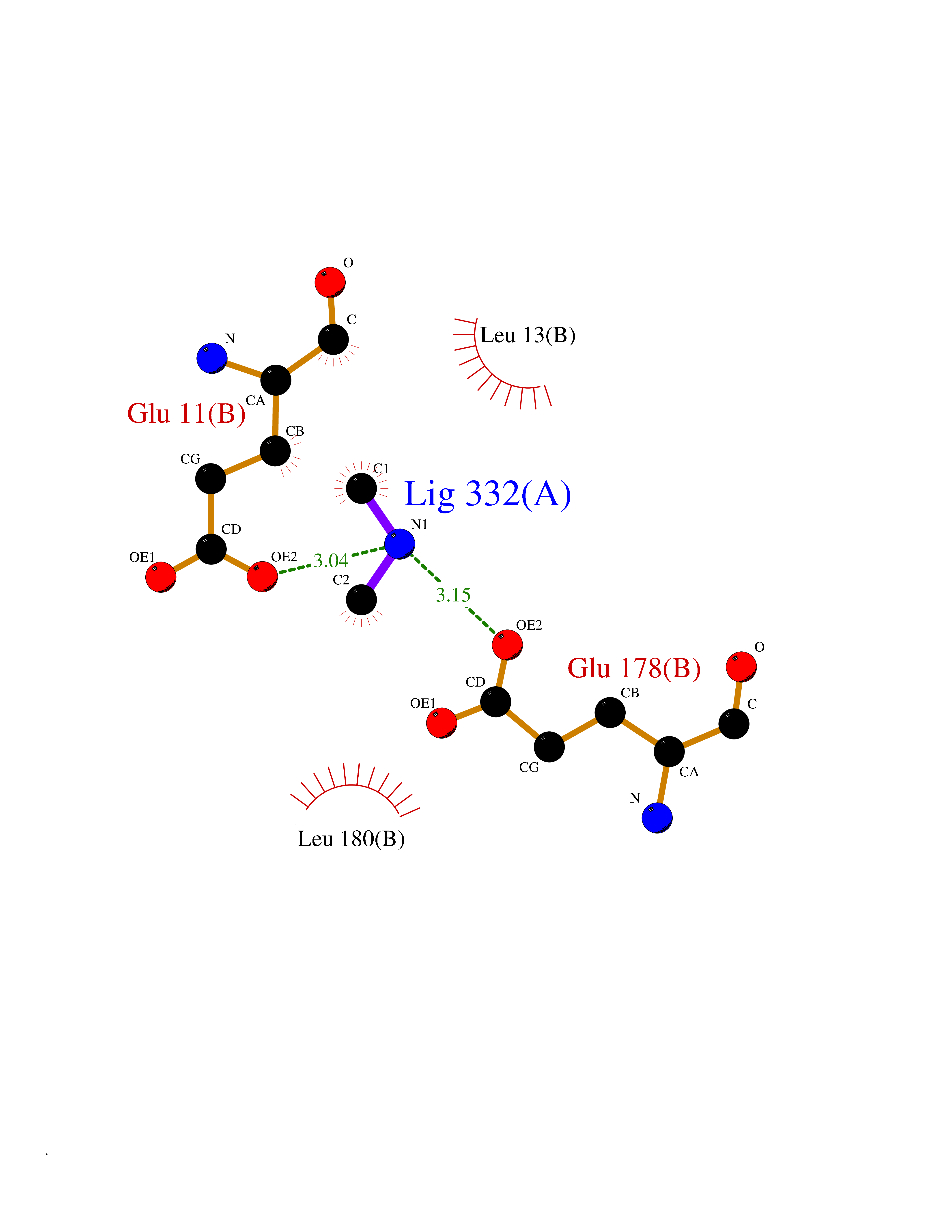 Ligplot Map 3D Binding mode Sequence HFFEGTEKLLEVWFSRQGSGDLRTIPRSEWDILLKDVQCSIISVTKTDKQEAYVLSESSMFVSKRRFILKTCGTTLLLKALVPLLKLARDYSGFDSIQSFFYSRKNFMKPSHQGYPHRNFQEEIEFLNAIFPNGAGYCMGRMNSDCWYLYTLDFRVISQPDQTLEILMSELDPAVMDQFYMKDGVTAKDVTRESGIRDLIPGSVIDATMFNPCGYSMNGMKSDGTYWTIAITPEPEFSYVSFETNLSQTSYDDLIRKVVEVFKPGKFVTTLFVNQSSKCPQKIEGFKRLDCQSAMFNDYNFVFTSFAKKQQ Hydrogen bonds contact Hydrophobic contact | ||||
| 65 | Sphingomyelin phosphodiesterase | 5I85 | 4.00 | |
Target general information Gen name SMPD1 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms ASM Protein family Acid sphingomyelinase family Biochemical class Hydrolase Function Acid sphingomyelin phosphodiesterase activity.Hydrolase activity, acting on glycosyl bonds.Sphingomyelin phosphodiesterase activity.Zinc ion binding. Related diseases Niemann-Pick disease A (NPDA) [MIM:257200]: An early-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Niemann-Pick disease type A is a primarily neurodegenerative disorder characterized by onset within the first year of life, intellectual disability, digestive disorders, failure to thrive, major hepatosplenomegaly, and severe neurologic symptoms. The severe neurological disorders and pulmonary infections lead to an early death, often around the age of four. Clinical features are variable. A phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. {ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1391960, ECO:0000269|PubMed:15221801, ECO:0000269|PubMed:15877209, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:1718266, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:2023926, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430884, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:8680412, ECO:0000269|PubMed:8693491, ECO:0000269|PubMed:9266408, ECO:0000269|PubMed:9660788}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Niemann-Pick disease B (NPDB) [MIM:607616]: A late-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Clinical signs involve only visceral organs. The most constant sign is hepatosplenomegaly which can be associated with pulmonary symptoms. Patients remain free of neurologic manifestations. However, a phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B. In Niemann-Pick disease type B, onset of the first symptoms occurs in early childhood and patients can survive into adulthood. {ECO:0000269|PubMed:12369017, ECO:0000269|PubMed:12556236, ECO:0000269|PubMed:1301192, ECO:0000269|PubMed:15241805, ECO:0000269|PubMed:16010684, ECO:0000269|PubMed:1618760, ECO:0000269|PubMed:16472269, ECO:0000269|PubMed:18815062, ECO:0000269|PubMed:1885770, ECO:0000269|PubMed:19050888, ECO:0000269|PubMed:19405096, ECO:0000269|PubMed:20386867, ECO:0000269|PubMed:21098024, ECO:0000269|PubMed:21621718, ECO:0000269|PubMed:22613662, ECO:0000269|PubMed:22818240, ECO:0000269|PubMed:23252888, ECO:0000269|PubMed:23430512, ECO:0000269|PubMed:25920558, ECO:0000269|PubMed:26084044, ECO:0000269|PubMed:26499107, ECO:0000269|PubMed:27338287, ECO:0000269|PubMed:27659707, ECO:0000269|PubMed:8051942, ECO:0000269|PubMed:8664904}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00381; DB12151; DB00477; DB01151; DB14009 Interacts with P55210 EC number 3.1.4.12; 3.1.4.3 Uniprot keywords 3D-structure; Alternative splicing; Direct protein sequencing; Disease variant; Disulfide bond; Glycoprotein; Glycosidase; Host-virus interaction; Hydrolase; Lipid droplet; Lipid metabolism; Lysosome; Metal-binding; Neurodegeneration; Niemann-Pick disease; Phosphoprotein; Proteomics identification; Reference proteome; Secreted; Signal; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 58913.8 Length 528 Aromaticity 0.11 Instability index 46.93 Isoelectric point 6.48 Charge (pH=7) -3.6 2D Binding mode Binding energy (Kcal/mol) -5.45  Molscript Map 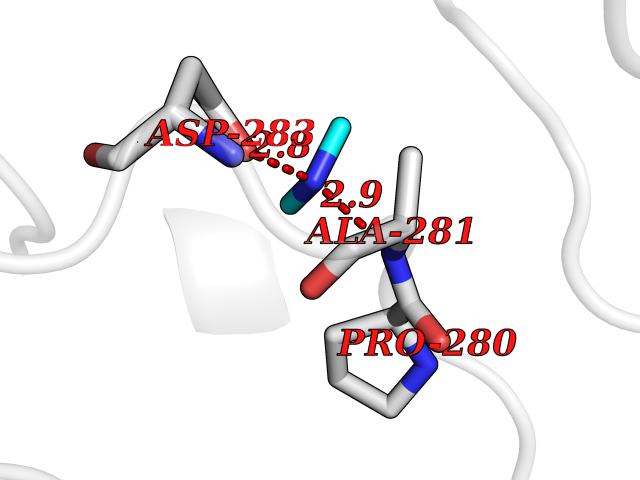 Pymol Map 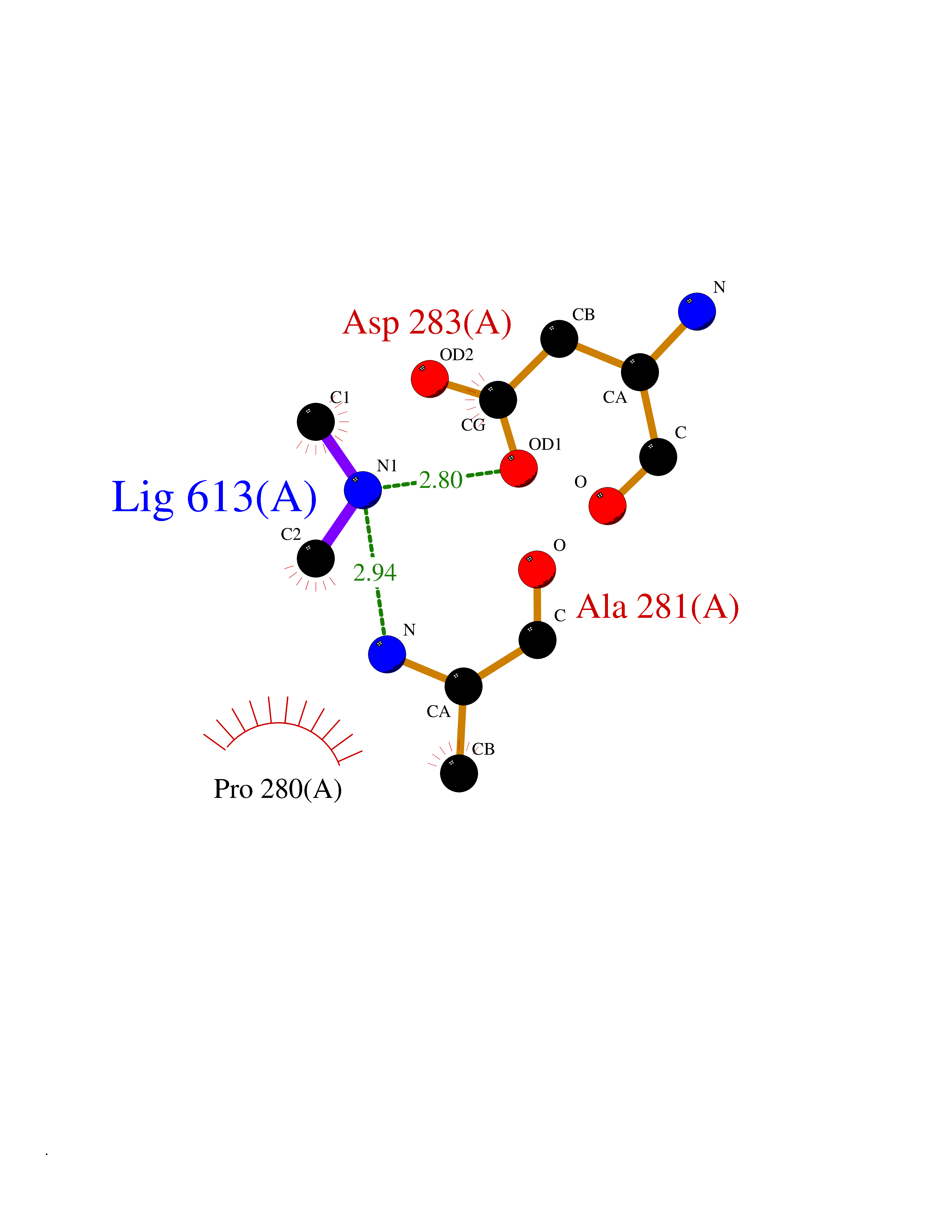 Ligplot Map 3D Binding mode Sequence WGNLTCPICKGLFTAINLGLKKEPNVARVGSVAIKLCNLLKIAPPAVCQSIVHLFEDDMVEVWRRSVLSPSEACGLLLGSTCGHWDIFSSWNISLPTVPKPPPKPPSPPAPGAPVSRILFLTDLHWDHDYLEGTDPDCADPLCCRRGSGLPPASRPGAGYWGEYSKCDLPLRTLESLLSGLGPAGPFDMVYWTGDIPAHDVWHQTRQDQLRALTTVTALVRKFLGPVPVYPAVGNHESTPVNSFPPPFIEGNHSSRWLYEAMAKAWEPWLPAEALRTLRIGGFYALSPYPGLRLISLNMNFCSRENFWLLINSTDPAGQLQWLVGELQAAEDRGDKVHIIGHIPPGHCLKSWSWNYYRIVARYENTLAAQFFGHTHVDEFEVFYDEETLSRPLAVAFLAPSATTYIGLNPGYRVYQIDGNYSGSSHVVLDHETYILNLTQANIPGAIPHWQLLYRARETYGLPNTLPTAWHNLVYRMRGDMQLFQTFWFLYHKGHPPSEPCGTPCRLATLCAQLSARADSPALCRHLM Hydrogen bonds contact Hydrophobic contact | ||||
| 66 | Histidine decarboxylase (HDC) | 4E1O | 4.00 | |
Target general information Gen name HDC Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Human histidine decarboxylase Protein family Group II decarboxylase family Biochemical class Carbon-carbon lyase Function Catalyzes the biosynthesis of histamine from histidine. Related diseases Corticosterone methyloxidase 1 deficiency (CMO-1 deficiency) [MIM:203400]: Autosomal recessive disorder of aldosterone biosynthesis. There are two biochemically different forms of selective aldosterone deficiency be termed corticosterone methyloxidase (CMO) deficiency type 1 and type 2. In CMO-1 deficiency, aldosterone is undetectable in plasma, while its immediate precursor, 18-hydroxycorticosterone, is low or normal. {ECO:0000269|PubMed:11238478, ECO:0000269|PubMed:8439335, ECO:0000269|PubMed:9177280}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Corticosterone methyloxidase 2 deficiency (CMO-2 deficiency) [MIM:610600]: Autosomal recessive disorder of aldosterone biosynthesis. In CMO-2 deficiency, aldosterone can be low or normal, but at the expense of increased secretion of 18-hydroxycorticosterone. Consequently, patients have a greatly increased ratio of 18-hydroxycorticosterone to aldosterone and a low ratio of corticosterone to 18-hydroxycorticosterone in serum. {ECO:0000269|PubMed:12788848, ECO:0000269|PubMed:1346492, ECO:0000269|PubMed:1594605, ECO:0000269|PubMed:9625333, ECO:0000269|PubMed:9814506}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00117; DB00114 Interacts with Q86UW9 EC number EC 4.1.1.22 Uniprot keywords 3D-structure; Alternative splicing; Catecholamine biosynthesis; Decarboxylase; Lyase; Proteomics identification; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A,B,C,D,E,F Molecular weight (Da) 107706 Length 956 Aromaticity 0.1 Instability index 55.17 Isoelectric point 6.23 Charge (pH=7) -9.63 2D Binding mode Binding energy (Kcal/mol) -5.45 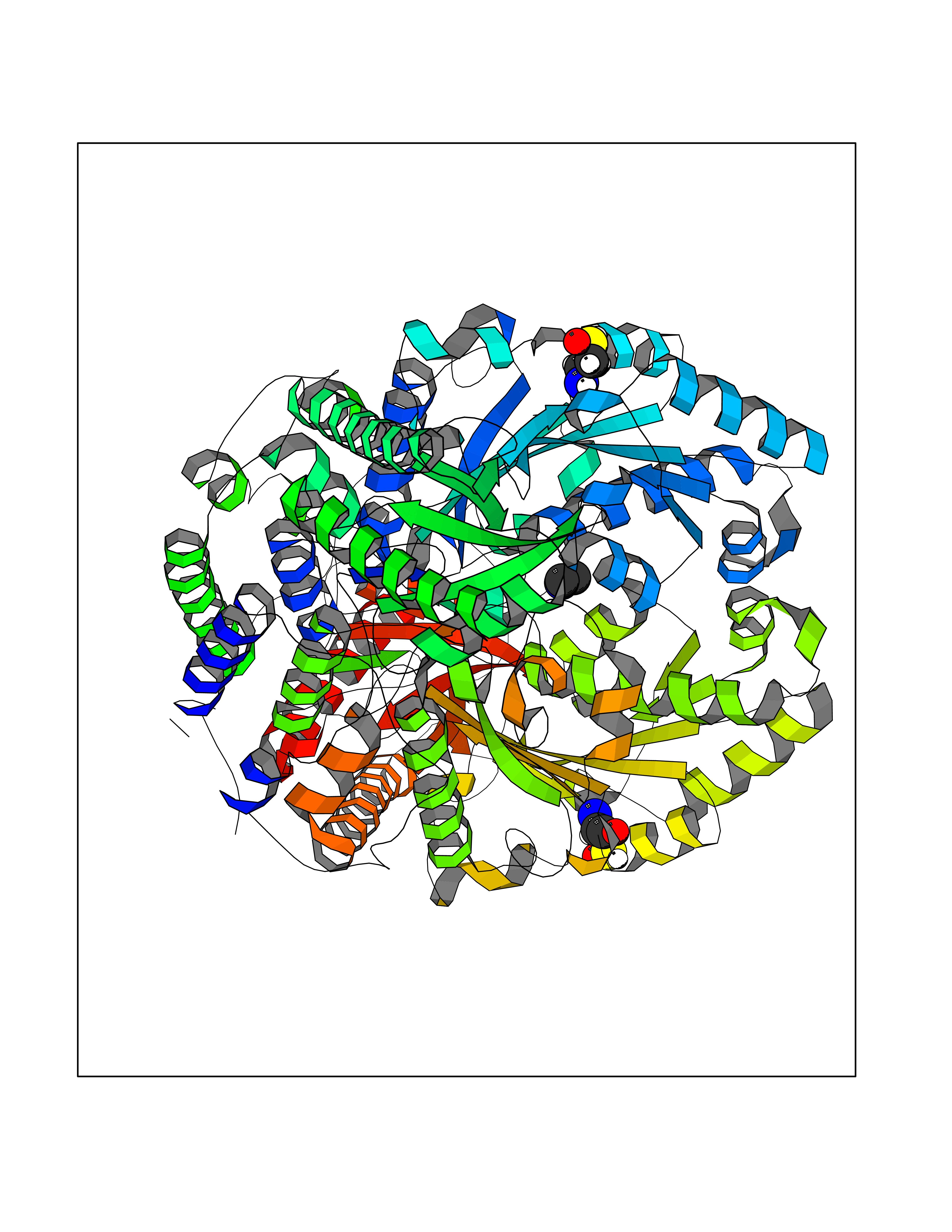 Molscript Map 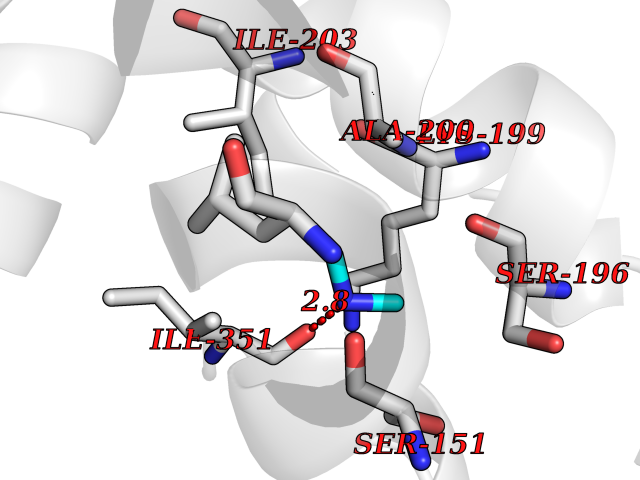 Pymol Map 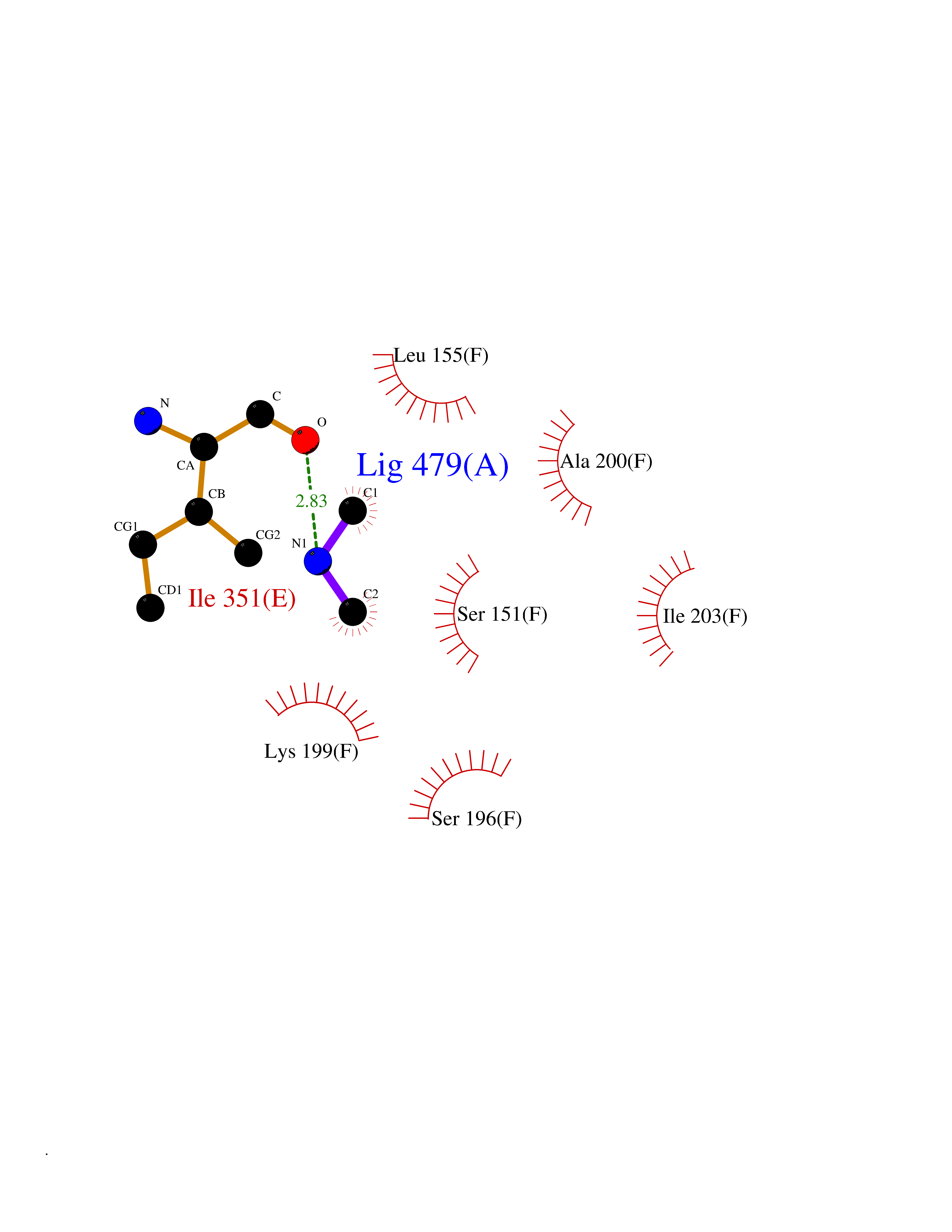 Ligplot Map 3D Binding mode Sequence GSMEPEEYRERGREMVDYICQYLSTVRERRVTPDVQPGYLRAQLPESAPEDPDSWDSIFGDIERIIMPGVVHWQSPHMHAYYPALTSWPSLLGDMLADAINCLGFTWASSPACTELEMNVMDWLAKMLGLPEHFLHHHPSSQGGGVLQSTVSESTLIALLAARKNKILEMKTSEPDADESSLNARLVAYASDQAHSSVEKAGLISLVKMKFLPVDDNFSLRGEALQKAIEEDKQRGLVPVFVCATLGTTGVCAFDXLSELGPICAREGLWLHIDAAYAGTAFLCPEFRGFLKGIEYADSFTFNPSKWMMVHFDCTGFWVKDKYKLQQTFSVNPIYLRHANSGVATDFMHWQIPLSRRFRSVKLWFVIRSFGVKNLQAHVRHGTEMAKYFESLVRNDPSFEIPAKRHLGLVVFRLKGPNSLTENVLKEIAKAGRLFLIPATIQDKLIIRFTVTSQFTTRDDILRDWNLIRDAATLILSQGSMEPEEYRERGREMVDYICQYLSTVRERRVTPDVQPGYLRAQLPESAPEDPDSWDSIFGDIERIIMPGVVHWQSPHMHAYYPALTSWPSLLGDMLADAINCLGFTWASSPACTELEMNVMDWLAKMLGLPEHFLHHHPSSQGGGVLQSTVSESTLIALLAARKNKILEMKTSEPDADESSLNARLVAYASDQAHSSVEKAGLISLVKMKFLPVDDNFSLRGEALQKAIEEDKQRGLVPVFVCATLGTTGVCAFDXLSELGPICAREGLWLHIDAAYAGTAFLCPEFRGFLKGIEYADSFTFNPSKWMMVHFDCTGFWVKDKYKLQQTFSVNPIYLRHANSGVATDFMHWQIPLSRRFRSVKLWFVIRSFGVKNLQAHVRHGTEMAKYFESLVRNDPSFEIPAKRHLGLVVFRLKGPNSLTENVLKEIAKAGRLFLIPATIQDKLIIRFTVTSQFTTRDDILRDWNLIRDAATLILSQ Hydrogen bonds contact Hydrophobic contact | ||||
| 67 | Deoxycytidine kinase (DCK) | 1P5Z | 4.00 | |
Target general information Gen name DCK Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms dCK Protein family DCK/DGK family Biochemical class Kinase Function Required for the phosphorylation of the deoxyribonucleosides deoxycytidine (dC), deoxyguanosine (dG) and deoxyadenosine (dA). Has broad substrate specificity, and does not display selectivity based on the chirality of the substrate. It is also an essential enzyme for the phosphorylation of numerous nucleoside analogs widely employed as antiviral and chemotherapeutic agents. Related diseases Microvascular complications of diabetes 5 (MVCD5) [MIM:612633]: Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Disease susceptibility is associated with variants affecting the gene represented in this entry. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. Drugs (DrugBank ID) DB02594; DB00242; DB00631; DB00987; DB01262; DB05494; DB00879; DB01073; DB00441; DB00709; DB01280; DB00642; DB04961; DB00943 Interacts with Q16854 EC number EC 2.7.1.74 Uniprot keywords 3D-structure; ATP-binding; Direct protein sequencing; Kinase; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Transferase Protein physicochemical properties Chain ID B Molecular weight (Da) 27128.5 Length 229 Aromaticity 0.13 Instability index 52.5 Isoelectric point 5.26 Charge (pH=7) -7.8 2D Binding mode Binding energy (Kcal/mol) -5.46 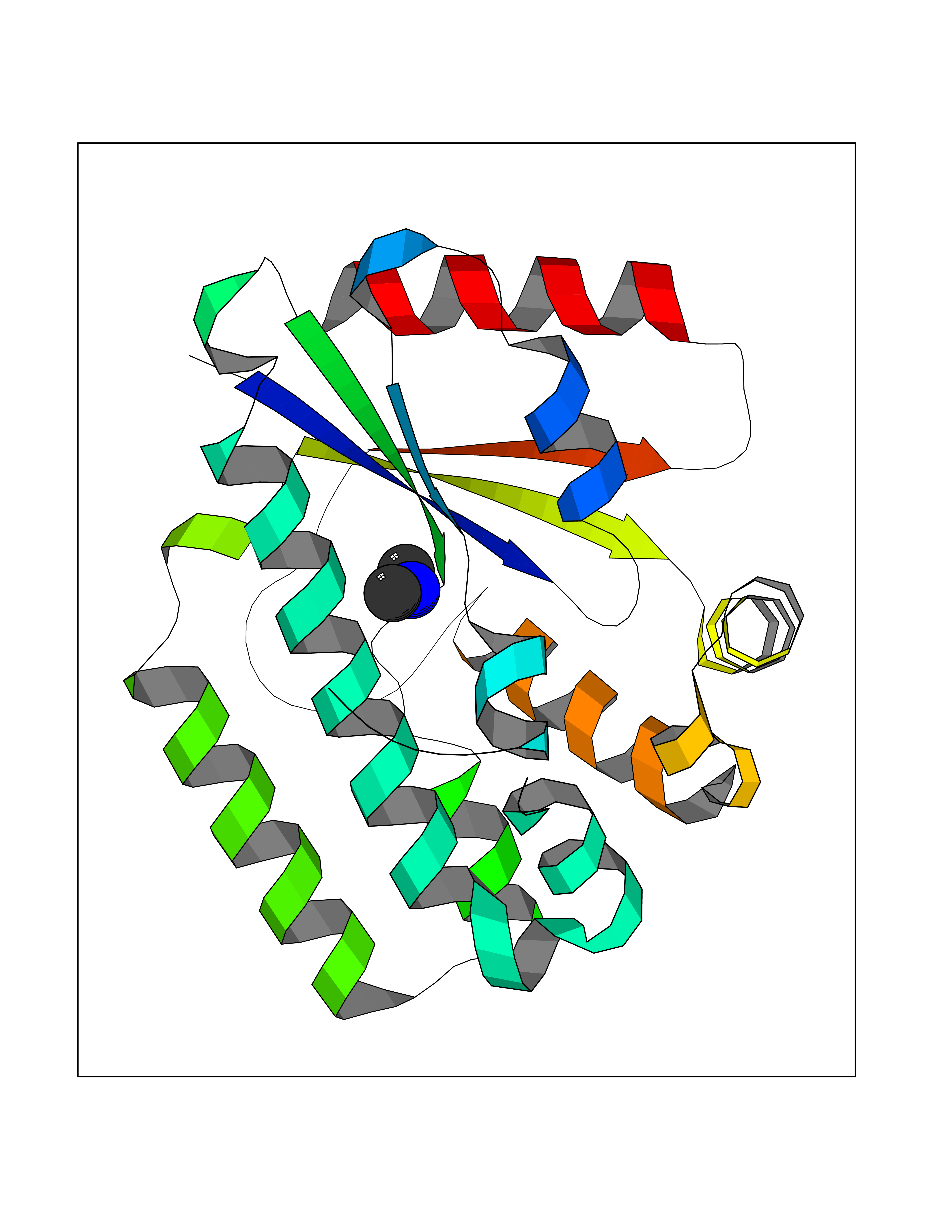 Molscript Map 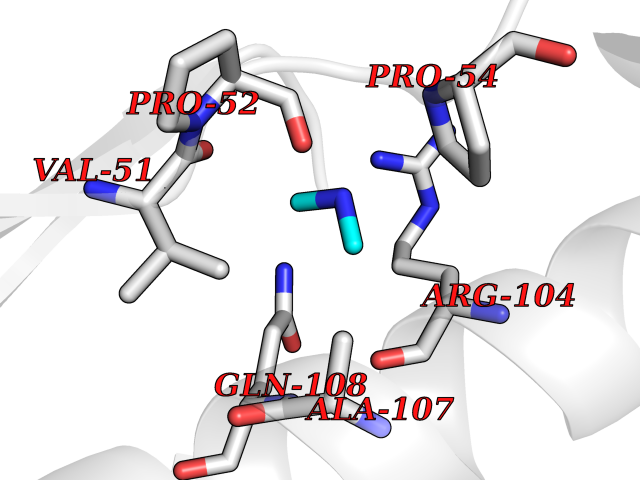 Pymol Map 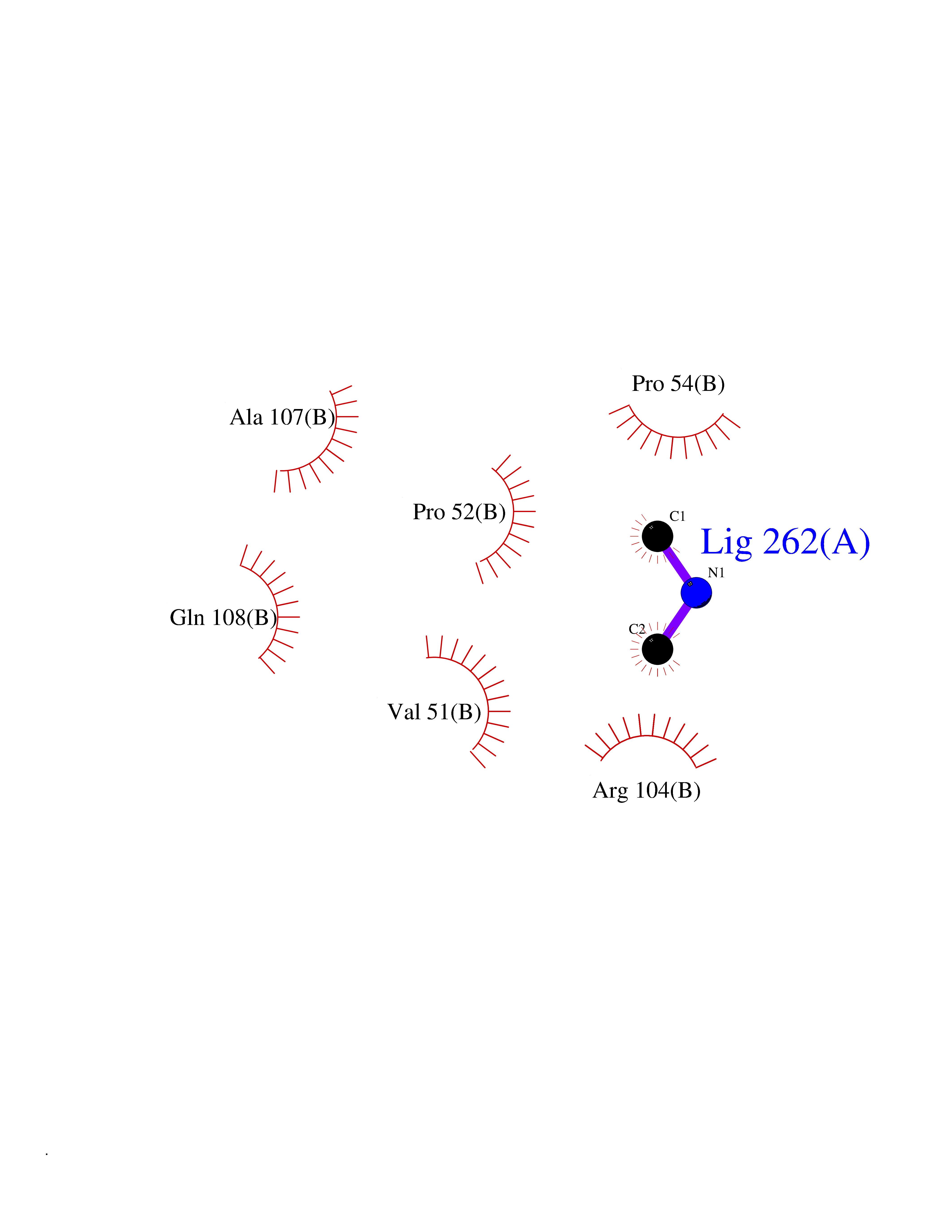 Ligplot Map 3D Binding mode Sequence RIKKISIEGNIAAGKSTFVNILKQLCEDWEVVPEPVARWCNVQSTNGGNVLQMMYEKPERWSFTFQTYACLSRIRAQLASLNGKLKDAEKPVLFFERSVYSDRYIFASNLYESECMNETEWTIYQDWHDWMNNQFGQSLELDGIIYLQATPETCLHRIYLRGRNEEQGIPLEYLEKLHYKHESWLLHRTLKTNFDYLQEVPILTLDVNEDFKDKYESLVEKVKEFLSTL Hydrogen bonds contact Hydrophobic contact | ||||
| 68 | Ribonucleoside-diphosphate reductase subunit M2 | 3OLJ | 4.00 | |
Target general information Gen name RRM2 Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms RR2 Protein family Ribonucleoside diphosphate reductase small chain family Biochemical class Oxidoreductase Function Metal ion binding.Ribonucleoside-diphosphate reductase activity, thioredoxin disulfide as acceptor. Related diseases Pyruvate kinase hyperactivity (PKHYP) [MIM:102900]: Autosomal dominant phenotype characterized by increase of red blood cell ATP. {ECO:0000269|PubMed:9090535}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Pyruvate kinase deficiency of red cells (PKRD) [MIM:266200]: A frequent cause of hereditary non-spherocytic hemolytic anemia. Clinically, pyruvate kinase-deficient patients suffer from a highly variable degree of chronic hemolysis, ranging from severe neonatal jaundice and fatal anemia at birth, severe transfusion-dependent chronic hemolysis, moderate hemolysis with exacerbation during infection, to a fully compensated hemolysis without apparent anemia. {ECO:0000269|PubMed:10087985, ECO:0000269|PubMed:10772876, ECO:0000269|PubMed:11328279, ECO:0000269|PubMed:11960989, ECO:0000269|PubMed:1536957, ECO:0000269|PubMed:1896471, ECO:0000269|PubMed:19085939, ECO:0000269|PubMed:2018831, ECO:0000269|PubMed:21794208, ECO:0000269|PubMed:7706479, ECO:0000269|PubMed:8161798, ECO:0000269|PubMed:8180378, ECO:0000269|PubMed:8476433, ECO:0000269|PubMed:8481523, ECO:0000269|PubMed:8483951, ECO:0000269|PubMed:8664896, ECO:0000269|PubMed:8807089, ECO:0000269|PubMed:9075576, ECO:0000269|PubMed:9482576, ECO:0000269|PubMed:9827908, ECO:0000269|PubMed:9886305, ECO:0000269|Ref.24}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB00242; DB05260; DB05801; DB05003; DB05428 Interacts with P41002; Q9UM11; P23921; O00560 EC number 1.17.4.1 Uniprot keywords 3D-structure; Alternative splicing; Cytoplasm; Deoxyribonucleotide synthesis; Iron; Metal-binding; Nucleus; Oxidoreductase; Phosphoprotein; Proteomics identification; Reference proteome; Ubl conjugation Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 33579.4 Length 286 Aromaticity 0.14 Instability index 43.7 Isoelectric point 5.12 Charge (pH=7) -12.86 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map 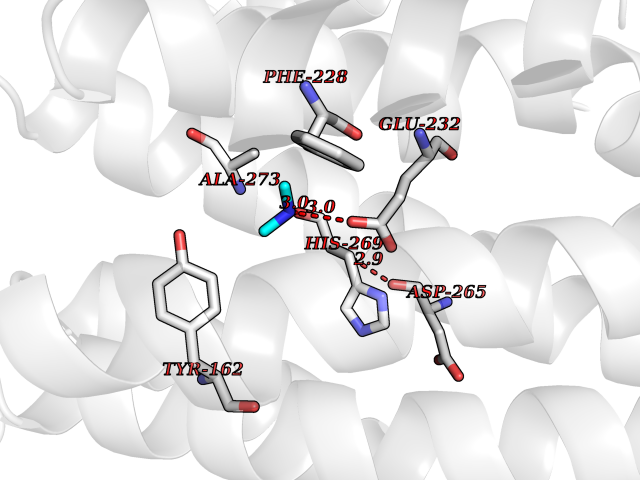 Pymol Map 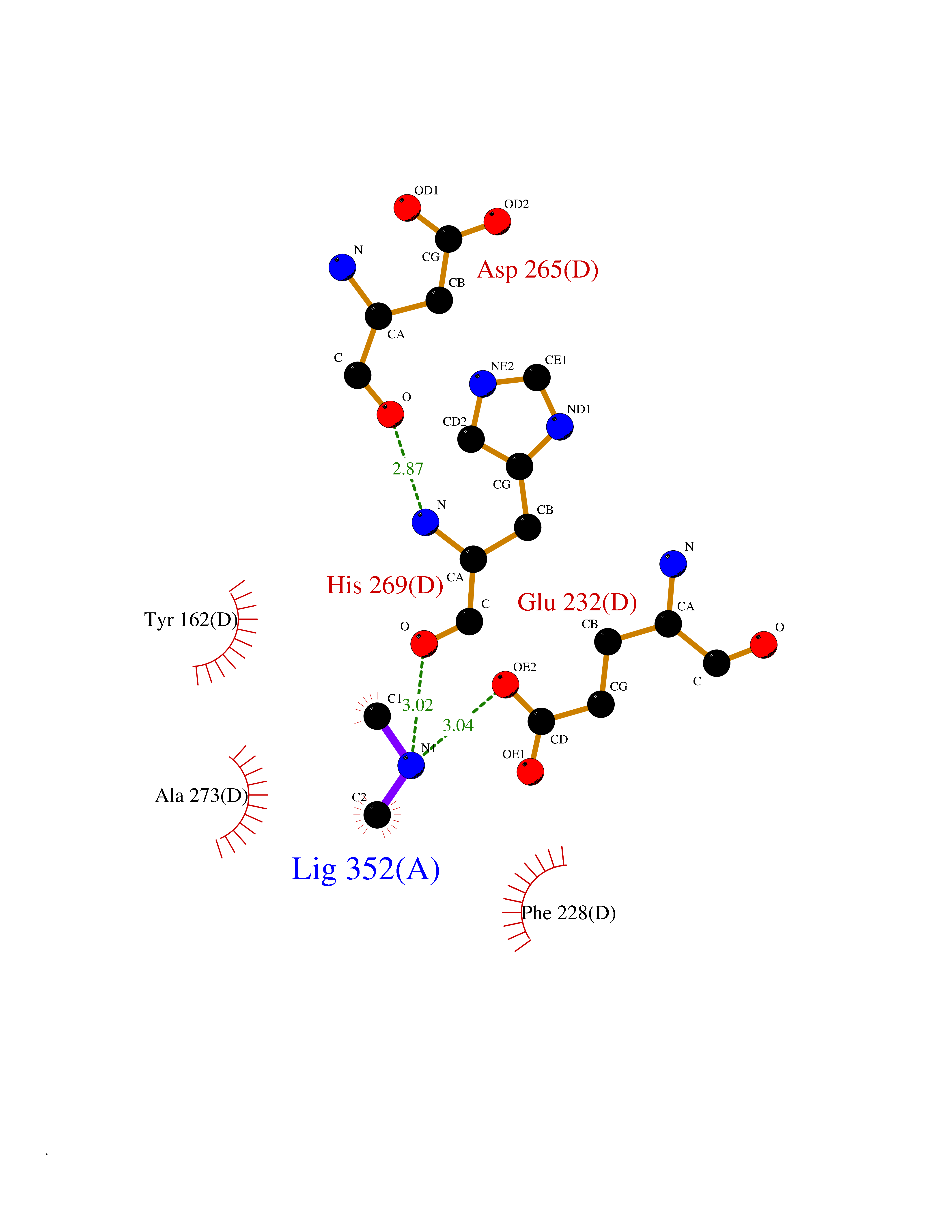 Ligplot Map 3D Binding mode Sequence MGVEDEPLLRENPRRFVIFPIEYHDIWQMYKKAEASFWTAEEVDLSKDIQHWESLKPEERYFISHVLAFFAASDGIVNENLVERFSQEVQITEARCFYGFQIAMENIHSEMYSLLIDTYIKDPKEREFLFNAIETMPCVKKKADWALRWIGDKEATYGERVVAFAAVEGIFFSGSFASIFWLKKRGLMPGLTFSNELISRDEGLHCDFACLMFKHLVHKPSEERVREIIINAVRIEQEFLTEALPVKLIGMNCTLMKQYIEFVADRLMLELGFSKVFRVENPFDFM Hydrogen bonds contact Hydrophobic contact | ||||
| 69 | 4-hydroxyphenylpyruvate dioxygenase | 3ISQ | 4.00 | |
Target general information Gen name HPD Organism Homo sapiens (Human) Uniprot ID TTD ID NA Synonyms PPD Protein family 4HPPD family Biochemical class Oxidoreductase Function 4-hydroxyphenylpyruvate dioxygenase activity.Metal ion binding. Related diseases Tyrosinemia 3 (TYRSN3) [MIM:276710]: An inborn error of metabolism characterized by elevations of tyrosine in the blood and urine, seizures and mild intellectual disability. {ECO:0000269|PubMed:10942115, ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Hawkinsinuria (HWKS) [MIM:140350]: An inborn error of tyrosine metabolism characterized by failure to thrive, persistent metabolic acidosis, fine and sparse hair, and excretion of the unusual cyclic amino acid metabolite, hawkinsin, in the urine. {ECO:0000269|PubMed:11073718}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02850; DB00348 Interacts with NA EC number 1.13.11.27 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Cytoplasm; Dioxygenase; Disease variant; Endoplasmic reticulum; Golgi apparatus; Intellectual disability; Iron; Membrane; Metal-binding; Oxidoreductase; Phenylalanine catabolism; Phosphoprotein; Proteomics identification; Reference proteome; Repeat; Tyrosine catabolism Protein physicochemical properties Chain ID A Molecular weight (Da) 43164.8 Length 376 Aromaticity 0.11 Instability index 32.38 Isoelectric point 6.73 Charge (pH=7) -1.04 2D Binding mode Binding energy (Kcal/mol) -5.45 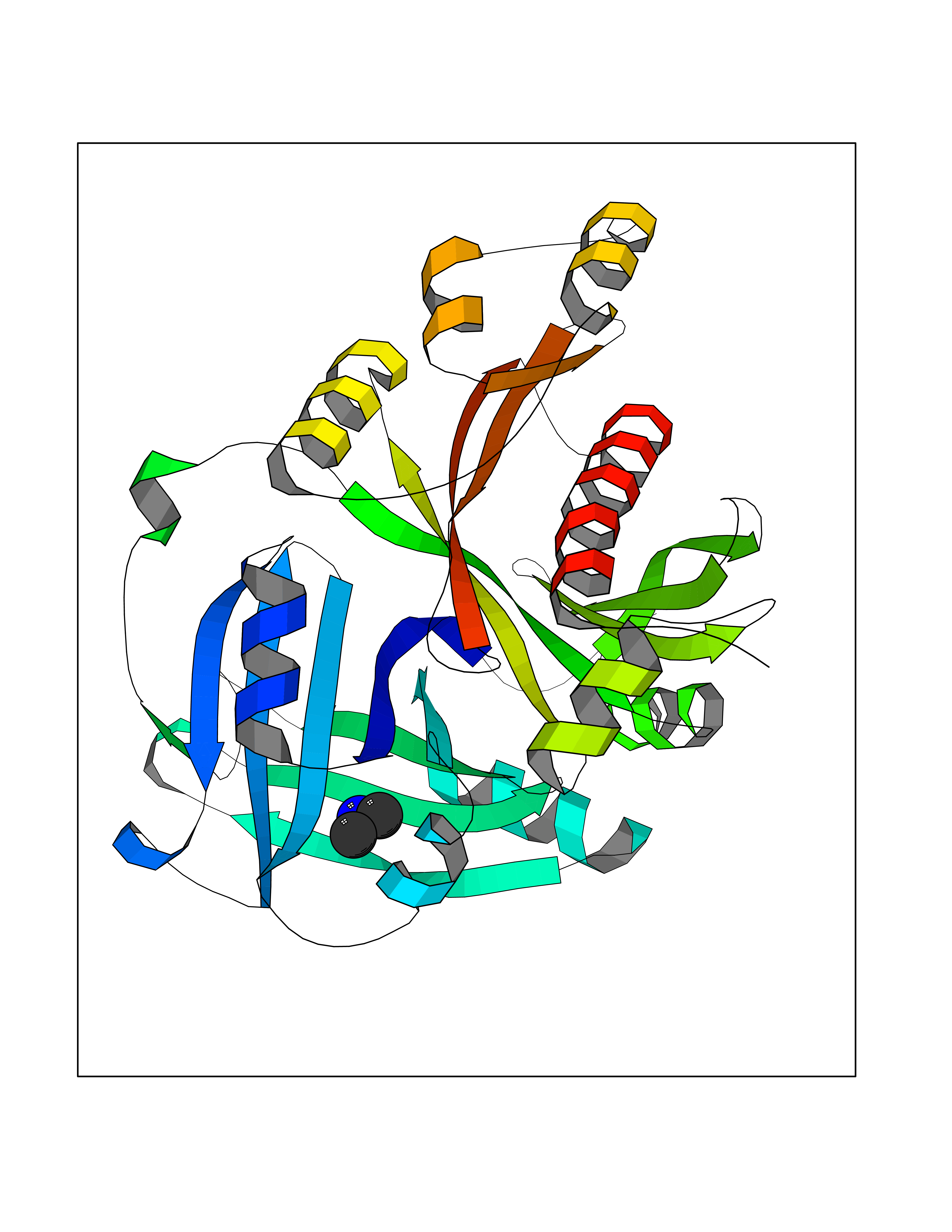 Molscript Map 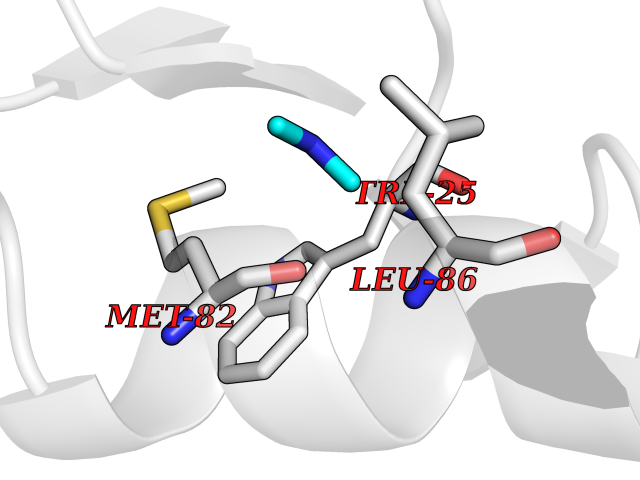 Pymol Map 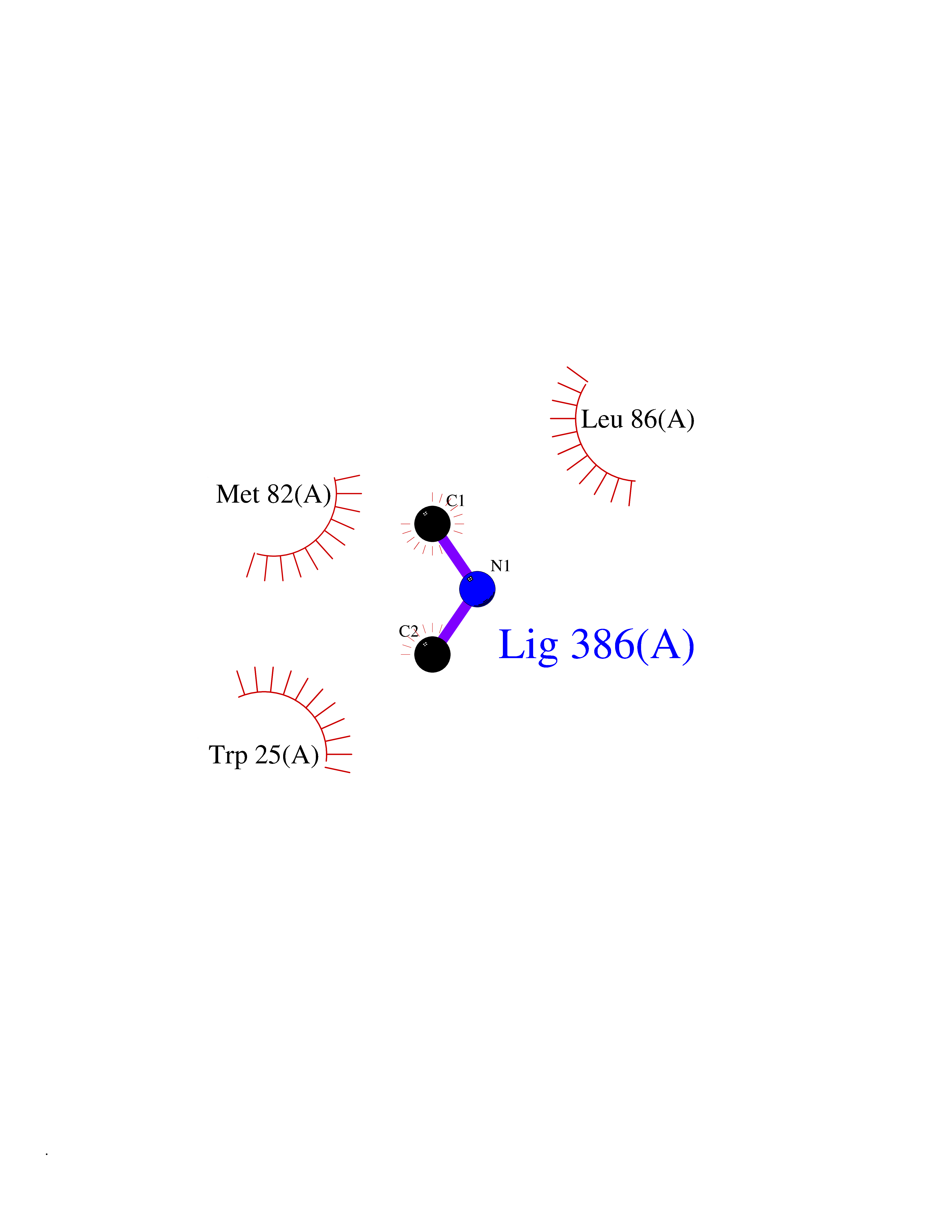 Ligplot Map 3D Binding mode Sequence AKPERGRFLHFHSVTFWVGNAKQAASFYCSKMGFEPLAYRGLETGSREVVSHVIKQGKIVFVLSSALNPWNKEMGDHLVKHGDGVKDIAFEVEDCDYIVQKARERGAKIMREPWVEQDKFGKVKFAVLQTYGDTTHTLVEKMNYIGQFLPGYEAPAFMDPLLPKLPKCSLEMIDHIVGNQPDQEMVSASEWYLKNLQFHRFWSVDDTQVHTEYSSLRSIVVANYEESIKMPINEPAPGKKKSQIQEYVDYNGGAGVQHIALKTEDIITAIRHLRERGLEFLSVPSTYYKQLREKLKTAKIKVKENIDALEELKILVDYDEKGYLLQIFTKPVQDRPTLFLEVIQRHNHQGFGAGNFNSLFKAFEEEQNLRGNLTNM Hydrogen bonds contact Hydrophobic contact | ||||
| 70 | Cystathionine gamma-lyase (CTH) | 3COG | 4.00 | |
Target general information Gen name CTH Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Gamma-cystathionase; Cysteine-protein sulfhydrase Protein family Trans-sulfuration enzymes family Biochemical class NA Function Catalyzes the last step in the trans-sulfuration pathway from methionine to cysteine. Has broad substrate specificity. Converts cystathionine to cysteine, ammonia and 2-oxobutanoate. Converts two cysteine molecules to lanthionine and hydrogen sulfide. Can also accept homocysteine as substrate. Specificity depends on the levels of the endogenous substrates. Generates the endogenous signaling molecule hydrogen sulfide (H2S), and so contributes to the regulation of blood pressure. Acts as a cysteine-protein sulfhydrase by mediating sulfhydration of target proteins: sulfhydration consists of converting -SH groups into -SSH on specific cysteine residues of target proteins such as GAPDH, PTPN1 and NF-kappa-B subunit RELA, thereby regulating their function. Related diseases Cystathioninuria (CSTNU) [MIM:219500]: Autosomal recessive phenotype characterized by abnormal accumulation of plasma cystathionine, leading to increased urinary excretion. {ECO:0000269|PubMed:12574942, ECO:0000269|PubMed:18476726}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB02328; DB03928; DB00151; DB04217; DB00114 Interacts with P32929; Q96NT3; Q96NT3-2; Q96HA8; Q6P9E2 EC number EC 4.4.1.1 Uniprot keywords 3D-structure; Alternative splicing; Amino-acid biosynthesis; Calmodulin-binding; Cysteine biosynthesis; Cytoplasm; Disease variant; Lipid metabolism; Lyase; Proteomics identification; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 86026 Length 782 Aromaticity 0.08 Instability index 32.4 Isoelectric point 6.27 Charge (pH=7) -9.46 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map 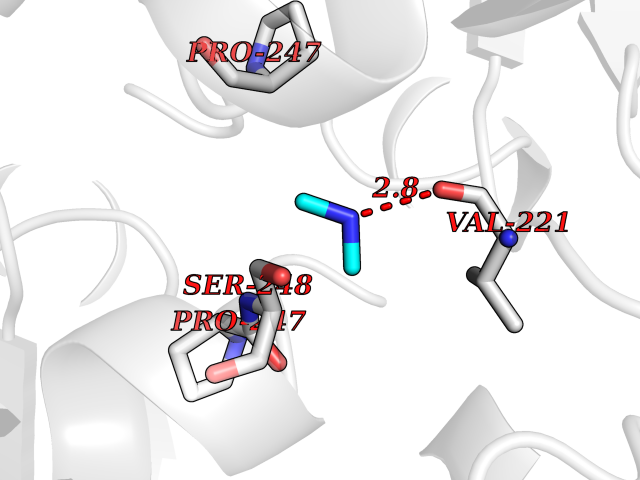 Pymol Map 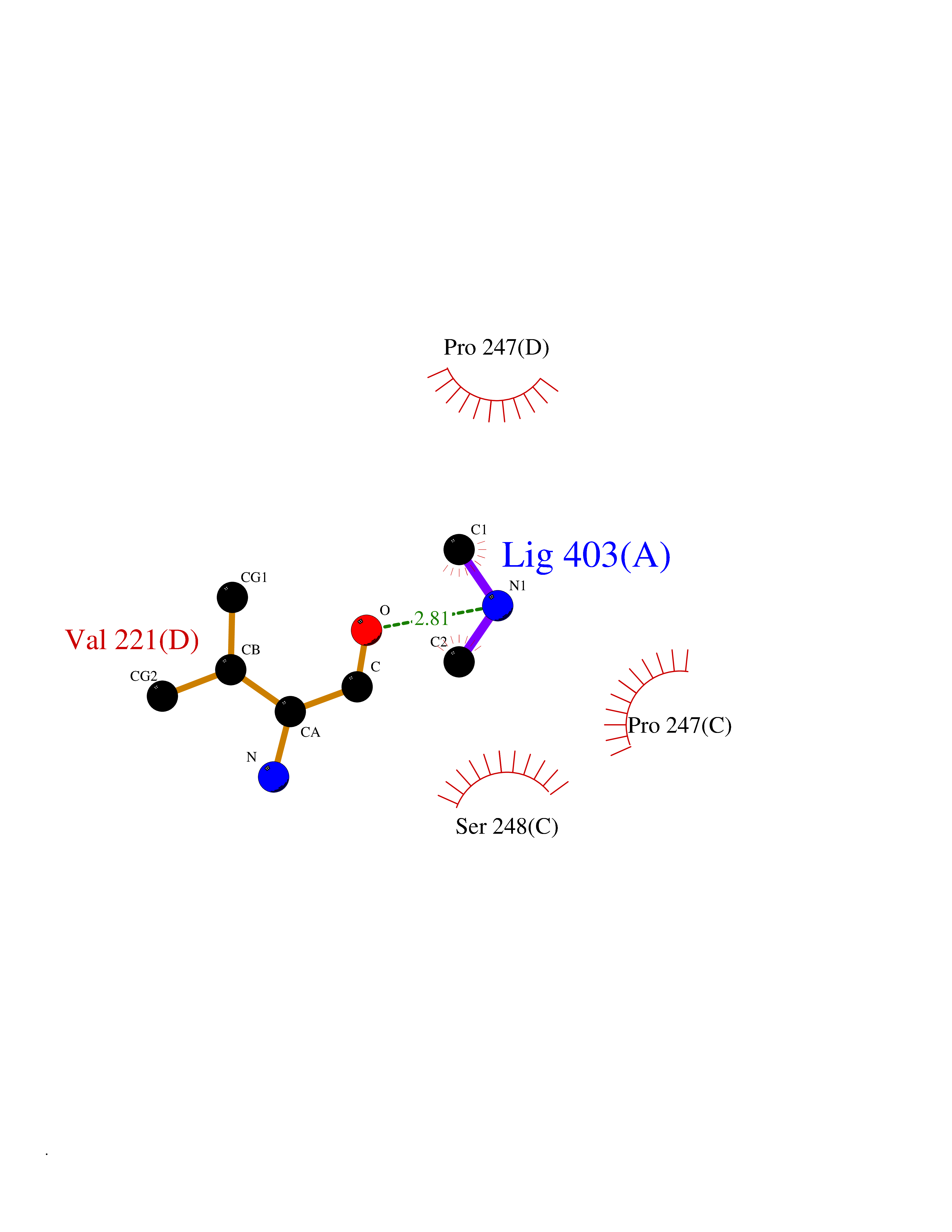 Ligplot Map 3D Binding mode Sequence GFLPHFQHFATQAIHVGQDPEQWTSRAVVPPISLSTTFKQGAPGQHSGFEYSRSGNPTRNCLEKAVAALDGAKYCLAFASGLAATVTITHLLKAGDQIICMDDVYGGTNRYFRQVASEFGLKISFVDCSKIKLLEAAITPETKLVWIETPTNPTQKVIDIEGCAHIVHKHGDIILVVDNTFMSPYFQRPLALGADISMYSATKYMNGHSDVVMGLVSVNCESLHNRLRFLQNSLGAVPSPIDCYLCNRGLKTLHVRMEKHFKNGMAVAQFLESNPWVEKVIYPGLPSHPQHELVKRQCTGCTGMVTFYIKGTLQHAEIFLKNLKLFTLAESLGGFESLAELPAIMTHASVLKNDRDVLGISDTLIRLSVGLEDEEDLLEDLDQALKAAHPPSGFLPHFQHFATQAIHVGQDPEQWTSRAVVPPISLSTTFKQGAPGQGFEYSRSGNPTRNCLEKAVAALDGAKYCLAFASGLAATVTITHLLKAGDQIICMDDVYGGTNRYFRQVASEFGLKISFVDCSKIKLLEAAITPETKLVWIETPTNPTQKVIDIEGCAHIVHKHGDIILVVDNTFMSPYFQRPLALGADISMYSATKYMNGHSDVVMGLVSVNCESLHNRLRFLQNSLGAVPSPIDCYLCNRGLKTLHVRMEKHFKNGMAVAQFLESNPWVEKVIYPGLPSHPQHELVKRQCTGCTGMVTFYIKGTLQHAEIFLKNLKLFTLAESLGGFESLAELPAIMTHASVLKNDRDVLGISDTLIRLSVGLEDEEDLLEDLDQALKAAHPPS Hydrogen bonds contact Hydrophobic contact | ||||
| 71 | Branched-chain-amino-acid transaminase 1 (BCAT1) | 2COI | 4.00 | |
Target general information Gen name BCAT1 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Protein ECA39; ECA39; Branched-chain-amino-acid aminotransferase, cytosolic; BCT1; BCAT(c) Protein family Class-IV pyridoxal-phosphate-dependent aminotransferase family Biochemical class Transaminase Function Catalyzes the first reaction in the catabolism of the essential branched chain amino acids leucine, isoleucine, and valine. Related diseases Charcot-Marie-Tooth disease, dominant intermediate C (CMTDIC) [MIM:608323]: A form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. The dominant intermediate type C is characterized by clinical and pathologic features intermediate between demyelinating and axonal peripheral neuropathies, and motor median nerve conduction velocities ranging from 25 to 45 m/sec. {ECO:0000269|PubMed:16429158}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Neurologic, endocrine, and pancreatic disease, multisystem, infantile-onset 2 (IMNEPD2) [MIM:619418]: An autosomal recessive disorder with variable clinical manifestations and severity. Main features include cholestatic hepatitis, poor feeding, poor overall growth, and hypoglycemia apparent from infancy. Most patients have variable global developmental delay, sensorineural deafness, retinal abnormalities with visual defects, and hypotonia. Some patients have endocrine abnormalities. Brain imaging often shows dysmyelination, thin corpus callosum, cerebral atrophy, and white matter abnormalities. Death in early childhood may occur. {ECO:0000269|PubMed:27633801, ECO:0000269|PubMed:29232904, ECO:0000269|PubMed:30304524}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Defects in YARS1 may be the cause of proximal-predominant motor neuropathy. Affected individuals may develop tremors, cramping of hands, asymmetric weakness in the upper and lower extremities, and present with elevated creatine kinase levels. {ECO:0000269|PubMed:36307205}. Drugs (DrugBank ID) DB00996; DB00142; DB00167; DB00149; DB07544; DB00114; DB00161 Interacts with P55212; O75190-2; O14645; P22607; P06396; O14901; P13473-2; O75400-2 EC number EC 2.6.1.42 Uniprot keywords 3D-structure; Acetylation; Alternative splicing; Amino-acid biosynthesis; Aminotransferase; Branched-chain amino acid biosynthesis; Cytoplasm; Lipid metabolism; Proteomics identification; Pyridoxal phosphate; Reference proteome; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 40123.7 Length 356 Aromaticity 0.1 Instability index 32.34 Isoelectric point 5.39 Charge (pH=7) -9 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map 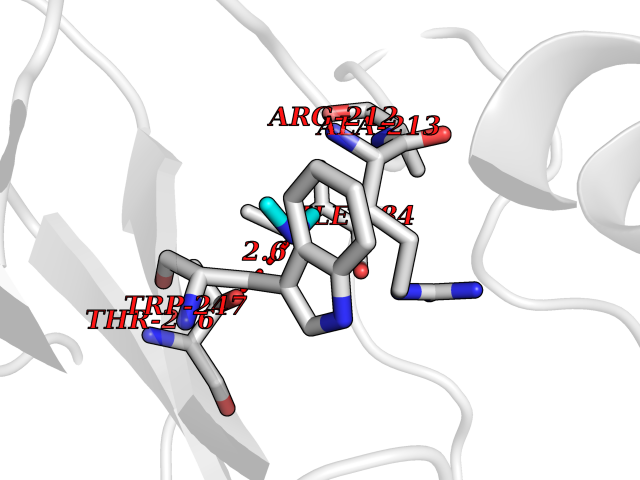 Pymol Map 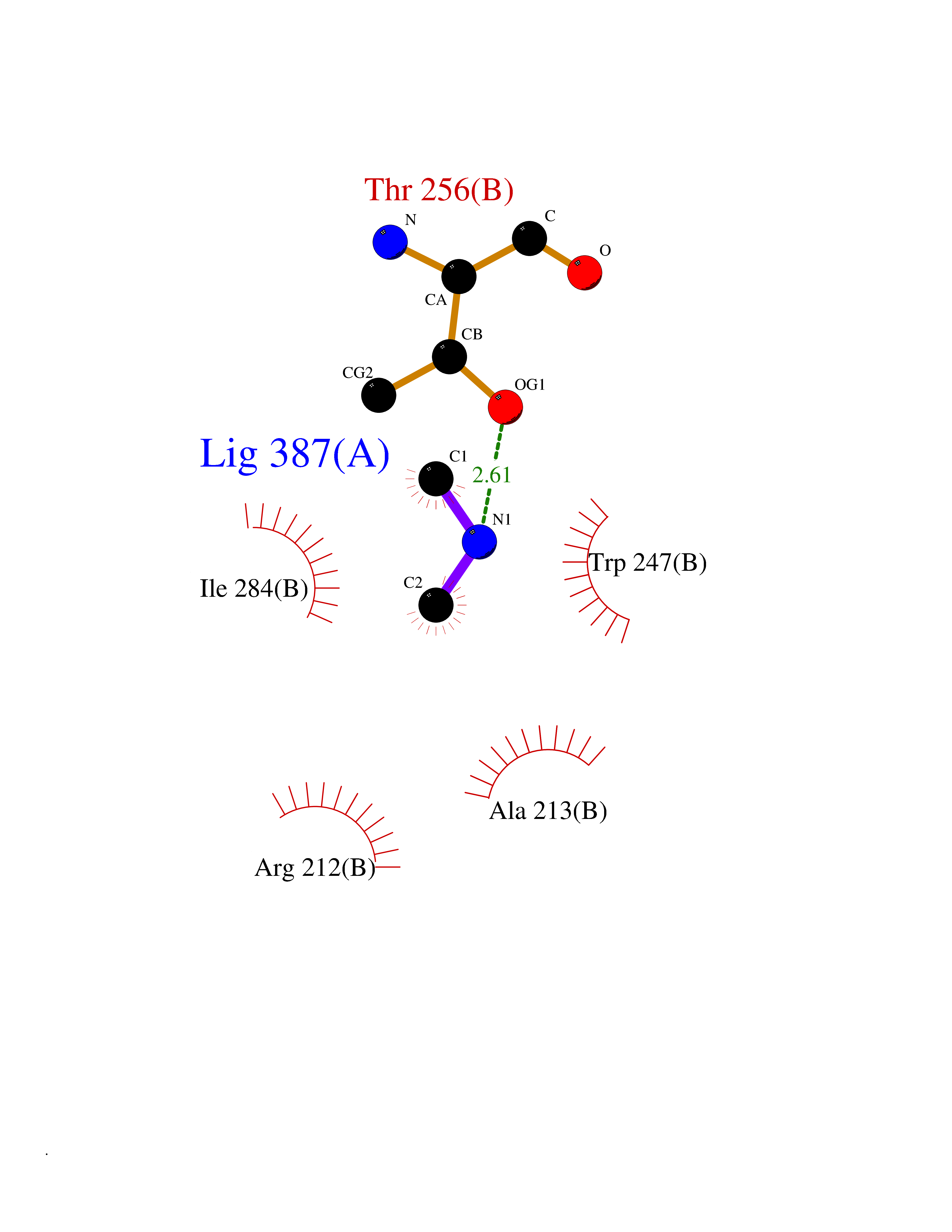 Ligplot Map 3D Binding mode Sequence TFKAKDLIVTPATILKEKPDPNLVFGTVFTDHMLTVEWSSEFGWEKPHIKPLQNLSLHPGSSALHYAVELFEGLKAFRGVDNKIRLFQPNLNMDRMYRSAVRATLPVFDKEELLECIQQLVKLDQEWVPYSTSASLYIRPTFIGTEPSLGVKKPTKALLFVLLSPVGPYFNPVSLWANPKYVRAWKGGTGDCKMGGNYGSSLFAQCEAVDNGCQQVLWLYGEDHQITEVGTMNLFLYWINEDGEEELATPPLDGIILPGVTRRCILDLAHQWGEFKVSERYLTMDDLTTALEGNRVREMFGSGTACVVCPVSDILYKGETIHIPTMENGPKLASRILSKLTDIQYGREERDWTIVL Hydrogen bonds contact Hydrophobic contact | ||||
| 72 | Oxygen-insensitive NAD(P)H nitroreductase | 1KQB | 4.00 | |
Target general information Gen name nfsB Organism Enterobacter cloacae Uniprot ID TTD ID NA Synonyms nfsI;nfnB Protein family Nitroreductase family Biochemical class Oxidoreductase Function Oxidoreductase activity. Related diseases A chromosomal aberration involving NFKB2 is found in a case of B-cell non Hodgkin lymphoma (B-NHL). Translocation t(10;14)(q24;q32) with IGHA1. The resulting oncogene is also called Lyt-10C alpha variant.; DISEASE: A chromosomal aberration involving NFKB2 is found in a cutaneous T-cell leukemia (C-TCL) cell line. This rearrangement produces the p80HT gene which codes for a truncated 80 kDa protein (p80HT).; DISEASE: In B-cell leukemia (B-CLL) cell line, LB40 and EB308, can be found after heterogeneous chromosomal aberrations, such as internal deletions.; DISEASE: Immunodeficiency, common variable, 10 (CVID10) [MIM:615577]: A primary immunodeficiency characterized by childhood-onset of recurrent infections, hypogammaglobulinemia, and decreased numbers of memory and marginal zone B-cells. Some patients may develop autoimmune features and have circulating autoantibodies. An unusual feature is central adrenal insufficiency. {ECO:0000269|PubMed:24140114, ECO:0000269|PubMed:25524009}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB03793; DB03247 Interacts with NA EC number 1.-.-.- Uniprot keywords 3D-structure; Flavoprotein; FMN; NAD; NADP; Oxidoreductase Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 47619.4 Length 432 Aromaticity 0.08 Instability index 38.43 Isoelectric point 5.52 Charge (pH=7) -12.98 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map 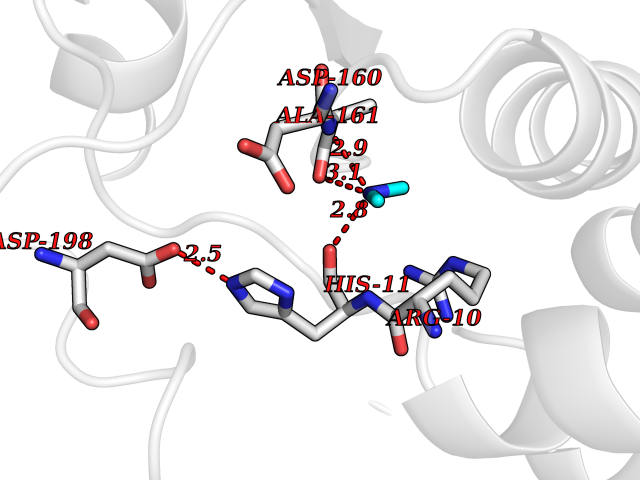 Pymol Map 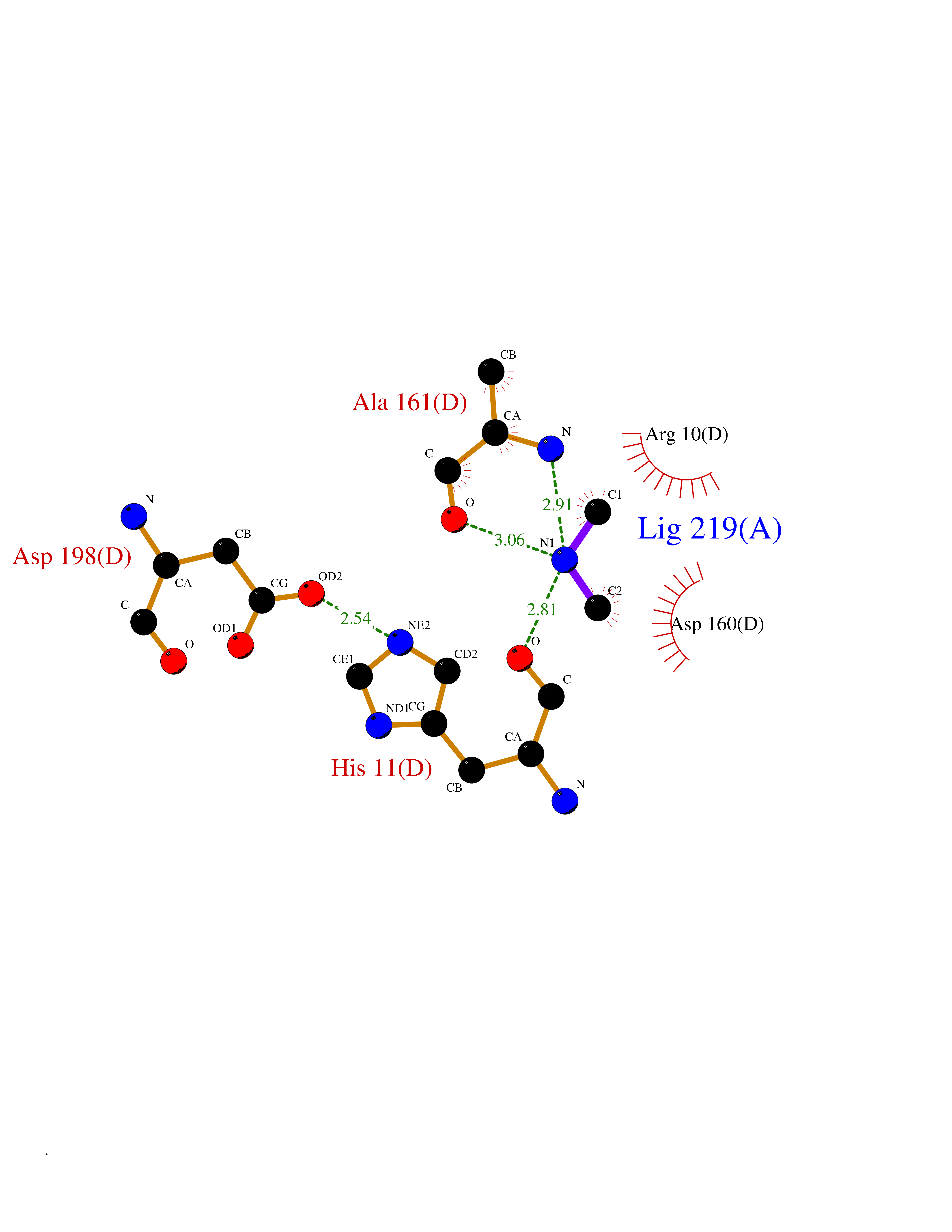 Ligplot Map 3D Binding mode Sequence DIISVALKRHSTKAFDASKKLTAEEAEKIKTLLQYSPSSTNSQPWHFIVASTEEGKARVAKSAAGTYVFNERKMLDASHVVVFCAKTAMDDAWLERVVDQEEADGRFNTPEAKAANHKGRTYFADMHRVDLKDDDQWMAKQVYLNVGNFLLGVGAMGLDAVPIEGFDAAILDEEFGLKEKGFTSLVVVPVGHHSVEDFNATLPKSRLPLSTIVTECDIISVALKRHSTKAFDASKKLTAEEAEKIKTLLQYSPSSTNSQPWHFIVASTEEGKARVAKSAAGTYVFNERKMLDASHVVVFCAKTAMDDAWLERVVDQEEADGRFNTPEAKAANHKGRTYFADMHRVDLKDDDQWMAKQVYLNVGNFLLGVGAMGLDAVPIEGFDAAILDEEFGLKEKGFTSLVVVPVGHHSVEDFNATLPKSRLPLSTIVTEC Hydrogen bonds contact Hydrophobic contact | ||||
| 73 | Dihydroorotate dehydrogenase (quinone), mitochondrial | 4CQ8 | 4.00 | |
Target general information Gen name PFF0160c Organism Plasmodium falciparum (isolate 3D7) Uniprot ID TTD ID NA Synonyms NA Protein family Dihydroorotate dehydrogenase family, Type 2 subfamily Biochemical class Oxidoreductase Function Dihydroorotate dehydrogenase activity. Related diseases Combined oxidative phosphorylation deficiency 33 (COXPD33) [MIM:617713]: An autosomal recessive disorder caused by multiple mitochondrial respiratory chain defects and impaired mitochondrial energy metabolism. Clinical manifestations are highly variable. Affected infants present with cardiomyopathy accompanied by multisystemic features involving liver, kidney, and brain. Death in infancy is observed in some patients. Children and adults present with myopathy and progressive external ophthalmoplegia. {ECO:0000269|PubMed:28942965}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB01117 Interacts with NA EC number 1.3.5.2 Uniprot keywords 3D-structure; Flavoprotein; FMN; Membrane; Mitochondrion; Mitochondrion inner membrane; Oxidoreductase; Pyrimidine biosynthesis; Reference proteome; Transit peptide; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID A,B Molecular weight (Da) 42573.5 Length 378 Aromaticity 0.1 Instability index 36.63 Isoelectric point 8.21 Charge (pH=7) 3.17 2D Binding mode Binding energy (Kcal/mol) -5.45 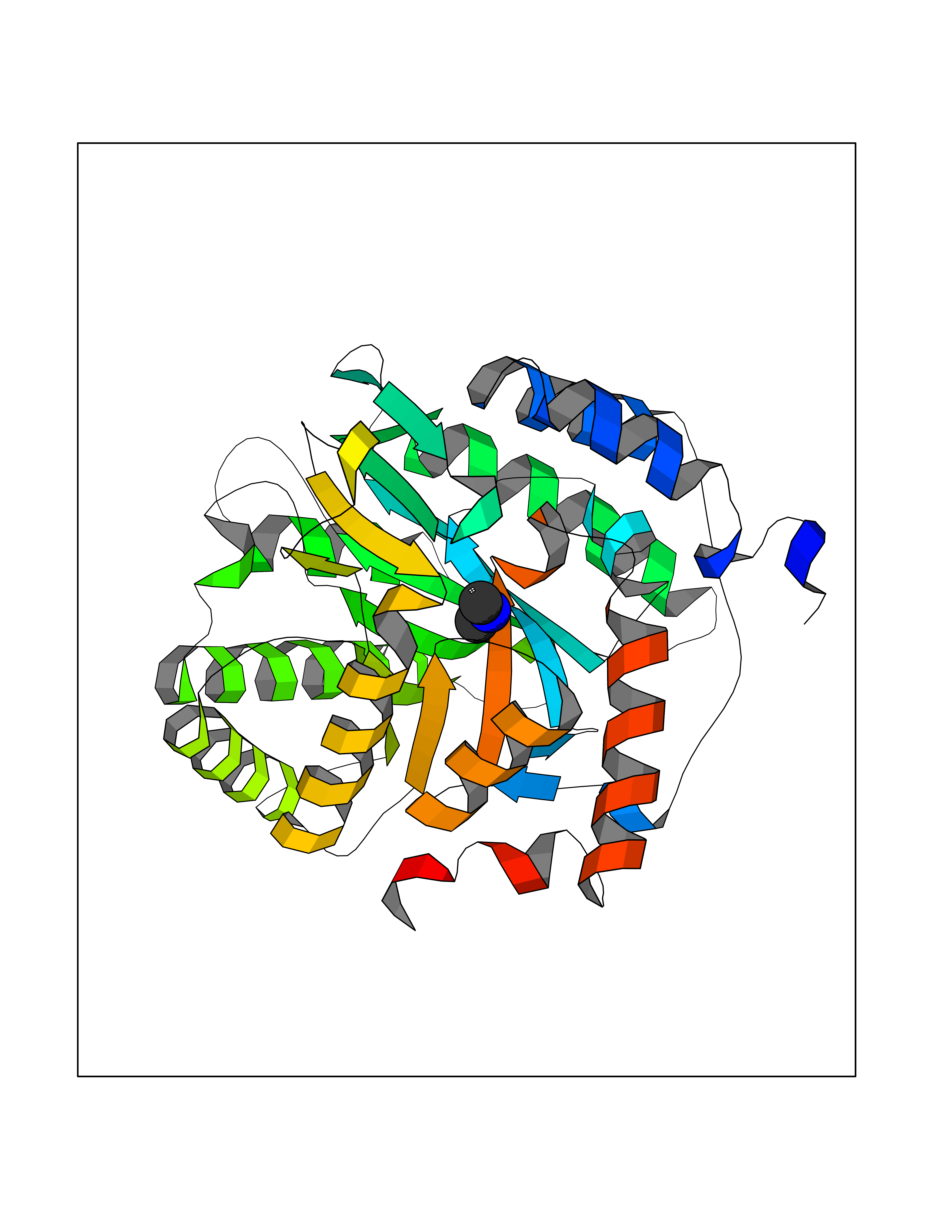 Molscript Map  Pymol Map  Ligplot Map 3D Binding mode Sequence ADPFESYNPEFFLYDIFLKFCLKYIDGEICHDLFLLLGKYNILPYDTSNDSIYACTNIKHLDFINPFGVAAGFDKNGVCIDSILKLGFSFIEIGTITPRGQTGNAKPRIFRDVESRSIINSCGFNNMGCDKVTENLILFRKRQEEDKLLSKHIVGVSIGKNKDTVNIVDDLKYCINKIGRYADYIAINVSSPNTPGLRDNQEAGKLKNIILSVKEEIDNLEKNNFLWFNTTKKKPLVFVKLAPDLNQEQKKEIADVLLETNIDGMIISNTTTQINDIKSFENKKGGVSGAKLKDISTKFICEMYNYTNKQIPIIASGGIFSGLDALEKIEAGASVCQLYSCLVFNGMKSAVQIKRELNHLLYQRGYYNLKEAIGRKHS Hydrogen bonds contact Hydrophobic contact | ||||
| 74 | Alr1529 protein | 1Z8H | 4.00 | |
Target general information Gen name alr1529 Organism Nostoc sp. (strain PCC 7120 / SAG 25.82 / UTEX 2576) Uniprot ID TTD ID NA Synonyms NA Protein family NA Biochemical class Hydrolase Function NA Related diseases Desbuquois dysplasia 1 (DBQD1) [MIM:251450]: A chondrodysplasia characterized by severe prenatal and postnatal growth retardation (less than -5 SD), joint laxity, short extremities, progressive scoliosis, round face, midface hypoplasia, prominent bulging eyes. The main radiologic features are short long bones with metaphyseal splay, a 'Swedish key' appearance of the proximal femur (exaggerated trochanter), and advance carpal and tarsal bone age. Two forms of Desbuquois dysplasia are distinguished on the basis of the presence or absence of characteristic hand anomalies: an extra ossification center distal to the second metacarpal, delta phalanx, bifid distal thumb phalanx, and phalangeal dislocations. {ECO:0000269|PubMed:19853239, ECO:0000269|PubMed:20425819, ECO:0000269|PubMed:21037275, ECO:0000269|PubMed:21412251, ECO:0000269|PubMed:21654728, ECO:0000269|PubMed:22539336}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Epiphyseal dysplasia, multiple, 7 (EDM7) [MIM:617719]: A form of multiple epiphyseal dysplasia, a generalized skeletal dysplasia associated with significant morbidity. Joint pain, joint deformity, waddling gait, and short stature are the main clinical signs and symptoms. Radiological examination of the skeleton shows delayed, irregular mineralization of the epiphyseal ossification centers and of the centers of the carpal and tarsal bones. Multiple epiphyseal dysplasia is broadly categorized into the more severe Fairbank and the milder Ribbing types. The Fairbank type is characterized by shortness of stature, short and stubby fingers, small epiphyses in several joints, including the knee, ankle, hand, and hip. The Ribbing type is confined predominantly to the hip joints and is characterized by hands that are normal and stature that is normal or near-normal. EDM7 inheritance is autosomal recessive. {ECO:0000269|PubMed:28742282}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number NA Uniprot keywords 3D-structure; Reference proteome Protein physicochemical properties Chain ID A,B,C,D Molecular weight (Da) 45603.2 Length 401 Aromaticity 0.11 Instability index 49.26 Isoelectric point 7.67 Charge (pH=7) 1.26 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map  Pymol Map 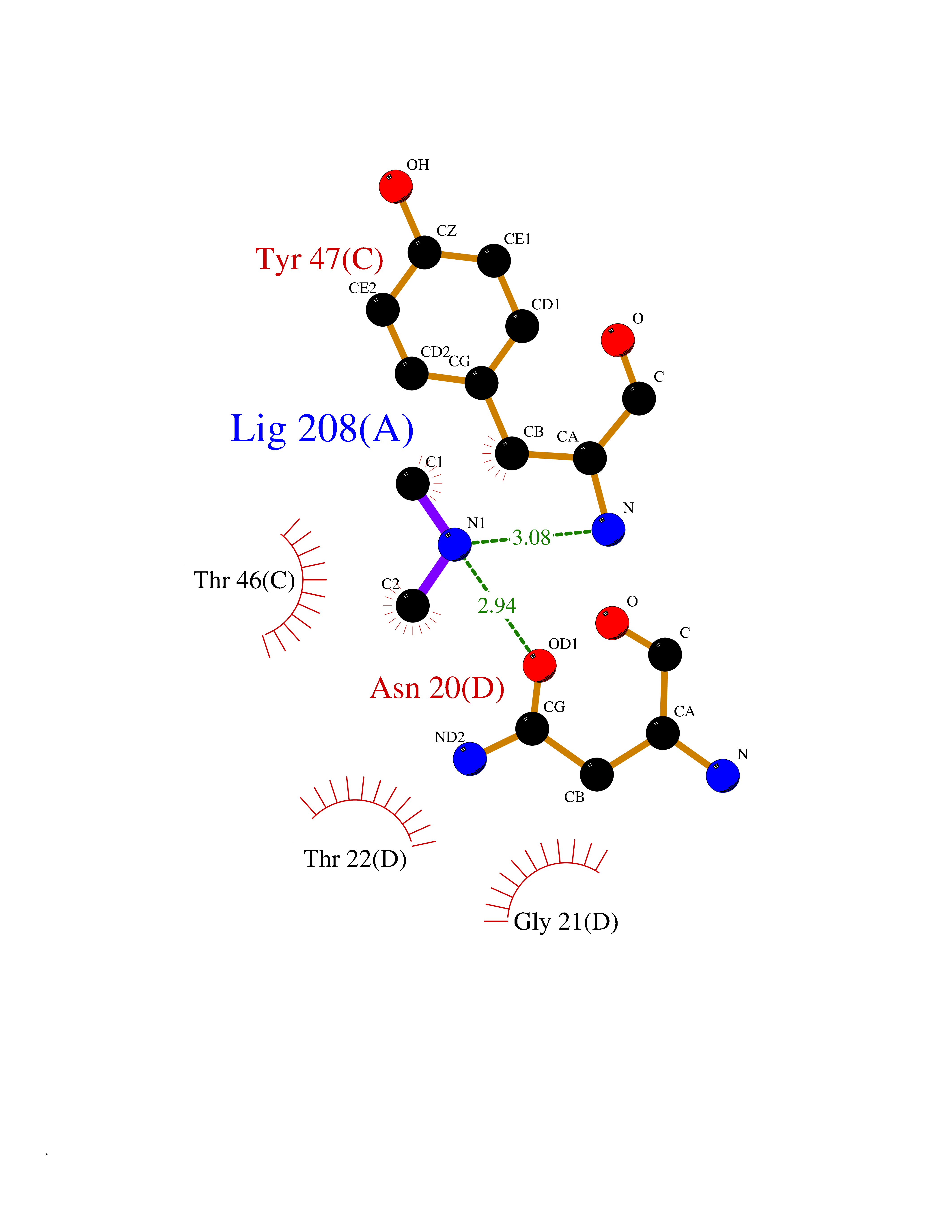 Ligplot Map 3D Binding mode Sequence KTQIRICFVGDSFVNGTGDPECLGWTGRVCVNANKKGYDVTYYNLGIRRDTSSDIAKRWLQEVSLRLHKEYNSLVVFSFGLNDTTLENGKPRVSIAETIKNTREILTQAKKLYPVLXISPAPYIEQQDPGRRRRTIDLSQQLALVCQDLDVPYLDVFPLLEKPSVWLHEAKANDGVHPQAGGYTEFARIVENWDAWLNWFRKTQIRICFVGDSFVNGTGDPECLGWTGRVCVNANKKGYDVTYYNLGIRRDTSSDIAKRWLQEVSLRLHKEYNSLVVFSFGLNDTTLENGKPRVSIAETIKNTREILTQAKKLYPVLXISPAPYIEQQDPGRRRRTIDLSQQLALVCQDLDVPYLDVFPLLEKPSVWLHEAKANDGVHPQAGGYTEFARIVENWDAWLNWF Hydrogen bonds contact Hydrophobic contact | ||||
| 75 | Kallikrein-5 (KLK5) | 6QFE | 4.00 | |
Target general information Gen name KLK5 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UNQ570/PRO1132; Stratum corneum tryptic enzyme; SCTE; Kallikrein-like protein 2; KLK-L2 Protein family Peptidase S1 family, Kallikrein subfamily Biochemical class Peptidase Function May be involved in desquamation. Related diseases Lipodystrophy, familial partial, 8 (FPLD8) [MIM:620679]: An autosomal dominant form of partial lipodystrophy, a disorder characterized by abnormal subcutaneous fat distribution. FPLD8 patients show selective loss of subcutaneous adipose tissue from the limbs, beginning around 13 to 15 years of age, and abnormal accumulation of subcutaneous adipose tissue in the dorsal neck and face, as well as in the posterior thoracic and abdominal regions. The disorder is associated with metabolic abnormalities, including diabetes mellitus and hyperlipidemia. {ECO:0000269|PubMed:27376152}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P20930; Q9NQG1 EC number EC 3.4.21.- Uniprot keywords 3D-structure; Disulfide bond; Glycoprotein; Hydrolase; Protease; Proteomics identification; Reference proteome; Secreted; Serine protease; Signal Protein physicochemical properties Chain ID A,B Molecular weight (Da) 50299.2 Length 454 Aromaticity 0.07 Instability index 40.74 Isoelectric point 9.25 Charge (pH=7) 23.09 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map 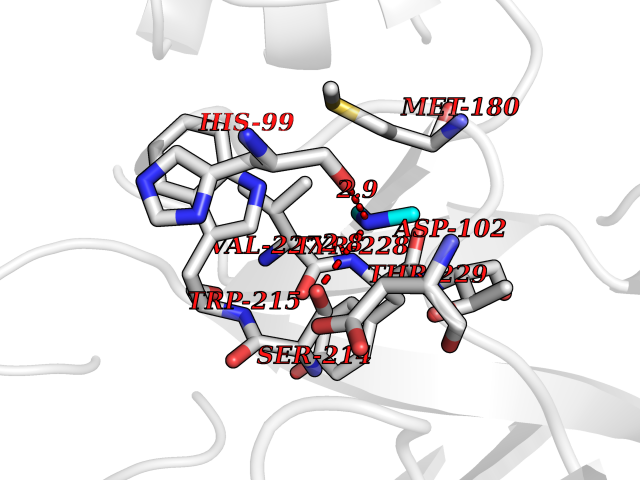 Pymol Map 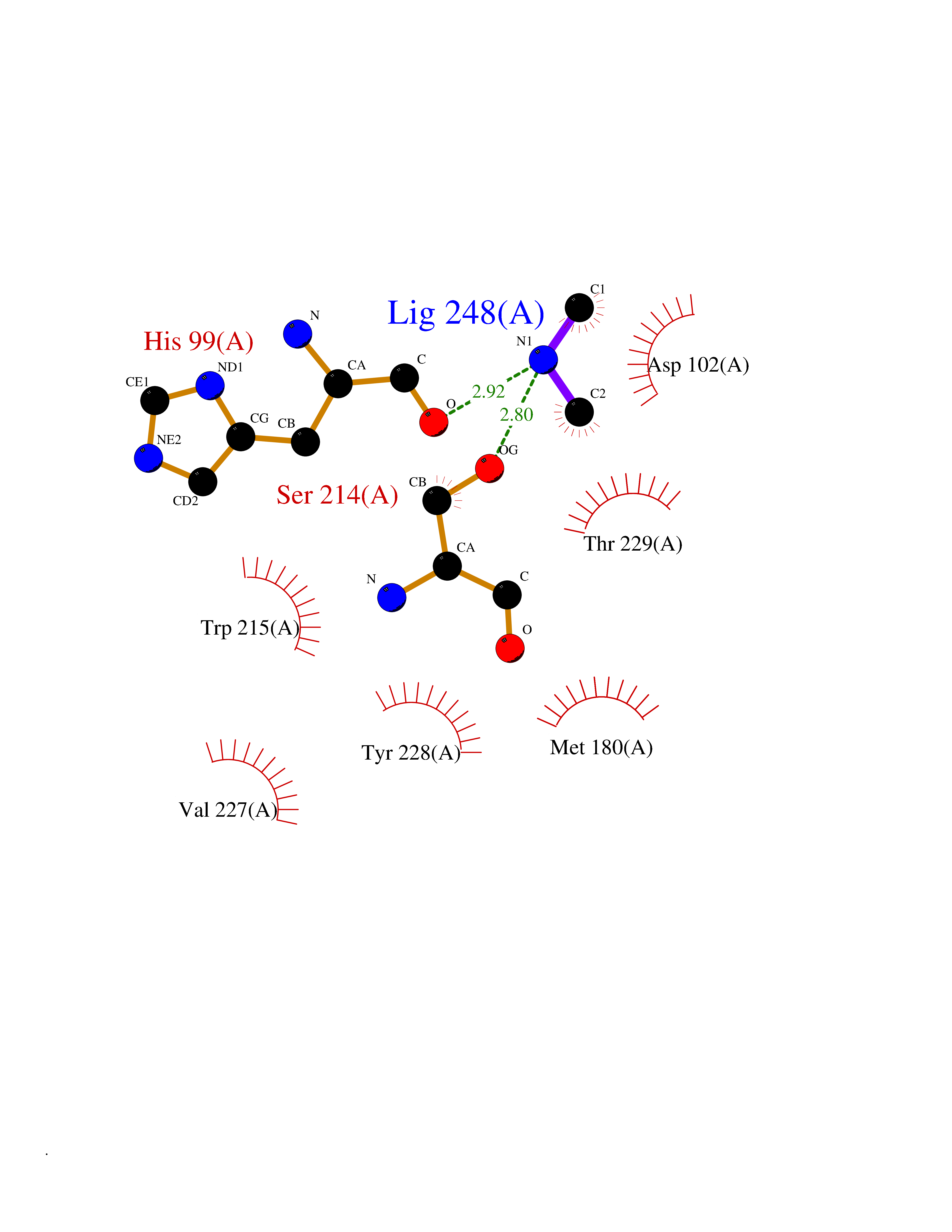 Ligplot Map 3D Binding mode Sequence IINGSDCDMHTQPWQAALLLRPNQLYCGAVLVHPQWLLTAAHCRKKVFRVRLGHYSLSPVYESGQQMFQGVKSIPHPGYSHPGHSNDLMLIKLNRRIRPTKDVRPINVSSHCPSAGTKCLVSGWGTTKSPQVHFPKVLQCLNISVLSQKRCEDAYPRQIDDTMFCAGDKAGRDSCQGDSGGPVVCNGSLQGLVSWGDYPCARPNRPGVYTNLCKFTKWIQETIQANSIINGSDCDMHTQPWQAALLLRPNQLYCGAVLVHPQWLLTAAHCRKKVFRVRLGHYSLSPVYESGQQMFQGVKSIPHPGYSHPGHSNDLMLIKLNRRIRPTKDVRPINVSSHCPSAGTKCLVSGWGTTKSPQVHFPKVLQCLNISVLSQKRCEDAYPRQIDDTMFCAGDKAGRDSCQGDSGGPVVCNGSLQGLVSWGDYPCARPNRPGVYTNLCKFTKWIQETIQANS Hydrogen bonds contact Hydrophobic contact | ||||
| 76 | MAPK-activated protein kinase 2 (MAPKAPK2) | 1NXK | 4.00 | |
Target general information Gen name MAPKAPK2 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms MK2; MK-2; MAPKactivated protein kinase 2; MAPKAPK-2; MAPKAP-K2; MAPKAP kinase 2; MAP kinaseactivated protein kinase 2; MAP kinase-activated protein kinase 2 Protein family Protein kinase superfamily, CAMK Ser/Thr protein kinase family Biochemical class Kinase Function Following stress, it is phosphorylated and activated by MAP kinase p38-alpha/MAPK14, leading to phosphorylation of substrates. Phosphorylates serine in the peptide sequence, Hyd-X-R-X(2)-S, where Hyd is a large hydrophobic residue. Phosphorylates ALOX5, CDC25B, CDC25C, CEP131, ELAVL1, HNRNPA0, HSP27/HSPB1, KRT18, KRT20, LIMK1, LSP1, PABPC1, PARN, PDE4A, RCSD1, RPS6KA3, TAB3 and TTP/ZFP36. Phosphorylates HSF1; leading to the interaction with HSP90 proteins and inhibiting HSF1 homotrimerization, DNA-binding and transactivation activities. Mediates phosphorylation of HSP27/HSPB1 in response to stress, leading to the dissociation of HSP27/HSPB1 from large small heat-shock protein (sHsps) oligomers and impairment of their chaperone activities and ability to protect against oxidative stress effectively. Involved in inflammatory response by regulating tumor necrosis factor (TNF) and IL6 production post-transcriptionally: acts by phosphorylating AU-rich elements (AREs)-binding proteins ELAVL1, HNRNPA0, PABPC1 and TTP/ZFP36, leading to the regulation of the stability and translation of TNF and IL6 mRNAs. Phosphorylation of TTP/ZFP36, a major post-transcriptional regulator of TNF, promotes its binding to 14-3-3 proteins and reduces its ARE mRNA affinity, leading to inhibition of dependent degradation of ARE-containing transcripts. Phosphorylates CEP131 in response to cellular stress induced by ultraviolet irradiation which promotes binding of CEP131 to 14-3-3 proteins and inhibits formation of novel centriolar satellites. Also involved in late G2/M checkpoint following DNA damage through a process of post-transcriptional mRNA stabilization: following DNA damage, relocalizes from nucleus to cytoplasm and phosphorylates HNRNPA0 and PARN, leading to stabilization of GADD45A mRNA. Involved in toll-like receptor signaling pathway (TLR) in dendritic cells: required for acute TLR-induced macropinocytosis by phosphorylating and activating RPS6KA3. Stress-activated serine/threonine-protein kinase involved in cytokine production, endocytosis, reorganization of the cytoskeleton, cell migration, cell cycle control, chromatin remodeling, DNA damage response and transcriptional regulation. Related diseases Erythrocytosis, familial, 4 (ECYT4) [MIM:611783]: An autosomal dominant disorder characterized by elevated serum hemoglobin and hematocrit, and normal platelet and leukocyte counts. {ECO:0000269|PubMed:18184961, ECO:0000269|PubMed:18378852, ECO:0000269|PubMed:19208626, ECO:0000269|PubMed:22367913}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07430; DB07431; DB07406; DB08358; DB07728; DB07234; DB02010 Interacts with Q00613; P04792; Q16539; Q9QWH1; P47811 EC number EC 2.7.11.1 Uniprot keywords 3D-structure; Alternative splicing; ATP-binding; Cytoplasm; DNA damage; Isopeptide bond; Kinase; Nucleotide-binding; Nucleus; Phosphoprotein; Proteomics identification; Reference proteome; Serine/threonine-protein kinase; Transferase; Ubl conjugation Protein physicochemical properties Chain ID A Molecular weight (Da) 32100.5 Length 290 Aromaticity 0.1 Instability index 43.53 Isoelectric point 8.76 Charge (pH=7) 5.44 2D Binding mode Binding energy (Kcal/mol) -5.46 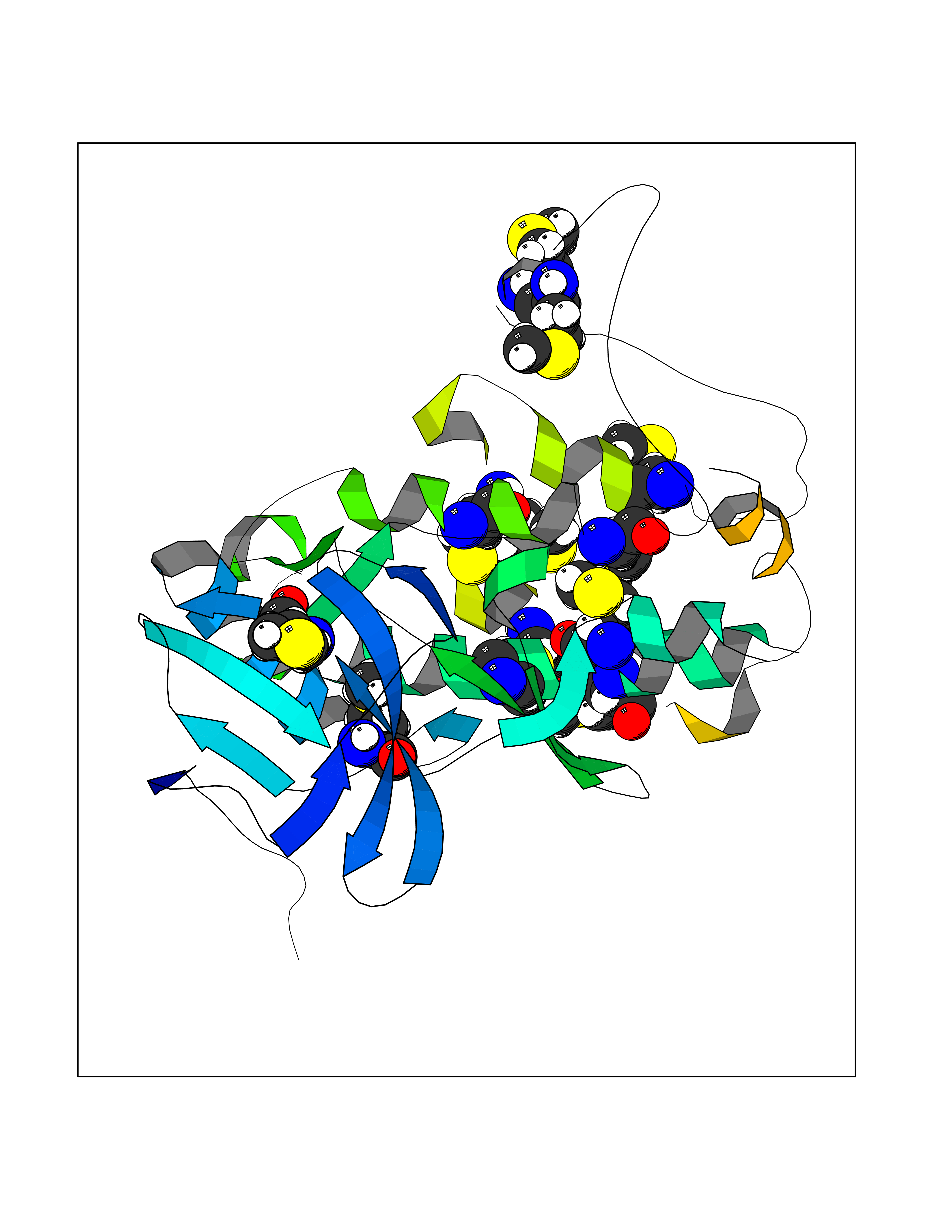 Molscript Map 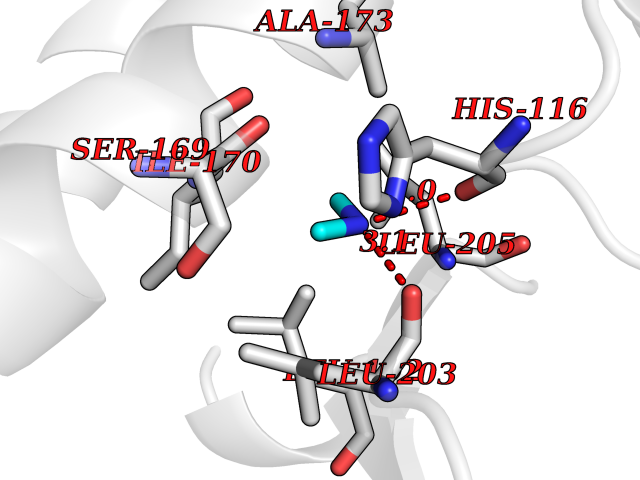 Pymol Map 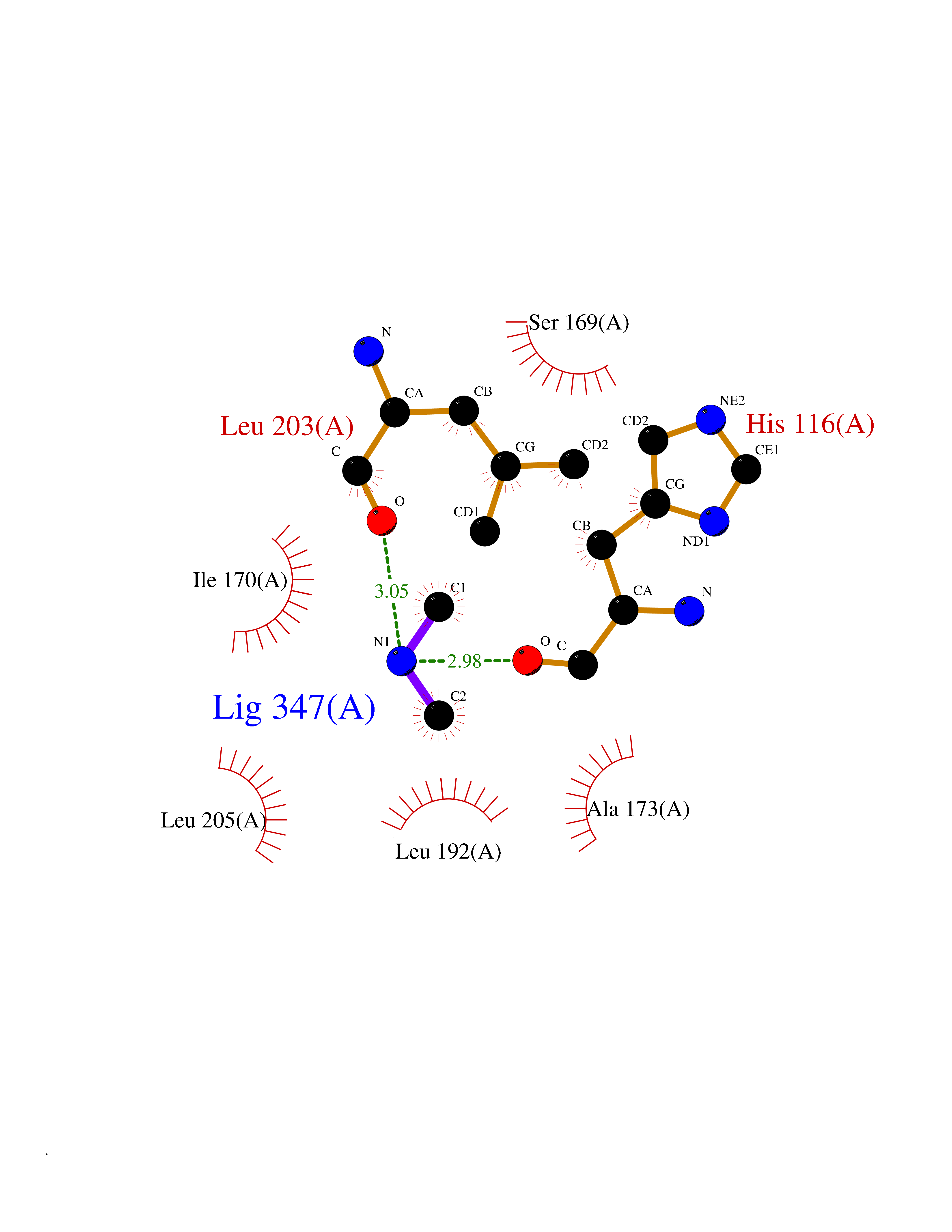 Ligplot Map 3D Binding mode Sequence QFPQFHVKSGLQIKKNAIIDDYKVTSQVLGLGINGKVLQIFNKRTQEKFALKXLQDCPKARREVELHWRASQCPHIVRIVDVYENLYAGRKCLLIVXECLDGGELFSRIQDRAFTEREASEIXKSIGEAIQYLHSINIAHRDVKPENLLYTSKRPNAILKLTDFGFAKETTPYYVAPEVLGPEKYDKSCDXWSLGVIXYILLCGYPPFYSNHGLAISPGXKTRIRXGQYEFPNPEWSEVSEEVKXLIRNLLKTEPTQRXTITEFXNHPWIXQSTKVPQTPLHTSRVLKED Hydrogen bonds contact Hydrophobic contact | ||||
| 77 | N-acetylmannosamine kinase (GNE) | 4ZHT | 4.00 | |
Target general information Gen name GNE Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms UDPGlcNAc2epimerase/ManAc kinase; GNE; Bifunctional UDPNacetylglucosamine 2epimerase/Nacetylmannosamine kinase Protein family UDP-N-acetylglucosamine 2-epimerase family; ROK (NagC/XylR) family Biochemical class Kinase Function Regulates and initiates biosynthesis of N- acetylneuraminic acid (NeuAc), a precursor of sialic acids. Plays an essential role in early development. Required for normal sialylation in hematopoietic cells. Sialylation is implicated in cell adhesion, signal transduction, tumorigenicity and metastatic behavior of malignant cells. {ECO:0000250, ECO:0000269|PubMed:10334995}. Related diseases Sialuria (SIALURIA) [MIM:269921]: In sialuria, free sialic acid accumulates in the cytoplasm and gram quantities of neuraminic acid are secreted in the urine. The metabolic defect involves lack of feedback inhibition of UDP-GlcNAc 2-epimerase by CMP-Neu5Ac, resulting in constitutive overproduction of free Neu5Ac. Clinical features include variable degrees of developmental delay, coarse facial features and hepatomegaly. Sialuria inheritance is autosomal dominant. {ECO:0000269|PubMed:10330343, ECO:0000269|PubMed:10356312, ECO:0000269|PubMed:11326336, ECO:0000269|PubMed:2808337}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Nonaka myopathy (NM) [MIM:605820]: An autosomal recessive myopathy characterized by early adult onset and progressive distal muscle weakness that preferentially affects the anterior tibial muscles, usually sparing the quadriceps femoris. Some individuals may have involvement of the upper limbs or proximal muscles. Muscle biopsy reveals presence of rimmed vacuoles. {ECO:0000269|PubMed:11528398, ECO:0000269|PubMed:11916006, ECO:0000269|PubMed:12177386, ECO:0000269|PubMed:12325084, ECO:0000269|PubMed:12409274, ECO:0000269|PubMed:12473753, ECO:0000269|PubMed:12473769, ECO:0000269|PubMed:12473780, ECO:0000269|PubMed:12497639, ECO:0000269|PubMed:12811782, ECO:0000269|PubMed:12913203, ECO:0000269|PubMed:14707127, ECO:0000269|PubMed:15146476, ECO:0000269|PubMed:16503651}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Thrombocytopenia 12 with or without myopathy (THC12) [MIM:620757]: A form of thrombocytopenia, a hematologic disorder defined by a decrease in the number of platelets in circulating blood, resulting in the potential for increased bleeding and decreased ability for clotting. THC12 is an autosomal recessive form manifesting from infancy or early childhood with bleeding episodes. Clinical features include petechiae, easy bruising, epistaxis, hematomas, menorrhagia, and increased bleeding after trauma or surgery. Rare patients may have thrombocytopenia without bleeding. Some affected individuals have myopathic features, usually apparent in the second or third decades of life. {ECO:0000269|PubMed:25257349, ECO:0000269|PubMed:30171045, ECO:0000269|PubMed:33198675, ECO:0000269|PubMed:34788986, ECO:0000269|PubMed:34858435, ECO:0000269|PubMed:35052006, ECO:0000269|PubMed:38237079}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with P12814; Q6UY14-3; Q969Y2; Q15323; P60370; P60409; P60410; P60411; Q9BQ66; P26371; Q9BYQ4; Q7Z3S9; O43597; Q6UY14-3; Q6P5X5; P27918; A8MQ03; Q16610; Q9UHF1; P28799; P49639; Q5T749; Q15323; O76011; Q6A162; P78385; P78386; O43790; Q07627; Q8IUG1; P60409; P60410; Q8IUC1; P60328; Q52LG2; Q3SY46; Q9BYP8; Q3LHN2; Q3SYF9; Q9BYR8; Q9BYR6; Q9BYQ7; Q9BYQ6; Q9BYR3; P26371; Q3LI64; Q3LI66; Q3LI67; Q9BYQ4; Q9BYQ3; Q9BYQ0; Q99750; Q8IV28; P0DPK4; O15496; O43609; O43610; P14373; Q8IWZ5; Q15654; O14817; Q2TAL6; Q9BRX9; O76024; Q9NZC7-5 EC number NA Uniprot keywords 3D-structure; Allosteric enzyme; Alternative splicing; ATP-binding; Cytoplasm; Disease variant; Hydrolase; Kinase; Metal-binding; Multifunctional enzyme; Nucleotide-binding; Phosphoprotein; Proteomics identification; Reference proteome; Transferase; Zinc Protein physicochemical properties Chain ID A Molecular weight (Da) 41589.2 Length 384 Aromaticity 0.07 Instability index 35.07 Isoelectric point 7.05 Charge (pH=7) 0.19 2D Binding mode Binding energy (Kcal/mol) -5.46 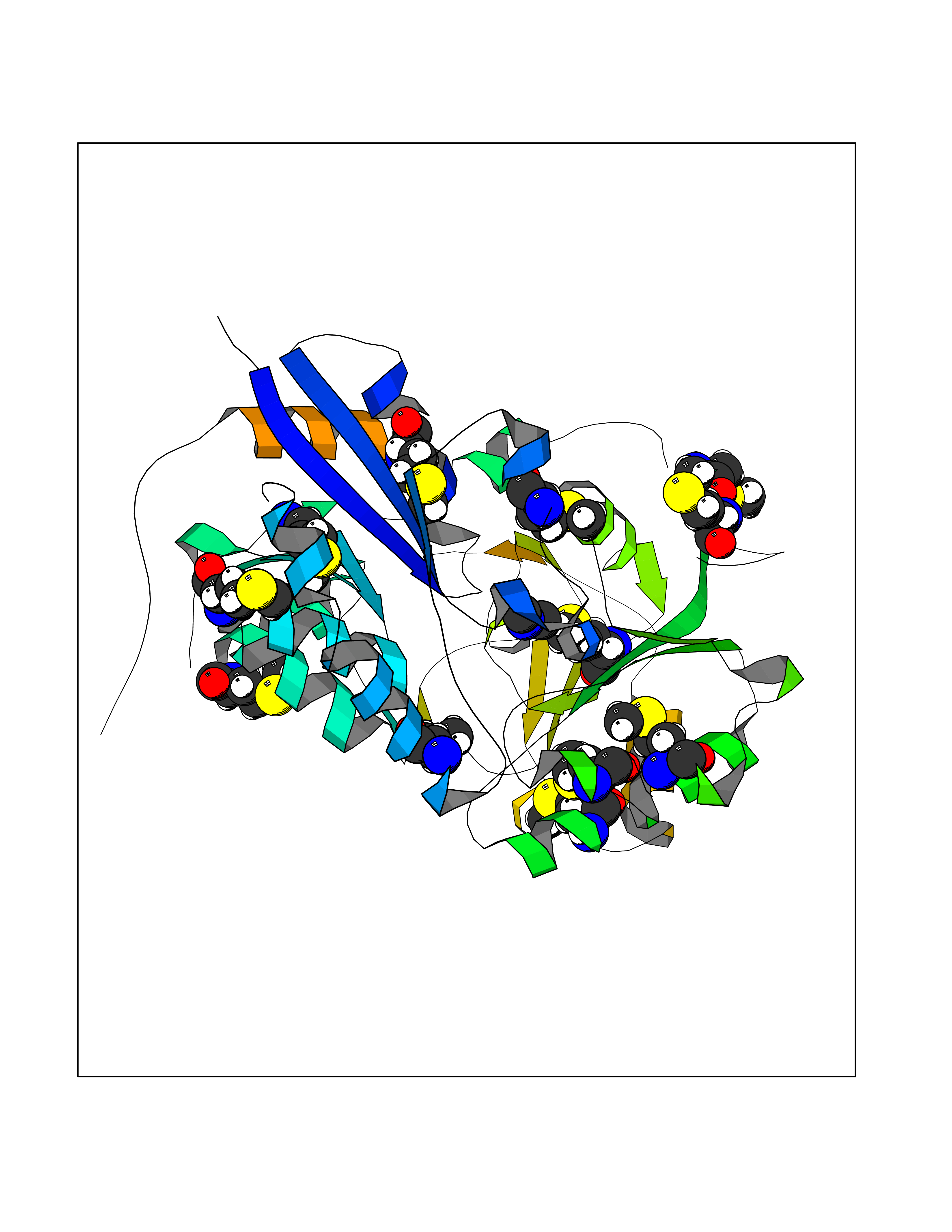 Molscript Map 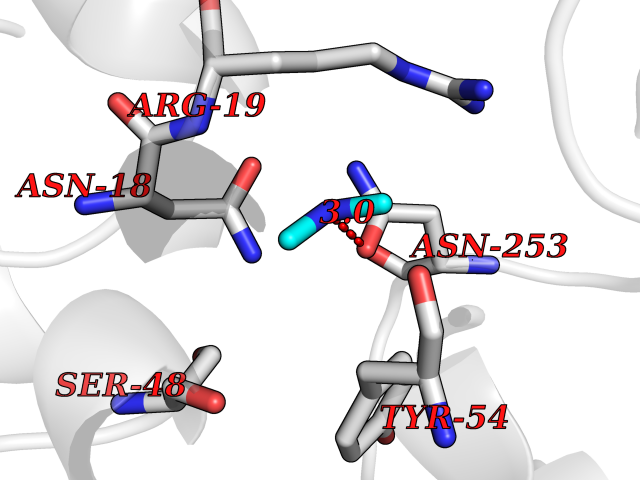 Pymol Map 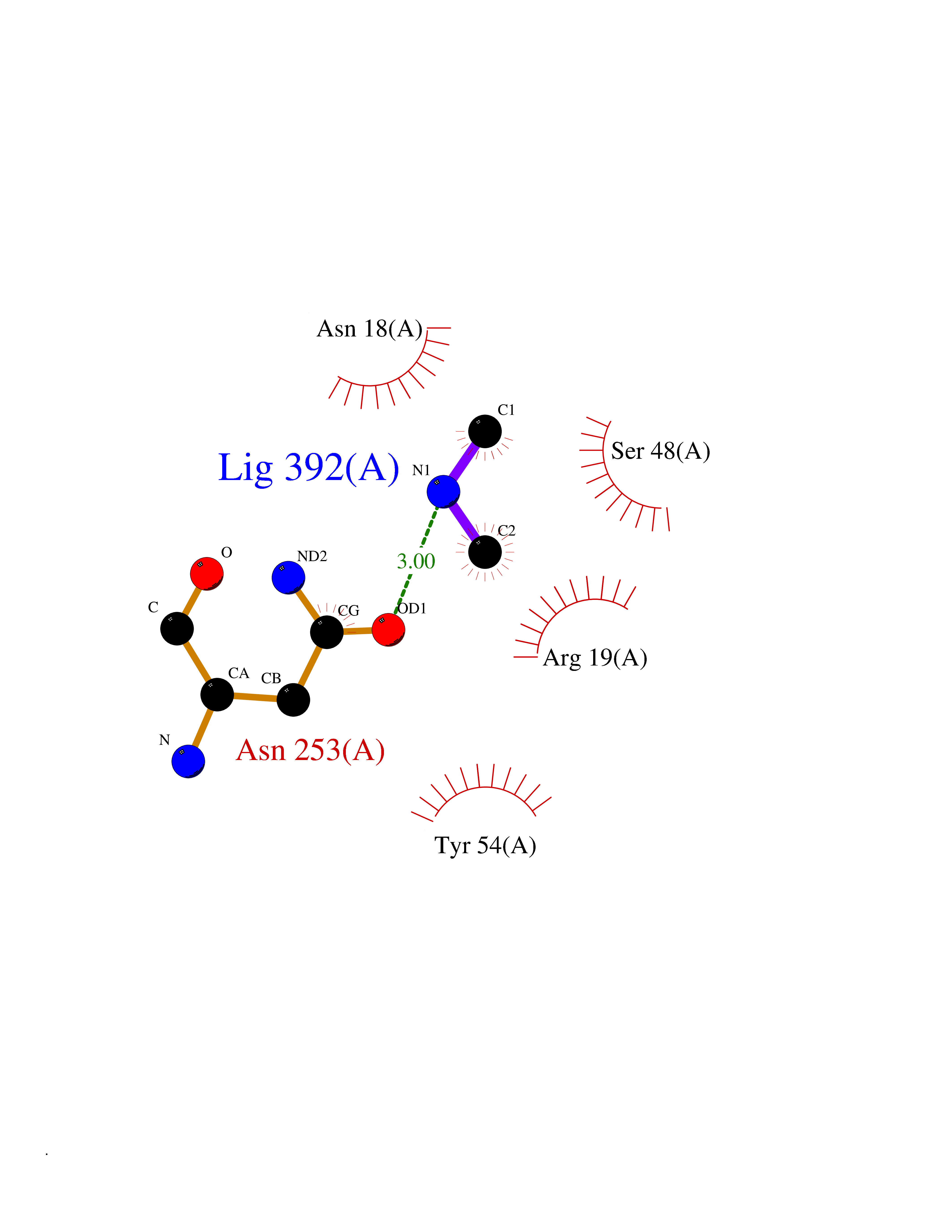 Ligplot Map 3D Binding mode Sequence NRKLRVCVATCNRADYSKLAPIXFGIKTEPEFFELDVVVLGSHLIDDYGNTYRXIEQDDFDINTRLHTIVRGEDEAAXVESVGLALVKLPDVLNRLKPDIXIVHGDRFDALALATSAALXNIRILHIEGGEVSGTIDDSIRHAITKLAHYHVCCTRSAEQHLISXCEDHDRILLAGCPSYDKLLSAKNKDYXSIIRXWLGDDVKSKDYIVALQHPVTTDIKHSIKXFELTLDALISFNKRTLVLFPNIDAGSKEXVRVXRKKGIEHHPNFRAVKHVPFDQFIQLVAHAGCXIGNSSCGVREVGAFGTPVINLGTRQIGRETGENVLHVRDADTQDKILQALHLQFGKQYPCSKIYGDGNAVPRILKFLKSIDLQEPLQKKFCFP Hydrogen bonds contact Hydrophobic contact | ||||
| 78 | Nicotinamide phosphoribosyltransferase (NAMPT) | 2E5D | 4.00 | |
Target general information Gen name NAMPT Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Visfatin; PreBcell colonyenhancing factor 1; PreB cellenhancing factor; Pre-B-cell colony-enhancing factor 1; Pre-B cell-enhancing factor; PBEF1; PBEF; Nampt; NAmPRTase Protein family NAPRTase family Biochemical class Glycosyltransferases Function It is the rate limiting component in the mammalian NAD biosynthesis pathway. The secreted form behaves both as a cytokine with immunomodulating properties and an adipokine with anti-diabetic properties, it has no enzymatic activity, partly because of lack of activation by ATP, which has a low level in extracellular space and plasma. Plays a role in the modulation of circadian clock function. NAMPT-dependent oscillatory production of NAD regulates oscillation of clock target gene expression by releasing the core clock component: CLOCK-ARNTL/BMAL1 heterodimer from NAD-dependent SIRT1-mediated suppression. Catalyzes the condensation of nicotinamide with 5-phosphoribosyl-1-pyrophosphate to yield nicotinamide mononucleotide, an intermediate in the biosynthesis of NAD. Related diseases Hemolytic anemia, non-spherocytic, due to glucose phosphate isomerase deficiency (HA-GPID) [MIM:613470]: A form of anemia in which there is no abnormal hemoglobin or spherocytosis. It is caused by glucose phosphate isomerase deficiency. {ECO:0000269|PubMed:28803808, ECO:0000269|PubMed:7989588, ECO:0000269|PubMed:8499925, ECO:0000269|PubMed:8822952, ECO:0000269|PubMed:8822954, ECO:0000269|PubMed:9446754, ECO:0000269|PubMed:9856489}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB12980; DB12731; DB05217 Interacts with P02792; Q01628; P03886; P43490; Q70CQ1-2 EC number EC 2.4.2.12 Uniprot keywords 3D-structure; Acetylation; Biological rhythms; Cytokine; Cytoplasm; Glycosyltransferase; Nucleus; Phosphoprotein; Proteomics identification; Pyridine nucleotide biosynthesis; Reference proteome; Secreted; Transferase Protein physicochemical properties Chain ID A,B Molecular weight (Da) 105483 Length 932 Aromaticity 0.11 Instability index 34.4 Isoelectric point 6.68 Charge (pH=7) -2.24 2D Binding mode Binding energy (Kcal/mol) -5.46 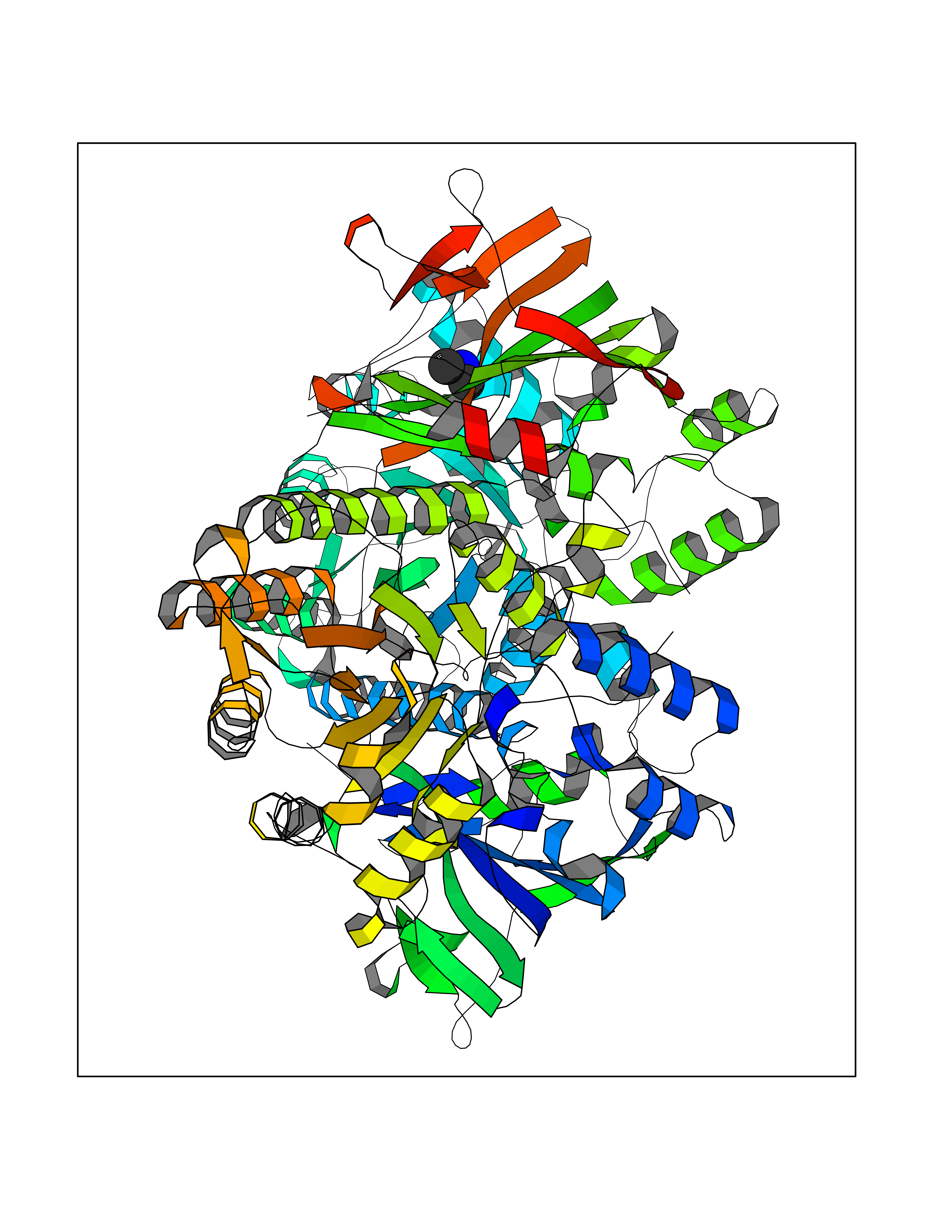 Molscript Map 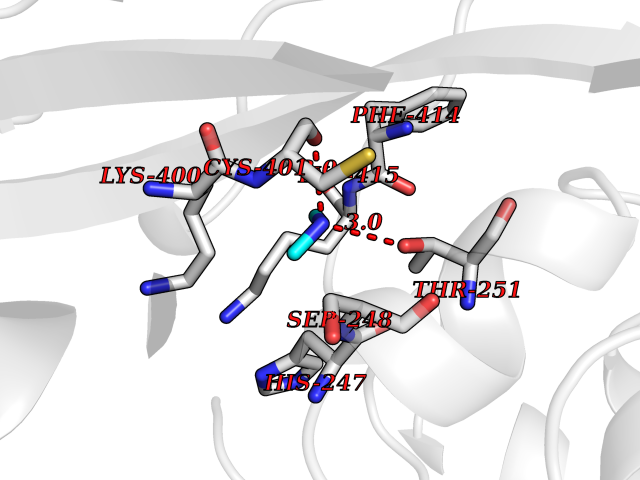 Pymol Map 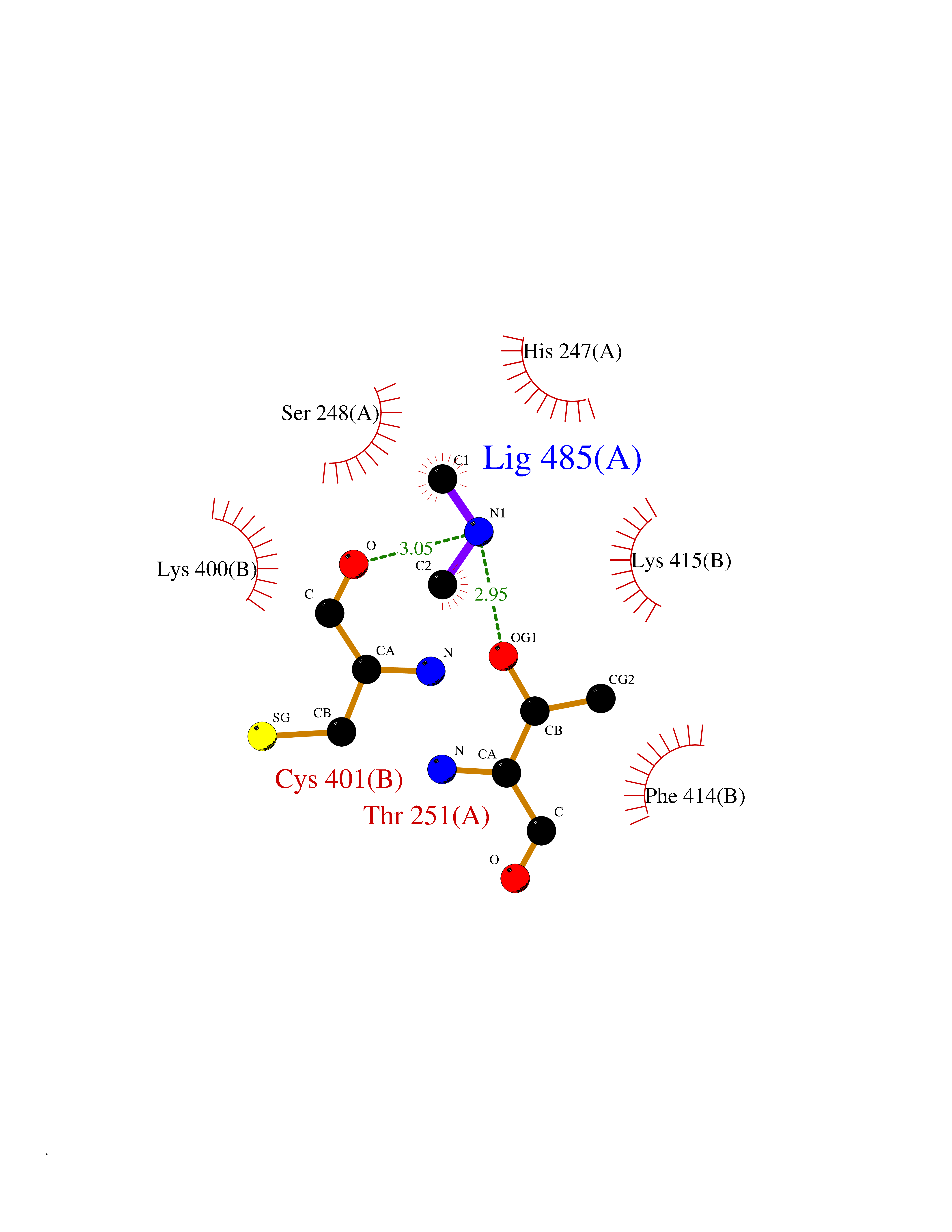 Ligplot Map 3D Binding mode Sequence EFNILLATDSYKVTHYKQYPPNTSKVYSYFECREKKYEETVFYGLQYILNKYLKGKVVTKEKIQEAKDVYKEHFQDDVFNEKGWNYILEKYDGHLPIEIKAVPEGFVIPRGNVLFTVENTDPECYWLTNWIETILVQSWYPITVATNSREQKKILAKYLLETSGNLDGLEYKLHDFGYRGVSSQETAGIGASAHLVNFKGTDTVAGLALIKKYYGTKDPVPGYSVPAAEHSTITAWGKDHEKDAFEHIVTQFSSVPVSVVSDSYDIYNACEKIWGEDLRHLIVSRSTQAPLIIRPDSGNPLDTVLKVLEILGKKFPVTENSKGYKLLPPYLRVIQGDGVDINTLQEIVEGMKQKMWSIENIAFGSGGGLLQKLTRDLLNCSFKCSYVVTNGLGINVFKDPVADPNKRSKKGRLSLHRTPAGNFVTLEEGKGDLEEYGQDLLHTVFKNGKVTKSYSFDEIRKNAQLNEFNILLATDSYKVTHYKQYPPNTSKVYSYFECREKKYEETVFYGLQYILNKYLKGKVVTKEKIQEAKDVYKEHFQDDVFNEKGWNYILEKYDGHLPIEIKAVPEGFVIPRGNVLFTVENTDPECYWLTNWIETILVQSWYPITVATNSREQKKILAKYLLETSGNLDGLEYKLHDFGYRGVSSQETAGIGASAHLVNFKGTDTVAGLALIKKYYGTKDPVPGYSVPAAEHSTITAWGKDHEKDAFEHIVTQFSSVPVSVVSDSYDIYNACEKIWGEDLRHLIVSRSTQAPLIIRPDSGNPLDTVLKVLEILGKKFPVTENSKGYKLLPPYLRVIQGDGVDINTLQEIVEGMKQKMWSIENIAFGSGGGLLQKLTRDLLNCSFKCSYVVTNGLGINVFKDPVADPNKRSKKGRLSLHRTPAGNFVTLEEGKGDLEEYGQDLLHTVFKNGKVTKSYSFDEIRKNAQLN Hydrogen bonds contact Hydrophobic contact | ||||
| 79 | Bacterial Cystathionine beta-lyase (Bact metC) | 4ITX | 4.00 | |
Target general information Gen name Bact metC Organism Escherichia coli (strain K12) Uniprot ID TTD ID Synonyms Cysteine-S-conjugate beta-lyase MetC; Cysteine lyase MetC; Cysteine desulfhydrase MetC; Cystathionine beta-lyase MetC; CL; CBL; Beta-cystathionase MetC; Bacterial CD Protein family Trans-sulfuration enzymes family Biochemical class Carbon-sulfur lyases Function Primarily catalyzes the cleavage of cystathionine to homocysteine, pyruvate and ammonia during methionine biosynthesis. Also exhibits cysteine desulfhydrase activity, producing sulfide from cysteine. In addition, under certain growth conditions, exhibits significant alanine racemase coactivity. Related diseases Coronary artery disease, autosomal dominant, 2 (ADCAD2) [MIM:610947]: A common heart disease characterized by reduced or absent blood flow in one or more of the arteries that encircle and supply the heart. Its most important complication is acute myocardial infarction. {ECO:0000269|PubMed:17332414, ECO:0000269|PubMed:23703864}. The disease is caused by variants affecting the gene represented in this entry.; DISEASE: Tooth agenesis, selective, 7 (STHAG7) [MIM:616724]: An autosomal dominant form of selective tooth agenesis, a common anomaly characterized by the congenital absence of one or more teeth. Selective tooth agenesis without associated systemic disorders has sometimes been divided into 2 types: oligodontia, defined as agenesis of 6 or more permanent teeth, and hypodontia, defined as agenesis of less than 6 teeth. The number in both cases does not include absence of third molars (wisdom teeth). {ECO:0000269|PubMed:26387593}. The disease is caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) NA Interacts with NA EC number EC 4.4.1.13 Uniprot keywords 3D-structure; Amino-acid biosynthesis; Cytoplasm; Direct protein sequencing; Lyase; Methionine biosynthesis; Pyridoxal phosphate; Reference proteome Protein physicochemical properties Chain ID A Molecular weight (Da) 42756.3 Length 391 Aromaticity 0.08 Instability index 27.11 Isoelectric point 6.01 Charge (pH=7) -6.11 2D Binding mode Binding energy (Kcal/mol) -5.46  Molscript Map 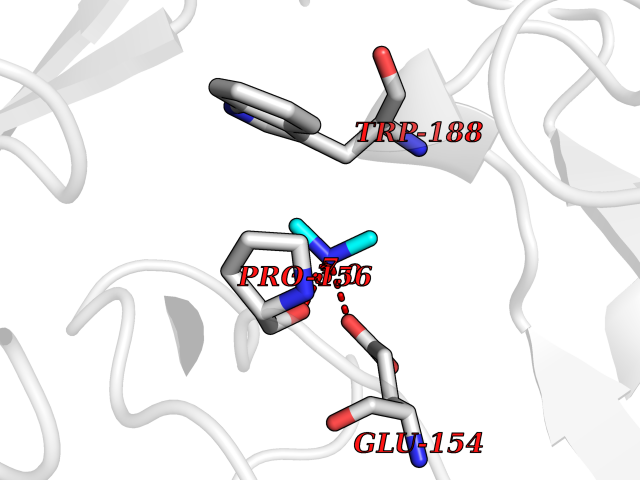 Pymol Map 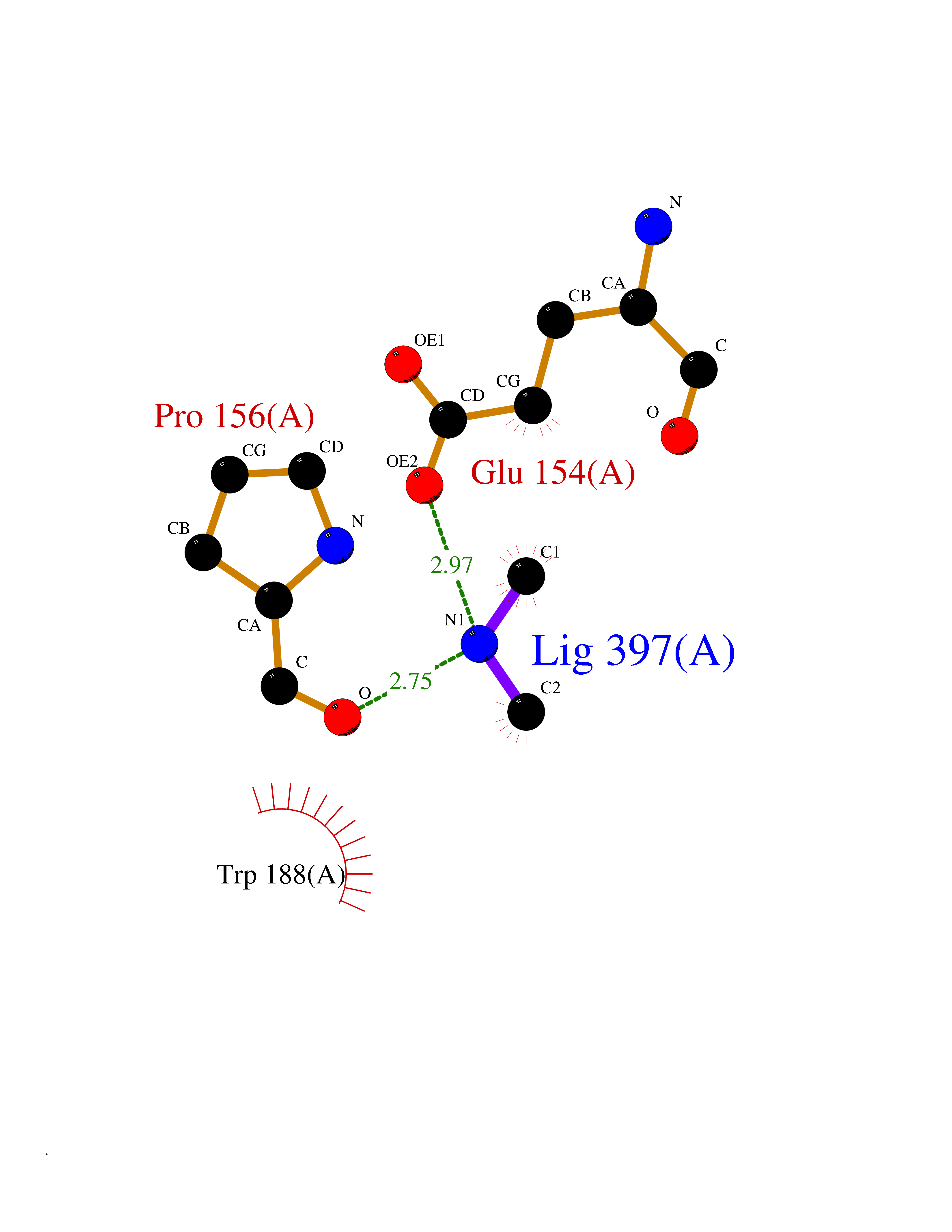 Ligplot Map 3D Binding mode Sequence KLDTQLVNAGRSKKYTLGAVNSVIQRASSLVFDSVEAKKHATRNRANGELFYGRRGTLTHFSLQQAMCELEGGAGCVLFPCGAAAVANSILAFIEQGDHVLMTNTAYESSQDFCSKILSKLGVTTSWFDPLIGADIVKHLQPNTKIVFLESPGSITMEVHDVPAIVAAVRSVVPDAIIMIDNTWAAGVLFKALDFGIDVSIQAATKYLVGHSDAMIGTAVCNARCWEQLRENAYLMGQMVDADTAYITSRGLRTLGVRLRQHHESSLKVAEWLAEHPQVARVNHPALPGSKGHEFWKRDFTGSSGLFSFVLKKKLNNEELANYLDNFSLFSMAYSWGGYESLILANQPEHIAAIRPQGEIDFSGTLIRLHIGLEDVDDLIADLDAGFARIV Hydrogen bonds contact Hydrophobic contact | ||||
| 80 | Tissue factor (F3) | 6R2W | 4.00 | |
Target general information Gen name F3 Organism Homo sapiens (Human) Uniprot ID TTD ID Synonyms Thromboplastin; TF; F3; Coagulation factor III; CD142 antigen Protein family Tissue factor family Biochemical class NA Function Initiates blood coagulation by forming a complex with circulating factor VII or VIIa. The [TF:VIIa] complex activates factors IX or X by specific limited protolysis. TF plays a role in normal hemostasis by initiating the cell-surface assemblyand propagation of the coagulation protease cascade. Related diseases Intellectual developmental disorder, autosomal recessive 54 (MRT54) [MIM:617028]: A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRT54 patients manifest intellectual disability, delayed speech and hyperactivity. {ECO:0000269|PubMed:27106596}. The disease may be caused by variants affecting the gene represented in this entry. Drugs (DrugBank ID) DB07207; DB07247; DB08232; DB06552; DB13150; DB00036; DB16732 Interacts with P55085; P08709; Q9UM19; Q92544 EC number NA Uniprot keywords 3D-structure; Alternative splicing; Blood coagulation; Disulfide bond; Glycoprotein; Hemostasis; Lipoprotein; Membrane; Palmitate; Proteomics identification; Reference proteome; Secreted; Signal; Transmembrane; Transmembrane helix Protein physicochemical properties Chain ID H Molecular weight (Da) 27479.3 Length 249 Aromaticity 0.09 Instability index 32.92 Isoelectric point 7.22 Charge (pH=7) 0.48 2D Binding mode Binding energy (Kcal/mol) -5.45  Molscript Map 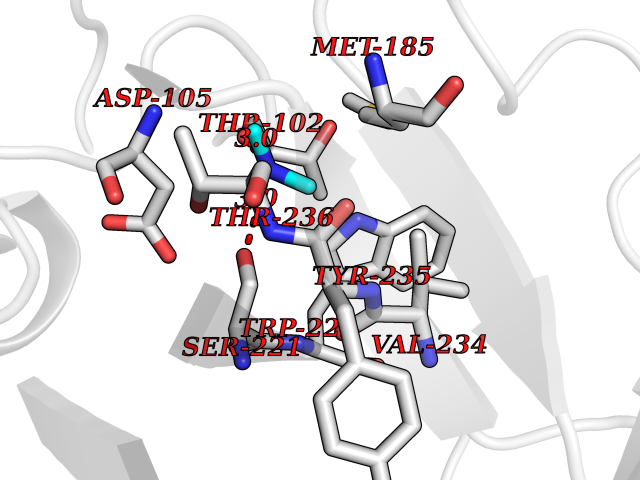 Pymol Map 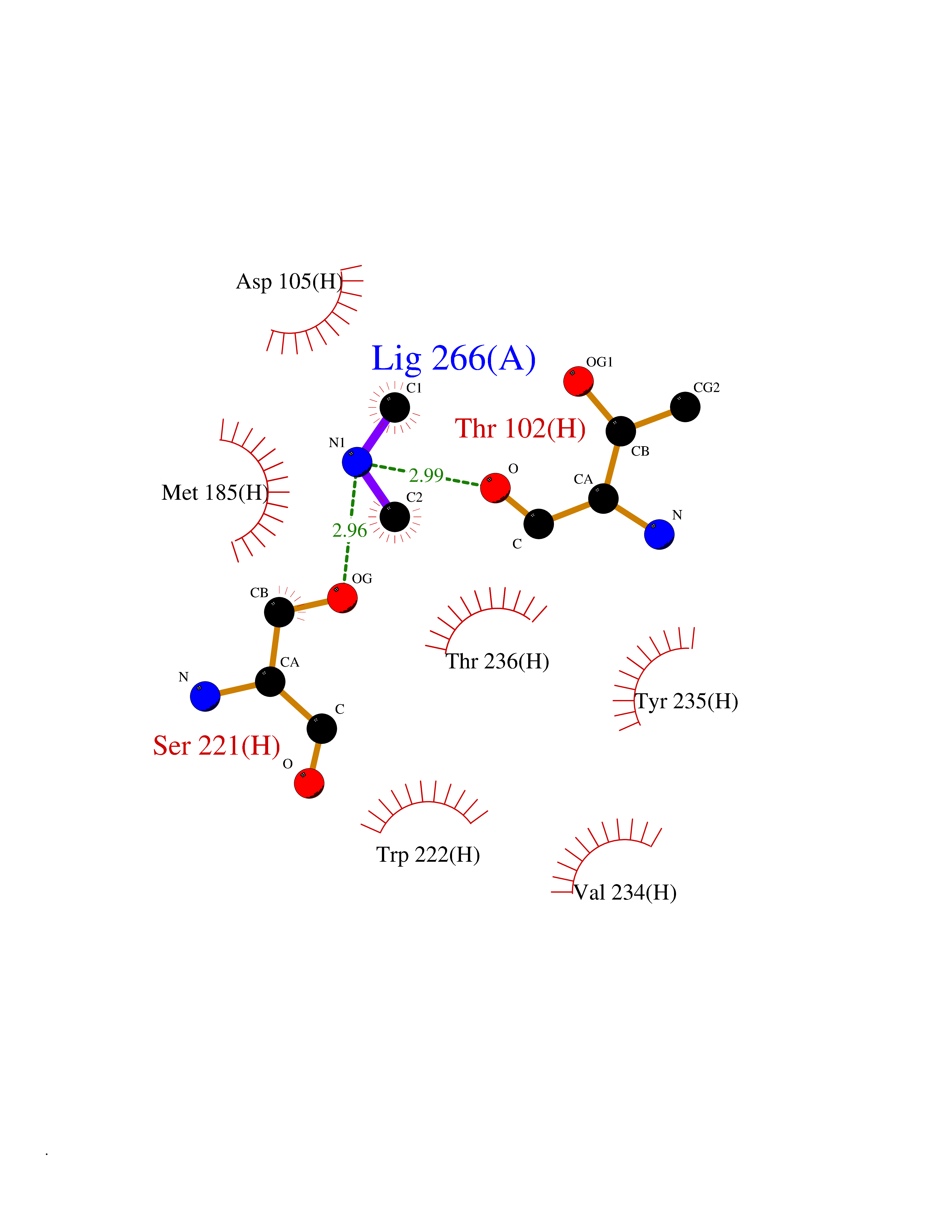 Ligplot Map 3D Binding mode Sequence IVGGKVCPKGECPWQVLLLVNGAQLCGGTLINTIWVVSAAHCFDKIKNWRNLIAVLGEHDLSEHDGDEQSRRVAQVIIPSTYVPGTTNHDIALLRLHQPVVLTDHVVPLCLPERTFSERTLAFVRFSLVSGWGQLLDRGATALELMVLNVPRVMTQDCEASYPGKITEYMFCAGYSDGSKDSCKGDSGGPHATHYRGTWYLTGIVSWGQGCATVGHFGVYTRVSQYIEWLQKLMRSEPRPGVLLRAPFP Hydrogen bonds contact Hydrophobic contact | ||||